User login
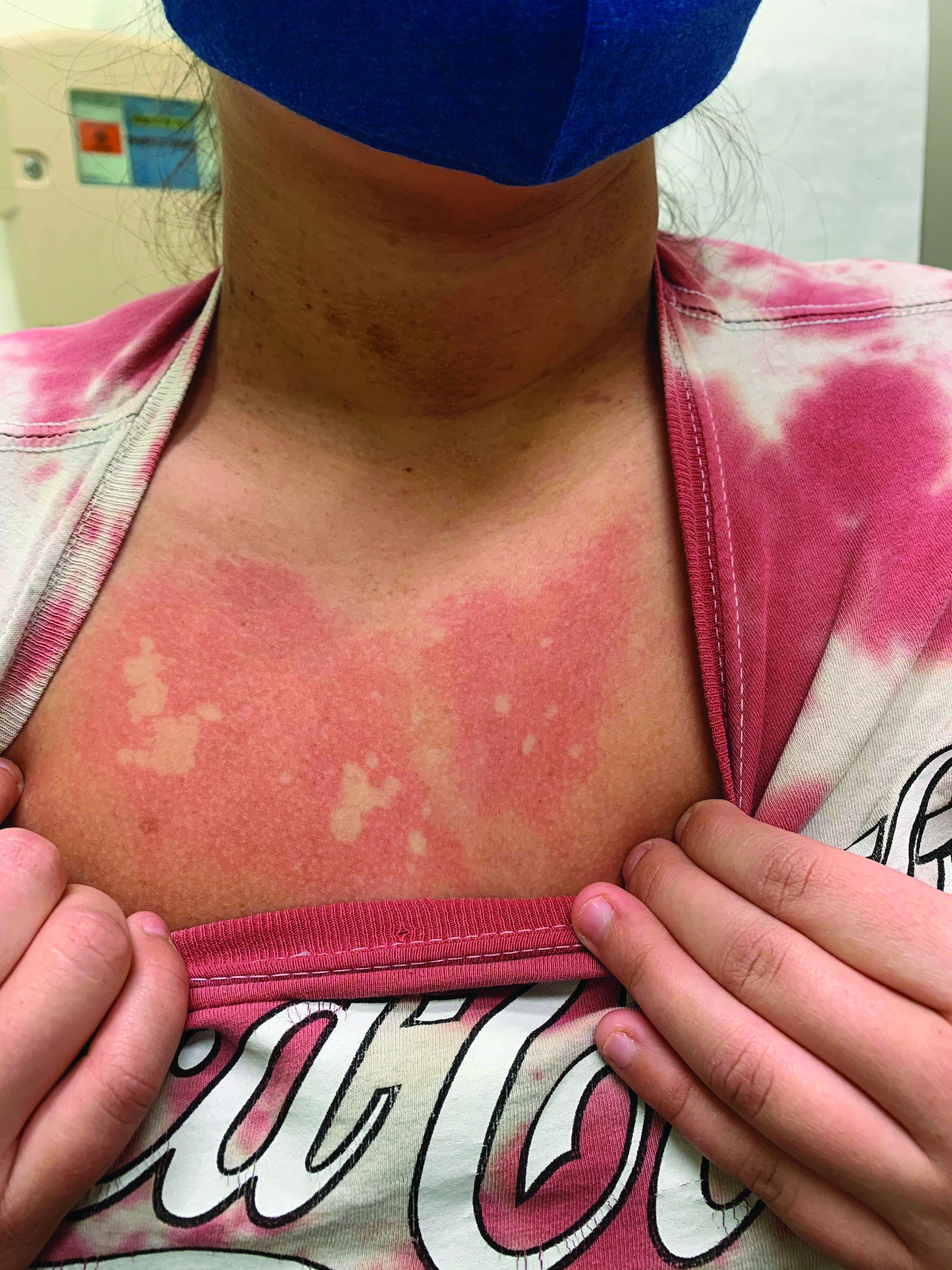
A girl presents with blotchy, slightly itchy spots on her chest, back
On close evaluation of the picture on her chest, she has pale macules and patches surrounded by erythematous ill-defined patches consistent with nevus anemicus. The findings of the picture raise the suspicion for neurofibromatosis, and it was recommended for her to be evaluated in person.
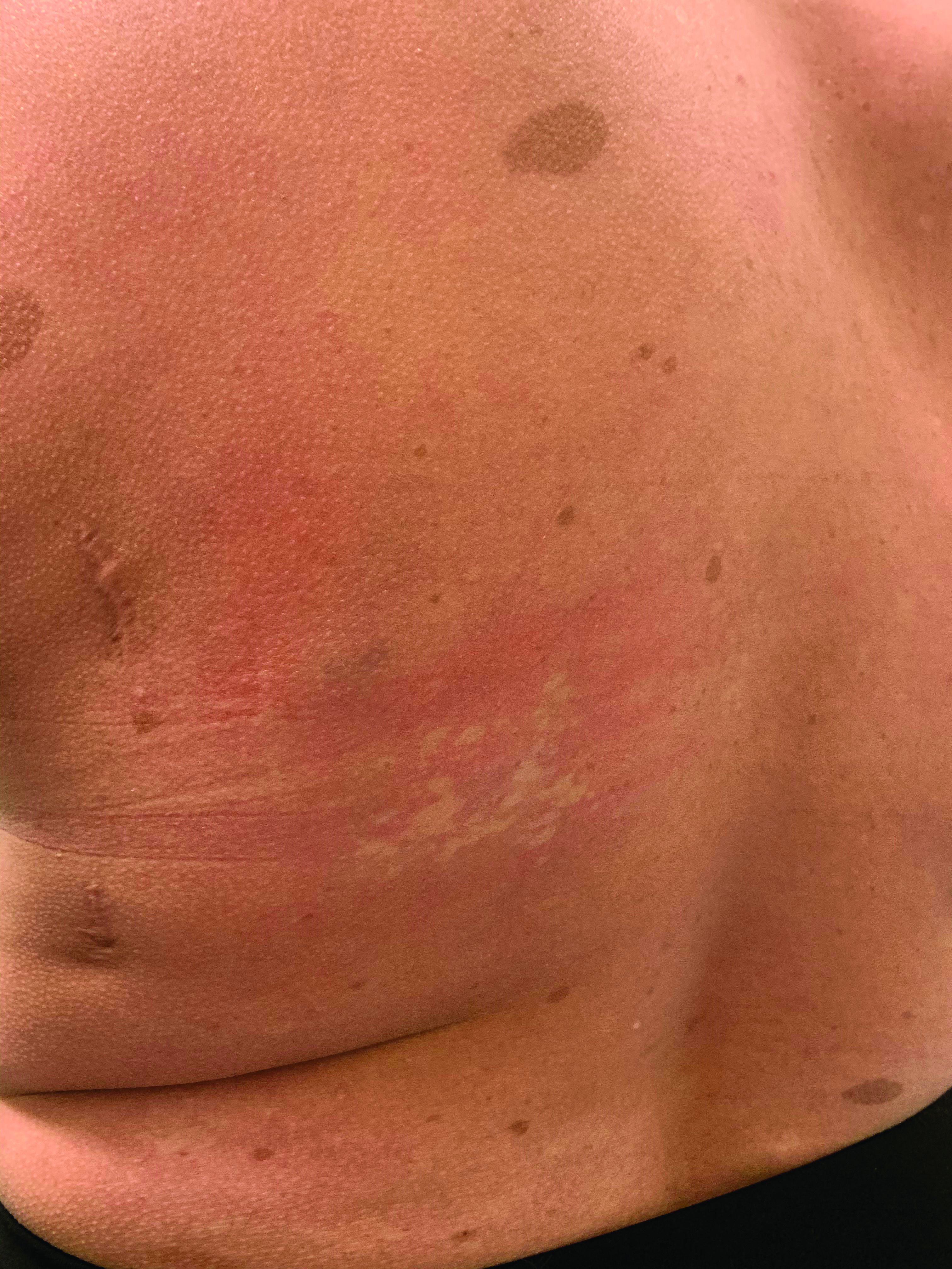
She comes several days later to the clinic. The caretaker, who is her aunt, reports she does not know much of the girl’s medical history as she recently moved from South America to live with her. The girl is a very nice and pleasant 8-year-old. She reports noticing the spots on her chest for about a year and that they seem to get a little itchier and more noticeable when she is hot or when she is running. She also reports increasing headaches for several months. She is being home schooled, and according to her aunt she is at par with her cousins who are about the same age. There is no history of seizures. She has had back surgery in the past. There is no history of hypertension. There is no family history of any genetic disorder or similar lesions.
On physical exam, her vital signs are normal, but her head circumference is over the 90th percentile. She is pleasant and interactive. On skin examination, she has slightly noticeable pale macules and patches on the chest and back that become more apparent after rubbing her skin. She has multiple light brown macules and oval patches on the chest, back, and neck. She has no axillary or inguinal freckling. She has scars on the back from her prior surgery.
As she was having worsening headaches, an MRI of the brain was ordered, which showed a left optic glioma. She was then referred to ophthalmology, neurology, and genetics.
Neurofibromatosis type 1 (NF1) is a common genetic autosomal dominant disorder cause by mutations on the NF1 gene on chromosome 17, which encodes for the protein neurofibromin. This protein works in the Ras-mitogen–activated protein kinase pathway as a negative regulator. Based on the National Institute of Health criteria, children need two or more of the following to be diagnosed with NF1: more than six café au lait macules larger than 5 mm in prepubescent children and 2.5 cm after puberty; axillary or inguinal freckling; two or more Lisch nodules; optic gliomas; two or more neurofibromas or one plexiform neurofibroma; or a first degree relative with a diagnosis of NF1. With these criteria, about 70% of the children can be diagnosed before the age of 1 year.1

Nevus anemicus is an uncommon birthmark, sometimes overlooked, that is characterized by pale, hypopigmented, well-defined macules and patches that do not turn red after trauma or changes in temperature. Nevus anemicus is usually localized on the torso but can be seen on the face, neck, and extremities. These lesions are present in 1%-2% of the general population. They are thought to occur because of increased sensitivity of the affected blood vessels to catecholamines, which causes permanent vasoconstriction, which leads to hypopigmentation on the area.2 These lesions are usually present at birth and have been described in patients with tuberous sclerosis, neurofibromatosis, and phakomatosis pigmentovascularis.
Recent studies of patients with neurofibromatosis and other RASopathies have noticed that nevus anemicus is present in about 8.8%-51% of the patients studied with a diagnosis NF1, compared with only 2% of the controls.3,4 The studies failed to report any cases of nevus anemicus in patients with other RASopathies associated with café au lait macules. Bulteel and colleagues recently reported two cases of non-NF1 RASopathies also associated with nevus anemicus in a patient with Legius syndrome and a patient with Noonan syndrome with multiple lentigines.5 The nevus anemicus was reported to occur most commonly on the anterior chest and be multiple, as seen in our patient.
The authors of the published studies advocate for the introduction of nevus anemicus as part of the diagnostic criteria for NF1, especially because it can be an early finding seen in babies, which can aid in early diagnosis of NF1.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no relevant financial disclosures. Email Dr. Matiz at [email protected].
References
1. Pediatrics. 2000 Mar. doi: 10.1542/peds.105.3.608.
2. Nevus Anemicus. StatPearls [Internet] (Treasure Island, Fla.: StatPearls Publishing; 2020 Jan).
3. J Am Acad Dermatol. 2013 Nov. doi: 10.1016/j.jaad.2013.06.039.
4. Pediatr Dermatol. 2015 May-Jun. doi: 10.1111/pde.12525.
5. JAAD Case Rep. 2018 Apr 5. doi: 10.1016/j.jdcr.2017.09.037.
On close evaluation of the picture on her chest, she has pale macules and patches surrounded by erythematous ill-defined patches consistent with nevus anemicus. The findings of the picture raise the suspicion for neurofibromatosis, and it was recommended for her to be evaluated in person.
She comes several days later to the clinic. The caretaker, who is her aunt, reports she does not know much of the girl’s medical history as she recently moved from South America to live with her. The girl is a very nice and pleasant 8-year-old. She reports noticing the spots on her chest for about a year and that they seem to get a little itchier and more noticeable when she is hot or when she is running. She also reports increasing headaches for several months. She is being home schooled, and according to her aunt she is at par with her cousins who are about the same age. There is no history of seizures. She has had back surgery in the past. There is no history of hypertension. There is no family history of any genetic disorder or similar lesions.
On physical exam, her vital signs are normal, but her head circumference is over the 90th percentile. She is pleasant and interactive. On skin examination, she has slightly noticeable pale macules and patches on the chest and back that become more apparent after rubbing her skin. She has multiple light brown macules and oval patches on the chest, back, and neck. She has no axillary or inguinal freckling. She has scars on the back from her prior surgery.
As she was having worsening headaches, an MRI of the brain was ordered, which showed a left optic glioma. She was then referred to ophthalmology, neurology, and genetics.
Neurofibromatosis type 1 (NF1) is a common genetic autosomal dominant disorder cause by mutations on the NF1 gene on chromosome 17, which encodes for the protein neurofibromin. This protein works in the Ras-mitogen–activated protein kinase pathway as a negative regulator. Based on the National Institute of Health criteria, children need two or more of the following to be diagnosed with NF1: more than six café au lait macules larger than 5 mm in prepubescent children and 2.5 cm after puberty; axillary or inguinal freckling; two or more Lisch nodules; optic gliomas; two or more neurofibromas or one plexiform neurofibroma; or a first degree relative with a diagnosis of NF1. With these criteria, about 70% of the children can be diagnosed before the age of 1 year.1
Nevus anemicus is an uncommon birthmark, sometimes overlooked, that is characterized by pale, hypopigmented, well-defined macules and patches that do not turn red after trauma or changes in temperature. Nevus anemicus is usually localized on the torso but can be seen on the face, neck, and extremities. These lesions are present in 1%-2% of the general population. They are thought to occur because of increased sensitivity of the affected blood vessels to catecholamines, which causes permanent vasoconstriction, which leads to hypopigmentation on the area.2 These lesions are usually present at birth and have been described in patients with tuberous sclerosis, neurofibromatosis, and phakomatosis pigmentovascularis.
Recent studies of patients with neurofibromatosis and other RASopathies have noticed that nevus anemicus is present in about 8.8%-51% of the patients studied with a diagnosis NF1, compared with only 2% of the controls.3,4 The studies failed to report any cases of nevus anemicus in patients with other RASopathies associated with café au lait macules. Bulteel and colleagues recently reported two cases of non-NF1 RASopathies also associated with nevus anemicus in a patient with Legius syndrome and a patient with Noonan syndrome with multiple lentigines.5 The nevus anemicus was reported to occur most commonly on the anterior chest and be multiple, as seen in our patient.
The authors of the published studies advocate for the introduction of nevus anemicus as part of the diagnostic criteria for NF1, especially because it can be an early finding seen in babies, which can aid in early diagnosis of NF1.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no relevant financial disclosures. Email Dr. Matiz at [email protected].
