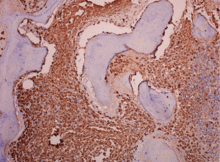
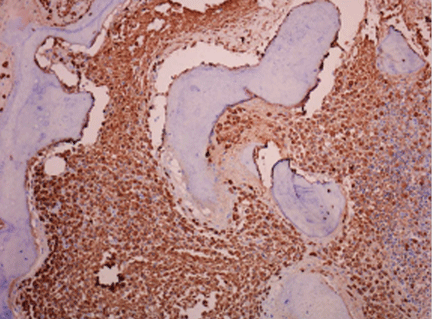
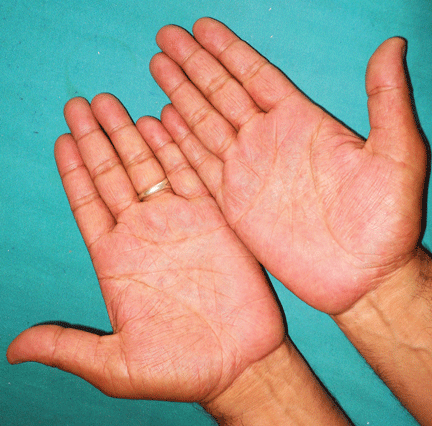
A 62-year-old woman has had flashing lights and floaters in her left eye with progressive loss of vision over the past month. She has not had recent trauma. She does not smoke.
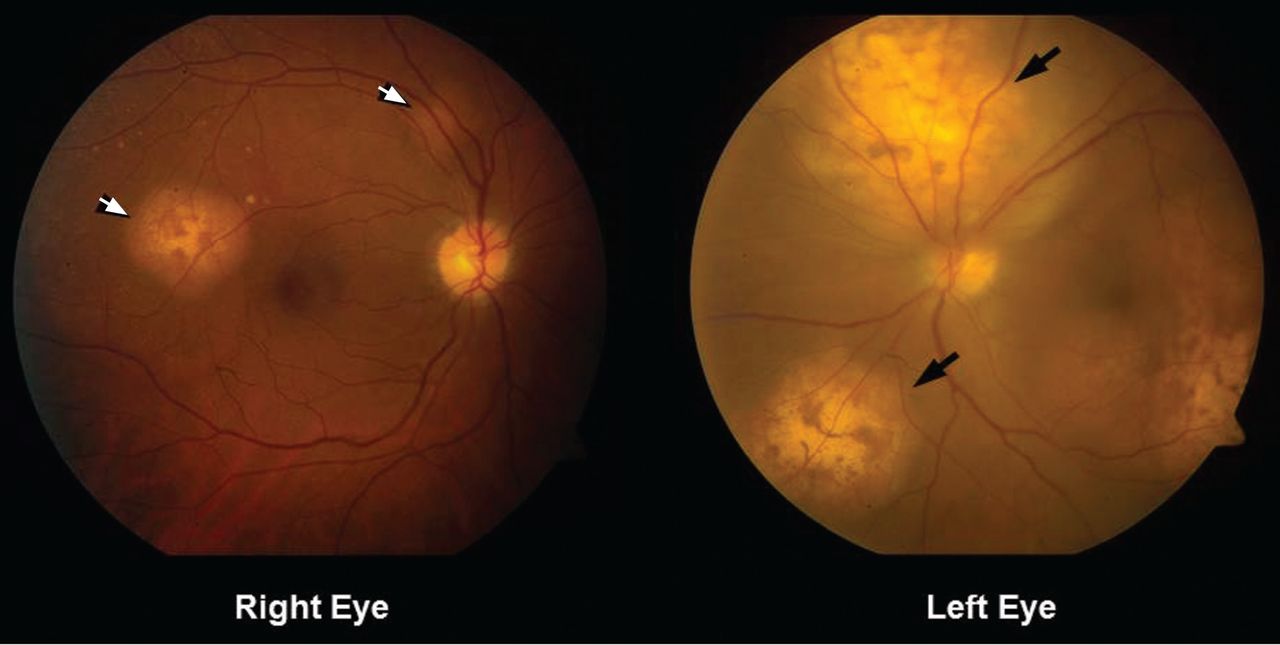
Figure 1. Funduscopy showed multiple lobulated, yellowish choroidal lesions in the posterior pole, with overlying subretinal fluid (arrows). Similar but smaller lesions were seen in the right eye (arrows).
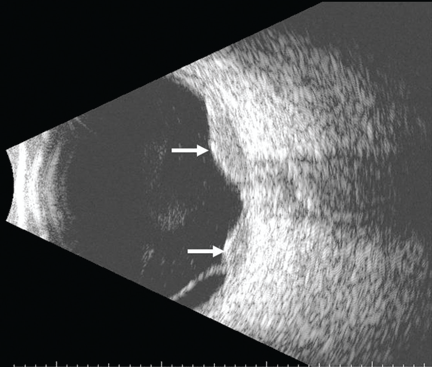
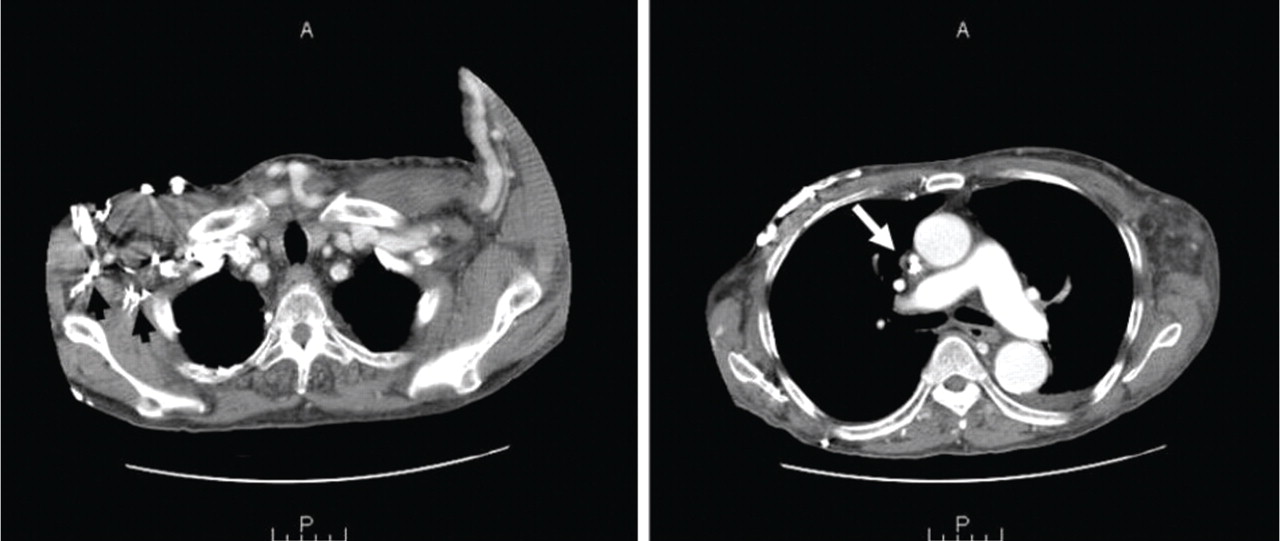
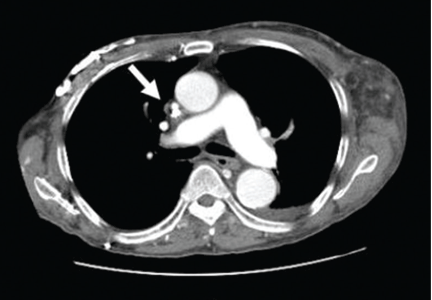
She was referred for an ophthalmologic evaluation. Her visual acuity was 20/20 in the right eye, but she could only count fingers with the left. The anterior segment appeared normal in both eyes. Funduscopic examination of the left eye revealed numerous lobulated, yellowish, choroidal lesions in the posterior pole with overlying subretinal fluid. The lesions involved the fovea, accounting for the poor visual acuity. There were two similar but smaller lesions in the right eye (Figure 1). Ultrasonography confirmed the choroidal location of the lesions (Figure 2).
Q: Which is the most likely diagnosis?
Retinal detachment
Choroidal melanoma
Uveitis
Uveal metastatic tumor
Figure 2. Ultrasonography of the left eye confirmed the choroidal location of the lesions noted on fundoscopy (arrows).
A: Uveal metastatic tumor is the correct diagnosis. Funduscopic findings of bilateral yellow choroidal lesions are consistent with metastatic cancer.
The patient was admitted to the hospital for a thorough evaluation. Computed tomography of the chest showed a 2.1-by-4.5-cm mass in the lower lobe of the left lung, highly suspicious for malignancy and associated with left hilar lymphadenopathy and right acute pulmonary embolism. Bronchoscopy showed an endobronchial tumor completely occluding the left lower lobe and the lingular orifices.
Pathologic specimens from the endobronchial tumor confirmed adenocarcinoma, consistent with a primary lung cancer.
THE OTHER DIAGNOSTIC CHOICES
Detachment or separation of the retina from the underlying pigment epithelium is one of the most commonly encountered eye emergencies.1 It requires urgent attention, since delay in treatment can cause permanent vision loss.
Retinal detachment differs from uveal metastatic tumor in that it presents and progresses rapidly. The common signs and symptoms are floaters in the center of the visual axis, a sensation of flashing lights (related to retinal traction), and, eventually, loss of vision. The detachment most often represents a break or tear (rhegmatogenous retinal detachment), but it is also a common sequela of neglected diabetic retinopathy. Exudative retinal detachment is usually secondary to uveal inflammation or a uveal tumor.
Choroidal melanoma, the most common primary intraocular malignancy, arises from melanocytes within the choroid. In most cases, it develops from preexisting melanocytic nevi.2 It may present as blurred vision, a paracentral scotoma, painless and progressive visual field loss, and floaters. Choroidal melanoma is usually pigmented (dark brown) and is invariably unilateral.
Uveitis is an inflammation of the uveal tract, which includes the iris, ciliary body, and choroid. It is classified as anterior, intermediate, or posterior uveitis or as panuveitis.3
Although flashing lights, floaters, and reduced vision can occur in uveitis, its other important presenting symptoms (ie, pain, redness, and photophobia) were absent in this patient. The absence of anterior chamber cells and corneal inflammatory deposits (keratic precipitates) also made uveitis less likely.4 However, granulomatous uveitis such as sarcoidosis can present as nodular thickening of the uvea, mimicking an intraocular tumor.5
THE MOST COMMON INTRAOCULAR MALIGNANCY
Uveal metastasis is the most common intraocular malignancy6 and is found on autopsy in up to 12% of people who die of cancer; it involves both eyes in 4.4% of cases. Multiple metastases are seen in one eye in up to 20% of cases.7
The tumors are most often in the choroid, probably because of its extensive blood supply. Breast cancer (in women) and lung cancer (in men) are the most common cancers with uveal metastasis.8 Uveal metastasis from cancers of the prostate, kidney, thyroid, and gastrointestinal tract and from lymphoma and leukemia is less common.8
Patients with choroidal metastasis can see flashing lights, floating spots, and distortion of their vision. In such patients, a careful history and physical examination can uncover signs and symptoms of the hidden cancer, especially of lung cancer.9
Once uveal metastasis is suspected, both eyes and orbits and the central nervous system should be examined, as this disease tends to present bilaterally and to involve the central nervous system.10 Uveal metastases respond to chemotherapy and radiotherapy, depending on the nature of the primary tumor. In general, treatment is based on the extent of the metastasis, prior treatments, and the patient’s overall functional status.
References
Hatten B, Browne V. Retinal detachment. Emerg Med J2011; 28:83.
Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol1997; 115:1537–1544.
Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol2005; 140:509–516.
Wertheim MS, Mathers WD, Planck SJ, et al. In vivo confocal microscopy of keratic precipitates. Arch Ophthalmol2004; 122:1773–1781.
Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina2001; 21:40–47.
Eliassi-Rad B, Albert DM, Green WR. Frequency of ocular metastases in patients dying of cancer in eye bank populations. Br J Ophthalmol1996; 80:125–128.
Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology1997; 104:1265–1276.
Herrag M, Lahmiti S, Yazidi AA, Le Lez ML, Diot P. Choroidal metastasis revealing a lung adenocarcinoma. Ann Thorac Surg2010; 89:1013–1014.
Kanthan GL, Jayamohan J, Yip D, Conway RM. Management of metastatic carcinoma of the uveal tract: an evidence-based analysis. Clin Exp Ophthalmol2007; 35:553–565.
A 62-year-old woman has had flashing lights and floaters in her left eye with progressive loss of vision over the past month. She has not had recent trauma. She does not smoke.
Figure 1. Funduscopy showed multiple lobulated, yellowish choroidal lesions in the posterior pole, with overlying subretinal fluid (arrows). Similar but smaller lesions were seen in the right eye (arrows).
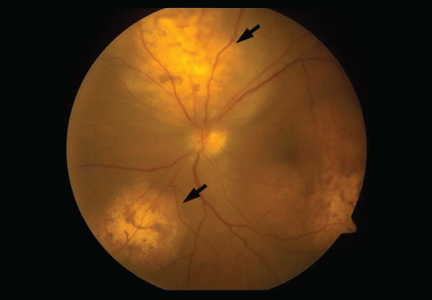
She was referred for an ophthalmologic evaluation. Her visual acuity was 20/20 in the right eye, but she could only count fingers with the left. The anterior segment appeared normal in both eyes. Funduscopic examination of the left eye revealed numerous lobulated, yellowish, choroidal lesions in the posterior pole with overlying subretinal fluid. The lesions involved the fovea, accounting for the poor visual acuity. There were two similar but smaller lesions in the right eye (Figure 1). Ultrasonography confirmed the choroidal location of the lesions (Figure 2).
Q: Which is the most likely diagnosis?
Retinal detachment
Choroidal melanoma
Uveitis
Uveal metastatic tumor
Figure 2. Ultrasonography of the left eye confirmed the choroidal location of the lesions noted on fundoscopy (arrows).
A: Uveal metastatic tumor is the correct diagnosis. Funduscopic findings of bilateral yellow choroidal lesions are consistent with metastatic cancer.
The patient was admitted to the hospital for a thorough evaluation. Computed tomography of the chest showed a 2.1-by-4.5-cm mass in the lower lobe of the left lung, highly suspicious for malignancy and associated with left hilar lymphadenopathy and right acute pulmonary embolism. Bronchoscopy showed an endobronchial tumor completely occluding the left lower lobe and the lingular orifices.
Pathologic specimens from the endobronchial tumor confirmed adenocarcinoma, consistent with a primary lung cancer.
THE OTHER DIAGNOSTIC CHOICES
Detachment or separation of the retina from the underlying pigment epithelium is one of the most commonly encountered eye emergencies.1 It requires urgent attention, since delay in treatment can cause permanent vision loss.
Retinal detachment differs from uveal metastatic tumor in that it presents and progresses rapidly. The common signs and symptoms are floaters in the center of the visual axis, a sensation of flashing lights (related to retinal traction), and, eventually, loss of vision. The detachment most often represents a break or tear (rhegmatogenous retinal detachment), but it is also a common sequela of neglected diabetic retinopathy. Exudative retinal detachment is usually secondary to uveal inflammation or a uveal tumor.
Choroidal melanoma, the most common primary intraocular malignancy, arises from melanocytes within the choroid. In most cases, it develops from preexisting melanocytic nevi.2 It may present as blurred vision, a paracentral scotoma, painless and progressive visual field loss, and floaters. Choroidal melanoma is usually pigmented (dark brown) and is invariably unilateral.
Uveitis is an inflammation of the uveal tract, which includes the iris, ciliary body, and choroid. It is classified as anterior, intermediate, or posterior uveitis or as panuveitis.3
Although flashing lights, floaters, and reduced vision can occur in uveitis, its other important presenting symptoms (ie, pain, redness, and photophobia) were absent in this patient. The absence of anterior chamber cells and corneal inflammatory deposits (keratic precipitates) also made uveitis less likely.4 However, granulomatous uveitis such as sarcoidosis can present as nodular thickening of the uvea, mimicking an intraocular tumor.5
THE MOST COMMON INTRAOCULAR MALIGNANCY
Uveal metastasis is the most common intraocular malignancy6 and is found on autopsy in up to 12% of people who die of cancer; it involves both eyes in 4.4% of cases. Multiple metastases are seen in one eye in up to 20% of cases.7
The tumors are most often in the choroid, probably because of its extensive blood supply. Breast cancer (in women) and lung cancer (in men) are the most common cancers with uveal metastasis.8 Uveal metastasis from cancers of the prostate, kidney, thyroid, and gastrointestinal tract and from lymphoma and leukemia is less common.8
Patients with choroidal metastasis can see flashing lights, floating spots, and distortion of their vision. In such patients, a careful history and physical examination can uncover signs and symptoms of the hidden cancer, especially of lung cancer.9
Once uveal metastasis is suspected, both eyes and orbits and the central nervous system should be examined, as this disease tends to present bilaterally and to involve the central nervous system.10 Uveal metastases respond to chemotherapy and radiotherapy, depending on the nature of the primary tumor. In general, treatment is based on the extent of the metastasis, prior treatments, and the patient’s overall functional status.
A 62-year-old woman has had flashing lights and floaters in her left eye with progressive loss of vision over the past month. She has not had recent trauma. She does not smoke.
Figure 1. Funduscopy showed multiple lobulated, yellowish choroidal lesions in the posterior pole, with overlying subretinal fluid (arrows). Similar but smaller lesions were seen in the right eye (arrows).
She was referred for an ophthalmologic evaluation. Her visual acuity was 20/20 in the right eye, but she could only count fingers with the left. The anterior segment appeared normal in both eyes. Funduscopic examination of the left eye revealed numerous lobulated, yellowish, choroidal lesions in the posterior pole with overlying subretinal fluid. The lesions involved the fovea, accounting for the poor visual acuity. There were two similar but smaller lesions in the right eye (Figure 1). Ultrasonography confirmed the choroidal location of the lesions (Figure 2).
Q: Which is the most likely diagnosis?
Retinal detachment
Choroidal melanoma
Uveitis
Uveal metastatic tumor
Figure 2. Ultrasonography of the left eye confirmed the choroidal location of the lesions noted on fundoscopy (arrows).
A: Uveal metastatic tumor is the correct diagnosis. Funduscopic findings of bilateral yellow choroidal lesions are consistent with metastatic cancer.
The patient was admitted to the hospital for a thorough evaluation. Computed tomography of the chest showed a 2.1-by-4.5-cm mass in the lower lobe of the left lung, highly suspicious for malignancy and associated with left hilar lymphadenopathy and right acute pulmonary embolism. Bronchoscopy showed an endobronchial tumor completely occluding the left lower lobe and the lingular orifices.
Pathologic specimens from the endobronchial tumor confirmed adenocarcinoma, consistent with a primary lung cancer.
THE OTHER DIAGNOSTIC CHOICES
Detachment or separation of the retina from the underlying pigment epithelium is one of the most commonly encountered eye emergencies.1 It requires urgent attention, since delay in treatment can cause permanent vision loss.
Retinal detachment differs from uveal metastatic tumor in that it presents and progresses rapidly. The common signs and symptoms are floaters in the center of the visual axis, a sensation of flashing lights (related to retinal traction), and, eventually, loss of vision. The detachment most often represents a break or tear (rhegmatogenous retinal detachment), but it is also a common sequela of neglected diabetic retinopathy. Exudative retinal detachment is usually secondary to uveal inflammation or a uveal tumor.
Choroidal melanoma, the most common primary intraocular malignancy, arises from melanocytes within the choroid. In most cases, it develops from preexisting melanocytic nevi.2 It may present as blurred vision, a paracentral scotoma, painless and progressive visual field loss, and floaters. Choroidal melanoma is usually pigmented (dark brown) and is invariably unilateral.
Uveitis is an inflammation of the uveal tract, which includes the iris, ciliary body, and choroid. It is classified as anterior, intermediate, or posterior uveitis or as panuveitis.3
Although flashing lights, floaters, and reduced vision can occur in uveitis, its other important presenting symptoms (ie, pain, redness, and photophobia) were absent in this patient. The absence of anterior chamber cells and corneal inflammatory deposits (keratic precipitates) also made uveitis less likely.4 However, granulomatous uveitis such as sarcoidosis can present as nodular thickening of the uvea, mimicking an intraocular tumor.5
THE MOST COMMON INTRAOCULAR MALIGNANCY
Uveal metastasis is the most common intraocular malignancy6 and is found on autopsy in up to 12% of people who die of cancer; it involves both eyes in 4.4% of cases. Multiple metastases are seen in one eye in up to 20% of cases.7
The tumors are most often in the choroid, probably because of its extensive blood supply. Breast cancer (in women) and lung cancer (in men) are the most common cancers with uveal metastasis.8 Uveal metastasis from cancers of the prostate, kidney, thyroid, and gastrointestinal tract and from lymphoma and leukemia is less common.8
Patients with choroidal metastasis can see flashing lights, floating spots, and distortion of their vision. In such patients, a careful history and physical examination can uncover signs and symptoms of the hidden cancer, especially of lung cancer.9
Once uveal metastasis is suspected, both eyes and orbits and the central nervous system should be examined, as this disease tends to present bilaterally and to involve the central nervous system.10 Uveal metastases respond to chemotherapy and radiotherapy, depending on the nature of the primary tumor. In general, treatment is based on the extent of the metastasis, prior treatments, and the patient’s overall functional status.
References
Hatten B, Browne V. Retinal detachment. Emerg Med J2011; 28:83.
Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol1997; 115:1537–1544.
Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol2005; 140:509–516.
Wertheim MS, Mathers WD, Planck SJ, et al. In vivo confocal microscopy of keratic precipitates. Arch Ophthalmol2004; 122:1773–1781.
Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina2001; 21:40–47.
Eliassi-Rad B, Albert DM, Green WR. Frequency of ocular metastases in patients dying of cancer in eye bank populations. Br J Ophthalmol1996; 80:125–128.
Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology1997; 104:1265–1276.
Herrag M, Lahmiti S, Yazidi AA, Le Lez ML, Diot P. Choroidal metastasis revealing a lung adenocarcinoma. Ann Thorac Surg2010; 89:1013–1014.
Kanthan GL, Jayamohan J, Yip D, Conway RM. Management of metastatic carcinoma of the uveal tract: an evidence-based analysis. Clin Exp Ophthalmol2007; 35:553–565.
References
Hatten B, Browne V. Retinal detachment. Emerg Med J2011; 28:83.
Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol1997; 115:1537–1544.
Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol2005; 140:509–516.
Wertheim MS, Mathers WD, Planck SJ, et al. In vivo confocal microscopy of keratic precipitates. Arch Ophthalmol2004; 122:1773–1781.
Desai UR, Tawansy KA, Joondeph BC, Schiffman RM. Choroidal granulomas in systemic sarcoidosis. Retina2001; 21:40–47.
Eliassi-Rad B, Albert DM, Green WR. Frequency of ocular metastases in patients dying of cancer in eye bank populations. Br J Ophthalmol1996; 80:125–128.
Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology1997; 104:1265–1276.
Herrag M, Lahmiti S, Yazidi AA, Le Lez ML, Diot P. Choroidal metastasis revealing a lung adenocarcinoma. Ann Thorac Surg2010; 89:1013–1014.
