User login
UPDATE: OVARIAN CANCER
- Risk of 3 types of ovarian cancer higher in women who have endometriosis
Janelle Yates, Senior Editor (March 2012) - Ovarian stimulation ups risk of ovarian tumors in later life
(Web exclusive, November 2011) - Can a novel risk-scoring system for ovarian cancer predict who is most likely to develop disease?
William H. Parker, MD (Examining the Evidence, July 2011) - Update on Ovarian Cancer
Nora Kizer, MD, MSCI; David G. Mutch, MD (July 2011)
Dr. Mutch reports receiving research support from the National Institutes of Health and the Gynecologic Oncology Group but no financial relationships relevant to this article. Dr. Novetsky reports no financial relationships relevant to this article.
To improve outcomes in women who have ovarian cancer—the deadliest gynecologic malignancy in the United States—we need to pursue a number of investigative approaches:
- We need to determine how to diagnose the disease in its early stages. At present, fewer than 20% of ovarian cancer cases are identified while disease is localized to the adnexae. Although a symptom index has been suggested as a useful tool to highlight women at risk for ovarian cancer, its appropriate implementation and effectiveness have yet to be determined.1 Moreover, the symptoms of ovarian cancer are vague and may not become apparent until after the disease has metastasized. Might screening trials detect ovarian cancer in its earlier stages? Are there harms involved in screening women with transvaginal ultrasonography (TVUS) and cancer antigen (CA) 125?
- We need new first-line agents to treat ovarian cancer. Traditional therapy is surgical cytoreduction followed by platinum-based chemotherapy. More recently, the addition of intraperitoneal chemotherapy has prolonged survival by approximately 16 months in women who have advanced disease.2 Despite this advance, the relapse rate remains high. What new therapies can we offer in addition to traditional platinum-based chemotherapy?
- Ovarian cancer recurs in most women, and the response to subsequent therapy is short-lived.3,4 Novel biologic agents may offer new hope as a means of treating recurrent disease with greater specificity and lower toxicity. Do any biologic agents increase survival and reduce the toxicity of treatment?
In this article, we highlight notable studies published in the past year that address these questions.
Screening for ovarian cancer is not useful in average-risk women
Buys SS, Partridge E, Black A, et al; PLCO Project Team. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Randomized Screening Trial. JAMA. 2011;305(22):2295–2303.
Gilbert L, Basso O, Sampalis J, et al; DOvE Study Group. Assessment of symptomatic women for early diagnosis of ovarian cancer: results from the prospective DOvE pilot project. Lancet Oncol. 2012;13(3):285–291.
The Prostate, Lung, Colorectal and Ovarian (PLCO) trial addresses the utility of screening for ovarian cancer using two readily available tests—TVUS and CA 125.
Effective screening for any disease requires the following:
- The disease must have a presymptomatic stage during which diagnosis leads to better outcomes (compared with waiting until the onset of symptoms)
- The screening test must be acceptable to the population in which it is used
- The test must lead to a reduction in morbidity and mortality that outweighs the harms of false-positive tests
- The benefits of the test must be achieved at an acceptable level of risk.5
The PLCO trial was designed to determine the effect of specific ovarian cancer screening tests on cause-specific mortality. Women aged 55 to 74 years were randomly assigned to annual screening with TVUS and CA 125 or to standard gynecologic care. A CA-125 level of 35 or higher was classified as abnormal, as were TVUS findings of enlarged ovaries or solid or papillary components. The trial had 88% power to detect a 35% reduction in ovarian cancer mortality using a one-side a of 0.05.
Early results demonstrated that a large number of surgeries would be required to detect one case of ovarian cancer. Mortality data from the study only recently matured.6
Details of the PLCO trial
Approximately 39,000 women were allocated to each arm of the PLCO trial and followed for 6 years. Ovarian cancer was diagnosed in 212 and 176 women in the screening and usual-care groups, respectively (relative risk, 1.21; 95% confidence interval [CI], 0.99–1.48). Equal percentages of women in each group were given a diagnosis of Stage III/IV cancer.
No survival benefit was seen in the screening group. Overall, 3,285 women had a false-positive screening test, with 33% undergoing surgery (21% surgical complication rate).
Barriers to effective screening
One of the major obstacles to the development of an accurate screening test for ovarian cancer has been the low prevalence of the disease. The relationship between sensitivity, specificity, prevalence, and positive predictive value (PPV) is demonstrated in the TABLE. With sensitivity of 100% and specificity of 95%, the PPV for ovarian cancer screening with TVUS and CA 125 is only 1%. Ovarian cancer has an annual prevalence of approximately 1 case in every 2,500 women.
Effect of disease prevalence on population-based screening
| Specificity of the screening test (%) | Positive predictive value (%) | |||
|---|---|---|---|---|
| 50% prevalence | 10% prevalence | 1% prevalence | Ovarian cancer (1 case in every 2,500 women) | |
| 90 | 91 | 53 | 9 | 0.4 |
| 95 | 95 | 69 | 17 | 0.8 |
| 99 | 99 | 92 | 50 | 4 |
| 99.9 | 99.9 | 99 | 91 | 29 |
Details of the trial by Gilbert and colleagues
In a recent pilot study, Gilbert and colleagues prospectively analyzed the utility of disseminating information about the symptoms of ovarian cancer to the general public. Following dissemination of information, women who were 50 years or older and who had experienced at least one ovarian cancer symptom longer than 2 weeks underwent CA-125 testing and TVUS. If both tests were normal, CA 125 was repeated; if it was normal again, the patient was discharged from the study.
Patients who had abnormal findings on either test repeated both screening tests, with additional testing performed as necessary. Outcomes of these women were compared with those of women who had been referred to the gynecologic oncology clinic.
Among 1,455 eligible patients, 11 cases of invasive ovarian cancer were diagnosed, four of which (36%) represented early-stage disease. Median CA-125 levels were lower in the study group. In addition, more women in the study group had early-stage disease that was completely resectable, compared with the clinic group, although this difference did not reach statistical significance.
Most cases of ovarian cancer in this study originated in the fallopian tube or peritoneum—not the ovary. In addition to the cases of ovarian cancer, 11 cases of uterine cancer were diagnosed. No patients underwent unwarranted major surgery, and none who were discharged from the study had a diagnosis of gynecologic cancer by 7 months of follow-up.
TVUS and CA 125 increase the number of cases of ovarian cancer that are diagnosed but do not provide a survival benefit. Identification of cases in the PLCO trial was, therefore, likely the result of lead-time bias—cases of ovarian cancer were detected sooner but not at an early stage.
Routine screening with TVUS and CA 125 are not recommended at this time for women at average risk for ovarian cancer. These and similar screening methods lead to a significant false-positive rate, with surgeries performed for benign indications and a high risk of surgical complications. Further studies to improve the sensitivity and specificity of these tests in women at average risk for ovarian cancer are ongoing.7
Women who have an elevated risk of ovarian cancer, such as women who carry the BRCA mutation or who have a family history of ovarian cancer, may benefit from routine screening because of increased disease prevalence in this population, although studies are needed to determine the best utilization of screening tests.
Women who have symptoms of ovarian cancer should undergo thorough evaluation that may lead to earlier diagnosis and improved outcomes.
Burger RA, Brady MF, Bookman MA, et al; Gynecologic Oncology Group. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483.
Perren TJ, Swart AM, Pfisterer J, et al; ICON7 Investigators. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496.
Epithelial ovarian cancer frequently expresses vascular endothelial growth factor (VEGF). Lower levels of VEGF are associated with decreased formation of new blood vessels and increased survival.
Bevacizumab is a monoclonal antibody that binds to VEGF and inhibits its biological activity. It has proved to be effective in the treatment of colorectal, lung, and brain cancers.
These two recent trials evaluated the benefit of adding bevacizumab to first-line chemotherapy for ovarian cancer. In the Gynecologic Oncology Group (GOG) trial (Protocol#218), investigators evaluated women with Stage III/IV ovarian cancer. The ICON7 trial also included women who had high-risk early-stage disease.
Both trials randomly assigned women to first-line chemotherapy with carboplatin and paclitaxel plus either bevacizumab or placebo. After completion of initial treatment, maintenance therapy continued for an additional 12 to 16 cycles.
In a third arm of GOG 218, bevacizumab was administered only during the six cycles of initial chemotherapy without any maintenance treatment.
Both trials were powered to detect an improvement in progression-free survival, not overall survival.
Findings of the trials
In GOG 218, approximately 600 women were allocated to each of its three arms. In ICON7, approximately 750 women were allocated to each of its two arms.
Both trials demonstrated a benefit when bevacizumab was added to initial treatment, followed by maintenance therapy. The hazard ratio for recurrent disease was 0.72 (95% CI, 0.63–0.82) and 0.81 (95% CI, 0.70–0.94) in the two trials, respectively, with increased progression-free survival of 1.7 to 3.8 months.
Neither study demonstrated a significant increase in overall survival. Women who did not receive bevacizumab maintenance therapy experienced no benefit from treatment.
No significant differences were observed between treatment groups. Bevacizumab was generally well tolerated. The gastrointestinal perforation rate with bevacizumab therapy ranged from 1% to 3%.
When it is administered with initial chemotherapy and continued as maintenance therapy, bevacizumab leads to overall improvement in progression-free survival without a significant increase in overall survival.
We need additional studies to identify molecular markers that will predict the response to bevacizumab and other biologic treatments and determine whether any subgroup of patients will experience greater benefit from the addition of bevacizumab to standard chemotherapy regimens. The findings of such trials will allow us to better tailor treatment to each ovarian cancer patient.
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392.
Approximately 15% of epithelial ovarian cancers demonstrate a mutation in one of the BRCA genes, which function as an important component of DNA repair. Another 35% acquire a similar phenotype due to somatic mutations or silencing of the BRCA genes.
The poly ADP ribose polymerase (PARP) protein plays a role in the repair of single-strand breaks. Tumors with the mutated BRCA phenotype are particularly sensitive to PARP inhibitors8 because PARP inhibition leads to double-strand DNA breaks that cannot be repaired in BRCA mutated tumors.9
A recent phase II trial in recurrent ovarian cancer demonstrated a nearly twofold response rate to olaparib, a PARP inhibitor, among women who had a known BRCA mutation.10
Details of the trial
This study by Ledermann and colleagues was designed to determine the effect of olaparib in all women who have ovarian cancer. It was designed as a randomized, double-blind, phase II trial. Women who had recurrent ovarian, fallopian-tube, or primary peritoneal cancer who were sensitive to platinum and had an objective response to their most recent chemotherapy were randomly assigned to oral olaparib (twice daily dosing) or placebo until such time as disease progressed. The trial had 80% power to detect a 25% decrease in the risk of progression in the olaparib group, with an a less than 0.20.
Two hundred sixty-five women were allocated to each of the two treatment arms. Women treated with olaparib had a risk of recurrence or death that was 35% (95% CI, 25%–49%) the risk among women treated with placebo; they also had a median progression-free survival that was 4 months longer. This response was seen in women with and without BRCA mutations.
Overall, olaparib was well tolerated, although women randomized to the olaparib group had a higher rate of moderate to severe side effects, mostly due to a higher rate of nausea, vomiting, fatigue, and anemia.
Maintenance therapy with oral olaparib significantly increases progression-free survival in women who have platinum-sensitive, recurrent ovarian cancer regardless of their BRCA-mutation status. No significant difference was seen in overall survival at an interim analysis.
Although olaparib is not FDA approved for treatment in patients, these results likely will renew interest in further studies to identify biomarkers to identify patients who are best suited for this treatment.
We want to hear from you! Tell us what you think.
1. Goff BA, Mandel LS, Drescher CW, et al. Development of an ovarian cancer symptom index: possibilities for earlier detection. Cancer. 2007;109(2):221-227.
2. Armstrong DK, Bundy B, Wenzel L, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34-43.
3. Parmar MKB, Ledermann JA, Colombo N, et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet. 2003;361(9375):2099-2106.
4. Pujade-Lauraine E, Wagner U, Aavall-Lundqvist E, et al. Pegylated liposomal Doxorubicin and Carboplatin compared with Paclitaxel and Carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol. 2010;28(20):3323-3329.
5. Mutch DG. Ovarian cancer: to screen or not to screen. Obstet Gynecol. 2009;113(4):772-774.
6. Partridge E, Kreimer AR, Greenlee RT, et al. Results from four rounds of ovarian cancer screening in a randomized trial. Obstet Gynecol. 2009;113(4):775-782.
7. Lu K, Skates S, Bevers T, et al. A prospective US ovarian cancer screening study using the risk of ovarian cancer algorithm (ROCA) [ASCO abstract 5003]. J Clin Oncol. 2010;28(15s):5003.-
8. Weberpals JI, Clark-Knowles KV, Vanderhyden BC. Sporadic epithelial ovarian cancer: clinical relevance of BRCA1 inhibition in the DNA damage and repair pathway. J Clin Oncol. 2008;26(19):3259-3267.-
9. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921.
10. Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852-861.

Akiva P. Novetsky, MD, MS
Dr. Novetsky is a Fellow in the Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, at Washington University School of Medicine in St. Louis, Missouri.
David G. Mutch, MD
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of the Division of Gynecologic Oncology at Washington University School of Medicine in St. Louis, Missouri.
Akiva P. Novetsky, MD, MS
Dr. Novetsky is a Fellow in the Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, at Washington University School of Medicine in St. Louis, Missouri.
David G. Mutch, MD
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of the Division of Gynecologic Oncology at Washington University School of Medicine in St. Louis, Missouri.
Akiva P. Novetsky, MD, MS
Dr. Novetsky is a Fellow in the Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, at Washington University School of Medicine in St. Louis, Missouri.
David G. Mutch, MD
Dr. Mutch is Ira C. and Judith Gall Professor of Obstetrics and Gynecology and Chief of the Division of Gynecologic Oncology at Washington University School of Medicine in St. Louis, Missouri.
- Risk of 3 types of ovarian cancer higher in women who have endometriosis
Janelle Yates, Senior Editor (March 2012) - Ovarian stimulation ups risk of ovarian tumors in later life
(Web exclusive, November 2011) - Can a novel risk-scoring system for ovarian cancer predict who is most likely to develop disease?
William H. Parker, MD (Examining the Evidence, July 2011) - Update on Ovarian Cancer
Nora Kizer, MD, MSCI; David G. Mutch, MD (July 2011)
Dr. Mutch reports receiving research support from the National Institutes of Health and the Gynecologic Oncology Group but no financial relationships relevant to this article. Dr. Novetsky reports no financial relationships relevant to this article.
To improve outcomes in women who have ovarian cancer—the deadliest gynecologic malignancy in the United States—we need to pursue a number of investigative approaches:
- We need to determine how to diagnose the disease in its early stages. At present, fewer than 20% of ovarian cancer cases are identified while disease is localized to the adnexae. Although a symptom index has been suggested as a useful tool to highlight women at risk for ovarian cancer, its appropriate implementation and effectiveness have yet to be determined.1 Moreover, the symptoms of ovarian cancer are vague and may not become apparent until after the disease has metastasized. Might screening trials detect ovarian cancer in its earlier stages? Are there harms involved in screening women with transvaginal ultrasonography (TVUS) and cancer antigen (CA) 125?
- We need new first-line agents to treat ovarian cancer. Traditional therapy is surgical cytoreduction followed by platinum-based chemotherapy. More recently, the addition of intraperitoneal chemotherapy has prolonged survival by approximately 16 months in women who have advanced disease.2 Despite this advance, the relapse rate remains high. What new therapies can we offer in addition to traditional platinum-based chemotherapy?
- Ovarian cancer recurs in most women, and the response to subsequent therapy is short-lived.3,4 Novel biologic agents may offer new hope as a means of treating recurrent disease with greater specificity and lower toxicity. Do any biologic agents increase survival and reduce the toxicity of treatment?
In this article, we highlight notable studies published in the past year that address these questions.
Screening for ovarian cancer is not useful in average-risk women
Buys SS, Partridge E, Black A, et al; PLCO Project Team. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Randomized Screening Trial. JAMA. 2011;305(22):2295–2303.
Gilbert L, Basso O, Sampalis J, et al; DOvE Study Group. Assessment of symptomatic women for early diagnosis of ovarian cancer: results from the prospective DOvE pilot project. Lancet Oncol. 2012;13(3):285–291.
The Prostate, Lung, Colorectal and Ovarian (PLCO) trial addresses the utility of screening for ovarian cancer using two readily available tests—TVUS and CA 125.
Effective screening for any disease requires the following:
- The disease must have a presymptomatic stage during which diagnosis leads to better outcomes (compared with waiting until the onset of symptoms)
- The screening test must be acceptable to the population in which it is used
- The test must lead to a reduction in morbidity and mortality that outweighs the harms of false-positive tests
- The benefits of the test must be achieved at an acceptable level of risk.5
The PLCO trial was designed to determine the effect of specific ovarian cancer screening tests on cause-specific mortality. Women aged 55 to 74 years were randomly assigned to annual screening with TVUS and CA 125 or to standard gynecologic care. A CA-125 level of 35 or higher was classified as abnormal, as were TVUS findings of enlarged ovaries or solid or papillary components. The trial had 88% power to detect a 35% reduction in ovarian cancer mortality using a one-side a of 0.05.
Early results demonstrated that a large number of surgeries would be required to detect one case of ovarian cancer. Mortality data from the study only recently matured.6
Details of the PLCO trial
Approximately 39,000 women were allocated to each arm of the PLCO trial and followed for 6 years. Ovarian cancer was diagnosed in 212 and 176 women in the screening and usual-care groups, respectively (relative risk, 1.21; 95% confidence interval [CI], 0.99–1.48). Equal percentages of women in each group were given a diagnosis of Stage III/IV cancer.
No survival benefit was seen in the screening group. Overall, 3,285 women had a false-positive screening test, with 33% undergoing surgery (21% surgical complication rate).
Barriers to effective screening
One of the major obstacles to the development of an accurate screening test for ovarian cancer has been the low prevalence of the disease. The relationship between sensitivity, specificity, prevalence, and positive predictive value (PPV) is demonstrated in the TABLE. With sensitivity of 100% and specificity of 95%, the PPV for ovarian cancer screening with TVUS and CA 125 is only 1%. Ovarian cancer has an annual prevalence of approximately 1 case in every 2,500 women.
Effect of disease prevalence on population-based screening
| Specificity of the screening test (%) | Positive predictive value (%) | |||
|---|---|---|---|---|
| 50% prevalence | 10% prevalence | 1% prevalence | Ovarian cancer (1 case in every 2,500 women) | |
| 90 | 91 | 53 | 9 | 0.4 |
| 95 | 95 | 69 | 17 | 0.8 |
| 99 | 99 | 92 | 50 | 4 |
| 99.9 | 99.9 | 99 | 91 | 29 |
Details of the trial by Gilbert and colleagues
In a recent pilot study, Gilbert and colleagues prospectively analyzed the utility of disseminating information about the symptoms of ovarian cancer to the general public. Following dissemination of information, women who were 50 years or older and who had experienced at least one ovarian cancer symptom longer than 2 weeks underwent CA-125 testing and TVUS. If both tests were normal, CA 125 was repeated; if it was normal again, the patient was discharged from the study.
Patients who had abnormal findings on either test repeated both screening tests, with additional testing performed as necessary. Outcomes of these women were compared with those of women who had been referred to the gynecologic oncology clinic.
Among 1,455 eligible patients, 11 cases of invasive ovarian cancer were diagnosed, four of which (36%) represented early-stage disease. Median CA-125 levels were lower in the study group. In addition, more women in the study group had early-stage disease that was completely resectable, compared with the clinic group, although this difference did not reach statistical significance.
Most cases of ovarian cancer in this study originated in the fallopian tube or peritoneum—not the ovary. In addition to the cases of ovarian cancer, 11 cases of uterine cancer were diagnosed. No patients underwent unwarranted major surgery, and none who were discharged from the study had a diagnosis of gynecologic cancer by 7 months of follow-up.
TVUS and CA 125 increase the number of cases of ovarian cancer that are diagnosed but do not provide a survival benefit. Identification of cases in the PLCO trial was, therefore, likely the result of lead-time bias—cases of ovarian cancer were detected sooner but not at an early stage.
Routine screening with TVUS and CA 125 are not recommended at this time for women at average risk for ovarian cancer. These and similar screening methods lead to a significant false-positive rate, with surgeries performed for benign indications and a high risk of surgical complications. Further studies to improve the sensitivity and specificity of these tests in women at average risk for ovarian cancer are ongoing.7
Women who have an elevated risk of ovarian cancer, such as women who carry the BRCA mutation or who have a family history of ovarian cancer, may benefit from routine screening because of increased disease prevalence in this population, although studies are needed to determine the best utilization of screening tests.
Women who have symptoms of ovarian cancer should undergo thorough evaluation that may lead to earlier diagnosis and improved outcomes.
Burger RA, Brady MF, Bookman MA, et al; Gynecologic Oncology Group. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483.
Perren TJ, Swart AM, Pfisterer J, et al; ICON7 Investigators. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496.
Epithelial ovarian cancer frequently expresses vascular endothelial growth factor (VEGF). Lower levels of VEGF are associated with decreased formation of new blood vessels and increased survival.
Bevacizumab is a monoclonal antibody that binds to VEGF and inhibits its biological activity. It has proved to be effective in the treatment of colorectal, lung, and brain cancers.
These two recent trials evaluated the benefit of adding bevacizumab to first-line chemotherapy for ovarian cancer. In the Gynecologic Oncology Group (GOG) trial (Protocol#218), investigators evaluated women with Stage III/IV ovarian cancer. The ICON7 trial also included women who had high-risk early-stage disease.
Both trials randomly assigned women to first-line chemotherapy with carboplatin and paclitaxel plus either bevacizumab or placebo. After completion of initial treatment, maintenance therapy continued for an additional 12 to 16 cycles.
In a third arm of GOG 218, bevacizumab was administered only during the six cycles of initial chemotherapy without any maintenance treatment.
Both trials were powered to detect an improvement in progression-free survival, not overall survival.
Findings of the trials
In GOG 218, approximately 600 women were allocated to each of its three arms. In ICON7, approximately 750 women were allocated to each of its two arms.
Both trials demonstrated a benefit when bevacizumab was added to initial treatment, followed by maintenance therapy. The hazard ratio for recurrent disease was 0.72 (95% CI, 0.63–0.82) and 0.81 (95% CI, 0.70–0.94) in the two trials, respectively, with increased progression-free survival of 1.7 to 3.8 months.
Neither study demonstrated a significant increase in overall survival. Women who did not receive bevacizumab maintenance therapy experienced no benefit from treatment.
No significant differences were observed between treatment groups. Bevacizumab was generally well tolerated. The gastrointestinal perforation rate with bevacizumab therapy ranged from 1% to 3%.
When it is administered with initial chemotherapy and continued as maintenance therapy, bevacizumab leads to overall improvement in progression-free survival without a significant increase in overall survival.
