User login
Toilet Seats and Dangerous Germs
In Search of a "Good Death"
An elderly patient, Mr. Jones, presents for his routine primary care office visit. His wife, who cares for him at home, reports that his dementia is growing worse. He is nonverbal, is having difficulty swallowing, and has lost 10% of his body weight in the past six months. He was recently hospitalized for treatment of aspiration pneumonia and experienced a marked decline during his hospital stay.
What action on the provider’s part would be most helpful for Mr. Jones and his family during this visit?
a) Order a swallow study and a chest x-ray.
b) Evaluate Mr. Jones for depression.
c) Help his family to plan for a comfortable death in the setting of their choosing.
The answer is, of course, “c.” Broaching this topic can be uncomfortable and time-consuming. But for a number of compelling reasons, providers should be communicating with their patients regarding their wishes in the final months of life.
Mr. Jones is at very high risk for dying in the coming six months; he qualifies for hospice care today. Timely hospice care will include physical and emotional support for his family and will allow Mr. Jones to die comfortably in the location that he and his family prefer.
Dementia and End-of-Life Care
Dementia was first identified as a terminal illness in 2000, when the American Medical Association (AMA) issued the Practical Guide for the Primary Care Physician on the Diagnosis, Management and Treatment of Dementia.1 This guide, in addition to more recently published literature,2,3,4 can assist primary care providers in diagnosing, managing, and treating patients with dementia from diagnosis to end of life. The AMA guide recommends that patients with dementia be offered comfort-focused care early in the course of their disease.1
Among recently admitted nursing home residents, it has been shown, about 48% have dementia.5,6 Even in this setting, patients with dementia are often not considered terminally ill.7 In one study of patients with advanced dementia who were admitted to a nursing home, only 1% were perceived by the facility staff to have a life expectancy of six months or less. In reality, 71% of those patients died within six months of admission.8 Alzheimer’s dementia, specifically, is the fifth leading cause of death among persons older than 65,9 yet even medical professionals often fail to recognize this condition as a terminal illness.
Although some 80% of Americans say they want to die at home, more than 70% die in a facility. Additionally, nearly 30% of Medicare enrollees are admitted to intensive care during their terminal hospital stay.10 Families of terminally ill patients, especially those with dementia, often make poor end-of-life decisions for several reasons: They do not understand the natural progression of the patient’s illness; they are unaware of the benefits and burdens of available treatments; and their decisions are often colored by the burden of guilt.11
Without essential conversations with the health care provider and an individualized plan in place, patients and their families will continue to seek help in emergency departments (EDs) and hospitals for treatment of possibly manageable symptoms: pain, fever, dyspnea, constipation.8 Many members of this vulnerable population will submit to aggressive medical interventions (eg, respirators, tube feeding, IV hydration, CPR12), when what they really require is high-touch comfort care.1
The Choice of Hospice
Hospice is a viable option for patients with dementia. Among family members of hospice care patients, 75% are very satisfied with that care.13 Yet according to 2008 statistics from the Hospice Association of America (HAA),14 only one in 10 patients who qualify for hospice care actually choose it. In one study, 10% of a group of recently admitted nursing home residents with dementia were perceived to have less than six months to live, but only 5.7% were referred to hospice.15 The HAA recommends that health care providers who care for terminally ill patients in clinical facilities open a dialogue with patients and family caregivers about the option of hospice.14
Once a patient is referred and accepted, the hospice team can educate the family about the benefits and burdens of end-of-life treatments. Together, they can formulate a plan and put into place resources that may be needed if the patient begins to deteriorate rapidly. Thanks to the Medicare Hospice Benefit16 (which provides specialized services in addition to members’ regular Medicare Part A benefits), oxygen can be ordered without the patient’s meeting oximetry specifications, and emergency medications may be kept in the patient’s home for future need. Patients who require inpatient hospice care will be transferred to a facility owned by or under contract with the Medicare-approved hospice program.
A Role in Providing End-of-Life Care
Primary care providers can play a significant role in improving quality of life for their end-stage patients. Optimal end-of-life care begins with information for patients and their caregivers about the expected progression of their illness. Family members may not know, for example, that dementia is considered a terminal illness.
During office visits with patients who have end-stage dementia and their family members, the provider should7,17-20:
• Review the expected course of dementia. Providers can help the family understand what to expect in the coming months. Hearing that the patient is in the late stage of the disease can be helpful to family members. Increased functional dependency (eg, dressing, bathing, toileting) and recurrent infections (with risk increased by dysphagia, apraxia, and reduced mobility) are likely.7,17
• Offer caregivers sources of emotional support. Families who receive such support are better able to provide care in the home, putting off the need for institutionalization. In addition to caregiver support groups, the Alzheimer’s Association offers a 24-hour helpline number: (800) 272-3900. The hospice team will also provide emotional and spiritual support.
• Remind caregivers that weight loss is expected in patients with end-stage dementia. Difficulty swallowing and other eating problems are common in patients with end-stage dementia. Tube feeding appears to offer neither survival advantage, nor improved nutritional status, nor improvement in quality of life in dementia patients, compared with hand feeding.18 Tube feeding has also been linked with increased risk of aspiration pneumonia. Yet surveyed hospital physicians often express the belief that feeding tubes have benefits not supported in evidence-based literature; they have also been shown to underestimate the 30-day mortality rate in dementia patients with feeding tubes.19 In some states, up to 44% of nursing home residents with dementia reportedly die with a feeding tube in place.7
Because the decision to implement enteral tube feeding is often based on emotional rather than factual data, it is important to discuss this practice with the family in advance of a crisis. Once hospitalized, patients with dementia are likely to be offered feeding tube insertion. The Alzheimer’s Association recommends a conscientious program of hand feeding rather than tube feeding.12 A brochure addressing this and other end-of-life decisions can be downloaded from the association’s Web site and shared with patients’ decision makers (www.alz.org/national/documents/brochure_endoflifedecisions.pdf).12
• Describe the burdens associated with hospitalization for patients with dementia. Adjusting to new routines and new caregivers who do not understand patients’ needs can trigger significant anxiety and accelerate their decline. Treatments that cause discomfort may agitate them. Patients with dementia who are hospitalized have been found to lose weight and experience loss of function in the activities of daily living—developments that are not reversed after hospital discharge.21
Nursing home residents may include a “do not hospitalize” order in their advance directives. For patients who would ordinarily be admitted for treatment of an infection, clinicians can consider less invasive therapy without moving the patient from where he or she lives. Oral antibiotics have been shown as effective as parenteral agents for treating infections in patients with dementia.21
• Explain why a do-not-resuscitate (DNR) order is advisable. The absence of a DNR directive is one of the strongest predictors of high utilization of medical care near the end of life.20 Contrary to perceptions families may have developed from watching television, the CPR survival rate for non–community-dwelling elderly persons is only 1% to 2%, and those who survive do so only briefly, if not with severe disability.22 Nor does CPR begin and end in the field; it is important for the provider to clarify that the CPR process starts at home but is concluded in the ED—or possibly in the ICU, with the patient on a ventilator. Thus, the choice of CPR should be portrayed as an option that is likely to be futile and that may actually increase a dying patient’s distress.
Many families struggle with the notion of withholding any intervention, no matter how small the potential benefit; they fear that the patient with a durable DNR order will receive limited care—or no care. It is important for the provider to clarify that “do not resuscitate” does not mean “do not treat.” In fact, families can still activate emergency medical services if they need help. But instead of performing CPR, the EMT will administer aggressive comfort measures (eg, pain management, hydration). Being with patients as natural death occurs may be the most important assistance family members can provide.
Conclusion
The health care provider plays an invaluable role in educating and supporting families who seek a good death for a loved one. Most elderly patients do not want aggressive end-of-life care; they and their families welcome discussions and strategies that will help them maintain control during the course of a terminal illness.
Many patients are often candidates for hospice care months sooner than clinicians realize. Early referral to hospice care will ensure that their physical needs—and the family’s emotional needs—are met in the optimal setting.
1. American Medical Association, Program on Aging and Community Health. Practical Guide for the Primary Care Physician on the Diagnosis, Management and Treatment of Dementia. Chicago, IL: American Medical Association; 2000.
2. Farias ST, Mungas D, Reed BR, et al. The measurement of everyday cognition (ECog): scale development and psychometric properties. Neuropsychology. 2008;22(4):531-544.
3. Boustani M, Peterson B, Hanson L, et al. Screening for dementia in primary care: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2003;138(11): 927-937.
4. US Preventive Services Task Force. Screening for dementia: recommendation and rationale. Ann Intern Med. 2003;138(11):925-926.
5. Magaziner J, German P, Zimmerman SI, et al; Epidemiology of Dementia in Nursing Homes Research Group. The prevalence of dementia in a statewide sample of new nursing home admissions aged 65 or older: diagnosis by expert panel. Gerontologist. 2000;40(6):663-672.
6. Jakob A, Busse A, Riedel-Heller SG, et al. Prevalence and incidence of dementia among nursing home residents and residents in homes for the aged in comparison to private homes [in German]. Z Gerontol Geriatr. 2002;35(5):474-481.
7. Sachs GA, Shega JW, Cox-Hayley D. Barriers to excellent end-of-life care for patients with dementia. J Gen Intern Med. 2004;19(10):1057-1063.
8. Mitchell SL, Kiely DK, Hamel MB. Dying with advanced dementia in the nursing home. Arch Intern Med. 2004;164(3):321-326.
9. Alzheimer’s Association. 2009 Alzheimer’s Disease Facts and Figures. www.alz.org/national/documents/report_alzfactsfigures2009.pdf. Accessed September 24, 2009.
10. Wennberg JE, Cooper MM. Dartmouth Atlas of Health Care. Washington, DC: AHA Press; 1999.
11. Forbes S, Bern-Klug M, Gessert C. End-of-life decision making for nursing home residents with dementia. J Nurs Scholarsh. 2000;32(3):251-258.
12. Alzheimer’s Association. End-of-Life Decisions: Honoring the Wishes of the Person With Alzheimer’s Disease. www.alz.org/national/docu ments/brochure_endoflifedecisions.pdf. Accessed September 23, 2009.
13. Mitchell SL, Kiely DK, Miller SC, et al. Hospice care for patients with dementia. J Pain Symptom Manage. 2007;34(1):7-16.
14. Hospice Association of America. Hospice facts and statistics (2008). www.nahc.org/facts/Hospice Stats08.pdf. Accessed October 26, 2009.
15. Mitchell SL, Morris JN, Park PS, Fried BE. Terminal care for persons with advanced dementia in the nursing home and home care settings. J Palliat Med. 2004;7(6):808-816.
16. Centers for Medicare and Medicaid Services, US Department of Health and Human Services. Medicare Hospice Benefits (2008). CMS Publication No. 02154. www.medicare.gov/publications/Pubs/pdf/02154.pdf. Accessed October 26, 2009.
17. Mitchell SL, Kiely DK, Hamel MB, et al. Estimating prognosis for nursing home residents with advanced dementia. JAMA. 2004;291(22):2734-2740.
18. Sampson EL, Candy B, Jones L. Enteral tube feeding for older people with advanced dementia. Cochrane Database Syst Rev. 2009;(2): CD007209.
19. Shega JW, Hougham GW, Stocking CB, et al. Barriers to limiting the practice of feeding tube placement in advanced dementia. J Palliat Med. 2003;6(6):885-893.
20. Haller IV, Gessert CE. Utilization of medical services at the end of life in older adults with cognitive impairment: focus on outliers. J Palliat Med. 2007;10(2):400-407.
21. Volicer L, McKee A, Hewitt S. Dementia. Neurol Clin. 2001;19(4):867-885.
22. Mohr M, Bömelburg K, Bahr J. Attempted CPR in nursing homes: life-saving at the end of life? [in German] Anasthesiol Intensivmed Notfallmed Schmerzther. 2001;36(9):566-572.
An elderly patient, Mr. Jones, presents for his routine primary care office visit. His wife, who cares for him at home, reports that his dementia is growing worse. He is nonverbal, is having difficulty swallowing, and has lost 10% of his body weight in the past six months. He was recently hospitalized for treatment of aspiration pneumonia and experienced a marked decline during his hospital stay.
What action on the provider’s part would be most helpful for Mr. Jones and his family during this visit?
a) Order a swallow study and a chest x-ray.
b) Evaluate Mr. Jones for depression.
c) Help his family to plan for a comfortable death in the setting of their choosing.
The answer is, of course, “c.” Broaching this topic can be uncomfortable and time-consuming. But for a number of compelling reasons, providers should be communicating with their patients regarding their wishes in the final months of life.
Mr. Jones is at very high risk for dying in the coming six months; he qualifies for hospice care today. Timely hospice care will include physical and emotional support for his family and will allow Mr. Jones to die comfortably in the location that he and his family prefer.
Dementia and End-of-Life Care
Dementia was first identified as a terminal illness in 2000, when the American Medical Association (AMA) issued the Practical Guide for the Primary Care Physician on the Diagnosis, Management and Treatment of Dementia.1 This guide, in addition to more recently published literature,2,3,4 can assist primary care providers in diagnosing, managing, and treating patients with dementia from diagnosis to end of life. The AMA guide recommends that patients with dementia be offered comfort-focused care early in the course of their disease.1
Among recently admitted nursing home residents, it has been shown, about 48% have dementia.5,6 Even in this setting, patients with dementia are often not considered terminally ill.7 In one study of patients with advanced dementia who were admitted to a nursing home, only 1% were perceived by the facility staff to have a life expectancy of six months or less. In reality, 71% of those patients died within six months of admission.8 Alzheimer’s dementia, specifically, is the fifth leading cause of death among persons older than 65,9 yet even medical professionals often fail to recognize this condition as a terminal illness.
Although some 80% of Americans say they want to die at home, more than 70% die in a facility. Additionally, nearly 30% of Medicare enrollees are admitted to intensive care during their terminal hospital stay.10 Families of terminally ill patients, especially those with dementia, often make poor end-of-life decisions for several reasons: They do not understand the natural progression of the patient’s illness; they are unaware of the benefits and burdens of available treatments; and their decisions are often colored by the burden of guilt.11
Without essential conversations with the health care provider and an individualized plan in place, patients and their families will continue to seek help in emergency departments (EDs) and hospitals for treatment of possibly manageable symptoms: pain, fever, dyspnea, constipation.8 Many members of this vulnerable population will submit to aggressive medical interventions (eg, respirators, tube feeding, IV hydration, CPR12), when what they really require is high-touch comfort care.1
The Choice of Hospice
Hospice is a viable option for patients with dementia. Among family members of hospice care patients, 75% are very satisfied with that care.13 Yet according to 2008 statistics from the Hospice Association of America (HAA),14 only one in 10 patients who qualify for hospice care actually choose it. In one study, 10% of a group of recently admitted nursing home residents with dementia were perceived to have less than six months to live, but only 5.7% were referred to hospice.15 The HAA recommends that health care providers who care for terminally ill patients in clinical facilities open a dialogue with patients and family caregivers about the option of hospice.14
Once a patient is referred and accepted, the hospice team can educate the family about the benefits and burdens of end-of-life treatments. Together, they can formulate a plan and put into place resources that may be needed if the patient begins to deteriorate rapidly. Thanks to the Medicare Hospice Benefit16 (which provides specialized services in addition to members’ regular Medicare Part A benefits), oxygen can be ordered without the patient’s meeting oximetry specifications, and emergency medications may be kept in the patient’s home for future need. Patients who require inpatient hospice care will be transferred to a facility owned by or under contract with the Medicare-approved hospice program.
A Role in Providing End-of-Life Care
Primary care providers can play a significant role in improving quality of life for their end-stage patients. Optimal end-of-life care begins with information for patients and their caregivers about the expected progression of their illness. Family members may not know, for example, that dementia is considered a terminal illness.
During office visits with patients who have end-stage dementia and their family members, the provider should7,17-20:
• Review the expected course of dementia. Providers can help the family understand what to expect in the coming months. Hearing that the patient is in the late stage of the disease can be helpful to family members. Increased functional dependency (eg, dressing, bathing, toileting) and recurrent infections (with risk increased by dysphagia, apraxia, and reduced mobility) are likely.7,17
• Offer caregivers sources of emotional support. Families who receive such support are better able to provide care in the home, putting off the need for institutionalization. In addition to caregiver support groups, the Alzheimer’s Association offers a 24-hour helpline number: (800) 272-3900. The hospice team will also provide emotional and spiritual support.
• Remind caregivers that weight loss is expected in patients with end-stage dementia. Difficulty swallowing and other eating problems are common in patients with end-stage dementia. Tube feeding appears to offer neither survival advantage, nor improved nutritional status, nor improvement in quality of life in dementia patients, compared with hand feeding.18 Tube feeding has also been linked with increased risk of aspiration pneumonia. Yet surveyed hospital physicians often express the belief that feeding tubes have benefits not supported in evidence-based literature; they have also been shown to underestimate the 30-day mortality rate in dementia patients with feeding tubes.19 In some states, up to 44% of nursing home residents with dementia reportedly die with a feeding tube in place.7
Because the decision to implement enteral tube feeding is often based on emotional rather than factual data, it is important to discuss this practice with the family in advance of a crisis. Once hospitalized, patients with dementia are likely to be offered feeding tube insertion. The Alzheimer’s Association recommends a conscientious program of hand feeding rather than tube feeding.12 A brochure addressing this and other end-of-life decisions can be downloaded from the association’s Web site and shared with patients’ decision makers (www.alz.org/national/documents/brochure_endoflifedecisions.pdf).12
• Describe the burdens associated with hospitalization for patients with dementia. Adjusting to new routines and new caregivers who do not understand patients’ needs can trigger significant anxiety and accelerate their decline. Treatments that cause discomfort may agitate them. Patients with dementia who are hospitalized have been found to lose weight and experience loss of function in the activities of daily living—developments that are not reversed after hospital discharge.21
Nursing home residents may include a “do not hospitalize” order in their advance directives. For patients who would ordinarily be admitted for treatment of an infection, clinicians can consider less invasive therapy without moving the patient from where he or she lives. Oral antibiotics have been shown as effective as parenteral agents for treating infections in patients with dementia.21
• Explain why a do-not-resuscitate (DNR) order is advisable. The absence of a DNR directive is one of the strongest predictors of high utilization of medical care near the end of life.20 Contrary to perceptions families may have developed from watching television, the CPR survival rate for non–community-dwelling elderly persons is only 1% to 2%, and those who survive do so only briefly, if not with severe disability.22 Nor does CPR begin and end in the field; it is important for the provider to clarify that the CPR process starts at home but is concluded in the ED—or possibly in the ICU, with the patient on a ventilator. Thus, the choice of CPR should be portrayed as an option that is likely to be futile and that may actually increase a dying patient’s distress.
Many families struggle with the notion of withholding any intervention, no matter how small the potential benefit; they fear that the patient with a durable DNR order will receive limited care—or no care. It is important for the provider to clarify that “do not resuscitate” does not mean “do not treat.” In fact, families can still activate emergency medical services if they need help. But instead of performing CPR, the EMT will administer aggressive comfort measures (eg, pain management, hydration). Being with patients as natural death occurs may be the most important assistance family members can provide.
Conclusion
The health care provider plays an invaluable role in educating and supporting families who seek a good death for a loved one. Most elderly patients do not want aggressive end-of-life care; they and their families welcome discussions and strategies that will help them maintain control during the course of a terminal illness.
Many patients are often candidates for hospice care months sooner than clinicians realize. Early referral to hospice care will ensure that their physical needs—and the family’s emotional needs—are met in the optimal setting.
An elderly patient, Mr. Jones, presents for his routine primary care office visit. His wife, who cares for him at home, reports that his dementia is growing worse. He is nonverbal, is having difficulty swallowing, and has lost 10% of his body weight in the past six months. He was recently hospitalized for treatment of aspiration pneumonia and experienced a marked decline during his hospital stay.
What action on the provider’s part would be most helpful for Mr. Jones and his family during this visit?
a) Order a swallow study and a chest x-ray.
b) Evaluate Mr. Jones for depression.
c) Help his family to plan for a comfortable death in the setting of their choosing.
The answer is, of course, “c.” Broaching this topic can be uncomfortable and time-consuming. But for a number of compelling reasons, providers should be communicating with their patients regarding their wishes in the final months of life.
Mr. Jones is at very high risk for dying in the coming six months; he qualifies for hospice care today. Timely hospice care will include physical and emotional support for his family and will allow Mr. Jones to die comfortably in the location that he and his family prefer.
Dementia and End-of-Life Care
Dementia was first identified as a terminal illness in 2000, when the American Medical Association (AMA) issued the Practical Guide for the Primary Care Physician on the Diagnosis, Management and Treatment of Dementia.1 This guide, in addition to more recently published literature,2,3,4 can assist primary care providers in diagnosing, managing, and treating patients with dementia from diagnosis to end of life. The AMA guide recommends that patients with dementia be offered comfort-focused care early in the course of their disease.1
Among recently admitted nursing home residents, it has been shown, about 48% have dementia.5,6 Even in this setting, patients with dementia are often not considered terminally ill.7 In one study of patients with advanced dementia who were admitted to a nursing home, only 1% were perceived by the facility staff to have a life expectancy of six months or less. In reality, 71% of those patients died within six months of admission.8 Alzheimer’s dementia, specifically, is the fifth leading cause of death among persons older than 65,9 yet even medical professionals often fail to recognize this condition as a terminal illness.
Although some 80% of Americans say they want to die at home, more than 70% die in a facility. Additionally, nearly 30% of Medicare enrollees are admitted to intensive care during their terminal hospital stay.10 Families of terminally ill patients, especially those with dementia, often make poor end-of-life decisions for several reasons: They do not understand the natural progression of the patient’s illness; they are unaware of the benefits and burdens of available treatments; and their decisions are often colored by the burden of guilt.11
Without essential conversations with the health care provider and an individualized plan in place, patients and their families will continue to seek help in emergency departments (EDs) and hospitals for treatment of possibly manageable symptoms: pain, fever, dyspnea, constipation.8 Many members of this vulnerable population will submit to aggressive medical interventions (eg, respirators, tube feeding, IV hydration, CPR12), when what they really require is high-touch comfort care.1
The Choice of Hospice
Hospice is a viable option for patients with dementia. Among family members of hospice care patients, 75% are very satisfied with that care.13 Yet according to 2008 statistics from the Hospice Association of America (HAA),14 only one in 10 patients who qualify for hospice care actually choose it. In one study, 10% of a group of recently admitted nursing home residents with dementia were perceived to have less than six months to live, but only 5.7% were referred to hospice.15 The HAA recommends that health care providers who care for terminally ill patients in clinical facilities open a dialogue with patients and family caregivers about the option of hospice.14
Once a patient is referred and accepted, the hospice team can educate the family about the benefits and burdens of end-of-life treatments. Together, they can formulate a plan and put into place resources that may be needed if the patient begins to deteriorate rapidly. Thanks to the Medicare Hospice Benefit16 (which provides specialized services in addition to members’ regular Medicare Part A benefits), oxygen can be ordered without the patient’s meeting oximetry specifications, and emergency medications may be kept in the patient’s home for future need. Patients who require inpatient hospice care will be transferred to a facility owned by or under contract with the Medicare-approved hospice program.
A Role in Providing End-of-Life Care
Primary care providers can play a significant role in improving quality of life for their end-stage patients. Optimal end-of-life care begins with information for patients and their caregivers about the expected progression of their illness. Family members may not know, for example, that dementia is considered a terminal illness.
During office visits with patients who have end-stage dementia and their family members, the provider should7,17-20:
• Review the expected course of dementia. Providers can help the family understand what to expect in the coming months. Hearing that the patient is in the late stage of the disease can be helpful to family members. Increased functional dependency (eg, dressing, bathing, toileting) and recurrent infections (with risk increased by dysphagia, apraxia, and reduced mobility) are likely.7,17
• Offer caregivers sources of emotional support. Families who receive such support are better able to provide care in the home, putting off the need for institutionalization. In addition to caregiver support groups, the Alzheimer’s Association offers a 24-hour helpline number: (800) 272-3900. The hospice team will also provide emotional and spiritual support.