Kanthan GL, Jayamohan J, Yip D, Conway RM. Management of metastatic carcinoma of the uveal tract: an evidence-based analysis. Clin Exp Ophthalmol2007; 35:553–565.
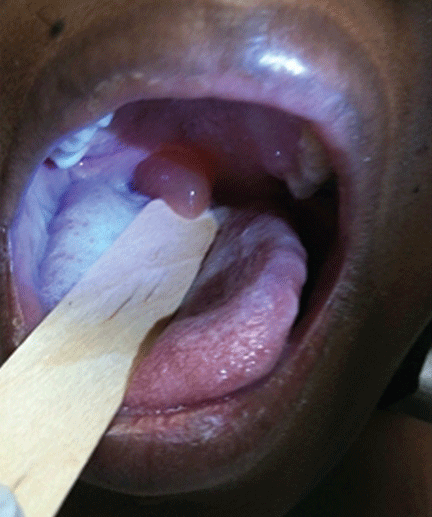
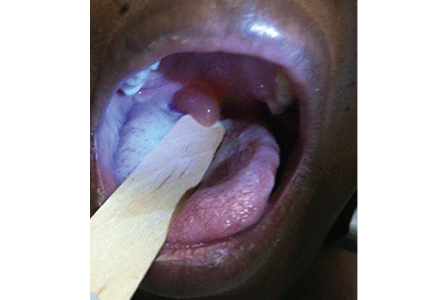
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
Hereditary angioedema
Infection causing epiglottitis masquerading as uvular swelling
Opioid-induced uvular hydrops
Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
Hereditary angioedema
Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
Saurabh Anil Pande, MD Albert Einstein Medical Center, Philadelphia, PA
Kanwal Raghav, MD The University of Texas MD Anderson Cancer Center, Houston, TX
Shivani Mehta, MD Hahnemann University Hospital, Philadelphia, PA
Gurinder Babbar, MBBS Philadelphia, PA
Saurabh Kandpal, MD Department of Hospital Medicine, Cleveland Clinic
Address: Saurabh Anil Pande, MD, Kraftsow Division Of Nephrology, Albert Einstein Medical Center, 5501 Old York Road, Philadelphia, PA 19131; e-mail [email protected]
Saurabh Anil Pande, MD Albert Einstein Medical Center, Philadelphia, PA
Kanwal Raghav, MD The University of Texas MD Anderson Cancer Center, Houston, TX
Shivani Mehta, MD Hahnemann University Hospital, Philadelphia, PA
Gurinder Babbar, MBBS Philadelphia, PA
Saurabh Kandpal, MD Department of Hospital Medicine, Cleveland Clinic
Address: Saurabh Anil Pande, MD, Kraftsow Division Of Nephrology, Albert Einstein Medical Center, 5501 Old York Road, Philadelphia, PA 19131; e-mail [email protected]
Author and Disclosure Information
Saurabh Anil Pande, MD Albert Einstein Medical Center, Philadelphia, PA
Kanwal Raghav, MD The University of Texas MD Anderson Cancer Center, Houston, TX
Shivani Mehta, MD Hahnemann University Hospital, Philadelphia, PA
Gurinder Babbar, MBBS Philadelphia, PA
Saurabh Kandpal, MD Department of Hospital Medicine, Cleveland Clinic
Address: Saurabh Anil Pande, MD, Kraftsow Division Of Nephrology, Albert Einstein Medical Center, 5501 Old York Road, Philadelphia, PA 19131; e-mail [email protected]
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
Hereditary angioedema
Infection causing epiglottitis masquerading as uvular swelling
Opioid-induced uvular hydrops
Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
Hereditary angioedema
Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
A serum thyroid-stimulating hormone level to rule out hypothyroidism
Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
Figure 1.
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
Hereditary angioedema
Infection causing epiglottitis masquerading as uvular swelling
Opioid-induced uvular hydrops
Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
Hereditary angioedema
Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
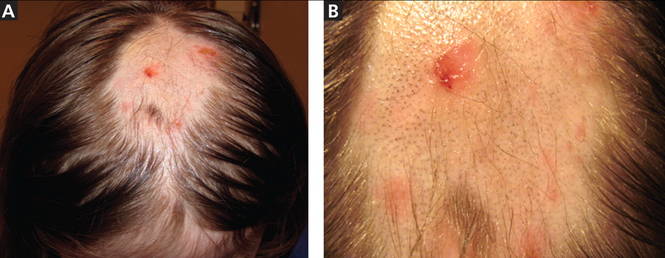
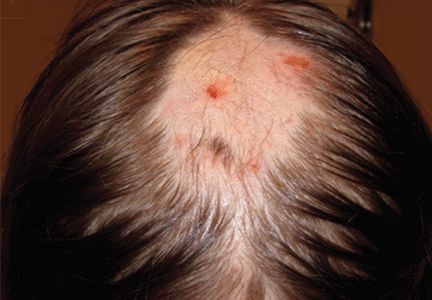
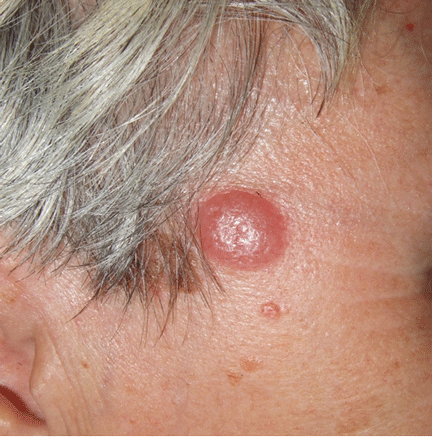
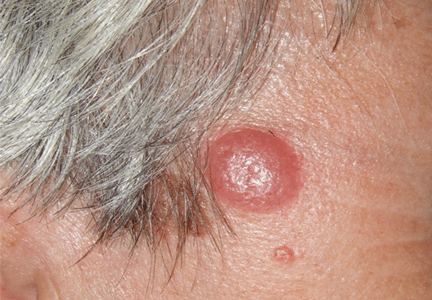
Figure 1. An irregular patch of alopecia (A) with small crusted areas. Close examination reveals broken hairs and areas of excoriation (B).
A 12-year-old girl has a large, irregular area of hair loss over the central frontoparietal scalp. Physical examination reveals scattered short hairs of varying lengths and a few small crusts throughout the area of alopecia (Figure 1). The remainder of the scalp appears normal.
Q: Which diagnosis is most likely?
Alopecia areata
Lichen planopilaris
Discoid lupus erythematosus
Trichotillomania
Follicular degeneration syndrome
A: The correct answer is trichotillomania, the compulsive pulling out of one’s own hair. Irregularly shaped areas of alopecia containing short hairs of varied lengths and excoriation should raise clinical suspicion of trichotillomania. Biopsy can confirm the diagnosis when follicles devoid of hair shafts, hemorrhage, and misshapen fragments of scalp hair (pigment casts) are seen.
DIAGNOSTIC CLUES
Trichotillomania may present as striking hair loss (alopecia) with an irregular pattern, often with sharp angles or scalloped borders.1 Short and broken hairs within involved areas are typically seen because regenerating hairs are too short to be grasped and pulled out.2 Although hair loss on the scalp may be most evident, hair loss on any hair-bearing area of the body may be noted, including eyebrows and eyelashes.
Family members and the affected individual are often aware of compulsive manipulation of hair.
Depression, anxiety, and other grooming behaviors such as skin-picking and nail-biting may be associated with trichotillomania. Affected individuals often feel a sense of gratification from pulling out hairs. Although systemic complications are rare, some individuals ingest the removed hairs (trichophagy), and the hairs may be caught in the gastric folds and eventually form a trichobezoar.3
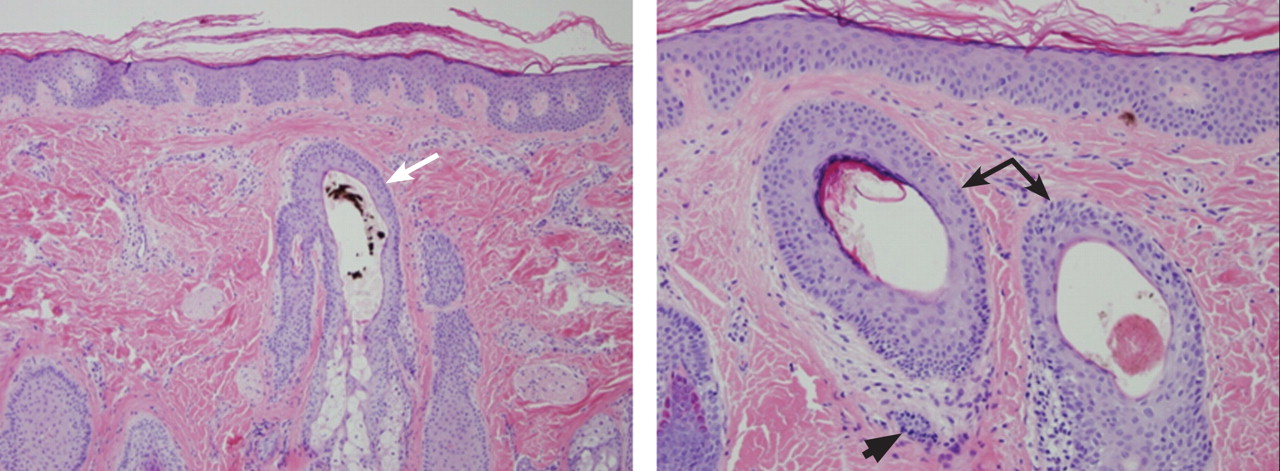
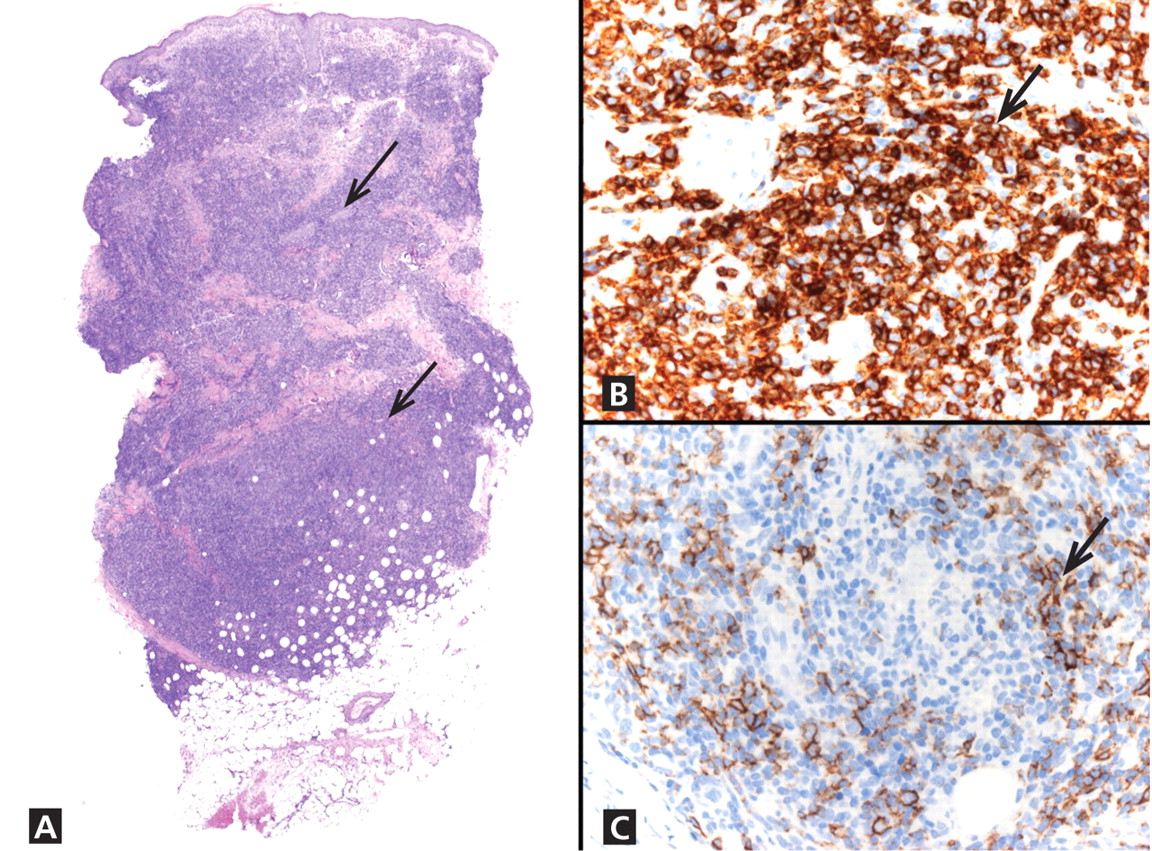
Figure 2. (Left) Biopsy reveals a hair follicle devoid of a normal hair shaft (white arrow) but instead containing pigmented hair fragments (hematoxylin and eosin, ×10). (Right) Also notable are hair follicles devoid of hair shafts (black arrows) and areas of sparse dermal inflammation (black arrowhead) (hematoxylin and eosin, ×20).
The diagnosis is usually based on clinical findings and by asking the patient about hair-pulling. Asking the patient if the habit is due to the feel of the hair, a need to calm himself or herself, or other factors may be revealing. The majority of cases can be diagnosed without biopsy. Biopsy from affected areas reveals changes related to trauma such as empty hair follicles, hemorrhage, and hair shaft fragments in the dermis2 (Figure 2). The number of catagen follicles is increased. Other causes of patchy alopecia are associated with different findings on biopsy.
Alopecia areata may be associated with an increased number of catagen hairs but is characterized by a peribulbar lymphocytic infiltrate.
Biopsy of lichen planopilaris typically reveals vacuolar changes along the dermal-follicular junction and necrotic keratinocytes.
Cutaneous lupus erythematosus is associated with thickening of the basement membrane zone, increased mucin in the dermis, follicular plugging by keratin, and vacuolar changes along the dermal-epidermal junction.
Biopsy of follicular degeneration syndrome exhibits premature desquamation of the internal root sheath as well as an increased number of fibrous tracts marking the sites of lost hairs.
The etiology of trichotillomania remains largely unknown, and the prognosis varies.4,5 There may be a family history, as there appears to be a genetic component to this disease. The disorder may also occur in the absence of external stressors.5
TREATMENT OPTIONS
Young children often develop trichotillomania that is transient in nature and most often does not require formal intervention. Older children may benefit from psychotherapy.5
Clomipramine (Anafranil) has been shown to be more effective than placebo.6 Selective serotonin reuptake inhibitors are no more effective than placebo.6,7 Pimozide (Orap), haloperidol (Haldol), and other agents have been reported to be of benefit in some instances. Although no large randomized clinical trials in children have been performed, N-acetylcysteine (Acetadote) seems to be a very promising form of therapy in adults.8 A multidisciplinary approach is usually helpful in finding the best treatment option for a particular patient.
References
Shah KN, Fried RG. Factitial dermatoses in children. Curr Opin Pediatr2006; 18:403–409.
Hautmann G, Hercogova J, Lotti T. Trichotillomania. J Am Acad Dermatol2002; 46:807–821.
Lynch KA, Feola PG, Guenther E. Gastric trichobezoar: an important cause of abdominal pain presenting to the pediatric emergency department. Pediatr Emerg Care2003; 19:343–347.
Franklin ME, Tolin DF, editors. In: Treating Trichotillomania: Cognitive-Behavioral Therapy for Hairpulling and Related Problems. New York, NY: Springer; 2007.
Duke DC, Keeley ML, Geffken GR, Storch EA. Trichotillomania: a current review. Clin Psychol Rev2010; 30:181–193.
Bloch MH, Landeros-Weisenberger A, Dombrowski P, et al. Systematic review: pharmacological and behavioral treatment for trichotillomania. Biol Psychiatry2007; 62:839–846.
Bloch MH. Trichotillomania across the life span. J Am Acad Child Adolesc Psychiatry2009; 48:879–883.
Grant JE, Odlaug BL, Kim SW. N-acetylcysteine, a glutamate modulator, in the treatment of trichotillomania: a double-blind, placebo-controlled study. Arch Gen Psychiatry2009; 66:756–763.
Figure 1. An irregular patch of alopecia (A) with small crusted areas. Close examination reveals broken hairs and areas of excoriation (B).
A 12-year-old girl has a large, irregular area of hair loss over the central frontoparietal scalp. Physical examination reveals scattered short hairs of varying lengths and a few small crusts throughout the area of alopecia (Figure 1). The remainder of the scalp appears normal.
Q: Which diagnosis is most likely?
Alopecia areata
Lichen planopilaris
Discoid lupus erythematosus
Trichotillomania
Follicular degeneration syndrome
A: The correct answer is trichotillomania, the compulsive pulling out of one’s own hair. Irregularly shaped areas of alopecia containing short hairs of varied lengths and excoriation should raise clinical suspicion of trichotillomania. Biopsy can confirm the diagnosis when follicles devoid of hair shafts, hemorrhage, and misshapen fragments of scalp hair (pigment casts) are seen.
DIAGNOSTIC CLUES
Trichotillomania may present as striking hair loss (alopecia) with an irregular pattern, often with sharp angles or scalloped borders.1 Short and broken hairs within involved areas are typically seen because regenerating hairs are too short to be grasped and pulled out.2 Although hair loss on the scalp may be most evident, hair loss on any hair-bearing area of the body may be noted, including eyebrows and eyelashes.
Family members and the affected individual are often aware of compulsive manipulation of hair.
Depression, anxiety, and other grooming behaviors such as skin-picking and nail-biting may be associated with trichotillomania. Affected individuals often feel a sense of gratification from pulling out hairs. Although systemic complications are rare, some individuals ingest the removed hairs (trichophagy), and the hairs may be caught in the gastric folds and eventually form a trichobezoar.3
Figure 2. (Left) Biopsy reveals a hair follicle devoid of a normal hair shaft (white arrow) but instead containing pigmented hair fragments (hematoxylin and eosin, ×10). (Right) Also notable are hair follicles devoid of hair shafts (black arrows) and areas of sparse dermal inflammation (black arrowhead) (hematoxylin and eosin, ×20).
The diagnosis is usually based on clinical findings and by asking the patient about hair-pulling. Asking the patient if the habit is due to the feel of the hair, a need to calm himself or herself, or other factors may be revealing. The majority of cases can be diagnosed without biopsy. Biopsy from affected areas reveals changes related to trauma such as empty hair follicles, hemorrhage, and hair shaft fragments in the dermis2 (Figure 2). The number of catagen follicles is increased. Other causes of patchy alopecia are associated with different findings on biopsy.
Alopecia areata may be associated with an increased number of catagen hairs but is characterized by a peribulbar lymphocytic infiltrate.
Biopsy of lichen planopilaris typically reveals vacuolar changes along the dermal-follicular junction and necrotic keratinocytes.
Cutaneous lupus erythematosus is associated with thickening of the basement membrane zone, increased mucin in the dermis, follicular plugging by keratin, and vacuolar changes along the dermal-epidermal junction.
Biopsy of follicular degeneration syndrome exhibits premature desquamation of the internal root sheath as well as an increased number of fibrous tracts marking the sites of lost hairs.
The etiology of trichotillomania remains largely unknown, and the prognosis varies.4,5 There may be a family history, as there appears to be a genetic component to this disease. The disorder may also occur in the absence of external stressors.5
TREATMENT OPTIONS
Young children often develop trichotillomania that is transient in nature and most often does not require formal intervention. Older children may benefit from psychotherapy.5
Clomipramine (Anafranil) has been shown to be more effective than placebo.6 Selective serotonin reuptake inhibitors are no more effective than placebo.6,7 Pimozide (Orap), haloperidol (Haldol), and other agents have been reported to be of benefit in some instances. Although no large randomized clinical trials in children have been performed, N-acetylcysteine (Acetadote) seems to be a very promising form of therapy in adults.8 A multidisciplinary approach is usually helpful in finding the best treatment option for a particular patient.
Figure 1. An irregular patch of alopecia (A) with small crusted areas. Close examination reveals broken hairs and areas of excoriation (B).
A 12-year-old girl has a large, irregular area of hair loss over the central frontoparietal scalp. Physical examination reveals scattered short hairs of varying lengths and a few small crusts throughout the area of alopecia (Figure 1). The remainder of the scalp appears normal.
Q: Which diagnosis is most likely?
Alopecia areata
Lichen planopilaris
Discoid lupus erythematosus
Trichotillomania
Follicular degeneration syndrome
A: The correct answer is trichotillomania, the compulsive pulling out of one’s own hair. Irregularly shaped areas of alopecia containing short hairs of varied lengths and excoriation should raise clinical suspicion of trichotillomania. Biopsy can confirm the diagnosis when follicles devoid of hair shafts, hemorrhage, and misshapen fragments of scalp hair (pigment casts) are seen.
DIAGNOSTIC CLUES
Trichotillomania may present as striking hair loss (alopecia) with an irregular pattern, often with sharp angles or scalloped borders.1 Short and broken hairs within involved areas are typically seen because regenerating hairs are too short to be grasped and pulled out.2 Although hair loss on the scalp may be most evident, hair loss on any hair-bearing area of the body may be noted, including eyebrows and eyelashes.
Family members and the affected individual are often aware of compulsive manipulation of hair.
Depression, anxiety, and other grooming behaviors such as skin-picking and nail-biting may be associated with trichotillomania. Affected individuals often feel a sense of gratification from pulling out hairs. Although systemic complications are rare, some individuals ingest the removed hairs (trichophagy), and the hairs may be caught in the gastric folds and eventually form a trichobezoar.3
Figure 2. (Left) Biopsy reveals a hair follicle devoid of a normal hair shaft (white arrow) but instead containing pigmented hair fragments (hematoxylin and eosin, ×10). (Right) Also notable are hair follicles devoid of hair shafts (black arrows) and areas of sparse dermal inflammation (black arrowhead) (hematoxylin and eosin, ×20).
The diagnosis is usually based on clinical findings and by asking the patient about hair-pulling. Asking the patient if the habit is due to the feel of the hair, a need to calm himself or herself, or other factors may be revealing. The majority of cases can be diagnosed without biopsy. Biopsy from affected areas reveals changes related to trauma such as empty hair follicles, hemorrhage, and hair shaft fragments in the dermis2 (Figure 2). The number of catagen follicles is increased. Other causes of patchy alopecia are associated with different findings on biopsy.
Alopecia areata may be associated with an increased number of catagen hairs but is characterized by a peribulbar lymphocytic infiltrate.
Biopsy of lichen planopilaris typically reveals vacuolar changes along the dermal-follicular junction and necrotic keratinocytes.
Cutaneous lupus erythematosus is associated with thickening of the basement membrane zone, increased mucin in the dermis, follicular plugging by keratin, and vacuolar changes along the dermal-epidermal junction.
Biopsy of follicular degeneration syndrome exhibits premature desquamation of the internal root sheath as well as an increased number of fibrous tracts marking the sites of lost hairs.
The etiology of trichotillomania remains largely unknown, and the prognosis varies.4,5 There may be a family history, as there appears to be a genetic component to this disease. The disorder may also occur in the absence of external stressors.5
TREATMENT OPTIONS
Young children often develop trichotillomania that is transient in nature and most often does not require formal intervention. Older children may benefit from psychotherapy.5
Clomipramine (Anafranil) has been shown to be more effective than placebo.6 Selective serotonin reuptake inhibitors are no more effective than placebo.6,7 Pimozide (Orap), haloperidol (Haldol), and other agents have been reported to be of benefit in some instances. Although no large randomized clinical trials in children have been performed, N-acetylcysteine (Acetadote) seems to be a very promising form of therapy in adults.8 A multidisciplinary approach is usually helpful in finding the best treatment option for a particular patient.
