User login
Are IV fluids better than oral rehydration for children with acute diarrhea and vomiting?
Intravenous fluid therapy (IVF) has a slightly lower failure rate than oral replacement therapy (ORT) in children with acute gastroenteritis, but the clinical significance is questionable. IVF takes longer to initiate than ORT and lengthens the hospital stay (strength of recommendation: B, meta-analysis of poor-to-moderate-quality trials).
Shorter hospital stay with oral replacement therapy
A 2006 systematic review compared ORT and IVF in 1811 children 0 to 18 years of age with viral gastroenteritis who were treated for failure to rehydrate in both outpatient and inpatient settings (18 randomized controlled trials [RCTs] of poor to moderate quality).1 The primary outcome was “continued failure to rehydrate,” which varied by study and included persistent vomiting, persistent dehydration, shock, or seizures.
Overall, the risk of failure to rehydrate was 4.9% for ORT and 1.3% for IVF (risk difference [RD]=4%; 95% confidence interval [CI], 1%-7%; number needed to treat [NNT]=25). The length of stay (24-hour observation unit or inpatient hospitalization) was shorter for ORT than IVF (6 studies, 526 patients; weighted mean difference (WMD)= −1.2 days; 95% CI, −2.38 to −0.02). Investigators found no difference in weight gain, hyponatremia, hypernatremia, duration of diarrhea, or total fluid intake at 24 hours.
ORT can be started more quickly than IVF
An RCT conducted in an urban emergency department evaluated ORT and IVF for 4 hours in 72 children ages 8 weeks to 3 years with moderate dehydration from viral gastroenteritis.2 This trial was included in the previously described review but evaluated additional outcomes: time required to initiate either ORT or IVF, improvement in symptoms at 2 hours, hospitalization rate, and preference for ORT in the future.
The authors also used a 10-point dehydration scoring system that included: decreased skin elasticity, capillary refill >2 seconds, general appearance, absence of tears, abnormal respirations, dry mucous membranes, sunken eyes, abnormal radial pulse, tachycardia >150 beats per minute, and decreased urine output. Details on the type of ORT or IVF were not reported.
ORT was initiated faster than IVF (mean difference [MD]=21 minutes; 95% CI, 10-32 minutes). No difference in improvement in dehydration scores was observed at 2 hours (ORT 78% vs IVF 80%; MD=1.2%; 95% CI, −20.5% to 18%). Nor was the hospitalization rate significantly different (IVF 48.7% and ORT 30.6%; MD=−18.1%; 95% CI, −40.1% to 4.1%). Most patients preferred to have the same therapy, whether ORT or IVF, with the next episode of gastroenteritis (61.3% vs 51.4%; MD=9.9%; 95% CI, −14 to 34).
1. Hartling L, Bellmare S, Wiebe N, et al. Oral versus intravenous rehydration for treating dehydration due to gastroenteritis in children. Cochrane Database Syst Rev. 2006;(3):CD004390.
2. Spandorfer PR, Alessandrini EA, Joffe MD, et al. Oral versus intravenous rehydration of moderately dehydrated children: a randomized, controlled trial. Pediatrics. 2005;115:295-301.
Intravenous fluid therapy (IVF) has a slightly lower failure rate than oral replacement therapy (ORT) in children with acute gastroenteritis, but the clinical significance is questionable. IVF takes longer to initiate than ORT and lengthens the hospital stay (strength of recommendation: B, meta-analysis of poor-to-moderate-quality trials).
Shorter hospital stay with oral replacement therapy
A 2006 systematic review compared ORT and IVF in 1811 children 0 to 18 years of age with viral gastroenteritis who were treated for failure to rehydrate in both outpatient and inpatient settings (18 randomized controlled trials [RCTs] of poor to moderate quality).1 The primary outcome was “continued failure to rehydrate,” which varied by study and included persistent vomiting, persistent dehydration, shock, or seizures.
Overall, the risk of failure to rehydrate was 4.9% for ORT and 1.3% for IVF (risk difference [RD]=4%; 95% confidence interval [CI], 1%-7%; number needed to treat [NNT]=25). The length of stay (24-hour observation unit or inpatient hospitalization) was shorter for ORT than IVF (6 studies, 526 patients; weighted mean difference (WMD)= −1.2 days; 95% CI, −2.38 to −0.02). Investigators found no difference in weight gain, hyponatremia, hypernatremia, duration of diarrhea, or total fluid intake at 24 hours.
ORT can be started more quickly than IVF
An RCT conducted in an urban emergency department evaluated ORT and IVF for 4 hours in 72 children ages 8 weeks to 3 years with moderate dehydration from viral gastroenteritis.2 This trial was included in the previously described review but evaluated additional outcomes: time required to initiate either ORT or IVF, improvement in symptoms at 2 hours, hospitalization rate, and preference for ORT in the future.
The authors also used a 10-point dehydration scoring system that included: decreased skin elasticity, capillary refill >2 seconds, general appearance, absence of tears, abnormal respirations, dry mucous membranes, sunken eyes, abnormal radial pulse, tachycardia >150 beats per minute, and decreased urine output. Details on the type of ORT or IVF were not reported.
ORT was initiated faster than IVF (mean difference [MD]=21 minutes; 95% CI, 10-32 minutes). No difference in improvement in dehydration scores was observed at 2 hours (ORT 78% vs IVF 80%; MD=1.2%; 95% CI, −20.5% to 18%). Nor was the hospitalization rate significantly different (IVF 48.7% and ORT 30.6%; MD=−18.1%; 95% CI, −40.1% to 4.1%). Most patients preferred to have the same therapy, whether ORT or IVF, with the next episode of gastroenteritis (61.3% vs 51.4%; MD=9.9%; 95% CI, −14 to 34).
Intravenous fluid therapy (IVF) has a slightly lower failure rate than oral replacement therapy (ORT) in children with acute gastroenteritis, but the clinical significance is questionable. IVF takes longer to initiate than ORT and lengthens the hospital stay (strength of recommendation: B, meta-analysis of poor-to-moderate-quality trials).
Shorter hospital stay with oral replacement therapy
A 2006 systematic review compared ORT and IVF in 1811 children 0 to 18 years of age with viral gastroenteritis who were treated for failure to rehydrate in both outpatient and inpatient settings (18 randomized controlled trials [RCTs] of poor to moderate quality).1 The primary outcome was “continued failure to rehydrate,” which varied by study and included persistent vomiting, persistent dehydration, shock, or seizures.
Overall, the risk of failure to rehydrate was 4.9% for ORT and 1.3% for IVF (risk difference [RD]=4%; 95% confidence interval [CI], 1%-7%; number needed to treat [NNT]=25). The length of stay (24-hour observation unit or inpatient hospitalization) was shorter for ORT than IVF (6 studies, 526 patients; weighted mean difference (WMD)= −1.2 days; 95% CI, −2.38 to −0.02). Investigators found no difference in weight gain, hyponatremia, hypernatremia, duration of diarrhea, or total fluid intake at 24 hours.
ORT can be started more quickly than IVF
An RCT conducted in an urban emergency department evaluated ORT and IVF for 4 hours in 72 children ages 8 weeks to 3 years with moderate dehydration from viral gastroenteritis.2 This trial was included in the previously described review but evaluated additional outcomes: time required to initiate either ORT or IVF, improvement in symptoms at 2 hours, hospitalization rate, and preference for ORT in the future.
The authors also used a 10-point dehydration scoring system that included: decreased skin elasticity, capillary refill >2 seconds, general appearance, absence of tears, abnormal respirations, dry mucous membranes, sunken eyes, abnormal radial pulse, tachycardia >150 beats per minute, and decreased urine output. Details on the type of ORT or IVF were not reported.
ORT was initiated faster than IVF (mean difference [MD]=21 minutes; 95% CI, 10-32 minutes). No difference in improvement in dehydration scores was observed at 2 hours (ORT 78% vs IVF 80%; MD=1.2%; 95% CI, −20.5% to 18%). Nor was the hospitalization rate significantly different (IVF 48.7% and ORT 30.6%; MD=−18.1%; 95% CI, −40.1% to 4.1%). Most patients preferred to have the same therapy, whether ORT or IVF, with the next episode of gastroenteritis (61.3% vs 51.4%; MD=9.9%; 95% CI, −14 to 34).
1. Hartling L, Bellmare S, Wiebe N, et al. Oral versus intravenous rehydration for treating dehydration due to gastroenteritis in children. Cochrane Database Syst Rev. 2006;(3):CD004390.
2. Spandorfer PR, Alessandrini EA, Joffe MD, et al. Oral versus intravenous rehydration of moderately dehydrated children: a randomized, controlled trial. Pediatrics. 2005;115:295-301.
1. Hartling L, Bellmare S, Wiebe N, et al. Oral versus intravenous rehydration for treating dehydration due to gastroenteritis in children. Cochrane Database Syst Rev. 2006;(3):CD004390.
2. Spandorfer PR, Alessandrini EA, Joffe MD, et al. Oral versus intravenous rehydration of moderately dehydrated children: a randomized, controlled trial. Pediatrics. 2005;115:295-301.
Evidence-based answers from the Family Physicians Inquiries Network

Do corticosteroids relieve Bell’s palsy?
Yes, but not severe disease. Corticosteroids likely improve facial motor function in adults with mild to moderate Bell’s palsy (strength of recommendation [SOR]: B, meta-analysis of heterogeneous randomized controlled trials [RCTs]). Corticosteroids are probably ineffective in treating cosmetically disabling or severe disease (SOR: A, meta-analysis and large RCT).
Improvement seen with corticosteroids in mild to moderate palsy
A 2010 Cochrane review of 8 RCTs (7 double-blind) compared corticosteroids with placebo in 1569 patients with Bell’s palsy, 24 months to 84 years of age.1 The definition of mild and moderate severity of symptoms differed across studies, as did corticosteroid doses. Only 6 trials required initiation of therapy within 3 days.
More patients in the corticosteroid group had completely recovered facial motor function at 6 months than patients taking placebo (77% vs 65%; 7 trials, 1507 patients; relative risk [RR]=0.71; 95% confidence interval [CI], 0.61-0.81; number needed to treat=10). Improvement in cosmetically disabling or severe disease wasn’t significant (5 trials, 668 patients; RR=0.97; 95% CI, 0.44-2.2).
Prednisolone with and without an antiviral reduces facial weakness
A 2012 prospective, randomized, double-blind, placebo-controlled, multicenter trial evaluated prednisolone (60 mg/day for 5 days, tapered for 5 days) in 829 adults, 18 to 75 years of age.2 Patients were randomized to one of 4 groups: placebo plus placebo, prednisolone plus placebo, valacyclovir plus placebo, and prednisolone plus valacyclovir. Facial function was assessed over 12 months using the Sunnybrook grading system (scored from 0 to 100; 0=complete paralysis, 100=normal function).
Compared to the groups not receiving any prednisolone, the 2 groups that received prednisolone, either with placebo or valacyclovir, had significantly less facial weakness at 12 months for both mild and moderate palsy (Sunnybrook scores <90: 184 patients; difference= −10.3%; 95% CI, −15.9 to −4.7; P<.001; Sunnybrook score <80: 134 patients; difference= −6.9%; 95% CI, −11.9 to −1.9; P=.01; Sunnybrook score <70: 98 patients; difference= −7.8%; 95% CI, −12.1 to −3.4; P<.001). Patients with severe disease (Sunnybrook score <50) didn’t show significant improvement (56 patients; difference= –2.9%; CI, −6.4 to 0.5; P=.10).
Guideline recommends corticosteroids for Bell’s palsy
The 2014 American Academy of Neurology evidence-based guideline reviewed all studies of the use of steroids in Bell’s palsy published after the original 2001 guideline.3 They found 2 high-quality RCTs, both of which are included in the 2010 Cochrane review. The 2014 guideline recommends corticosteroids for every patient who develops Bell’s palsy unless a medical contraindication exists (2 Class 1 studies [RCTs], Level A [must prescribe or offer]).
1. Salinas RA, Alvarez G, Daly F, et al. Corticosteroids for Bell’s palsy (idiopathic facial paralysis). Cochrane Database Syst Rev. 2010;(3):CD001942.
2. Berg T, Bylund N, Marsk E, et al. The effect of prednisolone on sequelae in Bell’s palsy. Arch Otolaryngol Head Neck Surg. 2012;138:445-449.
3. Gronseth G, Paduga R, American Academy of Neurology. Evidence-based guideline update: steroids and antivirals for Bell’s palsy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2012;79:2209-2213.
Yes, but not severe disease. Corticosteroids likely improve facial motor function in adults with mild to moderate Bell’s palsy (strength of recommendation [SOR]: B, meta-analysis of heterogeneous randomized controlled trials [RCTs]). Corticosteroids are probably ineffective in treating cosmetically disabling or severe disease (SOR: A, meta-analysis and large RCT).
Improvement seen with corticosteroids in mild to moderate palsy
A 2010 Cochrane review of 8 RCTs (7 double-blind) compared corticosteroids with placebo in 1569 patients with Bell’s palsy, 24 months to 84 years of age.1 The definition of mild and moderate severity of symptoms differed across studies, as did corticosteroid doses. Only 6 trials required initiation of therapy within 3 days.
