User login
Topical Cannabinoids in Dermatology
The prevalence of topical cannabinoids has risen sharply in recent years. Commercial advertisers promote their usage as a safe means to treat a multitude of skin disorders, including atopic dermatitis (AD), psoriasis, and acne. Topical compounds have garnered interest in laboratory studies, but the purchase of commercial formulations is limited to over-the-counter products from unregulated suppliers. In this article, we review the scientific evidence behind topical cannabinoids and evaluate their role in clinical dermatology.
Background
Cannabis is designated as a Schedule I drug, according to the Controlled Substances Act of 1970. This listing is given to substances with no therapeutic value and a high potential for abuse. However, as of 2017, 29 states and the District of Columbia have laws legalizing cannabis in some capacity. These regulations typically apply to medicinal use, though several states have now legalized recreational use.
Cannabinoids represent a broad class of chemical compounds derived from the cannabis plant. Originally, this class only comprised phytocannabinoids, cannabinoids produced by the cannabis plant. Tetrahydrocannabinol (THC) is the most well-known phytocannabinoid and leads to the psychoactive effects typically associated with cannabis use. Later investigation led to the discovery of endocannabinoids, cannabinoids that are naturally produced by human and animal bodies, as well as synthetic cannabinoids.1 Cannabidiol is a phytocannabinoid that has been investigated in neurologic and anti-inflammatory conditions.2-4
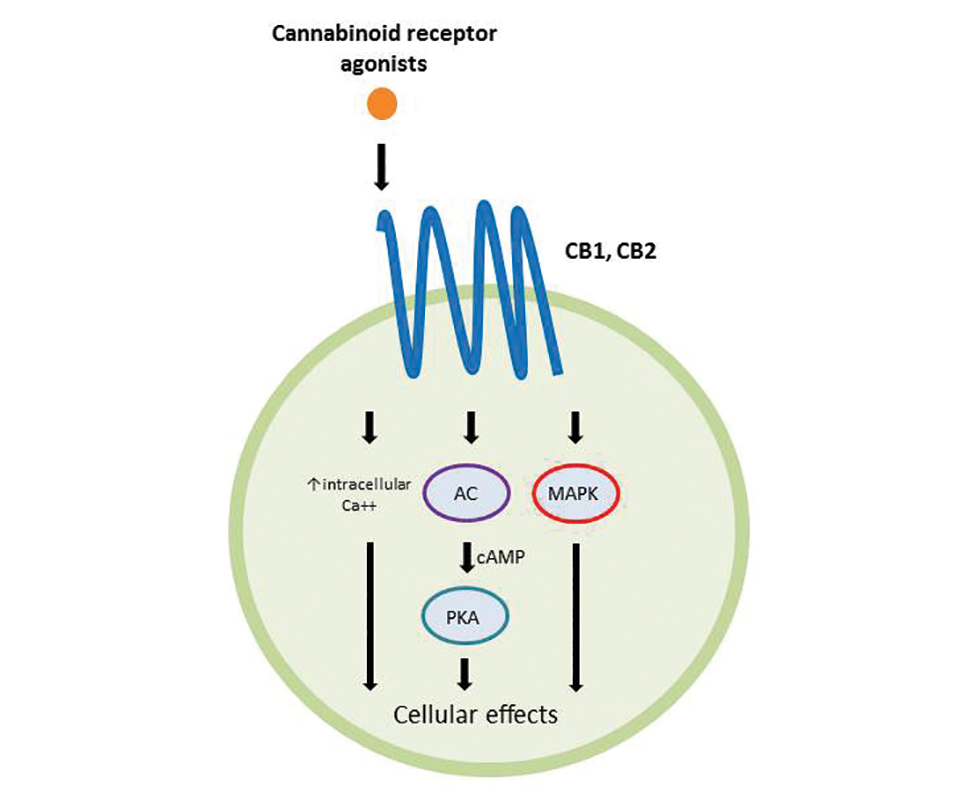
Cannabinoids act as agonists on 2 principal receptors— cannabinoid receptor type 1 (CB1) and cannabinoid receptor type 2 (CB2)—which are both G protein–coupled receptors (Figure).5 Both have distinct distributions throughout different organ systems, to which cannabinoids (eg, THC, cannabidiol, endocannabinoids) show differential binding.6,7 Importantly, the expression of CB1 and CB2 has been identified on sensory nerve fibers, inflammatory cells, and adnexal structures of human skin.8 Based on these associations, topical application of cannabinoids has become a modality of interest for dermatological disorders. These formulations aim to influence cutaneous morphology without producing psychoactive effects.
Topical Cannabinoids in Inflammatory Disorders
Atopic dermatitis has emerged as an active area of investigation for cannabinoid receptors and topical agonists (Table 1). In an animal model, Kim et al9 examined the effects of CB1 agonism on skin inflammation. Mice treated with topical CB1 agonists showed greater recovery of epidermal barrier function in acutely abrogated skin relative to those treated with a vehicle preparation. In addition, agonism of CB1 led to significant (P<.001) decreases in skin fold thickness among models of acute and chronic skin inflammation.9
Nam et al10 also examined the role of topical CB1 agonists in mice with induced AD-like symptoms. Relative to treatment with vehicle, CB1 agonists significantly reduced the recruitment of mast cells (P<.01) and lowered the blood concentration of histamine (P<.05). Given the noted decrease in the release of inflammatory mediators, the authors speculated that topical agonsim of CB1 may prove useful in several conditions related to mast cell activation, such as AD, contact dermatitis, and psoriasis.10
The anti-inflammatory properties of topical THC were evaluated by Gaffal et al.11 In a mouse model of allergic contact dermatitis, mice treated with topical THC showed decreases in myeloid immune cell infiltration, with these beneficial effects existing even in mice with deficient CB1 and CB2 receptors. These results support a potentially wide anti-inflammatory activity of topical THC.11
Topical Cannabinoids in Pain Management
The effects of smoked cannabis in treating pain have undergone thorough investigation over recent years. Benefits have been noted in treating neuropathic pain, particularly in human immunodeficiency virus–associated sensory neuropathy.12-15 Smoked cannabis also may provide value as a synergistic therapy with opioids, thereby allowing for lower opioid doses.16
In contrast, research into the relationship between topical application of cannabinoids and nociception remains in preliminary stages (Table 2). In a mouse model, Dogrul et al17 assessed the topical antinociceptive potential of a mixed CB1-CB2 agonist. Results showed significant (P<.01) and dose-dependent antinociceptive effects relative to treatment with a vehicle.17 In a related study, Yesilyurt et al18 evaluated whether a mixed CB1-CB2 agonist could enhance the antinociceptive effects of topical opioids. Among mice treated with the combination of a cannabinoid agonist and topical morphine, a significantly (P<.05) greater analgesic effect was demonstrated relative to topical morphine alone.18
Studies in humans have been far more limited. Phan et al19 conducted a small, nonrandomized, open-label trial of a topical cannabinoid cream in patients with facial postherpetic neuralgia. Of 8 patients treated, 5 noted a mean pain reduction of 87.8%. No comparison vehicle was used. Based on this narrow study design, it is difficult to extrapolate these positive results to a broader patient population.19
Commercial Products
Although preliminary models with topical cannabinoids have shown potential, large-scale clinical trials in humans have yet to be performed. Despite this lack of investigation, commercial formulations of topical cannabinoids are available to dermatology patients. These formulations are nonstandardized, and no safety data exists regarding their use. Topical cannabinoids on the market may contain various amounts of active ingredient and may be combined with a range of other compounds.
In dermatology offices, it is not uncommon for patients to express an intention to use topical cannabinoid products following their planned treatment or procedure. Patients also have been known to use topical cannabinoid products prior to dermatologic procedures, sometimes in place of an approved topical anesthetic, without consulting the physician performing the procedure. With interventions that lead to active areas of wound healing, the application of such products may increase the risk for contamination and infection. Therefore, patients should be counseled that the use of commercial topical cannabinoids could jeopardize the success of their planned procedure, put them at risk for infection, and possibly lead to systemic absorption and/or changes in wound-healing capacities.
Conclusion
Based on the results from recent animal models, cannabinoids may have a role in future treatment algorithms for several inflammatory conditions. However, current efficacy and safety data are almost entirely limited to preliminary animal studies in rodents. In addition, the formulation of topical cannabinoid products is nonstandardized and poorly regulated. As such, the present evidence does not support the use of topical cannabinoids in dermatology practices. Dermatologists should ask patients about the use of any cannabinoid products as part of a treatment program, especially given the unsubstantiated claims often made by unscrupulous advertisers. This issue highlights the need for further research and regulation.
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389-462.
- Giacoppo S, Galuppo M, Pollastro F, et al. A new formulation of cannabidiol in cream shows therapeutic effects in a mouse model of experimental autoimmune encephalomyelitis. Daru. 2015;23:48.
- Hammell DC, Zhang LP, Ma F, et al. Transdermal cannabidiol reduces inflammation and pain-related behaviours in a rat model of arthritis. Eur J Pain. 2016;20:936-948.
- Schicho R, Storr M. Topical and systemic cannabidiol improves trinitrobenzene sulfonic acid colitis in mice. Pharmacology. 2012;89:149-155.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161-202.
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199-215.
- Svizenska I, Dubovy P, Sulcova A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol Biochem Behav. 2008;90:501-511.
- Stander S, Schmelz M, Metze D, et al. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J Dermatol Sci. 2005;38:177-188.
- Kim HJ, Kim B, Park BM, et al. Topical cannabinoid receptor 1 agonist attenuates the cutaneous inflammatory responses in oxazolone-induced atopic dermatitis model. Int J Dermatol. 2015;54:E401-E408.
- Nam G, Jeong SK, Park BM, et al. Selective cannabinoid receptor-1 agonists regulate mast cell activation in an oxazolone-induced atopic dermatitis model. Ann Dermatol. 2016;28:22-29.
- Gaffal E, Cron M, Glodde N, et al. Anti-inflammatory activity of topical THC in DNFB-mediated mouse allergic contact dermatitis independent of CB1 and CB2 receptors. Allergy. 2013;68:994-1000.
- Abrams DI, Jay CA, Shade SB, et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68:515-521.
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology. 2009;34:672-680.
- Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14:136-148.
- Wilsey B, Marcotte T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9:506-521.
- Abrams DI, Couey P, Shade SB, et al. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther. 2011;90:844-851.
- Dogrul A, Gul H, Akar A, et al. Topical cannabinoid antinociception: synergy with spinal sites. Pain. 2003;105:11-16.
- Yesilyurt O, Dogrul A, Gul H, et al. Topical cannabinoid enhances topical morphine antinociception. Pain. 2003;105:303-308.
- Phan NQ, Siepmann D, Gralow I, et al. Adjuvant topical therapy with a cannabinoid receptor agonist in facial postherpetic neuralgia. J Dtsch Dermatol Ges. 2010;8:88-91.
The prevalence of topical cannabinoids has risen sharply in recent years. Commercial advertisers promote their usage as a safe means to treat a multitude of skin disorders, including atopic dermatitis (AD), psoriasis, and acne. Topical compounds have garnered interest in laboratory studies, but the purchase of commercial formulations is limited to over-the-counter products from unregulated suppliers. In this article, we review the scientific evidence behind topical cannabinoids and evaluate their role in clinical dermatology.
Background
Cannabis is designated as a Schedule I drug, according to the Controlled Substances Act of 1970. This listing is given to substances with no therapeutic value and a high potential for abuse. However, as of 2017, 29 states and the District of Columbia have laws legalizing cannabis in some capacity. These regulations typically apply to medicinal use, though several states have now legalized recreational use.
Cannabinoids represent a broad class of chemical compounds derived from the cannabis plant. Originally, this class only comprised phytocannabinoids, cannabinoids produced by the cannabis plant. Tetrahydrocannabinol (THC) is the most well-known phytocannabinoid and leads to the psychoactive effects typically associated with cannabis use. Later investigation led to the discovery of endocannabinoids, cannabinoids that are naturally produced by human and animal bodies, as well as synthetic cannabinoids.1 Cannabidiol is a phytocannabinoid that has been investigated in neurologic and anti-inflammatory conditions.2-4
Cannabinoids act as agonists on 2 principal receptors— cannabinoid receptor type 1 (CB1) and cannabinoid receptor type 2 (CB2)—which are both G protein–coupled receptors (Figure).5 Both have distinct distributions throughout different organ systems, to which cannabinoids (eg, THC, cannabidiol, endocannabinoids) show differential binding.6,7 Importantly, the expression of CB1 and CB2 has been identified on sensory nerve fibers, inflammatory cells, and adnexal structures of human skin.8 Based on these associations, topical application of cannabinoids has become a modality of interest for dermatological disorders. These formulations aim to influence cutaneous morphology without producing psychoactive effects.
Topical Cannabinoids in Inflammatory Disorders
Atopic dermatitis has emerged as an active area of investigation for cannabinoid receptors and topical agonists (Table 1). In an animal model, Kim et al9 examined the effects of CB1 agonism on skin inflammation. Mice treated with topical CB1 agonists showed greater recovery of epidermal barrier function in acutely abrogated skin relative to those treated with a vehicle preparation. In addition, agonism of CB1 led to significant (P<.001) decreases in skin fold thickness among models of acute and chronic skin inflammation.9
Nam et al10 also examined the role of topical CB1 agonists in mice with induced AD-like symptoms. Relative to treatment with vehicle, CB1 agonists significantly reduced the recruitment of mast cells (P<.01) and lowered the blood concentration of histamine (P<.05). Given the noted decrease in the release of inflammatory mediators, the authors speculated that topical agonsim of CB1 may prove useful in several conditions related to mast cell activation, such as AD, contact dermatitis, and psoriasis.10
The anti-inflammatory properties of topical THC were evaluated by Gaffal et al.11 In a mouse model of allergic contact dermatitis, mice treated with topical THC showed decreases in myeloid immune cell infiltration, with these beneficial effects existing even in mice with deficient CB1 and CB2 receptors. These results support a potentially wide anti-inflammatory activity of topical THC.11
Topical Cannabinoids in Pain Management
The effects of smoked cannabis in treating pain have undergone thorough investigation over recent years. Benefits have been noted in treating neuropathic pain, particularly in human immunodeficiency virus–associated sensory neuropathy.12-15 Smoked cannabis also may provide value as a synergistic therapy with opioids, thereby allowing for lower opioid doses.16
In contrast, research into the relationship between topical application of cannabinoids and nociception remains in preliminary stages (Table 2). In a mouse model, Dogrul et al17 assessed the topical antinociceptive potential of a mixed CB1-CB2 agonist. Results showed significant (P<.01) and dose-dependent antinociceptive effects relative to treatment with a vehicle.17 In a related study, Yesilyurt et al18 evaluated whether a mixed CB1-CB2 agonist could enhance the antinociceptive effects of topical opioids. Among mice treated with the combination of a cannabinoid agonist and topical morphine, a significantly (P<.05) greater analgesic effect was demonstrated relative to topical morphine alone.18
Studies in humans have been far more limited. Phan et al19 conducted a small, nonrandomized, open-label trial of a topical cannabinoid cream in patients with facial postherpetic neuralgia. Of 8 patients treated, 5 noted a mean pain reduction of 87.8%. No comparison vehicle was used. Based on this narrow study design, it is difficult to extrapolate these positive results to a broader patient population.19
Commercial Products
Although preliminary models with topical cannabinoids have shown potential, large-scale clinical trials in humans have yet to be performed. Despite this lack of investigation, commercial formulations of topical cannabinoids are available to dermatology patients. These formulations are nonstandardized, and no safety data exists regarding their use. Topical cannabinoids on the market may contain various amounts of active ingredient and may be combined with a range of other compounds.
In dermatology offices, it is not uncommon for patients to express an intention to use topical cannabinoid products following their planned treatment or procedure. Patients also have been known to use topical cannabinoid products prior to dermatologic procedures, sometimes in place of an approved topical anesthetic, without consulting the physician performing the procedure. With interventions that lead to active areas of wound healing, the application of such products may increase the risk for contamination and infection. Therefore, patients should be counseled that the use of commercial topical cannabinoids could jeopardize the success of their planned procedure, put them at risk for infection, and possibly lead to systemic absorption and/or changes in wound-healing capacities.
Conclusion
Based on the results from recent animal models, cannabinoids may have a role in future treatment algorithms for several inflammatory conditions. However, current efficacy and safety data are almost entirely limited to preliminary animal studies in rodents. In addition, the formulation of topical cannabinoid products is nonstandardized and poorly regulated. As such, the present evidence does not support the use of topical cannabinoids in dermatology practices. Dermatologists should ask patients about the use of any cannabinoid products as part of a treatment program, especially given the unsubstantiated claims often made by unscrupulous advertisers. This issue highlights the need for further research and regulation.
The prevalence of topical cannabinoids has risen sharply in recent years. Commercial advertisers promote their usage as a safe means to treat a multitude of skin disorders, including atopic dermatitis (AD), psoriasis, and acne. Topical compounds have garnered interest in laboratory studies, but the purchase of commercial formulations is limited to over-the-counter products from unregulated suppliers. In this article, we review the scientific evidence behind topical cannabinoids and evaluate their role in clinical dermatology.
Background
Cannabis is designated as a Schedule I drug, according to the Controlled Substances Act of 1970. This listing is given to substances with no therapeutic value and a high potential for abuse. However, as of 2017, 29 states and the District of Columbia have laws legalizing cannabis in some capacity. These regulations typically apply to medicinal use, though several states have now legalized recreational use.
Cannabinoids represent a broad class of chemical compounds derived from the cannabis plant. Originally, this class only comprised phytocannabinoids, cannabinoids produced by the cannabis plant. Tetrahydrocannabinol (THC) is the most well-known phytocannabinoid and leads to the psychoactive effects typically associated with cannabis use. Later investigation led to the discovery of endocannabinoids, cannabinoids that are naturally produced by human and animal bodies, as well as synthetic cannabinoids.1 Cannabidiol is a phytocannabinoid that has been investigated in neurologic and anti-inflammatory conditions.2-4
Cannabinoids act as agonists on 2 principal receptors— cannabinoid receptor type 1 (CB1) and cannabinoid receptor type 2 (CB2)—which are both G protein–coupled receptors (Figure).5 Both have distinct distributions throughout different organ systems, to which cannabinoids (eg, THC, cannabidiol, endocannabinoids) show differential binding.6,7 Importantly, the expression of CB1 and CB2 has been identified on sensory nerve fibers, inflammatory cells, and adnexal structures of human skin.8 Based on these associations, topical application of cannabinoids has become a modality of interest for dermatological disorders. These formulations aim to influence cutaneous morphology without producing psychoactive effects.
Topical Cannabinoids in Inflammatory Disorders
Atopic dermatitis has emerged as an active area of investigation for cannabinoid receptors and topical agonists (Table 1). In an animal model, Kim et al9 examined the effects of CB1 agonism on skin inflammation. Mice treated with topical CB1 agonists showed greater recovery of epidermal barrier function in acutely abrogated skin relative to those treated with a vehicle preparation. In addition, agonism of CB1 led to significant (P<.001) decreases in skin fold thickness among models of acute and chronic skin inflammation.9
Nam et al10 also examined the role of topical CB1 agonists in mice with induced AD-like symptoms. Relative to treatment with vehicle, CB1 agonists significantly reduced the recruitment of mast cells (P<.01) and lowered the blood concentration of histamine (P<.05). Given the noted decrease in the release of inflammatory mediators, the authors speculated that topical agonsim of CB1 may prove useful in several conditions related to mast cell activation, such as AD, contact dermatitis, and psoriasis.10
The anti-inflammatory properties of topical THC were evaluated by Gaffal et al.11 In a mouse model of allergic contact dermatitis, mice treated with topical THC showed decreases in myeloid immune cell infiltration, with these beneficial effects existing even in mice with deficient CB1 and CB2 receptors. These results support a potentially wide anti-inflammatory activity of topical THC.11
Topical Cannabinoids in Pain Management
The effects of smoked cannabis in treating pain have undergone thorough investigation over recent years. Benefits have been noted in treating neuropathic pain, particularly in human immunodeficiency virus–associated sensory neuropathy.12-15 Smoked cannabis also may provide value as a synergistic therapy with opioids, thereby allowing for lower opioid doses.16
In contrast, research into the relationship between topical application of cannabinoids and nociception remains in preliminary stages (Table 2). In a mouse model, Dogrul et al17 assessed the topical antinociceptive potential of a mixed CB1-CB2 agonist. Results showed significant (P<.01) and dose-dependent antinociceptive effects relative to treatment with a vehicle.17 In a related study, Yesilyurt et al18 evaluated whether a mixed CB1-CB2 agonist could enhance the antinociceptive effects of topical opioids. Among mice treated with the combination of a cannabinoid agonist and topical morphine, a significantly (P<.05) greater analgesic effect was demonstrated relative to topical morphine alone.18
Studies in humans have been far more limited. Phan et al19 conducted a small, nonrandomized, open-label trial of a topical cannabinoid cream in patients with facial postherpetic neuralgia. Of 8 patients treated, 5 noted a mean pain reduction of 87.8%. No comparison vehicle was used. Based on this narrow study design, it is difficult to extrapolate these positive results to a broader patient population.19
Commercial Products
Although preliminary models with topical cannabinoids have shown potential, large-scale clinical trials in humans have yet to be performed. Despite this lack of investigation, commercial formulations of topical cannabinoids are available to dermatology patients. These formulations are nonstandardized, and no safety data exists regarding their use. Topical cannabinoids on the market may contain various amounts of active ingredient and may be combined with a range of other compounds.
In dermatology offices, it is not uncommon for patients to express an intention to use topical cannabinoid products following their planned treatment or procedure. Patients also have been known to use topical cannabinoid products prior to dermatologic procedures, sometimes in place of an approved topical anesthetic, without consulting the physician performing the procedure. With interventions that lead to active areas of wound healing, the application of such products may increase the risk for contamination and infection. Therefore, patients should be counseled that the use of commercial topical cannabinoids could jeopardize the success of their planned procedure, put them at risk for infection, and possibly lead to systemic absorption and/or changes in wound-healing capacities.
Conclusion
Based on the results from recent animal models, cannabinoids may have a role in future treatment algorithms for several inflammatory conditions. However, current efficacy and safety data are almost entirely limited to preliminary animal studies in rodents. In addition, the formulation of topical cannabinoid products is nonstandardized and poorly regulated. As such, the present evidence does not support the use of topical cannabinoids in dermatology practices. Dermatologists should ask patients about the use of any cannabinoid products as part of a treatment program, especially given the unsubstantiated claims often made by unscrupulous advertisers. This issue highlights the need for further research and regulation.
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389-462.
- Giacoppo S, Galuppo M, Pollastro F, et al. A new formulation of cannabidiol in cream shows therapeutic effects in a mouse model of experimental autoimmune encephalomyelitis. Daru. 2015;23:48.
- Hammell DC, Zhang LP, Ma F, et al. Transdermal cannabidiol reduces inflammation and pain-related behaviours in a rat model of arthritis. Eur J Pain. 2016;20:936-948.
- Schicho R, Storr M. Topical and systemic cannabidiol improves trinitrobenzene sulfonic acid colitis in mice. Pharmacology. 2012;89:149-155.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161-202.
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199-215.
- Svizenska I, Dubovy P, Sulcova A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol Biochem Behav. 2008;90:501-511.
- Stander S, Schmelz M, Metze D, et al. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J Dermatol Sci. 2005;38:177-188.
- Kim HJ, Kim B, Park BM, et al. Topical cannabinoid receptor 1 agonist attenuates the cutaneous inflammatory responses in oxazolone-induced atopic dermatitis model. Int J Dermatol. 2015;54:E401-E408.
- Nam G, Jeong SK, Park BM, et al. Selective cannabinoid receptor-1 agonists regulate mast cell activation in an oxazolone-induced atopic dermatitis model. Ann Dermatol. 2016;28:22-29.
- Gaffal E, Cron M, Glodde N, et al. Anti-inflammatory activity of topical THC in DNFB-mediated mouse allergic contact dermatitis independent of CB1 and CB2 receptors. Allergy. 2013;68:994-1000.
- Abrams DI, Jay CA, Shade SB, et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68:515-521.
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology. 2009;34:672-680.
- Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14:136-148.
- Wilsey B, Marcotte T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9:506-521.
- Abrams DI, Couey P, Shade SB, et al. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther. 2011;90:844-851.
- Dogrul A, Gul H, Akar A, et al. Topical cannabinoid antinociception: synergy with spinal sites. Pain. 2003;105:11-16.
- Yesilyurt O, Dogrul A, Gul H, et al. Topical cannabinoid enhances topical morphine antinociception. Pain. 2003;105:303-308.
- Phan NQ, Siepmann D, Gralow I, et al. Adjuvant topical therapy with a cannabinoid receptor agonist in facial postherpetic neuralgia. J Dtsch Dermatol Ges. 2010;8:88-91.
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389-462.
- Giacoppo S, Galuppo M, Pollastro F, et al. A new formulation of cannabidiol in cream shows therapeutic effects in a mouse model of experimental autoimmune encephalomyelitis. Daru. 2015;23:48.
- Hammell DC, Zhang LP, Ma F, et al. Transdermal cannabidiol reduces inflammation and pain-related behaviours in a rat model of arthritis. Eur J Pain. 2016;20:936-948.
- Schicho R, Storr M. Topical and systemic cannabidiol improves trinitrobenzene sulfonic acid colitis in mice. Pharmacology. 2012;89:149-155.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161-202.
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199-215.
- Svizenska I, Dubovy P, Sulcova A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—a short review. Pharmacol Biochem Behav. 2008;90:501-511.
- Stander S, Schmelz M, Metze D, et al. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J Dermatol Sci. 2005;38:177-188.
- Kim HJ, Kim B, Park BM, et al. Topical cannabinoid receptor 1 agonist attenuates the cutaneous inflammatory responses in oxazolone-induced atopic dermatitis model. Int J Dermatol. 2015;54:E401-E408.
- Nam G, Jeong SK, Park BM, et al. Selective cannabinoid receptor-1 agonists regulate mast cell activation in an oxazolone-induced atopic dermatitis model. Ann Dermatol. 2016;28:22-29.
- Gaffal E, Cron M, Glodde N, et al. Anti-inflammatory activity of topical THC in DNFB-mediated mouse allergic contact dermatitis independent of CB1 and CB2 receptors. Allergy. 2013;68:994-1000.
- Abrams DI, Jay CA, Shade SB, et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68:515-521.
- Ellis RJ, Toperoff W, Vaida F, et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology. 2009;34:672-680.
- Wilsey B, Marcotte T, Deutsch R, et al. Low-dose vaporized cannabis significantly improves neuropathic pain. J Pain. 2013;14:136-148.
- Wilsey B, Marcotte T, Tsodikov A, et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. J Pain. 2008;9:506-521.
- Abrams DI, Couey P, Shade SB, et al. Cannabinoid-opioid interaction in chronic pain. Clin Pharmacol Ther. 2011;90:844-851.
- Dogrul A, Gul H, Akar A, et al. Topical cannabinoid antinociception: synergy with spinal sites. Pain. 2003;105:11-16.
- Yesilyurt O, Dogrul A, Gul H, et al. Topical cannabinoid enhances topical morphine antinociception. Pain. 2003;105:303-308.
- Phan NQ, Siepmann D, Gralow I, et al. Adjuvant topical therapy with a cannabinoid receptor agonist in facial postherpetic neuralgia. J Dtsch Dermatol Ges. 2010;8:88-91.
Practice Points
- Topical cannabinoids are advertised by companies as treatment options for numerous dermatologic conditions.
- Despite promising data in rodent models, there have been no rigorous studies to date confirming efficacy or safety in humans.
- Dermatologists should therefore inquire with patients about the use of any topical cannabinoid products, especially around the time of planned procedures, as they may affect treatment outcomes.

Systematic Review of Novel Synovial Fluid Markers and Polymerase Chain Reaction in the Diagnosis of Prosthetic Joint Infection
Take-Home Points
- Novel synovial markers and PCR have the potential to improve the detection of PJIs.
- 10Difficult-to-detect infections of prosthetic joints pose a diagnostic problem to surgeons and can lead to suboptimal outcomes.
- AD is a highly sensitive and specific synovial fluid marker for detecting PJIs.
- AD has shown promising results in detecting low virulence organisms.
- Studies are needed to determine how to best incorporate novel synovial markers and PCR to current diagnostic criteria in order to improve diagnostic accuracy.
Approximately 7 million Americans are living with a hip or knee replacement.1 According to projections, primary hip arthroplasties will increase by 174% and knee arthroplasties by 673% by 2030. Revision arthroplasties are projected to increase by 137% for hips and 601% for knees during the same time period.2 Infection and aseptic loosening are the most common causes of implant failure.3 The literature shows that infection is the most common cause of failure within 2 years after surgery and that aseptic loosening is the most common cause for late revision.3
Recent studies suggest that prosthetic joint infection (PJI) may be underreported because of difficulty making a diagnosis and that cases of aseptic loosening may in fact be attributable to infections with low-virulence organisms.2,3 These findings have led to new efforts to develop uniform criteria for diagnosing PJIs. In 2011, the Musculoskeletal Infection Society (MSIS) offered a new definition for PJI diagnosis, based on clinical and laboratory criteria, to increase the accuracy of PJI diagnosis.4 The MSIS committee acknowledged that PJI may be present even if these criteria are not met, particularly in the case of low-virulence organisms, as patients may not present with clinical signs of infection and may have normal inflammatory markers and joint aspirates. Reports of PJI cases misdiagnosed as aseptic loosening suggest that current screening and diagnostic tools are not sensitive enough to detect all infections and that PJI is likely underdiagnosed.
According to MSIS criteria, the diagnosis of PJI can be made when there is a sinus tract communicating with the prosthesis, when a pathogen is isolated by culture from 2 or more separate tissue or fluid samples obtained from the affected prosthetic joint, or when 4 of 6 criteria are met. The 6 criteria are (1) elevated serum erythrocyte sedimentation rate (ESR) (>30 mm/hour) and elevated C-reactive protein (CRP) level (>10 mg/L); (2) elevated synovial white blood cell (WBC) count (1100-4000 cells/μL); (3) elevated synovial polymorphonuclear leukocytes (>64%); (4) purulence in affected joint; (5) isolation of a microorganism in a culture of periprosthetic tissue or fluid; and (6) more than 5 neutrophils per high-power field in 5 high-power fields observed.
In this review article, we discuss recently developed novel synovial biomarkers and polymerase chain reaction (PCR) technologies that may help increase the sensitivity and specificity of diagnostic guidelines for PJI.
Methods
Using PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses), we performed a systematic review of specific synovial fluid markers and PCR used in PJI diagnosis. In May 2016, we searched the PubMed database for these criteria: ((((((PCR[Text Word]) OR IL-6[Text Word]) OR leukocyte esterase[Text Word]) OR alpha defensin[Text Word]) AND ((“infection/diagnosis”[MeSH Terms] OR “infection/surgery”[MeSH Terms])))) AND (prosthetic joint infection[MeSH Terms] OR periprosthetic joint infection[MeSH Terms]).
We included patients who had undergone total hip, knee, or shoulder arthroplasty (THA, TKA, TSA). Index tests were PCR and the synovial fluid markers α-defensin (AD), interleukin 6 (IL-6), and leukocyte esterase (LE). Reference tests included joint fluid/serum analysis or tissue analysis (ESR/CRP level, cell count, culture, frozen section), which defined the MSIS criteria for PJI. Primary outcomes of interest were sensitivity and specificity, and secondary outcomes of interest included positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (+LR), and negative likelihood ratio (–LR). Randomized controlled trials and controlled cohort studies in humans published within the past 10 years were included.
Results
Our full-text review yielded 15 papers that met our study inclusion criteria (Figure 1).
α-Defensin
One of the novel synovial biomarkers that has shown significant promise in diagnosing PJIs, even with difficult-to-detect organisms, is AD. 
AD has shown even more impressive results as a biomarker for PJI in the hip and knee, where infection with low virulence organism is less common. In 2014, Deirmengian and colleagues6 conducted a prospective clinical study of 149 patients who underwent revision THA or TKA for aseptic loosening (n = 112) or PJI (n = 37) as defined by MSIS criteria. Aseptic loosening was diagnosed when there was no identifiable reason for pain, and MSIS criteria were not met. Synovial fluid aspirates were collected before or during surgery. AD correctly identified 143 of the 149 patients with confirmed infection with sensitivity of 97.3% (95% confidence interval [CI], 85.8%-99.6%) and specificity of 95.5% (95% CI, 89.9%-98.5%). Similarly, Bingham and colleagues7 conducted a retrospective clinical study of 61 assays done on 57 patients who underwent revision arthroplasty for PJI as defined by MSIS criteria. Synovial fluid aspirates were collected before or during surgery. AD correctly identified all 19 PJIs with sensitivity of 100% (95% CI, 79%-100%) and specificity of 95% (95% CI, 83%-99%). Sensitivity and specificity of the AD assay more accurately predicted infection than synovial cell count or serum ESR/CRP level did.
These results are supported by another prospective study by Deirmengian and colleagues8 differentiating aseptic failures and PJIs in THA or TKA. The sensitivity and specificity of AD in diagnosing PJI were 100% (95% CI, 85.05%-100%). 
In a prospective study of 102 patients who underwent revision THA or TKA secondary to aseptic loosening or PJI, Frangiamore and colleagues9 also demonstrated the value of AD as a diagnostic for PJI in primary and revision hip and knee arthroplasty. 
Table 1 and Figure 2 provide a concise review of the findings of each study.
Interleukin 6
Another synovial fluid biomarker that has shown promise in PJI diagnosis is IL-6. In 2015, Frangiamore and colleagues10 conducted a prospective clinical study of 32 patients who underwent revision TSA. Synovial fluid aspiration was obtained before or during surgery. MSIS criteria were used to establish the diagnosis of PJI. IL-6 had sensitivity of 87% and specificity of 90%, with +LR of 8.45 and –LR of 0.15 in predicting PJI. Synovial fluid IL-6 had strong associations with frozen section histology and growth of P acnes. Frangiamore and colleagues10 recommended an ideal IL-6 cutoff of 359.1 pg/mL and reported that, though not as accurate as AD, synovial fluid IL-6 levels can help predict positive cultures in patients who undergo revision TSA.
Lenski and Scherer11 conducted another retrospective clinical study of the diagnostic value of IL-6 in PJI. 
Randau and colleagues12 conducted a prospective clinical study of 120 patients who presented with painful THA or TKA and underwent revision for PJI, aseptic failure, or aseptic revision without signs of infection or loosening. Synovial fluid aspirate was collected before or during surgery. 
Table 2 and Figure 3 provide a concise review of the findings of each study.
Leukocyte Esterase
LE strips are an inexpensive screening tool for PJI, according to some studies. In a prospective clinical study of 364 endoprosthetic joint (hip, knee, shoulder) interventions, Guenther and colleagues13 collected synovial fluid before surgery. Samples were tested with graded LE strips using PJI criteria set by the authors. Results were correlated with preoperative synovial fluid aspirations, serum CRP level, serum WBC count, and intraoperative histopathologic and microbiological findings. Whereas 293 (93.31%) of the 314 aseptic cases had negative test strip readings, 100% of the 50 infected cases were positive. LE had sensitivity of 100%, specificity of 96.5%, PPV of 82%, and NPV of 100%.
Wetters et al14 performed a prospective clinical study on 223 patients who underwent TKAs and THAs for suspected PJI based on having criteria defined by the authors of the study. Synovial fluid samples were collected either preoperatively or intraoperatively. 
Other authors have reported different findings that LE is an unreliable marker in PJI diagnosis. In one prospective clinical study of 85 patients who underwent primary or revision TSA, synovial fluid was collected during surgery.15 According to MSIS criteria, only 5 positive LE results predicted PJI among 21 primary and revision patients with positive cultures. Of the 7 revision patients who met the MSIS criteria for PJI, only 2 had a positive LE test. LE had sensitivity of 28.6%, specificity of 63.6%, PPV of 28.6%, and NPV of 87.5%. Six of the 7 revision patients grew P acnes. These results showed that LE was unreliable in detecting shoulder PJI.15
In another prospective clinical study, Tischler and colleagues16 enrolled 189 patients who underwent revision TKA or THA for aseptic failure or PJI as defined by the MSIS criteria. Synovial fluid was collected intraoperatively. 
Table 3 and Figure 4 provide a concise review of the findings of each study.
Polymerase Chain Reaction
Studies have found that PCR analysis of synovial fluid is effective in detecting bacteria on the surface of implants removed during revision arthroplasties. Comparison of the 16S rRNA gene sequences of bacterial genomes showed a diverse range of bacterial species within biofilms on the surface of clinical and subclinical infections.17 These findings, along with those of other studies, suggest that PCR analysis of synovial fluid is useful in diagnosing PJI and identifying organisms and their sensitivities to antibiotics.
Gallo and colleagues18 performed a prospective clinical study on 115 patients who underwent revision TKAs or THAs. Synovial fluid was collected intraoperatively. PCR assays targeting the 16S rDNA were carried out on 101 patients. PJIs were classified based on criteria of the authors of this study, of which there were 42. The sensitivity, specificity, PPV, NPV, +LR, and -LR for PCR were 71.4% (95% CI, 61.5%-75.5%), 97% (95% CI, 91.7%-99.1%), 92.6% (95% CI, 79.8%-97.9%), 86.5% (95% CI, 81.8%-88.4%), 23.6 (95% CI, 5.9%-93.8%), and 0.29 (95% CI, 0.17%-0.49%), respectively. Of note the most common organism detected in 42 PJIs was coagulase-negative Staphylococcus.
Marin and colleagues19 conducted a prospective study of 122 patients who underwent arthroplasty for suspected infection or aseptic loosening as defined by the authors’ clinicohistopathologic criteria. Synovial fluid and biopsy specimens were collected during surgery, and 40 patients met the infection criteria. The authors concluded that 16S PCR is more specific and has better PPV than culture does as one positive 16S PCR resulted in a specificity and PPV of PJI of 96.3% and 91.7%, respectively. However, they noted that culture was more sensitive in diagnosing PJI.
Jacovides and colleagues20 conducted a prospective study on 82 patients undergoing primary TKA, revision TKA, and revision THA.
The low PCR sensitivities reported in the literature were explained in a review by Hartley and Harris.21 They wrote that BR 16S rDNA and sequencing of PJI samples inherently have low sensitivity because of the contamination that can occur from the PCR reagents themselves or from sample mishandling. Techniques that address contaminant (extraneous DNA) removal, such as ultraviolet irradiation and DNase treatment, reduce Taq DNA polymerase activity, which reduces PCR sensitivity. 
Table 4 and Figure 5 provide a concise review of the findings of each study.
Discussion
Although there is no gold standard for the diagnosis of PJIs, several clinical and laboratory criteria guidelines are currently used to help clinicians diagnose infections of prosthetic joints. However, despite standardization of diagnostic criteria, PJI continue to be a diagnostic challenge.
AD is a highly sensitive and specific synovial fluid biomarker in detecting common PJIs. 

In summary, 5 AD studies5-9 had sensitivity ranging from 63% to 100% and specificity ranging from 95% to 100%; 3 IL-6 studies10-12 had sensitivity ranging from 46.8% to 90.9% and specificity ranging from 85.7% to 97.6%; 4 LE studies13-16 had sensitivity ranging from 28.6% to 100% and specificity ranging from 63.6% to 96.5%; and 3 PCR studies18-20 had sensitivity ranging from 67.1% to 95.7% and specificity ranging from 12.3% to 97.8%. Sensitivity and specificity were consistently higher for AD than for IL-6, LE, and PCR, though there was significant overlap, heterogeneity, and variation across all the included studies. 
Although the overall incidence of PJI is low, infected revisions remain a substantial financial burden to hospitals, as annual costs of infected revisions is estimated to exceed $1.62 billion by 2020.25 The usefulness of novel biomarkers and PCR in diagnosing PJI can be found in their ability to diagnose infections and facilitate appropriate early treatment. Several of these tests are readily available commercially and have the potential to be cost-effective diagnostic tools. The price to perform an AD test from Synovasure TM (Zimmer Biomet) ranges from $93 to $143. LE also provides an economic option for diagnosing PJI, as LE strips are commercially available for the cost of about 25 cents. PCR has also become an economic option, as costs can average $15.50 per sample extraction or PCR assay and $42.50 per amplicon sequence as reported in a study by Vandercam and colleagues.26 Future studies are needed to determine a diagnostic algorithm which incorporates these novel synovial markers to improve diagnostic accuracy of PJI in the most cost effective manner.
The current literature supports that AD can potentially be used to screen for PJI. Our findings suggest novel synovial fluid biomarkers may become of significant diagnostic use when combined with current laboratory and clinical diagnostic criteria. We recommend use of AD in cases in which pain, stiffness, and poor TJA outcome cannot be explained by errors in surgical technique, and infection is suspected despite MSIS criteria not being met.
The studies reviewed in this manuscript were limited in that none presented level I evidence (12 had level II evidence, and 3 had level III evidence), and there was significant heterogeneity (some studies used their own diagnostic standard, and others used the MSIS criteria). Larger scale prospective studies comparing serum ESR/CRP level and synovial fluid analysis to novel synovial markers are needed.
Am J Orthop. 2017;46(4):190-198. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Maradit Kremers H, Larson DR, Crowson CS, et al. Prevalence of total hip and knee replacement in the United States. J Bone Joint Surg Am. 2015;97(17):1386-1397.
2. Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am. 2007;89(4):780-785.
3. Sharkey PF, Lichstein PM, Shen C, Tokarski AT, Parvizi J. Why are total knee arthroplasties failing today—has anything changed after 10 years? J Arthroplasty. 2014;29(9):1774-1778.
4. Butler-Wu SM, Burns EM, Pottinger PS, et al. Optimization of periprosthetic culture for diagnosis of Propionibacterium acnes prosthetic joint infection. J Clin Microbiol. 2011;49(7):2490-2495.
5. Frangiamore SJ, Saleh A, Grosso MJ, et al. α-Defensin as a predictor of periprosthetic shoulder infection. J Shoulder Elbow Surg. 2015;24(7):1021-1027.
6. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Combined measurement of synovial fluid α-defensin and C-reactive protein levels: highly accurate for diagnosing periprosthetic joint infection. J Bone Joint Surg Am. 2014;96(17):1439-1445.
7. Bingham J, Clarke H, Spangehl M, Schwartz A, Beauchamp C, Goldberg B. The alpha defensin-1 biomarker assay can be used to evaluate the potentially infected total joint arthroplasty. Clin Orthop Relat Res. 2014;472(12):4006-4009.
8. Deirmengian C, Kardos K, Kilmartin P, et al. The alpha-defensin test for periprosthetic joint infection outperforms the leukocyte esterase test strip. Clin Orthop Relat Res. 2015;473(1):198-203.
9. Frangiamore SJ, Gajewski ND, Saleh A, Farias-Kovac M, Barsoum WK, Higuera CA. α-Defensin accuracy to diagnose periprosthetic joint infection—best available test? J Arthroplasty. 2016;31(2):456-460.
10. Frangiamore SJ, Saleh A, Kovac MF, et al. Synovial fluid interleukin-6 as a predictor of periprosthetic shoulder infection. J Bone Joint Surg Am. 2015;97(1):63-70.
11. Lenski M, Scherer MA. Synovial IL-6 as inflammatory marker in periprosthetic joint infections. J Arthroplasty. 2014;29(6):1105-1109.
12. Randau TM, Friedrich MJ, Wimmer MD, et al. Interleukin-6 in serum and in synovial fluid enhances the differentiation between periprosthetic joint infection and aseptic loosening. PLoS One. 2014;9(2):e89045.
13. Guenther D, Kokenge T, Jacobs O, et al. Excluding infections in arthroplasty using leucocyte esterase test. Int Orthop. 2014;38(11):2385-2390.
14. Wetters NG, Berend KR, Lombardi AV, Morris MJ, Tucker TL, Della Valle CJ. Leukocyte esterase reagent strips for the rapid diagnosis of periprosthetic joint infection. J Arthroplasty. 2012;27(8 suppl):8-11.
15. Nelson GN, Paxton ES, Narzikul A, Williams G, Lazarus MD, Abboud JA. Leukocyte esterase in the diagnosis of shoulder periprosthetic joint infection. J Shoulder Elbow Surg. 2015;24(9):1421-1426.
16. Tischler EH, Cavanaugh PK, Parvizi J. Leukocyte esterase strip test: matched for Musculoskeletal Infection Society criteria. J Bone Joint Surg Am. 2014;96(22):1917-1920.
17. Dempsey KE, Riggio MP, Lennon A, et al. Identification of bacteria on the surface of clinically infected and non-infected prosthetic hip joints removed during revision arthroplasties by 16S rRNA gene sequencing and by microbiological culture. Arthritis Res Ther. 2007;9(3):R46.
18. Gallo J, Kolar M, Dendis M, et al. Culture and PCR analysis of joint fluid in the diagnosis of prosthetic joint infection. New Microbiol. 2008;31(1):97-104.
19. Marin M, Garcia-Lechuz JM, Alonso P, et al. Role of universal 16S rRNA gene PCR and sequencing in diagnosis of prosthetic joint infection. J Clin Microbiol. 2012;50(3):583-589.
20. Jacovides CL, Kreft R, Adeli B, Hozack B, Ehrlich GD, Parvizi J. Successful identification of pathogens by polymerase chain reaction (PCR)-based electron spray ionization time-of-flight mass spectrometry (ESI-TOF-MS) in culture-negative periprosthetic joint infection. J Bone Joint Surg Am. 2012;94(24):2247-2254.
21. Hartley JC, Harris KA. Molecular techniques for diagnosing prosthetic joint infections. J Antimicrob Chemother. 2014;69(suppl 1):i21-i24.
22. Zappe B, Graf S, Ochsner PE, Zimmerli W, Sendi P. Propionibacterium spp. in prosthetic joint infections: a diagnostic challenge. Arch Orthop Trauma Surg. 2008;128(10):1039-1046.
23. Rasouli MR, Harandi AA, Adeli B, Purtill JJ, Parvizi J. Revision total knee arthroplasty: infection should be ruled out in all cases. J Arthroplasty. 2012;27(6):1239-1243.e1-e2.
24. Hunt RW, Bond MJ, Pater GD. Psychological responses to cancer: a case for cancer support groups. Community Health Stud. 1990;14(1):35-38.
25. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
26. Vandercam B, Jeumont S, Cornu O, et al. Amplification-based DNA analysis in the diagnosis of prosthetic joint infection. J Mol Diagn. 2008;10(6):537-543.
Take-Home Points
- Novel synovial markers and PCR have the potential to improve the detection of PJIs.
- 10Difficult-to-detect infections of prosthetic joints pose a diagnostic problem to surgeons and can lead to suboptimal outcomes.
- AD is a highly sensitive and specific synovial fluid marker for detecting PJIs.
- AD has shown promising results in detecting low virulence organisms.
- Studies are needed to determine how to best incorporate novel synovial markers and PCR to current diagnostic criteria in order to improve diagnostic accuracy.
Approximately 7 million Americans are living with a hip or knee replacement.1 According to projections, primary hip arthroplasties will increase by 174% and knee arthroplasties by 673% by 2030. Revision arthroplasties are projected to increase by 137% for hips and 601% for knees during the same time period.2 Infection and aseptic loosening are the most common causes of implant failure.3 The literature shows that infection is the most common cause of failure within 2 years after surgery and that aseptic loosening is the most common cause for late revision.3
Recent studies suggest that prosthetic joint infection (PJI) may be underreported because of difficulty making a diagnosis and that cases of aseptic loosening may in fact be attributable to infections with low-virulence organisms.2,3 These findings have led to new efforts to develop uniform criteria for diagnosing PJIs. In 2011, the Musculoskeletal Infection Society (MSIS) offered a new definition for PJI diagnosis, based on clinical and laboratory criteria, to increase the accuracy of PJI diagnosis.4 The MSIS committee acknowledged that PJI may be present even if these criteria are not met, particularly in the case of low-virulence organisms, as patients may not present with clinical signs of infection and may have normal inflammatory markers and joint aspirates. Reports of PJI cases misdiagnosed as aseptic loosening suggest that current screening and diagnostic tools are not sensitive enough to detect all infections and that PJI is likely underdiagnosed.
According to MSIS criteria, the diagnosis of PJI can be made when there is a sinus tract communicating with the prosthesis, when a pathogen is isolated by culture from 2 or more separate tissue or fluid samples obtained from the affected prosthetic joint, or when 4 of 6 criteria are met. The 6 criteria are (1) elevated serum erythrocyte sedimentation rate (ESR) (>30 mm/hour) and elevated C-reactive protein (CRP) level (>10 mg/L); (2) elevated synovial white blood cell (WBC) count (1100-4000 cells/μL); (3) elevated synovial polymorphonuclear leukocytes (>64%); (4) purulence in affected joint; (5) isolation of a microorganism in a culture of periprosthetic tissue or fluid; and (6) more than 5 neutrophils per high-power field in 5 high-power fields observed.
In this review article, we discuss recently developed novel synovial biomarkers and polymerase chain reaction (PCR) technologies that may help increase the sensitivity and specificity of diagnostic guidelines for PJI.
Methods
Using PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses), we performed a systematic review of specific synovial fluid markers and PCR used in PJI diagnosis. In May 2016, we searched the PubMed database for these criteria: ((((((PCR[Text Word]) OR IL-6[Text Word]) OR leukocyte esterase[Text Word]) OR alpha defensin[Text Word]) AND ((“infection/diagnosis”[MeSH Terms] OR “infection/surgery”[MeSH Terms])))) AND (prosthetic joint infection[MeSH Terms] OR periprosthetic joint infection[MeSH Terms]).
We included patients who had undergone total hip, knee, or shoulder arthroplasty (THA, TKA, TSA). Index tests were PCR and the synovial fluid markers α-defensin (AD), interleukin 6 (IL-6), and leukocyte esterase (LE). Reference tests included joint fluid/serum analysis or tissue analysis (ESR/CRP level, cell count, culture, frozen section), which defined the MSIS criteria for PJI. Primary outcomes of interest were sensitivity and specificity, and secondary outcomes of interest included positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (+LR), and negative likelihood ratio (–LR). Randomized controlled trials and controlled cohort studies in humans published within the past 10 years were included.
Results
Our full-text review yielded 15 papers that met our study inclusion criteria (Figure 1).
α-Defensin
One of the novel synovial biomarkers that has shown significant promise in diagnosing PJIs, even with difficult-to-detect organisms, is AD.
AD has shown even more impressive results as a biomarker for PJI in the hip and knee, where infection with low virulence organism is less common. In 2014, Deirmengian and colleagues6 conducted a prospective clinical study of 149 patients who underwent revision THA or TKA for aseptic loosening (n = 112) or PJI (n = 37) as defined by MSIS criteria. Aseptic loosening was diagnosed when there was no identifiable reason for pain, and MSIS criteria were not met. Synovial fluid aspirates were collected before or during surgery. AD correctly identified 143 of the 149 patients with confirmed infection with sensitivity of 97.3% (95% confidence interval [CI], 85.8%-99.6%) and specificity of 95.5% (95% CI, 89.9%-98.5%). Similarly, Bingham and colleagues7 conducted a retrospective clinical study of 61 assays done on 57 patients who underwent revision arthroplasty for PJI as defined by MSIS criteria. Synovial fluid aspirates were collected before or during surgery. AD correctly identified all 19 PJIs with sensitivity of 100% (95% CI, 79%-100%) and specificity of 95% (95% CI, 83%-99%). Sensitivity and specificity of the AD assay more accurately predicted infection than synovial cell count or serum ESR/CRP level did.
These results are supported by another prospective study by Deirmengian and colleagues8 differentiating aseptic failures and PJIs in THA or TKA. The sensitivity and specificity of AD in diagnosing PJI were 100% (95% CI, 85.05%-100%).
In a prospective study of 102 patients who underwent revision THA or TKA secondary to aseptic loosening or PJI, Frangiamore and colleagues9 also demonstrated the value of AD as a diagnostic for PJI in primary and revision hip and knee arthroplasty.
Table 1 and Figure 2 provide a concise review of the findings of each study.
Interleukin 6
Another synovial fluid biomarker that has shown promise in PJI diagnosis is IL-6. In 2015, Frangiamore and colleagues10 conducted a prospective clinical study of 32 patients who underwent revision TSA. Synovial fluid aspiration was obtained before or during surgery. MSIS criteria were used to establish the diagnosis of PJI. IL-6 had sensitivity of 87% and specificity of 90%, with +LR of 8.45 and –LR of 0.15 in predicting PJI. Synovial fluid IL-6 had strong associations with frozen section histology and growth of P acnes. Frangiamore and colleagues10 recommended an ideal IL-6 cutoff of 359.1 pg/mL and reported that, though not as accurate as AD, synovial fluid IL-6 levels can help predict positive cultures in patients who undergo revision TSA.
Lenski and Scherer11 conducted another retrospective clinical study of the diagnostic value of IL-6 in PJI.
Randau and colleagues12 conducted a prospective clinical study of 120 patients who presented with painful THA or TKA and underwent revision for PJI, aseptic failure, or aseptic revision without signs of infection or loosening. Synovial fluid aspirate was collected before or during surgery.
Table 2 and Figure 3 provide a concise review of the findings of each study.
Leukocyte Esterase
LE strips are an inexpensive screening tool for PJI, according to some studies. In a prospective clinical study of 364 endoprosthetic joint (hip, knee, shoulder) interventions, Guenther and colleagues13 collected synovial fluid before surgery. Samples were tested with graded LE strips using PJI criteria set by the authors. Results were correlated with preoperative synovial fluid aspirations, serum CRP level, serum WBC count, and intraoperative histopathologic and microbiological findings. Whereas 293 (93.31%) of the 314 aseptic cases had negative test strip readings, 100% of the 50 infected cases were positive. LE had sensitivity of 100%, specificity of 96.5%, PPV of 82%, and NPV of 100%.