References
Shah KN, Fried RG. Factitial dermatoses in children. Curr Opin Pediatr2006; 18:403–409.
Hautmann G, Hercogova J, Lotti T. Trichotillomania. J Am Acad Dermatol2002; 46:807–821.
Lynch KA, Feola PG, Guenther E. Gastric trichobezoar: an important cause of abdominal pain presenting to the pediatric emergency department. Pediatr Emerg Care2003; 19:343–347.
Franklin ME, Tolin DF, editors. In: Treating Trichotillomania: Cognitive-Behavioral Therapy for Hairpulling and Related Problems. New York, NY: Springer; 2007.
Duke DC, Keeley ML, Geffken GR, Storch EA. Trichotillomania: a current review. Clin Psychol Rev2010; 30:181–193.
Bloch MH, Landeros-Weisenberger A, Dombrowski P, et al. Systematic review: pharmacological and behavioral treatment for trichotillomania. Biol Psychiatry2007; 62:839–846.
Bloch MH. Trichotillomania across the life span. J Am Acad Child Adolesc Psychiatry2009; 48:879–883.
Grant JE, Odlaug BL, Kim SW. N-acetylcysteine, a glutamate modulator, in the treatment of trichotillomania: a double-blind, placebo-controlled study. Arch Gen Psychiatry2009; 66:756–763.
References
Shah KN, Fried RG. Factitial dermatoses in children. Curr Opin Pediatr2006; 18:403–409.
Hautmann G, Hercogova J, Lotti T. Trichotillomania. J Am Acad Dermatol2002; 46:807–821.
Lynch KA, Feola PG, Guenther E. Gastric trichobezoar: an important cause of abdominal pain presenting to the pediatric emergency department. Pediatr Emerg Care2003; 19:343–347.
Franklin ME, Tolin DF, editors. In: Treating Trichotillomania: Cognitive-Behavioral Therapy for Hairpulling and Related Problems. New York, NY: Springer; 2007.
Duke DC, Keeley ML, Geffken GR, Storch EA. Trichotillomania: a current review. Clin Psychol Rev2010; 30:181–193.
Bloch MH, Landeros-Weisenberger A, Dombrowski P, et al. Systematic review: pharmacological and behavioral treatment for trichotillomania. Biol Psychiatry2007; 62:839–846.
Bloch MH. Trichotillomania across the life span. J Am Acad Child Adolesc Psychiatry2009; 48:879–883.
Grant JE, Odlaug BL, Kim SW. N-acetylcysteine, a glutamate modulator, in the treatment of trichotillomania: a double-blind, placebo-controlled study. Arch Gen Psychiatry2009; 66:756–763.
A 50-year-old farmer reports having bilateral pleuritic chest pain for the past week. He was treated 25 years ago for brucellosis, with neither clinical nor radiologic lung involvement. He is a 30-pack-year smoker. He lives in a rural area. He reports no other symptoms.
Figure 2.
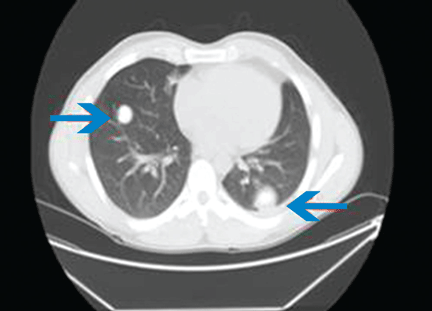
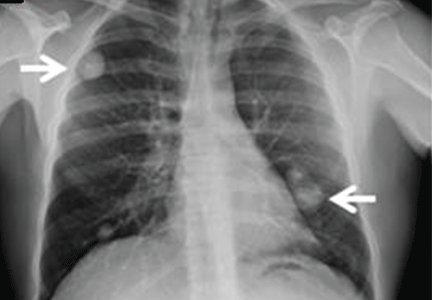
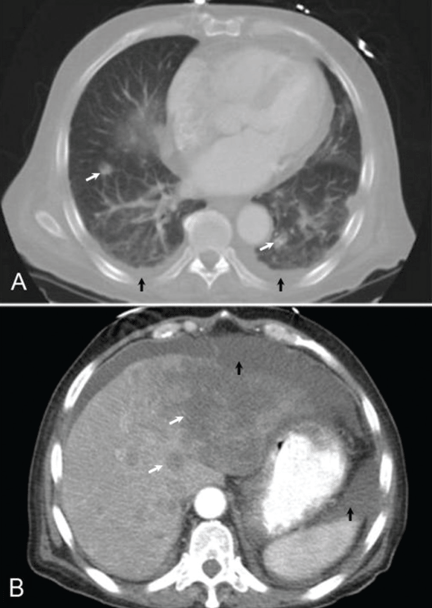
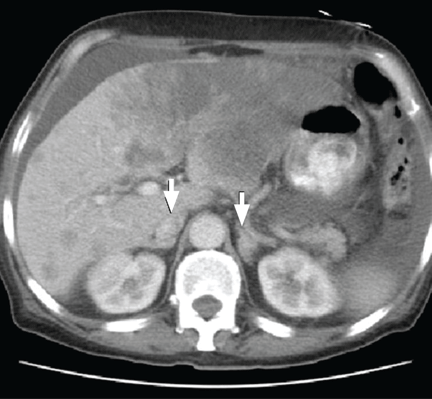
The physical examination is normal except for mild hepatomegaly. Laboratory tests (including transaminases) were normal, with the exception of the C-reactive protein level (7 mg/dL). Tumor markers, beta-2-microglobulin level, serologic tests for atypical bacteria and Brucella organisms, Mantoux test, protein electrophoresis, and tests for autoimmune antibodies were normal or negative. Echocardiography revealed no vegetations. However, chest radiography revealed multiple nodules in both lungs (Figure 1, arrows). Thoracic computed tomography showed well-defined nodules 2 to 3 cm in diameter suggestive of calcified granuloma (Figure 2, arrows).
Q: Which is the most likely diagnosis?
Pulmonary tuberculosis
Metastatic lung disease
Pulmonary brucellosis
Septic pulmonary emboli
Lymphoma
A: The most likely diagnosis is pulmonary brucellosis. The patient lives in a rural area where brucellosis is endemic, and his occupation has meant that he also has had decades of daily exposure to farm animals, mainly sheep.
Figure 3.
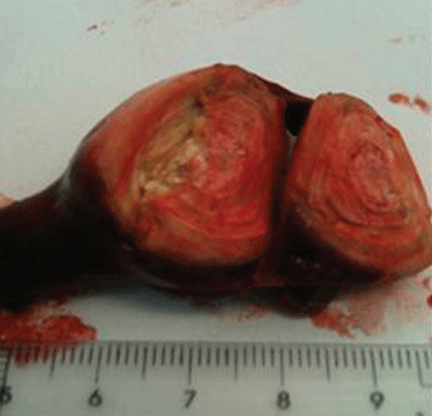
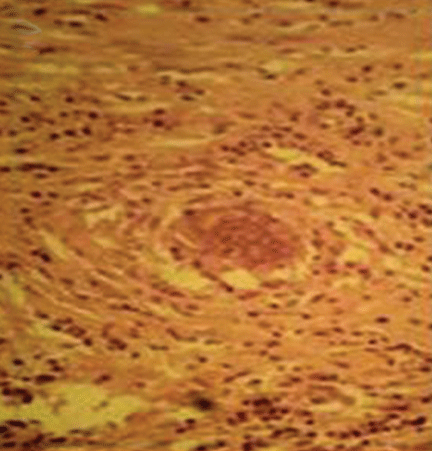
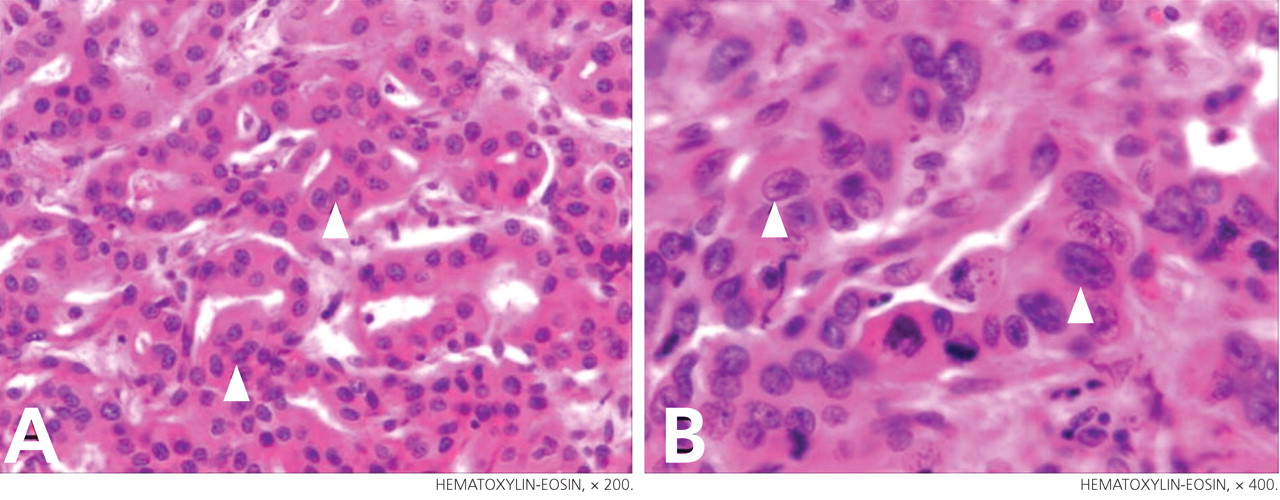
Lung biopsy specimens were obtained by minimally invasive thoracoscopy (Figure 3), and histologic study revealed noncaseating granulomas with central necrosis (Figure 4). Lastly, cultures of the resected nodule were positive for Brucella melitensis.
Figure 4.
Once the diagnosis of pulmonary brucellosis was made, the following treatment regimen was started: rifampicin 600 mg daily for 2 months, doxycycline 100 mg twice daily for 2 months, and intramuscular gentamicin 240 mg daily for 2 weeks. The chest pain gradually improved and resolved completely by 1 month after treatment was started; the lung lesions disappeared 8 weeks later. The patient remains disease-free at 6 months.
TYPICAL FEATURES OF BRUCELLOSIS
Brucellosis is a zoonotic disease transmitted to humans not only by ingestion of infected dairy products, but also by direct contact with infected animals or by inhalation of contaminated aerosols. This latter physiopathologic mechanism of acquiring the disease seems to be the most probable when the lungs are involved, 1 and it is common in people such as our patient, whose occupation exposes them to Brucella species.
Although brucellosis can initially present with mild respiratory tract symptoms, true pulmonary involvement (characterized by a more aggressive and prolonged course) is very uncommon, with a reported incidence of 1% to 7%.1,2 Respiratory involvement in brucellosis may appear as part of a systemic illness, as the presenting symptom of the disease, or even as a solitary abnormality on chest radiography.1 Bronchopneumonia, interstitial pneumonia, empyema, pleural effusion, paratracheal lymphadenopathy, and lung nodules have all been reported.2
Reinfection or a late relapse?
In our patient, a question was whether the second episode of brucellosis was a reinfection or a late relapse of the disease. Reinfection seemed the most feasible explanation, supported by his continuous occupational exposure, the properly treated first episode (rifampicin 600 mg daily and doxycycline 100 mg twice daily, both for 45 days), the long symptom-free period, and the fact that most relapses have been reported to occur during the first 6 months after therapy.3 However, late reactivation of an asymptomatic chronic lung infection was also possible, given the ability of Brucella species to survive inside the phagocytic mononuclear cells; brucellosis reactivation has been reported even 28 years after the first episode.4
DIAGNOSTIC CHALLENGES
The diagnosis of brucellosis with laboratory testing is challenging. The organism is difficult to isolate in sputum culture (only one case has been described until now),5 and serologic tests can be falsely negative, although this is rare.6,7 In fact, serologic testing in patients with focal brucellosis may be falsely negative when the serum agglutination test is performed,4,7 as could have occurred in our patient. In several studies, pleural fluid culture has been shown as a good method to isolate Brucella organisms,8 but biopsy is often the only way to establish the diagnosis.6
Complications of lung involvement in brucellosis are seldom severe and, when they appear, usually respond to the same treatment as for uncomplicated brucellosis.2
The combination of respiratory symptoms, epidemiologic risk factors, an endemic setting, and a history of a previous episode all raise clinical suspicion of brucellosis. If clinical suspicion is high, negative results of sputum, serology, or pleural fluid cultures should never rule out the disease; biopsy of the respiratory region affected is warranted.
References
Hatipoglu CA, Bilgin G, Tulek N, Kosar U. Pulmonary involvement in brucellosis. J Infect2005; 51:116–119.
Pappas G, Bosilkovski M, Akritidis N, Mastora M, Krteva L, Tsianos E. Brucellosis and the respiratory system. Clin Infect Dis2003; 37:e95–e99.
Ariza J, Corredoira J, Pallares R, et al. Characteristics of and risk factors for relapse of brucellosis in humans. Clin Infect Dis1995; 20:1241–1249.
Ögredici Ö, Erb S, Langer I, et al. Brucellosis reactivation after 28 years. Emerg Infect Dis2010; 16:2021–2022.
Gattas N, Loberant N, Rimon D. Miliary and reticulo-nodular pulmonary brucellosis. [in Hebrew]. Harefuah1998; 135:357–359,407.
Theegarten D, Albrecht S, Tötsch M, Teschler H, Neubauer H, Al Dahouk S. Brucellosis of the lung: case report and review of the literature. Virchows Arch2008; 452:97–101.
Celik AD, Yulugkural Z, Kilincer C, Hamamcioglu MK, Kuloglu F, Akata F. Negative serology: could exclude the diagnosis of brucellosis?Rheumatol Int2010; Epub ahead of print.
Kerem E, Diav O, Navon P, Branski D. Pleural fluid characteristics in pulmonary brucellosis. Thorax1994; 49:89–90.
Fernando Jaén Águila, MD, PhD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
José Antonio Vargas-Hitos, MD, PhD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
David Esteva Fernández, MD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
Juan Jiménez Alonso, MD, PhD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
Address: José Antonio Vargas-Hitos, MD, PhD, Department of Internal Medicine, Virgen de las Nieves University Hospital, 9th Floor, Av. Fuerzas Armadas Nº 2, 18014 Granada, Spain; e-mail [email protected]
Fernando Jaén Águila, MD, PhD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
José Antonio Vargas-Hitos, MD, PhD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
David Esteva Fernández, MD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
Juan Jiménez Alonso, MD, PhD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
Address: José Antonio Vargas-Hitos, MD, PhD, Department of Internal Medicine, Virgen de las Nieves University Hospital, 9th Floor, Av. Fuerzas Armadas Nº 2, 18014 Granada, Spain; e-mail [email protected]
Author and Disclosure Information
Fernando Jaén Águila, MD, PhD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
José Antonio Vargas-Hitos, MD, PhD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
David Esteva Fernández, MD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
Juan Jiménez Alonso, MD, PhD Department of Internal Medicine, Virgen de las Nieves University Hospital, Granada, Spain
Address: José Antonio Vargas-Hitos, MD, PhD, Department of Internal Medicine, Virgen de las Nieves University Hospital, 9th Floor, Av. Fuerzas Armadas Nº 2, 18014 Granada, Spain; e-mail [email protected]
A 50-year-old farmer reports having bilateral pleuritic chest pain for the past week. He was treated 25 years ago for brucellosis, with neither clinical nor radiologic lung involvement. He is a 30-pack-year smoker. He lives in a rural area. He reports no other symptoms.
Figure 2.
The physical examination is normal except for mild hepatomegaly. Laboratory tests (including transaminases) were normal, with the exception of the C-reactive protein level (7 mg/dL). Tumor markers, beta-2-microglobulin level, serologic tests for atypical bacteria and Brucella organisms, Mantoux test, protein electrophoresis, and tests for autoimmune antibodies were normal or negative. Echocardiography revealed no vegetations. However, chest radiography revealed multiple nodules in both lungs (Figure 1, arrows). Thoracic computed tomography showed well-defined nodules 2 to 3 cm in diameter suggestive of calcified granuloma (Figure 2, arrows).
Q: Which is the most likely diagnosis?
Pulmonary tuberculosis
Metastatic lung disease
Pulmonary brucellosis
Septic pulmonary emboli
Lymphoma
A: The most likely diagnosis is pulmonary brucellosis. The patient lives in a rural area where brucellosis is endemic, and his occupation has meant that he also has had decades of daily exposure to farm animals, mainly sheep.
Figure 3.
Lung biopsy specimens were obtained by minimally invasive thoracoscopy (Figure 3), and histologic study revealed noncaseating granulomas with central necrosis (Figure 4). Lastly, cultures of the resected nodule were positive for Brucella melitensis.
Figure 4.
Once the diagnosis of pulmonary brucellosis was made, the following treatment regimen was started: rifampicin 600 mg daily for 2 months, doxycycline 100 mg twice daily for 2 months, and intramuscular gentamicin 240 mg daily for 2 weeks. The chest pain gradually improved and resolved completely by 1 month after treatment was started; the lung lesions disappeared 8 weeks later. The patient remains disease-free at 6 months.
TYPICAL FEATURES OF BRUCELLOSIS
Brucellosis is a zoonotic disease transmitted to humans not only by ingestion of infected dairy products, but also by direct contact with infected animals or by inhalation of contaminated aerosols. This latter physiopathologic mechanism of acquiring the disease seems to be the most probable when the lungs are involved, 1 and it is common in people such as our patient, whose occupation exposes them to Brucella species.
Although brucellosis can initially present with mild respiratory tract symptoms, true pulmonary involvement (characterized by a more aggressive and prolonged course) is very uncommon, with a reported incidence of 1% to 7%.1,2 Respiratory involvement in brucellosis may appear as part of a systemic illness, as the presenting symptom of the disease, or even as a solitary abnormality on chest radiography.1 Bronchopneumonia, interstitial pneumonia, empyema, pleural effusion, paratracheal lymphadenopathy, and lung nodules have all been reported.2
Reinfection or a late relapse?
In our patient, a question was whether the second episode of brucellosis was a reinfection or a late relapse of the disease. Reinfection seemed the most feasible explanation, supported by his continuous occupational exposure, the properly treated first episode (rifampicin 600 mg daily and doxycycline 100 mg twice daily, both for 45 days), the long symptom-free period, and the fact that most relapses have been reported to occur during the first 6 months after therapy.3 However, late reactivation of an asymptomatic chronic lung infection was also possible, given the ability of Brucella species to survive inside the phagocytic mononuclear cells; brucellosis reactivation has been reported even 28 years after the first episode.4
DIAGNOSTIC CHALLENGES
The diagnosis of brucellosis with laboratory testing is challenging. The organism is difficult to isolate in sputum culture (only one case has been described until now),5 and serologic tests can be falsely negative, although this is rare.6,7 In fact, serologic testing in patients with focal brucellosis may be falsely negative when the serum agglutination test is performed,4,7 as could have occurred in our patient. In several studies, pleural fluid culture has been shown as a good method to isolate Brucella organisms,8 but biopsy is often the only way to establish the diagnosis.6
Complications of lung involvement in brucellosis are seldom severe and, when they appear, usually respond to the same treatment as for uncomplicated brucellosis.2
The combination of respiratory symptoms, epidemiologic risk factors, an endemic setting, and a history of a previous episode all raise clinical suspicion of brucellosis. If clinical suspicion is high, negative results of sputum, serology, or pleural fluid cultures should never rule out the disease; biopsy of the respiratory region affected is warranted.
Figure 1.
A 50-year-old farmer reports having bilateral pleuritic chest pain for the past week. He was treated 25 years ago for brucellosis, with neither clinical nor radiologic lung involvement. He is a 30-pack-year smoker. He lives in a rural area. He reports no other symptoms.
Figure 2.
The physical examination is normal except for mild hepatomegaly. Laboratory tests (including transaminases) were normal, with the exception of the C-reactive protein level (7 mg/dL). Tumor markers, beta-2-microglobulin level, serologic tests for atypical bacteria and Brucella organisms, Mantoux test, protein electrophoresis, and tests for autoimmune antibodies were normal or negative. Echocardiography revealed no vegetations. However, chest radiography revealed multiple nodules in both lungs (Figure 1, arrows). Thoracic computed tomography showed well-defined nodules 2 to 3 cm in diameter suggestive of calcified granuloma (Figure 2, arrows).
Q: Which is the most likely diagnosis?
Pulmonary tuberculosis
Metastatic lung disease
Pulmonary brucellosis
Septic pulmonary emboli
Lymphoma
A: The most likely diagnosis is pulmonary brucellosis. The patient lives in a rural area where brucellosis is endemic, and his occupation has meant that he also has had decades of daily exposure to farm animals, mainly sheep.
Figure 3.
Lung biopsy specimens were obtained by minimally invasive thoracoscopy (Figure 3), and histologic study revealed noncaseating granulomas with central necrosis (Figure 4). Lastly, cultures of the resected nodule were positive for Brucella melitensis.
Figure 4.
Once the diagnosis of pulmonary brucellosis was made, the following treatment regimen was started: rifampicin 600 mg daily for 2 months, doxycycline 100 mg twice daily for 2 months, and intramuscular gentamicin 240 mg daily for 2 weeks. The chest pain gradually improved and resolved completely by 1 month after treatment was started; the lung lesions disappeared 8 weeks later. The patient remains disease-free at 6 months.
TYPICAL FEATURES OF BRUCELLOSIS
Brucellosis is a zoonotic disease transmitted to humans not only by ingestion of infected dairy products, but also by direct contact with infected animals or by inhalation of contaminated aerosols. This latter physiopathologic mechanism of acquiring the disease seems to be the most probable when the lungs are involved, 1 and it is common in people such as our patient, whose occupation exposes them to Brucella species.
Although brucellosis can initially present with mild respiratory tract symptoms, true pulmonary involvement (characterized by a more aggressive and prolonged course) is very uncommon, with a reported incidence of 1% to 7%.1,2 Respiratory involvement in brucellosis may appear as part of a systemic illness, as the presenting symptom of the disease, or even as a solitary abnormality on chest radiography.1 Bronchopneumonia, interstitial pneumonia, empyema, pleural effusion, paratracheal lymphadenopathy, and lung nodules have all been reported.2
Reinfection or a late relapse?
In our patient, a question was whether the second episode of brucellosis was a reinfection or a late relapse of the disease. Reinfection seemed the most feasible explanation, supported by his continuous occupational exposure, the properly treated first episode (rifampicin 600 mg daily and doxycycline 100 mg twice daily, both for 45 days), the long symptom-free period, and the fact that most relapses have been reported to occur during the first 6 months after therapy.3 However, late reactivation of an asymptomatic chronic lung infection was also possible, given the ability of Brucella species to survive inside the phagocytic mononuclear cells; brucellosis reactivation has been reported even 28 years after the first episode.4
DIAGNOSTIC CHALLENGES
The diagnosis of brucellosis with laboratory testing is challenging. The organism is difficult to isolate in sputum culture (only one case has been described until now),5 and serologic tests can be falsely negative, although this is rare.6,7 In fact, serologic testing in patients with focal brucellosis may be falsely negative when the serum agglutination test is performed,4,7 as could have occurred in our patient. In several studies, pleural fluid culture has been shown as a good method to isolate Brucella organisms,8 but biopsy is often the only way to establish the diagnosis.6
Complications of lung involvement in brucellosis are seldom severe and, when they appear, usually respond to the same treatment as for uncomplicated brucellosis.2
The combination of respiratory symptoms, epidemiologic risk factors, an endemic setting, and a history of a previous episode all raise clinical suspicion of brucellosis. If clinical suspicion is high, negative results of sputum, serology, or pleural fluid cultures should never rule out the disease; biopsy of the respiratory region affected is warranted.
