User login
Comparing Depression Screening Tests in Older veterans
Nocardia Bacteremia Associated With Pulmonary Alveolar Proteinosis
Grand Rounds: Woman, 22, With Dizziness and Headache
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
Measuring Blood Ethanol: Can It Lead You Astray? (Maybe)
Meningitis: Current Evidence and Best Practice
Complications Associated With Use of Anterior Cruciate Ligament Fixation Devices
5 Points on Anterior Cruciate Ligament Injuries in Young Athletes
Pregnancy and epilepsy— managing both, in one patient
When a woman who has epilepsy is pregnant or planning for pregnancy, you face the challenge of balancing the benefits and teratogenic risks of her antiseizure medication. Here is help.
About 500,000 women of childbearing age in the United States suffer from epilepsy.1 For these patients and their physicians, family planning and pregnancy are complex and fraught with risk.
The dilemma
Infants born to women who have epilepsy have a twofold to threefold increased risk of congenital malformations, compared with infants born to women who do not have the disorder. The increased risk is mainly related to exposure to antiepileptic drugs (AEDs).2 Recent studies also suggest that children exposed to AEDs such as valproate, phenobarbital, and phenytoin in utero may have neurocognitive deficits, even when there are no major congenital malformations.1,3,4
Yet, discontinuing the drugs prior to conception or in early pregnancy is rarely a viable option. In one recent prospective study, convulsive seizures during the first trimester (the type and timing of seizure thought to have the most harmful effect on the developing fetus) were associated with malformations in 7.4% of pregnancies.2 Seizures also increase the risk of both fetal and maternal death, although the extent of that risk is unknown.5
- If feasible, women with epilepsy who are of childbearing age and taking phenobarbital, valproate, or topiramate should be switched to a safer antiepileptic drug (AED), such as lamotrigine, prior to pregnancy.
- Avoid topiramate in women with epilepsy of childbearing age. New human data show an increased risk of oral clefts, and the FDA recently placed topiramate in Pregnancy Category D.
- Avoid switching a pregnant patient to an AED that she has not taken before.
- Use the dosage of AED at which the patient is seizure-free prior to conception as a target level to adjust dosing during pregnancy.
- Start all women who have epilepsy and are of childbearing age on ≥0.4 mg folic acid daily prior to conception.
Ideally, pregnant women with epilepsy should be under the care of both an obstetrician experienced in high-risk pregnancy and a neurologist or epileptologist. In reality, those who live in areas with limited access to such specialized care or who have limited health coverage may be cared for throughout pregnancy by a generalist ObGyn. This evidence-based review was developed with that reality in mind.
Switching (or stopping) AEDs before conception
Changes in AEDs are rarely made after conception. Any switches that a patient may desire—from a potentially unsafe drug to a “safer” AED, for example—should be considered at least a year before she plans to conceive so that good seizure control can be achieved by then.
Begin by checking the serum drug level of the patient’s effective, yet potentially unsafe, antiseizure drug. That will allow you to determine the baseline therapeutic drug level and dosage at which the patient is seizure-free. Then add the second, safer AED and taper it up to its therapeutic dosage, guided by serum drug levels and the manufacturer’s recommended titration schedule. Once the new medication has reached the therapeutic serum level, begin titrating the older AED down. If the patient suffers a breakthrough seizure during the cross-taper, we recommend aborting the process and rapidly titrating the first drug back to the predetermined therapeutic level.
Is discontinuation of AED therapy advisable if a patient wants to become pregnant?
Stopping an AED is a clinical decision made by the treating physician in accordance with the patient’s wishes on a case-by-case basis and should be considered only when it is highly likely that seizures will not recur as a result. If the patient has a history of poorly controlled epilepsy despite adequate AED trials, or if she has a structural brain lesion, persistently abnormal electroencephalograms, or any other finding that suggests that she may have recurrent seizures, explain that the risk of discontinuing the medicine is greater than the risk of fetal exposure to an AED. It is also important to point out that more than 90% of women who have epilepsy have normal, healthy children14—and that there are other steps to take to mitigate risk.13
What to consider during the first trimester
Registries that aim to gather data on the outcomes of a large number of AED-exposed pregnancies are a source of reliable information regarding the risks associated with various antiseizure agents. The primary US-based registry is the AED Pregnancy Registry (http://aedpregnancyregistry.org). We recommend that physicians caring for pregnant women who have epilepsy encourage them to enroll early, before any prenatal tests are performed. Explain to your patient that by joining the registry, she will be helping others like her make informed decisions about prenatal care.
Prenatal testing. We also recommend that pregnant women who are taking an AED—particularly those taking a higher-risk drug such as valproate—undergo a detailed first-trimester ultrasonographic study between 16 and 20 weeks’ gestation. Amniocentesis should be avoided, if possible. If needed, however, amniotic alpha-fetoprotein levels may be determined for additional risk assessment.15
Medication changes. Once a woman is pregnant, stopping or switching AEDs requires a higher level of caution and is usually ill-advised. We generally avoid medication switches after conception. But if a patient explicitly requests a change to a “safer” agent, we may attempt a cross-taper, as we would before pregnancy. Evidence suggests, however, that it may be too late to avoid the risk of major congenital malformations, which typically develop very early in pregnancy.1,3
Avoid untried AEDs. We advise against changing a pregnant woman’s seizure medication to an agent she has not tried before, because of the risks of both common adverse effects, such as allergies, and rare idiosyncratic reactions leading to aplastic anemia and Stevens-Johnson syndrome.
For the developing fetus, newer drugs are safer
Newer-generation antiepileptic drugs (AEDs), which include lamotrigine, oxcarbazepine, topiramate, gabapentin, and levetiracetam, are not associated with an increased risk of major birth defects in the first year of life when they are used during the first trimester of pregnancy, according to a new cohort study from Denmark. The study, published in the May 18, 2011, issue of JAMA, includes data on 837,795 live-born infants in Denmark from January 1996 through September 2008. Individual-level information on the AEDs dispensed to mothers, the diagnosis of any birth defects, and potential confounders were ascertained from nationwide health registries.
Of the live births included in the study, 19,960 involved infants who had a diagnosis of a major birth defect (2.4%) during the first year of life. Among 1,532 pregnancies exposed to lamotrigine, oxcarbazepine, topiramate, gabapentin, or levetiracetam at any time during the first trimester, 49 infants (3.2%) had a major birth defect. In comparison, 19,911 infants (2.4%) of 836,263 unexposed pregnancies had a major birth defect. After adjustment for various variables, Ditte Molgaard-Nielsen, MSc, and Anders Hviid, MSc, DrMedSci, found no increased risk of major birth defects associated with use of the newer-generation AEDs, though exposure to gabapentin and levetiracetam during the first trimester was uncommon.
“Our study, to our knowledge, is the largest analytic cohort study on this topic and provides comprehensive safety information on a class of drugs commonly used during pregnancy,” write Molgaard-Nielsen and Hviid. “The use of lamotrigine and oxcarbazepine during the first trimester was not associated with moderate or greater risks of major birth defects like the older-generation antiepileptic drugs, but our study cannot exclude a minor excess in risk of major birth defects or risks of specific birth defects. Topiramate, gabapentin, and levetiracetam do not appear to be major teratogens, but our study cannot exclude minor to moderate risks of major birth defects,” the authors conclude.
Topiramate remains a category D drug
The findings of this cohort study do not change the fact that topiramate was recently designated as a Pregnancy Category D drug. The US Food and Drug Administration issued an alert on March 4, 2011, notifying health-care professionals and patients that the drug’s category had changed from C to D because of new evidence of an increased risk of oral clefts in infants exposed to the agent in utero. Pregnancy Category D means that there is positive evidence of human fetal risk based on human data but the potential benefits from use of the drug in pregnant women may be acceptable in certain situations despite its risks.
—Janelle Yates, Senior Editor
Reference
AED dosing throughout pregnancy
When seizures are well controlled prior to conception, they usually remain controlled during pregnancy, although both increases and decreases in seizure frequency have been reported.16 Seizure exacerbations are usually due to decreased AED levels; this may be the result of decreased plasma protein binding, decreased albumin concentration, or increased drug clearance,16 although stress, sleep deprivation, and noncompliance may be contributing factors, as well. The changes in pharmacokinetics make it imperative that seizure frequency as well as AED levels be carefully monitored throughout pregnancy.
Although detailed information about changes in serum levels of the newer AEDs during pregnancy is not available, it can be assumed that they will decline somewhat even if the dose remains the same. Carbamazepine has the least alteration in metabolism during pregnancy,17 whereas a widely disparate effect on lamotrigine metabolism during pregnancy has been noted. In some women, serum levels of lamotrigine have been shown to decrease by as much as 60% to 90% due to induction of UDP-glucuronosyltransferase (UGT) enzymes,18 the drug’s main metabolic enzymes. Increased clearance of lamotrigine typically occurs within the first several weeks of pregnancy and returns to baseline within 2 weeks after birth.
As a result, incremental dosing of lamotrigine is usually required early in pregnancy. In some cases, dramatic increases—several multiples of the preconception dosage—may be needed, followed by a rapid decrease after delivery.18
Monitoring drug levels
Our approach to monitoring AED levels in a pregnant woman who has epilepsy includes the following:
- Check levels at baseline—prior to conception whenever possible—and monthly throughout the pregnancy, with more frequent checks for women who have recurrent seizures and those who are taking lamotrigine
- Use the dosage at which the patient was seizure-free prior to conception as a target level during pregnancy
- Adjust the dosage as needed to maintain the preconception serum drug level.
Drug-specific considerations. Because phenytoin and valproate are highly protein-bound, we follow free levels during pregnancy rather than total levels alone. (If your facility is not equipped to track free drug levels, it is important to realize that total levels of these AEDs may not accurately reflect the drug level.) If your patient is taking phenytoin, and you’re unable to obtain this information, you can use the patient’s albumin level and the total phenytoin level to estimate the corrected level of the drug with the following formula:
| Corrected phenytoin = measured total level / [(0.2 × albumin level) + 0.1] |
Provide vitamin K augmentation late in pregnancy. In addition to routinely prescribing 4 mg/day of folic acid for pregnant women who have epilepsy, we recommend oral augmentation of vitamin K as another protective measure.
AEDs that induce hepatic CYP enzymes also induce vitamin K metabolism, thereby reducing the effectiveness of vitamin K-dependent clotting factors and predisposing newborns to hemorrhagic disease.13 It remains unclear whether only women who are taking CYP enzyme-inducing AEDs or all women taking AEDs should receive oral vitamin K supplementation in the last few weeks of pregnancy. We recommend oral vitamin K supplementation for all pregnant women who have epilepsy (phytonadione 10 mg/day), starting at 36 weeks’ gestation and continuing until delivery, despite the lack of a proven benefit, because it is safe and carries little, if any, risk.
An intramuscular injection of 1 mg of vitamin K is generally given to all newborns—regardless of whether the mother has epilepsy and takes an AED—to prevent hemorrhagic disease.13
Should women taking an AED breastfeed?
The advantages of breastfeeding are largely undisputed, but women being treated with an AED are generally concerned about the possibility of contaminated breast milk. Although antiepileptic agents such as gabapentin, lamotrigine, levetiracetam, and topiramate are excreted in breast milk in potentially clinically important amounts, no short-term adverse effects have been observed in nursing infants of women being treated with an AED.13 Little information is available regarding long-term effects, and the AAN and AES state that further study is needed. Nonetheless, breastfeeding is generally believed to be a relatively safe option for patients who have epilepsy who are being treated with an AED, and it is not contraindicated by AAN/AES guidelines.13
Indeed, pregnancy itself is relatively safe for women who have epilepsy. When you’re involved in their care, your awareness of the teratogenicity of various AEDs, the variables to consider in the management of epilepsy and pregnancy, and the steps to take to mitigate risk will help you maximize the chance of a positive outcome.
Read why it’s important to screen rigorously for malformations, and when cesarean delivery may be preferable to vaginal birth. See “Guidelines confirm safety of pregnancy in women who have epilepsy—with caveats,” by Janelle Yates (September 2009).
We want to hear from you! Tell us what you think.
1. Harden CL, Meador KJ, Pennell PB, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):133-141.
2. Sachdeo R. The evidence-based rationale for monotherapy in appropriate patients with epilepsy. Neurology. 2007;69(24 suppl 3):S1-S2.
3. Holmes GL, Harden C, Liporace J, et al. Postnatal concerns in children born to women with epilepsy. Epilepsy Behav. 2007;11(3):270-276.
4. Bromley RL, Baker GA, Meador KJ. Cognitive abilities and behaviour of children exposed to antiepileptic drugs in utero. Curr Opin Neurol. 2009;22(2):162-166.
5. Yerby MS, Kaplan P, Tran T. Risks and management of pregnancy in women with epilepsy. Cleve Clin J Med. 2004;71(suppl 2):S25-S37.
6. Samren EB, van Duijn CM, Koch S, et al. Maternal use of antiepileptic drugs and the risk of major congenital malformations: a joint European prospective study of human teratogenesis associated with maternal epilepsy. Epilepsia. 1997;38(9):981-990.
7. Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci USA. 2002;99(23):15089-15094.
8. Katz I, Kim J, Gale K, et al. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther. 2007;322(2):494-500.
9. Meador KJ, Baker GA, Finnell RH, et al. NEAD Study Group. In utero antiepileptic drug exposure: fetal death and malformations. Neurology. 2006;67(3):407-412.
10. Kaaja E, Kaaja R, Hiilesmaa V. Major malformations in offspring of women with epilepsy. Neurology. 2003;60(4):575-579.
11. Rasmussen MM, Clemmensen D. Folic acid supplementation in pregnant women. Dan Med Bull. 2010;57(1):A4134.-
12. Pittschieler S, Brezinka C, Jahn B, et al. Spontaneous abortion and the prophylactic effect of folic acid supplementation in epileptic women undergoing antiepileptic therapy. J Neurol. 2008;255(12):1926-1931.
13. Epilepsy Foundation. Pregnancy & parenting. http://www.epilepsyfoundation.org/living/women/pregnancy/weipregnancy.cfm Accessed April 27 2011.
14. Harden CL, Pennell PB, Koppel BS, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): vitamin K folic acid, blood levels, and breastfeeding: report of the Quality Standards Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):142-149.
15. Tettenborn B. Management of epilepsy in women of childbearing age: practice recommendations. CNS Drugs. 2006;20(5):373-387.
16. Harden CL, Hopp J, Ting TY, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):126-132.
17. Kennedy F, Morrow J, Hunt S, et al. PATH39 malformation risks of antiepileptic drugs in pregnancy: an update from the UK Epilepsy and Pregnancy Registry. J Neurol Neurosurg Psychiatry. 2010;81:e18. doi:10.1136/jnnp.2010.226340.7.-
18. Ohman I, Beck O, Vitols S, et al. Plasma concentrations of lamotrigine and its 2-N-glucuronide metabolite during pregnancy in women with epilepsy. Epilepsia. 2008;49(6):1075-1080.
19. Artama M, Auvinen A, Raudaskoski T, Isojarvi I, Isojarvi J. Antiepileptic drug use of women with epilepsy and congenital malformations in offspring. Neurology. 2005;64(11):1874-1878.
When a woman who has epilepsy is pregnant or planning for pregnancy, you face the challenge of balancing the benefits and teratogenic risks of her antiseizure medication. Here is help.
About 500,000 women of childbearing age in the United States suffer from epilepsy.1 For these patients and their physicians, family planning and pregnancy are complex and fraught with risk.
The dilemma
Infants born to women who have epilepsy have a twofold to threefold increased risk of congenital malformations, compared with infants born to women who do not have the disorder. The increased risk is mainly related to exposure to antiepileptic drugs (AEDs).2 Recent studies also suggest that children exposed to AEDs such as valproate, phenobarbital, and phenytoin in utero may have neurocognitive deficits, even when there are no major congenital malformations.1,3,4
Yet, discontinuing the drugs prior to conception or in early pregnancy is rarely a viable option. In one recent prospective study, convulsive seizures during the first trimester (the type and timing of seizure thought to have the most harmful effect on the developing fetus) were associated with malformations in 7.4% of pregnancies.2 Seizures also increase the risk of both fetal and maternal death, although the extent of that risk is unknown.5
- If feasible, women with epilepsy who are of childbearing age and taking phenobarbital, valproate, or topiramate should be switched to a safer antiepileptic drug (AED), such as lamotrigine, prior to pregnancy.
- Avoid topiramate in women with epilepsy of childbearing age. New human data show an increased risk of oral clefts, and the FDA recently placed topiramate in Pregnancy Category D.
- Avoid switching a pregnant patient to an AED that she has not taken before.
- Use the dosage of AED at which the patient is seizure-free prior to conception as a target level to adjust dosing during pregnancy.
- Start all women who have epilepsy and are of childbearing age on ≥0.4 mg folic acid daily prior to conception.
Ideally, pregnant women with epilepsy should be under the care of both an obstetrician experienced in high-risk pregnancy and a neurologist or epileptologist. In reality, those who live in areas with limited access to such specialized care or who have limited health coverage may be cared for throughout pregnancy by a generalist ObGyn. This evidence-based review was developed with that reality in mind.
Switching (or stopping) AEDs before conception
Changes in AEDs are rarely made after conception. Any switches that a patient may desire—from a potentially unsafe drug to a “safer” AED, for example—should be considered at least a year before she plans to conceive so that good seizure control can be achieved by then.
Begin by checking the serum drug level of the patient’s effective, yet potentially unsafe, antiseizure drug. That will allow you to determine the baseline therapeutic drug level and dosage at which the patient is seizure-free. Then add the second, safer AED and taper it up to its therapeutic dosage, guided by serum drug levels and the manufacturer’s recommended titration schedule. Once the new medication has reached the therapeutic serum level, begin titrating the older AED down. If the patient suffers a breakthrough seizure during the cross-taper, we recommend aborting the process and rapidly titrating the first drug back to the predetermined therapeutic level.
Is discontinuation of AED therapy advisable if a patient wants to become pregnant?
Stopping an AED is a clinical decision made by the treating physician in accordance with the patient’s wishes on a case-by-case basis and should be considered only when it is highly likely that seizures will not recur as a result. If the patient has a history of poorly controlled epilepsy despite adequate AED trials, or if she has a structural brain lesion, persistently abnormal electroencephalograms, or any other finding that suggests that she may have recurrent seizures, explain that the risk of discontinuing the medicine is greater than the risk of fetal exposure to an AED. It is also important to point out that more than 90% of women who have epilepsy have normal, healthy children14—and that there are other steps to take to mitigate risk.13
What to consider during the first trimester
Registries that aim to gather data on the outcomes of a large number of AED-exposed pregnancies are a source of reliable information regarding the risks associated with various antiseizure agents. The primary US-based registry is the AED Pregnancy Registry (http://aedpregnancyregistry.org). We recommend that physicians caring for pregnant women who have epilepsy encourage them to enroll early, before any prenatal tests are performed. Explain to your patient that by joining the registry, she will be helping others like her make informed decisions about prenatal care.
Prenatal testing. We also recommend that pregnant women who are taking an AED—particularly those taking a higher-risk drug such as valproate—undergo a detailed first-trimester ultrasonographic study between 16 and 20 weeks’ gestation. Amniocentesis should be avoided, if possible. If needed, however, amniotic alpha-fetoprotein levels may be determined for additional risk assessment.15
Medication changes. Once a woman is pregnant, stopping or switching AEDs requires a higher level of caution and is usually ill-advised. We generally avoid medication switches after conception. But if a patient explicitly requests a change to a “safer” agent, we may attempt a cross-taper, as we would before pregnancy. Evidence suggests, however, that it may be too late to avoid the risk of major congenital malformations, which typically develop very early in pregnancy.1,3
Avoid untried AEDs. We advise against changing a pregnant woman’s seizure medication to an agent she has not tried before, because of the risks of both common adverse effects, such as allergies, and rare idiosyncratic reactions leading to aplastic anemia and Stevens-Johnson syndrome.
For the developing fetus, newer drugs are safer
Newer-generation antiepileptic drugs (AEDs), which include lamotrigine, oxcarbazepine, topiramate, gabapentin, and levetiracetam, are not associated with an increased risk of major birth defects in the first year of life when they are used during the first trimester of pregnancy, according to a new cohort study from Denmark. The study, published in the May 18, 2011, issue of JAMA, includes data on 837,795 live-born infants in Denmark from January 1996 through September 2008. Individual-level information on the AEDs dispensed to mothers, the diagnosis of any birth defects, and potential confounders were ascertained from nationwide health registries.
Of the live births included in the study, 19,960 involved infants who had a diagnosis of a major birth defect (2.4%) during the first year of life. Among 1,532 pregnancies exposed to lamotrigine, oxcarbazepine, topiramate, gabapentin, or levetiracetam at any time during the first trimester, 49 infants (3.2%) had a major birth defect. In comparison, 19,911 infants (2.4%) of 836,263 unexposed pregnancies had a major birth defect. After adjustment for various variables, Ditte Molgaard-Nielsen, MSc, and Anders Hviid, MSc, DrMedSci, found no increased risk of major birth defects associated with use of the newer-generation AEDs, though exposure to gabapentin and levetiracetam during the first trimester was uncommon.
“Our study, to our knowledge, is the largest analytic cohort study on this topic and provides comprehensive safety information on a class of drugs commonly used during pregnancy,” write Molgaard-Nielsen and Hviid. “The use of lamotrigine and oxcarbazepine during the first trimester was not associated with moderate or greater risks of major birth defects like the older-generation antiepileptic drugs, but our study cannot exclude a minor excess in risk of major birth defects or risks of specific birth defects. Topiramate, gabapentin, and levetiracetam do not appear to be major teratogens, but our study cannot exclude minor to moderate risks of major birth defects,” the authors conclude.
Topiramate remains a category D drug
The findings of this cohort study do not change the fact that topiramate was recently designated as a Pregnancy Category D drug. The US Food and Drug Administration issued an alert on March 4, 2011, notifying health-care professionals and patients that the drug’s category had changed from C to D because of new evidence of an increased risk of oral clefts in infants exposed to the agent in utero. Pregnancy Category D means that there is positive evidence of human fetal risk based on human data but the potential benefits from use of the drug in pregnant women may be acceptable in certain situations despite its risks.