More patients in the corticosteroid group had completely recovered facial motor function at 6 months than patients taking placebo (77% vs 65%; 7 trials, 1507 patients; relative risk [RR]=0.71; 95% confidence interval [CI], 0.61-0.81; number needed to treat=10). Improvement in cosmetically disabling or severe disease wasn’t significant (5 trials, 668 patients; RR=0.97; 95% CI, 0.44-2.2).
Prednisolone with and without an antiviral reduces facial weakness
A 2012 prospective, randomized, double-blind, placebo-controlled, multicenter trial evaluated prednisolone (60 mg/day for 5 days, tapered for 5 days) in 829 adults, 18 to 75 years of age.2 Patients were randomized to one of 4 groups: placebo plus placebo, prednisolone plus placebo, valacyclovir plus placebo, and prednisolone plus valacyclovir. Facial function was assessed over 12 months using the Sunnybrook grading system (scored from 0 to 100; 0=complete paralysis, 100=normal function).
Compared to the groups not receiving any prednisolone, the 2 groups that received prednisolone, either with placebo or valacyclovir, had significantly less facial weakness at 12 months for both mild and moderate palsy (Sunnybrook scores <90: 184 patients; difference= −10.3%; 95% CI, −15.9 to −4.7; P<.001; Sunnybrook score <80: 134 patients; difference= −6.9%; 95% CI, −11.9 to −1.9; P=.01; Sunnybrook score <70: 98 patients; difference= −7.8%; 95% CI, −12.1 to −3.4; P<.001). Patients with severe disease (Sunnybrook score <50) didn’t show significant improvement (56 patients; difference= –2.9%; CI, −6.4 to 0.5; P=.10).
Guideline recommends corticosteroids for Bell’s palsy
The 2014 American Academy of Neurology evidence-based guideline reviewed all studies of the use of steroids in Bell’s palsy published after the original 2001 guideline.3 They found 2 high-quality RCTs, both of which are included in the 2010 Cochrane review. The 2014 guideline recommends corticosteroids for every patient who develops Bell’s palsy unless a medical contraindication exists (2 Class 1 studies [RCTs], Level A [must prescribe or offer]).
Yes, but not severe disease. Corticosteroids likely improve facial motor function in adults with mild to moderate Bell’s palsy (strength of recommendation [SOR]: B, meta-analysis of heterogeneous randomized controlled trials [RCTs]). Corticosteroids are probably ineffective in treating cosmetically disabling or severe disease (SOR: A, meta-analysis and large RCT).
Improvement seen with corticosteroids in mild to moderate palsy
A 2010 Cochrane review of 8 RCTs (7 double-blind) compared corticosteroids with placebo in 1569 patients with Bell’s palsy, 24 months to 84 years of age.1 The definition of mild and moderate severity of symptoms differed across studies, as did corticosteroid doses. Only 6 trials required initiation of therapy within 3 days.
More patients in the corticosteroid group had completely recovered facial motor function at 6 months than patients taking placebo (77% vs 65%; 7 trials, 1507 patients; relative risk [RR]=0.71; 95% confidence interval [CI], 0.61-0.81; number needed to treat=10). Improvement in cosmetically disabling or severe disease wasn’t significant (5 trials, 668 patients; RR=0.97; 95% CI, 0.44-2.2).
Prednisolone with and without an antiviral reduces facial weakness
A 2012 prospective, randomized, double-blind, placebo-controlled, multicenter trial evaluated prednisolone (60 mg/day for 5 days, tapered for 5 days) in 829 adults, 18 to 75 years of age.2 Patients were randomized to one of 4 groups: placebo plus placebo, prednisolone plus placebo, valacyclovir plus placebo, and prednisolone plus valacyclovir. Facial function was assessed over 12 months using the Sunnybrook grading system (scored from 0 to 100; 0=complete paralysis, 100=normal function).
Compared to the groups not receiving any prednisolone, the 2 groups that received prednisolone, either with placebo or valacyclovir, had significantly less facial weakness at 12 months for both mild and moderate palsy (Sunnybrook scores <90: 184 patients; difference= −10.3%; 95% CI, −15.9 to −4.7; P<.001; Sunnybrook score <80: 134 patients; difference= −6.9%; 95% CI, −11.9 to −1.9; P=.01; Sunnybrook score <70: 98 patients; difference= −7.8%; 95% CI, −12.1 to −3.4; P<.001). Patients with severe disease (Sunnybrook score <50) didn’t show significant improvement (56 patients; difference= –2.9%; CI, −6.4 to 0.5; P=.10).
Guideline recommends corticosteroids for Bell’s palsy
The 2014 American Academy of Neurology evidence-based guideline reviewed all studies of the use of steroids in Bell’s palsy published after the original 2001 guideline.3 They found 2 high-quality RCTs, both of which are included in the 2010 Cochrane review. The 2014 guideline recommends corticosteroids for every patient who develops Bell’s palsy unless a medical contraindication exists (2 Class 1 studies [RCTs], Level A [must prescribe or offer]).
1. Salinas RA, Alvarez G, Daly F, et al. Corticosteroids for Bell’s palsy (idiopathic facial paralysis). Cochrane Database Syst Rev. 2010;(3):CD001942.
2. Berg T, Bylund N, Marsk E, et al. The effect of prednisolone on sequelae in Bell’s palsy. Arch Otolaryngol Head Neck Surg. 2012;138:445-449.
3. Gronseth G, Paduga R, American Academy of Neurology. Evidence-based guideline update: steroids and antivirals for Bell’s palsy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2012;79:2209-2213.
1. Salinas RA, Alvarez G, Daly F, et al. Corticosteroids for Bell’s palsy (idiopathic facial paralysis). Cochrane Database Syst Rev. 2010;(3):CD001942.
2. Berg T, Bylund N, Marsk E, et al. The effect of prednisolone on sequelae in Bell’s palsy. Arch Otolaryngol Head Neck Surg. 2012;138:445-449.
3. Gronseth G, Paduga R, American Academy of Neurology. Evidence-based guideline update: steroids and antivirals for Bell’s palsy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2012;79:2209-2213.
Evidence-based answers from the Family Physicians Inquiries Network
Is there an increased risk of GI bleeds with SSRIs?
Yes. Selective serotonin reuptake inhibitors (SSRIs) are likely associated with a moderate increased risk of upper gastrointestinal (UGI) bleeding. Use of a nonsteroidal anti-inflammatory drug (NSAID) in combination with the SSRI appears to amplify the risk (strength of recommendation [SOR]: B, meta-analysis of cohort and case control studies).
The increased risk from SSRIs occurs within the first 7 to 28 days after exposure (SOR: B, retrospective study).
SSRIs raise bleeding risk; concurrent NSAIDs raise it more
A 2014 systematic review and meta-analysis of 19 case-control and cohort studies with a total of 446,949 patients investigated the risk of UGI bleeding in patients using SSRIs and NSAIDs.1 The studies, which included both inpatients and outpatients, were done in Europe and North America. Patients were at least 16 years old, but pooled demographics were not reported. Investigators compared SSRI use with or without concurrent NSAID use to placebo or no treatment.
SSRI use was associated with an increased risk of UGI bleeding in 15 case-control studies (393,268 patients; odds ratio [OR]=1.7; 95% confidence interval [CI], 1.4-1.9) and 4 cohort studies (53,681 patients; OR=1.7; 95% CI, 1.1-2.5). The simultaneous use of SSRIs and NSAIDs compared to nonuse of both medications was associated with a larger increase in bleeding risk (10 case-control studies, 223,336 patients; OR=4.3; 95% CI, 2.8-6.4).
The meta-analysis is limited by statistically significant heterogeneity in all of the pooled results and high risk of bias in 9 of the case-control studies and all of the cohort studies. There was no evidence of publication bias, however.
Bleeding risk rises 7 to 28 days after SSRI exposure
A 2014 case-crossover study of 5377 inpatients in Taiwan with a psychiatric diagnosis evaluated the risk of UGI bleeding within the first 28 days after SSRI exposure (SSRI-mediated inhibition of platelets occurs within the first 7 to 14 days).2 The average age of the patients was 58 years and 75% of the study population was male. Each patient served as his or her own control.
ORs were calculated to compare patients who were exposed to SSRIs only during 7-, 14-, and 21-day windows immediately before a UGI bleed to controls exposed to SSRIs only during the control periods before the 7-, 14-, and 21-day windows. The ORs were adjusted through multivariate analysis to account for 7 potential confounding factors.
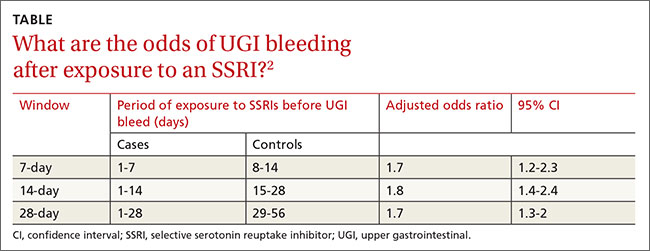
SSRI use was associated with an increased risk of UGI bleeding in 7-, 14-, and 21-day windows before the index event (TABLE2). An increased bleeding risk in the 14 days after SSRI initiation was observed in men (OR=2.4; 95% CI, 1.8-3.4) but not women (OR=1.0; 95% CI, 0.6-1.6). Increased bleeding risk in the 14 days after SSRI initiation was also observed in patients younger than 55 years (OR=2.1; 95% CI, 1.5-3.1), patients with a history of upper GI disease (OR=3.1; 95% CI, 1.7-6.0), and patients with no previous exposure to SSRIs (OR=2.6; 95% CI, 1.6-4.2).
This study didn’t account for SSRI indication as a potential confounder, and the study’s inclusion of inpatients, whose illnesses are typically more severe, may limit generalizability.
1. Anglin R, Yuan Y, Moayyedi P, et al. Risk of upper gastrointestinal bleeding with selective serotonin reuptake inhibitors with or without concurrent nonsteroidal anti-inflammatory use: a systematic review and meta-analysis. Am J Gastroenterol. 2014;109:811-819.
2. Wang YP, Chen YT, Tsai C, et al. Short-term use of serotonin reuptake inhibitors and risk of upper gastrointestinal bleeding. Am J Psychiatry. 2014;171:54-61.
Yes. Selective serotonin reuptake inhibitors (SSRIs) are likely associated with a moderate increased risk of upper gastrointestinal (UGI) bleeding. Use of a nonsteroidal anti-inflammatory drug (NSAID) in combination with the SSRI appears to amplify the risk (strength of recommendation [SOR]: B, meta-analysis of cohort and case control studies).
The increased risk from SSRIs occurs within the first 7 to 28 days after exposure (SOR: B, retrospective study).
SSRIs raise bleeding risk; concurrent NSAIDs raise it more
A 2014 systematic review and meta-analysis of 19 case-control and cohort studies with a total of 446,949 patients investigated the risk of UGI bleeding in patients using SSRIs and NSAIDs.1 The studies, which included both inpatients and outpatients, were done in Europe and North America. Patients were at least 16 years old, but pooled demographics were not reported. Investigators compared SSRI use with or without concurrent NSAID use to placebo or no treatment.
SSRI use was associated with an increased risk of UGI bleeding in 15 case-control studies (393,268 patients; odds ratio [OR]=1.7; 95% confidence interval [CI], 1.4-1.9) and 4 cohort studies (53,681 patients; OR=1.7; 95% CI, 1.1-2.5). The simultaneous use of SSRIs and NSAIDs compared to nonuse of both medications was associated with a larger increase in bleeding risk (10 case-control studies, 223,336 patients; OR=4.3; 95% CI, 2.8-6.4).
The meta-analysis is limited by statistically significant heterogeneity in all of the pooled results and high risk of bias in 9 of the case-control studies and all of the cohort studies. There was no evidence of publication bias, however.
Bleeding risk rises 7 to 28 days after SSRI exposure
A 2014 case-crossover study of 5377 inpatients in Taiwan with a psychiatric diagnosis evaluated the risk of UGI bleeding within the first 28 days after SSRI exposure (SSRI-mediated inhibition of platelets occurs within the first 7 to 14 days).2 The average age of the patients was 58 years and 75% of the study population was male. Each patient served as his or her own control.
ORs were calculated to compare patients who were exposed to SSRIs only during 7-, 14-, and 21-day windows immediately before a UGI bleed to controls exposed to SSRIs only during the control periods before the 7-, 14-, and 21-day windows. The ORs were adjusted through multivariate analysis to account for 7 potential confounding factors.
SSRI use was associated with an increased risk of UGI bleeding in 7-, 14-, and 21-day windows before the index event (TABLE2). An increased bleeding risk in the 14 days after SSRI initiation was observed in men (OR=2.4; 95% CI, 1.8-3.4) but not women (OR=1.0; 95% CI, 0.6-1.6). Increased bleeding risk in the 14 days after SSRI initiation was also observed in patients younger than 55 years (OR=2.1; 95% CI, 1.5-3.1), patients with a history of upper GI disease (OR=3.1; 95% CI, 1.7-6.0), and patients with no previous exposure to SSRIs (OR=2.6; 95% CI, 1.6-4.2).
This study didn’t account for SSRI indication as a potential confounder, and the study’s inclusion of inpatients, whose illnesses are typically more severe, may limit generalizability.
Yes. Selective serotonin reuptake inhibitors (SSRIs) are likely associated with a moderate increased risk of upper gastrointestinal (UGI) bleeding. Use of a nonsteroidal anti-inflammatory drug (NSAID) in combination with the SSRI appears to amplify the risk (strength of recommendation [SOR]: B, meta-analysis of cohort and case control studies).