We need additional studies to identify molecular markers that will predict the response to bevacizumab and other biologic treatments and determine whether any subgroup of patients will experience greater benefit from the addition of bevacizumab to standard chemotherapy regimens. The findings of such trials will allow us to better tailor treatment to each ovarian cancer patient.
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392.
Approximately 15% of epithelial ovarian cancers demonstrate a mutation in one of the BRCA genes, which function as an important component of DNA repair. Another 35% acquire a similar phenotype due to somatic mutations or silencing of the BRCA genes.
The poly ADP ribose polymerase (PARP) protein plays a role in the repair of single-strand breaks. Tumors with the mutated BRCA phenotype are particularly sensitive to PARP inhibitors8 because PARP inhibition leads to double-strand DNA breaks that cannot be repaired in BRCA mutated tumors.9
A recent phase II trial in recurrent ovarian cancer demonstrated a nearly twofold response rate to olaparib, a PARP inhibitor, among women who had a known BRCA mutation.10
Details of the trial
This study by Ledermann and colleagues was designed to determine the effect of olaparib in all women who have ovarian cancer. It was designed as a randomized, double-blind, phase II trial. Women who had recurrent ovarian, fallopian-tube, or primary peritoneal cancer who were sensitive to platinum and had an objective response to their most recent chemotherapy were randomly assigned to oral olaparib (twice daily dosing) or placebo until such time as disease progressed. The trial had 80% power to detect a 25% decrease in the risk of progression in the olaparib group, with an a less than 0.20.
Two hundred sixty-five women were allocated to each of the two treatment arms. Women treated with olaparib had a risk of recurrence or death that was 35% (95% CI, 25%–49%) the risk among women treated with placebo; they also had a median progression-free survival that was 4 months longer. This response was seen in women with and without BRCA mutations.
Overall, olaparib was well tolerated, although women randomized to the olaparib group had a higher rate of moderate to severe side effects, mostly due to a higher rate of nausea, vomiting, fatigue, and anemia.
Maintenance therapy with oral olaparib significantly increases progression-free survival in women who have platinum-sensitive, recurrent ovarian cancer regardless of their BRCA-mutation status. No significant difference was seen in overall survival at an interim analysis.
Although olaparib is not FDA approved for treatment in patients, these results likely will renew interest in further studies to identify biomarkers to identify patients who are best suited for this treatment.
We want to hear from you! Tell us what you think.
- Risk of 3 types of ovarian cancer higher in women who have endometriosis
Janelle Yates, Senior Editor (March 2012) - Ovarian stimulation ups risk of ovarian tumors in later life
(Web exclusive, November 2011) - Can a novel risk-scoring system for ovarian cancer predict who is most likely to develop disease?
William H. Parker, MD (Examining the Evidence, July 2011) - Update on Ovarian Cancer
Nora Kizer, MD, MSCI; David G. Mutch, MD (July 2011)
Dr. Mutch reports receiving research support from the National Institutes of Health and the Gynecologic Oncology Group but no financial relationships relevant to this article. Dr. Novetsky reports no financial relationships relevant to this article.
To improve outcomes in women who have ovarian cancer—the deadliest gynecologic malignancy in the United States—we need to pursue a number of investigative approaches:
- We need to determine how to diagnose the disease in its early stages. At present, fewer than 20% of ovarian cancer cases are identified while disease is localized to the adnexae. Although a symptom index has been suggested as a useful tool to highlight women at risk for ovarian cancer, its appropriate implementation and effectiveness have yet to be determined.1 Moreover, the symptoms of ovarian cancer are vague and may not become apparent until after the disease has metastasized. Might screening trials detect ovarian cancer in its earlier stages? Are there harms involved in screening women with transvaginal ultrasonography (TVUS) and cancer antigen (CA) 125?
- We need new first-line agents to treat ovarian cancer. Traditional therapy is surgical cytoreduction followed by platinum-based chemotherapy. More recently, the addition of intraperitoneal chemotherapy has prolonged survival by approximately 16 months in women who have advanced disease.2 Despite this advance, the relapse rate remains high. What new therapies can we offer in addition to traditional platinum-based chemotherapy?
- Ovarian cancer recurs in most women, and the response to subsequent therapy is short-lived.3,4 Novel biologic agents may offer new hope as a means of treating recurrent disease with greater specificity and lower toxicity. Do any biologic agents increase survival and reduce the toxicity of treatment?
In this article, we highlight notable studies published in the past year that address these questions.
Screening for ovarian cancer is not useful in average-risk women
Buys SS, Partridge E, Black A, et al; PLCO Project Team. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Randomized Screening Trial. JAMA. 2011;305(22):2295–2303.
Gilbert L, Basso O, Sampalis J, et al; DOvE Study Group. Assessment of symptomatic women for early diagnosis of ovarian cancer: results from the prospective DOvE pilot project. Lancet Oncol. 2012;13(3):285–291.
The Prostate, Lung, Colorectal and Ovarian (PLCO) trial addresses the utility of screening for ovarian cancer using two readily available tests—TVUS and CA 125.
Effective screening for any disease requires the following:
- The disease must have a presymptomatic stage during which diagnosis leads to better outcomes (compared with waiting until the onset of symptoms)
- The screening test must be acceptable to the population in which it is used
- The test must lead to a reduction in morbidity and mortality that outweighs the harms of false-positive tests
- The benefits of the test must be achieved at an acceptable level of risk.5
The PLCO trial was designed to determine the effect of specific ovarian cancer screening tests on cause-specific mortality. Women aged 55 to 74 years were randomly assigned to annual screening with TVUS and CA 125 or to standard gynecologic care. A CA-125 level of 35 or higher was classified as abnormal, as were TVUS findings of enlarged ovaries or solid or papillary components. The trial had 88% power to detect a 35% reduction in ovarian cancer mortality using a one-side a of 0.05.
Early results demonstrated that a large number of surgeries would be required to detect one case of ovarian cancer. Mortality data from the study only recently matured.6
Details of the PLCO trial
Approximately 39,000 women were allocated to each arm of the PLCO trial and followed for 6 years. Ovarian cancer was diagnosed in 212 and 176 women in the screening and usual-care groups, respectively (relative risk, 1.21; 95% confidence interval [CI], 0.99–1.48). Equal percentages of women in each group were given a diagnosis of Stage III/IV cancer.
No survival benefit was seen in the screening group. Overall, 3,285 women had a false-positive screening test, with 33% undergoing surgery (21% surgical complication rate).
Barriers to effective screening
One of the major obstacles to the development of an accurate screening test for ovarian cancer has been the low prevalence of the disease. The relationship between sensitivity, specificity, prevalence, and positive predictive value (PPV) is demonstrated in the TABLE. With sensitivity of 100% and specificity of 95%, the PPV for ovarian cancer screening with TVUS and CA 125 is only 1%. Ovarian cancer has an annual prevalence of approximately 1 case in every 2,500 women.
Effect of disease prevalence on population-based screening
| Specificity of the screening test (%) | Positive predictive value (%) | |||
|---|---|---|---|---|
| 50% prevalence | 10% prevalence | 1% prevalence | Ovarian cancer (1 case in every 2,500 women) | |
| 90 | 91 | 53 | 9 | 0.4 |
| 95 | 95 | 69 | 17 | 0.8 |
| 99 | 99 | 92 | 50 | 4 |
| 99.9 | 99.9 | 99 | 91 | 29 |
Details of the trial by Gilbert and colleagues
In a recent pilot study, Gilbert and colleagues prospectively analyzed the utility of disseminating information about the symptoms of ovarian cancer to the general public. Following dissemination of information, women who were 50 years or older and who had experienced at least one ovarian cancer symptom longer than 2 weeks underwent CA-125 testing and TVUS. If both tests were normal, CA 125 was repeated; if it was normal again, the patient was discharged from the study.
Patients who had abnormal findings on either test repeated both screening tests, with additional testing performed as necessary. Outcomes of these women were compared with those of women who had been referred to the gynecologic oncology clinic.
Among 1,455 eligible patients, 11 cases of invasive ovarian cancer were diagnosed, four of which (36%) represented early-stage disease. Median CA-125 levels were lower in the study group. In addition, more women in the study group had early-stage disease that was completely resectable, compared with the clinic group, although this difference did not reach statistical significance.
Most cases of ovarian cancer in this study originated in the fallopian tube or peritoneum—not the ovary. In addition to the cases of ovarian cancer, 11 cases of uterine cancer were diagnosed. No patients underwent unwarranted major surgery, and none who were discharged from the study had a diagnosis of gynecologic cancer by 7 months of follow-up.
TVUS and CA 125 increase the number of cases of ovarian cancer that are diagnosed but do not provide a survival benefit. Identification of cases in the PLCO trial was, therefore, likely the result of lead-time bias—cases of ovarian cancer were detected sooner but not at an early stage.
Routine screening with TVUS and CA 125 are not recommended at this time for women at average risk for ovarian cancer. These and similar screening methods lead to a significant false-positive rate, with surgeries performed for benign indications and a high risk of surgical complications. Further studies to improve the sensitivity and specificity of these tests in women at average risk for ovarian cancer are ongoing.7
Women who have an elevated risk of ovarian cancer, such as women who carry the BRCA mutation or who have a family history of ovarian cancer, may benefit from routine screening because of increased disease prevalence in this population, although studies are needed to determine the best utilization of screening tests.
Women who have symptoms of ovarian cancer should undergo thorough evaluation that may lead to earlier diagnosis and improved outcomes.
Burger RA, Brady MF, Bookman MA, et al; Gynecologic Oncology Group. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483.
Perren TJ, Swart AM, Pfisterer J, et al; ICON7 Investigators. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496.
Epithelial ovarian cancer frequently expresses vascular endothelial growth factor (VEGF). Lower levels of VEGF are associated with decreased formation of new blood vessels and increased survival.
Bevacizumab is a monoclonal antibody that binds to VEGF and inhibits its biological activity. It has proved to be effective in the treatment of colorectal, lung, and brain cancers.
These two recent trials evaluated the benefit of adding bevacizumab to first-line chemotherapy for ovarian cancer. In the Gynecologic Oncology Group (GOG) trial (Protocol#218), investigators evaluated women with Stage III/IV ovarian cancer. The ICON7 trial also included women who had high-risk early-stage disease.
Both trials randomly assigned women to first-line chemotherapy with carboplatin and paclitaxel plus either bevacizumab or placebo. After completion of initial treatment, maintenance therapy continued for an additional 12 to 16 cycles.
In a third arm of GOG 218, bevacizumab was administered only during the six cycles of initial chemotherapy without any maintenance treatment.
Both trials were powered to detect an improvement in progression-free survival, not overall survival.
Findings of the trials
In GOG 218, approximately 600 women were allocated to each of its three arms. In ICON7, approximately 750 women were allocated to each of its two arms.
Both trials demonstrated a benefit when bevacizumab was added to initial treatment, followed by maintenance therapy. The hazard ratio for recurrent disease was 0.72 (95% CI, 0.63–0.82) and 0.81 (95% CI, 0.70–0.94) in the two trials, respectively, with increased progression-free survival of 1.7 to 3.8 months.
Neither study demonstrated a significant increase in overall survival. Women who did not receive bevacizumab maintenance therapy experienced no benefit from treatment.
No significant differences were observed between treatment groups. Bevacizumab was generally well tolerated. The gastrointestinal perforation rate with bevacizumab therapy ranged from 1% to 3%.
When it is administered with initial chemotherapy and continued as maintenance therapy, bevacizumab leads to overall improvement in progression-free survival without a significant increase in overall survival.
We need additional studies to identify molecular markers that will predict the response to bevacizumab and other biologic treatments and determine whether any subgroup of patients will experience greater benefit from the addition of bevacizumab to standard chemotherapy regimens. The findings of such trials will allow us to better tailor treatment to each ovarian cancer patient.
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392.
Approximately 15% of epithelial ovarian cancers demonstrate a mutation in one of the BRCA genes, which function as an important component of DNA repair. Another 35% acquire a similar phenotype due to somatic mutations or silencing of the BRCA genes.
The poly ADP ribose polymerase (PARP) protein plays a role in the repair of single-strand breaks. Tumors with the mutated BRCA phenotype are particularly sensitive to PARP inhibitors8 because PARP inhibition leads to double-strand DNA breaks that cannot be repaired in BRCA mutated tumors.9
A recent phase II trial in recurrent ovarian cancer demonstrated a nearly twofold response rate to olaparib, a PARP inhibitor, among women who had a known BRCA mutation.10
Details of the trial
This study by Ledermann and colleagues was designed to determine the effect of olaparib in all women who have ovarian cancer. It was designed as a randomized, double-blind, phase II trial. Women who had recurrent ovarian, fallopian-tube, or primary peritoneal cancer who were sensitive to platinum and had an objective response to their most recent chemotherapy were randomly assigned to oral olaparib (twice daily dosing) or placebo until such time as disease progressed. The trial had 80% power to detect a 25% decrease in the risk of progression in the olaparib group, with an a less than 0.20.
Two hundred sixty-five women were allocated to each of the two treatment arms. Women treated with olaparib had a risk of recurrence or death that was 35% (95% CI, 25%–49%) the risk among women treated with placebo; they also had a median progression-free survival that was 4 months longer. This response was seen in women with and without BRCA mutations.
Overall, olaparib was well tolerated, although women randomized to the olaparib group had a higher rate of moderate to severe side effects, mostly due to a higher rate of nausea, vomiting, fatigue, and anemia.
Maintenance therapy with oral olaparib significantly increases progression-free survival in women who have platinum-sensitive, recurrent ovarian cancer regardless of their BRCA-mutation status. No significant difference was seen in overall survival at an interim analysis.
Although olaparib is not FDA approved for treatment in patients, these results likely will renew interest in further studies to identify biomarkers to identify patients who are best suited for this treatment.
We want to hear from you! Tell us what you think.
1. Goff BA, Mandel LS, Drescher CW, et al. Development of an ovarian cancer symptom index: possibilities for earlier detection. Cancer. 2007;109(2):221-227.
2. Armstrong DK, Bundy B, Wenzel L, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34-43.
3. Parmar MKB, Ledermann JA, Colombo N, et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet. 2003;361(9375):2099-2106.
4. Pujade-Lauraine E, Wagner U, Aavall-Lundqvist E, et al. Pegylated liposomal Doxorubicin and Carboplatin compared with Paclitaxel and Carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol. 2010;28(20):3323-3329.
5. Mutch DG. Ovarian cancer: to screen or not to screen. Obstet Gynecol. 2009;113(4):772-774.
6. Partridge E, Kreimer AR, Greenlee RT, et al. Results from four rounds of ovarian cancer screening in a randomized trial. Obstet Gynecol. 2009;113(4):775-782.
7. Lu K, Skates S, Bevers T, et al. A prospective US ovarian cancer screening study using the risk of ovarian cancer algorithm (ROCA) [ASCO abstract 5003]. J Clin Oncol. 2010;28(15s):5003.-
8. Weberpals JI, Clark-Knowles KV, Vanderhyden BC. Sporadic epithelial ovarian cancer: clinical relevance of BRCA1 inhibition in the DNA damage and repair pathway. J Clin Oncol. 2008;26(19):3259-3267.-
9. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921.
10. Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852-861.
1. Goff BA, Mandel LS, Drescher CW, et al. Development of an ovarian cancer symptom index: possibilities for earlier detection. Cancer. 2007;109(2):221-227.
2. Armstrong DK, Bundy B, Wenzel L, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34-43.
3. Parmar MKB, Ledermann JA, Colombo N, et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet. 2003;361(9375):2099-2106.
4. Pujade-Lauraine E, Wagner U, Aavall-Lundqvist E, et al. Pegylated liposomal Doxorubicin and Carboplatin compared with Paclitaxel and Carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol. 2010;28(20):3323-3329.
5. Mutch DG. Ovarian cancer: to screen or not to screen. Obstet Gynecol. 2009;113(4):772-774.
6. Partridge E, Kreimer AR, Greenlee RT, et al. Results from four rounds of ovarian cancer screening in a randomized trial. Obstet Gynecol. 2009;113(4):775-782.
7. Lu K, Skates S, Bevers T, et al. A prospective US ovarian cancer screening study using the risk of ovarian cancer algorithm (ROCA) [ASCO abstract 5003]. J Clin Oncol. 2010;28(15s):5003.-
8. Weberpals JI, Clark-Knowles KV, Vanderhyden BC. Sporadic epithelial ovarian cancer: clinical relevance of BRCA1 inhibition in the DNA damage and repair pathway. J Clin Oncol. 2008;26(19):3259-3267.-
9. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921.
10. Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852-861.
Cluster Headache: Hastening Diagnosis and Treatment
Headache is one of the health problems most commonly associated with significant morbidity, as well as considerable social and economic repercussions.1,2 Headaches are classified into three main types:
- Primary headaches, or headaches without known organic causes
- Secondary headaches, or headaches manifesting with symptoms due to organic causes; and
- Cranial neuralgias, facial pain, and other headaches.3
Cluster headache (CH) is a type of primary headache—one of the headache types encompassed by the term trigeminal autonomic cephalalgia.3 It is one of the most intense, excruciating headaches a patient can experience, and the diagnosis is often missed or delayed. Only 21% of patients receive a correct diagnosis of CH on first presentation, and the average patient visits three health care providers before the correct diagnosis is made.4,5 According to recently published results from the US Cluster Headache Survey,4 the diagnostic delay for CH averages five years or longer, limiting the patient’s access to correct treatment.
Patients with CH are prone to significant physical, social, and economic disability; most patients, for example, find it difficult to work during a CH period.1,2 Almost 20% of patients with CH report having lost a job because of their headaches, and about 8% are unemployed or on disability.4
Because of the pain severity and the associated impairment, the risk for suicide in the CH patient population is real.2,6,7 Jürgens et al7 report that 22% of patients with chronic CH and about 15% of those with episodic CH had suicidal tendencies; Rozen and Fishman4 report suicidal ideation in 55% of CH patients.
CHARACTERISTICS OF CLUSTER HEADACHE
As the name implies, attacks of this headache type tend to “cluster” together. In 85% to 90% of patients, CH is episodic, with cluster periods of headache attacks commonly lasting for one week to one year, and intervening remission periods that may last from one month to years.3,8 The remaining 10% to 15% of CH patients have the chronic CH type, in which cluster periods typically last for more than one year and are separated by remission periods lasting one month or less.3,8
Cluster headaches tend to occur in predictable patterns, often in the spring and fall.9-11 Most headaches begin between early evening and early morning, and patients are often awakened by CH during the night; according to responses in the US Cluster Headache Survey,4 onset times peak between midnight and 3 AM. Attacks can occur when the neck is rotated or flexed in specific ways; external pressure to the transverse processes of C4 or the nerve root of C2 can trigger a CH attack.12
Other CH triggers include alcohol (especially beer and red wine4,13), histamine, nitroglycerine, carbon dioxide, certain odors, and weather changes.3,4,11,14 Eighty percent or more of CH patients have a history of prolonged tobacco use, and at least 60% of CH patients who do not smoke were the children of smokers.15 No clear relationship has been found between CH and hormones.9,10
EPIDEMIOLOGY
Cluster headache is relatively rare, affecting about 0.1% of the population.8-10,16 Onset of the condition usually occurs between ages 20 and 40, and men are three to four times more likely to be affected than are women.3,16 A familial/genetic relationship may exist.10,17,18
DIAGNOSIS
Patient History
Diagnosis of headache relies heavily on the patient’s clinical history and physical exam.3,8,10 A detailed history should include the initial onset of CH, progression of the condition, and information about any precipitating event(s) and prodromal symptoms. Clinicians should document the pattern of pain by including specific information regarding its location, severity, quality, frequency, and duration. Of considerable value is the patient’s use of an accurate headache diary, which clinicians should encourage headache patients to maintain; in these, patients should be instructed to record the headache characteristics mentioned.3,8
Associated symptoms (assessed by conducting a complete review of systems), aggravating and alleviating factors, previous medical history, and psychosocial and family history are important in formulating the differential diagnosis, as misdiagnosis of CH is often related to inadequate history intake.13
Presentation
Cluster headaches share three main features: they are unilateral; they are associated with autonomic symptoms; and attacks tend to “cluster” in a circannual pattern (ie, clusters occurring at the same time of year) and/or circadian pattern (headache at the same time of day).8,19 The most common locations for cluster headaches are unilateral orbital, supraorbital, temporal, or a combination of these locations.3,8
About 30% of patients describe the pain as “stabbing,”3 and it is often compared to “a hot poker in the eye.” Pain peaks rapidly, usually within five to 10 minutes. It may radiate to the ipsilateral forehead, jaw, cheek, and/or teeth.3 Patients appear restless and agitated, unable to lie still.2,10 They often sit, holding their heads, and may pace the floor or bang their heads against the wall.
CH is associated with at least one of the following autonomic symptoms, occurring in the ipsilateral side of the head: conjunctival injection, nasal congestion, forehead and facial sweating, eyelid edema, lacrimation, rhinorrhea, ptosis, and miosis.3,13 Headaches may occur on one side of the head throughout one cluster episode, then shift to the contralateral side in subsequent periods.10 Aura occurs in 14% to 20% of patients,13,20-22 and nausea, as well as ipsilateral
visual, sensory, and speech/language disturbances have also been reported.3 Each CH attack lasts between 15 minutes and three hours, and attacks may range in frequency from one every other day to eight per day.3,13
Patients who have experienced at least five episodes of these headache symptoms, with severe pain in the specified areas and duration, accompanying autonomic symptoms, specified attack frequency, and symptoms not attributed to another disorder meet the diagnostic criteria for cluster headache given in the second edition of the International Classification of Headache Disorders (ICHD-II, 2004).3 The ICHD-II criteria, based on clinical and epidemiologic research, are recognized as a consensus guideline that is accepted worldwide to facilitate clinical practice.3 Patients who have experienced attacks fulfilling all but one of the ICHD-II criteria for CH are diagnosed with probable CH3 or cluster-like headache (CLH).23
Physical Examination
A thorough physical examination, including an investigation of the neurologic system, is essential to differentiate among primary, secondary, and other headache types. In the patient with CH, no neurologic deficits or deficits that suggest underlying disorders are usually found.3,10
Differential Diagnosis
In the evaluation of headache, it is important to differentiate CH from the other trigeminal autonomic cephalalgias: paroxysmal hemicrania (PH), short-lasting unilateral neuralgiform headache with conjunctival injection and tearing (SUNCT), and possibly hemicrania continua.3,24,25 As in CH, the pain of PH is severe, unilateral, and stabbing in quality; it, too, is associated with autonomic symptoms, often occurs at night, and can be episodic or chronic.3 However, PH headache lasts for only 2 to 30 minutes and can occur five times or more per day. Though difficult to distinguish from CH patients, those with PH usually respond to indomethacin, whereas those with CH ordinarily do not.3,8
As in patients with CH, those affected by SUNCT experience autonomic symptoms—most commonly, conjunctival injection and tearing.3,26 SUNCT differs from CH, however, in that the pain is moderate in severity, with a pulsating, burning, electric-like quality. Duration is much shorter, with episodes lasting between 5 seconds and 4 minutes.3,26
Hemicrania continua, though unilateral, is described as continuous and moderate in intensity. Like PH, it is also indomethacin-responsive.8,25,27
A broader differential diagnosis for CH, as detailed in the table,11,3,8,13,26,28 includes the other primary headaches: tension headache, migraine headache, and trigeminal neuralgia.3,26Tension headache, which affects 30% to 78% of the general population,3 is subdivided into infrequent episodic, frequent episodic, and chronic tension-type headache. Unlike CH, tension headache is mild to moderate in intensity and occurs bilaterally, with nonpulsating pressure or a tightening sensation. It is not aggravated by routine physical activity, nor is it associated with nausea, vomiting, or photophobia.