Wetters et al14 performed a prospective clinical study on 223 patients who underwent TKAs and THAs for suspected PJI based on having criteria defined by the authors of the study. Synovial fluid samples were collected either preoperatively or intraoperatively.
Other authors have reported different findings that LE is an unreliable marker in PJI diagnosis. In one prospective clinical study of 85 patients who underwent primary or revision TSA, synovial fluid was collected during surgery.15 According to MSIS criteria, only 5 positive LE results predicted PJI among 21 primary and revision patients with positive cultures. Of the 7 revision patients who met the MSIS criteria for PJI, only 2 had a positive LE test. LE had sensitivity of 28.6%, specificity of 63.6%, PPV of 28.6%, and NPV of 87.5%. Six of the 7 revision patients grew P acnes. These results showed that LE was unreliable in detecting shoulder PJI.15
In another prospective clinical study, Tischler and colleagues16 enrolled 189 patients who underwent revision TKA or THA for aseptic failure or PJI as defined by the MSIS criteria. Synovial fluid was collected intraoperatively.
Table 3 and Figure 4 provide a concise review of the findings of each study.
Polymerase Chain Reaction
Studies have found that PCR analysis of synovial fluid is effective in detecting bacteria on the surface of implants removed during revision arthroplasties. Comparison of the 16S rRNA gene sequences of bacterial genomes showed a diverse range of bacterial species within biofilms on the surface of clinical and subclinical infections.17 These findings, along with those of other studies, suggest that PCR analysis of synovial fluid is useful in diagnosing PJI and identifying organisms and their sensitivities to antibiotics.
Gallo and colleagues18 performed a prospective clinical study on 115 patients who underwent revision TKAs or THAs. Synovial fluid was collected intraoperatively. PCR assays targeting the 16S rDNA were carried out on 101 patients. PJIs were classified based on criteria of the authors of this study, of which there were 42. The sensitivity, specificity, PPV, NPV, +LR, and -LR for PCR were 71.4% (95% CI, 61.5%-75.5%), 97% (95% CI, 91.7%-99.1%), 92.6% (95% CI, 79.8%-97.9%), 86.5% (95% CI, 81.8%-88.4%), 23.6 (95% CI, 5.9%-93.8%), and 0.29 (95% CI, 0.17%-0.49%), respectively. Of note the most common organism detected in 42 PJIs was coagulase-negative Staphylococcus.
Marin and colleagues19 conducted a prospective study of 122 patients who underwent arthroplasty for suspected infection or aseptic loosening as defined by the authors’ clinicohistopathologic criteria. Synovial fluid and biopsy specimens were collected during surgery, and 40 patients met the infection criteria. The authors concluded that 16S PCR is more specific and has better PPV than culture does as one positive 16S PCR resulted in a specificity and PPV of PJI of 96.3% and 91.7%, respectively. However, they noted that culture was more sensitive in diagnosing PJI.
Jacovides and colleagues20 conducted a prospective study on 82 patients undergoing primary TKA, revision TKA, and revision THA.
The low PCR sensitivities reported in the literature were explained in a review by Hartley and Harris.21 They wrote that BR 16S rDNA and sequencing of PJI samples inherently have low sensitivity because of the contamination that can occur from the PCR reagents themselves or from sample mishandling. Techniques that address contaminant (extraneous DNA) removal, such as ultraviolet irradiation and DNase treatment, reduce Taq DNA polymerase activity, which reduces PCR sensitivity.
Table 4 and Figure 5 provide a concise review of the findings of each study.
Discussion
Although there is no gold standard for the diagnosis of PJIs, several clinical and laboratory criteria guidelines are currently used to help clinicians diagnose infections of prosthetic joints. However, despite standardization of diagnostic criteria, PJI continue to be a diagnostic challenge.
AD is a highly sensitive and specific synovial fluid biomarker in detecting common PJIs.
In summary, 5 AD studies5-9 had sensitivity ranging from 63% to 100% and specificity ranging from 95% to 100%; 3 IL-6 studies10-12 had sensitivity ranging from 46.8% to 90.9% and specificity ranging from 85.7% to 97.6%; 4 LE studies13-16 had sensitivity ranging from 28.6% to 100% and specificity ranging from 63.6% to 96.5%; and 3 PCR studies18-20 had sensitivity ranging from 67.1% to 95.7% and specificity ranging from 12.3% to 97.8%. Sensitivity and specificity were consistently higher for AD than for IL-6, LE, and PCR, though there was significant overlap, heterogeneity, and variation across all the included studies.
Although the overall incidence of PJI is low, infected revisions remain a substantial financial burden to hospitals, as annual costs of infected revisions is estimated to exceed $1.62 billion by 2020.25 The usefulness of novel biomarkers and PCR in diagnosing PJI can be found in their ability to diagnose infections and facilitate appropriate early treatment. Several of these tests are readily available commercially and have the potential to be cost-effective diagnostic tools. The price to perform an AD test from Synovasure TM (Zimmer Biomet) ranges from $93 to $143. LE also provides an economic option for diagnosing PJI, as LE strips are commercially available for the cost of about 25 cents. PCR has also become an economic option, as costs can average $15.50 per sample extraction or PCR assay and $42.50 per amplicon sequence as reported in a study by Vandercam and colleagues.26 Future studies are needed to determine a diagnostic algorithm which incorporates these novel synovial markers to improve diagnostic accuracy of PJI in the most cost effective manner.
The current literature supports that AD can potentially be used to screen for PJI. Our findings suggest novel synovial fluid biomarkers may become of significant diagnostic use when combined with current laboratory and clinical diagnostic criteria. We recommend use of AD in cases in which pain, stiffness, and poor TJA outcome cannot be explained by errors in surgical technique, and infection is suspected despite MSIS criteria not being met.
The studies reviewed in this manuscript were limited in that none presented level I evidence (12 had level II evidence, and 3 had level III evidence), and there was significant heterogeneity (some studies used their own diagnostic standard, and others used the MSIS criteria). Larger scale prospective studies comparing serum ESR/CRP level and synovial fluid analysis to novel synovial markers are needed.
Am J Orthop. 2017;46(4):190-198. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Novel synovial markers and PCR have the potential to improve the detection of PJIs.
- 10Difficult-to-detect infections of prosthetic joints pose a diagnostic problem to surgeons and can lead to suboptimal outcomes.
- AD is a highly sensitive and specific synovial fluid marker for detecting PJIs.
- AD has shown promising results in detecting low virulence organisms.
- Studies are needed to determine how to best incorporate novel synovial markers and PCR to current diagnostic criteria in order to improve diagnostic accuracy.
Approximately 7 million Americans are living with a hip or knee replacement.1 According to projections, primary hip arthroplasties will increase by 174% and knee arthroplasties by 673% by 2030. Revision arthroplasties are projected to increase by 137% for hips and 601% for knees during the same time period.2 Infection and aseptic loosening are the most common causes of implant failure.3 The literature shows that infection is the most common cause of failure within 2 years after surgery and that aseptic loosening is the most common cause for late revision.3
Recent studies suggest that prosthetic joint infection (PJI) may be underreported because of difficulty making a diagnosis and that cases of aseptic loosening may in fact be attributable to infections with low-virulence organisms.2,3 These findings have led to new efforts to develop uniform criteria for diagnosing PJIs. In 2011, the Musculoskeletal Infection Society (MSIS) offered a new definition for PJI diagnosis, based on clinical and laboratory criteria, to increase the accuracy of PJI diagnosis.4 The MSIS committee acknowledged that PJI may be present even if these criteria are not met, particularly in the case of low-virulence organisms, as patients may not present with clinical signs of infection and may have normal inflammatory markers and joint aspirates. Reports of PJI cases misdiagnosed as aseptic loosening suggest that current screening and diagnostic tools are not sensitive enough to detect all infections and that PJI is likely underdiagnosed.
According to MSIS criteria, the diagnosis of PJI can be made when there is a sinus tract communicating with the prosthesis, when a pathogen is isolated by culture from 2 or more separate tissue or fluid samples obtained from the affected prosthetic joint, or when 4 of 6 criteria are met. The 6 criteria are (1) elevated serum erythrocyte sedimentation rate (ESR) (>30 mm/hour) and elevated C-reactive protein (CRP) level (>10 mg/L); (2) elevated synovial white blood cell (WBC) count (1100-4000 cells/μL); (3) elevated synovial polymorphonuclear leukocytes (>64%); (4) purulence in affected joint; (5) isolation of a microorganism in a culture of periprosthetic tissue or fluid; and (6) more than 5 neutrophils per high-power field in 5 high-power fields observed.
In this review article, we discuss recently developed novel synovial biomarkers and polymerase chain reaction (PCR) technologies that may help increase the sensitivity and specificity of diagnostic guidelines for PJI.
Methods
Using PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses), we performed a systematic review of specific synovial fluid markers and PCR used in PJI diagnosis. In May 2016, we searched the PubMed database for these criteria: ((((((PCR[Text Word]) OR IL-6[Text Word]) OR leukocyte esterase[Text Word]) OR alpha defensin[Text Word]) AND ((“infection/diagnosis”[MeSH Terms] OR “infection/surgery”[MeSH Terms])))) AND (prosthetic joint infection[MeSH Terms] OR periprosthetic joint infection[MeSH Terms]).
We included patients who had undergone total hip, knee, or shoulder arthroplasty (THA, TKA, TSA). Index tests were PCR and the synovial fluid markers α-defensin (AD), interleukin 6 (IL-6), and leukocyte esterase (LE). Reference tests included joint fluid/serum analysis or tissue analysis (ESR/CRP level, cell count, culture, frozen section), which defined the MSIS criteria for PJI. Primary outcomes of interest were sensitivity and specificity, and secondary outcomes of interest included positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (+LR), and negative likelihood ratio (–LR). Randomized controlled trials and controlled cohort studies in humans published within the past 10 years were included.
Results
Our full-text review yielded 15 papers that met our study inclusion criteria (Figure 1).
α-Defensin
One of the novel synovial biomarkers that has shown significant promise in diagnosing PJIs, even with difficult-to-detect organisms, is AD.
AD has shown even more impressive results as a biomarker for PJI in the hip and knee, where infection with low virulence organism is less common. In 2014, Deirmengian and colleagues6 conducted a prospective clinical study of 149 patients who underwent revision THA or TKA for aseptic loosening (n = 112) or PJI (n = 37) as defined by MSIS criteria. Aseptic loosening was diagnosed when there was no identifiable reason for pain, and MSIS criteria were not met. Synovial fluid aspirates were collected before or during surgery. AD correctly identified 143 of the 149 patients with confirmed infection with sensitivity of 97.3% (95% confidence interval [CI], 85.8%-99.6%) and specificity of 95.5% (95% CI, 89.9%-98.5%). Similarly, Bingham and colleagues7 conducted a retrospective clinical study of 61 assays done on 57 patients who underwent revision arthroplasty for PJI as defined by MSIS criteria. Synovial fluid aspirates were collected before or during surgery. AD correctly identified all 19 PJIs with sensitivity of 100% (95% CI, 79%-100%) and specificity of 95% (95% CI, 83%-99%). Sensitivity and specificity of the AD assay more accurately predicted infection than synovial cell count or serum ESR/CRP level did.
These results are supported by another prospective study by Deirmengian and colleagues8 differentiating aseptic failures and PJIs in THA or TKA. The sensitivity and specificity of AD in diagnosing PJI were 100% (95% CI, 85.05%-100%).
In a prospective study of 102 patients who underwent revision THA or TKA secondary to aseptic loosening or PJI, Frangiamore and colleagues9 also demonstrated the value of AD as a diagnostic for PJI in primary and revision hip and knee arthroplasty.
Table 1 and Figure 2 provide a concise review of the findings of each study.
Interleukin 6
Another synovial fluid biomarker that has shown promise in PJI diagnosis is IL-6. In 2015, Frangiamore and colleagues10 conducted a prospective clinical study of 32 patients who underwent revision TSA. Synovial fluid aspiration was obtained before or during surgery. MSIS criteria were used to establish the diagnosis of PJI. IL-6 had sensitivity of 87% and specificity of 90%, with +LR of 8.45 and –LR of 0.15 in predicting PJI. Synovial fluid IL-6 had strong associations with frozen section histology and growth of P acnes. Frangiamore and colleagues10 recommended an ideal IL-6 cutoff of 359.1 pg/mL and reported that, though not as accurate as AD, synovial fluid IL-6 levels can help predict positive cultures in patients who undergo revision TSA.
Lenski and Scherer11 conducted another retrospective clinical study of the diagnostic value of IL-6 in PJI.
Randau and colleagues12 conducted a prospective clinical study of 120 patients who presented with painful THA or TKA and underwent revision for PJI, aseptic failure, or aseptic revision without signs of infection or loosening. Synovial fluid aspirate was collected before or during surgery.
Table 2 and Figure 3 provide a concise review of the findings of each study.
Leukocyte Esterase
LE strips are an inexpensive screening tool for PJI, according to some studies. In a prospective clinical study of 364 endoprosthetic joint (hip, knee, shoulder) interventions, Guenther and colleagues13 collected synovial fluid before surgery. Samples were tested with graded LE strips using PJI criteria set by the authors. Results were correlated with preoperative synovial fluid aspirations, serum CRP level, serum WBC count, and intraoperative histopathologic and microbiological findings. Whereas 293 (93.31%) of the 314 aseptic cases had negative test strip readings, 100% of the 50 infected cases were positive. LE had sensitivity of 100%, specificity of 96.5%, PPV of 82%, and NPV of 100%.
Wetters et al14 performed a prospective clinical study on 223 patients who underwent TKAs and THAs for suspected PJI based on having criteria defined by the authors of the study. Synovial fluid samples were collected either preoperatively or intraoperatively.
Other authors have reported different findings that LE is an unreliable marker in PJI diagnosis. In one prospective clinical study of 85 patients who underwent primary or revision TSA, synovial fluid was collected during surgery.15 According to MSIS criteria, only 5 positive LE results predicted PJI among 21 primary and revision patients with positive cultures. Of the 7 revision patients who met the MSIS criteria for PJI, only 2 had a positive LE test. LE had sensitivity of 28.6%, specificity of 63.6%, PPV of 28.6%, and NPV of 87.5%. Six of the 7 revision patients grew P acnes. These results showed that LE was unreliable in detecting shoulder PJI.15
In another prospective clinical study, Tischler and colleagues16 enrolled 189 patients who underwent revision TKA or THA for aseptic failure or PJI as defined by the MSIS criteria. Synovial fluid was collected intraoperatively.
Table 3 and Figure 4 provide a concise review of the findings of each study.
Polymerase Chain Reaction
Studies have found that PCR analysis of synovial fluid is effective in detecting bacteria on the surface of implants removed during revision arthroplasties. Comparison of the 16S rRNA gene sequences of bacterial genomes showed a diverse range of bacterial species within biofilms on the surface of clinical and subclinical infections.17 These findings, along with those of other studies, suggest that PCR analysis of synovial fluid is useful in diagnosing PJI and identifying organisms and their sensitivities to antibiotics.
Gallo and colleagues18 performed a prospective clinical study on 115 patients who underwent revision TKAs or THAs. Synovial fluid was collected intraoperatively. PCR assays targeting the 16S rDNA were carried out on 101 patients. PJIs were classified based on criteria of the authors of this study, of which there were 42. The sensitivity, specificity, PPV, NPV, +LR, and -LR for PCR were 71.4% (95% CI, 61.5%-75.5%), 97% (95% CI, 91.7%-99.1%), 92.6% (95% CI, 79.8%-97.9%), 86.5% (95% CI, 81.8%-88.4%), 23.6 (95% CI, 5.9%-93.8%), and 0.29 (95% CI, 0.17%-0.49%), respectively. Of note the most common organism detected in 42 PJIs was coagulase-negative Staphylococcus.
Marin and colleagues19 conducted a prospective study of 122 patients who underwent arthroplasty for suspected infection or aseptic loosening as defined by the authors’ clinicohistopathologic criteria. Synovial fluid and biopsy specimens were collected during surgery, and 40 patients met the infection criteria. The authors concluded that 16S PCR is more specific and has better PPV than culture does as one positive 16S PCR resulted in a specificity and PPV of PJI of 96.3% and 91.7%, respectively. However, they noted that culture was more sensitive in diagnosing PJI.
Jacovides and colleagues20 conducted a prospective study on 82 patients undergoing primary TKA, revision TKA, and revision THA.
The low PCR sensitivities reported in the literature were explained in a review by Hartley and Harris.21 They wrote that BR 16S rDNA and sequencing of PJI samples inherently have low sensitivity because of the contamination that can occur from the PCR reagents themselves or from sample mishandling. Techniques that address contaminant (extraneous DNA) removal, such as ultraviolet irradiation and DNase treatment, reduce Taq DNA polymerase activity, which reduces PCR sensitivity.
Table 4 and Figure 5 provide a concise review of the findings of each study.
Discussion
Although there is no gold standard for the diagnosis of PJIs, several clinical and laboratory criteria guidelines are currently used to help clinicians diagnose infections of prosthetic joints. However, despite standardization of diagnostic criteria, PJI continue to be a diagnostic challenge.
AD is a highly sensitive and specific synovial fluid biomarker in detecting common PJIs.
In summary, 5 AD studies5-9 had sensitivity ranging from 63% to 100% and specificity ranging from 95% to 100%; 3 IL-6 studies10-12 had sensitivity ranging from 46.8% to 90.9% and specificity ranging from 85.7% to 97.6%; 4 LE studies13-16 had sensitivity ranging from 28.6% to 100% and specificity ranging from 63.6% to 96.5%; and 3 PCR studies18-20 had sensitivity ranging from 67.1% to 95.7% and specificity ranging from 12.3% to 97.8%. Sensitivity and specificity were consistently higher for AD than for IL-6, LE, and PCR, though there was significant overlap, heterogeneity, and variation across all the included studies.
Although the overall incidence of PJI is low, infected revisions remain a substantial financial burden to hospitals, as annual costs of infected revisions is estimated to exceed $1.62 billion by 2020.25 The usefulness of novel biomarkers and PCR in diagnosing PJI can be found in their ability to diagnose infections and facilitate appropriate early treatment. Several of these tests are readily available commercially and have the potential to be cost-effective diagnostic tools. The price to perform an AD test from Synovasure TM (Zimmer Biomet) ranges from $93 to $143. LE also provides an economic option for diagnosing PJI, as LE strips are commercially available for the cost of about 25 cents. PCR has also become an economic option, as costs can average $15.50 per sample extraction or PCR assay and $42.50 per amplicon sequence as reported in a study by Vandercam and colleagues.26 Future studies are needed to determine a diagnostic algorithm which incorporates these novel synovial markers to improve diagnostic accuracy of PJI in the most cost effective manner.
The current literature supports that AD can potentially be used to screen for PJI. Our findings suggest novel synovial fluid biomarkers may become of significant diagnostic use when combined with current laboratory and clinical diagnostic criteria. We recommend use of AD in cases in which pain, stiffness, and poor TJA outcome cannot be explained by errors in surgical technique, and infection is suspected despite MSIS criteria not being met.
The studies reviewed in this manuscript were limited in that none presented level I evidence (12 had level II evidence, and 3 had level III evidence), and there was significant heterogeneity (some studies used their own diagnostic standard, and others used the MSIS criteria). Larger scale prospective studies comparing serum ESR/CRP level and synovial fluid analysis to novel synovial markers are needed.
Am J Orthop. 2017;46(4):190-198. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Maradit Kremers H, Larson DR, Crowson CS, et al. Prevalence of total hip and knee replacement in the United States. J Bone Joint Surg Am. 2015;97(17):1386-1397.
2. Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am. 2007;89(4):780-785.
3. Sharkey PF, Lichstein PM, Shen C, Tokarski AT, Parvizi J. Why are total knee arthroplasties failing today—has anything changed after 10 years? J Arthroplasty. 2014;29(9):1774-1778.
4. Butler-Wu SM, Burns EM, Pottinger PS, et al. Optimization of periprosthetic culture for diagnosis of Propionibacterium acnes prosthetic joint infection. J Clin Microbiol. 2011;49(7):2490-2495.
5. Frangiamore SJ, Saleh A, Grosso MJ, et al. α-Defensin as a predictor of periprosthetic shoulder infection. J Shoulder Elbow Surg. 2015;24(7):1021-1027.
6. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Combined measurement of synovial fluid α-defensin and C-reactive protein levels: highly accurate for diagnosing periprosthetic joint infection. J Bone Joint Surg Am. 2014;96(17):1439-1445.
7. Bingham J, Clarke H, Spangehl M, Schwartz A, Beauchamp C, Goldberg B. The alpha defensin-1 biomarker assay can be used to evaluate the potentially infected total joint arthroplasty. Clin Orthop Relat Res. 2014;472(12):4006-4009.
8. Deirmengian C, Kardos K, Kilmartin P, et al. The alpha-defensin test for periprosthetic joint infection outperforms the leukocyte esterase test strip. Clin Orthop Relat Res. 2015;473(1):198-203.
9. Frangiamore SJ, Gajewski ND, Saleh A, Farias-Kovac M, Barsoum WK, Higuera CA. α-Defensin accuracy to diagnose periprosthetic joint infection—best available test? J Arthroplasty. 2016;31(2):456-460.
10. Frangiamore SJ, Saleh A, Kovac MF, et al. Synovial fluid interleukin-6 as a predictor of periprosthetic shoulder infection. J Bone Joint Surg Am. 2015;97(1):63-70.
11. Lenski M, Scherer MA. Synovial IL-6 as inflammatory marker in periprosthetic joint infections. J Arthroplasty. 2014;29(6):1105-1109.
12. Randau TM, Friedrich MJ, Wimmer MD, et al. Interleukin-6 in serum and in synovial fluid enhances the differentiation between periprosthetic joint infection and aseptic loosening. PLoS One. 2014;9(2):e89045.
13. Guenther D, Kokenge T, Jacobs O, et al. Excluding infections in arthroplasty using leucocyte esterase test. Int Orthop. 2014;38(11):2385-2390.
14. Wetters NG, Berend KR, Lombardi AV, Morris MJ, Tucker TL, Della Valle CJ. Leukocyte esterase reagent strips for the rapid diagnosis of periprosthetic joint infection. J Arthroplasty. 2012;27(8 suppl):8-11.
15. Nelson GN, Paxton ES, Narzikul A, Williams G, Lazarus MD, Abboud JA. Leukocyte esterase in the diagnosis of shoulder periprosthetic joint infection. J Shoulder Elbow Surg. 2015;24(9):1421-1426.
16. Tischler EH, Cavanaugh PK, Parvizi J. Leukocyte esterase strip test: matched for Musculoskeletal Infection Society criteria. J Bone Joint Surg Am. 2014;96(22):1917-1920.
17. Dempsey KE, Riggio MP, Lennon A, et al. Identification of bacteria on the surface of clinically infected and non-infected prosthetic hip joints removed during revision arthroplasties by 16S rRNA gene sequencing and by microbiological culture. Arthritis Res Ther. 2007;9(3):R46.
18. Gallo J, Kolar M, Dendis M, et al. Culture and PCR analysis of joint fluid in the diagnosis of prosthetic joint infection. New Microbiol. 2008;31(1):97-104.
19. Marin M, Garcia-Lechuz JM, Alonso P, et al. Role of universal 16S rRNA gene PCR and sequencing in diagnosis of prosthetic joint infection. J Clin Microbiol. 2012;50(3):583-589.
20. Jacovides CL, Kreft R, Adeli B, Hozack B, Ehrlich GD, Parvizi J. Successful identification of pathogens by polymerase chain reaction (PCR)-based electron spray ionization time-of-flight mass spectrometry (ESI-TOF-MS) in culture-negative periprosthetic joint infection. J Bone Joint Surg Am. 2012;94(24):2247-2254.
21. Hartley JC, Harris KA. Molecular techniques for diagnosing prosthetic joint infections. J Antimicrob Chemother. 2014;69(suppl 1):i21-i24.
22. Zappe B, Graf S, Ochsner PE, Zimmerli W, Sendi P. Propionibacterium spp. in prosthetic joint infections: a diagnostic challenge. Arch Orthop Trauma Surg. 2008;128(10):1039-1046.
23. Rasouli MR, Harandi AA, Adeli B, Purtill JJ, Parvizi J. Revision total knee arthroplasty: infection should be ruled out in all cases. J Arthroplasty. 2012;27(6):1239-1243.e1-e2.
24. Hunt RW, Bond MJ, Pater GD. Psychological responses to cancer: a case for cancer support groups. Community Health Stud. 1990;14(1):35-38.
25. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
26. Vandercam B, Jeumont S, Cornu O, et al. Amplification-based DNA analysis in the diagnosis of prosthetic joint infection. J Mol Diagn. 2008;10(6):537-543.
1. Maradit Kremers H, Larson DR, Crowson CS, et al. Prevalence of total hip and knee replacement in the United States. J Bone Joint Surg Am. 2015;97(17):1386-1397.
2. Kurtz S, Ong K, Lau E, Mowat F, Halpern M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J Bone Joint Surg Am. 2007;89(4):780-785.
3. Sharkey PF, Lichstein PM, Shen C, Tokarski AT, Parvizi J. Why are total knee arthroplasties failing today—has anything changed after 10 years? J Arthroplasty. 2014;29(9):1774-1778.
4. Butler-Wu SM, Burns EM, Pottinger PS, et al. Optimization of periprosthetic culture for diagnosis of Propionibacterium acnes prosthetic joint infection. J Clin Microbiol. 2011;49(7):2490-2495.
5. Frangiamore SJ, Saleh A, Grosso MJ, et al. α-Defensin as a predictor of periprosthetic shoulder infection. J Shoulder Elbow Surg. 2015;24(7):1021-1027.
6. Deirmengian C, Kardos K, Kilmartin P, Cameron A, Schiller K, Parvizi J. Combined measurement of synovial fluid α-defensin and C-reactive protein levels: highly accurate for diagnosing periprosthetic joint infection. J Bone Joint Surg Am. 2014;96(17):1439-1445.
7. Bingham J, Clarke H, Spangehl M, Schwartz A, Beauchamp C, Goldberg B. The alpha defensin-1 biomarker assay can be used to evaluate the potentially infected total joint arthroplasty. Clin Orthop Relat Res. 2014;472(12):4006-4009.
8. Deirmengian C, Kardos K, Kilmartin P, et al. The alpha-defensin test for periprosthetic joint infection outperforms the leukocyte esterase test strip. Clin Orthop Relat Res. 2015;473(1):198-203.
9. Frangiamore SJ, Gajewski ND, Saleh A, Farias-Kovac M, Barsoum WK, Higuera CA. α-Defensin accuracy to diagnose periprosthetic joint infection—best available test? J Arthroplasty. 2016;31(2):456-460.
10. Frangiamore SJ, Saleh A, Kovac MF, et al. Synovial fluid interleukin-6 as a predictor of periprosthetic shoulder infection. J Bone Joint Surg Am. 2015;97(1):63-70.
11. Lenski M, Scherer MA. Synovial IL-6 as inflammatory marker in periprosthetic joint infections. J Arthroplasty. 2014;29(6):1105-1109.
12. Randau TM, Friedrich MJ, Wimmer MD, et al. Interleukin-6 in serum and in synovial fluid enhances the differentiation between periprosthetic joint infection and aseptic loosening. PLoS One. 2014;9(2):e89045.
13. Guenther D, Kokenge T, Jacobs O, et al. Excluding infections in arthroplasty using leucocyte esterase test. Int Orthop. 2014;38(11):2385-2390.
14. Wetters NG, Berend KR, Lombardi AV, Morris MJ, Tucker TL, Della Valle CJ. Leukocyte esterase reagent strips for the rapid diagnosis of periprosthetic joint infection. J Arthroplasty. 2012;27(8 suppl):8-11.
15. Nelson GN, Paxton ES, Narzikul A, Williams G, Lazarus MD, Abboud JA. Leukocyte esterase in the diagnosis of shoulder periprosthetic joint infection. J Shoulder Elbow Surg. 2015;24(9):1421-1426.
16. Tischler EH, Cavanaugh PK, Parvizi J. Leukocyte esterase strip test: matched for Musculoskeletal Infection Society criteria. J Bone Joint Surg Am. 2014;96(22):1917-1920.
17. Dempsey KE, Riggio MP, Lennon A, et al. Identification of bacteria on the surface of clinically infected and non-infected prosthetic hip joints removed during revision arthroplasties by 16S rRNA gene sequencing and by microbiological culture. Arthritis Res Ther. 2007;9(3):R46.
18. Gallo J, Kolar M, Dendis M, et al. Culture and PCR analysis of joint fluid in the diagnosis of prosthetic joint infection. New Microbiol. 2008;31(1):97-104.
19. Marin M, Garcia-Lechuz JM, Alonso P, et al. Role of universal 16S rRNA gene PCR and sequencing in diagnosis of prosthetic joint infection. J Clin Microbiol. 2012;50(3):583-589.
20. Jacovides CL, Kreft R, Adeli B, Hozack B, Ehrlich GD, Parvizi J. Successful identification of pathogens by polymerase chain reaction (PCR)-based electron spray ionization time-of-flight mass spectrometry (ESI-TOF-MS) in culture-negative periprosthetic joint infection. J Bone Joint Surg Am. 2012;94(24):2247-2254.
21. Hartley JC, Harris KA. Molecular techniques for diagnosing prosthetic joint infections. J Antimicrob Chemother. 2014;69(suppl 1):i21-i24.
22. Zappe B, Graf S, Ochsner PE, Zimmerli W, Sendi P. Propionibacterium spp. in prosthetic joint infections: a diagnostic challenge. Arch Orthop Trauma Surg. 2008;128(10):1039-1046.
23. Rasouli MR, Harandi AA, Adeli B, Purtill JJ, Parvizi J. Revision total knee arthroplasty: infection should be ruled out in all cases. J Arthroplasty. 2012;27(6):1239-1243.e1-e2.
24. Hunt RW, Bond MJ, Pater GD. Psychological responses to cancer: a case for cancer support groups. Community Health Stud. 1990;14(1):35-38.
25. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
26. Vandercam B, Jeumont S, Cornu O, et al. Amplification-based DNA analysis in the diagnosis of prosthetic joint infection. J Mol Diagn. 2008;10(6):537-543.
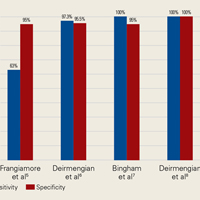
Traumatic Anterior Shoulder Instability: The US Military Experience
Take-Home Points
- Arthroscopic stabilization performed early results in better outcomes in patients with Bankart lesions.
- A subcritical level of bone loss of 13.5% has been shown to have a significant effect on outcomes, in addition to the established “critical amount”.
- Bone loss is a bipolar issue. Both sides must be considered in order to properly address shoulder instability.
- Off-track measurement has been shown to be even more positively predictive of outcomes than glenoid bone loss assessment.
- There are several bone loss management options including, the most common coracoid transfer, as well as distal tibial allograft and distal clavicular autograft.
Given its relatively young age, high activity level, and centralized medical care system, the US military population is ideal for studying traumatic anterior shoulder instability. There is a long history of military surgeons who have made significant contributions that have advanced our understanding of this pathology and its treatment and results. In this article, we describe the scope, treatment, and results of this pathology in the US military population.
Incidence and Pathology
At the United States Military Academy (USMA), Owens and colleagues1 studied the incidence of shoulder instability, including dislocation and subluxation, and found anterior instability events were far more common than in civilian populations. The incidence of shoulder instability was 0.08 per 1000 person-years in the general US population vs 1.69 per 1000 person-years in US military personnel. The factors associated with increased risk of shoulder instability injury in the military population were male sex, white race, junior enlisted rank, and age under 30 years. Owens and colleagues2 noted that subluxation accounted for almost 85% of the total anterior instability events. Owens and colleagues3 found the pathology in subluxation events was similar to that in full dislocations, with a soft-tissue anterior Bankart lesion and a Hill-Sachs lesion detected on magnetic resonance imaging in more than 90% of patients. In another study at the USMA, DeBerardino and colleagues4 noted that 97% of arthroscopically assessed shoulders in first-time dislocators involved complete detachment of the capsuloligamentous complex from the anterior glenoid rim and neck—a so-called Bankart lesion. Thus, in a military population, anterior instability resulting from subluxation or dislocation is a common finding that is often represented by a soft-tissue Bankart lesion and a Hill-Sachs defect.
Natural History of Traumatic Anterior Shoulder Instability in the Military
Several studies have evaluated the outcomes of nonoperative and operative treatment of shoulder instability. Although most have found better outcomes with operative intervention, Aronen and Regan5 reported good results (25% recurrence at nearly 3-year follow-up) with nonoperative treatment and adherence to a strict rehabilitation program. Most other comparative studies in this population have published contrary results. Wheeler and colleagues6 studied the natural history of anterior shoulder dislocations in a USMA cadet cohort and found recurrent instability after shoulder dislocation in 92% of cadets who had nonoperative treatment. Similarly, DeBerardino and colleagues4 found that, in the USMA, 90% of first-time traumatic anterior shoulder dislocations managed nonoperatively experienced recurrent instability. In a series of Army soldiers with shoulder instability, Bottoni and colleagues7 reported that 75% of nonoperatively managed patients had recurrent instability, and, of these, 67% progressed to surgical intervention. Nonoperative treatment for a first-time dislocation is still reasonable if a cadet or soldier needs to quickly return to functional duties. Athletes who develop shoulder instability during their playing season have been studied in a military population as well. In a multicenter study of service academy athletes with anterior instability, Dickens and colleagues8 found that, with conservative management and accelerated rehabilitation of in-season shoulder instability, 73% of athletes returned to sport by a mean of 5 days. However, the durability of this treatment should be questioned, as 64% later experienced recurrence.
Arthroscopic Stabilization of Acute Anterior Shoulder Dislocations
In an early series of cases of traumatic anterior shoulder instability in USMA cadets, Wheeler and colleagues6 found that, at 14 months, 78% of arthroscopically stabilized cases and 92% of nonoperatively treated cases were successful. Then, in the 1990s, DeBerardino and colleagues4 studied a series of young, active patients in the USMA and noted significantly better results with arthroscopic treatment, vs nonoperative treatment, at 2- to 5-year follow-up. Of the arthroscopically treated shoulders, 88% remained stable during the study and returned to preinjury activity levels, and 12% experienced recurrent instability (risk factors included 2+ sulcus sign, poor capsular labral tissue, and history of bilateral shoulder instability). In a long-term follow-up (mean, 11.7 years; range, 9.1-13.9 years) of the same cohort, Owens and colleagues9 found that 14% of patients available for follow-up had undergone revision stabilization surgery, and, of these, 21% reported experiencing subluxation events. The authors concluded that, in first-time dislocators in this active military population, acute arthroscopic Bankart repair resulted in excellent return to athletics and subjective function, and had acceptable recurrence and reoperation rates. Bottoni and colleagues,7 in a prospective, randomized evaluation of arthroscopic stabilization of acute, traumatic, first-time shoulder dislocations in the Army, noted an 89% success rate for arthroscopic treatment at an average follow-up of 36 months, with no recurrent instability. DeBerardino and colleagues10 compared West Point patients treated nonoperatively with those arthroscopically treated with staples, transglenoid sutures, or bioabsorbable anchors. Recurrence rates were 85% for nonoperative treatment, 22% for staples, 14% for transglenoid sutures, and 10% for bioabsorbable anchors.
Arthroscopic Versus Open Stabilization of Anterior Shoulder Instability
In a prospective, randomized clinical trial comparing open and arthroscopic shoulder stabilization for recurrent anterior instability in active-duty Army personnel, Bottoni and colleagues11 found comparable clinical outcomes. Stabilization surgery failed clinically in only 3 cases, 2 open and 1 arthroscopic. The authors concluded that arthroscopic stabilization can be safely performed for recurrent shoulder instability and that arthroscopic outcomes are similar to open outcomes. In a series of anterior shoulder subluxations in young athletes with Bankart lesions, Owens and colleagues12 found that open and arthroscopic stabilization performed early resulted in better outcomes, regardless of technique used. Recurrent subluxation occurred at a mean of 17 months in 3 of the 10 patients in the open group and 3 of the 9 patients in the arthroscopic group, for an overall recurrence rate of 31%. The authors concluded that, in this patient population with Bankart lesions caused by anterior subluxation events, surgery should be performed early.
Bone Lesions
Burkhart and De Beer13 first noted that bone loss has emerged as one of the most important considerations in the setting of shoulder instability in active patients. Other authors have found this to be true in military populations.14,15
The diagnosis of bone loss may include historical findings, such as increased number and ease of dislocations, as well as dislocation in lower positions of abduction. Physical examination findings may include apprehension in the midrange of motion. Advanced imaging, such as magnetic resonance arthrography, has since been validated as equivalent to 3-dimensional computed tomography (3-D CT) in determining glenoid bone loss.16 In 2007, Mologne and colleagues15 studied the amount of glenoid bone loss and the presence of fragmented bone or attritional bone loss and its effect on outcomes. They evaluated 21 patients who had arthroscopic treatment for anterior instability with anteroinferior glenoid bone loss between 20% and 30%. Average follow-up was 34 months. All patients received 3 or 4 anterior anchors. No patient with a bone fragment incorporated into the repair experienced recurrence or subluxation, whereas 30% of patients with attritional bone loss had recurrent instability.15
Classifying Bone Loss and Recognizing Its Effects
Burkhart and De Beer13 helped define the role and significance of bone loss in the setting of shoulder instability. They defined significant bone loss as an engaging Hill-Sachs lesion of the humerus in an abducted and externally rotated position or an “inverted pear” lesion of the glenoid. Overall analysis revealed recurrence in 4% of cases without significant bone loss and 65% of cases with significant bone loss. In a subanalysis of contact-sport athletes in the setting of bone loss, the failure rate increased to 89%, from 6.5%. Aiding in the quantitative assessment of glenoid bone loss, Itoi and colleagues17 showed that 21% glenoid bone loss resulted in instability that would not be corrected by a soft-tissue procedure alone. Bone loss of 20% to 25% has since been considered a “critical amount,” above which an arthroscopic Bankart has been questioned. More recently, several authors have shown that even less bone loss can have a significant effect on outcomes. Shaha and colleagues18 established that a subcritical level of bone loss (13.5%) on the anteroinferior glenoid resulted in clinical failure (as determined with the Western Ontario Shoulder Instability Index) even in cases in which frank recurrence or subluxation was avoided. It is thought that, in recurrent instability, glenoid bone loss incident rate is as high as 90%, and the corresponding percentage of patients with Hill-Sachs lesions is almost 100%.19,20 Thus, it is increasingly understood that bone loss is a bipolar issue and that both sides must be considered in order to properly address shoulder instability in this setting. In 2007, Yamamoto and colleagues21 introduced the glenoid track, a method for predicting whether a Hill-Sachs lesion will engage. Di Giacomo and colleagues22 refined the track concept to quantitatively determine which lesions will engage in the setting of both glenoid and humeral bone loss. Metzger and colleagues,23 confirming the track concept arthroscopically, found that manipulation with anesthesia and arthroscopic visualization was well predicted by preoperative track measurements, and thus these measurements can be a good guide for surgical management (Figures 1A, 1B). 

Strategies for Addressing Bone Loss in Anterior Shoulder Instability
Several approaches for managing bone loss in shoulder instability have been described—the most common being coracoid transfer (Latarjet procedure). Waterman and colleagues25 recently studied the effects of coracoid transfer, distal tibial allograft, and iliac crest augmentation on anterior shoulder instability in US military patients treated between 2006 and 2012. Of 64 patients who underwent a bone block procedure, 16 (25%) had a complication during short-term follow-up. Complications included neurologic injury, pain, infection, hardware failure, and recurrent instability. 

Conclusion
Traumatic anterior shoulder instability is a common pathology that continues to significantly challenge the readiness of the US military. Military surgeon-researchers have a long history of investigating approaches to the treatment of this pathology—applying good science to a large controlled population, using a single medical record, and demonstrating a commitment to return service members to the ready defense of the nation.
Am J Orthop. 2017;46(4):184-189. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Owens BD, Dawson L, Burks R, Cameron KL. Incidence of shoulder dislocation in the United States military: demographic considerations from a high-risk population. J Bone Joint Surg Am. 2009;91(4):791-796.
2. Owens BD, Duffey ML, Nelson BJ, DeBerardino TM, Taylor DC, Mountcastle SB. The incidence and characteristics of shoulder instability at the United States Military Academy. Am J Sports Med. 2007;35(7):1168-1173.
3. Owens BD, Nelson BJ, Duffey ML, et al. Pathoanatomy of first-time, traumatic, anterior glenohumeral subluxation events. J Bone Joint Surg Am. 2010;92(7):1605-1611.
4. DeBerardino TM, Arciero RA, Taylor DC, Uhorchak JM. Prospective evaluation of arthroscopic stabilization of acute, initial anterior shoulder dislocations in young athletes. Two- to five-year follow-up. Am J Sports Med. 2001;29(5):586-592.
5. Aronen JG, Regan K. Decreasing the incidence of recurrence of first time anterior shoulder dislocations with rehabilitation. Am J Sports Med. 1984;12(4):283-291.
6. Wheeler JH, Ryan JB, Arciero RA, Molinari RN. Arthroscopic versus nonoperative treatment of acute shoulder dislocations in young athletes. Arthroscopy. 1989;5(3):213-217.
7. Bottoni CR, Wilckens JH, DeBerardino TM, et al. A prospective, randomized evaluation of arthroscopic stabilization versus nonoperative treatment in patients with acute, traumatic, first-time shoulder dislocations. Am J Sports Med. 2002;30(4):576-580.
8. Dickens JF, Owens BD, Cameron KL, et al. Return to play and recurrent instability after in-season anterior shoulder instability: a prospective multicenter study. Am J Sports Med. 2014;42(12):2842-2850.
9. Owens BD, DeBerardino TM, Nelson BJ, et al. Long-term follow-up of acute arthroscopic Bankart repair for initial anterior shoulder dislocations in young athletes. Am J Sports Med. 2009;37(4):669-673.
10. DeBerardino TM, Arciero RA, Taylor DC. Arthroscopic stabilization of acute initial anterior shoulder dislocation: the West Point experience. J South Orthop Assoc. 1996;5(4):263-271.
11. Bottoni CR, Smith EL, Berkowitz MJ, Towle RB, Moore JH. Arthroscopic versus open shoulder stabilization for recurrent anterior instability: a prospective randomized clinical trial. Am J Sports Med. 2006;34(11):1730-1737.
12. Owens BD, Cameron KL, Peck KY, et al. Arthroscopic versus open stabilization for anterior shoulder subluxations. Orthop J Sports Med. 2015;3(1):2325967115571084.
13. Burkhart SS, De Beer JF. Traumatic glenohumeral bone defects and their relationship to failure of arthroscopic Bankart repairs: significance of the inverted-pear glenoid and the humeral engaging Hill-Sachs lesion. Arthroscopy. 2000;16(7):677-694.14. Shaha JS, Cook JB, Rowles DJ, Bottoni CR, Shaha SH, Tokish JM. Clinical validation of the glenoid track concept in anterior glenohumeral instability. J Bone Joint Surg Am. 2016;98(22):1918-1923.
15. Mologne TS, Provencher MT, Menzel KA, Vachon TA, Dewing CB. Arthroscopic stabilization in patients with an inverted pear glenoid: results in patients with bone loss of the anterior glenoid. Am J Sports Med. 2007;35(8):1276-1283.
16. Markenstein JE, Jaspars KC, van der Hulst VP, Willems WJ. The quantification of glenoid bone loss in anterior shoulder instability; MR-arthro compared to 3D-CT. Skeletal Radiol. 2014;43(4):475-483.
17. Itoi E, Lee SB, Berglund LJ, Berge LL, An KN. The effect of a glenoid defect on anteroinferior stability of the shoulder after Bankart repair: a cadaveric study. J Bone Joint Surg Am. 2000;82(1):35-46.
18. Shaha JS, Cook JB, Song DJ, et al. Redefining “critical” bone loss in shoulder instability: functional outcomes worsen with “subcritical” bone loss. Am J Sports Med. 2015;43(7):1719-1725.
19. Piasecki DP, Verma NN, Romeo AA, Levine WN, Bach BR Jr, Provencher MT. Glenoid bone deficiency in recurrent anterior shoulder instability: diagnosis and management. J Am Acad Orthop Surg. 2009;17(8):482-493.
20. Provencher MT, Frank RM, Leclere LE, et al. The Hill-Sachs lesion: diagnosis, classification, and management. J Am Acad Orthop Surg. 2012;20(4):242-252.
21. Yamamoto N, Itoi E, Abe H, et al. Contact between the glenoid and the humeral head in abduction, external rotation, and horizontal extension: a new concept of glenoid track. J Shoulder Elbow Surg. 2007;16(5):649-656.
22. Di Giacomo G, Itoi E, Burkhart SS. Evolving concept of bipolar bone loss and the Hill-Sachs lesion: from “engaging/non-engaging” lesion to “on-track/off-track” lesion. Arthroscopy. 2014;30(1):90-98.
23. Metzger PD, Barlow B, Leonardelli D, Peace W, Solomon DJ, Provencher MT. Clinical application of the “glenoid track” concept for defining humeral head engagement in anterior shoulder instability: a preliminary report. Orthop J Sports Med. 2013;1(2):2325967113496213.
24. Arciero RA, Parrino A, Bernhardson AS, et al. The effect of a combined glenoid and Hill-Sachs defect on glenohumeral stability: a biomechanical cadaveric study using 3-dimensional modeling of 142 patients. Am J Sports Med. 2015;43(6):1422-1429.
25. Waterman BR, Chandler PJ, Teague E, Provencher MT, Tokish JM, Pallis MP. Short-term outcomes of glenoid bone block augmentation for complex anterior shoulder instability in a high-risk population. Arthroscopy. 2016;32(9):1784-1790.
26. Schroder DT, Provencher MT, Mologne TS, Muldoon MP, Cox JS. The modified Bristow procedure for anterior shoulder instability: 26-year outcomes in Naval Academy midshipmen. Am J Sports Med. 2006;34(5):778-786.
27. Provencher MT, Frank RM, Golijanin P, et al. Distal tibia allograft glenoid reconstruction in recurrent anterior shoulder instability: clinical and radiographic outcomes. Arthroscopy. 2017;33(5):891-897.
28. Tokish JM, Fitzpatrick K, Cook JB, Mallon WJ. Arthroscopic distal clavicular autograft for treating shoulder instability with glenoid bone loss. Arthrosc Tech. 2014;3(4):e475-e481.
Take-Home Points
- Arthroscopic stabilization performed early results in better outcomes in patients with Bankart lesions.
- A subcritical level of bone loss of 13.5% has been shown to have a significant effect on outcomes, in addition to the established “critical amount”.
- Bone loss is a bipolar issue. Both sides must be considered in order to properly address shoulder instability.
- Off-track measurement has been shown to be even more positively predictive of outcomes than glenoid bone loss assessment.
- There are several bone loss management options including, the most common coracoid transfer, as well as distal tibial allograft and distal clavicular autograft.
Given its relatively young age, high activity level, and centralized medical care system, the US military population is ideal for studying traumatic anterior shoulder instability. There is a long history of military surgeons who have made significant contributions that have advanced our understanding of this pathology and its treatment and results. In this article, we describe the scope, treatment, and results of this pathology in the US military population.
Incidence and Pathology
At the United States Military Academy (USMA), Owens and colleagues1 studied the incidence of shoulder instability, including dislocation and subluxation, and found anterior instability events were far more common than in civilian populations. The incidence of shoulder instability was 0.08 per 1000 person-years in the general US population vs 1.69 per 1000 person-years in US military personnel. The factors associated with increased risk of shoulder instability injury in the military population were male sex, white race, junior enlisted rank, and age under 30 years. Owens and colleagues2 noted that subluxation accounted for almost 85% of the total anterior instability events. Owens and colleagues3 found the pathology in subluxation events was similar to that in full dislocations, with a soft-tissue anterior Bankart lesion and a Hill-Sachs lesion detected on magnetic resonance imaging in more than 90% of patients. In another study at the USMA, DeBerardino and colleagues4 noted that 97% of arthroscopically assessed shoulders in first-time dislocators involved complete detachment of the capsuloligamentous complex from the anterior glenoid rim and neck—a so-called Bankart lesion. Thus, in a military population, anterior instability resulting from subluxation or dislocation is a common finding that is often represented by a soft-tissue Bankart lesion and a Hill-Sachs defect.
Natural History of Traumatic Anterior Shoulder Instability in the Military
Several studies have evaluated the outcomes of nonoperative and operative treatment of shoulder instability. Although most have found better outcomes with operative intervention, Aronen and Regan5 reported good results (25% recurrence at nearly 3-year follow-up) with nonoperative treatment and adherence to a strict rehabilitation program. Most other comparative studies in this population have published contrary results. Wheeler and colleagues6 studied the natural history of anterior shoulder dislocations in a USMA cadet cohort and found recurrent instability after shoulder dislocation in 92% of cadets who had nonoperative treatment. Similarly, DeBerardino and colleagues4 found that, in the USMA, 90% of first-time traumatic anterior shoulder dislocations managed nonoperatively experienced recurrent instability. In a series of Army soldiers with shoulder instability, Bottoni and colleagues7 reported that 75% of nonoperatively managed patients had recurrent instability, and, of these, 67% progressed to surgical intervention. Nonoperative treatment for a first-time dislocation is still reasonable if a cadet or soldier needs to quickly return to functional duties. Athletes who develop shoulder instability during their playing season have been studied in a military population as well. In a multicenter study of service academy athletes with anterior instability, Dickens and colleagues8 found that, with conservative management and accelerated rehabilitation of in-season shoulder instability, 73% of athletes returned to sport by a mean of 5 days. However, the durability of this treatment should be questioned, as 64% later experienced recurrence.
Arthroscopic Stabilization of Acute Anterior Shoulder Dislocations
In an early series of cases of traumatic anterior shoulder instability in USMA cadets, Wheeler and colleagues6 found that, at 14 months, 78% of arthroscopically stabilized cases and 92% of nonoperatively treated cases were successful. Then, in the 1990s, DeBerardino and colleagues4 studied a series of young, active patients in the USMA and noted significantly better results with arthroscopic treatment, vs nonoperative treatment, at 2- to 5-year follow-up. Of the arthroscopically treated shoulders, 88% remained stable during the study and returned to preinjury activity levels, and 12% experienced recurrent instability (risk factors included 2+ sulcus sign, poor capsular labral tissue, and history of bilateral shoulder instability). In a long-term follow-up (mean, 11.7 years; range, 9.1-13.9 years) of the same cohort, Owens and colleagues9 found that 14% of patients available for follow-up had undergone revision stabilization surgery, and, of these, 21% reported experiencing subluxation events. The authors concluded that, in first-time dislocators in this active military population, acute arthroscopic Bankart repair resulted in excellent return to athletics and subjective function, and had acceptable recurrence and reoperation rates. Bottoni and colleagues,7 in a prospective, randomized evaluation of arthroscopic stabilization of acute, traumatic, first-time shoulder dislocations in the Army, noted an 89% success rate for arthroscopic treatment at an average follow-up of 36 months, with no recurrent instability. DeBerardino and colleagues10 compared West Point patients treated nonoperatively with those arthroscopically treated with staples, transglenoid sutures, or bioabsorbable anchors. Recurrence rates were 85% for nonoperative treatment, 22% for staples, 14% for transglenoid sutures, and 10% for bioabsorbable anchors.
Arthroscopic Versus Open Stabilization of Anterior Shoulder Instability
In a prospective, randomized clinical trial comparing open and arthroscopic shoulder stabilization for recurrent anterior instability in active-duty Army personnel, Bottoni and colleagues11 found comparable clinical outcomes. Stabilization surgery failed clinically in only 3 cases, 2 open and 1 arthroscopic. The authors concluded that arthroscopic stabilization can be safely performed for recurrent shoulder instability and that arthroscopic outcomes are similar to open outcomes. In a series of anterior shoulder subluxations in young athletes with Bankart lesions, Owens and colleagues12 found that open and arthroscopic stabilization performed early resulted in better outcomes, regardless of technique used. Recurrent subluxation occurred at a mean of 17 months in 3 of the 10 patients in the open group and 3 of the 9 patients in the arthroscopic group, for an overall recurrence rate of 31%. The authors concluded that, in this patient population with Bankart lesions caused by anterior subluxation events, surgery should be performed early.
Bone Lesions
Burkhart and De Beer13 first noted that bone loss has emerged as one of the most important considerations in the setting of shoulder instability in active patients. Other authors have found this to be true in military populations.14,15
The diagnosis of bone loss may include historical findings, such as increased number and ease of dislocations, as well as dislocation in lower positions of abduction. Physical examination findings may include apprehension in the midrange of motion. Advanced imaging, such as magnetic resonance arthrography, has since been validated as equivalent to 3-dimensional computed tomography (3-D CT) in determining glenoid bone loss.16 In 2007, Mologne and colleagues15 studied the amount of glenoid bone loss and the presence of fragmented bone or attritional bone loss and its effect on outcomes. They evaluated 21 patients who had arthroscopic treatment for anterior instability with anteroinferior glenoid bone loss between 20% and 30%. Average follow-up was 34 months. All patients received 3 or 4 anterior anchors. No patient with a bone fragment incorporated into the repair experienced recurrence or subluxation, whereas 30% of patients with attritional bone loss had recurrent instability.15
Classifying Bone Loss and Recognizing Its Effects
Burkhart and De Beer13 helped define the role and significance of bone loss in the setting of shoulder instability. They defined significant bone loss as an engaging Hill-Sachs lesion of the humerus in an abducted and externally rotated position or an “inverted pear” lesion of the glenoid. Overall analysis revealed recurrence in 4% of cases without significant bone loss and 65% of cases with significant bone loss. In a subanalysis of contact-sport athletes in the setting of bone loss, the failure rate increased to 89%, from 6.5%. Aiding in the quantitative assessment of glenoid bone loss, Itoi and colleagues17 showed that 21% glenoid bone loss resulted in instability that would not be corrected by a soft-tissue procedure alone. Bone loss of 20% to 25% has since been considered a “critical amount,” above which an arthroscopic Bankart has been questioned. More recently, several authors have shown that even less bone loss can have a significant effect on outcomes. Shaha and colleagues18 established that a subcritical level of bone loss (13.5%) on the anteroinferior glenoid resulted in clinical failure (as determined with the Western Ontario Shoulder Instability Index) even in cases in which frank recurrence or subluxation was avoided. It is thought that, in recurrent instability, glenoid bone loss incident rate is as high as 90%, and the corresponding percentage of patients with Hill-Sachs lesions is almost 100%.19,20 Thus, it is increasingly understood that bone loss is a bipolar issue and that both sides must be considered in order to properly address shoulder instability in this setting. In 2007, Yamamoto and colleagues21 introduced the glenoid track, a method for predicting whether a Hill-Sachs lesion will engage. Di Giacomo and colleagues22 refined the track concept to quantitatively determine which lesions will engage in the setting of both glenoid and humeral bone loss. Metzger and colleagues,23 confirming the track concept arthroscopically, found that manipulation with anesthesia and arthroscopic visualization was well predicted by preoperative track measurements, and thus these measurements can be a good guide for surgical management (Figures 1A, 1B).
