User login
Bilateral chylothorax in an AIDS patient with newly diagnosed Kaposi sarcoma
Kaposi sarcoma is an angioproliferative tumor that is associated with human herpes virus-8 (HHV-8). Mucocutaneous disease is the most common site for manifestation of AIDS-related Kaposi sarcoma, commonly affecting the lower extremities, oral mucosa, face, and genitalia. Pleural effusions can occur in 36%-60% of patients with Kaposi sarcoma, and it has been documented that chylothorax is a rare, but plausible presentation in patients with Kaposi sarcoma.1 We present here a case of bilateral chylothorax in a patient with AIDS-related Kaposi sarcoma.
Case presentation and summary
A 52-year-old MSM male with AIDS (CD4, <20 mm3; viral load, 58 copies/ml) presented to the emergency department with complaints of shortness of breath, productive cough, and diarrhea for 2 days prior to presentation. His medical history also included chronic obstructive pulmonary disease, coronary artery disease, and hyperlipidemia. The patient was not on HAART because of his history of noncompliance. The results of a chest X-ray and computed-tomography (CT) scan showed that the patient had bilateral pleural effusion and a spiculated 14-mm nodule in the left upper lobe.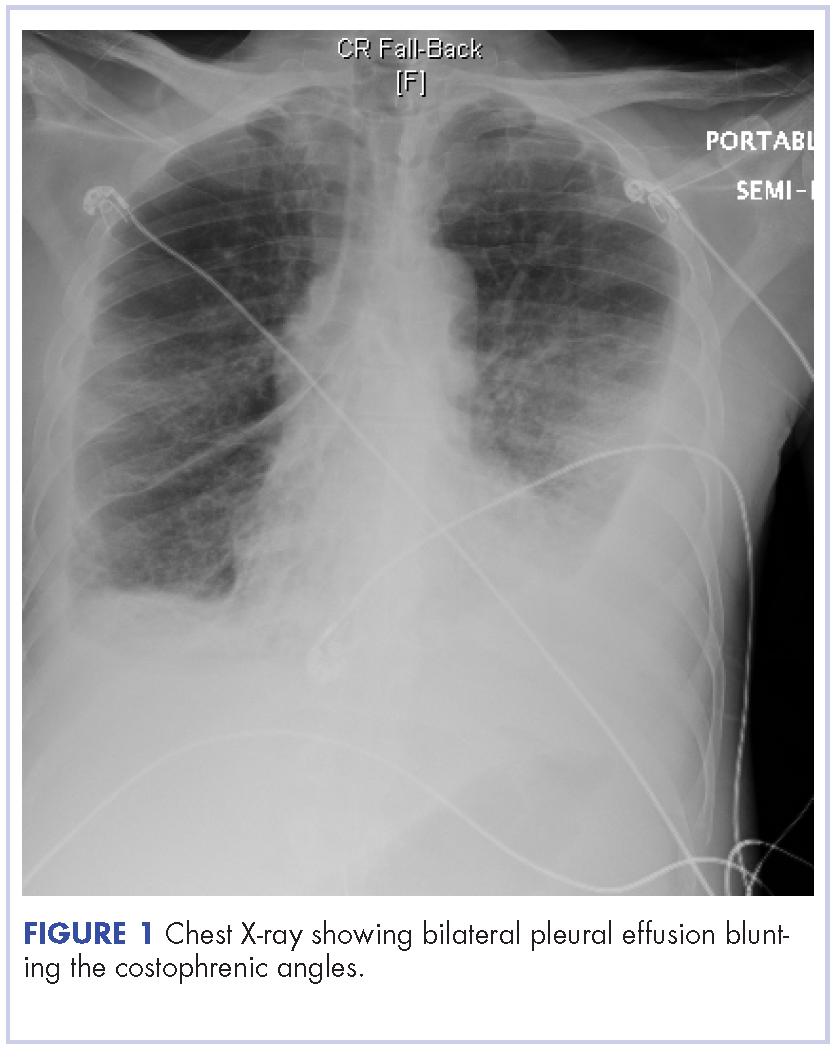
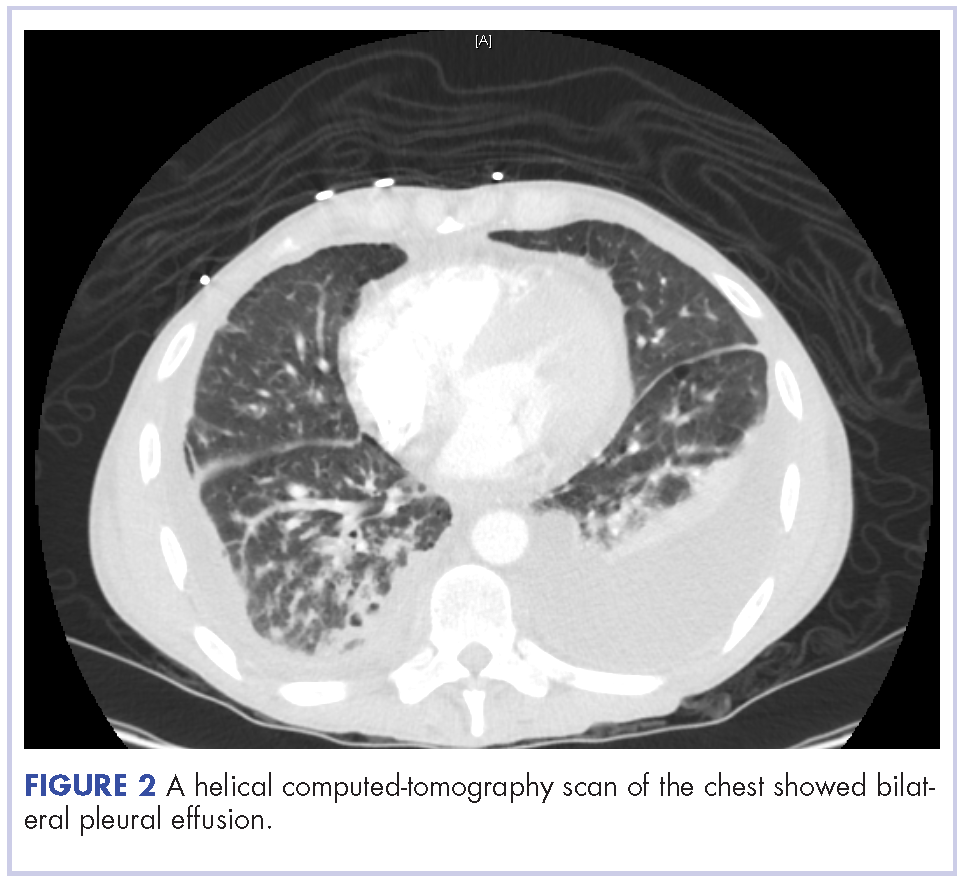
The patient underwent ultrasound-guided placement of a 12-French left-sided chest catheter, and a milky white fluid was aspirated from the left pleural space. Laboratory analysis of the pleural fluid confirmed an exudate with an elevated triglyceride level of 120 mg/dL (chylous, >110 mg/dL) indicating chylothorax.
On close physical examination, the patient was found to have multiple irregular plaques on the back and lower extremities. As described by dermatology, there was a violaceous indurated plaque on the left axillae, violaceous indurated plaques with superficial scale grouped on the left midlateral back, and hyperpigmented lichenified plaques and papules on bilateral shins, with some with plate-like scale. Two punch biopsies were taken of the skin lesions, which confirmed Kaposi sarcoma, plaque stage from the lesion biopsied on the back, and patch stage from the lesion biopsied in the left axilla. Cytology of the pleural fluid was negative for malignant cells. On review by the radiologist of the CT scan of the chest, there was no indication of gross distention of the thoracic duct. Treatment options were offered to the patient, and the patient was considering options for chemotherapy and home hospice given his advanced disease state at the time of discharge.
Discussion
Chylothorax occurs with a thoracic duct obstruction, which results in leakage of lymphatic fluid into the pleural cavity. The two leading causes of chylothorax are trauma and malignancy, with lymphoma being the most common cause of chylothorax among those with malignancy.2 Chylothorax, however, is a rare but documented complication of Kaposi sarcoma. Marais and colleagues reported the case of a 3-year-old HIV-positive patient with newly diagnosed Kaposi sarcoma who was found to have tumor infiltration in the thoracic duct leading to bilateral chylothorax.3 Maradona and colleagues described a 40-year-old man with AIDS-related Kaposi sarcoma who was found to have pleural and pericardial Kaposi sarcoma with chylothorax.4 Priest and colleagues wrote about a 32-year-old patient with AIDS with biopsy-proven Kaposi sarcoma who required multiple therapeutic thoracenteses for rapidly recurrent left chylothorax effusions.5
There are two leading discussions as to the pathophysiology of chylothorax that is related to Kaposi sarcoma: chylothorax developing secondary to metastatic disease or the development of chylothorax secondary to primary Kaposi sarcoma arising from the pleural region.6 One case report examined pleural and lung biopsies in a 34-year-old patient with AIDS-related Kaposi sarcoma that showed immunohistochemical staining that was suggestive of early-stage Kaposi sarcoma of lymphatic endothelial origin. The authors were attempting to illustrate that Kaposi sarcoma may have a stem-cell origin which can differentiate into lymph cells. Kontantinopoulos and colleagues postulated that in situ Kaposi sarcoma can arise from the lymphatic system with a resultant clinical presentation of chylothorax.7 The more mainstream thought however, is that chylothorax has been found to develop secondary to metastatic disease. The present case, therefore, illustrates an unusual presentation of cytology negative chylothorax in a patient with AIDS-related Kaposi sarcoma.
1. Sridar S, Garza EG, Cox J, Rumbak MJ. Serosanguineous pleural effusions in a patient with HIV and Kaposi sarcoma: pleuroscopic findings. J Bronchology Interv Pulmonol. 2011;18(4):337-339.
2. Light RW. Chylothorax and pseudochylothorax. In: Light RW, ed. Pleural diseases. 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2013:412-426.
3. Marais BJ, Pienaar J, Gie RP. Kaposi sarcoma with upper airway obstruction and bilateral chylothoraces. Pediatr Infect Dis J. 2003;22:926-928.
4. Maradona JA, Carton JA, Asensi V, Rodriguez-Guardado A. AIDS-related Kaposi sarcoma with chylothorax and pericardial involvement satisfactorily treated with liposomal doxorubicin. AIDS. 2002;16(5):806.
5. Priest ER, Weiss R. Chylothorax with Kaposi sarcoma. South Med J. 1991;84:806-807.
6. Pantanowitz L, Dezube BJ. Kaposi sarcoma in unusual locations. BMC Cancer. 2008;8:190.
7. Konstantinopoulos PA, Dezube BJ, Pantanowitz L. Morphologic and immunophenotypic evidence of in situ Kaposi sarcoma. BMC Clin Pathol. 2006;30:6:7.
Kaposi sarcoma is an angioproliferative tumor that is associated with human herpes virus-8 (HHV-8). Mucocutaneous disease is the most common site for manifestation of AIDS-related Kaposi sarcoma, commonly affecting the lower extremities, oral mucosa, face, and genitalia. Pleural effusions can occur in 36%-60% of patients with Kaposi sarcoma, and it has been documented that chylothorax is a rare, but plausible presentation in patients with Kaposi sarcoma.1 We present here a case of bilateral chylothorax in a patient with AIDS-related Kaposi sarcoma.
Case presentation and summary
A 52-year-old MSM male with AIDS (CD4, <20 mm3; viral load, 58 copies/ml) presented to the emergency department with complaints of shortness of breath, productive cough, and diarrhea for 2 days prior to presentation. His medical history also included chronic obstructive pulmonary disease, coronary artery disease, and hyperlipidemia. The patient was not on HAART because of his history of noncompliance. The results of a chest X-ray and computed-tomography (CT) scan showed that the patient had bilateral pleural effusion and a spiculated 14-mm nodule in the left upper lobe.
The patient underwent ultrasound-guided placement of a 12-French left-sided chest catheter, and a milky white fluid was aspirated from the left pleural space. Laboratory analysis of the pleural fluid confirmed an exudate with an elevated triglyceride level of 120 mg/dL (chylous, >110 mg/dL) indicating chylothorax.
On close physical examination, the patient was found to have multiple irregular plaques on the back and lower extremities. As described by dermatology, there was a violaceous indurated plaque on the left axillae, violaceous indurated plaques with superficial scale grouped on the left midlateral back, and hyperpigmented lichenified plaques and papules on bilateral shins, with some with plate-like scale. Two punch biopsies were taken of the skin lesions, which confirmed Kaposi sarcoma, plaque stage from the lesion biopsied on the back, and patch stage from the lesion biopsied in the left axilla. Cytology of the pleural fluid was negative for malignant cells. On review by the radiologist of the CT scan of the chest, there was no indication of gross distention of the thoracic duct. Treatment options were offered to the patient, and the patient was considering options for chemotherapy and home hospice given his advanced disease state at the time of discharge.
Discussion
Chylothorax occurs with a thoracic duct obstruction, which results in leakage of lymphatic fluid into the pleural cavity. The two leading causes of chylothorax are trauma and malignancy, with lymphoma being the most common cause of chylothorax among those with malignancy.2 Chylothorax, however, is a rare but documented complication of Kaposi sarcoma. Marais and colleagues reported the case of a 3-year-old HIV-positive patient with newly diagnosed Kaposi sarcoma who was found to have tumor infiltration in the thoracic duct leading to bilateral chylothorax.3 Maradona and colleagues described a 40-year-old man with AIDS-related Kaposi sarcoma who was found to have pleural and pericardial Kaposi sarcoma with chylothorax.4 Priest and colleagues wrote about a 32-year-old patient with AIDS with biopsy-proven Kaposi sarcoma who required multiple therapeutic thoracenteses for rapidly recurrent left chylothorax effusions.5
There are two leading discussions as to the pathophysiology of chylothorax that is related to Kaposi sarcoma: chylothorax developing secondary to metastatic disease or the development of chylothorax secondary to primary Kaposi sarcoma arising from the pleural region.6 One case report examined pleural and lung biopsies in a 34-year-old patient with AIDS-related Kaposi sarcoma that showed immunohistochemical staining that was suggestive of early-stage Kaposi sarcoma of lymphatic endothelial origin. The authors were attempting to illustrate that Kaposi sarcoma may have a stem-cell origin which can differentiate into lymph cells. Kontantinopoulos and colleagues postulated that in situ Kaposi sarcoma can arise from the lymphatic system with a resultant clinical presentation of chylothorax.7 The more mainstream thought however, is that chylothorax has been found to develop secondary to metastatic disease. The present case, therefore, illustrates an unusual presentation of cytology negative chylothorax in a patient with AIDS-related Kaposi sarcoma.
Kaposi sarcoma is an angioproliferative tumor that is associated with human herpes virus-8 (HHV-8). Mucocutaneous disease is the most common site for manifestation of AIDS-related Kaposi sarcoma, commonly affecting the lower extremities, oral mucosa, face, and genitalia. Pleural effusions can occur in 36%-60% of patients with Kaposi sarcoma, and it has been documented that chylothorax is a rare, but plausible presentation in patients with Kaposi sarcoma.1 We present here a case of bilateral chylothorax in a patient with AIDS-related Kaposi sarcoma.
Case presentation and summary
A 52-year-old MSM male with AIDS (CD4, <20 mm3; viral load, 58 copies/ml) presented to the emergency department with complaints of shortness of breath, productive cough, and diarrhea for 2 days prior to presentation. His medical history also included chronic obstructive pulmonary disease, coronary artery disease, and hyperlipidemia. The patient was not on HAART because of his history of noncompliance. The results of a chest X-ray and computed-tomography (CT) scan showed that the patient had bilateral pleural effusion and a spiculated 14-mm nodule in the left upper lobe.
The patient underwent ultrasound-guided placement of a 12-French left-sided chest catheter, and a milky white fluid was aspirated from the left pleural space. Laboratory analysis of the pleural fluid confirmed an exudate with an elevated triglyceride level of 120 mg/dL (chylous, >110 mg/dL) indicating chylothorax.
On close physical examination, the patient was found to have multiple irregular plaques on the back and lower extremities. As described by dermatology, there was a violaceous indurated plaque on the left axillae, violaceous indurated plaques with superficial scale grouped on the left midlateral back, and hyperpigmented lichenified plaques and papules on bilateral shins, with some with plate-like scale. Two punch biopsies were taken of the skin lesions, which confirmed Kaposi sarcoma, plaque stage from the lesion biopsied on the back, and patch stage from the lesion biopsied in the left axilla. Cytology of the pleural fluid was negative for malignant cells. On review by the radiologist of the CT scan of the chest, there was no indication of gross distention of the thoracic duct. Treatment options were offered to the patient, and the patient was considering options for chemotherapy and home hospice given his advanced disease state at the time of discharge.
Discussion
Chylothorax occurs with a thoracic duct obstruction, which results in leakage of lymphatic fluid into the pleural cavity. The two leading causes of chylothorax are trauma and malignancy, with lymphoma being the most common cause of chylothorax among those with malignancy.2 Chylothorax, however, is a rare but documented complication of Kaposi sarcoma. Marais and colleagues reported the case of a 3-year-old HIV-positive patient with newly diagnosed Kaposi sarcoma who was found to have tumor infiltration in the thoracic duct leading to bilateral chylothorax.3 Maradona and colleagues described a 40-year-old man with AIDS-related Kaposi sarcoma who was found to have pleural and pericardial Kaposi sarcoma with chylothorax.4 Priest and colleagues wrote about a 32-year-old patient with AIDS with biopsy-proven Kaposi sarcoma who required multiple therapeutic thoracenteses for rapidly recurrent left chylothorax effusions.5
There are two leading discussions as to the pathophysiology of chylothorax that is related to Kaposi sarcoma: chylothorax developing secondary to metastatic disease or the development of chylothorax secondary to primary Kaposi sarcoma arising from the pleural region.6 One case report examined pleural and lung biopsies in a 34-year-old patient with AIDS-related Kaposi sarcoma that showed immunohistochemical staining that was suggestive of early-stage Kaposi sarcoma of lymphatic endothelial origin. The authors were attempting to illustrate that Kaposi sarcoma may have a stem-cell origin which can differentiate into lymph cells. Kontantinopoulos and colleagues postulated that in situ Kaposi sarcoma can arise from the lymphatic system with a resultant clinical presentation of chylothorax.7 The more mainstream thought however, is that chylothorax has been found to develop secondary to metastatic disease. The present case, therefore, illustrates an unusual presentation of cytology negative chylothorax in a patient with AIDS-related Kaposi sarcoma.
1. Sridar S, Garza EG, Cox J, Rumbak MJ. Serosanguineous pleural effusions in a patient with HIV and Kaposi sarcoma: pleuroscopic findings. J Bronchology Interv Pulmonol. 2011;18(4):337-339.
2. Light RW. Chylothorax and pseudochylothorax. In: Light RW, ed. Pleural diseases. 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2013:412-426.
3. Marais BJ, Pienaar J, Gie RP. Kaposi sarcoma with upper airway obstruction and bilateral chylothoraces. Pediatr Infect Dis J. 2003;22:926-928.
4. Maradona JA, Carton JA, Asensi V, Rodriguez-Guardado A. AIDS-related Kaposi sarcoma with chylothorax and pericardial involvement satisfactorily treated with liposomal doxorubicin. AIDS. 2002;16(5):806.
5. Priest ER, Weiss R. Chylothorax with Kaposi sarcoma. South Med J. 1991;84:806-807.
6. Pantanowitz L, Dezube BJ. Kaposi sarcoma in unusual locations. BMC Cancer. 2008;8:190.
7. Konstantinopoulos PA, Dezube BJ, Pantanowitz L. Morphologic and immunophenotypic evidence of in situ Kaposi sarcoma. BMC Clin Pathol. 2006;30:6:7.
1. Sridar S, Garza EG, Cox J, Rumbak MJ. Serosanguineous pleural effusions in a patient with HIV and Kaposi sarcoma: pleuroscopic findings. J Bronchology Interv Pulmonol. 2011;18(4):337-339.
2. Light RW. Chylothorax and pseudochylothorax. In: Light RW, ed. Pleural diseases. 6th ed. Philadelphia: Lippincott Williams & Wilkins, 2013:412-426.
3. Marais BJ, Pienaar J, Gie RP. Kaposi sarcoma with upper airway obstruction and bilateral chylothoraces. Pediatr Infect Dis J. 2003;22:926-928.
4. Maradona JA, Carton JA, Asensi V, Rodriguez-Guardado A. AIDS-related Kaposi sarcoma with chylothorax and pericardial involvement satisfactorily treated with liposomal doxorubicin. AIDS. 2002;16(5):806.
5. Priest ER, Weiss R. Chylothorax with Kaposi sarcoma. South Med J. 1991;84:806-807.
6. Pantanowitz L, Dezube BJ. Kaposi sarcoma in unusual locations. BMC Cancer. 2008;8:190.
7. Konstantinopoulos PA, Dezube BJ, Pantanowitz L. Morphologic and immunophenotypic evidence of in situ Kaposi sarcoma. BMC Clin Pathol. 2006;30:6:7.
Metastatic Kaposi sarcoma with osseous involvement in a patient with AIDS
Kaposi sarcoma is an AIDS-defining illness associated with human herpes virus-8 (HHV-8) co-infection. It was described in 1872 by the Hungarian dermatologist Mortiz Kaposi, and was an isolated and sporadic occurrence before the emergence of HIV infection and AIDS.1 It was first affiliated as an AIDS-associated neoplasm in 1981.1 Kaposi sarcoma is a systemic disease that can present with cutaneous lesions with or without internal involvement. There are four subtypes: Classic, African endemic, AIDS-related (CD4 count, <200), and Kaposi sarcoma in iatrogenically immunosuppressed patients. The disease has the propensity to manifest in the skin and gastro-intestinal and respiratory tracts, and osseous involvement is rarely encountered. We present here the case of an AIDS-positive man with generalized bone pain as a result of metastasis from Kaposi sarcoma. Our discussion includes the epidemiological, clinical, pathological, and radiological facets of AIDS-related Kaposi sarcoma, and the anomaly of osseous involvement.
Case presentation and summary
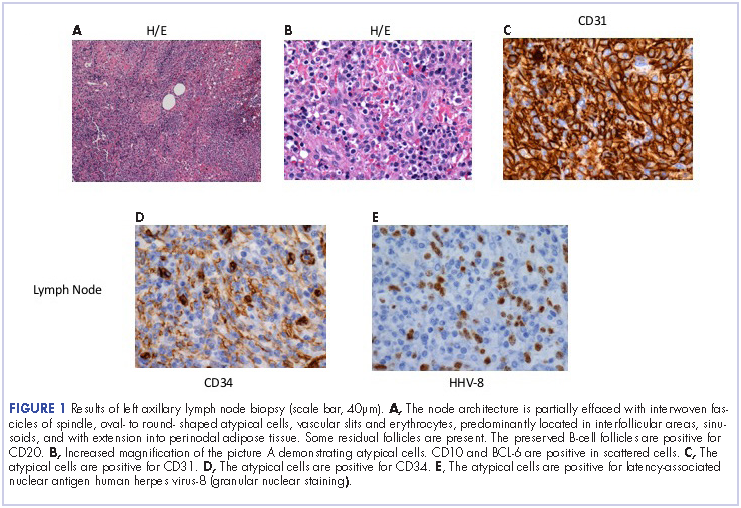
He restarted his previous HAART regimen in March 2016, and was subsequently started on chemotherapy with liposomal doxorubicin (50 mg [20 mg/m2] in 250 ml D5W IV every 2 weeks) because of his extensive disease.2 He completed 6 cycles by June 2016. However, he returned in July 2016 with worsening back pain. A repeat CT scan revealed significant improvement in the disseminated lymphadenopathy, but worsening osseous metastatic disease was seen in the lumbar, thoracic, and pelvic regions. A pelvic lytic lesion biopsy revealed Kaposi sarcoma; pathology showed spindle cells positive for CD34, CD31, and HHV-8 (Figure 2). The patient received palliative radiation to the spine, aiding in pain management and ambulatory dysfunction. He continued with his noncompliance with all medications and outpatient follow-ups, and succumbed to his disease burden.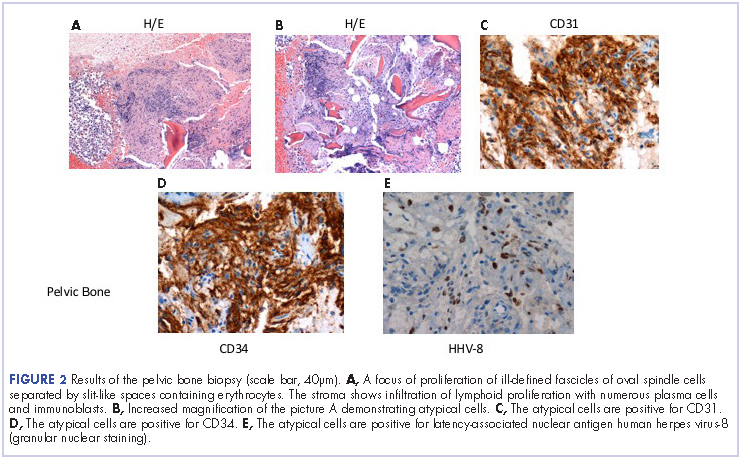
Discussion
Kaposi sarcoma is a low-grade mesenchymal tumor that involves the blood and lymphatic vessels.3 Its association with AIDS was revealed in the early 1980s at the start of the HIV epidemic in the United States. In 1994, Chang and colleagues discovered the association between Karposi sarcoma and HHV-8 by isolating DNA fragments of HHV in Kaposi sarcoma tumors from AIDS patients.4 The mode of transmission of HHV-8 has not been fully decoded. It has been presumed that adult homosexual contact continues to be an important route of transmission, inferring a common route of infection. In 1990, the overall risk of developing Kaposi sarcoma in AIDS patients was 20,000 times greater than it was in the general population, and 300 times greater than in other immunosuppressed patients.5 This suggests an increase in incidence, in direct relation, with a decrease in the CD4 count.
Kaposi sarcoma can present with a range of clinical features, from negligible cutaneous lesions to a hastily progressing neoplasm. Involvement in the musculoskeletal system is infrequent, but encountered increasingly in the AIDS-related subtype. Moreover, it is recurrently observed in the African population.6 In one of the largest reviews to date exploring Kaposi sarcoma involving the musculoskeletal system, Caponetti and colleagues observed the greatest osseous involvement distinctly in patients with CD4 and T-cell counts below 100 cells/mm3.6
Kaposi sarcoma musculoskeletal involvement, specifically bone, is atypical. If it does occur, it usually manifests as a result of contiguous invasion from an adjacent nonosseous lesion. Caponetti and colleagues that isolated osseous Kaposi sarcoma lesions (with no overlying skin lesion) were found to be more likely to be associated with AIDS in the review by Caponetti and colleagues.6 As in our patient, it is also typically a manifestation of more widely disseminated disease.7
Most of the osseous lytic lesions in AIDS patients are located in the axial skeleton. Radiological features of musculoskeletal Kaposi sarcoma are variable. As observed by Caponetti and colleagues, Kaposi sarcoma lesions can appear as a periosteal reaction, cortical erosions, osteolysis, or osseous destruction, with irregular-shaped cortical erosions being most typical.6 Despite their osteolytic features, Kaposi sarcoma lesions are often not visualized by conventional radiography.6 The preferred imaging for identification of lytic bone changes is CT (Figure 3). Magnetic resonance imaging can also help distinguish marrow abnormalities as well as adjacent soft tissues masses. Radiologically, Kaposi sarcoma osseous lesions have parallel features to bacillary angiomatosis, tuberculosis, or lymphoma.8 Therefore, biopsy of the lesion is essential in establishing the diagnosis of Kaposi sarcoma.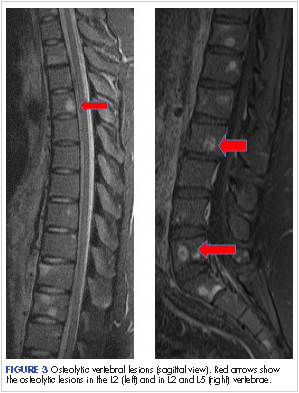
In theory, there should be clinical improvement in Kaposi sarcoma when immunity is restored. Cancers caused by the Epstein-Barr virus and Kaposi sarcoma-associated herpes virus may eventually also be preventable with vaccines.10
There is rarely bone involvement without the foreshadowing of a poor prognosis. Erroneous patient care may inevitably arise from Kaposi sarcoma in uncharacteristic sites. A differential of Kaposi sarcoma should be included if a patient with AIDS presents with osteolytic lesions on imaging. Biopsying the lesion cements the diagnosis and eliminates the possibility of mimicry conditions such as bacillary angiomatosis, benign vascular lesions, and angiosarcoma. As of today, a HAART regimen remains the standard initial care for patients with Kaposi sarcoma.
1. Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
2. Northfelt DW, Dezube BJ, Thommes JA, et al. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi sarcoma: results of a randomized phase III clinical trial. J Clin Oncol. 1998;16(7):2445-2451.
3. Restrepo CS, Martinez S, Lemos JA, et al. Imaging manifestations of Kaposi sarcoma. RadioGraphics. 2006;26:1169-1185.
4. Chang Y, Cesarman E, Pessin MS, et al. Identification of herpes virus-like DNA sequences in AIDS-associated Kaposi sarcoma. Science. 1994;266:1865-1869.
5. Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123-128.
6. Caponetti G, Dezube BJ, Restrepo CS, Pantanowitz I. Kaposi sarcoma of the musculoskeletal system: a review of 66 patients. Cancer. 2007;109(6):1040-1052.
7. Krishna G, Chitkara RK. Osseous Kaposi sarcoma. JAMA. 2003;286(9):1106.
8. Thanos L, Mylona S, Kalioras V, Pomoni M, Batakis N. Osseous Kaposi sarcoma in an HIV-positive patient. Skeletal Radiol. 2004;33(4):241-243.
9. Guiholt A, Dupin N, Marcelin AG, et al. Low T-cell response to human herpesvirus 8 in patients with AIDS-related and classic Kaposi sarcoma. J Infect Dis. 2006;194(8):1078-1088.
10. Gopal S, Achenbach CJ, Yanik EL, Dither DP, Eron JJ, Engels EA. Moving forward in HIV-associated cancer. J Clin Oncol. 2014;32(9):876-880.
Kaposi sarcoma is an AIDS-defining illness associated with human herpes virus-8 (HHV-8) co-infection. It was described in 1872 by the Hungarian dermatologist Mortiz Kaposi, and was an isolated and sporadic occurrence before the emergence of HIV infection and AIDS.1 It was first affiliated as an AIDS-associated neoplasm in 1981.1 Kaposi sarcoma is a systemic disease that can present with cutaneous lesions with or without internal involvement. There are four subtypes: Classic, African endemic, AIDS-related (CD4 count, <200), and Kaposi sarcoma in iatrogenically immunosuppressed patients. The disease has the propensity to manifest in the skin and gastro-intestinal and respiratory tracts, and osseous involvement is rarely encountered. We present here the case of an AIDS-positive man with generalized bone pain as a result of metastasis from Kaposi sarcoma. Our discussion includes the epidemiological, clinical, pathological, and radiological facets of AIDS-related Kaposi sarcoma, and the anomaly of osseous involvement.
Case presentation and summary
He restarted his previous HAART regimen in March 2016, and was subsequently started on chemotherapy with liposomal doxorubicin (50 mg [20 mg/m2] in 250 ml D5W IV every 2 weeks) because of his extensive disease.2 He completed 6 cycles by June 2016. However, he returned in July 2016 with worsening back pain. A repeat CT scan revealed significant improvement in the disseminated lymphadenopathy, but worsening osseous metastatic disease was seen in the lumbar, thoracic, and pelvic regions. A pelvic lytic lesion biopsy revealed Kaposi sarcoma; pathology showed spindle cells positive for CD34, CD31, and HHV-8 (Figure 2). The patient received palliative radiation to the spine, aiding in pain management and ambulatory dysfunction. He continued with his noncompliance with all medications and outpatient follow-ups, and succumbed to his disease burden.
Discussion
Kaposi sarcoma is a low-grade mesenchymal tumor that involves the blood and lymphatic vessels.3 Its association with AIDS was revealed in the early 1980s at the start of the HIV epidemic in the United States. In 1994, Chang and colleagues discovered the association between Karposi sarcoma and HHV-8 by isolating DNA fragments of HHV in Kaposi sarcoma tumors from AIDS patients.4 The mode of transmission of HHV-8 has not been fully decoded. It has been presumed that adult homosexual contact continues to be an important route of transmission, inferring a common route of infection. In 1990, the overall risk of developing Kaposi sarcoma in AIDS patients was 20,000 times greater than it was in the general population, and 300 times greater than in other immunosuppressed patients.5 This suggests an increase in incidence, in direct relation, with a decrease in the CD4 count.
Kaposi sarcoma can present with a range of clinical features, from negligible cutaneous lesions to a hastily progressing neoplasm. Involvement in the musculoskeletal system is infrequent, but encountered increasingly in the AIDS-related subtype. Moreover, it is recurrently observed in the African population.6 In one of the largest reviews to date exploring Kaposi sarcoma involving the musculoskeletal system, Caponetti and colleagues observed the greatest osseous involvement distinctly in patients with CD4 and T-cell counts below 100 cells/mm3.6
Kaposi sarcoma musculoskeletal involvement, specifically bone, is atypical. If it does occur, it usually manifests as a result of contiguous invasion from an adjacent nonosseous lesion. Caponetti and colleagues that isolated osseous Kaposi sarcoma lesions (with no overlying skin lesion) were found to be more likely to be associated with AIDS in the review by Caponetti and colleagues.6 As in our patient, it is also typically a manifestation of more widely disseminated disease.7
Most of the osseous lytic lesions in AIDS patients are located in the axial skeleton. Radiological features of musculoskeletal Kaposi sarcoma are variable. As observed by Caponetti and colleagues, Kaposi sarcoma lesions can appear as a periosteal reaction, cortical erosions, osteolysis, or osseous destruction, with irregular-shaped cortical erosions being most typical.6 Despite their osteolytic features, Kaposi sarcoma lesions are often not visualized by conventional radiography.6 The preferred imaging for identification of lytic bone changes is CT (Figure 3). Magnetic resonance imaging can also help distinguish marrow abnormalities as well as adjacent soft tissues masses. Radiologically, Kaposi sarcoma osseous lesions have parallel features to bacillary angiomatosis, tuberculosis, or lymphoma.8 Therefore, biopsy of the lesion is essential in establishing the diagnosis of Kaposi sarcoma.
In theory, there should be clinical improvement in Kaposi sarcoma when immunity is restored. Cancers caused by the Epstein-Barr virus and Kaposi sarcoma-associated herpes virus may eventually also be preventable with vaccines.10
There is rarely bone involvement without the foreshadowing of a poor prognosis. Erroneous patient care may inevitably arise from Kaposi sarcoma in uncharacteristic sites. A differential of Kaposi sarcoma should be included if a patient with AIDS presents with osteolytic lesions on imaging. Biopsying the lesion cements the diagnosis and eliminates the possibility of mimicry conditions such as bacillary angiomatosis, benign vascular lesions, and angiosarcoma. As of today, a HAART regimen remains the standard initial care for patients with Kaposi sarcoma.
Kaposi sarcoma is an AIDS-defining illness associated with human herpes virus-8 (HHV-8) co-infection. It was described in 1872 by the Hungarian dermatologist Mortiz Kaposi, and was an isolated and sporadic occurrence before the emergence of HIV infection and AIDS.1 It was first affiliated as an AIDS-associated neoplasm in 1981.1 Kaposi sarcoma is a systemic disease that can present with cutaneous lesions with or without internal involvement. There are four subtypes: Classic, African endemic, AIDS-related (CD4 count, <200), and Kaposi sarcoma in iatrogenically immunosuppressed patients. The disease has the propensity to manifest in the skin and gastro-intestinal and respiratory tracts, and osseous involvement is rarely encountered. We present here the case of an AIDS-positive man with generalized bone pain as a result of metastasis from Kaposi sarcoma. Our discussion includes the epidemiological, clinical, pathological, and radiological facets of AIDS-related Kaposi sarcoma, and the anomaly of osseous involvement.
Case presentation and summary
He restarted his previous HAART regimen in March 2016, and was subsequently started on chemotherapy with liposomal doxorubicin (50 mg [20 mg/m2] in 250 ml D5W IV every 2 weeks) because of his extensive disease.2 He completed 6 cycles by June 2016. However, he returned in July 2016 with worsening back pain. A repeat CT scan revealed significant improvement in the disseminated lymphadenopathy, but worsening osseous metastatic disease was seen in the lumbar, thoracic, and pelvic regions. A pelvic lytic lesion biopsy revealed Kaposi sarcoma; pathology showed spindle cells positive for CD34, CD31, and HHV-8 (Figure 2). The patient received palliative radiation to the spine, aiding in pain management and ambulatory dysfunction. He continued with his noncompliance with all medications and outpatient follow-ups, and succumbed to his disease burden.
