User login
CDC warns of enterovirus strain linked to polio-like condition
, according to a Health Network Alert advisory by the Centers for Disease Control and Prevention.
In August, health care providers and hospitals notified the CDC of an increase in severe respiratory illness in children who also tested positive for rhinovirus (RV) or enterovirus (EV). Additional testing revealed that some children were positive for EV-D68, which primarily causes acute respiratory illness. However, the virus has been associated with acute flaccid myelitis (AFM), a rare neurologic condition involving muscle weakness.
Also, in July and August 2022, surveillance networks reported an increase in EV-D68 activity compared with the same months in 2019, 2020, and 2021, the agency said in the alert. As of Aug. 30, the CDC has not received any reports of AFM beginning this year; however, spikes in EV-D68 typically come before cases of AFM, they said.
“Something we are always on the lookout for in the late summer and fall is AFM cases,” said Rick Malley, MD, of the division of infectious disease at Boston Children’s Hospital, in an interview with this news organization. “Unfortunately, we kind of expect them during enterovirus season,” he said. That season is thought to peak in the late summer and early fall.
Since the CDC began tracking AFM in August 2014, there have been 692 confirmed cases in the United States. AFM cases spiked in 2014, 2016, and 2018, mostly in young children. In 2021, there were 28 confirmed cases across 15 states. The CDC did not specify the age of those cases, but in 2018 – when EV-D68 most recently circulated at high levels – the median age of children who visited the emergency department or were hospitalized for EV-D68–associated respiratory illness was 3 years.
“[AFM] can be very severe and it can be very scary for the parents of children who have it,” Dr. Malley said, “but given the prevalence of enteroviruses in the community, you have to conclude it’s a relatively rare event in susceptible individuals. Why some get it and others don’t is unfortunately unclear at this moment.”
The CDC recommends that providers consider EV-D68 as a possible cause for acute, severe respiratory illness in children. If the cause of a respiratory illness in a severely ill patient is not clear, health professionals should test for RVs and EVs, if this is not already part of a typical diagnostic workflow, the agency said. Currently, there are no vaccines or specific treatments for RV or EV, and the CDC recommends supportive clinical management.
The advisory also urged providers to “strongly consider AFM in patients with acute flaccid limb weakness, especially after respiratory illness or fever, and between the months of August and November 2022.”
For any patient presenting with possible AFM, clinicians should collect samples from multiple sources, including cerebrospinal fluid, serum, stool, and a nasopharyngeal or oropharyngeal swab. Samples should be taken “as early as possible and preferably on the day of onset of limb weakness,” the alert said. There is currently no specific medicine for AFM, the agency said, though recommended interventions may vary for each patient.
A version of this article first appeared on Medscape.com.
, according to a Health Network Alert advisory by the Centers for Disease Control and Prevention.
In August, health care providers and hospitals notified the CDC of an increase in severe respiratory illness in children who also tested positive for rhinovirus (RV) or enterovirus (EV). Additional testing revealed that some children were positive for EV-D68, which primarily causes acute respiratory illness. However, the virus has been associated with acute flaccid myelitis (AFM), a rare neurologic condition involving muscle weakness.
Also, in July and August 2022, surveillance networks reported an increase in EV-D68 activity compared with the same months in 2019, 2020, and 2021, the agency said in the alert. As of Aug. 30, the CDC has not received any reports of AFM beginning this year; however, spikes in EV-D68 typically come before cases of AFM, they said.
“Something we are always on the lookout for in the late summer and fall is AFM cases,” said Rick Malley, MD, of the division of infectious disease at Boston Children’s Hospital, in an interview with this news organization. “Unfortunately, we kind of expect them during enterovirus season,” he said. That season is thought to peak in the late summer and early fall.
Since the CDC began tracking AFM in August 2014, there have been 692 confirmed cases in the United States. AFM cases spiked in 2014, 2016, and 2018, mostly in young children. In 2021, there were 28 confirmed cases across 15 states. The CDC did not specify the age of those cases, but in 2018 – when EV-D68 most recently circulated at high levels – the median age of children who visited the emergency department or were hospitalized for EV-D68–associated respiratory illness was 3 years.
“[AFM] can be very severe and it can be very scary for the parents of children who have it,” Dr. Malley said, “but given the prevalence of enteroviruses in the community, you have to conclude it’s a relatively rare event in susceptible individuals. Why some get it and others don’t is unfortunately unclear at this moment.”
The CDC recommends that providers consider EV-D68 as a possible cause for acute, severe respiratory illness in children. If the cause of a respiratory illness in a severely ill patient is not clear, health professionals should test for RVs and EVs, if this is not already part of a typical diagnostic workflow, the agency said. Currently, there are no vaccines or specific treatments for RV or EV, and the CDC recommends supportive clinical management.
The advisory also urged providers to “strongly consider AFM in patients with acute flaccid limb weakness, especially after respiratory illness or fever, and between the months of August and November 2022.”
For any patient presenting with possible AFM, clinicians should collect samples from multiple sources, including cerebrospinal fluid, serum, stool, and a nasopharyngeal or oropharyngeal swab. Samples should be taken “as early as possible and preferably on the day of onset of limb weakness,” the alert said. There is currently no specific medicine for AFM, the agency said, though recommended interventions may vary for each patient.
A version of this article first appeared on Medscape.com.
, according to a Health Network Alert advisory by the Centers for Disease Control and Prevention.
In August, health care providers and hospitals notified the CDC of an increase in severe respiratory illness in children who also tested positive for rhinovirus (RV) or enterovirus (EV). Additional testing revealed that some children were positive for EV-D68, which primarily causes acute respiratory illness. However, the virus has been associated with acute flaccid myelitis (AFM), a rare neurologic condition involving muscle weakness.
Also, in July and August 2022, surveillance networks reported an increase in EV-D68 activity compared with the same months in 2019, 2020, and 2021, the agency said in the alert. As of Aug. 30, the CDC has not received any reports of AFM beginning this year; however, spikes in EV-D68 typically come before cases of AFM, they said.
“Something we are always on the lookout for in the late summer and fall is AFM cases,” said Rick Malley, MD, of the division of infectious disease at Boston Children’s Hospital, in an interview with this news organization. “Unfortunately, we kind of expect them during enterovirus season,” he said. That season is thought to peak in the late summer and early fall.
Since the CDC began tracking AFM in August 2014, there have been 692 confirmed cases in the United States. AFM cases spiked in 2014, 2016, and 2018, mostly in young children. In 2021, there were 28 confirmed cases across 15 states. The CDC did not specify the age of those cases, but in 2018 – when EV-D68 most recently circulated at high levels – the median age of children who visited the emergency department or were hospitalized for EV-D68–associated respiratory illness was 3 years.
“[AFM] can be very severe and it can be very scary for the parents of children who have it,” Dr. Malley said, “but given the prevalence of enteroviruses in the community, you have to conclude it’s a relatively rare event in susceptible individuals. Why some get it and others don’t is unfortunately unclear at this moment.”
The CDC recommends that providers consider EV-D68 as a possible cause for acute, severe respiratory illness in children. If the cause of a respiratory illness in a severely ill patient is not clear, health professionals should test for RVs and EVs, if this is not already part of a typical diagnostic workflow, the agency said. Currently, there are no vaccines or specific treatments for RV or EV, and the CDC recommends supportive clinical management.
The advisory also urged providers to “strongly consider AFM in patients with acute flaccid limb weakness, especially after respiratory illness or fever, and between the months of August and November 2022.”
For any patient presenting with possible AFM, clinicians should collect samples from multiple sources, including cerebrospinal fluid, serum, stool, and a nasopharyngeal or oropharyngeal swab. Samples should be taken “as early as possible and preferably on the day of onset of limb weakness,” the alert said. There is currently no specific medicine for AFM, the agency said, though recommended interventions may vary for each patient.
A version of this article first appeared on Medscape.com.
Children and COVID: Weekly cases close out August with a second straight increase
New cases rose by 4.6% for the week of Aug. 26 to Sept. 1, following a week in which cases increased by almost 9%, as the second half of August basically reversed the two consecutive weeks of decreases during the first half of the month, based on the AAP/CHA data collected from state and territorial health departments.
Similar trends can be seen for emergency department visits, with the exception of children aged 0-11 years, whose ED visit rates have continued to fall since late July. Children aged 12-15, however, had a 7-day average of 4.4% of ED visits with diagnosed COVID on Aug. 25, compared with 3.1% for Aug. 12. Children aged 16-17 years were at 3.4% on Aug. 27, compared with 3.1% as late as Aug. 15, the Centers for Disease Control and Prevention reported.
Hospital admissions with confirmed COVID-19, reported only for children aged 0-17 years, also reflect the late-August trend of increased cases. New hospitalizations dropped from 0.46 per 100,000 population on July 30 to 0.40 per 100,000 on Aug. 19 but have since risen to 0.44 per 100,000 as of Aug. 27, the CDC said on its COVID Data Tracker.
