User login
FDA advises alternatives to paclitaxel-coated devices for PAD, pending review
“Alternative treatment options should generally be used for most patients,” rather than paclitaxel-coated balloons and stents for peripheral arterial disease (PAD), pending an ongoing safety review, according to the Food and Drug Administration.

The FDA conducted a preliminary analysis of long-term follow-up data (up to 5 years in some studies) of the pivotal premarket randomized trials for paclitaxel-coated products indicated for peripheral arterial disease (PAD). In a Letter to Healthcare providers issued March 15, the FDA reported that their preliminary review of these data found “a potentially concerning signal of increased long-term mortality in study subjects treated with paclitaxel-coated products, compared to patients treated with uncoated devices.”
The three trials (totaling 975 patients) that had 5-year follow-up data demonstrated an approximately 50% increased risk of mortality in subjects treated with paclitaxel-coated devices vs. those treated with control devices (20.1% vs. 13.4% crude risk of death at 5 years), according to the agency.
The FDA indicated that these data “should be interpreted with caution for several reasons.” They cited a large variability in the risk estimate of mortality because of the limited amount of long-term data and pointed out that the studies were not designed to be pooled. In addition, the specific cause and mechanism of the increased mortality was unknown.
The FDA also announced that they are planning on convening an Advisory Committee meeting of the Circulatory System Devices Panel to address this issue, including plausible mechanisms for this mortality effect, a re-examination of the benefit-risk profile, modifications of current and future clinical trials regarding these devices, and guidance to any regulatory action, as needed. The timing of this meeting is to be announced within the upcoming weeks.
The FDA letter further stated that the agency intends to conduct additional analyses “to determine whether the benefits continue to outweigh the risks for approved paclitaxel-coated balloons and paclitaxel-eluting stents when used in accordance with their indications for use.”
[email protected]
SOURCE: Food and Drug Administration Letter to Healthcare Providers. 2019 Mar 15.
“Alternative treatment options should generally be used for most patients,” rather than paclitaxel-coated balloons and stents for peripheral arterial disease (PAD), pending an ongoing safety review, according to the Food and Drug Administration.
The FDA conducted a preliminary analysis of long-term follow-up data (up to 5 years in some studies) of the pivotal premarket randomized trials for paclitaxel-coated products indicated for peripheral arterial disease (PAD). In a Letter to Healthcare providers issued March 15, the FDA reported that their preliminary review of these data found “a potentially concerning signal of increased long-term mortality in study subjects treated with paclitaxel-coated products, compared to patients treated with uncoated devices.”
The three trials (totaling 975 patients) that had 5-year follow-up data demonstrated an approximately 50% increased risk of mortality in subjects treated with paclitaxel-coated devices vs. those treated with control devices (20.1% vs. 13.4% crude risk of death at 5 years), according to the agency.
The FDA indicated that these data “should be interpreted with caution for several reasons.” They cited a large variability in the risk estimate of mortality because of the limited amount of long-term data and pointed out that the studies were not designed to be pooled. In addition, the specific cause and mechanism of the increased mortality was unknown.
The FDA also announced that they are planning on convening an Advisory Committee meeting of the Circulatory System Devices Panel to address this issue, including plausible mechanisms for this mortality effect, a re-examination of the benefit-risk profile, modifications of current and future clinical trials regarding these devices, and guidance to any regulatory action, as needed. The timing of this meeting is to be announced within the upcoming weeks.
The FDA letter further stated that the agency intends to conduct additional analyses “to determine whether the benefits continue to outweigh the risks for approved paclitaxel-coated balloons and paclitaxel-eluting stents when used in accordance with their indications for use.”
[email protected]
SOURCE: Food and Drug Administration Letter to Healthcare Providers. 2019 Mar 15.
“Alternative treatment options should generally be used for most patients,” rather than paclitaxel-coated balloons and stents for peripheral arterial disease (PAD), pending an ongoing safety review, according to the Food and Drug Administration.
The FDA conducted a preliminary analysis of long-term follow-up data (up to 5 years in some studies) of the pivotal premarket randomized trials for paclitaxel-coated products indicated for peripheral arterial disease (PAD). In a Letter to Healthcare providers issued March 15, the FDA reported that their preliminary review of these data found “a potentially concerning signal of increased long-term mortality in study subjects treated with paclitaxel-coated products, compared to patients treated with uncoated devices.”
The three trials (totaling 975 patients) that had 5-year follow-up data demonstrated an approximately 50% increased risk of mortality in subjects treated with paclitaxel-coated devices vs. those treated with control devices (20.1% vs. 13.4% crude risk of death at 5 years), according to the agency.
The FDA indicated that these data “should be interpreted with caution for several reasons.” They cited a large variability in the risk estimate of mortality because of the limited amount of long-term data and pointed out that the studies were not designed to be pooled. In addition, the specific cause and mechanism of the increased mortality was unknown.
The FDA also announced that they are planning on convening an Advisory Committee meeting of the Circulatory System Devices Panel to address this issue, including plausible mechanisms for this mortality effect, a re-examination of the benefit-risk profile, modifications of current and future clinical trials regarding these devices, and guidance to any regulatory action, as needed. The timing of this meeting is to be announced within the upcoming weeks.
The FDA letter further stated that the agency intends to conduct additional analyses “to determine whether the benefits continue to outweigh the risks for approved paclitaxel-coated balloons and paclitaxel-eluting stents when used in accordance with their indications for use.”
[email protected]
SOURCE: Food and Drug Administration Letter to Healthcare Providers. 2019 Mar 15.
Key clinical point: FDA advises that alternatives to paclitaxel-coated devices for PAD should be used for most patients.
Major finding: Five-year data demonstrated an approximately 50% increased risk of mortality in patients with paclitaxel-coated devices, compared with uncoated ones.
Study details: Preliminary FDA review of three trials with 975 patients.
Disclosures: Study is funded and performed by the FDA.
Source: Food and Drug Administration. Letter to Healthcare Providers. 2019 Mar 15.
Flu activity levels down, but outpatient visits highest since 1998-99
Influenza activity measures declined for a third consecutive week, but levels are higher than usual at this point in the flu season, according to the Centers for Disease Control and Prevention.
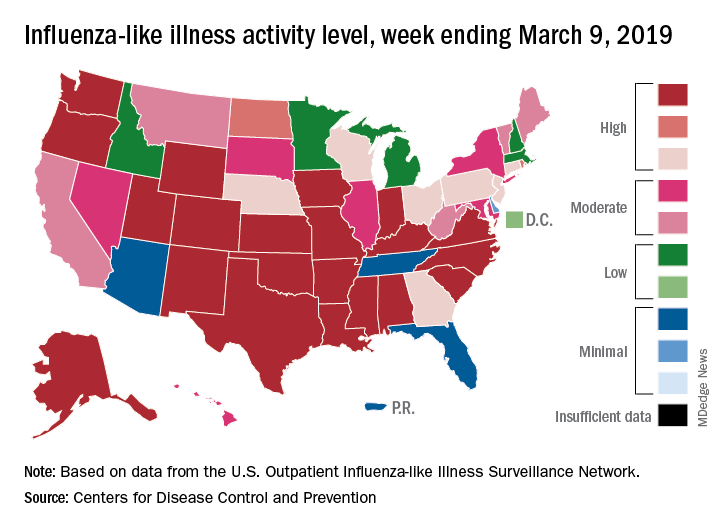
For the week ending March 9, an estimated 4.5% of outpatient visits were for influenza-like illness (ILI), which was down from 4.6% the previous week, the CDC’s influenza division reported March 15, but that is higher than the comparable week for any year since 1998-1999. During last year’s very severe flu season, the outpatient visit rate was just under 3.2% for the week ending March 10.