Discussion
Kaposi sarcoma is a low-grade mesenchymal tumor that involves the blood and lymphatic vessels.3 Its association with AIDS was revealed in the early 1980s at the start of the HIV epidemic in the United States. In 1994, Chang and colleagues discovered the association between Karposi sarcoma and HHV-8 by isolating DNA fragments of HHV in Kaposi sarcoma tumors from AIDS patients.4 The mode of transmission of HHV-8 has not been fully decoded. It has been presumed that adult homosexual contact continues to be an important route of transmission, inferring a common route of infection. In 1990, the overall risk of developing Kaposi sarcoma in AIDS patients was 20,000 times greater than it was in the general population, and 300 times greater than in other immunosuppressed patients.5 This suggests an increase in incidence, in direct relation, with a decrease in the CD4 count.
Kaposi sarcoma can present with a range of clinical features, from negligible cutaneous lesions to a hastily progressing neoplasm. Involvement in the musculoskeletal system is infrequent, but encountered increasingly in the AIDS-related subtype. Moreover, it is recurrently observed in the African population.6 In one of the largest reviews to date exploring Kaposi sarcoma involving the musculoskeletal system, Caponetti and colleagues observed the greatest osseous involvement distinctly in patients with CD4 and T-cell counts below 100 cells/mm3.6
Kaposi sarcoma musculoskeletal involvement, specifically bone, is atypical. If it does occur, it usually manifests as a result of contiguous invasion from an adjacent nonosseous lesion. Caponetti and colleagues that isolated osseous Kaposi sarcoma lesions (with no overlying skin lesion) were found to be more likely to be associated with AIDS in the review by Caponetti and colleagues.6 As in our patient, it is also typically a manifestation of more widely disseminated disease.7
Most of the osseous lytic lesions in AIDS patients are located in the axial skeleton. Radiological features of musculoskeletal Kaposi sarcoma are variable. As observed by Caponetti and colleagues, Kaposi sarcoma lesions can appear as a periosteal reaction, cortical erosions, osteolysis, or osseous destruction, with irregular-shaped cortical erosions being most typical.6 Despite their osteolytic features, Kaposi sarcoma lesions are often not visualized by conventional radiography.6 The preferred imaging for identification of lytic bone changes is CT (Figure 3). Magnetic resonance imaging can also help distinguish marrow abnormalities as well as adjacent soft tissues masses. Radiologically, Kaposi sarcoma osseous lesions have parallel features to bacillary angiomatosis, tuberculosis, or lymphoma.8 Therefore, biopsy of the lesion is essential in establishing the diagnosis of Kaposi sarcoma.
In theory, there should be clinical improvement in Kaposi sarcoma when immunity is restored. Cancers caused by the Epstein-Barr virus and Kaposi sarcoma-associated herpes virus may eventually also be preventable with vaccines.10
There is rarely bone involvement without the foreshadowing of a poor prognosis. Erroneous patient care may inevitably arise from Kaposi sarcoma in uncharacteristic sites. A differential of Kaposi sarcoma should be included if a patient with AIDS presents with osteolytic lesions on imaging. Biopsying the lesion cements the diagnosis and eliminates the possibility of mimicry conditions such as bacillary angiomatosis, benign vascular lesions, and angiosarcoma. As of today, a HAART regimen remains the standard initial care for patients with Kaposi sarcoma.
1. Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
2. Northfelt DW, Dezube BJ, Thommes JA, et al. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi sarcoma: results of a randomized phase III clinical trial. J Clin Oncol. 1998;16(7):2445-2451.
3. Restrepo CS, Martinez S, Lemos JA, et al. Imaging manifestations of Kaposi sarcoma. RadioGraphics. 2006;26:1169-1185.
4. Chang Y, Cesarman E, Pessin MS, et al. Identification of herpes virus-like DNA sequences in AIDS-associated Kaposi sarcoma. Science. 1994;266:1865-1869.
5. Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123-128.
6. Caponetti G, Dezube BJ, Restrepo CS, Pantanowitz I. Kaposi sarcoma of the musculoskeletal system: a review of 66 patients. Cancer. 2007;109(6):1040-1052.
7. Krishna G, Chitkara RK. Osseous Kaposi sarcoma. JAMA. 2003;286(9):1106.
8. Thanos L, Mylona S, Kalioras V, Pomoni M, Batakis N. Osseous Kaposi sarcoma in an HIV-positive patient. Skeletal Radiol. 2004;33(4):241-243.
9. Guiholt A, Dupin N, Marcelin AG, et al. Low T-cell response to human herpesvirus 8 in patients with AIDS-related and classic Kaposi sarcoma. J Infect Dis. 2006;194(8):1078-1088.
10. Gopal S, Achenbach CJ, Yanik EL, Dither DP, Eron JJ, Engels EA. Moving forward in HIV-associated cancer. J Clin Oncol. 2014;32(9):876-880.
1. Radu O, Pantanowitz L. Kaposi sarcoma. Arch Pathol Lab Med. 2013;137:289-294.
2. Northfelt DW, Dezube BJ, Thommes JA, et al. Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi sarcoma: results of a randomized phase III clinical trial. J Clin Oncol. 1998;16(7):2445-2451.
3. Restrepo CS, Martinez S, Lemos JA, et al. Imaging manifestations of Kaposi sarcoma. RadioGraphics. 2006;26:1169-1185.
4. Chang Y, Cesarman E, Pessin MS, et al. Identification of herpes virus-like DNA sequences in AIDS-associated Kaposi sarcoma. Science. 1994;266:1865-1869.
5. Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123-128.
6. Caponetti G, Dezube BJ, Restrepo CS, Pantanowitz I. Kaposi sarcoma of the musculoskeletal system: a review of 66 patients. Cancer. 2007;109(6):1040-1052.
7. Krishna G, Chitkara RK. Osseous Kaposi sarcoma. JAMA. 2003;286(9):1106.
8. Thanos L, Mylona S, Kalioras V, Pomoni M, Batakis N. Osseous Kaposi sarcoma in an HIV-positive patient. Skeletal Radiol. 2004;33(4):241-243.
9. Guiholt A, Dupin N, Marcelin AG, et al. Low T-cell response to human herpesvirus 8 in patients with AIDS-related and classic Kaposi sarcoma. J Infect Dis. 2006;194(8):1078-1088.
10. Gopal S, Achenbach CJ, Yanik EL, Dither DP, Eron JJ, Engels EA. Moving forward in HIV-associated cancer. J Clin Oncol. 2014;32(9):876-880.
A rare case of hypoglycemia induced by a classic gastrointestinal stromal tumor
Hypoglycemia, a frequently encountered medical emergency, is usually seen in patients with diabetes, most commonly as a result of iatrogenesis. However, it can also be encountered in nondiabetic patients. Various causes, such as pancreatic islet cell tumors producing insulin, primary or secondary adrenal insufficiency, advanced liver disease, pheochromocytoma and hypothyroidism, have been found to contribute to the condition in the nondiabetic population.1 In rare cases, an excessive production of insulin-like growth factor (IGF-2) – a condition known as nonislet cell tumor-induced hypoglycemia (NICTH) – has also been found to cause hypoglycemia. Hypoinsulinemic hypoglycemia, with low IGF-1 levels and an IGF-2-IgF1 ratio of greater than 10, is found to be suggestive of NICTH.
Case presentation and summary
An 81-year-old man with a history of diabetes mellitus, systolic heart failure, chronic kidney disease, and metastatic classical gastrointestinal spindle cell sarcoma presented to the emergency department with an acute change in mental status resulting from a new onset hypoglycemia. He was admitted, and during his hospital stay, he experienced severe hypoglycemic episodes with symptomatic presentations of diaphoresis on multiple occasions. A detailed history revealed that for diabetes, the patient had been on insulin for the first 12 years after his diagnosis, after which he was switched to metformin 500 mg twice daily for about 2 years, and as a satisfactory glycemic control was attained, eventually metformin had also been stopped 3 years prior to the current presentation.
The patient’s past medical records were obtained from the hospital at which he had been diagnosed gastrointestinal spindle cell sarcoma. Patient had not received treatment for the cancer as the disease was too widespread to be treated. The gastrointestinal spindle cell sarcoma, which had initially been surgically resected 7 years before the current presentation, had a recurrence 3 years later with abdominal and pulmonary metastasis, but no liver metastasis. No further intervention was carried out because the widely metastasized disease would not have benefited from any more surgical intervention and chemotherapy was not initiated because of the patient’s comorbid illnesses.
A blood sample drawn from the patient at the time of one hypoglycemic event, revealed low serum insulin <0.1 U/ml (normal, 2-19.6 U/ml); low C-peptide level, 0.59 ng/ml (0.8-3.85 ng/ml); low IGF-1, 16 ng/ml (5-4 ng/ml); and IGF-3, 0.9 ng/ml (2.2-4.5 ng/ml). IGF-2 levels were found to be markedly elevated at 945 ng/ml (47-350 ng/ml). The calculated IGF-2-IGF-1 ratio was 59.06 (normal, <10), suggesting NICTH as the etiology for the patient’s hypoglycemia.
The hypoglycemic episodes were initially treated with a continuous dextrose infusion followed by diazoxide treatment. However, diazoxide did not prevent his hypoglycemic episodes, so dexamethasone was considered as an alternative for his condition. The dexamethasone treatment resulted in the normalization of the patient’s serum glucose levels and resolution of his symptoms. The patient was discharged in a satisfactory state few days later and followed up thereafter. No recurrence of hypoglycemic episodes was found, and he was continued on dexamethasone therapy.
Discussion
Hypoglycemia due to NICTH is rare, with a prevalence of four times less than that of insulinoma.3 In most cases, NICTH occurs in patients with solid tumors of mesenchymal and epithelial origins such as hepatocellular carcinoma, gastric carcinoma or mesothelioma.4 In NICTH, the serum levels of insulin, C-peptide, and IGF-1 are usually decreased or undetectable. However, the circulating levels of total IGF2 may be increased, decreased, or normal. Concurrent normal to high morning cortisol and normal response to cosyntropin stimulation can rule out adrenal insufficiency and suggest NICTH. An IGF-2: IGF-1 ratio of >10 is considered to be clinically significant and highly suggestive of NICTH.5 Hypoglycemia in NICTH can be managed by administration of oral glucose, intravenous dextrose or glucagon. In some cases, diazoxide, a potent inhibitor of insulin secretion, has been found to be useful.6 Diazoxide directly inhibits the release of insulin through stimulation of adrenergic receptors and also has an extra pancreatic hyperglycemic effect, probably by inhibiting cyclic adenosine monophosphate phosphodiesterase, resulting in higher plasma levels of cyclic AMP and enhanced glycogenolysis.
Glucocorticoid therapy has been shown to suppress IGF-2 in a dose dependent manner and also by increasing gluconeogenesis.7 Surgical resection of the tumor whenever possible is the treatment of choice followed by radiotherapy and chemotherapy for inoperable disease and if successful, usually results in resolution of hypoglycemia. Imatinib, is the chemotherapeutic drug of choice for metastatic GIST, but many case reports have suggested worsening of hypoglycemia in advanced GIST with the use of the drug.8 The patient described in our report was not on any chemotherapy, hence hypoglycemia could not be attributed to it. On the basis of findings among 24 patients with GIST, Rikhof and colleagues have recommended monitoring plasma levels of pro-IGF-IIE to identify patients at high risk for developing hypoglycemia, especially those with progressive disease.9 Furthermore, over expression of IGF-2 as a predictor of potential relapse may be an area for potential research and further study.10
1. Marks V, Teale JD. Tumours producing hypoglycaemia. Diabetes Metab Rev. 1991;7:79-91.
2. Dutta P, Aggarwal A, Gogate Y, Nahar U, Shah VN, Singla M. Non-islet cell tumor-induced hypoglycemia: a report of five cases and brief review of the literature. Endocrinol Diabetes Metab Case Rep. 2013;2013:130046
3. de Groot JW, Rikhof B, van Doorn J, et al. Non-islet cell tumour-induced hypoglycaemia: a review of the literature including two new cases. Endocr Relat Cancer. 2007;14:979-93.
4. Fukuda I, Hizuka N, Ishikawa Y, et al. Clinical features of insulin-like growth factor II producing non-islet-cell tumor hypoglycemia
5. Marks V, Teale JD: Tumours producing hypoglycaemia. Endocr Relat Cancer. 1998;5:111-129.
6. Le Roith D. Tumor-induced hypoglycemia. N Engl J Med. 1999;341:757-758.
7. Teale JD, Marks V. Glucocorticoid therapy suppresses abnormal secretion of big IGF-II by non-islet cell tumours inducing hypoglycaemia (NICTH). Clin Endocrinol .1998;49:491-498.
8. Hamberg P, De Jong FA, Boonstra JG, et al. Non-islet-cell tumor induced hypoglycemia in patients with advanced gastrointestinal stromal tumor possibly worsened by imatinib. J Clin Oncol. 2006;24:e30-e31.
9. Rikhof B, van Doorn J, Suurmeijer AJ, et al. Insulin-like growth factors and insulin-like growth factor-binding proteins in relation to disease status and incidence of hypoglycaemia in patients with a gastrointestinal stromal tumour. Ann Oncol. 2009;20:1582-1588.
10. Braconi C, Bracci R, Bearzi I, et al. Insulin-like growth factor (IGF) 1 and 2 help to predict disease outcome in GIST patients. Ann Oncol. 2008;19:1293-1298.
Hypoglycemia, a frequently encountered medical emergency, is usually seen in patients with diabetes, most commonly as a result of iatrogenesis. However, it can also be encountered in nondiabetic patients. Various causes, such as pancreatic islet cell tumors producing insulin, primary or secondary adrenal insufficiency, advanced liver disease, pheochromocytoma and hypothyroidism, have been found to contribute to the condition in the nondiabetic population.1 In rare cases, an excessive production of insulin-like growth factor (IGF-2) – a condition known as nonislet cell tumor-induced hypoglycemia (NICTH) – has also been found to cause hypoglycemia. Hypoinsulinemic hypoglycemia, with low IGF-1 levels and an IGF-2-IgF1 ratio of greater than 10, is found to be suggestive of NICTH.
Case presentation and summary
An 81-year-old man with a history of diabetes mellitus, systolic heart failure, chronic kidney disease, and metastatic classical gastrointestinal spindle cell sarcoma presented to the emergency department with an acute change in mental status resulting from a new onset hypoglycemia. He was admitted, and during his hospital stay, he experienced severe hypoglycemic episodes with symptomatic presentations of diaphoresis on multiple occasions. A detailed history revealed that for diabetes, the patient had been on insulin for the first 12 years after his diagnosis, after which he was switched to metformin 500 mg twice daily for about 2 years, and as a satisfactory glycemic control was attained, eventually metformin had also been stopped 3 years prior to the current presentation.
The patient’s past medical records were obtained from the hospital at which he had been diagnosed gastrointestinal spindle cell sarcoma. Patient had not received treatment for the cancer as the disease was too widespread to be treated. The gastrointestinal spindle cell sarcoma, which had initially been surgically resected 7 years before the current presentation, had a recurrence 3 years later with abdominal and pulmonary metastasis, but no liver metastasis. No further intervention was carried out because the widely metastasized disease would not have benefited from any more surgical intervention and chemotherapy was not initiated because of the patient’s comorbid illnesses.
A blood sample drawn from the patient at the time of one hypoglycemic event, revealed low serum insulin <0.1 U/ml (normal, 2-19.6 U/ml); low C-peptide level, 0.59 ng/ml (0.8-3.85 ng/ml); low IGF-1, 16 ng/ml (5-4 ng/ml); and IGF-3, 0.9 ng/ml (2.2-4.5 ng/ml). IGF-2 levels were found to be markedly elevated at 945 ng/ml (47-350 ng/ml). The calculated IGF-2-IGF-1 ratio was 59.06 (normal, <10), suggesting NICTH as the etiology for the patient’s hypoglycemia.
The hypoglycemic episodes were initially treated with a continuous dextrose infusion followed by diazoxide treatment. However, diazoxide did not prevent his hypoglycemic episodes, so dexamethasone was considered as an alternative for his condition. The dexamethasone treatment resulted in the normalization of the patient’s serum glucose levels and resolution of his symptoms. The patient was discharged in a satisfactory state few days later and followed up thereafter. No recurrence of hypoglycemic episodes was found, and he was continued on dexamethasone therapy.
Discussion
Hypoglycemia due to NICTH is rare, with a prevalence of four times less than that of insulinoma.3 In most cases, NICTH occurs in patients with solid tumors of mesenchymal and epithelial origins such as hepatocellular carcinoma, gastric carcinoma or mesothelioma.4 In NICTH, the serum levels of insulin, C-peptide, and IGF-1 are usually decreased or undetectable. However, the circulating levels of total IGF2 may be increased, decreased, or normal. Concurrent normal to high morning cortisol and normal response to cosyntropin stimulation can rule out adrenal insufficiency and suggest NICTH. An IGF-2: IGF-1 ratio of >10 is considered to be clinically significant and highly suggestive of NICTH.5 Hypoglycemia in NICTH can be managed by administration of oral glucose, intravenous dextrose or glucagon. In some cases, diazoxide, a potent inhibitor of insulin secretion, has been found to be useful.6 Diazoxide directly inhibits the release of insulin through stimulation of adrenergic receptors and also has an extra pancreatic hyperglycemic effect, probably by inhibiting cyclic adenosine monophosphate phosphodiesterase, resulting in higher plasma levels of cyclic AMP and enhanced glycogenolysis.
Glucocorticoid therapy has been shown to suppress IGF-2 in a dose dependent manner and also by increasing gluconeogenesis.7 Surgical resection of the tumor whenever possible is the treatment of choice followed by radiotherapy and chemotherapy for inoperable disease and if successful, usually results in resolution of hypoglycemia. Imatinib, is the chemotherapeutic drug of choice for metastatic GIST, but many case reports have suggested worsening of hypoglycemia in advanced GIST with the use of the drug.8 The patient described in our report was not on any chemotherapy, hence hypoglycemia could not be attributed to it. On the basis of findings among 24 patients with GIST, Rikhof and colleagues have recommended monitoring plasma levels of pro-IGF-IIE to identify patients at high risk for developing hypoglycemia, especially those with progressive disease.9 Furthermore, over expression of IGF-2 as a predictor of potential relapse may be an area for potential research and further study.10
Hypoglycemia, a frequently encountered medical emergency, is usually seen in patients with diabetes, most commonly as a result of iatrogenesis. However, it can also be encountered in nondiabetic patients. Various causes, such as pancreatic islet cell tumors producing insulin, primary or secondary adrenal insufficiency, advanced liver disease, pheochromocytoma and hypothyroidism, have been found to contribute to the condition in the nondiabetic population.1 In rare cases, an excessive production of insulin-like growth factor (IGF-2) – a condition known as nonislet cell tumor-induced hypoglycemia (NICTH) – has also been found to cause hypoglycemia. Hypoinsulinemic hypoglycemia, with low IGF-1 levels and an IGF-2-IgF1 ratio of greater than 10, is found to be suggestive of NICTH.
Case presentation and summary
An 81-year-old man with a history of diabetes mellitus, systolic heart failure, chronic kidney disease, and metastatic classical gastrointestinal spindle cell sarcoma presented to the emergency department with an acute change in mental status resulting from a new onset hypoglycemia. He was admitted, and during his hospital stay, he experienced severe hypoglycemic episodes with symptomatic presentations of diaphoresis on multiple occasions. A detailed history revealed that for diabetes, the patient had been on insulin for the first 12 years after his diagnosis, after which he was switched to metformin 500 mg twice daily for about 2 years, and as a satisfactory glycemic control was attained, eventually metformin had also been stopped 3 years prior to the current presentation.
The patient’s past medical records were obtained from the hospital at which he had been diagnosed gastrointestinal spindle cell sarcoma. Patient had not received treatment for the cancer as the disease was too widespread to be treated. The gastrointestinal spindle cell sarcoma, which had initially been surgically resected 7 years before the current presentation, had a recurrence 3 years later with abdominal and pulmonary metastasis, but no liver metastasis. No further intervention was carried out because the widely metastasized disease would not have benefited from any more surgical intervention and chemotherapy was not initiated because of the patient’s comorbid illnesses.
A blood sample drawn from the patient at the time of one hypoglycemic event, revealed low serum insulin <0.1 U/ml (normal, 2-19.6 U/ml); low C-peptide level, 0.59 ng/ml (0.8-3.85 ng/ml); low IGF-1, 16 ng/ml (5-4 ng/ml); and IGF-3, 0.9 ng/ml (2.2-4.5 ng/ml). IGF-2 levels were found to be markedly elevated at 945 ng/ml (47-350 ng/ml). The calculated IGF-2-IGF-1 ratio was 59.06 (normal, <10), suggesting NICTH as the etiology for the patient’s hypoglycemia.
The hypoglycemic episodes were initially treated with a continuous dextrose infusion followed by diazoxide treatment. However, diazoxide did not prevent his hypoglycemic episodes, so dexamethasone was considered as an alternative for his condition. The dexamethasone treatment resulted in the normalization of the patient’s serum glucose levels and resolution of his symptoms. The patient was discharged in a satisfactory state few days later and followed up thereafter. No recurrence of hypoglycemic episodes was found, and he was continued on dexamethasone therapy.
Discussion
Hypoglycemia due to NICTH is rare, with a prevalence of four times less than that of insulinoma.3 In most cases, NICTH occurs in patients with solid tumors of mesenchymal and epithelial origins such as hepatocellular carcinoma, gastric carcinoma or mesothelioma.4 In NICTH, the serum levels of insulin, C-peptide, and IGF-1 are usually decreased or undetectable. However, the circulating levels of total IGF2 may be increased, decreased, or normal. Concurrent normal to high morning cortisol and normal response to cosyntropin stimulation can rule out adrenal insufficiency and suggest NICTH. An IGF-2: IGF-1 ratio of >10 is considered to be clinically significant and highly suggestive of NICTH.5 Hypoglycemia in NICTH can be managed by administration of oral glucose, intravenous dextrose or glucagon. In some cases, diazoxide, a potent inhibitor of insulin secretion, has been found to be useful.6 Diazoxide directly inhibits the release of insulin through stimulation of adrenergic receptors and also has an extra pancreatic hyperglycemic effect, probably by inhibiting cyclic adenosine monophosphate phosphodiesterase, resulting in higher plasma levels of cyclic AMP and enhanced glycogenolysis.
Glucocorticoid therapy has been shown to suppress IGF-2 in a dose dependent manner and also by increasing gluconeogenesis.7 Surgical resection of the tumor whenever possible is the treatment of choice followed by radiotherapy and chemotherapy for inoperable disease and if successful, usually results in resolution of hypoglycemia. Imatinib, is the chemotherapeutic drug of choice for metastatic GIST, but many case reports have suggested worsening of hypoglycemia in advanced GIST with the use of the drug.8 The patient described in our report was not on any chemotherapy, hence hypoglycemia could not be attributed to it. On the basis of findings among 24 patients with GIST, Rikhof and colleagues have recommended monitoring plasma levels of pro-IGF-IIE to identify patients at high risk for developing hypoglycemia, especially those with progressive disease.9 Furthermore, over expression of IGF-2 as a predictor of potential relapse may be an area for potential research and further study.10
1. Marks V, Teale JD. Tumours producing hypoglycaemia. Diabetes Metab Rev. 1991;7:79-91.
2. Dutta P, Aggarwal A, Gogate Y, Nahar U, Shah VN, Singla M. Non-islet cell tumor-induced hypoglycemia: a report of five cases and brief review of the literature. Endocrinol Diabetes Metab Case Rep. 2013;2013:130046
3. de Groot JW, Rikhof B, van Doorn J, et al. Non-islet cell tumour-induced hypoglycaemia: a review of the literature including two new cases. Endocr Relat Cancer. 2007;14:979-93.
4. Fukuda I, Hizuka N, Ishikawa Y, et al. Clinical features of insulin-like growth factor II producing non-islet-cell tumor hypoglycemia
5. Marks V, Teale JD: Tumours producing hypoglycaemia. Endocr Relat Cancer. 1998;5:111-129.
6. Le Roith D. Tumor-induced hypoglycemia. N Engl J Med. 1999;341:757-758.
7. Teale JD, Marks V. Glucocorticoid therapy suppresses abnormal secretion of big IGF-II by non-islet cell tumours inducing hypoglycaemia (NICTH). Clin Endocrinol .1998;49:491-498.
8. Hamberg P, De Jong FA, Boonstra JG, et al. Non-islet-cell tumor induced hypoglycemia in patients with advanced gastrointestinal stromal tumor possibly worsened by imatinib. J Clin Oncol. 2006;24:e30-e31.
9. Rikhof B, van Doorn J, Suurmeijer AJ, et al. Insulin-like growth factors and insulin-like growth factor-binding proteins in relation to disease status and incidence of hypoglycaemia in patients with a gastrointestinal stromal tumour. Ann Oncol. 2009;20:1582-1588.
10. Braconi C, Bracci R, Bearzi I, et al. Insulin-like growth factor (IGF) 1 and 2 help to predict disease outcome in GIST patients. Ann Oncol. 2008;19:1293-1298.
1. Marks V, Teale JD. Tumours producing hypoglycaemia. Diabetes Metab Rev. 1991;7:79-91.
2. Dutta P, Aggarwal A, Gogate Y, Nahar U, Shah VN, Singla M. Non-islet cell tumor-induced hypoglycemia: a report of five cases and brief review of the literature. Endocrinol Diabetes Metab Case Rep. 2013;2013:130046
3. de Groot JW, Rikhof B, van Doorn J, et al. Non-islet cell tumour-induced hypoglycaemia: a review of the literature including two new cases. Endocr Relat Cancer. 2007;14:979-93.
4. Fukuda I, Hizuka N, Ishikawa Y, et al. Clinical features of insulin-like growth factor II producing non-islet-cell tumor hypoglycemia
5. Marks V, Teale JD: Tumours producing hypoglycaemia. Endocr Relat Cancer. 1998;5:111-129.
6. Le Roith D. Tumor-induced hypoglycemia. N Engl J Med. 1999;341:757-758.
7. Teale JD, Marks V. Glucocorticoid therapy suppresses abnormal secretion of big IGF-II by non-islet cell tumours inducing hypoglycaemia (NICTH). Clin Endocrinol .1998;49:491-498.
8. Hamberg P, De Jong FA, Boonstra JG, et al. Non-islet-cell tumor induced hypoglycemia in patients with advanced gastrointestinal stromal tumor possibly worsened by imatinib. J Clin Oncol. 2006;24:e30-e31.
9. Rikhof B, van Doorn J, Suurmeijer AJ, et al. Insulin-like growth factors and insulin-like growth factor-binding proteins in relation to disease status and incidence of hypoglycaemia in patients with a gastrointestinal stromal tumour. Ann Oncol. 2009;20:1582-1588.
10. Braconi C, Bracci R, Bearzi I, et al. Insulin-like growth factor (IGF) 1 and 2 help to predict disease outcome in GIST patients. Ann Oncol. 2008;19:1293-1298.
Pregnant Patient Develops a Rare Case of Multiple Sclerosis
Pregnancy is generally found to offer a respite from multiple sclerosis (MS). Pregnant women rarely develop MS or to have relapses. But in a unique and challenging case, a woman in her 14th week of her second pregnancy developed signs and symptoms of tumefactive multiple sclerosis (TMS), a rare subtype of MS. The TMS was only one of several unexpected clinical puzzles, according to the clinicians reporting on the case.
The patient, who had been healthy, was admitted with acute onset of paresthesias and word-finding difficulty. She had just had a long drive from Florida, and the the clinicians first assumed that she was fatigued from the trip and from the pregnancy. A magnetic resonance imaging (MRI) scan of the brain, however, suggested an ischemic event.
While hospitalized, the patient’s condition rapidly worsened. More scan and test findings proved consistent with TMS. A repeat MRI scan showed interval progression with a growing tumefactive demyelinating lesion (TDL) with diffuse surrounding edema and new periventricular signal changes. Although rare, TDLs often represent fulminant forms of MS, the clinicians note. Because the lesions mimic strokes, tumors, and abscesses, diagnosis is difficult. Moreover, the gadolinium (which was avoided because it can cause birth defects) might have helped them visualize lesions sooner.
The patient was started on high-dose IV methylprednisolone and plasma exchange, but the response was mild. The poor response to both treatment modalities is infrequent in TMS, the clinicians say—yet another unforeseen obstacle.
In addition to counseling the patient about the usual protective effects of pregnancy, her clinicians counseled her “extensively” about natalizumab and the possible beneficial effects of disease-modifying therapies. But the patient made the difficult decision to terminate the pregnancy, in part because she felt it was better to focus on her existing child rather than on caring for 2 young children while having a chronic progressive disease with uncertain recovery.
Another surprise was in store. Within 12 hours after an uncomplicated dilatation and curettage, the patient was able to move her right arm. That “drastic improvement” was followed by moderate improvement in her right leg. Her “paradoxical” improvement after the termination might indicate a “different from expected” hormonal influence in the pathogenesis of TMS, the clinicians say, but more likely represents a delayed corroborating effect of steroids and plasma exchange.
In the following weeks, the patient’s recovery was “satisfying” with gradual improvement and partial return of expressive language. Eighteen months later, the patient was clinically stable on natalizumab.
Source:
Pakneshan S, Bernitsas E. BMJ Case Rep. 2017. pii: bcr-2017-219534.
doi: 10.1136/bcr-2017-219534.
Pregnancy is generally found to offer a respite from multiple sclerosis (MS). Pregnant women rarely develop MS or to have relapses. But in a unique and challenging case, a woman in her 14th week of her second pregnancy developed signs and symptoms of tumefactive multiple sclerosis (TMS), a rare subtype of MS. The TMS was only one of several unexpected clinical puzzles, according to the clinicians reporting on the case.
The patient, who had been healthy, was admitted with acute onset of paresthesias and word-finding difficulty. She had just had a long drive from Florida, and the the clinicians first assumed that she was fatigued from the trip and from the pregnancy. A magnetic resonance imaging (MRI) scan of the brain, however, suggested an ischemic event.
While hospitalized, the patient’s condition rapidly worsened. More scan and test findings proved consistent with TMS. A repeat MRI scan showed interval progression with a growing tumefactive demyelinating lesion (TDL) with diffuse surrounding edema and new periventricular signal changes. Although rare, TDLs often represent fulminant forms of MS, the clinicians note. Because the lesions mimic strokes, tumors, and abscesses, diagnosis is difficult. Moreover, the gadolinium (which was avoided because it can cause birth defects) might have helped them visualize lesions sooner.
The patient was started on high-dose IV methylprednisolone and plasma exchange, but the response was mild. The poor response to both treatment modalities is infrequent in TMS, the clinicians say—yet another unforeseen obstacle.
In addition to counseling the patient about the usual protective effects of pregnancy, her clinicians counseled her “extensively” about natalizumab and the possible beneficial effects of disease-modifying therapies. But the patient made the difficult decision to terminate the pregnancy, in part because she felt it was better to focus on her existing child rather than on caring for 2 young children while having a chronic progressive disease with uncertain recovery.
Another surprise was in store. Within 12 hours after an uncomplicated dilatation and curettage, the patient was able to move her right arm. That “drastic improvement” was followed by moderate improvement in her right leg. Her “paradoxical” improvement after the termination might indicate a “different from expected” hormonal influence in the pathogenesis of TMS, the clinicians say, but more likely represents a delayed corroborating effect of steroids and plasma exchange.
In the following weeks, the patient’s recovery was “satisfying” with gradual improvement and partial return of expressive language. Eighteen months later, the patient was clinically stable on natalizumab.
Source:
Pakneshan S, Bernitsas E. BMJ Case Rep. 2017. pii: bcr-2017-219534.
doi: 10.1136/bcr-2017-219534.
Pregnancy is generally found to offer a respite from multiple sclerosis (MS). Pregnant women rarely develop MS or to have relapses. But in a unique and challenging case, a woman in her 14th week of her second pregnancy developed signs and symptoms of tumefactive multiple sclerosis (TMS), a rare subtype of MS. The TMS was only one of several unexpected clinical puzzles, according to the clinicians reporting on the case.
The patient, who had been healthy, was admitted with acute onset of paresthesias and word-finding difficulty. She had just had a long drive from Florida, and the the clinicians first assumed that she was fatigued from the trip and from the pregnancy. A magnetic resonance imaging (MRI) scan of the brain, however, suggested an ischemic event.
While hospitalized, the patient’s condition rapidly worsened. More scan and test findings proved consistent with TMS. A repeat MRI scan showed interval progression with a growing tumefactive demyelinating lesion (TDL) with diffuse surrounding edema and new periventricular signal changes. Although rare, TDLs often represent fulminant forms of MS, the clinicians note. Because the lesions mimic strokes, tumors, and abscesses, diagnosis is difficult. Moreover, the gadolinium (which was avoided because it can cause birth defects) might have helped them visualize lesions sooner.
The patient was started on high-dose IV methylprednisolone and plasma exchange, but the response was mild. The poor response to both treatment modalities is infrequent in TMS, the clinicians say—yet another unforeseen obstacle.
In addition to counseling the patient about the usual protective effects of pregnancy, her clinicians counseled her “extensively” about natalizumab and the possible beneficial effects of disease-modifying therapies. But the patient made the difficult decision to terminate the pregnancy, in part because she felt it was better to focus on her existing child rather than on caring for 2 young children while having a chronic progressive disease with uncertain recovery.
Another surprise was in store. Within 12 hours after an uncomplicated dilatation and curettage, the patient was able to move her right arm. That “drastic improvement” was followed by moderate improvement in her right leg. Her “paradoxical” improvement after the termination might indicate a “different from expected” hormonal influence in the pathogenesis of TMS, the clinicians say, but more likely represents a delayed corroborating effect of steroids and plasma exchange.
In the following weeks, the patient’s recovery was “satisfying” with gradual improvement and partial return of expressive language. Eighteen months later, the patient was clinically stable on natalizumab.
Source:
Pakneshan S, Bernitsas E. BMJ Case Rep. 2017. pii: bcr-2017-219534.
doi: 10.1136/bcr-2017-219534.
Quality of Chronic Obstructive Pulmonary Disease-Related Health Care in Rural and Urban Veterans Affairs Clinics
Chronic obstructive pulmonary disease (COPD) affects between 11 and 24 million people in the U.S. and is the third leading cause of death in this country.1,2 Airflow obstruction on spirometry in addition to respiratory symptoms is required to establish a diagnosis of COPD.3,4 As many as 40% of patients with a clinical diagnosis of COPD have not had spirometry or have spirometry results inconsistent with the diagnosis of COPD.5,6 In addition to recommended spirometry, many patients with COPD do not receive other evidence-based therapies.7,8
About 50% of patients in the Minneapolis VA Health Care System (MVAHCS) receive care in its rural community-based outreach clinics (CBOCs). Data regarding the quality of general medical care between rural and urban populations are sparse; however, studies suggest that the quality of care delivered in rural clinics may be lower than the care provided in an urban setting.9-12 Care for patients with COPD in an urban setting is suboptimal with only 58% of patients receiving guideline-based care, and there are no comparative data for penetrance in the rural setting.8 Most published studies on patients with COPD treated in rural vs urban locations are outcomes studies that queried statewide or national registry data evaluating the frequency of emergency department (ED) visits or hospital admissions for COPD exacerbations, all-cause mortality, or COPD exacerbation-related mortality.13-18 There are no studies examining potential differences in the quality of health care received by patients with COPD in rural vs urban locations or whether these potential differences are associated with changes in health care utilization.
The authors sought to determine whether patients with the diagnosis of COPD treated in the MVAHCS and its 13 CBOCs receive similar quality of disease-related health care in rural vs urban primary care clinic locations. The authors hypothesized that patients who receive their primary care in rural clinics would be less likely to have had spirometry or to receive respiratory immunizations and short- or long-acting inhalers and that discrepancies would be associated with increased health care utilization in rural areas as measured by prescriptions for systemic corticosteroids, antibiotics, ED visits, or hospital admissions for COPD exacerbations.
Methods
The MVAHCS has 14 primary care locations; these locations were designated as rural or urban based on the Rural-Urban Commuting Area codes.19,20 There were 4 urban locations and 10 rural clinics; all rural clinics were farther than 40 miles from the main Minneapolis VAMC.
Patient Selection
The authors performed a retrospective chart review after receiving an institutional review board waiver for this quality assessment study. All patients who had a prior ICD-9 encounter diagnosis of COPD (codes: 491.0, 491.1, 491.2, 491.20, 491.21, 491.22, 491.8, 491.9, 492.0, 492.8, 494, 494.0, 494.1, 496) and who were seen in primary care during March 2015 were identified. Each subject’s first visit during that month was used as the start of the retrospective 1-year look-back period. All eligible subjects were sorted based on their rural or urban location and a randomly assigned number. Patients were then selected according to ascending numbers from each rural and urban clinic in proportion to the clinic’s representation among all eligible patients.
Outcomes
The primary outcomes—possible discrepancies in quality of health care for patients with COPD in rural vs urban primary care clinics—were assessed by (1) prior spirometry; (2) any prior pneumonia vaccination; (3) an influenza vaccination within the past year; (4) prescriptions within the past year for a short-acting beta agonist (SABA) metered-dose inhaler; and (5) prescriptions for a long-acting inhalers, including long-acting beta agonists (LABAs), long-acting muscarinic antagonists (LAMAs), or inhaled corticosteroids (ICSs).
Secondary outcomes included (1) an active prescription for home oxygen within the past calendar year; (2) health care utilization assessed via prescriptions for intermittent courses of oral corticosteroids; (3) prescriptions for respiratory antibiotics (macrolides, tetracyclines, fluoroquinolones) within the past year for COPD exacerbations; (4) ED visits; (5) hospital admissions (and need for mechanical ventilation) for COPD exacerbations within the past year; and (6) whether patients were seen by either VA or Non-VA pulmonology providers.