Strategies for Addressing Bone Loss in Anterior Shoulder Instability
Several approaches for managing bone loss in shoulder instability have been described—the most common being coracoid transfer (Latarjet procedure). Waterman and colleagues25 recently studied the effects of coracoid transfer, distal tibial allograft, and iliac crest augmentation on anterior shoulder instability in US military patients treated between 2006 and 2012. Of 64 patients who underwent a bone block procedure, 16 (25%) had a complication during short-term follow-up. Complications included neurologic injury, pain, infection, hardware failure, and recurrent instability.
Conclusion
Traumatic anterior shoulder instability is a common pathology that continues to significantly challenge the readiness of the US military. Military surgeon-researchers have a long history of investigating approaches to the treatment of this pathology—applying good science to a large controlled population, using a single medical record, and demonstrating a commitment to return service members to the ready defense of the nation.
Am J Orthop. 2017;46(4):184-189. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Arthroscopic stabilization performed early results in better outcomes in patients with Bankart lesions.
- A subcritical level of bone loss of 13.5% has been shown to have a significant effect on outcomes, in addition to the established “critical amount”.
- Bone loss is a bipolar issue. Both sides must be considered in order to properly address shoulder instability.
- Off-track measurement has been shown to be even more positively predictive of outcomes than glenoid bone loss assessment.
- There are several bone loss management options including, the most common coracoid transfer, as well as distal tibial allograft and distal clavicular autograft.
Given its relatively young age, high activity level, and centralized medical care system, the US military population is ideal for studying traumatic anterior shoulder instability. There is a long history of military surgeons who have made significant contributions that have advanced our understanding of this pathology and its treatment and results. In this article, we describe the scope, treatment, and results of this pathology in the US military population.
Incidence and Pathology
At the United States Military Academy (USMA), Owens and colleagues1 studied the incidence of shoulder instability, including dislocation and subluxation, and found anterior instability events were far more common than in civilian populations. The incidence of shoulder instability was 0.08 per 1000 person-years in the general US population vs 1.69 per 1000 person-years in US military personnel. The factors associated with increased risk of shoulder instability injury in the military population were male sex, white race, junior enlisted rank, and age under 30 years. Owens and colleagues2 noted that subluxation accounted for almost 85% of the total anterior instability events. Owens and colleagues3 found the pathology in subluxation events was similar to that in full dislocations, with a soft-tissue anterior Bankart lesion and a Hill-Sachs lesion detected on magnetic resonance imaging in more than 90% of patients. In another study at the USMA, DeBerardino and colleagues4 noted that 97% of arthroscopically assessed shoulders in first-time dislocators involved complete detachment of the capsuloligamentous complex from the anterior glenoid rim and neck—a so-called Bankart lesion. Thus, in a military population, anterior instability resulting from subluxation or dislocation is a common finding that is often represented by a soft-tissue Bankart lesion and a Hill-Sachs defect.
Natural History of Traumatic Anterior Shoulder Instability in the Military
Several studies have evaluated the outcomes of nonoperative and operative treatment of shoulder instability. Although most have found better outcomes with operative intervention, Aronen and Regan5 reported good results (25% recurrence at nearly 3-year follow-up) with nonoperative treatment and adherence to a strict rehabilitation program. Most other comparative studies in this population have published contrary results. Wheeler and colleagues6 studied the natural history of anterior shoulder dislocations in a USMA cadet cohort and found recurrent instability after shoulder dislocation in 92% of cadets who had nonoperative treatment. Similarly, DeBerardino and colleagues4 found that, in the USMA, 90% of first-time traumatic anterior shoulder dislocations managed nonoperatively experienced recurrent instability. In a series of Army soldiers with shoulder instability, Bottoni and colleagues7 reported that 75% of nonoperatively managed patients had recurrent instability, and, of these, 67% progressed to surgical intervention. Nonoperative treatment for a first-time dislocation is still reasonable if a cadet or soldier needs to quickly return to functional duties. Athletes who develop shoulder instability during their playing season have been studied in a military population as well. In a multicenter study of service academy athletes with anterior instability, Dickens and colleagues8 found that, with conservative management and accelerated rehabilitation of in-season shoulder instability, 73% of athletes returned to sport by a mean of 5 days. However, the durability of this treatment should be questioned, as 64% later experienced recurrence.
Arthroscopic Stabilization of Acute Anterior Shoulder Dislocations
In an early series of cases of traumatic anterior shoulder instability in USMA cadets, Wheeler and colleagues6 found that, at 14 months, 78% of arthroscopically stabilized cases and 92% of nonoperatively treated cases were successful. Then, in the 1990s, DeBerardino and colleagues4 studied a series of young, active patients in the USMA and noted significantly better results with arthroscopic treatment, vs nonoperative treatment, at 2- to 5-year follow-up. Of the arthroscopically treated shoulders, 88% remained stable during the study and returned to preinjury activity levels, and 12% experienced recurrent instability (risk factors included 2+ sulcus sign, poor capsular labral tissue, and history of bilateral shoulder instability). In a long-term follow-up (mean, 11.7 years; range, 9.1-13.9 years) of the same cohort, Owens and colleagues9 found that 14% of patients available for follow-up had undergone revision stabilization surgery, and, of these, 21% reported experiencing subluxation events. The authors concluded that, in first-time dislocators in this active military population, acute arthroscopic Bankart repair resulted in excellent return to athletics and subjective function, and had acceptable recurrence and reoperation rates. Bottoni and colleagues,7 in a prospective, randomized evaluation of arthroscopic stabilization of acute, traumatic, first-time shoulder dislocations in the Army, noted an 89% success rate for arthroscopic treatment at an average follow-up of 36 months, with no recurrent instability. DeBerardino and colleagues10 compared West Point patients treated nonoperatively with those arthroscopically treated with staples, transglenoid sutures, or bioabsorbable anchors. Recurrence rates were 85% for nonoperative treatment, 22% for staples, 14% for transglenoid sutures, and 10% for bioabsorbable anchors.
Arthroscopic Versus Open Stabilization of Anterior Shoulder Instability
In a prospective, randomized clinical trial comparing open and arthroscopic shoulder stabilization for recurrent anterior instability in active-duty Army personnel, Bottoni and colleagues11 found comparable clinical outcomes. Stabilization surgery failed clinically in only 3 cases, 2 open and 1 arthroscopic. The authors concluded that arthroscopic stabilization can be safely performed for recurrent shoulder instability and that arthroscopic outcomes are similar to open outcomes. In a series of anterior shoulder subluxations in young athletes with Bankart lesions, Owens and colleagues12 found that open and arthroscopic stabilization performed early resulted in better outcomes, regardless of technique used. Recurrent subluxation occurred at a mean of 17 months in 3 of the 10 patients in the open group and 3 of the 9 patients in the arthroscopic group, for an overall recurrence rate of 31%. The authors concluded that, in this patient population with Bankart lesions caused by anterior subluxation events, surgery should be performed early.
Bone Lesions
Burkhart and De Beer13 first noted that bone loss has emerged as one of the most important considerations in the setting of shoulder instability in active patients. Other authors have found this to be true in military populations.14,15
The diagnosis of bone loss may include historical findings, such as increased number and ease of dislocations, as well as dislocation in lower positions of abduction. Physical examination findings may include apprehension in the midrange of motion. Advanced imaging, such as magnetic resonance arthrography, has since been validated as equivalent to 3-dimensional computed tomography (3-D CT) in determining glenoid bone loss.16 In 2007, Mologne and colleagues15 studied the amount of glenoid bone loss and the presence of fragmented bone or attritional bone loss and its effect on outcomes. They evaluated 21 patients who had arthroscopic treatment for anterior instability with anteroinferior glenoid bone loss between 20% and 30%. Average follow-up was 34 months. All patients received 3 or 4 anterior anchors. No patient with a bone fragment incorporated into the repair experienced recurrence or subluxation, whereas 30% of patients with attritional bone loss had recurrent instability.15
Classifying Bone Loss and Recognizing Its Effects
Burkhart and De Beer13 helped define the role and significance of bone loss in the setting of shoulder instability. They defined significant bone loss as an engaging Hill-Sachs lesion of the humerus in an abducted and externally rotated position or an “inverted pear” lesion of the glenoid. Overall analysis revealed recurrence in 4% of cases without significant bone loss and 65% of cases with significant bone loss. In a subanalysis of contact-sport athletes in the setting of bone loss, the failure rate increased to 89%, from 6.5%. Aiding in the quantitative assessment of glenoid bone loss, Itoi and colleagues17 showed that 21% glenoid bone loss resulted in instability that would not be corrected by a soft-tissue procedure alone. Bone loss of 20% to 25% has since been considered a “critical amount,” above which an arthroscopic Bankart has been questioned. More recently, several authors have shown that even less bone loss can have a significant effect on outcomes. Shaha and colleagues18 established that a subcritical level of bone loss (13.5%) on the anteroinferior glenoid resulted in clinical failure (as determined with the Western Ontario Shoulder Instability Index) even in cases in which frank recurrence or subluxation was avoided. It is thought that, in recurrent instability, glenoid bone loss incident rate is as high as 90%, and the corresponding percentage of patients with Hill-Sachs lesions is almost 100%.19,20 Thus, it is increasingly understood that bone loss is a bipolar issue and that both sides must be considered in order to properly address shoulder instability in this setting. In 2007, Yamamoto and colleagues21 introduced the glenoid track, a method for predicting whether a Hill-Sachs lesion will engage. Di Giacomo and colleagues22 refined the track concept to quantitatively determine which lesions will engage in the setting of both glenoid and humeral bone loss. Metzger and colleagues,23 confirming the track concept arthroscopically, found that manipulation with anesthesia and arthroscopic visualization was well predicted by preoperative track measurements, and thus these measurements can be a good guide for surgical management (Figures 1A, 1B).
Strategies for Addressing Bone Loss in Anterior Shoulder Instability
Several approaches for managing bone loss in shoulder instability have been described—the most common being coracoid transfer (Latarjet procedure). Waterman and colleagues25 recently studied the effects of coracoid transfer, distal tibial allograft, and iliac crest augmentation on anterior shoulder instability in US military patients treated between 2006 and 2012. Of 64 patients who underwent a bone block procedure, 16 (25%) had a complication during short-term follow-up. Complications included neurologic injury, pain, infection, hardware failure, and recurrent instability.
Conclusion
Traumatic anterior shoulder instability is a common pathology that continues to significantly challenge the readiness of the US military. Military surgeon-researchers have a long history of investigating approaches to the treatment of this pathology—applying good science to a large controlled population, using a single medical record, and demonstrating a commitment to return service members to the ready defense of the nation.
Am J Orthop. 2017;46(4):184-189. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Owens BD, Dawson L, Burks R, Cameron KL. Incidence of shoulder dislocation in the United States military: demographic considerations from a high-risk population. J Bone Joint Surg Am. 2009;91(4):791-796.
2. Owens BD, Duffey ML, Nelson BJ, DeBerardino TM, Taylor DC, Mountcastle SB. The incidence and characteristics of shoulder instability at the United States Military Academy. Am J Sports Med. 2007;35(7):1168-1173.
3. Owens BD, Nelson BJ, Duffey ML, et al. Pathoanatomy of first-time, traumatic, anterior glenohumeral subluxation events. J Bone Joint Surg Am. 2010;92(7):1605-1611.
4. DeBerardino TM, Arciero RA, Taylor DC, Uhorchak JM. Prospective evaluation of arthroscopic stabilization of acute, initial anterior shoulder dislocations in young athletes. Two- to five-year follow-up. Am J Sports Med. 2001;29(5):586-592.
5. Aronen JG, Regan K. Decreasing the incidence of recurrence of first time anterior shoulder dislocations with rehabilitation. Am J Sports Med. 1984;12(4):283-291.
6. Wheeler JH, Ryan JB, Arciero RA, Molinari RN. Arthroscopic versus nonoperative treatment of acute shoulder dislocations in young athletes. Arthroscopy. 1989;5(3):213-217.
7. Bottoni CR, Wilckens JH, DeBerardino TM, et al. A prospective, randomized evaluation of arthroscopic stabilization versus nonoperative treatment in patients with acute, traumatic, first-time shoulder dislocations. Am J Sports Med. 2002;30(4):576-580.
8. Dickens JF, Owens BD, Cameron KL, et al. Return to play and recurrent instability after in-season anterior shoulder instability: a prospective multicenter study. Am J Sports Med. 2014;42(12):2842-2850.
9. Owens BD, DeBerardino TM, Nelson BJ, et al. Long-term follow-up of acute arthroscopic Bankart repair for initial anterior shoulder dislocations in young athletes. Am J Sports Med. 2009;37(4):669-673.
10. DeBerardino TM, Arciero RA, Taylor DC. Arthroscopic stabilization of acute initial anterior shoulder dislocation: the West Point experience. J South Orthop Assoc. 1996;5(4):263-271.
11. Bottoni CR, Smith EL, Berkowitz MJ, Towle RB, Moore JH. Arthroscopic versus open shoulder stabilization for recurrent anterior instability: a prospective randomized clinical trial. Am J Sports Med. 2006;34(11):1730-1737.
12. Owens BD, Cameron KL, Peck KY, et al. Arthroscopic versus open stabilization for anterior shoulder subluxations. Orthop J Sports Med. 2015;3(1):2325967115571084.
13. Burkhart SS, De Beer JF. Traumatic glenohumeral bone defects and their relationship to failure of arthroscopic Bankart repairs: significance of the inverted-pear glenoid and the humeral engaging Hill-Sachs lesion. Arthroscopy. 2000;16(7):677-694.14. Shaha JS, Cook JB, Rowles DJ, Bottoni CR, Shaha SH, Tokish JM. Clinical validation of the glenoid track concept in anterior glenohumeral instability. J Bone Joint Surg Am. 2016;98(22):1918-1923.
15. Mologne TS, Provencher MT, Menzel KA, Vachon TA, Dewing CB. Arthroscopic stabilization in patients with an inverted pear glenoid: results in patients with bone loss of the anterior glenoid. Am J Sports Med. 2007;35(8):1276-1283.
16. Markenstein JE, Jaspars KC, van der Hulst VP, Willems WJ. The quantification of glenoid bone loss in anterior shoulder instability; MR-arthro compared to 3D-CT. Skeletal Radiol. 2014;43(4):475-483.
17. Itoi E, Lee SB, Berglund LJ, Berge LL, An KN. The effect of a glenoid defect on anteroinferior stability of the shoulder after Bankart repair: a cadaveric study. J Bone Joint Surg Am. 2000;82(1):35-46.
18. Shaha JS, Cook JB, Song DJ, et al. Redefining “critical” bone loss in shoulder instability: functional outcomes worsen with “subcritical” bone loss. Am J Sports Med. 2015;43(7):1719-1725.
19. Piasecki DP, Verma NN, Romeo AA, Levine WN, Bach BR Jr, Provencher MT. Glenoid bone deficiency in recurrent anterior shoulder instability: diagnosis and management. J Am Acad Orthop Surg. 2009;17(8):482-493.
20. Provencher MT, Frank RM, Leclere LE, et al. The Hill-Sachs lesion: diagnosis, classification, and management. J Am Acad Orthop Surg. 2012;20(4):242-252.
21. Yamamoto N, Itoi E, Abe H, et al. Contact between the glenoid and the humeral head in abduction, external rotation, and horizontal extension: a new concept of glenoid track. J Shoulder Elbow Surg. 2007;16(5):649-656.
22. Di Giacomo G, Itoi E, Burkhart SS. Evolving concept of bipolar bone loss and the Hill-Sachs lesion: from “engaging/non-engaging” lesion to “on-track/off-track” lesion. Arthroscopy. 2014;30(1):90-98.
23. Metzger PD, Barlow B, Leonardelli D, Peace W, Solomon DJ, Provencher MT. Clinical application of the “glenoid track” concept for defining humeral head engagement in anterior shoulder instability: a preliminary report. Orthop J Sports Med. 2013;1(2):2325967113496213.
24. Arciero RA, Parrino A, Bernhardson AS, et al. The effect of a combined glenoid and Hill-Sachs defect on glenohumeral stability: a biomechanical cadaveric study using 3-dimensional modeling of 142 patients. Am J Sports Med. 2015;43(6):1422-1429.
25. Waterman BR, Chandler PJ, Teague E, Provencher MT, Tokish JM, Pallis MP. Short-term outcomes of glenoid bone block augmentation for complex anterior shoulder instability in a high-risk population. Arthroscopy. 2016;32(9):1784-1790.
26. Schroder DT, Provencher MT, Mologne TS, Muldoon MP, Cox JS. The modified Bristow procedure for anterior shoulder instability: 26-year outcomes in Naval Academy midshipmen. Am J Sports Med. 2006;34(5):778-786.
27. Provencher MT, Frank RM, Golijanin P, et al. Distal tibia allograft glenoid reconstruction in recurrent anterior shoulder instability: clinical and radiographic outcomes. Arthroscopy. 2017;33(5):891-897.
28. Tokish JM, Fitzpatrick K, Cook JB, Mallon WJ. Arthroscopic distal clavicular autograft for treating shoulder instability with glenoid bone loss. Arthrosc Tech. 2014;3(4):e475-e481.
1. Owens BD, Dawson L, Burks R, Cameron KL. Incidence of shoulder dislocation in the United States military: demographic considerations from a high-risk population. J Bone Joint Surg Am. 2009;91(4):791-796.
2. Owens BD, Duffey ML, Nelson BJ, DeBerardino TM, Taylor DC, Mountcastle SB. The incidence and characteristics of shoulder instability at the United States Military Academy. Am J Sports Med. 2007;35(7):1168-1173.
3. Owens BD, Nelson BJ, Duffey ML, et al. Pathoanatomy of first-time, traumatic, anterior glenohumeral subluxation events. J Bone Joint Surg Am. 2010;92(7):1605-1611.
4. DeBerardino TM, Arciero RA, Taylor DC, Uhorchak JM. Prospective evaluation of arthroscopic stabilization of acute, initial anterior shoulder dislocations in young athletes. Two- to five-year follow-up. Am J Sports Med. 2001;29(5):586-592.
5. Aronen JG, Regan K. Decreasing the incidence of recurrence of first time anterior shoulder dislocations with rehabilitation. Am J Sports Med. 1984;12(4):283-291.
6. Wheeler JH, Ryan JB, Arciero RA, Molinari RN. Arthroscopic versus nonoperative treatment of acute shoulder dislocations in young athletes. Arthroscopy. 1989;5(3):213-217.
7. Bottoni CR, Wilckens JH, DeBerardino TM, et al. A prospective, randomized evaluation of arthroscopic stabilization versus nonoperative treatment in patients with acute, traumatic, first-time shoulder dislocations. Am J Sports Med. 2002;30(4):576-580.
8. Dickens JF, Owens BD, Cameron KL, et al. Return to play and recurrent instability after in-season anterior shoulder instability: a prospective multicenter study. Am J Sports Med. 2014;42(12):2842-2850.
9. Owens BD, DeBerardino TM, Nelson BJ, et al. Long-term follow-up of acute arthroscopic Bankart repair for initial anterior shoulder dislocations in young athletes. Am J Sports Med. 2009;37(4):669-673.
10. DeBerardino TM, Arciero RA, Taylor DC. Arthroscopic stabilization of acute initial anterior shoulder dislocation: the West Point experience. J South Orthop Assoc. 1996;5(4):263-271.
11. Bottoni CR, Smith EL, Berkowitz MJ, Towle RB, Moore JH. Arthroscopic versus open shoulder stabilization for recurrent anterior instability: a prospective randomized clinical trial. Am J Sports Med. 2006;34(11):1730-1737.
12. Owens BD, Cameron KL, Peck KY, et al. Arthroscopic versus open stabilization for anterior shoulder subluxations. Orthop J Sports Med. 2015;3(1):2325967115571084.
13. Burkhart SS, De Beer JF. Traumatic glenohumeral bone defects and their relationship to failure of arthroscopic Bankart repairs: significance of the inverted-pear glenoid and the humeral engaging Hill-Sachs lesion. Arthroscopy. 2000;16(7):677-694.14. Shaha JS, Cook JB, Rowles DJ, Bottoni CR, Shaha SH, Tokish JM. Clinical validation of the glenoid track concept in anterior glenohumeral instability. J Bone Joint Surg Am. 2016;98(22):1918-1923.
15. Mologne TS, Provencher MT, Menzel KA, Vachon TA, Dewing CB. Arthroscopic stabilization in patients with an inverted pear glenoid: results in patients with bone loss of the anterior glenoid. Am J Sports Med. 2007;35(8):1276-1283.
16. Markenstein JE, Jaspars KC, van der Hulst VP, Willems WJ. The quantification of glenoid bone loss in anterior shoulder instability; MR-arthro compared to 3D-CT. Skeletal Radiol. 2014;43(4):475-483.
17. Itoi E, Lee SB, Berglund LJ, Berge LL, An KN. The effect of a glenoid defect on anteroinferior stability of the shoulder after Bankart repair: a cadaveric study. J Bone Joint Surg Am. 2000;82(1):35-46.
18. Shaha JS, Cook JB, Song DJ, et al. Redefining “critical” bone loss in shoulder instability: functional outcomes worsen with “subcritical” bone loss. Am J Sports Med. 2015;43(7):1719-1725.
19. Piasecki DP, Verma NN, Romeo AA, Levine WN, Bach BR Jr, Provencher MT. Glenoid bone deficiency in recurrent anterior shoulder instability: diagnosis and management. J Am Acad Orthop Surg. 2009;17(8):482-493.
20. Provencher MT, Frank RM, Leclere LE, et al. The Hill-Sachs lesion: diagnosis, classification, and management. J Am Acad Orthop Surg. 2012;20(4):242-252.
21. Yamamoto N, Itoi E, Abe H, et al. Contact between the glenoid and the humeral head in abduction, external rotation, and horizontal extension: a new concept of glenoid track. J Shoulder Elbow Surg. 2007;16(5):649-656.
22. Di Giacomo G, Itoi E, Burkhart SS. Evolving concept of bipolar bone loss and the Hill-Sachs lesion: from “engaging/non-engaging” lesion to “on-track/off-track” lesion. Arthroscopy. 2014;30(1):90-98.
23. Metzger PD, Barlow B, Leonardelli D, Peace W, Solomon DJ, Provencher MT. Clinical application of the “glenoid track” concept for defining humeral head engagement in anterior shoulder instability: a preliminary report. Orthop J Sports Med. 2013;1(2):2325967113496213.
24. Arciero RA, Parrino A, Bernhardson AS, et al. The effect of a combined glenoid and Hill-Sachs defect on glenohumeral stability: a biomechanical cadaveric study using 3-dimensional modeling of 142 patients. Am J Sports Med. 2015;43(6):1422-1429.
25. Waterman BR, Chandler PJ, Teague E, Provencher MT, Tokish JM, Pallis MP. Short-term outcomes of glenoid bone block augmentation for complex anterior shoulder instability in a high-risk population. Arthroscopy. 2016;32(9):1784-1790.
26. Schroder DT, Provencher MT, Mologne TS, Muldoon MP, Cox JS. The modified Bristow procedure for anterior shoulder instability: 26-year outcomes in Naval Academy midshipmen. Am J Sports Med. 2006;34(5):778-786.
27. Provencher MT, Frank RM, Golijanin P, et al. Distal tibia allograft glenoid reconstruction in recurrent anterior shoulder instability: clinical and radiographic outcomes. Arthroscopy. 2017;33(5):891-897.
28. Tokish JM, Fitzpatrick K, Cook JB, Mallon WJ. Arthroscopic distal clavicular autograft for treating shoulder instability with glenoid bone loss. Arthrosc Tech. 2014;3(4):e475-e481.
Bone Stress Injuries in the Military: Diagnosis, Management, and Prevention
Take-Home Points
- Stress injuries, specifically of the lower extremity, are very common in new military trainees.
- Stress injury can range from benign periosteal reaction to displaced fracture.
- Stress injury should be treated on a case-by-case basis, depending on the severity of injury, the location of the injury, and the likelihood of healing with nonoperative management.
- Modifiable risk factors such as nutritional status, training regiment, and even footwear should be investigated to determine potential causes of injury.
- Prevention is a crucial part of the treatment of these injuries, and early intervention such as careful pre-enrollment physicals and vitamin supplementation can be essential in lowering injury rates.
Bone stress injuries, which are common in military recruits, present in weight-bearing (WB) areas as indolent pain caused by repetitive stress and microtrauma. They were first reported in the metatarsals of Prussian soldiers in 1855.1 Today, stress injuries are increasingly common. One study estimated they account for 10% of patients seen by sports medicine practitioners.2 This injury most commonly affects military members, endurance athletes, and dancers.3-5 Specifically, the incidence of stress fractures in military members has been reported to range from 0.8% to 6.9% for men and from 3.4% to 21.0% for women.4 Because of repetitive vigorous lower extremity loading, stress fractures typically occur in the pelvis, femoral neck, tibial shaft, and metatarsals. Delayed diagnosis and the subsequent duration of treatment required for adequate healing can result in significant morbidity. In a 2009 to 2012 study of US military members, Waterman and colleagues6 found an incidence rate of 5.69 stress fractures per 1000 person-years. Fractures most frequently involved the tibia/fibula (2.26/1000), followed by the metatarsals (0.92/1000) and the femoral neck (0.49/1000).6 In addition, these injuries were most commonly encountered in new recruits, who were less accustomed to the high-volume, high-intensity training required during basic training.4,7 Enlisted junior service members have been reported to account for 77.5% of all stress fractures.6 Age under 20 years or over 40 years and white race have also been found to be risk factors for stress injury.6
The pathogenesis of stress injury is controversial. Stanitski and colleagues8 theorized that multiple submaximal mechanical insults create cumulative stress greater than bone capacity, eventually leading to fracture. Johnson9 conducted a biopsy study and postulated that an accelerated remodeling phase was responsible, whereas Friedenberg10 argued that stress injuries are a form of reduced healing, not an attempt to increase healing, caused by the absence of callous formation in the disease process.
Various other nonmodifiable and modifiable risk factors predispose military service members to stress injury. Nonmodifiable risk factors include sex, bone geometry, limb alignment, race, age, and anatomy. Lower extremity movement biomechanics resulting from dynamic limb alignment during activity may be important. Cameron and colleagues11 examined 1843 patients and found that those with knees in >5° of valgus or >5° of external rotation had higher injury rates. Although variables such as sex and limb alignment cannot be changed, proper identification of modifiable risk factors can assist with injury prevention, and nonmodifiable risk factors can help clinicians and researchers target injury prevention interventions to patients at highest risk.
Metabolic, hormonal, and nutritional status is crucial to overall bone health. Multiple studies have found that low body mass index (BMI) is a significant risk factor for stress fracture.7,12,13 Although low BMI is a concern, patients with abnormally high BMI may also be at increased risk for bone stress injury. In a recently released consensus statement on relative energy deficiency in sport (RED-S), the International Olympic Committee addressed the complex interplay of impairments in physiologic function—including metabolic rate, menstrual function, bone health, immunity, protein synthesis, and cardiovascular health—caused by relative energy deficiency.14 The committee stated that the cause of this syndrome is energy deficiency relative to the balance between dietary energy intake and energy expenditure required for health and activities of daily living, growth, and sporting activities. This finding reveals that conditions such as stress injury often may represent a much broader systemic deficit that may be influenced by a patient’s overall physiologic imbalance.
Diagnosis
History and Physical Examination
The onset of stress reaction typically is insidious, with the classic presentation being a new military recruit who is experiencing a sudden increase in pain during physical activity.15 Pain typically is initially present only during activity, and is relieved with rest, but with disease progression this evolves to pain at rest. It is crucial that the physician elicit the patient’s history of training and physical activity. Hsu and colleagues7 reported increased prevalence of overweight civilian recruits, indicating an increase in the number of new recruits having limited experience with the repetitive physical activity encountered in basic training. Stress injury should be suspected in the setting of worsening, indolent lower extremity pain that has been present for several days, especially in the higher-risk patient populations mentioned. Diet should be assessed, with specific attention given to the intake of fruits, vegetables, and foods high in vitamin D and calcium and, most important, the energy balance between intake and output.16 Special attention should also be given to female patients, who may experience the female athlete triad, a spectrum of low energy availability, menstrual dysfunction, and impaired bone turnover (high amount of resorption relative to formation). A key part of the RED-S consensus statement14 alerted healthcare providers that metabolic derangements do not solely affect female patients. These types of patients sustain a major insult to the homeostatic balance of the hormones that sustain adequate bone health. Beck and colleagues17 found that women with disrupted menstrual cycles are 2 to 4 times more likely to sustain a stress fracture than women without disrupted menstrual cycles, making this abnormality an important part of the history.
Examination should begin with careful evaluation of limb alignment and specific attention given to varus or valgus alignment of the knees.11 The feet should also be inspected, as pes planus or cavus foot may increase the risk of stress fracture.18 Identification of the area of maximal tenderness is important. The area in question may also be erythematous or warm secondary to the inflammatory response associated with attempted fracture healing. In chronic fractures in superficial areas such as the metatarsals, callus may be palpable. Although there are few specific tests for stress injury, pain may be reproducible with deep palpation and WB. 
Laboratory Testing
When a pathology is thought to have a nutritional or metabolic cause, particularly in a low-weight or underweight patient, a laboratory workup should be obtained. Specific laboratory tests that all patients should undergo are 25-hydroxyvitamin D3, complete blood cell count, and basic chemistry panel, including calcium and thyroid-stimulating hormone levels. Although not necessary for diagnosis, phosphate, parathyroid hormone, albumin, and prealbumin should also be considered. Females should undergo testing of follicle stimulating hormone, luteinizing hormone, estradiol, and testosterone and have a urine pregnancy test. In patients with signs of excessive cortisone, a dexamethasone suppression test can be administered.21 In males, low testosterone is a documented risk factor for stress injury.22
Imaging
Given their low cost and availability, plain radiographs typically are used for initial examination of a suspected stress injury. However, they often lack sensitivity, particularly in the early stages of stress fracture development (Figure 2). 
Management
Management of bone stress injury depends on many factors, including symptom duration, fracture location and severity, and risk of progression or nonunion (Table).13 
Pelvis
Pelvic stress fractures are rare and represent only 1.6% to 7.1% of all stress fractures.13,27,28 Given the low frequency, physicians must have a high index of suspicion to make the correct diagnosis. These fractures typically occur in marathon runners and other patients who present with persistent pain and a history of high levels of activity. As pelvic stress fractures typically involve the superior or inferior pubic rami, or sacrum, and are at low risk for nonunion,13 most are managed with nonoperative treatment and activity modification for 8 to 12 weeks.27
Femur
Femoral stress fractures are also relatively uncommon, accounting for about 10% of all stress fractures. Depending on their location, these fractures can be at high risk for progression, nonunion, and significant morbidity.29 Especially concerning are femoral neck stress fractures, which can involve either the tension side (lateral cortex) or the compression side (medial cortex) of the bone. Suspicion of a femoral neck stress fracture should prompt immediate NWB.5 Early recognition of these injuries is crucial because once displacement occurs, their complication and morbidity rates become high.13 Patients with compression-side fractures should undergo NWB treatment for 4 to 6 weeks and then slow progression to WB activity. Most return to light-impact activity by 3 to 4 months. By contrast, tension-side fractures are less likely to heal without operative intervention.11 All tension-side fractures (and any compression-side fractures >50% of the width of the femoral neck) should be treated with percutaneous placement of cannulated screws (Figure 3). 
Stress fractures of the femoral shaft are less common than those of the femoral neck and represent as little as 3% of all stress fractures.32 However, femoral shaft stress fractures are more common in military populations. In French military recruits, Niva and colleagues33 found an 18% incidence. Similar to femoral neck fractures, femoral shaft fractures typically are diagnosed with advanced imaging, though the fulcrum test and pain on WB can aid in the diagnosis.19 These injuries are often managed nonoperatively with NWB for a period. Weishaar and colleagues34 described US military cadets treated with progressive rehabilitation who returned to full activity within 12 weeks. Displaced femoral shaft fractures associated with bone stress injury are even less common, and should be managed operatively. Salminen and colleagues35 found an incidence of 1.5 fractures per 100,000 years of military service. Over a 20-year period, they surgically treated 10 of these fractures. Average time from intramedullary nailing to union was 3.5 months.
Tibia
The tibia is one of the more common locations for stress injury and fracture. In a prospective study with members of the military, Giladi and colleagues36 found that 71% of stress fractures were tibia fractures. In addition, a large study of 320 athletes with stress fractures found 49.1% in the tibia.37 Fractures typically are diaphyseal and transverse, usually occurring along the posteromedial cortex, where the bone experiences maximal compressive forces (Figure 4).5,13

Compression-side fractures often heal with nonoperative management, though healing may take several months. Swenson and colleagues40 studied the effects of pneumatic bracing on conservative management and return to play in athletes with tibial stress fractures. Patients with bracing returned to light activity within 7 days and full activity within 21 days, whereas those without bracing returned to light activity within 21 days and full activity within 77 days. Pulsed electromagnetic therapy is of controversial benefit in the management of these injuries. Rettig
Metatarsals
Stress fractures were first discovered by Briethaupt1 in the painful swollen feet of Prussian army members in 1855 and were initially named march fractures. Waterman and colleagues6 reported that metatarsal stress fractures accounted for 16% of all stress fractures in the US military between 2009 and 2012. The second metatarsal neck is the most common location for stress fractures, followed by the third and fourth metatarsals, with the fifth metatarsal being the least common.5 The second metatarsal is thought to sustain these injuries more often than the other metatarsals because of its relative lack of immobility. Donahue and Sharkey43 found that the dorsal aspect of the second metatarsal experiences twice the amount of strain experienced by the fifth metatarsal during gait, and that peak strain in the second metatarsal was further increased by simulated muscle fatigue. The risk of stress fracture can be additionally increased with use of minimalist footwear, as shown by Giuliani and colleagues,44 particularly in the absence of a progressive transition in gait and training volume with a change toward minimalist footwear. In patients with a suspected or confirmed fracture of the second, third, or fourth metatarsal, treatment typically is NWB and immobilization for at least 4 weeks.5 Fifth metatarsal stress injuries (Figure 2) typically are treated differently because of their higher risk of nonunion. Patients with a fifth metatarsal stress fracture complain of lateral midfoot pain with running and jumping. For those who present with this fracture early, acceptable treatment consists of 6 weeks of casting and NWB.5 In cases of failed nonoperative therapy, or presentation with radiographic evidence of nonunion, treatment should be intramedullary screw fixation, with bone graft supplementation based on surgeon preference. DeLee and colleagues45 reported on the results of 10 athletes with fifth metatarsal stress fractures treated with intramedullary screw fixation without bone grafting. All 10 experienced fracture union, at a mean of 7.5 weeks, and returned to sport within 8.5 weeks. One complication with this procedure is pain at the screw insertion site, but this can be successfully managed with footwear modification.45
Prevention
Proper identification of patients at high risk for stress injuries has the potential of reducing the incidence of these injuries. Lappe and colleagues46 prospectively examined female army recruits before and after 8 weeks of basic training and found that those who developed a stress fracture were more likely to have a smoking history, to drink more than 10 alcoholic beverages a week, to have a history of corticosteroid or depot medroxyprogesterone use, and to have lower body weight. In addition, the authors found that a history of prolonged exercise before enrollment was protective against fracture. This finding identifies the importance of having new recruits undergo risk factor screening, which could result in adjusting training regimens to try to reduce injury. The RED-S consensus statement14 offers a comprehensive description of the physiologic factors that can contribute to such injury. Similar to proper risk factor identification, implementation of proper exercise progression programs is a simple, modifiable method of limiting stress injuries. For new recruits or athletes who are resuming activity, injury can be effectively prevented by adjusting the frequency, duration, and intensity of training and the training loads used.47
Vitamin D and calcium supplementation is a simple intervention that can be helpful in injury prevention, and its use has very little downside. A double-blind study found a 20% lower incidence of stress fracture in female navy recruits who took 2000 mg of calcium and 800 IU of vitamin D as daily supplemention.48 Of importance, a meta-analysis of more than 65,000 patients found vitamin D supplementation was effective in reducing fracture risk only when combined with calcium, irrespective of age, sex, or prior fracture.49 In female patients with the female athlete triad, psychological counseling and nutritional consultation are essential in bone health maintenance and long-term prevention.50 Other therapies have been evaluated as well. Use of bisphosphonates is controversial for both treatment and prevention of stress fractures. In a randomized, double-blind study of the potential prophylactic effects of risedronate in 324 new infantry recruits, Milgrom and colleagues51 found no statistically significant differences in tibial, femoral, metatarsal, or total stress fracture incidence between the treatment and placebo groups. Therefore, bisphosphonates are seldom recommended as prevention or in primary management of stress fracture.
In addition to nutritional and pharmacologic therapy, activity modification may have a role in injury prevention. Gait retraining has been identified as a potential intervention for reducing stress fractures in patients with poor biomechanics.47 Crowell and Davis52 investigated the effect of gait retraining on the forces operating in the tibia in runners. After 1 month of gait retraining, tibial acceleration while running decreased by 50%, vertical force loading rate by 30%, and peak vertical force impact by 20%. Such studies indicate the importance of proper mechanics during repetitive activity, especially in patients not as accustomed to the rigorous training methods used with new military recruits. However, whether these reduced loads translate into reduced risk of stress fracture remains unclear. In addition, biomechanical shoe orthoses may lower the stress fracture risk in military recruits by reducing peak tibial strain.53 Warden and colleagues54 found a mechanical loading program was effective in enchaining the structural properties of bone in rats, leading the authors to hypothesize that a similar program aimed at modifying bone structure in humans could help prevent stress fracture. Although there have been no studies of such a strategy in humans, pretraining may be an area for future research, especially for military recruits.
Conclusion
Compared with the general population, members of the military (new recruits in particular) are at increased risk for bone stress injuries. Most of these injuries occur during basic training, when recruits significantly increase their repetitive physical activity. Although the exact pathophysiology of stress injury is debated, nutritional and metabolic abnormalities are contributors. The indolent nature of these injuries, and their high rate of false-negative plain radiographs, may result in a significant delay in diagnosis in the absence of advanced imaging studies. Although a majority of injuries heal with nonoperative management and NWB, several patterns, especially those on the tension side of the bone, are at high risk for progression to fracture and nonunion. These include lateral femoral cortex stress injuries and anterior tibial cortex fractures. There should be a low threshold for operative management in the setting of delayed union or failed nonoperative therapy. Of equal importance to orthopedic management of these injuries is the management of underlying systemic deficits, which may have subjected the patient to injury in the first place. Supplementation with vitamin D and calcium can be an important prophylaxis against stress injury. In addition, military recruits and athletes with underlying metabolic or hormonal deficiencies should receive proper attention with a focus on balancing energy intake and energy expenditure. Stress injury leading to fracture—increasingly common in military populations—often requires a multimodal approach for treatment and subsequent prevention.
Am J Orthop. 2017;46(4):176-183. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Briethaupt MD. Zur Pathologie des menschlichen Fusses [To the pathology of the human foot]. Med Zeitung. 1855;24:169-177.
2. Berger FH, de Jonge MC, Maas M. Stress fractures in the lower extremity. Eur J Radiol. 2007;62(1):16-26.
3. Almeida SA, Williams KM, Shaffer RA, Brodine SK. Epidemiological patterns of musculoskeletal injuries and physical training. Med Sci Sports Exerc. 1999;31(8):1176-1182.
4. Jones BH, Thacker SB, Gilchrist J, Kimsey CD, Sosin DM. Prevention of lower extremity stress fractures in athletes and soldiers: a systematic review. Epidemiol Rev. 2002;24(2):228-247.
5. Jacobs JM, Cameron KL, Bojescul JA. Lower extremity stress fractures in the military. Clin Sports Med. 2014;33(4):591-613.
6. Waterman BR, Gun B, Bader JO, Orr JD, Belmont PJ. Epidemiology of lower extremity stress fractures in the United States military. Mil Med. 2016;181(10):1308-1313.
7. Hsu LL, Nevin RL, Tobler SK, Rubertone MV. Trends in overweight and obesity among 18-year-old applicants to the United States military, 1993–2006. J Adolesc Health. 2007;41(6):610-612.
8. Stanitski CL, McMaster JH, Scranton PE. On the nature of stress fractures. Am J Sports Med. 1978;6(6):391-396.
9. Johnson LC. Histogenesis of stress fractures [annual lecture]. Washington, DC: Armed Forces Institute of Pathology; 1963.
10. Friedenberg ZB. Fatigue fractures of the tibia. Clin Orthop Relat Res. 1971;(76):111-115.
11. Cameron KL, Peck KY, Owens BD, et al. Biomechanical risk factors for lower extremity stress fracture. Orthop J Sports Med. 2013;1(4 suppl).
12. Knapik J, Montain S, McGraw S, Grier T, Ely M, Jones B. Stress fracture risk factors in basic combat training. Int J Sports Med. 2012;33(11):940-946.
13. Behrens SB, Deren ME, Matson A, Fadale PD, Monchik KO. Stress fractures of the pelvis and legs in athletes. Sports Health. 2013;5(2):165-174.
14. Mountjoy M, Sundgot-Borgen J, Burke L, et al. The IOC consensus statement: beyond the female athlete triad—relative energy deficiency in sport (RED-S). Br J Sports Med. 2014;48(7):491-497.
15. Maitra RS, Johnson DL. Stress fractures. Clinical history and physical examination. Clin Sports Med. 1997;16(2):259-274.
16. Nieves JW, Melsop K, Curtis M, et al. Nutritional factors that influence change in bone density and stress fracture risk among young female cross-country runners. PM R. 2010;2(8):740-750.
17. Beck BR, Matheson GO, Bergman G, et al. Do capacitively coupled electric fields accelerate tibial stress fracture healing? Am J Sports Med. 2008;36(3):545-553.
18. Simkin A, Leichter I, Giladi M, Stein M, Milgrom C. Combined effect of foot arch structure and an orthotic device on stress fractures. Foot Ankle. 1989;10(1):25-29.
19. Johnson AW, Weiss CB, Wheeler DL. Stress fractures of the femoral shaft in athletes—more common than expected: a new clinical test. Am J Sports Med. 1994;22(2):248-256.
20. Clement D, Ammann W, Taunton J, et al. Exercise-induced stress injuries to the femur. Int J Sports Med. 1993;14(6):347-352.
21. Wood PJ, Barth JH, Freedman DB, Perry L, Sheridan B. Evidence for the low dose dexamethasone suppression test to screen for Cushing’s syndrome—recommendations for a protocol for biochemistry laboratories. Ann Clin Biochem. 1997;34(pt 3):222-229.
22. Bennell K, Matheson G, Meeuwisse W, Brukner P. Risk factors for stress fractures. Sports Med. 1999;28(2):91-122.
23. Prather JL, Nusynowitz ML, Snowdy HA, Hughes AD, McCartney WH, Bagg RJ. Scintigraphic findings in stress fractures. J Bone Joint Surg Am. 1977;59(7):869-874.
24. Arendt EA, Griffiths HJ. The use of MR imaging in the assessment and clinical management of stress reactions of bone in high-performance athletes. Clin Sports Med. 1997;16(2):291-306.
25. Boden BP, Osbahr DC. High-risk stress fractures: evaluation and treatment. J Am Acad Orthop Surg. 2000;8(6):344-353.
26. Gaeta M, Minutoli F, Scribano E, et al. CT and MR imaging findings in athletes with early tibial stress injuries: comparison with bone scintigraphy findings and emphasis on cortical abnormalities. Radiology. 2005;235(2):553-561.
27. Matheson GO, Clement DB, Mckenzie DC, Taunton JE, Lloyd-Smith DR, Macintyre JG. Stress fractures in athletes. Am J Sports Med. 1987;15(1):46-58.
28. Iwamoto J, Takeda T. Stress fractures in athletes: review of 196 cases. J Orthop Sci. 2003;8(3):273-278.
29. Noakes TD, Smith JA, Lindenberg G, Wills CE. Pelvic stress fractures in long distance runners. Am J Sports Med. 1985;13(2):120-123.
30. Neubauer T, Brand J, Lidder S, Krawany M. Stress fractures of the femoral neck in runners: a review. Res Sports Med. 2016;24(3):283-297.
31. Evans JT, Guyver PM, Kassam AM, Hubble MJW. Displaced femoral neck stress fractures in Royal Marine recruits—management and results of operative treatment. J R Nav Med Serv. 2012;98(2):3-5.
32. Orava S. Stress fractures. Br J Sports Med. 1980;14(1):40-44.
33. Niva MH, Kiuru MJ, Haataja R, Pihlajamäki HK. Fatigue injuries of the femur. J Bone Joint Surg Br. 2005;87(10):1385-1390.
34. Weishaar MD, McMillian DJ, Moore JH. Identification and management of 2 femoral shaft stress injuries. J Orthop Sports Phys Ther. 2005;35(10):665-673.
35. Salminen ST, Pihlajamäki HK, Visuri TI, Böstman OM. Displaced fatigue fractures of the femoral shaft. Clin Orthop Relat Res. 2003;(409):250-259.
36. Giladi M, Ahronson Z, Stein M, Danon YL, Milgrom C. Unusual distribution and onset of stress fractures in soldiers. Clin Orthop Relat Res. 1985;(192):142-146.
37. Matheson GO, Clement DB, Mckenzie DC, Taunton JE, Lloyd-Smith DR, Macintyre JG. Stress fractures in athletes. Am J Sports Med. 1987;15(1):46-58.
38. Green NE, Rogers RA, Lipscomb AB. Nonunions of stress fractures of the tibia. Am J Sports Med. 1985;13(3):171-176.
39. Orava S, Hulkko A. Stress fracture of the mid-tibial shaft. Acta Orthop Scand. 1984;55(1):35-37.
40. Swenson EJ Jr, DeHaven KE, Sebastianelli WJ, Hanks G, Kalenak A, Lynch JM. The effect of a pneumatic leg brace on return to play in athletes with tibial stress fractures. Am J Sports Med. 1997;25(3):322-328.
41. Rettig AC, Shelbourne KD, McCarroll JR, Bisesi M, Watts J. The natural history and treatment of delayed union stress fractures of the anterior cortex of the tibia. Am J Sports Med. 1988;16(3):250-255.
42. Varner KE, Younas SA, Lintner DM, Marymont JV. Chronic anterior midtibial stress fractures in athletes treated with reamed intramedullary nailing. Am J Sports Med. 2005;33(7):1071-1076.
43. Donahue SW, Sharkey NA. Strains in the metatarsals during the stance phase of gait: implications for stress fractures. J Bone Joint Surg Am. 1999;81(9):1236-1244.
44. Giuliani J, Masini B, Alitz C, Owens BD. Barefoot-simulating footwear associated with metatarsal stress injury in 2 runners. Orthopedics. 2011;34(7):e320-e323.
45. DeLee JC, Evans JP, Julian J. Stress fracture of the fifth metatarsal. Am J Sports Med. 1983;11(5):349-353.
46. Lappe JM, Stegman MR, Recker RR. The impact of lifestyle factors on stress fractures in female army recruits. Osteoporos Int. 2001;12(1):35-42.
47. Friedl KE, Evans RK, Moran DS. Stress fracture and military medical readiness: bridging basic and applied research. Med Sci Sports Exerc. 2008;40(11 suppl):S609-S622.
48. Lappe J, Cullen D, Haynatzki G, Recker R, Ahlf R, Thompson K. Calcium and vitamin D supplementation decreases incidence of stress fractures in female navy recruits. J Bone Miner Res. 2008;23(5):741-749.
49. DIPART (Vitamin D Individual Patient Analysis of Randomized Trials) Group. Patient level pooled analysis of 68 500 patients from seven major vitamin D fracture trials in US and Europe. BMJ. 2010;340:b5463.
50. Duckham RL, Peirce N, Meyer C, Summers GD, Cameron N, Brooke-Wavell K. Risk factors for stress fracture in female endurance athletes: a cross-sectional study. BMJ Open. 2012;2(6).
51. Milgrom C, Finestone A, Novack V, et al. The effect of prophylactic treatment with risedronate on stress fracture incidence among infantry recruits. Bone. 2004;35(2):418-424.
52. Crowell HP, Davis IS. Gait retraining to reduce lower extremity loading in runners. Clin Biomech. 2011;26(1):78-83.
53. Ekenman I, Milgrom C, Finestone A, et al. The role of biomechanical shoe orthoses in tibial stress fracture prevention. Am J Sports Med. 2002;30(6):866-870.
54. Warden SJ, Hurst JA, Sanders MS, Turner CH, Burr DB, Li J. Bone adaptation to a mechanical loading program significantly increases skeletal fatigue resistance. J Bone Miner Res. 2005;20(5):809-816.
Take-Home Points
- Stress injuries, specifically of the lower extremity, are very common in new military trainees.
- Stress injury can range from benign periosteal reaction to displaced fracture.
- Stress injury should be treated on a case-by-case basis, depending on the severity of injury, the location of the injury, and the likelihood of healing with nonoperative management.
- Modifiable risk factors such as nutritional status, training regiment, and even footwear should be investigated to determine potential causes of injury.
- Prevention is a crucial part of the treatment of these injuries, and early intervention such as careful pre-enrollment physicals and vitamin supplementation can be essential in lowering injury rates.
Bone stress injuries, which are common in military recruits, present in weight-bearing (WB) areas as indolent pain caused by repetitive stress and microtrauma. They were first reported in the metatarsals of Prussian soldiers in 1855.1 Today, stress injuries are increasingly common. One study estimated they account for 10% of patients seen by sports medicine practitioners.2 This injury most commonly affects military members, endurance athletes, and dancers.3-5 Specifically, the incidence of stress fractures in military members has been reported to range from 0.8% to 6.9% for men and from 3.4% to 21.0% for women.4 Because of repetitive vigorous lower extremity loading, stress fractures typically occur in the pelvis, femoral neck, tibial shaft, and metatarsals. Delayed diagnosis and the subsequent duration of treatment required for adequate healing can result in significant morbidity. In a 2009 to 2012 study of US military members, Waterman and colleagues6 found an incidence rate of 5.69 stress fractures per 1000 person-years. Fractures most frequently involved the tibia/fibula (2.26/1000), followed by the metatarsals (0.92/1000) and the femoral neck (0.49/1000).6 In addition, these injuries were most commonly encountered in new recruits, who were less accustomed to the high-volume, high-intensity training required during basic training.4,7 Enlisted junior service members have been reported to account for 77.5% of all stress fractures.6 Age under 20 years or over 40 years and white race have also been found to be risk factors for stress injury.6
The pathogenesis of stress injury is controversial. Stanitski and colleagues8 theorized that multiple submaximal mechanical insults create cumulative stress greater than bone capacity, eventually leading to fracture. Johnson9 conducted a biopsy study and postulated that an accelerated remodeling phase was responsible, whereas Friedenberg10 argued that stress injuries are a form of reduced healing, not an attempt to increase healing, caused by the absence of callous formation in the disease process.
Various other nonmodifiable and modifiable risk factors predispose military service members to stress injury. Nonmodifiable risk factors include sex, bone geometry, limb alignment, race, age, and anatomy. Lower extremity movement biomechanics resulting from dynamic limb alignment during activity may be important. Cameron and colleagues11 examined 1843 patients and found that those with knees in >5° of valgus or >5° of external rotation had higher injury rates. Although variables such as sex and limb alignment cannot be changed, proper identification of modifiable risk factors can assist with injury prevention, and nonmodifiable risk factors can help clinicians and researchers target injury prevention interventions to patients at highest risk.
Metabolic, hormonal, and nutritional status is crucial to overall bone health. Multiple studies have found that low body mass index (BMI) is a significant risk factor for stress fracture.7,12,13 Although low BMI is a concern, patients with abnormally high BMI may also be at increased risk for bone stress injury. In a recently released consensus statement on relative energy deficiency in sport (RED-S), the International Olympic Committee addressed the complex interplay of impairments in physiologic function—including metabolic rate, menstrual function, bone health, immunity, protein synthesis, and cardiovascular health—caused by relative energy deficiency.14 The committee stated that the cause of this syndrome is energy deficiency relative to the balance between dietary energy intake and energy expenditure required for health and activities of daily living, growth, and sporting activities. This finding reveals that conditions such as stress injury often may represent a much broader systemic deficit that may be influenced by a patient’s overall physiologic imbalance.