References
Hatipoglu CA, Bilgin G, Tulek N, Kosar U. Pulmonary involvement in brucellosis. J Infect2005; 51:116–119.
Pappas G, Bosilkovski M, Akritidis N, Mastora M, Krteva L, Tsianos E. Brucellosis and the respiratory system. Clin Infect Dis2003; 37:e95–e99.
Ariza J, Corredoira J, Pallares R, et al. Characteristics of and risk factors for relapse of brucellosis in humans. Clin Infect Dis1995; 20:1241–1249.
Ögredici Ö, Erb S, Langer I, et al. Brucellosis reactivation after 28 years. Emerg Infect Dis2010; 16:2021–2022.
Gattas N, Loberant N, Rimon D. Miliary and reticulo-nodular pulmonary brucellosis. [in Hebrew]. Harefuah1998; 135:357–359,407.
Theegarten D, Albrecht S, Tötsch M, Teschler H, Neubauer H, Al Dahouk S. Brucellosis of the lung: case report and review of the literature. Virchows Arch2008; 452:97–101.
Celik AD, Yulugkural Z, Kilincer C, Hamamcioglu MK, Kuloglu F, Akata F. Negative serology: could exclude the diagnosis of brucellosis?Rheumatol Int2010; Epub ahead of print.
Kerem E, Diav O, Navon P, Branski D. Pleural fluid characteristics in pulmonary brucellosis. Thorax1994; 49:89–90.
References
Hatipoglu CA, Bilgin G, Tulek N, Kosar U. Pulmonary involvement in brucellosis. J Infect2005; 51:116–119.
Pappas G, Bosilkovski M, Akritidis N, Mastora M, Krteva L, Tsianos E. Brucellosis and the respiratory system. Clin Infect Dis2003; 37:e95–e99.
Ariza J, Corredoira J, Pallares R, et al. Characteristics of and risk factors for relapse of brucellosis in humans. Clin Infect Dis1995; 20:1241–1249.
Ögredici Ö, Erb S, Langer I, et al. Brucellosis reactivation after 28 years. Emerg Infect Dis2010; 16:2021–2022.
Gattas N, Loberant N, Rimon D. Miliary and reticulo-nodular pulmonary brucellosis. [in Hebrew]. Harefuah1998; 135:357–359,407.
Theegarten D, Albrecht S, Tötsch M, Teschler H, Neubauer H, Al Dahouk S. Brucellosis of the lung: case report and review of the literature. Virchows Arch2008; 452:97–101.
Celik AD, Yulugkural Z, Kilincer C, Hamamcioglu MK, Kuloglu F, Akata F. Negative serology: could exclude the diagnosis of brucellosis?Rheumatol Int2010; Epub ahead of print.
Kerem E, Diav O, Navon P, Branski D. Pleural fluid characteristics in pulmonary brucellosis. Thorax1994; 49:89–90.
A 64-year-old man with diabetes and hypertension presented with a 2-day history of sudden onset of acute pain and cyanosis on the sole of his right foot 4 days after undergoing cardiac catheterization and coronary angiography.
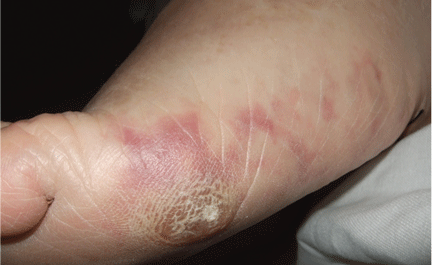
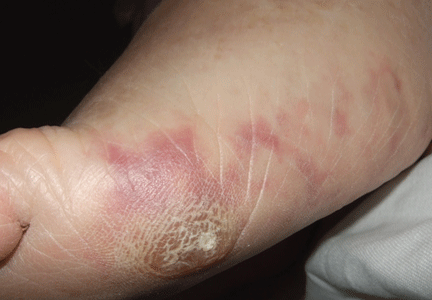
Figure1. Macular violaceous connecting rings in a net-like pattern compatible with livedo reticularis on the foot.
The physical examination revealed macular, violaceous, connecting rings in a net-like pattern that blanched with pressure and disappeared when the foot was elevated, a presentation compatible with livedo reticularis (Figure 1). Laboratory testing (complete blood cell count, biochemistry panel, coagulation test, and C-reactive protein test) was notable only for eosinophilia.
A few days later, the patient returned with abdominal pain, diarrhea, and acute renal injury with urinary eosinophils (7% of the white blood cells in the urine) and proteinuria.
Q: Which is the most likely diagnosis?
Infective endocarditis
Pernio (chilblain)
Cholesterol crystal embolism
Cutaneous small-vessel vasculitis
A: Cholesterol crystal embolism is the correct diagnosis.
Infective endocarditis is an infection of the endocardium, but systemic signs may be present, including cutaneous lesions such as Osler nodes (painful papules on the tips of the fingers and toes) and Janeway lesions (painless macules on the palms and soles). Histologic staining of skin biopsy specimens often shows vasculitis, occasionally with a positive Gram stain. Severe renal injury is not common, and the timing of the acute illness and skin lesion fits better with an acute embolic phenomenon.
Pernio is a form of cold injury, localized in peripheral parts of the body and occurring after exposure to cold temperatures in damp conditions. It usually manifests bilaterally as painful erythematous or purple lesions on the acral areas of the hands and feet, nose, ears, and, rarely, the thighs and buttocks. Pernio most commonly affects women between 20 and 40 years of age. It can be idiopathic or associated with a systemic disease such as systemic lupus erythematosus or Sjögren syndrome.
Cutaneous small-vessel vasculitis is a heterogeneous group of disorders with inflammation and damage of the blood vessels; it may be limited to the skin or it may involve multiple systems. Palpable or nonpalpable purpura and ulceration are common clinical findings, and histologic study shows an inflammatory infiltrate of vessel walls, fibrinoid necrosis, thrombosis, and extravasation of red blood cells.
While this patient’s problems are not consistent with small-vessel vasculitis, the single skin lesion, the timing after the catheterization, and the urinary eosinophils are best explained by cholesterol embolization.
CHOLESTEROL CRYSTAL EMBOLISM
Cholesterol crystal embolism is commonly iatrogenic, a complication of mechanical damage to the arterial walls from vascular surgery or invasive percutaneous procedures. Material dislodged from atheromatous arterial plaques can occlude the small vessels of the feet, producing this syndrome.
The onset of the clinical disease is often delayed for days to weeks after an angiographic procedure.1 Spontaneous hemorrhage, disruption of plaque, or drug therapy with an anticoagulant or a fibrinolytic can precipitate embolization of cholesterol crystals. The source of the emboli is atheromatous plaque in major blood vessels, particularly the abdominal aorta.
Many organs and systems can be affected
These emboli can affect many organs and systems: eg, the kidneys (causing hypertension and acute renal failure), the muscles (causing myalgias), gastrointestinal organs (causing bleeding, abdominal pain, and bowel infarction), the lungs (causing acute respiratory distress syndrome), the eyes (causing Hollenhorst plaques in retinal arteries), and the central nervous system (causing stroke, confusion, and delirium). Cardiac or central nervous system involvement is associated with a high risk of death.
After angiography, clinical manifestations of cholesterol embolization have been reported in 0.06% to 1.4% of patients,2,3 although the finding of cholesterol emboli is more common in autopsy studies.4
Recognizing skin signs is the key
Cutaneous abnormalities are usually the earliest and often the only clinical manifestation of this syndrome. Findings on the lower limbs include blue toes, cutaneous nodules, and livedo reticularis, affecting the feet and legs and sometimes extending upward to the trunk. Other findings include digital infarcts, ulceration, gangrene, purpura, cyanosis, and splinter hemorrhages in the nail bed.
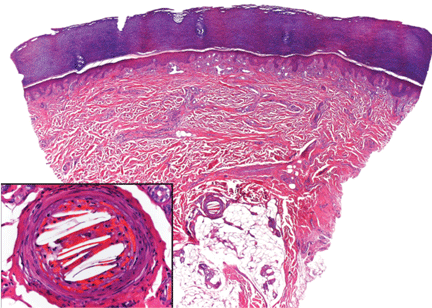
Figure 2. Skin biopsy showed needle-shaped clefts within the lumen of blood vessels, ie, dissolved cholesterol crystals obstructing small arteries.
In our patient, microscopic study of skin biopsy specimens showed needle-shaped clefts within the lumen of blood vessels—ie, dissolved cholesterol crystals obstructing small arteries (Figure 2).
Biopsy studies of skin lesions are positive in a high percentage of cases, showing dissolved cholesterol crystals within the lumen of blood vessels, especially in the small to large arteries and arterioles of the deep dermis or subcutaneous fat. Deep biopsies and carefully examination are necessary, as emboli tend to be patchily distributed. Early recognition of cutaneous clinical findings is essential to establish the proper diagnosis and treatment.
The diagnostic triad of this disease includes blue toe syndrome, acute or subacute renal failure, and a temporal relation with an inciting event (particularly angiography). But despite these diagnostic criteria,2 the diagnosis is often based on a combination of signs and symptoms specific to end-organ damage and a systemic inflammatory response.3
Histologic confirmation is considered essential to the diagnosis of cholesterol crystal embolism, and as the skin is the most accessible site, skin biopsy provides the best sample for histologic diagnosis.5
Postprocedural embolism of a blood clot, vasculitis, and infective endocarditis are the most important differential diagnoses.6,7
Treatment is supportive, preventive
Treatment is mainly supportive with hemodynamic monitoring, nutritional and metabolic support, mechanical ventilation, and dialysis if necessary. The underlying atherosclerotic disease should be treated aggressively. Prevention of another episode involves modification of traditional risk factors such as serum cholesterol, diabetes, hypertension, and smoking. Additional vascular surgery procedures should be avoided, as they can induce new episodes.
References
Donohue KG, Saap L, Falanga V. Cholesterol crystal embolization: an atherosclerotic disease with frequent and varied cutaneous manifestations. J Eur Acad Dermatol Venereol2003; 17:504–511.
Fukumoto Y, Tsutsui H, Tsuchihashi M, Masumoto A, Takeshita A; Cholesterol Embolism Study (CHEST) Investigators. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol2003; 42:211–216.
Johnson LW, Esente P, Giambartolomei A, et al. Peripheral vascular complications of coronary angioplasty by the femoral and brachial techniques. Cathet Cardiovasc Diagn1994; 31:165–172.
Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation2010; 122:631–641.
Jucgla A, Moreso F, Muniesa C, Moreno A, Vidaller A. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol2006; 55:786–793.
Maki T, Izumi C, Miyake M, et al. Cholesterol embolism after cardiac catheterization mimicking infective endocarditis. Intern Med2005; 44:1060–1063.
Salvador Arias-Santiago, MD, PhD Department of Dermatology, San Cecilio University Hospital, Department of Dermatology, Baza General Hospital, and Department of Histology, School of Medicine, Granada University, Granada, Spain
Jose Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Victor Carriel, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Jacinto Orgaz-Molina, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Miguel Gonález-Andrades, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Agustín Buendía-Eisman, MD, PhD Department of Dermatology, School of Medicine, Granada University, Granada, Spain
Miguel Alaminos, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Oloriz 16, Granada 18012, Spain; e-mail [email protected]
Salvador Arias-Santiago, MD, PhD Department of Dermatology, San Cecilio University Hospital, Department of Dermatology, Baza General Hospital, and Department of Histology, School of Medicine, Granada University, Granada, Spain
Jose Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Victor Carriel, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Jacinto Orgaz-Molina, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Miguel Gonález-Andrades, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Agustín Buendía-Eisman, MD, PhD Department of Dermatology, School of Medicine, Granada University, Granada, Spain
Miguel Alaminos, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Oloriz 16, Granada 18012, Spain; e-mail [email protected]
Author and Disclosure Information
Salvador Arias-Santiago, MD, PhD Department of Dermatology, San Cecilio University Hospital, Department of Dermatology, Baza General Hospital, and Department of Histology, School of Medicine, Granada University, Granada, Spain
Jose Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Victor Carriel, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Jacinto Orgaz-Molina, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Miguel Gonález-Andrades, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Agustín Buendía-Eisman, MD, PhD Department of Dermatology, School of Medicine, Granada University, Granada, Spain
Miguel Alaminos, MD, PhD Department of Histology, School of Medicine, Granada University, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Oloriz 16, Granada 18012, Spain; e-mail [email protected]
A 64-year-old man with diabetes and hypertension presented with a 2-day history of sudden onset of acute pain and cyanosis on the sole of his right foot 4 days after undergoing cardiac catheterization and coronary angiography.
Figure1. Macular violaceous connecting rings in a net-like pattern compatible with livedo reticularis on the foot.
The physical examination revealed macular, violaceous, connecting rings in a net-like pattern that blanched with pressure and disappeared when the foot was elevated, a presentation compatible with livedo reticularis (Figure 1). Laboratory testing (complete blood cell count, biochemistry panel, coagulation test, and C-reactive protein test) was notable only for eosinophilia.
A few days later, the patient returned with abdominal pain, diarrhea, and acute renal injury with urinary eosinophils (7% of the white blood cells in the urine) and proteinuria.
Q: Which is the most likely diagnosis?
Infective endocarditis
Pernio (chilblain)
Cholesterol crystal embolism
Cutaneous small-vessel vasculitis
A: Cholesterol crystal embolism is the correct diagnosis.
Infective endocarditis is an infection of the endocardium, but systemic signs may be present, including cutaneous lesions such as Osler nodes (painful papules on the tips of the fingers and toes) and Janeway lesions (painless macules on the palms and soles). Histologic staining of skin biopsy specimens often shows vasculitis, occasionally with a positive Gram stain. Severe renal injury is not common, and the timing of the acute illness and skin lesion fits better with an acute embolic phenomenon.
Pernio is a form of cold injury, localized in peripheral parts of the body and occurring after exposure to cold temperatures in damp conditions. It usually manifests bilaterally as painful erythematous or purple lesions on the acral areas of the hands and feet, nose, ears, and, rarely, the thighs and buttocks. Pernio most commonly affects women between 20 and 40 years of age. It can be idiopathic or associated with a systemic disease such as systemic lupus erythematosus or Sjögren syndrome.
Cutaneous small-vessel vasculitis is a heterogeneous group of disorders with inflammation and damage of the blood vessels; it may be limited to the skin or it may involve multiple systems. Palpable or nonpalpable purpura and ulceration are common clinical findings, and histologic study shows an inflammatory infiltrate of vessel walls, fibrinoid necrosis, thrombosis, and extravasation of red blood cells.
While this patient’s problems are not consistent with small-vessel vasculitis, the single skin lesion, the timing after the catheterization, and the urinary eosinophils are best explained by cholesterol embolization.
CHOLESTEROL CRYSTAL EMBOLISM
Cholesterol crystal embolism is commonly iatrogenic, a complication of mechanical damage to the arterial walls from vascular surgery or invasive percutaneous procedures. Material dislodged from atheromatous arterial plaques can occlude the small vessels of the feet, producing this syndrome.
The onset of the clinical disease is often delayed for days to weeks after an angiographic procedure.1 Spontaneous hemorrhage, disruption of plaque, or drug therapy with an anticoagulant or a fibrinolytic can precipitate embolization of cholesterol crystals. The source of the emboli is atheromatous plaque in major blood vessels, particularly the abdominal aorta.
Many organs and systems can be affected
These emboli can affect many organs and systems: eg, the kidneys (causing hypertension and acute renal failure), the muscles (causing myalgias), gastrointestinal organs (causing bleeding, abdominal pain, and bowel infarction), the lungs (causing acute respiratory distress syndrome), the eyes (causing Hollenhorst plaques in retinal arteries), and the central nervous system (causing stroke, confusion, and delirium). Cardiac or central nervous system involvement is associated with a high risk of death.
After angiography, clinical manifestations of cholesterol embolization have been reported in 0.06% to 1.4% of patients,2,3 although the finding of cholesterol emboli is more common in autopsy studies.4
Recognizing skin signs is the key
Cutaneous abnormalities are usually the earliest and often the only clinical manifestation of this syndrome. Findings on the lower limbs include blue toes, cutaneous nodules, and livedo reticularis, affecting the feet and legs and sometimes extending upward to the trunk. Other findings include digital infarcts, ulceration, gangrene, purpura, cyanosis, and splinter hemorrhages in the nail bed.
Figure 2. Skin biopsy showed needle-shaped clefts within the lumen of blood vessels, ie, dissolved cholesterol crystals obstructing small arteries.
In our patient, microscopic study of skin biopsy specimens showed needle-shaped clefts within the lumen of blood vessels—ie, dissolved cholesterol crystals obstructing small arteries (Figure 2).
Biopsy studies of skin lesions are positive in a high percentage of cases, showing dissolved cholesterol crystals within the lumen of blood vessels, especially in the small to large arteries and arterioles of the deep dermis or subcutaneous fat. Deep biopsies and carefully examination are necessary, as emboli tend to be patchily distributed. Early recognition of cutaneous clinical findings is essential to establish the proper diagnosis and treatment.
The diagnostic triad of this disease includes blue toe syndrome, acute or subacute renal failure, and a temporal relation with an inciting event (particularly angiography). But despite these diagnostic criteria,2 the diagnosis is often based on a combination of signs and symptoms specific to end-organ damage and a systemic inflammatory response.3
Histologic confirmation is considered essential to the diagnosis of cholesterol crystal embolism, and as the skin is the most accessible site, skin biopsy provides the best sample for histologic diagnosis.5
Postprocedural embolism of a blood clot, vasculitis, and infective endocarditis are the most important differential diagnoses.6,7
Treatment is supportive, preventive
Treatment is mainly supportive with hemodynamic monitoring, nutritional and metabolic support, mechanical ventilation, and dialysis if necessary. The underlying atherosclerotic disease should be treated aggressively. Prevention of another episode involves modification of traditional risk factors such as serum cholesterol, diabetes, hypertension, and smoking. Additional vascular surgery procedures should be avoided, as they can induce new episodes.
A 64-year-old man with diabetes and hypertension presented with a 2-day history of sudden onset of acute pain and cyanosis on the sole of his right foot 4 days after undergoing cardiac catheterization and coronary angiography.
Figure1. Macular violaceous connecting rings in a net-like pattern compatible with livedo reticularis on the foot.
The physical examination revealed macular, violaceous, connecting rings in a net-like pattern that blanched with pressure and disappeared when the foot was elevated, a presentation compatible with livedo reticularis (Figure 1). Laboratory testing (complete blood cell count, biochemistry panel, coagulation test, and C-reactive protein test) was notable only for eosinophilia.
A few days later, the patient returned with abdominal pain, diarrhea, and acute renal injury with urinary eosinophils (7% of the white blood cells in the urine) and proteinuria.
Q: Which is the most likely diagnosis?
Infective endocarditis
Pernio (chilblain)
Cholesterol crystal embolism
Cutaneous small-vessel vasculitis
A: Cholesterol crystal embolism is the correct diagnosis.
Infective endocarditis is an infection of the endocardium, but systemic signs may be present, including cutaneous lesions such as Osler nodes (painful papules on the tips of the fingers and toes) and Janeway lesions (painless macules on the palms and soles). Histologic staining of skin biopsy specimens often shows vasculitis, occasionally with a positive Gram stain. Severe renal injury is not common, and the timing of the acute illness and skin lesion fits better with an acute embolic phenomenon.
Pernio is a form of cold injury, localized in peripheral parts of the body and occurring after exposure to cold temperatures in damp conditions. It usually manifests bilaterally as painful erythematous or purple lesions on the acral areas of the hands and feet, nose, ears, and, rarely, the thighs and buttocks. Pernio most commonly affects women between 20 and 40 years of age. It can be idiopathic or associated with a systemic disease such as systemic lupus erythematosus or Sjögren syndrome.
Cutaneous small-vessel vasculitis is a heterogeneous group of disorders with inflammation and damage of the blood vessels; it may be limited to the skin or it may involve multiple systems. Palpable or nonpalpable purpura and ulceration are common clinical findings, and histologic study shows an inflammatory infiltrate of vessel walls, fibrinoid necrosis, thrombosis, and extravasation of red blood cells.
While this patient’s problems are not consistent with small-vessel vasculitis, the single skin lesion, the timing after the catheterization, and the urinary eosinophils are best explained by cholesterol embolization.
CHOLESTEROL CRYSTAL EMBOLISM
Cholesterol crystal embolism is commonly iatrogenic, a complication of mechanical damage to the arterial walls from vascular surgery or invasive percutaneous procedures. Material dislodged from atheromatous arterial plaques can occlude the small vessels of the feet, producing this syndrome.
The onset of the clinical disease is often delayed for days to weeks after an angiographic procedure.1 Spontaneous hemorrhage, disruption of plaque, or drug therapy with an anticoagulant or a fibrinolytic can precipitate embolization of cholesterol crystals. The source of the emboli is atheromatous plaque in major blood vessels, particularly the abdominal aorta.
Many organs and systems can be affected
These emboli can affect many organs and systems: eg, the kidneys (causing hypertension and acute renal failure), the muscles (causing myalgias), gastrointestinal organs (causing bleeding, abdominal pain, and bowel infarction), the lungs (causing acute respiratory distress syndrome), the eyes (causing Hollenhorst plaques in retinal arteries), and the central nervous system (causing stroke, confusion, and delirium). Cardiac or central nervous system involvement is associated with a high risk of death.
After angiography, clinical manifestations of cholesterol embolization have been reported in 0.06% to 1.4% of patients,2,3 although the finding of cholesterol emboli is more common in autopsy studies.4
Recognizing skin signs is the key
Cutaneous abnormalities are usually the earliest and often the only clinical manifestation of this syndrome. Findings on the lower limbs include blue toes, cutaneous nodules, and livedo reticularis, affecting the feet and legs and sometimes extending upward to the trunk. Other findings include digital infarcts, ulceration, gangrene, purpura, cyanosis, and splinter hemorrhages in the nail bed.
Figure 2. Skin biopsy showed needle-shaped clefts within the lumen of blood vessels, ie, dissolved cholesterol crystals obstructing small arteries.
In our patient, microscopic study of skin biopsy specimens showed needle-shaped clefts within the lumen of blood vessels—ie, dissolved cholesterol crystals obstructing small arteries (Figure 2).
Biopsy studies of skin lesions are positive in a high percentage of cases, showing dissolved cholesterol crystals within the lumen of blood vessels, especially in the small to large arteries and arterioles of the deep dermis or subcutaneous fat. Deep biopsies and carefully examination are necessary, as emboli tend to be patchily distributed. Early recognition of cutaneous clinical findings is essential to establish the proper diagnosis and treatment.