• Remind caregivers that weight loss is expected in patients with end-stage dementia. Difficulty swallowing and other eating problems are common in patients with end-stage dementia. Tube feeding appears to offer neither survival advantage, nor improved nutritional status, nor improvement in quality of life in dementia patients, compared with hand feeding.18 Tube feeding has also been linked with increased risk of aspiration pneumonia. Yet surveyed hospital physicians often express the belief that feeding tubes have benefits not supported in evidence-based literature; they have also been shown to underestimate the 30-day mortality rate in dementia patients with feeding tubes.19 In some states, up to 44% of nursing home residents with dementia reportedly die with a feeding tube in place.7
Because the decision to implement enteral tube feeding is often based on emotional rather than factual data, it is important to discuss this practice with the family in advance of a crisis. Once hospitalized, patients with dementia are likely to be offered feeding tube insertion. The Alzheimer’s Association recommends a conscientious program of hand feeding rather than tube feeding.12 A brochure addressing this and other end-of-life decisions can be downloaded from the association’s Web site and shared with patients’ decision makers (www.alz.org/national/documents/brochure_endoflifedecisions.pdf).12
• Describe the burdens associated with hospitalization for patients with dementia. Adjusting to new routines and new caregivers who do not understand patients’ needs can trigger significant anxiety and accelerate their decline. Treatments that cause discomfort may agitate them. Patients with dementia who are hospitalized have been found to lose weight and experience loss of function in the activities of daily living—developments that are not reversed after hospital discharge.21
Nursing home residents may include a “do not hospitalize” order in their advance directives. For patients who would ordinarily be admitted for treatment of an infection, clinicians can consider less invasive therapy without moving the patient from where he or she lives. Oral antibiotics have been shown as effective as parenteral agents for treating infections in patients with dementia.21
• Explain why a do-not-resuscitate (DNR) order is advisable. The absence of a DNR directive is one of the strongest predictors of high utilization of medical care near the end of life.20 Contrary to perceptions families may have developed from watching television, the CPR survival rate for non–community-dwelling elderly persons is only 1% to 2%, and those who survive do so only briefly, if not with severe disability.22 Nor does CPR begin and end in the field; it is important for the provider to clarify that the CPR process starts at home but is concluded in the ED—or possibly in the ICU, with the patient on a ventilator. Thus, the choice of CPR should be portrayed as an option that is likely to be futile and that may actually increase a dying patient’s distress.
Many families struggle with the notion of withholding any intervention, no matter how small the potential benefit; they fear that the patient with a durable DNR order will receive limited care—or no care. It is important for the provider to clarify that “do not resuscitate” does not mean “do not treat.” In fact, families can still activate emergency medical services if they need help. But instead of performing CPR, the EMT will administer aggressive comfort measures (eg, pain management, hydration). Being with patients as natural death occurs may be the most important assistance family members can provide.
Conclusion
The health care provider plays an invaluable role in educating and supporting families who seek a good death for a loved one. Most elderly patients do not want aggressive end-of-life care; they and their families welcome discussions and strategies that will help them maintain control during the course of a terminal illness.
Many patients are often candidates for hospice care months sooner than clinicians realize. Early referral to hospice care will ensure that their physical needs—and the family’s emotional needs—are met in the optimal setting.
1. American Medical Association, Program on Aging and Community Health. Practical Guide for the Primary Care Physician on the Diagnosis, Management and Treatment of Dementia. Chicago, IL: American Medical Association; 2000.
2. Farias ST, Mungas D, Reed BR, et al. The measurement of everyday cognition (ECog): scale development and psychometric properties. Neuropsychology. 2008;22(4):531-544.
3. Boustani M, Peterson B, Hanson L, et al. Screening for dementia in primary care: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2003;138(11): 927-937.
4. US Preventive Services Task Force. Screening for dementia: recommendation and rationale. Ann Intern Med. 2003;138(11):925-926.
5. Magaziner J, German P, Zimmerman SI, et al; Epidemiology of Dementia in Nursing Homes Research Group. The prevalence of dementia in a statewide sample of new nursing home admissions aged 65 or older: diagnosis by expert panel. Gerontologist. 2000;40(6):663-672.
6. Jakob A, Busse A, Riedel-Heller SG, et al. Prevalence and incidence of dementia among nursing home residents and residents in homes for the aged in comparison to private homes [in German]. Z Gerontol Geriatr. 2002;35(5):474-481.
7. Sachs GA, Shega JW, Cox-Hayley D. Barriers to excellent end-of-life care for patients with dementia. J Gen Intern Med. 2004;19(10):1057-1063.
8. Mitchell SL, Kiely DK, Hamel MB. Dying with advanced dementia in the nursing home. Arch Intern Med. 2004;164(3):321-326.
9. Alzheimer’s Association. 2009 Alzheimer’s Disease Facts and Figures. www.alz.org/national/documents/report_alzfactsfigures2009.pdf. Accessed September 24, 2009.
10. Wennberg JE, Cooper MM. Dartmouth Atlas of Health Care. Washington, DC: AHA Press; 1999.
11. Forbes S, Bern-Klug M, Gessert C. End-of-life decision making for nursing home residents with dementia. J Nurs Scholarsh. 2000;32(3):251-258.
12. Alzheimer’s Association. End-of-Life Decisions: Honoring the Wishes of the Person With Alzheimer’s Disease. www.alz.org/national/docu ments/brochure_endoflifedecisions.pdf. Accessed September 23, 2009.
13. Mitchell SL, Kiely DK, Miller SC, et al. Hospice care for patients with dementia. J Pain Symptom Manage. 2007;34(1):7-16.
14. Hospice Association of America. Hospice facts and statistics (2008). www.nahc.org/facts/Hospice Stats08.pdf. Accessed October 26, 2009.
15. Mitchell SL, Morris JN, Park PS, Fried BE. Terminal care for persons with advanced dementia in the nursing home and home care settings. J Palliat Med. 2004;7(6):808-816.
16. Centers for Medicare and Medicaid Services, US Department of Health and Human Services. Medicare Hospice Benefits (2008). CMS Publication No. 02154. www.medicare.gov/publications/Pubs/pdf/02154.pdf. Accessed October 26, 2009.
17. Mitchell SL, Kiely DK, Hamel MB, et al. Estimating prognosis for nursing home residents with advanced dementia. JAMA. 2004;291(22):2734-2740.
18. Sampson EL, Candy B, Jones L. Enteral tube feeding for older people with advanced dementia. Cochrane Database Syst Rev. 2009;(2): CD007209.
19. Shega JW, Hougham GW, Stocking CB, et al. Barriers to limiting the practice of feeding tube placement in advanced dementia. J Palliat Med. 2003;6(6):885-893.
20. Haller IV, Gessert CE. Utilization of medical services at the end of life in older adults with cognitive impairment: focus on outliers. J Palliat Med. 2007;10(2):400-407.
21. Volicer L, McKee A, Hewitt S. Dementia. Neurol Clin. 2001;19(4):867-885.
22. Mohr M, Bömelburg K, Bahr J. Attempted CPR in nursing homes: life-saving at the end of life? [in German] Anasthesiol Intensivmed Notfallmed Schmerzther. 2001;36(9):566-572.
1. American Medical Association, Program on Aging and Community Health. Practical Guide for the Primary Care Physician on the Diagnosis, Management and Treatment of Dementia. Chicago, IL: American Medical Association; 2000.
2. Farias ST, Mungas D, Reed BR, et al. The measurement of everyday cognition (ECog): scale development and psychometric properties. Neuropsychology. 2008;22(4):531-544.
3. Boustani M, Peterson B, Hanson L, et al. Screening for dementia in primary care: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2003;138(11): 927-937.
4. US Preventive Services Task Force. Screening for dementia: recommendation and rationale. Ann Intern Med. 2003;138(11):925-926.
5. Magaziner J, German P, Zimmerman SI, et al; Epidemiology of Dementia in Nursing Homes Research Group. The prevalence of dementia in a statewide sample of new nursing home admissions aged 65 or older: diagnosis by expert panel. Gerontologist. 2000;40(6):663-672.
6. Jakob A, Busse A, Riedel-Heller SG, et al. Prevalence and incidence of dementia among nursing home residents and residents in homes for the aged in comparison to private homes [in German]. Z Gerontol Geriatr. 2002;35(5):474-481.
7. Sachs GA, Shega JW, Cox-Hayley D. Barriers to excellent end-of-life care for patients with dementia. J Gen Intern Med. 2004;19(10):1057-1063.
8. Mitchell SL, Kiely DK, Hamel MB. Dying with advanced dementia in the nursing home. Arch Intern Med. 2004;164(3):321-326.
9. Alzheimer’s Association. 2009 Alzheimer’s Disease Facts and Figures. www.alz.org/national/documents/report_alzfactsfigures2009.pdf. Accessed September 24, 2009.
10. Wennberg JE, Cooper MM. Dartmouth Atlas of Health Care. Washington, DC: AHA Press; 1999.
11. Forbes S, Bern-Klug M, Gessert C. End-of-life decision making for nursing home residents with dementia. J Nurs Scholarsh. 2000;32(3):251-258.
12. Alzheimer’s Association. End-of-Life Decisions: Honoring the Wishes of the Person With Alzheimer’s Disease. www.alz.org/national/docu ments/brochure_endoflifedecisions.pdf. Accessed September 23, 2009.
13. Mitchell SL, Kiely DK, Miller SC, et al. Hospice care for patients with dementia. J Pain Symptom Manage. 2007;34(1):7-16.
14. Hospice Association of America. Hospice facts and statistics (2008). www.nahc.org/facts/Hospice Stats08.pdf. Accessed October 26, 2009.
15. Mitchell SL, Morris JN, Park PS, Fried BE. Terminal care for persons with advanced dementia in the nursing home and home care settings. J Palliat Med. 2004;7(6):808-816.
16. Centers for Medicare and Medicaid Services, US Department of Health and Human Services. Medicare Hospice Benefits (2008). CMS Publication No. 02154. www.medicare.gov/publications/Pubs/pdf/02154.pdf. Accessed October 26, 2009.
17. Mitchell SL, Kiely DK, Hamel MB, et al. Estimating prognosis for nursing home residents with advanced dementia. JAMA. 2004;291(22):2734-2740.
18. Sampson EL, Candy B, Jones L. Enteral tube feeding for older people with advanced dementia. Cochrane Database Syst Rev. 2009;(2): CD007209.
19. Shega JW, Hougham GW, Stocking CB, et al. Barriers to limiting the practice of feeding tube placement in advanced dementia. J Palliat Med. 2003;6(6):885-893.
20. Haller IV, Gessert CE. Utilization of medical services at the end of life in older adults with cognitive impairment: focus on outliers. J Palliat Med. 2007;10(2):400-407.
21. Volicer L, McKee A, Hewitt S. Dementia. Neurol Clin. 2001;19(4):867-885.
22. Mohr M, Bömelburg K, Bahr J. Attempted CPR in nursing homes: life-saving at the end of life? [in German] Anasthesiol Intensivmed Notfallmed Schmerzther. 2001;36(9):566-572.
Evaluation and Management of Headache in the Pediatric Patient
Multidrug-Resistant Gram-Negative Bacteria: Trends, Risk Factors, and Treatments
Clostridium difficile: An Old Player With a New Hand in the Game
Wound Management for Severe Open Fractures: Use of Antibiotic Bead Pouches and Vacuum-Assisted Closure
Acetabular Component Revision in Total Hip Arthroplasty. Part II: Management of Major Bone Loss and Pelvic Discontinuity
When starting an antidepressant, try either of these 2 drugs first
The authors report no financial relationships relevant to this article.
Meta-analysis of 117 high-quality studies found that sertraline and escitalopram are superior to other “new-generation” antidepressants.1
CASE: A woman with diabetes who is fatigued but cannot sleep
Mrs. D., 45 years old, has been your patient for several years. She has type 2 diabetes. On her latest visit, she reports a loss of energy and difficulty sleeping, and wonders if these symptoms could be related to the diabetes.
As you explore further and question Mrs. D. about her symptoms, she becomes tearful, and tells you she has episodes of sadness and no longer enjoys things the way she used to. Although she has no history of depression, when you suggest that her symptoms may be an indication of depression, she readily agrees.
You discuss treatment options, including antidepressants and psychotherapy. Mrs. D. decides to try medication. But with so many antidepressants on the market, how do you choose one?
Major depression is the fourth leading cause of disease globally, according to the World Health Organization.2 Depression is common in the United States as well, and primary care physicians, including ObGyns, are often the ones who are diagnosing and treating it. In fact, the US Preventive Services Task Force recently expanded its recommendation that primary care providers screen adults for depression, to include adolescents 12 to 18 years old.3 When depression is diagnosed, physicians must help patients decide on an initial treatment plan.
Not all antidepressants are equal
Options for initial treatment of unipolar major depression include psychotherapy and the use of an antidepressant. For mild and moderate depression, psychotherapy alone is as effective as medication. Combined psychotherapy and antidepressants are more effective than either treatment alone for all degrees of depression.4
The ideal medication for depression would be a drug with a high level of effectiveness and a low side-effect profile; until now, however, there has been little evidence to support one antidepressant over another. Previous meta-analyses have concluded that there are no significant differences in either efficacy or acceptability among the various second-generation antidepressants on the market.5,6 Therefore, physicians have historically made initial monotherapy treatment decisions based on side effects and cost.7,8 The meta-analysis we report here tells a different story, providing strong evidence that some antidepressants are more effective and better tolerated than others.
Two “best” drugs revealed
Cipriani and colleagues1 conducted a systematic review and multiple-treatments meta-analysis of 117 prospective randomized, controlled trials (RCTs). Taken together, the RCTs evaluated the comparative efficacy and acceptability of 12 second-generation antidepressants: bupropion, citalopram, duloxetine, escitalopram, fluoxetine, fluvoxamine, milnacipran, mirtazapine, paroxetine, reboxetine, sertraline, and venlafaxine.
The methodology of this meta-analysis differed from that of traditional meta-analyses by allowing the integration of data from both direct and indirect comparisons. (An indirect comparison is one in which drugs from different trials are assessed by combining the results of their effectiveness and comparing the combined finding with the effectiveness of a drug that all the trials have in common.) Previous studies, based only on direct comparison, yielded inconsistent results.
The studies included in this meta-analysis were all RCTs in which one of these 12 antidepressants was tested against one, or several, other second-generation antidepressants as monotherapy for the acute treatment phase of unipolar major depression. The authors excluded placebo-controlled trials in order to evaluate efficacy and acceptability of the study medications relative to other commonly used antidepressants. They defined acute treatment as 8 weeks of antidepressant therapy, with a range of 6 to 12 weeks. The primary outcomes studied were response to treatment and dropout rate.
Response to treatment (efficacy) was constructed as a Yes or No variable; a positive response was defined as a reduction of ≥50% in symptom score on either the Hamilton Depression Rating Scale or the Montgomery-Asberg Rating Scale, or a rating of “improved” or “very much improved” on the Clinical Global Impression scale at 8 weeks. Efficacy was calculated on an intention-to-treat basis; if data were missing for a participant, that person was classified as a nonresponder.
Dropout rate was used to represent acceptability, because the authors believed it to be a more clinically meaningful measure than either side effects or symptom scores. Comparative efficacy and acceptability were analyzed. Fluoxetine—the first of the secondgeneration antidepressants—was used as the reference medication. The FIGURE shows the outcomes for nine of the antidepressants, compared with those of fluoxetine. The other two antidepressants, milnacipran and reboxetine, were omitted because they are not available in the United States.
The overall meta-analysis included 25,928 individuals, with 24,595 in the efficacy analysis and 24,693 in the acceptability analysis. Nearly two thirds (64%) of the participants were women. The mean duration of follow-up was 8.1 weeks. The mean sample size per study was 110.
Studies of women with postpartum depression were excluded.
Escitalopram and sertraline stand out. Overall, escitalopram, mirtazapine, sertraline, and venlafaxine were significantly more efficacious than fluoxetine or the other medications. Bupropion, citalopram, escitalopram, and sertraline were better tolerated than the other antidepressants. Escitalopram and sertraline were found to have the best combination of efficacy and acceptability.
Efficacy results. Fifty-nine percent of participants responded to sertraline, versus a 52% response rate for fluoxetine (number needed to treat [NNT]=14). Similarly, 52% of participants responded to escitalopram, compared with 47% of those taking fluoxetine (NNT=20).
Acceptability results. In terms of the dropout rate, 28% of participants discontinued fluoxetine, versus 24% of patients taking sertraline. This means that 25 patients would need to be treated with sertraline, rather than fluoxetine, to avoid one discontinuation. In the comparison of fluoxetine versus escitalopram, 25% discontinued fluoxetine, compared with 24% who discontinued escitalopram.
The efficacy and acceptability of sertraline and escitalopram compared with other second-generation antidepressant medications follow similar trends.
Generic advantage. The investigators recommend sertraline as the best choice for an initial antidepressant because it is available in generic form and is therefore lower in cost. They further recommend that sertraline, instead of fluoxetine or placebo, be the new standard against which other antidepressants are compared.

FIGURE Sertraline and escitalopram come out on top in acceptability and efficacy
Researchers analyzed a number of second-generation antidepressants, using fluoxetine as the reference medication. Sertraline and escitalopram provided the best combination of efficacy and acceptability.1
Choice is now evidence-based
We now have solid evidence for choosing sertraline or escitalopram as the first medication to use when treating a patient with newly diagnosed depression. This represents a practice change because antidepressants that are less effective and less acceptable have been chosen more frequently than either of these medications. That conclusion is based on our analysis of the National Ambulatory Medical Care Survey database for outpatient and ambulatory clinic visits in 2005-2006 (the most recent data available). We conducted this analysis to determine which of the second-generation antidepressants were prescribed more often for initial monotherapy of major depression.
Our finding? An estimated 4 million patients 18 years and older given a diagnosis of depression in the course of the study year received new prescriptions for a single antidepressant. Six medications accounted for 90% of prescriptions, in this order:
- fluoxetine (Prozac)
- duloxetine (Cymbalta)
- escitalopram (Lexapro)
- paroxetine (Paxil)
- venlafaxine (Effexor)
- sertraline (Zoloft).
Sertraline and escitalopram, the drugs shown to be most effective and acceptable in the Cipriani meta-analysis, accounted for 11.8% and 14.5% of the prescriptions, respectively.
Caveats
This meta-analysis looked at the acute phase of treatment only
The results of this study are limited to initial therapy as measured at 8 weeks. Few long-term outcome data are available; response to initial therapy may not be a predictor of full remission or long-term success. Current guidelines suggest maintenance of the initial successful therapy, often with increasing intervals between visits, to prevent relapse.9
This study does not add new insight into long-term response rates. Nor does it deal with choice of a replacement or second antidepressant for nonresponders or those who cannot tolerate the initial drug.
What’s more, the study covers drug treatment alone, which may not be the best initial treatment for depression. Psychotherapy, in the form of cognitive behavioral therapy or interpersonal therapy, when available, is equally effective, has fewer potential physiologic side effects, and may produce longer-lasting results.10,11
Little is known about study design
The authors of this study had access only to limited information about inclusion criteria and the composition of initial study populations or settings. There is a difference between a trial designed to evaluate the “efficacy” of an intervention (i.e., “the beneficial and harmful effects of an intervention under controlled circumstances”) and the “effectiveness” of an intervention (i.e., the “beneficial and harmful effects of the intervention under usual circumstances”).12 It is not clear which of the 117 studies were efficacy studies and which were effectiveness studies. This may limit the overall generalizability of the study results to a primary care population.
Studies included in this meta-analysis were selected exclusively from published literature. There is some evidence of a bias toward the publication of studies that have yielded positive results, which may have the effect of overstating the effectiveness of a given antidepressant.13 However, we have no reason to believe that this bias would favor any particular drug.
Most of the included studies were sponsored by drug companies. Notably, pharmaceutical companies have the option of continuing to conduct trials of medications until a study results in a positive finding for their medication, with no penalty for the suppression of equivocal or negative results (negative publication bias). Under current FDA guidelines, there is little transparency for the consumer as to how many trials have been undertaken and the direction of the results, published or unpublished.14
We doubt that either publication bias or the design and sponsorship of the studies included in this meta-analysis present significant threats to the validity of these findings over other sources upon which guidelines rely, given that these issues are common to much of the research on pharmacotherapy. We also doubt that the compensation of the authors by pharmaceutical companies would bias the outcome of the study, in this instance. One of the authors (Furukawa) received compensation from Pfizer, the maker of Zoloft, which is also available as generic sertraline. None of the authors received compensation from Forest Pharmaceuticals, the maker of Lexapro (escitalopram).
No major barriers anticipated
Both sertraline and escitalopram are covered by most health insurers. As noted, sertraline is available in a generic formulation and is therefore much less expensive than escitalopram.
In a review of drug prices at www.pharmacychecker.com, we found that a prescription for a 3-month supply of Lexapro (10 mg) costs about $250. A 3-month supply of generic sertraline (100 mg) from the same sources costs approximately $35. Pfizer, maker of Zoloft, and Forest Pharmaceuticals, maker of Lexapro, both administer patient assistance programs to make these medications available to low-income, uninsured patients.
When you initiate an antidepressant for a patient who has not been treated for depression in the past, select either sertraline (Zoloft) or escitalopram (Lexapro).
Acknowledgment
Sofia Medvedev, PhD, of the University HealthSystem Consortium, Oak Brook, Ill., analyzed data from the National Ambulatory Medical Care Survey and the UHC Clinical Database as part of the development of the manuscript of this article.
1. Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373:746-758.
2. Murray CJ, Lopez AD. Global Burden of Disease. Cambridge, Mass: Harvard University Press; 1996.
3. Williams SB, O’Connor EA, Eder M, et al. Screening for child and adolescent depression in primary care settings: a systematic evidence review for the U.S. Preventive Services Task Force. Pediatrics. 2009;123:e716-e735.
4. Timonen M, Liukkonen T. Management of depression in adults. BMJ. 2008;336:435-439.
5. Gartlehner G, Hansen RA, Thieda P, et al. Comparative Effectiveness of Second-Generation Antidepressants in the Pharmacologic Treatment of Adult Depression. Comparative Effectiveness Review No. 7. (Prepared by RTI International–University of North Carolina Evidence-based Practice Center under Contract No. 290-02-0016.) Rockville, Md: Agency for Healthcare Research and Quality; January 2007. Available at: www.effectivehealthcare.ahrq.gov/reports/final.cfm. Accessed May 18, 2009.
6. Hansen RA, Gartlehner G, Lohr KN, et al. Efficacy and safety of second-generation antidepressants in the treatment of major depressive disorder. Ann Intern Med. 2005;143:415-426.
7. Adams SM, Miller KE, Zylstra RG. Pharmacologic management of adult depression. Am Fam Physician. 2008;77:785-792.
8. Qaseem A, Snow V, Denberg TD, et al. Using second-generation antidepressants to treat depressive disorders: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;149:725-733.
9. DeRubeis RJ, Hollon SD, Amsterdam JD, et al. Cognitive therapy vs medications in the treatment of moderate to severe depression. Arch Gen Psychiatry. 2005;62:409-416.
10. deMello MF, de Jesus MJ, Bacaltchuk J, et al. A systematic review of research findings on the efficacy of interpersonal therapy for depressive disorders. Eur Arch Psychiatry Clin Neurosci. 2005;255:75-82.
11. APA Practice Guidelines. Practice guideline for the treatment of patients with major depressive disorder. 2nd ed. Available at: www.psychiatryonline.com/content.aspx?aID=48727. Accessed October 15, 2009.
12. Sackett D. An introduction to performing therapeutic trials. In: Haynes RB, Sackett DL, Guyatt GH, Tugwell P. Clinical Epidemiology: How to Do Clinical Practice Research. 3rd ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2006.
13. Turner EH, Matthews AM, Linardatos E, et al. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358:252-260.
14. Mathew SJ, Charney DS. Publication bias and the efficacy of antidepressants. Am J Psychiatry. 2009;166:140-145.
The authors report no financial relationships relevant to this article.
Meta-analysis of 117 high-quality studies found that sertraline and escitalopram are superior to other “new-generation” antidepressants.1
CASE: A woman with diabetes who is fatigued but cannot sleep
Mrs. D., 45 years old, has been your patient for several years. She has type 2 diabetes. On her latest visit, she reports a loss of energy and difficulty sleeping, and wonders if these symptoms could be related to the diabetes.
As you explore further and question Mrs. D. about her symptoms, she becomes tearful, and tells you she has episodes of sadness and no longer enjoys things the way she used to. Although she has no history of depression, when you suggest that her symptoms may be an indication of depression, she readily agrees.
You discuss treatment options, including antidepressants and psychotherapy. Mrs. D. decides to try medication. But with so many antidepressants on the market, how do you choose one?
Major depression is the fourth leading cause of disease globally, according to the World Health Organization.2 Depression is common in the United States as well, and primary care physicians, including ObGyns, are often the ones who are diagnosing and treating it. In fact, the US Preventive Services Task Force recently expanded its recommendation that primary care providers screen adults for depression, to include adolescents 12 to 18 years old.3 When depression is diagnosed, physicians must help patients decide on an initial treatment plan.
Not all antidepressants are equal
Options for initial treatment of unipolar major depression include psychotherapy and the use of an antidepressant. For mild and moderate depression, psychotherapy alone is as effective as medication. Combined psychotherapy and antidepressants are more effective than either treatment alone for all degrees of depression.4
The ideal medication for depression would be a drug with a high level of effectiveness and a low side-effect profile; until now, however, there has been little evidence to support one antidepressant over another. Previous meta-analyses have concluded that there are no significant differences in either efficacy or acceptability among the various second-generation antidepressants on the market.5,6 Therefore, physicians have historically made initial monotherapy treatment decisions based on side effects and cost.7,8 The meta-analysis we report here tells a different story, providing strong evidence that some antidepressants are more effective and better tolerated than others.
