User login
A Toxic Fish Dinner
Emergent Management of Lightning Injuries
Current Management Options for Osteonecrosis of the Femoral Head: Part II, Operative Management
Occipitocervical Junction: Imaging, Pathology, Instrumentation
UPDATE ON PELVIC FLOOR DYSFUNCTION
Vulvar Pain Syndromes 3-Part Series
- Making the correct diagnosis
(September 2011) - A bounty of treatments-but not all of them are proven
(October 2011) - Provoked vestibulodynia
(Coming in November 2011)
Chronic pelvic pain: 11 critical questions about causes and care
Fred M. Howard, MD (August 2009)
Vague symptoms. Unexpected flares. Inconsistent manifestations. These characteristics can make diagnosis and treatment of chronic pelvic pain frustrating for both patient and physician. Most patients undergo myriad tests and studies to uncover the source of their pain—but a targeted pelvic exam may be all that is necessary to identify a prevalent but commonly overlooked cause of pelvic pain. Levator myalgia, myofascial pelvic pain syndrome, and pelvic floor spasm are all terms that describe a condition that may affect as many as 78% of women who are given a diagnosis of chronic pelvic pain.1 This syndrome may be represented by an array of symptoms, including pelvic pressure, dyspareunia, rectal discomfort, and irritative urinary symptoms such as spasms, frequency, and urgency. It is characterized by the presence of tight, band-like pelvic muscles that reproduce the patient’s pain when palpated.2
Diagnosis of this syndrome often surprises the patient. Although the concept of a muscle spasm is not foreign, the location is unexpected. Patients and physicians alike may forget that there is a large complex of muscles that completely lines the pelvic girdle. To complicate matters, the patient often associates the onset of her symptoms with an acute event such as a “bad” urinary tract infection or pelvic or vaginal surgery, which may divert attention from the musculature. Although a muscle spasm may be the cause of the patient’s pain, it’s important to realize that an underlying process may have triggered the original spasm. To provide effective treatment of pain, therefore, you must identify the fundamental cause, assuming that it is reversible, rather than focus exclusively on symptoms.
Although there are many therapeutic options for levator myalgia, an appraisal of the extensive literature on these medications is beyond the scope of this article. Rather, we will review alternative treatment modalities and summarize the results of five trials that explored physical therapy, trigger-point or chemodenervation injection, and neuromodulation (TABLE).
Weighing the nonpharmaceutical options for treatment
of myofascial pelvic pain
| Treatment | Pros | Cons |
|---|---|---|
| Physical therapy | Minimally invasive Moderate long-term success | Requires highly specialized therapist |
| Trigger-point injection | Minimally invasive Performed in clinic Immediate short-term success | Optimal injectable agent is unknown Botulinum toxin A lacks FDA approval for this indication Limited information on adverse events and long-term efficacy |
| Percutaneous tibial nerve stimulation | Minimally invasive Performed in clinic | Requires numerous office visits for treatment Lacks FDA approval for this indication Limited information on long-term efficacy |
| Sacral neuromodulation | Moderately invasive Permanent implant | Requires implantation in operating room Lacks FDA approval for this indication Limited information on long-term efficacy |
Pelvic myofascial therapy offers relief—but qualified therapists may be scarce
FitzGerald MP, Anderson RU, Potts J, et al; Urological Pelvic Pain Collaborative Research Network. Randomized multicenter feasibility trial of myofascial physical therapy for the treatment of urological chronic pelvic pain syndromes. J Urology. 2009;182(2):570–580.
Physical therapy of the pelvic floor—otherwise known as pelvic myofascial therapy—requires a therapist who is highly trained and specialized in this technique. It is more invasive than other forms of rehabilitative therapy because of the need to perform transvaginal maneuvers (FIGURE 1).
This pilot study by the Urological Pelvic Pain Collaborative Research Network evaluated the ability of patients to adhere to pelvic myofascial therapy, the response of their pain to therapy, and adverse events associated with manual therapy. It found that patients were willing to undergo the therapy, despite the invasive nature of the maneuvers, because it was significantly effective.
Details of the study
Patients (both men and women) were randomized to myofascial physical therapy or global therapeutic massage. Myofascial therapy consisted of internal or vaginal manipulation of the trigger-point muscle bundles and tissues of the pelvic floor. It also focused on muscles of the hip girdle and abdomen. The comparison group underwent traditional Western full-body massage. In both groups, treatment lasted 1 hour every week, and participants agreed to 10 full treatments.
Patients were eligible for the study if they experienced pelvic pain, urinary frequency, or bladder discomfort in the previous 6 months. In addition, an examiner must have been able to elicit tenderness upon palpation of the pelvic floor during examination. Patients were excluded if they showed signs of urinary tract infection or dysmenorrhea.
A total of 47 patients were randomized—24 to global massage and 23 to myofascial physical therapy. Overall, the myofascial group experienced a significantly higher rate of improvement in the global response at 12 weeks than did patients in the global-massage group (57% vs 21%; P=.03). Patients were willing to engage in myofascial pelvic therapy, and adverse events were minor.
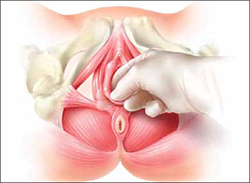
FIGURE 1 Transvaginal myofascial therapy
Physical therapy of the pelvic floor is more invasive than other forms of rehabilitative therapy because of the need to perform transvaginal maneuvers.
Need for specialized training may limit number of therapists
The randomized controlled study design renders these findings fairly reliable. Therapists were unmasked and aware of the treatment arms but were trained to make the different therapy sessions appear as similar as possible.
Although investigators were enthusiastic about their initial findings, additional studies are needed to validate the results. Moreover, these findings may be difficult to generalize because women who volunteer to participate in such a study may differ from the general population.
Nevertheless, patients who suffer from chronic pelvic pain may take heart that there is a nonpharmaceutical alternative to manage their symptoms, although availability is likely limited in many areas. Given the nature of the physical therapy required for this particular location of myofascial pain, specialized training is necessary for therapists. Despite motivated patients and well-informed providers, it may be difficult to find specialized therapists within local vicinities. Referrals to centers where this type of therapy is offered may be necessary.
Pelvic myofascial therapy is an effective and acceptable intervention for the treatment of levator myalgia.
The ideal agent for trigger-point injections remains a mystery
Langford CF, Udvari Nagy S, Ghoniem G M. Levator ani trigger point injections: An underutilized treatment for chronic pelvic pain. Neurourol Urodyn. 2007;26(1):59–62.
Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol. 2006;108(4):915–923.
Trigger points are discrete, tender areas within a ridge of contracted muscle. These points may cause focal pain or referred pain upon irritation of the muscle.2 Trigger-point injection therapy aims to anesthetize or relax these points by infiltrating the muscle with medications.
These two studies evaluated the value of trigger-point injections in the treatment of pelvic myofascial pain; they found that the injections provide relief, although the mechanism of action and the ideal agent remain to be determined.
Langford et al: Details of the study
In this prospective study, 18 women who had pelvic pain of at least 6 months’ duration and confirmed trigger points on examination underwent transvaginal injection of a solution of bupivacaine, lidocaine, and triamcinolone. They were assessed by questionnaire at baseline and 3 months after injection. Assessment included a visual analog scale for pain severity. Investigators defined success as a decrease in pain of 50% or more and global-satisfaction and global-cure visual scores of 60% or higher.
Thirteen of the 18 women (72.2%) improved after their first injection, with six women reporting a complete absence of pain. Overall, women reported significant decreases in pain and increases in the rates of satisfaction and cure, meeting the definition of success at 3 months after the injection.
Among the theories proposed to explain the mechanism of action of trigger-point injections are:
- disruption of reflex arcs within skeletal muscle
- release of endorphins
- mechanical changes in abnormally contracted muscle fibers.
This last theory highlights one of the limitations of this study—lack of a placebo arm. Could it be possible that the injection of any fluid produces the same effect?
This study was not designed to investigate the causal relationship between the injection of a particular solution and pain relief, but it does highlight the need for studies to clarify the mechanism of action, including use of a placebo. It also prompts questions about the duration of effect after a single injection.
Goal of chemodenervation is blocking of muscle activity
Botulinum toxin type A (Botox) blocks the release of acetylcholine from presynaptic neurons. The release of acetylcholine stimulates muscle contractions; therefore, blockage of its release reduces muscle activity. This type of chemodenervation has found widespread use, and botulinum toxin A now has approval from the Food and Drug Administration (FDA) for treatment of chronic migraine, limb spasticity, cervical dystonia, strabismus, hyperhidrosis, and facial cosmesis.3 Although it is not approved for pelvic floor levator spasm, its success in treating other myotonic disorders suggests that its application may be relevant.
Abbott et al: Details of the study
Abbott and colleagues performed a double-blind, randomized, controlled trial to compare injection of botulinum toxin A with injection of saline. They measured changes in the pain scale, quality of life, and vaginal pressure.
Women were eligible for the study if they had subjectively reported pelvic pain of more than 2 years’ duration and objective evidence of trigger points (on examination) and elevated vaginal resting pressure (by vaginal manometry). Neither the clinical research staff nor the patient knew the contents of the injections, but all women received a total of four—two at sites in the puborectalis muscle and two in the pubococcygeus muscle.
After periodic assessment by questionnaire and examination through 6 months after injection, no differences were found in the pain score or resting vaginal pressure between the group of women who received botulinum toxin A and the group who received placebo. However, each group experienced a significant reduction in pain and vaginal pressure, compared with baseline. And both groups reported improved quality of life, compared with baseline. Neither group reported voiding dysfunction.
These two studies support the use of trigger-point injection into pelvic floor muscles to reduce pelvic myofascial pain. The findings of Abbott and colleagues, in particular, suggest that the substance that is injected may not be as important as the actual needling of the muscle. Larger studies and comparisons between placebo, botulinum toxin A, and anesthetic solutions are needed to elucidate the therapeutic benefit of these particular medications.
Neuromodulation shows promise as treatment for pelvic myofascial pain
van Balken MR, Vandoninck V, Messelink, BJ, et al. Percutaneous tibial nerve stimulation as neuromodulative treatment of chronic pelvic pain. Eur Urol. 2003;43(2):158–163.
Zabihi N, Mourtzinos A, Maher MG, Raz S, Rodriguez LV. Short-term results of bilateral S2-S4 sacral neuromodulation for the treatment of refractory interstitial cystitis, painful bladder syndrome, and chronic pelvic pain. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19(4):553–557.
Neuromodulation is the science of using electrical impulses to alter neuronal activities. The exact mechanisms of action are unclear, but the technology has been utilized to control symptoms of overactive bladder and urinary retention caused by poor relaxation of the urethral and pelvic floor muscles. While studying the effects of sacral nerve root neuromodulation on the bladder, investigators noted improvements in other symptoms, such as pelvic pain.
Neuromodulation of the sacral nerve roots may be achieved by direct conduction of electrical impulses from a lead implanted in the sacrum (sacral neuromodulation) or by the retrograde conduction of these impulses through the posterior tibial nerve (percutaneous tibial nerve stimulation, or PTNS) (FIGURE 2). The tibial nerve arises from sacral nerves L5 to S3 and is one of the larger branches of the sciatic nerve.
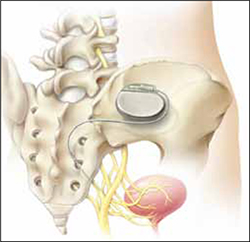
FIGURE 2 InterStim therapy
Stimulation of the sacral nerve has been used successfully to manage overactive bladder and urinary retention and may prove useful in the treatment of pelvic myofascial pain.
Van Balken et al: Details of the study
In this prospective observational study, 33 patients (both male and female) who had chronic pelvic pain by history and examination were treated with weekly, 30-minute outpatient sessions of PTNS for 12 weeks. Participants were asked to provide baseline pain scores and keep a diary of their pain. Quality-of-life questionnaires were also administered at baseline and at 12 weeks.
Investigators considered both subjective and objective success in their outcomes. If a patient elected to continue therapy, he or she was classified as a subjective success. Objective success required a decrease of at least 50% in the pain score. At the end of 12 weeks, although 33 patients (42%) wanted to continue therapy, only seven (21%) met the definition for objective success. Of those seven, six elected to continue therapy.
This study sheds light on a treatment modality that has not been studied adequately for the indication of pelvic pain but that may be promising in patients who have levator myalgia. Limitations of this study include the lack of a placebo arm, short-term outcome, and lack of localization of pain. Furthermore, although PTNS has FDA approval for treatment of urinary urgency, frequency, and urge incontinence, it is not approved for the treatment of pelvic pain. These preliminary findings demonstrate potential but, as with any new indication, long-term comparative studies are needed.
Zabihi et al: Details of the study
Patients in this retrospective study had a diagnosis of interstitial cystitis or chronic pelvic pain. Pelvic myofascial pain and trigger points were not required for eligibility. Thirty patients (21 women and nine men) had temporary placement of a lead containing four small electrodes along the S2 to S4 sacral nerve roots on both sides of the sacrum. They were then followed for a trial period of 2 to 4 weeks. To qualify for the final stage of the study, in which the leads were connected internally to a generator implanted in the buttocks, patients had to report improvement of at least 50% in their symptoms. If their improvement did not meet that threshold, the leads were removed.
Twenty-three patients (77%) met the criteria for permanent implantation. Of these patients, 42% reported improvement of more than 50% at 6 postoperative months. Quality-of-life scores also improved significantly.
Sacral neuromodulation is not FDA-approved for the treatment of chronic pelvic pain; further studies are needed before it can be recommended for this indication.
Neither of these studies required objective evidence of myofascial pain for inclusion. Therefore, although the benefits they demonstrated may be theorized to extend to the relief of myofascial pain, this fact cannot be corroborated.
We want to hear from you! Tell us what you think.
1. Bassaly R, Tidwell N, Bertolino S, Hoyte L, Downes K, Hart S. Myofascial pain and pelvic floor dysfunction in patients with interstitial cystitis. Int Urogynecol J. 2011;22(4):413-418.
2. Alvarez DJ, Rockwell PG. Trigger points: diagnosis and management. Am Fam Physician. 2002;65(4):653-660.
3. Allergan, Inc. Medication Guide: BOTOX. US Food and Drug Administration Web site. http://www.fda.gov/downloads/Drugs/DrugSafety/UCM176360.pdf. Published October 2010. Accessed August 30, 2011.

Amie Kawasaki, MD
Dr. Kawasaki is Fellow in the Division of Female Pelvic Medicine and Reconstructive Surgery, and Clinical Instructor of Obstetrics and Gynecology, at Duke University Medical Center in Durham, NC.

Cindy L. Amundsen, MD
Dr. Amundsen is Associate Professor and Fellowship Director of Female Pelvic Medicine and Reconstructive Surgery, Department of Obstetrics and Gynecology, at Duke University Medical Center in Durham, NC.
The authors report that they have no financial relationships relevant to this article.
Amie Kawasaki, MD
Dr. Kawasaki is Fellow in the Division of Female Pelvic Medicine and Reconstructive Surgery, and Clinical Instructor of Obstetrics and Gynecology, at Duke University Medical Center in Durham, NC.
Cindy L. Amundsen, MD
Dr. Amundsen is Associate Professor and Fellowship Director of Female Pelvic Medicine and Reconstructive Surgery, Department of Obstetrics and Gynecology, at Duke University Medical Center in Durham, NC.
The authors report that they have no financial relationships relevant to this article.
Amie Kawasaki, MD
Dr. Kawasaki is Fellow in the Division of Female Pelvic Medicine and Reconstructive Surgery, and Clinical Instructor of Obstetrics and Gynecology, at Duke University Medical Center in Durham, NC.
Cindy L. Amundsen, MD
Dr. Amundsen is Associate Professor and Fellowship Director of Female Pelvic Medicine and Reconstructive Surgery, Department of Obstetrics and Gynecology, at Duke University Medical Center in Durham, NC.
The authors report that they have no financial relationships relevant to this article.
Vulvar Pain Syndromes 3-Part Series
- Making the correct diagnosis
(September 2011) - A bounty of treatments-but not all of them are proven
(October 2011) - Provoked vestibulodynia
(Coming in November 2011)
Chronic pelvic pain: 11 critical questions about causes and care
Fred M. Howard, MD (August 2009)
Vague symptoms. Unexpected flares. Inconsistent manifestations. These characteristics can make diagnosis and treatment of chronic pelvic pain frustrating for both patient and physician. Most patients undergo myriad tests and studies to uncover the source of their pain—but a targeted pelvic exam may be all that is necessary to identify a prevalent but commonly overlooked cause of pelvic pain. Levator myalgia, myofascial pelvic pain syndrome, and pelvic floor spasm are all terms that describe a condition that may affect as many as 78% of women who are given a diagnosis of chronic pelvic pain.1 This syndrome may be represented by an array of symptoms, including pelvic pressure, dyspareunia, rectal discomfort, and irritative urinary symptoms such as spasms, frequency, and urgency. It is characterized by the presence of tight, band-like pelvic muscles that reproduce the patient’s pain when palpated.2
Diagnosis of this syndrome often surprises the patient. Although the concept of a muscle spasm is not foreign, the location is unexpected. Patients and physicians alike may forget that there is a large complex of muscles that completely lines the pelvic girdle. To complicate matters, the patient often associates the onset of her symptoms with an acute event such as a “bad” urinary tract infection or pelvic or vaginal surgery, which may divert attention from the musculature. Although a muscle spasm may be the cause of the patient’s pain, it’s important to realize that an underlying process may have triggered the original spasm. To provide effective treatment of pain, therefore, you must identify the fundamental cause, assuming that it is reversible, rather than focus exclusively on symptoms.
Although there are many therapeutic options for levator myalgia, an appraisal of the extensive literature on these medications is beyond the scope of this article. Rather, we will review alternative treatment modalities and summarize the results of five trials that explored physical therapy, trigger-point or chemodenervation injection, and neuromodulation (TABLE).
Weighing the nonpharmaceutical options for treatment
of myofascial pelvic pain
| Treatment | Pros | Cons |
|---|---|---|
| Physical therapy | Minimally invasive Moderate long-term success | Requires highly specialized therapist |
| Trigger-point injection | Minimally invasive Performed in clinic Immediate short-term success | Optimal injectable agent is unknown Botulinum toxin A lacks FDA approval for this indication Limited information on adverse events and long-term efficacy |
| Percutaneous tibial nerve stimulation | Minimally invasive Performed in clinic | Requires numerous office visits for treatment Lacks FDA approval for this indication Limited information on long-term efficacy |
| Sacral neuromodulation | Moderately invasive Permanent implant | Requires implantation in operating room Lacks FDA approval for this indication Limited information on long-term efficacy |
Pelvic myofascial therapy offers relief—but qualified therapists may be scarce
FitzGerald MP, Anderson RU, Potts J, et al; Urological Pelvic Pain Collaborative Research Network. Randomized multicenter feasibility trial of myofascial physical therapy for the treatment of urological chronic pelvic pain syndromes. J Urology. 2009;182(2):570–580.
Physical therapy of the pelvic floor—otherwise known as pelvic myofascial therapy—requires a therapist who is highly trained and specialized in this technique. It is more invasive than other forms of rehabilitative therapy because of the need to perform transvaginal maneuvers (FIGURE 1).
This pilot study by the Urological Pelvic Pain Collaborative Research Network evaluated the ability of patients to adhere to pelvic myofascial therapy, the response of their pain to therapy, and adverse events associated with manual therapy. It found that patients were willing to undergo the therapy, despite the invasive nature of the maneuvers, because it was significantly effective.
Details of the study
Patients (both men and women) were randomized to myofascial physical therapy or global therapeutic massage. Myofascial therapy consisted of internal or vaginal manipulation of the trigger-point muscle bundles and tissues of the pelvic floor. It also focused on muscles of the hip girdle and abdomen. The comparison group underwent traditional Western full-body massage. In both groups, treatment lasted 1 hour every week, and participants agreed to 10 full treatments.
Patients were eligible for the study if they experienced pelvic pain, urinary frequency, or bladder discomfort in the previous 6 months. In addition, an examiner must have been able to elicit tenderness upon palpation of the pelvic floor during examination. Patients were excluded if they showed signs of urinary tract infection or dysmenorrhea.
A total of 47 patients were randomized—24 to global massage and 23 to myofascial physical therapy. Overall, the myofascial group experienced a significantly higher rate of improvement in the global response at 12 weeks than did patients in the global-massage group (57% vs 21%; P=.03). Patients were willing to engage in myofascial pelvic therapy, and adverse events were minor.
FIGURE 1 Transvaginal myofascial therapy
Physical therapy of the pelvic floor is more invasive than other forms of rehabilitative therapy because of the need to perform transvaginal maneuvers.
Need for specialized training may limit number of therapists
The randomized controlled study design renders these findings fairly reliable. Therapists were unmasked and aware of the treatment arms but were trained to make the different therapy sessions appear as similar as possible.
Although investigators were enthusiastic about their initial findings, additional studies are needed to validate the results. Moreover, these findings may be difficult to generalize because women who volunteer to participate in such a study may differ from the general population.
Nevertheless, patients who suffer from chronic pelvic pain may take heart that there is a nonpharmaceutical alternative to manage their symptoms, although availability is likely limited in many areas. Given the nature of the physical therapy required for this particular location of myofascial pain, specialized training is necessary for therapists. Despite motivated patients and well-informed providers, it may be difficult to find specialized therapists within local vicinities. Referrals to centers where this type of therapy is offered may be necessary.
Pelvic myofascial therapy is an effective and acceptable intervention for the treatment of levator myalgia.
The ideal agent for trigger-point injections remains a mystery
Langford CF, Udvari Nagy S, Ghoniem G M. Levator ani trigger point injections: An underutilized treatment for chronic pelvic pain. Neurourol Urodyn. 2007;26(1):59–62.
Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol. 2006;108(4):915–923.
Trigger points are discrete, tender areas within a ridge of contracted muscle. These points may cause focal pain or referred pain upon irritation of the muscle.2 Trigger-point injection therapy aims to anesthetize or relax these points by infiltrating the muscle with medications.
These two studies evaluated the value of trigger-point injections in the treatment of pelvic myofascial pain; they found that the injections provide relief, although the mechanism of action and the ideal agent remain to be determined.
Langford et al: Details of the study
In this prospective study, 18 women who had pelvic pain of at least 6 months’ duration and confirmed trigger points on examination underwent transvaginal injection of a solution of bupivacaine, lidocaine, and triamcinolone. They were assessed by questionnaire at baseline and 3 months after injection. Assessment included a visual analog scale for pain severity. Investigators defined success as a decrease in pain of 50% or more and global-satisfaction and global-cure visual scores of 60% or higher.
Thirteen of the 18 women (72.2%) improved after their first injection, with six women reporting a complete absence of pain. Overall, women reported significant decreases in pain and increases in the rates of satisfaction and cure, meeting the definition of success at 3 months after the injection.
Among the theories proposed to explain the mechanism of action of trigger-point injections are:
- disruption of reflex arcs within skeletal muscle
- release of endorphins
- mechanical changes in abnormally contracted muscle fibers.
This last theory highlights one of the limitations of this study—lack of a placebo arm. Could it be possible that the injection of any fluid produces the same effect?
This study was not designed to investigate the causal relationship between the injection of a particular solution and pain relief, but it does highlight the need for studies to clarify the mechanism of action, including use of a placebo. It also prompts questions about the duration of effect after a single injection.
Goal of chemodenervation is blocking of muscle activity
Botulinum toxin type A (Botox) blocks the release of acetylcholine from presynaptic neurons. The release of acetylcholine stimulates muscle contractions; therefore, blockage of its release reduces muscle activity. This type of chemodenervation has found widespread use, and botulinum toxin A now has approval from the Food and Drug Administration (FDA) for treatment of chronic migraine, limb spasticity, cervical dystonia, strabismus, hyperhidrosis, and facial cosmesis.3 Although it is not approved for pelvic floor levator spasm, its success in treating other myotonic disorders suggests that its application may be relevant.
Abbott et al: Details of the study
Abbott and colleagues performed a double-blind, randomized, controlled trial to compare injection of botulinum toxin A with injection of saline. They measured changes in the pain scale, quality of life, and vaginal pressure.
