User login
Updates in the Management of Orthopedic Soft-Tissue Injuries Associated With Lower Extremity Trauma
UPDATE ON FERTILITY
Dr. Adamson reports that he receives research grants from LabCorp and Auxogyn, and is the founder and CEO of Advanced Reproductive Care. Dr. Abusief reports no financial relationships relevant to this article.
The field of reproductive endocrinology has advanced at warp speed over the past few decades—and shows no sign of stopping any time soon. In this article, we outline noteworthy developments of the past year:
- publication of two important Committee Opinions from the American Society for Reproductive Medicine (ASRM)—one of them on the need to reduce the rate of multiple gestation among women undergoing treatment for infertility and the other focusing on a method of achieving this goal: elective single embryo transfer
- two studies of vitrification for cryopreservation of embryos and oocytes
- a trio of investigations into the utility of anti-Müllerian hormone as a means of assessing ovarian reserve and reproductive potential.
Goal of non-ART infertility therapy should be to produce a single child
Practice Committee of the American Society for Reproductive Medicine. Multiple gestation associated with infertility therapy: an American Society for Reproductive Medicine Practice Committee opinion [published online ahead of print December 20, 2011]. Fertil Steril. doi:10.1016/j.fertnstert.2011.11.048.
The goal of infertility treatment is for each patient to have one healthy child at a time, according to a new Practice Committee Opinion from the American Society for Reproductive Medicine (ASRM).
In women who experience oligo-ovulation or anovulation, ovulation induction is typically offered. For ovulatory women who have unexplained or age-related infertility, the treatment often is controlled ovarian stimulation. Either intervention can lead to ovulation from multiple follicles and, ultimately, increase the risk of multiple gestation.
Multiple gestation increases maternal morbidity and both fetal and neonatal morbidity and mortality. Most of the poor perinatal outcomes relate directly to preterm birth. Treatment of women who have infertility, therefore, requires achieving a balance between two competing needs:
- maximizing the probability of pregnancy
- minimizing the risk of multiple (two fetuses or more) or high-order multiple (more than two fetuses) gestation.
Many multiple births are iatrogenic
Approximately 60% of twin births result from natural conception, 30% from ovulation induction and controlled ovarian stimulation, and 10% from assisted reproductive technologies (ART). For high-order multiple gestation, the figures are 20% for natural conception, 50% for ovulation induction and controlled ovarian stimulation, and 30% for ART. These statistics reveal that a very large percentage of multiple births are iatrogenic, with fertility treatment increasing the risk of twins by a factor of approximately 20 and the risk of high-order multiples by a factor of more than 100. The risk of monozygotic twinning also increases by a factor of 2 or 3 after ovulation induction, compared with natural conception.
Triplets should be a rarity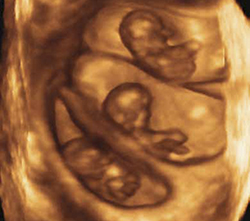
Three-dimensional sonogram of triplets.
Multiple gestation is expensive
The economic costs associated with excess perinatal and maternal morbidity are substantial. They include the immediate costs associated with maternal hospitalization and neonatal intensive care and lifetime costs associated with care for chronic illness, rehabilitation, and special education. Although these costs might be offset by the productivity of individuals, the overall benefit to society is clearly greater when a singleton is born. Personal and familial nonfinancial costs of morbidity and mortality can also be significant.
A sense of urgency on the part of the patient may contribute to an increased risk of multiple gestation by prompting more aggressive treatment. Other contributors include limited health coverage, which creates a personal financial burden, and inadequate patient education about the risks of multiple gestation.
Strategies for limiting the risk of multiple gestation
Appropriate treatment goals are the foundation of risk-reducing strategies. For example, ovulation induction in women who have oligo-ovulation or anovulation should aim toward producing a single oocyte. These women tend to respond to lower dosages of ovarian-stimulation drugs than are typically given. Therefore, women undergoing ovulation induction should receive a lower dosage of gonadotropins and be monitored very carefully for the number of developing follicles and ovarian hyperstimulation syndrome.
In contrast, the goal of controlled ovarian stimulation in ovulatory women who have unexplained or age-related subfertility is to stimulate the development and ovulation of more than one mature follicle to increase cycle fecundity.
Regrettably, efforts have failed to identify estradiol levels and the specific size and number of follicles that prevent multiple gestation. The most likely reason is that follicular size cannot accurately predict the maturity of the oocyte within—follicles as small as 10 mm sometimes yield mature and fertilizable oocytes. Moreover, the population that undergoes ovulation induction or controlled ovarian stimulation is very heterogenous. Therefore, it is not possible to propose valid guidelines to reduce the rate of multiple gestation.
Nevertheless, multiple gestation is sufficiently problematic that we recommend some strategies to reduce its incidence:
- Use low-dosage gonadotropin stimulation with careful monitoring, and limit the number of follicles that are roughly 15 mm or larger to two in patients 37 years of age or younger; three in patient 38 to 40 years old; and more in patients older than 40
- Develop specific cancellation criteria, which should be explained to and accepted by patients undergoing controlled ovarian stimulation. Gonadotropin-releasing hormone (GnRH) antagonists may be of benefit.1
- When clomiphene citrate stimulates the development of two or more mature follicles, outcomes do not differ from those obtained with controlled ovarian stimulation using gonadotropins and intrauterine insemination (IUI).2 Therefore, a reasonable strategy in many patients is to consider initiating treatment with clomiphene citrate and IUI and to proceed directly to in vitro fertilization (IVF) when treatment fails, thereby avoiding controlled ovarian stimulation altogether.3
- Pre-ovulatory ultrasonography-guided aspiration of excess follicles to reduce the risk of multiple gestation has potential benefit but needs further study.
Overall, regardless of the medication or regimen employed, it may not be possible to entirely eliminate the risk of multiple gestation associated with ovulation induction or controlled ovarian stimulation.
When to consider gestation reduction
High-order multifetal gestation reduction has been utilized as a strategy to reduce complications associated with ovulation induction and controlled ovarian stimulation, but use of this technology must be regarded as an adverse outcome of infertility treatment. Overall, data suggest that multifetal gestation reduction is associated with a reduced risk of prematurity, although its true benefit is difficult to elucidate due to potential bias in the interpretation of data. A small percentage of patients lose the entire pregnancy, and the procedure can present patients with a profound ethical dilemma and psychological trauma. Thorough counseling is imperative.
Despite feelings of loss and guilt that persist for a year or longer, most patients report that they would make the decision to undergo gestation reduction again if a similar situation arose in the future.4
The procedure should be performed only in a specialized center by an experienced practitioner.
When performing ovulation induction and controlled ovarian stimulation, use the lowest dose of drug necessary to obtain a single mature follicle in anovulatory women, two follicles in young ovulatory women, and three follicles in women 38 to 40 years old. Because of the high risk of multiple gestation associated with controlled ovarian stimulation followed by IUI, consider moving directly to IVF after use of clomiphene citrate and IUI.
Practice Committee of the Society for Assisted Reproductive Technology and Practice Committee of the American Society for Reproductive Medicine. Elective single-embryo transfer [published online ahead of print December 22, 2011]. Fertil Steril. doi:10.1016/j.fertnstert.2011.11.050.
As IVF implantation rates have improved, the practice of transferring multiple embryos has resulted in a much-increased pregnancy rate but also a high percentage of multiple gestations. Elective single embryo transfer (eSET) has been advocated as the only effective means to avoid multiple pregnancy in IVF cycles, but there is significant concern that it might ultimately reduce the pregnancy rate.
ASRM recently published a Practice Committee Opinion that offers guidance for patient selection and describes barriers to eSET. Patient selection is critical.
Utilization of eSET in the United States has increased over the past decade but still lags behind other countries. Use of double embryo transfer (DET) has increased, significantly reducing the likelihood of high-order multiple pregnancies associated with ART but producing no change in the twin pregnancy rate (FIGURE). Randomized, controlled trials and other studies have demonstrated that the cumulative pregnancy rate per retrieval is no different for eSET followed by frozen embryo transfer than it is for DET in properly selected patients.
Most transfers involve two embryos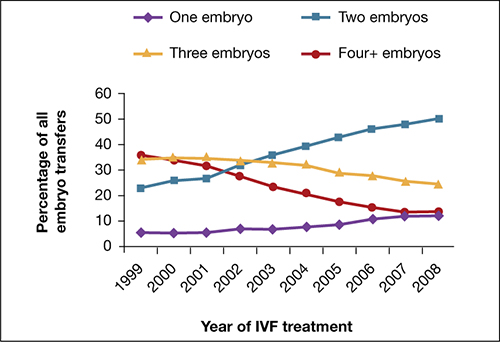
Percentage of transfer of one, two, three, or four or more embryos among all in vitro fertilization cycles performed in the United States, 1999–2008.
SOURCE: ASRM. Reproduced with permission.eSET is most appropriate for women who have a good prognosis:
- age younger than 35 years
- >1 top-quality embryo available for transfer
- first or second treatment cycle
- prior successful IVF
- recipients of embryos from donated eggs.
Women 35 to 40 years old can be considered for eSET if they have top-quality, blastocyst-stage embryos available for transfer.
Barriers to eSET include a lack of provider and patient education about it, financial considerations, embryo selection, and successful cryopreservation. When insurance coverage or refund guarantees are available, patient acceptance of eSET increases.
Elective single embryo transfer is the only ART embryo transfer strategy that will reduce the twin pregnancy rate. However, it is not a good approach for all patients and must be carefully utilized in selected patients who have a good prognosis.
Vitrification for cryopreservation of embryos appears to be superior to slow freezing
Leibo S, Pool T. The principal variables of cryopreservation: solutions, temperatures, and rate changes. Fertil Steril. 2011;96(2):269–276.
Cobo A, Diaz C. Clinical application of oocyte vitrification: a systematic review and meta-analysis of randomized controlled trials. Fertil Steril. 2011;96(2):277–285.
Cryopreservation is a method by which cells are suspended in a solution of salts and low-molecular-weight organic compound, cooled to subzero temperatures (approximately –196°C) in liquid nitrogen, stored, and then rewarmed. Cryopreservation has become a major component of the practice of assisted reproduction, with more than 37,000 pregnancies produced from cryopreserved embryos from 2005 through 2009 in the United States alone.5,6
Standard (slow) freezing methods for embryo cryopreservation involve suspension of the embryos in a 10% solution of propylene glycol supplemented with 3.4% sucrose, cooling them to –35°C at a rate of 0.3°C/min, submerging them in liquid nitrogen for storage, and rewarming the frozen embryos at a rate of approximately 300°C/min to thaw them.5
A major advance in the science of cryopreservation is the use of vitrification, a method of freezing in which the embryos are equilibrated with a 10% or 15% solution of cryoprotectant and then exposed briefly (30–60 seconds) to a 20% to 40% solution of cryoprotectant to achieve relative cellular dehydration. The embryos are then placed in a storage container and submerged in liquid nitrogen. During vitrification, embryos can be cooled at a rate exceeding 1,000°C/min. Vitrified embryos are stored at approximately –196°C and thawed in ultra-rapid fashion.
The development of vitrification methods has significantly advanced the technology of oocyte cryopreservation, which has been utilized for:
- preservation of fertility in cancer patients
- social reasons (e.g., lack of a partner)
- egg-donation programs
- minimization of the risk of ovarian hyperstimulation syndrome
- storage of surplus eggs when embryo cryopreservation is not feasible.
Cobo and Diaz recently conducted a systematic review and meta-analysis of randomized, controlled trials of oocyte vitrification. They found that the potential for fertilization, embryogenesis, and pregnancy from oocytes that had undergone vitrification and warming was not significantly different from the potential for fresh oocytes and was better than the potential for oocytes that had undergone freezing and thawing from standard freezing cycles.
Although the findings of the meta-analysis were limited by the small number of studies and possible selection bias, an increasing body of evidence supports the use of vitrification for cryopreservation of oocytes. Large-scale controlled trials are needed. Until they are performed, the findings of the meta-analysis should be interpreted with caution.
Newer ultra-rapid freezing of oocytes and embryos using vitrification appears to produce results that are superior to those obtained with traditional slow freezing. Large randomized, controlled trials are needed to confirm the improved efficacy of vitrification.
Anti-Müllerian hormone is an informative test of ovarian reserve—but lacks a nod from the FDA
Buyuk E, Seifer D, Younger J, Grazi R, Lieman H. Random anti-Mullerian hormone (AMH) is a predictor of ovarian response in women with elevated baseline early follicular follicle-stimulating hormone levels. Fertil Steril. 2011;95(7):2369–2372.
Li H, Yeung PW, Lau E, Ho PC, Ng EH. Evaluating the performance of serum anti-Müllerian hormone concentration in predicting the live birth rate of controlled ovarian stimulation and intrauterine insemination. Fertil Steril. 2010;94(6):2177–2181.
Lee J, Kim S, Jee B, Suh C, Kim KC, Moon SY. Anti-Müllerian hormone as a predictor of controlled ovarian hyperstimulation outcome: comparison of two commercial immunoassay kits. Fertil Steril. 2011;95(8):2602–2604.
Although it is well understood that both the quantity and quality of oocytes decline with age, the assessment of ovarian reserve continues to be a clinical challenge. Accurate evaluation can predict a woman’s response to infertility treatment, including IVF, and estimate her chance of conception. Noninvasive tests of ovarian reserve are a critical component of any evaluation of fertility. Although a woman’s age is the single most important historical factor in the assessment of reproductive capacity, there is significant variation in ovarian aging among women.
Historically, age, antral follicle count (AFC), and measurement of cycle day 3 follicle-stimulating hormone (FSH) and estradiol (E2) levels have been the most widely used measures of ovarian reserve, but mounting evidence suggests that assessment of the anti-Müllerian hormone (AMH) level may be even more informative.
AMH, also known as Müllerian-inhibiting substance, is a dimeric glycoprotein. A member of the transforming growth factor–ß family, AMH is closely related to inhibin and activin and is secreted by granulosa cells of preantral and small antral follicles in post-pubertal females.7 AMH aids in the coordination of ovarian follicular development by inhibiting recruitment of additional primordial follicles and decreasing the sensitivity of preantral and small antral follicles to FSH.8,9
AMH levels, measurable in serum, decline with age and are undetectable after menopause.10 Unlike FSH, which fluctuates during the menstrual cycle, AMH exhibits minimal intercycle and intracycle variation. The AMH level remains stable in women taking oral contraceptives and even in women who are pregnant.11
AMH is independently and significantly correlated with the ovarian response to gonadotropin therapy, with decreased levels of AMH associated with a poor response, and increased levels associated with a strong response.12 In the first cycle of IVF, an elevated AMH level has been associated with excessive response to gonadotropins and an increased risk of ovarian hyperstimulation syndrome (OHSS), independent of age and the presence of polycystic ovary syndrome.12
In a recent study of women who had an elevated FSH level and were undergoing IVF, the AMH level was strongly associated with the number of oocytes retrieved.13 Women who had an elevated FSH level but a serum AMH level of 0.6 ng/mL or above had a greater number of oocytes and day-3 embryos retrieved; they also had a lower cancellation rate than women who had a lower AMH level.13
Although no single test can predict the outcome of treatment for infertility, AMH concentrations are significantly higher in women who have a live birth (from the first cycle of stimulated IUI or after three cycles) than in women who do not.14
Two ELISA kits, one value?
Two types of enzyme-linked immunosorbent assay (ELISA) kits are commercially available for measurement of the AMH level: one from Immunotech Beckman Coulter and the other from Diagnostic Systems Laboratories. Neither kit has been approved for clinical use by the US Food and Drug Administration.
Studies comparing the values obtained using each kit have been inconsistent, generating controversy about the measurement of AMH. A recent study of women who were undergoing controlled ovarian stimulation found that the AMH levels obtained by the two kits were similar and significantly correlated with each other.15 In that study, the AMH level was measured on the day before gonadotropin administration or on the day of oocyte retrieval.15 In addition, the AMH concentrations measured by both kits were significantly associated with age, basal FSH levels, AFC, and the outcome of controlled ovarian stimulation.15 The authors concluded:
- The two commercially available kits provide reliable and similar results.
- The AMH level measured by either kit can predict the outcome of controlled ovarian stimulation, with similar reference values.15
Measurement of the AMH level can be an informative aspect of the evaluation of a patient’s fertility, as well as a valuable tool in the assessment of ovarian reserve. The AMH level can also help clinicians identify the appropriate dose of gonadotropins and predict which patients might be likely to over- or under-respond to stimulation—ultimately reducing the length and cost of treatment. Knowledge of the patient’s AMH level might inform pretreatment counseling and help women achieve reasonable expectations.
AMH is a useful test to help predict a patient’s response to ovarian stimulation and her chances of achieving pregnancy. However, AMH is only one measure of ovarian reserve and should not be used alone as a reason to exclude patients from treatment. In our practice, we use the AMH level along with cycle day 3 antral follicle count and FSH and estradiol levels.
We want to hear from you! Tell us what you think.
1. Ragni G, Caliari I, Nicolosi AE, Arnoldi M, Somigliana E, Crosignani PG. Preventing high-order multiple pregnancies during controlled ovarian hyperstimulation and intrauterine insemination: 3 years’ experience using low-dose recombinant follicle-stimulating hormone and gonadotropin-releasing hormone antagonists. Fertil Steril. 2006;85(3):619-624.
2. Ghesquiere SL, Castelain EG, Spiessens C, et al. Relationship between follicle number and (multiple) live birth rate after controlled ovarian hyperstimulation and intrauterine insemination. Am J Obstet Gynecol. 2007;197(6):589.e1-5.
3. Reindollar RH, Regan MM, Neumann PJ, et al. A randomized clinical trial to evaluate optimal treatment for unexplained infertility: the fast track and standard treatment (FASTT) trial. Fertil Steril. 2010;94(3):888-899.
4. Schreiner-Engel P, Walther VN, Mindes J, et al. First-trimester multifetal pregnancy reduction: acute and persistent psychologic reactions. Am J Obstet Gynecol. 1995;172(2 Pt 1):541.-
5. Leibo S, Pool T. The principal variables of cryopreservation: solutions temperatures, and rate changes. Fertil Steril. 2011;96(2):269-276.
6. Society for Assisted Reproductive Technology; American Society for Reproductive Medicine. Assisted reproductive technology in the United States: 2001 results generated from the American Society for Reproductive Medicine/Society for Assisted Reproductive Technology registry. Fertil Steril. 2007;87(6):1253-1266.
7. Vigier JA, Picard JY, Tran D, Legeai L, Josso N. Production of anti-Müllerian hormone: another homology between Sertoli and granulosa cells. Endocrinology. 1984;114(4):1315-1320.
8. Durlinger All, Gruijters MJG, Kramer P, et al. Anti-Müllerian hormone attenuates the effects of FSH on follicle development in the mouse ovary. Endocrinology. 2001;142(11):4891-4899.
9. Salmon NA, Handyside AH, Joyce IM. Oocyte regulation and anti-Müllerian hormone expression in granulosa cells during ovarian follicle development in mice. Dev Biol. 2004;266(1):201-208.
10. Shin SY, Lee JR, Noh GW, et al. Analysis of serum levels of anti-Müllerian hormone, inhibin B, insulin-like growth factor-I, insulin-like growth factor binding protein-3, and follicle-stimulating hormone with respect to age and menopausal status. J Korean Med Sci. 2008;23(1):104-110.
11. Streuli I, Fraisse T, Chapron C, Bijaoui G, Bischof P, de Ziegler D. Clinical uses of anti-Müllerian hormone assays: pitfalls and promises. Fertil Steril. 2009;91(1):226-230.
12. Nardo L, Gelbaya T, Wilkinson H, et al. Circulating basal anti-Müllerian hormone levels as predictor of ovarian response in women undergoing ovarian stimulation for in vitro fertilization. Fertil Steril. 2009;92(5):1586-1593.
13. Buyuk E, Seifer D, Younger J, Grazi R, Lieman H. Random anti-Müllerian hormone (AMH) is a predictor of ovarian response in women with elevated baseline early follicular follicle-stimulating hormone levels. Fertil Steril. 2011;95(7):2369-2372.
14. Li HW, Yeung PW, Lau E&, Ho PC, Ng EH. Evaluating the performance of serum anti-Müllerian hormone concentration in predicting the live birth rate of controlled ovarian stimulation and intrauterine insemination. Fertil Steril. 2010;94(6):2177-2181.
15. Lee J, Kim S, Jee B, Suh C, Kim KC, Moon SY. Anti- Müllerian hormone as a predictor of controlled ovarian hyperstimulation outcome: comparison of two commercial immunoassay kits. Fertil Steril. 2011;95(8):2602-2604.

David G. Adamson, MD
Dr. Adamson is Director of Fertility Physicians of Northern California in Palo Alto and San Jose; Adjunct Clinical Professor at Stanford University School of Medicine; Associate Clinical Professor at the University of California, San Francisco, School of Medicine; and Past President of the American Society for Reproductive Medicine.
Mary E. Abusief, MD
Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility at Fertility Physicians of Northern California in Palo Alto and San Jose, California.
David G. Adamson, MD
Dr. Adamson is Director of Fertility Physicians of Northern California in Palo Alto and San Jose; Adjunct Clinical Professor at Stanford University School of Medicine; Associate Clinical Professor at the University of California, San Francisco, School of Medicine; and Past President of the American Society for Reproductive Medicine.
Mary E. Abusief, MD
Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility at Fertility Physicians of Northern California in Palo Alto and San Jose, California.
David G. Adamson, MD
Dr. Adamson is Director of Fertility Physicians of Northern California in Palo Alto and San Jose; Adjunct Clinical Professor at Stanford University School of Medicine; Associate Clinical Professor at the University of California, San Francisco, School of Medicine; and Past President of the American Society for Reproductive Medicine.
Mary E. Abusief, MD
Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility at Fertility Physicians of Northern California in Palo Alto and San Jose, California.
Dr. Adamson reports that he receives research grants from LabCorp and Auxogyn, and is the founder and CEO of Advanced Reproductive Care. Dr. Abusief reports no financial relationships relevant to this article.
The field of reproductive endocrinology has advanced at warp speed over the past few decades—and shows no sign of stopping any time soon. In this article, we outline noteworthy developments of the past year:
- publication of two important Committee Opinions from the American Society for Reproductive Medicine (ASRM)—one of them on the need to reduce the rate of multiple gestation among women undergoing treatment for infertility and the other focusing on a method of achieving this goal: elective single embryo transfer
- two studies of vitrification for cryopreservation of embryos and oocytes
- a trio of investigations into the utility of anti-Müllerian hormone as a means of assessing ovarian reserve and reproductive potential.
Goal of non-ART infertility therapy should be to produce a single child
Practice Committee of the American Society for Reproductive Medicine. Multiple gestation associated with infertility therapy: an American Society for Reproductive Medicine Practice Committee opinion [published online ahead of print December 20, 2011]. Fertil Steril. doi:10.1016/j.fertnstert.2011.11.048.
The goal of infertility treatment is for each patient to have one healthy child at a time, according to a new Practice Committee Opinion from the American Society for Reproductive Medicine (ASRM).
In women who experience oligo-ovulation or anovulation, ovulation induction is typically offered. For ovulatory women who have unexplained or age-related infertility, the treatment often is controlled ovarian stimulation. Either intervention can lead to ovulation from multiple follicles and, ultimately, increase the risk of multiple gestation.