—Janelle Yates, Senior Editor
Reference
AED dosing throughout pregnancy
When seizures are well controlled prior to conception, they usually remain controlled during pregnancy, although both increases and decreases in seizure frequency have been reported.16 Seizure exacerbations are usually due to decreased AED levels; this may be the result of decreased plasma protein binding, decreased albumin concentration, or increased drug clearance,16 although stress, sleep deprivation, and noncompliance may be contributing factors, as well. The changes in pharmacokinetics make it imperative that seizure frequency as well as AED levels be carefully monitored throughout pregnancy.
Although detailed information about changes in serum levels of the newer AEDs during pregnancy is not available, it can be assumed that they will decline somewhat even if the dose remains the same. Carbamazepine has the least alteration in metabolism during pregnancy,17 whereas a widely disparate effect on lamotrigine metabolism during pregnancy has been noted. In some women, serum levels of lamotrigine have been shown to decrease by as much as 60% to 90% due to induction of UDP-glucuronosyltransferase (UGT) enzymes,18 the drug’s main metabolic enzymes. Increased clearance of lamotrigine typically occurs within the first several weeks of pregnancy and returns to baseline within 2 weeks after birth.
As a result, incremental dosing of lamotrigine is usually required early in pregnancy. In some cases, dramatic increases—several multiples of the preconception dosage—may be needed, followed by a rapid decrease after delivery.18
Monitoring drug levels
Our approach to monitoring AED levels in a pregnant woman who has epilepsy includes the following:
- Check levels at baseline—prior to conception whenever possible—and monthly throughout the pregnancy, with more frequent checks for women who have recurrent seizures and those who are taking lamotrigine
- Use the dosage at which the patient was seizure-free prior to conception as a target level during pregnancy
- Adjust the dosage as needed to maintain the preconception serum drug level.
Drug-specific considerations. Because phenytoin and valproate are highly protein-bound, we follow free levels during pregnancy rather than total levels alone. (If your facility is not equipped to track free drug levels, it is important to realize that total levels of these AEDs may not accurately reflect the drug level.) If your patient is taking phenytoin, and you’re unable to obtain this information, you can use the patient’s albumin level and the total phenytoin level to estimate the corrected level of the drug with the following formula:
| Corrected phenytoin = measured total level / [(0.2 × albumin level) + 0.1] |
Provide vitamin K augmentation late in pregnancy. In addition to routinely prescribing 4 mg/day of folic acid for pregnant women who have epilepsy, we recommend oral augmentation of vitamin K as another protective measure.
AEDs that induce hepatic CYP enzymes also induce vitamin K metabolism, thereby reducing the effectiveness of vitamin K-dependent clotting factors and predisposing newborns to hemorrhagic disease.13 It remains unclear whether only women who are taking CYP enzyme-inducing AEDs or all women taking AEDs should receive oral vitamin K supplementation in the last few weeks of pregnancy. We recommend oral vitamin K supplementation for all pregnant women who have epilepsy (phytonadione 10 mg/day), starting at 36 weeks’ gestation and continuing until delivery, despite the lack of a proven benefit, because it is safe and carries little, if any, risk.
An intramuscular injection of 1 mg of vitamin K is generally given to all newborns—regardless of whether the mother has epilepsy and takes an AED—to prevent hemorrhagic disease.13
Should women taking an AED breastfeed?
The advantages of breastfeeding are largely undisputed, but women being treated with an AED are generally concerned about the possibility of contaminated breast milk. Although antiepileptic agents such as gabapentin, lamotrigine, levetiracetam, and topiramate are excreted in breast milk in potentially clinically important amounts, no short-term adverse effects have been observed in nursing infants of women being treated with an AED.13 Little information is available regarding long-term effects, and the AAN and AES state that further study is needed. Nonetheless, breastfeeding is generally believed to be a relatively safe option for patients who have epilepsy who are being treated with an AED, and it is not contraindicated by AAN/AES guidelines.13
Indeed, pregnancy itself is relatively safe for women who have epilepsy. When you’re involved in their care, your awareness of the teratogenicity of various AEDs, the variables to consider in the management of epilepsy and pregnancy, and the steps to take to mitigate risk will help you maximize the chance of a positive outcome.
Read why it’s important to screen rigorously for malformations, and when cesarean delivery may be preferable to vaginal birth. See “Guidelines confirm safety of pregnancy in women who have epilepsy—with caveats,” by Janelle Yates (September 2009).
We want to hear from you! Tell us what you think.
When a woman who has epilepsy is pregnant or planning for pregnancy, you face the challenge of balancing the benefits and teratogenic risks of her antiseizure medication. Here is help.
About 500,000 women of childbearing age in the United States suffer from epilepsy.1 For these patients and their physicians, family planning and pregnancy are complex and fraught with risk.
The dilemma
Infants born to women who have epilepsy have a twofold to threefold increased risk of congenital malformations, compared with infants born to women who do not have the disorder. The increased risk is mainly related to exposure to antiepileptic drugs (AEDs).2 Recent studies also suggest that children exposed to AEDs such as valproate, phenobarbital, and phenytoin in utero may have neurocognitive deficits, even when there are no major congenital malformations.1,3,4
Yet, discontinuing the drugs prior to conception or in early pregnancy is rarely a viable option. In one recent prospective study, convulsive seizures during the first trimester (the type and timing of seizure thought to have the most harmful effect on the developing fetus) were associated with malformations in 7.4% of pregnancies.2 Seizures also increase the risk of both fetal and maternal death, although the extent of that risk is unknown.5
- If feasible, women with epilepsy who are of childbearing age and taking phenobarbital, valproate, or topiramate should be switched to a safer antiepileptic drug (AED), such as lamotrigine, prior to pregnancy.
- Avoid topiramate in women with epilepsy of childbearing age. New human data show an increased risk of oral clefts, and the FDA recently placed topiramate in Pregnancy Category D.
- Avoid switching a pregnant patient to an AED that she has not taken before.
- Use the dosage of AED at which the patient is seizure-free prior to conception as a target level to adjust dosing during pregnancy.
- Start all women who have epilepsy and are of childbearing age on ≥0.4 mg folic acid daily prior to conception.
Ideally, pregnant women with epilepsy should be under the care of both an obstetrician experienced in high-risk pregnancy and a neurologist or epileptologist. In reality, those who live in areas with limited access to such specialized care or who have limited health coverage may be cared for throughout pregnancy by a generalist ObGyn. This evidence-based review was developed with that reality in mind.
Switching (or stopping) AEDs before conception
Changes in AEDs are rarely made after conception. Any switches that a patient may desire—from a potentially unsafe drug to a “safer” AED, for example—should be considered at least a year before she plans to conceive so that good seizure control can be achieved by then.
Begin by checking the serum drug level of the patient’s effective, yet potentially unsafe, antiseizure drug. That will allow you to determine the baseline therapeutic drug level and dosage at which the patient is seizure-free. Then add the second, safer AED and taper it up to its therapeutic dosage, guided by serum drug levels and the manufacturer’s recommended titration schedule. Once the new medication has reached the therapeutic serum level, begin titrating the older AED down. If the patient suffers a breakthrough seizure during the cross-taper, we recommend aborting the process and rapidly titrating the first drug back to the predetermined therapeutic level.
Is discontinuation of AED therapy advisable if a patient wants to become pregnant?
Stopping an AED is a clinical decision made by the treating physician in accordance with the patient’s wishes on a case-by-case basis and should be considered only when it is highly likely that seizures will not recur as a result. If the patient has a history of poorly controlled epilepsy despite adequate AED trials, or if she has a structural brain lesion, persistently abnormal electroencephalograms, or any other finding that suggests that she may have recurrent seizures, explain that the risk of discontinuing the medicine is greater than the risk of fetal exposure to an AED. It is also important to point out that more than 90% of women who have epilepsy have normal, healthy children14—and that there are other steps to take to mitigate risk.13
What to consider during the first trimester
Registries that aim to gather data on the outcomes of a large number of AED-exposed pregnancies are a source of reliable information regarding the risks associated with various antiseizure agents. The primary US-based registry is the AED Pregnancy Registry (http://aedpregnancyregistry.org). We recommend that physicians caring for pregnant women who have epilepsy encourage them to enroll early, before any prenatal tests are performed. Explain to your patient that by joining the registry, she will be helping others like her make informed decisions about prenatal care.
Prenatal testing. We also recommend that pregnant women who are taking an AED—particularly those taking a higher-risk drug such as valproate—undergo a detailed first-trimester ultrasonographic study between 16 and 20 weeks’ gestation. Amniocentesis should be avoided, if possible. If needed, however, amniotic alpha-fetoprotein levels may be determined for additional risk assessment.15
Medication changes. Once a woman is pregnant, stopping or switching AEDs requires a higher level of caution and is usually ill-advised. We generally avoid medication switches after conception. But if a patient explicitly requests a change to a “safer” agent, we may attempt a cross-taper, as we would before pregnancy. Evidence suggests, however, that it may be too late to avoid the risk of major congenital malformations, which typically develop very early in pregnancy.1,3
Avoid untried AEDs. We advise against changing a pregnant woman’s seizure medication to an agent she has not tried before, because of the risks of both common adverse effects, such as allergies, and rare idiosyncratic reactions leading to aplastic anemia and Stevens-Johnson syndrome.
For the developing fetus, newer drugs are safer
Newer-generation antiepileptic drugs (AEDs), which include lamotrigine, oxcarbazepine, topiramate, gabapentin, and levetiracetam, are not associated with an increased risk of major birth defects in the first year of life when they are used during the first trimester of pregnancy, according to a new cohort study from Denmark. The study, published in the May 18, 2011, issue of JAMA, includes data on 837,795 live-born infants in Denmark from January 1996 through September 2008. Individual-level information on the AEDs dispensed to mothers, the diagnosis of any birth defects, and potential confounders were ascertained from nationwide health registries.
Of the live births included in the study, 19,960 involved infants who had a diagnosis of a major birth defect (2.4%) during the first year of life. Among 1,532 pregnancies exposed to lamotrigine, oxcarbazepine, topiramate, gabapentin, or levetiracetam at any time during the first trimester, 49 infants (3.2%) had a major birth defect. In comparison, 19,911 infants (2.4%) of 836,263 unexposed pregnancies had a major birth defect. After adjustment for various variables, Ditte Molgaard-Nielsen, MSc, and Anders Hviid, MSc, DrMedSci, found no increased risk of major birth defects associated with use of the newer-generation AEDs, though exposure to gabapentin and levetiracetam during the first trimester was uncommon.
“Our study, to our knowledge, is the largest analytic cohort study on this topic and provides comprehensive safety information on a class of drugs commonly used during pregnancy,” write Molgaard-Nielsen and Hviid. “The use of lamotrigine and oxcarbazepine during the first trimester was not associated with moderate or greater risks of major birth defects like the older-generation antiepileptic drugs, but our study cannot exclude a minor excess in risk of major birth defects or risks of specific birth defects. Topiramate, gabapentin, and levetiracetam do not appear to be major teratogens, but our study cannot exclude minor to moderate risks of major birth defects,” the authors conclude.
Topiramate remains a category D drug
The findings of this cohort study do not change the fact that topiramate was recently designated as a Pregnancy Category D drug. The US Food and Drug Administration issued an alert on March 4, 2011, notifying health-care professionals and patients that the drug’s category had changed from C to D because of new evidence of an increased risk of oral clefts in infants exposed to the agent in utero. Pregnancy Category D means that there is positive evidence of human fetal risk based on human data but the potential benefits from use of the drug in pregnant women may be acceptable in certain situations despite its risks.
—Janelle Yates, Senior Editor
Reference
AED dosing throughout pregnancy
When seizures are well controlled prior to conception, they usually remain controlled during pregnancy, although both increases and decreases in seizure frequency have been reported.16 Seizure exacerbations are usually due to decreased AED levels; this may be the result of decreased plasma protein binding, decreased albumin concentration, or increased drug clearance,16 although stress, sleep deprivation, and noncompliance may be contributing factors, as well. The changes in pharmacokinetics make it imperative that seizure frequency as well as AED levels be carefully monitored throughout pregnancy.
Although detailed information about changes in serum levels of the newer AEDs during pregnancy is not available, it can be assumed that they will decline somewhat even if the dose remains the same. Carbamazepine has the least alteration in metabolism during pregnancy,17 whereas a widely disparate effect on lamotrigine metabolism during pregnancy has been noted. In some women, serum levels of lamotrigine have been shown to decrease by as much as 60% to 90% due to induction of UDP-glucuronosyltransferase (UGT) enzymes,18 the drug’s main metabolic enzymes. Increased clearance of lamotrigine typically occurs within the first several weeks of pregnancy and returns to baseline within 2 weeks after birth.
As a result, incremental dosing of lamotrigine is usually required early in pregnancy. In some cases, dramatic increases—several multiples of the preconception dosage—may be needed, followed by a rapid decrease after delivery.18
Monitoring drug levels
Our approach to monitoring AED levels in a pregnant woman who has epilepsy includes the following:
- Check levels at baseline—prior to conception whenever possible—and monthly throughout the pregnancy, with more frequent checks for women who have recurrent seizures and those who are taking lamotrigine
- Use the dosage at which the patient was seizure-free prior to conception as a target level during pregnancy
- Adjust the dosage as needed to maintain the preconception serum drug level.
Drug-specific considerations. Because phenytoin and valproate are highly protein-bound, we follow free levels during pregnancy rather than total levels alone. (If your facility is not equipped to track free drug levels, it is important to realize that total levels of these AEDs may not accurately reflect the drug level.) If your patient is taking phenytoin, and you’re unable to obtain this information, you can use the patient’s albumin level and the total phenytoin level to estimate the corrected level of the drug with the following formula:
| Corrected phenytoin = measured total level / [(0.2 × albumin level) + 0.1] |
Provide vitamin K augmentation late in pregnancy. In addition to routinely prescribing 4 mg/day of folic acid for pregnant women who have epilepsy, we recommend oral augmentation of vitamin K as another protective measure.
AEDs that induce hepatic CYP enzymes also induce vitamin K metabolism, thereby reducing the effectiveness of vitamin K-dependent clotting factors and predisposing newborns to hemorrhagic disease.13 It remains unclear whether only women who are taking CYP enzyme-inducing AEDs or all women taking AEDs should receive oral vitamin K supplementation in the last few weeks of pregnancy. We recommend oral vitamin K supplementation for all pregnant women who have epilepsy (phytonadione 10 mg/day), starting at 36 weeks’ gestation and continuing until delivery, despite the lack of a proven benefit, because it is safe and carries little, if any, risk.
An intramuscular injection of 1 mg of vitamin K is generally given to all newborns—regardless of whether the mother has epilepsy and takes an AED—to prevent hemorrhagic disease.13
Should women taking an AED breastfeed?
The advantages of breastfeeding are largely undisputed, but women being treated with an AED are generally concerned about the possibility of contaminated breast milk. Although antiepileptic agents such as gabapentin, lamotrigine, levetiracetam, and topiramate are excreted in breast milk in potentially clinically important amounts, no short-term adverse effects have been observed in nursing infants of women being treated with an AED.13 Little information is available regarding long-term effects, and the AAN and AES state that further study is needed. Nonetheless, breastfeeding is generally believed to be a relatively safe option for patients who have epilepsy who are being treated with an AED, and it is not contraindicated by AAN/AES guidelines.13
Indeed, pregnancy itself is relatively safe for women who have epilepsy. When you’re involved in their care, your awareness of the teratogenicity of various AEDs, the variables to consider in the management of epilepsy and pregnancy, and the steps to take to mitigate risk will help you maximize the chance of a positive outcome.
Read why it’s important to screen rigorously for malformations, and when cesarean delivery may be preferable to vaginal birth. See “Guidelines confirm safety of pregnancy in women who have epilepsy—with caveats,” by Janelle Yates (September 2009).
We want to hear from you! Tell us what you think.
1. Harden CL, Meador KJ, Pennell PB, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):133-141.
2. Sachdeo R. The evidence-based rationale for monotherapy in appropriate patients with epilepsy. Neurology. 2007;69(24 suppl 3):S1-S2.
3. Holmes GL, Harden C, Liporace J, et al. Postnatal concerns in children born to women with epilepsy. Epilepsy Behav. 2007;11(3):270-276.
4. Bromley RL, Baker GA, Meador KJ. Cognitive abilities and behaviour of children exposed to antiepileptic drugs in utero. Curr Opin Neurol. 2009;22(2):162-166.
5. Yerby MS, Kaplan P, Tran T. Risks and management of pregnancy in women with epilepsy. Cleve Clin J Med. 2004;71(suppl 2):S25-S37.
6. Samren EB, van Duijn CM, Koch S, et al. Maternal use of antiepileptic drugs and the risk of major congenital malformations: a joint European prospective study of human teratogenesis associated with maternal epilepsy. Epilepsia. 1997;38(9):981-990.
7. Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci USA. 2002;99(23):15089-15094.
8. Katz I, Kim J, Gale K, et al. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther. 2007;322(2):494-500.
9. Meador KJ, Baker GA, Finnell RH, et al. NEAD Study Group. In utero antiepileptic drug exposure: fetal death and malformations. Neurology. 2006;67(3):407-412.
10. Kaaja E, Kaaja R, Hiilesmaa V. Major malformations in offspring of women with epilepsy. Neurology. 2003;60(4):575-579.
11. Rasmussen MM, Clemmensen D. Folic acid supplementation in pregnant women. Dan Med Bull. 2010;57(1):A4134.-
12. Pittschieler S, Brezinka C, Jahn B, et al. Spontaneous abortion and the prophylactic effect of folic acid supplementation in epileptic women undergoing antiepileptic therapy. J Neurol. 2008;255(12):1926-1931.
13. Epilepsy Foundation. Pregnancy & parenting. http://www.epilepsyfoundation.org/living/women/pregnancy/weipregnancy.cfm Accessed April 27 2011.
14. Harden CL, Pennell PB, Koppel BS, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): vitamin K folic acid, blood levels, and breastfeeding: report of the Quality Standards Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):142-149.
15. Tettenborn B. Management of epilepsy in women of childbearing age: practice recommendations. CNS Drugs. 2006;20(5):373-387.
16. Harden CL, Hopp J, Ting TY, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):126-132.
17. Kennedy F, Morrow J, Hunt S, et al. PATH39 malformation risks of antiepileptic drugs in pregnancy: an update from the UK Epilepsy and Pregnancy Registry. J Neurol Neurosurg Psychiatry. 2010;81:e18. doi:10.1136/jnnp.2010.226340.7.-
18. Ohman I, Beck O, Vitols S, et al. Plasma concentrations of lamotrigine and its 2-N-glucuronide metabolite during pregnancy in women with epilepsy. Epilepsia. 2008;49(6):1075-1080.
19. Artama M, Auvinen A, Raudaskoski T, Isojarvi I, Isojarvi J. Antiepileptic drug use of women with epilepsy and congenital malformations in offspring. Neurology. 2005;64(11):1874-1878.
1. Harden CL, Meador KJ, Pennell PB, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): teratogenesis and perinatal outcomes: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):133-141.
2. Sachdeo R. The evidence-based rationale for monotherapy in appropriate patients with epilepsy. Neurology. 2007;69(24 suppl 3):S1-S2.
3. Holmes GL, Harden C, Liporace J, et al. Postnatal concerns in children born to women with epilepsy. Epilepsy Behav. 2007;11(3):270-276.
4. Bromley RL, Baker GA, Meador KJ. Cognitive abilities and behaviour of children exposed to antiepileptic drugs in utero. Curr Opin Neurol. 2009;22(2):162-166.
5. Yerby MS, Kaplan P, Tran T. Risks and management of pregnancy in women with epilepsy. Cleve Clin J Med. 2004;71(suppl 2):S25-S37.
6. Samren EB, van Duijn CM, Koch S, et al. Maternal use of antiepileptic drugs and the risk of major congenital malformations: a joint European prospective study of human teratogenesis associated with maternal epilepsy. Epilepsia. 1997;38(9):981-990.
7. Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci USA. 2002;99(23):15089-15094.
8. Katz I, Kim J, Gale K, et al. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther. 2007;322(2):494-500.
9. Meador KJ, Baker GA, Finnell RH, et al. NEAD Study Group. In utero antiepileptic drug exposure: fetal death and malformations. Neurology. 2006;67(3):407-412.
10. Kaaja E, Kaaja R, Hiilesmaa V. Major malformations in offspring of women with epilepsy. Neurology. 2003;60(4):575-579.
11. Rasmussen MM, Clemmensen D. Folic acid supplementation in pregnant women. Dan Med Bull. 2010;57(1):A4134.-
12. Pittschieler S, Brezinka C, Jahn B, et al. Spontaneous abortion and the prophylactic effect of folic acid supplementation in epileptic women undergoing antiepileptic therapy. J Neurol. 2008;255(12):1926-1931.
13. Epilepsy Foundation. Pregnancy & parenting. http://www.epilepsyfoundation.org/living/women/pregnancy/weipregnancy.cfm Accessed April 27 2011.
14. Harden CL, Pennell PB, Koppel BS, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): vitamin K folic acid, blood levels, and breastfeeding: report of the Quality Standards Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):142-149.
15. Tettenborn B. Management of epilepsy in women of childbearing age: practice recommendations. CNS Drugs. 2006;20(5):373-387.
16. Harden CL, Hopp J, Ting TY, et al. American Academy of Neurology; American Epilepsy Society. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency: report of the Quality Standards Subcommittee and Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and American Epilepsy Society. Neurology. 2009;73(2):126-132.
17. Kennedy F, Morrow J, Hunt S, et al. PATH39 malformation risks of antiepileptic drugs in pregnancy: an update from the UK Epilepsy and Pregnancy Registry. J Neurol Neurosurg Psychiatry. 2010;81:e18. doi:10.1136/jnnp.2010.226340.7.-
18. Ohman I, Beck O, Vitols S, et al. Plasma concentrations of lamotrigine and its 2-N-glucuronide metabolite during pregnancy in women with epilepsy. Epilepsia. 2008;49(6):1075-1080.
19. Artama M, Auvinen A, Raudaskoski T, Isojarvi I, Isojarvi J. Antiepileptic drug use of women with epilepsy and congenital malformations in offspring. Neurology. 2005;64(11):1874-1878.
UPDATE ON INFECTIOUS DISEASE
- New group B strep guidelines clarify management of key groups
Janelle Yates (March 2011) - Does third-trimester screening accurately predict GBS in labor?