Initial vaccinations, meanwhile, have declined since early August for all children, according to a separate report from the AAP. A look at CDC data for two specific days – the first and last Mondays of the month – shows that those aged under 5 received 12,982 doses on Aug. 1, compared with 5,824 doses on Aug. 29. Over that same time, initial vaccinations in 5- to 11-year-olds went from 9,058 to 2,879, while among those aged 12-17 they dropped from 4,245 to 1,226.
Cumulatively, 5.5% of all children under age 5 had received at least one dose and 1.3% were fully vaccinated by Aug. 30, compared with 38.1% and 30.7%, respectively, of those aged 5-11 and 70.7% and 60.5% of 12- to 17-year-olds, the CDC said.
New cases rose by 4.6% for the week of Aug. 26 to Sept. 1, following a week in which cases increased by almost 9%, as the second half of August basically reversed the two consecutive weeks of decreases during the first half of the month, based on the AAP/CHA data collected from state and territorial health departments.
Similar trends can be seen for emergency department visits, with the exception of children aged 0-11 years, whose ED visit rates have continued to fall since late July. Children aged 12-15, however, had a 7-day average of 4.4% of ED visits with diagnosed COVID on Aug. 25, compared with 3.1% for Aug. 12. Children aged 16-17 years were at 3.4% on Aug. 27, compared with 3.1% as late as Aug. 15, the Centers for Disease Control and Prevention reported.
Hospital admissions with confirmed COVID-19, reported only for children aged 0-17 years, also reflect the late-August trend of increased cases. New hospitalizations dropped from 0.46 per 100,000 population on July 30 to 0.40 per 100,000 on Aug. 19 but have since risen to 0.44 per 100,000 as of Aug. 27, the CDC said on its COVID Data Tracker.
Initial vaccinations, meanwhile, have declined since early August for all children, according to a separate report from the AAP. A look at CDC data for two specific days – the first and last Mondays of the month – shows that those aged under 5 received 12,982 doses on Aug. 1, compared with 5,824 doses on Aug. 29. Over that same time, initial vaccinations in 5- to 11-year-olds went from 9,058 to 2,879, while among those aged 12-17 they dropped from 4,245 to 1,226.
Cumulatively, 5.5% of all children under age 5 had received at least one dose and 1.3% were fully vaccinated by Aug. 30, compared with 38.1% and 30.7%, respectively, of those aged 5-11 and 70.7% and 60.5% of 12- to 17-year-olds, the CDC said.
New cases rose by 4.6% for the week of Aug. 26 to Sept. 1, following a week in which cases increased by almost 9%, as the second half of August basically reversed the two consecutive weeks of decreases during the first half of the month, based on the AAP/CHA data collected from state and territorial health departments.
Similar trends can be seen for emergency department visits, with the exception of children aged 0-11 years, whose ED visit rates have continued to fall since late July. Children aged 12-15, however, had a 7-day average of 4.4% of ED visits with diagnosed COVID on Aug. 25, compared with 3.1% for Aug. 12. Children aged 16-17 years were at 3.4% on Aug. 27, compared with 3.1% as late as Aug. 15, the Centers for Disease Control and Prevention reported.
Hospital admissions with confirmed COVID-19, reported only for children aged 0-17 years, also reflect the late-August trend of increased cases. New hospitalizations dropped from 0.46 per 100,000 population on July 30 to 0.40 per 100,000 on Aug. 19 but have since risen to 0.44 per 100,000 as of Aug. 27, the CDC said on its COVID Data Tracker.
Initial vaccinations, meanwhile, have declined since early August for all children, according to a separate report from the AAP. A look at CDC data for two specific days – the first and last Mondays of the month – shows that those aged under 5 received 12,982 doses on Aug. 1, compared with 5,824 doses on Aug. 29. Over that same time, initial vaccinations in 5- to 11-year-olds went from 9,058 to 2,879, while among those aged 12-17 they dropped from 4,245 to 1,226.
Cumulatively, 5.5% of all children under age 5 had received at least one dose and 1.3% were fully vaccinated by Aug. 30, compared with 38.1% and 30.7%, respectively, of those aged 5-11 and 70.7% and 60.5% of 12- to 17-year-olds, the CDC said.
FDA okays spesolimab, first treatment for generalized pustular psoriasis
The U.S. Food and Drug Administration has approved the biologic agent spesolimab (Spevigo) for the treatment of flares in adults with generalized pustular psoriasis (GPP), the company that manufactures the drug has announced.
Until this approval, “there were no FDA-approved options to treat patients experiencing a GPP flare,” Mark Lebwohl, MD, principal investigator in the pivotal spesolimab trial, told this news organization. The approval “is a turning point for dermatologists and clinicians who treat patients living with this devastating and debilitating disease,” he said. Treatment with spesolimab “rapidly improves the clinical symptoms of GPP flares and will greatly improve our ability to help our patients manage painful flares,” noted Dr. Lebwohl, dean of clinical therapeutics and professor of dermatology, Icahn School of Medicine at Mount Sinai, New York.
Spesolimab, manufactured by Boehringer Ingelheim, is a novel, selective monoclonal antibody that blocks interleukin-36 signaling known to be involved in GPP. It received priority review and had orphan drug and breakthrough therapy designation.
GPP affects an estimated 1 of every 10,000 people in the United States.
Though rare, GPP is a potentially life-threatening disease that is distinct from plaque psoriasis. GPP is caused by the accumulation of neutrophils in the skin. Throughout the course of the disease, patients may suffer recurring episodes of widespread eruptions of painful, sterile pustules across all parts of the body.
Spesolimab was evaluated in a global, 12-week, placebo-controlled clinical trial that involved 53 adults experiencing a GPP flare. After 1 week, significantly more patients treated with spesolimab than placebo showed no visible pustules (54% vs 6%), according to the company.
The most common adverse reactions, seen in at least 5% of patients treated with spesolimab, were asthenia and fatigue; nausea and vomiting; headache; pruritus and prurigo; hematoma and bruising at the infusion site; and urinary tract infection.
Dr. Lebwohl is a paid consultant to Boehringer Ingelheim.
A version of this article first appeared on Medscape.com.
This article was updated 9/6/22.
The U.S. Food and Drug Administration has approved the biologic agent spesolimab (Spevigo) for the treatment of flares in adults with generalized pustular psoriasis (GPP), the company that manufactures the drug has announced.
Until this approval, “there were no FDA-approved options to treat patients experiencing a GPP flare,” Mark Lebwohl, MD, principal investigator in the pivotal spesolimab trial, told this news organization. The approval “is a turning point for dermatologists and clinicians who treat patients living with this devastating and debilitating disease,” he said. Treatment with spesolimab “rapidly improves the clinical symptoms of GPP flares and will greatly improve our ability to help our patients manage painful flares,” noted Dr. Lebwohl, dean of clinical therapeutics and professor of dermatology, Icahn School of Medicine at Mount Sinai, New York.
Spesolimab, manufactured by Boehringer Ingelheim, is a novel, selective monoclonal antibody that blocks interleukin-36 signaling known to be involved in GPP. It received priority review and had orphan drug and breakthrough therapy designation.
GPP affects an estimated 1 of every 10,000 people in the United States.
Though rare, GPP is a potentially life-threatening disease that is distinct from plaque psoriasis. GPP is caused by the accumulation of neutrophils in the skin. Throughout the course of the disease, patients may suffer recurring episodes of widespread eruptions of painful, sterile pustules across all parts of the body.
Spesolimab was evaluated in a global, 12-week, placebo-controlled clinical trial that involved 53 adults experiencing a GPP flare. After 1 week, significantly more patients treated with spesolimab than placebo showed no visible pustules (54% vs 6%), according to the company.
The most common adverse reactions, seen in at least 5% of patients treated with spesolimab, were asthenia and fatigue; nausea and vomiting; headache; pruritus and prurigo; hematoma and bruising at the infusion site; and urinary tract infection.
Dr. Lebwohl is a paid consultant to Boehringer Ingelheim.
A version of this article first appeared on Medscape.com.
This article was updated 9/6/22.
The U.S. Food and Drug Administration has approved the biologic agent spesolimab (Spevigo) for the treatment of flares in adults with generalized pustular psoriasis (GPP), the company that manufactures the drug has announced.
Until this approval, “there were no FDA-approved options to treat patients experiencing a GPP flare,” Mark Lebwohl, MD, principal investigator in the pivotal spesolimab trial, told this news organization. The approval “is a turning point for dermatologists and clinicians who treat patients living with this devastating and debilitating disease,” he said. Treatment with spesolimab “rapidly improves the clinical symptoms of GPP flares and will greatly improve our ability to help our patients manage painful flares,” noted Dr. Lebwohl, dean of clinical therapeutics and professor of dermatology, Icahn School of Medicine at Mount Sinai, New York.