Although the number of states at level 10 on the CDC’s 1-10 scale remained at 21, the activity map actually looks more red than last week since Rhode Island and West Virgina were replaced by the much larger states of Iowa and Washington. The number of states in the high range (8-10), did go down from 32 to 30, data from the CDC’s Outpatient ILI Surveillance Network show.
Of those four deaths, only one occurred during the most recent reporting week, the CDC said.
Influenza activity measures declined for a third consecutive week, but levels are higher than usual at this point in the flu season, according to the Centers for Disease Control and Prevention.
For the week ending March 9, an estimated 4.5% of outpatient visits were for influenza-like illness (ILI), which was down from 4.6% the previous week, the CDC’s influenza division reported March 15, but that is higher than the comparable week for any year since 1998-1999. During last year’s very severe flu season, the outpatient visit rate was just under 3.2% for the week ending March 10.
Although the number of states at level 10 on the CDC’s 1-10 scale remained at 21, the activity map actually looks more red than last week since Rhode Island and West Virgina were replaced by the much larger states of Iowa and Washington. The number of states in the high range (8-10), did go down from 32 to 30, data from the CDC’s Outpatient ILI Surveillance Network show.
Of those four deaths, only one occurred during the most recent reporting week, the CDC said.
Influenza activity measures declined for a third consecutive week, but levels are higher than usual at this point in the flu season, according to the Centers for Disease Control and Prevention.
For the week ending March 9, an estimated 4.5% of outpatient visits were for influenza-like illness (ILI), which was down from 4.6% the previous week, the CDC’s influenza division reported March 15, but that is higher than the comparable week for any year since 1998-1999. During last year’s very severe flu season, the outpatient visit rate was just under 3.2% for the week ending March 10.
Although the number of states at level 10 on the CDC’s 1-10 scale remained at 21, the activity map actually looks more red than last week since Rhode Island and West Virgina were replaced by the much larger states of Iowa and Washington. The number of states in the high range (8-10), did go down from 32 to 30, data from the CDC’s Outpatient ILI Surveillance Network show.
Of those four deaths, only one occurred during the most recent reporting week, the CDC said.
Measles now confirmed in 12 states
The number of new measles cases was down by more than half last week, but another state has been added to the list of those with reported cases in 2019, according to the Centers for Disease Control and Prevention.
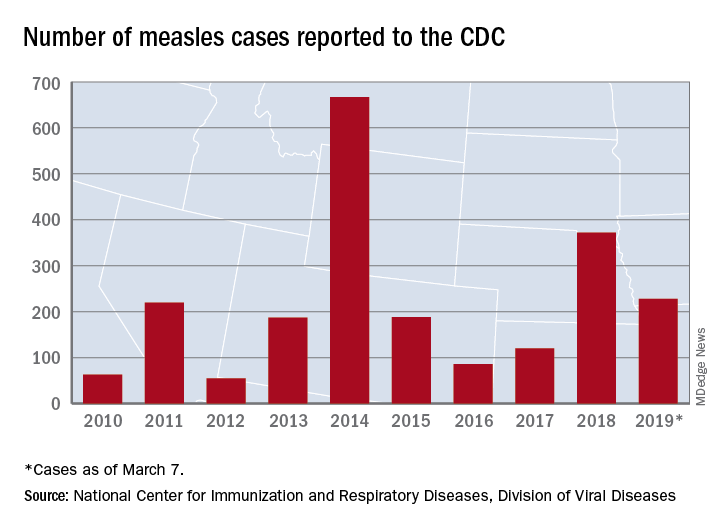
The total for the year is 228 cases, which moves 2019 ahead of 2011 for third place over the last decade, the CDC reported March 11. Going back even further in time, the 206 measles cases reported through January and February is the highest 2-month total in a quarter of a century, the Washington Post said.
New Hampshire became the 12th and latest state to report a case of measles this year, joining California, Colorado, Connecticut, Georgia, Illinois, Kentucky, New Jersey, New York, Oregon, Texas, and Washington. California’s situation is now considered an outbreak (defined as three or more cases), but one of the three outbreaks in New York has been taken off the list, so total outbreaks for 2019 remain at six, the CDC said.
For the third consecutive week, New York City produced the most measles cases, with Brooklyn’s Williamsburg neighborhood recording 11 of the U.S. total of 21. The outbreak in King County, Wash., – totaling 70 cases this year – may be winding down as only one new case was reported last week, and no new cases are being investigated, the county’s public health service reported.
The number of new measles cases was down by more than half last week, but another state has been added to the list of those with reported cases in 2019, according to the Centers for Disease Control and Prevention.
The total for the year is 228 cases, which moves 2019 ahead of 2011 for third place over the last decade, the CDC reported March 11. Going back even further in time, the 206 measles cases reported through January and February is the highest 2-month total in a quarter of a century, the Washington Post said.
New Hampshire became the 12th and latest state to report a case of measles this year, joining California, Colorado, Connecticut, Georgia, Illinois, Kentucky, New Jersey, New York, Oregon, Texas, and Washington. California’s situation is now considered an outbreak (defined as three or more cases), but one of the three outbreaks in New York has been taken off the list, so total outbreaks for 2019 remain at six, the CDC said.
For the third consecutive week, New York City produced the most measles cases, with Brooklyn’s Williamsburg neighborhood recording 11 of the U.S. total of 21. The outbreak in King County, Wash., – totaling 70 cases this year – may be winding down as only one new case was reported last week, and no new cases are being investigated, the county’s public health service reported.
The number of new measles cases was down by more than half last week, but another state has been added to the list of those with reported cases in 2019, according to the Centers for Disease Control and Prevention.
The total for the year is 228 cases, which moves 2019 ahead of 2011 for third place over the last decade, the CDC reported March 11. Going back even further in time, the 206 measles cases reported through January and February is the highest 2-month total in a quarter of a century, the Washington Post said.
New Hampshire became the 12th and latest state to report a case of measles this year, joining California, Colorado, Connecticut, Georgia, Illinois, Kentucky, New Jersey, New York, Oregon, Texas, and Washington. California’s situation is now considered an outbreak (defined as three or more cases), but one of the three outbreaks in New York has been taken off the list, so total outbreaks for 2019 remain at six, the CDC said.
For the third consecutive week, New York City produced the most measles cases, with Brooklyn’s Williamsburg neighborhood recording 11 of the U.S. total of 21. The outbreak in King County, Wash., – totaling 70 cases this year – may be winding down as only one new case was reported last week, and no new cases are being investigated, the county’s public health service reported.
FDA approves Tecentriq plus Abraxane in breast cancer
The Food and Drug Administration has granted accelerated approval for the combination of atezolizumab (Tecentriq) plus nanoparticle albumin–bound paclitaxel (nab-paclitaxel; Abraxane) for the treatment of adults with unresectable locally advanced or metastatic programmed death-ligand 1 (PD-L1)–positive triple-negative breast cancer (TNBC).