Janelle Yates (Web exclusive, March 2011) - Does the rate of postcesarean maternal infection vary by uterine closure technique?
Vincenzo Berghella, MD (Examining the Evidence, February 2011) - Is there a link between cerebral palsy and chorioamnionitis?
Errol R. Norwitz, MD, PhD (Examining the Evidence, December 2010)
Dr. Tita reports receiving research support from the NIH. Dr. Subramaniam and Dr. Andrews report no financial relationships relevant to this article.
Despite continuing advances in obstetric and neonatal care, including the use of antimicrobials, infection remains a major cause of maternal and perinatal morbidity and death. Indeed, infection is among the top five causes of maternal death in the United States, with 10% to 15% of deaths directly linked to it. Maternal and fetal infections are also a common cause of perinatal death. Clearly, interventions to prevent infection or minimize its effect during pregnancy and postpartum are a priority.
This article focuses on three notable developments of the past year:
- release of revised guidelines on the prevention of perinatal group B streptococcal disease (GBS)
- publication of surveillance data on 2009 influenza A(H1N1) among pregnant women, which reveals the life-threatening nature of the flu in this population
- publication of a Committee Opinion from ACOG on the timing of antimicrobial prophylaxis for cesarean delivery, in which administration within 60 minutes of the start of the procedure is recommended.
There’s room for improvement in GBS screening and prophylaxis,
says CDC
Verani JR, McGee L, Schrag SJ, National Center for Immunization and Respiratory Diseases. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC, 2010. MMWR Recomm Rep. 2010;59(RR-10):1–36. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5910a1.htm.
The latest revision of CDC guidelines on screening and prophylaxis for perinatal group B streptococcal disease were developed by a multidisciplinary working group representing several professional organizations, including ACOG. Information that has come to light since the most recent guidelines were released in 2002 was incorporated, and areas that have seen suboptimal implementation or interpretation were addressed here as well.
Although the use of prophylactic antibiotics during labor reduces the incidence of invasive GBS during the first week of life, GBS remains a leading cause of neonatal morbidity and death in the United States and elsewhere. Universal screening and intrapartum antibiotic prophylaxis for women who test positive for GBS remain the cornerstone of prevention of early-onset neonatal GBS disease. Penicillin G is still the agent of choice, and ampicillin is an acceptable alternative. When prophylaxis is warranted, intravenous antibiotic administration at least 4 hours before delivery is recommended.
Other recommendations from the CDC include:
- Women who have GBS bacteriuria (≥104 colony-forming units/mL) any time during pregnancy, and women who had a previous infant with invasive GBS disease, should receive intrapartum antibiotic prophylaxis. In these cases, third-trimester screening for GBS is unnecessary.
- All other women should be screened at 35 to 37 weeks’ gestation for rectovaginal GBS colonization.
- At the onset of labor or rupture of membranes, antibiotic prophylaxis should be given to all women who tested positive for GBS—with the exception of women who are undergoing cesarean delivery, at any gestational age, with intact membranes.
- If GBS status is unknown, intrapartum prophylaxis should be given for gestational ages below 37 weeks, membrane rupture lasting 18 hours or longer, or temperature of 100.4°F (38.0°C) or above.
- Health-care providers should inform women of their GBS screening results and the recommended interventions.
Some updates to the guidelines clarify areas in which there was some confusion. For example, the guidelines delineate that women who have preterm labor or preterm premature rupture of the membranes (PPROM) should be screened for GBS and started on prophylaxis immediately, unless they have received a negative screen within the preceding 5 weeks. Antibiotics should be discontinued if a woman who has intact membranes is found not to be in true labor or if the GBS culture is negative.
For the woman who has PPROM, antibiotics to prolong latency are sufficient, provided they include adequate GBS coverage; otherwise, GBS prophylaxis is warranted for as long as 48 hours.
In addition:
- Women who are allergic to penicillin and who have no history of anaphylaxis, angioedema, respiratory distress, or urticaria in response to penicillin or a cephalosporin, should be given cefazolin for GBS prophylaxis. Women at risk of anaphylaxis should undergo antimicrobial susceptibility testing. For these women, clindamycin is acceptable if the GBS isolate is susceptible to both clindamycin and erythromycin or resistant to erythromycin with negative inducible clindamycin resistance. Otherwise, vancomycin is recommended. Erythromycin is not acceptable because of a high prevalence of resistance.
- The dosage of penicillin is 5 million U initially, followed by 2.5 million to 3 million U every 4 hours.
- If a laboratory providing nucleic acid amplification testing (NAAT) is available, intrapartum testing for women who have unknown GBS status and no intrapartum risk factors (i.e., gestational age <37 weeks, membrane rupture for 18 hours or longer, or temperature ≥100.4°F) is an option.
The implementation of CDC guidelines in 2002 had a salutary impact on the burden of neonatal invasive GBS disease. The 2010 updated guidelines address areas of suboptimal compliance. Health institutions should determine where changes need to be made to better adhere to these guidelines, which are readily available online.1
Be vigilant for influenza among your pregnant patients—
and take necessary action
Siston AM, Rasmussen SA, Honein MA, et al, for the Pandemic H1N1 Influenza in Pregnancy Working Group. Pandemic 2009 influenza A(H1N1) virus illness among pregnant women in the United States. JAMA. 2010;303(15):1517–1525.
The recent H1N1 pandemic highlighted the status of influenza as a major public health problem, not only among children and the elderly but especially among pregnant women. This report by Siston and colleagues describes the US experience of the pandemic. To ascertain the severity of infection during pregnancy, the authors analyzed data from 788 pregnant women who developed symptoms of H1N1 infection between April and August 2009. These women were identified through the CDC national surveillance system. Data from an additional 165 women who developed symptoms through December 2011 and who were admitted to an ICU because of influenza were also analyzed.
Siston and colleagues found a high case-fatality rate (5%) among pregnant women who had influenza. Almost one fourth (22.6%) of women who were hospitalized with influenza were admitted to an ICU because of severe illness.
The timing of antiviral treatment influenced the course of the illness. For example, women who received antiviral treatment within 2 days of the onset of symptoms had a significantly lower risk of death (0.5%), compared with women who received treatment within 3 to 4 days (5%) and with women who were treated after 4 days (27%). Women who were not treated at all also had an elevated risk of ICU admission and death, although that risk was not as high as it was among women treated 4 days or longer after the onset of symptoms. This finding suggests that severity of illness may play a role in determining who receives antiviral treatment.
The high incidence of severe morbidity and death among pregnant women who contract H1N1 influenza is consistent with the findings of several studies, suggesting that pregnant women are especially vulnerable to the virus. Although Siston and colleagues focused on pandemic influenza, which may be more severe than seasonal flu, their data and other studies suggest that pregnant women have increased susceptibility to influenza-like illness during regular flu season (October to May).
More than 10% of pregnant women may have confirmed influenza during flu season, making it one of the most common infections during pregnancy. Therefore, during flu season, providers should maintain a high index of suspicion for viral infection. Antiviral treatment—typically oseltamivir (75 mg orally twice daily) or zanamivir (5 mg inhaled twice daily) for 5 days—should be administered promptly, ideally within 2 days of the onset of symptoms.
If a pregnant woman is exposed to influenza, 10 days of prophylactic antiviral therapy (75 mg oral oseltamivir or 10 mg inhaled zanamivir daily) is indicated.
The CDC makes recommendations annually about which antivirals to use. Oseltamivir and zanamivir are preferred because they cover both types of human influenza (A and B), and 99% of circulating influenza viruses are susceptible to them.
ACOG recently emphasized the high-risk nature of influenza during pregnancy and urged universal vaccination of women who will be pregnant or postpartum during the flu season as “an integral element of prenatal care.”1
ACOG: Give prophylactic antimicrobials before the incision
in cesarean delivery
ACOG committee opinion no. 465: Antimicrobial prophylaxis for cesarean delivery: timing of administration. Obstet Gynecol. 2010;116(3):791–792.
The use of antimicrobial prophylaxis for cesarean delivery is associated with a reduction of 50% or more in the rates of postcesarean infection and severe adverse outcomes, including maternal death. However, there has been some controversy surrounding the question of timing of antimicrobial administration. Should the drugs be administered at the time the cord is clamped or prior to the cesarean skin incision? And, if the latter, just how long before the incision should antimicrobials be given?
ACOG weighed in on this question in September 2010 in a Committee Opinion based on a review of data. It recommended that, whenever feasible, antimicrobials should be administered within 60 minutes before the start of the procedure.
In the past, antimicrobial administration at the time of cord clamping was proposed to reduce fetal exposure and prevent the masking of neonatal infection (falsely negative culture results). However, the data ACOG reviewed from randomized, clinical trials indicate that pre-incision antimicrobials may further reduce the risk of infection (including endometritis and wound infection) without apparent perinatal harm.
First-generation cephalosporins (commonly, 1 g of cefazolin) remain the antibiotic of choice, but the combination of clindamycin and gentamicin was suggested as an acceptable alternative for women who are allergic to penicillin.
ACOG reviewed studies by Thigpen and colleagues and Sullivan and coworkers, as well as a meta-analysis by Costantine and associates.2-4 The relatively small size of these studies and the mixed results of other studies point to the need for further investigation, a fact acknowledged by ACOG. Nevertheless, the committee concluded that “preoperative administration significantly reduces endometritis and total maternal infectious morbidity, compared with administration of antimicrobials after umbilical cord clamping.”
Antimicrobial prophylaxis for surgical-site infection is a well-established practice for both obstetric and gynecologic surgery. Cesarean delivery accounts for one third (more than 1.3 million) of all births in the United States each year and carries a risk of infection at least five times higher than that associated with vaginal delivery. Given the rising incidence of cesarean delivery in the United States and elsewhere, the prevention of postcesarean infection is a priority.
This ACOG Committee Opinion should prompt providers who continue to administer antibiotics after cord clamping to reevaluate the practice.
Even so, given the relative paucity of randomized trials addressing this issue and the mixed results from individual studies, it is essential that we continue to monitor the effectiveness and safety of pre-incision prophylaxis.
Larger follow-up studies to further evaluate resistance profiles and the effects of pre-incision administration on the newborn should be undertaken in concert with implementation of this recommendation.
We want to hear from you! Tell us what you think.
1. ACOG committee opinion#468: Influenza vaccination during pregnancy. Obstet Gynecol. 2010;116(4):1006-1007.
2. Thigpen BD, Hood WA, Chauhan S, et al. Timing of prophylactice antibiotic administration in the uninfected laboring gravida: a randomized clinical trial. Am J Obstet Gynecol. 2005;192(6):1864-1871.
3. Sullivan SA, Smith T, Chang E, Hulsey T, Vandorsten JP, Soper D. Administration of cefazolin prior to skin incision is superior to cefazolin at cord clamping in preventing postcesarean infectious morbidity: a randomized, controlled trial. Am J Obstet Gynecol. 2007;196(5):455.e1-5.
4. Costantine MM, Rahman M, Ghulmiyah L, et al. Timing of perioperative antibiotics for cesarean delivery: a meta-analysis. Am J Obstet Gynecol. 2008;199(3):301.e1-6.

Alan T.N. Tita, MD, PhD
Dr. Tita is Associate Professor of Obstetrics and Gynecology at the University of Alabama in Birmingham, Ala.
Akila Subramaniam, MD
Dr. Subramaniam is a Resident in the Department of Obstetrics and Gynecology at the University of Alabama in Birmingham, Ala.
William W. Andrews, MD, PhD
Dr. Andrews is Professor and Chairman of Obstetrics and Gynecology at the University of Alabama in Birmingham, Ala.
Alan T.N. Tita, MD, PhD
Dr. Tita is Associate Professor of Obstetrics and Gynecology at the University of Alabama in Birmingham, Ala.
Akila Subramaniam, MD
Dr. Subramaniam is a Resident in the Department of Obstetrics and Gynecology at the University of Alabama in Birmingham, Ala.
William W. Andrews, MD, PhD
Dr. Andrews is Professor and Chairman of Obstetrics and Gynecology at the University of Alabama in Birmingham, Ala.
Alan T.N. Tita, MD, PhD
Dr. Tita is Associate Professor of Obstetrics and Gynecology at the University of Alabama in Birmingham, Ala.
Akila Subramaniam, MD
Dr. Subramaniam is a Resident in the Department of Obstetrics and Gynecology at the University of Alabama in Birmingham, Ala.
William W. Andrews, MD, PhD
Dr. Andrews is Professor and Chairman of Obstetrics and Gynecology at the University of Alabama in Birmingham, Ala.
- New group B strep guidelines clarify management of key groups
Janelle Yates (March 2011) - Does third-trimester screening accurately predict GBS in labor?
Janelle Yates (Web exclusive, March 2011) - Does the rate of postcesarean maternal infection vary by uterine closure technique?
Vincenzo Berghella, MD (Examining the Evidence, February 2011) - Is there a link between cerebral palsy and chorioamnionitis?
Errol R. Norwitz, MD, PhD (Examining the Evidence, December 2010)
Dr. Tita reports receiving research support from the NIH. Dr. Subramaniam and Dr. Andrews report no financial relationships relevant to this article.
Despite continuing advances in obstetric and neonatal care, including the use of antimicrobials, infection remains a major cause of maternal and perinatal morbidity and death. Indeed, infection is among the top five causes of maternal death in the United States, with 10% to 15% of deaths directly linked to it. Maternal and fetal infections are also a common cause of perinatal death. Clearly, interventions to prevent infection or minimize its effect during pregnancy and postpartum are a priority.
This article focuses on three notable developments of the past year:
- release of revised guidelines on the prevention of perinatal group B streptococcal disease (GBS)
- publication of surveillance data on 2009 influenza A(H1N1) among pregnant women, which reveals the life-threatening nature of the flu in this population
- publication of a Committee Opinion from ACOG on the timing of antimicrobial prophylaxis for cesarean delivery, in which administration within 60 minutes of the start of the procedure is recommended.
There’s room for improvement in GBS screening and prophylaxis,
says CDC
Verani JR, McGee L, Schrag SJ, National Center for Immunization and Respiratory Diseases. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC, 2010. MMWR Recomm Rep. 2010;59(RR-10):1–36. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5910a1.htm.
The latest revision of CDC guidelines on screening and prophylaxis for perinatal group B streptococcal disease were developed by a multidisciplinary working group representing several professional organizations, including ACOG. Information that has come to light since the most recent guidelines were released in 2002 was incorporated, and areas that have seen suboptimal implementation or interpretation were addressed here as well.
Although the use of prophylactic antibiotics during labor reduces the incidence of invasive GBS during the first week of life, GBS remains a leading cause of neonatal morbidity and death in the United States and elsewhere. Universal screening and intrapartum antibiotic prophylaxis for women who test positive for GBS remain the cornerstone of prevention of early-onset neonatal GBS disease. Penicillin G is still the agent of choice, and ampicillin is an acceptable alternative. When prophylaxis is warranted, intravenous antibiotic administration at least 4 hours before delivery is recommended.
Other recommendations from the CDC include:
- Women who have GBS bacteriuria (≥104 colony-forming units/mL) any time during pregnancy, and women who had a previous infant with invasive GBS disease, should receive intrapartum antibiotic prophylaxis. In these cases, third-trimester screening for GBS is unnecessary.
- All other women should be screened at 35 to 37 weeks’ gestation for rectovaginal GBS colonization.
- At the onset of labor or rupture of membranes, antibiotic prophylaxis should be given to all women who tested positive for GBS—with the exception of women who are undergoing cesarean delivery, at any gestational age, with intact membranes.
- If GBS status is unknown, intrapartum prophylaxis should be given for gestational ages below 37 weeks, membrane rupture lasting 18 hours or longer, or temperature of 100.4°F (38.0°C) or above.
- Health-care providers should inform women of their GBS screening results and the recommended interventions.
Some updates to the guidelines clarify areas in which there was some confusion. For example, the guidelines delineate that women who have preterm labor or preterm premature rupture of the membranes (PPROM) should be screened for GBS and started on prophylaxis immediately, unless they have received a negative screen within the preceding 5 weeks. Antibiotics should be discontinued if a woman who has intact membranes is found not to be in true labor or if the GBS culture is negative.
For the woman who has PPROM, antibiotics to prolong latency are sufficient, provided they include adequate GBS coverage; otherwise, GBS prophylaxis is warranted for as long as 48 hours.
In addition:
- Women who are allergic to penicillin and who have no history of anaphylaxis, angioedema, respiratory distress, or urticaria in response to penicillin or a cephalosporin, should be given cefazolin for GBS prophylaxis. Women at risk of anaphylaxis should undergo antimicrobial susceptibility testing. For these women, clindamycin is acceptable if the GBS isolate is susceptible to both clindamycin and erythromycin or resistant to erythromycin with negative inducible clindamycin resistance. Otherwise, vancomycin is recommended. Erythromycin is not acceptable because of a high prevalence of resistance.
- The dosage of penicillin is 5 million U initially, followed by 2.5 million to 3 million U every 4 hours.
- If a laboratory providing nucleic acid amplification testing (NAAT) is available, intrapartum testing for women who have unknown GBS status and no intrapartum risk factors (i.e., gestational age <37 weeks, membrane rupture for 18 hours or longer, or temperature ≥100.4°F) is an option.
The implementation of CDC guidelines in 2002 had a salutary impact on the burden of neonatal invasive GBS disease. The 2010 updated guidelines address areas of suboptimal compliance. Health institutions should determine where changes need to be made to better adhere to these guidelines, which are readily available online.1
Be vigilant for influenza among your pregnant patients—
and take necessary action
Siston AM, Rasmussen SA, Honein MA, et al, for the Pandemic H1N1 Influenza in Pregnancy Working Group. Pandemic 2009 influenza A(H1N1) virus illness among pregnant women in the United States. JAMA. 2010;303(15):1517–1525.
The recent H1N1 pandemic highlighted the status of influenza as a major public health problem, not only among children and the elderly but especially among pregnant women. This report by Siston and colleagues describes the US experience of the pandemic. To ascertain the severity of infection during pregnancy, the authors analyzed data from 788 pregnant women who developed symptoms of H1N1 infection between April and August 2009. These women were identified through the CDC national surveillance system. Data from an additional 165 women who developed symptoms through December 2011 and who were admitted to an ICU because of influenza were also analyzed.
Siston and colleagues found a high case-fatality rate (5%) among pregnant women who had influenza. Almost one fourth (22.6%) of women who were hospitalized with influenza were admitted to an ICU because of severe illness.
The timing of antiviral treatment influenced the course of the illness. For example, women who received antiviral treatment within 2 days of the onset of symptoms had a significantly lower risk of death (0.5%), compared with women who received treatment within 3 to 4 days (5%) and with women who were treated after 4 days (27%). Women who were not treated at all also had an elevated risk of ICU admission and death, although that risk was not as high as it was among women treated 4 days or longer after the onset of symptoms. This finding suggests that severity of illness may play a role in determining who receives antiviral treatment.
The high incidence of severe morbidity and death among pregnant women who contract H1N1 influenza is consistent with the findings of several studies, suggesting that pregnant women are especially vulnerable to the virus. Although Siston and colleagues focused on pandemic influenza, which may be more severe than seasonal flu, their data and other studies suggest that pregnant women have increased susceptibility to influenza-like illness during regular flu season (October to May).
More than 10% of pregnant women may have confirmed influenza during flu season, making it one of the most common infections during pregnancy. Therefore, during flu season, providers should maintain a high index of suspicion for viral infection. Antiviral treatment—typically oseltamivir (75 mg orally twice daily) or zanamivir (5 mg inhaled twice daily) for 5 days—should be administered promptly, ideally within 2 days of the onset of symptoms.
If a pregnant woman is exposed to influenza, 10 days of prophylactic antiviral therapy (75 mg oral oseltamivir or 10 mg inhaled zanamivir daily) is indicated.
The CDC makes recommendations annually about which antivirals to use. Oseltamivir and zanamivir are preferred because they cover both types of human influenza (A and B), and 99% of circulating influenza viruses are susceptible to them.
ACOG recently emphasized the high-risk nature of influenza during pregnancy and urged universal vaccination of women who will be pregnant or postpartum during the flu season as “an integral element of prenatal care.”1
ACOG: Give prophylactic antimicrobials before the incision
in cesarean delivery
ACOG committee opinion no. 465: Antimicrobial prophylaxis for cesarean delivery: timing of administration. Obstet Gynecol. 2010;116(3):791–792.
The use of antimicrobial prophylaxis for cesarean delivery is associated with a reduction of 50% or more in the rates of postcesarean infection and severe adverse outcomes, including maternal death. However, there has been some controversy surrounding the question of timing of antimicrobial administration. Should the drugs be administered at the time the cord is clamped or prior to the cesarean skin incision? And, if the latter, just how long before the incision should antimicrobials be given?
ACOG weighed in on this question in September 2010 in a Committee Opinion based on a review of data. It recommended that, whenever feasible, antimicrobials should be administered within 60 minutes before the start of the procedure.
In the past, antimicrobial administration at the time of cord clamping was proposed to reduce fetal exposure and prevent the masking of neonatal infection (falsely negative culture results). However, the data ACOG reviewed from randomized, clinical trials indicate that pre-incision antimicrobials may further reduce the risk of infection (including endometritis and wound infection) without apparent perinatal harm.
First-generation cephalosporins (commonly, 1 g of cefazolin) remain the antibiotic of choice, but the combination of clindamycin and gentamicin was suggested as an acceptable alternative for women who are allergic to penicillin.
ACOG reviewed studies by Thigpen and colleagues and Sullivan and coworkers, as well as a meta-analysis by Costantine and associates.2-4 The relatively small size of these studies and the mixed results of other studies point to the need for further investigation, a fact acknowledged by ACOG. Nevertheless, the committee concluded that “preoperative administration significantly reduces endometritis and total maternal infectious morbidity, compared with administration of antimicrobials after umbilical cord clamping.”
Antimicrobial prophylaxis for surgical-site infection is a well-established practice for both obstetric and gynecologic surgery. Cesarean delivery accounts for one third (more than 1.3 million) of all births in the United States each year and carries a risk of infection at least five times higher than that associated with vaginal delivery. Given the rising incidence of cesarean delivery in the United States and elsewhere, the prevention of postcesarean infection is a priority.
