User login
FDA approves Mavenclad for treatment of relapsing MS
including relapsing/remitting and active secondary progressive disease.

The drug’s manufacturer, EMD Serono, said in a press release that cladribine is the first short-course oral therapy for such patients, and its use is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS. Cladribine is not recommended for use in patients with clinically isolated syndrome.
The agency’s decision is based on results from a clinical trial of 1,326 patients with relapsing MS who had experienced at least one relapse in the previous 12 months. Patients who received cladribine had significantly fewer relapses than did those who received placebo; the progression of disability was also significantly reduced in the cladribine group, compared with placebo, according to the FDA’s announcement.
The most common adverse events associated with cladribine include upper respiratory tract infections, headache, and decreased lymphocyte counts. In addition, the medication must be dispensed with a patient medication guide because the label includes a boxed warning for increased risk of malignancy and fetal harm. Other warnings include a risk for decreased lymphocyte count, hematologic toxicity and bone marrow suppression, and graft-versus-host-disease.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis. The approval of Mavenclad represents an additional option for patients who have tried another treatment without success,” Billy Dunn, MD, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in the announcement.
The approved dose of cladribine is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg per year, each consisting of 2 treatment weeks. Additional courses of cladribine are not to be administered because retreatment with cladribine during years 3 and 4 may further increase the risk of malignancy. The safety and efficacy of reinitiating cladribine more than 2 years after completing two treatment courses has not been studied, according to EMD Serono.
Cladribine is approved in more than 50 other countries and was approved for use in the European Union in August 2017.
including relapsing/remitting and active secondary progressive disease.
The drug’s manufacturer, EMD Serono, said in a press release that cladribine is the first short-course oral therapy for such patients, and its use is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS. Cladribine is not recommended for use in patients with clinically isolated syndrome.
The agency’s decision is based on results from a clinical trial of 1,326 patients with relapsing MS who had experienced at least one relapse in the previous 12 months. Patients who received cladribine had significantly fewer relapses than did those who received placebo; the progression of disability was also significantly reduced in the cladribine group, compared with placebo, according to the FDA’s announcement.
The most common adverse events associated with cladribine include upper respiratory tract infections, headache, and decreased lymphocyte counts. In addition, the medication must be dispensed with a patient medication guide because the label includes a boxed warning for increased risk of malignancy and fetal harm. Other warnings include a risk for decreased lymphocyte count, hematologic toxicity and bone marrow suppression, and graft-versus-host-disease.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis. The approval of Mavenclad represents an additional option for patients who have tried another treatment without success,” Billy Dunn, MD, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in the announcement.
The approved dose of cladribine is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg per year, each consisting of 2 treatment weeks. Additional courses of cladribine are not to be administered because retreatment with cladribine during years 3 and 4 may further increase the risk of malignancy. The safety and efficacy of reinitiating cladribine more than 2 years after completing two treatment courses has not been studied, according to EMD Serono.
Cladribine is approved in more than 50 other countries and was approved for use in the European Union in August 2017.
including relapsing/remitting and active secondary progressive disease.
The drug’s manufacturer, EMD Serono, said in a press release that cladribine is the first short-course oral therapy for such patients, and its use is generally recommended for patients who have had an inadequate response to, or are unable to tolerate, an alternate drug indicated for the treatment of MS. Cladribine is not recommended for use in patients with clinically isolated syndrome.
The agency’s decision is based on results from a clinical trial of 1,326 patients with relapsing MS who had experienced at least one relapse in the previous 12 months. Patients who received cladribine had significantly fewer relapses than did those who received placebo; the progression of disability was also significantly reduced in the cladribine group, compared with placebo, according to the FDA’s announcement.
The most common adverse events associated with cladribine include upper respiratory tract infections, headache, and decreased lymphocyte counts. In addition, the medication must be dispensed with a patient medication guide because the label includes a boxed warning for increased risk of malignancy and fetal harm. Other warnings include a risk for decreased lymphocyte count, hematologic toxicity and bone marrow suppression, and graft-versus-host-disease.
“We are committed to supporting the development of safe and effective treatments for patients with multiple sclerosis. The approval of Mavenclad represents an additional option for patients who have tried another treatment without success,” Billy Dunn, MD, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in the announcement.
The approved dose of cladribine is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg per year, each consisting of 2 treatment weeks. Additional courses of cladribine are not to be administered because retreatment with cladribine during years 3 and 4 may further increase the risk of malignancy. The safety and efficacy of reinitiating cladribine more than 2 years after completing two treatment courses has not been studied, according to EMD Serono.
Cladribine is approved in more than 50 other countries and was approved for use in the European Union in August 2017.