This conditional approval is granted to medicines that fill an unmet medical need for serious or life-threatening diseases or conditions, but the FDA may require confirmatory trials to provide verification and description of clinical benefit to allow continued approval.
The approval is based on the phase 3 IMpassion130 trial (NCT02425891), which enrolled 902 patients with unresectable, locally advanced or metastatic TNBC who had not received prior lines of chemo for metastatic disease, according to Genentech.
The multicenter, randomized, double-blind study has been evaluating the drug combination’s efficacy, safety, and pharmacokinetics. Compared with placebo plus nab-paclitaxel, atezolizumab/nab-paclitaxel demonstrated significantly superior progression-free survival (median PFS, 7.4 months vs. 4.8 months; hazard ratio, 0.60; 95% confidence interval, 0.48-0.77; P less than .0001).
The overall survival data for the intention-to-treat population remains immature, but further data will be shared with the FDA in the future, according to Genentech.
No new safety signals were seen in the atezolizumab/nab-paclitaxel arm, and the combination’s safety appeared consistent with the known safety profiles of each medicine individually.
The most common grade 3-4 events (occurring in more than 2% of patients) in the combination arm included low red blood cells, low white blood cells, feeling tired, low blood potassium level, and pneumonia.
The most common side effects (occurring in more than 20% of patients) in the combination arm included hair loss, tingling, nausea, diarrhea, headache, low red blood cells, low white blood cells, and decreased appetite.
Atezolizumab is a monoclonal antibody that binds to the PD-L1 receptor, which could possibly lead to the reactivation of T cells; however, atezolizumab also may interact with other cells in the body. Nab-paclitaxel is an injectable suspension of the common chemotherapy drug.
The Food and Drug Administration has granted accelerated approval for the combination of atezolizumab (Tecentriq) plus nanoparticle albumin–bound paclitaxel (nab-paclitaxel; Abraxane) for the treatment of adults with unresectable locally advanced or metastatic programmed death-ligand 1 (PD-L1)–positive triple-negative breast cancer (TNBC).
This conditional approval is granted to medicines that fill an unmet medical need for serious or life-threatening diseases or conditions, but the FDA may require confirmatory trials to provide verification and description of clinical benefit to allow continued approval.
The approval is based on the phase 3 IMpassion130 trial (NCT02425891), which enrolled 902 patients with unresectable, locally advanced or metastatic TNBC who had not received prior lines of chemo for metastatic disease, according to Genentech.
The multicenter, randomized, double-blind study has been evaluating the drug combination’s efficacy, safety, and pharmacokinetics. Compared with placebo plus nab-paclitaxel, atezolizumab/nab-paclitaxel demonstrated significantly superior progression-free survival (median PFS, 7.4 months vs. 4.8 months; hazard ratio, 0.60; 95% confidence interval, 0.48-0.77; P less than .0001).
The overall survival data for the intention-to-treat population remains immature, but further data will be shared with the FDA in the future, according to Genentech.
No new safety signals were seen in the atezolizumab/nab-paclitaxel arm, and the combination’s safety appeared consistent with the known safety profiles of each medicine individually.
The most common grade 3-4 events (occurring in more than 2% of patients) in the combination arm included low red blood cells, low white blood cells, feeling tired, low blood potassium level, and pneumonia.
The most common side effects (occurring in more than 20% of patients) in the combination arm included hair loss, tingling, nausea, diarrhea, headache, low red blood cells, low white blood cells, and decreased appetite.
Atezolizumab is a monoclonal antibody that binds to the PD-L1 receptor, which could possibly lead to the reactivation of T cells; however, atezolizumab also may interact with other cells in the body. Nab-paclitaxel is an injectable suspension of the common chemotherapy drug.
The Food and Drug Administration has granted accelerated approval for the combination of atezolizumab (Tecentriq) plus nanoparticle albumin–bound paclitaxel (nab-paclitaxel; Abraxane) for the treatment of adults with unresectable locally advanced or metastatic programmed death-ligand 1 (PD-L1)–positive triple-negative breast cancer (TNBC).
This conditional approval is granted to medicines that fill an unmet medical need for serious or life-threatening diseases or conditions, but the FDA may require confirmatory trials to provide verification and description of clinical benefit to allow continued approval.
The approval is based on the phase 3 IMpassion130 trial (NCT02425891), which enrolled 902 patients with unresectable, locally advanced or metastatic TNBC who had not received prior lines of chemo for metastatic disease, according to Genentech.
The multicenter, randomized, double-blind study has been evaluating the drug combination’s efficacy, safety, and pharmacokinetics. Compared with placebo plus nab-paclitaxel, atezolizumab/nab-paclitaxel demonstrated significantly superior progression-free survival (median PFS, 7.4 months vs. 4.8 months; hazard ratio, 0.60; 95% confidence interval, 0.48-0.77; P less than .0001).
The overall survival data for the intention-to-treat population remains immature, but further data will be shared with the FDA in the future, according to Genentech.
No new safety signals were seen in the atezolizumab/nab-paclitaxel arm, and the combination’s safety appeared consistent with the known safety profiles of each medicine individually.
The most common grade 3-4 events (occurring in more than 2% of patients) in the combination arm included low red blood cells, low white blood cells, feeling tired, low blood potassium level, and pneumonia.
The most common side effects (occurring in more than 20% of patients) in the combination arm included hair loss, tingling, nausea, diarrhea, headache, low red blood cells, low white blood cells, and decreased appetite.
Atezolizumab is a monoclonal antibody that binds to the PD-L1 receptor, which could possibly lead to the reactivation of T cells; however, atezolizumab also may interact with other cells in the body. Nab-paclitaxel is an injectable suspension of the common chemotherapy drug.
Flu activity down for a second straight week
A second straight week of reduced influenza activity suggests that the 2018-2019 flu season is on the decline, according to the most recent data from the Centers for Disease Control and Prevention.
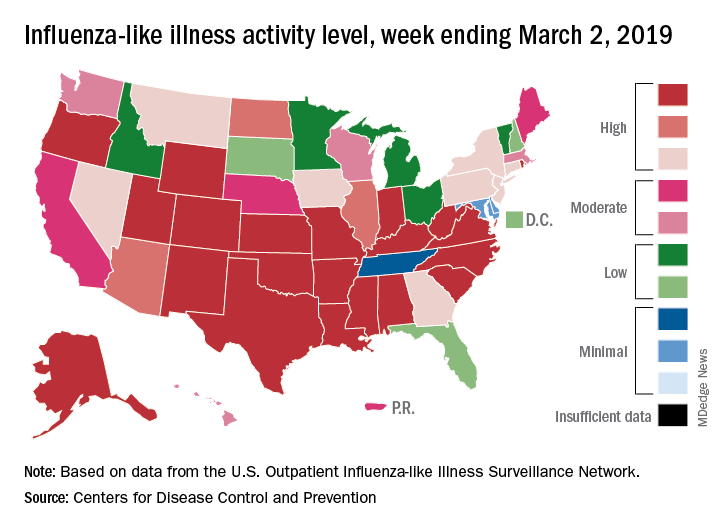
The proportion of outpatient visits for influenza-like illness (ILI) was 4.7% during the week ending March 2, which means that, thanks to a revision of the number for the previous week (Feb. 23) from 5.0% down to 4.9%, there have been two straight weeks of declines since outpatient visits reached a season-high 5.0% for the week ending Feb. 16, the CDC’s influenza division said March 8. The national baseline level is 2.2%.
