User login
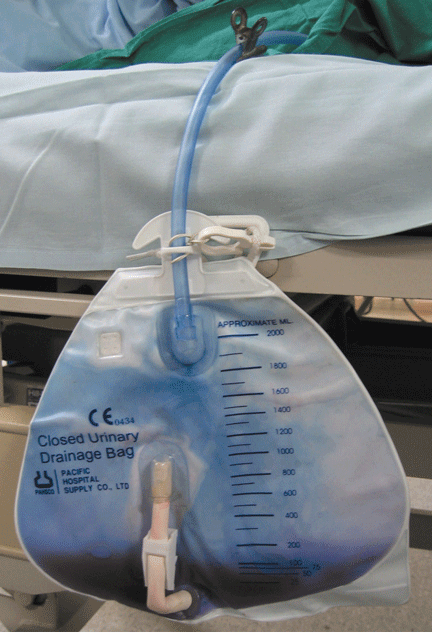
Purple urine in a woman with chronic kidney disease
On examination, her blood pressure is 138/70 mm Hg, respiratory rate 18 breaths/minute, and heart rate 80 beats/minute. Her abdomen is soft. She has never undergone abdominal surgery. She is not taking any medications that may have caused urine discoloration.
Plain radiography of the abdomen reveals no abnormal gas. Laboratory test results are as follows:
- White blood cell count 15.1 × 109/L (reference range 4–11), with 85% neutrophils (reference range 39.5%–74%)
- C-reactive protein 6.27 mg/dL (reference range 0.0–1.0)
- Blood urea nitrogen 54 mg/dL (reference range 8–25)
- Serum creatinine 2.5 mg/dL (0.70–1.40)
- Estimated glomerular filtration rate 20 mL/min/1.73 m2 (< 60 is sufficient for the diagnosis of chronic kidney disease)
- Liver function tests are normal
- Urine pH 8.0 (4.80–7.80); urine is positive for nitrates and for marked pyuria and bacteriuria.
Urine culture yields more than 100,000 colony-forming units of Pseudomonas aeruginosa, Morganella morganii, and Proteus vulgaris. These results and the patient’s presentation point to a diagnosis of purple urine bag syndrome. After placement of a new urinary catheter and 7 days of intravenous ciprofloxacin (Cipro) 250 mg every 12 hours, the color of her urine returns to normal.
PURPLE URINE BAG SYNDROME
Purple urine bag syndrome is rare, and catheter-associated urinary tract infection is the main cause.1 However, it has also been associated with intestinal intussusception.2 In our patient, the examination and radiography ruled out intussusception.
Factors reported to be involved in the development of this syndrome include older age, female sex, chronic constipation, chronic urinary catheterization, alkaline (common) or acidic (uncommon) urine, and a higher bacterial load in the urine.3,4
The pathogenesis of purple-colored urine4,5 is thought to start with the metabolism of dietary tryptophan by intestinal bacteria to indole. Indole is then absorbed into the portal circulation and is converted to indoxyl sulfate, which is excreted into the urine. In vitro experiments4,5 have shown that certain bacteria in the urine produce indoxyl sulfatase and indoxyl phosphatase, which break down indoxyl sulfate to indoxyl. Indoxyl can then be converted to indigo or indirubin in alkaline5 or acidic4 urine. When blue indigo and red indirubin mix together, the result is purple.4,5
Bacteria that possess indoxyl sulfatase or indoxyl phosphatase include P aeruginosa, M morganii, P vulgaris, Escherichia coli, and Providencia species.5,6 However, not all bacteria of the same species produce the enzymes required for the formation of purple urine.5 This may explain the rarity of this syndrome despite the common occurrence of urinary tract infection in patients with risk factors for purple urine bag syndrome.
CHRONIC KIDNEY DISEASE: A POTENTIAL RISK FACTOR
Chronic kidney disease was shown to be a risk factor for purple urine bag syndrome in a small cohort study of Taiwanese patients.7 The serum and urine levels of indoxyl sulfate increased markedly in patients who had chronic kidney disease or who were undergoing dialysis because of impaired renal clearance.6 Furthermore, indoxyl sulfate, which plays an important role in this syndrome, is also cytotoxic and may increase the rate of renal failure in uremic rats.4
Although purple urine itself is usually considered benign,3 it should prompt an evaluation for urinary tract infection, especially in patients with kidney disease. Failure to treat the underlying infection can lead to septicemia or Fourier gangrene.1,3
- Tasi YM, Huang MS, Yang CJ, Yeh SM, Liu CC. Purple urine bag syndrome, not always a benign process. Am J Emerg Med 2009; 27:895–897.
- Pillai RN, Clavijo J, Narayanan M, Zaman K. An association of purple urine bag syndrome with intussusception. Urology 2007; 70:812.e1–812.e2.
- Pillai BP, Chong VH, Yong AM. Purple urine bag syndrome. Singapore Med J 2009; 50:e193–e194.
- Bar-Or D, Rael LT, Bar-Or R, Craun ML, Statz J, Garrett RE. Mass spectrometry analysis of urine and catheter of a patient with purple urinary bag syndrome. Clin Chim Acta 2007; 378:216–218.
- Dealler SF, Hawkey PM, Millar MR. Enzymatic degradation of urinary indoxyl sulfate by Providencia stuartii and Klebsiella pneumoniae causes the purple urine bag syndrome. J Clin Microbiol 1988; 26:2152–2156.
- Wang IK, Ho DR, Chang HY, Lin CL, Chuang FR. Purple urine bag syndrome in a hemodialysis patient. Intern Med 2005; 44:859–861.
- Yang CJ, Lu PL, Chen TC, et al. Chronic kidney disease is a potential risk factor for the development of purple urine bag syndrome. J Am Geriatr Soc 2009; 57:1937–1938.
On examination, her blood pressure is 138/70 mm Hg, respiratory rate 18 breaths/minute, and heart rate 80 beats/minute. Her abdomen is soft. She has never undergone abdominal surgery. She is not taking any medications that may have caused urine discoloration.
Plain radiography of the abdomen reveals no abnormal gas. Laboratory test results are as follows:
- White blood cell count 15.1 × 109/L (reference range 4–11), with 85% neutrophils (reference range 39.5%–74%)
- C-reactive protein 6.27 mg/dL (reference range 0.0–1.0)
- Blood urea nitrogen 54 mg/dL (reference range 8–25)
- Serum creatinine 2.5 mg/dL (0.70–1.40)
- Estimated glomerular filtration rate 20 mL/min/1.73 m2 (< 60 is sufficient for the diagnosis of chronic kidney disease)
- Liver function tests are normal
- Urine pH 8.0 (4.80–7.80); urine is positive for nitrates and for marked pyuria and bacteriuria.
Urine culture yields more than 100,000 colony-forming units of Pseudomonas aeruginosa, Morganella morganii, and Proteus vulgaris. These results and the patient’s presentation point to a diagnosis of purple urine bag syndrome. After placement of a new urinary catheter and 7 days of intravenous ciprofloxacin (Cipro) 250 mg every 12 hours, the color of her urine returns to normal.
PURPLE URINE BAG SYNDROME
Purple urine bag syndrome is rare, and catheter-associated urinary tract infection is the main cause.1 However, it has also been associated with intestinal intussusception.2 In our patient, the examination and radiography ruled out intussusception.
Factors reported to be involved in the development of this syndrome include older age, female sex, chronic constipation, chronic urinary catheterization, alkaline (common) or acidic (uncommon) urine, and a higher bacterial load in the urine.3,4
The pathogenesis of purple-colored urine4,5 is thought to start with the metabolism of dietary tryptophan by intestinal bacteria to indole. Indole is then absorbed into the portal circulation and is converted to indoxyl sulfate, which is excreted into the urine. In vitro experiments4,5 have shown that certain bacteria in the urine produce indoxyl sulfatase and indoxyl phosphatase, which break down indoxyl sulfate to indoxyl. Indoxyl can then be converted to indigo or indirubin in alkaline5 or acidic4 urine. When blue indigo and red indirubin mix together, the result is purple.4,5
Bacteria that possess indoxyl sulfatase or indoxyl phosphatase include P aeruginosa, M morganii, P vulgaris, Escherichia coli, and Providencia species.5,6 However, not all bacteria of the same species produce the enzymes required for the formation of purple urine.5 This may explain the rarity of this syndrome despite the common occurrence of urinary tract infection in patients with risk factors for purple urine bag syndrome.
CHRONIC KIDNEY DISEASE: A POTENTIAL RISK FACTOR
Chronic kidney disease was shown to be a risk factor for purple urine bag syndrome in a small cohort study of Taiwanese patients.7 The serum and urine levels of indoxyl sulfate increased markedly in patients who had chronic kidney disease or who were undergoing dialysis because of impaired renal clearance.6 Furthermore, indoxyl sulfate, which plays an important role in this syndrome, is also cytotoxic and may increase the rate of renal failure in uremic rats.4
Although purple urine itself is usually considered benign,3 it should prompt an evaluation for urinary tract infection, especially in patients with kidney disease. Failure to treat the underlying infection can lead to septicemia or Fourier gangrene.1,3
On examination, her blood pressure is 138/70 mm Hg, respiratory rate 18 breaths/minute, and heart rate 80 beats/minute. Her abdomen is soft. She has never undergone abdominal surgery. She is not taking any medications that may have caused urine discoloration.
Plain radiography of the abdomen reveals no abnormal gas. Laboratory test results are as follows:
- White blood cell count 15.1 × 109/L (reference range 4–11), with 85% neutrophils (reference range 39.5%–74%)
- C-reactive protein 6.27 mg/dL (reference range 0.0–1.0)
- Blood urea nitrogen 54 mg/dL (reference range 8–25)
- Serum creatinine 2.5 mg/dL (0.70–1.40)
- Estimated glomerular filtration rate 20 mL/min/1.73 m2 (< 60 is sufficient for the diagnosis of chronic kidney disease)
- Liver function tests are normal
- Urine pH 8.0 (4.80–7.80); urine is positive for nitrates and for marked pyuria and bacteriuria.
Urine culture yields more than 100,000 colony-forming units of Pseudomonas aeruginosa, Morganella morganii, and Proteus vulgaris. These results and the patient’s presentation point to a diagnosis of purple urine bag syndrome. After placement of a new urinary catheter and 7 days of intravenous ciprofloxacin (Cipro) 250 mg every 12 hours, the color of her urine returns to normal.
PURPLE URINE BAG SYNDROME
Purple urine bag syndrome is rare, and catheter-associated urinary tract infection is the main cause.1 However, it has also been associated with intestinal intussusception.2 In our patient, the examination and radiography ruled out intussusception.
Factors reported to be involved in the development of this syndrome include older age, female sex, chronic constipation, chronic urinary catheterization, alkaline (common) or acidic (uncommon) urine, and a higher bacterial load in the urine.3,4
The pathogenesis of purple-colored urine4,5 is thought to start with the metabolism of dietary tryptophan by intestinal bacteria to indole. Indole is then absorbed into the portal circulation and is converted to indoxyl sulfate, which is excreted into the urine. In vitro experiments4,5 have shown that certain bacteria in the urine produce indoxyl sulfatase and indoxyl phosphatase, which break down indoxyl sulfate to indoxyl. Indoxyl can then be converted to indigo or indirubin in alkaline5 or acidic4 urine. When blue indigo and red indirubin mix together, the result is purple.4,5
Bacteria that possess indoxyl sulfatase or indoxyl phosphatase include P aeruginosa, M morganii, P vulgaris, Escherichia coli, and Providencia species.5,6 However, not all bacteria of the same species produce the enzymes required for the formation of purple urine.5 This may explain the rarity of this syndrome despite the common occurrence of urinary tract infection in patients with risk factors for purple urine bag syndrome.
CHRONIC KIDNEY DISEASE: A POTENTIAL RISK FACTOR
Chronic kidney disease was shown to be a risk factor for purple urine bag syndrome in a small cohort study of Taiwanese patients.7 The serum and urine levels of indoxyl sulfate increased markedly in patients who had chronic kidney disease or who were undergoing dialysis because of impaired renal clearance.6 Furthermore, indoxyl sulfate, which plays an important role in this syndrome, is also cytotoxic and may increase the rate of renal failure in uremic rats.4
Although purple urine itself is usually considered benign,3 it should prompt an evaluation for urinary tract infection, especially in patients with kidney disease. Failure to treat the underlying infection can lead to septicemia or Fourier gangrene.1,3
- Tasi YM, Huang MS, Yang CJ, Yeh SM, Liu CC. Purple urine bag syndrome, not always a benign process. Am J Emerg Med 2009; 27:895–897.
- Pillai RN, Clavijo J, Narayanan M, Zaman K. An association of purple urine bag syndrome with intussusception. Urology 2007; 70:812.e1–812.e2.
- Pillai BP, Chong VH, Yong AM. Purple urine bag syndrome. Singapore Med J 2009; 50:e193–e194.
- Bar-Or D, Rael LT, Bar-Or R, Craun ML, Statz J, Garrett RE. Mass spectrometry analysis of urine and catheter of a patient with purple urinary bag syndrome. Clin Chim Acta 2007; 378:216–218.
- Dealler SF, Hawkey PM, Millar MR. Enzymatic degradation of urinary indoxyl sulfate by Providencia stuartii and Klebsiella pneumoniae causes the purple urine bag syndrome. J Clin Microbiol 1988; 26:2152–2156.
- Wang IK, Ho DR, Chang HY, Lin CL, Chuang FR. Purple urine bag syndrome in a hemodialysis patient. Intern Med 2005; 44:859–861.
- Yang CJ, Lu PL, Chen TC, et al. Chronic kidney disease is a potential risk factor for the development of purple urine bag syndrome. J Am Geriatr Soc 2009; 57:1937–1938.
- Tasi YM, Huang MS, Yang CJ, Yeh SM, Liu CC. Purple urine bag syndrome, not always a benign process. Am J Emerg Med 2009; 27:895–897.
- Pillai RN, Clavijo J, Narayanan M, Zaman K. An association of purple urine bag syndrome with intussusception. Urology 2007; 70:812.e1–812.e2.
- Pillai BP, Chong VH, Yong AM. Purple urine bag syndrome. Singapore Med J 2009; 50:e193–e194.
- Bar-Or D, Rael LT, Bar-Or R, Craun ML, Statz J, Garrett RE. Mass spectrometry analysis of urine and catheter of a patient with purple urinary bag syndrome. Clin Chim Acta 2007; 378:216–218.
- Dealler SF, Hawkey PM, Millar MR. Enzymatic degradation of urinary indoxyl sulfate by Providencia stuartii and Klebsiella pneumoniae causes the purple urine bag syndrome. J Clin Microbiol 1988; 26:2152–2156.
- Wang IK, Ho DR, Chang HY, Lin CL, Chuang FR. Purple urine bag syndrome in a hemodialysis patient. Intern Med 2005; 44:859–861.
- Yang CJ, Lu PL, Chen TC, et al. Chronic kidney disease is a potential risk factor for the development of purple urine bag syndrome. J Am Geriatr Soc 2009; 57:1937–1938.
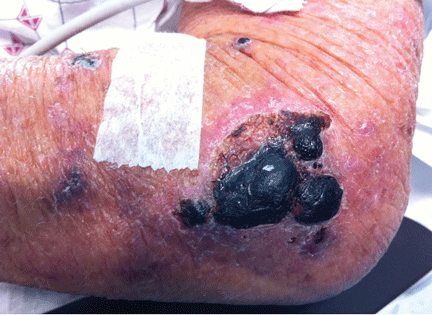
Purpuric lesion on the elbow
A 75-year-old man was admitted to the hospital with new-onset atrial fibrillation. He underwent rate control, and a heparin infusion was started. Warfarin (Coumadin) 10 mg was added on the second hospital day. Two days later, the heparin infusion was discontinued when the international normalized ratio (INR) was in the therapeutic range.
Q: Which is the most likely diagnosis?
- Pyoderma gangrenosum
- Cutaneous vasculitis
- Warfarin-induced skin necrosis
- Ecthyma gangrenosum
- Dermatitis herpetiformis
A: The most likely diagnosis is warfarin-induced skin necrosis, a rare paradoxical complication that occurs in 0.01% to 0.1% of patients receiving this drug.1 Microthrombosis leads to necrosis of the skin and subcutaneous tissues, arising within 2 to 10 days after the start of anticoagulation therapy, although in rare cases it can occur months to years later.2,3
The most common risk factors include the unopposed use of warfarin (ie, unopposed by heparin at the start of therapy), using higher doses of warfarin during the initiation of anticoagulation, and inadequate overlap with an effective parenteral anticoagulant. Patients with protein C or S deficiency, heparin-induced thrombocytopenia,4 resistance to activated protein C, antithrombin deficiency, and lupus anticoagulant have also been reported to be at risk.
The most common sites affected are areas with high subcutaneous fat content, such as the abdomen, thighs, breasts, and buttocks. Skin presentations can vary from dermal plaques to petechial lesions, which rapidly progress to well-demarcated, bluish-black, painful lesions and eventually to hemorrhagic bullae and necrosis.1
At the start of warfarin therapy, the levels of protein C and factor VII (with half-lives of 5 to 8 hours) fall faster than those of other vitamin-K-dependent factors (ie, factors II, IX, and X). This causes a transient imbalance in procoagulant and anticoagulant pathways favoring thrombosis of the microvasculature, with resulting necrosis. Patients with hereditary protein C deficiency are at higher risk.5
Histologic review of lesions often shows venous thrombosis and diffuse necrosis of the dermis and subcutaneous tissue.2
Promptly stopping the warfarin and choosing alternative anticoagulation may help prevent further progression of this condition. Wound care, debridement, and sometimes skin grafting may be necessary, depending on the extent of the lesions. A rechallenge with warfarin is often difficult, but cases have been reported in which treatment was resumed without adverse consequences.6 Avoiding large loading doses of warfarin, gradually increasing doses over an extended period (about 10 days),3 and starting warfarin with a heparin bridge for at least 5 days (which was not done in this patient) would prevent the condition.
Early recognition, differentiation, and diagnosis are essential to minimize morbidity and to prevent death.
CASE CONTINUED
Warfarin was discontinued once the patient developed the skin lesions. He received vitamin K and fresh frozen plasma to normalize his INR, and he was started on a heparin infusion, after which the lesions began to heal. The patient refused a skin biopsy. Platelet counts remained stable during his hospital course. Protein C levels were not checked, given his recent use of warfarin. He was started on dabigatran (Pradaxa) and was discharged a week later.
THE OTHER DIAGNOSTIC CHOICES
Pyoderma gangrenosum is an uncommon ulcerative skin condition often associated with autoimmune disease. It usually starts at the site of a minor injury, more commonly on the legs, and gradually progresses to a painful ulcer.
Cutaneous vasculitis is an inflammation of small blood vessels characterized by palpable purpura. The lesions can resemble urticaria, petechia, or erythema multiforme. It is commonly associated with infection, drug therapy, inflammatory disease, and malignancy.
Ecthyma gangrenosum is an infection of skin caused by Pseudomonas aeruginosa. Usually, it presents as hemorrhagic pustules or infarct-like areas with surrounding erythema that evolve into necrotic ulcers surrounded by erythema.
Dermatitis herpetiformis is a chronic skin condition, presenting with fluid-filled blisters and commonly involving the neck, back, scalp, and elbows. This condition is associated with celiac disease, and the lesions are extremely pruritic.
- Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol 2009; 61:325–332.
- Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy 2006; 26:1175–1179.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg 2000; 87:266–272.
- Warkentin TE, Sikov WM, Lillicrap DP. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. Am J Hematol 1999; 62:44–48.
- Ad-El DD, Meirovitz A, Weinberg A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg 2000; 53:624–626.
- Jillella AP, Lutcher CL. Reinstituting warfarin in patients who develop warfarin skin necrosis. Am J Hematol 1996; 52:117–119.
A 75-year-old man was admitted to the hospital with new-onset atrial fibrillation. He underwent rate control, and a heparin infusion was started. Warfarin (Coumadin) 10 mg was added on the second hospital day. Two days later, the heparin infusion was discontinued when the international normalized ratio (INR) was in the therapeutic range.