References
1. Pediatrics. 2000 Mar. doi: 10.1542/peds.105.3.608.
2. Nevus Anemicus. StatPearls [Internet] (Treasure Island, Fla.: StatPearls Publishing; 2020 Jan).
3. J Am Acad Dermatol. 2013 Nov. doi: 10.1016/j.jaad.2013.06.039.
4. Pediatr Dermatol. 2015 May-Jun. doi: 10.1111/pde.12525.
5. JAAD Case Rep. 2018 Apr 5. doi: 10.1016/j.jdcr.2017.09.037.
On close evaluation of the picture on her chest, she has pale macules and patches surrounded by erythematous ill-defined patches consistent with nevus anemicus. The findings of the picture raise the suspicion for neurofibromatosis, and it was recommended for her to be evaluated in person.
She comes several days later to the clinic. The caretaker, who is her aunt, reports she does not know much of the girl’s medical history as she recently moved from South America to live with her. The girl is a very nice and pleasant 8-year-old. She reports noticing the spots on her chest for about a year and that they seem to get a little itchier and more noticeable when she is hot or when she is running. She also reports increasing headaches for several months. She is being home schooled, and according to her aunt she is at par with her cousins who are about the same age. There is no history of seizures. She has had back surgery in the past. There is no history of hypertension. There is no family history of any genetic disorder or similar lesions.
On physical exam, her vital signs are normal, but her head circumference is over the 90th percentile. She is pleasant and interactive. On skin examination, she has slightly noticeable pale macules and patches on the chest and back that become more apparent after rubbing her skin. She has multiple light brown macules and oval patches on the chest, back, and neck. She has no axillary or inguinal freckling. She has scars on the back from her prior surgery.
As she was having worsening headaches, an MRI of the brain was ordered, which showed a left optic glioma. She was then referred to ophthalmology, neurology, and genetics.
Neurofibromatosis type 1 (NF1) is a common genetic autosomal dominant disorder cause by mutations on the NF1 gene on chromosome 17, which encodes for the protein neurofibromin. This protein works in the Ras-mitogen–activated protein kinase pathway as a negative regulator. Based on the National Institute of Health criteria, children need two or more of the following to be diagnosed with NF1: more than six café au lait macules larger than 5 mm in prepubescent children and 2.5 cm after puberty; axillary or inguinal freckling; two or more Lisch nodules; optic gliomas; two or more neurofibromas or one plexiform neurofibroma; or a first degree relative with a diagnosis of NF1. With these criteria, about 70% of the children can be diagnosed before the age of 1 year.1
Nevus anemicus is an uncommon birthmark, sometimes overlooked, that is characterized by pale, hypopigmented, well-defined macules and patches that do not turn red after trauma or changes in temperature. Nevus anemicus is usually localized on the torso but can be seen on the face, neck, and extremities. These lesions are present in 1%-2% of the general population. They are thought to occur because of increased sensitivity of the affected blood vessels to catecholamines, which causes permanent vasoconstriction, which leads to hypopigmentation on the area.2 These lesions are usually present at birth and have been described in patients with tuberous sclerosis, neurofibromatosis, and phakomatosis pigmentovascularis.
Recent studies of patients with neurofibromatosis and other RASopathies have noticed that nevus anemicus is present in about 8.8%-51% of the patients studied with a diagnosis NF1, compared with only 2% of the controls.3,4 The studies failed to report any cases of nevus anemicus in patients with other RASopathies associated with café au lait macules. Bulteel and colleagues recently reported two cases of non-NF1 RASopathies also associated with nevus anemicus in a patient with Legius syndrome and a patient with Noonan syndrome with multiple lentigines.5 The nevus anemicus was reported to occur most commonly on the anterior chest and be multiple, as seen in our patient.
The authors of the published studies advocate for the introduction of nevus anemicus as part of the diagnostic criteria for NF1, especially because it can be an early finding seen in babies, which can aid in early diagnosis of NF1.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no relevant financial disclosures. Email Dr. Matiz at [email protected].
References
1. Pediatrics. 2000 Mar. doi: 10.1542/peds.105.3.608.
2. Nevus Anemicus. StatPearls [Internet] (Treasure Island, Fla.: StatPearls Publishing; 2020 Jan).
3. J Am Acad Dermatol. 2013 Nov. doi: 10.1016/j.jaad.2013.06.039.
4. Pediatr Dermatol. 2015 May-Jun. doi: 10.1111/pde.12525.
5. JAAD Case Rep. 2018 Apr 5. doi: 10.1016/j.jdcr.2017.09.037.
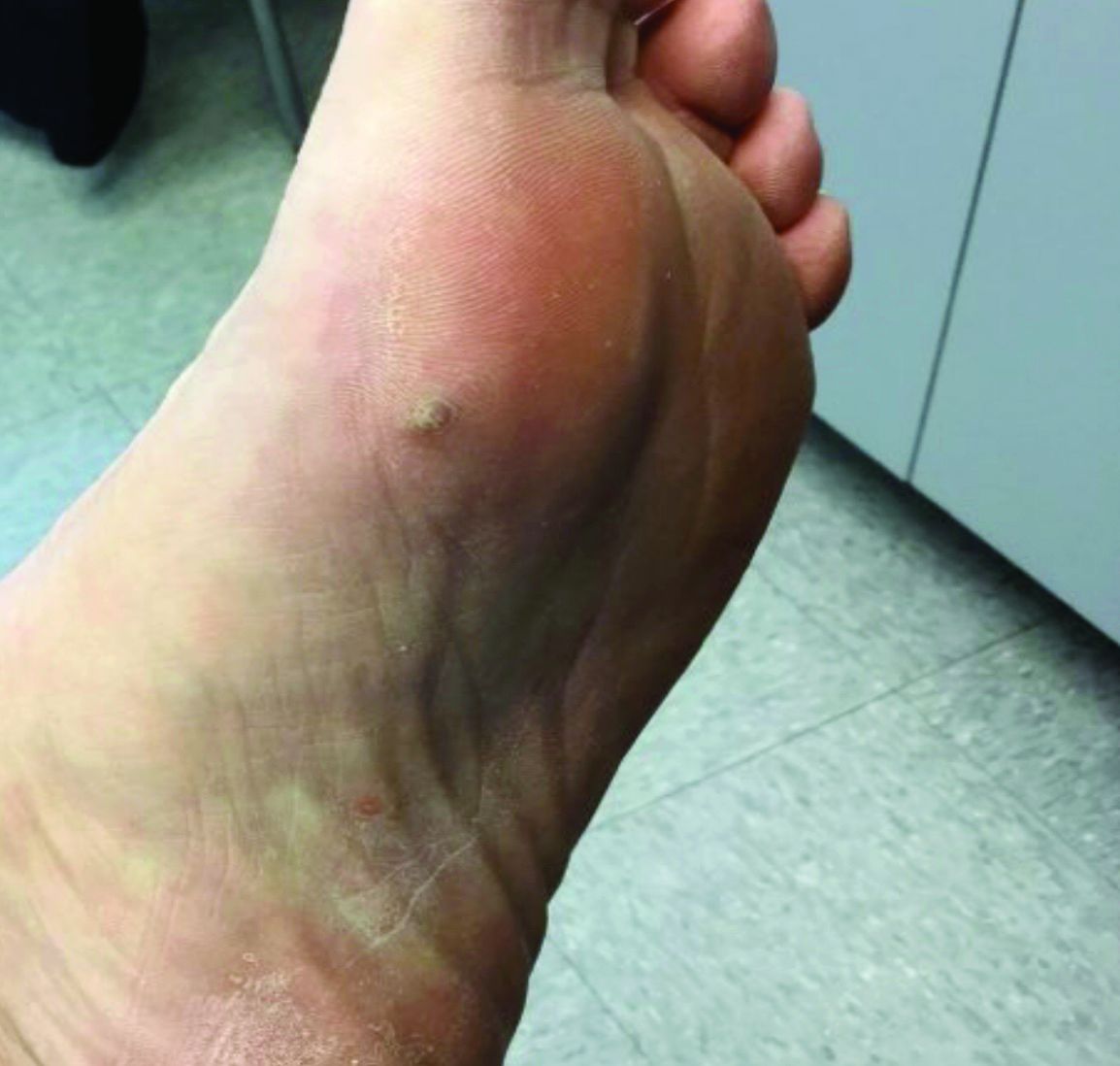
A 70-year-old presented with a 3-week history of asymptomatic violaceous papules on his feet
and named the condition multiple benign pigmented hemorrhagic sarcoma. The disease emerged again at the onset of the AIDS epidemic among homosexual men. There are five variants: HIV/AIDS–related KS, classic KS, African cutaneous KS, African lymphadenopathic KS, and immunosuppression-associated KS (from immunosuppressive therapy or malignancies such as lymphoma).
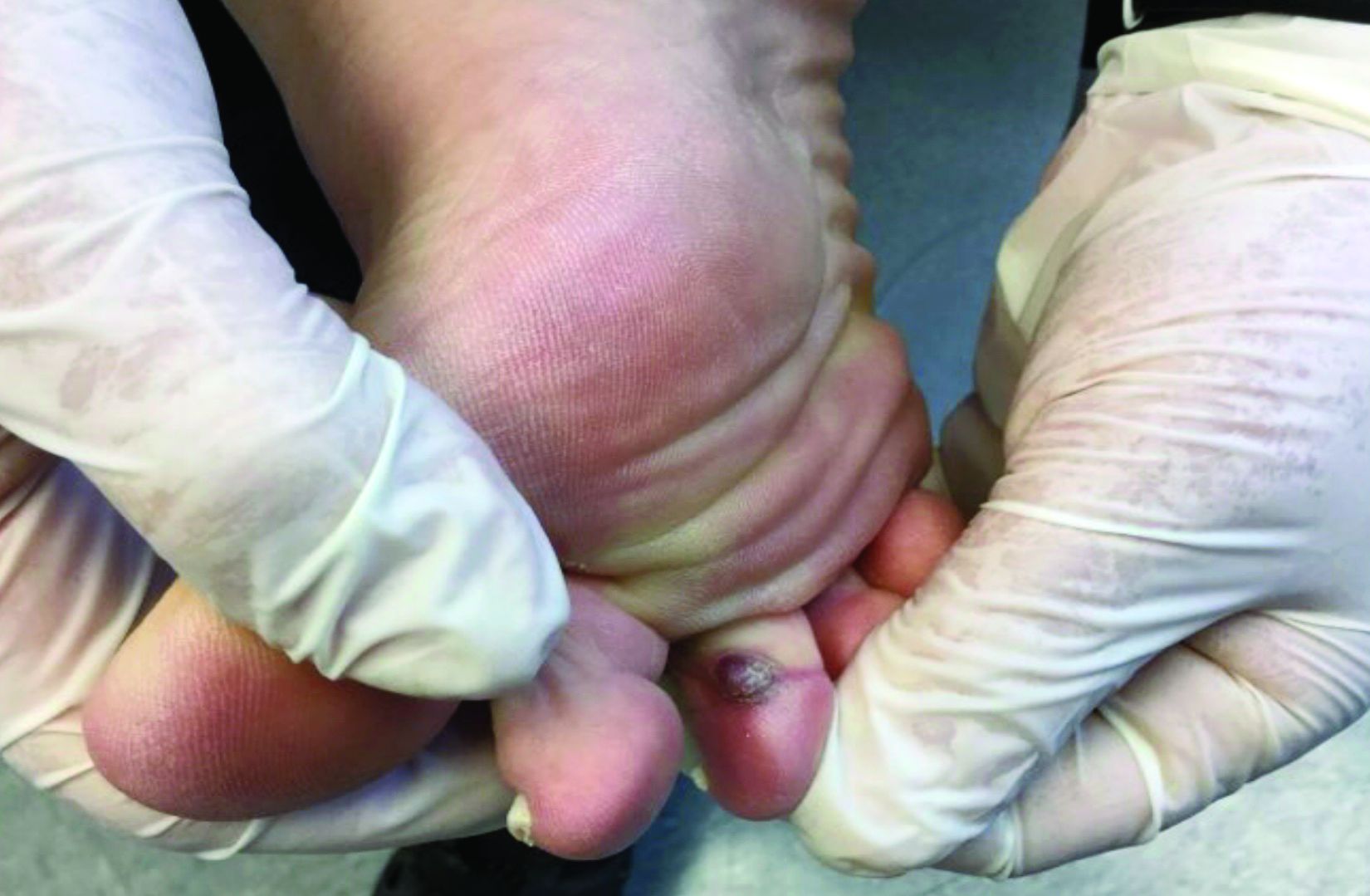
KS is caused by human herpes virus type 8 (HHV-8). Patients with KS have an increased risk of developing other malignancies such as lymphomas, leukemia, and myeloma. This patient exhibited classic KS.
The various forms of KS may appear different clinically. The lesions may appear as erythematous macules, small violaceous papules, large plaques, or ulcerated nodules. In classic KS, violaceous to bluish-black macules evolve to papules or plaques. Lesions are generally asymptomatic. The most common locations are the toes and soles, although other areas may be affected. Any mucocutaneous surface can be involved. The most common areas of internal involvement are the gastrointestinal system and lymphatics.
Histology reveals angular vessels lined by atypical cells. An associated inflammatory infiltrate containing plasma cells may be present in the upper dermis and perivascular areas. Nodules and plaques reveal a spindle cell neoplasm pattern. Lesions will stain positive for HHV-8.
In patients with HIV/AIDS–related KS, highly active antiretroviral therapy is the most important and beneficial treatment. Since the introduction of HAART, the incidence of KS has greatly decreased. However, there are a proportion of HIV/AIDS–associated Kaposi’s sarcoma patients with well-controlled HIV and undetectable viral loads who require further treatment.
Lesions may spontaneously resolve on their own. Other treatment methods include: cryotherapy, topical alitretinoin (9-cis-retinoic acid), intralesional interferon-alpha or vinblastine, superficial radiotherapy, liposomal doxorubicin, daunorubicin or paclitaxel. Small lesions that are asymptomatic may be monitored.
This patient had no internal involvement and responded well to cryotherapy.
This case and photo were provided by Dr. Bilu Martin.

Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
and named the condition multiple benign pigmented hemorrhagic sarcoma. The disease emerged again at the onset of the AIDS epidemic among homosexual men. There are five variants: HIV/AIDS–related KS, classic KS, African cutaneous KS, African lymphadenopathic KS, and immunosuppression-associated KS (from immunosuppressive therapy or malignancies such as lymphoma).
KS is caused by human herpes virus type 8 (HHV-8). Patients with KS have an increased risk of developing other malignancies such as lymphomas, leukemia, and myeloma. This patient exhibited classic KS.
The various forms of KS may appear different clinically. The lesions may appear as erythematous macules, small violaceous papules, large plaques, or ulcerated nodules. In classic KS, violaceous to bluish-black macules evolve to papules or plaques. Lesions are generally asymptomatic. The most common locations are the toes and soles, although other areas may be affected. Any mucocutaneous surface can be involved. The most common areas of internal involvement are the gastrointestinal system and lymphatics.
Histology reveals angular vessels lined by atypical cells. An associated inflammatory infiltrate containing plasma cells may be present in the upper dermis and perivascular areas. Nodules and plaques reveal a spindle cell neoplasm pattern. Lesions will stain positive for HHV-8.