This marks the second 2-week drop in ILI visits for the 2018-2019 season, as there was similar dip in the beginning of January before activity started rising again.
This compares with 24 the week before; 32 states were in the high range of 8-10, compared with the 33 reported last week, based on data from the Outpatient ILI Surveillance Network.
There were nine flu-related pediatric deaths reported during the week, with three occurring in the week ending March 2. To underscore the preliminary nature of these data, one of the deaths reported this week occurred in 2016. A total of 64 deaths in children have been associated with influenza so far for the 2018-2019 season, and the total for the 2015-2016 season is now 95, the CDC said.
A second straight week of reduced influenza activity suggests that the 2018-2019 flu season is on the decline, according to the most recent data from the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 4.7% during the week ending March 2, which means that, thanks to a revision of the number for the previous week (Feb. 23) from 5.0% down to 4.9%, there have been two straight weeks of declines since outpatient visits reached a season-high 5.0% for the week ending Feb. 16, the CDC’s influenza division said March 8. The national baseline level is 2.2%.
This marks the second 2-week drop in ILI visits for the 2018-2019 season, as there was similar dip in the beginning of January before activity started rising again.
This compares with 24 the week before; 32 states were in the high range of 8-10, compared with the 33 reported last week, based on data from the Outpatient ILI Surveillance Network.
There were nine flu-related pediatric deaths reported during the week, with three occurring in the week ending March 2. To underscore the preliminary nature of these data, one of the deaths reported this week occurred in 2016. A total of 64 deaths in children have been associated with influenza so far for the 2018-2019 season, and the total for the 2015-2016 season is now 95, the CDC said.
A second straight week of reduced influenza activity suggests that the 2018-2019 flu season is on the decline, according to the most recent data from the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 4.7% during the week ending March 2, which means that, thanks to a revision of the number for the previous week (Feb. 23) from 5.0% down to 4.9%, there have been two straight weeks of declines since outpatient visits reached a season-high 5.0% for the week ending Feb. 16, the CDC’s influenza division said March 8. The national baseline level is 2.2%.
This marks the second 2-week drop in ILI visits for the 2018-2019 season, as there was similar dip in the beginning of January before activity started rising again.
This compares with 24 the week before; 32 states were in the high range of 8-10, compared with the 33 reported last week, based on data from the Outpatient ILI Surveillance Network.
There were nine flu-related pediatric deaths reported during the week, with three occurring in the week ending March 2. To underscore the preliminary nature of these data, one of the deaths reported this week occurred in 2016. A total of 64 deaths in children have been associated with influenza so far for the 2018-2019 season, and the total for the 2015-2016 season is now 95, the CDC said.
FDA approves liquid colchicine for gout
for prophylaxis of gout flares in adults, according to a statement from Romeg Therapeutics.
Colchicine has been used in capsule and tablet forms to treat this form of arthritis for decades. An advantage to the new formulation is that it allows physicians to “easily make dose adjustments,” according to the statement.
“Existing therapies do not adequately address the physician’s need to adjust dosages of colchicine to manage the toxicity profile for patients with renal and liver impairments, side effects, common drug-to-drug interactions, and age-related health disorders,” said Naomi Vishnupad, PhD, chief scientific officer of Romeg Therapeutics, in the statement.
According to the prescribing information for the drug on the FDA website, this formulation is indicated for prophylaxis rather than acute treatment of gout flares because the safety profile of acute treatment with it has not yet been studied. It is contraindicated in patients with hepatic and/or renal impairment. Gastrointestinal symptoms were the most commonly reported adverse reactions.
The drug is expected to be available this summer.
for prophylaxis of gout flares in adults, according to a statement from Romeg Therapeutics.
Colchicine has been used in capsule and tablet forms to treat this form of arthritis for decades. An advantage to the new formulation is that it allows physicians to “easily make dose adjustments,” according to the statement.
“Existing therapies do not adequately address the physician’s need to adjust dosages of colchicine to manage the toxicity profile for patients with renal and liver impairments, side effects, common drug-to-drug interactions, and age-related health disorders,” said Naomi Vishnupad, PhD, chief scientific officer of Romeg Therapeutics, in the statement.
According to the prescribing information for the drug on the FDA website, this formulation is indicated for prophylaxis rather than acute treatment of gout flares because the safety profile of acute treatment with it has not yet been studied. It is contraindicated in patients with hepatic and/or renal impairment. Gastrointestinal symptoms were the most commonly reported adverse reactions.
The drug is expected to be available this summer.
for prophylaxis of gout flares in adults, according to a statement from Romeg Therapeutics.
Colchicine has been used in capsule and tablet forms to treat this form of arthritis for decades. An advantage to the new formulation is that it allows physicians to “easily make dose adjustments,” according to the statement.
“Existing therapies do not adequately address the physician’s need to adjust dosages of colchicine to manage the toxicity profile for patients with renal and liver impairments, side effects, common drug-to-drug interactions, and age-related health disorders,” said Naomi Vishnupad, PhD, chief scientific officer of Romeg Therapeutics, in the statement.
According to the prescribing information for the drug on the FDA website, this formulation is indicated for prophylaxis rather than acute treatment of gout flares because the safety profile of acute treatment with it has not yet been studied. It is contraindicated in patients with hepatic and/or renal impairment. Gastrointestinal symptoms were the most commonly reported adverse reactions.
The drug is expected to be available this summer.
Measles cases jumped 30% last week
according to the Centers for Disease Control and Prevention.

Those new cases represent a 30% increase in measles cases for the year, bringing the total to 206 reported to the CDC through Feb. 28. After just 2 months, 2019 has had more cases than all but 3 other years over the last decade, CDC data show.
The 11th state to report a case of measles this year is New Jersey, which joins California, Colorado, Connecticut, Georgia, Illinois (one outbreak), Kentucky, New York (three outbreaks), Oregon, Texas (one outbreak), and Washington (one outbreak), the CDC said.
The outbreak in Washington (4 new cases/70 for the year) had been the largest, but the majority of the new cases over the last 2 weeks have occurred in New York City, specifically Brooklyn, which reported 30 cases last week and 17 of the 32 new U.S. cases the week before.
Most of the 120 cases reported in the borough since the beginning of its outbreak in October of 2018 “have involved members of the Jewish Orthodox community. The initial child with measles was unvaccinated and acquired measles on a visit to Israel, where a large outbreak of the disease is occurring. Since then, there have been additional people from Brooklyn and Queens who were unvaccinated and acquired measles while in Israel. People who did not travel were also infected in Brooklyn or Rockland County,” the CDC said.
according to the Centers for Disease Control and Prevention.
Those new cases represent a 30% increase in measles cases for the year, bringing the total to 206 reported to the CDC through Feb. 28. After just 2 months, 2019 has had more cases than all but 3 other years over the last decade, CDC data show.
The 11th state to report a case of measles this year is New Jersey, which joins California, Colorado, Connecticut, Georgia, Illinois (one outbreak), Kentucky, New York (three outbreaks), Oregon, Texas (one outbreak), and Washington (one outbreak), the CDC said.