Q: Which is the most likely diagnosis?
- Pyoderma gangrenosum
- Cutaneous vasculitis
- Warfarin-induced skin necrosis
- Ecthyma gangrenosum
- Dermatitis herpetiformis
A: The most likely diagnosis is warfarin-induced skin necrosis, a rare paradoxical complication that occurs in 0.01% to 0.1% of patients receiving this drug.1 Microthrombosis leads to necrosis of the skin and subcutaneous tissues, arising within 2 to 10 days after the start of anticoagulation therapy, although in rare cases it can occur months to years later.2,3
The most common risk factors include the unopposed use of warfarin (ie, unopposed by heparin at the start of therapy), using higher doses of warfarin during the initiation of anticoagulation, and inadequate overlap with an effective parenteral anticoagulant. Patients with protein C or S deficiency, heparin-induced thrombocytopenia,4 resistance to activated protein C, antithrombin deficiency, and lupus anticoagulant have also been reported to be at risk.
The most common sites affected are areas with high subcutaneous fat content, such as the abdomen, thighs, breasts, and buttocks. Skin presentations can vary from dermal plaques to petechial lesions, which rapidly progress to well-demarcated, bluish-black, painful lesions and eventually to hemorrhagic bullae and necrosis.1
At the start of warfarin therapy, the levels of protein C and factor VII (with half-lives of 5 to 8 hours) fall faster than those of other vitamin-K-dependent factors (ie, factors II, IX, and X). This causes a transient imbalance in procoagulant and anticoagulant pathways favoring thrombosis of the microvasculature, with resulting necrosis. Patients with hereditary protein C deficiency are at higher risk.5
Histologic review of lesions often shows venous thrombosis and diffuse necrosis of the dermis and subcutaneous tissue.2
Promptly stopping the warfarin and choosing alternative anticoagulation may help prevent further progression of this condition. Wound care, debridement, and sometimes skin grafting may be necessary, depending on the extent of the lesions. A rechallenge with warfarin is often difficult, but cases have been reported in which treatment was resumed without adverse consequences.6 Avoiding large loading doses of warfarin, gradually increasing doses over an extended period (about 10 days),3 and starting warfarin with a heparin bridge for at least 5 days (which was not done in this patient) would prevent the condition.
Early recognition, differentiation, and diagnosis are essential to minimize morbidity and to prevent death.
CASE CONTINUED
Warfarin was discontinued once the patient developed the skin lesions. He received vitamin K and fresh frozen plasma to normalize his INR, and he was started on a heparin infusion, after which the lesions began to heal. The patient refused a skin biopsy. Platelet counts remained stable during his hospital course. Protein C levels were not checked, given his recent use of warfarin. He was started on dabigatran (Pradaxa) and was discharged a week later.
THE OTHER DIAGNOSTIC CHOICES
Pyoderma gangrenosum is an uncommon ulcerative skin condition often associated with autoimmune disease. It usually starts at the site of a minor injury, more commonly on the legs, and gradually progresses to a painful ulcer.
Cutaneous vasculitis is an inflammation of small blood vessels characterized by palpable purpura. The lesions can resemble urticaria, petechia, or erythema multiforme. It is commonly associated with infection, drug therapy, inflammatory disease, and malignancy.
Ecthyma gangrenosum is an infection of skin caused by Pseudomonas aeruginosa. Usually, it presents as hemorrhagic pustules or infarct-like areas with surrounding erythema that evolve into necrotic ulcers surrounded by erythema.
Dermatitis herpetiformis is a chronic skin condition, presenting with fluid-filled blisters and commonly involving the neck, back, scalp, and elbows. This condition is associated with celiac disease, and the lesions are extremely pruritic.
A 75-year-old man was admitted to the hospital with new-onset atrial fibrillation. He underwent rate control, and a heparin infusion was started. Warfarin (Coumadin) 10 mg was added on the second hospital day. Two days later, the heparin infusion was discontinued when the international normalized ratio (INR) was in the therapeutic range.
Q: Which is the most likely diagnosis?
- Pyoderma gangrenosum
- Cutaneous vasculitis
- Warfarin-induced skin necrosis
- Ecthyma gangrenosum
- Dermatitis herpetiformis
A: The most likely diagnosis is warfarin-induced skin necrosis, a rare paradoxical complication that occurs in 0.01% to 0.1% of patients receiving this drug.1 Microthrombosis leads to necrosis of the skin and subcutaneous tissues, arising within 2 to 10 days after the start of anticoagulation therapy, although in rare cases it can occur months to years later.2,3
The most common risk factors include the unopposed use of warfarin (ie, unopposed by heparin at the start of therapy), using higher doses of warfarin during the initiation of anticoagulation, and inadequate overlap with an effective parenteral anticoagulant. Patients with protein C or S deficiency, heparin-induced thrombocytopenia,4 resistance to activated protein C, antithrombin deficiency, and lupus anticoagulant have also been reported to be at risk.
The most common sites affected are areas with high subcutaneous fat content, such as the abdomen, thighs, breasts, and buttocks. Skin presentations can vary from dermal plaques to petechial lesions, which rapidly progress to well-demarcated, bluish-black, painful lesions and eventually to hemorrhagic bullae and necrosis.1
At the start of warfarin therapy, the levels of protein C and factor VII (with half-lives of 5 to 8 hours) fall faster than those of other vitamin-K-dependent factors (ie, factors II, IX, and X). This causes a transient imbalance in procoagulant and anticoagulant pathways favoring thrombosis of the microvasculature, with resulting necrosis. Patients with hereditary protein C deficiency are at higher risk.5
Histologic review of lesions often shows venous thrombosis and diffuse necrosis of the dermis and subcutaneous tissue.2
Promptly stopping the warfarin and choosing alternative anticoagulation may help prevent further progression of this condition. Wound care, debridement, and sometimes skin grafting may be necessary, depending on the extent of the lesions. A rechallenge with warfarin is often difficult, but cases have been reported in which treatment was resumed without adverse consequences.6 Avoiding large loading doses of warfarin, gradually increasing doses over an extended period (about 10 days),3 and starting warfarin with a heparin bridge for at least 5 days (which was not done in this patient) would prevent the condition.
Early recognition, differentiation, and diagnosis are essential to minimize morbidity and to prevent death.
CASE CONTINUED
Warfarin was discontinued once the patient developed the skin lesions. He received vitamin K and fresh frozen plasma to normalize his INR, and he was started on a heparin infusion, after which the lesions began to heal. The patient refused a skin biopsy. Platelet counts remained stable during his hospital course. Protein C levels were not checked, given his recent use of warfarin. He was started on dabigatran (Pradaxa) and was discharged a week later.
THE OTHER DIAGNOSTIC CHOICES
Pyoderma gangrenosum is an uncommon ulcerative skin condition often associated with autoimmune disease. It usually starts at the site of a minor injury, more commonly on the legs, and gradually progresses to a painful ulcer.
Cutaneous vasculitis is an inflammation of small blood vessels characterized by palpable purpura. The lesions can resemble urticaria, petechia, or erythema multiforme. It is commonly associated with infection, drug therapy, inflammatory disease, and malignancy.
Ecthyma gangrenosum is an infection of skin caused by Pseudomonas aeruginosa. Usually, it presents as hemorrhagic pustules or infarct-like areas with surrounding erythema that evolve into necrotic ulcers surrounded by erythema.
Dermatitis herpetiformis is a chronic skin condition, presenting with fluid-filled blisters and commonly involving the neck, back, scalp, and elbows. This condition is associated with celiac disease, and the lesions are extremely pruritic.
- Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol 2009; 61:325–332.
- Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy 2006; 26:1175–1179.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg 2000; 87:266–272.
- Warkentin TE, Sikov WM, Lillicrap DP. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. Am J Hematol 1999; 62:44–48.
- Ad-El DD, Meirovitz A, Weinberg A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg 2000; 53:624–626.
- Jillella AP, Lutcher CL. Reinstituting warfarin in patients who develop warfarin skin necrosis. Am J Hematol 1996; 52:117–119.
- Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol 2009; 61:325–332.
- Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy 2006; 26:1175–1179.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg 2000; 87:266–272.
- Warkentin TE, Sikov WM, Lillicrap DP. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. Am J Hematol 1999; 62:44–48.
- Ad-El DD, Meirovitz A, Weinberg A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg 2000; 53:624–626.
- Jillella AP, Lutcher CL. Reinstituting warfarin in patients who develop warfarin skin necrosis. Am J Hematol 1996; 52:117–119.
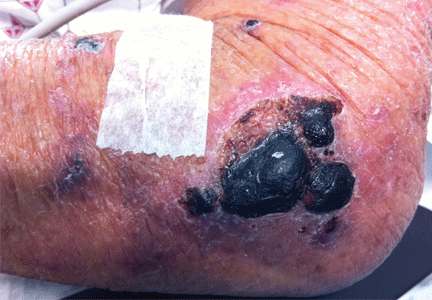
A 26-year-old woman with a lump in her chest
A 26-year-old Filipino woman presented for evaluation of sternal pain associated with a palpable mass that she had noticed 8 months earlier. She had no history of significant medical illness. She had recently immigrated to El Paso, TX, from the Philippines.
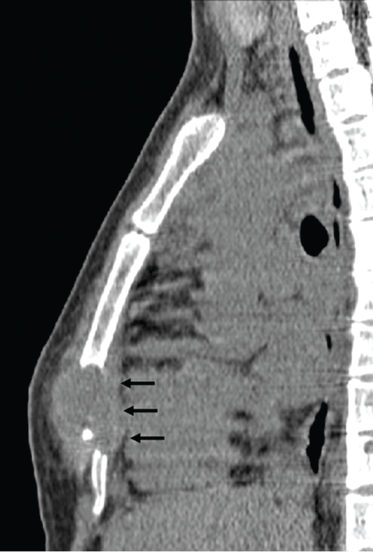
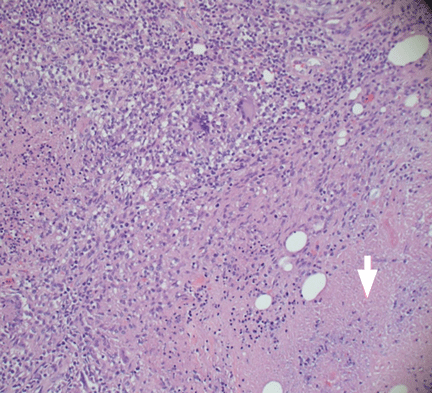
Q: Which is the most likely diagnosis?
- Plasmacytoma
- Chondrosarcoma
- Extrapulmonary tuberculosis
- Lymphoma
- Metastatic breast cancer
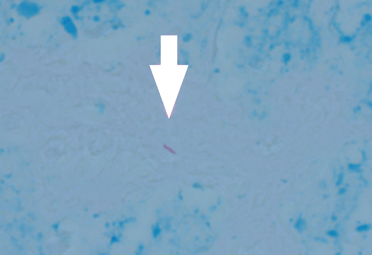
EXTRAPULMONARY TUBERCULOSIS
Extrapulmonary tuberculosis accounts for about 20% of all cases of tuberculosis.1
Risk factors for tuberculosis include advanced age, immunosuppression (eg, as occurs in HIV infection), organ transplantation, and therapy with a tumor necrosis factor alpha inhibitor.1–4 Risk factors unique to extrapulmonary tuberculosis infection include female sex and non-Hispanic black ethnicity.2 Because of the high prevalence of tuberculosis in certain parts of the world, obtaining a travel or residence history is an essential part of the clinical evaluation.
Skeletal tuberculosis accounts for 11% to 27% of extrapulmonary cases and, by extrapolation, 2% to 5% of all cases of tuberculosis.1–3 Although the spine is the site most commonly involved, any bone may be affected. When the chest wall is involved, the most common locations are the margin of the sternum and along rib shafts.5
Most patients present with pain and swelling. The presence of constitutional symptoms is variable, occurring in about one-third of patients.6 Classically, the lesion of tuberculous osteomyelitis is described as a “cold abscess,” as it is characterized by swelling and erythema with little or no warmth. Spontaneous drainage and sinus tract formation may occur.5
The differential diagnosis of tuberculous osteomyelitis includes pyogenic bacterial infection, atypical bacterial infection (nocardia, meliodosis, brucellosis), fungal infection (coccidioidomycosis, histoplasmosis, blastomycosis), and metastatic and primary bone malignancies. Diagnosis requires a high index of suspicion, biopsy for histopathologic examination, acid-fast staining, and mycobacterial culture.7
Patients generally respond well to 6 months of a standard four-drug regimen for tuberculosis. Surgery is indicated for abscess drainage, debridement of infected tissue, spine stabilization, and relief of spinal cord compression.5
Our patient had complete resolution of her sternal mass with drug therapy alone.
- Peto HM, Pratt RH, Harrington TA, LoBue PA, Armstrong LR. Epidemiology of extrapulmonary tuberculosis in the United States, 1993–2006. Clin Infect Dis 2009; 49:1350–1357.
- Yang Z, Kong Y, Wilson F, et al. Identification of risk factors for extrapulmonary tuberculosis. Clin Infect Dis 2004; 38:199–205.
- Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345:1098–1104.
- Alagarsamy S, Dhand S, Aung S, Wolff M, Bahrain M. Sternal tuberculosis: a rare case mimicking sarcoma and review of the literature. Infect Dis Clin Pract 2009; 17:138–143.
- Morris BS, Maheshwari M, Chalwa A. Chest wall tuberculosis: a review of CT appearances. Br J Radiol 2004; 77:449–457.
- Sandher DS, Al-Jibury M, Paton RW, Ormerod LP. Bone and joint tuberculosis: cases in Blackburn between 1988 and 2005. J Bone Joint Surg Br 2007; 89:1379–1381.
- Centers for Disease Control and Prevention (CDC). Case definitions for infectious conditions under public health surveillance. http://cdc.gov/mmwr/preview/mmwrhtml/00047449.htm. Accessed October 6, 2011.
A 26-year-old Filipino woman presented for evaluation of sternal pain associated with a palpable mass that she had noticed 8 months earlier. She had no history of significant medical illness. She had recently immigrated to El Paso, TX, from the Philippines.
Q: Which is the most likely diagnosis?
- Plasmacytoma
- Chondrosarcoma
- Extrapulmonary tuberculosis
- Lymphoma
- Metastatic breast cancer
EXTRAPULMONARY TUBERCULOSIS
Extrapulmonary tuberculosis accounts for about 20% of all cases of tuberculosis.1
Risk factors for tuberculosis include advanced age, immunosuppression (eg, as occurs in HIV infection), organ transplantation, and therapy with a tumor necrosis factor alpha inhibitor.1–4 Risk factors unique to extrapulmonary tuberculosis infection include female sex and non-Hispanic black ethnicity.2 Because of the high prevalence of tuberculosis in certain parts of the world, obtaining a travel or residence history is an essential part of the clinical evaluation.
Skeletal tuberculosis accounts for 11% to 27% of extrapulmonary cases and, by extrapolation, 2% to 5% of all cases of tuberculosis.1–3 Although the spine is the site most commonly involved, any bone may be affected. When the chest wall is involved, the most common locations are the margin of the sternum and along rib shafts.5
Most patients present with pain and swelling. The presence of constitutional symptoms is variable, occurring in about one-third of patients.6 Classically, the lesion of tuberculous osteomyelitis is described as a “cold abscess,” as it is characterized by swelling and erythema with little or no warmth. Spontaneous drainage and sinus tract formation may occur.5
The differential diagnosis of tuberculous osteomyelitis includes pyogenic bacterial infection, atypical bacterial infection (nocardia, meliodosis, brucellosis), fungal infection (coccidioidomycosis, histoplasmosis, blastomycosis), and metastatic and primary bone malignancies. Diagnosis requires a high index of suspicion, biopsy for histopathologic examination, acid-fast staining, and mycobacterial culture.7
Patients generally respond well to 6 months of a standard four-drug regimen for tuberculosis. Surgery is indicated for abscess drainage, debridement of infected tissue, spine stabilization, and relief of spinal cord compression.5
Our patient had complete resolution of her sternal mass with drug therapy alone.
A 26-year-old Filipino woman presented for evaluation of sternal pain associated with a palpable mass that she had noticed 8 months earlier. She had no history of significant medical illness. She had recently immigrated to El Paso, TX, from the Philippines.
Q: Which is the most likely diagnosis?
- Plasmacytoma
- Chondrosarcoma
- Extrapulmonary tuberculosis
- Lymphoma
- Metastatic breast cancer
EXTRAPULMONARY TUBERCULOSIS
Extrapulmonary tuberculosis accounts for about 20% of all cases of tuberculosis.1
Risk factors for tuberculosis include advanced age, immunosuppression (eg, as occurs in HIV infection), organ transplantation, and therapy with a tumor necrosis factor alpha inhibitor.1–4 Risk factors unique to extrapulmonary tuberculosis infection include female sex and non-Hispanic black ethnicity.2 Because of the high prevalence of tuberculosis in certain parts of the world, obtaining a travel or residence history is an essential part of the clinical evaluation.
Skeletal tuberculosis accounts for 11% to 27% of extrapulmonary cases and, by extrapolation, 2% to 5% of all cases of tuberculosis.1–3 Although the spine is the site most commonly involved, any bone may be affected. When the chest wall is involved, the most common locations are the margin of the sternum and along rib shafts.5
Most patients present with pain and swelling. The presence of constitutional symptoms is variable, occurring in about one-third of patients.6 Classically, the lesion of tuberculous osteomyelitis is described as a “cold abscess,” as it is characterized by swelling and erythema with little or no warmth. Spontaneous drainage and sinus tract formation may occur.5
The differential diagnosis of tuberculous osteomyelitis includes pyogenic bacterial infection, atypical bacterial infection (nocardia, meliodosis, brucellosis), fungal infection (coccidioidomycosis, histoplasmosis, blastomycosis), and metastatic and primary bone malignancies. Diagnosis requires a high index of suspicion, biopsy for histopathologic examination, acid-fast staining, and mycobacterial culture.7
Patients generally respond well to 6 months of a standard four-drug regimen for tuberculosis. Surgery is indicated for abscess drainage, debridement of infected tissue, spine stabilization, and relief of spinal cord compression.5
Our patient had complete resolution of her sternal mass with drug therapy alone.
- Peto HM, Pratt RH, Harrington TA, LoBue PA, Armstrong LR. Epidemiology of extrapulmonary tuberculosis in the United States, 1993–2006. Clin Infect Dis 2009; 49:1350–1357.
- Yang Z, Kong Y, Wilson F, et al. Identification of risk factors for extrapulmonary tuberculosis. Clin Infect Dis 2004; 38:199–205.
- Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345:1098–1104.
- Alagarsamy S, Dhand S, Aung S, Wolff M, Bahrain M. Sternal tuberculosis: a rare case mimicking sarcoma and review of the literature. Infect Dis Clin Pract 2009; 17:138–143.
- Morris BS, Maheshwari M, Chalwa A. Chest wall tuberculosis: a review of CT appearances. Br J Radiol 2004; 77:449–457.
- Sandher DS, Al-Jibury M, Paton RW, Ormerod LP. Bone and joint tuberculosis: cases in Blackburn between 1988 and 2005. J Bone Joint Surg Br 2007; 89:1379–1381.
- Centers for Disease Control and Prevention (CDC). Case definitions for infectious conditions under public health surveillance. http://cdc.gov/mmwr/preview/mmwrhtml/00047449.htm. Accessed October 6, 2011.
- Peto HM, Pratt RH, Harrington TA, LoBue PA, Armstrong LR. Epidemiology of extrapulmonary tuberculosis in the United States, 1993–2006. Clin Infect Dis 2009; 49:1350–1357.
- Yang Z, Kong Y, Wilson F, et al. Identification of risk factors for extrapulmonary tuberculosis. Clin Infect Dis 2004; 38:199–205.
- Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001; 345:1098–1104.
- Alagarsamy S, Dhand S, Aung S, Wolff M, Bahrain M. Sternal tuberculosis: a rare case mimicking sarcoma and review of the literature. Infect Dis Clin Pract 2009; 17:138–143.