The increased risk from SSRIs occurs within the first 7 to 28 days after exposure (SOR: B, retrospective study).
SSRIs raise bleeding risk; concurrent NSAIDs raise it more
A 2014 systematic review and meta-analysis of 19 case-control and cohort studies with a total of 446,949 patients investigated the risk of UGI bleeding in patients using SSRIs and NSAIDs.1 The studies, which included both inpatients and outpatients, were done in Europe and North America. Patients were at least 16 years old, but pooled demographics were not reported. Investigators compared SSRI use with or without concurrent NSAID use to placebo or no treatment.
SSRI use was associated with an increased risk of UGI bleeding in 15 case-control studies (393,268 patients; odds ratio [OR]=1.7; 95% confidence interval [CI], 1.4-1.9) and 4 cohort studies (53,681 patients; OR=1.7; 95% CI, 1.1-2.5). The simultaneous use of SSRIs and NSAIDs compared to nonuse of both medications was associated with a larger increase in bleeding risk (10 case-control studies, 223,336 patients; OR=4.3; 95% CI, 2.8-6.4).
The meta-analysis is limited by statistically significant heterogeneity in all of the pooled results and high risk of bias in 9 of the case-control studies and all of the cohort studies. There was no evidence of publication bias, however.
Bleeding risk rises 7 to 28 days after SSRI exposure
A 2014 case-crossover study of 5377 inpatients in Taiwan with a psychiatric diagnosis evaluated the risk of UGI bleeding within the first 28 days after SSRI exposure (SSRI-mediated inhibition of platelets occurs within the first 7 to 14 days).2 The average age of the patients was 58 years and 75% of the study population was male. Each patient served as his or her own control.
ORs were calculated to compare patients who were exposed to SSRIs only during 7-, 14-, and 21-day windows immediately before a UGI bleed to controls exposed to SSRIs only during the control periods before the 7-, 14-, and 21-day windows. The ORs were adjusted through multivariate analysis to account for 7 potential confounding factors.
SSRI use was associated with an increased risk of UGI bleeding in 7-, 14-, and 21-day windows before the index event (TABLE2). An increased bleeding risk in the 14 days after SSRI initiation was observed in men (OR=2.4; 95% CI, 1.8-3.4) but not women (OR=1.0; 95% CI, 0.6-1.6). Increased bleeding risk in the 14 days after SSRI initiation was also observed in patients younger than 55 years (OR=2.1; 95% CI, 1.5-3.1), patients with a history of upper GI disease (OR=3.1; 95% CI, 1.7-6.0), and patients with no previous exposure to SSRIs (OR=2.6; 95% CI, 1.6-4.2).
This study didn’t account for SSRI indication as a potential confounder, and the study’s inclusion of inpatients, whose illnesses are typically more severe, may limit generalizability.
1. Anglin R, Yuan Y, Moayyedi P, et al. Risk of upper gastrointestinal bleeding with selective serotonin reuptake inhibitors with or without concurrent nonsteroidal anti-inflammatory use: a systematic review and meta-analysis. Am J Gastroenterol. 2014;109:811-819.
2. Wang YP, Chen YT, Tsai C, et al. Short-term use of serotonin reuptake inhibitors and risk of upper gastrointestinal bleeding. Am J Psychiatry. 2014;171:54-61.
1. Anglin R, Yuan Y, Moayyedi P, et al. Risk of upper gastrointestinal bleeding with selective serotonin reuptake inhibitors with or without concurrent nonsteroidal anti-inflammatory use: a systematic review and meta-analysis. Am J Gastroenterol. 2014;109:811-819.
2. Wang YP, Chen YT, Tsai C, et al. Short-term use of serotonin reuptake inhibitors and risk of upper gastrointestinal bleeding. Am J Psychiatry. 2014;171:54-61.
Evidence-based answers from the Family Physicians Inquiries Network
What is the optimal duration of PPI therapy for healing an ulcer?
It depends on the type of ulcer. For Helicobacter pylori-associated peptic ulcers, 7-day treatment with a proton pump inhibitor (PPI) plus 2 antibiotics heals more than 90% of ulcers and is as effective as the same regimen followed by 2 to 4 additional weeks of PPI therapy (strength of recommendation [SOR]: A, meta-analysis of randomized controlled trials [RCTs]).
For peptic ulcers associated with nonsteroidal anti-inflammatory drugs (NSAIDs), 8 weeks of PPI treatment is better than 4 weeks in the case of gastric ulcers, but no more effective than 4 weeks for duodenal ulcers. (SOR: A, meta-analysis of RCTs).
For gastric ulcers resulting from endoscopic submucosal dissection, 4 weeks of PPI therapy is as effective as 8 weeks, but both regimens leave nearly a third of ulcers unhealed (SOR: B, single RCT).
For H pylori ulcers, 7 days of therapy does the trick
A 2005 meta-analysis of 6 RCTs with 862 patients compared 7 days of triple therapy with a PPI and 2 antibiotics with the same regimen followed by 2 to 4 additional weeks of PPI therapy.1 One RCT studied both duodenal and gastric ulcers; the remaining 5 assessed only duodenal ulcers. Investigators included only studies that clearly identified both H pylori eradication and ulcer healing as treatment goals and specified the number of patients treated, the number who experienced successful healing, endoscopic ulcer confirmation, and no concurrent NSAID use.
Triple therapy regimens comprised either omeprazole or esomeprazole 20 mg twice daily plus clarithromycin and either metronidazole, amoxicillin, or tinidazole for 7 days. In all studies, patients randomly assigned to receive an additional 2 to 4 weeks of PPI treatment were given omeprazole 20 mg/d.
Mean ulcer healing rates were 91% (95% confidence interval [CI], 87%-95%) for 7 days of PPI triple therapy compared with 92% (95% CI, 89%-96%) when PPI treatment was extended for an additional 2 to 4 weeks (odds ratio=1.1; 95% CI, 0.71-1.7).
Longer PPI therapy works better for NSAID-associated gastric ulcers
A 1998 meta-analysis examined 2 large RCTs that evaluated healing rates of NSAID-associated ulcers at 4 weeks and 8 weeks in 656 patients with gastric or duodenal ulcers who were treated with omeprazole 20 mg/d or 40 mg/d.2 Patients had ulcers 3 mm or larger or more than 10 erosions in the stomach or duodenum. Gastric ulcers outnumbered duodenal ulcers 2 to 1. Patients had taken continuous therapeutic doses of NSAIDs for at least 5 days per week during 2 weeks in the month preceding PPI therapy; about half were H pylori-positive.
For gastric ulcers, treatment success at 8 weeks was significantly higher at both PPI doses than at 4 weeks. The 208 patients taking the 20-mg dose showed 67% treatment success at 4 weeks and 83% at 8 weeks (P=.001). The 212 patients taking 40 mg had 67% treatment success at 4 weeks and 82% at 8 weeks (P=.002).
Duodenal ulcers showed no difference in healing at 4 and 8 weeks at either PPI dose. The 20-mg dose (116 patients) produced 84% treatment success at 4 weeks compared with 93% at 8 weeks (P=.2), and the 40-mg dose (120 patients) showed 86% treatment success at 4 weeks compared with 88% at 8 weeks (P=.8).
Procedure-induced ulcers respond similarly to 4- and 8-week regimens
A 2014 RCT assessed the effect of 4 and 8 weeks of PPI treatment on healing of gastric ulcers resulting from endoscopic submucosal dissection (ESD), a procedure used to treat early gastric cancer or adenoma that leaves a large ulcer at the site.3 The study randomly assigned 84 patients to treatment with lansoprazole 30 mg/d for 4 or 8 weeks after undergoing ESD. Exclusion criteria included NSAID use or ingestion of mucosal protective agents within 4 weeks of the procedure, illness that might influence PPI effects, history of gastric surgery, and pregnancy or breastfeeding.
All patients underwent endoscopy the day after ESD and again at 8 weeks. Ulcer dimension (mm2) was determined by multiplying the longest diameter by the diameter perpendicular to the longest diameter. The ulcer reduction ratio, an assessment of healing, was determined by dividing the ulcer dimension at 8 weeks after ESD by the initial ulcer dimension.
No significant difference was observed in the 4-week and 8-week groups in terms of ulcer healing (68% vs 69%, respectively; P=.93) or the ulcer reduction ratio (0.0081 vs 0.0037, respectively; P=.15).
1. Gisbert JP, Pajares JM. Systematic review and meta-analysis: is 1-week proton pump inhibitor-based triple therapy sufficient to heal peptic ulcer? Aliment Pharmacol Ther. 2005;21:795-804.
2. Yeomans ND. New data on healing of nonsteroidal anti-inflammatory drug-associated ulcers and erosions. Omeprazole NSAID Steering Committee. Am J Med. 1998;104:56S-61S.
3. Park JH, Baek EK, Choi CH, et al. Comparison of the efficacy of 4- and 8-week lansoprazole treatment for ESD-induced gastric ulcers: a randomized, prospective, controlled study. Surg Endosc. 2014;28:235-241.
It depends on the type of ulcer. For Helicobacter pylori-associated peptic ulcers, 7-day treatment with a proton pump inhibitor (PPI) plus 2 antibiotics heals more than 90% of ulcers and is as effective as the same regimen followed by 2 to 4 additional weeks of PPI therapy (strength of recommendation [SOR]: A, meta-analysis of randomized controlled trials [RCTs]).
For peptic ulcers associated with nonsteroidal anti-inflammatory drugs (NSAIDs), 8 weeks of PPI treatment is better than 4 weeks in the case of gastric ulcers, but no more effective than 4 weeks for duodenal ulcers. (SOR: A, meta-analysis of RCTs).
For gastric ulcers resulting from endoscopic submucosal dissection, 4 weeks of PPI therapy is as effective as 8 weeks, but both regimens leave nearly a third of ulcers unhealed (SOR: B, single RCT).
For H pylori ulcers, 7 days of therapy does the trick
A 2005 meta-analysis of 6 RCTs with 862 patients compared 7 days of triple therapy with a PPI and 2 antibiotics with the same regimen followed by 2 to 4 additional weeks of PPI therapy.1 One RCT studied both duodenal and gastric ulcers; the remaining 5 assessed only duodenal ulcers. Investigators included only studies that clearly identified both H pylori eradication and ulcer healing as treatment goals and specified the number of patients treated, the number who experienced successful healing, endoscopic ulcer confirmation, and no concurrent NSAID use.
Triple therapy regimens comprised either omeprazole or esomeprazole 20 mg twice daily plus clarithromycin and either metronidazole, amoxicillin, or tinidazole for 7 days. In all studies, patients randomly assigned to receive an additional 2 to 4 weeks of PPI treatment were given omeprazole 20 mg/d.
Mean ulcer healing rates were 91% (95% confidence interval [CI], 87%-95%) for 7 days of PPI triple therapy compared with 92% (95% CI, 89%-96%) when PPI treatment was extended for an additional 2 to 4 weeks (odds ratio=1.1; 95% CI, 0.71-1.7).
Longer PPI therapy works better for NSAID-associated gastric ulcers
A 1998 meta-analysis examined 2 large RCTs that evaluated healing rates of NSAID-associated ulcers at 4 weeks and 8 weeks in 656 patients with gastric or duodenal ulcers who were treated with omeprazole 20 mg/d or 40 mg/d.2 Patients had ulcers 3 mm or larger or more than 10 erosions in the stomach or duodenum. Gastric ulcers outnumbered duodenal ulcers 2 to 1. Patients had taken continuous therapeutic doses of NSAIDs for at least 5 days per week during 2 weeks in the month preceding PPI therapy; about half were H pylori-positive.
For gastric ulcers, treatment success at 8 weeks was significantly higher at both PPI doses than at 4 weeks. The 208 patients taking the 20-mg dose showed 67% treatment success at 4 weeks and 83% at 8 weeks (P=.001). The 212 patients taking 40 mg had 67% treatment success at 4 weeks and 82% at 8 weeks (P=.002).
Duodenal ulcers showed no difference in healing at 4 and 8 weeks at either PPI dose. The 20-mg dose (116 patients) produced 84% treatment success at 4 weeks compared with 93% at 8 weeks (P=.2), and the 40-mg dose (120 patients) showed 86% treatment success at 4 weeks compared with 88% at 8 weeks (P=.8).
Procedure-induced ulcers respond similarly to 4- and 8-week regimens
A 2014 RCT assessed the effect of 4 and 8 weeks of PPI treatment on healing of gastric ulcers resulting from endoscopic submucosal dissection (ESD), a procedure used to treat early gastric cancer or adenoma that leaves a large ulcer at the site.3 The study randomly assigned 84 patients to treatment with lansoprazole 30 mg/d for 4 or 8 weeks after undergoing ESD. Exclusion criteria included NSAID use or ingestion of mucosal protective agents within 4 weeks of the procedure, illness that might influence PPI effects, history of gastric surgery, and pregnancy or breastfeeding.
All patients underwent endoscopy the day after ESD and again at 8 weeks. Ulcer dimension (mm2) was determined by multiplying the longest diameter by the diameter perpendicular to the longest diameter. The ulcer reduction ratio, an assessment of healing, was determined by dividing the ulcer dimension at 8 weeks after ESD by the initial ulcer dimension.