Data Collection
Patients’ demographic data and comorbidities were collected via chart review. A 1-year prescription medication list was obtained by an electronic database search of the MVAHCS electronic medical record (EMR). Additional antibiotics and corticosteroid prescriptions for COPD exacerbations paid for by the VA but filled at a local pharmacy were manually searched from a separate database to supplement the electronic prescription list. Comparison of the electronic prescription list and pharmacy records in 25% of patients found 100% concordance in the prescription lists. The investigator manually reviewed and extracted the following data from the EMR, scanned-in records, and a Midwest VA COPD registry database: most recent spirometry results; immunization status for influenza in the past year; prior pneumonia vaccination; home oxygen prescription; whether the patient received respiratory antibiotic or intermittent oral corticosteroid treatment for COPD exacerbations; whether the patient had a ED visit or hospital admission for COPD exacerbation with or without need for mechanical ventilation; and whether the patient had been seen by a pulmonology provider. The investigator reviewed all primary care provider notes in the past year for documentation of non-VA ED visits or hospitalizations that were not present in the EMR, Midwest VA COPD registry database, or scanned patient records.
Data Analysis
Results are described as mean ± standard deviation, median (interquartile range) or proportion, expressed as a percentage as appropriate for the level of measurement and distribution. The proportions meeting the COPD quality of health care outcomes in the urban and rural groups were compared using a chi-square test of proportions, and 95% confidence intervals (CI) on the differences were estimated. Samples of 400 patients each from the rural and urban groups were estimated to provide a 95% 2-sided CI on the differences of about ± 0.05 (5%), assuming the proportion meeting the quality of care outcomes in the urban group would be at least 0.8 (80%).
Results
The authors identified 1,538 patients with a previous encounter diagnosis of COPD who were seen in a primary care clinic in the MVAHCS in March of 2015. The authors reviewed the medical records of 801 randomly selected patients: 400 rural clinic patients and 401 urban clinic patients. Demographic characteristics and major comorbidities of rural and urban patients were similar except more rural patients were white, and fewer had a record of obstructive sleep apnea, alcoholism, or addictive disorders (Table 1). Prescriptions for common chronic medical conditions were similar for rural and urban groups, including medications for depression (31% vs 33%) or diabetes mellitus (25% vs 28%). In patients who had spirometry, the severity of COPD, as assessed by mean forced expiratory volume (FEV1), was similar between rural and urban patients (2.06 L vs 2.10 L). 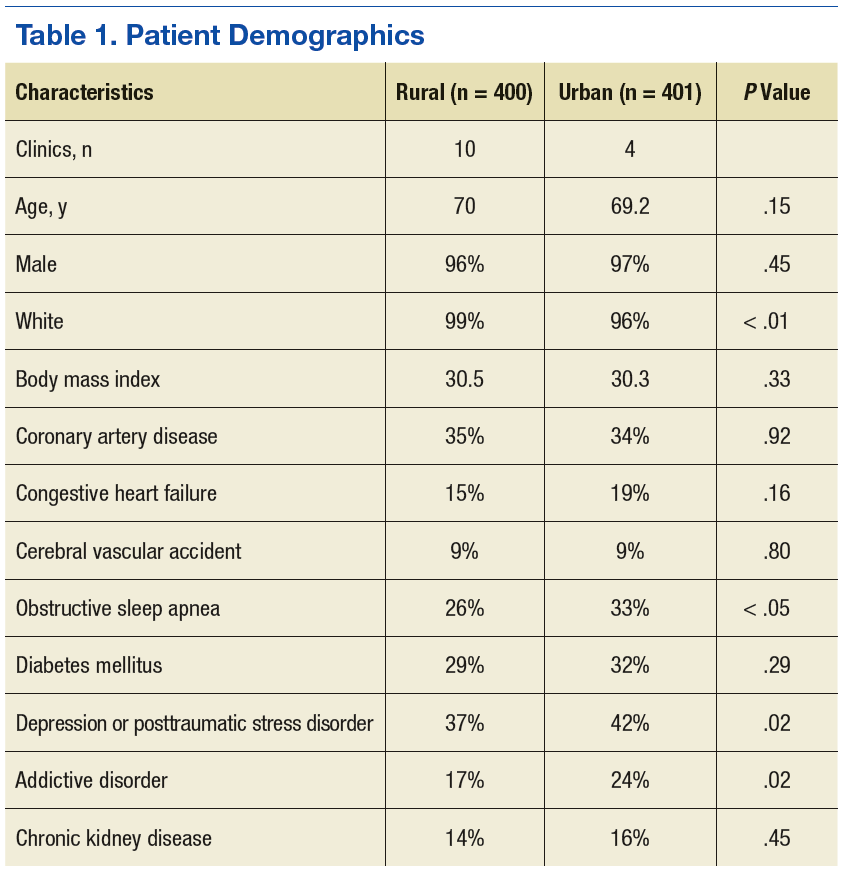
Quality of COPD Care
Spirometry was documented in fewer rural clinic patients than in urban clinic patients (51% vs 82%; difference 31%, 95% CI: 25% to 37%) (Table 2).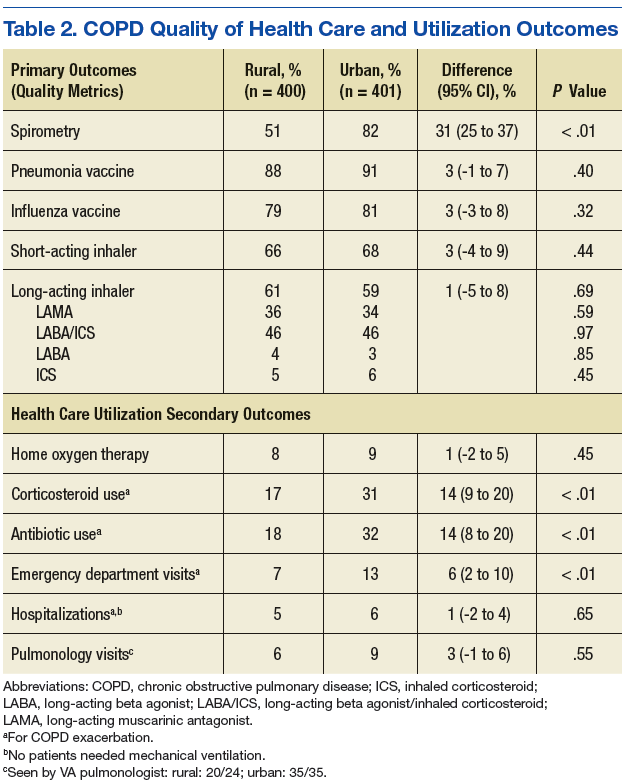
COPD Outcomes
Home oxygen prescription rates were similar for rural and urban clinic patients (8% vs 9%; difference 1%, 95% CI: -2% to 5%). Rural patients received fewer prescriptions for intermittent oral corticosteroids (17% vs 31%; difference 14%, 95% CI: 9% to 20%) and antibiotics for COPD exacerbations (18% vs 32%; difference 14%, 95% CI: 8% to 20%). Rural patients had fewer ED visits for COPD exacerbations (7% vs 13%; difference 6%, 95% CI: 2% to 10%), and similar admission rates for COPD exacerbations (5% vs 6%; difference 1%, 95% CI: -2% to 4%). Of the few patients hospitalized for COPD exacerbations, none required mechanical ventilation. There was no significant difference in the number of rural vs urban patients seen by a pulmonologist in the calendar year of the study (6% vs 9%; difference 3%, 95% CI:-1% to 6%), with the majority seen by VA providers: 20/24 rural patients and 35/35 urban patients.
Discussion
Fewer rural patients had prior spirometry; otherwise, the COPD-related quality metrics were similar between rural and urban patient groups in the MVAHCS, including immunizations for pneumonia and influenza, and prescribing rates for short- and long-acting inhaler therapy. Despite the similarity in these COPD quality measures, rural clinic patients seemed to have less health care utilization related to COPD exacerbations.
Spirometry with airflow obstruction in the presence of respiratory symptoms is required for accurate diagnosis of COPD.3,4 Spirometry has been available at the MVAHCS hospital-based clinic for years. Efforts to address this disparity led to implementation of on-site spirometry at all rural and urban clinics about 2 years prior to the patient enrollment visit date for the study. Fewer rural patients had spirometry, which is possibly from prior disparity in resources; yet rates of spirometry in all patients with a clinical diagnosis of COPD in the MVAHCS are higher (rural 51%, urban 82%) than previously reported. A nationwide study of 94,000 veterans with recent clinical diagnosis of COPD found only 37% had spirometry within 2.5 years of diagnosis,21 and another non-VA study (n = 553) showed only 31% of patients discharged from a hospital with a diagnosis of COPD exacerbation had spirometry performed within a 8-year period prior to hospitalization.22
Annual influenza vaccines are recommended for everyone aged > 6 months, and the pneumonia vaccine is recommended for all patients with COPD in order to reduce the risk of COPD exacerbations and pneumonias.23,24 The rates of vaccination at MVAHCS rural and urban clinics for both influenza (79% vs 81%) and pneumococcus (88% vs 91%) are higher than previously published studies of patients with COPD for influenza vaccination (30%-51%) and pneumonia vaccination (21%-51%) and did not differ between rural and urban clinics.7,25-28 The observed high vaccination rates may be due to EMR prompts and requirements to document vaccination status and offer recommended vaccinations.
Long-acting inhalers have been shown to reduce rates of COPD exacerbations and improve patients’ quality of life.29 The authors found no disparity in the prescription rate of short- or long-acting inhalers between rural and urban patients, and no difference in the severity of COPD, as indicated by FEV1, that might influence prescription rates.
The authors attempted to evaluate health care resource utilization as an indicator of health care quality and outcomes. Based on previous reports, the authors expected to find lower quality of care and increased utilization in rural patients. Previous studies have shown rural patients can be more symptomatic with a higher body mass index, airflow obstruction, dyspnea, and exercise capacity index (BODE index) than are patients in urban settings.16,30 Statewide and national registry data have shown rural patients have higher rates of primary care visits, ER visits, and hospitalizations for COPD exacerbations.
Rural patients also have been shown to have higher mortality rates and were more likely to be in a long-term care center and less likely to have home care or palliative care than were their urban counterparts.13-18,31 If the severity of illness is similar in rural and urban areas, higher health care utilization related to COPD would suggest that patients in rural settings may be receiving inferior quality of health care. The authors could not find any previous reports of the quality of COPD care delivered in rural vs urban settings.
In this study the only difference in quality of care was the lower proportion of rural patients with a record of spirometry that is needed to confirm the diagnosis. The observed differences in the quality of care measures wouldn’t be expected to lead to large differences in the outcome measures. Contrary to the literature and the observed similarity in quality of care, rural patients had better COPD outcomes perhaps due to unmeasured differences in risk or failure to capture medical visits outside of the VA system. The severity of COPD based on FEV1 and concurrent diagnoses, such as heart failure, did not suggest that rural patients in this comparison had a higher burden of illness or risk of poor COPD outcomes.
More than 70,000 patients spanning a large geographic region receive primary care at MVAHCS, which provides comparable care to all COPD patients, regardless of location, by using the same EMR system, providing evidence-based order sets for disease management, proactively offering on-site and remote COPD case managers for high-risk patients, and more recently, implementing on-site spirometry testing in all clinics. This approach as opposed to the traditional outreach clinic model may in part explain the similarity in quality of care in urban and rural clinics that was not reported in previous studies.
Limitations
This study was performed retrospectively, increasing the potential of missing data, especially from outside the VA health care system. Patients were not randomly assigned to rural or urban clinics, so differences in patient characteristics could exist. Alcohol and addictive disorders were more common in urban patients, which might affect adherence to prescribed medications. In addition, lower rate of obstructive sleep apnea was found in the rural population, which has been linked to increased airway inflammation and COPD exacerbations resulting in hospitalization.32,33 Mortality was not assessed as all patients were alive and seen in clinic at time of enrollment.
The authors were not able to record a patients’ residence in a long-term care facility or institutionalization, use of home care, or palliative care services due to limitations in the EMR system. Whether patients received comanaged primary care or underwent pulmonary rehabilitation could not be obtained from the EMR. Most patients never had lung volumes or diffusion capacity and thus were not included. The authors could not report whether inhaler therapy was appropriate compared with the Global Initiative for Chronic Obstructive Lund Disease(GOLD)severity score because most of the spirometry was done at a discordant time to when inhaler therapy was assessed, and new GOLD guidelines include patient symptoms that were not reliably recorded in the EMR. Last, the authors had hoped to include smoking status and cessation practices as part of the quality measures, but due to significant variability in patients’ documented smoking status in the same period, the data were deemed unreliable.
Conclusion
No disparities were found between rural and urban clinics in the quality of health care for patients with COPD in the MVAHCS except that fewer rural patients had prior spirometry; a difference that is likely due to the fact that only recently has spirometry been implemented in the MVAHCS rural clinics. Overall the quality of COPD care was high and above the previously reported rates. Further larger studies of rural and urban quality of health care for patients with COPD are needed in other VA and non-VA systems to determine whether disparities exist and whether they are associated with clinical outcomes, including ED visits, hospitalizations, and mortality.
1. Blackwell DL, Lucas JW, Clarke TC. Summary health statistics for U.S. adults: national health interview survey, 2012. Vital Health Stat 10. 2014(260):1-161.
2. Xu J, Murphy SL, Kochanek KD, Bastian BA. Deaths: final data for 2013. Natl Vital Stat Rep. 2016;64(2):1-119.
3. Celli BR, MacNee W, Force AET. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932-946.
4. Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS; GOLD Scientific Committee. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163(5):1256-1276.
5. Ghattas C, Dai A, Gemmel DJ, Awad MH. Over diagnosis of chronic obstructive pulmonary disease in an underserved patient population. Int J Chron Obstruct Pulmon Dis. 2013;8:545-549.
6. Zwar NA, Marks GB, Hermiz O, et al. Predictors of accuracy of diagnosis of chronic obstructive pulmonary disease in general practice. Med J Aust. 2011;195(4):168-171.
7. Lopez-Campos JL, Abad Arranz M, Calero-Acuña C, et al. Guideline adherence in outpatient clinics for chronic obstructive pulmonary disease: results from a clinical audit. PLoS One. 2016;11(3):e0151896.
8. McGlynn EA, Asch SM, Adams J, et al. The quality of health care delivered to adults in the United States. N Engl J Med. 2003;348(26):2635-2645.
9. Spoont M, Greer N, Su J, Fitzgerald P, Rutks I, Wilt TJ. Rural vs. Urban Ambulatory Health Care: A Systematic Review. Washington, DC: U.S. Department of Veteran Affairs; 2011.
10. Weeks WB, Wallace AE, Wang S, Lee A, Kazis LE. Rural-urban disparities in health-related quality of life within disease categories of Veterans. J Rural Health. 2006;22(3):204-211.
11. Wallace AE, Weeks WB, Wang S, Lee AF, Kazis LE. Rural and urban disparities in health-related quality of life among veterans with psychiatric disorders. Psychiatr Serv. 2006;57(6):851-856.
12. Meit M, Knudson A, Gilbert T, et al; Rural Health Reform Policy Research Center. The 2014 update of the rural-urban chartbook. https://ruralhealth.und .edu/projects/health-reform-policy-research-center/pdf/2014-rural-urban-chartbook-update.pdf. Published October 2014. Accessed April 18, 2017.
13. Jackson BE, Suzuki S, Coultas D, et al. Safety-net facilities and hospitalization rates of chronic obstructive pulmonary disease: a cross-sectional analysis of the 2007 Texas Health Care Information Council inpatient data. Int J Chron Obstruct Pulmon Dis. 2011;6:563-571.
14. Jackson BE, Suzuki S, Lo K, et al. Geographic disparity in COPD hospitalization rates among the Texas population. Respir Med. 2011;105(5):734-739.15. Skinner HG, Blanchard J, Elixhauser A. Trends in emergency department visits, 2006-2011: statistical brief #179. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb179-Emergency-Department-Trends.pdf. Published September 2014. Accessed May 9, 2017.
16. Jackson BE, Coultas DB, Suzuki S, Singh KP, Bae S. Rural-urban disparities in quality of life among patients with COPD. J Rural Health. 2013;29(suppl 1):62S-69S.
17. Singh GK, Siahpush M. Widening rural-urban disparities in life expectancy, U.S., 1969-2009. Am J Prev Med. 2014;46(2):e19-e29.
18. Abrams TE, Vaughan-Sarrazin M, Fan VS, Kaboli PJ. Geographic isolation and the risk for chronic obstructive pulmonary disease-related mortality: a cohort study. Ann Intern Med. 2011;155(2):80-86.
19. Hart LG, Larson EH, Lishner DM. Rural definitions for health policy and research. Am J Public Health. 2005;95(7):1149-1155.
20. U.S. Department of Agriculture, Economic Research Service. Rural-urban commuting area codes. https://www.ers.usda.gov/data-products/rural-urban-commuting-area-codes.aspx#U20K1F50H0A. Updated October 12, 2016. Accessed May 9, 2017.
21. Joo MJ, Lee TA, Weiss KB. Geographic variation of spirometry use in newly diagnosed COPD. Chest. 2008;134(1):38-45.
22. Damarla M, Celli BR, Mullerova HX, Pinto-Plata VM. Discrepancy in the use of confirmatory tests in patients hospitalized with the diagnosis of chronic obstructive pulmonary disease or congestive heart failure. Respir Care. 2006;51(10):1120-1124.
23. Grohskopf LA, Sokolow LZ, Olsen SJ, Bresee JS, Broder KR, Karron RA. Prevention and control of influenza with vaccines: recommendations of the advisory committee on immunization practices, United States, 2015-16 influenza season. MMWR Morb Mortal Wkly Rep. 2015;64(30):818-825.
24. Kim DK, Bridges CB, Harriman KH; Advisory Committee on Immunization Practices. Advisory committee on immunization practices recommended immunization schedule for adults aged 19 years or older: United States, 2016. Ann Intern Med. 2016;164(3):184-194.
25. Cimen P, Unlu M, Kirakli C, et al. Should patients with COPD be vaccinated? Respir Care. 2015;60(2):239-243.
26. Mowls DS, Cheruvu VK, Zullo MD. Influenza vaccination in adults with chronic obstructive pulmonary disease: the impact of a diagnostic breathing test on vaccination rates PLoS One. 2013;8(6):e67600.
27. Shoup JA, Madrid C, Koehler C, et al. Effectiveness and cost of influenza vaccine reminders for adults with asthma or chronic obstructive pulmonary disease. Am J Manag Care. 2015;21(7):e405-e413.
28. Arinez-Fernandez MC, Carrasco-Garrido P, Garcia-Carballo M, Hernandez-Barrera V, de Miguel AG, Jimenez-Garcia R. Determinants of pneumococcal vaccination among patients with chronic obstructive pulmonary disease in Spain. Hum Vaccin. 2006;2(3):99-104.29. Qaseem A, Wilt TJ, Weinberger SE, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med. 2011;155(3):179-191.
30. Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(10):1005-1012.
31. Goodridge D, Lawson J, Rennie D, Marciniuk D. Rural/urban differences in health care utilization and place of death for persons with respiratory illness in the last year of life. Rural Remote Health. 2010;10(2):1349.
32. Wang Y, Hu K, Liu K, et al. Obstructive sleep apnea exacerbates airway inflammation in patients with chronic obstructive pulmonary disease. Sleep Med. 2015;16(9):1123-1130.
33. Marin JM, Soriano JB, Carrizo SJ, Boldova A, Celli BR. Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am J Respir Crit Care Med. 2010;182(3):325-331
Chronic obstructive pulmonary disease (COPD) affects between 11 and 24 million people in the U.S. and is the third leading cause of death in this country.1,2 Airflow obstruction on spirometry in addition to respiratory symptoms is required to establish a diagnosis of COPD.3,4 As many as 40% of patients with a clinical diagnosis of COPD have not had spirometry or have spirometry results inconsistent with the diagnosis of COPD.5,6 In addition to recommended spirometry, many patients with COPD do not receive other evidence-based therapies.7,8
About 50% of patients in the Minneapolis VA Health Care System (MVAHCS) receive care in its rural community-based outreach clinics (CBOCs). Data regarding the quality of general medical care between rural and urban populations are sparse; however, studies suggest that the quality of care delivered in rural clinics may be lower than the care provided in an urban setting.9-12 Care for patients with COPD in an urban setting is suboptimal with only 58% of patients receiving guideline-based care, and there are no comparative data for penetrance in the rural setting.8 Most published studies on patients with COPD treated in rural vs urban locations are outcomes studies that queried statewide or national registry data evaluating the frequency of emergency department (ED) visits or hospital admissions for COPD exacerbations, all-cause mortality, or COPD exacerbation-related mortality.13-18 There are no studies examining potential differences in the quality of health care received by patients with COPD in rural vs urban locations or whether these potential differences are associated with changes in health care utilization.
The authors sought to determine whether patients with the diagnosis of COPD treated in the MVAHCS and its 13 CBOCs receive similar quality of disease-related health care in rural vs urban primary care clinic locations. The authors hypothesized that patients who receive their primary care in rural clinics would be less likely to have had spirometry or to receive respiratory immunizations and short- or long-acting inhalers and that discrepancies would be associated with increased health care utilization in rural areas as measured by prescriptions for systemic corticosteroids, antibiotics, ED visits, or hospital admissions for COPD exacerbations.
Methods
The MVAHCS has 14 primary care locations; these locations were designated as rural or urban based on the Rural-Urban Commuting Area codes.19,20 There were 4 urban locations and 10 rural clinics; all rural clinics were farther than 40 miles from the main Minneapolis VAMC.
Patient Selection
The authors performed a retrospective chart review after receiving an institutional review board waiver for this quality assessment study. All patients who had a prior ICD-9 encounter diagnosis of COPD (codes: 491.0, 491.1, 491.2, 491.20, 491.21, 491.22, 491.8, 491.9, 492.0, 492.8, 494, 494.0, 494.1, 496) and who were seen in primary care during March 2015 were identified. Each subject’s first visit during that month was used as the start of the retrospective 1-year look-back period. All eligible subjects were sorted based on their rural or urban location and a randomly assigned number. Patients were then selected according to ascending numbers from each rural and urban clinic in proportion to the clinic’s representation among all eligible patients.
Outcomes
The primary outcomes—possible discrepancies in quality of health care for patients with COPD in rural vs urban primary care clinics—were assessed by (1) prior spirometry; (2) any prior pneumonia vaccination; (3) an influenza vaccination within the past year; (4) prescriptions within the past year for a short-acting beta agonist (SABA) metered-dose inhaler; and (5) prescriptions for a long-acting inhalers, including long-acting beta agonists (LABAs), long-acting muscarinic antagonists (LAMAs), or inhaled corticosteroids (ICSs).
Secondary outcomes included (1) an active prescription for home oxygen within the past calendar year; (2) health care utilization assessed via prescriptions for intermittent courses of oral corticosteroids; (3) prescriptions for respiratory antibiotics (macrolides, tetracyclines, fluoroquinolones) within the past year for COPD exacerbations; (4) ED visits; (5) hospital admissions (and need for mechanical ventilation) for COPD exacerbations within the past year; and (6) whether patients were seen by either VA or Non-VA pulmonology providers.
Data Collection
Patients’ demographic data and comorbidities were collected via chart review. A 1-year prescription medication list was obtained by an electronic database search of the MVAHCS electronic medical record (EMR). Additional antibiotics and corticosteroid prescriptions for COPD exacerbations paid for by the VA but filled at a local pharmacy were manually searched from a separate database to supplement the electronic prescription list. Comparison of the electronic prescription list and pharmacy records in 25% of patients found 100% concordance in the prescription lists. The investigator manually reviewed and extracted the following data from the EMR, scanned-in records, and a Midwest VA COPD registry database: most recent spirometry results; immunization status for influenza in the past year; prior pneumonia vaccination; home oxygen prescription; whether the patient received respiratory antibiotic or intermittent oral corticosteroid treatment for COPD exacerbations; whether the patient had a ED visit or hospital admission for COPD exacerbation with or without need for mechanical ventilation; and whether the patient had been seen by a pulmonology provider. The investigator reviewed all primary care provider notes in the past year for documentation of non-VA ED visits or hospitalizations that were not present in the EMR, Midwest VA COPD registry database, or scanned patient records.
Data Analysis
Results are described as mean ± standard deviation, median (interquartile range) or proportion, expressed as a percentage as appropriate for the level of measurement and distribution. The proportions meeting the COPD quality of health care outcomes in the urban and rural groups were compared using a chi-square test of proportions, and 95% confidence intervals (CI) on the differences were estimated. Samples of 400 patients each from the rural and urban groups were estimated to provide a 95% 2-sided CI on the differences of about ± 0.05 (5%), assuming the proportion meeting the quality of care outcomes in the urban group would be at least 0.8 (80%).
Results
The authors identified 1,538 patients with a previous encounter diagnosis of COPD who were seen in a primary care clinic in the MVAHCS in March of 2015. The authors reviewed the medical records of 801 randomly selected patients: 400 rural clinic patients and 401 urban clinic patients. Demographic characteristics and major comorbidities of rural and urban patients were similar except more rural patients were white, and fewer had a record of obstructive sleep apnea, alcoholism, or addictive disorders (Table 1). Prescriptions for common chronic medical conditions were similar for rural and urban groups, including medications for depression (31% vs 33%) or diabetes mellitus (25% vs 28%). In patients who had spirometry, the severity of COPD, as assessed by mean forced expiratory volume (FEV1), was similar between rural and urban patients (2.06 L vs 2.10 L).
Quality of COPD Care
Spirometry was documented in fewer rural clinic patients than in urban clinic patients (51% vs 82%; difference 31%, 95% CI: 25% to 37%) (Table 2).
COPD Outcomes
Home oxygen prescription rates were similar for rural and urban clinic patients (8% vs 9%; difference 1%, 95% CI: -2% to 5%). Rural patients received fewer prescriptions for intermittent oral corticosteroids (17% vs 31%; difference 14%, 95% CI: 9% to 20%) and antibiotics for COPD exacerbations (18% vs 32%; difference 14%, 95% CI: 8% to 20%). Rural patients had fewer ED visits for COPD exacerbations (7% vs 13%; difference 6%, 95% CI: 2% to 10%), and similar admission rates for COPD exacerbations (5% vs 6%; difference 1%, 95% CI: -2% to 4%). Of the few patients hospitalized for COPD exacerbations, none required mechanical ventilation. There was no significant difference in the number of rural vs urban patients seen by a pulmonologist in the calendar year of the study (6% vs 9%; difference 3%, 95% CI:-1% to 6%), with the majority seen by VA providers: 20/24 rural patients and 35/35 urban patients.
Discussion
Fewer rural patients had prior spirometry; otherwise, the COPD-related quality metrics were similar between rural and urban patient groups in the MVAHCS, including immunizations for pneumonia and influenza, and prescribing rates for short- and long-acting inhaler therapy. Despite the similarity in these COPD quality measures, rural clinic patients seemed to have less health care utilization related to COPD exacerbations.
Spirometry with airflow obstruction in the presence of respiratory symptoms is required for accurate diagnosis of COPD.3,4 Spirometry has been available at the MVAHCS hospital-based clinic for years. Efforts to address this disparity led to implementation of on-site spirometry at all rural and urban clinics about 2 years prior to the patient enrollment visit date for the study. Fewer rural patients had spirometry, which is possibly from prior disparity in resources; yet rates of spirometry in all patients with a clinical diagnosis of COPD in the MVAHCS are higher (rural 51%, urban 82%) than previously reported. A nationwide study of 94,000 veterans with recent clinical diagnosis of COPD found only 37% had spirometry within 2.5 years of diagnosis,21 and another non-VA study (n = 553) showed only 31% of patients discharged from a hospital with a diagnosis of COPD exacerbation had spirometry performed within a 8-year period prior to hospitalization.22
Annual influenza vaccines are recommended for everyone aged > 6 months, and the pneumonia vaccine is recommended for all patients with COPD in order to reduce the risk of COPD exacerbations and pneumonias.23,24 The rates of vaccination at MVAHCS rural and urban clinics for both influenza (79% vs 81%) and pneumococcus (88% vs 91%) are higher than previously published studies of patients with COPD for influenza vaccination (30%-51%) and pneumonia vaccination (21%-51%) and did not differ between rural and urban clinics.7,25-28 The observed high vaccination rates may be due to EMR prompts and requirements to document vaccination status and offer recommended vaccinations.
Long-acting inhalers have been shown to reduce rates of COPD exacerbations and improve patients’ quality of life.29 The authors found no disparity in the prescription rate of short- or long-acting inhalers between rural and urban patients, and no difference in the severity of COPD, as indicated by FEV1, that might influence prescription rates.
The authors attempted to evaluate health care resource utilization as an indicator of health care quality and outcomes. Based on previous reports, the authors expected to find lower quality of care and increased utilization in rural patients. Previous studies have shown rural patients can be more symptomatic with a higher body mass index, airflow obstruction, dyspnea, and exercise capacity index (BODE index) than are patients in urban settings.16,30 Statewide and national registry data have shown rural patients have higher rates of primary care visits, ER visits, and hospitalizations for COPD exacerbations.
Rural patients also have been shown to have higher mortality rates and were more likely to be in a long-term care center and less likely to have home care or palliative care than were their urban counterparts.13-18,31 If the severity of illness is similar in rural and urban areas, higher health care utilization related to COPD would suggest that patients in rural settings may be receiving inferior quality of health care. The authors could not find any previous reports of the quality of COPD care delivered in rural vs urban settings.
In this study the only difference in quality of care was the lower proportion of rural patients with a record of spirometry that is needed to confirm the diagnosis. The observed differences in the quality of care measures wouldn’t be expected to lead to large differences in the outcome measures. Contrary to the literature and the observed similarity in quality of care, rural patients had better COPD outcomes perhaps due to unmeasured differences in risk or failure to capture medical visits outside of the VA system. The severity of COPD based on FEV1 and concurrent diagnoses, such as heart failure, did not suggest that rural patients in this comparison had a higher burden of illness or risk of poor COPD outcomes.
More than 70,000 patients spanning a large geographic region receive primary care at MVAHCS, which provides comparable care to all COPD patients, regardless of location, by using the same EMR system, providing evidence-based order sets for disease management, proactively offering on-site and remote COPD case managers for high-risk patients, and more recently, implementing on-site spirometry testing in all clinics. This approach as opposed to the traditional outreach clinic model may in part explain the similarity in quality of care in urban and rural clinics that was not reported in previous studies.
Limitations
This study was performed retrospectively, increasing the potential of missing data, especially from outside the VA health care system. Patients were not randomly assigned to rural or urban clinics, so differences in patient characteristics could exist. Alcohol and addictive disorders were more common in urban patients, which might affect adherence to prescribed medications. In addition, lower rate of obstructive sleep apnea was found in the rural population, which has been linked to increased airway inflammation and COPD exacerbations resulting in hospitalization.32,33 Mortality was not assessed as all patients were alive and seen in clinic at time of enrollment.
The authors were not able to record a patients’ residence in a long-term care facility or institutionalization, use of home care, or palliative care services due to limitations in the EMR system. Whether patients received comanaged primary care or underwent pulmonary rehabilitation could not be obtained from the EMR. Most patients never had lung volumes or diffusion capacity and thus were not included. The authors could not report whether inhaler therapy was appropriate compared with the Global Initiative for Chronic Obstructive Lund Disease(GOLD)severity score because most of the spirometry was done at a discordant time to when inhaler therapy was assessed, and new GOLD guidelines include patient symptoms that were not reliably recorded in the EMR. Last, the authors had hoped to include smoking status and cessation practices as part of the quality measures, but due to significant variability in patients’ documented smoking status in the same period, the data were deemed unreliable.
Conclusion
No disparities were found between rural and urban clinics in the quality of health care for patients with COPD in the MVAHCS except that fewer rural patients had prior spirometry; a difference that is likely due to the fact that only recently has spirometry been implemented in the MVAHCS rural clinics. Overall the quality of COPD care was high and above the previously reported rates. Further larger studies of rural and urban quality of health care for patients with COPD are needed in other VA and non-VA systems to determine whether disparities exist and whether they are associated with clinical outcomes, including ED visits, hospitalizations, and mortality.
Chronic obstructive pulmonary disease (COPD) affects between 11 and 24 million people in the U.S. and is the third leading cause of death in this country.1,2 Airflow obstruction on spirometry in addition to respiratory symptoms is required to establish a diagnosis of COPD.3,4 As many as 40% of patients with a clinical diagnosis of COPD have not had spirometry or have spirometry results inconsistent with the diagnosis of COPD.5,6 In addition to recommended spirometry, many patients with COPD do not receive other evidence-based therapies.7,8
About 50% of patients in the Minneapolis VA Health Care System (MVAHCS) receive care in its rural community-based outreach clinics (CBOCs). Data regarding the quality of general medical care between rural and urban populations are sparse; however, studies suggest that the quality of care delivered in rural clinics may be lower than the care provided in an urban setting.9-12 Care for patients with COPD in an urban setting is suboptimal with only 58% of patients receiving guideline-based care, and there are no comparative data for penetrance in the rural setting.8 Most published studies on patients with COPD treated in rural vs urban locations are outcomes studies that queried statewide or national registry data evaluating the frequency of emergency department (ED) visits or hospital admissions for COPD exacerbations, all-cause mortality, or COPD exacerbation-related mortality.13-18 There are no studies examining potential differences in the quality of health care received by patients with COPD in rural vs urban locations or whether these potential differences are associated with changes in health care utilization.
The authors sought to determine whether patients with the diagnosis of COPD treated in the MVAHCS and its 13 CBOCs receive similar quality of disease-related health care in rural vs urban primary care clinic locations. The authors hypothesized that patients who receive their primary care in rural clinics would be less likely to have had spirometry or to receive respiratory immunizations and short- or long-acting inhalers and that discrepancies would be associated with increased health care utilization in rural areas as measured by prescriptions for systemic corticosteroids, antibiotics, ED visits, or hospital admissions for COPD exacerbations.
Methods
The MVAHCS has 14 primary care locations; these locations were designated as rural or urban based on the Rural-Urban Commuting Area codes.19,20 There were 4 urban locations and 10 rural clinics; all rural clinics were farther than 40 miles from the main Minneapolis VAMC.
Patient Selection
The authors performed a retrospective chart review after receiving an institutional review board waiver for this quality assessment study. All patients who had a prior ICD-9 encounter diagnosis of COPD (codes: 491.0, 491.1, 491.2, 491.20, 491.21, 491.22, 491.8, 491.9, 492.0, 492.8, 494, 494.0, 494.1, 496) and who were seen in primary care during March 2015 were identified. Each subject’s first visit during that month was used as the start of the retrospective 1-year look-back period. All eligible subjects were sorted based on their rural or urban location and a randomly assigned number. Patients were then selected according to ascending numbers from each rural and urban clinic in proportion to the clinic’s representation among all eligible patients.
Outcomes
The primary outcomes—possible discrepancies in quality of health care for patients with COPD in rural vs urban primary care clinics—were assessed by (1) prior spirometry; (2) any prior pneumonia vaccination; (3) an influenza vaccination within the past year; (4) prescriptions within the past year for a short-acting beta agonist (SABA) metered-dose inhaler; and (5) prescriptions for a long-acting inhalers, including long-acting beta agonists (LABAs), long-acting muscarinic antagonists (LAMAs), or inhaled corticosteroids (ICSs).
Secondary outcomes included (1) an active prescription for home oxygen within the past calendar year; (2) health care utilization assessed via prescriptions for intermittent courses of oral corticosteroids; (3) prescriptions for respiratory antibiotics (macrolides, tetracyclines, fluoroquinolones) within the past year for COPD exacerbations; (4) ED visits; (5) hospital admissions (and need for mechanical ventilation) for COPD exacerbations within the past year; and (6) whether patients were seen by either VA or Non-VA pulmonology providers.
Data Collection
Patients’ demographic data and comorbidities were collected via chart review. A 1-year prescription medication list was obtained by an electronic database search of the MVAHCS electronic medical record (EMR). Additional antibiotics and corticosteroid prescriptions for COPD exacerbations paid for by the VA but filled at a local pharmacy were manually searched from a separate database to supplement the electronic prescription list. Comparison of the electronic prescription list and pharmacy records in 25% of patients found 100% concordance in the prescription lists. The investigator manually reviewed and extracted the following data from the EMR, scanned-in records, and a Midwest VA COPD registry database: most recent spirometry results; immunization status for influenza in the past year; prior pneumonia vaccination; home oxygen prescription; whether the patient received respiratory antibiotic or intermittent oral corticosteroid treatment for COPD exacerbations; whether the patient had a ED visit or hospital admission for COPD exacerbation with or without need for mechanical ventilation; and whether the patient had been seen by a pulmonology provider. The investigator reviewed all primary care provider notes in the past year for documentation of non-VA ED visits or hospitalizations that were not present in the EMR, Midwest VA COPD registry database, or scanned patient records.
Data Analysis
Results are described as mean ± standard deviation, median (interquartile range) or proportion, expressed as a percentage as appropriate for the level of measurement and distribution. The proportions meeting the COPD quality of health care outcomes in the urban and rural groups were compared using a chi-square test of proportions, and 95% confidence intervals (CI) on the differences were estimated. Samples of 400 patients each from the rural and urban groups were estimated to provide a 95% 2-sided CI on the differences of about ± 0.05 (5%), assuming the proportion meeting the quality of care outcomes in the urban group would be at least 0.8 (80%).