2018-2019 flu season: Going but not gone yet
The 2018-2019 flu season again showed real signs of ending as influenza activity levels dropped during the week ending March 23, according to the Centers for Disease Control and Prevention.
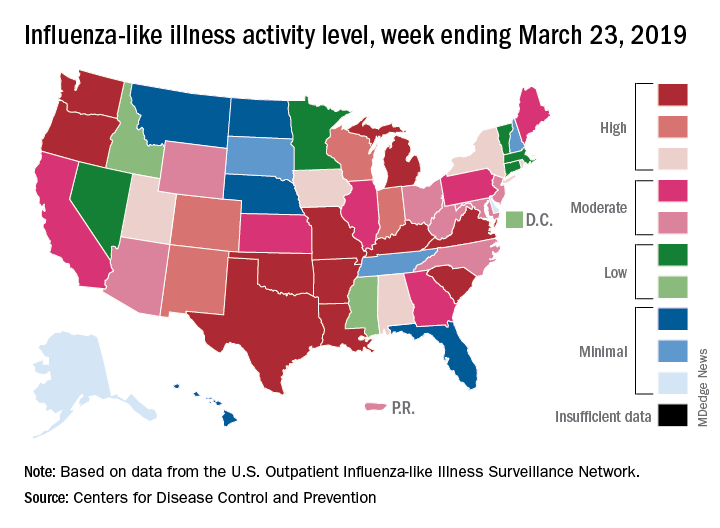
Despite those declines, however, current levels of influenza-like illness (ILI) activity are still elevated enough that the CDC issued a health advisory on March 28 to inform clinicians about the “increasing proportion of activity due to influenza A(H3N2) viruses, continued circulation of influenza A(H1N1) viruses, and low levels of influenza B viruses.”
The CDC’s weekly flu report, released March 29, does show that the overall burden is improving. The national proportion of outpatient visits for ILI dropped from 4.3% for the week ending March 16 to 3.8% for the latest reporting week, the CDC’s influenza division reported. The figure for March 16 was originally reported to be 4.4% but was revised in the new report.
The length of this years’ flu season, when measured as the number of weeks at or above the baseline level of 2.2%, is now 18 weeks. By this measure, the last five seasons have averaged 16 weeks, the CDC noted.
Influenza was considered widespread in 34 states and Puerto Rico for the week ending March 23, down from 44 states the previous week. The number of states at the highest level of ILI activity on the CDC’s 1-10 scale dropped from 20 to 11, and those in the high range (8-10) dropped from 26 to 20, data from the CDC’s Outpatient ILI Surveillance Network show.
There was one flu-related pediatric death during the week of March 23 but none reported from earlier weeks, which brings the total to 77 for the 2018-2019 season, the CDC said.
The 2018-2019 flu season again showed real signs of ending as influenza activity levels dropped during the week ending March 23, according to the Centers for Disease Control and Prevention.
Despite those declines, however, current levels of influenza-like illness (ILI) activity are still elevated enough that the CDC issued a health advisory on March 28 to inform clinicians about the “increasing proportion of activity due to influenza A(H3N2) viruses, continued circulation of influenza A(H1N1) viruses, and low levels of influenza B viruses.”
The CDC’s weekly flu report, released March 29, does show that the overall burden is improving. The national proportion of outpatient visits for ILI dropped from 4.3% for the week ending March 16 to 3.8% for the latest reporting week, the CDC’s influenza division reported. The figure for March 16 was originally reported to be 4.4% but was revised in the new report.
The length of this years’ flu season, when measured as the number of weeks at or above the baseline level of 2.2%, is now 18 weeks. By this measure, the last five seasons have averaged 16 weeks, the CDC noted.
Influenza was considered widespread in 34 states and Puerto Rico for the week ending March 23, down from 44 states the previous week. The number of states at the highest level of ILI activity on the CDC’s 1-10 scale dropped from 20 to 11, and those in the high range (8-10) dropped from 26 to 20, data from the CDC’s Outpatient ILI Surveillance Network show.
There was one flu-related pediatric death during the week of March 23 but none reported from earlier weeks, which brings the total to 77 for the 2018-2019 season, the CDC said.
The 2018-2019 flu season again showed real signs of ending as influenza activity levels dropped during the week ending March 23, according to the Centers for Disease Control and Prevention.
Despite those declines, however, current levels of influenza-like illness (ILI) activity are still elevated enough that the CDC issued a health advisory on March 28 to inform clinicians about the “increasing proportion of activity due to influenza A(H3N2) viruses, continued circulation of influenza A(H1N1) viruses, and low levels of influenza B viruses.”