Two “best” drugs revealed
Cipriani and colleagues1 conducted a systematic review and multiple-treatments meta-analysis of 117 prospective randomized, controlled trials (RCTs). Taken together, the RCTs evaluated the comparative efficacy and acceptability of 12 second-generation antidepressants: bupropion, citalopram, duloxetine, escitalopram, fluoxetine, fluvoxamine, milnacipran, mirtazapine, paroxetine, reboxetine, sertraline, and venlafaxine.
The methodology of this meta-analysis differed from that of traditional meta-analyses by allowing the integration of data from both direct and indirect comparisons. (An indirect comparison is one in which drugs from different trials are assessed by combining the results of their effectiveness and comparing the combined finding with the effectiveness of a drug that all the trials have in common.) Previous studies, based only on direct comparison, yielded inconsistent results.
The studies included in this meta-analysis were all RCTs in which one of these 12 antidepressants was tested against one, or several, other second-generation antidepressants as monotherapy for the acute treatment phase of unipolar major depression. The authors excluded placebo-controlled trials in order to evaluate efficacy and acceptability of the study medications relative to other commonly used antidepressants. They defined acute treatment as 8 weeks of antidepressant therapy, with a range of 6 to 12 weeks. The primary outcomes studied were response to treatment and dropout rate.
Response to treatment (efficacy) was constructed as a Yes or No variable; a positive response was defined as a reduction of ≥50% in symptom score on either the Hamilton Depression Rating Scale or the Montgomery-Asberg Rating Scale, or a rating of “improved” or “very much improved” on the Clinical Global Impression scale at 8 weeks. Efficacy was calculated on an intention-to-treat basis; if data were missing for a participant, that person was classified as a nonresponder.
Dropout rate was used to represent acceptability, because the authors believed it to be a more clinically meaningful measure than either side effects or symptom scores. Comparative efficacy and acceptability were analyzed. Fluoxetine—the first of the secondgeneration antidepressants—was used as the reference medication. The FIGURE shows the outcomes for nine of the antidepressants, compared with those of fluoxetine. The other two antidepressants, milnacipran and reboxetine, were omitted because they are not available in the United States.
The overall meta-analysis included 25,928 individuals, with 24,595 in the efficacy analysis and 24,693 in the acceptability analysis. Nearly two thirds (64%) of the participants were women. The mean duration of follow-up was 8.1 weeks. The mean sample size per study was 110.
Studies of women with postpartum depression were excluded.
Escitalopram and sertraline stand out. Overall, escitalopram, mirtazapine, sertraline, and venlafaxine were significantly more efficacious than fluoxetine or the other medications. Bupropion, citalopram, escitalopram, and sertraline were better tolerated than the other antidepressants. Escitalopram and sertraline were found to have the best combination of efficacy and acceptability.
Efficacy results. Fifty-nine percent of participants responded to sertraline, versus a 52% response rate for fluoxetine (number needed to treat [NNT]=14). Similarly, 52% of participants responded to escitalopram, compared with 47% of those taking fluoxetine (NNT=20).
Acceptability results. In terms of the dropout rate, 28% of participants discontinued fluoxetine, versus 24% of patients taking sertraline. This means that 25 patients would need to be treated with sertraline, rather than fluoxetine, to avoid one discontinuation. In the comparison of fluoxetine versus escitalopram, 25% discontinued fluoxetine, compared with 24% who discontinued escitalopram.
The efficacy and acceptability of sertraline and escitalopram compared with other second-generation antidepressant medications follow similar trends.
Generic advantage. The investigators recommend sertraline as the best choice for an initial antidepressant because it is available in generic form and is therefore lower in cost. They further recommend that sertraline, instead of fluoxetine or placebo, be the new standard against which other antidepressants are compared.
FIGURE Sertraline and escitalopram come out on top in acceptability and efficacy
Researchers analyzed a number of second-generation antidepressants, using fluoxetine as the reference medication. Sertraline and escitalopram provided the best combination of efficacy and acceptability.1
Choice is now evidence-based
We now have solid evidence for choosing sertraline or escitalopram as the first medication to use when treating a patient with newly diagnosed depression. This represents a practice change because antidepressants that are less effective and less acceptable have been chosen more frequently than either of these medications. That conclusion is based on our analysis of the National Ambulatory Medical Care Survey database for outpatient and ambulatory clinic visits in 2005-2006 (the most recent data available). We conducted this analysis to determine which of the second-generation antidepressants were prescribed more often for initial monotherapy of major depression.
Our finding? An estimated 4 million patients 18 years and older given a diagnosis of depression in the course of the study year received new prescriptions for a single antidepressant. Six medications accounted for 90% of prescriptions, in this order:
- fluoxetine (Prozac)
- duloxetine (Cymbalta)
- escitalopram (Lexapro)
- paroxetine (Paxil)
- venlafaxine (Effexor)
- sertraline (Zoloft).
Sertraline and escitalopram, the drugs shown to be most effective and acceptable in the Cipriani meta-analysis, accounted for 11.8% and 14.5% of the prescriptions, respectively.
Caveats
This meta-analysis looked at the acute phase of treatment only
The results of this study are limited to initial therapy as measured at 8 weeks. Few long-term outcome data are available; response to initial therapy may not be a predictor of full remission or long-term success. Current guidelines suggest maintenance of the initial successful therapy, often with increasing intervals between visits, to prevent relapse.9
This study does not add new insight into long-term response rates. Nor does it deal with choice of a replacement or second antidepressant for nonresponders or those who cannot tolerate the initial drug.
What’s more, the study covers drug treatment alone, which may not be the best initial treatment for depression. Psychotherapy, in the form of cognitive behavioral therapy or interpersonal therapy, when available, is equally effective, has fewer potential physiologic side effects, and may produce longer-lasting results.10,11
Little is known about study design
The authors of this study had access only to limited information about inclusion criteria and the composition of initial study populations or settings. There is a difference between a trial designed to evaluate the “efficacy” of an intervention (i.e., “the beneficial and harmful effects of an intervention under controlled circumstances”) and the “effectiveness” of an intervention (i.e., the “beneficial and harmful effects of the intervention under usual circumstances”).12 It is not clear which of the 117 studies were efficacy studies and which were effectiveness studies. This may limit the overall generalizability of the study results to a primary care population.
Studies included in this meta-analysis were selected exclusively from published literature. There is some evidence of a bias toward the publication of studies that have yielded positive results, which may have the effect of overstating the effectiveness of a given antidepressant.13 However, we have no reason to believe that this bias would favor any particular drug.
Most of the included studies were sponsored by drug companies. Notably, pharmaceutical companies have the option of continuing to conduct trials of medications until a study results in a positive finding for their medication, with no penalty for the suppression of equivocal or negative results (negative publication bias). Under current FDA guidelines, there is little transparency for the consumer as to how many trials have been undertaken and the direction of the results, published or unpublished.14
We doubt that either publication bias or the design and sponsorship of the studies included in this meta-analysis present significant threats to the validity of these findings over other sources upon which guidelines rely, given that these issues are common to much of the research on pharmacotherapy. We also doubt that the compensation of the authors by pharmaceutical companies would bias the outcome of the study, in this instance. One of the authors (Furukawa) received compensation from Pfizer, the maker of Zoloft, which is also available as generic sertraline. None of the authors received compensation from Forest Pharmaceuticals, the maker of Lexapro (escitalopram).
No major barriers anticipated
Both sertraline and escitalopram are covered by most health insurers. As noted, sertraline is available in a generic formulation and is therefore much less expensive than escitalopram.
In a review of drug prices at www.pharmacychecker.com, we found that a prescription for a 3-month supply of Lexapro (10 mg) costs about $250. A 3-month supply of generic sertraline (100 mg) from the same sources costs approximately $35. Pfizer, maker of Zoloft, and Forest Pharmaceuticals, maker of Lexapro, both administer patient assistance programs to make these medications available to low-income, uninsured patients.
When you initiate an antidepressant for a patient who has not been treated for depression in the past, select either sertraline (Zoloft) or escitalopram (Lexapro).
Acknowledgment
Sofia Medvedev, PhD, of the University HealthSystem Consortium, Oak Brook, Ill., analyzed data from the National Ambulatory Medical Care Survey and the UHC Clinical Database as part of the development of the manuscript of this article.
The authors report no financial relationships relevant to this article.
Meta-analysis of 117 high-quality studies found that sertraline and escitalopram are superior to other “new-generation” antidepressants.1
CASE: A woman with diabetes who is fatigued but cannot sleep
Mrs. D., 45 years old, has been your patient for several years. She has type 2 diabetes. On her latest visit, she reports a loss of energy and difficulty sleeping, and wonders if these symptoms could be related to the diabetes.
As you explore further and question Mrs. D. about her symptoms, she becomes tearful, and tells you she has episodes of sadness and no longer enjoys things the way she used to. Although she has no history of depression, when you suggest that her symptoms may be an indication of depression, she readily agrees.
You discuss treatment options, including antidepressants and psychotherapy. Mrs. D. decides to try medication. But with so many antidepressants on the market, how do you choose one?
Major depression is the fourth leading cause of disease globally, according to the World Health Organization.2 Depression is common in the United States as well, and primary care physicians, including ObGyns, are often the ones who are diagnosing and treating it. In fact, the US Preventive Services Task Force recently expanded its recommendation that primary care providers screen adults for depression, to include adolescents 12 to 18 years old.3 When depression is diagnosed, physicians must help patients decide on an initial treatment plan.
Not all antidepressants are equal
Options for initial treatment of unipolar major depression include psychotherapy and the use of an antidepressant. For mild and moderate depression, psychotherapy alone is as effective as medication. Combined psychotherapy and antidepressants are more effective than either treatment alone for all degrees of depression.4
The ideal medication for depression would be a drug with a high level of effectiveness and a low side-effect profile; until now, however, there has been little evidence to support one antidepressant over another. Previous meta-analyses have concluded that there are no significant differences in either efficacy or acceptability among the various second-generation antidepressants on the market.5,6 Therefore, physicians have historically made initial monotherapy treatment decisions based on side effects and cost.7,8 The meta-analysis we report here tells a different story, providing strong evidence that some antidepressants are more effective and better tolerated than others.
Two “best” drugs revealed
Cipriani and colleagues1 conducted a systematic review and multiple-treatments meta-analysis of 117 prospective randomized, controlled trials (RCTs). Taken together, the RCTs evaluated the comparative efficacy and acceptability of 12 second-generation antidepressants: bupropion, citalopram, duloxetine, escitalopram, fluoxetine, fluvoxamine, milnacipran, mirtazapine, paroxetine, reboxetine, sertraline, and venlafaxine.
The methodology of this meta-analysis differed from that of traditional meta-analyses by allowing the integration of data from both direct and indirect comparisons. (An indirect comparison is one in which drugs from different trials are assessed by combining the results of their effectiveness and comparing the combined finding with the effectiveness of a drug that all the trials have in common.) Previous studies, based only on direct comparison, yielded inconsistent results.
The studies included in this meta-analysis were all RCTs in which one of these 12 antidepressants was tested against one, or several, other second-generation antidepressants as monotherapy for the acute treatment phase of unipolar major depression. The authors excluded placebo-controlled trials in order to evaluate efficacy and acceptability of the study medications relative to other commonly used antidepressants. They defined acute treatment as 8 weeks of antidepressant therapy, with a range of 6 to 12 weeks. The primary outcomes studied were response to treatment and dropout rate.
Response to treatment (efficacy) was constructed as a Yes or No variable; a positive response was defined as a reduction of ≥50% in symptom score on either the Hamilton Depression Rating Scale or the Montgomery-Asberg Rating Scale, or a rating of “improved” or “very much improved” on the Clinical Global Impression scale at 8 weeks. Efficacy was calculated on an intention-to-treat basis; if data were missing for a participant, that person was classified as a nonresponder.
Dropout rate was used to represent acceptability, because the authors believed it to be a more clinically meaningful measure than either side effects or symptom scores. Comparative efficacy and acceptability were analyzed. Fluoxetine—the first of the secondgeneration antidepressants—was used as the reference medication. The FIGURE shows the outcomes for nine of the antidepressants, compared with those of fluoxetine. The other two antidepressants, milnacipran and reboxetine, were omitted because they are not available in the United States.
The overall meta-analysis included 25,928 individuals, with 24,595 in the efficacy analysis and 24,693 in the acceptability analysis. Nearly two thirds (64%) of the participants were women. The mean duration of follow-up was 8.1 weeks. The mean sample size per study was 110.
Studies of women with postpartum depression were excluded.
Escitalopram and sertraline stand out. Overall, escitalopram, mirtazapine, sertraline, and venlafaxine were significantly more efficacious than fluoxetine or the other medications. Bupropion, citalopram, escitalopram, and sertraline were better tolerated than the other antidepressants. Escitalopram and sertraline were found to have the best combination of efficacy and acceptability.
Efficacy results. Fifty-nine percent of participants responded to sertraline, versus a 52% response rate for fluoxetine (number needed to treat [NNT]=14). Similarly, 52% of participants responded to escitalopram, compared with 47% of those taking fluoxetine (NNT=20).
Acceptability results. In terms of the dropout rate, 28% of participants discontinued fluoxetine, versus 24% of patients taking sertraline. This means that 25 patients would need to be treated with sertraline, rather than fluoxetine, to avoid one discontinuation. In the comparison of fluoxetine versus escitalopram, 25% discontinued fluoxetine, compared with 24% who discontinued escitalopram.
The efficacy and acceptability of sertraline and escitalopram compared with other second-generation antidepressant medications follow similar trends.
Generic advantage. The investigators recommend sertraline as the best choice for an initial antidepressant because it is available in generic form and is therefore lower in cost. They further recommend that sertraline, instead of fluoxetine or placebo, be the new standard against which other antidepressants are compared.
FIGURE Sertraline and escitalopram come out on top in acceptability and efficacy
Researchers analyzed a number of second-generation antidepressants, using fluoxetine as the reference medication. Sertraline and escitalopram provided the best combination of efficacy and acceptability.1
Choice is now evidence-based
We now have solid evidence for choosing sertraline or escitalopram as the first medication to use when treating a patient with newly diagnosed depression. This represents a practice change because antidepressants that are less effective and less acceptable have been chosen more frequently than either of these medications. That conclusion is based on our analysis of the National Ambulatory Medical Care Survey database for outpatient and ambulatory clinic visits in 2005-2006 (the most recent data available). We conducted this analysis to determine which of the second-generation antidepressants were prescribed more often for initial monotherapy of major depression.
Our finding? An estimated 4 million patients 18 years and older given a diagnosis of depression in the course of the study year received new prescriptions for a single antidepressant. Six medications accounted for 90% of prescriptions, in this order:
- fluoxetine (Prozac)
- duloxetine (Cymbalta)
- escitalopram (Lexapro)
- paroxetine (Paxil)
- venlafaxine (Effexor)
- sertraline (Zoloft).
Sertraline and escitalopram, the drugs shown to be most effective and acceptable in the Cipriani meta-analysis, accounted for 11.8% and 14.5% of the prescriptions, respectively.
Caveats
This meta-analysis looked at the acute phase of treatment only
The results of this study are limited to initial therapy as measured at 8 weeks. Few long-term outcome data are available; response to initial therapy may not be a predictor of full remission or long-term success. Current guidelines suggest maintenance of the initial successful therapy, often with increasing intervals between visits, to prevent relapse.9
This study does not add new insight into long-term response rates. Nor does it deal with choice of a replacement or second antidepressant for nonresponders or those who cannot tolerate the initial drug.
What’s more, the study covers drug treatment alone, which may not be the best initial treatment for depression. Psychotherapy, in the form of cognitive behavioral therapy or interpersonal therapy, when available, is equally effective, has fewer potential physiologic side effects, and may produce longer-lasting results.10,11
Little is known about study design
The authors of this study had access only to limited information about inclusion criteria and the composition of initial study populations or settings. There is a difference between a trial designed to evaluate the “efficacy” of an intervention (i.e., “the beneficial and harmful effects of an intervention under controlled circumstances”) and the “effectiveness” of an intervention (i.e., the “beneficial and harmful effects of the intervention under usual circumstances”).12 It is not clear which of the 117 studies were efficacy studies and which were effectiveness studies. This may limit the overall generalizability of the study results to a primary care population.
Studies included in this meta-analysis were selected exclusively from published literature. There is some evidence of a bias toward the publication of studies that have yielded positive results, which may have the effect of overstating the effectiveness of a given antidepressant.13 However, we have no reason to believe that this bias would favor any particular drug.
Most of the included studies were sponsored by drug companies. Notably, pharmaceutical companies have the option of continuing to conduct trials of medications until a study results in a positive finding for their medication, with no penalty for the suppression of equivocal or negative results (negative publication bias). Under current FDA guidelines, there is little transparency for the consumer as to how many trials have been undertaken and the direction of the results, published or unpublished.14
We doubt that either publication bias or the design and sponsorship of the studies included in this meta-analysis present significant threats to the validity of these findings over other sources upon which guidelines rely, given that these issues are common to much of the research on pharmacotherapy. We also doubt that the compensation of the authors by pharmaceutical companies would bias the outcome of the study, in this instance. One of the authors (Furukawa) received compensation from Pfizer, the maker of Zoloft, which is also available as generic sertraline. None of the authors received compensation from Forest Pharmaceuticals, the maker of Lexapro (escitalopram).
No major barriers anticipated
Both sertraline and escitalopram are covered by most health insurers. As noted, sertraline is available in a generic formulation and is therefore much less expensive than escitalopram.
In a review of drug prices at www.pharmacychecker.com, we found that a prescription for a 3-month supply of Lexapro (10 mg) costs about $250. A 3-month supply of generic sertraline (100 mg) from the same sources costs approximately $35. Pfizer, maker of Zoloft, and Forest Pharmaceuticals, maker of Lexapro, both administer patient assistance programs to make these medications available to low-income, uninsured patients.
When you initiate an antidepressant for a patient who has not been treated for depression in the past, select either sertraline (Zoloft) or escitalopram (Lexapro).
Acknowledgment
Sofia Medvedev, PhD, of the University HealthSystem Consortium, Oak Brook, Ill., analyzed data from the National Ambulatory Medical Care Survey and the UHC Clinical Database as part of the development of the manuscript of this article.
1. Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373:746-758.
2. Murray CJ, Lopez AD. Global Burden of Disease. Cambridge, Mass: Harvard University Press; 1996.
3. Williams SB, O’Connor EA, Eder M, et al. Screening for child and adolescent depression in primary care settings: a systematic evidence review for the U.S. Preventive Services Task Force. Pediatrics. 2009;123:e716-e735.
4. Timonen M, Liukkonen T. Management of depression in adults. BMJ. 2008;336:435-439.
5. Gartlehner G, Hansen RA, Thieda P, et al. Comparative Effectiveness of Second-Generation Antidepressants in the Pharmacologic Treatment of Adult Depression. Comparative Effectiveness Review No. 7. (Prepared by RTI International–University of North Carolina Evidence-based Practice Center under Contract No. 290-02-0016.) Rockville, Md: Agency for Healthcare Research and Quality; January 2007. Available at: www.effectivehealthcare.ahrq.gov/reports/final.cfm. Accessed May 18, 2009.
6. Hansen RA, Gartlehner G, Lohr KN, et al. Efficacy and safety of second-generation antidepressants in the treatment of major depressive disorder. Ann Intern Med. 2005;143:415-426.
7. Adams SM, Miller KE, Zylstra RG. Pharmacologic management of adult depression. Am Fam Physician. 2008;77:785-792.
8. Qaseem A, Snow V, Denberg TD, et al. Using second-generation antidepressants to treat depressive disorders: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;149:725-733.
9. DeRubeis RJ, Hollon SD, Amsterdam JD, et al. Cognitive therapy vs medications in the treatment of moderate to severe depression. Arch Gen Psychiatry. 2005;62:409-416.
10. deMello MF, de Jesus MJ, Bacaltchuk J, et al. A systematic review of research findings on the efficacy of interpersonal therapy for depressive disorders. Eur Arch Psychiatry Clin Neurosci. 2005;255:75-82.
11. APA Practice Guidelines. Practice guideline for the treatment of patients with major depressive disorder. 2nd ed. Available at: www.psychiatryonline.com/content.aspx?aID=48727. Accessed October 15, 2009.
12. Sackett D. An introduction to performing therapeutic trials. In: Haynes RB, Sackett DL, Guyatt GH, Tugwell P. Clinical Epidemiology: How to Do Clinical Practice Research. 3rd ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2006.
13. Turner EH, Matthews AM, Linardatos E, et al. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358:252-260.
14. Mathew SJ, Charney DS. Publication bias and the efficacy of antidepressants. Am J Psychiatry. 2009;166:140-145.
1. Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373:746-758.
2. Murray CJ, Lopez AD. Global Burden of Disease. Cambridge, Mass: Harvard University Press; 1996.
3. Williams SB, O’Connor EA, Eder M, et al. Screening for child and adolescent depression in primary care settings: a systematic evidence review for the U.S. Preventive Services Task Force. Pediatrics. 2009;123:e716-e735.
4. Timonen M, Liukkonen T. Management of depression in adults. BMJ. 2008;336:435-439.
5. Gartlehner G, Hansen RA, Thieda P, et al. Comparative Effectiveness of Second-Generation Antidepressants in the Pharmacologic Treatment of Adult Depression. Comparative Effectiveness Review No. 7. (Prepared by RTI International–University of North Carolina Evidence-based Practice Center under Contract No. 290-02-0016.) Rockville, Md: Agency for Healthcare Research and Quality; January 2007. Available at: www.effectivehealthcare.ahrq.gov/reports/final.cfm. Accessed May 18, 2009.
6. Hansen RA, Gartlehner G, Lohr KN, et al. Efficacy and safety of second-generation antidepressants in the treatment of major depressive disorder. Ann Intern Med. 2005;143:415-426.
7. Adams SM, Miller KE, Zylstra RG. Pharmacologic management of adult depression. Am Fam Physician. 2008;77:785-792.
8. Qaseem A, Snow V, Denberg TD, et al. Using second-generation antidepressants to treat depressive disorders: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;149:725-733.
9. DeRubeis RJ, Hollon SD, Amsterdam JD, et al. Cognitive therapy vs medications in the treatment of moderate to severe depression. Arch Gen Psychiatry. 2005;62:409-416.
10. deMello MF, de Jesus MJ, Bacaltchuk J, et al. A systematic review of research findings on the efficacy of interpersonal therapy for depressive disorders. Eur Arch Psychiatry Clin Neurosci. 2005;255:75-82.
11. APA Practice Guidelines. Practice guideline for the treatment of patients with major depressive disorder. 2nd ed. Available at: www.psychiatryonline.com/content.aspx?aID=48727. Accessed October 15, 2009.
12. Sackett D. An introduction to performing therapeutic trials. In: Haynes RB, Sackett DL, Guyatt GH, Tugwell P. Clinical Epidemiology: How to Do Clinical Practice Research. 3rd ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2006.
13. Turner EH, Matthews AM, Linardatos E, et al. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358:252-260.
14. Mathew SJ, Charney DS. Publication bias and the efficacy of antidepressants. Am J Psychiatry. 2009;166:140-145.
OSTEOPOROSIS
The author reports that he serves on the advisory boards of Amgen, Boehringer Ingelheim, Depomed, Eli Lilly, and Novo Nordisk. He is a speaker for the Alliance for Bone Health, Eli Lilly, and Warner Chilcott.
Some hormonal contraceptives may affect bone density
Berenson AB, Rahman M, Breitkopf CR, Bi LX. Effects of depot medroxyprogesterone acetate and 20-microgram oral contraceptives on bone mineral density. Obstet Gynecol. 2008;112:788–799.
American College of Obstetricians and Gynecologists. ACOG Committee Opinion. No. 415. September 2008. Depot medroxyprogesterone acetate and bone effects. Obstet Gynecol. 2008;112:727–730.
A woman’s contraceptive choice may affect her bone mineral density (BMD)—particularly if she chooses depot medroxyprogesterone acetate (DMPA) or a very-low-dose oral contraceptive (OC) as her method.
In the case of DMPA, studies have shown that its use for 2 years significantly impairs BMD at the hip and spine, regardless of the patient’s age, although BMD usually rebounds after discontinuation of the drug.1
In the case of very-low-dose OCs, there is evidence that young women who use a pill that contains only 20 μg of ethinyl estradiol have a lower increase in BMD than do women the same age who do not use hormonal contraception. (OCs that contain a higher dosage of ethinyl estradiol have not been shown to hamper BMD.) A study by Polatti and colleagues reported a 7.8% increase in BMD over 5 years among women 19 to 22 years old who did not use OCs, compared with no change in BMD among women who used OCs containing 20 μg of ethinyl estradiol.2
DMPA, BMD, and the FDA
The deleterious effect of DMPA on BMD is particularly relevant in perimenopausal women, who have already begun to experience the age-related decline in BMD that starts around the age of 30. The effect is also troubling in adolescents, who normally experience a large accretion of bone during the teen and early adult years.