The outbreak in Washington (4 new cases/70 for the year) had been the largest, but the majority of the new cases over the last 2 weeks have occurred in New York City, specifically Brooklyn, which reported 30 cases last week and 17 of the 32 new U.S. cases the week before.
Most of the 120 cases reported in the borough since the beginning of its outbreak in October of 2018 “have involved members of the Jewish Orthodox community. The initial child with measles was unvaccinated and acquired measles on a visit to Israel, where a large outbreak of the disease is occurring. Since then, there have been additional people from Brooklyn and Queens who were unvaccinated and acquired measles while in Israel. People who did not travel were also infected in Brooklyn or Rockland County,” the CDC said.
according to the Centers for Disease Control and Prevention.
Those new cases represent a 30% increase in measles cases for the year, bringing the total to 206 reported to the CDC through Feb. 28. After just 2 months, 2019 has had more cases than all but 3 other years over the last decade, CDC data show.
The 11th state to report a case of measles this year is New Jersey, which joins California, Colorado, Connecticut, Georgia, Illinois (one outbreak), Kentucky, New York (three outbreaks), Oregon, Texas (one outbreak), and Washington (one outbreak), the CDC said.
The outbreak in Washington (4 new cases/70 for the year) had been the largest, but the majority of the new cases over the last 2 weeks have occurred in New York City, specifically Brooklyn, which reported 30 cases last week and 17 of the 32 new U.S. cases the week before.
Most of the 120 cases reported in the borough since the beginning of its outbreak in October of 2018 “have involved members of the Jewish Orthodox community. The initial child with measles was unvaccinated and acquired measles on a visit to Israel, where a large outbreak of the disease is occurring. Since then, there have been additional people from Brooklyn and Queens who were unvaccinated and acquired measles while in Israel. People who did not travel were also infected in Brooklyn or Rockland County,” the CDC said.
Priority review granted to lenalidomide for FL, MZL
The Food and Drug Administration has granted priority review to a supplemental new drug application (sNDA) for lenalidomide (Revlimid).
Celgene is seeking approval for lenalidomide in combination with rituximab to treat patients with previously treated follicular lymphoma (FL) or marginal zone lymphoma (MZL).
The FDA plans to make a decision on the sNDA by June 27, 2019.
The FDA aims to take action on a priority review application within 6 months of receiving it rather than the standard 10 months. The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The sNDA for lenalidomide is supported by the phase 3 AUGMENT study (NCT01938001) in which researchers compared rituximab plus lenalidomide to rituximab plus placebo in patients with relapsed/refractory FL or MZL.
Results from AUGMENT were presented at the 2018 annual meeting of the American Society of Hematology (Blood 2018 Nov 29;132:445).
According to the ASH abstract, the trial included 358 patients who were randomized to receive rituximab plus lenalidomide (n = 178) or rituximab plus placebo (n = 180).
At a median follow-up of 28.3 months, the overall response rate was 78% in the lenalidomide arm and 53% in the placebo arm (P less than .0001). The complete response rate was 34% and 18%, respectively (P = .001).
The median progression-free survival was 39.4 months in the lenalidomide arm and 14.1 months in the placebo arm. Overall survival data were not mature, but there were 16 deaths reported in the lenalidomide arm and 26 deaths in the placebo arm.
Treatment-emergent adverse events that were more common in the lenalidomide arm than the placebo arm included infections, cutaneous reactions, constipation, thrombocytopenia, and tumor flare reaction.
The Food and Drug Administration has granted priority review to a supplemental new drug application (sNDA) for lenalidomide (Revlimid).
Celgene is seeking approval for lenalidomide in combination with rituximab to treat patients with previously treated follicular lymphoma (FL) or marginal zone lymphoma (MZL).
The FDA plans to make a decision on the sNDA by June 27, 2019.
The FDA aims to take action on a priority review application within 6 months of receiving it rather than the standard 10 months. The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The sNDA for lenalidomide is supported by the phase 3 AUGMENT study (NCT01938001) in which researchers compared rituximab plus lenalidomide to rituximab plus placebo in patients with relapsed/refractory FL or MZL.
Results from AUGMENT were presented at the 2018 annual meeting of the American Society of Hematology (Blood 2018 Nov 29;132:445).
According to the ASH abstract, the trial included 358 patients who were randomized to receive rituximab plus lenalidomide (n = 178) or rituximab plus placebo (n = 180).
At a median follow-up of 28.3 months, the overall response rate was 78% in the lenalidomide arm and 53% in the placebo arm (P less than .0001). The complete response rate was 34% and 18%, respectively (P = .001).
The median progression-free survival was 39.4 months in the lenalidomide arm and 14.1 months in the placebo arm. Overall survival data were not mature, but there were 16 deaths reported in the lenalidomide arm and 26 deaths in the placebo arm.
Treatment-emergent adverse events that were more common in the lenalidomide arm than the placebo arm included infections, cutaneous reactions, constipation, thrombocytopenia, and tumor flare reaction.
The Food and Drug Administration has granted priority review to a supplemental new drug application (sNDA) for lenalidomide (Revlimid).
Celgene is seeking approval for lenalidomide in combination with rituximab to treat patients with previously treated follicular lymphoma (FL) or marginal zone lymphoma (MZL).
The FDA plans to make a decision on the sNDA by June 27, 2019.
The FDA aims to take action on a priority review application within 6 months of receiving it rather than the standard 10 months. The FDA grants priority review to applications for products that are expected to provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The sNDA for lenalidomide is supported by the phase 3 AUGMENT study (NCT01938001) in which researchers compared rituximab plus lenalidomide to rituximab plus placebo in patients with relapsed/refractory FL or MZL.
Results from AUGMENT were presented at the 2018 annual meeting of the American Society of Hematology (Blood 2018 Nov 29;132:445).
According to the ASH abstract, the trial included 358 patients who were randomized to receive rituximab plus lenalidomide (n = 178) or rituximab plus placebo (n = 180).
At a median follow-up of 28.3 months, the overall response rate was 78% in the lenalidomide arm and 53% in the placebo arm (P less than .0001). The complete response rate was 34% and 18%, respectively (P = .001).
The median progression-free survival was 39.4 months in the lenalidomide arm and 14.1 months in the placebo arm. Overall survival data were not mature, but there were 16 deaths reported in the lenalidomide arm and 26 deaths in the placebo arm.
Treatment-emergent adverse events that were more common in the lenalidomide arm than the placebo arm included infections, cutaneous reactions, constipation, thrombocytopenia, and tumor flare reaction.
Flu season shows signs of peaking
The 2018-2019 flu season may have peaked as the major nationwide measure of influenza activity held steady for the week ending Feb. 23, according to the Centers for Disease Control and Prevention. The proportion of outpatient visits for influenza-like illness (ILI) was 5.0% for the most recent reporting week, the CDC’s influenza division said in its March 1 report. The previous week’s outpatient visit rate, originally reported as 5.1%, was revised this week to 5.0% as well, suggesting that flu activity is no longer increasing.