The diagnostic triad of this disease includes blue toe syndrome, acute or subacute renal failure, and a temporal relation with an inciting event (particularly angiography). But despite these diagnostic criteria,2 the diagnosis is often based on a combination of signs and symptoms specific to end-organ damage and a systemic inflammatory response.3
Histologic confirmation is considered essential to the diagnosis of cholesterol crystal embolism, and as the skin is the most accessible site, skin biopsy provides the best sample for histologic diagnosis.5
Postprocedural embolism of a blood clot, vasculitis, and infective endocarditis are the most important differential diagnoses.6,7
Treatment is supportive, preventive
Treatment is mainly supportive with hemodynamic monitoring, nutritional and metabolic support, mechanical ventilation, and dialysis if necessary. The underlying atherosclerotic disease should be treated aggressively. Prevention of another episode involves modification of traditional risk factors such as serum cholesterol, diabetes, hypertension, and smoking. Additional vascular surgery procedures should be avoided, as they can induce new episodes.
References
Donohue KG, Saap L, Falanga V. Cholesterol crystal embolization: an atherosclerotic disease with frequent and varied cutaneous manifestations. J Eur Acad Dermatol Venereol2003; 17:504–511.
Fukumoto Y, Tsutsui H, Tsuchihashi M, Masumoto A, Takeshita A; Cholesterol Embolism Study (CHEST) Investigators. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol2003; 42:211–216.
Johnson LW, Esente P, Giambartolomei A, et al. Peripheral vascular complications of coronary angioplasty by the femoral and brachial techniques. Cathet Cardiovasc Diagn1994; 31:165–172.
Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation2010; 122:631–641.
Jucgla A, Moreso F, Muniesa C, Moreno A, Vidaller A. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol2006; 55:786–793.
Maki T, Izumi C, Miyake M, et al. Cholesterol embolism after cardiac catheterization mimicking infective endocarditis. Intern Med2005; 44:1060–1063.
Donohue KG, Saap L, Falanga V. Cholesterol crystal embolization: an atherosclerotic disease with frequent and varied cutaneous manifestations. J Eur Acad Dermatol Venereol2003; 17:504–511.
Fukumoto Y, Tsutsui H, Tsuchihashi M, Masumoto A, Takeshita A; Cholesterol Embolism Study (CHEST) Investigators. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol2003; 42:211–216.
Johnson LW, Esente P, Giambartolomei A, et al. Peripheral vascular complications of coronary angioplasty by the femoral and brachial techniques. Cathet Cardiovasc Diagn1994; 31:165–172.
Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation2010; 122:631–641.
Jucgla A, Moreso F, Muniesa C, Moreno A, Vidaller A. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol2006; 55:786–793.
Maki T, Izumi C, Miyake M, et al. Cholesterol embolism after cardiac catheterization mimicking infective endocarditis. Intern Med2005; 44:1060–1063.
A 60-year-old man presented with progressive swelling of his face and neck, which had begun 2 weeks earlier. He denied any headache, lightheadedness, blurry vision, syncope, or change in his cognitive or memory function. A review of symptoms was unremarkable.
The patient had hypertension and end-stage renal disease, for which he was receiving hemodialysis via a catheter tunneled into his right internal jugular vein. He had undergone multiple unsuccessful attempts to create an arteriovenous fistula over the previous 2 years.
Figure 1. Swollen face, congested conjunctivae, and multiple dilated tortuous veins on the chest and abdominal walls.
On physical examination, his vital signs were normal. Swelling of the face and fullness in his neck with bilateral congested conjunctivae were noted. His jugular venous pressure was elevated at 10 cm. Multiple dilated tortuous veins were noticed on his upper chest and across his abdominal wall (Figure 1). The rest of the examination was unremarkable.
Doppler ultrasonography revealed chronic thrombosis and reverse flow in the right internal jugular vein and reverse flow in the right subclavian vein. These findings were consistent with central venous thrombosis and superior vena cava (SVC) syndrome.
Figure 2. Multiple collateral veins in the upper chest (black arrows, left panel), stenosis of the superior vena cava, and the intraluminal catheter in place (right panel).
Computed tomography (CT) of the chest showed significant stenosis of the right innominate vein and SVC due to thrombosis. Numerous collateral veins were seen in the neck and across both shoulders (Figure 2).
Diagnosis: SVC syndrome secondary to intravascular thrombosis related to his central venous dialysis catheter.
SVC SYNDROME
The SVC is the major drainage vessel for venous blood from the head, neck, upper extremities, and upper thorax. Obstruction to its flow increases venous pressure, which results in interstitial edema and retrograde collateral flow.1
More than 80% of cases of SVC syndrome are caused by malignant lung tumors and lymphoma.
Nonmalignant causes include mediastinal fibrosis; vascular diseases (eg, aortic aneurysm, large-vessel vasculitis); infections such as histoplasmosis, tuberculosis, syphilis, and actinomycosis; benign mediastinal tumors such as teratoma, cystic hygroma, thymoma, and dermoid cyst; and thrombosis from central venous catheters, pacemaker leads, and guidewires.2–6 A recent report suggests that benign causes may now account for up to 40% of cases as a result of a rise in the use of indwelling central venous catheters and cardiac pacemakers during the past 2 decades, resulting in a higher incidence of SVC thrombosis.7
An obstructed SVC initiates collateral venous return to the heart from the upper half of the body through different pathways. The most important pathway is the azygos venous system, which includes the azygos vein. Occlusion of the SVC at the level of the azygos vein contributes to the appearance of collateral veins on the chest and abdominal walls, and venous blood flows via these collaterals into the inferior vena cava.1,8,9
Different presentations
The diagnosis of SVC syndrome is often made on clinical grounds alone, ie, the combination of the clinical presentation and, often, a thoracic malignancy or contributing factors such as a central catheter.1
With slowly progressive obstruction of the SVC, the most common presenting symptoms include swelling of the face, neck, and both arms. On the other hand, adequate collateral drainage may develop,1 and patients may have minimal symptoms.
However, a rapid onset of SVC syndrome in the absence of collateral circulation will cause a more dramatic and life-threatening presentation, often with neurologic and respiratory sequelae such as cerebral and laryngeal edema and respiratory embarrassment, which were not present in our patient’s case.1,10–15 These serious complications are rare and are considered an acute emergency. In these cases, special attention to airway, breathing, and circulation (the “ABCs”) is essential, and endovascular repairs and stenting or open surgical reconstruction and alternate approaches for renal replacement therapy should be considered.1,12,13,15
CT is diagnostic and provides accurate information about the location of the obstruction and about other critical surrounding structures such as the lungs, mediastinum, and adjacent blood vessels.1,7,10,11 Our patient’s CT scan confirmed a significant stenosis of the SVC due to thrombosis, with no compression coming from the lungs or mediastinal structures.
Thrombolytic therapy in acute cases
In cases of acute thrombosis (with symptom onset less than 2 days previously), thrombolytic therapy followed by anticoagulation is recommended and may both cause the symptoms to regress within several days and allow the central catheter to be kept in.16 However, thrombolytic therapy is less effective in chronic thrombosis (with onset of symptoms more than 10 days previously).16
Vascular or surgical intervention is often needed to treat SVC syndrome related to dialysis access.
Most experts recommend anticoagulation after thrombosis to prevent disease progression and recurrence, although the benefit of either short-term or long-term anticoagulation therapy for this syndrome is unclear.16
Recommended treatments for cancer-related SVC syndrome include chemotherapy and radiation to shrink the tumor that is causing the obstruction. Tissue diagnosis is often necessary to direct treatment decisions.1 However, percutaneous angioplasty and the use of intravenous stents are becoming increasingly common and are simple, safe, and effective in rapidly relieving SVC syndrome caused by malignant diseases.1 A bypass of the SVC may be indicated in some cases.1 Adjunctive therapies include diuretics, corticosteroids, thrombolytics, anticoagulation, and elevating the head of the patient’s bed.1
CASE CONTINUED
Our patient was started on heparin intravenously for 7 days and long-term oral anticoagulant therapy with warfarin (Coumadin) to continue as long as the catheter was in place, with a target international normalized ratio between 2 and 2.5. He required no other interventions, and his dialysis catheter remained functioning. He was monitored in the hospital for 2 weeks, during which his symptoms gradually improved, with noticeable resolution of his facial swelling.
He was discharged home to continue on an oral anticoagulant and was then followed to monitor for a reappearance of the symptoms (which would force the removal of the catheter), and to pursue possible percutaneous angioplasty, stenting, or surgical reconstruction of the SVC if needed.
References
Wilson LD, Detterbeck FC, Yahalom J. Clinical practice. Superior vena cava syndrome with malignant causes. N Engl J Med2007; 356:1862–1869.
Parish JM, Marschke RF, Dines DE, Lee RE. Etiologic considerations in superior vena cava syndrome. Mayo Clin Proc1981; 56:407–413.
Markman M. Diagnosis and management of superior vena cava syndrome. Cleve Clin J Med1999; 66:59–61.
Khanna S, Sniderman K, Simons M, Besley M, Uldall R. Superior vena cava stenosis associated with hemodialysis catheters. Am J Kidney Dis1993; 21:278–281.
Bertrand M, Presant CA, Klein L, Scott E. Iatrogenic superior vena cava syndrome. A new entity. Cancer1984; 54:376–378.
Rice TW, Rodriguez RM, Light RW. The superior vena cava syndrome: clinical characteristics and evolving etiology. Medicine (Baltimore)2006; 85:37–42.
Plekker D, Ellis T, Irusen EM, Bolliger CT, Diacon AH. Clinical and radiological grading of superior vena cava obstruction. Respiration2008; 76:69–75.
Sheth S, Ebert MD, Fishman EK. Superior vena cava obstruction evaluation with MDCT. AJR Am J Roentgenol2010; 194:W336–W346.
DeMichele A, Glick J. Cancer-related emergencies. In:Lenhard R, Osteen R, Gansler T, eds. Clinical Oncology. Atlanta, GA: American Cancer Society; 2001:733–764.
Chen JC, Bongard F, Klein SR. A contemporary perspective on superior vena cava syndrome. Am J Surg1990; 160:207–211.
Sheikh MA, Fernandez BB, Gray BH, Graham LM, Carman TL. Endovascular stenting of nonmalignant superior vena cava syndrome. Catheter Cardiovasc Interv2005; 65:405–411.
Flinterman LE, Van Der Meer FJ, Rosendaal FR, Doggen CJ. Current perspective of venous thrombosis in the upper extremity. J Thromb Haemost2008; 6:1262–1266.
Greenberg S, Kosinski R, Daniels J. Treatment of superior vena cava thrombosis with recombinant tissue type plasminogen activator. Chest1991; 99:1298–1301.
Molhem A, Sabry A, Bawadekji H, Al Saran K. Superior vena cava syndrome in hemodialysis patient. Saudi J Kidney Dis Transpl2011; 22:381–386.
Akoglu H, Yilmaz R, Peynircioglu B, et al. A rare complication of hemodialysis catheters: superior vena cava syndrome. Hemodial Int2007; 11:385–391.
Khaldoon Shaheen, MD Department of Medicine, Case Western Reserve University/St. Vincent Charity Medical Center, Cleveland, OH
M. Chadi Alraies, MD, FACP Clinical Assistant Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Staff, Department of Hospital Medicine, Cleveland Clinic
Address: Khaldoon Shaheen, MD, Department of Internal Medicine, St. Vincent Charity Medical Center, 2351 East 22nd Street, Cleveland, OH 44115; e-mail [email protected]
Khaldoon Shaheen, MD Department of Medicine, Case Western Reserve University/St. Vincent Charity Medical Center, Cleveland, OH
M. Chadi Alraies, MD, FACP Clinical Assistant Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Staff, Department of Hospital Medicine, Cleveland Clinic
Address: Khaldoon Shaheen, MD, Department of Internal Medicine, St. Vincent Charity Medical Center, 2351 East 22nd Street, Cleveland, OH 44115; e-mail [email protected]
Author and Disclosure Information
Khaldoon Shaheen, MD Department of Medicine, Case Western Reserve University/St. Vincent Charity Medical Center, Cleveland, OH
M. Chadi Alraies, MD, FACP Clinical Assistant Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Staff, Department of Hospital Medicine, Cleveland Clinic
Address: Khaldoon Shaheen, MD, Department of Internal Medicine, St. Vincent Charity Medical Center, 2351 East 22nd Street, Cleveland, OH 44115; e-mail [email protected]
A 60-year-old man presented with progressive swelling of his face and neck, which had begun 2 weeks earlier. He denied any headache, lightheadedness, blurry vision, syncope, or change in his cognitive or memory function. A review of symptoms was unremarkable.
The patient had hypertension and end-stage renal disease, for which he was receiving hemodialysis via a catheter tunneled into his right internal jugular vein. He had undergone multiple unsuccessful attempts to create an arteriovenous fistula over the previous 2 years.
Figure 1. Swollen face, congested conjunctivae, and multiple dilated tortuous veins on the chest and abdominal walls.
On physical examination, his vital signs were normal. Swelling of the face and fullness in his neck with bilateral congested conjunctivae were noted. His jugular venous pressure was elevated at 10 cm. Multiple dilated tortuous veins were noticed on his upper chest and across his abdominal wall (Figure 1). The rest of the examination was unremarkable.
Doppler ultrasonography revealed chronic thrombosis and reverse flow in the right internal jugular vein and reverse flow in the right subclavian vein. These findings were consistent with central venous thrombosis and superior vena cava (SVC) syndrome.
Figure 2. Multiple collateral veins in the upper chest (black arrows, left panel), stenosis of the superior vena cava, and the intraluminal catheter in place (right panel).
Computed tomography (CT) of the chest showed significant stenosis of the right innominate vein and SVC due to thrombosis. Numerous collateral veins were seen in the neck and across both shoulders (Figure 2).
Diagnosis: SVC syndrome secondary to intravascular thrombosis related to his central venous dialysis catheter.
SVC SYNDROME
The SVC is the major drainage vessel for venous blood from the head, neck, upper extremities, and upper thorax. Obstruction to its flow increases venous pressure, which results in interstitial edema and retrograde collateral flow.1
More than 80% of cases of SVC syndrome are caused by malignant lung tumors and lymphoma.
Nonmalignant causes include mediastinal fibrosis; vascular diseases (eg, aortic aneurysm, large-vessel vasculitis); infections such as histoplasmosis, tuberculosis, syphilis, and actinomycosis; benign mediastinal tumors such as teratoma, cystic hygroma, thymoma, and dermoid cyst; and thrombosis from central venous catheters, pacemaker leads, and guidewires.2–6 A recent report suggests that benign causes may now account for up to 40% of cases as a result of a rise in the use of indwelling central venous catheters and cardiac pacemakers during the past 2 decades, resulting in a higher incidence of SVC thrombosis.7
An obstructed SVC initiates collateral venous return to the heart from the upper half of the body through different pathways. The most important pathway is the azygos venous system, which includes the azygos vein. Occlusion of the SVC at the level of the azygos vein contributes to the appearance of collateral veins on the chest and abdominal walls, and venous blood flows via these collaterals into the inferior vena cava.1,8,9
Different presentations
The diagnosis of SVC syndrome is often made on clinical grounds alone, ie, the combination of the clinical presentation and, often, a thoracic malignancy or contributing factors such as a central catheter.1
With slowly progressive obstruction of the SVC, the most common presenting symptoms include swelling of the face, neck, and both arms. On the other hand, adequate collateral drainage may develop,1 and patients may have minimal symptoms.
However, a rapid onset of SVC syndrome in the absence of collateral circulation will cause a more dramatic and life-threatening presentation, often with neurologic and respiratory sequelae such as cerebral and laryngeal edema and respiratory embarrassment, which were not present in our patient’s case.1,10–15 These serious complications are rare and are considered an acute emergency. In these cases, special attention to airway, breathing, and circulation (the “ABCs”) is essential, and endovascular repairs and stenting or open surgical reconstruction and alternate approaches for renal replacement therapy should be considered.1,12,13,15
CT is diagnostic and provides accurate information about the location of the obstruction and about other critical surrounding structures such as the lungs, mediastinum, and adjacent blood vessels.1,7,10,11 Our patient’s CT scan confirmed a significant stenosis of the SVC due to thrombosis, with no compression coming from the lungs or mediastinal structures.
Thrombolytic therapy in acute cases
In cases of acute thrombosis (with symptom onset less than 2 days previously), thrombolytic therapy followed by anticoagulation is recommended and may both cause the symptoms to regress within several days and allow the central catheter to be kept in.16 However, thrombolytic therapy is less effective in chronic thrombosis (with onset of symptoms more than 10 days previously).16
Vascular or surgical intervention is often needed to treat SVC syndrome related to dialysis access.
Most experts recommend anticoagulation after thrombosis to prevent disease progression and recurrence, although the benefit of either short-term or long-term anticoagulation therapy for this syndrome is unclear.16
Recommended treatments for cancer-related SVC syndrome include chemotherapy and radiation to shrink the tumor that is causing the obstruction. Tissue diagnosis is often necessary to direct treatment decisions.1 However, percutaneous angioplasty and the use of intravenous stents are becoming increasingly common and are simple, safe, and effective in rapidly relieving SVC syndrome caused by malignant diseases.1 A bypass of the SVC may be indicated in some cases.1 Adjunctive therapies include diuretics, corticosteroids, thrombolytics, anticoagulation, and elevating the head of the patient’s bed.1
CASE CONTINUED
Our patient was started on heparin intravenously for 7 days and long-term oral anticoagulant therapy with warfarin (Coumadin) to continue as long as the catheter was in place, with a target international normalized ratio between 2 and 2.5. He required no other interventions, and his dialysis catheter remained functioning. He was monitored in the hospital for 2 weeks, during which his symptoms gradually improved, with noticeable resolution of his facial swelling.
He was discharged home to continue on an oral anticoagulant and was then followed to monitor for a reappearance of the symptoms (which would force the removal of the catheter), and to pursue possible percutaneous angioplasty, stenting, or surgical reconstruction of the SVC if needed.
A 60-year-old man presented with progressive swelling of his face and neck, which had begun 2 weeks earlier. He denied any headache, lightheadedness, blurry vision, syncope, or change in his cognitive or memory function. A review of symptoms was unremarkable.
The patient had hypertension and end-stage renal disease, for which he was receiving hemodialysis via a catheter tunneled into his right internal jugular vein. He had undergone multiple unsuccessful attempts to create an arteriovenous fistula over the previous 2 years.
Figure 1. Swollen face, congested conjunctivae, and multiple dilated tortuous veins on the chest and abdominal walls.
On physical examination, his vital signs were normal. Swelling of the face and fullness in his neck with bilateral congested conjunctivae were noted. His jugular venous pressure was elevated at 10 cm. Multiple dilated tortuous veins were noticed on his upper chest and across his abdominal wall (Figure 1). The rest of the examination was unremarkable.
Doppler ultrasonography revealed chronic thrombosis and reverse flow in the right internal jugular vein and reverse flow in the right subclavian vein. These findings were consistent with central venous thrombosis and superior vena cava (SVC) syndrome.
Figure 2. Multiple collateral veins in the upper chest (black arrows, left panel), stenosis of the superior vena cava, and the intraluminal catheter in place (right panel).
Computed tomography (CT) of the chest showed significant stenosis of the right innominate vein and SVC due to thrombosis. Numerous collateral veins were seen in the neck and across both shoulders (Figure 2).
Diagnosis: SVC syndrome secondary to intravascular thrombosis related to his central venous dialysis catheter.
SVC SYNDROME
The SVC is the major drainage vessel for venous blood from the head, neck, upper extremities, and upper thorax. Obstruction to its flow increases venous pressure, which results in interstitial edema and retrograde collateral flow.1
More than 80% of cases of SVC syndrome are caused by malignant lung tumors and lymphoma.
Nonmalignant causes include mediastinal fibrosis; vascular diseases (eg, aortic aneurysm, large-vessel vasculitis); infections such as histoplasmosis, tuberculosis, syphilis, and actinomycosis; benign mediastinal tumors such as teratoma, cystic hygroma, thymoma, and dermoid cyst; and thrombosis from central venous catheters, pacemaker leads, and guidewires.2–6 A recent report suggests that benign causes may now account for up to 40% of cases as a result of a rise in the use of indwelling central venous catheters and cardiac pacemakers during the past 2 decades, resulting in a higher incidence of SVC thrombosis.7
An obstructed SVC initiates collateral venous return to the heart from the upper half of the body through different pathways. The most important pathway is the azygos venous system, which includes the azygos vein. Occlusion of the SVC at the level of the azygos vein contributes to the appearance of collateral veins on the chest and abdominal walls, and venous blood flows via these collaterals into the inferior vena cava.1,8,9
Different presentations
The diagnosis of SVC syndrome is often made on clinical grounds alone, ie, the combination of the clinical presentation and, often, a thoracic malignancy or contributing factors such as a central catheter.1
With slowly progressive obstruction of the SVC, the most common presenting symptoms include swelling of the face, neck, and both arms. On the other hand, adequate collateral drainage may develop,1 and patients may have minimal symptoms.
However, a rapid onset of SVC syndrome in the absence of collateral circulation will cause a more dramatic and life-threatening presentation, often with neurologic and respiratory sequelae such as cerebral and laryngeal edema and respiratory embarrassment, which were not present in our patient’s case.1,10–15 These serious complications are rare and are considered an acute emergency. In these cases, special attention to airway, breathing, and circulation (the “ABCs”) is essential, and endovascular repairs and stenting or open surgical reconstruction and alternate approaches for renal replacement therapy should be considered.1,12,13,15
CT is diagnostic and provides accurate information about the location of the obstruction and about other critical surrounding structures such as the lungs, mediastinum, and adjacent blood vessels.1,7,10,11 Our patient’s CT scan confirmed a significant stenosis of the SVC due to thrombosis, with no compression coming from the lungs or mediastinal structures.
Thrombolytic therapy in acute cases
In cases of acute thrombosis (with symptom onset less than 2 days previously), thrombolytic therapy followed by anticoagulation is recommended and may both cause the symptoms to regress within several days and allow the central catheter to be kept in.16 However, thrombolytic therapy is less effective in chronic thrombosis (with onset of symptoms more than 10 days previously).16
Vascular or surgical intervention is often needed to treat SVC syndrome related to dialysis access.
Most experts recommend anticoagulation after thrombosis to prevent disease progression and recurrence, although the benefit of either short-term or long-term anticoagulation therapy for this syndrome is unclear.16
Recommended treatments for cancer-related SVC syndrome include chemotherapy and radiation to shrink the tumor that is causing the obstruction. Tissue diagnosis is often necessary to direct treatment decisions.1 However, percutaneous angioplasty and the use of intravenous stents are becoming increasingly common and are simple, safe, and effective in rapidly relieving SVC syndrome caused by malignant diseases.1 A bypass of the SVC may be indicated in some cases.1 Adjunctive therapies include diuretics, corticosteroids, thrombolytics, anticoagulation, and elevating the head of the patient’s bed.1
CASE CONTINUED
Our patient was started on heparin intravenously for 7 days and long-term oral anticoagulant therapy with warfarin (Coumadin) to continue as long as the catheter was in place, with a target international normalized ratio between 2 and 2.5. He required no other interventions, and his dialysis catheter remained functioning. He was monitored in the hospital for 2 weeks, during which his symptoms gradually improved, with noticeable resolution of his facial swelling.
He was discharged home to continue on an oral anticoagulant and was then followed to monitor for a reappearance of the symptoms (which would force the removal of the catheter), and to pursue possible percutaneous angioplasty, stenting, or surgical reconstruction of the SVC if needed.