In 2004, the US Food and Drug Administration (FDA) added a boxed warning to DMPA labeling advising women to limit their use of the drug to 2 years. Since that time, other studies have found that BMD increases to a greater degree among past users of DMPA than among never users—suggesting that DMPA-related bone loss is reversible.
ACOG: DMPA can be used longer than 2 years in some
In September 2008, the American College of Obstetricians and Gynecologists (ACOG) released a committee opinion acknowledging the association between DMPA and BMD loss. The committee pointed out, however, that “current evidence suggests that partial or full recovery of BMD occurs at the spine and at least partial recovery occurs at the hip after discontinuation of DMPA” ( FIGURE ).
The ACOG opinion also noted that, “given the efficacy of DMPA, particularly for populations such as adolescents, for whom contraceptive adherence can be challenging, or for those who feel they could not comply with a daily contraceptive method or a method that must be used with each act of intercourse, the possible adverse effects of DMPA must be balanced against the significant personal and public health impact of unintended pregnancy.”
The committee recommended that, despite concerns about bone loss, practitioners should not hesitate to prescribe DMPA. Nor should they limit its use to 2 consecutive years or perform BMD monitoring solely in response to DMPA use. “Any observed short-term loss in BMD associated with DMPA use may be recovered and is unlikely to place a woman at risk of fracture during use or in later years,” the committee opinion noted.
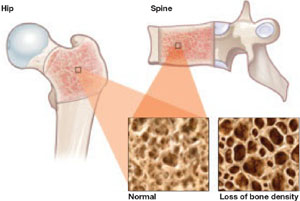
FIGURE DMPA-related bone loss is largely reversible
Depot medroxyprogesterone acetate (DMPA) is associated with a loss of bone mineral density at the hip and spine. Once the drug is discontinued, however, bone density appears to recover at least partially at both sites.
Study explores bone gains after DMPA
Berenson and associates measured BMD every 6 months for as long as 3 years in 703 white, African-American, and Hispanic women who used an OC, DMPA, or nonhormonal contraception. They also measured BMD for up to 2 additional years in 68 women who discontinued DMPA. They found no differences between races—although they did find the expected DMPA-associated bone loss.
They also found a small amount of bone loss (0.5% at the lumbar spine and 1.3% at the femoral neck) in users of very-low-dose OCs (20 μg ethinyl estradiol), compared with a gain of 1.9% at the lumbar spine and 0.6% at the femoral neck in nonusers of hormonal contraception.
Women who made a transition from DMPA to a very-low-dose OC recovered bone mass slowly. After DMPA was discontinued, women who selected nonhormonal contraception recovered BMD (4.9% at the spine, 3.2% at the femoral neck)—unlike those who chose a very-low-dose OC, who regained BMD at the spine (2.3%) but not the femoral neck (–0.7%).
Use of very-low-dose oral contraception (20 μg ethinyl estradiol) may lead to a small amount of bone loss or failure of bone accretion. Use of depot medroxyprogesterone acetate (DMPA) is associated with a greater degree of bone loss, but this loss is largely reversible at the spine. Use of a 20-μg oral contraceptive (OC) after discontinuation of DMPA may slow bone recovery.
As ACOG has indicated, concerns about skeletal health should not influence the decision to initiate or continue DMPA. Likewise, such concerns should not lead to restrictions on the use of OCs in teens or adult women. However, clinicians may wish to take bone effects into consideration when choosing the estrogen dosage of OCs for women younger than 30 who have yet to attain peak bone mass.
Denosumab nears FDA approval for treatment of osteoporosis
Cummings SR, San Martin J, McClung MR, et al; FREEDOM trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–765.
Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res. 2009 [Epub ahead of print].
Miller PD, Bolognese MA, Lewiecki EM, et al, for the Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone. 2008;43:222–229.
An FDA panel advising the Division of Reproductive and Urologic Products voted to approve denosumab (proposed brand name: Prolia) as a treatment for osteoporosis. The drug is a fully human monoclonal antibody to the receptor activator of nuclear factor-B ligand (RANKL), a cytokine that is essential for the formation, function, and survival of osteoclasts. By binding RANKL, denosumab prevents interaction between RANKL and its receptor, RANK, on osteoclasts and osteoclast precursors and thereby inhibits osteoclast-mediated bone resorption. Its effects are reversible.
Trial: Denosumab versus placebo
Cummings and colleagues reported the findings of the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial, which involved 7,868 women 60 to 90 years old who had a BMD T-score between –2.5 and –4.0 at the lumbar spine or total hip. Participants were randomly assigned to receive 60 mg of denosumab or placebo subcutaneously every 6 months for 36 months. The primary endpoint was new vertebral fracture. Secondary endpoints included nonvertebral and hip fractures.
Findings included:
- Denosumab reduced the risk of new, radiographically detected vertebral fracture, with a cumulative incidence of 2.3%, versus 7.2% in the placebo group (risk ratio, 0.32; P<.001).
- Denosumab reduced the risk of hip fracture, with a cumulative incidence of 0.7% in the denosumab group, versus 1.2% in the placebo group (hazard ratio, 0.60; P=.04).
- Denosumab reduced the risk of nonvertebral fracture, with a cumulative incidence of 6.5% in the denosumab group, versus 8.0% in the placebo group (hazard ratio, 0.80; P=.01).
- There was no increase in the risk of cancer, infection, cardiovascular disease, delayed fracture healing, or hypocalcemia, and no cases of osteonecrosis of the jaw or adverse reaction to injection of the drug.
How does denosumab compare with alendronate?
Kendler and associates explored a clinically relevant question: What are the effects of switching from a bisphosphonate (in this case, alendronate) to denosumab?
They studied 504 postmenopausal women who were at least 55 years old, had a T-score between –2 and –4, and had been taking alendronate for at least 6 months. These women were randomized in double-blinded, double-dummy fashion to 60 mg of subcutaneous denosumab or a continuation of 70 mg of oral alendronate. Follow-up was 12 months.
Findings included:
- Among the women making a transition to denosumab, total hip BMD increased by 1.9% at 12 months, versus 1.05% in women continuing on alendronate (P<.0001).
- Women making a transition to denosumab also gained significantly more BMD at 12 months at the lumbar spine, femoral neck, and the distal third of the radius (all P<.0125).
- The transition to denosumab reduced bone turnover to a greater degree than did continuing alendronate.
What happens when denosumab is discontinued?
Miller and colleagues randomized postmenopausal women who had a lumbar spine T-score of –1.8 to –4.0 or a proximal femur T-score of –1.8 to –3.5 to one of the following arms:
- denosumab every 3 months (6, 14, or 30 mg)
- denosumab every 6 months (14, 60, 100, or 210 mg)
- open-label oral alendronate every week (70 mg)
- placebo.
After 24 months, women taking denosumab either:
- continued treatment at 60 mg every 6 months for an additional 24 months
- discontinued therapy
- discontinued treatment for 12 months and then reinitiated denosumab at 60 mg every 6 months for an additional 12 months.
The placebo cohort was maintained throughout this period.
Continuous, long-term denosumab increased BMD at the lumbar spine (9.4% to 11.8%) and total hip (4.0% to 6.1%). In contrast, discontinuation of denosumab was associated with a decrease in BMD of 6.6% at the lumbar spine and 5.3% at the total hip within the first 12 months. Retreatment with denosumab increased lumbar spine BMD by 9% from the original baseline values. Levels of bone turnover markers increased upon discontinuation of denosumab and decreased with retreatment. Adverse events occurred at similar rates in all treatment groups.
Among postmenopausal women who have low bone mineral density (BMD), long-term treatment with denosumab leads to gains in BMD and a reduction of markers of bone turnover. These effects are fully reversible upon discontinuation of the drug, but reoccur when treatment is restored.
In addition, switching from alendronate to denosumab produces a greater reduction in bone turnover than does continuation on the bisphosphonate.
Denosumab is a safe, extremely potent agent that will undoubtedly find a place in the ObGyn armamentarium, where it will join bisphosphonates, selective estrogen receptor modulators, and hormone therapy. The long dosing interval (every 6 months) should help increase compliance.
TSEC, a novel compound, enters development
Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrogen complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril. 2009;92:1045–1052.
Although raloxifene is the only selective estrogen receptor modulator (SERM) approved by the FDA for prevention and treatment of osteoporosis, numerous other SERMs have been explored for this application. One new category of drug is the tissue selective estrogen complex (TSEC), which pairs a SERM with an estrogen. This was previously attempted unsuccessfully with raloxifene and 17β-estradiol. The ideal SERM–estrogen combination would have the positive attributes of both classes of drugs with fewer, or none, of the undesired effects. An appropriate TSEC would therefore alleviate hot flushes, treat vulvar and vaginal atrophy, and protect against bone loss without stimulating the endometrium and increasing the risk of breast cancer.
One third-generation SERM that has been investigated for its utility in this regard is bazedoxifene (BZA), which produced endometrial thickness and amenorrhea rates comparable to those of placebo. It also produced greater gains in BMD at the lumbar spine in a 2-year, randomized, double-blind, placebo-controlled trial.3 The incidence of new vertebral fracture was significantly lower with BZA than with placebo in a study of postmenopausal women with osteoporosis.4
Given these favorable data, a TSEC containing BZA and conjugated equine estrogen was designed as a potential new comprehensive menopausal therapy.
Substudies suggest bazedoxifene has promise
Lindsay and associates reported on two osteoporosis substudies of the Selective estrogen Menopause and Response to Therapy (SMART) Trial. The main study, which evaluated the incidence of endometrial hyperplasia at 12 months, will be reported elsewhere; it was a multicenter, double-blind, randomized, placebo-controlled phase 3 trial that enrolled 3,397 women.
Substudy I involved women who were more than 5 years postmenopausal, and Substudy II included women who were between 1 and 5 years postmenopausal. Eligible screened subjects were randomly assigned to one of the following treatment groups:
- bazedoxifene (10, 20, or 40 mg), each with conjugated equine estrogen (0.625 or 0.45 mg)
- raloxifene (60 mg)
- placebo.
To maintain blinding, the combination of BZA and conjugated equine estrogen was provided as a single, encapsulated tablet to match the placebo, as was raloxifene. Subjects were directed to take one tablet orally at approximately the same time each day for 2 years. The primary outcome for both substudies was a change in BMD at the lumbar spine, as measured by dual-energy x-ray absorptiometry.
In both substudies, BMD increased to a greater degree at the lumbar spine and total hip at all BZA-estrogen dosages than with placebo, and it increased to a greater degree at the lumbar spine at most BZA-estrogen dosages, compared with raloxifene.
Osteocalcin and N-telopeptide significantly decreased at all BZA-estrogen dosages, compared with placebo, and at most BZA-estrogen dosages, compared with raloxifene.
Bazedoxifene is a potential therapeutic agent for menopausal women that may protect the endometrium while preventing bone loss in a population at higher risk of osteoporosis. It is not yet approved for this indication; further investigation is needed.
The combination of an estrogen and a selective estrogen receptor modulator to potentially relieve vasomotor symptoms, prevent vulvovaginal atrophy, and preserve bone mass without stimulating the endometrium or increasing the risk of breast cancer or venous thromboembolism is very exciting—and just might revolutionize treatment of menopausal women
Bolognese M, Krege JH, Utian WH, et al. Effects of arzoxifene on bone mineral density and endometrium in postmenopausal women with normal or low bone mass. J Clin Endocrinol Metab. 2009;94:2284–2289.
The benzothiophene derivative arzoxifene, which has been in development for the prevention and treatment of osteoporosis, as well as for reduction of the risk of invasive breast cancer in postmenopausal women, has been pulled from the regulatory approval process by its manufacturer, Eli Lilly.
This move is somewhat surprising because, in a phase 3 trial, arzoxifene significantly increased bone mineral density at the lumbar spine (2.9%) and total hip (2.2%), compared with placebo. It also decreased levels of biochemical markers of bone metabolism.
In the trial, Bolognese and associates randomly assigned 331 postmenopausal women who had normal or low bone mass to receive 20 mg of arzoxifene or placebo daily for 2 years.
The trial also found that changes in breast density were neutral or slightly decreased in the arzoxifene group, and there was no evidence of endometrial hyperplasia or carcinoma, based on central review of baseline and follow-up endometrial biopsies. Nor were there any significant differences between groups in endometrial thickness, as assessed by transvaginal ultrasonography, or in the incidence of uterine polyps, vaginal bleeding, and hot flushes.
Nevertheless, Lilly issued a press release in late August announcing that arzoxifene would not be submitted to the FDA for regulatory review.5 It appears that, although initial results from the “pivotal” 5-year, phase 3 GJAD “Generations” trial indicated that the drug had met the primary endpoints of significantly reducing the risk of vertebral fracture and invasive breast cancer in postmenopausal women, “the study failed to demonstrate a statistically significant difference in key secondary efficacy endpoints, such as nonvertebral fractures, clinical vertebral fractures, cardiovascular events, and cognitive function, compared with placebo.”
The release went on to say: “In addition, certain adverse events, including venous thromboembolic events, hot flushes, and gynecological-related events, were reported more frequently in the arzoxifene group, compared with placebo.”5
Arzoxifene therefore joins an expanding list of selective estrogen receptor modulators (SERMs) that did not make it out of clinical trials: idoxifene, droloxifene, levormeloxifene, and lasofoxifene (approved in Europe, however), to name a few. The fall of arzoxifene again underscores the notion that “not all SERMs are created equal.”6
1. Scholes D, LaCroix AZ, Ichikawa LE, Barlow WE, Ott SM. Change in bone mineral density among adolescent women using and discontinuing depot medroxyprogesterone acetate contraception. Arch Pediatr Adolesc Med. 2005;159:139-144.
2. Polatti F, Perotti F, Filippa N, Gallina D, Nappi RE. Bone mass and long-term monophasic oral contraceptive treatment in young women. Contraception. 1995;51:221-224.
3. Miller PD, Chines AA, Christiansen C, et al. Effects of bazedoxifene on BMD and bone turnover in postmenopausal women: 2-year results of a randomized, double-blind, placebo-, and active-controlled study. J Bone Miner Res. 2008;23:525-535.
4. Silverman SL, Christiansen C, Genant HK, et al. Efficacy of bazedoxifene in reducing new vertebral fracture risk in postmenopausal women with osteoporosis: results from a 3-year, randomized, placebo- and active-controlled clinical trial. J Bone Miner Res. 2008;23:1923-1934.
5. Based on preliminary phase III GHAD study results, Lilly concludes arzoxifene’s clinical profile does not support regulatory submission [press release]. Available at http://newsroom.lilly.com/releasedetail.cfm?ReleaseID=403905. Accessed October 2, 2009.
6. Goldstein SR. Not all SERMs are created equal. Menopause. 2006;13:325-327.
The author reports that he serves on the advisory boards of Amgen, Boehringer Ingelheim, Depomed, Eli Lilly, and Novo Nordisk. He is a speaker for the Alliance for Bone Health, Eli Lilly, and Warner Chilcott.
Some hormonal contraceptives may affect bone density
Berenson AB, Rahman M, Breitkopf CR, Bi LX. Effects of depot medroxyprogesterone acetate and 20-microgram oral contraceptives on bone mineral density. Obstet Gynecol. 2008;112:788–799.
American College of Obstetricians and Gynecologists. ACOG Committee Opinion. No. 415. September 2008. Depot medroxyprogesterone acetate and bone effects. Obstet Gynecol. 2008;112:727–730.
A woman’s contraceptive choice may affect her bone mineral density (BMD)—particularly if she chooses depot medroxyprogesterone acetate (DMPA) or a very-low-dose oral contraceptive (OC) as her method.
In the case of DMPA, studies have shown that its use for 2 years significantly impairs BMD at the hip and spine, regardless of the patient’s age, although BMD usually rebounds after discontinuation of the drug.1
In the case of very-low-dose OCs, there is evidence that young women who use a pill that contains only 20 μg of ethinyl estradiol have a lower increase in BMD than do women the same age who do not use hormonal contraception. (OCs that contain a higher dosage of ethinyl estradiol have not been shown to hamper BMD.) A study by Polatti and colleagues reported a 7.8% increase in BMD over 5 years among women 19 to 22 years old who did not use OCs, compared with no change in BMD among women who used OCs containing 20 μg of ethinyl estradiol.2
DMPA, BMD, and the FDA
The deleterious effect of DMPA on BMD is particularly relevant in perimenopausal women, who have already begun to experience the age-related decline in BMD that starts around the age of 30. The effect is also troubling in adolescents, who normally experience a large accretion of bone during the teen and early adult years.
In 2004, the US Food and Drug Administration (FDA) added a boxed warning to DMPA labeling advising women to limit their use of the drug to 2 years. Since that time, other studies have found that BMD increases to a greater degree among past users of DMPA than among never users—suggesting that DMPA-related bone loss is reversible.
ACOG: DMPA can be used longer than 2 years in some
In September 2008, the American College of Obstetricians and Gynecologists (ACOG) released a committee opinion acknowledging the association between DMPA and BMD loss. The committee pointed out, however, that “current evidence suggests that partial or full recovery of BMD occurs at the spine and at least partial recovery occurs at the hip after discontinuation of DMPA” ( FIGURE ).
The ACOG opinion also noted that, “given the efficacy of DMPA, particularly for populations such as adolescents, for whom contraceptive adherence can be challenging, or for those who feel they could not comply with a daily contraceptive method or a method that must be used with each act of intercourse, the possible adverse effects of DMPA must be balanced against the significant personal and public health impact of unintended pregnancy.”
The committee recommended that, despite concerns about bone loss, practitioners should not hesitate to prescribe DMPA. Nor should they limit its use to 2 consecutive years or perform BMD monitoring solely in response to DMPA use. “Any observed short-term loss in BMD associated with DMPA use may be recovered and is unlikely to place a woman at risk of fracture during use or in later years,” the committee opinion noted.
FIGURE DMPA-related bone loss is largely reversible
Depot medroxyprogesterone acetate (DMPA) is associated with a loss of bone mineral density at the hip and spine. Once the drug is discontinued, however, bone density appears to recover at least partially at both sites.
Study explores bone gains after DMPA
Berenson and associates measured BMD every 6 months for as long as 3 years in 703 white, African-American, and Hispanic women who used an OC, DMPA, or nonhormonal contraception. They also measured BMD for up to 2 additional years in 68 women who discontinued DMPA. They found no differences between races—although they did find the expected DMPA-associated bone loss.
They also found a small amount of bone loss (0.5% at the lumbar spine and 1.3% at the femoral neck) in users of very-low-dose OCs (20 μg ethinyl estradiol), compared with a gain of 1.9% at the lumbar spine and 0.6% at the femoral neck in nonusers of hormonal contraception.
Women who made a transition from DMPA to a very-low-dose OC recovered bone mass slowly. After DMPA was discontinued, women who selected nonhormonal contraception recovered BMD (4.9% at the spine, 3.2% at the femoral neck)—unlike those who chose a very-low-dose OC, who regained BMD at the spine (2.3%) but not the femoral neck (–0.7%).
Use of very-low-dose oral contraception (20 μg ethinyl estradiol) may lead to a small amount of bone loss or failure of bone accretion. Use of depot medroxyprogesterone acetate (DMPA) is associated with a greater degree of bone loss, but this loss is largely reversible at the spine. Use of a 20-μg oral contraceptive (OC) after discontinuation of DMPA may slow bone recovery.
As ACOG has indicated, concerns about skeletal health should not influence the decision to initiate or continue DMPA. Likewise, such concerns should not lead to restrictions on the use of OCs in teens or adult women. However, clinicians may wish to take bone effects into consideration when choosing the estrogen dosage of OCs for women younger than 30 who have yet to attain peak bone mass.
Denosumab nears FDA approval for treatment of osteoporosis
Cummings SR, San Martin J, McClung MR, et al; FREEDOM trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–765.
Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res. 2009 [Epub ahead of print].
Miller PD, Bolognese MA, Lewiecki EM, et al, for the Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone. 2008;43:222–229.
An FDA panel advising the Division of Reproductive and Urologic Products voted to approve denosumab (proposed brand name: Prolia) as a treatment for osteoporosis. The drug is a fully human monoclonal antibody to the receptor activator of nuclear factor-B ligand (RANKL), a cytokine that is essential for the formation, function, and survival of osteoclasts. By binding RANKL, denosumab prevents interaction between RANKL and its receptor, RANK, on osteoclasts and osteoclast precursors and thereby inhibits osteoclast-mediated bone resorption. Its effects are reversible.
Trial: Denosumab versus placebo
Cummings and colleagues reported the findings of the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial, which involved 7,868 women 60 to 90 years old who had a BMD T-score between –2.5 and –4.0 at the lumbar spine or total hip. Participants were randomly assigned to receive 60 mg of denosumab or placebo subcutaneously every 6 months for 36 months. The primary endpoint was new vertebral fracture. Secondary endpoints included nonvertebral and hip fractures.
Findings included:
- Denosumab reduced the risk of new, radiographically detected vertebral fracture, with a cumulative incidence of 2.3%, versus 7.2% in the placebo group (risk ratio, 0.32; P<.001).
- Denosumab reduced the risk of hip fracture, with a cumulative incidence of 0.7% in the denosumab group, versus 1.2% in the placebo group (hazard ratio, 0.60; P=.04).
- Denosumab reduced the risk of nonvertebral fracture, with a cumulative incidence of 6.5% in the denosumab group, versus 8.0% in the placebo group (hazard ratio, 0.80; P=.01).
- There was no increase in the risk of cancer, infection, cardiovascular disease, delayed fracture healing, or hypocalcemia, and no cases of osteonecrosis of the jaw or adverse reaction to injection of the drug.
How does denosumab compare with alendronate?
Kendler and associates explored a clinically relevant question: What are the effects of switching from a bisphosphonate (in this case, alendronate) to denosumab?
They studied 504 postmenopausal women who were at least 55 years old, had a T-score between –2 and –4, and had been taking alendronate for at least 6 months. These women were randomized in double-blinded, double-dummy fashion to 60 mg of subcutaneous denosumab or a continuation of 70 mg of oral alendronate. Follow-up was 12 months.
Findings included:
- Among the women making a transition to denosumab, total hip BMD increased by 1.9% at 12 months, versus 1.05% in women continuing on alendronate (P<.0001).
- Women making a transition to denosumab also gained significantly more BMD at 12 months at the lumbar spine, femoral neck, and the distal third of the radius (all P<.0125).
- The transition to denosumab reduced bone turnover to a greater degree than did continuing alendronate.
What happens when denosumab is discontinued?
Miller and colleagues randomized postmenopausal women who had a lumbar spine T-score of –1.8 to –4.0 or a proximal femur T-score of –1.8 to –3.5 to one of the following arms:
- denosumab every 3 months (6, 14, or 30 mg)
- denosumab every 6 months (14, 60, 100, or 210 mg)
- open-label oral alendronate every week (70 mg)
- placebo.
After 24 months, women taking denosumab either:
- continued treatment at 60 mg every 6 months for an additional 24 months
- discontinued therapy
- discontinued treatment for 12 months and then reinitiated denosumab at 60 mg every 6 months for an additional 12 months.
The placebo cohort was maintained throughout this period.
Continuous, long-term denosumab increased BMD at the lumbar spine (9.4% to 11.8%) and total hip (4.0% to 6.1%). In contrast, discontinuation of denosumab was associated with a decrease in BMD of 6.6% at the lumbar spine and 5.3% at the total hip within the first 12 months. Retreatment with denosumab increased lumbar spine BMD by 9% from the original baseline values. Levels of bone turnover markers increased upon discontinuation of denosumab and decreased with retreatment. Adverse events occurred at similar rates in all treatment groups.
Among postmenopausal women who have low bone mineral density (BMD), long-term treatment with denosumab leads to gains in BMD and a reduction of markers of bone turnover. These effects are fully reversible upon discontinuation of the drug, but reoccur when treatment is restored.
In addition, switching from alendronate to denosumab produces a greater reduction in bone turnover than does continuation on the bisphosphonate.
Denosumab is a safe, extremely potent agent that will undoubtedly find a place in the ObGyn armamentarium, where it will join bisphosphonates, selective estrogen receptor modulators, and hormone therapy. The long dosing interval (every 6 months) should help increase compliance.
TSEC, a novel compound, enters development
Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrogen complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril. 2009;92:1045–1052.
Although raloxifene is the only selective estrogen receptor modulator (SERM) approved by the FDA for prevention and treatment of osteoporosis, numerous other SERMs have been explored for this application. One new category of drug is the tissue selective estrogen complex (TSEC), which pairs a SERM with an estrogen. This was previously attempted unsuccessfully with raloxifene and 17β-estradiol. The ideal SERM–estrogen combination would have the positive attributes of both classes of drugs with fewer, or none, of the undesired effects. An appropriate TSEC would therefore alleviate hot flushes, treat vulvar and vaginal atrophy, and protect against bone loss without stimulating the endometrium and increasing the risk of breast cancer.