The CDC’s weekly flu report, released March 29, does show that the overall burden is improving. The national proportion of outpatient visits for ILI dropped from 4.3% for the week ending March 16 to 3.8% for the latest reporting week, the CDC’s influenza division reported. The figure for March 16 was originally reported to be 4.4% but was revised in the new report.
The length of this years’ flu season, when measured as the number of weeks at or above the baseline level of 2.2%, is now 18 weeks. By this measure, the last five seasons have averaged 16 weeks, the CDC noted.
Influenza was considered widespread in 34 states and Puerto Rico for the week ending March 23, down from 44 states the previous week. The number of states at the highest level of ILI activity on the CDC’s 1-10 scale dropped from 20 to 11, and those in the high range (8-10) dropped from 26 to 20, data from the CDC’s Outpatient ILI Surveillance Network show.
There was one flu-related pediatric death during the week of March 23 but none reported from earlier weeks, which brings the total to 77 for the 2018-2019 season, the CDC said.
Cimzia becomes first FDA-approved treatment for nonradiographic axial spondyloarthritis
, with objective evidence of inflammation, making it the first treatment approved by the agency for the condition.
![]()
The FDA approved the tumor necrosis factor inhibitor based on results from a randomized clinical trial in 317 adult patients with nonradiographic axial spondyloarthritis (nr-axSpA) who had elevated C-reactive protein levels and/or sacroiliitis (inflammation of the sacroiliac joints) on MRI.
The trial entailed 52 weeks of double-blind therapy with certolizumab at a starting dose of 400 mg on weeks 0, 2, and 4 followed by 200 mg every 2 weeks, or placebo. The Ankylosing Spondylitis Disease Activity Score Major Improvement rate, defined as at least a 2-point improvement from baseline, was 47% in the active treatment arm, compared with 7% on placebo. The Assessment in Ankylosing Spondylitis International Society 40% response rate, a more patient-reported outcome measure, was 57% in the certolizumab group and 16% in controls (Arthritis Rheumatol. 2019 March 8. doi: 10.1002/art.40866).
The overall safety profile observed in the Cimzia treatment group was consistent with the known safety profile of certolizumab.
Cimzia was first approved in 2008 and has FDA-approved indications for adult patients with Crohn’s disease, moderate to severe rheumatoid arthritis, active ankylosing spondylitis and moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
, with objective evidence of inflammation, making it the first treatment approved by the agency for the condition.
![]()
The FDA approved the tumor necrosis factor inhibitor based on results from a randomized clinical trial in 317 adult patients with nonradiographic axial spondyloarthritis (nr-axSpA) who had elevated C-reactive protein levels and/or sacroiliitis (inflammation of the sacroiliac joints) on MRI.
The trial entailed 52 weeks of double-blind therapy with certolizumab at a starting dose of 400 mg on weeks 0, 2, and 4 followed by 200 mg every 2 weeks, or placebo. The Ankylosing Spondylitis Disease Activity Score Major Improvement rate, defined as at least a 2-point improvement from baseline, was 47% in the active treatment arm, compared with 7% on placebo. The Assessment in Ankylosing Spondylitis International Society 40% response rate, a more patient-reported outcome measure, was 57% in the certolizumab group and 16% in controls (Arthritis Rheumatol. 2019 March 8. doi: 10.1002/art.40866).
The overall safety profile observed in the Cimzia treatment group was consistent with the known safety profile of certolizumab.
Cimzia was first approved in 2008 and has FDA-approved indications for adult patients with Crohn’s disease, moderate to severe rheumatoid arthritis, active ankylosing spondylitis and moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
, with objective evidence of inflammation, making it the first treatment approved by the agency for the condition.
![]()
The FDA approved the tumor necrosis factor inhibitor based on results from a randomized clinical trial in 317 adult patients with nonradiographic axial spondyloarthritis (nr-axSpA) who had elevated C-reactive protein levels and/or sacroiliitis (inflammation of the sacroiliac joints) on MRI.
The trial entailed 52 weeks of double-blind therapy with certolizumab at a starting dose of 400 mg on weeks 0, 2, and 4 followed by 200 mg every 2 weeks, or placebo. The Ankylosing Spondylitis Disease Activity Score Major Improvement rate, defined as at least a 2-point improvement from baseline, was 47% in the active treatment arm, compared with 7% on placebo. The Assessment in Ankylosing Spondylitis International Society 40% response rate, a more patient-reported outcome measure, was 57% in the certolizumab group and 16% in controls (Arthritis Rheumatol. 2019 March 8. doi: 10.1002/art.40866).