Results
The authors identified 1,538 patients with a previous encounter diagnosis of COPD who were seen in a primary care clinic in the MVAHCS in March of 2015. The authors reviewed the medical records of 801 randomly selected patients: 400 rural clinic patients and 401 urban clinic patients. Demographic characteristics and major comorbidities of rural and urban patients were similar except more rural patients were white, and fewer had a record of obstructive sleep apnea, alcoholism, or addictive disorders (Table 1). Prescriptions for common chronic medical conditions were similar for rural and urban groups, including medications for depression (31% vs 33%) or diabetes mellitus (25% vs 28%). In patients who had spirometry, the severity of COPD, as assessed by mean forced expiratory volume (FEV1), was similar between rural and urban patients (2.06 L vs 2.10 L).
Quality of COPD Care
Spirometry was documented in fewer rural clinic patients than in urban clinic patients (51% vs 82%; difference 31%, 95% CI: 25% to 37%) (Table 2).
COPD Outcomes
Home oxygen prescription rates were similar for rural and urban clinic patients (8% vs 9%; difference 1%, 95% CI: -2% to 5%). Rural patients received fewer prescriptions for intermittent oral corticosteroids (17% vs 31%; difference 14%, 95% CI: 9% to 20%) and antibiotics for COPD exacerbations (18% vs 32%; difference 14%, 95% CI: 8% to 20%). Rural patients had fewer ED visits for COPD exacerbations (7% vs 13%; difference 6%, 95% CI: 2% to 10%), and similar admission rates for COPD exacerbations (5% vs 6%; difference 1%, 95% CI: -2% to 4%). Of the few patients hospitalized for COPD exacerbations, none required mechanical ventilation. There was no significant difference in the number of rural vs urban patients seen by a pulmonologist in the calendar year of the study (6% vs 9%; difference 3%, 95% CI:-1% to 6%), with the majority seen by VA providers: 20/24 rural patients and 35/35 urban patients.
Discussion
Fewer rural patients had prior spirometry; otherwise, the COPD-related quality metrics were similar between rural and urban patient groups in the MVAHCS, including immunizations for pneumonia and influenza, and prescribing rates for short- and long-acting inhaler therapy. Despite the similarity in these COPD quality measures, rural clinic patients seemed to have less health care utilization related to COPD exacerbations.
Spirometry with airflow obstruction in the presence of respiratory symptoms is required for accurate diagnosis of COPD.3,4 Spirometry has been available at the MVAHCS hospital-based clinic for years. Efforts to address this disparity led to implementation of on-site spirometry at all rural and urban clinics about 2 years prior to the patient enrollment visit date for the study. Fewer rural patients had spirometry, which is possibly from prior disparity in resources; yet rates of spirometry in all patients with a clinical diagnosis of COPD in the MVAHCS are higher (rural 51%, urban 82%) than previously reported. A nationwide study of 94,000 veterans with recent clinical diagnosis of COPD found only 37% had spirometry within 2.5 years of diagnosis,21 and another non-VA study (n = 553) showed only 31% of patients discharged from a hospital with a diagnosis of COPD exacerbation had spirometry performed within a 8-year period prior to hospitalization.22
Annual influenza vaccines are recommended for everyone aged > 6 months, and the pneumonia vaccine is recommended for all patients with COPD in order to reduce the risk of COPD exacerbations and pneumonias.23,24 The rates of vaccination at MVAHCS rural and urban clinics for both influenza (79% vs 81%) and pneumococcus (88% vs 91%) are higher than previously published studies of patients with COPD for influenza vaccination (30%-51%) and pneumonia vaccination (21%-51%) and did not differ between rural and urban clinics.7,25-28 The observed high vaccination rates may be due to EMR prompts and requirements to document vaccination status and offer recommended vaccinations.
Long-acting inhalers have been shown to reduce rates of COPD exacerbations and improve patients’ quality of life.29 The authors found no disparity in the prescription rate of short- or long-acting inhalers between rural and urban patients, and no difference in the severity of COPD, as indicated by FEV1, that might influence prescription rates.
The authors attempted to evaluate health care resource utilization as an indicator of health care quality and outcomes. Based on previous reports, the authors expected to find lower quality of care and increased utilization in rural patients. Previous studies have shown rural patients can be more symptomatic with a higher body mass index, airflow obstruction, dyspnea, and exercise capacity index (BODE index) than are patients in urban settings.16,30 Statewide and national registry data have shown rural patients have higher rates of primary care visits, ER visits, and hospitalizations for COPD exacerbations.
Rural patients also have been shown to have higher mortality rates and were more likely to be in a long-term care center and less likely to have home care or palliative care than were their urban counterparts.13-18,31 If the severity of illness is similar in rural and urban areas, higher health care utilization related to COPD would suggest that patients in rural settings may be receiving inferior quality of health care. The authors could not find any previous reports of the quality of COPD care delivered in rural vs urban settings.
In this study the only difference in quality of care was the lower proportion of rural patients with a record of spirometry that is needed to confirm the diagnosis. The observed differences in the quality of care measures wouldn’t be expected to lead to large differences in the outcome measures. Contrary to the literature and the observed similarity in quality of care, rural patients had better COPD outcomes perhaps due to unmeasured differences in risk or failure to capture medical visits outside of the VA system. The severity of COPD based on FEV1 and concurrent diagnoses, such as heart failure, did not suggest that rural patients in this comparison had a higher burden of illness or risk of poor COPD outcomes.
More than 70,000 patients spanning a large geographic region receive primary care at MVAHCS, which provides comparable care to all COPD patients, regardless of location, by using the same EMR system, providing evidence-based order sets for disease management, proactively offering on-site and remote COPD case managers for high-risk patients, and more recently, implementing on-site spirometry testing in all clinics. This approach as opposed to the traditional outreach clinic model may in part explain the similarity in quality of care in urban and rural clinics that was not reported in previous studies.
Limitations
This study was performed retrospectively, increasing the potential of missing data, especially from outside the VA health care system. Patients were not randomly assigned to rural or urban clinics, so differences in patient characteristics could exist. Alcohol and addictive disorders were more common in urban patients, which might affect adherence to prescribed medications. In addition, lower rate of obstructive sleep apnea was found in the rural population, which has been linked to increased airway inflammation and COPD exacerbations resulting in hospitalization.32,33 Mortality was not assessed as all patients were alive and seen in clinic at time of enrollment.
The authors were not able to record a patients’ residence in a long-term care facility or institutionalization, use of home care, or palliative care services due to limitations in the EMR system. Whether patients received comanaged primary care or underwent pulmonary rehabilitation could not be obtained from the EMR. Most patients never had lung volumes or diffusion capacity and thus were not included. The authors could not report whether inhaler therapy was appropriate compared with the Global Initiative for Chronic Obstructive Lund Disease(GOLD)severity score because most of the spirometry was done at a discordant time to when inhaler therapy was assessed, and new GOLD guidelines include patient symptoms that were not reliably recorded in the EMR. Last, the authors had hoped to include smoking status and cessation practices as part of the quality measures, but due to significant variability in patients’ documented smoking status in the same period, the data were deemed unreliable.
Conclusion
No disparities were found between rural and urban clinics in the quality of health care for patients with COPD in the MVAHCS except that fewer rural patients had prior spirometry; a difference that is likely due to the fact that only recently has spirometry been implemented in the MVAHCS rural clinics. Overall the quality of COPD care was high and above the previously reported rates. Further larger studies of rural and urban quality of health care for patients with COPD are needed in other VA and non-VA systems to determine whether disparities exist and whether they are associated with clinical outcomes, including ED visits, hospitalizations, and mortality.
1. Blackwell DL, Lucas JW, Clarke TC. Summary health statistics for U.S. adults: national health interview survey, 2012. Vital Health Stat 10. 2014(260):1-161.
2. Xu J, Murphy SL, Kochanek KD, Bastian BA. Deaths: final data for 2013. Natl Vital Stat Rep. 2016;64(2):1-119.
3. Celli BR, MacNee W, Force AET. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932-946.
4. Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS; GOLD Scientific Committee. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163(5):1256-1276.
5. Ghattas C, Dai A, Gemmel DJ, Awad MH. Over diagnosis of chronic obstructive pulmonary disease in an underserved patient population. Int J Chron Obstruct Pulmon Dis. 2013;8:545-549.
6. Zwar NA, Marks GB, Hermiz O, et al. Predictors of accuracy of diagnosis of chronic obstructive pulmonary disease in general practice. Med J Aust. 2011;195(4):168-171.
7. Lopez-Campos JL, Abad Arranz M, Calero-Acuña C, et al. Guideline adherence in outpatient clinics for chronic obstructive pulmonary disease: results from a clinical audit. PLoS One. 2016;11(3):e0151896.
8. McGlynn EA, Asch SM, Adams J, et al. The quality of health care delivered to adults in the United States. N Engl J Med. 2003;348(26):2635-2645.
9. Spoont M, Greer N, Su J, Fitzgerald P, Rutks I, Wilt TJ. Rural vs. Urban Ambulatory Health Care: A Systematic Review. Washington, DC: U.S. Department of Veteran Affairs; 2011.
10. Weeks WB, Wallace AE, Wang S, Lee A, Kazis LE. Rural-urban disparities in health-related quality of life within disease categories of Veterans. J Rural Health. 2006;22(3):204-211.
11. Wallace AE, Weeks WB, Wang S, Lee AF, Kazis LE. Rural and urban disparities in health-related quality of life among veterans with psychiatric disorders. Psychiatr Serv. 2006;57(6):851-856.
12. Meit M, Knudson A, Gilbert T, et al; Rural Health Reform Policy Research Center. The 2014 update of the rural-urban chartbook. https://ruralhealth.und .edu/projects/health-reform-policy-research-center/pdf/2014-rural-urban-chartbook-update.pdf. Published October 2014. Accessed April 18, 2017.
13. Jackson BE, Suzuki S, Coultas D, et al. Safety-net facilities and hospitalization rates of chronic obstructive pulmonary disease: a cross-sectional analysis of the 2007 Texas Health Care Information Council inpatient data. Int J Chron Obstruct Pulmon Dis. 2011;6:563-571.
14. Jackson BE, Suzuki S, Lo K, et al. Geographic disparity in COPD hospitalization rates among the Texas population. Respir Med. 2011;105(5):734-739.15. Skinner HG, Blanchard J, Elixhauser A. Trends in emergency department visits, 2006-2011: statistical brief #179. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb179-Emergency-Department-Trends.pdf. Published September 2014. Accessed May 9, 2017.
16. Jackson BE, Coultas DB, Suzuki S, Singh KP, Bae S. Rural-urban disparities in quality of life among patients with COPD. J Rural Health. 2013;29(suppl 1):62S-69S.
17. Singh GK, Siahpush M. Widening rural-urban disparities in life expectancy, U.S., 1969-2009. Am J Prev Med. 2014;46(2):e19-e29.
18. Abrams TE, Vaughan-Sarrazin M, Fan VS, Kaboli PJ. Geographic isolation and the risk for chronic obstructive pulmonary disease-related mortality: a cohort study. Ann Intern Med. 2011;155(2):80-86.
19. Hart LG, Larson EH, Lishner DM. Rural definitions for health policy and research. Am J Public Health. 2005;95(7):1149-1155.
20. U.S. Department of Agriculture, Economic Research Service. Rural-urban commuting area codes. https://www.ers.usda.gov/data-products/rural-urban-commuting-area-codes.aspx#U20K1F50H0A. Updated October 12, 2016. Accessed May 9, 2017.
21. Joo MJ, Lee TA, Weiss KB. Geographic variation of spirometry use in newly diagnosed COPD. Chest. 2008;134(1):38-45.
22. Damarla M, Celli BR, Mullerova HX, Pinto-Plata VM. Discrepancy in the use of confirmatory tests in patients hospitalized with the diagnosis of chronic obstructive pulmonary disease or congestive heart failure. Respir Care. 2006;51(10):1120-1124.
23. Grohskopf LA, Sokolow LZ, Olsen SJ, Bresee JS, Broder KR, Karron RA. Prevention and control of influenza with vaccines: recommendations of the advisory committee on immunization practices, United States, 2015-16 influenza season. MMWR Morb Mortal Wkly Rep. 2015;64(30):818-825.
24. Kim DK, Bridges CB, Harriman KH; Advisory Committee on Immunization Practices. Advisory committee on immunization practices recommended immunization schedule for adults aged 19 years or older: United States, 2016. Ann Intern Med. 2016;164(3):184-194.
25. Cimen P, Unlu M, Kirakli C, et al. Should patients with COPD be vaccinated? Respir Care. 2015;60(2):239-243.
26. Mowls DS, Cheruvu VK, Zullo MD. Influenza vaccination in adults with chronic obstructive pulmonary disease: the impact of a diagnostic breathing test on vaccination rates PLoS One. 2013;8(6):e67600.
27. Shoup JA, Madrid C, Koehler C, et al. Effectiveness and cost of influenza vaccine reminders for adults with asthma or chronic obstructive pulmonary disease. Am J Manag Care. 2015;21(7):e405-e413.
28. Arinez-Fernandez MC, Carrasco-Garrido P, Garcia-Carballo M, Hernandez-Barrera V, de Miguel AG, Jimenez-Garcia R. Determinants of pneumococcal vaccination among patients with chronic obstructive pulmonary disease in Spain. Hum Vaccin. 2006;2(3):99-104.29. Qaseem A, Wilt TJ, Weinberger SE, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med. 2011;155(3):179-191.
30. Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(10):1005-1012.
31. Goodridge D, Lawson J, Rennie D, Marciniuk D. Rural/urban differences in health care utilization and place of death for persons with respiratory illness in the last year of life. Rural Remote Health. 2010;10(2):1349.
32. Wang Y, Hu K, Liu K, et al. Obstructive sleep apnea exacerbates airway inflammation in patients with chronic obstructive pulmonary disease. Sleep Med. 2015;16(9):1123-1130.
33. Marin JM, Soriano JB, Carrizo SJ, Boldova A, Celli BR. Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am J Respir Crit Care Med. 2010;182(3):325-331
1. Blackwell DL, Lucas JW, Clarke TC. Summary health statistics for U.S. adults: national health interview survey, 2012. Vital Health Stat 10. 2014(260):1-161.
2. Xu J, Murphy SL, Kochanek KD, Bastian BA. Deaths: final data for 2013. Natl Vital Stat Rep. 2016;64(2):1-119.
3. Celli BR, MacNee W, Force AET. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932-946.
4. Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS; GOLD Scientific Committee. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163(5):1256-1276.
5. Ghattas C, Dai A, Gemmel DJ, Awad MH. Over diagnosis of chronic obstructive pulmonary disease in an underserved patient population. Int J Chron Obstruct Pulmon Dis. 2013;8:545-549.
6. Zwar NA, Marks GB, Hermiz O, et al. Predictors of accuracy of diagnosis of chronic obstructive pulmonary disease in general practice. Med J Aust. 2011;195(4):168-171.
7. Lopez-Campos JL, Abad Arranz M, Calero-Acuña C, et al. Guideline adherence in outpatient clinics for chronic obstructive pulmonary disease: results from a clinical audit. PLoS One. 2016;11(3):e0151896.
8. McGlynn EA, Asch SM, Adams J, et al. The quality of health care delivered to adults in the United States. N Engl J Med. 2003;348(26):2635-2645.
9. Spoont M, Greer N, Su J, Fitzgerald P, Rutks I, Wilt TJ. Rural vs. Urban Ambulatory Health Care: A Systematic Review. Washington, DC: U.S. Department of Veteran Affairs; 2011.
10. Weeks WB, Wallace AE, Wang S, Lee A, Kazis LE. Rural-urban disparities in health-related quality of life within disease categories of Veterans. J Rural Health. 2006;22(3):204-211.
11. Wallace AE, Weeks WB, Wang S, Lee AF, Kazis LE. Rural and urban disparities in health-related quality of life among veterans with psychiatric disorders. Psychiatr Serv. 2006;57(6):851-856.
12. Meit M, Knudson A, Gilbert T, et al; Rural Health Reform Policy Research Center. The 2014 update of the rural-urban chartbook. https://ruralhealth.und .edu/projects/health-reform-policy-research-center/pdf/2014-rural-urban-chartbook-update.pdf. Published October 2014. Accessed April 18, 2017.
13. Jackson BE, Suzuki S, Coultas D, et al. Safety-net facilities and hospitalization rates of chronic obstructive pulmonary disease: a cross-sectional analysis of the 2007 Texas Health Care Information Council inpatient data. Int J Chron Obstruct Pulmon Dis. 2011;6:563-571.
14. Jackson BE, Suzuki S, Lo K, et al. Geographic disparity in COPD hospitalization rates among the Texas population. Respir Med. 2011;105(5):734-739.15. Skinner HG, Blanchard J, Elixhauser A. Trends in emergency department visits, 2006-2011: statistical brief #179. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb179-Emergency-Department-Trends.pdf. Published September 2014. Accessed May 9, 2017.
16. Jackson BE, Coultas DB, Suzuki S, Singh KP, Bae S. Rural-urban disparities in quality of life among patients with COPD. J Rural Health. 2013;29(suppl 1):62S-69S.
17. Singh GK, Siahpush M. Widening rural-urban disparities in life expectancy, U.S., 1969-2009. Am J Prev Med. 2014;46(2):e19-e29.
18. Abrams TE, Vaughan-Sarrazin M, Fan VS, Kaboli PJ. Geographic isolation and the risk for chronic obstructive pulmonary disease-related mortality: a cohort study. Ann Intern Med. 2011;155(2):80-86.
19. Hart LG, Larson EH, Lishner DM. Rural definitions for health policy and research. Am J Public Health. 2005;95(7):1149-1155.
20. U.S. Department of Agriculture, Economic Research Service. Rural-urban commuting area codes. https://www.ers.usda.gov/data-products/rural-urban-commuting-area-codes.aspx#U20K1F50H0A. Updated October 12, 2016. Accessed May 9, 2017.
21. Joo MJ, Lee TA, Weiss KB. Geographic variation of spirometry use in newly diagnosed COPD. Chest. 2008;134(1):38-45.
22. Damarla M, Celli BR, Mullerova HX, Pinto-Plata VM. Discrepancy in the use of confirmatory tests in patients hospitalized with the diagnosis of chronic obstructive pulmonary disease or congestive heart failure. Respir Care. 2006;51(10):1120-1124.
23. Grohskopf LA, Sokolow LZ, Olsen SJ, Bresee JS, Broder KR, Karron RA. Prevention and control of influenza with vaccines: recommendations of the advisory committee on immunization practices, United States, 2015-16 influenza season. MMWR Morb Mortal Wkly Rep. 2015;64(30):818-825.
24. Kim DK, Bridges CB, Harriman KH; Advisory Committee on Immunization Practices. Advisory committee on immunization practices recommended immunization schedule for adults aged 19 years or older: United States, 2016. Ann Intern Med. 2016;164(3):184-194.
25. Cimen P, Unlu M, Kirakli C, et al. Should patients with COPD be vaccinated? Respir Care. 2015;60(2):239-243.
26. Mowls DS, Cheruvu VK, Zullo MD. Influenza vaccination in adults with chronic obstructive pulmonary disease: the impact of a diagnostic breathing test on vaccination rates PLoS One. 2013;8(6):e67600.
27. Shoup JA, Madrid C, Koehler C, et al. Effectiveness and cost of influenza vaccine reminders for adults with asthma or chronic obstructive pulmonary disease. Am J Manag Care. 2015;21(7):e405-e413.
28. Arinez-Fernandez MC, Carrasco-Garrido P, Garcia-Carballo M, Hernandez-Barrera V, de Miguel AG, Jimenez-Garcia R. Determinants of pneumococcal vaccination among patients with chronic obstructive pulmonary disease in Spain. Hum Vaccin. 2006;2(3):99-104.29. Qaseem A, Wilt TJ, Weinberger SE, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med. 2011;155(3):179-191.
30. Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(10):1005-1012.
31. Goodridge D, Lawson J, Rennie D, Marciniuk D. Rural/urban differences in health care utilization and place of death for persons with respiratory illness in the last year of life. Rural Remote Health. 2010;10(2):1349.
32. Wang Y, Hu K, Liu K, et al. Obstructive sleep apnea exacerbates airway inflammation in patients with chronic obstructive pulmonary disease. Sleep Med. 2015;16(9):1123-1130.
33. Marin JM, Soriano JB, Carrizo SJ, Boldova A, Celli BR. Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am J Respir Crit Care Med. 2010;182(3):325-331
Cutaneous Myoepithelial Carcinoma With Disseminated Metastases
Cutaneous myoepithelial tumors are rare neoplasms but are being increasingly recognized and reported in the literature.1-7 Myoepithelial tumors are related to benign mixed tumors of the skin but lack the epithelial ductules that are present in mixed tumors. Cutaneous myoepithelial tumors may show a variety of architectural, cytological, and stromal features. Their immunophenotype usually is characterized by coexpression of an epithelial marker (eg, keratin, epithelial membrane antigen [EMA]) and S-100 protein; they also may express a variety of other myoepithelial markers, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin.7 EWS RNA binding protein 1 (EWSR1) and pleomorphic adenoma gene 1 (PLAG1) gene rearrangement has been detected in subsets of these tumors on in situ hybridization.8-10
Malignant myoepithelial tumors of the skin, also referred to as cutaneous myoepithelial carcinomas, are exceedingly rare. Including the current case, a search of PubMed articles indexed for MEDLINE and Google Scholar using the terms myoepithelial carcinoma and cutaneous revealed 12 cases that have been reported in the literature (Table).1-7,11-13 These tumors often occur in the head and neck areas and the lower extremities and display a bimodal age distribution, generally occurring in patients younger than 21 years and older than 50 years of age; they also show a slight female predominance. Available follow-up data from the literature have shown local recurrence or metastasis in 3 cases3,4,6; however, in one case the metastatic focus was not histologically identified.4 Cutaneous myoepithelial carcinoma presenting with metastatic disease further limits treatment options. Here, we describe a case of metastatic cutaneous myoepithelial carcinoma in a 47-year-old man, a rare example of cutaneous myoepithelial carcinoma with histologically documented metastatic disease at the initial presentation.
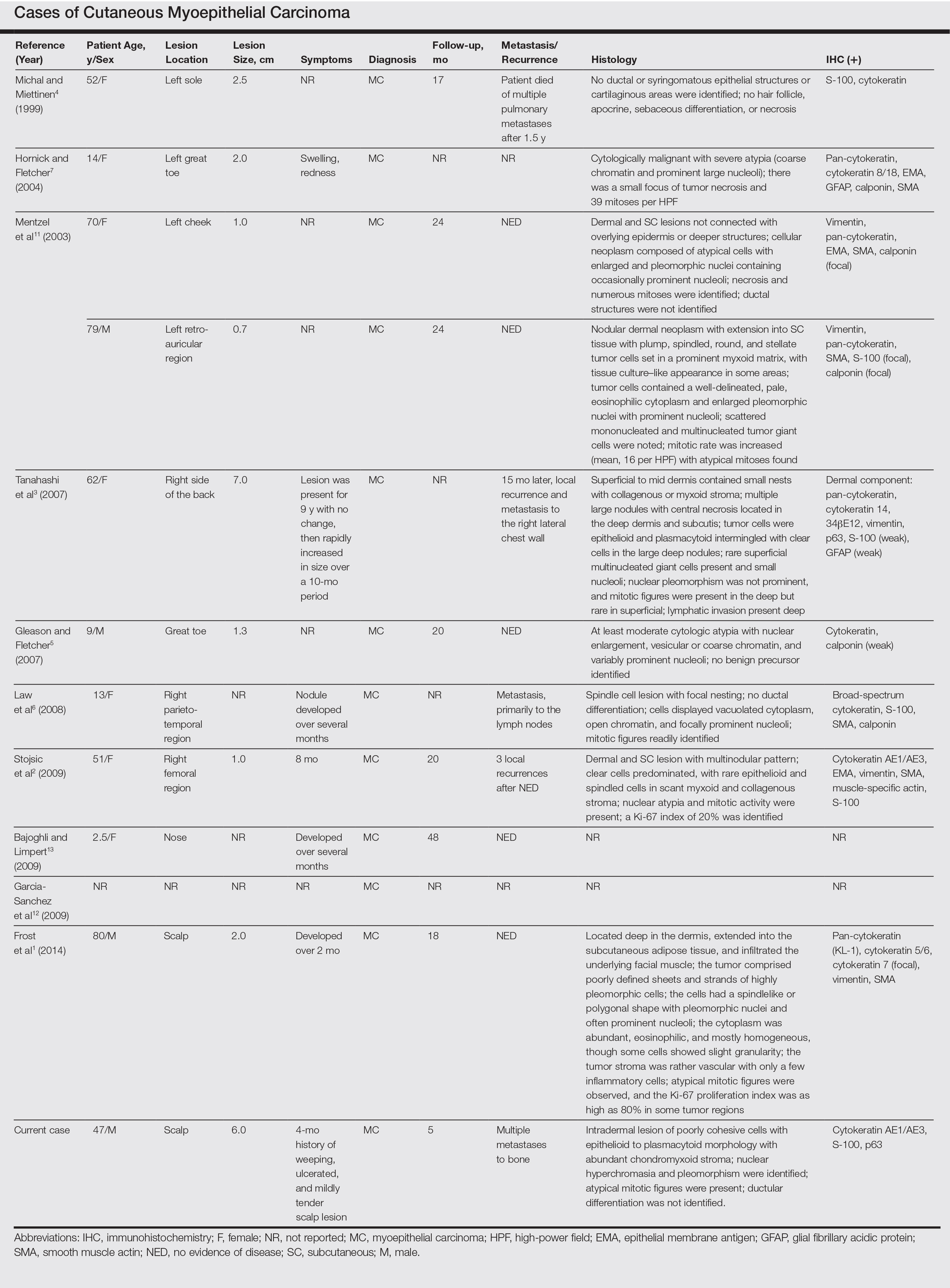
Case Report
A 47-year-old man who underwent a renal transplant 19 years prior presented with a weeping, ulcerated, mildly tender lesion on the scalp of 4 months’ duration with neck and back pain of 3 months’ duration. Physical examination demonstrated a 6-cm area of ulceration on the anterior crown of the scalp with adjacent enlarged keratoacanthomalike craters and satellite nodules (Figure 1). He was previously diagnosed with basal cell carcinoma (BCC) of the scalp at an outside institution 4 years prior and was treated with radiation therapy. The prior scalp biopsy for BCC diagnosis was unavailable for review. The patient had a history of chronic eczematous dermatitis in the waistband area that had been present for 19 years and another BCC with nodular and infiltrative patterns on the left helix. Of note, he also had been taking long-term immunosuppressant medications (ie, cyclosporine, azathioprine) for maintenance following the renal transplant.
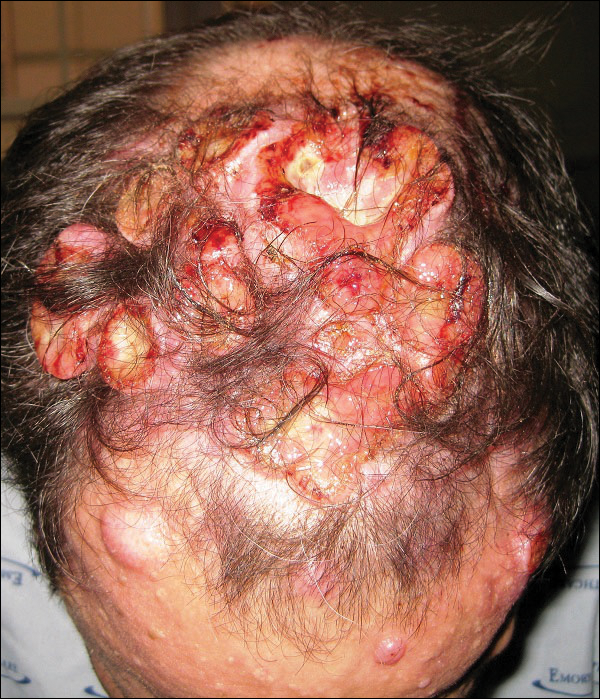
Because of the extensive ulceration of the primary lesion, a shave biopsy of the scalp was performed on an adjacent satellite nodule. Histopathologic findings showed an intradermal neoplasm characterized by poorly cohesive cells exhibiting epithelioid to plasmacytoid morphologic features surrounded by abundant chondromyxoid stroma. Ductular differentiation was not identified (Figure 2A). The neoplastic cells displayed hyperchromatic nuclei with marked nuclear pleomorphism and atypical mitotic figures (Figure 2B). On immunohistochemistry the tumor cells stained positive for cytokeratin AE1/AE3 (Figure 3), S-100 protein (Figure 4), and p63, and were negative for calponin, desmin, melan-A, cytokeratin 7, and brachyury (Figure 5).
Radiographic imaging was performed due to the patient’s history of neck and back pain. Magnetic resonance imaging showed innumerable slightly expansile, T1-hypointense, T2-hyperintense, and robustly enhancing lesions involving the cervical, thoracic, lumbar, and sacral spine, as well as the thoracic ribs and bilateral iliac bones. There was no evidence of soft tissue tumor around the bone lesions. Ventral cervical spinal cord compression was detected at the C4 vertebra, causing a symptomatic radiculopathy; however, due to widely metastatic disease, the patient was not considered appropriate for neurosurgical intervention of the compression. Computerized tomography of the chest, abdomen, and pelvis did not identify any visceral source of malignancy, though multiple bilaterally enlarged cervical lymph nodes were identified on magnetic resonance imaging.
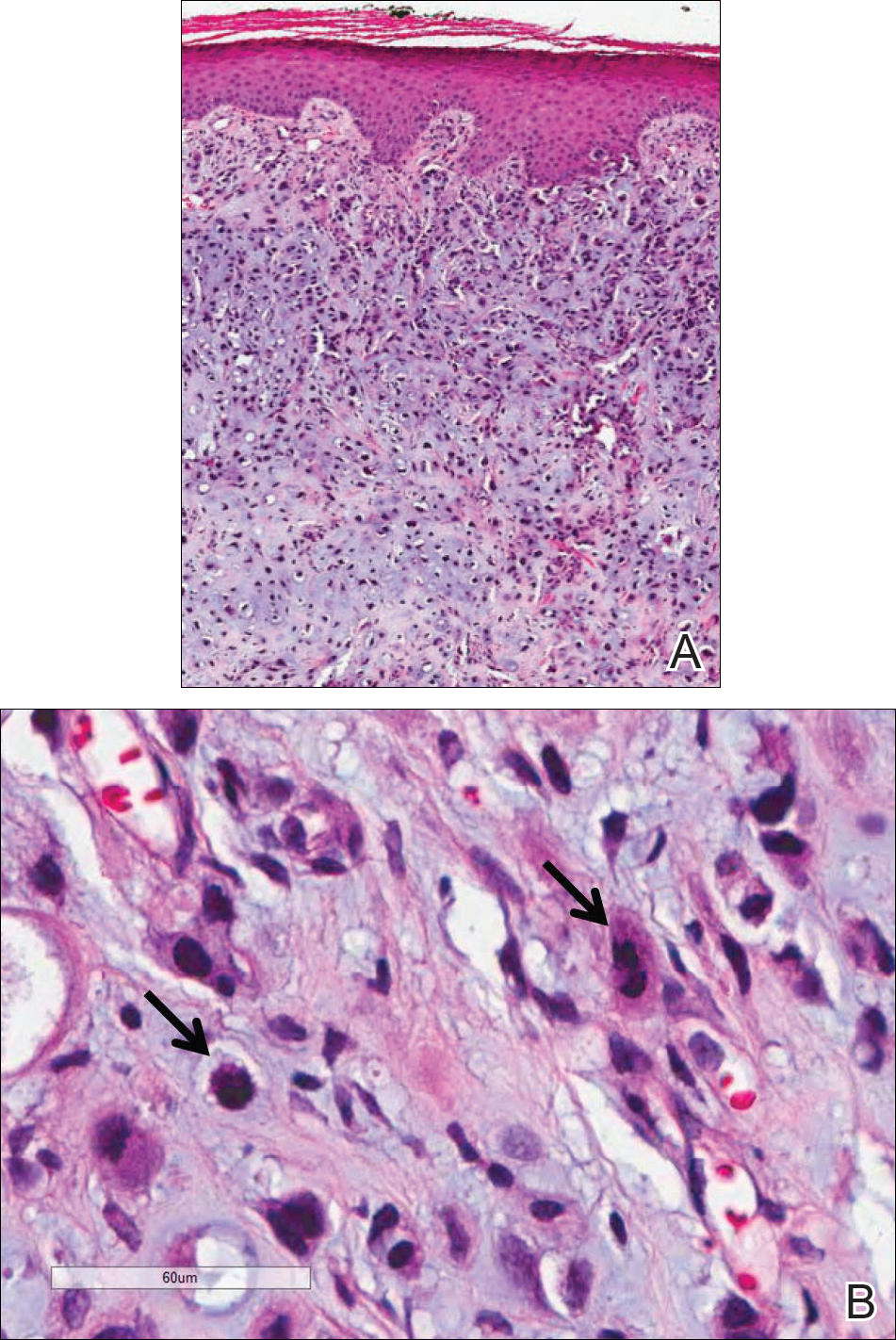
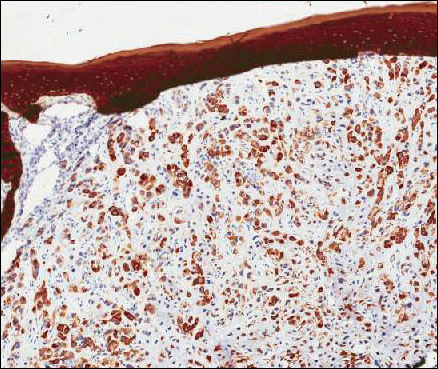
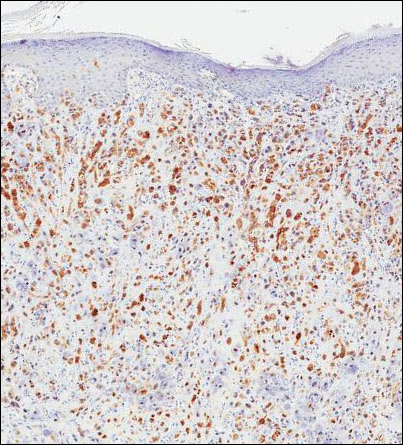

Fine needle aspiration of a left iliac bone lesion demonstrated neoplastic cells and chondromyxoid stroma essentially identical to the features shown in the skin biopsy (Figure 6). Given the morphologic features of the tumor and coexpression of cytokeratin and S-100 protein, the findings were interpreted as primary cutaneous myoepithelial carcinoma with disseminated metastatic lesions. The patient began treatment with carboplatin and paclitaxel chemotherapy. To combat the symptomatic bone pain and upper extremity radiculopathy, palliative radiation was administered to the cervical spine, lumbar spine, and right sacrum (30 Gy to each site in 10 fractions at 3 Gy per fraction). Despite the attempted chemotherapy and radiation, the patient continued to decline, and after 2 months, he elected to pursue palliative care. The patient died after 3 months in palliative care (5 months after the initial presentation).
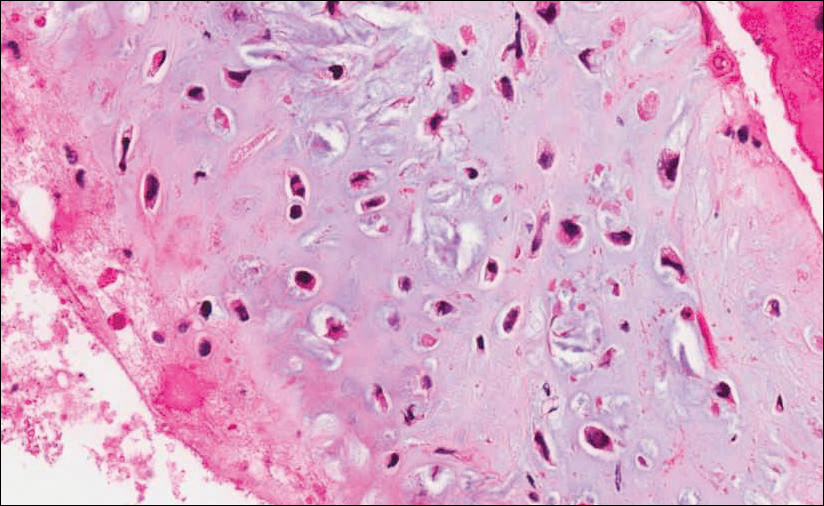
Comment
Myoepithelial cells normally surround ducts in secretory organs, such as the breasts, salivary glands, and cutaneous sweat glands. Myoepithelial neoplasms are well recognized in the salivary glands14,15; however, myoepithelial neoplasms also can arise in other sites, including the soft tissue4,5,16-18 and skin.1-3,7,11,19,20 Myoepithelioma of soft tissue was first described by Burke et al21 in 1995 and later described in the skin by Fernandez-Figueras et al22 in 1998. Since then, diagnostic criteria for cutaneous myoepithelial neoplasms have evolved, suggesting a spectrum of disease rather than a single distinct entity.11 Most often, cutaneous myoepithelial carcinomas arise as soft nodular lesions in the head and neck areas or extremities of adults. The nodules typically are nontender and range in size from 0.5 to 18.0 cm. Our review of the literature revealed 11 additional cases of cutaneous myoepithelial carcinomas have been reported, ranging in size from 0.7 to 7.0 cm (Table). In our case, the main lesion was 6 cm, mildly tender, ulcerated, and accompanied by satellite nodules.