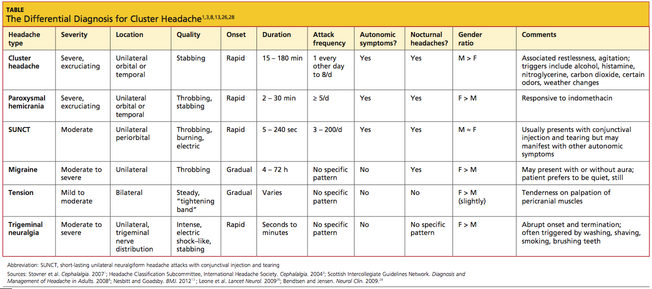
Migraine headache, also a more common primary headache type than CH,3 occurs unilaterally, is moderate to severe in intensity, and is often described as throbbing. More gradual than CH in onset, migraine is often associated with nausea, vomiting, photophobia, phonophobia, and/or visual aura. Migraine headache lacks the ipsilateral autonomic manifestations of CH, and migraineurs prefer to rest or sleep—in contrast to the extreme restlessness or agitation seen in CH patients.3
Also like CH, trigeminal neuralgia is unilateral with a trigeminal nerve distribution, and the pain can be severe and stabbing.3 However, trigeminal neuralgia lacks the autonomic symptoms associated with CH, and the pain lasts from only seconds to minutes. This headache type is often triggered by washing, shaving, or brushing teeth.3
It is also critical to exclude secondary headaches, especially those with serious causes, including meningitis, subarachnoid hemorrhage, epidural or subdural hematoma, glaucoma, tumors, temporal arteritis, or purulent sinusitis.27 Red flags associated with these conditions are:
- A complaint of the patient’s “worst headache ever” (thunderclap headache)
- First severe headache
- A subacute headache worsening over days or weeks
- An abnormal neurologic examination
- Fever or other unexplained systemic signs
- A headache preceded by vomiting
- Headache that is induced by bending, lifting, or coughing
- Headache that disturbs the patient’s sleep or presents immediately upon awakening
- History of known systemic illness
- Headache onset after age 55; and
- Pain associated with local tenderness, for example, near the temporal artery.27
DIAGNOSTIC TESTING
Since, by definition, primary headaches are those without underlying organic causes, diagnostic tests and neuroimaging studies are generally not recommended,10,29 especially when the patient history and presentation confirm the required ICHD-II diagnostic criteria. However, neuroimaging is often recommended for a patient with CH or CH-like presentations.8,23,30-32
In a literature review published in 2006, Detsky et al30 examined the correlation between clinical features of headache (as described in the ICHD-II criteria) and intracranial abnormality (as found on CT or MRI). They found an increased risk for serious intracranial abnormalities among study subjects with cluster-type headache. In any patient with chronic headache, they found, abnormal neurologic findings on physical exam represent the greatest predictor for intracranial pathology.30
In a similar study of 1,872 consecutive patients with nonacute headaches who underwent CT or MRI, one of 20 patients with CH was found to have a pituitary adenoma.33 When Favier et al34 reviewed 31 cases of trigeminal autonomic cephalalgia (TAC), including 10 with atypical symptoms, they found that even typical TAC can result from underlying pathologies with rare warning signs and symptoms. In some patients, neuroimaging study results were normal on initial diagnosis, but pathologies were discovered later after symptoms worsened or treatments ceased to be effective, prompting further imaging studies.
In a review of case studies of patients with CLH, Mainardi et al23 found that of 38 patients who fulfilled the ICHD-II criteria for CH, 12 patients (31.6%) had vascular pathologies, 12 (31.6%) had tumors, and five (13.2%) had inflammatory or infectious pathologies. The researchers recommended that all patients with symptoms of CH or CLH undergo cerebral MRI with contrast medium, even though the yield for abnormal findings would likely be low.23 Wilbrink et al32 also found a wide range of pathologies without typical warning signs or symptoms among 56 case studies of TAC and TAC-like syndromes—and recommended that all such patients be considered candidates for neuroimaging.
Recommendations from both the Scottish Intercollegiate Guidelines Network (SIGN)8 and the Taiwan Headache Society treatment guidelines31 include neuroimaging of patients with CH or CLH.
MANAGEMENT
Clinicians may wish to consider referring patients to a neurologist at the initial diagnosis of CH. Patients with atypical symptomatology or neurologic abnormalities, and those who respond insufficiently to treatment warrant a neurology referral for further investigation.
The two treatment strategies for CH are first, symptomatic treatment for acute attacks; and second, intervention to prevent or reduce further attacks and to shorten the cluster period.8,11,14,35,36
Acute Treatment
Acute symptomatic treatment is aimed at aborting the pain within 15 to 30 minutes from headache onset.14,35,36 Currently, it is generally accepted that 100% oxygen and parenteral triptans (5-HT1B/1D, not through the alimentary tract) are considered first-line treatment options.8,11,13,14,35,36
For the majority of patients (particularly those with episodic CH), inhaled normobaric oxygen effectively relieves CH pain within 15 minutes.14,37-39 Oxygen is administered at 6 to 12 L/min with a nonrebreather mask for at least 15 to 20 minutes.8,14,13 Although associated adverse events are rare, oxygen is inconvenient to transport, and it poses a fire hazard. Additionally, high-flow oxygen is contraindicated in patients with chronic obstructive pulmonary disease, as these patients depend on the hypoxic drive and run the risk of respiratory depression.14,40,41
Triptans, too, have been found effective in the acute treatment of CH; administration by subcutaneous injection or intranasal delivery is considered more effective than the oral route due to faster onset of action,11,14,35 and oxygen use may enhance triptans’ efficacy.38 In two 2010 reviews of the relevant literature, subcutaneous sumatriptan, dosed at either 6 mg or 12 mg, provided effective pain relief within 15 minutes for most patients, with no statistical between-dosage differences.11,35 The most common adverse effects were injection-site reactions, nausea, vomiting, dizziness, fatigue, and paresthesias.35
Intranasal zolmitriptan (5 mg and 10 mg) and intranasal sumatriptan 20 mg were also found effective, with significant pain relief within 30 minutes. Bad taste is a common complaint.35 Of note, both sumatriptan and zolmitriptan are contraindicated in patients with cardiovascular or cerebrovascular disease.14
In an older study of efficacy, safety, and tolerability of subcutaneous sumatriptan, almost 70% of patients averaging between one and six CH attacks per day were found to be using more than the 12-mg maximum recommended daily dosage—as much as 36 mg in a 24-hour period.42 Nevertheless, the researchers concluded that subcutaneous sumatriptan was effective and well tolerated without decreased efficacy over one year in patients with CH.
Ergotamine, once commonly used for the acute treatment of CH, has fallen out of favor in recent years due to its vasoconstrictive effects and serious adverse effects profile.14,35 Dihydroergotamine (DHE) is most effective when administered by IV (though not easily accessible for an acute attack); however, evidence regarding its efficacy and tolerability in other forms is insufficient to recommend DHE for acute CH therapy.14,35
Intranasal lidocaine, somatostatin by infusion, and subcutaneous octreotide are considered second-line treatment choices for patients who are resistant to first-line therapies or who cannot tolerate them.14,35,43,44
Cluster Headache Prophylaxis
A CH period can last for weeks to months. Prophylactic modalities, which are intended to shorten this period and to reduce the frequency and severity of headache attacks, are categorized into transitional and maintenance prophylaxis treatments.14,35
Transitional prophylaxis, a shorter course of treatment, is often started with maintenance prophylaxis (which is used throughout each cluster period) to hasten the response to the maintenance treatment. Corticosteroids are commonly used as a transitional treatment modality. In prednisone use, a starting dose of at least 40 mg/d by mouth is often required to provide benefit.14 The peak dose is usually given for three to 10 days, then gradually tapered over the succeeding 10 to 30 days. Headache recurrence is common during the prednisone taper. Ergotamine tartrate and DHE are also used as transitional prophylaxis treatment for CH.14
Verapamil is considered the maintenance prophylaxis drug of choice due to its efficacy and safety.14,35,45 The dosage required for adequate response ranges from 200 mg to 960 mg/d, in divided doses or in extended-release formulation. Most patients respond to daily doses between 200 mg and 480 mg.14,46,47 Constipation is the most common adverse effect. Slow titration and frequent ECG monitoring, particularly when dosing is increased, are necessary to avoid heart block, bradycardia, hypotension, and peripheral edema.13
Lithium is often used as second-line therapy for maintenance prophylaxis, possibly in combination with verapamil or topiramate, to improve pain control.35 Lithium carbonate, given at a dosage of 600 to 900 mg/d to maintain a serum level of 0.4 to 0.8 mEq/L, and topiramate, at dosages ranging from 50 to 200 mg/d, may be needed to achieve an adequate response.14
In at least one small study, melatonin (10 mg/d) has been associated with CH remission in 50% of treated patients.14,48 It may be used in combination with other prophylactic medications.14
Among numerous other agents that have been used for CH prophylaxis, neither sodium valproate, sumatriptan, cimetidine/chlorpheniramine, misoprostol, nor oxygen is recommended for prevention of CH.35
Narcotics
Because of its excruciating pain, CH has been referred to as “suicide headache.”2,6,7,10,13 Acute and prophylactic treatments for CH will likely reduce the number of headache attacks; however, with CH attacks as frequent as eight times per day, these treatments may not be adequate.24,49
The use of any narcotic is not ideal due to its potential for addiction, and it may cause medication-overuse headache. Furthermore, in oral form, a narcotic may not relieve CH pain quickly enough. Low-dose levomethadone is an opioid that has been used prophylactically with some success in patients with chronic CH.24,49 However, the primary care provider whose CH patient finds pain control inadequate should refer to a neurologist or a pain management specialist for evaluation—and possibly for treatment with an invasive procedure.
Invasive Procedures
Greater occipital nerve block has shown promise in the treatment of CH.50,51 In a small, double-blind study, patients with episodic or chronic CH were randomized to receive a suboccipital injection, of either combined long- and rapid-acting betamethasone or saline (placebo), in the area of the greater occipital nerve.50 Eighty-five percent of treated patients were free of headache attacks within 72 hours, compared with none in the control group. Use of lidocaine with triamcinolone was found somewhat less effective.51
Occipital nerve stimulation has also shown promise for patients with chronic CH who become resistant or are unresponsive to conventional treatments, or who cannot tolerate them.14,24 It appears to induce gradual neuromodulation, with gradual benefits (after six to 30 months). Deep brain stimulation (ie, of the posterior hypothalamus), delivered via implanted electrodes, and other procedures have produced results ranging from “excellent” to “transient remission,” reducing the use of ablative surgeries.14,52 Because invasive modalities carry a risk for serious adverse effects,14 their use should be reserved for a select patient population.
PATIENT/FAMILY EDUCATION
Patients with CH need to be educated regarding the nature, signs and symptoms, and triggers of CH. The indications for acute and prophylactic treatments and the adverse effects associated with each therapy must also be reviewed. Clear follow-up instructions are essential, including what conditions warrant further evaluation: worsening of the condition, changes in symptoms (impaired alertness, vision, movement, or sensation; onset of seizures), or treatment failure.
CONCLUSION
Cluster headache, a relatively uncommon primary headache that can cause excruciating and debilitating pain, is often misdiagnosed and inappropriately treated, with serious physical, social, and economic ramifications. This headache type is unilateral, associated with autonomic symptoms, and characterized by clustering of headache/remission periods in a circannual and/or circadian pattern. Diagnosis is made through the health history and physical exam, based on criteria from the ICHD-II. Neuroimaging may not be necessary, but given the evidence that CH and TAC are often associated with serious underlying pathology, MRI with contrast should be considered, and consultation with a neurologist at initial diagnosis is recommended.
Treatment is aimed at aborting the pain within 15 to 30 minutes of an acute headache attack and preventing further episodes through transitional and maintenance prophylaxis. Newer invasive options that are showing great promise may be considered for a select patient population. Clinicians should involve patients in treatment decisions that will address their individual needs, improving function and optimizing outcomes.
1. Stovner LJ, Hagen H, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193-210.
2. Vaughan R. My own private purgatory: how cluster headaches affect my life. Headache. 2008;48(10):1541-1543.
3. Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders. 2nd ed. Cephalalgia. 2004;24 suppl 1:9-160.
4. Rozen TD, Fishman RS. Cluster headache in the United States of America: demographics, clinical characteristics, triggers, suicidality, and personal burden. Headache. 2012;52(1): 99-113.
5. Bahra A, Goadsby PJ. Diagnostic delays and mis-management in cluster headache. Acta Neurol Scand. 2004;109(3):175-179.
6. Dousset V, Laporte A, Legoff M, et al. Validation of a brief self-administered questionnaire for cluster headache screening in a tertiary center. Headache. 2009;49(1):64-70.
7. Jürgens TP, Gaul C, Lindwurm A, et al. Impairment in episodic and chronic cluster headache. Cephalalgia. 2011;31(6):671-682.
8. Scottish Intercollegiate Guidelines Network (SIGN). Diagnosis and management of headache in adults: a national clinical guideline. Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN). 2008;1-81. (SIGN publication; no. 107). www.sign.ac.uk/guide lines/fulltext/107/index.html. Accessed May 8, 2012.
9. Bahra A, May A, Goadsby PJ. Cluster headache: a prospective clinical study with diagnostic implications. Neurology. 2002;58(3): 354-361.
10. May A. Cluster headache: pathogenesis, diagnosis, and management. Lancet. 2005; 366(9488):843-855.
11. Law S, Derry S, Moore RA. Triptans for acute cluster headache. Cochrane Database Syst Rev. 2010 Apr 14;(4):CD008042.
12. Rozen TD. Trigeminal autonomic cephalalgias. Neurol Clin. 2009;27(2):537-556.
13. Nesbitt AD, Goadsby PJ. Cluster headache. BMJ. 2012;344:e2407. doi: 10.1136/bmj
.e2407.
14. Ashkenazi A, Schwedt T. Cluster headache: acute and prophylactic therapy. Headache. 2011;51(2):272-286.
15. Rozen TD. Cluster headache as the result of secondhand cigarette smoke exposure during childhood. Headache. 2010;50(1):130-132.
16. Fischera M, Marziniak M, Gralow I, Evers S. The incidence and prevalence of cluster headache: a meta-analysis of population-based studies. Cephalalgia. 2008;28(6):614-618.
17. De Simone R, Fiorillo C, Bonuso S, Castaldo G. A cluster headache family with possible autosomal recessive inheritance. Neurology. 2003;61(4):578-579.
18. Leone M, Russell MB, Rigamonti A, et al. Increased familial risk of cluster headache. Neurology. 2001;56(9):1233-1236.
19. Matharu MS, Boes CJ, Goadsby PJ. Management of trigeminal autonomic cephalgias and hemicrania continua. Drugs. 2003;63(16): 1637-1677.
20. Evans RW, Krymchantowski AV. Cluster and other nonmigraine primary headaches with aura. Headache. 2011;51(4):604-608.
21. Rozen TD. Cluster headache with aura. Curr Pain Headache Rep. 2011;15(2):98-100.
22. Silberstein SD, Niknam R, Rozen TD, Young WB. Cluster headache with aura. Neurology. 2000;54(1):219-221.
23. Mainardi F, Trucco M, Maggioni F, et al. Cluster-like headache: a comprehensive reappraisal. Cephalalgia. 2010;30(4):399-412.
24. Magis D, Bruno MA, Fumal A, et al. Central modulation in cluster headache patients treated with occipital nerve stimulation: an FDG-PET study. BMC Neurol. 2011;11:25.
25. Goadsby PJ, Lipton RB. A review of paroxysmal hemicranias, SUNCT syndrome and other short-lasting headaches with autonomic feature, including new cases. Brain. 1997; 120(pt 1):193-209.
26. Leone M, Bussone G. Pathophysiology of trigeminal autonomic cephalalgias. Lancet Neurol. 2009;8(8):755-764.
27. Goadsby PJ, Raskin NH. Chapter 14. Headache. In: Longo DL, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York: McGraw-Hill; 2011:112-128.
28. Bendtsen L, Jensen R. Tension-type headache. Neurol Clin. 2009;27(2):525-535.
29. van Kleef M, Lataster A, Narouze S, et al. Evidenced-based interventional pain medicine according to clinical diagnoses. 2. Cluster headache. Pain Pract. 2009;9(6):435-442.
30. Detsky ME, McDonald DR, Baerlocher MO, et al. Does this patient with headache have a migraine or need neuroimaging? JAMA. 2006; 296(10):1274-1283.
31. Treatment Guideline Subcommittee of the Taiwan Headache Society. Neuroimaging guidelines in nonacute headaches [in Chinese]. Acta Neurol Taiwan. 2010;19(2):137-144.
32. Wilbrink LA, Ferrari MD, Kruit MC, Haan J. Neuroimaging in trigeminal autonomic cephalgias: when, how, and of what? Curr Opin Neurol. 2009;22(3):247-253.
33. Sempere AP, Porta-Etessam J, Medrano V, et al. Neuroimaging in the evaluation of patients with non-acute headache. Cephalalgia. 2005;25(1):30-35.
34. Favier I, van Vliet J, Roon K, et al. Trigeminal autonomic cephalgias due to structural lesions: a review of 31 cases. Arch Neurol. 2007;64(1):25-31.
35. Francis GJ, Becker WJ, Pringsheim TM. Acute and preventive pharmacologic treatment of cluster headache. Neurology. 2010; 75(5):463-473.
36. Bennett MH, French C, Schnabel A, et al. Normobaric and hyperbaric oxygen therapy for migraine and cluster headache. Cochrane Database Syst Rev. 2008 Jul 16;(3):CD005219.
37. Rozen TD. Inhaled oxygen for cluster headache: efficacy, mechanism of action, utilization, and economics. Curr Pain Headache Rep. 2012 Jan 29. [Epub ahead of print]
38. Rozen TD, Fishman RS. Inhaled oxygen and cluster headache sufferers in the United States: use, efficacy and economics: results from the United States Cluster Headache Survey. Headache. 2011;51(2):191-200.
39. Kudrow L. Response of cluster headache attacks to oxygen inhalation. Headache. 1981; 21:1-4.
40. Cohen AS, Burns B, Goadsby PJ. High-flow oxygen for treatment of cluster headache: a randomized trial. JAMA. 2009;302(22):2451-2457.
41. Mahadevan SV. Emergency airway management. In: Auerbach PS. Wilderness Medicine. 6th ed. Stanford, CA: Elsevier; 2011.
42. Göbel H, Lindner V, Heinze A, et al. Acute therapy for cluster headache with sumatriptan: findings of a one-year long-term study. Neurology. 1998;51(3):908-911.
43. Sicuteri F, Geppetti P, Marabini S, Lembeck F. Pain relief by somatostatin in attacks of cluster headache. Pain. 1984;184:359-365.
44. Matharu MS, Levy MJ, Meeran K, Goadsby PJ. Subcutaneous octreotide in cluster headache: randomized placebo-controlled double-blind crossover study. Ann Neurol. 2004;56(4): 488-494.
45. Leone M, D’Amico D, Frediani F, et al. Verapamil in the prophylaxis of episodic cluster headache: a double-blind study versus placebo. Neurology. 2000;54(6):1382-1385.
46. Tfelt-Hansen P, Tfelt-Hansen J. Verapamil for cluster headache: clinical pharmacology and possible mode of action. Headache. 2009;49(1): 117-125.
47. Blau JN, Engel HO. Individualizing treatment with verapamil for cluster headache patients. Headache. 2004;44(10):1013-1018.
48. Leone M, D’Amico D, Moschiano F, et al. Melatonin versus placebo in the prophylaxis of cluster headache: a double-blind pilot study with parallel groups. Cephalalgia. 1996;16(7): 494-496.
49. Sprenger T, Seifert CL, Miederer M, et al. Successful prophylactic treatment of chronic cluster headache with low-dose levomethadone. J Neurol. 2008;255(11):1832-1833.
50. Ambrosini A, Vandenheede M, Rossi P, et al. Suboccipital injection with a mixture of rapid- and long-acting steroids in cluster headache: a double-blind placebo-controlled study. Pain. 2005;118(1-2):92-96.
51. Peres MF, Stiles MA, Siow HC, et al. Greater occipital nerve blockade for cluster headache. Cephalalgia. 2002;22(7):520-522.
52. Leone M, Proietti Cecchini A, Franzini A, et al. Lessons from 8 years’ experience of hypothalamic stimulation in cluster headache. Cephalalgia. 2008;28(7):787-797.
Headache is one of the health problems most commonly associated with significant morbidity, as well as considerable social and economic repercussions.1,2 Headaches are classified into three main types:
- Primary headaches, or headaches without known organic causes
- Secondary headaches, or headaches manifesting with symptoms due to organic causes; and
- Cranial neuralgias, facial pain, and other headaches.3
Cluster headache (CH) is a type of primary headache—one of the headache types encompassed by the term trigeminal autonomic cephalalgia.3 It is one of the most intense, excruciating headaches a patient can experience, and the diagnosis is often missed or delayed. Only 21% of patients receive a correct diagnosis of CH on first presentation, and the average patient visits three health care providers before the correct diagnosis is made.4,5 According to recently published results from the US Cluster Headache Survey,4 the diagnostic delay for CH averages five years or longer, limiting the patient’s access to correct treatment.
Patients with CH are prone to significant physical, social, and economic disability; most patients, for example, find it difficult to work during a CH period.1,2 Almost 20% of patients with CH report having lost a job because of their headaches, and about 8% are unemployed or on disability.4
Because of the pain severity and the associated impairment, the risk for suicide in the CH patient population is real.2,6,7 Jürgens et al7 report that 22% of patients with chronic CH and about 15% of those with episodic CH had suicidal tendencies; Rozen and Fishman4 report suicidal ideation in 55% of CH patients.