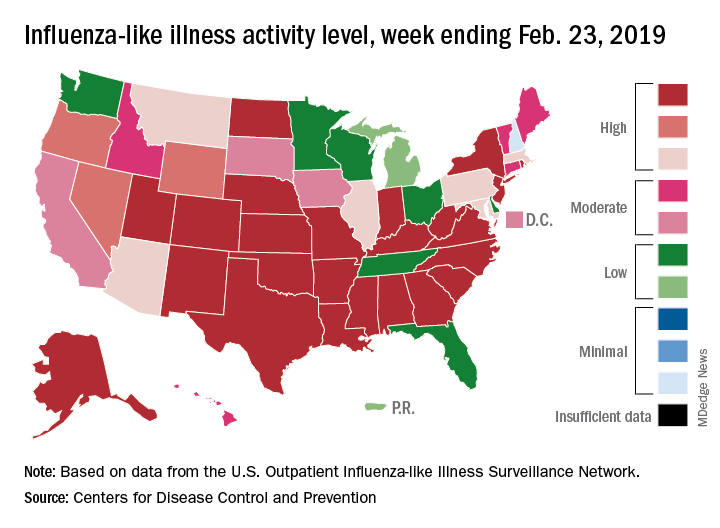
Activity at the state level was more mixed. The number of states at level 10 on the CDC’s 1-10 scale of ILI activity stayed at 24 as Indiana and North Dakota replaced Tennessee and Wyoming, but the number of states in the high range (8-10) of the activity scale increased from 30 to 33, CDC data show.
The signs of plateauing ILI activity did not, however, extend to flu-related deaths, with 15 reported among children – the highest weekly number for the 2018-2019 season, although 11 actually occurred in previous weeks – during the week ending Feb. 23 and 289 deaths among all ages for the week ending Feb. 16, which is already more than the 268 listed the week before despite less complete reporting (82% vs. 97%), the CDC reported. Total flu-related deaths in children are now up to 56, compared with 138 at the corresponding point in the 2017-2018 season.
The 2018-2019 flu season may have peaked as the major nationwide measure of influenza activity held steady for the week ending Feb. 23, according to the Centers for Disease Control and Prevention. The proportion of outpatient visits for influenza-like illness (ILI) was 5.0% for the most recent reporting week, the CDC’s influenza division said in its March 1 report. The previous week’s outpatient visit rate, originally reported as 5.1%, was revised this week to 5.0% as well, suggesting that flu activity is no longer increasing.
Activity at the state level was more mixed. The number of states at level 10 on the CDC’s 1-10 scale of ILI activity stayed at 24 as Indiana and North Dakota replaced Tennessee and Wyoming, but the number of states in the high range (8-10) of the activity scale increased from 30 to 33, CDC data show.
The signs of plateauing ILI activity did not, however, extend to flu-related deaths, with 15 reported among children – the highest weekly number for the 2018-2019 season, although 11 actually occurred in previous weeks – during the week ending Feb. 23 and 289 deaths among all ages for the week ending Feb. 16, which is already more than the 268 listed the week before despite less complete reporting (82% vs. 97%), the CDC reported. Total flu-related deaths in children are now up to 56, compared with 138 at the corresponding point in the 2017-2018 season.
The 2018-2019 flu season may have peaked as the major nationwide measure of influenza activity held steady for the week ending Feb. 23, according to the Centers for Disease Control and Prevention. The proportion of outpatient visits for influenza-like illness (ILI) was 5.0% for the most recent reporting week, the CDC’s influenza division said in its March 1 report. The previous week’s outpatient visit rate, originally reported as 5.1%, was revised this week to 5.0% as well, suggesting that flu activity is no longer increasing.
Activity at the state level was more mixed. The number of states at level 10 on the CDC’s 1-10 scale of ILI activity stayed at 24 as Indiana and North Dakota replaced Tennessee and Wyoming, but the number of states in the high range (8-10) of the activity scale increased from 30 to 33, CDC data show.
The signs of plateauing ILI activity did not, however, extend to flu-related deaths, with 15 reported among children – the highest weekly number for the 2018-2019 season, although 11 actually occurred in previous weeks – during the week ending Feb. 23 and 289 deaths among all ages for the week ending Feb. 16, which is already more than the 268 listed the week before despite less complete reporting (82% vs. 97%), the CDC reported. Total flu-related deaths in children are now up to 56, compared with 138 at the corresponding point in the 2017-2018 season.
Selinexor hits FDA stumbling block
Karyopharm Therapeutics must finish a randomized phase 3 trial of selinexor plus dexamethasone before the Food and Drug Administration will proceed with a safety and tolerability assessment for the first-in-class multiple myeloma drug.
By an 8-5 vote, the FDA Oncologic Drugs Advisory Committee said that data from STORM 2, Karyopharm’s single-arm phase 2b trial, didn’t sufficiently show that selinexor exerted any significant benefit over dexamethasone alone, used because the company claims it potentiates selinexor’s action.
Committee members also expressed concerns about the drug’s challenging adverse event profile. In STORM Part 2, 60% of patients experienced serious treatment-emergent adverse events and 10 died from them.
“This trial design is not adequate to assess tolerability and efficacy,” and move the drug along, said Christian S. Hinrichs, MD, of the National Cancer Institute. For that to happen, “we’d be looking for several things. We’d be looking for a subset of patients who benefited profoundly, which could be somewhat compelling despite a lower overall response rate. Next we might be looking for durable response, and here we see 4-month responses. And finally, what we look for in a single-arm trial is a really favorable side effect profile, like we see in checkpoint inhibitors. That is clearly not the case with this drug. So, on the basis of both the trial design and the results, I find it hard to conclude that these data allow for an adequate assessment that safety and efficacy are proven.”
The decision came despite the pleas of 15 patients and one patient advocate who said the drug improved clinical status and quality of life, and even extended life beyond what anyone expected. However, several committee members noted that Karyopharm paid for speakers’ travel and that patients who had negative experiences would probably be too sick to attend.
Selinexor is a completely new therapeutic option for relapsed multiple myeloma patients. It is a twice-weekly, oral tablet that inhibits nuclear export protein Exportin 1 (XPO1), which regulates the localization of tumor suppressor proteins and is associated with poor prognosis. Aberrant XPO1 expression causes tumor suppressors to locate away from their targets, allowing tumors to grow. Inhibiting it with selinexor blocks signal transduction pathways, interrupting tumor cell proliferation and inducing apoptosis while sparing normal cells.
Karyopharm is seeking approval of selinexor in combination with low-dose dexamethasone for the treatment of patients with relapsed/refractory multiple myeloma who have received at least three prior therapies and whose disease is refractory to at least one proteasome inhibitor, at least one immunomodulatory imide drug, and an anti-CD38 monoclonal antibody.
This disease is referred to as “triple-class refractory” multiple myeloma. At this stage, patients have exhausted every effective treatment option and are faced with the choice of supportive care or recycling previously successful drugs. Their median overall survival time is 3-5 months.
Karyopharm submitted its the New Drug Application using the Accelerated Approval pathway, arguing that the drug meets an unmet medical need and can be approved on surrogate endpoints – in this case, overall response rate.
The modified intent-to-treat analysis comprised 122 patients. The overall response rate was 25.4% with a median response duration of 4.4 months. Two patients had a complete response; six had a very good partial response; and 23 had a partial response.
Some committee members, however, said it would be impossible to tease out how much of the response could be due to the co-administration of 20 mg dexamethasone with each dose. In a phase 1 dose-ranging study of selinexor as monotherapy, it produced only one partial response in 56 patients. And, FDA pointed out, historical studies have shown response rates of 10%-27% for high-dose dexamethasone.