No significant difference was observed in the 4-week and 8-week groups in terms of ulcer healing (68% vs 69%, respectively; P=.93) or the ulcer reduction ratio (0.0081 vs 0.0037, respectively; P=.15).
It depends on the type of ulcer. For Helicobacter pylori-associated peptic ulcers, 7-day treatment with a proton pump inhibitor (PPI) plus 2 antibiotics heals more than 90% of ulcers and is as effective as the same regimen followed by 2 to 4 additional weeks of PPI therapy (strength of recommendation [SOR]: A, meta-analysis of randomized controlled trials [RCTs]).
For peptic ulcers associated with nonsteroidal anti-inflammatory drugs (NSAIDs), 8 weeks of PPI treatment is better than 4 weeks in the case of gastric ulcers, but no more effective than 4 weeks for duodenal ulcers. (SOR: A, meta-analysis of RCTs).
For gastric ulcers resulting from endoscopic submucosal dissection, 4 weeks of PPI therapy is as effective as 8 weeks, but both regimens leave nearly a third of ulcers unhealed (SOR: B, single RCT).
For H pylori ulcers, 7 days of therapy does the trick
A 2005 meta-analysis of 6 RCTs with 862 patients compared 7 days of triple therapy with a PPI and 2 antibiotics with the same regimen followed by 2 to 4 additional weeks of PPI therapy.1 One RCT studied both duodenal and gastric ulcers; the remaining 5 assessed only duodenal ulcers. Investigators included only studies that clearly identified both H pylori eradication and ulcer healing as treatment goals and specified the number of patients treated, the number who experienced successful healing, endoscopic ulcer confirmation, and no concurrent NSAID use.
Triple therapy regimens comprised either omeprazole or esomeprazole 20 mg twice daily plus clarithromycin and either metronidazole, amoxicillin, or tinidazole for 7 days. In all studies, patients randomly assigned to receive an additional 2 to 4 weeks of PPI treatment were given omeprazole 20 mg/d.
Mean ulcer healing rates were 91% (95% confidence interval [CI], 87%-95%) for 7 days of PPI triple therapy compared with 92% (95% CI, 89%-96%) when PPI treatment was extended for an additional 2 to 4 weeks (odds ratio=1.1; 95% CI, 0.71-1.7).
Longer PPI therapy works better for NSAID-associated gastric ulcers
A 1998 meta-analysis examined 2 large RCTs that evaluated healing rates of NSAID-associated ulcers at 4 weeks and 8 weeks in 656 patients with gastric or duodenal ulcers who were treated with omeprazole 20 mg/d or 40 mg/d.2 Patients had ulcers 3 mm or larger or more than 10 erosions in the stomach or duodenum. Gastric ulcers outnumbered duodenal ulcers 2 to 1. Patients had taken continuous therapeutic doses of NSAIDs for at least 5 days per week during 2 weeks in the month preceding PPI therapy; about half were H pylori-positive.
For gastric ulcers, treatment success at 8 weeks was significantly higher at both PPI doses than at 4 weeks. The 208 patients taking the 20-mg dose showed 67% treatment success at 4 weeks and 83% at 8 weeks (P=.001). The 212 patients taking 40 mg had 67% treatment success at 4 weeks and 82% at 8 weeks (P=.002).
Duodenal ulcers showed no difference in healing at 4 and 8 weeks at either PPI dose. The 20-mg dose (116 patients) produced 84% treatment success at 4 weeks compared with 93% at 8 weeks (P=.2), and the 40-mg dose (120 patients) showed 86% treatment success at 4 weeks compared with 88% at 8 weeks (P=.8).
Procedure-induced ulcers respond similarly to 4- and 8-week regimens
A 2014 RCT assessed the effect of 4 and 8 weeks of PPI treatment on healing of gastric ulcers resulting from endoscopic submucosal dissection (ESD), a procedure used to treat early gastric cancer or adenoma that leaves a large ulcer at the site.3 The study randomly assigned 84 patients to treatment with lansoprazole 30 mg/d for 4 or 8 weeks after undergoing ESD. Exclusion criteria included NSAID use or ingestion of mucosal protective agents within 4 weeks of the procedure, illness that might influence PPI effects, history of gastric surgery, and pregnancy or breastfeeding.
All patients underwent endoscopy the day after ESD and again at 8 weeks. Ulcer dimension (mm2) was determined by multiplying the longest diameter by the diameter perpendicular to the longest diameter. The ulcer reduction ratio, an assessment of healing, was determined by dividing the ulcer dimension at 8 weeks after ESD by the initial ulcer dimension.
No significant difference was observed in the 4-week and 8-week groups in terms of ulcer healing (68% vs 69%, respectively; P=.93) or the ulcer reduction ratio (0.0081 vs 0.0037, respectively; P=.15).
1. Gisbert JP, Pajares JM. Systematic review and meta-analysis: is 1-week proton pump inhibitor-based triple therapy sufficient to heal peptic ulcer? Aliment Pharmacol Ther. 2005;21:795-804.
2. Yeomans ND. New data on healing of nonsteroidal anti-inflammatory drug-associated ulcers and erosions. Omeprazole NSAID Steering Committee. Am J Med. 1998;104:56S-61S.
3. Park JH, Baek EK, Choi CH, et al. Comparison of the efficacy of 4- and 8-week lansoprazole treatment for ESD-induced gastric ulcers: a randomized, prospective, controlled study. Surg Endosc. 2014;28:235-241.
1. Gisbert JP, Pajares JM. Systematic review and meta-analysis: is 1-week proton pump inhibitor-based triple therapy sufficient to heal peptic ulcer? Aliment Pharmacol Ther. 2005;21:795-804.
2. Yeomans ND. New data on healing of nonsteroidal anti-inflammatory drug-associated ulcers and erosions. Omeprazole NSAID Steering Committee. Am J Med. 1998;104:56S-61S.
3. Park JH, Baek EK, Choi CH, et al. Comparison of the efficacy of 4- and 8-week lansoprazole treatment for ESD-induced gastric ulcers: a randomized, prospective, controlled study. Surg Endosc. 2014;28:235-241.
Evidence-based answers from the Family Physicians Inquiries Network
Is prazosin effective for PTSD-associated nightmares?
Yes. Prazosin has been shown to reduce both frequency and severity of nightmares in patients who meet diagnostic criteria for post-traumatic stress disorder (PTSD) (strength of recommendation: A, systematic review of randomized, controlled trials [RCTs]).
Patients who meet PTSD criteria show best response
A 2012 systematic review of prazosin (1-16 mg) for PTSD included 21 studies (4 RCTs, 4 open-label case series, 4 retrospective case series, and 9 case reports) with 285 patients, 85% of whom were combat veterans.1 All the studies were limited by small sample sizes and a lack of demographic diversity.
To measure prazosin’s effect on nightmares, the studies used the Clinician-Administered PTSD Scale (CAPS-B2), scored from 0 to 8, which sums the frequency of nightmares (0=none in the past week, 4=daily nightmares) and the intensity of distressing dreams (0=none, 4=incapacitating distress).
The 3 highest-quality RCTs used similar methods and included only 63 patients who met diagnostic criteria for PTSD. Each found statistically significant reductions in nightmares among patients taking prazosin compared with placebo (CAPS-B2 improvements of 3.3, 3.3, and 1.5 for prazosin vs 0.4, 0.9, and 0 for placebo; P<.05 for all comparisons).
In the fourth RCT, comprised of 50 patients, only 58% of participants met full clinical diagnostic criteria for PTSD. The primary outcome was the number of recalled nightmares, which didn’t show a statistically significant decrease in the prazosin group compared with placebo (decrease in mean weekly nightmares of 0.7 with prazosin vs an increase of 0.1 with placebo).
Prazosin provides significant relief in small study of combat veterans
A 2013 RCT evaluated the effect of prazosin on nightmares in 67 soldiers with combat PTSD.2 All patients met criteria for PTSD as outlined in the Diagnostic and Statistical Manual of Mental Disorders, 4th edition. Men received doses titrated to a mean of 4 mg in the morning and 15.6 mg at bedtime; women received a mean of 1.7 mg in the morning and 7 mg at bedtime.
After 15 weeks, the CAPS-B2 score decreased by 3.1 for prazosin compared with 1.2 for placebo (P<.05).
1. Kung S, Espinel Z, Lapid M. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87:890-900.
2. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170:1003-1010.
Yes. Prazosin has been shown to reduce both frequency and severity of nightmares in patients who meet diagnostic criteria for post-traumatic stress disorder (PTSD) (strength of recommendation: A, systematic review of randomized, controlled trials [RCTs]).
Patients who meet PTSD criteria show best response
A 2012 systematic review of prazosin (1-16 mg) for PTSD included 21 studies (4 RCTs, 4 open-label case series, 4 retrospective case series, and 9 case reports) with 285 patients, 85% of whom were combat veterans.1 All the studies were limited by small sample sizes and a lack of demographic diversity.
To measure prazosin’s effect on nightmares, the studies used the Clinician-Administered PTSD Scale (CAPS-B2), scored from 0 to 8, which sums the frequency of nightmares (0=none in the past week, 4=daily nightmares) and the intensity of distressing dreams (0=none, 4=incapacitating distress).
The 3 highest-quality RCTs used similar methods and included only 63 patients who met diagnostic criteria for PTSD. Each found statistically significant reductions in nightmares among patients taking prazosin compared with placebo (CAPS-B2 improvements of 3.3, 3.3, and 1.5 for prazosin vs 0.4, 0.9, and 0 for placebo; P<.05 for all comparisons).
In the fourth RCT, comprised of 50 patients, only 58% of participants met full clinical diagnostic criteria for PTSD. The primary outcome was the number of recalled nightmares, which didn’t show a statistically significant decrease in the prazosin group compared with placebo (decrease in mean weekly nightmares of 0.7 with prazosin vs an increase of 0.1 with placebo).
Prazosin provides significant relief in small study of combat veterans
A 2013 RCT evaluated the effect of prazosin on nightmares in 67 soldiers with combat PTSD.2 All patients met criteria for PTSD as outlined in the Diagnostic and Statistical Manual of Mental Disorders, 4th edition. Men received doses titrated to a mean of 4 mg in the morning and 15.6 mg at bedtime; women received a mean of 1.7 mg in the morning and 7 mg at bedtime.
After 15 weeks, the CAPS-B2 score decreased by 3.1 for prazosin compared with 1.2 for placebo (P<.05).
Yes. Prazosin has been shown to reduce both frequency and severity of nightmares in patients who meet diagnostic criteria for post-traumatic stress disorder (PTSD) (strength of recommendation: A, systematic review of randomized, controlled trials [RCTs]).
Patients who meet PTSD criteria show best response
A 2012 systematic review of prazosin (1-16 mg) for PTSD included 21 studies (4 RCTs, 4 open-label case series, 4 retrospective case series, and 9 case reports) with 285 patients, 85% of whom were combat veterans.1 All the studies were limited by small sample sizes and a lack of demographic diversity.
To measure prazosin’s effect on nightmares, the studies used the Clinician-Administered PTSD Scale (CAPS-B2), scored from 0 to 8, which sums the frequency of nightmares (0=none in the past week, 4=daily nightmares) and the intensity of distressing dreams (0=none, 4=incapacitating distress).
The 3 highest-quality RCTs used similar methods and included only 63 patients who met diagnostic criteria for PTSD. Each found statistically significant reductions in nightmares among patients taking prazosin compared with placebo (CAPS-B2 improvements of 3.3, 3.3, and 1.5 for prazosin vs 0.4, 0.9, and 0 for placebo; P<.05 for all comparisons).
In the fourth RCT, comprised of 50 patients, only 58% of participants met full clinical diagnostic criteria for PTSD. The primary outcome was the number of recalled nightmares, which didn’t show a statistically significant decrease in the prazosin group compared with placebo (decrease in mean weekly nightmares of 0.7 with prazosin vs an increase of 0.1 with placebo).
Prazosin provides significant relief in small study of combat veterans
A 2013 RCT evaluated the effect of prazosin on nightmares in 67 soldiers with combat PTSD.2 All patients met criteria for PTSD as outlined in the Diagnostic and Statistical Manual of Mental Disorders, 4th edition. Men received doses titrated to a mean of 4 mg in the morning and 15.6 mg at bedtime; women received a mean of 1.7 mg in the morning and 7 mg at bedtime.
After 15 weeks, the CAPS-B2 score decreased by 3.1 for prazosin compared with 1.2 for placebo (P<.05).
1. Kung S, Espinel Z, Lapid M. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87:890-900.
2. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170:1003-1010.
1. Kung S, Espinel Z, Lapid M. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87:890-900.
2. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170:1003-1010.
Evidence-based answers from the Family Physicians Inquiries Network
Which is better for IBS pain in women—antispasmodics or antidepressants?
It’s unclear which therapy is more effective because the evidence is insufficient. What is known is that tricyclic antidepressants, peppermint oil, and antispasmodics all have been shown superior to placebo for treating abdominal pain in female patients with irritable bowel syndrome (IBS) (strength of recommendation: A, meta-analyses).