In patients with HIV/AIDS–related KS, highly active antiretroviral therapy is the most important and beneficial treatment. Since the introduction of HAART, the incidence of KS has greatly decreased. However, there are a proportion of HIV/AIDS–associated Kaposi’s sarcoma patients with well-controlled HIV and undetectable viral loads who require further treatment.
Lesions may spontaneously resolve on their own. Other treatment methods include: cryotherapy, topical alitretinoin (9-cis-retinoic acid), intralesional interferon-alpha or vinblastine, superficial radiotherapy, liposomal doxorubicin, daunorubicin or paclitaxel. Small lesions that are asymptomatic may be monitored.
This patient had no internal involvement and responded well to cryotherapy.
This case and photo were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
and named the condition multiple benign pigmented hemorrhagic sarcoma. The disease emerged again at the onset of the AIDS epidemic among homosexual men. There are five variants: HIV/AIDS–related KS, classic KS, African cutaneous KS, African lymphadenopathic KS, and immunosuppression-associated KS (from immunosuppressive therapy or malignancies such as lymphoma).
KS is caused by human herpes virus type 8 (HHV-8). Patients with KS have an increased risk of developing other malignancies such as lymphomas, leukemia, and myeloma. This patient exhibited classic KS.
The various forms of KS may appear different clinically. The lesions may appear as erythematous macules, small violaceous papules, large plaques, or ulcerated nodules. In classic KS, violaceous to bluish-black macules evolve to papules or plaques. Lesions are generally asymptomatic. The most common locations are the toes and soles, although other areas may be affected. Any mucocutaneous surface can be involved. The most common areas of internal involvement are the gastrointestinal system and lymphatics.
Histology reveals angular vessels lined by atypical cells. An associated inflammatory infiltrate containing plasma cells may be present in the upper dermis and perivascular areas. Nodules and plaques reveal a spindle cell neoplasm pattern. Lesions will stain positive for HHV-8.
In patients with HIV/AIDS–related KS, highly active antiretroviral therapy is the most important and beneficial treatment. Since the introduction of HAART, the incidence of KS has greatly decreased. However, there are a proportion of HIV/AIDS–associated Kaposi’s sarcoma patients with well-controlled HIV and undetectable viral loads who require further treatment.
Lesions may spontaneously resolve on their own. Other treatment methods include: cryotherapy, topical alitretinoin (9-cis-retinoic acid), intralesional interferon-alpha or vinblastine, superficial radiotherapy, liposomal doxorubicin, daunorubicin or paclitaxel. Small lesions that are asymptomatic may be monitored.
This patient had no internal involvement and responded well to cryotherapy.
This case and photo were provided by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
An 11-year-old female with a 3-year history of alopecia
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.

Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.

Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at [email protected].
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at [email protected].
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at [email protected].
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
An 11-year-old female is seen in clinic with a 3-year history of alopecia. The patient recently immigrated to the United States from Afghanistan. Prior to immigrating, she was evaluated for "scarring alopecia" and had been treated with oral and topical steroids as well as oral and topical antifungals. When active, she had itching and tenderness. She is not actively losing any hair at this time, but she has not regrown any of her hair. The patient has no family members with alopecia. She reports some burning pain and itching of her scalp, and denies any muscle pain or weakness or sun sensitivity.
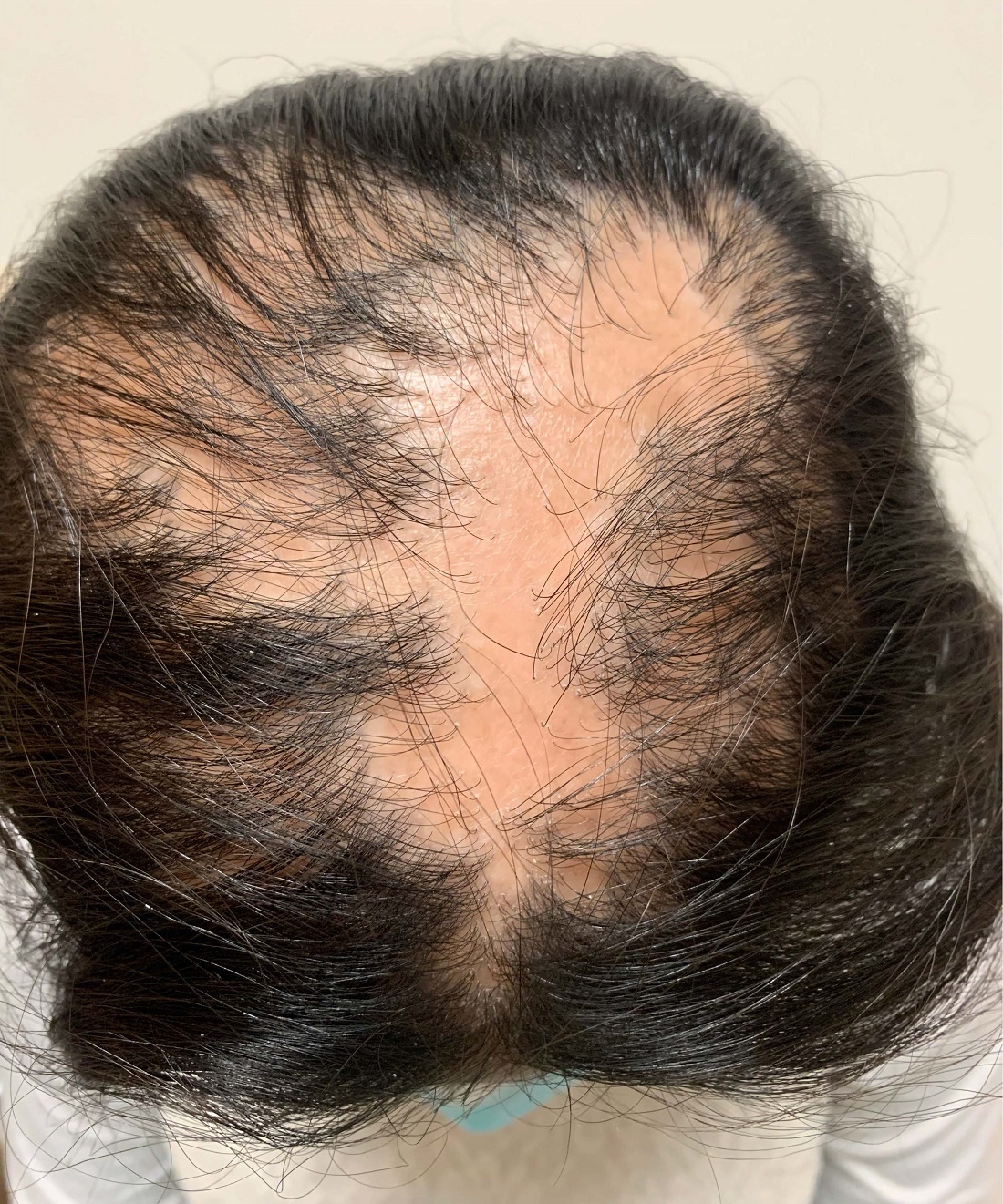
On physical exam, you see 50% loss of hair on the superior scalp with preservation of the anterior hair line. Patches of hair can be seen throughout, with segments of smooth-skinned alopecia, without pustules. There is a loss of the follicle pattern in scarred areas, and magnification or "dermoscopy" shows perifollicular erythema and scaling at the border of the affected scalp. Labs are all within normal limits. Bacterial and fungal cultures of the scalp do not grow organisms.
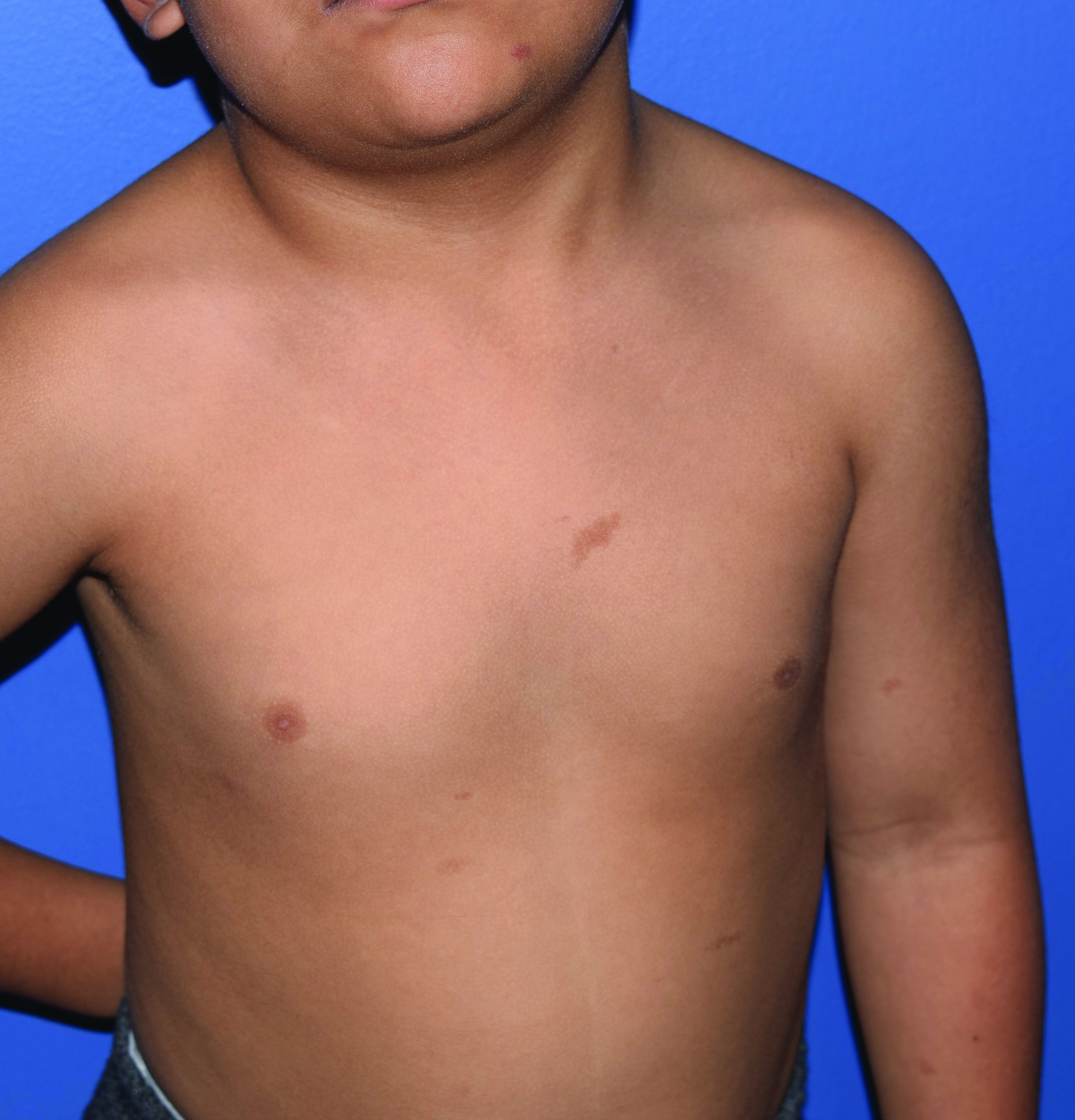
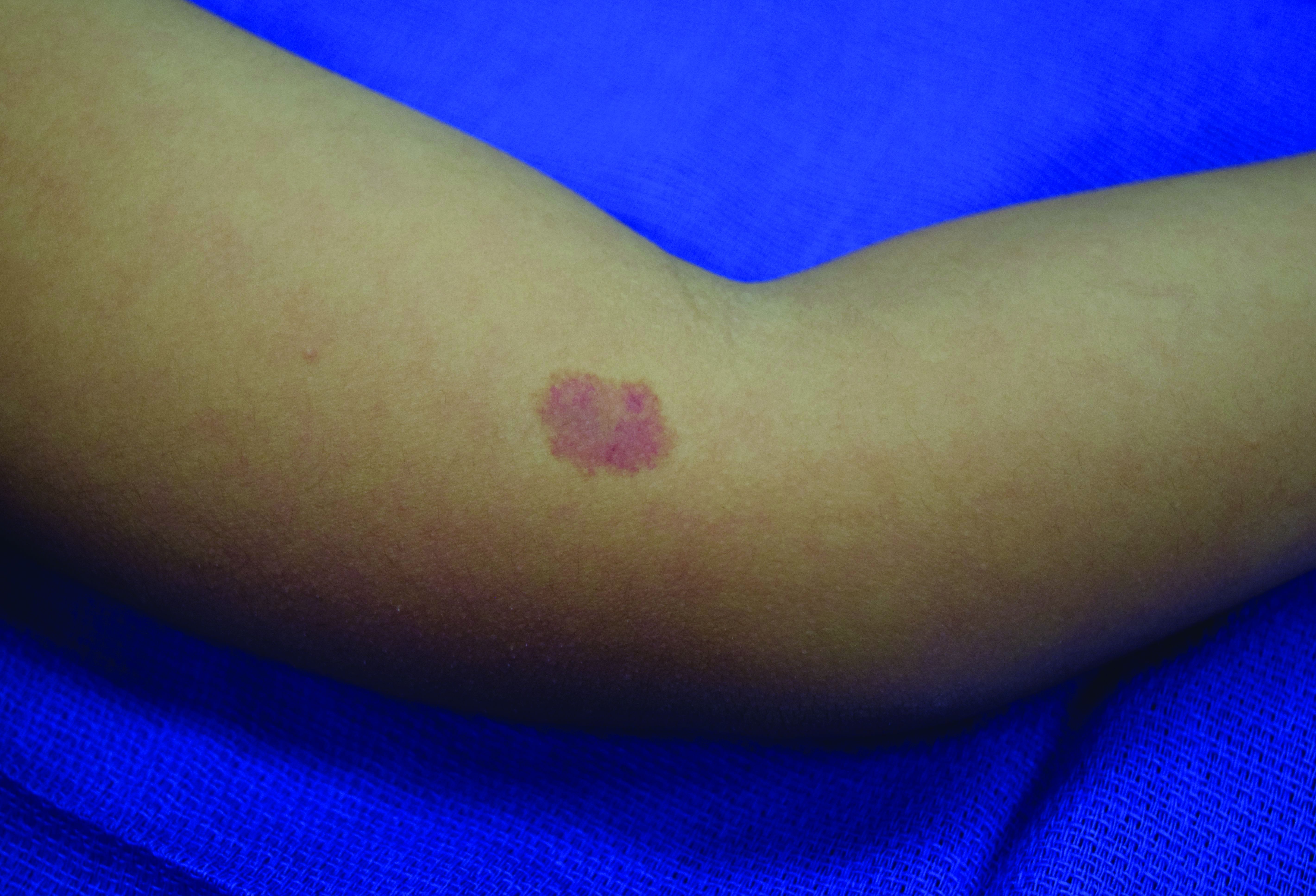
A 4-year-old presented to our pediatric dermatology clinic for evaluation of asymptomatic "brown spots."
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1

In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4

It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.

Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4
It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.
Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4
It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.
Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
A teen presents with a severe, tender rash on the extremities
“There’s rue for you, and here’s some for me; we may call it herb of grace o’ Sundays. O, you must wear your rue with a difference.”