Histologically, cutaneous myoepithelial tumors typically are well-defined, dermal-based nodules with no connection to the overlying epidermis. Similar to myoepithelial tumors of other sites, they can be diagnostically challenging due to the heterogeneity of both their architectural and cytological features. The presence of a chondromyxoid or hyalinized stroma is common but not always present. Neoplastic myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show growth patterns in clusters, cords, glands, or sheets. Focal epithelial cells can be present. Although benign myoepithelial neoplasms with overt ductal differentiation are consistent with cutaneous mixed tumors (chondroid syringomas), those without ducts are characterized as myoepitheliomas. It is uncertain if cases with only focal ductal differentiation should be classified as mixed tumors or as myoepitheliomas. Malignant myoepithelial tumors show infiltrative borders, nuclear pleomorphism, coarse nuclear chromatin, prominent nucleoli, and increased mitotic activity. A 2003 study by Hornick and Fletcher16 found that cytologic atypia was the primary predictor of malignant behavior for myoepithelial neoplasms of the soft tissue.
Despite a wide variety of expression patterns, immunohistochemistry is critical in demonstrating myoepithelial differentiation and establishing a diagnosis of a myoepithelial neoplasm. Most cases display coexpression of epithelial markers, including keratins and/or EMA as well as S-100 protein. Myogenic markers also may be variably expressed; however, the absence of myogenic markers does not exclude the diagnosis of a myoepithelial tumor. Commonly expressed epithelial markers are cytokeratin AE1/AE3, cytokeratin 8/18, and EMA, while commonly expressed myogenic markers include muscle specific actin and smooth muscle actin.5,7,11,19 Myoepithelial tumors also may express calponin, p63, and glial fibrillary acidic protein.16
Molecular studies also can aid in the diagnosis of myoepithelial tumors. A study by Antonescu et al8 demonstrated EWSR1 gene rearrangement in 45% (30/66) of extrasalivary myoepithelial tumors and the absence of EWSR1 gene rearrangement in salivary gland myoepithelial tumors. The authors also showed that EWSR1-negative tumors were more likely to be superficially located, display ductal differentiation, and possess a benign clinical course.8 In another study, Bahrami et al23 suggested that a subset of mixed tumors, specifically those with tubuloductal differentiation, are genetically linked to salivary gland pleomorphic adenomas, which was achieved by the coexpression of the PLAG1 protein and PLAG1 gene rearrangement on immunohistochemistry and fluorescence in situ hybridization (FISH), respectively. Of the 19 cases evaluated, 11 (58%) expressed nuclear staining for PLAG1 immunohistochemistry; 8 of those 11 showed positive gene rearrangement for PLAG1 using FISH. These findings raise the possibility that cutaneous mixed tumors may be more closely related to those of the salivary glands, while deep myoepithelial tumors that lack ductal differentiation may represent a distinct group. Similar to the study by Antonescu et al,8 Flucke et al10 investigated EWSR1 gene rearrangement but limited their sample to cutaneous tumors, including myoepitheliomas, mixed tumors, and myoepithelial carcinoma. The authors found that 44% of cases (7/16) expressed EWSR1; this expression suggests that cutaneous myoepithelial tumors may have a genetic relationship to their soft tissue, bone, and visceral counterparts.10
Myoepithelial tumors display a broad spectrum of morphologic features; however, one of the most common growth patterns is that of oval to round cells forming cords and chains in a chondromyxoid stroma. As such, the histopathologic differential diagnosis for myoepithelial tumors includes other epithelioid or round-cell neoplasms with similar growth patterns including extraskeletal myxoid chondrosarcoma (EMC), ossifying fibromyxoid tumor of soft parts, and extra-axial soft tissue chordoma. Extraskeletal myxoid chondrosarcoma bears the closest similarity to myoepithelial tumors both histologically and by ancillary studies. It typically possesses cords or chains of small round tumor cells set in a chondromyxoid or myxoid background. In contrast to myoepithelial tumors, which typically have more abundant cytoplasm and can show at least focal areas of spindle cell growth, the cells of EMC are more uniform, small, round cells with relatively scant cytoplasm. Extraskeletal myxoid chondrosarcomas lack the typical myoepithelial coexpression of cytokeratin and S-100 protein, with a minority of EMCs expressing S-100 protein but rarely cytokeratin. Most cases of EMC possess a balanced t(9;22) translocation involving the EWSR1 gene,24 a finding that could lead to confusion with soft tissue myoepithelial tumors, which also may show EWSR1 rearrangement on FISH. Ossifying fibromyxoid tumor of soft parts is also composed of round cells arranged in cords in a myxoid or fibrous stroma; the majority of cases also display a peripheral rim of mature bone, a feature that is not typically seen in myoepithelial tumors. Similar to myoepithelial tumors, ossifying fibromyxoid tumor of soft parts often is positive for S-100 protein; however, it rarely is positive for cytokeratins. Ossifying fibromyxoid tumor of soft parts has been shown to have a rearrangement of the PHD finger protein 1 (PHF1) gene in approximately half of cases, a molecular finding that has not been reported for myoepithelial tumors.25 Finally, extra-axial soft tissue chordomas, though quite rare, may possess striking similarities to myoepithelial tumors both histopathologically and immunohistochemically. Chordomas are composed of epithelioid cells arranged in nests, nodules, and chains with a variably myxoid background. A variable amount of cells with bubbly cytoplasm (known as physaliphorous cells) can be seen. High mitotic activity is not a characteristic feature in chordomas. They classically coexpress cytokeratins and S-100 protein, similar to myoepithelial tumors.
Because cutaneous myoepithelial tumors are relatively rare, a well-defined standard of care for treatment is lacking. Surgical excision is the primary treatment method in most reported cases in the literature.17,19 Miller et al29 reported the successful treatment of recurrent cutaneous myoepitheliomas with Mohs micrographic surgery. Chemotherapy may be useful in the setting of metastatic myoepithelial carcinomas in adults, but reported results are inconsistent.30,31 Radiation treatment of recurrent or metastatic disease has not been shown to be effective. A study of children treated with surgical resection and chemotherapy using ifosfamide, cisplatin, and etoposide followed by radiation therapy showed positive results.32
Our case highlights several challenges that may arise in establishing a diagnosis of cutaneous myoepithelial carcinoma with disseminated metastases. The diagnostic difficulty in our case was compounded by the advanced nature of the lesion at the time of presentation. Given the rarity of metastatic cutaneous myoepithelial carcinomas and the lack of a prior primary diagnosis of a malignant myoepithelioma, the index of suspicion for this entity was not high. A report of myoepithelial carcinoma of the parotid gland metastatic to the skin has been reported,33 but in the absence of salivary gland involvement or other visceral lesions, metastasis from any source other than our patient’s cutaneous scalp lesion is unlikely. The histopathologic features in combination with the characteristic immunophenotype, unique clinical setting, and radiographic findings were essential to arriving at the correct diagnosis. Unlike previously reported metastatic lesions, our case is unique in that metastatic lesions were identified at the time of initial clinical presentation.
Conclusion
Cutaneous myoepithelial carcinomas are exceedingly rare tumors with a wide range of histopathologic and immunohistochemical findings. In challenging cases, studies for EWSR1 or PLAG1 gene rearrangement can be helpful. Furthermore, this case illustrates the potential for widespread dissemination of myoepithelial carcinomas requiring clinical evaluation and imaging studies to exclude metastatic lesions.
- Frost MW, Steiniche T, Damsgaard TE, et al. Primary cutaneous myoepithelial carcinoma: a case report and review of the literature. APMIS. 2014;122:369-379.
- Stojsic Z, Brasanac D, Boricic I, et al. Clear cell myoepithelial carcinoma of the skin. a case report. J Cutan Pathol. 2009;36:680-683.
- Tanahashi J, Kashima K, Daa T, et al. A case of cutaneous myoepithelial carcinoma. J Cutan Pathol. 2007;34:648-653.
- Michal M, Miettinen M. Myoepitheliomas of the skin and soft tissues. report of 12 cases. Virchows Arch. 1999;434:393-400.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Law RM, Viglione MP, Barrett TL. Metastatic myoepithelial carcinoma in a child. J Cutan Pathol. 2008;35:779-781.
- Hornick JL, Fletcher CD. Cutaneous myoepithelioma: a clinicopathologic and immunohistochemical study of 14 cases. Hum Pathol. 2004;35:14-24.
- Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. a molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114-1124.
- Antonescu CR, Zhang L, Shao SY, et al. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer. 2013;52:675-682.
- Flucke U, Palmedo G, Blankenhorn N, et al. EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: a study of 18 cases. Mod Pathol. 2011;24:1444-1450.
- Mentzel T, Requena L, Kaddu S, et al. Cutaneous myoepithelial neoplasms: clinicopathologic and immunohistochemical study of 20 cases suggesting a continuous spectrum ranging from benign mixed tumor of the skin to cutaneous myoepithelioma and myoepithelial carcinoma. J Cutan Pathol. 2003;30:294-302.
- Garcia-Sanchez S, Elices M, Nieto S. Cutaneous myoepithelial carcinoma (malignant myoepithelial tumor of skin). Virchows Archiv. 2009;455(suppl 1):1-482.
- Bajoghli A, Limpert J. Treatment of cutaneous malignant myoepithelioma on the nasal ala using Mohs micrographic surgery in a two and a half year old child. J Invest Dermatol. 2009;129:S44.
- Prasad AR, Savera AT, Gown AM, et al. The myoepithelial immunophenotype in 135 benign and malignant salivary gland tumors other than pleomorphic adenoma. Arch Pathol Lab Med. 1999;123:801-806.
- Savera AT, Sloman A, Huvos AG, et al. Myoepithelial carcinoma of the salivary glands. a clinicopathologic study of 25 patients. Am J Surg Pathol. 2000;24:761-774.
- Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2003;27:1183-1196.
- Kilpatrick SE, Hitchcock MG, Kraus MD, et al. Mixed tumors and myoepitheliomas of soft tissue: a clinicopathologic study of 19 cases with a unifying concept. Am J Surg Pathol. 1997;21:13-22.
- Neto AG, Pineda-Daboin K, Luna MA. Myoepithelioma of the soft tissue of the head and neck: a case report and review of the literature. Head Neck. 2004;26:470-473.
- Kutzner H, Mentzel T, Kaddu S, et al. Cutaneous myoepithelioma: an under-recognized cutaneous neoplasm composed of myoepithelial cells. Am J Surg Pathol. 2001;25:348-355.
- Dix BT, Hentges MJ, Saltrick KR, et al. Cutaneous myoepithelioma in the foot: case report. Foot Ankle Spec. 2013;6:239-241.
- Burke T, Sahin A, Johnson DE, et al. Myoepithelioma of the retroperitoneum. Ultrastruct Pathol. 1995;19:269-274.
- Fernandez-Figueras MT, Puig L, Trias I, et al. Benign myoepithelioma of the skin. Am J Dermatopathol. 1998;20:208-212.
- Bahrami A, Dalton JD, Krane JF, et al. A subset of cutaneous and soft tissue mixed tumors are genetically linked to their salivary gland counterpart. Genes Chromosomes Cancer. 2012;51:140-148.
- Panagopoulos I, Mertens F, Isaksson M, et al. Molecular genetic characterization of the EWS/CHN and RBP56/CHN fusion genes in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2002;35:340-352.
- Graham RP, Weiss SW, Sukov WR, et al. PHF1 rearrangements in ossifying fibromyxoid tumors of soft parts: a fluorescence in situ hybridization study of 41 cases with emphasis on the malignant variant. Am J Surg Pathol. 2013;37:1751-1755.
- Dabska M. Parachordoma: a new clinicopathologic entity. Cancer. 1977;40:1586-1592.
- Fletcher CDM, Mertens F, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002.
- Lauer SR, Edgar MA, Gardner JM, et al. Soft tissue chordomas: a clinicopathologic analysis of 11 cases. Am J Surg Pathol. 2013;37:719-726.
- Miller TD, McCalmont T, Tope WD. Recurrent cutaneous myoepithelioma treated using Mohs micrographic surgery: case report and review of the literature. Dermatol Surg. 2009;35:139-143.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Noronha V, Cooper DL, Higgins SA, et al. Metastatic myoepithelial carcinoma of the vulva treated with carboplatin and paclitaxel. Lancet Oncol. 2006;7:270-271.
- Bisogno G, Tagarelli A, Schiavetti A, et al. Myoepithelial carcinoma treatment in children: a report from the TREP project. Pediatr Blood Cancer. 2014;61:643-646.
- He DQ, Hua CG, Tang XF, et al. Cutaneous metastasis from a parotid myoepithelial carcinoma: a case report and review of the literature. J Cutan Pathol. 2008;35:1138-1143.
Cutaneous myoepithelial tumors are rare neoplasms but are being increasingly recognized and reported in the literature.1-7 Myoepithelial tumors are related to benign mixed tumors of the skin but lack the epithelial ductules that are present in mixed tumors. Cutaneous myoepithelial tumors may show a variety of architectural, cytological, and stromal features. Their immunophenotype usually is characterized by coexpression of an epithelial marker (eg, keratin, epithelial membrane antigen [EMA]) and S-100 protein; they also may express a variety of other myoepithelial markers, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin.7 EWS RNA binding protein 1 (EWSR1) and pleomorphic adenoma gene 1 (PLAG1) gene rearrangement has been detected in subsets of these tumors on in situ hybridization.8-10
Malignant myoepithelial tumors of the skin, also referred to as cutaneous myoepithelial carcinomas, are exceedingly rare. Including the current case, a search of PubMed articles indexed for MEDLINE and Google Scholar using the terms myoepithelial carcinoma and cutaneous revealed 12 cases that have been reported in the literature (Table).1-7,11-13 These tumors often occur in the head and neck areas and the lower extremities and display a bimodal age distribution, generally occurring in patients younger than 21 years and older than 50 years of age; they also show a slight female predominance. Available follow-up data from the literature have shown local recurrence or metastasis in 3 cases3,4,6; however, in one case the metastatic focus was not histologically identified.4 Cutaneous myoepithelial carcinoma presenting with metastatic disease further limits treatment options. Here, we describe a case of metastatic cutaneous myoepithelial carcinoma in a 47-year-old man, a rare example of cutaneous myoepithelial carcinoma with histologically documented metastatic disease at the initial presentation.
Case Report
A 47-year-old man who underwent a renal transplant 19 years prior presented with a weeping, ulcerated, mildly tender lesion on the scalp of 4 months’ duration with neck and back pain of 3 months’ duration. Physical examination demonstrated a 6-cm area of ulceration on the anterior crown of the scalp with adjacent enlarged keratoacanthomalike craters and satellite nodules (Figure 1). He was previously diagnosed with basal cell carcinoma (BCC) of the scalp at an outside institution 4 years prior and was treated with radiation therapy. The prior scalp biopsy for BCC diagnosis was unavailable for review. The patient had a history of chronic eczematous dermatitis in the waistband area that had been present for 19 years and another BCC with nodular and infiltrative patterns on the left helix. Of note, he also had been taking long-term immunosuppressant medications (ie, cyclosporine, azathioprine) for maintenance following the renal transplant.
Because of the extensive ulceration of the primary lesion, a shave biopsy of the scalp was performed on an adjacent satellite nodule. Histopathologic findings showed an intradermal neoplasm characterized by poorly cohesive cells exhibiting epithelioid to plasmacytoid morphologic features surrounded by abundant chondromyxoid stroma. Ductular differentiation was not identified (Figure 2A). The neoplastic cells displayed hyperchromatic nuclei with marked nuclear pleomorphism and atypical mitotic figures (Figure 2B). On immunohistochemistry the tumor cells stained positive for cytokeratin AE1/AE3 (Figure 3), S-100 protein (Figure 4), and p63, and were negative for calponin, desmin, melan-A, cytokeratin 7, and brachyury (Figure 5).
Radiographic imaging was performed due to the patient’s history of neck and back pain. Magnetic resonance imaging showed innumerable slightly expansile, T1-hypointense, T2-hyperintense, and robustly enhancing lesions involving the cervical, thoracic, lumbar, and sacral spine, as well as the thoracic ribs and bilateral iliac bones. There was no evidence of soft tissue tumor around the bone lesions. Ventral cervical spinal cord compression was detected at the C4 vertebra, causing a symptomatic radiculopathy; however, due to widely metastatic disease, the patient was not considered appropriate for neurosurgical intervention of the compression. Computerized tomography of the chest, abdomen, and pelvis did not identify any visceral source of malignancy, though multiple bilaterally enlarged cervical lymph nodes were identified on magnetic resonance imaging.
Fine needle aspiration of a left iliac bone lesion demonstrated neoplastic cells and chondromyxoid stroma essentially identical to the features shown in the skin biopsy (Figure 6). Given the morphologic features of the tumor and coexpression of cytokeratin and S-100 protein, the findings were interpreted as primary cutaneous myoepithelial carcinoma with disseminated metastatic lesions. The patient began treatment with carboplatin and paclitaxel chemotherapy. To combat the symptomatic bone pain and upper extremity radiculopathy, palliative radiation was administered to the cervical spine, lumbar spine, and right sacrum (30 Gy to each site in 10 fractions at 3 Gy per fraction). Despite the attempted chemotherapy and radiation, the patient continued to decline, and after 2 months, he elected to pursue palliative care. The patient died after 3 months in palliative care (5 months after the initial presentation).
Comment
Myoepithelial cells normally surround ducts in secretory organs, such as the breasts, salivary glands, and cutaneous sweat glands. Myoepithelial neoplasms are well recognized in the salivary glands14,15; however, myoepithelial neoplasms also can arise in other sites, including the soft tissue4,5,16-18 and skin.1-3,7,11,19,20 Myoepithelioma of soft tissue was first described by Burke et al21 in 1995 and later described in the skin by Fernandez-Figueras et al22 in 1998. Since then, diagnostic criteria for cutaneous myoepithelial neoplasms have evolved, suggesting a spectrum of disease rather than a single distinct entity.11 Most often, cutaneous myoepithelial carcinomas arise as soft nodular lesions in the head and neck areas or extremities of adults. The nodules typically are nontender and range in size from 0.5 to 18.0 cm. Our review of the literature revealed 11 additional cases of cutaneous myoepithelial carcinomas have been reported, ranging in size from 0.7 to 7.0 cm (Table). In our case, the main lesion was 6 cm, mildly tender, ulcerated, and accompanied by satellite nodules.
Histologically, cutaneous myoepithelial tumors typically are well-defined, dermal-based nodules with no connection to the overlying epidermis. Similar to myoepithelial tumors of other sites, they can be diagnostically challenging due to the heterogeneity of both their architectural and cytological features. The presence of a chondromyxoid or hyalinized stroma is common but not always present. Neoplastic myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show growth patterns in clusters, cords, glands, or sheets. Focal epithelial cells can be present. Although benign myoepithelial neoplasms with overt ductal differentiation are consistent with cutaneous mixed tumors (chondroid syringomas), those without ducts are characterized as myoepitheliomas. It is uncertain if cases with only focal ductal differentiation should be classified as mixed tumors or as myoepitheliomas. Malignant myoepithelial tumors show infiltrative borders, nuclear pleomorphism, coarse nuclear chromatin, prominent nucleoli, and increased mitotic activity. A 2003 study by Hornick and Fletcher16 found that cytologic atypia was the primary predictor of malignant behavior for myoepithelial neoplasms of the soft tissue.
Despite a wide variety of expression patterns, immunohistochemistry is critical in demonstrating myoepithelial differentiation and establishing a diagnosis of a myoepithelial neoplasm. Most cases display coexpression of epithelial markers, including keratins and/or EMA as well as S-100 protein. Myogenic markers also may be variably expressed; however, the absence of myogenic markers does not exclude the diagnosis of a myoepithelial tumor. Commonly expressed epithelial markers are cytokeratin AE1/AE3, cytokeratin 8/18, and EMA, while commonly expressed myogenic markers include muscle specific actin and smooth muscle actin.5,7,11,19 Myoepithelial tumors also may express calponin, p63, and glial fibrillary acidic protein.16
Molecular studies also can aid in the diagnosis of myoepithelial tumors. A study by Antonescu et al8 demonstrated EWSR1 gene rearrangement in 45% (30/66) of extrasalivary myoepithelial tumors and the absence of EWSR1 gene rearrangement in salivary gland myoepithelial tumors. The authors also showed that EWSR1-negative tumors were more likely to be superficially located, display ductal differentiation, and possess a benign clinical course.8 In another study, Bahrami et al23 suggested that a subset of mixed tumors, specifically those with tubuloductal differentiation, are genetically linked to salivary gland pleomorphic adenomas, which was achieved by the coexpression of the PLAG1 protein and PLAG1 gene rearrangement on immunohistochemistry and fluorescence in situ hybridization (FISH), respectively. Of the 19 cases evaluated, 11 (58%) expressed nuclear staining for PLAG1 immunohistochemistry; 8 of those 11 showed positive gene rearrangement for PLAG1 using FISH. These findings raise the possibility that cutaneous mixed tumors may be more closely related to those of the salivary glands, while deep myoepithelial tumors that lack ductal differentiation may represent a distinct group. Similar to the study by Antonescu et al,8 Flucke et al10 investigated EWSR1 gene rearrangement but limited their sample to cutaneous tumors, including myoepitheliomas, mixed tumors, and myoepithelial carcinoma. The authors found that 44% of cases (7/16) expressed EWSR1; this expression suggests that cutaneous myoepithelial tumors may have a genetic relationship to their soft tissue, bone, and visceral counterparts.10
Myoepithelial tumors display a broad spectrum of morphologic features; however, one of the most common growth patterns is that of oval to round cells forming cords and chains in a chondromyxoid stroma. As such, the histopathologic differential diagnosis for myoepithelial tumors includes other epithelioid or round-cell neoplasms with similar growth patterns including extraskeletal myxoid chondrosarcoma (EMC), ossifying fibromyxoid tumor of soft parts, and extra-axial soft tissue chordoma. Extraskeletal myxoid chondrosarcoma bears the closest similarity to myoepithelial tumors both histologically and by ancillary studies. It typically possesses cords or chains of small round tumor cells set in a chondromyxoid or myxoid background. In contrast to myoepithelial tumors, which typically have more abundant cytoplasm and can show at least focal areas of spindle cell growth, the cells of EMC are more uniform, small, round cells with relatively scant cytoplasm. Extraskeletal myxoid chondrosarcomas lack the typical myoepithelial coexpression of cytokeratin and S-100 protein, with a minority of EMCs expressing S-100 protein but rarely cytokeratin. Most cases of EMC possess a balanced t(9;22) translocation involving the EWSR1 gene,24 a finding that could lead to confusion with soft tissue myoepithelial tumors, which also may show EWSR1 rearrangement on FISH. Ossifying fibromyxoid tumor of soft parts is also composed of round cells arranged in cords in a myxoid or fibrous stroma; the majority of cases also display a peripheral rim of mature bone, a feature that is not typically seen in myoepithelial tumors. Similar to myoepithelial tumors, ossifying fibromyxoid tumor of soft parts often is positive for S-100 protein; however, it rarely is positive for cytokeratins. Ossifying fibromyxoid tumor of soft parts has been shown to have a rearrangement of the PHD finger protein 1 (PHF1) gene in approximately half of cases, a molecular finding that has not been reported for myoepithelial tumors.25 Finally, extra-axial soft tissue chordomas, though quite rare, may possess striking similarities to myoepithelial tumors both histopathologically and immunohistochemically. Chordomas are composed of epithelioid cells arranged in nests, nodules, and chains with a variably myxoid background. A variable amount of cells with bubbly cytoplasm (known as physaliphorous cells) can be seen. High mitotic activity is not a characteristic feature in chordomas. They classically coexpress cytokeratins and S-100 protein, similar to myoepithelial tumors.
Because cutaneous myoepithelial tumors are relatively rare, a well-defined standard of care for treatment is lacking. Surgical excision is the primary treatment method in most reported cases in the literature.17,19 Miller et al29 reported the successful treatment of recurrent cutaneous myoepitheliomas with Mohs micrographic surgery. Chemotherapy may be useful in the setting of metastatic myoepithelial carcinomas in adults, but reported results are inconsistent.30,31 Radiation treatment of recurrent or metastatic disease has not been shown to be effective. A study of children treated with surgical resection and chemotherapy using ifosfamide, cisplatin, and etoposide followed by radiation therapy showed positive results.32
Our case highlights several challenges that may arise in establishing a diagnosis of cutaneous myoepithelial carcinoma with disseminated metastases. The diagnostic difficulty in our case was compounded by the advanced nature of the lesion at the time of presentation. Given the rarity of metastatic cutaneous myoepithelial carcinomas and the lack of a prior primary diagnosis of a malignant myoepithelioma, the index of suspicion for this entity was not high. A report of myoepithelial carcinoma of the parotid gland metastatic to the skin has been reported,33 but in the absence of salivary gland involvement or other visceral lesions, metastasis from any source other than our patient’s cutaneous scalp lesion is unlikely. The histopathologic features in combination with the characteristic immunophenotype, unique clinical setting, and radiographic findings were essential to arriving at the correct diagnosis. Unlike previously reported metastatic lesions, our case is unique in that metastatic lesions were identified at the time of initial clinical presentation.
Conclusion
Cutaneous myoepithelial carcinomas are exceedingly rare tumors with a wide range of histopathologic and immunohistochemical findings. In challenging cases, studies for EWSR1 or PLAG1 gene rearrangement can be helpful. Furthermore, this case illustrates the potential for widespread dissemination of myoepithelial carcinomas requiring clinical evaluation and imaging studies to exclude metastatic lesions.
Cutaneous myoepithelial tumors are rare neoplasms but are being increasingly recognized and reported in the literature.1-7 Myoepithelial tumors are related to benign mixed tumors of the skin but lack the epithelial ductules that are present in mixed tumors. Cutaneous myoepithelial tumors may show a variety of architectural, cytological, and stromal features. Their immunophenotype usually is characterized by coexpression of an epithelial marker (eg, keratin, epithelial membrane antigen [EMA]) and S-100 protein; they also may express a variety of other myoepithelial markers, including keratins, smooth muscle actin, calponin, glial fibrillary acidic protein, p63, and desmin.7 EWS RNA binding protein 1 (EWSR1) and pleomorphic adenoma gene 1 (PLAG1) gene rearrangement has been detected in subsets of these tumors on in situ hybridization.8-10
Malignant myoepithelial tumors of the skin, also referred to as cutaneous myoepithelial carcinomas, are exceedingly rare. Including the current case, a search of PubMed articles indexed for MEDLINE and Google Scholar using the terms myoepithelial carcinoma and cutaneous revealed 12 cases that have been reported in the literature (Table).1-7,11-13 These tumors often occur in the head and neck areas and the lower extremities and display a bimodal age distribution, generally occurring in patients younger than 21 years and older than 50 years of age; they also show a slight female predominance. Available follow-up data from the literature have shown local recurrence or metastasis in 3 cases3,4,6; however, in one case the metastatic focus was not histologically identified.4 Cutaneous myoepithelial carcinoma presenting with metastatic disease further limits treatment options. Here, we describe a case of metastatic cutaneous myoepithelial carcinoma in a 47-year-old man, a rare example of cutaneous myoepithelial carcinoma with histologically documented metastatic disease at the initial presentation.
Case Report
A 47-year-old man who underwent a renal transplant 19 years prior presented with a weeping, ulcerated, mildly tender lesion on the scalp of 4 months’ duration with neck and back pain of 3 months’ duration. Physical examination demonstrated a 6-cm area of ulceration on the anterior crown of the scalp with adjacent enlarged keratoacanthomalike craters and satellite nodules (Figure 1). He was previously diagnosed with basal cell carcinoma (BCC) of the scalp at an outside institution 4 years prior and was treated with radiation therapy. The prior scalp biopsy for BCC diagnosis was unavailable for review. The patient had a history of chronic eczematous dermatitis in the waistband area that had been present for 19 years and another BCC with nodular and infiltrative patterns on the left helix. Of note, he also had been taking long-term immunosuppressant medications (ie, cyclosporine, azathioprine) for maintenance following the renal transplant.
Because of the extensive ulceration of the primary lesion, a shave biopsy of the scalp was performed on an adjacent satellite nodule. Histopathologic findings showed an intradermal neoplasm characterized by poorly cohesive cells exhibiting epithelioid to plasmacytoid morphologic features surrounded by abundant chondromyxoid stroma. Ductular differentiation was not identified (Figure 2A). The neoplastic cells displayed hyperchromatic nuclei with marked nuclear pleomorphism and atypical mitotic figures (Figure 2B). On immunohistochemistry the tumor cells stained positive for cytokeratin AE1/AE3 (Figure 3), S-100 protein (Figure 4), and p63, and were negative for calponin, desmin, melan-A, cytokeratin 7, and brachyury (Figure 5).
Radiographic imaging was performed due to the patient’s history of neck and back pain. Magnetic resonance imaging showed innumerable slightly expansile, T1-hypointense, T2-hyperintense, and robustly enhancing lesions involving the cervical, thoracic, lumbar, and sacral spine, as well as the thoracic ribs and bilateral iliac bones. There was no evidence of soft tissue tumor around the bone lesions. Ventral cervical spinal cord compression was detected at the C4 vertebra, causing a symptomatic radiculopathy; however, due to widely metastatic disease, the patient was not considered appropriate for neurosurgical intervention of the compression. Computerized tomography of the chest, abdomen, and pelvis did not identify any visceral source of malignancy, though multiple bilaterally enlarged cervical lymph nodes were identified on magnetic resonance imaging.
Fine needle aspiration of a left iliac bone lesion demonstrated neoplastic cells and chondromyxoid stroma essentially identical to the features shown in the skin biopsy (Figure 6). Given the morphologic features of the tumor and coexpression of cytokeratin and S-100 protein, the findings were interpreted as primary cutaneous myoepithelial carcinoma with disseminated metastatic lesions. The patient began treatment with carboplatin and paclitaxel chemotherapy. To combat the symptomatic bone pain and upper extremity radiculopathy, palliative radiation was administered to the cervical spine, lumbar spine, and right sacrum (30 Gy to each site in 10 fractions at 3 Gy per fraction). Despite the attempted chemotherapy and radiation, the patient continued to decline, and after 2 months, he elected to pursue palliative care. The patient died after 3 months in palliative care (5 months after the initial presentation).
Comment
Myoepithelial cells normally surround ducts in secretory organs, such as the breasts, salivary glands, and cutaneous sweat glands. Myoepithelial neoplasms are well recognized in the salivary glands14,15; however, myoepithelial neoplasms also can arise in other sites, including the soft tissue4,5,16-18 and skin.1-3,7,11,19,20 Myoepithelioma of soft tissue was first described by Burke et al21 in 1995 and later described in the skin by Fernandez-Figueras et al22 in 1998. Since then, diagnostic criteria for cutaneous myoepithelial neoplasms have evolved, suggesting a spectrum of disease rather than a single distinct entity.11 Most often, cutaneous myoepithelial carcinomas arise as soft nodular lesions in the head and neck areas or extremities of adults. The nodules typically are nontender and range in size from 0.5 to 18.0 cm. Our review of the literature revealed 11 additional cases of cutaneous myoepithelial carcinomas have been reported, ranging in size from 0.7 to 7.0 cm (Table). In our case, the main lesion was 6 cm, mildly tender, ulcerated, and accompanied by satellite nodules.
Histologically, cutaneous myoepithelial tumors typically are well-defined, dermal-based nodules with no connection to the overlying epidermis. Similar to myoepithelial tumors of other sites, they can be diagnostically challenging due to the heterogeneity of both their architectural and cytological features. The presence of a chondromyxoid or hyalinized stroma is common but not always present. Neoplastic myoepithelial cells can exhibit spindled, epithelioid, plasmacytoid, or clear cell morphologic features and show growth patterns in clusters, cords, glands, or sheets. Focal epithelial cells can be present. Although benign myoepithelial neoplasms with overt ductal differentiation are consistent with cutaneous mixed tumors (chondroid syringomas), those without ducts are characterized as myoepitheliomas. It is uncertain if cases with only focal ductal differentiation should be classified as mixed tumors or as myoepitheliomas. Malignant myoepithelial tumors show infiltrative borders, nuclear pleomorphism, coarse nuclear chromatin, prominent nucleoli, and increased mitotic activity. A 2003 study by Hornick and Fletcher16 found that cytologic atypia was the primary predictor of malignant behavior for myoepithelial neoplasms of the soft tissue.
Despite a wide variety of expression patterns, immunohistochemistry is critical in demonstrating myoepithelial differentiation and establishing a diagnosis of a myoepithelial neoplasm. Most cases display coexpression of epithelial markers, including keratins and/or EMA as well as S-100 protein. Myogenic markers also may be variably expressed; however, the absence of myogenic markers does not exclude the diagnosis of a myoepithelial tumor. Commonly expressed epithelial markers are cytokeratin AE1/AE3, cytokeratin 8/18, and EMA, while commonly expressed myogenic markers include muscle specific actin and smooth muscle actin.5,7,11,19 Myoepithelial tumors also may express calponin, p63, and glial fibrillary acidic protein.16
Molecular studies also can aid in the diagnosis of myoepithelial tumors. A study by Antonescu et al8 demonstrated EWSR1 gene rearrangement in 45% (30/66) of extrasalivary myoepithelial tumors and the absence of EWSR1 gene rearrangement in salivary gland myoepithelial tumors. The authors also showed that EWSR1-negative tumors were more likely to be superficially located, display ductal differentiation, and possess a benign clinical course.8 In another study, Bahrami et al23 suggested that a subset of mixed tumors, specifically those with tubuloductal differentiation, are genetically linked to salivary gland pleomorphic adenomas, which was achieved by the coexpression of the PLAG1 protein and PLAG1 gene rearrangement on immunohistochemistry and fluorescence in situ hybridization (FISH), respectively. Of the 19 cases evaluated, 11 (58%) expressed nuclear staining for PLAG1 immunohistochemistry; 8 of those 11 showed positive gene rearrangement for PLAG1 using FISH. These findings raise the possibility that cutaneous mixed tumors may be more closely related to those of the salivary glands, while deep myoepithelial tumors that lack ductal differentiation may represent a distinct group. Similar to the study by Antonescu et al,8 Flucke et al10 investigated EWSR1 gene rearrangement but limited their sample to cutaneous tumors, including myoepitheliomas, mixed tumors, and myoepithelial carcinoma. The authors found that 44% of cases (7/16) expressed EWSR1; this expression suggests that cutaneous myoepithelial tumors may have a genetic relationship to their soft tissue, bone, and visceral counterparts.10
Myoepithelial tumors display a broad spectrum of morphologic features; however, one of the most common growth patterns is that of oval to round cells forming cords and chains in a chondromyxoid stroma. As such, the histopathologic differential diagnosis for myoepithelial tumors includes other epithelioid or round-cell neoplasms with similar growth patterns including extraskeletal myxoid chondrosarcoma (EMC), ossifying fibromyxoid tumor of soft parts, and extra-axial soft tissue chordoma. Extraskeletal myxoid chondrosarcoma bears the closest similarity to myoepithelial tumors both histologically and by ancillary studies. It typically possesses cords or chains of small round tumor cells set in a chondromyxoid or myxoid background. In contrast to myoepithelial tumors, which typically have more abundant cytoplasm and can show at least focal areas of spindle cell growth, the cells of EMC are more uniform, small, round cells with relatively scant cytoplasm. Extraskeletal myxoid chondrosarcomas lack the typical myoepithelial coexpression of cytokeratin and S-100 protein, with a minority of EMCs expressing S-100 protein but rarely cytokeratin. Most cases of EMC possess a balanced t(9;22) translocation involving the EWSR1 gene,24 a finding that could lead to confusion with soft tissue myoepithelial tumors, which also may show EWSR1 rearrangement on FISH. Ossifying fibromyxoid tumor of soft parts is also composed of round cells arranged in cords in a myxoid or fibrous stroma; the majority of cases also display a peripheral rim of mature bone, a feature that is not typically seen in myoepithelial tumors. Similar to myoepithelial tumors, ossifying fibromyxoid tumor of soft parts often is positive for S-100 protein; however, it rarely is positive for cytokeratins. Ossifying fibromyxoid tumor of soft parts has been shown to have a rearrangement of the PHD finger protein 1 (PHF1) gene in approximately half of cases, a molecular finding that has not been reported for myoepithelial tumors.25 Finally, extra-axial soft tissue chordomas, though quite rare, may possess striking similarities to myoepithelial tumors both histopathologically and immunohistochemically. Chordomas are composed of epithelioid cells arranged in nests, nodules, and chains with a variably myxoid background. A variable amount of cells with bubbly cytoplasm (known as physaliphorous cells) can be seen. High mitotic activity is not a characteristic feature in chordomas. They classically coexpress cytokeratins and S-100 protein, similar to myoepithelial tumors.