This ACOG Committee Opinion should prompt providers who continue to administer antibiotics after cord clamping to reevaluate the practice.
Even so, given the relative paucity of randomized trials addressing this issue and the mixed results from individual studies, it is essential that we continue to monitor the effectiveness and safety of pre-incision prophylaxis.
Larger follow-up studies to further evaluate resistance profiles and the effects of pre-incision administration on the newborn should be undertaken in concert with implementation of this recommendation.
We want to hear from you! Tell us what you think.
- New group B strep guidelines clarify management of key groups
Janelle Yates (March 2011) - Does third-trimester screening accurately predict GBS in labor?
Janelle Yates (Web exclusive, March 2011) - Does the rate of postcesarean maternal infection vary by uterine closure technique?
Vincenzo Berghella, MD (Examining the Evidence, February 2011) - Is there a link between cerebral palsy and chorioamnionitis?
Errol R. Norwitz, MD, PhD (Examining the Evidence, December 2010)
Dr. Tita reports receiving research support from the NIH. Dr. Subramaniam and Dr. Andrews report no financial relationships relevant to this article.
Despite continuing advances in obstetric and neonatal care, including the use of antimicrobials, infection remains a major cause of maternal and perinatal morbidity and death. Indeed, infection is among the top five causes of maternal death in the United States, with 10% to 15% of deaths directly linked to it. Maternal and fetal infections are also a common cause of perinatal death. Clearly, interventions to prevent infection or minimize its effect during pregnancy and postpartum are a priority.
This article focuses on three notable developments of the past year:
- release of revised guidelines on the prevention of perinatal group B streptococcal disease (GBS)
- publication of surveillance data on 2009 influenza A(H1N1) among pregnant women, which reveals the life-threatening nature of the flu in this population
- publication of a Committee Opinion from ACOG on the timing of antimicrobial prophylaxis for cesarean delivery, in which administration within 60 minutes of the start of the procedure is recommended.
There’s room for improvement in GBS screening and prophylaxis,
says CDC
Verani JR, McGee L, Schrag SJ, National Center for Immunization and Respiratory Diseases. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC, 2010. MMWR Recomm Rep. 2010;59(RR-10):1–36. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5910a1.htm.
The latest revision of CDC guidelines on screening and prophylaxis for perinatal group B streptococcal disease were developed by a multidisciplinary working group representing several professional organizations, including ACOG. Information that has come to light since the most recent guidelines were released in 2002 was incorporated, and areas that have seen suboptimal implementation or interpretation were addressed here as well.
Although the use of prophylactic antibiotics during labor reduces the incidence of invasive GBS during the first week of life, GBS remains a leading cause of neonatal morbidity and death in the United States and elsewhere. Universal screening and intrapartum antibiotic prophylaxis for women who test positive for GBS remain the cornerstone of prevention of early-onset neonatal GBS disease. Penicillin G is still the agent of choice, and ampicillin is an acceptable alternative. When prophylaxis is warranted, intravenous antibiotic administration at least 4 hours before delivery is recommended.
Other recommendations from the CDC include:
- Women who have GBS bacteriuria (≥104 colony-forming units/mL) any time during pregnancy, and women who had a previous infant with invasive GBS disease, should receive intrapartum antibiotic prophylaxis. In these cases, third-trimester screening for GBS is unnecessary.
- All other women should be screened at 35 to 37 weeks’ gestation for rectovaginal GBS colonization.
- At the onset of labor or rupture of membranes, antibiotic prophylaxis should be given to all women who tested positive for GBS—with the exception of women who are undergoing cesarean delivery, at any gestational age, with intact membranes.
- If GBS status is unknown, intrapartum prophylaxis should be given for gestational ages below 37 weeks, membrane rupture lasting 18 hours or longer, or temperature of 100.4°F (38.0°C) or above.
- Health-care providers should inform women of their GBS screening results and the recommended interventions.
Some updates to the guidelines clarify areas in which there was some confusion. For example, the guidelines delineate that women who have preterm labor or preterm premature rupture of the membranes (PPROM) should be screened for GBS and started on prophylaxis immediately, unless they have received a negative screen within the preceding 5 weeks. Antibiotics should be discontinued if a woman who has intact membranes is found not to be in true labor or if the GBS culture is negative.
For the woman who has PPROM, antibiotics to prolong latency are sufficient, provided they include adequate GBS coverage; otherwise, GBS prophylaxis is warranted for as long as 48 hours.
In addition:
- Women who are allergic to penicillin and who have no history of anaphylaxis, angioedema, respiratory distress, or urticaria in response to penicillin or a cephalosporin, should be given cefazolin for GBS prophylaxis. Women at risk of anaphylaxis should undergo antimicrobial susceptibility testing. For these women, clindamycin is acceptable if the GBS isolate is susceptible to both clindamycin and erythromycin or resistant to erythromycin with negative inducible clindamycin resistance. Otherwise, vancomycin is recommended. Erythromycin is not acceptable because of a high prevalence of resistance.
- The dosage of penicillin is 5 million U initially, followed by 2.5 million to 3 million U every 4 hours.
- If a laboratory providing nucleic acid amplification testing (NAAT) is available, intrapartum testing for women who have unknown GBS status and no intrapartum risk factors (i.e., gestational age <37 weeks, membrane rupture for 18 hours or longer, or temperature ≥100.4°F) is an option.
The implementation of CDC guidelines in 2002 had a salutary impact on the burden of neonatal invasive GBS disease. The 2010 updated guidelines address areas of suboptimal compliance. Health institutions should determine where changes need to be made to better adhere to these guidelines, which are readily available online.1
Be vigilant for influenza among your pregnant patients—
and take necessary action
Siston AM, Rasmussen SA, Honein MA, et al, for the Pandemic H1N1 Influenza in Pregnancy Working Group. Pandemic 2009 influenza A(H1N1) virus illness among pregnant women in the United States. JAMA. 2010;303(15):1517–1525.
The recent H1N1 pandemic highlighted the status of influenza as a major public health problem, not only among children and the elderly but especially among pregnant women. This report by Siston and colleagues describes the US experience of the pandemic. To ascertain the severity of infection during pregnancy, the authors analyzed data from 788 pregnant women who developed symptoms of H1N1 infection between April and August 2009. These women were identified through the CDC national surveillance system. Data from an additional 165 women who developed symptoms through December 2011 and who were admitted to an ICU because of influenza were also analyzed.
Siston and colleagues found a high case-fatality rate (5%) among pregnant women who had influenza. Almost one fourth (22.6%) of women who were hospitalized with influenza were admitted to an ICU because of severe illness.
The timing of antiviral treatment influenced the course of the illness. For example, women who received antiviral treatment within 2 days of the onset of symptoms had a significantly lower risk of death (0.5%), compared with women who received treatment within 3 to 4 days (5%) and with women who were treated after 4 days (27%). Women who were not treated at all also had an elevated risk of ICU admission and death, although that risk was not as high as it was among women treated 4 days or longer after the onset of symptoms. This finding suggests that severity of illness may play a role in determining who receives antiviral treatment.
The high incidence of severe morbidity and death among pregnant women who contract H1N1 influenza is consistent with the findings of several studies, suggesting that pregnant women are especially vulnerable to the virus. Although Siston and colleagues focused on pandemic influenza, which may be more severe than seasonal flu, their data and other studies suggest that pregnant women have increased susceptibility to influenza-like illness during regular flu season (October to May).
More than 10% of pregnant women may have confirmed influenza during flu season, making it one of the most common infections during pregnancy. Therefore, during flu season, providers should maintain a high index of suspicion for viral infection. Antiviral treatment—typically oseltamivir (75 mg orally twice daily) or zanamivir (5 mg inhaled twice daily) for 5 days—should be administered promptly, ideally within 2 days of the onset of symptoms.
If a pregnant woman is exposed to influenza, 10 days of prophylactic antiviral therapy (75 mg oral oseltamivir or 10 mg inhaled zanamivir daily) is indicated.
The CDC makes recommendations annually about which antivirals to use. Oseltamivir and zanamivir are preferred because they cover both types of human influenza (A and B), and 99% of circulating influenza viruses are susceptible to them.
ACOG recently emphasized the high-risk nature of influenza during pregnancy and urged universal vaccination of women who will be pregnant or postpartum during the flu season as “an integral element of prenatal care.”1
ACOG: Give prophylactic antimicrobials before the incision
in cesarean delivery
ACOG committee opinion no. 465: Antimicrobial prophylaxis for cesarean delivery: timing of administration. Obstet Gynecol. 2010;116(3):791–792.
The use of antimicrobial prophylaxis for cesarean delivery is associated with a reduction of 50% or more in the rates of postcesarean infection and severe adverse outcomes, including maternal death. However, there has been some controversy surrounding the question of timing of antimicrobial administration. Should the drugs be administered at the time the cord is clamped or prior to the cesarean skin incision? And, if the latter, just how long before the incision should antimicrobials be given?
ACOG weighed in on this question in September 2010 in a Committee Opinion based on a review of data. It recommended that, whenever feasible, antimicrobials should be administered within 60 minutes before the start of the procedure.
In the past, antimicrobial administration at the time of cord clamping was proposed to reduce fetal exposure and prevent the masking of neonatal infection (falsely negative culture results). However, the data ACOG reviewed from randomized, clinical trials indicate that pre-incision antimicrobials may further reduce the risk of infection (including endometritis and wound infection) without apparent perinatal harm.
First-generation cephalosporins (commonly, 1 g of cefazolin) remain the antibiotic of choice, but the combination of clindamycin and gentamicin was suggested as an acceptable alternative for women who are allergic to penicillin.
ACOG reviewed studies by Thigpen and colleagues and Sullivan and coworkers, as well as a meta-analysis by Costantine and associates.2-4 The relatively small size of these studies and the mixed results of other studies point to the need for further investigation, a fact acknowledged by ACOG. Nevertheless, the committee concluded that “preoperative administration significantly reduces endometritis and total maternal infectious morbidity, compared with administration of antimicrobials after umbilical cord clamping.”
Antimicrobial prophylaxis for surgical-site infection is a well-established practice for both obstetric and gynecologic surgery. Cesarean delivery accounts for one third (more than 1.3 million) of all births in the United States each year and carries a risk of infection at least five times higher than that associated with vaginal delivery. Given the rising incidence of cesarean delivery in the United States and elsewhere, the prevention of postcesarean infection is a priority.
This ACOG Committee Opinion should prompt providers who continue to administer antibiotics after cord clamping to reevaluate the practice.
Even so, given the relative paucity of randomized trials addressing this issue and the mixed results from individual studies, it is essential that we continue to monitor the effectiveness and safety of pre-incision prophylaxis.
Larger follow-up studies to further evaluate resistance profiles and the effects of pre-incision administration on the newborn should be undertaken in concert with implementation of this recommendation.
We want to hear from you! Tell us what you think.
1. ACOG committee opinion#468: Influenza vaccination during pregnancy. Obstet Gynecol. 2010;116(4):1006-1007.
2. Thigpen BD, Hood WA, Chauhan S, et al. Timing of prophylactice antibiotic administration in the uninfected laboring gravida: a randomized clinical trial. Am J Obstet Gynecol. 2005;192(6):1864-1871.
3. Sullivan SA, Smith T, Chang E, Hulsey T, Vandorsten JP, Soper D. Administration of cefazolin prior to skin incision is superior to cefazolin at cord clamping in preventing postcesarean infectious morbidity: a randomized, controlled trial. Am J Obstet Gynecol. 2007;196(5):455.e1-5.
4. Costantine MM, Rahman M, Ghulmiyah L, et al. Timing of perioperative antibiotics for cesarean delivery: a meta-analysis. Am J Obstet Gynecol. 2008;199(3):301.e1-6.
1. ACOG committee opinion#468: Influenza vaccination during pregnancy. Obstet Gynecol. 2010;116(4):1006-1007.
2. Thigpen BD, Hood WA, Chauhan S, et al. Timing of prophylactice antibiotic administration in the uninfected laboring gravida: a randomized clinical trial. Am J Obstet Gynecol. 2005;192(6):1864-1871.
3. Sullivan SA, Smith T, Chang E, Hulsey T, Vandorsten JP, Soper D. Administration of cefazolin prior to skin incision is superior to cefazolin at cord clamping in preventing postcesarean infectious morbidity: a randomized, controlled trial. Am J Obstet Gynecol. 2007;196(5):455.e1-5.
4. Costantine MM, Rahman M, Ghulmiyah L, et al. Timing of perioperative antibiotics for cesarean delivery: a meta-analysis. Am J Obstet Gynecol. 2008;199(3):301.e1-6.
Sexual Pain Disorders in Women
Though common, sexual pain disorders in women are often difficult to identify and treat because of the complexity of potential underlying causes. This article will define and describe these conditions in an effort to provide evaluation and treatment strategies for the primary care provider.
SEXUAL PAIN DISORDERS
Dyspareunia is defined as genital pain that may occur before, during, or after vaginal penetration, thus interfering with sexual intercourse and causing marked personal distress and/or interpersonal difficulty.1 Categorization of the condition as lifelong (primary) or acquired (secondary), generalized or situational, may indicate possible underlying causes. Physiological, psychological, or combined factors may be at play. It should be noted that, according to the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DMS-IV-TR),1 painful penetration is not diagnosed as dyspareunia if it is solely the result of lack of lubrication, the physiological effect of a medication, or attributable to the patient’s general medical condition.
Painful intercourse is a prevalent symptom among sexually active women. Between 8% and 21% of women in the general population and 10% of those ages 57 to 85 have been estimated to experience significant dyspareunia.2,3 For some women, sexual pain leads to avoidance of sexual intercourse or contact. Other women remain sexually active despite persistently painful intercourse.
Transient pain during intercourse is predictable in certain situations, such as times of stress, with frequent intercourse (eg, in attempts to conceive), intercourse after a prolonged hiatus from it, or hymenal rupture at coitarche. Dyspareunia, particularly deep dyspareunia, may occur with midcycle intercourse due to normal local inflammation that occurs with ovulation (mittelschmerz).
A second sexual pain disorder experienced by women is vaginismus, which is defined as recurrent or persistent involuntary contraction or spasm of the musculature of the outer third of the vagina (the perineal and levator muscles) that interferes with vaginal penetration, whether associated with sexual intercourse, speculum insertion, or tampon use.1,4-6 Vaginismus may be a cause or a consequence of dyspareunia. This condition is perhaps best described as pelvic floor motor or muscular instability,7 because it can be characterized by:
1) Hypertonicity (reduced ability to relax)
2) Hypocontractility (reduced ability to contract) or
3) Resting muscular instability (as measured by electromyelography [EMG]).8
According to DSM-IV-TR criteria,1 vaginismus also causes marked personal distress or interpersonal difficulty and is not due exclusively to the direct physiological effects of a general medical condition. Vaginismus may occur as a result of psychological factors, such as fear of penetration, or as a conditioned response to pain.
Dyspareunia and Vaginismus: A Clinical Case
Anna is a 30-year-old woman who has been married for three years. She reports that initially, intercourse with her husband was satisfying and pleasurable. She desired sex and was easily aroused and orgasmic. However, when she began to experience a burning pain during intercourse, she tried at first to ignore it, hoping that it would go away on its own. Anticipating pain, she started to avoid sex. When she finally saw her clinician, she was given a diagnosis of candidiasis and responded well to medication.
Although the physical cause of Anna’s dyspareunia was resolved, she continued to be tense when she anticipated an opportunity for sex, associating it with the prior painful experiences. When the couple did attempt vaginal intercourse, penetration was difficult and painful in a different way; it seemed as if the opening to her vagina was closing up, that it was becoming too tight. Anna felt frustrated with herself and her body and worried that her husband would give up on her.
She returned to her clinician, who referred her to a sex therapist. Anna was anxious and skeptical at first but also highly motivated to resolve the pain and resume her previously satisfying sex life. The therapist explained the connection between Anna’s original experience of dyspareunia and the anxiety/avoidance pattern that had developed, causing her pelvic floor muscles to overcontract involuntarily and to make penetration both difficult and uncomfortable. Even though the original physical source of painful intercourse was resolved, Anna had gone on to develop anticipatory anxiety, triggering secondary vaginismus—an automatic, protective response against anticipated pain.
A brief course of cognitive behavioral sex therapy helped Anna to resolve her anxiety, relax her pelvic floor muscles, and gradually return to satisfying sex.
CAUSES OF DYSPAREUNIA
There is a wide and confusing spectrum of causes of primary and secondary dyspareunia. Primary dyspareunia, the less common condition, is associated with imperforate or microperforate hymen, congenital vulvovaginal abnormalities, and acquired vulvovaginal abnormalities resulting from genital surgery, modification, or infundibulation.9
Secondary dyspareunia, or painful intercourse that occurs after a history of pain-free sex, is most often caused by either vaginal atrophy or vulvodynia. Vaginal atrophy, resulting from low circulating estrogen, may cause symptoms of itching, dryness, and irritation; it is experienced by about 40% of postmenopausal women,10 as well as many premenopausal women who take low-dose estrogen oral contraceptives.11-13
Vulvodynia, which affects 16% of women, refers to vulvar pain, usually described as a burning or cutting sensation, with no significant physical changes seen on examination.7,8,14 Vulvodynia may be localized or generalized to the entire vulva and provoked or unprovoked. Vestibulodynia, which is a subtype of vulvodynia, refers to unexplained pain in the vestibule of the vaginal introitus; the only physical finding may be erythema. Provoked vestibulodynia is the most common form of introital pain, affecting 3% to 15% of women.15-17
Both vulvodynia and vestibulodynia have been explained as neurosensory disorders that are triggered by stimuli such as repeated vaginal infections or long-term use of low-dose estrogen oral contraceptives.12 Other less common vulvar conditions that cause dyspareunia include noninfectious skin conditions, such as dermatitis and lichenoid conditions.18 Many women also experience painful vulvar or vaginal irritation due to sensitivity to spermicide on condoms; latex allergy; and/or ingredients in some lubricants, soaps, body perfumes, detergents, or douches.18,19 Less common vulvovaginal conditions associated with dyspareunia include anomalies, infection, radiation effects, and scarring injuries (see Figure 120).
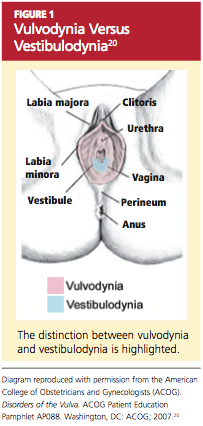
In some cases, dyspareunia is caused by conditions involving the uterus, ovaries, bladder, or rectum. Uterine conditions include myomas, adenomyosis, endometriosis, retroversion, or retroflexion.21,22 In the case of prolapsed uterus, contact with the cervix or uterus may result in what is described by the patient as “an electric shock” or “stabbing” or “shooting” pain.23
Ovarian cysts, intermittent ovarian torsion, and hydrosalpinx can each cause painful intercourse. Associated bladder and urinary tract conditions include urinary tract infection; urethral caruncle or diverticulum; urethral atrophy resulting from low circulating estrogen in women of any age; and interstitial cystitis, which is usually associated with pain after intercourse and/or during the luteal phase of the menstrual cycle.22,24 Anorectal inflammatory conditions, such as lichen sclerosis, Crohn’s disease, and severe constipation with or without hemorrhoids, may be causes of dyspareunia. In rare instances, dyspareunia may be a sign of a tumor in the urogenital or lower GI tract.25,26
Dyspareunia may be indicative of pelvic adhesions resulting from surgery or inflammatory conditions, such as current or past pelvic inflammatory disease.22 The majority (50% to 80%) of women with endometriosis complain of pain with deep thrusting during coitus,27 which is most severe before menstruation and in sexual positions in which the woman has less control over the depth and force of penetration.
Musculoskeletal conditions associated with dyspareunia include scoliosis, levator ani muscle myalgia, and levator ani/pelvic floor musculature spasm.28,29 These may be primary or secondary to another condition that causes deep dyspareunia and may involve pain in the lower back, sacroiliac joint, piriformis, or obturator internus.28
Neurologic conditions, such as pudendal neuralgia and sciatica, can also be associated with pelvic floor injuries during childbirth, nerve entrapment, or straddle injuries.12 Genital piercing and cutting can lead to painful neuromas, neurofibromas, and neuralgia.30
Finally, whether or not identifiable physical causes exist, the experience of pain can be triggered or exacerbated by psychosocial factors. When vaginal penetration has been painful, a pattern of anticipatory anxiety and fear is common. Certain cognitive styles are also associated with dyspareunia. Examples include hypervigilance (“I’m just waiting for it to start hurting”) and catastrophizing (“This hurts so much that I’m going to have to give up having sex altogether”).31,32
Generalized anxiety and somatization, defined as the tendency to experience a variety of physical symptoms without physical origin in reaction to stress, may predispose a woman to experience dyspareunia.1,31 Relationship distress is another related psychosocial factor.33 Even in relationships that are for the most part positive, a woman’s reluctance to communicate her sexual needs may result in foreplay and stimulation that are inadequate to achieve arousal and lubrication.
EVALUATION OF THE PATIENT
Any woman who reports pain during intercourse should undergo a careful history and physical exam directed toward detection of signs of vaginal atrophy (especially women who are menopausal or taking oral estrogen-containing hormonal contraceptives), vulvodynia, vestibulodynia, and other physical and psychological conditions that can cause sexual pain.
A sexual pain history should be taken, focusing on pain experienced with intercourse and/or with tampon or speculum insertion. Pain assessment includes location, intensity, quality (burning, shooting, or dull pain), and duration of the pain. The clinician should ask whether penetrative sex has ever been pain-free to assess for a lifelong or situational condition. Pain frequency, kinds of associated activities, and factors that exacerbate or alleviate the symptom of pain should be clarified. Assessment should include questions about relationship distress; sexual pain can exert negative impact on the relationship, which can then exacerbate sexual pain by impairing arousal. It is important to ascertain whether pain occurs or has occurred with all of the patient’s sexual partners.
The patient should be asked what she identifies as the source of her pain. What treatments have been tried, whether they have been helpful, and what treatments the patient thinks might be effective should all be addressed. The International Pelvic Pain Society34 offers a comprehensive pelvic pain assessment form (see box, “Resources for Clinicians and Patients,” below), which includes the short-form McGill Pain Questionnaire35 for describing and documenting characteristics of pain.