Multiple gestation increases maternal morbidity and both fetal and neonatal morbidity and mortality. Most of the poor perinatal outcomes relate directly to preterm birth. Treatment of women who have infertility, therefore, requires achieving a balance between two competing needs:
- maximizing the probability of pregnancy
- minimizing the risk of multiple (two fetuses or more) or high-order multiple (more than two fetuses) gestation.
Many multiple births are iatrogenic
Approximately 60% of twin births result from natural conception, 30% from ovulation induction and controlled ovarian stimulation, and 10% from assisted reproductive technologies (ART). For high-order multiple gestation, the figures are 20% for natural conception, 50% for ovulation induction and controlled ovarian stimulation, and 30% for ART. These statistics reveal that a very large percentage of multiple births are iatrogenic, with fertility treatment increasing the risk of twins by a factor of approximately 20 and the risk of high-order multiples by a factor of more than 100. The risk of monozygotic twinning also increases by a factor of 2 or 3 after ovulation induction, compared with natural conception.
Triplets should be a rarity
Three-dimensional sonogram of triplets.
Multiple gestation is expensive
The economic costs associated with excess perinatal and maternal morbidity are substantial. They include the immediate costs associated with maternal hospitalization and neonatal intensive care and lifetime costs associated with care for chronic illness, rehabilitation, and special education. Although these costs might be offset by the productivity of individuals, the overall benefit to society is clearly greater when a singleton is born. Personal and familial nonfinancial costs of morbidity and mortality can also be significant.
A sense of urgency on the part of the patient may contribute to an increased risk of multiple gestation by prompting more aggressive treatment. Other contributors include limited health coverage, which creates a personal financial burden, and inadequate patient education about the risks of multiple gestation.
Strategies for limiting the risk of multiple gestation
Appropriate treatment goals are the foundation of risk-reducing strategies. For example, ovulation induction in women who have oligo-ovulation or anovulation should aim toward producing a single oocyte. These women tend to respond to lower dosages of ovarian-stimulation drugs than are typically given. Therefore, women undergoing ovulation induction should receive a lower dosage of gonadotropins and be monitored very carefully for the number of developing follicles and ovarian hyperstimulation syndrome.
In contrast, the goal of controlled ovarian stimulation in ovulatory women who have unexplained or age-related subfertility is to stimulate the development and ovulation of more than one mature follicle to increase cycle fecundity.
Regrettably, efforts have failed to identify estradiol levels and the specific size and number of follicles that prevent multiple gestation. The most likely reason is that follicular size cannot accurately predict the maturity of the oocyte within—follicles as small as 10 mm sometimes yield mature and fertilizable oocytes. Moreover, the population that undergoes ovulation induction or controlled ovarian stimulation is very heterogenous. Therefore, it is not possible to propose valid guidelines to reduce the rate of multiple gestation.
Nevertheless, multiple gestation is sufficiently problematic that we recommend some strategies to reduce its incidence:
- Use low-dosage gonadotropin stimulation with careful monitoring, and limit the number of follicles that are roughly 15 mm or larger to two in patients 37 years of age or younger; three in patient 38 to 40 years old; and more in patients older than 40
- Develop specific cancellation criteria, which should be explained to and accepted by patients undergoing controlled ovarian stimulation. Gonadotropin-releasing hormone (GnRH) antagonists may be of benefit.1
- When clomiphene citrate stimulates the development of two or more mature follicles, outcomes do not differ from those obtained with controlled ovarian stimulation using gonadotropins and intrauterine insemination (IUI).2 Therefore, a reasonable strategy in many patients is to consider initiating treatment with clomiphene citrate and IUI and to proceed directly to in vitro fertilization (IVF) when treatment fails, thereby avoiding controlled ovarian stimulation altogether.3
- Pre-ovulatory ultrasonography-guided aspiration of excess follicles to reduce the risk of multiple gestation has potential benefit but needs further study.
Overall, regardless of the medication or regimen employed, it may not be possible to entirely eliminate the risk of multiple gestation associated with ovulation induction or controlled ovarian stimulation.
When to consider gestation reduction
High-order multifetal gestation reduction has been utilized as a strategy to reduce complications associated with ovulation induction and controlled ovarian stimulation, but use of this technology must be regarded as an adverse outcome of infertility treatment. Overall, data suggest that multifetal gestation reduction is associated with a reduced risk of prematurity, although its true benefit is difficult to elucidate due to potential bias in the interpretation of data. A small percentage of patients lose the entire pregnancy, and the procedure can present patients with a profound ethical dilemma and psychological trauma. Thorough counseling is imperative.
Despite feelings of loss and guilt that persist for a year or longer, most patients report that they would make the decision to undergo gestation reduction again if a similar situation arose in the future.4
The procedure should be performed only in a specialized center by an experienced practitioner.
When performing ovulation induction and controlled ovarian stimulation, use the lowest dose of drug necessary to obtain a single mature follicle in anovulatory women, two follicles in young ovulatory women, and three follicles in women 38 to 40 years old. Because of the high risk of multiple gestation associated with controlled ovarian stimulation followed by IUI, consider moving directly to IVF after use of clomiphene citrate and IUI.
Practice Committee of the Society for Assisted Reproductive Technology and Practice Committee of the American Society for Reproductive Medicine. Elective single-embryo transfer [published online ahead of print December 22, 2011]. Fertil Steril. doi:10.1016/j.fertnstert.2011.11.050.
As IVF implantation rates have improved, the practice of transferring multiple embryos has resulted in a much-increased pregnancy rate but also a high percentage of multiple gestations. Elective single embryo transfer (eSET) has been advocated as the only effective means to avoid multiple pregnancy in IVF cycles, but there is significant concern that it might ultimately reduce the pregnancy rate.
ASRM recently published a Practice Committee Opinion that offers guidance for patient selection and describes barriers to eSET. Patient selection is critical.
Utilization of eSET in the United States has increased over the past decade but still lags behind other countries. Use of double embryo transfer (DET) has increased, significantly reducing the likelihood of high-order multiple pregnancies associated with ART but producing no change in the twin pregnancy rate (FIGURE). Randomized, controlled trials and other studies have demonstrated that the cumulative pregnancy rate per retrieval is no different for eSET followed by frozen embryo transfer than it is for DET in properly selected patients.
Most transfers involve two embryos
Percentage of transfer of one, two, three, or four or more embryos among all in vitro fertilization cycles performed in the United States, 1999–2008.
SOURCE: ASRM. Reproduced with permission.eSET is most appropriate for women who have a good prognosis:
- age younger than 35 years
- >1 top-quality embryo available for transfer
- first or second treatment cycle
- prior successful IVF
- recipients of embryos from donated eggs.
Women 35 to 40 years old can be considered for eSET if they have top-quality, blastocyst-stage embryos available for transfer.
Barriers to eSET include a lack of provider and patient education about it, financial considerations, embryo selection, and successful cryopreservation. When insurance coverage or refund guarantees are available, patient acceptance of eSET increases.
Elective single embryo transfer is the only ART embryo transfer strategy that will reduce the twin pregnancy rate. However, it is not a good approach for all patients and must be carefully utilized in selected patients who have a good prognosis.
Vitrification for cryopreservation of embryos appears to be superior to slow freezing
Leibo S, Pool T. The principal variables of cryopreservation: solutions, temperatures, and rate changes. Fertil Steril. 2011;96(2):269–276.
Cobo A, Diaz C. Clinical application of oocyte vitrification: a systematic review and meta-analysis of randomized controlled trials. Fertil Steril. 2011;96(2):277–285.
Cryopreservation is a method by which cells are suspended in a solution of salts and low-molecular-weight organic compound, cooled to subzero temperatures (approximately –196°C) in liquid nitrogen, stored, and then rewarmed. Cryopreservation has become a major component of the practice of assisted reproduction, with more than 37,000 pregnancies produced from cryopreserved embryos from 2005 through 2009 in the United States alone.5,6
Standard (slow) freezing methods for embryo cryopreservation involve suspension of the embryos in a 10% solution of propylene glycol supplemented with 3.4% sucrose, cooling them to –35°C at a rate of 0.3°C/min, submerging them in liquid nitrogen for storage, and rewarming the frozen embryos at a rate of approximately 300°C/min to thaw them.5
A major advance in the science of cryopreservation is the use of vitrification, a method of freezing in which the embryos are equilibrated with a 10% or 15% solution of cryoprotectant and then exposed briefly (30–60 seconds) to a 20% to 40% solution of cryoprotectant to achieve relative cellular dehydration. The embryos are then placed in a storage container and submerged in liquid nitrogen. During vitrification, embryos can be cooled at a rate exceeding 1,000°C/min. Vitrified embryos are stored at approximately –196°C and thawed in ultra-rapid fashion.
The development of vitrification methods has significantly advanced the technology of oocyte cryopreservation, which has been utilized for:
- preservation of fertility in cancer patients
- social reasons (e.g., lack of a partner)
- egg-donation programs
- minimization of the risk of ovarian hyperstimulation syndrome
- storage of surplus eggs when embryo cryopreservation is not feasible.
Cobo and Diaz recently conducted a systematic review and meta-analysis of randomized, controlled trials of oocyte vitrification. They found that the potential for fertilization, embryogenesis, and pregnancy from oocytes that had undergone vitrification and warming was not significantly different from the potential for fresh oocytes and was better than the potential for oocytes that had undergone freezing and thawing from standard freezing cycles.
Although the findings of the meta-analysis were limited by the small number of studies and possible selection bias, an increasing body of evidence supports the use of vitrification for cryopreservation of oocytes. Large-scale controlled trials are needed. Until they are performed, the findings of the meta-analysis should be interpreted with caution.
Newer ultra-rapid freezing of oocytes and embryos using vitrification appears to produce results that are superior to those obtained with traditional slow freezing. Large randomized, controlled trials are needed to confirm the improved efficacy of vitrification.
Anti-Müllerian hormone is an informative test of ovarian reserve—but lacks a nod from the FDA
Buyuk E, Seifer D, Younger J, Grazi R, Lieman H. Random anti-Mullerian hormone (AMH) is a predictor of ovarian response in women with elevated baseline early follicular follicle-stimulating hormone levels. Fertil Steril. 2011;95(7):2369–2372.
Li H, Yeung PW, Lau E, Ho PC, Ng EH. Evaluating the performance of serum anti-Müllerian hormone concentration in predicting the live birth rate of controlled ovarian stimulation and intrauterine insemination. Fertil Steril. 2010;94(6):2177–2181.
Lee J, Kim S, Jee B, Suh C, Kim KC, Moon SY. Anti-Müllerian hormone as a predictor of controlled ovarian hyperstimulation outcome: comparison of two commercial immunoassay kits. Fertil Steril. 2011;95(8):2602–2604.
Although it is well understood that both the quantity and quality of oocytes decline with age, the assessment of ovarian reserve continues to be a clinical challenge. Accurate evaluation can predict a woman’s response to infertility treatment, including IVF, and estimate her chance of conception. Noninvasive tests of ovarian reserve are a critical component of any evaluation of fertility. Although a woman’s age is the single most important historical factor in the assessment of reproductive capacity, there is significant variation in ovarian aging among women.
Historically, age, antral follicle count (AFC), and measurement of cycle day 3 follicle-stimulating hormone (FSH) and estradiol (E2) levels have been the most widely used measures of ovarian reserve, but mounting evidence suggests that assessment of the anti-Müllerian hormone (AMH) level may be even more informative.
AMH, also known as Müllerian-inhibiting substance, is a dimeric glycoprotein. A member of the transforming growth factor–ß family, AMH is closely related to inhibin and activin and is secreted by granulosa cells of preantral and small antral follicles in post-pubertal females.7 AMH aids in the coordination of ovarian follicular development by inhibiting recruitment of additional primordial follicles and decreasing the sensitivity of preantral and small antral follicles to FSH.8,9
AMH levels, measurable in serum, decline with age and are undetectable after menopause.10 Unlike FSH, which fluctuates during the menstrual cycle, AMH exhibits minimal intercycle and intracycle variation. The AMH level remains stable in women taking oral contraceptives and even in women who are pregnant.11
AMH is independently and significantly correlated with the ovarian response to gonadotropin therapy, with decreased levels of AMH associated with a poor response, and increased levels associated with a strong response.12 In the first cycle of IVF, an elevated AMH level has been associated with excessive response to gonadotropins and an increased risk of ovarian hyperstimulation syndrome (OHSS), independent of age and the presence of polycystic ovary syndrome.12
In a recent study of women who had an elevated FSH level and were undergoing IVF, the AMH level was strongly associated with the number of oocytes retrieved.13 Women who had an elevated FSH level but a serum AMH level of 0.6 ng/mL or above had a greater number of oocytes and day-3 embryos retrieved; they also had a lower cancellation rate than women who had a lower AMH level.13
Although no single test can predict the outcome of treatment for infertility, AMH concentrations are significantly higher in women who have a live birth (from the first cycle of stimulated IUI or after three cycles) than in women who do not.14
Two ELISA kits, one value?
Two types of enzyme-linked immunosorbent assay (ELISA) kits are commercially available for measurement of the AMH level: one from Immunotech Beckman Coulter and the other from Diagnostic Systems Laboratories. Neither kit has been approved for clinical use by the US Food and Drug Administration.
Studies comparing the values obtained using each kit have been inconsistent, generating controversy about the measurement of AMH. A recent study of women who were undergoing controlled ovarian stimulation found that the AMH levels obtained by the two kits were similar and significantly correlated with each other.15 In that study, the AMH level was measured on the day before gonadotropin administration or on the day of oocyte retrieval.15 In addition, the AMH concentrations measured by both kits were significantly associated with age, basal FSH levels, AFC, and the outcome of controlled ovarian stimulation.15 The authors concluded:
- The two commercially available kits provide reliable and similar results.
- The AMH level measured by either kit can predict the outcome of controlled ovarian stimulation, with similar reference values.15
Measurement of the AMH level can be an informative aspect of the evaluation of a patient’s fertility, as well as a valuable tool in the assessment of ovarian reserve. The AMH level can also help clinicians identify the appropriate dose of gonadotropins and predict which patients might be likely to over- or under-respond to stimulation—ultimately reducing the length and cost of treatment. Knowledge of the patient’s AMH level might inform pretreatment counseling and help women achieve reasonable expectations.
AMH is a useful test to help predict a patient’s response to ovarian stimulation and her chances of achieving pregnancy. However, AMH is only one measure of ovarian reserve and should not be used alone as a reason to exclude patients from treatment. In our practice, we use the AMH level along with cycle day 3 antral follicle count and FSH and estradiol levels.
We want to hear from you! Tell us what you think.
Dr. Adamson reports that he receives research grants from LabCorp and Auxogyn, and is the founder and CEO of Advanced Reproductive Care. Dr. Abusief reports no financial relationships relevant to this article.
The field of reproductive endocrinology has advanced at warp speed over the past few decades—and shows no sign of stopping any time soon. In this article, we outline noteworthy developments of the past year:
- publication of two important Committee Opinions from the American Society for Reproductive Medicine (ASRM)—one of them on the need to reduce the rate of multiple gestation among women undergoing treatment for infertility and the other focusing on a method of achieving this goal: elective single embryo transfer
- two studies of vitrification for cryopreservation of embryos and oocytes
- a trio of investigations into the utility of anti-Müllerian hormone as a means of assessing ovarian reserve and reproductive potential.
Goal of non-ART infertility therapy should be to produce a single child
Practice Committee of the American Society for Reproductive Medicine. Multiple gestation associated with infertility therapy: an American Society for Reproductive Medicine Practice Committee opinion [published online ahead of print December 20, 2011]. Fertil Steril. doi:10.1016/j.fertnstert.2011.11.048.
The goal of infertility treatment is for each patient to have one healthy child at a time, according to a new Practice Committee Opinion from the American Society for Reproductive Medicine (ASRM).
In women who experience oligo-ovulation or anovulation, ovulation induction is typically offered. For ovulatory women who have unexplained or age-related infertility, the treatment often is controlled ovarian stimulation. Either intervention can lead to ovulation from multiple follicles and, ultimately, increase the risk of multiple gestation.
Multiple gestation increases maternal morbidity and both fetal and neonatal morbidity and mortality. Most of the poor perinatal outcomes relate directly to preterm birth. Treatment of women who have infertility, therefore, requires achieving a balance between two competing needs:
- maximizing the probability of pregnancy
- minimizing the risk of multiple (two fetuses or more) or high-order multiple (more than two fetuses) gestation.
Many multiple births are iatrogenic
Approximately 60% of twin births result from natural conception, 30% from ovulation induction and controlled ovarian stimulation, and 10% from assisted reproductive technologies (ART). For high-order multiple gestation, the figures are 20% for natural conception, 50% for ovulation induction and controlled ovarian stimulation, and 30% for ART. These statistics reveal that a very large percentage of multiple births are iatrogenic, with fertility treatment increasing the risk of twins by a factor of approximately 20 and the risk of high-order multiples by a factor of more than 100. The risk of monozygotic twinning also increases by a factor of 2 or 3 after ovulation induction, compared with natural conception.
Triplets should be a rarity
Three-dimensional sonogram of triplets.
Multiple gestation is expensive
The economic costs associated with excess perinatal and maternal morbidity are substantial. They include the immediate costs associated with maternal hospitalization and neonatal intensive care and lifetime costs associated with care for chronic illness, rehabilitation, and special education. Although these costs might be offset by the productivity of individuals, the overall benefit to society is clearly greater when a singleton is born. Personal and familial nonfinancial costs of morbidity and mortality can also be significant.
A sense of urgency on the part of the patient may contribute to an increased risk of multiple gestation by prompting more aggressive treatment. Other contributors include limited health coverage, which creates a personal financial burden, and inadequate patient education about the risks of multiple gestation.
Strategies for limiting the risk of multiple gestation
Appropriate treatment goals are the foundation of risk-reducing strategies. For example, ovulation induction in women who have oligo-ovulation or anovulation should aim toward producing a single oocyte. These women tend to respond to lower dosages of ovarian-stimulation drugs than are typically given. Therefore, women undergoing ovulation induction should receive a lower dosage of gonadotropins and be monitored very carefully for the number of developing follicles and ovarian hyperstimulation syndrome.
In contrast, the goal of controlled ovarian stimulation in ovulatory women who have unexplained or age-related subfertility is to stimulate the development and ovulation of more than one mature follicle to increase cycle fecundity.
Regrettably, efforts have failed to identify estradiol levels and the specific size and number of follicles that prevent multiple gestation. The most likely reason is that follicular size cannot accurately predict the maturity of the oocyte within—follicles as small as 10 mm sometimes yield mature and fertilizable oocytes. Moreover, the population that undergoes ovulation induction or controlled ovarian stimulation is very heterogenous. Therefore, it is not possible to propose valid guidelines to reduce the rate of multiple gestation.
Nevertheless, multiple gestation is sufficiently problematic that we recommend some strategies to reduce its incidence:
- Use low-dosage gonadotropin stimulation with careful monitoring, and limit the number of follicles that are roughly 15 mm or larger to two in patients 37 years of age or younger; three in patient 38 to 40 years old; and more in patients older than 40
- Develop specific cancellation criteria, which should be explained to and accepted by patients undergoing controlled ovarian stimulation. Gonadotropin-releasing hormone (GnRH) antagonists may be of benefit.1
- When clomiphene citrate stimulates the development of two or more mature follicles, outcomes do not differ from those obtained with controlled ovarian stimulation using gonadotropins and intrauterine insemination (IUI).2 Therefore, a reasonable strategy in many patients is to consider initiating treatment with clomiphene citrate and IUI and to proceed directly to in vitro fertilization (IVF) when treatment fails, thereby avoiding controlled ovarian stimulation altogether.3
- Pre-ovulatory ultrasonography-guided aspiration of excess follicles to reduce the risk of multiple gestation has potential benefit but needs further study.
Overall, regardless of the medication or regimen employed, it may not be possible to entirely eliminate the risk of multiple gestation associated with ovulation induction or controlled ovarian stimulation.
When to consider gestation reduction
High-order multifetal gestation reduction has been utilized as a strategy to reduce complications associated with ovulation induction and controlled ovarian stimulation, but use of this technology must be regarded as an adverse outcome of infertility treatment. Overall, data suggest that multifetal gestation reduction is associated with a reduced risk of prematurity, although its true benefit is difficult to elucidate due to potential bias in the interpretation of data. A small percentage of patients lose the entire pregnancy, and the procedure can present patients with a profound ethical dilemma and psychological trauma. Thorough counseling is imperative.
Despite feelings of loss and guilt that persist for a year or longer, most patients report that they would make the decision to undergo gestation reduction again if a similar situation arose in the future.4
The procedure should be performed only in a specialized center by an experienced practitioner.
When performing ovulation induction and controlled ovarian stimulation, use the lowest dose of drug necessary to obtain a single mature follicle in anovulatory women, two follicles in young ovulatory women, and three follicles in women 38 to 40 years old. Because of the high risk of multiple gestation associated with controlled ovarian stimulation followed by IUI, consider moving directly to IVF after use of clomiphene citrate and IUI.
Practice Committee of the Society for Assisted Reproductive Technology and Practice Committee of the American Society for Reproductive Medicine. Elective single-embryo transfer [published online ahead of print December 22, 2011]. Fertil Steril. doi:10.1016/j.fertnstert.2011.11.050.
As IVF implantation rates have improved, the practice of transferring multiple embryos has resulted in a much-increased pregnancy rate but also a high percentage of multiple gestations. Elective single embryo transfer (eSET) has been advocated as the only effective means to avoid multiple pregnancy in IVF cycles, but there is significant concern that it might ultimately reduce the pregnancy rate.
ASRM recently published a Practice Committee Opinion that offers guidance for patient selection and describes barriers to eSET. Patient selection is critical.
Utilization of eSET in the United States has increased over the past decade but still lags behind other countries. Use of double embryo transfer (DET) has increased, significantly reducing the likelihood of high-order multiple pregnancies associated with ART but producing no change in the twin pregnancy rate (FIGURE). Randomized, controlled trials and other studies have demonstrated that the cumulative pregnancy rate per retrieval is no different for eSET followed by frozen embryo transfer than it is for DET in properly selected patients.
Most transfers involve two embryos
Percentage of transfer of one, two, three, or four or more embryos among all in vitro fertilization cycles performed in the United States, 1999–2008.
SOURCE: ASRM. Reproduced with permission.eSET is most appropriate for women who have a good prognosis:
- age younger than 35 years
- >1 top-quality embryo available for transfer
- first or second treatment cycle
- prior successful IVF
- recipients of embryos from donated eggs.
Women 35 to 40 years old can be considered for eSET if they have top-quality, blastocyst-stage embryos available for transfer.
Barriers to eSET include a lack of provider and patient education about it, financial considerations, embryo selection, and successful cryopreservation. When insurance coverage or refund guarantees are available, patient acceptance of eSET increases.
Elective single embryo transfer is the only ART embryo transfer strategy that will reduce the twin pregnancy rate. However, it is not a good approach for all patients and must be carefully utilized in selected patients who have a good prognosis.
Vitrification for cryopreservation of embryos appears to be superior to slow freezing
Leibo S, Pool T. The principal variables of cryopreservation: solutions, temperatures, and rate changes. Fertil Steril. 2011;96(2):269–276.