Women were eligible for the study if they had subjectively reported pelvic pain of more than 2 years’ duration and objective evidence of trigger points (on examination) and elevated vaginal resting pressure (by vaginal manometry). Neither the clinical research staff nor the patient knew the contents of the injections, but all women received a total of four—two at sites in the puborectalis muscle and two in the pubococcygeus muscle.
After periodic assessment by questionnaire and examination through 6 months after injection, no differences were found in the pain score or resting vaginal pressure between the group of women who received botulinum toxin A and the group who received placebo. However, each group experienced a significant reduction in pain and vaginal pressure, compared with baseline. And both groups reported improved quality of life, compared with baseline. Neither group reported voiding dysfunction.
These two studies support the use of trigger-point injection into pelvic floor muscles to reduce pelvic myofascial pain. The findings of Abbott and colleagues, in particular, suggest that the substance that is injected may not be as important as the actual needling of the muscle. Larger studies and comparisons between placebo, botulinum toxin A, and anesthetic solutions are needed to elucidate the therapeutic benefit of these particular medications.
Neuromodulation shows promise as treatment for pelvic myofascial pain
van Balken MR, Vandoninck V, Messelink, BJ, et al. Percutaneous tibial nerve stimulation as neuromodulative treatment of chronic pelvic pain. Eur Urol. 2003;43(2):158–163.
Zabihi N, Mourtzinos A, Maher MG, Raz S, Rodriguez LV. Short-term results of bilateral S2-S4 sacral neuromodulation for the treatment of refractory interstitial cystitis, painful bladder syndrome, and chronic pelvic pain. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19(4):553–557.
Neuromodulation is the science of using electrical impulses to alter neuronal activities. The exact mechanisms of action are unclear, but the technology has been utilized to control symptoms of overactive bladder and urinary retention caused by poor relaxation of the urethral and pelvic floor muscles. While studying the effects of sacral nerve root neuromodulation on the bladder, investigators noted improvements in other symptoms, such as pelvic pain.
Neuromodulation of the sacral nerve roots may be achieved by direct conduction of electrical impulses from a lead implanted in the sacrum (sacral neuromodulation) or by the retrograde conduction of these impulses through the posterior tibial nerve (percutaneous tibial nerve stimulation, or PTNS) (FIGURE 2). The tibial nerve arises from sacral nerves L5 to S3 and is one of the larger branches of the sciatic nerve.
FIGURE 2 InterStim therapy
Stimulation of the sacral nerve has been used successfully to manage overactive bladder and urinary retention and may prove useful in the treatment of pelvic myofascial pain.
Van Balken et al: Details of the study
In this prospective observational study, 33 patients (both male and female) who had chronic pelvic pain by history and examination were treated with weekly, 30-minute outpatient sessions of PTNS for 12 weeks. Participants were asked to provide baseline pain scores and keep a diary of their pain. Quality-of-life questionnaires were also administered at baseline and at 12 weeks.
Investigators considered both subjective and objective success in their outcomes. If a patient elected to continue therapy, he or she was classified as a subjective success. Objective success required a decrease of at least 50% in the pain score. At the end of 12 weeks, although 33 patients (42%) wanted to continue therapy, only seven (21%) met the definition for objective success. Of those seven, six elected to continue therapy.
This study sheds light on a treatment modality that has not been studied adequately for the indication of pelvic pain but that may be promising in patients who have levator myalgia. Limitations of this study include the lack of a placebo arm, short-term outcome, and lack of localization of pain. Furthermore, although PTNS has FDA approval for treatment of urinary urgency, frequency, and urge incontinence, it is not approved for the treatment of pelvic pain. These preliminary findings demonstrate potential but, as with any new indication, long-term comparative studies are needed.
Zabihi et al: Details of the study
Patients in this retrospective study had a diagnosis of interstitial cystitis or chronic pelvic pain. Pelvic myofascial pain and trigger points were not required for eligibility. Thirty patients (21 women and nine men) had temporary placement of a lead containing four small electrodes along the S2 to S4 sacral nerve roots on both sides of the sacrum. They were then followed for a trial period of 2 to 4 weeks. To qualify for the final stage of the study, in which the leads were connected internally to a generator implanted in the buttocks, patients had to report improvement of at least 50% in their symptoms. If their improvement did not meet that threshold, the leads were removed.
Twenty-three patients (77%) met the criteria for permanent implantation. Of these patients, 42% reported improvement of more than 50% at 6 postoperative months. Quality-of-life scores also improved significantly.
Sacral neuromodulation is not FDA-approved for the treatment of chronic pelvic pain; further studies are needed before it can be recommended for this indication.
Neither of these studies required objective evidence of myofascial pain for inclusion. Therefore, although the benefits they demonstrated may be theorized to extend to the relief of myofascial pain, this fact cannot be corroborated.
We want to hear from you! Tell us what you think.
Vulvar Pain Syndromes 3-Part Series
- Making the correct diagnosis
(September 2011) - A bounty of treatments-but not all of them are proven
(October 2011) - Provoked vestibulodynia
(Coming in November 2011)
Chronic pelvic pain: 11 critical questions about causes and care
Fred M. Howard, MD (August 2009)
Vague symptoms. Unexpected flares. Inconsistent manifestations. These characteristics can make diagnosis and treatment of chronic pelvic pain frustrating for both patient and physician. Most patients undergo myriad tests and studies to uncover the source of their pain—but a targeted pelvic exam may be all that is necessary to identify a prevalent but commonly overlooked cause of pelvic pain. Levator myalgia, myofascial pelvic pain syndrome, and pelvic floor spasm are all terms that describe a condition that may affect as many as 78% of women who are given a diagnosis of chronic pelvic pain.1 This syndrome may be represented by an array of symptoms, including pelvic pressure, dyspareunia, rectal discomfort, and irritative urinary symptoms such as spasms, frequency, and urgency. It is characterized by the presence of tight, band-like pelvic muscles that reproduce the patient’s pain when palpated.2
Diagnosis of this syndrome often surprises the patient. Although the concept of a muscle spasm is not foreign, the location is unexpected. Patients and physicians alike may forget that there is a large complex of muscles that completely lines the pelvic girdle. To complicate matters, the patient often associates the onset of her symptoms with an acute event such as a “bad” urinary tract infection or pelvic or vaginal surgery, which may divert attention from the musculature. Although a muscle spasm may be the cause of the patient’s pain, it’s important to realize that an underlying process may have triggered the original spasm. To provide effective treatment of pain, therefore, you must identify the fundamental cause, assuming that it is reversible, rather than focus exclusively on symptoms.
Although there are many therapeutic options for levator myalgia, an appraisal of the extensive literature on these medications is beyond the scope of this article. Rather, we will review alternative treatment modalities and summarize the results of five trials that explored physical therapy, trigger-point or chemodenervation injection, and neuromodulation (TABLE).
Weighing the nonpharmaceutical options for treatment
of myofascial pelvic pain
| Treatment | Pros | Cons |
|---|---|---|
| Physical therapy | Minimally invasive Moderate long-term success | Requires highly specialized therapist |
| Trigger-point injection | Minimally invasive Performed in clinic Immediate short-term success | Optimal injectable agent is unknown Botulinum toxin A lacks FDA approval for this indication Limited information on adverse events and long-term efficacy |
| Percutaneous tibial nerve stimulation | Minimally invasive Performed in clinic | Requires numerous office visits for treatment Lacks FDA approval for this indication Limited information on long-term efficacy |
| Sacral neuromodulation | Moderately invasive Permanent implant | Requires implantation in operating room Lacks FDA approval for this indication Limited information on long-term efficacy |
Pelvic myofascial therapy offers relief—but qualified therapists may be scarce
FitzGerald MP, Anderson RU, Potts J, et al; Urological Pelvic Pain Collaborative Research Network. Randomized multicenter feasibility trial of myofascial physical therapy for the treatment of urological chronic pelvic pain syndromes. J Urology. 2009;182(2):570–580.
Physical therapy of the pelvic floor—otherwise known as pelvic myofascial therapy—requires a therapist who is highly trained and specialized in this technique. It is more invasive than other forms of rehabilitative therapy because of the need to perform transvaginal maneuvers (FIGURE 1).
This pilot study by the Urological Pelvic Pain Collaborative Research Network evaluated the ability of patients to adhere to pelvic myofascial therapy, the response of their pain to therapy, and adverse events associated with manual therapy. It found that patients were willing to undergo the therapy, despite the invasive nature of the maneuvers, because it was significantly effective.
Details of the study
Patients (both men and women) were randomized to myofascial physical therapy or global therapeutic massage. Myofascial therapy consisted of internal or vaginal manipulation of the trigger-point muscle bundles and tissues of the pelvic floor. It also focused on muscles of the hip girdle and abdomen. The comparison group underwent traditional Western full-body massage. In both groups, treatment lasted 1 hour every week, and participants agreed to 10 full treatments.
Patients were eligible for the study if they experienced pelvic pain, urinary frequency, or bladder discomfort in the previous 6 months. In addition, an examiner must have been able to elicit tenderness upon palpation of the pelvic floor during examination. Patients were excluded if they showed signs of urinary tract infection or dysmenorrhea.
A total of 47 patients were randomized—24 to global massage and 23 to myofascial physical therapy. Overall, the myofascial group experienced a significantly higher rate of improvement in the global response at 12 weeks than did patients in the global-massage group (57% vs 21%; P=.03). Patients were willing to engage in myofascial pelvic therapy, and adverse events were minor.
FIGURE 1 Transvaginal myofascial therapy
Physical therapy of the pelvic floor is more invasive than other forms of rehabilitative therapy because of the need to perform transvaginal maneuvers.
Need for specialized training may limit number of therapists
The randomized controlled study design renders these findings fairly reliable. Therapists were unmasked and aware of the treatment arms but were trained to make the different therapy sessions appear as similar as possible.
Although investigators were enthusiastic about their initial findings, additional studies are needed to validate the results. Moreover, these findings may be difficult to generalize because women who volunteer to participate in such a study may differ from the general population.
Nevertheless, patients who suffer from chronic pelvic pain may take heart that there is a nonpharmaceutical alternative to manage their symptoms, although availability is likely limited in many areas. Given the nature of the physical therapy required for this particular location of myofascial pain, specialized training is necessary for therapists. Despite motivated patients and well-informed providers, it may be difficult to find specialized therapists within local vicinities. Referrals to centers where this type of therapy is offered may be necessary.
Pelvic myofascial therapy is an effective and acceptable intervention for the treatment of levator myalgia.
The ideal agent for trigger-point injections remains a mystery
Langford CF, Udvari Nagy S, Ghoniem G M. Levator ani trigger point injections: An underutilized treatment for chronic pelvic pain. Neurourol Urodyn. 2007;26(1):59–62.
Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol. 2006;108(4):915–923.
Trigger points are discrete, tender areas within a ridge of contracted muscle. These points may cause focal pain or referred pain upon irritation of the muscle.2 Trigger-point injection therapy aims to anesthetize or relax these points by infiltrating the muscle with medications.
These two studies evaluated the value of trigger-point injections in the treatment of pelvic myofascial pain; they found that the injections provide relief, although the mechanism of action and the ideal agent remain to be determined.
Langford et al: Details of the study
In this prospective study, 18 women who had pelvic pain of at least 6 months’ duration and confirmed trigger points on examination underwent transvaginal injection of a solution of bupivacaine, lidocaine, and triamcinolone. They were assessed by questionnaire at baseline and 3 months after injection. Assessment included a visual analog scale for pain severity. Investigators defined success as a decrease in pain of 50% or more and global-satisfaction and global-cure visual scores of 60% or higher.
Thirteen of the 18 women (72.2%) improved after their first injection, with six women reporting a complete absence of pain. Overall, women reported significant decreases in pain and increases in the rates of satisfaction and cure, meeting the definition of success at 3 months after the injection.
Among the theories proposed to explain the mechanism of action of trigger-point injections are:
- disruption of reflex arcs within skeletal muscle
- release of endorphins
- mechanical changes in abnormally contracted muscle fibers.
This last theory highlights one of the limitations of this study—lack of a placebo arm. Could it be possible that the injection of any fluid produces the same effect?
This study was not designed to investigate the causal relationship between the injection of a particular solution and pain relief, but it does highlight the need for studies to clarify the mechanism of action, including use of a placebo. It also prompts questions about the duration of effect after a single injection.
Goal of chemodenervation is blocking of muscle activity
Botulinum toxin type A (Botox) blocks the release of acetylcholine from presynaptic neurons. The release of acetylcholine stimulates muscle contractions; therefore, blockage of its release reduces muscle activity. This type of chemodenervation has found widespread use, and botulinum toxin A now has approval from the Food and Drug Administration (FDA) for treatment of chronic migraine, limb spasticity, cervical dystonia, strabismus, hyperhidrosis, and facial cosmesis.3 Although it is not approved for pelvic floor levator spasm, its success in treating other myotonic disorders suggests that its application may be relevant.
Abbott et al: Details of the study
Abbott and colleagues performed a double-blind, randomized, controlled trial to compare injection of botulinum toxin A with injection of saline. They measured changes in the pain scale, quality of life, and vaginal pressure.
Women were eligible for the study if they had subjectively reported pelvic pain of more than 2 years’ duration and objective evidence of trigger points (on examination) and elevated vaginal resting pressure (by vaginal manometry). Neither the clinical research staff nor the patient knew the contents of the injections, but all women received a total of four—two at sites in the puborectalis muscle and two in the pubococcygeus muscle.
After periodic assessment by questionnaire and examination through 6 months after injection, no differences were found in the pain score or resting vaginal pressure between the group of women who received botulinum toxin A and the group who received placebo. However, each group experienced a significant reduction in pain and vaginal pressure, compared with baseline. And both groups reported improved quality of life, compared with baseline. Neither group reported voiding dysfunction.
These two studies support the use of trigger-point injection into pelvic floor muscles to reduce pelvic myofascial pain. The findings of Abbott and colleagues, in particular, suggest that the substance that is injected may not be as important as the actual needling of the muscle. Larger studies and comparisons between placebo, botulinum toxin A, and anesthetic solutions are needed to elucidate the therapeutic benefit of these particular medications.
Neuromodulation shows promise as treatment for pelvic myofascial pain
van Balken MR, Vandoninck V, Messelink, BJ, et al. Percutaneous tibial nerve stimulation as neuromodulative treatment of chronic pelvic pain. Eur Urol. 2003;43(2):158–163.
Zabihi N, Mourtzinos A, Maher MG, Raz S, Rodriguez LV. Short-term results of bilateral S2-S4 sacral neuromodulation for the treatment of refractory interstitial cystitis, painful bladder syndrome, and chronic pelvic pain. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19(4):553–557.
Neuromodulation is the science of using electrical impulses to alter neuronal activities. The exact mechanisms of action are unclear, but the technology has been utilized to control symptoms of overactive bladder and urinary retention caused by poor relaxation of the urethral and pelvic floor muscles. While studying the effects of sacral nerve root neuromodulation on the bladder, investigators noted improvements in other symptoms, such as pelvic pain.
Neuromodulation of the sacral nerve roots may be achieved by direct conduction of electrical impulses from a lead implanted in the sacrum (sacral neuromodulation) or by the retrograde conduction of these impulses through the posterior tibial nerve (percutaneous tibial nerve stimulation, or PTNS) (FIGURE 2). The tibial nerve arises from sacral nerves L5 to S3 and is one of the larger branches of the sciatic nerve.
FIGURE 2 InterStim therapy
Stimulation of the sacral nerve has been used successfully to manage overactive bladder and urinary retention and may prove useful in the treatment of pelvic myofascial pain.
Van Balken et al: Details of the study
In this prospective observational study, 33 patients (both male and female) who had chronic pelvic pain by history and examination were treated with weekly, 30-minute outpatient sessions of PTNS for 12 weeks. Participants were asked to provide baseline pain scores and keep a diary of their pain. Quality-of-life questionnaires were also administered at baseline and at 12 weeks.
Investigators considered both subjective and objective success in their outcomes. If a patient elected to continue therapy, he or she was classified as a subjective success. Objective success required a decrease of at least 50% in the pain score. At the end of 12 weeks, although 33 patients (42%) wanted to continue therapy, only seven (21%) met the definition for objective success. Of those seven, six elected to continue therapy.
This study sheds light on a treatment modality that has not been studied adequately for the indication of pelvic pain but that may be promising in patients who have levator myalgia. Limitations of this study include the lack of a placebo arm, short-term outcome, and lack of localization of pain. Furthermore, although PTNS has FDA approval for treatment of urinary urgency, frequency, and urge incontinence, it is not approved for the treatment of pelvic pain. These preliminary findings demonstrate potential but, as with any new indication, long-term comparative studies are needed.
Zabihi et al: Details of the study
Patients in this retrospective study had a diagnosis of interstitial cystitis or chronic pelvic pain. Pelvic myofascial pain and trigger points were not required for eligibility. Thirty patients (21 women and nine men) had temporary placement of a lead containing four small electrodes along the S2 to S4 sacral nerve roots on both sides of the sacrum. They were then followed for a trial period of 2 to 4 weeks. To qualify for the final stage of the study, in which the leads were connected internally to a generator implanted in the buttocks, patients had to report improvement of at least 50% in their symptoms. If their improvement did not meet that threshold, the leads were removed.
Twenty-three patients (77%) met the criteria for permanent implantation. Of these patients, 42% reported improvement of more than 50% at 6 postoperative months. Quality-of-life scores also improved significantly.
Sacral neuromodulation is not FDA-approved for the treatment of chronic pelvic pain; further studies are needed before it can be recommended for this indication.
Neither of these studies required objective evidence of myofascial pain for inclusion. Therefore, although the benefits they demonstrated may be theorized to extend to the relief of myofascial pain, this fact cannot be corroborated.
We want to hear from you! Tell us what you think.
1. Bassaly R, Tidwell N, Bertolino S, Hoyte L, Downes K, Hart S. Myofascial pain and pelvic floor dysfunction in patients with interstitial cystitis. Int Urogynecol J. 2011;22(4):413-418.
2. Alvarez DJ, Rockwell PG. Trigger points: diagnosis and management. Am Fam Physician. 2002;65(4):653-660.
3. Allergan, Inc. Medication Guide: BOTOX. US Food and Drug Administration Web site. http://www.fda.gov/downloads/Drugs/DrugSafety/UCM176360.pdf. Published October 2010. Accessed August 30, 2011.
1. Bassaly R, Tidwell N, Bertolino S, Hoyte L, Downes K, Hart S. Myofascial pain and pelvic floor dysfunction in patients with interstitial cystitis. Int Urogynecol J. 2011;22(4):413-418.
2. Alvarez DJ, Rockwell PG. Trigger points: diagnosis and management. Am Fam Physician. 2002;65(4):653-660.
3. Allergan, Inc. Medication Guide: BOTOX. US Food and Drug Administration Web site. http://www.fda.gov/downloads/Drugs/DrugSafety/UCM176360.pdf. Published October 2010. Accessed August 30, 2011.
Is private ObGyn practice on its way out?
- Medicare and Medicaid are on the brink of insolvency, and you’re not just a bystander
Robert L. Barbieri, MD (Editorial, October 2011) - 14 questions (and answers) about health reform and you
Lucia DiVenere, ACOG Director of Government Affairs, with OBG MANAGEMENT Senior Editor Janelle Yates (June 2010)
In the 18 months since the Patient Protection and Affordable Care Act—otherwise known as ACA, or health-care reform—was signed into law by President Barack Obama, the outlook for private practice, in any specialty, has dimmed. As a recent report on the ramifications of the ACA put it:
The imperative to care for more patients, to provide higher perceived quality, at less cost, with increased reporting and tracking demands, in an environment of high potential liability and problematic reimbursement, will put additional stress on physicians, particularly those in private practice.1
To explore the impact of these stresses on ObGyns specifically, the editors of OBG Management invited Lucia DiVenere, Senior Director of Government Affairs for the American Congress of Obstetricians and Gynecologists (ACOG), to talk about the outlook for private practice in the coming years. In the exchange, Ms. DiVenere discusses the short- and long-term effects of the ACA, the ways in which ObGyn practice is (or is not) evolving, the challenge of making the switch to electronic health records (EHRs), the reasons ACOG opposed the ACA, and other issues related to the current practice environment. In addition, two ObGyns in private practice describe the many challenges they face (see “The view from private practice,” pages 44 and 52).
What nonlegislative forces have affected private practice?
OBG Management: A recent report on the ramifications of the ACA argues that formal reform was inevitable. It also asserts that private practice was subject to many negative pressures long before health-care reform was passed.1 Do you agree?
Ms. DiVenere: Our nation’s health-care system is always evolving, and over the past decade, we’ve seen a clear trend toward system integration—that is, larger, physician-led group practices and hospital employment of physicians.
Looking at physicians as a whole, the percentage who practice solo or in two-physician practices fell from 40.7% in 1996–97 to 32.5% by 2004–05, according to a 2007 survey.2 And the American Medical Association (AMA) reported that the percentage of physicians “with an ownership stake in their practice declined from 61.6% to 54.4% as more physicians opted for employment. Both the trends away from solo and two-physician practices and toward employment were more pronounced for specialists and older physicians.”2
OBG Management: What has the trend been for ObGyns, specifically?
Ms. DiVenere: The ObGyn specialty employs the group practice model—health care delivered by three or more physicians—more frequently than other specialties do, largely because of the support it provides for 24–7 OB call schedules.
As for private practice, ObGyns have been moving away from it for 10 years or longer. Data from a 1991 ACOG survey shows that 77% of respondents were in private practice; by 2003, that percentage had fallen to 70%.3 In 2003, ObGyns in private practice tended to be older (median age: 47 years) than their salaried colleagues (median: 42 years) and were more likely to be male (87% vs 77%).3 The demographic change toward women ObGyns may add to this trend line.
OBG Management: What economic forces have shaped practice paradigms in ObGyn?
Ms. DiVenere: Median expenses for private practices have been steadily rising in relation to revenues—from 52% in 1990 to 71% in 2002—making it difficult for practices to remain solvent.4 In addition, a 2011 survey from Medscape reveals that ObGyns in solo practice earn $15,000 to $25,000 less annually than their employed colleagues.5
ObGyns who have made the switch from private to hospital practice, or who have become ObGyn hospitalists, often point to the difficulties of maintaining a solvent private practice, especially given the push toward electronic health records (EHRs) and increasing regulatory and administrative burdens. These and other issues contribute to rising practice costs and increasing demands on an ObGyn’s time and attention.
“I still love what I do”
I’ve been in solo ObGyn practice for 10 years. Before that, I worked 10 years for two medical groups—that makes 20 years of medical practice. I entered medicine late after teaching school for 10 years.
Most of my patients used to have union jobs and were employed by the steel mills in south Chicago and Northwest Indiana or in construction or manufacturing. One of the benefits of a union job was good insurance. As the economy began to sour, those mills changed hands and are now owned largely by foreign companies. Wages were cut dramatically, and insurance benefits are now “bare bones.” I continue to see my patients regardless of their circumstances.
Most maternity benefits require a hefty out-of-pocket expense. Around here, the doctor gets stuck with the deductible and, consequently, ends up doing lots of free deliveries. I haven’t figured it out yet, but I’m willing to bet that I lose money on OB.
Most patients realize that it’s tough to run a business on a declining revenue stream and are grateful that I take care of them. I’ve treated many of their family members, delivered their babies, provided primary care, done prolapse repairs on mom and grandmom. I know everybody by name—that’s the school teacher in me. I still feel honored to do what I do, but it isn’t easy. The other docs who cover for me on my rare days off, for CME, tell me I have “a nice practice.” That’s why I do it, for the good, salt-of-the-earth folks who would like to pay their bills if times were better.
Medicine is changing quickly. It has taken time to learn the electronic health record (EHR) at the local hospital. Every documentation takes longer. I now spend more time at the computer desk than with patients on hospital rounds. I have read about accountable care organizations and being “enabled,” but the next round of payment cuts will likely kill private practice.
I have Indiana University medical students come and rotate with me, and I try to be as upbeat as I can. The students tell me that my office is the one everyone wants to rotate through. I used to hope that someone might come back and join me here—but maybe the young people have it right. They won’t live with a pager all the time. They won’t do call. To them, medicine will be a job. They will be “providers.”