- Morris BS, Maheshwari M, Chalwa A. Chest wall tuberculosis: a review of CT appearances. Br J Radiol 2004; 77:449–457.
- Sandher DS, Al-Jibury M, Paton RW, Ormerod LP. Bone and joint tuberculosis: cases in Blackburn between 1988 and 2005. J Bone Joint Surg Br 2007; 89:1379–1381.
- Centers for Disease Control and Prevention (CDC). Case definitions for infectious conditions under public health surveillance. http://cdc.gov/mmwr/preview/mmwrhtml/00047449.htm. Accessed October 6, 2011.
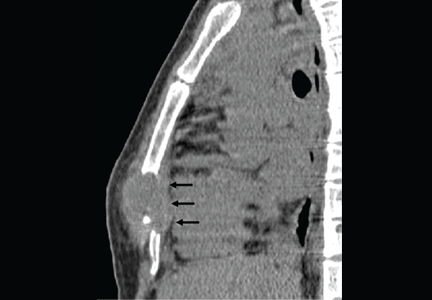
High creatinine 6 months after renal transplant
A 42-year-old man presented with acute renal failure with a serum creatinine of 6.07 mg/dL (baseline 2.0 mg/dL) 6 months after receiving a kidney transplant from a deceased donor. He was asymptomatic, had no previous symptoms of transplant rejection, and was compliant with his immunosuppressive regimen. The physical examination and the rest of the laboratory workup were normal.
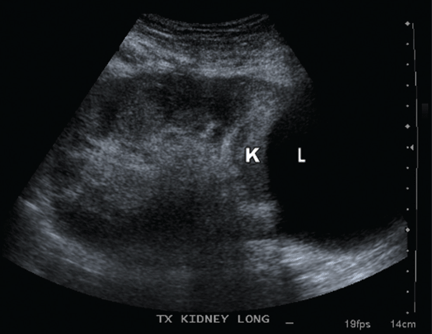
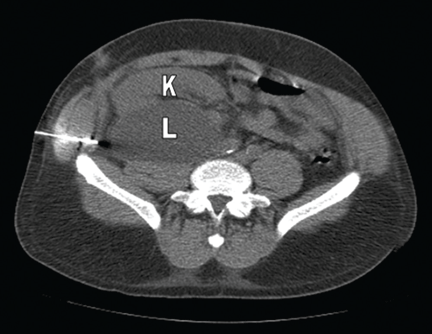
Q: Which is the most likely diagnosis?
- A lymphocele
- A hematoma
- A urinoma
- A perirenal abscess
- A simple renal cyst
A: A lymphocele is the most likely diagnosis. A lymphocele— a collection of lymph without an epithelial lining—develops in as many as 20% of kidney transplant recipients.1 Many causative factors have been proposed, including leakage of lymph from recipient lymphatic channels,2 use of diuretics,2 obesity,3 kidney biopsy,4 acute rejection,3 and the use of sirolimus5 (Rapamune) and high-dose corticosteroids.6 Some believe that lymphoceles may also arise from severed lymphatic vessels of the donor-kidney allograft.7
Ultrasonography can usually distinguish a lymphocele from other fluid collections on the basis of fluid appearance, shape, and position. In most cases, the lymphocele is adjacent to the lower pole and medial to the allograft, and appears anechoic on ultrasonography, with a thin, distinct wall. The typical features on analysis of aspirated fluid—ie, a creatinine level approximately the same as in the serum, a low protein value, and a high lymphocyte count compared with serum values—confirm the diagnosis of lymphocele.
A hematoma can occur in any location and have a heterogeneous appearance, as it contains both clotted (echogenic) and unclotted (anechoic) blood. They are usually seen within the first 1 to 2 weeks after surgery and may also develop after trauma or renal biopsy.
A urinoma is a collection of urine outside the bladder, resulting from a ureteral leak. They are predominantly anechoic, with an often indistinct wall. If there is a clinical suspicion, the diagnosis can be confirmed on aspiration by a high creatinine level in the fluid compared with the serum value.
A perirenal abscess commonly presents with pain, fever, and a complex fluid collection on ultrasonography, sometimes with an air fluid level. Aspiration of purulent fluid confirms the diagnosis.
A simple renal cyst appears within or protruding from the renal parenchyma as a spherical or eggshaped fluid-filled sac with an anechoic lumen and no measurable wall thickness.
SYMPTOMS AND MANAGEMENT
Lymphoceles are mostly inconsequential but can cause renal failure by compressing the ureter, renal vessels, or renal allograft. Other manifestations may include pain and swelling at the kidney allograft site, wound drainage, unilateral lower-extremity edema, deep vein thrombosis due to compression of iliac veins,8 urinary urgency or frequency due to extrinsic bladder compression, and urinary retention.9
If the lymphocele is clinically significant, percutaneous drainage guided by ultrasonography is recommended as the initial curative procedure.10 Sclerotherapy with different chemical agents is effective, but success depends on the size of the lymphocele cavity.11
If these conservative therapies fail, lymphocele unroofing into the peritoneal cavity is needed. This is accomplished by laparoscopy12,13 or open surgery. Although laparoscopic drainage is considered the procedure of choice, open surgery may be required for multiloculated lymphoceles and those adjacent to vital structures.14,15
Kidneys are the most commonly transplanted solid organs. Every year, about 16,000 kidney transplantations are performed in the United States. It is common for the primary care physician to initially see these patients in cases of associated complications. Internists must be aware of the common causes of acute renal failure in this population, eg, acute rejection, drug toxicity, and obstruction. Lymphoceles are an important cause of renal failure due to obstruction. Early recognition and appropriate treatment of this complication can improve the outcome of the allograft.
- O’neill WC, Baumgarten DA. Ultrasonography in renal transplantation. Am J Kidney Dis 2002; 39:663–678.
- Braun WE, Banowsky LH, Straffon RA, et al. Lymphocytes associated with renal transplantation. Report of 15 cases and review of the literature. Am J Med 1974; 57:714–729.
- Goel M, Flechner SM, Zhou L, et al. The influence of various maintenance immunosuppressive drugs on lymphocele formation and treatment after kidney transplantation. J Urol 2004; 171:1788–1792.
- Mundy AR, Podesta ML, Bewick M, Rudge CJ, Ellis FG. The urological complications of 1000 renal transplants. Br J Urol 1981; 53:397–402.
- Giessing M, Fischer TJ, Deger S, et al. Increased frequency of lymphoceles under treatment with sirolimus following renal transplantation: a single center experience. Transplant Proc 2002; 34:1815–1816.
- Amante AJ, Kahan BD. Technical complications of renal transplantation. Surg Clin North Am 1994; 74:1117–1131.
- Saidi RF, Wertheim JA, Ko DS, et al. Impact of donor kidney recovery method on lymphatic complications in kidney transplantation. Transplant Proc 2008; 40:1054–1055.
- Iwan-Zietek I, Zietek Z, Sulikowski T, et al. Minimally invasive methods for the treatment of lymphocele after kidney transplantation. Transplant Proc 2009; 41:3073–3076.
- Hwang EC, Kang TW, Koh YS, et al. Post-transplant lymphocele: an unusual cause of acute urinary retention mimicking urethral injury. Int J Urol 2006; 13:468–470.
- Zietek Z, Sulikowski T, Tejchman K, et al. Lymphocele after kidney transplantation. Transplant Proc 2007; 39:2744–2747.
- Mahrer A, Ramchandani P, Trerotola SO, Shlansky-Goldberg RD, Itkin M. Sclerotherapy in the management of postoperative lymphocele. J Vasc Interv Radiol 2010; 21:1050–1053.
- Risaliti A, Corno V, Donini A, et al. Laparoscopic treatment of symptomatic lymphoceles after kidney transplantation. Surg Endosc 2000; 14:293–295.
- Ostrowski M, Lubikowski J, Kowalczyk M, Power J. Laparoscopic lymphocele drainage after renal transplantation. Ann Transplant 2000; 5:25–27.
- Fuller TF, Kang SM, Hirose R, Feng S, Stock PG, Freise CE. Management of lymphoceles after renal transplantation: laparoscopic versus open drainage. J Urol 2003; 169:2022–2025.
- Hsu TH, Gill IS, Grune MT, et al. Laparoscopic lymphocelectomy: a multi-institutional analysis. J Urol 2000; 163:1096–1098.
A 42-year-old man presented with acute renal failure with a serum creatinine of 6.07 mg/dL (baseline 2.0 mg/dL) 6 months after receiving a kidney transplant from a deceased donor. He was asymptomatic, had no previous symptoms of transplant rejection, and was compliant with his immunosuppressive regimen. The physical examination and the rest of the laboratory workup were normal.
Q: Which is the most likely diagnosis?
- A lymphocele
- A hematoma
- A urinoma
- A perirenal abscess
- A simple renal cyst
A: A lymphocele is the most likely diagnosis. A lymphocele— a collection of lymph without an epithelial lining—develops in as many as 20% of kidney transplant recipients.1 Many causative factors have been proposed, including leakage of lymph from recipient lymphatic channels,2 use of diuretics,2 obesity,3 kidney biopsy,4 acute rejection,3 and the use of sirolimus5 (Rapamune) and high-dose corticosteroids.6 Some believe that lymphoceles may also arise from severed lymphatic vessels of the donor-kidney allograft.7
Ultrasonography can usually distinguish a lymphocele from other fluid collections on the basis of fluid appearance, shape, and position. In most cases, the lymphocele is adjacent to the lower pole and medial to the allograft, and appears anechoic on ultrasonography, with a thin, distinct wall. The typical features on analysis of aspirated fluid—ie, a creatinine level approximately the same as in the serum, a low protein value, and a high lymphocyte count compared with serum values—confirm the diagnosis of lymphocele.
A hematoma can occur in any location and have a heterogeneous appearance, as it contains both clotted (echogenic) and unclotted (anechoic) blood. They are usually seen within the first 1 to 2 weeks after surgery and may also develop after trauma or renal biopsy.
A urinoma is a collection of urine outside the bladder, resulting from a ureteral leak. They are predominantly anechoic, with an often indistinct wall. If there is a clinical suspicion, the diagnosis can be confirmed on aspiration by a high creatinine level in the fluid compared with the serum value.
A perirenal abscess commonly presents with pain, fever, and a complex fluid collection on ultrasonography, sometimes with an air fluid level. Aspiration of purulent fluid confirms the diagnosis.
A simple renal cyst appears within or protruding from the renal parenchyma as a spherical or eggshaped fluid-filled sac with an anechoic lumen and no measurable wall thickness.
SYMPTOMS AND MANAGEMENT
Lymphoceles are mostly inconsequential but can cause renal failure by compressing the ureter, renal vessels, or renal allograft. Other manifestations may include pain and swelling at the kidney allograft site, wound drainage, unilateral lower-extremity edema, deep vein thrombosis due to compression of iliac veins,8 urinary urgency or frequency due to extrinsic bladder compression, and urinary retention.9
If the lymphocele is clinically significant, percutaneous drainage guided by ultrasonography is recommended as the initial curative procedure.10 Sclerotherapy with different chemical agents is effective, but success depends on the size of the lymphocele cavity.11
If these conservative therapies fail, lymphocele unroofing into the peritoneal cavity is needed. This is accomplished by laparoscopy12,13 or open surgery. Although laparoscopic drainage is considered the procedure of choice, open surgery may be required for multiloculated lymphoceles and those adjacent to vital structures.14,15
Kidneys are the most commonly transplanted solid organs. Every year, about 16,000 kidney transplantations are performed in the United States. It is common for the primary care physician to initially see these patients in cases of associated complications. Internists must be aware of the common causes of acute renal failure in this population, eg, acute rejection, drug toxicity, and obstruction. Lymphoceles are an important cause of renal failure due to obstruction. Early recognition and appropriate treatment of this complication can improve the outcome of the allograft.
A 42-year-old man presented with acute renal failure with a serum creatinine of 6.07 mg/dL (baseline 2.0 mg/dL) 6 months after receiving a kidney transplant from a deceased donor. He was asymptomatic, had no previous symptoms of transplant rejection, and was compliant with his immunosuppressive regimen. The physical examination and the rest of the laboratory workup were normal.
Q: Which is the most likely diagnosis?
- A lymphocele
- A hematoma
- A urinoma
- A perirenal abscess
- A simple renal cyst
A: A lymphocele is the most likely diagnosis. A lymphocele— a collection of lymph without an epithelial lining—develops in as many as 20% of kidney transplant recipients.1 Many causative factors have been proposed, including leakage of lymph from recipient lymphatic channels,2 use of diuretics,2 obesity,3 kidney biopsy,4 acute rejection,3 and the use of sirolimus5 (Rapamune) and high-dose corticosteroids.6 Some believe that lymphoceles may also arise from severed lymphatic vessels of the donor-kidney allograft.7
Ultrasonography can usually distinguish a lymphocele from other fluid collections on the basis of fluid appearance, shape, and position. In most cases, the lymphocele is adjacent to the lower pole and medial to the allograft, and appears anechoic on ultrasonography, with a thin, distinct wall. The typical features on analysis of aspirated fluid—ie, a creatinine level approximately the same as in the serum, a low protein value, and a high lymphocyte count compared with serum values—confirm the diagnosis of lymphocele.
A hematoma can occur in any location and have a heterogeneous appearance, as it contains both clotted (echogenic) and unclotted (anechoic) blood. They are usually seen within the first 1 to 2 weeks after surgery and may also develop after trauma or renal biopsy.
A urinoma is a collection of urine outside the bladder, resulting from a ureteral leak. They are predominantly anechoic, with an often indistinct wall. If there is a clinical suspicion, the diagnosis can be confirmed on aspiration by a high creatinine level in the fluid compared with the serum value.
A perirenal abscess commonly presents with pain, fever, and a complex fluid collection on ultrasonography, sometimes with an air fluid level. Aspiration of purulent fluid confirms the diagnosis.
A simple renal cyst appears within or protruding from the renal parenchyma as a spherical or eggshaped fluid-filled sac with an anechoic lumen and no measurable wall thickness.
SYMPTOMS AND MANAGEMENT
Lymphoceles are mostly inconsequential but can cause renal failure by compressing the ureter, renal vessels, or renal allograft. Other manifestations may include pain and swelling at the kidney allograft site, wound drainage, unilateral lower-extremity edema, deep vein thrombosis due to compression of iliac veins,8 urinary urgency or frequency due to extrinsic bladder compression, and urinary retention.9
If the lymphocele is clinically significant, percutaneous drainage guided by ultrasonography is recommended as the initial curative procedure.10 Sclerotherapy with different chemical agents is effective, but success depends on the size of the lymphocele cavity.11
If these conservative therapies fail, lymphocele unroofing into the peritoneal cavity is needed. This is accomplished by laparoscopy12,13 or open surgery. Although laparoscopic drainage is considered the procedure of choice, open surgery may be required for multiloculated lymphoceles and those adjacent to vital structures.14,15
Kidneys are the most commonly transplanted solid organs. Every year, about 16,000 kidney transplantations are performed in the United States. It is common for the primary care physician to initially see these patients in cases of associated complications. Internists must be aware of the common causes of acute renal failure in this population, eg, acute rejection, drug toxicity, and obstruction. Lymphoceles are an important cause of renal failure due to obstruction. Early recognition and appropriate treatment of this complication can improve the outcome of the allograft.
- O’neill WC, Baumgarten DA. Ultrasonography in renal transplantation. Am J Kidney Dis 2002; 39:663–678.
- Braun WE, Banowsky LH, Straffon RA, et al. Lymphocytes associated with renal transplantation. Report of 15 cases and review of the literature. Am J Med 1974; 57:714–729.
- Goel M, Flechner SM, Zhou L, et al. The influence of various maintenance immunosuppressive drugs on lymphocele formation and treatment after kidney transplantation. J Urol 2004; 171:1788–1792.
- Mundy AR, Podesta ML, Bewick M, Rudge CJ, Ellis FG. The urological complications of 1000 renal transplants. Br J Urol 1981; 53:397–402.
- Giessing M, Fischer TJ, Deger S, et al. Increased frequency of lymphoceles under treatment with sirolimus following renal transplantation: a single center experience. Transplant Proc 2002; 34:1815–1816.
- Amante AJ, Kahan BD. Technical complications of renal transplantation. Surg Clin North Am 1994; 74:1117–1131.
- Saidi RF, Wertheim JA, Ko DS, et al. Impact of donor kidney recovery method on lymphatic complications in kidney transplantation. Transplant Proc 2008; 40:1054–1055.
- Iwan-Zietek I, Zietek Z, Sulikowski T, et al. Minimally invasive methods for the treatment of lymphocele after kidney transplantation. Transplant Proc 2009; 41:3073–3076.
- Hwang EC, Kang TW, Koh YS, et al. Post-transplant lymphocele: an unusual cause of acute urinary retention mimicking urethral injury. Int J Urol 2006; 13:468–470.
- Zietek Z, Sulikowski T, Tejchman K, et al. Lymphocele after kidney transplantation. Transplant Proc 2007; 39:2744–2747.
- Mahrer A, Ramchandani P, Trerotola SO, Shlansky-Goldberg RD, Itkin M. Sclerotherapy in the management of postoperative lymphocele. J Vasc Interv Radiol 2010; 21:1050–1053.
- Risaliti A, Corno V, Donini A, et al. Laparoscopic treatment of symptomatic lymphoceles after kidney transplantation. Surg Endosc 2000; 14:293–295.
- Ostrowski M, Lubikowski J, Kowalczyk M, Power J. Laparoscopic lymphocele drainage after renal transplantation. Ann Transplant 2000; 5:25–27.
- Fuller TF, Kang SM, Hirose R, Feng S, Stock PG, Freise CE. Management of lymphoceles after renal transplantation: laparoscopic versus open drainage. J Urol 2003; 169:2022–2025.
- Hsu TH, Gill IS, Grune MT, et al. Laparoscopic lymphocelectomy: a multi-institutional analysis. J Urol 2000; 163:1096–1098.
- O’neill WC, Baumgarten DA. Ultrasonography in renal transplantation. Am J Kidney Dis 2002; 39:663–678.
- Braun WE, Banowsky LH, Straffon RA, et al. Lymphocytes associated with renal transplantation. Report of 15 cases and review of the literature. Am J Med 1974; 57:714–729.
- Goel M, Flechner SM, Zhou L, et al. The influence of various maintenance immunosuppressive drugs on lymphocele formation and treatment after kidney transplantation. J Urol 2004; 171:1788–1792.
- Mundy AR, Podesta ML, Bewick M, Rudge CJ, Ellis FG. The urological complications of 1000 renal transplants. Br J Urol 1981; 53:397–402.
- Giessing M, Fischer TJ, Deger S, et al. Increased frequency of lymphoceles under treatment with sirolimus following renal transplantation: a single center experience. Transplant Proc 2002; 34:1815–1816.
- Amante AJ, Kahan BD. Technical complications of renal transplantation. Surg Clin North Am 1994; 74:1117–1131.
- Saidi RF, Wertheim JA, Ko DS, et al. Impact of donor kidney recovery method on lymphatic complications in kidney transplantation. Transplant Proc 2008; 40:1054–1055.
- Iwan-Zietek I, Zietek Z, Sulikowski T, et al. Minimally invasive methods for the treatment of lymphocele after kidney transplantation. Transplant Proc 2009; 41:3073–3076.
- Hwang EC, Kang TW, Koh YS, et al. Post-transplant lymphocele: an unusual cause of acute urinary retention mimicking urethral injury. Int J Urol 2006; 13:468–470.
- Zietek Z, Sulikowski T, Tejchman K, et al. Lymphocele after kidney transplantation. Transplant Proc 2007; 39:2744–2747.
- Mahrer A, Ramchandani P, Trerotola SO, Shlansky-Goldberg RD, Itkin M. Sclerotherapy in the management of postoperative lymphocele. J Vasc Interv Radiol 2010; 21:1050–1053.
- Risaliti A, Corno V, Donini A, et al. Laparoscopic treatment of symptomatic lymphoceles after kidney transplantation. Surg Endosc 2000; 14:293–295.