Spesolimab, manufactured by Boehringer Ingelheim, is a novel, selective monoclonal antibody that blocks interleukin-36 signaling known to be involved in GPP. It received priority review and had orphan drug and breakthrough therapy designation.
GPP affects an estimated 1 of every 10,000 people in the United States.
Though rare, GPP is a potentially life-threatening disease that is distinct from plaque psoriasis. GPP is caused by the accumulation of neutrophils in the skin. Throughout the course of the disease, patients may suffer recurring episodes of widespread eruptions of painful, sterile pustules across all parts of the body.
Spesolimab was evaluated in a global, 12-week, placebo-controlled clinical trial that involved 53 adults experiencing a GPP flare. After 1 week, significantly more patients treated with spesolimab than placebo showed no visible pustules (54% vs 6%), according to the company.
The most common adverse reactions, seen in at least 5% of patients treated with spesolimab, were asthenia and fatigue; nausea and vomiting; headache; pruritus and prurigo; hematoma and bruising at the infusion site; and urinary tract infection.
Dr. Lebwohl is a paid consultant to Boehringer Ingelheim.
A version of this article first appeared on Medscape.com.
This article was updated 9/6/22.
CDC gives final approval to Omicron COVID-19 vaccine boosters
The Centers for Disease Control and Prevention on Sept. 1 approved the use of vaccines designed to target both Omicron and the older variants of the coronavirus, a step that may aid a goal of a widespread immunization campaign before winter arrives in the United States.
The CDC’s Advisory Committee on Immunization Practices voted 13-1 on two separate questions. One sought the panel’s backing for the use of a single dose of a new version of the Pfizer COVID-19 vaccines for people aged 12 and older. The second question dealt with a single dose of the reworked Moderna vaccine for people aged 18 and older.
The federal government wants to speed use of revamped COVID-19 shots, which the Food and Drug Administration on Sept. 1 cleared for use in the United States. Hours later, CDC Director Rochelle Walensky, MD, agreed with the panel’s recommendation.
“The updated COVID-19 boosters are formulated to better protect against the most recently circulating COVID-19 variant,” Dr. Walensky said in a statement. “They can help restore protection that has waned since previous vaccination and were designed to provide broader protection against newer variants. This recommendation followed a comprehensive scientific evaluation and robust scientific discussion. If you are eligible, there is no bad time to get your COVID-19 booster and I strongly encourage you to receive it.”
The FDA vote on Aug. 31 expanded the emergency use authorization EUA for both Moderna and Pfizer’s original COVID-19 vaccines. The new products are also called “updated boosters.” Both contain two mRNA components of SARS-CoV-2 virus, one of the original strain and another that is found in the BA.4 and BA.5 strains of the Omicron variant, the FDA said.
Basically, the FDA cleared the way for these new boosters after it relied heavily on results of certain blood tests that suggested an immune response boost from the new formulas, plus 18 months of mostly safe use of the original versions of the shots.
What neither the FDA nor the CDC has, however, is evidence from studies in humans on how well these new vaccines work or whether they are as safe as the originals. But the FDA did consider clinical evidence for the older shots and results from studies on the new boosters that were done in mice.
ACIP Committee member Pablo Sanchez, MD, of Ohio State University was the sole “no” vote on each question.
“It’s a new vaccine, it’s a new platform. There’s a lot of hesitancy already. We need the human data,” Dr. Sanchez said.
Dr. Sanchez did not doubt that the newer versions of the vaccine would prove safe.
“I personally am in the age group where I’m at high risk and I’m almost sure that I will receive it,” Dr. Sanchez said. “I just feel that this was a bit premature, and I wish that we had seen that data. Having said that, I am comfortable that the vaccine will likely be safe like the others.”
Dr. Sanchez was not alone in raising concerns about backing new COVID-19 shots for which there is not direct clinical evidence from human studies.
Committee member Sarah Long, MD, of Drexel University in Philadelphia, said during the discussion she would “reluctantly” vote in favor of the updated vaccines. She said she believes they will have the potential to reduce hospitalizations and even deaths, even with questions remaining about the data.
Dr. Long joined other committee members in pointing to the approach to updating flu vaccines as a model. In an attempt to keep ahead of influenza, companies seek to defeat new strains through tweaks to their FDA-approved vaccines. There is not much clinical information available about these revised products, Dr. Long said. She compared it to remodeling an existing home.
“It is the same scaffolding, part of the same roof, we’re just putting in some dormers and windows,” with the revisions to the flu vaccine, she said.
Earlier in the day, committee member Jamie Loehr, MD, of Cayuga Family Medicine in Ithaca, N.Y., also used changes to the annual flu shots as the model for advancing COVID-19 shots.
“So after thinking about it, I am comfortable even though we don’t have human data,” he said.
There were several questions during the meeting about why the FDA had not convened a meeting of its Vaccines and Related Biological Products Advisory Committee (regarding these specific bivalent vaccines). Typically, the FDA committee of advisers considers new vaccines before the agency authorizes their use. In this case, however, the agency acted on its own.
The FDA said the committee considered the new, bivalent COVID-19 boosters in earlier meetings and that was enough outside feedback.
But holding a meeting of advisers on these specific products could have helped build public confidence in these medicines, Dorit Reiss, PhD, of the University of California Hastings College of Law, said during the public comment session of the CDC advisers’ meeting.
“We could wish the vaccines were more effective against infection, but they’re safe and they prevent hospitalization and death,” she said.
The Department of Health and Human Services anticipated the backing of ACIP. The Administration for Strategic Preparedness and Response on Aug. 31 began distributing “millions of doses of the updated booster to tens of thousands of sites nationwide,” Jason Roos, PhD, chief operating officer for HHS Coordination Operations and Response Element, wrote in a blog.
“These boosters will be available at tens of thousands of vaccination sites ... including local pharmacies, their physicians’ offices, and vaccine centers operated by state and local health officials,”Dr. Roos wrote.
A version of this article first appeared on WebMD.com.
The Centers for Disease Control and Prevention on Sept. 1 approved the use of vaccines designed to target both Omicron and the older variants of the coronavirus, a step that may aid a goal of a widespread immunization campaign before winter arrives in the United States.
The CDC’s Advisory Committee on Immunization Practices voted 13-1 on two separate questions. One sought the panel’s backing for the use of a single dose of a new version of the Pfizer COVID-19 vaccines for people aged 12 and older. The second question dealt with a single dose of the reworked Moderna vaccine for people aged 18 and older.
The federal government wants to speed use of revamped COVID-19 shots, which the Food and Drug Administration on Sept. 1 cleared for use in the United States. Hours later, CDC Director Rochelle Walensky, MD, agreed with the panel’s recommendation.
“The updated COVID-19 boosters are formulated to better protect against the most recently circulating COVID-19 variant,” Dr. Walensky said in a statement. “They can help restore protection that has waned since previous vaccination and were designed to provide broader protection against newer variants. This recommendation followed a comprehensive scientific evaluation and robust scientific discussion. If you are eligible, there is no bad time to get your COVID-19 booster and I strongly encourage you to receive it.”
The FDA vote on Aug. 31 expanded the emergency use authorization EUA for both Moderna and Pfizer’s original COVID-19 vaccines. The new products are also called “updated boosters.” Both contain two mRNA components of SARS-CoV-2 virus, one of the original strain and another that is found in the BA.4 and BA.5 strains of the Omicron variant, the FDA said.
Basically, the FDA cleared the way for these new boosters after it relied heavily on results of certain blood tests that suggested an immune response boost from the new formulas, plus 18 months of mostly safe use of the original versions of the shots.
What neither the FDA nor the CDC has, however, is evidence from studies in humans on how well these new vaccines work or whether they are as safe as the originals. But the FDA did consider clinical evidence for the older shots and results from studies on the new boosters that were done in mice.
ACIP Committee member Pablo Sanchez, MD, of Ohio State University was the sole “no” vote on each question.
“It’s a new vaccine, it’s a new platform. There’s a lot of hesitancy already. We need the human data,” Dr. Sanchez said.
Dr. Sanchez did not doubt that the newer versions of the vaccine would prove safe.
“I personally am in the age group where I’m at high risk and I’m almost sure that I will receive it,” Dr. Sanchez said. “I just feel that this was a bit premature, and I wish that we had seen that data. Having said that, I am comfortable that the vaccine will likely be safe like the others.”
Dr. Sanchez was not alone in raising concerns about backing new COVID-19 shots for which there is not direct clinical evidence from human studies.
Committee member Sarah Long, MD, of Drexel University in Philadelphia, said during the discussion she would “reluctantly” vote in favor of the updated vaccines. She said she believes they will have the potential to reduce hospitalizations and even deaths, even with questions remaining about the data.
Dr. Long joined other committee members in pointing to the approach to updating flu vaccines as a model. In an attempt to keep ahead of influenza, companies seek to defeat new strains through tweaks to their FDA-approved vaccines. There is not much clinical information available about these revised products, Dr. Long said. She compared it to remodeling an existing home.