A detailed sexual function history begins with an assessment of sexual function in each domain (desire, arousal, orgasm).36 Other sexual problems, such as vaginal dryness, lack of desire or arousal, inability to experience orgasm, and forced or coerced sex should be identified. The clinician ascertains distress related to the patient’s sexual symptoms (eg, “How bothered are you by the pain you experience with intercourse?”). Information about her partner’s sexual functioning can be relevant, since delayed ejaculation may involve prolonged, ultimately painful intercourse.37 Commonly used objective assessment instruments for female sexual function include the Female Sexual Function Index (FSFI),38 the Sexual Function Questionnaire,39 and the Female Sexual Distress Scale (FSDS).40
A complete evaluation includes relevant medical, surgical, obstetric, and social history, along with a review of systems (see Table 1,34,38-40).

Medication History
A thorough assessment identifies OTC and prescription medications, including contraceptive methods. Long-term use of oral combined contraceptives has been associated with vaginal atrophy and vulvodynia.12 Clarifying whether the patient is using condoms with nonoxynol 9 is important, since this spermicide can cause painful irritation in some women.19 An allergy or sensitivity to latex in condoms or to chemicals in some lubricants can also cause irritation.
Physical Examination
The vulva should be inspected for signs of pallor, loss or thinning of hair, and clitoral shrinkage or shrinkage of the labia minora or majora. These can be signs of atrophy due to low estrogen. The vulva is also examined for skin changes, including inflammation, excoriation, scarring, fissuring, laceration, inability to retract the clitoral hood or complete loss of the clitoris by overlying tissue, and trauma.18 Additional assessment is needed for women with infundibulation in cases of female circumcision (also referred to as genital mutilation), as practiced in parts of the Middle East and Africa.30,41 A water-based lubricant, ideally warmed, can be used at the introitus and on the outer speculum blades to minimize discomfort during the exam without interfering with cytology, if a Pap smear is also being performed.42
For women with introital pain, a “cotton swab test” is performed to quantify, localize, and map the pain (see Table 2,28,43).

The hymen is inspected and its morphology documented, particularly in the woman or girl with recent coitarche. The vagina is inspected for presence of a septum.14 Signs of atrophy include lack of lubrication, easy bleeding, fissures, discomfort even with use of a small speculum, and petechiae.18 Atrophic mucosa will appear flat and pale, with no evidence of normal rugae. The tissue’s elasticity will have decreased.
If there is suspicion of pelvic organ prolapse, vaginitis, or sexually transmitted infection, further assessments are made. Using one intravaginal finger if two fingers are painful, the clinician systematically palpates the urethra, bladder, bilateral fornices, posterior fornix, cervix, adnexa, and uterus in an attempt to reproduce the pain experienced during intercourse.29 Uterine size, contour, mobility, and any nodularity or irregularity are all noted.
During the pelvic examination, the clinician uses a hand mirror to show the patient the depth of penetration with the examining finger, a speculum, or a vaginal dilator—demonstrating the diameter of the introitus and vaginal canal, if possible. This is a valuable opportunity to educate the patient regarding her genital anatomy in addition to any physical findings that might explain her pain.
A rectovaginal examination is important for identifying constipation, hemorrhoids, or nodules along the rectovaginal septum or uterosacral ligaments, possibly indicating endometriosis.21,22 In patients with anal fissures, tensing of the anal sphincter and pelvic floor muscles to slow defecation (or to restrict the diameter of the feces) can result in a paradoxical contraction of the puborectalis portion of the levator ani muscles.28 This creates a vicious cycle of pelvic floor muscle spasm.
Pelvic floor function and strength are systematically evaluated using the Modified Oxford Scale44 or the Brink Scoring System.45 Scoliosis and hip height are assessed by asking the patient to bend over and touch her toes. Curvature of the spine or unequal hip height may indicate musculoskeletal problems that can cause or contribute to pelvic pain during intercourse.
Laboratory Workup
The vaginal maturation index18,46 may be assessed by taking a specimen for wet prep in the office setting or by sending a fixed smear on a slide or in a liquid cytologic preparation to a cytopathology laboratory. Vaginal pH, in combination with a wet prep, can be obtained to help assess for bacterial vaginosis or vaginal candidiasis. Laboratory tests are not regularly required, however, to diagnose and treat dyspareunia. Nevertheless, dyspareunia of musculoskeletal origin may require diagnostic neuromuscular evaluation, including EMG, ultrasonography, and manual assessment.28 These are commonly performed by a physical therapist with expertise in pelvic physical therapy and women’s health.
TREATMENT/MANAGEMENT
Treatment begins with psychosexual education and counseling, in which underlying causes are addressed. The clinician educates the patient about the prevalence and common causes of painful intercourse, assessing what the patient has already done to alleviate the pain and reinforcing constructive and positive behaviors. It is therapeutic to validate the patient’s experience of pain and to explain that both pain and sexuality are mind and body processes. Even if the pain is due to a physical cause, it can have psychologic aspects or sequelae. The clinician normalizes the fact that pain associated with sex can cause anxiety and fear about participating in intercourse and that these are natural, instinctive reactions to pain.47
The patient is given an explanation of any physical and psychologic findings that might be contributing to her pain. This approach gives the patient hope for improvement with education and treatment. The emphasis is on the aspects of her sexual functioning that are still intact and positive features in her sexual history or relationship. A central goal is to minimize self-blame, hopelessness, and anxiety.
The patient is counseled about the need for adequate foreplay and stimulation to promote arousal and lubrication. Masturbation (alone and/or in conjunction with partnered sex) can be discussed as a way to increase vaginal lubrication, blood flow, and comfort with genital touch.
Another helpful strategy is to give the patient permission to take a hiatus from sexual intercourse.47 Women experiencing genital pain often avoid nonsexual affection and other sexual activities with their partners, fearing that they will lead to painful intercourse. In such cases, the woman is encouraged to form an explicit agreement with her partner that they will not have intercourse until she has made progress at managing her pain. The goal is to create greater openness to nonsexual affection and nonpenetrative sexual activity.
The clinician may suggest sensate focus exercises48,49 (see Table 3,4,48,49). In cases of vaginal atrophy and/or dryness, vaginal moisturizers and lubricants can help. Products containing perfumes, warming or tingling agents, parabens, and other chemicals can be irritating in some women.18

For the premenopausal patient using a low-dose oral contraceptive, the clinician’s instinct may be to provide a higher-dose estrogen pill. This may actually be counterproductive, however, because oral estrogen promotes synthesis in the liver of sex hormone–binding globulin, which then reduces circulating estrogen and can exacerbate the problem.50 Although the vast majority of women who use estrogen-containing oral contraceptives do not appear to develop symptomatic vulvovaginal atrophy, women who do experience bothersome atrophic symptoms may benefit from switching to an alternative contraceptive method (eg, an intrauterine device). Menopausal women may benefit from a topical estrogen product in the form of a vaginal ring, cream, or tablet.18,51,52
For vulvodynia, medical therapies such as topical anesthetics and centrally acting medications are under investigation. For cases of provoked vestibulodynia that do not respond to first-line treatment, patients may benefit from pelvic floor physical therapy, cognitive behavioral therapy aimed at pain reduction combined with sex therapy, or vestibulectomy.53,54
Addressing Bowel and Bladder Concerns
The patient with chronic constipation must be educated about the importance of adequate hydration and nutrition for bowel regularity. She is counseled about avoidance of straining or prolonged sitting during defecation and is offered primary treatment for hemorrhoids. A history of rectal bleeding, particularly in the absence of hemorrhoids, may be indicative of an anorectal or a colon malignancy, and the patient with such a history should be referred for further evaluation. The International Foundation for Functional Gastric Disorders (see “Resources” box) provides useful information for patients regarding constipation and associated pelvic floor disorders.
Urinary voiding prior to and following coitus or oral sex may reduce bladder discomfort and infection. Use of a dental dam for oral sex and good oral hygiene with regular dental care may also reduce the incidence of irritation and infection. Educational resources to address bladder function and incontinence can be found in the “Resources” box.
Treatment for Vaginismus
Vaginismus can be treated with desensitization techniques, including relaxation training, dilator therapy, pelvic floor therapy, and cognitive behavioral sex therapy, which teaches the patient to relax the introital muscles and give her the experience of controlled, pain-free penetration.55,56 A fundamental technique of relaxation training is deep breathing. Understanding the mechanics of breathing is important for physiologic quieting of the autonomic nervous system and facilitating relaxation of the pelvic floor.57
Dilator therapy can be used for treatment of vaginismus and vaginal stenosis, although in the presence of adhesions or a septum, lysis may be required first. A vaginal dilator can be inserted for approximately 15 minutes daily. Once the dilator passes easily with no painful stretching sensation, the patient should move to the next size larger dilator. The dilator can also be moved slowly and gently in and out and from side to side.
Topical lidocaine (5%) gel or ointment can facilitate use of the dilator. The patient applies a small amount to the site of vulvar pain (commonly the posterior fourchette of the introitus) with the tip of a cotton swab 15 to 30 minutes before inserting the dilator.
Some vaginal dilators may be obtained directly by the patient (see “Resources” box below), while others require a prescription. Graduated or vibrating dildos can be obtained at retail outlets selling sexual products. A clean finger can also be used for dilation.
Pelvic Floor Therapy
Several modalities offered by specialized physical therapists are used to improve pelvic and vulvovaginal blood flow and control over the pelvic musculature. This may help to alleviate pain with intercourse, particularly in patients with signs of pelvic floor weakness, poor muscle control, or instability. Modalities include pelvic floor exercises and manual therapy techniques (eg, transvaginal trigger point therapy, transvaginal and/or transrectal massage, dry needling of a trigger point).4,58 Surface electrical stimulation of the pelvic floor musculature is also used to decrease pain and muscle spasm.59
Kegel exercises60 (ie, tensing and relaxing the pelvic floor muscles) may help a patient gain control over both contraction and relaxation of the pelvic muscles.60,61 However, if palpation of the pelvic floor reveals tight, inelastic, and dense tissue, assessment and grading of muscle strength may be misleading. For example, the pelvic floor may demonstrate a state of constant contraction (hypertonicity or a “short pelvic floor”).62 Attempts at voluntary contraction of a muscle in this state may be perceived and graded incorrectly as weak.
Traditional Kegel exercises are contraindicated in women with muscle hypertonicity and an inability to relax these muscles. Some practitioners use the term “reverse Kegel” to describe active, bearing-down exercises that increase pelvic floor relaxation. Biofeedback training (surface EMG and/or rehabilitative ultrasound imaging) can be useful to reeducate resting muscle tone of the pelvic floor. Breathing, tactile, verbal, and imagery cues are essential.61,62
Cognitive Behavioral Sex Therapy
This specialized form of psychotherapy helps patients identify cognitive, emotional, and relationship factors that contribute to their pain. Patients learn coping strategies, including relaxation techniques and modification of thoughts, feelings, and behaviors to reduce anxiety, tension, and pain. As pain management improves, the focus shifts to enhancing sexual functioning, including restarting the sexual relationship if sex has been avoided because of pain.63
WHEN AND WHERE TO REFER
Referral is appropriate if the patient’s condition worsens, is unresponsive to therapy, or requires specialized evaluation and treatment. Sex therapy, psychotherapy, or couples counseling may be indicated in more complex cases in which a sexual disorder and significant psychologic and/or relationship problems coexist. Examples may include an unresolved history of sexual abuse, clinical anxiety or depression, sexual phobia or aversion, and general relationship distress. To locate qualified sex therapists, see the “Resources” box.
Physical therapy may be prescribed if the patient demonstrates or reports persistent pain or lack of improvement with initial pelvic floor therapy, an inability to use a dilator on her own, or increased pain or perceived tightness while using a dilator; or if she develops pelvic or perineal pain at rest. To locate specially trained physical therapists, see the “Resources” box.
CONCLUSION
Though commonly seen in primary care, sexual pain disorders in women are often difficult to diagnose and treat because of the confusing array of possible contributory factors. This article presents an overview of possible presentations, causes, diagnostic strategies, and treatment options, integrating evidence-based approaches from the fields of medicine, psychology, and physical therapy.
REFERENCES
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DMS-IV-TR). Washington, DC: American Psychiatric Association; 2000:554-558.
2. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8): 762-774.
3. Latthe P, Latthe M, Say L, et al. WHO systematic review of prevalence of chronic pelvic pain: a neglected reproductive health morbidity. BMC Public Health. 2006;6:177-183.
4. Simons DG, Travell JG, Simons LS, Cummings BD. Myofascial Pain and Dysfunction: The Trigger Point Manual. 2nd ed. Baltimore, MD: Lippincott Williams & Wilkins; 1999:vol 2, chap 6:110-131.
5. Perry CP. Vulvodynia. In: Howard FM, Perry CP, Carter JE, El-Minawi AM, eds. Pelvic Pain: Diagnosis and Management. Philadelphia, PA: Lippincott Williams & Wilkins, 2000:204-211.
6. Glazer HI, Jantos M, Hartmann EH, Swencioniswom C. Electromyographic comparisons of the pelvic floor in women with dysesthetic vulvodynia and asymptomatic women. J Reprod Med. 1998;43(11):959-962.
7. Binik YM. The DSM diagnostic criteria for vaginismus. Arch Sex Behav. 2010;39(2):278-291.
8. Haefner HK. Report of the International Society for the Study of Vulvovaginal Disease terminology and classification of vulvodynia. J Low Genit Tract Dis. 2007;11(1):48-49.
9. Huffman JW. Dyspareunia of vulvo-vaginal origin: causes and management. Postgrad Med. 1983;73(2):287-296.
10. Krychman ML. Vaginal estrogens for treatment of dyspareunia. J Sex Med. 2010 Nov 22;8:666-674. [Epub ahead of print]
11. Kao A, Binik YM, Kapuscinski A, Khalifé S. Dyspareunia in postmenopausal women: a critical review. Pain Res Manag. 2008;13(3):243-254.
12. Steege JF, Zolnoun DA. Evaluation and treatment of dyspareunia. Obstet Gynecol. 2009;113 (5):1124-1136.
13. Lüdicke F, Johannisson E, Helmerhorst FM, et al. Effect of a combined oral contraceptive containing 3 mg of drospirenone and 30 microg of ethinyl estradiol on the human endometrium. Fertil Steril. 2001;76(1):102-107.
14. Strauhal MJ, Frahm J, Morrison P, et al. Vulvar pain: a comprehensive review. J Womens Health Phys Ther. 2007;31(3):6-22.
15. Harlow BL, Stewart EG. A population-based assessment of chronic unexplained vulvar pain: have we underestimated the prevalence of vulvodynia? J Am Med Womens Assoc. 2003;58(2): 82-88.
16. Goetsch MF. Vulvar vestibulitis: prevalence and historic features in a general gynecologic practice population. Am J Obstetr Gynecol. 1991; 164(6 pt 1):1609-1616.
17. Danielsson I, Sjöberg I, Stenlund H, Wikman M. Prevalence and incidence of prolonged and severe dyspareunia in women: results from a population study. Scand J Public Health. 2003;31 (2):113-118.
18. Mac Bride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc. 2010;85(1): 87-94.
19. Barditch-Crovo P, Witter F, Hamzeh F, et al. Quantitation of vaginally administered nonoxynol-9 in premenopausal women. Contraception. 1997;55(4):261-263.
20. American College of Obstetricians and Gynecologists. Disorders of the Vulva. ACOG Patient Education Pamphlet AP088. Washington, DC: American College of Obstetricians and Gynecologists; 2007.
21. Ferrero S, Abbamonte LH, Giordano M, et al. Uterine myomas, dyspareunia, and sexual function. Fertil Steril. 2006;86(5):1504-1510.
22. Walid MS, Heaton RL. Dyspareunia: a complex problem requiring a selective approach. Sex Health. 2009;6(3):250-253.
23. Sobghol SS, Alizadeli Charndabee SM. Rate and related factors of dyspareunia in reproductive age women: a cross-sectional study. Int J Impot Res. 2007;19(1):88-94.
24. Ferrero S, Ragni N, Remorgida V. Deep dyspareunia: causes, treatments, and results. Curr Opin Obstet Gynecol. 2008;20(4):394-399.
25. Kim IY, Sadeghi F, Slawin KM. Dyspareunia: an unusual presentation of leiomyoma of the bladder. Rev Urol. 2001;3(3):152-154.
26. Akbulut S, Cakabay B, Sezgin A, Ozmen C. A rare cause of severe dyspareunia: a case report and literature review. Arch Gynecol Obstet. 2009 Apr 26. [Epub ahead of print.]
27. Ferrero S, Esposito F, Abbamonte LH, et al. Quality of sex life in women with endometriosis and deep dyspareunia. Fertil Steril. 2005;83(3): 573-579.
28. Fisher KA. Management of dyspareunia and associated levator ani muscle overactivity. Phys Ther. 2007;87(7):935-941.
29. Bø K, Sherburn M. Evaluation of female pelvic-floor muscle function and strength. Phys Ther. 2005;85(3):269-282.
30. Royal College of Obstetricians and Gynaecologists. Female Genital Mutilation and its Management. Green-top Guideline No 53 (May 2009). www.rcog.org.uk/files/rcog-corp/Green Top53FemaleGenitalMutilation.pdf. Accessed April 4, 2011.
31. Payne KA, Binik YM, Amsel R, Khalifé S. When sex hurts, anxiety and fear orient attention towards pain. Eur J Pain. 2005;9(4):427-436.
32. Pukall CF, Binik YM, Khalifé S, et al. Vestibular tactile and pain thresholds in women with vulvar vestibulitis syndrome. Pain. 2002;96(1-2):163-175.
33. Meana M, Binik YM, Khalifé S, Cohen D. Psychosocial correlates of pain attributions in women with dyspareunia. Psychosomatics. 1999:40(6): 497-502.
34. Pelvic Pain Assessment Form (2008). International Pelvic Pain Society. www.pelvicpain.org/pdf/History_and_Physical_Form/IPPS-H&PformR-MSW.pdf. Accessed April 11, 2011.
35. Melzack R. The short-form McGill Pain Questionnaire. Pain. 1987;30(2):191-197.
36. Basson R. Sexuality and sexual disorders in women. In: Clinical Updates in Women’s Health Care Monograph. Washington, DC: American College of Obstetricians and Gynecologists. 2003;2(2):1-94.
37. Oberg K, Sjögren Fugl-Meyer K. On Swedish women’s distressing sexual dysfunctions: some concomitant conditions and life satisfaction. J Sex Med. 2005;2(2):169-180.
38. Rosen R, Brown C, Heiman J, et al. The Female Sexual Function Index (FSFI): a multidimensional self-report instrument for the assessment of female sexual function. J Sex Marital Ther. 2000;26(2):191-208.
39. Quirk FH, Heiman JR, Rosen RC, et al. Development of a sexual function questionnaire for clinical trials of female sexual dysfunction. J Womens Health Gend Based Med. 2002;11(3): 277-289.
40. Derogatis LR, Rosen R, Leiblum S, et al. The Female Sexual Distress Scale (FSDS): Initial validation of a standardized scale for assessment of sexually related personal distress in women. J Sex Marital Ther. 2002;28(4):317-330.
41. van Lankveld JJ, Granot M, Weijmar Schultz WC, et al. Women’s sexual pain disorders. J Sex Med. 2010;7(1 pt 2):615-631.
42. Hathaway JK, Pathak PK, Maney R. Is liquid-based Pap testing affected by water-based lubricant? Obstet Gynecol. 2006;107(1):66-70.
43. Bergeron S, Binik YM, Khalifé S, et al. Vulvar vestibulitis syndrome: reliability of diagnosis and evaluation of current diagnostic criteria. Obstet Gynecol. 2001;98(1):45-51.
44. Laycock J, Holmes DM. The place of physiotherapy in the management of pelvic floor dysfunction. Obstet Gynaecol. 2003;5:194-199.
45. Brink CA, Sampselle CM, Wells TJ, et al. A digital test for pelvic muscle strength in older women with urinary incontinence. Nurs Res. 1989;38(4):196-199.
46. Hess R, Austin RM, Dillon S, et al. Vaginal maturation index self-sample collection in mid-life women: acceptability and correlation with physician-collected samples. Menopause. 2008; 15(4 pt 1):726-729.
47. Haefner HK, Collins ME, Davis GD, et al. The vulvodynia guideline. J Low Genit Tract Dis. 2005; 9(1):40-51.
48. Albaugh JA, Kellogg-Spadt S. Sensate focus and its role in treating sexual dysfunction. Urol Nurs. 2002;22(6):402-403.
49. Masters WH, Johnson VE. Human Sexual Inadequacy. New York, NY: Ishi Press; 2010: 67-75.
50. Panzer C, Wise S, Fantini G, et al. Impact of oral contraceptives on sex hormone–binding globulin and androgen levels: a retrospective study in women with sexual dysfunction. J Sex Med. 2006;3(1):104-113.
51. Suckling J, Kennedy R, Lethaby A. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev. 2006; (4):CD001500.
52. Johnston SL, Farrell SA, Bouchard C, et al; SOGC joint Committee–Clinical Practice Gynaecology and Urogynaecology. The detection and management of vaginal atrophy. J Obstet Gynaecol Can. 2004;26(5):503-515.
53. Bergeron S, Khalifé S, Glazer HI, Binik YM. Surgical and behavioral treatments for vestibulodynia: two-and-one-half year follow-up and predictors of outcome. Obstet Gynecol. 2008;111(1): 159-166.
54. Pukall CF, Smith KB, Chamberlain SM. Provoked vestibulodynia. Womens Health (Lond Engl). 2007;3(5):583-592.
55. van Lankveld JJ, ter Kuile MM, de Groot HE, et al. Cognitive-behavioral therapy for women with lifelong vaginismus: a randomized waiting-list controlled trial of efficacy. J Consult Clin Psychol. 2006;74(1):168-178.
56. McGuire H, Hawton K. Interventions for vaginismus. Cochrane Database Syst Rev. 2003;(1): CD001760.
57. Calais-Germain B. Analysis of the principal types of breathing. In: Calais-Germain B. Anatomy of Breathing. New York, NY: Eastland Press; 2006:133-158.
58. Carter J. Abdominal wall and pelvic myofascial trigger points. In: Howard FM, Perry P, Carter J, El-Minawi AM. Pelvic Pain: Diagnosis and Management. Baltimore, MD: Lippincott Williams & Wilkins; 2000:315-358.
