User login
Safe Use of Buprenorphine/Naloxone in a Veteran With Acute Hepatitis C Virus Infection
September 2011

Disaster Preparedness 10 Years After 9/11:
The Experts Weigh In
Current Management Options for Osteonecrosis of the Femoral Head: Part 1, Diagnosis and Nonoperative Management
UPDATE ON SEXUAL DYSFUNCTION
- Cancer survivors have many complaints not addressed by their physicians
Janelle Yates, Senior Editor (Web Exclusive, July 2011) - How to talk to patients about sex
Barbara S. Levy, MD (Web Audio, September 2010) - Sexual dysfunction: What can you do for your patients?
Barbara S. Levy, MD (Update, September 2010)
Sexual dysfunction is common among women in the United States. One recent study put the prevalence of distressing sexual dysfunction at 22.2%.1 When cancer enters the picture, that percentage rises—dramatically. A 2010 survey from the Lance Armstrong Foundation found that 46% of people affected by cancer report problems with sex after treatment.2
In this article, I highlight three recent studies that explore the sexual effects of cancer and its treatment:
- a prospective cohort study showing that a majority of women treated for breast cancer experience sexual dysfunction afterward
- two longitudinal studies of women affected by gynecologic cancer, which show significant disruption of sexual function in the short and long term.
Sexual function deteriorates in many women after they are treated for breast Ca
Panjari M, Bell RJ, Davis SR. Sexual function after breast cancer. J Sex Med. 2011;8(1):294–302.
According to this prospective cohort study from Australia, a majority of women report significant sexual dysfunction after treatment for breast cancer—even when their sexual function was good, and satisfying, at the time of diagnosis (TABLE).
The Health and Wellbeing after Breast Cancer Study enrolled 1,684 Australian women within 12 months of their first diagnosis of invasive breast cancer. Each woman completed a questionnaire at the time of enrollment, and will complete annual follow-up questionnaires for 5 years to assess the impact of invasive breast cancer on physical, psychological, and socioeconomic wellbeing. Embedded within the 12-month questionnaire was the validated Menopause-Specific Quality of Life Questionnaire (MENQOL), which was used in this study to explore the sexual consequences of the diagnosis and treatment of invasive breast cancer.
Of the initial cohort, 1,011 women completed the 12-month questionnaire. These were women younger than 70 years who had a sexual partner and no evidence of active breast cancer. The authors describe the women in this cohort as representative of all women in Victoria, Australia, who have a new diagnosis of invasive breast cancer, in regard to both age (mean, 59 ± 11 years) and the stage of tumor (stage I, 48%; stage II–IV, 52%) at diagnosis.
Of this group, 70% were treated with lumpectomy and radiation therapy, and 30% were treated with mastectomy (2.6% with bilateral mastectomy). Of the women who underwent mastectomy, 9.6% had reconstructive surgery during the first year after diagnosis.
Forty-nine percent of women were treated with tamoxifen, and 28.2% were treated with an aromatase inhibitor.
After breast Ca, women experience low desire and less frequent sexual activity – as well as distress over both outcomes
| Symptom | Yes | No |
|---|---|---|
| Decreased desire | 71.7% | 19.7% |
| Decreased sexual activity | 72.5% | 21.1% |
| Distressed by sexual function | 49.1% | 8.1% |
| Seeking increase in desire | 64.1% | 19.9% |
| Source: Panjari et al. | ||
More than two thirds of women reported sexual dysfunction 12 months after treatment
At baseline, 83% of women described their prediagnosis sexual function as good and satisfying. Twelve months later, 70% reported significant sexual dysfunction, and 77% reported vasomotor symptoms.
Women who reported new-onset sexual dysfunction were more likely to:
- have become menopausal since diagnosis
- experience hot flashes or night sweats
- be treated with an aromatase inhibitor.
There was no association between sexual dysfunction and stage of disease at diagnosis; type of surgery (lumpectomy or mastectomy); breast reconstruction; lymphedema; or axillary dissection.
Vasomotor symptoms in women taking endocrine therapy were associated with sexual dysfunction
Further analysis demonstrated that, among women who experienced vasomotor symptoms, those taking an aromatase inhibitor were more than three times as likely to report sexual dysfunction (odds ratio [OR], 3.49; 95% confidence interval [CI], 1.72–7.09), compared with women who were not on endocrine therapy—and those taking tamoxifen were almost twice as likely to report sexual dysfunction (OR, 1.73; 95% CI, 1.04–2.89). Chemotherapy was not independently related to sexual dysfunction.
In summary: 70% of women who were free of breast cancer 1 year after enrollment reported bothersome sexual consequences of their disease and its treatment; 77% reported vasomotor symptoms. Women who were rendered menopausal and those who experienced vasomotor symptoms while taking an aromatase inhibitor were at high risk of sexual dysfunction.
Be aware of the side effects of breast cancer and its treatment, and not only prepare your patients for the likely consequences but also make yourself knowledgeable about strategies to ameliorate their vaginal dryness and to improve elasticity and arousal for them.
Proactive stretching, use of vaginal dilators and topical oils, and, most important, psychological strategies to help your patients and their partners adjust to the inevitable physical changes will go a long way toward improving their sexual experiences.
Katie* and Julie* tell typical stories of deep dissatisfaction with the health system after their cancer treatment
Katie: “I wasn’t prepared”
When my doctor told me I had locally advanced breast cancer 3 years ago, when I was 50, I wasn’t that surprised by the cancer diagnosis (I have a strong family history of breast cancer)—only by the fact that the tumor had developed so fast since my previous mammogram 15 months earlier. As treatment, I underwent neoadjuvant chemotherapy and bilateral mastectomy (I had the unaffected breast removed as a preventive measure). I also had breast reconstruction and started taking an aromatase inhibitor.
At the time of my diagnosis, I lost all desire for sexual intimacy—no big surprise there. But even after my treatment was over, my desire did not return. Part of the problem was the fact that chemotherapy rendered me menopausal, and the aromatase inhibitor I was taking compounded the menopausal experience. Quite suddenly, I was experiencing hot flashes, vaginal dryness and itching, pain during intercourse, severe bone and joint aches, weight gain (particularly around my abdomen), and general lethargy.
No one in my family had ever mentioned these effects of cancer. And none of my doctors prepared me, either. In fact, when I raised the subject, they seemed genuinely surprised! They offered no remedy other than a recommendation to apply a “moisturizer”—but they gave no details about what kind or how to use it. My oncologist did say that local estrogen would help relieve the pain of intercourse—but then she recommended strongly against it because my cancer was hormone-receptor positive.
My plastic surgeon did a much better job of explaining the effects and outcome of reconstruction than any of my other physicians, including my ObGyn, did of preparing me for menopause and sexual dysfunction.
All of my physicians strike me as caring, sensitive people, but their underlying attitude, as I perceive it, is that I should be grateful to be alive. In their view, it seems, enjoyment of sex is icing on the cake and, quite frankly, I am plenty lucky to have the cake. My oncologist even told me to let her know if I started “feeling better and having fewer hot flashes” so that she could perform ovarian ablation (and start the whole cycle over again). I was struck by how matter of factly she gave this advice, as though quality of life counts for nothing.
Three years into my postcancer life, I can say I have “adjusted” to my problems rather than overcome them. I am still taking an aromatase inhibitor. Sex is still slightly painful; I still struggle with vaginal dryness; and I sometimes feel like an old woman because of my bone and joint stiffness and pain. I did find out about an over-the-counter vaginal suppository, made with vitamin E and coconut oils, from another breast cancer survivor. And I switched from one aromatase inhibitor to another in an attempt to alleviate my achy joints. It helped.
I am grateful for my life—very much so—and for the expertise of my physicians, who helped to save it. But I wish they had prepared me better for the aftermath of cancer treatment. And I wish there were more remedies for women like me, who cannot take hormones.
Julie: “I’m on my own”
It was more than a surprise when my new doctor told me I had cancer. Until then, I had avoided doctors. That attitude can mean a premature death sentence when it comes to cervical cancer. It was a pretty awful realization that I could have avoided the drastic measures it took to save my life if I had just gotten annual Pap smears and exams. I was 39 at the time of diagnosis.
But after all the surgery and chemo and radiation were finished, the message I received was essentially: “OK, you’re good, for now. Just come back every few months for a check-up.”
What about the aftermath of all that treatment? What about the other aspects of the experience? I found that my doctors had very little to offer outside of surgery and drugs and the quick advice to get counseling or some other support services.
“I’m on my own” is what I’ve been telling concerned folks who ask how I’m doing. I am truly grateful for the skill, medicine, and machinery that made the killing of an invasive tumor possible. But I’m on my own when it comes to finding or inventing ways to cope with the new challenges of a pelvic area damaged by radiation and detoxing from the heavy metal—platinum—that was an ingredient in the chemo I received.
My partner and I have had to be persistently creative, careful, delicate, uncritical, and extremely patient with each other to bring about the return of a “normal” sex life. We have been successful, for the most part, but there is also a slightly new definition of what “normal” is for us. Our latest triumph is that we no longer have to use copious amounts of lubricant to engage in intercourse. Sex is no longer painful, as the vaginal tissues have been slowly, patiently engaged on a regularly scheduled basis. Can you imagine sex on a schedule? Neither could I, but that is what we found worked from as early as 1 week postradiation. As it turns out, this was good advice—really, the only advice I got when it came to the practicalities of restoring function, but it required a fair amount of tweaking and personalization as well.
Another big change in perception that I had to accept as part of my new norm is learning to talk about my most personal areas in a matter of fact way.
The cancer conveyor-belt approach to treatment is a very streamlined, well-run system. I’ve been impressed with the expertise, efficiency, and demeanor of all the professionals I have encountered. Everyone—even receptionists—has been helpful and empathetic, especially my own ObGyn, who has hugged me and cried with me and offered to put herself on the line for me and speak out to the media when I had no health insurance. But for most patients post-treatment, we figuratively walk off a cliff and find ourselves in new territory without any network or structure like we experienced during the “war” on our cancer. This new territory is a place of possibility within the health-care field—one I hope is developing now.

Dr. Barbara S. Levy asks: How do we respond? We physicians are so focused on treating or curing disease that we often lose sight of the woman who has the disease.
Katie’s case is much too common. Women are often reluctant to address their sexuality with us—especially when we have been dismissive. We must recognize this important aspect of quality of life and relationships and be prepared to raise the issue with our patients before they begin therapy.
Educating ourselves and then our patients about strategies to reduce the impact of treatment and menopause on sexual function is the first step. Acknowledging and validating their concerns and being able to offer practical steps to preserve healthy sexual function is something, I think, that all ObGyns should be able to do. Strategies to maintain vulvovaginal elasticity include avoidance of soaps and drying chemicals, daily perineal and vaginal stretching—either manually, with a dilator, or via frequent sexual intercourse. And topical lubricants and moisturizing agents should be recommended to maintain vaginal pH and reduce the dryness, itching, and overall dysesthesia.
As the studies highlighted in this Update on Sexual Dysfunction demonstrate, the sexual consequences of radical hysterectomy are significant. Julie also became menopausal as a result of treatment—yet no one prepared her for the symptoms she would experience, and no member of her treatment team helped her understand the likely impact of therapy. Radiation oncologists do address the need for vaginal dilators or daily intercourse to maintain depth and caliber of the vagina during and after therapy, but they are less likely to prepare a patient for menopausal symptoms.
If our patients are to have an optimal experience, we need to provide coordinated, cross-disciplinary care that includes management not only of immediate side effects of treatment but also psychosocial and long-term hormonal and sexual sequelae of therapy. Julie charted her own course with the help of a very dedicated and sensitive partner to successfully overcome the negative effects of radiation, chemotherapy, surgery, and menopause on her sexuality.
Gynecologic cancer disrupts sexual function, over the short term and the long term
Jensen PT, Groenvold M, Klee MC, Thranov I, Petersen MA, Machin D. Early-stage cervical carcinoma, radical hysterectomy, and sexual function: a longitudinal study. Cancer. 2004;100(1):97–106.
Vaz AF, Pinto-Neto AM, Conde DM, et al. Quality of life and menopausal and sexual symptoms in gynecologic cancer survivors: a cohort study. Menopause. 2011;18(6):662–669.
These two studies explored sexual function after treatment for gynecologic cancer. The investigators found significant disruption of function.
Jensen et al: Radical hysterectomy for cervical Ca damages sexual function significantly
This was a prospective cohort study of 173 women who had early-stage cervical carcinoma and who underwent radical hysterectomy with pelvic lymphadenectomy (all of them node-negative). A validated questionnaire was administered six times, from 5 weeks to 24 months after surgery. An age-matched group of women without cancer was used for comparison. At the 12-month follow-up, patients were asked to report their sexual function at baseline and compare it to their current status. Overall, the women had a higher level of dissatisfaction with their sexual experiences 12 months after surgery than at baseline.
Details of the study
Women in the Jensen study were 23 to 75 years old (median age, 42.7 years), and 93% were sexually active at the time of diagnosis, reporting an average of one to two sexual activities in a week. Forty-six women (25%) were postmenopausal at diagnosis, compared with 34% of the control group. Only 8% of patients were using HT at study entry, compared with 25% of women in the control group. By 12 months after surgery, however, 25% of gynecologic cancer patients were taking systemic HT.
Findings included low libido and other ills
Severe lack of lubrication and low or no sexual desire were reported by cancer patients throughout the first 2 years after surgery. Patients also reported severe problems achieving orgasm as long as 6 months after surgery, as well as reduced vaginal size; both problems rendered their sexual experiences unsatisfactory. Nevertheless, 40% of patients reported at least some sexual activity by 5 weeks after surgery. By 6 months after surgery, there were almost as many sexually active women among the patient group as there were in the control group. However, at 18 months after treatment, patients reported less interest in intimacy—among both themselves and their partners—than among women in the control group. Overall, women treated for cervical cancer had a higher level of dissatisfaction with their sexual experiences 12 months after surgery than they did before diagnosis.
Although 91% of women who were sexually active before surgery resumed intercourse within 12 months, the frequency of sexual activity declined from one to two times per week to three to four times per month. Major long-term changes occurred in regard to libido (interest in sexual relations), arousal (vaginal lubrication), and vaginal size. Although dyspareunia was a significant problem 5 weeks to 3 months after surgery, it resolved within 1 to 2 years.
Jensen and colleagues concluded that radical hysterectomy for treatment of early-stage cervical cancer has significant negative effects on sexual function in the short and long term. They postulated a neurogenic basis for the sexual complaints and discussed both histologic and clinical studies to support this hypothesis.
They also emphasized the need to discuss the risks and management of sexual dysfunction with patients before and after surgery. Better management of the psychosocial consequences of a cancer diagnosis and the physical effects of radical hysterectomy may help avoid the negative experiences that were reported in this study.
Vaz et al: Rate of dyspareunia was high among women treated for endometrial or cervical Ca
Investigators followed 107 women from initial consultation for radiation therapy through 3 years post-treatment. Although a significant percentage (50%) of the cohort was lost to follow-up—many due to death or tumor recurrence—50% of those who remained reported dyspareunia 3 years after radiotherapy.
Women in this study were 21 to 75 years old (median age, 60 years) and had cervical or endometrial carcinoma. Eighty-nine women (83%) received external pelvic radiation as well as brachytherapy. Before beginning radiation therapy, 37.4% of the cohort underwent surgery for treatment of their cancer. Sixty-four percent of the cohort had stage III or IV disease.
At enrollment, 50% of women reported having a life partner, 82% were postmenopausal, and 11.2% were taking HT. However, only 21.5% of women reported sexual activity. The authors opine that this low rate of sexual activity may have been due to recent surgery, bleeding, or pain related to cancer.
All women were offered “standard” interventions for their dyspareunia, including the use of vaginal dilators twice daily for 2 years, as well as the use of vaginal lubricants. Patients who experienced menopausal symptoms—hot flashes, decreased libido, dyspareunia—were referred to a menopause outpatient clinic.
Before treatment, 20% of women reported dyspareunia. Three years later, 44% of patients reported sexual activity, but 50% had dyspareunia. Twenty-one percent reported lower sexual interest relative to baseline, 8% reported vaginal dryness, and 21% reported vasomotor symptoms. Although there was a trend toward increasing sexual activity with decreasing vaginal dryness, the rise in dyspareunia from 20% to 50% over 3 years is troubling.
Radical hysterectomy and radiation therapy to the pelvis cause neurovascular disruption and sexual consequences quite similar to those found after radical prostate surgery. Sexual arousal and orgasm are dependent on both the parasympathetic and sympathetic nerves supplying the pelvis. these nerves are disrupted in Frankenhauser’s plexus during the parametrial dissection of radical hysterectomy and lie clearly within the radiation treatment field.
Dilators and lubricants may be useful in minimizing actual shrinkage of the vagina. However, the elasticity of vaginal tissues, vasodilation during arousal, and transudation across the vaginal wall may all be lost or significantly compromised.
Advise your patients of the potential for sexual side effects of cervical and endometrial cancer treatment before they undergo that therapy. Proactive management of some of the expected problems, such as reduced elasticity and lubrication, as well as treatment for arousal dysfunction (perhaps with the same pharmacotherapeutic agents that provide improvement for many men after radical prostatectomy), may help your patients avoid the distress and disappointment they often experience after successful treatment of their cancers.
When a patient has undergone treatment for cancer,
ask about her sexual function
A symposium on sexual health yields recommendations for your broader care of cancer patients
How likely are you to encounter a cancer survivor in your practice?
Very.
According to the Centers for Disease Control and Prevention (CDC), there were 6.3 million female cancer survivors in the United States as of 2007—and that number has likely increased by a million or more.3 In fact, the number of cancer survivors is expected to double by 2016.
How likely is that cancer survivor to have sexual dysfunction?
Highly.
According to a 2010 survey by the Lance Armstrong Foundation, 46% of cancer survivors report problems with sexual functioning after treatment—and that’s probably a conservative figure, given that 64% of all people with cancer have a malignancy that directly affects sexual organs.2
One more question for you to ponder: How likely is the cancer patient’s sexual dysfunction to go unaddressed?
Extremely.
According to speakers at the Cancer Survivorship and Sexual Health Symposium, held June 17–19, 2011, in Washington, DC, cancer survivors are ill prepared for many of the symptoms of sexual dysfunction that develop after treatment, and many physicians fail to address this dimension of their health.
The symposium, sponsored by the International Society for Sexual Medicine and the Sexual Medicine Society of North America, was organized to address these gaps in care. During the 3-day conference, speakers from oncology, gynecology, mental health, urology, and other specialties presented data and described their experience managing cancer patients. They also offered recommendations for clinicians:
- Talk about it. Address the “highly prevalent but commonly ignored” adverse sexual effects of malignancy and its treatment. Ask: “How has cancer affected your sex life?”
- Try to prevent it. Consider nerve-sparing strategies during radical hysterectomy, radical trachelectomy, and clitoral preservation, which may lead to improved sexual function
- Encourage and support use of dilators. Advise women who have gynecologic cancer to use dilators to maintain vaginal patency, and be aware that compliance is linked to support from a health-care provider
- Encourage sexual activity, which can help preserve function
- Consider local estrogen. When it is appropriate, prescribe vaginal estrogen, which is minimally absorbed, to reduce vaginal symptoms of menopause. (The safety of local estrogen remains in question for women who have breast cancer.)
- Check for problems at each follow-up appointment, and be prepared to explain function and treatment options more than once
- Promote female genital blood flow. For example, it may be appropriate to begin sexual rehabilitation, such as use of vaginal dilators, during treatment
- Consider referral to a sexual rehabilitation program that includes medical and psychological approaches
- Build a network of psychologists, sex therapists, and other professionals who can assist you in managing your patients’ complaints.
“Discomfort around human sexuality is the main reason the issue doesn’t get raised by health-care providers,” said symposium speaker Sharon L. Bober, PhD, of the Dana-Farber Cancer Institute in Boston. “No one wants to initiate the conversation.” Dr. Bober emphasized the importance of asking about sexual function when a cancer survivor presents for care. “A majority of cancer patients in the community don’t hear this question from their providers.”
—Janelle Yates, Senior Editor
We want to hear from you! Tell us what you think.
1. Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol. 2008;112(5):970-978.
2. Rechis R, Boerner L. How cancer has affected post-treatment survivors: a LIVESTRONG report. Austin, Tex: Lance Armstrong Foundation 2011;13-
3. Centers for Disease Control and Prevention. Cancer survivors—United States, 2007. MMWR. 2011;60(9):269-272
| ObGyn Stacy T. Lindau, MD, shares her expertise in managing the sexual concerns of cancer survivors |

Barbara S. Levy, MD
Dr. Levy is Medical Director of Women’s Health, Franciscan Health System, Tacoma, Wash. She serves on the OBG Management Board of Editors.
The author reports no financial relationships relevant to this article.
| ObGyn Stacy T. Lindau, MD, shares her expertise in managing the sexual concerns of cancer survivors |
Barbara S. Levy, MD
Dr. Levy is Medical Director of Women’s Health, Franciscan Health System, Tacoma, Wash. She serves on the OBG Management Board of Editors.
The author reports no financial relationships relevant to this article.
| ObGyn Stacy T. Lindau, MD, shares her expertise in managing the sexual concerns of cancer survivors |
Barbara S. Levy, MD
Dr. Levy is Medical Director of Women’s Health, Franciscan Health System, Tacoma, Wash. She serves on the OBG Management Board of Editors.
The author reports no financial relationships relevant to this article.
- Cancer survivors have many complaints not addressed by their physicians
Janelle Yates, Senior Editor (Web Exclusive, July 2011) - How to talk to patients about sex
Barbara S. Levy, MD (Web Audio, September 2010) - Sexual dysfunction: What can you do for your patients?
Barbara S. Levy, MD (Update, September 2010)
Sexual dysfunction is common among women in the United States. One recent study put the prevalence of distressing sexual dysfunction at 22.2%.1 When cancer enters the picture, that percentage rises—dramatically. A 2010 survey from the Lance Armstrong Foundation found that 46% of people affected by cancer report problems with sex after treatment.2
In this article, I highlight three recent studies that explore the sexual effects of cancer and its treatment:
- a prospective cohort study showing that a majority of women treated for breast cancer experience sexual dysfunction afterward
- two longitudinal studies of women affected by gynecologic cancer, which show significant disruption of sexual function in the short and long term.
Sexual function deteriorates in many women after they are treated for breast Ca
Panjari M, Bell RJ, Davis SR. Sexual function after breast cancer. J Sex Med. 2011;8(1):294–302.
According to this prospective cohort study from Australia, a majority of women report significant sexual dysfunction after treatment for breast cancer—even when their sexual function was good, and satisfying, at the time of diagnosis (TABLE).
The Health and Wellbeing after Breast Cancer Study enrolled 1,684 Australian women within 12 months of their first diagnosis of invasive breast cancer. Each woman completed a questionnaire at the time of enrollment, and will complete annual follow-up questionnaires for 5 years to assess the impact of invasive breast cancer on physical, psychological, and socioeconomic wellbeing. Embedded within the 12-month questionnaire was the validated Menopause-Specific Quality of Life Questionnaire (MENQOL), which was used in this study to explore the sexual consequences of the diagnosis and treatment of invasive breast cancer.
Of the initial cohort, 1,011 women completed the 12-month questionnaire. These were women younger than 70 years who had a sexual partner and no evidence of active breast cancer. The authors describe the women in this cohort as representative of all women in Victoria, Australia, who have a new diagnosis of invasive breast cancer, in regard to both age (mean, 59 ± 11 years) and the stage of tumor (stage I, 48%; stage II–IV, 52%) at diagnosis.
Of this group, 70% were treated with lumpectomy and radiation therapy, and 30% were treated with mastectomy (2.6% with bilateral mastectomy). Of the women who underwent mastectomy, 9.6% had reconstructive surgery during the first year after diagnosis.
Forty-nine percent of women were treated with tamoxifen, and 28.2% were treated with an aromatase inhibitor.
After breast Ca, women experience low desire and less frequent sexual activity – as well as distress over both outcomes
| Symptom | Yes | No |
|---|---|---|
| Decreased desire | 71.7% | 19.7% |
| Decreased sexual activity | 72.5% | 21.1% |
| Distressed by sexual function | 49.1% | 8.1% |
| Seeking increase in desire | 64.1% | 19.9% |
| Source: Panjari et al. | ||
More than two thirds of women reported sexual dysfunction 12 months after treatment
At baseline, 83% of women described their prediagnosis sexual function as good and satisfying. Twelve months later, 70% reported significant sexual dysfunction, and 77% reported vasomotor symptoms.
Women who reported new-onset sexual dysfunction were more likely to:
- have become menopausal since diagnosis
- experience hot flashes or night sweats
- be treated with an aromatase inhibitor.
There was no association between sexual dysfunction and stage of disease at diagnosis; type of surgery (lumpectomy or mastectomy); breast reconstruction; lymphedema; or axillary dissection.
Vasomotor symptoms in women taking endocrine therapy were associated with sexual dysfunction
Further analysis demonstrated that, among women who experienced vasomotor symptoms, those taking an aromatase inhibitor were more than three times as likely to report sexual dysfunction (odds ratio [OR], 3.49; 95% confidence interval [CI], 1.72–7.09), compared with women who were not on endocrine therapy—and those taking tamoxifen were almost twice as likely to report sexual dysfunction (OR, 1.73; 95% CI, 1.04–2.89). Chemotherapy was not independently related to sexual dysfunction.
In summary: 70% of women who were free of breast cancer 1 year after enrollment reported bothersome sexual consequences of their disease and its treatment; 77% reported vasomotor symptoms. Women who were rendered menopausal and those who experienced vasomotor symptoms while taking an aromatase inhibitor were at high risk of sexual dysfunction.
