User login
FDA approves anifrolumab (Saphnelo) as first new lupus treatment in more than 10 years
Anifrolumab, an inhibitor of type 1 interferons, received approval from the Food and Drug Administration for the treatment of adults with moderate to severe systemic lupus erythematosus (SLE) who are receiving standard therapy, according to a statement released Aug. 2 from its manufacturer, AstraZeneca.

Anifrolumab will be marketed as Saphnelo. It is a fully human monoclonal antibody against subunit 1 of the type 1 interferon receptor, and its approval represents the only new treatment approved for patients with SLE in a decade. The recommended dosage is 300 mg as an intravenous infusion over a 30-minute period every 4 weeks, according to its prescribing information, and it will be sold in a single-dose vial containing 300 mg/2 mL (150 mg/mL).
Increased type I interferon (IFN) signaling is associated with increased disease activity in patients with SLE, and the option of a type I IFN receptor antagonist may allow physicians to treat patients with fewer corticosteroids, according to the statement.
The approval was based on data from three trials. The TULIP (Treatment of Uncontrolled Lupus via the Interferon Pathway) phase 3 research included two randomized, double-blind, placebo-controlled studies, TULIP-1 and TULIP-2. The TULIP trials each enrolled seropositive patients with moderate to severe active disease despite standard-of-care therapy (SOC), which included oral corticosteroids, antimalarials, and immunosuppressants (methotrexate, azathioprine, or mycophenolate mofetil). All patients met American College of Rheumatology criteria and had an SLE Disease Activity Index (SLEDAI)-2K of 6 or greater, as well as British Isles Lupus Assessment Group (BILAG) index scoring showing one or more organ systems with grade A involvement or two or more with grade B. Both trials required stable SOC therapy throughout the study except for mandatory attempts at oral corticosteroid tapering for patients who were receiving 10 mg/day or more of prednisone or its equivalent at study entry.
TULIP-1 failed to meet its primary endpoint of SLE Responder Index (SRI) at 52 weeks, but investigators determined after the trial that some patients taking anifrolumab had been inappropriately labeled as nonresponders because the trial automatically required any patient who used a restricted drug, including NSAIDs, to be classified as a nonresponder even if they used the medication for something unrelated to SLE. When these rules were amended in a post hoc analysis, differences between the groups treated with anifrolumab and placebo widened in secondary endpoints for oral corticosteroid dose reduction, Cutaneous Lupus Erythematosus Disease Activity Severity Index response, and BILAG-Based Composite Lupus Assessment (BICLA) response.
The TULIP-2 trial included 362 patients who received a fixed dose of 300 mg anifrolumab or a placebo intravenously every 4 weeks for 48 weeks. In this study, anifrolumab patients showed significant improvement in disease activity on the BICLA scale, compared with placebo patients. The BICLA response was 47.8% in patients taking anifrolumab and 31.5% in placebo-treated patients (P = .001).
In the MUSE phase 2 trial, 305 adults with SLE were randomized to a fixed-dose intravenous infusion of 300 mg or 1,000 mg of anifrolumab or a placebo every 4 weeks, plus SOC, for 48 weeks. Patients in this study showed significant improvement on either dose, compared with placebo.
The results from the MUSE trial were published online in Arthritis & Rheumatology Nov. 7, 2016, followed by the TULIP-1 trial in The Lancet Rheumatology Nov. 11, 2019, and the TULIP-2 trial in the New England Journal of Medicine Jan. 16, 2020.
The most common treatment-related adverse events in all three studies were nasopharyngitis, upper respiratory tract infection, bronchitis, infusion-related reactions, herpes zoster, and cough. Infusion-related reactions in the trials were similar in anifrolumab and placebo patients, and included headache, nausea, vomiting, fatigue, and dizziness.
Anifrolumab has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus and is not recommended for these patients, according to the statement.
AstraZeneca said in its statement that anifrolumab is also under regulatory review in Japan and the European Union, and it continues to evaluate anifrolumab in patients with SLE in a long-term extension phase 3 trial and a phase 3 trial assessing subcutaneous delivery. The company said it “is exploring the potential of Saphnelo in a variety of diseases where type I IFN plays a key role, including lupus nephritis, cutaneous lupus erythematosus, and myositis.”
Anifrolumab, an inhibitor of type 1 interferons, received approval from the Food and Drug Administration for the treatment of adults with moderate to severe systemic lupus erythematosus (SLE) who are receiving standard therapy, according to a statement released Aug. 2 from its manufacturer, AstraZeneca.
Anifrolumab will be marketed as Saphnelo. It is a fully human monoclonal antibody against subunit 1 of the type 1 interferon receptor, and its approval represents the only new treatment approved for patients with SLE in a decade. The recommended dosage is 300 mg as an intravenous infusion over a 30-minute period every 4 weeks, according to its prescribing information, and it will be sold in a single-dose vial containing 300 mg/2 mL (150 mg/mL).
Increased type I interferon (IFN) signaling is associated with increased disease activity in patients with SLE, and the option of a type I IFN receptor antagonist may allow physicians to treat patients with fewer corticosteroids, according to the statement.
The approval was based on data from three trials. The TULIP (Treatment of Uncontrolled Lupus via the Interferon Pathway) phase 3 research included two randomized, double-blind, placebo-controlled studies, TULIP-1 and TULIP-2. The TULIP trials each enrolled seropositive patients with moderate to severe active disease despite standard-of-care therapy (SOC), which included oral corticosteroids, antimalarials, and immunosuppressants (methotrexate, azathioprine, or mycophenolate mofetil). All patients met American College of Rheumatology criteria and had an SLE Disease Activity Index (SLEDAI)-2K of 6 or greater, as well as British Isles Lupus Assessment Group (BILAG) index scoring showing one or more organ systems with grade A involvement or two or more with grade B. Both trials required stable SOC therapy throughout the study except for mandatory attempts at oral corticosteroid tapering for patients who were receiving 10 mg/day or more of prednisone or its equivalent at study entry.
TULIP-1 failed to meet its primary endpoint of SLE Responder Index (SRI) at 52 weeks, but investigators determined after the trial that some patients taking anifrolumab had been inappropriately labeled as nonresponders because the trial automatically required any patient who used a restricted drug, including NSAIDs, to be classified as a nonresponder even if they used the medication for something unrelated to SLE. When these rules were amended in a post hoc analysis, differences between the groups treated with anifrolumab and placebo widened in secondary endpoints for oral corticosteroid dose reduction, Cutaneous Lupus Erythematosus Disease Activity Severity Index response, and BILAG-Based Composite Lupus Assessment (BICLA) response.
The TULIP-2 trial included 362 patients who received a fixed dose of 300 mg anifrolumab or a placebo intravenously every 4 weeks for 48 weeks. In this study, anifrolumab patients showed significant improvement in disease activity on the BICLA scale, compared with placebo patients. The BICLA response was 47.8% in patients taking anifrolumab and 31.5% in placebo-treated patients (P = .001).
In the MUSE phase 2 trial, 305 adults with SLE were randomized to a fixed-dose intravenous infusion of 300 mg or 1,000 mg of anifrolumab or a placebo every 4 weeks, plus SOC, for 48 weeks. Patients in this study showed significant improvement on either dose, compared with placebo.
The results from the MUSE trial were published online in Arthritis & Rheumatology Nov. 7, 2016, followed by the TULIP-1 trial in The Lancet Rheumatology Nov. 11, 2019, and the TULIP-2 trial in the New England Journal of Medicine Jan. 16, 2020.
The most common treatment-related adverse events in all three studies were nasopharyngitis, upper respiratory tract infection, bronchitis, infusion-related reactions, herpes zoster, and cough. Infusion-related reactions in the trials were similar in anifrolumab and placebo patients, and included headache, nausea, vomiting, fatigue, and dizziness.
Anifrolumab has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus and is not recommended for these patients, according to the statement.
AstraZeneca said in its statement that anifrolumab is also under regulatory review in Japan and the European Union, and it continues to evaluate anifrolumab in patients with SLE in a long-term extension phase 3 trial and a phase 3 trial assessing subcutaneous delivery. The company said it “is exploring the potential of Saphnelo in a variety of diseases where type I IFN plays a key role, including lupus nephritis, cutaneous lupus erythematosus, and myositis.”
Anifrolumab, an inhibitor of type 1 interferons, received approval from the Food and Drug Administration for the treatment of adults with moderate to severe systemic lupus erythematosus (SLE) who are receiving standard therapy, according to a statement released Aug. 2 from its manufacturer, AstraZeneca.
Anifrolumab will be marketed as Saphnelo. It is a fully human monoclonal antibody against subunit 1 of the type 1 interferon receptor, and its approval represents the only new treatment approved for patients with SLE in a decade. The recommended dosage is 300 mg as an intravenous infusion over a 30-minute period every 4 weeks, according to its prescribing information, and it will be sold in a single-dose vial containing 300 mg/2 mL (150 mg/mL).
Increased type I interferon (IFN) signaling is associated with increased disease activity in patients with SLE, and the option of a type I IFN receptor antagonist may allow physicians to treat patients with fewer corticosteroids, according to the statement.
The approval was based on data from three trials. The TULIP (Treatment of Uncontrolled Lupus via the Interferon Pathway) phase 3 research included two randomized, double-blind, placebo-controlled studies, TULIP-1 and TULIP-2. The TULIP trials each enrolled seropositive patients with moderate to severe active disease despite standard-of-care therapy (SOC), which included oral corticosteroids, antimalarials, and immunosuppressants (methotrexate, azathioprine, or mycophenolate mofetil). All patients met American College of Rheumatology criteria and had an SLE Disease Activity Index (SLEDAI)-2K of 6 or greater, as well as British Isles Lupus Assessment Group (BILAG) index scoring showing one or more organ systems with grade A involvement or two or more with grade B. Both trials required stable SOC therapy throughout the study except for mandatory attempts at oral corticosteroid tapering for patients who were receiving 10 mg/day or more of prednisone or its equivalent at study entry.
TULIP-1 failed to meet its primary endpoint of SLE Responder Index (SRI) at 52 weeks, but investigators determined after the trial that some patients taking anifrolumab had been inappropriately labeled as nonresponders because the trial automatically required any patient who used a restricted drug, including NSAIDs, to be classified as a nonresponder even if they used the medication for something unrelated to SLE. When these rules were amended in a post hoc analysis, differences between the groups treated with anifrolumab and placebo widened in secondary endpoints for oral corticosteroid dose reduction, Cutaneous Lupus Erythematosus Disease Activity Severity Index response, and BILAG-Based Composite Lupus Assessment (BICLA) response.
The TULIP-2 trial included 362 patients who received a fixed dose of 300 mg anifrolumab or a placebo intravenously every 4 weeks for 48 weeks. In this study, anifrolumab patients showed significant improvement in disease activity on the BICLA scale, compared with placebo patients. The BICLA response was 47.8% in patients taking anifrolumab and 31.5% in placebo-treated patients (P = .001).
In the MUSE phase 2 trial, 305 adults with SLE were randomized to a fixed-dose intravenous infusion of 300 mg or 1,000 mg of anifrolumab or a placebo every 4 weeks, plus SOC, for 48 weeks. Patients in this study showed significant improvement on either dose, compared with placebo.
The results from the MUSE trial were published online in Arthritis & Rheumatology Nov. 7, 2016, followed by the TULIP-1 trial in The Lancet Rheumatology Nov. 11, 2019, and the TULIP-2 trial in the New England Journal of Medicine Jan. 16, 2020.
The most common treatment-related adverse events in all three studies were nasopharyngitis, upper respiratory tract infection, bronchitis, infusion-related reactions, herpes zoster, and cough. Infusion-related reactions in the trials were similar in anifrolumab and placebo patients, and included headache, nausea, vomiting, fatigue, and dizziness.
Anifrolumab has not been evaluated in patients with severe active lupus nephritis or severe active central nervous system lupus and is not recommended for these patients, according to the statement.
AstraZeneca said in its statement that anifrolumab is also under regulatory review in Japan and the European Union, and it continues to evaluate anifrolumab in patients with SLE in a long-term extension phase 3 trial and a phase 3 trial assessing subcutaneous delivery. The company said it “is exploring the potential of Saphnelo in a variety of diseases where type I IFN plays a key role, including lupus nephritis, cutaneous lupus erythematosus, and myositis.”
Children and COVID: Vaccinations, new cases both rising
COVID-19 vaccine initiations rose in U.S. children for the second consecutive week, but new pediatric cases jumped by 64% in just 1 week, according to new data.
the American Academy of Pediatrics and the Children’s Hospital Association said in their weekly COVID-19 report.
“After decreases in weekly reported cases over the past couple of months, in July we have seen steady increases in cases added to the cumulative total,” the AAP noted. In this latest reversal of COVID fortunes, the steady increase in new cases is in its fourth consecutive week since hitting a low of 8,447 in late June.
As of July 22, the total number of reported cases was over 4.12 million in 49 states, the District of Columbia, New York City, Puerto Rico, and Guam, and there have been 349 deaths in children in the 46 jurisdictions reporting age distributions of COVID-19 deaths, the AAP and CHA said in their report.
Meanwhile, over 9.3 million children received at least one dose of COVID vaccine as of July 26, according to the Centers for Disease Control and Prevention.
Vaccine initiation rose for the second week in a row after falling for several weeks as 301,000 children aged 12-15 years and almost 115,000 children aged 16-17 got their first dose during the week ending July 26. Children aged 12-15 represented 14.1% (up from 13.5% a week before) of all first vaccinations and 16- to 17-year-olds were 5.4% (up from 5.1%) of all vaccine initiators, according to the CDC’s COVID Data Tracker.
Just over 37% of all 12- to 15-year-olds have received at least one dose of the Pfizer-BioNTech vaccine since the CDC approved its use for children under age 16 in May, and almost 28% are fully vaccinated. Use in children aged 16-17 started earlier (December 2020), and 48% of that age group have received a first dose and over 39% have completed the vaccine regimen, the CDC said.
COVID-19 vaccine initiations rose in U.S. children for the second consecutive week, but new pediatric cases jumped by 64% in just 1 week, according to new data.
the American Academy of Pediatrics and the Children’s Hospital Association said in their weekly COVID-19 report.
“After decreases in weekly reported cases over the past couple of months, in July we have seen steady increases in cases added to the cumulative total,” the AAP noted. In this latest reversal of COVID fortunes, the steady increase in new cases is in its fourth consecutive week since hitting a low of 8,447 in late June.
As of July 22, the total number of reported cases was over 4.12 million in 49 states, the District of Columbia, New York City, Puerto Rico, and Guam, and there have been 349 deaths in children in the 46 jurisdictions reporting age distributions of COVID-19 deaths, the AAP and CHA said in their report.
Meanwhile, over 9.3 million children received at least one dose of COVID vaccine as of July 26, according to the Centers for Disease Control and Prevention.
Vaccine initiation rose for the second week in a row after falling for several weeks as 301,000 children aged 12-15 years and almost 115,000 children aged 16-17 got their first dose during the week ending July 26. Children aged 12-15 represented 14.1% (up from 13.5% a week before) of all first vaccinations and 16- to 17-year-olds were 5.4% (up from 5.1%) of all vaccine initiators, according to the CDC’s COVID Data Tracker.
Just over 37% of all 12- to 15-year-olds have received at least one dose of the Pfizer-BioNTech vaccine since the CDC approved its use for children under age 16 in May, and almost 28% are fully vaccinated. Use in children aged 16-17 started earlier (December 2020), and 48% of that age group have received a first dose and over 39% have completed the vaccine regimen, the CDC said.
COVID-19 vaccine initiations rose in U.S. children for the second consecutive week, but new pediatric cases jumped by 64% in just 1 week, according to new data.
the American Academy of Pediatrics and the Children’s Hospital Association said in their weekly COVID-19 report.
“After decreases in weekly reported cases over the past couple of months, in July we have seen steady increases in cases added to the cumulative total,” the AAP noted. In this latest reversal of COVID fortunes, the steady increase in new cases is in its fourth consecutive week since hitting a low of 8,447 in late June.
As of July 22, the total number of reported cases was over 4.12 million in 49 states, the District of Columbia, New York City, Puerto Rico, and Guam, and there have been 349 deaths in children in the 46 jurisdictions reporting age distributions of COVID-19 deaths, the AAP and CHA said in their report.
Meanwhile, over 9.3 million children received at least one dose of COVID vaccine as of July 26, according to the Centers for Disease Control and Prevention.
Vaccine initiation rose for the second week in a row after falling for several weeks as 301,000 children aged 12-15 years and almost 115,000 children aged 16-17 got their first dose during the week ending July 26. Children aged 12-15 represented 14.1% (up from 13.5% a week before) of all first vaccinations and 16- to 17-year-olds were 5.4% (up from 5.1%) of all vaccine initiators, according to the CDC’s COVID Data Tracker.
Just over 37% of all 12- to 15-year-olds have received at least one dose of the Pfizer-BioNTech vaccine since the CDC approved its use for children under age 16 in May, and almost 28% are fully vaccinated. Use in children aged 16-17 started earlier (December 2020), and 48% of that age group have received a first dose and over 39% have completed the vaccine regimen, the CDC said.
CDC panel updates info on rare side effect after J&J vaccine
Despite recent reports of Guillain-Barré Syndrome (GBS) after the Johnson & Johnson vaccine,

The company also presented new data suggesting that the shots generate strong immune responses against circulating variants and that antibodies generated by the vaccine stay elevated for at least 8 months.
Members of the Advisory Committee on Immunization Practices (ACIP) did not vote, but discussed and affirmed their support for recent decisions by the Food and Drug Administration and CDC to update patient information about the very low risk of GBS that appears to be associated with the vaccine, but to continue offering the vaccine to people in the United States.
The Johnson & Johnson shot has been a minor player in the U.S. vaccination campaign, accounting for less than 4% of all vaccine doses given in this country. Still, the single-dose inoculation, which doesn’t require ultra-cold storage, has been important for reaching people in rural areas, through mobile clinics, at colleges and primary care offices, and in vulnerable populations – those who are incarcerated or homeless.
The FDA says it has received reports of 100 cases of GBS after the Johnson & Johnson vaccine in its Vaccine Adverse Event Reporting System database through the end of June. The cases are still under investigation.
To date, more than 12 million doses of the vaccine have been administered, making the rate of GBS 8.1 cases for every million doses administered.
Although it is still extremely rare, that’s above the expected background rate of GBS of 1.6 cases for every million people, said Grace Lee, MD, a Stanford, Calif., pediatrician who chairs the ACIP’s Vaccine Safety Technical Work Group.
So far, most GBS cases (61%) have been among men. The midpoint age of the cases was 57 years. The average time to onset was 14 days, and 98% of cases occurred within 42 days of the shot. Facial paralysis has been associated with an estimated 30%-50% of cases. One person, who had heart failure, high blood pressure, and diabetes, has died.
Still, the benefits of the vaccine far outweigh its risks. For every million doses given to people over age 50, the vaccine prevents nearly 7,500 COVID-19 hospitalizations and nearly 100 deaths in women, and more than 13,000 COVID-19 hospitalizations and more than 2,400 deaths in men.
Rates of GBS after the mRNA vaccines made by Pfizer and Moderna were around 1 case for every 1 million doses given, which is not above the rate that would be expected without vaccination.
The link to the Johnson & Johnson vaccine prompted the FDA to add a warning to the vaccine’s patient safety information on July 12.
Also in July, the European Medicines Agency recommended a similar warning for the product information of the AstraZeneca vaccine Vaxzevria, which relies on similar technology.
Good against variants
Johnson & Johnson also presented new information showing its vaccine maintained high levels of neutralizing antibodies against four of the so-called “variants of concern” – Alpha, Gamma, Beta, and Delta. The protection generated by the vaccine lasted for at least 8 months after the shot, the company said.
“We’re still learning about the duration of protection and the breadth of coverage against this evolving variant landscape for each of the authorized vaccines,” said Mathai Mammen, MD, PhD, global head of research and development at Janssen, the company that makes the vaccine for J&J.
The company also said that its vaccine generated very strong T-cell responses. T cells destroy infected cells and, along with antibodies, are an important part of the body’s immune response.
Antibody levels and T-cell responses are markers for immunity. Measuring these levels isn’t the same as proving that shots can fend off an infection.
It’s still unclear exactly which component of the immune response is most important for fighting off COVID-19.
Dr. Mammen said the companies are still gathering that clinical data, and would present it soon.
“We will have a better view of the clinical efficacy in the coming weeks,” he said.
A version of this article first appeared on Medscape.com.
Despite recent reports of Guillain-Barré Syndrome (GBS) after the Johnson & Johnson vaccine,
The company also presented new data suggesting that the shots generate strong immune responses against circulating variants and that antibodies generated by the vaccine stay elevated for at least 8 months.
Members of the Advisory Committee on Immunization Practices (ACIP) did not vote, but discussed and affirmed their support for recent decisions by the Food and Drug Administration and CDC to update patient information about the very low risk of GBS that appears to be associated with the vaccine, but to continue offering the vaccine to people in the United States.
The Johnson & Johnson shot has been a minor player in the U.S. vaccination campaign, accounting for less than 4% of all vaccine doses given in this country. Still, the single-dose inoculation, which doesn’t require ultra-cold storage, has been important for reaching people in rural areas, through mobile clinics, at colleges and primary care offices, and in vulnerable populations – those who are incarcerated or homeless.
The FDA says it has received reports of 100 cases of GBS after the Johnson & Johnson vaccine in its Vaccine Adverse Event Reporting System database through the end of June. The cases are still under investigation.
To date, more than 12 million doses of the vaccine have been administered, making the rate of GBS 8.1 cases for every million doses administered.
Although it is still extremely rare, that’s above the expected background rate of GBS of 1.6 cases for every million people, said Grace Lee, MD, a Stanford, Calif., pediatrician who chairs the ACIP’s Vaccine Safety Technical Work Group.
So far, most GBS cases (61%) have been among men. The midpoint age of the cases was 57 years. The average time to onset was 14 days, and 98% of cases occurred within 42 days of the shot. Facial paralysis has been associated with an estimated 30%-50% of cases. One person, who had heart failure, high blood pressure, and diabetes, has died.
Still, the benefits of the vaccine far outweigh its risks. For every million doses given to people over age 50, the vaccine prevents nearly 7,500 COVID-19 hospitalizations and nearly 100 deaths in women, and more than 13,000 COVID-19 hospitalizations and more than 2,400 deaths in men.
Rates of GBS after the mRNA vaccines made by Pfizer and Moderna were around 1 case for every 1 million doses given, which is not above the rate that would be expected without vaccination.
The link to the Johnson & Johnson vaccine prompted the FDA to add a warning to the vaccine’s patient safety information on July 12.
Also in July, the European Medicines Agency recommended a similar warning for the product information of the AstraZeneca vaccine Vaxzevria, which relies on similar technology.
Good against variants
Johnson & Johnson also presented new information showing its vaccine maintained high levels of neutralizing antibodies against four of the so-called “variants of concern” – Alpha, Gamma, Beta, and Delta. The protection generated by the vaccine lasted for at least 8 months after the shot, the company said.
“We’re still learning about the duration of protection and the breadth of coverage against this evolving variant landscape for each of the authorized vaccines,” said Mathai Mammen, MD, PhD, global head of research and development at Janssen, the company that makes the vaccine for J&J.
The company also said that its vaccine generated very strong T-cell responses. T cells destroy infected cells and, along with antibodies, are an important part of the body’s immune response.
Antibody levels and T-cell responses are markers for immunity. Measuring these levels isn’t the same as proving that shots can fend off an infection.
It’s still unclear exactly which component of the immune response is most important for fighting off COVID-19.
Dr. Mammen said the companies are still gathering that clinical data, and would present it soon.
“We will have a better view of the clinical efficacy in the coming weeks,” he said.
A version of this article first appeared on Medscape.com.
Despite recent reports of Guillain-Barré Syndrome (GBS) after the Johnson & Johnson vaccine,
The company also presented new data suggesting that the shots generate strong immune responses against circulating variants and that antibodies generated by the vaccine stay elevated for at least 8 months.
Members of the Advisory Committee on Immunization Practices (ACIP) did not vote, but discussed and affirmed their support for recent decisions by the Food and Drug Administration and CDC to update patient information about the very low risk of GBS that appears to be associated with the vaccine, but to continue offering the vaccine to people in the United States.
The Johnson & Johnson shot has been a minor player in the U.S. vaccination campaign, accounting for less than 4% of all vaccine doses given in this country. Still, the single-dose inoculation, which doesn’t require ultra-cold storage, has been important for reaching people in rural areas, through mobile clinics, at colleges and primary care offices, and in vulnerable populations – those who are incarcerated or homeless.
The FDA says it has received reports of 100 cases of GBS after the Johnson & Johnson vaccine in its Vaccine Adverse Event Reporting System database through the end of June. The cases are still under investigation.
To date, more than 12 million doses of the vaccine have been administered, making the rate of GBS 8.1 cases for every million doses administered.
Although it is still extremely rare, that’s above the expected background rate of GBS of 1.6 cases for every million people, said Grace Lee, MD, a Stanford, Calif., pediatrician who chairs the ACIP’s Vaccine Safety Technical Work Group.
So far, most GBS cases (61%) have been among men. The midpoint age of the cases was 57 years. The average time to onset was 14 days, and 98% of cases occurred within 42 days of the shot. Facial paralysis has been associated with an estimated 30%-50% of cases. One person, who had heart failure, high blood pressure, and diabetes, has died.
Still, the benefits of the vaccine far outweigh its risks. For every million doses given to people over age 50, the vaccine prevents nearly 7,500 COVID-19 hospitalizations and nearly 100 deaths in women, and more than 13,000 COVID-19 hospitalizations and more than 2,400 deaths in men.
Rates of GBS after the mRNA vaccines made by Pfizer and Moderna were around 1 case for every 1 million doses given, which is not above the rate that would be expected without vaccination.
The link to the Johnson & Johnson vaccine prompted the FDA to add a warning to the vaccine’s patient safety information on July 12.
Also in July, the European Medicines Agency recommended a similar warning for the product information of the AstraZeneca vaccine Vaxzevria, which relies on similar technology.
Good against variants
Johnson & Johnson also presented new information showing its vaccine maintained high levels of neutralizing antibodies against four of the so-called “variants of concern” – Alpha, Gamma, Beta, and Delta. The protection generated by the vaccine lasted for at least 8 months after the shot, the company said.
“We’re still learning about the duration of protection and the breadth of coverage against this evolving variant landscape for each of the authorized vaccines,” said Mathai Mammen, MD, PhD, global head of research and development at Janssen, the company that makes the vaccine for J&J.
The company also said that its vaccine generated very strong T-cell responses. T cells destroy infected cells and, along with antibodies, are an important part of the body’s immune response.
Antibody levels and T-cell responses are markers for immunity. Measuring these levels isn’t the same as proving that shots can fend off an infection.
It’s still unclear exactly which component of the immune response is most important for fighting off COVID-19.
Dr. Mammen said the companies are still gathering that clinical data, and would present it soon.
“We will have a better view of the clinical efficacy in the coming weeks,” he said.
A version of this article first appeared on Medscape.com.
FDA approves intravenous immunoglobulin for dermatomyositis
, according to a statement from manufacturer Octapharma USA.

Dermatomyositis is a rare, idiopathic autoimmune disorder that affects approximately 10 out of every million people in the United States, mainly adults in their late 40s to early 60s, according to the company, but children aged 5-15 years can be affected. The disease is characterized by skin rashes, chronic muscle inflammation, progressive muscle weakness, and risk for mortality that is three times higher than for the general population.
There are no previously approved treatments for dermatomyositis prior to Octagam 10%, which also is indicated for chronic immune thrombocytopenic purpura in adults.
The approval for dermatomyositis was based on the results of a phase 3 randomized, double-blind, placebo-controlled clinical trial (the ProDERM trial) that included 95 adult patients at 36 sites worldwide, with 17 sites in the United States. In the trial, 78.7% of patients with dermatomyositis who were randomized to receive 2 g/kg of Octagam 10% every 4 weeks showed response at 16 weeks, compared with 43.8% of patients who received placebo. Response was based on the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria. Placebo patients who switched to intravenous immunoglobulin (IVIG) during a trial extension had response rates at week 40 similar to the original patients at week 16.
“The study gives clinicians much more confidence in the efficacy and safety of intravenous immunoglobulin and provides valuable information about what type of patient is best suited for the treatment,” Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh and a member of the ProDERM study Steering Committee, said in the Octapharma statement.
Safety and tolerability were similar to profiles seen with other IVIG medications, according to the statement. The medication does carry a boxed warning from its chronic ITP approval, cautioning about the potential for thrombosis, renal dysfunction, and acute renal failure.
The most common adverse reactions reported by dermatomyositis patients in the ProDERM trial were headache, fever, nausea, vomiting, increased blood pressure, chills, musculoskeletal pain, increased heart rate, dyspnea, and reactions at the infusion sites.
Read the full prescribing information here.
, according to a statement from manufacturer Octapharma USA.
Dermatomyositis is a rare, idiopathic autoimmune disorder that affects approximately 10 out of every million people in the United States, mainly adults in their late 40s to early 60s, according to the company, but children aged 5-15 years can be affected. The disease is characterized by skin rashes, chronic muscle inflammation, progressive muscle weakness, and risk for mortality that is three times higher than for the general population.
There are no previously approved treatments for dermatomyositis prior to Octagam 10%, which also is indicated for chronic immune thrombocytopenic purpura in adults.
The approval for dermatomyositis was based on the results of a phase 3 randomized, double-blind, placebo-controlled clinical trial (the ProDERM trial) that included 95 adult patients at 36 sites worldwide, with 17 sites in the United States. In the trial, 78.7% of patients with dermatomyositis who were randomized to receive 2 g/kg of Octagam 10% every 4 weeks showed response at 16 weeks, compared with 43.8% of patients who received placebo. Response was based on the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria. Placebo patients who switched to intravenous immunoglobulin (IVIG) during a trial extension had response rates at week 40 similar to the original patients at week 16.
“The study gives clinicians much more confidence in the efficacy and safety of intravenous immunoglobulin and provides valuable information about what type of patient is best suited for the treatment,” Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh and a member of the ProDERM study Steering Committee, said in the Octapharma statement.
Safety and tolerability were similar to profiles seen with other IVIG medications, according to the statement. The medication does carry a boxed warning from its chronic ITP approval, cautioning about the potential for thrombosis, renal dysfunction, and acute renal failure.
The most common adverse reactions reported by dermatomyositis patients in the ProDERM trial were headache, fever, nausea, vomiting, increased blood pressure, chills, musculoskeletal pain, increased heart rate, dyspnea, and reactions at the infusion sites.
Read the full prescribing information here.
, according to a statement from manufacturer Octapharma USA.
Dermatomyositis is a rare, idiopathic autoimmune disorder that affects approximately 10 out of every million people in the United States, mainly adults in their late 40s to early 60s, according to the company, but children aged 5-15 years can be affected. The disease is characterized by skin rashes, chronic muscle inflammation, progressive muscle weakness, and risk for mortality that is three times higher than for the general population.
There are no previously approved treatments for dermatomyositis prior to Octagam 10%, which also is indicated for chronic immune thrombocytopenic purpura in adults.
The approval for dermatomyositis was based on the results of a phase 3 randomized, double-blind, placebo-controlled clinical trial (the ProDERM trial) that included 95 adult patients at 36 sites worldwide, with 17 sites in the United States. In the trial, 78.7% of patients with dermatomyositis who were randomized to receive 2 g/kg of Octagam 10% every 4 weeks showed response at 16 weeks, compared with 43.8% of patients who received placebo. Response was based on the 2016 American College of Rheumatology/European Alliance of Associations for Rheumatology myositis response criteria. Placebo patients who switched to intravenous immunoglobulin (IVIG) during a trial extension had response rates at week 40 similar to the original patients at week 16.
“The study gives clinicians much more confidence in the efficacy and safety of intravenous immunoglobulin and provides valuable information about what type of patient is best suited for the treatment,” Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh and a member of the ProDERM study Steering Committee, said in the Octapharma statement.
Safety and tolerability were similar to profiles seen with other IVIG medications, according to the statement. The medication does carry a boxed warning from its chronic ITP approval, cautioning about the potential for thrombosis, renal dysfunction, and acute renal failure.
The most common adverse reactions reported by dermatomyositis patients in the ProDERM trial were headache, fever, nausea, vomiting, increased blood pressure, chills, musculoskeletal pain, increased heart rate, dyspnea, and reactions at the infusion sites.
Read the full prescribing information here.
Children and COVID: New vaccinations increase as cases continue to climb
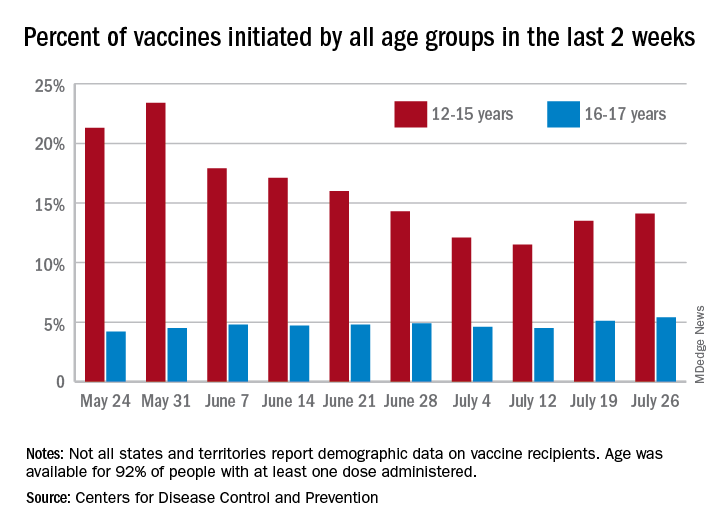
Children aged 12-15 years represented 13.5% of all first vaccinations received during the 2 weeks ending July 19, compared with 11.5% for the 2 weeks ending July 12, marking the first increase since the end of May. First vaccinations in 16- and 17-year-olds, who make up a much smaller share of the U.S. population, also went up, topping 5%, the Centers for Disease Control and Prevention said in its COVID Data Tracker.
The total number of vaccine initiations was almost 250,000 for the week ending July 19, after dropping to a low of 201,000 the previous week. Before that, first vaccinations had fallen in 5 of the previous 6 weeks, going from 1.4 million on May 24 to 307,000 on July 5, the CDC said.
New cases of COVID-19, unfortunately, continued to follow the trend among the larger population: As of July 15, weekly cases in children were up by 179% since dropping to 8,400 on June 24, the American Academy of Pediatrics and the Children’s Hospital Association said in a joint report. The 23,551 new cases in children for the week ending July 15 were 15.9% of all cases reported.
With those new cases, the total number of children infected with COVID-19 comes to almost 4.1 million since the start of the pandemic, the AAP and CHA said. The CDC data indicate that just over 5.35 million children aged 12-15 years and 3.53 million 16- and 17-year-olds have received at least one dose of the COVID-19 vaccine and that 6.8 million children aged 12-17 are fully vaccinated.
Fully vaccinated children represent 26.4% of all 12- to 15-year-olds and 38.3% of the 16- 17-year-olds as of July 19. The corresponding numbers for those who have received at least one dose are 35.2% (ages 12-15) and 46.8% (16-17), the CDC said.
The AAP recently recommended in-person learning with universal masking in schools this fall “because a significant portion of the student population is not yet eligible for vaccines. ... Many schools will not have a system to monitor vaccine status of students, teachers and staff, and some communities overall have low vaccination uptake where the virus may be circulating more prominently.”
Children aged 12-15 years represented 13.5% of all first vaccinations received during the 2 weeks ending July 19, compared with 11.5% for the 2 weeks ending July 12, marking the first increase since the end of May. First vaccinations in 16- and 17-year-olds, who make up a much smaller share of the U.S. population, also went up, topping 5%, the Centers for Disease Control and Prevention said in its COVID Data Tracker.
The total number of vaccine initiations was almost 250,000 for the week ending July 19, after dropping to a low of 201,000 the previous week. Before that, first vaccinations had fallen in 5 of the previous 6 weeks, going from 1.4 million on May 24 to 307,000 on July 5, the CDC said.
New cases of COVID-19, unfortunately, continued to follow the trend among the larger population: As of July 15, weekly cases in children were up by 179% since dropping to 8,400 on June 24, the American Academy of Pediatrics and the Children’s Hospital Association said in a joint report. The 23,551 new cases in children for the week ending July 15 were 15.9% of all cases reported.
With those new cases, the total number of children infected with COVID-19 comes to almost 4.1 million since the start of the pandemic, the AAP and CHA said. The CDC data indicate that just over 5.35 million children aged 12-15 years and 3.53 million 16- and 17-year-olds have received at least one dose of the COVID-19 vaccine and that 6.8 million children aged 12-17 are fully vaccinated.
Fully vaccinated children represent 26.4% of all 12- to 15-year-olds and 38.3% of the 16- 17-year-olds as of July 19. The corresponding numbers for those who have received at least one dose are 35.2% (ages 12-15) and 46.8% (16-17), the CDC said.
The AAP recently recommended in-person learning with universal masking in schools this fall “because a significant portion of the student population is not yet eligible for vaccines. ... Many schools will not have a system to monitor vaccine status of students, teachers and staff, and some communities overall have low vaccination uptake where the virus may be circulating more prominently.”
Children aged 12-15 years represented 13.5% of all first vaccinations received during the 2 weeks ending July 19, compared with 11.5% for the 2 weeks ending July 12, marking the first increase since the end of May. First vaccinations in 16- and 17-year-olds, who make up a much smaller share of the U.S. population, also went up, topping 5%, the Centers for Disease Control and Prevention said in its COVID Data Tracker.
The total number of vaccine initiations was almost 250,000 for the week ending July 19, after dropping to a low of 201,000 the previous week. Before that, first vaccinations had fallen in 5 of the previous 6 weeks, going from 1.4 million on May 24 to 307,000 on July 5, the CDC said.
New cases of COVID-19, unfortunately, continued to follow the trend among the larger population: As of July 15, weekly cases in children were up by 179% since dropping to 8,400 on June 24, the American Academy of Pediatrics and the Children’s Hospital Association said in a joint report. The 23,551 new cases in children for the week ending July 15 were 15.9% of all cases reported.
With those new cases, the total number of children infected with COVID-19 comes to almost 4.1 million since the start of the pandemic, the AAP and CHA said. The CDC data indicate that just over 5.35 million children aged 12-15 years and 3.53 million 16- and 17-year-olds have received at least one dose of the COVID-19 vaccine and that 6.8 million children aged 12-17 are fully vaccinated.
Fully vaccinated children represent 26.4% of all 12- to 15-year-olds and 38.3% of the 16- 17-year-olds as of July 19. The corresponding numbers for those who have received at least one dose are 35.2% (ages 12-15) and 46.8% (16-17), the CDC said.
The AAP recently recommended in-person learning with universal masking in schools this fall “because a significant portion of the student population is not yet eligible for vaccines. ... Many schools will not have a system to monitor vaccine status of students, teachers and staff, and some communities overall have low vaccination uptake where the virus may be circulating more prominently.”
Children and COVID: New vaccinations drop as the case count rises
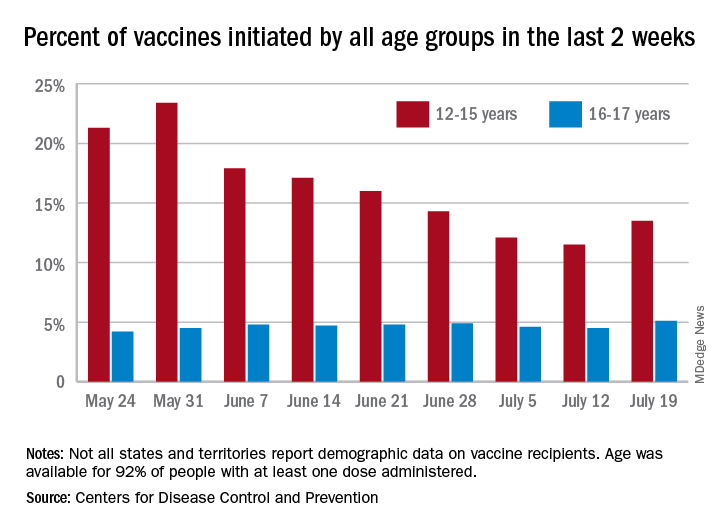
With only a quarter of all children aged 12-15 years fully vaccinated against COVID-19, first vaccinations continued to drop and new cases for all children rose for the second consecutive week.
Just under 25% of children aged 12-15 had completed the vaccine regimen as of July 12, and just over one-third (33.5%) had received at least one dose. Meanwhile, that age group represented 11.5% of people who initiated vaccination during the 2 weeks ending July 12, down from 12.1% a week earlier, the Centers for Disease Control and Prevention said. The total number of new vaccinations for the week ending July 12 was just over 201,000, compared with 307,000 for the previous week.
New cases of COVID-19, however, were on the rise in children. The 19,000 new cases reported for the week ending July 8 were up from 12,000 a week earlier and 8,000 the week before that, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
That report also shows that children made up 22.3% of all new cases during the week of July 2-8, compared with 16.8% the previous week, and that there were nine deaths in children that same week, the most since March. COVID-related deaths among children total 344 in the 46 jurisdictions (43 states, New York City, Puerto Rico, and Guam) that are reporting such data by age. “It is not possible to standardize more detailed age ranges for children based on what is publicly available from the states,” the two groups noted.
Such data are available from the CDC’s COVID Data Tracker, however, and they show that children aged 16-17 years, who became eligible for COVID vaccination before the younger age group, are further ahead in the process. Among the older children, almost 46% had gotten at least one dose and 37% were fully vaccinated by July 12.
With only a quarter of all children aged 12-15 years fully vaccinated against COVID-19, first vaccinations continued to drop and new cases for all children rose for the second consecutive week.
Just under 25% of children aged 12-15 had completed the vaccine regimen as of July 12, and just over one-third (33.5%) had received at least one dose. Meanwhile, that age group represented 11.5% of people who initiated vaccination during the 2 weeks ending July 12, down from 12.1% a week earlier, the Centers for Disease Control and Prevention said. The total number of new vaccinations for the week ending July 12 was just over 201,000, compared with 307,000 for the previous week.
New cases of COVID-19, however, were on the rise in children. The 19,000 new cases reported for the week ending July 8 were up from 12,000 a week earlier and 8,000 the week before that, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
That report also shows that children made up 22.3% of all new cases during the week of July 2-8, compared with 16.8% the previous week, and that there were nine deaths in children that same week, the most since March. COVID-related deaths among children total 344 in the 46 jurisdictions (43 states, New York City, Puerto Rico, and Guam) that are reporting such data by age. “It is not possible to standardize more detailed age ranges for children based on what is publicly available from the states,” the two groups noted.
Such data are available from the CDC’s COVID Data Tracker, however, and they show that children aged 16-17 years, who became eligible for COVID vaccination before the younger age group, are further ahead in the process. Among the older children, almost 46% had gotten at least one dose and 37% were fully vaccinated by July 12.
With only a quarter of all children aged 12-15 years fully vaccinated against COVID-19, first vaccinations continued to drop and new cases for all children rose for the second consecutive week.
Just under 25% of children aged 12-15 had completed the vaccine regimen as of July 12, and just over one-third (33.5%) had received at least one dose. Meanwhile, that age group represented 11.5% of people who initiated vaccination during the 2 weeks ending July 12, down from 12.1% a week earlier, the Centers for Disease Control and Prevention said. The total number of new vaccinations for the week ending July 12 was just over 201,000, compared with 307,000 for the previous week.
New cases of COVID-19, however, were on the rise in children. The 19,000 new cases reported for the week ending July 8 were up from 12,000 a week earlier and 8,000 the week before that, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
That report also shows that children made up 22.3% of all new cases during the week of July 2-8, compared with 16.8% the previous week, and that there were nine deaths in children that same week, the most since March. COVID-related deaths among children total 344 in the 46 jurisdictions (43 states, New York City, Puerto Rico, and Guam) that are reporting such data by age. “It is not possible to standardize more detailed age ranges for children based on what is publicly available from the states,” the two groups noted.
Such data are available from the CDC’s COVID Data Tracker, however, and they show that children aged 16-17 years, who became eligible for COVID vaccination before the younger age group, are further ahead in the process. Among the older children, almost 46% had gotten at least one dose and 37% were fully vaccinated by July 12.
Delta becomes dominant coronavirus variant in U.S.
The contagious Delta variant has become the dominant form of the coronavirus in the United States, now accounting for more than 51% of COVID-19 cases in the country, according to new CDC data to updated on July 6.
The variant, also known as B.1.617.2 and first detected in India, makes up more than 80% of new cases in some Midwestern states, including Iowa, Kansas, and Missouri. Delta also accounts for 74% of cases in Western states such as Colorado and Utah and 59% of cases in Southern states such as Louisiana and Texas.
Communities with low vaccination rates are bearing the brunt of new Delta cases. Public health experts are urging those who are unvaccinated to get a shot to protect themselves and their communities against future surges.
“Right now we have two Americas: the vaccinated and the unvaccinated,” Paul Offit, MD, an infectious disease specialist at Children’s Hospital of Philadelphia, told NPR.
“We’re feeling pretty good right now because it’s the summer,” he said. “But come winter, if we still have a significant percentage of the population that is unvaccinated, we’re going to see this virus surge again.”
So far, COVID-19 vaccines appear to protect people against the Delta variant. But health officials are watching other variants that could evade vaccine protection and lead to major outbreaks this year.
For instance, certain mutations in the Epsilon variant may allow it to evade the immunity from past infections and current COVID-19 vaccines, according to a new study published July 1 in the Science. The variant, also known as B.1.427/B.1.429 and first identified in California, has now been reported in 34 countries and could become widespread in the United States.
Researchers from the University of Washington and clinics in Switzerland tested the variant in blood samples from vaccinated people, as well as those who were previously infected with COVID-19. They found that the neutralizing power was reduced by about 2 to 3½ times.
The research team also visualized the variant and found that three mutations on Epsilon’s spike protein allow the virus to escape certain antibodies and lower the strength of vaccines.
Epsilon “relies on an indirect and unusual neutralization-escape strategy,” they wrote, saying that understanding these escape routes could help scientists track new variants, curb the pandemic, and create booster shots.
In Australia, for instance, public health officials have detected the Lambda variant, which could be more infectious than the Delta variant and resistant to vaccines, according to Sky News.
A hotel quarantine program in New South Wales identified the variant in someone who had returned from travel, the news outlet reported. Also known as C.37, Lambda was named a “variant of interest” by the World Health Organization in June.
Lambda was first identified in Peru in December and now accounts for more than 80% of the country’s cases, according to the Financial Times. It has since been found in 27 countries, including the U.S., U.K., and Germany.
The variant has seven mutations on the spike protein that allow the virus to infect human cells, the news outlet reported. One mutation is like another mutation on the Delta variant, which could make it more contagious.
In a preprint study published July 1, researchers at the University of Chile at Santiago found that Lambda is better able to escape antibodies created by the CoronaVac vaccine made by Sinovac in China. In the paper, which hasn’t yet been peer-reviewed, researchers tested blood samples from local health care workers in Santiago who had received two doses of the vaccine.
“Our data revealed that the spike protein ... carries mutations conferring increased infectivity and the ability to escape from neutralizing antibodies,” they wrote.
The research team urged countries to continue testing for contagious variants, even in areas with high vaccination rates, so scientists can identify mutations quickly and analyze whether new variants can escape vaccines.
“The world has to get its act together,” Saad Omer, PhD, director of the Yale Institute for Global Health, told NPR. “Otherwise yet another, potentially more dangerous, variant could emerge.”
A version of this article first appeared on WebMD.com.
The contagious Delta variant has become the dominant form of the coronavirus in the United States, now accounting for more than 51% of COVID-19 cases in the country, according to new CDC data to updated on July 6.
The variant, also known as B.1.617.2 and first detected in India, makes up more than 80% of new cases in some Midwestern states, including Iowa, Kansas, and Missouri. Delta also accounts for 74% of cases in Western states such as Colorado and Utah and 59% of cases in Southern states such as Louisiana and Texas.
Communities with low vaccination rates are bearing the brunt of new Delta cases. Public health experts are urging those who are unvaccinated to get a shot to protect themselves and their communities against future surges.
“Right now we have two Americas: the vaccinated and the unvaccinated,” Paul Offit, MD, an infectious disease specialist at Children’s Hospital of Philadelphia, told NPR.
“We’re feeling pretty good right now because it’s the summer,” he said. “But come winter, if we still have a significant percentage of the population that is unvaccinated, we’re going to see this virus surge again.”
So far, COVID-19 vaccines appear to protect people against the Delta variant. But health officials are watching other variants that could evade vaccine protection and lead to major outbreaks this year.
For instance, certain mutations in the Epsilon variant may allow it to evade the immunity from past infections and current COVID-19 vaccines, according to a new study published July 1 in the Science. The variant, also known as B.1.427/B.1.429 and first identified in California, has now been reported in 34 countries and could become widespread in the United States.
Researchers from the University of Washington and clinics in Switzerland tested the variant in blood samples from vaccinated people, as well as those who were previously infected with COVID-19. They found that the neutralizing power was reduced by about 2 to 3½ times.
The research team also visualized the variant and found that three mutations on Epsilon’s spike protein allow the virus to escape certain antibodies and lower the strength of vaccines.
Epsilon “relies on an indirect and unusual neutralization-escape strategy,” they wrote, saying that understanding these escape routes could help scientists track new variants, curb the pandemic, and create booster shots.
In Australia, for instance, public health officials have detected the Lambda variant, which could be more infectious than the Delta variant and resistant to vaccines, according to Sky News.
A hotel quarantine program in New South Wales identified the variant in someone who had returned from travel, the news outlet reported. Also known as C.37, Lambda was named a “variant of interest” by the World Health Organization in June.
Lambda was first identified in Peru in December and now accounts for more than 80% of the country’s cases, according to the Financial Times. It has since been found in 27 countries, including the U.S., U.K., and Germany.
The variant has seven mutations on the spike protein that allow the virus to infect human cells, the news outlet reported. One mutation is like another mutation on the Delta variant, which could make it more contagious.
In a preprint study published July 1, researchers at the University of Chile at Santiago found that Lambda is better able to escape antibodies created by the CoronaVac vaccine made by Sinovac in China. In the paper, which hasn’t yet been peer-reviewed, researchers tested blood samples from local health care workers in Santiago who had received two doses of the vaccine.
“Our data revealed that the spike protein ... carries mutations conferring increased infectivity and the ability to escape from neutralizing antibodies,” they wrote.
The research team urged countries to continue testing for contagious variants, even in areas with high vaccination rates, so scientists can identify mutations quickly and analyze whether new variants can escape vaccines.
“The world has to get its act together,” Saad Omer, PhD, director of the Yale Institute for Global Health, told NPR. “Otherwise yet another, potentially more dangerous, variant could emerge.”
A version of this article first appeared on WebMD.com.
The contagious Delta variant has become the dominant form of the coronavirus in the United States, now accounting for more than 51% of COVID-19 cases in the country, according to new CDC data to updated on July 6.
The variant, also known as B.1.617.2 and first detected in India, makes up more than 80% of new cases in some Midwestern states, including Iowa, Kansas, and Missouri. Delta also accounts for 74% of cases in Western states such as Colorado and Utah and 59% of cases in Southern states such as Louisiana and Texas.
Communities with low vaccination rates are bearing the brunt of new Delta cases. Public health experts are urging those who are unvaccinated to get a shot to protect themselves and their communities against future surges.
“Right now we have two Americas: the vaccinated and the unvaccinated,” Paul Offit, MD, an infectious disease specialist at Children’s Hospital of Philadelphia, told NPR.
“We’re feeling pretty good right now because it’s the summer,” he said. “But come winter, if we still have a significant percentage of the population that is unvaccinated, we’re going to see this virus surge again.”
So far, COVID-19 vaccines appear to protect people against the Delta variant. But health officials are watching other variants that could evade vaccine protection and lead to major outbreaks this year.
For instance, certain mutations in the Epsilon variant may allow it to evade the immunity from past infections and current COVID-19 vaccines, according to a new study published July 1 in the Science. The variant, also known as B.1.427/B.1.429 and first identified in California, has now been reported in 34 countries and could become widespread in the United States.
Researchers from the University of Washington and clinics in Switzerland tested the variant in blood samples from vaccinated people, as well as those who were previously infected with COVID-19. They found that the neutralizing power was reduced by about 2 to 3½ times.
The research team also visualized the variant and found that three mutations on Epsilon’s spike protein allow the virus to escape certain antibodies and lower the strength of vaccines.
Epsilon “relies on an indirect and unusual neutralization-escape strategy,” they wrote, saying that understanding these escape routes could help scientists track new variants, curb the pandemic, and create booster shots.
In Australia, for instance, public health officials have detected the Lambda variant, which could be more infectious than the Delta variant and resistant to vaccines, according to Sky News.
A hotel quarantine program in New South Wales identified the variant in someone who had returned from travel, the news outlet reported. Also known as C.37, Lambda was named a “variant of interest” by the World Health Organization in June.
Lambda was first identified in Peru in December and now accounts for more than 80% of the country’s cases, according to the Financial Times. It has since been found in 27 countries, including the U.S., U.K., and Germany.
The variant has seven mutations on the spike protein that allow the virus to infect human cells, the news outlet reported. One mutation is like another mutation on the Delta variant, which could make it more contagious.
In a preprint study published July 1, researchers at the University of Chile at Santiago found that Lambda is better able to escape antibodies created by the CoronaVac vaccine made by Sinovac in China. In the paper, which hasn’t yet been peer-reviewed, researchers tested blood samples from local health care workers in Santiago who had received two doses of the vaccine.
“Our data revealed that the spike protein ... carries mutations conferring increased infectivity and the ability to escape from neutralizing antibodies,” they wrote.
The research team urged countries to continue testing for contagious variants, even in areas with high vaccination rates, so scientists can identify mutations quickly and analyze whether new variants can escape vaccines.
“The world has to get its act together,” Saad Omer, PhD, director of the Yale Institute for Global Health, told NPR. “Otherwise yet another, potentially more dangerous, variant could emerge.”
A version of this article first appeared on WebMD.com.
Small uptick in children’s COVID vaccinations can’t change overall decline
The weekly number of 12- to 15-year-olds receiving a first dose of COVID-19 vaccine rose slightly, but the age group’s share of all first vaccinations continues to drop, according to data from the Centers for Disease Control and Prevention.

the CDC reported on its COVID Data Tracker site.
As of July 5, not quite one-third (32.2%) of 12- to 15-year-olds had received at least one dose of the vaccine and 23.4% were fully vaccinated. For those aged 16-17 years, 44.5% have gotten at least one dose and 35.9% are fully vaccinated. Total numbers of fully vaccinated individuals in each age group are 4.9 million (12-15) and 3.4 million (16-17), the CDC said.
Looking at another measure, percentage of all vaccines initiated by each age group over the previous 14 days, shows that the decline has not stopped for those aged 12-15. They represented 12.1% of all first vaccines administered during the 2 weeks ending July 4, compared with 14.3% on June 28 and 23.4% (the highest proportion reached) on May 30. The 16- and 17-year olds were at 4.6% on July 4, but that figure has only ranged from 4.2% to 4.9% since late May, based on CDC data.
The numbers for full vaccination follow a similar trajectory. Children aged 12-15 represented 12.1% of all those completing the vaccine regimen over the 2 weeks ending July 4, down from 16.7% a week earlier (June 28) and from a high of 21.5% for the 2 weeks ending June 21. Full vaccination for 16- and 17-year-olds matched their pattern for first doses: nothing lower than 4.2% or higher than 4.6%, the COVID Data Tracker shows.
The weekly number of 12- to 15-year-olds receiving a first dose of COVID-19 vaccine rose slightly, but the age group’s share of all first vaccinations continues to drop, according to data from the Centers for Disease Control and Prevention.
the CDC reported on its COVID Data Tracker site.
As of July 5, not quite one-third (32.2%) of 12- to 15-year-olds had received at least one dose of the vaccine and 23.4% were fully vaccinated. For those aged 16-17 years, 44.5% have gotten at least one dose and 35.9% are fully vaccinated. Total numbers of fully vaccinated individuals in each age group are 4.9 million (12-15) and 3.4 million (16-17), the CDC said.
Looking at another measure, percentage of all vaccines initiated by each age group over the previous 14 days, shows that the decline has not stopped for those aged 12-15. They represented 12.1% of all first vaccines administered during the 2 weeks ending July 4, compared with 14.3% on June 28 and 23.4% (the highest proportion reached) on May 30. The 16- and 17-year olds were at 4.6% on July 4, but that figure has only ranged from 4.2% to 4.9% since late May, based on CDC data.
The numbers for full vaccination follow a similar trajectory. Children aged 12-15 represented 12.1% of all those completing the vaccine regimen over the 2 weeks ending July 4, down from 16.7% a week earlier (June 28) and from a high of 21.5% for the 2 weeks ending June 21. Full vaccination for 16- and 17-year-olds matched their pattern for first doses: nothing lower than 4.2% or higher than 4.6%, the COVID Data Tracker shows.
The weekly number of 12- to 15-year-olds receiving a first dose of COVID-19 vaccine rose slightly, but the age group’s share of all first vaccinations continues to drop, according to data from the Centers for Disease Control and Prevention.
the CDC reported on its COVID Data Tracker site.
As of July 5, not quite one-third (32.2%) of 12- to 15-year-olds had received at least one dose of the vaccine and 23.4% were fully vaccinated. For those aged 16-17 years, 44.5% have gotten at least one dose and 35.9% are fully vaccinated. Total numbers of fully vaccinated individuals in each age group are 4.9 million (12-15) and 3.4 million (16-17), the CDC said.
Looking at another measure, percentage of all vaccines initiated by each age group over the previous 14 days, shows that the decline has not stopped for those aged 12-15. They represented 12.1% of all first vaccines administered during the 2 weeks ending July 4, compared with 14.3% on June 28 and 23.4% (the highest proportion reached) on May 30. The 16- and 17-year olds were at 4.6% on July 4, but that figure has only ranged from 4.2% to 4.9% since late May, based on CDC data.
The numbers for full vaccination follow a similar trajectory. Children aged 12-15 represented 12.1% of all those completing the vaccine regimen over the 2 weeks ending July 4, down from 16.7% a week earlier (June 28) and from a high of 21.5% for the 2 weeks ending June 21. Full vaccination for 16- and 17-year-olds matched their pattern for first doses: nothing lower than 4.2% or higher than 4.6%, the COVID Data Tracker shows.
New COVID-19 vaccinations decline again in 12- to 15-year-olds
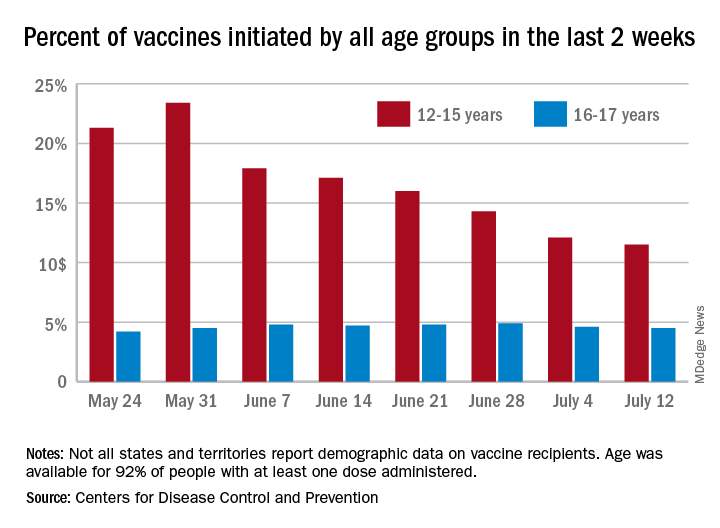
Even though less than 21% of all children aged 12-15 years are fully vaccinated against COVID-19, the number seeking first vaccinations continues to decline, according to data from the Centers for Disease Control and Prevention.

and 462,000 during the week ending June 14. Collectively, 30.2% of 12- to 15-year-olds have gotten at least one dose of vaccine so far and 20.7% are now fully vaccinated, the CDC said on its COVID Data Tracker site.
Among children aged 16-17 years, who were able to start the vaccination process earlier, 42.9% have received at least one dose and 34.0% have completed the COVID-19 vaccine regimen. Vaccine initiation – measured as the proportion of all individuals getting a first shot over the previous 2 weeks – has been consistently around 4.8% during the month of June for this age group but has dropped from 17.9% on June 7 to 14.3% on June 28 for those aged 12-15, the CDC data show.
Looking at the same measure for vaccine completion, 16.7% of all those who reached full vaccination status in the 14 days ending June 28 were 12- to 15-years-olds, down from 21.5% on June 21 and 19.6% on June 14. The numbers for those aged 15-16 were, respectively, 4.6%, 4.5%, and 4.2%, the CDC reported.
Fortunately, in the wake of recent vaccination trends, new cases of COVID-19 in children were down to their lowest level – just 8,447 for the week ending June 24 – since May of 2020, according to a new report from the American Academy of Pediatrics and the Children’s Hospital Association.
New cases had been well over 15,000 the previous week (June 17), following weeks of 14,000 (June 10) and 16,000 (June 3) new cases, so the latest drop down to just four digits represents a 1-week decline of over 46% in the 49 states (excluding New York) that are reporting age distribution, along with the District of Columbia, New York City, Puerto Rico, and Guam.
The cumulative number of child COVID-19 cases in those jurisdictions is about 4.03 million since the beginning of the pandemic, which represents 14.2% of all cases in the United States. At the state level, the cumulative rate of cases in children is highest in Vermont (22.7%) and lowest in Florida (8.9%), which uses an age range of 0-14 years for children, compared with 0-17 or 0-19 for most states, the AAP and CHA said.
Severe illness has been rare in children, which is reflected in the proportion of children among all hospitalizations, 2.2% in 24 jurisdictions, and the proportion of deaths, 0.06% in 46 jurisdictions, since the start of the pandemic, the AAP and CHA said, with a total of 336 COVID-19–related deaths reported.
Even though less than 21% of all children aged 12-15 years are fully vaccinated against COVID-19, the number seeking first vaccinations continues to decline, according to data from the Centers for Disease Control and Prevention.
and 462,000 during the week ending June 14. Collectively, 30.2% of 12- to 15-year-olds have gotten at least one dose of vaccine so far and 20.7% are now fully vaccinated, the CDC said on its COVID Data Tracker site.
Among children aged 16-17 years, who were able to start the vaccination process earlier, 42.9% have received at least one dose and 34.0% have completed the COVID-19 vaccine regimen. Vaccine initiation – measured as the proportion of all individuals getting a first shot over the previous 2 weeks – has been consistently around 4.8% during the month of June for this age group but has dropped from 17.9% on June 7 to 14.3% on June 28 for those aged 12-15, the CDC data show.
Looking at the same measure for vaccine completion, 16.7% of all those who reached full vaccination status in the 14 days ending June 28 were 12- to 15-years-olds, down from 21.5% on June 21 and 19.6% on June 14. The numbers for those aged 15-16 were, respectively, 4.6%, 4.5%, and 4.2%, the CDC reported.
Fortunately, in the wake of recent vaccination trends, new cases of COVID-19 in children were down to their lowest level – just 8,447 for the week ending June 24 – since May of 2020, according to a new report from the American Academy of Pediatrics and the Children’s Hospital Association.
New cases had been well over 15,000 the previous week (June 17), following weeks of 14,000 (June 10) and 16,000 (June 3) new cases, so the latest drop down to just four digits represents a 1-week decline of over 46% in the 49 states (excluding New York) that are reporting age distribution, along with the District of Columbia, New York City, Puerto Rico, and Guam.
The cumulative number of child COVID-19 cases in those jurisdictions is about 4.03 million since the beginning of the pandemic, which represents 14.2% of all cases in the United States. At the state level, the cumulative rate of cases in children is highest in Vermont (22.7%) and lowest in Florida (8.9%), which uses an age range of 0-14 years for children, compared with 0-17 or 0-19 for most states, the AAP and CHA said.
Severe illness has been rare in children, which is reflected in the proportion of children among all hospitalizations, 2.2% in 24 jurisdictions, and the proportion of deaths, 0.06% in 46 jurisdictions, since the start of the pandemic, the AAP and CHA said, with a total of 336 COVID-19–related deaths reported.
Even though less than 21% of all children aged 12-15 years are fully vaccinated against COVID-19, the number seeking first vaccinations continues to decline, according to data from the Centers for Disease Control and Prevention.
and 462,000 during the week ending June 14. Collectively, 30.2% of 12- to 15-year-olds have gotten at least one dose of vaccine so far and 20.7% are now fully vaccinated, the CDC said on its COVID Data Tracker site.
Among children aged 16-17 years, who were able to start the vaccination process earlier, 42.9% have received at least one dose and 34.0% have completed the COVID-19 vaccine regimen. Vaccine initiation – measured as the proportion of all individuals getting a first shot over the previous 2 weeks – has been consistently around 4.8% during the month of June for this age group but has dropped from 17.9% on June 7 to 14.3% on June 28 for those aged 12-15, the CDC data show.
Looking at the same measure for vaccine completion, 16.7% of all those who reached full vaccination status in the 14 days ending June 28 were 12- to 15-years-olds, down from 21.5% on June 21 and 19.6% on June 14. The numbers for those aged 15-16 were, respectively, 4.6%, 4.5%, and 4.2%, the CDC reported.
Fortunately, in the wake of recent vaccination trends, new cases of COVID-19 in children were down to their lowest level – just 8,447 for the week ending June 24 – since May of 2020, according to a new report from the American Academy of Pediatrics and the Children’s Hospital Association.
New cases had been well over 15,000 the previous week (June 17), following weeks of 14,000 (June 10) and 16,000 (June 3) new cases, so the latest drop down to just four digits represents a 1-week decline of over 46% in the 49 states (excluding New York) that are reporting age distribution, along with the District of Columbia, New York City, Puerto Rico, and Guam.
The cumulative number of child COVID-19 cases in those jurisdictions is about 4.03 million since the beginning of the pandemic, which represents 14.2% of all cases in the United States. At the state level, the cumulative rate of cases in children is highest in Vermont (22.7%) and lowest in Florida (8.9%), which uses an age range of 0-14 years for children, compared with 0-17 or 0-19 for most states, the AAP and CHA said.
Severe illness has been rare in children, which is reflected in the proportion of children among all hospitalizations, 2.2% in 24 jurisdictions, and the proportion of deaths, 0.06% in 46 jurisdictions, since the start of the pandemic, the AAP and CHA said, with a total of 336 COVID-19–related deaths reported.
Children and COVID: Vaccination trends beginning to diverge
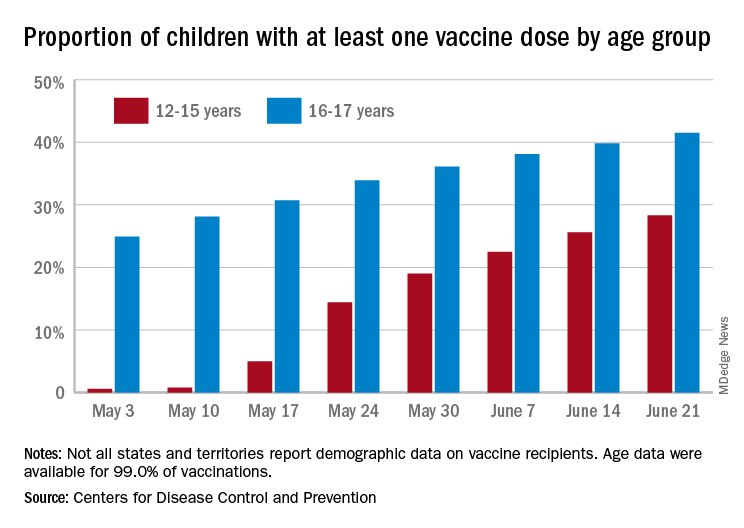
As more adolescents became eligible for a second dose of the Pfizer vaccine since it received approval from the Food and Drug Administration in mid-May, the share of 12- to 15-year-olds considered fully vaccinated rose from 11.4% on June 14 to 17.8% on June 28, an increase of 56%, the CDC’s COVID Data Tracker indicated June 22.
For children aged 16-17 years, who have been receiving the vaccine since early April, full vaccination rose by 9.6% in that same week, going from 29.1% on June 14 to 31.9% on June 21. The cumulative numbers for first vaccinations are higher, of course, but are rising more slowly in both age groups: 41.5% of those aged 16-17 had received at least one dose by June 21 (up by 4.3%), with the 12- to 15-year-olds at 28.3% (up by 10.5%), based on the CDC data.
Limiting the time frame to just the last 2 weeks, however, shows the opposite of rising among the younger children. During the 2 weeks ending June 7, 17.9% of those initiating a first dose were 12-15 years old, but that 2-week figure slipped to 17.1% as of June 14 and was down to 16.0% on June 21. The older group was slow but steady over that time: 4.8%, 4.7%, and 4.8%, the CDC said. To give those figures some context, those aged 25-39 years represented 23.7% of past-2-week initiations on June 7 and 24.3% on June 21.
Although no COVID-19 vaccine has been approved for children under 12 years, about 0.4% of that age group – just over 167,000 children – have received a first dose and almost 91,000 are fully vaccinated, according to CDC data.
As more adolescents became eligible for a second dose of the Pfizer vaccine since it received approval from the Food and Drug Administration in mid-May, the share of 12- to 15-year-olds considered fully vaccinated rose from 11.4% on June 14 to 17.8% on June 28, an increase of 56%, the CDC’s COVID Data Tracker indicated June 22.
For children aged 16-17 years, who have been receiving the vaccine since early April, full vaccination rose by 9.6% in that same week, going from 29.1% on June 14 to 31.9% on June 21. The cumulative numbers for first vaccinations are higher, of course, but are rising more slowly in both age groups: 41.5% of those aged 16-17 had received at least one dose by June 21 (up by 4.3%), with the 12- to 15-year-olds at 28.3% (up by 10.5%), based on the CDC data.
Limiting the time frame to just the last 2 weeks, however, shows the opposite of rising among the younger children. During the 2 weeks ending June 7, 17.9% of those initiating a first dose were 12-15 years old, but that 2-week figure slipped to 17.1% as of June 14 and was down to 16.0% on June 21. The older group was slow but steady over that time: 4.8%, 4.7%, and 4.8%, the CDC said. To give those figures some context, those aged 25-39 years represented 23.7% of past-2-week initiations on June 7 and 24.3% on June 21.
Although no COVID-19 vaccine has been approved for children under 12 years, about 0.4% of that age group – just over 167,000 children – have received a first dose and almost 91,000 are fully vaccinated, according to CDC data.
As more adolescents became eligible for a second dose of the Pfizer vaccine since it received approval from the Food and Drug Administration in mid-May, the share of 12- to 15-year-olds considered fully vaccinated rose from 11.4% on June 14 to 17.8% on June 28, an increase of 56%, the CDC’s COVID Data Tracker indicated June 22.
For children aged 16-17 years, who have been receiving the vaccine since early April, full vaccination rose by 9.6% in that same week, going from 29.1% on June 14 to 31.9% on June 21. The cumulative numbers for first vaccinations are higher, of course, but are rising more slowly in both age groups: 41.5% of those aged 16-17 had received at least one dose by June 21 (up by 4.3%), with the 12- to 15-year-olds at 28.3% (up by 10.5%), based on the CDC data.
Limiting the time frame to just the last 2 weeks, however, shows the opposite of rising among the younger children. During the 2 weeks ending June 7, 17.9% of those initiating a first dose were 12-15 years old, but that 2-week figure slipped to 17.1% as of June 14 and was down to 16.0% on June 21. The older group was slow but steady over that time: 4.8%, 4.7%, and 4.8%, the CDC said. To give those figures some context, those aged 25-39 years represented 23.7% of past-2-week initiations on June 7 and 24.3% on June 21.
Although no COVID-19 vaccine has been approved for children under 12 years, about 0.4% of that age group – just over 167,000 children – have received a first dose and almost 91,000 are fully vaccinated, according to CDC data.