- Ostrowski M, Lubikowski J, Kowalczyk M, Power J. Laparoscopic lymphocele drainage after renal transplantation. Ann Transplant 2000; 5:25–27.
- Fuller TF, Kang SM, Hirose R, Feng S, Stock PG, Freise CE. Management of lymphoceles after renal transplantation: laparoscopic versus open drainage. J Urol 2003; 169:2022–2025.
- Hsu TH, Gill IS, Grune MT, et al. Laparoscopic lymphocelectomy: a multi-institutional analysis. J Urol 2000; 163:1096–1098.

Chest pain followed by sudden collapse
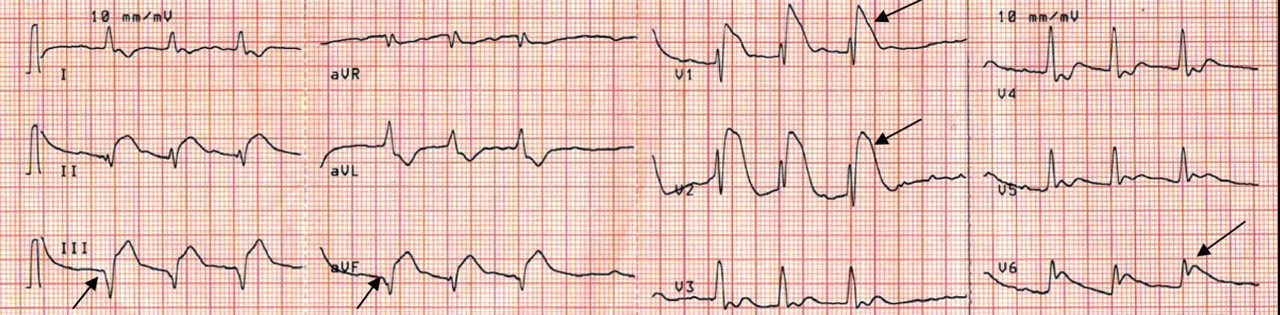
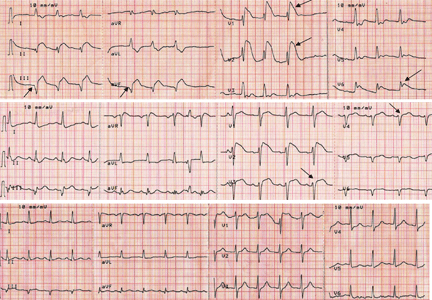
Q: Given what we know so far, what is the most likely cause of the ST segment elevation in leads V1 and V2?
- Brugada syndrome
- Pulmonary embolism
- Right ventricular injury
- Anterior myocardial infarction
A: The correct answer is right ventricular injury (discussed below).
Brugada syndrome is a genetic disorder caused by a mutation in the cardiac sodium channel gene. It is characterized by a pronounced elevation of the J point, a coved-type ST segment elevation in leads V1 and V2, and a propensity to develop malignant ventricular arrhythmias and sudden cardiac death.
In this patient, the pattern of ST segment elevation in leads V1 and V2 may be falsely interpreted as the classic type 1 Brugada electrocardiographic pattern. However, the classic type 1 Brugada electrocardiogram is characterized by a coved ST elevation followed by a negative T wave.1 The absence of T-wave inversion following ST segment elevation in this patient excluded Brugada syndrome. Moreover, the main presentation in patients with Brugada syndrome is either syncopy or sudden cardiac death.
Pulmonary embolism can present with various electrocardiographic patterns. ST segment elevation in the antroseptal leads is an extremely rare sign and has been demonstrated in a few reports.2,3 Pulmonary embolism can also present with abnormal Q waves in leads III and aVF but not in lead II.4 The initial electrocardiographic rhythm in patients who present with cardiac arrest is usually pulseless electrical activity; however, the combination of increased right ventricular oxygen consumption due to increased right ventricular afterload and right ventricular hypoperfusion due to hypotension can lead to right ventricular ischemia and subsequent arrhythmias. Mittal and Arora5 described a case of submassive pulmonary embolism with right ventricular infarction presenting with sustained ventricular tachycardia.
The prognosis is usually poor in patients with cardiac arrest due to pulmonary embolism, which is usually caused by a massive embolus and usually necessitates thrombolytic therapy.
In the patient described here, pulmonary embolism was part of the differential diagnosis, given the presence of ST segment elevation in leads V1 and V2 in the context of the clinical scenario. However, the restoration of spontaneous circulation without any specific treatment for pulmonary embolism and the normal oxygenation after cardiac arrest excluded pulmonary embolism.
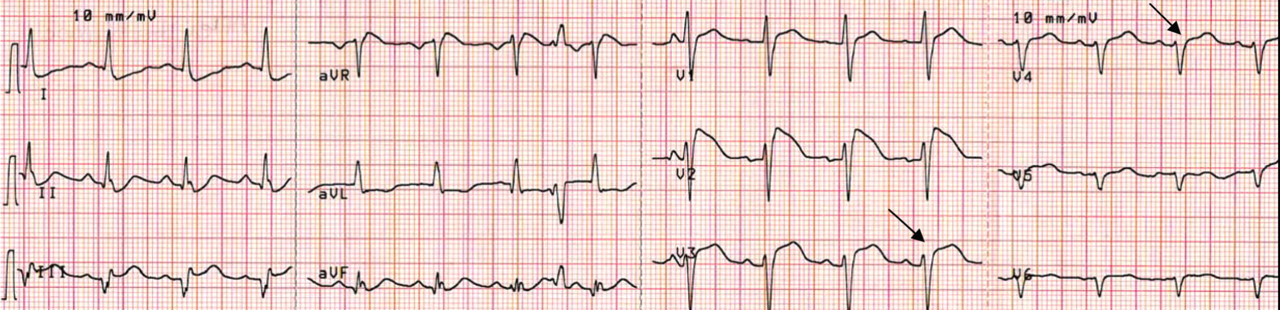
Right ventricular myocardial injury is important to recognize for therapeutic and prognostic reasons. It is usually associated with inferior infarction because it is typically secondary to an acute occlusion of the proximal right coronary artery proximal to the take-off of the right ventricular marginal branch. In the described scenario, the presence of ST segment elevation and Q waves in the inferior leads together with reciprocal ST segment depression in leads I and aVL represents an inferior myocardial infarct. ST segment elevation in the right precordial leads V3R and V4R is a marker for right ventricular injury—especially in V4R, in which it is a powerful predictor of right ventricular involvement. ST segment elevation in leads V1 and V2 is not usually demonstrated in patients with right ventricular injury because the electrical current of injury from the left ventricle inferior myocardial infarction dominates the right ventricular electrical forces, blocking the appearance of ST segment elevation in these leads.6 Data from the Hirulog and Early Reperfusion or Occlusion-2 trial showed that ST segment elevation of 1 mm or greater in lead V1 is associated with an increased risk of death in patients with acute inferior myocardial infarction.7 Furthermore, the presence of ST-segment elevation in lead V6 in patients with acute Q-wave inferior myocardial infarction, as evident in the first electrocardiogram, is associated with larger infarct size and a greater incidence of major arrhythmias.8
DETERMINING THE CULPRIT VESSEL
In the scenario described here, differentiating between right ventricular injury and anterior myocardial infarction is important to determine the culprit vessel.
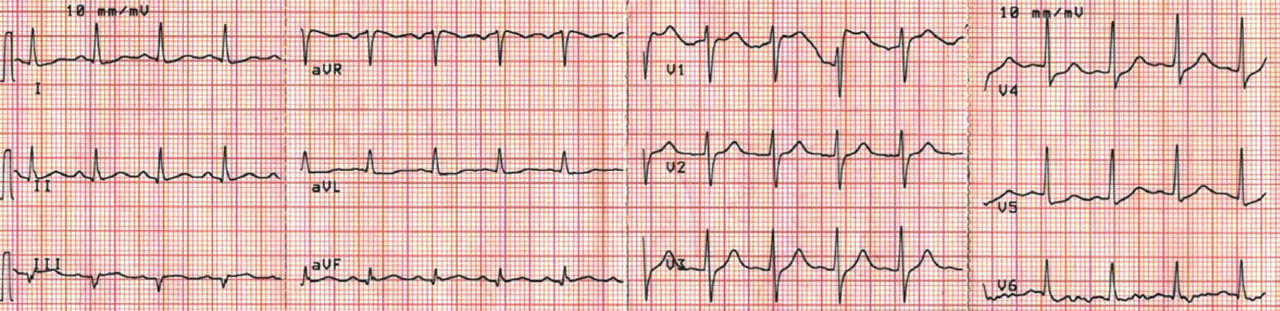
CASE CONCLUDED
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- Livaditis IG, Paraschos M, Dimopoulos K. Massive pulmonary embolism with ST elevation in leads V1–V3 and successful thrombolysis with tenecteplase. Heart 2004; 90:e41.
- Falterman TJ, Martinez JA, Daberkow D, Weiss LD. Pulmonary embolism with ST segment elevation in leads V1 to V4: case report and review of the literature regarding electrocardiographic changes in acute pulmonary embolism. J Emerg Med 2001; 21:255–261.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Mittal SR, Arora H. Pulmonary embolism with isolated right ventricular infarction. Indian Heart J 2001; 53:218–220.
- Geft IL, Shah PK, Rodriguez L, et al. ST elevations in leads V1 to V5 may be caused by right coronary artery occlusion and acute right ventricular infarction. Am J Cardiol 1984; 53:991–996.
- Wong CK, Gao W, Stewart RA, et al; Hirulog and Early Reperfusion or Occlusion-2 Investigators. Prognostic value of lead V1 ST elevation during acute inferior myocardial infarction. Circulation 2010; 122:463–469.
- Tsuka Y, Sugiura T, Hatada K, Abe Y, Takahashi N, Iwasaka T. Clinical characteristics of ST-segment elevation in lead V6 in patients with Q-wave acute inferior wall myocardial infarction. Coron Artery Dis 1999; 10:465–469.
Q: Given what we know so far, what is the most likely cause of the ST segment elevation in leads V1 and V2?
- Brugada syndrome
- Pulmonary embolism
- Right ventricular injury
- Anterior myocardial infarction
A: The correct answer is right ventricular injury (discussed below).
Brugada syndrome is a genetic disorder caused by a mutation in the cardiac sodium channel gene. It is characterized by a pronounced elevation of the J point, a coved-type ST segment elevation in leads V1 and V2, and a propensity to develop malignant ventricular arrhythmias and sudden cardiac death.
In this patient, the pattern of ST segment elevation in leads V1 and V2 may be falsely interpreted as the classic type 1 Brugada electrocardiographic pattern. However, the classic type 1 Brugada electrocardiogram is characterized by a coved ST elevation followed by a negative T wave.1 The absence of T-wave inversion following ST segment elevation in this patient excluded Brugada syndrome. Moreover, the main presentation in patients with Brugada syndrome is either syncopy or sudden cardiac death.
Pulmonary embolism can present with various electrocardiographic patterns. ST segment elevation in the antroseptal leads is an extremely rare sign and has been demonstrated in a few reports.2,3 Pulmonary embolism can also present with abnormal Q waves in leads III and aVF but not in lead II.4 The initial electrocardiographic rhythm in patients who present with cardiac arrest is usually pulseless electrical activity; however, the combination of increased right ventricular oxygen consumption due to increased right ventricular afterload and right ventricular hypoperfusion due to hypotension can lead to right ventricular ischemia and subsequent arrhythmias. Mittal and Arora5 described a case of submassive pulmonary embolism with right ventricular infarction presenting with sustained ventricular tachycardia.
The prognosis is usually poor in patients with cardiac arrest due to pulmonary embolism, which is usually caused by a massive embolus and usually necessitates thrombolytic therapy.
In the patient described here, pulmonary embolism was part of the differential diagnosis, given the presence of ST segment elevation in leads V1 and V2 in the context of the clinical scenario. However, the restoration of spontaneous circulation without any specific treatment for pulmonary embolism and the normal oxygenation after cardiac arrest excluded pulmonary embolism.
Right ventricular myocardial injury is important to recognize for therapeutic and prognostic reasons. It is usually associated with inferior infarction because it is typically secondary to an acute occlusion of the proximal right coronary artery proximal to the take-off of the right ventricular marginal branch. In the described scenario, the presence of ST segment elevation and Q waves in the inferior leads together with reciprocal ST segment depression in leads I and aVL represents an inferior myocardial infarct. ST segment elevation in the right precordial leads V3R and V4R is a marker for right ventricular injury—especially in V4R, in which it is a powerful predictor of right ventricular involvement. ST segment elevation in leads V1 and V2 is not usually demonstrated in patients with right ventricular injury because the electrical current of injury from the left ventricle inferior myocardial infarction dominates the right ventricular electrical forces, blocking the appearance of ST segment elevation in these leads.6 Data from the Hirulog and Early Reperfusion or Occlusion-2 trial showed that ST segment elevation of 1 mm or greater in lead V1 is associated with an increased risk of death in patients with acute inferior myocardial infarction.7 Furthermore, the presence of ST-segment elevation in lead V6 in patients with acute Q-wave inferior myocardial infarction, as evident in the first electrocardiogram, is associated with larger infarct size and a greater incidence of major arrhythmias.8
DETERMINING THE CULPRIT VESSEL
In the scenario described here, differentiating between right ventricular injury and anterior myocardial infarction is important to determine the culprit vessel.
CASE CONCLUDED
Q: Given what we know so far, what is the most likely cause of the ST segment elevation in leads V1 and V2?
- Brugada syndrome
- Pulmonary embolism
- Right ventricular injury
- Anterior myocardial infarction
A: The correct answer is right ventricular injury (discussed below).
Brugada syndrome is a genetic disorder caused by a mutation in the cardiac sodium channel gene. It is characterized by a pronounced elevation of the J point, a coved-type ST segment elevation in leads V1 and V2, and a propensity to develop malignant ventricular arrhythmias and sudden cardiac death.
In this patient, the pattern of ST segment elevation in leads V1 and V2 may be falsely interpreted as the classic type 1 Brugada electrocardiographic pattern. However, the classic type 1 Brugada electrocardiogram is characterized by a coved ST elevation followed by a negative T wave.1 The absence of T-wave inversion following ST segment elevation in this patient excluded Brugada syndrome. Moreover, the main presentation in patients with Brugada syndrome is either syncopy or sudden cardiac death.
Pulmonary embolism can present with various electrocardiographic patterns. ST segment elevation in the antroseptal leads is an extremely rare sign and has been demonstrated in a few reports.2,3 Pulmonary embolism can also present with abnormal Q waves in leads III and aVF but not in lead II.4 The initial electrocardiographic rhythm in patients who present with cardiac arrest is usually pulseless electrical activity; however, the combination of increased right ventricular oxygen consumption due to increased right ventricular afterload and right ventricular hypoperfusion due to hypotension can lead to right ventricular ischemia and subsequent arrhythmias. Mittal and Arora5 described a case of submassive pulmonary embolism with right ventricular infarction presenting with sustained ventricular tachycardia.
The prognosis is usually poor in patients with cardiac arrest due to pulmonary embolism, which is usually caused by a massive embolus and usually necessitates thrombolytic therapy.
In the patient described here, pulmonary embolism was part of the differential diagnosis, given the presence of ST segment elevation in leads V1 and V2 in the context of the clinical scenario. However, the restoration of spontaneous circulation without any specific treatment for pulmonary embolism and the normal oxygenation after cardiac arrest excluded pulmonary embolism.
Right ventricular myocardial injury is important to recognize for therapeutic and prognostic reasons. It is usually associated with inferior infarction because it is typically secondary to an acute occlusion of the proximal right coronary artery proximal to the take-off of the right ventricular marginal branch. In the described scenario, the presence of ST segment elevation and Q waves in the inferior leads together with reciprocal ST segment depression in leads I and aVL represents an inferior myocardial infarct. ST segment elevation in the right precordial leads V3R and V4R is a marker for right ventricular injury—especially in V4R, in which it is a powerful predictor of right ventricular involvement. ST segment elevation in leads V1 and V2 is not usually demonstrated in patients with right ventricular injury because the electrical current of injury from the left ventricle inferior myocardial infarction dominates the right ventricular electrical forces, blocking the appearance of ST segment elevation in these leads.6 Data from the Hirulog and Early Reperfusion or Occlusion-2 trial showed that ST segment elevation of 1 mm or greater in lead V1 is associated with an increased risk of death in patients with acute inferior myocardial infarction.7 Furthermore, the presence of ST-segment elevation in lead V6 in patients with acute Q-wave inferior myocardial infarction, as evident in the first electrocardiogram, is associated with larger infarct size and a greater incidence of major arrhythmias.8
DETERMINING THE CULPRIT VESSEL
In the scenario described here, differentiating between right ventricular injury and anterior myocardial infarction is important to determine the culprit vessel.
CASE CONCLUDED
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- Livaditis IG, Paraschos M, Dimopoulos K. Massive pulmonary embolism with ST elevation in leads V1–V3 and successful thrombolysis with tenecteplase. Heart 2004; 90:e41.
- Falterman TJ, Martinez JA, Daberkow D, Weiss LD. Pulmonary embolism with ST segment elevation in leads V1 to V4: case report and review of the literature regarding electrocardiographic changes in acute pulmonary embolism. J Emerg Med 2001; 21:255–261.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Mittal SR, Arora H. Pulmonary embolism with isolated right ventricular infarction. Indian Heart J 2001; 53:218–220.
- Geft IL, Shah PK, Rodriguez L, et al. ST elevations in leads V1 to V5 may be caused by right coronary artery occlusion and acute right ventricular infarction. Am J Cardiol 1984; 53:991–996.
- Wong CK, Gao W, Stewart RA, et al; Hirulog and Early Reperfusion or Occlusion-2 Investigators. Prognostic value of lead V1 ST elevation during acute inferior myocardial infarction. Circulation 2010; 122:463–469.
- Tsuka Y, Sugiura T, Hatada K, Abe Y, Takahashi N, Iwasaka T. Clinical characteristics of ST-segment elevation in lead V6 in patients with Q-wave acute inferior wall myocardial infarction. Coron Artery Dis 1999; 10:465–469.
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111:659–670.
- Livaditis IG, Paraschos M, Dimopoulos K. Massive pulmonary embolism with ST elevation in leads V1–V3 and successful thrombolysis with tenecteplase. Heart 2004; 90:e41.
- Falterman TJ, Martinez JA, Daberkow D, Weiss LD. Pulmonary embolism with ST segment elevation in leads V1 to V4: case report and review of the literature regarding electrocardiographic changes in acute pulmonary embolism. J Emerg Med 2001; 21:255–261.
- Sreeram N, Cheriex EC, Smeets JL, Gorgels AP, Wellens HJ. Value of the 12-lead electrocardiogram at hospital admission in the diagnosis of pulmonary embolism. Am J Cardiol 1994; 73:298–303.
- Mittal SR, Arora H. Pulmonary embolism with isolated right ventricular infarction. Indian Heart J 2001; 53:218–220.
- Geft IL, Shah PK, Rodriguez L, et al. ST elevations in leads V1 to V5 may be caused by right coronary artery occlusion and acute right ventricular infarction. Am J Cardiol 1984; 53:991–996.
- Wong CK, Gao W, Stewart RA, et al; Hirulog and Early Reperfusion or Occlusion-2 Investigators. Prognostic value of lead V1 ST elevation during acute inferior myocardial infarction. Circulation 2010; 122:463–469.
- Tsuka Y, Sugiura T, Hatada K, Abe Y, Takahashi N, Iwasaka T. Clinical characteristics of ST-segment elevation in lead V6 in patients with Q-wave acute inferior wall myocardial infarction. Coron Artery Dis 1999; 10:465–469.