— Ophelia in Hamlet by William Shakespeare
The patient was admitted to the hospital for IV fluids, pain control, and observation. The following day she admitted using the leaves of a plant on the trail as a bug repellent, as one time was taught by her grandfather. She rubbed some of the leaves on the brother as well. The grandfather shared some pictures of the bushes, and the plant was identified as Ruta graveolens.
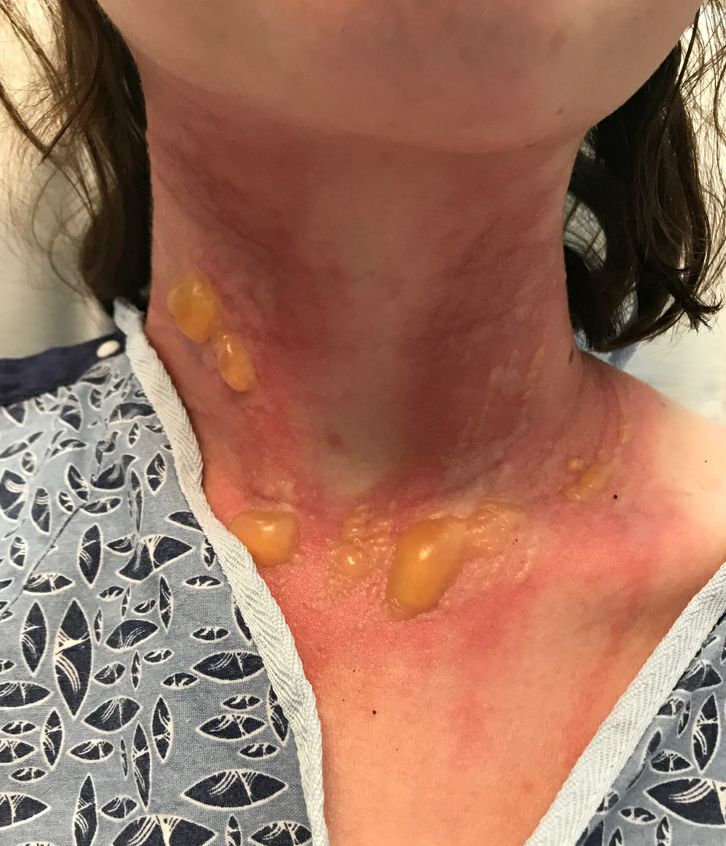
The blisters were deroofed, cleaned with saline, and wrapped with triamcinolone ointment and petrolatum. The patient was also started on a prednisone taper and received analgesics for the severe pain.
Ruta graveolens also known as common rue or herb of grace, is an ornamental plant from the Rutaceae family. This plant is also used as a medicinal herb, condiment, and as an insect repellent. If ingested in large doses, it can cause severe abdominal pain and vomiting. It also can be hepatotoxic.
The herb contains furocumarines, such as 8-methoxypsoralen and 5-methoxypsoralen and furoquinoline alkaloids. These chemicals when exposed to UVA radiation cause cell injury and inflammation of the skin. This is considered a phototoxic reaction of the skin, compared with allergic reactions, such as poison ivy dermatitis, which need a prior sensitization to the allergen for the T cells to be activated and cause injury in the skin. Other common plants and fruits that can cause phytophotodermatitis include citrus fruits, figs, carrots, celery, parsnips, parsley, and other wildflowers like hogweed.
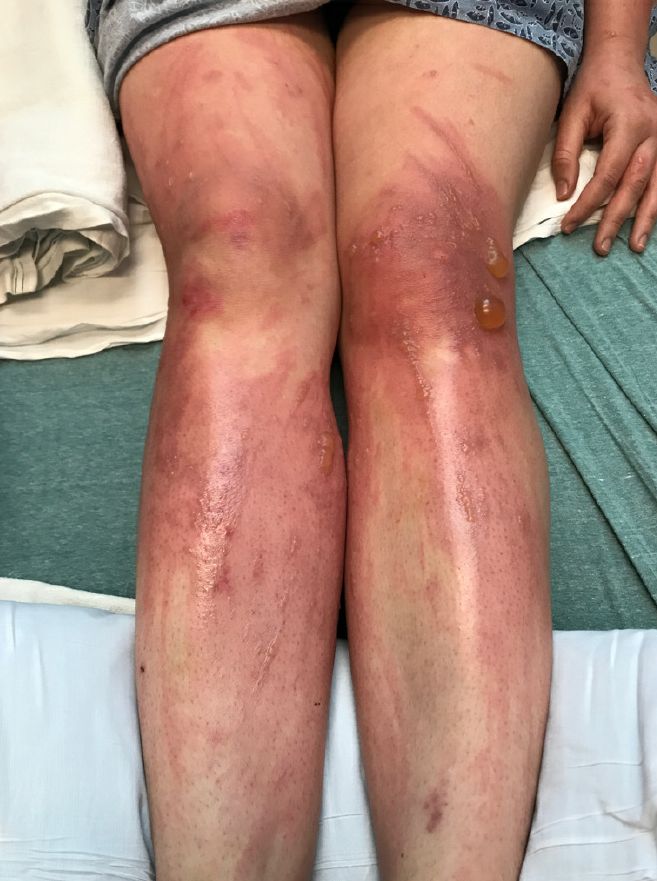
Depending on the degree of injury, the patients can be treated with topical corticosteroids, petrolatum wraps, and pain control. In severe cases like our patient, systemic prednisone may help stop the progression of the lesions and help with the inflammation. Skin hyperpigmentation after the initial injury may take months to clear, and some patient can develop scars.
The differential diagnosis should include severe bullous contact dermatitis like exposure to urushiol in poison ivy; second- and third-degree burns; severe medications reactions such Stevens-Johnson syndrome or toxic epidermal necrolysis, and inmunobullous diseases such as bullous lupus erythematosus, pemphigus vulgaris, or bullous pemphigoid. If there is no history of exposure or there are any other systemic symptoms, consider performing a skin biopsy of one of the lesions.
In this patient’s case, the history of exposure and skin findings helped the dermatologist on call make the right diagnosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
References
J Burn Care Res. 2018 Oct 23;39(6):1064-6.
Dermatitis. 2007 Mar;18(1):52-5.
BMJ Case Rep. 2015 Dec 23;2015:bcr2015213388.
“There’s rue for you, and here’s some for me; we may call it herb of grace o’ Sundays. O, you must wear your rue with a difference.”
— Ophelia in Hamlet by William Shakespeare
The patient was admitted to the hospital for IV fluids, pain control, and observation. The following day she admitted using the leaves of a plant on the trail as a bug repellent, as one time was taught by her grandfather. She rubbed some of the leaves on the brother as well. The grandfather shared some pictures of the bushes, and the plant was identified as Ruta graveolens.
The blisters were deroofed, cleaned with saline, and wrapped with triamcinolone ointment and petrolatum. The patient was also started on a prednisone taper and received analgesics for the severe pain.
Ruta graveolens also known as common rue or herb of grace, is an ornamental plant from the Rutaceae family. This plant is also used as a medicinal herb, condiment, and as an insect repellent. If ingested in large doses, it can cause severe abdominal pain and vomiting. It also can be hepatotoxic.
The herb contains furocumarines, such as 8-methoxypsoralen and 5-methoxypsoralen and furoquinoline alkaloids. These chemicals when exposed to UVA radiation cause cell injury and inflammation of the skin. This is considered a phototoxic reaction of the skin, compared with allergic reactions, such as poison ivy dermatitis, which need a prior sensitization to the allergen for the T cells to be activated and cause injury in the skin. Other common plants and fruits that can cause phytophotodermatitis include citrus fruits, figs, carrots, celery, parsnips, parsley, and other wildflowers like hogweed.
Depending on the degree of injury, the patients can be treated with topical corticosteroids, petrolatum wraps, and pain control. In severe cases like our patient, systemic prednisone may help stop the progression of the lesions and help with the inflammation. Skin hyperpigmentation after the initial injury may take months to clear, and some patient can develop scars.
The differential diagnosis should include severe bullous contact dermatitis like exposure to urushiol in poison ivy; second- and third-degree burns; severe medications reactions such Stevens-Johnson syndrome or toxic epidermal necrolysis, and inmunobullous diseases such as bullous lupus erythematosus, pemphigus vulgaris, or bullous pemphigoid. If there is no history of exposure or there are any other systemic symptoms, consider performing a skin biopsy of one of the lesions.
In this patient’s case, the history of exposure and skin findings helped the dermatologist on call make the right diagnosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
References
J Burn Care Res. 2018 Oct 23;39(6):1064-6.
Dermatitis. 2007 Mar;18(1):52-5.
BMJ Case Rep. 2015 Dec 23;2015:bcr2015213388.
“There’s rue for you, and here’s some for me; we may call it herb of grace o’ Sundays. O, you must wear your rue with a difference.”
— Ophelia in Hamlet by William Shakespeare
The patient was admitted to the hospital for IV fluids, pain control, and observation. The following day she admitted using the leaves of a plant on the trail as a bug repellent, as one time was taught by her grandfather. She rubbed some of the leaves on the brother as well. The grandfather shared some pictures of the bushes, and the plant was identified as Ruta graveolens.
The blisters were deroofed, cleaned with saline, and wrapped with triamcinolone ointment and petrolatum. The patient was also started on a prednisone taper and received analgesics for the severe pain.
Ruta graveolens also known as common rue or herb of grace, is an ornamental plant from the Rutaceae family. This plant is also used as a medicinal herb, condiment, and as an insect repellent. If ingested in large doses, it can cause severe abdominal pain and vomiting. It also can be hepatotoxic.
The herb contains furocumarines, such as 8-methoxypsoralen and 5-methoxypsoralen and furoquinoline alkaloids. These chemicals when exposed to UVA radiation cause cell injury and inflammation of the skin. This is considered a phototoxic reaction of the skin, compared with allergic reactions, such as poison ivy dermatitis, which need a prior sensitization to the allergen for the T cells to be activated and cause injury in the skin. Other common plants and fruits that can cause phytophotodermatitis include citrus fruits, figs, carrots, celery, parsnips, parsley, and other wildflowers like hogweed.
Depending on the degree of injury, the patients can be treated with topical corticosteroids, petrolatum wraps, and pain control. In severe cases like our patient, systemic prednisone may help stop the progression of the lesions and help with the inflammation. Skin hyperpigmentation after the initial injury may take months to clear, and some patient can develop scars.
The differential diagnosis should include severe bullous contact dermatitis like exposure to urushiol in poison ivy; second- and third-degree burns; severe medications reactions such Stevens-Johnson syndrome or toxic epidermal necrolysis, and inmunobullous diseases such as bullous lupus erythematosus, pemphigus vulgaris, or bullous pemphigoid. If there is no history of exposure or there are any other systemic symptoms, consider performing a skin biopsy of one of the lesions.
In this patient’s case, the history of exposure and skin findings helped the dermatologist on call make the right diagnosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at [email protected].
References
J Burn Care Res. 2018 Oct 23;39(6):1064-6.
Dermatitis. 2007 Mar;18(1):52-5.
BMJ Case Rep. 2015 Dec 23;2015:bcr2015213388.
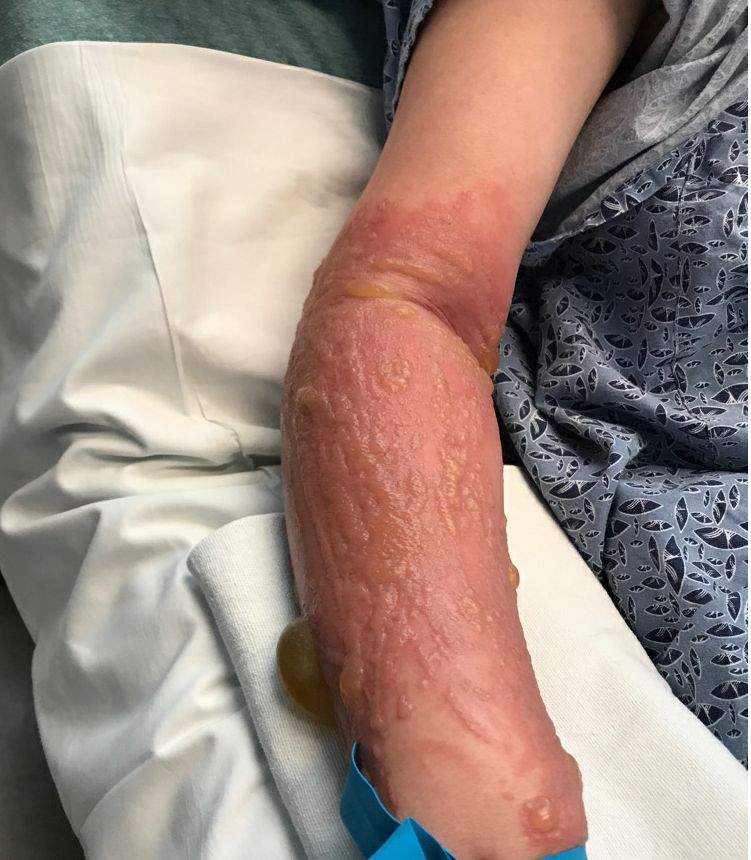
She started taking lithium for depression and anxiety 3 weeks prior to her developing the rash. She denies taking any other medications, supplements, or recreational drugs.
She denied any prior history of photosensitivity, no history of mouth ulcers, joint pain, muscle weakness, hair loss, or any other symptoms.
Besides her brother, there are no other affected family members, and no history of immune bullous disorders or other skin conditions.
On physical exam, the girl appears in a lot of pain and is uncomfortable. The skin is red and hot, and there are tense bullae on the neck, arms, and legs. There are no ocular or mucosal lesions.