Cobo A, Diaz C. Clinical application of oocyte vitrification: a systematic review and meta-analysis of randomized controlled trials. Fertil Steril. 2011;96(2):277–285.
Cryopreservation is a method by which cells are suspended in a solution of salts and low-molecular-weight organic compound, cooled to subzero temperatures (approximately –196°C) in liquid nitrogen, stored, and then rewarmed. Cryopreservation has become a major component of the practice of assisted reproduction, with more than 37,000 pregnancies produced from cryopreserved embryos from 2005 through 2009 in the United States alone.5,6
Standard (slow) freezing methods for embryo cryopreservation involve suspension of the embryos in a 10% solution of propylene glycol supplemented with 3.4% sucrose, cooling them to –35°C at a rate of 0.3°C/min, submerging them in liquid nitrogen for storage, and rewarming the frozen embryos at a rate of approximately 300°C/min to thaw them.5
A major advance in the science of cryopreservation is the use of vitrification, a method of freezing in which the embryos are equilibrated with a 10% or 15% solution of cryoprotectant and then exposed briefly (30–60 seconds) to a 20% to 40% solution of cryoprotectant to achieve relative cellular dehydration. The embryos are then placed in a storage container and submerged in liquid nitrogen. During vitrification, embryos can be cooled at a rate exceeding 1,000°C/min. Vitrified embryos are stored at approximately –196°C and thawed in ultra-rapid fashion.
The development of vitrification methods has significantly advanced the technology of oocyte cryopreservation, which has been utilized for:
- preservation of fertility in cancer patients
- social reasons (e.g., lack of a partner)
- egg-donation programs
- minimization of the risk of ovarian hyperstimulation syndrome
- storage of surplus eggs when embryo cryopreservation is not feasible.
Cobo and Diaz recently conducted a systematic review and meta-analysis of randomized, controlled trials of oocyte vitrification. They found that the potential for fertilization, embryogenesis, and pregnancy from oocytes that had undergone vitrification and warming was not significantly different from the potential for fresh oocytes and was better than the potential for oocytes that had undergone freezing and thawing from standard freezing cycles.
Although the findings of the meta-analysis were limited by the small number of studies and possible selection bias, an increasing body of evidence supports the use of vitrification for cryopreservation of oocytes. Large-scale controlled trials are needed. Until they are performed, the findings of the meta-analysis should be interpreted with caution.
Newer ultra-rapid freezing of oocytes and embryos using vitrification appears to produce results that are superior to those obtained with traditional slow freezing. Large randomized, controlled trials are needed to confirm the improved efficacy of vitrification.
Anti-Müllerian hormone is an informative test of ovarian reserve—but lacks a nod from the FDA
Buyuk E, Seifer D, Younger J, Grazi R, Lieman H. Random anti-Mullerian hormone (AMH) is a predictor of ovarian response in women with elevated baseline early follicular follicle-stimulating hormone levels. Fertil Steril. 2011;95(7):2369–2372.
Li H, Yeung PW, Lau E, Ho PC, Ng EH. Evaluating the performance of serum anti-Müllerian hormone concentration in predicting the live birth rate of controlled ovarian stimulation and intrauterine insemination. Fertil Steril. 2010;94(6):2177–2181.
Lee J, Kim S, Jee B, Suh C, Kim KC, Moon SY. Anti-Müllerian hormone as a predictor of controlled ovarian hyperstimulation outcome: comparison of two commercial immunoassay kits. Fertil Steril. 2011;95(8):2602–2604.
Although it is well understood that both the quantity and quality of oocytes decline with age, the assessment of ovarian reserve continues to be a clinical challenge. Accurate evaluation can predict a woman’s response to infertility treatment, including IVF, and estimate her chance of conception. Noninvasive tests of ovarian reserve are a critical component of any evaluation of fertility. Although a woman’s age is the single most important historical factor in the assessment of reproductive capacity, there is significant variation in ovarian aging among women.
Historically, age, antral follicle count (AFC), and measurement of cycle day 3 follicle-stimulating hormone (FSH) and estradiol (E2) levels have been the most widely used measures of ovarian reserve, but mounting evidence suggests that assessment of the anti-Müllerian hormone (AMH) level may be even more informative.
AMH, also known as Müllerian-inhibiting substance, is a dimeric glycoprotein. A member of the transforming growth factor–ß family, AMH is closely related to inhibin and activin and is secreted by granulosa cells of preantral and small antral follicles in post-pubertal females.7 AMH aids in the coordination of ovarian follicular development by inhibiting recruitment of additional primordial follicles and decreasing the sensitivity of preantral and small antral follicles to FSH.8,9
AMH levels, measurable in serum, decline with age and are undetectable after menopause.10 Unlike FSH, which fluctuates during the menstrual cycle, AMH exhibits minimal intercycle and intracycle variation. The AMH level remains stable in women taking oral contraceptives and even in women who are pregnant.11
AMH is independently and significantly correlated with the ovarian response to gonadotropin therapy, with decreased levels of AMH associated with a poor response, and increased levels associated with a strong response.12 In the first cycle of IVF, an elevated AMH level has been associated with excessive response to gonadotropins and an increased risk of ovarian hyperstimulation syndrome (OHSS), independent of age and the presence of polycystic ovary syndrome.12
In a recent study of women who had an elevated FSH level and were undergoing IVF, the AMH level was strongly associated with the number of oocytes retrieved.13 Women who had an elevated FSH level but a serum AMH level of 0.6 ng/mL or above had a greater number of oocytes and day-3 embryos retrieved; they also had a lower cancellation rate than women who had a lower AMH level.13
Although no single test can predict the outcome of treatment for infertility, AMH concentrations are significantly higher in women who have a live birth (from the first cycle of stimulated IUI or after three cycles) than in women who do not.14
Two ELISA kits, one value?
Two types of enzyme-linked immunosorbent assay (ELISA) kits are commercially available for measurement of the AMH level: one from Immunotech Beckman Coulter and the other from Diagnostic Systems Laboratories. Neither kit has been approved for clinical use by the US Food and Drug Administration.
Studies comparing the values obtained using each kit have been inconsistent, generating controversy about the measurement of AMH. A recent study of women who were undergoing controlled ovarian stimulation found that the AMH levels obtained by the two kits were similar and significantly correlated with each other.15 In that study, the AMH level was measured on the day before gonadotropin administration or on the day of oocyte retrieval.15 In addition, the AMH concentrations measured by both kits were significantly associated with age, basal FSH levels, AFC, and the outcome of controlled ovarian stimulation.15 The authors concluded:
- The two commercially available kits provide reliable and similar results.
- The AMH level measured by either kit can predict the outcome of controlled ovarian stimulation, with similar reference values.15
Measurement of the AMH level can be an informative aspect of the evaluation of a patient’s fertility, as well as a valuable tool in the assessment of ovarian reserve. The AMH level can also help clinicians identify the appropriate dose of gonadotropins and predict which patients might be likely to over- or under-respond to stimulation—ultimately reducing the length and cost of treatment. Knowledge of the patient’s AMH level might inform pretreatment counseling and help women achieve reasonable expectations.
AMH is a useful test to help predict a patient’s response to ovarian stimulation and her chances of achieving pregnancy. However, AMH is only one measure of ovarian reserve and should not be used alone as a reason to exclude patients from treatment. In our practice, we use the AMH level along with cycle day 3 antral follicle count and FSH and estradiol levels.
We want to hear from you! Tell us what you think.
1. Ragni G, Caliari I, Nicolosi AE, Arnoldi M, Somigliana E, Crosignani PG. Preventing high-order multiple pregnancies during controlled ovarian hyperstimulation and intrauterine insemination: 3 years’ experience using low-dose recombinant follicle-stimulating hormone and gonadotropin-releasing hormone antagonists. Fertil Steril. 2006;85(3):619-624.
2. Ghesquiere SL, Castelain EG, Spiessens C, et al. Relationship between follicle number and (multiple) live birth rate after controlled ovarian hyperstimulation and intrauterine insemination. Am J Obstet Gynecol. 2007;197(6):589.e1-5.
3. Reindollar RH, Regan MM, Neumann PJ, et al. A randomized clinical trial to evaluate optimal treatment for unexplained infertility: the fast track and standard treatment (FASTT) trial. Fertil Steril. 2010;94(3):888-899.
4. Schreiner-Engel P, Walther VN, Mindes J, et al. First-trimester multifetal pregnancy reduction: acute and persistent psychologic reactions. Am J Obstet Gynecol. 1995;172(2 Pt 1):541.-
5. Leibo S, Pool T. The principal variables of cryopreservation: solutions temperatures, and rate changes. Fertil Steril. 2011;96(2):269-276.
6. Society for Assisted Reproductive Technology; American Society for Reproductive Medicine. Assisted reproductive technology in the United States: 2001 results generated from the American Society for Reproductive Medicine/Society for Assisted Reproductive Technology registry. Fertil Steril. 2007;87(6):1253-1266.
7. Vigier JA, Picard JY, Tran D, Legeai L, Josso N. Production of anti-Müllerian hormone: another homology between Sertoli and granulosa cells. Endocrinology. 1984;114(4):1315-1320.
8. Durlinger All, Gruijters MJG, Kramer P, et al. Anti-Müllerian hormone attenuates the effects of FSH on follicle development in the mouse ovary. Endocrinology. 2001;142(11):4891-4899.
9. Salmon NA, Handyside AH, Joyce IM. Oocyte regulation and anti-Müllerian hormone expression in granulosa cells during ovarian follicle development in mice. Dev Biol. 2004;266(1):201-208.
10. Shin SY, Lee JR, Noh GW, et al. Analysis of serum levels of anti-Müllerian hormone, inhibin B, insulin-like growth factor-I, insulin-like growth factor binding protein-3, and follicle-stimulating hormone with respect to age and menopausal status. J Korean Med Sci. 2008;23(1):104-110.
11. Streuli I, Fraisse T, Chapron C, Bijaoui G, Bischof P, de Ziegler D. Clinical uses of anti-Müllerian hormone assays: pitfalls and promises. Fertil Steril. 2009;91(1):226-230.
12. Nardo L, Gelbaya T, Wilkinson H, et al. Circulating basal anti-Müllerian hormone levels as predictor of ovarian response in women undergoing ovarian stimulation for in vitro fertilization. Fertil Steril. 2009;92(5):1586-1593.
13. Buyuk E, Seifer D, Younger J, Grazi R, Lieman H. Random anti-Müllerian hormone (AMH) is a predictor of ovarian response in women with elevated baseline early follicular follicle-stimulating hormone levels. Fertil Steril. 2011;95(7):2369-2372.
14. Li HW, Yeung PW, Lau E&, Ho PC, Ng EH. Evaluating the performance of serum anti-Müllerian hormone concentration in predicting the live birth rate of controlled ovarian stimulation and intrauterine insemination. Fertil Steril. 2010;94(6):2177-2181.
15. Lee J, Kim S, Jee B, Suh C, Kim KC, Moon SY. Anti- Müllerian hormone as a predictor of controlled ovarian hyperstimulation outcome: comparison of two commercial immunoassay kits. Fertil Steril. 2011;95(8):2602-2604.
1. Ragni G, Caliari I, Nicolosi AE, Arnoldi M, Somigliana E, Crosignani PG. Preventing high-order multiple pregnancies during controlled ovarian hyperstimulation and intrauterine insemination: 3 years’ experience using low-dose recombinant follicle-stimulating hormone and gonadotropin-releasing hormone antagonists. Fertil Steril. 2006;85(3):619-624.
2. Ghesquiere SL, Castelain EG, Spiessens C, et al. Relationship between follicle number and (multiple) live birth rate after controlled ovarian hyperstimulation and intrauterine insemination. Am J Obstet Gynecol. 2007;197(6):589.e1-5.
3. Reindollar RH, Regan MM, Neumann PJ, et al. A randomized clinical trial to evaluate optimal treatment for unexplained infertility: the fast track and standard treatment (FASTT) trial. Fertil Steril. 2010;94(3):888-899.
4. Schreiner-Engel P, Walther VN, Mindes J, et al. First-trimester multifetal pregnancy reduction: acute and persistent psychologic reactions. Am J Obstet Gynecol. 1995;172(2 Pt 1):541.-
5. Leibo S, Pool T. The principal variables of cryopreservation: solutions temperatures, and rate changes. Fertil Steril. 2011;96(2):269-276.
6. Society for Assisted Reproductive Technology; American Society for Reproductive Medicine. Assisted reproductive technology in the United States: 2001 results generated from the American Society for Reproductive Medicine/Society for Assisted Reproductive Technology registry. Fertil Steril. 2007;87(6):1253-1266.
7. Vigier JA, Picard JY, Tran D, Legeai L, Josso N. Production of anti-Müllerian hormone: another homology between Sertoli and granulosa cells. Endocrinology. 1984;114(4):1315-1320.
8. Durlinger All, Gruijters MJG, Kramer P, et al. Anti-Müllerian hormone attenuates the effects of FSH on follicle development in the mouse ovary. Endocrinology. 2001;142(11):4891-4899.
9. Salmon NA, Handyside AH, Joyce IM. Oocyte regulation and anti-Müllerian hormone expression in granulosa cells during ovarian follicle development in mice. Dev Biol. 2004;266(1):201-208.
10. Shin SY, Lee JR, Noh GW, et al. Analysis of serum levels of anti-Müllerian hormone, inhibin B, insulin-like growth factor-I, insulin-like growth factor binding protein-3, and follicle-stimulating hormone with respect to age and menopausal status. J Korean Med Sci. 2008;23(1):104-110.
11. Streuli I, Fraisse T, Chapron C, Bijaoui G, Bischof P, de Ziegler D. Clinical uses of anti-Müllerian hormone assays: pitfalls and promises. Fertil Steril. 2009;91(1):226-230.
12. Nardo L, Gelbaya T, Wilkinson H, et al. Circulating basal anti-Müllerian hormone levels as predictor of ovarian response in women undergoing ovarian stimulation for in vitro fertilization. Fertil Steril. 2009;92(5):1586-1593.
13. Buyuk E, Seifer D, Younger J, Grazi R, Lieman H. Random anti-Müllerian hormone (AMH) is a predictor of ovarian response in women with elevated baseline early follicular follicle-stimulating hormone levels. Fertil Steril. 2011;95(7):2369-2372.
14. Li HW, Yeung PW, Lau E&, Ho PC, Ng EH. Evaluating the performance of serum anti-Müllerian hormone concentration in predicting the live birth rate of controlled ovarian stimulation and intrauterine insemination. Fertil Steril. 2010;94(6):2177-2181.
15. Lee J, Kim S, Jee B, Suh C, Kim KC, Moon SY. Anti- Müllerian hormone as a predictor of controlled ovarian hyperstimulation outcome: comparison of two commercial immunoassay kits. Fertil Steril. 2011;95(8):2602-2604.
Appendicitis Review
Appendicitis is a transmural inflammatory process and a common cause of an acute abdomen. Inflammation that leads to perforation of the appendix, which is associated with increased morbidity and mortality, warrants prompt diagnosis. Etiology, clinical presentation, diagnostic studies, and the management of confirmed appendicitis will be addressed here.
Frequently, the etiology of appendicitis is luminal obstruction by a fecalith (the result of inspissated fecal material and inorganic salts1), but the condition may also result from parasites, a malignancy, a foreign body, or fibrosis.1-3 In some instances, lymphoid hyperplasia, resulting from a viral or bacterial infection, has been targeted as the cause of luminal obstruction.1,4 Nevertheless, in one-third to one-half of patients, obstruction is not evident as a precipitating factor in the development of appendicitis. In such cases, the basis for the inflammation is unknown.5
As the obstructed appendix becomes congested, the intraluminal pressure and venous pressure increase, leading to stasis and ischemia.1,5-8 The appendix becomes engorged with secretions. At this stage, the condition is considered uncomplicated, but if an inflamed appendix becomes gangrenous or perforates, the condition is then referred to as complicated appendicitis. Complicated appendicitis allows for invasion by intestinal bacteria of the abdominal cavity, potentially leading to peritonitis, septicemia, abscess, or fistula formation.5,9
Conventional teaching supports the concept that uncomplicated appendicitis, unless treated surgically, eventually evolves into complicated appendicitis.10 Recent research refutes this assumption, however, as different etiologies may be associated with differences in progression10-12; whether uncomplicated and complicated appendicitis are attributable to different etiologies is a question requiring further research. Irrespective of the natural progression of the disease, the current standard of care for appendicitis is still an appendectomy.13 In US hospitals in 2007 (the most recent year for which data are available), appendectomy was performed on 326,000 patients, or 10.9 patients per 10,000 population.14
EPIDEMIOLOGY
Appendicitis is most frequently seen in the second decade of life and occurs slightly more often in males than in females.2,15 Furthermore, according to data reported to the National Hospital Discharge Survey (1970 to 2004), the rate of nonperforated appendicitis is much higher in men than in women.12 In appendicitis, the risk for rupture is small within the first 36 hours of symptom onset. Beyond that point, there is a 5% increased risk for rupture with each ensuing 12-hour period.16
In neonates and infants, appendicitis is rare.3 In children younger than 3 years, however, the rate of perforation is 80% to 100%.3,17,18 This high rate may be explained by the very young child’s limited ability to articulate his or her symptoms, or by caregiver reports that are typically limited to irritability or change in diet.3,17,19 According to Marudanayagam et al,2 who performed a retrospective study of 2,660 appendectomies during a six-year period, the perforation rate declined from 23.4% in patients age 10 or younger to 6.9% in those in their 20s, then rose steadily to more than 50% in patients 70 or older.
PATIENT EVALUATION
In most cases, a diagnosis of appendicitis can be made with a careful history, systematic physical exam, and a limited number of laboratory tests without special diagnostic modalities.13 The presence of symptoms and signs may help to rule in a diagnosis of appendicitis, but the absence of clinical findings often does not exclude its possibility.16 While adult and pediatric patients with appendicitis share many clinical findings (see Table 13,8,13,18), the occurrence rate of the various findings may differ among patient populations.3,15

The median time from onset of symptoms until the patient presents for a medical evaluation averages 24 hours or less.16 Diagnosis in patients at extremes of age often proves more difficult than in other patients.20 Thus, a high level of suspicion must be maintained in these patient populations.
The Symptom History
The appendix is located in the posteromedial wall of the cecum, approximately 3 cm below the ileocecal valve.1 Initial pain perceived around the umbilicus represents a referred pain resulting from the visceral innervation of the midgut.20 As the inflammatory process within the appendix advances, the pain localizes to the anatomical position of the right lower quadrant (RLQ), with involvement of the surrounding parietal peritoneum.20(McBurney’s point, at the junction of the lateral and middle thirds of a line extending from the anterior superior iliac spine to the umbilicus, was noted as the point of maximal tenderness to palpation in acute appendicitis by Charles McBurney in the late 1800s.21)
This progression of symptoms, first recognized by John Benjamin Murphy in 1904, is considered a more reliable indicator of appendicitis than RLQ pain alone3,22; in one large retrospective study, this migratory pain had the highest positive predictive value for pediatric and adult patients (94.2% and 89.6%, respectively).15 However, migration of pain occurs in only 50% to 60% of patients, and therefore may not be helpful.1,23
According to results from other studies, unfortunately, this progression of symptoms is not often present in pediatric patients.17 The somatic RLQ pain is continuous and more severe than is the early visceral periumbilical pain.1 Since the anatomic position of the appendix can vary, a number of patients do not necessarily present with pain in the RLQ but elsewhere.24
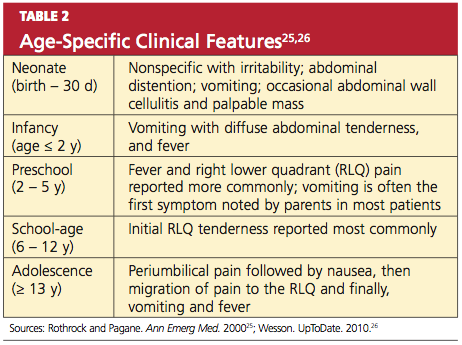
Certain clinical findings appear to be relatively age-dependent (see Table 225,26).
Classic findings in the adult diagnosed with appendicitis, as described by Becker et al,27 begin with periumbilical pain, then nausea, followed by migration of the pain to the RLQ, then vomiting and fever. Abdominal pain and anorexia are the most common presenting symptoms.20 Nausea and vomiting that begin after the onset of abdominal pain are typical; in isolation, however, these manifestations have weak diagnostic predictability for appendicitis.28 In adults, if nausea and vomiting precede abdominal pain, consideration should be given to a diagnosis of gastroenteritis rather than appendicitis.29
Among patients who are pregnant or elderly, RLQ pain remains a significant historical finding.30 In the pregnant woman, a diagnosis of appendicitis is often overlooked because of the discomforts common to pregnancy and the expanding gravid uterus.31 Elderly patients often present with vague or atypical symptoms, such as mild pain.20 In these patient populations, the diagnosis of appendicitis is often delayed.
In addition to obtaining a thorough history of the presentation of pain, it is important to conduct a complete review of the gastrointestinal, genitourinary, pulmonary, musculoskeletal, neurologic, and reproductive systems for possible alternate etiologies.
Physical Examination
The number of physical findings varies among patients who present with appendicitis3,8,20 (see Table 33,8,20). A thorough physical examination is thus required to help the clinician exclude other diseases and establish the diagnosis of appendicitis. It is important to tailor the exam according to the patient’s age and developmental stage.19
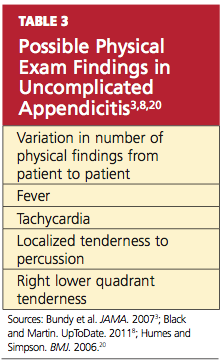
The cooperation of children undergoing the physical examination for appendicitis may vary. It may be helpful to instruct a young child to “show me with your finger where it hurts the most.”3 However, Bundy et al3 report that the presence of RLQ tenderness on palpation is of minimal value in children; rather, fever is the single most useful sign among pediatric patients and conversely, its absence reduces the risk.
Tachycardia is associated with risk for rupture.16,20 In the elderly patient, fever (> 38°C) is also strongly correlated with an increased risk for rupture.30
To alleviate pain, a patient with appendicitis may maintain the hips and knees in a slightly flexed position. While asking distracting questions, the examiner should observe the patient’s facial expressions to detect involuntary guarding.3 RLQ tenderness to percussion is often positive.
The patient may experience tenderness on palpation of the posterior abdominal wall (K sign) or right-side flank tenderness.24 Increased pain with coughing (Dunphy’s sign) or firm percussion of the heel (the heel jar test) may be elicited.8,25 A number of additional peritoneal signs, resulting from an inflamed appendix, may occur (see Table 43,8,20), but examination techniques that elicit these signs should be minimized so as to not cause the patient any unnecessary pain.

Depending on the location of the appendix, rectal and vaginal exams may yield normal findings or may elicit tenderness.32 The rectal examination should be performed with considerable care, using the smallest digit possible for an adequate assessment, especially in the younger patient.33
Several scoring systems have been designed for adults and children with suspected appendicitis, using findings from the history, the physical exam, and laboratory testing (see Table 5,34 for example). Despite their protocol-based approach, the scoring systems have yielded mixed results in clinical practice,34-36 and there is no scoring system for evaluation of the pregnant patient.37 Neither has there been any recommendation for or endorsement of a diagnostic guideline from any medical or professional organization.38 Thus, clinical gestalt is usually relied upon instead.