Diagnosis
History and Physical Examination
The onset of stress reaction typically is insidious, with the classic presentation being a new military recruit who is experiencing a sudden increase in pain during physical activity.15 Pain typically is initially present only during activity, and is relieved with rest, but with disease progression this evolves to pain at rest. It is crucial that the physician elicit the patient’s history of training and physical activity. Hsu and colleagues7 reported increased prevalence of overweight civilian recruits, indicating an increase in the number of new recruits having limited experience with the repetitive physical activity encountered in basic training. Stress injury should be suspected in the setting of worsening, indolent lower extremity pain that has been present for several days, especially in the higher-risk patient populations mentioned. Diet should be assessed, with specific attention given to the intake of fruits, vegetables, and foods high in vitamin D and calcium and, most important, the energy balance between intake and output.16 Special attention should also be given to female patients, who may experience the female athlete triad, a spectrum of low energy availability, menstrual dysfunction, and impaired bone turnover (high amount of resorption relative to formation). A key part of the RED-S consensus statement14 alerted healthcare providers that metabolic derangements do not solely affect female patients. These types of patients sustain a major insult to the homeostatic balance of the hormones that sustain adequate bone health. Beck and colleagues17 found that women with disrupted menstrual cycles are 2 to 4 times more likely to sustain a stress fracture than women without disrupted menstrual cycles, making this abnormality an important part of the history.
Examination should begin with careful evaluation of limb alignment and specific attention given to varus or valgus alignment of the knees.11 The feet should also be inspected, as pes planus or cavus foot may increase the risk of stress fracture.18 Identification of the area of maximal tenderness is important. The area in question may also be erythematous or warm secondary to the inflammatory response associated with attempted fracture healing. In chronic fractures in superficial areas such as the metatarsals, callus may be palpable. Although there are few specific tests for stress injury, pain may be reproducible with deep palpation and WB.
Laboratory Testing
When a pathology is thought to have a nutritional or metabolic cause, particularly in a low-weight or underweight patient, a laboratory workup should be obtained. Specific laboratory tests that all patients should undergo are 25-hydroxyvitamin D3, complete blood cell count, and basic chemistry panel, including calcium and thyroid-stimulating hormone levels. Although not necessary for diagnosis, phosphate, parathyroid hormone, albumin, and prealbumin should also be considered. Females should undergo testing of follicle stimulating hormone, luteinizing hormone, estradiol, and testosterone and have a urine pregnancy test. In patients with signs of excessive cortisone, a dexamethasone suppression test can be administered.21 In males, low testosterone is a documented risk factor for stress injury.22
Imaging
Given their low cost and availability, plain radiographs typically are used for initial examination of a suspected stress injury. However, they often lack sensitivity, particularly in the early stages of stress fracture development (Figure 2).
Management
Management of bone stress injury depends on many factors, including symptom duration, fracture location and severity, and risk of progression or nonunion (Table).13
Pelvis
Pelvic stress fractures are rare and represent only 1.6% to 7.1% of all stress fractures.13,27,28 Given the low frequency, physicians must have a high index of suspicion to make the correct diagnosis. These fractures typically occur in marathon runners and other patients who present with persistent pain and a history of high levels of activity. As pelvic stress fractures typically involve the superior or inferior pubic rami, or sacrum, and are at low risk for nonunion,13 most are managed with nonoperative treatment and activity modification for 8 to 12 weeks.27
Femur
Femoral stress fractures are also relatively uncommon, accounting for about 10% of all stress fractures. Depending on their location, these fractures can be at high risk for progression, nonunion, and significant morbidity.29 Especially concerning are femoral neck stress fractures, which can involve either the tension side (lateral cortex) or the compression side (medial cortex) of the bone. Suspicion of a femoral neck stress fracture should prompt immediate NWB.5 Early recognition of these injuries is crucial because once displacement occurs, their complication and morbidity rates become high.13 Patients with compression-side fractures should undergo NWB treatment for 4 to 6 weeks and then slow progression to WB activity. Most return to light-impact activity by 3 to 4 months. By contrast, tension-side fractures are less likely to heal without operative intervention.11 All tension-side fractures (and any compression-side fractures >50% of the width of the femoral neck) should be treated with percutaneous placement of cannulated screws (Figure 3).
Stress fractures of the femoral shaft are less common than those of the femoral neck and represent as little as 3% of all stress fractures.32 However, femoral shaft stress fractures are more common in military populations. In French military recruits, Niva and colleagues33 found an 18% incidence. Similar to femoral neck fractures, femoral shaft fractures typically are diagnosed with advanced imaging, though the fulcrum test and pain on WB can aid in the diagnosis.19 These injuries are often managed nonoperatively with NWB for a period. Weishaar and colleagues34 described US military cadets treated with progressive rehabilitation who returned to full activity within 12 weeks. Displaced femoral shaft fractures associated with bone stress injury are even less common, and should be managed operatively. Salminen and colleagues35 found an incidence of 1.5 fractures per 100,000 years of military service. Over a 20-year period, they surgically treated 10 of these fractures. Average time from intramedullary nailing to union was 3.5 months.
Tibia
The tibia is one of the more common locations for stress injury and fracture. In a prospective study with members of the military, Giladi and colleagues36 found that 71% of stress fractures were tibia fractures. In addition, a large study of 320 athletes with stress fractures found 49.1% in the tibia.37 Fractures typically are diaphyseal and transverse, usually occurring along the posteromedial cortex, where the bone experiences maximal compressive forces (Figure 4).5,13
Compression-side fractures often heal with nonoperative management, though healing may take several months. Swenson and colleagues40 studied the effects of pneumatic bracing on conservative management and return to play in athletes with tibial stress fractures. Patients with bracing returned to light activity within 7 days and full activity within 21 days, whereas those without bracing returned to light activity within 21 days and full activity within 77 days. Pulsed electromagnetic therapy is of controversial benefit in the management of these injuries. Rettig
Metatarsals
Stress fractures were first discovered by Briethaupt1 in the painful swollen feet of Prussian army members in 1855 and were initially named march fractures. Waterman and colleagues6 reported that metatarsal stress fractures accounted for 16% of all stress fractures in the US military between 2009 and 2012. The second metatarsal neck is the most common location for stress fractures, followed by the third and fourth metatarsals, with the fifth metatarsal being the least common.5 The second metatarsal is thought to sustain these injuries more often than the other metatarsals because of its relative lack of immobility. Donahue and Sharkey43 found that the dorsal aspect of the second metatarsal experiences twice the amount of strain experienced by the fifth metatarsal during gait, and that peak strain in the second metatarsal was further increased by simulated muscle fatigue. The risk of stress fracture can be additionally increased with use of minimalist footwear, as shown by Giuliani and colleagues,44 particularly in the absence of a progressive transition in gait and training volume with a change toward minimalist footwear. In patients with a suspected or confirmed fracture of the second, third, or fourth metatarsal, treatment typically is NWB and immobilization for at least 4 weeks.5 Fifth metatarsal stress injuries (Figure 2) typically are treated differently because of their higher risk of nonunion. Patients with a fifth metatarsal stress fracture complain of lateral midfoot pain with running and jumping. For those who present with this fracture early, acceptable treatment consists of 6 weeks of casting and NWB.5 In cases of failed nonoperative therapy, or presentation with radiographic evidence of nonunion, treatment should be intramedullary screw fixation, with bone graft supplementation based on surgeon preference. DeLee and colleagues45 reported on the results of 10 athletes with fifth metatarsal stress fractures treated with intramedullary screw fixation without bone grafting. All 10 experienced fracture union, at a mean of 7.5 weeks, and returned to sport within 8.5 weeks. One complication with this procedure is pain at the screw insertion site, but this can be successfully managed with footwear modification.45
Prevention
Proper identification of patients at high risk for stress injuries has the potential of reducing the incidence of these injuries. Lappe and colleagues46 prospectively examined female army recruits before and after 8 weeks of basic training and found that those who developed a stress fracture were more likely to have a smoking history, to drink more than 10 alcoholic beverages a week, to have a history of corticosteroid or depot medroxyprogesterone use, and to have lower body weight. In addition, the authors found that a history of prolonged exercise before enrollment was protective against fracture. This finding identifies the importance of having new recruits undergo risk factor screening, which could result in adjusting training regimens to try to reduce injury. The RED-S consensus statement14 offers a comprehensive description of the physiologic factors that can contribute to such injury. Similar to proper risk factor identification, implementation of proper exercise progression programs is a simple, modifiable method of limiting stress injuries. For new recruits or athletes who are resuming activity, injury can be effectively prevented by adjusting the frequency, duration, and intensity of training and the training loads used.47
Vitamin D and calcium supplementation is a simple intervention that can be helpful in injury prevention, and its use has very little downside. A double-blind study found a 20% lower incidence of stress fracture in female navy recruits who took 2000 mg of calcium and 800 IU of vitamin D as daily supplemention.48 Of importance, a meta-analysis of more than 65,000 patients found vitamin D supplementation was effective in reducing fracture risk only when combined with calcium, irrespective of age, sex, or prior fracture.49 In female patients with the female athlete triad, psychological counseling and nutritional consultation are essential in bone health maintenance and long-term prevention.50 Other therapies have been evaluated as well. Use of bisphosphonates is controversial for both treatment and prevention of stress fractures. In a randomized, double-blind study of the potential prophylactic effects of risedronate in 324 new infantry recruits, Milgrom and colleagues51 found no statistically significant differences in tibial, femoral, metatarsal, or total stress fracture incidence between the treatment and placebo groups. Therefore, bisphosphonates are seldom recommended as prevention or in primary management of stress fracture.
In addition to nutritional and pharmacologic therapy, activity modification may have a role in injury prevention. Gait retraining has been identified as a potential intervention for reducing stress fractures in patients with poor biomechanics.47 Crowell and Davis52 investigated the effect of gait retraining on the forces operating in the tibia in runners. After 1 month of gait retraining, tibial acceleration while running decreased by 50%, vertical force loading rate by 30%, and peak vertical force impact by 20%. Such studies indicate the importance of proper mechanics during repetitive activity, especially in patients not as accustomed to the rigorous training methods used with new military recruits. However, whether these reduced loads translate into reduced risk of stress fracture remains unclear. In addition, biomechanical shoe orthoses may lower the stress fracture risk in military recruits by reducing peak tibial strain.53 Warden and colleagues54 found a mechanical loading program was effective in enchaining the structural properties of bone in rats, leading the authors to hypothesize that a similar program aimed at modifying bone structure in humans could help prevent stress fracture. Although there have been no studies of such a strategy in humans, pretraining may be an area for future research, especially for military recruits.
Conclusion
Compared with the general population, members of the military (new recruits in particular) are at increased risk for bone stress injuries. Most of these injuries occur during basic training, when recruits significantly increase their repetitive physical activity. Although the exact pathophysiology of stress injury is debated, nutritional and metabolic abnormalities are contributors. The indolent nature of these injuries, and their high rate of false-negative plain radiographs, may result in a significant delay in diagnosis in the absence of advanced imaging studies. Although a majority of injuries heal with nonoperative management and NWB, several patterns, especially those on the tension side of the bone, are at high risk for progression to fracture and nonunion. These include lateral femoral cortex stress injuries and anterior tibial cortex fractures. There should be a low threshold for operative management in the setting of delayed union or failed nonoperative therapy. Of equal importance to orthopedic management of these injuries is the management of underlying systemic deficits, which may have subjected the patient to injury in the first place. Supplementation with vitamin D and calcium can be an important prophylaxis against stress injury. In addition, military recruits and athletes with underlying metabolic or hormonal deficiencies should receive proper attention with a focus on balancing energy intake and energy expenditure. Stress injury leading to fracture—increasingly common in military populations—often requires a multimodal approach for treatment and subsequent prevention.
Am J Orthop. 2017;46(4):176-183. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Stress injuries, specifically of the lower extremity, are very common in new military trainees.
- Stress injury can range from benign periosteal reaction to displaced fracture.
- Stress injury should be treated on a case-by-case basis, depending on the severity of injury, the location of the injury, and the likelihood of healing with nonoperative management.
- Modifiable risk factors such as nutritional status, training regiment, and even footwear should be investigated to determine potential causes of injury.
- Prevention is a crucial part of the treatment of these injuries, and early intervention such as careful pre-enrollment physicals and vitamin supplementation can be essential in lowering injury rates.
Bone stress injuries, which are common in military recruits, present in weight-bearing (WB) areas as indolent pain caused by repetitive stress and microtrauma. They were first reported in the metatarsals of Prussian soldiers in 1855.1 Today, stress injuries are increasingly common. One study estimated they account for 10% of patients seen by sports medicine practitioners.2 This injury most commonly affects military members, endurance athletes, and dancers.3-5 Specifically, the incidence of stress fractures in military members has been reported to range from 0.8% to 6.9% for men and from 3.4% to 21.0% for women.4 Because of repetitive vigorous lower extremity loading, stress fractures typically occur in the pelvis, femoral neck, tibial shaft, and metatarsals. Delayed diagnosis and the subsequent duration of treatment required for adequate healing can result in significant morbidity. In a 2009 to 2012 study of US military members, Waterman and colleagues6 found an incidence rate of 5.69 stress fractures per 1000 person-years. Fractures most frequently involved the tibia/fibula (2.26/1000), followed by the metatarsals (0.92/1000) and the femoral neck (0.49/1000).6 In addition, these injuries were most commonly encountered in new recruits, who were less accustomed to the high-volume, high-intensity training required during basic training.4,7 Enlisted junior service members have been reported to account for 77.5% of all stress fractures.6 Age under 20 years or over 40 years and white race have also been found to be risk factors for stress injury.6
The pathogenesis of stress injury is controversial. Stanitski and colleagues8 theorized that multiple submaximal mechanical insults create cumulative stress greater than bone capacity, eventually leading to fracture. Johnson9 conducted a biopsy study and postulated that an accelerated remodeling phase was responsible, whereas Friedenberg10 argued that stress injuries are a form of reduced healing, not an attempt to increase healing, caused by the absence of callous formation in the disease process.
Various other nonmodifiable and modifiable risk factors predispose military service members to stress injury. Nonmodifiable risk factors include sex, bone geometry, limb alignment, race, age, and anatomy. Lower extremity movement biomechanics resulting from dynamic limb alignment during activity may be important. Cameron and colleagues11 examined 1843 patients and found that those with knees in >5° of valgus or >5° of external rotation had higher injury rates. Although variables such as sex and limb alignment cannot be changed, proper identification of modifiable risk factors can assist with injury prevention, and nonmodifiable risk factors can help clinicians and researchers target injury prevention interventions to patients at highest risk.
Metabolic, hormonal, and nutritional status is crucial to overall bone health. Multiple studies have found that low body mass index (BMI) is a significant risk factor for stress fracture.7,12,13 Although low BMI is a concern, patients with abnormally high BMI may also be at increased risk for bone stress injury. In a recently released consensus statement on relative energy deficiency in sport (RED-S), the International Olympic Committee addressed the complex interplay of impairments in physiologic function—including metabolic rate, menstrual function, bone health, immunity, protein synthesis, and cardiovascular health—caused by relative energy deficiency.14 The committee stated that the cause of this syndrome is energy deficiency relative to the balance between dietary energy intake and energy expenditure required for health and activities of daily living, growth, and sporting activities. This finding reveals that conditions such as stress injury often may represent a much broader systemic deficit that may be influenced by a patient’s overall physiologic imbalance.
Diagnosis
History and Physical Examination
The onset of stress reaction typically is insidious, with the classic presentation being a new military recruit who is experiencing a sudden increase in pain during physical activity.15 Pain typically is initially present only during activity, and is relieved with rest, but with disease progression this evolves to pain at rest. It is crucial that the physician elicit the patient’s history of training and physical activity. Hsu and colleagues7 reported increased prevalence of overweight civilian recruits, indicating an increase in the number of new recruits having limited experience with the repetitive physical activity encountered in basic training. Stress injury should be suspected in the setting of worsening, indolent lower extremity pain that has been present for several days, especially in the higher-risk patient populations mentioned. Diet should be assessed, with specific attention given to the intake of fruits, vegetables, and foods high in vitamin D and calcium and, most important, the energy balance between intake and output.16 Special attention should also be given to female patients, who may experience the female athlete triad, a spectrum of low energy availability, menstrual dysfunction, and impaired bone turnover (high amount of resorption relative to formation). A key part of the RED-S consensus statement14 alerted healthcare providers that metabolic derangements do not solely affect female patients. These types of patients sustain a major insult to the homeostatic balance of the hormones that sustain adequate bone health. Beck and colleagues17 found that women with disrupted menstrual cycles are 2 to 4 times more likely to sustain a stress fracture than women without disrupted menstrual cycles, making this abnormality an important part of the history.
Examination should begin with careful evaluation of limb alignment and specific attention given to varus or valgus alignment of the knees.11 The feet should also be inspected, as pes planus or cavus foot may increase the risk of stress fracture.18 Identification of the area of maximal tenderness is important. The area in question may also be erythematous or warm secondary to the inflammatory response associated with attempted fracture healing. In chronic fractures in superficial areas such as the metatarsals, callus may be palpable. Although there are few specific tests for stress injury, pain may be reproducible with deep palpation and WB.
Laboratory Testing
When a pathology is thought to have a nutritional or metabolic cause, particularly in a low-weight or underweight patient, a laboratory workup should be obtained. Specific laboratory tests that all patients should undergo are 25-hydroxyvitamin D3, complete blood cell count, and basic chemistry panel, including calcium and thyroid-stimulating hormone levels. Although not necessary for diagnosis, phosphate, parathyroid hormone, albumin, and prealbumin should also be considered. Females should undergo testing of follicle stimulating hormone, luteinizing hormone, estradiol, and testosterone and have a urine pregnancy test. In patients with signs of excessive cortisone, a dexamethasone suppression test can be administered.21 In males, low testosterone is a documented risk factor for stress injury.22
Imaging
Given their low cost and availability, plain radiographs typically are used for initial examination of a suspected stress injury. However, they often lack sensitivity, particularly in the early stages of stress fracture development (Figure 2).
Management
Management of bone stress injury depends on many factors, including symptom duration, fracture location and severity, and risk of progression or nonunion (Table).13
Pelvis
Pelvic stress fractures are rare and represent only 1.6% to 7.1% of all stress fractures.13,27,28 Given the low frequency, physicians must have a high index of suspicion to make the correct diagnosis. These fractures typically occur in marathon runners and other patients who present with persistent pain and a history of high levels of activity. As pelvic stress fractures typically involve the superior or inferior pubic rami, or sacrum, and are at low risk for nonunion,13 most are managed with nonoperative treatment and activity modification for 8 to 12 weeks.27
Femur
Femoral stress fractures are also relatively uncommon, accounting for about 10% of all stress fractures. Depending on their location, these fractures can be at high risk for progression, nonunion, and significant morbidity.29 Especially concerning are femoral neck stress fractures, which can involve either the tension side (lateral cortex) or the compression side (medial cortex) of the bone. Suspicion of a femoral neck stress fracture should prompt immediate NWB.5 Early recognition of these injuries is crucial because once displacement occurs, their complication and morbidity rates become high.13 Patients with compression-side fractures should undergo NWB treatment for 4 to 6 weeks and then slow progression to WB activity. Most return to light-impact activity by 3 to 4 months. By contrast, tension-side fractures are less likely to heal without operative intervention.11 All tension-side fractures (and any compression-side fractures >50% of the width of the femoral neck) should be treated with percutaneous placement of cannulated screws (Figure 3).
Stress fractures of the femoral shaft are less common than those of the femoral neck and represent as little as 3% of all stress fractures.32 However, femoral shaft stress fractures are more common in military populations. In French military recruits, Niva and colleagues33 found an 18% incidence. Similar to femoral neck fractures, femoral shaft fractures typically are diagnosed with advanced imaging, though the fulcrum test and pain on WB can aid in the diagnosis.19 These injuries are often managed nonoperatively with NWB for a period. Weishaar and colleagues34 described US military cadets treated with progressive rehabilitation who returned to full activity within 12 weeks. Displaced femoral shaft fractures associated with bone stress injury are even less common, and should be managed operatively. Salminen and colleagues35 found an incidence of 1.5 fractures per 100,000 years of military service. Over a 20-year period, they surgically treated 10 of these fractures. Average time from intramedullary nailing to union was 3.5 months.
Tibia
The tibia is one of the more common locations for stress injury and fracture. In a prospective study with members of the military, Giladi and colleagues36 found that 71% of stress fractures were tibia fractures. In addition, a large study of 320 athletes with stress fractures found 49.1% in the tibia.37 Fractures typically are diaphyseal and transverse, usually occurring along the posteromedial cortex, where the bone experiences maximal compressive forces (Figure 4).5,13
Compression-side fractures often heal with nonoperative management, though healing may take several months. Swenson and colleagues40 studied the effects of pneumatic bracing on conservative management and return to play in athletes with tibial stress fractures. Patients with bracing returned to light activity within 7 days and full activity within 21 days, whereas those without bracing returned to light activity within 21 days and full activity within 77 days. Pulsed electromagnetic therapy is of controversial benefit in the management of these injuries. Rettig
Metatarsals
Stress fractures were first discovered by Briethaupt1 in the painful swollen feet of Prussian army members in 1855 and were initially named march fractures. Waterman and colleagues6 reported that metatarsal stress fractures accounted for 16% of all stress fractures in the US military between 2009 and 2012. The second metatarsal neck is the most common location for stress fractures, followed by the third and fourth metatarsals, with the fifth metatarsal being the least common.5 The second metatarsal is thought to sustain these injuries more often than the other metatarsals because of its relative lack of immobility. Donahue and Sharkey43 found that the dorsal aspect of the second metatarsal experiences twice the amount of strain experienced by the fifth metatarsal during gait, and that peak strain in the second metatarsal was further increased by simulated muscle fatigue. The risk of stress fracture can be additionally increased with use of minimalist footwear, as shown by Giuliani and colleagues,44 particularly in the absence of a progressive transition in gait and training volume with a change toward minimalist footwear. In patients with a suspected or confirmed fracture of the second, third, or fourth metatarsal, treatment typically is NWB and immobilization for at least 4 weeks.5 Fifth metatarsal stress injuries (Figure 2) typically are treated differently because of their higher risk of nonunion. Patients with a fifth metatarsal stress fracture complain of lateral midfoot pain with running and jumping. For those who present with this fracture early, acceptable treatment consists of 6 weeks of casting and NWB.5 In cases of failed nonoperative therapy, or presentation with radiographic evidence of nonunion, treatment should be intramedullary screw fixation, with bone graft supplementation based on surgeon preference. DeLee and colleagues45 reported on the results of 10 athletes with fifth metatarsal stress fractures treated with intramedullary screw fixation without bone grafting. All 10 experienced fracture union, at a mean of 7.5 weeks, and returned to sport within 8.5 weeks. One complication with this procedure is pain at the screw insertion site, but this can be successfully managed with footwear modification.45
Prevention
Proper identification of patients at high risk for stress injuries has the potential of reducing the incidence of these injuries. Lappe and colleagues46 prospectively examined female army recruits before and after 8 weeks of basic training and found that those who developed a stress fracture were more likely to have a smoking history, to drink more than 10 alcoholic beverages a week, to have a history of corticosteroid or depot medroxyprogesterone use, and to have lower body weight. In addition, the authors found that a history of prolonged exercise before enrollment was protective against fracture. This finding identifies the importance of having new recruits undergo risk factor screening, which could result in adjusting training regimens to try to reduce injury. The RED-S consensus statement14 offers a comprehensive description of the physiologic factors that can contribute to such injury. Similar to proper risk factor identification, implementation of proper exercise progression programs is a simple, modifiable method of limiting stress injuries. For new recruits or athletes who are resuming activity, injury can be effectively prevented by adjusting the frequency, duration, and intensity of training and the training loads used.47
Vitamin D and calcium supplementation is a simple intervention that can be helpful in injury prevention, and its use has very little downside. A double-blind study found a 20% lower incidence of stress fracture in female navy recruits who took 2000 mg of calcium and 800 IU of vitamin D as daily supplemention.48 Of importance, a meta-analysis of more than 65,000 patients found vitamin D supplementation was effective in reducing fracture risk only when combined with calcium, irrespective of age, sex, or prior fracture.49 In female patients with the female athlete triad, psychological counseling and nutritional consultation are essential in bone health maintenance and long-term prevention.50 Other therapies have been evaluated as well. Use of bisphosphonates is controversial for both treatment and prevention of stress fractures. In a randomized, double-blind study of the potential prophylactic effects of risedronate in 324 new infantry recruits, Milgrom and colleagues51 found no statistically significant differences in tibial, femoral, metatarsal, or total stress fracture incidence between the treatment and placebo groups. Therefore, bisphosphonates are seldom recommended as prevention or in primary management of stress fracture.
In addition to nutritional and pharmacologic therapy, activity modification may have a role in injury prevention. Gait retraining has been identified as a potential intervention for reducing stress fractures in patients with poor biomechanics.47 Crowell and Davis52 investigated the effect of gait retraining on the forces operating in the tibia in runners. After 1 month of gait retraining, tibial acceleration while running decreased by 50%, vertical force loading rate by 30%, and peak vertical force impact by 20%. Such studies indicate the importance of proper mechanics during repetitive activity, especially in patients not as accustomed to the rigorous training methods used with new military recruits. However, whether these reduced loads translate into reduced risk of stress fracture remains unclear. In addition, biomechanical shoe orthoses may lower the stress fracture risk in military recruits by reducing peak tibial strain.53 Warden and colleagues54 found a mechanical loading program was effective in enchaining the structural properties of bone in rats, leading the authors to hypothesize that a similar program aimed at modifying bone structure in humans could help prevent stress fracture. Although there have been no studies of such a strategy in humans, pretraining may be an area for future research, especially for military recruits.
Conclusion
Compared with the general population, members of the military (new recruits in particular) are at increased risk for bone stress injuries. Most of these injuries occur during basic training, when recruits significantly increase their repetitive physical activity. Although the exact pathophysiology of stress injury is debated, nutritional and metabolic abnormalities are contributors. The indolent nature of these injuries, and their high rate of false-negative plain radiographs, may result in a significant delay in diagnosis in the absence of advanced imaging studies. Although a majority of injuries heal with nonoperative management and NWB, several patterns, especially those on the tension side of the bone, are at high risk for progression to fracture and nonunion. These include lateral femoral cortex stress injuries and anterior tibial cortex fractures. There should be a low threshold for operative management in the setting of delayed union or failed nonoperative therapy. Of equal importance to orthopedic management of these injuries is the management of underlying systemic deficits, which may have subjected the patient to injury in the first place. Supplementation with vitamin D and calcium can be an important prophylaxis against stress injury. In addition, military recruits and athletes with underlying metabolic or hormonal deficiencies should receive proper attention with a focus on balancing energy intake and energy expenditure. Stress injury leading to fracture—increasingly common in military populations—often requires a multimodal approach for treatment and subsequent prevention.
Am J Orthop. 2017;46(4):176-183. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Briethaupt MD. Zur Pathologie des menschlichen Fusses [To the pathology of the human foot]. Med Zeitung. 1855;24:169-177.
2. Berger FH, de Jonge MC, Maas M. Stress fractures in the lower extremity. Eur J Radiol. 2007;62(1):16-26.
3. Almeida SA, Williams KM, Shaffer RA, Brodine SK. Epidemiological patterns of musculoskeletal injuries and physical training. Med Sci Sports Exerc. 1999;31(8):1176-1182.
4. Jones BH, Thacker SB, Gilchrist J, Kimsey CD, Sosin DM. Prevention of lower extremity stress fractures in athletes and soldiers: a systematic review. Epidemiol Rev. 2002;24(2):228-247.
5. Jacobs JM, Cameron KL, Bojescul JA. Lower extremity stress fractures in the military. Clin Sports Med. 2014;33(4):591-613.
6. Waterman BR, Gun B, Bader JO, Orr JD, Belmont PJ. Epidemiology of lower extremity stress fractures in the United States military. Mil Med. 2016;181(10):1308-1313.
7. Hsu LL, Nevin RL, Tobler SK, Rubertone MV. Trends in overweight and obesity among 18-year-old applicants to the United States military, 1993–2006. J Adolesc Health. 2007;41(6):610-612.
8. Stanitski CL, McMaster JH, Scranton PE. On the nature of stress fractures. Am J Sports Med. 1978;6(6):391-396.
9. Johnson LC. Histogenesis of stress fractures [annual lecture]. Washington, DC: Armed Forces Institute of Pathology; 1963.
10. Friedenberg ZB. Fatigue fractures of the tibia. Clin Orthop Relat Res. 1971;(76):111-115.
11. Cameron KL, Peck KY, Owens BD, et al. Biomechanical risk factors for lower extremity stress fracture. Orthop J Sports Med. 2013;1(4 suppl).
12. Knapik J, Montain S, McGraw S, Grier T, Ely M, Jones B. Stress fracture risk factors in basic combat training. Int J Sports Med. 2012;33(11):940-946.
13. Behrens SB, Deren ME, Matson A, Fadale PD, Monchik KO. Stress fractures of the pelvis and legs in athletes. Sports Health. 2013;5(2):165-174.
14. Mountjoy M, Sundgot-Borgen J, Burke L, et al. The IOC consensus statement: beyond the female athlete triad—relative energy deficiency in sport (RED-S). Br J Sports Med. 2014;48(7):491-497.
15. Maitra RS, Johnson DL. Stress fractures. Clinical history and physical examination. Clin Sports Med. 1997;16(2):259-274.
16. Nieves JW, Melsop K, Curtis M, et al. Nutritional factors that influence change in bone density and stress fracture risk among young female cross-country runners. PM R. 2010;2(8):740-750.
17. Beck BR, Matheson GO, Bergman G, et al. Do capacitively coupled electric fields accelerate tibial stress fracture healing? Am J Sports Med. 2008;36(3):545-553.
18. Simkin A, Leichter I, Giladi M, Stein M, Milgrom C. Combined effect of foot arch structure and an orthotic device on stress fractures. Foot Ankle. 1989;10(1):25-29.
19. Johnson AW, Weiss CB, Wheeler DL. Stress fractures of the femoral shaft in athletes—more common than expected: a new clinical test. Am J Sports Med. 1994;22(2):248-256.
20. Clement D, Ammann W, Taunton J, et al. Exercise-induced stress injuries to the femur. Int J Sports Med. 1993;14(6):347-352.
21. Wood PJ, Barth JH, Freedman DB, Perry L, Sheridan B. Evidence for the low dose dexamethasone suppression test to screen for Cushing’s syndrome—recommendations for a protocol for biochemistry laboratories. Ann Clin Biochem. 1997;34(pt 3):222-229.
22. Bennell K, Matheson G, Meeuwisse W, Brukner P. Risk factors for stress fractures. Sports Med. 1999;28(2):91-122.
23. Prather JL, Nusynowitz ML, Snowdy HA, Hughes AD, McCartney WH, Bagg RJ. Scintigraphic findings in stress fractures. J Bone Joint Surg Am. 1977;59(7):869-874.
24. Arendt EA, Griffiths HJ. The use of MR imaging in the assessment and clinical management of stress reactions of bone in high-performance athletes. Clin Sports Med. 1997;16(2):291-306.
25. Boden BP, Osbahr DC. High-risk stress fractures: evaluation and treatment. J Am Acad Orthop Surg. 2000;8(6):344-353.
26. Gaeta M, Minutoli F, Scribano E, et al. CT and MR imaging findings in athletes with early tibial stress injuries: comparison with bone scintigraphy findings and emphasis on cortical abnormalities. Radiology. 2005;235(2):553-561.
27. Matheson GO, Clement DB, Mckenzie DC, Taunton JE, Lloyd-Smith DR, Macintyre JG. Stress fractures in athletes. Am J Sports Med. 1987;15(1):46-58.
28. Iwamoto J, Takeda T. Stress fractures in athletes: review of 196 cases. J Orthop Sci. 2003;8(3):273-278.
29. Noakes TD, Smith JA, Lindenberg G, Wills CE. Pelvic stress fractures in long distance runners. Am J Sports Med. 1985;13(2):120-123.
30. Neubauer T, Brand J, Lidder S, Krawany M. Stress fractures of the femoral neck in runners: a review. Res Sports Med. 2016;24(3):283-297.
31. Evans JT, Guyver PM, Kassam AM, Hubble MJW. Displaced femoral neck stress fractures in Royal Marine recruits—management and results of operative treatment. J R Nav Med Serv. 2012;98(2):3-5.
32. Orava S. Stress fractures. Br J Sports Med. 1980;14(1):40-44.
33. Niva MH, Kiuru MJ, Haataja R, Pihlajamäki HK. Fatigue injuries of the femur. J Bone Joint Surg Br. 2005;87(10):1385-1390.
34. Weishaar MD, McMillian DJ, Moore JH. Identification and management of 2 femoral shaft stress injuries. J Orthop Sports Phys Ther. 2005;35(10):665-673.
35. Salminen ST, Pihlajamäki HK, Visuri TI, Böstman OM. Displaced fatigue fractures of the femoral shaft. Clin Orthop Relat Res. 2003;(409):250-259.
36. Giladi M, Ahronson Z, Stein M, Danon YL, Milgrom C. Unusual distribution and onset of stress fractures in soldiers. Clin Orthop Relat Res. 1985;(192):142-146.
37. Matheson GO, Clement DB, Mckenzie DC, Taunton JE, Lloyd-Smith DR, Macintyre JG. Stress fractures in athletes. Am J Sports Med. 1987;15(1):46-58.
38. Green NE, Rogers RA, Lipscomb AB. Nonunions of stress fractures of the tibia. Am J Sports Med. 1985;13(3):171-176.
39. Orava S, Hulkko A. Stress fracture of the mid-tibial shaft. Acta Orthop Scand. 1984;55(1):35-37.
40. Swenson EJ Jr, DeHaven KE, Sebastianelli WJ, Hanks G, Kalenak A, Lynch JM. The effect of a pneumatic leg brace on return to play in athletes with tibial stress fractures. Am J Sports Med. 1997;25(3):322-328.
41. Rettig AC, Shelbourne KD, McCarroll JR, Bisesi M, Watts J. The natural history and treatment of delayed union stress fractures of the anterior cortex of the tibia. Am J Sports Med. 1988;16(3):250-255.
42. Varner KE, Younas SA, Lintner DM, Marymont JV. Chronic anterior midtibial stress fractures in athletes treated with reamed intramedullary nailing. Am J Sports Med. 2005;33(7):1071-1076.
43. Donahue SW, Sharkey NA. Strains in the metatarsals during the stance phase of gait: implications for stress fractures. J Bone Joint Surg Am. 1999;81(9):1236-1244.
44. Giuliani J, Masini B, Alitz C, Owens BD. Barefoot-simulating footwear associated with metatarsal stress injury in 2 runners. Orthopedics. 2011;34(7):e320-e323.
45. DeLee JC, Evans JP, Julian J. Stress fracture of the fifth metatarsal. Am J Sports Med. 1983;11(5):349-353.
46. Lappe JM, Stegman MR, Recker RR. The impact of lifestyle factors on stress fractures in female army recruits. Osteoporos Int. 2001;12(1):35-42.
47. Friedl KE, Evans RK, Moran DS. Stress fracture and military medical readiness: bridging basic and applied research. Med Sci Sports Exerc. 2008;40(11 suppl):S609-S622.
48. Lappe J, Cullen D, Haynatzki G, Recker R, Ahlf R, Thompson K. Calcium and vitamin D supplementation decreases incidence of stress fractures in female navy recruits. J Bone Miner Res. 2008;23(5):741-749.
49. DIPART (Vitamin D Individual Patient Analysis of Randomized Trials) Group. Patient level pooled analysis of 68 500 patients from seven major vitamin D fracture trials in US and Europe. BMJ. 2010;340:b5463.
50. Duckham RL, Peirce N, Meyer C, Summers GD, Cameron N, Brooke-Wavell K. Risk factors for stress fracture in female endurance athletes: a cross-sectional study. BMJ Open. 2012;2(6).
51. Milgrom C, Finestone A, Novack V, et al. The effect of prophylactic treatment with risedronate on stress fracture incidence among infantry recruits. Bone. 2004;35(2):418-424.
52. Crowell HP, Davis IS. Gait retraining to reduce lower extremity loading in runners. Clin Biomech. 2011;26(1):78-83.
53. Ekenman I, Milgrom C, Finestone A, et al. The role of biomechanical shoe orthoses in tibial stress fracture prevention. Am J Sports Med. 2002;30(6):866-870.
54. Warden SJ, Hurst JA, Sanders MS, Turner CH, Burr DB, Li J. Bone adaptation to a mechanical loading program significantly increases skeletal fatigue resistance. J Bone Miner Res. 2005;20(5):809-816.
1. Briethaupt MD. Zur Pathologie des menschlichen Fusses [To the pathology of the human foot]. Med Zeitung. 1855;24:169-177.
2. Berger FH, de Jonge MC, Maas M. Stress fractures in the lower extremity. Eur J Radiol. 2007;62(1):16-26.
3. Almeida SA, Williams KM, Shaffer RA, Brodine SK. Epidemiological patterns of musculoskeletal injuries and physical training. Med Sci Sports Exerc. 1999;31(8):1176-1182.
4. Jones BH, Thacker SB, Gilchrist J, Kimsey CD, Sosin DM. Prevention of lower extremity stress fractures in athletes and soldiers: a systematic review. Epidemiol Rev. 2002;24(2):228-247.
5. Jacobs JM, Cameron KL, Bojescul JA. Lower extremity stress fractures in the military. Clin Sports Med. 2014;33(4):591-613.
6. Waterman BR, Gun B, Bader JO, Orr JD, Belmont PJ. Epidemiology of lower extremity stress fractures in the United States military. Mil Med. 2016;181(10):1308-1313.
7. Hsu LL, Nevin RL, Tobler SK, Rubertone MV. Trends in overweight and obesity among 18-year-old applicants to the United States military, 1993–2006. J Adolesc Health. 2007;41(6):610-612.
8. Stanitski CL, McMaster JH, Scranton PE. On the nature of stress fractures. Am J Sports Med. 1978;6(6):391-396.
9. Johnson LC. Histogenesis of stress fractures [annual lecture]. Washington, DC: Armed Forces Institute of Pathology; 1963.
10. Friedenberg ZB. Fatigue fractures of the tibia. Clin Orthop Relat Res. 1971;(76):111-115.
11. Cameron KL, Peck KY, Owens BD, et al. Biomechanical risk factors for lower extremity stress fracture. Orthop J Sports Med. 2013;1(4 suppl).
12. Knapik J, Montain S, McGraw S, Grier T, Ely M, Jones B. Stress fracture risk factors in basic combat training. Int J Sports Med. 2012;33(11):940-946.
13. Behrens SB, Deren ME, Matson A, Fadale PD, Monchik KO. Stress fractures of the pelvis and legs in athletes. Sports Health. 2013;5(2):165-174.
14. Mountjoy M, Sundgot-Borgen J, Burke L, et al. The IOC consensus statement: beyond the female athlete triad—relative energy deficiency in sport (RED-S). Br J Sports Med. 2014;48(7):491-497.
15. Maitra RS, Johnson DL. Stress fractures. Clinical history and physical examination. Clin Sports Med. 1997;16(2):259-274.
16. Nieves JW, Melsop K, Curtis M, et al. Nutritional factors that influence change in bone density and stress fracture risk among young female cross-country runners. PM R. 2010;2(8):740-750.
17. Beck BR, Matheson GO, Bergman G, et al. Do capacitively coupled electric fields accelerate tibial stress fracture healing? Am J Sports Med. 2008;36(3):545-553.
18. Simkin A, Leichter I, Giladi M, Stein M, Milgrom C. Combined effect of foot arch structure and an orthotic device on stress fractures. Foot Ankle. 1989;10(1):25-29.
19. Johnson AW, Weiss CB, Wheeler DL. Stress fractures of the femoral shaft in athletes—more common than expected: a new clinical test. Am J Sports Med. 1994;22(2):248-256.
20. Clement D, Ammann W, Taunton J, et al. Exercise-induced stress injuries to the femur. Int J Sports Med. 1993;14(6):347-352.
21. Wood PJ, Barth JH, Freedman DB, Perry L, Sheridan B. Evidence for the low dose dexamethasone suppression test to screen for Cushing’s syndrome—recommendations for a protocol for biochemistry laboratories. Ann Clin Biochem. 1997;34(pt 3):222-229.
22. Bennell K, Matheson G, Meeuwisse W, Brukner P. Risk factors for stress fractures. Sports Med. 1999;28(2):91-122.
23. Prather JL, Nusynowitz ML, Snowdy HA, Hughes AD, McCartney WH, Bagg RJ. Scintigraphic findings in stress fractures. J Bone Joint Surg Am. 1977;59(7):869-874.
24. Arendt EA, Griffiths HJ. The use of MR imaging in the assessment and clinical management of stress reactions of bone in high-performance athletes. Clin Sports Med. 1997;16(2):291-306.
25. Boden BP, Osbahr DC. High-risk stress fractures: evaluation and treatment. J Am Acad Orthop Surg. 2000;8(6):344-353.
26. Gaeta M, Minutoli F, Scribano E, et al. CT and MR imaging findings in athletes with early tibial stress injuries: comparison with bone scintigraphy findings and emphasis on cortical abnormalities. Radiology. 2005;235(2):553-561.
27. Matheson GO, Clement DB, Mckenzie DC, Taunton JE, Lloyd-Smith DR, Macintyre JG. Stress fractures in athletes. Am J Sports Med. 1987;15(1):46-58.
28. Iwamoto J, Takeda T. Stress fractures in athletes: review of 196 cases. J Orthop Sci. 2003;8(3):273-278.
29. Noakes TD, Smith JA, Lindenberg G, Wills CE. Pelvic stress fractures in long distance runners. Am J Sports Med. 1985;13(2):120-123.
30. Neubauer T, Brand J, Lidder S, Krawany M. Stress fractures of the femoral neck in runners: a review. Res Sports Med. 2016;24(3):283-297.
31. Evans JT, Guyver PM, Kassam AM, Hubble MJW. Displaced femoral neck stress fractures in Royal Marine recruits—management and results of operative treatment. J R Nav Med Serv. 2012;98(2):3-5.
32. Orava S. Stress fractures. Br J Sports Med. 1980;14(1):40-44.
33. Niva MH, Kiuru MJ, Haataja R, Pihlajamäki HK. Fatigue injuries of the femur. J Bone Joint Surg Br. 2005;87(10):1385-1390.
34. Weishaar MD, McMillian DJ, Moore JH. Identification and management of 2 femoral shaft stress injuries. J Orthop Sports Phys Ther. 2005;35(10):665-673.
35. Salminen ST, Pihlajamäki HK, Visuri TI, Böstman OM. Displaced fatigue fractures of the femoral shaft. Clin Orthop Relat Res. 2003;(409):250-259.
36. Giladi M, Ahronson Z, Stein M, Danon YL, Milgrom C. Unusual distribution and onset of stress fractures in soldiers. Clin Orthop Relat Res. 1985;(192):142-146.
37. Matheson GO, Clement DB, Mckenzie DC, Taunton JE, Lloyd-Smith DR, Macintyre JG. Stress fractures in athletes. Am J Sports Med. 1987;15(1):46-58.
38. Green NE, Rogers RA, Lipscomb AB. Nonunions of stress fractures of the tibia. Am J Sports Med. 1985;13(3):171-176.
39. Orava S, Hulkko A. Stress fracture of the mid-tibial shaft. Acta Orthop Scand. 1984;55(1):35-37.
40. Swenson EJ Jr, DeHaven KE, Sebastianelli WJ, Hanks G, Kalenak A, Lynch JM. The effect of a pneumatic leg brace on return to play in athletes with tibial stress fractures. Am J Sports Med. 1997;25(3):322-328.
41. Rettig AC, Shelbourne KD, McCarroll JR, Bisesi M, Watts J. The natural history and treatment of delayed union stress fractures of the anterior cortex of the tibia. Am J Sports Med. 1988;16(3):250-255.
42. Varner KE, Younas SA, Lintner DM, Marymont JV. Chronic anterior midtibial stress fractures in athletes treated with reamed intramedullary nailing. Am J Sports Med. 2005;33(7):1071-1076.
43. Donahue SW, Sharkey NA. Strains in the metatarsals during the stance phase of gait: implications for stress fractures. J Bone Joint Surg Am. 1999;81(9):1236-1244.
44. Giuliani J, Masini B, Alitz C, Owens BD. Barefoot-simulating footwear associated with metatarsal stress injury in 2 runners. Orthopedics. 2011;34(7):e320-e323.
45. DeLee JC, Evans JP, Julian J. Stress fracture of the fifth metatarsal. Am J Sports Med. 1983;11(5):349-353.
46. Lappe JM, Stegman MR, Recker RR. The impact of lifestyle factors on stress fractures in female army recruits. Osteoporos Int. 2001;12(1):35-42.
47. Friedl KE, Evans RK, Moran DS. Stress fracture and military medical readiness: bridging basic and applied research. Med Sci Sports Exerc. 2008;40(11 suppl):S609-S622.
48. Lappe J, Cullen D, Haynatzki G, Recker R, Ahlf R, Thompson K. Calcium and vitamin D supplementation decreases incidence of stress fractures in female navy recruits. J Bone Miner Res. 2008;23(5):741-749.
49. DIPART (Vitamin D Individual Patient Analysis of Randomized Trials) Group. Patient level pooled analysis of 68 500 patients from seven major vitamin D fracture trials in US and Europe. BMJ. 2010;340:b5463.
50. Duckham RL, Peirce N, Meyer C, Summers GD, Cameron N, Brooke-Wavell K. Risk factors for stress fracture in female endurance athletes: a cross-sectional study. BMJ Open. 2012;2(6).
51. Milgrom C, Finestone A, Novack V, et al. The effect of prophylactic treatment with risedronate on stress fracture incidence among infantry recruits. Bone. 2004;35(2):418-424.
52. Crowell HP, Davis IS. Gait retraining to reduce lower extremity loading in runners. Clin Biomech. 2011;26(1):78-83.
53. Ekenman I, Milgrom C, Finestone A, et al. The role of biomechanical shoe orthoses in tibial stress fracture prevention. Am J Sports Med. 2002;30(6):866-870.
54. Warden SJ, Hurst JA, Sanders MS, Turner CH, Burr DB, Li J. Bone adaptation to a mechanical loading program significantly increases skeletal fatigue resistance. J Bone Miner Res. 2005;20(5):809-816.
Applying Military Strategy to Complex Knee Reconstruction: Tips for Planning and Executing Advanced Surgery
Take-Home Points
- Thorough preoperative planning is imperative and inclusive of history, physical examination, radiographs, and MRI and potentially CT scan.
- Plan carefully for needed graft sources (autografts and allografts).
- Rehabilitation starts preoperatively and a detailed individualized plan is often warranted.
- Indicated ligamentous repair or augmented repair with reconstruction is more likely to succeed when performed within 2 weeks of injury.
- Complex combined knee restoration surgery can be safely performed in an outpatient setting.
Complex combined knee restoration surgery can be safely performed in an outpatient setting. The term complex knee restoration is used to describe management of knee injuries that are more involved—that is, there is damage to the menisci, cartilage, ligaments, and bones. Management entails not only determining the best treatment options but navigating the more complex logistics of making sure all necessary grafts (fresh and frozen allografts and autografts), implants, and instrumentation are readily available as these cases come to fruition.
The military healthcare paradigm often involves the added logistics of transporting the service member to the correct military treatment facility at the correct time and ensuring the patient’s work-up is complete before he or she arrives for the complex knee restoration. Such cases require significant rehabilitation and time away from family and work, so anything that reduces the morbidity of the surgical undertaking and the overall “morbidity footprint” of time away and that helps the patient return to normal function are value-added and worthy of our attention and diligence in developing an efficient system for managing complex cases.
The globally integrated military healthcare system that is in place has matured over the past decades to allow for the significant majority of the necessary preoperative work-up to be performed at a soldier’s current duty station, wherever in the world that may be, under the guidance of local healthcare providers with specific inputs from the knee restoration surgeon who eventually receives the patient for the planned surgical intervention.
Algorithm for Knee Restoration Planning
Alignment Issues
The first task is to confirm the realignment indication. Realignment may be performed with a proximal opening-wedge medial tibial osteotomy (OWMTO), a distal opening-wedge lateral femoral osteotomy (OWLFO), or a tibial tubercle osteotomy (TTO).1 Given the reproducible clinical improvement achieved and the robust nature of the fixation, these osteotomies are often the first surgical step in complex knee restorations.2 The final determination, made by the surgeon in consultation with the patient, is whether to perform the indicated osteotomy alone or in combination with the rest of the planned restoration surgery. In the vast majority of cases I have managed over the past 2 decades, I have performed the entire knee restoration in a single operation.3 Within the past 5 years, combining the procedures has become even more feasible with the important progress made in multimodal pain management and with the close collaboration of anesthesiologists.4
Meniscus and Cartilage Status
The integration status of meniscus and cartilage within the medial and lateral tibiofemoral compartments is crucial to the comprehensive restoration plan. In fact, the success of the restoration can be said to be dependent on the functional status and health of meniscus and cartilage—which either succeed together or fail apart.
Important covariables are age, prior surgical interventions, activity level expected or allowed after surgery, and size, location, and depth of cartilage injury.5 Whether a cartilage injury is monopolar or bipolar is determined with advanced imaging (magnetic resonance imaging [MRI], computed tomography [CT], weight-bearing radiography) along with analysis of a thorough history (including a review of prior operative reports and arthroscopic images) and a knee examination. Bipolar injuries that involve the condyle and juxtaposed plateau often bode poorly for good clinical outcomes—compared with unipolar lesions, which usually involve the condylar surfaces in isolation. The same thinking regarding the patellofemoral compartment is appropriate. Cartilage lesions that involve the juxtaposed surfaces of the patellar and trochlear groove do poorer than isolated lesions, which are more amenable to cartilage restoration options. The literature on potential cartilage restoration options for the patella and trochlea is expanding. I use the 3-dimensional cartilage restoration option of a fresh patellar osteochondral allograft (OCA) for high-grade cartilage lesions thought to be clinically significant. Other options, such as microfracture, cell-based cartilage restoration, and Osteochondral Autograft Transfer System (Arthrex) procedures (from the thinner condylar cartilage), have varied in their outcomes for patellar lesions. According to more recent literature and a review of my clinical results, fresh patellar OCAs are a good option for patellar lesions.6 Similarly, trochlear lesions can be managed with microfracture, cell-based therapies, or fresh OCAs, depending on surgeon preference.
Functional total or subtotal meniscectomies are often best managed with meniscal allograft transplantation (MAT). An intact or replaced medial or lateral meniscus works synergistically with any planned anterior cruciate ligament (ACL) reconstruction. Again, the adage that meniscus and cartilage succeed together or fail apart is appropriate when planning complex knee restoration. Signs of extrusion or joint-space narrowing and root avulsion or significant loss of meniscal tissue, visualized on MRI or on prior surgical images, often help substantiate a MAT plan. MAT has had the best long-term results when performed in compartments with cartilage damage limited to grade I and grade II changes, in stable knees, and in knees that can be concurrently stabilized.5 Technological advances have increased the value of MAT by limiting the morbidity of the operation and thus allowing for other surgery to be performed concomitantly and safely as part of comprehensive knee restoration. Over the past 20 years, I have arthroscopically performed MAT with bone plugs for medial and lateral procedures, and my results with active-duty soldiers have been promising, paralleling the clinic success reported in the literature.5 Alignment must be considered when performing MAT or cartilage restoration. If the addition of meniscal transplantation or cartilage restoration leaves the knee with residual malalignment of 6° or more, corrective osteotomy is performed.
My view and practice have been to plan for an unloading chondroprotective osteotomy. The goal is a balanced mechanical axis, whether achieved with mere joint-space restoration or with an osteotomy added.
Ligament Status
A comprehensive plan for establishing ligamentous stability is paramount to the overall clinical success of complex knee restorations. Meniscus and cartilage restoration efforts are wasted if clinically significant ligamentous laxity is not concomitantly treated with reconstruction surgery. Revision ACL surgery is by far the most commonly performed surgery in complex knee cases. Diligence in interpreting advanced MRI and physical examination findings is required to make sure there are no concomitant patholaxities in the medial, lateral, posterior, posteromedial, and posterolateral ligamentous complexes. Appropriate ligamentous reconstruction is warranted to maximize clinical results in complex knee restorations. Such cases more commonly require allograft tissue, as the availability of autograft tissue is the limiting issue with 2 or more ligament reconstructions. Military treatment facilities, in which comprehensive knee restorations are performed, have soft-tissue allografts on hand at all times. Having tissue readily available makes it less imperative to determine the most appropriate combined ligamentous reconstruction surgery before the patient arrives—a process that is often difficult. This situation is in contradistinction to the need for specific matched-for-size allograft frozen meniscus and fresh cartilage tissues, both of which require tissue-form procurement in advance of planned restoration surgery.
Rehabilitation Plan
The rehabilitation plan is driven by the part of the complex knee restoration that demands the most caution with respect to weight-bearing and range of motion (ROM) during the first 6 weeks after surgery. The most limiting restorative surgeries involve meniscus and cartilage. Recent clinical trial results support weight-bearing soon after tibial osteotomy performed in the absence of meniscus and cartilage restoration that would otherwise limit weight-bearing for 6 weeks.7 Therefore, most of these complex knee restorations are appropriately managed with a hinged brace locked in extension for toe-touch weight-bearing ambulation, with ROM usually limited to 0° to 90° during the first 6 weeks. Quadriceps rehabilitation with straight-leg raises and isometric contractions is prescribed with a focus on maintaining full extension as the default resting knee position until normalized resting quadriceps tone returns. Full weight-bearing and advancement to full flexion are routinely allowed by 6 weeks.
Case Report
A 41-year-old male service member who was overseas was referred to my clinic for high tibial osteotomy consideration and possible revision ACL reconstruction. His symptoms were medial pain, recurrent instability, and patellofemoral crepitance. Three years earlier, he underwent autograft transtibial ACL reconstruction with significant débridement of the medial meniscus. Before his trip to the United States, I asked that new MRI scans, full-length standing hip–knee–ankle bilateral alignment radiographs, and a 4-view weight-bearing knee series (including a posteroanterior Rosenberg view) be obtained and sent for my review (Figure 1). 