Antispasmodics and tricyclics alleviate abdominal pain
A 2011 Cochrane review of 56 randomized controlled trials (RCTs) with 3725 patients compared bulking agents, antispasmodics, or antidepressants with placebo for treating IBS.1 The pooled results from 13 RCTs with 1392 patients (65% female, mean age 45 years) showed that more patients had improved abdominal pain with antispasmodics than placebo over treatment periods varying from 6 days to 6 months (58% vs 46%; relative risk [RR]=1.3; 95% confidence interval [CI], 1.1-1.6; number needed to treat [NNT]=7).
The clinical relevance of the antispasmodic data is limited because the antispasmodics found effective for abdominal pain aren’t available in the United States. The pooled results from 8 RCTs with 517 patients (72% female, mean age 40) demonstrated greater improvement of abdominal pain with tricyclic and selective serotonin reuptake inhibitor antidepressants than placebo over 6 to 12 weeks (54% vs 37%; RR=1.5; 95% CI, 1.1–2.1; NNT=5). However, subgroup analysis found a statistically significant benefit for tricyclic antidepressants (4 trials; N=320; RR=1.3; 95% CI, 1.0-1.6) but no benefit for SSRIs (4 trials; N=197; RR=2.3; 95% CI, 0.79-6.7).
Effective antispasmodics aren’t available in the United States
A 2012 meta-analysis of 23 RCTs with 2585 patients examined the effect of antispasmodic agents, alone or in combination, to treat IBS.2 Pooled results from 13 RCTs with 2394 patients (69% female, ages 16 years or older) favored treatment with antispasmodics over placebo for abdominal pain (odds ratio [OR]=1.5; 95% CI, 1.3-1.8). No difference in adverse events was found between antispasmodics and placebo (9 trials; N=2239; OR=0.74; 95% CI, 0.54-0.98). The antispasmodics found effective for abdominal pain in this meta-analysis aren’t available in the United States.
Peppermint oil helps, but can cause heartburn
A 2013 meta-analysis of 9 RCTs with 726 patients compared various doses of enteric-coated peppermint oil with placebo over a minimum of 2 weeks’ treatment.3 Five RCTs with 357 patients (62% female, 13.4% children) demonstrated improvement of abdominal pain in 57% of patients taking peppermint oil compared with 27% receiving placebo (RR=2.1; 95% CI, 1.6-2.8; NNT=4 at 2 to 8 weeks). No statistically significant heterogeneity was identified among the treatment groups.
Pooled analysis found that peppermint oil patients were more likely than placebo patients to experience an adverse event (7 trials; N=474; 22% vs 13%; RR=1.7; 95% CI, 1.3-2.4), but that the events were generally mild and transient. The most frequently reported adverse event was heartburn.
1. Ruepert L, Quartero AO, de Wit NJ, et al. Bulking agents, antispasmodics, and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.
2. Martinez-Vasquez MA, Vasquez-Elizondro G, Gonzalez-Gonzalez JA, et al. Effect of antispasmodic agents, alone or in combination, in the treatment of irritable bowel syndrome: systematic review and meta-analysis. Rev Gastroenterol Mexico. 2012;77:82-90.
3. Khanna R, MacDonald JK, Levesque BG. Peppermint oil for the treatment of irritable bowel syndrome: a systematic review and meta-analysis. J Clin Gastroenterol. 2014;48:505-512.
It’s unclear which therapy is more effective because the evidence is insufficient. What is known is that tricyclic antidepressants, peppermint oil, and antispasmodics all have been shown superior to placebo for treating abdominal pain in female patients with irritable bowel syndrome (IBS) (strength of recommendation: A, meta-analyses).
Antispasmodics and tricyclics alleviate abdominal pain
A 2011 Cochrane review of 56 randomized controlled trials (RCTs) with 3725 patients compared bulking agents, antispasmodics, or antidepressants with placebo for treating IBS.1 The pooled results from 13 RCTs with 1392 patients (65% female, mean age 45 years) showed that more patients had improved abdominal pain with antispasmodics than placebo over treatment periods varying from 6 days to 6 months (58% vs 46%; relative risk [RR]=1.3; 95% confidence interval [CI], 1.1-1.6; number needed to treat [NNT]=7).
The clinical relevance of the antispasmodic data is limited because the antispasmodics found effective for abdominal pain aren’t available in the United States. The pooled results from 8 RCTs with 517 patients (72% female, mean age 40) demonstrated greater improvement of abdominal pain with tricyclic and selective serotonin reuptake inhibitor antidepressants than placebo over 6 to 12 weeks (54% vs 37%; RR=1.5; 95% CI, 1.1–2.1; NNT=5). However, subgroup analysis found a statistically significant benefit for tricyclic antidepressants (4 trials; N=320; RR=1.3; 95% CI, 1.0-1.6) but no benefit for SSRIs (4 trials; N=197; RR=2.3; 95% CI, 0.79-6.7).
Effective antispasmodics aren’t available in the United States
A 2012 meta-analysis of 23 RCTs with 2585 patients examined the effect of antispasmodic agents, alone or in combination, to treat IBS.2 Pooled results from 13 RCTs with 2394 patients (69% female, ages 16 years or older) favored treatment with antispasmodics over placebo for abdominal pain (odds ratio [OR]=1.5; 95% CI, 1.3-1.8). No difference in adverse events was found between antispasmodics and placebo (9 trials; N=2239; OR=0.74; 95% CI, 0.54-0.98). The antispasmodics found effective for abdominal pain in this meta-analysis aren’t available in the United States.
Peppermint oil helps, but can cause heartburn
A 2013 meta-analysis of 9 RCTs with 726 patients compared various doses of enteric-coated peppermint oil with placebo over a minimum of 2 weeks’ treatment.3 Five RCTs with 357 patients (62% female, 13.4% children) demonstrated improvement of abdominal pain in 57% of patients taking peppermint oil compared with 27% receiving placebo (RR=2.1; 95% CI, 1.6-2.8; NNT=4 at 2 to 8 weeks). No statistically significant heterogeneity was identified among the treatment groups.
Pooled analysis found that peppermint oil patients were more likely than placebo patients to experience an adverse event (7 trials; N=474; 22% vs 13%; RR=1.7; 95% CI, 1.3-2.4), but that the events were generally mild and transient. The most frequently reported adverse event was heartburn.
It’s unclear which therapy is more effective because the evidence is insufficient. What is known is that tricyclic antidepressants, peppermint oil, and antispasmodics all have been shown superior to placebo for treating abdominal pain in female patients with irritable bowel syndrome (IBS) (strength of recommendation: A, meta-analyses).
Antispasmodics and tricyclics alleviate abdominal pain
A 2011 Cochrane review of 56 randomized controlled trials (RCTs) with 3725 patients compared bulking agents, antispasmodics, or antidepressants with placebo for treating IBS.1 The pooled results from 13 RCTs with 1392 patients (65% female, mean age 45 years) showed that more patients had improved abdominal pain with antispasmodics than placebo over treatment periods varying from 6 days to 6 months (58% vs 46%; relative risk [RR]=1.3; 95% confidence interval [CI], 1.1-1.6; number needed to treat [NNT]=7).
The clinical relevance of the antispasmodic data is limited because the antispasmodics found effective for abdominal pain aren’t available in the United States. The pooled results from 8 RCTs with 517 patients (72% female, mean age 40) demonstrated greater improvement of abdominal pain with tricyclic and selective serotonin reuptake inhibitor antidepressants than placebo over 6 to 12 weeks (54% vs 37%; RR=1.5; 95% CI, 1.1–2.1; NNT=5). However, subgroup analysis found a statistically significant benefit for tricyclic antidepressants (4 trials; N=320; RR=1.3; 95% CI, 1.0-1.6) but no benefit for SSRIs (4 trials; N=197; RR=2.3; 95% CI, 0.79-6.7).
Effective antispasmodics aren’t available in the United States
A 2012 meta-analysis of 23 RCTs with 2585 patients examined the effect of antispasmodic agents, alone or in combination, to treat IBS.2 Pooled results from 13 RCTs with 2394 patients (69% female, ages 16 years or older) favored treatment with antispasmodics over placebo for abdominal pain (odds ratio [OR]=1.5; 95% CI, 1.3-1.8). No difference in adverse events was found between antispasmodics and placebo (9 trials; N=2239; OR=0.74; 95% CI, 0.54-0.98). The antispasmodics found effective for abdominal pain in this meta-analysis aren’t available in the United States.
Peppermint oil helps, but can cause heartburn
A 2013 meta-analysis of 9 RCTs with 726 patients compared various doses of enteric-coated peppermint oil with placebo over a minimum of 2 weeks’ treatment.3 Five RCTs with 357 patients (62% female, 13.4% children) demonstrated improvement of abdominal pain in 57% of patients taking peppermint oil compared with 27% receiving placebo (RR=2.1; 95% CI, 1.6-2.8; NNT=4 at 2 to 8 weeks). No statistically significant heterogeneity was identified among the treatment groups.
Pooled analysis found that peppermint oil patients were more likely than placebo patients to experience an adverse event (7 trials; N=474; 22% vs 13%; RR=1.7; 95% CI, 1.3-2.4), but that the events were generally mild and transient. The most frequently reported adverse event was heartburn.
1. Ruepert L, Quartero AO, de Wit NJ, et al. Bulking agents, antispasmodics, and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.
2. Martinez-Vasquez MA, Vasquez-Elizondro G, Gonzalez-Gonzalez JA, et al. Effect of antispasmodic agents, alone or in combination, in the treatment of irritable bowel syndrome: systematic review and meta-analysis. Rev Gastroenterol Mexico. 2012;77:82-90.
3. Khanna R, MacDonald JK, Levesque BG. Peppermint oil for the treatment of irritable bowel syndrome: a systematic review and meta-analysis. J Clin Gastroenterol. 2014;48:505-512.
1. Ruepert L, Quartero AO, de Wit NJ, et al. Bulking agents, antispasmodics, and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460.
2. Martinez-Vasquez MA, Vasquez-Elizondro G, Gonzalez-Gonzalez JA, et al. Effect of antispasmodic agents, alone or in combination, in the treatment of irritable bowel syndrome: systematic review and meta-analysis. Rev Gastroenterol Mexico. 2012;77:82-90.
3. Khanna R, MacDonald JK, Levesque BG. Peppermint oil for the treatment of irritable bowel syndrome: a systematic review and meta-analysis. J Clin Gastroenterol. 2014;48:505-512.
Evidence-based answers from the Family Physicians Inquiries Network
Are overweight children likely to become overweight adults?
Yes. Overweight children (body mass index [BMI] ≥85th to <95th percentile) are likely to become overweight or obese adults with a BMI ≥25 (strength of recommendation [SOR]: B, systematic review of high-quality prospective longitudinal studies).
Obese adolescents are significantly more likely to develop severe obesity than normal weight or overweight adolescents (SOR: B, prospective cohort study). (See “Definition of terms” below.)
The trend to overweight and obesity in adulthood is clear
A systematic review of 20 prospective and 5 retrospective trials tracked 179,303 overweight and obese children into adulthood.1 Investigators included studies for evaluation if they were written in English, prospective or retrospective longitudinal in design, described at least one anthropometric measurement, and included odds ratios or risk ratios in the results. The results were not pooled because of heterogeneity among studies.
In high-quality trials, the percentages of overweight or obese children and adolescents who became overweight or obese adults varied: overweight children (76% to 83%), obese children (18% to 60%), overweight adolescents (22% to 58%) and obese adolescents (24% to 90%). Limitations of the review included an inadequate description of the anthropometric measurement protocol, use of self-reported weight and height, and the fact that all studies were conducted in high-income countries.
Obesity in adolescence often progresses to severe obesity later on
A prospective cohort trial followed 8834 nonobese and obese individuals, ages 12 to 21, for 13 years to assess risk of adult obesity.2 Patients were drawn from the National Longitudinal Study of Adolescent Health, which is a representative sample of United States schools from 1994 to 1995 with respect to region, urbanicity, school size, school type, and ethnicity.
Researchers observed a total of 703 incident cases of severe obesity in adulthood, indicating a total incidence rate of 7.9% (95% confidence interval [CI], 7.4%-8.5%). Obese adolescents were significantly more likely to develop severe obesity than nonobese adolescents who were normal weight or overweight (hazard ratio [HR]=16; 95% CI, 12-21).
A significant proportion of obese adolescents became severely obese by their early 30s, with an incidence of 37% in men (95% CI, 31%-44%) and 51% in women (95% CI, 45%-58%). Black women had the highest incidence at 52% (95% CI, 41%-64%). Fewer than 5% of patients (across sex and race) who were normal weight in adolescence became severely obese in adulthood.
Normal weight: Body mass index (BMI) ≥5th to <85th percentile for individuals <20 years old or BMI ≥18.5 to <25 for individuals >20 years.
Overweight: BMI ≥85th to <95th percentile or BMI ≥25 to <30.
Obesity: BMI ≥95th to <120% of 95th percentile or BMI ≥30 to <40.
Severe obesity: BMI ≥120% of 95th percentile; BMI ≥40.
1. Singh AS, Mulder C, Twisk WR, et al. Tracking of childhood overweight into adulthood: a systemic review of the literature. Obes Rev. 2008;9:474-488.
2. The NS, Suchindran C, North KE, et al. Association of adolescent obesity with risk of severe obesity in adulthood. JAMA. 2010;304:2042-2047.
Yes. Overweight children (body mass index [BMI] ≥85th to <95th percentile) are likely to become overweight or obese adults with a BMI ≥25 (strength of recommendation [SOR]: B, systematic review of high-quality prospective longitudinal studies).