A 48-year-old woman with an ecchymotic rash
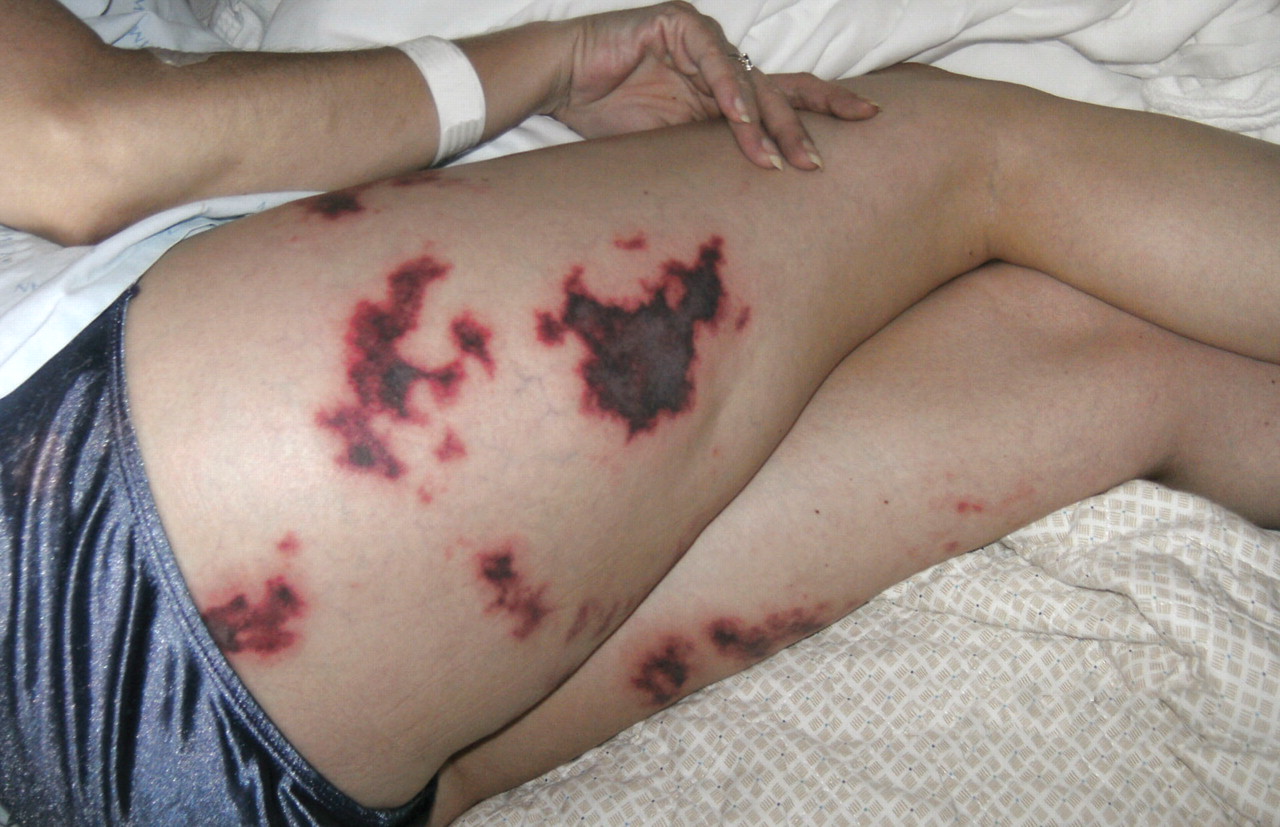
She had no constitutional symptoms and no history of venous thromboembolism, stroke, pregnancy loss, recent anticoagulation, or endovascular procedures.
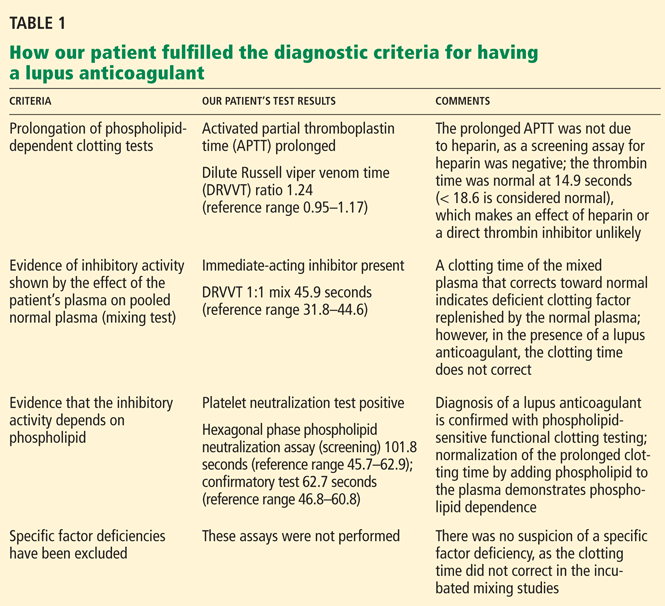
Q: What is the most likely diagnosis?
- Chronic meningococcemia
- Cholesterol embolism
- Antiphospholipid syndrome
- Cryoglobulinemic vasculitis
- Heterozygous protein C deficiency
A: The most likely diagnosis is skin necrosis due to intravascular thrombosis, consistent with antiphospholipid syndrome. By clinical and laboratory criteria, the patient has systemic lupus erythematosus. Pain and swelling in multiple joints is indicative of polyarthritis associated with lupus. Retesting 12 weeks later again detected lupus anticoagulant, confirming the diagnosis of antiphospholipid syndrome.2
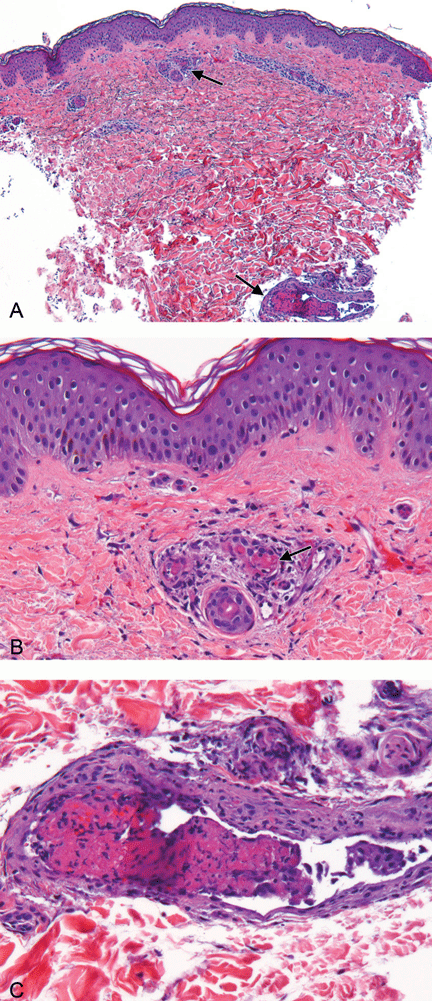
In the hospital, the patient was started on unfractionated heparin, later switched to warfarin. Her skin lesions gradually cleared, her pain diminished significantly, and no new lesions appeared after the start of anticoagulation therapy. For her lupus, she was started on hydroxychloroquine (Plaquenil), which has been suggested to also have an adjuvant antithrombotic role in antiphospholipid syndrome.2 On a follow-up visit 3 months later, she was doing well.
MORE ABOUT ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome is termed primary when no underlying disease is identified, and secondary when it occurs in conjunction with an autoimmune rheumatologic disease, an infection, malignancy, or certain drugs.3 It is the most common cause of acquired thrombophilia.4 Arterial or venous thromboses and recurrent miscarriages are salient clinical features.
Laboratory abnormalities include the presence of a lupus anticoagulant and anticardiolipin and beta-2-glycoprotein 1 antibodies.
Skin manifestations include livedo reticularis, purpuric macular lesions, atrophie blanche, cutaneous infarcts, ulceration, and painful nodules.5 Livedo reticularis, a violaceous, lace-like cutaneous discoloration, is the most commonly described skin lesion, present in 20% to 50% of cases.5,6 Cutaneous necrosis may involve the legs, face, and ears, or it may be generalized.6
The prothrombotic state is believed to be immune-mediated, with complement activation.2 Endothelial cells and monocytes are activated by antiphospholipid antibodies with activity against beta-2-glycoprotein 1, resulting in up-regulation of tissue factor and in platelet activation.2 Histopathologic examination reveals noninflammatory vascular thromboses with endothelial damage.5
Although antiphospholipid syndrome seems to be immune-mediated, immunosuppressive therapy has not proved very effective,3 and anticoagulation is the recommended treatment.3,7
THE OTHER DIAGNOSTIC POSSIBILITIES
Chronic meningococcemia, sometimes associated with terminal complement deficiency, is associated with a petechial rash in 50% to 80% of cases. The rash can become confluent, resulting in hemorrhagic patches with central necrosis, resembling the lesions in our patient.
However, these skin lesions are due to thrombi in the dermal vessels, associated with leukocytoclastic vasculitis. These dermatopathologic changes were not seen in our patient. Moreover, meningococci were not identified in blood cultures or in the luminal thrombi and vessel walls.
Cholesterol embolism occurs when cholesterol crystals break off from severely atherosclerotic plaques, either spontaneously or after local trauma induced by angiography or aortic injury. The crystals shower downstream through the arterial system, often immediately occluding arterioles 100 to 200 μm in diameter.
Our patient had no such history, and the skin biopsy did not show the characteristic “cholesterol clefts”—biconvex, needle-shaped clefts left by the dissolved crystals of cholesterol within the occluded vessels.
Cryoglobulinemic vasculitis is an immune-complex-mediated condition involving small- to medium-size vessels, often associated with hepatitis C virus infection. Skin lesions appear in dependent areas and include erythematous macules and purpuric papules.
Cryoglobulins were not detected in our patient’s sera, nor did the skin biopsy indicate the typical leukocytoclastic vasculitis seen in this condition.
Heterozygous protein C deficiency causes venous thromboembolism and warfarin-induced skin necrosis. Spontaneous thrombosis of cutaneous arterioles (as in our patient) is not a usual manifestation. Also, our patient had normal protein C levels and no history of warfarin use before the skin lesions developed.
Acknowledgment: The authors are grateful to Dr. Judith Drazba, PhD, of Research Core Services (Imaging) at Cleveland Clinic for help in the preparation of the photomicrographs.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Myones BL, McCurdy D. The antiphospholipid syndrome: immunologic and clinical aspects. Clinical spectrum and treatment. J Rheumatol Suppl 2000; 58:20–28.
- Bick RL, Baker WF. Antiphospholipid syndrome and thrombosis. Semin Thromb Hemost 1999; 25:333–350.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol 1997; 36:970–982.
- Nahass GT. Antiphospholipid antibodies and the antiphospholipid antibody syndrome. J Am Acad Dermatol 1997; 36:149–168.
- Petri M. Pathogenesis and treatment of the antiphospholipid antibody syndrome. Med Clin North Am 1997; 81:151–177.
She had no constitutional symptoms and no history of venous thromboembolism, stroke, pregnancy loss, recent anticoagulation, or endovascular procedures.
Q: What is the most likely diagnosis?
- Chronic meningococcemia
- Cholesterol embolism
- Antiphospholipid syndrome
- Cryoglobulinemic vasculitis
- Heterozygous protein C deficiency
A: The most likely diagnosis is skin necrosis due to intravascular thrombosis, consistent with antiphospholipid syndrome. By clinical and laboratory criteria, the patient has systemic lupus erythematosus. Pain and swelling in multiple joints is indicative of polyarthritis associated with lupus. Retesting 12 weeks later again detected lupus anticoagulant, confirming the diagnosis of antiphospholipid syndrome.2
In the hospital, the patient was started on unfractionated heparin, later switched to warfarin. Her skin lesions gradually cleared, her pain diminished significantly, and no new lesions appeared after the start of anticoagulation therapy. For her lupus, she was started on hydroxychloroquine (Plaquenil), which has been suggested to also have an adjuvant antithrombotic role in antiphospholipid syndrome.2 On a follow-up visit 3 months later, she was doing well.
MORE ABOUT ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome is termed primary when no underlying disease is identified, and secondary when it occurs in conjunction with an autoimmune rheumatologic disease, an infection, malignancy, or certain drugs.3 It is the most common cause of acquired thrombophilia.4 Arterial or venous thromboses and recurrent miscarriages are salient clinical features.
Laboratory abnormalities include the presence of a lupus anticoagulant and anticardiolipin and beta-2-glycoprotein 1 antibodies.
Skin manifestations include livedo reticularis, purpuric macular lesions, atrophie blanche, cutaneous infarcts, ulceration, and painful nodules.5 Livedo reticularis, a violaceous, lace-like cutaneous discoloration, is the most commonly described skin lesion, present in 20% to 50% of cases.5,6 Cutaneous necrosis may involve the legs, face, and ears, or it may be generalized.6
The prothrombotic state is believed to be immune-mediated, with complement activation.2 Endothelial cells and monocytes are activated by antiphospholipid antibodies with activity against beta-2-glycoprotein 1, resulting in up-regulation of tissue factor and in platelet activation.2 Histopathologic examination reveals noninflammatory vascular thromboses with endothelial damage.5
Although antiphospholipid syndrome seems to be immune-mediated, immunosuppressive therapy has not proved very effective,3 and anticoagulation is the recommended treatment.3,7
THE OTHER DIAGNOSTIC POSSIBILITIES
Chronic meningococcemia, sometimes associated with terminal complement deficiency, is associated with a petechial rash in 50% to 80% of cases. The rash can become confluent, resulting in hemorrhagic patches with central necrosis, resembling the lesions in our patient.
However, these skin lesions are due to thrombi in the dermal vessels, associated with leukocytoclastic vasculitis. These dermatopathologic changes were not seen in our patient. Moreover, meningococci were not identified in blood cultures or in the luminal thrombi and vessel walls.
Cholesterol embolism occurs when cholesterol crystals break off from severely atherosclerotic plaques, either spontaneously or after local trauma induced by angiography or aortic injury. The crystals shower downstream through the arterial system, often immediately occluding arterioles 100 to 200 μm in diameter.
Our patient had no such history, and the skin biopsy did not show the characteristic “cholesterol clefts”—biconvex, needle-shaped clefts left by the dissolved crystals of cholesterol within the occluded vessels.
Cryoglobulinemic vasculitis is an immune-complex-mediated condition involving small- to medium-size vessels, often associated with hepatitis C virus infection. Skin lesions appear in dependent areas and include erythematous macules and purpuric papules.
Cryoglobulins were not detected in our patient’s sera, nor did the skin biopsy indicate the typical leukocytoclastic vasculitis seen in this condition.
Heterozygous protein C deficiency causes venous thromboembolism and warfarin-induced skin necrosis. Spontaneous thrombosis of cutaneous arterioles (as in our patient) is not a usual manifestation. Also, our patient had normal protein C levels and no history of warfarin use before the skin lesions developed.
Acknowledgment: The authors are grateful to Dr. Judith Drazba, PhD, of Research Core Services (Imaging) at Cleveland Clinic for help in the preparation of the photomicrographs.
She had no constitutional symptoms and no history of venous thromboembolism, stroke, pregnancy loss, recent anticoagulation, or endovascular procedures.
Q: What is the most likely diagnosis?
- Chronic meningococcemia
- Cholesterol embolism
- Antiphospholipid syndrome
- Cryoglobulinemic vasculitis
- Heterozygous protein C deficiency
A: The most likely diagnosis is skin necrosis due to intravascular thrombosis, consistent with antiphospholipid syndrome. By clinical and laboratory criteria, the patient has systemic lupus erythematosus. Pain and swelling in multiple joints is indicative of polyarthritis associated with lupus. Retesting 12 weeks later again detected lupus anticoagulant, confirming the diagnosis of antiphospholipid syndrome.2
In the hospital, the patient was started on unfractionated heparin, later switched to warfarin. Her skin lesions gradually cleared, her pain diminished significantly, and no new lesions appeared after the start of anticoagulation therapy. For her lupus, she was started on hydroxychloroquine (Plaquenil), which has been suggested to also have an adjuvant antithrombotic role in antiphospholipid syndrome.2 On a follow-up visit 3 months later, she was doing well.
MORE ABOUT ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome is termed primary when no underlying disease is identified, and secondary when it occurs in conjunction with an autoimmune rheumatologic disease, an infection, malignancy, or certain drugs.3 It is the most common cause of acquired thrombophilia.4 Arterial or venous thromboses and recurrent miscarriages are salient clinical features.
Laboratory abnormalities include the presence of a lupus anticoagulant and anticardiolipin and beta-2-glycoprotein 1 antibodies.
Skin manifestations include livedo reticularis, purpuric macular lesions, atrophie blanche, cutaneous infarcts, ulceration, and painful nodules.5 Livedo reticularis, a violaceous, lace-like cutaneous discoloration, is the most commonly described skin lesion, present in 20% to 50% of cases.5,6 Cutaneous necrosis may involve the legs, face, and ears, or it may be generalized.6
The prothrombotic state is believed to be immune-mediated, with complement activation.2 Endothelial cells and monocytes are activated by antiphospholipid antibodies with activity against beta-2-glycoprotein 1, resulting in up-regulation of tissue factor and in platelet activation.2 Histopathologic examination reveals noninflammatory vascular thromboses with endothelial damage.5
Although antiphospholipid syndrome seems to be immune-mediated, immunosuppressive therapy has not proved very effective,3 and anticoagulation is the recommended treatment.3,7
THE OTHER DIAGNOSTIC POSSIBILITIES
Chronic meningococcemia, sometimes associated with terminal complement deficiency, is associated with a petechial rash in 50% to 80% of cases. The rash can become confluent, resulting in hemorrhagic patches with central necrosis, resembling the lesions in our patient.
However, these skin lesions are due to thrombi in the dermal vessels, associated with leukocytoclastic vasculitis. These dermatopathologic changes were not seen in our patient. Moreover, meningococci were not identified in blood cultures or in the luminal thrombi and vessel walls.
Cholesterol embolism occurs when cholesterol crystals break off from severely atherosclerotic plaques, either spontaneously or after local trauma induced by angiography or aortic injury. The crystals shower downstream through the arterial system, often immediately occluding arterioles 100 to 200 μm in diameter.
Our patient had no such history, and the skin biopsy did not show the characteristic “cholesterol clefts”—biconvex, needle-shaped clefts left by the dissolved crystals of cholesterol within the occluded vessels.
Cryoglobulinemic vasculitis is an immune-complex-mediated condition involving small- to medium-size vessels, often associated with hepatitis C virus infection. Skin lesions appear in dependent areas and include erythematous macules and purpuric papules.
Cryoglobulins were not detected in our patient’s sera, nor did the skin biopsy indicate the typical leukocytoclastic vasculitis seen in this condition.
Heterozygous protein C deficiency causes venous thromboembolism and warfarin-induced skin necrosis. Spontaneous thrombosis of cutaneous arterioles (as in our patient) is not a usual manifestation. Also, our patient had normal protein C levels and no history of warfarin use before the skin lesions developed.
Acknowledgment: The authors are grateful to Dr. Judith Drazba, PhD, of Research Core Services (Imaging) at Cleveland Clinic for help in the preparation of the photomicrographs.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Myones BL, McCurdy D. The antiphospholipid syndrome: immunologic and clinical aspects. Clinical spectrum and treatment. J Rheumatol Suppl 2000; 58:20–28.
- Bick RL, Baker WF. Antiphospholipid syndrome and thrombosis. Semin Thromb Hemost 1999; 25:333–350.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol 1997; 36:970–982.
- Nahass GT. Antiphospholipid antibodies and the antiphospholipid antibody syndrome. J Am Acad Dermatol 1997; 36:149–168.
- Petri M. Pathogenesis and treatment of the antiphospholipid antibody syndrome. Med Clin North Am 1997; 81:151–177.
- Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. On behalf of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the ISTH. Thromb Haemost 1995; 74:1185–1190.
- Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet 2010; 376:1498–1509.
- Myones BL, McCurdy D. The antiphospholipid syndrome: immunologic and clinical aspects. Clinical spectrum and treatment. J Rheumatol Suppl 2000; 58:20–28.
- Bick RL, Baker WF. Antiphospholipid syndrome and thrombosis. Semin Thromb Hemost 1999; 25:333–350.