“It is the same scaffolding, part of the same roof, we’re just putting in some dormers and windows,” with the revisions to the flu vaccine, she said.
Earlier in the day, committee member Jamie Loehr, MD, of Cayuga Family Medicine in Ithaca, N.Y., also used changes to the annual flu shots as the model for advancing COVID-19 shots.
“So after thinking about it, I am comfortable even though we don’t have human data,” he said.
There were several questions during the meeting about why the FDA had not convened a meeting of its Vaccines and Related Biological Products Advisory Committee (regarding these specific bivalent vaccines). Typically, the FDA committee of advisers considers new vaccines before the agency authorizes their use. In this case, however, the agency acted on its own.
The FDA said the committee considered the new, bivalent COVID-19 boosters in earlier meetings and that was enough outside feedback.
But holding a meeting of advisers on these specific products could have helped build public confidence in these medicines, Dorit Reiss, PhD, of the University of California Hastings College of Law, said during the public comment session of the CDC advisers’ meeting.
“We could wish the vaccines were more effective against infection, but they’re safe and they prevent hospitalization and death,” she said.
The Department of Health and Human Services anticipated the backing of ACIP. The Administration for Strategic Preparedness and Response on Aug. 31 began distributing “millions of doses of the updated booster to tens of thousands of sites nationwide,” Jason Roos, PhD, chief operating officer for HHS Coordination Operations and Response Element, wrote in a blog.
“These boosters will be available at tens of thousands of vaccination sites ... including local pharmacies, their physicians’ offices, and vaccine centers operated by state and local health officials,”Dr. Roos wrote.
A version of this article first appeared on WebMD.com.
The Centers for Disease Control and Prevention on Sept. 1 approved the use of vaccines designed to target both Omicron and the older variants of the coronavirus, a step that may aid a goal of a widespread immunization campaign before winter arrives in the United States.
The CDC’s Advisory Committee on Immunization Practices voted 13-1 on two separate questions. One sought the panel’s backing for the use of a single dose of a new version of the Pfizer COVID-19 vaccines for people aged 12 and older. The second question dealt with a single dose of the reworked Moderna vaccine for people aged 18 and older.
The federal government wants to speed use of revamped COVID-19 shots, which the Food and Drug Administration on Sept. 1 cleared for use in the United States. Hours later, CDC Director Rochelle Walensky, MD, agreed with the panel’s recommendation.
“The updated COVID-19 boosters are formulated to better protect against the most recently circulating COVID-19 variant,” Dr. Walensky said in a statement. “They can help restore protection that has waned since previous vaccination and were designed to provide broader protection against newer variants. This recommendation followed a comprehensive scientific evaluation and robust scientific discussion. If you are eligible, there is no bad time to get your COVID-19 booster and I strongly encourage you to receive it.”
The FDA vote on Aug. 31 expanded the emergency use authorization EUA for both Moderna and Pfizer’s original COVID-19 vaccines. The new products are also called “updated boosters.” Both contain two mRNA components of SARS-CoV-2 virus, one of the original strain and another that is found in the BA.4 and BA.5 strains of the Omicron variant, the FDA said.
Basically, the FDA cleared the way for these new boosters after it relied heavily on results of certain blood tests that suggested an immune response boost from the new formulas, plus 18 months of mostly safe use of the original versions of the shots.
What neither the FDA nor the CDC has, however, is evidence from studies in humans on how well these new vaccines work or whether they are as safe as the originals. But the FDA did consider clinical evidence for the older shots and results from studies on the new boosters that were done in mice.
ACIP Committee member Pablo Sanchez, MD, of Ohio State University was the sole “no” vote on each question.
“It’s a new vaccine, it’s a new platform. There’s a lot of hesitancy already. We need the human data,” Dr. Sanchez said.
Dr. Sanchez did not doubt that the newer versions of the vaccine would prove safe.
“I personally am in the age group where I’m at high risk and I’m almost sure that I will receive it,” Dr. Sanchez said. “I just feel that this was a bit premature, and I wish that we had seen that data. Having said that, I am comfortable that the vaccine will likely be safe like the others.”
Dr. Sanchez was not alone in raising concerns about backing new COVID-19 shots for which there is not direct clinical evidence from human studies.
Committee member Sarah Long, MD, of Drexel University in Philadelphia, said during the discussion she would “reluctantly” vote in favor of the updated vaccines. She said she believes they will have the potential to reduce hospitalizations and even deaths, even with questions remaining about the data.
Dr. Long joined other committee members in pointing to the approach to updating flu vaccines as a model. In an attempt to keep ahead of influenza, companies seek to defeat new strains through tweaks to their FDA-approved vaccines. There is not much clinical information available about these revised products, Dr. Long said. She compared it to remodeling an existing home.
“It is the same scaffolding, part of the same roof, we’re just putting in some dormers and windows,” with the revisions to the flu vaccine, she said.
Earlier in the day, committee member Jamie Loehr, MD, of Cayuga Family Medicine in Ithaca, N.Y., also used changes to the annual flu shots as the model for advancing COVID-19 shots.
“So after thinking about it, I am comfortable even though we don’t have human data,” he said.
There were several questions during the meeting about why the FDA had not convened a meeting of its Vaccines and Related Biological Products Advisory Committee (regarding these specific bivalent vaccines). Typically, the FDA committee of advisers considers new vaccines before the agency authorizes their use. In this case, however, the agency acted on its own.
The FDA said the committee considered the new, bivalent COVID-19 boosters in earlier meetings and that was enough outside feedback.
But holding a meeting of advisers on these specific products could have helped build public confidence in these medicines, Dorit Reiss, PhD, of the University of California Hastings College of Law, said during the public comment session of the CDC advisers’ meeting.
“We could wish the vaccines were more effective against infection, but they’re safe and they prevent hospitalization and death,” she said.
The Department of Health and Human Services anticipated the backing of ACIP. The Administration for Strategic Preparedness and Response on Aug. 31 began distributing “millions of doses of the updated booster to tens of thousands of sites nationwide,” Jason Roos, PhD, chief operating officer for HHS Coordination Operations and Response Element, wrote in a blog.
“These boosters will be available at tens of thousands of vaccination sites ... including local pharmacies, their physicians’ offices, and vaccine centers operated by state and local health officials,”Dr. Roos wrote.
A version of this article first appeared on WebMD.com.
Intera Oncology recalls hepatic artery infusion pumps for possible life-threatening issue
Although no injuries or deaths related to the pump malfunction have been reported yet, the U.S. Food and Drug Administration has deemed the recall Class I, the most serious category that indicates the device could cause injury or death.
Intera Oncology initiated the recall in July following reports from clinicians that the pumps, which are implanted to deliver chemotherapy to treat liver tumors, were delivering medications faster than expected. A fast flow rate can lead to life-threatening hematologic toxicity, neurotoxicity, or death. It also means patients will run out of medication too soon, potentially leading to disease progression or death.
The FDA notice states the company has advised customers to continue to monitor flow rate as per standard refill procedure as well as monitor for liver toxicity to adjust dosing as per standard protocols.
The company also said to consider pump replacement if altered flow can’t be adequately managed by dosing adjustments or having patients come in for medication refills and to verify the flow rate sooner than every 2 weeks if the pump appears to be flowing more than 15% outside its labeled specification.
Questions about the recall can be directed to Intera Oncology at (800) 660-2660 or support@interaoncol.
A version of this article first appeared on Medscape.com.
Although no injuries or deaths related to the pump malfunction have been reported yet, the U.S. Food and Drug Administration has deemed the recall Class I, the most serious category that indicates the device could cause injury or death.
Intera Oncology initiated the recall in July following reports from clinicians that the pumps, which are implanted to deliver chemotherapy to treat liver tumors, were delivering medications faster than expected. A fast flow rate can lead to life-threatening hematologic toxicity, neurotoxicity, or death. It also means patients will run out of medication too soon, potentially leading to disease progression or death.
The FDA notice states the company has advised customers to continue to monitor flow rate as per standard refill procedure as well as monitor for liver toxicity to adjust dosing as per standard protocols.
The company also said to consider pump replacement if altered flow can’t be adequately managed by dosing adjustments or having patients come in for medication refills and to verify the flow rate sooner than every 2 weeks if the pump appears to be flowing more than 15% outside its labeled specification.
Questions about the recall can be directed to Intera Oncology at (800) 660-2660 or support@interaoncol.
A version of this article first appeared on Medscape.com.
Although no injuries or deaths related to the pump malfunction have been reported yet, the U.S. Food and Drug Administration has deemed the recall Class I, the most serious category that indicates the device could cause injury or death.