One third-generation SERM that has been investigated for its utility in this regard is bazedoxifene (BZA), which produced endometrial thickness and amenorrhea rates comparable to those of placebo. It also produced greater gains in BMD at the lumbar spine in a 2-year, randomized, double-blind, placebo-controlled trial.3 The incidence of new vertebral fracture was significantly lower with BZA than with placebo in a study of postmenopausal women with osteoporosis.4
Given these favorable data, a TSEC containing BZA and conjugated equine estrogen was designed as a potential new comprehensive menopausal therapy.
Substudies suggest bazedoxifene has promise
Lindsay and associates reported on two osteoporosis substudies of the Selective estrogen Menopause and Response to Therapy (SMART) Trial. The main study, which evaluated the incidence of endometrial hyperplasia at 12 months, will be reported elsewhere; it was a multicenter, double-blind, randomized, placebo-controlled phase 3 trial that enrolled 3,397 women.
Substudy I involved women who were more than 5 years postmenopausal, and Substudy II included women who were between 1 and 5 years postmenopausal. Eligible screened subjects were randomly assigned to one of the following treatment groups:
- bazedoxifene (10, 20, or 40 mg), each with conjugated equine estrogen (0.625 or 0.45 mg)
- raloxifene (60 mg)
- placebo.
To maintain blinding, the combination of BZA and conjugated equine estrogen was provided as a single, encapsulated tablet to match the placebo, as was raloxifene. Subjects were directed to take one tablet orally at approximately the same time each day for 2 years. The primary outcome for both substudies was a change in BMD at the lumbar spine, as measured by dual-energy x-ray absorptiometry.
In both substudies, BMD increased to a greater degree at the lumbar spine and total hip at all BZA-estrogen dosages than with placebo, and it increased to a greater degree at the lumbar spine at most BZA-estrogen dosages, compared with raloxifene.
Osteocalcin and N-telopeptide significantly decreased at all BZA-estrogen dosages, compared with placebo, and at most BZA-estrogen dosages, compared with raloxifene.
Bazedoxifene is a potential therapeutic agent for menopausal women that may protect the endometrium while preventing bone loss in a population at higher risk of osteoporosis. It is not yet approved for this indication; further investigation is needed.
The combination of an estrogen and a selective estrogen receptor modulator to potentially relieve vasomotor symptoms, prevent vulvovaginal atrophy, and preserve bone mass without stimulating the endometrium or increasing the risk of breast cancer or venous thromboembolism is very exciting—and just might revolutionize treatment of menopausal women
Bolognese M, Krege JH, Utian WH, et al. Effects of arzoxifene on bone mineral density and endometrium in postmenopausal women with normal or low bone mass. J Clin Endocrinol Metab. 2009;94:2284–2289.
The benzothiophene derivative arzoxifene, which has been in development for the prevention and treatment of osteoporosis, as well as for reduction of the risk of invasive breast cancer in postmenopausal women, has been pulled from the regulatory approval process by its manufacturer, Eli Lilly.
This move is somewhat surprising because, in a phase 3 trial, arzoxifene significantly increased bone mineral density at the lumbar spine (2.9%) and total hip (2.2%), compared with placebo. It also decreased levels of biochemical markers of bone metabolism.
In the trial, Bolognese and associates randomly assigned 331 postmenopausal women who had normal or low bone mass to receive 20 mg of arzoxifene or placebo daily for 2 years.
The trial also found that changes in breast density were neutral or slightly decreased in the arzoxifene group, and there was no evidence of endometrial hyperplasia or carcinoma, based on central review of baseline and follow-up endometrial biopsies. Nor were there any significant differences between groups in endometrial thickness, as assessed by transvaginal ultrasonography, or in the incidence of uterine polyps, vaginal bleeding, and hot flushes.
Nevertheless, Lilly issued a press release in late August announcing that arzoxifene would not be submitted to the FDA for regulatory review.5 It appears that, although initial results from the “pivotal” 5-year, phase 3 GJAD “Generations” trial indicated that the drug had met the primary endpoints of significantly reducing the risk of vertebral fracture and invasive breast cancer in postmenopausal women, “the study failed to demonstrate a statistically significant difference in key secondary efficacy endpoints, such as nonvertebral fractures, clinical vertebral fractures, cardiovascular events, and cognitive function, compared with placebo.”
The release went on to say: “In addition, certain adverse events, including venous thromboembolic events, hot flushes, and gynecological-related events, were reported more frequently in the arzoxifene group, compared with placebo.”5
Arzoxifene therefore joins an expanding list of selective estrogen receptor modulators (SERMs) that did not make it out of clinical trials: idoxifene, droloxifene, levormeloxifene, and lasofoxifene (approved in Europe, however), to name a few. The fall of arzoxifene again underscores the notion that “not all SERMs are created equal.”6
The author reports that he serves on the advisory boards of Amgen, Boehringer Ingelheim, Depomed, Eli Lilly, and Novo Nordisk. He is a speaker for the Alliance for Bone Health, Eli Lilly, and Warner Chilcott.
Some hormonal contraceptives may affect bone density
Berenson AB, Rahman M, Breitkopf CR, Bi LX. Effects of depot medroxyprogesterone acetate and 20-microgram oral contraceptives on bone mineral density. Obstet Gynecol. 2008;112:788–799.
American College of Obstetricians and Gynecologists. ACOG Committee Opinion. No. 415. September 2008. Depot medroxyprogesterone acetate and bone effects. Obstet Gynecol. 2008;112:727–730.
A woman’s contraceptive choice may affect her bone mineral density (BMD)—particularly if she chooses depot medroxyprogesterone acetate (DMPA) or a very-low-dose oral contraceptive (OC) as her method.
In the case of DMPA, studies have shown that its use for 2 years significantly impairs BMD at the hip and spine, regardless of the patient’s age, although BMD usually rebounds after discontinuation of the drug.1
In the case of very-low-dose OCs, there is evidence that young women who use a pill that contains only 20 μg of ethinyl estradiol have a lower increase in BMD than do women the same age who do not use hormonal contraception. (OCs that contain a higher dosage of ethinyl estradiol have not been shown to hamper BMD.) A study by Polatti and colleagues reported a 7.8% increase in BMD over 5 years among women 19 to 22 years old who did not use OCs, compared with no change in BMD among women who used OCs containing 20 μg of ethinyl estradiol.2
DMPA, BMD, and the FDA
The deleterious effect of DMPA on BMD is particularly relevant in perimenopausal women, who have already begun to experience the age-related decline in BMD that starts around the age of 30. The effect is also troubling in adolescents, who normally experience a large accretion of bone during the teen and early adult years.
In 2004, the US Food and Drug Administration (FDA) added a boxed warning to DMPA labeling advising women to limit their use of the drug to 2 years. Since that time, other studies have found that BMD increases to a greater degree among past users of DMPA than among never users—suggesting that DMPA-related bone loss is reversible.
ACOG: DMPA can be used longer than 2 years in some
In September 2008, the American College of Obstetricians and Gynecologists (ACOG) released a committee opinion acknowledging the association between DMPA and BMD loss. The committee pointed out, however, that “current evidence suggests that partial or full recovery of BMD occurs at the spine and at least partial recovery occurs at the hip after discontinuation of DMPA” ( FIGURE ).
The ACOG opinion also noted that, “given the efficacy of DMPA, particularly for populations such as adolescents, for whom contraceptive adherence can be challenging, or for those who feel they could not comply with a daily contraceptive method or a method that must be used with each act of intercourse, the possible adverse effects of DMPA must be balanced against the significant personal and public health impact of unintended pregnancy.”
The committee recommended that, despite concerns about bone loss, practitioners should not hesitate to prescribe DMPA. Nor should they limit its use to 2 consecutive years or perform BMD monitoring solely in response to DMPA use. “Any observed short-term loss in BMD associated with DMPA use may be recovered and is unlikely to place a woman at risk of fracture during use or in later years,” the committee opinion noted.
FIGURE DMPA-related bone loss is largely reversible
Depot medroxyprogesterone acetate (DMPA) is associated with a loss of bone mineral density at the hip and spine. Once the drug is discontinued, however, bone density appears to recover at least partially at both sites.
Study explores bone gains after DMPA
Berenson and associates measured BMD every 6 months for as long as 3 years in 703 white, African-American, and Hispanic women who used an OC, DMPA, or nonhormonal contraception. They also measured BMD for up to 2 additional years in 68 women who discontinued DMPA. They found no differences between races—although they did find the expected DMPA-associated bone loss.
They also found a small amount of bone loss (0.5% at the lumbar spine and 1.3% at the femoral neck) in users of very-low-dose OCs (20 μg ethinyl estradiol), compared with a gain of 1.9% at the lumbar spine and 0.6% at the femoral neck in nonusers of hormonal contraception.
Women who made a transition from DMPA to a very-low-dose OC recovered bone mass slowly. After DMPA was discontinued, women who selected nonhormonal contraception recovered BMD (4.9% at the spine, 3.2% at the femoral neck)—unlike those who chose a very-low-dose OC, who regained BMD at the spine (2.3%) but not the femoral neck (–0.7%).
Use of very-low-dose oral contraception (20 μg ethinyl estradiol) may lead to a small amount of bone loss or failure of bone accretion. Use of depot medroxyprogesterone acetate (DMPA) is associated with a greater degree of bone loss, but this loss is largely reversible at the spine. Use of a 20-μg oral contraceptive (OC) after discontinuation of DMPA may slow bone recovery.
As ACOG has indicated, concerns about skeletal health should not influence the decision to initiate or continue DMPA. Likewise, such concerns should not lead to restrictions on the use of OCs in teens or adult women. However, clinicians may wish to take bone effects into consideration when choosing the estrogen dosage of OCs for women younger than 30 who have yet to attain peak bone mass.
Denosumab nears FDA approval for treatment of osteoporosis
Cummings SR, San Martin J, McClung MR, et al; FREEDOM trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–765.
Kendler DL, Roux C, Benhamou CL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res. 2009 [Epub ahead of print].
Miller PD, Bolognese MA, Lewiecki EM, et al, for the Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone. 2008;43:222–229.
An FDA panel advising the Division of Reproductive and Urologic Products voted to approve denosumab (proposed brand name: Prolia) as a treatment for osteoporosis. The drug is a fully human monoclonal antibody to the receptor activator of nuclear factor-B ligand (RANKL), a cytokine that is essential for the formation, function, and survival of osteoclasts. By binding RANKL, denosumab prevents interaction between RANKL and its receptor, RANK, on osteoclasts and osteoclast precursors and thereby inhibits osteoclast-mediated bone resorption. Its effects are reversible.
Trial: Denosumab versus placebo
Cummings and colleagues reported the findings of the Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial, which involved 7,868 women 60 to 90 years old who had a BMD T-score between –2.5 and –4.0 at the lumbar spine or total hip. Participants were randomly assigned to receive 60 mg of denosumab or placebo subcutaneously every 6 months for 36 months. The primary endpoint was new vertebral fracture. Secondary endpoints included nonvertebral and hip fractures.
Findings included:
- Denosumab reduced the risk of new, radiographically detected vertebral fracture, with a cumulative incidence of 2.3%, versus 7.2% in the placebo group (risk ratio, 0.32; P<.001).
- Denosumab reduced the risk of hip fracture, with a cumulative incidence of 0.7% in the denosumab group, versus 1.2% in the placebo group (hazard ratio, 0.60; P=.04).
- Denosumab reduced the risk of nonvertebral fracture, with a cumulative incidence of 6.5% in the denosumab group, versus 8.0% in the placebo group (hazard ratio, 0.80; P=.01).
- There was no increase in the risk of cancer, infection, cardiovascular disease, delayed fracture healing, or hypocalcemia, and no cases of osteonecrosis of the jaw or adverse reaction to injection of the drug.
How does denosumab compare with alendronate?
Kendler and associates explored a clinically relevant question: What are the effects of switching from a bisphosphonate (in this case, alendronate) to denosumab?
They studied 504 postmenopausal women who were at least 55 years old, had a T-score between –2 and –4, and had been taking alendronate for at least 6 months. These women were randomized in double-blinded, double-dummy fashion to 60 mg of subcutaneous denosumab or a continuation of 70 mg of oral alendronate. Follow-up was 12 months.
Findings included:
- Among the women making a transition to denosumab, total hip BMD increased by 1.9% at 12 months, versus 1.05% in women continuing on alendronate (P<.0001).
- Women making a transition to denosumab also gained significantly more BMD at 12 months at the lumbar spine, femoral neck, and the distal third of the radius (all P<.0125).
- The transition to denosumab reduced bone turnover to a greater degree than did continuing alendronate.
What happens when denosumab is discontinued?
Miller and colleagues randomized postmenopausal women who had a lumbar spine T-score of –1.8 to –4.0 or a proximal femur T-score of –1.8 to –3.5 to one of the following arms:
- denosumab every 3 months (6, 14, or 30 mg)
- denosumab every 6 months (14, 60, 100, or 210 mg)
- open-label oral alendronate every week (70 mg)
- placebo.
After 24 months, women taking denosumab either:
- continued treatment at 60 mg every 6 months for an additional 24 months
- discontinued therapy
- discontinued treatment for 12 months and then reinitiated denosumab at 60 mg every 6 months for an additional 12 months.
The placebo cohort was maintained throughout this period.
Continuous, long-term denosumab increased BMD at the lumbar spine (9.4% to 11.8%) and total hip (4.0% to 6.1%). In contrast, discontinuation of denosumab was associated with a decrease in BMD of 6.6% at the lumbar spine and 5.3% at the total hip within the first 12 months. Retreatment with denosumab increased lumbar spine BMD by 9% from the original baseline values. Levels of bone turnover markers increased upon discontinuation of denosumab and decreased with retreatment. Adverse events occurred at similar rates in all treatment groups.
Among postmenopausal women who have low bone mineral density (BMD), long-term treatment with denosumab leads to gains in BMD and a reduction of markers of bone turnover. These effects are fully reversible upon discontinuation of the drug, but reoccur when treatment is restored.
In addition, switching from alendronate to denosumab produces a greater reduction in bone turnover than does continuation on the bisphosphonate.
Denosumab is a safe, extremely potent agent that will undoubtedly find a place in the ObGyn armamentarium, where it will join bisphosphonates, selective estrogen receptor modulators, and hormone therapy. The long dosing interval (every 6 months) should help increase compliance.
TSEC, a novel compound, enters development
Lindsay R, Gallagher JC, Kagan R, Pickar JH, Constantine G. Efficacy of tissue-selective estrogen complex of bazedoxifene/conjugated estrogens for osteoporosis prevention in at-risk postmenopausal women. Fertil Steril. 2009;92:1045–1052.
Although raloxifene is the only selective estrogen receptor modulator (SERM) approved by the FDA for prevention and treatment of osteoporosis, numerous other SERMs have been explored for this application. One new category of drug is the tissue selective estrogen complex (TSEC), which pairs a SERM with an estrogen. This was previously attempted unsuccessfully with raloxifene and 17β-estradiol. The ideal SERM–estrogen combination would have the positive attributes of both classes of drugs with fewer, or none, of the undesired effects. An appropriate TSEC would therefore alleviate hot flushes, treat vulvar and vaginal atrophy, and protect against bone loss without stimulating the endometrium and increasing the risk of breast cancer.
One third-generation SERM that has been investigated for its utility in this regard is bazedoxifene (BZA), which produced endometrial thickness and amenorrhea rates comparable to those of placebo. It also produced greater gains in BMD at the lumbar spine in a 2-year, randomized, double-blind, placebo-controlled trial.3 The incidence of new vertebral fracture was significantly lower with BZA than with placebo in a study of postmenopausal women with osteoporosis.4
Given these favorable data, a TSEC containing BZA and conjugated equine estrogen was designed as a potential new comprehensive menopausal therapy.
Substudies suggest bazedoxifene has promise
Lindsay and associates reported on two osteoporosis substudies of the Selective estrogen Menopause and Response to Therapy (SMART) Trial. The main study, which evaluated the incidence of endometrial hyperplasia at 12 months, will be reported elsewhere; it was a multicenter, double-blind, randomized, placebo-controlled phase 3 trial that enrolled 3,397 women.
Substudy I involved women who were more than 5 years postmenopausal, and Substudy II included women who were between 1 and 5 years postmenopausal. Eligible screened subjects were randomly assigned to one of the following treatment groups:
- bazedoxifene (10, 20, or 40 mg), each with conjugated equine estrogen (0.625 or 0.45 mg)
- raloxifene (60 mg)
- placebo.
To maintain blinding, the combination of BZA and conjugated equine estrogen was provided as a single, encapsulated tablet to match the placebo, as was raloxifene. Subjects were directed to take one tablet orally at approximately the same time each day for 2 years. The primary outcome for both substudies was a change in BMD at the lumbar spine, as measured by dual-energy x-ray absorptiometry.
In both substudies, BMD increased to a greater degree at the lumbar spine and total hip at all BZA-estrogen dosages than with placebo, and it increased to a greater degree at the lumbar spine at most BZA-estrogen dosages, compared with raloxifene.
Osteocalcin and N-telopeptide significantly decreased at all BZA-estrogen dosages, compared with placebo, and at most BZA-estrogen dosages, compared with raloxifene.
Bazedoxifene is a potential therapeutic agent for menopausal women that may protect the endometrium while preventing bone loss in a population at higher risk of osteoporosis. It is not yet approved for this indication; further investigation is needed.
The combination of an estrogen and a selective estrogen receptor modulator to potentially relieve vasomotor symptoms, prevent vulvovaginal atrophy, and preserve bone mass without stimulating the endometrium or increasing the risk of breast cancer or venous thromboembolism is very exciting—and just might revolutionize treatment of menopausal women
Bolognese M, Krege JH, Utian WH, et al. Effects of arzoxifene on bone mineral density and endometrium in postmenopausal women with normal or low bone mass. J Clin Endocrinol Metab. 2009;94:2284–2289.
The benzothiophene derivative arzoxifene, which has been in development for the prevention and treatment of osteoporosis, as well as for reduction of the risk of invasive breast cancer in postmenopausal women, has been pulled from the regulatory approval process by its manufacturer, Eli Lilly.
This move is somewhat surprising because, in a phase 3 trial, arzoxifene significantly increased bone mineral density at the lumbar spine (2.9%) and total hip (2.2%), compared with placebo. It also decreased levels of biochemical markers of bone metabolism.
In the trial, Bolognese and associates randomly assigned 331 postmenopausal women who had normal or low bone mass to receive 20 mg of arzoxifene or placebo daily for 2 years.
The trial also found that changes in breast density were neutral or slightly decreased in the arzoxifene group, and there was no evidence of endometrial hyperplasia or carcinoma, based on central review of baseline and follow-up endometrial biopsies. Nor were there any significant differences between groups in endometrial thickness, as assessed by transvaginal ultrasonography, or in the incidence of uterine polyps, vaginal bleeding, and hot flushes.
Nevertheless, Lilly issued a press release in late August announcing that arzoxifene would not be submitted to the FDA for regulatory review.5 It appears that, although initial results from the “pivotal” 5-year, phase 3 GJAD “Generations” trial indicated that the drug had met the primary endpoints of significantly reducing the risk of vertebral fracture and invasive breast cancer in postmenopausal women, “the study failed to demonstrate a statistically significant difference in key secondary efficacy endpoints, such as nonvertebral fractures, clinical vertebral fractures, cardiovascular events, and cognitive function, compared with placebo.”
The release went on to say: “In addition, certain adverse events, including venous thromboembolic events, hot flushes, and gynecological-related events, were reported more frequently in the arzoxifene group, compared with placebo.”5
Arzoxifene therefore joins an expanding list of selective estrogen receptor modulators (SERMs) that did not make it out of clinical trials: idoxifene, droloxifene, levormeloxifene, and lasofoxifene (approved in Europe, however), to name a few. The fall of arzoxifene again underscores the notion that “not all SERMs are created equal.”6
1. Scholes D, LaCroix AZ, Ichikawa LE, Barlow WE, Ott SM. Change in bone mineral density among adolescent women using and discontinuing depot medroxyprogesterone acetate contraception. Arch Pediatr Adolesc Med. 2005;159:139-144.
2. Polatti F, Perotti F, Filippa N, Gallina D, Nappi RE. Bone mass and long-term monophasic oral contraceptive treatment in young women. Contraception. 1995;51:221-224.
3. Miller PD, Chines AA, Christiansen C, et al. Effects of bazedoxifene on BMD and bone turnover in postmenopausal women: 2-year results of a randomized, double-blind, placebo-, and active-controlled study. J Bone Miner Res. 2008;23:525-535.
4. Silverman SL, Christiansen C, Genant HK, et al. Efficacy of bazedoxifene in reducing new vertebral fracture risk in postmenopausal women with osteoporosis: results from a 3-year, randomized, placebo- and active-controlled clinical trial. J Bone Miner Res. 2008;23:1923-1934.
5. Based on preliminary phase III GHAD study results, Lilly concludes arzoxifene’s clinical profile does not support regulatory submission [press release]. Available at http://newsroom.lilly.com/releasedetail.cfm?ReleaseID=403905. Accessed October 2, 2009.
6. Goldstein SR. Not all SERMs are created equal. Menopause. 2006;13:325-327.
1. Scholes D, LaCroix AZ, Ichikawa LE, Barlow WE, Ott SM. Change in bone mineral density among adolescent women using and discontinuing depot medroxyprogesterone acetate contraception. Arch Pediatr Adolesc Med. 2005;159:139-144.
2. Polatti F, Perotti F, Filippa N, Gallina D, Nappi RE. Bone mass and long-term monophasic oral contraceptive treatment in young women. Contraception. 1995;51:221-224.
3. Miller PD, Chines AA, Christiansen C, et al. Effects of bazedoxifene on BMD and bone turnover in postmenopausal women: 2-year results of a randomized, double-blind, placebo-, and active-controlled study. J Bone Miner Res. 2008;23:525-535.
4. Silverman SL, Christiansen C, Genant HK, et al. Efficacy of bazedoxifene in reducing new vertebral fracture risk in postmenopausal women with osteoporosis: results from a 3-year, randomized, placebo- and active-controlled clinical trial. J Bone Miner Res. 2008;23:1923-1934.
5. Based on preliminary phase III GHAD study results, Lilly concludes arzoxifene’s clinical profile does not support regulatory submission [press release]. Available at http://newsroom.lilly.com/releasedetail.cfm?ReleaseID=403905. Accessed October 2, 2009.
6. Goldstein SR. Not all SERMs are created equal. Menopause. 2006;13:325-327.
Can progesterone prevent prematurity—dependably?
CASE: Patient worries about recurrent preterm birth
Ms. Jones is 13 weeks into her fourth pregnancy when she arrives at your office for her first prenatal visit. Her obstetric history is significant. In 2003, her first pregnancy was complicated by preterm labor at 25 weeks, preterm premature rupture of membranes at 26 weeks, and spontaneous vaginal delivery at 27 weeks. The infant experienced respiratory distress syndrome, bronchopulmonary dysplasia, necrotizing enterocolitis, and grade III intraventricular hemorrhage, and she was given a diagnosis of mild cerebral palsy at age 3.
Two years later, the patient’s second pregnancy was complicated by preterm labor at 22 weeks and spontaneous vaginal delivery at 23 weeks, with an Apgar score of 3, 1, and 0. The infant did not survive.
In 2007, Ms. Jones was given a diagnosis of missed abortion at 8 weeks’ gestation and underwent dilation and curettage.
Today, she asks what you plan to do to optimize the outcome of her current pregnancy. Her risk of preterm birth is significantly higher than that of the general population, which is 12.7%.
What can you offer to her?
Progesterone supplementation is the best option for Ms. Jones. Data accumulating over the past 30 years suggest that progesterone reduces the likelihood of preterm birth in women who have a history of spontaneous preterm birth. In fact, a cumulative meta-analysis noted that evidence of progesterone’s benefit is striking enough that “statistical uncertainty” is not a valid reason for forgoing its use.1
This article describes what’s been learned about progesterone supplementation to reduce preterm birth—specifically, the patients likely to benefit, the various formulations available, and the data on long-term outcomes—with an eye toward helping you weigh its utility in your practice.
The article focuses on four vulnerable populations:
- Women who have a history of preterm birth. Data suggest these patients are likely to benefit from progesterone.
- Women carrying a multiple gestation. Progesterone does not appear to prevent preterm birth in this group.
- Women who have a short cervix. Some data are promising. Further study is needed.
- Women who experience preterm labor. Data are promising, but preliminary.
Despite decades of research, initiative, and medical advances, the rate of preterm birth continues to rise, affecting one of every eight infants born in the United States—more than 500,000 babies each year. The impact of preterm birth is enormous, with implications that span from the immediate to the long-term.
In 2001, preterm birth surpassed birth defects as the leading cause of neonatal mortality. It is also the leading cause of infant mortality among African Americans and the second leading overall cause of all infant mortality.