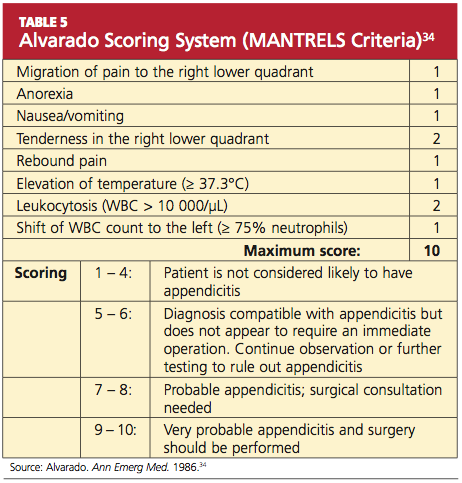
Conditions to Rule Out
The patient with abdominal pain and suspected appendicitis should be evaluated for other causes during the physical examination (see Table 62,8,39-42). In addition to investigation for other abdominal etiologies, auscultation to the heart and lungs and an assessment of the peripheral vasculature are imperative. Auscultation of the lungs is important to rule out a right lower lobe pneumonia that may generate referred pain to the RLQ due to a shared T9 dermatome distribution.20,25
In males, the patient with abdominal pain should be assessed for a testicular etiology, and a pelvic examination is indicated in any female with abdominal pain, to rule out a gynecologic origin.1,3 In the infant with suspected appendicitis, a diagnosis of Hirschsprung’s disease (a congenital obstruction of the colon) should also be considered.17
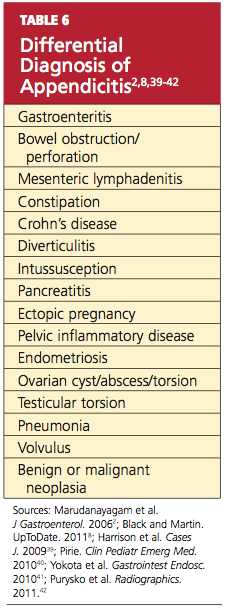
LABORATORY WORK-UP
Based on the patient’s history and physical exam findings, certain laboratory and imaging studies can be useful in confirming the diagnosis of appendicitis. A white blood cell (WBC) count with differential is helpful in both diagnosis and exclusion of appendicitis: Appendicitis often leads to moderate leukocytosis (WBC, 10,000 to 20,000/μL) with neutrophilia.13 Similarly, the finding of a normal or low WBC and absent left shift helps to rule out appendicitis.43
A C-reactive protein (CRP) value greater than 3.0 mg/dL, when combined with moderate leukocytosis, may increase the likelihood of appendicitis and rule out other conditions (eg, gastroenteritis, mesenteric adenitis, pelvic inflammatory disease).15,44 Additionally, an elevated CRP may be sensitive (83% to > 90%) for detecting appendiceal perforation and abscess formation.44 The role of cytokine levels, such as interleukin-6 (IL-6) and IL-10, may be helpful but remain under investigation and are not typically used in the diagnosis of appendicitis.45
Because negative findings in the β-hCG rule out intrauterine or ectopic pregnancy, this test should be ordered for all women capable of pregnancy who present with acute abdominal pain.20 Urinalysis may be indicated to exclude abdominal pain of urinary tract etiology.3,46
Imaging Studies
Not all patients with a presumptive diagnosis of appendicitis require imaging. Such studies can be foregone in patients with low clinical suspicion for appendicitis, although they should be instructed to return if the pain worsens, changes, or does not resolve. Likewise, patients with a high clinical suspicion for appendicitis may be referred to a surgeon as early as possible (without imaging).13
In children, however, the classic clinical and laboratory findings are often less reliable in diagnosing appendicitis. Positive results on CT or ultrasound—that is, inflammation and distention of the appendix or free fluid in the abdomen—are associated with confirmed appendicitis more than 90% of the time.15
CT and ultrasound are currently considered the imaging studies of choice.13 Of the two, multidetector CT is more accurate for detecting inflammation of the appendix (sensitivity, 98.5%; specificity, 98%; 99.5% negative predictive value),47 especially in the obese patient.48 While CT use has increased, the overall negative appendectomy rate was similar in some clinical trials with or without CT use.49,50 Additionally, the cost, availability, length of test, and radiation exposure associated with CT have raised concern about this imaging choice.
Ultrasound is useful to confirm appendicitis, particularly in patients with limited abdominal fat, but it has limitations in ruling out the condition.51 These include its operator-dependent nature, limited ability to allow visualization of the appendix in obese patients, and lack of sensitivity in cases in which the appendix is perforated or only the distal tip is involved.7
Plain radiographs are not used to diagnose appendicitis, although they may be helpful to evaluate patients with atypical symptoms1 or to rule out other causes of abdominal pain. For example, a chest x-ray may be used to rule out pneumonia or to look for free air under the diaphragm, suggesting a different etiology.20,25
Imaging studies can be helpful when differentiating between complicated versus uncomplicated appendicitis and ruling out other causes of the acute abdomen (eg, gastroenteritis, diverticulitis, pelvic inflammatory disease). Alternatively, watchful observation is essential until the diagnosis becomes clearer or exploratory laparoscopic surgeries have been used to evaluate the acute abdomen.52
MANAGEMENT OF APPENDICITIS
Appendectomy remains the standard of care for appendicitis.13,53,54 While the clinical presentation often dictates what surgical approach should be taken, up to 76% of appendectomies are performed using a laparoscopic procedure rather than open surgery.55
Patients with uncomplicated appendicitis should be given nothing by mouth, but adequate hydration should be provided with IV fluids. IV analgesia should be considered if pain is causing distress to the patient. Current evidence suggests that administration of opioids does not alter the clinician’s diagnostic accuracy.56
The treatment of a patient with complicated appendicitis who is hemodynamically stable is less clear. The conventional treatment is antibiotics and drainage, followed by appendectomy at a later date6; this procedure is referred to as interval appendectomy. Some authorities suggest that in cases of appendicitis resolved with antibiotics, interval appendectomy should no longer be recommended.57,58 In 2011, Blakely et al59 reported that in children with perforated appendicitis, early surgery results in reduced recovery time and fewer adverse events, compared with delayed appendectomy.
Preoperative antibiotics have demonstrated efficacy in decreasing postoperative wound infections; the timing of antibiotic administration is critical to its efficacy.60,61 The first dose should be given within 60 minutes before the incision is made to achieve adequate antibiotic serum and tissue levels. The antibiotic should be discontinued 24 hours after the surgery has been completed.60,62
The agent selected for antibiotic prophylaxis should be effective against the most likely infecting organism.17,61,62 In a patient with uncomplicated appendicitis, the antibiotic of choice should be effective against gram-negative bacilli, such as Escherichia coli and Bacteroides fragilis.46,61,62 A single dose of cefoxitin, cefotetan, cefotaxime, or ampicillin/sulbactam is typically prescribed to prevent postsurgical site infections in patients with uncomplicated appendicitis (Table 762-65). For β-lactam–allergic patients, an alternative antibiotic regimen is metronidazole with an aminoglycoside.61,62
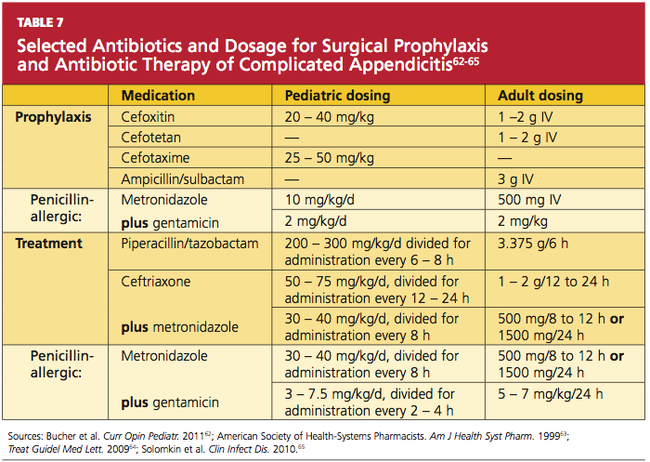
In Lieu of Surgery
As an alternative to surgery, several randomized studies have suggested that antibiotics alone can be used to treat uncomplicated appendicitis.66-68 Recent evidence suggests that a nonsurgical antibiotic approach may result in significant cost savings,69 attributable to eliminating surgery and a reduced risk for complications. Of additional benefit is eliminating surgery-associated morbidity and mortality.
Because design limitations lessen the reliability of the studies cited, however, appendectomy is still preferred, based on the current evidence.53,54 More studies are needed to determine the efficacy of antibiotic therapy alone, with consideration of the surgical risks associated with appendectomy.
POSTOPERATIVE CARE
Adequate pain control, advancement of diet, and monitoring for development of complications constitute typical postoperative care. Complications of appendectomy include both short- and long-term risks (eg, infection, adhesions, obstruction) associated with any surgical intervention.
CONCLUSION
Primary care providers should be well versed in identifying the symptoms and signs of appendicitis. In cases with equivocal findings, imaging studies and/or laboratory tests should be ordered to help confirm the diagnosis. The standard of care is appendectomy; therefore, a surgical consult is needed. Recent evidence suggests that a nonsurgical, antibiotic approach in the treatment of uncomplicated appendicitis may be beneficial. However, large, randomized trials with children enrolled, clear inclusion criteria, and outcome reporting with an intention-to-treat basis will help validate this approach as an alternative to current practice.
1. Birnbaum BA, Wilson SR. Appendicitis at the millennium. Radiology. 2000;215(2):337-348.
2. Marudanayagam R, Williams GT, Rees BI. Review of the pathological results of 2660 appendicectomy specimens. J Gastroenterol. 2006;41(8):745-749.
3. Bundy DG, Byerley JS, Liles EA, et al. Does this child have appendicitis? JAMA. 2007;298(4):438-451.
4. Alder AC, Fomby TB, Woodward WA, et al. Association of viral infection and appendicitis. Arch Surg. 2010;145(1):63-71.
5. Rubin R. The gastrointestinal tract. In: Rubin R, Strayer DS, eds. Rubin’s Pathology: Clinicopathologic Foundations of Medicine. 6th ed. Lippincott, Williams and Wilkins; 2012: 671.
6. McQuaid KR. Gastrointestinal disorders. In: McPhee SJ, Papadakis MA, eds. 2011 Current Medical Diagnosis and Treatment. 50th ed. McGraw Hill; 2011: 606-608.
7. Brennan GD. Pediatric appendicitis: pathophysiology and appropriate use of diagnostic imaging. CJEM. 2006;8(6):425-432.
8. Black C, Martin R. Acute appendicitis in adults: clinical manifestations and diagnosis (2011). www .uptodate.com/contents/acute-appendicitis-in-adults-clinical-manifestations-and-diagnosis. Accessed December 14, 2011.
9. Pittman-Waller VA, Myers JG, Stewart RM, et al. Appendicitis: why so complicated? Analysis of 5755 consecutive appendectomies. Am Surg. 2000; 6(66):548-554.
10. Mazuski JE, Solomkin JS. Intra-abdominal infections. Surg Clin North Am. 2009;89(2):421-437.
11. Andersson RE. The natural history and traditional management of appendicitis revisited: spontaneous resolution and predominance of prehospital perforations imply that a correct diagnosis is more important than an early diagnosis. World J Surg. 2007;31(1):86-92.
12. Livingston EH, Woodward WA, Sarosi GA, Haley RW. Disconnect between incidence of nonperforated and perforated appendicitis: implications for pathophysiology and management. Ann Surg. 2007;245(6):886-892.
13. Howell JM, Eddy OL, Lukens TW, et al; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of emergency department patients with suspected appendicitis. Ann Emerg Med. 2010; 55(1):71-116.
14. CDC. Number, rate, and standard error of all-listed surgical and nonsurgical procedures for discharges from short-stay hospitals, by selected procedure categories: United States, 2007. www.cdc
.gov/nchs/data/nhds/4procedures/2009 pro4_num berrate.pdf. Accessed December 14, 2011.
15. Lee SL, Ho HS. Acute appendicitis: is there a difference between children and adults? Am Surg. 2006;5(72):409-413.
16. Bickell NA, Aufses AH Jr, Rojas M, Bodian C. How time affects the risk of rupture in appendicitis. J Am Coll Surg. 2006;202(3):401-406.
17. Morrow SE, Newman KD. Current management of appendicitis. Semin Pediatr Surg. 2007; 16(1):34-40.
18. Nance ML, Adamson WT, Hedrick HL. Appendicitis in the young child: a continuing diagnostic challenge. Pediatr Emerg Care. 2000; 16(3):160-162.
19. Feinberg AN, Feinberg LA. The gastrointestinal tract, liver, gallbladder, and pancreas. In: Greydanus D, Feinberg A, Patel D, Homnick D, eds. The Pediatic Diagnostic Examination. McGraw-Hill Professional; 2008:267.
20. Humes DJ, Simpson J. Acute appendicitis. BMJ. 2006;333(7567):530-534.
21. Yale SH, Musana KA. Charles Heber McBurney (1845-1913). Clin Med Res. 2005;3(3):187-189.
22. Murphy JB. Two thousand operations for appendicitis with deductions from his personal experience. Am J Med Sci. 1904;128:187-211.
23. Andersson RE. Meta-analysis of the clinical and laboratory diagnosis of appendicitis. Br J Surg. 2004;91(1):28-37.
24. Wani I. K-sign in retrocaecal appendicitis: a case series. Cases J. 2009;2:157.
25. Rothrock SG, Pagane J. Acute appendicitis in children: emergency department diagnosis and management. Ann Emerg Med. 2000;36(1):39-51.
26. Wesson DE. Acute appendicitis in children: clinical manifestations and diagnosis (2010). www .uptodate.com/contents/acute-appendicitis-in-chil dren-clinical-manifestations-and-diagnosis? source=related_link. Accessed December 14, 2011.
27. Becker T, Kharbanda A, Bachur R. Atypical clinical features of pediatric appendicitis. Acad Emerg Med. 2007;14(2):124-129.
28. Laméris W, van Randen A, Go P, et al. Single and combined diagnostic value of clinical features and laboratory tests in acute appendicitis. Acad Emerg Med. 2009;16(9):835-842.
29. McCollough M, Sharieff G. Abdominal pain in children. Pediatr Clin N Am. 2006;53:107-137.
30. Sheu BF, Chiu TF, Chen JC, et al. Risk factors associated with perforated appendicitis in elderly patients presenting with signs and symptoms of acute appendicitis. ANZ J Surg. 2007;77(8):662-666.
31. Borst AR. Acute appendicitis: pregnancy complicates this diagnosis. JAAPA. 2007; 20(12):36-38.
32. Sedlak M, Wagner OJ, Wild B, et al. Is there still a role for rectal examination in suspected appendicitis in adults? Am J Emerg Med. 2008;26 (3):359-377.
33. Ylitalo AW. Digital rectal examination. http://emedicine.medscape.com/article/1948001-overview#a1. Accessed December 14, 2011.
34. Alvarado A. A practical score for the early diagnosis of acute appendicitis. Ann Emerg Med. 1986;15(5):557-564.
35. Schneider C, Kharbanda A, Bachur R. Evaluating appendicitis scoring systems using a prospective pediatric cohort. Ann Emerg Med. 2007; 49(6):778-784.
36. Kharbanda AB, Taylor GA, Fishman SJ, Bachur RG. A clinical decision rule to identify children at low risk for appendicitis. Pediatrics. 2005; 116(3):709–16.
37. Brown JJ, Wilson C, Coleman S, Joypaul BV. Appendicitis in pregnancy. Colorectal Dis. 2009; 11(2):116-122.
38. Richardson E, Paulson C, Hitchcock K. Clinical inquiries. History, exam, and labs: is one enough to diagnose acute adult appendicitis? J Fam Practice. 2007;56(6):474-476.
39. Harrison S, Mahawar K, Brown D, et al. Acute appendicitis presenting as small bowel obstruction: two case reports. Cases J. 2009;2:9106.
40. Pirie J. Management of constipation in the emergency department. Clin Pediatr Emerg Med. 2010;11(3):182-188.
41. Yokota S, Togashi K, Kasahara N, et al. Crohn’s disease confined to the appendix. Gastrointest Endosc. 2010;72(5):1063-1064.
42. Purysko AS, Remer EM, Filho HM, et al. Beyond appendicitis: common and uncommon gastrointestinal causes of right lower quadrant abdominal pain at multidetector CT. Radiographics. 2011;31(4):927-947.
43. Wang LT, Prentiss KA, Simon JZ, et al. The use of white blood cell count and left shift in the diagnosis of appendicitis in children. Pediatr Emerg Care. 2007;23(2):69-76.
44. Kwan KY, Nager AL. Diagnosing pediatric appendicitis: usefulness of laboratory markers. Am J Emerg Med. 2010;28(9):1009-1015.
45. Yildirim O, Solak C, Koçer B, et al. The role of serum inflammatory markers in acute appendicitis and their success in preventing negative laparotomy. J Invest Surg. 2006;19(6):345-352.
46. Spirt MJ. Complicated intra-abdominal infections: a focus on appendicitis and diverticulitis. Postgrad Med. 2010;122(1):39-51.
47. Pickhardt PJ, Lawrence EM, Pooler D, Bruce RJ. Diagnostic performance of multidetector computed tomography for suspected acute appendicitis. Ann Intern Med. 2011:154(12):789-796.
48. Coursey CA, Nelson RC, Moreno RD, et al. Appendicitis, body mass index, and CT: is CT more valuable for obese patients than thin patients? Am Surg. 2011;77(4):471-475.
49. Petrosyan M, Estrada J, Chan S, et al. CT scan in patients with suspected appendicitis: clinical implications for the acute care surgeon. Eur Surg Res. 2008;40(2):211-219.
50. Huynh V, Lalezarzadeh F, Lawandy S, et al. Abdominal computed tomography in the evaluation of acute and perforated appendicitis in the community setting. Am Surg. 2007;73(10):1002-1005.
51. Fox JC, Solley M, Anderson CL, et al. Prospective evaluation of emergency physician performed bedside ultrasound to detect acute appendicitis. Eur J Emerg Med. 2008;15(2):80-85.
52. Al-Mulhim AS, Nasser MA, Abdullah MM, et al. Emergency laparoscopy for acute abdominal conditions: a prospective study. J Laparoendosc Adv Surg Tech A. 2008;18(4):599-602.
53. Varadhan KK, Humes DJ, Neal KR, Lobo DN. Antibiotic therapy versus appendectomy for acute appendicitis: a meta-analysis. World J Surg. 2010;34(2):199-209.
54. Fitzmaurice GJ, McWilliams B, Hurreiz H, Epanomeritakis E. Antibiotics versus appendectomy in the management of acute appendicitis: a review of the current evidence. Can J Surg. 2011; 54(5):307-314.
55. Ingraham AM, Cohen ME, Bilimoria KY, et al. Comparison of outcomes after laparoscopic versus open appendectomy for acute appendicitis at 222 ACS NSQIP hospitals. Surgery. 2010;148(4): 625-635.
56. Manterola C, Vial M, Moraga J, Astudillo P. Analgesia in patients with acute abdominal pain. Cochrane Database Syst Rev. 2011;(1):CD005660.
57. Puapong D, Lee SL, Haigh PI, et al. Routine interval appendectomy in children is not indicated. J Pediatr Surg. 2007;42(9):1500-1503.
58. Deakin DE, Ahmed I. Interval appendectomy after resolution of adult inflammatory appendix mass: is it necessary? Surgeon. 2007;5(1):45-50.
59. Blakely ML, Williams R, Dassinger MS, et al. Early vs interval appendectomy for children with perforated appendicitis. Arch Surg. 2011;146 (6):660-665.
60. Salkind AR, Rao KC. Antibiotic prophylaxis to prevent surgical site infections. Am Fam Physician. 2011;83(5):585-590.
61. James M. Antibiotics and perioperative infections. Best Pract Res Clin Anaesthesiol. 2008; 22(3):571-584.
62. Bucher BT, Warner BW, Dillon PA. Antibiotic prophylaxis and the prevention of surgical site infection. Curr Opin Pediatr. 2011;23(3):334-338.
63. American Society of Health-Systems Pharmacists. ASHP therapeutic guidelines on antimicrobial prophylaxis in surgery. Am J Health Syst Pharm. 1999;56(18):1839-1888.
64. Antimicrobial prophylaxis for surgery. Treat Guidel Med Lett. 2009;7(82):47-52.
65. Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133-164.
66. Vons C, Barry C, Maitre S, et al. Amoxicillin plus clavulanic acid versus appendicectomy for treatment of acute uncomplicated appendicitis: an open-label, non-inferiority, randomized controlled trial. Lancet. 2011;377(9777):1573-1579.
67. Hansson J, Körner U, Khorram-Manesh A, et al. Randomized clinical trial of antibiotic therapy versus appendicectomy as primary treatment of acute appendicitis in unselected patients. Br J Surg. 2009;96(5):473-481.
68. Styrud J, Eriksson S, Nilsson I, et al. Appendectomy versus antibiotic treatment in acute appendicitis: a prospective multicenter randomized control trial. World J Surg. 2006;30(6):1033-1037.
69. Sakorafas GH, Mastoraki A, Lappas C, et al. Conservative treatment of acute appendicitis: heresy or an effective and acceptable alternative to surgery? Eur J Gastroenterol Hepatol. 2011;23 (2):121-127.
Appendicitis is a transmural inflammatory process and a common cause of an acute abdomen. Inflammation that leads to perforation of the appendix, which is associated with increased morbidity and mortality, warrants prompt diagnosis. Etiology, clinical presentation, diagnostic studies, and the management of confirmed appendicitis will be addressed here.
Frequently, the etiology of appendicitis is luminal obstruction by a fecalith (the result of inspissated fecal material and inorganic salts1), but the condition may also result from parasites, a malignancy, a foreign body, or fibrosis.1-3 In some instances, lymphoid hyperplasia, resulting from a viral or bacterial infection, has been targeted as the cause of luminal obstruction.1,4 Nevertheless, in one-third to one-half of patients, obstruction is not evident as a precipitating factor in the development of appendicitis. In such cases, the basis for the inflammation is unknown.5
As the obstructed appendix becomes congested, the intraluminal pressure and venous pressure increase, leading to stasis and ischemia.1,5-8 The appendix becomes engorged with secretions. At this stage, the condition is considered uncomplicated, but if an inflamed appendix becomes gangrenous or perforates, the condition is then referred to as complicated appendicitis. Complicated appendicitis allows for invasion by intestinal bacteria of the abdominal cavity, potentially leading to peritonitis, septicemia, abscess, or fistula formation.5,9
Conventional teaching supports the concept that uncomplicated appendicitis, unless treated surgically, eventually evolves into complicated appendicitis.10 Recent research refutes this assumption, however, as different etiologies may be associated with differences in progression10-12; whether uncomplicated and complicated appendicitis are attributable to different etiologies is a question requiring further research. Irrespective of the natural progression of the disease, the current standard of care for appendicitis is still an appendectomy.13 In US hospitals in 2007 (the most recent year for which data are available), appendectomy was performed on 326,000 patients, or 10.9 patients per 10,000 population.14
EPIDEMIOLOGY
Appendicitis is most frequently seen in the second decade of life and occurs slightly more often in males than in females.2,15 Furthermore, according to data reported to the National Hospital Discharge Survey (1970 to 2004), the rate of nonperforated appendicitis is much higher in men than in women.12 In appendicitis, the risk for rupture is small within the first 36 hours of symptom onset. Beyond that point, there is a 5% increased risk for rupture with each ensuing 12-hour period.16
In neonates and infants, appendicitis is rare.3 In children younger than 3 years, however, the rate of perforation is 80% to 100%.3,17,18 This high rate may be explained by the very young child’s limited ability to articulate his or her symptoms, or by caregiver reports that are typically limited to irritability or change in diet.3,17,19 According to Marudanayagam et al,2 who performed a retrospective study of 2,660 appendectomies during a six-year period, the perforation rate declined from 23.4% in patients age 10 or younger to 6.9% in those in their 20s, then rose steadily to more than 50% in patients 70 or older.