References
Wilson LD, Detterbeck FC, Yahalom J. Clinical practice. Superior vena cava syndrome with malignant causes. N Engl J Med2007; 356:1862–1869.
Parish JM, Marschke RF, Dines DE, Lee RE. Etiologic considerations in superior vena cava syndrome. Mayo Clin Proc1981; 56:407–413.
Markman M. Diagnosis and management of superior vena cava syndrome. Cleve Clin J Med1999; 66:59–61.
Khanna S, Sniderman K, Simons M, Besley M, Uldall R. Superior vena cava stenosis associated with hemodialysis catheters. Am J Kidney Dis1993; 21:278–281.
Bertrand M, Presant CA, Klein L, Scott E. Iatrogenic superior vena cava syndrome. A new entity. Cancer1984; 54:376–378.
Rice TW, Rodriguez RM, Light RW. The superior vena cava syndrome: clinical characteristics and evolving etiology. Medicine (Baltimore)2006; 85:37–42.
Plekker D, Ellis T, Irusen EM, Bolliger CT, Diacon AH. Clinical and radiological grading of superior vena cava obstruction. Respiration2008; 76:69–75.
Sheth S, Ebert MD, Fishman EK. Superior vena cava obstruction evaluation with MDCT. AJR Am J Roentgenol2010; 194:W336–W346.
DeMichele A, Glick J. Cancer-related emergencies. In:Lenhard R, Osteen R, Gansler T, eds. Clinical Oncology. Atlanta, GA: American Cancer Society; 2001:733–764.
Chen JC, Bongard F, Klein SR. A contemporary perspective on superior vena cava syndrome. Am J Surg1990; 160:207–211.
Sheikh MA, Fernandez BB, Gray BH, Graham LM, Carman TL. Endovascular stenting of nonmalignant superior vena cava syndrome. Catheter Cardiovasc Interv2005; 65:405–411.
Flinterman LE, Van Der Meer FJ, Rosendaal FR, Doggen CJ. Current perspective of venous thrombosis in the upper extremity. J Thromb Haemost2008; 6:1262–1266.
Greenberg S, Kosinski R, Daniels J. Treatment of superior vena cava thrombosis with recombinant tissue type plasminogen activator. Chest1991; 99:1298–1301.
Molhem A, Sabry A, Bawadekji H, Al Saran K. Superior vena cava syndrome in hemodialysis patient. Saudi J Kidney Dis Transpl2011; 22:381–386.
Akoglu H, Yilmaz R, Peynircioglu B, et al. A rare complication of hemodialysis catheters: superior vena cava syndrome. Hemodial Int2007; 11:385–391.
References
Wilson LD, Detterbeck FC, Yahalom J. Clinical practice. Superior vena cava syndrome with malignant causes. N Engl J Med2007; 356:1862–1869.
Parish JM, Marschke RF, Dines DE, Lee RE. Etiologic considerations in superior vena cava syndrome. Mayo Clin Proc1981; 56:407–413.
Markman M. Diagnosis and management of superior vena cava syndrome. Cleve Clin J Med1999; 66:59–61.
Khanna S, Sniderman K, Simons M, Besley M, Uldall R. Superior vena cava stenosis associated with hemodialysis catheters. Am J Kidney Dis1993; 21:278–281.
Bertrand M, Presant CA, Klein L, Scott E. Iatrogenic superior vena cava syndrome. A new entity. Cancer1984; 54:376–378.
Rice TW, Rodriguez RM, Light RW. The superior vena cava syndrome: clinical characteristics and evolving etiology. Medicine (Baltimore)2006; 85:37–42.
Plekker D, Ellis T, Irusen EM, Bolliger CT, Diacon AH. Clinical and radiological grading of superior vena cava obstruction. Respiration2008; 76:69–75.
Sheth S, Ebert MD, Fishman EK. Superior vena cava obstruction evaluation with MDCT. AJR Am J Roentgenol2010; 194:W336–W346.
DeMichele A, Glick J. Cancer-related emergencies. In:Lenhard R, Osteen R, Gansler T, eds. Clinical Oncology. Atlanta, GA: American Cancer Society; 2001:733–764.
Chen JC, Bongard F, Klein SR. A contemporary perspective on superior vena cava syndrome. Am J Surg1990; 160:207–211.
Sheikh MA, Fernandez BB, Gray BH, Graham LM, Carman TL. Endovascular stenting of nonmalignant superior vena cava syndrome. Catheter Cardiovasc Interv2005; 65:405–411.
Flinterman LE, Van Der Meer FJ, Rosendaal FR, Doggen CJ. Current perspective of venous thrombosis in the upper extremity. J Thromb Haemost2008; 6:1262–1266.
Greenberg S, Kosinski R, Daniels J. Treatment of superior vena cava thrombosis with recombinant tissue type plasminogen activator. Chest1991; 99:1298–1301.
Molhem A, Sabry A, Bawadekji H, Al Saran K. Superior vena cava syndrome in hemodialysis patient. Saudi J Kidney Dis Transpl2011; 22:381–386.
Akoglu H, Yilmaz R, Peynircioglu B, et al. A rare complication of hemodialysis catheters: superior vena cava syndrome. Hemodial Int2007; 11:385–391.
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).
Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).
One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.
References
Teodosio C, García-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol2010; 125:719–726.
van de Loosdrecht AA, Alhan C, Béné MC, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica2009; 94:1124–1134.
Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol2003; 162:737–746.
Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res2001; 25:603–625.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest2007; 37:435–453.
Horny HP, Metcalfe DD, Bennet JM, et al. Mastocytosis. In:Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:54–63.
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am2006; 26:465–485.
González de Olano D, de la Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy2007; 37:1547–1555.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood2006; 108:2366–2372.
Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol2009; 124:514–521.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets2006; 5:61–77.
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).
Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).
One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).
Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).
One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.
References
Teodosio C, García-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol2010; 125:719–726.
van de Loosdrecht AA, Alhan C, Béné MC, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica2009; 94:1124–1134.
Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol2003; 162:737–746.
Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res2001; 25:603–625.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest2007; 37:435–453.
Horny HP, Metcalfe DD, Bennet JM, et al. Mastocytosis. In:Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:54–63.
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am2006; 26:465–485.
González de Olano D, de la Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy2007; 37:1547–1555.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood2006; 108:2366–2372.
Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol2009; 124:514–521.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets2006; 5:61–77.
References
Teodosio C, García-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol2010; 125:719–726.
van de Loosdrecht AA, Alhan C, Béné MC, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica2009; 94:1124–1134.
Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol2003; 162:737–746.
Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res2001; 25:603–625.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest2007; 37:435–453.
Horny HP, Metcalfe DD, Bennet JM, et al. Mastocytosis. In:Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:54–63.
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am2006; 26:465–485.
González de Olano D, de la Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy2007; 37:1547–1555.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood2006; 108:2366–2372.
Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol2009; 124:514–521.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets2006; 5:61–77.
Figure 1. Erythema of both hands.
A 42-year-old businessman presented with recurrent redness, swelling, warmth, and burning pain on both hands and both feet for the preceding year (Figure 1). His symptoms were worse during heat exposure or physical effort and were relieved by immersing his hands and feet in cold water and by elevating his limbs. He had no other local or systemic symptoms, and he had not been on any medications. His medical history was noncontributory, and there was no family history of similar illness.
Figure 2. Blanching of erythema with pressure.
The erythema blanched when pressure was applied (Figure 2). The affected areas were slightly tender. Other areas of his skin and mucosae were normal. Neurologic examination and examination of other systems were normal. Results of laboratory testing (complete blood cell count, biochemistry panel, antinuclear antibody test) and gastrointestinal endoscopy were normal.
Q: What is the diagnosis?
Fabry disease
Peripheral neuropathy
Polycythemia
Primary idiopathic erythromelalgia
Erythrodysesthesia syndrome
A: The correct diagnosis is primary idiopathic erythromelalgia.
Erythromelalgia is a relatively rare clinical condition of uncertain etiology, characterized by the triad of episodic redness, warmth, and burning pain in the extremities.1,2
The condition has primary (ie, no underlying cause is found) and secondary forms.1,3 The primary form may be inherited in an autosomal dominant manner (in which case the symptoms begin in childhood), or it may be idiopathic.4 On the other hand, erythromelalgia can also be secondary to polycythemia vera and other myeloproliferative disorders, connective tissue disorders, neuropathies, spinal cord diseases, carcinoma of the colon and thyroid, and astrocytomas.1–6
The common pathologic mechanism of erythromelalgia is thought to be microvascular arteriovenous shunting.5 A mutation in the voltage-gated sodium channel alpha subunit NaV1.7 may result in primary erythromelalgia. Small-fiber neuropathy2 can manifest as erythromelalgia and may respond to steroids.
The symptoms can be intermittent or, in rare cases, constant.3 The lower limbs are more commonly involved than the upper ones.3 Involvement is often bilateral and symmetric.3 The symptoms may worsen at night, after alcohol consumption, with higher environmental temperature, and with moderate exercise.3 In rare cases, ulceration and gangrene may occur. Patients may get relief by cooling the affected areas.2
DIAGNOSIS BY EXCLUSION OR BASED ON THE PRESENTATION
The diagnosis of erythromelalgia is based on a detailed history and physical examination during a painful episode.2,3 Because the condition is intermittent, only about two-thirds of patients have abnormal findings on physical examination at the time of presentation; in such cases, the diagnosis is based on the history alone.2
Testing is needed to exclude other diagnoses and to determine the cause of secondary erythromelalgia. Histopathologic study of lesions is not helpful, as the features are nonspecific and hence nondiagnostic.2
In our patient, the typical clinical features and the lack of an obvious cause on diagnostic testing confirmed the diagnosis.
THE DIFFERENTIAL DIAGNOSIS
Other conditions can be ruled out by clinical features and laboratory testing.
Fabry disease causes paresthesia and burning pain in the extremities but not erythema. Characteristic dark red keratotic papules are seen all over the body (angiokeratoma corporis diffusum). It is often associated with progressive renal insufficiency.
Peripheral neuropathy of varying causes may also cause pain in the extremities but not erythema. Neurologic examination and a nerve conduction velocity study can resolve the diagnostic problem. Clinical features and laboratory testing often help to pinpoint the cause of neuropathy.
Polycythemia may cause erythema in the hands, feet, and face and mucosal engorgement. Telangiectasia, petechiae, and cyanosis may also occur. Our patient’s normal complete blood cell count excluded this condition.
Erythrodysesthesia syndrome, typically caused by chemotherapeutic drugs, was not included in the differential diagnosis since our patient had taken no medications.
Other causes of palmar erythema to rule out, depending on the patient’s presentation, may include thyrotoxicosis, chronic febrile illness, leukemia, hepatic insufficiency, chronic alcoholism, and rheumatoid arthritis.
TREATMENT AND PROGNOSIS
There is no definitive therapy for erythromelalgia.4 Treatment is often difficult and needs a multidisciplinary approach. Simple measures such as cooling2 (eg, applying cold towels, immersion in cool water, walking on cold floors) or elevating the affected extremity often relieve symptoms. Patients should avoid precipitating factors such as warmth, dependency of extremities, exercise, tight footwear, and alcohol intake.
If there is an underlying disease, treating the disease may also alleviate the symptoms.7 Aspirin2,7 is the therapy of choice for erythromelalgia in patients with an underlying myeloproliferative disorder, and some authors have advocated it for all patients with erythromelalgia unless there is a contraindication.7
Other possible first-line treatments include the synthetic prostaglandin E1 analogue misoprostol (Cytotec) and prostacyclins. Gabapentin (Neurontin), serotonin reuptake inhibitors such as sertaline (Zoloft) and venlafaxine (Effexor), and intravenous nitroprusside (Nitro-press) are considered second-line drugs.7
Surgical sympathectomy8 has also been tried, with variable results.
Outcomes in patients with erythromelalgia
In a case series from Mayo Clinic,2 approximately equal numbers of patients with erythromelalgia became worse, stayed the same, or got better, and the disease resolved in 10% over a mean of 8.7 years.
THE OUTCOME IN OUR PATIENT
We advised our patient to avoid strenuous activity in a warm environment and to work in cooler areas as much as possible. We told him to wrap his affected extremities with cold towels during attacks, and we prescribed aspirin (650 mg/day) for 3 months. The treatment did not cure his condition, but his symptoms lessened within 2 months. We later referred him to a pain clinic.
References
Kalgaard OM, Seem E, Kvernebo K. Erythromelalgia: a clinical study of 87 cases. Intern Med1997; 242:191–197.
Davis MD, O’Fallon WM, Rogers RS, Rooke TW. Natural history of erythromelalgia: presentation and outcome in 168 patients. Arch Dermatol2000; 136:330–336.
Galimberti D, Pontón A, Rubio L, et al. A case of primary erythromelalgia. J Eur Acad Dermatol Venereol2009; 23:1338–1339.
Mørk C, Kvernebo K, Asker CL, Salerud EG. Reduced skin capillary density during attacks of erythromelalgia implies arteriovenous shunting as pathogenetic mechanism. J Invest Dermatol2002; 119:949–953.
James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed.Philadelphia, PA: Saunders Elsevier; 2006.
Mørk C, Kvernebo K. Erythromelalgia. In:Lebwohl MG, Heymann WR, Berth-Jones J, Koulson Y. editors. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 3rd ed.Philadelphia, PA: Saunders Elsevier; 2010:236–238.
Seishima M, Kanoh H, Izumi T, et al. A refractory case of secondary erythermalgia successfully treated with lumbar sympathetic ganglion block. Br J Dermatol2000; 143:868–872.
Sudip Kumar Ghosh, MD, DNB Assistant Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Debabrata Bandyopadhyay, MD Professor, Department of Dermatology, Venereology, and Leprosy, Calcutta Medical College, Kolkata, India
Loknath Ghoshal, MD Resident Medical Officer-Cum-Clinical Tutor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Address: Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Road, Kolkata 700004, West Bengal, India; e-mail [email protected]
Sudip Kumar Ghosh, MD, DNB Assistant Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Debabrata Bandyopadhyay, MD Professor, Department of Dermatology, Venereology, and Leprosy, Calcutta Medical College, Kolkata, India
Loknath Ghoshal, MD Resident Medical Officer-Cum-Clinical Tutor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Address: Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Road, Kolkata 700004, West Bengal, India; e-mail [email protected]
Author and Disclosure Information
Sudip Kumar Ghosh, MD, DNB Assistant Professor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Debabrata Bandyopadhyay, MD Professor, Department of Dermatology, Venereology, and Leprosy, Calcutta Medical College, Kolkata, India
Loknath Ghoshal, MD Resident Medical Officer-Cum-Clinical Tutor, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, Kolkata, India
Address: Sudip Kumar Ghosh, MD, DNB, Department of Dermatology, Venereology, and Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Road, Kolkata 700004, West Bengal, India; e-mail [email protected]
Figure 1. Erythema of both hands.
A 42-year-old businessman presented with recurrent redness, swelling, warmth, and burning pain on both hands and both feet for the preceding year (Figure 1). His symptoms were worse during heat exposure or physical effort and were relieved by immersing his hands and feet in cold water and by elevating his limbs. He had no other local or systemic symptoms, and he had not been on any medications. His medical history was noncontributory, and there was no family history of similar illness.
Figure 2. Blanching of erythema with pressure.
The erythema blanched when pressure was applied (Figure 2). The affected areas were slightly tender. Other areas of his skin and mucosae were normal. Neurologic examination and examination of other systems were normal. Results of laboratory testing (complete blood cell count, biochemistry panel, antinuclear antibody test) and gastrointestinal endoscopy were normal.
Q: What is the diagnosis?
Fabry disease
Peripheral neuropathy
Polycythemia
Primary idiopathic erythromelalgia
Erythrodysesthesia syndrome
A: The correct diagnosis is primary idiopathic erythromelalgia.
Erythromelalgia is a relatively rare clinical condition of uncertain etiology, characterized by the triad of episodic redness, warmth, and burning pain in the extremities.1,2
The condition has primary (ie, no underlying cause is found) and secondary forms.1,3 The primary form may be inherited in an autosomal dominant manner (in which case the symptoms begin in childhood), or it may be idiopathic.4 On the other hand, erythromelalgia can also be secondary to polycythemia vera and other myeloproliferative disorders, connective tissue disorders, neuropathies, spinal cord diseases, carcinoma of the colon and thyroid, and astrocytomas.1–6
The common pathologic mechanism of erythromelalgia is thought to be microvascular arteriovenous shunting.5 A mutation in the voltage-gated sodium channel alpha subunit NaV1.7 may result in primary erythromelalgia. Small-fiber neuropathy2 can manifest as erythromelalgia and may respond to steroids.
The symptoms can be intermittent or, in rare cases, constant.3 The lower limbs are more commonly involved than the upper ones.3 Involvement is often bilateral and symmetric.3 The symptoms may worsen at night, after alcohol consumption, with higher environmental temperature, and with moderate exercise.3 In rare cases, ulceration and gangrene may occur. Patients may get relief by cooling the affected areas.2
DIAGNOSIS BY EXCLUSION OR BASED ON THE PRESENTATION
The diagnosis of erythromelalgia is based on a detailed history and physical examination during a painful episode.2,3 Because the condition is intermittent, only about two-thirds of patients have abnormal findings on physical examination at the time of presentation; in such cases, the diagnosis is based on the history alone.2
Testing is needed to exclude other diagnoses and to determine the cause of secondary erythromelalgia. Histopathologic study of lesions is not helpful, as the features are nonspecific and hence nondiagnostic.2
In our patient, the typical clinical features and the lack of an obvious cause on diagnostic testing confirmed the diagnosis.
THE DIFFERENTIAL DIAGNOSIS
Other conditions can be ruled out by clinical features and laboratory testing.
Fabry disease causes paresthesia and burning pain in the extremities but not erythema. Characteristic dark red keratotic papules are seen all over the body (angiokeratoma corporis diffusum). It is often associated with progressive renal insufficiency.
Peripheral neuropathy of varying causes may also cause pain in the extremities but not erythema. Neurologic examination and a nerve conduction velocity study can resolve the diagnostic problem. Clinical features and laboratory testing often help to pinpoint the cause of neuropathy.
Polycythemia may cause erythema in the hands, feet, and face and mucosal engorgement. Telangiectasia, petechiae, and cyanosis may also occur. Our patient’s normal complete blood cell count excluded this condition.
Erythrodysesthesia syndrome, typically caused by chemotherapeutic drugs, was not included in the differential diagnosis since our patient had taken no medications.
Other causes of palmar erythema to rule out, depending on the patient’s presentation, may include thyrotoxicosis, chronic febrile illness, leukemia, hepatic insufficiency, chronic alcoholism, and rheumatoid arthritis.
TREATMENT AND PROGNOSIS
There is no definitive therapy for erythromelalgia.4 Treatment is often difficult and needs a multidisciplinary approach. Simple measures such as cooling2 (eg, applying cold towels, immersion in cool water, walking on cold floors) or elevating the affected extremity often relieve symptoms. Patients should avoid precipitating factors such as warmth, dependency of extremities, exercise, tight footwear, and alcohol intake.
If there is an underlying disease, treating the disease may also alleviate the symptoms.7 Aspirin2,7 is the therapy of choice for erythromelalgia in patients with an underlying myeloproliferative disorder, and some authors have advocated it for all patients with erythromelalgia unless there is a contraindication.7
Other possible first-line treatments include the synthetic prostaglandin E1 analogue misoprostol (Cytotec) and prostacyclins. Gabapentin (Neurontin), serotonin reuptake inhibitors such as sertaline (Zoloft) and venlafaxine (Effexor), and intravenous nitroprusside (Nitro-press) are considered second-line drugs.7
Surgical sympathectomy8 has also been tried, with variable results.
Outcomes in patients with erythromelalgia
In a case series from Mayo Clinic,2 approximately equal numbers of patients with erythromelalgia became worse, stayed the same, or got better, and the disease resolved in 10% over a mean of 8.7 years.
THE OUTCOME IN OUR PATIENT
We advised our patient to avoid strenuous activity in a warm environment and to work in cooler areas as much as possible. We told him to wrap his affected extremities with cold towels during attacks, and we prescribed aspirin (650 mg/day) for 3 months. The treatment did not cure his condition, but his symptoms lessened within 2 months. We later referred him to a pain clinic.
Figure 1. Erythema of both hands.
A 42-year-old businessman presented with recurrent redness, swelling, warmth, and burning pain on both hands and both feet for the preceding year (Figure 1). His symptoms were worse during heat exposure or physical effort and were relieved by immersing his hands and feet in cold water and by elevating his limbs. He had no other local or systemic symptoms, and he had not been on any medications. His medical history was noncontributory, and there was no family history of similar illness.
Figure 2. Blanching of erythema with pressure.
The erythema blanched when pressure was applied (Figure 2). The affected areas were slightly tender. Other areas of his skin and mucosae were normal. Neurologic examination and examination of other systems were normal. Results of laboratory testing (complete blood cell count, biochemistry panel, antinuclear antibody test) and gastrointestinal endoscopy were normal.
Q: What is the diagnosis?
Fabry disease
Peripheral neuropathy
Polycythemia
Primary idiopathic erythromelalgia
Erythrodysesthesia syndrome
A: The correct diagnosis is primary idiopathic erythromelalgia.
Erythromelalgia is a relatively rare clinical condition of uncertain etiology, characterized by the triad of episodic redness, warmth, and burning pain in the extremities.1,2
The condition has primary (ie, no underlying cause is found) and secondary forms.1,3 The primary form may be inherited in an autosomal dominant manner (in which case the symptoms begin in childhood), or it may be idiopathic.4 On the other hand, erythromelalgia can also be secondary to polycythemia vera and other myeloproliferative disorders, connective tissue disorders, neuropathies, spinal cord diseases, carcinoma of the colon and thyroid, and astrocytomas.1–6
The common pathologic mechanism of erythromelalgia is thought to be microvascular arteriovenous shunting.5 A mutation in the voltage-gated sodium channel alpha subunit NaV1.7 may result in primary erythromelalgia. Small-fiber neuropathy2 can manifest as erythromelalgia and may respond to steroids.
The symptoms can be intermittent or, in rare cases, constant.3 The lower limbs are more commonly involved than the upper ones.3 Involvement is often bilateral and symmetric.3 The symptoms may worsen at night, after alcohol consumption, with higher environmental temperature, and with moderate exercise.3 In rare cases, ulceration and gangrene may occur. Patients may get relief by cooling the affected areas.2
DIAGNOSIS BY EXCLUSION OR BASED ON THE PRESENTATION
The diagnosis of erythromelalgia is based on a detailed history and physical examination during a painful episode.2,3 Because the condition is intermittent, only about two-thirds of patients have abnormal findings on physical examination at the time of presentation; in such cases, the diagnosis is based on the history alone.2
Testing is needed to exclude other diagnoses and to determine the cause of secondary erythromelalgia. Histopathologic study of lesions is not helpful, as the features are nonspecific and hence nondiagnostic.2
In our patient, the typical clinical features and the lack of an obvious cause on diagnostic testing confirmed the diagnosis.
THE DIFFERENTIAL DIAGNOSIS
Other conditions can be ruled out by clinical features and laboratory testing.
Fabry disease causes paresthesia and burning pain in the extremities but not erythema. Characteristic dark red keratotic papules are seen all over the body (angiokeratoma corporis diffusum). It is often associated with progressive renal insufficiency.