59. Gentilcore-Saulnier E, McLean L, Goldfinger C, et al. Pelvic floor muscle assessment outcomes in women with and without provoked vestibulodynia and the impact of a physical therapy program. J Sex Med. 2010;7(2 pt 2):1003-1022.
60. Kegel AH. Physiologic therapy for urinary stress incontinence. JAMA. 1951;146(10):915-917.
61. Marques A, Stothers L, Macnab A. The status of pelvic floor muscle training for women. Can Urol Assoc J. 2010;4(6):419-424.
62. FitzGerald MP, Kotarinos R. Rehabilitation of the short pelvic floor. I: Background and patient evaluation. Int Urogynecol J Pelvic Floor Dysfunct. 2003;14(4):261-268.
63. Binik Y, Bergeron S, Khalifé S. Dyspareunia and vaginismus: So-called pain disorders. In: Leiblum S, ed. Principles and Practice of Sex Therapy. 3rd ed. New York: Guilford Press; 2007: 124-156.
Though common, sexual pain disorders in women are often difficult to identify and treat because of the complexity of potential underlying causes. This article will define and describe these conditions in an effort to provide evaluation and treatment strategies for the primary care provider.
SEXUAL PAIN DISORDERS
Dyspareunia is defined as genital pain that may occur before, during, or after vaginal penetration, thus interfering with sexual intercourse and causing marked personal distress and/or interpersonal difficulty.1 Categorization of the condition as lifelong (primary) or acquired (secondary), generalized or situational, may indicate possible underlying causes. Physiological, psychological, or combined factors may be at play. It should be noted that, according to the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DMS-IV-TR),1 painful penetration is not diagnosed as dyspareunia if it is solely the result of lack of lubrication, the physiological effect of a medication, or attributable to the patient’s general medical condition.
Painful intercourse is a prevalent symptom among sexually active women. Between 8% and 21% of women in the general population and 10% of those ages 57 to 85 have been estimated to experience significant dyspareunia.2,3 For some women, sexual pain leads to avoidance of sexual intercourse or contact. Other women remain sexually active despite persistently painful intercourse.
Transient pain during intercourse is predictable in certain situations, such as times of stress, with frequent intercourse (eg, in attempts to conceive), intercourse after a prolonged hiatus from it, or hymenal rupture at coitarche. Dyspareunia, particularly deep dyspareunia, may occur with midcycle intercourse due to normal local inflammation that occurs with ovulation (mittelschmerz).
A second sexual pain disorder experienced by women is vaginismus, which is defined as recurrent or persistent involuntary contraction or spasm of the musculature of the outer third of the vagina (the perineal and levator muscles) that interferes with vaginal penetration, whether associated with sexual intercourse, speculum insertion, or tampon use.1,4-6 Vaginismus may be a cause or a consequence of dyspareunia. This condition is perhaps best described as pelvic floor motor or muscular instability,7 because it can be characterized by:
1) Hypertonicity (reduced ability to relax)
2) Hypocontractility (reduced ability to contract) or
3) Resting muscular instability (as measured by electromyelography [EMG]).8
According to DSM-IV-TR criteria,1 vaginismus also causes marked personal distress or interpersonal difficulty and is not due exclusively to the direct physiological effects of a general medical condition. Vaginismus may occur as a result of psychological factors, such as fear of penetration, or as a conditioned response to pain.
Dyspareunia and Vaginismus: A Clinical Case
Anna is a 30-year-old woman who has been married for three years. She reports that initially, intercourse with her husband was satisfying and pleasurable. She desired sex and was easily aroused and orgasmic. However, when she began to experience a burning pain during intercourse, she tried at first to ignore it, hoping that it would go away on its own. Anticipating pain, she started to avoid sex. When she finally saw her clinician, she was given a diagnosis of candidiasis and responded well to medication.
Although the physical cause of Anna’s dyspareunia was resolved, she continued to be tense when she anticipated an opportunity for sex, associating it with the prior painful experiences. When the couple did attempt vaginal intercourse, penetration was difficult and painful in a different way; it seemed as if the opening to her vagina was closing up, that it was becoming too tight. Anna felt frustrated with herself and her body and worried that her husband would give up on her.
She returned to her clinician, who referred her to a sex therapist. Anna was anxious and skeptical at first but also highly motivated to resolve the pain and resume her previously satisfying sex life. The therapist explained the connection between Anna’s original experience of dyspareunia and the anxiety/avoidance pattern that had developed, causing her pelvic floor muscles to overcontract involuntarily and to make penetration both difficult and uncomfortable. Even though the original physical source of painful intercourse was resolved, Anna had gone on to develop anticipatory anxiety, triggering secondary vaginismus—an automatic, protective response against anticipated pain.
A brief course of cognitive behavioral sex therapy helped Anna to resolve her anxiety, relax her pelvic floor muscles, and gradually return to satisfying sex.
CAUSES OF DYSPAREUNIA
There is a wide and confusing spectrum of causes of primary and secondary dyspareunia. Primary dyspareunia, the less common condition, is associated with imperforate or microperforate hymen, congenital vulvovaginal abnormalities, and acquired vulvovaginal abnormalities resulting from genital surgery, modification, or infundibulation.9
Secondary dyspareunia, or painful intercourse that occurs after a history of pain-free sex, is most often caused by either vaginal atrophy or vulvodynia. Vaginal atrophy, resulting from low circulating estrogen, may cause symptoms of itching, dryness, and irritation; it is experienced by about 40% of postmenopausal women,10 as well as many premenopausal women who take low-dose estrogen oral contraceptives.11-13
Vulvodynia, which affects 16% of women, refers to vulvar pain, usually described as a burning or cutting sensation, with no significant physical changes seen on examination.7,8,14 Vulvodynia may be localized or generalized to the entire vulva and provoked or unprovoked. Vestibulodynia, which is a subtype of vulvodynia, refers to unexplained pain in the vestibule of the vaginal introitus; the only physical finding may be erythema. Provoked vestibulodynia is the most common form of introital pain, affecting 3% to 15% of women.15-17
Both vulvodynia and vestibulodynia have been explained as neurosensory disorders that are triggered by stimuli such as repeated vaginal infections or long-term use of low-dose estrogen oral contraceptives.12 Other less common vulvar conditions that cause dyspareunia include noninfectious skin conditions, such as dermatitis and lichenoid conditions.18 Many women also experience painful vulvar or vaginal irritation due to sensitivity to spermicide on condoms; latex allergy; and/or ingredients in some lubricants, soaps, body perfumes, detergents, or douches.18,19 Less common vulvovaginal conditions associated with dyspareunia include anomalies, infection, radiation effects, and scarring injuries (see Figure 120).
In some cases, dyspareunia is caused by conditions involving the uterus, ovaries, bladder, or rectum. Uterine conditions include myomas, adenomyosis, endometriosis, retroversion, or retroflexion.21,22 In the case of prolapsed uterus, contact with the cervix or uterus may result in what is described by the patient as “an electric shock” or “stabbing” or “shooting” pain.23
Ovarian cysts, intermittent ovarian torsion, and hydrosalpinx can each cause painful intercourse. Associated bladder and urinary tract conditions include urinary tract infection; urethral caruncle or diverticulum; urethral atrophy resulting from low circulating estrogen in women of any age; and interstitial cystitis, which is usually associated with pain after intercourse and/or during the luteal phase of the menstrual cycle.22,24 Anorectal inflammatory conditions, such as lichen sclerosis, Crohn’s disease, and severe constipation with or without hemorrhoids, may be causes of dyspareunia. In rare instances, dyspareunia may be a sign of a tumor in the urogenital or lower GI tract.25,26
Dyspareunia may be indicative of pelvic adhesions resulting from surgery or inflammatory conditions, such as current or past pelvic inflammatory disease.22 The majority (50% to 80%) of women with endometriosis complain of pain with deep thrusting during coitus,27 which is most severe before menstruation and in sexual positions in which the woman has less control over the depth and force of penetration.
Musculoskeletal conditions associated with dyspareunia include scoliosis, levator ani muscle myalgia, and levator ani/pelvic floor musculature spasm.28,29 These may be primary or secondary to another condition that causes deep dyspareunia and may involve pain in the lower back, sacroiliac joint, piriformis, or obturator internus.28
Neurologic conditions, such as pudendal neuralgia and sciatica, can also be associated with pelvic floor injuries during childbirth, nerve entrapment, or straddle injuries.12 Genital piercing and cutting can lead to painful neuromas, neurofibromas, and neuralgia.30
Finally, whether or not identifiable physical causes exist, the experience of pain can be triggered or exacerbated by psychosocial factors. When vaginal penetration has been painful, a pattern of anticipatory anxiety and fear is common. Certain cognitive styles are also associated with dyspareunia. Examples include hypervigilance (“I’m just waiting for it to start hurting”) and catastrophizing (“This hurts so much that I’m going to have to give up having sex altogether”).31,32
Generalized anxiety and somatization, defined as the tendency to experience a variety of physical symptoms without physical origin in reaction to stress, may predispose a woman to experience dyspareunia.1,31 Relationship distress is another related psychosocial factor.33 Even in relationships that are for the most part positive, a woman’s reluctance to communicate her sexual needs may result in foreplay and stimulation that are inadequate to achieve arousal and lubrication.
EVALUATION OF THE PATIENT
Any woman who reports pain during intercourse should undergo a careful history and physical exam directed toward detection of signs of vaginal atrophy (especially women who are menopausal or taking oral estrogen-containing hormonal contraceptives), vulvodynia, vestibulodynia, and other physical and psychological conditions that can cause sexual pain.
A sexual pain history should be taken, focusing on pain experienced with intercourse and/or with tampon or speculum insertion. Pain assessment includes location, intensity, quality (burning, shooting, or dull pain), and duration of the pain. The clinician should ask whether penetrative sex has ever been pain-free to assess for a lifelong or situational condition. Pain frequency, kinds of associated activities, and factors that exacerbate or alleviate the symptom of pain should be clarified. Assessment should include questions about relationship distress; sexual pain can exert negative impact on the relationship, which can then exacerbate sexual pain by impairing arousal. It is important to ascertain whether pain occurs or has occurred with all of the patient’s sexual partners.
The patient should be asked what she identifies as the source of her pain. What treatments have been tried, whether they have been helpful, and what treatments the patient thinks might be effective should all be addressed. The International Pelvic Pain Society34 offers a comprehensive pelvic pain assessment form (see box, “Resources for Clinicians and Patients,” below), which includes the short-form McGill Pain Questionnaire35 for describing and documenting characteristics of pain.
A detailed sexual function history begins with an assessment of sexual function in each domain (desire, arousal, orgasm).36 Other sexual problems, such as vaginal dryness, lack of desire or arousal, inability to experience orgasm, and forced or coerced sex should be identified. The clinician ascertains distress related to the patient’s sexual symptoms (eg, “How bothered are you by the pain you experience with intercourse?”). Information about her partner’s sexual functioning can be relevant, since delayed ejaculation may involve prolonged, ultimately painful intercourse.37 Commonly used objective assessment instruments for female sexual function include the Female Sexual Function Index (FSFI),38 the Sexual Function Questionnaire,39 and the Female Sexual Distress Scale (FSDS).40
A complete evaluation includes relevant medical, surgical, obstetric, and social history, along with a review of systems (see Table 1,34,38-40).
Medication History
A thorough assessment identifies OTC and prescription medications, including contraceptive methods. Long-term use of oral combined contraceptives has been associated with vaginal atrophy and vulvodynia.12 Clarifying whether the patient is using condoms with nonoxynol 9 is important, since this spermicide can cause painful irritation in some women.19 An allergy or sensitivity to latex in condoms or to chemicals in some lubricants can also cause irritation.
Physical Examination
The vulva should be inspected for signs of pallor, loss or thinning of hair, and clitoral shrinkage or shrinkage of the labia minora or majora. These can be signs of atrophy due to low estrogen. The vulva is also examined for skin changes, including inflammation, excoriation, scarring, fissuring, laceration, inability to retract the clitoral hood or complete loss of the clitoris by overlying tissue, and trauma.18 Additional assessment is needed for women with infundibulation in cases of female circumcision (also referred to as genital mutilation), as practiced in parts of the Middle East and Africa.30,41 A water-based lubricant, ideally warmed, can be used at the introitus and on the outer speculum blades to minimize discomfort during the exam without interfering with cytology, if a Pap smear is also being performed.42
For women with introital pain, a “cotton swab test” is performed to quantify, localize, and map the pain (see Table 2,28,43).
The hymen is inspected and its morphology documented, particularly in the woman or girl with recent coitarche. The vagina is inspected for presence of a septum.14 Signs of atrophy include lack of lubrication, easy bleeding, fissures, discomfort even with use of a small speculum, and petechiae.18 Atrophic mucosa will appear flat and pale, with no evidence of normal rugae. The tissue’s elasticity will have decreased.
If there is suspicion of pelvic organ prolapse, vaginitis, or sexually transmitted infection, further assessments are made. Using one intravaginal finger if two fingers are painful, the clinician systematically palpates the urethra, bladder, bilateral fornices, posterior fornix, cervix, adnexa, and uterus in an attempt to reproduce the pain experienced during intercourse.29 Uterine size, contour, mobility, and any nodularity or irregularity are all noted.
During the pelvic examination, the clinician uses a hand mirror to show the patient the depth of penetration with the examining finger, a speculum, or a vaginal dilator—demonstrating the diameter of the introitus and vaginal canal, if possible. This is a valuable opportunity to educate the patient regarding her genital anatomy in addition to any physical findings that might explain her pain.
A rectovaginal examination is important for identifying constipation, hemorrhoids, or nodules along the rectovaginal septum or uterosacral ligaments, possibly indicating endometriosis.21,22 In patients with anal fissures, tensing of the anal sphincter and pelvic floor muscles to slow defecation (or to restrict the diameter of the feces) can result in a paradoxical contraction of the puborectalis portion of the levator ani muscles.28 This creates a vicious cycle of pelvic floor muscle spasm.
Pelvic floor function and strength are systematically evaluated using the Modified Oxford Scale44 or the Brink Scoring System.45 Scoliosis and hip height are assessed by asking the patient to bend over and touch her toes. Curvature of the spine or unequal hip height may indicate musculoskeletal problems that can cause or contribute to pelvic pain during intercourse.
Laboratory Workup
The vaginal maturation index18,46 may be assessed by taking a specimen for wet prep in the office setting or by sending a fixed smear on a slide or in a liquid cytologic preparation to a cytopathology laboratory. Vaginal pH, in combination with a wet prep, can be obtained to help assess for bacterial vaginosis or vaginal candidiasis. Laboratory tests are not regularly required, however, to diagnose and treat dyspareunia. Nevertheless, dyspareunia of musculoskeletal origin may require diagnostic neuromuscular evaluation, including EMG, ultrasonography, and manual assessment.28 These are commonly performed by a physical therapist with expertise in pelvic physical therapy and women’s health.
TREATMENT/MANAGEMENT
Treatment begins with psychosexual education and counseling, in which underlying causes are addressed. The clinician educates the patient about the prevalence and common causes of painful intercourse, assessing what the patient has already done to alleviate the pain and reinforcing constructive and positive behaviors. It is therapeutic to validate the patient’s experience of pain and to explain that both pain and sexuality are mind and body processes. Even if the pain is due to a physical cause, it can have psychologic aspects or sequelae. The clinician normalizes the fact that pain associated with sex can cause anxiety and fear about participating in intercourse and that these are natural, instinctive reactions to pain.47
The patient is given an explanation of any physical and psychologic findings that might be contributing to her pain. This approach gives the patient hope for improvement with education and treatment. The emphasis is on the aspects of her sexual functioning that are still intact and positive features in her sexual history or relationship. A central goal is to minimize self-blame, hopelessness, and anxiety.
The patient is counseled about the need for adequate foreplay and stimulation to promote arousal and lubrication. Masturbation (alone and/or in conjunction with partnered sex) can be discussed as a way to increase vaginal lubrication, blood flow, and comfort with genital touch.
Another helpful strategy is to give the patient permission to take a hiatus from sexual intercourse.47 Women experiencing genital pain often avoid nonsexual affection and other sexual activities with their partners, fearing that they will lead to painful intercourse. In such cases, the woman is encouraged to form an explicit agreement with her partner that they will not have intercourse until she has made progress at managing her pain. The goal is to create greater openness to nonsexual affection and nonpenetrative sexual activity.
The clinician may suggest sensate focus exercises48,49 (see Table 3,4,48,49). In cases of vaginal atrophy and/or dryness, vaginal moisturizers and lubricants can help. Products containing perfumes, warming or tingling agents, parabens, and other chemicals can be irritating in some women.18
For the premenopausal patient using a low-dose oral contraceptive, the clinician’s instinct may be to provide a higher-dose estrogen pill. This may actually be counterproductive, however, because oral estrogen promotes synthesis in the liver of sex hormone–binding globulin, which then reduces circulating estrogen and can exacerbate the problem.50 Although the vast majority of women who use estrogen-containing oral contraceptives do not appear to develop symptomatic vulvovaginal atrophy, women who do experience bothersome atrophic symptoms may benefit from switching to an alternative contraceptive method (eg, an intrauterine device). Menopausal women may benefit from a topical estrogen product in the form of a vaginal ring, cream, or tablet.18,51,52
For vulvodynia, medical therapies such as topical anesthetics and centrally acting medications are under investigation. For cases of provoked vestibulodynia that do not respond to first-line treatment, patients may benefit from pelvic floor physical therapy, cognitive behavioral therapy aimed at pain reduction combined with sex therapy, or vestibulectomy.53,54
Addressing Bowel and Bladder Concerns
The patient with chronic constipation must be educated about the importance of adequate hydration and nutrition for bowel regularity. She is counseled about avoidance of straining or prolonged sitting during defecation and is offered primary treatment for hemorrhoids. A history of rectal bleeding, particularly in the absence of hemorrhoids, may be indicative of an anorectal or a colon malignancy, and the patient with such a history should be referred for further evaluation. The International Foundation for Functional Gastric Disorders (see “Resources” box) provides useful information for patients regarding constipation and associated pelvic floor disorders.
Urinary voiding prior to and following coitus or oral sex may reduce bladder discomfort and infection. Use of a dental dam for oral sex and good oral hygiene with regular dental care may also reduce the incidence of irritation and infection. Educational resources to address bladder function and incontinence can be found in the “Resources” box.
Treatment for Vaginismus
Vaginismus can be treated with desensitization techniques, including relaxation training, dilator therapy, pelvic floor therapy, and cognitive behavioral sex therapy, which teaches the patient to relax the introital muscles and give her the experience of controlled, pain-free penetration.55,56 A fundamental technique of relaxation training is deep breathing. Understanding the mechanics of breathing is important for physiologic quieting of the autonomic nervous system and facilitating relaxation of the pelvic floor.57
Dilator therapy can be used for treatment of vaginismus and vaginal stenosis, although in the presence of adhesions or a septum, lysis may be required first. A vaginal dilator can be inserted for approximately 15 minutes daily. Once the dilator passes easily with no painful stretching sensation, the patient should move to the next size larger dilator. The dilator can also be moved slowly and gently in and out and from side to side.
Topical lidocaine (5%) gel or ointment can facilitate use of the dilator. The patient applies a small amount to the site of vulvar pain (commonly the posterior fourchette of the introitus) with the tip of a cotton swab 15 to 30 minutes before inserting the dilator.
Some vaginal dilators may be obtained directly by the patient (see “Resources” box below), while others require a prescription. Graduated or vibrating dildos can be obtained at retail outlets selling sexual products. A clean finger can also be used for dilation.
Pelvic Floor Therapy
Several modalities offered by specialized physical therapists are used to improve pelvic and vulvovaginal blood flow and control over the pelvic musculature. This may help to alleviate pain with intercourse, particularly in patients with signs of pelvic floor weakness, poor muscle control, or instability. Modalities include pelvic floor exercises and manual therapy techniques (eg, transvaginal trigger point therapy, transvaginal and/or transrectal massage, dry needling of a trigger point).4,58 Surface electrical stimulation of the pelvic floor musculature is also used to decrease pain and muscle spasm.59
Kegel exercises60 (ie, tensing and relaxing the pelvic floor muscles) may help a patient gain control over both contraction and relaxation of the pelvic muscles.60,61 However, if palpation of the pelvic floor reveals tight, inelastic, and dense tissue, assessment and grading of muscle strength may be misleading. For example, the pelvic floor may demonstrate a state of constant contraction (hypertonicity or a “short pelvic floor”).62 Attempts at voluntary contraction of a muscle in this state may be perceived and graded incorrectly as weak.
Traditional Kegel exercises are contraindicated in women with muscle hypertonicity and an inability to relax these muscles. Some practitioners use the term “reverse Kegel” to describe active, bearing-down exercises that increase pelvic floor relaxation. Biofeedback training (surface EMG and/or rehabilitative ultrasound imaging) can be useful to reeducate resting muscle tone of the pelvic floor. Breathing, tactile, verbal, and imagery cues are essential.61,62
Cognitive Behavioral Sex Therapy
This specialized form of psychotherapy helps patients identify cognitive, emotional, and relationship factors that contribute to their pain. Patients learn coping strategies, including relaxation techniques and modification of thoughts, feelings, and behaviors to reduce anxiety, tension, and pain. As pain management improves, the focus shifts to enhancing sexual functioning, including restarting the sexual relationship if sex has been avoided because of pain.63
WHEN AND WHERE TO REFER
Referral is appropriate if the patient’s condition worsens, is unresponsive to therapy, or requires specialized evaluation and treatment. Sex therapy, psychotherapy, or couples counseling may be indicated in more complex cases in which a sexual disorder and significant psychologic and/or relationship problems coexist. Examples may include an unresolved history of sexual abuse, clinical anxiety or depression, sexual phobia or aversion, and general relationship distress. To locate qualified sex therapists, see the “Resources” box.
Physical therapy may be prescribed if the patient demonstrates or reports persistent pain or lack of improvement with initial pelvic floor therapy, an inability to use a dilator on her own, or increased pain or perceived tightness while using a dilator; or if she develops pelvic or perineal pain at rest. To locate specially trained physical therapists, see the “Resources” box.