Review of the patient’s detailed preoperative imaging work-up and electronic medical record (available through the military’s healthcare system) made it clear that far more surgical intervention was needed than originally assumed. A significant full-thickness chondral lesion of the patella and a subtotal medial meniscectomy would necessitate patellar cartilage restoration and medial MAT in addition to the high tibial osteotomy and revision ACL reconstruction.
Had this patient been sent through the military medical evacuation system, he would have had to make 2 overseas trips—one trip for preoperative evaluation and advanced imaging, whereby he would have been placed on a match list and had to wait for a requested meniscal allograft and an appropriate graft for his patella, and the other trip for his complex surgery. Fortunately, the military’s integrated healthcare network with true 2-way communication and the collaborative use of integrated electronic medical records proved extremely valuable in making management of this complex knee restoration as efficient as possible. From the perspective of the soldier and his military unit, only 1 big overseas trip was needed; from the perspective of the military healthcare system, responsible use of healthcare personnel and monetary resources and well-planned complex knee restoration surgery saved a knee and allowed a soldier-athlete to rejoin the fields of friendly strife.
This patient had undergone functional complete medial meniscectomy and had significant medial compartment pain, varus alignment, and minimal medial joint-space narrowing (assumed grossly intact cartilage about plateau and condyle), plus patellofemoral pain and crepitance with a large high-grade posttraumatic patellar chondral lesion with normal patellofemoral alignment. He also had an isolated failed ACL graft from prior ACL reconstruction. The previous hardware placement was analyzed, and it was determined that the femoral interference screw could be left in place and that the tibial interference screw most likely would be removed. The mechanical axis determined from the bilateral long-leg standing images dictated a need for proximal OWMTO for correction up to 8° to allow the axis to cross the center of the knee. The 8° correction is the measured correction needed to move the axis from its pass through the medial compartment to a more balanced position across the middle of the knee.
The overall plan encompassed major concomitant corrective and restorative surgery: tibial osteotomy, medial MAT, revision ACL reconstruction, and fresh mega-patellar OCA. Once the frozen meniscus and eventually the fresh patella (both matched for size) were obtained, arrangements for the patient’s trip for the complex surgery were finalized.
Surgery was started with brief arthroscopic evaluation to confirm the overall appropriateness of the planned procedure and to determine if any other minor deficiencies would warrant operative intervention. Once confirmed, the restoration proceeded as planned. The OWMTO was performed with a PEEK (polyetheretherketone) wedge implant (iBalance; Arthrex) followed by arthroscopic preparation for medial MAT with removal of any meniscal remnants and placement of passing sutures (Figure 2A).


When the arthroscopic portion of the surgery was finished, a medial parapatellar arthrotomy was made to allow the patella to be inverted and complete fresh mega-patellar OCA placement (Figure 4).


The knee was placed in a ROM brace locked in full extension. The patient was able to do straight-leg raises and calf pumps in the recovery room and was discharged home with a saphenous nerve block and an iPACK (Interspace between the Popliteal Artery and the Capsule of the posterior Knee) nerve block in place. Home-based therapy was started immediately. After the patient’s first postoperative visit, formal therapy (discussed earlier) was initiated (Figure 6).

Discussion
All-inside GraftLink ACL reconstruction with cortical suspensory fixation appears well suited to combined medial and lateral MAT and/or cartilage restoration—whether it be large fresh OCA combined with medial MAT (as in this patient’s case) or another form of cartilage restoration. Arthroscopic MAT with anatomically fashioned and placed bone plugs minimizes the morbidity within the notch footprints and allows for discrete revision socket formation for both femoral and tibial ACL graft placement. In this case, preparation for the medial MAT and ACL sockets was followed by MAT/ACL construct implantation and secure fixation. The arthrotomy was thereby minimized and placed to allow for efficient mega-patellar OCA graft placement.
Over the past decade, I have performed similar concomitant procedures using the same surgical principles that allow for efficient and reproducible complex knee restoration (Figure 7).
Although use of an algorithm for the management of complex knee restorations is not universally feasible, I offer guidelines for complex knee injuries:
- At each decision point, determine whether the knee and the patient can withstand the planned surgical intervention.
- After deciding to proceed with knee restoration, list the meniscus, cartilage, and ligament injuries that must be addressed.
- Determine which repairs (meniscus, cartilage, ligament) are warranted. Repairs generally are best performed within a period of 7 to 14 days.
- Determine which ligament injuries warrant reconstruction. Allograft tissue typically is used for multiligament reconstruction.
- Rank-order the ligament reconstruction requirements. It is fine to proceed with all of the reconstructions if the case is moving smoothly, if there are no developing tourniquet-time issues, and if the soft-tissue envelope is responding as expected.
- Consider autograft and/or allograft tissue needs for concomitant or staged meniscus and cartilage restoration options/requirements.
Am J Orthop. 2017;46(4):170-175, 202. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Uquillas C, Rossy W, Nathasingh CK, Strauss E, Jazrawi L, Gonzalez-Lomas G. Osteotomies about the knee: AAOS exhibit selection. J Bone Joint Surg Am. 2014;96(24):e199.
2. Mehl J, Paul J, Feucht MJ, et al. ACL deficiency and varus osteoarthritis: high tibial osteotomy alone or combined with ACL reconstruction? Arch Orthop Trauma Surg. 2017;137(2):233-240.
3. Scordino LE, DeBerardino TM. Surgical treatment of osteoarthritis in the middle-aged athlete: new horizons in high tibial osteotomies. Sports Med Arthrosc. 2013;21(1):47-51.
4. Ferrari D, Lopes TJ, França PF, Azevedo FM, Pappas E. Outpatient versus inpatient anterior cruciate ligament reconstruction: a systematic review with meta-analysis. Knee. 2017;24(2):197-206.
5. Weber AE, Gitelis ME, McCarthy MA, Yanke AB, Cole BJ. Malalignment: a requirement for cartilage and organ restoration. Sports Med Arthrosc. 2016;24(2):e14-e22.
6. Prince MR, King AH, Stuart MJ, Dahm DL, Krych AJ. Treatment of patellofemoral cartilage lesions in the young, active patient. J Knee Surg. 2015;28(4):285-295.
7. Scordino LE, DeBerardino TM. Surgical treatment of osteoarthritis in the middle-aged athlete: new horizons in high tibial osteotomies. Sports Med Arthrosc. 2013;21(1):47-51.
Take-Home Points
- Thorough preoperative planning is imperative and inclusive of history, physical examination, radiographs, and MRI and potentially CT scan.
- Plan carefully for needed graft sources (autografts and allografts).
- Rehabilitation starts preoperatively and a detailed individualized plan is often warranted.
- Indicated ligamentous repair or augmented repair with reconstruction is more likely to succeed when performed within 2 weeks of injury.
- Complex combined knee restoration surgery can be safely performed in an outpatient setting.
Complex combined knee restoration surgery can be safely performed in an outpatient setting. The term complex knee restoration is used to describe management of knee injuries that are more involved—that is, there is damage to the menisci, cartilage, ligaments, and bones. Management entails not only determining the best treatment options but navigating the more complex logistics of making sure all necessary grafts (fresh and frozen allografts and autografts), implants, and instrumentation are readily available as these cases come to fruition.
The military healthcare paradigm often involves the added logistics of transporting the service member to the correct military treatment facility at the correct time and ensuring the patient’s work-up is complete before he or she arrives for the complex knee restoration. Such cases require significant rehabilitation and time away from family and work, so anything that reduces the morbidity of the surgical undertaking and the overall “morbidity footprint” of time away and that helps the patient return to normal function are value-added and worthy of our attention and diligence in developing an efficient system for managing complex cases.
The globally integrated military healthcare system that is in place has matured over the past decades to allow for the significant majority of the necessary preoperative work-up to be performed at a soldier’s current duty station, wherever in the world that may be, under the guidance of local healthcare providers with specific inputs from the knee restoration surgeon who eventually receives the patient for the planned surgical intervention.
Algorithm for Knee Restoration Planning
Alignment Issues
The first task is to confirm the realignment indication. Realignment may be performed with a proximal opening-wedge medial tibial osteotomy (OWMTO), a distal opening-wedge lateral femoral osteotomy (OWLFO), or a tibial tubercle osteotomy (TTO).1 Given the reproducible clinical improvement achieved and the robust nature of the fixation, these osteotomies are often the first surgical step in complex knee restorations.2 The final determination, made by the surgeon in consultation with the patient, is whether to perform the indicated osteotomy alone or in combination with the rest of the planned restoration surgery. In the vast majority of cases I have managed over the past 2 decades, I have performed the entire knee restoration in a single operation.3 Within the past 5 years, combining the procedures has become even more feasible with the important progress made in multimodal pain management and with the close collaboration of anesthesiologists.4
Meniscus and Cartilage Status
The integration status of meniscus and cartilage within the medial and lateral tibiofemoral compartments is crucial to the comprehensive restoration plan. In fact, the success of the restoration can be said to be dependent on the functional status and health of meniscus and cartilage—which either succeed together or fail apart.
Important covariables are age, prior surgical interventions, activity level expected or allowed after surgery, and size, location, and depth of cartilage injury.5 Whether a cartilage injury is monopolar or bipolar is determined with advanced imaging (magnetic resonance imaging [MRI], computed tomography [CT], weight-bearing radiography) along with analysis of a thorough history (including a review of prior operative reports and arthroscopic images) and a knee examination. Bipolar injuries that involve the condyle and juxtaposed plateau often bode poorly for good clinical outcomes—compared with unipolar lesions, which usually involve the condylar surfaces in isolation. The same thinking regarding the patellofemoral compartment is appropriate. Cartilage lesions that involve the juxtaposed surfaces of the patellar and trochlear groove do poorer than isolated lesions, which are more amenable to cartilage restoration options. The literature on potential cartilage restoration options for the patella and trochlea is expanding. I use the 3-dimensional cartilage restoration option of a fresh patellar osteochondral allograft (OCA) for high-grade cartilage lesions thought to be clinically significant. Other options, such as microfracture, cell-based cartilage restoration, and Osteochondral Autograft Transfer System (Arthrex) procedures (from the thinner condylar cartilage), have varied in their outcomes for patellar lesions. According to more recent literature and a review of my clinical results, fresh patellar OCAs are a good option for patellar lesions.6 Similarly, trochlear lesions can be managed with microfracture, cell-based therapies, or fresh OCAs, depending on surgeon preference.
Functional total or subtotal meniscectomies are often best managed with meniscal allograft transplantation (MAT). An intact or replaced medial or lateral meniscus works synergistically with any planned anterior cruciate ligament (ACL) reconstruction. Again, the adage that meniscus and cartilage succeed together or fail apart is appropriate when planning complex knee restoration. Signs of extrusion or joint-space narrowing and root avulsion or significant loss of meniscal tissue, visualized on MRI or on prior surgical images, often help substantiate a MAT plan. MAT has had the best long-term results when performed in compartments with cartilage damage limited to grade I and grade II changes, in stable knees, and in knees that can be concurrently stabilized.5 Technological advances have increased the value of MAT by limiting the morbidity of the operation and thus allowing for other surgery to be performed concomitantly and safely as part of comprehensive knee restoration. Over the past 20 years, I have arthroscopically performed MAT with bone plugs for medial and lateral procedures, and my results with active-duty soldiers have been promising, paralleling the clinic success reported in the literature.5 Alignment must be considered when performing MAT or cartilage restoration. If the addition of meniscal transplantation or cartilage restoration leaves the knee with residual malalignment of 6° or more, corrective osteotomy is performed.
My view and practice have been to plan for an unloading chondroprotective osteotomy. The goal is a balanced mechanical axis, whether achieved with mere joint-space restoration or with an osteotomy added.
Ligament Status
A comprehensive plan for establishing ligamentous stability is paramount to the overall clinical success of complex knee restorations. Meniscus and cartilage restoration efforts are wasted if clinically significant ligamentous laxity is not concomitantly treated with reconstruction surgery. Revision ACL surgery is by far the most commonly performed surgery in complex knee cases. Diligence in interpreting advanced MRI and physical examination findings is required to make sure there are no concomitant patholaxities in the medial, lateral, posterior, posteromedial, and posterolateral ligamentous complexes. Appropriate ligamentous reconstruction is warranted to maximize clinical results in complex knee restorations. Such cases more commonly require allograft tissue, as the availability of autograft tissue is the limiting issue with 2 or more ligament reconstructions. Military treatment facilities, in which comprehensive knee restorations are performed, have soft-tissue allografts on hand at all times. Having tissue readily available makes it less imperative to determine the most appropriate combined ligamentous reconstruction surgery before the patient arrives—a process that is often difficult. This situation is in contradistinction to the need for specific matched-for-size allograft frozen meniscus and fresh cartilage tissues, both of which require tissue-form procurement in advance of planned restoration surgery.
Rehabilitation Plan
The rehabilitation plan is driven by the part of the complex knee restoration that demands the most caution with respect to weight-bearing and range of motion (ROM) during the first 6 weeks after surgery. The most limiting restorative surgeries involve meniscus and cartilage. Recent clinical trial results support weight-bearing soon after tibial osteotomy performed in the absence of meniscus and cartilage restoration that would otherwise limit weight-bearing for 6 weeks.7 Therefore, most of these complex knee restorations are appropriately managed with a hinged brace locked in extension for toe-touch weight-bearing ambulation, with ROM usually limited to 0° to 90° during the first 6 weeks. Quadriceps rehabilitation with straight-leg raises and isometric contractions is prescribed with a focus on maintaining full extension as the default resting knee position until normalized resting quadriceps tone returns. Full weight-bearing and advancement to full flexion are routinely allowed by 6 weeks.
Case Report
A 41-year-old male service member who was overseas was referred to my clinic for high tibial osteotomy consideration and possible revision ACL reconstruction. His symptoms were medial pain, recurrent instability, and patellofemoral crepitance. Three years earlier, he underwent autograft transtibial ACL reconstruction with significant débridement of the medial meniscus. Before his trip to the United States, I asked that new MRI scans, full-length standing hip–knee–ankle bilateral alignment radiographs, and a 4-view weight-bearing knee series (including a posteroanterior Rosenberg view) be obtained and sent for my review (Figure 1).
Review of the patient’s detailed preoperative imaging work-up and electronic medical record (available through the military’s healthcare system) made it clear that far more surgical intervention was needed than originally assumed. A significant full-thickness chondral lesion of the patella and a subtotal medial meniscectomy would necessitate patellar cartilage restoration and medial MAT in addition to the high tibial osteotomy and revision ACL reconstruction.
Had this patient been sent through the military medical evacuation system, he would have had to make 2 overseas trips—one trip for preoperative evaluation and advanced imaging, whereby he would have been placed on a match list and had to wait for a requested meniscal allograft and an appropriate graft for his patella, and the other trip for his complex surgery. Fortunately, the military’s integrated healthcare network with true 2-way communication and the collaborative use of integrated electronic medical records proved extremely valuable in making management of this complex knee restoration as efficient as possible. From the perspective of the soldier and his military unit, only 1 big overseas trip was needed; from the perspective of the military healthcare system, responsible use of healthcare personnel and monetary resources and well-planned complex knee restoration surgery saved a knee and allowed a soldier-athlete to rejoin the fields of friendly strife.
This patient had undergone functional complete medial meniscectomy and had significant medial compartment pain, varus alignment, and minimal medial joint-space narrowing (assumed grossly intact cartilage about plateau and condyle), plus patellofemoral pain and crepitance with a large high-grade posttraumatic patellar chondral lesion with normal patellofemoral alignment. He also had an isolated failed ACL graft from prior ACL reconstruction. The previous hardware placement was analyzed, and it was determined that the femoral interference screw could be left in place and that the tibial interference screw most likely would be removed. The mechanical axis determined from the bilateral long-leg standing images dictated a need for proximal OWMTO for correction up to 8° to allow the axis to cross the center of the knee. The 8° correction is the measured correction needed to move the axis from its pass through the medial compartment to a more balanced position across the middle of the knee.
The overall plan encompassed major concomitant corrective and restorative surgery: tibial osteotomy, medial MAT, revision ACL reconstruction, and fresh mega-patellar OCA. Once the frozen meniscus and eventually the fresh patella (both matched for size) were obtained, arrangements for the patient’s trip for the complex surgery were finalized.
Surgery was started with brief arthroscopic evaluation to confirm the overall appropriateness of the planned procedure and to determine if any other minor deficiencies would warrant operative intervention. Once confirmed, the restoration proceeded as planned. The OWMTO was performed with a PEEK (polyetheretherketone) wedge implant (iBalance; Arthrex) followed by arthroscopic preparation for medial MAT with removal of any meniscal remnants and placement of passing sutures (Figure 2A).
When the arthroscopic portion of the surgery was finished, a medial parapatellar arthrotomy was made to allow the patella to be inverted and complete fresh mega-patellar OCA placement (Figure 4).
The knee was placed in a ROM brace locked in full extension. The patient was able to do straight-leg raises and calf pumps in the recovery room and was discharged home with a saphenous nerve block and an iPACK (Interspace between the Popliteal Artery and the Capsule of the posterior Knee) nerve block in place. Home-based therapy was started immediately. After the patient’s first postoperative visit, formal therapy (discussed earlier) was initiated (Figure 6).
Discussion
All-inside GraftLink ACL reconstruction with cortical suspensory fixation appears well suited to combined medial and lateral MAT and/or cartilage restoration—whether it be large fresh OCA combined with medial MAT (as in this patient’s case) or another form of cartilage restoration. Arthroscopic MAT with anatomically fashioned and placed bone plugs minimizes the morbidity within the notch footprints and allows for discrete revision socket formation for both femoral and tibial ACL graft placement. In this case, preparation for the medial MAT and ACL sockets was followed by MAT/ACL construct implantation and secure fixation. The arthrotomy was thereby minimized and placed to allow for efficient mega-patellar OCA graft placement.
Over the past decade, I have performed similar concomitant procedures using the same surgical principles that allow for efficient and reproducible complex knee restoration (Figure 7).
Although use of an algorithm for the management of complex knee restorations is not universally feasible, I offer guidelines for complex knee injuries:
- At each decision point, determine whether the knee and the patient can withstand the planned surgical intervention.
- After deciding to proceed with knee restoration, list the meniscus, cartilage, and ligament injuries that must be addressed.
- Determine which repairs (meniscus, cartilage, ligament) are warranted. Repairs generally are best performed within a period of 7 to 14 days.
- Determine which ligament injuries warrant reconstruction. Allograft tissue typically is used for multiligament reconstruction.
- Rank-order the ligament reconstruction requirements. It is fine to proceed with all of the reconstructions if the case is moving smoothly, if there are no developing tourniquet-time issues, and if the soft-tissue envelope is responding as expected.
- Consider autograft and/or allograft tissue needs for concomitant or staged meniscus and cartilage restoration options/requirements.
Am J Orthop. 2017;46(4):170-175, 202. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Thorough preoperative planning is imperative and inclusive of history, physical examination, radiographs, and MRI and potentially CT scan.
- Plan carefully for needed graft sources (autografts and allografts).
- Rehabilitation starts preoperatively and a detailed individualized plan is often warranted.
- Indicated ligamentous repair or augmented repair with reconstruction is more likely to succeed when performed within 2 weeks of injury.
- Complex combined knee restoration surgery can be safely performed in an outpatient setting.
Complex combined knee restoration surgery can be safely performed in an outpatient setting. The term complex knee restoration is used to describe management of knee injuries that are more involved—that is, there is damage to the menisci, cartilage, ligaments, and bones. Management entails not only determining the best treatment options but navigating the more complex logistics of making sure all necessary grafts (fresh and frozen allografts and autografts), implants, and instrumentation are readily available as these cases come to fruition.
The military healthcare paradigm often involves the added logistics of transporting the service member to the correct military treatment facility at the correct time and ensuring the patient’s work-up is complete before he or she arrives for the complex knee restoration. Such cases require significant rehabilitation and time away from family and work, so anything that reduces the morbidity of the surgical undertaking and the overall “morbidity footprint” of time away and that helps the patient return to normal function are value-added and worthy of our attention and diligence in developing an efficient system for managing complex cases.
The globally integrated military healthcare system that is in place has matured over the past decades to allow for the significant majority of the necessary preoperative work-up to be performed at a soldier’s current duty station, wherever in the world that may be, under the guidance of local healthcare providers with specific inputs from the knee restoration surgeon who eventually receives the patient for the planned surgical intervention.
Algorithm for Knee Restoration Planning
Alignment Issues
The first task is to confirm the realignment indication. Realignment may be performed with a proximal opening-wedge medial tibial osteotomy (OWMTO), a distal opening-wedge lateral femoral osteotomy (OWLFO), or a tibial tubercle osteotomy (TTO).1 Given the reproducible clinical improvement achieved and the robust nature of the fixation, these osteotomies are often the first surgical step in complex knee restorations.2 The final determination, made by the surgeon in consultation with the patient, is whether to perform the indicated osteotomy alone or in combination with the rest of the planned restoration surgery. In the vast majority of cases I have managed over the past 2 decades, I have performed the entire knee restoration in a single operation.3 Within the past 5 years, combining the procedures has become even more feasible with the important progress made in multimodal pain management and with the close collaboration of anesthesiologists.4
Meniscus and Cartilage Status
The integration status of meniscus and cartilage within the medial and lateral tibiofemoral compartments is crucial to the comprehensive restoration plan. In fact, the success of the restoration can be said to be dependent on the functional status and health of meniscus and cartilage—which either succeed together or fail apart.
Important covariables are age, prior surgical interventions, activity level expected or allowed after surgery, and size, location, and depth of cartilage injury.5 Whether a cartilage injury is monopolar or bipolar is determined with advanced imaging (magnetic resonance imaging [MRI], computed tomography [CT], weight-bearing radiography) along with analysis of a thorough history (including a review of prior operative reports and arthroscopic images) and a knee examination. Bipolar injuries that involve the condyle and juxtaposed plateau often bode poorly for good clinical outcomes—compared with unipolar lesions, which usually involve the condylar surfaces in isolation. The same thinking regarding the patellofemoral compartment is appropriate. Cartilage lesions that involve the juxtaposed surfaces of the patellar and trochlear groove do poorer than isolated lesions, which are more amenable to cartilage restoration options. The literature on potential cartilage restoration options for the patella and trochlea is expanding. I use the 3-dimensional cartilage restoration option of a fresh patellar osteochondral allograft (OCA) for high-grade cartilage lesions thought to be clinically significant. Other options, such as microfracture, cell-based cartilage restoration, and Osteochondral Autograft Transfer System (Arthrex) procedures (from the thinner condylar cartilage), have varied in their outcomes for patellar lesions. According to more recent literature and a review of my clinical results, fresh patellar OCAs are a good option for patellar lesions.6 Similarly, trochlear lesions can be managed with microfracture, cell-based therapies, or fresh OCAs, depending on surgeon preference.
Functional total or subtotal meniscectomies are often best managed with meniscal allograft transplantation (MAT). An intact or replaced medial or lateral meniscus works synergistically with any planned anterior cruciate ligament (ACL) reconstruction. Again, the adage that meniscus and cartilage succeed together or fail apart is appropriate when planning complex knee restoration. Signs of extrusion or joint-space narrowing and root avulsion or significant loss of meniscal tissue, visualized on MRI or on prior surgical images, often help substantiate a MAT plan. MAT has had the best long-term results when performed in compartments with cartilage damage limited to grade I and grade II changes, in stable knees, and in knees that can be concurrently stabilized.5 Technological advances have increased the value of MAT by limiting the morbidity of the operation and thus allowing for other surgery to be performed concomitantly and safely as part of comprehensive knee restoration. Over the past 20 years, I have arthroscopically performed MAT with bone plugs for medial and lateral procedures, and my results with active-duty soldiers have been promising, paralleling the clinic success reported in the literature.5 Alignment must be considered when performing MAT or cartilage restoration. If the addition of meniscal transplantation or cartilage restoration leaves the knee with residual malalignment of 6° or more, corrective osteotomy is performed.
My view and practice have been to plan for an unloading chondroprotective osteotomy. The goal is a balanced mechanical axis, whether achieved with mere joint-space restoration or with an osteotomy added.
Ligament Status
A comprehensive plan for establishing ligamentous stability is paramount to the overall clinical success of complex knee restorations. Meniscus and cartilage restoration efforts are wasted if clinically significant ligamentous laxity is not concomitantly treated with reconstruction surgery. Revision ACL surgery is by far the most commonly performed surgery in complex knee cases. Diligence in interpreting advanced MRI and physical examination findings is required to make sure there are no concomitant patholaxities in the medial, lateral, posterior, posteromedial, and posterolateral ligamentous complexes. Appropriate ligamentous reconstruction is warranted to maximize clinical results in complex knee restorations. Such cases more commonly require allograft tissue, as the availability of autograft tissue is the limiting issue with 2 or more ligament reconstructions. Military treatment facilities, in which comprehensive knee restorations are performed, have soft-tissue allografts on hand at all times. Having tissue readily available makes it less imperative to determine the most appropriate combined ligamentous reconstruction surgery before the patient arrives—a process that is often difficult. This situation is in contradistinction to the need for specific matched-for-size allograft frozen meniscus and fresh cartilage tissues, both of which require tissue-form procurement in advance of planned restoration surgery.
Rehabilitation Plan
The rehabilitation plan is driven by the part of the complex knee restoration that demands the most caution with respect to weight-bearing and range of motion (ROM) during the first 6 weeks after surgery. The most limiting restorative surgeries involve meniscus and cartilage. Recent clinical trial results support weight-bearing soon after tibial osteotomy performed in the absence of meniscus and cartilage restoration that would otherwise limit weight-bearing for 6 weeks.7 Therefore, most of these complex knee restorations are appropriately managed with a hinged brace locked in extension for toe-touch weight-bearing ambulation, with ROM usually limited to 0° to 90° during the first 6 weeks. Quadriceps rehabilitation with straight-leg raises and isometric contractions is prescribed with a focus on maintaining full extension as the default resting knee position until normalized resting quadriceps tone returns. Full weight-bearing and advancement to full flexion are routinely allowed by 6 weeks.
Case Report
A 41-year-old male service member who was overseas was referred to my clinic for high tibial osteotomy consideration and possible revision ACL reconstruction. His symptoms were medial pain, recurrent instability, and patellofemoral crepitance. Three years earlier, he underwent autograft transtibial ACL reconstruction with significant débridement of the medial meniscus. Before his trip to the United States, I asked that new MRI scans, full-length standing hip–knee–ankle bilateral alignment radiographs, and a 4-view weight-bearing knee series (including a posteroanterior Rosenberg view) be obtained and sent for my review (Figure 1).
Review of the patient’s detailed preoperative imaging work-up and electronic medical record (available through the military’s healthcare system) made it clear that far more surgical intervention was needed than originally assumed. A significant full-thickness chondral lesion of the patella and a subtotal medial meniscectomy would necessitate patellar cartilage restoration and medial MAT in addition to the high tibial osteotomy and revision ACL reconstruction.
Had this patient been sent through the military medical evacuation system, he would have had to make 2 overseas trips—one trip for preoperative evaluation and advanced imaging, whereby he would have been placed on a match list and had to wait for a requested meniscal allograft and an appropriate graft for his patella, and the other trip for his complex surgery. Fortunately, the military’s integrated healthcare network with true 2-way communication and the collaborative use of integrated electronic medical records proved extremely valuable in making management of this complex knee restoration as efficient as possible. From the perspective of the soldier and his military unit, only 1 big overseas trip was needed; from the perspective of the military healthcare system, responsible use of healthcare personnel and monetary resources and well-planned complex knee restoration surgery saved a knee and allowed a soldier-athlete to rejoin the fields of friendly strife.
This patient had undergone functional complete medial meniscectomy and had significant medial compartment pain, varus alignment, and minimal medial joint-space narrowing (assumed grossly intact cartilage about plateau and condyle), plus patellofemoral pain and crepitance with a large high-grade posttraumatic patellar chondral lesion with normal patellofemoral alignment. He also had an isolated failed ACL graft from prior ACL reconstruction. The previous hardware placement was analyzed, and it was determined that the femoral interference screw could be left in place and that the tibial interference screw most likely would be removed. The mechanical axis determined from the bilateral long-leg standing images dictated a need for proximal OWMTO for correction up to 8° to allow the axis to cross the center of the knee. The 8° correction is the measured correction needed to move the axis from its pass through the medial compartment to a more balanced position across the middle of the knee.
The overall plan encompassed major concomitant corrective and restorative surgery: tibial osteotomy, medial MAT, revision ACL reconstruction, and fresh mega-patellar OCA. Once the frozen meniscus and eventually the fresh patella (both matched for size) were obtained, arrangements for the patient’s trip for the complex surgery were finalized.
Surgery was started with brief arthroscopic evaluation to confirm the overall appropriateness of the planned procedure and to determine if any other minor deficiencies would warrant operative intervention. Once confirmed, the restoration proceeded as planned. The OWMTO was performed with a PEEK (polyetheretherketone) wedge implant (iBalance; Arthrex) followed by arthroscopic preparation for medial MAT with removal of any meniscal remnants and placement of passing sutures (Figure 2A).
When the arthroscopic portion of the surgery was finished, a medial parapatellar arthrotomy was made to allow the patella to be inverted and complete fresh mega-patellar OCA placement (Figure 4).
The knee was placed in a ROM brace locked in full extension. The patient was able to do straight-leg raises and calf pumps in the recovery room and was discharged home with a saphenous nerve block and an iPACK (Interspace between the Popliteal Artery and the Capsule of the posterior Knee) nerve block in place. Home-based therapy was started immediately. After the patient’s first postoperative visit, formal therapy (discussed earlier) was initiated (Figure 6).
Discussion
All-inside GraftLink ACL reconstruction with cortical suspensory fixation appears well suited to combined medial and lateral MAT and/or cartilage restoration—whether it be large fresh OCA combined with medial MAT (as in this patient’s case) or another form of cartilage restoration. Arthroscopic MAT with anatomically fashioned and placed bone plugs minimizes the morbidity within the notch footprints and allows for discrete revision socket formation for both femoral and tibial ACL graft placement. In this case, preparation for the medial MAT and ACL sockets was followed by MAT/ACL construct implantation and secure fixation. The arthrotomy was thereby minimized and placed to allow for efficient mega-patellar OCA graft placement.
Over the past decade, I have performed similar concomitant procedures using the same surgical principles that allow for efficient and reproducible complex knee restoration (Figure 7).
Although use of an algorithm for the management of complex knee restorations is not universally feasible, I offer guidelines for complex knee injuries:
- At each decision point, determine whether the knee and the patient can withstand the planned surgical intervention.
- After deciding to proceed with knee restoration, list the meniscus, cartilage, and ligament injuries that must be addressed.
- Determine which repairs (meniscus, cartilage, ligament) are warranted. Repairs generally are best performed within a period of 7 to 14 days.
- Determine which ligament injuries warrant reconstruction. Allograft tissue typically is used for multiligament reconstruction.
- Rank-order the ligament reconstruction requirements. It is fine to proceed with all of the reconstructions if the case is moving smoothly, if there are no developing tourniquet-time issues, and if the soft-tissue envelope is responding as expected.
- Consider autograft and/or allograft tissue needs for concomitant or staged meniscus and cartilage restoration options/requirements.
Am J Orthop. 2017;46(4):170-175, 202. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Uquillas C, Rossy W, Nathasingh CK, Strauss E, Jazrawi L, Gonzalez-Lomas G. Osteotomies about the knee: AAOS exhibit selection. J Bone Joint Surg Am. 2014;96(24):e199.
2. Mehl J, Paul J, Feucht MJ, et al. ACL deficiency and varus osteoarthritis: high tibial osteotomy alone or combined with ACL reconstruction? Arch Orthop Trauma Surg. 2017;137(2):233-240.
3. Scordino LE, DeBerardino TM. Surgical treatment of osteoarthritis in the middle-aged athlete: new horizons in high tibial osteotomies. Sports Med Arthrosc. 2013;21(1):47-51.
4. Ferrari D, Lopes TJ, França PF, Azevedo FM, Pappas E. Outpatient versus inpatient anterior cruciate ligament reconstruction: a systematic review with meta-analysis. Knee. 2017;24(2):197-206.
5. Weber AE, Gitelis ME, McCarthy MA, Yanke AB, Cole BJ. Malalignment: a requirement for cartilage and organ restoration. Sports Med Arthrosc. 2016;24(2):e14-e22.
6. Prince MR, King AH, Stuart MJ, Dahm DL, Krych AJ. Treatment of patellofemoral cartilage lesions in the young, active patient. J Knee Surg. 2015;28(4):285-295.
7. Scordino LE, DeBerardino TM. Surgical treatment of osteoarthritis in the middle-aged athlete: new horizons in high tibial osteotomies. Sports Med Arthrosc. 2013;21(1):47-51.
1. Uquillas C, Rossy W, Nathasingh CK, Strauss E, Jazrawi L, Gonzalez-Lomas G. Osteotomies about the knee: AAOS exhibit selection. J Bone Joint Surg Am. 2014;96(24):e199.
2. Mehl J, Paul J, Feucht MJ, et al. ACL deficiency and varus osteoarthritis: high tibial osteotomy alone or combined with ACL reconstruction? Arch Orthop Trauma Surg. 2017;137(2):233-240.
3. Scordino LE, DeBerardino TM. Surgical treatment of osteoarthritis in the middle-aged athlete: new horizons in high tibial osteotomies. Sports Med Arthrosc. 2013;21(1):47-51.
4. Ferrari D, Lopes TJ, França PF, Azevedo FM, Pappas E. Outpatient versus inpatient anterior cruciate ligament reconstruction: a systematic review with meta-analysis. Knee. 2017;24(2):197-206.
5. Weber AE, Gitelis ME, McCarthy MA, Yanke AB, Cole BJ. Malalignment: a requirement for cartilage and organ restoration. Sports Med Arthrosc. 2016;24(2):e14-e22.
6. Prince MR, King AH, Stuart MJ, Dahm DL, Krych AJ. Treatment of patellofemoral cartilage lesions in the young, active patient. J Knee Surg. 2015;28(4):285-295.
7. Scordino LE, DeBerardino TM. Surgical treatment of osteoarthritis in the middle-aged athlete: new horizons in high tibial osteotomies. Sports Med Arthrosc. 2013;21(1):47-51.
Zollinger-Ellison Syndrome: Not Your Average Peptic Ulcer Disease
IN THIS ARTICLE
- Diagnostic criteria
- Pharmacologic management
- Patient education
A more severe variant of peptic ulcer disease, Zollinger-Ellison syndrome (ZES) is a rare, chronic, and potentially life-threatening ulcerative disorder. Because the syndrome can be easily misdiagnosed based on clinical presentation alone, primary care clinicians need to be aware of its diagnostic features and know when referral to a gastroenterologist is necessary. Clinicians should suspect ZES in patients with peptic ulcer disease that is refractory to traditional medications.
Caused by a gastrin-secreting neuroendocrine tumor of the pancreas or duodenum called a gastrinoma, ZES can be benign or malignant. It typically manifests in white men ages 30 to 50.1 Due to the significant number of patients treated for a benign cause of peptic ulcer disease (eg, Helicobacter pylori or NSAID-induced ulcers) who are never tested for ZES, the exact incidence is difficult to determine.2 However, it is estimated that approximately 0.1 to 3 people per million develop the disease annually.3
PATHOPHYSIOLOGY
Approximately 80% of gastrinomas occur in the “gastrinoma triangle,” outlined by the hepatic portal vein, neck and body of the pancreas, and latter two-thirds of the duodenum (see Figure).1,4,5 Most gastrinomas involved in ZES occur sporadically, but there is a hereditary component associated with multiple endocrine neoplasia type 1 (MEN1), an autosomal dominant disorder.4
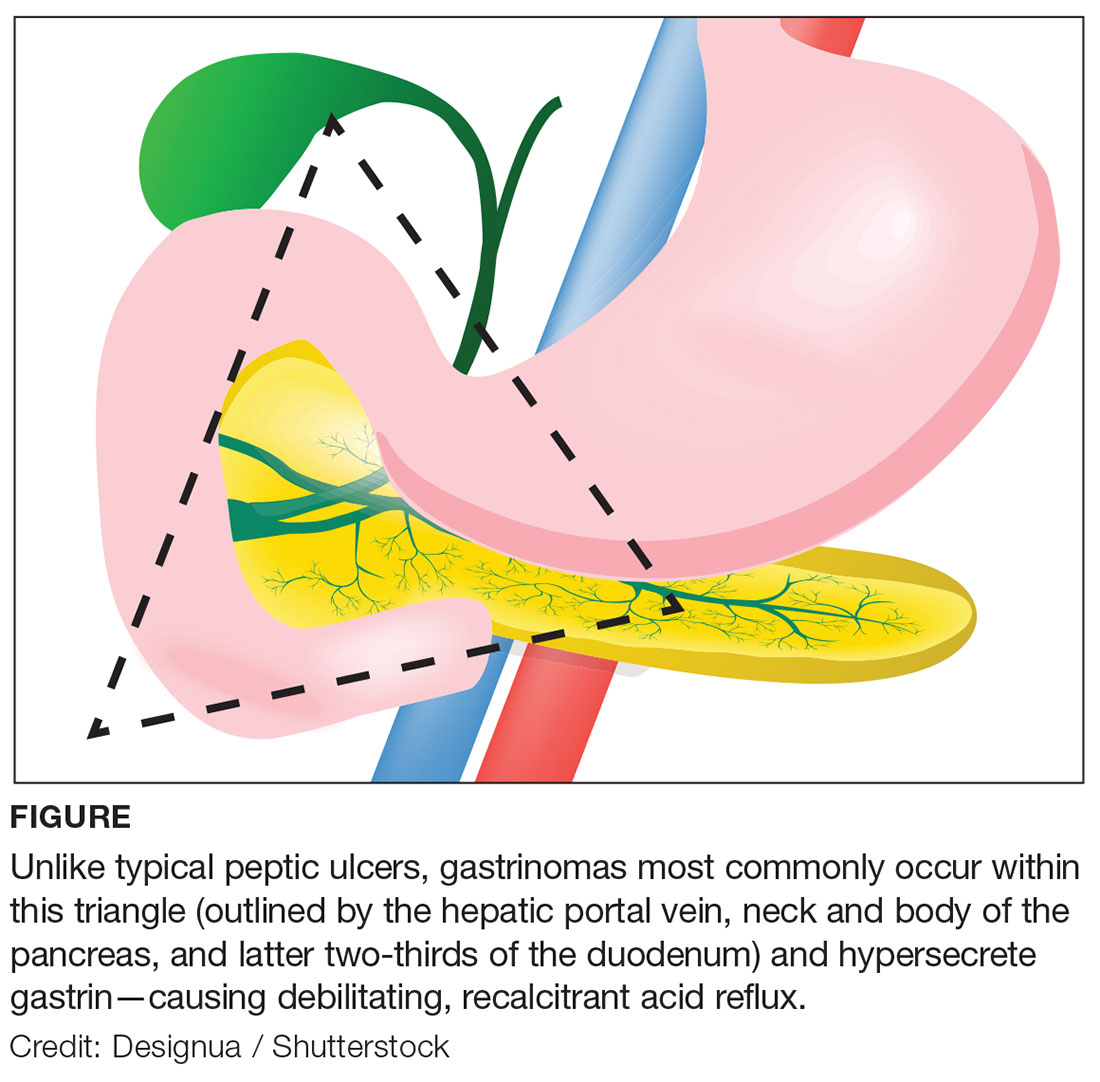
The overproduction and secretion of gastrin by the gastrinoma stimulates hypersecretion of hydrochloric acid.4 This is distinguished from high gastrin levels in the setting of fasting hypochlorhydria or achlorhydria, which may be caused by chronic atrophic gastritis, proton pump inhibitor (PPI) use, or pernicious anemia.5 The chronic hypersecretion of acid causes ulcerations to form. Most commonly, a single ulcer forms in the first portion of the duodenum.3
CLINICAL PRESENTATION
Patients with ZES often report vague abdominal pain that may mimic peptic ulcer disease on initial presentation. The widespread use of PPIs can mask symptoms, and one-fourth of patients present with no abdominal pain at all.6 Patients may also present with
The physical exam may be within normal limits, and no physical finding is considered pathognomonic for ZES. Findings may include epigastric tenderness; pallor, due to an ulcer-related anemia or GI bleed; jaundice, if there is liver involvement; and esophageal or dental erosions, due to excessive acid.8
DIAGNOSIS
Patients with symptoms refractory to medical management should be referred to a specialist for further testing. Once a patient is referred, a gastroenterologist will perform lab tests and imaging studies. In order to be diagnosed with ZES, the patient must exhibit an acidic environment with a pH less than 2 and an inappropriate release of gastrin with a basal acid output greater than 15 mEq/h (or > 5 mEq/h in a patient with prior acid reduction surgery).5,6
Fasting serum gastrin (FSG) is the initial study of choice, followed by a secretin-stimulating test when necessary.9 Diagnosis is established by an FSG level greater than 100 pg/mL; if more than 10-fold the normal level, no further testing is needed. However, results often range from 100 to 1,000 pg/mL.6,10 At these values, further testing with secretin stimulation is warranted.9 The test is performed with an IV injection of secretin, and blood samples are obtained to measure serum gastrin levels.10 An increase greater than 100 pg/mL is considered positive; one greater than 200 pg/mL is diagnostic.3
Once lab tests have been performed, a series of imaging studies are indicated. Endoscopy is used to identify active ulcers and erosions due to long-term acid secretion.3 CT, MRI, and somatostatin receptor scintigraphy (a specialized form of imaging that is the study of choice for localizing gastrinomas) are performed to localize primary tumors and identify any metastatic disease that may be present.10 Finally, after lab tests and imaging studies have been completed, genetic screening for MEN1 is used to determine if the patient has a sporadic or hereditary gastrinoma.3
MANAGEMENT
Once ZES has been diagnosed, the specialist will refer the patient for surgical opinion. The main objectives of surgery are to determine whether the tumor is malignant via biopsy, and to resect the tumor to suppress the acid hypersecretion, if indicated in the absence of liver metastasis and large pancreatic tumor size. Medical management should begin immediately to prevent any further damage from prolonged gastric hypersecretion.1
Pharmacologic options include PPIs, H2-receptor antagonists, and somatostatin analogues; PPIs are considered firstline therapy. Many patients with ZES require a higher dosage than is needed with typical GERD (60-100 mg/d vs 20-40 mg/d). Somatostatin analogues can be used in conjunction with PPIs and have been shown to inhibit tumor growth in patients with malignant ZES.1
Once a ZES diagnosis has been made, the tumor(s) resected (if appropriate), and vagotomy considered or performed, patients will need routine follow-up with their gastroenterologist and their primary care provider, who can manage medications and recommend any lifestyle changes.5
PROGNOSIS
The most important prognostic factor of patients with ZES is whether the gastrinoma is benign or malignant. There are two patterns: aggressive disease (25%) and nonaggressive disease (75%).5 At diagnosis, 40% to 70% of patients with sporadic ZES present with lymph node metastases, and 20% to 40% present with liver metastases. Patients with liver metastases have a 10-year survival rate of 30%, compared to a 15-year survival rate of 83% in patients without liver metastases.11,12
Along with the tumor itself, another prognostic factor to consider is the FSG level at diagnosis. Patients with higher FSG levels have decreased five- and 10-year survival rates compared to patients with lower FSG values. The 10-year survival rate for patients with a lower FSG value (0-499 pg/mL) is 86%, while the 10-year survival rate for those with a greater FSG value (> 1,000 pg/mL) is 73%.11,12 Overall, the prognosis is good for patients with ZES. The 10-year survival rate is high, and management is possible with medications and surgical resection of the gastrinoma.
PATIENT EDUCATION
Once patients are diagnosed, treatment with PPIs is typically lifelong unless they are considered cured by surgical resection. Patients need to understand that compliance is necessary to properly manage symptoms; certain foods, alcohol, and tobacco can affect the condition, and lifestyle modifications should be made, as they would with typical GERD or peptic ulcer disease.
CONCLUSION
ZES is frequently overlooked, and patients often continue to experience unresolved symptoms related to hypergastrinemia. Due to its complexity and ability to mimic other disorders—as well as the implications of duodenal versus pancreatic location, and other disorders of the kidney or endocrine system suggestive of MEN1—ZES should be ruled out in any patient with unexplained persistent GERD, peptic ulcer disease, elevated FSG, chronic diarrhea, and/or abdominal pain.5
The gastrinoma itself is a well-differentiated and slow-growing tumor in the majority of cases, making the prognosis for ZES favorable for long-term survival. Proper pharmacologic management is instrumental for controlling symptoms and decreasing acid production. Surgical resection offers patients the best chance for a complete cure. Clinicians and patients should be well educated about ZES in order to successfully manage the disorder.
1. Tomassetti P, Campana D, Piscitelli L, et al. Treatment of Zollinger-Ellison syndrome. World J Gastroenterol. 2005; 11(35):5423-5432.
2. Metz DC. Diagnosis of the Zollinger-Ellison syndrome. Clin Gastroenterol Hepatol. 2016;10(2):126-130.
3. Epelboym I, Mazeh H. Zollinger-Ellison syndrome: classical considerations and current controversies. Oncologist. 2014; 19(1):44-50.
4. Papadakis M, McPhee S, Rabow M. Current Medical Diagnosis and Treatment 2014. New York, NY: McGraw-Hill Education; 2014:600-601.
5. Feldman M, Friedman LS, Lawrence BJ. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Philadelphia, PA: Saunders/Elsevier; 2016:511-515.
6. Ito T, Cadiot G, Jensen RT. Diagnosis of Zollinger-Ellison syndrome: increasingly difficult. World J Gastroenterol. 2012; 18(39):5495-5503.
7. Blonski WC, Katzka DA, Lichtenstein GR, Metz DC. Idiopathic gastric acid hypersecretion presenting as a diarrheal disorder and mimicking both Zollinger-Ellison syndrome and Crohn’s disease. Eur J Gastroenterol Hepatol. 2005;17(4):441-444.
8. Roy PK. Zollinger-Ellison syndrome clinical presentation. http://emedicine.medscape.com/article/183555-clinical#b4. Accessed June 14, 2017.
9. Berna MJ, Hoffmann KM, Serrano J, et al. Serum gastrin in Zollinger-Ellison syndrome: I. prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2229 cases from the literature. Medicine (Baltimore). 2006;85(6):295-330.
10. Moore AR, Varro A, Pritchard M. Zollinger-Ellison syndrome. Gastrointestinal Nursing. 2012;10(5):44-49.
11. Weber HC, Venzon DJ, Lin JT, et al. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology. 1995;108(6):1637-1649.
12. Berger AC, Gibril F, Venzon DJ, et al. Prognostic value of initial fasting serum gastrin levels in patients with Zollinger-Ellison syndrome. J Clin Oncol. 2001;19(12):3051-3057.
IN THIS ARTICLE
- Diagnostic criteria
- Pharmacologic management
- Patient education
A more severe variant of peptic ulcer disease, Zollinger-Ellison syndrome (ZES) is a rare, chronic, and potentially life-threatening ulcerative disorder. Because the syndrome can be easily misdiagnosed based on clinical presentation alone, primary care clinicians need to be aware of its diagnostic features and know when referral to a gastroenterologist is necessary. Clinicians should suspect ZES in patients with peptic ulcer disease that is refractory to traditional medications.
Caused by a gastrin-secreting neuroendocrine tumor of the pancreas or duodenum called a gastrinoma, ZES can be benign or malignant. It typically manifests in white men ages 30 to 50.1 Due to the significant number of patients treated for a benign cause of peptic ulcer disease (eg, Helicobacter pylori or NSAID-induced ulcers) who are never tested for ZES, the exact incidence is difficult to determine.2 However, it is estimated that approximately 0.1 to 3 people per million develop the disease annually.3
PATHOPHYSIOLOGY
Approximately 80% of gastrinomas occur in the “gastrinoma triangle,” outlined by the hepatic portal vein, neck and body of the pancreas, and latter two-thirds of the duodenum (see Figure).1,4,5 Most gastrinomas involved in ZES occur sporadically, but there is a hereditary component associated with multiple endocrine neoplasia type 1 (MEN1), an autosomal dominant disorder.4
The overproduction and secretion of gastrin by the gastrinoma stimulates hypersecretion of hydrochloric acid.4 This is distinguished from high gastrin levels in the setting of fasting hypochlorhydria or achlorhydria, which may be caused by chronic atrophic gastritis, proton pump inhibitor (PPI) use, or pernicious anemia.5 The chronic hypersecretion of acid causes ulcerations to form. Most commonly, a single ulcer forms in the first portion of the duodenum.3
CLINICAL PRESENTATION
Patients with ZES often report vague abdominal pain that may mimic peptic ulcer disease on initial presentation. The widespread use of PPIs can mask symptoms, and one-fourth of patients present with no abdominal pain at all.6 Patients may also present with
The physical exam may be within normal limits, and no physical finding is considered pathognomonic for ZES. Findings may include epigastric tenderness; pallor, due to an ulcer-related anemia or GI bleed; jaundice, if there is liver involvement; and esophageal or dental erosions, due to excessive acid.8
DIAGNOSIS
Patients with symptoms refractory to medical management should be referred to a specialist for further testing. Once a patient is referred, a gastroenterologist will perform lab tests and imaging studies. In order to be diagnosed with ZES, the patient must exhibit an acidic environment with a pH less than 2 and an inappropriate release of gastrin with a basal acid output greater than 15 mEq/h (or > 5 mEq/h in a patient with prior acid reduction surgery).5,6
Fasting serum gastrin (FSG) is the initial study of choice, followed by a secretin-stimulating test when necessary.9 Diagnosis is established by an FSG level greater than 100 pg/mL; if more than 10-fold the normal level, no further testing is needed. However, results often range from 100 to 1,000 pg/mL.6,10 At these values, further testing with secretin stimulation is warranted.9 The test is performed with an IV injection of secretin, and blood samples are obtained to measure serum gastrin levels.10 An increase greater than 100 pg/mL is considered positive; one greater than 200 pg/mL is diagnostic.3
Once lab tests have been performed, a series of imaging studies are indicated. Endoscopy is used to identify active ulcers and erosions due to long-term acid secretion.3 CT, MRI, and somatostatin receptor scintigraphy (a specialized form of imaging that is the study of choice for localizing gastrinomas) are performed to localize primary tumors and identify any metastatic disease that may be present.10 Finally, after lab tests and imaging studies have been completed, genetic screening for MEN1 is used to determine if the patient has a sporadic or hereditary gastrinoma.3
MANAGEMENT
Once ZES has been diagnosed, the specialist will refer the patient for surgical opinion. The main objectives of surgery are to determine whether the tumor is malignant via biopsy, and to resect the tumor to suppress the acid hypersecretion, if indicated in the absence of liver metastasis and large pancreatic tumor size. Medical management should begin immediately to prevent any further damage from prolonged gastric hypersecretion.1
Pharmacologic options include PPIs, H2-receptor antagonists, and somatostatin analogues; PPIs are considered firstline therapy. Many patients with ZES require a higher dosage than is needed with typical GERD (60-100 mg/d vs 20-40 mg/d). Somatostatin analogues can be used in conjunction with PPIs and have been shown to inhibit tumor growth in patients with malignant ZES.1
Once a ZES diagnosis has been made, the tumor(s) resected (if appropriate), and vagotomy considered or performed, patients will need routine follow-up with their gastroenterologist and their primary care provider, who can manage medications and recommend any lifestyle changes.5
PROGNOSIS
The most important prognostic factor of patients with ZES is whether the gastrinoma is benign or malignant. There are two patterns: aggressive disease (25%) and nonaggressive disease (75%).5 At diagnosis, 40% to 70% of patients with sporadic ZES present with lymph node metastases, and 20% to 40% present with liver metastases. Patients with liver metastases have a 10-year survival rate of 30%, compared to a 15-year survival rate of 83% in patients without liver metastases.11,12
Along with the tumor itself, another prognostic factor to consider is the FSG level at diagnosis. Patients with higher FSG levels have decreased five- and 10-year survival rates compared to patients with lower FSG values. The 10-year survival rate for patients with a lower FSG value (0-499 pg/mL) is 86%, while the 10-year survival rate for those with a greater FSG value (> 1,000 pg/mL) is 73%.11,12 Overall, the prognosis is good for patients with ZES. The 10-year survival rate is high, and management is possible with medications and surgical resection of the gastrinoma.
PATIENT EDUCATION
Once patients are diagnosed, treatment with PPIs is typically lifelong unless they are considered cured by surgical resection. Patients need to understand that compliance is necessary to properly manage symptoms; certain foods, alcohol, and tobacco can affect the condition, and lifestyle modifications should be made, as they would with typical GERD or peptic ulcer disease.
CONCLUSION
ZES is frequently overlooked, and patients often continue to experience unresolved symptoms related to hypergastrinemia. Due to its complexity and ability to mimic other disorders—as well as the implications of duodenal versus pancreatic location, and other disorders of the kidney or endocrine system suggestive of MEN1—ZES should be ruled out in any patient with unexplained persistent GERD, peptic ulcer disease, elevated FSG, chronic diarrhea, and/or abdominal pain.5
The gastrinoma itself is a well-differentiated and slow-growing tumor in the majority of cases, making the prognosis for ZES favorable for long-term survival. Proper pharmacologic management is instrumental for controlling symptoms and decreasing acid production. Surgical resection offers patients the best chance for a complete cure. Clinicians and patients should be well educated about ZES in order to successfully manage the disorder.
IN THIS ARTICLE
- Diagnostic criteria
- Pharmacologic management
- Patient education
A more severe variant of peptic ulcer disease, Zollinger-Ellison syndrome (ZES) is a rare, chronic, and potentially life-threatening ulcerative disorder. Because the syndrome can be easily misdiagnosed based on clinical presentation alone, primary care clinicians need to be aware of its diagnostic features and know when referral to a gastroenterologist is necessary. Clinicians should suspect ZES in patients with peptic ulcer disease that is refractory to traditional medications.