Be aware of the side effects of breast cancer and its treatment, and not only prepare your patients for the likely consequences but also make yourself knowledgeable about strategies to ameliorate their vaginal dryness and to improve elasticity and arousal for them.
Proactive stretching, use of vaginal dilators and topical oils, and, most important, psychological strategies to help your patients and their partners adjust to the inevitable physical changes will go a long way toward improving their sexual experiences.
Katie* and Julie* tell typical stories of deep dissatisfaction with the health system after their cancer treatment
Katie: “I wasn’t prepared”
When my doctor told me I had locally advanced breast cancer 3 years ago, when I was 50, I wasn’t that surprised by the cancer diagnosis (I have a strong family history of breast cancer)—only by the fact that the tumor had developed so fast since my previous mammogram 15 months earlier. As treatment, I underwent neoadjuvant chemotherapy and bilateral mastectomy (I had the unaffected breast removed as a preventive measure). I also had breast reconstruction and started taking an aromatase inhibitor.
At the time of my diagnosis, I lost all desire for sexual intimacy—no big surprise there. But even after my treatment was over, my desire did not return. Part of the problem was the fact that chemotherapy rendered me menopausal, and the aromatase inhibitor I was taking compounded the menopausal experience. Quite suddenly, I was experiencing hot flashes, vaginal dryness and itching, pain during intercourse, severe bone and joint aches, weight gain (particularly around my abdomen), and general lethargy.
No one in my family had ever mentioned these effects of cancer. And none of my doctors prepared me, either. In fact, when I raised the subject, they seemed genuinely surprised! They offered no remedy other than a recommendation to apply a “moisturizer”—but they gave no details about what kind or how to use it. My oncologist did say that local estrogen would help relieve the pain of intercourse—but then she recommended strongly against it because my cancer was hormone-receptor positive.
My plastic surgeon did a much better job of explaining the effects and outcome of reconstruction than any of my other physicians, including my ObGyn, did of preparing me for menopause and sexual dysfunction.
All of my physicians strike me as caring, sensitive people, but their underlying attitude, as I perceive it, is that I should be grateful to be alive. In their view, it seems, enjoyment of sex is icing on the cake and, quite frankly, I am plenty lucky to have the cake. My oncologist even told me to let her know if I started “feeling better and having fewer hot flashes” so that she could perform ovarian ablation (and start the whole cycle over again). I was struck by how matter of factly she gave this advice, as though quality of life counts for nothing.
Three years into my postcancer life, I can say I have “adjusted” to my problems rather than overcome them. I am still taking an aromatase inhibitor. Sex is still slightly painful; I still struggle with vaginal dryness; and I sometimes feel like an old woman because of my bone and joint stiffness and pain. I did find out about an over-the-counter vaginal suppository, made with vitamin E and coconut oils, from another breast cancer survivor. And I switched from one aromatase inhibitor to another in an attempt to alleviate my achy joints. It helped.
I am grateful for my life—very much so—and for the expertise of my physicians, who helped to save it. But I wish they had prepared me better for the aftermath of cancer treatment. And I wish there were more remedies for women like me, who cannot take hormones.
Julie: “I’m on my own”
It was more than a surprise when my new doctor told me I had cancer. Until then, I had avoided doctors. That attitude can mean a premature death sentence when it comes to cervical cancer. It was a pretty awful realization that I could have avoided the drastic measures it took to save my life if I had just gotten annual Pap smears and exams. I was 39 at the time of diagnosis.
But after all the surgery and chemo and radiation were finished, the message I received was essentially: “OK, you’re good, for now. Just come back every few months for a check-up.”
What about the aftermath of all that treatment? What about the other aspects of the experience? I found that my doctors had very little to offer outside of surgery and drugs and the quick advice to get counseling or some other support services.
“I’m on my own” is what I’ve been telling concerned folks who ask how I’m doing. I am truly grateful for the skill, medicine, and machinery that made the killing of an invasive tumor possible. But I’m on my own when it comes to finding or inventing ways to cope with the new challenges of a pelvic area damaged by radiation and detoxing from the heavy metal—platinum—that was an ingredient in the chemo I received.
My partner and I have had to be persistently creative, careful, delicate, uncritical, and extremely patient with each other to bring about the return of a “normal” sex life. We have been successful, for the most part, but there is also a slightly new definition of what “normal” is for us. Our latest triumph is that we no longer have to use copious amounts of lubricant to engage in intercourse. Sex is no longer painful, as the vaginal tissues have been slowly, patiently engaged on a regularly scheduled basis. Can you imagine sex on a schedule? Neither could I, but that is what we found worked from as early as 1 week postradiation. As it turns out, this was good advice—really, the only advice I got when it came to the practicalities of restoring function, but it required a fair amount of tweaking and personalization as well.
Another big change in perception that I had to accept as part of my new norm is learning to talk about my most personal areas in a matter of fact way.
The cancer conveyor-belt approach to treatment is a very streamlined, well-run system. I’ve been impressed with the expertise, efficiency, and demeanor of all the professionals I have encountered. Everyone—even receptionists—has been helpful and empathetic, especially my own ObGyn, who has hugged me and cried with me and offered to put herself on the line for me and speak out to the media when I had no health insurance. But for most patients post-treatment, we figuratively walk off a cliff and find ourselves in new territory without any network or structure like we experienced during the “war” on our cancer. This new territory is a place of possibility within the health-care field—one I hope is developing now.
Dr. Barbara S. Levy asks: How do we respond? We physicians are so focused on treating or curing disease that we often lose sight of the woman who has the disease.
Katie’s case is much too common. Women are often reluctant to address their sexuality with us—especially when we have been dismissive. We must recognize this important aspect of quality of life and relationships and be prepared to raise the issue with our patients before they begin therapy.
Educating ourselves and then our patients about strategies to reduce the impact of treatment and menopause on sexual function is the first step. Acknowledging and validating their concerns and being able to offer practical steps to preserve healthy sexual function is something, I think, that all ObGyns should be able to do. Strategies to maintain vulvovaginal elasticity include avoidance of soaps and drying chemicals, daily perineal and vaginal stretching—either manually, with a dilator, or via frequent sexual intercourse. And topical lubricants and moisturizing agents should be recommended to maintain vaginal pH and reduce the dryness, itching, and overall dysesthesia.
As the studies highlighted in this Update on Sexual Dysfunction demonstrate, the sexual consequences of radical hysterectomy are significant. Julie also became menopausal as a result of treatment—yet no one prepared her for the symptoms she would experience, and no member of her treatment team helped her understand the likely impact of therapy. Radiation oncologists do address the need for vaginal dilators or daily intercourse to maintain depth and caliber of the vagina during and after therapy, but they are less likely to prepare a patient for menopausal symptoms.
If our patients are to have an optimal experience, we need to provide coordinated, cross-disciplinary care that includes management not only of immediate side effects of treatment but also psychosocial and long-term hormonal and sexual sequelae of therapy. Julie charted her own course with the help of a very dedicated and sensitive partner to successfully overcome the negative effects of radiation, chemotherapy, surgery, and menopause on her sexuality.
Gynecologic cancer disrupts sexual function, over the short term and the long term
Jensen PT, Groenvold M, Klee MC, Thranov I, Petersen MA, Machin D. Early-stage cervical carcinoma, radical hysterectomy, and sexual function: a longitudinal study. Cancer. 2004;100(1):97–106.
Vaz AF, Pinto-Neto AM, Conde DM, et al. Quality of life and menopausal and sexual symptoms in gynecologic cancer survivors: a cohort study. Menopause. 2011;18(6):662–669.
These two studies explored sexual function after treatment for gynecologic cancer. The investigators found significant disruption of function.
Jensen et al: Radical hysterectomy for cervical Ca damages sexual function significantly
This was a prospective cohort study of 173 women who had early-stage cervical carcinoma and who underwent radical hysterectomy with pelvic lymphadenectomy (all of them node-negative). A validated questionnaire was administered six times, from 5 weeks to 24 months after surgery. An age-matched group of women without cancer was used for comparison. At the 12-month follow-up, patients were asked to report their sexual function at baseline and compare it to their current status. Overall, the women had a higher level of dissatisfaction with their sexual experiences 12 months after surgery than at baseline.
Details of the study
Women in the Jensen study were 23 to 75 years old (median age, 42.7 years), and 93% were sexually active at the time of diagnosis, reporting an average of one to two sexual activities in a week. Forty-six women (25%) were postmenopausal at diagnosis, compared with 34% of the control group. Only 8% of patients were using HT at study entry, compared with 25% of women in the control group. By 12 months after surgery, however, 25% of gynecologic cancer patients were taking systemic HT.
Findings included low libido and other ills
Severe lack of lubrication and low or no sexual desire were reported by cancer patients throughout the first 2 years after surgery. Patients also reported severe problems achieving orgasm as long as 6 months after surgery, as well as reduced vaginal size; both problems rendered their sexual experiences unsatisfactory. Nevertheless, 40% of patients reported at least some sexual activity by 5 weeks after surgery. By 6 months after surgery, there were almost as many sexually active women among the patient group as there were in the control group. However, at 18 months after treatment, patients reported less interest in intimacy—among both themselves and their partners—than among women in the control group. Overall, women treated for cervical cancer had a higher level of dissatisfaction with their sexual experiences 12 months after surgery than they did before diagnosis.
Although 91% of women who were sexually active before surgery resumed intercourse within 12 months, the frequency of sexual activity declined from one to two times per week to three to four times per month. Major long-term changes occurred in regard to libido (interest in sexual relations), arousal (vaginal lubrication), and vaginal size. Although dyspareunia was a significant problem 5 weeks to 3 months after surgery, it resolved within 1 to 2 years.
Jensen and colleagues concluded that radical hysterectomy for treatment of early-stage cervical cancer has significant negative effects on sexual function in the short and long term. They postulated a neurogenic basis for the sexual complaints and discussed both histologic and clinical studies to support this hypothesis.
They also emphasized the need to discuss the risks and management of sexual dysfunction with patients before and after surgery. Better management of the psychosocial consequences of a cancer diagnosis and the physical effects of radical hysterectomy may help avoid the negative experiences that were reported in this study.
Vaz et al: Rate of dyspareunia was high among women treated for endometrial or cervical Ca
Investigators followed 107 women from initial consultation for radiation therapy through 3 years post-treatment. Although a significant percentage (50%) of the cohort was lost to follow-up—many due to death or tumor recurrence—50% of those who remained reported dyspareunia 3 years after radiotherapy.
Women in this study were 21 to 75 years old (median age, 60 years) and had cervical or endometrial carcinoma. Eighty-nine women (83%) received external pelvic radiation as well as brachytherapy. Before beginning radiation therapy, 37.4% of the cohort underwent surgery for treatment of their cancer. Sixty-four percent of the cohort had stage III or IV disease.
At enrollment, 50% of women reported having a life partner, 82% were postmenopausal, and 11.2% were taking HT. However, only 21.5% of women reported sexual activity. The authors opine that this low rate of sexual activity may have been due to recent surgery, bleeding, or pain related to cancer.
All women were offered “standard” interventions for their dyspareunia, including the use of vaginal dilators twice daily for 2 years, as well as the use of vaginal lubricants. Patients who experienced menopausal symptoms—hot flashes, decreased libido, dyspareunia—were referred to a menopause outpatient clinic.
Before treatment, 20% of women reported dyspareunia. Three years later, 44% of patients reported sexual activity, but 50% had dyspareunia. Twenty-one percent reported lower sexual interest relative to baseline, 8% reported vaginal dryness, and 21% reported vasomotor symptoms. Although there was a trend toward increasing sexual activity with decreasing vaginal dryness, the rise in dyspareunia from 20% to 50% over 3 years is troubling.
Radical hysterectomy and radiation therapy to the pelvis cause neurovascular disruption and sexual consequences quite similar to those found after radical prostate surgery. Sexual arousal and orgasm are dependent on both the parasympathetic and sympathetic nerves supplying the pelvis. these nerves are disrupted in Frankenhauser’s plexus during the parametrial dissection of radical hysterectomy and lie clearly within the radiation treatment field.
Dilators and lubricants may be useful in minimizing actual shrinkage of the vagina. However, the elasticity of vaginal tissues, vasodilation during arousal, and transudation across the vaginal wall may all be lost or significantly compromised.
Advise your patients of the potential for sexual side effects of cervical and endometrial cancer treatment before they undergo that therapy. Proactive management of some of the expected problems, such as reduced elasticity and lubrication, as well as treatment for arousal dysfunction (perhaps with the same pharmacotherapeutic agents that provide improvement for many men after radical prostatectomy), may help your patients avoid the distress and disappointment they often experience after successful treatment of their cancers.
When a patient has undergone treatment for cancer,
ask about her sexual function
A symposium on sexual health yields recommendations for your broader care of cancer patients
How likely are you to encounter a cancer survivor in your practice?
Very.
According to the Centers for Disease Control and Prevention (CDC), there were 6.3 million female cancer survivors in the United States as of 2007—and that number has likely increased by a million or more.3 In fact, the number of cancer survivors is expected to double by 2016.
How likely is that cancer survivor to have sexual dysfunction?
Highly.
According to a 2010 survey by the Lance Armstrong Foundation, 46% of cancer survivors report problems with sexual functioning after treatment—and that’s probably a conservative figure, given that 64% of all people with cancer have a malignancy that directly affects sexual organs.2
One more question for you to ponder: How likely is the cancer patient’s sexual dysfunction to go unaddressed?
Extremely.
According to speakers at the Cancer Survivorship and Sexual Health Symposium, held June 17–19, 2011, in Washington, DC, cancer survivors are ill prepared for many of the symptoms of sexual dysfunction that develop after treatment, and many physicians fail to address this dimension of their health.
The symposium, sponsored by the International Society for Sexual Medicine and the Sexual Medicine Society of North America, was organized to address these gaps in care. During the 3-day conference, speakers from oncology, gynecology, mental health, urology, and other specialties presented data and described their experience managing cancer patients. They also offered recommendations for clinicians:
- Talk about it. Address the “highly prevalent but commonly ignored” adverse sexual effects of malignancy and its treatment. Ask: “How has cancer affected your sex life?”
- Try to prevent it. Consider nerve-sparing strategies during radical hysterectomy, radical trachelectomy, and clitoral preservation, which may lead to improved sexual function
- Encourage and support use of dilators. Advise women who have gynecologic cancer to use dilators to maintain vaginal patency, and be aware that compliance is linked to support from a health-care provider
- Encourage sexual activity, which can help preserve function
- Consider local estrogen. When it is appropriate, prescribe vaginal estrogen, which is minimally absorbed, to reduce vaginal symptoms of menopause. (The safety of local estrogen remains in question for women who have breast cancer.)
- Check for problems at each follow-up appointment, and be prepared to explain function and treatment options more than once
- Promote female genital blood flow. For example, it may be appropriate to begin sexual rehabilitation, such as use of vaginal dilators, during treatment
- Consider referral to a sexual rehabilitation program that includes medical and psychological approaches
- Build a network of psychologists, sex therapists, and other professionals who can assist you in managing your patients’ complaints.
“Discomfort around human sexuality is the main reason the issue doesn’t get raised by health-care providers,” said symposium speaker Sharon L. Bober, PhD, of the Dana-Farber Cancer Institute in Boston. “No one wants to initiate the conversation.” Dr. Bober emphasized the importance of asking about sexual function when a cancer survivor presents for care. “A majority of cancer patients in the community don’t hear this question from their providers.”
—Janelle Yates, Senior Editor
We want to hear from you! Tell us what you think.
- Cancer survivors have many complaints not addressed by their physicians
Janelle Yates, Senior Editor (Web Exclusive, July 2011) - How to talk to patients about sex
Barbara S. Levy, MD (Web Audio, September 2010) - Sexual dysfunction: What can you do for your patients?
Barbara S. Levy, MD (Update, September 2010)
Sexual dysfunction is common among women in the United States. One recent study put the prevalence of distressing sexual dysfunction at 22.2%.1 When cancer enters the picture, that percentage rises—dramatically. A 2010 survey from the Lance Armstrong Foundation found that 46% of people affected by cancer report problems with sex after treatment.2
In this article, I highlight three recent studies that explore the sexual effects of cancer and its treatment:
- a prospective cohort study showing that a majority of women treated for breast cancer experience sexual dysfunction afterward
- two longitudinal studies of women affected by gynecologic cancer, which show significant disruption of sexual function in the short and long term.
Sexual function deteriorates in many women after they are treated for breast Ca
Panjari M, Bell RJ, Davis SR. Sexual function after breast cancer. J Sex Med. 2011;8(1):294–302.
According to this prospective cohort study from Australia, a majority of women report significant sexual dysfunction after treatment for breast cancer—even when their sexual function was good, and satisfying, at the time of diagnosis (TABLE).
The Health and Wellbeing after Breast Cancer Study enrolled 1,684 Australian women within 12 months of their first diagnosis of invasive breast cancer. Each woman completed a questionnaire at the time of enrollment, and will complete annual follow-up questionnaires for 5 years to assess the impact of invasive breast cancer on physical, psychological, and socioeconomic wellbeing. Embedded within the 12-month questionnaire was the validated Menopause-Specific Quality of Life Questionnaire (MENQOL), which was used in this study to explore the sexual consequences of the diagnosis and treatment of invasive breast cancer.
Of the initial cohort, 1,011 women completed the 12-month questionnaire. These were women younger than 70 years who had a sexual partner and no evidence of active breast cancer. The authors describe the women in this cohort as representative of all women in Victoria, Australia, who have a new diagnosis of invasive breast cancer, in regard to both age (mean, 59 ± 11 years) and the stage of tumor (stage I, 48%; stage II–IV, 52%) at diagnosis.
Of this group, 70% were treated with lumpectomy and radiation therapy, and 30% were treated with mastectomy (2.6% with bilateral mastectomy). Of the women who underwent mastectomy, 9.6% had reconstructive surgery during the first year after diagnosis.
Forty-nine percent of women were treated with tamoxifen, and 28.2% were treated with an aromatase inhibitor.
After breast Ca, women experience low desire and less frequent sexual activity – as well as distress over both outcomes
| Symptom | Yes | No |
|---|---|---|
| Decreased desire | 71.7% | 19.7% |
| Decreased sexual activity | 72.5% | 21.1% |
| Distressed by sexual function | 49.1% | 8.1% |
| Seeking increase in desire | 64.1% | 19.9% |
| Source: Panjari et al. | ||
More than two thirds of women reported sexual dysfunction 12 months after treatment
At baseline, 83% of women described their prediagnosis sexual function as good and satisfying. Twelve months later, 70% reported significant sexual dysfunction, and 77% reported vasomotor symptoms.
Women who reported new-onset sexual dysfunction were more likely to:
- have become menopausal since diagnosis
- experience hot flashes or night sweats
- be treated with an aromatase inhibitor.
There was no association between sexual dysfunction and stage of disease at diagnosis; type of surgery (lumpectomy or mastectomy); breast reconstruction; lymphedema; or axillary dissection.
Vasomotor symptoms in women taking endocrine therapy were associated with sexual dysfunction
Further analysis demonstrated that, among women who experienced vasomotor symptoms, those taking an aromatase inhibitor were more than three times as likely to report sexual dysfunction (odds ratio [OR], 3.49; 95% confidence interval [CI], 1.72–7.09), compared with women who were not on endocrine therapy—and those taking tamoxifen were almost twice as likely to report sexual dysfunction (OR, 1.73; 95% CI, 1.04–2.89). Chemotherapy was not independently related to sexual dysfunction.
In summary: 70% of women who were free of breast cancer 1 year after enrollment reported bothersome sexual consequences of their disease and its treatment; 77% reported vasomotor symptoms. Women who were rendered menopausal and those who experienced vasomotor symptoms while taking an aromatase inhibitor were at high risk of sexual dysfunction.
Be aware of the side effects of breast cancer and its treatment, and not only prepare your patients for the likely consequences but also make yourself knowledgeable about strategies to ameliorate their vaginal dryness and to improve elasticity and arousal for them.
Proactive stretching, use of vaginal dilators and topical oils, and, most important, psychological strategies to help your patients and their partners adjust to the inevitable physical changes will go a long way toward improving their sexual experiences.
Katie* and Julie* tell typical stories of deep dissatisfaction with the health system after their cancer treatment
Katie: “I wasn’t prepared”
When my doctor told me I had locally advanced breast cancer 3 years ago, when I was 50, I wasn’t that surprised by the cancer diagnosis (I have a strong family history of breast cancer)—only by the fact that the tumor had developed so fast since my previous mammogram 15 months earlier. As treatment, I underwent neoadjuvant chemotherapy and bilateral mastectomy (I had the unaffected breast removed as a preventive measure). I also had breast reconstruction and started taking an aromatase inhibitor.
At the time of my diagnosis, I lost all desire for sexual intimacy—no big surprise there. But even after my treatment was over, my desire did not return. Part of the problem was the fact that chemotherapy rendered me menopausal, and the aromatase inhibitor I was taking compounded the menopausal experience. Quite suddenly, I was experiencing hot flashes, vaginal dryness and itching, pain during intercourse, severe bone and joint aches, weight gain (particularly around my abdomen), and general lethargy.
No one in my family had ever mentioned these effects of cancer. And none of my doctors prepared me, either. In fact, when I raised the subject, they seemed genuinely surprised! They offered no remedy other than a recommendation to apply a “moisturizer”—but they gave no details about what kind or how to use it. My oncologist did say that local estrogen would help relieve the pain of intercourse—but then she recommended strongly against it because my cancer was hormone-receptor positive.
My plastic surgeon did a much better job of explaining the effects and outcome of reconstruction than any of my other physicians, including my ObGyn, did of preparing me for menopause and sexual dysfunction.
All of my physicians strike me as caring, sensitive people, but their underlying attitude, as I perceive it, is that I should be grateful to be alive. In their view, it seems, enjoyment of sex is icing on the cake and, quite frankly, I am plenty lucky to have the cake. My oncologist even told me to let her know if I started “feeling better and having fewer hot flashes” so that she could perform ovarian ablation (and start the whole cycle over again). I was struck by how matter of factly she gave this advice, as though quality of life counts for nothing.
Three years into my postcancer life, I can say I have “adjusted” to my problems rather than overcome them. I am still taking an aromatase inhibitor. Sex is still slightly painful; I still struggle with vaginal dryness; and I sometimes feel like an old woman because of my bone and joint stiffness and pain. I did find out about an over-the-counter vaginal suppository, made with vitamin E and coconut oils, from another breast cancer survivor. And I switched from one aromatase inhibitor to another in an attempt to alleviate my achy joints. It helped.
I am grateful for my life—very much so—and for the expertise of my physicians, who helped to save it. But I wish they had prepared me better for the aftermath of cancer treatment. And I wish there were more remedies for women like me, who cannot take hormones.
Julie: “I’m on my own”
It was more than a surprise when my new doctor told me I had cancer. Until then, I had avoided doctors. That attitude can mean a premature death sentence when it comes to cervical cancer. It was a pretty awful realization that I could have avoided the drastic measures it took to save my life if I had just gotten annual Pap smears and exams. I was 39 at the time of diagnosis.
But after all the surgery and chemo and radiation were finished, the message I received was essentially: “OK, you’re good, for now. Just come back every few months for a check-up.”
What about the aftermath of all that treatment? What about the other aspects of the experience? I found that my doctors had very little to offer outside of surgery and drugs and the quick advice to get counseling or some other support services.
“I’m on my own” is what I’ve been telling concerned folks who ask how I’m doing. I am truly grateful for the skill, medicine, and machinery that made the killing of an invasive tumor possible. But I’m on my own when it comes to finding or inventing ways to cope with the new challenges of a pelvic area damaged by radiation and detoxing from the heavy metal—platinum—that was an ingredient in the chemo I received.
My partner and I have had to be persistently creative, careful, delicate, uncritical, and extremely patient with each other to bring about the return of a “normal” sex life. We have been successful, for the most part, but there is also a slightly new definition of what “normal” is for us. Our latest triumph is that we no longer have to use copious amounts of lubricant to engage in intercourse. Sex is no longer painful, as the vaginal tissues have been slowly, patiently engaged on a regularly scheduled basis. Can you imagine sex on a schedule? Neither could I, but that is what we found worked from as early as 1 week postradiation. As it turns out, this was good advice—really, the only advice I got when it came to the practicalities of restoring function, but it required a fair amount of tweaking and personalization as well.
Another big change in perception that I had to accept as part of my new norm is learning to talk about my most personal areas in a matter of fact way.
The cancer conveyor-belt approach to treatment is a very streamlined, well-run system. I’ve been impressed with the expertise, efficiency, and demeanor of all the professionals I have encountered. Everyone—even receptionists—has been helpful and empathetic, especially my own ObGyn, who has hugged me and cried with me and offered to put herself on the line for me and speak out to the media when I had no health insurance. But for most patients post-treatment, we figuratively walk off a cliff and find ourselves in new territory without any network or structure like we experienced during the “war” on our cancer. This new territory is a place of possibility within the health-care field—one I hope is developing now.
Dr. Barbara S. Levy asks: How do we respond? We physicians are so focused on treating or curing disease that we often lose sight of the woman who has the disease.
Katie’s case is much too common. Women are often reluctant to address their sexuality with us—especially when we have been dismissive. We must recognize this important aspect of quality of life and relationships and be prepared to raise the issue with our patients before they begin therapy.
Educating ourselves and then our patients about strategies to reduce the impact of treatment and menopause on sexual function is the first step. Acknowledging and validating their concerns and being able to offer practical steps to preserve healthy sexual function is something, I think, that all ObGyns should be able to do. Strategies to maintain vulvovaginal elasticity include avoidance of soaps and drying chemicals, daily perineal and vaginal stretching—either manually, with a dilator, or via frequent sexual intercourse. And topical lubricants and moisturizing agents should be recommended to maintain vaginal pH and reduce the dryness, itching, and overall dysesthesia.