The outlook for babies who survive preterm birth is concerning, as well. One of every five children who have mental retardation was born preterm, as was one of every three children who have vision impairment, and roughly one of every two children who have cerebral palsy. Low-birth-weight babies are commonly born preterm and face an increased risk of cardiovascular disease (including myocardial infarction, stroke, and hypertension), diabetes, and, possibly, cancer as adults.
Preterm birth not only affects the health of the baby and the family, but has long-term health and economic implications for society, costing at least $26 billion a year.26
POPULATION 1: Women who have a history of preterm birth
Women who have already delivered preterm face an elevated risk of doing so in any subsequent pregnancy ( TABLE 1 ). Three recent double-blind, randomized, controlled trials explored the efficacy of progesterone in the prevention of recurrent preterm birth.2-4 All three trials enrolled women at high risk of preterm birth; two included only women who had a history of spontaneous preterm birth, and 90% of the participants of the third trial had such a history as their risk factor.
The trials involved three different formulations of progesterone:
- intramuscular injection of 250 mg of 17α-hydroxyprogesterone caproate
- 100-mg vaginal suppository of progesterone
- 90 mg of vaginal progesterone gel (Prochieve 8% / Crinone 8%).
Meta-analyses of all studies, including these three, found that the risk of recurrent preterm birth can be reduced by as much as 40% to 55% and low birth weight by 50% using progesterone.5,6
TABLE 1
A woman who gives birth prematurely once likely will the next time
| Source | Gestational age at first delivery | Relative risk of recurrent preterm birth (95% confidence interval) |
|---|---|---|
| Maternal–Fetal Medicine Units Network30 | 2.5 (1.9–3.2) | |
| Missouri database, 1989–199731 | 3.6 (3.2–4.0) | |
| University of Texas Southwestern Medical Center, 1988–199932 | 5.9 (4.5–7.0) | |
| Denmark, 1982–198733 | 32–36 weeks | 4.8 (3.9–6.0) |
| Denmark, 1982–1987,33 Maternal–Fetal Medicine Units Network30 | 6.0 (4.1–8.8) | |
| Maternal–Fetal Medicine Units Network30 | 10.6 (2.9–38.3) |
Details of the trials
Meis and colleagues conducted a multicenter trial of 463 pregnant women who had a documented history of spontaneous preterm delivery.2 Starting between 16 and 20 weeks’ gestation, participants were randomized in a 2:1 ratio to weekly injection of 250 mg of 17α-hydroxyprogesterone caproate or an inert oil placebo, with injections continuing until delivery or 36 weeks’ gestation.
Among the findings:
- Treatment with progesterone significantly reduced the risk of delivery at less than 37 weeks’ gestation, with an incidence of 36.3% in the progesterone group versus 54.9% in the placebo group (relative risk [RR], 0.66; 95% confidence interval [CI], 0.54–0.81).
- Progesterone reduced the risk of delivery at less than 35 weeks’ gestation, with an incidence of 20.6% in the progesterone group versus 30.7% in the placebo group (RR, 0.67; 95% CI, 0.48–0.93).
- Progesterone reduced the risk of delivery at less than 32 weeks’ gestation, with an incidence of 11.4% in the progesterone group versus 19.6% in the placebo group (RR, 0.58; 95% CI, 0.37–0.91).
- Progesterone was effective in African Americans and non–African Americans.
- Infants of women treated with progesterone had significantly lower rates of necrotizing enterocolitis and intraventricular hemorrhage and less need for supplemental oxygen.
O’Brien and associates studied 659 pregnant women who had a history of spontaneous preterm birth.4 Participants were randomly assigned to receive daily treatment with progesterone vaginal gel or placebo, starting between 18 and 22.9 weeks’ gestation and continuing until delivery, 37 weeks’ gestation, or premature rupture of membranes. The gel was administered in the morning.
In this trial, progesterone did not decrease the rate of preterm birth at 32 weeks’ gestation or less (10% in the progesterone group versus 11.3% in the placebo group; odds ratio, 0.9; 95% CI, 0.52–1.56).
It is unclear whether the formulation, timing, or dosage was responsible for the different outcomes in these trials ( TABLE 2) .
TABLE 2
5 Progesterone formulations have been tested for the prevention of preterm birth
| Formulation | Dosage | Administration | Dosing schedule | Gestational age at initiation | Gestational age at completion |
|---|---|---|---|---|---|
| 17α-Hydroxyprogesterone caproate2 | 250 mg | Intramuscular | Weekly | 16.0–20.0 weeks | 36.9 weeks |
| Progesterone3 | 100 mg | Vaginal suppository | Daily at bedtime | 24 weeks | 34 weeks |
| Progesterone14 | 200 mg | Vaginal suppository | Daily at bedtime | 24 weeks | 34 weeks |
| Prochieve 8%/Crinone 8%4 | 90 mg | Vaginal suppository bioadhesive formulation/gel | Every morning | 18.0–22.9 weeks | 37 weeks |
| Progesterone19 | 400 mg | Vaginal suppository | Daily | After arrest of preterm labor | Delivery |
In this population, the number needed to treat is low
At least five strong meta-analyses have explored the prevention of recurrent preterm birth.1,7-10 These analyses demonstrate that progesterone supplementation significantly reduces the incidence of low birth weight and preterm birth. In some cases, it also reduces the rate of respiratory distress syndrome and intraventricular hemorrhage.
Based on these data, Petrini and associates calculated that, if all pregnant women who had a history of spontaneous preterm birth had been offered progesterone in 2002, 10,000 preterm births could have been prevented.11
The number needed to treat (NNT) to avoid one preterm birth was eight for 17α-hydroxyprogesterone caproate and 10 using another progesterone formulation. The NNT to prevent low birth weight was 12.
To put these figures in context, consider the use of low-dose aspirin to prevent stroke, which has a NNT of 102, and the use of a β-blocker to prevent cardiac death in patients who have suffered a myocardial infarction, which carries a NNT of 42.
POPULATION 2: Women who are carrying a multiple gestation
Given the success of progesterone in preventing recurrent preterm birth, it was a matter of time before investigators began to consider its use in another high-risk group: women carrying a multiple gestation. In two double-blind, placebo-controlled trials—one from the United States and the other from the United Kingdom—17α-hydroxyprogesterone caproate or placebo was given, starting between 16 and 20 weeks’ gestation in women who were carrying a twin or triplet gestation.12,13 Neither trial demonstrated a benefit for the use of progesterone in this population.
The etiology of preterm birth is likely different in women with a previous preterm birth than it is in women carrying a multiple gestation. The former are more likely to have an inflammatory, immunologic, or infectious process that leads to recurrent preterm birth, whereas women carrying multiples are thought to be at risk of preterm birth by virtue of the “stretch hypothesis”—the theory that the uterus is stretched excessively, leading to an earlier trigger of labor. Women who had a history of preterm birth and who were carrying a multiple gestation were eligible for these trials.
In the US trial, progesterone failed to reduce the rate of preterm birth in women who were carrying twins or triplets.13 This lack of benefit was seen regardless of whether conception was spontaneous or the result of assisted reproductive technologies, whether placentation was dichorionic or monochorionic, and regardless of the cutoff for gestational age. On average, the women in this trial delivered at 34.8 weeks, compared with a national average of 35.2 weeks for women carrying twins.13
Similar findings were reported from the UK trial, which enrolled 500 women carrying a twin gestation who were randomized to daily vaginal progesterone gel (90 mg) or placebo from 24 to 34 weeks’ gestation.12
A meta-analysis of the three trials that included multiple gestation12-14 found progesterone to have no benefit in women carrying twins. The pooled odds ratio of the effect of progesterone on preterm delivery or intrauterine death before 34 weeks’ gestation was 1.16 (95% CI, 0.89–1.51).12
Progesterone is a familiar player in the ObGyn specialty. In its natural form, the steroid hormone is produced by the corpus luteum to promote pregnancy.
In target cells, progesterone binds to its receptor and forms a transcription factor. It also can be active independent of nuclear receptors, which may explain why it remains effective even when circulating concentrations are high, suggesting that its action may be local and not systemic.
Progesterone exerts biologic effects on the myometrium, chorioamniotic membranes, and cervix.
Myometrial effects include:
- a decrease in the conduction of contractions
- a reduction of spontaneous muscle activity
- a decrease in the number of oxytocin receptors
- prevention of the formation of gap junctions
- a rise in the threshold for stimulation.
Progesterone decreases myometrial estrogen responsiveness by inhibiting estrogen-receptor expression and appears to maintain uterine quiescence by limiting the production of prostaglandins and inhibiting the expression of contraction-associated protein genes, including gap junctions, ion channels, oxytocin, and prostaglandin receptors within the myometrium.27 Some investigators have suggested that progesterone prevents preterm birth predominantly by virtue of its anti- inflammatory properties28 and ability to prevent cervical ripening.29
The 17α-hydroxyprogesterone form of the hormone also affects salivary concentrations of estriol. In a secondary analysis, the ratio of salivary estriol to progesterone increased as pregnancy progressed among women who received placebo, but remained flat among women treated with 17α-hydroxyprogesterone.2 One theory is that labor may be triggered by an increase in the activity of estriol, compared with progesterone.
It also is notable that estriol concentrations in the mother’s blood and saliva derive mainly from the fetus and placenta (from the fetus’ production of cortisol), suggesting that the action of 17α-hydroxyprogesterone acetate may also affect the feto-placental unit.
POPULATION 3: Women who have a short cervix
Because women who have a short cervix have a heightened risk of spontaneous preterm delivery, the utility of progesterone in prolonging gestation was explored in this population—with less than definitive results. An editorial accompanying the main study of this issue concluded that it is too early to recommend use of progesterone in women who have a short cervix.15
Progesterone was effective overall, but not in subgroup analysis
Iams and associates expertly delineated the risk of spontaneous preterm birth in the setting of a shortened cervix at 24 weeks’ gestation. They found a cervical length of about 12 mm to be at the first centile, with a relative risk of preterm birth of 14.16 (Compare this with the average cervical length of 36 to 44 mm at 24 weeks.)
Fonseca and colleagues then explored the benefit of progesterone therapy in preventing preterm birth in women who had a shortened cervical length between 20 and 25 weeks’ gestation.14 They screened more than 24,000 women and found 413 who had a cervical length of less than 15 mm. Of these women, 250 were randomized to micronized progesterone (200 mg in a vaginal suppository), starting at 24 weeks. This was twice the dosage given in the Brazilian trial involving women who had a history of preterm birth,3 but the authors thought that women who had a short cervix were at higher risk of preterm birth and, therefore, needed a higher dosage of progesterone. Although all women in this trial had a short cervix, the population overall was more heterogeneous than in other trials, including women who had a history of preterm birth (30% of participants) and women carrying a multiple gestation (19% of participants).
Progesterone reduced the risk of preterm birth in the overall cohort, with 19% of the women who received progesterone delivering preterm, versus 34% of those who received placebo (odd ratio [OR], 0.56; 95% CI, 0.36–0.86). Progesterone did not reduce the rate of perinatal mortality or neonatal morbidity. A subgroup analysis of only the nulli parous women was conducted, given that 30% of the study population had a history of preterm birth. That analysis showed no benefit.
DeFranco and associates17 published a secondary analysis of 46 women from a large randomized trial4 who had a cervical length of less than 28 mm. Of these women, 19 received progesterone and 27 received placebo. Of the 19 who received progesterone, 15 had a history of preterm birth. Of the 27 who received placebo, 22 had such a history. The authors found that progesterone significantly reduced preterm birth at less than 37, 35, and 32 weeks. However, again because of the small sample size and the inclusion of women with a history of preterm birth, these findings are not definitive.
Randomized trials designed to test the effect of progesterone in women who have a short cervix are called for. Numerous studies are under way.15
POPULATION 4: Women who experience preterm labor
Two recent trials explored the use of progesterone in this context. In one, progesterone was administered during the episode of preterm labor; in the other, it was given after successful tocolysis.
In the first trial, Facchinetti and colleagues studied 60 women who were pregnant with a singleton fetus and who were in active preterm labor.18 These women were randomly assigned to 341 mg of intramuscular 17α-hydroxyprogesterone caproate or placebo twice weekly, with cervical length monitored weekly. Women in the progesterone group were less likely to deliver by 7 or 21 days, and their cervical length was longer at both time points.
Borna and Sahabi evaluated use of progesterone as maintenance therapy after successful tocolysis.19 Seventy women were randomly assigned to progesterone (400-mg suppository) or no treatment. Women who received progesterone had a longer latency period (36 versus 24 days; P=.03), less respiratory distress (11% versus 36%), and a lower rate of low birth weight (27% versus 52%) than did women receiving no treatment.
What are the long-term effects of progesterone exposure?
Therapeutic interventions during pregnancy affect two people—one of them during a period of intense development that can have a lifelong impact. Although studies of progesterone to prevent preterm birth involve administration of the hormone after 16 weeks—well beyond the major period of organogenesis—concerns about potential teratogenic and other long-term effects have been raised. It is notable that progesterone has been widely used for decades during the first trimester—the period of organogenesis—in women who have a poor pregnancy history and early loss, to treat the “luteal phase defect.”
A Cochrane review of 14 studies of progesterone in the prevention of stillbirth and miscarriage20 and a systematic review of 14 cohort and case-control studies21 involving first-trimester exposure found no harm related to progesterone use. These findings are consistent with those of a meta-analysis by Coomarasamy and colleagues, which also found no harm related to the use of progesterone.1
Numerous studies have explored the long-term effects of progesterone on offspring, including a review of outcomes of pregnancies treated before 199022 and data from animal studies.23 Children from a trial by Meis22 were followed up at around 4 years of age to assess any differences in physical health and the achievement of developmental milestones between children who were exposed to progesterone and those who were not.24 Investigators used the Ages and Stages Questionnaire score, assessment of developmental milestones, and physical exams to evaluate the 348 children. No differences were seen in height, weight, and head-circumference percentiles; achievement of developmental milestones; gender roles; and physical health.
In a systematic review of 11 trials (2,425 women and 3,187 infants) involving the use of progesterone to prevent preterm birth in high-risk women—including those who had a history of preterm birth, those carrying a multiple gestation, and those with a short cervix—Dodd and colleagues found mixed results.10 Progesterone reduced the rate of preterm birth before 34 weeks’ gestation in women who had a history of preterm birth, as well as in those who had a short cervix, but no improvement was seen in women carrying a multiple gestation.
A cumulative meta-analysis by Coomarasamy and colleagues found that progestogens significantly reduce the rate of preterm birth, a benefit that was evident beginning in 1975.1
The most recent Committee Opinion from the American College of Obstetricians and Gynecologists25 concludes that “it is important to offer progesterone for pregnancy prolongation to only women with a documented history of a previous spontaneous birth at less than 37 weeks of gestation.” The opinion also takes into account the findings of the Fonseca trial in regard to women who have a short cervix,14 and concludes that “progesterone supplementation may be considered for use in asymptomatic women with a short cervix.”
Trials of other high-risk groups, including women who have a positive fibronectin test, bleeding, or iatrogenic preterm labor, are needed. The fact that progesterone supplementation is not universally effective in women who have a history of preterm birth suggests that not all pathways leading to preterm birth are ameliorated by progesterone therapy. Given the many similarities between women who have a history of preterm birth and women who have a short cervix, evidence may ultimately be available to support the benefits of progesterone in both situations. However, the lack of a benefit in women carrying a multiple gestation likely reflects the different underlying mechanism in that group.
CASE: RESOLVED
You discuss with Ms. Jones the options available to reduce the likelihood of recurrent preterm birth. She opts for progesterone supplementation, which is initiated at 16 weeks’ gestation, with no restrictions on activity. A sonogram at 18 weeks reveals normal anatomy and a cervical length of 4 cm.
At 22 weeks’ gestation, Ms. Jones visits the labor and delivery unit complaining of leaking fluid. You perform a sterile speculum exam, which is negative, monitor her for several hours, and send her home.
At 26 weeks, the patient experiences contractions and is again evaluated. An examination reveals the cervix to be long and closed. After prolonged monitoring, Ms. Jones is again sent home.
At 37 weeks’ gestation, the patient reports another episode of leaking fluid. This time, a sterile speculum exam is positive, and you begin induction of labor.
Labor proceeds smoothly, and Ms. Jones delivers a 3,100-g infant. The newborn has an Apgar score of 8 and 9 at 1 and 5 minutes, respectively.
1. Coomarasamy A, Thangaratinam S, Gee H, Khan KS. Progesterone for the prevention of preterm birth: a critical evaluation of evidence. Eur J Obstet Gynecol Reprod Biol. 2006;129:111-118.
2. Meis PJ, Klebanoff M, Thom E, et al. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N Engl J Med. 2003;348:2379-2385.
3. da Fonseca EB, Bittar RE, Carvalho MH, Zugaib M. Prophylactic administration of progesterone by vaginal suppository to reduce the incidence of spontaneous preterm birth in women at increased risk: a randomized placebo-controlled double-blind study. Am J Obstet Gynecol. 2003;188:419-424.
4. O’Brien JM, Adair CD, Lewis DF, et al. Progesterone vaginal gel for the reduction of recurrent preterm birth: primary results from a randomized, double-blind, placebo-controlled trial. Ultrasound Obstet Gynecol. 2007;30:687-696.
5. Sanchez-Ramos L, Kaunitz AM, Delke I. Progestational agents to prevent preterm birth: a meta-analysis of randomized controlled trials. Obstet Gynecol. 2005;105:273-279.
6. Dodd JM, Flenady V, Cincotta R, Crowther CA. Prenatal administration of progesterone for preventing preterm birth. Cochrane Database Syst Rev. 2006;(1):CD004947.-
7. Keirse MJ. Progestogen administration in pregnancy may prevent preterm delivery. Br J Obstet Gynaecol. 1990;97:149-154.
8. Dodd JM, Crowther CA, Cincotta R, Flenady V, Robinson JS. Progesterone supplementation for preventing preterm birth: a systematic review and meta-analysis. Acta Obstet Gynecol Scand. 2005;84:526-533.
9. Mackenzie R, Walker M, Armson A, Hannah ME. Progesterone for the prevention of preterm birth among women at increased risk: a systematic review and meta-analysis of randomized controlled trials. Am J Obstet Gynecol. 2006;194:1234-1242.
10. Dodd JM, Flenady VJ, Cincotta R, Crowther CA. Progesterone for the prevention of preterm birth: a systematic review. Obstet Gynecol. 2008;112:127-134.
11. Petrini JR, Callaghan WM, Klebanoff M, et al. Estimated effect of 17 alpha-hydroxyprogesterone caproate on preterm birth in the United States. Obstet Gynecol. 2005;105:267-272.
12. Norman JE, Mackenzie F, Owen P, et al. Progesterone for the prevention of preterm birth in twin pregnancy (STOPPIT): a randomised, double-blind, placebo-controlled study and meta-analysis. Lancet. 2009;373:2034-2040.
13. Rouse DJ, Caritis SN, Peaceman AM, et al. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. A trial of 17 alpha-hydroxyprogesterone caproate to prevent prematurity in twins. N Engl J Med. 2007;357:454-461.
14. Fonseca EB, Celik E, Parra M, Singh M, Nicolaides KH. Fetal Medicine Foundation Second Trimester Screening Group. Progesterone and the risk of preterm birth among women with a short cervix. N Engl J Med. 2007;357:462-469.
15. Thornton JG. Progesterone and preterm labor—still no definite answers. N Engl J Med. 2007;357:499-501.
16. Iams JD, Goldenberg RL, Meis PJ, et al. The length of the cervix and the risk of spontaneous premature delivery. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. N Engl J Med. 1996;334:567-572.
17. DeFranco EA, O’Brien JM, Adair CD, et al. Vaginal progesterone is associated with a decrease in risk for early preterm birth and improved neonatal outcome in women with a short cervix: a secondary analysis from a randomized, double-blind, placebo-controlled trial. Ultrasound Obstet Gynecol. 2007;30:697-705.
18. Facchinetti F, Paganelli S, Comitini G, Dante G, Volpe A. Cervical length changes during preterm cervical ripening: effects of 17-alpha-hydroxyprogesterone caproate. Am J Obstet Gynecol. 2007;196:453.e1-453.e4.
19. Borna S, Sahabi N. Progesterone for maintenance tocolytic therapy after threatened preterm labour: a randomised controlled trial. Aust N Z J Obstet Gynaecol. 2008;48:58-63.
20. Oates-Whitehead RM, Haas DM, Carrier JA. Progestogen for preventing miscarriage. Cochrane Database Syst Rev. 2003;(4):CD003511.-
21. Raman-Wilms L, Tseng AL, Wighardt S, Einarson TR, Koren G. Fetal genital effects of first trimester sex hormone exposure: a meta-analysis. Obstet Gynecol. 1995;85:141-149.
22. Meis PJ. Society for Maternal–Fetal Medicine. 17 Hydroxyprogesterone for the prevention of preterm delivery. Obstet Gynecol. 2005;105(5 Pt 1):1128-1135.
23. Christian MS, Brent RL, Calda P. Embryo–fetal toxicity signals for 17alpha-hydroxyprogesterone caproate in high-risk pregnancies: a review of the non-clinical literature for embryo–fetal toxicity with progestins. J Matern Fetal Neonatal Med. 2007;20:89-112.
24. Northen AT, Norman GS, Anderson K, et al. National Institute of Child Health and Human Development (NICHD) Maternal–Fetal Medicine Units (MFMU) Network. Follow-up of children exposed in utero to 17 alpha-hydroxyprogesterone caproate compared with placebo. Obstet Gynecol. 2007;110:865-872.
25. American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 419, October 2008. Use of progesterone to reduce preterm birth. Washington, DC: ACOG; 2008.
26. Preterm birth costs US $26 billion a year [press release]. Washington, DC: National Academies. July 13, 2006. Available at: http://www8.nationalacademies.org/onpinews/newsitem.aspx?RecordID=11622. Accessed October 5, 2009.
27. Norwitz ER. A blood test to predict preterm birth: don’t mess with maternal–fetal stress. J Clin Endocrinol Metab. 2009;94:1886-1889.
28. Elovitz MA, Gonzalez J. Medroxyprogesterone acetate modulates the immune response in the uterus, cervix and placenta in a mouse model of preterm birth. J Matern Fetal Neonatal Med. 2008;21:223-230.
29. Xu H, Gonzalez JM, Ofori E, Elovitz MA. Preventing cervical ripening: the primary mechanism by which progestational agents prevent preterm birth? Am J Obstet Gynecol. 2008;198:314.e1-314.e8.
30. Mercer BM, Goldenberg RL, Moawad AH, et al. The preterm prediction study: effect of gestational age and cause of preterm birth on subsequent obstetric outcome. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. Am J Obstet Gynecol. 1999;181(5 Pt 1):1216-1221.
31. Ananth CV, Getahun D, Peltier MR, Salihu HM, Vintzileos AM. Recurrence of spontaneous versus medically indicated preterm birth. Am J Obstet Gynecol. 2006;195:643-650.
32. Bloom SL, Yost NP, McIntire DD, Leveno KJ. Recurrence of preterm birth in singleton and twin pregnancies. Obstet Gynecol. 2001;98:379-385.
33. Kristensen J, Langhoff-Roos J, Kristensen FB. Implications of idiopathic preterm delivery for previous and subsequent pregnancies. Obstet Gynecol. 1995;86:800-804.
CASE: Patient worries about recurrent preterm birth
Ms. Jones is 13 weeks into her fourth pregnancy when she arrives at your office for her first prenatal visit. Her obstetric history is significant. In 2003, her first pregnancy was complicated by preterm labor at 25 weeks, preterm premature rupture of membranes at 26 weeks, and spontaneous vaginal delivery at 27 weeks. The infant experienced respiratory distress syndrome, bronchopulmonary dysplasia, necrotizing enterocolitis, and grade III intraventricular hemorrhage, and she was given a diagnosis of mild cerebral palsy at age 3.
Two years later, the patient’s second pregnancy was complicated by preterm labor at 22 weeks and spontaneous vaginal delivery at 23 weeks, with an Apgar score of 3, 1, and 0. The infant did not survive.
In 2007, Ms. Jones was given a diagnosis of missed abortion at 8 weeks’ gestation and underwent dilation and curettage.
Today, she asks what you plan to do to optimize the outcome of her current pregnancy. Her risk of preterm birth is significantly higher than that of the general population, which is 12.7%.
What can you offer to her?
Progesterone supplementation is the best option for Ms. Jones. Data accumulating over the past 30 years suggest that progesterone reduces the likelihood of preterm birth in women who have a history of spontaneous preterm birth. In fact, a cumulative meta-analysis noted that evidence of progesterone’s benefit is striking enough that “statistical uncertainty” is not a valid reason for forgoing its use.1
This article describes what’s been learned about progesterone supplementation to reduce preterm birth—specifically, the patients likely to benefit, the various formulations available, and the data on long-term outcomes—with an eye toward helping you weigh its utility in your practice.