A 31-year-old with a 3-week history of a waxing and waning, mildly pruritic eruption on his neck, chest, and back
Prurigo pigmentosa is an inflammatory disorder of uncertain etiology characterized by the eruption of erythematous, markedly pruritic, urticaria-like papules and vesicles on the posterior neck, mid- to upper back, and chest. Crops of papules appear rapidly and then involute within days, leaving behind postinflammatory hyperpigmentation in a netlike configuration. New papules may appear prior to resolution of hyperpigmented macules, resulting in a mixed presentation of erythematous papules overlying reticulated hyperpigmentation.1

The condition was initially described in Japanese individuals, and to date, most cases have occurred in this population.2 However, the incidence of prurigo pigmentosa is increasing worldwide, including in the United States, which has led to the identification of several metabolic risk factors including diabetes mellitus, fasting, and dieting, with the common etiologic endpoint of ketosis.3With the increasing popularity of diets with strict carbohydrate limits, often with the goal of ketosis, dermatologists should be aware of the clinical appearance and common history of this rash to facilitate prompt diagnosis and treatment.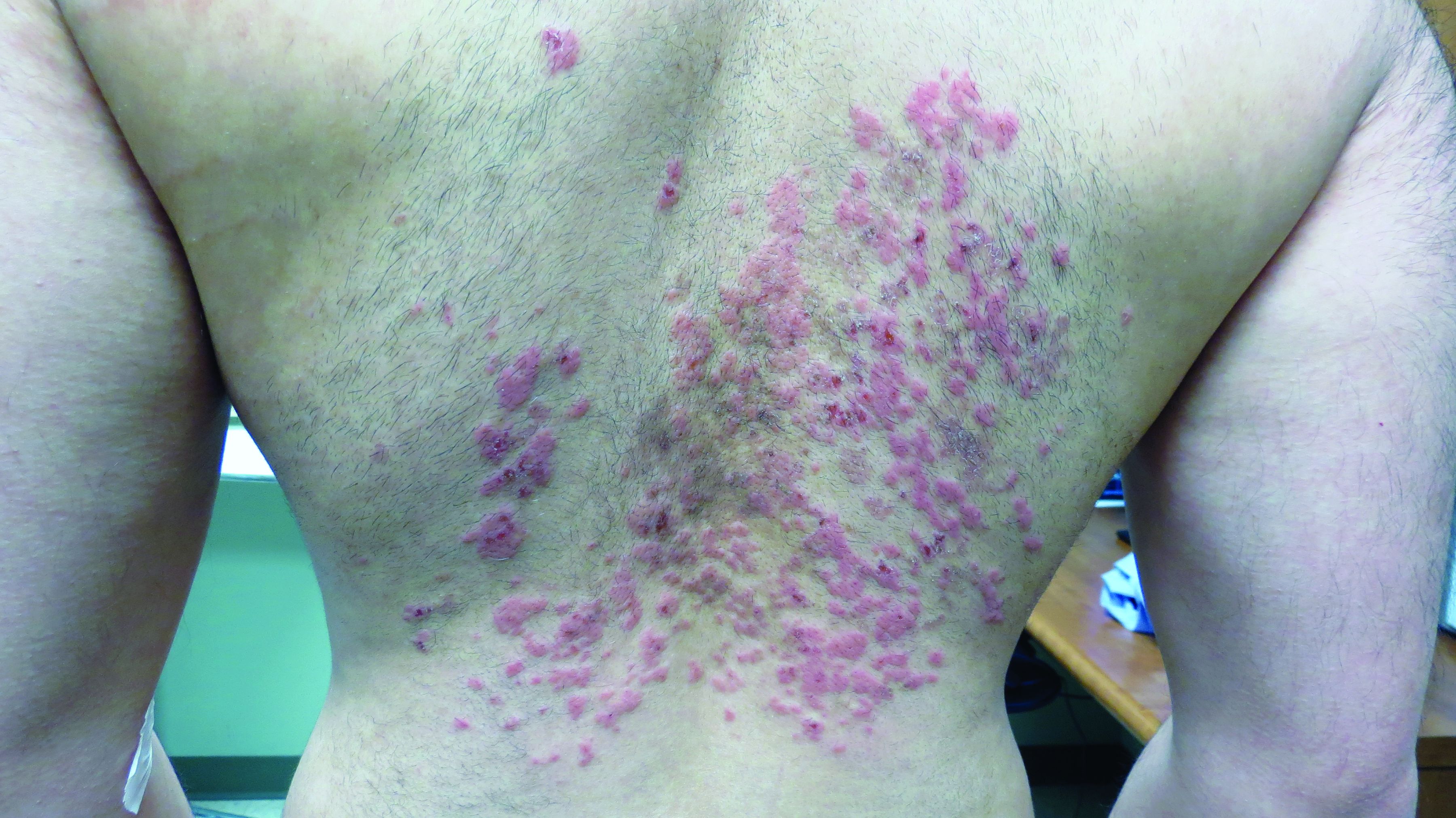
Clinical exam with appropriate history is usually sufficient for diagnosis. However, biopsy with histopathologic analysis can be utilized to confirm atypical cases. Histopathologic findings depend on the stage of the lesion biopsied. The earliest finding is a shallow perivascular neutrophilic infiltrate, neutrophil exocytosis, and epidermal and superficial dermal edema. As lesions progress, the prominent findings include epidermal vesiculation with necrotic keratinocytes and a lichenoid infiltrate dominated by lymphocytes and eosinophils. In the final stages, lesions demonstrate variable parakeratosis and acanthosis, as well as prominent dermal melanophagia.1
Treatment of prurigo pigmentosa includes modification of the patient’s underlying health issues to avoid ketosis, and in the case of diet-induced ketosis, reinstitution of a more balanced diet with sufficient carbohydrates. In the case of the patient presented here, rash resolved 1 week following instruction to include more carbohydrates in his diet. For recalcitrant cases or those without a clear precipitating factor, the addition of oral antibiotics is often helpful. Tetracyclines or dapsone are typically employed, usually in courses of 1-2 months.3,4
Dr. Johnson is a PGY-4 dermatology resident at Carilion Clinic in Roanoke, Va. He provided the case and photos. Donna Bilu Martin, MD, is the editor of the column.
References
1. Boer A et al. Am J Dermatopathol. 2003 Apr;25(2):117-292.
2. Satter E et al. J Cutan Pathol. 2016 Oct;43(10):809-14.
3. Alshaya M et al. JAAD Case Rep. 2019 Jun 8;5(6):504-7.
4. Hartman M et al. Cutis. 2019 Mar;103(3):E10-3.
Prurigo pigmentosa is an inflammatory disorder of uncertain etiology characterized by the eruption of erythematous, markedly pruritic, urticaria-like papules and vesicles on the posterior neck, mid- to upper back, and chest. Crops of papules appear rapidly and then involute within days, leaving behind postinflammatory hyperpigmentation in a netlike configuration. New papules may appear prior to resolution of hyperpigmented macules, resulting in a mixed presentation of erythematous papules overlying reticulated hyperpigmentation.1
The condition was initially described in Japanese individuals, and to date, most cases have occurred in this population.2 However, the incidence of prurigo pigmentosa is increasing worldwide, including in the United States, which has led to the identification of several metabolic risk factors including diabetes mellitus, fasting, and dieting, with the common etiologic endpoint of ketosis.3With the increasing popularity of diets with strict carbohydrate limits, often with the goal of ketosis, dermatologists should be aware of the clinical appearance and common history of this rash to facilitate prompt diagnosis and treatment.
Clinical exam with appropriate history is usually sufficient for diagnosis. However, biopsy with histopathologic analysis can be utilized to confirm atypical cases. Histopathologic findings depend on the stage of the lesion biopsied. The earliest finding is a shallow perivascular neutrophilic infiltrate, neutrophil exocytosis, and epidermal and superficial dermal edema. As lesions progress, the prominent findings include epidermal vesiculation with necrotic keratinocytes and a lichenoid infiltrate dominated by lymphocytes and eosinophils. In the final stages, lesions demonstrate variable parakeratosis and acanthosis, as well as prominent dermal melanophagia.1
Treatment of prurigo pigmentosa includes modification of the patient’s underlying health issues to avoid ketosis, and in the case of diet-induced ketosis, reinstitution of a more balanced diet with sufficient carbohydrates. In the case of the patient presented here, rash resolved 1 week following instruction to include more carbohydrates in his diet. For recalcitrant cases or those without a clear precipitating factor, the addition of oral antibiotics is often helpful. Tetracyclines or dapsone are typically employed, usually in courses of 1-2 months.3,4
Dr. Johnson is a PGY-4 dermatology resident at Carilion Clinic in Roanoke, Va. He provided the case and photos. Donna Bilu Martin, MD, is the editor of the column.
References
1. Boer A et al. Am J Dermatopathol. 2003 Apr;25(2):117-292.
2. Satter E et al. J Cutan Pathol. 2016 Oct;43(10):809-14.
3. Alshaya M et al. JAAD Case Rep. 2019 Jun 8;5(6):504-7.
4. Hartman M et al. Cutis. 2019 Mar;103(3):E10-3.
Prurigo pigmentosa is an inflammatory disorder of uncertain etiology characterized by the eruption of erythematous, markedly pruritic, urticaria-like papules and vesicles on the posterior neck, mid- to upper back, and chest. Crops of papules appear rapidly and then involute within days, leaving behind postinflammatory hyperpigmentation in a netlike configuration. New papules may appear prior to resolution of hyperpigmented macules, resulting in a mixed presentation of erythematous papules overlying reticulated hyperpigmentation.1
The condition was initially described in Japanese individuals, and to date, most cases have occurred in this population.2 However, the incidence of prurigo pigmentosa is increasing worldwide, including in the United States, which has led to the identification of several metabolic risk factors including diabetes mellitus, fasting, and dieting, with the common etiologic endpoint of ketosis.3With the increasing popularity of diets with strict carbohydrate limits, often with the goal of ketosis, dermatologists should be aware of the clinical appearance and common history of this rash to facilitate prompt diagnosis and treatment.
Clinical exam with appropriate history is usually sufficient for diagnosis. However, biopsy with histopathologic analysis can be utilized to confirm atypical cases. Histopathologic findings depend on the stage of the lesion biopsied. The earliest finding is a shallow perivascular neutrophilic infiltrate, neutrophil exocytosis, and epidermal and superficial dermal edema. As lesions progress, the prominent findings include epidermal vesiculation with necrotic keratinocytes and a lichenoid infiltrate dominated by lymphocytes and eosinophils. In the final stages, lesions demonstrate variable parakeratosis and acanthosis, as well as prominent dermal melanophagia.1
Treatment of prurigo pigmentosa includes modification of the patient’s underlying health issues to avoid ketosis, and in the case of diet-induced ketosis, reinstitution of a more balanced diet with sufficient carbohydrates. In the case of the patient presented here, rash resolved 1 week following instruction to include more carbohydrates in his diet. For recalcitrant cases or those without a clear precipitating factor, the addition of oral antibiotics is often helpful. Tetracyclines or dapsone are typically employed, usually in courses of 1-2 months.3,4
Dr. Johnson is a PGY-4 dermatology resident at Carilion Clinic in Roanoke, Va. He provided the case and photos. Donna Bilu Martin, MD, is the editor of the column.
References
1. Boer A et al. Am J Dermatopathol. 2003 Apr;25(2):117-292.
2. Satter E et al. J Cutan Pathol. 2016 Oct;43(10):809-14.
3. Alshaya M et al. JAAD Case Rep. 2019 Jun 8;5(6):504-7.
4. Hartman M et al. Cutis. 2019 Mar;103(3):E10-3.
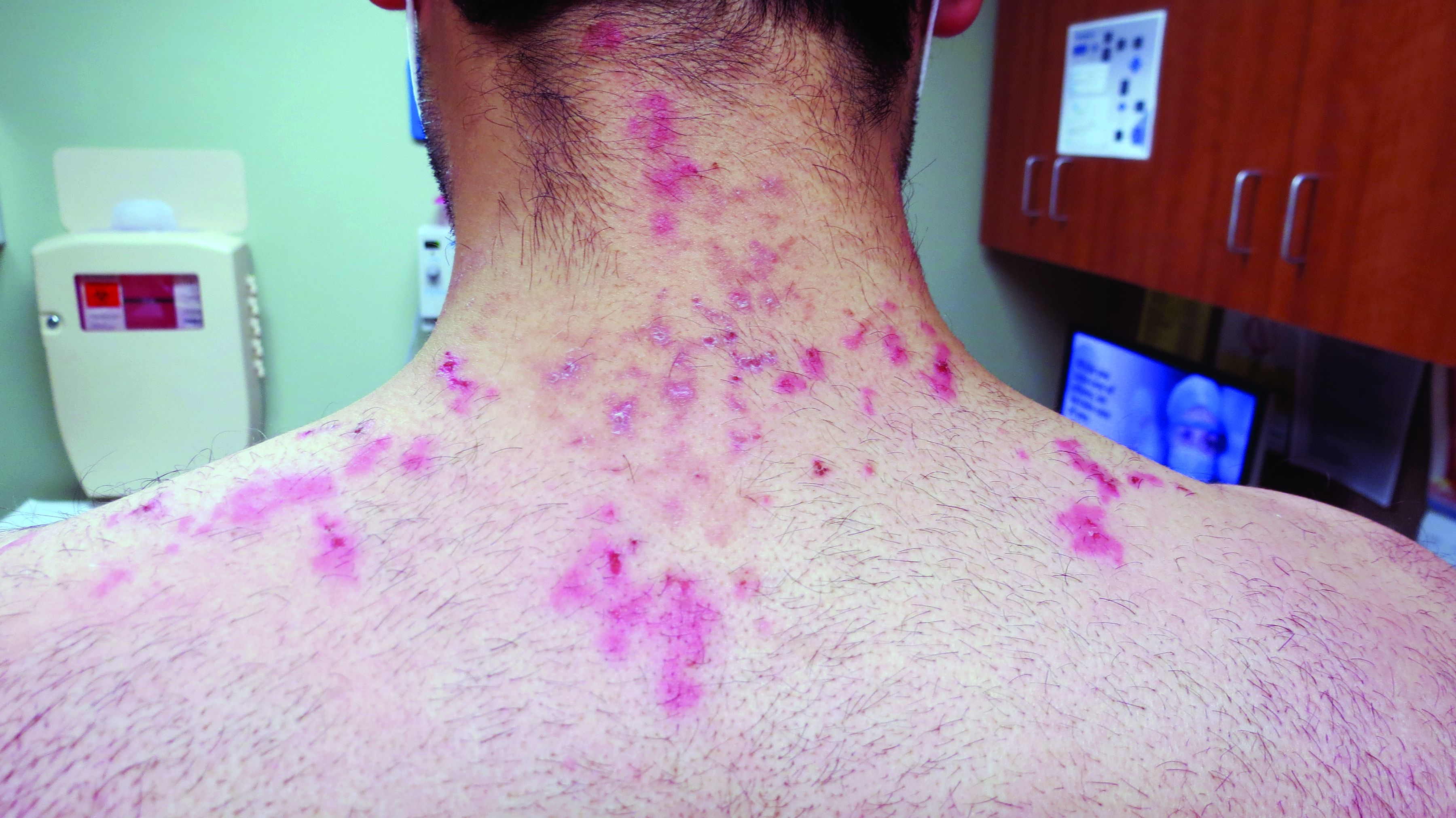
A teen girl presents with a pinkish-red bump on her right leg
This atypical lesion might warrant a biopsy. However, upon closer examination, you can appreciate a small papule with a whitish center, at the inferior margin of the tumor (6 o’clock), and another flat-topped papule with a white center several centimeters inferior-lateral to the lesion, both consistent with molluscum lesions. Therefore, the tumor is consistent with a giant molluscum contagiosum.
Molluscum contagiosum is a cutaneous viral infection caused by the poxvirus, which commonly affects children. It can spread easily by direct physical contact, fomites, and autoinoculation.1 It usually presents with skin-colored or pink pearly dome-shaped papules with central umbilication that can occur anywhere on the face or body. The skin lesions can be asymptomatic or pruritic. When the size of the molluscum is 0.5 cm or more in diameter, it is considered a giant molluscum. Atypical size and appearance may be seen in patients with altered or impaired immunity such as those with HIV.2,3 Giant molluscum has been reported in immunocompetent patients as well.4,5
The diagnosis of molluscum contagiosum usually is made clinically. Our patient had typically appearing molluscum lesions approximate to the larger lesion of concern. She was overall healthy without any history of impaired immunity so no further work-up was pursued. However, a biopsy of the skin lesion may be considered if the diagnosis is unclear.