As the studies highlighted in this Update on Sexual Dysfunction demonstrate, the sexual consequences of radical hysterectomy are significant. Julie also became menopausal as a result of treatment—yet no one prepared her for the symptoms she would experience, and no member of her treatment team helped her understand the likely impact of therapy. Radiation oncologists do address the need for vaginal dilators or daily intercourse to maintain depth and caliber of the vagina during and after therapy, but they are less likely to prepare a patient for menopausal symptoms.
If our patients are to have an optimal experience, we need to provide coordinated, cross-disciplinary care that includes management not only of immediate side effects of treatment but also psychosocial and long-term hormonal and sexual sequelae of therapy. Julie charted her own course with the help of a very dedicated and sensitive partner to successfully overcome the negative effects of radiation, chemotherapy, surgery, and menopause on her sexuality.
Gynecologic cancer disrupts sexual function, over the short term and the long term
Jensen PT, Groenvold M, Klee MC, Thranov I, Petersen MA, Machin D. Early-stage cervical carcinoma, radical hysterectomy, and sexual function: a longitudinal study. Cancer. 2004;100(1):97–106.
Vaz AF, Pinto-Neto AM, Conde DM, et al. Quality of life and menopausal and sexual symptoms in gynecologic cancer survivors: a cohort study. Menopause. 2011;18(6):662–669.
These two studies explored sexual function after treatment for gynecologic cancer. The investigators found significant disruption of function.
Jensen et al: Radical hysterectomy for cervical Ca damages sexual function significantly
This was a prospective cohort study of 173 women who had early-stage cervical carcinoma and who underwent radical hysterectomy with pelvic lymphadenectomy (all of them node-negative). A validated questionnaire was administered six times, from 5 weeks to 24 months after surgery. An age-matched group of women without cancer was used for comparison. At the 12-month follow-up, patients were asked to report their sexual function at baseline and compare it to their current status. Overall, the women had a higher level of dissatisfaction with their sexual experiences 12 months after surgery than at baseline.
Details of the study
Women in the Jensen study were 23 to 75 years old (median age, 42.7 years), and 93% were sexually active at the time of diagnosis, reporting an average of one to two sexual activities in a week. Forty-six women (25%) were postmenopausal at diagnosis, compared with 34% of the control group. Only 8% of patients were using HT at study entry, compared with 25% of women in the control group. By 12 months after surgery, however, 25% of gynecologic cancer patients were taking systemic HT.
Findings included low libido and other ills
Severe lack of lubrication and low or no sexual desire were reported by cancer patients throughout the first 2 years after surgery. Patients also reported severe problems achieving orgasm as long as 6 months after surgery, as well as reduced vaginal size; both problems rendered their sexual experiences unsatisfactory. Nevertheless, 40% of patients reported at least some sexual activity by 5 weeks after surgery. By 6 months after surgery, there were almost as many sexually active women among the patient group as there were in the control group. However, at 18 months after treatment, patients reported less interest in intimacy—among both themselves and their partners—than among women in the control group. Overall, women treated for cervical cancer had a higher level of dissatisfaction with their sexual experiences 12 months after surgery than they did before diagnosis.
Although 91% of women who were sexually active before surgery resumed intercourse within 12 months, the frequency of sexual activity declined from one to two times per week to three to four times per month. Major long-term changes occurred in regard to libido (interest in sexual relations), arousal (vaginal lubrication), and vaginal size. Although dyspareunia was a significant problem 5 weeks to 3 months after surgery, it resolved within 1 to 2 years.
Jensen and colleagues concluded that radical hysterectomy for treatment of early-stage cervical cancer has significant negative effects on sexual function in the short and long term. They postulated a neurogenic basis for the sexual complaints and discussed both histologic and clinical studies to support this hypothesis.
They also emphasized the need to discuss the risks and management of sexual dysfunction with patients before and after surgery. Better management of the psychosocial consequences of a cancer diagnosis and the physical effects of radical hysterectomy may help avoid the negative experiences that were reported in this study.
Vaz et al: Rate of dyspareunia was high among women treated for endometrial or cervical Ca
Investigators followed 107 women from initial consultation for radiation therapy through 3 years post-treatment. Although a significant percentage (50%) of the cohort was lost to follow-up—many due to death or tumor recurrence—50% of those who remained reported dyspareunia 3 years after radiotherapy.
Women in this study were 21 to 75 years old (median age, 60 years) and had cervical or endometrial carcinoma. Eighty-nine women (83%) received external pelvic radiation as well as brachytherapy. Before beginning radiation therapy, 37.4% of the cohort underwent surgery for treatment of their cancer. Sixty-four percent of the cohort had stage III or IV disease.
At enrollment, 50% of women reported having a life partner, 82% were postmenopausal, and 11.2% were taking HT. However, only 21.5% of women reported sexual activity. The authors opine that this low rate of sexual activity may have been due to recent surgery, bleeding, or pain related to cancer.
All women were offered “standard” interventions for their dyspareunia, including the use of vaginal dilators twice daily for 2 years, as well as the use of vaginal lubricants. Patients who experienced menopausal symptoms—hot flashes, decreased libido, dyspareunia—were referred to a menopause outpatient clinic.
Before treatment, 20% of women reported dyspareunia. Three years later, 44% of patients reported sexual activity, but 50% had dyspareunia. Twenty-one percent reported lower sexual interest relative to baseline, 8% reported vaginal dryness, and 21% reported vasomotor symptoms. Although there was a trend toward increasing sexual activity with decreasing vaginal dryness, the rise in dyspareunia from 20% to 50% over 3 years is troubling.
Radical hysterectomy and radiation therapy to the pelvis cause neurovascular disruption and sexual consequences quite similar to those found after radical prostate surgery. Sexual arousal and orgasm are dependent on both the parasympathetic and sympathetic nerves supplying the pelvis. these nerves are disrupted in Frankenhauser’s plexus during the parametrial dissection of radical hysterectomy and lie clearly within the radiation treatment field.
Dilators and lubricants may be useful in minimizing actual shrinkage of the vagina. However, the elasticity of vaginal tissues, vasodilation during arousal, and transudation across the vaginal wall may all be lost or significantly compromised.
Advise your patients of the potential for sexual side effects of cervical and endometrial cancer treatment before they undergo that therapy. Proactive management of some of the expected problems, such as reduced elasticity and lubrication, as well as treatment for arousal dysfunction (perhaps with the same pharmacotherapeutic agents that provide improvement for many men after radical prostatectomy), may help your patients avoid the distress and disappointment they often experience after successful treatment of their cancers.
When a patient has undergone treatment for cancer,
ask about her sexual function
A symposium on sexual health yields recommendations for your broader care of cancer patients
How likely are you to encounter a cancer survivor in your practice?
Very.
According to the Centers for Disease Control and Prevention (CDC), there were 6.3 million female cancer survivors in the United States as of 2007—and that number has likely increased by a million or more.3 In fact, the number of cancer survivors is expected to double by 2016.
How likely is that cancer survivor to have sexual dysfunction?
Highly.
According to a 2010 survey by the Lance Armstrong Foundation, 46% of cancer survivors report problems with sexual functioning after treatment—and that’s probably a conservative figure, given that 64% of all people with cancer have a malignancy that directly affects sexual organs.2
One more question for you to ponder: How likely is the cancer patient’s sexual dysfunction to go unaddressed?
Extremely.
According to speakers at the Cancer Survivorship and Sexual Health Symposium, held June 17–19, 2011, in Washington, DC, cancer survivors are ill prepared for many of the symptoms of sexual dysfunction that develop after treatment, and many physicians fail to address this dimension of their health.
The symposium, sponsored by the International Society for Sexual Medicine and the Sexual Medicine Society of North America, was organized to address these gaps in care. During the 3-day conference, speakers from oncology, gynecology, mental health, urology, and other specialties presented data and described their experience managing cancer patients. They also offered recommendations for clinicians:
- Talk about it. Address the “highly prevalent but commonly ignored” adverse sexual effects of malignancy and its treatment. Ask: “How has cancer affected your sex life?”
- Try to prevent it. Consider nerve-sparing strategies during radical hysterectomy, radical trachelectomy, and clitoral preservation, which may lead to improved sexual function
- Encourage and support use of dilators. Advise women who have gynecologic cancer to use dilators to maintain vaginal patency, and be aware that compliance is linked to support from a health-care provider
- Encourage sexual activity, which can help preserve function
- Consider local estrogen. When it is appropriate, prescribe vaginal estrogen, which is minimally absorbed, to reduce vaginal symptoms of menopause. (The safety of local estrogen remains in question for women who have breast cancer.)
- Check for problems at each follow-up appointment, and be prepared to explain function and treatment options more than once
- Promote female genital blood flow. For example, it may be appropriate to begin sexual rehabilitation, such as use of vaginal dilators, during treatment
- Consider referral to a sexual rehabilitation program that includes medical and psychological approaches
- Build a network of psychologists, sex therapists, and other professionals who can assist you in managing your patients’ complaints.
“Discomfort around human sexuality is the main reason the issue doesn’t get raised by health-care providers,” said symposium speaker Sharon L. Bober, PhD, of the Dana-Farber Cancer Institute in Boston. “No one wants to initiate the conversation.” Dr. Bober emphasized the importance of asking about sexual function when a cancer survivor presents for care. “A majority of cancer patients in the community don’t hear this question from their providers.”
—Janelle Yates, Senior Editor
We want to hear from you! Tell us what you think.
1. Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol. 2008;112(5):970-978.
2. Rechis R, Boerner L. How cancer has affected post-treatment survivors: a LIVESTRONG report. Austin, Tex: Lance Armstrong Foundation 2011;13-
3. Centers for Disease Control and Prevention. Cancer survivors—United States, 2007. MMWR. 2011;60(9):269-272
1. Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol. 2008;112(5):970-978.
2. Rechis R, Boerner L. How cancer has affected post-treatment survivors: a LIVESTRONG report. Austin, Tex: Lance Armstrong Foundation 2011;13-
3. Centers for Disease Control and Prevention. Cancer survivors—United States, 2007. MMWR. 2011;60(9):269-272
Vulvar pain syndromes: Making the correct diagnosis
Although the incidence of vulvar pain has increased over the past decade—thanks to both greater awareness and increasing numbers of affected women—the phenomenon is not a recent development. As early as 1874, T. Galliard Thomas wrote, “[T]his disorder, although fortunately not very frequent, is by no means very rare.”1 He went on to express “surprise” that it had not been “more generally and fully described.”
Despite the focus Thomas directed to the issue, vulvar pain did not get much attention until the 21st century, when a number of studies began to gauge its prevalence. For example, in a study in Boston of about 5,000 women, the lifetime prevalence of chronic vulvar pain was 16%.2 And in a study in Texas, the prevalence of vulvar pain in an urban, largely minority population was estimated to be 11%.3 The Boston study also reported that “nearly 40% of women chose not to seek treatment, and, of those who did, 60% saw three or more doctors, many of whom could not provide a diagnosis.”2
Clearly, there is a need for comprehensive information on vulvar pain and its causes, symptoms, diagnosis, and treatment. To address the lack of guidance, OBG Management Contributing Editor Neal M. Lonky, MD, assembled a panel of experts on vulvar pain syndromes and invited them to share their considerable knowledge. The ensuing discussion, presented in three parts, offers a gold mine of information.
In this opening article, the panel focuses on causes, symptomatology, and diagnosis of this common complaint. In Part 2, which will appear in the October issue of this journal, the focus is the bounty of treatment options. Part 3 follows in November, when the discussion shifts to vestibulodynia.
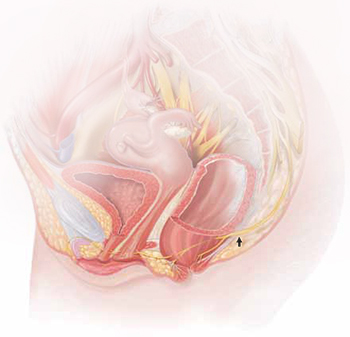
The lower vagina and vulva are richly supplied with peripheral nerves and are, therefore, sensitive to pain, particularly the region of the hymeneal ring. Although the pudendal nerve (arrow) courses through the area, it is an uncommon source of vulvar pain.
Common diagnoses—and misdiagnoses
Dr. Lonky: What are the most common diagnoses when vulvar pain is the complaint?
Dr. Gunter: The most common cause of chronic vulvar pain is vulvodynia, although lichen simplex chronicus, chronic yeast infections, and non-neoplastic epithelial disorders, such as lichen sclerosus and lichen planus, can also produce irritation and pain. In postmenopausal women, atrophic vaginitis can also cause a burning pain, although symptoms are typically more vaginal than vulvar. Yeast and lichen simplex chronicus typically produce itching, although sometimes they can present with irritation and pain, so they must be considered in the differential diagnosis. It is important to remember that many women with vulvodynia have used multiple topical agents and may have developed complex hygiene rituals in an attempt to treat their symptoms, which can result in a secondary lichen simplex chronicus.
That said, there is a high frequency of misdiagnosis with yeast. For example, in a study by Nyirjesy and colleagues, two thirds of women who were referred to a tertiary clinic for chronic vulvovaginal candidiasis were found to have a noninfectious entity instead—most commonly lichen simplex chronicus and vulvodynia.4
Dr. Edwards: The most common “diagnosis” for vulvar pain is vulvodynia. However, the definition of vulvodynia is pain—i.e., burning, rawness, irritation, soreness, aching, or stabbing or stinging sensations—in the absence of skin disease, infection, or specific neurologic disease. Therefore, even though the usual cause of vulvar pain is vulvodynia, it is a diagnosis of exclusion, and skin disease, infection, and neurologic disease must be ruled out.
In regard to infection, Candida albicans and bacterial vaginosis (BV) are usually the first conditions that are considered when a patient complains of vulvar pain, but they are not common causes of vulvar pain and are never causes of chronic vulvar pain. Very rarely they may cause recurrent pain that clears, at least briefly, with treatment.
Candida albicans is usually primarily pruritic, and BV produces discharge and odor, sometimes with minor symptoms. Non-albicans Candida (e.g., Candida glabrata) is nearly always asymptomatic, but it occasionally causes irritation and burning.
Group B streptococcus is another infectious entity that very, very occasionally causes irritation and dyspareunia but is usually only a colonizer.
Herpes simplex virus is a cause of recurrent but not chronic pain.
Chronic pain is more likely to be caused by skin disease than by infection. Lichen simplex chronicus causes itching; any pain is due to erosions from scratching.
Dr. Haefner: Several other infectious conditions or their treatments can cause vulvar pain. For example, herpes (particularly primary herpes infection) is classically associated with vulvar pain. The pain is so great that, at times, the patient requires admission for pain control. Surprisingly, despite the known pain of herpes, approximately 80% of patients who have it are unaware of their diagnosis.
Although condyloma is generally a painless condition, many patients complain of pain following treatment for it, whether treatment involves topical medications or laser surgery.
Chancroid is a painful vulvar ulcer. Trichomonas can sometimes be associated with vulvar pain.
Dr. Lonky: What terminology do we use when we discuss vulvar pain?
Dr. Haefner: The current terminology used to describe vulvar pain was published in 2004, after years of debate over nomenclature within the International Society for the Study of Vulvovaginal Disease.5 The terminology lists two major categories of vulvar pain:
- pain related to a specific disorder. This category encompasses numerous conditions that feature an abnormal appearance of the vulva (Table 1).
TABLE 1
Terminology and classification of vulvar pain from the International Society for the Study of Vulvovaginal Disease
|
| SOURCE: Moyal-Barracco and Lynch.5 Reproduced with permission from the Journal of Reproductive Medicine. |
- vulvodynia, in which the vulva appears normal, other than occasional erythema, which is most prominent at the duct openings (vestibular ducts—Bartholin’s and Skene’s).
As for vulvar pain, there are two major forms:
- hyperalgesia (a low threshold for pain)
- allodynia (pain in response to light touch).
Some diseases that are associated with vulvar pain do not qualify for the diagnosis of vulvodynia (Table 2) because they are associated with an abnormal appearance of the vulva.
TABLE 2
Conditions other than vulvodynia that are associated with vulvar pain
| Acute irritant contact dermatitis (e.g., erosion due to podofilox, imiquimod, cantharidin, fluorouracil, or podophyllin toxin) |
| Aphthous ulcer |
| Atrophy |
| Bartholin’s abscess |
| Candidiasis |
| Carcinoma |
| Chronic irritant contact dermatitis |
| Endometriosis |
| Herpes (simplex and zoster) |
| Immunobullous diseases (including cicatricial pemphigoid, pemphigus vulgaris, linear immunoglobulin A disease, etc.) |
| Lichen planus |
| Lichen sclerosus |
| Podophyllin overdose (see above) |
| Prolapsed urethra |
| Sjögren’s syndrome |
| Trauma |
| Trichomoniasis |
| Vulvar intraepithelial neoplasia |
What needs to be ruled out for a diagnosis of vulvodynia?
Dr. Lonky: What skin diseases need to be ruled out before vulvodynia can be diagnosed?
Dr. Edwards: Skin diseases that affect the vulva are usually pruritic—pain is a later sign. Lichen simplex chronicus (also known as eczema) is pruritus caused by any irritant; any pain that arises is produced by visible excoriations from scratching.
Lichen sclerosus manifests as white epithelium that has a crinkling, shiny, or waxy texture. It can produce pain, especially dyspareunia. The pain is caused by erosions that arise from fragility and introital narrowing and inelasticity.
Vulvovaginal lichen planus is usually erosive and preferentially affects mucous membranes, especially the vestibule; it sometimes affects the vagina and mouth, as well.
Desquamative inflammatory vaginitis is most likely a skin disease that affects only the vagina. It involves introital redness and a clinically and microscopically purulent vaginal discharge that also reveals parabasal cells and absent lactobacilli.
Dr. Lonky: You mentioned that neurologic diseases can sometimes cause vulvar pain. Which ones?
Dr. Edwards: Pudendal neuralgia, diabetic neuropathy, and post-herpetic neuralgia are the most common specific neurologic causes of vulvar pain. Multiple sclerosis can also produce pain syndromes. Post-herpetic neuralgia follows herpes zoster—not herpes simplex—virus infection.
Dr. Lonky: Any other conditions that can cause vulvar pain?
Dr. Haefner: Aphthous ulcers are common and are often flared by stress.
Non-neoplastic epithelial disorders are also seen frequently in health-care providers’ offices; many patients who experience them report pain on the vulva.
It is always important to consider cancer when a patient has an abnormal vulvar appearance and pain that has persisted despite treatment.
What are the most common vulvar pain syndromes?
Dr. Lonky: If you were to rank vulvar pain syndromes according to their prevalence, what would the most common syndromes be?
Dr. Gunter: Given the misdiagnosis of many women, who are told they have chronic yeast infection, as I mentioned, it’s hard to know which vulvar pain syndromes are most prevalent. I suspect that lichen simplex chronicus is most common, followed by vulvodynia, with chronic yeast infection a distant third.
My experience reflects what Nyirjesy and colleagues4 found: 65% to 75% of women referred to my clinic with chronic yeast actually have lichen simplex chronicus or vulvodynia. In postmenopausal women, atrophic vaginitis is also a consideration; it’s becoming more common now that the use of systemic hormone replacement therapy is decreasing.
Dr. Lonky: What about subsets of vulvodynia? Which ones are most common?
Dr. Edwards: There is good evidence of marked overlap among subsets of vulvodynia. The vast majority of women who have vulvodynia experience primarily provoked vestibular pain, regardless of age. However, I find that almost all patients also report pain that extends beyond the vestibule at times, as well as occasional unprovoked pain.
The diagnosis requires the exclusion of other causes of vulvar pain, and the subset is identified by the location of pain (that is, is it strictly localized or generalized or even migratory?) and its provoked or unprovoked nature.
Localized clitoral pain and vulvar pain localized to one side of the vulva are extremely uncommon, but they do occur. And although I rarely encounter teenagers and prepubertal children who have vulvodynia, I do have patients in both age groups who have vulvodynia.
Dr. Lonky: Are there racial differences in the prevalence of vulvodynia?
Dr. Edwards: Although several good studies show that women of African descent and white patients are equally likely to experience vulvodynia, the vast majority (99%) of my patients who have vulvodynia are white. My patients of African descent consult me primarily for itching or discharge.
My local demographics prevent me from judging the likelihood of Asians having vulvodynia, and our Hispanic population has limited access to health care.
In general, I don’t think that demographics are useful in making the diagnosis of vulvodynia.
Do women who have vulvar pain tend to have comorbidities?
Dr. Lonky: Do your patients who have vulvodynia or another vulvar pain syndrome tend to have comorbidities? If so, is this information helpful in establishing the diagnosis and planning therapy?
Dr. Haefner: Women who have vulvodynia often have other medical problems as well. In my practice, when new patients who have vulvodynia complete their intake survey, they often report a history of headache, irritable bowel syndrome, interstitial cystitis, fibromyalgia,6 chronic fatigue syndrome, back pain, and temporomandibular joint (TMJ) disorder. These comorbidities are not particularly helpful in establishing the diagnosis of vulvodynia, but they are an important consideration when choosing therapy for the patient. Often, the medications chosen to treat one condition will also benefit another condition. However, it’s important to check for potential interactions between drugs before prescribing a new treatment.
Dr. Gunter: A significant number of women who have vulvodynia also have other chronic pain syndromes. For example, the incidence of bladder pain syndrome–interstitial cystitis is 68% to 82% among women who have vulvodynia, compared with a baseline rate among all women of 6% to 11%.7-10 The rate of irritable bowel syndrome is more than doubled among women who have vulvodynia, compared with the general population (27% versus 12%).8 Another common comorbidity, hypertonic somatic dysfunction of the pelvic floor, is identified in 10% to 90% of women who have chronic vulvar pain.8,11,12 These women also have a higher incidence of nongenital pain syndromes, such as fibromyalgia, migraine, and TMJ dysfunction, than the general population, as Dr. Haefner noted.8,12,13
Many studies have evaluated psychological and emotional contributions to chronic vulvar pain. Pain and depression are intimately related—the incidence of depression among all people who experience chronic pain ranges from 27% to 54%, compared with 5% to 17% among the general population.14-16 The relationship is complex because chronic illness in general is associated with depression. Nevertheless, several studies have noted an increase in anxiety, stress, and depression among women who have vulvodynia.17-19
I screen every patient for depression using a Patient Health Questionnaire (PHQ-9); I also screen for anxiety. I find that a significant percentage of patients in my clinic are depressed or have an anxiety disorder. Failure to address these comorbidities makes treatment very difficult. I typically prescribe citalopram (Celexa), although there is some question whether it can safely be combined with a tricyclic antidepressant. We also offer stress-reduction classes, teach every patient the value of diaphragmatic breathing, offer mind-body classes for anxiety and stress, and provide intensive programs where the patient can learn important self-care skills, such as pacing (spacing activities throughout the day in a manner that avoids aggravating the pain), and address her anxiety and stress in a more guided manner. We also have a psychologist who specializes in pain for any patient who may need one-on-one counseling.
Dr. Edwards: The presence of comorbidities is somewhat useful in making the diagnosis of vulvodynia. I question my diagnosis, in fact, when a patient who has vulvodynia does not have headaches, low energy, depression, anxiety, irritable bowel syndrome, constipation, fibromyalgia, chronic fatigue, sensitivity to medications, TMJ dysfunction, or urinary symptoms.
How common is pudendal neuralgia?
Dr. Lonky: How prevalent is a finding of pudendal neuralgia?
Dr. Edwards: The prevalence and incidence of pudendal neuralgia are not known. Those who specialize in this condition think it is relatively common. I do not identify or suspect it very often. Its definitive diagnosis and management are outside the purview of the general gynecologist, but the general gynecologist should recognize the symptoms of pudendal neuralgia and refer the patient for evaluation and therapy.
Dr. Lonky: What are those symptoms?
Dr. Haefner: Pudendal neuralgia often occurs following trauma to the pudendal nerve. The pudendal nerve arises from sacral nerves, generally sacral nerves 2 to 4. Several tests can be utilized to diagnose this condition, including quantitative sensor tests, pudendal nerve motor latency tests, electromyography (EMG), and pudendal nerve blocks.20
Nantes Criteria allow for making a diagnosis of pudendal neuralgia (Table 3).21
TABLE 3
Nantes Criteria for pudendal neuralgia by pudendal nerve entrapment
Essential criteria
|
Complementary diagnostic criteria
|
Exclusion criteria
|
Associated signs not excluding the diagnosis
|
| SOURCE: Labat et al.21 Reproduced with permission from Neurology and Urodynamics. |
Initial treatments for pudendal neuralgia should be conservative. Treatments consist of lifestyle changes to prevent flare of disease. Physical therapy, medical management, nerve blocks, and alternative treatments may be beneficial.
Pudendal nerve entrapment is often exacerbated by sitting (not on a toilet seat, however) and is reduced in a standing position. It tends to increase in intensity throughout the day.22 The final treatment for pudendal nerve entrapment is surgery if the nerve is compressed. By this time, the generalist is not generally the provider who performs the surgery.
Dr. Gunter: I believe pudendal neuralgia is sometimes overdiagnosed. EMG studies of the pudendal nerve, often touted as a diagnostic tool, are unreliable (they can be abnormal after vaginal delivery or vaginal hysterectomy, for example). In my experience, bilateral pain is less likely to be pudendal neuralgia; spontaneous bilateral compression neuropathy at exactly the same level is not a common phenomenon in chronic pain.
I reserve the diagnosis of pudendal neuralgia for women who have allodynia in the distribution of the pudendal nerve with severe pain on sitting, and who have exquisite tenderness when pressure is applied over the pudendal nerve (at the level of the ischial spine on vaginal examination). Typically, the vaginal sidewall on the affected side is very sensitive to light touch. I do see pudendal nerve pain after vaginal surgery when there has been some compromise of the pudendal nerve or the sacral plexus. This is typically unilateral pain.