CHARACTERISTICS OF CLUSTER HEADACHE
As the name implies, attacks of this headache type tend to “cluster” together. In 85% to 90% of patients, CH is episodic, with cluster periods of headache attacks commonly lasting for one week to one year, and intervening remission periods that may last from one month to years.3,8 The remaining 10% to 15% of CH patients have the chronic CH type, in which cluster periods typically last for more than one year and are separated by remission periods lasting one month or less.3,8
Cluster headaches tend to occur in predictable patterns, often in the spring and fall.9-11 Most headaches begin between early evening and early morning, and patients are often awakened by CH during the night; according to responses in the US Cluster Headache Survey,4 onset times peak between midnight and 3 AM. Attacks can occur when the neck is rotated or flexed in specific ways; external pressure to the transverse processes of C4 or the nerve root of C2 can trigger a CH attack.12
Other CH triggers include alcohol (especially beer and red wine4,13), histamine, nitroglycerine, carbon dioxide, certain odors, and weather changes.3,4,11,14 Eighty percent or more of CH patients have a history of prolonged tobacco use, and at least 60% of CH patients who do not smoke were the children of smokers.15 No clear relationship has been found between CH and hormones.9,10
EPIDEMIOLOGY
Cluster headache is relatively rare, affecting about 0.1% of the population.8-10,16 Onset of the condition usually occurs between ages 20 and 40, and men are three to four times more likely to be affected than are women.3,16 A familial/genetic relationship may exist.10,17,18
DIAGNOSIS
Patient History
Diagnosis of headache relies heavily on the patient’s clinical history and physical exam.3,8,10 A detailed history should include the initial onset of CH, progression of the condition, and information about any precipitating event(s) and prodromal symptoms. Clinicians should document the pattern of pain by including specific information regarding its location, severity, quality, frequency, and duration. Of considerable value is the patient’s use of an accurate headache diary, which clinicians should encourage headache patients to maintain; in these, patients should be instructed to record the headache characteristics mentioned.3,8
Associated symptoms (assessed by conducting a complete review of systems), aggravating and alleviating factors, previous medical history, and psychosocial and family history are important in formulating the differential diagnosis, as misdiagnosis of CH is often related to inadequate history intake.13
Presentation
Cluster headaches share three main features: they are unilateral; they are associated with autonomic symptoms; and attacks tend to “cluster” in a circannual pattern (ie, clusters occurring at the same time of year) and/or circadian pattern (headache at the same time of day).8,19 The most common locations for cluster headaches are unilateral orbital, supraorbital, temporal, or a combination of these locations.3,8
About 30% of patients describe the pain as “stabbing,”3 and it is often compared to “a hot poker in the eye.” Pain peaks rapidly, usually within five to 10 minutes. It may radiate to the ipsilateral forehead, jaw, cheek, and/or teeth.3 Patients appear restless and agitated, unable to lie still.2,10 They often sit, holding their heads, and may pace the floor or bang their heads against the wall.
CH is associated with at least one of the following autonomic symptoms, occurring in the ipsilateral side of the head: conjunctival injection, nasal congestion, forehead and facial sweating, eyelid edema, lacrimation, rhinorrhea, ptosis, and miosis.3,13 Headaches may occur on one side of the head throughout one cluster episode, then shift to the contralateral side in subsequent periods.10 Aura occurs in 14% to 20% of patients,13,20-22 and nausea, as well as ipsilateral
visual, sensory, and speech/language disturbances have also been reported.3 Each CH attack lasts between 15 minutes and three hours, and attacks may range in frequency from one every other day to eight per day.3,13
Patients who have experienced at least five episodes of these headache symptoms, with severe pain in the specified areas and duration, accompanying autonomic symptoms, specified attack frequency, and symptoms not attributed to another disorder meet the diagnostic criteria for cluster headache given in the second edition of the International Classification of Headache Disorders (ICHD-II, 2004).3 The ICHD-II criteria, based on clinical and epidemiologic research, are recognized as a consensus guideline that is accepted worldwide to facilitate clinical practice.3 Patients who have experienced attacks fulfilling all but one of the ICHD-II criteria for CH are diagnosed with probable CH3 or cluster-like headache (CLH).23
Physical Examination
A thorough physical examination, including an investigation of the neurologic system, is essential to differentiate among primary, secondary, and other headache types. In the patient with CH, no neurologic deficits or deficits that suggest underlying disorders are usually found.3,10
Differential Diagnosis
In the evaluation of headache, it is important to differentiate CH from the other trigeminal autonomic cephalalgias: paroxysmal hemicrania (PH), short-lasting unilateral neuralgiform headache with conjunctival injection and tearing (SUNCT), and possibly hemicrania continua.3,24,25 As in CH, the pain of PH is severe, unilateral, and stabbing in quality; it, too, is associated with autonomic symptoms, often occurs at night, and can be episodic or chronic.3 However, PH headache lasts for only 2 to 30 minutes and can occur five times or more per day. Though difficult to distinguish from CH patients, those with PH usually respond to indomethacin, whereas those with CH ordinarily do not.3,8
As in patients with CH, those affected by SUNCT experience autonomic symptoms—most commonly, conjunctival injection and tearing.3,26 SUNCT differs from CH, however, in that the pain is moderate in severity, with a pulsating, burning, electric-like quality. Duration is much shorter, with episodes lasting between 5 seconds and 4 minutes.3,26
Hemicrania continua, though unilateral, is described as continuous and moderate in intensity. Like PH, it is also indomethacin-responsive.8,25,27
A broader differential diagnosis for CH, as detailed in the table,11,3,8,13,26,28 includes the other primary headaches: tension headache, migraine headache, and trigeminal neuralgia.3,26Tension headache, which affects 30% to 78% of the general population,3 is subdivided into infrequent episodic, frequent episodic, and chronic tension-type headache. Unlike CH, tension headache is mild to moderate in intensity and occurs bilaterally, with nonpulsating pressure or a tightening sensation. It is not aggravated by routine physical activity, nor is it associated with nausea, vomiting, or photophobia.
Migraine headache, also a more common primary headache type than CH,3 occurs unilaterally, is moderate to severe in intensity, and is often described as throbbing. More gradual than CH in onset, migraine is often associated with nausea, vomiting, photophobia, phonophobia, and/or visual aura. Migraine headache lacks the ipsilateral autonomic manifestations of CH, and migraineurs prefer to rest or sleep—in contrast to the extreme restlessness or agitation seen in CH patients.3
Also like CH, trigeminal neuralgia is unilateral with a trigeminal nerve distribution, and the pain can be severe and stabbing.3 However, trigeminal neuralgia lacks the autonomic symptoms associated with CH, and the pain lasts from only seconds to minutes. This headache type is often triggered by washing, shaving, or brushing teeth.3
It is also critical to exclude secondary headaches, especially those with serious causes, including meningitis, subarachnoid hemorrhage, epidural or subdural hematoma, glaucoma, tumors, temporal arteritis, or purulent sinusitis.27 Red flags associated with these conditions are:
- A complaint of the patient’s “worst headache ever” (thunderclap headache)
- First severe headache
- A subacute headache worsening over days or weeks
- An abnormal neurologic examination
- Fever or other unexplained systemic signs
- A headache preceded by vomiting
- Headache that is induced by bending, lifting, or coughing
- Headache that disturbs the patient’s sleep or presents immediately upon awakening
- History of known systemic illness
- Headache onset after age 55; and
- Pain associated with local tenderness, for example, near the temporal artery.27
DIAGNOSTIC TESTING
Since, by definition, primary headaches are those without underlying organic causes, diagnostic tests and neuroimaging studies are generally not recommended,10,29 especially when the patient history and presentation confirm the required ICHD-II diagnostic criteria. However, neuroimaging is often recommended for a patient with CH or CH-like presentations.8,23,30-32
In a literature review published in 2006, Detsky et al30 examined the correlation between clinical features of headache (as described in the ICHD-II criteria) and intracranial abnormality (as found on CT or MRI). They found an increased risk for serious intracranial abnormalities among study subjects with cluster-type headache. In any patient with chronic headache, they found, abnormal neurologic findings on physical exam represent the greatest predictor for intracranial pathology.30
In a similar study of 1,872 consecutive patients with nonacute headaches who underwent CT or MRI, one of 20 patients with CH was found to have a pituitary adenoma.33 When Favier et al34 reviewed 31 cases of trigeminal autonomic cephalalgia (TAC), including 10 with atypical symptoms, they found that even typical TAC can result from underlying pathologies with rare warning signs and symptoms. In some patients, neuroimaging study results were normal on initial diagnosis, but pathologies were discovered later after symptoms worsened or treatments ceased to be effective, prompting further imaging studies.
In a review of case studies of patients with CLH, Mainardi et al23 found that of 38 patients who fulfilled the ICHD-II criteria for CH, 12 patients (31.6%) had vascular pathologies, 12 (31.6%) had tumors, and five (13.2%) had inflammatory or infectious pathologies. The researchers recommended that all patients with symptoms of CH or CLH undergo cerebral MRI with contrast medium, even though the yield for abnormal findings would likely be low.23 Wilbrink et al32 also found a wide range of pathologies without typical warning signs or symptoms among 56 case studies of TAC and TAC-like syndromes—and recommended that all such patients be considered candidates for neuroimaging.
Recommendations from both the Scottish Intercollegiate Guidelines Network (SIGN)8 and the Taiwan Headache Society treatment guidelines31 include neuroimaging of patients with CH or CLH.
MANAGEMENT
Clinicians may wish to consider referring patients to a neurologist at the initial diagnosis of CH. Patients with atypical symptomatology or neurologic abnormalities, and those who respond insufficiently to treatment warrant a neurology referral for further investigation.
The two treatment strategies for CH are first, symptomatic treatment for acute attacks; and second, intervention to prevent or reduce further attacks and to shorten the cluster period.8,11,14,35,36
Acute Treatment
Acute symptomatic treatment is aimed at aborting the pain within 15 to 30 minutes from headache onset.14,35,36 Currently, it is generally accepted that 100% oxygen and parenteral triptans (5-HT1B/1D, not through the alimentary tract) are considered first-line treatment options.8,11,13,14,35,36
For the majority of patients (particularly those with episodic CH), inhaled normobaric oxygen effectively relieves CH pain within 15 minutes.14,37-39 Oxygen is administered at 6 to 12 L/min with a nonrebreather mask for at least 15 to 20 minutes.8,14,13 Although associated adverse events are rare, oxygen is inconvenient to transport, and it poses a fire hazard. Additionally, high-flow oxygen is contraindicated in patients with chronic obstructive pulmonary disease, as these patients depend on the hypoxic drive and run the risk of respiratory depression.14,40,41
Triptans, too, have been found effective in the acute treatment of CH; administration by subcutaneous injection or intranasal delivery is considered more effective than the oral route due to faster onset of action,11,14,35 and oxygen use may enhance triptans’ efficacy.38 In two 2010 reviews of the relevant literature, subcutaneous sumatriptan, dosed at either 6 mg or 12 mg, provided effective pain relief within 15 minutes for most patients, with no statistical between-dosage differences.11,35 The most common adverse effects were injection-site reactions, nausea, vomiting, dizziness, fatigue, and paresthesias.35
Intranasal zolmitriptan (5 mg and 10 mg) and intranasal sumatriptan 20 mg were also found effective, with significant pain relief within 30 minutes. Bad taste is a common complaint.35 Of note, both sumatriptan and zolmitriptan are contraindicated in patients with cardiovascular or cerebrovascular disease.14
In an older study of efficacy, safety, and tolerability of subcutaneous sumatriptan, almost 70% of patients averaging between one and six CH attacks per day were found to be using more than the 12-mg maximum recommended daily dosage—as much as 36 mg in a 24-hour period.42 Nevertheless, the researchers concluded that subcutaneous sumatriptan was effective and well tolerated without decreased efficacy over one year in patients with CH.
Ergotamine, once commonly used for the acute treatment of CH, has fallen out of favor in recent years due to its vasoconstrictive effects and serious adverse effects profile.14,35 Dihydroergotamine (DHE) is most effective when administered by IV (though not easily accessible for an acute attack); however, evidence regarding its efficacy and tolerability in other forms is insufficient to recommend DHE for acute CH therapy.14,35
Intranasal lidocaine, somatostatin by infusion, and subcutaneous octreotide are considered second-line treatment choices for patients who are resistant to first-line therapies or who cannot tolerate them.14,35,43,44
Cluster Headache Prophylaxis
A CH period can last for weeks to months. Prophylactic modalities, which are intended to shorten this period and to reduce the frequency and severity of headache attacks, are categorized into transitional and maintenance prophylaxis treatments.14,35
Transitional prophylaxis, a shorter course of treatment, is often started with maintenance prophylaxis (which is used throughout each cluster period) to hasten the response to the maintenance treatment. Corticosteroids are commonly used as a transitional treatment modality. In prednisone use, a starting dose of at least 40 mg/d by mouth is often required to provide benefit.14 The peak dose is usually given for three to 10 days, then gradually tapered over the succeeding 10 to 30 days. Headache recurrence is common during the prednisone taper. Ergotamine tartrate and DHE are also used as transitional prophylaxis treatment for CH.14
Verapamil is considered the maintenance prophylaxis drug of choice due to its efficacy and safety.14,35,45 The dosage required for adequate response ranges from 200 mg to 960 mg/d, in divided doses or in extended-release formulation. Most patients respond to daily doses between 200 mg and 480 mg.14,46,47 Constipation is the most common adverse effect. Slow titration and frequent ECG monitoring, particularly when dosing is increased, are necessary to avoid heart block, bradycardia, hypotension, and peripheral edema.13
Lithium is often used as second-line therapy for maintenance prophylaxis, possibly in combination with verapamil or topiramate, to improve pain control.35 Lithium carbonate, given at a dosage of 600 to 900 mg/d to maintain a serum level of 0.4 to 0.8 mEq/L, and topiramate, at dosages ranging from 50 to 200 mg/d, may be needed to achieve an adequate response.14
In at least one small study, melatonin (10 mg/d) has been associated with CH remission in 50% of treated patients.14,48 It may be used in combination with other prophylactic medications.14
Among numerous other agents that have been used for CH prophylaxis, neither sodium valproate, sumatriptan, cimetidine/chlorpheniramine, misoprostol, nor oxygen is recommended for prevention of CH.35
Narcotics
Because of its excruciating pain, CH has been referred to as “suicide headache.”2,6,7,10,13 Acute and prophylactic treatments for CH will likely reduce the number of headache attacks; however, with CH attacks as frequent as eight times per day, these treatments may not be adequate.24,49
The use of any narcotic is not ideal due to its potential for addiction, and it may cause medication-overuse headache. Furthermore, in oral form, a narcotic may not relieve CH pain quickly enough. Low-dose levomethadone is an opioid that has been used prophylactically with some success in patients with chronic CH.24,49 However, the primary care provider whose CH patient finds pain control inadequate should refer to a neurologist or a pain management specialist for evaluation—and possibly for treatment with an invasive procedure.
Invasive Procedures
Greater occipital nerve block has shown promise in the treatment of CH.50,51 In a small, double-blind study, patients with episodic or chronic CH were randomized to receive a suboccipital injection, of either combined long- and rapid-acting betamethasone or saline (placebo), in the area of the greater occipital nerve.50 Eighty-five percent of treated patients were free of headache attacks within 72 hours, compared with none in the control group. Use of lidocaine with triamcinolone was found somewhat less effective.51
Occipital nerve stimulation has also shown promise for patients with chronic CH who become resistant or are unresponsive to conventional treatments, or who cannot tolerate them.14,24 It appears to induce gradual neuromodulation, with gradual benefits (after six to 30 months). Deep brain stimulation (ie, of the posterior hypothalamus), delivered via implanted electrodes, and other procedures have produced results ranging from “excellent” to “transient remission,” reducing the use of ablative surgeries.14,52 Because invasive modalities carry a risk for serious adverse effects,14 their use should be reserved for a select patient population.
PATIENT/FAMILY EDUCATION
Patients with CH need to be educated regarding the nature, signs and symptoms, and triggers of CH. The indications for acute and prophylactic treatments and the adverse effects associated with each therapy must also be reviewed. Clear follow-up instructions are essential, including what conditions warrant further evaluation: worsening of the condition, changes in symptoms (impaired alertness, vision, movement, or sensation; onset of seizures), or treatment failure.
CONCLUSION
Cluster headache, a relatively uncommon primary headache that can cause excruciating and debilitating pain, is often misdiagnosed and inappropriately treated, with serious physical, social, and economic ramifications. This headache type is unilateral, associated with autonomic symptoms, and characterized by clustering of headache/remission periods in a circannual and/or circadian pattern. Diagnosis is made through the health history and physical exam, based on criteria from the ICHD-II. Neuroimaging may not be necessary, but given the evidence that CH and TAC are often associated with serious underlying pathology, MRI with contrast should be considered, and consultation with a neurologist at initial diagnosis is recommended.
Treatment is aimed at aborting the pain within 15 to 30 minutes of an acute headache attack and preventing further episodes through transitional and maintenance prophylaxis. Newer invasive options that are showing great promise may be considered for a select patient population. Clinicians should involve patients in treatment decisions that will address their individual needs, improving function and optimizing outcomes.
Headache is one of the health problems most commonly associated with significant morbidity, as well as considerable social and economic repercussions.1,2 Headaches are classified into three main types:
- Primary headaches, or headaches without known organic causes
- Secondary headaches, or headaches manifesting with symptoms due to organic causes; and
- Cranial neuralgias, facial pain, and other headaches.3
Cluster headache (CH) is a type of primary headache—one of the headache types encompassed by the term trigeminal autonomic cephalalgia.3 It is one of the most intense, excruciating headaches a patient can experience, and the diagnosis is often missed or delayed. Only 21% of patients receive a correct diagnosis of CH on first presentation, and the average patient visits three health care providers before the correct diagnosis is made.4,5 According to recently published results from the US Cluster Headache Survey,4 the diagnostic delay for CH averages five years or longer, limiting the patient’s access to correct treatment.
Patients with CH are prone to significant physical, social, and economic disability; most patients, for example, find it difficult to work during a CH period.1,2 Almost 20% of patients with CH report having lost a job because of their headaches, and about 8% are unemployed or on disability.4
Because of the pain severity and the associated impairment, the risk for suicide in the CH patient population is real.2,6,7 Jürgens et al7 report that 22% of patients with chronic CH and about 15% of those with episodic CH had suicidal tendencies; Rozen and Fishman4 report suicidal ideation in 55% of CH patients.
CHARACTERISTICS OF CLUSTER HEADACHE
As the name implies, attacks of this headache type tend to “cluster” together. In 85% to 90% of patients, CH is episodic, with cluster periods of headache attacks commonly lasting for one week to one year, and intervening remission periods that may last from one month to years.3,8 The remaining 10% to 15% of CH patients have the chronic CH type, in which cluster periods typically last for more than one year and are separated by remission periods lasting one month or less.3,8
Cluster headaches tend to occur in predictable patterns, often in the spring and fall.9-11 Most headaches begin between early evening and early morning, and patients are often awakened by CH during the night; according to responses in the US Cluster Headache Survey,4 onset times peak between midnight and 3 AM. Attacks can occur when the neck is rotated or flexed in specific ways; external pressure to the transverse processes of C4 or the nerve root of C2 can trigger a CH attack.12
Other CH triggers include alcohol (especially beer and red wine4,13), histamine, nitroglycerine, carbon dioxide, certain odors, and weather changes.3,4,11,14 Eighty percent or more of CH patients have a history of prolonged tobacco use, and at least 60% of CH patients who do not smoke were the children of smokers.15 No clear relationship has been found between CH and hormones.9,10
EPIDEMIOLOGY
Cluster headache is relatively rare, affecting about 0.1% of the population.8-10,16 Onset of the condition usually occurs between ages 20 and 40, and men are three to four times more likely to be affected than are women.3,16 A familial/genetic relationship may exist.10,17,18
DIAGNOSIS
Patient History
Diagnosis of headache relies heavily on the patient’s clinical history and physical exam.3,8,10 A detailed history should include the initial onset of CH, progression of the condition, and information about any precipitating event(s) and prodromal symptoms. Clinicians should document the pattern of pain by including specific information regarding its location, severity, quality, frequency, and duration. Of considerable value is the patient’s use of an accurate headache diary, which clinicians should encourage headache patients to maintain; in these, patients should be instructed to record the headache characteristics mentioned.3,8
Associated symptoms (assessed by conducting a complete review of systems), aggravating and alleviating factors, previous medical history, and psychosocial and family history are important in formulating the differential diagnosis, as misdiagnosis of CH is often related to inadequate history intake.13
Presentation
Cluster headaches share three main features: they are unilateral; they are associated with autonomic symptoms; and attacks tend to “cluster” in a circannual pattern (ie, clusters occurring at the same time of year) and/or circadian pattern (headache at the same time of day).8,19 The most common locations for cluster headaches are unilateral orbital, supraorbital, temporal, or a combination of these locations.3,8
About 30% of patients describe the pain as “stabbing,”3 and it is often compared to “a hot poker in the eye.” Pain peaks rapidly, usually within five to 10 minutes. It may radiate to the ipsilateral forehead, jaw, cheek, and/or teeth.3 Patients appear restless and agitated, unable to lie still.2,10 They often sit, holding their heads, and may pace the floor or bang their heads against the wall.
CH is associated with at least one of the following autonomic symptoms, occurring in the ipsilateral side of the head: conjunctival injection, nasal congestion, forehead and facial sweating, eyelid edema, lacrimation, rhinorrhea, ptosis, and miosis.3,13 Headaches may occur on one side of the head throughout one cluster episode, then shift to the contralateral side in subsequent periods.10 Aura occurs in 14% to 20% of patients,13,20-22 and nausea, as well as ipsilateral
visual, sensory, and speech/language disturbances have also been reported.3 Each CH attack lasts between 15 minutes and three hours, and attacks may range in frequency from one every other day to eight per day.3,13
Patients who have experienced at least five episodes of these headache symptoms, with severe pain in the specified areas and duration, accompanying autonomic symptoms, specified attack frequency, and symptoms not attributed to another disorder meet the diagnostic criteria for cluster headache given in the second edition of the International Classification of Headache Disorders (ICHD-II, 2004).3 The ICHD-II criteria, based on clinical and epidemiologic research, are recognized as a consensus guideline that is accepted worldwide to facilitate clinical practice.3 Patients who have experienced attacks fulfilling all but one of the ICHD-II criteria for CH are diagnosed with probable CH3 or cluster-like headache (CLH).23
Physical Examination
A thorough physical examination, including an investigation of the neurologic system, is essential to differentiate among primary, secondary, and other headache types. In the patient with CH, no neurologic deficits or deficits that suggest underlying disorders are usually found.3,10
Differential Diagnosis
In the evaluation of headache, it is important to differentiate CH from the other trigeminal autonomic cephalalgias: paroxysmal hemicrania (PH), short-lasting unilateral neuralgiform headache with conjunctival injection and tearing (SUNCT), and possibly hemicrania continua.3,24,25 As in CH, the pain of PH is severe, unilateral, and stabbing in quality; it, too, is associated with autonomic symptoms, often occurs at night, and can be episodic or chronic.3 However, PH headache lasts for only 2 to 30 minutes and can occur five times or more per day. Though difficult to distinguish from CH patients, those with PH usually respond to indomethacin, whereas those with CH ordinarily do not.3,8
As in patients with CH, those affected by SUNCT experience autonomic symptoms—most commonly, conjunctival injection and tearing.3,26 SUNCT differs from CH, however, in that the pain is moderate in severity, with a pulsating, burning, electric-like quality. Duration is much shorter, with episodes lasting between 5 seconds and 4 minutes.3,26
Hemicrania continua, though unilateral, is described as continuous and moderate in intensity. Like PH, it is also indomethacin-responsive.8,25,27
A broader differential diagnosis for CH, as detailed in the table,11,3,8,13,26,28 includes the other primary headaches: tension headache, migraine headache, and trigeminal neuralgia.3,26Tension headache, which affects 30% to 78% of the general population,3 is subdivided into infrequent episodic, frequent episodic, and chronic tension-type headache. Unlike CH, tension headache is mild to moderate in intensity and occurs bilaterally, with nonpulsating pressure or a tightening sensation. It is not aggravated by routine physical activity, nor is it associated with nausea, vomiting, or photophobia.