PATIENT EVALUATION
In most cases, a diagnosis of appendicitis can be made with a careful history, systematic physical exam, and a limited number of laboratory tests without special diagnostic modalities.13 The presence of symptoms and signs may help to rule in a diagnosis of appendicitis, but the absence of clinical findings often does not exclude its possibility.16 While adult and pediatric patients with appendicitis share many clinical findings (see Table 13,8,13,18), the occurrence rate of the various findings may differ among patient populations.3,15
The median time from onset of symptoms until the patient presents for a medical evaluation averages 24 hours or less.16 Diagnosis in patients at extremes of age often proves more difficult than in other patients.20 Thus, a high level of suspicion must be maintained in these patient populations.
The Symptom History
The appendix is located in the posteromedial wall of the cecum, approximately 3 cm below the ileocecal valve.1 Initial pain perceived around the umbilicus represents a referred pain resulting from the visceral innervation of the midgut.20 As the inflammatory process within the appendix advances, the pain localizes to the anatomical position of the right lower quadrant (RLQ), with involvement of the surrounding parietal peritoneum.20(McBurney’s point, at the junction of the lateral and middle thirds of a line extending from the anterior superior iliac spine to the umbilicus, was noted as the point of maximal tenderness to palpation in acute appendicitis by Charles McBurney in the late 1800s.21)
This progression of symptoms, first recognized by John Benjamin Murphy in 1904, is considered a more reliable indicator of appendicitis than RLQ pain alone3,22; in one large retrospective study, this migratory pain had the highest positive predictive value for pediatric and adult patients (94.2% and 89.6%, respectively).15 However, migration of pain occurs in only 50% to 60% of patients, and therefore may not be helpful.1,23
According to results from other studies, unfortunately, this progression of symptoms is not often present in pediatric patients.17 The somatic RLQ pain is continuous and more severe than is the early visceral periumbilical pain.1 Since the anatomic position of the appendix can vary, a number of patients do not necessarily present with pain in the RLQ but elsewhere.24
Certain clinical findings appear to be relatively age-dependent (see Table 225,26).
Classic findings in the adult diagnosed with appendicitis, as described by Becker et al,27 begin with periumbilical pain, then nausea, followed by migration of the pain to the RLQ, then vomiting and fever. Abdominal pain and anorexia are the most common presenting symptoms.20 Nausea and vomiting that begin after the onset of abdominal pain are typical; in isolation, however, these manifestations have weak diagnostic predictability for appendicitis.28 In adults, if nausea and vomiting precede abdominal pain, consideration should be given to a diagnosis of gastroenteritis rather than appendicitis.29
Among patients who are pregnant or elderly, RLQ pain remains a significant historical finding.30 In the pregnant woman, a diagnosis of appendicitis is often overlooked because of the discomforts common to pregnancy and the expanding gravid uterus.31 Elderly patients often present with vague or atypical symptoms, such as mild pain.20 In these patient populations, the diagnosis of appendicitis is often delayed.
In addition to obtaining a thorough history of the presentation of pain, it is important to conduct a complete review of the gastrointestinal, genitourinary, pulmonary, musculoskeletal, neurologic, and reproductive systems for possible alternate etiologies.
Physical Examination
The number of physical findings varies among patients who present with appendicitis3,8,20 (see Table 33,8,20). A thorough physical examination is thus required to help the clinician exclude other diseases and establish the diagnosis of appendicitis. It is important to tailor the exam according to the patient’s age and developmental stage.19
The cooperation of children undergoing the physical examination for appendicitis may vary. It may be helpful to instruct a young child to “show me with your finger where it hurts the most.”3 However, Bundy et al3 report that the presence of RLQ tenderness on palpation is of minimal value in children; rather, fever is the single most useful sign among pediatric patients and conversely, its absence reduces the risk.
Tachycardia is associated with risk for rupture.16,20 In the elderly patient, fever (> 38°C) is also strongly correlated with an increased risk for rupture.30
To alleviate pain, a patient with appendicitis may maintain the hips and knees in a slightly flexed position. While asking distracting questions, the examiner should observe the patient’s facial expressions to detect involuntary guarding.3 RLQ tenderness to percussion is often positive.
The patient may experience tenderness on palpation of the posterior abdominal wall (K sign) or right-side flank tenderness.24 Increased pain with coughing (Dunphy’s sign) or firm percussion of the heel (the heel jar test) may be elicited.8,25 A number of additional peritoneal signs, resulting from an inflamed appendix, may occur (see Table 43,8,20), but examination techniques that elicit these signs should be minimized so as to not cause the patient any unnecessary pain.
Depending on the location of the appendix, rectal and vaginal exams may yield normal findings or may elicit tenderness.32 The rectal examination should be performed with considerable care, using the smallest digit possible for an adequate assessment, especially in the younger patient.33
Several scoring systems have been designed for adults and children with suspected appendicitis, using findings from the history, the physical exam, and laboratory testing (see Table 5,34 for example). Despite their protocol-based approach, the scoring systems have yielded mixed results in clinical practice,34-36 and there is no scoring system for evaluation of the pregnant patient.37 Neither has there been any recommendation for or endorsement of a diagnostic guideline from any medical or professional organization.38 Thus, clinical gestalt is usually relied upon instead.
Conditions to Rule Out
The patient with abdominal pain and suspected appendicitis should be evaluated for other causes during the physical examination (see Table 62,8,39-42). In addition to investigation for other abdominal etiologies, auscultation to the heart and lungs and an assessment of the peripheral vasculature are imperative. Auscultation of the lungs is important to rule out a right lower lobe pneumonia that may generate referred pain to the RLQ due to a shared T9 dermatome distribution.20,25
In males, the patient with abdominal pain should be assessed for a testicular etiology, and a pelvic examination is indicated in any female with abdominal pain, to rule out a gynecologic origin.1,3 In the infant with suspected appendicitis, a diagnosis of Hirschsprung’s disease (a congenital obstruction of the colon) should also be considered.17
LABORATORY WORK-UP
Based on the patient’s history and physical exam findings, certain laboratory and imaging studies can be useful in confirming the diagnosis of appendicitis. A white blood cell (WBC) count with differential is helpful in both diagnosis and exclusion of appendicitis: Appendicitis often leads to moderate leukocytosis (WBC, 10,000 to 20,000/μL) with neutrophilia.13 Similarly, the finding of a normal or low WBC and absent left shift helps to rule out appendicitis.43
A C-reactive protein (CRP) value greater than 3.0 mg/dL, when combined with moderate leukocytosis, may increase the likelihood of appendicitis and rule out other conditions (eg, gastroenteritis, mesenteric adenitis, pelvic inflammatory disease).15,44 Additionally, an elevated CRP may be sensitive (83% to > 90%) for detecting appendiceal perforation and abscess formation.44 The role of cytokine levels, such as interleukin-6 (IL-6) and IL-10, may be helpful but remain under investigation and are not typically used in the diagnosis of appendicitis.45
Because negative findings in the β-hCG rule out intrauterine or ectopic pregnancy, this test should be ordered for all women capable of pregnancy who present with acute abdominal pain.20 Urinalysis may be indicated to exclude abdominal pain of urinary tract etiology.3,46
Imaging Studies
Not all patients with a presumptive diagnosis of appendicitis require imaging. Such studies can be foregone in patients with low clinical suspicion for appendicitis, although they should be instructed to return if the pain worsens, changes, or does not resolve. Likewise, patients with a high clinical suspicion for appendicitis may be referred to a surgeon as early as possible (without imaging).13
In children, however, the classic clinical and laboratory findings are often less reliable in diagnosing appendicitis. Positive results on CT or ultrasound—that is, inflammation and distention of the appendix or free fluid in the abdomen—are associated with confirmed appendicitis more than 90% of the time.15
CT and ultrasound are currently considered the imaging studies of choice.13 Of the two, multidetector CT is more accurate for detecting inflammation of the appendix (sensitivity, 98.5%; specificity, 98%; 99.5% negative predictive value),47 especially in the obese patient.48 While CT use has increased, the overall negative appendectomy rate was similar in some clinical trials with or without CT use.49,50 Additionally, the cost, availability, length of test, and radiation exposure associated with CT have raised concern about this imaging choice.
Ultrasound is useful to confirm appendicitis, particularly in patients with limited abdominal fat, but it has limitations in ruling out the condition.51 These include its operator-dependent nature, limited ability to allow visualization of the appendix in obese patients, and lack of sensitivity in cases in which the appendix is perforated or only the distal tip is involved.7
Plain radiographs are not used to diagnose appendicitis, although they may be helpful to evaluate patients with atypical symptoms1 or to rule out other causes of abdominal pain. For example, a chest x-ray may be used to rule out pneumonia or to look for free air under the diaphragm, suggesting a different etiology.20,25
Imaging studies can be helpful when differentiating between complicated versus uncomplicated appendicitis and ruling out other causes of the acute abdomen (eg, gastroenteritis, diverticulitis, pelvic inflammatory disease). Alternatively, watchful observation is essential until the diagnosis becomes clearer or exploratory laparoscopic surgeries have been used to evaluate the acute abdomen.52
MANAGEMENT OF APPENDICITIS
Appendectomy remains the standard of care for appendicitis.13,53,54 While the clinical presentation often dictates what surgical approach should be taken, up to 76% of appendectomies are performed using a laparoscopic procedure rather than open surgery.55
Patients with uncomplicated appendicitis should be given nothing by mouth, but adequate hydration should be provided with IV fluids. IV analgesia should be considered if pain is causing distress to the patient. Current evidence suggests that administration of opioids does not alter the clinician’s diagnostic accuracy.56
The treatment of a patient with complicated appendicitis who is hemodynamically stable is less clear. The conventional treatment is antibiotics and drainage, followed by appendectomy at a later date6; this procedure is referred to as interval appendectomy. Some authorities suggest that in cases of appendicitis resolved with antibiotics, interval appendectomy should no longer be recommended.57,58 In 2011, Blakely et al59 reported that in children with perforated appendicitis, early surgery results in reduced recovery time and fewer adverse events, compared with delayed appendectomy.
Preoperative antibiotics have demonstrated efficacy in decreasing postoperative wound infections; the timing of antibiotic administration is critical to its efficacy.60,61 The first dose should be given within 60 minutes before the incision is made to achieve adequate antibiotic serum and tissue levels. The antibiotic should be discontinued 24 hours after the surgery has been completed.60,62
The agent selected for antibiotic prophylaxis should be effective against the most likely infecting organism.17,61,62 In a patient with uncomplicated appendicitis, the antibiotic of choice should be effective against gram-negative bacilli, such as Escherichia coli and Bacteroides fragilis.46,61,62 A single dose of cefoxitin, cefotetan, cefotaxime, or ampicillin/sulbactam is typically prescribed to prevent postsurgical site infections in patients with uncomplicated appendicitis (Table 762-65). For β-lactam–allergic patients, an alternative antibiotic regimen is metronidazole with an aminoglycoside.61,62
In Lieu of Surgery
As an alternative to surgery, several randomized studies have suggested that antibiotics alone can be used to treat uncomplicated appendicitis.66-68 Recent evidence suggests that a nonsurgical antibiotic approach may result in significant cost savings,69 attributable to eliminating surgery and a reduced risk for complications. Of additional benefit is eliminating surgery-associated morbidity and mortality.
Because design limitations lessen the reliability of the studies cited, however, appendectomy is still preferred, based on the current evidence.53,54 More studies are needed to determine the efficacy of antibiotic therapy alone, with consideration of the surgical risks associated with appendectomy.
POSTOPERATIVE CARE
Adequate pain control, advancement of diet, and monitoring for development of complications constitute typical postoperative care. Complications of appendectomy include both short- and long-term risks (eg, infection, adhesions, obstruction) associated with any surgical intervention.
CONCLUSION
Primary care providers should be well versed in identifying the symptoms and signs of appendicitis. In cases with equivocal findings, imaging studies and/or laboratory tests should be ordered to help confirm the diagnosis. The standard of care is appendectomy; therefore, a surgical consult is needed. Recent evidence suggests that a nonsurgical, antibiotic approach in the treatment of uncomplicated appendicitis may be beneficial. However, large, randomized trials with children enrolled, clear inclusion criteria, and outcome reporting with an intention-to-treat basis will help validate this approach as an alternative to current practice.
Appendicitis is a transmural inflammatory process and a common cause of an acute abdomen. Inflammation that leads to perforation of the appendix, which is associated with increased morbidity and mortality, warrants prompt diagnosis. Etiology, clinical presentation, diagnostic studies, and the management of confirmed appendicitis will be addressed here.
Frequently, the etiology of appendicitis is luminal obstruction by a fecalith (the result of inspissated fecal material and inorganic salts1), but the condition may also result from parasites, a malignancy, a foreign body, or fibrosis.1-3 In some instances, lymphoid hyperplasia, resulting from a viral or bacterial infection, has been targeted as the cause of luminal obstruction.1,4 Nevertheless, in one-third to one-half of patients, obstruction is not evident as a precipitating factor in the development of appendicitis. In such cases, the basis for the inflammation is unknown.5
As the obstructed appendix becomes congested, the intraluminal pressure and venous pressure increase, leading to stasis and ischemia.1,5-8 The appendix becomes engorged with secretions. At this stage, the condition is considered uncomplicated, but if an inflamed appendix becomes gangrenous or perforates, the condition is then referred to as complicated appendicitis. Complicated appendicitis allows for invasion by intestinal bacteria of the abdominal cavity, potentially leading to peritonitis, septicemia, abscess, or fistula formation.5,9
Conventional teaching supports the concept that uncomplicated appendicitis, unless treated surgically, eventually evolves into complicated appendicitis.10 Recent research refutes this assumption, however, as different etiologies may be associated with differences in progression10-12; whether uncomplicated and complicated appendicitis are attributable to different etiologies is a question requiring further research. Irrespective of the natural progression of the disease, the current standard of care for appendicitis is still an appendectomy.13 In US hospitals in 2007 (the most recent year for which data are available), appendectomy was performed on 326,000 patients, or 10.9 patients per 10,000 population.14
EPIDEMIOLOGY
Appendicitis is most frequently seen in the second decade of life and occurs slightly more often in males than in females.2,15 Furthermore, according to data reported to the National Hospital Discharge Survey (1970 to 2004), the rate of nonperforated appendicitis is much higher in men than in women.12 In appendicitis, the risk for rupture is small within the first 36 hours of symptom onset. Beyond that point, there is a 5% increased risk for rupture with each ensuing 12-hour period.16
In neonates and infants, appendicitis is rare.3 In children younger than 3 years, however, the rate of perforation is 80% to 100%.3,17,18 This high rate may be explained by the very young child’s limited ability to articulate his or her symptoms, or by caregiver reports that are typically limited to irritability or change in diet.3,17,19 According to Marudanayagam et al,2 who performed a retrospective study of 2,660 appendectomies during a six-year period, the perforation rate declined from 23.4% in patients age 10 or younger to 6.9% in those in their 20s, then rose steadily to more than 50% in patients 70 or older.
PATIENT EVALUATION
In most cases, a diagnosis of appendicitis can be made with a careful history, systematic physical exam, and a limited number of laboratory tests without special diagnostic modalities.13 The presence of symptoms and signs may help to rule in a diagnosis of appendicitis, but the absence of clinical findings often does not exclude its possibility.16 While adult and pediatric patients with appendicitis share many clinical findings (see Table 13,8,13,18), the occurrence rate of the various findings may differ among patient populations.3,15
The median time from onset of symptoms until the patient presents for a medical evaluation averages 24 hours or less.16 Diagnosis in patients at extremes of age often proves more difficult than in other patients.20 Thus, a high level of suspicion must be maintained in these patient populations.
The Symptom History
The appendix is located in the posteromedial wall of the cecum, approximately 3 cm below the ileocecal valve.1 Initial pain perceived around the umbilicus represents a referred pain resulting from the visceral innervation of the midgut.20 As the inflammatory process within the appendix advances, the pain localizes to the anatomical position of the right lower quadrant (RLQ), with involvement of the surrounding parietal peritoneum.20(McBurney’s point, at the junction of the lateral and middle thirds of a line extending from the anterior superior iliac spine to the umbilicus, was noted as the point of maximal tenderness to palpation in acute appendicitis by Charles McBurney in the late 1800s.21)
This progression of symptoms, first recognized by John Benjamin Murphy in 1904, is considered a more reliable indicator of appendicitis than RLQ pain alone3,22; in one large retrospective study, this migratory pain had the highest positive predictive value for pediatric and adult patients (94.2% and 89.6%, respectively).15 However, migration of pain occurs in only 50% to 60% of patients, and therefore may not be helpful.1,23
According to results from other studies, unfortunately, this progression of symptoms is not often present in pediatric patients.17 The somatic RLQ pain is continuous and more severe than is the early visceral periumbilical pain.1 Since the anatomic position of the appendix can vary, a number of patients do not necessarily present with pain in the RLQ but elsewhere.24
Certain clinical findings appear to be relatively age-dependent (see Table 225,26).
Classic findings in the adult diagnosed with appendicitis, as described by Becker et al,27 begin with periumbilical pain, then nausea, followed by migration of the pain to the RLQ, then vomiting and fever. Abdominal pain and anorexia are the most common presenting symptoms.20 Nausea and vomiting that begin after the onset of abdominal pain are typical; in isolation, however, these manifestations have weak diagnostic predictability for appendicitis.28 In adults, if nausea and vomiting precede abdominal pain, consideration should be given to a diagnosis of gastroenteritis rather than appendicitis.29
Among patients who are pregnant or elderly, RLQ pain remains a significant historical finding.30 In the pregnant woman, a diagnosis of appendicitis is often overlooked because of the discomforts common to pregnancy and the expanding gravid uterus.31 Elderly patients often present with vague or atypical symptoms, such as mild pain.20 In these patient populations, the diagnosis of appendicitis is often delayed.
In addition to obtaining a thorough history of the presentation of pain, it is important to conduct a complete review of the gastrointestinal, genitourinary, pulmonary, musculoskeletal, neurologic, and reproductive systems for possible alternate etiologies.
Physical Examination
The number of physical findings varies among patients who present with appendicitis3,8,20 (see Table 33,8,20). A thorough physical examination is thus required to help the clinician exclude other diseases and establish the diagnosis of appendicitis. It is important to tailor the exam according to the patient’s age and developmental stage.19
The cooperation of children undergoing the physical examination for appendicitis may vary. It may be helpful to instruct a young child to “show me with your finger where it hurts the most.”3 However, Bundy et al3 report that the presence of RLQ tenderness on palpation is of minimal value in children; rather, fever is the single most useful sign among pediatric patients and conversely, its absence reduces the risk.
Tachycardia is associated with risk for rupture.16,20 In the elderly patient, fever (> 38°C) is also strongly correlated with an increased risk for rupture.30
To alleviate pain, a patient with appendicitis may maintain the hips and knees in a slightly flexed position. While asking distracting questions, the examiner should observe the patient’s facial expressions to detect involuntary guarding.3 RLQ tenderness to percussion is often positive.
The patient may experience tenderness on palpation of the posterior abdominal wall (K sign) or right-side flank tenderness.24 Increased pain with coughing (Dunphy’s sign) or firm percussion of the heel (the heel jar test) may be elicited.8,25 A number of additional peritoneal signs, resulting from an inflamed appendix, may occur (see Table 43,8,20), but examination techniques that elicit these signs should be minimized so as to not cause the patient any unnecessary pain.
Depending on the location of the appendix, rectal and vaginal exams may yield normal findings or may elicit tenderness.32 The rectal examination should be performed with considerable care, using the smallest digit possible for an adequate assessment, especially in the younger patient.33
Several scoring systems have been designed for adults and children with suspected appendicitis, using findings from the history, the physical exam, and laboratory testing (see Table 5,34 for example). Despite their protocol-based approach, the scoring systems have yielded mixed results in clinical practice,34-36 and there is no scoring system for evaluation of the pregnant patient.37 Neither has there been any recommendation for or endorsement of a diagnostic guideline from any medical or professional organization.38 Thus, clinical gestalt is usually relied upon instead.
Conditions to Rule Out
The patient with abdominal pain and suspected appendicitis should be evaluated for other causes during the physical examination (see Table 62,8,39-42). In addition to investigation for other abdominal etiologies, auscultation to the heart and lungs and an assessment of the peripheral vasculature are imperative. Auscultation of the lungs is important to rule out a right lower lobe pneumonia that may generate referred pain to the RLQ due to a shared T9 dermatome distribution.20,25
In males, the patient with abdominal pain should be assessed for a testicular etiology, and a pelvic examination is indicated in any female with abdominal pain, to rule out a gynecologic origin.1,3 In the infant with suspected appendicitis, a diagnosis of Hirschsprung’s disease (a congenital obstruction of the colon) should also be considered.17
LABORATORY WORK-UP
Based on the patient’s history and physical exam findings, certain laboratory and imaging studies can be useful in confirming the diagnosis of appendicitis. A white blood cell (WBC) count with differential is helpful in both diagnosis and exclusion of appendicitis: Appendicitis often leads to moderate leukocytosis (WBC, 10,000 to 20,000/μL) with neutrophilia.13 Similarly, the finding of a normal or low WBC and absent left shift helps to rule out appendicitis.43
A C-reactive protein (CRP) value greater than 3.0 mg/dL, when combined with moderate leukocytosis, may increase the likelihood of appendicitis and rule out other conditions (eg, gastroenteritis, mesenteric adenitis, pelvic inflammatory disease).15,44 Additionally, an elevated CRP may be sensitive (83% to > 90%) for detecting appendiceal perforation and abscess formation.44 The role of cytokine levels, such as interleukin-6 (IL-6) and IL-10, may be helpful but remain under investigation and are not typically used in the diagnosis of appendicitis.45
Because negative findings in the β-hCG rule out intrauterine or ectopic pregnancy, this test should be ordered for all women capable of pregnancy who present with acute abdominal pain.20 Urinalysis may be indicated to exclude abdominal pain of urinary tract etiology.3,46
Imaging Studies
Not all patients with a presumptive diagnosis of appendicitis require imaging. Such studies can be foregone in patients with low clinical suspicion for appendicitis, although they should be instructed to return if the pain worsens, changes, or does not resolve. Likewise, patients with a high clinical suspicion for appendicitis may be referred to a surgeon as early as possible (without imaging).13
In children, however, the classic clinical and laboratory findings are often less reliable in diagnosing appendicitis. Positive results on CT or ultrasound—that is, inflammation and distention of the appendix or free fluid in the abdomen—are associated with confirmed appendicitis more than 90% of the time.15
CT and ultrasound are currently considered the imaging studies of choice.13 Of the two, multidetector CT is more accurate for detecting inflammation of the appendix (sensitivity, 98.5%; specificity, 98%; 99.5% negative predictive value),47 especially in the obese patient.48 While CT use has increased, the overall negative appendectomy rate was similar in some clinical trials with or without CT use.49,50 Additionally, the cost, availability, length of test, and radiation exposure associated with CT have raised concern about this imaging choice.