The overall safety profile observed in the Cimzia treatment group was consistent with the known safety profile of certolizumab.
Cimzia was first approved in 2008 and has FDA-approved indications for adult patients with Crohn’s disease, moderate to severe rheumatoid arthritis, active ankylosing spondylitis and moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
FDA panel calls for changes to breast implant rupture screening
A Food and Drug Administration advisory panel urged the agency to switch its recommended screening method for silent breast implant ruptures from MRI to ultrasound and to push the first screening examination back from the current 3 years post implant to 5 years.
Members of the FDA’s General and Plastic Surgery Advisory Panel also made suggestions to the FDA regarding how it might improve communication about the risks of breast implants to the public in general and to people considering implants in particular.
The panel also discussed the sort of safety and efficacy assessments the FDA should require for acellular dermal matrix (ADM), also known as mesh, to add the material’s label for use during breast reconstruction or implant augmentation. Surgeons have used mesh routinely as a surgical aid at other body sites, such as the abdomen. Although ADM is now also widely used during breast surgery, it has never undergone testing or labeling for use in that setting.
The FDA convened the advisory committee meeting largely to assess and discuss data and concerns about two recently appreciated complications of breast implant placement – breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL) and a still poorly defined and described constellation of autoimmune and rheumatoid-like symptoms reported anecdotally by some breast implant recipients called Breast Implant Illness (BII). But agency officials asked the panel to also address these other issues related to the safety of breast implants and implant surgery.
The revised screening recommendations were primarily a response to a lack of compliance with current FDA recommendations to screen for breast implant rupture with MRI starting 3 years after placement and then every 2 years.

The problem is that a screening MRI costs about $1,500-$2,000 and is generally not covered by insurance when done for this purpose, although it is often covered when used to investigate a suspected rupture. The result is that less than 5% of implanted patients comply with the recommended screening schedule, noted committee chair Frank R. Lewis Jr., MD, executive director, emeritus, of the American Board of Surgery in Philadelphia.
“Effectively it’s a useless recommendation,” he said. “Ultrasound is far easier, quicker, and cheaper” and seems effective for screening.
The advisory panel recommended starting ultrasound screening 5 years after implantation, based on MRI screening data showing that virtually all ruptures don’t occur until after 5 years, and then following with ultrasound screening every 3 years after that. The panel recommended using MRI when the ultrasound result is equivocal or when the patient has symptoms suggesting rupture.

After hearing testimony during the sessions from several dozen women who told horror stories of the complications they experienced from breast implants, panel member Karen E. Burke, MD, PhD, spoke for many on the panel when she said “no doubt patients feel that the informed consent process failed them, that they were not aware of the risks.”
Dr. Burke suggested that patients must be informed so that they realize that breast implants are not static objects that will always sit unchanged in their body for the rest of their lives, that certain factors such as allergy or family history of tissue disease might predispose them to autoimmune-type reactions and that the diverse symptoms described for BII are possible sequelae.
A black box warning for the potential of developing anaplastic large-cell lymphoma should also go into the label, said Dr. Burke, a dermatologist who practices in New York City.
Dr. Lewis ridiculed the information booklets that implant manufacturers currently provide for patients as too long and dense. “They were not constructed to inform patients in the best way; they were constructed to provide legal protection.” He called for creating a two- or three-page list of potential adverse effects and points to consider.
Other panel members suggested public service advertisements similar to what is used to inform consumers about the risk from cigarettes. Dr. Burke recommended getting the word out about BII to other medical specialties that are more likely to see affected patients first, such as rheumatologists, immunologists, and dermatologists. She vowed to speak about these complications at an upcoming meeting of the American Academy of Dermatology. But other panel members noted that BII right now remains without any official medical definition nor clear causal link to breast implants.

The question of exactly what safety and efficacy data the FDA might require from manufacturers seeking a breast surgery indication for ADM was less clear.
Binita Ashar, MD, director of the FDA’s Division of Surgical Devices, highlighted the agency’s dilemma about considering data for a breast surgery indication. “The challenge for us is that we can’t expect a control arm because everyone today is using” mesh, she explained. “We’re looking for guidance on how to understand the risk-to-benefit profile” of ADM.
A plastic surgeon on the advisory panel, Pierre M. Chevray, MD, PhD, from Houston Methodist Hospital summarized the way ADM mesh reached its current niche in routine, U.S. breast surgery.