Dr. Lonky: Thank you all. We’ll continue our discussion, with a focus on treatment, in the October 2011 issue.
- Part 2: A bounty of treatment options
(October 2011) - Part 3: Vestibulodynia
(November 2011)
We want to hear from you! Tell us what you think.
1. Thomas TG. Practical Treatise on the Diseases of Women. Philadelphia Pa: Henry C. Lea; 1874.
2. Harlow BL, Stewart EG. A population-based assessment of chronic unexplained vulvar pain: have we underestimated the prevalence of vulvodynia? J Am Med Womens Assoc. 2003;58(2):82-88.
3. Lavy RJ, Hynan LS, Haley RW. Prevalence of vulvar pain in an urban minority population. J Reprod Med. 2007;52(1):59-62.
4. Nyirjesy P, Peyton C, Weitz MV, Mathew L, Culhane JF. Causes of chronic vaginitis: analysis of a prospective database of affected women. Obstet Gynecol. 2006;108(5):1185-1191.
5. Moyal-Barracco M, Lynch PJ. 2003 ISSVD terminology and classification of vulvodynia: a historical perspective. J Reprod Med. 2004;49(10):772-777.
6. Yunas MB. Fibromyalgia and overlapping disorders: the unifying concept of central sensitivity syndromes. Semin Arthritis Rheum. 2007;36(6):339-356.
7. Kahn BS, Tatro C, Parsons CL, Willems JJ. Prevalence of interstitial cystitis in vulvodynia patients detected by bladder potassium sensitivity. J Sex Med. 2010;7(2 Pt 2):996-1002.
8. Arnold JD, Bachman GS, Rosen R, Kelly S, Rhoads GG. Vulvodynia: characteristics and associations with comorbidities and quality of life. Obstet Gynecol. 2006;107(3):617-624.
9. Parsons CL, Dell J, Stanford EJ, et al. The prevalence of interstitial cystitis in gynecologic patients with pelvic pain, as detected by intravesical potassium sensitivity. Am J Obstet Gynecol. 2002;187(5):1395-1400.
10. Clemens JQ, Meenan RT, O’Keefe Rosetti MC, et al. Prevalence of interstitial cystitis symptoms in a managed care population. J Urol. 2005;174(2):576-580.
11. Engman M, Lindehammar H, Wijma B. Surface electromyography diagnostics in women with partial vaginismus with or without vulvar vestibulitis and in asymptomatic women. J Psychosom Obstet Gynecol. 2004;25(3-4):281-294.
12. Gunter J. Vulvodynia: new thoughts on a devastating condition. Obstet Gynecol Surv. 2007;62(12):812-819.
13. Gordon AS, Panahlan-Jand M, McComb F, Melegari C, Sharp S. Characteristics of women with vulvar pain disorders: a Web-based survey. J Sex Marital Ther. 2003;29(suppl 1):45.-
14. Whitten CE, Cristobal K. Chronic pain is a chronic condition not just a symptom. Permanente J. 2005;9(3):43.-
15. Manchikanti L, Fellows B, Pampati V, et al. Comparison of psychological status of chronic pain patients and the general population. Pain Physician. 2002;5(1):40-48.
16. Banks SM, Kerns RD. Explaining the high rates of depression in chronic pain: a diathesis-stress framework. Psychological Bulletin. 1996;119(1):95-110.
17. Sadownik LA. Clinical correlates of vulvodynia patients. A prospective study of 300 patients. J Reprod Med. 2000;5:40-48.Editor found in PubMed: Sadownik LA. Clinical profile of vulvodynia patients. A prospective study of 300 patients. J Reprod Med. 2000;45(8):679–684. Could not find the citation listed. Please confirm.
18. Reed BD, Haefner HK, Punch MR, Roth RS, Gorenflo DW, Gillespie BW. Psychosocial and sexual functioning in women with vulvodynia and chronic pelvic pain. A comparative evaluation. J Reprod Med. 2000;45(8):624-632.
19. Landry T, Bergeron S. Biopsychosocial factors associated with dyspareunia in a community sample of adolescent girls. Arch Sex Behav. 2011;June 22.
20. Goldstein A, Pukall C, Goldstein I. When Sex Hurts: A Woman’s Guide to Banishing Sexual Pain. Cambridge Mass: Da Capo Lifelong Books; 2011;117-126.
21. Labat JJ, Riant T, Robert R, Amarenco G, Lefaucheur JP, Rigaud J. Diagnostic criteria for pudendal neuralgia by pudendal nerve entrapment (Nantes criteria). Neurol Urodyn. 2008;27(4):306-310.
22. Popeney C, Answell V, Renney K. Pudendal entrapment as an etiology of chronic perineal pain: Diagnosis and treatment. Neurol Urodyn. 2007;26(6):820-827.

| Neal M. Lonky, MD, MPH, moderator of this discussion, is Clinical Professor of Obstetrics and Gynecology at the University of California–Irvine and a member of the Board of Directors of Southern California Permanente Medical Group. He serves as an OBG Management Contributing Editor. |

| Libby Edwards, MD, is Adjunct Clinical Associate Professor of Dermatology at the University of North Carolina in Chapel Hill, NC, and Chief of Dermatology at Carolinas Medical Center in Charlotte, NC. Dr. Edwards is a Past President and Past Secretary General of the International Society for the Study of Vulvovaginal Disease. |

| Jennifer Gunter, MD, is Director of Pelvic Pain and Vulvovaginal Disorders for Kaiser Permanente in San Francisco, Calif. |

| Hope K. Haefner, MD, is Professor of Obstetrics and Gynecology at the University of Michigan Hospitals and Co-Director of the University of Michigan Center for Vulvar Diseases in Ann Arbor, Mich. |
|
| Neal M. Lonky, MD, MPH, moderator of this discussion, is Clinical Professor of Obstetrics and Gynecology at the University of California–Irvine and a member of the Board of Directors of Southern California Permanente Medical Group. He serves as an OBG Management Contributing Editor. |
|
| Libby Edwards, MD, is Adjunct Clinical Associate Professor of Dermatology at the University of North Carolina in Chapel Hill, NC, and Chief of Dermatology at Carolinas Medical Center in Charlotte, NC. Dr. Edwards is a Past President and Past Secretary General of the International Society for the Study of Vulvovaginal Disease. |
|
| Jennifer Gunter, MD, is Director of Pelvic Pain and Vulvovaginal Disorders for Kaiser Permanente in San Francisco, Calif. |
|
| Hope K. Haefner, MD, is Professor of Obstetrics and Gynecology at the University of Michigan Hospitals and Co-Director of the University of Michigan Center for Vulvar Diseases in Ann Arbor, Mich. |
|
| Neal M. Lonky, MD, MPH, moderator of this discussion, is Clinical Professor of Obstetrics and Gynecology at the University of California–Irvine and a member of the Board of Directors of Southern California Permanente Medical Group. He serves as an OBG Management Contributing Editor. |
|
| Libby Edwards, MD, is Adjunct Clinical Associate Professor of Dermatology at the University of North Carolina in Chapel Hill, NC, and Chief of Dermatology at Carolinas Medical Center in Charlotte, NC. Dr. Edwards is a Past President and Past Secretary General of the International Society for the Study of Vulvovaginal Disease. |
|
| Jennifer Gunter, MD, is Director of Pelvic Pain and Vulvovaginal Disorders for Kaiser Permanente in San Francisco, Calif. |
|
| Hope K. Haefner, MD, is Professor of Obstetrics and Gynecology at the University of Michigan Hospitals and Co-Director of the University of Michigan Center for Vulvar Diseases in Ann Arbor, Mich. |
Although the incidence of vulvar pain has increased over the past decade—thanks to both greater awareness and increasing numbers of affected women—the phenomenon is not a recent development. As early as 1874, T. Galliard Thomas wrote, “[T]his disorder, although fortunately not very frequent, is by no means very rare.”1 He went on to express “surprise” that it had not been “more generally and fully described.”
Despite the focus Thomas directed to the issue, vulvar pain did not get much attention until the 21st century, when a number of studies began to gauge its prevalence. For example, in a study in Boston of about 5,000 women, the lifetime prevalence of chronic vulvar pain was 16%.2 And in a study in Texas, the prevalence of vulvar pain in an urban, largely minority population was estimated to be 11%.3 The Boston study also reported that “nearly 40% of women chose not to seek treatment, and, of those who did, 60% saw three or more doctors, many of whom could not provide a diagnosis.”2
Clearly, there is a need for comprehensive information on vulvar pain and its causes, symptoms, diagnosis, and treatment. To address the lack of guidance, OBG Management Contributing Editor Neal M. Lonky, MD, assembled a panel of experts on vulvar pain syndromes and invited them to share their considerable knowledge. The ensuing discussion, presented in three parts, offers a gold mine of information.
In this opening article, the panel focuses on causes, symptomatology, and diagnosis of this common complaint. In Part 2, which will appear in the October issue of this journal, the focus is the bounty of treatment options. Part 3 follows in November, when the discussion shifts to vestibulodynia.
The lower vagina and vulva are richly supplied with peripheral nerves and are, therefore, sensitive to pain, particularly the region of the hymeneal ring. Although the pudendal nerve (arrow) courses through the area, it is an uncommon source of vulvar pain.
Common diagnoses—and misdiagnoses
Dr. Lonky: What are the most common diagnoses when vulvar pain is the complaint?
Dr. Gunter: The most common cause of chronic vulvar pain is vulvodynia, although lichen simplex chronicus, chronic yeast infections, and non-neoplastic epithelial disorders, such as lichen sclerosus and lichen planus, can also produce irritation and pain. In postmenopausal women, atrophic vaginitis can also cause a burning pain, although symptoms are typically more vaginal than vulvar. Yeast and lichen simplex chronicus typically produce itching, although sometimes they can present with irritation and pain, so they must be considered in the differential diagnosis. It is important to remember that many women with vulvodynia have used multiple topical agents and may have developed complex hygiene rituals in an attempt to treat their symptoms, which can result in a secondary lichen simplex chronicus.
That said, there is a high frequency of misdiagnosis with yeast. For example, in a study by Nyirjesy and colleagues, two thirds of women who were referred to a tertiary clinic for chronic vulvovaginal candidiasis were found to have a noninfectious entity instead—most commonly lichen simplex chronicus and vulvodynia.4
Dr. Edwards: The most common “diagnosis” for vulvar pain is vulvodynia. However, the definition of vulvodynia is pain—i.e., burning, rawness, irritation, soreness, aching, or stabbing or stinging sensations—in the absence of skin disease, infection, or specific neurologic disease. Therefore, even though the usual cause of vulvar pain is vulvodynia, it is a diagnosis of exclusion, and skin disease, infection, and neurologic disease must be ruled out.
In regard to infection, Candida albicans and bacterial vaginosis (BV) are usually the first conditions that are considered when a patient complains of vulvar pain, but they are not common causes of vulvar pain and are never causes of chronic vulvar pain. Very rarely they may cause recurrent pain that clears, at least briefly, with treatment.
Candida albicans is usually primarily pruritic, and BV produces discharge and odor, sometimes with minor symptoms. Non-albicans Candida (e.g., Candida glabrata) is nearly always asymptomatic, but it occasionally causes irritation and burning.
Group B streptococcus is another infectious entity that very, very occasionally causes irritation and dyspareunia but is usually only a colonizer.
Herpes simplex virus is a cause of recurrent but not chronic pain.
Chronic pain is more likely to be caused by skin disease than by infection. Lichen simplex chronicus causes itching; any pain is due to erosions from scratching.
Dr. Haefner: Several other infectious conditions or their treatments can cause vulvar pain. For example, herpes (particularly primary herpes infection) is classically associated with vulvar pain. The pain is so great that, at times, the patient requires admission for pain control. Surprisingly, despite the known pain of herpes, approximately 80% of patients who have it are unaware of their diagnosis.
Although condyloma is generally a painless condition, many patients complain of pain following treatment for it, whether treatment involves topical medications or laser surgery.
Chancroid is a painful vulvar ulcer. Trichomonas can sometimes be associated with vulvar pain.
Dr. Lonky: What terminology do we use when we discuss vulvar pain?
Dr. Haefner: The current terminology used to describe vulvar pain was published in 2004, after years of debate over nomenclature within the International Society for the Study of Vulvovaginal Disease.5 The terminology lists two major categories of vulvar pain:
- pain related to a specific disorder. This category encompasses numerous conditions that feature an abnormal appearance of the vulva (Table 1).
TABLE 1
Terminology and classification of vulvar pain from the International Society for the Study of Vulvovaginal Disease
|
| SOURCE: Moyal-Barracco and Lynch.5 Reproduced with permission from the Journal of Reproductive Medicine. |
- vulvodynia, in which the vulva appears normal, other than occasional erythema, which is most prominent at the duct openings (vestibular ducts—Bartholin’s and Skene’s).
As for vulvar pain, there are two major forms:
- hyperalgesia (a low threshold for pain)
- allodynia (pain in response to light touch).
Some diseases that are associated with vulvar pain do not qualify for the diagnosis of vulvodynia (Table 2) because they are associated with an abnormal appearance of the vulva.
TABLE 2
Conditions other than vulvodynia that are associated with vulvar pain
| Acute irritant contact dermatitis (e.g., erosion due to podofilox, imiquimod, cantharidin, fluorouracil, or podophyllin toxin) |
| Aphthous ulcer |
| Atrophy |
| Bartholin’s abscess |
| Candidiasis |
| Carcinoma |
| Chronic irritant contact dermatitis |
| Endometriosis |
| Herpes (simplex and zoster) |
| Immunobullous diseases (including cicatricial pemphigoid, pemphigus vulgaris, linear immunoglobulin A disease, etc.) |
| Lichen planus |
| Lichen sclerosus |
| Podophyllin overdose (see above) |
| Prolapsed urethra |
| Sjögren’s syndrome |
| Trauma |
| Trichomoniasis |
| Vulvar intraepithelial neoplasia |
What needs to be ruled out for a diagnosis of vulvodynia?
Dr. Lonky: What skin diseases need to be ruled out before vulvodynia can be diagnosed?
Dr. Edwards: Skin diseases that affect the vulva are usually pruritic—pain is a later sign. Lichen simplex chronicus (also known as eczema) is pruritus caused by any irritant; any pain that arises is produced by visible excoriations from scratching.
Lichen sclerosus manifests as white epithelium that has a crinkling, shiny, or waxy texture. It can produce pain, especially dyspareunia. The pain is caused by erosions that arise from fragility and introital narrowing and inelasticity.
Vulvovaginal lichen planus is usually erosive and preferentially affects mucous membranes, especially the vestibule; it sometimes affects the vagina and mouth, as well.
Desquamative inflammatory vaginitis is most likely a skin disease that affects only the vagina. It involves introital redness and a clinically and microscopically purulent vaginal discharge that also reveals parabasal cells and absent lactobacilli.
Dr. Lonky: You mentioned that neurologic diseases can sometimes cause vulvar pain. Which ones?
Dr. Edwards: Pudendal neuralgia, diabetic neuropathy, and post-herpetic neuralgia are the most common specific neurologic causes of vulvar pain. Multiple sclerosis can also produce pain syndromes. Post-herpetic neuralgia follows herpes zoster—not herpes simplex—virus infection.
Dr. Lonky: Any other conditions that can cause vulvar pain?
Dr. Haefner: Aphthous ulcers are common and are often flared by stress.
Non-neoplastic epithelial disorders are also seen frequently in health-care providers’ offices; many patients who experience them report pain on the vulva.
It is always important to consider cancer when a patient has an abnormal vulvar appearance and pain that has persisted despite treatment.
What are the most common vulvar pain syndromes?
Dr. Lonky: If you were to rank vulvar pain syndromes according to their prevalence, what would the most common syndromes be?
Dr. Gunter: Given the misdiagnosis of many women, who are told they have chronic yeast infection, as I mentioned, it’s hard to know which vulvar pain syndromes are most prevalent. I suspect that lichen simplex chronicus is most common, followed by vulvodynia, with chronic yeast infection a distant third.
My experience reflects what Nyirjesy and colleagues4 found: 65% to 75% of women referred to my clinic with chronic yeast actually have lichen simplex chronicus or vulvodynia. In postmenopausal women, atrophic vaginitis is also a consideration; it’s becoming more common now that the use of systemic hormone replacement therapy is decreasing.
Dr. Lonky: What about subsets of vulvodynia? Which ones are most common?
Dr. Edwards: There is good evidence of marked overlap among subsets of vulvodynia. The vast majority of women who have vulvodynia experience primarily provoked vestibular pain, regardless of age. However, I find that almost all patients also report pain that extends beyond the vestibule at times, as well as occasional unprovoked pain.
The diagnosis requires the exclusion of other causes of vulvar pain, and the subset is identified by the location of pain (that is, is it strictly localized or generalized or even migratory?) and its provoked or unprovoked nature.
Localized clitoral pain and vulvar pain localized to one side of the vulva are extremely uncommon, but they do occur. And although I rarely encounter teenagers and prepubertal children who have vulvodynia, I do have patients in both age groups who have vulvodynia.
Dr. Lonky: Are there racial differences in the prevalence of vulvodynia?
Dr. Edwards: Although several good studies show that women of African descent and white patients are equally likely to experience vulvodynia, the vast majority (99%) of my patients who have vulvodynia are white. My patients of African descent consult me primarily for itching or discharge.
My local demographics prevent me from judging the likelihood of Asians having vulvodynia, and our Hispanic population has limited access to health care.
In general, I don’t think that demographics are useful in making the diagnosis of vulvodynia.
Do women who have vulvar pain tend to have comorbidities?
Dr. Lonky: Do your patients who have vulvodynia or another vulvar pain syndrome tend to have comorbidities? If so, is this information helpful in establishing the diagnosis and planning therapy?
Dr. Haefner: Women who have vulvodynia often have other medical problems as well. In my practice, when new patients who have vulvodynia complete their intake survey, they often report a history of headache, irritable bowel syndrome, interstitial cystitis, fibromyalgia,6 chronic fatigue syndrome, back pain, and temporomandibular joint (TMJ) disorder. These comorbidities are not particularly helpful in establishing the diagnosis of vulvodynia, but they are an important consideration when choosing therapy for the patient. Often, the medications chosen to treat one condition will also benefit another condition. However, it’s important to check for potential interactions between drugs before prescribing a new treatment.
Dr. Gunter: A significant number of women who have vulvodynia also have other chronic pain syndromes. For example, the incidence of bladder pain syndrome–interstitial cystitis is 68% to 82% among women who have vulvodynia, compared with a baseline rate among all women of 6% to 11%.7-10 The rate of irritable bowel syndrome is more than doubled among women who have vulvodynia, compared with the general population (27% versus 12%).8 Another common comorbidity, hypertonic somatic dysfunction of the pelvic floor, is identified in 10% to 90% of women who have chronic vulvar pain.8,11,12 These women also have a higher incidence of nongenital pain syndromes, such as fibromyalgia, migraine, and TMJ dysfunction, than the general population, as Dr. Haefner noted.8,12,13
Many studies have evaluated psychological and emotional contributions to chronic vulvar pain. Pain and depression are intimately related—the incidence of depression among all people who experience chronic pain ranges from 27% to 54%, compared with 5% to 17% among the general population.14-16 The relationship is complex because chronic illness in general is associated with depression. Nevertheless, several studies have noted an increase in anxiety, stress, and depression among women who have vulvodynia.17-19
I screen every patient for depression using a Patient Health Questionnaire (PHQ-9); I also screen for anxiety. I find that a significant percentage of patients in my clinic are depressed or have an anxiety disorder. Failure to address these comorbidities makes treatment very difficult. I typically prescribe citalopram (Celexa), although there is some question whether it can safely be combined with a tricyclic antidepressant. We also offer stress-reduction classes, teach every patient the value of diaphragmatic breathing, offer mind-body classes for anxiety and stress, and provide intensive programs where the patient can learn important self-care skills, such as pacing (spacing activities throughout the day in a manner that avoids aggravating the pain), and address her anxiety and stress in a more guided manner. We also have a psychologist who specializes in pain for any patient who may need one-on-one counseling.
Dr. Edwards: The presence of comorbidities is somewhat useful in making the diagnosis of vulvodynia. I question my diagnosis, in fact, when a patient who has vulvodynia does not have headaches, low energy, depression, anxiety, irritable bowel syndrome, constipation, fibromyalgia, chronic fatigue, sensitivity to medications, TMJ dysfunction, or urinary symptoms.
How common is pudendal neuralgia?
Dr. Lonky: How prevalent is a finding of pudendal neuralgia?
Dr. Edwards: The prevalence and incidence of pudendal neuralgia are not known. Those who specialize in this condition think it is relatively common. I do not identify or suspect it very often. Its definitive diagnosis and management are outside the purview of the general gynecologist, but the general gynecologist should recognize the symptoms of pudendal neuralgia and refer the patient for evaluation and therapy.
Dr. Lonky: What are those symptoms?
Dr. Haefner: Pudendal neuralgia often occurs following trauma to the pudendal nerve. The pudendal nerve arises from sacral nerves, generally sacral nerves 2 to 4. Several tests can be utilized to diagnose this condition, including quantitative sensor tests, pudendal nerve motor latency tests, electromyography (EMG), and pudendal nerve blocks.20
Nantes Criteria allow for making a diagnosis of pudendal neuralgia (Table 3).21
TABLE 3
Nantes Criteria for pudendal neuralgia by pudendal nerve entrapment
Essential criteria
|
Complementary diagnostic criteria
|
Exclusion criteria
|
Associated signs not excluding the diagnosis
|
| SOURCE: Labat et al.21 Reproduced with permission from Neurology and Urodynamics. |
Initial treatments for pudendal neuralgia should be conservative. Treatments consist of lifestyle changes to prevent flare of disease. Physical therapy, medical management, nerve blocks, and alternative treatments may be beneficial.
Pudendal nerve entrapment is often exacerbated by sitting (not on a toilet seat, however) and is reduced in a standing position. It tends to increase in intensity throughout the day.22 The final treatment for pudendal nerve entrapment is surgery if the nerve is compressed. By this time, the generalist is not generally the provider who performs the surgery.
Dr. Gunter: I believe pudendal neuralgia is sometimes overdiagnosed. EMG studies of the pudendal nerve, often touted as a diagnostic tool, are unreliable (they can be abnormal after vaginal delivery or vaginal hysterectomy, for example). In my experience, bilateral pain is less likely to be pudendal neuralgia; spontaneous bilateral compression neuropathy at exactly the same level is not a common phenomenon in chronic pain.
I reserve the diagnosis of pudendal neuralgia for women who have allodynia in the distribution of the pudendal nerve with severe pain on sitting, and who have exquisite tenderness when pressure is applied over the pudendal nerve (at the level of the ischial spine on vaginal examination). Typically, the vaginal sidewall on the affected side is very sensitive to light touch. I do see pudendal nerve pain after vaginal surgery when there has been some compromise of the pudendal nerve or the sacral plexus. This is typically unilateral pain.
Dr. Lonky: Thank you all. We’ll continue our discussion, with a focus on treatment, in the October 2011 issue.
- Part 2: A bounty of treatment options
(October 2011) - Part 3: Vestibulodynia
(November 2011)
We want to hear from you! Tell us what you think.
Although the incidence of vulvar pain has increased over the past decade—thanks to both greater awareness and increasing numbers of affected women—the phenomenon is not a recent development. As early as 1874, T. Galliard Thomas wrote, “[T]his disorder, although fortunately not very frequent, is by no means very rare.”1 He went on to express “surprise” that it had not been “more generally and fully described.”
Despite the focus Thomas directed to the issue, vulvar pain did not get much attention until the 21st century, when a number of studies began to gauge its prevalence. For example, in a study in Boston of about 5,000 women, the lifetime prevalence of chronic vulvar pain was 16%.2 And in a study in Texas, the prevalence of vulvar pain in an urban, largely minority population was estimated to be 11%.3 The Boston study also reported that “nearly 40% of women chose not to seek treatment, and, of those who did, 60% saw three or more doctors, many of whom could not provide a diagnosis.”2
Clearly, there is a need for comprehensive information on vulvar pain and its causes, symptoms, diagnosis, and treatment. To address the lack of guidance, OBG Management Contributing Editor Neal M. Lonky, MD, assembled a panel of experts on vulvar pain syndromes and invited them to share their considerable knowledge. The ensuing discussion, presented in three parts, offers a gold mine of information.
In this opening article, the panel focuses on causes, symptomatology, and diagnosis of this common complaint. In Part 2, which will appear in the October issue of this journal, the focus is the bounty of treatment options. Part 3 follows in November, when the discussion shifts to vestibulodynia.
The lower vagina and vulva are richly supplied with peripheral nerves and are, therefore, sensitive to pain, particularly the region of the hymeneal ring. Although the pudendal nerve (arrow) courses through the area, it is an uncommon source of vulvar pain.
Common diagnoses—and misdiagnoses
Dr. Lonky: What are the most common diagnoses when vulvar pain is the complaint?
Dr. Gunter: The most common cause of chronic vulvar pain is vulvodynia, although lichen simplex chronicus, chronic yeast infections, and non-neoplastic epithelial disorders, such as lichen sclerosus and lichen planus, can also produce irritation and pain. In postmenopausal women, atrophic vaginitis can also cause a burning pain, although symptoms are typically more vaginal than vulvar. Yeast and lichen simplex chronicus typically produce itching, although sometimes they can present with irritation and pain, so they must be considered in the differential diagnosis. It is important to remember that many women with vulvodynia have used multiple topical agents and may have developed complex hygiene rituals in an attempt to treat their symptoms, which can result in a secondary lichen simplex chronicus.