However, those in favor of the drug pointed out that the STORM patients were steroid-refractory, and that a 25% response rate would be unlikely on low-dose dexamethasone alone. This is proof of the company’s claim that the steroid works synergistically with selinexor, they said.
These members also pointed out that even a few years ago, there simply were no patients like the STORM cohort. Only recently have these patients lived long enough to develop resistance against all therapeutic lines, so it’s unrealistic to use historical data to judge what a reasonable response rate looks like in this situation.
Committee members also choked on STORM’s adverse event (AE) profile. All patients experienced at least one treatment-emergent AE, and 60% had at least one serious AE. Most (88.6%) required a dose modification due to an AE, and 28.5% discontinued due to one. The most common AEs were thrombocytopenia, anemia, nausea, fatigue, and decreased appetite. The company said these were “typically reversible and manageable with dose reductions.”
Additionally, there were 23 deaths in the trial. About half (13) were due to disease progression, but the remainder were due to a fatal treatment-emergent AE. Two of these (one pneumonia and one sepsis) were directly due to selinexor, the company said.
Despite the committee’s concerns, 16 of the 17 speakers described positive experiences with selinexor. They universally acknowledged that “it’s a hard drug to take,” and that side effects need to be managed proactively. But they also said, universally, that the drug has brought them additional months of good-quality life, decreased lengthy hospital stays, enabled them to participate in important family events, and even travel. Some also expressed the hope that selinexor would be a bridge drug, decreasing their disease burden enough that they could qualify for other clinical trials of new investigational drugs.
Only Stephanie Fox-Rawlings, PhD, of the National Center for Health Research, urged a delay. “Even if these adverse events are manageable, they harm patients’ quality of life,” she said. “This may be acceptable to some, but if the drug can’t provide a meaningful benefit then they are not worth it and in this clinical trial there was no improvement noted in quality of life. This drug has serious risks and we don’t know if it works.”
Dr. Fox said she was “very glad” that Karyopharm has completed recruitment for its phase 3 randomized study, dubbed BOSTON. BOSTON will assign active patients to once-weekly 100 mg selinexor plus weight-dosed bortezomib, plus twice-weekly 20 mg dexamethasone. The comparator group will receive weight-based bortezomib twice a week and 20 mg dexamethasone four times a week. Patients who progress can cross over to the active arm. The company hopes for even better results, saying that the proteasome inhibitor has also shown a synergistic effect with selinexor. Results are expected in 2020.
“The BOSTON study doesn’t solve anything,” retorted committee member David Harrington, PhD, emeritus professor of biostatistics at the Dana-Farber Cancer Institute. “It’s a different clinical profile, different dosing, a different combination of agents, and it doesn’t isolate the single-arm activity of selinexor.”
Karyopharm Therapeutics must finish a randomized phase 3 trial of selinexor plus dexamethasone before the Food and Drug Administration will proceed with a safety and tolerability assessment for the first-in-class multiple myeloma drug.
By an 8-5 vote, the FDA Oncologic Drugs Advisory Committee said that data from STORM 2, Karyopharm’s single-arm phase 2b trial, didn’t sufficiently show that selinexor exerted any significant benefit over dexamethasone alone, used because the company claims it potentiates selinexor’s action.
Committee members also expressed concerns about the drug’s challenging adverse event profile. In STORM Part 2, 60% of patients experienced serious treatment-emergent adverse events and 10 died from them.
“This trial design is not adequate to assess tolerability and efficacy,” and move the drug along, said Christian S. Hinrichs, MD, of the National Cancer Institute. For that to happen, “we’d be looking for several things. We’d be looking for a subset of patients who benefited profoundly, which could be somewhat compelling despite a lower overall response rate. Next we might be looking for durable response, and here we see 4-month responses. And finally, what we look for in a single-arm trial is a really favorable side effect profile, like we see in checkpoint inhibitors. That is clearly not the case with this drug. So, on the basis of both the trial design and the results, I find it hard to conclude that these data allow for an adequate assessment that safety and efficacy are proven.”
The decision came despite the pleas of 15 patients and one patient advocate who said the drug improved clinical status and quality of life, and even extended life beyond what anyone expected. However, several committee members noted that Karyopharm paid for speakers’ travel and that patients who had negative experiences would probably be too sick to attend.
Selinexor is a completely new therapeutic option for relapsed multiple myeloma patients. It is a twice-weekly, oral tablet that inhibits nuclear export protein Exportin 1 (XPO1), which regulates the localization of tumor suppressor proteins and is associated with poor prognosis. Aberrant XPO1 expression causes tumor suppressors to locate away from their targets, allowing tumors to grow. Inhibiting it with selinexor blocks signal transduction pathways, interrupting tumor cell proliferation and inducing apoptosis while sparing normal cells.
Karyopharm is seeking approval of selinexor in combination with low-dose dexamethasone for the treatment of patients with relapsed/refractory multiple myeloma who have received at least three prior therapies and whose disease is refractory to at least one proteasome inhibitor, at least one immunomodulatory imide drug, and an anti-CD38 monoclonal antibody.
This disease is referred to as “triple-class refractory” multiple myeloma. At this stage, patients have exhausted every effective treatment option and are faced with the choice of supportive care or recycling previously successful drugs. Their median overall survival time is 3-5 months.
Karyopharm submitted its the New Drug Application using the Accelerated Approval pathway, arguing that the drug meets an unmet medical need and can be approved on surrogate endpoints – in this case, overall response rate.
The modified intent-to-treat analysis comprised 122 patients. The overall response rate was 25.4% with a median response duration of 4.4 months. Two patients had a complete response; six had a very good partial response; and 23 had a partial response.
Some committee members, however, said it would be impossible to tease out how much of the response could be due to the co-administration of 20 mg dexamethasone with each dose. In a phase 1 dose-ranging study of selinexor as monotherapy, it produced only one partial response in 56 patients. And, FDA pointed out, historical studies have shown response rates of 10%-27% for high-dose dexamethasone.
However, those in favor of the drug pointed out that the STORM patients were steroid-refractory, and that a 25% response rate would be unlikely on low-dose dexamethasone alone. This is proof of the company’s claim that the steroid works synergistically with selinexor, they said.
These members also pointed out that even a few years ago, there simply were no patients like the STORM cohort. Only recently have these patients lived long enough to develop resistance against all therapeutic lines, so it’s unrealistic to use historical data to judge what a reasonable response rate looks like in this situation.
Committee members also choked on STORM’s adverse event (AE) profile. All patients experienced at least one treatment-emergent AE, and 60% had at least one serious AE. Most (88.6%) required a dose modification due to an AE, and 28.5% discontinued due to one. The most common AEs were thrombocytopenia, anemia, nausea, fatigue, and decreased appetite. The company said these were “typically reversible and manageable with dose reductions.”
Additionally, there were 23 deaths in the trial. About half (13) were due to disease progression, but the remainder were due to a fatal treatment-emergent AE. Two of these (one pneumonia and one sepsis) were directly due to selinexor, the company said.
Despite the committee’s concerns, 16 of the 17 speakers described positive experiences with selinexor. They universally acknowledged that “it’s a hard drug to take,” and that side effects need to be managed proactively. But they also said, universally, that the drug has brought them additional months of good-quality life, decreased lengthy hospital stays, enabled them to participate in important family events, and even travel. Some also expressed the hope that selinexor would be a bridge drug, decreasing their disease burden enough that they could qualify for other clinical trials of new investigational drugs.