Obese adolescents are significantly more likely to develop severe obesity than normal weight or overweight adolescents (SOR: B, prospective cohort study). (See “Definition of terms” below.)
The trend to overweight and obesity in adulthood is clear
A systematic review of 20 prospective and 5 retrospective trials tracked 179,303 overweight and obese children into adulthood.1 Investigators included studies for evaluation if they were written in English, prospective or retrospective longitudinal in design, described at least one anthropometric measurement, and included odds ratios or risk ratios in the results. The results were not pooled because of heterogeneity among studies.
In high-quality trials, the percentages of overweight or obese children and adolescents who became overweight or obese adults varied: overweight children (76% to 83%), obese children (18% to 60%), overweight adolescents (22% to 58%) and obese adolescents (24% to 90%). Limitations of the review included an inadequate description of the anthropometric measurement protocol, use of self-reported weight and height, and the fact that all studies were conducted in high-income countries.
Obesity in adolescence often progresses to severe obesity later on
A prospective cohort trial followed 8834 nonobese and obese individuals, ages 12 to 21, for 13 years to assess risk of adult obesity.2 Patients were drawn from the National Longitudinal Study of Adolescent Health, which is a representative sample of United States schools from 1994 to 1995 with respect to region, urbanicity, school size, school type, and ethnicity.
Researchers observed a total of 703 incident cases of severe obesity in adulthood, indicating a total incidence rate of 7.9% (95% confidence interval [CI], 7.4%-8.5%). Obese adolescents were significantly more likely to develop severe obesity than nonobese adolescents who were normal weight or overweight (hazard ratio [HR]=16; 95% CI, 12-21).
A significant proportion of obese adolescents became severely obese by their early 30s, with an incidence of 37% in men (95% CI, 31%-44%) and 51% in women (95% CI, 45%-58%). Black women had the highest incidence at 52% (95% CI, 41%-64%). Fewer than 5% of patients (across sex and race) who were normal weight in adolescence became severely obese in adulthood.
Normal weight: Body mass index (BMI) ≥5th to <85th percentile for individuals <20 years old or BMI ≥18.5 to <25 for individuals >20 years.
Overweight: BMI ≥85th to <95th percentile or BMI ≥25 to <30.
Obesity: BMI ≥95th to <120% of 95th percentile or BMI ≥30 to <40.
Severe obesity: BMI ≥120% of 95th percentile; BMI ≥40.
Yes. Overweight children (body mass index [BMI] ≥85th to <95th percentile) are likely to become overweight or obese adults with a BMI ≥25 (strength of recommendation [SOR]: B, systematic review of high-quality prospective longitudinal studies).
Obese adolescents are significantly more likely to develop severe obesity than normal weight or overweight adolescents (SOR: B, prospective cohort study). (See “Definition of terms” below.)
The trend to overweight and obesity in adulthood is clear
A systematic review of 20 prospective and 5 retrospective trials tracked 179,303 overweight and obese children into adulthood.1 Investigators included studies for evaluation if they were written in English, prospective or retrospective longitudinal in design, described at least one anthropometric measurement, and included odds ratios or risk ratios in the results. The results were not pooled because of heterogeneity among studies.
In high-quality trials, the percentages of overweight or obese children and adolescents who became overweight or obese adults varied: overweight children (76% to 83%), obese children (18% to 60%), overweight adolescents (22% to 58%) and obese adolescents (24% to 90%). Limitations of the review included an inadequate description of the anthropometric measurement protocol, use of self-reported weight and height, and the fact that all studies were conducted in high-income countries.
Obesity in adolescence often progresses to severe obesity later on
A prospective cohort trial followed 8834 nonobese and obese individuals, ages 12 to 21, for 13 years to assess risk of adult obesity.2 Patients were drawn from the National Longitudinal Study of Adolescent Health, which is a representative sample of United States schools from 1994 to 1995 with respect to region, urbanicity, school size, school type, and ethnicity.
Researchers observed a total of 703 incident cases of severe obesity in adulthood, indicating a total incidence rate of 7.9% (95% confidence interval [CI], 7.4%-8.5%). Obese adolescents were significantly more likely to develop severe obesity than nonobese adolescents who were normal weight or overweight (hazard ratio [HR]=16; 95% CI, 12-21).
A significant proportion of obese adolescents became severely obese by their early 30s, with an incidence of 37% in men (95% CI, 31%-44%) and 51% in women (95% CI, 45%-58%). Black women had the highest incidence at 52% (95% CI, 41%-64%). Fewer than 5% of patients (across sex and race) who were normal weight in adolescence became severely obese in adulthood.
Normal weight: Body mass index (BMI) ≥5th to <85th percentile for individuals <20 years old or BMI ≥18.5 to <25 for individuals >20 years.
Overweight: BMI ≥85th to <95th percentile or BMI ≥25 to <30.
Obesity: BMI ≥95th to <120% of 95th percentile or BMI ≥30 to <40.
Severe obesity: BMI ≥120% of 95th percentile; BMI ≥40.
1. Singh AS, Mulder C, Twisk WR, et al. Tracking of childhood overweight into adulthood: a systemic review of the literature. Obes Rev. 2008;9:474-488.
2. The NS, Suchindran C, North KE, et al. Association of adolescent obesity with risk of severe obesity in adulthood. JAMA. 2010;304:2042-2047.
1. Singh AS, Mulder C, Twisk WR, et al. Tracking of childhood overweight into adulthood: a systemic review of the literature. Obes Rev. 2008;9:474-488.
2. The NS, Suchindran C, North KE, et al. Association of adolescent obesity with risk of severe obesity in adulthood. JAMA. 2010;304:2042-2047.
Evidence-based answers from the Family Physicians Inquiries Network
What treatments best prevent chronic tension headaches?
Biofeedback and tricyclic antidepressants appear to be effective as prophylactic treatment for chronic tension headaches, although tricyclic antidepressants have more adverse effects (strength of recommendation [SOR]: B, meta-analysis of randomized controlled trials [RCTs] and pre-post trials).
Acupuncture shows limited evidence for effectiveness after 20 to 25 weeks of treatment (SOR: B, meta-analysis of RCTs).
Biofeedback gets results without adverse effects
A 2008 meta-analysis of 53 trials (32 RCTs, 21 pre-post; 1532 patients, 72% female, average age 36 years) examined the effectiveness of biofeedback for tension-type headaches (TTH) with a mean duration of headache symptoms of 14 years.1 The mean duration of treatment was fewer than 10 hours over 11 sessions. Control groups included placebo, relaxation, pharmacotherapy, cognitive therapy, and physical therapy.
Biofeedback reduced headache pain (measured in structured headache diaries by frequency, duration, and intensity) more than controls (weighted mean difference [WMD]=0.73; 95% confidence interval [CI], 0.61-0.84; WMD effect sizes >0.8 are considered large, 0.6-0.8 are moderate, and 0.2-0.6 are small).
Eighteen trials (15 RCTs, 3 pre-post; 736 patients) included follow-up analysis ranging from 3 to 60 months (mean 15 months), during which biofeedback showed a moderate effect size compared with controls (WMD=0.62; 95% CI, 0.53-0.72). Biofeedback plus relaxation produced a larger effect than controls (6 RCTs, 3 pre-post; 124 patients; WMD=0.98; 95% CI, 0.69-1.3). No adverse effects were reported.
Tricyclics help, too, but have adverse effects
A 2010 meta-analysis of 37 RCTs examined the effectiveness of tricyclic antidepressants as prophylactic treatment for headaches in 3176 patients.2 Seventeen RCTs (1275 patients, 73% women, average age 40 years) evaluated tension headaches, and 15 of them included only patients who had headaches for more than 14 days per month (the other 20 RCTs, with 1901 patients, studied migraine headaches).
Control groups included placebo, selective serotonin reuptake inhibitors (SSRIs), tetracyclics, topiramate, dihydroergotamine, spinal manipulation, and cognitive behavioral therapy, but only placebo (8 RCTs included) and SSRIs (4 RCTs included) had more than 2 RCTs evaluated. Variables included headache days per month, headache intensity (measured by scales that varied among trials), and headache index (calculated by multiplying intensity by frequency).
In 8 trials with 574 patients, the standard mean difference (SMD) headache index improved for tricyclics compared with placebo over a treatment duration averaging 10 weeks (WMD=−0.99; 95% CI, −1.7 to −0.32). However, the tricyclic arm reported more adverse effects such as drowsiness, dizziness, dry mouth, and abdominal complaints (relative risk [RR]=1.9; 95% CI, 1.2-3.0).
Tricyclics and SSRIs reduced the frequency of TTH equally (4 trials; N=479; standardized mean difference=−0.80; 95% CI, −1.63 to 0.02) but tricyclics were more likely to reduce intensity by 50% (RR=1.7; 95% CI, 1.3-2.2).
Headaches decline after acupuncture, not during
A 2008 meta-analysis of 5 RCTs (838 patients, 68% female) evaluated the effectiveness of acupuncture compared with sham acupuncture for treating episodic and chronic TTH.3 Separate data on efficacy for each subtype was not provided. Selection of acupuncture points varied among the trials, and treatment durations ranged from 3 to 8 weeks.
Headache days per month didn’t decline during treatment (WMD=−2.9; 95% CI, −7.5 to 1.6), but were significantly decreased on follow-up at 20 to 25 weeks (WMD=−1.8; 95% CI, −3.0 to −0.64).
1. Netoriuc Y, Rief W, Martin A. Meta-analysis of biofeedback for tension-type headache: efficacy, specificity and treatment moderators. J Consult Clin Psychol. 2008;76:379-396.
2. Jackson JL, Shimeall W, Sessums L, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222.
3. Davis MA, Kononowech RW, Rolin SA, et al. Acupuncture for tension-type headache: a meta-analysis of randomized, controlled trials. J Pain. 2008;9:667-677.
Biofeedback and tricyclic antidepressants appear to be effective as prophylactic treatment for chronic tension headaches, although tricyclic antidepressants have more adverse effects (strength of recommendation [SOR]: B, meta-analysis of randomized controlled trials [RCTs] and pre-post trials).
Acupuncture shows limited evidence for effectiveness after 20 to 25 weeks of treatment (SOR: B, meta-analysis of RCTs).
Biofeedback gets results without adverse effects
A 2008 meta-analysis of 53 trials (32 RCTs, 21 pre-post; 1532 patients, 72% female, average age 36 years) examined the effectiveness of biofeedback for tension-type headaches (TTH) with a mean duration of headache symptoms of 14 years.1 The mean duration of treatment was fewer than 10 hours over 11 sessions. Control groups included placebo, relaxation, pharmacotherapy, cognitive therapy, and physical therapy.
Biofeedback reduced headache pain (measured in structured headache diaries by frequency, duration, and intensity) more than controls (weighted mean difference [WMD]=0.73; 95% confidence interval [CI], 0.61-0.84; WMD effect sizes >0.8 are considered large, 0.6-0.8 are moderate, and 0.2-0.6 are small).
Eighteen trials (15 RCTs, 3 pre-post; 736 patients) included follow-up analysis ranging from 3 to 60 months (mean 15 months), during which biofeedback showed a moderate effect size compared with controls (WMD=0.62; 95% CI, 0.53-0.72). Biofeedback plus relaxation produced a larger effect than controls (6 RCTs, 3 pre-post; 124 patients; WMD=0.98; 95% CI, 0.69-1.3). No adverse effects were reported.
Tricyclics help, too, but have adverse effects
A 2010 meta-analysis of 37 RCTs examined the effectiveness of tricyclic antidepressants as prophylactic treatment for headaches in 3176 patients.2 Seventeen RCTs (1275 patients, 73% women, average age 40 years) evaluated tension headaches, and 15 of them included only patients who had headaches for more than 14 days per month (the other 20 RCTs, with 1901 patients, studied migraine headaches).
Control groups included placebo, selective serotonin reuptake inhibitors (SSRIs), tetracyclics, topiramate, dihydroergotamine, spinal manipulation, and cognitive behavioral therapy, but only placebo (8 RCTs included) and SSRIs (4 RCTs included) had more than 2 RCTs evaluated. Variables included headache days per month, headache intensity (measured by scales that varied among trials), and headache index (calculated by multiplying intensity by frequency).
In 8 trials with 574 patients, the standard mean difference (SMD) headache index improved for tricyclics compared with placebo over a treatment duration averaging 10 weeks (WMD=−0.99; 95% CI, −1.7 to −0.32). However, the tricyclic arm reported more adverse effects such as drowsiness, dizziness, dry mouth, and abdominal complaints (relative risk [RR]=1.9; 95% CI, 1.2-3.0).
Tricyclics and SSRIs reduced the frequency of TTH equally (4 trials; N=479; standardized mean difference=−0.80; 95% CI, −1.63 to 0.02) but tricyclics were more likely to reduce intensity by 50% (RR=1.7; 95% CI, 1.3-2.2).
Headaches decline after acupuncture, not during
A 2008 meta-analysis of 5 RCTs (838 patients, 68% female) evaluated the effectiveness of acupuncture compared with sham acupuncture for treating episodic and chronic TTH.3 Separate data on efficacy for each subtype was not provided. Selection of acupuncture points varied among the trials, and treatment durations ranged from 3 to 8 weeks.