That said, there is a high frequency of misdiagnosis with yeast. For example, in a study by Nyirjesy and colleagues, two thirds of women who were referred to a tertiary clinic for chronic vulvovaginal candidiasis were found to have a noninfectious entity instead—most commonly lichen simplex chronicus and vulvodynia.4
Dr. Edwards: The most common “diagnosis” for vulvar pain is vulvodynia. However, the definition of vulvodynia is pain—i.e., burning, rawness, irritation, soreness, aching, or stabbing or stinging sensations—in the absence of skin disease, infection, or specific neurologic disease. Therefore, even though the usual cause of vulvar pain is vulvodynia, it is a diagnosis of exclusion, and skin disease, infection, and neurologic disease must be ruled out.
In regard to infection, Candida albicans and bacterial vaginosis (BV) are usually the first conditions that are considered when a patient complains of vulvar pain, but they are not common causes of vulvar pain and are never causes of chronic vulvar pain. Very rarely they may cause recurrent pain that clears, at least briefly, with treatment.
Candida albicans is usually primarily pruritic, and BV produces discharge and odor, sometimes with minor symptoms. Non-albicans Candida (e.g., Candida glabrata) is nearly always asymptomatic, but it occasionally causes irritation and burning.
Group B streptococcus is another infectious entity that very, very occasionally causes irritation and dyspareunia but is usually only a colonizer.
Herpes simplex virus is a cause of recurrent but not chronic pain.
Chronic pain is more likely to be caused by skin disease than by infection. Lichen simplex chronicus causes itching; any pain is due to erosions from scratching.
Dr. Haefner: Several other infectious conditions or their treatments can cause vulvar pain. For example, herpes (particularly primary herpes infection) is classically associated with vulvar pain. The pain is so great that, at times, the patient requires admission for pain control. Surprisingly, despite the known pain of herpes, approximately 80% of patients who have it are unaware of their diagnosis.
Although condyloma is generally a painless condition, many patients complain of pain following treatment for it, whether treatment involves topical medications or laser surgery.
Chancroid is a painful vulvar ulcer. Trichomonas can sometimes be associated with vulvar pain.
Dr. Lonky: What terminology do we use when we discuss vulvar pain?
Dr. Haefner: The current terminology used to describe vulvar pain was published in 2004, after years of debate over nomenclature within the International Society for the Study of Vulvovaginal Disease.5 The terminology lists two major categories of vulvar pain:
- pain related to a specific disorder. This category encompasses numerous conditions that feature an abnormal appearance of the vulva (Table 1).
TABLE 1
Terminology and classification of vulvar pain from the International Society for the Study of Vulvovaginal Disease
|
| SOURCE: Moyal-Barracco and Lynch.5 Reproduced with permission from the Journal of Reproductive Medicine. |
- vulvodynia, in which the vulva appears normal, other than occasional erythema, which is most prominent at the duct openings (vestibular ducts—Bartholin’s and Skene’s).
As for vulvar pain, there are two major forms:
- hyperalgesia (a low threshold for pain)
- allodynia (pain in response to light touch).
Some diseases that are associated with vulvar pain do not qualify for the diagnosis of vulvodynia (Table 2) because they are associated with an abnormal appearance of the vulva.
TABLE 2
Conditions other than vulvodynia that are associated with vulvar pain
| Acute irritant contact dermatitis (e.g., erosion due to podofilox, imiquimod, cantharidin, fluorouracil, or podophyllin toxin) |
| Aphthous ulcer |
| Atrophy |
| Bartholin’s abscess |
| Candidiasis |
| Carcinoma |
| Chronic irritant contact dermatitis |
| Endometriosis |
| Herpes (simplex and zoster) |
| Immunobullous diseases (including cicatricial pemphigoid, pemphigus vulgaris, linear immunoglobulin A disease, etc.) |
| Lichen planus |
| Lichen sclerosus |
| Podophyllin overdose (see above) |
| Prolapsed urethra |
| Sjögren’s syndrome |
| Trauma |
| Trichomoniasis |
| Vulvar intraepithelial neoplasia |
What needs to be ruled out for a diagnosis of vulvodynia?
Dr. Lonky: What skin diseases need to be ruled out before vulvodynia can be diagnosed?
Dr. Edwards: Skin diseases that affect the vulva are usually pruritic—pain is a later sign. Lichen simplex chronicus (also known as eczema) is pruritus caused by any irritant; any pain that arises is produced by visible excoriations from scratching.
Lichen sclerosus manifests as white epithelium that has a crinkling, shiny, or waxy texture. It can produce pain, especially dyspareunia. The pain is caused by erosions that arise from fragility and introital narrowing and inelasticity.
Vulvovaginal lichen planus is usually erosive and preferentially affects mucous membranes, especially the vestibule; it sometimes affects the vagina and mouth, as well.
Desquamative inflammatory vaginitis is most likely a skin disease that affects only the vagina. It involves introital redness and a clinically and microscopically purulent vaginal discharge that also reveals parabasal cells and absent lactobacilli.
Dr. Lonky: You mentioned that neurologic diseases can sometimes cause vulvar pain. Which ones?
Dr. Edwards: Pudendal neuralgia, diabetic neuropathy, and post-herpetic neuralgia are the most common specific neurologic causes of vulvar pain. Multiple sclerosis can also produce pain syndromes. Post-herpetic neuralgia follows herpes zoster—not herpes simplex—virus infection.
Dr. Lonky: Any other conditions that can cause vulvar pain?
Dr. Haefner: Aphthous ulcers are common and are often flared by stress.
Non-neoplastic epithelial disorders are also seen frequently in health-care providers’ offices; many patients who experience them report pain on the vulva.
It is always important to consider cancer when a patient has an abnormal vulvar appearance and pain that has persisted despite treatment.
What are the most common vulvar pain syndromes?
Dr. Lonky: If you were to rank vulvar pain syndromes according to their prevalence, what would the most common syndromes be?
Dr. Gunter: Given the misdiagnosis of many women, who are told they have chronic yeast infection, as I mentioned, it’s hard to know which vulvar pain syndromes are most prevalent. I suspect that lichen simplex chronicus is most common, followed by vulvodynia, with chronic yeast infection a distant third.
My experience reflects what Nyirjesy and colleagues4 found: 65% to 75% of women referred to my clinic with chronic yeast actually have lichen simplex chronicus or vulvodynia. In postmenopausal women, atrophic vaginitis is also a consideration; it’s becoming more common now that the use of systemic hormone replacement therapy is decreasing.
Dr. Lonky: What about subsets of vulvodynia? Which ones are most common?
Dr. Edwards: There is good evidence of marked overlap among subsets of vulvodynia. The vast majority of women who have vulvodynia experience primarily provoked vestibular pain, regardless of age. However, I find that almost all patients also report pain that extends beyond the vestibule at times, as well as occasional unprovoked pain.
The diagnosis requires the exclusion of other causes of vulvar pain, and the subset is identified by the location of pain (that is, is it strictly localized or generalized or even migratory?) and its provoked or unprovoked nature.
Localized clitoral pain and vulvar pain localized to one side of the vulva are extremely uncommon, but they do occur. And although I rarely encounter teenagers and prepubertal children who have vulvodynia, I do have patients in both age groups who have vulvodynia.
Dr. Lonky: Are there racial differences in the prevalence of vulvodynia?
Dr. Edwards: Although several good studies show that women of African descent and white patients are equally likely to experience vulvodynia, the vast majority (99%) of my patients who have vulvodynia are white. My patients of African descent consult me primarily for itching or discharge.
My local demographics prevent me from judging the likelihood of Asians having vulvodynia, and our Hispanic population has limited access to health care.
In general, I don’t think that demographics are useful in making the diagnosis of vulvodynia.
Do women who have vulvar pain tend to have comorbidities?
Dr. Lonky: Do your patients who have vulvodynia or another vulvar pain syndrome tend to have comorbidities? If so, is this information helpful in establishing the diagnosis and planning therapy?
Dr. Haefner: Women who have vulvodynia often have other medical problems as well. In my practice, when new patients who have vulvodynia complete their intake survey, they often report a history of headache, irritable bowel syndrome, interstitial cystitis, fibromyalgia,6 chronic fatigue syndrome, back pain, and temporomandibular joint (TMJ) disorder. These comorbidities are not particularly helpful in establishing the diagnosis of vulvodynia, but they are an important consideration when choosing therapy for the patient. Often, the medications chosen to treat one condition will also benefit another condition. However, it’s important to check for potential interactions between drugs before prescribing a new treatment.
Dr. Gunter: A significant number of women who have vulvodynia also have other chronic pain syndromes. For example, the incidence of bladder pain syndrome–interstitial cystitis is 68% to 82% among women who have vulvodynia, compared with a baseline rate among all women of 6% to 11%.7-10 The rate of irritable bowel syndrome is more than doubled among women who have vulvodynia, compared with the general population (27% versus 12%).8 Another common comorbidity, hypertonic somatic dysfunction of the pelvic floor, is identified in 10% to 90% of women who have chronic vulvar pain.8,11,12 These women also have a higher incidence of nongenital pain syndromes, such as fibromyalgia, migraine, and TMJ dysfunction, than the general population, as Dr. Haefner noted.8,12,13
Many studies have evaluated psychological and emotional contributions to chronic vulvar pain. Pain and depression are intimately related—the incidence of depression among all people who experience chronic pain ranges from 27% to 54%, compared with 5% to 17% among the general population.14-16 The relationship is complex because chronic illness in general is associated with depression. Nevertheless, several studies have noted an increase in anxiety, stress, and depression among women who have vulvodynia.17-19
I screen every patient for depression using a Patient Health Questionnaire (PHQ-9); I also screen for anxiety. I find that a significant percentage of patients in my clinic are depressed or have an anxiety disorder. Failure to address these comorbidities makes treatment very difficult. I typically prescribe citalopram (Celexa), although there is some question whether it can safely be combined with a tricyclic antidepressant. We also offer stress-reduction classes, teach every patient the value of diaphragmatic breathing, offer mind-body classes for anxiety and stress, and provide intensive programs where the patient can learn important self-care skills, such as pacing (spacing activities throughout the day in a manner that avoids aggravating the pain), and address her anxiety and stress in a more guided manner. We also have a psychologist who specializes in pain for any patient who may need one-on-one counseling.
Dr. Edwards: The presence of comorbidities is somewhat useful in making the diagnosis of vulvodynia. I question my diagnosis, in fact, when a patient who has vulvodynia does not have headaches, low energy, depression, anxiety, irritable bowel syndrome, constipation, fibromyalgia, chronic fatigue, sensitivity to medications, TMJ dysfunction, or urinary symptoms.
How common is pudendal neuralgia?
Dr. Lonky: How prevalent is a finding of pudendal neuralgia?
Dr. Edwards: The prevalence and incidence of pudendal neuralgia are not known. Those who specialize in this condition think it is relatively common. I do not identify or suspect it very often. Its definitive diagnosis and management are outside the purview of the general gynecologist, but the general gynecologist should recognize the symptoms of pudendal neuralgia and refer the patient for evaluation and therapy.
Dr. Lonky: What are those symptoms?
Dr. Haefner: Pudendal neuralgia often occurs following trauma to the pudendal nerve. The pudendal nerve arises from sacral nerves, generally sacral nerves 2 to 4. Several tests can be utilized to diagnose this condition, including quantitative sensor tests, pudendal nerve motor latency tests, electromyography (EMG), and pudendal nerve blocks.20
Nantes Criteria allow for making a diagnosis of pudendal neuralgia (Table 3).21
TABLE 3
Nantes Criteria for pudendal neuralgia by pudendal nerve entrapment
Essential criteria
|
Complementary diagnostic criteria
|
Exclusion criteria
|
Associated signs not excluding the diagnosis
|
| SOURCE: Labat et al.21 Reproduced with permission from Neurology and Urodynamics. |
Initial treatments for pudendal neuralgia should be conservative. Treatments consist of lifestyle changes to prevent flare of disease. Physical therapy, medical management, nerve blocks, and alternative treatments may be beneficial.
Pudendal nerve entrapment is often exacerbated by sitting (not on a toilet seat, however) and is reduced in a standing position. It tends to increase in intensity throughout the day.22 The final treatment for pudendal nerve entrapment is surgery if the nerve is compressed. By this time, the generalist is not generally the provider who performs the surgery.
Dr. Gunter: I believe pudendal neuralgia is sometimes overdiagnosed. EMG studies of the pudendal nerve, often touted as a diagnostic tool, are unreliable (they can be abnormal after vaginal delivery or vaginal hysterectomy, for example). In my experience, bilateral pain is less likely to be pudendal neuralgia; spontaneous bilateral compression neuropathy at exactly the same level is not a common phenomenon in chronic pain.
I reserve the diagnosis of pudendal neuralgia for women who have allodynia in the distribution of the pudendal nerve with severe pain on sitting, and who have exquisite tenderness when pressure is applied over the pudendal nerve (at the level of the ischial spine on vaginal examination). Typically, the vaginal sidewall on the affected side is very sensitive to light touch. I do see pudendal nerve pain after vaginal surgery when there has been some compromise of the pudendal nerve or the sacral plexus. This is typically unilateral pain.
Dr. Lonky: Thank you all. We’ll continue our discussion, with a focus on treatment, in the October 2011 issue.
- Part 2: A bounty of treatment options
(October 2011) - Part 3: Vestibulodynia
(November 2011)
We want to hear from you! Tell us what you think.
1. Thomas TG. Practical Treatise on the Diseases of Women. Philadelphia Pa: Henry C. Lea; 1874.
2. Harlow BL, Stewart EG. A population-based assessment of chronic unexplained vulvar pain: have we underestimated the prevalence of vulvodynia? J Am Med Womens Assoc. 2003;58(2):82-88.
3. Lavy RJ, Hynan LS, Haley RW. Prevalence of vulvar pain in an urban minority population. J Reprod Med. 2007;52(1):59-62.
4. Nyirjesy P, Peyton C, Weitz MV, Mathew L, Culhane JF. Causes of chronic vaginitis: analysis of a prospective database of affected women. Obstet Gynecol. 2006;108(5):1185-1191.
5. Moyal-Barracco M, Lynch PJ. 2003 ISSVD terminology and classification of vulvodynia: a historical perspective. J Reprod Med. 2004;49(10):772-777.
6. Yunas MB. Fibromyalgia and overlapping disorders: the unifying concept of central sensitivity syndromes. Semin Arthritis Rheum. 2007;36(6):339-356.
7. Kahn BS, Tatro C, Parsons CL, Willems JJ. Prevalence of interstitial cystitis in vulvodynia patients detected by bladder potassium sensitivity. J Sex Med. 2010;7(2 Pt 2):996-1002.
8. Arnold JD, Bachman GS, Rosen R, Kelly S, Rhoads GG. Vulvodynia: characteristics and associations with comorbidities and quality of life. Obstet Gynecol. 2006;107(3):617-624.
9. Parsons CL, Dell J, Stanford EJ, et al. The prevalence of interstitial cystitis in gynecologic patients with pelvic pain, as detected by intravesical potassium sensitivity. Am J Obstet Gynecol. 2002;187(5):1395-1400.
10. Clemens JQ, Meenan RT, O’Keefe Rosetti MC, et al. Prevalence of interstitial cystitis symptoms in a managed care population. J Urol. 2005;174(2):576-580.
11. Engman M, Lindehammar H, Wijma B. Surface electromyography diagnostics in women with partial vaginismus with or without vulvar vestibulitis and in asymptomatic women. J Psychosom Obstet Gynecol. 2004;25(3-4):281-294.
12. Gunter J. Vulvodynia: new thoughts on a devastating condition. Obstet Gynecol Surv. 2007;62(12):812-819.
13. Gordon AS, Panahlan-Jand M, McComb F, Melegari C, Sharp S. Characteristics of women with vulvar pain disorders: a Web-based survey. J Sex Marital Ther. 2003;29(suppl 1):45.-
14. Whitten CE, Cristobal K. Chronic pain is a chronic condition not just a symptom. Permanente J. 2005;9(3):43.-
15. Manchikanti L, Fellows B, Pampati V, et al. Comparison of psychological status of chronic pain patients and the general population. Pain Physician. 2002;5(1):40-48.
16. Banks SM, Kerns RD. Explaining the high rates of depression in chronic pain: a diathesis-stress framework. Psychological Bulletin. 1996;119(1):95-110.
17. Sadownik LA. Clinical correlates of vulvodynia patients. A prospective study of 300 patients. J Reprod Med. 2000;5:40-48.Editor found in PubMed: Sadownik LA. Clinical profile of vulvodynia patients. A prospective study of 300 patients. J Reprod Med. 2000;45(8):679–684. Could not find the citation listed. Please confirm.
18. Reed BD, Haefner HK, Punch MR, Roth RS, Gorenflo DW, Gillespie BW. Psychosocial and sexual functioning in women with vulvodynia and chronic pelvic pain. A comparative evaluation. J Reprod Med. 2000;45(8):624-632.
19. Landry T, Bergeron S. Biopsychosocial factors associated with dyspareunia in a community sample of adolescent girls. Arch Sex Behav. 2011;June 22.
20. Goldstein A, Pukall C, Goldstein I. When Sex Hurts: A Woman’s Guide to Banishing Sexual Pain. Cambridge Mass: Da Capo Lifelong Books; 2011;117-126.
21. Labat JJ, Riant T, Robert R, Amarenco G, Lefaucheur JP, Rigaud J. Diagnostic criteria for pudendal neuralgia by pudendal nerve entrapment (Nantes criteria). Neurol Urodyn. 2008;27(4):306-310.
22. Popeney C, Answell V, Renney K. Pudendal entrapment as an etiology of chronic perineal pain: Diagnosis and treatment. Neurol Urodyn. 2007;26(6):820-827.
1. Thomas TG. Practical Treatise on the Diseases of Women. Philadelphia Pa: Henry C. Lea; 1874.
2. Harlow BL, Stewart EG. A population-based assessment of chronic unexplained vulvar pain: have we underestimated the prevalence of vulvodynia? J Am Med Womens Assoc. 2003;58(2):82-88.
3. Lavy RJ, Hynan LS, Haley RW. Prevalence of vulvar pain in an urban minority population. J Reprod Med. 2007;52(1):59-62.
4. Nyirjesy P, Peyton C, Weitz MV, Mathew L, Culhane JF. Causes of chronic vaginitis: analysis of a prospective database of affected women. Obstet Gynecol. 2006;108(5):1185-1191.
5. Moyal-Barracco M, Lynch PJ. 2003 ISSVD terminology and classification of vulvodynia: a historical perspective. J Reprod Med. 2004;49(10):772-777.
6. Yunas MB. Fibromyalgia and overlapping disorders: the unifying concept of central sensitivity syndromes. Semin Arthritis Rheum. 2007;36(6):339-356.
7. Kahn BS, Tatro C, Parsons CL, Willems JJ. Prevalence of interstitial cystitis in vulvodynia patients detected by bladder potassium sensitivity. J Sex Med. 2010;7(2 Pt 2):996-1002.
8. Arnold JD, Bachman GS, Rosen R, Kelly S, Rhoads GG. Vulvodynia: characteristics and associations with comorbidities and quality of life. Obstet Gynecol. 2006;107(3):617-624.
9. Parsons CL, Dell J, Stanford EJ, et al. The prevalence of interstitial cystitis in gynecologic patients with pelvic pain, as detected by intravesical potassium sensitivity. Am J Obstet Gynecol. 2002;187(5):1395-1400.
10. Clemens JQ, Meenan RT, O’Keefe Rosetti MC, et al. Prevalence of interstitial cystitis symptoms in a managed care population. J Urol. 2005;174(2):576-580.
11. Engman M, Lindehammar H, Wijma B. Surface electromyography diagnostics in women with partial vaginismus with or without vulvar vestibulitis and in asymptomatic women. J Psychosom Obstet Gynecol. 2004;25(3-4):281-294.
12. Gunter J. Vulvodynia: new thoughts on a devastating condition. Obstet Gynecol Surv. 2007;62(12):812-819.
13. Gordon AS, Panahlan-Jand M, McComb F, Melegari C, Sharp S. Characteristics of women with vulvar pain disorders: a Web-based survey. J Sex Marital Ther. 2003;29(suppl 1):45.-
14. Whitten CE, Cristobal K. Chronic pain is a chronic condition not just a symptom. Permanente J. 2005;9(3):43.-
15. Manchikanti L, Fellows B, Pampati V, et al. Comparison of psychological status of chronic pain patients and the general population. Pain Physician. 2002;5(1):40-48.
16. Banks SM, Kerns RD. Explaining the high rates of depression in chronic pain: a diathesis-stress framework. Psychological Bulletin. 1996;119(1):95-110.
17. Sadownik LA. Clinical correlates of vulvodynia patients. A prospective study of 300 patients. J Reprod Med. 2000;5:40-48.Editor found in PubMed: Sadownik LA. Clinical profile of vulvodynia patients. A prospective study of 300 patients. J Reprod Med. 2000;45(8):679–684. Could not find the citation listed. Please confirm.
18. Reed BD, Haefner HK, Punch MR, Roth RS, Gorenflo DW, Gillespie BW. Psychosocial and sexual functioning in women with vulvodynia and chronic pelvic pain. A comparative evaluation. J Reprod Med. 2000;45(8):624-632.
19. Landry T, Bergeron S. Biopsychosocial factors associated with dyspareunia in a community sample of adolescent girls. Arch Sex Behav. 2011;June 22.
20. Goldstein A, Pukall C, Goldstein I. When Sex Hurts: A Woman’s Guide to Banishing Sexual Pain. Cambridge Mass: Da Capo Lifelong Books; 2011;117-126.
21. Labat JJ, Riant T, Robert R, Amarenco G, Lefaucheur JP, Rigaud J. Diagnostic criteria for pudendal neuralgia by pudendal nerve entrapment (Nantes criteria). Neurol Urodyn. 2008;27(4):306-310.
22. Popeney C, Answell V, Renney K. Pudendal entrapment as an etiology of chronic perineal pain: Diagnosis and treatment. Neurol Urodyn. 2007;26(6):820-827.
IN THIS ARTICLE
Autosomal Dominant Polycystic Kidney Disease
Twice as common as autism and half as well-known,1 autosomal polycystic kidney disease (ADPKD) occurs in one in 400 to one in 1,000 people.2 It is an inherited progressive genetic disorder that causes hypertension and decreased renal function and, over time, can lead to kidney failure. Two polycstin genes that code for ADPKD, PKD1 and PKD2, were identified in 1994 and 1996, respectively.3,4 Awareness and understanding of the genes responsible for ADPKD have increased clinicians’ ability to identify at-risk patients and to slow or alter the course of the disease.
Case Presentation
A 45-year-old black man presents to your office with severe, nonradiating back pain and new-onset hypertension. Regarding the pain, he stated, “I turned around to see who kicked me, but no one was there.” When the pain began, he went to see the nurse at the school where he is employed, and she found that his blood pressure was high at 162/90 mm Hg. Although the patient’s back pain is resolving, he is very concerned about his blood pressure, since he has never had a high reading before.
He is the baseball coach and physical education teacher at the local high school and is in excellent physical condition as a result of his professional interaction with teenagers every day. He does not smoke or use any illicit drugs but does admit to occasional alcohol consumption. His medical history is significant only for occasional broken fingers and twisted ankles, all occurring while he was engaged in sports.
His family history includes one brother without medical problems, a brother and a sister with hypertension, a sister with diabetes and obesity, and a brother with a congenital abnormality that required a living donor kidney transplant at age 17 (the father served as donor). No family-wide workup has ever been done because no one practitioner has ever made a connection among these conditions and considered a diagnosis of ADPKD.
The patient’s blood pressure in the office is 172/92 mm Hg while sitting and 166/88 mm Hg while standing. He is somewhat sore with a localized spasm in the lumbar-sacral area but no radiation of pain. The patient has trouble touching his toes but reports that he can never touch his toes. His straight leg lift is negative. The rest of his physical exam is noncontributory.
What should be the next step in this patient’s workup?
PATHOPHYSIOLOGY
ADPKD is a progressive expansion of numerous fluid-filled cysts that result in massive enlargement of the kidneys.5 Less than 5% of all nephrons become cystic; however, the average volume of a polycystic kidney is 1,000 mL (normal, 300 mL), that is, the volume of a standard-sized pineapple. Even with this significant enlargement, a decline in the glomerular filtration rate (GFR) is not usually seen initially. Each cyst is derived from a single hyperproliferative epithelial cell. Increased cellular proliferation, followed by fluid secretion and alterations in the extracellular matrix, cause an outpouching from the parent nephron, which eventually detaches from the parent nephron and continues to enlarge and autonomously secrete fluid.6,7
PKD1 and PKD2 are two genes responsible for ADPKD that have been isolated so far. Since there are families carrying neither the PKD1 nor the PKD2 gene that still have an inherited type of ADPKD, there is suspicion that at least one more PKD gene, not yet isolated, exists.8 It is also possible that other genetic or environmental factors may be at play.9,10
In 1994, the PKD1 gene was isolated on chromosome 16,3 and it was found to code for polycystin 1. A lack of polycystin 1 causes an abnormality in the Na+/K(+)-ATPase pumps, leading to abnormal sodium reabsorption.11 How and why this happens is not quite clear. However, the hypertension that is a key objective finding in patients with ADPKD is thought to result from this pump abnormality.
PKD2 is found on the long arm of chromosome 4 and codes for polycystin 2.4 Polycystin 2 is an amino acid that is responsible for voltage-activated cellular calcium channels,5 again explaining the hypertension so commonly seen in the course of ADPKD. ADPKD-associated hypertension may be present as early as the teenage years.12
EPIDEMIOLOGY
More than 85% of ADPKD cases are associated with PKD1, and this form is called polycystic kidney disease 1 (PKD 1), the more aggressive form of the disease.13,14 PKD 2 (the form associated with the gene PKD2), though less common, is also likely to progress to end-stage renal disease (ESRD), but at a later age (median age of 74 years, compared with 54 in patients with PKD 1).14 ADPKD accounts for about 5% of cases of ESRD in North America,9 but for most patients, presentation and decreased renal function do not occur until the 40s.15 However, patients with the risk factors listed in Table 15,16-19 are likely to experience a more rapid and aggressive form of the disease.