Ultrasound is useful to confirm appendicitis, particularly in patients with limited abdominal fat, but it has limitations in ruling out the condition.51 These include its operator-dependent nature, limited ability to allow visualization of the appendix in obese patients, and lack of sensitivity in cases in which the appendix is perforated or only the distal tip is involved.7
Plain radiographs are not used to diagnose appendicitis, although they may be helpful to evaluate patients with atypical symptoms1 or to rule out other causes of abdominal pain. For example, a chest x-ray may be used to rule out pneumonia or to look for free air under the diaphragm, suggesting a different etiology.20,25
Imaging studies can be helpful when differentiating between complicated versus uncomplicated appendicitis and ruling out other causes of the acute abdomen (eg, gastroenteritis, diverticulitis, pelvic inflammatory disease). Alternatively, watchful observation is essential until the diagnosis becomes clearer or exploratory laparoscopic surgeries have been used to evaluate the acute abdomen.52
MANAGEMENT OF APPENDICITIS
Appendectomy remains the standard of care for appendicitis.13,53,54 While the clinical presentation often dictates what surgical approach should be taken, up to 76% of appendectomies are performed using a laparoscopic procedure rather than open surgery.55
Patients with uncomplicated appendicitis should be given nothing by mouth, but adequate hydration should be provided with IV fluids. IV analgesia should be considered if pain is causing distress to the patient. Current evidence suggests that administration of opioids does not alter the clinician’s diagnostic accuracy.56
The treatment of a patient with complicated appendicitis who is hemodynamically stable is less clear. The conventional treatment is antibiotics and drainage, followed by appendectomy at a later date6; this procedure is referred to as interval appendectomy. Some authorities suggest that in cases of appendicitis resolved with antibiotics, interval appendectomy should no longer be recommended.57,58 In 2011, Blakely et al59 reported that in children with perforated appendicitis, early surgery results in reduced recovery time and fewer adverse events, compared with delayed appendectomy.
Preoperative antibiotics have demonstrated efficacy in decreasing postoperative wound infections; the timing of antibiotic administration is critical to its efficacy.60,61 The first dose should be given within 60 minutes before the incision is made to achieve adequate antibiotic serum and tissue levels. The antibiotic should be discontinued 24 hours after the surgery has been completed.60,62
The agent selected for antibiotic prophylaxis should be effective against the most likely infecting organism.17,61,62 In a patient with uncomplicated appendicitis, the antibiotic of choice should be effective against gram-negative bacilli, such as Escherichia coli and Bacteroides fragilis.46,61,62 A single dose of cefoxitin, cefotetan, cefotaxime, or ampicillin/sulbactam is typically prescribed to prevent postsurgical site infections in patients with uncomplicated appendicitis (Table 762-65). For β-lactam–allergic patients, an alternative antibiotic regimen is metronidazole with an aminoglycoside.61,62
In Lieu of Surgery
As an alternative to surgery, several randomized studies have suggested that antibiotics alone can be used to treat uncomplicated appendicitis.66-68 Recent evidence suggests that a nonsurgical antibiotic approach may result in significant cost savings,69 attributable to eliminating surgery and a reduced risk for complications. Of additional benefit is eliminating surgery-associated morbidity and mortality.
Because design limitations lessen the reliability of the studies cited, however, appendectomy is still preferred, based on the current evidence.53,54 More studies are needed to determine the efficacy of antibiotic therapy alone, with consideration of the surgical risks associated with appendectomy.
POSTOPERATIVE CARE
Adequate pain control, advancement of diet, and monitoring for development of complications constitute typical postoperative care. Complications of appendectomy include both short- and long-term risks (eg, infection, adhesions, obstruction) associated with any surgical intervention.
CONCLUSION
Primary care providers should be well versed in identifying the symptoms and signs of appendicitis. In cases with equivocal findings, imaging studies and/or laboratory tests should be ordered to help confirm the diagnosis. The standard of care is appendectomy; therefore, a surgical consult is needed. Recent evidence suggests that a nonsurgical, antibiotic approach in the treatment of uncomplicated appendicitis may be beneficial. However, large, randomized trials with children enrolled, clear inclusion criteria, and outcome reporting with an intention-to-treat basis will help validate this approach as an alternative to current practice.
1. Birnbaum BA, Wilson SR. Appendicitis at the millennium. Radiology. 2000;215(2):337-348.
2. Marudanayagam R, Williams GT, Rees BI. Review of the pathological results of 2660 appendicectomy specimens. J Gastroenterol. 2006;41(8):745-749.
3. Bundy DG, Byerley JS, Liles EA, et al. Does this child have appendicitis? JAMA. 2007;298(4):438-451.
4. Alder AC, Fomby TB, Woodward WA, et al. Association of viral infection and appendicitis. Arch Surg. 2010;145(1):63-71.
5. Rubin R. The gastrointestinal tract. In: Rubin R, Strayer DS, eds. Rubin’s Pathology: Clinicopathologic Foundations of Medicine. 6th ed. Lippincott, Williams and Wilkins; 2012: 671.
6. McQuaid KR. Gastrointestinal disorders. In: McPhee SJ, Papadakis MA, eds. 2011 Current Medical Diagnosis and Treatment. 50th ed. McGraw Hill; 2011: 606-608.
7. Brennan GD. Pediatric appendicitis: pathophysiology and appropriate use of diagnostic imaging. CJEM. 2006;8(6):425-432.
8. Black C, Martin R. Acute appendicitis in adults: clinical manifestations and diagnosis (2011). www .uptodate.com/contents/acute-appendicitis-in-adults-clinical-manifestations-and-diagnosis. Accessed December 14, 2011.
9. Pittman-Waller VA, Myers JG, Stewart RM, et al. Appendicitis: why so complicated? Analysis of 5755 consecutive appendectomies. Am Surg. 2000; 6(66):548-554.
10. Mazuski JE, Solomkin JS. Intra-abdominal infections. Surg Clin North Am. 2009;89(2):421-437.
11. Andersson RE. The natural history and traditional management of appendicitis revisited: spontaneous resolution and predominance of prehospital perforations imply that a correct diagnosis is more important than an early diagnosis. World J Surg. 2007;31(1):86-92.
12. Livingston EH, Woodward WA, Sarosi GA, Haley RW. Disconnect between incidence of nonperforated and perforated appendicitis: implications for pathophysiology and management. Ann Surg. 2007;245(6):886-892.
13. Howell JM, Eddy OL, Lukens TW, et al; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of emergency department patients with suspected appendicitis. Ann Emerg Med. 2010; 55(1):71-116.
14. CDC. Number, rate, and standard error of all-listed surgical and nonsurgical procedures for discharges from short-stay hospitals, by selected procedure categories: United States, 2007. www.cdc
.gov/nchs/data/nhds/4procedures/2009 pro4_num berrate.pdf. Accessed December 14, 2011.
15. Lee SL, Ho HS. Acute appendicitis: is there a difference between children and adults? Am Surg. 2006;5(72):409-413.
16. Bickell NA, Aufses AH Jr, Rojas M, Bodian C. How time affects the risk of rupture in appendicitis. J Am Coll Surg. 2006;202(3):401-406.
17. Morrow SE, Newman KD. Current management of appendicitis. Semin Pediatr Surg. 2007; 16(1):34-40.
18. Nance ML, Adamson WT, Hedrick HL. Appendicitis in the young child: a continuing diagnostic challenge. Pediatr Emerg Care. 2000; 16(3):160-162.
19. Feinberg AN, Feinberg LA. The gastrointestinal tract, liver, gallbladder, and pancreas. In: Greydanus D, Feinberg A, Patel D, Homnick D, eds. The Pediatic Diagnostic Examination. McGraw-Hill Professional; 2008:267.
20. Humes DJ, Simpson J. Acute appendicitis. BMJ. 2006;333(7567):530-534.
21. Yale SH, Musana KA. Charles Heber McBurney (1845-1913). Clin Med Res. 2005;3(3):187-189.
22. Murphy JB. Two thousand operations for appendicitis with deductions from his personal experience. Am J Med Sci. 1904;128:187-211.
23. Andersson RE. Meta-analysis of the clinical and laboratory diagnosis of appendicitis. Br J Surg. 2004;91(1):28-37.
24. Wani I. K-sign in retrocaecal appendicitis: a case series. Cases J. 2009;2:157.
25. Rothrock SG, Pagane J. Acute appendicitis in children: emergency department diagnosis and management. Ann Emerg Med. 2000;36(1):39-51.
26. Wesson DE. Acute appendicitis in children: clinical manifestations and diagnosis (2010). www .uptodate.com/contents/acute-appendicitis-in-chil dren-clinical-manifestations-and-diagnosis? source=related_link. Accessed December 14, 2011.
27. Becker T, Kharbanda A, Bachur R. Atypical clinical features of pediatric appendicitis. Acad Emerg Med. 2007;14(2):124-129.
28. Laméris W, van Randen A, Go P, et al. Single and combined diagnostic value of clinical features and laboratory tests in acute appendicitis. Acad Emerg Med. 2009;16(9):835-842.
29. McCollough M, Sharieff G. Abdominal pain in children. Pediatr Clin N Am. 2006;53:107-137.
30. Sheu BF, Chiu TF, Chen JC, et al. Risk factors associated with perforated appendicitis in elderly patients presenting with signs and symptoms of acute appendicitis. ANZ J Surg. 2007;77(8):662-666.
31. Borst AR. Acute appendicitis: pregnancy complicates this diagnosis. JAAPA. 2007; 20(12):36-38.
32. Sedlak M, Wagner OJ, Wild B, et al. Is there still a role for rectal examination in suspected appendicitis in adults? Am J Emerg Med. 2008;26 (3):359-377.
33. Ylitalo AW. Digital rectal examination. http://emedicine.medscape.com/article/1948001-overview#a1. Accessed December 14, 2011.
34. Alvarado A. A practical score for the early diagnosis of acute appendicitis. Ann Emerg Med. 1986;15(5):557-564.
35. Schneider C, Kharbanda A, Bachur R. Evaluating appendicitis scoring systems using a prospective pediatric cohort. Ann Emerg Med. 2007; 49(6):778-784.
36. Kharbanda AB, Taylor GA, Fishman SJ, Bachur RG. A clinical decision rule to identify children at low risk for appendicitis. Pediatrics. 2005; 116(3):709–16.
37. Brown JJ, Wilson C, Coleman S, Joypaul BV. Appendicitis in pregnancy. Colorectal Dis. 2009; 11(2):116-122.
38. Richardson E, Paulson C, Hitchcock K. Clinical inquiries. History, exam, and labs: is one enough to diagnose acute adult appendicitis? J Fam Practice. 2007;56(6):474-476.
39. Harrison S, Mahawar K, Brown D, et al. Acute appendicitis presenting as small bowel obstruction: two case reports. Cases J. 2009;2:9106.
40. Pirie J. Management of constipation in the emergency department. Clin Pediatr Emerg Med. 2010;11(3):182-188.
41. Yokota S, Togashi K, Kasahara N, et al. Crohn’s disease confined to the appendix. Gastrointest Endosc. 2010;72(5):1063-1064.
42. Purysko AS, Remer EM, Filho HM, et al. Beyond appendicitis: common and uncommon gastrointestinal causes of right lower quadrant abdominal pain at multidetector CT. Radiographics. 2011;31(4):927-947.
43. Wang LT, Prentiss KA, Simon JZ, et al. The use of white blood cell count and left shift in the diagnosis of appendicitis in children. Pediatr Emerg Care. 2007;23(2):69-76.
44. Kwan KY, Nager AL. Diagnosing pediatric appendicitis: usefulness of laboratory markers. Am J Emerg Med. 2010;28(9):1009-1015.
45. Yildirim O, Solak C, Koçer B, et al. The role of serum inflammatory markers in acute appendicitis and their success in preventing negative laparotomy. J Invest Surg. 2006;19(6):345-352.
46. Spirt MJ. Complicated intra-abdominal infections: a focus on appendicitis and diverticulitis. Postgrad Med. 2010;122(1):39-51.
47. Pickhardt PJ, Lawrence EM, Pooler D, Bruce RJ. Diagnostic performance of multidetector computed tomography for suspected acute appendicitis. Ann Intern Med. 2011:154(12):789-796.
48. Coursey CA, Nelson RC, Moreno RD, et al. Appendicitis, body mass index, and CT: is CT more valuable for obese patients than thin patients? Am Surg. 2011;77(4):471-475.
49. Petrosyan M, Estrada J, Chan S, et al. CT scan in patients with suspected appendicitis: clinical implications for the acute care surgeon. Eur Surg Res. 2008;40(2):211-219.
50. Huynh V, Lalezarzadeh F, Lawandy S, et al. Abdominal computed tomography in the evaluation of acute and perforated appendicitis in the community setting. Am Surg. 2007;73(10):1002-1005.
51. Fox JC, Solley M, Anderson CL, et al. Prospective evaluation of emergency physician performed bedside ultrasound to detect acute appendicitis. Eur J Emerg Med. 2008;15(2):80-85.
52. Al-Mulhim AS, Nasser MA, Abdullah MM, et al. Emergency laparoscopy for acute abdominal conditions: a prospective study. J Laparoendosc Adv Surg Tech A. 2008;18(4):599-602.
53. Varadhan KK, Humes DJ, Neal KR, Lobo DN. Antibiotic therapy versus appendectomy for acute appendicitis: a meta-analysis. World J Surg. 2010;34(2):199-209.
54. Fitzmaurice GJ, McWilliams B, Hurreiz H, Epanomeritakis E. Antibiotics versus appendectomy in the management of acute appendicitis: a review of the current evidence. Can J Surg. 2011; 54(5):307-314.
55. Ingraham AM, Cohen ME, Bilimoria KY, et al. Comparison of outcomes after laparoscopic versus open appendectomy for acute appendicitis at 222 ACS NSQIP hospitals. Surgery. 2010;148(4): 625-635.
56. Manterola C, Vial M, Moraga J, Astudillo P. Analgesia in patients with acute abdominal pain. Cochrane Database Syst Rev. 2011;(1):CD005660.
57. Puapong D, Lee SL, Haigh PI, et al. Routine interval appendectomy in children is not indicated. J Pediatr Surg. 2007;42(9):1500-1503.
58. Deakin DE, Ahmed I. Interval appendectomy after resolution of adult inflammatory appendix mass: is it necessary? Surgeon. 2007;5(1):45-50.
59. Blakely ML, Williams R, Dassinger MS, et al. Early vs interval appendectomy for children with perforated appendicitis. Arch Surg. 2011;146 (6):660-665.
60. Salkind AR, Rao KC. Antibiotic prophylaxis to prevent surgical site infections. Am Fam Physician. 2011;83(5):585-590.
61. James M. Antibiotics and perioperative infections. Best Pract Res Clin Anaesthesiol. 2008; 22(3):571-584.
62. Bucher BT, Warner BW, Dillon PA. Antibiotic prophylaxis and the prevention of surgical site infection. Curr Opin Pediatr. 2011;23(3):334-338.
63. American Society of Health-Systems Pharmacists. ASHP therapeutic guidelines on antimicrobial prophylaxis in surgery. Am J Health Syst Pharm. 1999;56(18):1839-1888.
64. Antimicrobial prophylaxis for surgery. Treat Guidel Med Lett. 2009;7(82):47-52.
65. Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133-164.
66. Vons C, Barry C, Maitre S, et al. Amoxicillin plus clavulanic acid versus appendicectomy for treatment of acute uncomplicated appendicitis: an open-label, non-inferiority, randomized controlled trial. Lancet. 2011;377(9777):1573-1579.
67. Hansson J, Körner U, Khorram-Manesh A, et al. Randomized clinical trial of antibiotic therapy versus appendicectomy as primary treatment of acute appendicitis in unselected patients. Br J Surg. 2009;96(5):473-481.
68. Styrud J, Eriksson S, Nilsson I, et al. Appendectomy versus antibiotic treatment in acute appendicitis: a prospective multicenter randomized control trial. World J Surg. 2006;30(6):1033-1037.
69. Sakorafas GH, Mastoraki A, Lappas C, et al. Conservative treatment of acute appendicitis: heresy or an effective and acceptable alternative to surgery? Eur J Gastroenterol Hepatol. 2011;23 (2):121-127.
1. Birnbaum BA, Wilson SR. Appendicitis at the millennium. Radiology. 2000;215(2):337-348.
2. Marudanayagam R, Williams GT, Rees BI. Review of the pathological results of 2660 appendicectomy specimens. J Gastroenterol. 2006;41(8):745-749.
3. Bundy DG, Byerley JS, Liles EA, et al. Does this child have appendicitis? JAMA. 2007;298(4):438-451.
4. Alder AC, Fomby TB, Woodward WA, et al. Association of viral infection and appendicitis. Arch Surg. 2010;145(1):63-71.
5. Rubin R. The gastrointestinal tract. In: Rubin R, Strayer DS, eds. Rubin’s Pathology: Clinicopathologic Foundations of Medicine. 6th ed. Lippincott, Williams and Wilkins; 2012: 671.
6. McQuaid KR. Gastrointestinal disorders. In: McPhee SJ, Papadakis MA, eds. 2011 Current Medical Diagnosis and Treatment. 50th ed. McGraw Hill; 2011: 606-608.
7. Brennan GD. Pediatric appendicitis: pathophysiology and appropriate use of diagnostic imaging. CJEM. 2006;8(6):425-432.
8. Black C, Martin R. Acute appendicitis in adults: clinical manifestations and diagnosis (2011). www .uptodate.com/contents/acute-appendicitis-in-adults-clinical-manifestations-and-diagnosis. Accessed December 14, 2011.
9. Pittman-Waller VA, Myers JG, Stewart RM, et al. Appendicitis: why so complicated? Analysis of 5755 consecutive appendectomies. Am Surg. 2000; 6(66):548-554.
10. Mazuski JE, Solomkin JS. Intra-abdominal infections. Surg Clin North Am. 2009;89(2):421-437.
11. Andersson RE. The natural history and traditional management of appendicitis revisited: spontaneous resolution and predominance of prehospital perforations imply that a correct diagnosis is more important than an early diagnosis. World J Surg. 2007;31(1):86-92.
12. Livingston EH, Woodward WA, Sarosi GA, Haley RW. Disconnect between incidence of nonperforated and perforated appendicitis: implications for pathophysiology and management. Ann Surg. 2007;245(6):886-892.
13. Howell JM, Eddy OL, Lukens TW, et al; American College of Emergency Physicians. Clinical policy: critical issues in the evaluation and management of emergency department patients with suspected appendicitis. Ann Emerg Med. 2010; 55(1):71-116.
14. CDC. Number, rate, and standard error of all-listed surgical and nonsurgical procedures for discharges from short-stay hospitals, by selected procedure categories: United States, 2007. www.cdc
.gov/nchs/data/nhds/4procedures/2009 pro4_num berrate.pdf. Accessed December 14, 2011.
15. Lee SL, Ho HS. Acute appendicitis: is there a difference between children and adults? Am Surg. 2006;5(72):409-413.
16. Bickell NA, Aufses AH Jr, Rojas M, Bodian C. How time affects the risk of rupture in appendicitis. J Am Coll Surg. 2006;202(3):401-406.
17. Morrow SE, Newman KD. Current management of appendicitis. Semin Pediatr Surg. 2007; 16(1):34-40.
18. Nance ML, Adamson WT, Hedrick HL. Appendicitis in the young child: a continuing diagnostic challenge. Pediatr Emerg Care. 2000; 16(3):160-162.
19. Feinberg AN, Feinberg LA. The gastrointestinal tract, liver, gallbladder, and pancreas. In: Greydanus D, Feinberg A, Patel D, Homnick D, eds. The Pediatic Diagnostic Examination. McGraw-Hill Professional; 2008:267.
20. Humes DJ, Simpson J. Acute appendicitis. BMJ. 2006;333(7567):530-534.
21. Yale SH, Musana KA. Charles Heber McBurney (1845-1913). Clin Med Res. 2005;3(3):187-189.
22. Murphy JB. Two thousand operations for appendicitis with deductions from his personal experience. Am J Med Sci. 1904;128:187-211.
23. Andersson RE. Meta-analysis of the clinical and laboratory diagnosis of appendicitis. Br J Surg. 2004;91(1):28-37.
24. Wani I. K-sign in retrocaecal appendicitis: a case series. Cases J. 2009;2:157.
25. Rothrock SG, Pagane J. Acute appendicitis in children: emergency department diagnosis and management. Ann Emerg Med. 2000;36(1):39-51.
26. Wesson DE. Acute appendicitis in children: clinical manifestations and diagnosis (2010). www .uptodate.com/contents/acute-appendicitis-in-chil dren-clinical-manifestations-and-diagnosis? source=related_link. Accessed December 14, 2011.
27. Becker T, Kharbanda A, Bachur R. Atypical clinical features of pediatric appendicitis. Acad Emerg Med. 2007;14(2):124-129.
28. Laméris W, van Randen A, Go P, et al. Single and combined diagnostic value of clinical features and laboratory tests in acute appendicitis. Acad Emerg Med. 2009;16(9):835-842.
29. McCollough M, Sharieff G. Abdominal pain in children. Pediatr Clin N Am. 2006;53:107-137.
30. Sheu BF, Chiu TF, Chen JC, et al. Risk factors associated with perforated appendicitis in elderly patients presenting with signs and symptoms of acute appendicitis. ANZ J Surg. 2007;77(8):662-666.
31. Borst AR. Acute appendicitis: pregnancy complicates this diagnosis. JAAPA. 2007; 20(12):36-38.
32. Sedlak M, Wagner OJ, Wild B, et al. Is there still a role for rectal examination in suspected appendicitis in adults? Am J Emerg Med. 2008;26 (3):359-377.
33. Ylitalo AW. Digital rectal examination. http://emedicine.medscape.com/article/1948001-overview#a1. Accessed December 14, 2011.
34. Alvarado A. A practical score for the early diagnosis of acute appendicitis. Ann Emerg Med. 1986;15(5):557-564.
35. Schneider C, Kharbanda A, Bachur R. Evaluating appendicitis scoring systems using a prospective pediatric cohort. Ann Emerg Med. 2007; 49(6):778-784.
36. Kharbanda AB, Taylor GA, Fishman SJ, Bachur RG. A clinical decision rule to identify children at low risk for appendicitis. Pediatrics. 2005; 116(3):709–16.
37. Brown JJ, Wilson C, Coleman S, Joypaul BV. Appendicitis in pregnancy. Colorectal Dis. 2009; 11(2):116-122.
38. Richardson E, Paulson C, Hitchcock K. Clinical inquiries. History, exam, and labs: is one enough to diagnose acute adult appendicitis? J Fam Practice. 2007;56(6):474-476.
39. Harrison S, Mahawar K, Brown D, et al. Acute appendicitis presenting as small bowel obstruction: two case reports. Cases J. 2009;2:9106.
40. Pirie J. Management of constipation in the emergency department. Clin Pediatr Emerg Med. 2010;11(3):182-188.
41. Yokota S, Togashi K, Kasahara N, et al. Crohn’s disease confined to the appendix. Gastrointest Endosc. 2010;72(5):1063-1064.