CONCLUSION
Though commonly seen in primary care, sexual pain disorders in women are often difficult to diagnose and treat because of the confusing array of possible contributory factors. This article presents an overview of possible presentations, causes, diagnostic strategies, and treatment options, integrating evidence-based approaches from the fields of medicine, psychology, and physical therapy.
REFERENCES
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DMS-IV-TR). Washington, DC: American Psychiatric Association; 2000:554-558.
2. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8): 762-774.
3. Latthe P, Latthe M, Say L, et al. WHO systematic review of prevalence of chronic pelvic pain: a neglected reproductive health morbidity. BMC Public Health. 2006;6:177-183.
4. Simons DG, Travell JG, Simons LS, Cummings BD. Myofascial Pain and Dysfunction: The Trigger Point Manual. 2nd ed. Baltimore, MD: Lippincott Williams & Wilkins; 1999:vol 2, chap 6:110-131.
5. Perry CP. Vulvodynia. In: Howard FM, Perry CP, Carter JE, El-Minawi AM, eds. Pelvic Pain: Diagnosis and Management. Philadelphia, PA: Lippincott Williams & Wilkins, 2000:204-211.
6. Glazer HI, Jantos M, Hartmann EH, Swencioniswom C. Electromyographic comparisons of the pelvic floor in women with dysesthetic vulvodynia and asymptomatic women. J Reprod Med. 1998;43(11):959-962.
7. Binik YM. The DSM diagnostic criteria for vaginismus. Arch Sex Behav. 2010;39(2):278-291.
8. Haefner HK. Report of the International Society for the Study of Vulvovaginal Disease terminology and classification of vulvodynia. J Low Genit Tract Dis. 2007;11(1):48-49.
9. Huffman JW. Dyspareunia of vulvo-vaginal origin: causes and management. Postgrad Med. 1983;73(2):287-296.
10. Krychman ML. Vaginal estrogens for treatment of dyspareunia. J Sex Med. 2010 Nov 22;8:666-674. [Epub ahead of print]
11. Kao A, Binik YM, Kapuscinski A, Khalifé S. Dyspareunia in postmenopausal women: a critical review. Pain Res Manag. 2008;13(3):243-254.
12. Steege JF, Zolnoun DA. Evaluation and treatment of dyspareunia. Obstet Gynecol. 2009;113 (5):1124-1136.
13. Lüdicke F, Johannisson E, Helmerhorst FM, et al. Effect of a combined oral contraceptive containing 3 mg of drospirenone and 30 microg of ethinyl estradiol on the human endometrium. Fertil Steril. 2001;76(1):102-107.
14. Strauhal MJ, Frahm J, Morrison P, et al. Vulvar pain: a comprehensive review. J Womens Health Phys Ther. 2007;31(3):6-22.
15. Harlow BL, Stewart EG. A population-based assessment of chronic unexplained vulvar pain: have we underestimated the prevalence of vulvodynia? J Am Med Womens Assoc. 2003;58(2): 82-88.
16. Goetsch MF. Vulvar vestibulitis: prevalence and historic features in a general gynecologic practice population. Am J Obstetr Gynecol. 1991; 164(6 pt 1):1609-1616.
17. Danielsson I, Sjöberg I, Stenlund H, Wikman M. Prevalence and incidence of prolonged and severe dyspareunia in women: results from a population study. Scand J Public Health. 2003;31 (2):113-118.
18. Mac Bride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc. 2010;85(1): 87-94.
19. Barditch-Crovo P, Witter F, Hamzeh F, et al. Quantitation of vaginally administered nonoxynol-9 in premenopausal women. Contraception. 1997;55(4):261-263.
20. American College of Obstetricians and Gynecologists. Disorders of the Vulva. ACOG Patient Education Pamphlet AP088. Washington, DC: American College of Obstetricians and Gynecologists; 2007.
21. Ferrero S, Abbamonte LH, Giordano M, et al. Uterine myomas, dyspareunia, and sexual function. Fertil Steril. 2006;86(5):1504-1510.
22. Walid MS, Heaton RL. Dyspareunia: a complex problem requiring a selective approach. Sex Health. 2009;6(3):250-253.
23. Sobghol SS, Alizadeli Charndabee SM. Rate and related factors of dyspareunia in reproductive age women: a cross-sectional study. Int J Impot Res. 2007;19(1):88-94.
24. Ferrero S, Ragni N, Remorgida V. Deep dyspareunia: causes, treatments, and results. Curr Opin Obstet Gynecol. 2008;20(4):394-399.
25. Kim IY, Sadeghi F, Slawin KM. Dyspareunia: an unusual presentation of leiomyoma of the bladder. Rev Urol. 2001;3(3):152-154.
26. Akbulut S, Cakabay B, Sezgin A, Ozmen C. A rare cause of severe dyspareunia: a case report and literature review. Arch Gynecol Obstet. 2009 Apr 26. [Epub ahead of print.]
27. Ferrero S, Esposito F, Abbamonte LH, et al. Quality of sex life in women with endometriosis and deep dyspareunia. Fertil Steril. 2005;83(3): 573-579.
28. Fisher KA. Management of dyspareunia and associated levator ani muscle overactivity. Phys Ther. 2007;87(7):935-941.
29. Bø K, Sherburn M. Evaluation of female pelvic-floor muscle function and strength. Phys Ther. 2005;85(3):269-282.
30. Royal College of Obstetricians and Gynaecologists. Female Genital Mutilation and its Management. Green-top Guideline No 53 (May 2009). www.rcog.org.uk/files/rcog-corp/Green Top53FemaleGenitalMutilation.pdf. Accessed April 4, 2011.
31. Payne KA, Binik YM, Amsel R, Khalifé S. When sex hurts, anxiety and fear orient attention towards pain. Eur J Pain. 2005;9(4):427-436.
32. Pukall CF, Binik YM, Khalifé S, et al. Vestibular tactile and pain thresholds in women with vulvar vestibulitis syndrome. Pain. 2002;96(1-2):163-175.
33. Meana M, Binik YM, Khalifé S, Cohen D. Psychosocial correlates of pain attributions in women with dyspareunia. Psychosomatics. 1999:40(6): 497-502.
34. Pelvic Pain Assessment Form (2008). International Pelvic Pain Society. www.pelvicpain.org/pdf/History_and_Physical_Form/IPPS-H&PformR-MSW.pdf. Accessed April 11, 2011.
35. Melzack R. The short-form McGill Pain Questionnaire. Pain. 1987;30(2):191-197.
36. Basson R. Sexuality and sexual disorders in women. In: Clinical Updates in Women’s Health Care Monograph. Washington, DC: American College of Obstetricians and Gynecologists. 2003;2(2):1-94.
37. Oberg K, Sjögren Fugl-Meyer K. On Swedish women’s distressing sexual dysfunctions: some concomitant conditions and life satisfaction. J Sex Med. 2005;2(2):169-180.
38. Rosen R, Brown C, Heiman J, et al. The Female Sexual Function Index (FSFI): a multidimensional self-report instrument for the assessment of female sexual function. J Sex Marital Ther. 2000;26(2):191-208.
39. Quirk FH, Heiman JR, Rosen RC, et al. Development of a sexual function questionnaire for clinical trials of female sexual dysfunction. J Womens Health Gend Based Med. 2002;11(3): 277-289.
40. Derogatis LR, Rosen R, Leiblum S, et al. The Female Sexual Distress Scale (FSDS): Initial validation of a standardized scale for assessment of sexually related personal distress in women. J Sex Marital Ther. 2002;28(4):317-330.
41. van Lankveld JJ, Granot M, Weijmar Schultz WC, et al. Women’s sexual pain disorders. J Sex Med. 2010;7(1 pt 2):615-631.
42. Hathaway JK, Pathak PK, Maney R. Is liquid-based Pap testing affected by water-based lubricant? Obstet Gynecol. 2006;107(1):66-70.
43. Bergeron S, Binik YM, Khalifé S, et al. Vulvar vestibulitis syndrome: reliability of diagnosis and evaluation of current diagnostic criteria. Obstet Gynecol. 2001;98(1):45-51.
44. Laycock J, Holmes DM. The place of physiotherapy in the management of pelvic floor dysfunction. Obstet Gynaecol. 2003;5:194-199.
45. Brink CA, Sampselle CM, Wells TJ, et al. A digital test for pelvic muscle strength in older women with urinary incontinence. Nurs Res. 1989;38(4):196-199.
46. Hess R, Austin RM, Dillon S, et al. Vaginal maturation index self-sample collection in mid-life women: acceptability and correlation with physician-collected samples. Menopause. 2008; 15(4 pt 1):726-729.
47. Haefner HK, Collins ME, Davis GD, et al. The vulvodynia guideline. J Low Genit Tract Dis. 2005; 9(1):40-51.
48. Albaugh JA, Kellogg-Spadt S. Sensate focus and its role in treating sexual dysfunction. Urol Nurs. 2002;22(6):402-403.
49. Masters WH, Johnson VE. Human Sexual Inadequacy. New York, NY: Ishi Press; 2010: 67-75.
50. Panzer C, Wise S, Fantini G, et al. Impact of oral contraceptives on sex hormone–binding globulin and androgen levels: a retrospective study in women with sexual dysfunction. J Sex Med. 2006;3(1):104-113.
51. Suckling J, Kennedy R, Lethaby A. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev. 2006; (4):CD001500.
52. Johnston SL, Farrell SA, Bouchard C, et al; SOGC joint Committee–Clinical Practice Gynaecology and Urogynaecology. The detection and management of vaginal atrophy. J Obstet Gynaecol Can. 2004;26(5):503-515.
53. Bergeron S, Khalifé S, Glazer HI, Binik YM. Surgical and behavioral treatments for vestibulodynia: two-and-one-half year follow-up and predictors of outcome. Obstet Gynecol. 2008;111(1): 159-166.
54. Pukall CF, Smith KB, Chamberlain SM. Provoked vestibulodynia. Womens Health (Lond Engl). 2007;3(5):583-592.
55. van Lankveld JJ, ter Kuile MM, de Groot HE, et al. Cognitive-behavioral therapy for women with lifelong vaginismus: a randomized waiting-list controlled trial of efficacy. J Consult Clin Psychol. 2006;74(1):168-178.
56. McGuire H, Hawton K. Interventions for vaginismus. Cochrane Database Syst Rev. 2003;(1): CD001760.
57. Calais-Germain B. Analysis of the principal types of breathing. In: Calais-Germain B. Anatomy of Breathing. New York, NY: Eastland Press; 2006:133-158.
58. Carter J. Abdominal wall and pelvic myofascial trigger points. In: Howard FM, Perry P, Carter J, El-Minawi AM. Pelvic Pain: Diagnosis and Management. Baltimore, MD: Lippincott Williams & Wilkins; 2000:315-358.
59. Gentilcore-Saulnier E, McLean L, Goldfinger C, et al. Pelvic floor muscle assessment outcomes in women with and without provoked vestibulodynia and the impact of a physical therapy program. J Sex Med. 2010;7(2 pt 2):1003-1022.
60. Kegel AH. Physiologic therapy for urinary stress incontinence. JAMA. 1951;146(10):915-917.
61. Marques A, Stothers L, Macnab A. The status of pelvic floor muscle training for women. Can Urol Assoc J. 2010;4(6):419-424.
62. FitzGerald MP, Kotarinos R. Rehabilitation of the short pelvic floor. I: Background and patient evaluation. Int Urogynecol J Pelvic Floor Dysfunct. 2003;14(4):261-268.
63. Binik Y, Bergeron S, Khalifé S. Dyspareunia and vaginismus: So-called pain disorders. In: Leiblum S, ed. Principles and Practice of Sex Therapy. 3rd ed. New York: Guilford Press; 2007: 124-156.
Though common, sexual pain disorders in women are often difficult to identify and treat because of the complexity of potential underlying causes. This article will define and describe these conditions in an effort to provide evaluation and treatment strategies for the primary care provider.
SEXUAL PAIN DISORDERS
Dyspareunia is defined as genital pain that may occur before, during, or after vaginal penetration, thus interfering with sexual intercourse and causing marked personal distress and/or interpersonal difficulty.1 Categorization of the condition as lifelong (primary) or acquired (secondary), generalized or situational, may indicate possible underlying causes. Physiological, psychological, or combined factors may be at play. It should be noted that, according to the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DMS-IV-TR),1 painful penetration is not diagnosed as dyspareunia if it is solely the result of lack of lubrication, the physiological effect of a medication, or attributable to the patient’s general medical condition.
Painful intercourse is a prevalent symptom among sexually active women. Between 8% and 21% of women in the general population and 10% of those ages 57 to 85 have been estimated to experience significant dyspareunia.2,3 For some women, sexual pain leads to avoidance of sexual intercourse or contact. Other women remain sexually active despite persistently painful intercourse.
Transient pain during intercourse is predictable in certain situations, such as times of stress, with frequent intercourse (eg, in attempts to conceive), intercourse after a prolonged hiatus from it, or hymenal rupture at coitarche. Dyspareunia, particularly deep dyspareunia, may occur with midcycle intercourse due to normal local inflammation that occurs with ovulation (mittelschmerz).
A second sexual pain disorder experienced by women is vaginismus, which is defined as recurrent or persistent involuntary contraction or spasm of the musculature of the outer third of the vagina (the perineal and levator muscles) that interferes with vaginal penetration, whether associated with sexual intercourse, speculum insertion, or tampon use.1,4-6 Vaginismus may be a cause or a consequence of dyspareunia. This condition is perhaps best described as pelvic floor motor or muscular instability,7 because it can be characterized by:
1) Hypertonicity (reduced ability to relax)
2) Hypocontractility (reduced ability to contract) or
3) Resting muscular instability (as measured by electromyelography [EMG]).8
According to DSM-IV-TR criteria,1 vaginismus also causes marked personal distress or interpersonal difficulty and is not due exclusively to the direct physiological effects of a general medical condition. Vaginismus may occur as a result of psychological factors, such as fear of penetration, or as a conditioned response to pain.
Dyspareunia and Vaginismus: A Clinical Case
Anna is a 30-year-old woman who has been married for three years. She reports that initially, intercourse with her husband was satisfying and pleasurable. She desired sex and was easily aroused and orgasmic. However, when she began to experience a burning pain during intercourse, she tried at first to ignore it, hoping that it would go away on its own. Anticipating pain, she started to avoid sex. When she finally saw her clinician, she was given a diagnosis of candidiasis and responded well to medication.
Although the physical cause of Anna’s dyspareunia was resolved, she continued to be tense when she anticipated an opportunity for sex, associating it with the prior painful experiences. When the couple did attempt vaginal intercourse, penetration was difficult and painful in a different way; it seemed as if the opening to her vagina was closing up, that it was becoming too tight. Anna felt frustrated with herself and her body and worried that her husband would give up on her.
She returned to her clinician, who referred her to a sex therapist. Anna was anxious and skeptical at first but also highly motivated to resolve the pain and resume her previously satisfying sex life. The therapist explained the connection between Anna’s original experience of dyspareunia and the anxiety/avoidance pattern that had developed, causing her pelvic floor muscles to overcontract involuntarily and to make penetration both difficult and uncomfortable. Even though the original physical source of painful intercourse was resolved, Anna had gone on to develop anticipatory anxiety, triggering secondary vaginismus—an automatic, protective response against anticipated pain.
A brief course of cognitive behavioral sex therapy helped Anna to resolve her anxiety, relax her pelvic floor muscles, and gradually return to satisfying sex.
CAUSES OF DYSPAREUNIA
There is a wide and confusing spectrum of causes of primary and secondary dyspareunia. Primary dyspareunia, the less common condition, is associated with imperforate or microperforate hymen, congenital vulvovaginal abnormalities, and acquired vulvovaginal abnormalities resulting from genital surgery, modification, or infundibulation.9
Secondary dyspareunia, or painful intercourse that occurs after a history of pain-free sex, is most often caused by either vaginal atrophy or vulvodynia. Vaginal atrophy, resulting from low circulating estrogen, may cause symptoms of itching, dryness, and irritation; it is experienced by about 40% of postmenopausal women,10 as well as many premenopausal women who take low-dose estrogen oral contraceptives.11-13
Vulvodynia, which affects 16% of women, refers to vulvar pain, usually described as a burning or cutting sensation, with no significant physical changes seen on examination.7,8,14 Vulvodynia may be localized or generalized to the entire vulva and provoked or unprovoked. Vestibulodynia, which is a subtype of vulvodynia, refers to unexplained pain in the vestibule of the vaginal introitus; the only physical finding may be erythema. Provoked vestibulodynia is the most common form of introital pain, affecting 3% to 15% of women.15-17
Both vulvodynia and vestibulodynia have been explained as neurosensory disorders that are triggered by stimuli such as repeated vaginal infections or long-term use of low-dose estrogen oral contraceptives.12 Other less common vulvar conditions that cause dyspareunia include noninfectious skin conditions, such as dermatitis and lichenoid conditions.18 Many women also experience painful vulvar or vaginal irritation due to sensitivity to spermicide on condoms; latex allergy; and/or ingredients in some lubricants, soaps, body perfumes, detergents, or douches.18,19 Less common vulvovaginal conditions associated with dyspareunia include anomalies, infection, radiation effects, and scarring injuries (see Figure 120).
In some cases, dyspareunia is caused by conditions involving the uterus, ovaries, bladder, or rectum. Uterine conditions include myomas, adenomyosis, endometriosis, retroversion, or retroflexion.21,22 In the case of prolapsed uterus, contact with the cervix or uterus may result in what is described by the patient as “an electric shock” or “stabbing” or “shooting” pain.23
Ovarian cysts, intermittent ovarian torsion, and hydrosalpinx can each cause painful intercourse. Associated bladder and urinary tract conditions include urinary tract infection; urethral caruncle or diverticulum; urethral atrophy resulting from low circulating estrogen in women of any age; and interstitial cystitis, which is usually associated with pain after intercourse and/or during the luteal phase of the menstrual cycle.22,24 Anorectal inflammatory conditions, such as lichen sclerosis, Crohn’s disease, and severe constipation with or without hemorrhoids, may be causes of dyspareunia. In rare instances, dyspareunia may be a sign of a tumor in the urogenital or lower GI tract.25,26
Dyspareunia may be indicative of pelvic adhesions resulting from surgery or inflammatory conditions, such as current or past pelvic inflammatory disease.22 The majority (50% to 80%) of women with endometriosis complain of pain with deep thrusting during coitus,27 which is most severe before menstruation and in sexual positions in which the woman has less control over the depth and force of penetration.
Musculoskeletal conditions associated with dyspareunia include scoliosis, levator ani muscle myalgia, and levator ani/pelvic floor musculature spasm.28,29 These may be primary or secondary to another condition that causes deep dyspareunia and may involve pain in the lower back, sacroiliac joint, piriformis, or obturator internus.28
Neurologic conditions, such as pudendal neuralgia and sciatica, can also be associated with pelvic floor injuries during childbirth, nerve entrapment, or straddle injuries.12 Genital piercing and cutting can lead to painful neuromas, neurofibromas, and neuralgia.30
Finally, whether or not identifiable physical causes exist, the experience of pain can be triggered or exacerbated by psychosocial factors. When vaginal penetration has been painful, a pattern of anticipatory anxiety and fear is common. Certain cognitive styles are also associated with dyspareunia. Examples include hypervigilance (“I’m just waiting for it to start hurting”) and catastrophizing (“This hurts so much that I’m going to have to give up having sex altogether”).31,32
Generalized anxiety and somatization, defined as the tendency to experience a variety of physical symptoms without physical origin in reaction to stress, may predispose a woman to experience dyspareunia.1,31 Relationship distress is another related psychosocial factor.33 Even in relationships that are for the most part positive, a woman’s reluctance to communicate her sexual needs may result in foreplay and stimulation that are inadequate to achieve arousal and lubrication.
EVALUATION OF THE PATIENT
Any woman who reports pain during intercourse should undergo a careful history and physical exam directed toward detection of signs of vaginal atrophy (especially women who are menopausal or taking oral estrogen-containing hormonal contraceptives), vulvodynia, vestibulodynia, and other physical and psychological conditions that can cause sexual pain.
A sexual pain history should be taken, focusing on pain experienced with intercourse and/or with tampon or speculum insertion. Pain assessment includes location, intensity, quality (burning, shooting, or dull pain), and duration of the pain. The clinician should ask whether penetrative sex has ever been pain-free to assess for a lifelong or situational condition. Pain frequency, kinds of associated activities, and factors that exacerbate or alleviate the symptom of pain should be clarified. Assessment should include questions about relationship distress; sexual pain can exert negative impact on the relationship, which can then exacerbate sexual pain by impairing arousal. It is important to ascertain whether pain occurs or has occurred with all of the patient’s sexual partners.
The patient should be asked what she identifies as the source of her pain. What treatments have been tried, whether they have been helpful, and what treatments the patient thinks might be effective should all be addressed. The International Pelvic Pain Society34 offers a comprehensive pelvic pain assessment form (see box, “Resources for Clinicians and Patients,” below), which includes the short-form McGill Pain Questionnaire35 for describing and documenting characteristics of pain.
A detailed sexual function history begins with an assessment of sexual function in each domain (desire, arousal, orgasm).36 Other sexual problems, such as vaginal dryness, lack of desire or arousal, inability to experience orgasm, and forced or coerced sex should be identified. The clinician ascertains distress related to the patient’s sexual symptoms (eg, “How bothered are you by the pain you experience with intercourse?”). Information about her partner’s sexual functioning can be relevant, since delayed ejaculation may involve prolonged, ultimately painful intercourse.37 Commonly used objective assessment instruments for female sexual function include the Female Sexual Function Index (FSFI),38 the Sexual Function Questionnaire,39 and the Female Sexual Distress Scale (FSDS).40
A complete evaluation includes relevant medical, surgical, obstetric, and social history, along with a review of systems (see Table 1,34,38-40).
Medication History
A thorough assessment identifies OTC and prescription medications, including contraceptive methods. Long-term use of oral combined contraceptives has been associated with vaginal atrophy and vulvodynia.12 Clarifying whether the patient is using condoms with nonoxynol 9 is important, since this spermicide can cause painful irritation in some women.19 An allergy or sensitivity to latex in condoms or to chemicals in some lubricants can also cause irritation.