Caused by a gastrin-secreting neuroendocrine tumor of the pancreas or duodenum called a gastrinoma, ZES can be benign or malignant. It typically manifests in white men ages 30 to 50.1 Due to the significant number of patients treated for a benign cause of peptic ulcer disease (eg, Helicobacter pylori or NSAID-induced ulcers) who are never tested for ZES, the exact incidence is difficult to determine.2 However, it is estimated that approximately 0.1 to 3 people per million develop the disease annually.3
PATHOPHYSIOLOGY
Approximately 80% of gastrinomas occur in the “gastrinoma triangle,” outlined by the hepatic portal vein, neck and body of the pancreas, and latter two-thirds of the duodenum (see Figure).1,4,5 Most gastrinomas involved in ZES occur sporadically, but there is a hereditary component associated with multiple endocrine neoplasia type 1 (MEN1), an autosomal dominant disorder.4
The overproduction and secretion of gastrin by the gastrinoma stimulates hypersecretion of hydrochloric acid.4 This is distinguished from high gastrin levels in the setting of fasting hypochlorhydria or achlorhydria, which may be caused by chronic atrophic gastritis, proton pump inhibitor (PPI) use, or pernicious anemia.5 The chronic hypersecretion of acid causes ulcerations to form. Most commonly, a single ulcer forms in the first portion of the duodenum.3
CLINICAL PRESENTATION
Patients with ZES often report vague abdominal pain that may mimic peptic ulcer disease on initial presentation. The widespread use of PPIs can mask symptoms, and one-fourth of patients present with no abdominal pain at all.6 Patients may also present with
The physical exam may be within normal limits, and no physical finding is considered pathognomonic for ZES. Findings may include epigastric tenderness; pallor, due to an ulcer-related anemia or GI bleed; jaundice, if there is liver involvement; and esophageal or dental erosions, due to excessive acid.8
DIAGNOSIS
Patients with symptoms refractory to medical management should be referred to a specialist for further testing. Once a patient is referred, a gastroenterologist will perform lab tests and imaging studies. In order to be diagnosed with ZES, the patient must exhibit an acidic environment with a pH less than 2 and an inappropriate release of gastrin with a basal acid output greater than 15 mEq/h (or > 5 mEq/h in a patient with prior acid reduction surgery).5,6
Fasting serum gastrin (FSG) is the initial study of choice, followed by a secretin-stimulating test when necessary.9 Diagnosis is established by an FSG level greater than 100 pg/mL; if more than 10-fold the normal level, no further testing is needed. However, results often range from 100 to 1,000 pg/mL.6,10 At these values, further testing with secretin stimulation is warranted.9 The test is performed with an IV injection of secretin, and blood samples are obtained to measure serum gastrin levels.10 An increase greater than 100 pg/mL is considered positive; one greater than 200 pg/mL is diagnostic.3
Once lab tests have been performed, a series of imaging studies are indicated. Endoscopy is used to identify active ulcers and erosions due to long-term acid secretion.3 CT, MRI, and somatostatin receptor scintigraphy (a specialized form of imaging that is the study of choice for localizing gastrinomas) are performed to localize primary tumors and identify any metastatic disease that may be present.10 Finally, after lab tests and imaging studies have been completed, genetic screening for MEN1 is used to determine if the patient has a sporadic or hereditary gastrinoma.3
MANAGEMENT
Once ZES has been diagnosed, the specialist will refer the patient for surgical opinion. The main objectives of surgery are to determine whether the tumor is malignant via biopsy, and to resect the tumor to suppress the acid hypersecretion, if indicated in the absence of liver metastasis and large pancreatic tumor size. Medical management should begin immediately to prevent any further damage from prolonged gastric hypersecretion.1
Pharmacologic options include PPIs, H2-receptor antagonists, and somatostatin analogues; PPIs are considered firstline therapy. Many patients with ZES require a higher dosage than is needed with typical GERD (60-100 mg/d vs 20-40 mg/d). Somatostatin analogues can be used in conjunction with PPIs and have been shown to inhibit tumor growth in patients with malignant ZES.1
Once a ZES diagnosis has been made, the tumor(s) resected (if appropriate), and vagotomy considered or performed, patients will need routine follow-up with their gastroenterologist and their primary care provider, who can manage medications and recommend any lifestyle changes.5
PROGNOSIS
The most important prognostic factor of patients with ZES is whether the gastrinoma is benign or malignant. There are two patterns: aggressive disease (25%) and nonaggressive disease (75%).5 At diagnosis, 40% to 70% of patients with sporadic ZES present with lymph node metastases, and 20% to 40% present with liver metastases. Patients with liver metastases have a 10-year survival rate of 30%, compared to a 15-year survival rate of 83% in patients without liver metastases.11,12
Along with the tumor itself, another prognostic factor to consider is the FSG level at diagnosis. Patients with higher FSG levels have decreased five- and 10-year survival rates compared to patients with lower FSG values. The 10-year survival rate for patients with a lower FSG value (0-499 pg/mL) is 86%, while the 10-year survival rate for those with a greater FSG value (> 1,000 pg/mL) is 73%.11,12 Overall, the prognosis is good for patients with ZES. The 10-year survival rate is high, and management is possible with medications and surgical resection of the gastrinoma.
PATIENT EDUCATION
Once patients are diagnosed, treatment with PPIs is typically lifelong unless they are considered cured by surgical resection. Patients need to understand that compliance is necessary to properly manage symptoms; certain foods, alcohol, and tobacco can affect the condition, and lifestyle modifications should be made, as they would with typical GERD or peptic ulcer disease.
CONCLUSION
ZES is frequently overlooked, and patients often continue to experience unresolved symptoms related to hypergastrinemia. Due to its complexity and ability to mimic other disorders—as well as the implications of duodenal versus pancreatic location, and other disorders of the kidney or endocrine system suggestive of MEN1—ZES should be ruled out in any patient with unexplained persistent GERD, peptic ulcer disease, elevated FSG, chronic diarrhea, and/or abdominal pain.5
The gastrinoma itself is a well-differentiated and slow-growing tumor in the majority of cases, making the prognosis for ZES favorable for long-term survival. Proper pharmacologic management is instrumental for controlling symptoms and decreasing acid production. Surgical resection offers patients the best chance for a complete cure. Clinicians and patients should be well educated about ZES in order to successfully manage the disorder.
1. Tomassetti P, Campana D, Piscitelli L, et al. Treatment of Zollinger-Ellison syndrome. World J Gastroenterol. 2005; 11(35):5423-5432.
2. Metz DC. Diagnosis of the Zollinger-Ellison syndrome. Clin Gastroenterol Hepatol. 2016;10(2):126-130.
3. Epelboym I, Mazeh H. Zollinger-Ellison syndrome: classical considerations and current controversies. Oncologist. 2014; 19(1):44-50.
4. Papadakis M, McPhee S, Rabow M. Current Medical Diagnosis and Treatment 2014. New York, NY: McGraw-Hill Education; 2014:600-601.
5. Feldman M, Friedman LS, Lawrence BJ. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Philadelphia, PA: Saunders/Elsevier; 2016:511-515.
6. Ito T, Cadiot G, Jensen RT. Diagnosis of Zollinger-Ellison syndrome: increasingly difficult. World J Gastroenterol. 2012; 18(39):5495-5503.
7. Blonski WC, Katzka DA, Lichtenstein GR, Metz DC. Idiopathic gastric acid hypersecretion presenting as a diarrheal disorder and mimicking both Zollinger-Ellison syndrome and Crohn’s disease. Eur J Gastroenterol Hepatol. 2005;17(4):441-444.
8. Roy PK. Zollinger-Ellison syndrome clinical presentation. http://emedicine.medscape.com/article/183555-clinical#b4. Accessed June 14, 2017.
9. Berna MJ, Hoffmann KM, Serrano J, et al. Serum gastrin in Zollinger-Ellison syndrome: I. prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2229 cases from the literature. Medicine (Baltimore). 2006;85(6):295-330.
10. Moore AR, Varro A, Pritchard M. Zollinger-Ellison syndrome. Gastrointestinal Nursing. 2012;10(5):44-49.
11. Weber HC, Venzon DJ, Lin JT, et al. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology. 1995;108(6):1637-1649.
12. Berger AC, Gibril F, Venzon DJ, et al. Prognostic value of initial fasting serum gastrin levels in patients with Zollinger-Ellison syndrome. J Clin Oncol. 2001;19(12):3051-3057.
1. Tomassetti P, Campana D, Piscitelli L, et al. Treatment of Zollinger-Ellison syndrome. World J Gastroenterol. 2005; 11(35):5423-5432.
2. Metz DC. Diagnosis of the Zollinger-Ellison syndrome. Clin Gastroenterol Hepatol. 2016;10(2):126-130.
3. Epelboym I, Mazeh H. Zollinger-Ellison syndrome: classical considerations and current controversies. Oncologist. 2014; 19(1):44-50.
4. Papadakis M, McPhee S, Rabow M. Current Medical Diagnosis and Treatment 2014. New York, NY: McGraw-Hill Education; 2014:600-601.
5. Feldman M, Friedman LS, Lawrence BJ. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Philadelphia, PA: Saunders/Elsevier; 2016:511-515.
6. Ito T, Cadiot G, Jensen RT. Diagnosis of Zollinger-Ellison syndrome: increasingly difficult. World J Gastroenterol. 2012; 18(39):5495-5503.
7. Blonski WC, Katzka DA, Lichtenstein GR, Metz DC. Idiopathic gastric acid hypersecretion presenting as a diarrheal disorder and mimicking both Zollinger-Ellison syndrome and Crohn’s disease. Eur J Gastroenterol Hepatol. 2005;17(4):441-444.
8. Roy PK. Zollinger-Ellison syndrome clinical presentation. http://emedicine.medscape.com/article/183555-clinical#b4. Accessed June 14, 2017.
9. Berna MJ, Hoffmann KM, Serrano J, et al. Serum gastrin in Zollinger-Ellison syndrome: I. prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2229 cases from the literature. Medicine (Baltimore). 2006;85(6):295-330.
10. Moore AR, Varro A, Pritchard M. Zollinger-Ellison syndrome. Gastrointestinal Nursing. 2012;10(5):44-49.
11. Weber HC, Venzon DJ, Lin JT, et al. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology. 1995;108(6):1637-1649.
12. Berger AC, Gibril F, Venzon DJ, et al. Prognostic value of initial fasting serum gastrin levels in patients with Zollinger-Ellison syndrome. J Clin Oncol. 2001;19(12):3051-3057.
2017 Update on infectious disease
In this Update we review the results of 4 recent investigations that have important implications:
- the first analysis of the US Zika Virus Infection in Pregnancy Registry
- a study revealing an improved antibiotic regimen to prevent postcesarean infection
- an important new methodology for reducing the rate of perinatal transmission of hepatitis B virus (HBV) infection
- the risks and benefits of combination antiretroviral therapy (ART) in pregnancy.
Zika virus-associated birth defect rates similar regardless of symptom presence; first-trimester exposure has highest rate of anomalies
Honein MA, Dawson AL, Petersen EE, et al; US Zika Pregnancy Registry Collaboration. Birth defects among fetuses and infants of US women with evidence of possible Zika virus infection during pregnancy. JAMA. 2017;317(1):59-68.
Honein and colleagues provide a summary of the data from the US Zika Virus in Pregnancy Registry (a collaboration between the Centers for Disease Control and Prevention and state and local health departments), estimating the proportion of fetuses and infants with birth defects based on maternal symptoms of Zika virus infection and trimester of possible infection.
Related article:
Zika virus: Counseling considerations for this emerging perinatal threat
Details of the study
The authors evaluated the outcomes of 442 women who had laboratory evidence of a possible Zika virus infection during pregnancy. Overall, 26 infants (6%; 95% confidence interval (CI), 4%-8%) had evidence of birth defects related to the Zika virus. Of note, abnormalities were detected in 16 of the 271 children (6%; 95% CI, 4%-9%) born to women who were asymptomatic and 10 of 167 (6%; 95% CI, 3%-11%) children delivered to women with symptomatic infections.
The most common birth defect was microcephaly, although other serious central nervous system abnormalities were noted as well. Nine of 85 women (11%; 95% CI, 6%-19%) who had exposure only during the first trimester had infants with birth defects. There were no documented abnormalities in infants born to mothers who developed Zika virus infection only in the second or third trimester.
Related article:
Zika virus update: A rapidly moving target
Key study findings
This article is important for several reasons. First, the authors describe the largest series of pregnant women in the United States with Zika virus infection. All of these patients developed Zika virus infection as a result of foreign travel or exposure to sexual partners who had traveled to Zika virus endemic areas. Second, the authors confirmed findings that previously had been based only on mathematical models rather than on actual case series. Specifically, they demonstrated that the risk of a serious birth defect following first-trimester exposure to Zika virus infection was approximately 11%, with a 95% CI that extended from 6% to 19%. Finally, Honein and colleagues highlighted the key fact that the risk of a serious birth defect was comparable in mothers who had either an asymptomatic or a symptomatic infection, a finding that seems somewhat counterintuitive.
This study's critical observations are a "call to action" for clinicians who provide prenatal care.1,2 Proactive steps include:
- For patients considering pregnancy, strongly advise against travel to any area of the world where Zika virus is endemic until an effective vaccine is available to protect against this infection.
- For any woman with a newly diagnosed pregnancy, ask about travel to an endemic area.
- Inquire also about a pregnant woman's exposure to partners who live in, or who have traveled to, areas of the world where Zika virus infection is endemic.
- Be aware that both asymptomatic and symptomatic infection in the first trimester of pregnancy pose a grave risk to the fetus.
- Recognize that, although microcephaly is the principal abnormality associated with Zika virus infection, other central nervous system anomalies also may occur in these children. These include ventriculomegaly, subcortical calcifications, abnormalities of the corpus callosum, cerebral atrophy, and cerebellar abnormalities. In addition, infected infants may have arthrogryposis.
- Finally, as Honein and colleagues noted, laboratory testing for Zika virus infection is imperfect. In the early stages of infection or exposure, testing for Zika virus infection by polymerase chain reaction (PCR) in both serum and urine is the preferred test. After a period of 2 weeks, the preferred laboratory test is an immunoglobulin M (IgM) assay. Positive tests on the IgM assay must be confirmed by the plaque neutralization reduction test--a very important test for differentiating Zika virus infection from infection caused by other arboviruses, such as those that cause dengue fever and chikungunya.
Read about prophylaxis for postcesarean infection
Two antibiotics before cesarean delivery reduce infection rates further than one agent
Tita AT, Szychowski JM, Boggess K, et al; for the C/SOAP Trial Consortium. Adjunctive azithromycin prophylaxis for cesarean delivery. N Engl J Med. 2016;375(13):1231-1241.
Tita and colleagues reported the results of a multicenter trial that was designed to assess whether a combination of 2 antibiotics, including one that specifically targets ureaplasma species, provided more effective prophylaxis against postcesarean infection than single-agent prophylaxis.
Details of the study
The Cesarean Section Optimal Antibiotic Prophylaxis (C/SOAP) trial was conducted at 14 centers in the United States and included 2,013 women who were at least at 24 weeks' gestation and who had a cesarean delivery during labor or after membrane rupture.
The authors randomly assigned 1,019 women to receive 500 mg of intravenous azithromycin plus conventional single-agent prophylaxis (usually cefazolin) and 994 women to receive a placebo plus conventional prophylaxis. The primary outcome was the composite of endometritis, wound infection, or other infection occurring within 6 weeks.
The authors observed that the primary outcome occurred in 62 women (6.1%) who received azithromycin plus conventional prophylaxis and in 119 women (12%) who received only single-agent prophylaxis. The relative risk of developing a postoperative infection was 0.51 in women who received the combined therapy. There were significant differences between the 2 groups in both the rates of endometritis (3.8% vs 6.1%, P = .02) and wound infection (2.4% vs 6.6%, P<.001). There were no differences between the groups in the frequency of the secondary neonatal composite outcome, which included neonatal death and serious neonatal complications.
Related article:
Preventing infection after cesarean delivery: 5 more evidence-based measures to consider
Efficacy of dual-agent prophylaxis
At present, the standard of care is to administer prophylactic antibiotics to all women having cesarean delivery, including women having a scheduled cesarean in the absence of labor or ruptured membranes. Multiple studies have shown clearly that prophylaxis reduces the frequency of endometritis and, in high-risk patient populations, wound infection, and that prophylaxis is most beneficial when administered prior to the time the surgical incision is made. The most commonly used drug for prophylaxis is cefazolin, a first-generation cephalosporin. The usual recommended dose is 2 g, administered immediately prior to surgery.3,4
Although most centers in the United States traditionally have used just a single antibiotic for prophylaxis, selected recent reports indicate that expanding the spectrum of activity of prophylactic antibiotics can result in additional beneficial effects. Specifically, Tita and colleagues evaluated an indigent patient population with an inherently high rate of postoperative infection.5 They showed that adding azithromycin 500 mg to cefazolin significantly reduced the rate of postcesarean endometritis. In a follow-up report from the same institution, Tita and colleagues demonstrated that adding azithromycin also significantly reduced the frequency of wound infection.6 Of note, in both these investigations, the antibiotics were administered after cord clamping. In a subsequent report, Ward and Duff showed that the combination of azithromycin plus cefazolin administered preoperatively resulted in a combined rate of endometritis and wound infection that was less than 3%.7
Related article:
Preventing infection after cesarean delivery: Evidence-based guidance
C/SOAP trial confirmed lower infection rates with combined regimen
Results of the present study confirm the findings of these 3 investigations. The trial included a large sample size. The study was carefully designed, and the end points were clearly defined. It included only patients at increased risk for postoperative infection by virtue of being in labor or having ruptured membranes at the time of cesarean delivery. Patients who received standard prophylaxis, usually cefazolin, plus azithromycin had a significantly lower risk of postcesarean endometritis and wound infection compared with patients who received a single antibiotic. The overall risk of infection was reduced by an impressive 50%.
Based on the results of the C/SOAP trial, considered in conjunction with the 3 previously cited investigations,5-7 we believe that the standard approach to antibiotic prophylaxis should be to administer both cefazolin, in a dose of 2 g, plus azithromycin, in a dose of 500 mg, prior to surgery. Cefazolin can be administered as an intravenous bolus; azithromycin should be administered as a continuous infusion over a 60-minute period prior to surgery. Clinicians may anticipate very low rates of both endometritis and wound infection with this regimen.
Read about reducing HBV transmission
Tenofovir treatment in pregnant women with HBV reduces vertical transmission
Pan CQ, Duan Z, Dai E, et al; China Study Group for the Mother-to-Child Transmission of Hepatitis B. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med. 2016;374(24):2324-2334.
A multicenter, open-label, randomized, parallel-group investigation was conducted from March 2012 to June 2013 at academic tertiary care centers in 5 geographic regions of China. Two hundred mothers, who were positive for both hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg) and who had HBV DNA concentrations of 200,000 IU/mL or greater, were randomly assigned in a 1:1 ratio to either tenofovir or to usual treatment. Exclusion criteria were coexistent viral infections or medical conditions, renal failure, laboratory abnormalities, fetal deformities, and use of many medications.
Related article:
5 ways to reduce infection risk during pregnancy
Details of the study
Women in the active treatment group received tenofovir 300 mg by mouth daily from 30 to 32 weeks' gestation until postpartum week 4. Patients were monitored every 4 weeks in the antepartum period for adverse events and laboratory abnormalities. In the postpartum period, mother-infant dyads were evaluated at weeks 4, 12, 24, and 28.
Primary outcomes were the rates of mother-to-child transmission and birth defects with, or without, tenofovir exposure. Secondary outcomes were the percentage of mothers who had an HBV DNA serum concentration of less than 200,000 IU/mL at delivery and the percentage of mothers with HBeAg or HBsAg loss or seroconversion at postpartum week 28. Safety outcomes included the adverse event profile of tenofovir in mothers and safety events in the mother-infant dyads. These outcomes encompassed all adverse events and drug discontinuations in patients who received at least one dose of tenofovir.
Sixty-eight percent of mothers in the tenofovir group, compared with 2% of mothers in the control group, had HBV levels less than 200,000 IU/mL at delivery (P<.001). The rate of mother-to-child HBV transmission at postpartum week 28 was lower in the tenofovir group. In the intention-to-treat analysis, the rate was 5% (95% CI, 1-10; 5 of 97 infants) in the tenofovir group versus 18% (95% CI, 10-26; 18 of 100 infants) in the control group (P = .007). In the per-protocol analysis, the rate was 0% (95% CI, 0-3; 0 of 92 infants) in the tenofovir group versus 7% (95% CI, 2-12; 6 of 88 infants) in the control group (P = .01). Maternal and infant safety profiles were similar between the 2 groups, with the exception of elevated creatinine kinase and alanine aminotransferase levels in mothers treated with tenofovir. Maternal HBV serologic titers did not differ significantly between the 2 groups.
Study strengths and limitations
This study's strengths include a multicenter, randomized controlled design, with strict inclusion and exclusion criteria. The results are clinically relevant and of global impact, with potential to decrease morbidity and mortality from HBV infection in children born to infected mothers.
A limitation, however, is that the study was probably underpowered to detect small differences in the rate of birth defects between the tenofovir and usual-care treatment groups. Additionally, some patients ceased taking tenofovir in the postpartum time period. Abrupt cessation may be associated with acute, severe HBV exacerbation.
HBV is a serious infection that can lead to liver failure and cirrhosis. HBV infection is most likely to have long-term sequelae if acquired in the perinatal period. If untreated, chronic HBV infection will develop in 80% to 90% of infants born to mothers positive for HBeAg. Current immunoprophylaxis for at-risk neonates is postnatal HBV vaccine in combination with hepatitis B immune globulin. Unfortunately, this immunoprophylaxis fails in 10% to 30% of infants born to mothers with an HBV DNA level of greater than 6 log 10 copies/mL. Thus, the observations of Pan and colleagues are welcome findings.
Based on the results of this study, we recommend the use of tenofovir to decrease HBV transmission during pregnancy for women with high viral loads.
Benefits of ART for reducing mother-to-baby HIV transmission outweigh higher risk of adverse outcomes
Fowler MG, Qin M, Fiscus SA, et al; IMPAACT 1077BF/1077FF PROMISE Study Team. Benefits and risks of antiretroviral therapy for perinatal HIV prevention. N Engl J Med. 2016;375(18):1726-1737.
Part of the larger PROMISE (Promoting Maternal and Infant Survival Everywhere) trial, a study by Fowler and colleagues compared the relative efficacy and safety of various proven ART strategies for prevention of mother-to-child transmission of HIV infection in women with relatively high CD4 counts.
Details of the study
The trial was conducted at 14 sites in 7 countries. Patients were stratified according to HBV coinfection status and country of origin. The primary efficacy outcome was frequency of early infant HIV infection.
Women were randomly assigned to 1 of 3 treatment categories:
- zidovudine alone (zidovudine plus a single intrapartum dose of nevirapine, followed by 6 to 14 days of tenofovir plus emtricitabine postpartum)
- zidovudine-based ART (zidovudine in combination with lamivudine and lopinavir-ritonavir)
- tenofovir-based ART (tenofovir in combination with emtricitabine and lopinavir-ritonavir).
All regimens were continued through 6 to 14 days postpartum. All infants received nevirapine at birth and in the immediate postpartum period.
Two trial periods. During period 1 (April 2011-September 2012), safety data on tenofovir in pregnancy were limited. Women without HBV coinfection were assigned only to zidovudine alone or zidovudine-based ART. During period 2 (October 2012-October 2014), since more information about tenofovir use in pregnancy was available, the study protocol was modified to allow women to be assigned to any of the 3 regimens, regardless of their HBV status.
Inclusion criteria were as follows: CD4 count of at least 350 cells/mm3 (or country-specific threshold for initiating triple-drug ART, if that threshold was higher), gestation of at least 14 weeks and not in labor, no previous use of triple-drug ART, no clinical or immune-related indication for triple-drug ART, hemoglobin level of at least 6.5 g/dL, an absolute neutrophil count of at least 750 cells/mm3, an alanine aminotransferase level of less than 2.5 times the upper limit of normal range, an estimated creatinine clearance of greater than 60 mL/min, and no serious pregnancy complications. Patients were excluded if they had active tuberculosis, HBV infection requiring treatment, a structural or conduction heart defect, or a fetus with a serious congenital malformation.
Primary outcomes. The primary efficacy outcome was early infant HIV infection, defined as a positive infant HIV nucleic acid test result at birth or at 1 week postpartum. The primary safety outcome was a composite of adverse events.
Adverse events in mothers were defined as hematologic abnormalities, abnormal blood chemical values, or abnormal signs/symptoms during pregnancy through 1 week postpartum. Severe pregnancy composite outcomes were low birth weight (<2,500 g), preterm delivery before 37 weeks' gestation, spontaneous abortion (<20 weeks), stillbirth (≥20 weeks), or congenital anomaly. Adverse events in infants were defined as death from any cause, hematologic abnormalities or abnormal blood chemical values, and abnormal signs/symptoms through 1 week postpartum.
A total of 3,490 mother-infant sets were included in the analysis (2,261 during trial period 1 and 1,229 during trial period 2). Baseline maternal characteristics were well balanced between groups. Most women were African, young (median age, 26 years), and asymptomatic.
Related article:
2016 Update on infectious disease
Study results
The combined maternal ART-treated groups had significantly lower rates of early transmission of HIV infection compared with the zidovudine-alone group (0.5% vs 1.8%, -1.3 percentage points; CI, -2.1 to -0.4). The zidovudine-based ART-treated group had a significantly higher rate of infant HIV-free survival through postpartum week 1 than did the zidovudine-alone group (P = .001) or the tenofovir-based ART group (P = .002).
When examining trial periods 1 and 2 combined, the zidovudine-based ART group experienced significantly higher rates of any adverse event than those receiving zidovudine alone (21.1% vs 17.3%, P = .008) and higher rates of abnormal blood chemical values (5.8% vs 1.3%, P<.001). During period 2 alone, the tenofovir-based ART group had significantly higher rates of abnormal blood chemical values than did the zidovudine-alone group (2.9% vs 0.8%, P = .03). There were no significant differences between the 2 ART treatment groups. No maternal deaths occurred during the study, and the trial-drug discontinuation rate was low (2%-5%) and did not vary among the 3 groups.
During trial periods 1 and 2, the zidovudine-based ART group had significantly higher rates of adverse pregnancy outcomes than did the zidovudine-alone group (40% vs 27.5%, P<.001). These included low birth weight less than 2,500 g (23% vs 12%) and preterm delivery before 37 weeks (20.5% vs 13.1%). During trial period 2, the tenofovir-based ART group had significantly higher rates of adverse pregnancy outcomes than did the zidovudine-alone group (34.7% vs 27.2%, P = .04). There were no significant differences for any outcome between the 2 ART-treated groups, and there were no significant differences in stillbirth or spontaneous abortion and congenital anomalies among the 3 groups.
Regarding severe pregnancy outcomes, there were no significant differences (composite or individual) between the zidovudine-based ART group and the zidovudine-alone group. The tenofovir-based ART group experienced significantly higher rates of composite severe adverse pregnancy outcomes compared with the zidovudine-based ART group (9.2% vs 4.3%, P = .02), and very preterm birth before 34 weeks (6.0% vs 2.6%, P = .04).
Infant safety outcomes were also examined. There were no significant differences for composite or individual adverse neonatal outcomes other than death. The tenofovir-based ART group experienced a significantly higher rate of infant death than did the zidovudine-based ART group (4.4% vs 0.6%, P<.001). However, a post hoc analysis suggested that extreme prematurity contributed to the infant mortality.
Limitations of the study
This study had minor limitations. It divided patients into only 2 major categories with respect to gestational age--more than or less than 34 weeks. Some maternal medical conditions, such as malaria, were not controlled for. In addition, breastfeeding and formula feeding were combined for analysis, and we know that breastfeeding would inherently confer a higher risk of HIV transmission.
Nevertheless, this study was thoughtfully designed and carefully conducted, and the results are of significant global impact.
Although antenatal ART was associated with a higher risk of adverse maternal and neonatal outcomes when compared with zidovudine alone, these risks are outweighed by the benefit of significantly lower rates of early HIV transmission. Therefore, women who meet the World Health Organization's (WHO) eligibility criteria should be treated with combination ART during pregnancy. The WHO major eligibility criteria for ART during pregnancy are:
- CD4 count of ≤350 cells/mm3, irrespective of clinical staging
- clinical stage 3 or stage 4 disease, irrespective of CD4 cell count.
The WHO recommends starting ART at 14 weeks' gestation.8
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Chelliah A, Duff P. Zika virus: counseling considerations for this emerging perinatal threat. OBG Manag. 2016;28(3):28-34.
- Chelliah A, Duff P. Zika virus update: a rapidly moving target. OBG Manag. 2016;28(8):17-26.
- Patrick KE, Deatsman SL, Duff P. Preventing infection after cesarean delivery: evidence-based guidance. OBG Manag. 2016;28(11):41-47.
- Patrick KE, Deatsman SL, Duff P. Preventing infection after cesarean delivery: 5 more evidenced-based methods to consider. OBG Manag. 2016;28(12):18-22.
- Tita AT, Hauth JC, Grimes A, Owen J, Stamm AM, Andrews WW. Decreasing incidence of postcesarean endometritis with extended-spectrum antibiotic prophylaxis. Obstet Gynecol. 2008;111(1):51-56.
- Tita AT, Owen J, Stamm AM, Grimes A, Hauth JC, Andrews WW. Impact of extended-spectrum antibiotic prophylaxis on incidence of postcesarean surgical wound infection. Am J Obstet Gynecol. 2008;199(3):303.e1-e3.
- Ward E, Duff P. A comparison of 3 antibiotic regimens for prevention of postcesarean endometritis: an historical cohort study. Am J Obstet Gynecol. 2016;214(6):751.e1-e4.
- New guidance on prevention of mother-to-child transmission of HIV and infant feeding in the context of HIV. World Health Organization website. http://www.who.int/hiv/pub/mtct/PMTCTfactsheet/en/. Published July 20, 2010. Accessed June 16, 2017.

Dr. Jackson is a third-year Maternal-Fetal Medicine Fellow in the Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.

Dr. Duff is Associate Dean for Student Affairs and Professor of Obstetrics and Gynecology in the Division of Maternal-Fetal Medicine, University of Florida College of Medicine.
The authors report no financial relationships relevant to this article.
Dr. Jackson is a third-year Maternal-Fetal Medicine Fellow in the Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
Dr. Duff is Associate Dean for Student Affairs and Professor of Obstetrics and Gynecology in the Division of Maternal-Fetal Medicine, University of Florida College of Medicine.
The authors report no financial relationships relevant to this article.
Dr. Jackson is a third-year Maternal-Fetal Medicine Fellow in the Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
Dr. Duff is Associate Dean for Student Affairs and Professor of Obstetrics and Gynecology in the Division of Maternal-Fetal Medicine, University of Florida College of Medicine.
The authors report no financial relationships relevant to this article.
In this Update we review the results of 4 recent investigations that have important implications:
- the first analysis of the US Zika Virus Infection in Pregnancy Registry
- a study revealing an improved antibiotic regimen to prevent postcesarean infection
- an important new methodology for reducing the rate of perinatal transmission of hepatitis B virus (HBV) infection
- the risks and benefits of combination antiretroviral therapy (ART) in pregnancy.
Zika virus-associated birth defect rates similar regardless of symptom presence; first-trimester exposure has highest rate of anomalies
Honein MA, Dawson AL, Petersen EE, et al; US Zika Pregnancy Registry Collaboration. Birth defects among fetuses and infants of US women with evidence of possible Zika virus infection during pregnancy. JAMA. 2017;317(1):59-68.
Honein and colleagues provide a summary of the data from the US Zika Virus in Pregnancy Registry (a collaboration between the Centers for Disease Control and Prevention and state and local health departments), estimating the proportion of fetuses and infants with birth defects based on maternal symptoms of Zika virus infection and trimester of possible infection.
Related article:
Zika virus: Counseling considerations for this emerging perinatal threat
Details of the study
The authors evaluated the outcomes of 442 women who had laboratory evidence of a possible Zika virus infection during pregnancy. Overall, 26 infants (6%; 95% confidence interval (CI), 4%-8%) had evidence of birth defects related to the Zika virus. Of note, abnormalities were detected in 16 of the 271 children (6%; 95% CI, 4%-9%) born to women who were asymptomatic and 10 of 167 (6%; 95% CI, 3%-11%) children delivered to women with symptomatic infections.
The most common birth defect was microcephaly, although other serious central nervous system abnormalities were noted as well. Nine of 85 women (11%; 95% CI, 6%-19%) who had exposure only during the first trimester had infants with birth defects. There were no documented abnormalities in infants born to mothers who developed Zika virus infection only in the second or third trimester.
Related article:
Zika virus update: A rapidly moving target
Key study findings
This article is important for several reasons. First, the authors describe the largest series of pregnant women in the United States with Zika virus infection. All of these patients developed Zika virus infection as a result of foreign travel or exposure to sexual partners who had traveled to Zika virus endemic areas. Second, the authors confirmed findings that previously had been based only on mathematical models rather than on actual case series. Specifically, they demonstrated that the risk of a serious birth defect following first-trimester exposure to Zika virus infection was approximately 11%, with a 95% CI that extended from 6% to 19%. Finally, Honein and colleagues highlighted the key fact that the risk of a serious birth defect was comparable in mothers who had either an asymptomatic or a symptomatic infection, a finding that seems somewhat counterintuitive.
This study's critical observations are a "call to action" for clinicians who provide prenatal care.1,2 Proactive steps include:
- For patients considering pregnancy, strongly advise against travel to any area of the world where Zika virus is endemic until an effective vaccine is available to protect against this infection.
- For any woman with a newly diagnosed pregnancy, ask about travel to an endemic area.
- Inquire also about a pregnant woman's exposure to partners who live in, or who have traveled to, areas of the world where Zika virus infection is endemic.
- Be aware that both asymptomatic and symptomatic infection in the first trimester of pregnancy pose a grave risk to the fetus.
- Recognize that, although microcephaly is the principal abnormality associated with Zika virus infection, other central nervous system anomalies also may occur in these children. These include ventriculomegaly, subcortical calcifications, abnormalities of the corpus callosum, cerebral atrophy, and cerebellar abnormalities. In addition, infected infants may have arthrogryposis.
- Finally, as Honein and colleagues noted, laboratory testing for Zika virus infection is imperfect. In the early stages of infection or exposure, testing for Zika virus infection by polymerase chain reaction (PCR) in both serum and urine is the preferred test. After a period of 2 weeks, the preferred laboratory test is an immunoglobulin M (IgM) assay. Positive tests on the IgM assay must be confirmed by the plaque neutralization reduction test--a very important test for differentiating Zika virus infection from infection caused by other arboviruses, such as those that cause dengue fever and chikungunya.
Read about prophylaxis for postcesarean infection
Two antibiotics before cesarean delivery reduce infection rates further than one agent
Tita AT, Szychowski JM, Boggess K, et al; for the C/SOAP Trial Consortium. Adjunctive azithromycin prophylaxis for cesarean delivery. N Engl J Med. 2016;375(13):1231-1241.
Tita and colleagues reported the results of a multicenter trial that was designed to assess whether a combination of 2 antibiotics, including one that specifically targets ureaplasma species, provided more effective prophylaxis against postcesarean infection than single-agent prophylaxis.
Details of the study
The Cesarean Section Optimal Antibiotic Prophylaxis (C/SOAP) trial was conducted at 14 centers in the United States and included 2,013 women who were at least at 24 weeks' gestation and who had a cesarean delivery during labor or after membrane rupture.
The authors randomly assigned 1,019 women to receive 500 mg of intravenous azithromycin plus conventional single-agent prophylaxis (usually cefazolin) and 994 women to receive a placebo plus conventional prophylaxis. The primary outcome was the composite of endometritis, wound infection, or other infection occurring within 6 weeks.
The authors observed that the primary outcome occurred in 62 women (6.1%) who received azithromycin plus conventional prophylaxis and in 119 women (12%) who received only single-agent prophylaxis. The relative risk of developing a postoperative infection was 0.51 in women who received the combined therapy. There were significant differences between the 2 groups in both the rates of endometritis (3.8% vs 6.1%, P = .02) and wound infection (2.4% vs 6.6%, P<.001). There were no differences between the groups in the frequency of the secondary neonatal composite outcome, which included neonatal death and serious neonatal complications.
Related article:
Preventing infection after cesarean delivery: 5 more evidence-based measures to consider
Efficacy of dual-agent prophylaxis
At present, the standard of care is to administer prophylactic antibiotics to all women having cesarean delivery, including women having a scheduled cesarean in the absence of labor or ruptured membranes. Multiple studies have shown clearly that prophylaxis reduces the frequency of endometritis and, in high-risk patient populations, wound infection, and that prophylaxis is most beneficial when administered prior to the time the surgical incision is made. The most commonly used drug for prophylaxis is cefazolin, a first-generation cephalosporin. The usual recommended dose is 2 g, administered immediately prior to surgery.3,4
Although most centers in the United States traditionally have used just a single antibiotic for prophylaxis, selected recent reports indicate that expanding the spectrum of activity of prophylactic antibiotics can result in additional beneficial effects. Specifically, Tita and colleagues evaluated an indigent patient population with an inherently high rate of postoperative infection.5 They showed that adding azithromycin 500 mg to cefazolin significantly reduced the rate of postcesarean endometritis. In a follow-up report from the same institution, Tita and colleagues demonstrated that adding azithromycin also significantly reduced the frequency of wound infection.6 Of note, in both these investigations, the antibiotics were administered after cord clamping. In a subsequent report, Ward and Duff showed that the combination of azithromycin plus cefazolin administered preoperatively resulted in a combined rate of endometritis and wound infection that was less than 3%.7
Related article:
Preventing infection after cesarean delivery: Evidence-based guidance
C/SOAP trial confirmed lower infection rates with combined regimen
Results of the present study confirm the findings of these 3 investigations. The trial included a large sample size. The study was carefully designed, and the end points were clearly defined. It included only patients at increased risk for postoperative infection by virtue of being in labor or having ruptured membranes at the time of cesarean delivery. Patients who received standard prophylaxis, usually cefazolin, plus azithromycin had a significantly lower risk of postcesarean endometritis and wound infection compared with patients who received a single antibiotic. The overall risk of infection was reduced by an impressive 50%.
Based on the results of the C/SOAP trial, considered in conjunction with the 3 previously cited investigations,5-7 we believe that the standard approach to antibiotic prophylaxis should be to administer both cefazolin, in a dose of 2 g, plus azithromycin, in a dose of 500 mg, prior to surgery. Cefazolin can be administered as an intravenous bolus; azithromycin should be administered as a continuous infusion over a 60-minute period prior to surgery. Clinicians may anticipate very low rates of both endometritis and wound infection with this regimen.
Read about reducing HBV transmission
Tenofovir treatment in pregnant women with HBV reduces vertical transmission
Pan CQ, Duan Z, Dai E, et al; China Study Group for the Mother-to-Child Transmission of Hepatitis B. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med. 2016;374(24):2324-2334.
A multicenter, open-label, randomized, parallel-group investigation was conducted from March 2012 to June 2013 at academic tertiary care centers in 5 geographic regions of China. Two hundred mothers, who were positive for both hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg) and who had HBV DNA concentrations of 200,000 IU/mL or greater, were randomly assigned in a 1:1 ratio to either tenofovir or to usual treatment. Exclusion criteria were coexistent viral infections or medical conditions, renal failure, laboratory abnormalities, fetal deformities, and use of many medications.
Related article:
5 ways to reduce infection risk during pregnancy
Details of the study
Women in the active treatment group received tenofovir 300 mg by mouth daily from 30 to 32 weeks' gestation until postpartum week 4. Patients were monitored every 4 weeks in the antepartum period for adverse events and laboratory abnormalities. In the postpartum period, mother-infant dyads were evaluated at weeks 4, 12, 24, and 28.
Primary outcomes were the rates of mother-to-child transmission and birth defects with, or without, tenofovir exposure. Secondary outcomes were the percentage of mothers who had an HBV DNA serum concentration of less than 200,000 IU/mL at delivery and the percentage of mothers with HBeAg or HBsAg loss or seroconversion at postpartum week 28. Safety outcomes included the adverse event profile of tenofovir in mothers and safety events in the mother-infant dyads. These outcomes encompassed all adverse events and drug discontinuations in patients who received at least one dose of tenofovir.
Sixty-eight percent of mothers in the tenofovir group, compared with 2% of mothers in the control group, had HBV levels less than 200,000 IU/mL at delivery (P<.001). The rate of mother-to-child HBV transmission at postpartum week 28 was lower in the tenofovir group. In the intention-to-treat analysis, the rate was 5% (95% CI, 1-10; 5 of 97 infants) in the tenofovir group versus 18% (95% CI, 10-26; 18 of 100 infants) in the control group (P = .007). In the per-protocol analysis, the rate was 0% (95% CI, 0-3; 0 of 92 infants) in the tenofovir group versus 7% (95% CI, 2-12; 6 of 88 infants) in the control group (P = .01). Maternal and infant safety profiles were similar between the 2 groups, with the exception of elevated creatinine kinase and alanine aminotransferase levels in mothers treated with tenofovir. Maternal HBV serologic titers did not differ significantly between the 2 groups.
Study strengths and limitations
This study's strengths include a multicenter, randomized controlled design, with strict inclusion and exclusion criteria. The results are clinically relevant and of global impact, with potential to decrease morbidity and mortality from HBV infection in children born to infected mothers.
A limitation, however, is that the study was probably underpowered to detect small differences in the rate of birth defects between the tenofovir and usual-care treatment groups. Additionally, some patients ceased taking tenofovir in the postpartum time period. Abrupt cessation may be associated with acute, severe HBV exacerbation.
HBV is a serious infection that can lead to liver failure and cirrhosis. HBV infection is most likely to have long-term sequelae if acquired in the perinatal period. If untreated, chronic HBV infection will develop in 80% to 90% of infants born to mothers positive for HBeAg. Current immunoprophylaxis for at-risk neonates is postnatal HBV vaccine in combination with hepatitis B immune globulin. Unfortunately, this immunoprophylaxis fails in 10% to 30% of infants born to mothers with an HBV DNA level of greater than 6 log 10 copies/mL. Thus, the observations of Pan and colleagues are welcome findings.
Based on the results of this study, we recommend the use of tenofovir to decrease HBV transmission during pregnancy for women with high viral loads.
Benefits of ART for reducing mother-to-baby HIV transmission outweigh higher risk of adverse outcomes
Fowler MG, Qin M, Fiscus SA, et al; IMPAACT 1077BF/1077FF PROMISE Study Team. Benefits and risks of antiretroviral therapy for perinatal HIV prevention. N Engl J Med. 2016;375(18):1726-1737.
Part of the larger PROMISE (Promoting Maternal and Infant Survival Everywhere) trial, a study by Fowler and colleagues compared the relative efficacy and safety of various proven ART strategies for prevention of mother-to-child transmission of HIV infection in women with relatively high CD4 counts.
Details of the study
The trial was conducted at 14 sites in 7 countries. Patients were stratified according to HBV coinfection status and country of origin. The primary efficacy outcome was frequency of early infant HIV infection.
Women were randomly assigned to 1 of 3 treatment categories:
- zidovudine alone (zidovudine plus a single intrapartum dose of nevirapine, followed by 6 to 14 days of tenofovir plus emtricitabine postpartum)
- zidovudine-based ART (zidovudine in combination with lamivudine and lopinavir-ritonavir)
- tenofovir-based ART (tenofovir in combination with emtricitabine and lopinavir-ritonavir).
All regimens were continued through 6 to 14 days postpartum. All infants received nevirapine at birth and in the immediate postpartum period.
Two trial periods. During period 1 (April 2011-September 2012), safety data on tenofovir in pregnancy were limited. Women without HBV coinfection were assigned only to zidovudine alone or zidovudine-based ART. During period 2 (October 2012-October 2014), since more information about tenofovir use in pregnancy was available, the study protocol was modified to allow women to be assigned to any of the 3 regimens, regardless of their HBV status.
Inclusion criteria were as follows: CD4 count of at least 350 cells/mm3 (or country-specific threshold for initiating triple-drug ART, if that threshold was higher), gestation of at least 14 weeks and not in labor, no previous use of triple-drug ART, no clinical or immune-related indication for triple-drug ART, hemoglobin level of at least 6.5 g/dL, an absolute neutrophil count of at least 750 cells/mm3, an alanine aminotransferase level of less than 2.5 times the upper limit of normal range, an estimated creatinine clearance of greater than 60 mL/min, and no serious pregnancy complications. Patients were excluded if they had active tuberculosis, HBV infection requiring treatment, a structural or conduction heart defect, or a fetus with a serious congenital malformation.
Primary outcomes. The primary efficacy outcome was early infant HIV infection, defined as a positive infant HIV nucleic acid test result at birth or at 1 week postpartum. The primary safety outcome was a composite of adverse events.
Adverse events in mothers were defined as hematologic abnormalities, abnormal blood chemical values, or abnormal signs/symptoms during pregnancy through 1 week postpartum. Severe pregnancy composite outcomes were low birth weight (<2,500 g), preterm delivery before 37 weeks' gestation, spontaneous abortion (<20 weeks), stillbirth (≥20 weeks), or congenital anomaly. Adverse events in infants were defined as death from any cause, hematologic abnormalities or abnormal blood chemical values, and abnormal signs/symptoms through 1 week postpartum.
A total of 3,490 mother-infant sets were included in the analysis (2,261 during trial period 1 and 1,229 during trial period 2). Baseline maternal characteristics were well balanced between groups. Most women were African, young (median age, 26 years), and asymptomatic.
Related article:
2016 Update on infectious disease
Study results
The combined maternal ART-treated groups had significantly lower rates of early transmission of HIV infection compared with the zidovudine-alone group (0.5% vs 1.8%, -1.3 percentage points; CI, -2.1 to -0.4). The zidovudine-based ART-treated group had a significantly higher rate of infant HIV-free survival through postpartum week 1 than did the zidovudine-alone group (P = .001) or the tenofovir-based ART group (P = .002).
When examining trial periods 1 and 2 combined, the zidovudine-based ART group experienced significantly higher rates of any adverse event than those receiving zidovudine alone (21.1% vs 17.3%, P = .008) and higher rates of abnormal blood chemical values (5.8% vs 1.3%, P<.001). During period 2 alone, the tenofovir-based ART group had significantly higher rates of abnormal blood chemical values than did the zidovudine-alone group (2.9% vs 0.8%, P = .03). There were no significant differences between the 2 ART treatment groups. No maternal deaths occurred during the study, and the trial-drug discontinuation rate was low (2%-5%) and did not vary among the 3 groups.
During trial periods 1 and 2, the zidovudine-based ART group had significantly higher rates of adverse pregnancy outcomes than did the zidovudine-alone group (40% vs 27.5%, P<.001). These included low birth weight less than 2,500 g (23% vs 12%) and preterm delivery before 37 weeks (20.5% vs 13.1%). During trial period 2, the tenofovir-based ART group had significantly higher rates of adverse pregnancy outcomes than did the zidovudine-alone group (34.7% vs 27.2%, P = .04). There were no significant differences for any outcome between the 2 ART-treated groups, and there were no significant differences in stillbirth or spontaneous abortion and congenital anomalies among the 3 groups.
Regarding severe pregnancy outcomes, there were no significant differences (composite or individual) between the zidovudine-based ART group and the zidovudine-alone group. The tenofovir-based ART group experienced significantly higher rates of composite severe adverse pregnancy outcomes compared with the zidovudine-based ART group (9.2% vs 4.3%, P = .02), and very preterm birth before 34 weeks (6.0% vs 2.6%, P = .04).
Infant safety outcomes were also examined. There were no significant differences for composite or individual adverse neonatal outcomes other than death. The tenofovir-based ART group experienced a significantly higher rate of infant death than did the zidovudine-based ART group (4.4% vs 0.6%, P<.001). However, a post hoc analysis suggested that extreme prematurity contributed to the infant mortality.
Limitations of the study
This study had minor limitations. It divided patients into only 2 major categories with respect to gestational age--more than or less than 34 weeks. Some maternal medical conditions, such as malaria, were not controlled for. In addition, breastfeeding and formula feeding were combined for analysis, and we know that breastfeeding would inherently confer a higher risk of HIV transmission.
Nevertheless, this study was thoughtfully designed and carefully conducted, and the results are of significant global impact.
Although antenatal ART was associated with a higher risk of adverse maternal and neonatal outcomes when compared with zidovudine alone, these risks are outweighed by the benefit of significantly lower rates of early HIV transmission. Therefore, women who meet the World Health Organization's (WHO) eligibility criteria should be treated with combination ART during pregnancy. The WHO major eligibility criteria for ART during pregnancy are:
- CD4 count of ≤350 cells/mm3, irrespective of clinical staging
- clinical stage 3 or stage 4 disease, irrespective of CD4 cell count.
The WHO recommends starting ART at 14 weeks' gestation.8
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
In this Update we review the results of 4 recent investigations that have important implications:
- the first analysis of the US Zika Virus Infection in Pregnancy Registry
- a study revealing an improved antibiotic regimen to prevent postcesarean infection
- an important new methodology for reducing the rate of perinatal transmission of hepatitis B virus (HBV) infection
- the risks and benefits of combination antiretroviral therapy (ART) in pregnancy.
Zika virus-associated birth defect rates similar regardless of symptom presence; first-trimester exposure has highest rate of anomalies
Honein MA, Dawson AL, Petersen EE, et al; US Zika Pregnancy Registry Collaboration. Birth defects among fetuses and infants of US women with evidence of possible Zika virus infection during pregnancy. JAMA. 2017;317(1):59-68.
Honein and colleagues provide a summary of the data from the US Zika Virus in Pregnancy Registry (a collaboration between the Centers for Disease Control and Prevention and state and local health departments), estimating the proportion of fetuses and infants with birth defects based on maternal symptoms of Zika virus infection and trimester of possible infection.
Related article:
Zika virus: Counseling considerations for this emerging perinatal threat
Details of the study
The authors evaluated the outcomes of 442 women who had laboratory evidence of a possible Zika virus infection during pregnancy. Overall, 26 infants (6%; 95% confidence interval (CI), 4%-8%) had evidence of birth defects related to the Zika virus. Of note, abnormalities were detected in 16 of the 271 children (6%; 95% CI, 4%-9%) born to women who were asymptomatic and 10 of 167 (6%; 95% CI, 3%-11%) children delivered to women with symptomatic infections.
The most common birth defect was microcephaly, although other serious central nervous system abnormalities were noted as well. Nine of 85 women (11%; 95% CI, 6%-19%) who had exposure only during the first trimester had infants with birth defects. There were no documented abnormalities in infants born to mothers who developed Zika virus infection only in the second or third trimester.
Related article:
Zika virus update: A rapidly moving target
Key study findings
This article is important for several reasons. First, the authors describe the largest series of pregnant women in the United States with Zika virus infection. All of these patients developed Zika virus infection as a result of foreign travel or exposure to sexual partners who had traveled to Zika virus endemic areas. Second, the authors confirmed findings that previously had been based only on mathematical models rather than on actual case series. Specifically, they demonstrated that the risk of a serious birth defect following first-trimester exposure to Zika virus infection was approximately 11%, with a 95% CI that extended from 6% to 19%. Finally, Honein and colleagues highlighted the key fact that the risk of a serious birth defect was comparable in mothers who had either an asymptomatic or a symptomatic infection, a finding that seems somewhat counterintuitive.
This study's critical observations are a "call to action" for clinicians who provide prenatal care.1,2 Proactive steps include:
- For patients considering pregnancy, strongly advise against travel to any area of the world where Zika virus is endemic until an effective vaccine is available to protect against this infection.
- For any woman with a newly diagnosed pregnancy, ask about travel to an endemic area.
- Inquire also about a pregnant woman's exposure to partners who live in, or who have traveled to, areas of the world where Zika virus infection is endemic.
- Be aware that both asymptomatic and symptomatic infection in the first trimester of pregnancy pose a grave risk to the fetus.
- Recognize that, although microcephaly is the principal abnormality associated with Zika virus infection, other central nervous system anomalies also may occur in these children. These include ventriculomegaly, subcortical calcifications, abnormalities of the corpus callosum, cerebral atrophy, and cerebellar abnormalities. In addition, infected infants may have arthrogryposis.
- Finally, as Honein and colleagues noted, laboratory testing for Zika virus infection is imperfect. In the early stages of infection or exposure, testing for Zika virus infection by polymerase chain reaction (PCR) in both serum and urine is the preferred test. After a period of 2 weeks, the preferred laboratory test is an immunoglobulin M (IgM) assay. Positive tests on the IgM assay must be confirmed by the plaque neutralization reduction test--a very important test for differentiating Zika virus infection from infection caused by other arboviruses, such as those that cause dengue fever and chikungunya.
Read about prophylaxis for postcesarean infection
Two antibiotics before cesarean delivery reduce infection rates further than one agent
Tita AT, Szychowski JM, Boggess K, et al; for the C/SOAP Trial Consortium. Adjunctive azithromycin prophylaxis for cesarean delivery. N Engl J Med. 2016;375(13):1231-1241.
Tita and colleagues reported the results of a multicenter trial that was designed to assess whether a combination of 2 antibiotics, including one that specifically targets ureaplasma species, provided more effective prophylaxis against postcesarean infection than single-agent prophylaxis.
Details of the study
The Cesarean Section Optimal Antibiotic Prophylaxis (C/SOAP) trial was conducted at 14 centers in the United States and included 2,013 women who were at least at 24 weeks' gestation and who had a cesarean delivery during labor or after membrane rupture.