Only Stephanie Fox-Rawlings, PhD, of the National Center for Health Research, urged a delay. “Even if these adverse events are manageable, they harm patients’ quality of life,” she said. “This may be acceptable to some, but if the drug can’t provide a meaningful benefit then they are not worth it and in this clinical trial there was no improvement noted in quality of life. This drug has serious risks and we don’t know if it works.”
Dr. Fox said she was “very glad” that Karyopharm has completed recruitment for its phase 3 randomized study, dubbed BOSTON. BOSTON will assign active patients to once-weekly 100 mg selinexor plus weight-dosed bortezomib, plus twice-weekly 20 mg dexamethasone. The comparator group will receive weight-based bortezomib twice a week and 20 mg dexamethasone four times a week. Patients who progress can cross over to the active arm. The company hopes for even better results, saying that the proteasome inhibitor has also shown a synergistic effect with selinexor. Results are expected in 2020.
“The BOSTON study doesn’t solve anything,” retorted committee member David Harrington, PhD, emeritus professor of biostatistics at the Dana-Farber Cancer Institute. “It’s a different clinical profile, different dosing, a different combination of agents, and it doesn’t isolate the single-arm activity of selinexor.”
Karyopharm Therapeutics must finish a randomized phase 3 trial of selinexor plus dexamethasone before the Food and Drug Administration will proceed with a safety and tolerability assessment for the first-in-class multiple myeloma drug.
By an 8-5 vote, the FDA Oncologic Drugs Advisory Committee said that data from STORM 2, Karyopharm’s single-arm phase 2b trial, didn’t sufficiently show that selinexor exerted any significant benefit over dexamethasone alone, used because the company claims it potentiates selinexor’s action.
Committee members also expressed concerns about the drug’s challenging adverse event profile. In STORM Part 2, 60% of patients experienced serious treatment-emergent adverse events and 10 died from them.
“This trial design is not adequate to assess tolerability and efficacy,” and move the drug along, said Christian S. Hinrichs, MD, of the National Cancer Institute. For that to happen, “we’d be looking for several things. We’d be looking for a subset of patients who benefited profoundly, which could be somewhat compelling despite a lower overall response rate. Next we might be looking for durable response, and here we see 4-month responses. And finally, what we look for in a single-arm trial is a really favorable side effect profile, like we see in checkpoint inhibitors. That is clearly not the case with this drug. So, on the basis of both the trial design and the results, I find it hard to conclude that these data allow for an adequate assessment that safety and efficacy are proven.”
The decision came despite the pleas of 15 patients and one patient advocate who said the drug improved clinical status and quality of life, and even extended life beyond what anyone expected. However, several committee members noted that Karyopharm paid for speakers’ travel and that patients who had negative experiences would probably be too sick to attend.
Selinexor is a completely new therapeutic option for relapsed multiple myeloma patients. It is a twice-weekly, oral tablet that inhibits nuclear export protein Exportin 1 (XPO1), which regulates the localization of tumor suppressor proteins and is associated with poor prognosis. Aberrant XPO1 expression causes tumor suppressors to locate away from their targets, allowing tumors to grow. Inhibiting it with selinexor blocks signal transduction pathways, interrupting tumor cell proliferation and inducing apoptosis while sparing normal cells.
Karyopharm is seeking approval of selinexor in combination with low-dose dexamethasone for the treatment of patients with relapsed/refractory multiple myeloma who have received at least three prior therapies and whose disease is refractory to at least one proteasome inhibitor, at least one immunomodulatory imide drug, and an anti-CD38 monoclonal antibody.
This disease is referred to as “triple-class refractory” multiple myeloma. At this stage, patients have exhausted every effective treatment option and are faced with the choice of supportive care or recycling previously successful drugs. Their median overall survival time is 3-5 months.
Karyopharm submitted its the New Drug Application using the Accelerated Approval pathway, arguing that the drug meets an unmet medical need and can be approved on surrogate endpoints – in this case, overall response rate.
The modified intent-to-treat analysis comprised 122 patients. The overall response rate was 25.4% with a median response duration of 4.4 months. Two patients had a complete response; six had a very good partial response; and 23 had a partial response.
Some committee members, however, said it would be impossible to tease out how much of the response could be due to the co-administration of 20 mg dexamethasone with each dose. In a phase 1 dose-ranging study of selinexor as monotherapy, it produced only one partial response in 56 patients. And, FDA pointed out, historical studies have shown response rates of 10%-27% for high-dose dexamethasone.
However, those in favor of the drug pointed out that the STORM patients were steroid-refractory, and that a 25% response rate would be unlikely on low-dose dexamethasone alone. This is proof of the company’s claim that the steroid works synergistically with selinexor, they said.
These members also pointed out that even a few years ago, there simply were no patients like the STORM cohort. Only recently have these patients lived long enough to develop resistance against all therapeutic lines, so it’s unrealistic to use historical data to judge what a reasonable response rate looks like in this situation.
Committee members also choked on STORM’s adverse event (AE) profile. All patients experienced at least one treatment-emergent AE, and 60% had at least one serious AE. Most (88.6%) required a dose modification due to an AE, and 28.5% discontinued due to one. The most common AEs were thrombocytopenia, anemia, nausea, fatigue, and decreased appetite. The company said these were “typically reversible and manageable with dose reductions.”
Additionally, there were 23 deaths in the trial. About half (13) were due to disease progression, but the remainder were due to a fatal treatment-emergent AE. Two of these (one pneumonia and one sepsis) were directly due to selinexor, the company said.
Despite the committee’s concerns, 16 of the 17 speakers described positive experiences with selinexor. They universally acknowledged that “it’s a hard drug to take,” and that side effects need to be managed proactively. But they also said, universally, that the drug has brought them additional months of good-quality life, decreased lengthy hospital stays, enabled them to participate in important family events, and even travel. Some also expressed the hope that selinexor would be a bridge drug, decreasing their disease burden enough that they could qualify for other clinical trials of new investigational drugs.
Only Stephanie Fox-Rawlings, PhD, of the National Center for Health Research, urged a delay. “Even if these adverse events are manageable, they harm patients’ quality of life,” she said. “This may be acceptable to some, but if the drug can’t provide a meaningful benefit then they are not worth it and in this clinical trial there was no improvement noted in quality of life. This drug has serious risks and we don’t know if it works.”
Dr. Fox said she was “very glad” that Karyopharm has completed recruitment for its phase 3 randomized study, dubbed BOSTON. BOSTON will assign active patients to once-weekly 100 mg selinexor plus weight-dosed bortezomib, plus twice-weekly 20 mg dexamethasone. The comparator group will receive weight-based bortezomib twice a week and 20 mg dexamethasone four times a week. Patients who progress can cross over to the active arm. The company hopes for even better results, saying that the proteasome inhibitor has also shown a synergistic effect with selinexor. Results are expected in 2020.
“The BOSTON study doesn’t solve anything,” retorted committee member David Harrington, PhD, emeritus professor of biostatistics at the Dana-Farber Cancer Institute. “It’s a different clinical profile, different dosing, a different combination of agents, and it doesn’t isolate the single-arm activity of selinexor.”