Because cutaneous myoepithelial tumors are relatively rare, a well-defined standard of care for treatment is lacking. Surgical excision is the primary treatment method in most reported cases in the literature.17,19 Miller et al29 reported the successful treatment of recurrent cutaneous myoepitheliomas with Mohs micrographic surgery. Chemotherapy may be useful in the setting of metastatic myoepithelial carcinomas in adults, but reported results are inconsistent.30,31 Radiation treatment of recurrent or metastatic disease has not been shown to be effective. A study of children treated with surgical resection and chemotherapy using ifosfamide, cisplatin, and etoposide followed by radiation therapy showed positive results.32
Our case highlights several challenges that may arise in establishing a diagnosis of cutaneous myoepithelial carcinoma with disseminated metastases. The diagnostic difficulty in our case was compounded by the advanced nature of the lesion at the time of presentation. Given the rarity of metastatic cutaneous myoepithelial carcinomas and the lack of a prior primary diagnosis of a malignant myoepithelioma, the index of suspicion for this entity was not high. A report of myoepithelial carcinoma of the parotid gland metastatic to the skin has been reported,33 but in the absence of salivary gland involvement or other visceral lesions, metastasis from any source other than our patient’s cutaneous scalp lesion is unlikely. The histopathologic features in combination with the characteristic immunophenotype, unique clinical setting, and radiographic findings were essential to arriving at the correct diagnosis. Unlike previously reported metastatic lesions, our case is unique in that metastatic lesions were identified at the time of initial clinical presentation.
Conclusion
Cutaneous myoepithelial carcinomas are exceedingly rare tumors with a wide range of histopathologic and immunohistochemical findings. In challenging cases, studies for EWSR1 or PLAG1 gene rearrangement can be helpful. Furthermore, this case illustrates the potential for widespread dissemination of myoepithelial carcinomas requiring clinical evaluation and imaging studies to exclude metastatic lesions.
- Frost MW, Steiniche T, Damsgaard TE, et al. Primary cutaneous myoepithelial carcinoma: a case report and review of the literature. APMIS. 2014;122:369-379.
- Stojsic Z, Brasanac D, Boricic I, et al. Clear cell myoepithelial carcinoma of the skin. a case report. J Cutan Pathol. 2009;36:680-683.
- Tanahashi J, Kashima K, Daa T, et al. A case of cutaneous myoepithelial carcinoma. J Cutan Pathol. 2007;34:648-653.
- Michal M, Miettinen M. Myoepitheliomas of the skin and soft tissues. report of 12 cases. Virchows Arch. 1999;434:393-400.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Law RM, Viglione MP, Barrett TL. Metastatic myoepithelial carcinoma in a child. J Cutan Pathol. 2008;35:779-781.
- Hornick JL, Fletcher CD. Cutaneous myoepithelioma: a clinicopathologic and immunohistochemical study of 14 cases. Hum Pathol. 2004;35:14-24.
- Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. a molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114-1124.
- Antonescu CR, Zhang L, Shao SY, et al. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer. 2013;52:675-682.
- Flucke U, Palmedo G, Blankenhorn N, et al. EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: a study of 18 cases. Mod Pathol. 2011;24:1444-1450.
- Mentzel T, Requena L, Kaddu S, et al. Cutaneous myoepithelial neoplasms: clinicopathologic and immunohistochemical study of 20 cases suggesting a continuous spectrum ranging from benign mixed tumor of the skin to cutaneous myoepithelioma and myoepithelial carcinoma. J Cutan Pathol. 2003;30:294-302.
- Garcia-Sanchez S, Elices M, Nieto S. Cutaneous myoepithelial carcinoma (malignant myoepithelial tumor of skin). Virchows Archiv. 2009;455(suppl 1):1-482.
- Bajoghli A, Limpert J. Treatment of cutaneous malignant myoepithelioma on the nasal ala using Mohs micrographic surgery in a two and a half year old child. J Invest Dermatol. 2009;129:S44.
- Prasad AR, Savera AT, Gown AM, et al. The myoepithelial immunophenotype in 135 benign and malignant salivary gland tumors other than pleomorphic adenoma. Arch Pathol Lab Med. 1999;123:801-806.
- Savera AT, Sloman A, Huvos AG, et al. Myoepithelial carcinoma of the salivary glands. a clinicopathologic study of 25 patients. Am J Surg Pathol. 2000;24:761-774.
- Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2003;27:1183-1196.
- Kilpatrick SE, Hitchcock MG, Kraus MD, et al. Mixed tumors and myoepitheliomas of soft tissue: a clinicopathologic study of 19 cases with a unifying concept. Am J Surg Pathol. 1997;21:13-22.
- Neto AG, Pineda-Daboin K, Luna MA. Myoepithelioma of the soft tissue of the head and neck: a case report and review of the literature. Head Neck. 2004;26:470-473.
- Kutzner H, Mentzel T, Kaddu S, et al. Cutaneous myoepithelioma: an under-recognized cutaneous neoplasm composed of myoepithelial cells. Am J Surg Pathol. 2001;25:348-355.
- Dix BT, Hentges MJ, Saltrick KR, et al. Cutaneous myoepithelioma in the foot: case report. Foot Ankle Spec. 2013;6:239-241.
- Burke T, Sahin A, Johnson DE, et al. Myoepithelioma of the retroperitoneum. Ultrastruct Pathol. 1995;19:269-274.
- Fernandez-Figueras MT, Puig L, Trias I, et al. Benign myoepithelioma of the skin. Am J Dermatopathol. 1998;20:208-212.
- Bahrami A, Dalton JD, Krane JF, et al. A subset of cutaneous and soft tissue mixed tumors are genetically linked to their salivary gland counterpart. Genes Chromosomes Cancer. 2012;51:140-148.
- Panagopoulos I, Mertens F, Isaksson M, et al. Molecular genetic characterization of the EWS/CHN and RBP56/CHN fusion genes in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2002;35:340-352.
- Graham RP, Weiss SW, Sukov WR, et al. PHF1 rearrangements in ossifying fibromyxoid tumors of soft parts: a fluorescence in situ hybridization study of 41 cases with emphasis on the malignant variant. Am J Surg Pathol. 2013;37:1751-1755.
- Dabska M. Parachordoma: a new clinicopathologic entity. Cancer. 1977;40:1586-1592.
- Fletcher CDM, Mertens F, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002.
- Lauer SR, Edgar MA, Gardner JM, et al. Soft tissue chordomas: a clinicopathologic analysis of 11 cases. Am J Surg Pathol. 2013;37:719-726.
- Miller TD, McCalmont T, Tope WD. Recurrent cutaneous myoepithelioma treated using Mohs micrographic surgery: case report and review of the literature. Dermatol Surg. 2009;35:139-143.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Noronha V, Cooper DL, Higgins SA, et al. Metastatic myoepithelial carcinoma of the vulva treated with carboplatin and paclitaxel. Lancet Oncol. 2006;7:270-271.
- Bisogno G, Tagarelli A, Schiavetti A, et al. Myoepithelial carcinoma treatment in children: a report from the TREP project. Pediatr Blood Cancer. 2014;61:643-646.
- He DQ, Hua CG, Tang XF, et al. Cutaneous metastasis from a parotid myoepithelial carcinoma: a case report and review of the literature. J Cutan Pathol. 2008;35:1138-1143.
- Frost MW, Steiniche T, Damsgaard TE, et al. Primary cutaneous myoepithelial carcinoma: a case report and review of the literature. APMIS. 2014;122:369-379.
- Stojsic Z, Brasanac D, Boricic I, et al. Clear cell myoepithelial carcinoma of the skin. a case report. J Cutan Pathol. 2009;36:680-683.
- Tanahashi J, Kashima K, Daa T, et al. A case of cutaneous myoepithelial carcinoma. J Cutan Pathol. 2007;34:648-653.
- Michal M, Miettinen M. Myoepitheliomas of the skin and soft tissues. report of 12 cases. Virchows Arch. 1999;434:393-400.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Law RM, Viglione MP, Barrett TL. Metastatic myoepithelial carcinoma in a child. J Cutan Pathol. 2008;35:779-781.
- Hornick JL, Fletcher CD. Cutaneous myoepithelioma: a clinicopathologic and immunohistochemical study of 14 cases. Hum Pathol. 2004;35:14-24.
- Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. a molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114-1124.
- Antonescu CR, Zhang L, Shao SY, et al. Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer. 2013;52:675-682.
- Flucke U, Palmedo G, Blankenhorn N, et al. EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: a study of 18 cases. Mod Pathol. 2011;24:1444-1450.
- Mentzel T, Requena L, Kaddu S, et al. Cutaneous myoepithelial neoplasms: clinicopathologic and immunohistochemical study of 20 cases suggesting a continuous spectrum ranging from benign mixed tumor of the skin to cutaneous myoepithelioma and myoepithelial carcinoma. J Cutan Pathol. 2003;30:294-302.
- Garcia-Sanchez S, Elices M, Nieto S. Cutaneous myoepithelial carcinoma (malignant myoepithelial tumor of skin). Virchows Archiv. 2009;455(suppl 1):1-482.
- Bajoghli A, Limpert J. Treatment of cutaneous malignant myoepithelioma on the nasal ala using Mohs micrographic surgery in a two and a half year old child. J Invest Dermatol. 2009;129:S44.
- Prasad AR, Savera AT, Gown AM, et al. The myoepithelial immunophenotype in 135 benign and malignant salivary gland tumors other than pleomorphic adenoma. Arch Pathol Lab Med. 1999;123:801-806.
- Savera AT, Sloman A, Huvos AG, et al. Myoepithelial carcinoma of the salivary glands. a clinicopathologic study of 25 patients. Am J Surg Pathol. 2000;24:761-774.
- Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2003;27:1183-1196.
- Kilpatrick SE, Hitchcock MG, Kraus MD, et al. Mixed tumors and myoepitheliomas of soft tissue: a clinicopathologic study of 19 cases with a unifying concept. Am J Surg Pathol. 1997;21:13-22.
- Neto AG, Pineda-Daboin K, Luna MA. Myoepithelioma of the soft tissue of the head and neck: a case report and review of the literature. Head Neck. 2004;26:470-473.
- Kutzner H, Mentzel T, Kaddu S, et al. Cutaneous myoepithelioma: an under-recognized cutaneous neoplasm composed of myoepithelial cells. Am J Surg Pathol. 2001;25:348-355.
- Dix BT, Hentges MJ, Saltrick KR, et al. Cutaneous myoepithelioma in the foot: case report. Foot Ankle Spec. 2013;6:239-241.
- Burke T, Sahin A, Johnson DE, et al. Myoepithelioma of the retroperitoneum. Ultrastruct Pathol. 1995;19:269-274.
- Fernandez-Figueras MT, Puig L, Trias I, et al. Benign myoepithelioma of the skin. Am J Dermatopathol. 1998;20:208-212.
- Bahrami A, Dalton JD, Krane JF, et al. A subset of cutaneous and soft tissue mixed tumors are genetically linked to their salivary gland counterpart. Genes Chromosomes Cancer. 2012;51:140-148.
- Panagopoulos I, Mertens F, Isaksson M, et al. Molecular genetic characterization of the EWS/CHN and RBP56/CHN fusion genes in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2002;35:340-352.
- Graham RP, Weiss SW, Sukov WR, et al. PHF1 rearrangements in ossifying fibromyxoid tumors of soft parts: a fluorescence in situ hybridization study of 41 cases with emphasis on the malignant variant. Am J Surg Pathol. 2013;37:1751-1755.
- Dabska M. Parachordoma: a new clinicopathologic entity. Cancer. 1977;40:1586-1592.
- Fletcher CDM, Mertens F, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002.
- Lauer SR, Edgar MA, Gardner JM, et al. Soft tissue chordomas: a clinicopathologic analysis of 11 cases. Am J Surg Pathol. 2013;37:719-726.
- Miller TD, McCalmont T, Tope WD. Recurrent cutaneous myoepithelioma treated using Mohs micrographic surgery: case report and review of the literature. Dermatol Surg. 2009;35:139-143.
- Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813-1824.
- Noronha V, Cooper DL, Higgins SA, et al. Metastatic myoepithelial carcinoma of the vulva treated with carboplatin and paclitaxel. Lancet Oncol. 2006;7:270-271.
- Bisogno G, Tagarelli A, Schiavetti A, et al. Myoepithelial carcinoma treatment in children: a report from the TREP project. Pediatr Blood Cancer. 2014;61:643-646.
- He DQ, Hua CG, Tang XF, et al. Cutaneous metastasis from a parotid myoepithelial carcinoma: a case report and review of the literature. J Cutan Pathol. 2008;35:1138-1143.
Practice Points
- Cutaneous myoepithelial carcinoma is a rare malignant adnexal neoplasm with metastatic potential that can present in the skin.
- Cutaneous myoepithelial carcinoma is a tumor that can occasionally show EWSR1 gene rearrangement.
- Excision with negative margins and close follow-up is recommended for cutaneous myoepithelial carcinoma.
Open Navicular Dislocation With Midfoot Dissociation in a 45-Year-Old Man
Take-Home Points
- Stability of the foot is dependent on both the medial and lateral longitudinal columns; injuries to a single column alone are extremely rare.
- Midfoot fractures that are recognized and treated early have generally favorable outcomes compared to those identified in a delayed fashion.
- The most frequent complication of navicular dislocation is AVN, which is said to occur in as many as 25% of cases.
- Many specialists agree that navicular dislocations are best treated with open reduction.
- Ultimately, the goals of surgical intervention are to minimize pain and to establish stability of the plantigrade foot.
Traumatic dislocation of the tarsal navicular (especially without a navicular body fracture) is uncommon.1 The regional anatomy and ligamentous architecture confer stability to the midfoot.2-6 Navicular dislocation is part of a complex disruption involving structures in the adjacent column.6
Navicular dislocation has been associated with several bony and soft-tissue injury patterns, including comminuted intra-articular fracture of the calcaneus and associated calcaneocuboid joint subluxation; fracture and subluxation of the calcaneocuboid joint; fracture-dislocation of the calcaneocuboid joint with fractures of the third and fourth metatarsals; and a combination of fractures of the intermediate cuneiform, the second through fourth metatarsals, and the cuboid.4–11 In this article, we report a case of open complete navicular dislocation with talar head fracture and associated subtalar and calcaneocuboid subluxations in a 45-year-old man. The injury was managed with open reduction and stabilization with Kirschner wires (K-wires), which later required naviculocuneiform and intercuneiform fusion for posttraumatic avascular necrosis (AVN). The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 45-year-old man sustained blunt trauma to the right foot in a high-speed head-on collision. He was hemodynamically stable with isolated complaints of pain in the foot. Physical examination revealed a grossly open 10-cm wound extending from the heel pad medially to the dorsal surface of the navicular. The navicular was clearly visible through the wound.
Plain radiographs of the foot showed complete medial dislocation of the navicular with complete disruption of all 3 naviculocuneiform joints (Figures 1A-1C). 
On day of presentation, the patient was taken to the operating room for irrigation, débridement, reduction of the joints, and primary closure of the right foot wound. Minimal contamination was noted. Attempted gentle reduction maneuvers included distraction, adduction, and pronation of the forefoot with concomitant lateral pressure on the navicular. 

An especially prominent medial navicular was noted on postreduction films. Initially, this suggested inadequate reduction of the naviculocuneiform joints, but, on close radiographic examination of each naviculocuneiform joint and imaging of the contralateral foot, we determined that the prominence represented a type III accessory navicular, also known as a cornuate navicular. Contralateral imaging showed an identical and asymptomatic medial prominence.
After surgery, the patient was made non-weight-bearing in a splint, received intravenous antibiotics for 48 hours, and was discharged shortly thereafter. Radiographs at 3 and 6 weeks after injury showed maintenance of the reduction. K-wires were removed at 6 weeks. The patient was advanced to partial weight-bearing at 6 weeks and to full weight-bearing at 3 months.
Over succeeding months, the patient developed pain accompanied by significant midfoot deformity and was found to have navicular collapse consistent with AVN and posttraumatic arthritis (Figures 4A, 4B). 
Twenty-four months after fusion, the patient was fully ambulatory with no significant discomfort or disability. 
Discussion
The naviculocuneiform joints are important for the dissipation of loading stresses on the midfoot but provide little motion. The plantar and dorsal ligaments are thick structures that stabilize these joints, predisposing the navicular to fracture rather than isolated dislocation. The stability of the foot is dependent on both the medial and lateral longitudinal columns, and it is thought impossible to injure one column without disrupting the other.6 Several patterns of associated lateral column disruptions have been documented, including 3 cases similar to our patient’s, involving navicular dislocation with associated calcaneocuboid joint injuries.5,6,10
Authors have proposed several mechanisms accounting for navicular dislocations. In the setting of acute trauma, the navicular displaces dorsally as the result of forefoot plantar flexion and axial loading.4 A severe abduction/pronation injury leading to a midtarsal dislocation followed by a spontaneous reduction can force the navicular to dislocate medially.6 This disruption of the naviculocuneiform joint and concurrent “nutcracker” injury to the lateral column can produce an associated disruption of the calcaneocuboid joint.6 Depending on the direction of the deforming force, the forefoot can dislocate superolaterally if the force is plantar or inferolaterally if the force is dorsal. The remaining soft-tissue attachments help determine the position of the navicular. A third postulated mechanism involves a complex wringing injury to the forefoot.10Most specialists agree that navicular dislocations are best treated with open reduction.4,6 The goal of surgical intervention is to establish a stable plantigrade foot and to minimize pain. The current literature supports using either wires or screws to maintain reduction of midfoot injuries. Wires can be used for both talonavicular and naviculocuneiform fixation. Screws can be placed across the naviculocuneiform joints, as there is little normal physiologic motion through these joints.4 The talonavicular joint and the cuboid-metatarsal joints provide most of the motion in the midfoot and should not be readily fused.5 Stabilization of both columns is considered necessary to avoid complications such as subluxation and midfoot deformity.Given the postreduction stability of the lateral column in the present case, bicolumnar stabilization was not considered necessary. It is possible that subsequent collapse of the midfoot may have been attenuated in the presence of lateral fixation, but this would not necessarily have prevented complications of AVN.
Midfoot fractures that are recognized and treated early have generally favorable outcomes,5-11 though chronic pain and subsequent deformity are not uncommon. Perhaps the most frequently reported complication of navicular dislocation is AVN, which is thought to occur in approximately 25% of cases.12 AVN is a well-recognized complication of hindfoot and midfoot trauma. In the tarsal navicular, blood supply to the central-third watershed region is marginal. Small branches of the posterior tibial and dorsalis pedis arteries that supply the medial and lateral areas are readily injured. Not surprisingly, the risk for AVN is high when the dislocated bone is severely displaced.6 In some circumstances, the shared blood supply of the posterior tibialis may be the only remaining osseous supply. The tendon and its soft-tissue attachments should therefore be carefully monitored during dissection and reduction.6 In most cases, AVN of the foot manifests clinically within the first 10 months after injury, as was the case with our patient.13 AVN can result in the Charcot-like collapse of the medial column, leading to progressive midfoot plantar deformities.4 Variations of midfoot fusion are often required.4,6AVN may be difficult to differentiate from posttraumatic arthritis. These conditions can have similar clinical presentations and appearances on plain radiographs. In such situations, magnetic resonance imaging or bone scintigraphy may determine the diagnosis. Damage to the articular surface at time of injury and residual articular displacement, instability, and joint subluxation after injury are considered risk factors for the development of posttraumatic arthritis in the foot and ankle.14 Reports suggest that the severity of the damage to the articular surface is directly proportional to the degree of arthritis.14 Such damage may not be initially visible, especially in axial impaction injuries, but latent deterioration of the articular surface can occur.15 For patients with significant dislocations of the naviculocuneiform joints, some authors advocate primary and early fusion15 instead of the more conservative approach used here. Primary fusions are argued to have minimal deleterious effects on function, secondary to the absence of normal physiologic motion through the affected joints.15 However, there is relatively little published evidence on long-term outcomes in primary versus secondary naviculocuneiform fusions.
Successful treatment of midfoot fractures and dislocations requires intimate knowledge of foot and ankle anatomy and mechanics. Surgeons must be able to anticipate, identify, and counsel patients about acute and delayed complications in these already challenging injuries.
Am J Orthop. 2017;46(3):E186-E189. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Main BJ, Jowett RL. Injuries of the midtarsal joint. J Bone Jt Surg Br. 1975;57(1):89-97.
2. Pinney SJ, Sangeorzan BJ. Fractures of the tarsal bones. Orthop Clin North Am. 2001;32(1):21-33.
3. Vaishya R, Patrick JH. Isolated dorsal fracture-dislocation of tarsal navicular. Injury. 1991;22(1):47-48.
4. Early JS. Fractures and dislocations of the midfoot and forefoot. In: Bucholz WB, Heckman JD, Court-Brown C, et al, eds. Rockwood & Green’s Fractures in Adults. 6th ed. Philadelphia, PA: Lippincott; 2005:2337-2401.
5. Rao H. Complete open dislocation of the navicular: a case report. J Foot Ankle Surg. 2012;51(2):209-211.
6. Dhillon MS, Nagi ON. Total dislocation of the navicular: are they ever isolated injuries? J Bone Joint Surg Br. 1999;81(5):881-885.
7. Kollmannsberger A, De Boer P. Isolated calcaneo-cuboid dislocation: brief report. J Bone Joint Surg Br. 1989;71(2):323.
8. Randall RL, Hall RJ, Slabaugh P. An unusual midfoot dislocation: a case report. Am J Orthop. 1997;26(7):494-496.
9. Ruthman JC, Meyn NP. Isolated plantar midtarsal dislocation. Am J Emerg Med. 1988;6(6):599-601.
10. Pathria MN, Rosenstein A, Bjorkengren AG, Gershuni D, Resnick D. Isolated dislocation of the tarsal navicular: a case report. Foot Ankle. 1988;9(3):146-149.
11. Puente CA, Alaez JP, Marti DG. Tarsal fracture dislocation with plantar dislocation of the navicular: a case study. Foot Ankle Int. 1996;17(2):111-113.
12. Davis AT, Dann A, Kuldjanov D. Complete medial dislocation of the tarsal navicular without fracture: report of a rare injury. J Foot Ankle Surg. 2013;52(3):393-396.
13. Buchan CA, Pearce DH, Lau J, White LW. Imaging of postoperative avascular necrosis of the ankle and foot. Semin Musculoskelet Radiol. 2012;16(3):192-204.
14. Olson SA, Furman B, Guilak F. Joint injury and post-traumatic arthritis. HSS J. 2012;8(1):23-25.
15. Grambart S, Patel S, Schuberth JM. Naviculocuneiform dislocations treated with immediate arthrodesis: a report of 2 cases. J Foot Ankle Surg. 2005;44(3):228-235.
Take-Home Points
- Stability of the foot is dependent on both the medial and lateral longitudinal columns; injuries to a single column alone are extremely rare.
- Midfoot fractures that are recognized and treated early have generally favorable outcomes compared to those identified in a delayed fashion.
- The most frequent complication of navicular dislocation is AVN, which is said to occur in as many as 25% of cases.
- Many specialists agree that navicular dislocations are best treated with open reduction.
- Ultimately, the goals of surgical intervention are to minimize pain and to establish stability of the plantigrade foot.
Traumatic dislocation of the tarsal navicular (especially without a navicular body fracture) is uncommon.1 The regional anatomy and ligamentous architecture confer stability to the midfoot.2-6 Navicular dislocation is part of a complex disruption involving structures in the adjacent column.6
Navicular dislocation has been associated with several bony and soft-tissue injury patterns, including comminuted intra-articular fracture of the calcaneus and associated calcaneocuboid joint subluxation; fracture and subluxation of the calcaneocuboid joint; fracture-dislocation of the calcaneocuboid joint with fractures of the third and fourth metatarsals; and a combination of fractures of the intermediate cuneiform, the second through fourth metatarsals, and the cuboid.4–11 In this article, we report a case of open complete navicular dislocation with talar head fracture and associated subtalar and calcaneocuboid subluxations in a 45-year-old man. The injury was managed with open reduction and stabilization with Kirschner wires (K-wires), which later required naviculocuneiform and intercuneiform fusion for posttraumatic avascular necrosis (AVN). The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 45-year-old man sustained blunt trauma to the right foot in a high-speed head-on collision. He was hemodynamically stable with isolated complaints of pain in the foot. Physical examination revealed a grossly open 10-cm wound extending from the heel pad medially to the dorsal surface of the navicular. The navicular was clearly visible through the wound.
Plain radiographs of the foot showed complete medial dislocation of the navicular with complete disruption of all 3 naviculocuneiform joints (Figures 1A-1C).
On day of presentation, the patient was taken to the operating room for irrigation, débridement, reduction of the joints, and primary closure of the right foot wound. Minimal contamination was noted. Attempted gentle reduction maneuvers included distraction, adduction, and pronation of the forefoot with concomitant lateral pressure on the navicular.
An especially prominent medial navicular was noted on postreduction films. Initially, this suggested inadequate reduction of the naviculocuneiform joints, but, on close radiographic examination of each naviculocuneiform joint and imaging of the contralateral foot, we determined that the prominence represented a type III accessory navicular, also known as a cornuate navicular. Contralateral imaging showed an identical and asymptomatic medial prominence.
After surgery, the patient was made non-weight-bearing in a splint, received intravenous antibiotics for 48 hours, and was discharged shortly thereafter. Radiographs at 3 and 6 weeks after injury showed maintenance of the reduction. K-wires were removed at 6 weeks. The patient was advanced to partial weight-bearing at 6 weeks and to full weight-bearing at 3 months.
Over succeeding months, the patient developed pain accompanied by significant midfoot deformity and was found to have navicular collapse consistent with AVN and posttraumatic arthritis (Figures 4A, 4B).
Twenty-four months after fusion, the patient was fully ambulatory with no significant discomfort or disability.
Discussion
The naviculocuneiform joints are important for the dissipation of loading stresses on the midfoot but provide little motion. The plantar and dorsal ligaments are thick structures that stabilize these joints, predisposing the navicular to fracture rather than isolated dislocation. The stability of the foot is dependent on both the medial and lateral longitudinal columns, and it is thought impossible to injure one column without disrupting the other.6 Several patterns of associated lateral column disruptions have been documented, including 3 cases similar to our patient’s, involving navicular dislocation with associated calcaneocuboid joint injuries.5,6,10
Authors have proposed several mechanisms accounting for navicular dislocations. In the setting of acute trauma, the navicular displaces dorsally as the result of forefoot plantar flexion and axial loading.4 A severe abduction/pronation injury leading to a midtarsal dislocation followed by a spontaneous reduction can force the navicular to dislocate medially.6 This disruption of the naviculocuneiform joint and concurrent “nutcracker” injury to the lateral column can produce an associated disruption of the calcaneocuboid joint.6 Depending on the direction of the deforming force, the forefoot can dislocate superolaterally if the force is plantar or inferolaterally if the force is dorsal. The remaining soft-tissue attachments help determine the position of the navicular. A third postulated mechanism involves a complex wringing injury to the forefoot.10Most specialists agree that navicular dislocations are best treated with open reduction.4,6 The goal of surgical intervention is to establish a stable plantigrade foot and to minimize pain. The current literature supports using either wires or screws to maintain reduction of midfoot injuries. Wires can be used for both talonavicular and naviculocuneiform fixation. Screws can be placed across the naviculocuneiform joints, as there is little normal physiologic motion through these joints.4 The talonavicular joint and the cuboid-metatarsal joints provide most of the motion in the midfoot and should not be readily fused.5 Stabilization of both columns is considered necessary to avoid complications such as subluxation and midfoot deformity.Given the postreduction stability of the lateral column in the present case, bicolumnar stabilization was not considered necessary. It is possible that subsequent collapse of the midfoot may have been attenuated in the presence of lateral fixation, but this would not necessarily have prevented complications of AVN.
Midfoot fractures that are recognized and treated early have generally favorable outcomes,5-11 though chronic pain and subsequent deformity are not uncommon. Perhaps the most frequently reported complication of navicular dislocation is AVN, which is thought to occur in approximately 25% of cases.12 AVN is a well-recognized complication of hindfoot and midfoot trauma. In the tarsal navicular, blood supply to the central-third watershed region is marginal. Small branches of the posterior tibial and dorsalis pedis arteries that supply the medial and lateral areas are readily injured. Not surprisingly, the risk for AVN is high when the dislocated bone is severely displaced.6 In some circumstances, the shared blood supply of the posterior tibialis may be the only remaining osseous supply. The tendon and its soft-tissue attachments should therefore be carefully monitored during dissection and reduction.6 In most cases, AVN of the foot manifests clinically within the first 10 months after injury, as was the case with our patient.13 AVN can result in the Charcot-like collapse of the medial column, leading to progressive midfoot plantar deformities.4 Variations of midfoot fusion are often required.4,6AVN may be difficult to differentiate from posttraumatic arthritis. These conditions can have similar clinical presentations and appearances on plain radiographs. In such situations, magnetic resonance imaging or bone scintigraphy may determine the diagnosis. Damage to the articular surface at time of injury and residual articular displacement, instability, and joint subluxation after injury are considered risk factors for the development of posttraumatic arthritis in the foot and ankle.14 Reports suggest that the severity of the damage to the articular surface is directly proportional to the degree of arthritis.14 Such damage may not be initially visible, especially in axial impaction injuries, but latent deterioration of the articular surface can occur.15 For patients with significant dislocations of the naviculocuneiform joints, some authors advocate primary and early fusion15 instead of the more conservative approach used here. Primary fusions are argued to have minimal deleterious effects on function, secondary to the absence of normal physiologic motion through the affected joints.15 However, there is relatively little published evidence on long-term outcomes in primary versus secondary naviculocuneiform fusions.
Successful treatment of midfoot fractures and dislocations requires intimate knowledge of foot and ankle anatomy and mechanics. Surgeons must be able to anticipate, identify, and counsel patients about acute and delayed complications in these already challenging injuries.
Am J Orthop. 2017;46(3):E186-E189. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Stability of the foot is dependent on both the medial and lateral longitudinal columns; injuries to a single column alone are extremely rare.
- Midfoot fractures that are recognized and treated early have generally favorable outcomes compared to those identified in a delayed fashion.
- The most frequent complication of navicular dislocation is AVN, which is said to occur in as many as 25% of cases.
- Many specialists agree that navicular dislocations are best treated with open reduction.
- Ultimately, the goals of surgical intervention are to minimize pain and to establish stability of the plantigrade foot.
Traumatic dislocation of the tarsal navicular (especially without a navicular body fracture) is uncommon.1 The regional anatomy and ligamentous architecture confer stability to the midfoot.2-6 Navicular dislocation is part of a complex disruption involving structures in the adjacent column.6
Navicular dislocation has been associated with several bony and soft-tissue injury patterns, including comminuted intra-articular fracture of the calcaneus and associated calcaneocuboid joint subluxation; fracture and subluxation of the calcaneocuboid joint; fracture-dislocation of the calcaneocuboid joint with fractures of the third and fourth metatarsals; and a combination of fractures of the intermediate cuneiform, the second through fourth metatarsals, and the cuboid.4–11 In this article, we report a case of open complete navicular dislocation with talar head fracture and associated subtalar and calcaneocuboid subluxations in a 45-year-old man. The injury was managed with open reduction and stabilization with Kirschner wires (K-wires), which later required naviculocuneiform and intercuneiform fusion for posttraumatic avascular necrosis (AVN). The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 45-year-old man sustained blunt trauma to the right foot in a high-speed head-on collision. He was hemodynamically stable with isolated complaints of pain in the foot. Physical examination revealed a grossly open 10-cm wound extending from the heel pad medially to the dorsal surface of the navicular. The navicular was clearly visible through the wound.
Plain radiographs of the foot showed complete medial dislocation of the navicular with complete disruption of all 3 naviculocuneiform joints (Figures 1A-1C).
On day of presentation, the patient was taken to the operating room for irrigation, débridement, reduction of the joints, and primary closure of the right foot wound. Minimal contamination was noted. Attempted gentle reduction maneuvers included distraction, adduction, and pronation of the forefoot with concomitant lateral pressure on the navicular.
An especially prominent medial navicular was noted on postreduction films. Initially, this suggested inadequate reduction of the naviculocuneiform joints, but, on close radiographic examination of each naviculocuneiform joint and imaging of the contralateral foot, we determined that the prominence represented a type III accessory navicular, also known as a cornuate navicular. Contralateral imaging showed an identical and asymptomatic medial prominence.
After surgery, the patient was made non-weight-bearing in a splint, received intravenous antibiotics for 48 hours, and was discharged shortly thereafter. Radiographs at 3 and 6 weeks after injury showed maintenance of the reduction. K-wires were removed at 6 weeks. The patient was advanced to partial weight-bearing at 6 weeks and to full weight-bearing at 3 months.
Over succeeding months, the patient developed pain accompanied by significant midfoot deformity and was found to have navicular collapse consistent with AVN and posttraumatic arthritis (Figures 4A, 4B).
Twenty-four months after fusion, the patient was fully ambulatory with no significant discomfort or disability.
Discussion
The naviculocuneiform joints are important for the dissipation of loading stresses on the midfoot but provide little motion. The plantar and dorsal ligaments are thick structures that stabilize these joints, predisposing the navicular to fracture rather than isolated dislocation. The stability of the foot is dependent on both the medial and lateral longitudinal columns, and it is thought impossible to injure one column without disrupting the other.6 Several patterns of associated lateral column disruptions have been documented, including 3 cases similar to our patient’s, involving navicular dislocation with associated calcaneocuboid joint injuries.5,6,10
Authors have proposed several mechanisms accounting for navicular dislocations. In the setting of acute trauma, the navicular displaces dorsally as the result of forefoot plantar flexion and axial loading.4 A severe abduction/pronation injury leading to a midtarsal dislocation followed by a spontaneous reduction can force the navicular to dislocate medially.6 This disruption of the naviculocuneiform joint and concurrent “nutcracker” injury to the lateral column can produce an associated disruption of the calcaneocuboid joint.6 Depending on the direction of the deforming force, the forefoot can dislocate superolaterally if the force is plantar or inferolaterally if the force is dorsal. The remaining soft-tissue attachments help determine the position of the navicular. A third postulated mechanism involves a complex wringing injury to the forefoot.10Most specialists agree that navicular dislocations are best treated with open reduction.4,6 The goal of surgical intervention is to establish a stable plantigrade foot and to minimize pain. The current literature supports using either wires or screws to maintain reduction of midfoot injuries. Wires can be used for both talonavicular and naviculocuneiform fixation. Screws can be placed across the naviculocuneiform joints, as there is little normal physiologic motion through these joints.4 The talonavicular joint and the cuboid-metatarsal joints provide most of the motion in the midfoot and should not be readily fused.5 Stabilization of both columns is considered necessary to avoid complications such as subluxation and midfoot deformity.Given the postreduction stability of the lateral column in the present case, bicolumnar stabilization was not considered necessary. It is possible that subsequent collapse of the midfoot may have been attenuated in the presence of lateral fixation, but this would not necessarily have prevented complications of AVN.
Midfoot fractures that are recognized and treated early have generally favorable outcomes,5-11 though chronic pain and subsequent deformity are not uncommon. Perhaps the most frequently reported complication of navicular dislocation is AVN, which is thought to occur in approximately 25% of cases.12 AVN is a well-recognized complication of hindfoot and midfoot trauma. In the tarsal navicular, blood supply to the central-third watershed region is marginal. Small branches of the posterior tibial and dorsalis pedis arteries that supply the medial and lateral areas are readily injured. Not surprisingly, the risk for AVN is high when the dislocated bone is severely displaced.6 In some circumstances, the shared blood supply of the posterior tibialis may be the only remaining osseous supply. The tendon and its soft-tissue attachments should therefore be carefully monitored during dissection and reduction.6 In most cases, AVN of the foot manifests clinically within the first 10 months after injury, as was the case with our patient.13 AVN can result in the Charcot-like collapse of the medial column, leading to progressive midfoot plantar deformities.4 Variations of midfoot fusion are often required.4,6AVN may be difficult to differentiate from posttraumatic arthritis. These conditions can have similar clinical presentations and appearances on plain radiographs. In such situations, magnetic resonance imaging or bone scintigraphy may determine the diagnosis. Damage to the articular surface at time of injury and residual articular displacement, instability, and joint subluxation after injury are considered risk factors for the development of posttraumatic arthritis in the foot and ankle.14 Reports suggest that the severity of the damage to the articular surface is directly proportional to the degree of arthritis.14 Such damage may not be initially visible, especially in axial impaction injuries, but latent deterioration of the articular surface can occur.15 For patients with significant dislocations of the naviculocuneiform joints, some authors advocate primary and early fusion15 instead of the more conservative approach used here. Primary fusions are argued to have minimal deleterious effects on function, secondary to the absence of normal physiologic motion through the affected joints.15 However, there is relatively little published evidence on long-term outcomes in primary versus secondary naviculocuneiform fusions.
Successful treatment of midfoot fractures and dislocations requires intimate knowledge of foot and ankle anatomy and mechanics. Surgeons must be able to anticipate, identify, and counsel patients about acute and delayed complications in these already challenging injuries.
Am J Orthop. 2017;46(3):E186-E189. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Main BJ, Jowett RL. Injuries of the midtarsal joint. J Bone Jt Surg Br. 1975;57(1):89-97.
2. Pinney SJ, Sangeorzan BJ. Fractures of the tarsal bones. Orthop Clin North Am. 2001;32(1):21-33.
3. Vaishya R, Patrick JH. Isolated dorsal fracture-dislocation of tarsal navicular. Injury. 1991;22(1):47-48.
4. Early JS. Fractures and dislocations of the midfoot and forefoot. In: Bucholz WB, Heckman JD, Court-Brown C, et al, eds. Rockwood & Green’s Fractures in Adults. 6th ed. Philadelphia, PA: Lippincott; 2005:2337-2401.