Intera Oncology initiated the recall in July following reports from clinicians that the pumps, which are implanted to deliver chemotherapy to treat liver tumors, were delivering medications faster than expected. A fast flow rate can lead to life-threatening hematologic toxicity, neurotoxicity, or death. It also means patients will run out of medication too soon, potentially leading to disease progression or death.
The FDA notice states the company has advised customers to continue to monitor flow rate as per standard refill procedure as well as monitor for liver toxicity to adjust dosing as per standard protocols.
The company also said to consider pump replacement if altered flow can’t be adequately managed by dosing adjustments or having patients come in for medication refills and to verify the flow rate sooner than every 2 weeks if the pump appears to be flowing more than 15% outside its labeled specification.
Questions about the recall can be directed to Intera Oncology at (800) 660-2660 or support@interaoncol.
A version of this article first appeared on Medscape.com.
Children and COVID: New cases increase; hospital admissions could follow
New cases of COVID-19 in children were up again after 2 weeks of declines, and preliminary data suggest that hospitalizations may be on the rise as well.
, based on data collected by the American Academy of Pediatrics and the Children’s Hospital Association from state and territorial health departments.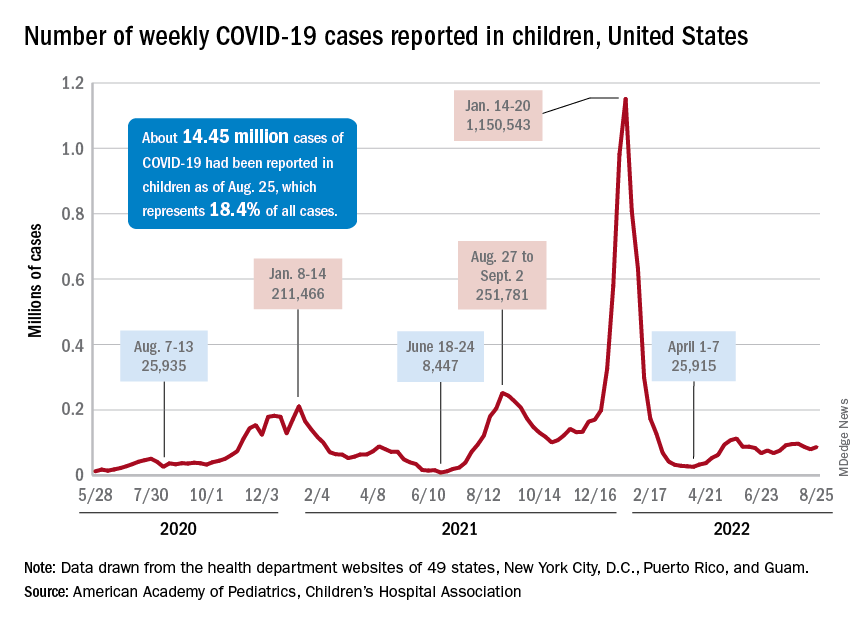
A similar increase seems to be reflected by hospital-level data. The latest 7-day (Aug. 21-27) average is 305 new admissions with diagnosed COVID per day among children aged 0-17 years, compared with 290 per day for the week of Aug. 14-20, the Centers for Disease Control and Prevention reported, while also noting the potential for reporting delays in the most recent 7-day period.
Daily hospital admissions for COVID had been headed downward through the first half of August, falling from 0.46 per 100,000 population at the end of July to 0.40 on Aug. 19, the CDC said on its COVID Data Tracker. Since then, however, admissions have gone the other way, with the preliminary nature of the latest data suggesting that the numbers will be even higher as more hospitals report over the next few days.
Vaccine initiations continue to fall
Initiations among school-age children have fallen for 3 consecutive weeks since Aug. 3, when numbers receiving their first vaccinations reached late-summer highs for those aged 5-11 and 12-17 years. Children under age 5, included in the CDC data for the first time on Aug. 11 as separate groups – under 2 years and 2-4 years – have had vaccine initiations drop by 8.0% and 19.8% over the 2 following weeks, the CDC said.
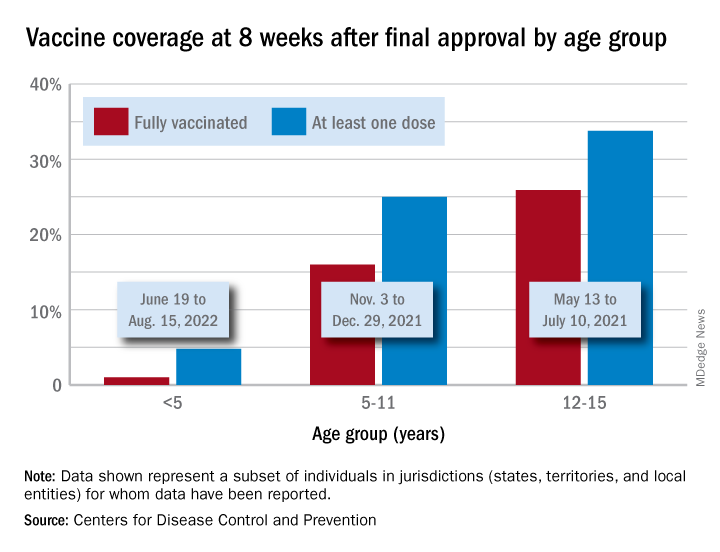
Through their first 8 weeks of vaccine eligibility (June 19 to Aug. 15), 4.8% of children under 5 years of age had received a first vaccination and 1.0% were fully vaccinated. For the two other age groups (5-11 and 12-15) who became eligible after the very first emergency authorization back in 2020, the respective proportions were 25.0% and 16.0% (5-11) and 33.8% and 26.1% (12-15) through the first 8 weeks, according to CDC data.
New cases of COVID-19 in children were up again after 2 weeks of declines, and preliminary data suggest that hospitalizations may be on the rise as well.
, based on data collected by the American Academy of Pediatrics and the Children’s Hospital Association from state and territorial health departments.
A similar increase seems to be reflected by hospital-level data. The latest 7-day (Aug. 21-27) average is 305 new admissions with diagnosed COVID per day among children aged 0-17 years, compared with 290 per day for the week of Aug. 14-20, the Centers for Disease Control and Prevention reported, while also noting the potential for reporting delays in the most recent 7-day period.
Daily hospital admissions for COVID had been headed downward through the first half of August, falling from 0.46 per 100,000 population at the end of July to 0.40 on Aug. 19, the CDC said on its COVID Data Tracker. Since then, however, admissions have gone the other way, with the preliminary nature of the latest data suggesting that the numbers will be even higher as more hospitals report over the next few days.
Vaccine initiations continue to fall
Initiations among school-age children have fallen for 3 consecutive weeks since Aug. 3, when numbers receiving their first vaccinations reached late-summer highs for those aged 5-11 and 12-17 years. Children under age 5, included in the CDC data for the first time on Aug. 11 as separate groups – under 2 years and 2-4 years – have had vaccine initiations drop by 8.0% and 19.8% over the 2 following weeks, the CDC said.
Through their first 8 weeks of vaccine eligibility (June 19 to Aug. 15), 4.8% of children under 5 years of age had received a first vaccination and 1.0% were fully vaccinated. For the two other age groups (5-11 and 12-15) who became eligible after the very first emergency authorization back in 2020, the respective proportions were 25.0% and 16.0% (5-11) and 33.8% and 26.1% (12-15) through the first 8 weeks, according to CDC data.
New cases of COVID-19 in children were up again after 2 weeks of declines, and preliminary data suggest that hospitalizations may be on the rise as well.
, based on data collected by the American Academy of Pediatrics and the Children’s Hospital Association from state and territorial health departments.
A similar increase seems to be reflected by hospital-level data. The latest 7-day (Aug. 21-27) average is 305 new admissions with diagnosed COVID per day among children aged 0-17 years, compared with 290 per day for the week of Aug. 14-20, the Centers for Disease Control and Prevention reported, while also noting the potential for reporting delays in the most recent 7-day period.
Daily hospital admissions for COVID had been headed downward through the first half of August, falling from 0.46 per 100,000 population at the end of July to 0.40 on Aug. 19, the CDC said on its COVID Data Tracker. Since then, however, admissions have gone the other way, with the preliminary nature of the latest data suggesting that the numbers will be even higher as more hospitals report over the next few days.
Vaccine initiations continue to fall
Initiations among school-age children have fallen for 3 consecutive weeks since Aug. 3, when numbers receiving their first vaccinations reached late-summer highs for those aged 5-11 and 12-17 years. Children under age 5, included in the CDC data for the first time on Aug. 11 as separate groups – under 2 years and 2-4 years – have had vaccine initiations drop by 8.0% and 19.8% over the 2 following weeks, the CDC said.
Through their first 8 weeks of vaccine eligibility (June 19 to Aug. 15), 4.8% of children under 5 years of age had received a first vaccination and 1.0% were fully vaccinated. For the two other age groups (5-11 and 12-15) who became eligible after the very first emergency authorization back in 2020, the respective proportions were 25.0% and 16.0% (5-11) and 33.8% and 26.1% (12-15) through the first 8 weeks, according to CDC data.