- Gibson GE, Su WP, Pittelkow MR. Antiphospholipid syndrome and the skin. J Am Acad Dermatol 1997; 36:970–982.
- Nahass GT. Antiphospholipid antibodies and the antiphospholipid antibody syndrome. J Am Acad Dermatol 1997; 36:149–168.
- Petri M. Pathogenesis and treatment of the antiphospholipid antibody syndrome. Med Clin North Am 1997; 81:151–177.
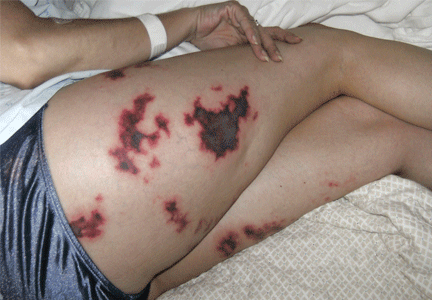
Diffuse reticulonodular infiltrates
A 69-year-old woman presented to the hospital with a 3-month history of fever of unknown origin and dyspnea. Her medical history included diabetes mellitus and, many years ago, partially treated latent tuberculosis infection and pancreatic cancer, treated with Whipple surgery and chemotherapy.
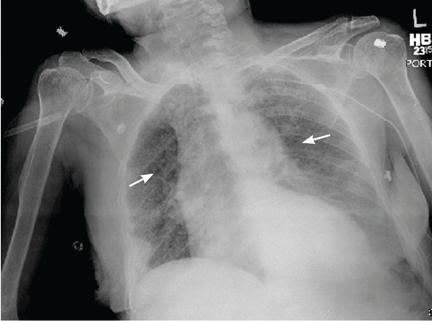
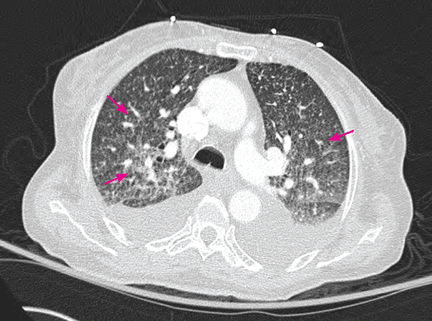
Q: What is the most likely diagnosis?
- Sarcoidosis of the lungs and central nervous system
- Hypersensitivity pneumonitis
- Miliary tuberculosis
- Cancer with pulmonary and brain metastases
- Disseminated fungal infection
A: The correct diagnosis is miliary tuberculosis, ie, progressive and widely disseminated hematogenous tuberculosis infection. Granulomas involving multiple organs suggested a broad differential diagnosis. The negative workup for infectious disease initially supported sarcoidosis by exclusion, but her condition failed to respond to steroid treatment. Multiple organ involvement is atypical for hypersensitivity pneumonitis, and antibody panels were negative. The absence of malignant cells in multiple biopsy specimens made metastasis unlikely.
Her remote history of partially treated latent tuberculosis infection raised our clinical suspicion and prompted mycobacterial antibiotic coverage. Three weeks after the initial sample collection, results from an independent laboratory revealed the presence of acid-fast bacilli in cultures of bronchoalveolar lavage fluid, cerebrospinal fluid, and blood, which were confirmed to be M tuberculosis. Despite treatment, the patient died of multiple organ failure.
Tuberculosis is rare in the United States, with 11,181 reported cases in 2010.1 Miliary tuberculosis is associated with malnutrition, HIV infection, AIDS, alcoholism, diabetes, chronic kidney failure, immunosuppressive drugs, and organ transplantation.2 Acid-fast bacilli smears and cultures confirm the diagnosis but have low sensitivity.3 Granulomas characterize miliary tuberculosis histopathologically. They may be present in sarcoidosis, hypersensitivity pneumonitis, and fungal infection but are not specific for these conditions. Tuberculin skin testing2 and interferon-gamma-release assay can be falsely negative in immunosuppressed patients,4 highlighting the emphasis on clinical suspicion. The estimated death rate is 20%,3,5 with central nervous system involvement being an independent predictor.5 Empiric therapy should not be delayed, as culture results may not be available for 6 to 8 weeks.
- US Centers for Disease Control and Prevention (CDC). Trends in tuberculosis—United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:333–337.
- Sharma SK, Mohan A, Sharma A, Mitra DK. Miliary tuberculosis: new insights into an old disease. Lancet Infect Dis 2005; 5:415–430.
- Maartens G, Willcox PA, Benatar SR. Miliary tuberculosis: rapid diagnosis, hematologic abnormalities, and outcome in 109 treated adults. Am J Med 1990; 89:291–296.
- Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K; IGRA Expert Committee. Updated guidelines for using Interferon gamma release assays to detect Mycobacterium tuberculosis infection—United States, 2010. MMWR Recomm Rep 2010; 59:1–25.
- Kim JH, Langston AA, Gallis HA. Miliary tuberculosis: epidemiology, clinical manifestations, diagnosis, and outcome. Rev Infect Dis 1990; 12:583–590.
A 69-year-old woman presented to the hospital with a 3-month history of fever of unknown origin and dyspnea. Her medical history included diabetes mellitus and, many years ago, partially treated latent tuberculosis infection and pancreatic cancer, treated with Whipple surgery and chemotherapy.
Q: What is the most likely diagnosis?
- Sarcoidosis of the lungs and central nervous system
- Hypersensitivity pneumonitis
- Miliary tuberculosis
- Cancer with pulmonary and brain metastases
- Disseminated fungal infection
A: The correct diagnosis is miliary tuberculosis, ie, progressive and widely disseminated hematogenous tuberculosis infection. Granulomas involving multiple organs suggested a broad differential diagnosis. The negative workup for infectious disease initially supported sarcoidosis by exclusion, but her condition failed to respond to steroid treatment. Multiple organ involvement is atypical for hypersensitivity pneumonitis, and antibody panels were negative. The absence of malignant cells in multiple biopsy specimens made metastasis unlikely.
Her remote history of partially treated latent tuberculosis infection raised our clinical suspicion and prompted mycobacterial antibiotic coverage. Three weeks after the initial sample collection, results from an independent laboratory revealed the presence of acid-fast bacilli in cultures of bronchoalveolar lavage fluid, cerebrospinal fluid, and blood, which were confirmed to be M tuberculosis. Despite treatment, the patient died of multiple organ failure.
Tuberculosis is rare in the United States, with 11,181 reported cases in 2010.1 Miliary tuberculosis is associated with malnutrition, HIV infection, AIDS, alcoholism, diabetes, chronic kidney failure, immunosuppressive drugs, and organ transplantation.2 Acid-fast bacilli smears and cultures confirm the diagnosis but have low sensitivity.3 Granulomas characterize miliary tuberculosis histopathologically. They may be present in sarcoidosis, hypersensitivity pneumonitis, and fungal infection but are not specific for these conditions. Tuberculin skin testing2 and interferon-gamma-release assay can be falsely negative in immunosuppressed patients,4 highlighting the emphasis on clinical suspicion. The estimated death rate is 20%,3,5 with central nervous system involvement being an independent predictor.5 Empiric therapy should not be delayed, as culture results may not be available for 6 to 8 weeks.
A 69-year-old woman presented to the hospital with a 3-month history of fever of unknown origin and dyspnea. Her medical history included diabetes mellitus and, many years ago, partially treated latent tuberculosis infection and pancreatic cancer, treated with Whipple surgery and chemotherapy.
Q: What is the most likely diagnosis?
- Sarcoidosis of the lungs and central nervous system
- Hypersensitivity pneumonitis
- Miliary tuberculosis
- Cancer with pulmonary and brain metastases
- Disseminated fungal infection
A: The correct diagnosis is miliary tuberculosis, ie, progressive and widely disseminated hematogenous tuberculosis infection. Granulomas involving multiple organs suggested a broad differential diagnosis. The negative workup for infectious disease initially supported sarcoidosis by exclusion, but her condition failed to respond to steroid treatment. Multiple organ involvement is atypical for hypersensitivity pneumonitis, and antibody panels were negative. The absence of malignant cells in multiple biopsy specimens made metastasis unlikely.
Her remote history of partially treated latent tuberculosis infection raised our clinical suspicion and prompted mycobacterial antibiotic coverage. Three weeks after the initial sample collection, results from an independent laboratory revealed the presence of acid-fast bacilli in cultures of bronchoalveolar lavage fluid, cerebrospinal fluid, and blood, which were confirmed to be M tuberculosis. Despite treatment, the patient died of multiple organ failure.
Tuberculosis is rare in the United States, with 11,181 reported cases in 2010.1 Miliary tuberculosis is associated with malnutrition, HIV infection, AIDS, alcoholism, diabetes, chronic kidney failure, immunosuppressive drugs, and organ transplantation.2 Acid-fast bacilli smears and cultures confirm the diagnosis but have low sensitivity.3 Granulomas characterize miliary tuberculosis histopathologically. They may be present in sarcoidosis, hypersensitivity pneumonitis, and fungal infection but are not specific for these conditions. Tuberculin skin testing2 and interferon-gamma-release assay can be falsely negative in immunosuppressed patients,4 highlighting the emphasis on clinical suspicion. The estimated death rate is 20%,3,5 with central nervous system involvement being an independent predictor.5 Empiric therapy should not be delayed, as culture results may not be available for 6 to 8 weeks.
- US Centers for Disease Control and Prevention (CDC). Trends in tuberculosis—United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:333–337.
- Sharma SK, Mohan A, Sharma A, Mitra DK. Miliary tuberculosis: new insights into an old disease. Lancet Infect Dis 2005; 5:415–430.
- Maartens G, Willcox PA, Benatar SR. Miliary tuberculosis: rapid diagnosis, hematologic abnormalities, and outcome in 109 treated adults. Am J Med 1990; 89:291–296.
- Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K; IGRA Expert Committee. Updated guidelines for using Interferon gamma release assays to detect Mycobacterium tuberculosis infection—United States, 2010. MMWR Recomm Rep 2010; 59:1–25.
- Kim JH, Langston AA, Gallis HA. Miliary tuberculosis: epidemiology, clinical manifestations, diagnosis, and outcome. Rev Infect Dis 1990; 12:583–590.
- US Centers for Disease Control and Prevention (CDC). Trends in tuberculosis—United States, 2010. MMWR Morb Mortal Wkly Rep 2011; 60:333–337.
- Sharma SK, Mohan A, Sharma A, Mitra DK. Miliary tuberculosis: new insights into an old disease. Lancet Infect Dis 2005; 5:415–430.
- Maartens G, Willcox PA, Benatar SR. Miliary tuberculosis: rapid diagnosis, hematologic abnormalities, and outcome in 109 treated adults. Am J Med 1990; 89:291–296.
- Mazurek GH, Jereb J, Vernon A, LoBue P, Goldberg S, Castro K; IGRA Expert Committee. Updated guidelines for using Interferon gamma release assays to detect Mycobacterium tuberculosis infection—United States, 2010. MMWR Recomm Rep 2010; 59:1–25.
- Kim JH, Langston AA, Gallis HA. Miliary tuberculosis: epidemiology, clinical manifestations, diagnosis, and outcome. Rev Infect Dis 1990; 12:583–590.
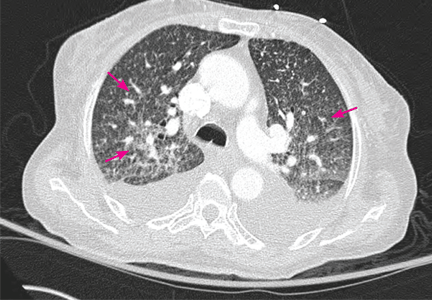
Cytomegalovirus colitis
A 21-year-old woman with Crohn disease presented to the hospital after 5 days of diffuse abdominal pain, nausea, vomiting, and watery diarrhea despite taking azathioprine (Imuran) 100 mg daily as maintenance therapy. She had been hospitalized 2 weeks previously at another hospital for a Crohn disease flare, which was treated with intravenous methylprednisolone (Solu-Medrol).
On admission to our hospital, her temperature was 38.4°C (101.1°F), heart rate 78 per minute, respiratory rate 18 per minute, blood pressure 110/50 mm Hg, and oxygen saturation 98% while breathing room air. She had diffuse abdominal tenderness without rebound tenderness.
Because Clostridium difficile has a high prevalence in our hospital, treament for C difficile diarrhea was started empirically directly upon hospital admission; it was stopped 48 hours later when stool cultures came back negative for C difficile.
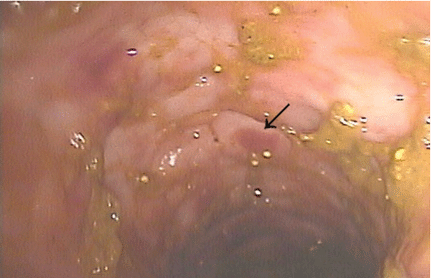
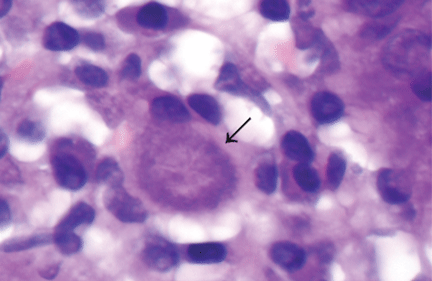
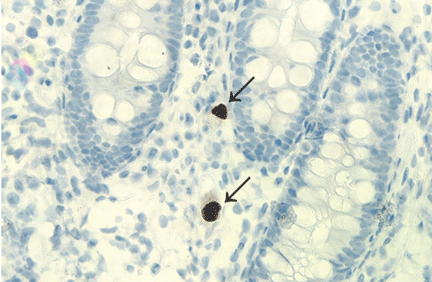
COLITIS AND CYTOMEGALOVIRUS INFECTION
CMV colitis is common in patients with inflammatory bowel disease (ie, Crohn disease or ulcerative colitis) who are on long-term immunosuppressive therapy. Heightened suspicion for it is needed when treating patients with inflammatory bowel disease, as they tend to present with atypical symptoms and signs.
It is also important to keep a wide differential diagnosis in mind, as acute fever and diarrhea in patients with inflammatory bowel disease are not always related to the underlying disease. In these patients, a variety of diagnostic tests may be necessary to exclude an opportunistic infection and an unrelated intercurrent illness.
Human CMV is a member of the family of herpes viruses, which persist for life after a primary infection. In exacerbations of inflammatory bowel disease, it is not clear whether CMV is a nonpathogenic bystander or a true pathogen.1 Most CMV infections in patients with inflammatory bowel disease are due to reactivation of the virus, as levels of inflammatory cytokines such as tumor necrosis factor are increased in the intestinal mucosa in active inflammatory bowel disease, and these cytokines are known to trigger reactivation.2
In patients with chronic inflammatory bowel disease, CMV colitis usually presents with abdominal pain, diarrhea, intestinal bleeding, and fever. The gold standard for diagnosis is immunohistochemical testing of colon biopsy samples using monoclonal antibodies against CMV. Owl’s eye inclusion bodies on histopathologic sections are highly specific for CMV infection. Other diagnostic studies include endoscopy and serologic testing.
The gastrointestinal tract is thought to contain latent CMV after a primary infection, and long-term treatment with immunomodulatory drugs such as azathioprine and corticosteroids can cause local reactivation of the latent virus.3
CMV infection in patients with inflammatory bowel disease is associated with poor outcomes, such as the need for colectomy.4 The prevalence of CMV infection in patients with inflammatory bowel disease has been reported as 5% to 36%, and higher in patients with disease refractory to steroid therapy.1,5
When a patient with inflammatory bowel disease is diagnosed with CMV infection, the immunomodulatory drugs should be stopped and the corticosteroids should be tapered to the lowest possible dose. Treatment of the infection is intravenous ganciclovir at 5 mg per kilogram of body weight twice daily for 14 days, followed by oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks.
After receiving intravenous ganciclovir for 14 days, our patient received oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks. Her azathioprine was stopped while she was taking the antivirals, and it was resumed the day after she completed the course of valacyclovir.
The response to treatment is monitored with a cytomegalovirus pp 65 antigenemia assay. Immunomodulatory therapy can be reintroduced slowly if needed.
- Kandiel A, Lashner B. Cytomegalovirus colitis complicating inflammatory bowel disease. Am J Gastroenterol 2006; 101:2857–2865.
- Söderberg-Nauclér C, Fish KN, Nelson JA. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J Clin Invest 1997; 100:3154–3163.
- Goodgame RW. Gastrointestinal cytomegalovirus disease. Ann Intern Med 1993; 119:924–935.
- Cottone M, Pietrosi G, Martorana G, et al. Prevalence of cytomegalovirus infection in severe refractory ulcerative and Crohn’s colitis. Am J Gastroenterol 2001; 96:773–775.
- Kishore J, Ghoshal U, Ghoshal UC, et al. Infection with cytomegalovirus in patients with inflammatory bowel disease: prevalence, clinical significance, and outcome. J Med Microbiol 2004; 53:1155–1160.
A 21-year-old woman with Crohn disease presented to the hospital after 5 days of diffuse abdominal pain, nausea, vomiting, and watery diarrhea despite taking azathioprine (Imuran) 100 mg daily as maintenance therapy. She had been hospitalized 2 weeks previously at another hospital for a Crohn disease flare, which was treated with intravenous methylprednisolone (Solu-Medrol).
On admission to our hospital, her temperature was 38.4°C (101.1°F), heart rate 78 per minute, respiratory rate 18 per minute, blood pressure 110/50 mm Hg, and oxygen saturation 98% while breathing room air. She had diffuse abdominal tenderness without rebound tenderness.
Because Clostridium difficile has a high prevalence in our hospital, treament for C difficile diarrhea was started empirically directly upon hospital admission; it was stopped 48 hours later when stool cultures came back negative for C difficile.
COLITIS AND CYTOMEGALOVIRUS INFECTION
CMV colitis is common in patients with inflammatory bowel disease (ie, Crohn disease or ulcerative colitis) who are on long-term immunosuppressive therapy. Heightened suspicion for it is needed when treating patients with inflammatory bowel disease, as they tend to present with atypical symptoms and signs.
It is also important to keep a wide differential diagnosis in mind, as acute fever and diarrhea in patients with inflammatory bowel disease are not always related to the underlying disease. In these patients, a variety of diagnostic tests may be necessary to exclude an opportunistic infection and an unrelated intercurrent illness.
Human CMV is a member of the family of herpes viruses, which persist for life after a primary infection. In exacerbations of inflammatory bowel disease, it is not clear whether CMV is a nonpathogenic bystander or a true pathogen.1 Most CMV infections in patients with inflammatory bowel disease are due to reactivation of the virus, as levels of inflammatory cytokines such as tumor necrosis factor are increased in the intestinal mucosa in active inflammatory bowel disease, and these cytokines are known to trigger reactivation.2
In patients with chronic inflammatory bowel disease, CMV colitis usually presents with abdominal pain, diarrhea, intestinal bleeding, and fever. The gold standard for diagnosis is immunohistochemical testing of colon biopsy samples using monoclonal antibodies against CMV. Owl’s eye inclusion bodies on histopathologic sections are highly specific for CMV infection. Other diagnostic studies include endoscopy and serologic testing.
The gastrointestinal tract is thought to contain latent CMV after a primary infection, and long-term treatment with immunomodulatory drugs such as azathioprine and corticosteroids can cause local reactivation of the latent virus.3
CMV infection in patients with inflammatory bowel disease is associated with poor outcomes, such as the need for colectomy.4 The prevalence of CMV infection in patients with inflammatory bowel disease has been reported as 5% to 36%, and higher in patients with disease refractory to steroid therapy.1,5
When a patient with inflammatory bowel disease is diagnosed with CMV infection, the immunomodulatory drugs should be stopped and the corticosteroids should be tapered to the lowest possible dose. Treatment of the infection is intravenous ganciclovir at 5 mg per kilogram of body weight twice daily for 14 days, followed by oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks.
After receiving intravenous ganciclovir for 14 days, our patient received oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks. Her azathioprine was stopped while she was taking the antivirals, and it was resumed the day after she completed the course of valacyclovir.