The article focuses on four vulnerable populations:
- Women who have a history of preterm birth. Data suggest these patients are likely to benefit from progesterone.
- Women carrying a multiple gestation. Progesterone does not appear to prevent preterm birth in this group.
- Women who have a short cervix. Some data are promising. Further study is needed.
- Women who experience preterm labor. Data are promising, but preliminary.
Despite decades of research, initiative, and medical advances, the rate of preterm birth continues to rise, affecting one of every eight infants born in the United States—more than 500,000 babies each year. The impact of preterm birth is enormous, with implications that span from the immediate to the long-term.
In 2001, preterm birth surpassed birth defects as the leading cause of neonatal mortality. It is also the leading cause of infant mortality among African Americans and the second leading overall cause of all infant mortality.
The outlook for babies who survive preterm birth is concerning, as well. One of every five children who have mental retardation was born preterm, as was one of every three children who have vision impairment, and roughly one of every two children who have cerebral palsy. Low-birth-weight babies are commonly born preterm and face an increased risk of cardiovascular disease (including myocardial infarction, stroke, and hypertension), diabetes, and, possibly, cancer as adults.
Preterm birth not only affects the health of the baby and the family, but has long-term health and economic implications for society, costing at least $26 billion a year.26
POPULATION 1: Women who have a history of preterm birth
Women who have already delivered preterm face an elevated risk of doing so in any subsequent pregnancy ( TABLE 1 ). Three recent double-blind, randomized, controlled trials explored the efficacy of progesterone in the prevention of recurrent preterm birth.2-4 All three trials enrolled women at high risk of preterm birth; two included only women who had a history of spontaneous preterm birth, and 90% of the participants of the third trial had such a history as their risk factor.
The trials involved three different formulations of progesterone:
- intramuscular injection of 250 mg of 17α-hydroxyprogesterone caproate
- 100-mg vaginal suppository of progesterone
- 90 mg of vaginal progesterone gel (Prochieve 8% / Crinone 8%).
Meta-analyses of all studies, including these three, found that the risk of recurrent preterm birth can be reduced by as much as 40% to 55% and low birth weight by 50% using progesterone.5,6
TABLE 1
A woman who gives birth prematurely once likely will the next time
| Source | Gestational age at first delivery | Relative risk of recurrent preterm birth (95% confidence interval) |
|---|---|---|
| Maternal–Fetal Medicine Units Network30 | 2.5 (1.9–3.2) | |
| Missouri database, 1989–199731 | 3.6 (3.2–4.0) | |
| University of Texas Southwestern Medical Center, 1988–199932 | 5.9 (4.5–7.0) | |
| Denmark, 1982–198733 | 32–36 weeks | 4.8 (3.9–6.0) |
| Denmark, 1982–1987,33 Maternal–Fetal Medicine Units Network30 | 6.0 (4.1–8.8) | |
| Maternal–Fetal Medicine Units Network30 | 10.6 (2.9–38.3) |
Details of the trials
Meis and colleagues conducted a multicenter trial of 463 pregnant women who had a documented history of spontaneous preterm delivery.2 Starting between 16 and 20 weeks’ gestation, participants were randomized in a 2:1 ratio to weekly injection of 250 mg of 17α-hydroxyprogesterone caproate or an inert oil placebo, with injections continuing until delivery or 36 weeks’ gestation.
Among the findings:
- Treatment with progesterone significantly reduced the risk of delivery at less than 37 weeks’ gestation, with an incidence of 36.3% in the progesterone group versus 54.9% in the placebo group (relative risk [RR], 0.66; 95% confidence interval [CI], 0.54–0.81).
- Progesterone reduced the risk of delivery at less than 35 weeks’ gestation, with an incidence of 20.6% in the progesterone group versus 30.7% in the placebo group (RR, 0.67; 95% CI, 0.48–0.93).
- Progesterone reduced the risk of delivery at less than 32 weeks’ gestation, with an incidence of 11.4% in the progesterone group versus 19.6% in the placebo group (RR, 0.58; 95% CI, 0.37–0.91).
- Progesterone was effective in African Americans and non–African Americans.
- Infants of women treated with progesterone had significantly lower rates of necrotizing enterocolitis and intraventricular hemorrhage and less need for supplemental oxygen.
O’Brien and associates studied 659 pregnant women who had a history of spontaneous preterm birth.4 Participants were randomly assigned to receive daily treatment with progesterone vaginal gel or placebo, starting between 18 and 22.9 weeks’ gestation and continuing until delivery, 37 weeks’ gestation, or premature rupture of membranes. The gel was administered in the morning.
In this trial, progesterone did not decrease the rate of preterm birth at 32 weeks’ gestation or less (10% in the progesterone group versus 11.3% in the placebo group; odds ratio, 0.9; 95% CI, 0.52–1.56).
It is unclear whether the formulation, timing, or dosage was responsible for the different outcomes in these trials ( TABLE 2) .
TABLE 2
5 Progesterone formulations have been tested for the prevention of preterm birth
| Formulation | Dosage | Administration | Dosing schedule | Gestational age at initiation | Gestational age at completion |
|---|---|---|---|---|---|
| 17α-Hydroxyprogesterone caproate2 | 250 mg | Intramuscular | Weekly | 16.0–20.0 weeks | 36.9 weeks |
| Progesterone3 | 100 mg | Vaginal suppository | Daily at bedtime | 24 weeks | 34 weeks |
| Progesterone14 | 200 mg | Vaginal suppository | Daily at bedtime | 24 weeks | 34 weeks |
| Prochieve 8%/Crinone 8%4 | 90 mg | Vaginal suppository bioadhesive formulation/gel | Every morning | 18.0–22.9 weeks | 37 weeks |
| Progesterone19 | 400 mg | Vaginal suppository | Daily | After arrest of preterm labor | Delivery |
In this population, the number needed to treat is low
At least five strong meta-analyses have explored the prevention of recurrent preterm birth.1,7-10 These analyses demonstrate that progesterone supplementation significantly reduces the incidence of low birth weight and preterm birth. In some cases, it also reduces the rate of respiratory distress syndrome and intraventricular hemorrhage.
Based on these data, Petrini and associates calculated that, if all pregnant women who had a history of spontaneous preterm birth had been offered progesterone in 2002, 10,000 preterm births could have been prevented.11
The number needed to treat (NNT) to avoid one preterm birth was eight for 17α-hydroxyprogesterone caproate and 10 using another progesterone formulation. The NNT to prevent low birth weight was 12.
To put these figures in context, consider the use of low-dose aspirin to prevent stroke, which has a NNT of 102, and the use of a β-blocker to prevent cardiac death in patients who have suffered a myocardial infarction, which carries a NNT of 42.
POPULATION 2: Women who are carrying a multiple gestation
Given the success of progesterone in preventing recurrent preterm birth, it was a matter of time before investigators began to consider its use in another high-risk group: women carrying a multiple gestation. In two double-blind, placebo-controlled trials—one from the United States and the other from the United Kingdom—17α-hydroxyprogesterone caproate or placebo was given, starting between 16 and 20 weeks’ gestation in women who were carrying a twin or triplet gestation.12,13 Neither trial demonstrated a benefit for the use of progesterone in this population.
The etiology of preterm birth is likely different in women with a previous preterm birth than it is in women carrying a multiple gestation. The former are more likely to have an inflammatory, immunologic, or infectious process that leads to recurrent preterm birth, whereas women carrying multiples are thought to be at risk of preterm birth by virtue of the “stretch hypothesis”—the theory that the uterus is stretched excessively, leading to an earlier trigger of labor. Women who had a history of preterm birth and who were carrying a multiple gestation were eligible for these trials.
In the US trial, progesterone failed to reduce the rate of preterm birth in women who were carrying twins or triplets.13 This lack of benefit was seen regardless of whether conception was spontaneous or the result of assisted reproductive technologies, whether placentation was dichorionic or monochorionic, and regardless of the cutoff for gestational age. On average, the women in this trial delivered at 34.8 weeks, compared with a national average of 35.2 weeks for women carrying twins.13
Similar findings were reported from the UK trial, which enrolled 500 women carrying a twin gestation who were randomized to daily vaginal progesterone gel (90 mg) or placebo from 24 to 34 weeks’ gestation.12
A meta-analysis of the three trials that included multiple gestation12-14 found progesterone to have no benefit in women carrying twins. The pooled odds ratio of the effect of progesterone on preterm delivery or intrauterine death before 34 weeks’ gestation was 1.16 (95% CI, 0.89–1.51).12
Progesterone is a familiar player in the ObGyn specialty. In its natural form, the steroid hormone is produced by the corpus luteum to promote pregnancy.
In target cells, progesterone binds to its receptor and forms a transcription factor. It also can be active independent of nuclear receptors, which may explain why it remains effective even when circulating concentrations are high, suggesting that its action may be local and not systemic.
Progesterone exerts biologic effects on the myometrium, chorioamniotic membranes, and cervix.
Myometrial effects include:
- a decrease in the conduction of contractions
- a reduction of spontaneous muscle activity
- a decrease in the number of oxytocin receptors
- prevention of the formation of gap junctions
- a rise in the threshold for stimulation.
Progesterone decreases myometrial estrogen responsiveness by inhibiting estrogen-receptor expression and appears to maintain uterine quiescence by limiting the production of prostaglandins and inhibiting the expression of contraction-associated protein genes, including gap junctions, ion channels, oxytocin, and prostaglandin receptors within the myometrium.27 Some investigators have suggested that progesterone prevents preterm birth predominantly by virtue of its anti- inflammatory properties28 and ability to prevent cervical ripening.29
The 17α-hydroxyprogesterone form of the hormone also affects salivary concentrations of estriol. In a secondary analysis, the ratio of salivary estriol to progesterone increased as pregnancy progressed among women who received placebo, but remained flat among women treated with 17α-hydroxyprogesterone.2 One theory is that labor may be triggered by an increase in the activity of estriol, compared with progesterone.
It also is notable that estriol concentrations in the mother’s blood and saliva derive mainly from the fetus and placenta (from the fetus’ production of cortisol), suggesting that the action of 17α-hydroxyprogesterone acetate may also affect the feto-placental unit.
POPULATION 3: Women who have a short cervix
Because women who have a short cervix have a heightened risk of spontaneous preterm delivery, the utility of progesterone in prolonging gestation was explored in this population—with less than definitive results. An editorial accompanying the main study of this issue concluded that it is too early to recommend use of progesterone in women who have a short cervix.15
Progesterone was effective overall, but not in subgroup analysis
Iams and associates expertly delineated the risk of spontaneous preterm birth in the setting of a shortened cervix at 24 weeks’ gestation. They found a cervical length of about 12 mm to be at the first centile, with a relative risk of preterm birth of 14.16 (Compare this with the average cervical length of 36 to 44 mm at 24 weeks.)
Fonseca and colleagues then explored the benefit of progesterone therapy in preventing preterm birth in women who had a shortened cervical length between 20 and 25 weeks’ gestation.14 They screened more than 24,000 women and found 413 who had a cervical length of less than 15 mm. Of these women, 250 were randomized to micronized progesterone (200 mg in a vaginal suppository), starting at 24 weeks. This was twice the dosage given in the Brazilian trial involving women who had a history of preterm birth,3 but the authors thought that women who had a short cervix were at higher risk of preterm birth and, therefore, needed a higher dosage of progesterone. Although all women in this trial had a short cervix, the population overall was more heterogeneous than in other trials, including women who had a history of preterm birth (30% of participants) and women carrying a multiple gestation (19% of participants).
Progesterone reduced the risk of preterm birth in the overall cohort, with 19% of the women who received progesterone delivering preterm, versus 34% of those who received placebo (odd ratio [OR], 0.56; 95% CI, 0.36–0.86). Progesterone did not reduce the rate of perinatal mortality or neonatal morbidity. A subgroup analysis of only the nulli parous women was conducted, given that 30% of the study population had a history of preterm birth. That analysis showed no benefit.
DeFranco and associates17 published a secondary analysis of 46 women from a large randomized trial4 who had a cervical length of less than 28 mm. Of these women, 19 received progesterone and 27 received placebo. Of the 19 who received progesterone, 15 had a history of preterm birth. Of the 27 who received placebo, 22 had such a history. The authors found that progesterone significantly reduced preterm birth at less than 37, 35, and 32 weeks. However, again because of the small sample size and the inclusion of women with a history of preterm birth, these findings are not definitive.
Randomized trials designed to test the effect of progesterone in women who have a short cervix are called for. Numerous studies are under way.15
POPULATION 4: Women who experience preterm labor
Two recent trials explored the use of progesterone in this context. In one, progesterone was administered during the episode of preterm labor; in the other, it was given after successful tocolysis.
In the first trial, Facchinetti and colleagues studied 60 women who were pregnant with a singleton fetus and who were in active preterm labor.18 These women were randomly assigned to 341 mg of intramuscular 17α-hydroxyprogesterone caproate or placebo twice weekly, with cervical length monitored weekly. Women in the progesterone group were less likely to deliver by 7 or 21 days, and their cervical length was longer at both time points.
Borna and Sahabi evaluated use of progesterone as maintenance therapy after successful tocolysis.19 Seventy women were randomly assigned to progesterone (400-mg suppository) or no treatment. Women who received progesterone had a longer latency period (36 versus 24 days; P=.03), less respiratory distress (11% versus 36%), and a lower rate of low birth weight (27% versus 52%) than did women receiving no treatment.
What are the long-term effects of progesterone exposure?
Therapeutic interventions during pregnancy affect two people—one of them during a period of intense development that can have a lifelong impact. Although studies of progesterone to prevent preterm birth involve administration of the hormone after 16 weeks—well beyond the major period of organogenesis—concerns about potential teratogenic and other long-term effects have been raised. It is notable that progesterone has been widely used for decades during the first trimester—the period of organogenesis—in women who have a poor pregnancy history and early loss, to treat the “luteal phase defect.”
A Cochrane review of 14 studies of progesterone in the prevention of stillbirth and miscarriage20 and a systematic review of 14 cohort and case-control studies21 involving first-trimester exposure found no harm related to progesterone use. These findings are consistent with those of a meta-analysis by Coomarasamy and colleagues, which also found no harm related to the use of progesterone.1
Numerous studies have explored the long-term effects of progesterone on offspring, including a review of outcomes of pregnancies treated before 199022 and data from animal studies.23 Children from a trial by Meis22 were followed up at around 4 years of age to assess any differences in physical health and the achievement of developmental milestones between children who were exposed to progesterone and those who were not.24 Investigators used the Ages and Stages Questionnaire score, assessment of developmental milestones, and physical exams to evaluate the 348 children. No differences were seen in height, weight, and head-circumference percentiles; achievement of developmental milestones; gender roles; and physical health.
In a systematic review of 11 trials (2,425 women and 3,187 infants) involving the use of progesterone to prevent preterm birth in high-risk women—including those who had a history of preterm birth, those carrying a multiple gestation, and those with a short cervix—Dodd and colleagues found mixed results.10 Progesterone reduced the rate of preterm birth before 34 weeks’ gestation in women who had a history of preterm birth, as well as in those who had a short cervix, but no improvement was seen in women carrying a multiple gestation.
A cumulative meta-analysis by Coomarasamy and colleagues found that progestogens significantly reduce the rate of preterm birth, a benefit that was evident beginning in 1975.1
The most recent Committee Opinion from the American College of Obstetricians and Gynecologists25 concludes that “it is important to offer progesterone for pregnancy prolongation to only women with a documented history of a previous spontaneous birth at less than 37 weeks of gestation.” The opinion also takes into account the findings of the Fonseca trial in regard to women who have a short cervix,14 and concludes that “progesterone supplementation may be considered for use in asymptomatic women with a short cervix.”
Trials of other high-risk groups, including women who have a positive fibronectin test, bleeding, or iatrogenic preterm labor, are needed. The fact that progesterone supplementation is not universally effective in women who have a history of preterm birth suggests that not all pathways leading to preterm birth are ameliorated by progesterone therapy. Given the many similarities between women who have a history of preterm birth and women who have a short cervix, evidence may ultimately be available to support the benefits of progesterone in both situations. However, the lack of a benefit in women carrying a multiple gestation likely reflects the different underlying mechanism in that group.
CASE: RESOLVED
You discuss with Ms. Jones the options available to reduce the likelihood of recurrent preterm birth. She opts for progesterone supplementation, which is initiated at 16 weeks’ gestation, with no restrictions on activity. A sonogram at 18 weeks reveals normal anatomy and a cervical length of 4 cm.
At 22 weeks’ gestation, Ms. Jones visits the labor and delivery unit complaining of leaking fluid. You perform a sterile speculum exam, which is negative, monitor her for several hours, and send her home.
At 26 weeks, the patient experiences contractions and is again evaluated. An examination reveals the cervix to be long and closed. After prolonged monitoring, Ms. Jones is again sent home.
At 37 weeks’ gestation, the patient reports another episode of leaking fluid. This time, a sterile speculum exam is positive, and you begin induction of labor.
Labor proceeds smoothly, and Ms. Jones delivers a 3,100-g infant. The newborn has an Apgar score of 8 and 9 at 1 and 5 minutes, respectively.
CASE: Patient worries about recurrent preterm birth
Ms. Jones is 13 weeks into her fourth pregnancy when she arrives at your office for her first prenatal visit. Her obstetric history is significant. In 2003, her first pregnancy was complicated by preterm labor at 25 weeks, preterm premature rupture of membranes at 26 weeks, and spontaneous vaginal delivery at 27 weeks. The infant experienced respiratory distress syndrome, bronchopulmonary dysplasia, necrotizing enterocolitis, and grade III intraventricular hemorrhage, and she was given a diagnosis of mild cerebral palsy at age 3.
Two years later, the patient’s second pregnancy was complicated by preterm labor at 22 weeks and spontaneous vaginal delivery at 23 weeks, with an Apgar score of 3, 1, and 0. The infant did not survive.
In 2007, Ms. Jones was given a diagnosis of missed abortion at 8 weeks’ gestation and underwent dilation and curettage.
Today, she asks what you plan to do to optimize the outcome of her current pregnancy. Her risk of preterm birth is significantly higher than that of the general population, which is 12.7%.
What can you offer to her?
Progesterone supplementation is the best option for Ms. Jones. Data accumulating over the past 30 years suggest that progesterone reduces the likelihood of preterm birth in women who have a history of spontaneous preterm birth. In fact, a cumulative meta-analysis noted that evidence of progesterone’s benefit is striking enough that “statistical uncertainty” is not a valid reason for forgoing its use.1
This article describes what’s been learned about progesterone supplementation to reduce preterm birth—specifically, the patients likely to benefit, the various formulations available, and the data on long-term outcomes—with an eye toward helping you weigh its utility in your practice.
The article focuses on four vulnerable populations:
- Women who have a history of preterm birth. Data suggest these patients are likely to benefit from progesterone.
- Women carrying a multiple gestation. Progesterone does not appear to prevent preterm birth in this group.
- Women who have a short cervix. Some data are promising. Further study is needed.
- Women who experience preterm labor. Data are promising, but preliminary.
Despite decades of research, initiative, and medical advances, the rate of preterm birth continues to rise, affecting one of every eight infants born in the United States—more than 500,000 babies each year. The impact of preterm birth is enormous, with implications that span from the immediate to the long-term.
In 2001, preterm birth surpassed birth defects as the leading cause of neonatal mortality. It is also the leading cause of infant mortality among African Americans and the second leading overall cause of all infant mortality.
The outlook for babies who survive preterm birth is concerning, as well. One of every five children who have mental retardation was born preterm, as was one of every three children who have vision impairment, and roughly one of every two children who have cerebral palsy. Low-birth-weight babies are commonly born preterm and face an increased risk of cardiovascular disease (including myocardial infarction, stroke, and hypertension), diabetes, and, possibly, cancer as adults.
Preterm birth not only affects the health of the baby and the family, but has long-term health and economic implications for society, costing at least $26 billion a year.26
POPULATION 1: Women who have a history of preterm birth
Women who have already delivered preterm face an elevated risk of doing so in any subsequent pregnancy ( TABLE 1 ). Three recent double-blind, randomized, controlled trials explored the efficacy of progesterone in the prevention of recurrent preterm birth.2-4 All three trials enrolled women at high risk of preterm birth; two included only women who had a history of spontaneous preterm birth, and 90% of the participants of the third trial had such a history as their risk factor.
The trials involved three different formulations of progesterone:
- intramuscular injection of 250 mg of 17α-hydroxyprogesterone caproate
- 100-mg vaginal suppository of progesterone
- 90 mg of vaginal progesterone gel (Prochieve 8% / Crinone 8%).
Meta-analyses of all studies, including these three, found that the risk of recurrent preterm birth can be reduced by as much as 40% to 55% and low birth weight by 50% using progesterone.5,6
TABLE 1
A woman who gives birth prematurely once likely will the next time
| Source | Gestational age at first delivery | Relative risk of recurrent preterm birth (95% confidence interval) |
|---|---|---|
| Maternal–Fetal Medicine Units Network30 | 2.5 (1.9–3.2) | |
| Missouri database, 1989–199731 | 3.6 (3.2–4.0) | |
| University of Texas Southwestern Medical Center, 1988–199932 | 5.9 (4.5–7.0) | |
| Denmark, 1982–198733 | 32–36 weeks | 4.8 (3.9–6.0) |
| Denmark, 1982–1987,33 Maternal–Fetal Medicine Units Network30 | 6.0 (4.1–8.8) | |
| Maternal–Fetal Medicine Units Network30 | 10.6 (2.9–38.3) |
Details of the trials
Meis and colleagues conducted a multicenter trial of 463 pregnant women who had a documented history of spontaneous preterm delivery.2 Starting between 16 and 20 weeks’ gestation, participants were randomized in a 2:1 ratio to weekly injection of 250 mg of 17α-hydroxyprogesterone caproate or an inert oil placebo, with injections continuing until delivery or 36 weeks’ gestation.
Among the findings:
- Treatment with progesterone significantly reduced the risk of delivery at less than 37 weeks’ gestation, with an incidence of 36.3% in the progesterone group versus 54.9% in the placebo group (relative risk [RR], 0.66; 95% confidence interval [CI], 0.54–0.81).
- Progesterone reduced the risk of delivery at less than 35 weeks’ gestation, with an incidence of 20.6% in the progesterone group versus 30.7% in the placebo group (RR, 0.67; 95% CI, 0.48–0.93).
- Progesterone reduced the risk of delivery at less than 32 weeks’ gestation, with an incidence of 11.4% in the progesterone group versus 19.6% in the placebo group (RR, 0.58; 95% CI, 0.37–0.91).
- Progesterone was effective in African Americans and non–African Americans.
- Infants of women treated with progesterone had significantly lower rates of necrotizing enterocolitis and intraventricular hemorrhage and less need for supplemental oxygen.
O’Brien and associates studied 659 pregnant women who had a history of spontaneous preterm birth.4 Participants were randomly assigned to receive daily treatment with progesterone vaginal gel or placebo, starting between 18 and 22.9 weeks’ gestation and continuing until delivery, 37 weeks’ gestation, or premature rupture of membranes. The gel was administered in the morning.
In this trial, progesterone did not decrease the rate of preterm birth at 32 weeks’ gestation or less (10% in the progesterone group versus 11.3% in the placebo group; odds ratio, 0.9; 95% CI, 0.52–1.56).
It is unclear whether the formulation, timing, or dosage was responsible for the different outcomes in these trials ( TABLE 2) .
TABLE 2
5 Progesterone formulations have been tested for the prevention of preterm birth
| Formulation | Dosage | Administration | Dosing schedule | Gestational age at initiation | Gestational age at completion |
|---|---|---|---|---|---|
| 17α-Hydroxyprogesterone caproate2 | 250 mg | Intramuscular | Weekly | 16.0–20.0 weeks | 36.9 weeks |
| Progesterone3 | 100 mg | Vaginal suppository | Daily at bedtime | 24 weeks | 34 weeks |
| Progesterone14 | 200 mg | Vaginal suppository | Daily at bedtime | 24 weeks | 34 weeks |
| Prochieve 8%/Crinone 8%4 | 90 mg | Vaginal suppository bioadhesive formulation/gel | Every morning | 18.0–22.9 weeks | 37 weeks |
| Progesterone19 | 400 mg | Vaginal suppository | Daily | After arrest of preterm labor | Delivery |
In this population, the number needed to treat is low
At least five strong meta-analyses have explored the prevention of recurrent preterm birth.1,7-10 These analyses demonstrate that progesterone supplementation significantly reduces the incidence of low birth weight and preterm birth. In some cases, it also reduces the rate of respiratory distress syndrome and intraventricular hemorrhage.
Based on these data, Petrini and associates calculated that, if all pregnant women who had a history of spontaneous preterm birth had been offered progesterone in 2002, 10,000 preterm births could have been prevented.11
The number needed to treat (NNT) to avoid one preterm birth was eight for 17α-hydroxyprogesterone caproate and 10 using another progesterone formulation. The NNT to prevent low birth weight was 12.