42. Purysko AS, Remer EM, Filho HM, et al. Beyond appendicitis: common and uncommon gastrointestinal causes of right lower quadrant abdominal pain at multidetector CT. Radiographics. 2011;31(4):927-947.
43. Wang LT, Prentiss KA, Simon JZ, et al. The use of white blood cell count and left shift in the diagnosis of appendicitis in children. Pediatr Emerg Care. 2007;23(2):69-76.
44. Kwan KY, Nager AL. Diagnosing pediatric appendicitis: usefulness of laboratory markers. Am J Emerg Med. 2010;28(9):1009-1015.
45. Yildirim O, Solak C, Koçer B, et al. The role of serum inflammatory markers in acute appendicitis and their success in preventing negative laparotomy. J Invest Surg. 2006;19(6):345-352.
46. Spirt MJ. Complicated intra-abdominal infections: a focus on appendicitis and diverticulitis. Postgrad Med. 2010;122(1):39-51.
47. Pickhardt PJ, Lawrence EM, Pooler D, Bruce RJ. Diagnostic performance of multidetector computed tomography for suspected acute appendicitis. Ann Intern Med. 2011:154(12):789-796.
48. Coursey CA, Nelson RC, Moreno RD, et al. Appendicitis, body mass index, and CT: is CT more valuable for obese patients than thin patients? Am Surg. 2011;77(4):471-475.
49. Petrosyan M, Estrada J, Chan S, et al. CT scan in patients with suspected appendicitis: clinical implications for the acute care surgeon. Eur Surg Res. 2008;40(2):211-219.
50. Huynh V, Lalezarzadeh F, Lawandy S, et al. Abdominal computed tomography in the evaluation of acute and perforated appendicitis in the community setting. Am Surg. 2007;73(10):1002-1005.
51. Fox JC, Solley M, Anderson CL, et al. Prospective evaluation of emergency physician performed bedside ultrasound to detect acute appendicitis. Eur J Emerg Med. 2008;15(2):80-85.
52. Al-Mulhim AS, Nasser MA, Abdullah MM, et al. Emergency laparoscopy for acute abdominal conditions: a prospective study. J Laparoendosc Adv Surg Tech A. 2008;18(4):599-602.
53. Varadhan KK, Humes DJ, Neal KR, Lobo DN. Antibiotic therapy versus appendectomy for acute appendicitis: a meta-analysis. World J Surg. 2010;34(2):199-209.
54. Fitzmaurice GJ, McWilliams B, Hurreiz H, Epanomeritakis E. Antibiotics versus appendectomy in the management of acute appendicitis: a review of the current evidence. Can J Surg. 2011; 54(5):307-314.
55. Ingraham AM, Cohen ME, Bilimoria KY, et al. Comparison of outcomes after laparoscopic versus open appendectomy for acute appendicitis at 222 ACS NSQIP hospitals. Surgery. 2010;148(4): 625-635.
56. Manterola C, Vial M, Moraga J, Astudillo P. Analgesia in patients with acute abdominal pain. Cochrane Database Syst Rev. 2011;(1):CD005660.
57. Puapong D, Lee SL, Haigh PI, et al. Routine interval appendectomy in children is not indicated. J Pediatr Surg. 2007;42(9):1500-1503.
58. Deakin DE, Ahmed I. Interval appendectomy after resolution of adult inflammatory appendix mass: is it necessary? Surgeon. 2007;5(1):45-50.
59. Blakely ML, Williams R, Dassinger MS, et al. Early vs interval appendectomy for children with perforated appendicitis. Arch Surg. 2011;146 (6):660-665.
60. Salkind AR, Rao KC. Antibiotic prophylaxis to prevent surgical site infections. Am Fam Physician. 2011;83(5):585-590.
61. James M. Antibiotics and perioperative infections. Best Pract Res Clin Anaesthesiol. 2008; 22(3):571-584.
62. Bucher BT, Warner BW, Dillon PA. Antibiotic prophylaxis and the prevention of surgical site infection. Curr Opin Pediatr. 2011;23(3):334-338.
63. American Society of Health-Systems Pharmacists. ASHP therapeutic guidelines on antimicrobial prophylaxis in surgery. Am J Health Syst Pharm. 1999;56(18):1839-1888.
64. Antimicrobial prophylaxis for surgery. Treat Guidel Med Lett. 2009;7(82):47-52.
65. Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133-164.
66. Vons C, Barry C, Maitre S, et al. Amoxicillin plus clavulanic acid versus appendicectomy for treatment of acute uncomplicated appendicitis: an open-label, non-inferiority, randomized controlled trial. Lancet. 2011;377(9777):1573-1579.
67. Hansson J, Körner U, Khorram-Manesh A, et al. Randomized clinical trial of antibiotic therapy versus appendicectomy as primary treatment of acute appendicitis in unselected patients. Br J Surg. 2009;96(5):473-481.
68. Styrud J, Eriksson S, Nilsson I, et al. Appendectomy versus antibiotic treatment in acute appendicitis: a prospective multicenter randomized control trial. World J Surg. 2006;30(6):1033-1037.
69. Sakorafas GH, Mastoraki A, Lappas C, et al. Conservative treatment of acute appendicitis: heresy or an effective and acceptable alternative to surgery? Eur J Gastroenterol Hepatol. 2011;23 (2):121-127.
Myocardial Stiffness: Important Predictor of Survival in CHF
Effect of a VA Chronic Disease Management Initiative on Hospitalizations for Ambulatory Care Sensitive Conditions
Rocky Mountain Spotted Fever Presenting as Febrile Pancytopenia
Cover

Tres Pasitos: "Three Little Steps" to Aldicarb Poisoning
Rhabdomyolysis: Evaluation and Emergent Management
2012 Update on Obstetrics
The authors report no financial relationships relevant to this article.
The year that has followed our inaugural “Update on Obstetrics” [OBG Management, January 2011, available at www.obgmanagement.com] saw a resurgence of interest in a number of aspects of obstetric care. We want to highlight four of them in this Update because we think they are particularly important—given the attention they’ve received in the medical literature and in the consumer media:
- the ever-increasing cesarean delivery rate
- home birth
- postpartum hemorrhage
- measurement of cervical length and the use of progesterone.
Taming the cesarean delivery rate—how can we
accomplish this?
No one should be surprised to learn that the cesarean delivery rate increased nearly sevenfold from 1970 to 2011—from a rate of approximately 5% in 1970 to nearly 35%. Recall that, in the 1990s, the US Public Health Service proposed, as part of Healthy People 2010, a target cesarean rate of 15%, with a vaginal birth after cesarean (VBAC) rate of 60%. Today, the cesarean delivery rate is, as we said, nearly 35% and the VBAC rate is less than 10%.
Many factors have been cited for the rise, including:
- the obesity pandemic
- delaying childbearing
- increasing use of assisted reproduction
- multiple gestation (although the incidence of higher-order multiple gestations is now decreasing, the rate of twin births remains quite high relative to past decades).
So, how did this happen? And what can we do?
For one, VBAC is not likely to gain in popularity. More than 60% of US hospitals that provide OB services handle a volume of fewer than 1,000 deliveries a year. Such low volumes generally will not be able to support (either with dollars or staffing) the resources needed to safely provide VBAC.
Other options have been proposed: Loosen the guidelines for VBAC, change the personnel requirements, gather community groups of doctors, attorneys, and patients to agree on guidelines that, if followed, would protect physicians from being sued1—and so on. The medicolegal reality, however, is that these options have not been shown to be viable. We have concluded that increasing VBAC utilization is not the answer. Rather, addressing ways to prevent primary cesarean delivery holds the most promise for, ultimately, reducing the current rising trend.
On a positive note: The most recent data available from the National Center for Health Statistics suggest that the cesarean delivery rate has dropped slightly: from 32.9% in 2009 to 32.8% in 2010. The drop is truly slight; we’ll watch with interest to see if a trend has begun.
Considering that the cesarean delivery rate in 1970 was 5%, and that the dictum at the time was “once a section, always a section,” it seems clear (to us, at least) that the solution to this problem lies in preventing first cesarean deliveries. How can the specialty and, in some ways, you, in your practice, work toward this goal? Here are possible strategies:
- Eliminate elective inductions of labor when the modified Bishop score is less than 8
- Return to defining “post-term” as 42—not 41—completed weeks’ gestation
- Eliminate all elective inductions before 39 weeks’ gestation
- Provide better and more standardized training of physicians in the interpretation of fetal heart-rate tracings
- Improve communication between obstetricians and anesthesiologists in regard to managing pain during labor
- Institute mandatory review of all cesarean deliveries that are performed in the latent phase of labor and all so-called “stat cesareans”
- Readjust the compensation scale for physicians and hospitals in such a way that successful vaginal delivery is rewarded.
Even if all these measures were implemented, we think it’s unlikely that we will ever see a 5% cesarean delivery rate again—although probably for good reason. But even a return to a more manageable 20% rate seems a reasonable goal.
Home birth: Consider where you stand
We suppose that one way to avoid cesarean delivery would be to deliver at home. The topic, and practice, of home birth has mushroomed in the past few years—for a number of social and economic reasons, probably. It seems to us that there are a few basic issues that must be addressed, however, before it’s possible to come to grips with home birth in the 21st century in an enlightened way:
- In 1935, the maternal mortality rate approached 500 to 600 for every 100,000 births; most of those deaths occurred at home. In 2009, the maternal mortality rate was approximately 8 for every 100,000 births. Both rates are very low, but the difference would be significant to the 492 to 592 women who met a potentially preventable death.
- Methods of identifying who might be an appropriate candidate for a home birth are, at best, imprecise.
- Infrastructure for rapidly transporting mother and baby to a hospital if matters go awry is inadequate.
Although evidence is limited, what data there are suggest that one significant outcome—neonatal death—occurs with higher frequency among home births than among hospital births, even after correcting for anomalies (odds ratio, 2.9 [95% confidence interval, 1.3–6.2]).2 Although women who delivered at home did have fewer episiotomies, fewer third- and fourth-degree perineal lacerations, fewer operative deliveries (vaginal and cesarean) and a lower rate of infection, those reductions seem inconsequential compared to the death of a newborn….
Bottom line? Home birth is legal; home birth may be appropriate for some women who are at low risk and willing to accept a legitimate amount of personal risk; and you, as an OB, are in no way required to participate in or endorse the practice.
Many institutions have addressed this matter by developing a family-centered health care model for obstetrics—so-called hospitals within hospitals—that allow for a less interventionist approach to childbirth within the safety net of a hospital facility, should unforeseeable complications arise. Consider your interest in affiliating with such a facility, based on your acceptance of the practice of home birth and your comfort with being part of this approach.
Formal, systematic planning is key to managing
postpartum hemorrhage
A question for mothers-to-be: What could be worse than having a cesarean delivery in your home?
Answer: Having an associated postpartum bleed.
Perhaps that isn’t the most elegant segue, but postpartum hemorrhage is a significant problem that remains a major contributor to maternal mortality in the United States. And, in fact, a prolonged and unsuccessful labor—the kind that could present to your hospital from an outside birthing facility or home—necessitating a cesarean delivery is a set-up for postpartum hemorrhage.
One of the tenets of emergency management in obstetrics is the three-pronged preparedness of 1) risk identification, 2) foreseeability, and 3) having a plan for taking action. Of late, many institutions have begun to develop a formal plan for managing OB emergencies—in particular, postpartum hemorrhage.
(A note about the potential role of interventional radiology in the management of postpartum hemorrhage: Our experience is limited, but we conjecture that, in most US hospitals that provide OB services, mobilizing an interventional radiology group in an emergency isn’t feasible. That makes it essential to have established medical and surgical management guidelines for such cases.)
To establish a plan on labor and delivery for managing postpartum bleeds, we recommend the following steps and direct you to ACOG’s “Practice Bulletin#76” for more specific information3:
- Establish a list of conditions that predispose a woman to postpartum hemorrhage and post that list throughout labor and delivery to heighten the awareness of team members
- Establish protocols for pharmacotherapeutic intervention—including oxytocin, methylergonovine, misoprostol, and prostaglandin F2a, with dosage and frequency guidelines and algorithms for use—and have those protocols readily available on labor and delivery, either on-line or posted
- Establish an “all-hands-on-deck” protocol for surgical emergencies—actual or potential—that includes what personnel to call and in what order to call them
- Use simulation to practice the all-hands-on-deck protocol and evaluate team and individual performance in managing hemorrhage
- Establish blood product replacement protocols, including order sets for products that are linked to particular diagnoses (e.g., typing and cross-matching for patients coming in to deliver who have a diagnosis of placenta previa; adding products such as fresh frozen plasma and platelets for patients who have complicating diagnoses, such as suspected placenta accreta or severe preeclampsia).
To prevent preterm birth: Cervical length screening
and progesterone
Preterm birth accounts for almost 13% of births in the United States, with spontaneous preterm labor and preterm rupture of membranes accounting for approximately 80% of those cases.4 Once preterm labor has begun, little in the way of successful intervention is possible, beyond short-term prolongation of pregnancy with tocolytic agents to allow for corticosteroid administration. Studies in recent years have, therefore, moved the focus back on prevention, using the same treatments that were used 60 years ago—progesterone supplementation and cerclage—with the addition of transvaginal ultrasonography (US) screening for cervical length.
Several large, randomized trials have examined the use of intramuscular injection or vaginal delivery of progesterone to prevent preterm birth in patients who are at high risk of preterm birth based on their obstetric history.5,6 Both 17a-hydroxyprogesterone caproate and vaginal progesterone suppositories are associated with a significant reduction in the risk of preterm birth in singleton pregnancies. ACOG reconfirmed the value of this finding in a 2011 Committee Opinion, which recommended the use of progesterone supplementation in singleton pregnancies in which there is a history of preterm labor or preterm rupture of membranes.7
There is mounting evidence that cervical length is inversely related to risk of preterm birth.The real question, however, is: What should be done about transvaginal cervical length: Should we be screening, or not? As recently as 2009, a Cochrane Review did not advocate universal screening for cervical length as a predictor for preterm birth4—despite mounting evidence that cervical length is inversely related to risk of preterm birth, with progressively shorter length (starting at <25 mm) associated with significantly higher risk of preterm birth.8,9 Keeping in mind that the decision to screen depends on your ability to treat the condition for which you are screening, what was needed was proof that intervention works.
2011 brought two studies that recommend screening for cervical length based on a successful reduction in preterm birth with a specific intervention. A large, randomized trial of vaginal progesterone gel for the prevention of preterm birth used universal screening for shortened cervical length (10 to 20 mm) as the criterion for randomization to treatment or placebo. The investigators demonstrated a 45% reduction in preterm birth of less than 33 weeks in the treatment arm.10
An interesting aspect of this study: The reduction in preterm birth was not, in fact, seen in patients who had a history of preterm birth, suggesting that this may be a different patient population that benefits from vaginal progesterone.
On the other hand, a recent meta-analysis concluded that patients who meet the criteria of 1) cervical length less than 25 mm and 2) a history of prior spontaneous preterm birth experience a significant reduction in preterm birth and a reduction in perinatal morbidity and mortality if they have cervical cerclage placed.11
Although these publications lead us to hope that there may be some benefit from preventive intervention for preterm birth, the question of how to screen for, and prevent, spontaneous preterm birth remains somewhat nebulous: It hasn’t been determined which patient population will benefit from which combination of screening and intervention. Larger trials for specific populations are still needed.
This is what we know, for now:
- Women who have a history of spontaneous preterm birth should have a thorough evaluation of their OB history to determine possible modifiable risk factors (e.g., smoking, short inter-pregnancy interval) and to determine, as definitively as possible, the likely cause of that preterm birth
- Women who have a singleton pregnancy and a history of either spontaneous preterm labor or preterm rupture of membranes can be offered progesterone supplementation as intramuscular 17a-hydroxyprogesterone or a vaginal preparation to reduce their risk of preterm birth
- Women who have an asymptomatic shortening of the cervix, as measured on transvaginal US at 18 to 24 weeks’ gestation, can be offered vaginal progesterone to reduce their risk of preterm birth
- Women who have a history of preterm birth and cervical shortening may see a reduction in their risk of preterm birth from cerclage placement
- The use of screening for cervical length or progesterone supplementation, or both, in a multiple gestation pregnancy are not recommended because their benefit in this population has not been demonstrated.
Until we fully understand the various etiologic pathways of spontaneous preterm birth, we won’t have a one-size-fits-all solution to this major cause of perinatal morbidity and mortality.
We want to hear from you! Tell us what you think.
1. Scott JR. Vaginal birth after cesarean delivery; a common sense approach. Obstet Gynecol. 2011;118(2 Pt 1):342-350.
2. American College of Obstetricians and Gynecologists Committee on Obstetric Practice. ACOG Committee Opinion No. 476: Planned home birth. Obstet Gynecol. 2011;117(2 Pt 1):425-428.
3. American College of Obstetricians and Gynecologists Committee on Practice Bulletins. ACOG Practice Bulletin No. 76: Postpartum hemorrhage. Obstet Gynecol. 2006;108(4):1039-1047.
4. Berghella V, Baxter JK, Hendrix NW. Cervical assessment by ultrasound for preventing preterm delivery (Review). Cochrane Database Syst Rev. 2009;(3):CD007235.-
5. da Fonseca EB, Bittar RE, Carvalho MH, Zugaib M. Prophylactic administration of progesterone by vaginal suppository to reduce the incidence of spontaneous preterm birth in women at increased risk: a randomized placebo-controlled double-blind study. Am J Obstet Gynecol. 2003;188(2):419-424.
6. Meis PJ, Klebanoff M, Thom E, et al. National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N Engl J Med. 2003;348(24):2379-2385.
7. American College of Obstetricians and Gynecologists Committee on Obstetric Practice. ACOG Committee Opinion No. 419: Use of progesterone to reduce preterm birth. Obstet Gynecol. 2008;112(4):963-935.
8. Owen J, Yost N, Berghella V, et al:. National Institute of Child Health and Human Development, Maternal-Fetal Medicine Units Network. Mid-trimester endovaginal sonography in women at high risk for spontaneous preterm birth. JAMA. 2001;286(11):1340-1348.
9. Durnwald CP, Walker H, Lundy JC, Iams JD. Rates of recurrent preterm birth by obstetrical history and cervical length. Am J Obstet Gynecol. 2005;193(3):1170-1174.
10. Hassan SS, Romero R, Vidyadhari D, et al. PREGNANT Trial. Vaginal progesterone reduces the rate of preterm birth in women with a sonographic short cervix: a multicenter randomized, double-blind, placebo-controlled trial. Ultrasound Obstet Gynecol. 2011;38(1):18-31.
11. Berghella V, Rafael TJ, Szychowski M, Rust OA, Owen J. Cerclage for short cervix on ultrasonography in women with singleton gestations and previous preterm birth. Obstet Gynecol. 2011;117(3):663-771

John T. Repke, MD
Dr. Repke is University Professor and Chairman, Department of Obstetrics and Gynecology, at Penn State University College of Medicine, and Obstetrician-Gynecologist-in-Chief at the Milton S. Hershey Medical Center in Hershey, Pa. He serves on the OBG Management Board of Editors.
Jaimey M. Pauli, MD
Dr. Pauli is Assistant Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, at Penn State University College of Medicine and the Milton S. Hershey Medical Center in Hershey, Pa.
John T. Repke, MD
Dr. Repke is University Professor and Chairman, Department of Obstetrics and Gynecology, at Penn State University College of Medicine, and Obstetrician-Gynecologist-in-Chief at the Milton S. Hershey Medical Center in Hershey, Pa. He serves on the OBG Management Board of Editors.
Jaimey M. Pauli, MD
Dr. Pauli is Assistant Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, at Penn State University College of Medicine and the Milton S. Hershey Medical Center in Hershey, Pa.
John T. Repke, MD
Dr. Repke is University Professor and Chairman, Department of Obstetrics and Gynecology, at Penn State University College of Medicine, and Obstetrician-Gynecologist-in-Chief at the Milton S. Hershey Medical Center in Hershey, Pa. He serves on the OBG Management Board of Editors.
Jaimey M. Pauli, MD
Dr. Pauli is Assistant Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, at Penn State University College of Medicine and the Milton S. Hershey Medical Center in Hershey, Pa.
The authors report no financial relationships relevant to this article.
The year that has followed our inaugural “Update on Obstetrics” [OBG Management, January 2011, available at www.obgmanagement.com] saw a resurgence of interest in a number of aspects of obstetric care. We want to highlight four of them in this Update because we think they are particularly important—given the attention they’ve received in the medical literature and in the consumer media:
- the ever-increasing cesarean delivery rate
- home birth
- postpartum hemorrhage
- measurement of cervical length and the use of progesterone.
Taming the cesarean delivery rate—how can we
accomplish this?
No one should be surprised to learn that the cesarean delivery rate increased nearly sevenfold from 1970 to 2011—from a rate of approximately 5% in 1970 to nearly 35%. Recall that, in the 1990s, the US Public Health Service proposed, as part of Healthy People 2010, a target cesarean rate of 15%, with a vaginal birth after cesarean (VBAC) rate of 60%. Today, the cesarean delivery rate is, as we said, nearly 35% and the VBAC rate is less than 10%.
Many factors have been cited for the rise, including:
- the obesity pandemic
- delaying childbearing
- increasing use of assisted reproduction
- multiple gestation (although the incidence of higher-order multiple gestations is now decreasing, the rate of twin births remains quite high relative to past decades).
So, how did this happen? And what can we do?
For one, VBAC is not likely to gain in popularity. More than 60% of US hospitals that provide OB services handle a volume of fewer than 1,000 deliveries a year. Such low volumes generally will not be able to support (either with dollars or staffing) the resources needed to safely provide VBAC.
Other options have been proposed: Loosen the guidelines for VBAC, change the personnel requirements, gather community groups of doctors, attorneys, and patients to agree on guidelines that, if followed, would protect physicians from being sued1—and so on. The medicolegal reality, however, is that these options have not been shown to be viable. We have concluded that increasing VBAC utilization is not the answer. Rather, addressing ways to prevent primary cesarean delivery holds the most promise for, ultimately, reducing the current rising trend.
On a positive note: The most recent data available from the National Center for Health Statistics suggest that the cesarean delivery rate has dropped slightly: from 32.9% in 2009 to 32.8% in 2010. The drop is truly slight; we’ll watch with interest to see if a trend has begun.
Considering that the cesarean delivery rate in 1970 was 5%, and that the dictum at the time was “once a section, always a section,” it seems clear (to us, at least) that the solution to this problem lies in preventing first cesarean deliveries. How can the specialty and, in some ways, you, in your practice, work toward this goal? Here are possible strategies:
- Eliminate elective inductions of labor when the modified Bishop score is less than 8
- Return to defining “post-term” as 42—not 41—completed weeks’ gestation
- Eliminate all elective inductions before 39 weeks’ gestation
- Provide better and more standardized training of physicians in the interpretation of fetal heart-rate tracings
- Improve communication between obstetricians and anesthesiologists in regard to managing pain during labor
- Institute mandatory review of all cesarean deliveries that are performed in the latent phase of labor and all so-called “stat cesareans”
- Readjust the compensation scale for physicians and hospitals in such a way that successful vaginal delivery is rewarded.