Migraine headache, also a more common primary headache type than CH,3 occurs unilaterally, is moderate to severe in intensity, and is often described as throbbing. More gradual than CH in onset, migraine is often associated with nausea, vomiting, photophobia, phonophobia, and/or visual aura. Migraine headache lacks the ipsilateral autonomic manifestations of CH, and migraineurs prefer to rest or sleep—in contrast to the extreme restlessness or agitation seen in CH patients.3
Also like CH, trigeminal neuralgia is unilateral with a trigeminal nerve distribution, and the pain can be severe and stabbing.3 However, trigeminal neuralgia lacks the autonomic symptoms associated with CH, and the pain lasts from only seconds to minutes. This headache type is often triggered by washing, shaving, or brushing teeth.3
It is also critical to exclude secondary headaches, especially those with serious causes, including meningitis, subarachnoid hemorrhage, epidural or subdural hematoma, glaucoma, tumors, temporal arteritis, or purulent sinusitis.27 Red flags associated with these conditions are:
- A complaint of the patient’s “worst headache ever” (thunderclap headache)
- First severe headache
- A subacute headache worsening over days or weeks
- An abnormal neurologic examination
- Fever or other unexplained systemic signs
- A headache preceded by vomiting
- Headache that is induced by bending, lifting, or coughing
- Headache that disturbs the patient’s sleep or presents immediately upon awakening
- History of known systemic illness
- Headache onset after age 55; and
- Pain associated with local tenderness, for example, near the temporal artery.27
DIAGNOSTIC TESTING
Since, by definition, primary headaches are those without underlying organic causes, diagnostic tests and neuroimaging studies are generally not recommended,10,29 especially when the patient history and presentation confirm the required ICHD-II diagnostic criteria. However, neuroimaging is often recommended for a patient with CH or CH-like presentations.8,23,30-32
In a literature review published in 2006, Detsky et al30 examined the correlation between clinical features of headache (as described in the ICHD-II criteria) and intracranial abnormality (as found on CT or MRI). They found an increased risk for serious intracranial abnormalities among study subjects with cluster-type headache. In any patient with chronic headache, they found, abnormal neurologic findings on physical exam represent the greatest predictor for intracranial pathology.30
In a similar study of 1,872 consecutive patients with nonacute headaches who underwent CT or MRI, one of 20 patients with CH was found to have a pituitary adenoma.33 When Favier et al34 reviewed 31 cases of trigeminal autonomic cephalalgia (TAC), including 10 with atypical symptoms, they found that even typical TAC can result from underlying pathologies with rare warning signs and symptoms. In some patients, neuroimaging study results were normal on initial diagnosis, but pathologies were discovered later after symptoms worsened or treatments ceased to be effective, prompting further imaging studies.
In a review of case studies of patients with CLH, Mainardi et al23 found that of 38 patients who fulfilled the ICHD-II criteria for CH, 12 patients (31.6%) had vascular pathologies, 12 (31.6%) had tumors, and five (13.2%) had inflammatory or infectious pathologies. The researchers recommended that all patients with symptoms of CH or CLH undergo cerebral MRI with contrast medium, even though the yield for abnormal findings would likely be low.23 Wilbrink et al32 also found a wide range of pathologies without typical warning signs or symptoms among 56 case studies of TAC and TAC-like syndromes—and recommended that all such patients be considered candidates for neuroimaging.
Recommendations from both the Scottish Intercollegiate Guidelines Network (SIGN)8 and the Taiwan Headache Society treatment guidelines31 include neuroimaging of patients with CH or CLH.
MANAGEMENT
Clinicians may wish to consider referring patients to a neurologist at the initial diagnosis of CH. Patients with atypical symptomatology or neurologic abnormalities, and those who respond insufficiently to treatment warrant a neurology referral for further investigation.
The two treatment strategies for CH are first, symptomatic treatment for acute attacks; and second, intervention to prevent or reduce further attacks and to shorten the cluster period.8,11,14,35,36
Acute Treatment
Acute symptomatic treatment is aimed at aborting the pain within 15 to 30 minutes from headache onset.14,35,36 Currently, it is generally accepted that 100% oxygen and parenteral triptans (5-HT1B/1D, not through the alimentary tract) are considered first-line treatment options.8,11,13,14,35,36
For the majority of patients (particularly those with episodic CH), inhaled normobaric oxygen effectively relieves CH pain within 15 minutes.14,37-39 Oxygen is administered at 6 to 12 L/min with a nonrebreather mask for at least 15 to 20 minutes.8,14,13 Although associated adverse events are rare, oxygen is inconvenient to transport, and it poses a fire hazard. Additionally, high-flow oxygen is contraindicated in patients with chronic obstructive pulmonary disease, as these patients depend on the hypoxic drive and run the risk of respiratory depression.14,40,41
Triptans, too, have been found effective in the acute treatment of CH; administration by subcutaneous injection or intranasal delivery is considered more effective than the oral route due to faster onset of action,11,14,35 and oxygen use may enhance triptans’ efficacy.38 In two 2010 reviews of the relevant literature, subcutaneous sumatriptan, dosed at either 6 mg or 12 mg, provided effective pain relief within 15 minutes for most patients, with no statistical between-dosage differences.11,35 The most common adverse effects were injection-site reactions, nausea, vomiting, dizziness, fatigue, and paresthesias.35
Intranasal zolmitriptan (5 mg and 10 mg) and intranasal sumatriptan 20 mg were also found effective, with significant pain relief within 30 minutes. Bad taste is a common complaint.35 Of note, both sumatriptan and zolmitriptan are contraindicated in patients with cardiovascular or cerebrovascular disease.14
In an older study of efficacy, safety, and tolerability of subcutaneous sumatriptan, almost 70% of patients averaging between one and six CH attacks per day were found to be using more than the 12-mg maximum recommended daily dosage—as much as 36 mg in a 24-hour period.42 Nevertheless, the researchers concluded that subcutaneous sumatriptan was effective and well tolerated without decreased efficacy over one year in patients with CH.
Ergotamine, once commonly used for the acute treatment of CH, has fallen out of favor in recent years due to its vasoconstrictive effects and serious adverse effects profile.14,35 Dihydroergotamine (DHE) is most effective when administered by IV (though not easily accessible for an acute attack); however, evidence regarding its efficacy and tolerability in other forms is insufficient to recommend DHE for acute CH therapy.14,35
Intranasal lidocaine, somatostatin by infusion, and subcutaneous octreotide are considered second-line treatment choices for patients who are resistant to first-line therapies or who cannot tolerate them.14,35,43,44
Cluster Headache Prophylaxis
A CH period can last for weeks to months. Prophylactic modalities, which are intended to shorten this period and to reduce the frequency and severity of headache attacks, are categorized into transitional and maintenance prophylaxis treatments.14,35
Transitional prophylaxis, a shorter course of treatment, is often started with maintenance prophylaxis (which is used throughout each cluster period) to hasten the response to the maintenance treatment. Corticosteroids are commonly used as a transitional treatment modality. In prednisone use, a starting dose of at least 40 mg/d by mouth is often required to provide benefit.14 The peak dose is usually given for three to 10 days, then gradually tapered over the succeeding 10 to 30 days. Headache recurrence is common during the prednisone taper. Ergotamine tartrate and DHE are also used as transitional prophylaxis treatment for CH.14
Verapamil is considered the maintenance prophylaxis drug of choice due to its efficacy and safety.14,35,45 The dosage required for adequate response ranges from 200 mg to 960 mg/d, in divided doses or in extended-release formulation. Most patients respond to daily doses between 200 mg and 480 mg.14,46,47 Constipation is the most common adverse effect. Slow titration and frequent ECG monitoring, particularly when dosing is increased, are necessary to avoid heart block, bradycardia, hypotension, and peripheral edema.13
Lithium is often used as second-line therapy for maintenance prophylaxis, possibly in combination with verapamil or topiramate, to improve pain control.35 Lithium carbonate, given at a dosage of 600 to 900 mg/d to maintain a serum level of 0.4 to 0.8 mEq/L, and topiramate, at dosages ranging from 50 to 200 mg/d, may be needed to achieve an adequate response.14
In at least one small study, melatonin (10 mg/d) has been associated with CH remission in 50% of treated patients.14,48 It may be used in combination with other prophylactic medications.14
Among numerous other agents that have been used for CH prophylaxis, neither sodium valproate, sumatriptan, cimetidine/chlorpheniramine, misoprostol, nor oxygen is recommended for prevention of CH.35
Narcotics
Because of its excruciating pain, CH has been referred to as “suicide headache.”2,6,7,10,13 Acute and prophylactic treatments for CH will likely reduce the number of headache attacks; however, with CH attacks as frequent as eight times per day, these treatments may not be adequate.24,49
The use of any narcotic is not ideal due to its potential for addiction, and it may cause medication-overuse headache. Furthermore, in oral form, a narcotic may not relieve CH pain quickly enough. Low-dose levomethadone is an opioid that has been used prophylactically with some success in patients with chronic CH.24,49 However, the primary care provider whose CH patient finds pain control inadequate should refer to a neurologist or a pain management specialist for evaluation—and possibly for treatment with an invasive procedure.
Invasive Procedures
Greater occipital nerve block has shown promise in the treatment of CH.50,51 In a small, double-blind study, patients with episodic or chronic CH were randomized to receive a suboccipital injection, of either combined long- and rapid-acting betamethasone or saline (placebo), in the area of the greater occipital nerve.50 Eighty-five percent of treated patients were free of headache attacks within 72 hours, compared with none in the control group. Use of lidocaine with triamcinolone was found somewhat less effective.51
Occipital nerve stimulation has also shown promise for patients with chronic CH who become resistant or are unresponsive to conventional treatments, or who cannot tolerate them.14,24 It appears to induce gradual neuromodulation, with gradual benefits (after six to 30 months). Deep brain stimulation (ie, of the posterior hypothalamus), delivered via implanted electrodes, and other procedures have produced results ranging from “excellent” to “transient remission,” reducing the use of ablative surgeries.14,52 Because invasive modalities carry a risk for serious adverse effects,14 their use should be reserved for a select patient population.
PATIENT/FAMILY EDUCATION
Patients with CH need to be educated regarding the nature, signs and symptoms, and triggers of CH. The indications for acute and prophylactic treatments and the adverse effects associated with each therapy must also be reviewed. Clear follow-up instructions are essential, including what conditions warrant further evaluation: worsening of the condition, changes in symptoms (impaired alertness, vision, movement, or sensation; onset of seizures), or treatment failure.
CONCLUSION
Cluster headache, a relatively uncommon primary headache that can cause excruciating and debilitating pain, is often misdiagnosed and inappropriately treated, with serious physical, social, and economic ramifications. This headache type is unilateral, associated with autonomic symptoms, and characterized by clustering of headache/remission periods in a circannual and/or circadian pattern. Diagnosis is made through the health history and physical exam, based on criteria from the ICHD-II. Neuroimaging may not be necessary, but given the evidence that CH and TAC are often associated with serious underlying pathology, MRI with contrast should be considered, and consultation with a neurologist at initial diagnosis is recommended.
Treatment is aimed at aborting the pain within 15 to 30 minutes of an acute headache attack and preventing further episodes through transitional and maintenance prophylaxis. Newer invasive options that are showing great promise may be considered for a select patient population. Clinicians should involve patients in treatment decisions that will address their individual needs, improving function and optimizing outcomes.
1. Stovner LJ, Hagen H, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193-210.
2. Vaughan R. My own private purgatory: how cluster headaches affect my life. Headache. 2008;48(10):1541-1543.
3. Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders. 2nd ed. Cephalalgia. 2004;24 suppl 1:9-160.
4. Rozen TD, Fishman RS. Cluster headache in the United States of America: demographics, clinical characteristics, triggers, suicidality, and personal burden. Headache. 2012;52(1): 99-113.
5. Bahra A, Goadsby PJ. Diagnostic delays and mis-management in cluster headache. Acta Neurol Scand. 2004;109(3):175-179.
6. Dousset V, Laporte A, Legoff M, et al. Validation of a brief self-administered questionnaire for cluster headache screening in a tertiary center. Headache. 2009;49(1):64-70.
7. Jürgens TP, Gaul C, Lindwurm A, et al. Impairment in episodic and chronic cluster headache. Cephalalgia. 2011;31(6):671-682.
8. Scottish Intercollegiate Guidelines Network (SIGN). Diagnosis and management of headache in adults: a national clinical guideline. Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN). 2008;1-81. (SIGN publication; no. 107). www.sign.ac.uk/guide lines/fulltext/107/index.html. Accessed May 8, 2012.
9. Bahra A, May A, Goadsby PJ. Cluster headache: a prospective clinical study with diagnostic implications. Neurology. 2002;58(3): 354-361.
10. May A. Cluster headache: pathogenesis, diagnosis, and management. Lancet. 2005; 366(9488):843-855.
11. Law S, Derry S, Moore RA. Triptans for acute cluster headache. Cochrane Database Syst Rev. 2010 Apr 14;(4):CD008042.
12. Rozen TD. Trigeminal autonomic cephalalgias. Neurol Clin. 2009;27(2):537-556.
13. Nesbitt AD, Goadsby PJ. Cluster headache. BMJ. 2012;344:e2407. doi: 10.1136/bmj
.e2407.
14. Ashkenazi A, Schwedt T. Cluster headache: acute and prophylactic therapy. Headache. 2011;51(2):272-286.
15. Rozen TD. Cluster headache as the result of secondhand cigarette smoke exposure during childhood. Headache. 2010;50(1):130-132.
16. Fischera M, Marziniak M, Gralow I, Evers S. The incidence and prevalence of cluster headache: a meta-analysis of population-based studies. Cephalalgia. 2008;28(6):614-618.
17. De Simone R, Fiorillo C, Bonuso S, Castaldo G. A cluster headache family with possible autosomal recessive inheritance. Neurology. 2003;61(4):578-579.
18. Leone M, Russell MB, Rigamonti A, et al. Increased familial risk of cluster headache. Neurology. 2001;56(9):1233-1236.
19. Matharu MS, Boes CJ, Goadsby PJ. Management of trigeminal autonomic cephalgias and hemicrania continua. Drugs. 2003;63(16): 1637-1677.
20. Evans RW, Krymchantowski AV. Cluster and other nonmigraine primary headaches with aura. Headache. 2011;51(4):604-608.
21. Rozen TD. Cluster headache with aura. Curr Pain Headache Rep. 2011;15(2):98-100.
22. Silberstein SD, Niknam R, Rozen TD, Young WB. Cluster headache with aura. Neurology. 2000;54(1):219-221.
23. Mainardi F, Trucco M, Maggioni F, et al. Cluster-like headache: a comprehensive reappraisal. Cephalalgia. 2010;30(4):399-412.
24. Magis D, Bruno MA, Fumal A, et al. Central modulation in cluster headache patients treated with occipital nerve stimulation: an FDG-PET study. BMC Neurol. 2011;11:25.
25. Goadsby PJ, Lipton RB. A review of paroxysmal hemicranias, SUNCT syndrome and other short-lasting headaches with autonomic feature, including new cases. Brain. 1997; 120(pt 1):193-209.
26. Leone M, Bussone G. Pathophysiology of trigeminal autonomic cephalalgias. Lancet Neurol. 2009;8(8):755-764.
27. Goadsby PJ, Raskin NH. Chapter 14. Headache. In: Longo DL, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York: McGraw-Hill; 2011:112-128.
28. Bendtsen L, Jensen R. Tension-type headache. Neurol Clin. 2009;27(2):525-535.
29. van Kleef M, Lataster A, Narouze S, et al. Evidenced-based interventional pain medicine according to clinical diagnoses. 2. Cluster headache. Pain Pract. 2009;9(6):435-442.
30. Detsky ME, McDonald DR, Baerlocher MO, et al. Does this patient with headache have a migraine or need neuroimaging? JAMA. 2006; 296(10):1274-1283.
31. Treatment Guideline Subcommittee of the Taiwan Headache Society. Neuroimaging guidelines in nonacute headaches [in Chinese]. Acta Neurol Taiwan. 2010;19(2):137-144.
32. Wilbrink LA, Ferrari MD, Kruit MC, Haan J. Neuroimaging in trigeminal autonomic cephalgias: when, how, and of what? Curr Opin Neurol. 2009;22(3):247-253.
33. Sempere AP, Porta-Etessam J, Medrano V, et al. Neuroimaging in the evaluation of patients with non-acute headache. Cephalalgia. 2005;25(1):30-35.
34. Favier I, van Vliet J, Roon K, et al. Trigeminal autonomic cephalgias due to structural lesions: a review of 31 cases. Arch Neurol. 2007;64(1):25-31.
35. Francis GJ, Becker WJ, Pringsheim TM. Acute and preventive pharmacologic treatment of cluster headache. Neurology. 2010; 75(5):463-473.
36. Bennett MH, French C, Schnabel A, et al. Normobaric and hyperbaric oxygen therapy for migraine and cluster headache. Cochrane Database Syst Rev. 2008 Jul 16;(3):CD005219.
37. Rozen TD. Inhaled oxygen for cluster headache: efficacy, mechanism of action, utilization, and economics. Curr Pain Headache Rep. 2012 Jan 29. [Epub ahead of print]
38. Rozen TD, Fishman RS. Inhaled oxygen and cluster headache sufferers in the United States: use, efficacy and economics: results from the United States Cluster Headache Survey. Headache. 2011;51(2):191-200.
39. Kudrow L. Response of cluster headache attacks to oxygen inhalation. Headache. 1981; 21:1-4.
40. Cohen AS, Burns B, Goadsby PJ. High-flow oxygen for treatment of cluster headache: a randomized trial. JAMA. 2009;302(22):2451-2457.
41. Mahadevan SV. Emergency airway management. In: Auerbach PS. Wilderness Medicine. 6th ed. Stanford, CA: Elsevier; 2011.
42. Göbel H, Lindner V, Heinze A, et al. Acute therapy for cluster headache with sumatriptan: findings of a one-year long-term study. Neurology. 1998;51(3):908-911.
43. Sicuteri F, Geppetti P, Marabini S, Lembeck F. Pain relief by somatostatin in attacks of cluster headache. Pain. 1984;184:359-365.
44. Matharu MS, Levy MJ, Meeran K, Goadsby PJ. Subcutaneous octreotide in cluster headache: randomized placebo-controlled double-blind crossover study. Ann Neurol. 2004;56(4): 488-494.
45. Leone M, D’Amico D, Frediani F, et al. Verapamil in the prophylaxis of episodic cluster headache: a double-blind study versus placebo. Neurology. 2000;54(6):1382-1385.
46. Tfelt-Hansen P, Tfelt-Hansen J. Verapamil for cluster headache: clinical pharmacology and possible mode of action. Headache. 2009;49(1): 117-125.
47. Blau JN, Engel HO. Individualizing treatment with verapamil for cluster headache patients. Headache. 2004;44(10):1013-1018.
48. Leone M, D’Amico D, Moschiano F, et al. Melatonin versus placebo in the prophylaxis of cluster headache: a double-blind pilot study with parallel groups. Cephalalgia. 1996;16(7): 494-496.
49. Sprenger T, Seifert CL, Miederer M, et al. Successful prophylactic treatment of chronic cluster headache with low-dose levomethadone. J Neurol. 2008;255(11):1832-1833.
50. Ambrosini A, Vandenheede M, Rossi P, et al. Suboccipital injection with a mixture of rapid- and long-acting steroids in cluster headache: a double-blind placebo-controlled study. Pain. 2005;118(1-2):92-96.
51. Peres MF, Stiles MA, Siow HC, et al. Greater occipital nerve blockade for cluster headache. Cephalalgia. 2002;22(7):520-522.
52. Leone M, Proietti Cecchini A, Franzini A, et al. Lessons from 8 years’ experience of hypothalamic stimulation in cluster headache. Cephalalgia. 2008;28(7):787-797.
1. Stovner LJ, Hagen H, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007;27(3):193-210.
2. Vaughan R. My own private purgatory: how cluster headaches affect my life. Headache. 2008;48(10):1541-1543.
3. Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders. 2nd ed. Cephalalgia. 2004;24 suppl 1:9-160.
4. Rozen TD, Fishman RS. Cluster headache in the United States of America: demographics, clinical characteristics, triggers, suicidality, and personal burden. Headache. 2012;52(1): 99-113.
5. Bahra A, Goadsby PJ. Diagnostic delays and mis-management in cluster headache. Acta Neurol Scand. 2004;109(3):175-179.
6. Dousset V, Laporte A, Legoff M, et al. Validation of a brief self-administered questionnaire for cluster headache screening in a tertiary center. Headache. 2009;49(1):64-70.
7. Jürgens TP, Gaul C, Lindwurm A, et al. Impairment in episodic and chronic cluster headache. Cephalalgia. 2011;31(6):671-682.
8. Scottish Intercollegiate Guidelines Network (SIGN). Diagnosis and management of headache in adults: a national clinical guideline. Edinburgh (Scotland): Scottish Intercollegiate Guidelines Network (SIGN). 2008;1-81. (SIGN publication; no. 107). www.sign.ac.uk/guide lines/fulltext/107/index.html. Accessed May 8, 2012.
9. Bahra A, May A, Goadsby PJ. Cluster headache: a prospective clinical study with diagnostic implications. Neurology. 2002;58(3): 354-361.
10. May A. Cluster headache: pathogenesis, diagnosis, and management. Lancet. 2005; 366(9488):843-855.
11. Law S, Derry S, Moore RA. Triptans for acute cluster headache. Cochrane Database Syst Rev. 2010 Apr 14;(4):CD008042.
12. Rozen TD. Trigeminal autonomic cephalalgias. Neurol Clin. 2009;27(2):537-556.
13. Nesbitt AD, Goadsby PJ. Cluster headache. BMJ. 2012;344:e2407. doi: 10.1136/bmj
.e2407.