To put these figures in context, consider the use of low-dose aspirin to prevent stroke, which has a NNT of 102, and the use of a β-blocker to prevent cardiac death in patients who have suffered a myocardial infarction, which carries a NNT of 42.
POPULATION 2: Women who are carrying a multiple gestation
Given the success of progesterone in preventing recurrent preterm birth, it was a matter of time before investigators began to consider its use in another high-risk group: women carrying a multiple gestation. In two double-blind, placebo-controlled trials—one from the United States and the other from the United Kingdom—17α-hydroxyprogesterone caproate or placebo was given, starting between 16 and 20 weeks’ gestation in women who were carrying a twin or triplet gestation.12,13 Neither trial demonstrated a benefit for the use of progesterone in this population.
The etiology of preterm birth is likely different in women with a previous preterm birth than it is in women carrying a multiple gestation. The former are more likely to have an inflammatory, immunologic, or infectious process that leads to recurrent preterm birth, whereas women carrying multiples are thought to be at risk of preterm birth by virtue of the “stretch hypothesis”—the theory that the uterus is stretched excessively, leading to an earlier trigger of labor. Women who had a history of preterm birth and who were carrying a multiple gestation were eligible for these trials.
In the US trial, progesterone failed to reduce the rate of preterm birth in women who were carrying twins or triplets.13 This lack of benefit was seen regardless of whether conception was spontaneous or the result of assisted reproductive technologies, whether placentation was dichorionic or monochorionic, and regardless of the cutoff for gestational age. On average, the women in this trial delivered at 34.8 weeks, compared with a national average of 35.2 weeks for women carrying twins.13
Similar findings were reported from the UK trial, which enrolled 500 women carrying a twin gestation who were randomized to daily vaginal progesterone gel (90 mg) or placebo from 24 to 34 weeks’ gestation.12
A meta-analysis of the three trials that included multiple gestation12-14 found progesterone to have no benefit in women carrying twins. The pooled odds ratio of the effect of progesterone on preterm delivery or intrauterine death before 34 weeks’ gestation was 1.16 (95% CI, 0.89–1.51).12
Progesterone is a familiar player in the ObGyn specialty. In its natural form, the steroid hormone is produced by the corpus luteum to promote pregnancy.
In target cells, progesterone binds to its receptor and forms a transcription factor. It also can be active independent of nuclear receptors, which may explain why it remains effective even when circulating concentrations are high, suggesting that its action may be local and not systemic.
Progesterone exerts biologic effects on the myometrium, chorioamniotic membranes, and cervix.
Myometrial effects include:
- a decrease in the conduction of contractions
- a reduction of spontaneous muscle activity
- a decrease in the number of oxytocin receptors
- prevention of the formation of gap junctions
- a rise in the threshold for stimulation.
Progesterone decreases myometrial estrogen responsiveness by inhibiting estrogen-receptor expression and appears to maintain uterine quiescence by limiting the production of prostaglandins and inhibiting the expression of contraction-associated protein genes, including gap junctions, ion channels, oxytocin, and prostaglandin receptors within the myometrium.27 Some investigators have suggested that progesterone prevents preterm birth predominantly by virtue of its anti- inflammatory properties28 and ability to prevent cervical ripening.29
The 17α-hydroxyprogesterone form of the hormone also affects salivary concentrations of estriol. In a secondary analysis, the ratio of salivary estriol to progesterone increased as pregnancy progressed among women who received placebo, but remained flat among women treated with 17α-hydroxyprogesterone.2 One theory is that labor may be triggered by an increase in the activity of estriol, compared with progesterone.
It also is notable that estriol concentrations in the mother’s blood and saliva derive mainly from the fetus and placenta (from the fetus’ production of cortisol), suggesting that the action of 17α-hydroxyprogesterone acetate may also affect the feto-placental unit.
POPULATION 3: Women who have a short cervix
Because women who have a short cervix have a heightened risk of spontaneous preterm delivery, the utility of progesterone in prolonging gestation was explored in this population—with less than definitive results. An editorial accompanying the main study of this issue concluded that it is too early to recommend use of progesterone in women who have a short cervix.15
Progesterone was effective overall, but not in subgroup analysis
Iams and associates expertly delineated the risk of spontaneous preterm birth in the setting of a shortened cervix at 24 weeks’ gestation. They found a cervical length of about 12 mm to be at the first centile, with a relative risk of preterm birth of 14.16 (Compare this with the average cervical length of 36 to 44 mm at 24 weeks.)
Fonseca and colleagues then explored the benefit of progesterone therapy in preventing preterm birth in women who had a shortened cervical length between 20 and 25 weeks’ gestation.14 They screened more than 24,000 women and found 413 who had a cervical length of less than 15 mm. Of these women, 250 were randomized to micronized progesterone (200 mg in a vaginal suppository), starting at 24 weeks. This was twice the dosage given in the Brazilian trial involving women who had a history of preterm birth,3 but the authors thought that women who had a short cervix were at higher risk of preterm birth and, therefore, needed a higher dosage of progesterone. Although all women in this trial had a short cervix, the population overall was more heterogeneous than in other trials, including women who had a history of preterm birth (30% of participants) and women carrying a multiple gestation (19% of participants).
Progesterone reduced the risk of preterm birth in the overall cohort, with 19% of the women who received progesterone delivering preterm, versus 34% of those who received placebo (odd ratio [OR], 0.56; 95% CI, 0.36–0.86). Progesterone did not reduce the rate of perinatal mortality or neonatal morbidity. A subgroup analysis of only the nulli parous women was conducted, given that 30% of the study population had a history of preterm birth. That analysis showed no benefit.
DeFranco and associates17 published a secondary analysis of 46 women from a large randomized trial4 who had a cervical length of less than 28 mm. Of these women, 19 received progesterone and 27 received placebo. Of the 19 who received progesterone, 15 had a history of preterm birth. Of the 27 who received placebo, 22 had such a history. The authors found that progesterone significantly reduced preterm birth at less than 37, 35, and 32 weeks. However, again because of the small sample size and the inclusion of women with a history of preterm birth, these findings are not definitive.
Randomized trials designed to test the effect of progesterone in women who have a short cervix are called for. Numerous studies are under way.15
POPULATION 4: Women who experience preterm labor
Two recent trials explored the use of progesterone in this context. In one, progesterone was administered during the episode of preterm labor; in the other, it was given after successful tocolysis.
In the first trial, Facchinetti and colleagues studied 60 women who were pregnant with a singleton fetus and who were in active preterm labor.18 These women were randomly assigned to 341 mg of intramuscular 17α-hydroxyprogesterone caproate or placebo twice weekly, with cervical length monitored weekly. Women in the progesterone group were less likely to deliver by 7 or 21 days, and their cervical length was longer at both time points.
Borna and Sahabi evaluated use of progesterone as maintenance therapy after successful tocolysis.19 Seventy women were randomly assigned to progesterone (400-mg suppository) or no treatment. Women who received progesterone had a longer latency period (36 versus 24 days; P=.03), less respiratory distress (11% versus 36%), and a lower rate of low birth weight (27% versus 52%) than did women receiving no treatment.
What are the long-term effects of progesterone exposure?
Therapeutic interventions during pregnancy affect two people—one of them during a period of intense development that can have a lifelong impact. Although studies of progesterone to prevent preterm birth involve administration of the hormone after 16 weeks—well beyond the major period of organogenesis—concerns about potential teratogenic and other long-term effects have been raised. It is notable that progesterone has been widely used for decades during the first trimester—the period of organogenesis—in women who have a poor pregnancy history and early loss, to treat the “luteal phase defect.”
A Cochrane review of 14 studies of progesterone in the prevention of stillbirth and miscarriage20 and a systematic review of 14 cohort and case-control studies21 involving first-trimester exposure found no harm related to progesterone use. These findings are consistent with those of a meta-analysis by Coomarasamy and colleagues, which also found no harm related to the use of progesterone.1
Numerous studies have explored the long-term effects of progesterone on offspring, including a review of outcomes of pregnancies treated before 199022 and data from animal studies.23 Children from a trial by Meis22 were followed up at around 4 years of age to assess any differences in physical health and the achievement of developmental milestones between children who were exposed to progesterone and those who were not.24 Investigators used the Ages and Stages Questionnaire score, assessment of developmental milestones, and physical exams to evaluate the 348 children. No differences were seen in height, weight, and head-circumference percentiles; achievement of developmental milestones; gender roles; and physical health.
In a systematic review of 11 trials (2,425 women and 3,187 infants) involving the use of progesterone to prevent preterm birth in high-risk women—including those who had a history of preterm birth, those carrying a multiple gestation, and those with a short cervix—Dodd and colleagues found mixed results.10 Progesterone reduced the rate of preterm birth before 34 weeks’ gestation in women who had a history of preterm birth, as well as in those who had a short cervix, but no improvement was seen in women carrying a multiple gestation.
A cumulative meta-analysis by Coomarasamy and colleagues found that progestogens significantly reduce the rate of preterm birth, a benefit that was evident beginning in 1975.1
The most recent Committee Opinion from the American College of Obstetricians and Gynecologists25 concludes that “it is important to offer progesterone for pregnancy prolongation to only women with a documented history of a previous spontaneous birth at less than 37 weeks of gestation.” The opinion also takes into account the findings of the Fonseca trial in regard to women who have a short cervix,14 and concludes that “progesterone supplementation may be considered for use in asymptomatic women with a short cervix.”
Trials of other high-risk groups, including women who have a positive fibronectin test, bleeding, or iatrogenic preterm labor, are needed. The fact that progesterone supplementation is not universally effective in women who have a history of preterm birth suggests that not all pathways leading to preterm birth are ameliorated by progesterone therapy. Given the many similarities between women who have a history of preterm birth and women who have a short cervix, evidence may ultimately be available to support the benefits of progesterone in both situations. However, the lack of a benefit in women carrying a multiple gestation likely reflects the different underlying mechanism in that group.
CASE: RESOLVED
You discuss with Ms. Jones the options available to reduce the likelihood of recurrent preterm birth. She opts for progesterone supplementation, which is initiated at 16 weeks’ gestation, with no restrictions on activity. A sonogram at 18 weeks reveals normal anatomy and a cervical length of 4 cm.
At 22 weeks’ gestation, Ms. Jones visits the labor and delivery unit complaining of leaking fluid. You perform a sterile speculum exam, which is negative, monitor her for several hours, and send her home.
At 26 weeks, the patient experiences contractions and is again evaluated. An examination reveals the cervix to be long and closed. After prolonged monitoring, Ms. Jones is again sent home.
At 37 weeks’ gestation, the patient reports another episode of leaking fluid. This time, a sterile speculum exam is positive, and you begin induction of labor.
Labor proceeds smoothly, and Ms. Jones delivers a 3,100-g infant. The newborn has an Apgar score of 8 and 9 at 1 and 5 minutes, respectively.
1. Coomarasamy A, Thangaratinam S, Gee H, Khan KS. Progesterone for the prevention of preterm birth: a critical evaluation of evidence. Eur J Obstet Gynecol Reprod Biol. 2006;129:111-118.
2. Meis PJ, Klebanoff M, Thom E, et al. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N Engl J Med. 2003;348:2379-2385.
3. da Fonseca EB, Bittar RE, Carvalho MH, Zugaib M. Prophylactic administration of progesterone by vaginal suppository to reduce the incidence of spontaneous preterm birth in women at increased risk: a randomized placebo-controlled double-blind study. Am J Obstet Gynecol. 2003;188:419-424.
4. O’Brien JM, Adair CD, Lewis DF, et al. Progesterone vaginal gel for the reduction of recurrent preterm birth: primary results from a randomized, double-blind, placebo-controlled trial. Ultrasound Obstet Gynecol. 2007;30:687-696.
5. Sanchez-Ramos L, Kaunitz AM, Delke I. Progestational agents to prevent preterm birth: a meta-analysis of randomized controlled trials. Obstet Gynecol. 2005;105:273-279.
6. Dodd JM, Flenady V, Cincotta R, Crowther CA. Prenatal administration of progesterone for preventing preterm birth. Cochrane Database Syst Rev. 2006;(1):CD004947.-
7. Keirse MJ. Progestogen administration in pregnancy may prevent preterm delivery. Br J Obstet Gynaecol. 1990;97:149-154.
8. Dodd JM, Crowther CA, Cincotta R, Flenady V, Robinson JS. Progesterone supplementation for preventing preterm birth: a systematic review and meta-analysis. Acta Obstet Gynecol Scand. 2005;84:526-533.
9. Mackenzie R, Walker M, Armson A, Hannah ME. Progesterone for the prevention of preterm birth among women at increased risk: a systematic review and meta-analysis of randomized controlled trials. Am J Obstet Gynecol. 2006;194:1234-1242.
10. Dodd JM, Flenady VJ, Cincotta R, Crowther CA. Progesterone for the prevention of preterm birth: a systematic review. Obstet Gynecol. 2008;112:127-134.
11. Petrini JR, Callaghan WM, Klebanoff M, et al. Estimated effect of 17 alpha-hydroxyprogesterone caproate on preterm birth in the United States. Obstet Gynecol. 2005;105:267-272.
12. Norman JE, Mackenzie F, Owen P, et al. Progesterone for the prevention of preterm birth in twin pregnancy (STOPPIT): a randomised, double-blind, placebo-controlled study and meta-analysis. Lancet. 2009;373:2034-2040.
13. Rouse DJ, Caritis SN, Peaceman AM, et al. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. A trial of 17 alpha-hydroxyprogesterone caproate to prevent prematurity in twins. N Engl J Med. 2007;357:454-461.
14. Fonseca EB, Celik E, Parra M, Singh M, Nicolaides KH. Fetal Medicine Foundation Second Trimester Screening Group. Progesterone and the risk of preterm birth among women with a short cervix. N Engl J Med. 2007;357:462-469.
15. Thornton JG. Progesterone and preterm labor—still no definite answers. N Engl J Med. 2007;357:499-501.
16. Iams JD, Goldenberg RL, Meis PJ, et al. The length of the cervix and the risk of spontaneous premature delivery. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. N Engl J Med. 1996;334:567-572.
17. DeFranco EA, O’Brien JM, Adair CD, et al. Vaginal progesterone is associated with a decrease in risk for early preterm birth and improved neonatal outcome in women with a short cervix: a secondary analysis from a randomized, double-blind, placebo-controlled trial. Ultrasound Obstet Gynecol. 2007;30:697-705.
18. Facchinetti F, Paganelli S, Comitini G, Dante G, Volpe A. Cervical length changes during preterm cervical ripening: effects of 17-alpha-hydroxyprogesterone caproate. Am J Obstet Gynecol. 2007;196:453.e1-453.e4.
19. Borna S, Sahabi N. Progesterone for maintenance tocolytic therapy after threatened preterm labour: a randomised controlled trial. Aust N Z J Obstet Gynaecol. 2008;48:58-63.
20. Oates-Whitehead RM, Haas DM, Carrier JA. Progestogen for preventing miscarriage. Cochrane Database Syst Rev. 2003;(4):CD003511.-
21. Raman-Wilms L, Tseng AL, Wighardt S, Einarson TR, Koren G. Fetal genital effects of first trimester sex hormone exposure: a meta-analysis. Obstet Gynecol. 1995;85:141-149.
22. Meis PJ. Society for Maternal–Fetal Medicine. 17 Hydroxyprogesterone for the prevention of preterm delivery. Obstet Gynecol. 2005;105(5 Pt 1):1128-1135.
23. Christian MS, Brent RL, Calda P. Embryo–fetal toxicity signals for 17alpha-hydroxyprogesterone caproate in high-risk pregnancies: a review of the non-clinical literature for embryo–fetal toxicity with progestins. J Matern Fetal Neonatal Med. 2007;20:89-112.
24. Northen AT, Norman GS, Anderson K, et al. National Institute of Child Health and Human Development (NICHD) Maternal–Fetal Medicine Units (MFMU) Network. Follow-up of children exposed in utero to 17 alpha-hydroxyprogesterone caproate compared with placebo. Obstet Gynecol. 2007;110:865-872.
25. American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 419, October 2008. Use of progesterone to reduce preterm birth. Washington, DC: ACOG; 2008.
26. Preterm birth costs US $26 billion a year [press release]. Washington, DC: National Academies. July 13, 2006. Available at: http://www8.nationalacademies.org/onpinews/newsitem.aspx?RecordID=11622. Accessed October 5, 2009.
27. Norwitz ER. A blood test to predict preterm birth: don’t mess with maternal–fetal stress. J Clin Endocrinol Metab. 2009;94:1886-1889.
28. Elovitz MA, Gonzalez J. Medroxyprogesterone acetate modulates the immune response in the uterus, cervix and placenta in a mouse model of preterm birth. J Matern Fetal Neonatal Med. 2008;21:223-230.
29. Xu H, Gonzalez JM, Ofori E, Elovitz MA. Preventing cervical ripening: the primary mechanism by which progestational agents prevent preterm birth? Am J Obstet Gynecol. 2008;198:314.e1-314.e8.
30. Mercer BM, Goldenberg RL, Moawad AH, et al. The preterm prediction study: effect of gestational age and cause of preterm birth on subsequent obstetric outcome. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. Am J Obstet Gynecol. 1999;181(5 Pt 1):1216-1221.
31. Ananth CV, Getahun D, Peltier MR, Salihu HM, Vintzileos AM. Recurrence of spontaneous versus medically indicated preterm birth. Am J Obstet Gynecol. 2006;195:643-650.
32. Bloom SL, Yost NP, McIntire DD, Leveno KJ. Recurrence of preterm birth in singleton and twin pregnancies. Obstet Gynecol. 2001;98:379-385.
33. Kristensen J, Langhoff-Roos J, Kristensen FB. Implications of idiopathic preterm delivery for previous and subsequent pregnancies. Obstet Gynecol. 1995;86:800-804.
1. Coomarasamy A, Thangaratinam S, Gee H, Khan KS. Progesterone for the prevention of preterm birth: a critical evaluation of evidence. Eur J Obstet Gynecol Reprod Biol. 2006;129:111-118.
2. Meis PJ, Klebanoff M, Thom E, et al. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N Engl J Med. 2003;348:2379-2385.
3. da Fonseca EB, Bittar RE, Carvalho MH, Zugaib M. Prophylactic administration of progesterone by vaginal suppository to reduce the incidence of spontaneous preterm birth in women at increased risk: a randomized placebo-controlled double-blind study. Am J Obstet Gynecol. 2003;188:419-424.
4. O’Brien JM, Adair CD, Lewis DF, et al. Progesterone vaginal gel for the reduction of recurrent preterm birth: primary results from a randomized, double-blind, placebo-controlled trial. Ultrasound Obstet Gynecol. 2007;30:687-696.
5. Sanchez-Ramos L, Kaunitz AM, Delke I. Progestational agents to prevent preterm birth: a meta-analysis of randomized controlled trials. Obstet Gynecol. 2005;105:273-279.
6. Dodd JM, Flenady V, Cincotta R, Crowther CA. Prenatal administration of progesterone for preventing preterm birth. Cochrane Database Syst Rev. 2006;(1):CD004947.-
7. Keirse MJ. Progestogen administration in pregnancy may prevent preterm delivery. Br J Obstet Gynaecol. 1990;97:149-154.
8. Dodd JM, Crowther CA, Cincotta R, Flenady V, Robinson JS. Progesterone supplementation for preventing preterm birth: a systematic review and meta-analysis. Acta Obstet Gynecol Scand. 2005;84:526-533.
9. Mackenzie R, Walker M, Armson A, Hannah ME. Progesterone for the prevention of preterm birth among women at increased risk: a systematic review and meta-analysis of randomized controlled trials. Am J Obstet Gynecol. 2006;194:1234-1242.
10. Dodd JM, Flenady VJ, Cincotta R, Crowther CA. Progesterone for the prevention of preterm birth: a systematic review. Obstet Gynecol. 2008;112:127-134.
11. Petrini JR, Callaghan WM, Klebanoff M, et al. Estimated effect of 17 alpha-hydroxyprogesterone caproate on preterm birth in the United States. Obstet Gynecol. 2005;105:267-272.
12. Norman JE, Mackenzie F, Owen P, et al. Progesterone for the prevention of preterm birth in twin pregnancy (STOPPIT): a randomised, double-blind, placebo-controlled study and meta-analysis. Lancet. 2009;373:2034-2040.
13. Rouse DJ, Caritis SN, Peaceman AM, et al. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. A trial of 17 alpha-hydroxyprogesterone caproate to prevent prematurity in twins. N Engl J Med. 2007;357:454-461.
14. Fonseca EB, Celik E, Parra M, Singh M, Nicolaides KH. Fetal Medicine Foundation Second Trimester Screening Group. Progesterone and the risk of preterm birth among women with a short cervix. N Engl J Med. 2007;357:462-469.
15. Thornton JG. Progesterone and preterm labor—still no definite answers. N Engl J Med. 2007;357:499-501.
16. Iams JD, Goldenberg RL, Meis PJ, et al. The length of the cervix and the risk of spontaneous premature delivery. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. N Engl J Med. 1996;334:567-572.
17. DeFranco EA, O’Brien JM, Adair CD, et al. Vaginal progesterone is associated with a decrease in risk for early preterm birth and improved neonatal outcome in women with a short cervix: a secondary analysis from a randomized, double-blind, placebo-controlled trial. Ultrasound Obstet Gynecol. 2007;30:697-705.
18. Facchinetti F, Paganelli S, Comitini G, Dante G, Volpe A. Cervical length changes during preterm cervical ripening: effects of 17-alpha-hydroxyprogesterone caproate. Am J Obstet Gynecol. 2007;196:453.e1-453.e4.
19. Borna S, Sahabi N. Progesterone for maintenance tocolytic therapy after threatened preterm labour: a randomised controlled trial. Aust N Z J Obstet Gynaecol. 2008;48:58-63.
20. Oates-Whitehead RM, Haas DM, Carrier JA. Progestogen for preventing miscarriage. Cochrane Database Syst Rev. 2003;(4):CD003511.-
21. Raman-Wilms L, Tseng AL, Wighardt S, Einarson TR, Koren G. Fetal genital effects of first trimester sex hormone exposure: a meta-analysis. Obstet Gynecol. 1995;85:141-149.
22. Meis PJ. Society for Maternal–Fetal Medicine. 17 Hydroxyprogesterone for the prevention of preterm delivery. Obstet Gynecol. 2005;105(5 Pt 1):1128-1135.
23. Christian MS, Brent RL, Calda P. Embryo–fetal toxicity signals for 17alpha-hydroxyprogesterone caproate in high-risk pregnancies: a review of the non-clinical literature for embryo–fetal toxicity with progestins. J Matern Fetal Neonatal Med. 2007;20:89-112.
24. Northen AT, Norman GS, Anderson K, et al. National Institute of Child Health and Human Development (NICHD) Maternal–Fetal Medicine Units (MFMU) Network. Follow-up of children exposed in utero to 17 alpha-hydroxyprogesterone caproate compared with placebo. Obstet Gynecol. 2007;110:865-872.
25. American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 419, October 2008. Use of progesterone to reduce preterm birth. Washington, DC: ACOG; 2008.
26. Preterm birth costs US $26 billion a year [press release]. Washington, DC: National Academies. July 13, 2006. Available at: http://www8.nationalacademies.org/onpinews/newsitem.aspx?RecordID=11622. Accessed October 5, 2009.
27. Norwitz ER. A blood test to predict preterm birth: don’t mess with maternal–fetal stress. J Clin Endocrinol Metab. 2009;94:1886-1889.
28. Elovitz MA, Gonzalez J. Medroxyprogesterone acetate modulates the immune response in the uterus, cervix and placenta in a mouse model of preterm birth. J Matern Fetal Neonatal Med. 2008;21:223-230.
29. Xu H, Gonzalez JM, Ofori E, Elovitz MA. Preventing cervical ripening: the primary mechanism by which progestational agents prevent preterm birth? Am J Obstet Gynecol. 2008;198:314.e1-314.e8.
30. Mercer BM, Goldenberg RL, Moawad AH, et al. The preterm prediction study: effect of gestational age and cause of preterm birth on subsequent obstetric outcome. National Institute of Child Health and Human Development Maternal–Fetal Medicine Units Network. Am J Obstet Gynecol. 1999;181(5 Pt 1):1216-1221.
31. Ananth CV, Getahun D, Peltier MR, Salihu HM, Vintzileos AM. Recurrence of spontaneous versus medically indicated preterm birth. Am J Obstet Gynecol. 2006;195:643-650.
32. Bloom SL, Yost NP, McIntire DD, Leveno KJ. Recurrence of preterm birth in singleton and twin pregnancies. Obstet Gynecol. 2001;98:379-385.
33. Kristensen J, Langhoff-Roos J, Kristensen FB. Implications of idiopathic preterm delivery for previous and subsequent pregnancies. Obstet Gynecol. 1995;86:800-804.