Peripheral neuropathy of varying causes may also cause pain in the extremities but not erythema. Neurologic examination and a nerve conduction velocity study can resolve the diagnostic problem. Clinical features and laboratory testing often help to pinpoint the cause of neuropathy.
Polycythemia may cause erythema in the hands, feet, and face and mucosal engorgement. Telangiectasia, petechiae, and cyanosis may also occur. Our patient’s normal complete blood cell count excluded this condition.
Erythrodysesthesia syndrome, typically caused by chemotherapeutic drugs, was not included in the differential diagnosis since our patient had taken no medications.
Other causes of palmar erythema to rule out, depending on the patient’s presentation, may include thyrotoxicosis, chronic febrile illness, leukemia, hepatic insufficiency, chronic alcoholism, and rheumatoid arthritis.
TREATMENT AND PROGNOSIS
There is no definitive therapy for erythromelalgia.4 Treatment is often difficult and needs a multidisciplinary approach. Simple measures such as cooling2 (eg, applying cold towels, immersion in cool water, walking on cold floors) or elevating the affected extremity often relieve symptoms. Patients should avoid precipitating factors such as warmth, dependency of extremities, exercise, tight footwear, and alcohol intake.
If there is an underlying disease, treating the disease may also alleviate the symptoms.7 Aspirin2,7 is the therapy of choice for erythromelalgia in patients with an underlying myeloproliferative disorder, and some authors have advocated it for all patients with erythromelalgia unless there is a contraindication.7
Other possible first-line treatments include the synthetic prostaglandin E1 analogue misoprostol (Cytotec) and prostacyclins. Gabapentin (Neurontin), serotonin reuptake inhibitors such as sertaline (Zoloft) and venlafaxine (Effexor), and intravenous nitroprusside (Nitro-press) are considered second-line drugs.7
Surgical sympathectomy8 has also been tried, with variable results.
Outcomes in patients with erythromelalgia
In a case series from Mayo Clinic,2 approximately equal numbers of patients with erythromelalgia became worse, stayed the same, or got better, and the disease resolved in 10% over a mean of 8.7 years.
THE OUTCOME IN OUR PATIENT
We advised our patient to avoid strenuous activity in a warm environment and to work in cooler areas as much as possible. We told him to wrap his affected extremities with cold towels during attacks, and we prescribed aspirin (650 mg/day) for 3 months. The treatment did not cure his condition, but his symptoms lessened within 2 months. We later referred him to a pain clinic.
References
Kalgaard OM, Seem E, Kvernebo K. Erythromelalgia: a clinical study of 87 cases. Intern Med1997; 242:191–197.
Davis MD, O’Fallon WM, Rogers RS, Rooke TW. Natural history of erythromelalgia: presentation and outcome in 168 patients. Arch Dermatol2000; 136:330–336.
Galimberti D, Pontón A, Rubio L, et al. A case of primary erythromelalgia. J Eur Acad Dermatol Venereol2009; 23:1338–1339.
Mørk C, Kvernebo K, Asker CL, Salerud EG. Reduced skin capillary density during attacks of erythromelalgia implies arteriovenous shunting as pathogenetic mechanism. J Invest Dermatol2002; 119:949–953.
James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed.Philadelphia, PA: Saunders Elsevier; 2006.
Mørk C, Kvernebo K. Erythromelalgia. In:Lebwohl MG, Heymann WR, Berth-Jones J, Koulson Y. editors. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 3rd ed.Philadelphia, PA: Saunders Elsevier; 2010:236–238.
Seishima M, Kanoh H, Izumi T, et al. A refractory case of secondary erythermalgia successfully treated with lumbar sympathetic ganglion block. Br J Dermatol2000; 143:868–872.
References
Kalgaard OM, Seem E, Kvernebo K. Erythromelalgia: a clinical study of 87 cases. Intern Med1997; 242:191–197.
Davis MD, O’Fallon WM, Rogers RS, Rooke TW. Natural history of erythromelalgia: presentation and outcome in 168 patients. Arch Dermatol2000; 136:330–336.
Galimberti D, Pontón A, Rubio L, et al. A case of primary erythromelalgia. J Eur Acad Dermatol Venereol2009; 23:1338–1339.
Mørk C, Kvernebo K, Asker CL, Salerud EG. Reduced skin capillary density during attacks of erythromelalgia implies arteriovenous shunting as pathogenetic mechanism. J Invest Dermatol2002; 119:949–953.
James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 10th ed.Philadelphia, PA: Saunders Elsevier; 2006.
Mørk C, Kvernebo K. Erythromelalgia. In:Lebwohl MG, Heymann WR, Berth-Jones J, Koulson Y. editors. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 3rd ed.Philadelphia, PA: Saunders Elsevier; 2010:236–238.
Seishima M, Kanoh H, Izumi T, et al. A refractory case of secondary erythermalgia successfully treated with lumbar sympathetic ganglion block. Br J Dermatol2000; 143:868–872.
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Figure 1. The patient has hyperpigmentation of the skin creases on the palms, as well as on the lips and the lower gum.
At the time of presentation, he is hypotensive, with a blood pressure of 72/68 mm Hg, an irregular heart rate at 105/minute, and hyperpigmention of the gums, lips, and skin creases in his palms (Figure 1). The rest of the examination is normal.
Initial laboratory values are as follows:
White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
Mild anemia, with a hemoglobin of 116 g/L (140–175)
Activated partial thromboplastin time 59.9 sec (23.0–32.4)
Serum sodium 135 mmol/L (136–142)
Serum potassium 4.6 mmol/L (3.5–5.0)
Aspartate aminotransferase 58 U/L (10–30)
Alanine aminotransferase 16 U/L (10–40)
Alkaline phosphatase 328 U/L (30–120)
Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Figure 2. Computed tomography of the chest shows metastases in the lungs (A, white arrows) and liver (B, white arrows), bilateral pleural effusion (A, black arrows) and ascites (B, black arrows).
Echocardiography shows loculated fluids posterior to the left ventricle with no tamponade. Based on this finding, computed tomography (CT) of the chest is performed and demonstrates multiple small nodules in the lung parenchyma bilaterally, a finding consistent with metastatic disease with no visualized primary lung tumor mass. The same study also identifies multiple hypodense hepatic lesions with ascites surrounding the liver, another finding consistent with metastatic disease(Figure 2). Despite aggressive volume repletion, the patient remains hypotensive and symptomatic.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
Serum parathyroid-hormone-related protein
Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
Serum thyrotropin level
Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
Figure 3. Studies of biopsy samples confirm metastatic, poorly differentiated adenocarcinoma in the liver. The neoplastic cells form ill-defined, gland-like structures (arrowheads, panel A). The cells have atypical nuclei with abundant eosinophilic cytoplasm, and abnormal mitotic figures are present (arrowheads, panel B). Further immunoperoxidase staining was as follows: cytokeratin-7-positive; cytokeratin-20-positive; hepatocyte-specific-antigen-negative; TTF1-negative. These staining patterns indicated cholangiocarcinoma or pancreatic adeno-carcinoma as the possible primary tumor.
Liver biopsy confirmed metastatic, poorly differentiated adenocarcinoma, with cholangiocarcinoma and pancreatic adenocarcinoma possible primary tumors (Figure 3). The level of the tumor marker CA 19-9 was elevated at 4,628 U/mL (reference range 0–35), whereas levels of the markers CEA, CA-125, and prostate-specific antigen were normal.
Figure 4. Computed tomography of the abdomen showed enlarged adrenal glands (arrows).
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
References
Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol2009; 160:233–237.
Oelkers W. Adrenal insufficiency. N Engl J Med1996; 335:1206–1212.
Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer1987; 60:103–107.
Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev1949; 29:132.
Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician2003; 68:1955–1960.
Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol2000; 143:91–97.
Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer1990; 65:177–179.
Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet1981; 152:607–610.
Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J1978; 1:1591–1592.
Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer1984; 54:552–557.
Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol2010; 2010. pii:749239.
Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J2008; 32:1047–1052.
Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J2009; 102:665–667.
Khaldoon Shaheen, MD Department of Medicine, Case Western Reserve University–St. Vincent Charity Medical Center, Cleveland, OH
Abdul Hamid Alraiyes, MD, FCCP Department of Pulmonary, Critical Care, and Environmental Medicine, Tulane University Health Sciences Center, New Orleans, LA
M. Motaz Baibars, MD, FACP Department of Medicine, Case Western Reserve University–St. Vincent Charity Medical Center, Cleveland, OH
M. Chadi Alraies, MD, FACP Clinical Assistant Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Staff, Department of Hospital Medicine, Cleveland Clinic
Khaldoon Shaheen, MD Department of Medicine, Case Western Reserve University–St. Vincent Charity Medical Center, Cleveland, OH
Abdul Hamid Alraiyes, MD, FCCP Department of Pulmonary, Critical Care, and Environmental Medicine, Tulane University Health Sciences Center, New Orleans, LA
M. Motaz Baibars, MD, FACP Department of Medicine, Case Western Reserve University–St. Vincent Charity Medical Center, Cleveland, OH
M. Chadi Alraies, MD, FACP Clinical Assistant Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Staff, Department of Hospital Medicine, Cleveland Clinic
Khaldoon Shaheen, MD Department of Medicine, Case Western Reserve University–St. Vincent Charity Medical Center, Cleveland, OH
Abdul Hamid Alraiyes, MD, FCCP Department of Pulmonary, Critical Care, and Environmental Medicine, Tulane University Health Sciences Center, New Orleans, LA
M. Motaz Baibars, MD, FACP Department of Medicine, Case Western Reserve University–St. Vincent Charity Medical Center, Cleveland, OH
M. Chadi Alraies, MD, FACP Clinical Assistant Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Staff, Department of Hospital Medicine, Cleveland Clinic
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Figure 1. The patient has hyperpigmentation of the skin creases on the palms, as well as on the lips and the lower gum.
At the time of presentation, he is hypotensive, with a blood pressure of 72/68 mm Hg, an irregular heart rate at 105/minute, and hyperpigmention of the gums, lips, and skin creases in his palms (Figure 1). The rest of the examination is normal.
Initial laboratory values are as follows:
White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
Mild anemia, with a hemoglobin of 116 g/L (140–175)
Activated partial thromboplastin time 59.9 sec (23.0–32.4)
Serum sodium 135 mmol/L (136–142)
Serum potassium 4.6 mmol/L (3.5–5.0)
Aspartate aminotransferase 58 U/L (10–30)
Alanine aminotransferase 16 U/L (10–40)
Alkaline phosphatase 328 U/L (30–120)
Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Figure 2. Computed tomography of the chest shows metastases in the lungs (A, white arrows) and liver (B, white arrows), bilateral pleural effusion (A, black arrows) and ascites (B, black arrows).
Echocardiography shows loculated fluids posterior to the left ventricle with no tamponade. Based on this finding, computed tomography (CT) of the chest is performed and demonstrates multiple small nodules in the lung parenchyma bilaterally, a finding consistent with metastatic disease with no visualized primary lung tumor mass. The same study also identifies multiple hypodense hepatic lesions with ascites surrounding the liver, another finding consistent with metastatic disease(Figure 2). Despite aggressive volume repletion, the patient remains hypotensive and symptomatic.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
Serum parathyroid-hormone-related protein
Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
Serum thyrotropin level
Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
Figure 3. Studies of biopsy samples confirm metastatic, poorly differentiated adenocarcinoma in the liver. The neoplastic cells form ill-defined, gland-like structures (arrowheads, panel A). The cells have atypical nuclei with abundant eosinophilic cytoplasm, and abnormal mitotic figures are present (arrowheads, panel B). Further immunoperoxidase staining was as follows: cytokeratin-7-positive; cytokeratin-20-positive; hepatocyte-specific-antigen-negative; TTF1-negative. These staining patterns indicated cholangiocarcinoma or pancreatic adeno-carcinoma as the possible primary tumor.
Liver biopsy confirmed metastatic, poorly differentiated adenocarcinoma, with cholangiocarcinoma and pancreatic adenocarcinoma possible primary tumors (Figure 3). The level of the tumor marker CA 19-9 was elevated at 4,628 U/mL (reference range 0–35), whereas levels of the markers CEA, CA-125, and prostate-specific antigen were normal.
Figure 4. Computed tomography of the abdomen showed enlarged adrenal glands (arrows).
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
A 65-year-old man presents with a 2-month history of generalized weakness, dizziness, and blurred vision. His symptoms began gradually and have been progressing over the last few weeks, so that they now affect his ability to perform normal daily activities.
He has lost 20 lb and has become anorectic. He has no fever, night sweats, headache, cough, hemoptysis, or dyspnea. He has no history of abdominal pain, changes in bowel habits, nausea, vomiting, or urinary symptoms. He was admitted 6 weeks ago for the same symptoms; he was treated for hypotension and received intravenous (IV) fluids and electrolyte supplements for dehydration.
He has a history of hypertension, stroke, vascular dementia, and atrial fibrillation. He is taking warfarin (Coumadin), extended-release diltiazem (Cardizem), simvastatin (Zocor), and donepezil (Aricept). He underwent right hemicolectomy 5 years ago for a large tubular adenoma with high-grade dysplasia in the cecum.
Figure 1. The patient has hyperpigmentation of the skin creases on the palms, as well as on the lips and the lower gum.
At the time of presentation, he is hypotensive, with a blood pressure of 72/68 mm Hg, an irregular heart rate at 105/minute, and hyperpigmention of the gums, lips, and skin creases in his palms (Figure 1). The rest of the examination is normal.
Initial laboratory values are as follows:
White blood cell count 7.4 × 109/L (reference range 4.5–11.0), with a normal differential
Mild anemia, with a hemoglobin of 116 g/L (140–175)
Activated partial thromboplastin time 59.9 sec (23.0–32.4)
Serum sodium 135 mmol/L (136–142)
Serum potassium 4.6 mmol/L (3.5–5.0)
Aspartate aminotransferase 58 U/L (10–30)
Alanine aminotransferase 16 U/L (10–40)
Alkaline phosphatase 328 U/L (30–120)
Urea, creatinine, and corrected calcium are normal.
Electrocardiography shows atrial fibrillation with low-voltage QRS complexes. Chest radiography is normal. A stool test is negative for occult blood. A workup for sepsis is negative.
Figure 2. Computed tomography of the chest shows metastases in the lungs (A, white arrows) and liver (B, white arrows), bilateral pleural effusion (A, black arrows) and ascites (B, black arrows).
Echocardiography shows loculated fluids posterior to the left ventricle with no tamponade. Based on this finding, computed tomography (CT) of the chest is performed and demonstrates multiple small nodules in the lung parenchyma bilaterally, a finding consistent with metastatic disease with no visualized primary lung tumor mass. The same study also identifies multiple hypodense hepatic lesions with ascites surrounding the liver, another finding consistent with metastatic disease(Figure 2). Despite aggressive volume repletion, the patient remains hypotensive and symptomatic.
Q: Which is the appropriate test at this point to determine the cause of the hypotension?
Serum parathyroid-hormone-related protein
Baseline serum cortisol, plasma adrenocorticotropic hormone (ACTH) levels, and an ACTH stimulation test with cosyntropin (Cortrosyn)
Serum thyrotropin level
Aspiration biopsy of subcutaneous fat with Congo red and immunostaining
Late-night salivary cortisol
A: The correct next step is to measure baseline serum cortisol, to test ACTH levels, and to order an ACTH stimulation test with cosyntropin.
Primary adrenocortical insufficiency should be considered in patients with metastatic malignancy who present with peripheral vascular collapse, particularly when it is associated with cutaneous hyperpigmentation, chronic malaise, fatigue, weakness, anorexia, weight loss, hypoglycemia, and electrolyte disturbances such as hyponatremia and hyperkalemia.
Checking the baseline serum cortisol and ACTH levels and cosyntropin stimulation testing are vital steps in making an early diagnosis of primary adrenocortical insufficiency. Inappropriately low serum cortisol is highly suggestive of primary adrenal insufficiency, especially if accompanied by simultaneous elevation of the plasma ACTH level. The result of the ACTH stimulation test with cosyntropin is often confirmatory.
Measuring the serum parathyroid-hormone-related protein level is not indicated, since the patient has a normal corrected calcium. Patients with ectopic Cushing syndrome may present with weight loss due to underlying malignancy, but the presence of hypotension and a lack of hypokalemia makes such a diagnosis unlikely, and, therefore, measurement of late-night salivary cortisol is not the best answer. Amyloidosis, hypothyroidism, or hyperthyroidism are unlikely to have this patient’s presentation.
RESULTS OF FURTHER EVALUATION
Our patient’s ACTH serum level was elevated, and an ACTH stimulation test with cosyntropin confirmed the diagnosis of primary adrenal insufficiency.
Figure 3. Studies of biopsy samples confirm metastatic, poorly differentiated adenocarcinoma in the liver. The neoplastic cells form ill-defined, gland-like structures (arrowheads, panel A). The cells have atypical nuclei with abundant eosinophilic cytoplasm, and abnormal mitotic figures are present (arrowheads, panel B). Further immunoperoxidase staining was as follows: cytokeratin-7-positive; cytokeratin-20-positive; hepatocyte-specific-antigen-negative; TTF1-negative. These staining patterns indicated cholangiocarcinoma or pancreatic adeno-carcinoma as the possible primary tumor.
Liver biopsy confirmed metastatic, poorly differentiated adenocarcinoma, with cholangiocarcinoma and pancreatic adenocarcinoma possible primary tumors (Figure 3). The level of the tumor marker CA 19-9 was elevated at 4,628 U/mL (reference range 0–35), whereas levels of the markers CEA, CA-125, and prostate-specific antigen were normal.
Figure 4. Computed tomography of the abdomen showed enlarged adrenal glands (arrows).
CT of the abdomen failed to demonstrate primary tumors, but both adrenal glands were enlarged, likely from metastasis (Figure 4). His hypotension responded to treatment with hydrocortisone and fludrocortisone, and his symptoms resolved. No further testing or therapy was directed to the primary occult malignancy, as it was considered advanced. The prognosis was discussed with the patient, and he deferred any further management and was discharged to hospice care. He died a few months later.
PRIMARY ADRENOCORTICAL INSUFFICIENCY
Primary adrenocortical insufficiency is an uncommon disorder caused by destruction or dysfunction of the adrenal cortices. It is characterized by chronic deficiency of cortisol, aldosterone, and adrenal androgens. In the United States, nearly 6 million people are considered to have undiagnosed adrenal insufficiency, which is clinically significant only during times of physiologic stress.1
Primary adrenocortical insufficiency affects men and women equally. However, the idiopathic autoimmune form of adrenal insufficiency (Addison disease) is two to three times more common in women than in men.
If the condition is undiagnosed or ineffectively treated, the risk of significant morbidity and death is high. Symptoms and signs are nonspecific, and the onset is insidious.
Almost all patients with primary adrenal insufficiency have malaise, fatigue, anorexia, and weight loss. Vomiting, abdominal pain, and fever are more common during an adrenal crisis, when a patient with subclinical disease is subjected to major stress. Postural dizziness or syncope is a common result of volume depletion and hypotension.2–4 It is commonly accompanied by hyponatremia and hyperkalemia.
Hyperpigmentation is the most characteristic physical finding and is caused by an ACTH-mediated increase in melanin content in the skin.2,4,5 The resulting brown hyperpigmentation is most obvious in areas exposed to sunlight (face, neck, backs of hands), and in areas exposed to chronic friction or pressure, such as the elbows, knees, knuckles, waist, and shoulders (brassiere straps).4 Pigmentation is also prominent in the palmar creases, areolae, axillae, perineum, surgical scars, and umbilicus. Other patterns of hyperpigmentation are patchy pigmentation on the inner surface of lips, the buccal mucosa, under the tongue, and on the hard palate.3,5 The hyperpigmentation begins to fade within several days and largely disappears after a few months of adequate glucocorticoid therapy.4
In the United States, 80% of cases of primary adrenocortical insufficiency are caused by autoimmune adrenal destruction. The remainder are caused by infectious diseases (eg, tuberculosis, fungal infection, cytomegalovirus infection, and Mycobacterium aviumintracellulare infection in the context of human immunodeficiency virus infection), by infiltration of the adrenal glands by metastatic cancer, by adrenal hemorrhage, or by drugs such as ketoconazole, fluconazole (Diflucan), metyrapone (Metopirone), mitotane (Lysodren), and etomidate (Amidate).4,6
Adrenal metastatic disease
Infiltration of the adrenal glands by metastatic cancer is not uncommon, probably because of their rich sinusoidal blood supply, and the adrenals are the fourth most common site of metastasis. Common primary tumors are lung, breast, melanoma, gastric, esophageal, and colorectal cancers, while metastasis due to an undetermined primary tumor is the least common.7
Clinically evident adrenal insufficiency produced by metastatic carcinoma is uncommon because most of the adrenal cortex must be destroyed before hypofunction becomes evident.7–9
Malignancy rarely presents first as adrenal insufficiency caused by metastatic infiltration.10
Hormonal therapy may significantly improve symptoms and quality of life in patients with metastatic adrenal insufficiency.8,11
DIAGNOSIS AND MANAGEMENT
Once primary adrenal insufficiency is suspected, prompt diagnosis and treatment are essential. A low plasma cortisol level (< 3 μg/dL) at 8 am is highly suggestive of adrenal insufficiency if exposure to exogenous glucocorticoids has been excluded (including oral, inhaled, and injected),12,13 especially if accompanied by simultaneous elevation of the plasma ACTH level (usually > 200 pg/mL). An 8 am cortisol concentration above 15 μg/dL makes adrenal insufficiency highly unlikely, but levels between 3 and 15 μg/dL are nondiagnostic and need to be further evaluated by an ACTH stimulation test with cosyntropin.4,7
Imaging in primary adrenal insufficiency may be considered when the condition is not clearly autoimmune.14 Abdominal CT is the ideal imaging test for detecting abnormal adrenal glands. CT shows small, noncalcified adrenals in autoimmune Addison disease. It demonstrates enlarged adrenals in about 85% of cases caused by metastatic or granulomatous disease; and calcification is noted in cases of tuberculous adrenal disease.4
Management involves treating the underlying cause and starting hormone replacement therapy. Hormonal therapy consists of corticosteroids and mineralocorticoids; hydrocortisone is the drug of choice and is usually given with fludrocortisone acetate, which has a potent sodium-retaining effect. In the presence of a stressor (fever, surgery, severe illness), the dose of hydrocortisone should be doubled (> 50 mg hydrocortisone per day) for at least 3 to 5 days.2,4
References
Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol2009; 160:233–237.
Oelkers W. Adrenal insufficiency. N Engl J Med1996; 335:1206–1212.
Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer1987; 60:103–107.
Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev1949; 29:132.
Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician2003; 68:1955–1960.
Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol2000; 143:91–97.
Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer1990; 65:177–179.
Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet1981; 152:607–610.
Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J1978; 1:1591–1592.
Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer1984; 54:552–557.
Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol2010; 2010. pii:749239.
Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J2008; 32:1047–1052.
Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J2009; 102:665–667.
References
Erichsen MM, Løvås K, Fougner KJ, et al. Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. Eur J Endocrinol2009; 160:233–237.
Oelkers W. Adrenal insufficiency. N Engl J Med1996; 335:1206–1212.
Redman BG, Pazdur R, Zingas AP, Loredo R. Prospective evaluation of adrenal insufficiency in patients with adrenal metastasis. Cancer1987; 60:103–107.