COVID to blame as U.S. life expectancy falls
All 50 states and the District of Columbia saw drops in life expectancy, according to the report from the Centers for Disease Control and Prevention’s National Center for Health Statistics.
The declines were mostly because of COVID-19 and “unintentional injuries,” such as drug overdoses.
The overall drop took national life expectancy from 78.8 years in 2019 to 77 years in 2020, the first year of the pandemic, ABC News reported.
States in the West and Northwest generally had higher life expectancy, with states in the South having the lowest.
Hawaii had the highest life expectancy at 80.7 years. It was followed by Washington, Minnesota, California, and Massachusetts. Mississippi had the lowest at 71.9 years, the figures show. The others in the bottom five were West Virginia, Louisiana, Alabama, and Kentucky.
In 2020, COVID-19 was the third-highest cause of death, leading to more than 350,000, the CDC reported earlier this year. At the same time, more people are dying annually from drug overdoses. A record 83,500 fatal overdoses were reported in 2020.
A version of this article first appeared on WebMD.com.
All 50 states and the District of Columbia saw drops in life expectancy, according to the report from the Centers for Disease Control and Prevention’s National Center for Health Statistics.
The declines were mostly because of COVID-19 and “unintentional injuries,” such as drug overdoses.
The overall drop took national life expectancy from 78.8 years in 2019 to 77 years in 2020, the first year of the pandemic, ABC News reported.
States in the West and Northwest generally had higher life expectancy, with states in the South having the lowest.
Hawaii had the highest life expectancy at 80.7 years. It was followed by Washington, Minnesota, California, and Massachusetts. Mississippi had the lowest at 71.9 years, the figures show. The others in the bottom five were West Virginia, Louisiana, Alabama, and Kentucky.
In 2020, COVID-19 was the third-highest cause of death, leading to more than 350,000, the CDC reported earlier this year. At the same time, more people are dying annually from drug overdoses. A record 83,500 fatal overdoses were reported in 2020.
A version of this article first appeared on WebMD.com.
All 50 states and the District of Columbia saw drops in life expectancy, according to the report from the Centers for Disease Control and Prevention’s National Center for Health Statistics.
The declines were mostly because of COVID-19 and “unintentional injuries,” such as drug overdoses.
The overall drop took national life expectancy from 78.8 years in 2019 to 77 years in 2020, the first year of the pandemic, ABC News reported.
States in the West and Northwest generally had higher life expectancy, with states in the South having the lowest.
Hawaii had the highest life expectancy at 80.7 years. It was followed by Washington, Minnesota, California, and Massachusetts. Mississippi had the lowest at 71.9 years, the figures show. The others in the bottom five were West Virginia, Louisiana, Alabama, and Kentucky.
In 2020, COVID-19 was the third-highest cause of death, leading to more than 350,000, the CDC reported earlier this year. At the same time, more people are dying annually from drug overdoses. A record 83,500 fatal overdoses were reported in 2020.
A version of this article first appeared on WebMD.com.
Children and COVID: New cases fall again, ED rates rebound for some
The 7-day average percentage of ED visits with diagnosed COVID, which had reached a post-Omicron high of 3.5% in late July for those aged 12-15, began to fall and was down to 3.0% on Aug. 12. That trend reversed, however, and the rate was up to 3.6% on Aug. 19, the last date for which data are available from the Centers for Disease Control and Prevention.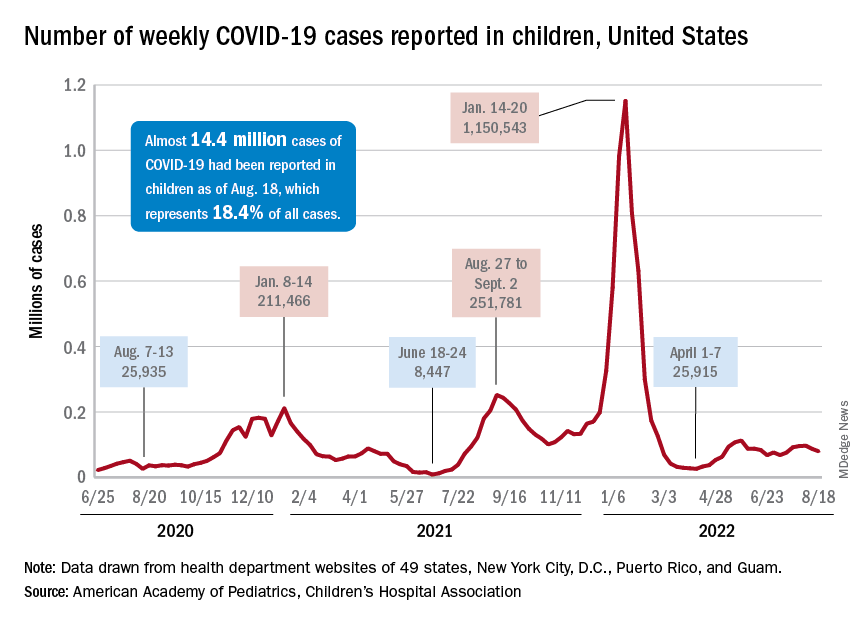
That change of COVID fortunes cannot yet be seen for all children. The 7-day average ED visit rate for those aged 0-11 years peaked at 6.8% during the last week of July and has continued to fall, dropping from 5.7% on Aug. 12 to 5.1% on Aug. 19. Children aged 16-17 years seem to be taking a middle path: Their ED-visit rate declined from late July into mid-August but held steady over the last week, according to the CDC’s COVID Data Tracker.
There is a hint of the same trend regarding new admissions among children aged 0-17 years. The national rate, which had declined in recent weeks, ticked up from 0.42 to 0.43 new admissions per 100,000 population over the last week of available data, the CDC said.
Weekly cases fall below 80,000
New cases in general were down by 8.5% from the previous week, dropping from 87,902 for the week of Aug. 5-11 to 79,525 for Aug. 12-18. That marked the second straight week with fewer cases after a 4-week period that saw weekly totals increase from almost 68,000 to nearly 97,000, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
The AAP and CHA put the cumulative number of child COVID-19 cases at just under 14.4 million since the pandemic began, which represents 18.4% of cases among all ages. The CDC estimates that there have been almost 14.7 million cases in children aged 0-17 years, as well as 1,750 deaths, of which 14 were reported in the last week (Aug. 16-22).
The CDC age subgroups indicate that children aged 0-4 years have experienced fewer cases (2.9 million) than children aged 5-11 years (5.6 million cases) and 12-15 (3.0 million cases) but more deaths: 548 so far, versus 432 for 5- to 11-year-olds and 437 for 12- to 15-year-olds, the COVID Data Tracker shows. Those aged 0-4 make up 6% of the total U.S. population, compared with 8.7% and 5.1%, respectively, for the older children.
Most younger children still not vaccinated
Although it may not qualify as a big push to vaccinate children before the start of the new school year, first-time vaccinations did rise somewhat in late July and August for children aged 5-17 years. Among children younger than 5 years, though, initial doses of the vaccine fell during the second full week of August, especially in 2- to 4-year-olds, based on the CDC data.
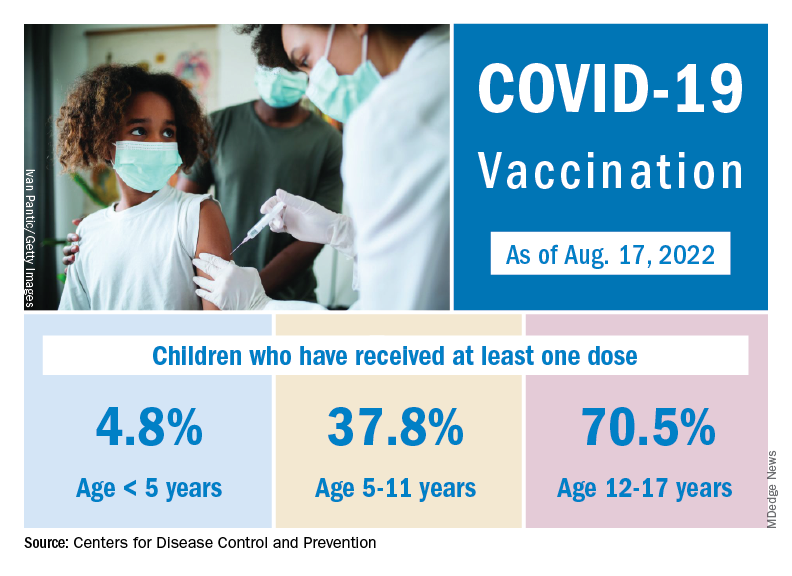
Through almost 2 months of vaccine eligibility, 4.8% of children under age 5 have received at least one dose and 0.9% are fully vaccinated as of Aug. 17. The current rates are 37.8% (one dose) and 30.4% (completed) for those aged 5-11 and 70.5% and 60.3% for 12- to 17-year-olds.