14. Ashkenazi A, Schwedt T. Cluster headache: acute and prophylactic therapy. Headache. 2011;51(2):272-286.
15. Rozen TD. Cluster headache as the result of secondhand cigarette smoke exposure during childhood. Headache. 2010;50(1):130-132.
16. Fischera M, Marziniak M, Gralow I, Evers S. The incidence and prevalence of cluster headache: a meta-analysis of population-based studies. Cephalalgia. 2008;28(6):614-618.
17. De Simone R, Fiorillo C, Bonuso S, Castaldo G. A cluster headache family with possible autosomal recessive inheritance. Neurology. 2003;61(4):578-579.
18. Leone M, Russell MB, Rigamonti A, et al. Increased familial risk of cluster headache. Neurology. 2001;56(9):1233-1236.
19. Matharu MS, Boes CJ, Goadsby PJ. Management of trigeminal autonomic cephalgias and hemicrania continua. Drugs. 2003;63(16): 1637-1677.
20. Evans RW, Krymchantowski AV. Cluster and other nonmigraine primary headaches with aura. Headache. 2011;51(4):604-608.
21. Rozen TD. Cluster headache with aura. Curr Pain Headache Rep. 2011;15(2):98-100.
22. Silberstein SD, Niknam R, Rozen TD, Young WB. Cluster headache with aura. Neurology. 2000;54(1):219-221.
23. Mainardi F, Trucco M, Maggioni F, et al. Cluster-like headache: a comprehensive reappraisal. Cephalalgia. 2010;30(4):399-412.
24. Magis D, Bruno MA, Fumal A, et al. Central modulation in cluster headache patients treated with occipital nerve stimulation: an FDG-PET study. BMC Neurol. 2011;11:25.
25. Goadsby PJ, Lipton RB. A review of paroxysmal hemicranias, SUNCT syndrome and other short-lasting headaches with autonomic feature, including new cases. Brain. 1997; 120(pt 1):193-209.
26. Leone M, Bussone G. Pathophysiology of trigeminal autonomic cephalalgias. Lancet Neurol. 2009;8(8):755-764.
27. Goadsby PJ, Raskin NH. Chapter 14. Headache. In: Longo DL, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine. 18th ed. New York: McGraw-Hill; 2011:112-128.
28. Bendtsen L, Jensen R. Tension-type headache. Neurol Clin. 2009;27(2):525-535.
29. van Kleef M, Lataster A, Narouze S, et al. Evidenced-based interventional pain medicine according to clinical diagnoses. 2. Cluster headache. Pain Pract. 2009;9(6):435-442.
30. Detsky ME, McDonald DR, Baerlocher MO, et al. Does this patient with headache have a migraine or need neuroimaging? JAMA. 2006; 296(10):1274-1283.
31. Treatment Guideline Subcommittee of the Taiwan Headache Society. Neuroimaging guidelines in nonacute headaches [in Chinese]. Acta Neurol Taiwan. 2010;19(2):137-144.
32. Wilbrink LA, Ferrari MD, Kruit MC, Haan J. Neuroimaging in trigeminal autonomic cephalgias: when, how, and of what? Curr Opin Neurol. 2009;22(3):247-253.
33. Sempere AP, Porta-Etessam J, Medrano V, et al. Neuroimaging in the evaluation of patients with non-acute headache. Cephalalgia. 2005;25(1):30-35.
34. Favier I, van Vliet J, Roon K, et al. Trigeminal autonomic cephalgias due to structural lesions: a review of 31 cases. Arch Neurol. 2007;64(1):25-31.
35. Francis GJ, Becker WJ, Pringsheim TM. Acute and preventive pharmacologic treatment of cluster headache. Neurology. 2010; 75(5):463-473.
36. Bennett MH, French C, Schnabel A, et al. Normobaric and hyperbaric oxygen therapy for migraine and cluster headache. Cochrane Database Syst Rev. 2008 Jul 16;(3):CD005219.
37. Rozen TD. Inhaled oxygen for cluster headache: efficacy, mechanism of action, utilization, and economics. Curr Pain Headache Rep. 2012 Jan 29. [Epub ahead of print]
38. Rozen TD, Fishman RS. Inhaled oxygen and cluster headache sufferers in the United States: use, efficacy and economics: results from the United States Cluster Headache Survey. Headache. 2011;51(2):191-200.
39. Kudrow L. Response of cluster headache attacks to oxygen inhalation. Headache. 1981; 21:1-4.
40. Cohen AS, Burns B, Goadsby PJ. High-flow oxygen for treatment of cluster headache: a randomized trial. JAMA. 2009;302(22):2451-2457.
41. Mahadevan SV. Emergency airway management. In: Auerbach PS. Wilderness Medicine. 6th ed. Stanford, CA: Elsevier; 2011.
42. Göbel H, Lindner V, Heinze A, et al. Acute therapy for cluster headache with sumatriptan: findings of a one-year long-term study. Neurology. 1998;51(3):908-911.
43. Sicuteri F, Geppetti P, Marabini S, Lembeck F. Pain relief by somatostatin in attacks of cluster headache. Pain. 1984;184:359-365.
44. Matharu MS, Levy MJ, Meeran K, Goadsby PJ. Subcutaneous octreotide in cluster headache: randomized placebo-controlled double-blind crossover study. Ann Neurol. 2004;56(4): 488-494.
45. Leone M, D’Amico D, Frediani F, et al. Verapamil in the prophylaxis of episodic cluster headache: a double-blind study versus placebo. Neurology. 2000;54(6):1382-1385.
46. Tfelt-Hansen P, Tfelt-Hansen J. Verapamil for cluster headache: clinical pharmacology and possible mode of action. Headache. 2009;49(1): 117-125.
47. Blau JN, Engel HO. Individualizing treatment with verapamil for cluster headache patients. Headache. 2004;44(10):1013-1018.
48. Leone M, D’Amico D, Moschiano F, et al. Melatonin versus placebo in the prophylaxis of cluster headache: a double-blind pilot study with parallel groups. Cephalalgia. 1996;16(7): 494-496.
49. Sprenger T, Seifert CL, Miederer M, et al. Successful prophylactic treatment of chronic cluster headache with low-dose levomethadone. J Neurol. 2008;255(11):1832-1833.
50. Ambrosini A, Vandenheede M, Rossi P, et al. Suboccipital injection with a mixture of rapid- and long-acting steroids in cluster headache: a double-blind placebo-controlled study. Pain. 2005;118(1-2):92-96.
51. Peres MF, Stiles MA, Siow HC, et al. Greater occipital nerve blockade for cluster headache. Cephalalgia. 2002;22(7):520-522.
52. Leone M, Proietti Cecchini A, Franzini A, et al. Lessons from 8 years’ experience of hypothalamic stimulation in cluster headache. Cephalalgia. 2008;28(7):787-797.
Ménière Disease and the Elderly
Burn Scar Carcinoma: Patients With Marjolin's Ulcer
The Effects of a Glipizide Formulary Conversion
Amyotrophic Lateral Sclerosis, Nutrition, and Feeding Tube Placement
.
.
.
Grand Rounds: Man, 65, With Heart Failure Symptoms
A black man, age 65, with no known history of cardiopulmonary disease presented with acute-onset exertional dyspnea and lower extremity edema. He also reported an episode of syncope, as well as occasional dizziness and abdominal bloating. He said he experienced exertional dyspnea while doing a routine step aerobic exercise. His exercise regimen included distance walking, yoga, and aerobics four to five days per week.
The patient’s medical history was remarkable for a single episode of a bleeding ulcer in previous years, low back pain, shoulder pain, and a septic arthritic hip. His social history was negative for use of tobacco, alcohol, or illegal drugs. He was married and had two biological daughters with fairly unremarkable medical histories. The patient had earned a master’s degree, worked full-time in the insurance business, and was an avid worldwide traveler. He reported diminished quality of life as a result of his acute-onset heart failure symptoms, which reduced his ability to exercise routinely, work full-time, or travel.
The patient’s sudden experience of exertional dyspnea prompted him to visit his primary care provider, who ordered an ECG that demonstrated low voltage patterns and a first-degree atrioventricular (AV) block. Subsequent stress echocardiography showed generalized thickening of the left ventricular myocardium. Posterior wall thickness measured 1.7 cm (normal range, 0.6 to 1.1 cm), septal thickness measured 1.9 cm (normal, 0.6 to 1.1 cm), and ejection fraction was 65%. The stress echocardiogram also showed a speckling pattern (brightly scattered spots) on the myocardium.
Although stress echocardiography results were negative for ischemic disease, the patient did experience dyspnea during the exam. He underwent cardiac catheterization, which indicated normal coronary arteries.
Additional diagnostic studies included cardiac MRI with and without contrast, which showed nulling of the heart muscle and delayed patchy hyperenhancement; this suggested myocardial tissue abnormality as result of amyloid fibril deposition.1 Both pulmonary and tricuspid aortic valves were normal, with no evidence of stenosis. No regional wall motion abnormalities were noted.
Laboratory findings during the work-up were lipid panel, unremarkable; complete blood count (CBC), mild anemia and leukopenia; and urinalysis, positive for proteinuria. Brain natriuretic peptide (BNP) was measured at 686 pg/mL (normal, 0.0 to 100 pg/mL), indicating moderate heart failure. A peripheral blood smear was negative for monoclonal plasma cells.
The patient’s physical exam was unremarkable except for 2+ pedal edema bilaterally. In consideration of normal coronary arteries on cardiac catheterization, the patient’s heart failure symptoms, and stress echocardiography abnormalities, a heart biopsy was ordered. An endomyocardial biopsy with Congo Red stain demonstrated an apple-green birefringent pattern viewed under high-definition polarized light microscope, which was consistent with amyloid deposition.2
The patient was given a diagnosis of primary amyloidosis by his local cardiologist despite negative findings on the peripheral blood smear for monoclonal plasma cells (which are typically found in primary amyloidosis).3 He presented to an institution well-known for its expertise in amyloidosis, for a second opinion. There, the diagnosis was negated, based on reevaluation of the patient’s previous heart specimen through immunohistochemical studies. These studies were positive for serum amyloid P, which is suggestive of transthyretin (TTR) or familial amyloidosis.4 Genetic testing revealed a familial amyloidosis DNA sequence analysis with the Val122Ile variant (ie, isoleucine for valine at position 1225). With the correct diagnosis confirmed, the patient was referred to another highly regarded institution to begin a work-up for cardiac transplantation. Meanwhile, he was cautiously treated with the loop diuretic furosemide to manage his shortness of breath and peripheral edema.
Fifteen months later (13 weeks after being listed for transplant), the patient underwent successful cardiac transplantation.
On pathologic review of the patient’s extricated heart, the myocardium was found to be grossly thickened (see figure, above) and weighed 540 g; the average adult heart weighs 300 to 350 g, depending on the patient’s size.6 Congo Red staining showed extensive amyloid deposits with infiltration throughout the myocardium.
Ninety percent of the amyloid deposits were interstitial, 5% were in the vessels, and 5% were noted in a nodular pattern. The left ventricular cavity showed dilated and thickened walls. Intramural and extramural blood vessels were infiltrated with amyloid as well.
Six months after transplantation, the patient underwent diagnostic testing to assess the function and structure of his new heart. Cardiac catheterization was negative for coronary artery disease. Thirteen months posttransplantation, endomyocardial biopsy with Congo Red stain was negative for amyloid deposition or organ rejection.
About 24 months posttransplantation, the patient was taking tacrolimus, pravastatin, pantoprazole, dapsone, propanolol, colchicine, and donepezil. Stress echocardiography demonstrated normal right and left ventricular systolic function; no wall-motion abnormalities or left ventricular hypertrophy were detected, and the right atrium was of normal size. There was abnormal structural enlargement of the left atrium at the site of anastamosis—a common finding in cardiac transplant patients. The aortic, tricuspid, and mitral valves were all normal.
At that time, it was decided not to repeat endomyocardial biopsy because of normal results on molecular expression testing (a noninvasive technique called AlloMap®7-9), which is performed to assess for heart transplant rejection. The patient’s lipid panel remained within normal limits. CBC indicated persistent anemia and leukopenia. Urine protein and BNP test results were not available.
Since undergoing cardiac transplantation, the patient has resumed his normal routine activities, including some type of exercise five days per week. He said his diet is maintained in moderation. He denied shortness of breath, chest pain, dizziness, or edema. He has returned to full-time employment and has vacationed in Croatia, Italy, and Central America.
DISCUSSION
Familial amyloidosis is an autosomal dominant disease characterized by the production of mutated proteins, most commonly ATTR. Presence of the ATTR Val 122Ile allele has been reported in 3.9% of all black Americans, and in one study, 23% of black Americans diagnosed with cardiac amyloidosis at autopsy were heterozygous for this variant allele.10-12 ATTR Val122Ile usually manifests in the fifth
or sixth decade of life with its characteristic presentation of infiltrative/restrictive cardiomyopathy,13 resulting in heart failure and sometimes peripheral neuropathy.10,11,14
Pathophysiology
In patients with ATTR Val122Ile, cardiomyopathy results from the deposition of mutant protein fibrils in the cardiac muscle, leading to restricted heart wall motion10,15 and stiffening of the cardiac ventricles, with subsequent disruption of the diastolic filling properties of the cardiac muscle.16 Fluid overload and heart failure follow.3,10,17 The atrium of the heart dilates, and the walls of the ventricles become thickened and fibrous.18 Liepnieks and Benson6 reported that the cadaver heart of one Val122Ile patient infiltrated with amyloid protein fibrils weighed 725 g—more than double the weight of an average adult heart.
In patients with cardiac amyloidosis, ECG can detect arrhythmia, and echocardiography shows cardiac enlargement; however, as in the case patient, cardiac catheterization shows normal coronary arteries.15,16,19,20 Thus, previously healthy patients who present with heart failure and negative results on cardiac catheterization should undergo further work-up for cardiac amyloidosis.19
Amyloidosis affects all populations globally.10 In systemic amyloidosis, amyloid releases into the plasma, infiltrating and impairing multiple organs. Poor survival has been reported in patients with heart failure symptoms resulting from amyloid deposition.20,21
Types of Amyloidosis
Primary amyloidosis, the most common of the three amyloidosis types, can be systemic or localized.22 It occurs when protein fibrils, developed from immunoglobin light chains or monoclonal plasma cells and measuring 7 to 10 nm in diameter, adhere to the heart, kidneys, peripheral nerves, eyes, and other organs.5,11,20,23,24 Known for its relation to multiple myeloma,19 primary amyloidosis is associated with a poor prognosis.3,10
Secondary amyloidosis results from a chronic inflammatory disorder, such as rheumatoid arthritis or ankylosing spondylitis—conditions that trigger the production of amyloid proteins.3,10 This type has also been associated with substance abuse and AIDS.23
Familial or hereditary amyloidosis, according to Benson,10 is a group of diseases, each resulting from mutation in a specific protein. In the United States, the most common type of familial amyloidosis is ATTR.11 More than 100 mutant types of ATTR proteins have been identified, each involving a specific nationality or group of nationalities.10,11,23
Mutant ATTR amyloid, when deposited in specific organs, leads to their dysfunction and ultimate failure.6 ATTR may affect the cardiac, gastric, renal, ophthalmic, or nervous system. Depending on the ATTR variant, the resulting clinical features are age- and time-dependent, with onset most common between the third and fifth decade of life.10
Prevalence
The prevalence of ATTR Val 122Ile amyloidosis is reportedly high in West Africa, and in the US African-American population (3.9%).4,11,14,25,26 In a study conducted at a county hospital in Indianapolis, 3% of black newborns were found positive for ATTR Val122Ile through DNA sampling of umbilical cord blood.25 These statistics are of concern, as ATTR amyloidosis could be a significant health concern in a patient population that is already medically underserved.
Yamashita et al25 estimate that 1.35 million Americans of African-American descent may be affected by ATTR Val122Ile and vulnerable to restrictive cardiomyopathy–related heart failure and death. At the very least, this disorder can impair quality of life, especially in the presence of other comorbid conditions.
Clinical Presentation
The presence of exertional syncope at presentation is ominous, as it may be a marker of severe restrictive cardiomyopathy, postural hypotension due to excessive diuresis or autonomic neuropathy, ventricular arrhythmias from localized hypoperfusion, and rarely from cardiac tamponade due to pericardial involvement.27 Despite widespread involvement of the conduction system in specimens at autopsy, high-grade IV block is unusual.3
Diagnostic Studies
ECG. Both ECG and Holter monitoring can detect the arrhythmias and conduction disturbances (eg, first-degree AV block, low voltage patterns) associated with cardiac amyloidosis. Patients often experience syncopal and near-syncopal episodes as a result of conduction disturbances.28 Patients with conditions such as cardiac amyloidosis who present with severe heart failure are at high risk for sudden death secondary to conduction disturbances. Many have benefited from implanted defibrillators.29
Echocardiography. In patients with ATTR Val122Ile cardiac amyloidosis, echocardiography reveals thickened ventricular walls (ie, measuring ≥ 15 mm; normal, ≤ 11 mm).19 Amyloid-restrictive cardiomyopathy is associated with a marked dissociation between short- and long-axis systolic function, in cases in which left ventricular ejection fraction is normal.30
Echocardiography may demonstrate the characteristic specular or granular sparkling appearance that signifies advanced disease.15,21 Only a minority have this pattern in the myocardium, however, and changes in echocardiographic technology have made this finding less noticeable.30
MRI. Among more recently used diagnostic studies, cardiac magnetic resonance (CMR) has been reported to demonstrate late gadolinium enhancement (LGE) in perhaps 80% of patients with familial amyloidosis and cardiac involvement (as determined through biopsy and Congo Red stain). LGE-CMR shows darkening of the cardiac tissue, a common occurrence in amyloidosis.31
LGE is associated with increased thickness of both left and right ventricles, lower ECG voltage patterns, elevated BNP, and elevated troponin T.31,32 Globally, LGE is associated with the worst prognosis in patients with cardiac amyloidosis. Use of LGE-CMR testing can help facilitate early detection of cardiac amyloidosis in patients who may be vulnerable to cardiac damage.31
Cardiac catheterization. In patients with cardiac amyloidosis, cardiac catheterization usually shows normal coronary arteries.3
Diagnosis
Early diagnosis of ATTR cardiac amyloidosis is crucial to the patient’s survival; it should be ruled out in any African-American patient with unexplained heart failure and echocardiography showing increased wall thickness with a nondilated left ventricular cavity. Additional clues include significant proteinuria, hepatomegaly disproportionate to the degree of heart failure, or corresponding neuropathy. Known family history of the disease, along with variant type, allows for a prompt and correct diagnosis.10
It has been reported that most clinicians who encounter heart failure, particularly in black patients, do not consider amyloidosis in the differential diagnosis, because of the high prevalence of hypertension and congestive heart failure in this population.10,15,19 As a result, amyloidosis often goes undiagnosed.19
Findings of enlarged and thickened cardiac walls on echocardiography but normal coronary arteries on cardiac catheterization should alert the treating clinician to further work-up for cardiac amyloidosis.19 In such a patient, according to Kristen et al,21 endomyocardial biopsy with Congo Red staining is the gold standard for diagnosis of amyloidosis.
In ATTR Val122Ile familial amyloidosis, it is unclear whether patients who are homozygous for the disease present with symptoms at earlier onset with more progressive illness or die sooner than those who are heterozygous.33 Nevertheless, once the diagnosis is confirmed, it is important to determine the patient’s specific variant type by DNA testing so that appropriate treatment can be initiated and the patient’s prognosis evaluated.5,10,13
Treatment
For familial amyloidosis in general, some researchers advocate liver transplantation to remove the source of mutant amyloid protein and stop all deposition of amyloid fibrils; this procedure can be followed later by transplantation of other affected organs (including the heart).5,23 Maurer et al34 have reported improved one-year survival rates among patients with ATTR amyloidosis who underwent both cardiac and liver transplantation: 75%, versus 23% in patients who did not receive transplanted organs.
Management of cardiac amyloidosis usually requires a twofold approach: treating associated congestive heart failure, and preventing further deposition of amyloid.24 In the case patient (as in most patients with ATTR amyloidosis), heart transplantation was deemed the only life-sustaining treatment option.11,19,33
Pharmacotherapeutic options are limited for patients with ATTR Val122Ile familial amyloidosis. Conventional heart failure agents (eg, ACE inhibitors, angiotensin receptor blockers, digoxin, β-blockers, calcium channel blockers) can exacerbate heart failure symptoms, leading to a potentially life-threatening arrhythmia.3,11,19,24,35 Amyloid fibrils bind to digitalis, increasing susceptibility to digitalis toxicity; and to nifedipine, causing hemodynamic deterioration. Verapamil should be avoided, as it may induce severe left ventricular dysfunction. ACE inhibitors often provoke profound hypotension in primary amyloidosis.24,35
Diuretics, too (eg, furosemide, as was prescribed for the case patient), must be used with caution.3 These agents have been used to treat fluid overload and the resulting peripheral edema and shortness of breath found in ATTR Val122Ile patients who experience heart failure.36 According to Dubrey et al,5 cautious use of diuretics is necessary for management of heart failure symptoms in these patients.
Because the risk for intracardiac thrombus is high, anticoagulation (using agents other than β-blockers or calcium channel blockers) should be implemented unless compelling risks are involved.11,24 Amiodarone is relatively well tolerated for ventricular tachydysrhythmias and in atrial fibrillation if the goal is maintaining sinus rhythm.37
Regarding heart transplantation in patients with familial amyloidosis, Jacob et al33 hypothesize that since mutant amyloid protein is synthesized by the liver, it would take approximately 50 years for a transplanted heart to become affected by amyloid deposition. In a 59-year-old Afro-Caribbean man with familial amyloidosis who underwent cardiac transplantation, Hamour et al11 reported that the donor heart remained amyloid-free three years posttransplantation, as demonstrated by serial cardiac biopsy.
On the Horizon
Clinical trials are now under way to examine pharmacotherapeutic options for patients with ATTR amyloidosis. Now being examined in clinical trials, for example, is Fx-1006A, a drug that stabilizes ATTR and prevents the misfolding of the amyloid protein fibril, in turn preventing it from binding to the target organ.38 Similarly, ALN-TTR, a drug believed to prevent disease manifestation and possibly facilitate disease regression, is being investigated in early human trials.39
Additionally, the use of genetic testing is recommended in at-risk individuals to identify the TTR gene. Affected patients may benefit from prophylactic medical management, which would halt amyloidogenesis of TTR—and possibly treat the condition as well.35 Pharmacotherapeutic agents like diflunisal, an NSAID, antagonize the aggregation of TTR protein and hinder formation of the amyloid fibrils.40
CONCLUSION
ATTR Val122Ile familial amyloidosis is a rare disorder that causes abnormal synthesis of amyloid protein in the liver, which then infiltrates the cardiac structure, leading to restrictive cardiomyopathy and progressive heart failure. Patients who present with symptoms of heart failure, cardiac enlargement on echocardiography, and a finding of granular speckling patterns, though not specific on echocardiography, should prompt the health care provider to refer the patient to a cardiologist familiar with cardiac amyloidosis for further work-up.