What’s the treatment plan?
Treatment may not be necessary for molluscum contagiosum because it is often self-limited in immunocompetent children, although it can take many months to years to resolve. Treatment may be considered to reduce autoinoculation or risk of transmission because of close contact to others, to alleviate discomfort, including itching, to reduce cosmetic concerns and to prevent secondary infection.6

The most common treatments for molluscum contagiosum are cantharidin or cryotherapy. Other treatment available include topical retinoids, immunomodulators such as cimetidine, or antivirals such as cidofovir.1 Lesions with or without treatment may exhibit the BOTE (beginning of the end) sign, which is an apparent worsening associated with the body’s immune response to the molluscum virus and generally indicates imminent resolution.
What’s the differential diagnosis?
The differential diagnosis for giant molluscum contagiosum includes epidermal inclusion cyst, skin tag, pilomatrixoma, and amelanotic melanoma.
Epidermal inclusion cyst typically presents as a firm, mobile nodule under the skin with central punctum, which can enlarge and become inflamed. It can be painful, especially when infected. Definitive treatment is surgical excision because it rarely resolves spontaneously.
Skin tags, also known as acrochordons, are benign skin-colored papules most often found in the skin folds. People with obesity and type 2 diabetes are at higher risk for skin tags. Skin tags may be treated with cryotherapy, surgical excision, or ligation.
Pilomatrixoma is a benign skin tumor derived from hair matrix cells. It is usually a nontender, firm, skin-colored or red-purple subcutaneous nodule that may have calcifications. Treatment is surgical excision.
Amelanotic melanoma is a melanoma with little or no pigment and can present as a skin- or red-colored nodule. While these are quite uncommon, recognition that many pediatric melanomas present as amelanotic lesions makes it important to consider this in the differential diagnosis of growing papules and nodules.7 Treatment and prognosis is similar to that of pigmented melanoma, but as it is often clinically challenging to diagnose because of atypical features, it may be detected in more advanced stages.
Our patient underwent cryotherapy with liquid nitrogen to the nodule given the large size of the lesion, with resolution without recurrence.
Dr. Lee is a pediatric dermatology research fellow in the division of pediatric and adolescent dermatology at the University of California, San Diego and Rady Children’s Hospital–San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Neither Dr. Lee nor Dr. Eichenfield had any relevant financial disclosures. Email them at [email protected].
References
1. Recent Pat Inflamm Allergy Drug Discov. 2017. doi: 10.2174/1872213X11666170518114456.
2. J Epidemiol Glob Health. 2013 Dec. doi: 10.1016/j.jegh.2013.06.002.
3. Trop Doct. 2015 Apr. doi: 10.1177/0049475514568133.
4. J Pak Med Assoc. 2013 Jun;63(6):778-9.
5. Dermatol Pract Concept. 2016 Jul. doi: 10.5826/dpc.0603a15.
6 Molluscum Contagiosum, in “Red Book: 2018 Report of the Committee on Infectious Diseases,” 31st ed. (Itasca, Ill.: American Academy of Pediatrics, 2018, pp. 565-66).
7. J Am Acad Dermatol. 2013 Jun. doi: 10.1016/j.jaad.2012.12.953.
This atypical lesion might warrant a biopsy. However, upon closer examination, you can appreciate a small papule with a whitish center, at the inferior margin of the tumor (6 o’clock), and another flat-topped papule with a white center several centimeters inferior-lateral to the lesion, both consistent with molluscum lesions. Therefore, the tumor is consistent with a giant molluscum contagiosum.
Molluscum contagiosum is a cutaneous viral infection caused by the poxvirus, which commonly affects children. It can spread easily by direct physical contact, fomites, and autoinoculation.1 It usually presents with skin-colored or pink pearly dome-shaped papules with central umbilication that can occur anywhere on the face or body. The skin lesions can be asymptomatic or pruritic. When the size of the molluscum is 0.5 cm or more in diameter, it is considered a giant molluscum. Atypical size and appearance may be seen in patients with altered or impaired immunity such as those with HIV.2,3 Giant molluscum has been reported in immunocompetent patients as well.4,5
The diagnosis of molluscum contagiosum usually is made clinically. Our patient had typically appearing molluscum lesions approximate to the larger lesion of concern. She was overall healthy without any history of impaired immunity so no further work-up was pursued. However, a biopsy of the skin lesion may be considered if the diagnosis is unclear.
What’s the treatment plan?
Treatment may not be necessary for molluscum contagiosum because it is often self-limited in immunocompetent children, although it can take many months to years to resolve. Treatment may be considered to reduce autoinoculation or risk of transmission because of close contact to others, to alleviate discomfort, including itching, to reduce cosmetic concerns and to prevent secondary infection.6
The most common treatments for molluscum contagiosum are cantharidin or cryotherapy. Other treatment available include topical retinoids, immunomodulators such as cimetidine, or antivirals such as cidofovir.1 Lesions with or without treatment may exhibit the BOTE (beginning of the end) sign, which is an apparent worsening associated with the body’s immune response to the molluscum virus and generally indicates imminent resolution.
What’s the differential diagnosis?
The differential diagnosis for giant molluscum contagiosum includes epidermal inclusion cyst, skin tag, pilomatrixoma, and amelanotic melanoma.
Epidermal inclusion cyst typically presents as a firm, mobile nodule under the skin with central punctum, which can enlarge and become inflamed. It can be painful, especially when infected. Definitive treatment is surgical excision because it rarely resolves spontaneously.
Skin tags, also known as acrochordons, are benign skin-colored papules most often found in the skin folds. People with obesity and type 2 diabetes are at higher risk for skin tags. Skin tags may be treated with cryotherapy, surgical excision, or ligation.
Pilomatrixoma is a benign skin tumor derived from hair matrix cells. It is usually a nontender, firm, skin-colored or red-purple subcutaneous nodule that may have calcifications. Treatment is surgical excision.
Amelanotic melanoma is a melanoma with little or no pigment and can present as a skin- or red-colored nodule. While these are quite uncommon, recognition that many pediatric melanomas present as amelanotic lesions makes it important to consider this in the differential diagnosis of growing papules and nodules.7 Treatment and prognosis is similar to that of pigmented melanoma, but as it is often clinically challenging to diagnose because of atypical features, it may be detected in more advanced stages.
Our patient underwent cryotherapy with liquid nitrogen to the nodule given the large size of the lesion, with resolution without recurrence.
Dr. Lee is a pediatric dermatology research fellow in the division of pediatric and adolescent dermatology at the University of California, San Diego and Rady Children’s Hospital–San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Neither Dr. Lee nor Dr. Eichenfield had any relevant financial disclosures. Email them at [email protected].
References
1. Recent Pat Inflamm Allergy Drug Discov. 2017. doi: 10.2174/1872213X11666170518114456.
2. J Epidemiol Glob Health. 2013 Dec. doi: 10.1016/j.jegh.2013.06.002.
3. Trop Doct. 2015 Apr. doi: 10.1177/0049475514568133.
4. J Pak Med Assoc. 2013 Jun;63(6):778-9.
5. Dermatol Pract Concept. 2016 Jul. doi: 10.5826/dpc.0603a15.
6 Molluscum Contagiosum, in “Red Book: 2018 Report of the Committee on Infectious Diseases,” 31st ed. (Itasca, Ill.: American Academy of Pediatrics, 2018, pp. 565-66).
7. J Am Acad Dermatol. 2013 Jun. doi: 10.1016/j.jaad.2012.12.953.
This atypical lesion might warrant a biopsy. However, upon closer examination, you can appreciate a small papule with a whitish center, at the inferior margin of the tumor (6 o’clock), and another flat-topped papule with a white center several centimeters inferior-lateral to the lesion, both consistent with molluscum lesions. Therefore, the tumor is consistent with a giant molluscum contagiosum.
Molluscum contagiosum is a cutaneous viral infection caused by the poxvirus, which commonly affects children. It can spread easily by direct physical contact, fomites, and autoinoculation.1 It usually presents with skin-colored or pink pearly dome-shaped papules with central umbilication that can occur anywhere on the face or body. The skin lesions can be asymptomatic or pruritic. When the size of the molluscum is 0.5 cm or more in diameter, it is considered a giant molluscum. Atypical size and appearance may be seen in patients with altered or impaired immunity such as those with HIV.2,3 Giant molluscum has been reported in immunocompetent patients as well.4,5
The diagnosis of molluscum contagiosum usually is made clinically. Our patient had typically appearing molluscum lesions approximate to the larger lesion of concern. She was overall healthy without any history of impaired immunity so no further work-up was pursued. However, a biopsy of the skin lesion may be considered if the diagnosis is unclear.
What’s the treatment plan?
Treatment may not be necessary for molluscum contagiosum because it is often self-limited in immunocompetent children, although it can take many months to years to resolve. Treatment may be considered to reduce autoinoculation or risk of transmission because of close contact to others, to alleviate discomfort, including itching, to reduce cosmetic concerns and to prevent secondary infection.6
The most common treatments for molluscum contagiosum are cantharidin or cryotherapy. Other treatment available include topical retinoids, immunomodulators such as cimetidine, or antivirals such as cidofovir.1 Lesions with or without treatment may exhibit the BOTE (beginning of the end) sign, which is an apparent worsening associated with the body’s immune response to the molluscum virus and generally indicates imminent resolution.
What’s the differential diagnosis?
The differential diagnosis for giant molluscum contagiosum includes epidermal inclusion cyst, skin tag, pilomatrixoma, and amelanotic melanoma.
Epidermal inclusion cyst typically presents as a firm, mobile nodule under the skin with central punctum, which can enlarge and become inflamed. It can be painful, especially when infected. Definitive treatment is surgical excision because it rarely resolves spontaneously.
Skin tags, also known as acrochordons, are benign skin-colored papules most often found in the skin folds. People with obesity and type 2 diabetes are at higher risk for skin tags. Skin tags may be treated with cryotherapy, surgical excision, or ligation.
Pilomatrixoma is a benign skin tumor derived from hair matrix cells. It is usually a nontender, firm, skin-colored or red-purple subcutaneous nodule that may have calcifications. Treatment is surgical excision.
Amelanotic melanoma is a melanoma with little or no pigment and can present as a skin- or red-colored nodule. While these are quite uncommon, recognition that many pediatric melanomas present as amelanotic lesions makes it important to consider this in the differential diagnosis of growing papules and nodules.7 Treatment and prognosis is similar to that of pigmented melanoma, but as it is often clinically challenging to diagnose because of atypical features, it may be detected in more advanced stages.
Our patient underwent cryotherapy with liquid nitrogen to the nodule given the large size of the lesion, with resolution without recurrence.
Dr. Lee is a pediatric dermatology research fellow in the division of pediatric and adolescent dermatology at the University of California, San Diego and Rady Children’s Hospital–San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Neither Dr. Lee nor Dr. Eichenfield had any relevant financial disclosures. Email them at [email protected].
References
1. Recent Pat Inflamm Allergy Drug Discov. 2017. doi: 10.2174/1872213X11666170518114456.
2. J Epidemiol Glob Health. 2013 Dec. doi: 10.1016/j.jegh.2013.06.002.
3. Trop Doct. 2015 Apr. doi: 10.1177/0049475514568133.
4. J Pak Med Assoc. 2013 Jun;63(6):778-9.
5. Dermatol Pract Concept. 2016 Jul. doi: 10.5826/dpc.0603a15.
6 Molluscum Contagiosum, in “Red Book: 2018 Report of the Committee on Infectious Diseases,” 31st ed. (Itasca, Ill.: American Academy of Pediatrics, 2018, pp. 565-66).
7. J Am Acad Dermatol. 2013 Jun. doi: 10.1016/j.jaad.2012.12.953.
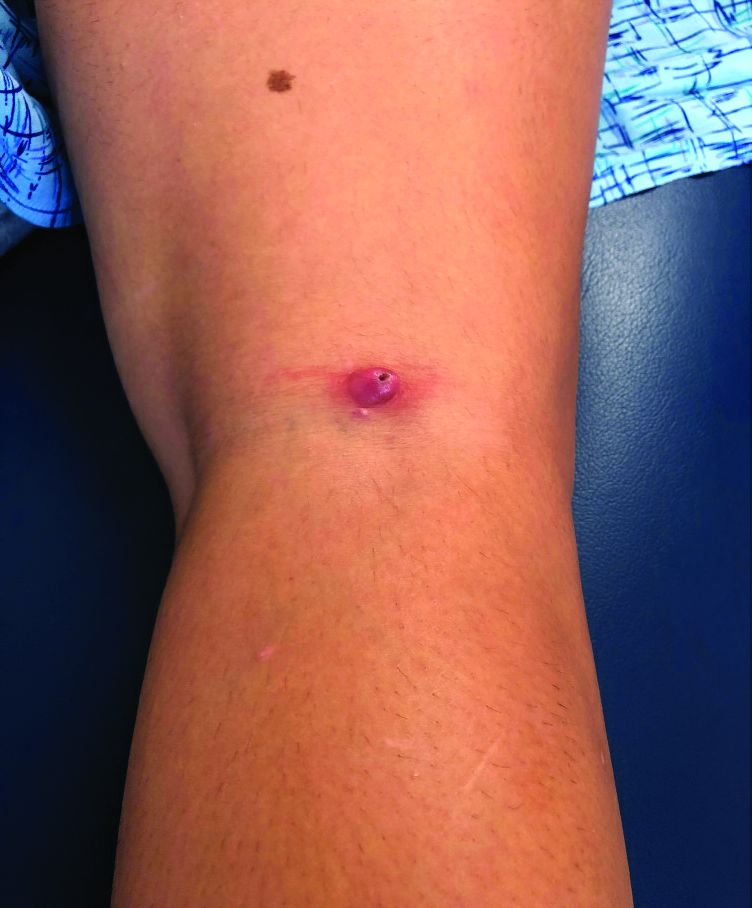
A woman with an asymptomatic eruption on her palms after exposure to water
This eruption can be accompanied by a mild burning or tingling sensation, which will subside with the rest of the symptoms in minutes to hours after drying.1
AWP is most frequently associated with cystic fibrosis (CF).2 It can be observed in up to 80% of CF patients and is considered a clinical sign of the disease. AWP can be present in CF carriers to a lesser extent,2,4 and has also been associated with focal hyperhidrosis, atopic dermatitis, Raynaud phenomenon, and COX-2 inhibitor use.5
While a definitive cause is unknown, it is thought that AWP is caused by dysregulation of sweat glands in the palms through increased expression of aquaporin, a protein crucial in the transport of water between cells.3
AWP is quite rare and benign in nature. However, because of its strong association with CF, genetic screening should be considered in asymptomatic patients. Our patient had been screened in the past and is not a CF carrier. Often, the itching or burning associated with CF is mild and easily controlled. The patient was placed on low dose isotretinoin for treatment of her acne. Interestingly, the patient claimed her eruption no longer appeared after starting isotretinoin therapy. To our knowledge, this is the first reported case of AWP resolving with isotretinoin use.