The 7-day average percentage of ED visits with diagnosed COVID, which had reached a post-Omicron high of 3.5% in late July for those aged 12-15, began to fall and was down to 3.0% on Aug. 12. That trend reversed, however, and the rate was up to 3.6% on Aug. 19, the last date for which data are available from the Centers for Disease Control and Prevention.
That change of COVID fortunes cannot yet be seen for all children. The 7-day average ED visit rate for those aged 0-11 years peaked at 6.8% during the last week of July and has continued to fall, dropping from 5.7% on Aug. 12 to 5.1% on Aug. 19. Children aged 16-17 years seem to be taking a middle path: Their ED-visit rate declined from late July into mid-August but held steady over the last week, according to the CDC’s COVID Data Tracker.
There is a hint of the same trend regarding new admissions among children aged 0-17 years. The national rate, which had declined in recent weeks, ticked up from 0.42 to 0.43 new admissions per 100,000 population over the last week of available data, the CDC said.
Weekly cases fall below 80,000
New cases in general were down by 8.5% from the previous week, dropping from 87,902 for the week of Aug. 5-11 to 79,525 for Aug. 12-18. That marked the second straight week with fewer cases after a 4-week period that saw weekly totals increase from almost 68,000 to nearly 97,000, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
The AAP and CHA put the cumulative number of child COVID-19 cases at just under 14.4 million since the pandemic began, which represents 18.4% of cases among all ages. The CDC estimates that there have been almost 14.7 million cases in children aged 0-17 years, as well as 1,750 deaths, of which 14 were reported in the last week (Aug. 16-22).
The CDC age subgroups indicate that children aged 0-4 years have experienced fewer cases (2.9 million) than children aged 5-11 years (5.6 million cases) and 12-15 (3.0 million cases) but more deaths: 548 so far, versus 432 for 5- to 11-year-olds and 437 for 12- to 15-year-olds, the COVID Data Tracker shows. Those aged 0-4 make up 6% of the total U.S. population, compared with 8.7% and 5.1%, respectively, for the older children.
Most younger children still not vaccinated
Although it may not qualify as a big push to vaccinate children before the start of the new school year, first-time vaccinations did rise somewhat in late July and August for children aged 5-17 years. Among children younger than 5 years, though, initial doses of the vaccine fell during the second full week of August, especially in 2- to 4-year-olds, based on the CDC data.
Through almost 2 months of vaccine eligibility, 4.8% of children under age 5 have received at least one dose and 0.9% are fully vaccinated as of Aug. 17. The current rates are 37.8% (one dose) and 30.4% (completed) for those aged 5-11 and 70.5% and 60.3% for 12- to 17-year-olds.
The 7-day average percentage of ED visits with diagnosed COVID, which had reached a post-Omicron high of 3.5% in late July for those aged 12-15, began to fall and was down to 3.0% on Aug. 12. That trend reversed, however, and the rate was up to 3.6% on Aug. 19, the last date for which data are available from the Centers for Disease Control and Prevention.
That change of COVID fortunes cannot yet be seen for all children. The 7-day average ED visit rate for those aged 0-11 years peaked at 6.8% during the last week of July and has continued to fall, dropping from 5.7% on Aug. 12 to 5.1% on Aug. 19. Children aged 16-17 years seem to be taking a middle path: Their ED-visit rate declined from late July into mid-August but held steady over the last week, according to the CDC’s COVID Data Tracker.
There is a hint of the same trend regarding new admissions among children aged 0-17 years. The national rate, which had declined in recent weeks, ticked up from 0.42 to 0.43 new admissions per 100,000 population over the last week of available data, the CDC said.
Weekly cases fall below 80,000
New cases in general were down by 8.5% from the previous week, dropping from 87,902 for the week of Aug. 5-11 to 79,525 for Aug. 12-18. That marked the second straight week with fewer cases after a 4-week period that saw weekly totals increase from almost 68,000 to nearly 97,000, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
The AAP and CHA put the cumulative number of child COVID-19 cases at just under 14.4 million since the pandemic began, which represents 18.4% of cases among all ages. The CDC estimates that there have been almost 14.7 million cases in children aged 0-17 years, as well as 1,750 deaths, of which 14 were reported in the last week (Aug. 16-22).
The CDC age subgroups indicate that children aged 0-4 years have experienced fewer cases (2.9 million) than children aged 5-11 years (5.6 million cases) and 12-15 (3.0 million cases) but more deaths: 548 so far, versus 432 for 5- to 11-year-olds and 437 for 12- to 15-year-olds, the COVID Data Tracker shows. Those aged 0-4 make up 6% of the total U.S. population, compared with 8.7% and 5.1%, respectively, for the older children.
Most younger children still not vaccinated
Although it may not qualify as a big push to vaccinate children before the start of the new school year, first-time vaccinations did rise somewhat in late July and August for children aged 5-17 years. Among children younger than 5 years, though, initial doses of the vaccine fell during the second full week of August, especially in 2- to 4-year-olds, based on the CDC data.
Through almost 2 months of vaccine eligibility, 4.8% of children under age 5 have received at least one dose and 0.9% are fully vaccinated as of Aug. 17. The current rates are 37.8% (one dose) and 30.4% (completed) for those aged 5-11 and 70.5% and 60.3% for 12- to 17-year-olds.
FDA approves first gene therapy, betibeglogene autotemcel (Zynteglo), for beta-thalassemia

Betibeglogene autotemcel, a one-time gene therapy, represents a potential cure in which functional copies of the mutated gene are inserted into patients’ hematopoietic stem cells via a replication-defective lentivirus.
“Today’s approval is an important advance in the treatment of beta-thalassemia, particularly in individuals who require ongoing red blood cell transfusions,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in an FDA press release. “Given the potential health complications associated with this serious disease, this action highlights the FDA’s continued commitment to supporting development of innovative therapies for patients who have limited treatment options.”
The approval was based on phase 3 trials, in which 89% of 41 patients aged 4-34 years who received the therapy maintained normal or near-normal hemoglobin levels and didn’t need transfusions for at least a year. The patients were as young as age 4, maker Bluebird Bio said in a press release.
FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee unanimously recommended approval in June. The gene therapy had been approved in Europe, where it carried a price tag of about $1.8 million, but Bluebird pulled it from the market in 2021 because of problems with reimbursement.
“The decision to discontinue operations in Europe resulted from prolonged negotiations with European payers and challenges to achieving appropriate value recognition and market access,” the company said in a Securities and Exchange Commission filing.
The projected price in the United States is even higher: $2.1 million.
But the Institute for Clinical and Economic Review, an influential Boston-based nonprofit organization that specializes in medical cost-effectiveness analyses, concluded in June that, “given the high annual costs of standard care ... this new treatment meets commonly accepted value thresholds at an anticipated price of $2.1 million,” particularly with Bluebird’s proposal to pay back 80% of the cost if patients need a transfusion within 5 years.
The company is planning an October 2022 launch and estimates the U.S. market for betibeglogene autotemcel to be about 1,500 patients.
Adverse events in studies were “infrequent and consisted primarily of nonserious infusion-related reactions,” such as abdominal pain, hot flush, dyspnea, tachycardia, noncardiac chest pain, and cytopenias, including thrombocytopenia, leukopenia, and neutropenia. One case of thrombocytopenia was considered serious but resolved, according to the company.
Most of the serious adverse events were related to hematopoietic stem cell collection and the busulfan conditioning regimen. Insertional oncogenesis and/or cancer have been reported with Bluebird’s other gene therapy products, but no cases have been associated with betibeglogene autotemcel.
A version of this article first appeared on Medscape.com.
Betibeglogene autotemcel, a one-time gene therapy, represents a potential cure in which functional copies of the mutated gene are inserted into patients’ hematopoietic stem cells via a replication-defective lentivirus.
“Today’s approval is an important advance in the treatment of beta-thalassemia, particularly in individuals who require ongoing red blood cell transfusions,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in an FDA press release. “Given the potential health complications associated with this serious disease, this action highlights the FDA’s continued commitment to supporting development of innovative therapies for patients who have limited treatment options.”
The approval was based on phase 3 trials, in which 89% of 41 patients aged 4-34 years who received the therapy maintained normal or near-normal hemoglobin levels and didn’t need transfusions for at least a year. The patients were as young as age 4, maker Bluebird Bio said in a press release.
FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee unanimously recommended approval in June. The gene therapy had been approved in Europe, where it carried a price tag of about $1.8 million, but Bluebird pulled it from the market in 2021 because of problems with reimbursement.
“The decision to discontinue operations in Europe resulted from prolonged negotiations with European payers and challenges to achieving appropriate value recognition and market access,” the company said in a Securities and Exchange Commission filing.