Even with the same germline mutation in a family with this inherited disease, the severity of ADPKD among family members is quite variable; this is true even in the case of twins.9,10,20 Since the age and symptoms at presentation can vary so greatly, a uniform method of identifying patients with ADPKD, along with staging, was needed. Most patients do not undergo genetic testing (ie, DNA linkage or gene-based direct sequencing9) for a diagnosis of ADPKD or to differentiate between the PKD 1 and PKD 2 disease forms unless they are participating in a research study. Diagnostic criteria were needed that were applicable for any type of ADPKD.
In 2009, the University of Toronto’s Division of Nephrology convened experts in the fields of nephrology and radiology to reach a consensus on standardized ultrasonographic diagnostic criteria.21 They formulated definitions based on a study of 948 individuals who were at risk for either PKD 1 or PKD 2 (see Table 221). The specificity and sensitivity of the resulting criteria range from 82% to 100%, making it possible to standardize the care and classification of renal patients worldwide.
Since family members with the same genotypes can experience very divergent disease manifestations, the two-hit hypothesis has been developed.22 In simple terms, it proposes that after the germline mutation (PKD1 or PKD2), there is a second somatic mutation that leads to progressive cyst formation; when the number and size of cysts increase, the patient starts to experience symptoms of ADPKD.22
Age at presentation can be quite variable, as can the presenting symptoms. Most patients with PKD 1 present in their 50s, with 54 being the average age in US patients.14 The most common presenting symptom is flank or back pain.2,5 The pain is due to the massive enlargement of the kidneys, causing a stretching of the kidney capsule and leading to a chronic, dull and persistent pain in the low back. Severe pain, sharp and cutting, occurs when one of the cysts hemorrhages; to some patients, the pain resembles a quick, powerful “kick in the back.” Hematuria can occur following cyst hemorrhage; depending on the location of the cyst that burst within the kidney (ie, how close it is to the collecting system) and how large it is, the amount and color of the hematuria can be impressive.
ADPKD is more common in men than women, and cyst rupture can be precipitated by trauma or lifting heavy objects. Cyst hemorrhage can turn the urine bright red, which is especially frightening to the male patient. Hematuria is often the key presenting symptom in patients who will be diagnosed with ADPKD-induced hypertension.
Besides hematuria, other common manifestations of ADPKD include:
• Hypertension (60% of affected patients, which increases to 100% by the time ESRD develops)
• Extrarenal cysts (100% of affected patients)
• Urinary tract infections
• Nephrolithiasis (20% of affected patients)
• Proteinuria, occasionally (18% of affected patients).2,5,23
Among these manifestations, those most commonly attributed to a diagnosis of ADPKD are hypertension, kidney stones, and urinary tract or kidney infections. Since isolated proteinuria is unusual in patients with ADPKD, it is recommended that another cause of kidney disease be explored in patients with this presentation.24
Extrarenal manifestations of cyst development are common, eventually occurring in all patients as they age. Hepatic cysts are universal in patients with ADPKD by age 30, although hepatic function is preserved. There may be a mild elevation in the alkaline phosphatase level in patients with ADPKD, resulting from the presence of hepatic cysts. Cysts may also be found in the pancreas, spleen, thyroid, and epididymis.5,25 Some patients may complain of dyspnea, pain, early satiety, or lower extremity edema as a result of enlarged cyst.
The Case Patient
Because you recently attended a lecture about ADPKD, you are aware that flank pain in men with hypertension is indicative of ADPKD until proven otherwise. Believing that this patient’s hypertension is renal in origin, you order an abdominal ultrasound. You also order a comprehensive metabolic panel and a complete blood count. The patient’s GFR is measured at 89 mL/min (indicative of stage 2 kidney disease). Other results are shown in Table 3.
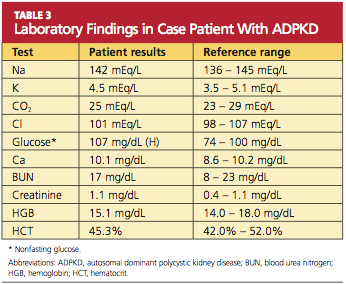
The very broad differential includes essential hypertension, hypertension resulting from intake of “power drinks” or salt in an athlete, illicit use of medications (including steroids), herniated disc leading to transient hypertension, and urinary tract infection or sexually transmitted disease. All of this is moot when the ultrasound shows both kidneys measuring greater than 15 cm, with four distinct cysts on the right kidney and three distinct cysts on the left.
You explain to the patient that ADPKD is a genetic disease and that he and his siblings each had a certain chance of inheriting it. Although different presentations may occur (“congenital” polycystic kidney disease, hypertension, or obesity), they all must undergo ultrasonographic screening for ADPKD. You add that although ADPKD is a genetic disease, it can also be diagnosed in different members of the same family at different ages.
TREATMENT
The goal of treatment for the patient with ADPKD is to slow cyst development and the natural course of the disease. If this can be achieved, the need for dialysis or kidney transplantation may be postponed for a number of years. Because cyst growth causes an elevation in renin and activates the angiotensin II renin system26 (see figure,24), an ACE inhibitor is the most effective treatment to lower blood pressure and thus slow the progression of ADPKD. Most patients with ADPKD are started on an ACE inhibitor at an early age to slow the rate of disease progression.27,28 Several studies are under way to determine the best antihypertensive medication and the optimal age for initiating treatment.29,30
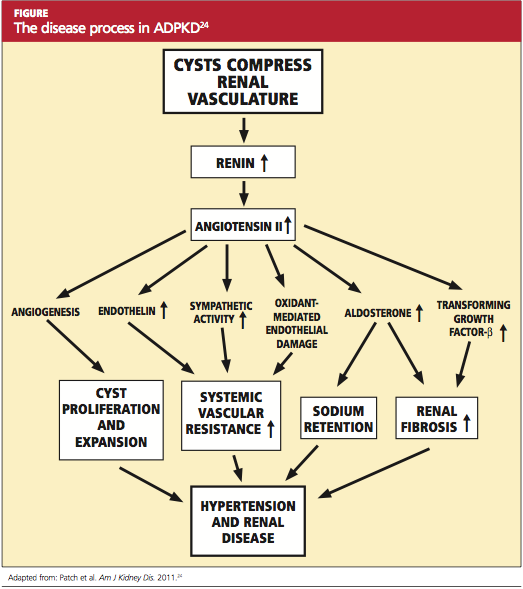
Lipid screening and treatment for dyslipidemia are important23 because ADPKD can lead to a reduction in kidney function, resulting in chronic kidney disease (CKD). CKD is considered a coronary heart disease risk equivalent, and most professionals will treat the patient with ADPKD for hyperlipidemia.23,31 While there are no data showing that statin use will reduce the incidence of ESRD or delay the need for dialysis or kidney transplantation in patients with ADPKD, the beneficial effects of good renal blood flow and endothelial function have been noted.32,33
One of the most common and significant complications in ADPKD is intracranial hemorrhage resulting from a ruptured cerebral aneurysm. In the younger adult, the incidence of cerebral aneurysm is 4%, but incidence increases to 10% in patients older than 65.34-36 Family clusters of aneurysms have been reported.37 If an intracranial aneurysm is found in the family history, the risk of an aneurysm in another family member increases to 22%.38
Since rupture of an intracranial hemorrhage is associated with a 30-day mortality rate of 50% and 80% morbidity,5,38 standard of care for patients with ADPKD includes CT or magnetic resonance angiographic (MRA) screening in the asymptomatic patient with a positive family history.34,38 If an aneurysm is found, the lifetime chance of rupture is 50%, although the risk is greater in the case of an aneurysm larger than 10 mm.5
As in all patients with kidney disease, left ventricular hypertrophy is common among patients with ADPKD.23,28,39
The Case Patient
The patient is started on an ACE inhibitor, scheduled for fasting lipid screening, and referred to a nephrology practice for disease management. As research and investigation of possible treatment options for ADPKD are ongoing, the patient may benefit from participating in a new research protocol.
Because the patient’s family has no history of cerebral aneurysm, CT/MRA screening is not required. A discussion of the pros and cons of genetic testing for the entire family, including nieces and nephews, is initiated. The patient and his family are referred to a genetic counselor to decide whether the benefit of early treatment for hypertension outweighs the risk of carrying a diagnosis of ADPKD for his younger relatives, who may later seek health insurance coverage.
NATURAL PROGRESSION OF ADPKD
Hypertension and cyst formation will continue as the patient ages. The natural progression of ADPKD is to renal failure with renal replacement therapy (dialysis or organ transplantation) as treatment options. If the progression of ADPKD can be slowed through pharmacotherapy, the patient may live for many years without needing dialysis. This ideal can be accomplished only through aggressive hypertension control, which should be started in the teenage years.23,30,31
Suggestions to increase fluid consumption and to limit the use of NSAIDs, contrast dye, and MRI with gadolinium are appropriate. It is rare for hypertension to be diagnosed before some organ damage has already occurred.12 Often the patient’s renal function, as determined by measuring the GFR, remains stable until the patient reaches his or her 40s.40 However, kidney damage often begins before any detectable change in GFR. Once the GFR does start to decline, the average decrease is 4.4 to 5.9 mL/min/1.73m2 each year.41
MANAGEMENT CONSIDERATIONS
For ESRD Organ Transplantation
Kidney transplantation—the only curative treatment for ADPKD—can be offered to patients once the GFR falls below 20 mL/min. However, the patient with ADPKD can experience kidney enlargement to such an extent that introducing a third kidney into the limited abdominal space becomes technically difficult. Although nephrectomy is avoided whenever possible, there are cases in which there is no alternative.42
In addition to space concerns, recurrent urinary tract infections, chronic pain, renal cell carcinoma, chronic hematuria, or chronic cyst infections can necessitate a nephrectomy.43,44 A laparoscopic approach with decompression of cysts or removal of only one kidney is preferred.43,45 If removal of both kidneys is required before a transplant, the patient must be maintained on dialysis until after transplantation. Since the transplant waiting list can exceed seven years in some areas, most patients arrange for a willing live donor before agreeing to a bilateral nephrectomy.46,47
Dialysis
Either peritoneal dialysis (PD) or hemodialysis (HD) can be offered to patients with severe ADPKD. Depending on the size of the native kidneys and the history of previous abdominal surgery, PD often offers a better chance of survival in these patients, particularly compared with patients who have ESRD associated with other causes.48
For management of the patient with ADPKD who receives PD, it can be difficult to differentiate between the pain of a cyst and the pain of a peritoneal infection. Generally, cyst rupture is accompanied by hematuria; and peritonitis, by cloudy fluid.5 Management provided by an experienced nephrologist and PD nurse is vital.
In ADPKD patients who undergo HD, too, survival is better than in patients who have ESRD with other causes49,50; five-year survival can be as high as 10% to 15%.51 This is likely due to the lower incidence of coronary artery disease in the ADPKD population, compared with patients who have ESRD associated with other chronic diseases.49
FUTURE TRENDS AND ONGOING TRIALS
HALT PKD29,30 is an NIH-funded, double-blind study to determine whether adding an angiotensin receptor blocker (ARB) to standard ACE inhibitor therapy results in a more significant decrease in the progression of renal cysts. The rationale for this is that the ARB is expected to block the renin-angiotensin-aldosterone system in the kidney. Use of ACE inhibitor monotherapy versus ARB/ACE inhibitor therapy is being compared in two study arms: patients between ages 15 and 49 with a GFR of 60 mL/min or greater; and patients between ages 18 and 64 with a GFR of 25 to 60 mL/min.29 To date, preliminary results indicate no benefit in adding the second medication.49
The TEMPO Trial52 is a multicenter, double-blind study looking at the effect of tolvaptan on renal cyst growth. Tolvaptan is a potent vasopressin receptor antagonist, and in vitro evidence has shown that intracellular cyclic adenosine monophosphate (cAMP) plays a large role in the development of cysts in patients with ADPKD. If it is possible to block the cAMP that is causing cyst growth, progression of ADPKD should slow.53,54 Only short-term effects of tolvaptan use are currently known.55
High Water Intake to Slow Progression of Polycystic Kidney Disease56 is an open-label, nonrandomized trial in which patients drink a minimum of
3 L of water. Previously, a small study showed that an increase in fluid intake partially suppresses the urine osmolality and the serum antidiuretic hormone (ADH) levels.57 According to this theory, increasing water intake to greater than 3 L/d may result in complete suppression of ADH and cAMP. This is a small study (n = 20),56 since patients with ADPKD are likely to have urinary concentrating defects, and hyponatremia is a concern is in these patients.58
Sirolimus and ADPKD59 is an open-label randomized study to see whether sirolimus (also known as rapamycin) can reduce cyst growth. Originally, it was noted that posttransplant ADPKD patients underwent a regression of both liver and kidney cysts when they were taking sirolimus, and a preliminary crossover study was done.60 However, preliminary results at 18 months showed no difference in renal growth or cyst growth but did show kidney damage as determined by an increase of proteinuria in the treatment group.59 The study is still in progress.
Somatostatin in Polycystic Kidney61 is a long-term (three-year) study following patients who agreed to participate in a randomized, double-blind protocol; in it, an intramuscular injection of either an octreotide (ie, somatastatin) or placebo was administered every four weeks for one year in an effort to reduce the size of kidney and liver cysts.62 At one year, the quality of life in the treatment group was rated better, as measured by pain reduction and improved physical activity. Cyst growth in the treatment group was smaller for both the kidney and liver. However, the GFR decreased to the same degree in both groups.62
CONCLUSION
ADPKD is a common, often overlooked genetic disease that is a cause of hypertension. ADPKD’s presenting symptoms of flank pain, back pain, and/or hematuria often bring the patient to the provider, but a high index of suspicion must be maintained to diagnose these patients at an early age. Due to the variable presentation even within affected families, many patients do not realize that their family carries the PKD gene.
While genetic testing is available, ultrasound is a quick, relatively inexpensive, and easy method to screen for this diagnosis. The progression of ADPKD to ESRD, requiring dialysis or organ transplantation, can be slowed with early and aggressive treatment of hypertension. As with all patients affected by renal impairment, suggestions for patients with ADPKD to avoid use of NSAIDs, contrast dye, and gadolinium-enhanced MRI are appropriate. The primary care PA or NP is in an appropriate position to see to this.
REFERENCES
1. Newschaffer CJ, Falb MD, Gurney JG. National autism prevalence trends from United States special education data. Pediatrics. 2005;115 (3):e277-e282.
2. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287-1301.
3. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16: European Polycystic Kidney Disease Consortium. Cell. 1994;77(6):881-894.
4. Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996; 272(5266):1339-1342.
5. Chapman AB. Polycystic and other cystic kidney diseases. In: Greenberg A, ed. Primer on Kidney Diseases: Expert Consult. 5th ed. National Kidney Foundation; 2009:345-353.
6. Fischer E, Legue E, Doyen A, et al. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006;38(1):21-23.
7. Murcia NS, Sweeney WE Jr, Avner ED. New insights into the molecular pathophysiology of polycystic kidney disease. Kidney Int. 1999;55 (4):1187-1197.
8. Paterson AD, Pei Y. Is there a third gene for autosomal dominant polycystic kidney disease? Kidney Int. 1998;54(5):1759-1761.
9. Pei Y. Practical genetics for autosomal dominant polycystic kidney disease. Nephron Clin Pract. 2011;118(1):c19-c30.
10. Tan YC, Blumenfeld J, Rennert H. Autosomal dominant polycystic kidney disease: genetics, mutations and microRNAs. Biochim Biophys Acta. 2011 Mar 16 [Epub ahead of print].
11. Avner ED, Sweeney WE Jr, Nelson WJ. Abnormal sodium pump distribution during renal tubulogenesis in congenital murine polycystic kidney disease. Proc Natl Acad Sci U S A. 1992;89(16):7447-7451.
12. Torra R, Badenas C, Darnell A, et al. Linkage, clinical features, and prognosis of autosomal dominant polycystic kidney disease types 1 and 2. J Am Soc Nephrol. 1996;7(10):2142-2151.
13. Davies F, Coles GA, Harper PS, et al. Polycystic kidney disease re-evaluated: a population-based study. Q J Med. 1991;79(290):477-485.
14. Hateboer N, v Dijk MA, Bogdanova N, et al; European PKD1-PKD2 Study Group. Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet. 1999;353(9147):103-107.
15. Peters DJ, Breuning MH. Autosomal dominant polycystic kidney disease: modification of disease progression. Lancet. 2001;358(9291):1439-1444.
16. Dicks E, Ravani P, Langman D, et al. Incident renal events and risk factors in autosomal dominant polycystic kidney disease: a population- and family-based cohort followed for 22 years. Clin J Am Soc Nephrol. 2006;1(4):710-717.
17. Fick-Brosnahan GM, Belz MM, McFann KK, etc. Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease: a longitudinal study. Am J Kidney Dis. 2002;39(6):1127-1134.
18. Fick-Brosnahan GM, Tran ZV, Johnson AM, et al. Progression of autosomal-dominant polycystic kidney disease in children. Kidney Int. 2001;59(5):1654-1662.
19. Johnson AM, Gabow PA. Identification of patients with autosomal dominant polycystic kidney disease at highest risk for end-stage renal disease. J Am Soc Nephrol. 1997;8(10): 1560-1567.
20. Risk D. Autosomal dominant polycystic kidney disease. Presented at: National Kidney Foundation, Spring Clinical Meetings; April 28, 2011; Las Vegas, NV.
21. Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205-212.
22. Watnick T, Germino GG. Molecular basis of autosomal dominant polycystic kidney disease. Semin Nephrol. 1999;19(4):327-343.
23. Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol. 2009;5(4):221-228.
24. Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis. 2011;57(6):856-862.
25. Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17(2):173-180.
26. Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med. 1990;323(16):1091-1096.
27. Jafar TH, Stark PC, Schmid CH, et al. The effect of angiotensin-converting-enzyme inhibitors on progression of advanced polycystic kidney disease. Kidney Int. 2005;67(1):265-271.
28. Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009;20(9):1888-1893.
29. National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH, sponsor. HALT PKD (Halt Progression of Polycystic Kidney Disease): Efficacy of Aggressive Renin-Angiotensin-Aldosterone Axis Blockade in Preventing/Slowing Renal Function Decline in ADPKD. www2.niddk.nih.gov/NR/rdonlyres/175578F6-62B4-429A-9BBF-96CCEC2FFB3A/0/KUHHALT PKDPROTOCOL9107.pdf. Accessed July 22, 2011.
30. Chapman AB, Torres VE, Perrone RD, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol. 2010;5(1):102-109.
31. Taylor M, Johnson AM, Tison M, et al. Earlier diagnosis of autosomal dominant polycystic kidney disease: importance of family history and implications for cardiovascular and renal complications. Am J Kidney Dis. 2005;46(3):415-423.
32. Namli S, Oflaz H, Turgut F, et al. Improvement of endothelial dysfunction with simvastatin in patients with autosomal dominant polycystic kidney disease. Ren Fail. 2007;29(1):55-59.
33. Bremmer MS, Jacobs SC. Renal artery embolization for the symptomatic treatment of adult polycystic kidney disease. Nat Clin Pract Nephrol. 2008;4(5):236-237.
34. Chapman AB, Rubinstein D, Hughes R, et al. Intracranial aneurysms in autosomal dominant polycystic kidney disease. N Engl J Med. 1992; 327(13):916-920.
35. Schievink WI, Torres VE, Piepgras DG, Wiebers DO. Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1992;3(1):88-95.
36. Fick GM, Gabow PA. Hereditary and acquired cystic disease of the kidney. Kidney Int. 1994;46(4):951-964.
37. Watson ML. Complications of polycystic kidney disease. Kidney Int. 1997;51(1):353-365.
38. Huston J 3rd, Torres VE, Sulivan PP, et al. Value of magnetic resonance angiography for the detection of intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;3(12):1871-1877.
39. Longenecker JC, Coresh J, Powe NR, et al. Traditional cardiovascular disease risk factors in dialysis patients compared with the general population: the CHOICE Study. J Am Soc Nephrol. 2002;13(7):1918-1927.
40. Meijer E, Rook M, Tent H, et al. Early renal abnormalities in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2010; 5(6):1091-1098.
41. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149-168.
42. Tabibi A, Simforoosh N, Abadpour P, et al. Concomitant nephrectomy of massively enlarged kidneys and renal transplantation in autosomal dominant polycystic kidney disease. Transplant Proc. 2005;37(7):2939-2940.
43. Dunn MD, Portis AJ, Elbahnasy AM, et al. Laparoscopic nephrectomy in patients with end-stage renal disease and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000;35(4):720-725.
44. Sulikowski T, Tejchman K, Zietek Z, et al. Experience with autosomal dominant polycystic kidney disease in patients before and after renal transplantation: a 7-year observation. Transplant Proc. 2009;41(1):177-180.
45. Desai MR, Nandkishore SK, Ganpule A, Thimmegowda M. Pretransplant laparoscopic nephrectomy in adult polycystic kidney disease: a single centre experience. BJU Int. 2008;101 (1):94-97.
46. Glassman DT, Nipkow L, Bartlett ST, Jacobs SC. Bilateral nephrectomy with concomitant renal graft transplantation for autosomal dominant polycystic kidney disease. J Urol. 2000;164 (3 pt 1):661-664.
47. Fuller TF, Brennan TV, Feng S, et al. End stage polycystic kidney disease: indications and timing of native nephrectomy relative to kidney transplantation. J Urol. 2005;174(6):2284-2288.
48. Abbott KC, Agodoa LY. Polycystic kidney disease at end-stage renal disease in the United States: patient characteristics and survival. Clin Nephrol. 2002;57(3):208-214.
49. Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis. 2001;38(4):777-784.
50. Batista PB, Lopes AA, Costa FA. Association between attributed cause of end-stage renal disease and risk of death in Brazilian patients receiving renal replacement therapy. Ren Fail. 2005;27(6):651-656.
51. Pirson Y, Christophe JL, Goffin E. Outcome of renal replacement therapy in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 1996;11 suppl 6:24-28.
52. Torres VE, Meijer E, Bae KT, et al. Rationale and design of the TEMPO (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes) 3-4 Study. Am J Kidney Dis. 2011;57(5):692-699.
53. Calvet JP. Strategies to inhibit cyst formation in ADPKD. Clin J Am Soc Nephrol. 2008;3 (4):1205-1211.
54. Grantham JJ. Lillian Jean Kaplan International Prize for advancement in the understanding of polycystic kidney disease. Understanding polycystic kidney disease: a systems biology approach. Kidney Int. 2003;64(4):1157-1162.
55. Irazabal MV, Torres VE, Hogan MC, et al. Short-term effects of tolvaptan on renal function and volume in patients with Autosomal Dominant Polycystic Kidney Disease. Kidney Int. 2011 May 4 [Epub ahead of print].
56. New York University, sponsor. High Water Intake to Slow Progression of Polycystic Kidney Disease. http://clinicaltrials.gov/ct2/show/NCT00784030. Accessed July 22, 2011.
57. Wang CJ, Creed C, Winklhofer FT, Grantham JJ. Water prescription in autosomal dominant polycystic kidney disease: a pilot study. Clin J Am Soc Nephrol. 2011;6(1):192-197.
58. Grampsas SA, Chandhoke Ps, Fan J, et al. Anatomic and metabolic risk factors for nephrolithiasis in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000;36(1):53-57.
59. Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363(9):820-829.
60. Perico N, Antiga L, Caroli A, et al. Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol. 2010;21(6):1031-1040.
61. Mario Negri Institute for Pharmacological Research, sponsor. Somatostatin in Polycystic Kidney: a Long-term Three Year Follow up Study. http://clinicaltrials.gov/ct2/show/NCT00309283. Accessed July 22, 2011.
62. Hogan MC, Masyuk TV, Page LJ, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010; 21(6):1052-1061.
Twice as common as autism and half as well-known,1 autosomal polycystic kidney disease (ADPKD) occurs in one in 400 to one in 1,000 people.2 It is an inherited progressive genetic disorder that causes hypertension and decreased renal function and, over time, can lead to kidney failure. Two polycstin genes that code for ADPKD, PKD1 and PKD2, were identified in 1994 and 1996, respectively.3,4 Awareness and understanding of the genes responsible for ADPKD have increased clinicians’ ability to identify at-risk patients and to slow or alter the course of the disease.
Case Presentation
A 45-year-old black man presents to your office with severe, nonradiating back pain and new-onset hypertension. Regarding the pain, he stated, “I turned around to see who kicked me, but no one was there.” When the pain began, he went to see the nurse at the school where he is employed, and she found that his blood pressure was high at 162/90 mm Hg. Although the patient’s back pain is resolving, he is very concerned about his blood pressure, since he has never had a high reading before.
He is the baseball coach and physical education teacher at the local high school and is in excellent physical condition as a result of his professional interaction with teenagers every day. He does not smoke or use any illicit drugs but does admit to occasional alcohol consumption. His medical history is significant only for occasional broken fingers and twisted ankles, all occurring while he was engaged in sports.
His family history includes one brother without medical problems, a brother and a sister with hypertension, a sister with diabetes and obesity, and a brother with a congenital abnormality that required a living donor kidney transplant at age 17 (the father served as donor). No family-wide workup has ever been done because no one practitioner has ever made a connection among these conditions and considered a diagnosis of ADPKD.