This case and photo were submitted by Mr. Birk, University of Texas, Austin, Texas; and Dr. Mamelak, Sanova Dermatology, in Austin. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at MDedge.com/Dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Katz M, Ramot Y. CMAJ. 2015 Dec 8;187(18):E515.
2. Tolland JP et al. Dermatology. 2010;221(4):326-30.
3. Kabashima K et al. J Am Acad Dermatol. 2008 Aug;59(2 Suppl 1):S28-32.
4. Gild R et al. Br J Dermatol. 2010 Nov;163(5):1082-4.
5. Glatz M and Muellegger RR. BMJ Case Rep. 2014. doi: 10.1136/bcr-2014-203929.
This eruption can be accompanied by a mild burning or tingling sensation, which will subside with the rest of the symptoms in minutes to hours after drying.1
AWP is most frequently associated with cystic fibrosis (CF).2 It can be observed in up to 80% of CF patients and is considered a clinical sign of the disease. AWP can be present in CF carriers to a lesser extent,2,4 and has also been associated with focal hyperhidrosis, atopic dermatitis, Raynaud phenomenon, and COX-2 inhibitor use.5
While a definitive cause is unknown, it is thought that AWP is caused by dysregulation of sweat glands in the palms through increased expression of aquaporin, a protein crucial in the transport of water between cells.3
AWP is quite rare and benign in nature. However, because of its strong association with CF, genetic screening should be considered in asymptomatic patients. Our patient had been screened in the past and is not a CF carrier. Often, the itching or burning associated with CF is mild and easily controlled. The patient was placed on low dose isotretinoin for treatment of her acne. Interestingly, the patient claimed her eruption no longer appeared after starting isotretinoin therapy. To our knowledge, this is the first reported case of AWP resolving with isotretinoin use.
This case and photo were submitted by Mr. Birk, University of Texas, Austin, Texas; and Dr. Mamelak, Sanova Dermatology, in Austin. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at MDedge.com/Dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Katz M, Ramot Y. CMAJ. 2015 Dec 8;187(18):E515.
2. Tolland JP et al. Dermatology. 2010;221(4):326-30.
3. Kabashima K et al. J Am Acad Dermatol. 2008 Aug;59(2 Suppl 1):S28-32.
4. Gild R et al. Br J Dermatol. 2010 Nov;163(5):1082-4.
5. Glatz M and Muellegger RR. BMJ Case Rep. 2014. doi: 10.1136/bcr-2014-203929.
This eruption can be accompanied by a mild burning or tingling sensation, which will subside with the rest of the symptoms in minutes to hours after drying.1
AWP is most frequently associated with cystic fibrosis (CF).2 It can be observed in up to 80% of CF patients and is considered a clinical sign of the disease. AWP can be present in CF carriers to a lesser extent,2,4 and has also been associated with focal hyperhidrosis, atopic dermatitis, Raynaud phenomenon, and COX-2 inhibitor use.5
While a definitive cause is unknown, it is thought that AWP is caused by dysregulation of sweat glands in the palms through increased expression of aquaporin, a protein crucial in the transport of water between cells.3
AWP is quite rare and benign in nature. However, because of its strong association with CF, genetic screening should be considered in asymptomatic patients. Our patient had been screened in the past and is not a CF carrier. Often, the itching or burning associated with CF is mild and easily controlled. The patient was placed on low dose isotretinoin for treatment of her acne. Interestingly, the patient claimed her eruption no longer appeared after starting isotretinoin therapy. To our knowledge, this is the first reported case of AWP resolving with isotretinoin use.
This case and photo were submitted by Mr. Birk, University of Texas, Austin, Texas; and Dr. Mamelak, Sanova Dermatology, in Austin. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at MDedge.com/Dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Katz M, Ramot Y. CMAJ. 2015 Dec 8;187(18):E515.
2. Tolland JP et al. Dermatology. 2010;221(4):326-30.
3. Kabashima K et al. J Am Acad Dermatol. 2008 Aug;59(2 Suppl 1):S28-32.
4. Gild R et al. Br J Dermatol. 2010 Nov;163(5):1082-4.
5. Glatz M and Muellegger RR. BMJ Case Rep. 2014. doi: 10.1136/bcr-2014-203929.
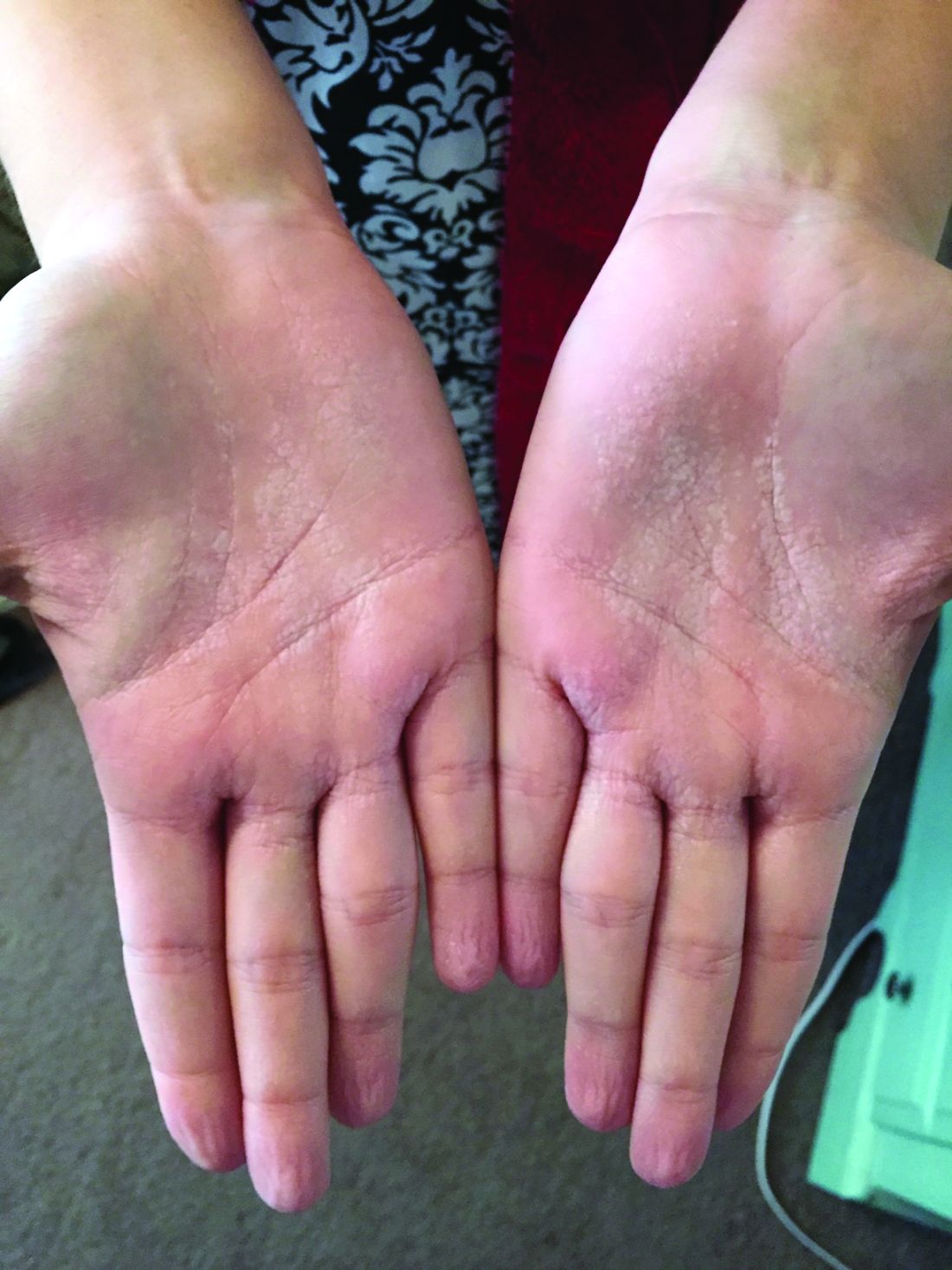
A 36-year-old presents with a mildly pruritic rash consisting of pink papules on his hand
. MG is a dermatophytic folliculitis that classically presents as folliculocentric plaque, in which there are papules, pustules, and nodules, usually found on the lower leg and almost exclusively in adults.1 Wrists are commonly affected as well.

MG is typically caused by mechanical disruption of hair follicles that allows fungi to penetrate deep into dermal tissue.2 Quite often, the source of infection is typically the patient’s skin or nails. Associated risk factors include longstanding fungal infection, shaving or other cutaneous trauma, topical steroids, and immunosuppressive therapy.3,4 Although MG can be caused by other fungal species, it is most often caused by Trichophyton rubrum or Trichophyton tonsurans.1 There are two types of MG, the perifollicular papular form, which is localized and typically occurs in healthy individuals, and the deep subcutaneous plaque or nodular forms that usually occur in immunocompromised individuals.5
MG is an important clinical manifestation to be familiar with because of the increase in the numbers of solid-organ transplants and patients on immunosuppressive therapies. These patients are highly predisposed to opportunistic infections with aggressive clinical courses and will usually require prolonged treatment as relapses are common.3,5

Tissue culture and skin biopsy are often needed to establish the diagnosis. If a topical antifungal has been used, KOH (potassium hydroxide) and culture may be negative. This patient’s tissue culture was positive for T. rubrum. The histopathology revealed hyperkeratosis and acanthosis with focal parakeratosis and a lymphohistiocytic infiltrate in the dermis. On PAS (Periodic acid–Schiff ) stain, PAS-positive hyphae were identified in the keratin layer, confirming a diagnosis of tinea infection.
First line treatment includes systemic antifungals such as griseofulvin, ketoconazole, itraconazole, and terbinafine. Duration of therapy is typically 4-8 weeks or until all lesions are cleared.3,5
This case and photo were submitted by Mr. Hakimi of University of California San Diego School of Medicine and Dr. Sateesh of San Diego Family Dermatology. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1.“Fitzpatrick’s Dermatology in General Medicine” (New York: McGraw-Hill Medical, 2012).
2. Bonifaz A et al. Gac Med Mex. Sep-Oct 2008;144(5):427-33.
3. Romero FA et al. Transpl Infect Dis. 2011 Aug;13(4):424-3. doi:10.1111/j.1399-3062.2010.00596.x
4. Chou WY, Hsu CJ. Medicine (Baltimore). 2016 Jan;95(2):e2245. doi: 10.1097/MD.0000000000002245.
5. Ilkit M et al. Med Mycol. 2102 Jul;50(5):449-57.
. MG is a dermatophytic folliculitis that classically presents as folliculocentric plaque, in which there are papules, pustules, and nodules, usually found on the lower leg and almost exclusively in adults.1 Wrists are commonly affected as well.
MG is typically caused by mechanical disruption of hair follicles that allows fungi to penetrate deep into dermal tissue.2 Quite often, the source of infection is typically the patient’s skin or nails. Associated risk factors include longstanding fungal infection, shaving or other cutaneous trauma, topical steroids, and immunosuppressive therapy.3,4 Although MG can be caused by other fungal species, it is most often caused by Trichophyton rubrum or Trichophyton tonsurans.1 There are two types of MG, the perifollicular papular form, which is localized and typically occurs in healthy individuals, and the deep subcutaneous plaque or nodular forms that usually occur in immunocompromised individuals.5
MG is an important clinical manifestation to be familiar with because of the increase in the numbers of solid-organ transplants and patients on immunosuppressive therapies. These patients are highly predisposed to opportunistic infections with aggressive clinical courses and will usually require prolonged treatment as relapses are common.3,5
Tissue culture and skin biopsy are often needed to establish the diagnosis. If a topical antifungal has been used, KOH (potassium hydroxide) and culture may be negative. This patient’s tissue culture was positive for T. rubrum. The histopathology revealed hyperkeratosis and acanthosis with focal parakeratosis and a lymphohistiocytic infiltrate in the dermis. On PAS (Periodic acid–Schiff ) stain, PAS-positive hyphae were identified in the keratin layer, confirming a diagnosis of tinea infection.
First line treatment includes systemic antifungals such as griseofulvin, ketoconazole, itraconazole, and terbinafine. Duration of therapy is typically 4-8 weeks or until all lesions are cleared.3,5
This case and photo were submitted by Mr. Hakimi of University of California San Diego School of Medicine and Dr. Sateesh of San Diego Family Dermatology. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1.“Fitzpatrick’s Dermatology in General Medicine” (New York: McGraw-Hill Medical, 2012).
2. Bonifaz A et al. Gac Med Mex. Sep-Oct 2008;144(5):427-33.
3. Romero FA et al. Transpl Infect Dis. 2011 Aug;13(4):424-3. doi:10.1111/j.1399-3062.2010.00596.x
4. Chou WY, Hsu CJ. Medicine (Baltimore). 2016 Jan;95(2):e2245. doi: 10.1097/MD.0000000000002245.
5. Ilkit M et al. Med Mycol. 2102 Jul;50(5):449-57.
. MG is a dermatophytic folliculitis that classically presents as folliculocentric plaque, in which there are papules, pustules, and nodules, usually found on the lower leg and almost exclusively in adults.1 Wrists are commonly affected as well.
MG is typically caused by mechanical disruption of hair follicles that allows fungi to penetrate deep into dermal tissue.2 Quite often, the source of infection is typically the patient’s skin or nails. Associated risk factors include longstanding fungal infection, shaving or other cutaneous trauma, topical steroids, and immunosuppressive therapy.3,4 Although MG can be caused by other fungal species, it is most often caused by Trichophyton rubrum or Trichophyton tonsurans.1 There are two types of MG, the perifollicular papular form, which is localized and typically occurs in healthy individuals, and the deep subcutaneous plaque or nodular forms that usually occur in immunocompromised individuals.5
MG is an important clinical manifestation to be familiar with because of the increase in the numbers of solid-organ transplants and patients on immunosuppressive therapies. These patients are highly predisposed to opportunistic infections with aggressive clinical courses and will usually require prolonged treatment as relapses are common.3,5
Tissue culture and skin biopsy are often needed to establish the diagnosis. If a topical antifungal has been used, KOH (potassium hydroxide) and culture may be negative. This patient’s tissue culture was positive for T. rubrum. The histopathology revealed hyperkeratosis and acanthosis with focal parakeratosis and a lymphohistiocytic infiltrate in the dermis. On PAS (Periodic acid–Schiff ) stain, PAS-positive hyphae were identified in the keratin layer, confirming a diagnosis of tinea infection.
First line treatment includes systemic antifungals such as griseofulvin, ketoconazole, itraconazole, and terbinafine. Duration of therapy is typically 4-8 weeks or until all lesions are cleared.3,5
This case and photo were submitted by Mr. Hakimi of University of California San Diego School of Medicine and Dr. Sateesh of San Diego Family Dermatology. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1.“Fitzpatrick’s Dermatology in General Medicine” (New York: McGraw-Hill Medical, 2012).