Physical Examination
The vulva should be inspected for signs of pallor, loss or thinning of hair, and clitoral shrinkage or shrinkage of the labia minora or majora. These can be signs of atrophy due to low estrogen. The vulva is also examined for skin changes, including inflammation, excoriation, scarring, fissuring, laceration, inability to retract the clitoral hood or complete loss of the clitoris by overlying tissue, and trauma.18 Additional assessment is needed for women with infundibulation in cases of female circumcision (also referred to as genital mutilation), as practiced in parts of the Middle East and Africa.30,41 A water-based lubricant, ideally warmed, can be used at the introitus and on the outer speculum blades to minimize discomfort during the exam without interfering with cytology, if a Pap smear is also being performed.42
For women with introital pain, a “cotton swab test” is performed to quantify, localize, and map the pain (see Table 2,28,43).
The hymen is inspected and its morphology documented, particularly in the woman or girl with recent coitarche. The vagina is inspected for presence of a septum.14 Signs of atrophy include lack of lubrication, easy bleeding, fissures, discomfort even with use of a small speculum, and petechiae.18 Atrophic mucosa will appear flat and pale, with no evidence of normal rugae. The tissue’s elasticity will have decreased.
If there is suspicion of pelvic organ prolapse, vaginitis, or sexually transmitted infection, further assessments are made. Using one intravaginal finger if two fingers are painful, the clinician systematically palpates the urethra, bladder, bilateral fornices, posterior fornix, cervix, adnexa, and uterus in an attempt to reproduce the pain experienced during intercourse.29 Uterine size, contour, mobility, and any nodularity or irregularity are all noted.
During the pelvic examination, the clinician uses a hand mirror to show the patient the depth of penetration with the examining finger, a speculum, or a vaginal dilator—demonstrating the diameter of the introitus and vaginal canal, if possible. This is a valuable opportunity to educate the patient regarding her genital anatomy in addition to any physical findings that might explain her pain.
A rectovaginal examination is important for identifying constipation, hemorrhoids, or nodules along the rectovaginal septum or uterosacral ligaments, possibly indicating endometriosis.21,22 In patients with anal fissures, tensing of the anal sphincter and pelvic floor muscles to slow defecation (or to restrict the diameter of the feces) can result in a paradoxical contraction of the puborectalis portion of the levator ani muscles.28 This creates a vicious cycle of pelvic floor muscle spasm.
Pelvic floor function and strength are systematically evaluated using the Modified Oxford Scale44 or the Brink Scoring System.45 Scoliosis and hip height are assessed by asking the patient to bend over and touch her toes. Curvature of the spine or unequal hip height may indicate musculoskeletal problems that can cause or contribute to pelvic pain during intercourse.
Laboratory Workup
The vaginal maturation index18,46 may be assessed by taking a specimen for wet prep in the office setting or by sending a fixed smear on a slide or in a liquid cytologic preparation to a cytopathology laboratory. Vaginal pH, in combination with a wet prep, can be obtained to help assess for bacterial vaginosis or vaginal candidiasis. Laboratory tests are not regularly required, however, to diagnose and treat dyspareunia. Nevertheless, dyspareunia of musculoskeletal origin may require diagnostic neuromuscular evaluation, including EMG, ultrasonography, and manual assessment.28 These are commonly performed by a physical therapist with expertise in pelvic physical therapy and women’s health.
TREATMENT/MANAGEMENT
Treatment begins with psychosexual education and counseling, in which underlying causes are addressed. The clinician educates the patient about the prevalence and common causes of painful intercourse, assessing what the patient has already done to alleviate the pain and reinforcing constructive and positive behaviors. It is therapeutic to validate the patient’s experience of pain and to explain that both pain and sexuality are mind and body processes. Even if the pain is due to a physical cause, it can have psychologic aspects or sequelae. The clinician normalizes the fact that pain associated with sex can cause anxiety and fear about participating in intercourse and that these are natural, instinctive reactions to pain.47
The patient is given an explanation of any physical and psychologic findings that might be contributing to her pain. This approach gives the patient hope for improvement with education and treatment. The emphasis is on the aspects of her sexual functioning that are still intact and positive features in her sexual history or relationship. A central goal is to minimize self-blame, hopelessness, and anxiety.
The patient is counseled about the need for adequate foreplay and stimulation to promote arousal and lubrication. Masturbation (alone and/or in conjunction with partnered sex) can be discussed as a way to increase vaginal lubrication, blood flow, and comfort with genital touch.
Another helpful strategy is to give the patient permission to take a hiatus from sexual intercourse.47 Women experiencing genital pain often avoid nonsexual affection and other sexual activities with their partners, fearing that they will lead to painful intercourse. In such cases, the woman is encouraged to form an explicit agreement with her partner that they will not have intercourse until she has made progress at managing her pain. The goal is to create greater openness to nonsexual affection and nonpenetrative sexual activity.
The clinician may suggest sensate focus exercises48,49 (see Table 3,4,48,49). In cases of vaginal atrophy and/or dryness, vaginal moisturizers and lubricants can help. Products containing perfumes, warming or tingling agents, parabens, and other chemicals can be irritating in some women.18
For the premenopausal patient using a low-dose oral contraceptive, the clinician’s instinct may be to provide a higher-dose estrogen pill. This may actually be counterproductive, however, because oral estrogen promotes synthesis in the liver of sex hormone–binding globulin, which then reduces circulating estrogen and can exacerbate the problem.50 Although the vast majority of women who use estrogen-containing oral contraceptives do not appear to develop symptomatic vulvovaginal atrophy, women who do experience bothersome atrophic symptoms may benefit from switching to an alternative contraceptive method (eg, an intrauterine device). Menopausal women may benefit from a topical estrogen product in the form of a vaginal ring, cream, or tablet.18,51,52
For vulvodynia, medical therapies such as topical anesthetics and centrally acting medications are under investigation. For cases of provoked vestibulodynia that do not respond to first-line treatment, patients may benefit from pelvic floor physical therapy, cognitive behavioral therapy aimed at pain reduction combined with sex therapy, or vestibulectomy.53,54
Addressing Bowel and Bladder Concerns
The patient with chronic constipation must be educated about the importance of adequate hydration and nutrition for bowel regularity. She is counseled about avoidance of straining or prolonged sitting during defecation and is offered primary treatment for hemorrhoids. A history of rectal bleeding, particularly in the absence of hemorrhoids, may be indicative of an anorectal or a colon malignancy, and the patient with such a history should be referred for further evaluation. The International Foundation for Functional Gastric Disorders (see “Resources” box) provides useful information for patients regarding constipation and associated pelvic floor disorders.
Urinary voiding prior to and following coitus or oral sex may reduce bladder discomfort and infection. Use of a dental dam for oral sex and good oral hygiene with regular dental care may also reduce the incidence of irritation and infection. Educational resources to address bladder function and incontinence can be found in the “Resources” box.
Treatment for Vaginismus
Vaginismus can be treated with desensitization techniques, including relaxation training, dilator therapy, pelvic floor therapy, and cognitive behavioral sex therapy, which teaches the patient to relax the introital muscles and give her the experience of controlled, pain-free penetration.55,56 A fundamental technique of relaxation training is deep breathing. Understanding the mechanics of breathing is important for physiologic quieting of the autonomic nervous system and facilitating relaxation of the pelvic floor.57
Dilator therapy can be used for treatment of vaginismus and vaginal stenosis, although in the presence of adhesions or a septum, lysis may be required first. A vaginal dilator can be inserted for approximately 15 minutes daily. Once the dilator passes easily with no painful stretching sensation, the patient should move to the next size larger dilator. The dilator can also be moved slowly and gently in and out and from side to side.
Topical lidocaine (5%) gel or ointment can facilitate use of the dilator. The patient applies a small amount to the site of vulvar pain (commonly the posterior fourchette of the introitus) with the tip of a cotton swab 15 to 30 minutes before inserting the dilator.
Some vaginal dilators may be obtained directly by the patient (see “Resources” box below), while others require a prescription. Graduated or vibrating dildos can be obtained at retail outlets selling sexual products. A clean finger can also be used for dilation.
Pelvic Floor Therapy
Several modalities offered by specialized physical therapists are used to improve pelvic and vulvovaginal blood flow and control over the pelvic musculature. This may help to alleviate pain with intercourse, particularly in patients with signs of pelvic floor weakness, poor muscle control, or instability. Modalities include pelvic floor exercises and manual therapy techniques (eg, transvaginal trigger point therapy, transvaginal and/or transrectal massage, dry needling of a trigger point).4,58 Surface electrical stimulation of the pelvic floor musculature is also used to decrease pain and muscle spasm.59
Kegel exercises60 (ie, tensing and relaxing the pelvic floor muscles) may help a patient gain control over both contraction and relaxation of the pelvic muscles.60,61 However, if palpation of the pelvic floor reveals tight, inelastic, and dense tissue, assessment and grading of muscle strength may be misleading. For example, the pelvic floor may demonstrate a state of constant contraction (hypertonicity or a “short pelvic floor”).62 Attempts at voluntary contraction of a muscle in this state may be perceived and graded incorrectly as weak.
Traditional Kegel exercises are contraindicated in women with muscle hypertonicity and an inability to relax these muscles. Some practitioners use the term “reverse Kegel” to describe active, bearing-down exercises that increase pelvic floor relaxation. Biofeedback training (surface EMG and/or rehabilitative ultrasound imaging) can be useful to reeducate resting muscle tone of the pelvic floor. Breathing, tactile, verbal, and imagery cues are essential.61,62
Cognitive Behavioral Sex Therapy
This specialized form of psychotherapy helps patients identify cognitive, emotional, and relationship factors that contribute to their pain. Patients learn coping strategies, including relaxation techniques and modification of thoughts, feelings, and behaviors to reduce anxiety, tension, and pain. As pain management improves, the focus shifts to enhancing sexual functioning, including restarting the sexual relationship if sex has been avoided because of pain.63
WHEN AND WHERE TO REFER
Referral is appropriate if the patient’s condition worsens, is unresponsive to therapy, or requires specialized evaluation and treatment. Sex therapy, psychotherapy, or couples counseling may be indicated in more complex cases in which a sexual disorder and significant psychologic and/or relationship problems coexist. Examples may include an unresolved history of sexual abuse, clinical anxiety or depression, sexual phobia or aversion, and general relationship distress. To locate qualified sex therapists, see the “Resources” box.
Physical therapy may be prescribed if the patient demonstrates or reports persistent pain or lack of improvement with initial pelvic floor therapy, an inability to use a dilator on her own, or increased pain or perceived tightness while using a dilator; or if she develops pelvic or perineal pain at rest. To locate specially trained physical therapists, see the “Resources” box.
CONCLUSION
Though commonly seen in primary care, sexual pain disorders in women are often difficult to diagnose and treat because of the confusing array of possible contributory factors. This article presents an overview of possible presentations, causes, diagnostic strategies, and treatment options, integrating evidence-based approaches from the fields of medicine, psychology, and physical therapy.
REFERENCES
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DMS-IV-TR). Washington, DC: American Psychiatric Association; 2000:554-558.
2. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8): 762-774.
3. Latthe P, Latthe M, Say L, et al. WHO systematic review of prevalence of chronic pelvic pain: a neglected reproductive health morbidity. BMC Public Health. 2006;6:177-183.
4. Simons DG, Travell JG, Simons LS, Cummings BD. Myofascial Pain and Dysfunction: The Trigger Point Manual. 2nd ed. Baltimore, MD: Lippincott Williams & Wilkins; 1999:vol 2, chap 6:110-131.
5. Perry CP. Vulvodynia. In: Howard FM, Perry CP, Carter JE, El-Minawi AM, eds. Pelvic Pain: Diagnosis and Management. Philadelphia, PA: Lippincott Williams & Wilkins, 2000:204-211.
6. Glazer HI, Jantos M, Hartmann EH, Swencioniswom C. Electromyographic comparisons of the pelvic floor in women with dysesthetic vulvodynia and asymptomatic women. J Reprod Med. 1998;43(11):959-962.
7. Binik YM. The DSM diagnostic criteria for vaginismus. Arch Sex Behav. 2010;39(2):278-291.
8. Haefner HK. Report of the International Society for the Study of Vulvovaginal Disease terminology and classification of vulvodynia. J Low Genit Tract Dis. 2007;11(1):48-49.
9. Huffman JW. Dyspareunia of vulvo-vaginal origin: causes and management. Postgrad Med. 1983;73(2):287-296.
10. Krychman ML. Vaginal estrogens for treatment of dyspareunia. J Sex Med. 2010 Nov 22;8:666-674. [Epub ahead of print]
11. Kao A, Binik YM, Kapuscinski A, Khalifé S. Dyspareunia in postmenopausal women: a critical review. Pain Res Manag. 2008;13(3):243-254.
12. Steege JF, Zolnoun DA. Evaluation and treatment of dyspareunia. Obstet Gynecol. 2009;113 (5):1124-1136.
13. Lüdicke F, Johannisson E, Helmerhorst FM, et al. Effect of a combined oral contraceptive containing 3 mg of drospirenone and 30 microg of ethinyl estradiol on the human endometrium. Fertil Steril. 2001;76(1):102-107.
14. Strauhal MJ, Frahm J, Morrison P, et al. Vulvar pain: a comprehensive review. J Womens Health Phys Ther. 2007;31(3):6-22.
15. Harlow BL, Stewart EG. A population-based assessment of chronic unexplained vulvar pain: have we underestimated the prevalence of vulvodynia? J Am Med Womens Assoc. 2003;58(2): 82-88.
16. Goetsch MF. Vulvar vestibulitis: prevalence and historic features in a general gynecologic practice population. Am J Obstetr Gynecol. 1991; 164(6 pt 1):1609-1616.
17. Danielsson I, Sjöberg I, Stenlund H, Wikman M. Prevalence and incidence of prolonged and severe dyspareunia in women: results from a population study. Scand J Public Health. 2003;31 (2):113-118.
18. Mac Bride MB, Rhodes DJ, Shuster LT. Vulvovaginal atrophy. Mayo Clin Proc. 2010;85(1): 87-94.
19. Barditch-Crovo P, Witter F, Hamzeh F, et al. Quantitation of vaginally administered nonoxynol-9 in premenopausal women. Contraception. 1997;55(4):261-263.
20. American College of Obstetricians and Gynecologists. Disorders of the Vulva. ACOG Patient Education Pamphlet AP088. Washington, DC: American College of Obstetricians and Gynecologists; 2007.
21. Ferrero S, Abbamonte LH, Giordano M, et al. Uterine myomas, dyspareunia, and sexual function. Fertil Steril. 2006;86(5):1504-1510.
22. Walid MS, Heaton RL. Dyspareunia: a complex problem requiring a selective approach. Sex Health. 2009;6(3):250-253.
23. Sobghol SS, Alizadeli Charndabee SM. Rate and related factors of dyspareunia in reproductive age women: a cross-sectional study. Int J Impot Res. 2007;19(1):88-94.
24. Ferrero S, Ragni N, Remorgida V. Deep dyspareunia: causes, treatments, and results. Curr Opin Obstet Gynecol. 2008;20(4):394-399.
25. Kim IY, Sadeghi F, Slawin KM. Dyspareunia: an unusual presentation of leiomyoma of the bladder. Rev Urol. 2001;3(3):152-154.
26. Akbulut S, Cakabay B, Sezgin A, Ozmen C. A rare cause of severe dyspareunia: a case report and literature review. Arch Gynecol Obstet. 2009 Apr 26. [Epub ahead of print.]
27. Ferrero S, Esposito F, Abbamonte LH, et al. Quality of sex life in women with endometriosis and deep dyspareunia. Fertil Steril. 2005;83(3): 573-579.
28. Fisher KA. Management of dyspareunia and associated levator ani muscle overactivity. Phys Ther. 2007;87(7):935-941.
29. Bø K, Sherburn M. Evaluation of female pelvic-floor muscle function and strength. Phys Ther. 2005;85(3):269-282.
30. Royal College of Obstetricians and Gynaecologists. Female Genital Mutilation and its Management. Green-top Guideline No 53 (May 2009). www.rcog.org.uk/files/rcog-corp/Green Top53FemaleGenitalMutilation.pdf. Accessed April 4, 2011.
31. Payne KA, Binik YM, Amsel R, Khalifé S. When sex hurts, anxiety and fear orient attention towards pain. Eur J Pain. 2005;9(4):427-436.
32. Pukall CF, Binik YM, Khalifé S, et al. Vestibular tactile and pain thresholds in women with vulvar vestibulitis syndrome. Pain. 2002;96(1-2):163-175.
33. Meana M, Binik YM, Khalifé S, Cohen D. Psychosocial correlates of pain attributions in women with dyspareunia. Psychosomatics. 1999:40(6): 497-502.
34. Pelvic Pain Assessment Form (2008). International Pelvic Pain Society. www.pelvicpain.org/pdf/History_and_Physical_Form/IPPS-H&PformR-MSW.pdf. Accessed April 11, 2011.
35. Melzack R. The short-form McGill Pain Questionnaire. Pain. 1987;30(2):191-197.
36. Basson R. Sexuality and sexual disorders in women. In: Clinical Updates in Women’s Health Care Monograph. Washington, DC: American College of Obstetricians and Gynecologists. 2003;2(2):1-94.
37. Oberg K, Sjögren Fugl-Meyer K. On Swedish women’s distressing sexual dysfunctions: some concomitant conditions and life satisfaction. J Sex Med. 2005;2(2):169-180.
38. Rosen R, Brown C, Heiman J, et al. The Female Sexual Function Index (FSFI): a multidimensional self-report instrument for the assessment of female sexual function. J Sex Marital Ther. 2000;26(2):191-208.
39. Quirk FH, Heiman JR, Rosen RC, et al. Development of a sexual function questionnaire for clinical trials of female sexual dysfunction. J Womens Health Gend Based Med. 2002;11(3): 277-289.
40. Derogatis LR, Rosen R, Leiblum S, et al. The Female Sexual Distress Scale (FSDS): Initial validation of a standardized scale for assessment of sexually related personal distress in women. J Sex Marital Ther. 2002;28(4):317-330.
41. van Lankveld JJ, Granot M, Weijmar Schultz WC, et al. Women’s sexual pain disorders. J Sex Med. 2010;7(1 pt 2):615-631.
42. Hathaway JK, Pathak PK, Maney R. Is liquid-based Pap testing affected by water-based lubricant? Obstet Gynecol. 2006;107(1):66-70.
43. Bergeron S, Binik YM, Khalifé S, et al. Vulvar vestibulitis syndrome: reliability of diagnosis and evaluation of current diagnostic criteria. Obstet Gynecol. 2001;98(1):45-51.
44. Laycock J, Holmes DM. The place of physiotherapy in the management of pelvic floor dysfunction. Obstet Gynaecol. 2003;5:194-199.
45. Brink CA, Sampselle CM, Wells TJ, et al. A digital test for pelvic muscle strength in older women with urinary incontinence. Nurs Res. 1989;38(4):196-199.
46. Hess R, Austin RM, Dillon S, et al. Vaginal maturation index self-sample collection in mid-life women: acceptability and correlation with physician-collected samples. Menopause. 2008; 15(4 pt 1):726-729.
47. Haefner HK, Collins ME, Davis GD, et al. The vulvodynia guideline. J Low Genit Tract Dis. 2005; 9(1):40-51.
48. Albaugh JA, Kellogg-Spadt S. Sensate focus and its role in treating sexual dysfunction. Urol Nurs. 2002;22(6):402-403.
49. Masters WH, Johnson VE. Human Sexual Inadequacy. New York, NY: Ishi Press; 2010: 67-75.
50. Panzer C, Wise S, Fantini G, et al. Impact of oral contraceptives on sex hormone–binding globulin and androgen levels: a retrospective study in women with sexual dysfunction. J Sex Med. 2006;3(1):104-113.
51. Suckling J, Kennedy R, Lethaby A. Local oestrogen for vaginal atrophy in postmenopausal women. Cochrane Database Syst Rev. 2006; (4):CD001500.
52. Johnston SL, Farrell SA, Bouchard C, et al; SOGC joint Committee–Clinical Practice Gynaecology and Urogynaecology. The detection and management of vaginal atrophy. J Obstet Gynaecol Can. 2004;26(5):503-515.
53. Bergeron S, Khalifé S, Glazer HI, Binik YM. Surgical and behavioral treatments for vestibulodynia: two-and-one-half year follow-up and predictors of outcome. Obstet Gynecol. 2008;111(1): 159-166.
54. Pukall CF, Smith KB, Chamberlain SM. Provoked vestibulodynia. Womens Health (Lond Engl). 2007;3(5):583-592.
55. van Lankveld JJ, ter Kuile MM, de Groot HE, et al. Cognitive-behavioral therapy for women with lifelong vaginismus: a randomized waiting-list controlled trial of efficacy. J Consult Clin Psychol. 2006;74(1):168-178.
56. McGuire H, Hawton K. Interventions for vaginismus. Cochrane Database Syst Rev. 2003;(1): CD001760.
57. Calais-Germain B. Analysis of the principal types of breathing. In: Calais-Germain B. Anatomy of Breathing. New York, NY: Eastland Press; 2006:133-158.
58. Carter J. Abdominal wall and pelvic myofascial trigger points. In: Howard FM, Perry P, Carter J, El-Minawi AM. Pelvic Pain: Diagnosis and Management. Baltimore, MD: Lippincott Williams & Wilkins; 2000:315-358.
59. Gentilcore-Saulnier E, McLean L, Goldfinger C, et al. Pelvic floor muscle assessment outcomes in women with and without provoked vestibulodynia and the impact of a physical therapy program. J Sex Med. 2010;7(2 pt 2):1003-1022.
60. Kegel AH. Physiologic therapy for urinary stress incontinence. JAMA. 1951;146(10):915-917.
61. Marques A, Stothers L, Macnab A. The status of pelvic floor muscle training for women. Can Urol Assoc J. 2010;4(6):419-424.
62. FitzGerald MP, Kotarinos R. Rehabilitation of the short pelvic floor. I: Background and patient evaluation. Int Urogynecol J Pelvic Floor Dysfunct. 2003;14(4):261-268.
63. Binik Y, Bergeron S, Khalifé S. Dyspareunia and vaginismus: So-called pain disorders. In: Leiblum S, ed. Principles and Practice of Sex Therapy. 3rd ed. New York: Guilford Press; 2007: 124-156.