The authors randomly assigned 1,019 women to receive 500 mg of intravenous azithromycin plus conventional single-agent prophylaxis (usually cefazolin) and 994 women to receive a placebo plus conventional prophylaxis. The primary outcome was the composite of endometritis, wound infection, or other infection occurring within 6 weeks.
The authors observed that the primary outcome occurred in 62 women (6.1%) who received azithromycin plus conventional prophylaxis and in 119 women (12%) who received only single-agent prophylaxis. The relative risk of developing a postoperative infection was 0.51 in women who received the combined therapy. There were significant differences between the 2 groups in both the rates of endometritis (3.8% vs 6.1%, P = .02) and wound infection (2.4% vs 6.6%, P<.001). There were no differences between the groups in the frequency of the secondary neonatal composite outcome, which included neonatal death and serious neonatal complications.
Related article:
Preventing infection after cesarean delivery: 5 more evidence-based measures to consider
Efficacy of dual-agent prophylaxis
At present, the standard of care is to administer prophylactic antibiotics to all women having cesarean delivery, including women having a scheduled cesarean in the absence of labor or ruptured membranes. Multiple studies have shown clearly that prophylaxis reduces the frequency of endometritis and, in high-risk patient populations, wound infection, and that prophylaxis is most beneficial when administered prior to the time the surgical incision is made. The most commonly used drug for prophylaxis is cefazolin, a first-generation cephalosporin. The usual recommended dose is 2 g, administered immediately prior to surgery.3,4
Although most centers in the United States traditionally have used just a single antibiotic for prophylaxis, selected recent reports indicate that expanding the spectrum of activity of prophylactic antibiotics can result in additional beneficial effects. Specifically, Tita and colleagues evaluated an indigent patient population with an inherently high rate of postoperative infection.5 They showed that adding azithromycin 500 mg to cefazolin significantly reduced the rate of postcesarean endometritis. In a follow-up report from the same institution, Tita and colleagues demonstrated that adding azithromycin also significantly reduced the frequency of wound infection.6 Of note, in both these investigations, the antibiotics were administered after cord clamping. In a subsequent report, Ward and Duff showed that the combination of azithromycin plus cefazolin administered preoperatively resulted in a combined rate of endometritis and wound infection that was less than 3%.7
Related article:
Preventing infection after cesarean delivery: Evidence-based guidance
C/SOAP trial confirmed lower infection rates with combined regimen
Results of the present study confirm the findings of these 3 investigations. The trial included a large sample size. The study was carefully designed, and the end points were clearly defined. It included only patients at increased risk for postoperative infection by virtue of being in labor or having ruptured membranes at the time of cesarean delivery. Patients who received standard prophylaxis, usually cefazolin, plus azithromycin had a significantly lower risk of postcesarean endometritis and wound infection compared with patients who received a single antibiotic. The overall risk of infection was reduced by an impressive 50%.
Based on the results of the C/SOAP trial, considered in conjunction with the 3 previously cited investigations,5-7 we believe that the standard approach to antibiotic prophylaxis should be to administer both cefazolin, in a dose of 2 g, plus azithromycin, in a dose of 500 mg, prior to surgery. Cefazolin can be administered as an intravenous bolus; azithromycin should be administered as a continuous infusion over a 60-minute period prior to surgery. Clinicians may anticipate very low rates of both endometritis and wound infection with this regimen.
Read about reducing HBV transmission
Tenofovir treatment in pregnant women with HBV reduces vertical transmission
Pan CQ, Duan Z, Dai E, et al; China Study Group for the Mother-to-Child Transmission of Hepatitis B. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med. 2016;374(24):2324-2334.
A multicenter, open-label, randomized, parallel-group investigation was conducted from March 2012 to June 2013 at academic tertiary care centers in 5 geographic regions of China. Two hundred mothers, who were positive for both hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg) and who had HBV DNA concentrations of 200,000 IU/mL or greater, were randomly assigned in a 1:1 ratio to either tenofovir or to usual treatment. Exclusion criteria were coexistent viral infections or medical conditions, renal failure, laboratory abnormalities, fetal deformities, and use of many medications.
Related article:
5 ways to reduce infection risk during pregnancy
Details of the study
Women in the active treatment group received tenofovir 300 mg by mouth daily from 30 to 32 weeks' gestation until postpartum week 4. Patients were monitored every 4 weeks in the antepartum period for adverse events and laboratory abnormalities. In the postpartum period, mother-infant dyads were evaluated at weeks 4, 12, 24, and 28.
Primary outcomes were the rates of mother-to-child transmission and birth defects with, or without, tenofovir exposure. Secondary outcomes were the percentage of mothers who had an HBV DNA serum concentration of less than 200,000 IU/mL at delivery and the percentage of mothers with HBeAg or HBsAg loss or seroconversion at postpartum week 28. Safety outcomes included the adverse event profile of tenofovir in mothers and safety events in the mother-infant dyads. These outcomes encompassed all adverse events and drug discontinuations in patients who received at least one dose of tenofovir.
Sixty-eight percent of mothers in the tenofovir group, compared with 2% of mothers in the control group, had HBV levels less than 200,000 IU/mL at delivery (P<.001). The rate of mother-to-child HBV transmission at postpartum week 28 was lower in the tenofovir group. In the intention-to-treat analysis, the rate was 5% (95% CI, 1-10; 5 of 97 infants) in the tenofovir group versus 18% (95% CI, 10-26; 18 of 100 infants) in the control group (P = .007). In the per-protocol analysis, the rate was 0% (95% CI, 0-3; 0 of 92 infants) in the tenofovir group versus 7% (95% CI, 2-12; 6 of 88 infants) in the control group (P = .01). Maternal and infant safety profiles were similar between the 2 groups, with the exception of elevated creatinine kinase and alanine aminotransferase levels in mothers treated with tenofovir. Maternal HBV serologic titers did not differ significantly between the 2 groups.
Study strengths and limitations
This study's strengths include a multicenter, randomized controlled design, with strict inclusion and exclusion criteria. The results are clinically relevant and of global impact, with potential to decrease morbidity and mortality from HBV infection in children born to infected mothers.
A limitation, however, is that the study was probably underpowered to detect small differences in the rate of birth defects between the tenofovir and usual-care treatment groups. Additionally, some patients ceased taking tenofovir in the postpartum time period. Abrupt cessation may be associated with acute, severe HBV exacerbation.
HBV is a serious infection that can lead to liver failure and cirrhosis. HBV infection is most likely to have long-term sequelae if acquired in the perinatal period. If untreated, chronic HBV infection will develop in 80% to 90% of infants born to mothers positive for HBeAg. Current immunoprophylaxis for at-risk neonates is postnatal HBV vaccine in combination with hepatitis B immune globulin. Unfortunately, this immunoprophylaxis fails in 10% to 30% of infants born to mothers with an HBV DNA level of greater than 6 log 10 copies/mL. Thus, the observations of Pan and colleagues are welcome findings.
Based on the results of this study, we recommend the use of tenofovir to decrease HBV transmission during pregnancy for women with high viral loads.
Benefits of ART for reducing mother-to-baby HIV transmission outweigh higher risk of adverse outcomes
Fowler MG, Qin M, Fiscus SA, et al; IMPAACT 1077BF/1077FF PROMISE Study Team. Benefits and risks of antiretroviral therapy for perinatal HIV prevention. N Engl J Med. 2016;375(18):1726-1737.
Part of the larger PROMISE (Promoting Maternal and Infant Survival Everywhere) trial, a study by Fowler and colleagues compared the relative efficacy and safety of various proven ART strategies for prevention of mother-to-child transmission of HIV infection in women with relatively high CD4 counts.
Details of the study
The trial was conducted at 14 sites in 7 countries. Patients were stratified according to HBV coinfection status and country of origin. The primary efficacy outcome was frequency of early infant HIV infection.
Women were randomly assigned to 1 of 3 treatment categories:
- zidovudine alone (zidovudine plus a single intrapartum dose of nevirapine, followed by 6 to 14 days of tenofovir plus emtricitabine postpartum)
- zidovudine-based ART (zidovudine in combination with lamivudine and lopinavir-ritonavir)
- tenofovir-based ART (tenofovir in combination with emtricitabine and lopinavir-ritonavir).
All regimens were continued through 6 to 14 days postpartum. All infants received nevirapine at birth and in the immediate postpartum period.
Two trial periods. During period 1 (April 2011-September 2012), safety data on tenofovir in pregnancy were limited. Women without HBV coinfection were assigned only to zidovudine alone or zidovudine-based ART. During period 2 (October 2012-October 2014), since more information about tenofovir use in pregnancy was available, the study protocol was modified to allow women to be assigned to any of the 3 regimens, regardless of their HBV status.
Inclusion criteria were as follows: CD4 count of at least 350 cells/mm3 (or country-specific threshold for initiating triple-drug ART, if that threshold was higher), gestation of at least 14 weeks and not in labor, no previous use of triple-drug ART, no clinical or immune-related indication for triple-drug ART, hemoglobin level of at least 6.5 g/dL, an absolute neutrophil count of at least 750 cells/mm3, an alanine aminotransferase level of less than 2.5 times the upper limit of normal range, an estimated creatinine clearance of greater than 60 mL/min, and no serious pregnancy complications. Patients were excluded if they had active tuberculosis, HBV infection requiring treatment, a structural or conduction heart defect, or a fetus with a serious congenital malformation.
Primary outcomes. The primary efficacy outcome was early infant HIV infection, defined as a positive infant HIV nucleic acid test result at birth or at 1 week postpartum. The primary safety outcome was a composite of adverse events.
Adverse events in mothers were defined as hematologic abnormalities, abnormal blood chemical values, or abnormal signs/symptoms during pregnancy through 1 week postpartum. Severe pregnancy composite outcomes were low birth weight (<2,500 g), preterm delivery before 37 weeks' gestation, spontaneous abortion (<20 weeks), stillbirth (≥20 weeks), or congenital anomaly. Adverse events in infants were defined as death from any cause, hematologic abnormalities or abnormal blood chemical values, and abnormal signs/symptoms through 1 week postpartum.
A total of 3,490 mother-infant sets were included in the analysis (2,261 during trial period 1 and 1,229 during trial period 2). Baseline maternal characteristics were well balanced between groups. Most women were African, young (median age, 26 years), and asymptomatic.
Related article:
2016 Update on infectious disease
Study results
The combined maternal ART-treated groups had significantly lower rates of early transmission of HIV infection compared with the zidovudine-alone group (0.5% vs 1.8%, -1.3 percentage points; CI, -2.1 to -0.4). The zidovudine-based ART-treated group had a significantly higher rate of infant HIV-free survival through postpartum week 1 than did the zidovudine-alone group (P = .001) or the tenofovir-based ART group (P = .002).
When examining trial periods 1 and 2 combined, the zidovudine-based ART group experienced significantly higher rates of any adverse event than those receiving zidovudine alone (21.1% vs 17.3%, P = .008) and higher rates of abnormal blood chemical values (5.8% vs 1.3%, P<.001). During period 2 alone, the tenofovir-based ART group had significantly higher rates of abnormal blood chemical values than did the zidovudine-alone group (2.9% vs 0.8%, P = .03). There were no significant differences between the 2 ART treatment groups. No maternal deaths occurred during the study, and the trial-drug discontinuation rate was low (2%-5%) and did not vary among the 3 groups.
During trial periods 1 and 2, the zidovudine-based ART group had significantly higher rates of adverse pregnancy outcomes than did the zidovudine-alone group (40% vs 27.5%, P<.001). These included low birth weight less than 2,500 g (23% vs 12%) and preterm delivery before 37 weeks (20.5% vs 13.1%). During trial period 2, the tenofovir-based ART group had significantly higher rates of adverse pregnancy outcomes than did the zidovudine-alone group (34.7% vs 27.2%, P = .04). There were no significant differences for any outcome between the 2 ART-treated groups, and there were no significant differences in stillbirth or spontaneous abortion and congenital anomalies among the 3 groups.
Regarding severe pregnancy outcomes, there were no significant differences (composite or individual) between the zidovudine-based ART group and the zidovudine-alone group. The tenofovir-based ART group experienced significantly higher rates of composite severe adverse pregnancy outcomes compared with the zidovudine-based ART group (9.2% vs 4.3%, P = .02), and very preterm birth before 34 weeks (6.0% vs 2.6%, P = .04).
Infant safety outcomes were also examined. There were no significant differences for composite or individual adverse neonatal outcomes other than death. The tenofovir-based ART group experienced a significantly higher rate of infant death than did the zidovudine-based ART group (4.4% vs 0.6%, P<.001). However, a post hoc analysis suggested that extreme prematurity contributed to the infant mortality.
Limitations of the study
This study had minor limitations. It divided patients into only 2 major categories with respect to gestational age--more than or less than 34 weeks. Some maternal medical conditions, such as malaria, were not controlled for. In addition, breastfeeding and formula feeding were combined for analysis, and we know that breastfeeding would inherently confer a higher risk of HIV transmission.
Nevertheless, this study was thoughtfully designed and carefully conducted, and the results are of significant global impact.
Although antenatal ART was associated with a higher risk of adverse maternal and neonatal outcomes when compared with zidovudine alone, these risks are outweighed by the benefit of significantly lower rates of early HIV transmission. Therefore, women who meet the World Health Organization's (WHO) eligibility criteria should be treated with combination ART during pregnancy. The WHO major eligibility criteria for ART during pregnancy are:
- CD4 count of ≤350 cells/mm3, irrespective of clinical staging
- clinical stage 3 or stage 4 disease, irrespective of CD4 cell count.
The WHO recommends starting ART at 14 weeks' gestation.8
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Chelliah A, Duff P. Zika virus: counseling considerations for this emerging perinatal threat. OBG Manag. 2016;28(3):28-34.
- Chelliah A, Duff P. Zika virus update: a rapidly moving target. OBG Manag. 2016;28(8):17-26.
- Patrick KE, Deatsman SL, Duff P. Preventing infection after cesarean delivery: evidence-based guidance. OBG Manag. 2016;28(11):41-47.
- Patrick KE, Deatsman SL, Duff P. Preventing infection after cesarean delivery: 5 more evidenced-based methods to consider. OBG Manag. 2016;28(12):18-22.
- Tita AT, Hauth JC, Grimes A, Owen J, Stamm AM, Andrews WW. Decreasing incidence of postcesarean endometritis with extended-spectrum antibiotic prophylaxis. Obstet Gynecol. 2008;111(1):51-56.
- Tita AT, Owen J, Stamm AM, Grimes A, Hauth JC, Andrews WW. Impact of extended-spectrum antibiotic prophylaxis on incidence of postcesarean surgical wound infection. Am J Obstet Gynecol. 2008;199(3):303.e1-e3.
- Ward E, Duff P. A comparison of 3 antibiotic regimens for prevention of postcesarean endometritis: an historical cohort study. Am J Obstet Gynecol. 2016;214(6):751.e1-e4.
- New guidance on prevention of mother-to-child transmission of HIV and infant feeding in the context of HIV. World Health Organization website. http://www.who.int/hiv/pub/mtct/PMTCTfactsheet/en/. Published July 20, 2010. Accessed June 16, 2017.
- Chelliah A, Duff P. Zika virus: counseling considerations for this emerging perinatal threat. OBG Manag. 2016;28(3):28-34.
- Chelliah A, Duff P. Zika virus update: a rapidly moving target. OBG Manag. 2016;28(8):17-26.
- Patrick KE, Deatsman SL, Duff P. Preventing infection after cesarean delivery: evidence-based guidance. OBG Manag. 2016;28(11):41-47.
- Patrick KE, Deatsman SL, Duff P. Preventing infection after cesarean delivery: 5 more evidenced-based methods to consider. OBG Manag. 2016;28(12):18-22.
- Tita AT, Hauth JC, Grimes A, Owen J, Stamm AM, Andrews WW. Decreasing incidence of postcesarean endometritis with extended-spectrum antibiotic prophylaxis. Obstet Gynecol. 2008;111(1):51-56.
- Tita AT, Owen J, Stamm AM, Grimes A, Hauth JC, Andrews WW. Impact of extended-spectrum antibiotic prophylaxis on incidence of postcesarean surgical wound infection. Am J Obstet Gynecol. 2008;199(3):303.e1-e3.
- Ward E, Duff P. A comparison of 3 antibiotic regimens for prevention of postcesarean endometritis: an historical cohort study. Am J Obstet Gynecol. 2016;214(6):751.e1-e4.
- New guidance on prevention of mother-to-child transmission of HIV and infant feeding in the context of HIV. World Health Organization website. http://www.who.int/hiv/pub/mtct/PMTCTfactsheet/en/. Published July 20, 2010. Accessed June 16, 2017.
Molecular Markers and Targeted Therapies in the Management of Non-Small Cell Lung Cancer
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5 BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
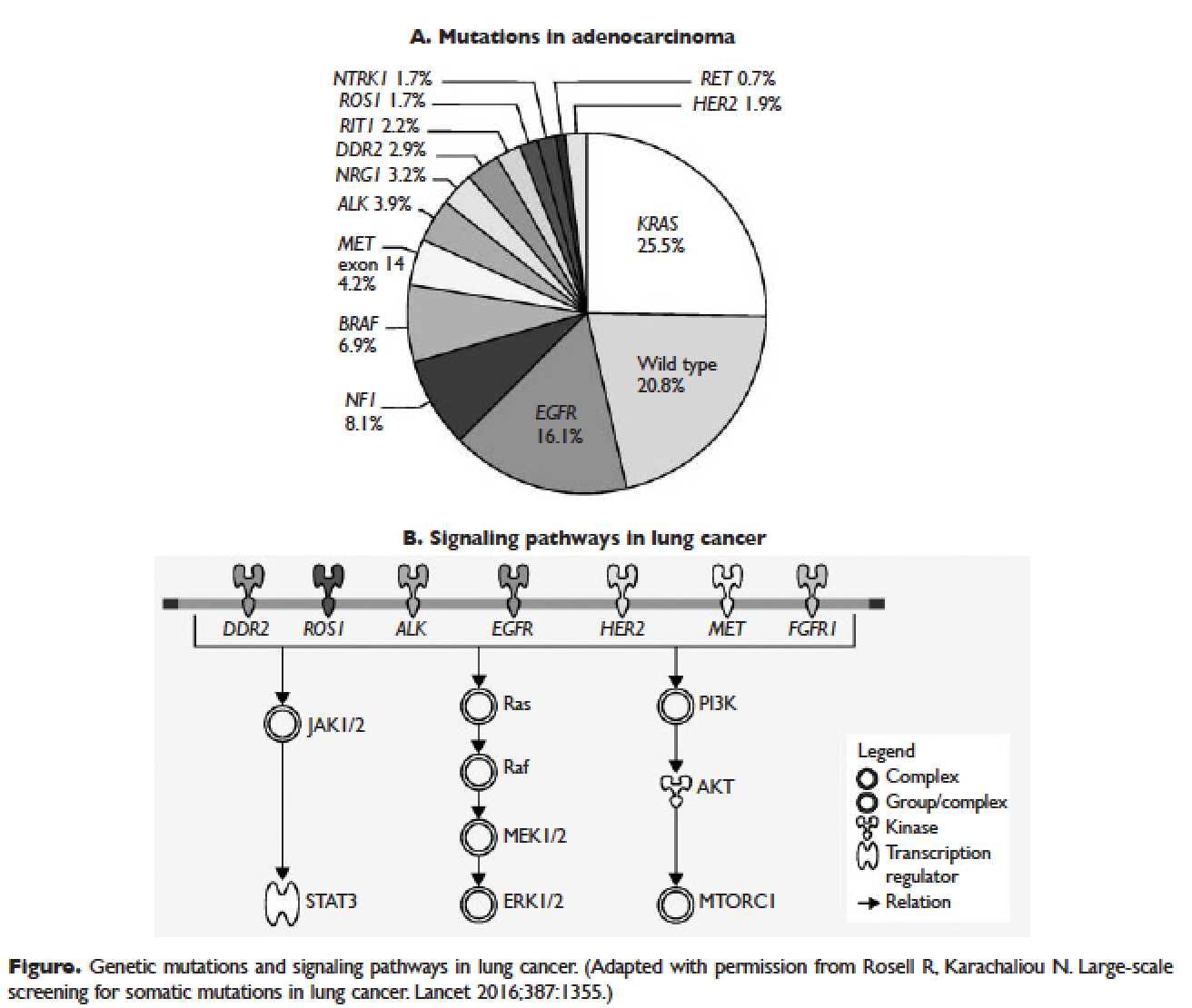
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
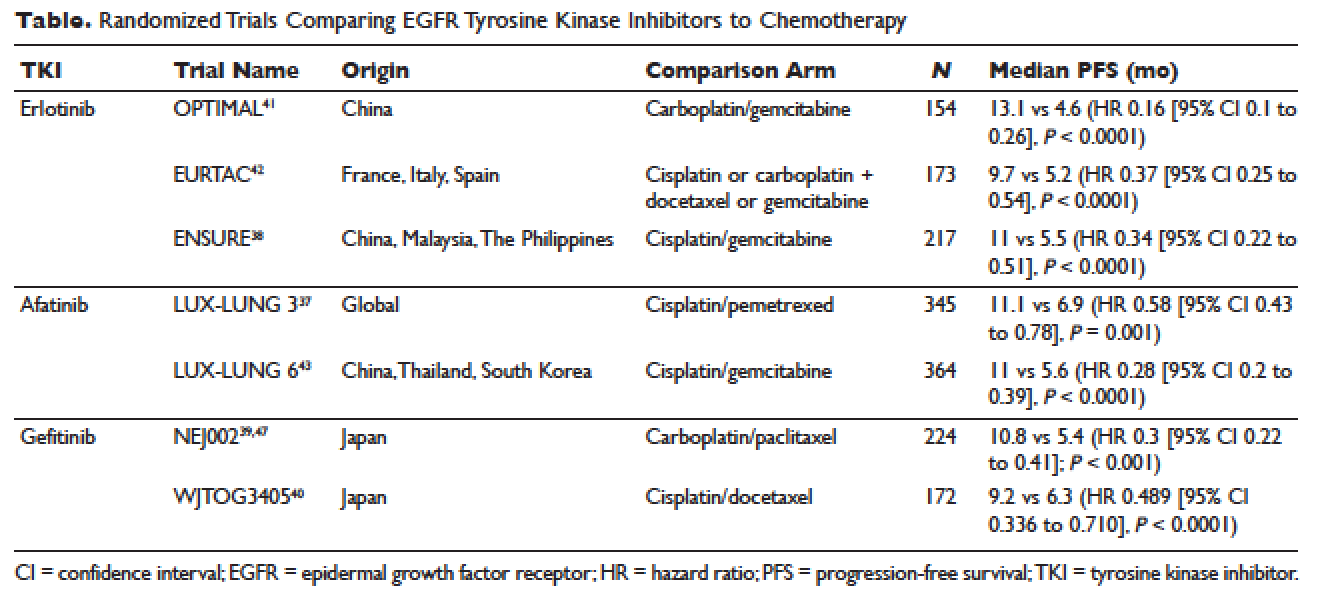
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62 ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5 BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62 ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
INTRODUCTION
Lung cancer is the second most common type of cancer in the United States, with 222,500 estimated new cases in 2017, according to the American Cancer Society.1 However, it is by far the number one cause of death due to cancer, with an estimated 155,870 lung cancer–related deaths occurring in 2017, which is higher than the number of deaths due to breast cancer, prostate cancer, and colorectal cancer combined.1,2 Despite slightly decreasing incidence and mortality over the past decade, largely due to smoking cessation, the 5-year survival rate of lung cancer remains dismal at approximately 18%.2–4
Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases.4 Traditionally, it is further divided based on histology: adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and not otherwise specified.5 Chemotherapy had been the cornerstone of treatment for stage IV NSCLC. It is not target-specific and is most effective against rapidly growing cells. Common adverse effects include alopecia, nausea/vomiting, myelosuppression, cardiotoxicity, neuropathy, and nephrotoxicity. However, this paradigm has shifted following the discovery of mutations of the epidermal growth factor receptor (EGFR) gene as an oncogenic driver that confers sensitivity to small molecule tyrosine kinase inhibitors (TKIs) targeting EGFR.6 The EGFR inhibitors are given orally and have a spectrum of toxicities (eg, such as rash, diarrhea, and elevated transaminases) different from that of systemic chemotherapy, which is often administered intravenously. Following the discovery of EGFR mutations, rearrangements of the anaplastic lymphoma kinase (ALK) gene7 and ROS1 gene8 were identified as targetable driver mutations in NSCLC. The frequency of both rearrangements is lower than that of EGFR mutations. Additionally, BRAF V600E mutation has been identified in NSCLC.9–12 This activation mutation is commonly seen in melanoma. Agents that have already been approved for the treatment of melanoma with the BRAF V600E mutation are being tested in NSCLC patients with this mutation.13–16
Given the effectiveness and tolerability of targeted therapy, identifying this distinct molecular subset of NSCLC patients is critical in treatment. Currently, molecular testing is mandatory in all stage IV patients with non-squamous cell carcinoma, as a preponderance of patients with driver mutations have this histology subtype.5,17–19 For patients with squamous cell carcinoma, molecular testing should be considered if the biopsy specimen is small, there is mixed histology, or the patient is a nonsmoker.5,20 Several techniques are commonly utilized in detecting these genetic alterations. EGFR mutation can be detected by polymerase chain reaction (PCR), ALK or ROS1 rearrangement can be detected by fluorescence in-situ hybridization (FISH), and immunohistochemistry (IHC) can also be used to detect ALK rearrangement. The current guideline is to use comprehensive genomic profiling to capture all the potential molecular targets simultaneously instead of running stepwise tests just for EGFR, ALK, and ROS1.5 BRAF V600E mutation,13–16 MET exon 14 skipping mutation,21–24 RET rearrangements,25–27 and HER2 mutations28–30 are among the emergent genetic alterations with various responses to targeted therapy.31 Some of these targeted agents have been approved for other types of malignancy, and others are still in the development phase.
Several initiatives worldwide have reported better outcomes of patients with driver mutations treated with targeted therapy. For instance, the Lung Cancer Mutation Consortium in the United States demonstrated that the median survival of patients without driver mutations, with drivers mutations but not treated with targeted therapy, and with driver mutations and treated with targeted therapy was 2.08 years, 2.38 years, and 3.49 years, respectively.32 The French Cooperative Thoracic Intergroup-French National Cancer Institute demonstrated that the median survival for patients with driver mutations versus those without driver mutations was 16.5 months versus 11.8 months.33 The Spanish Lung Cancer Group demonstrated that the overall survival (OS) for patients with EGFR mutations treated with erlotinib was 27 months.34 The mutations in lung cancer, their frequencies, and the downstream signaling pathways are depicted in the Figure.35
In this article, we discuss targeted therapy for patients with EGFR mutations, ALK rearrangements, ROS1 rearrangements, and BRAF V600E mutation. We also discuss the management of patients with EGFR mutations who develop a secondary mutation after TKI therapy. Almost all of the targeted agents discussed herein have been approved by the US Food and Drug Administration (FDA), so they are considered standard of care. All available phase 3 trials pertinent to these targeted therapies are included in the discussion.
EGFR MUTATIONS
CASE PRESENTATION 1
A 54-year-old Caucasian man who is a former smoker with a 10 pack-year history and past medical history of hypertension and dyslipidemia presents with progressive dyspnea for several weeks. A chest x-ray shows moderate pleural effusion on the left side with possible mass-like opacity on the left upper lung field. An ultrasound-guided thoracentesis is performed and cytology is positive for adenocarcinoma of likely pulmonary origin. Staging workup including positron emission tomography (PET)/computed tomography (CT) and magnetic resonance imaging of the brain with and without contrast is done. PET/CT shows a 5.5-cm mass in the left upper lobe of the lung with high fluorodeoxyglucose (FDG) uptake, several 1- to 2-cm mediastinal lymph nodes with moderate FDG uptake, and small pleural effusion on both sides with moderate FDG uptake. MRI-brain is negative for malignancy. The patient subsequently undergoes a CT-guided biopsy of the lung mass, which shows moderately differentiated adenocarcinoma. Comprehensive molecular profiling reveals EGFR L858R mutation only. The patient now presents for the initial consultation. Of note, his Eastern Cooperative Oncology Group performance status is 1.
What is the next step in the management of this patient?
FIRST-LINE TKI FOR SENSITIZING EGFR MUTATIONS
The 2 most common EGFR mutations are deletions in exon 19 and substitution of arginine for leucine in exon 21 (L858R), found in approximately 45% and 40% of patients with EGFR mutations, respectively.36 Both mutations are sensitive to EGFR TKIs. The benefit may be greater in patients with exon 19 deletions as compared to exon 21 L858R substitution,37,38 but this has not been demonstrated consistently in clinical trials.39-43 In the United States, EGFR mutations are found in approximately 10% of patients with NSCLC, while the incidence can be as high as 50% in Asia.44 Even though the cobas EGFR mutation test is the companion diagnostic approved by the US FDA, a positive test result from any laboratory with the Clinical Laboratory Improvement Amendments (CLIA) certificate should prompt the use of an EGFR TKI as the initial treatment.
Three EGFR TKIs that have been approved as first-line therapy in the United States are available: erlotinib, afatinib, and gefitinib.5 Both erlotinib and gefitinib are considered first-generation TKIs. They have higher binding affinity for the 2 common EGFR mutations than wild-type EGFR. In addition, they reversibly bind to the intracellular tyrosine kinase domain, resulting in inhibition of autophosphorylation of the tyrosine residues. Afatinib, a second-generation and irreversible TKI, targets EGFR (HER1) as well as HER2 and HER4.45
The superior efficacy of the EGFR TKIs over platinum doublet chemotherapy in treatment-naïve patients with EGFR mutations has been demonstrated in 7 randomized trials to date (Table).46 Erlotinib was the TKI arm for the OPTIMAL,41 EURTAC,42 and ENSURE trials;38 afatinib was the TKI arm for LUX-LUNG 337 and 6;43 gefitinib was the TKI arm for NEJ00239,47 and WJTOG3405.40 A meta-analysis of these 7 trials by Lee et al showed that progression-free survival (PFS) was significantly prolonged by EGFR TKIs (hazard ratio [HR] 0.37 [95% confidence interval {CI} 0.32 to 0.42]).46 For instance, in the EURTAC trial, median PFS was 9.7 months for patients treated with erlotinib as compared to 5.2 months for patients treated with platinum/gemcitabine or platinum/docetaxel.42 In this meta-analysis, prespecified subgroups included age, sex, ethnicity, smoking status, performance status, tumor histology, and EGFR mutation subtype. The superior outcome with TKIs was observed in all subgroups. Furthermore, patients with exon 19 deletions, nonsmokers, and women had even better outcomes.46
Erlotinib is the most commonly used TKI in the United States largely because gefitinib was off the market for some time until it was re-approved by the FDA in 2015. Interestingly, this “re-approval” was not based on either 1 of the 2 prospective trials (NEJ00239,47 and WJTOG340540), but rather was based on an exploratory analysis of the IPASS trial48,49 as well as a prospective phase 4, single-arm trial in Europe (IFUM).50 The superior efficacy of gefitinib over carboplatin/paclitaxel among patients with EGFR mutations in the IPASS trial was confirmed by blind independent central review, with longer PFS (HR 0.54 [95% CI 0.38 to 0.79] P = 0.0012) and higher objective response rate (ORR; odds ratio 3 [95% CI 1.63 to 5.54], P = 0.0004).49
CASE 1 CONTINUED
Based on the EGFR L858R mutation status, the patient is started on erlotinib. He is quite happy that he does not need intravenous chemotherapy but wants to know what toxicities he might potentially have with erlotinib.
What are the common adverse effects (AEs) of EGFR TKIs? How are AEs of TKIs managed?
Safety Profile
The important toxicities associated with EGFR TKIs are rash, gastrointestinal toxicity, hepatic toxicity, and pulmonary toxicity. Rash is an AE specific to all agents blocking the EGFR pathway, including small molecules and monoclonal antibodies such as cetuximab. The epidermis has a high level of expression of EGFR, which contributes to this toxicity.51 Rash usually presents as dry skin or acneiform eruption. Prophylactic treatment with oral tetracyclines and topical corticosteroids is generally recommended upon initiation of TKI therapy. Diarrhea is the most prevalent gastrointestinal toxicity. All patients starting treatment should be given prescriptions to manage diarrhea such as loperamide and be advised to call when it occurs. Hepatic toxicity is often manifested as elevated transaminases or bilirubin. Interstitial lung disease (ILD) is a rare but potentially fatal pulmonary toxicity.
Rash of any grade was reported in 49.2% of patients treated with erlotinib in clinical trials, while grade 3 rash occurred in 6% of patients and no grade 4 was reported. Diarrhea of any grade was reported in 20.3% of patients, grade 3 diarrhea occurred in 1.8%, and no grade 4 was reported. Grade 2 and 3 alanine aminotransferase (ALT) elevations were seen in 2% and 1% of patients, respectively. Grade 2 and 3 bilirubin elevations were seen in 4% and less than 1% of patients, respectively. The incidence of serious ILD-like events was less than 1%.52
Afatinib is associated with higher incidences of rash and diarrhea. Specifically, diarrhea and rash of all grades were reported in 96% and 90% of patients treated with afatinib, respectively. Paronychia of all grades occurred in 58% of patients. Elevated ALT of all grades was seen in 11% of patients. Approximately 1.5% of patients treated with afatinib across clinical trials had ILD or ILD-like AEs.53
Gefitinib, the most commonly used TKI outside United States, has a toxicity profile similar to erlotinib, except for hepatic toxicity. For instance, rash of all grades occurred in 47% of patients, diarrhea of all grades occurred in 29% of patients, and ILD or ILD-like AEs occurred in 1.3% of patients across clinical trials. In comparison, elevated ALT and aspartate aminotransferase (AST) of all grades was seen in 38% and 40% of patients, respectively.54 Therefore, close monitoring of liver function is clinically warranted. In particular, patients need to be advised to avoid concomitant use of herbal supplements, a common practice in Asian countries.
CASE 1 CONTINUED
The patient does well while on erlotinib at 150 mg orally once daily for about 8 months, until he develops increasing abdominal pain. A CT scan of the abdomen and pelvis with contrast shows a new 8-cm right adrenal mass. Additionally, a repeat CT scan of the chest with contrast shows a stable lung mass but enlarging mediastinal lymphadenopathy.
How would you manage the patient at this point?
MANAGEMENT OF T790M MUTATION AFTER PROGRESSION ON FIRST-LINE EGFR TKIS
As mentioned above, the median PFS of patients with EGFR mutations treated with 1 of the 3 TKIs is around 9 to 13 months.46 Of the various resistance mechanisms that have been described, the T790M mutation is found in approximately 60% of patients who progress after treatment with first-line TKIs.55,56 Other mechanisms, such as HER2 amplification, MET amplification, or rarely small cell transformation, have been reported.56 The first- and second-generation EGFR TKIs function by binding to the ATP-binding domain of mutated EGFR, leading to inhibition of the downstream signaling pathways (Figure, part B) and ultimately cell death.35 The T790M mutation hinders the interaction between the ATP-binding domain of EGFR kinase and TKIs, resulting in treatment resistance and disease progression.57,58
Osimertinib is a third-generation irreversible EGFR TKI with activity against both sensitizing EGFR and resistant T790M mutations. It has low affinity for wide-type EGFR as well as insulin receptor and insulin-like growth factor receptor.59 Osimertinib has been fully approved for NSCLC patients with EGFR mutations who have progressed on first-line EGFR TKIs with the development of T790M mutation. An international phase 3 trial (AURA3) randomly assigned 419 patients in a 2:1 ratio to either osimertinib or platinum/pemetrexed. Eligible patients all had the documented EGFR mutations and disease progression after first-line EGFR TKIs. Central confirmation of the T790M mutation was required. Median PFS by investigator assessment, the trial’s primary end point, was 10.1 months for osimertinib versus 4.4 months for chemotherapy (HR 0.3 [95% CI 0.23 to 0.41]; P < 0.001). ORR was 71% for osimertinib versus 31% for chemotherapy (HR 5.39 [95% CI 3.47 to 8.48], P < 0.001). A total of 144 patients with stable and asymptomatic brain metastases were also eligible. Median PFS for this subset of patients treated with osimertinib and chemotherapy was 8.5 months and 4.2 months, respectively (HR 0.32 [95% CI 0.21 to 0.49]). In the AURA3 trial, osimertinib was better tolerated than chemotherapy, with 23% of patients treated with osimertinib experiencing grade 3 or 4 AEs as compared to 47% of chemotherapy-treated patients. The most common AEs of any grade were diarrhea (41%), rash (34%), dry skin (23%), and paronychia (22%).60
For the case patient, a reasonable approach would be to obtain a tissue biopsy of the adrenal mass and more importantly to check for the T790M mutation. Similar to the companion diagnostic for EGFR mutations, the cobas EGFR mutation test v2 is the FDA-approved test for T790M. However, if this resistance mutation is detected by any CLIA-certified laboratories, osimertinib should be the recommended treatment option. If tissue biopsy is not feasible, plasma-based testing should be considered. A blood-based companion diagnostic also is FDA approved.
ALK REARRANGEMENTS
CASE 2 PRESENTATION
A 42-year-old Korean woman who is a non-smoker with no significant past medical history presents with fatigue, unintentional weight loss of 20 lb in the past 4 months, and vague abdominal pain. A CT can of the abdomen and pelvis without contrast shows multiple foci in the liver and an indeterminate nodule in the right lung base. She subsequently undergoes PET/CT, which confirms multiple liver nodules/masses ranging from 1 to 3 cm with moderate FDG uptake. In addition, there is a 3.5-cm pleura-based lung mass on the right side with moderate FDG uptake. MRI-brain with and without contrast is negative for malignancy. A CT-guided biopsy of 1 of the liver masses is ordered and pathology returns positive for poorly differentiated adenocarcinoma consistent with lung primary. Molecular analysis reveals an echinoderm microtubule-associated protein-like 4 (EML4)-ALK rearrangement. She is placed on crizotinib by an outside oncologist and after about 3 weeks of therapy is doing well. She is now in your clinic for a second opinion. She says that some of her friends told her about another medication called ceritinib and was wondering if she would need to switch her cancer treatment.
How would you respond to this patient’s inquiry?
FIRST-LINE TKIS FOR ALK REARRANGEMENTS
ALK rearrangements are found in 2% to 7% of NSCLC, with EML4-ALK being the most prevalent fusion variant.61 The inversion of chromosome 2p leads to the fusion of the EML4 gene and the ALK gene, which causes the constitutive activation of the fusion protein and ultimately increased transformation and tumorigenicity.7,61 Patients harboring ALK rearrangements tend to be non-smokers. Adenocarcinoma, especially signet ring cell subtype, is the predominant histology. Compared to EGFR mutations, patients with ALK mutations are significantly younger and more likely to be men.62 ALK rearrangements can be detected by either FISH or IHC, and most next-generation sequencing (NGS) panels have the ability to identify this driver mutation.
Crizotinib is the first approved ALK inhibitor for the treatment of NSCLC in this molecular subset of patients.63 PROFILE 1014 is a phase 3 randomized trial that compared crizotinib with chemotherapy containing platinum/pemetrexed for up to 6 cycles. Crossover to crizotinib was allowed for patients with disease progression on chemotherapy. The primary end point was PFS by independent radiologic review. The crizotinib arm demonstrated superior PFS (10.9 months versus 7 months; HR 0.45 [95% CI 0.35 to 0.6], P < 0.001) and ORR (74% versus 45%, P < 0.001). Median survival was not reached in either arm (HR 0.82 [95% CI 0.54 to 1.26], P = 0.36).64 Based on this international trial, crizotinib is considered standard of care in the United States for treatment-naïve patients with advanced NSCLC harboring ALK rearrangements. The current recommended dose is 250 mg orally twice daily. Common treatment-related AEs of all grades include vision disorder (62%), nausea (53%), diarrhea (43%), vomiting (40%), edema (28%), and constipation (27%).65 PROFILE 1007 compared crizotinib with pemetrexed or docetaxel in ALK-rearranged NSCLC patients with prior exposure to 1 platinum-based chemotherapy. The median PFS was 7.7 months for crizotinib as compared to 3 months for chemotherapy (HR 0.49 [95% CI 0.37 to 0.64], P < 0.001). The response rates were 65% and 20% for crizotinib and chemotherapy, respectively (P < 0.001).66 In other countries, crizotinib following 1 prior platinum-based regimen may be considered standard of care based on this trial.
Ceritinib is an oral second-generation ALK inhibitor that is 20 times more potent than crizotinib based on enzymatic assays.67 It also targets ROS1 and insulin-like growth factor 1 receptor but not c-MET. It was first approved by the FDA in April 2014 for metastatic ALK-rearranged NSCLC following crizotinib.68 In May 2017, the FDA granted approval of ceritinib for treatment-naïve patients. This decision was based on the results of the ASCEND-4 trial, a randomized phase 3 trial assessing the efficacy and safety of ceritinib over chemotherapy in the first-line setting. The trial assigned 376 patients to either ceritinib at 750 mg once daily or platinum/pemetrexed for 4 cycles followed by maintenance pemetrexed. Median PFS was 16.6 months for ceritinib versus 8.1 months for chemotherapy (HR 0.55 [95% CI 0.42 to 0.73]; P < 0.00001).69 Toxicities of ceritinib are not negligible, with gastrointestinal toxicity being the most prevalent. For instance, diarrhea, nausea, vomiting, abdominal pain, and constipation of all grades were seen in 86%, 80%, 60%, 54%, and 29% of patients, respectively. Furthermore, fatigue and decreased appetite occurred in 52% and 34% of patients, respectively. In terms of laboratory abnormalities, 84% of patients experienced decreased hemoglobin of all grades; 80% increased ALT; 75% increased AST; 58% increased creatinine; 49% increased glucose; 36% decreased phosphate; and 28% increased lipase. Due to these AEs, the incidence of dose reduction was about 58% and the median onset was around 7 weeks.70
Alectinib is another oral second-generation ALK inhibitor that was approved by the FDA in December 2015 for the treatment of NSCLC patients with ALK rearrangements who have progressed on or are intolerant to crizotinib.71 Its indication will soon be broadened to the first-line setting based on the ALEX trial.72 Alectinib is a potent and highly selective TKI of ALK73 with activity against known resistant mutations to crizotinib.74,75 It also inhibits RET but not ROS1 or c-MET.76 ALEX, a randomized phase 3 study, compared alectinib with crizotinib in treatment-naïve patients with NSCLC harboring ALK rearrangements. The trial enrolled 303 patients and the median follow-up was approximately 18 months. The alectinib arm (600 mg twice daily) demonstrated significantly higher PFS by investigator-assessment, the trial’s primary end point. The 12-month event-free survival was 68.4% (95% CI 61% to 75.9%) versus 48.7% (95% CI 40.4% to 56.9%) for alectinib and crizotinib, respectively (HR 0.47 [95% CI 0.34 to 0.65], P < 0.001). The median PFS was not reached in the alectinib arm (95% CI 17.7 months to not estimable) as compared to 11.1 months in the crizotinib arm (95% CI 9.1 to 13.1 months).72 Alectinib is generally well tolerated. Common AEs of all grades include fatigue (41%), constipation (34%), edema (30%), and myalgia (29%). As alectinib can cause anemia, lymphopenia, hepatic toxicity, increased creatine phosphokinase, hyperglycemia, electrolyte abnormalities, and increased creatinine, periodic monitoring of these laboratory values is important, although most of these abnormalities are grade 1 or 2.77
Brigatinib, another oral second-generation ALK inhibitor, was granted accelerated approval by the FDA in April 2017 for ALK-rearranged and crizotinib-resistant NSCLC based on the ALTA trial. This randomized phase 2 study of brigatinib showed an ORR by investigator assessment of 54% (97.5% CI 43% to 65%) in the 180 mg once daily arm with lead-in of 90 mg once daily for 7 days. Median PFS was 12.9 months (95% CI 11.1 months to not reached [NR]).78 Currently, a phase 3 study of brigatinib versus crizotinib in ALK inhibitor–naïve patients is recruiting participants (ALTA-1L). It will be interesting to see if brigatinib can achieve a front-line indication.
Starting the case patient on crizotinib is well within the treatment guidelines. One may consider ceritinib or alectinib in the first-line setting, but both TKIs can be reserved upon disease progression. We would recommend a repeat biopsy at that point to look for resistant mechanisms, as certain secondary ALK mutations may be rescued by certain next-generation ALK inhibitors. For instance, the F1174V mutation has been reported to confer resistance to ceritinib but sensitivity to alectinib, while the opposite is true for I1171T. The G1202R mutation is resistant to ceritinib, alectinib, and brigatinib, but lorlatinib, a third-generation ALK inhibitor, has shown activity against this mutation.79 Furthermore, brain metastasis represents a treatment challenge for patients with ALK rearrangements. It is also an efficacy measure of next-generation ALK inhibitors, all of which have demonstrated better central nervous system activity than crizotinib.69,78,80 If the case patient were found to have brain metastasis at the initial diagnosis, either ceritinib or alectinib would be a reasonable choice since crizotinib has limited penetration of blood-brain barrier.81
ROS1 REARRANGEMENTS
CASE PRESENTATION 3
A 66-year-old Chinese woman who is a non-smoker with a past medical history of hypertension and hypothyroidism presents to the emergency department for worsening lower back pain. Initial workup includes x-ray of the lumbar spine followed by MRI with contrast, which shows a soft tissue mass at L3-4 without cord compression. CT of the chest, abdomen, and pelvis with contrast shows a 7-cm right hilar mass, bilateral small lung nodules, mediastinal lymphadenopathy, and multiple lytic lesions in ribs, lumbar spine, and pelvis. MRI-brain with and without contrast is negative for malignancy. She undergoes endo-bronchial ultrasound and biopsy of the right hilar mass, which shows poorly differentiated adenocarcinoma. While waiting for the result of the molecular analysis, the patient undergoes palliative radiation therapy to L2-5 with good pain relief. She is discharged from the hospital and presents to your clinic for follow up. Molecular analysis now reveals ROS1 rearrangement with CD74-ROS1 fusion.
What treatment plan should be put in place for this patient?
FIRST-LINE THERAPY FOR ROS1 REARRANGEMENTS
Approximately 2.4% of lung adenocarcinomas harbor ROS1 rearrangements.82 This distinct genetic alteration occurs more frequently in NSCLC patients who are younger, female, and never-smokers, and who have adenocarcinomas.8 It has been shown that ROS1 rearrangements rarely overlap with other genetic alterations including KRAS mutations, EGFR mutations, and ALK rearrangements.83 As a receptor tyrosine kinase, ROS1 is similar to ALK and insulin receptor family members.84 Crizotinib, which targets ALK, ROS1, and c-MET, was approved by the FDA on March 11, 2016, for the treatment of metastatic ROS1-rearranged NSCLC.85 The approval was based on a phase 2 expansion cohort of the original phase 1 study. Among 50 US patients enrolled in this expansion cohort, 3 had complete responses and 33 had partial responses with ORR of 72% (95% CI 58% to 84%). Median PFS was 19.2 months (95% CI 14.4 months to NR) and median duration of response (DOR) was 17.6 months (95% CI 14.5 months to NR).86 During longer follow-up, independent radiology review confirmed high ORR of 66% and median DOR of 18.3 months.85
Interestingly, no companion diagnostic assay has been approved for the detection of ROS1 rearrangements with the approval of crizotinib. In the United States, break apart FISH is the most common detection method. In fact, in the above mentioned phase 2 study, ROS1 rearrangements were detected in 49 out of 50 patients by this method.86 FISH can be technically challenging when dealing with high volume and multiple targets. Reverse transcriptase-PCR is another detection method, but it requires knowledge of the fusion partners. To date, at least 14 ROS1 fusion partners have been reported, with CD74 being the most common.87 NGS with appropriate design and validation can also be used to detect ROS1 rearrangements.
For the case patient, the recommendation would be to start her on crizotinib at 250 mg twice daily. Monitoring for vision disturbance, gastrointestinal complaints, and edema is warranted. Because the estimated onset of response is around 7.9 weeks,86 plans should be made to repeat her scans in approximately 2 months.
BRAF V600E MUTATIONS
CASE PRESENTATION 4
A 71-year-old Caucasian man with a past medical history of hypertension, dyslipidemia, and ischemic cerebrovascular accident without residual deficits was diagnosed with stage IV adenocarcinoma of the lung about 8 months ago. He has a 40 pack-year smoking history and quit smoking when he was diagnosed with lung cancer. His disease burden involved a large mediastinal mass, scattered pleural nodules, multiple lymphadenopathy, and several soft tissue masses. His outside oncologist started him on chemotherapy containing carboplatin and pemetrexed for 6 cycles followed by maintenance pemetrexed. The most recent restaging scans show disease progression with enlarging soft tissue masses and several new lytic bone lesions. MRI-brain with and without contrast shows 2 subcentimeter enhancing lesions. He transferred care to you approximately 4 weeks ago. You ordered a repeat biopsy of 1 of the enlarging soft tissue masses. Molecular analysis revealed BRAF V600E mutation. In the interim, he underwent stereotactic radiosurgery for the 2 brain lesions without any complications. The patient is now in your clinic for follow up.
What would be your recommended systemic treatment?
TARGETED THERAPIES FOR BRAF V600E MUTATION
BRAF mutations were first recognized as activating mutations in advanced melanomas, with BRAF V600E, resulting from the substitution of glutamic acid for valine at amino acid 600, being the most common. BRAF plays an important role in the mitogen-activated protein kinase (MAPK) signaling pathway. Briefly, the activation of MAPK pathway occurs upon ligand binding of receptor tyrosine kinases, which then involves RAS/BRAF/MEK/ERK in a stepwise manner, ultimately leading to cell survival. BRAF mutations have been increasingly recognized also as driver mutations in NSCLC.9–12 They can be detected by PCR or NGS method. The characteristics of NSCLC patients harboring BRAF mutations have been described by various groups.9–12 For instance, 1 case series showed that the incidence was 2.2% among patients with advanced lung adenocarcinoma; 50% of mutations were V600E, while G469A and D594G accounted for the remaining 39% and 11% of patients, respectively. All patients were either current or former smokers. The median OS of patients with BRAF mutations in this case series was NR, while it was 37 months for patients with EGFR mutations (P = 0.73) and NR for patients with ALK rearrangements (P = 0.64).9
For patients with BRAF V600E–mutant NSCLC who have progressed on platinum-based chemotherapy, the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) may represent a new treatment paradigm. This was illustrated in a phase 2, nonrandomized, open-label study. A total of 57 patients were enrolled and 36 patients (63.2% [95% CI 49.3% to 75.6%]) achieved an overall response by investigator assessment, the trial’s primary end point. Disease control rate was 78.9% (95% CI 66.1% to 88.6%), with 4% complete response, 60% partial response, and 16% stable disease. PFS was 9.7 months (95% CI [6.9 to 19.6 months]). The safety profile was comparable to what had been observed in patients with melanoma treated with this regimen. More specifically, 56% of patients on this trial reported serious AEs, including pyrexia (16%), anemia (5%), confusional state (4%), decreased appetite (4%), hemoptysis (4%), hypercalcemia (4%), nausea (4%), and cutaneous squamous cell carcinoma (4%). In addition, neutropenia (9%) and hyponatremia (7%) were the most common grade 3-4 AEs.16
The case patient has experienced disease progression after 1 line of platinum-based chemotherapy, so the combination of dabrafenib and trametinib would be a robust systemic treatment option. dabrafenib as a single agent has also been studied in BRAF V600E–mutant NSCLC in a phase 2 trial. The overall response by investigator assessment among 84 patients was 33% (95% CI 23% to 45%).14 Vemurafenib, another oral BRAF TKI, has demonstrated efficacy for NSCLC patients harboring BRAF V600E mutation. In the cohort of 20 patients with NSCLC, the response rate was 42% (95% CI 20% to 67%) and median PFS was 7.3 months (95% CI 3.5 to 10.8 months).13 Patients with non-V600E mutations have shown variable responses to targeted therapies. MEK TKIs may be considered in this setting; however, the details of this discussion are beyond the scope of this review.
CONCLUSION
The management of advanced NSCLC with driver mutations has seen revolutionary changes over the past decade. Tremendous research has been done in order to first understand the molecular pathogenesis of NSCLC and then discover driver mutations that would lead to development of targeted therapies with clinically significant efficacy as well as tolerability. More recently, increasing efforts have focused on how to conquer acquired resistance in patients with disease progression after first-line TKIs. The field of EGFR-mutant NSCLC has set a successful example, but the work is nowhere near finished. The goals are to search for more driver mutations and to design agents that could potentially block cell survival signals once and for all.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30.
- Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol 2016;893:1–19.
- Alberg AJ, Brock MV, Ford JG, et al. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013;143:e1S–29S.
- Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-3013, based on November 2015 SEER data submission, posted to the SEER website, April 2016. Bethesda (MD): National Cancer Institute; 2016.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Non-Small Cell Lung Cancer: 1–190.
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–39.
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–6.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancer. J Clin Oncol 2012;30:863–70.
- Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–51.
- Kinno T, Tsuta K, Shiraishi K, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol 2014;25:138–42.
- Litvak AM, Paik PK, Woo KM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol 2014;9:1669–74.
- Villaruz LC, Socinski MA, Abberbock S, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer 2015;121:448–56.
- Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–36.
- Planchard D, Kim TM, Mazieres J, et al. DaBRAFenib in patients with BRAF V600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17:642–50.
- Gautschi O, Milia J, Cabarrou B, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol 2015;10:1451–7.
- Planchard D, Besse B, Groen HJ, et al. DaBRAFenib plus trametinib in patients with previously treated BRAF V600E-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol 2016;17:984–93.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. J Thorac Oncol 2013;8:823–59.
- Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer and Association for Molecular Pathology. Arch Pathol Lab Med 2013;137:828–60.
- Leighl NB, Rekhtman N, Biermann WA, et al. Molecular testing for selection of patients with lung cancer for epidermal growth factor receptor and anaplastic lymphoma kinase tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology guideline. J Clin Oncol 2014;32:3673–9.