2. Bonifaz A et al. Gac Med Mex. Sep-Oct 2008;144(5):427-33.
3. Romero FA et al. Transpl Infect Dis. 2011 Aug;13(4):424-3. doi:10.1111/j.1399-3062.2010.00596.x
4. Chou WY, Hsu CJ. Medicine (Baltimore). 2016 Jan;95(2):e2245. doi: 10.1097/MD.0000000000002245.
5. Ilkit M et al. Med Mycol. 2102 Jul;50(5):449-57.
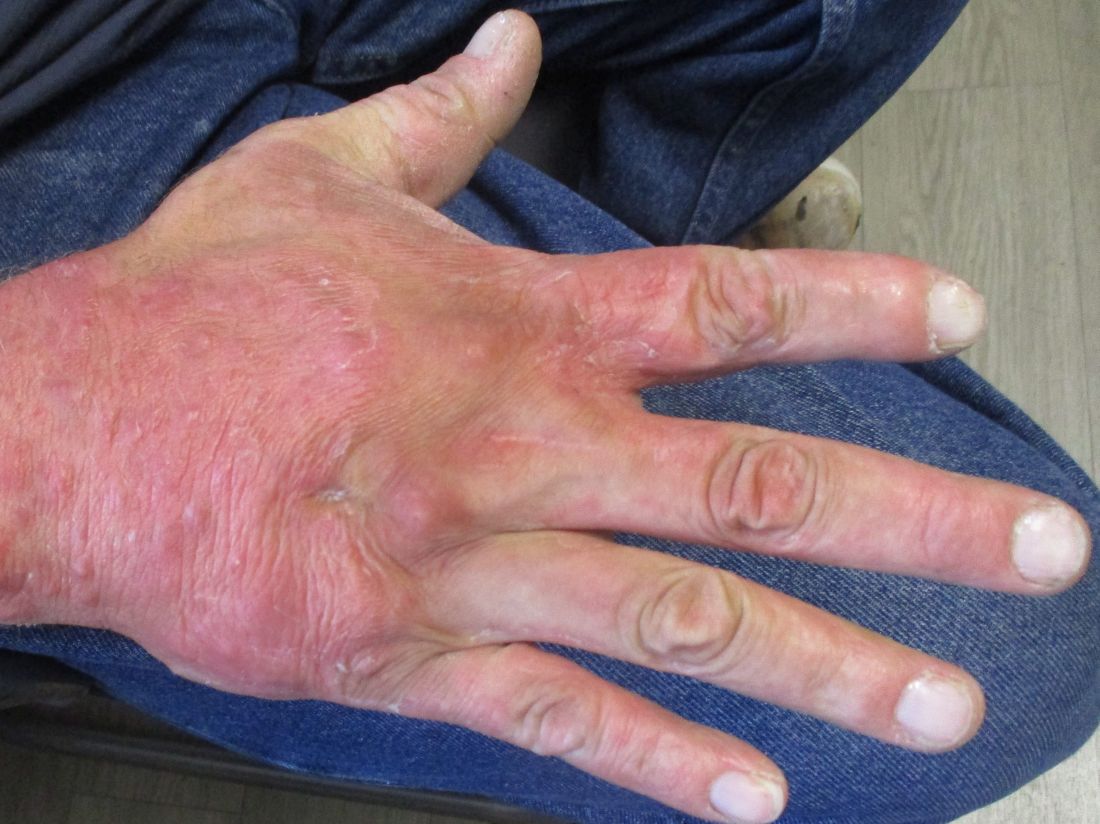
A 4-year-old with a lesion on her cheek, which grew and became firmer over two months
The patient was diagnosed with idiopathic facial aseptic granuloma (IFAG) based on the clinical findings, as well as the associated history of chalazia and erythematous papules seen in childhood rosacea.
She was treated with several months of azithromycin, sulfur wash, and metronidazole cream with improvement of some of the smaller lesions but no change on the larger nodules. Later she was treated with oral and topical ivermectin with no improvement. Some of the nodules slowly resolved except for the larger lesion on the right cheek. She was later treated with a 6-week course of clarithromycin with partial improvement of the nodule. The lesion resolved after 2 months of stopping clarithromycin.
IFAG is a rare condition seen in prepubescent children. The etiology of this condition is not well understood and is thought to be on the spectrum of childhood rosacea.1 From several recent reports, IFAG usually is seen in children with associated conditions including chalazia, conjunctivitis, blepharitis, and telangiectasias, which can be seen in patients with rosacea. These associated findings suggest the possibility of IFAG being a form of granulomatous rosacea in children.
This condition presents in childhood between the ages of 8 months and 13 years. Most of the cases occur in toddlers, and girls appear to be more affected than boys. The lesions appear as pink, rubbery, nontender, nonfluctuant nodules on the cheeks, which can be single or multiple. A large prospective study in 30 children demonstrated that more 70% of the lesions cultured were negative for bacteria. Histologic analysis of some of the lesions showed a chronic dermal lymphohistiocytic granulomatous perifollicular infiltrate with numerous foreign body–type giant cells.2
The differential diagnosis of these lesions should include infectious pyodermas such as mycobacterial infections, cutaneous leishmaniasis, and botryomycosis; deep fungal infections such as sporotrichosis, coccidioidomycosis, and cryptococcosis; childhood nodulocystic acne; pilomatrixoma; epidermoid cyst; vascular tumors or malformations; and leukemia cutis.3
The diagnosis is usually clinical but in atypical cases a skin biopsy with tissue cultures should be performed. The decision to biopsy these lesions will need to be done in a one by one basis, as a biopsy may leave scaring on the area affected.
It has been postulated that a color Doppler ultrasound of the lesion may be a helpful ancillary study. Echographic findings show a well demarcated solid-cystic, hypoechoic dermal lesion, the largest axis of which lies parallel to the skin surface. The lesion lacks calcium deposits. Other findings include increased echogenicity of the underlaying hypodermis. The findings may vary depending on the stage of the lesion.4
The course of the condition may last on average months to years. Some lesions resolve spontaneously and others may respond to courses of oral antibiotics such as clarithromycin, azithromycin, or ivermectin. In our patient, several lesions improved with oral antibiotics, but the larger lesions were more persistent and resolved after a year.
The lesions usually resolve without scarring. In those patients with associated rosacea, maintenance topical treatments may be warranted and also may need follow-up with ophthalmology because they tend to commonly have ocular rosacea as well.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. Pediatr Dermatol. 2013 Jan-Feb;30(1):109-11.
2. Br J Dermatol. 2007 Apr;156(4):705-8.
3. Pediatr Dermatol. 2018 Jul;35(4):490-3.
4. Actas Dermosifiliogr. 2019 Oct;110(8):637-41.
The patient was diagnosed with idiopathic facial aseptic granuloma (IFAG) based on the clinical findings, as well as the associated history of chalazia and erythematous papules seen in childhood rosacea.
She was treated with several months of azithromycin, sulfur wash, and metronidazole cream with improvement of some of the smaller lesions but no change on the larger nodules. Later she was treated with oral and topical ivermectin with no improvement. Some of the nodules slowly resolved except for the larger lesion on the right cheek. She was later treated with a 6-week course of clarithromycin with partial improvement of the nodule. The lesion resolved after 2 months of stopping clarithromycin.
IFAG is a rare condition seen in prepubescent children. The etiology of this condition is not well understood and is thought to be on the spectrum of childhood rosacea.1 From several recent reports, IFAG usually is seen in children with associated conditions including chalazia, conjunctivitis, blepharitis, and telangiectasias, which can be seen in patients with rosacea. These associated findings suggest the possibility of IFAG being a form of granulomatous rosacea in children.
This condition presents in childhood between the ages of 8 months and 13 years. Most of the cases occur in toddlers, and girls appear to be more affected than boys. The lesions appear as pink, rubbery, nontender, nonfluctuant nodules on the cheeks, which can be single or multiple. A large prospective study in 30 children demonstrated that more 70% of the lesions cultured were negative for bacteria. Histologic analysis of some of the lesions showed a chronic dermal lymphohistiocytic granulomatous perifollicular infiltrate with numerous foreign body–type giant cells.2
The differential diagnosis of these lesions should include infectious pyodermas such as mycobacterial infections, cutaneous leishmaniasis, and botryomycosis; deep fungal infections such as sporotrichosis, coccidioidomycosis, and cryptococcosis; childhood nodulocystic acne; pilomatrixoma; epidermoid cyst; vascular tumors or malformations; and leukemia cutis.3
The diagnosis is usually clinical but in atypical cases a skin biopsy with tissue cultures should be performed. The decision to biopsy these lesions will need to be done in a one by one basis, as a biopsy may leave scaring on the area affected.
It has been postulated that a color Doppler ultrasound of the lesion may be a helpful ancillary study. Echographic findings show a well demarcated solid-cystic, hypoechoic dermal lesion, the largest axis of which lies parallel to the skin surface. The lesion lacks calcium deposits. Other findings include increased echogenicity of the underlaying hypodermis. The findings may vary depending on the stage of the lesion.4
The course of the condition may last on average months to years. Some lesions resolve spontaneously and others may respond to courses of oral antibiotics such as clarithromycin, azithromycin, or ivermectin. In our patient, several lesions improved with oral antibiotics, but the larger lesions were more persistent and resolved after a year.
The lesions usually resolve without scarring. In those patients with associated rosacea, maintenance topical treatments may be warranted and also may need follow-up with ophthalmology because they tend to commonly have ocular rosacea as well.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. Pediatr Dermatol. 2013 Jan-Feb;30(1):109-11.
2. Br J Dermatol. 2007 Apr;156(4):705-8.
3. Pediatr Dermatol. 2018 Jul;35(4):490-3.
4. Actas Dermosifiliogr. 2019 Oct;110(8):637-41.
The patient was diagnosed with idiopathic facial aseptic granuloma (IFAG) based on the clinical findings, as well as the associated history of chalazia and erythematous papules seen in childhood rosacea.
She was treated with several months of azithromycin, sulfur wash, and metronidazole cream with improvement of some of the smaller lesions but no change on the larger nodules. Later she was treated with oral and topical ivermectin with no improvement. Some of the nodules slowly resolved except for the larger lesion on the right cheek. She was later treated with a 6-week course of clarithromycin with partial improvement of the nodule. The lesion resolved after 2 months of stopping clarithromycin.
IFAG is a rare condition seen in prepubescent children. The etiology of this condition is not well understood and is thought to be on the spectrum of childhood rosacea.1 From several recent reports, IFAG usually is seen in children with associated conditions including chalazia, conjunctivitis, blepharitis, and telangiectasias, which can be seen in patients with rosacea. These associated findings suggest the possibility of IFAG being a form of granulomatous rosacea in children.
This condition presents in childhood between the ages of 8 months and 13 years. Most of the cases occur in toddlers, and girls appear to be more affected than boys. The lesions appear as pink, rubbery, nontender, nonfluctuant nodules on the cheeks, which can be single or multiple. A large prospective study in 30 children demonstrated that more 70% of the lesions cultured were negative for bacteria. Histologic analysis of some of the lesions showed a chronic dermal lymphohistiocytic granulomatous perifollicular infiltrate with numerous foreign body–type giant cells.2
The differential diagnosis of these lesions should include infectious pyodermas such as mycobacterial infections, cutaneous leishmaniasis, and botryomycosis; deep fungal infections such as sporotrichosis, coccidioidomycosis, and cryptococcosis; childhood nodulocystic acne; pilomatrixoma; epidermoid cyst; vascular tumors or malformations; and leukemia cutis.3
The diagnosis is usually clinical but in atypical cases a skin biopsy with tissue cultures should be performed. The decision to biopsy these lesions will need to be done in a one by one basis, as a biopsy may leave scaring on the area affected.
It has been postulated that a color Doppler ultrasound of the lesion may be a helpful ancillary study. Echographic findings show a well demarcated solid-cystic, hypoechoic dermal lesion, the largest axis of which lies parallel to the skin surface. The lesion lacks calcium deposits. Other findings include increased echogenicity of the underlaying hypodermis. The findings may vary depending on the stage of the lesion.4
The course of the condition may last on average months to years. Some lesions resolve spontaneously and others may respond to courses of oral antibiotics such as clarithromycin, azithromycin, or ivermectin. In our patient, several lesions improved with oral antibiotics, but the larger lesions were more persistent and resolved after a year.
The lesions usually resolve without scarring. In those patients with associated rosacea, maintenance topical treatments may be warranted and also may need follow-up with ophthalmology because they tend to commonly have ocular rosacea as well.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She said she had no relevant financial disclosures. Email her at [email protected].
References
1. Pediatr Dermatol. 2013 Jan-Feb;30(1):109-11.
2. Br J Dermatol. 2007 Apr;156(4):705-8.
3. Pediatr Dermatol. 2018 Jul;35(4):490-3.
4. Actas Dermosifiliogr. 2019 Oct;110(8):637-41.
A 4-year-old female is brought to our pediatric dermatology clinic for evaluation of a persistent lesion on the cheek.
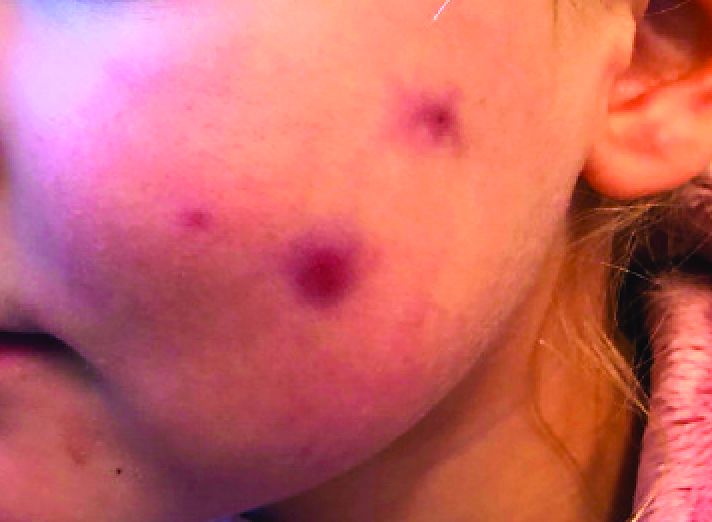
The mother of the child reports that the lesion started as a small "bug bite" and then started growing and getting firmer for the past 2 months. The girl has developed other smaller red, pimple-like lesions on the cheeks and one of them is starting to increase in size.
She denies any tenderness on the area or any purulent discharge. She has had no fevers, chills, weight loss, nose bleeds, fatigue, or any other symptoms. The mother has not noted any changes on the child's body odor, any rapid growth, or hair on her axillary or pubic area. She was treated with three different courses of oral antibiotics including cephalexin, trimethoprim/sulfamethoxazole, and clindamycin, as well as topical mupirocin, with no improvement.
Her past medical history is significant for several episodes of eyelid cysts that were treated with warm compresses and topical erythromycin ointment. The family history is significant for the father having severe acne as a teenager. She has two cats, she has not traveled, and she has an older sister who has no lesions.
On physical examination she is a lovely 4-year-old female in no acute distress. Her height is on the 70th percentile and weight on the 40th percentile for her age. Her blood pressure is 95/84 with a heart rate of 96. On skin examination she has several pink macules and papules on her bilateral cheeks. On the left cheek there are two pink nodules: One is 1 cm, and the other is 7 mm. The nodules are not tender. There is no warmth, fluctuance, or discharge from the lesions.
She has no cervical lymphadenopathy. She has no axillary or pubic hair. She is Tanner stage I.