The patient’s blood pressure in the office is 172/92 mm Hg while sitting and 166/88 mm Hg while standing. He is somewhat sore with a localized spasm in the lumbar-sacral area but no radiation of pain. The patient has trouble touching his toes but reports that he can never touch his toes. His straight leg lift is negative. The rest of his physical exam is noncontributory.
What should be the next step in this patient’s workup?
PATHOPHYSIOLOGY
ADPKD is a progressive expansion of numerous fluid-filled cysts that result in massive enlargement of the kidneys.5 Less than 5% of all nephrons become cystic; however, the average volume of a polycystic kidney is 1,000 mL (normal, 300 mL), that is, the volume of a standard-sized pineapple. Even with this significant enlargement, a decline in the glomerular filtration rate (GFR) is not usually seen initially. Each cyst is derived from a single hyperproliferative epithelial cell. Increased cellular proliferation, followed by fluid secretion and alterations in the extracellular matrix, cause an outpouching from the parent nephron, which eventually detaches from the parent nephron and continues to enlarge and autonomously secrete fluid.6,7
PKD1 and PKD2 are two genes responsible for ADPKD that have been isolated so far. Since there are families carrying neither the PKD1 nor the PKD2 gene that still have an inherited type of ADPKD, there is suspicion that at least one more PKD gene, not yet isolated, exists.8 It is also possible that other genetic or environmental factors may be at play.9,10
In 1994, the PKD1 gene was isolated on chromosome 16,3 and it was found to code for polycystin 1. A lack of polycystin 1 causes an abnormality in the Na+/K(+)-ATPase pumps, leading to abnormal sodium reabsorption.11 How and why this happens is not quite clear. However, the hypertension that is a key objective finding in patients with ADPKD is thought to result from this pump abnormality.
PKD2 is found on the long arm of chromosome 4 and codes for polycystin 2.4 Polycystin 2 is an amino acid that is responsible for voltage-activated cellular calcium channels,5 again explaining the hypertension so commonly seen in the course of ADPKD. ADPKD-associated hypertension may be present as early as the teenage years.12
EPIDEMIOLOGY
More than 85% of ADPKD cases are associated with PKD1, and this form is called polycystic kidney disease 1 (PKD 1), the more aggressive form of the disease.13,14 PKD 2 (the form associated with the gene PKD2), though less common, is also likely to progress to end-stage renal disease (ESRD), but at a later age (median age of 74 years, compared with 54 in patients with PKD 1).14 ADPKD accounts for about 5% of cases of ESRD in North America,9 but for most patients, presentation and decreased renal function do not occur until the 40s.15 However, patients with the risk factors listed in Table 15,16-19 are likely to experience a more rapid and aggressive form of the disease.
Even with the same germline mutation in a family with this inherited disease, the severity of ADPKD among family members is quite variable; this is true even in the case of twins.9,10,20 Since the age and symptoms at presentation can vary so greatly, a uniform method of identifying patients with ADPKD, along with staging, was needed. Most patients do not undergo genetic testing (ie, DNA linkage or gene-based direct sequencing9) for a diagnosis of ADPKD or to differentiate between the PKD 1 and PKD 2 disease forms unless they are participating in a research study. Diagnostic criteria were needed that were applicable for any type of ADPKD.
In 2009, the University of Toronto’s Division of Nephrology convened experts in the fields of nephrology and radiology to reach a consensus on standardized ultrasonographic diagnostic criteria.21 They formulated definitions based on a study of 948 individuals who were at risk for either PKD 1 or PKD 2 (see Table 221). The specificity and sensitivity of the resulting criteria range from 82% to 100%, making it possible to standardize the care and classification of renal patients worldwide.
Since family members with the same genotypes can experience very divergent disease manifestations, the two-hit hypothesis has been developed.22 In simple terms, it proposes that after the germline mutation (PKD1 or PKD2), there is a second somatic mutation that leads to progressive cyst formation; when the number and size of cysts increase, the patient starts to experience symptoms of ADPKD.22
Age at presentation can be quite variable, as can the presenting symptoms. Most patients with PKD 1 present in their 50s, with 54 being the average age in US patients.14 The most common presenting symptom is flank or back pain.2,5 The pain is due to the massive enlargement of the kidneys, causing a stretching of the kidney capsule and leading to a chronic, dull and persistent pain in the low back. Severe pain, sharp and cutting, occurs when one of the cysts hemorrhages; to some patients, the pain resembles a quick, powerful “kick in the back.” Hematuria can occur following cyst hemorrhage; depending on the location of the cyst that burst within the kidney (ie, how close it is to the collecting system) and how large it is, the amount and color of the hematuria can be impressive.
ADPKD is more common in men than women, and cyst rupture can be precipitated by trauma or lifting heavy objects. Cyst hemorrhage can turn the urine bright red, which is especially frightening to the male patient. Hematuria is often the key presenting symptom in patients who will be diagnosed with ADPKD-induced hypertension.
Besides hematuria, other common manifestations of ADPKD include:
• Hypertension (60% of affected patients, which increases to 100% by the time ESRD develops)
• Extrarenal cysts (100% of affected patients)
• Urinary tract infections
• Nephrolithiasis (20% of affected patients)
• Proteinuria, occasionally (18% of affected patients).2,5,23
Among these manifestations, those most commonly attributed to a diagnosis of ADPKD are hypertension, kidney stones, and urinary tract or kidney infections. Since isolated proteinuria is unusual in patients with ADPKD, it is recommended that another cause of kidney disease be explored in patients with this presentation.24
Extrarenal manifestations of cyst development are common, eventually occurring in all patients as they age. Hepatic cysts are universal in patients with ADPKD by age 30, although hepatic function is preserved. There may be a mild elevation in the alkaline phosphatase level in patients with ADPKD, resulting from the presence of hepatic cysts. Cysts may also be found in the pancreas, spleen, thyroid, and epididymis.5,25 Some patients may complain of dyspnea, pain, early satiety, or lower extremity edema as a result of enlarged cyst.
The Case Patient
Because you recently attended a lecture about ADPKD, you are aware that flank pain in men with hypertension is indicative of ADPKD until proven otherwise. Believing that this patient’s hypertension is renal in origin, you order an abdominal ultrasound. You also order a comprehensive metabolic panel and a complete blood count. The patient’s GFR is measured at 89 mL/min (indicative of stage 2 kidney disease). Other results are shown in Table 3.
The very broad differential includes essential hypertension, hypertension resulting from intake of “power drinks” or salt in an athlete, illicit use of medications (including steroids), herniated disc leading to transient hypertension, and urinary tract infection or sexually transmitted disease. All of this is moot when the ultrasound shows both kidneys measuring greater than 15 cm, with four distinct cysts on the right kidney and three distinct cysts on the left.
You explain to the patient that ADPKD is a genetic disease and that he and his siblings each had a certain chance of inheriting it. Although different presentations may occur (“congenital” polycystic kidney disease, hypertension, or obesity), they all must undergo ultrasonographic screening for ADPKD. You add that although ADPKD is a genetic disease, it can also be diagnosed in different members of the same family at different ages.
TREATMENT
The goal of treatment for the patient with ADPKD is to slow cyst development and the natural course of the disease. If this can be achieved, the need for dialysis or kidney transplantation may be postponed for a number of years. Because cyst growth causes an elevation in renin and activates the angiotensin II renin system26 (see figure,24), an ACE inhibitor is the most effective treatment to lower blood pressure and thus slow the progression of ADPKD. Most patients with ADPKD are started on an ACE inhibitor at an early age to slow the rate of disease progression.27,28 Several studies are under way to determine the best antihypertensive medication and the optimal age for initiating treatment.29,30
Lipid screening and treatment for dyslipidemia are important23 because ADPKD can lead to a reduction in kidney function, resulting in chronic kidney disease (CKD). CKD is considered a coronary heart disease risk equivalent, and most professionals will treat the patient with ADPKD for hyperlipidemia.23,31 While there are no data showing that statin use will reduce the incidence of ESRD or delay the need for dialysis or kidney transplantation in patients with ADPKD, the beneficial effects of good renal blood flow and endothelial function have been noted.32,33
One of the most common and significant complications in ADPKD is intracranial hemorrhage resulting from a ruptured cerebral aneurysm. In the younger adult, the incidence of cerebral aneurysm is 4%, but incidence increases to 10% in patients older than 65.34-36 Family clusters of aneurysms have been reported.37 If an intracranial aneurysm is found in the family history, the risk of an aneurysm in another family member increases to 22%.38
Since rupture of an intracranial hemorrhage is associated with a 30-day mortality rate of 50% and 80% morbidity,5,38 standard of care for patients with ADPKD includes CT or magnetic resonance angiographic (MRA) screening in the asymptomatic patient with a positive family history.34,38 If an aneurysm is found, the lifetime chance of rupture is 50%, although the risk is greater in the case of an aneurysm larger than 10 mm.5
As in all patients with kidney disease, left ventricular hypertrophy is common among patients with ADPKD.23,28,39
The Case Patient
The patient is started on an ACE inhibitor, scheduled for fasting lipid screening, and referred to a nephrology practice for disease management. As research and investigation of possible treatment options for ADPKD are ongoing, the patient may benefit from participating in a new research protocol.
Because the patient’s family has no history of cerebral aneurysm, CT/MRA screening is not required. A discussion of the pros and cons of genetic testing for the entire family, including nieces and nephews, is initiated. The patient and his family are referred to a genetic counselor to decide whether the benefit of early treatment for hypertension outweighs the risk of carrying a diagnosis of ADPKD for his younger relatives, who may later seek health insurance coverage.
NATURAL PROGRESSION OF ADPKD
Hypertension and cyst formation will continue as the patient ages. The natural progression of ADPKD is to renal failure with renal replacement therapy (dialysis or organ transplantation) as treatment options. If the progression of ADPKD can be slowed through pharmacotherapy, the patient may live for many years without needing dialysis. This ideal can be accomplished only through aggressive hypertension control, which should be started in the teenage years.23,30,31
Suggestions to increase fluid consumption and to limit the use of NSAIDs, contrast dye, and MRI with gadolinium are appropriate. It is rare for hypertension to be diagnosed before some organ damage has already occurred.12 Often the patient’s renal function, as determined by measuring the GFR, remains stable until the patient reaches his or her 40s.40 However, kidney damage often begins before any detectable change in GFR. Once the GFR does start to decline, the average decrease is 4.4 to 5.9 mL/min/1.73m2 each year.41
MANAGEMENT CONSIDERATIONS
For ESRD Organ Transplantation
Kidney transplantation—the only curative treatment for ADPKD—can be offered to patients once the GFR falls below 20 mL/min. However, the patient with ADPKD can experience kidney enlargement to such an extent that introducing a third kidney into the limited abdominal space becomes technically difficult. Although nephrectomy is avoided whenever possible, there are cases in which there is no alternative.42
In addition to space concerns, recurrent urinary tract infections, chronic pain, renal cell carcinoma, chronic hematuria, or chronic cyst infections can necessitate a nephrectomy.43,44 A laparoscopic approach with decompression of cysts or removal of only one kidney is preferred.43,45 If removal of both kidneys is required before a transplant, the patient must be maintained on dialysis until after transplantation. Since the transplant waiting list can exceed seven years in some areas, most patients arrange for a willing live donor before agreeing to a bilateral nephrectomy.46,47
Dialysis
Either peritoneal dialysis (PD) or hemodialysis (HD) can be offered to patients with severe ADPKD. Depending on the size of the native kidneys and the history of previous abdominal surgery, PD often offers a better chance of survival in these patients, particularly compared with patients who have ESRD associated with other causes.48
For management of the patient with ADPKD who receives PD, it can be difficult to differentiate between the pain of a cyst and the pain of a peritoneal infection. Generally, cyst rupture is accompanied by hematuria; and peritonitis, by cloudy fluid.5 Management provided by an experienced nephrologist and PD nurse is vital.
In ADPKD patients who undergo HD, too, survival is better than in patients who have ESRD with other causes49,50; five-year survival can be as high as 10% to 15%.51 This is likely due to the lower incidence of coronary artery disease in the ADPKD population, compared with patients who have ESRD associated with other chronic diseases.49
FUTURE TRENDS AND ONGOING TRIALS
HALT PKD29,30 is an NIH-funded, double-blind study to determine whether adding an angiotensin receptor blocker (ARB) to standard ACE inhibitor therapy results in a more significant decrease in the progression of renal cysts. The rationale for this is that the ARB is expected to block the renin-angiotensin-aldosterone system in the kidney. Use of ACE inhibitor monotherapy versus ARB/ACE inhibitor therapy is being compared in two study arms: patients between ages 15 and 49 with a GFR of 60 mL/min or greater; and patients between ages 18 and 64 with a GFR of 25 to 60 mL/min.29 To date, preliminary results indicate no benefit in adding the second medication.49
The TEMPO Trial52 is a multicenter, double-blind study looking at the effect of tolvaptan on renal cyst growth. Tolvaptan is a potent vasopressin receptor antagonist, and in vitro evidence has shown that intracellular cyclic adenosine monophosphate (cAMP) plays a large role in the development of cysts in patients with ADPKD. If it is possible to block the cAMP that is causing cyst growth, progression of ADPKD should slow.53,54 Only short-term effects of tolvaptan use are currently known.55
High Water Intake to Slow Progression of Polycystic Kidney Disease56 is an open-label, nonrandomized trial in which patients drink a minimum of
3 L of water. Previously, a small study showed that an increase in fluid intake partially suppresses the urine osmolality and the serum antidiuretic hormone (ADH) levels.57 According to this theory, increasing water intake to greater than 3 L/d may result in complete suppression of ADH and cAMP. This is a small study (n = 20),56 since patients with ADPKD are likely to have urinary concentrating defects, and hyponatremia is a concern is in these patients.58
Sirolimus and ADPKD59 is an open-label randomized study to see whether sirolimus (also known as rapamycin) can reduce cyst growth. Originally, it was noted that posttransplant ADPKD patients underwent a regression of both liver and kidney cysts when they were taking sirolimus, and a preliminary crossover study was done.60 However, preliminary results at 18 months showed no difference in renal growth or cyst growth but did show kidney damage as determined by an increase of proteinuria in the treatment group.59 The study is still in progress.
Somatostatin in Polycystic Kidney61 is a long-term (three-year) study following patients who agreed to participate in a randomized, double-blind protocol; in it, an intramuscular injection of either an octreotide (ie, somatastatin) or placebo was administered every four weeks for one year in an effort to reduce the size of kidney and liver cysts.62 At one year, the quality of life in the treatment group was rated better, as measured by pain reduction and improved physical activity. Cyst growth in the treatment group was smaller for both the kidney and liver. However, the GFR decreased to the same degree in both groups.62
CONCLUSION
ADPKD is a common, often overlooked genetic disease that is a cause of hypertension. ADPKD’s presenting symptoms of flank pain, back pain, and/or hematuria often bring the patient to the provider, but a high index of suspicion must be maintained to diagnose these patients at an early age. Due to the variable presentation even within affected families, many patients do not realize that their family carries the PKD gene.
While genetic testing is available, ultrasound is a quick, relatively inexpensive, and easy method to screen for this diagnosis. The progression of ADPKD to ESRD, requiring dialysis or organ transplantation, can be slowed with early and aggressive treatment of hypertension. As with all patients affected by renal impairment, suggestions for patients with ADPKD to avoid use of NSAIDs, contrast dye, and gadolinium-enhanced MRI are appropriate. The primary care PA or NP is in an appropriate position to see to this.
REFERENCES
1. Newschaffer CJ, Falb MD, Gurney JG. National autism prevalence trends from United States special education data. Pediatrics. 2005;115 (3):e277-e282.
2. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287-1301.
3. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16: European Polycystic Kidney Disease Consortium. Cell. 1994;77(6):881-894.
4. Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996; 272(5266):1339-1342.
5. Chapman AB. Polycystic and other cystic kidney diseases. In: Greenberg A, ed. Primer on Kidney Diseases: Expert Consult. 5th ed. National Kidney Foundation; 2009:345-353.
6. Fischer E, Legue E, Doyen A, et al. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006;38(1):21-23.
7. Murcia NS, Sweeney WE Jr, Avner ED. New insights into the molecular pathophysiology of polycystic kidney disease. Kidney Int. 1999;55 (4):1187-1197.
8. Paterson AD, Pei Y. Is there a third gene for autosomal dominant polycystic kidney disease? Kidney Int. 1998;54(5):1759-1761.
9. Pei Y. Practical genetics for autosomal dominant polycystic kidney disease. Nephron Clin Pract. 2011;118(1):c19-c30.
10. Tan YC, Blumenfeld J, Rennert H. Autosomal dominant polycystic kidney disease: genetics, mutations and microRNAs. Biochim Biophys Acta. 2011 Mar 16 [Epub ahead of print].
11. Avner ED, Sweeney WE Jr, Nelson WJ. Abnormal sodium pump distribution during renal tubulogenesis in congenital murine polycystic kidney disease. Proc Natl Acad Sci U S A. 1992;89(16):7447-7451.
12. Torra R, Badenas C, Darnell A, et al. Linkage, clinical features, and prognosis of autosomal dominant polycystic kidney disease types 1 and 2. J Am Soc Nephrol. 1996;7(10):2142-2151.
13. Davies F, Coles GA, Harper PS, et al. Polycystic kidney disease re-evaluated: a population-based study. Q J Med. 1991;79(290):477-485.
14. Hateboer N, v Dijk MA, Bogdanova N, et al; European PKD1-PKD2 Study Group. Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet. 1999;353(9147):103-107.
15. Peters DJ, Breuning MH. Autosomal dominant polycystic kidney disease: modification of disease progression. Lancet. 2001;358(9291):1439-1444.
16. Dicks E, Ravani P, Langman D, et al. Incident renal events and risk factors in autosomal dominant polycystic kidney disease: a population- and family-based cohort followed for 22 years. Clin J Am Soc Nephrol. 2006;1(4):710-717.
17. Fick-Brosnahan GM, Belz MM, McFann KK, etc. Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease: a longitudinal study. Am J Kidney Dis. 2002;39(6):1127-1134.
18. Fick-Brosnahan GM, Tran ZV, Johnson AM, et al. Progression of autosomal-dominant polycystic kidney disease in children. Kidney Int. 2001;59(5):1654-1662.
19. Johnson AM, Gabow PA. Identification of patients with autosomal dominant polycystic kidney disease at highest risk for end-stage renal disease. J Am Soc Nephrol. 1997;8(10): 1560-1567.
20. Risk D. Autosomal dominant polycystic kidney disease. Presented at: National Kidney Foundation, Spring Clinical Meetings; April 28, 2011; Las Vegas, NV.
21. Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205-212.
22. Watnick T, Germino GG. Molecular basis of autosomal dominant polycystic kidney disease. Semin Nephrol. 1999;19(4):327-343.
23. Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol. 2009;5(4):221-228.
24. Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis. 2011;57(6):856-862.
25. Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17(2):173-180.
26. Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med. 1990;323(16):1091-1096.
27. Jafar TH, Stark PC, Schmid CH, et al. The effect of angiotensin-converting-enzyme inhibitors on progression of advanced polycystic kidney disease. Kidney Int. 2005;67(1):265-271.
28. Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009;20(9):1888-1893.
29. National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH, sponsor. HALT PKD (Halt Progression of Polycystic Kidney Disease): Efficacy of Aggressive Renin-Angiotensin-Aldosterone Axis Blockade in Preventing/Slowing Renal Function Decline in ADPKD. www2.niddk.nih.gov/NR/rdonlyres/175578F6-62B4-429A-9BBF-96CCEC2FFB3A/0/KUHHALT PKDPROTOCOL9107.pdf. Accessed July 22, 2011.
30. Chapman AB, Torres VE, Perrone RD, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol. 2010;5(1):102-109.
31. Taylor M, Johnson AM, Tison M, et al. Earlier diagnosis of autosomal dominant polycystic kidney disease: importance of family history and implications for cardiovascular and renal complications. Am J Kidney Dis. 2005;46(3):415-423.
32. Namli S, Oflaz H, Turgut F, et al. Improvement of endothelial dysfunction with simvastatin in patients with autosomal dominant polycystic kidney disease. Ren Fail. 2007;29(1):55-59.
33. Bremmer MS, Jacobs SC. Renal artery embolization for the symptomatic treatment of adult polycystic kidney disease. Nat Clin Pract Nephrol. 2008;4(5):236-237.
34. Chapman AB, Rubinstein D, Hughes R, et al. Intracranial aneurysms in autosomal dominant polycystic kidney disease. N Engl J Med. 1992; 327(13):916-920.
35. Schievink WI, Torres VE, Piepgras DG, Wiebers DO. Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1992;3(1):88-95.
36. Fick GM, Gabow PA. Hereditary and acquired cystic disease of the kidney. Kidney Int. 1994;46(4):951-964.
37. Watson ML. Complications of polycystic kidney disease. Kidney Int. 1997;51(1):353-365.
38. Huston J 3rd, Torres VE, Sulivan PP, et al. Value of magnetic resonance angiography for the detection of intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;3(12):1871-1877.
39. Longenecker JC, Coresh J, Powe NR, et al. Traditional cardiovascular disease risk factors in dialysis patients compared with the general population: the CHOICE Study. J Am Soc Nephrol. 2002;13(7):1918-1927.
40. Meijer E, Rook M, Tent H, et al. Early renal abnormalities in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2010; 5(6):1091-1098.
41. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149-168.
42. Tabibi A, Simforoosh N, Abadpour P, et al. Concomitant nephrectomy of massively enlarged kidneys and renal transplantation in autosomal dominant polycystic kidney disease. Transplant Proc. 2005;37(7):2939-2940.
43. Dunn MD, Portis AJ, Elbahnasy AM, et al. Laparoscopic nephrectomy in patients with end-stage renal disease and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000;35(4):720-725.
44. Sulikowski T, Tejchman K, Zietek Z, et al. Experience with autosomal dominant polycystic kidney disease in patients before and after renal transplantation: a 7-year observation. Transplant Proc. 2009;41(1):177-180.
45. Desai MR, Nandkishore SK, Ganpule A, Thimmegowda M. Pretransplant laparoscopic nephrectomy in adult polycystic kidney disease: a single centre experience. BJU Int. 2008;101 (1):94-97.
46. Glassman DT, Nipkow L, Bartlett ST, Jacobs SC. Bilateral nephrectomy with concomitant renal graft transplantation for autosomal dominant polycystic kidney disease. J Urol. 2000;164 (3 pt 1):661-664.
47. Fuller TF, Brennan TV, Feng S, et al. End stage polycystic kidney disease: indications and timing of native nephrectomy relative to kidney transplantation. J Urol. 2005;174(6):2284-2288.
48. Abbott KC, Agodoa LY. Polycystic kidney disease at end-stage renal disease in the United States: patient characteristics and survival. Clin Nephrol. 2002;57(3):208-214.
49. Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis. 2001;38(4):777-784.
50. Batista PB, Lopes AA, Costa FA. Association between attributed cause of end-stage renal disease and risk of death in Brazilian patients receiving renal replacement therapy. Ren Fail. 2005;27(6):651-656.
51. Pirson Y, Christophe JL, Goffin E. Outcome of renal replacement therapy in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 1996;11 suppl 6:24-28.
52. Torres VE, Meijer E, Bae KT, et al. Rationale and design of the TEMPO (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes) 3-4 Study. Am J Kidney Dis. 2011;57(5):692-699.
53. Calvet JP. Strategies to inhibit cyst formation in ADPKD. Clin J Am Soc Nephrol. 2008;3 (4):1205-1211.
54. Grantham JJ. Lillian Jean Kaplan International Prize for advancement in the understanding of polycystic kidney disease. Understanding polycystic kidney disease: a systems biology approach. Kidney Int. 2003;64(4):1157-1162.
55. Irazabal MV, Torres VE, Hogan MC, et al. Short-term effects of tolvaptan on renal function and volume in patients with Autosomal Dominant Polycystic Kidney Disease. Kidney Int. 2011 May 4 [Epub ahead of print].
56. New York University, sponsor. High Water Intake to Slow Progression of Polycystic Kidney Disease. http://clinicaltrials.gov/ct2/show/NCT00784030. Accessed July 22, 2011.
57. Wang CJ, Creed C, Winklhofer FT, Grantham JJ. Water prescription in autosomal dominant polycystic kidney disease: a pilot study. Clin J Am Soc Nephrol. 2011;6(1):192-197.
58. Grampsas SA, Chandhoke Ps, Fan J, et al. Anatomic and metabolic risk factors for nephrolithiasis in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000;36(1):53-57.
59. Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363(9):820-829.
60. Perico N, Antiga L, Caroli A, et al. Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol. 2010;21(6):1031-1040.
61. Mario Negri Institute for Pharmacological Research, sponsor. Somatostatin in Polycystic Kidney: a Long-term Three Year Follow up Study. http://clinicaltrials.gov/ct2/show/NCT00309283. Accessed July 22, 2011.
62. Hogan MC, Masyuk TV, Page LJ, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010; 21(6):1052-1061.
Twice as common as autism and half as well-known,1 autosomal polycystic kidney disease (ADPKD) occurs in one in 400 to one in 1,000 people.2 It is an inherited progressive genetic disorder that causes hypertension and decreased renal function and, over time, can lead to kidney failure. Two polycstin genes that code for ADPKD, PKD1 and PKD2, were identified in 1994 and 1996, respectively.3,4 Awareness and understanding of the genes responsible for ADPKD have increased clinicians’ ability to identify at-risk patients and to slow or alter the course of the disease.
Case Presentation
A 45-year-old black man presents to your office with severe, nonradiating back pain and new-onset hypertension. Regarding the pain, he stated, “I turned around to see who kicked me, but no one was there.” When the pain began, he went to see the nurse at the school where he is employed, and she found that his blood pressure was high at 162/90 mm Hg. Although the patient’s back pain is resolving, he is very concerned about his blood pressure, since he has never had a high reading before.