Even if all these measures were implemented, we think it’s unlikely that we will ever see a 5% cesarean delivery rate again—although probably for good reason. But even a return to a more manageable 20% rate seems a reasonable goal.
Home birth: Consider where you stand
We suppose that one way to avoid cesarean delivery would be to deliver at home. The topic, and practice, of home birth has mushroomed in the past few years—for a number of social and economic reasons, probably. It seems to us that there are a few basic issues that must be addressed, however, before it’s possible to come to grips with home birth in the 21st century in an enlightened way:
- In 1935, the maternal mortality rate approached 500 to 600 for every 100,000 births; most of those deaths occurred at home. In 2009, the maternal mortality rate was approximately 8 for every 100,000 births. Both rates are very low, but the difference would be significant to the 492 to 592 women who met a potentially preventable death.
- Methods of identifying who might be an appropriate candidate for a home birth are, at best, imprecise.
- Infrastructure for rapidly transporting mother and baby to a hospital if matters go awry is inadequate.
Although evidence is limited, what data there are suggest that one significant outcome—neonatal death—occurs with higher frequency among home births than among hospital births, even after correcting for anomalies (odds ratio, 2.9 [95% confidence interval, 1.3–6.2]).2 Although women who delivered at home did have fewer episiotomies, fewer third- and fourth-degree perineal lacerations, fewer operative deliveries (vaginal and cesarean) and a lower rate of infection, those reductions seem inconsequential compared to the death of a newborn….
Bottom line? Home birth is legal; home birth may be appropriate for some women who are at low risk and willing to accept a legitimate amount of personal risk; and you, as an OB, are in no way required to participate in or endorse the practice.
Many institutions have addressed this matter by developing a family-centered health care model for obstetrics—so-called hospitals within hospitals—that allow for a less interventionist approach to childbirth within the safety net of a hospital facility, should unforeseeable complications arise. Consider your interest in affiliating with such a facility, based on your acceptance of the practice of home birth and your comfort with being part of this approach.
Formal, systematic planning is key to managing
postpartum hemorrhage
A question for mothers-to-be: What could be worse than having a cesarean delivery in your home?
Answer: Having an associated postpartum bleed.
Perhaps that isn’t the most elegant segue, but postpartum hemorrhage is a significant problem that remains a major contributor to maternal mortality in the United States. And, in fact, a prolonged and unsuccessful labor—the kind that could present to your hospital from an outside birthing facility or home—necessitating a cesarean delivery is a set-up for postpartum hemorrhage.
One of the tenets of emergency management in obstetrics is the three-pronged preparedness of 1) risk identification, 2) foreseeability, and 3) having a plan for taking action. Of late, many institutions have begun to develop a formal plan for managing OB emergencies—in particular, postpartum hemorrhage.
(A note about the potential role of interventional radiology in the management of postpartum hemorrhage: Our experience is limited, but we conjecture that, in most US hospitals that provide OB services, mobilizing an interventional radiology group in an emergency isn’t feasible. That makes it essential to have established medical and surgical management guidelines for such cases.)
To establish a plan on labor and delivery for managing postpartum bleeds, we recommend the following steps and direct you to ACOG’s “Practice Bulletin#76” for more specific information3:
- Establish a list of conditions that predispose a woman to postpartum hemorrhage and post that list throughout labor and delivery to heighten the awareness of team members
- Establish protocols for pharmacotherapeutic intervention—including oxytocin, methylergonovine, misoprostol, and prostaglandin F2a, with dosage and frequency guidelines and algorithms for use—and have those protocols readily available on labor and delivery, either on-line or posted
- Establish an “all-hands-on-deck” protocol for surgical emergencies—actual or potential—that includes what personnel to call and in what order to call them
- Use simulation to practice the all-hands-on-deck protocol and evaluate team and individual performance in managing hemorrhage
- Establish blood product replacement protocols, including order sets for products that are linked to particular diagnoses (e.g., typing and cross-matching for patients coming in to deliver who have a diagnosis of placenta previa; adding products such as fresh frozen plasma and platelets for patients who have complicating diagnoses, such as suspected placenta accreta or severe preeclampsia).
To prevent preterm birth: Cervical length screening
and progesterone
Preterm birth accounts for almost 13% of births in the United States, with spontaneous preterm labor and preterm rupture of membranes accounting for approximately 80% of those cases.4 Once preterm labor has begun, little in the way of successful intervention is possible, beyond short-term prolongation of pregnancy with tocolytic agents to allow for corticosteroid administration. Studies in recent years have, therefore, moved the focus back on prevention, using the same treatments that were used 60 years ago—progesterone supplementation and cerclage—with the addition of transvaginal ultrasonography (US) screening for cervical length.
Several large, randomized trials have examined the use of intramuscular injection or vaginal delivery of progesterone to prevent preterm birth in patients who are at high risk of preterm birth based on their obstetric history.5,6 Both 17a-hydroxyprogesterone caproate and vaginal progesterone suppositories are associated with a significant reduction in the risk of preterm birth in singleton pregnancies. ACOG reconfirmed the value of this finding in a 2011 Committee Opinion, which recommended the use of progesterone supplementation in singleton pregnancies in which there is a history of preterm labor or preterm rupture of membranes.7
There is mounting evidence that cervical length is inversely related to risk of preterm birth.The real question, however, is: What should be done about transvaginal cervical length: Should we be screening, or not? As recently as 2009, a Cochrane Review did not advocate universal screening for cervical length as a predictor for preterm birth4—despite mounting evidence that cervical length is inversely related to risk of preterm birth, with progressively shorter length (starting at <25 mm) associated with significantly higher risk of preterm birth.8,9 Keeping in mind that the decision to screen depends on your ability to treat the condition for which you are screening, what was needed was proof that intervention works.
2011 brought two studies that recommend screening for cervical length based on a successful reduction in preterm birth with a specific intervention. A large, randomized trial of vaginal progesterone gel for the prevention of preterm birth used universal screening for shortened cervical length (10 to 20 mm) as the criterion for randomization to treatment or placebo. The investigators demonstrated a 45% reduction in preterm birth of less than 33 weeks in the treatment arm.10
An interesting aspect of this study: The reduction in preterm birth was not, in fact, seen in patients who had a history of preterm birth, suggesting that this may be a different patient population that benefits from vaginal progesterone.
On the other hand, a recent meta-analysis concluded that patients who meet the criteria of 1) cervical length less than 25 mm and 2) a history of prior spontaneous preterm birth experience a significant reduction in preterm birth and a reduction in perinatal morbidity and mortality if they have cervical cerclage placed.11
Although these publications lead us to hope that there may be some benefit from preventive intervention for preterm birth, the question of how to screen for, and prevent, spontaneous preterm birth remains somewhat nebulous: It hasn’t been determined which patient population will benefit from which combination of screening and intervention. Larger trials for specific populations are still needed.
This is what we know, for now:
- Women who have a history of spontaneous preterm birth should have a thorough evaluation of their OB history to determine possible modifiable risk factors (e.g., smoking, short inter-pregnancy interval) and to determine, as definitively as possible, the likely cause of that preterm birth
- Women who have a singleton pregnancy and a history of either spontaneous preterm labor or preterm rupture of membranes can be offered progesterone supplementation as intramuscular 17a-hydroxyprogesterone or a vaginal preparation to reduce their risk of preterm birth
- Women who have an asymptomatic shortening of the cervix, as measured on transvaginal US at 18 to 24 weeks’ gestation, can be offered vaginal progesterone to reduce their risk of preterm birth
- Women who have a history of preterm birth and cervical shortening may see a reduction in their risk of preterm birth from cerclage placement
- The use of screening for cervical length or progesterone supplementation, or both, in a multiple gestation pregnancy are not recommended because their benefit in this population has not been demonstrated.
Until we fully understand the various etiologic pathways of spontaneous preterm birth, we won’t have a one-size-fits-all solution to this major cause of perinatal morbidity and mortality.
We want to hear from you! Tell us what you think.
The authors report no financial relationships relevant to this article.
The year that has followed our inaugural “Update on Obstetrics” [OBG Management, January 2011, available at www.obgmanagement.com] saw a resurgence of interest in a number of aspects of obstetric care. We want to highlight four of them in this Update because we think they are particularly important—given the attention they’ve received in the medical literature and in the consumer media:
- the ever-increasing cesarean delivery rate
- home birth
- postpartum hemorrhage
- measurement of cervical length and the use of progesterone.
Taming the cesarean delivery rate—how can we
accomplish this?
No one should be surprised to learn that the cesarean delivery rate increased nearly sevenfold from 1970 to 2011—from a rate of approximately 5% in 1970 to nearly 35%. Recall that, in the 1990s, the US Public Health Service proposed, as part of Healthy People 2010, a target cesarean rate of 15%, with a vaginal birth after cesarean (VBAC) rate of 60%. Today, the cesarean delivery rate is, as we said, nearly 35% and the VBAC rate is less than 10%.
Many factors have been cited for the rise, including:
- the obesity pandemic
- delaying childbearing
- increasing use of assisted reproduction
- multiple gestation (although the incidence of higher-order multiple gestations is now decreasing, the rate of twin births remains quite high relative to past decades).
So, how did this happen? And what can we do?
For one, VBAC is not likely to gain in popularity. More than 60% of US hospitals that provide OB services handle a volume of fewer than 1,000 deliveries a year. Such low volumes generally will not be able to support (either with dollars or staffing) the resources needed to safely provide VBAC.
Other options have been proposed: Loosen the guidelines for VBAC, change the personnel requirements, gather community groups of doctors, attorneys, and patients to agree on guidelines that, if followed, would protect physicians from being sued1—and so on. The medicolegal reality, however, is that these options have not been shown to be viable. We have concluded that increasing VBAC utilization is not the answer. Rather, addressing ways to prevent primary cesarean delivery holds the most promise for, ultimately, reducing the current rising trend.
On a positive note: The most recent data available from the National Center for Health Statistics suggest that the cesarean delivery rate has dropped slightly: from 32.9% in 2009 to 32.8% in 2010. The drop is truly slight; we’ll watch with interest to see if a trend has begun.
Considering that the cesarean delivery rate in 1970 was 5%, and that the dictum at the time was “once a section, always a section,” it seems clear (to us, at least) that the solution to this problem lies in preventing first cesarean deliveries. How can the specialty and, in some ways, you, in your practice, work toward this goal? Here are possible strategies:
- Eliminate elective inductions of labor when the modified Bishop score is less than 8
- Return to defining “post-term” as 42—not 41—completed weeks’ gestation
- Eliminate all elective inductions before 39 weeks’ gestation
- Provide better and more standardized training of physicians in the interpretation of fetal heart-rate tracings
- Improve communication between obstetricians and anesthesiologists in regard to managing pain during labor
- Institute mandatory review of all cesarean deliveries that are performed in the latent phase of labor and all so-called “stat cesareans”
- Readjust the compensation scale for physicians and hospitals in such a way that successful vaginal delivery is rewarded.
Even if all these measures were implemented, we think it’s unlikely that we will ever see a 5% cesarean delivery rate again—although probably for good reason. But even a return to a more manageable 20% rate seems a reasonable goal.
Home birth: Consider where you stand
We suppose that one way to avoid cesarean delivery would be to deliver at home. The topic, and practice, of home birth has mushroomed in the past few years—for a number of social and economic reasons, probably. It seems to us that there are a few basic issues that must be addressed, however, before it’s possible to come to grips with home birth in the 21st century in an enlightened way:
- In 1935, the maternal mortality rate approached 500 to 600 for every 100,000 births; most of those deaths occurred at home. In 2009, the maternal mortality rate was approximately 8 for every 100,000 births. Both rates are very low, but the difference would be significant to the 492 to 592 women who met a potentially preventable death.
- Methods of identifying who might be an appropriate candidate for a home birth are, at best, imprecise.
- Infrastructure for rapidly transporting mother and baby to a hospital if matters go awry is inadequate.
Although evidence is limited, what data there are suggest that one significant outcome—neonatal death—occurs with higher frequency among home births than among hospital births, even after correcting for anomalies (odds ratio, 2.9 [95% confidence interval, 1.3–6.2]).2 Although women who delivered at home did have fewer episiotomies, fewer third- and fourth-degree perineal lacerations, fewer operative deliveries (vaginal and cesarean) and a lower rate of infection, those reductions seem inconsequential compared to the death of a newborn….
Bottom line? Home birth is legal; home birth may be appropriate for some women who are at low risk and willing to accept a legitimate amount of personal risk; and you, as an OB, are in no way required to participate in or endorse the practice.
Many institutions have addressed this matter by developing a family-centered health care model for obstetrics—so-called hospitals within hospitals—that allow for a less interventionist approach to childbirth within the safety net of a hospital facility, should unforeseeable complications arise. Consider your interest in affiliating with such a facility, based on your acceptance of the practice of home birth and your comfort with being part of this approach.
Formal, systematic planning is key to managing
postpartum hemorrhage
A question for mothers-to-be: What could be worse than having a cesarean delivery in your home?
Answer: Having an associated postpartum bleed.
Perhaps that isn’t the most elegant segue, but postpartum hemorrhage is a significant problem that remains a major contributor to maternal mortality in the United States. And, in fact, a prolonged and unsuccessful labor—the kind that could present to your hospital from an outside birthing facility or home—necessitating a cesarean delivery is a set-up for postpartum hemorrhage.
One of the tenets of emergency management in obstetrics is the three-pronged preparedness of 1) risk identification, 2) foreseeability, and 3) having a plan for taking action. Of late, many institutions have begun to develop a formal plan for managing OB emergencies—in particular, postpartum hemorrhage.
(A note about the potential role of interventional radiology in the management of postpartum hemorrhage: Our experience is limited, but we conjecture that, in most US hospitals that provide OB services, mobilizing an interventional radiology group in an emergency isn’t feasible. That makes it essential to have established medical and surgical management guidelines for such cases.)
To establish a plan on labor and delivery for managing postpartum bleeds, we recommend the following steps and direct you to ACOG’s “Practice Bulletin#76” for more specific information3:
- Establish a list of conditions that predispose a woman to postpartum hemorrhage and post that list throughout labor and delivery to heighten the awareness of team members
- Establish protocols for pharmacotherapeutic intervention—including oxytocin, methylergonovine, misoprostol, and prostaglandin F2a, with dosage and frequency guidelines and algorithms for use—and have those protocols readily available on labor and delivery, either on-line or posted
- Establish an “all-hands-on-deck” protocol for surgical emergencies—actual or potential—that includes what personnel to call and in what order to call them
- Use simulation to practice the all-hands-on-deck protocol and evaluate team and individual performance in managing hemorrhage
- Establish blood product replacement protocols, including order sets for products that are linked to particular diagnoses (e.g., typing and cross-matching for patients coming in to deliver who have a diagnosis of placenta previa; adding products such as fresh frozen plasma and platelets for patients who have complicating diagnoses, such as suspected placenta accreta or severe preeclampsia).
To prevent preterm birth: Cervical length screening
and progesterone
Preterm birth accounts for almost 13% of births in the United States, with spontaneous preterm labor and preterm rupture of membranes accounting for approximately 80% of those cases.4 Once preterm labor has begun, little in the way of successful intervention is possible, beyond short-term prolongation of pregnancy with tocolytic agents to allow for corticosteroid administration. Studies in recent years have, therefore, moved the focus back on prevention, using the same treatments that were used 60 years ago—progesterone supplementation and cerclage—with the addition of transvaginal ultrasonography (US) screening for cervical length.
Several large, randomized trials have examined the use of intramuscular injection or vaginal delivery of progesterone to prevent preterm birth in patients who are at high risk of preterm birth based on their obstetric history.5,6 Both 17a-hydroxyprogesterone caproate and vaginal progesterone suppositories are associated with a significant reduction in the risk of preterm birth in singleton pregnancies. ACOG reconfirmed the value of this finding in a 2011 Committee Opinion, which recommended the use of progesterone supplementation in singleton pregnancies in which there is a history of preterm labor or preterm rupture of membranes.7
There is mounting evidence that cervical length is inversely related to risk of preterm birth.The real question, however, is: What should be done about transvaginal cervical length: Should we be screening, or not? As recently as 2009, a Cochrane Review did not advocate universal screening for cervical length as a predictor for preterm birth4—despite mounting evidence that cervical length is inversely related to risk of preterm birth, with progressively shorter length (starting at <25 mm) associated with significantly higher risk of preterm birth.8,9 Keeping in mind that the decision to screen depends on your ability to treat the condition for which you are screening, what was needed was proof that intervention works.
2011 brought two studies that recommend screening for cervical length based on a successful reduction in preterm birth with a specific intervention. A large, randomized trial of vaginal progesterone gel for the prevention of preterm birth used universal screening for shortened cervical length (10 to 20 mm) as the criterion for randomization to treatment or placebo. The investigators demonstrated a 45% reduction in preterm birth of less than 33 weeks in the treatment arm.10
An interesting aspect of this study: The reduction in preterm birth was not, in fact, seen in patients who had a history of preterm birth, suggesting that this may be a different patient population that benefits from vaginal progesterone.
On the other hand, a recent meta-analysis concluded that patients who meet the criteria of 1) cervical length less than 25 mm and 2) a history of prior spontaneous preterm birth experience a significant reduction in preterm birth and a reduction in perinatal morbidity and mortality if they have cervical cerclage placed.11
Although these publications lead us to hope that there may be some benefit from preventive intervention for preterm birth, the question of how to screen for, and prevent, spontaneous preterm birth remains somewhat nebulous: It hasn’t been determined which patient population will benefit from which combination of screening and intervention. Larger trials for specific populations are still needed.
This is what we know, for now:
- Women who have a history of spontaneous preterm birth should have a thorough evaluation of their OB history to determine possible modifiable risk factors (e.g., smoking, short inter-pregnancy interval) and to determine, as definitively as possible, the likely cause of that preterm birth
- Women who have a singleton pregnancy and a history of either spontaneous preterm labor or preterm rupture of membranes can be offered progesterone supplementation as intramuscular 17a-hydroxyprogesterone or a vaginal preparation to reduce their risk of preterm birth
- Women who have an asymptomatic shortening of the cervix, as measured on transvaginal US at 18 to 24 weeks’ gestation, can be offered vaginal progesterone to reduce their risk of preterm birth
- Women who have a history of preterm birth and cervical shortening may see a reduction in their risk of preterm birth from cerclage placement
- The use of screening for cervical length or progesterone supplementation, or both, in a multiple gestation pregnancy are not recommended because their benefit in this population has not been demonstrated.
Until we fully understand the various etiologic pathways of spontaneous preterm birth, we won’t have a one-size-fits-all solution to this major cause of perinatal morbidity and mortality.
We want to hear from you! Tell us what you think.
1. Scott JR. Vaginal birth after cesarean delivery; a common sense approach. Obstet Gynecol. 2011;118(2 Pt 1):342-350.
2. American College of Obstetricians and Gynecologists Committee on Obstetric Practice. ACOG Committee Opinion No. 476: Planned home birth. Obstet Gynecol. 2011;117(2 Pt 1):425-428.
3. American College of Obstetricians and Gynecologists Committee on Practice Bulletins. ACOG Practice Bulletin No. 76: Postpartum hemorrhage. Obstet Gynecol. 2006;108(4):1039-1047.
4. Berghella V, Baxter JK, Hendrix NW. Cervical assessment by ultrasound for preventing preterm delivery (Review). Cochrane Database Syst Rev. 2009;(3):CD007235.-
5. da Fonseca EB, Bittar RE, Carvalho MH, Zugaib M. Prophylactic administration of progesterone by vaginal suppository to reduce the incidence of spontaneous preterm birth in women at increased risk: a randomized placebo-controlled double-blind study. Am J Obstet Gynecol. 2003;188(2):419-424.
6. Meis PJ, Klebanoff M, Thom E, et al. National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N Engl J Med. 2003;348(24):2379-2385.
7. American College of Obstetricians and Gynecologists Committee on Obstetric Practice. ACOG Committee Opinion No. 419: Use of progesterone to reduce preterm birth. Obstet Gynecol. 2008;112(4):963-935.
8. Owen J, Yost N, Berghella V, et al:. National Institute of Child Health and Human Development, Maternal-Fetal Medicine Units Network. Mid-trimester endovaginal sonography in women at high risk for spontaneous preterm birth. JAMA. 2001;286(11):1340-1348.
9. Durnwald CP, Walker H, Lundy JC, Iams JD. Rates of recurrent preterm birth by obstetrical history and cervical length. Am J Obstet Gynecol. 2005;193(3):1170-1174.
10. Hassan SS, Romero R, Vidyadhari D, et al. PREGNANT Trial. Vaginal progesterone reduces the rate of preterm birth in women with a sonographic short cervix: a multicenter randomized, double-blind, placebo-controlled trial. Ultrasound Obstet Gynecol. 2011;38(1):18-31.
11. Berghella V, Rafael TJ, Szychowski M, Rust OA, Owen J. Cerclage for short cervix on ultrasonography in women with singleton gestations and previous preterm birth. Obstet Gynecol. 2011;117(3):663-771
1. Scott JR. Vaginal birth after cesarean delivery; a common sense approach. Obstet Gynecol. 2011;118(2 Pt 1):342-350.
2. American College of Obstetricians and Gynecologists Committee on Obstetric Practice. ACOG Committee Opinion No. 476: Planned home birth. Obstet Gynecol. 2011;117(2 Pt 1):425-428.
3. American College of Obstetricians and Gynecologists Committee on Practice Bulletins. ACOG Practice Bulletin No. 76: Postpartum hemorrhage. Obstet Gynecol. 2006;108(4):1039-1047.
4. Berghella V, Baxter JK, Hendrix NW. Cervical assessment by ultrasound for preventing preterm delivery (Review). Cochrane Database Syst Rev. 2009;(3):CD007235.-
5. da Fonseca EB, Bittar RE, Carvalho MH, Zugaib M. Prophylactic administration of progesterone by vaginal suppository to reduce the incidence of spontaneous preterm birth in women at increased risk: a randomized placebo-controlled double-blind study. Am J Obstet Gynecol. 2003;188(2):419-424.
6. Meis PJ, Klebanoff M, Thom E, et al. National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N Engl J Med. 2003;348(24):2379-2385.
7. American College of Obstetricians and Gynecologists Committee on Obstetric Practice. ACOG Committee Opinion No. 419: Use of progesterone to reduce preterm birth. Obstet Gynecol. 2008;112(4):963-935.
8. Owen J, Yost N, Berghella V, et al:. National Institute of Child Health and Human Development, Maternal-Fetal Medicine Units Network. Mid-trimester endovaginal sonography in women at high risk for spontaneous preterm birth. JAMA. 2001;286(11):1340-1348.
9. Durnwald CP, Walker H, Lundy JC, Iams JD. Rates of recurrent preterm birth by obstetrical history and cervical length. Am J Obstet Gynecol. 2005;193(3):1170-1174.
10. Hassan SS, Romero R, Vidyadhari D, et al. PREGNANT Trial. Vaginal progesterone reduces the rate of preterm birth in women with a sonographic short cervix: a multicenter randomized, double-blind, placebo-controlled trial. Ultrasound Obstet Gynecol. 2011;38(1):18-31.
11. Berghella V, Rafael TJ, Szychowski M, Rust OA, Owen J. Cerclage for short cervix on ultrasonography in women with singleton gestations and previous preterm birth. Obstet Gynecol. 2011;117(3):663-771