Headache days per month didn’t decline during treatment (WMD=−2.9; 95% CI, −7.5 to 1.6), but were significantly decreased on follow-up at 20 to 25 weeks (WMD=−1.8; 95% CI, −3.0 to −0.64).
Biofeedback and tricyclic antidepressants appear to be effective as prophylactic treatment for chronic tension headaches, although tricyclic antidepressants have more adverse effects (strength of recommendation [SOR]: B, meta-analysis of randomized controlled trials [RCTs] and pre-post trials).
Acupuncture shows limited evidence for effectiveness after 20 to 25 weeks of treatment (SOR: B, meta-analysis of RCTs).
Biofeedback gets results without adverse effects
A 2008 meta-analysis of 53 trials (32 RCTs, 21 pre-post; 1532 patients, 72% female, average age 36 years) examined the effectiveness of biofeedback for tension-type headaches (TTH) with a mean duration of headache symptoms of 14 years.1 The mean duration of treatment was fewer than 10 hours over 11 sessions. Control groups included placebo, relaxation, pharmacotherapy, cognitive therapy, and physical therapy.
Biofeedback reduced headache pain (measured in structured headache diaries by frequency, duration, and intensity) more than controls (weighted mean difference [WMD]=0.73; 95% confidence interval [CI], 0.61-0.84; WMD effect sizes >0.8 are considered large, 0.6-0.8 are moderate, and 0.2-0.6 are small).
Eighteen trials (15 RCTs, 3 pre-post; 736 patients) included follow-up analysis ranging from 3 to 60 months (mean 15 months), during which biofeedback showed a moderate effect size compared with controls (WMD=0.62; 95% CI, 0.53-0.72). Biofeedback plus relaxation produced a larger effect than controls (6 RCTs, 3 pre-post; 124 patients; WMD=0.98; 95% CI, 0.69-1.3). No adverse effects were reported.
Tricyclics help, too, but have adverse effects
A 2010 meta-analysis of 37 RCTs examined the effectiveness of tricyclic antidepressants as prophylactic treatment for headaches in 3176 patients.2 Seventeen RCTs (1275 patients, 73% women, average age 40 years) evaluated tension headaches, and 15 of them included only patients who had headaches for more than 14 days per month (the other 20 RCTs, with 1901 patients, studied migraine headaches).
Control groups included placebo, selective serotonin reuptake inhibitors (SSRIs), tetracyclics, topiramate, dihydroergotamine, spinal manipulation, and cognitive behavioral therapy, but only placebo (8 RCTs included) and SSRIs (4 RCTs included) had more than 2 RCTs evaluated. Variables included headache days per month, headache intensity (measured by scales that varied among trials), and headache index (calculated by multiplying intensity by frequency).
In 8 trials with 574 patients, the standard mean difference (SMD) headache index improved for tricyclics compared with placebo over a treatment duration averaging 10 weeks (WMD=−0.99; 95% CI, −1.7 to −0.32). However, the tricyclic arm reported more adverse effects such as drowsiness, dizziness, dry mouth, and abdominal complaints (relative risk [RR]=1.9; 95% CI, 1.2-3.0).
Tricyclics and SSRIs reduced the frequency of TTH equally (4 trials; N=479; standardized mean difference=−0.80; 95% CI, −1.63 to 0.02) but tricyclics were more likely to reduce intensity by 50% (RR=1.7; 95% CI, 1.3-2.2).
Headaches decline after acupuncture, not during
A 2008 meta-analysis of 5 RCTs (838 patients, 68% female) evaluated the effectiveness of acupuncture compared with sham acupuncture for treating episodic and chronic TTH.3 Separate data on efficacy for each subtype was not provided. Selection of acupuncture points varied among the trials, and treatment durations ranged from 3 to 8 weeks.
Headache days per month didn’t decline during treatment (WMD=−2.9; 95% CI, −7.5 to 1.6), but were significantly decreased on follow-up at 20 to 25 weeks (WMD=−1.8; 95% CI, −3.0 to −0.64).
1. Netoriuc Y, Rief W, Martin A. Meta-analysis of biofeedback for tension-type headache: efficacy, specificity and treatment moderators. J Consult Clin Psychol. 2008;76:379-396.
2. Jackson JL, Shimeall W, Sessums L, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222.
3. Davis MA, Kononowech RW, Rolin SA, et al. Acupuncture for tension-type headache: a meta-analysis of randomized, controlled trials. J Pain. 2008;9:667-677.
1. Netoriuc Y, Rief W, Martin A. Meta-analysis of biofeedback for tension-type headache: efficacy, specificity and treatment moderators. J Consult Clin Psychol. 2008;76:379-396.
2. Jackson JL, Shimeall W, Sessums L, et al. Tricyclic antidepressants and headaches: systematic review and meta-analysis. BMJ. 2010;341:c5222.
3. Davis MA, Kononowech RW, Rolin SA, et al. Acupuncture for tension-type headache: a meta-analysis of randomized, controlled trials. J Pain. 2008;9:667-677.
Evidence-based answers from the Family Physicians Inquiries Network
Does team-based care improve outcomes for patients with chronic diseases?
Yes, team-based care appears to lower systolic blood pressure (SBP) by 5 to 11 mm Hg and diastolic blood pressure (DBP) by 2 to 6 mm Hg in patients with hypertension and improve lipid control in patients with diabetes (strength of recommendation: C, disease-oriented outcomes).
Team-based care lowers BP in hypertensive patients
A systematic review evaluated 80 trials (total N not defined), which included randomized controlled trials (RCTs) and quasi-experimental trials, to compare blood pressure control in hypertensive patients who received team-based care with that of patients who received usual care.1
Team-based care was defined as adding new staff or changing the roles of existing staff to provide process support and share responsibility for hypertension care with a primary care provider. Examples included using staff to help with medication management, active patient follow-up, adherence, and self-management support. The mean duration of the interventions was 12 months.
The intervention group showed greater reductions in SBP (44 trials; 5.4 mm Hg; interquartile interval [IQI]=2.0-7.2) and DBP (38 trials; 1.8 mm Hg; IQI=0.7-3.2) compared with usual care.
Free medication, care involving nurses, pharmacists lead to lower BP
Another meta-analysis examined 37 RCTs (total N not provided) comparing blood pressure control in hypertensive patients who received team-based care with patients who received usual care.2 The meta-analysis divided the studies by specific types of team-based interventions and analyzed the effect of each type on blood pressure control. It also analyzed studies based on what kind of health care professionals were involved in the intervention.
The largest absolute changes in both SBP and DBP were observed with the following interventions, compared with the control group: free medication (3 trials; SBP reduction (SBPR)=−11 mm Hg; interquartile range [IQR]=−15 to −9.1; DBP reduction [DBPR]=−6.4 mm Hg; IQR=−8.7 to −3.9); pharmacist recommending medication to physician (15 trials; SBPR=−9.3 mm Hg; IQR=−15 to −5.0; DBPR=−3.6 mm Hg; IQR=−7.0 to −1.0); education about BP medications (23 trials; SBPR=−8.8 mm Hg; IQR=−12 to −4.3; DBPR=−3.6 mm Hg; IQR=−7.0 to −1.0); and pharmacist-performed intervention (22 trials; SBPR=−8.4 mm Hg; IQR=−12 to −4; DBPR=−3.3 mm Hg; IQR=−6.9 to −0.90).
Patients who underwent team-based care interventions had greater SBP control (defined as <140, or <130 in patients with diabetes mellitus or chronic kidney disease) than the control group in trials involving nurses (8 trials; odds ratio [OR]=1.7; 95% confidence interval [CI], 1.5-1.9), trials conducted in community pharmacies (5 trials; OR=2.9; 95% CI, 1.8-4.6), and trials incorporating pharmacists into primary care clinics (9 trials; OR=2.2; 95% CI, 1.8-2.7).
Team-based care improves lipid control
An RCT of 6963 patients with type 2 diabetes mellitus in 9 clinics examined the proportion of patients receiving team-based care that achieved a target low-density lipoprotein (LDL) of ≤100 mg/dL compared with patients receiving usual care.3
Clinics were randomized to participate in team-based care (defined as a physician-pharmacist team in which the pharmacist reviewed the medical charts of patients with elevated LDL and then developed individualized, evidence-based treatment recommendations) or usual care, which involved access to a disease management program providing automated quality reporting, benchmarking, and care opportunity decision support.
Over 2 years, the team-based care model had significantly more patients with a lower LDL, more patients at LDL goal, and more patients on lipid-lowering medication (TABLE3).
1. Proia KK, Thota AB, Njie GJ, et al. Team-based care and improved blood pressure control: a community guide systematic review. Am J Prev Med. 2014;47:86-99.
2. Carter BL, Rogers M, Daly J, et al. The potency of team-based care interventions for hypertension: a meta-analysis. Arch Intern Med. 2009;169:1748-1755.
3. Pape GA, Hunt JS, Butler KL, et al. Team-based care approach to cholesterol management in diabetes mellitus. Arch Intern Med. 2011;171:1480-1486.
Yes, team-based care appears to lower systolic blood pressure (SBP) by 5 to 11 mm Hg and diastolic blood pressure (DBP) by 2 to 6 mm Hg in patients with hypertension and improve lipid control in patients with diabetes (strength of recommendation: C, disease-oriented outcomes).
Team-based care lowers BP in hypertensive patients
A systematic review evaluated 80 trials (total N not defined), which included randomized controlled trials (RCTs) and quasi-experimental trials, to compare blood pressure control in hypertensive patients who received team-based care with that of patients who received usual care.1
Team-based care was defined as adding new staff or changing the roles of existing staff to provide process support and share responsibility for hypertension care with a primary care provider. Examples included using staff to help with medication management, active patient follow-up, adherence, and self-management support. The mean duration of the interventions was 12 months.
The intervention group showed greater reductions in SBP (44 trials; 5.4 mm Hg; interquartile interval [IQI]=2.0-7.2) and DBP (38 trials; 1.8 mm Hg; IQI=0.7-3.2) compared with usual care.
Free medication, care involving nurses, pharmacists lead to lower BP
Another meta-analysis examined 37 RCTs (total N not provided) comparing blood pressure control in hypertensive patients who received team-based care with patients who received usual care.2 The meta-analysis divided the studies by specific types of team-based interventions and analyzed the effect of each type on blood pressure control. It also analyzed studies based on what kind of health care professionals were involved in the intervention.
The largest absolute changes in both SBP and DBP were observed with the following interventions, compared with the control group: free medication (3 trials; SBP reduction (SBPR)=−11 mm Hg; interquartile range [IQR]=−15 to −9.1; DBP reduction [DBPR]=−6.4 mm Hg; IQR=−8.7 to −3.9); pharmacist recommending medication to physician (15 trials; SBPR=−9.3 mm Hg; IQR=−15 to −5.0; DBPR=−3.6 mm Hg; IQR=−7.0 to −1.0); education about BP medications (23 trials; SBPR=−8.8 mm Hg; IQR=−12 to −4.3; DBPR=−3.6 mm Hg; IQR=−7.0 to −1.0); and pharmacist-performed intervention (22 trials; SBPR=−8.4 mm Hg; IQR=−12 to −4; DBPR=−3.3 mm Hg; IQR=−6.9 to −0.90).
Patients who underwent team-based care interventions had greater SBP control (defined as <140, or <130 in patients with diabetes mellitus or chronic kidney disease) than the control group in trials involving nurses (8 trials; odds ratio [OR]=1.7; 95% confidence interval [CI], 1.5-1.9), trials conducted in community pharmacies (5 trials; OR=2.9; 95% CI, 1.8-4.6), and trials incorporating pharmacists into primary care clinics (9 trials; OR=2.2; 95% CI, 1.8-2.7).
Team-based care improves lipid control
An RCT of 6963 patients with type 2 diabetes mellitus in 9 clinics examined the proportion of patients receiving team-based care that achieved a target low-density lipoprotein (LDL) of ≤100 mg/dL compared with patients receiving usual care.3
Clinics were randomized to participate in team-based care (defined as a physician-pharmacist team in which the pharmacist reviewed the medical charts of patients with elevated LDL and then developed individualized, evidence-based treatment recommendations) or usual care, which involved access to a disease management program providing automated quality reporting, benchmarking, and care opportunity decision support.
Over 2 years, the team-based care model had significantly more patients with a lower LDL, more patients at LDL goal, and more patients on lipid-lowering medication (TABLE3).
Yes, team-based care appears to lower systolic blood pressure (SBP) by 5 to 11 mm Hg and diastolic blood pressure (DBP) by 2 to 6 mm Hg in patients with hypertension and improve lipid control in patients with diabetes (strength of recommendation: C, disease-oriented outcomes).
Team-based care lowers BP in hypertensive patients
A systematic review evaluated 80 trials (total N not defined), which included randomized controlled trials (RCTs) and quasi-experimental trials, to compare blood pressure control in hypertensive patients who received team-based care with that of patients who received usual care.1
Team-based care was defined as adding new staff or changing the roles of existing staff to provide process support and share responsibility for hypertension care with a primary care provider. Examples included using staff to help with medication management, active patient follow-up, adherence, and self-management support. The mean duration of the interventions was 12 months.
The intervention group showed greater reductions in SBP (44 trials; 5.4 mm Hg; interquartile interval [IQI]=2.0-7.2) and DBP (38 trials; 1.8 mm Hg; IQI=0.7-3.2) compared with usual care.