5. Rao H. Complete open dislocation of the navicular: a case report. J Foot Ankle Surg. 2012;51(2):209-211.
6. Dhillon MS, Nagi ON. Total dislocation of the navicular: are they ever isolated injuries? J Bone Joint Surg Br. 1999;81(5):881-885.
7. Kollmannsberger A, De Boer P. Isolated calcaneo-cuboid dislocation: brief report. J Bone Joint Surg Br. 1989;71(2):323.
8. Randall RL, Hall RJ, Slabaugh P. An unusual midfoot dislocation: a case report. Am J Orthop. 1997;26(7):494-496.
9. Ruthman JC, Meyn NP. Isolated plantar midtarsal dislocation. Am J Emerg Med. 1988;6(6):599-601.
10. Pathria MN, Rosenstein A, Bjorkengren AG, Gershuni D, Resnick D. Isolated dislocation of the tarsal navicular: a case report. Foot Ankle. 1988;9(3):146-149.
11. Puente CA, Alaez JP, Marti DG. Tarsal fracture dislocation with plantar dislocation of the navicular: a case study. Foot Ankle Int. 1996;17(2):111-113.
12. Davis AT, Dann A, Kuldjanov D. Complete medial dislocation of the tarsal navicular without fracture: report of a rare injury. J Foot Ankle Surg. 2013;52(3):393-396.
13. Buchan CA, Pearce DH, Lau J, White LW. Imaging of postoperative avascular necrosis of the ankle and foot. Semin Musculoskelet Radiol. 2012;16(3):192-204.
14. Olson SA, Furman B, Guilak F. Joint injury and post-traumatic arthritis. HSS J. 2012;8(1):23-25.
15. Grambart S, Patel S, Schuberth JM. Naviculocuneiform dislocations treated with immediate arthrodesis: a report of 2 cases. J Foot Ankle Surg. 2005;44(3):228-235.
1. Main BJ, Jowett RL. Injuries of the midtarsal joint. J Bone Jt Surg Br. 1975;57(1):89-97.
2. Pinney SJ, Sangeorzan BJ. Fractures of the tarsal bones. Orthop Clin North Am. 2001;32(1):21-33.
3. Vaishya R, Patrick JH. Isolated dorsal fracture-dislocation of tarsal navicular. Injury. 1991;22(1):47-48.
4. Early JS. Fractures and dislocations of the midfoot and forefoot. In: Bucholz WB, Heckman JD, Court-Brown C, et al, eds. Rockwood & Green’s Fractures in Adults. 6th ed. Philadelphia, PA: Lippincott; 2005:2337-2401.
5. Rao H. Complete open dislocation of the navicular: a case report. J Foot Ankle Surg. 2012;51(2):209-211.
6. Dhillon MS, Nagi ON. Total dislocation of the navicular: are they ever isolated injuries? J Bone Joint Surg Br. 1999;81(5):881-885.
7. Kollmannsberger A, De Boer P. Isolated calcaneo-cuboid dislocation: brief report. J Bone Joint Surg Br. 1989;71(2):323.
8. Randall RL, Hall RJ, Slabaugh P. An unusual midfoot dislocation: a case report. Am J Orthop. 1997;26(7):494-496.
9. Ruthman JC, Meyn NP. Isolated plantar midtarsal dislocation. Am J Emerg Med. 1988;6(6):599-601.
10. Pathria MN, Rosenstein A, Bjorkengren AG, Gershuni D, Resnick D. Isolated dislocation of the tarsal navicular: a case report. Foot Ankle. 1988;9(3):146-149.
11. Puente CA, Alaez JP, Marti DG. Tarsal fracture dislocation with plantar dislocation of the navicular: a case study. Foot Ankle Int. 1996;17(2):111-113.
12. Davis AT, Dann A, Kuldjanov D. Complete medial dislocation of the tarsal navicular without fracture: report of a rare injury. J Foot Ankle Surg. 2013;52(3):393-396.
13. Buchan CA, Pearce DH, Lau J, White LW. Imaging of postoperative avascular necrosis of the ankle and foot. Semin Musculoskelet Radiol. 2012;16(3):192-204.
14. Olson SA, Furman B, Guilak F. Joint injury and post-traumatic arthritis. HSS J. 2012;8(1):23-25.
15. Grambart S, Patel S, Schuberth JM. Naviculocuneiform dislocations treated with immediate arthrodesis: a report of 2 cases. J Foot Ankle Surg. 2005;44(3):228-235.
Rare Dual Lesion: Extraskeletal Osteosarcoma Developing Within a Simple Lipoma
Take-Home Points
- Rare and histologically indistinguishable from osteosarcoma of bone.
- Most common presentation is an enlarging mass in the thigh or buttock.
- Secondary extraosseous osteosarcoma usually arises in the field of prior external beam radiation or brachytherapy.
- Radiographic pattern of mineralization is central amorphous or cloudlike.
- On cross sectional imaging, the soft-tissue mass is separate from the underlying bone and periosteum.
Aside from multiple myeloma, osteosarcoma is the most common primary malignancy of bone, but extraosseous osteosarcoma is rare and accounts for only 1% of soft-tissue sarcomas and only 4% of all osteosarcomas.1-3 Benign mesenchymal tumors, such as lipomas, are common, and they are estimated to outnumber their malignant counterparts by more than a factor of 100. However, the true ratio is unknown, as many clinically benign lipomas are not biopsied.4 Conventional lipoma is the most common lipoma and is biologically indolent. Conventional lipoma generally does not transform biologically into a more aggressive type of neoplasm—unlike atypical lipomatous tumors, which may demonstrate this type of evolution with multiple local recurrences.
This article is the first report of a case of radiation-associated extraosseous osteosarcoma that developed within a benign conventional lipoma. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
In March 2013, a 72-year-old woman presented to a general surgeon with a right thigh mass of several weeks’ duration. The patient, who had a remote history of thyroid carcinoma, underwent thyroidectomy in 1991, excision of melanoma of the chest in 1998, and resection and adjuvant external beam radiotherapy (30 fractions) for Merkel cell carcinoma of the right proximal lateral leg (malignancy images unavailable) at an outside institution in 2003. Regional lymph node dissection at the time was negative. The patient remained disease-free the next 10 years. On presentation, magnetic resonance imaging (MRI) showed a 2.2-cm mass encircled by a tumor of lipomatous tissue within the vastus intermedius muscle, adjacent to but separate from the right distal femur (Figures 1A-1C). 
Physical examination revealed marked ecchymosis of the left groin at the access site for embolization as well as massive ecchymosis and swelling along the right distal thigh, medial knee, and medial lower leg. The neurovascular structures were intact with full motor function and sensation distally, as well as normal distal pulses. No inguinal adenopathy was identified. The proximal portion of the prior radiation tattoo was at the inferior extent of the lesion on MRI.
The patient was treated with doxorubicin and ifosfamide (2 cycles) while waiting for the hematoma to shrink. Contrast-enhanced MRI showed a 2.2-cm enhancing mass with isointense T1 signal and heterogeneously hyperintense STIR (short tau inversion recovery) signal surrounded by a circumscribed nonenhancing lipomatous tumor within the vastus intermedius muscle, adjacent to the distal femoral cortex. There was no invasion of the bone, and a fat plane between the enhancing mass and the femoral cortex was identified (Figures 2A-2E). 


After 3 cycles of neoadjuvant chemotherapy with doxorubicin and ifosfamide, MRI showed a marked reduction in hematoma size, to 2.4 cm × 0.7 cm × 3.2 cm (estimated volume, ~3 mL), from 10 cm × 3.4 cm × 7.3 cm (estimated volume, ~130 mL), so the decision was made to proceed with surgery, excising the hematoma and sarcoma separately. First, wide resection of the hematoma yielded a 7-cm × 4-cm resection specimen with negative margins on frozen section. Subsequently, definitive radical resection of the tumor with wide margins yielded a 13-cm × 9-cm × 4-cm specimen. The resection specimen contained an intramuscular, mobile, encapsulated 2.0-cm × 1.5-cm × 1.0-cm mass with 2 components. The first was a tan-white solid mass containing thin deposits of calcified matrix, and the second, which surrounded the first, was composed of well-circumscribed soft yellow lobulated adipose tissue (Figure 5). 

After surgery, the patient’s dermatologist performed a shave biopsy of a lentiginous lesion anterior to the knee. Subsequently, the patient began having increasing knee pain and developed, on the lower extremity, small areas of erythema that were attributed to mild cellulitis. Four months after surgery, emergent contrast-enhanced MRI showed enhancement of thickened synovium of the knee joint (Figure 7). 
Since the lavage, the patient remained in good condition. There was no evidence of local recurrence on contrast-enhanced MRI (Figure 8), or metastases the first year, and she remained clinically free of disease the first 22 months of follow-up.
Discussion
Extraosseous osteosarcoma, typically a high-grade malignant neoplasm of the soft tissues that produces osteoid or cartilaginous matrix, is histologically indistinguishable from osteosarcoma of bone. 
Conventional lipoma, the most common subtype of lipoma, is a benign mesenchymal tumor. Other subtypes are hibernoma, fibrolipoma, angiolipoma, myelolipoma, spindle-cell lipoma, pleomorphic lipoma, and atypical lipomatous tumor.7 Atypical lipomatous tumor and well-differentiated liposarcoma are distinguished from each other by location: The World Health Organization recommends the term atypical lipomatous tumor for tumors that arise in the extremities and trunk lesions and well-differentiated liposarcoma for neoplasms that develop in the retroperitoneum, peritoneum, mediastinum, spermatic cord, and thoracic cavity.8 On PET, hypermetabolic activity is nonspecific and can be seen in malignant tumors and some benign reactive processes, such as evolving heterotopic ossification. However, simple lipomas, including those with mature ossification or dystrophic calcification, do not manifest increased FDG avidity.9
We are not aware of any published cases of extraosseous osteosarcoma arising within a conventional lipoma. A limited number of cases of coexisting conventional lipoma and spindle-cell lipoma or liposarcoma have been reported.10-13 Retroperitoneal liposarcoma with areas of dedifferentiation into osteosarcoma has also been described.14 Development of malignant fibrous histiocytoma and liposarcoma have also been reported within intraosseous lipomas.15 One theory is based on premalignancy as a biological concept as opposed to a morphologic one. In other words, lesions that may be considered morphologically benign may already have the biological phenotype for malignancy that is not yet reflected morphologically.16 However, it has been suggested that such findings may instead result from initial sampling error or histologic misdiagnosis.17,18There is a spectrum of findings on imaging studies of extraosseous osteosarcoma. Plain radiographs show a soft-tissue density with variable degrees of central calcification that reflects mineralization of deposited neoplastic bone. The pattern of calcification is characteristically amorphous or cloudlike, as opposed to the ring-and-arc observed in cartilage matrix. On CT, the soft-tissue mass of extraosseous osteosarcoma is separate from the underlying bone and periosteum—a defining characteristic that distinguishes it from conventional intramedullary and juxtacortical osteosarcoma.6 The central pattern of amorphous calcification helps to differentiate extraosseous osteosarcoma from heterotopic ossification, which characteristically demonstrates zonation, with trabecular architecture and mature cortical bone peripherally.1 Enhancement of extraskeletal osteosarcoma tends to be heterogeneous and depends on the quantity of necrosis. Extraskeletal osteosarcoma tends to be isointense on T1-weighted MRI and mildly hyperintense on T2-weighted MRI.1,6 Areas of very low signal intensity on both T1- and T2-weighted MRI may reflect mineralization.19 If intratumoral hemorrhage has occurred, there may be signal intensity of blood products of various ages.1,3 Tumors with abundant hemorrhage can be mistaken for hematoma. FDG-PET radiotracer accumulation tends to be intense peripherally with variable central activity depending on quantity of necrosis and hemorrhage.1The radiologic differential diagnosis includes myositis ossificans, chondrosseous lipoma, parosteal lipoma (ossifying variant), liposarcoma with metaplastic bone, dedifferentiated liposarcoma with osteosarcoma or chondrosarcoma component, and malignant mesenchymoma. Other common soft-tissue sarcomas, such as fibrosarcoma, leiomyosarcoma, and pleomorphic undifferentiated sarcoma, are excluded by the presence of fat within the tumor. The radiographic pattern of osteoid matrix produced by the tumor in our patient may be seen in heterotopic ossification, but the absence of mature ossification with zonation was evidence against heterotopic ossification, and microscopically it was neoplastic rather than reactive osteoid. In addition, it is possible that, because of the small size of the soft-tissue component, it was difficult to appreciate the less mature osteoid matrix peripherally. The lack of characteristic rings and arcs helps exclude benign and malignant cartilage containing neoplasms. Malignant mesenchymoma is a diagnosis of exclusion, and such tumors are usually better classified as sarcomas that have undergone heterologous differentiation.
The histologic diagnosis of extraosseous osteosarcoma requires identification of malignant mesenchymal cells that secrete neoplastic osteoid that may or may not mineralize. It is important to exclude the possibility that the malignant bone-forming tumor is part of a different type of sarcoma, the most common being dedifferentiated liposarcoma. Immunohistochemistry can be helpful in this situation, as dedifferentiated liposarcomas demonstrate nuclear expression of MDM2, CDK4, and p16, a constellation of findings rare in conventional and extraosseous osteosarcoma.20-23 Osteosarcoma has not previously been reported as arising in a lipoma; in our patient’s case, we excluded the possibility that the fatty component represented an underlying atypical lipomatous tumor/well-differentiated or dedifferentiated liposarcoma on the basis of morphology and lack of expression of MDM2, CDK4, and p16.
Although histologically identical to osteosarcoma of bone, extraosseous osteosarcoma is treated differently because of its relatively decreased chemosensitivity and radiosensitivity. Treatment tends to be focused on limb-sparing wide local excision, and local recurrence complicates about 50% of cases.1 Neoadjuvant or adjuvant treatment with radiation or chemotherapy is often provided.6 Platinum and doxorubicin chemotherapeutic agents, which are first-line treatments for osteosarcoma of bone, tend to be less effective in extraosseous osteosarcoma, and ifosfamide is more often used instead.5
Primary extraosseous osteosarcoma classically has a poor prognosis, with 2- to 3-year mortality of 50%, and prognosis tends to be worse for secondary radiation-induced sarcomas than for primary sarcomas.2,6 However, with there being improved treatment protocols involving surgery and chemoradiation, more recent 5-year survival rates without metastatic disease are between 60% and 80%, though there is no definite consensus regarding the optimal systemic therapy regimen.1,24 In a 2014 review of 53 patients who presented with localized disease, Choi and colleagues25 identified a 3-year cumulative 39% incidence of death caused by disease, and in 2016 Sio and colleagues26 reported that 55% of patients, most of whom had stage 3 disease, were alive at median follow-up of 45 months. Similar to osteosarcoma of bone, metastases may develop up to 10 years after primary treatment and are most commonly to the lung (80%-88%). Because extraosseous osteosarcoma is rare, no definite prognostic factors have been determined, but metastases at presentation and large tumor size (>5 cm) likely portend a worse prognosis.2,3,27 Fibroblastic and chondroblastic subtypes may have a slightly better prognosis.6,28
Conclusion
Extraosseous osteosarcoma is a rare malignancy that should be considered in the appropriate clinical and imaging scenario. This article is the first report of a case of a radiation-associated extraosseous osteosarcoma that developed within a lipoma with preoperative and postoperative multimodality imaging.
Am J Orthop. 2017;46(3):E200-E206. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Mc Auley G, Jagannathan J, O’Regan K, et al. Extraskeletal osteosarcoma: spectrum of imaging findings. AJR Am J Roentgenol. 2012;198(1):W31-W37.
2. Vikram S, Salih S, Krishnan A, et al. Radiation-induced extra-osseous osteosarcoma—a case report and review of literature. Indian J Surg Oncol. 2013;4(4):374-377.
3. Rosenberg AE. Extraskeletal osteosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon, France: IARC; 2013:161-162.
4. Ramnani BG, Kumar A, Chandak S, Ranjan A, Patel MK. Clinicopathological profile of benign soft tissue tumours: a study in a tertiary care hospital in Western India. J Clin Diagn Res. 2014;8(10):FC01-FC04.
5. Ahmad SA, Patel SR, Ballo MT, et al. Extraosseous osteosarcoma: response to treatment and long-term outcome. J Clin Oncol. 2002;20(2):521-527.
6. Mavrogenis AF, Papadogeorgou E, Papagelopoulos PJ. Extraskeletal osteosarcoma: a case report. Acta Orthop Traumatol Turc. 2012;46(3):215-219.
7. Morell N, Quinn RH. Lipoma. orthoinfo.aaos.org/topic.cfm?topic=a00631. Published 2012. Accessed December 28, 2014.
8. Kransdorf MJ, Bancroft LW, Peterson JJ, Murphey MD, Foster WC, Temple HT. Imaging of fatty tumors: distinction of lipoma and well-differentiated liposarcoma. Radiology. 2002;224(1):99-104.
9. Suzuki R, Watanabe H, Yanagawa T, et al. PET evaluation of fatty tumors in the extremity: possibility of using the standardized uptake value (SUV) to differentiate benign tumors from liposarcoma. Ann Nucl Med. 2005;19(8):661-670.
10. Laliotis A, De Bree E, Vasilaki S, Papadakis M, Melissas J. Co-existence of intramuscular spindle cell lipoma with an intramuscular ordinary lipoma: report of a case. Pol J Pathol. 2013;64(3):224-227.
11. Wright C. Liposarcoma arising in a simple lipoma. J Pathol Bacteriol. 1948;60:483-487.
12. Sampson CC, Saunders EH, Green WE, Laurey JR. Liposarcoma developing in a lipoma. Arch Pathol. 1960;69:506-510.
13. Sternberg SS. Liposarcoma arising within a subcutaneous lipoma. Cancer. 1952;5(5):975-978.
14. Ho L, Wassef H, Chang D, Boswell W, Henderson R, Seto J. Liposarcoma of the retroperitoneum with dedifferentiation to osteosarcoma: a case report. Clin Nucl Med. 2011;36(5):400-402.
15. Milgram JW. Malignant transformation in bone lipomas. Skeletal Radiol. 1990;19(5):347-352.
16. Mentzel T. Biological continuum of benign, atypical, and malignant mesenchymal neoplasms—does it exist? J Pathol. 2000;190(5):523-525.
17. Murphey MD, Carroll JF, Flemming DJ, Pope TL, Gannon FH, Kransdorf MJ. From the archives of the AFIP: benign musculoskeletal lipomatous lesions. Radiographics. 2004;24(5):1433-1466.
18. Zornig C, Schröder S. Does malignant transformation of benign soft-tissue tumours occur? A clinicomorphological study of ten initially misdiagnosed soft-tissue sarcomas. J Cancer Res Clin Oncol. 1992;118(2):166-169.
19. Dönmez FY, Tüzün U, Başaran C, Tunaci M, Bilgiç B, Acunaş G. MRI findings in parosteal osteosarcoma: correlation with histopathology. Diagn Interv Radiol. 2008;14(3):142-152.
20. Mariño-Enriquez A, Hornick JL, Dal Cin P, Cibas ES, Qian X. Dedifferentiated liposarcoma and pleomorphic liposarcoma: a comparative study of cytomorphology and MDM2/CDK4 expression on fine-needle aspiration. Cancer Cytopathol. 2014;122(2):128-137.
21. Yoshida A, Ushiku T, Motoi T, et al. MDM2 and CDK4 immunohistochemical coexpression in high-grade osteosarcoma: correlation with a dedifferentiated subtype. Am J Surg Pathol. 2012;36(3):423-431.
22. Thway K, Flora R, Shah C, Olmos D, Fisher C. Diagnostic utility of p16, CDK4, and MDM2 as an immunohistochemical panel in distinguishing well-differentiated and dedifferentiated liposarcomas from other adipocytic tumors. Am J Surg Pathol. 2012;36(3):462-469.
23. Lokka S, Scheel AH, Dango S, et al. Challenging dedifferentiated liposarcoma identified by MDM2-amplification, a report of two cases. BMC Clin Pathol. 2014;14:36.
24. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015.
25. Choi LE, Healey JH, Kuk D, Brennan MF. Analysis of outcomes in extraskeletal osteosarcoma: a review of fifty-three cases. J Bone Joint Surg Am. 2014;96(1):e2.
26. Sio TT, Vu CC, Sohawon S, et al. Extraskeletal osteosarcoma: an international Rare Cancer Network study. Am J Clin Oncol. 2016;39(1):32-36.
27. Bane BL, Evans HL, Ro JY, et al. Extraskeletal osteosarcoma. A clinicopathologic review of 26 cases. Cancer. 1990;65(12):2762-2770.
28. Lee JS, Fetsch JF, Wasdhal DA, Lee BP, Pritchard DJ, Nascimento AG. A review of 40 patients with extraskeletal osteosarcoma. Cancer. 1995;76(11):2253-2259.
Take-Home Points
- Rare and histologically indistinguishable from osteosarcoma of bone.
- Most common presentation is an enlarging mass in the thigh or buttock.
- Secondary extraosseous osteosarcoma usually arises in the field of prior external beam radiation or brachytherapy.
- Radiographic pattern of mineralization is central amorphous or cloudlike.
- On cross sectional imaging, the soft-tissue mass is separate from the underlying bone and periosteum.
Aside from multiple myeloma, osteosarcoma is the most common primary malignancy of bone, but extraosseous osteosarcoma is rare and accounts for only 1% of soft-tissue sarcomas and only 4% of all osteosarcomas.1-3 Benign mesenchymal tumors, such as lipomas, are common, and they are estimated to outnumber their malignant counterparts by more than a factor of 100. However, the true ratio is unknown, as many clinically benign lipomas are not biopsied.4 Conventional lipoma is the most common lipoma and is biologically indolent. Conventional lipoma generally does not transform biologically into a more aggressive type of neoplasm—unlike atypical lipomatous tumors, which may demonstrate this type of evolution with multiple local recurrences.
This article is the first report of a case of radiation-associated extraosseous osteosarcoma that developed within a benign conventional lipoma. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
In March 2013, a 72-year-old woman presented to a general surgeon with a right thigh mass of several weeks’ duration. The patient, who had a remote history of thyroid carcinoma, underwent thyroidectomy in 1991, excision of melanoma of the chest in 1998, and resection and adjuvant external beam radiotherapy (30 fractions) for Merkel cell carcinoma of the right proximal lateral leg (malignancy images unavailable) at an outside institution in 2003. Regional lymph node dissection at the time was negative. The patient remained disease-free the next 10 years. On presentation, magnetic resonance imaging (MRI) showed a 2.2-cm mass encircled by a tumor of lipomatous tissue within the vastus intermedius muscle, adjacent to but separate from the right distal femur (Figures 1A-1C).
Physical examination revealed marked ecchymosis of the left groin at the access site for embolization as well as massive ecchymosis and swelling along the right distal thigh, medial knee, and medial lower leg. The neurovascular structures were intact with full motor function and sensation distally, as well as normal distal pulses. No inguinal adenopathy was identified. The proximal portion of the prior radiation tattoo was at the inferior extent of the lesion on MRI.
The patient was treated with doxorubicin and ifosfamide (2 cycles) while waiting for the hematoma to shrink. Contrast-enhanced MRI showed a 2.2-cm enhancing mass with isointense T1 signal and heterogeneously hyperintense STIR (short tau inversion recovery) signal surrounded by a circumscribed nonenhancing lipomatous tumor within the vastus intermedius muscle, adjacent to the distal femoral cortex. There was no invasion of the bone, and a fat plane between the enhancing mass and the femoral cortex was identified (Figures 2A-2E).
After 3 cycles of neoadjuvant chemotherapy with doxorubicin and ifosfamide, MRI showed a marked reduction in hematoma size, to 2.4 cm × 0.7 cm × 3.2 cm (estimated volume, ~3 mL), from 10 cm × 3.4 cm × 7.3 cm (estimated volume, ~130 mL), so the decision was made to proceed with surgery, excising the hematoma and sarcoma separately. First, wide resection of the hematoma yielded a 7-cm × 4-cm resection specimen with negative margins on frozen section. Subsequently, definitive radical resection of the tumor with wide margins yielded a 13-cm × 9-cm × 4-cm specimen. The resection specimen contained an intramuscular, mobile, encapsulated 2.0-cm × 1.5-cm × 1.0-cm mass with 2 components. The first was a tan-white solid mass containing thin deposits of calcified matrix, and the second, which surrounded the first, was composed of well-circumscribed soft yellow lobulated adipose tissue (Figure 5).
After surgery, the patient’s dermatologist performed a shave biopsy of a lentiginous lesion anterior to the knee. Subsequently, the patient began having increasing knee pain and developed, on the lower extremity, small areas of erythema that were attributed to mild cellulitis. Four months after surgery, emergent contrast-enhanced MRI showed enhancement of thickened synovium of the knee joint (Figure 7).
Since the lavage, the patient remained in good condition. There was no evidence of local recurrence on contrast-enhanced MRI (Figure 8), or metastases the first year, and she remained clinically free of disease the first 22 months of follow-up.
Discussion
Extraosseous osteosarcoma, typically a high-grade malignant neoplasm of the soft tissues that produces osteoid or cartilaginous matrix, is histologically indistinguishable from osteosarcoma of bone.
Conventional lipoma, the most common subtype of lipoma, is a benign mesenchymal tumor. Other subtypes are hibernoma, fibrolipoma, angiolipoma, myelolipoma, spindle-cell lipoma, pleomorphic lipoma, and atypical lipomatous tumor.7 Atypical lipomatous tumor and well-differentiated liposarcoma are distinguished from each other by location: The World Health Organization recommends the term atypical lipomatous tumor for tumors that arise in the extremities and trunk lesions and well-differentiated liposarcoma for neoplasms that develop in the retroperitoneum, peritoneum, mediastinum, spermatic cord, and thoracic cavity.8 On PET, hypermetabolic activity is nonspecific and can be seen in malignant tumors and some benign reactive processes, such as evolving heterotopic ossification. However, simple lipomas, including those with mature ossification or dystrophic calcification, do not manifest increased FDG avidity.9
We are not aware of any published cases of extraosseous osteosarcoma arising within a conventional lipoma. A limited number of cases of coexisting conventional lipoma and spindle-cell lipoma or liposarcoma have been reported.10-13 Retroperitoneal liposarcoma with areas of dedifferentiation into osteosarcoma has also been described.14 Development of malignant fibrous histiocytoma and liposarcoma have also been reported within intraosseous lipomas.15 One theory is based on premalignancy as a biological concept as opposed to a morphologic one. In other words, lesions that may be considered morphologically benign may already have the biological phenotype for malignancy that is not yet reflected morphologically.16 However, it has been suggested that such findings may instead result from initial sampling error or histologic misdiagnosis.17,18There is a spectrum of findings on imaging studies of extraosseous osteosarcoma. Plain radiographs show a soft-tissue density with variable degrees of central calcification that reflects mineralization of deposited neoplastic bone. The pattern of calcification is characteristically amorphous or cloudlike, as opposed to the ring-and-arc observed in cartilage matrix. On CT, the soft-tissue mass of extraosseous osteosarcoma is separate from the underlying bone and periosteum—a defining characteristic that distinguishes it from conventional intramedullary and juxtacortical osteosarcoma.6 The central pattern of amorphous calcification helps to differentiate extraosseous osteosarcoma from heterotopic ossification, which characteristically demonstrates zonation, with trabecular architecture and mature cortical bone peripherally.1 Enhancement of extraskeletal osteosarcoma tends to be heterogeneous and depends on the quantity of necrosis. Extraskeletal osteosarcoma tends to be isointense on T1-weighted MRI and mildly hyperintense on T2-weighted MRI.1,6 Areas of very low signal intensity on both T1- and T2-weighted MRI may reflect mineralization.19 If intratumoral hemorrhage has occurred, there may be signal intensity of blood products of various ages.1,3 Tumors with abundant hemorrhage can be mistaken for hematoma. FDG-PET radiotracer accumulation tends to be intense peripherally with variable central activity depending on quantity of necrosis and hemorrhage.1The radiologic differential diagnosis includes myositis ossificans, chondrosseous lipoma, parosteal lipoma (ossifying variant), liposarcoma with metaplastic bone, dedifferentiated liposarcoma with osteosarcoma or chondrosarcoma component, and malignant mesenchymoma. Other common soft-tissue sarcomas, such as fibrosarcoma, leiomyosarcoma, and pleomorphic undifferentiated sarcoma, are excluded by the presence of fat within the tumor. The radiographic pattern of osteoid matrix produced by the tumor in our patient may be seen in heterotopic ossification, but the absence of mature ossification with zonation was evidence against heterotopic ossification, and microscopically it was neoplastic rather than reactive osteoid. In addition, it is possible that, because of the small size of the soft-tissue component, it was difficult to appreciate the less mature osteoid matrix peripherally. The lack of characteristic rings and arcs helps exclude benign and malignant cartilage containing neoplasms. Malignant mesenchymoma is a diagnosis of exclusion, and such tumors are usually better classified as sarcomas that have undergone heterologous differentiation.
The histologic diagnosis of extraosseous osteosarcoma requires identification of malignant mesenchymal cells that secrete neoplastic osteoid that may or may not mineralize. It is important to exclude the possibility that the malignant bone-forming tumor is part of a different type of sarcoma, the most common being dedifferentiated liposarcoma. Immunohistochemistry can be helpful in this situation, as dedifferentiated liposarcomas demonstrate nuclear expression of MDM2, CDK4, and p16, a constellation of findings rare in conventional and extraosseous osteosarcoma.20-23 Osteosarcoma has not previously been reported as arising in a lipoma; in our patient’s case, we excluded the possibility that the fatty component represented an underlying atypical lipomatous tumor/well-differentiated or dedifferentiated liposarcoma on the basis of morphology and lack of expression of MDM2, CDK4, and p16.
Although histologically identical to osteosarcoma of bone, extraosseous osteosarcoma is treated differently because of its relatively decreased chemosensitivity and radiosensitivity. Treatment tends to be focused on limb-sparing wide local excision, and local recurrence complicates about 50% of cases.1 Neoadjuvant or adjuvant treatment with radiation or chemotherapy is often provided.6 Platinum and doxorubicin chemotherapeutic agents, which are first-line treatments for osteosarcoma of bone, tend to be less effective in extraosseous osteosarcoma, and ifosfamide is more often used instead.5
Primary extraosseous osteosarcoma classically has a poor prognosis, with 2- to 3-year mortality of 50%, and prognosis tends to be worse for secondary radiation-induced sarcomas than for primary sarcomas.2,6 However, with there being improved treatment protocols involving surgery and chemoradiation, more recent 5-year survival rates without metastatic disease are between 60% and 80%, though there is no definite consensus regarding the optimal systemic therapy regimen.1,24 In a 2014 review of 53 patients who presented with localized disease, Choi and colleagues25 identified a 3-year cumulative 39% incidence of death caused by disease, and in 2016 Sio and colleagues26 reported that 55% of patients, most of whom had stage 3 disease, were alive at median follow-up of 45 months. Similar to osteosarcoma of bone, metastases may develop up to 10 years after primary treatment and are most commonly to the lung (80%-88%). Because extraosseous osteosarcoma is rare, no definite prognostic factors have been determined, but metastases at presentation and large tumor size (>5 cm) likely portend a worse prognosis.2,3,27 Fibroblastic and chondroblastic subtypes may have a slightly better prognosis.6,28
Conclusion
Extraosseous osteosarcoma is a rare malignancy that should be considered in the appropriate clinical and imaging scenario. This article is the first report of a case of a radiation-associated extraosseous osteosarcoma that developed within a lipoma with preoperative and postoperative multimodality imaging.
Am J Orthop. 2017;46(3):E200-E206. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Rare and histologically indistinguishable from osteosarcoma of bone.
- Most common presentation is an enlarging mass in the thigh or buttock.
- Secondary extraosseous osteosarcoma usually arises in the field of prior external beam radiation or brachytherapy.
- Radiographic pattern of mineralization is central amorphous or cloudlike.
- On cross sectional imaging, the soft-tissue mass is separate from the underlying bone and periosteum.
Aside from multiple myeloma, osteosarcoma is the most common primary malignancy of bone, but extraosseous osteosarcoma is rare and accounts for only 1% of soft-tissue sarcomas and only 4% of all osteosarcomas.1-3 Benign mesenchymal tumors, such as lipomas, are common, and they are estimated to outnumber their malignant counterparts by more than a factor of 100. However, the true ratio is unknown, as many clinically benign lipomas are not biopsied.4 Conventional lipoma is the most common lipoma and is biologically indolent. Conventional lipoma generally does not transform biologically into a more aggressive type of neoplasm—unlike atypical lipomatous tumors, which may demonstrate this type of evolution with multiple local recurrences.
This article is the first report of a case of radiation-associated extraosseous osteosarcoma that developed within a benign conventional lipoma. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
In March 2013, a 72-year-old woman presented to a general surgeon with a right thigh mass of several weeks’ duration. The patient, who had a remote history of thyroid carcinoma, underwent thyroidectomy in 1991, excision of melanoma of the chest in 1998, and resection and adjuvant external beam radiotherapy (30 fractions) for Merkel cell carcinoma of the right proximal lateral leg (malignancy images unavailable) at an outside institution in 2003. Regional lymph node dissection at the time was negative. The patient remained disease-free the next 10 years. On presentation, magnetic resonance imaging (MRI) showed a 2.2-cm mass encircled by a tumor of lipomatous tissue within the vastus intermedius muscle, adjacent to but separate from the right distal femur (Figures 1A-1C).
Physical examination revealed marked ecchymosis of the left groin at the access site for embolization as well as massive ecchymosis and swelling along the right distal thigh, medial knee, and medial lower leg. The neurovascular structures were intact with full motor function and sensation distally, as well as normal distal pulses. No inguinal adenopathy was identified. The proximal portion of the prior radiation tattoo was at the inferior extent of the lesion on MRI.
The patient was treated with doxorubicin and ifosfamide (2 cycles) while waiting for the hematoma to shrink. Contrast-enhanced MRI showed a 2.2-cm enhancing mass with isointense T1 signal and heterogeneously hyperintense STIR (short tau inversion recovery) signal surrounded by a circumscribed nonenhancing lipomatous tumor within the vastus intermedius muscle, adjacent to the distal femoral cortex. There was no invasion of the bone, and a fat plane between the enhancing mass and the femoral cortex was identified (Figures 2A-2E).
After 3 cycles of neoadjuvant chemotherapy with doxorubicin and ifosfamide, MRI showed a marked reduction in hematoma size, to 2.4 cm × 0.7 cm × 3.2 cm (estimated volume, ~3 mL), from 10 cm × 3.4 cm × 7.3 cm (estimated volume, ~130 mL), so the decision was made to proceed with surgery, excising the hematoma and sarcoma separately. First, wide resection of the hematoma yielded a 7-cm × 4-cm resection specimen with negative margins on frozen section. Subsequently, definitive radical resection of the tumor with wide margins yielded a 13-cm × 9-cm × 4-cm specimen. The resection specimen contained an intramuscular, mobile, encapsulated 2.0-cm × 1.5-cm × 1.0-cm mass with 2 components. The first was a tan-white solid mass containing thin deposits of calcified matrix, and the second, which surrounded the first, was composed of well-circumscribed soft yellow lobulated adipose tissue (Figure 5).
After surgery, the patient’s dermatologist performed a shave biopsy of a lentiginous lesion anterior to the knee. Subsequently, the patient began having increasing knee pain and developed, on the lower extremity, small areas of erythema that were attributed to mild cellulitis. Four months after surgery, emergent contrast-enhanced MRI showed enhancement of thickened synovium of the knee joint (Figure 7).
Since the lavage, the patient remained in good condition. There was no evidence of local recurrence on contrast-enhanced MRI (Figure 8), or metastases the first year, and she remained clinically free of disease the first 22 months of follow-up.
Discussion
Extraosseous osteosarcoma, typically a high-grade malignant neoplasm of the soft tissues that produces osteoid or cartilaginous matrix, is histologically indistinguishable from osteosarcoma of bone.