I may not have practiced in the golden age of medicine, but at least I feel that I had an impact on the lives of the families I have been honored to serve. I still love what I do—it’s just getting harder to justify doing it.
—Mary Vanko, MD
Munster, Ind.
What are the short-term effects of formal reform?
OBG Management: What effect has health-care reform had so far?
Ms. DiVenere: In 2010, twice as many physician practices were bought by hospitals and health systems as in 2009. We can’t conclude that the 2010 law is responsible for these changes, but we know that the architects of health reform were aware of this trend, believed it was beneficial, and looked for ways to encourage it, including through development of accountable care organizations (ACOs), which give hospitals a new and potentially lucrative reason to purchase private practices.
OBG Management: What exactly is an ACO?
Ms. DiVenere: An ACO consists of aligned providers—most likely, large multispecialty groups, often affiliated with the same hospital—who agree to manage patients for a set fee, sharing the risk and potential profit. ACOs are required to have shared governance, which gives them the authority to impose standards for practice, reporting, and compensation—including rewards and penalties—across a group of physicians.
Each ACO must sign a 3-year contract with the US Department of Health and Human Services (HHS) and include a sufficient number of primary care professionals to care for at least 5,000 beneficiaries. ACOs will be evaluated by quality-performance measures to be determined by the Secretary of HHS.
What aspects of ACA will have the biggest impact?
OBG Management: What provision of the new law will have the most direct impact on ObGyns in private practice?
Ms. DiVenere: All ObGyns may benefit from the guarantee of insurance coverage for our patients for maternity and preventive care. And all ObGyns should join ACOG’s fight to repeal the Independent Payment Advisory Board, which may hold enormous power to cut physician reimbursement.
Here’s the quote all ObGyns—especially those in private practice—should read, from an article written by President Obama’s health-reform deputies:
To realize the full benefits of the Affordable Care Act, physicians will need to embrace rather than resist change. The economic forces put in motion by the Act are likely to lead to vertical organization of providers and accelerate physician employment by hospitals and aggregation into larger physician groups. The most successful physicians will be those who most effectively collaborate with other providers to improve outcomes, care productivity, and patient experience.6
OBG Management: Do these health-reform deputies offer any concrete vision of how this change will be achieved?
Ms. DiVenere: They detail what physicians need to do, and how they should change the way they practice, under the Act. For example, to meet the increasing demand for health care, they recommend that practices:
- “Redesign care to include a team of nonphysician providers, such as nurse practitioners, physician assistants, care coordinators, and dieticians”
- “Develop approaches to engage and monitor patients outside of the office.”6
And to meet the requirements for payment reform, information transparency, and quality, they suggest that practices:
- “Focus care around exceptional patient experience and shared clinical outcome goals”
- “Engage in shared decision-making discussions regarding treatment goals and approaches”
- “Proactively manage preventive care”
- “Establish teams to take part in bundled payments and incentive programs”
- “Expand use of electronic health records”
- “Collaborate with hospitals to dramatically reduce readmissions and hospital-acquired infections”
- “Incorporate patient-centered outcomes research to tailor care.”6
To capture value, the authors recommend that practices “redesign medical office processes to capture savings from administrative simplification.”6
OBG Management: Do they propose any method for implementing these changes?
Ms. DiVenere: The White House is hoping that ACOs can lead. Medicare ACOs will attempt to accomplish these changes by managing hospital and physician services, prompting physicians and hospitals to change how they are both clinically organized and paid for services—this change, in particular, is considered by some to be essential to improving the quality and efficiency of health care.
The new Centers for Medicaid and Medicare Services (CMS) Innovation Center, created by the ACA, is given broad authority to test, evaluate, and adopt systems that foster patient-centered care, improve quality, and contain the costs of Medicare, Medicaid, and the Children’s Health Insurance Program (CHIP). The law specifically guides the Innovation Center to look for ways to encourage physicians to transition from fee-for-service to salary-based payment.
We can safely assume that the experts behind these provisions believe that large, hospital-centered systems or large, physician-led groups can better serve the needs of patients and our health-care system in general than can small private practices. Today, 24% of ObGyns run solo practices, and 27% are in single-specialty practices. Health-care reform can mean big changes for them.
Is the EHR a realistic goal for private practices?
OBG Management: How does the push for EHRs affect physicians in private practice?
Ms. DiVenere: Very profoundly. From the point of view of an ObGyn in private practice, EHRs offer a number of benefits. They can:
- help make sense of our increasingly fragmented health-care system
- improve patient safety
- increase efficiency
- reduce paperwork.
In addition, insurers may save by reducing unnecessary tests, and patients can certainly benefit from better coordination and documentation of care. These advantages don’t necessarily translate into savings or revenue for physician practices, however. Many ObGyns—especially those in solo or small practices—don’t feel confident making such a large capital investment. In fact, only about one third of ObGyn practices have an EHR.
OBG Management: Is it primarily cost that deters ObGyns from adopting EHRs?
Ms. DiVenere: That, and the fact that EHR systems are not yet fully interoperable across small practices, insurers, and government agencies. The initial cost of purchasing an EHR system for a small practice is about $50,000 per physician, and there are ongoing costs in staff training and hardware and software updates. A steep learning curve means fewer patients can be seen in an hour. It can take a practice months—even years—for physicians to return to their previous level of productivity. That’s a lot to ask a busy practicing physician to take on.
OBG Management: Is there any way around the push for EHRs?
Ms. DiVenere: Congress wants to move us to full adoption of health information technology (HIT). Under health-care reform, beginning in 2013, all health insurance plans must comply with a uniform standard for electronic transactions, including eligibility verification and health claim status.
In 2014, uniform standards must:
- allow automatic reconciliation of electronic funds transfers and HIPAA payment and remittance
- use standardized and consistent methods of health plan enrollment and editing of claims
- use unique health plan identifiers to simplify and improve routing of health-care transactions
- use standardized claims attachments
- improve practice data collection and evaluation.
Uniformity and standardization can help address one of the major roadblocks to physician adoption of HIT. Still, it’s little wonder that median expenses for private practices have been steadily rising in relation to revenues.
So far, the cons of an EHR outweigh the pros
My private practice made the transition to electronic health records (EHRs) about 4 months ago. We have discovered that EHRs do have a number of positive characteristics:
- Prescriptions are completely legible and can be sent directly to the pharmacy
- The staff no longer needs to search for charts
- If test results have been downloaded, they can be quickly accessed.
However, EHRs also require a lot of time to learn how to use them properly. And the problems don’t end there. For example, instead of looking at a patient’s face when taking a history, we now look at the monitor.
In addition, the templates have many data fields that auto-populate as “normal.” There is an illusion that a thorough history and physical were performed—so it requires a lot of time meticulously reviewing each chart to make sure that it is accurate. One must always be aware of the potential for insurance fraud and the medicolegal risk of documenting something as normal when it isn’t.
Ordering labs is cumbersome because each test must be handled separately, and the terminology does not always match the options at our contracted laboratories. We spend a lot of time searching for each lab that is ordered.
To achieve “meaningful use” of the EHR, certain parameters must be met at every single visit. The medication list must be reviewed (even if the patient takes no medications), and there must be a notation that cervical and breast cancer screening have been ordered, even though recent Pap smear and mammogram results are included in the chart. Regardless of the patient’s age or situation, the issues of contraception, sexually transmitted disease, tobacco use, and domestic violence must be addressed.
So when a 65-year-old woman presents with postmenopausal bleeding, I have to comment on these issues or delete them from the report. I have to provide the same documentation when she returns the next week for a biopsy and the week after that when she returns for the results.
It has become impossible to see patients in the time frame I have used for the past 24 years, and my patients and staff remain frustrated. I am always behind schedule, and I fear that the computer gets more attention than my patients do during the office visit.
—Mark A. Firestone, MD
Aventura, Fla
Is there a physician shortage in ObGyn?
OBG Management: There has been a lot of attention focused on the shortage of physicians in this country. How severe is the shortage likely to be in the specialty of ObGyn?
Ms. DiVenere: William F. Rayburn, MD, MBA, from the University of New Mexico Health Sciences Center and School of Medicine in Albuquerque, has provided ACOG with important work on this topic. Dr. Rayburn is Randolph V. Seligman Professor and Chair in the Department of Obstetrics and Gynecology at that institution. His report7 shows that the gap between the supply of ObGyns and the demand for women’s health care is widening.
Data from this report point to a shortage of 9,000 to 14,000 ObGyns in 20 years. After 2030, the ObGyn shortage may be even more pronounced, as the population of women is projected to increase 36% by 2050, while the number of ObGyns remains constant.
OBG Management: Would the incorporation of more midlevel providers—that is, nurse midwives, physician assistants, and others—ease some of the strains on the ObGyn workforce in general and on private practice specifically?
Ms. DiVenere: That’s very possible and is one of the reasons ACOG encourages greater use of collaborative care. It’s certainly true that the ObGyn specialty is historically comfortable with collaboration.
In 2008, the average ObGyn practice employed 2.6 nonphysician clinicians, certified nurse midwives (CNMs), physician assistants, or nurse practitioners. ACOG strongly supports collaborative practice between ObGyns and qualified midwives (CNMs, certified midwives) and other nonphysician clinicians.
In a recent issue of Obstetrics & Gynecology, ACOG’s Immediate Past President Richard N. Waldman, MD, and Holly Powell Kennedy, CNM, PhD, president of the American College of Nurse Midwives, wrote about the importance of collaborative practice, which can “increase efficiency, improve clinical outcomes, and enhance provider satisfaction.”8 The September 2011 issue of Obstetrics & Gynecology highlights four real-life stories of successful collaborative practice.
Because of our commitment to the benefits of collaborative practice, ACOG supported a provision in the ACA that increases payment for CNMs. Beginning this year, the Medicare program will reimburse CNMs at 100% of the physician payment rate for the same services. Before this law, CNMs were paid at 65% of a physician’s rate for the same services. ACOG’s leadership believes that better reimbursement for CNMs will help ObGyn practices and improve care.
Although ACOG supported that provision in the law, we did not support passage of the ACA. ACOG’s Executive Board carefully considered all elements of the bill and decided that, although it included many provisions that are helpful to our patients, it also included many harmful provisions for our members. We felt that the promise of the women’s health provisions could be realized only if the legislation worked for practicing physicians, too.
Why wasn’t the SGR formula abolished?
OBG Management: What is the effect, for physicians in general and ObGyns in private practice specifically, of the failure to fix the sustainable growth rate (SGR) formula and implement liability reform in the ACA?
Ms. DiVenere: The fact that we have no fix for SGR and no medical liability reform is a disgrace. And the absence of solutions to these two issues is one of the main reasons ACOG decided not to support passage of the ACA. Our Executive Board was very clear in telling Congress that we can’t build a reformed health-care system on broken payment and liability systems—it just won’t work.
ObGyns themselves are the first to point out this fact. ObGyns listed medical liability (65.3%) and financial viability of their practice (44.3%) as their top two professional concerns in a 2008 ACOG survey.9
For all ObGyns, these two continuing problems make practice more difficult every day—problems felt most acutely by ObGyns in private practice. Employed physicians often come under a hospital’s or health system’s liability policy and often don’t pay their own premiums. Employed physicians usually are on salary, sheltering them from the vagaries of Congressional action, or inaction, on looming double-digit Medicare physician payment cuts under the SGR.
Some legislators, and some ObGyns, think Medicare doesn’t apply to us. But today, 92% of ObGyns participate in the Medicare program and 63% accept all Medicare patients. This fact reflects ObGyn training and commitment to serve as lifelong principal-care physicians for women, including women who have disabilities. Fifty-six percent of all Medicare beneficiaries are women. With the Baby Boomer generation transitioning to Medicare, and with shortages in primary care physicians, it is likely that ObGyns will become more involved in delivering health care for this population.
Medicare physician payments matter to ObGyns beyond the Medicare program, too, as Tricare and private payers often follow Medicare payment and coverage policies. As a specialty, we have much at stake in ensuring a stable Medicare system for years to come, starting with an improved physician payment system. [Editor’s note: See Dr. Robert L. Barbieri’s editorial on the subject.]
OBG Management: What is ACOG doing to improve this system?
Ms. DiVenere: ACOG is urging the US Congress to ensure that a better system adheres to the following priniciples:
- Medicare payments should fairly and accurately reflect the cost of care. In the final 2011 Medicare physician fee schedule, CMS is proposing to reduce the physician work value for ObGyn care to women by 11% below what is paid to other physicians for similar men’s services—exactly the opposite of what should be done to encourage good care coordination, and in direct contradiction to recommendations by the Resource-Based Relative Value Scale Update Committee (RUC). Medicare payments to obstetricians are already well below the cost of maternity care; no further cuts should be allowed for this care.
- A new payment system should be as simple, coordinated, and transparent as possible and recognize that there is no one-size-fits-all model. A new Medicare system should coordinate closely with other governmental and nongovernmental programs to ensure that information technology is interoperable, that quality measurement relies on high-quality, risk-adjusted data, and to guard against new and special systems that apply to only one program or may only be workable for one type of specialty or only certain types of diseases and conditions. ObGyns often see relatively few Medicare patients, and unique Medicare requirements can pose significant administrative challenges and inefficiencies to ObGyn participation.
- Congress should encourage, and remove barriers to, ObGyn and physician development of ACOs, medical homes for women, and other innovative models. Proposed rules on the Medicare Shared Savings Program allowing for expedited antitrust review should be extended to ACOs and other physician-led models of care that do not participate in the Medicare Shared Savings Program. These models should also recognize the dual role ObGyns may play, as both primary and specialty care providers.
- Congress should repeal the Independent Medicare Payment Advisory Board. Leaving Medicare payment decisions in the hands of an unelected, unaccountable body with minimal congressional oversight is bad for all physicians and for our patients.

The outlook for private practice, in any specialty, has dimmed. Says ACOG’s Lucia DiVenere: “Median expenses for private practices have been steadily rising in relationship to revenues—from 52% in 1990 to 71% in 2002—making it difficult for practices to remain solvent.”
Is private practice doomed?
OBG Management: In your opinion, over the long term, is health reform a positive or a negative for ObGyns in private practice?
Ms. DiVenere: On balance, I believe that the health reform law is a positive for our patients, and that fact may lead to an eventual positive for its Fellows, ACOG hopes. What it may mean for ObGyns in private practice, though, is more troubling.
The law has many intended purposes: 1) cover the uninsured, 2) tilt our health-care system toward primary care and use of nonphysician providers, and 3) push practices toward integration with hospitals and health systems and other paths to physician employment. Support for continuation or growth in any type of physician private practice is hard to find in the ACA.
OBG Management: What changes are in the pipeline?
Ms. DiVenere: Under the ACA, by 2013, the Secretary of HHS, with input from stakeholders, will set up a Physician Compare Web site, modeled after the program that exists for hospitals, using data from the Physician Quality Reporting Initiative (PQRI). Data on this site would be made public on January 1, 2013, comparing physicians in terms of quality of care and patient experience.
By law, these data are intended to be statistically valid and risk-adjusted; each physician must have time to review his or her information before it becomes publicly available; data must ensure appropriate attribution of care when multiple providers are involved; and the Secretary of HHS must give physicians timely performance feedback.
Data elements—to the extent that scientifically sound measures exist—will include:
- quality, patient satisfaction, and health outcomes information on Medicare physicians
- physician care coordination and risk-adjusted resource use
- safety, effectiveness, and timeliness of care.
Physicians who successfully interact with this program will likely be those who have a robust EHR system.
If this and other elements of the Affordable Care Act become real, however, we’ll likely see a fundamental shift in the kinds of settings in which ObGyns and other physicians opt to practice.
We want to hear from you! Tell us what you think.
1. Physicians Foundation. Health Reform and the Decline of Physician Private Practice. Boston Mass: Physicians Foundation; 2010.
2. American Medical Association. Health Care Trends 2008. http://www.ama-assn.org/resources/doc/clrpd/2008-trends.pdf. Accessed September 1 2011.
3. American College of Obstetricians and Gynecologists. Profile of ObGyn Practice. Washington DC: ACOG; 2003.
4. American College of Obstetricians and Gynecologists. Financial Trends in ObGyn Practice 1990–2002. Washington, DC: ACOG; 2004:6.
5. Medscape Physician Compensation Report: 2011 Results. . Accessed September 1 2011.
6. Kocher R, Emanuel EJ, DeParle NA. The Affordable Care Act and the future of clinical medicine: the opportunities and challenges. Ann Intern Med. 2010;153(8):536-539.
7. American College of Obstetricians and Gynecologists. The Obstetrician-Gynecologist Workforce in the United States: Facts Figures, and Implications 2011. Washington, DC: ACOG; 2011.
8. Waldman RN, Kennedy HP. Collaborative practice between obstetricians and midwives. Obstet Gynecol. 2011;118(3):503-504.
9. American College of Obstetricians and Gynecologists. Socioeconomic Survey of ACOG Fellows. Summary of Results. Washington DC: 2008:1.
| Ms. DiVenere talks about a fundamental shift in the way you’ll work |
Lucia DiVenere
ACOG Senior Director of Government Affairs, with OBG Management Senior Editor Janelle Yates

Lucia DiVenere
| Ms. DiVenere talks about a fundamental shift in the way you’ll work |
Lucia DiVenere
ACOG Senior Director of Government Affairs, with OBG Management Senior Editor Janelle Yates
Lucia DiVenere
| Ms. DiVenere talks about a fundamental shift in the way you’ll work |
Lucia DiVenere
ACOG Senior Director of Government Affairs, with OBG Management Senior Editor Janelle Yates
Lucia DiVenere
- Medicare and Medicaid are on the brink of insolvency, and you’re not just a bystander
Robert L. Barbieri, MD (Editorial, October 2011) - 14 questions (and answers) about health reform and you
Lucia DiVenere, ACOG Director of Government Affairs, with OBG MANAGEMENT Senior Editor Janelle Yates (June 2010)
In the 18 months since the Patient Protection and Affordable Care Act—otherwise known as ACA, or health-care reform—was signed into law by President Barack Obama, the outlook for private practice, in any specialty, has dimmed. As a recent report on the ramifications of the ACA put it:
The imperative to care for more patients, to provide higher perceived quality, at less cost, with increased reporting and tracking demands, in an environment of high potential liability and problematic reimbursement, will put additional stress on physicians, particularly those in private practice.1
To explore the impact of these stresses on ObGyns specifically, the editors of OBG Management invited Lucia DiVenere, Senior Director of Government Affairs for the American Congress of Obstetricians and Gynecologists (ACOG), to talk about the outlook for private practice in the coming years. In the exchange, Ms. DiVenere discusses the short- and long-term effects of the ACA, the ways in which ObGyn practice is (or is not) evolving, the challenge of making the switch to electronic health records (EHRs), the reasons ACOG opposed the ACA, and other issues related to the current practice environment. In addition, two ObGyns in private practice describe the many challenges they face (see “The view from private practice,” pages 44 and 52).
What nonlegislative forces have affected private practice?
OBG Management: A recent report on the ramifications of the ACA argues that formal reform was inevitable. It also asserts that private practice was subject to many negative pressures long before health-care reform was passed.1 Do you agree?
Ms. DiVenere: Our nation’s health-care system is always evolving, and over the past decade, we’ve seen a clear trend toward system integration—that is, larger, physician-led group practices and hospital employment of physicians.
Looking at physicians as a whole, the percentage who practice solo or in two-physician practices fell from 40.7% in 1996–97 to 32.5% by 2004–05, according to a 2007 survey.2 And the American Medical Association (AMA) reported that the percentage of physicians “with an ownership stake in their practice declined from 61.6% to 54.4% as more physicians opted for employment. Both the trends away from solo and two-physician practices and toward employment were more pronounced for specialists and older physicians.”2
OBG Management: What has the trend been for ObGyns, specifically?
Ms. DiVenere: The ObGyn specialty employs the group practice model—health care delivered by three or more physicians—more frequently than other specialties do, largely because of the support it provides for 24–7 OB call schedules.
As for private practice, ObGyns have been moving away from it for 10 years or longer. Data from a 1991 ACOG survey shows that 77% of respondents were in private practice; by 2003, that percentage had fallen to 70%.3 In 2003, ObGyns in private practice tended to be older (median age: 47 years) than their salaried colleagues (median: 42 years) and were more likely to be male (87% vs 77%).3 The demographic change toward women ObGyns may add to this trend line.
OBG Management: What economic forces have shaped practice paradigms in ObGyn?
Ms. DiVenere: Median expenses for private practices have been steadily rising in relation to revenues—from 52% in 1990 to 71% in 2002—making it difficult for practices to remain solvent.4 In addition, a 2011 survey from Medscape reveals that ObGyns in solo practice earn $15,000 to $25,000 less annually than their employed colleagues.5
ObGyns who have made the switch from private to hospital practice, or who have become ObGyn hospitalists, often point to the difficulties of maintaining a solvent private practice, especially given the push toward electronic health records (EHRs) and increasing regulatory and administrative burdens. These and other issues contribute to rising practice costs and increasing demands on an ObGyn’s time and attention.
“I still love what I do”
I’ve been in solo ObGyn practice for 10 years. Before that, I worked 10 years for two medical groups—that makes 20 years of medical practice. I entered medicine late after teaching school for 10 years.
Most of my patients used to have union jobs and were employed by the steel mills in south Chicago and Northwest Indiana or in construction or manufacturing. One of the benefits of a union job was good insurance. As the economy began to sour, those mills changed hands and are now owned largely by foreign companies. Wages were cut dramatically, and insurance benefits are now “bare bones.” I continue to see my patients regardless of their circumstances.
Most maternity benefits require a hefty out-of-pocket expense. Around here, the doctor gets stuck with the deductible and, consequently, ends up doing lots of free deliveries. I haven’t figured it out yet, but I’m willing to bet that I lose money on OB.
Most patients realize that it’s tough to run a business on a declining revenue stream and are grateful that I take care of them. I’ve treated many of their family members, delivered their babies, provided primary care, done prolapse repairs on mom and grandmom. I know everybody by name—that’s the school teacher in me. I still feel honored to do what I do, but it isn’t easy. The other docs who cover for me on my rare days off, for CME, tell me I have “a nice practice.” That’s why I do it, for the good, salt-of-the-earth folks who would like to pay their bills if times were better.
Medicine is changing quickly. It has taken time to learn the electronic health record (EHR) at the local hospital. Every documentation takes longer. I now spend more time at the computer desk than with patients on hospital rounds. I have read about accountable care organizations and being “enabled,” but the next round of payment cuts will likely kill private practice.
I have Indiana University medical students come and rotate with me, and I try to be as upbeat as I can. The students tell me that my office is the one everyone wants to rotate through. I used to hope that someone might come back and join me here—but maybe the young people have it right. They won’t live with a pager all the time. They won’t do call. To them, medicine will be a job. They will be “providers.”
I may not have practiced in the golden age of medicine, but at least I feel that I had an impact on the lives of the families I have been honored to serve. I still love what I do—it’s just getting harder to justify doing it.
—Mary Vanko, MD
Munster, Ind.
What are the short-term effects of formal reform?
OBG Management: What effect has health-care reform had so far?
Ms. DiVenere: In 2010, twice as many physician practices were bought by hospitals and health systems as in 2009. We can’t conclude that the 2010 law is responsible for these changes, but we know that the architects of health reform were aware of this trend, believed it was beneficial, and looked for ways to encourage it, including through development of accountable care organizations (ACOs), which give hospitals a new and potentially lucrative reason to purchase private practices.
OBG Management: What exactly is an ACO?
Ms. DiVenere: An ACO consists of aligned providers—most likely, large multispecialty groups, often affiliated with the same hospital—who agree to manage patients for a set fee, sharing the risk and potential profit. ACOs are required to have shared governance, which gives them the authority to impose standards for practice, reporting, and compensation—including rewards and penalties—across a group of physicians.
Each ACO must sign a 3-year contract with the US Department of Health and Human Services (HHS) and include a sufficient number of primary care professionals to care for at least 5,000 beneficiaries. ACOs will be evaluated by quality-performance measures to be determined by the Secretary of HHS.
What aspects of ACA will have the biggest impact?
OBG Management: What provision of the new law will have the most direct impact on ObGyns in private practice?
Ms. DiVenere: All ObGyns may benefit from the guarantee of insurance coverage for our patients for maternity and preventive care. And all ObGyns should join ACOG’s fight to repeal the Independent Payment Advisory Board, which may hold enormous power to cut physician reimbursement.
Here’s the quote all ObGyns—especially those in private practice—should read, from an article written by President Obama’s health-reform deputies:
To realize the full benefits of the Affordable Care Act, physicians will need to embrace rather than resist change. The economic forces put in motion by the Act are likely to lead to vertical organization of providers and accelerate physician employment by hospitals and aggregation into larger physician groups. The most successful physicians will be those who most effectively collaborate with other providers to improve outcomes, care productivity, and patient experience.6
OBG Management: Do these health-reform deputies offer any concrete vision of how this change will be achieved?
Ms. DiVenere: They detail what physicians need to do, and how they should change the way they practice, under the Act. For example, to meet the increasing demand for health care, they recommend that practices:
- “Redesign care to include a team of nonphysician providers, such as nurse practitioners, physician assistants, care coordinators, and dieticians”
- “Develop approaches to engage and monitor patients outside of the office.”6
And to meet the requirements for payment reform, information transparency, and quality, they suggest that practices:
- “Focus care around exceptional patient experience and shared clinical outcome goals”
- “Engage in shared decision-making discussions regarding treatment goals and approaches”
- “Proactively manage preventive care”
- “Establish teams to take part in bundled payments and incentive programs”
- “Expand use of electronic health records”
- “Collaborate with hospitals to dramatically reduce readmissions and hospital-acquired infections”
- “Incorporate patient-centered outcomes research to tailor care.”6
To capture value, the authors recommend that practices “redesign medical office processes to capture savings from administrative simplification.”6
OBG Management: Do they propose any method for implementing these changes?
Ms. DiVenere: The White House is hoping that ACOs can lead. Medicare ACOs will attempt to accomplish these changes by managing hospital and physician services, prompting physicians and hospitals to change how they are both clinically organized and paid for services—this change, in particular, is considered by some to be essential to improving the quality and efficiency of health care.
The new Centers for Medicaid and Medicare Services (CMS) Innovation Center, created by the ACA, is given broad authority to test, evaluate, and adopt systems that foster patient-centered care, improve quality, and contain the costs of Medicare, Medicaid, and the Children’s Health Insurance Program (CHIP). The law specifically guides the Innovation Center to look for ways to encourage physicians to transition from fee-for-service to salary-based payment.
We can safely assume that the experts behind these provisions believe that large, hospital-centered systems or large, physician-led groups can better serve the needs of patients and our health-care system in general than can small private practices. Today, 24% of ObGyns run solo practices, and 27% are in single-specialty practices. Health-care reform can mean big changes for them.
Is the EHR a realistic goal for private practices?
OBG Management: How does the push for EHRs affect physicians in private practice?
Ms. DiVenere: Very profoundly. From the point of view of an ObGyn in private practice, EHRs offer a number of benefits. They can:
- help make sense of our increasingly fragmented health-care system
- improve patient safety
- increase efficiency
- reduce paperwork.
In addition, insurers may save by reducing unnecessary tests, and patients can certainly benefit from better coordination and documentation of care. These advantages don’t necessarily translate into savings or revenue for physician practices, however. Many ObGyns—especially those in solo or small practices—don’t feel confident making such a large capital investment. In fact, only about one third of ObGyn practices have an EHR.
OBG Management: Is it primarily cost that deters ObGyns from adopting EHRs?
Ms. DiVenere: That, and the fact that EHR systems are not yet fully interoperable across small practices, insurers, and government agencies. The initial cost of purchasing an EHR system for a small practice is about $50,000 per physician, and there are ongoing costs in staff training and hardware and software updates. A steep learning curve means fewer patients can be seen in an hour. It can take a practice months—even years—for physicians to return to their previous level of productivity. That’s a lot to ask a busy practicing physician to take on.
OBG Management: Is there any way around the push for EHRs?
Ms. DiVenere: Congress wants to move us to full adoption of health information technology (HIT). Under health-care reform, beginning in 2013, all health insurance plans must comply with a uniform standard for electronic transactions, including eligibility verification and health claim status.
In 2014, uniform standards must:
- allow automatic reconciliation of electronic funds transfers and HIPAA payment and remittance
- use standardized and consistent methods of health plan enrollment and editing of claims
- use unique health plan identifiers to simplify and improve routing of health-care transactions
- use standardized claims attachments
- improve practice data collection and evaluation.
Uniformity and standardization can help address one of the major roadblocks to physician adoption of HIT. Still, it’s little wonder that median expenses for private practices have been steadily rising in relation to revenues.
So far, the cons of an EHR outweigh the pros
My private practice made the transition to electronic health records (EHRs) about 4 months ago. We have discovered that EHRs do have a number of positive characteristics:
- Prescriptions are completely legible and can be sent directly to the pharmacy
- The staff no longer needs to search for charts
- If test results have been downloaded, they can be quickly accessed.
However, EHRs also require a lot of time to learn how to use them properly. And the problems don’t end there. For example, instead of looking at a patient’s face when taking a history, we now look at the monitor.
In addition, the templates have many data fields that auto-populate as “normal.” There is an illusion that a thorough history and physical were performed—so it requires a lot of time meticulously reviewing each chart to make sure that it is accurate. One must always be aware of the potential for insurance fraud and the medicolegal risk of documenting something as normal when it isn’t.
Ordering labs is cumbersome because each test must be handled separately, and the terminology does not always match the options at our contracted laboratories. We spend a lot of time searching for each lab that is ordered.
To achieve “meaningful use” of the EHR, certain parameters must be met at every single visit. The medication list must be reviewed (even if the patient takes no medications), and there must be a notation that cervical and breast cancer screening have been ordered, even though recent Pap smear and mammogram results are included in the chart. Regardless of the patient’s age or situation, the issues of contraception, sexually transmitted disease, tobacco use, and domestic violence must be addressed.
So when a 65-year-old woman presents with postmenopausal bleeding, I have to comment on these issues or delete them from the report. I have to provide the same documentation when she returns the next week for a biopsy and the week after that when she returns for the results.
It has become impossible to see patients in the time frame I have used for the past 24 years, and my patients and staff remain frustrated. I am always behind schedule, and I fear that the computer gets more attention than my patients do during the office visit.
—Mark A. Firestone, MD
Aventura, Fla
Is there a physician shortage in ObGyn?
OBG Management: There has been a lot of attention focused on the shortage of physicians in this country. How severe is the shortage likely to be in the specialty of ObGyn?
Ms. DiVenere: William F. Rayburn, MD, MBA, from the University of New Mexico Health Sciences Center and School of Medicine in Albuquerque, has provided ACOG with important work on this topic. Dr. Rayburn is Randolph V. Seligman Professor and Chair in the Department of Obstetrics and Gynecology at that institution. His report7 shows that the gap between the supply of ObGyns and the demand for women’s health care is widening.
Data from this report point to a shortage of 9,000 to 14,000 ObGyns in 20 years. After 2030, the ObGyn shortage may be even more pronounced, as the population of women is projected to increase 36% by 2050, while the number of ObGyns remains constant.
OBG Management: Would the incorporation of more midlevel providers—that is, nurse midwives, physician assistants, and others—ease some of the strains on the ObGyn workforce in general and on private practice specifically?
Ms. DiVenere: That’s very possible and is one of the reasons ACOG encourages greater use of collaborative care. It’s certainly true that the ObGyn specialty is historically comfortable with collaboration.
In 2008, the average ObGyn practice employed 2.6 nonphysician clinicians, certified nurse midwives (CNMs), physician assistants, or nurse practitioners. ACOG strongly supports collaborative practice between ObGyns and qualified midwives (CNMs, certified midwives) and other nonphysician clinicians.
In a recent issue of Obstetrics & Gynecology, ACOG’s Immediate Past President Richard N. Waldman, MD, and Holly Powell Kennedy, CNM, PhD, president of the American College of Nurse Midwives, wrote about the importance of collaborative practice, which can “increase efficiency, improve clinical outcomes, and enhance provider satisfaction.”8 The September 2011 issue of Obstetrics & Gynecology highlights four real-life stories of successful collaborative practice.
Because of our commitment to the benefits of collaborative practice, ACOG supported a provision in the ACA that increases payment for CNMs. Beginning this year, the Medicare program will reimburse CNMs at 100% of the physician payment rate for the same services. Before this law, CNMs were paid at 65% of a physician’s rate for the same services. ACOG’s leadership believes that better reimbursement for CNMs will help ObGyn practices and improve care.
Although ACOG supported that provision in the law, we did not support passage of the ACA. ACOG’s Executive Board carefully considered all elements of the bill and decided that, although it included many provisions that are helpful to our patients, it also included many harmful provisions for our members. We felt that the promise of the women’s health provisions could be realized only if the legislation worked for practicing physicians, too.
Why wasn’t the SGR formula abolished?
OBG Management: What is the effect, for physicians in general and ObGyns in private practice specifically, of the failure to fix the sustainable growth rate (SGR) formula and implement liability reform in the ACA?
Ms. DiVenere: The fact that we have no fix for SGR and no medical liability reform is a disgrace. And the absence of solutions to these two issues is one of the main reasons ACOG decided not to support passage of the ACA. Our Executive Board was very clear in telling Congress that we can’t build a reformed health-care system on broken payment and liability systems—it just won’t work.
ObGyns themselves are the first to point out this fact. ObGyns listed medical liability (65.3%) and financial viability of their practice (44.3%) as their top two professional concerns in a 2008 ACOG survey.9
For all ObGyns, these two continuing problems make practice more difficult every day—problems felt most acutely by ObGyns in private practice. Employed physicians often come under a hospital’s or health system’s liability policy and often don’t pay their own premiums. Employed physicians usually are on salary, sheltering them from the vagaries of Congressional action, or inaction, on looming double-digit Medicare physician payment cuts under the SGR.
Some legislators, and some ObGyns, think Medicare doesn’t apply to us. But today, 92% of ObGyns participate in the Medicare program and 63% accept all Medicare patients. This fact reflects ObGyn training and commitment to serve as lifelong principal-care physicians for women, including women who have disabilities. Fifty-six percent of all Medicare beneficiaries are women. With the Baby Boomer generation transitioning to Medicare, and with shortages in primary care physicians, it is likely that ObGyns will become more involved in delivering health care for this population.
Medicare physician payments matter to ObGyns beyond the Medicare program, too, as Tricare and private payers often follow Medicare payment and coverage policies. As a specialty, we have much at stake in ensuring a stable Medicare system for years to come, starting with an improved physician payment system. [Editor’s note: See Dr. Robert L. Barbieri’s editorial on the subject.]
OBG Management: What is ACOG doing to improve this system?
Ms. DiVenere: ACOG is urging the US Congress to ensure that a better system adheres to the following priniciples:
- Medicare payments should fairly and accurately reflect the cost of care. In the final 2011 Medicare physician fee schedule, CMS is proposing to reduce the physician work value for ObGyn care to women by 11% below what is paid to other physicians for similar men’s services—exactly the opposite of what should be done to encourage good care coordination, and in direct contradiction to recommendations by the Resource-Based Relative Value Scale Update Committee (RUC). Medicare payments to obstetricians are already well below the cost of maternity care; no further cuts should be allowed for this care.
- A new payment system should be as simple, coordinated, and transparent as possible and recognize that there is no one-size-fits-all model. A new Medicare system should coordinate closely with other governmental and nongovernmental programs to ensure that information technology is interoperable, that quality measurement relies on high-quality, risk-adjusted data, and to guard against new and special systems that apply to only one program or may only be workable for one type of specialty or only certain types of diseases and conditions. ObGyns often see relatively few Medicare patients, and unique Medicare requirements can pose significant administrative challenges and inefficiencies to ObGyn participation.
- Congress should encourage, and remove barriers to, ObGyn and physician development of ACOs, medical homes for women, and other innovative models. Proposed rules on the Medicare Shared Savings Program allowing for expedited antitrust review should be extended to ACOs and other physician-led models of care that do not participate in the Medicare Shared Savings Program. These models should also recognize the dual role ObGyns may play, as both primary and specialty care providers.
- Congress should repeal the Independent Medicare Payment Advisory Board. Leaving Medicare payment decisions in the hands of an unelected, unaccountable body with minimal congressional oversight is bad for all physicians and for our patients.
The outlook for private practice, in any specialty, has dimmed. Says ACOG’s Lucia DiVenere: “Median expenses for private practices have been steadily rising in relationship to revenues—from 52% in 1990 to 71% in 2002—making it difficult for practices to remain solvent.”
Is private practice doomed?
OBG Management: In your opinion, over the long term, is health reform a positive or a negative for ObGyns in private practice?
Ms. DiVenere: On balance, I believe that the health reform law is a positive for our patients, and that fact may lead to an eventual positive for its Fellows, ACOG hopes. What it may mean for ObGyns in private practice, though, is more troubling.
The law has many intended purposes: 1) cover the uninsured, 2) tilt our health-care system toward primary care and use of nonphysician providers, and 3) push practices toward integration with hospitals and health systems and other paths to physician employment. Support for continuation or growth in any type of physician private practice is hard to find in the ACA.
OBG Management: What changes are in the pipeline?
Ms. DiVenere: Under the ACA, by 2013, the Secretary of HHS, with input from stakeholders, will set up a Physician Compare Web site, modeled after the program that exists for hospitals, using data from the Physician Quality Reporting Initiative (PQRI). Data on this site would be made public on January 1, 2013, comparing physicians in terms of quality of care and patient experience.
By law, these data are intended to be statistically valid and risk-adjusted; each physician must have time to review his or her information before it becomes publicly available; data must ensure appropriate attribution of care when multiple providers are involved; and the Secretary of HHS must give physicians timely performance feedback.
Data elements—to the extent that scientifically sound measures exist—will include:
- quality, patient satisfaction, and health outcomes information on Medicare physicians
- physician care coordination and risk-adjusted resource use
- safety, effectiveness, and timeliness of care.
Physicians who successfully interact with this program will likely be those who have a robust EHR system.
If this and other elements of the Affordable Care Act become real, however, we’ll likely see a fundamental shift in the kinds of settings in which ObGyns and other physicians opt to practice.
We want to hear from you! Tell us what you think.
- Medicare and Medicaid are on the brink of insolvency, and you’re not just a bystander
Robert L. Barbieri, MD (Editorial, October 2011) - 14 questions (and answers) about health reform and you
Lucia DiVenere, ACOG Director of Government Affairs, with OBG MANAGEMENT Senior Editor Janelle Yates (June 2010)
In the 18 months since the Patient Protection and Affordable Care Act—otherwise known as ACA, or health-care reform—was signed into law by President Barack Obama, the outlook for private practice, in any specialty, has dimmed. As a recent report on the ramifications of the ACA put it:
The imperative to care for more patients, to provide higher perceived quality, at less cost, with increased reporting and tracking demands, in an environment of high potential liability and problematic reimbursement, will put additional stress on physicians, particularly those in private practice.1
To explore the impact of these stresses on ObGyns specifically, the editors of OBG Management invited Lucia DiVenere, Senior Director of Government Affairs for the American Congress of Obstetricians and Gynecologists (ACOG), to talk about the outlook for private practice in the coming years. In the exchange, Ms. DiVenere discusses the short- and long-term effects of the ACA, the ways in which ObGyn practice is (or is not) evolving, the challenge of making the switch to electronic health records (EHRs), the reasons ACOG opposed the ACA, and other issues related to the current practice environment. In addition, two ObGyns in private practice describe the many challenges they face (see “The view from private practice,” pages 44 and 52).
What nonlegislative forces have affected private practice?
OBG Management: A recent report on the ramifications of the ACA argues that formal reform was inevitable. It also asserts that private practice was subject to many negative pressures long before health-care reform was passed.1 Do you agree?
Ms. DiVenere: Our nation’s health-care system is always evolving, and over the past decade, we’ve seen a clear trend toward system integration—that is, larger, physician-led group practices and hospital employment of physicians.
Looking at physicians as a whole, the percentage who practice solo or in two-physician practices fell from 40.7% in 1996–97 to 32.5% by 2004–05, according to a 2007 survey.2 And the American Medical Association (AMA) reported that the percentage of physicians “with an ownership stake in their practice declined from 61.6% to 54.4% as more physicians opted for employment. Both the trends away from solo and two-physician practices and toward employment were more pronounced for specialists and older physicians.”2
OBG Management: What has the trend been for ObGyns, specifically?
Ms. DiVenere: The ObGyn specialty employs the group practice model—health care delivered by three or more physicians—more frequently than other specialties do, largely because of the support it provides for 24–7 OB call schedules.
As for private practice, ObGyns have been moving away from it for 10 years or longer. Data from a 1991 ACOG survey shows that 77% of respondents were in private practice; by 2003, that percentage had fallen to 70%.3 In 2003, ObGyns in private practice tended to be older (median age: 47 years) than their salaried colleagues (median: 42 years) and were more likely to be male (87% vs 77%).3 The demographic change toward women ObGyns may add to this trend line.
OBG Management: What economic forces have shaped practice paradigms in ObGyn?
Ms. DiVenere: Median expenses for private practices have been steadily rising in relation to revenues—from 52% in 1990 to 71% in 2002—making it difficult for practices to remain solvent.4 In addition, a 2011 survey from Medscape reveals that ObGyns in solo practice earn $15,000 to $25,000 less annually than their employed colleagues.5
ObGyns who have made the switch from private to hospital practice, or who have become ObGyn hospitalists, often point to the difficulties of maintaining a solvent private practice, especially given the push toward electronic health records (EHRs) and increasing regulatory and administrative burdens. These and other issues contribute to rising practice costs and increasing demands on an ObGyn’s time and attention.
“I still love what I do”
I’ve been in solo ObGyn practice for 10 years. Before that, I worked 10 years for two medical groups—that makes 20 years of medical practice. I entered medicine late after teaching school for 10 years.
Most of my patients used to have union jobs and were employed by the steel mills in south Chicago and Northwest Indiana or in construction or manufacturing. One of the benefits of a union job was good insurance. As the economy began to sour, those mills changed hands and are now owned largely by foreign companies. Wages were cut dramatically, and insurance benefits are now “bare bones.” I continue to see my patients regardless of their circumstances.
Most maternity benefits require a hefty out-of-pocket expense. Around here, the doctor gets stuck with the deductible and, consequently, ends up doing lots of free deliveries. I haven’t figured it out yet, but I’m willing to bet that I lose money on OB.
Most patients realize that it’s tough to run a business on a declining revenue stream and are grateful that I take care of them. I’ve treated many of their family members, delivered their babies, provided primary care, done prolapse repairs on mom and grandmom. I know everybody by name—that’s the school teacher in me. I still feel honored to do what I do, but it isn’t easy. The other docs who cover for me on my rare days off, for CME, tell me I have “a nice practice.” That’s why I do it, for the good, salt-of-the-earth folks who would like to pay their bills if times were better.
Medicine is changing quickly. It has taken time to learn the electronic health record (EHR) at the local hospital. Every documentation takes longer. I now spend more time at the computer desk than with patients on hospital rounds. I have read about accountable care organizations and being “enabled,” but the next round of payment cuts will likely kill private practice.
I have Indiana University medical students come and rotate with me, and I try to be as upbeat as I can. The students tell me that my office is the one everyone wants to rotate through. I used to hope that someone might come back and join me here—but maybe the young people have it right. They won’t live with a pager all the time. They won’t do call. To them, medicine will be a job. They will be “providers.”
I may not have practiced in the golden age of medicine, but at least I feel that I had an impact on the lives of the families I have been honored to serve. I still love what I do—it’s just getting harder to justify doing it.
—Mary Vanko, MD
Munster, Ind.
What are the short-term effects of formal reform?
OBG Management: What effect has health-care reform had so far?
Ms. DiVenere: In 2010, twice as many physician practices were bought by hospitals and health systems as in 2009. We can’t conclude that the 2010 law is responsible for these changes, but we know that the architects of health reform were aware of this trend, believed it was beneficial, and looked for ways to encourage it, including through development of accountable care organizations (ACOs), which give hospitals a new and potentially lucrative reason to purchase private practices.
OBG Management: What exactly is an ACO?
Ms. DiVenere: An ACO consists of aligned providers—most likely, large multispecialty groups, often affiliated with the same hospital—who agree to manage patients for a set fee, sharing the risk and potential profit. ACOs are required to have shared governance, which gives them the authority to impose standards for practice, reporting, and compensation—including rewards and penalties—across a group of physicians.
Each ACO must sign a 3-year contract with the US Department of Health and Human Services (HHS) and include a sufficient number of primary care professionals to care for at least 5,000 beneficiaries. ACOs will be evaluated by quality-performance measures to be determined by the Secretary of HHS.
What aspects of ACA will have the biggest impact?
OBG Management: What provision of the new law will have the most direct impact on ObGyns in private practice?
Ms. DiVenere: All ObGyns may benefit from the guarantee of insurance coverage for our patients for maternity and preventive care. And all ObGyns should join ACOG’s fight to repeal the Independent Payment Advisory Board, which may hold enormous power to cut physician reimbursement.
Here’s the quote all ObGyns—especially those in private practice—should read, from an article written by President Obama’s health-reform deputies:
To realize the full benefits of the Affordable Care Act, physicians will need to embrace rather than resist change. The economic forces put in motion by the Act are likely to lead to vertical organization of providers and accelerate physician employment by hospitals and aggregation into larger physician groups. The most successful physicians will be those who most effectively collaborate with other providers to improve outcomes, care productivity, and patient experience.6
OBG Management: Do these health-reform deputies offer any concrete vision of how this change will be achieved?
Ms. DiVenere: They detail what physicians need to do, and how they should change the way they practice, under the Act. For example, to meet the increasing demand for health care, they recommend that practices:
- “Redesign care to include a team of nonphysician providers, such as nurse practitioners, physician assistants, care coordinators, and dieticians”
- “Develop approaches to engage and monitor patients outside of the office.”6
And to meet the requirements for payment reform, information transparency, and quality, they suggest that practices:
- “Focus care around exceptional patient experience and shared clinical outcome goals”
- “Engage in shared decision-making discussions regarding treatment goals and approaches”
- “Proactively manage preventive care”
- “Establish teams to take part in bundled payments and incentive programs”
- “Expand use of electronic health records”
- “Collaborate with hospitals to dramatically reduce readmissions and hospital-acquired infections”
- “Incorporate patient-centered outcomes research to tailor care.”6
To capture value, the authors recommend that practices “redesign medical office processes to capture savings from administrative simplification.”6
OBG Management: Do they propose any method for implementing these changes?
Ms. DiVenere: The White House is hoping that ACOs can lead. Medicare ACOs will attempt to accomplish these changes by managing hospital and physician services, prompting physicians and hospitals to change how they are both clinically organized and paid for services—this change, in particular, is considered by some to be essential to improving the quality and efficiency of health care.
The new Centers for Medicaid and Medicare Services (CMS) Innovation Center, created by the ACA, is given broad authority to test, evaluate, and adopt systems that foster patient-centered care, improve quality, and contain the costs of Medicare, Medicaid, and the Children’s Health Insurance Program (CHIP). The law specifically guides the Innovation Center to look for ways to encourage physicians to transition from fee-for-service to salary-based payment.
We can safely assume that the experts behind these provisions believe that large, hospital-centered systems or large, physician-led groups can better serve the needs of patients and our health-care system in general than can small private practices. Today, 24% of ObGyns run solo practices, and 27% are in single-specialty practices. Health-care reform can mean big changes for them.
Is the EHR a realistic goal for private practices?
OBG Management: How does the push for EHRs affect physicians in private practice?
Ms. DiVenere: Very profoundly. From the point of view of an ObGyn in private practice, EHRs offer a number of benefits. They can:
- help make sense of our increasingly fragmented health-care system
- improve patient safety
- increase efficiency
- reduce paperwork.
In addition, insurers may save by reducing unnecessary tests, and patients can certainly benefit from better coordination and documentation of care. These advantages don’t necessarily translate into savings or revenue for physician practices, however. Many ObGyns—especially those in solo or small practices—don’t feel confident making such a large capital investment. In fact, only about one third of ObGyn practices have an EHR.
OBG Management: Is it primarily cost that deters ObGyns from adopting EHRs?
Ms. DiVenere: That, and the fact that EHR systems are not yet fully interoperable across small practices, insurers, and government agencies. The initial cost of purchasing an EHR system for a small practice is about $50,000 per physician, and there are ongoing costs in staff training and hardware and software updates. A steep learning curve means fewer patients can be seen in an hour. It can take a practice months—even years—for physicians to return to their previous level of productivity. That’s a lot to ask a busy practicing physician to take on.
OBG Management: Is there any way around the push for EHRs?
Ms. DiVenere: Congress wants to move us to full adoption of health information technology (HIT). Under health-care reform, beginning in 2013, all health insurance plans must comply with a uniform standard for electronic transactions, including eligibility verification and health claim status.
In 2014, uniform standards must:
- allow automatic reconciliation of electronic funds transfers and HIPAA payment and remittance
- use standardized and consistent methods of health plan enrollment and editing of claims
- use unique health plan identifiers to simplify and improve routing of health-care transactions
- use standardized claims attachments
- improve practice data collection and evaluation.
Uniformity and standardization can help address one of the major roadblocks to physician adoption of HIT. Still, it’s little wonder that median expenses for private practices have been steadily rising in relation to revenues.
So far, the cons of an EHR outweigh the pros
My private practice made the transition to electronic health records (EHRs) about 4 months ago. We have discovered that EHRs do have a number of positive characteristics:
- Prescriptions are completely legible and can be sent directly to the pharmacy
- The staff no longer needs to search for charts
- If test results have been downloaded, they can be quickly accessed.
However, EHRs also require a lot of time to learn how to use them properly. And the problems don’t end there. For example, instead of looking at a patient’s face when taking a history, we now look at the monitor.
In addition, the templates have many data fields that auto-populate as “normal.” There is an illusion that a thorough history and physical were performed—so it requires a lot of time meticulously reviewing each chart to make sure that it is accurate. One must always be aware of the potential for insurance fraud and the medicolegal risk of documenting something as normal when it isn’t.
Ordering labs is cumbersome because each test must be handled separately, and the terminology does not always match the options at our contracted laboratories. We spend a lot of time searching for each lab that is ordered.
To achieve “meaningful use” of the EHR, certain parameters must be met at every single visit. The medication list must be reviewed (even if the patient takes no medications), and there must be a notation that cervical and breast cancer screening have been ordered, even though recent Pap smear and mammogram results are included in the chart. Regardless of the patient’s age or situation, the issues of contraception, sexually transmitted disease, tobacco use, and domestic violence must be addressed.
So when a 65-year-old woman presents with postmenopausal bleeding, I have to comment on these issues or delete them from the report. I have to provide the same documentation when she returns the next week for a biopsy and the week after that when she returns for the results.
It has become impossible to see patients in the time frame I have used for the past 24 years, and my patients and staff remain frustrated. I am always behind schedule, and I fear that the computer gets more attention than my patients do during the office visit.
—Mark A. Firestone, MD
Aventura, Fla
Is there a physician shortage in ObGyn?
OBG Management: There has been a lot of attention focused on the shortage of physicians in this country. How severe is the shortage likely to be in the specialty of ObGyn?
Ms. DiVenere: William F. Rayburn, MD, MBA, from the University of New Mexico Health Sciences Center and School of Medicine in Albuquerque, has provided ACOG with important work on this topic. Dr. Rayburn is Randolph V. Seligman Professor and Chair in the Department of Obstetrics and Gynecology at that institution. His report7 shows that the gap between the supply of ObGyns and the demand for women’s health care is widening.
Data from this report point to a shortage of 9,000 to 14,000 ObGyns in 20 years. After 2030, the ObGyn shortage may be even more pronounced, as the population of women is projected to increase 36% by 2050, while the number of ObGyns remains constant.
OBG Management: Would the incorporation of more midlevel providers—that is, nurse midwives, physician assistants, and others—ease some of the strains on the ObGyn workforce in general and on private practice specifically?
Ms. DiVenere: That’s very possible and is one of the reasons ACOG encourages greater use of collaborative care. It’s certainly true that the ObGyn specialty is historically comfortable with collaboration.
In 2008, the average ObGyn practice employed 2.6 nonphysician clinicians, certified nurse midwives (CNMs), physician assistants, or nurse practitioners. ACOG strongly supports collaborative practice between ObGyns and qualified midwives (CNMs, certified midwives) and other nonphysician clinicians.
In a recent issue of Obstetrics & Gynecology, ACOG’s Immediate Past President Richard N. Waldman, MD, and Holly Powell Kennedy, CNM, PhD, president of the American College of Nurse Midwives, wrote about the importance of collaborative practice, which can “increase efficiency, improve clinical outcomes, and enhance provider satisfaction.”8 The September 2011 issue of Obstetrics & Gynecology highlights four real-life stories of successful collaborative practice.
Because of our commitment to the benefits of collaborative practice, ACOG supported a provision in the ACA that increases payment for CNMs. Beginning this year, the Medicare program will reimburse CNMs at 100% of the physician payment rate for the same services. Before this law, CNMs were paid at 65% of a physician’s rate for the same services. ACOG’s leadership believes that better reimbursement for CNMs will help ObGyn practices and improve care.
Although ACOG supported that provision in the law, we did not support passage of the ACA. ACOG’s Executive Board carefully considered all elements of the bill and decided that, although it included many provisions that are helpful to our patients, it also included many harmful provisions for our members. We felt that the promise of the women’s health provisions could be realized only if the legislation worked for practicing physicians, too.
Why wasn’t the SGR formula abolished?
OBG Management: What is the effect, for physicians in general and ObGyns in private practice specifically, of the failure to fix the sustainable growth rate (SGR) formula and implement liability reform in the ACA?
Ms. DiVenere: The fact that we have no fix for SGR and no medical liability reform is a disgrace. And the absence of solutions to these two issues is one of the main reasons ACOG decided not to support passage of the ACA. Our Executive Board was very clear in telling Congress that we can’t build a reformed health-care system on broken payment and liability systems—it just won’t work.
ObGyns themselves are the first to point out this fact. ObGyns listed medical liability (65.3%) and financial viability of their practice (44.3%) as their top two professional concerns in a 2008 ACOG survey.9
For all ObGyns, these two continuing problems make practice more difficult every day—problems felt most acutely by ObGyns in private practice. Employed physicians often come under a hospital’s or health system’s liability policy and often don’t pay their own premiums. Employed physicians usually are on salary, sheltering them from the vagaries of Congressional action, or inaction, on looming double-digit Medicare physician payment cuts under the SGR.
Some legislators, and some ObGyns, think Medicare doesn’t apply to us. But today, 92% of ObGyns participate in the Medicare program and 63% accept all Medicare patients. This fact reflects ObGyn training and commitment to serve as lifelong principal-care physicians for women, including women who have disabilities. Fifty-six percent of all Medicare beneficiaries are women. With the Baby Boomer generation transitioning to Medicare, and with shortages in primary care physicians, it is likely that ObGyns will become more involved in delivering health care for this population.
Medicare physician payments matter to ObGyns beyond the Medicare program, too, as Tricare and private payers often follow Medicare payment and coverage policies. As a specialty, we have much at stake in ensuring a stable Medicare system for years to come, starting with an improved physician payment system. [Editor’s note: See Dr. Robert L. Barbieri’s editorial on the subject.]
OBG Management: What is ACOG doing to improve this system?
Ms. DiVenere: ACOG is urging the US Congress to ensure that a better system adheres to the following priniciples:
- Medicare payments should fairly and accurately reflect the cost of care. In the final 2011 Medicare physician fee schedule, CMS is proposing to reduce the physician work value for ObGyn care to women by 11% below what is paid to other physicians for similar men’s services—exactly the opposite of what should be done to encourage good care coordination, and in direct contradiction to recommendations by the Resource-Based Relative Value Scale Update Committee (RUC). Medicare payments to obstetricians are already well below the cost of maternity care; no further cuts should be allowed for this care.
- A new payment system should be as simple, coordinated, and transparent as possible and recognize that there is no one-size-fits-all model. A new Medicare system should coordinate closely with other governmental and nongovernmental programs to ensure that information technology is interoperable, that quality measurement relies on high-quality, risk-adjusted data, and to guard against new and special systems that apply to only one program or may only be workable for one type of specialty or only certain types of diseases and conditions. ObGyns often see relatively few Medicare patients, and unique Medicare requirements can pose significant administrative challenges and inefficiencies to ObGyn participation.
- Congress should encourage, and remove barriers to, ObGyn and physician development of ACOs, medical homes for women, and other innovative models. Proposed rules on the Medicare Shared Savings Program allowing for expedited antitrust review should be extended to ACOs and other physician-led models of care that do not participate in the Medicare Shared Savings Program. These models should also recognize the dual role ObGyns may play, as both primary and specialty care providers.
- Congress should repeal the Independent Medicare Payment Advisory Board. Leaving Medicare payment decisions in the hands of an unelected, unaccountable body with minimal congressional oversight is bad for all physicians and for our patients.
The outlook for private practice, in any specialty, has dimmed. Says ACOG’s Lucia DiVenere: “Median expenses for private practices have been steadily rising in relationship to revenues—from 52% in 1990 to 71% in 2002—making it difficult for practices to remain solvent.”
Is private practice doomed?
OBG Management: In your opinion, over the long term, is health reform a positive or a negative for ObGyns in private practice?
Ms. DiVenere: On balance, I believe that the health reform law is a positive for our patients, and that fact may lead to an eventual positive for its Fellows, ACOG hopes. What it may mean for ObGyns in private practice, though, is more troubling.
The law has many intended purposes: 1) cover the uninsured, 2) tilt our health-care system toward primary care and use of nonphysician providers, and 3) push practices toward integration with hospitals and health systems and other paths to physician employment. Support for continuation or growth in any type of physician private practice is hard to find in the ACA.
OBG Management: What changes are in the pipeline?
Ms. DiVenere: Under the ACA, by 2013, the Secretary of HHS, with input from stakeholders, will set up a Physician Compare Web site, modeled after the program that exists for hospitals, using data from the Physician Quality Reporting Initiative (PQRI). Data on this site would be made public on January 1, 2013, comparing physicians in terms of quality of care and patient experience.
By law, these data are intended to be statistically valid and risk-adjusted; each physician must have time to review his or her information before it becomes publicly available; data must ensure appropriate attribution of care when multiple providers are involved; and the Secretary of HHS must give physicians timely performance feedback.
Data elements—to the extent that scientifically sound measures exist—will include:
- quality, patient satisfaction, and health outcomes information on Medicare physicians
- physician care coordination and risk-adjusted resource use
- safety, effectiveness, and timeliness of care.
Physicians who successfully interact with this program will likely be those who have a robust EHR system.
If this and other elements of the Affordable Care Act become real, however, we’ll likely see a fundamental shift in the kinds of settings in which ObGyns and other physicians opt to practice.
We want to hear from you! Tell us what you think.
1. Physicians Foundation. Health Reform and the Decline of Physician Private Practice. Boston Mass: Physicians Foundation; 2010.
2. American Medical Association. Health Care Trends 2008. http://www.ama-assn.org/resources/doc/clrpd/2008-trends.pdf. Accessed September 1 2011.
3. American College of Obstetricians and Gynecologists. Profile of ObGyn Practice. Washington DC: ACOG; 2003.
4. American College of Obstetricians and Gynecologists. Financial Trends in ObGyn Practice 1990–2002. Washington, DC: ACOG; 2004:6.
5. Medscape Physician Compensation Report: 2011 Results. . Accessed September 1 2011.
6. Kocher R, Emanuel EJ, DeParle NA. The Affordable Care Act and the future of clinical medicine: the opportunities and challenges. Ann Intern Med. 2010;153(8):536-539.
7. American College of Obstetricians and Gynecologists. The Obstetrician-Gynecologist Workforce in the United States: Facts Figures, and Implications 2011. Washington, DC: ACOG; 2011.
8. Waldman RN, Kennedy HP. Collaborative practice between obstetricians and midwives. Obstet Gynecol. 2011;118(3):503-504.
9. American College of Obstetricians and Gynecologists. Socioeconomic Survey of ACOG Fellows. Summary of Results. Washington DC: 2008:1.
1. Physicians Foundation. Health Reform and the Decline of Physician Private Practice. Boston Mass: Physicians Foundation; 2010.
2. American Medical Association. Health Care Trends 2008. http://www.ama-assn.org/resources/doc/clrpd/2008-trends.pdf. Accessed September 1 2011.
3. American College of Obstetricians and Gynecologists. Profile of ObGyn Practice. Washington DC: ACOG; 2003.
4. American College of Obstetricians and Gynecologists. Financial Trends in ObGyn Practice 1990–2002. Washington, DC: ACOG; 2004:6.
5. Medscape Physician Compensation Report: 2011 Results. . Accessed September 1 2011.
6. Kocher R, Emanuel EJ, DeParle NA. The Affordable Care Act and the future of clinical medicine: the opportunities and challenges. Ann Intern Med. 2010;153(8):536-539.
7. American College of Obstetricians and Gynecologists. The Obstetrician-Gynecologist Workforce in the United States: Facts Figures, and Implications 2011. Washington, DC: ACOG; 2011.
8. Waldman RN, Kennedy HP. Collaborative practice between obstetricians and midwives. Obstet Gynecol. 2011;118(3):503-504.
9. American College of Obstetricians and Gynecologists. Socioeconomic Survey of ACOG Fellows. Summary of Results. Washington DC: 2008:1.
Organic Erectile Dysfunction
Erectile dysfunction (ED) is defined as the recurrent inability, of three months’ duration or longer, to attain or maintain an erection sufficient for satisfactory sexual performance.1,2 It is classified as either psychogenic or organic; organic ED will be addressed here. Up to 80% of cases of organic ED can be further categorized into vascular, neurogenic, anatomic, or hormonal subtypes,2,3 with many affected patients vulnerable to potentially serious comorbidities and risk factors.
In the early 1990s, the NIH reported that nearly 30 million US men were affected by ED.2 However, increased public awareness of the condition, beginning with the availability of effective oral medications in the late 1990s, may have led to increased numbers of reported cases—and of men seeking treatment.
COMMON CONDITION, COMMONLY UNTREATED
According to results from the 2001-2002 National Health and Nutrition Examination Survey,4,5 ED affects about 8% of US men: 4% of those in their 50s, 17% of those in their 60s, 47% of men older than 65, and 78% of men 75 or older. More than 27,000 men from Europe and North and South America (ages 20 to 75) were interviewed for the 2004 Men’s Attitudes to Life Events and Sexuality (MALES) study; findings included an overall prevalence for ED of 16%, and 22% among men from the United States.6 See information regarding self-reported ED prevalence in Table 1.7
An important finding from the MALES study was that only 58% of respondents with self-reported ED had sought medical treatment for the condition.6 Some men may be reluctant to broach the subject with a health care provider because they consider ED a purely sexual problem—whereas the etiology is most commonly vascular.8 Thus, it is important for health care providers to include the sexual history as a routine component in the wellness exam for all patients, and to be aware of the potential presentation in patients with organic ED, appropriate work-up, and physical examination.
Additionally, ED can be a wake-up call for men at risk for cardiovascular disease (CVD); ED occurs at a mean time of five years before a cardiovascular event.9 Therefore, in addition to counseling patients with ED regarding treatment and prevention strategies, clinicians should be prepared to identify and address the risk factors and medical comorbidities associated with ED.
The Mechanisms of Erection
When sexual stimulation occurs, parasympathetic activity increases production of the nucleotide cyclic guanosine monophosphate (cGMP), resulting in relaxation of cavernosal smooth muscle and an influx of blood into the penis. This filling action produces expansion of the sinusoidal spaces in the penis, compressing venous channels and thereby preventing outflow of blood to allow maintenance of a rigid erection.10,11
Nitric oxide is needed for vasodilation of the corpora cavernosa. Inhibition of the enzyme phosphodiesterase type 5 (PDE-5) allows increased production and accumulation of penile cGMP. This results in relaxation of the smooth-muscle cells and improved erectile function.1,10,11
PRESENTATION, HISTORY, AND TYPES OF ORGANIC ED
In the primary care setting, the opportunity to assess for and address erectile function generally presents itself at the patient’s annual wellness visit—but could easily go unmentioned if the health care provider does not initiate the discussion. Direct questioning during the review of systems may ease any anxiety for the patient and open up a dialogue regarding other problems or medical conditions. (See box, “How and What To Ask,”12)
Providers can also assess the patient’s risk factors for ED and its associated comorbid conditions (see Table 21,13,14). The patient’s medical history may hold important clues to a diagnosis of organic ED.
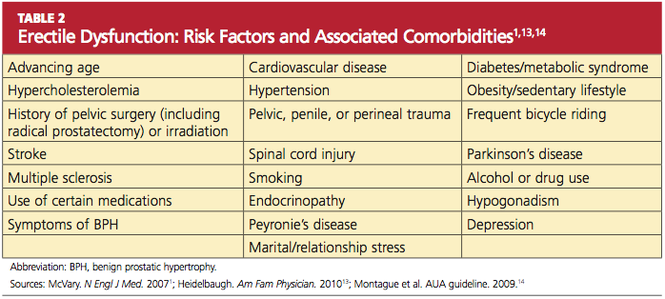
Because certain vasculopathies, including diabetes, are known to accelerate the process of endothelial dysfunction, inquiries should be made into the sexual history of men with these disorders, regardless of age.10 Marked hyperlipidemia, uncontrolled hypertension, atherosclerosis, and other manifestations of CVD can impair erectile as well as endothelial function, leading to vascular ED.
The potential for atherosclerosis in the penile arteries (which are significantly narrower than the carotid, coronary, or femoral arteries) should prompt the primary care provider to evaluate the patient’s risk for vascular disease and its sequelae in the heart as well.8 (Of note, in one study of metabolic syndrome and ED, ED was found to be a predictor for impaired elasticity in the large arteries, independent of cardiovascular risk factors.15) In addition to damaging the small blood vessels and the endothelium, diabetes and other components of the metabolic syndrome contribute to ED by affecting cavernous nerve terminals and smooth muscle.1,14
Lifestyle factors, such as overconsumption of alcohol, smoking, recreational drug use, obesity, and inactivity can contribute to decreased libido and ED.14 Cigarette smoking can cause vasoconstriction and penile venous leak, resulting in poor erectile quality or even loss of erection. While small amounts of alcohol can improve libido and erection due to its anti-anxiety and vasodilatory effects, “more is not better” in this case, as higher doses result in sedation and decreased libido. Chronic alcoholism results in hypogonadism and polyneuropathy.16
Neurogenic ED can result from abnormalities at the level of the brain, the spinal cord, or the pelvis. Previous pelvic surgery, such as prostatectomy or colectomy, can disrupt the cavernous or pudendal nerves. Spinal cord injury, Parkinson’s disease, multiple sclerosis, diabetes with peripheral neuropathy, or a history of trauma can all result in neurogenic ED.16
Additionally, there is evidence of perineal compression from frequent bicycle riding leading to vascular, endothelial, and neurogenic dysfunction in men, with increased incidence of ED.17
Age alone is a risk factor for ED,7,18 as sexual function declines even in healthy men as they age. Penile sensitivity decreases, and there is increased latency between sexual stimulation and resulting erection, as well as between ejaculations. Ejaculations are less forceful, with less ejaculatory volume.16 Other factors that may affect the degree of ED include a history of stroke or pelvic surgery, especially in high-risk cardiac patients.19
Though not the focus of this article, the potential for psychogenic ED should be mentioned, as it warrants screening with a complete psychosocial history to rule out this etiology. The interview should allow time for the patient to discuss current or past relationship difficulties, marital conflict, life stressors, depression, and anxiety. Whatever the etiology of ED, the primary care provider should always elicit a sexual history, including number of sexual partners, current relationship status (ie, committed or casual), and incidence of ED with specific partners.20 The effects of ED on these relationships should be discussed.
A medication history is important for the ED workup. Antidepressants, especially the SSRIs, and certain hypertensives can affect the libido, arousal, and orgasm.21 See Table 31,13,22 for commonly used agents that can affect erectile function and sexual satisfaction.
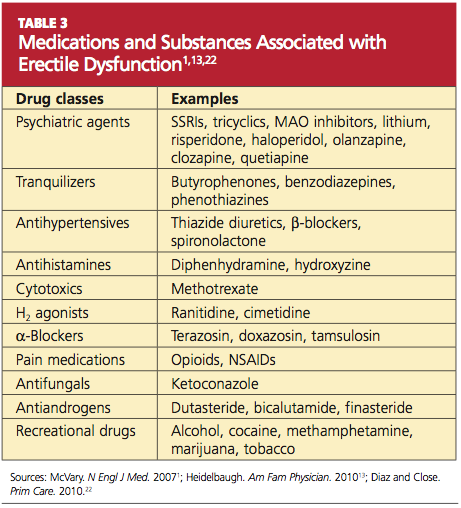
Previous Medical History
The patient should be asked whether he has had any previous testing or treatment for ED. Was any therapy used effective? Did he experience any adverse effects during treatment? If treatment results were unsatisfactory, he should be questioned to ensure that he was using treatment correctly. Commonly, patients do not understand that sexual stimulation is necessary to achieve an erection when they are being treated with an oral PDE-5 inhibitor.23
PHYSICAL EXAMINATION
The clinician who performs the physical examination should take into account the psychogenic, neurogenic, and vasculogenic causes of ED. The genitourinary examination alone is not sufficient; abdominal, vascular, and neurologic exams must be included. The patient should be assessed for body habitus, secondary sex characteristics (including ruling out gynecomastia), and lower extremity pulses.22
The genitourinary exam should include testicular, rectal, and penile examination to rule out physical abnormalities. Small testicles (< 5 mL) or testicular atrophy on palpation should lead the examiner to consider testosterone testing24,25 (see “Diagnostic Testing,” below). A digital rectal examination may reveal an enlarged prostate (this finding, in the presence of lower urinary tract symptoms, has been associated with ED26). Palpation of the penis for plaques and questioning about pain with erection or deviation of the erect penis can lead to a diagnosis of Peyronie’s disease27; if this is the case, the patient can be referred to urology for treatment.
The vascular system should be assessed by listening for abdominal or femoral bruits and the lower extremities checked for appropriate hair growth, skin color, and distal pulses. The examiner should note any scars indicating a history of bypass surgery, which may have been overlooked during the surgical history.
DIAGNOSTIC TESTING
A fasting glucose level and a lipid profile should be routinely obtained because of the links between ED and diabetes and CVD.28,29 A chemistry panel and complete blood count, if one has not been performed recently, can be used to assess the patient’s general health, with a thyroid-stimulating hormone (TSH) level tested in men who report fatigue or weakness.
Men who are older than 50 should be considered for prostate-specific antigen (PSA) testing. Testosterone levels (free and total) are generally reserved for patients with small or atrophic testicles25 or for those who report decreased libido with fatigue or other symptoms of hypogonadism, including depression and decreased muscle strength, muscle mass, or vitality.22,24,25,30 If testosterone levels are ordered, the patient should be counseled to have samples drawn in the early morning to avoid the influence of daily hormonal fluctuations. In patients found to have low testosterone levels (ie, < 300 ng/dL,13 occurring in 12.5% to 35% of men with ED24), prolactin testing may be considered next; in patients with ED, hypoprolactinemia is associated with dyslipidemia, metabolic syndrome, and other risk factors for cardiovascular events.29,31,32
Imaging Studies
A number of imaging studies, once frequently used in evaluation for ED, are considered outdated and are no longer performed. However, Doppler blood flow analysis or ultrasonographic penile blood flow studies can be useful for identifying and evaluating the specific causes of vasculogenic ED, including arterial insufficiency and veno-occlusive disease.33,34 Other conditions associated with ED, such as Peyronie’s disease and priapism, can also be identified through these modalities.33
TREATMENT
The PDE-5 Inhibitors
While historically not the first treatment available for ED, medical therapy is certainly the most popular option for men with ED. The oral PDE-5 inhibitors first became available in the 1990s. All three such agents currently available in the US—sildenafil, tadalafil, and vardenafil—have been demonstrated effective in improving and maintaining erections,35-38 but their pharmacokinetics differ,39 and few head-to-head studies have been conducted that demonstrate greater efficacy of one agent over another.39,40 It has been suggested that patients try all three medications to determine preference and satisfaction.39
Qaseem et al24 report that PDE-5 inhibitors demonstrated clinical benefit regardless of baseline severity of dysfunction or ED cause, including diabetes and depression. These medications require sexual stimulation to be effective and typically are effective about one hour after dosing.23 Tadalafil is remarkable for the length of its half-life, which makes it appropriate at 2.5- or 5.0-mg doses for once-daily dosing41; it is also noted for a relatively low incidence of associated flushing,42 but back pain is more common among tadalafil users than among men using sildenafil or vardenafil.11,39,43
In addition to back pain and myalgia, the most common adverse effects associated with PDE-5 inhibitor use are dyspepsia, flushing, headache, and nasal congestion.42 Because absorption of sildenafil is reduced by food intake, and vardenafil44 to a lesser extent, these agents are best avoided in close proximity to meals. Absorption of tadalafil is not affected by food.45 Higher doses of sildenafil and vardenafil were associated with greater benefit in treating ED.24,46
Note: All three PDE-5 inhibitors are contraindicated in men taking long-acting nitrates or nitroglycerin.24 Their effectiveness may be potentiated by use of erythromycin, ketoconazole, and HIV protease inhibitors.39 Information about dosing options and adverse effects can be found in Table 411.

Testosterone
Hypogonadism, though found in only 5% of men with ED,47 may be an indication for testosterone replacement therapy, and testosterone use has been found to improve libido in men with sexual dysfunction.48 However, the authors of the American College of Physicians clinical practice guidelines24 found insufficient evidence that testosterone replacement improved erections or quality of sexual intercourse. There was also insufficient evidence that testosterone plus sildenafil was more effective than sildenafil alone.24,49,50
Additional Pharmacologic Considerations
A careful review of the medication history for the patient with ED may lead the astute clinician to consider whether a change in current prescriptions might improve the man’s sexual function. Among the commonly used agents listed in Table 3, those with the greatest apparent impact on erections are thiazide diuretics and β-blockers, SSRIs, and tricyclic antidepressants.1,4
For a man who is interested in preserving potency, the clinician should select medications with the least effect on erectile function—for instance, an ACE inhibitor1 rather than a thiazide or β-blocker for the patient with hypertension. In the patient affected by depression, a trial of bupropion, substituted for or added to his current antidepressant, may be helpful.51 Medical therapies for hyperglycemia, hyperlipidemia, and hypertension should be considered if warranted, as should cardiovascular screening with exercise stress testing. (See box, “ED, CAD, and PDE-5 Inhibitors”52)

Alternative and Nonpharmacologic Options
A number of herbal products have been touted to improve sexual function. The FDA53 has issued press releases warning consumers about these and other alternatives to prescription medications. According to the FDA, many “sexual enhancement” products have been found to contain active ingredients resembling those found in one or more PDE-5 inhibitors. These products are in violation of federal law, and consumers are advised to avoid supplements that claim to produce effects similar to those of prescription drugs.53
By contrast, one potentially beneficial supplement currently under study is L-citrulline. As the body converts oral L-citrulline to L-arginine, vasodilation and endothelial function improve, often leading to resolution of mild ED. In a small, single-blind placebo-controlled study, patients who took L-citrulline (1.5 g/d in divided doses) reported improvement in hardness scale grading and increased satisfaction regarding sexual function. No adverse effects were reported, making L-citrulline a possible option for patients who hesitate to use a PDE-5 inhibitor.54
For patients with lack of response or contraindications to accepted medical therapy, other options include alprostadil intraurethral suppositories, penile prosthetic implants, vacuum erection devices, vascular surgery, yohimbine and other herbal therapies, and trazodone.1,14,55-59 These methods may require detailed counseling. Primary care providers who are unfamiliar with these modalities or reluctant to introduce them may wish to refer patients to urology.
PATIENT EDUCATION
Aggressive reduction in cardiovascular risks with smoking cessation, weight loss, and physical activity should be encouraged. In a study of obese men with ED (but without diabetes, hypertension, or hyperlipidemia), Esposito et al60 found that reducing caloric intake and increasing physical activity was associated with improved sexual function in about one-third of the subjects. Bicycle riders can decrease risks of perineal compression with newer seat designs.17
Smoking cessation alone has been shown to improve erectile function at one-year follow-up in at least 25% of men who stopped smoking, compared with none of the men who continued to smoke.61 Patients can help preserve endothelial function with efforts to maintain normal blood pressure and glycemic control.
Researchers are currently looking at occupational exposures as a cause of ED. Exposure to plastics containing bisphenol-A, for example, has been linked to decreased libido.62
CONCLUSION
In addition to addressing erectile dysfunction as a sexual problem, the health care provider should recognize its association with CVD, diabetes, depression, and other serious illnesses. However, it is also important to open a dialogue about sexual function to encourage the patient to discuss the problem of ED. Timely diagnosis and treatment of these conditions may be of even greater benefit to the patient than the effective therapies available for ED itself.
REFERENCES
1. McVary KT. Clinical practice: erectile dysfunction. N Engl J Med. 2007;357(24):2472-2481.
2. NIH Consensus Conference. Impotence: NIH Consensus Development Panel on Impotence. JAMA. 1993;270(1):83-90.
3. Hafez ES, Hafez SD. Erectile dysfunction: anatomical parameters, etiology, diagnosis, and therapy. Arch Androl. 2005;51(1):15-31.
4. Francis ME, Kusek JW, Nyberg LM, Eggers PW. The contribution of common medical conditions and drug exposures to erectile dysfunction in adult males. J Urol. 2007;178(2):591-596.
5. Saigal CS, Wessells H, Pace J, et al. Predictors and prevalence of erectile dysfunction in a racially diverse population. Arch Intern Med. 2006;166 (2):207-212.
6. Rosen RC, Fisher WA, Eardley I, et al. The multinational Men’s Attitudes to Life Events and Sexuality (MALES) study. I. Prevalence of erectile dysfunction and related health concerns in the general population. Curr Med Res Opin. 2004;20 (5):607-617.
7. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8): 762-774.
8. Batty GD, Li Q, Czernichow S, et al. Erectile dysfunction and later cardiovascular disease in men with type 2 diabetes: prospective cohort study based on the ADVANCE (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified-Release Controlled Evaluation) trial. J Am Coll Cardiol. 2010;56(23): 1908-1913.
9. Hodges LD, Kirby M, Solanki J, et al. The temporal relationship between erectile dysfunction and cardiovascular disease. Int J Clin Pract. 2007; 61(12):2019-2025.
10. Miner MM, Kuritzky L. Erectile dysfunction: a sentinel marker for cardiovascular disease in primary care. Cleve Clin J Med. 2007;74 suppl 3: S30-S37.
11. McCullough AR. An update on the PDE-5 inhibitors (PDE-5i). J Androl. 2003;24(6 suppl): S52-S58.
12. Rosen RC, Cappelleri JC, Smith MD, et al. Development and evaluation of an abridged, 5-item version of the International Index of Erectile Function (IIEF-5) as a diagnostic tool for erectile dysfunction. Int J Impot Res. 1999;11(6):
319-326.
13. Heidelbaugh JJ. Management of erectile dysfunction. Am Fam Physician. 2010;81(3):305-312.
14. Montague DK, Jarow JP, Broderick GA, et al; Erectile Dysfunction Guideline Update Panel. AUA guideline on the management of erectile dysfunction: an update (2006). American Urological Association Education and Research, Inc. www.auanet.org/content/guidelines-and-quality- care/clinical-guidelines.cfm?sub=ed. Accessed August 8, 2011.
15. Pohjantahti-Maaroos H, Palomäki A. Comparison of metabolic syndrome subjects with and without erectile dysfunction: levels of circulating oxidised LDL and arterial elasticity. Int J Clin Pract. 2011;65(3):274-280.
16. Lue TF. Male sexual dysfunction. In: Tanagho EA, McAninch JW, eds. Smith’s General Urology. 16th ed. San Francisco, CA: McGraw-Hill; 2004: 592-596.
17. Sommer F, Goldstein I, Korda JB. Bicycle riding and erectile dysfunction: a review. J Sex Med. 2010;7(7):2346-2358.
18. Feldman HA, Johannes CB, Derby CA, et al. Erectile dysfunction and coronary risk factors: prospective results from the Massachusetts male aging study. Prev Med. 2000;30(4):328-338.
19. Böhm M, Baumhäkel M, Probstfield JL, et al. Sexual function, satisfaction, and association of erectile dysfunction with cardiovascular disease and risk factors in cardiovascular high-risk patients: substudy of the ONgoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial/Telmisartan Randomized AssessmeNT Study in ACE-INtolerant Subjects with Cardiovascular Disease (ONTARGET/TRANSCEND). Am Heart J. 2007;154(1):94-101.
20. Athanasiadis L, Papaharitou S, Salpiggidis G, et al. Educating physicians to treat erectile dysfunction patients: development and evaluation of a course on communication and management strategies. J Sex Med. 2006;3(1):47-55.
21. Kennedy SH, Rizvi S. Sexual dysfunction, depression, and the impact of antidepressants.
J Clin Psychopharmacol. 2009;29(2):157-164.
22. Diaz VA Jr, Close JD. Male sexual dysfunction. Prim Care. 2010;37(3):473-489, vii-viii.
23. Carson CC, Lue TF. Phosphodiesterase type 5 inhibitors for erectile dysfunction. BJU Int. 2005; 96(3):257-280.
24. Qaseem A, Snow V, Denberg TD, et al. Hormonal testing and pharmacologic treatment of erectile dysfunction: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2009;151(9):630-649.
25. Dandona P, Rosenberg MT. A practical guide to male hypogonadism in the primary care setting. Int J Clin Pract. 2010;64(6):682-696.
26. Paick SH, Meehan A, Lee M, et al. The relationship among lower urinary tract symptoms, prostate specific antigen and erectile dysfunction in men with benign prostatic hyperplasia: results from the Proscar Long-Term Efficacy and Safety Study. J Urol. 2005;173(3):903-907.
27. Smith JF, Walsh TJ, Lue TF. Peyronie’s disease: a critical appraisal of current diagnosis and treatment. Int J Impot Res. 2008;20(5):445-459.
28. Gazzaruso C, Coppola A, Giustina A. Erectile dysfunction and coronary artery disease in patients with diabetes. Curr Diabetes Rev. 2011; 7(2):143-147.
29. Vlachopoulos C, Ioakeimidis N, Terentes-Printzios D, Stefanidis C. The triad: erectile dysfunction—endothelial dysfunction—cardiovascular disease. Curr Pharm Des. 2008;14(35): 3700-3704.
30. Makhlouf AA, Mohamed MA, Seftel AD, Niedergerger C. Hypogonadism is associated with overt depression symptoms in men with erectile dysfunction. Int J Impot Res. 2008;20(2): 157-161.
31. Corona G, Rastrelli G, Boddi V, et al. Prolactin levels independently predict major cardiovascular events in patients with erectile dysfunction. Int J Androl. 2011;34(3):217-224.
32. Vlachopoulos C, Rokkas K, Ioakeimidis N, Stefanadis C. Inflammation, metabolic syndrome, erectile dysfunction, and coronary artery disease: common links. Eur Urol. 2007;52(6):1590-1600.
33. LeRoy TJ, Broderick GA. Doppler blood flow analysis of erectile function: who, when, and how. Urol Clin North Am. 2011;38(2):147-154.
34. Aversa A, Sarteschi LM. J Sex Med. The role of penile color-duplex ultrasound for the evaluation of erectile dysfunction. J Sex Med. 2007;4(5): 1437-1447.
35. Fink HA, MacDonald R, Rutks IR, et al. Sildenafil for male erectile dysfunction: a systematic review and meta-analysis. Arch Intern Med. 2002; 16(12):1349-1360.
36. Perimenis P, Roumeguere T, Heidler H, et al. Evaluation of patient expectations and treatment satisfaction after 1-year tadalafil therapy for erectile dysfunction: the DETECT study. J Sex Med. 2009;6(1):257-267.
37. Goldstein I, Kim E, Steers WD, et al. Efficacy and safety of tadalafil in men with erectile dysfunction with a high prevalence of comorbid conditions: results from MOMENTUS: multiple observations in men with erectile dysfunction in National Tadalafil Study in the US. J Sex Med. 2007;4(1):166-175.
38. Van Ahlen H, Zumbé J, Stauch K, Hanisch JU. The Real-Life Safety and Efficacy of vardenafil (REALISE) study: results in men from Europe and overseas with erectile dysfunction and cardiovascular or metabolic conditions. J Sex Med. 2010; 7(9):3161-3169.
39. Raheem AA, Kell P. Patient preference and satisfaction in erectile dysfunction therapy: a comparison of the three phosphodiesterase-5 inhibitors sildenafil, vardenafil and tadalafil. Patient Prefer Adherence. 2009;3:99-104.
40. Jannini EA, Isidori AM, Gravina GL, et al. The ENDOTRIAL study: a spontaneous, open-label, randomized, multicenter, crossover study on the efficacy of sildenafil, tadalafil, and vardenafil in the treatment of erectile dysfunction. J Sex Med. 2009;6(9):2547-2560.
41. Washington SL 3rd, Shindel AW. A once-daily dose of tadalafil for erectile dysfunction: compliance and efficacy. Drug Des Devel Ther. 2010 Sep 7;4:159-171.
42. Brock G, Glina S, Moncada I, et al. Likelihood of tadalafil-associated adverse events in integrated multiclinical trial database: classification tree analysis in men with erectile dysfunction. Urology. 2009;73(4):756-761.
43. Montorsi F, Avera A, Mocada I, et al. A randomized, double-blind, placebo-controlled, parallel study to assess the efficacy and safety of once-a-day tadalafil in men with erectile dysfunction who are naïve to PDE5 inhibitors. J Sex Med. 2011 Jun 27 [Epub ahead of print].
44. Rajagopalan P, Mazzu A, Xia C, et al. Effect of high-fat breakfast and moderate-fat evening meal on the pharmacokinetics of vardenafil, an oral phosphodiesterase-5 inhibitor for the treatment of erectile dysfunction. J Clin Pharmacol. 2003;43 (3):260-267.
45. Eardley I, Donatucci C, Corbin J, et al. Pharmacotherapy for erectile dysfunction. J Sex Med. 2010;7(1 pt 2):524-540.
46. Loran OB, Ströberg P, Lee SW, et al. Sildenafil citrate 100 mg starting dose in men with erectile dysfunction in an international, double-blind, placebo-controlled study: effect on the sexual experience and reducing feelings of anxiety about the next intercourse attempt. J Sex Med. 2009; 6(10):2826-2835.
47. Mihkail N. Does testosterone have a role in erectile function? Am J Med. 2006;119(5):373-382.
48. Boloña ER, Uraga MV, Haddad RM, et al. Testosterone use in men with sexual dysfunction: a systematic review and meta-analysis of randomized placebo-controlled trials. Mayo Clin Proc. 2007;82(1):20-28.
49. Shabsigh R, Kaufman JM, Steidle C, Padma-Nathan H. Randomized study of testosterone gel as adjunctive therapy to sildenafil in hypogonadal men with erectile dysfunction who do not respond to sildenafil alone. J Urol. 2004;172(2): 658-663.
50. Aversa A, Isidori AM, Spera G, et al. Androgens improve cavernous vasodilation and response to sildenafil in patients with erectile dysfunction. Clin Endocrinol (Oxf). 2003;58(5):632-638.
51. Safarinejad MR. The effects of the adjunctive bupropion on male sexual dysfunction induced by a selective serotonin reuptake inhibitor: a double-blind placebo-controlled and randomized study. BJU Int. 2010;106(6):840-847.
52. Gazzaruso C, Solerte SB, Pujia A, et al. Erectile dysfunction as a predictor of cardiovascular events and death in diabetic patients with angiographically proven asymptomatic coronary artery disease: a potential protective role for statins and 5-phosphodiesterase inhibitors. J Am Coll Cardiol. 2008;51(21):2040-2044.
53. FDA. Tainted sexual enhancement products. www.fda.gov/Drugs/ResourcesForYou/Consumers/BuyingUsing MedicineSafely/MedicationHealthFraud/ucm234539.htm. Accessed August 8, 2011.
54. Cormio L, De Siati M, Lorusso F, et al. Oral L-citrulline supplementation improves erection hardness in men with mild erectile dysfunction. Urology. 2011;77(1):119-122.
55. Padma-Nathan H, Hellstrom WJ, Kaiser FE, et al. Treatment of men with erectile dysfunction with transurethral alprostadil: Medicated Urethral System for Erection (MUSE) Study Group. N Engl J Med. 1997;33(1)6:1-17.
56. Wolter CE, Hellstrom JG. The hydrophilic-coated penile prosthesis: 1-year experience. J Sex Med. 2004:1(2):221-224.
57. Yuan J, Hoang AN, Romero CA, et al. Vacuum therapy in erectile dysfunction: science and clinical evidence. Int J Impot Res. 2010;22(4):211-219.
58. Lebret T, Hervé JM, Gorny P, et al. Efficacy and safety of a novel combination of L-arginine glutamate and yohimbine hydrochloride: a new oral therapy for erectile dysfunction. Eur Urol. 2002; 41(6):608-613.
59. Fink HA, MacDonald R, Rutks IR, Wilt TJ. Trazodone for erectile dysfunction: a systematic review and meta-analysis. BJU Int. 2003;92(4): 441-446.
60. Esposito K, Giugliano F, Di Palo C, et al. Effect of lifestyle changes on erectile dysfunction in obese men: a randomized controlled trial. JAMA. 2004;291(24):2978-2984.
61. Pourmand G, Alidaee M, Rasuli S, et al. Do cigarette smokers with erectile dysfunction benefit from stopping? A prospective study. BJU Int. 2004;94(9):1310-1313.
62. Li D, Zhou Z, Qing D, et al. Occupational exposure to bisphenol-A (BPA) and the risk of self-reported male sexual dysfunction. Hum Reprod. 2010;25(2):519-527.
Erectile dysfunction (ED) is defined as the recurrent inability, of three months’ duration or longer, to attain or maintain an erection sufficient for satisfactory sexual performance.1,2 It is classified as either psychogenic or organic; organic ED will be addressed here. Up to 80% of cases of organic ED can be further categorized into vascular, neurogenic, anatomic, or hormonal subtypes,2,3 with many affected patients vulnerable to potentially serious comorbidities and risk factors.
In the early 1990s, the NIH reported that nearly 30 million US men were affected by ED.2 However, increased public awareness of the condition, beginning with the availability of effective oral medications in the late 1990s, may have led to increased numbers of reported cases—and of men seeking treatment.
COMMON CONDITION, COMMONLY UNTREATED
According to results from the 2001-2002 National Health and Nutrition Examination Survey,4,5 ED affects about 8% of US men: 4% of those in their 50s, 17% of those in their 60s, 47% of men older than 65, and 78% of men 75 or older. More than 27,000 men from Europe and North and South America (ages 20 to 75) were interviewed for the 2004 Men’s Attitudes to Life Events and Sexuality (MALES) study; findings included an overall prevalence for ED of 16%, and 22% among men from the United States.6 See information regarding self-reported ED prevalence in Table 1.7
An important finding from the MALES study was that only 58% of respondents with self-reported ED had sought medical treatment for the condition.6 Some men may be reluctant to broach the subject with a health care provider because they consider ED a purely sexual problem—whereas the etiology is most commonly vascular.8 Thus, it is important for health care providers to include the sexual history as a routine component in the wellness exam for all patients, and to be aware of the potential presentation in patients with organic ED, appropriate work-up, and physical examination.
Additionally, ED can be a wake-up call for men at risk for cardiovascular disease (CVD); ED occurs at a mean time of five years before a cardiovascular event.9 Therefore, in addition to counseling patients with ED regarding treatment and prevention strategies, clinicians should be prepared to identify and address the risk factors and medical comorbidities associated with ED.
The Mechanisms of Erection
When sexual stimulation occurs, parasympathetic activity increases production of the nucleotide cyclic guanosine monophosphate (cGMP), resulting in relaxation of cavernosal smooth muscle and an influx of blood into the penis. This filling action produces expansion of the sinusoidal spaces in the penis, compressing venous channels and thereby preventing outflow of blood to allow maintenance of a rigid erection.10,11
Nitric oxide is needed for vasodilation of the corpora cavernosa. Inhibition of the enzyme phosphodiesterase type 5 (PDE-5) allows increased production and accumulation of penile cGMP. This results in relaxation of the smooth-muscle cells and improved erectile function.1,10,11
PRESENTATION, HISTORY, AND TYPES OF ORGANIC ED
In the primary care setting, the opportunity to assess for and address erectile function generally presents itself at the patient’s annual wellness visit—but could easily go unmentioned if the health care provider does not initiate the discussion. Direct questioning during the review of systems may ease any anxiety for the patient and open up a dialogue regarding other problems or medical conditions. (See box, “How and What To Ask,”12)
Providers can also assess the patient’s risk factors for ED and its associated comorbid conditions (see Table 21,13,14). The patient’s medical history may hold important clues to a diagnosis of organic ED.
Because certain vasculopathies, including diabetes, are known to accelerate the process of endothelial dysfunction, inquiries should be made into the sexual history of men with these disorders, regardless of age.10 Marked hyperlipidemia, uncontrolled hypertension, atherosclerosis, and other manifestations of CVD can impair erectile as well as endothelial function, leading to vascular ED.
The potential for atherosclerosis in the penile arteries (which are significantly narrower than the carotid, coronary, or femoral arteries) should prompt the primary care provider to evaluate the patient’s risk for vascular disease and its sequelae in the heart as well.8 (Of note, in one study of metabolic syndrome and ED, ED was found to be a predictor for impaired elasticity in the large arteries, independent of cardiovascular risk factors.15) In addition to damaging the small blood vessels and the endothelium, diabetes and other components of the metabolic syndrome contribute to ED by affecting cavernous nerve terminals and smooth muscle.1,14
Lifestyle factors, such as overconsumption of alcohol, smoking, recreational drug use, obesity, and inactivity can contribute to decreased libido and ED.14 Cigarette smoking can cause vasoconstriction and penile venous leak, resulting in poor erectile quality or even loss of erection. While small amounts of alcohol can improve libido and erection due to its anti-anxiety and vasodilatory effects, “more is not better” in this case, as higher doses result in sedation and decreased libido. Chronic alcoholism results in hypogonadism and polyneuropathy.16
Neurogenic ED can result from abnormalities at the level of the brain, the spinal cord, or the pelvis. Previous pelvic surgery, such as prostatectomy or colectomy, can disrupt the cavernous or pudendal nerves. Spinal cord injury, Parkinson’s disease, multiple sclerosis, diabetes with peripheral neuropathy, or a history of trauma can all result in neurogenic ED.16
Additionally, there is evidence of perineal compression from frequent bicycle riding leading to vascular, endothelial, and neurogenic dysfunction in men, with increased incidence of ED.17
Age alone is a risk factor for ED,7,18 as sexual function declines even in healthy men as they age. Penile sensitivity decreases, and there is increased latency between sexual stimulation and resulting erection, as well as between ejaculations. Ejaculations are less forceful, with less ejaculatory volume.16 Other factors that may affect the degree of ED include a history of stroke or pelvic surgery, especially in high-risk cardiac patients.19
Though not the focus of this article, the potential for psychogenic ED should be mentioned, as it warrants screening with a complete psychosocial history to rule out this etiology. The interview should allow time for the patient to discuss current or past relationship difficulties, marital conflict, life stressors, depression, and anxiety. Whatever the etiology of ED, the primary care provider should always elicit a sexual history, including number of sexual partners, current relationship status (ie, committed or casual), and incidence of ED with specific partners.20 The effects of ED on these relationships should be discussed.
A medication history is important for the ED workup. Antidepressants, especially the SSRIs, and certain hypertensives can affect the libido, arousal, and orgasm.21 See Table 31,13,22 for commonly used agents that can affect erectile function and sexual satisfaction.
Previous Medical History
The patient should be asked whether he has had any previous testing or treatment for ED. Was any therapy used effective? Did he experience any adverse effects during treatment? If treatment results were unsatisfactory, he should be questioned to ensure that he was using treatment correctly. Commonly, patients do not understand that sexual stimulation is necessary to achieve an erection when they are being treated with an oral PDE-5 inhibitor.23
PHYSICAL EXAMINATION
The clinician who performs the physical examination should take into account the psychogenic, neurogenic, and vasculogenic causes of ED. The genitourinary examination alone is not sufficient; abdominal, vascular, and neurologic exams must be included. The patient should be assessed for body habitus, secondary sex characteristics (including ruling out gynecomastia), and lower extremity pulses.22
The genitourinary exam should include testicular, rectal, and penile examination to rule out physical abnormalities. Small testicles (< 5 mL) or testicular atrophy on palpation should lead the examiner to consider testosterone testing24,25 (see “Diagnostic Testing,” below). A digital rectal examination may reveal an enlarged prostate (this finding, in the presence of lower urinary tract symptoms, has been associated with ED26). Palpation of the penis for plaques and questioning about pain with erection or deviation of the erect penis can lead to a diagnosis of Peyronie’s disease27; if this is the case, the patient can be referred to urology for treatment.
The vascular system should be assessed by listening for abdominal or femoral bruits and the lower extremities checked for appropriate hair growth, skin color, and distal pulses. The examiner should note any scars indicating a history of bypass surgery, which may have been overlooked during the surgical history.
DIAGNOSTIC TESTING
A fasting glucose level and a lipid profile should be routinely obtained because of the links between ED and diabetes and CVD.28,29 A chemistry panel and complete blood count, if one has not been performed recently, can be used to assess the patient’s general health, with a thyroid-stimulating hormone (TSH) level tested in men who report fatigue or weakness.
Men who are older than 50 should be considered for prostate-specific antigen (PSA) testing. Testosterone levels (free and total) are generally reserved for patients with small or atrophic testicles25 or for those who report decreased libido with fatigue or other symptoms of hypogonadism, including depression and decreased muscle strength, muscle mass, or vitality.22,24,25,30 If testosterone levels are ordered, the patient should be counseled to have samples drawn in the early morning to avoid the influence of daily hormonal fluctuations. In patients found to have low testosterone levels (ie, < 300 ng/dL,13 occurring in 12.5% to 35% of men with ED24), prolactin testing may be considered next; in patients with ED, hypoprolactinemia is associated with dyslipidemia, metabolic syndrome, and other risk factors for cardiovascular events.29,31,32
Imaging Studies
A number of imaging studies, once frequently used in evaluation for ED, are considered outdated and are no longer performed. However, Doppler blood flow analysis or ultrasonographic penile blood flow studies can be useful for identifying and evaluating the specific causes of vasculogenic ED, including arterial insufficiency and veno-occlusive disease.33,34 Other conditions associated with ED, such as Peyronie’s disease and priapism, can also be identified through these modalities.33
TREATMENT
The PDE-5 Inhibitors
While historically not the first treatment available for ED, medical therapy is certainly the most popular option for men with ED. The oral PDE-5 inhibitors first became available in the 1990s. All three such agents currently available in the US—sildenafil, tadalafil, and vardenafil—have been demonstrated effective in improving and maintaining erections,35-38 but their pharmacokinetics differ,39 and few head-to-head studies have been conducted that demonstrate greater efficacy of one agent over another.39,40 It has been suggested that patients try all three medications to determine preference and satisfaction.39
Qaseem et al24 report that PDE-5 inhibitors demonstrated clinical benefit regardless of baseline severity of dysfunction or ED cause, including diabetes and depression. These medications require sexual stimulation to be effective and typically are effective about one hour after dosing.23 Tadalafil is remarkable for the length of its half-life, which makes it appropriate at 2.5- or 5.0-mg doses for once-daily dosing41; it is also noted for a relatively low incidence of associated flushing,42 but back pain is more common among tadalafil users than among men using sildenafil or vardenafil.11,39,43
In addition to back pain and myalgia, the most common adverse effects associated with PDE-5 inhibitor use are dyspepsia, flushing, headache, and nasal congestion.42 Because absorption of sildenafil is reduced by food intake, and vardenafil44 to a lesser extent, these agents are best avoided in close proximity to meals. Absorption of tadalafil is not affected by food.45 Higher doses of sildenafil and vardenafil were associated with greater benefit in treating ED.24,46
Note: All three PDE-5 inhibitors are contraindicated in men taking long-acting nitrates or nitroglycerin.24 Their effectiveness may be potentiated by use of erythromycin, ketoconazole, and HIV protease inhibitors.39 Information about dosing options and adverse effects can be found in Table 411.
Testosterone
Hypogonadism, though found in only 5% of men with ED,47 may be an indication for testosterone replacement therapy, and testosterone use has been found to improve libido in men with sexual dysfunction.48 However, the authors of the American College of Physicians clinical practice guidelines24 found insufficient evidence that testosterone replacement improved erections or quality of sexual intercourse. There was also insufficient evidence that testosterone plus sildenafil was more effective than sildenafil alone.24,49,50
Additional Pharmacologic Considerations
A careful review of the medication history for the patient with ED may lead the astute clinician to consider whether a change in current prescriptions might improve the man’s sexual function. Among the commonly used agents listed in Table 3, those with the greatest apparent impact on erections are thiazide diuretics and β-blockers, SSRIs, and tricyclic antidepressants.1,4
For a man who is interested in preserving potency, the clinician should select medications with the least effect on erectile function—for instance, an ACE inhibitor1 rather than a thiazide or β-blocker for the patient with hypertension. In the patient affected by depression, a trial of bupropion, substituted for or added to his current antidepressant, may be helpful.51 Medical therapies for hyperglycemia, hyperlipidemia, and hypertension should be considered if warranted, as should cardiovascular screening with exercise stress testing. (See box, “ED, CAD, and PDE-5 Inhibitors”52)
Alternative and Nonpharmacologic Options
A number of herbal products have been touted to improve sexual function. The FDA53 has issued press releases warning consumers about these and other alternatives to prescription medications. According to the FDA, many “sexual enhancement” products have been found to contain active ingredients resembling those found in one or more PDE-5 inhibitors. These products are in violation of federal law, and consumers are advised to avoid supplements that claim to produce effects similar to those of prescription drugs.53
By contrast, one potentially beneficial supplement currently under study is L-citrulline. As the body converts oral L-citrulline to L-arginine, vasodilation and endothelial function improve, often leading to resolution of mild ED. In a small, single-blind placebo-controlled study, patients who took L-citrulline (1.5 g/d in divided doses) reported improvement in hardness scale grading and increased satisfaction regarding sexual function. No adverse effects were reported, making L-citrulline a possible option for patients who hesitate to use a PDE-5 inhibitor.54
For patients with lack of response or contraindications to accepted medical therapy, other options include alprostadil intraurethral suppositories, penile prosthetic implants, vacuum erection devices, vascular surgery, yohimbine and other herbal therapies, and trazodone.1,14,55-59 These methods may require detailed counseling. Primary care providers who are unfamiliar with these modalities or reluctant to introduce them may wish to refer patients to urology.
PATIENT EDUCATION
Aggressive reduction in cardiovascular risks with smoking cessation, weight loss, and physical activity should be encouraged. In a study of obese men with ED (but without diabetes, hypertension, or hyperlipidemia), Esposito et al60 found that reducing caloric intake and increasing physical activity was associated with improved sexual function in about one-third of the subjects. Bicycle riders can decrease risks of perineal compression with newer seat designs.17
Smoking cessation alone has been shown to improve erectile function at one-year follow-up in at least 25% of men who stopped smoking, compared with none of the men who continued to smoke.61 Patients can help preserve endothelial function with efforts to maintain normal blood pressure and glycemic control.
Researchers are currently looking at occupational exposures as a cause of ED. Exposure to plastics containing bisphenol-A, for example, has been linked to decreased libido.62
CONCLUSION
In addition to addressing erectile dysfunction as a sexual problem, the health care provider should recognize its association with CVD, diabetes, depression, and other serious illnesses. However, it is also important to open a dialogue about sexual function to encourage the patient to discuss the problem of ED. Timely diagnosis and treatment of these conditions may be of even greater benefit to the patient than the effective therapies available for ED itself.
REFERENCES
1. McVary KT. Clinical practice: erectile dysfunction. N Engl J Med. 2007;357(24):2472-2481.
2. NIH Consensus Conference. Impotence: NIH Consensus Development Panel on Impotence. JAMA. 1993;270(1):83-90.
3. Hafez ES, Hafez SD. Erectile dysfunction: anatomical parameters, etiology, diagnosis, and therapy. Arch Androl. 2005;51(1):15-31.
4. Francis ME, Kusek JW, Nyberg LM, Eggers PW. The contribution of common medical conditions and drug exposures to erectile dysfunction in adult males. J Urol. 2007;178(2):591-596.
5. Saigal CS, Wessells H, Pace J, et al. Predictors and prevalence of erectile dysfunction in a racially diverse population. Arch Intern Med. 2006;166 (2):207-212.
6. Rosen RC, Fisher WA, Eardley I, et al. The multinational Men’s Attitudes to Life Events and Sexuality (MALES) study. I. Prevalence of erectile dysfunction and related health concerns in the general population. Curr Med Res Opin. 2004;20 (5):607-617.
7. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8): 762-774.
8. Batty GD, Li Q, Czernichow S, et al. Erectile dysfunction and later cardiovascular disease in men with type 2 diabetes: prospective cohort study based on the ADVANCE (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified-Release Controlled Evaluation) trial. J Am Coll Cardiol. 2010;56(23): 1908-1913.
9. Hodges LD, Kirby M, Solanki J, et al. The temporal relationship between erectile dysfunction and cardiovascular disease. Int J Clin Pract. 2007; 61(12):2019-2025.
10. Miner MM, Kuritzky L. Erectile dysfunction: a sentinel marker for cardiovascular disease in primary care. Cleve Clin J Med. 2007;74 suppl 3: S30-S37.
11. McCullough AR. An update on the PDE-5 inhibitors (PDE-5i). J Androl. 2003;24(6 suppl): S52-S58.
12. Rosen RC, Cappelleri JC, Smith MD, et al. Development and evaluation of an abridged, 5-item version of the International Index of Erectile Function (IIEF-5) as a diagnostic tool for erectile dysfunction. Int J Impot Res. 1999;11(6):
319-326.
13. Heidelbaugh JJ. Management of erectile dysfunction. Am Fam Physician. 2010;81(3):305-312.
14. Montague DK, Jarow JP, Broderick GA, et al; Erectile Dysfunction Guideline Update Panel. AUA guideline on the management of erectile dysfunction: an update (2006). American Urological Association Education and Research, Inc. www.auanet.org/content/guidelines-and-quality- care/clinical-guidelines.cfm?sub=ed. Accessed August 8, 2011.
15. Pohjantahti-Maaroos H, Palomäki A. Comparison of metabolic syndrome subjects with and without erectile dysfunction: levels of circulating oxidised LDL and arterial elasticity. Int J Clin Pract. 2011;65(3):274-280.
16. Lue TF. Male sexual dysfunction. In: Tanagho EA, McAninch JW, eds. Smith’s General Urology. 16th ed. San Francisco, CA: McGraw-Hill; 2004: 592-596.
17. Sommer F, Goldstein I, Korda JB. Bicycle riding and erectile dysfunction: a review. J Sex Med. 2010;7(7):2346-2358.
18. Feldman HA, Johannes CB, Derby CA, et al. Erectile dysfunction and coronary risk factors: prospective results from the Massachusetts male aging study. Prev Med. 2000;30(4):328-338.
19. Böhm M, Baumhäkel M, Probstfield JL, et al. Sexual function, satisfaction, and association of erectile dysfunction with cardiovascular disease and risk factors in cardiovascular high-risk patients: substudy of the ONgoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial/Telmisartan Randomized AssessmeNT Study in ACE-INtolerant Subjects with Cardiovascular Disease (ONTARGET/TRANSCEND). Am Heart J. 2007;154(1):94-101.
20. Athanasiadis L, Papaharitou S, Salpiggidis G, et al. Educating physicians to treat erectile dysfunction patients: development and evaluation of a course on communication and management strategies. J Sex Med. 2006;3(1):47-55.
21. Kennedy SH, Rizvi S. Sexual dysfunction, depression, and the impact of antidepressants.
J Clin Psychopharmacol. 2009;29(2):157-164.
22. Diaz VA Jr, Close JD. Male sexual dysfunction. Prim Care. 2010;37(3):473-489, vii-viii.
23. Carson CC, Lue TF. Phosphodiesterase type 5 inhibitors for erectile dysfunction. BJU Int. 2005; 96(3):257-280.
24. Qaseem A, Snow V, Denberg TD, et al. Hormonal testing and pharmacologic treatment of erectile dysfunction: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2009;151(9):630-649.
25. Dandona P, Rosenberg MT. A practical guide to male hypogonadism in the primary care setting. Int J Clin Pract. 2010;64(6):682-696.
26. Paick SH, Meehan A, Lee M, et al. The relationship among lower urinary tract symptoms, prostate specific antigen and erectile dysfunction in men with benign prostatic hyperplasia: results from the Proscar Long-Term Efficacy and Safety Study. J Urol. 2005;173(3):903-907.
27. Smith JF, Walsh TJ, Lue TF. Peyronie’s disease: a critical appraisal of current diagnosis and treatment. Int J Impot Res. 2008;20(5):445-459.
28. Gazzaruso C, Coppola A, Giustina A. Erectile dysfunction and coronary artery disease in patients with diabetes. Curr Diabetes Rev. 2011; 7(2):143-147.
29. Vlachopoulos C, Ioakeimidis N, Terentes-Printzios D, Stefanidis C. The triad: erectile dysfunction—endothelial dysfunction—cardiovascular disease. Curr Pharm Des. 2008;14(35): 3700-3704.
30. Makhlouf AA, Mohamed MA, Seftel AD, Niedergerger C. Hypogonadism is associated with overt depression symptoms in men with erectile dysfunction. Int J Impot Res. 2008;20(2): 157-161.
31. Corona G, Rastrelli G, Boddi V, et al. Prolactin levels independently predict major cardiovascular events in patients with erectile dysfunction. Int J Androl. 2011;34(3):217-224.
32. Vlachopoulos C, Rokkas K, Ioakeimidis N, Stefanadis C. Inflammation, metabolic syndrome, erectile dysfunction, and coronary artery disease: common links. Eur Urol. 2007;52(6):1590-1600.
33. LeRoy TJ, Broderick GA. Doppler blood flow analysis of erectile function: who, when, and how. Urol Clin North Am. 2011;38(2):147-154.
34. Aversa A, Sarteschi LM. J Sex Med. The role of penile color-duplex ultrasound for the evaluation of erectile dysfunction. J Sex Med. 2007;4(5): 1437-1447.
35. Fink HA, MacDonald R, Rutks IR, et al. Sildenafil for male erectile dysfunction: a systematic review and meta-analysis. Arch Intern Med. 2002; 16(12):1349-1360.
36. Perimenis P, Roumeguere T, Heidler H, et al. Evaluation of patient expectations and treatment satisfaction after 1-year tadalafil therapy for erectile dysfunction: the DETECT study. J Sex Med. 2009;6(1):257-267.
37. Goldstein I, Kim E, Steers WD, et al. Efficacy and safety of tadalafil in men with erectile dysfunction with a high prevalence of comorbid conditions: results from MOMENTUS: multiple observations in men with erectile dysfunction in National Tadalafil Study in the US. J Sex Med. 2007;4(1):166-175.
38. Van Ahlen H, Zumbé J, Stauch K, Hanisch JU. The Real-Life Safety and Efficacy of vardenafil (REALISE) study: results in men from Europe and overseas with erectile dysfunction and cardiovascular or metabolic conditions. J Sex Med. 2010; 7(9):3161-3169.
39. Raheem AA, Kell P. Patient preference and satisfaction in erectile dysfunction therapy: a comparison of the three phosphodiesterase-5 inhibitors sildenafil, vardenafil and tadalafil. Patient Prefer Adherence. 2009;3:99-104.
40. Jannini EA, Isidori AM, Gravina GL, et al. The ENDOTRIAL study: a spontaneous, open-label, randomized, multicenter, crossover study on the efficacy of sildenafil, tadalafil, and vardenafil in the treatment of erectile dysfunction. J Sex Med. 2009;6(9):2547-2560.
41. Washington SL 3rd, Shindel AW. A once-daily dose of tadalafil for erectile dysfunction: compliance and efficacy. Drug Des Devel Ther. 2010 Sep 7;4:159-171.
42. Brock G, Glina S, Moncada I, et al. Likelihood of tadalafil-associated adverse events in integrated multiclinical trial database: classification tree analysis in men with erectile dysfunction. Urology. 2009;73(4):756-761.
43. Montorsi F, Avera A, Mocada I, et al. A randomized, double-blind, placebo-controlled, parallel study to assess the efficacy and safety of once-a-day tadalafil in men with erectile dysfunction who are naïve to PDE5 inhibitors. J Sex Med. 2011 Jun 27 [Epub ahead of print].
44. Rajagopalan P, Mazzu A, Xia C, et al. Effect of high-fat breakfast and moderate-fat evening meal on the pharmacokinetics of vardenafil, an oral phosphodiesterase-5 inhibitor for the treatment of erectile dysfunction. J Clin Pharmacol. 2003;43 (3):260-267.
45. Eardley I, Donatucci C, Corbin J, et al. Pharmacotherapy for erectile dysfunction. J Sex Med. 2010;7(1 pt 2):524-540.
46. Loran OB, Ströberg P, Lee SW, et al. Sildenafil citrate 100 mg starting dose in men with erectile dysfunction in an international, double-blind, placebo-controlled study: effect on the sexual experience and reducing feelings of anxiety about the next intercourse attempt. J Sex Med. 2009; 6(10):2826-2835.
47. Mihkail N. Does testosterone have a role in erectile function? Am J Med. 2006;119(5):373-382.
48. Boloña ER, Uraga MV, Haddad RM, et al. Testosterone use in men with sexual dysfunction: a systematic review and meta-analysis of randomized placebo-controlled trials. Mayo Clin Proc. 2007;82(1):20-28.
49. Shabsigh R, Kaufman JM, Steidle C, Padma-Nathan H. Randomized study of testosterone gel as adjunctive therapy to sildenafil in hypogonadal men with erectile dysfunction who do not respond to sildenafil alone. J Urol. 2004;172(2): 658-663.
50. Aversa A, Isidori AM, Spera G, et al. Androgens improve cavernous vasodilation and response to sildenafil in patients with erectile dysfunction. Clin Endocrinol (Oxf). 2003;58(5):632-638.
51. Safarinejad MR. The effects of the adjunctive bupropion on male sexual dysfunction induced by a selective serotonin reuptake inhibitor: a double-blind placebo-controlled and randomized study. BJU Int. 2010;106(6):840-847.
52. Gazzaruso C, Solerte SB, Pujia A, et al. Erectile dysfunction as a predictor of cardiovascular events and death in diabetic patients with angiographically proven asymptomatic coronary artery disease: a potential protective role for statins and 5-phosphodiesterase inhibitors. J Am Coll Cardiol. 2008;51(21):2040-2044.
53. FDA. Tainted sexual enhancement products. www.fda.gov/Drugs/ResourcesForYou/Consumers/BuyingUsing MedicineSafely/MedicationHealthFraud/ucm234539.htm. Accessed August 8, 2011.
54. Cormio L, De Siati M, Lorusso F, et al. Oral L-citrulline supplementation improves erection hardness in men with mild erectile dysfunction. Urology. 2011;77(1):119-122.
55. Padma-Nathan H, Hellstrom WJ, Kaiser FE, et al. Treatment of men with erectile dysfunction with transurethral alprostadil: Medicated Urethral System for Erection (MUSE) Study Group. N Engl J Med. 1997;33(1)6:1-17.
56. Wolter CE, Hellstrom JG. The hydrophilic-coated penile prosthesis: 1-year experience. J Sex Med. 2004:1(2):221-224.
57. Yuan J, Hoang AN, Romero CA, et al. Vacuum therapy in erectile dysfunction: science and clinical evidence. Int J Impot Res. 2010;22(4):211-219.
58. Lebret T, Hervé JM, Gorny P, et al. Efficacy and safety of a novel combination of L-arginine glutamate and yohimbine hydrochloride: a new oral therapy for erectile dysfunction. Eur Urol. 2002; 41(6):608-613.
59. Fink HA, MacDonald R, Rutks IR, Wilt TJ. Trazodone for erectile dysfunction: a systematic review and meta-analysis. BJU Int. 2003;92(4): 441-446.
60. Esposito K, Giugliano F, Di Palo C, et al. Effect of lifestyle changes on erectile dysfunction in obese men: a randomized controlled trial. JAMA. 2004;291(24):2978-2984.
61. Pourmand G, Alidaee M, Rasuli S, et al. Do cigarette smokers with erectile dysfunction benefit from stopping? A prospective study. BJU Int. 2004;94(9):1310-1313.
62. Li D, Zhou Z, Qing D, et al. Occupational exposure to bisphenol-A (BPA) and the risk of self-reported male sexual dysfunction. Hum Reprod. 2010;25(2):519-527.
Erectile dysfunction (ED) is defined as the recurrent inability, of three months’ duration or longer, to attain or maintain an erection sufficient for satisfactory sexual performance.1,2 It is classified as either psychogenic or organic; organic ED will be addressed here. Up to 80% of cases of organic ED can be further categorized into vascular, neurogenic, anatomic, or hormonal subtypes,2,3 with many affected patients vulnerable to potentially serious comorbidities and risk factors.
In the early 1990s, the NIH reported that nearly 30 million US men were affected by ED.2 However, increased public awareness of the condition, beginning with the availability of effective oral medications in the late 1990s, may have led to increased numbers of reported cases—and of men seeking treatment.
COMMON CONDITION, COMMONLY UNTREATED
According to results from the 2001-2002 National Health and Nutrition Examination Survey,4,5 ED affects about 8% of US men: 4% of those in their 50s, 17% of those in their 60s, 47% of men older than 65, and 78% of men 75 or older. More than 27,000 men from Europe and North and South America (ages 20 to 75) were interviewed for the 2004 Men’s Attitudes to Life Events and Sexuality (MALES) study; findings included an overall prevalence for ED of 16%, and 22% among men from the United States.6 See information regarding self-reported ED prevalence in Table 1.7
An important finding from the MALES study was that only 58% of respondents with self-reported ED had sought medical treatment for the condition.6 Some men may be reluctant to broach the subject with a health care provider because they consider ED a purely sexual problem—whereas the etiology is most commonly vascular.8 Thus, it is important for health care providers to include the sexual history as a routine component in the wellness exam for all patients, and to be aware of the potential presentation in patients with organic ED, appropriate work-up, and physical examination.
Additionally, ED can be a wake-up call for men at risk for cardiovascular disease (CVD); ED occurs at a mean time of five years before a cardiovascular event.9 Therefore, in addition to counseling patients with ED regarding treatment and prevention strategies, clinicians should be prepared to identify and address the risk factors and medical comorbidities associated with ED.
The Mechanisms of Erection
When sexual stimulation occurs, parasympathetic activity increases production of the nucleotide cyclic guanosine monophosphate (cGMP), resulting in relaxation of cavernosal smooth muscle and an influx of blood into the penis. This filling action produces expansion of the sinusoidal spaces in the penis, compressing venous channels and thereby preventing outflow of blood to allow maintenance of a rigid erection.10,11
Nitric oxide is needed for vasodilation of the corpora cavernosa. Inhibition of the enzyme phosphodiesterase type 5 (PDE-5) allows increased production and accumulation of penile cGMP. This results in relaxation of the smooth-muscle cells and improved erectile function.1,10,11
PRESENTATION, HISTORY, AND TYPES OF ORGANIC ED
In the primary care setting, the opportunity to assess for and address erectile function generally presents itself at the patient’s annual wellness visit—but could easily go unmentioned if the health care provider does not initiate the discussion. Direct questioning during the review of systems may ease any anxiety for the patient and open up a dialogue regarding other problems or medical conditions. (See box, “How and What To Ask,”12)
Providers can also assess the patient’s risk factors for ED and its associated comorbid conditions (see Table 21,13,14). The patient’s medical history may hold important clues to a diagnosis of organic ED.
Because certain vasculopathies, including diabetes, are known to accelerate the process of endothelial dysfunction, inquiries should be made into the sexual history of men with these disorders, regardless of age.10 Marked hyperlipidemia, uncontrolled hypertension, atherosclerosis, and other manifestations of CVD can impair erectile as well as endothelial function, leading to vascular ED.
The potential for atherosclerosis in the penile arteries (which are significantly narrower than the carotid, coronary, or femoral arteries) should prompt the primary care provider to evaluate the patient’s risk for vascular disease and its sequelae in the heart as well.8 (Of note, in one study of metabolic syndrome and ED, ED was found to be a predictor for impaired elasticity in the large arteries, independent of cardiovascular risk factors.15) In addition to damaging the small blood vessels and the endothelium, diabetes and other components of the metabolic syndrome contribute to ED by affecting cavernous nerve terminals and smooth muscle.1,14
Lifestyle factors, such as overconsumption of alcohol, smoking, recreational drug use, obesity, and inactivity can contribute to decreased libido and ED.14 Cigarette smoking can cause vasoconstriction and penile venous leak, resulting in poor erectile quality or even loss of erection. While small amounts of alcohol can improve libido and erection due to its anti-anxiety and vasodilatory effects, “more is not better” in this case, as higher doses result in sedation and decreased libido. Chronic alcoholism results in hypogonadism and polyneuropathy.16
Neurogenic ED can result from abnormalities at the level of the brain, the spinal cord, or the pelvis. Previous pelvic surgery, such as prostatectomy or colectomy, can disrupt the cavernous or pudendal nerves. Spinal cord injury, Parkinson’s disease, multiple sclerosis, diabetes with peripheral neuropathy, or a history of trauma can all result in neurogenic ED.16
Additionally, there is evidence of perineal compression from frequent bicycle riding leading to vascular, endothelial, and neurogenic dysfunction in men, with increased incidence of ED.17
Age alone is a risk factor for ED,7,18 as sexual function declines even in healthy men as they age. Penile sensitivity decreases, and there is increased latency between sexual stimulation and resulting erection, as well as between ejaculations. Ejaculations are less forceful, with less ejaculatory volume.16 Other factors that may affect the degree of ED include a history of stroke or pelvic surgery, especially in high-risk cardiac patients.19
Though not the focus of this article, the potential for psychogenic ED should be mentioned, as it warrants screening with a complete psychosocial history to rule out this etiology. The interview should allow time for the patient to discuss current or past relationship difficulties, marital conflict, life stressors, depression, and anxiety. Whatever the etiology of ED, the primary care provider should always elicit a sexual history, including number of sexual partners, current relationship status (ie, committed or casual), and incidence of ED with specific partners.20 The effects of ED on these relationships should be discussed.
A medication history is important for the ED workup. Antidepressants, especially the SSRIs, and certain hypertensives can affect the libido, arousal, and orgasm.21 See Table 31,13,22 for commonly used agents that can affect erectile function and sexual satisfaction.
Previous Medical History
The patient should be asked whether he has had any previous testing or treatment for ED. Was any therapy used effective? Did he experience any adverse effects during treatment? If treatment results were unsatisfactory, he should be questioned to ensure that he was using treatment correctly. Commonly, patients do not understand that sexual stimulation is necessary to achieve an erection when they are being treated with an oral PDE-5 inhibitor.23
PHYSICAL EXAMINATION
The clinician who performs the physical examination should take into account the psychogenic, neurogenic, and vasculogenic causes of ED. The genitourinary examination alone is not sufficient; abdominal, vascular, and neurologic exams must be included. The patient should be assessed for body habitus, secondary sex characteristics (including ruling out gynecomastia), and lower extremity pulses.22
The genitourinary exam should include testicular, rectal, and penile examination to rule out physical abnormalities. Small testicles (< 5 mL) or testicular atrophy on palpation should lead the examiner to consider testosterone testing24,25 (see “Diagnostic Testing,” below). A digital rectal examination may reveal an enlarged prostate (this finding, in the presence of lower urinary tract symptoms, has been associated with ED26). Palpation of the penis for plaques and questioning about pain with erection or deviation of the erect penis can lead to a diagnosis of Peyronie’s disease27; if this is the case, the patient can be referred to urology for treatment.
The vascular system should be assessed by listening for abdominal or femoral bruits and the lower extremities checked for appropriate hair growth, skin color, and distal pulses. The examiner should note any scars indicating a history of bypass surgery, which may have been overlooked during the surgical history.
DIAGNOSTIC TESTING
A fasting glucose level and a lipid profile should be routinely obtained because of the links between ED and diabetes and CVD.28,29 A chemistry panel and complete blood count, if one has not been performed recently, can be used to assess the patient’s general health, with a thyroid-stimulating hormone (TSH) level tested in men who report fatigue or weakness.
Men who are older than 50 should be considered for prostate-specific antigen (PSA) testing. Testosterone levels (free and total) are generally reserved for patients with small or atrophic testicles25 or for those who report decreased libido with fatigue or other symptoms of hypogonadism, including depression and decreased muscle strength, muscle mass, or vitality.22,24,25,30 If testosterone levels are ordered, the patient should be counseled to have samples drawn in the early morning to avoid the influence of daily hormonal fluctuations. In patients found to have low testosterone levels (ie, < 300 ng/dL,13 occurring in 12.5% to 35% of men with ED24), prolactin testing may be considered next; in patients with ED, hypoprolactinemia is associated with dyslipidemia, metabolic syndrome, and other risk factors for cardiovascular events.29,31,32
Imaging Studies
A number of imaging studies, once frequently used in evaluation for ED, are considered outdated and are no longer performed. However, Doppler blood flow analysis or ultrasonographic penile blood flow studies can be useful for identifying and evaluating the specific causes of vasculogenic ED, including arterial insufficiency and veno-occlusive disease.33,34 Other conditions associated with ED, such as Peyronie’s disease and priapism, can also be identified through these modalities.33
TREATMENT
The PDE-5 Inhibitors
While historically not the first treatment available for ED, medical therapy is certainly the most popular option for men with ED. The oral PDE-5 inhibitors first became available in the 1990s. All three such agents currently available in the US—sildenafil, tadalafil, and vardenafil—have been demonstrated effective in improving and maintaining erections,35-38 but their pharmacokinetics differ,39 and few head-to-head studies have been conducted that demonstrate greater efficacy of one agent over another.39,40 It has been suggested that patients try all three medications to determine preference and satisfaction.39
Qaseem et al24 report that PDE-5 inhibitors demonstrated clinical benefit regardless of baseline severity of dysfunction or ED cause, including diabetes and depression. These medications require sexual stimulation to be effective and typically are effective about one hour after dosing.23 Tadalafil is remarkable for the length of its half-life, which makes it appropriate at 2.5- or 5.0-mg doses for once-daily dosing41; it is also noted for a relatively low incidence of associated flushing,42 but back pain is more common among tadalafil users than among men using sildenafil or vardenafil.11,39,43
In addition to back pain and myalgia, the most common adverse effects associated with PDE-5 inhibitor use are dyspepsia, flushing, headache, and nasal congestion.42 Because absorption of sildenafil is reduced by food intake, and vardenafil44 to a lesser extent, these agents are best avoided in close proximity to meals. Absorption of tadalafil is not affected by food.45 Higher doses of sildenafil and vardenafil were associated with greater benefit in treating ED.24,46
Note: All three PDE-5 inhibitors are contraindicated in men taking long-acting nitrates or nitroglycerin.24 Their effectiveness may be potentiated by use of erythromycin, ketoconazole, and HIV protease inhibitors.39 Information about dosing options and adverse effects can be found in Table 411.
Testosterone
Hypogonadism, though found in only 5% of men with ED,47 may be an indication for testosterone replacement therapy, and testosterone use has been found to improve libido in men with sexual dysfunction.48 However, the authors of the American College of Physicians clinical practice guidelines24 found insufficient evidence that testosterone replacement improved erections or quality of sexual intercourse. There was also insufficient evidence that testosterone plus sildenafil was more effective than sildenafil alone.24,49,50
Additional Pharmacologic Considerations
A careful review of the medication history for the patient with ED may lead the astute clinician to consider whether a change in current prescriptions might improve the man’s sexual function. Among the commonly used agents listed in Table 3, those with the greatest apparent impact on erections are thiazide diuretics and β-blockers, SSRIs, and tricyclic antidepressants.1,4
For a man who is interested in preserving potency, the clinician should select medications with the least effect on erectile function—for instance, an ACE inhibitor1 rather than a thiazide or β-blocker for the patient with hypertension. In the patient affected by depression, a trial of bupropion, substituted for or added to his current antidepressant, may be helpful.51 Medical therapies for hyperglycemia, hyperlipidemia, and hypertension should be considered if warranted, as should cardiovascular screening with exercise stress testing. (See box, “ED, CAD, and PDE-5 Inhibitors”52)
Alternative and Nonpharmacologic Options
A number of herbal products have been touted to improve sexual function. The FDA53 has issued press releases warning consumers about these and other alternatives to prescription medications. According to the FDA, many “sexual enhancement” products have been found to contain active ingredients resembling those found in one or more PDE-5 inhibitors. These products are in violation of federal law, and consumers are advised to avoid supplements that claim to produce effects similar to those of prescription drugs.53
By contrast, one potentially beneficial supplement currently under study is L-citrulline. As the body converts oral L-citrulline to L-arginine, vasodilation and endothelial function improve, often leading to resolution of mild ED. In a small, single-blind placebo-controlled study, patients who took L-citrulline (1.5 g/d in divided doses) reported improvement in hardness scale grading and increased satisfaction regarding sexual function. No adverse effects were reported, making L-citrulline a possible option for patients who hesitate to use a PDE-5 inhibitor.54
For patients with lack of response or contraindications to accepted medical therapy, other options include alprostadil intraurethral suppositories, penile prosthetic implants, vacuum erection devices, vascular surgery, yohimbine and other herbal therapies, and trazodone.1,14,55-59 These methods may require detailed counseling. Primary care providers who are unfamiliar with these modalities or reluctant to introduce them may wish to refer patients to urology.
PATIENT EDUCATION
Aggressive reduction in cardiovascular risks with smoking cessation, weight loss, and physical activity should be encouraged. In a study of obese men with ED (but without diabetes, hypertension, or hyperlipidemia), Esposito et al60 found that reducing caloric intake and increasing physical activity was associated with improved sexual function in about one-third of the subjects. Bicycle riders can decrease risks of perineal compression with newer seat designs.17
Smoking cessation alone has been shown to improve erectile function at one-year follow-up in at least 25% of men who stopped smoking, compared with none of the men who continued to smoke.61 Patients can help preserve endothelial function with efforts to maintain normal blood pressure and glycemic control.
Researchers are currently looking at occupational exposures as a cause of ED. Exposure to plastics containing bisphenol-A, for example, has been linked to decreased libido.62
CONCLUSION
In addition to addressing erectile dysfunction as a sexual problem, the health care provider should recognize its association with CVD, diabetes, depression, and other serious illnesses. However, it is also important to open a dialogue about sexual function to encourage the patient to discuss the problem of ED. Timely diagnosis and treatment of these conditions may be of even greater benefit to the patient than the effective therapies available for ED itself.
REFERENCES
1. McVary KT. Clinical practice: erectile dysfunction. N Engl J Med. 2007;357(24):2472-2481.
2. NIH Consensus Conference. Impotence: NIH Consensus Development Panel on Impotence. JAMA. 1993;270(1):83-90.
3. Hafez ES, Hafez SD. Erectile dysfunction: anatomical parameters, etiology, diagnosis, and therapy. Arch Androl. 2005;51(1):15-31.
4. Francis ME, Kusek JW, Nyberg LM, Eggers PW. The contribution of common medical conditions and drug exposures to erectile dysfunction in adult males. J Urol. 2007;178(2):591-596.
5. Saigal CS, Wessells H, Pace J, et al. Predictors and prevalence of erectile dysfunction in a racially diverse population. Arch Intern Med. 2006;166 (2):207-212.
6. Rosen RC, Fisher WA, Eardley I, et al. The multinational Men’s Attitudes to Life Events and Sexuality (MALES) study. I. Prevalence of erectile dysfunction and related health concerns in the general population. Curr Med Res Opin. 2004;20 (5):607-617.
7. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8): 762-774.
8. Batty GD, Li Q, Czernichow S, et al. Erectile dysfunction and later cardiovascular disease in men with type 2 diabetes: prospective cohort study based on the ADVANCE (Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified-Release Controlled Evaluation) trial. J Am Coll Cardiol. 2010;56(23): 1908-1913.
9. Hodges LD, Kirby M, Solanki J, et al. The temporal relationship between erectile dysfunction and cardiovascular disease. Int J Clin Pract. 2007; 61(12):2019-2025.
10. Miner MM, Kuritzky L. Erectile dysfunction: a sentinel marker for cardiovascular disease in primary care. Cleve Clin J Med. 2007;74 suppl 3: S30-S37.
11. McCullough AR. An update on the PDE-5 inhibitors (PDE-5i). J Androl. 2003;24(6 suppl): S52-S58.
12. Rosen RC, Cappelleri JC, Smith MD, et al. Development and evaluation of an abridged, 5-item version of the International Index of Erectile Function (IIEF-5) as a diagnostic tool for erectile dysfunction. Int J Impot Res. 1999;11(6):
319-326.
13. Heidelbaugh JJ. Management of erectile dysfunction. Am Fam Physician. 2010;81(3):305-312.
14. Montague DK, Jarow JP, Broderick GA, et al; Erectile Dysfunction Guideline Update Panel. AUA guideline on the management of erectile dysfunction: an update (2006). American Urological Association Education and Research, Inc. www.auanet.org/content/guidelines-and-quality- care/clinical-guidelines.cfm?sub=ed. Accessed August 8, 2011.
15. Pohjantahti-Maaroos H, Palomäki A. Comparison of metabolic syndrome subjects with and without erectile dysfunction: levels of circulating oxidised LDL and arterial elasticity. Int J Clin Pract. 2011;65(3):274-280.
16. Lue TF. Male sexual dysfunction. In: Tanagho EA, McAninch JW, eds. Smith’s General Urology. 16th ed. San Francisco, CA: McGraw-Hill; 2004: 592-596.
17. Sommer F, Goldstein I, Korda JB. Bicycle riding and erectile dysfunction: a review. J Sex Med. 2010;7(7):2346-2358.
18. Feldman HA, Johannes CB, Derby CA, et al. Erectile dysfunction and coronary risk factors: prospective results from the Massachusetts male aging study. Prev Med. 2000;30(4):328-338.
19. Böhm M, Baumhäkel M, Probstfield JL, et al. Sexual function, satisfaction, and association of erectile dysfunction with cardiovascular disease and risk factors in cardiovascular high-risk patients: substudy of the ONgoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial/Telmisartan Randomized AssessmeNT Study in ACE-INtolerant Subjects with Cardiovascular Disease (ONTARGET/TRANSCEND). Am Heart J. 2007;154(1):94-101.
20. Athanasiadis L, Papaharitou S, Salpiggidis G, et al. Educating physicians to treat erectile dysfunction patients: development and evaluation of a course on communication and management strategies. J Sex Med. 2006;3(1):47-55.
21. Kennedy SH, Rizvi S. Sexual dysfunction, depression, and the impact of antidepressants.
J Clin Psychopharmacol. 2009;29(2):157-164.
22. Diaz VA Jr, Close JD. Male sexual dysfunction. Prim Care. 2010;37(3):473-489, vii-viii.
23. Carson CC, Lue TF. Phosphodiesterase type 5 inhibitors for erectile dysfunction. BJU Int. 2005; 96(3):257-280.
24. Qaseem A, Snow V, Denberg TD, et al. Hormonal testing and pharmacologic treatment of erectile dysfunction: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2009;151(9):630-649.
25. Dandona P, Rosenberg MT. A practical guide to male hypogonadism in the primary care setting. Int J Clin Pract. 2010;64(6):682-696.
26. Paick SH, Meehan A, Lee M, et al. The relationship among lower urinary tract symptoms, prostate specific antigen and erectile dysfunction in men with benign prostatic hyperplasia: results from the Proscar Long-Term Efficacy and Safety Study. J Urol. 2005;173(3):903-907.
27. Smith JF, Walsh TJ, Lue TF. Peyronie’s disease: a critical appraisal of current diagnosis and treatment. Int J Impot Res. 2008;20(5):445-459.
28. Gazzaruso C, Coppola A, Giustina A. Erectile dysfunction and coronary artery disease in patients with diabetes. Curr Diabetes Rev. 2011; 7(2):143-147.
29. Vlachopoulos C, Ioakeimidis N, Terentes-Printzios D, Stefanidis C. The triad: erectile dysfunction—endothelial dysfunction—cardiovascular disease. Curr Pharm Des. 2008;14(35): 3700-3704.
30. Makhlouf AA, Mohamed MA, Seftel AD, Niedergerger C. Hypogonadism is associated with overt depression symptoms in men with erectile dysfunction. Int J Impot Res. 2008;20(2): 157-161.
31. Corona G, Rastrelli G, Boddi V, et al. Prolactin levels independently predict major cardiovascular events in patients with erectile dysfunction. Int J Androl. 2011;34(3):217-224.
32. Vlachopoulos C, Rokkas K, Ioakeimidis N, Stefanadis C. Inflammation, metabolic syndrome, erectile dysfunction, and coronary artery disease: common links. Eur Urol. 2007;52(6):1590-1600.
33. LeRoy TJ, Broderick GA. Doppler blood flow analysis of erectile function: who, when, and how. Urol Clin North Am. 2011;38(2):147-154.
34. Aversa A, Sarteschi LM. J Sex Med. The role of penile color-duplex ultrasound for the evaluation of erectile dysfunction. J Sex Med. 2007;4(5): 1437-1447.
35. Fink HA, MacDonald R, Rutks IR, et al. Sildenafil for male erectile dysfunction: a systematic review and meta-analysis. Arch Intern Med. 2002; 16(12):1349-1360.
36. Perimenis P, Roumeguere T, Heidler H, et al. Evaluation of patient expectations and treatment satisfaction after 1-year tadalafil therapy for erectile dysfunction: the DETECT study. J Sex Med. 2009;6(1):257-267.
37. Goldstein I, Kim E, Steers WD, et al. Efficacy and safety of tadalafil in men with erectile dysfunction with a high prevalence of comorbid conditions: results from MOMENTUS: multiple observations in men with erectile dysfunction in National Tadalafil Study in the US. J Sex Med. 2007;4(1):166-175.
38. Van Ahlen H, Zumbé J, Stauch K, Hanisch JU. The Real-Life Safety and Efficacy of vardenafil (REALISE) study: results in men from Europe and overseas with erectile dysfunction and cardiovascular or metabolic conditions. J Sex Med. 2010; 7(9):3161-3169.
39. Raheem AA, Kell P. Patient preference and satisfaction in erectile dysfunction therapy: a comparison of the three phosphodiesterase-5 inhibitors sildenafil, vardenafil and tadalafil. Patient Prefer Adherence. 2009;3:99-104.
40. Jannini EA, Isidori AM, Gravina GL, et al. The ENDOTRIAL study: a spontaneous, open-label, randomized, multicenter, crossover study on the efficacy of sildenafil, tadalafil, and vardenafil in the treatment of erectile dysfunction. J Sex Med. 2009;6(9):2547-2560.
41. Washington SL 3rd, Shindel AW. A once-daily dose of tadalafil for erectile dysfunction: compliance and efficacy. Drug Des Devel Ther. 2010 Sep 7;4:159-171.
42. Brock G, Glina S, Moncada I, et al. Likelihood of tadalafil-associated adverse events in integrated multiclinical trial database: classification tree analysis in men with erectile dysfunction. Urology. 2009;73(4):756-761.
43. Montorsi F, Avera A, Mocada I, et al. A randomized, double-blind, placebo-controlled, parallel study to assess the efficacy and safety of once-a-day tadalafil in men with erectile dysfunction who are naïve to PDE5 inhibitors. J Sex Med. 2011 Jun 27 [Epub ahead of print].
44. Rajagopalan P, Mazzu A, Xia C, et al. Effect of high-fat breakfast and moderate-fat evening meal on the pharmacokinetics of vardenafil, an oral phosphodiesterase-5 inhibitor for the treatment of erectile dysfunction. J Clin Pharmacol. 2003;43 (3):260-267.
45. Eardley I, Donatucci C, Corbin J, et al. Pharmacotherapy for erectile dysfunction. J Sex Med. 2010;7(1 pt 2):524-540.
46. Loran OB, Ströberg P, Lee SW, et al. Sildenafil citrate 100 mg starting dose in men with erectile dysfunction in an international, double-blind, placebo-controlled study: effect on the sexual experience and reducing feelings of anxiety about the next intercourse attempt. J Sex Med. 2009; 6(10):2826-2835.
47. Mihkail N. Does testosterone have a role in erectile function? Am J Med. 2006;119(5):373-382.
48. Boloña ER, Uraga MV, Haddad RM, et al. Testosterone use in men with sexual dysfunction: a systematic review and meta-analysis of randomized placebo-controlled trials. Mayo Clin Proc. 2007;82(1):20-28.
49. Shabsigh R, Kaufman JM, Steidle C, Padma-Nathan H. Randomized study of testosterone gel as adjunctive therapy to sildenafil in hypogonadal men with erectile dysfunction who do not respond to sildenafil alone. J Urol. 2004;172(2): 658-663.
50. Aversa A, Isidori AM, Spera G, et al. Androgens improve cavernous vasodilation and response to sildenafil in patients with erectile dysfunction. Clin Endocrinol (Oxf). 2003;58(5):632-638.
51. Safarinejad MR. The effects of the adjunctive bupropion on male sexual dysfunction induced by a selective serotonin reuptake inhibitor: a double-blind placebo-controlled and randomized study. BJU Int. 2010;106(6):840-847.
52. Gazzaruso C, Solerte SB, Pujia A, et al. Erectile dysfunction as a predictor of cardiovascular events and death in diabetic patients with angiographically proven asymptomatic coronary artery disease: a potential protective role for statins and 5-phosphodiesterase inhibitors. J Am Coll Cardiol. 2008;51(21):2040-2044.
53. FDA. Tainted sexual enhancement products. www.fda.gov/Drugs/ResourcesForYou/Consumers/BuyingUsing MedicineSafely/MedicationHealthFraud/ucm234539.htm. Accessed August 8, 2011.
54. Cormio L, De Siati M, Lorusso F, et al. Oral L-citrulline supplementation improves erection hardness in men with mild erectile dysfunction. Urology. 2011;77(1):119-122.
55. Padma-Nathan H, Hellstrom WJ, Kaiser FE, et al. Treatment of men with erectile dysfunction with transurethral alprostadil: Medicated Urethral System for Erection (MUSE) Study Group. N Engl J Med. 1997;33(1)6:1-17.
56. Wolter CE, Hellstrom JG. The hydrophilic-coated penile prosthesis: 1-year experience. J Sex Med. 2004:1(2):221-224.
57. Yuan J, Hoang AN, Romero CA, et al. Vacuum therapy in erectile dysfunction: science and clinical evidence. Int J Impot Res. 2010;22(4):211-219.
58. Lebret T, Hervé JM, Gorny P, et al. Efficacy and safety of a novel combination of L-arginine glutamate and yohimbine hydrochloride: a new oral therapy for erectile dysfunction. Eur Urol. 2002; 41(6):608-613.
59. Fink HA, MacDonald R, Rutks IR, Wilt TJ. Trazodone for erectile dysfunction: a systematic review and meta-analysis. BJU Int. 2003;92(4): 441-446.
60. Esposito K, Giugliano F, Di Palo C, et al. Effect of lifestyle changes on erectile dysfunction in obese men: a randomized controlled trial. JAMA. 2004;291(24):2978-2984.
61. Pourmand G, Alidaee M, Rasuli S, et al. Do cigarette smokers with erectile dysfunction benefit from stopping? A prospective study. BJU Int. 2004;94(9):1310-1313.
62. Li D, Zhou Z, Qing D, et al. Occupational exposure to bisphenol-A (BPA) and the risk of self-reported male sexual dysfunction. Hum Reprod. 2010;25(2):519-527.