Berger M., Hypofunction of the adrenal cortex in infancy. Manit Med Rev1949; 29:132.
Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, café au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician2003; 68:1955–1960.
Lutz A, Stojkovic M, Schmidt M, Arlt W, Allolio B, Reincke M. Adrenocortical function in patients with macrometastases of the adrenal gland. Eur J Endocrinol2000; 143:91–97.
Kung AW, Pun KK, Lam K, Wang C, Leung CY. Addisonian crisis as presenting feature in malignancies. Cancer1990; 65:177–179.
Cedermark BJ, Sjöberg HE. The clinical significance of metastases to the adrenal glands. Surg Gynecol Obstet1981; 152:607–610.
Rosenthal FD, Davies MK, Burden AC. Malignant disease presenting as Addison’s disease. Br Med J1978; 1:1591–1592.
Seidenwurm DJ, Elmer EB, Kaplan LM, Williams EK, Morris DG, Hoffman AR. Metastases to the adrenal glands and the development of Addison’s disease. Cancer1984; 54:552–557.
Santiago AH, Ratzan S. Acute adrenal crisis in an asthmatic child treated with inhaled fluticasone proprionate. Int J Pediatr Endocrinol2010; 2010. pii:749239.
Holme J, Tomlinson JW, Stockley RA, Stewart PM, Barlow N, Sullivan AL. Adrenal suppression in bronchiectasis and the impact of inhaled corticosteroids. Eur Respir J2008; 32:1047–1052.
Mohammad K, Sadikot RT. Adrenal insufficiency as a presenting manifestation of nonsmall cell lung cancer. South Med J2009; 102:665–667.
Figure 1. A firm, pink nodule, 10 mm in diameter, with surrounding telangiectasias, at the site of a previous mosquito bite.
An otherwise healthy 54-year-old woman presented with a 3-month history of an asymptomatic lesion on the face, at the site of a previous mosquito bite. Physical examination revealed a firm, pink nodule 10 mm in diameter (Figure 1). Telangiectasias were visible, more clearly by dermoscopy, but other features of a basal cell carcinoma were absent. No other skin problem was noted, and no lymphadenopathy was detected.
Figure 2. In A, dense chronic inflammation (arrows) is noted in the upper and deeper dermis (hematoxylin-eosin, × 4). Immunohistochemical staining shows, in B, CD20 B lymphocytes (arrow) with a central predominance (× 40); in C, a smaller number of CD3 T lymphocytes (arrow) are present at the periphery of rudimentary germinal centers (× 40).
Histologic study of a biopsy specimen noted a dense dermal inflammatory infiltrate consisting of B lymphocytes (CD20+) with a smaller number of T lymphocytes (CD3+) arranged in several germinal centers, with an admixture of plasma cells and eosinophils (Figure 2). No clonal population was identified by gene-rearrangement studies. Borrelia burgdorferi infection was ruled out.
Q: Which is the most likely diagnosis?
Basal cell carcinoma
Squamous cell carcinoma
Lymphocytoma cutis
Amelanotic melanoma
Pyogenic granuloma
A: The correct answer is lymphocytoma cutis. The differential diagnosis of a pink papule on the face of a middle-aged person includes nonmelanoma skin cancer, lymphoma, lymphocytoma cutis, metastatic disease, certain infections, Jessner lymphocytic infiltrate, connective tissue disease, and some adnexal tumors. Histologic study is a useful diagnostic aid in this context.
Basal cell carcinoma is the most common cutaneous malignant neoplasm, and although these tumors rarely metastasize, they are capable of gross tissue destruction, particularly those lesions arising on the face. Clinically, this tumor presents as a shiny, pearly nodule with telangiectasias on the surface, as in our patient, but skin biopsy shows large basaloid lobules of varying shape and size forming a relatively circumscribed mass with a “palisade” around the rim of the lobule.
Squamous cell carcinoma manifests as shallow ulcers, often with a keratinous crust and elevated, indurate borders, but also as plaques or nodules. The clinical diagnosis should be confirmed with skin biopsy, which reveals atypical keratinocytes extending from the epidermis to the dermis with dyskeratosis, intercellular bridges, variable central keratinization, and horn pearl formation, depending on the differentiation of the tumor.
Amelanotic melanoma is nonpigmented and appears as a pink nodule mimicking basal cell carcinoma or squamous cell carcinoma. Histologic study is necessary for the diagnosis, and shows an atypical proliferation of melanocytic cells in the epidermis and dermis.
Pyogenic granuloma is a very common benign vascular lesion considered to be a hyperplastic process or a vascular neoplasm. The lesion typically presents as a red or bluish papule or polyp that bleeds easily, and a reddish homogeneous area surrounded by a white “collarette” is found in most cases. Histologic features of an early lesion resemble granulation tissue and include lobules of capillaries and venules that often radiate from larger, more central vessels.
LYMPHOCYTOMA CUTIS: KEY FEATURES
Lymphocytoma cutis (pseudolymphoma) is a benign reactive polyclonal and inflammatory disorder that most frequently includes B lymphocytes, with a smaller population of T lymphocytes. It infiltrates the skin and resembles rudimentary germinal follicles, as in the present case. The lesion usually presents as an asymptomatic red-brown or violet papule or nodule, 3 mm to 5 cm in diameter, most often on the face, chest, or upper extremities.1 The lesion may be solitary, as in our patient, but lesions may also be grouped or numerous and widespread. It is three times more common in women than in men. It may resolve spontaneously, but it may also recur.
In Europe, lymphocytoma cutis occurs most often in B burgdorferi infection after a tick bite. Lymphocytoma cutis occurs in 1.3% of cases of B burgdorferi infection,2 although other infectious, physical, or chemical agents may produce the same reaction pattern. Tattooing (particularly red areas), acupuncture, vaccination, arthropod reactions, hyposensitization antigen reaction, and ingestion of drug have been implicated in this form of lymphoid hyperplasia.3,4
DIAGNOSTIC CHALLENGES
Lymphocytoma cutis can be challenging to diagnose, and although it can be suspected clinically, incisional biopsy is usually necessary in order to differentiate it from cutaneous B lymphoma.5
The infiltrate is predominantly nodular (> 90%) and located in the upper and mid dermis (“top heavy”) in lymphocytoma cutis, whereas it can be nodular or diffuse in cutaneous B lymphoma, with sharply demarcated borders that are convex rather than concave. Lymphoid follicles with germinal centers are sometimes present, and the interfollicular cellular population is polymorphic in lymphocytoma cutis (lymphocytes, plasma cells, histiocytes, eosinophils). In lymphocytoma cutis, cells express the phenotype of mature B lymphocytes (CD20, CD79a) and show regular and sharply demarcated networks of CD21+ follicular dendritic cells, whereas in cutaneous B lymphoma these networks are irregular. Light chains are usually polyclonal, although monoclonal populations of B cell in cases of cutaneous lymphocytoma cutis have been described. Extracutaneous involvement is possible in cutaneous B lymphoma but is usually absent in lymphocytoma cutis.
Lymphocytoma cutis typically involutes over a period of months, even with no treatment, as it did in our patient. Otherwise, there are different therapeutic options, including intralesional and topical corticosteroids, surgery, and cryosurgery.6 Photodynamic therapy with delta-aminolevulinic acid is an effective and safe modality for the treatment of lymphocytoma cutis and may be cosmetically beneficial.7
Albrecht S, Hofstadter S, Artsob H, Chaban O, From L. Lymphadenosis benigna cutis resulting from Borrelia infection (Borrelia lymphocytoma). J Am Acad Dermatol1991; 24:621–625.
Peretz E, Grunwald MH, Cagnano E, Halevy S. Follicular B-cell pseudolymphoma. Australas J Dermatol2000; 41:48–49.
Hermes B, Haas N, Grabbe J, Czarnetzki BM. Foreign-body granuloma and IgE-pseudolymphoma after multiple bee stings. Br J Dermatol1994; 130:780–784.
Kerl H, Fink-Puches R, Cerroni L. Diagnostic criteria of primary cutaneous B-cell lymphomas and pseudolymphomas. Keio J Med2001; 50:269–273.
Kuflik AS, Schwartz RA. Lymphocytoma cutis: a series of five patients successfully treated with cryosurgery. J Am Acad Dermatol1992; 26:449–452.
Takeda H, Kaneko T, Harada K, Matsuzaki Y, Nakano H, Hanada K. Successful treatment of lymphadenosis benigna cutis with topical photodynamic therapy with delta-aminolevulinic acid. Dermatology2005; 211:264–266.
Salvador Arias-Santiago, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
José Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Antonio Cutando, PhD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Agustín Buendía-Eisman, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Ramón Naranjo-Sintes, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Antonio Campos, PhD Department of Histology, School of Medicine, Granada, Spain
Miguel Alaminos-Mingorance Department of Histology, School of Medicine, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Olóriz 16, Granada 18012, Spain; e-mail [email protected]
Salvador Arias-Santiago, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
José Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Antonio Cutando, PhD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Agustín Buendía-Eisman, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Ramón Naranjo-Sintes, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Antonio Campos, PhD Department of Histology, School of Medicine, Granada, Spain
Miguel Alaminos-Mingorance Department of Histology, School of Medicine, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Olóriz 16, Granada 18012, Spain; e-mail [email protected]
Author and Disclosure Information
Salvador Arias-Santiago, MD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
José Aneiros-Fernández, MD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Antonio Cutando, PhD Department of Pathology, San Cecilio University Hospital, Granada, Spain
Agustín Buendía-Eisman, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Ramón Naranjo-Sintes, PhD Department of Dermatology, San Cecilio University Hospital, Granada, Spain
Antonio Campos, PhD Department of Histology, School of Medicine, Granada, Spain
Miguel Alaminos-Mingorance Department of Histology, School of Medicine, Granada, Spain
Address: Salvador Arias-Santiago, MD, Department of Dermatology, San Cecilio University Hospital, Av Dr. Olóriz 16, Granada 18012, Spain; e-mail [email protected]
Figure 1. A firm, pink nodule, 10 mm in diameter, with surrounding telangiectasias, at the site of a previous mosquito bite.
An otherwise healthy 54-year-old woman presented with a 3-month history of an asymptomatic lesion on the face, at the site of a previous mosquito bite. Physical examination revealed a firm, pink nodule 10 mm in diameter (Figure 1). Telangiectasias were visible, more clearly by dermoscopy, but other features of a basal cell carcinoma were absent. No other skin problem was noted, and no lymphadenopathy was detected.
Figure 2. In A, dense chronic inflammation (arrows) is noted in the upper and deeper dermis (hematoxylin-eosin, × 4). Immunohistochemical staining shows, in B, CD20 B lymphocytes (arrow) with a central predominance (× 40); in C, a smaller number of CD3 T lymphocytes (arrow) are present at the periphery of rudimentary germinal centers (× 40).
Histologic study of a biopsy specimen noted a dense dermal inflammatory infiltrate consisting of B lymphocytes (CD20+) with a smaller number of T lymphocytes (CD3+) arranged in several germinal centers, with an admixture of plasma cells and eosinophils (Figure 2). No clonal population was identified by gene-rearrangement studies. Borrelia burgdorferi infection was ruled out.
Q: Which is the most likely diagnosis?
Basal cell carcinoma
Squamous cell carcinoma
Lymphocytoma cutis
Amelanotic melanoma
Pyogenic granuloma
A: The correct answer is lymphocytoma cutis. The differential diagnosis of a pink papule on the face of a middle-aged person includes nonmelanoma skin cancer, lymphoma, lymphocytoma cutis, metastatic disease, certain infections, Jessner lymphocytic infiltrate, connective tissue disease, and some adnexal tumors. Histologic study is a useful diagnostic aid in this context.
Basal cell carcinoma is the most common cutaneous malignant neoplasm, and although these tumors rarely metastasize, they are capable of gross tissue destruction, particularly those lesions arising on the face. Clinically, this tumor presents as a shiny, pearly nodule with telangiectasias on the surface, as in our patient, but skin biopsy shows large basaloid lobules of varying shape and size forming a relatively circumscribed mass with a “palisade” around the rim of the lobule.
Squamous cell carcinoma manifests as shallow ulcers, often with a keratinous crust and elevated, indurate borders, but also as plaques or nodules. The clinical diagnosis should be confirmed with skin biopsy, which reveals atypical keratinocytes extending from the epidermis to the dermis with dyskeratosis, intercellular bridges, variable central keratinization, and horn pearl formation, depending on the differentiation of the tumor.
Amelanotic melanoma is nonpigmented and appears as a pink nodule mimicking basal cell carcinoma or squamous cell carcinoma. Histologic study is necessary for the diagnosis, and shows an atypical proliferation of melanocytic cells in the epidermis and dermis.
Pyogenic granuloma is a very common benign vascular lesion considered to be a hyperplastic process or a vascular neoplasm. The lesion typically presents as a red or bluish papule or polyp that bleeds easily, and a reddish homogeneous area surrounded by a white “collarette” is found in most cases. Histologic features of an early lesion resemble granulation tissue and include lobules of capillaries and venules that often radiate from larger, more central vessels.
LYMPHOCYTOMA CUTIS: KEY FEATURES
Lymphocytoma cutis (pseudolymphoma) is a benign reactive polyclonal and inflammatory disorder that most frequently includes B lymphocytes, with a smaller population of T lymphocytes. It infiltrates the skin and resembles rudimentary germinal follicles, as in the present case. The lesion usually presents as an asymptomatic red-brown or violet papule or nodule, 3 mm to 5 cm in diameter, most often on the face, chest, or upper extremities.1 The lesion may be solitary, as in our patient, but lesions may also be grouped or numerous and widespread. It is three times more common in women than in men. It may resolve spontaneously, but it may also recur.
In Europe, lymphocytoma cutis occurs most often in B burgdorferi infection after a tick bite. Lymphocytoma cutis occurs in 1.3% of cases of B burgdorferi infection,2 although other infectious, physical, or chemical agents may produce the same reaction pattern. Tattooing (particularly red areas), acupuncture, vaccination, arthropod reactions, hyposensitization antigen reaction, and ingestion of drug have been implicated in this form of lymphoid hyperplasia.3,4
DIAGNOSTIC CHALLENGES
Lymphocytoma cutis can be challenging to diagnose, and although it can be suspected clinically, incisional biopsy is usually necessary in order to differentiate it from cutaneous B lymphoma.5
The infiltrate is predominantly nodular (> 90%) and located in the upper and mid dermis (“top heavy”) in lymphocytoma cutis, whereas it can be nodular or diffuse in cutaneous B lymphoma, with sharply demarcated borders that are convex rather than concave. Lymphoid follicles with germinal centers are sometimes present, and the interfollicular cellular population is polymorphic in lymphocytoma cutis (lymphocytes, plasma cells, histiocytes, eosinophils). In lymphocytoma cutis, cells express the phenotype of mature B lymphocytes (CD20, CD79a) and show regular and sharply demarcated networks of CD21+ follicular dendritic cells, whereas in cutaneous B lymphoma these networks are irregular. Light chains are usually polyclonal, although monoclonal populations of B cell in cases of cutaneous lymphocytoma cutis have been described. Extracutaneous involvement is possible in cutaneous B lymphoma but is usually absent in lymphocytoma cutis.
Lymphocytoma cutis typically involutes over a period of months, even with no treatment, as it did in our patient. Otherwise, there are different therapeutic options, including intralesional and topical corticosteroids, surgery, and cryosurgery.6 Photodynamic therapy with delta-aminolevulinic acid is an effective and safe modality for the treatment of lymphocytoma cutis and may be cosmetically beneficial.7
Figure 1. A firm, pink nodule, 10 mm in diameter, with surrounding telangiectasias, at the site of a previous mosquito bite.
An otherwise healthy 54-year-old woman presented with a 3-month history of an asymptomatic lesion on the face, at the site of a previous mosquito bite. Physical examination revealed a firm, pink nodule 10 mm in diameter (Figure 1). Telangiectasias were visible, more clearly by dermoscopy, but other features of a basal cell carcinoma were absent. No other skin problem was noted, and no lymphadenopathy was detected.
Figure 2. In A, dense chronic inflammation (arrows) is noted in the upper and deeper dermis (hematoxylin-eosin, × 4). Immunohistochemical staining shows, in B, CD20 B lymphocytes (arrow) with a central predominance (× 40); in C, a smaller number of CD3 T lymphocytes (arrow) are present at the periphery of rudimentary germinal centers (× 40).
Histologic study of a biopsy specimen noted a dense dermal inflammatory infiltrate consisting of B lymphocytes (CD20+) with a smaller number of T lymphocytes (CD3+) arranged in several germinal centers, with an admixture of plasma cells and eosinophils (Figure 2). No clonal population was identified by gene-rearrangement studies. Borrelia burgdorferi infection was ruled out.
Q: Which is the most likely diagnosis?
Basal cell carcinoma
Squamous cell carcinoma
Lymphocytoma cutis
Amelanotic melanoma
Pyogenic granuloma
A: The correct answer is lymphocytoma cutis. The differential diagnosis of a pink papule on the face of a middle-aged person includes nonmelanoma skin cancer, lymphoma, lymphocytoma cutis, metastatic disease, certain infections, Jessner lymphocytic infiltrate, connective tissue disease, and some adnexal tumors. Histologic study is a useful diagnostic aid in this context.
Basal cell carcinoma is the most common cutaneous malignant neoplasm, and although these tumors rarely metastasize, they are capable of gross tissue destruction, particularly those lesions arising on the face. Clinically, this tumor presents as a shiny, pearly nodule with telangiectasias on the surface, as in our patient, but skin biopsy shows large basaloid lobules of varying shape and size forming a relatively circumscribed mass with a “palisade” around the rim of the lobule.
Squamous cell carcinoma manifests as shallow ulcers, often with a keratinous crust and elevated, indurate borders, but also as plaques or nodules. The clinical diagnosis should be confirmed with skin biopsy, which reveals atypical keratinocytes extending from the epidermis to the dermis with dyskeratosis, intercellular bridges, variable central keratinization, and horn pearl formation, depending on the differentiation of the tumor.
Amelanotic melanoma is nonpigmented and appears as a pink nodule mimicking basal cell carcinoma or squamous cell carcinoma. Histologic study is necessary for the diagnosis, and shows an atypical proliferation of melanocytic cells in the epidermis and dermis.
Pyogenic granuloma is a very common benign vascular lesion considered to be a hyperplastic process or a vascular neoplasm. The lesion typically presents as a red or bluish papule or polyp that bleeds easily, and a reddish homogeneous area surrounded by a white “collarette” is found in most cases. Histologic features of an early lesion resemble granulation tissue and include lobules of capillaries and venules that often radiate from larger, more central vessels.
LYMPHOCYTOMA CUTIS: KEY FEATURES
Lymphocytoma cutis (pseudolymphoma) is a benign reactive polyclonal and inflammatory disorder that most frequently includes B lymphocytes, with a smaller population of T lymphocytes. It infiltrates the skin and resembles rudimentary germinal follicles, as in the present case. The lesion usually presents as an asymptomatic red-brown or violet papule or nodule, 3 mm to 5 cm in diameter, most often on the face, chest, or upper extremities.1 The lesion may be solitary, as in our patient, but lesions may also be grouped or numerous and widespread. It is three times more common in women than in men. It may resolve spontaneously, but it may also recur.
In Europe, lymphocytoma cutis occurs most often in B burgdorferi infection after a tick bite. Lymphocytoma cutis occurs in 1.3% of cases of B burgdorferi infection,2 although other infectious, physical, or chemical agents may produce the same reaction pattern. Tattooing (particularly red areas), acupuncture, vaccination, arthropod reactions, hyposensitization antigen reaction, and ingestion of drug have been implicated in this form of lymphoid hyperplasia.3,4
DIAGNOSTIC CHALLENGES
Lymphocytoma cutis can be challenging to diagnose, and although it can be suspected clinically, incisional biopsy is usually necessary in order to differentiate it from cutaneous B lymphoma.5
The infiltrate is predominantly nodular (> 90%) and located in the upper and mid dermis (“top heavy”) in lymphocytoma cutis, whereas it can be nodular or diffuse in cutaneous B lymphoma, with sharply demarcated borders that are convex rather than concave. Lymphoid follicles with germinal centers are sometimes present, and the interfollicular cellular population is polymorphic in lymphocytoma cutis (lymphocytes, plasma cells, histiocytes, eosinophils). In lymphocytoma cutis, cells express the phenotype of mature B lymphocytes (CD20, CD79a) and show regular and sharply demarcated networks of CD21+ follicular dendritic cells, whereas in cutaneous B lymphoma these networks are irregular. Light chains are usually polyclonal, although monoclonal populations of B cell in cases of cutaneous lymphocytoma cutis have been described. Extracutaneous involvement is possible in cutaneous B lymphoma but is usually absent in lymphocytoma cutis.
Lymphocytoma cutis typically involutes over a period of months, even with no treatment, as it did in our patient. Otherwise, there are different therapeutic options, including intralesional and topical corticosteroids, surgery, and cryosurgery.6 Photodynamic therapy with delta-aminolevulinic acid is an effective and safe modality for the treatment of lymphocytoma cutis and may be cosmetically beneficial.7
Albrecht S, Hofstadter S, Artsob H, Chaban O, From L. Lymphadenosis benigna cutis resulting from Borrelia infection (Borrelia lymphocytoma). J Am Acad Dermatol1991; 24:621–625.
Peretz E, Grunwald MH, Cagnano E, Halevy S. Follicular B-cell pseudolymphoma. Australas J Dermatol2000; 41:48–49.
Hermes B, Haas N, Grabbe J, Czarnetzki BM. Foreign-body granuloma and IgE-pseudolymphoma after multiple bee stings. Br J Dermatol1994; 130:780–784.
Kerl H, Fink-Puches R, Cerroni L. Diagnostic criteria of primary cutaneous B-cell lymphomas and pseudolymphomas. Keio J Med2001; 50:269–273.
Kuflik AS, Schwartz RA. Lymphocytoma cutis: a series of five patients successfully treated with cryosurgery. J Am Acad Dermatol1992; 26:449–452.
Takeda H, Kaneko T, Harada K, Matsuzaki Y, Nakano H, Hanada K. Successful treatment of lymphadenosis benigna cutis with topical photodynamic therapy with delta-aminolevulinic acid. Dermatology2005; 211:264–266.
Albrecht S, Hofstadter S, Artsob H, Chaban O, From L. Lymphadenosis benigna cutis resulting from Borrelia infection (Borrelia lymphocytoma). J Am Acad Dermatol1991; 24:621–625.
Peretz E, Grunwald MH, Cagnano E, Halevy S. Follicular B-cell pseudolymphoma. Australas J Dermatol2000; 41:48–49.
Hermes B, Haas N, Grabbe J, Czarnetzki BM. Foreign-body granuloma and IgE-pseudolymphoma after multiple bee stings. Br J Dermatol1994; 130:780–784.
Kerl H, Fink-Puches R, Cerroni L. Diagnostic criteria of primary cutaneous B-cell lymphomas and pseudolymphomas. Keio J Med2001; 50:269–273.
Kuflik AS, Schwartz RA. Lymphocytoma cutis: a series of five patients successfully treated with cryosurgery. J Am Acad Dermatol1992; 26:449–452.
Takeda H, Kaneko T, Harada K, Matsuzaki Y, Nakano H, Hanada K. Successful treatment of lymphadenosis benigna cutis with topical photodynamic therapy with delta-aminolevulinic acid. Dermatology2005; 211:264–266.