The response to treatment is monitored with a cytomegalovirus pp 65 antigenemia assay. Immunomodulatory therapy can be reintroduced slowly if needed.
A 21-year-old woman with Crohn disease presented to the hospital after 5 days of diffuse abdominal pain, nausea, vomiting, and watery diarrhea despite taking azathioprine (Imuran) 100 mg daily as maintenance therapy. She had been hospitalized 2 weeks previously at another hospital for a Crohn disease flare, which was treated with intravenous methylprednisolone (Solu-Medrol).
On admission to our hospital, her temperature was 38.4°C (101.1°F), heart rate 78 per minute, respiratory rate 18 per minute, blood pressure 110/50 mm Hg, and oxygen saturation 98% while breathing room air. She had diffuse abdominal tenderness without rebound tenderness.
Because Clostridium difficile has a high prevalence in our hospital, treament for C difficile diarrhea was started empirically directly upon hospital admission; it was stopped 48 hours later when stool cultures came back negative for C difficile.
COLITIS AND CYTOMEGALOVIRUS INFECTION
CMV colitis is common in patients with inflammatory bowel disease (ie, Crohn disease or ulcerative colitis) who are on long-term immunosuppressive therapy. Heightened suspicion for it is needed when treating patients with inflammatory bowel disease, as they tend to present with atypical symptoms and signs.
It is also important to keep a wide differential diagnosis in mind, as acute fever and diarrhea in patients with inflammatory bowel disease are not always related to the underlying disease. In these patients, a variety of diagnostic tests may be necessary to exclude an opportunistic infection and an unrelated intercurrent illness.
Human CMV is a member of the family of herpes viruses, which persist for life after a primary infection. In exacerbations of inflammatory bowel disease, it is not clear whether CMV is a nonpathogenic bystander or a true pathogen.1 Most CMV infections in patients with inflammatory bowel disease are due to reactivation of the virus, as levels of inflammatory cytokines such as tumor necrosis factor are increased in the intestinal mucosa in active inflammatory bowel disease, and these cytokines are known to trigger reactivation.2
In patients with chronic inflammatory bowel disease, CMV colitis usually presents with abdominal pain, diarrhea, intestinal bleeding, and fever. The gold standard for diagnosis is immunohistochemical testing of colon biopsy samples using monoclonal antibodies against CMV. Owl’s eye inclusion bodies on histopathologic sections are highly specific for CMV infection. Other diagnostic studies include endoscopy and serologic testing.
The gastrointestinal tract is thought to contain latent CMV after a primary infection, and long-term treatment with immunomodulatory drugs such as azathioprine and corticosteroids can cause local reactivation of the latent virus.3
CMV infection in patients with inflammatory bowel disease is associated with poor outcomes, such as the need for colectomy.4 The prevalence of CMV infection in patients with inflammatory bowel disease has been reported as 5% to 36%, and higher in patients with disease refractory to steroid therapy.1,5
When a patient with inflammatory bowel disease is diagnosed with CMV infection, the immunomodulatory drugs should be stopped and the corticosteroids should be tapered to the lowest possible dose. Treatment of the infection is intravenous ganciclovir at 5 mg per kilogram of body weight twice daily for 14 days, followed by oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks.
After receiving intravenous ganciclovir for 14 days, our patient received oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks. Her azathioprine was stopped while she was taking the antivirals, and it was resumed the day after she completed the course of valacyclovir.
The response to treatment is monitored with a cytomegalovirus pp 65 antigenemia assay. Immunomodulatory therapy can be reintroduced slowly if needed.
- Kandiel A, Lashner B. Cytomegalovirus colitis complicating inflammatory bowel disease. Am J Gastroenterol 2006; 101:2857–2865.
- Söderberg-Nauclér C, Fish KN, Nelson JA. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J Clin Invest 1997; 100:3154–3163.
- Goodgame RW. Gastrointestinal cytomegalovirus disease. Ann Intern Med 1993; 119:924–935.
- Cottone M, Pietrosi G, Martorana G, et al. Prevalence of cytomegalovirus infection in severe refractory ulcerative and Crohn’s colitis. Am J Gastroenterol 2001; 96:773–775.
- Kishore J, Ghoshal U, Ghoshal UC, et al. Infection with cytomegalovirus in patients with inflammatory bowel disease: prevalence, clinical significance, and outcome. J Med Microbiol 2004; 53:1155–1160.
- Kandiel A, Lashner B. Cytomegalovirus colitis complicating inflammatory bowel disease. Am J Gastroenterol 2006; 101:2857–2865.
- Söderberg-Nauclér C, Fish KN, Nelson JA. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J Clin Invest 1997; 100:3154–3163.
- Goodgame RW. Gastrointestinal cytomegalovirus disease. Ann Intern Med 1993; 119:924–935.
- Cottone M, Pietrosi G, Martorana G, et al. Prevalence of cytomegalovirus infection in severe refractory ulcerative and Crohn’s colitis. Am J Gastroenterol 2001; 96:773–775.
- Kishore J, Ghoshal U, Ghoshal UC, et al. Infection with cytomegalovirus in patients with inflammatory bowel disease: prevalence, clinical significance, and outcome. J Med Microbiol 2004; 53:1155–1160.
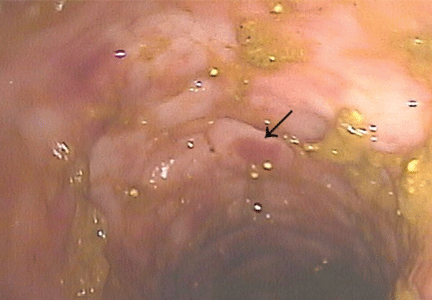
Presumed premature ventricular contractions
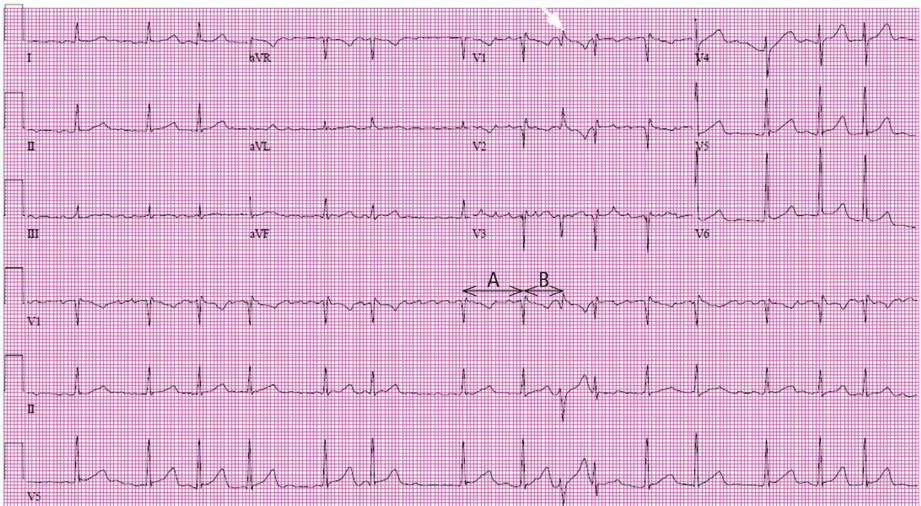
What is the diagnosis? Is a cardiology consult warranted?
AN ABERRANT CONDUCTION PATTERN
The finding seen in this electrocardiogram is known as the Ashman phenomenon, an aberrant conduction pattern seen in atrial dysrhythmias, mainly atrial fibrillation, atrial tachycardia, and atrial ectopy, when a relatively long cycle is followed by a relatively short cycle. The beat terminating the short cycle often has the morphology of right bundle branch block.
This pattern was first described by Gouaux and Ashman in 1947; however, the aberrant conduction of supraventricular impulses was first described by Lewis in 1910.1,2
Ashman phenomenon and right bundle branch block
The three criteria for the diagnosis of right bundle branch block in adults are:
- A QRS duration of 120 msec or more
- An rsr', rsR', or rSR' in leads V1 or V2 (the R' or r' deflection is usually wider than the initial R wave)
- The duration of the S wave in I and V6 is usually greater than that of the R wave or is greater than 40 msec.3
Variation in the heart rate (due to atrial fibrillation in this patient) affects the width of the QRS interval; the refractory period of a cycle is influenced by the RR interval of the previous cycle. Therefore, if after a long cycle with a consequent long refractory period, a shorter cycle follows, then the beat terminating the short cycle is likely to be aberrantly conducted because one of the bundle branches is still in the refractory period. Because the refractory period for the right bundle branch is longer than that of the left bundle branch, the right bundle branch block pattern is more common.4
In our patient’s tracing (Figure 1), the aberrantly conducted beat has the shortest coupling intervals of any of the conducted beats on the tracing. Although the RR interval preceding the short cycle is not the longest on this tracing, it is moderately long, and so the refractory period of the right bundle branch is moderately long.
The Ashman pattern vs ventricular premature beat
Atrial arrhythmias cause a variation in the refractory period of the bundle branches and the ventricular conduction system, and this explains why the Ashman phenomenon occurs more often in this setting. It is important to distinguish the aberrant conduction seen in the Ashman phenomenon, which electrophysiologically is restricted to the His-Purkinje system, from premature ventricular complexes and ventricular tachycardia.
The current criteria used to distinguish the Ashman phenomenon were described by Fisch5,6:
- A relatively long cycle immediately preceding the cycle terminated by the aberrant QRS complex: a short-long-short interval is even more likely to initiate aberration. The aberration can be left or right bundle branch block, or both, even in the same patient.
- Right bundle branch block morphology, with normal orientation of the initial QRS vector. Concealed perpetuation of the aberration is possible, and so a series of wide QRS supraventricular beats is possible.
- Irregular coupling of aberrant QRS complexes.
- Lack of a fully compensatory pause.
In Figure 1, the second aberrantly conducted beat is not as aberrant as the first, even though it is even more premature than the first. This can be explained because the refractory period of the right bundle branch has now shortened.
Also, the mechanism of aberrancy of the second beat may be partly the result of concealed perpetuation, ie, incomplete penetration of the His bundle depolarizations in either direction with secondary abnormalities of antegrade or retrograde conduction. This pattern is not directly reflected on the surface electrocardiogram but can be detected on intracardiac electrophysiologic studies.7 In concealed perpetuation, instead of inducing tachycardia, the extra stimuli are followed by pauses that exceed the tachycardia cycle length.8
Treated by managing the atrial arrhythmia
There is no specific treatment for the aberrant cycles. Rather, treatment is directed at the atrial arrhythmia.9 Adequate control of the underlying process and the atrial tachyarrhythmia itself is important. In our patient, control of the exacerbation of chronic obstructive pulmonary disease and of the heart rate improved the ventricular response to atrial fibrillation.
- Fisch C, Knoebel SB. Vagaries of acceleration dependent aberration. Br Heart J 1992; 67:16–24.
- Gouaux JL, Ashman R. Auricular fibrillation with aberration simulating ventricular paroxysmal tachycardia. Am Heart J 1947; 34:366–373.
- Surawicz B, Childers R, Deal BJ, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part III: intraventricular conduction disturbances: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:976–981.
- Antunes E, Brugada J, Steurer G, Andries E, Brugada P. The differential diagnosis of a regular tachycardia with a wide QRS complex on the 12-lead ECG: ventricular tachycardia, supraventricular tachycardia with aberrant intraventricular conduction, and supraventricular tachycardia with anterograde conduction over an accessory pathway. Pacing Clin Electrophysiol 1994; 17:1515–1524.
- Fisch C, Knoebel SB, eds. Clinical Electrocardiography of Arrhythmias. Armonk, NY: Futura Publishing Company, 2000:407.
- Gulamhusein S, Yee R, Ko PT, Klein GJ. Electrocardiographic criteria for differentiating aberrancy and ventricular extrasystole in chronic atrial fibrillation: validation by intracardiac recordings. J Electrocardiol 1985; 18:41–50.
- Josephson ME. Miscellaneous phenomena related to atrioventricular conduction. In: Clinical Cardiac Electrophysiology: Techniques and Interpretations. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2002:140–154.
- Josephson ME. Recurrent ventricular tachycardia. In: Clinical Cardiac Electrophysiology: Techniques and Interpretations. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2002:425–610.
- Hope RR, Lazzara R, Scherlag BJ. The induction of ventricular arrhythmias in acute myocardial ischemia by atrial pacing with long-short cycle sequences. Chest 1977; 71:651–658.
What is the diagnosis? Is a cardiology consult warranted?
AN ABERRANT CONDUCTION PATTERN
The finding seen in this electrocardiogram is known as the Ashman phenomenon, an aberrant conduction pattern seen in atrial dysrhythmias, mainly atrial fibrillation, atrial tachycardia, and atrial ectopy, when a relatively long cycle is followed by a relatively short cycle. The beat terminating the short cycle often has the morphology of right bundle branch block.
This pattern was first described by Gouaux and Ashman in 1947; however, the aberrant conduction of supraventricular impulses was first described by Lewis in 1910.1,2
Ashman phenomenon and right bundle branch block
The three criteria for the diagnosis of right bundle branch block in adults are:
- A QRS duration of 120 msec or more
- An rsr', rsR', or rSR' in leads V1 or V2 (the R' or r' deflection is usually wider than the initial R wave)
- The duration of the S wave in I and V6 is usually greater than that of the R wave or is greater than 40 msec.3
Variation in the heart rate (due to atrial fibrillation in this patient) affects the width of the QRS interval; the refractory period of a cycle is influenced by the RR interval of the previous cycle. Therefore, if after a long cycle with a consequent long refractory period, a shorter cycle follows, then the beat terminating the short cycle is likely to be aberrantly conducted because one of the bundle branches is still in the refractory period. Because the refractory period for the right bundle branch is longer than that of the left bundle branch, the right bundle branch block pattern is more common.4
In our patient’s tracing (Figure 1), the aberrantly conducted beat has the shortest coupling intervals of any of the conducted beats on the tracing. Although the RR interval preceding the short cycle is not the longest on this tracing, it is moderately long, and so the refractory period of the right bundle branch is moderately long.
The Ashman pattern vs ventricular premature beat
Atrial arrhythmias cause a variation in the refractory period of the bundle branches and the ventricular conduction system, and this explains why the Ashman phenomenon occurs more often in this setting. It is important to distinguish the aberrant conduction seen in the Ashman phenomenon, which electrophysiologically is restricted to the His-Purkinje system, from premature ventricular complexes and ventricular tachycardia.
The current criteria used to distinguish the Ashman phenomenon were described by Fisch5,6:
- A relatively long cycle immediately preceding the cycle terminated by the aberrant QRS complex: a short-long-short interval is even more likely to initiate aberration. The aberration can be left or right bundle branch block, or both, even in the same patient.
- Right bundle branch block morphology, with normal orientation of the initial QRS vector. Concealed perpetuation of the aberration is possible, and so a series of wide QRS supraventricular beats is possible.
- Irregular coupling of aberrant QRS complexes.
- Lack of a fully compensatory pause.
In Figure 1, the second aberrantly conducted beat is not as aberrant as the first, even though it is even more premature than the first. This can be explained because the refractory period of the right bundle branch has now shortened.
Also, the mechanism of aberrancy of the second beat may be partly the result of concealed perpetuation, ie, incomplete penetration of the His bundle depolarizations in either direction with secondary abnormalities of antegrade or retrograde conduction. This pattern is not directly reflected on the surface electrocardiogram but can be detected on intracardiac electrophysiologic studies.7 In concealed perpetuation, instead of inducing tachycardia, the extra stimuli are followed by pauses that exceed the tachycardia cycle length.8
Treated by managing the atrial arrhythmia
There is no specific treatment for the aberrant cycles. Rather, treatment is directed at the atrial arrhythmia.9 Adequate control of the underlying process and the atrial tachyarrhythmia itself is important. In our patient, control of the exacerbation of chronic obstructive pulmonary disease and of the heart rate improved the ventricular response to atrial fibrillation.
What is the diagnosis? Is a cardiology consult warranted?
AN ABERRANT CONDUCTION PATTERN
The finding seen in this electrocardiogram is known as the Ashman phenomenon, an aberrant conduction pattern seen in atrial dysrhythmias, mainly atrial fibrillation, atrial tachycardia, and atrial ectopy, when a relatively long cycle is followed by a relatively short cycle. The beat terminating the short cycle often has the morphology of right bundle branch block.
This pattern was first described by Gouaux and Ashman in 1947; however, the aberrant conduction of supraventricular impulses was first described by Lewis in 1910.1,2
Ashman phenomenon and right bundle branch block
The three criteria for the diagnosis of right bundle branch block in adults are:
- A QRS duration of 120 msec or more
- An rsr', rsR', or rSR' in leads V1 or V2 (the R' or r' deflection is usually wider than the initial R wave)
- The duration of the S wave in I and V6 is usually greater than that of the R wave or is greater than 40 msec.3
Variation in the heart rate (due to atrial fibrillation in this patient) affects the width of the QRS interval; the refractory period of a cycle is influenced by the RR interval of the previous cycle. Therefore, if after a long cycle with a consequent long refractory period, a shorter cycle follows, then the beat terminating the short cycle is likely to be aberrantly conducted because one of the bundle branches is still in the refractory period. Because the refractory period for the right bundle branch is longer than that of the left bundle branch, the right bundle branch block pattern is more common.4
In our patient’s tracing (Figure 1), the aberrantly conducted beat has the shortest coupling intervals of any of the conducted beats on the tracing. Although the RR interval preceding the short cycle is not the longest on this tracing, it is moderately long, and so the refractory period of the right bundle branch is moderately long.
The Ashman pattern vs ventricular premature beat
Atrial arrhythmias cause a variation in the refractory period of the bundle branches and the ventricular conduction system, and this explains why the Ashman phenomenon occurs more often in this setting. It is important to distinguish the aberrant conduction seen in the Ashman phenomenon, which electrophysiologically is restricted to the His-Purkinje system, from premature ventricular complexes and ventricular tachycardia.
The current criteria used to distinguish the Ashman phenomenon were described by Fisch5,6:
- A relatively long cycle immediately preceding the cycle terminated by the aberrant QRS complex: a short-long-short interval is even more likely to initiate aberration. The aberration can be left or right bundle branch block, or both, even in the same patient.
- Right bundle branch block morphology, with normal orientation of the initial QRS vector. Concealed perpetuation of the aberration is possible, and so a series of wide QRS supraventricular beats is possible.
- Irregular coupling of aberrant QRS complexes.
- Lack of a fully compensatory pause.
In Figure 1, the second aberrantly conducted beat is not as aberrant as the first, even though it is even more premature than the first. This can be explained because the refractory period of the right bundle branch has now shortened.
Also, the mechanism of aberrancy of the second beat may be partly the result of concealed perpetuation, ie, incomplete penetration of the His bundle depolarizations in either direction with secondary abnormalities of antegrade or retrograde conduction. This pattern is not directly reflected on the surface electrocardiogram but can be detected on intracardiac electrophysiologic studies.7 In concealed perpetuation, instead of inducing tachycardia, the extra stimuli are followed by pauses that exceed the tachycardia cycle length.8
Treated by managing the atrial arrhythmia
There is no specific treatment for the aberrant cycles. Rather, treatment is directed at the atrial arrhythmia.9 Adequate control of the underlying process and the atrial tachyarrhythmia itself is important. In our patient, control of the exacerbation of chronic obstructive pulmonary disease and of the heart rate improved the ventricular response to atrial fibrillation.
- Fisch C, Knoebel SB. Vagaries of acceleration dependent aberration. Br Heart J 1992; 67:16–24.
- Gouaux JL, Ashman R. Auricular fibrillation with aberration simulating ventricular paroxysmal tachycardia. Am Heart J 1947; 34:366–373.
- Surawicz B, Childers R, Deal BJ, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part III: intraventricular conduction disturbances: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:976–981.
- Antunes E, Brugada J, Steurer G, Andries E, Brugada P. The differential diagnosis of a regular tachycardia with a wide QRS complex on the 12-lead ECG: ventricular tachycardia, supraventricular tachycardia with aberrant intraventricular conduction, and supraventricular tachycardia with anterograde conduction over an accessory pathway. Pacing Clin Electrophysiol 1994; 17:1515–1524.