Diagnosed patients must undergo genetic testing to determine the specific variant type so that prompt treatment can be initiated. In patients with ATTR Val122Ile familial amyloidosis, the treatment of choice is cardiac transplantation. Although the mutant amyloid protein continues to be synthesized in the liver, the donor heart is unlikely to become affected by this substance for many years. Appropriately treated patients can maintain good quality of life, free of heart failure.
REFERENCES
1. Lim RP, Srichai MB, Lee VS. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. AJR Am J Roentgenol. 2007;188 (6):1675-1681.
2. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-104.
3. Kendall H. Cardiac amyloidosis. Crit Care Nurse. 2010;30(2):16-23.
4. Eriksson M, Büttener J, Todorov T, et al. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with AATR amyloid. Am J Surg Pathol. 2009;33(1):58-65.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
6. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277-282.
7. XDx Expression Diagnostics. Allomap®: molecular expression testing (2004). www.allomap.com. Accessed May 14, 2012.
8. Mandras SA, Crespo J, Patel HM. Innovative application of immunologic principles in heart transplantation. Ochsner J. 2010;10(4):231-235.
9. Yamani MH, Taylor DO, Rodriguez R, et al. Transplant vasculopathy is associated with increased AlloMap gene expression score. J Heart Lung Transplant. 2007;26(4):403-406.
10. Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17(6):909-927.
11. Hamour IM, Lachmann HJ, Goodman HJ, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056-1059.
12. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466-473.
13. Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52(4):347-361.
14. Askanas V, Engel WK, McFerrin J, Vattemi G. Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology. 2003;61(2):257-260.
15. Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidoses associated with the transthyretin lle122 mutation in a Caucasian family. Amyloid. 2001;8(4):263-269.
16. Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24(3): 214-220.
17. Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10 (8):iii23-iii33.
18. Bruce J. Getting to the heart of cardiomyopathies. Nursing. 2005;35(8):44-47.
19. Falk RH. The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis. 2002;12(1):141-143.
20. Piper C, Butz T, Farr M, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue Doppler echocardiography, and myocardial biopsy. Amyloid. 2010;17(1):1-9.
21. Kristen AV, Meyer FJ, Perz JB, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 suppl):S151-S155.
22. Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-183.
23. Picken MM. New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr Opin Nephrol Hypertens. 2007; 16(3):196-203.
24. Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079-1085.
25. Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile 122 allele frequency in an African-American population. Amyloid. 2005;12(2):127-130.
26. Benson MD, Yazaki M, Magy N. Laboratory assessment of transthyretin amyloidosis. Clin Chem Lab Med. 2002;40(12):1262-1265.
27. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80(9):1242-1245.
28. Correia MJ, Coutinho CA, Conceiçao I, et al. Role of heart rate variability in the assessment of autonomic dysfunction in type I familial amyloidotic polyneuropathy. Folia Cardiol. 2005;12(suppl C):459-462.
29. Kadish A, Mehra M. Heart failure devices: Implantable cardioverter-defibrillators and biventricular pacing therapy. Circulation. 2005; 111(24):3327-3335.
30. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43(3):410-415.
31. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155-164.
32. Fealey ME, Edwards WD, Buadi FK, et al. Echocardiographic features of cardiac amyloidosis presenting as endomyocardial disease in a 54-year-old male. J Cardiol. 2009;54(1):162-166.
33. Jacob EK, Edwards WD, Zucker M, et al. Homozygous transthyretin mutation in an African American male. J Mol Diagn. 2007; 9(1):127-131.
34. Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539-545.
35. Buxbaum J, Alexander A, Koziol J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African-Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864-870.
36. Rose BD, Colucci WS. Use of diuretics in heart failure (2010). www.uptodate.com/con tents/use-of-diuretics-in-patients-with-heart-failure. Accessed May 14, 2012.
37. Trappe H-J. Treating critical supraventricular and ventricular arrhythmias. J Emerg Trauma Shock. 2010;3(2):143-152.
38. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219-3230.
39. Alnylam Pharmaceuticals. TTR Amyloidosis: ALN-TTR (2011). www.alnylam.com/Programs-and-Pipeline/Programs/index.php. Accessed May 14, 2012.
40. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin: potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355-374.
A black man, age 65, with no known history of cardiopulmonary disease presented with acute-onset exertional dyspnea and lower extremity edema. He also reported an episode of syncope, as well as occasional dizziness and abdominal bloating. He said he experienced exertional dyspnea while doing a routine step aerobic exercise. His exercise regimen included distance walking, yoga, and aerobics four to five days per week.
The patient’s medical history was remarkable for a single episode of a bleeding ulcer in previous years, low back pain, shoulder pain, and a septic arthritic hip. His social history was negative for use of tobacco, alcohol, or illegal drugs. He was married and had two biological daughters with fairly unremarkable medical histories. The patient had earned a master’s degree, worked full-time in the insurance business, and was an avid worldwide traveler. He reported diminished quality of life as a result of his acute-onset heart failure symptoms, which reduced his ability to exercise routinely, work full-time, or travel.
The patient’s sudden experience of exertional dyspnea prompted him to visit his primary care provider, who ordered an ECG that demonstrated low voltage patterns and a first-degree atrioventricular (AV) block. Subsequent stress echocardiography showed generalized thickening of the left ventricular myocardium. Posterior wall thickness measured 1.7 cm (normal range, 0.6 to 1.1 cm), septal thickness measured 1.9 cm (normal, 0.6 to 1.1 cm), and ejection fraction was 65%. The stress echocardiogram also showed a speckling pattern (brightly scattered spots) on the myocardium.
Although stress echocardiography results were negative for ischemic disease, the patient did experience dyspnea during the exam. He underwent cardiac catheterization, which indicated normal coronary arteries.
Additional diagnostic studies included cardiac MRI with and without contrast, which showed nulling of the heart muscle and delayed patchy hyperenhancement; this suggested myocardial tissue abnormality as result of amyloid fibril deposition.1 Both pulmonary and tricuspid aortic valves were normal, with no evidence of stenosis. No regional wall motion abnormalities were noted.
Laboratory findings during the work-up were lipid panel, unremarkable; complete blood count (CBC), mild anemia and leukopenia; and urinalysis, positive for proteinuria. Brain natriuretic peptide (BNP) was measured at 686 pg/mL (normal, 0.0 to 100 pg/mL), indicating moderate heart failure. A peripheral blood smear was negative for monoclonal plasma cells.
The patient’s physical exam was unremarkable except for 2+ pedal edema bilaterally. In consideration of normal coronary arteries on cardiac catheterization, the patient’s heart failure symptoms, and stress echocardiography abnormalities, a heart biopsy was ordered. An endomyocardial biopsy with Congo Red stain demonstrated an apple-green birefringent pattern viewed under high-definition polarized light microscope, which was consistent with amyloid deposition.2
The patient was given a diagnosis of primary amyloidosis by his local cardiologist despite negative findings on the peripheral blood smear for monoclonal plasma cells (which are typically found in primary amyloidosis).3 He presented to an institution well-known for its expertise in amyloidosis, for a second opinion. There, the diagnosis was negated, based on reevaluation of the patient’s previous heart specimen through immunohistochemical studies. These studies were positive for serum amyloid P, which is suggestive of transthyretin (TTR) or familial amyloidosis.4 Genetic testing revealed a familial amyloidosis DNA sequence analysis with the Val122Ile variant (ie, isoleucine for valine at position 1225). With the correct diagnosis confirmed, the patient was referred to another highly regarded institution to begin a work-up for cardiac transplantation. Meanwhile, he was cautiously treated with the loop diuretic furosemide to manage his shortness of breath and peripheral edema.
Fifteen months later (13 weeks after being listed for transplant), the patient underwent successful cardiac transplantation.
On pathologic review of the patient’s extricated heart, the myocardium was found to be grossly thickened (see figure, above) and weighed 540 g; the average adult heart weighs 300 to 350 g, depending on the patient’s size.6 Congo Red staining showed extensive amyloid deposits with infiltration throughout the myocardium.
Ninety percent of the amyloid deposits were interstitial, 5% were in the vessels, and 5% were noted in a nodular pattern. The left ventricular cavity showed dilated and thickened walls. Intramural and extramural blood vessels were infiltrated with amyloid as well.
Six months after transplantation, the patient underwent diagnostic testing to assess the function and structure of his new heart. Cardiac catheterization was negative for coronary artery disease. Thirteen months posttransplantation, endomyocardial biopsy with Congo Red stain was negative for amyloid deposition or organ rejection.
About 24 months posttransplantation, the patient was taking tacrolimus, pravastatin, pantoprazole, dapsone, propanolol, colchicine, and donepezil. Stress echocardiography demonstrated normal right and left ventricular systolic function; no wall-motion abnormalities or left ventricular hypertrophy were detected, and the right atrium was of normal size. There was abnormal structural enlargement of the left atrium at the site of anastamosis—a common finding in cardiac transplant patients. The aortic, tricuspid, and mitral valves were all normal.
At that time, it was decided not to repeat endomyocardial biopsy because of normal results on molecular expression testing (a noninvasive technique called AlloMap®7-9), which is performed to assess for heart transplant rejection. The patient’s lipid panel remained within normal limits. CBC indicated persistent anemia and leukopenia. Urine protein and BNP test results were not available.
Since undergoing cardiac transplantation, the patient has resumed his normal routine activities, including some type of exercise five days per week. He said his diet is maintained in moderation. He denied shortness of breath, chest pain, dizziness, or edema. He has returned to full-time employment and has vacationed in Croatia, Italy, and Central America.
DISCUSSION
Familial amyloidosis is an autosomal dominant disease characterized by the production of mutated proteins, most commonly ATTR. Presence of the ATTR Val 122Ile allele has been reported in 3.9% of all black Americans, and in one study, 23% of black Americans diagnosed with cardiac amyloidosis at autopsy were heterozygous for this variant allele.10-12 ATTR Val122Ile usually manifests in the fifth
or sixth decade of life with its characteristic presentation of infiltrative/restrictive cardiomyopathy,13 resulting in heart failure and sometimes peripheral neuropathy.10,11,14
Pathophysiology
In patients with ATTR Val122Ile, cardiomyopathy results from the deposition of mutant protein fibrils in the cardiac muscle, leading to restricted heart wall motion10,15 and stiffening of the cardiac ventricles, with subsequent disruption of the diastolic filling properties of the cardiac muscle.16 Fluid overload and heart failure follow.3,10,17 The atrium of the heart dilates, and the walls of the ventricles become thickened and fibrous.18 Liepnieks and Benson6 reported that the cadaver heart of one Val122Ile patient infiltrated with amyloid protein fibrils weighed 725 g—more than double the weight of an average adult heart.
In patients with cardiac amyloidosis, ECG can detect arrhythmia, and echocardiography shows cardiac enlargement; however, as in the case patient, cardiac catheterization shows normal coronary arteries.15,16,19,20 Thus, previously healthy patients who present with heart failure and negative results on cardiac catheterization should undergo further work-up for cardiac amyloidosis.19
Amyloidosis affects all populations globally.10 In systemic amyloidosis, amyloid releases into the plasma, infiltrating and impairing multiple organs. Poor survival has been reported in patients with heart failure symptoms resulting from amyloid deposition.20,21
Types of Amyloidosis
Primary amyloidosis, the most common of the three amyloidosis types, can be systemic or localized.22 It occurs when protein fibrils, developed from immunoglobin light chains or monoclonal plasma cells and measuring 7 to 10 nm in diameter, adhere to the heart, kidneys, peripheral nerves, eyes, and other organs.5,11,20,23,24 Known for its relation to multiple myeloma,19 primary amyloidosis is associated with a poor prognosis.3,10
Secondary amyloidosis results from a chronic inflammatory disorder, such as rheumatoid arthritis or ankylosing spondylitis—conditions that trigger the production of amyloid proteins.3,10 This type has also been associated with substance abuse and AIDS.23
Familial or hereditary amyloidosis, according to Benson,10 is a group of diseases, each resulting from mutation in a specific protein. In the United States, the most common type of familial amyloidosis is ATTR.11 More than 100 mutant types of ATTR proteins have been identified, each involving a specific nationality or group of nationalities.10,11,23
Mutant ATTR amyloid, when deposited in specific organs, leads to their dysfunction and ultimate failure.6 ATTR may affect the cardiac, gastric, renal, ophthalmic, or nervous system. Depending on the ATTR variant, the resulting clinical features are age- and time-dependent, with onset most common between the third and fifth decade of life.10
Prevalence
The prevalence of ATTR Val 122Ile amyloidosis is reportedly high in West Africa, and in the US African-American population (3.9%).4,11,14,25,26 In a study conducted at a county hospital in Indianapolis, 3% of black newborns were found positive for ATTR Val122Ile through DNA sampling of umbilical cord blood.25 These statistics are of concern, as ATTR amyloidosis could be a significant health concern in a patient population that is already medically underserved.
Yamashita et al25 estimate that 1.35 million Americans of African-American descent may be affected by ATTR Val122Ile and vulnerable to restrictive cardiomyopathy–related heart failure and death. At the very least, this disorder can impair quality of life, especially in the presence of other comorbid conditions.
Clinical Presentation
The presence of exertional syncope at presentation is ominous, as it may be a marker of severe restrictive cardiomyopathy, postural hypotension due to excessive diuresis or autonomic neuropathy, ventricular arrhythmias from localized hypoperfusion, and rarely from cardiac tamponade due to pericardial involvement.27 Despite widespread involvement of the conduction system in specimens at autopsy, high-grade IV block is unusual.3
Diagnostic Studies
ECG. Both ECG and Holter monitoring can detect the arrhythmias and conduction disturbances (eg, first-degree AV block, low voltage patterns) associated with cardiac amyloidosis. Patients often experience syncopal and near-syncopal episodes as a result of conduction disturbances.28 Patients with conditions such as cardiac amyloidosis who present with severe heart failure are at high risk for sudden death secondary to conduction disturbances. Many have benefited from implanted defibrillators.29
Echocardiography. In patients with ATTR Val122Ile cardiac amyloidosis, echocardiography reveals thickened ventricular walls (ie, measuring ≥ 15 mm; normal, ≤ 11 mm).19 Amyloid-restrictive cardiomyopathy is associated with a marked dissociation between short- and long-axis systolic function, in cases in which left ventricular ejection fraction is normal.30
Echocardiography may demonstrate the characteristic specular or granular sparkling appearance that signifies advanced disease.15,21 Only a minority have this pattern in the myocardium, however, and changes in echocardiographic technology have made this finding less noticeable.30
MRI. Among more recently used diagnostic studies, cardiac magnetic resonance (CMR) has been reported to demonstrate late gadolinium enhancement (LGE) in perhaps 80% of patients with familial amyloidosis and cardiac involvement (as determined through biopsy and Congo Red stain). LGE-CMR shows darkening of the cardiac tissue, a common occurrence in amyloidosis.31
LGE is associated with increased thickness of both left and right ventricles, lower ECG voltage patterns, elevated BNP, and elevated troponin T.31,32 Globally, LGE is associated with the worst prognosis in patients with cardiac amyloidosis. Use of LGE-CMR testing can help facilitate early detection of cardiac amyloidosis in patients who may be vulnerable to cardiac damage.31
Cardiac catheterization. In patients with cardiac amyloidosis, cardiac catheterization usually shows normal coronary arteries.3
Diagnosis
Early diagnosis of ATTR cardiac amyloidosis is crucial to the patient’s survival; it should be ruled out in any African-American patient with unexplained heart failure and echocardiography showing increased wall thickness with a nondilated left ventricular cavity. Additional clues include significant proteinuria, hepatomegaly disproportionate to the degree of heart failure, or corresponding neuropathy. Known family history of the disease, along with variant type, allows for a prompt and correct diagnosis.10
It has been reported that most clinicians who encounter heart failure, particularly in black patients, do not consider amyloidosis in the differential diagnosis, because of the high prevalence of hypertension and congestive heart failure in this population.10,15,19 As a result, amyloidosis often goes undiagnosed.19
Findings of enlarged and thickened cardiac walls on echocardiography but normal coronary arteries on cardiac catheterization should alert the treating clinician to further work-up for cardiac amyloidosis.19 In such a patient, according to Kristen et al,21 endomyocardial biopsy with Congo Red staining is the gold standard for diagnosis of amyloidosis.
In ATTR Val122Ile familial amyloidosis, it is unclear whether patients who are homozygous for the disease present with symptoms at earlier onset with more progressive illness or die sooner than those who are heterozygous.33 Nevertheless, once the diagnosis is confirmed, it is important to determine the patient’s specific variant type by DNA testing so that appropriate treatment can be initiated and the patient’s prognosis evaluated.5,10,13
Treatment
For familial amyloidosis in general, some researchers advocate liver transplantation to remove the source of mutant amyloid protein and stop all deposition of amyloid fibrils; this procedure can be followed later by transplantation of other affected organs (including the heart).5,23 Maurer et al34 have reported improved one-year survival rates among patients with ATTR amyloidosis who underwent both cardiac and liver transplantation: 75%, versus 23% in patients who did not receive transplanted organs.
Management of cardiac amyloidosis usually requires a twofold approach: treating associated congestive heart failure, and preventing further deposition of amyloid.24 In the case patient (as in most patients with ATTR amyloidosis), heart transplantation was deemed the only life-sustaining treatment option.11,19,33
Pharmacotherapeutic options are limited for patients with ATTR Val122Ile familial amyloidosis. Conventional heart failure agents (eg, ACE inhibitors, angiotensin receptor blockers, digoxin, β-blockers, calcium channel blockers) can exacerbate heart failure symptoms, leading to a potentially life-threatening arrhythmia.3,11,19,24,35 Amyloid fibrils bind to digitalis, increasing susceptibility to digitalis toxicity; and to nifedipine, causing hemodynamic deterioration. Verapamil should be avoided, as it may induce severe left ventricular dysfunction. ACE inhibitors often provoke profound hypotension in primary amyloidosis.24,35
Diuretics, too (eg, furosemide, as was prescribed for the case patient), must be used with caution.3 These agents have been used to treat fluid overload and the resulting peripheral edema and shortness of breath found in ATTR Val122Ile patients who experience heart failure.36 According to Dubrey et al,5 cautious use of diuretics is necessary for management of heart failure symptoms in these patients.
Because the risk for intracardiac thrombus is high, anticoagulation (using agents other than β-blockers or calcium channel blockers) should be implemented unless compelling risks are involved.11,24 Amiodarone is relatively well tolerated for ventricular tachydysrhythmias and in atrial fibrillation if the goal is maintaining sinus rhythm.37
Regarding heart transplantation in patients with familial amyloidosis, Jacob et al33 hypothesize that since mutant amyloid protein is synthesized by the liver, it would take approximately 50 years for a transplanted heart to become affected by amyloid deposition. In a 59-year-old Afro-Caribbean man with familial amyloidosis who underwent cardiac transplantation, Hamour et al11 reported that the donor heart remained amyloid-free three years posttransplantation, as demonstrated by serial cardiac biopsy.
On the Horizon
Clinical trials are now under way to examine pharmacotherapeutic options for patients with ATTR amyloidosis. Now being examined in clinical trials, for example, is Fx-1006A, a drug that stabilizes ATTR and prevents the misfolding of the amyloid protein fibril, in turn preventing it from binding to the target organ.38 Similarly, ALN-TTR, a drug believed to prevent disease manifestation and possibly facilitate disease regression, is being investigated in early human trials.39
Additionally, the use of genetic testing is recommended in at-risk individuals to identify the TTR gene. Affected patients may benefit from prophylactic medical management, which would halt amyloidogenesis of TTR—and possibly treat the condition as well.35 Pharmacotherapeutic agents like diflunisal, an NSAID, antagonize the aggregation of TTR protein and hinder formation of the amyloid fibrils.40
CONCLUSION
ATTR Val122Ile familial amyloidosis is a rare disorder that causes abnormal synthesis of amyloid protein in the liver, which then infiltrates the cardiac structure, leading to restrictive cardiomyopathy and progressive heart failure. Patients who present with symptoms of heart failure, cardiac enlargement on echocardiography, and a finding of granular speckling patterns, though not specific on echocardiography, should prompt the health care provider to refer the patient to a cardiologist familiar with cardiac amyloidosis for further work-up.
Diagnosed patients must undergo genetic testing to determine the specific variant type so that prompt treatment can be initiated. In patients with ATTR Val122Ile familial amyloidosis, the treatment of choice is cardiac transplantation. Although the mutant amyloid protein continues to be synthesized in the liver, the donor heart is unlikely to become affected by this substance for many years. Appropriately treated patients can maintain good quality of life, free of heart failure.
REFERENCES
1. Lim RP, Srichai MB, Lee VS. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. AJR Am J Roentgenol. 2007;188 (6):1675-1681.
2. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-104.
3. Kendall H. Cardiac amyloidosis. Crit Care Nurse. 2010;30(2):16-23.
4. Eriksson M, Büttener J, Todorov T, et al. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with AATR amyloid. Am J Surg Pathol. 2009;33(1):58-65.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
6. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277-282.
7. XDx Expression Diagnostics. Allomap®: molecular expression testing (2004). www.allomap.com. Accessed May 14, 2012.
8. Mandras SA, Crespo J, Patel HM. Innovative application of immunologic principles in heart transplantation. Ochsner J. 2010;10(4):231-235.
9. Yamani MH, Taylor DO, Rodriguez R, et al. Transplant vasculopathy is associated with increased AlloMap gene expression score. J Heart Lung Transplant. 2007;26(4):403-406.
10. Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17(6):909-927.
11. Hamour IM, Lachmann HJ, Goodman HJ, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056-1059.
12. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466-473.
13. Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52(4):347-361.
14. Askanas V, Engel WK, McFerrin J, Vattemi G. Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology. 2003;61(2):257-260.
15. Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidoses associated with the transthyretin lle122 mutation in a Caucasian family. Amyloid. 2001;8(4):263-269.
16. Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24(3): 214-220.
17. Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10 (8):iii23-iii33.
18. Bruce J. Getting to the heart of cardiomyopathies. Nursing. 2005;35(8):44-47.
19. Falk RH. The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis. 2002;12(1):141-143.
20. Piper C, Butz T, Farr M, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue Doppler echocardiography, and myocardial biopsy. Amyloid. 2010;17(1):1-9.
21. Kristen AV, Meyer FJ, Perz JB, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 suppl):S151-S155.
22. Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-183.
23. Picken MM. New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr Opin Nephrol Hypertens. 2007; 16(3):196-203.
24. Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079-1085.
25. Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile 122 allele frequency in an African-American population. Amyloid. 2005;12(2):127-130.
26. Benson MD, Yazaki M, Magy N. Laboratory assessment of transthyretin amyloidosis. Clin Chem Lab Med. 2002;40(12):1262-1265.
27. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80(9):1242-1245.
28. Correia MJ, Coutinho CA, Conceiçao I, et al. Role of heart rate variability in the assessment of autonomic dysfunction in type I familial amyloidotic polyneuropathy. Folia Cardiol. 2005;12(suppl C):459-462.
29. Kadish A, Mehra M. Heart failure devices: Implantable cardioverter-defibrillators and biventricular pacing therapy. Circulation. 2005; 111(24):3327-3335.
30. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43(3):410-415.
31. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155-164.
32. Fealey ME, Edwards WD, Buadi FK, et al. Echocardiographic features of cardiac amyloidosis presenting as endomyocardial disease in a 54-year-old male. J Cardiol. 2009;54(1):162-166.
33. Jacob EK, Edwards WD, Zucker M, et al. Homozygous transthyretin mutation in an African American male. J Mol Diagn. 2007; 9(1):127-131.
34. Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539-545.
35. Buxbaum J, Alexander A, Koziol J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African-Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864-870.
36. Rose BD, Colucci WS. Use of diuretics in heart failure (2010). www.uptodate.com/con tents/use-of-diuretics-in-patients-with-heart-failure. Accessed May 14, 2012.
37. Trappe H-J. Treating critical supraventricular and ventricular arrhythmias. J Emerg Trauma Shock. 2010;3(2):143-152.
38. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219-3230.
39. Alnylam Pharmaceuticals. TTR Amyloidosis: ALN-TTR (2011). www.alnylam.com/Programs-and-Pipeline/Programs/index.php. Accessed May 14, 2012.
40. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin: potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355-374.
A black man, age 65, with no known history of cardiopulmonary disease presented with acute-onset exertional dyspnea and lower extremity edema. He also reported an episode of syncope, as well as occasional dizziness and abdominal bloating. He said he experienced exertional dyspnea while doing a routine step aerobic exercise. His exercise regimen included distance walking, yoga, and aerobics four to five days per week.
The patient’s medical history was remarkable for a single episode of a bleeding ulcer in previous years, low back pain, shoulder pain, and a septic arthritic hip. His social history was negative for use of tobacco, alcohol, or illegal drugs. He was married and had two biological daughters with fairly unremarkable medical histories. The patient had earned a master’s degree, worked full-time in the insurance business, and was an avid worldwide traveler. He reported diminished quality of life as a result of his acute-onset heart failure symptoms, which reduced his ability to exercise routinely, work full-time, or travel.
The patient’s sudden experience of exertional dyspnea prompted him to visit his primary care provider, who ordered an ECG that demonstrated low voltage patterns and a first-degree atrioventricular (AV) block. Subsequent stress echocardiography showed generalized thickening of the left ventricular myocardium. Posterior wall thickness measured 1.7 cm (normal range, 0.6 to 1.1 cm), septal thickness measured 1.9 cm (normal, 0.6 to 1.1 cm), and ejection fraction was 65%. The stress echocardiogram also showed a speckling pattern (brightly scattered spots) on the myocardium.
Although stress echocardiography results were negative for ischemic disease, the patient did experience dyspnea during the exam. He underwent cardiac catheterization, which indicated normal coronary arteries.
Additional diagnostic studies included cardiac MRI with and without contrast, which showed nulling of the heart muscle and delayed patchy hyperenhancement; this suggested myocardial tissue abnormality as result of amyloid fibril deposition.1 Both pulmonary and tricuspid aortic valves were normal, with no evidence of stenosis. No regional wall motion abnormalities were noted.
Laboratory findings during the work-up were lipid panel, unremarkable; complete blood count (CBC), mild anemia and leukopenia; and urinalysis, positive for proteinuria. Brain natriuretic peptide (BNP) was measured at 686 pg/mL (normal, 0.0 to 100 pg/mL), indicating moderate heart failure. A peripheral blood smear was negative for monoclonal plasma cells.
The patient’s physical exam was unremarkable except for 2+ pedal edema bilaterally. In consideration of normal coronary arteries on cardiac catheterization, the patient’s heart failure symptoms, and stress echocardiography abnormalities, a heart biopsy was ordered. An endomyocardial biopsy with Congo Red stain demonstrated an apple-green birefringent pattern viewed under high-definition polarized light microscope, which was consistent with amyloid deposition.2
The patient was given a diagnosis of primary amyloidosis by his local cardiologist despite negative findings on the peripheral blood smear for monoclonal plasma cells (which are typically found in primary amyloidosis).3 He presented to an institution well-known for its expertise in amyloidosis, for a second opinion. There, the diagnosis was negated, based on reevaluation of the patient’s previous heart specimen through immunohistochemical studies. These studies were positive for serum amyloid P, which is suggestive of transthyretin (TTR) or familial amyloidosis.4 Genetic testing revealed a familial amyloidosis DNA sequence analysis with the Val122Ile variant (ie, isoleucine for valine at position 1225). With the correct diagnosis confirmed, the patient was referred to another highly regarded institution to begin a work-up for cardiac transplantation. Meanwhile, he was cautiously treated with the loop diuretic furosemide to manage his shortness of breath and peripheral edema.
Fifteen months later (13 weeks after being listed for transplant), the patient underwent successful cardiac transplantation.
On pathologic review of the patient’s extricated heart, the myocardium was found to be grossly thickened (see figure, above) and weighed 540 g; the average adult heart weighs 300 to 350 g, depending on the patient’s size.6 Congo Red staining showed extensive amyloid deposits with infiltration throughout the myocardium.
Ninety percent of the amyloid deposits were interstitial, 5% were in the vessels, and 5% were noted in a nodular pattern. The left ventricular cavity showed dilated and thickened walls. Intramural and extramural blood vessels were infiltrated with amyloid as well.
Six months after transplantation, the patient underwent diagnostic testing to assess the function and structure of his new heart. Cardiac catheterization was negative for coronary artery disease. Thirteen months posttransplantation, endomyocardial biopsy with Congo Red stain was negative for amyloid deposition or organ rejection.
About 24 months posttransplantation, the patient was taking tacrolimus, pravastatin, pantoprazole, dapsone, propanolol, colchicine, and donepezil. Stress echocardiography demonstrated normal right and left ventricular systolic function; no wall-motion abnormalities or left ventricular hypertrophy were detected, and the right atrium was of normal size. There was abnormal structural enlargement of the left atrium at the site of anastamosis—a common finding in cardiac transplant patients. The aortic, tricuspid, and mitral valves were all normal.
At that time, it was decided not to repeat endomyocardial biopsy because of normal results on molecular expression testing (a noninvasive technique called AlloMap®7-9), which is performed to assess for heart transplant rejection. The patient’s lipid panel remained within normal limits. CBC indicated persistent anemia and leukopenia. Urine protein and BNP test results were not available.
Since undergoing cardiac transplantation, the patient has resumed his normal routine activities, including some type of exercise five days per week. He said his diet is maintained in moderation. He denied shortness of breath, chest pain, dizziness, or edema. He has returned to full-time employment and has vacationed in Croatia, Italy, and Central America.
DISCUSSION
Familial amyloidosis is an autosomal dominant disease characterized by the production of mutated proteins, most commonly ATTR. Presence of the ATTR Val 122Ile allele has been reported in 3.9% of all black Americans, and in one study, 23% of black Americans diagnosed with cardiac amyloidosis at autopsy were heterozygous for this variant allele.10-12 ATTR Val122Ile usually manifests in the fifth
or sixth decade of life with its characteristic presentation of infiltrative/restrictive cardiomyopathy,13 resulting in heart failure and sometimes peripheral neuropathy.10,11,14
Pathophysiology
In patients with ATTR Val122Ile, cardiomyopathy results from the deposition of mutant protein fibrils in the cardiac muscle, leading to restricted heart wall motion10,15 and stiffening of the cardiac ventricles, with subsequent disruption of the diastolic filling properties of the cardiac muscle.16 Fluid overload and heart failure follow.3,10,17 The atrium of the heart dilates, and the walls of the ventricles become thickened and fibrous.18 Liepnieks and Benson6 reported that the cadaver heart of one Val122Ile patient infiltrated with amyloid protein fibrils weighed 725 g—more than double the weight of an average adult heart.
In patients with cardiac amyloidosis, ECG can detect arrhythmia, and echocardiography shows cardiac enlargement; however, as in the case patient, cardiac catheterization shows normal coronary arteries.15,16,19,20 Thus, previously healthy patients who present with heart failure and negative results on cardiac catheterization should undergo further work-up for cardiac amyloidosis.19
Amyloidosis affects all populations globally.10 In systemic amyloidosis, amyloid releases into the plasma, infiltrating and impairing multiple organs. Poor survival has been reported in patients with heart failure symptoms resulting from amyloid deposition.20,21
Types of Amyloidosis
Primary amyloidosis, the most common of the three amyloidosis types, can be systemic or localized.22 It occurs when protein fibrils, developed from immunoglobin light chains or monoclonal plasma cells and measuring 7 to 10 nm in diameter, adhere to the heart, kidneys, peripheral nerves, eyes, and other organs.5,11,20,23,24 Known for its relation to multiple myeloma,19 primary amyloidosis is associated with a poor prognosis.3,10
Secondary amyloidosis results from a chronic inflammatory disorder, such as rheumatoid arthritis or ankylosing spondylitis—conditions that trigger the production of amyloid proteins.3,10 This type has also been associated with substance abuse and AIDS.23
Familial or hereditary amyloidosis, according to Benson,10 is a group of diseases, each resulting from mutation in a specific protein. In the United States, the most common type of familial amyloidosis is ATTR.11 More than 100 mutant types of ATTR proteins have been identified, each involving a specific nationality or group of nationalities.10,11,23
Mutant ATTR amyloid, when deposited in specific organs, leads to their dysfunction and ultimate failure.6 ATTR may affect the cardiac, gastric, renal, ophthalmic, or nervous system. Depending on the ATTR variant, the resulting clinical features are age- and time-dependent, with onset most common between the third and fifth decade of life.10
Prevalence
The prevalence of ATTR Val 122Ile amyloidosis is reportedly high in West Africa, and in the US African-American population (3.9%).4,11,14,25,26 In a study conducted at a county hospital in Indianapolis, 3% of black newborns were found positive for ATTR Val122Ile through DNA sampling of umbilical cord blood.25 These statistics are of concern, as ATTR amyloidosis could be a significant health concern in a patient population that is already medically underserved.
Yamashita et al25 estimate that 1.35 million Americans of African-American descent may be affected by ATTR Val122Ile and vulnerable to restrictive cardiomyopathy–related heart failure and death. At the very least, this disorder can impair quality of life, especially in the presence of other comorbid conditions.
Clinical Presentation
The presence of exertional syncope at presentation is ominous, as it may be a marker of severe restrictive cardiomyopathy, postural hypotension due to excessive diuresis or autonomic neuropathy, ventricular arrhythmias from localized hypoperfusion, and rarely from cardiac tamponade due to pericardial involvement.27 Despite widespread involvement of the conduction system in specimens at autopsy, high-grade IV block is unusual.3
Diagnostic Studies
ECG. Both ECG and Holter monitoring can detect the arrhythmias and conduction disturbances (eg, first-degree AV block, low voltage patterns) associated with cardiac amyloidosis. Patients often experience syncopal and near-syncopal episodes as a result of conduction disturbances.28 Patients with conditions such as cardiac amyloidosis who present with severe heart failure are at high risk for sudden death secondary to conduction disturbances. Many have benefited from implanted defibrillators.29
Echocardiography. In patients with ATTR Val122Ile cardiac amyloidosis, echocardiography reveals thickened ventricular walls (ie, measuring ≥ 15 mm; normal, ≤ 11 mm).19 Amyloid-restrictive cardiomyopathy is associated with a marked dissociation between short- and long-axis systolic function, in cases in which left ventricular ejection fraction is normal.30
Echocardiography may demonstrate the characteristic specular or granular sparkling appearance that signifies advanced disease.15,21 Only a minority have this pattern in the myocardium, however, and changes in echocardiographic technology have made this finding less noticeable.30
MRI. Among more recently used diagnostic studies, cardiac magnetic resonance (CMR) has been reported to demonstrate late gadolinium enhancement (LGE) in perhaps 80% of patients with familial amyloidosis and cardiac involvement (as determined through biopsy and Congo Red stain). LGE-CMR shows darkening of the cardiac tissue, a common occurrence in amyloidosis.31
LGE is associated with increased thickness of both left and right ventricles, lower ECG voltage patterns, elevated BNP, and elevated troponin T.31,32 Globally, LGE is associated with the worst prognosis in patients with cardiac amyloidosis. Use of LGE-CMR testing can help facilitate early detection of cardiac amyloidosis in patients who may be vulnerable to cardiac damage.31
Cardiac catheterization. In patients with cardiac amyloidosis, cardiac catheterization usually shows normal coronary arteries.3
Diagnosis
Early diagnosis of ATTR cardiac amyloidosis is crucial to the patient’s survival; it should be ruled out in any African-American patient with unexplained heart failure and echocardiography showing increased wall thickness with a nondilated left ventricular cavity. Additional clues include significant proteinuria, hepatomegaly disproportionate to the degree of heart failure, or corresponding neuropathy. Known family history of the disease, along with variant type, allows for a prompt and correct diagnosis.10
It has been reported that most clinicians who encounter heart failure, particularly in black patients, do not consider amyloidosis in the differential diagnosis, because of the high prevalence of hypertension and congestive heart failure in this population.10,15,19 As a result, amyloidosis often goes undiagnosed.19
Findings of enlarged and thickened cardiac walls on echocardiography but normal coronary arteries on cardiac catheterization should alert the treating clinician to further work-up for cardiac amyloidosis.19 In such a patient, according to Kristen et al,21 endomyocardial biopsy with Congo Red staining is the gold standard for diagnosis of amyloidosis.
In ATTR Val122Ile familial amyloidosis, it is unclear whether patients who are homozygous for the disease present with symptoms at earlier onset with more progressive illness or die sooner than those who are heterozygous.33 Nevertheless, once the diagnosis is confirmed, it is important to determine the patient’s specific variant type by DNA testing so that appropriate treatment can be initiated and the patient’s prognosis evaluated.5,10,13
Treatment
For familial amyloidosis in general, some researchers advocate liver transplantation to remove the source of mutant amyloid protein and stop all deposition of amyloid fibrils; this procedure can be followed later by transplantation of other affected organs (including the heart).5,23 Maurer et al34 have reported improved one-year survival rates among patients with ATTR amyloidosis who underwent both cardiac and liver transplantation: 75%, versus 23% in patients who did not receive transplanted organs.
Management of cardiac amyloidosis usually requires a twofold approach: treating associated congestive heart failure, and preventing further deposition of amyloid.24 In the case patient (as in most patients with ATTR amyloidosis), heart transplantation was deemed the only life-sustaining treatment option.11,19,33
Pharmacotherapeutic options are limited for patients with ATTR Val122Ile familial amyloidosis. Conventional heart failure agents (eg, ACE inhibitors, angiotensin receptor blockers, digoxin, β-blockers, calcium channel blockers) can exacerbate heart failure symptoms, leading to a potentially life-threatening arrhythmia.3,11,19,24,35 Amyloid fibrils bind to digitalis, increasing susceptibility to digitalis toxicity; and to nifedipine, causing hemodynamic deterioration. Verapamil should be avoided, as it may induce severe left ventricular dysfunction. ACE inhibitors often provoke profound hypotension in primary amyloidosis.24,35
Diuretics, too (eg, furosemide, as was prescribed for the case patient), must be used with caution.3 These agents have been used to treat fluid overload and the resulting peripheral edema and shortness of breath found in ATTR Val122Ile patients who experience heart failure.36 According to Dubrey et al,5 cautious use of diuretics is necessary for management of heart failure symptoms in these patients.
Because the risk for intracardiac thrombus is high, anticoagulation (using agents other than β-blockers or calcium channel blockers) should be implemented unless compelling risks are involved.11,24 Amiodarone is relatively well tolerated for ventricular tachydysrhythmias and in atrial fibrillation if the goal is maintaining sinus rhythm.37
Regarding heart transplantation in patients with familial amyloidosis, Jacob et al33 hypothesize that since mutant amyloid protein is synthesized by the liver, it would take approximately 50 years for a transplanted heart to become affected by amyloid deposition. In a 59-year-old Afro-Caribbean man with familial amyloidosis who underwent cardiac transplantation, Hamour et al11 reported that the donor heart remained amyloid-free three years posttransplantation, as demonstrated by serial cardiac biopsy.
On the Horizon
Clinical trials are now under way to examine pharmacotherapeutic options for patients with ATTR amyloidosis. Now being examined in clinical trials, for example, is Fx-1006A, a drug that stabilizes ATTR and prevents the misfolding of the amyloid protein fibril, in turn preventing it from binding to the target organ.38 Similarly, ALN-TTR, a drug believed to prevent disease manifestation and possibly facilitate disease regression, is being investigated in early human trials.39
Additionally, the use of genetic testing is recommended in at-risk individuals to identify the TTR gene. Affected patients may benefit from prophylactic medical management, which would halt amyloidogenesis of TTR—and possibly treat the condition as well.35 Pharmacotherapeutic agents like diflunisal, an NSAID, antagonize the aggregation of TTR protein and hinder formation of the amyloid fibrils.40
CONCLUSION
ATTR Val122Ile familial amyloidosis is a rare disorder that causes abnormal synthesis of amyloid protein in the liver, which then infiltrates the cardiac structure, leading to restrictive cardiomyopathy and progressive heart failure. Patients who present with symptoms of heart failure, cardiac enlargement on echocardiography, and a finding of granular speckling patterns, though not specific on echocardiography, should prompt the health care provider to refer the patient to a cardiologist familiar with cardiac amyloidosis for further work-up.
Diagnosed patients must undergo genetic testing to determine the specific variant type so that prompt treatment can be initiated. In patients with ATTR Val122Ile familial amyloidosis, the treatment of choice is cardiac transplantation. Although the mutant amyloid protein continues to be synthesized in the liver, the donor heart is unlikely to become affected by this substance for many years. Appropriately treated patients can maintain good quality of life, free of heart failure.
REFERENCES
1. Lim RP, Srichai MB, Lee VS. Non-ischemic causes of delayed myocardial hyperenhancement on MRI. AJR Am J Roentgenol. 2007;188 (6):1675-1681.
2. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid. 2010;17(3-4):101-104.
3. Kendall H. Cardiac amyloidosis. Crit Care Nurse. 2010;30(2):16-23.
4. Eriksson M, Büttener J, Todorov T, et al. Prevalence of germline mutations in the TTR gene in a consecutive series of surgical pathology specimens with AATR amyloid. Am J Surg Pathol. 2009;33(1):58-65.
5. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84.
6. Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277-282.
7. XDx Expression Diagnostics. Allomap®: molecular expression testing (2004). www.allomap.com. Accessed May 14, 2012.
8. Mandras SA, Crespo J, Patel HM. Innovative application of immunologic principles in heart transplantation. Ochsner J. 2010;10(4):231-235.
9. Yamani MH, Taylor DO, Rodriguez R, et al. Transplant vasculopathy is associated with increased AlloMap gene expression score. J Heart Lung Transplant. 2007;26(4):403-406.
10. Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17(6):909-927.
11. Hamour IM, Lachmann HJ, Goodman HJ, et al. Heart transplantation for homozygous familial transthyretin (TTR) V122I cardiac amyloidosis. Am J Transplant. 2008;8(5):1056-1059.
12. Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466-473.
13. Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52(4):347-361.
14. Askanas V, Engel WK, McFerrin J, Vattemi G. Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology. 2003;61(2):257-260.
15. Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidoses associated with the transthyretin lle122 mutation in a Caucasian family. Amyloid. 2001;8(4):263-269.
16. Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opin Cardiol. 2009;24(3): 214-220.
17. Nihoyannopoulos P, Dawson D. Restrictive cardiomyopathies. Eur J Echocardiogr. 2009;10 (8):iii23-iii33.
18. Bruce J. Getting to the heart of cardiomyopathies. Nursing. 2005;35(8):44-47.
19. Falk RH. The neglected entity of familial cardiac amyloidosis in African Americans. Ethn Dis. 2002;12(1):141-143.
20. Piper C, Butz T, Farr M, et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue Doppler echocardiography, and myocardial biopsy. Amyloid. 2010;17(1):1-9.
21. Kristen AV, Meyer FJ, Perz JB, et al. Risk stratification in cardiac amyloidosis: novel approaches. Transplantation. 2005;80(1 suppl):S151-S155.
22. Westermark P, Benson MD, Buxbaum JN, et al. A primer of amyloid nomenclature. Amyloid. 2007;14(3):179-183.
23. Picken MM. New insights into systemic amyloidosis: the importance of diagnosis of specific type. Curr Opin Nephrol Hypertens. 2007; 16(3):196-203.
24. Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124(9):1079-1085.
25. Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile 122 allele frequency in an African-American population. Amyloid. 2005;12(2):127-130.
26. Benson MD, Yazaki M, Magy N. Laboratory assessment of transthyretin amyloidosis. Clin Chem Lab Med. 2002;40(12):1262-1265.
27. Chamarthi B, Dubrey SW, Cha K, et al. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol. 1997;80(9):1242-1245.
28. Correia MJ, Coutinho CA, Conceiçao I, et al. Role of heart rate variability in the assessment of autonomic dysfunction in type I familial amyloidotic polyneuropathy. Folia Cardiol. 2005;12(suppl C):459-462.
29. Kadish A, Mehra M. Heart failure devices: Implantable cardioverter-defibrillators and biventricular pacing therapy. Circulation. 2005; 111(24):3327-3335.
30. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43(3):410-415.
31. Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3(2):155-164.
32. Fealey ME, Edwards WD, Buadi FK, et al. Echocardiographic features of cardiac amyloidosis presenting as endomyocardial disease in a 54-year-old male. J Cardiol. 2009;54(1):162-166.
33. Jacob EK, Edwards WD, Zucker M, et al. Homozygous transthyretin mutation in an African American male. J Mol Diagn. 2007; 9(1):127-131.
34. Maurer MS, Raina A, Hesdorffer C, et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation. 2007;83(5):539-545.
35. Buxbaum J, Alexander A, Koziol J, et al. Significance of the amyloidogenic transthyretin Val 122 Ile allele in African-Americans in the Arteriosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Am Heart J. 2010;159(5):864-870.
36. Rose BD, Colucci WS. Use of diuretics in heart failure (2010). www.uptodate.com/con tents/use-of-diuretics-in-patients-with-heart-failure. Accessed May 14, 2012.
37. Trappe H-J. Treating critical supraventricular and ventricular arrhythmias. J Emerg Trauma Shock. 2010;3(2):143-152.
38. Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219-3230.
39. Alnylam Pharmaceuticals. TTR Amyloidosis: ALN-TTR (2011). www.alnylam.com/Programs-and-Pipeline/Programs/index.php. Accessed May 14, 2012.
40. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin: potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355-374.