He is the baseball coach and physical education teacher at the local high school and is in excellent physical condition as a result of his professional interaction with teenagers every day. He does not smoke or use any illicit drugs but does admit to occasional alcohol consumption. His medical history is significant only for occasional broken fingers and twisted ankles, all occurring while he was engaged in sports.
His family history includes one brother without medical problems, a brother and a sister with hypertension, a sister with diabetes and obesity, and a brother with a congenital abnormality that required a living donor kidney transplant at age 17 (the father served as donor). No family-wide workup has ever been done because no one practitioner has ever made a connection among these conditions and considered a diagnosis of ADPKD.
The patient’s blood pressure in the office is 172/92 mm Hg while sitting and 166/88 mm Hg while standing. He is somewhat sore with a localized spasm in the lumbar-sacral area but no radiation of pain. The patient has trouble touching his toes but reports that he can never touch his toes. His straight leg lift is negative. The rest of his physical exam is noncontributory.
What should be the next step in this patient’s workup?
PATHOPHYSIOLOGY
ADPKD is a progressive expansion of numerous fluid-filled cysts that result in massive enlargement of the kidneys.5 Less than 5% of all nephrons become cystic; however, the average volume of a polycystic kidney is 1,000 mL (normal, 300 mL), that is, the volume of a standard-sized pineapple. Even with this significant enlargement, a decline in the glomerular filtration rate (GFR) is not usually seen initially. Each cyst is derived from a single hyperproliferative epithelial cell. Increased cellular proliferation, followed by fluid secretion and alterations in the extracellular matrix, cause an outpouching from the parent nephron, which eventually detaches from the parent nephron and continues to enlarge and autonomously secrete fluid.6,7
PKD1 and PKD2 are two genes responsible for ADPKD that have been isolated so far. Since there are families carrying neither the PKD1 nor the PKD2 gene that still have an inherited type of ADPKD, there is suspicion that at least one more PKD gene, not yet isolated, exists.8 It is also possible that other genetic or environmental factors may be at play.9,10
In 1994, the PKD1 gene was isolated on chromosome 16,3 and it was found to code for polycystin 1. A lack of polycystin 1 causes an abnormality in the Na+/K(+)-ATPase pumps, leading to abnormal sodium reabsorption.11 How and why this happens is not quite clear. However, the hypertension that is a key objective finding in patients with ADPKD is thought to result from this pump abnormality.
PKD2 is found on the long arm of chromosome 4 and codes for polycystin 2.4 Polycystin 2 is an amino acid that is responsible for voltage-activated cellular calcium channels,5 again explaining the hypertension so commonly seen in the course of ADPKD. ADPKD-associated hypertension may be present as early as the teenage years.12
EPIDEMIOLOGY
More than 85% of ADPKD cases are associated with PKD1, and this form is called polycystic kidney disease 1 (PKD 1), the more aggressive form of the disease.13,14 PKD 2 (the form associated with the gene PKD2), though less common, is also likely to progress to end-stage renal disease (ESRD), but at a later age (median age of 74 years, compared with 54 in patients with PKD 1).14 ADPKD accounts for about 5% of cases of ESRD in North America,9 but for most patients, presentation and decreased renal function do not occur until the 40s.15 However, patients with the risk factors listed in Table 15,16-19 are likely to experience a more rapid and aggressive form of the disease.
Even with the same germline mutation in a family with this inherited disease, the severity of ADPKD among family members is quite variable; this is true even in the case of twins.9,10,20 Since the age and symptoms at presentation can vary so greatly, a uniform method of identifying patients with ADPKD, along with staging, was needed. Most patients do not undergo genetic testing (ie, DNA linkage or gene-based direct sequencing9) for a diagnosis of ADPKD or to differentiate between the PKD 1 and PKD 2 disease forms unless they are participating in a research study. Diagnostic criteria were needed that were applicable for any type of ADPKD.
In 2009, the University of Toronto’s Division of Nephrology convened experts in the fields of nephrology and radiology to reach a consensus on standardized ultrasonographic diagnostic criteria.21 They formulated definitions based on a study of 948 individuals who were at risk for either PKD 1 or PKD 2 (see Table 221). The specificity and sensitivity of the resulting criteria range from 82% to 100%, making it possible to standardize the care and classification of renal patients worldwide.
Since family members with the same genotypes can experience very divergent disease manifestations, the two-hit hypothesis has been developed.22 In simple terms, it proposes that after the germline mutation (PKD1 or PKD2), there is a second somatic mutation that leads to progressive cyst formation; when the number and size of cysts increase, the patient starts to experience symptoms of ADPKD.22
Age at presentation can be quite variable, as can the presenting symptoms. Most patients with PKD 1 present in their 50s, with 54 being the average age in US patients.14 The most common presenting symptom is flank or back pain.2,5 The pain is due to the massive enlargement of the kidneys, causing a stretching of the kidney capsule and leading to a chronic, dull and persistent pain in the low back. Severe pain, sharp and cutting, occurs when one of the cysts hemorrhages; to some patients, the pain resembles a quick, powerful “kick in the back.” Hematuria can occur following cyst hemorrhage; depending on the location of the cyst that burst within the kidney (ie, how close it is to the collecting system) and how large it is, the amount and color of the hematuria can be impressive.
ADPKD is more common in men than women, and cyst rupture can be precipitated by trauma or lifting heavy objects. Cyst hemorrhage can turn the urine bright red, which is especially frightening to the male patient. Hematuria is often the key presenting symptom in patients who will be diagnosed with ADPKD-induced hypertension.
Besides hematuria, other common manifestations of ADPKD include:
• Hypertension (60% of affected patients, which increases to 100% by the time ESRD develops)
• Extrarenal cysts (100% of affected patients)
• Urinary tract infections
• Nephrolithiasis (20% of affected patients)
• Proteinuria, occasionally (18% of affected patients).2,5,23
Among these manifestations, those most commonly attributed to a diagnosis of ADPKD are hypertension, kidney stones, and urinary tract or kidney infections. Since isolated proteinuria is unusual in patients with ADPKD, it is recommended that another cause of kidney disease be explored in patients with this presentation.24
Extrarenal manifestations of cyst development are common, eventually occurring in all patients as they age. Hepatic cysts are universal in patients with ADPKD by age 30, although hepatic function is preserved. There may be a mild elevation in the alkaline phosphatase level in patients with ADPKD, resulting from the presence of hepatic cysts. Cysts may also be found in the pancreas, spleen, thyroid, and epididymis.5,25 Some patients may complain of dyspnea, pain, early satiety, or lower extremity edema as a result of enlarged cyst.
The Case Patient
Because you recently attended a lecture about ADPKD, you are aware that flank pain in men with hypertension is indicative of ADPKD until proven otherwise. Believing that this patient’s hypertension is renal in origin, you order an abdominal ultrasound. You also order a comprehensive metabolic panel and a complete blood count. The patient’s GFR is measured at 89 mL/min (indicative of stage 2 kidney disease). Other results are shown in Table 3.
The very broad differential includes essential hypertension, hypertension resulting from intake of “power drinks” or salt in an athlete, illicit use of medications (including steroids), herniated disc leading to transient hypertension, and urinary tract infection or sexually transmitted disease. All of this is moot when the ultrasound shows both kidneys measuring greater than 15 cm, with four distinct cysts on the right kidney and three distinct cysts on the left.
You explain to the patient that ADPKD is a genetic disease and that he and his siblings each had a certain chance of inheriting it. Although different presentations may occur (“congenital” polycystic kidney disease, hypertension, or obesity), they all must undergo ultrasonographic screening for ADPKD. You add that although ADPKD is a genetic disease, it can also be diagnosed in different members of the same family at different ages.
TREATMENT
The goal of treatment for the patient with ADPKD is to slow cyst development and the natural course of the disease. If this can be achieved, the need for dialysis or kidney transplantation may be postponed for a number of years. Because cyst growth causes an elevation in renin and activates the angiotensin II renin system26 (see figure,24), an ACE inhibitor is the most effective treatment to lower blood pressure and thus slow the progression of ADPKD. Most patients with ADPKD are started on an ACE inhibitor at an early age to slow the rate of disease progression.27,28 Several studies are under way to determine the best antihypertensive medication and the optimal age for initiating treatment.29,30
Lipid screening and treatment for dyslipidemia are important23 because ADPKD can lead to a reduction in kidney function, resulting in chronic kidney disease (CKD). CKD is considered a coronary heart disease risk equivalent, and most professionals will treat the patient with ADPKD for hyperlipidemia.23,31 While there are no data showing that statin use will reduce the incidence of ESRD or delay the need for dialysis or kidney transplantation in patients with ADPKD, the beneficial effects of good renal blood flow and endothelial function have been noted.32,33
One of the most common and significant complications in ADPKD is intracranial hemorrhage resulting from a ruptured cerebral aneurysm. In the younger adult, the incidence of cerebral aneurysm is 4%, but incidence increases to 10% in patients older than 65.34-36 Family clusters of aneurysms have been reported.37 If an intracranial aneurysm is found in the family history, the risk of an aneurysm in another family member increases to 22%.38
Since rupture of an intracranial hemorrhage is associated with a 30-day mortality rate of 50% and 80% morbidity,5,38 standard of care for patients with ADPKD includes CT or magnetic resonance angiographic (MRA) screening in the asymptomatic patient with a positive family history.34,38 If an aneurysm is found, the lifetime chance of rupture is 50%, although the risk is greater in the case of an aneurysm larger than 10 mm.5
As in all patients with kidney disease, left ventricular hypertrophy is common among patients with ADPKD.23,28,39
The Case Patient
The patient is started on an ACE inhibitor, scheduled for fasting lipid screening, and referred to a nephrology practice for disease management. As research and investigation of possible treatment options for ADPKD are ongoing, the patient may benefit from participating in a new research protocol.
Because the patient’s family has no history of cerebral aneurysm, CT/MRA screening is not required. A discussion of the pros and cons of genetic testing for the entire family, including nieces and nephews, is initiated. The patient and his family are referred to a genetic counselor to decide whether the benefit of early treatment for hypertension outweighs the risk of carrying a diagnosis of ADPKD for his younger relatives, who may later seek health insurance coverage.
NATURAL PROGRESSION OF ADPKD
Hypertension and cyst formation will continue as the patient ages. The natural progression of ADPKD is to renal failure with renal replacement therapy (dialysis or organ transplantation) as treatment options. If the progression of ADPKD can be slowed through pharmacotherapy, the patient may live for many years without needing dialysis. This ideal can be accomplished only through aggressive hypertension control, which should be started in the teenage years.23,30,31
Suggestions to increase fluid consumption and to limit the use of NSAIDs, contrast dye, and MRI with gadolinium are appropriate. It is rare for hypertension to be diagnosed before some organ damage has already occurred.12 Often the patient’s renal function, as determined by measuring the GFR, remains stable until the patient reaches his or her 40s.40 However, kidney damage often begins before any detectable change in GFR. Once the GFR does start to decline, the average decrease is 4.4 to 5.9 mL/min/1.73m2 each year.41
MANAGEMENT CONSIDERATIONS
For ESRD Organ Transplantation
Kidney transplantation—the only curative treatment for ADPKD—can be offered to patients once the GFR falls below 20 mL/min. However, the patient with ADPKD can experience kidney enlargement to such an extent that introducing a third kidney into the limited abdominal space becomes technically difficult. Although nephrectomy is avoided whenever possible, there are cases in which there is no alternative.42
In addition to space concerns, recurrent urinary tract infections, chronic pain, renal cell carcinoma, chronic hematuria, or chronic cyst infections can necessitate a nephrectomy.43,44 A laparoscopic approach with decompression of cysts or removal of only one kidney is preferred.43,45 If removal of both kidneys is required before a transplant, the patient must be maintained on dialysis until after transplantation. Since the transplant waiting list can exceed seven years in some areas, most patients arrange for a willing live donor before agreeing to a bilateral nephrectomy.46,47
Dialysis
Either peritoneal dialysis (PD) or hemodialysis (HD) can be offered to patients with severe ADPKD. Depending on the size of the native kidneys and the history of previous abdominal surgery, PD often offers a better chance of survival in these patients, particularly compared with patients who have ESRD associated with other causes.48
For management of the patient with ADPKD who receives PD, it can be difficult to differentiate between the pain of a cyst and the pain of a peritoneal infection. Generally, cyst rupture is accompanied by hematuria; and peritonitis, by cloudy fluid.5 Management provided by an experienced nephrologist and PD nurse is vital.
In ADPKD patients who undergo HD, too, survival is better than in patients who have ESRD with other causes49,50; five-year survival can be as high as 10% to 15%.51 This is likely due to the lower incidence of coronary artery disease in the ADPKD population, compared with patients who have ESRD associated with other chronic diseases.49
FUTURE TRENDS AND ONGOING TRIALS
HALT PKD29,30 is an NIH-funded, double-blind study to determine whether adding an angiotensin receptor blocker (ARB) to standard ACE inhibitor therapy results in a more significant decrease in the progression of renal cysts. The rationale for this is that the ARB is expected to block the renin-angiotensin-aldosterone system in the kidney. Use of ACE inhibitor monotherapy versus ARB/ACE inhibitor therapy is being compared in two study arms: patients between ages 15 and 49 with a GFR of 60 mL/min or greater; and patients between ages 18 and 64 with a GFR of 25 to 60 mL/min.29 To date, preliminary results indicate no benefit in adding the second medication.49
The TEMPO Trial52 is a multicenter, double-blind study looking at the effect of tolvaptan on renal cyst growth. Tolvaptan is a potent vasopressin receptor antagonist, and in vitro evidence has shown that intracellular cyclic adenosine monophosphate (cAMP) plays a large role in the development of cysts in patients with ADPKD. If it is possible to block the cAMP that is causing cyst growth, progression of ADPKD should slow.53,54 Only short-term effects of tolvaptan use are currently known.55
High Water Intake to Slow Progression of Polycystic Kidney Disease56 is an open-label, nonrandomized trial in which patients drink a minimum of
3 L of water. Previously, a small study showed that an increase in fluid intake partially suppresses the urine osmolality and the serum antidiuretic hormone (ADH) levels.57 According to this theory, increasing water intake to greater than 3 L/d may result in complete suppression of ADH and cAMP. This is a small study (n = 20),56 since patients with ADPKD are likely to have urinary concentrating defects, and hyponatremia is a concern is in these patients.58
Sirolimus and ADPKD59 is an open-label randomized study to see whether sirolimus (also known as rapamycin) can reduce cyst growth. Originally, it was noted that posttransplant ADPKD patients underwent a regression of both liver and kidney cysts when they were taking sirolimus, and a preliminary crossover study was done.60 However, preliminary results at 18 months showed no difference in renal growth or cyst growth but did show kidney damage as determined by an increase of proteinuria in the treatment group.59 The study is still in progress.
Somatostatin in Polycystic Kidney61 is a long-term (three-year) study following patients who agreed to participate in a randomized, double-blind protocol; in it, an intramuscular injection of either an octreotide (ie, somatastatin) or placebo was administered every four weeks for one year in an effort to reduce the size of kidney and liver cysts.62 At one year, the quality of life in the treatment group was rated better, as measured by pain reduction and improved physical activity. Cyst growth in the treatment group was smaller for both the kidney and liver. However, the GFR decreased to the same degree in both groups.62
CONCLUSION
ADPKD is a common, often overlooked genetic disease that is a cause of hypertension. ADPKD’s presenting symptoms of flank pain, back pain, and/or hematuria often bring the patient to the provider, but a high index of suspicion must be maintained to diagnose these patients at an early age. Due to the variable presentation even within affected families, many patients do not realize that their family carries the PKD gene.
While genetic testing is available, ultrasound is a quick, relatively inexpensive, and easy method to screen for this diagnosis. The progression of ADPKD to ESRD, requiring dialysis or organ transplantation, can be slowed with early and aggressive treatment of hypertension. As with all patients affected by renal impairment, suggestions for patients with ADPKD to avoid use of NSAIDs, contrast dye, and gadolinium-enhanced MRI are appropriate. The primary care PA or NP is in an appropriate position to see to this.
REFERENCES
1. Newschaffer CJ, Falb MD, Gurney JG. National autism prevalence trends from United States special education data. Pediatrics. 2005;115 (3):e277-e282.
2. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287-1301.
3. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16: European Polycystic Kidney Disease Consortium. Cell. 1994;77(6):881-894.
4. Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996; 272(5266):1339-1342.
5. Chapman AB. Polycystic and other cystic kidney diseases. In: Greenberg A, ed. Primer on Kidney Diseases: Expert Consult. 5th ed. National Kidney Foundation; 2009:345-353.
6. Fischer E, Legue E, Doyen A, et al. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006;38(1):21-23.
7. Murcia NS, Sweeney WE Jr, Avner ED. New insights into the molecular pathophysiology of polycystic kidney disease. Kidney Int. 1999;55 (4):1187-1197.
8. Paterson AD, Pei Y. Is there a third gene for autosomal dominant polycystic kidney disease? Kidney Int. 1998;54(5):1759-1761.
9. Pei Y. Practical genetics for autosomal dominant polycystic kidney disease. Nephron Clin Pract. 2011;118(1):c19-c30.
10. Tan YC, Blumenfeld J, Rennert H. Autosomal dominant polycystic kidney disease: genetics, mutations and microRNAs. Biochim Biophys Acta. 2011 Mar 16 [Epub ahead of print].
11. Avner ED, Sweeney WE Jr, Nelson WJ. Abnormal sodium pump distribution during renal tubulogenesis in congenital murine polycystic kidney disease. Proc Natl Acad Sci U S A. 1992;89(16):7447-7451.
12. Torra R, Badenas C, Darnell A, et al. Linkage, clinical features, and prognosis of autosomal dominant polycystic kidney disease types 1 and 2. J Am Soc Nephrol. 1996;7(10):2142-2151.
13. Davies F, Coles GA, Harper PS, et al. Polycystic kidney disease re-evaluated: a population-based study. Q J Med. 1991;79(290):477-485.
14. Hateboer N, v Dijk MA, Bogdanova N, et al; European PKD1-PKD2 Study Group. Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet. 1999;353(9147):103-107.
15. Peters DJ, Breuning MH. Autosomal dominant polycystic kidney disease: modification of disease progression. Lancet. 2001;358(9291):1439-1444.
16. Dicks E, Ravani P, Langman D, et al. Incident renal events and risk factors in autosomal dominant polycystic kidney disease: a population- and family-based cohort followed for 22 years. Clin J Am Soc Nephrol. 2006;1(4):710-717.
17. Fick-Brosnahan GM, Belz MM, McFann KK, etc. Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease: a longitudinal study. Am J Kidney Dis. 2002;39(6):1127-1134.
18. Fick-Brosnahan GM, Tran ZV, Johnson AM, et al. Progression of autosomal-dominant polycystic kidney disease in children. Kidney Int. 2001;59(5):1654-1662.
19. Johnson AM, Gabow PA. Identification of patients with autosomal dominant polycystic kidney disease at highest risk for end-stage renal disease. J Am Soc Nephrol. 1997;8(10): 1560-1567.
20. Risk D. Autosomal dominant polycystic kidney disease. Presented at: National Kidney Foundation, Spring Clinical Meetings; April 28, 2011; Las Vegas, NV.
21. Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20(1):205-212.
22. Watnick T, Germino GG. Molecular basis of autosomal dominant polycystic kidney disease. Semin Nephrol. 1999;19(4):327-343.
23. Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol. 2009;5(4):221-228.
24. Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis. 2011;57(6):856-862.
25. Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17(2):173-180.
26. Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med. 1990;323(16):1091-1096.
27. Jafar TH, Stark PC, Schmid CH, et al. The effect of angiotensin-converting-enzyme inhibitors on progression of advanced polycystic kidney disease. Kidney Int. 2005;67(1):265-271.
28. Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2009;20(9):1888-1893.
29. National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH, sponsor. HALT PKD (Halt Progression of Polycystic Kidney Disease): Efficacy of Aggressive Renin-Angiotensin-Aldosterone Axis Blockade in Preventing/Slowing Renal Function Decline in ADPKD. www2.niddk.nih.gov/NR/rdonlyres/175578F6-62B4-429A-9BBF-96CCEC2FFB3A/0/KUHHALT PKDPROTOCOL9107.pdf. Accessed July 22, 2011.
30. Chapman AB, Torres VE, Perrone RD, et al. The HALT polycystic kidney disease trials: design and implementation. Clin J Am Soc Nephrol. 2010;5(1):102-109.
31. Taylor M, Johnson AM, Tison M, et al. Earlier diagnosis of autosomal dominant polycystic kidney disease: importance of family history and implications for cardiovascular and renal complications. Am J Kidney Dis. 2005;46(3):415-423.
32. Namli S, Oflaz H, Turgut F, et al. Improvement of endothelial dysfunction with simvastatin in patients with autosomal dominant polycystic kidney disease. Ren Fail. 2007;29(1):55-59.
33. Bremmer MS, Jacobs SC. Renal artery embolization for the symptomatic treatment of adult polycystic kidney disease. Nat Clin Pract Nephrol. 2008;4(5):236-237.
34. Chapman AB, Rubinstein D, Hughes R, et al. Intracranial aneurysms in autosomal dominant polycystic kidney disease. N Engl J Med. 1992; 327(13):916-920.
35. Schievink WI, Torres VE, Piepgras DG, Wiebers DO. Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1992;3(1):88-95.
36. Fick GM, Gabow PA. Hereditary and acquired cystic disease of the kidney. Kidney Int. 1994;46(4):951-964.
37. Watson ML. Complications of polycystic kidney disease. Kidney Int. 1997;51(1):353-365.
38. Huston J 3rd, Torres VE, Sulivan PP, et al. Value of magnetic resonance angiography for the detection of intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1993;3(12):1871-1877.
39. Longenecker JC, Coresh J, Powe NR, et al. Traditional cardiovascular disease risk factors in dialysis patients compared with the general population: the CHOICE Study. J Am Soc Nephrol. 2002;13(7):1918-1927.
40. Meijer E, Rook M, Tent H, et al. Early renal abnormalities in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2010; 5(6):1091-1098.
41. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149-168.
42. Tabibi A, Simforoosh N, Abadpour P, et al. Concomitant nephrectomy of massively enlarged kidneys and renal transplantation in autosomal dominant polycystic kidney disease. Transplant Proc. 2005;37(7):2939-2940.
43. Dunn MD, Portis AJ, Elbahnasy AM, et al. Laparoscopic nephrectomy in patients with end-stage renal disease and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000;35(4):720-725.
44. Sulikowski T, Tejchman K, Zietek Z, et al. Experience with autosomal dominant polycystic kidney disease in patients before and after renal transplantation: a 7-year observation. Transplant Proc. 2009;41(1):177-180.
45. Desai MR, Nandkishore SK, Ganpule A, Thimmegowda M. Pretransplant laparoscopic nephrectomy in adult polycystic kidney disease: a single centre experience. BJU Int. 2008;101 (1):94-97.
46. Glassman DT, Nipkow L, Bartlett ST, Jacobs SC. Bilateral nephrectomy with concomitant renal graft transplantation for autosomal dominant polycystic kidney disease. J Urol. 2000;164 (3 pt 1):661-664.
47. Fuller TF, Brennan TV, Feng S, et al. End stage polycystic kidney disease: indications and timing of native nephrectomy relative to kidney transplantation. J Urol. 2005;174(6):2284-2288.
48. Abbott KC, Agodoa LY. Polycystic kidney disease at end-stage renal disease in the United States: patient characteristics and survival. Clin Nephrol. 2002;57(3):208-214.
49. Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis. 2001;38(4):777-784.
50. Batista PB, Lopes AA, Costa FA. Association between attributed cause of end-stage renal disease and risk of death in Brazilian patients receiving renal replacement therapy. Ren Fail. 2005;27(6):651-656.
51. Pirson Y, Christophe JL, Goffin E. Outcome of renal replacement therapy in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 1996;11 suppl 6:24-28.
52. Torres VE, Meijer E, Bae KT, et al. Rationale and design of the TEMPO (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes) 3-4 Study. Am J Kidney Dis. 2011;57(5):692-699.
53. Calvet JP. Strategies to inhibit cyst formation in ADPKD. Clin J Am Soc Nephrol. 2008;3 (4):1205-1211.
54. Grantham JJ. Lillian Jean Kaplan International Prize for advancement in the understanding of polycystic kidney disease. Understanding polycystic kidney disease: a systems biology approach. Kidney Int. 2003;64(4):1157-1162.
55. Irazabal MV, Torres VE, Hogan MC, et al. Short-term effects of tolvaptan on renal function and volume in patients with Autosomal Dominant Polycystic Kidney Disease. Kidney Int. 2011 May 4 [Epub ahead of print].
56. New York University, sponsor. High Water Intake to Slow Progression of Polycystic Kidney Disease. http://clinicaltrials.gov/ct2/show/NCT00784030. Accessed July 22, 2011.
57. Wang CJ, Creed C, Winklhofer FT, Grantham JJ. Water prescription in autosomal dominant polycystic kidney disease: a pilot study. Clin J Am Soc Nephrol. 2011;6(1):192-197.
58. Grampsas SA, Chandhoke Ps, Fan J, et al. Anatomic and metabolic risk factors for nephrolithiasis in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000;36(1):53-57.
59. Serra AL, Poster D, Kistler AD, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010; 363(9):820-829.
60. Perico N, Antiga L, Caroli A, et al. Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol. 2010;21(6):1031-1040.
61. Mario Negri Institute for Pharmacological Research, sponsor. Somatostatin in Polycystic Kidney: a Long-term Three Year Follow up Study. http://clinicaltrials.gov/ct2/show/NCT00309283. Accessed July 22, 2011.
62. Hogan MC, Masyuk TV, Page LJ, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010; 21(6):1052-1061.