- Fisch C, Knoebel SB, eds. Clinical Electrocardiography of Arrhythmias. Armonk, NY: Futura Publishing Company, 2000:407.
- Gulamhusein S, Yee R, Ko PT, Klein GJ. Electrocardiographic criteria for differentiating aberrancy and ventricular extrasystole in chronic atrial fibrillation: validation by intracardiac recordings. J Electrocardiol 1985; 18:41–50.
- Josephson ME. Miscellaneous phenomena related to atrioventricular conduction. In: Clinical Cardiac Electrophysiology: Techniques and Interpretations. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2002:140–154.
- Josephson ME. Recurrent ventricular tachycardia. In: Clinical Cardiac Electrophysiology: Techniques and Interpretations. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2002:425–610.
- Hope RR, Lazzara R, Scherlag BJ. The induction of ventricular arrhythmias in acute myocardial ischemia by atrial pacing with long-short cycle sequences. Chest 1977; 71:651–658.
- Fisch C, Knoebel SB. Vagaries of acceleration dependent aberration. Br Heart J 1992; 67:16–24.
- Gouaux JL, Ashman R. Auricular fibrillation with aberration simulating ventricular paroxysmal tachycardia. Am Heart J 1947; 34:366–373.
- Surawicz B, Childers R, Deal BJ, et al; American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram: part III: intraventricular conduction disturbances: a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society. Endorsed by the International Society for Computerized Electrocardiology. J Am Coll Cardiol 2009; 53:976–981.
- Antunes E, Brugada J, Steurer G, Andries E, Brugada P. The differential diagnosis of a regular tachycardia with a wide QRS complex on the 12-lead ECG: ventricular tachycardia, supraventricular tachycardia with aberrant intraventricular conduction, and supraventricular tachycardia with anterograde conduction over an accessory pathway. Pacing Clin Electrophysiol 1994; 17:1515–1524.
- Fisch C, Knoebel SB, eds. Clinical Electrocardiography of Arrhythmias. Armonk, NY: Futura Publishing Company, 2000:407.
- Gulamhusein S, Yee R, Ko PT, Klein GJ. Electrocardiographic criteria for differentiating aberrancy and ventricular extrasystole in chronic atrial fibrillation: validation by intracardiac recordings. J Electrocardiol 1985; 18:41–50.
- Josephson ME. Miscellaneous phenomena related to atrioventricular conduction. In: Clinical Cardiac Electrophysiology: Techniques and Interpretations. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2002:140–154.
- Josephson ME. Recurrent ventricular tachycardia. In: Clinical Cardiac Electrophysiology: Techniques and Interpretations. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2002:425–610.
- Hope RR, Lazzara R, Scherlag BJ. The induction of ventricular arrhythmias in acute myocardial ischemia by atrial pacing with long-short cycle sequences. Chest 1977; 71:651–658.
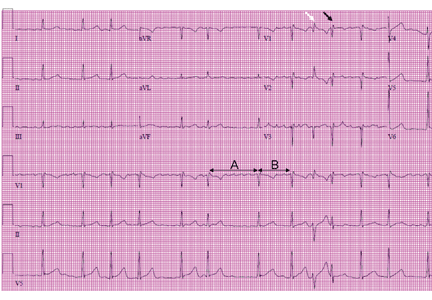
A less common source of dyspnea in scleroderma
A 48-year-old man reports progressive exercise intolerance, shortness of breath, fatigue, and melena over the past month. He has a long history of Raynaud phenomenon, and 5 months ago he developed severe sclerodactyly in both hands, diagnosed as limited cutaneous systemic sclerosis (scleroderma).
He has no chest pain, swelling of the lower limbs, change in weight, cough, fever, chills, or sick contacts, and he has not traveled recently.
His symptoms began as fatigue and shortness of breath, which worsened until he began having episodes of abdominal pain with melena and dizzy spills, although he never passed out.
He is currently taking long-term low-dose prednisone and mycophenolate mofetil (Cell-Cept) for the systemic sclerosis, and omeprazole (Prilosec) for gastroesophageal reflux. His father had lupus, and his grandmother had colon cancer.
An outpatient workup for sclerosis-related lung and heart involvement is negative. The workup includes computed tomography of the chest, pulmonary function tests, and Doppler echocardiography.
He is afebrile, with a blood pressure of 105/60 mm Hg and a pulse of 98. His cardiopulmonary examination results are normal. He has mild epigastric tenderness without rebound or guarding. His hemoglobin concentration at the time of hospital admission is 7.8 g/dL, down from 14.5 g/dL recorded when limited cutaneous systemic sclerosis was diagnosed. Iron studies reveal iron deficiency.
The antral ectasia is treated with argon plasma coagulation during the endoscopic examination.
Afterward, the patient's hemoglobin stabilizes, and the melena resolves. He is discharged on an oral proton pump inhibitor, with instructions to follow up for another endoscopic session in 1 month.
GASTROINTESTINAL FEATURES OF SYSTEMIC SCLEROSIS
Sclerodermal disorders have diverse manifestations that always include characteristic cutaneous signs. While there are several well-recognized symptomatic conditions commonly associated with scleroderma, attention must also be paid to the less common causes of these symptoms. Scleroderma has gastrointestinal complications that can easily be missed and may not respond to immunomodulatory or proton pump inhibitor therapy: complications can include esophageal dysmotility, hypomotility, gastric paresis, reflux esophagitis, strictures, drug-related ulcer, malabsorption, bacterial overgrowth, and pseudo-obstruction.1
This patient had an underrecognized cause of dyspnea in the setting of systemic sclerosis. Vascular symptoms of limited cutaneous systemic sclerosis are typically attributed to Raynaud phenomenon; gastrointestinal symptoms are typically attributed to esophageal dysmotility; and associated dyspnea is often considered to represent pulmonary or cardiac involvement of the sclerosis. However, gastric antral vascular ectasia should be considered in any patient with scleroderma and evidence of anemia.
The prevalence of gastric antral vascular ectasia in patients with systemic sclerosis is estimated to be about 6%.2–4 It is a relatively rare cause of upper gastrointestinal blood loss that can be clinically silent until the patient develops severe iron deficiency anemia and symptoms of dyspnea, fatigue, or congestive heart failure.
Gastric antral vascular ectasia in scleroderma usually presents as iron deficiency anemia, and only presents overtly as hematemesis or melena 10% to 14% of the time.4 Because of the often occult nature of the bleeding, the condition may be clinically silent in the early phase. Symptoms of shortness of breath and fatigue may not develop until the anemia worsens rapidly or becomes severe. Anemia is present in almost all cases of gastric antral vascular ectasia (96% to 100%) and should be a strong clinical clue for early endoscopic evaluation in patients with scleroderma, especially if there is already suspicion of upper gastrointestinal bleeding.2–5
The distinctive endoscopic streaky pattern of ectasia along the stomach antrum seen in gastric antral vascular ectasia is called “watermelon stomach”4,5 because the striped pattern recalls the stripes of a watermelon. The endoscopic appearance can vary, however, from the watermelon pattern to a coalescence of angiodysplastic lesions termed “honeycomb stomach,” which can easily be mistaken for antral gastritis.4,5 Therefore, biopsy often serves to confirm the diagnosis, with histologic features including dilated mucosal capillaries with focal fibrin thrombosis and fibromuscular hyperplasia of the lamina propria.
Gastric antral vascular ectasia often requires multiple transfusions of red blood cells, as well as repeated treatments with endoscopic argon plasma coagulation, whereby ionized argon gas is used to conduct an electric current that coagulates the surface of the mucosa to a few millimeters depth.4–6
A knowledge of the association between scleroderma and gastric antral vascular ectasia can lead to earlier recognition and treatment and can avoid unnecessary testing and complications of severe anemia.
- Forbes A, Marie I. Gastrointestinal complications: the most frequent internal complications of systemic sclerosis. Rheumatology (Oxford) 2009; 48(suppl 3):iii36–iii39.
- Ingraham KM, O’Brien MS, Shenin M, Derk CT, Steen VD. Gastric antral vascular ectasia in systemic sclerosis: demographics and disease predictors. J Rheumatol 2010; 37:603–607.
- Watson M, Hally RJ, McCue PA, Varga J, Jiménez SA. Gastric antral vascular ectasia (watermelon stomach) in patients with systemic sclerosis. Arthritis Rheum 1996; 39:341–346.
- Marie I, Ducrotte P, Antonietti M, Herve S, Levesque H. Watermelon stomach in systemic sclerosis: its incidence and management. Aliment Pharmacol Ther 2008; 28:412–421.
- Selinger CP, Ang YS. Gastric antral vascular ectasia (GAVE): an update on clinical presentation, pathophysiology and treatment. Digestion 2008; 77:131–137.
- Chaves DM, Sakai P, Oliveira CV, Cheng S, Ishioka S. Watermelon stomach: clinical aspects and treatment with argon plasma coagulation. Arq Gastroenterol 2006; 43:191–195.
A 48-year-old man reports progressive exercise intolerance, shortness of breath, fatigue, and melena over the past month. He has a long history of Raynaud phenomenon, and 5 months ago he developed severe sclerodactyly in both hands, diagnosed as limited cutaneous systemic sclerosis (scleroderma).
He has no chest pain, swelling of the lower limbs, change in weight, cough, fever, chills, or sick contacts, and he has not traveled recently.
His symptoms began as fatigue and shortness of breath, which worsened until he began having episodes of abdominal pain with melena and dizzy spills, although he never passed out.
He is currently taking long-term low-dose prednisone and mycophenolate mofetil (Cell-Cept) for the systemic sclerosis, and omeprazole (Prilosec) for gastroesophageal reflux. His father had lupus, and his grandmother had colon cancer.
An outpatient workup for sclerosis-related lung and heart involvement is negative. The workup includes computed tomography of the chest, pulmonary function tests, and Doppler echocardiography.
He is afebrile, with a blood pressure of 105/60 mm Hg and a pulse of 98. His cardiopulmonary examination results are normal. He has mild epigastric tenderness without rebound or guarding. His hemoglobin concentration at the time of hospital admission is 7.8 g/dL, down from 14.5 g/dL recorded when limited cutaneous systemic sclerosis was diagnosed. Iron studies reveal iron deficiency.
The antral ectasia is treated with argon plasma coagulation during the endoscopic examination.
Afterward, the patient's hemoglobin stabilizes, and the melena resolves. He is discharged on an oral proton pump inhibitor, with instructions to follow up for another endoscopic session in 1 month.
GASTROINTESTINAL FEATURES OF SYSTEMIC SCLEROSIS
Sclerodermal disorders have diverse manifestations that always include characteristic cutaneous signs. While there are several well-recognized symptomatic conditions commonly associated with scleroderma, attention must also be paid to the less common causes of these symptoms. Scleroderma has gastrointestinal complications that can easily be missed and may not respond to immunomodulatory or proton pump inhibitor therapy: complications can include esophageal dysmotility, hypomotility, gastric paresis, reflux esophagitis, strictures, drug-related ulcer, malabsorption, bacterial overgrowth, and pseudo-obstruction.1
This patient had an underrecognized cause of dyspnea in the setting of systemic sclerosis. Vascular symptoms of limited cutaneous systemic sclerosis are typically attributed to Raynaud phenomenon; gastrointestinal symptoms are typically attributed to esophageal dysmotility; and associated dyspnea is often considered to represent pulmonary or cardiac involvement of the sclerosis. However, gastric antral vascular ectasia should be considered in any patient with scleroderma and evidence of anemia.
The prevalence of gastric antral vascular ectasia in patients with systemic sclerosis is estimated to be about 6%.2–4 It is a relatively rare cause of upper gastrointestinal blood loss that can be clinically silent until the patient develops severe iron deficiency anemia and symptoms of dyspnea, fatigue, or congestive heart failure.
Gastric antral vascular ectasia in scleroderma usually presents as iron deficiency anemia, and only presents overtly as hematemesis or melena 10% to 14% of the time.4 Because of the often occult nature of the bleeding, the condition may be clinically silent in the early phase. Symptoms of shortness of breath and fatigue may not develop until the anemia worsens rapidly or becomes severe. Anemia is present in almost all cases of gastric antral vascular ectasia (96% to 100%) and should be a strong clinical clue for early endoscopic evaluation in patients with scleroderma, especially if there is already suspicion of upper gastrointestinal bleeding.2–5
The distinctive endoscopic streaky pattern of ectasia along the stomach antrum seen in gastric antral vascular ectasia is called “watermelon stomach”4,5 because the striped pattern recalls the stripes of a watermelon. The endoscopic appearance can vary, however, from the watermelon pattern to a coalescence of angiodysplastic lesions termed “honeycomb stomach,” which can easily be mistaken for antral gastritis.4,5 Therefore, biopsy often serves to confirm the diagnosis, with histologic features including dilated mucosal capillaries with focal fibrin thrombosis and fibromuscular hyperplasia of the lamina propria.
Gastric antral vascular ectasia often requires multiple transfusions of red blood cells, as well as repeated treatments with endoscopic argon plasma coagulation, whereby ionized argon gas is used to conduct an electric current that coagulates the surface of the mucosa to a few millimeters depth.4–6
A knowledge of the association between scleroderma and gastric antral vascular ectasia can lead to earlier recognition and treatment and can avoid unnecessary testing and complications of severe anemia.
A 48-year-old man reports progressive exercise intolerance, shortness of breath, fatigue, and melena over the past month. He has a long history of Raynaud phenomenon, and 5 months ago he developed severe sclerodactyly in both hands, diagnosed as limited cutaneous systemic sclerosis (scleroderma).
He has no chest pain, swelling of the lower limbs, change in weight, cough, fever, chills, or sick contacts, and he has not traveled recently.
His symptoms began as fatigue and shortness of breath, which worsened until he began having episodes of abdominal pain with melena and dizzy spills, although he never passed out.
He is currently taking long-term low-dose prednisone and mycophenolate mofetil (Cell-Cept) for the systemic sclerosis, and omeprazole (Prilosec) for gastroesophageal reflux. His father had lupus, and his grandmother had colon cancer.
An outpatient workup for sclerosis-related lung and heart involvement is negative. The workup includes computed tomography of the chest, pulmonary function tests, and Doppler echocardiography.
He is afebrile, with a blood pressure of 105/60 mm Hg and a pulse of 98. His cardiopulmonary examination results are normal. He has mild epigastric tenderness without rebound or guarding. His hemoglobin concentration at the time of hospital admission is 7.8 g/dL, down from 14.5 g/dL recorded when limited cutaneous systemic sclerosis was diagnosed. Iron studies reveal iron deficiency.
The antral ectasia is treated with argon plasma coagulation during the endoscopic examination.
Afterward, the patient's hemoglobin stabilizes, and the melena resolves. He is discharged on an oral proton pump inhibitor, with instructions to follow up for another endoscopic session in 1 month.
GASTROINTESTINAL FEATURES OF SYSTEMIC SCLEROSIS
Sclerodermal disorders have diverse manifestations that always include characteristic cutaneous signs. While there are several well-recognized symptomatic conditions commonly associated with scleroderma, attention must also be paid to the less common causes of these symptoms. Scleroderma has gastrointestinal complications that can easily be missed and may not respond to immunomodulatory or proton pump inhibitor therapy: complications can include esophageal dysmotility, hypomotility, gastric paresis, reflux esophagitis, strictures, drug-related ulcer, malabsorption, bacterial overgrowth, and pseudo-obstruction.1
This patient had an underrecognized cause of dyspnea in the setting of systemic sclerosis. Vascular symptoms of limited cutaneous systemic sclerosis are typically attributed to Raynaud phenomenon; gastrointestinal symptoms are typically attributed to esophageal dysmotility; and associated dyspnea is often considered to represent pulmonary or cardiac involvement of the sclerosis. However, gastric antral vascular ectasia should be considered in any patient with scleroderma and evidence of anemia.
The prevalence of gastric antral vascular ectasia in patients with systemic sclerosis is estimated to be about 6%.2–4 It is a relatively rare cause of upper gastrointestinal blood loss that can be clinically silent until the patient develops severe iron deficiency anemia and symptoms of dyspnea, fatigue, or congestive heart failure.
Gastric antral vascular ectasia in scleroderma usually presents as iron deficiency anemia, and only presents overtly as hematemesis or melena 10% to 14% of the time.4 Because of the often occult nature of the bleeding, the condition may be clinically silent in the early phase. Symptoms of shortness of breath and fatigue may not develop until the anemia worsens rapidly or becomes severe. Anemia is present in almost all cases of gastric antral vascular ectasia (96% to 100%) and should be a strong clinical clue for early endoscopic evaluation in patients with scleroderma, especially if there is already suspicion of upper gastrointestinal bleeding.2–5
The distinctive endoscopic streaky pattern of ectasia along the stomach antrum seen in gastric antral vascular ectasia is called “watermelon stomach”4,5 because the striped pattern recalls the stripes of a watermelon. The endoscopic appearance can vary, however, from the watermelon pattern to a coalescence of angiodysplastic lesions termed “honeycomb stomach,” which can easily be mistaken for antral gastritis.4,5 Therefore, biopsy often serves to confirm the diagnosis, with histologic features including dilated mucosal capillaries with focal fibrin thrombosis and fibromuscular hyperplasia of the lamina propria.
Gastric antral vascular ectasia often requires multiple transfusions of red blood cells, as well as repeated treatments with endoscopic argon plasma coagulation, whereby ionized argon gas is used to conduct an electric current that coagulates the surface of the mucosa to a few millimeters depth.4–6
A knowledge of the association between scleroderma and gastric antral vascular ectasia can lead to earlier recognition and treatment and can avoid unnecessary testing and complications of severe anemia.
- Forbes A, Marie I. Gastrointestinal complications: the most frequent internal complications of systemic sclerosis. Rheumatology (Oxford) 2009; 48(suppl 3):iii36–iii39.
- Ingraham KM, O’Brien MS, Shenin M, Derk CT, Steen VD. Gastric antral vascular ectasia in systemic sclerosis: demographics and disease predictors. J Rheumatol 2010; 37:603–607.
- Watson M, Hally RJ, McCue PA, Varga J, Jiménez SA. Gastric antral vascular ectasia (watermelon stomach) in patients with systemic sclerosis. Arthritis Rheum 1996; 39:341–346.
- Marie I, Ducrotte P, Antonietti M, Herve S, Levesque H. Watermelon stomach in systemic sclerosis: its incidence and management. Aliment Pharmacol Ther 2008; 28:412–421.
- Selinger CP, Ang YS. Gastric antral vascular ectasia (GAVE): an update on clinical presentation, pathophysiology and treatment. Digestion 2008; 77:131–137.
- Chaves DM, Sakai P, Oliveira CV, Cheng S, Ishioka S. Watermelon stomach: clinical aspects and treatment with argon plasma coagulation. Arq Gastroenterol 2006; 43:191–195.
- Forbes A, Marie I. Gastrointestinal complications: the most frequent internal complications of systemic sclerosis. Rheumatology (Oxford) 2009; 48(suppl 3):iii36–iii39.
- Ingraham KM, O’Brien MS, Shenin M, Derk CT, Steen VD. Gastric antral vascular ectasia in systemic sclerosis: demographics and disease predictors. J Rheumatol 2010; 37:603–607.
- Watson M, Hally RJ, McCue PA, Varga J, Jiménez SA. Gastric antral vascular ectasia (watermelon stomach) in patients with systemic sclerosis. Arthritis Rheum 1996; 39:341–346.
- Marie I, Ducrotte P, Antonietti M, Herve S, Levesque H. Watermelon stomach in systemic sclerosis: its incidence and management. Aliment Pharmacol Ther 2008; 28:412–421.
- Selinger CP, Ang YS. Gastric antral vascular ectasia (GAVE): an update on clinical presentation, pathophysiology and treatment. Digestion 2008; 77:131–137.
- Chaves DM, Sakai P, Oliveira CV, Cheng S, Ishioka S. Watermelon stomach: clinical aspects and treatment with argon plasma coagulation. Arq Gastroenterol 2006; 43:191–195.