Conventional lipoma, the most common subtype of lipoma, is a benign mesenchymal tumor. Other subtypes are hibernoma, fibrolipoma, angiolipoma, myelolipoma, spindle-cell lipoma, pleomorphic lipoma, and atypical lipomatous tumor.7 Atypical lipomatous tumor and well-differentiated liposarcoma are distinguished from each other by location: The World Health Organization recommends the term atypical lipomatous tumor for tumors that arise in the extremities and trunk lesions and well-differentiated liposarcoma for neoplasms that develop in the retroperitoneum, peritoneum, mediastinum, spermatic cord, and thoracic cavity.8 On PET, hypermetabolic activity is nonspecific and can be seen in malignant tumors and some benign reactive processes, such as evolving heterotopic ossification. However, simple lipomas, including those with mature ossification or dystrophic calcification, do not manifest increased FDG avidity.9
We are not aware of any published cases of extraosseous osteosarcoma arising within a conventional lipoma. A limited number of cases of coexisting conventional lipoma and spindle-cell lipoma or liposarcoma have been reported.10-13 Retroperitoneal liposarcoma with areas of dedifferentiation into osteosarcoma has also been described.14 Development of malignant fibrous histiocytoma and liposarcoma have also been reported within intraosseous lipomas.15 One theory is based on premalignancy as a biological concept as opposed to a morphologic one. In other words, lesions that may be considered morphologically benign may already have the biological phenotype for malignancy that is not yet reflected morphologically.16 However, it has been suggested that such findings may instead result from initial sampling error or histologic misdiagnosis.17,18There is a spectrum of findings on imaging studies of extraosseous osteosarcoma. Plain radiographs show a soft-tissue density with variable degrees of central calcification that reflects mineralization of deposited neoplastic bone. The pattern of calcification is characteristically amorphous or cloudlike, as opposed to the ring-and-arc observed in cartilage matrix. On CT, the soft-tissue mass of extraosseous osteosarcoma is separate from the underlying bone and periosteum—a defining characteristic that distinguishes it from conventional intramedullary and juxtacortical osteosarcoma.6 The central pattern of amorphous calcification helps to differentiate extraosseous osteosarcoma from heterotopic ossification, which characteristically demonstrates zonation, with trabecular architecture and mature cortical bone peripherally.1 Enhancement of extraskeletal osteosarcoma tends to be heterogeneous and depends on the quantity of necrosis. Extraskeletal osteosarcoma tends to be isointense on T1-weighted MRI and mildly hyperintense on T2-weighted MRI.1,6 Areas of very low signal intensity on both T1- and T2-weighted MRI may reflect mineralization.19 If intratumoral hemorrhage has occurred, there may be signal intensity of blood products of various ages.1,3 Tumors with abundant hemorrhage can be mistaken for hematoma. FDG-PET radiotracer accumulation tends to be intense peripherally with variable central activity depending on quantity of necrosis and hemorrhage.1The radiologic differential diagnosis includes myositis ossificans, chondrosseous lipoma, parosteal lipoma (ossifying variant), liposarcoma with metaplastic bone, dedifferentiated liposarcoma with osteosarcoma or chondrosarcoma component, and malignant mesenchymoma. Other common soft-tissue sarcomas, such as fibrosarcoma, leiomyosarcoma, and pleomorphic undifferentiated sarcoma, are excluded by the presence of fat within the tumor. The radiographic pattern of osteoid matrix produced by the tumor in our patient may be seen in heterotopic ossification, but the absence of mature ossification with zonation was evidence against heterotopic ossification, and microscopically it was neoplastic rather than reactive osteoid. In addition, it is possible that, because of the small size of the soft-tissue component, it was difficult to appreciate the less mature osteoid matrix peripherally. The lack of characteristic rings and arcs helps exclude benign and malignant cartilage containing neoplasms. Malignant mesenchymoma is a diagnosis of exclusion, and such tumors are usually better classified as sarcomas that have undergone heterologous differentiation.
The histologic diagnosis of extraosseous osteosarcoma requires identification of malignant mesenchymal cells that secrete neoplastic osteoid that may or may not mineralize. It is important to exclude the possibility that the malignant bone-forming tumor is part of a different type of sarcoma, the most common being dedifferentiated liposarcoma. Immunohistochemistry can be helpful in this situation, as dedifferentiated liposarcomas demonstrate nuclear expression of MDM2, CDK4, and p16, a constellation of findings rare in conventional and extraosseous osteosarcoma.20-23 Osteosarcoma has not previously been reported as arising in a lipoma; in our patient’s case, we excluded the possibility that the fatty component represented an underlying atypical lipomatous tumor/well-differentiated or dedifferentiated liposarcoma on the basis of morphology and lack of expression of MDM2, CDK4, and p16.
Although histologically identical to osteosarcoma of bone, extraosseous osteosarcoma is treated differently because of its relatively decreased chemosensitivity and radiosensitivity. Treatment tends to be focused on limb-sparing wide local excision, and local recurrence complicates about 50% of cases.1 Neoadjuvant or adjuvant treatment with radiation or chemotherapy is often provided.6 Platinum and doxorubicin chemotherapeutic agents, which are first-line treatments for osteosarcoma of bone, tend to be less effective in extraosseous osteosarcoma, and ifosfamide is more often used instead.5
Primary extraosseous osteosarcoma classically has a poor prognosis, with 2- to 3-year mortality of 50%, and prognosis tends to be worse for secondary radiation-induced sarcomas than for primary sarcomas.2,6 However, with there being improved treatment protocols involving surgery and chemoradiation, more recent 5-year survival rates without metastatic disease are between 60% and 80%, though there is no definite consensus regarding the optimal systemic therapy regimen.1,24 In a 2014 review of 53 patients who presented with localized disease, Choi and colleagues25 identified a 3-year cumulative 39% incidence of death caused by disease, and in 2016 Sio and colleagues26 reported that 55% of patients, most of whom had stage 3 disease, were alive at median follow-up of 45 months. Similar to osteosarcoma of bone, metastases may develop up to 10 years after primary treatment and are most commonly to the lung (80%-88%). Because extraosseous osteosarcoma is rare, no definite prognostic factors have been determined, but metastases at presentation and large tumor size (>5 cm) likely portend a worse prognosis.2,3,27 Fibroblastic and chondroblastic subtypes may have a slightly better prognosis.6,28
Conclusion
Extraosseous osteosarcoma is a rare malignancy that should be considered in the appropriate clinical and imaging scenario. This article is the first report of a case of a radiation-associated extraosseous osteosarcoma that developed within a lipoma with preoperative and postoperative multimodality imaging.
Am J Orthop. 2017;46(3):E200-E206. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Mc Auley G, Jagannathan J, O’Regan K, et al. Extraskeletal osteosarcoma: spectrum of imaging findings. AJR Am J Roentgenol. 2012;198(1):W31-W37.
2. Vikram S, Salih S, Krishnan A, et al. Radiation-induced extra-osseous osteosarcoma—a case report and review of literature. Indian J Surg Oncol. 2013;4(4):374-377.
3. Rosenberg AE. Extraskeletal osteosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon, France: IARC; 2013:161-162.
4. Ramnani BG, Kumar A, Chandak S, Ranjan A, Patel MK. Clinicopathological profile of benign soft tissue tumours: a study in a tertiary care hospital in Western India. J Clin Diagn Res. 2014;8(10):FC01-FC04.
5. Ahmad SA, Patel SR, Ballo MT, et al. Extraosseous osteosarcoma: response to treatment and long-term outcome. J Clin Oncol. 2002;20(2):521-527.
6. Mavrogenis AF, Papadogeorgou E, Papagelopoulos PJ. Extraskeletal osteosarcoma: a case report. Acta Orthop Traumatol Turc. 2012;46(3):215-219.
7. Morell N, Quinn RH. Lipoma. orthoinfo.aaos.org/topic.cfm?topic=a00631. Published 2012. Accessed December 28, 2014.
8. Kransdorf MJ, Bancroft LW, Peterson JJ, Murphey MD, Foster WC, Temple HT. Imaging of fatty tumors: distinction of lipoma and well-differentiated liposarcoma. Radiology. 2002;224(1):99-104.
9. Suzuki R, Watanabe H, Yanagawa T, et al. PET evaluation of fatty tumors in the extremity: possibility of using the standardized uptake value (SUV) to differentiate benign tumors from liposarcoma. Ann Nucl Med. 2005;19(8):661-670.
10. Laliotis A, De Bree E, Vasilaki S, Papadakis M, Melissas J. Co-existence of intramuscular spindle cell lipoma with an intramuscular ordinary lipoma: report of a case. Pol J Pathol. 2013;64(3):224-227.
11. Wright C. Liposarcoma arising in a simple lipoma. J Pathol Bacteriol. 1948;60:483-487.
12. Sampson CC, Saunders EH, Green WE, Laurey JR. Liposarcoma developing in a lipoma. Arch Pathol. 1960;69:506-510.
13. Sternberg SS. Liposarcoma arising within a subcutaneous lipoma. Cancer. 1952;5(5):975-978.
14. Ho L, Wassef H, Chang D, Boswell W, Henderson R, Seto J. Liposarcoma of the retroperitoneum with dedifferentiation to osteosarcoma: a case report. Clin Nucl Med. 2011;36(5):400-402.
15. Milgram JW. Malignant transformation in bone lipomas. Skeletal Radiol. 1990;19(5):347-352.
16. Mentzel T. Biological continuum of benign, atypical, and malignant mesenchymal neoplasms—does it exist? J Pathol. 2000;190(5):523-525.
17. Murphey MD, Carroll JF, Flemming DJ, Pope TL, Gannon FH, Kransdorf MJ. From the archives of the AFIP: benign musculoskeletal lipomatous lesions. Radiographics. 2004;24(5):1433-1466.
18. Zornig C, Schröder S. Does malignant transformation of benign soft-tissue tumours occur? A clinicomorphological study of ten initially misdiagnosed soft-tissue sarcomas. J Cancer Res Clin Oncol. 1992;118(2):166-169.
19. Dönmez FY, Tüzün U, Başaran C, Tunaci M, Bilgiç B, Acunaş G. MRI findings in parosteal osteosarcoma: correlation with histopathology. Diagn Interv Radiol. 2008;14(3):142-152.
20. Mariño-Enriquez A, Hornick JL, Dal Cin P, Cibas ES, Qian X. Dedifferentiated liposarcoma and pleomorphic liposarcoma: a comparative study of cytomorphology and MDM2/CDK4 expression on fine-needle aspiration. Cancer Cytopathol. 2014;122(2):128-137.
21. Yoshida A, Ushiku T, Motoi T, et al. MDM2 and CDK4 immunohistochemical coexpression in high-grade osteosarcoma: correlation with a dedifferentiated subtype. Am J Surg Pathol. 2012;36(3):423-431.
22. Thway K, Flora R, Shah C, Olmos D, Fisher C. Diagnostic utility of p16, CDK4, and MDM2 as an immunohistochemical panel in distinguishing well-differentiated and dedifferentiated liposarcomas from other adipocytic tumors. Am J Surg Pathol. 2012;36(3):462-469.
23. Lokka S, Scheel AH, Dango S, et al. Challenging dedifferentiated liposarcoma identified by MDM2-amplification, a report of two cases. BMC Clin Pathol. 2014;14:36.
24. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015.
25. Choi LE, Healey JH, Kuk D, Brennan MF. Analysis of outcomes in extraskeletal osteosarcoma: a review of fifty-three cases. J Bone Joint Surg Am. 2014;96(1):e2.
26. Sio TT, Vu CC, Sohawon S, et al. Extraskeletal osteosarcoma: an international Rare Cancer Network study. Am J Clin Oncol. 2016;39(1):32-36.
27. Bane BL, Evans HL, Ro JY, et al. Extraskeletal osteosarcoma. A clinicopathologic review of 26 cases. Cancer. 1990;65(12):2762-2770.
28. Lee JS, Fetsch JF, Wasdhal DA, Lee BP, Pritchard DJ, Nascimento AG. A review of 40 patients with extraskeletal osteosarcoma. Cancer. 1995;76(11):2253-2259.
1. Mc Auley G, Jagannathan J, O’Regan K, et al. Extraskeletal osteosarcoma: spectrum of imaging findings. AJR Am J Roentgenol. 2012;198(1):W31-W37.
2. Vikram S, Salih S, Krishnan A, et al. Radiation-induced extra-osseous osteosarcoma—a case report and review of literature. Indian J Surg Oncol. 2013;4(4):374-377.
3. Rosenberg AE. Extraskeletal osteosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyon, France: IARC; 2013:161-162.
4. Ramnani BG, Kumar A, Chandak S, Ranjan A, Patel MK. Clinicopathological profile of benign soft tissue tumours: a study in a tertiary care hospital in Western India. J Clin Diagn Res. 2014;8(10):FC01-FC04.
5. Ahmad SA, Patel SR, Ballo MT, et al. Extraosseous osteosarcoma: response to treatment and long-term outcome. J Clin Oncol. 2002;20(2):521-527.
6. Mavrogenis AF, Papadogeorgou E, Papagelopoulos PJ. Extraskeletal osteosarcoma: a case report. Acta Orthop Traumatol Turc. 2012;46(3):215-219.
7. Morell N, Quinn RH. Lipoma. orthoinfo.aaos.org/topic.cfm?topic=a00631. Published 2012. Accessed December 28, 2014.
8. Kransdorf MJ, Bancroft LW, Peterson JJ, Murphey MD, Foster WC, Temple HT. Imaging of fatty tumors: distinction of lipoma and well-differentiated liposarcoma. Radiology. 2002;224(1):99-104.
9. Suzuki R, Watanabe H, Yanagawa T, et al. PET evaluation of fatty tumors in the extremity: possibility of using the standardized uptake value (SUV) to differentiate benign tumors from liposarcoma. Ann Nucl Med. 2005;19(8):661-670.
10. Laliotis A, De Bree E, Vasilaki S, Papadakis M, Melissas J. Co-existence of intramuscular spindle cell lipoma with an intramuscular ordinary lipoma: report of a case. Pol J Pathol. 2013;64(3):224-227.
11. Wright C. Liposarcoma arising in a simple lipoma. J Pathol Bacteriol. 1948;60:483-487.
12. Sampson CC, Saunders EH, Green WE, Laurey JR. Liposarcoma developing in a lipoma. Arch Pathol. 1960;69:506-510.
13. Sternberg SS. Liposarcoma arising within a subcutaneous lipoma. Cancer. 1952;5(5):975-978.
14. Ho L, Wassef H, Chang D, Boswell W, Henderson R, Seto J. Liposarcoma of the retroperitoneum with dedifferentiation to osteosarcoma: a case report. Clin Nucl Med. 2011;36(5):400-402.
15. Milgram JW. Malignant transformation in bone lipomas. Skeletal Radiol. 1990;19(5):347-352.
16. Mentzel T. Biological continuum of benign, atypical, and malignant mesenchymal neoplasms—does it exist? J Pathol. 2000;190(5):523-525.
17. Murphey MD, Carroll JF, Flemming DJ, Pope TL, Gannon FH, Kransdorf MJ. From the archives of the AFIP: benign musculoskeletal lipomatous lesions. Radiographics. 2004;24(5):1433-1466.
18. Zornig C, Schröder S. Does malignant transformation of benign soft-tissue tumours occur? A clinicomorphological study of ten initially misdiagnosed soft-tissue sarcomas. J Cancer Res Clin Oncol. 1992;118(2):166-169.
19. Dönmez FY, Tüzün U, Başaran C, Tunaci M, Bilgiç B, Acunaş G. MRI findings in parosteal osteosarcoma: correlation with histopathology. Diagn Interv Radiol. 2008;14(3):142-152.
20. Mariño-Enriquez A, Hornick JL, Dal Cin P, Cibas ES, Qian X. Dedifferentiated liposarcoma and pleomorphic liposarcoma: a comparative study of cytomorphology and MDM2/CDK4 expression on fine-needle aspiration. Cancer Cytopathol. 2014;122(2):128-137.
21. Yoshida A, Ushiku T, Motoi T, et al. MDM2 and CDK4 immunohistochemical coexpression in high-grade osteosarcoma: correlation with a dedifferentiated subtype. Am J Surg Pathol. 2012;36(3):423-431.
22. Thway K, Flora R, Shah C, Olmos D, Fisher C. Diagnostic utility of p16, CDK4, and MDM2 as an immunohistochemical panel in distinguishing well-differentiated and dedifferentiated liposarcomas from other adipocytic tumors. Am J Surg Pathol. 2012;36(3):462-469.
23. Lokka S, Scheel AH, Dango S, et al. Challenging dedifferentiated liposarcoma identified by MDM2-amplification, a report of two cases. BMC Clin Pathol. 2014;14:36.
24. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015.
25. Choi LE, Healey JH, Kuk D, Brennan MF. Analysis of outcomes in extraskeletal osteosarcoma: a review of fifty-three cases. J Bone Joint Surg Am. 2014;96(1):e2.
26. Sio TT, Vu CC, Sohawon S, et al. Extraskeletal osteosarcoma: an international Rare Cancer Network study. Am J Clin Oncol. 2016;39(1):32-36.
27. Bane BL, Evans HL, Ro JY, et al. Extraskeletal osteosarcoma. A clinicopathologic review of 26 cases. Cancer. 1990;65(12):2762-2770.
28. Lee JS, Fetsch JF, Wasdhal DA, Lee BP, Pritchard DJ, Nascimento AG. A review of 40 patients with extraskeletal osteosarcoma. Cancer. 1995;76(11):2253-2259.
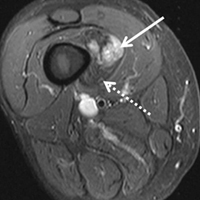
Leukocytoclastic Vasculitis Resolution With Topical Dapsone
Leukocytoclastic vasculitis (LCV) is a disease characterized by inflammation of small vessels with characteristic clinical findings of petechiae and palpable purpura.1 Numerous etiologies have been described, but the disease commonly remains idiopathic.2,3 Leukocytoclastic vasculitis often spontaneously resolves within weeks and requires only symptomatic treatment. Chronic or severe disease can require systemic medical treatment with agents such as colchicine, dapsone, and corticosteroids. These agents are effective but carry risks of serious side effects.4,5 These side effects and/or medical contraindications prevent some patients from taking systemic medications for LCV. We present a case of LCV that resolved after treatment with topical dapsone, highlighting a potential new treatment ofLCV with a markedly better side-effect profile.
Case Report
A 60-year-old woman with recent upper respiratory tract and sinus infections presented to our dermatology clinic with painful palpable purpura on the bilateral shins, thighs, and dorsal aspects of the feet of several months’ duration (Figure, A). Her primary care provider initiated treatment with amoxicillin and doxycycline for the infections. When the rash developed approximately 1.5 weeks following initiation of her symptoms, the patient was referred to the dermatology and rheumatology departments at our institution. The treating dermatologist (M.B.T.) obtained a 4-mm punch biopsy from the right lower leg and LCV was shown on histology. The patient completed a 14-day course of doxycycline and amoxicillin without resolution of the eruption. After an extensive investigation, the treating rheumatologist concluded that the LCV was idiopathic or secondary to an infection or drug exposure. The rheumatologist started the patient on oral prednisone for the chronic symptomatic LCV, but she was intolerant of this medication and discontinued it after 1 week. Our dermatology clinic started her on triamcinolone cream 0.1% twice daily, but she continued to experience new and worsening lesions. At her follow-up appointment 1 month later, triamcinolone cream was discontinued and dapsone gel 5% twice daily was started. She experienced resolution of her previously recalcitrant LCV within 3 weeks (Figure, B).
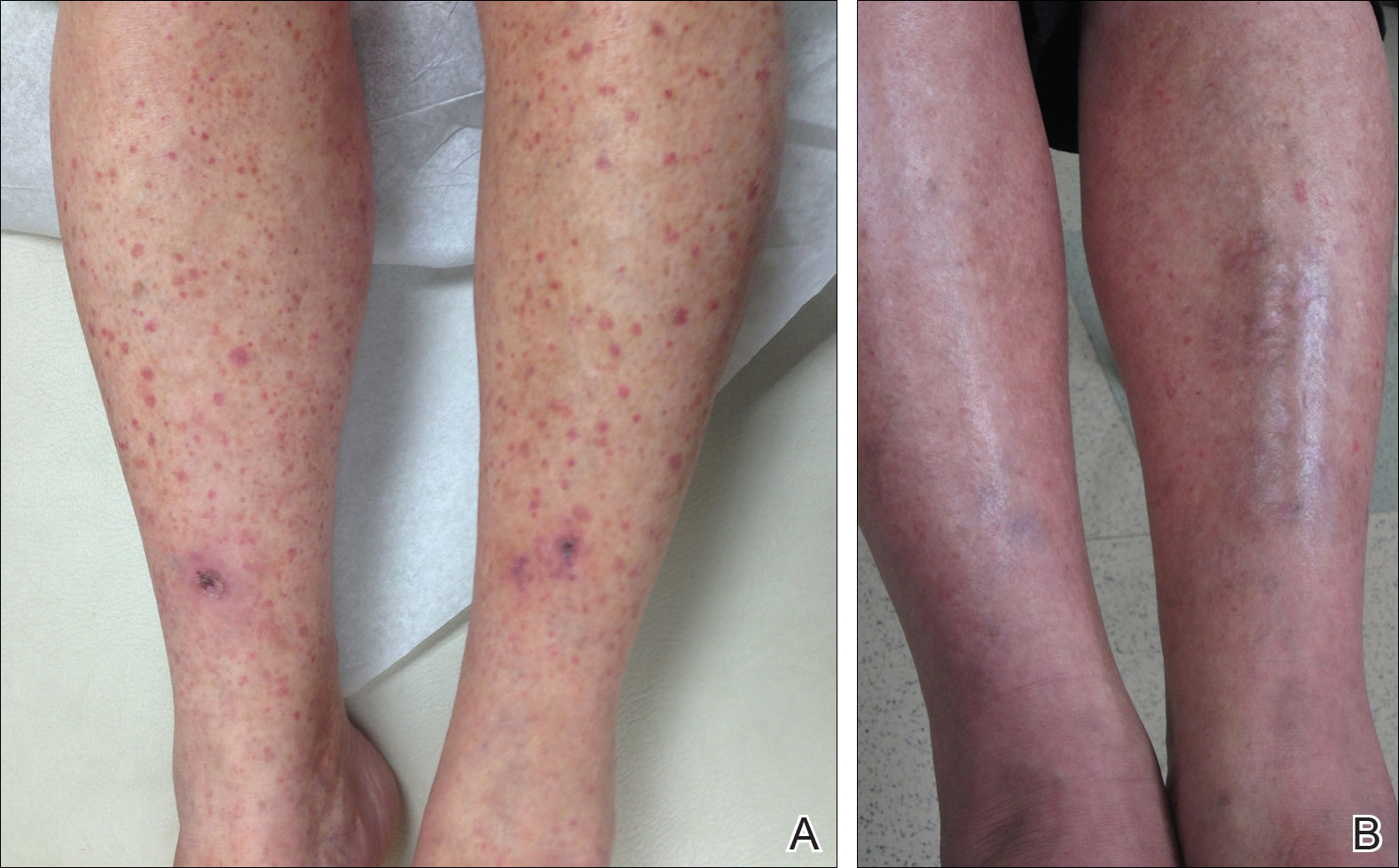
Comment
Established therapies for LCV carry serious side-effect profiles, which can preclude their use.5 Therefore, a topical therapeutic alternative for LCV would be ideal. Systemic prednisone is the first-line therapy for chronic and/or symptomatic LCV, but its side effects include suppression of the hypothalamic-pituitary-adrenal axis, immunosuppression, osteonecrosis, and glucose intolerance.5 Colchicine therapy carries risks for blood dyscrasia, immunosuppression, and gastrointestinal tract upset. Systemic dapsone also is an effective therapy for chronic and/or symptomatic LCV.5,6 However, systemic dapsone requires glucose-6-phosphate dehydrogenase deficiency screening and routine monitoring of blood counts, and it also carries the risk for serious adverse effects including neuropathy, blood dyscrasia, and hypersensitivity syndrome.5,6 Topical dapsone may provide similar efficacy with far fewer adverse effects and has proven to be a safe treatment of acne, even when used in patients with glucose-6-phosphate dehydrogenase deficiency. It displays low systemic absorption and does not accumulate over time once a steady state is reached.7 It also has been shown to be beneficial in other vasculopathies such as erythema elevatum diutinum and in other neutrophilic inflammatory disorders such as pyoderma gangrenosum.8,9 A case of methemoglobinemia due to topical dapsone has been reported.10 Although this effect is rare, clinicians should be aware of such adverse effects when using medications for off-label purposes.
Leukocytoclastic vasculitis can spontaneously resolve; however, our patient’s disease was chronic for several months, and she continued to develop new lesions without signs of resolution. After initiating topical dapsone, she experienced resolution within 3 weeks.
Conclusion
Topical dapsone is a novel approach for treating LCV. Given this drug’s favorable side-effect profile compared to the currently available therapeutic alternatives, we believe it is a reasonable option in select patients. Further investigation is needed to prove its efficacy, but it could be an ideal alternative for patients with contraindications to traditional therapies and/or for those unable to tolerate systemic therapy.
- Koutkia P, Mylonakis E, Rounds S, et al. Leucocytoclastic vasculitis: an update for the clinician. Scand J Rheumatol. 2001;30:315-322.
- Af Ekenstam E, Callen JP. Cutaneous leukocytoclastic vasculitis. clinical and laboratory features of 82 patients seen in private practice. Arch Dermatol. 1984;120:484-489.
- Gyselbrecht L, de Keyser F, Ongenae K, et al. Etiological factors and underlying conditions in patientswith leucocytoclastic vasculitis. Clin Exp Rheumatol. 1996;14:665-668.
- Sais G, Vidaller A, Jucglà A, et al. Colchicine in the treatment of cutaneous leukocytoclastic vasculitis. results of a prospective, randomized controlled trial. Arch Dermatol. 1995;131:1399-1402.
- Sunderkotter C, Bonsmann G, Sindrilaru A, et al. Management of leukocytoclastic vasculitis: clinical review. J Dermatol Treat. 2005;16:193-206.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol. 2001;45:420-434.
- Stotland M, Shalita AR, Kissling RF. Dapsone 5% gel: a review of its efficacy and safety in the treatment of acne vulgaris. Am J Clin Dermatol. 2009;10:221-227.
- Frieling GW, Williams NL, Lim SJ, et al. Novel use of topical dapsone 5% gel for erythema elevatum diutinum: safer and effective. J Drugs Dermatol. 2013;12:481-484.
- Handler MZ, Hamilton H, Aires D. Treatment of peristomal pyoderma gangrenosum with topical crushed dapsone. J Drugs Dermatol. 2011;10:1059-1061.
- Swartzentruber GS, Yanta JH, Pizon AF. Methemoglobi-nemia as a complication of topical dapsone. N Engl J Med. 2015;372:491-492.
Leukocytoclastic vasculitis (LCV) is a disease characterized by inflammation of small vessels with characteristic clinical findings of petechiae and palpable purpura.1 Numerous etiologies have been described, but the disease commonly remains idiopathic.2,3 Leukocytoclastic vasculitis often spontaneously resolves within weeks and requires only symptomatic treatment. Chronic or severe disease can require systemic medical treatment with agents such as colchicine, dapsone, and corticosteroids. These agents are effective but carry risks of serious side effects.4,5 These side effects and/or medical contraindications prevent some patients from taking systemic medications for LCV. We present a case of LCV that resolved after treatment with topical dapsone, highlighting a potential new treatment ofLCV with a markedly better side-effect profile.
Case Report
A 60-year-old woman with recent upper respiratory tract and sinus infections presented to our dermatology clinic with painful palpable purpura on the bilateral shins, thighs, and dorsal aspects of the feet of several months’ duration (Figure, A). Her primary care provider initiated treatment with amoxicillin and doxycycline for the infections. When the rash developed approximately 1.5 weeks following initiation of her symptoms, the patient was referred to the dermatology and rheumatology departments at our institution. The treating dermatologist (M.B.T.) obtained a 4-mm punch biopsy from the right lower leg and LCV was shown on histology. The patient completed a 14-day course of doxycycline and amoxicillin without resolution of the eruption. After an extensive investigation, the treating rheumatologist concluded that the LCV was idiopathic or secondary to an infection or drug exposure. The rheumatologist started the patient on oral prednisone for the chronic symptomatic LCV, but she was intolerant of this medication and discontinued it after 1 week. Our dermatology clinic started her on triamcinolone cream 0.1% twice daily, but she continued to experience new and worsening lesions. At her follow-up appointment 1 month later, triamcinolone cream was discontinued and dapsone gel 5% twice daily was started. She experienced resolution of her previously recalcitrant LCV within 3 weeks (Figure, B).
Comment
Established therapies for LCV carry serious side-effect profiles, which can preclude their use.5 Therefore, a topical therapeutic alternative for LCV would be ideal. Systemic prednisone is the first-line therapy for chronic and/or symptomatic LCV, but its side effects include suppression of the hypothalamic-pituitary-adrenal axis, immunosuppression, osteonecrosis, and glucose intolerance.5 Colchicine therapy carries risks for blood dyscrasia, immunosuppression, and gastrointestinal tract upset. Systemic dapsone also is an effective therapy for chronic and/or symptomatic LCV.5,6 However, systemic dapsone requires glucose-6-phosphate dehydrogenase deficiency screening and routine monitoring of blood counts, and it also carries the risk for serious adverse effects including neuropathy, blood dyscrasia, and hypersensitivity syndrome.5,6 Topical dapsone may provide similar efficacy with far fewer adverse effects and has proven to be a safe treatment of acne, even when used in patients with glucose-6-phosphate dehydrogenase deficiency. It displays low systemic absorption and does not accumulate over time once a steady state is reached.7 It also has been shown to be beneficial in other vasculopathies such as erythema elevatum diutinum and in other neutrophilic inflammatory disorders such as pyoderma gangrenosum.8,9 A case of methemoglobinemia due to topical dapsone has been reported.10 Although this effect is rare, clinicians should be aware of such adverse effects when using medications for off-label purposes.
Leukocytoclastic vasculitis can spontaneously resolve; however, our patient’s disease was chronic for several months, and she continued to develop new lesions without signs of resolution. After initiating topical dapsone, she experienced resolution within 3 weeks.
Conclusion
Topical dapsone is a novel approach for treating LCV. Given this drug’s favorable side-effect profile compared to the currently available therapeutic alternatives, we believe it is a reasonable option in select patients. Further investigation is needed to prove its efficacy, but it could be an ideal alternative for patients with contraindications to traditional therapies and/or for those unable to tolerate systemic therapy.
Leukocytoclastic vasculitis (LCV) is a disease characterized by inflammation of small vessels with characteristic clinical findings of petechiae and palpable purpura.1 Numerous etiologies have been described, but the disease commonly remains idiopathic.2,3 Leukocytoclastic vasculitis often spontaneously resolves within weeks and requires only symptomatic treatment. Chronic or severe disease can require systemic medical treatment with agents such as colchicine, dapsone, and corticosteroids. These agents are effective but carry risks of serious side effects.4,5 These side effects and/or medical contraindications prevent some patients from taking systemic medications for LCV. We present a case of LCV that resolved after treatment with topical dapsone, highlighting a potential new treatment ofLCV with a markedly better side-effect profile.
Case Report
A 60-year-old woman with recent upper respiratory tract and sinus infections presented to our dermatology clinic with painful palpable purpura on the bilateral shins, thighs, and dorsal aspects of the feet of several months’ duration (Figure, A). Her primary care provider initiated treatment with amoxicillin and doxycycline for the infections. When the rash developed approximately 1.5 weeks following initiation of her symptoms, the patient was referred to the dermatology and rheumatology departments at our institution. The treating dermatologist (M.B.T.) obtained a 4-mm punch biopsy from the right lower leg and LCV was shown on histology. The patient completed a 14-day course of doxycycline and amoxicillin without resolution of the eruption. After an extensive investigation, the treating rheumatologist concluded that the LCV was idiopathic or secondary to an infection or drug exposure. The rheumatologist started the patient on oral prednisone for the chronic symptomatic LCV, but she was intolerant of this medication and discontinued it after 1 week. Our dermatology clinic started her on triamcinolone cream 0.1% twice daily, but she continued to experience new and worsening lesions. At her follow-up appointment 1 month later, triamcinolone cream was discontinued and dapsone gel 5% twice daily was started. She experienced resolution of her previously recalcitrant LCV within 3 weeks (Figure, B).
Comment
Established therapies for LCV carry serious side-effect profiles, which can preclude their use.5 Therefore, a topical therapeutic alternative for LCV would be ideal. Systemic prednisone is the first-line therapy for chronic and/or symptomatic LCV, but its side effects include suppression of the hypothalamic-pituitary-adrenal axis, immunosuppression, osteonecrosis, and glucose intolerance.5 Colchicine therapy carries risks for blood dyscrasia, immunosuppression, and gastrointestinal tract upset. Systemic dapsone also is an effective therapy for chronic and/or symptomatic LCV.5,6 However, systemic dapsone requires glucose-6-phosphate dehydrogenase deficiency screening and routine monitoring of blood counts, and it also carries the risk for serious adverse effects including neuropathy, blood dyscrasia, and hypersensitivity syndrome.5,6 Topical dapsone may provide similar efficacy with far fewer adverse effects and has proven to be a safe treatment of acne, even when used in patients with glucose-6-phosphate dehydrogenase deficiency. It displays low systemic absorption and does not accumulate over time once a steady state is reached.7 It also has been shown to be beneficial in other vasculopathies such as erythema elevatum diutinum and in other neutrophilic inflammatory disorders such as pyoderma gangrenosum.8,9 A case of methemoglobinemia due to topical dapsone has been reported.10 Although this effect is rare, clinicians should be aware of such adverse effects when using medications for off-label purposes.
Leukocytoclastic vasculitis can spontaneously resolve; however, our patient’s disease was chronic for several months, and she continued to develop new lesions without signs of resolution. After initiating topical dapsone, she experienced resolution within 3 weeks.
Conclusion
Topical dapsone is a novel approach for treating LCV. Given this drug’s favorable side-effect profile compared to the currently available therapeutic alternatives, we believe it is a reasonable option in select patients. Further investigation is needed to prove its efficacy, but it could be an ideal alternative for patients with contraindications to traditional therapies and/or for those unable to tolerate systemic therapy.
- Koutkia P, Mylonakis E, Rounds S, et al. Leucocytoclastic vasculitis: an update for the clinician. Scand J Rheumatol. 2001;30:315-322.
- Af Ekenstam E, Callen JP. Cutaneous leukocytoclastic vasculitis. clinical and laboratory features of 82 patients seen in private practice. Arch Dermatol. 1984;120:484-489.
- Gyselbrecht L, de Keyser F, Ongenae K, et al. Etiological factors and underlying conditions in patientswith leucocytoclastic vasculitis. Clin Exp Rheumatol. 1996;14:665-668.
- Sais G, Vidaller A, Jucglà A, et al. Colchicine in the treatment of cutaneous leukocytoclastic vasculitis. results of a prospective, randomized controlled trial. Arch Dermatol. 1995;131:1399-1402.
- Sunderkotter C, Bonsmann G, Sindrilaru A, et al. Management of leukocytoclastic vasculitis: clinical review. J Dermatol Treat. 2005;16:193-206.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol. 2001;45:420-434.
- Stotland M, Shalita AR, Kissling RF. Dapsone 5% gel: a review of its efficacy and safety in the treatment of acne vulgaris. Am J Clin Dermatol. 2009;10:221-227.
- Frieling GW, Williams NL, Lim SJ, et al. Novel use of topical dapsone 5% gel for erythema elevatum diutinum: safer and effective. J Drugs Dermatol. 2013;12:481-484.
- Handler MZ, Hamilton H, Aires D. Treatment of peristomal pyoderma gangrenosum with topical crushed dapsone. J Drugs Dermatol. 2011;10:1059-1061.
- Swartzentruber GS, Yanta JH, Pizon AF. Methemoglobi-nemia as a complication of topical dapsone. N Engl J Med. 2015;372:491-492.
- Koutkia P, Mylonakis E, Rounds S, et al. Leucocytoclastic vasculitis: an update for the clinician. Scand J Rheumatol. 2001;30:315-322.
- Af Ekenstam E, Callen JP. Cutaneous leukocytoclastic vasculitis. clinical and laboratory features of 82 patients seen in private practice. Arch Dermatol. 1984;120:484-489.
- Gyselbrecht L, de Keyser F, Ongenae K, et al. Etiological factors and underlying conditions in patientswith leucocytoclastic vasculitis. Clin Exp Rheumatol. 1996;14:665-668.
- Sais G, Vidaller A, Jucglà A, et al. Colchicine in the treatment of cutaneous leukocytoclastic vasculitis. results of a prospective, randomized controlled trial. Arch Dermatol. 1995;131:1399-1402.
- Sunderkotter C, Bonsmann G, Sindrilaru A, et al. Management of leukocytoclastic vasculitis: clinical review. J Dermatol Treat. 2005;16:193-206.
- Zhu YI, Stiller MJ. Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol. 2001;45:420-434.
- Stotland M, Shalita AR, Kissling RF. Dapsone 5% gel: a review of its efficacy and safety in the treatment of acne vulgaris. Am J Clin Dermatol. 2009;10:221-227.
- Frieling GW, Williams NL, Lim SJ, et al. Novel use of topical dapsone 5% gel for erythema elevatum diutinum: safer and effective. J Drugs Dermatol. 2013;12:481-484.
- Handler MZ, Hamilton H, Aires D. Treatment of peristomal pyoderma gangrenosum with topical crushed dapsone. J Drugs Dermatol. 2011;10:1059-1061.
- Swartzentruber GS, Yanta JH, Pizon AF. Methemoglobi-nemia as a complication of topical dapsone. N Engl J Med. 2015;372:491-492.
Practice Points
- Leukocytoclastic vasculitis is characterized by inflammation of small vessels with characteristic clinical findings of petechiae and palpable purpura.
- Leukocytoclastic vasculitis often spontaneously resolves within weeks and requires only symptomatic treatment, but chronic or severe disease can require systemic medical treatment with agents such as colchicine, dapsone, and corticosteroids.
Muckle-Wells Syndrome in the Setting of Basal Cell Nevus Syndrome
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.

The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
Practice Points
- An urticarial rash occurring in childhood with symptoms of fever, joint pain, and swelling along with visual symptoms should prompt consideration of a cryopyrin-associated periodic syndrome.
- Histopathology shows a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. This atypical urticaria contrasts with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria.