User login
FDA OKs new drug for triple-negative breast cancer
The US Food and Drug Administration (FDA) granted accelerated approval to sacituzumab govitecan (Trodelvy, Immunomedics) for the treatment of metastatic triple-negative breast cancer (TNBC).
Eligible patients must have received at least two prior therapies.
TNBC is so-called because it lacks the three cellular targets present in more common forms of breast cancer. It is usually treated with chemotherapy.
Sacituzumab govitecan offers a new approach – and it has a target.
Given intravenously, the new drug is an antibody-drug conjugate in which SN-38, an active metabolite of the chemotherapy drug irinotecan (multiple brands), is coupled to a monoclonal antibody that targets an antigen that has high expression in TNBC and induces cancer cell growth.
“Metastatic triple-negative breast cancer is an aggressive form of breast cancer with limited treatment options,” observed Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research in a press statement. “There is intense interest in finding new medications” for this patient population, he added.
The new approval is based on safety and efficacy results from a phase 1/2 clinical trial of 108 patients (median age, 56 years) who had received at least two prior treatments for metastatic disease.
The overall response rate was 33% (n = 36), including three complete responses. Median duration of response was 7.7 months. Of responders, 55.6% maintained their response for at least 6 months and 16.7% for at least 12 months.
Median progression-free survival was 5.5 months, and median overall survival was 13.0 months.
The study data were published last year in the New England Journal of Medicine.
“It’s not every day that we see this sort of clinical activity in this aggressive subtype of breast cancer,” said senior study author Kevin Kalinsky, MD, in an interview at that time. He is a medical oncologist at New York–Presbyterian Hospital and Columbia University Medical Center in New York City.
The most common side effects of the new therapy were nausea, neutropenia, diarrhea, fatigue, anemia, vomiting, alopecia, constipation, decreased appetite, rash, and abdominal pain.
No peripheral neuropathy of grade 3 or higher was reported.
In the study, patients received sacituzumab govitecan intravenously (10 mg/kg body weight) on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity.
The 108 participants received a mean 18.7 doses of sacituzumab govitecan, or 9.6 cycles. The median duration of exposure was 5.1 months.
Three patients discontinued treatment because of adverse events, and two patients discontinued because of drug-related events.
The prescribing information includes a boxed warning regarding the risks of severe neutropenia and severe diarrhea. Blood cell counts should be monitored during treatment and granulocyte-colony stimulating factor (G-CSF) therapy should be considered. Anti-infective treatment should be initiated in the event of febrile neutropenia. Patients with reduced uridine diphosphate-glucuronosyltransferase 1A1 (UGT1A1) activity are at increased risk for neutropenia following initiation of treatment.
The new drug can also cause hypersensitivity reactions including severe anaphylactic reactions.
Women who are pregnant should not take sacituzumab govitecan.
This article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) granted accelerated approval to sacituzumab govitecan (Trodelvy, Immunomedics) for the treatment of metastatic triple-negative breast cancer (TNBC).
Eligible patients must have received at least two prior therapies.
TNBC is so-called because it lacks the three cellular targets present in more common forms of breast cancer. It is usually treated with chemotherapy.
Sacituzumab govitecan offers a new approach – and it has a target.
Given intravenously, the new drug is an antibody-drug conjugate in which SN-38, an active metabolite of the chemotherapy drug irinotecan (multiple brands), is coupled to a monoclonal antibody that targets an antigen that has high expression in TNBC and induces cancer cell growth.
“Metastatic triple-negative breast cancer is an aggressive form of breast cancer with limited treatment options,” observed Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research in a press statement. “There is intense interest in finding new medications” for this patient population, he added.
The new approval is based on safety and efficacy results from a phase 1/2 clinical trial of 108 patients (median age, 56 years) who had received at least two prior treatments for metastatic disease.
The overall response rate was 33% (n = 36), including three complete responses. Median duration of response was 7.7 months. Of responders, 55.6% maintained their response for at least 6 months and 16.7% for at least 12 months.
Median progression-free survival was 5.5 months, and median overall survival was 13.0 months.
The study data were published last year in the New England Journal of Medicine.
“It’s not every day that we see this sort of clinical activity in this aggressive subtype of breast cancer,” said senior study author Kevin Kalinsky, MD, in an interview at that time. He is a medical oncologist at New York–Presbyterian Hospital and Columbia University Medical Center in New York City.
The most common side effects of the new therapy were nausea, neutropenia, diarrhea, fatigue, anemia, vomiting, alopecia, constipation, decreased appetite, rash, and abdominal pain.
No peripheral neuropathy of grade 3 or higher was reported.
In the study, patients received sacituzumab govitecan intravenously (10 mg/kg body weight) on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity.
The 108 participants received a mean 18.7 doses of sacituzumab govitecan, or 9.6 cycles. The median duration of exposure was 5.1 months.
Three patients discontinued treatment because of adverse events, and two patients discontinued because of drug-related events.
The prescribing information includes a boxed warning regarding the risks of severe neutropenia and severe diarrhea. Blood cell counts should be monitored during treatment and granulocyte-colony stimulating factor (G-CSF) therapy should be considered. Anti-infective treatment should be initiated in the event of febrile neutropenia. Patients with reduced uridine diphosphate-glucuronosyltransferase 1A1 (UGT1A1) activity are at increased risk for neutropenia following initiation of treatment.
The new drug can also cause hypersensitivity reactions including severe anaphylactic reactions.
Women who are pregnant should not take sacituzumab govitecan.
This article first appeared on Medscape.com.
The US Food and Drug Administration (FDA) granted accelerated approval to sacituzumab govitecan (Trodelvy, Immunomedics) for the treatment of metastatic triple-negative breast cancer (TNBC).
Eligible patients must have received at least two prior therapies.
TNBC is so-called because it lacks the three cellular targets present in more common forms of breast cancer. It is usually treated with chemotherapy.
Sacituzumab govitecan offers a new approach – and it has a target.
Given intravenously, the new drug is an antibody-drug conjugate in which SN-38, an active metabolite of the chemotherapy drug irinotecan (multiple brands), is coupled to a monoclonal antibody that targets an antigen that has high expression in TNBC and induces cancer cell growth.
“Metastatic triple-negative breast cancer is an aggressive form of breast cancer with limited treatment options,” observed Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research in a press statement. “There is intense interest in finding new medications” for this patient population, he added.
The new approval is based on safety and efficacy results from a phase 1/2 clinical trial of 108 patients (median age, 56 years) who had received at least two prior treatments for metastatic disease.
The overall response rate was 33% (n = 36), including three complete responses. Median duration of response was 7.7 months. Of responders, 55.6% maintained their response for at least 6 months and 16.7% for at least 12 months.
Median progression-free survival was 5.5 months, and median overall survival was 13.0 months.
The study data were published last year in the New England Journal of Medicine.
“It’s not every day that we see this sort of clinical activity in this aggressive subtype of breast cancer,” said senior study author Kevin Kalinsky, MD, in an interview at that time. He is a medical oncologist at New York–Presbyterian Hospital and Columbia University Medical Center in New York City.
The most common side effects of the new therapy were nausea, neutropenia, diarrhea, fatigue, anemia, vomiting, alopecia, constipation, decreased appetite, rash, and abdominal pain.
No peripheral neuropathy of grade 3 or higher was reported.
In the study, patients received sacituzumab govitecan intravenously (10 mg/kg body weight) on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity.
The 108 participants received a mean 18.7 doses of sacituzumab govitecan, or 9.6 cycles. The median duration of exposure was 5.1 months.
Three patients discontinued treatment because of adverse events, and two patients discontinued because of drug-related events.
The prescribing information includes a boxed warning regarding the risks of severe neutropenia and severe diarrhea. Blood cell counts should be monitored during treatment and granulocyte-colony stimulating factor (G-CSF) therapy should be considered. Anti-infective treatment should be initiated in the event of febrile neutropenia. Patients with reduced uridine diphosphate-glucuronosyltransferase 1A1 (UGT1A1) activity are at increased risk for neutropenia following initiation of treatment.
The new drug can also cause hypersensitivity reactions including severe anaphylactic reactions.
Women who are pregnant should not take sacituzumab govitecan.
This article first appeared on Medscape.com.
FDA approves ibrutinib-rituximab combo for newly diagnosed CLL, SLL in adults
The Food and Drug Administration has expanded the indication for ibrutinib (Imbruvica) to allow its combination with rituximab for frontline treatment of chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) in adults.

The approval, announced April 21, was based on findings from the randomized, controlled, open-label, phase 3 E1912 trial of 529 patients, which demonstrated significantly improved progression-free survival (PFS) among those who received ibrutinib plus rituximab, compared with those who received fludarabine, cyclophosphamide, and rituximab (FCR) (87% vs. 75%; hazard ratio, 0.34). Median PFS was not reached in either arm after a median follow-up of 37 months.
E1912 was the first study to show superiority of a chemotherapy-free regimen over FCR chemoimmunotherapy, considered the gold standard for newly diagnosed CLL and SLL for the past 2 decades.
The recommended dosage for the newly approved combination is a once-daily 420-mg dose of ibrutinib taken with a glass of water, with rituximab initiation in the second cycle at doses of 50 mg/m2 on day 1, 325 mg/m2 on day 2, and 500 mg/m2 on days 1-5 of subsequent cycles for a total of six cycles.
The Food and Drug Administration has expanded the indication for ibrutinib (Imbruvica) to allow its combination with rituximab for frontline treatment of chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) in adults.
The approval, announced April 21, was based on findings from the randomized, controlled, open-label, phase 3 E1912 trial of 529 patients, which demonstrated significantly improved progression-free survival (PFS) among those who received ibrutinib plus rituximab, compared with those who received fludarabine, cyclophosphamide, and rituximab (FCR) (87% vs. 75%; hazard ratio, 0.34). Median PFS was not reached in either arm after a median follow-up of 37 months.
E1912 was the first study to show superiority of a chemotherapy-free regimen over FCR chemoimmunotherapy, considered the gold standard for newly diagnosed CLL and SLL for the past 2 decades.
The recommended dosage for the newly approved combination is a once-daily 420-mg dose of ibrutinib taken with a glass of water, with rituximab initiation in the second cycle at doses of 50 mg/m2 on day 1, 325 mg/m2 on day 2, and 500 mg/m2 on days 1-5 of subsequent cycles for a total of six cycles.
The Food and Drug Administration has expanded the indication for ibrutinib (Imbruvica) to allow its combination with rituximab for frontline treatment of chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) in adults.
The approval, announced April 21, was based on findings from the randomized, controlled, open-label, phase 3 E1912 trial of 529 patients, which demonstrated significantly improved progression-free survival (PFS) among those who received ibrutinib plus rituximab, compared with those who received fludarabine, cyclophosphamide, and rituximab (FCR) (87% vs. 75%; hazard ratio, 0.34). Median PFS was not reached in either arm after a median follow-up of 37 months.
E1912 was the first study to show superiority of a chemotherapy-free regimen over FCR chemoimmunotherapy, considered the gold standard for newly diagnosed CLL and SLL for the past 2 decades.
The recommended dosage for the newly approved combination is a once-daily 420-mg dose of ibrutinib taken with a glass of water, with rituximab initiation in the second cycle at doses of 50 mg/m2 on day 1, 325 mg/m2 on day 2, and 500 mg/m2 on days 1-5 of subsequent cycles for a total of six cycles.
FDA authorizes first COVID-19 test kit with home collection option
a reissue of the emergency use authorization allowing for testing of samples self-collected by patients at home with the Pixel by LabCorp COVID-19 RT-PCR Test.

The reissued authorization allows for testing of a sample taken from the nose by way of a self-collection kit that contains nasal swabs and saline, according to the FDA press release. After self-swabbing, users should send the samples in an insulated package to a LabCorp laboratory for testing. LabCorp intends to make the Pixel test available to consumers in most states, accessible through doctors’ orders.
The Pixel test includes a specific Q-tip–style cotton swab for patients to use to collect their samples, the FDA noted. Because of concerns with sterility and cross-reactivity caused by inherent genetic material in cotton swabs, generic cotton swabs should not be used as a substitute. The FDA will work with test developers to determine if generic cotton swabs can be used safely and effectively with other tests.
“Throughout this pandemic we have been facilitating test development to ensure patients’ access to accurate diagnostics, which includes supporting the development of reliable and accurate at-home sample collection options. ... [The FDA] worked with LabCorp to ensure the data demonstrated from at-home patient sample collection is as safe and accurate as sample collection at a doctor’s office, hospital, or other testing site. With this action, there is now a convenient and reliable option for patient sample collection from the comfort and safety of their home,” FDA Commissioner Stephen M. Hahn, MD, said in the press release.
a reissue of the emergency use authorization allowing for testing of samples self-collected by patients at home with the Pixel by LabCorp COVID-19 RT-PCR Test.
The reissued authorization allows for testing of a sample taken from the nose by way of a self-collection kit that contains nasal swabs and saline, according to the FDA press release. After self-swabbing, users should send the samples in an insulated package to a LabCorp laboratory for testing. LabCorp intends to make the Pixel test available to consumers in most states, accessible through doctors’ orders.
The Pixel test includes a specific Q-tip–style cotton swab for patients to use to collect their samples, the FDA noted. Because of concerns with sterility and cross-reactivity caused by inherent genetic material in cotton swabs, generic cotton swabs should not be used as a substitute. The FDA will work with test developers to determine if generic cotton swabs can be used safely and effectively with other tests.
“Throughout this pandemic we have been facilitating test development to ensure patients’ access to accurate diagnostics, which includes supporting the development of reliable and accurate at-home sample collection options. ... [The FDA] worked with LabCorp to ensure the data demonstrated from at-home patient sample collection is as safe and accurate as sample collection at a doctor’s office, hospital, or other testing site. With this action, there is now a convenient and reliable option for patient sample collection from the comfort and safety of their home,” FDA Commissioner Stephen M. Hahn, MD, said in the press release.
a reissue of the emergency use authorization allowing for testing of samples self-collected by patients at home with the Pixel by LabCorp COVID-19 RT-PCR Test.
The reissued authorization allows for testing of a sample taken from the nose by way of a self-collection kit that contains nasal swabs and saline, according to the FDA press release. After self-swabbing, users should send the samples in an insulated package to a LabCorp laboratory for testing. LabCorp intends to make the Pixel test available to consumers in most states, accessible through doctors’ orders.
The Pixel test includes a specific Q-tip–style cotton swab for patients to use to collect their samples, the FDA noted. Because of concerns with sterility and cross-reactivity caused by inherent genetic material in cotton swabs, generic cotton swabs should not be used as a substitute. The FDA will work with test developers to determine if generic cotton swabs can be used safely and effectively with other tests.
“Throughout this pandemic we have been facilitating test development to ensure patients’ access to accurate diagnostics, which includes supporting the development of reliable and accurate at-home sample collection options. ... [The FDA] worked with LabCorp to ensure the data demonstrated from at-home patient sample collection is as safe and accurate as sample collection at a doctor’s office, hospital, or other testing site. With this action, there is now a convenient and reliable option for patient sample collection from the comfort and safety of their home,” FDA Commissioner Stephen M. Hahn, MD, said in the press release.
FDA approves first targeted drug for bile duct cancer
The U.S. Food and Drug Administration (FDA) has granted accelerated approval of a new targeted therapy for use in some patients with cholangiocarcinoma, a rare cancer of the bile ducts.
The product is pemigatinib (Pemazyre, Incyte), an oral kinase inhibitor.
It was approved specifically for use in patients with advanced cholangiocarcinoma who have received prior treatment and who have tumors that have a fusion or other rearrangement of the fibroblast growth factor receptor 2 (FGFR2) gene.
These FGFR2 genetic abnormalities are found in about 9% to 14% of patients with cholangiocarcinoma, notes the FDA.
At diagnosis, a majority of patients with cholangiocarcinoma have advanced disease, meaning that the disease is no longer treatable with surgery, the agency also notes. Until now, a combination of chemotherapy drugs has been the standard initial treatment.
Now the subgroup of patients with FGFR2 tumors have the option of a targeted therapy.
“Although cholangiocarcinoma is considered a rare disease, it has been on the rise over the past three decades,” commented Ghassan Abou-Alfa, MD, of Memorial Sloan Kettering Cancer Center, New York City, in a press release. “It is encouraging to have a new targeted treatment option for patients who historically have had limited options after first-line chemotherapy or surgery, in which relapse rates remain high.”
The accelerated approval was based on the overall response rate (ORR) and duration of response in an open-label clinical trial that involved 107 patients (the FIGHT-202 study).
As a condition of the accelerated approval, the manufacturer will complete and submit the results of a randomized trial demonstrating an improvement in progression-free survival or overall survival, the FDA noted.
Results from open-label clinical trial
The FIGHT-202 study enrolled 107 patients with locally advanced or metastatic cholangiocarcinoma who had received prior treatment and who had tumors with an FGFR2 fusion or rearrangement.
All patients received pemigatinib once a day for 14 days, followed by 7 days off, in 21-day cycles until the disease progressed or the patient experienced an unreasonable level of side effects. Patients underwent scanning every 8 weeks to assess ORR.
The ORR was 36% (38 of 107 patients), which included 2.8% of patients with a complete response and 33% with partial response.
Among the 38 patients who had a response, 24 patients (63%) had a response that lasted 6 months or longer, and 7 patients (18%) had a response that lasted 12 months or longer.
“With pemigatinib, we considered the observed efficacy results to be clinically meaningful and the overall risk to benefit assessment for patients with tumors harboring FGFR2 gene fusions and other rearrangements to be favorable, particularly when we considered that these patients have no other good options following first-line treatment with chemotherapy,” commented Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The most common adverse reactions, which occurred in 20% or more of patients who received pemigatinib, were electrolyte disorders (hyperphosphatemia and hypophosphatemia), alopecia, diarrhea, nail toxicity, fatigue, dysgeusia, nausea, constipation, stomatitis, dry eye, dry mouth, decreased appetite, vomiting, joint pain, abdominal pain, back pain, and dry skin. Ocular toxicity was seen rarely, the agency notes.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration (FDA) has granted accelerated approval of a new targeted therapy for use in some patients with cholangiocarcinoma, a rare cancer of the bile ducts.
The product is pemigatinib (Pemazyre, Incyte), an oral kinase inhibitor.
It was approved specifically for use in patients with advanced cholangiocarcinoma who have received prior treatment and who have tumors that have a fusion or other rearrangement of the fibroblast growth factor receptor 2 (FGFR2) gene.
These FGFR2 genetic abnormalities are found in about 9% to 14% of patients with cholangiocarcinoma, notes the FDA.
At diagnosis, a majority of patients with cholangiocarcinoma have advanced disease, meaning that the disease is no longer treatable with surgery, the agency also notes. Until now, a combination of chemotherapy drugs has been the standard initial treatment.
Now the subgroup of patients with FGFR2 tumors have the option of a targeted therapy.
“Although cholangiocarcinoma is considered a rare disease, it has been on the rise over the past three decades,” commented Ghassan Abou-Alfa, MD, of Memorial Sloan Kettering Cancer Center, New York City, in a press release. “It is encouraging to have a new targeted treatment option for patients who historically have had limited options after first-line chemotherapy or surgery, in which relapse rates remain high.”
The accelerated approval was based on the overall response rate (ORR) and duration of response in an open-label clinical trial that involved 107 patients (the FIGHT-202 study).
As a condition of the accelerated approval, the manufacturer will complete and submit the results of a randomized trial demonstrating an improvement in progression-free survival or overall survival, the FDA noted.
Results from open-label clinical trial
The FIGHT-202 study enrolled 107 patients with locally advanced or metastatic cholangiocarcinoma who had received prior treatment and who had tumors with an FGFR2 fusion or rearrangement.
All patients received pemigatinib once a day for 14 days, followed by 7 days off, in 21-day cycles until the disease progressed or the patient experienced an unreasonable level of side effects. Patients underwent scanning every 8 weeks to assess ORR.
The ORR was 36% (38 of 107 patients), which included 2.8% of patients with a complete response and 33% with partial response.
Among the 38 patients who had a response, 24 patients (63%) had a response that lasted 6 months or longer, and 7 patients (18%) had a response that lasted 12 months or longer.
“With pemigatinib, we considered the observed efficacy results to be clinically meaningful and the overall risk to benefit assessment for patients with tumors harboring FGFR2 gene fusions and other rearrangements to be favorable, particularly when we considered that these patients have no other good options following first-line treatment with chemotherapy,” commented Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The most common adverse reactions, which occurred in 20% or more of patients who received pemigatinib, were electrolyte disorders (hyperphosphatemia and hypophosphatemia), alopecia, diarrhea, nail toxicity, fatigue, dysgeusia, nausea, constipation, stomatitis, dry eye, dry mouth, decreased appetite, vomiting, joint pain, abdominal pain, back pain, and dry skin. Ocular toxicity was seen rarely, the agency notes.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration (FDA) has granted accelerated approval of a new targeted therapy for use in some patients with cholangiocarcinoma, a rare cancer of the bile ducts.
The product is pemigatinib (Pemazyre, Incyte), an oral kinase inhibitor.
It was approved specifically for use in patients with advanced cholangiocarcinoma who have received prior treatment and who have tumors that have a fusion or other rearrangement of the fibroblast growth factor receptor 2 (FGFR2) gene.
These FGFR2 genetic abnormalities are found in about 9% to 14% of patients with cholangiocarcinoma, notes the FDA.
At diagnosis, a majority of patients with cholangiocarcinoma have advanced disease, meaning that the disease is no longer treatable with surgery, the agency also notes. Until now, a combination of chemotherapy drugs has been the standard initial treatment.
Now the subgroup of patients with FGFR2 tumors have the option of a targeted therapy.
“Although cholangiocarcinoma is considered a rare disease, it has been on the rise over the past three decades,” commented Ghassan Abou-Alfa, MD, of Memorial Sloan Kettering Cancer Center, New York City, in a press release. “It is encouraging to have a new targeted treatment option for patients who historically have had limited options after first-line chemotherapy or surgery, in which relapse rates remain high.”
The accelerated approval was based on the overall response rate (ORR) and duration of response in an open-label clinical trial that involved 107 patients (the FIGHT-202 study).
As a condition of the accelerated approval, the manufacturer will complete and submit the results of a randomized trial demonstrating an improvement in progression-free survival or overall survival, the FDA noted.
Results from open-label clinical trial
The FIGHT-202 study enrolled 107 patients with locally advanced or metastatic cholangiocarcinoma who had received prior treatment and who had tumors with an FGFR2 fusion or rearrangement.
All patients received pemigatinib once a day for 14 days, followed by 7 days off, in 21-day cycles until the disease progressed or the patient experienced an unreasonable level of side effects. Patients underwent scanning every 8 weeks to assess ORR.
The ORR was 36% (38 of 107 patients), which included 2.8% of patients with a complete response and 33% with partial response.
Among the 38 patients who had a response, 24 patients (63%) had a response that lasted 6 months or longer, and 7 patients (18%) had a response that lasted 12 months or longer.
“With pemigatinib, we considered the observed efficacy results to be clinically meaningful and the overall risk to benefit assessment for patients with tumors harboring FGFR2 gene fusions and other rearrangements to be favorable, particularly when we considered that these patients have no other good options following first-line treatment with chemotherapy,” commented Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The most common adverse reactions, which occurred in 20% or more of patients who received pemigatinib, were electrolyte disorders (hyperphosphatemia and hypophosphatemia), alopecia, diarrhea, nail toxicity, fatigue, dysgeusia, nausea, constipation, stomatitis, dry eye, dry mouth, decreased appetite, vomiting, joint pain, abdominal pain, back pain, and dry skin. Ocular toxicity was seen rarely, the agency notes.
This article first appeared on Medscape.com.
FDA approves first new breast cancer drug with international group
The U.S. Food and Drug Administration has approved the oral therapy tucatinib (Tukysa, Seattle Genetics) for the treatment of advanced HER2-positive breast cancer. This is the first new drug approved under Project Orbis, an international collaboration.
Tucatinib, which is a small-molecule tyrosine kinase inhibitor, is approved in combination with trastuzumab and capecitabine to treat patients who have received one or more prior treatments for advanced disease.
The FDA collaborated with the regulatory authorities of Australia, Canada, Singapore, and Switzerland on this review. However, only the FDA has approved tucatinib; the application is still under review at the other agencies.
While working with Project Orbis in 2019, the FDA granted an accelerated, conditional approval to a drug combination that included previously approved agents.
“The FDA’s Project Orbis provides a framework for concurrent submission and review of oncology drug applications among the FDA’s international collaborators,” said Richard Pazdur, MD, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in a statement.
Collaboration among regulators may allow patients with cancer to receive earlier access to products in other countries where there may be significant delays in regulatory submissions, according to the FDA.
The new drug is a “valuable addition” to the roster of treatments for advanced HER2-positive breast cancer, said study investigator Eric Winer, MD, Dana-Farber Cancer Institute, Boston, Massachusetts, in a company press statement.
“With highly significant and clinically important results for overall and progression-free survival, the addition of [tucatinib] to trastuzumab and capecitabine has the potential to become a standard of care for people with HER2-positive metastatic breast cancer after having received one or more previous anti-HER2 therapies in the metastatic setting,” he said.
The new approval is based on safety and efficacy results from the phase 2 HER2CLIMB trial that enrolled 612 patients with HER2-positive unresectable locally advanced or metastatic breast cancer who had previously received, either separately or in combination, trastuzumab, pertuzumab, and ado-trastuzumab emtansine.
Nearly half (48%) of patients in the study had brain metastases at the start of the trial. The primary outcome measure was progression-free survival (PFS). All patients received trastuzumab and capecitabine and were randomly assigned to either tucatinib or placebo.
Median PFS in the tucatinib patient group was 7.8 months, compared with 5.6 months in the placebo group. The PFS results in the subgroup of patients with brain metastases were nearly the same.
Median overall survival in the tucatinib patient group was 21.9 months versus 17.4 months in the placebo group.
The new drug is a rare success in the treatment of breast cancer brain metastases, said Jawad Fares, MD, of Northwestern University, Chicago, Illinois, who spoke to Medscape Medical News when the phase 3 trial data were first presented at the 2019 San Antonio Breast Cancer Symposium.
“Outcomes in the field have been pretty dismal,” summarized Fares, who was not involved in the study.
The results of the HER2CLIMB study, which was funded by Seattle Genetics, were published in the New England Journal of Medicine last year.
According to the FDA, common side effects with tucatinib were diarrhea, palmar-plantar erythrodysesthesia syndrome, nausea, fatigue, hepatotoxicity, vomiting, stomatitis, decreased appetite, abdominal pain, headache, anemia, and rash.
Tucatinib can cause serious side effects, including diarrhea associated with dehydration, acute kidney injury, and death. Health care professionals should start antidiarrheals as clinically indicated if diarrhea occurs and should interrupt treatment or reduce the dosage. Tucatinib can also cause severe hepatotoxicity; patients should be monitored with liver tests.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration has approved the oral therapy tucatinib (Tukysa, Seattle Genetics) for the treatment of advanced HER2-positive breast cancer. This is the first new drug approved under Project Orbis, an international collaboration.
Tucatinib, which is a small-molecule tyrosine kinase inhibitor, is approved in combination with trastuzumab and capecitabine to treat patients who have received one or more prior treatments for advanced disease.
The FDA collaborated with the regulatory authorities of Australia, Canada, Singapore, and Switzerland on this review. However, only the FDA has approved tucatinib; the application is still under review at the other agencies.
While working with Project Orbis in 2019, the FDA granted an accelerated, conditional approval to a drug combination that included previously approved agents.
“The FDA’s Project Orbis provides a framework for concurrent submission and review of oncology drug applications among the FDA’s international collaborators,” said Richard Pazdur, MD, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in a statement.
Collaboration among regulators may allow patients with cancer to receive earlier access to products in other countries where there may be significant delays in regulatory submissions, according to the FDA.
The new drug is a “valuable addition” to the roster of treatments for advanced HER2-positive breast cancer, said study investigator Eric Winer, MD, Dana-Farber Cancer Institute, Boston, Massachusetts, in a company press statement.
“With highly significant and clinically important results for overall and progression-free survival, the addition of [tucatinib] to trastuzumab and capecitabine has the potential to become a standard of care for people with HER2-positive metastatic breast cancer after having received one or more previous anti-HER2 therapies in the metastatic setting,” he said.
The new approval is based on safety and efficacy results from the phase 2 HER2CLIMB trial that enrolled 612 patients with HER2-positive unresectable locally advanced or metastatic breast cancer who had previously received, either separately or in combination, trastuzumab, pertuzumab, and ado-trastuzumab emtansine.
Nearly half (48%) of patients in the study had brain metastases at the start of the trial. The primary outcome measure was progression-free survival (PFS). All patients received trastuzumab and capecitabine and were randomly assigned to either tucatinib or placebo.
Median PFS in the tucatinib patient group was 7.8 months, compared with 5.6 months in the placebo group. The PFS results in the subgroup of patients with brain metastases were nearly the same.
Median overall survival in the tucatinib patient group was 21.9 months versus 17.4 months in the placebo group.
The new drug is a rare success in the treatment of breast cancer brain metastases, said Jawad Fares, MD, of Northwestern University, Chicago, Illinois, who spoke to Medscape Medical News when the phase 3 trial data were first presented at the 2019 San Antonio Breast Cancer Symposium.
“Outcomes in the field have been pretty dismal,” summarized Fares, who was not involved in the study.
The results of the HER2CLIMB study, which was funded by Seattle Genetics, were published in the New England Journal of Medicine last year.
According to the FDA, common side effects with tucatinib were diarrhea, palmar-plantar erythrodysesthesia syndrome, nausea, fatigue, hepatotoxicity, vomiting, stomatitis, decreased appetite, abdominal pain, headache, anemia, and rash.
Tucatinib can cause serious side effects, including diarrhea associated with dehydration, acute kidney injury, and death. Health care professionals should start antidiarrheals as clinically indicated if diarrhea occurs and should interrupt treatment or reduce the dosage. Tucatinib can also cause severe hepatotoxicity; patients should be monitored with liver tests.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration has approved the oral therapy tucatinib (Tukysa, Seattle Genetics) for the treatment of advanced HER2-positive breast cancer. This is the first new drug approved under Project Orbis, an international collaboration.
Tucatinib, which is a small-molecule tyrosine kinase inhibitor, is approved in combination with trastuzumab and capecitabine to treat patients who have received one or more prior treatments for advanced disease.
The FDA collaborated with the regulatory authorities of Australia, Canada, Singapore, and Switzerland on this review. However, only the FDA has approved tucatinib; the application is still under review at the other agencies.
While working with Project Orbis in 2019, the FDA granted an accelerated, conditional approval to a drug combination that included previously approved agents.
“The FDA’s Project Orbis provides a framework for concurrent submission and review of oncology drug applications among the FDA’s international collaborators,” said Richard Pazdur, MD, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in a statement.
Collaboration among regulators may allow patients with cancer to receive earlier access to products in other countries where there may be significant delays in regulatory submissions, according to the FDA.
The new drug is a “valuable addition” to the roster of treatments for advanced HER2-positive breast cancer, said study investigator Eric Winer, MD, Dana-Farber Cancer Institute, Boston, Massachusetts, in a company press statement.
“With highly significant and clinically important results for overall and progression-free survival, the addition of [tucatinib] to trastuzumab and capecitabine has the potential to become a standard of care for people with HER2-positive metastatic breast cancer after having received one or more previous anti-HER2 therapies in the metastatic setting,” he said.
The new approval is based on safety and efficacy results from the phase 2 HER2CLIMB trial that enrolled 612 patients with HER2-positive unresectable locally advanced or metastatic breast cancer who had previously received, either separately or in combination, trastuzumab, pertuzumab, and ado-trastuzumab emtansine.
Nearly half (48%) of patients in the study had brain metastases at the start of the trial. The primary outcome measure was progression-free survival (PFS). All patients received trastuzumab and capecitabine and were randomly assigned to either tucatinib or placebo.
Median PFS in the tucatinib patient group was 7.8 months, compared with 5.6 months in the placebo group. The PFS results in the subgroup of patients with brain metastases were nearly the same.
Median overall survival in the tucatinib patient group was 21.9 months versus 17.4 months in the placebo group.
The new drug is a rare success in the treatment of breast cancer brain metastases, said Jawad Fares, MD, of Northwestern University, Chicago, Illinois, who spoke to Medscape Medical News when the phase 3 trial data were first presented at the 2019 San Antonio Breast Cancer Symposium.
“Outcomes in the field have been pretty dismal,” summarized Fares, who was not involved in the study.
The results of the HER2CLIMB study, which was funded by Seattle Genetics, were published in the New England Journal of Medicine last year.
According to the FDA, common side effects with tucatinib were diarrhea, palmar-plantar erythrodysesthesia syndrome, nausea, fatigue, hepatotoxicity, vomiting, stomatitis, decreased appetite, abdominal pain, headache, anemia, and rash.
Tucatinib can cause serious side effects, including diarrhea associated with dehydration, acute kidney injury, and death. Health care professionals should start antidiarrheals as clinically indicated if diarrhea occurs and should interrupt treatment or reduce the dosage. Tucatinib can also cause severe hepatotoxicity; patients should be monitored with liver tests.
This article first appeared on Medscape.com.
2019-2020 flu season ends with ‘very high’ activity in New Jersey
The 2019-2020 flu season is ending, but not without a revised map to reflect the COVID-induced new world order.
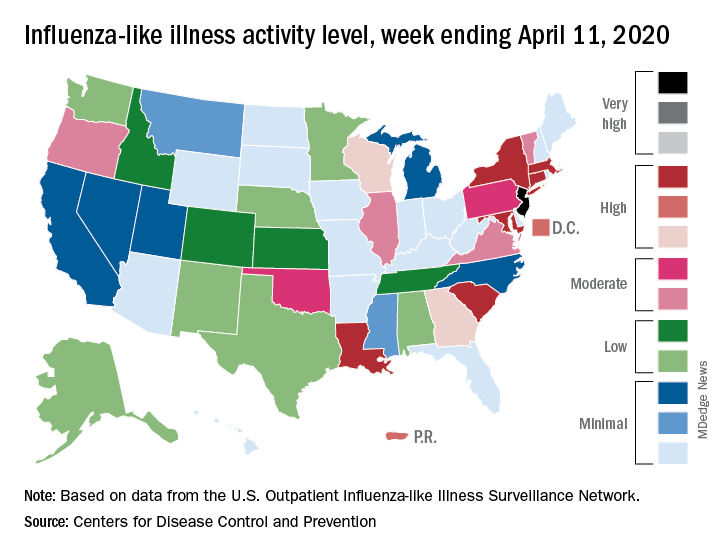
For the week ending April 11, those additions encompass only New Jersey at level 13 and New York City at level 12, the CDC reported April 17.
Eight states, plus the District of Columbia and Puerto Rico, were in the “high” range of flu activity, which runs from level 8 to level 10, for the same week. Those eight states included Connecticut, Georgia, Louisiana, Maryland, Massachusetts, New York, South Carolina, and Wisconsin.
The CDC’s influenza division included this note with its latest FluView report: “The COVID-19 pandemic is affecting healthcare seeking behavior. The number of persons and their reasons for seeking care in the outpatient and ED settings is changing. These changes impact data from ILINet [Outpatient Influenza-like Illness Surveillance Network] in ways that are difficult to differentiate from changes in illness levels, therefore ILINet data should be interpreted with caution.”
Outpatient visits for influenza-like illness made up 2.9% of all visits to health care providers for the week ending April 11, which is the 23rd consecutive week that it’s been at or above the national baseline level of 2.4%. Twenty-three weeks is longer than this has occurred during any flu season since the CDC started setting a baseline in 2007, according to ILINet data.
Mortality from pneumonia and influenza, at 11.7%, was well above the epidemic threshold of 7.0%, although, again, pneumonia mortality “is being driven primarily by an increase in non-influenza pneumonia deaths due to COVID-19,” the CDC wrote.
The total number of influenza-related deaths in children, with reports of two more added this week, is 168 for the season – higher than two of the last three seasons: 144 in 2018-2019, 188 in 2017-2018, and 110 in 2016-2017, according to the CDC.
The 2019-2020 flu season is ending, but not without a revised map to reflect the COVID-induced new world order.
For the week ending April 11, those additions encompass only New Jersey at level 13 and New York City at level 12, the CDC reported April 17.
Eight states, plus the District of Columbia and Puerto Rico, were in the “high” range of flu activity, which runs from level 8 to level 10, for the same week. Those eight states included Connecticut, Georgia, Louisiana, Maryland, Massachusetts, New York, South Carolina, and Wisconsin.
The CDC’s influenza division included this note with its latest FluView report: “The COVID-19 pandemic is affecting healthcare seeking behavior. The number of persons and their reasons for seeking care in the outpatient and ED settings is changing. These changes impact data from ILINet [Outpatient Influenza-like Illness Surveillance Network] in ways that are difficult to differentiate from changes in illness levels, therefore ILINet data should be interpreted with caution.”
Outpatient visits for influenza-like illness made up 2.9% of all visits to health care providers for the week ending April 11, which is the 23rd consecutive week that it’s been at or above the national baseline level of 2.4%. Twenty-three weeks is longer than this has occurred during any flu season since the CDC started setting a baseline in 2007, according to ILINet data.
Mortality from pneumonia and influenza, at 11.7%, was well above the epidemic threshold of 7.0%, although, again, pneumonia mortality “is being driven primarily by an increase in non-influenza pneumonia deaths due to COVID-19,” the CDC wrote.
The total number of influenza-related deaths in children, with reports of two more added this week, is 168 for the season – higher than two of the last three seasons: 144 in 2018-2019, 188 in 2017-2018, and 110 in 2016-2017, according to the CDC.
The 2019-2020 flu season is ending, but not without a revised map to reflect the COVID-induced new world order.
For the week ending April 11, those additions encompass only New Jersey at level 13 and New York City at level 12, the CDC reported April 17.
Eight states, plus the District of Columbia and Puerto Rico, were in the “high” range of flu activity, which runs from level 8 to level 10, for the same week. Those eight states included Connecticut, Georgia, Louisiana, Maryland, Massachusetts, New York, South Carolina, and Wisconsin.
The CDC’s influenza division included this note with its latest FluView report: “The COVID-19 pandemic is affecting healthcare seeking behavior. The number of persons and their reasons for seeking care in the outpatient and ED settings is changing. These changes impact data from ILINet [Outpatient Influenza-like Illness Surveillance Network] in ways that are difficult to differentiate from changes in illness levels, therefore ILINet data should be interpreted with caution.”
Outpatient visits for influenza-like illness made up 2.9% of all visits to health care providers for the week ending April 11, which is the 23rd consecutive week that it’s been at or above the national baseline level of 2.4%. Twenty-three weeks is longer than this has occurred during any flu season since the CDC started setting a baseline in 2007, according to ILINet data.
Mortality from pneumonia and influenza, at 11.7%, was well above the epidemic threshold of 7.0%, although, again, pneumonia mortality “is being driven primarily by an increase in non-influenza pneumonia deaths due to COVID-19,” the CDC wrote.
The total number of influenza-related deaths in children, with reports of two more added this week, is 168 for the season – higher than two of the last three seasons: 144 in 2018-2019, 188 in 2017-2018, and 110 in 2016-2017, according to the CDC.
Mitomycin approved for low-grade upper tract urothelial cancer
pyelocalyceal

“This is the first approval specifically for patients with low-grade [upper tract urothelial cancer] and provides an option for some patients who may otherwise require a nephroureterectomy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
“Due to substantial treatment challenges associated with the complex anatomy of the upper urinary tract, many patients need to be treated with radical surgery – usually complete removal of the affected kidney, ureter, and bladder cuff," Dr. Pazdur added. "Jelmyto gives patients, for the first time, an alternative treatment option for low-grade [upper tract urothelial cancer].”
The FDA’s approval of mitomycin is based on results from the phase 3 OLYMPUS trial (NCT02793128). This ongoing, single-arm trial enrolled 71 patients with treatment-naive or recurrent low-grade noninvasive upper tract urothelial cancer with at least one measurable papillary tumor located above the ureteropelvic junction. Patients with larger tumors were allowed to have prior tumor debulking.
The patients received mitomycin weekly for 6 weeks at the recommended dose of 4 mg/mL, instilled via ureteral catheter or nephrostomy tube, with the total instillation volume based on volumetric measurements using pyelography, not exceeding 15 mL (60 mg mitomycin).
Patients who achieved a complete response at 3 months could receive monthly instillations up to a maximum of 11 additional instillations.
At 3 months, 41 patients (58%) achieved a complete response (CR). At 12 months after CR determination, 19 patients were still in CR, and 7 patients had documented recurrences. The median duration of CR was not reached.
The most common adverse events (occurring in at least 20% of patients) were ureteric obstruction, flank pain, urinary tract infection, hematuria, renal dysfunction, fatigue, nausea, abdominal pain, dysuria, and vomiting. Ureteric obstruction occurred in 58% of patients, and 88% of those patients required ureteral stent placement.
In all, 23% of patients discontinued mitomycin due to adverse events, and 34% had dose interruptions due to adverse events.
The approval of mitomycin was granted to UroGen Pharma. The FDA granted the application priority review, fast track designation, and breakthrough therapy designation.
The full prescribing information for mitomycin is available for download from the FDA website.
pyelocalyceal
“This is the first approval specifically for patients with low-grade [upper tract urothelial cancer] and provides an option for some patients who may otherwise require a nephroureterectomy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
“Due to substantial treatment challenges associated with the complex anatomy of the upper urinary tract, many patients need to be treated with radical surgery – usually complete removal of the affected kidney, ureter, and bladder cuff," Dr. Pazdur added. "Jelmyto gives patients, for the first time, an alternative treatment option for low-grade [upper tract urothelial cancer].”
The FDA’s approval of mitomycin is based on results from the phase 3 OLYMPUS trial (NCT02793128). This ongoing, single-arm trial enrolled 71 patients with treatment-naive or recurrent low-grade noninvasive upper tract urothelial cancer with at least one measurable papillary tumor located above the ureteropelvic junction. Patients with larger tumors were allowed to have prior tumor debulking.
The patients received mitomycin weekly for 6 weeks at the recommended dose of 4 mg/mL, instilled via ureteral catheter or nephrostomy tube, with the total instillation volume based on volumetric measurements using pyelography, not exceeding 15 mL (60 mg mitomycin).
Patients who achieved a complete response at 3 months could receive monthly instillations up to a maximum of 11 additional instillations.
At 3 months, 41 patients (58%) achieved a complete response (CR). At 12 months after CR determination, 19 patients were still in CR, and 7 patients had documented recurrences. The median duration of CR was not reached.
The most common adverse events (occurring in at least 20% of patients) were ureteric obstruction, flank pain, urinary tract infection, hematuria, renal dysfunction, fatigue, nausea, abdominal pain, dysuria, and vomiting. Ureteric obstruction occurred in 58% of patients, and 88% of those patients required ureteral stent placement.
In all, 23% of patients discontinued mitomycin due to adverse events, and 34% had dose interruptions due to adverse events.
The approval of mitomycin was granted to UroGen Pharma. The FDA granted the application priority review, fast track designation, and breakthrough therapy designation.
The full prescribing information for mitomycin is available for download from the FDA website.
pyelocalyceal
“This is the first approval specifically for patients with low-grade [upper tract urothelial cancer] and provides an option for some patients who may otherwise require a nephroureterectomy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
“Due to substantial treatment challenges associated with the complex anatomy of the upper urinary tract, many patients need to be treated with radical surgery – usually complete removal of the affected kidney, ureter, and bladder cuff," Dr. Pazdur added. "Jelmyto gives patients, for the first time, an alternative treatment option for low-grade [upper tract urothelial cancer].”
The FDA’s approval of mitomycin is based on results from the phase 3 OLYMPUS trial (NCT02793128). This ongoing, single-arm trial enrolled 71 patients with treatment-naive or recurrent low-grade noninvasive upper tract urothelial cancer with at least one measurable papillary tumor located above the ureteropelvic junction. Patients with larger tumors were allowed to have prior tumor debulking.
The patients received mitomycin weekly for 6 weeks at the recommended dose of 4 mg/mL, instilled via ureteral catheter or nephrostomy tube, with the total instillation volume based on volumetric measurements using pyelography, not exceeding 15 mL (60 mg mitomycin).
Patients who achieved a complete response at 3 months could receive monthly instillations up to a maximum of 11 additional instillations.
At 3 months, 41 patients (58%) achieved a complete response (CR). At 12 months after CR determination, 19 patients were still in CR, and 7 patients had documented recurrences. The median duration of CR was not reached.
The most common adverse events (occurring in at least 20% of patients) were ureteric obstruction, flank pain, urinary tract infection, hematuria, renal dysfunction, fatigue, nausea, abdominal pain, dysuria, and vomiting. Ureteric obstruction occurred in 58% of patients, and 88% of those patients required ureteral stent placement.
In all, 23% of patients discontinued mitomycin due to adverse events, and 34% had dose interruptions due to adverse events.
The approval of mitomycin was granted to UroGen Pharma. The FDA granted the application priority review, fast track designation, and breakthrough therapy designation.
The full prescribing information for mitomycin is available for download from the FDA website.
FROM FDA
COVID-19: When health care personnel become patients
according to the Centers for Disease Control and Prevention.
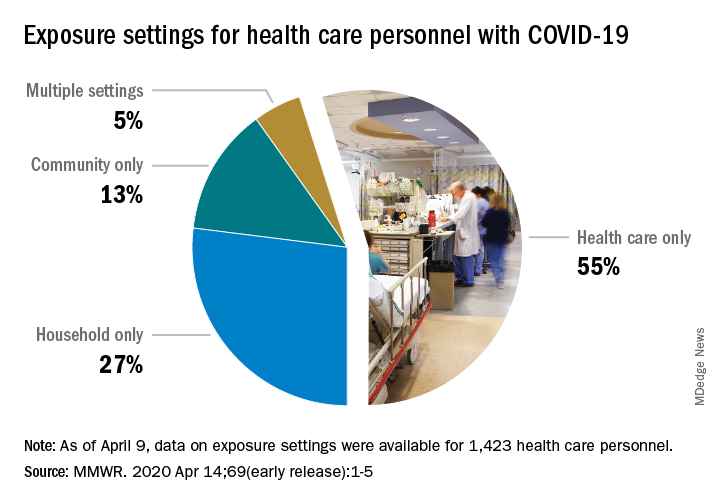
That number, however, is probably an underestimation because health care personnel (HCP) status was available for just over 49,000 of the 315,000 COVID-19 cases reported to the CDC as of April 9. Of the cases with known HCP status, 9,282 (19%) were health care personnel, Matthew J. Stuckey, PhD, and the CDC’s COVID-19 Response Team said.
“The number of cases in HCP reported here must be considered a lower bound because additional cases likely have gone unidentified or unreported,” they said.
The median age of the nearly 9,300 HCP with COVID-19 was 42 years, and the majority (55%) were aged 16-44 years; another 21% were 45-54, 18% were 55-64, and 6% were age 65 and over. The oldest group, however, represented 10 of the 27 known HCP deaths, the investigators reported in the Morbidity and Mortality Weekly Report.
The majority of infected HCP (55%) reported exposure to a COVID-19 patient in the health care setting, but “there were also known exposures in households and in the community, highlighting the potential for exposure in multiple settings, especially as community transmission increases,” the response team said.
Since “contact tracing after recognized occupational exposures likely will fail to identify many HCP at risk for developing COVID-19,” other measures will probably be needed to “reduce the risk for infected HCP transmitting the virus to colleagues and patients,” they added.
HCP with COVID-19 were less likely to be hospitalized (8%-10%) than the overall population (21%-31%), which “might reflect the younger median age … of HCP patients, compared with that of reported COVID-19 patients overall, as well as prioritization of HCP for testing, which might identify less-severe illness,” the investigators suggested.
The prevalence of underlying conditions in HCP patients, 38%, was the same as all patients with COVID-19, and 92% of the HCP patients presented with fever, cough, or shortness of breath. Two-thirds of all HCP reported muscle aches, and 65% reported headache, the CDC response team noted.
“It is critical to make every effort to ensure the health and safety of this essential national workforce of approximately 18 million HCP, both at work and in the community,” they wrote.
SOURCE: Stuckey MJ et al. MMWR. Apr 14;69(early release):1-5.
according to the Centers for Disease Control and Prevention.
That number, however, is probably an underestimation because health care personnel (HCP) status was available for just over 49,000 of the 315,000 COVID-19 cases reported to the CDC as of April 9. Of the cases with known HCP status, 9,282 (19%) were health care personnel, Matthew J. Stuckey, PhD, and the CDC’s COVID-19 Response Team said.
“The number of cases in HCP reported here must be considered a lower bound because additional cases likely have gone unidentified or unreported,” they said.
The median age of the nearly 9,300 HCP with COVID-19 was 42 years, and the majority (55%) were aged 16-44 years; another 21% were 45-54, 18% were 55-64, and 6% were age 65 and over. The oldest group, however, represented 10 of the 27 known HCP deaths, the investigators reported in the Morbidity and Mortality Weekly Report.
The majority of infected HCP (55%) reported exposure to a COVID-19 patient in the health care setting, but “there were also known exposures in households and in the community, highlighting the potential for exposure in multiple settings, especially as community transmission increases,” the response team said.
Since “contact tracing after recognized occupational exposures likely will fail to identify many HCP at risk for developing COVID-19,” other measures will probably be needed to “reduce the risk for infected HCP transmitting the virus to colleagues and patients,” they added.
HCP with COVID-19 were less likely to be hospitalized (8%-10%) than the overall population (21%-31%), which “might reflect the younger median age … of HCP patients, compared with that of reported COVID-19 patients overall, as well as prioritization of HCP for testing, which might identify less-severe illness,” the investigators suggested.
The prevalence of underlying conditions in HCP patients, 38%, was the same as all patients with COVID-19, and 92% of the HCP patients presented with fever, cough, or shortness of breath. Two-thirds of all HCP reported muscle aches, and 65% reported headache, the CDC response team noted.
“It is critical to make every effort to ensure the health and safety of this essential national workforce of approximately 18 million HCP, both at work and in the community,” they wrote.
SOURCE: Stuckey MJ et al. MMWR. Apr 14;69(early release):1-5.
according to the Centers for Disease Control and Prevention.
That number, however, is probably an underestimation because health care personnel (HCP) status was available for just over 49,000 of the 315,000 COVID-19 cases reported to the CDC as of April 9. Of the cases with known HCP status, 9,282 (19%) were health care personnel, Matthew J. Stuckey, PhD, and the CDC’s COVID-19 Response Team said.
“The number of cases in HCP reported here must be considered a lower bound because additional cases likely have gone unidentified or unreported,” they said.
The median age of the nearly 9,300 HCP with COVID-19 was 42 years, and the majority (55%) were aged 16-44 years; another 21% were 45-54, 18% were 55-64, and 6% were age 65 and over. The oldest group, however, represented 10 of the 27 known HCP deaths, the investigators reported in the Morbidity and Mortality Weekly Report.
The majority of infected HCP (55%) reported exposure to a COVID-19 patient in the health care setting, but “there were also known exposures in households and in the community, highlighting the potential for exposure in multiple settings, especially as community transmission increases,” the response team said.
Since “contact tracing after recognized occupational exposures likely will fail to identify many HCP at risk for developing COVID-19,” other measures will probably be needed to “reduce the risk for infected HCP transmitting the virus to colleagues and patients,” they added.
HCP with COVID-19 were less likely to be hospitalized (8%-10%) than the overall population (21%-31%), which “might reflect the younger median age … of HCP patients, compared with that of reported COVID-19 patients overall, as well as prioritization of HCP for testing, which might identify less-severe illness,” the investigators suggested.
The prevalence of underlying conditions in HCP patients, 38%, was the same as all patients with COVID-19, and 92% of the HCP patients presented with fever, cough, or shortness of breath. Two-thirds of all HCP reported muscle aches, and 65% reported headache, the CDC response team noted.
“It is critical to make every effort to ensure the health and safety of this essential national workforce of approximately 18 million HCP, both at work and in the community,” they wrote.
SOURCE: Stuckey MJ et al. MMWR. Apr 14;69(early release):1-5.
FROM THE MMWR
FDA approves emergency use of saliva test to detect COVID-19
As the race to develop rapid testing for COVID-19 expands, the Food and Drug Administration has granted emergency approval for an approach that uses saliva as the primary test biomaterial.
According to a document provided to the FDA, the Rutgers Clinical Genomics Laboratory TaqPath SARS-CoV-2 Assay is intended for the qualitative detection of nucleic acid from SARS-CoV-2 in oropharyngeal (throat) swab, nasopharyngeal swab, anterior nasal swab, mid-turbinate nasal swab from individuals suspected of COVID-19 by their health care clinicians. To expand on this assay, Rutgers University–based RUCDR Infinite Biologics developed a saliva collection method in partnership with Spectrum Solutions and Accurate Diagnostic Labs.
The document states that Samples are transported for RNA extraction and are tested within 48 hours of collection. In saliva samples obtained from 60 patients evaluated by the researchers, all were in agreement with the presence of COVID-19.

“If shown to be as accurate as nasopharyngeal and oropharyngeal samples, saliva as a biomatrix offers the advantage of not generating aerosols or creating as many respiratory droplets during specimen procurement, therefore decreasing the risk of transmission to the health care worker doing the testing,” said Matthew P. Cheng, MDCM, of the division of infectious diseases at McGill University Health Centre, Montreal, who was not involved in development of the test but who has written about diagnostic testing for the virus.
“Also, it may be easy enough for patients to do saliva self-collection at home. However, it is important to note that SARS-CoV-2 tests on saliva have not yet undergone the more rigorous evaluation of full FDA authorization, and saliva is not a preferred specimen type of the FDA nor the [Centers for Disease Control and Prevention] for respiratory virus testing.”

In a prepared statement, Andrew I. Brooks, PhD, chief operating officer at RUCDR Infinite Biologics, said the saliva collection method enables clinicians to preserve personal protective equipment for use in patient care instead of testing. “We can significantly increase the number of people tested each and every day as self-collection of saliva is quicker and more scalable than swab collections,” he said. “All of this combined will have a tremendous impact on testing in New Jersey and across the United States.”
The tests are currently available to the RWJBarnabas Health network, based in West Orange, N.J., which has partnered with Rutgers University.
As the race to develop rapid testing for COVID-19 expands, the Food and Drug Administration has granted emergency approval for an approach that uses saliva as the primary test biomaterial.
According to a document provided to the FDA, the Rutgers Clinical Genomics Laboratory TaqPath SARS-CoV-2 Assay is intended for the qualitative detection of nucleic acid from SARS-CoV-2 in oropharyngeal (throat) swab, nasopharyngeal swab, anterior nasal swab, mid-turbinate nasal swab from individuals suspected of COVID-19 by their health care clinicians. To expand on this assay, Rutgers University–based RUCDR Infinite Biologics developed a saliva collection method in partnership with Spectrum Solutions and Accurate Diagnostic Labs.
The document states that Samples are transported for RNA extraction and are tested within 48 hours of collection. In saliva samples obtained from 60 patients evaluated by the researchers, all were in agreement with the presence of COVID-19.
“If shown to be as accurate as nasopharyngeal and oropharyngeal samples, saliva as a biomatrix offers the advantage of not generating aerosols or creating as many respiratory droplets during specimen procurement, therefore decreasing the risk of transmission to the health care worker doing the testing,” said Matthew P. Cheng, MDCM, of the division of infectious diseases at McGill University Health Centre, Montreal, who was not involved in development of the test but who has written about diagnostic testing for the virus.
“Also, it may be easy enough for patients to do saliva self-collection at home. However, it is important to note that SARS-CoV-2 tests on saliva have not yet undergone the more rigorous evaluation of full FDA authorization, and saliva is not a preferred specimen type of the FDA nor the [Centers for Disease Control and Prevention] for respiratory virus testing.”
In a prepared statement, Andrew I. Brooks, PhD, chief operating officer at RUCDR Infinite Biologics, said the saliva collection method enables clinicians to preserve personal protective equipment for use in patient care instead of testing. “We can significantly increase the number of people tested each and every day as self-collection of saliva is quicker and more scalable than swab collections,” he said. “All of this combined will have a tremendous impact on testing in New Jersey and across the United States.”
The tests are currently available to the RWJBarnabas Health network, based in West Orange, N.J., which has partnered with Rutgers University.
As the race to develop rapid testing for COVID-19 expands, the Food and Drug Administration has granted emergency approval for an approach that uses saliva as the primary test biomaterial.
According to a document provided to the FDA, the Rutgers Clinical Genomics Laboratory TaqPath SARS-CoV-2 Assay is intended for the qualitative detection of nucleic acid from SARS-CoV-2 in oropharyngeal (throat) swab, nasopharyngeal swab, anterior nasal swab, mid-turbinate nasal swab from individuals suspected of COVID-19 by their health care clinicians. To expand on this assay, Rutgers University–based RUCDR Infinite Biologics developed a saliva collection method in partnership with Spectrum Solutions and Accurate Diagnostic Labs.
The document states that Samples are transported for RNA extraction and are tested within 48 hours of collection. In saliva samples obtained from 60 patients evaluated by the researchers, all were in agreement with the presence of COVID-19.
“If shown to be as accurate as nasopharyngeal and oropharyngeal samples, saliva as a biomatrix offers the advantage of not generating aerosols or creating as many respiratory droplets during specimen procurement, therefore decreasing the risk of transmission to the health care worker doing the testing,” said Matthew P. Cheng, MDCM, of the division of infectious diseases at McGill University Health Centre, Montreal, who was not involved in development of the test but who has written about diagnostic testing for the virus.
“Also, it may be easy enough for patients to do saliva self-collection at home. However, it is important to note that SARS-CoV-2 tests on saliva have not yet undergone the more rigorous evaluation of full FDA authorization, and saliva is not a preferred specimen type of the FDA nor the [Centers for Disease Control and Prevention] for respiratory virus testing.”
In a prepared statement, Andrew I. Brooks, PhD, chief operating officer at RUCDR Infinite Biologics, said the saliva collection method enables clinicians to preserve personal protective equipment for use in patient care instead of testing. “We can significantly increase the number of people tested each and every day as self-collection of saliva is quicker and more scalable than swab collections,” he said. “All of this combined will have a tremendous impact on testing in New Jersey and across the United States.”
The tests are currently available to the RWJBarnabas Health network, based in West Orange, N.J., which has partnered with Rutgers University.
Hypertension goes unmedicated in 40% of adults
Roughly 30% of adults in the United States had hypertension in 2017, and just under 60% of those adults reported using antihypertensive medication, according to the Centers for Disease Control and Prevention.
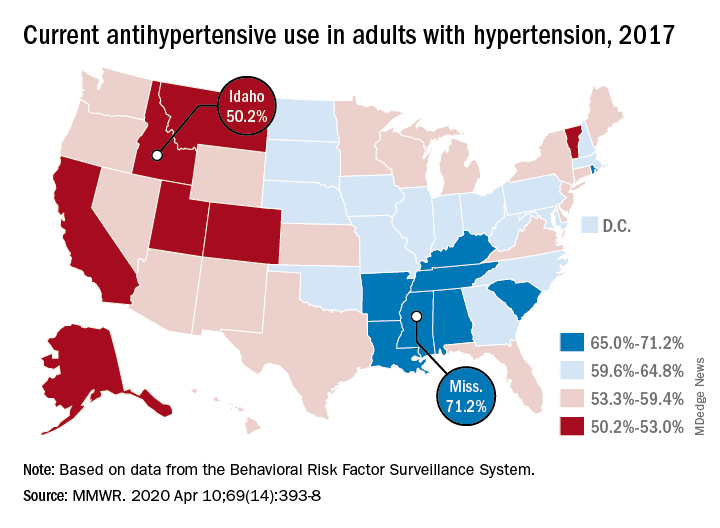
There is, however, quite a bit of variation from those age-standardized national figures when state-level data are considered.
In Alabama and West Virginia, the prevalence of hypertension in 2017 was 38.6%, the highest in the country, with Arkansas (38.5%) and Mississippi (38.2%) not far behind. Meanwhile, Minnesota came in with a lowest-in-the-nation rate of 24.3%, which was nearly matched by Colorado at 24.8%, Claudine M. Samanic, PhD, and associates wrote in the MMWR.
There was also a considerable gap between the states in hypertensive adults’ self-reported use of antihypertensive drugs, which was generally higher in the states with a greater prevalence of disease, they noted.
Adults in Mississippi were the most likely (71.2%) to be taking medication, along with those in Alabama (70.5%) and Arkansas (69.3%). Idaho occupied the other end of the scale with a rate of 50.2%, while Montana and Vermont were slightly better at 51.7%, based on survey data from the Behavioral Risk Factor Surveillance System.
“Prevalence of antihypertensive medication use was higher in older age groups, highest among blacks, and higher among women [64.0%] than men [56.7%]. This overall gender difference has been reported previously, but the reasons are unclear,” wrote Dr. Samanic and associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion.
The BRFSS data for 2017 are based on based on interviews with 450,016 adults. Respondents were asked, “Have you ever been told by a doctor, nurse, or other health professional that you have high blood pressure?” and were considered to have hypertension if they answered yes.
SOURCE: Samanic CM et al. MMWR. 2020 Apr 10;69(14):393-8.
Roughly 30% of adults in the United States had hypertension in 2017, and just under 60% of those adults reported using antihypertensive medication, according to the Centers for Disease Control and Prevention.
There is, however, quite a bit of variation from those age-standardized national figures when state-level data are considered.
In Alabama and West Virginia, the prevalence of hypertension in 2017 was 38.6%, the highest in the country, with Arkansas (38.5%) and Mississippi (38.2%) not far behind. Meanwhile, Minnesota came in with a lowest-in-the-nation rate of 24.3%, which was nearly matched by Colorado at 24.8%, Claudine M. Samanic, PhD, and associates wrote in the MMWR.
There was also a considerable gap between the states in hypertensive adults’ self-reported use of antihypertensive drugs, which was generally higher in the states with a greater prevalence of disease, they noted.
Adults in Mississippi were the most likely (71.2%) to be taking medication, along with those in Alabama (70.5%) and Arkansas (69.3%). Idaho occupied the other end of the scale with a rate of 50.2%, while Montana and Vermont were slightly better at 51.7%, based on survey data from the Behavioral Risk Factor Surveillance System.
“Prevalence of antihypertensive medication use was higher in older age groups, highest among blacks, and higher among women [64.0%] than men [56.7%]. This overall gender difference has been reported previously, but the reasons are unclear,” wrote Dr. Samanic and associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion.
The BRFSS data for 2017 are based on based on interviews with 450,016 adults. Respondents were asked, “Have you ever been told by a doctor, nurse, or other health professional that you have high blood pressure?” and were considered to have hypertension if they answered yes.
SOURCE: Samanic CM et al. MMWR. 2020 Apr 10;69(14):393-8.
Roughly 30% of adults in the United States had hypertension in 2017, and just under 60% of those adults reported using antihypertensive medication, according to the Centers for Disease Control and Prevention.
There is, however, quite a bit of variation from those age-standardized national figures when state-level data are considered.
In Alabama and West Virginia, the prevalence of hypertension in 2017 was 38.6%, the highest in the country, with Arkansas (38.5%) and Mississippi (38.2%) not far behind. Meanwhile, Minnesota came in with a lowest-in-the-nation rate of 24.3%, which was nearly matched by Colorado at 24.8%, Claudine M. Samanic, PhD, and associates wrote in the MMWR.
There was also a considerable gap between the states in hypertensive adults’ self-reported use of antihypertensive drugs, which was generally higher in the states with a greater prevalence of disease, they noted.
Adults in Mississippi were the most likely (71.2%) to be taking medication, along with those in Alabama (70.5%) and Arkansas (69.3%). Idaho occupied the other end of the scale with a rate of 50.2%, while Montana and Vermont were slightly better at 51.7%, based on survey data from the Behavioral Risk Factor Surveillance System.
“Prevalence of antihypertensive medication use was higher in older age groups, highest among blacks, and higher among women [64.0%] than men [56.7%]. This overall gender difference has been reported previously, but the reasons are unclear,” wrote Dr. Samanic and associates at the CDC’s National Center for Chronic Disease Prevention and Health Promotion.
The BRFSS data for 2017 are based on based on interviews with 450,016 adults. Respondents were asked, “Have you ever been told by a doctor, nurse, or other health professional that you have high blood pressure?” and were considered to have hypertension if they answered yes.
SOURCE: Samanic CM et al. MMWR. 2020 Apr 10;69(14):393-8.
FROM THE MMWR