- Paik PK, Varghese AM, Sima CS, et al. Response to erlotinib in patients with EGFR mutant advanced non-small cell lung cancers with a squamous or squamous-like component. Mol Cancer Ther 2012;11:2535–40.
- Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015;5:842–9.
- Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age, and stage-dependent MET genomic amplification, and c-MET overexpression. J Clin Oncol 2016;34:721–30.
- Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol 2016;11:1493–502.
- Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017;103:27–37.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 2013;3:630–5.
- Lin JJ, Kennedy E, Sequist LV, et al. Clinical activity of alectinib in advanced RET-rearranged non-small cell lung cancer. J Thorac Oncol 2016;11:2027–32.
- Drilon A, Rekhtman N, Arcila M, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol 2016;17:1653–60.
- Cappuzzo F, Bemis L, Varella-Garcia M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med 2006;354:2619–21.
- Mazieres J. Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targted drugs: results from the European EUHER2 cohort. Annal Oncol 2016;27:281–6.
- Ou SH, Schrock AB, Bocharov EV, et al. HER2 transmembrane (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol 2017;12:446–57.
- Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emergent therapies. Cancer Discov 2017;7:596–609.
- Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006.
- Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016;387:1415–26.
- Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958–67.
- Rosell R, Karachaliou N, et al. Large-scale screening for somatic mutations in lung cancer. Lancet 2016;387:1354–6.
- Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005;97:339–46.
- Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34.
- Wu YL, Chou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol 2015;26:1883–9.
- Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Eng J Med 2010;362:2380–8.
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
- Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
- Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46.
- Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:213–22.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol 2009;10:432–3.
- Nelson V, Ziehr J, Aqulnik M, et al. Afatinib: emerging next-generation tyrosine kinase inhibitor for NSCLC. Onco Targets Ther 2013;5:135–43.
- Lee CK, Wu YL, Ding PN, et al. Impact of specific epidermal growth factor receptor (EGFR) mutations and clinical characteristics on outcomes after treatment with EGFR tyrosine kinase inhibitors versus chemotherapy in EGFR-mutant lung cancer: a meta-analysis. J Clin Oncol 2015;33:1958–65.
- Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013;24:54–9.
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57.
- Wu YL, Saijo N, Thongprasert S, et al. Efficacy according to blind independent central review: post-hoc analyses from the phase III, randomized, multicenter, IPASS study of first-line gefitinib versus carboplatin/paclitaxel in Asian patients with EFGR mutation-positive advanced NSCLC. Lung Cancer 2017;104:119–25.
- Douillard JY, Ostoros G, Cobo M, et al. First-line gefitinib in Caucasian EGFR-mutation positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 2014;110:55–62.
- Hu JC, Sadeghi P, Pinter-Brown LC, et al. Cutaneous side effects of epidermal growth factor receptor inhibitors: clinical presentation, pathogenesis, and management. J Am Acad Dermatol 2007;56:317–26.
- Tarceva [package insert]. South San Francisco (CA): Genentech, Inc; 2010. www.accessdata.fda.gov/drugsatfda_docs/label/2010/021743s14s16lbl.pdf. Accessed April 23, 2017.
- Gilotrif [package insert.] Ridgefield (CT): Boehringer Ingelheim, Inc; 2013. www.accessdata.fda.gov/drugsatfda_docs/label/2013/201292s000lbl.pdf. Accessed April 23, 2017.
- Iressa [package insert]. Wilmington (DE): AstraZeneca, Inc; 2015. Error! Hyperlink reference not valid. Accessed April 23, 2017.
- Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–22.
- Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR mutant lung cancers. Clin Cancer Res 2013;19:2240–7.
- Yun CH, Mengwasser KE, Tom AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5.
- Sos ML, Rode HB, Heynck S, et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res 2010;70:868–74.
- Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T190M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61.
- Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40.
- Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med 2010;363:1693–703.
- Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–53.
- Kazandjian D, Blumenthal GM, Chen HY, et al. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014;19:e5–11.
- Solomon BJ, Mok T, Kim DW, et al. First-ling crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371:2167–77.
- Xalkori [package insert]. New York: Pfizer, Inc; 2011. www.accessdata.fda.gov/drugsatfda_docs/label/2012/202570s002lbl.pdf. Accessed April 23, 2017.
- Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368:2385–94.
- Marsilje TH, Pei W, Chen B, et al. Synthesis, structure-activity relationships and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90.
- Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res 2015;21:2436–9.
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017;389:917–29.
- Zykadia [package insert]. East Hanover (NJ): Novartis Pharmaceuticals Corporation, Inc; 2016. www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/zykadia.pdf. Accessed April 23, 2017.
- Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res 2016;22:5171–6.
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. New Engl J Med 2017 June 6 [Epub ahead of print].
- Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem 2012;20:1271–80.
- Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
- Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett 2014;351:215–21.
- Kodama T, Tsukaguchi T, Satoh T, et al. Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol Cancer Ther 2014;13:2910–8.
- Alecensa [package insert]. South San Francisco (CA): Genentech, Inc; 2015. www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed April 23, 2017.
- Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol 2017 May 5 [Epub ahead of print].
- Zhu V, Ou SH. Safety of alectinib for the treatment of metastatic ALK-rearranged non-small cell lung cancer. Expert Opin Drug Saf 2017;16:509–14.
- Gadgeel SM, Shaw AT, Govindan R, et al. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small cell lung cancer. J Clin Oncol 2016;34:4079–85.
- Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol 2011;29:e443–5.
- Zhu Q, Zhan P, Zhang X, et al. Clinicopathologic characteristics of patients with ROS1 fusion gene in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res 2015;4:300–9.
- Lin JJ, Ritterhouse LL, Ali SM, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol 2017;12:872–7.
- Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta 2009;1795:37–52.
- Kazandjian D, Blumenthal G, Luo L, et al. Benefit-Risk summary of crizotinib for the treatment of patients with ROS1 alteration-positive metastatic NSCLC. Oncologist 2016;21:974–80.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014;371:1963–71.
- Zhu VW, Upadhyay D, Schrock AB, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer 2016;97:48–50.
Targeted Therapy and Immunotherapy in the Treatment of Metastatic Cutaneous Melanoma
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.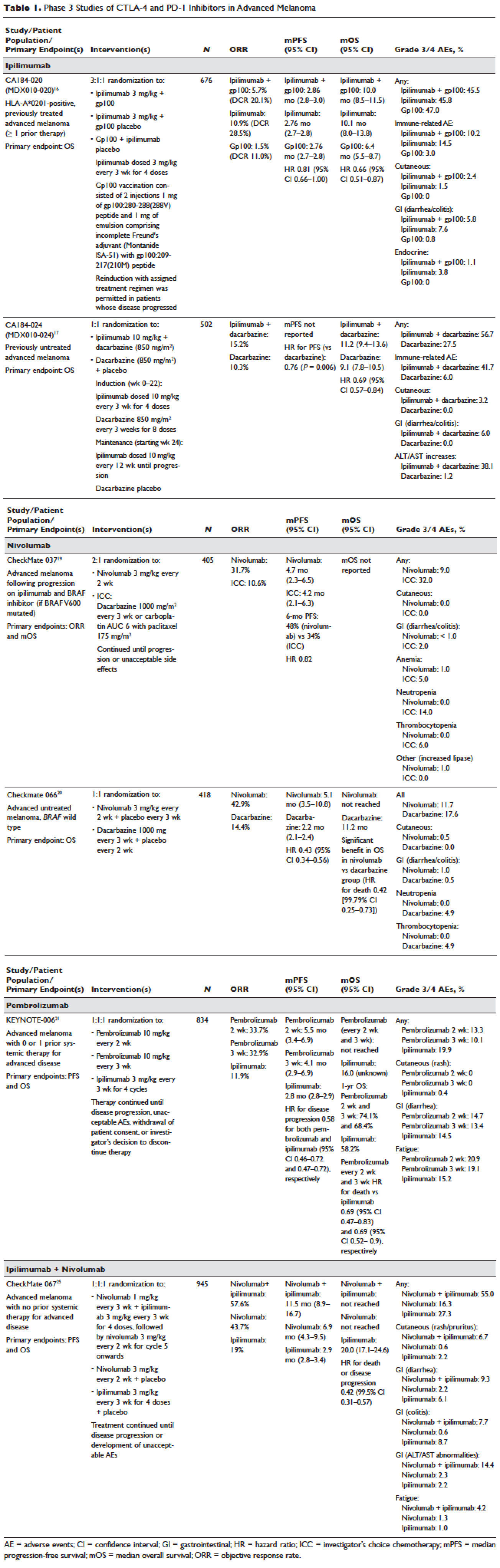
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56 BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4 NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7 NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
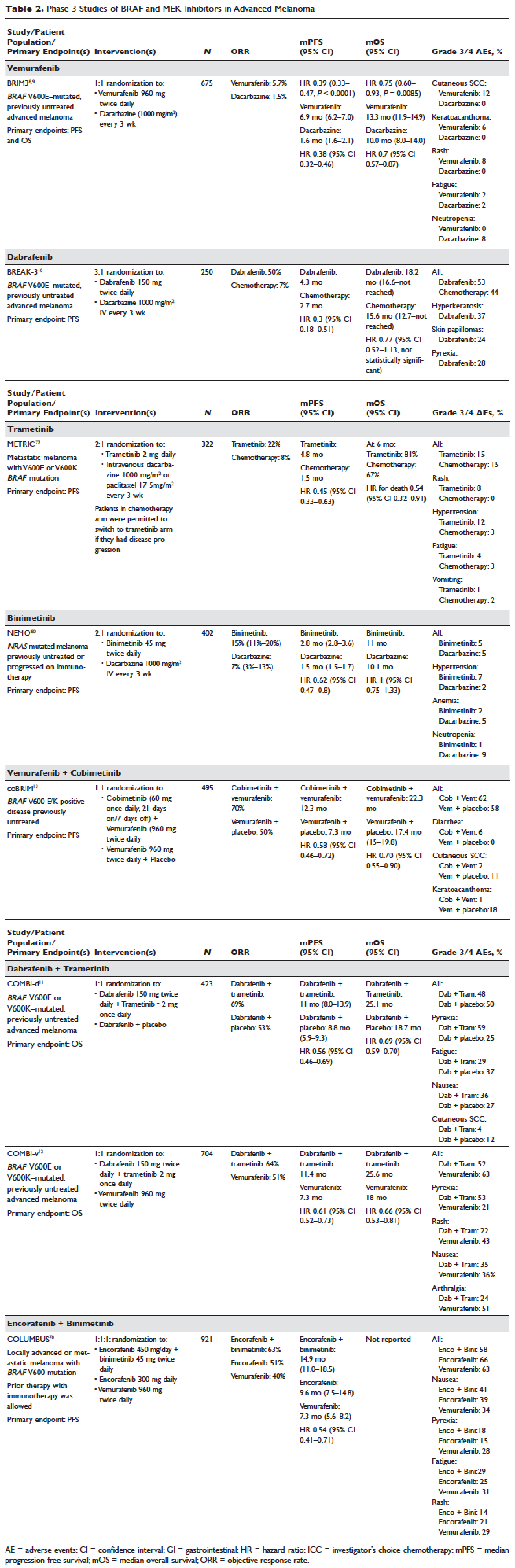
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56 BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4 NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7 NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
INTRODUCTION
The incidence of cutaneous melanoma has increased over the past 2 decades, with SEER estimates indicating that the number of new cases of melanoma diagnosed annually rose from 38,300 in 1996 to 76,000 in 2016.1 Among persons younger than 50 years, the incidence is higher in females, and younger women (aged 15–39 years) are especially vulnerable.2 Among persons older than 50, melanoma incidence in men is nearly twice that of women, in whom melanomas are often thicker and often associated with worse outcomes.1,2 Approximately 85% of melanomas are diagnosed at early stages when surgery is curative, but the lifetime probability of developing invasive disease is 3% in men and 2% in women.
Prior to the advent of effective immunotherapies and targeted therapies, melanoma was often managed with chemotherapy, which had dismal response rates and commensurately poor outcomes. Advances in the understanding of the molecular etiopathogenesis and immune escape responses of cutaneous metastatic melanoma have transformed therapeutic approaches. Specifically, improved understanding of the genetic mutations driving melanoma tumorigenesis coupled with insights into mechanisms of tumor-mediated immune evasion resulted in development of inhibitors of mitogen-activated protein kinases (MAPK; BRAF and MEK) along with inhibitors of negative regulatory immune checkpoints (cytotoxic T lymphocyte–associated antigen 4 [CTLA-4] and programmed cell death-1 [PD-1]). In this review, we discuss the role of immune therapy, targeted therapy, and combinations of these in the treatment of metastatic cutaneous melanoma. We limit the immuno-therapy discussion to approved CTLA-4/PD-1 inhibitors and the targeted therapy discussion to approved BRAF/NRAS/MEK inhibitors and do not discuss non-checkpoint immunotherapies including cytokines (HD IL-2), vaccines, or adoptive T-cell approaches. Interested readers are directed to other excellent works covering these important topics.26–29
DEVELOPMENT OF TARGETED AND NOVEL IMMUNE THERAPIES
For many years the degree of ultraviolet (UV) light exposure was considered the sole major risk factor for melanoma oncogenesis, even though its mechanism was largely unknown.3 However, clinical observations regarding the occurrence of melanoma on less exposed areas (trunk and limbs) in individuals with intermittent sun exposure led to the proposition that melanomas that arose in younger patients with intermittent sun exposure were distinct from melanomas that arose in older patients in association with markers of chronic sun exposure—the “divergent pathway” hypothesis.3 Critical to this understanding were whole-exome sequencing data from multiple groups, including The Cancer Genome Atlas, that identified patterns of mutations in oncogenic drivers that were distinct in patients with and without chronically sun-damaged (CSD) skin.4–7 It is now clear that based on its association with CSD skin, melanoma can be subclassified into CSD or non-CSD melanoma. CSD and non-CSD melanoma have distinct clinico-pathological characteristics and are associated with different driver mutations. CSD melanomas typically arise in older patients on sun-exposed areas (head/neck, dorsal surfaces of distal extremities) and are associated with particular driver mutations (BRAF non-V600E, NRAS, NF1, or KIT) and genetic signatures of UV-induced DNA damage (G > T [UVA] or C > T [UVB]) transitions. Conversely, non-CSD melanomas typically arise in younger (< 55 years) patients on intermittently sun-exposed areas (trunk, proximal extremities) and are associated with BRAF V600E/K driver mutations and often lack genetic signatures of UV mutagenesis.
Identification of driver mutations in components of the MAPK pathway, including BRAF and NRAS, facilitated the development of targeted inhibitors. The BRAF inhibitors vemurafenib and dabrafenib have been shown in pivotal phase 3 studies to significantly improve overall and progression-free survival in patients with metastatic melanoma compared with chemotherapy and garnered regulatory approval (vemurafenib, BRIM-3;8,9 dabrafenib, BREAK-310). Concomitant MEK and BRAF inhibition extends the duration of benefit by preventing downstream kinase activation in the MAPK pathway. Notably, concomitant MEK inhibition alters the side-effect profile of BRAF inhibitors, with reduced incidence of keratoacanthomas and cutaneous squamous cell carcinomas that are attributable to on-target, off-tumor effects of BRAF inhibitors. Combined BRAF and MEK inhibition (vemurafenib/cobimetinib and dabrafenib/trametinib) further improved overall and progression-free survival compared to single-agent BRAF inhibition in phase 3 studies (COMBI-d,11 COMBI-v,12 and coBRIM13). Although often deep, the responses seen with the use of targeted kinase inhibitors are not often durable, with the vast majority of patients progressing after 12 to 15 months of therapy.In parallel, work primarily done in murine models of chronic viral infection uncovered the role played by co-inhibitory or co-excitatory immune checkpoints in mediating T-cell immune responses. These efforts clarified that tumor-mediated immune suppression primarily occurs through enhancement of inhibitory signals via the negative T-cell immune checkpoints CTLA-4 or PD-1.14,15 Blockade of negative T-cell immune checkpoints resulted in activation of the adaptive immune system, resulting in durable anti-tumor responses as demonstrated in studies of the CTLA-4 inhibitor ipilimumab (CA184-02016 and CA184-02417) and the PD-1 inhibitors nivolumab (CA209-003,18 CheckMate 037,19 and CheckMate 06620) and pembrolizumab (KEYNOTE-00121 and KEYNOTE-00622). Compared to the deep but short-lived responses seen with targeted kinase inhibitors, patients treated with CTLA-4 or PD-1 immune checkpoint blockade often developed durable responses that persisted even after completion of therapy. Combined CTLA-4 and PD-1 blockade results in greater magnitude of response with proportionately increased toxicity.23–25
IMMUNOTHERAPY
CTLA-4 AND PD-1 IMMUNE CHECKPOINT INHIBITORS
The novel success of immunotherapy in recent decades is largely attributable to improved understanding of adaptive immune physiology, specifically T-cell activation and regulation. T-cell activation requires 2 independent signaling events: it is initiated upon recognition of the antigen-MHC class II-receptor complex on antigen-presenting cells (APC), and requires a secondary co-stimulatory interaction of CD80/CD86 (B7.1/B7.2) on APCs and CD28 molecule on T-cells; without this second event, T-cells enter an anergic state.30–32 Upon successful signaling and co-stimulation, newly activated T-cells upregulate CTLA-4, which can bind to B7 molecules with a nearly 100-fold greater affinity than CD28.33,34 Unlike CD28, CTLA-4 engagement negatively regulates T-cell activation. The opposing signals produced by CD28 and CTLA-4 are integrated by the T-cell to determine eventual response to activation, and provide a means by which T-cell activation is homeostatically regulated to prevent exaggerated physiologic immune responses.35 It was hypothesized that CTLA-4 blockade would permit T-cell activation, which is thwarted in the tumor microenvironment by tumor-mediated CTLA-4 engagement, thereby unleashing an anti-tumor immune response.36
PD-1 is a member of the CD28 and CTLA-4 immunoglobulin super family and, similar to CTLA-4, binds activated T-cells. PD-1 has 2 ligands on activated T-cells: PD-L1 and PD-L2.37 PD-L1 is constitutively expressed by a variety of immune and non-immune cells, particularly in inflammatory environments including tumor microenvironments, in response to the release of inflammatory cytokines such as interferon (IFN)-γ.37,38 Conversely, PD-L2 is only minimally expressed constitutively, although its expression on immune and non-immune cells can be induced by similar cues from inflammatory microenvironments. PD-L1 and PD-L2 cross-compete for binding to PD-1, with PD-L2 exhibiting 2- to 6-fold greater relative affinity than PD-L1.39 PD-L1/PD-1 binding results in phosphorylation of 2 tyrosinases in the intracellular portion of PD-1, which contains immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM). PD-1 ITSM subsequently recruits either of 2 SH2-domain–containing protein tyrosine phosphatases: SHP-1 and SHP-2. SHP-2 signaling suppresses PI3K/Akt activation, down-regulates Bcl-xL, and suppresses expression of multiple transcription factors that mediate T-cell effector function including GATA-3, Eomes, and T-bet.40–42 The net effect of PD-L1/PD-1 engagement is to suppress T-cell proliferation, cytokine production, cytolytic function, and survival. Unlike CTLA-4, which primarily affects the priming phase of naive T-cell activation, PD-1 chiefly regulates the effector phase of T-cell function. Furthermore, because PD-L1/PD-L2 expression is limited to inflammatory microenvironments, the effects of PD-1 are less generalized than those of CTLA-4.
SINGLE AGENT ACTIVITY OF CTLA-4 AND PD-1 INHIBITORS
Ipilimumab (MDX-010) is a human IgG1 monoclonal antibody shown to inhibit CTLA-4.43 Early studies tested different formulations (transfectoma-derived and hybridoma-derived), doses, and schedules of ipilimumab primarily in patients with advanced refractory melanoma.44–46 Although responses were infrequent, responding patients experienced durable remissions at 1- and 2-year time points. Notably, in a foreshadowing of changes to response criteria used to evaluate these agents, several treated patients who initially had radiographically stable disease upon completion of therapy subsequently experienced a gradual decline in tumor burden.
Ipilimumab was subsequently evaluated in 2 phase 3 trials. The first study (MDX010-020/CA184-020), which involved 676 HLA-A*0201–positive patients with advanced melanoma, compared ipilimumab 3 mg/kg every 3 weeks for 4 doses either singly or in combination with gp100 vaccine with a gp100-only control arm.16 Ipilimumab administration resulted in objective responses in 11% of patients and improved progression-free and overall survival compared to gp100 alone. Of note, ipilimumab monotherapy was superior to ipilimumab/gp100 combination, possibly related to timing of vaccine in relation to ipilimumab. A confirmatory study (CA184-024) compared a higher dose of ipilimumab (10 mg/kg) in combination with dacarbazine to dacarbazine monotherapy in previously untreated melanoma and was positive.17 Given the lack of augmented efficacy with the higher (10 mg/kg) dose, ipilimumab received regulatory approval in 2011 for the treatment of melanoma at the lower dose: 3 mg/kg administered every 3 weeks for 4 doses (Table 1). Survival data was strikingly similar to patterns observed in prior phase 2 studies, with survival curves plateauing after 2 years at 23.5% to 28.5% of treated patients. Pooled survival data from prospective and retrospective studies of ipilimumab corroborate the plateau of 22% (26% treated; 20% untreated) reached at year 3 regardless of prior therapy or ipilimumab dose, underscoring the durability of long-term survival in ipilimumab-treated patients.47 Ipilimumab administration resulted in an unusual spectrum of toxicities including diarrhea, rash, hepatitis, and hypophysitis (termed immune-related adverse events, or irAEs) in up to a third of patients.
Pembrolizumab and nivolumab are humanized IgG4 monoclonal antibodies that target the PD-1 receptor found on activated T cells, B cells, and myeloid cells. Pembrolizumab and nivolumab are engineered similarly: by immunizing transgenic mice with recombinant human PD-1-Fc protein and subsequently screening murine splenic cells fused with myeloma cells for hybridomas producing antibodies reactive to PD-1-Fc.48,49 Unlike IgG1, the IgG4 moiety neither engages Fc receptors nor activates complement, avoiding cytotoxic effects of the antibody upon binding to the T cells that it is intended to activate. Both pembrolizumab and nivolumab bind PD-1 with high affinity and specificity, effectively inhibiting the interaction between PD-1 and ligands PD-L1 and PD-L2.
Nivolumab was first studied in a phase 1 study (CA209-003) of 296 patients with advanced cancers who received 1, 3, or 10 mg/kg administered every 2 weeks.18 Histologies tested included melanoma, non–small-cell lung cancer (NSCLC), renal-cell cancer (RCC), castration-resistant prostate cancer (CRPC), and colorectal cancer (CRC). Responses were seen in melanoma and RCC and unusually in NSCLC, including in both squamous and non-squamous tumors. Objective responses were noted in 41% of the 107 melanoma patients treated at 3 mg/kg. Survival was improved, with 1- and 2-year survival rates of 62% and 43% at extended follow up.50
Subsequently, nivolumab was compared to chemotherapy in a pair of phase 3 studies involving both previously untreated (Checkmate 066) and ipilimumab/BRAF inhibitor–refractory (CheckMate 037) patients.19,20 In both studies, nivolumab produced durable responses in 32% to 34% of patients and improved survival over chemotherapy. Compared to ipilimumab, the incidence of irAEs was much lower with nivolumab. The depth and magnitude of responses observed led to regulatory approval for nivolumab in both indications (untreated and ipilimumab/BRAF inhibitor–treated melanoma) in 2014. Data from both studies are summarized in Table 1.
Pembrolizumab was first evaluated in a phase 1 study of 30 patients with a variety of solid organ malignancies in which no dose-limiting toxicities were observed and no defined maximal tolerated dose was reached.51 Per protocol, maximal administered dose was 10 mg/kg every 2 weeks. Following startling responses including 2 complete responses of long duration, pembrolizumab was evaluated in a large phase 1 study (KEYNOTE-001) of 1260 patients that evaluated 3 doses (10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, and 2 mg/kg every 3 weeks) in separate melanoma and NSCLC substudies.21 Both ipilimumab-naïve and ipilimumab-treated patients were enrolled in the melanoma substudy. Objective responses were seen in 38% ofpatients across all 3 dosing schedules and were similar in both ipilimumab-naïve and ipilimumab-treated patients. Similar to nivolumab, most responders experienced durable remissions.
Pembrolizumab was subsequently compared to ipilimumab in untreated patients (KEYNOTE-006) in which patients were randomly assigned to receive either ipilimumab or pembrolizumab at 1 of 2 doses: 10 mg/kg every 2 weeks and pembrolizumab 10 mg/kg every 3 weeks.22 Response rates were greater with pembrolizumab than ipilimumab, with commensurately greater 1-year survival rates. Rates of treatment-related adverse events requiring discontinuation of study drug were much lower with pembrolizumab than ipilimumab. This trial was instrumental in proving the superior profile of pembrolizumab over ipilimumab. The US Food and Drug Administration (FDA) granted pembrolizumab accelerated approval for second-line treatment of melanoma in 2014, and updated this to include a first-line indication in 2015 (Table 1).
EFFICACY OF COMBINED CTLA-4 AND PD-1 INHIBITION
Preclinical studies demonstrated that PD-1 blockade was more effective than CTLA-4 blockade and combination PD-1/CTLA-4 blockade was synergistic, with complete rejection of tumors in approximately half of the treated animals.14 This hypothesis was evaluated in a phase 1 study that explored both concurrent and sequential combinations of ipilimumab and nivolumab along with increasing doses of both agents in PD-1/CTLA-4–naïve advanced melanoma.23 Responses were greater in the concurrent arm (40%) than in the sequential arm (20%) across dose-levels with a small fraction of patients treated in the concurrent arm experiencing a profound reduction (80%) in tumor burden.
The superiority of ipilimumab/nivolumab combination to ipilimumab monotherapy was demonstrated in a randomized blinded phase 2 study (CheckMate 069).24 Of the 4 different ipilimumab/nivolumab doses explored in the phase 1 study (3 mg/kg and 0.3 mg/kg, 3 mg/kg and 1 mg/kg, 1 mg/kg and 3 mg/kg, 3 mg/kg and 3 mg/kg), ipilimumab 3 mg/kg and nivolumab 1 mg/kg (followed by nivolumab 3 mg/kg) was compared to ipilimumab and nivolumab-matched placebo. Responses were significantly greater with dual PD-1/CTLA-4 blockade compared to CTLA-4 blockade alone (59% versus 11%). Concurrently, a 3-arm randomized phase 3 study compared the same dose of ipilimumab/nivolumab to ipilimumab and nivolumab in previously untreated advanced melanoma (CheckMate 067).25 Similar to CheckMate 069, CheckMate 067 demonstrated that ipilimumab/nivolumab combination resulted in more profound responses (58%) than either ipilimumab (19%) or nivolumab (44%) alone. Toxicity, primarily diarrhea, fatigue, pruritus, and rash, was considerable in the combination arm (55% grade 3/4 adverse events) and resulted in treatment discontinuation in 30% of patients. The profound and durable responses observed led to accelerated approval of ipilimumab/nivolumab combination in 2015 (Table 1).
Efforts to improve the toxicity/benefit ratio of ipilimumab/nivolumab combination have centered around studying lower doses and/or extended dosing schedules of ipilimumab, including ipilimumab 1 mg/kg every 6 or 12 weeks with nivolumab dosed at 3 mg/kg every 2 weeks or 480 mg every 4 weeks. Promising data from a first-line study in NSCLC (CheckMate 012) support the evaluation of nivolumab in combination with lower-dosed ipilimumab (1 mg/kg every 6 or 12 weeks).52 This approach is being tested against platinum doublet chemotherapy in a confirmatory phase 3 study in NSCLC (CheckMate 227).
TARGETED THERAPY
MAPK KINASE PATHWAY IN MELANOMA TUMORIGENESIS
The MAPK pathway mediates cellular responses to growth signals. RAF kinases are central mediators in the MAPK pathway and exert their effect primarily through MEK phosphorylation and activation following dimerization (hetero- or homo-) of RAF molecules. As a result, RAF is integral to multiple cellular processes, including transcriptional regulation, cellular differentiation, and cell proliferation. MAPK pathway activation is a common event in many cancers, primarily due to activating mutations in BRAF or RAS. Alternatively, MAPK pathway activation can occur in the absence of activating mutations in BRAF or NRAS through down-regulation of MAPK pathway inhibitory proteins (RAF-1 inhibitory protein or SPRY-2), C-MET overexpression, or activating mutations in non-BRAF/NRAS kinases including CRAF, HRAS, and NRAS.53,54
Somatic point mutations in BRAF are frequently observed (37%–50%) in malignant melanomas and at lower frequency in a range of human cancers including NSCLC, colorectal cancer, papillary thyroid cancer, ovarian cancer, glioma, and gastrointestinal stromal tumor.6,55,56 BRAF mutations in melanoma typically occur within the activation segment of the kinase domain (exon 15). Between 80% and 90% of activating mutations result in an amino acid substitution of glutamate (E) for valine (V) at position 600: V600E.57,58 V600E mutations are true oncogenic drivers, resulting in increased kinase activity with demonstrable transformational capacity in vitro. BRAF mutations are usually mutually exclusive, with tumors typically containing no other driver mutations in NRAS, KIT, NF1, or other genes.
NRAS mutations are less common than BRAF mutations, having a reported frequency of 13% to 25% in melanoma.4 NRAS mutations generally occur within the P-loop region of the G domain (exon 2), or less commonly in the switch II region of the G domain (exon 3). Most NRAS exon 2 mutations comprise amino acid substitutions at position 61 from glutamine (Q) to arginine (R; 35%), lysine (K; 34%) and less often to glutamate (E), leucine (L), or proline (P). Preclinical data suggest that NRAS mutations paradoxically stimulate the MAPK pathway and thus enhance tumor growth in vitro.59,60 Several important phenotypic differences distinguish NRAS- from BRAF-mutated melanoma. NRAS-mutated tumors are typically associated with increasing age and CSD skin, while BRAF-mutated tumors arise in younger patients in non-CSD skin. A large population-based study suggested that NRAS-mutated melanomas were associated with mitoses and lower tumor infiltrating lymphocytes (TIL) grade, and arose in anatomic sites other than the head/neck, while BRAF-mutated tumors were associated with mitoses and superficial spreading histology.61 Although the lower TIL grade seen with NRAS-mutated melanomas suggests a more immunosuppressed microenvironment and argues for poorer responses to immune therapies, clinical studies comparing responses to immunotherapies in various categories of driver mutations provide conflicting results for the prognostic role of NRAS mutations in relation to immune checkpoint blockade and other immune therapies.62–64
NF1 represents the third known driver in cutaneous melanoma, with mutations reported in 12% of cases.6,7 NF1 encodes neurofibromin, which has GTPase activity and regulates RAS proteins; NF1 loss results in increased RAS.65 Unlike BRAF or NRAS, which are usually mutually exclusive, NF1 mutations in melanoma can occur singly or in combination with either BRAF or NRAS mutations. In these settings, NF1 mutations are associated with RAS activation, MEK-dependence, and resistance to RAF inhibition.66
MAPK PATHWAY INHIBITION SINGLY AND IN COMBINATION
Although multiple MEK 1/2 inhibitors (AS703026, AZD8330/ARRY-704, AZD6244, CH5126766, CI-1040, GSK1120212, PD0325901, RDEA119, and XL518) and RAF inhibitors (ARQ 680, GDC-0879, GSK2118436, PLX4032, RAF265, sorafenib, XL281/BMS-908662) were developed, the initial evaluation of MAPK pathway inhibitors in advanced human cancers began with CI-1040. Preclinical data suggested that CI-1040 potently and selectively inhibited both MEK1 and MEK2, but phase 1 and 2 human trial results were disappointing, likely because these trials were not selectively enriched for NRAS/BRAF–mutated tumors or cancers in which these oncogenic mutations were most commonly detected, such as melanoma.67,68 The subsequent evaluation of selumetinib (AZD6244/ARRY-142886) in a phase 2 study was also negative. Although investigators enrolled a presumably enriched population (cutaneous melanoma), the incidence of NRAS/BRAF–mutated tumors was not ascertained to determine this, but rather assumed, which led to a discrepancy between the assumed (prestudy) and observed (on-study) proportions of BRAF/NRAS mutations that was not accounted for in power calculations.69,70 Lessons learned from these earlier misadventures informed the current paradigm of targeted therapy development: (1) identification of a highly specific and potent inhibitor through high-throughput screening; (2) establishment of maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D) in unselected patients; (3) confirmation of RP2D in selected tumor types enriched for target of interest; and (4) confirmatory study against standard comparator to seek regulatory approval.
Vemurafenib and dabrafenib were evaluated in this tiered fashion in phase 1 dose-finding studies comprising unselected patients, followed by phase 2 studies in advanced BRAF V600E–mutated melanoma. Both were subsequently evaluated in randomized phase 3 trials (vemurafenib, BRIM-38; dabrafenib, BREAK-310) that compared them with dacarbazine (1000 mg/m2 intravenously every 3 weeks) in the treatment of advanced BRAF V600E–mutated melanoma. Response kinetics for both agents were remarkably similar: single-agent BRAF inhibitors resulted in rapid (time to response 2–3 months), profound (approximately 50% objective responses) reductions in tumor burden that lasted 6 to 7 months. Adverse events common to both agents included rash, fatigue, and arthralgia, although clinically significant photosensitivity was more common with vemurafenib and clinically significant pyrexia was more common with dabrafenib. Class-specific adverse events included the development of cutaneous squamous-cell carcinomas and keratoacanthomas secondary to paradoxical activation of MAPK pathway signaling either through activating mutations in HRAS or mutations or amplifications in receptor tyrosine kinases upstream of BRAF, resulting in elevated levels of RAS–guanosine triphosphate complexes.71 Results of these studies resulted in regulatory approval of single-agent BRAF inhibitors for the treatment of BRAF V600E (and later V600K)–mutated melanoma (vemurafenib in 2011; dabrafenib in 2013). Details regarding trial populations, study interventions, efficacy, and adverse events are summarized in Table 2.
Responses to BRAF inhibitors are typically profound but temporary. Mechanisms of acquired resistance are diverse and include reactivation of MAPK pathway–dependent signaling (RAS activation or increased RAF expression), and development of MAPK pathway–independent signaling (COT overexpression; increased PI3K or AKT signaling) that permits bypass of inhibited BRAF signaling within the MAPK pathway.72–76 These findings suggested that upfront inhibition of both MEK and mutant BRAF may produce more durable responses than BRAF inhibition alone. Three pivotal phase 3 studies established the superiority of combination BRAF and MEK inhibition over BRAF inhibition alone: COMBI-d11 (dabrafenib/trametinib versus dabrafenib/placebo), COMBI-v12 (dabrafenib/trametinib versus vemurafenib), and coBRIM13 (vemurafenib/cobimetinib versus vemurafenib/placebo). As expected, compared to BRAF inhibitor monotherapy, combination BRAF and MEK inhibition produced greater responses and improved progression-free and overall survival (Table 2). Interestingly, the rate of cutaneous squamous-cell carcinomas was much lower with combination therapy, reflecting the more profound degree of MAPK pathway inhibition achieved with combination BRAF and MEK inhibition. Based on these results, FDA approval was granted for both dabrafenib/trametinib and vemurafenib/cobimetinib combinations in 2015. Although the dabrafenib/trametinib combination was only approved in 2015, trametinib had independently gained FDA approval in 2013 for the treatment of BRAF V600E/K–mutated melanoma on the basis of the phase 3 METRIC study.77
Encorafenib (LGX818) and binimetinib (MEK162, ARRY-162, ARRY-438162) are new BRAF and MEK inhibitors currently being evaluated in clinical trials. Encorafenib/binimetinib combination was first evaluated in a phase 3 study (COLUMBUS) that compared it with vemurafenib monotherapy in BRAF-mutant melanoma.78 Unsurprisingly, encorafenib/binimetinib combination produced greater and more durable responses compared to vemurafenib monotherapy. The median progression-free survival of the encorafenib/binimetinib combination (14.9 months) was greater than vemurafenib monotherapy (7.3 months) in this study, and intriguingly greater than that seen with vemurafenib/cobimetinib (coBRIM 9.9 months) and dabrafenib/trametinib (COMBI-d 9.3 months; COMBI-v 11.4 months). Of note, although encorafenib has an IC50 midway between dabrafenib and vemurafenib in cell-free assays (0.8 nM dabrafenib, 4 nM encorafenib, and 31 nM vemurafenib), it has an extremely slower off-rate from BRAF V600E, which results in significantly greater target inhibition in cells following drug wash-out.79 This may account for the significantly greater clinical benefit seen with encorafenib/binimetinib in clinical trials. Final study data are eagerly awaited. Regulatory approval has been sought, and is pending at this time.
Binimetinib has been compared to dacarbazine in a phase 3 study (NEMO) of patients with NRAS-mutant melanoma, most of whom had been previously treated with immunotherapy.80 Response rates were low in both arms, although slightly greater with binimetinib than dacarbazine (15% versus 9%), commensurate with a modest improvement in progression-free survival. FDA approval has been sought and remains pending at this time.
KIT INHIBITION SINGLY AND IN COMBINATION
The KIT receptor protein tyrosine kinase is a transmembrane protein consisting of extracellular and intracellular domains. Activating KIT mutations occur in 2% to 8% of all melanoma patients and may be found in all melanoma subtypes but are commonest in acral melanomas (10%–20%) and mucosal melanomas (15%–20%). Activating KIT mutations primarily occur in exons 11 and 13, which code for the juxtamembrane and kinase domains, respectively.5,81–83
Imatinib mesylate is a tyrosine kinase inhibitor of the 2-phenyl amino pyrimidine class that occupies the tyrosine kinase active site with resultant blocking of tyrosine kinase activity. Imatinib mesylate is known to block KIT and has been extensively studied in patients with gastrointestinal stromal tumors (GIST), 80% of whom harbor KIT mutations, in both the adjuvant and the metastatic settings. In melanoma, imatinib mesylate was studied in a Chinese open-label, phase 2 study of imatinib mesylate monotherapy in metastatic melanoma harboring KIT mutation or amplification; 25% of the study patients had mucosal disease and the rest had cutaneous disease, with acral involvement in 50% of all patients.84 Overall response rate was 23%, while 51% of patients remained alive at 1 year with no differences in response rate and/or survival being noted between patients with either KIT mutations or amplifications. In a separate study of imatinib mesylate at 400 mg daily or 400 mg twice daily in Caucasian patients with KIT-mutated/amplified melanoma, similar response and survival rates were reported, although patients with KIT mutations did nonsignificantly better than those with KIT amplifications.85
Other novel studies evaluating KIT inhibitors include KIT inhibition in combination with the VEGF inhibitor bevacizumab and a study of selective BCR-ABL kinase inhibitor nilotinib in imatinib-resistant melanoma. In the former phase 1/2 study, Flaherty and colleagues studied imatinib 800 mg daily and bevacizumab at 10 mg/kg every 2 weeks in 63 patients with advanced tumors, including 23 with metastatic melanoma. Although the combination was relatively nontoxic, no significant efficacy signal was seen and further accrual to the phase 2 portion was halted after the first stage was completed.86 Nilotinib is a BCR-ABL1 tyrosine kinase inhibitor intelligently designed based on the structure of the ABL-imatinib complex that is 10 to 30 times more potent than imatinib in inhibiting BCR-ABL1 tyrosine kinase activity. Nilotinib is approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML), with reported efficacy in patients with central nervous system (CNS) involvement.87,88 Nilotinib has been studied in a single study of KIT-mutated/amplified melanoma that included patients with imatinib-resistance and those with treated CNS disease. Nilotinib appeared to be active in imatinib-resistant melanoma, although no responses were seen in the CNS disease cohort.89 Overall, the response rates observed with KIT inhibition in melanoma are much lower than those observed in CML and GIST.
CONCLUSION AND FUTURE DIRECTIONS
Prior to 2011, the only approved agents for the treatment of advanced melanoma were dacarbazine and high-dose interleukin-2. Since 2011, drug approvals in melanoma have proceeded at a frenetic pace unmatched in any other disease. The primary events underlying this are advances in our understanding of the gene mutation landscape driving melanoma tumorigenesis, accompanied by insights into the means by which tumors circumvent the induction of effective anti-tumor T-cell responses. These insights have resulted in the development of inhibitors targeting MAPK pathway kinases BRAF, MEK, and NRAS), KIT, and regulatory immune checkpoints (CTLA-4 and PD-1). Although BRAF/MEK inhibition results in profound reductions and even occasional complete responses in patients, these responses are typically short lived, rarely lasting more than 9 to 11 months; the encorafenib/binimetinib combination may improve that duration marginally. However, the signature therapeutic advance in melanoma of the past decade is immunotherapy, particularly the development of inhibitors of CTLA-4 and PD-1 immune checkpoints. With these agents, significant proportions of treated patients remain free of progression off-therapy (ipilimumab 23%; nivolumab 34%; pembrolizumab 35%; ipilimumab/nivolumab 64%), and some patients can be successfully re-induced after delayed progression. Separately, the high response rates observed with the use of KIT inhibitors in CML and GIST have not been observed in KIT mutated/amplified melanoma and development of agents in this space has been limited. The challenges ahead center around identifying predictive biomarkers and circumventing primary or acquired resistance, with the eventual goal of producing durable remissions in the majority of treated patients.
Our improved understanding of the mechanisms of acquired resistance to BRAF/MEK inhibitors suggests that anti-tumor activity may be achieved by targeting multiple pathways, possibly with combination regimens comprising other inhibitors and/or immunotherapy. Preclinical data supports the use of combination strategies targeting both ERK and PI3K/mTOR to circumvent acquired resistance.90 Ongoing studies are evaluating combinations with biguanides (metformin: NCT02143050 and NCT01638676; phenformin: NCT03026517), HSP90 inhibitors (XL888: NCT02721459; AT13387: NCT02097225), and decitabine (NCT01876641).
One complexity affecting management of resistance in the targeted therapy landscape remains tumor heterogeneity, particularly intra- and intertumoral heterogeneity, which may explain the apparent contradiction between continued efficacy of BRAF inhibitors in BRAF-resistant tumors and preclinical data predicting slower progression of resistant tumors on cessation of BRAF inhibitors.91–94 These data provide a rationale to investigate intermittent dosing regimens with BRAF/MEK inhibitors; several studies exploring this approach are ongoing (NCT01894672 and NCT02583516).
Given the specificity, adaptability, and memory response associated with immunotherapy, it is likely that these agents will be used to treat the majority of patients regardless of mutational status. Hence, identifying predictive biomarkers of response to immune checkpoint inhibitors is vital. The presence of CD8+ T-cell infiltrate and IFN-γ gene signature, which indicate an “inflamed” tumor microenvironment, are highly predictive of clinical benefit from PD-1 inhibitors.95,96 However, not all PD-1 responders have “inflamed” tumor microenvironments, and not all patients with an “inflamed” tumor microenvironment respond to immune checkpoint inhibitors. The complexity of the immune system is reflected in the multiple non-redundant immunologic pathways, both positive and negative, with checkpoints and ligands that emerge dynamically in response to treatment. Given the dynamic nature of the immune response, it is unlikely that any single immunologic biomarker identified pre-treatment will be completely predictive. Rather, the complexity of the biomarker approach must match the complexity of the immune response elicited, and will likely incorporate multifarious elements including CD8+ T-cell infiltrate, IFN-γ gene signature, and additional elements including microbiome, genetic polymorphisms, and tumor mutation load. The goal is to use multiple markers to guide development of combinations and then, depending on initial response, to examine tumors for alterations to guide decisions about additional treatment(s) to improve responses, with the eventual goal being durable clinical responses for all patients.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
- Guy GP, Thomas CC, Thompson T, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6.
- Anderson WF, Pfeiffer RM, Tucker MA, Rosenberg PS. Divergent cancer pathways for early-onset and late-onset cutaneous malignant melanoma. Cancer 2009;115:4176–85.
- Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47.
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–6.
- Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–63.
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–96.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 2014;15:323–32.
- Hauschild A, Grob J-J, Demidov LV, et al. DaBRAFenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380:358–65.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014;371:1877–88.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined daBRAFenib and trametinib. N Engl J Med 2015;372:30–9.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76.
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010;107:4275–80.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23.
- Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54.
- Weber JS, Angelo SP D’, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2015;16:375–84.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013;369:134–44.
- Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32.
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33.
- Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015;372:2006–17.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34.
- Eggermont AMM. Therapeutic vaccines in solid tumours: can they be harmful? Eur J Cancer 2009;45:2087–90.
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012;4:127ps8.
- Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014;192:5451–8.
- Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62–8.
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992;356:607–9.
- Greenfield EA, Nguyen KA, Kuchroo VK. CD28/B7 costimulation: a review. Crit Rev Immunol 1998;18:389–418.
- Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med 2006;355:973–5.
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001;19:565–94.
- Collins AV, Brodie DW, Gilbert RJC, et al. The interaction properties of costimulatory molecules revisited. Immunity 2002;17:201–10.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995;182:459–65.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734–6.
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704.
- Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 2002;169:5538–45.
- Youngnak P, Kozono Y, Kozono H, et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun 2003;307:672–7.
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 2004;173:945–54.
- Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 2005;25:9543–53.
- Riley JL. PD-1 signaling in primary T cells. Immunol Rev 2009;229:114–25.
- Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 2013;1291:1–13.
- Weber JS, O’Day S, Urba W, et al. Phase I/II study of ipilimumab for patients with metastatic melanoma. J Clin Oncol 2008;26:5950–6.
- Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A 2008;105:3005–10.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 2010;11:155–64.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–94.
- Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014;2:846–56.
- Poole RM. Pembrolizumab: first global approval. Drugs 2014;74:1973–81.
- Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30.
- Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–93.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 2017;18:31–41.
- Dhomen N, Marais R. BRAF signaling and targeted therapies in melanoma. Hematol Oncol Clin North Am 2009;23:529–45, ix.
- Satyamoorthy K, Li G, Gerrero MR, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res 2003;63:756–9.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54.
- Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–14.
- Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010;8:67.
- Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS ONE 2012;7:e35309.
- Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464:431–5.
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010;464:427–30.
- Thomas NE, Edmiston SN, Alexander A, et al. Association between NRAS and BRAF mutational status and melanoma-specific survival among patients with higher-risk primary melanoma. JAMA Oncology 2015;1:359–68.
- Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 2012;35:66–72.
- Johnson DB, Lovly CM, Flavin M, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95.
- Johnson DB, Frampton GM, Rioth MJ, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–67.
- Kiuru M, Busam KJ. The NF1 gene in tumor syndromes and melanoma. Lab Invest 2017;97:146–57.
- Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 2014;74:2340–50.
- Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol 2005;23:5281–93.
- Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62.
- Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res 2012;18:555–67.
- Davar D, Kirkwood JM. CCR 20th anniversary commentary: MAPK/ERK pathway inhibition in melanoma-kinase inhibition redux. Clin Cancer Res 2015;21:5412–4.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15.
- Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010;468:968–72.
- Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7.
- Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93.
- Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014;4:94–109.
- Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14.
- Dummer R, Ascierto PA, Gogas HJ, et al. Results of COLUMBUS Part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Presented at Society for Melanoma Research 2016 Congress. November 6-9, 2016. Boston (MA).
- Adelmann CH, Ching G, Du L, et al. Comparative profiles of BRAF inhibitors: the paradox index as a predictor of clinical toxicity. Oncotarget 2016;7:30453–60.
- Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2017;18:435–45.
- Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–93.
- Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res 2008;14:6821–8.
- Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res 2010;23:210–5.
- Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 2011;29:2904–9.
- Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 2013;31:3182–90.
- Flaherty KT, Hamilton BK, Rosen MA, et al. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015;20:952–9.
- Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013;27:107–12.
- Reinwald M, Schleyer E, Kiewe P, et al. Efficacy and pharmacologic data of second-generation tyrosine kinase inhibitor nilotinib in BCR-ABL-positive leukemia patients with central nervous system relapse after allogeneic stem cell transplantation. Biomed Res Int 2014;2014:637059.
- Carvajal RD, Lawrence DP, Weber JS, et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin Cancer Res 2015;21:2289–96.
- Carlino MS, Todd JR, Gowrishankar K, et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol 2014;8:544–54.
- Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther 2013;12:1332–42.
- Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer 2014;120:3142–53.
- Thakur M Das, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–5.
- Thakur M Das, Stuart DD. Molecular pathways: response and resistance to BRAF and MEK inhibitors in BRAF(V600E) tumors. Clin Cancer Res 2014;20:1074–80.
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71.
- Ayers M, Lunceford J, Nebozhyn M, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunotherapy Cancer 2015;3(Suppl 2):P80.
Adjuvant Chemotherapy in the Treatment of Colon Cancer
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
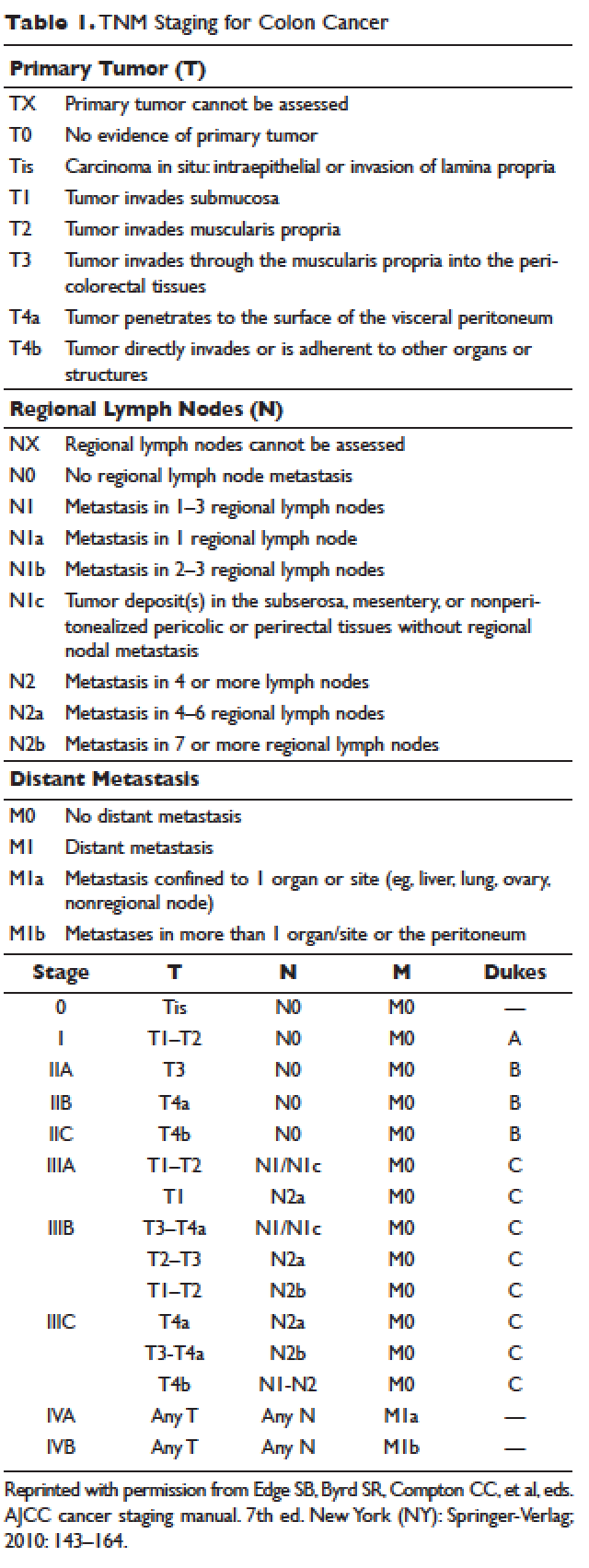
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.

SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
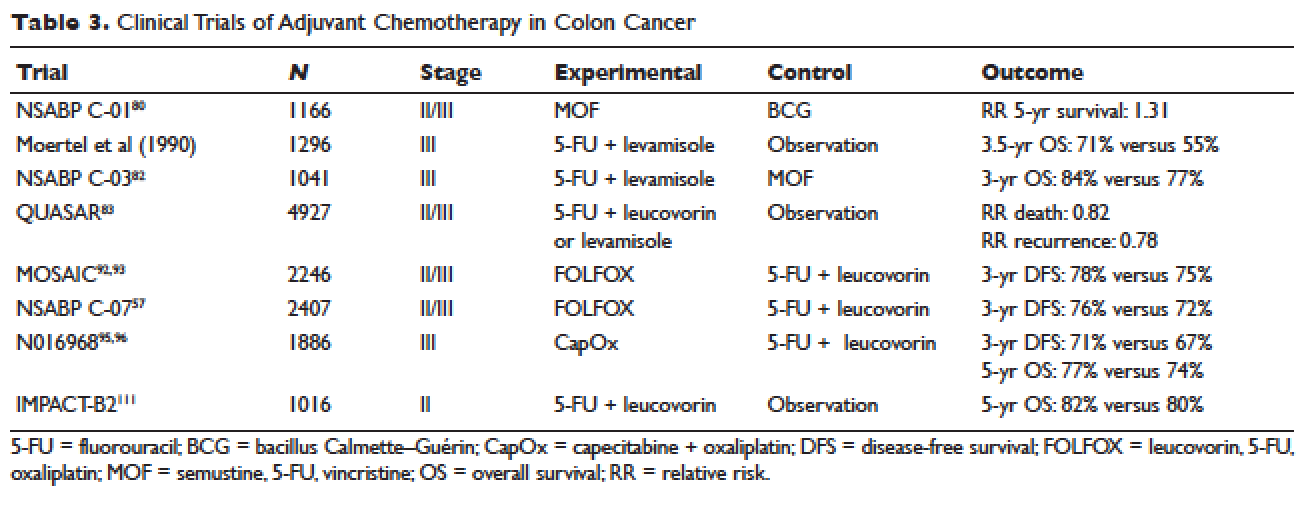
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.
SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.
SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.