The projected price in the United States is even higher: $2.1 million.
But the Institute for Clinical and Economic Review, an influential Boston-based nonprofit organization that specializes in medical cost-effectiveness analyses, concluded in June that, “given the high annual costs of standard care ... this new treatment meets commonly accepted value thresholds at an anticipated price of $2.1 million,” particularly with Bluebird’s proposal to pay back 80% of the cost if patients need a transfusion within 5 years.
The company is planning an October 2022 launch and estimates the U.S. market for betibeglogene autotemcel to be about 1,500 patients.
Adverse events in studies were “infrequent and consisted primarily of nonserious infusion-related reactions,” such as abdominal pain, hot flush, dyspnea, tachycardia, noncardiac chest pain, and cytopenias, including thrombocytopenia, leukopenia, and neutropenia. One case of thrombocytopenia was considered serious but resolved, according to the company.
Most of the serious adverse events were related to hematopoietic stem cell collection and the busulfan conditioning regimen. Insertional oncogenesis and/or cancer have been reported with Bluebird’s other gene therapy products, but no cases have been associated with betibeglogene autotemcel.
A version of this article first appeared on Medscape.com.
Betibeglogene autotemcel, a one-time gene therapy, represents a potential cure in which functional copies of the mutated gene are inserted into patients’ hematopoietic stem cells via a replication-defective lentivirus.
“Today’s approval is an important advance in the treatment of beta-thalassemia, particularly in individuals who require ongoing red blood cell transfusions,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in an FDA press release. “Given the potential health complications associated with this serious disease, this action highlights the FDA’s continued commitment to supporting development of innovative therapies for patients who have limited treatment options.”
The approval was based on phase 3 trials, in which 89% of 41 patients aged 4-34 years who received the therapy maintained normal or near-normal hemoglobin levels and didn’t need transfusions for at least a year. The patients were as young as age 4, maker Bluebird Bio said in a press release.
FDA’s Cellular, Tissue, and Gene Therapies Advisory Committee unanimously recommended approval in June. The gene therapy had been approved in Europe, where it carried a price tag of about $1.8 million, but Bluebird pulled it from the market in 2021 because of problems with reimbursement.
“The decision to discontinue operations in Europe resulted from prolonged negotiations with European payers and challenges to achieving appropriate value recognition and market access,” the company said in a Securities and Exchange Commission filing.
The projected price in the United States is even higher: $2.1 million.
But the Institute for Clinical and Economic Review, an influential Boston-based nonprofit organization that specializes in medical cost-effectiveness analyses, concluded in June that, “given the high annual costs of standard care ... this new treatment meets commonly accepted value thresholds at an anticipated price of $2.1 million,” particularly with Bluebird’s proposal to pay back 80% of the cost if patients need a transfusion within 5 years.
The company is planning an October 2022 launch and estimates the U.S. market for betibeglogene autotemcel to be about 1,500 patients.
Adverse events in studies were “infrequent and consisted primarily of nonserious infusion-related reactions,” such as abdominal pain, hot flush, dyspnea, tachycardia, noncardiac chest pain, and cytopenias, including thrombocytopenia, leukopenia, and neutropenia. One case of thrombocytopenia was considered serious but resolved, according to the company.
Most of the serious adverse events were related to hematopoietic stem cell collection and the busulfan conditioning regimen. Insertional oncogenesis and/or cancer have been reported with Bluebird’s other gene therapy products, but no cases have been associated with betibeglogene autotemcel.
A version of this article first appeared on Medscape.com.
FDA approves adalimumab-bwwd biosimilar (Hadlima) in high-concentration form
The U.S. Food and Drug Administration today approved a citrate-free, high-concentration formulation of adalimumab-bwwd (Hadlima), the manufacturer, Samsung Bioepis, and its commercialization partner Organon said in an announcement.
Hadlima is a biosimilar of the tumor necrosis factor inhibitor reference product adalimumab (Humira).
Hadlima was first approved in July 2019 in a citrated, 50-mg/mL formulation. The new citrate-free, 100-mg/mL version will be available in prefilled syringe and autoinjector options.
The 100-mg/mL formulation is indicated for the same seven conditions as its 50-mg/mL counterpart: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn’s disease, and ulcerative colitis.
The approval was based on clinical data from a randomized, single-blind, two-arm, parallel group, single-dose study that compared the pharmacokinetics, safety, tolerability, and immunogenicity of the 100-mg/mL and 50-mg/mL formulations of Hadlima in healthy volunteers.
Both low- and high-concentration formulations of Humira are currently marketed in the United States. Organon said that it expects to market Hadlima in the United States on or after July 1, 2023, in accordance with a licensing agreement with AbbVie.
The prescribing information for Hadlima includes specific warnings and areas of concern. The drug should not be administered to individuals who are known to be hypersensitive to adalimumab. The drug may lower the ability of the immune system to fight infections and may increase risk of infections, including serious infections leading to hospitalization or death, such as tuberculosis, bacterial sepsis, invasive fungal infections (such as histoplasmosis), and infections attributable to other opportunistic pathogens.
A test for latent TB infection should be given before administration, and treatment of TB should begin before administration of Hadlima.
Patients taking Hadlima should not take a live vaccine.
The most common adverse effects (incidence > 10%) include infections (for example, upper respiratory infections, sinusitis), injection site reactions, headache, and rash.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration today approved a citrate-free, high-concentration formulation of adalimumab-bwwd (Hadlima), the manufacturer, Samsung Bioepis, and its commercialization partner Organon said in an announcement.
Hadlima is a biosimilar of the tumor necrosis factor inhibitor reference product adalimumab (Humira).
Hadlima was first approved in July 2019 in a citrated, 50-mg/mL formulation. The new citrate-free, 100-mg/mL version will be available in prefilled syringe and autoinjector options.
The 100-mg/mL formulation is indicated for the same seven conditions as its 50-mg/mL counterpart: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn’s disease, and ulcerative colitis.
The approval was based on clinical data from a randomized, single-blind, two-arm, parallel group, single-dose study that compared the pharmacokinetics, safety, tolerability, and immunogenicity of the 100-mg/mL and 50-mg/mL formulations of Hadlima in healthy volunteers.
Both low- and high-concentration formulations of Humira are currently marketed in the United States. Organon said that it expects to market Hadlima in the United States on or after July 1, 2023, in accordance with a licensing agreement with AbbVie.
The prescribing information for Hadlima includes specific warnings and areas of concern. The drug should not be administered to individuals who are known to be hypersensitive to adalimumab. The drug may lower the ability of the immune system to fight infections and may increase risk of infections, including serious infections leading to hospitalization or death, such as tuberculosis, bacterial sepsis, invasive fungal infections (such as histoplasmosis), and infections attributable to other opportunistic pathogens.
A test for latent TB infection should be given before administration, and treatment of TB should begin before administration of Hadlima.
Patients taking Hadlima should not take a live vaccine.
The most common adverse effects (incidence > 10%) include infections (for example, upper respiratory infections, sinusitis), injection site reactions, headache, and rash.
A version of this article first appeared on Medscape.com.
The U.S. Food and Drug Administration today approved a citrate-free, high-concentration formulation of adalimumab-bwwd (Hadlima), the manufacturer, Samsung Bioepis, and its commercialization partner Organon said in an announcement.
Hadlima is a biosimilar of the tumor necrosis factor inhibitor reference product adalimumab (Humira).
Hadlima was first approved in July 2019 in a citrated, 50-mg/mL formulation. The new citrate-free, 100-mg/mL version will be available in prefilled syringe and autoinjector options.
The 100-mg/mL formulation is indicated for the same seven conditions as its 50-mg/mL counterpart: rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, plaque psoriasis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn’s disease, and ulcerative colitis.
The approval was based on clinical data from a randomized, single-blind, two-arm, parallel group, single-dose study that compared the pharmacokinetics, safety, tolerability, and immunogenicity of the 100-mg/mL and 50-mg/mL formulations of Hadlima in healthy volunteers.
Both low- and high-concentration formulations of Humira are currently marketed in the United States. Organon said that it expects to market Hadlima in the United States on or after July 1, 2023, in accordance with a licensing agreement with AbbVie.
The prescribing information for Hadlima includes specific warnings and areas of concern. The drug should not be administered to individuals who are known to be hypersensitive to adalimumab. The drug may lower the ability of the immune system to fight infections and may increase risk of infections, including serious infections leading to hospitalization or death, such as tuberculosis, bacterial sepsis, invasive fungal infections (such as histoplasmosis), and infections attributable to other opportunistic pathogens.
A test for latent TB infection should be given before administration, and treatment of TB should begin before administration of Hadlima.
Patients taking Hadlima should not take a live vaccine.
The most common adverse effects (incidence > 10%) include infections (for example, upper respiratory infections, sinusitis), injection site reactions, headache, and rash.
A version of this article first appeared on Medscape.com.