Free medication, care involving nurses, pharmacists lead to lower BP
Another meta-analysis examined 37 RCTs (total N not provided) comparing blood pressure control in hypertensive patients who received team-based care with patients who received usual care.2 The meta-analysis divided the studies by specific types of team-based interventions and analyzed the effect of each type on blood pressure control. It also analyzed studies based on what kind of health care professionals were involved in the intervention.
The largest absolute changes in both SBP and DBP were observed with the following interventions, compared with the control group: free medication (3 trials; SBP reduction (SBPR)=−11 mm Hg; interquartile range [IQR]=−15 to −9.1; DBP reduction [DBPR]=−6.4 mm Hg; IQR=−8.7 to −3.9); pharmacist recommending medication to physician (15 trials; SBPR=−9.3 mm Hg; IQR=−15 to −5.0; DBPR=−3.6 mm Hg; IQR=−7.0 to −1.0); education about BP medications (23 trials; SBPR=−8.8 mm Hg; IQR=−12 to −4.3; DBPR=−3.6 mm Hg; IQR=−7.0 to −1.0); and pharmacist-performed intervention (22 trials; SBPR=−8.4 mm Hg; IQR=−12 to −4; DBPR=−3.3 mm Hg; IQR=−6.9 to −0.90).
Patients who underwent team-based care interventions had greater SBP control (defined as <140, or <130 in patients with diabetes mellitus or chronic kidney disease) than the control group in trials involving nurses (8 trials; odds ratio [OR]=1.7; 95% confidence interval [CI], 1.5-1.9), trials conducted in community pharmacies (5 trials; OR=2.9; 95% CI, 1.8-4.6), and trials incorporating pharmacists into primary care clinics (9 trials; OR=2.2; 95% CI, 1.8-2.7).
Team-based care improves lipid control
An RCT of 6963 patients with type 2 diabetes mellitus in 9 clinics examined the proportion of patients receiving team-based care that achieved a target low-density lipoprotein (LDL) of ≤100 mg/dL compared with patients receiving usual care.3
Clinics were randomized to participate in team-based care (defined as a physician-pharmacist team in which the pharmacist reviewed the medical charts of patients with elevated LDL and then developed individualized, evidence-based treatment recommendations) or usual care, which involved access to a disease management program providing automated quality reporting, benchmarking, and care opportunity decision support.
Over 2 years, the team-based care model had significantly more patients with a lower LDL, more patients at LDL goal, and more patients on lipid-lowering medication (TABLE3).
1. Proia KK, Thota AB, Njie GJ, et al. Team-based care and improved blood pressure control: a community guide systematic review. Am J Prev Med. 2014;47:86-99.
2. Carter BL, Rogers M, Daly J, et al. The potency of team-based care interventions for hypertension: a meta-analysis. Arch Intern Med. 2009;169:1748-1755.
3. Pape GA, Hunt JS, Butler KL, et al. Team-based care approach to cholesterol management in diabetes mellitus. Arch Intern Med. 2011;171:1480-1486.
1. Proia KK, Thota AB, Njie GJ, et al. Team-based care and improved blood pressure control: a community guide systematic review. Am J Prev Med. 2014;47:86-99.
2. Carter BL, Rogers M, Daly J, et al. The potency of team-based care interventions for hypertension: a meta-analysis. Arch Intern Med. 2009;169:1748-1755.
3. Pape GA, Hunt JS, Butler KL, et al. Team-based care approach to cholesterol management in diabetes mellitus. Arch Intern Med. 2011;171:1480-1486.
Evidence-based answers from the Family Physicians Inquiries Network
Do trigger point injections effectively treat fibromyalgia?
Possibly. Trigger point injections appear effective in reducing pain and increasing pressure thresholds in patients with fibromyalgia and myofascial trigger points (strength of recommendation [SOR]: B, small randomized controlled trials [RCTs]).
Consensus guidelines suggest that trigger point injections may have a role in the treatment of fibromyalgia (SOR: C, expert opinion).
Active injections produce sustained improvement
A 2011 double-blind RCT randomized 68 female patients with both fibromyalgia and myofascial trigger points to either active trigger point injections with 1 mL 0.5% bupivacaine or placebo-like needle penetration with no medication to an area near the trigger point.1 Patients were evaluated for both local and generalized fibromyalgia symptoms at 4 and 8 days (trial period) and after 30 days (follow-up). Injections occurred on Days 1 and 4, with an option of additional injections on Days 8 and 11.
Compared to baseline (7 days before the injection), patients receiving active trigger point injections had decreased myofascial pain episodes 7 days after the injection (5.6 vs 0.97 episodes; P<.001), decreased pain intensity (62 vs 19/100 mm Visual Analog Scale score; P<.001), and increased pressure threshold at the trigger point (1.5 vs 2.9 kg/cm2; P<.0001), whereas the control group showed no differences.
During Days 1 to 8, patients receiving active trigger point injections required less acetaminophen (0.2 vs 2.7 tablets/d; P<.0001). At Day 8, no patients in the active trigger point injection group requested additional injections, whereas all the patients in the control group requested an injection (P<.0001).
At Day 8, patients also had significantly decreased intensity of fibromyalgia pain, fewer tender points, and higher tender point pressure thresholds; none of these differences were statistically significant in the placebo injection group (data presented graphically). The improvements persisted at 30 days of follow-up (data presented graphically).
Small study shows improvement with injections after 2 weeks
An uncontrolled prospective before-after study in 1996 evaluated the effectiveness of 0.5% lidocaine trigger point injections in 9 patients with myofascial trigger points plus fibromyalgia compared with 9 patients with myofascial trigger points alone.2
Immediately after injection, patients with fibromyalgia had a nonsignificant worsening in pain intensity (pain scale 8.1 to 8.4/10; P>.1), but there was a significant improvement at 2 weeks (5.9; P<.01). The pressure threshold also decreased initially (1.7 to 1.4 kg/cm2; P>.1), but significantly increased at 2 weeks (2.4 kg/cm2; P<.01). In comparison, patients without fibromyalgia showed immediate improvement in all domains, which persisted at 2 weeks (P<.01).
What the guidelines say
Recent Canadian Fibromyalgia Guidelines discuss trigger point injections in the section on “off-label” medications, stating that they “may have some place in treatment of fibromyalgia.”3
1. Affaitati G, Costantini R, Fabrizio A, et al. Effects of treatment of peripheral pain generators in fibromyalgia patients. Eur J Pain. 2011;15:61-69.
2. Hong CZ, Hsueh TC. Difference in pain relief after trigger point injections in myofascial pain patients with and without fibromyalgia. Arch Phys Med Rehabil. 1996;77:1161-1166.
3. Fitzcharles MA, Ste-Marie PA, Goldenberg DL, et al. 2012. Canadian Guidelines for the diagnosis and management of fibromyalgia syndrome: executive summary. Pain Res Manag. 2013;18:119-126.
Possibly. Trigger point injections appear effective in reducing pain and increasing pressure thresholds in patients with fibromyalgia and myofascial trigger points (strength of recommendation [SOR]: B, small randomized controlled trials [RCTs]).
Consensus guidelines suggest that trigger point injections may have a role in the treatment of fibromyalgia (SOR: C, expert opinion).
Active injections produce sustained improvement
A 2011 double-blind RCT randomized 68 female patients with both fibromyalgia and myofascial trigger points to either active trigger point injections with 1 mL 0.5% bupivacaine or placebo-like needle penetration with no medication to an area near the trigger point.1 Patients were evaluated for both local and generalized fibromyalgia symptoms at 4 and 8 days (trial period) and after 30 days (follow-up). Injections occurred on Days 1 and 4, with an option of additional injections on Days 8 and 11.
Compared to baseline (7 days before the injection), patients receiving active trigger point injections had decreased myofascial pain episodes 7 days after the injection (5.6 vs 0.97 episodes; P<.001), decreased pain intensity (62 vs 19/100 mm Visual Analog Scale score; P<.001), and increased pressure threshold at the trigger point (1.5 vs 2.9 kg/cm2; P<.0001), whereas the control group showed no differences.
During Days 1 to 8, patients receiving active trigger point injections required less acetaminophen (0.2 vs 2.7 tablets/d; P<.0001). At Day 8, no patients in the active trigger point injection group requested additional injections, whereas all the patients in the control group requested an injection (P<.0001).
At Day 8, patients also had significantly decreased intensity of fibromyalgia pain, fewer tender points, and higher tender point pressure thresholds; none of these differences were statistically significant in the placebo injection group (data presented graphically). The improvements persisted at 30 days of follow-up (data presented graphically).
Small study shows improvement with injections after 2 weeks
An uncontrolled prospective before-after study in 1996 evaluated the effectiveness of 0.5% lidocaine trigger point injections in 9 patients with myofascial trigger points plus fibromyalgia compared with 9 patients with myofascial trigger points alone.2
Immediately after injection, patients with fibromyalgia had a nonsignificant worsening in pain intensity (pain scale 8.1 to 8.4/10; P>.1), but there was a significant improvement at 2 weeks (5.9; P<.01). The pressure threshold also decreased initially (1.7 to 1.4 kg/cm2; P>.1), but significantly increased at 2 weeks (2.4 kg/cm2; P<.01). In comparison, patients without fibromyalgia showed immediate improvement in all domains, which persisted at 2 weeks (P<.01).
What the guidelines say
Recent Canadian Fibromyalgia Guidelines discuss trigger point injections in the section on “off-label” medications, stating that they “may have some place in treatment of fibromyalgia.”3
Possibly. Trigger point injections appear effective in reducing pain and increasing pressure thresholds in patients with fibromyalgia and myofascial trigger points (strength of recommendation [SOR]: B, small randomized controlled trials [RCTs]).
Consensus guidelines suggest that trigger point injections may have a role in the treatment of fibromyalgia (SOR: C, expert opinion).
Active injections produce sustained improvement
A 2011 double-blind RCT randomized 68 female patients with both fibromyalgia and myofascial trigger points to either active trigger point injections with 1 mL 0.5% bupivacaine or placebo-like needle penetration with no medication to an area near the trigger point.1 Patients were evaluated for both local and generalized fibromyalgia symptoms at 4 and 8 days (trial period) and after 30 days (follow-up). Injections occurred on Days 1 and 4, with an option of additional injections on Days 8 and 11.
Compared to baseline (7 days before the injection), patients receiving active trigger point injections had decreased myofascial pain episodes 7 days after the injection (5.6 vs 0.97 episodes; P<.001), decreased pain intensity (62 vs 19/100 mm Visual Analog Scale score; P<.001), and increased pressure threshold at the trigger point (1.5 vs 2.9 kg/cm2; P<.0001), whereas the control group showed no differences.
During Days 1 to 8, patients receiving active trigger point injections required less acetaminophen (0.2 vs 2.7 tablets/d; P<.0001). At Day 8, no patients in the active trigger point injection group requested additional injections, whereas all the patients in the control group requested an injection (P<.0001).
At Day 8, patients also had significantly decreased intensity of fibromyalgia pain, fewer tender points, and higher tender point pressure thresholds; none of these differences were statistically significant in the placebo injection group (data presented graphically). The improvements persisted at 30 days of follow-up (data presented graphically).
Small study shows improvement with injections after 2 weeks
An uncontrolled prospective before-after study in 1996 evaluated the effectiveness of 0.5% lidocaine trigger point injections in 9 patients with myofascial trigger points plus fibromyalgia compared with 9 patients with myofascial trigger points alone.2
Immediately after injection, patients with fibromyalgia had a nonsignificant worsening in pain intensity (pain scale 8.1 to 8.4/10; P>.1), but there was a significant improvement at 2 weeks (5.9; P<.01). The pressure threshold also decreased initially (1.7 to 1.4 kg/cm2; P>.1), but significantly increased at 2 weeks (2.4 kg/cm2; P<.01). In comparison, patients without fibromyalgia showed immediate improvement in all domains, which persisted at 2 weeks (P<.01).
What the guidelines say
Recent Canadian Fibromyalgia Guidelines discuss trigger point injections in the section on “off-label” medications, stating that they “may have some place in treatment of fibromyalgia.”3
1. Affaitati G, Costantini R, Fabrizio A, et al. Effects of treatment of peripheral pain generators in fibromyalgia patients. Eur J Pain. 2011;15:61-69.
2. Hong CZ, Hsueh TC. Difference in pain relief after trigger point injections in myofascial pain patients with and without fibromyalgia. Arch Phys Med Rehabil. 1996;77:1161-1166.
3. Fitzcharles MA, Ste-Marie PA, Goldenberg DL, et al. 2012. Canadian Guidelines for the diagnosis and management of fibromyalgia syndrome: executive summary. Pain Res Manag. 2013;18:119-126.
1. Affaitati G, Costantini R, Fabrizio A, et al. Effects of treatment of peripheral pain generators in fibromyalgia patients. Eur J Pain. 2011;15:61-69.
2. Hong CZ, Hsueh TC. Difference in pain relief after trigger point injections in myofascial pain patients with and without fibromyalgia. Arch Phys Med Rehabil. 1996;77:1161-1166.
3. Fitzcharles MA, Ste-Marie PA, Goldenberg DL, et al. 2012. Canadian Guidelines for the diagnosis and management of fibromyalgia syndrome: executive summary. Pain Res Manag. 2013;18:119-126.
Evidence-based answers from the Family Physicians Inquiries Network