About 20 years ago, plastic surgeons began using mesh during implant surgery to improve eventual breast cosmesis. Surgeons began to wrap the implant in mesh and then attached the mesh to the pectoral muscle so that the implant could go on top of the muscle and not beneath it. It greatly diminished capsular contraction around the implant over time, reduced the risk for implant movement, and allowed for more natural positioning of the breast with the implant inside, he said.

Another factor in the growing use of mesh was heavy promotion by manufacturers to a generation of plastic surgeons, Dr. Chevray said. But use of ADM may also lead to a slightly increased rate of seromas and infections.
“The benefit from mesh is hard to prove and is questionable” because it largely depends on a subjective assessment by a surgeon or patient, Dr. Chevray said. “The cost [of ADM] is substantial, but no data have shown that outcomes are better” with its use. Despite that, “nearly every surgeon uses mesh” these days, he noted.
A Food and Drug Administration advisory panel urged the agency to switch its recommended screening method for silent breast implant ruptures from MRI to ultrasound and to push the first screening examination back from the current 3 years post implant to 5 years.
Members of the FDA’s General and Plastic Surgery Advisory Panel also made suggestions to the FDA regarding how it might improve communication about the risks of breast implants to the public in general and to people considering implants in particular.
The panel also discussed the sort of safety and efficacy assessments the FDA should require for acellular dermal matrix (ADM), also known as mesh, to add the material’s label for use during breast reconstruction or implant augmentation. Surgeons have used mesh routinely as a surgical aid at other body sites, such as the abdomen. Although ADM is now also widely used during breast surgery, it has never undergone testing or labeling for use in that setting.
The FDA convened the advisory committee meeting largely to assess and discuss data and concerns about two recently appreciated complications of breast implant placement – breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL) and a still poorly defined and described constellation of autoimmune and rheumatoid-like symptoms reported anecdotally by some breast implant recipients called Breast Implant Illness (BII). But agency officials asked the panel to also address these other issues related to the safety of breast implants and implant surgery.
The revised screening recommendations were primarily a response to a lack of compliance with current FDA recommendations to screen for breast implant rupture with MRI starting 3 years after placement and then every 2 years.
The problem is that a screening MRI costs about $1,500-$2,000 and is generally not covered by insurance when done for this purpose, although it is often covered when used to investigate a suspected rupture. The result is that less than 5% of implanted patients comply with the recommended screening schedule, noted committee chair Frank R. Lewis Jr., MD, executive director, emeritus, of the American Board of Surgery in Philadelphia.
“Effectively it’s a useless recommendation,” he said. “Ultrasound is far easier, quicker, and cheaper” and seems effective for screening.
The advisory panel recommended starting ultrasound screening 5 years after implantation, based on MRI screening data showing that virtually all ruptures don’t occur until after 5 years, and then following with ultrasound screening every 3 years after that. The panel recommended using MRI when the ultrasound result is equivocal or when the patient has symptoms suggesting rupture.
After hearing testimony during the sessions from several dozen women who told horror stories of the complications they experienced from breast implants, panel member Karen E. Burke, MD, PhD, spoke for many on the panel when she said “no doubt patients feel that the informed consent process failed them, that they were not aware of the risks.”
Dr. Burke suggested that patients must be informed so that they realize that breast implants are not static objects that will always sit unchanged in their body for the rest of their lives, that certain factors such as allergy or family history of tissue disease might predispose them to autoimmune-type reactions and that the diverse symptoms described for BII are possible sequelae.
A black box warning for the potential of developing anaplastic large-cell lymphoma should also go into the label, said Dr. Burke, a dermatologist who practices in New York City.
Dr. Lewis ridiculed the information booklets that implant manufacturers currently provide for patients as too long and dense. “They were not constructed to inform patients in the best way; they were constructed to provide legal protection.” He called for creating a two- or three-page list of potential adverse effects and points to consider.
Other panel members suggested public service advertisements similar to what is used to inform consumers about the risk from cigarettes. Dr. Burke recommended getting the word out about BII to other medical specialties that are more likely to see affected patients first, such as rheumatologists, immunologists, and dermatologists. She vowed to speak about these complications at an upcoming meeting of the American Academy of Dermatology. But other panel members noted that BII right now remains without any official medical definition nor clear causal link to breast implants.
The question of exactly what safety and efficacy data the FDA might require from manufacturers seeking a breast surgery indication for ADM was less clear.
Binita Ashar, MD, director of the FDA’s Division of Surgical Devices, highlighted the agency’s dilemma about considering data for a breast surgery indication. “The challenge for us is that we can’t expect a control arm because everyone today is using” mesh, she explained. “We’re looking for guidance on how to understand the risk-to-benefit profile” of ADM.
A plastic surgeon on the advisory panel, Pierre M. Chevray, MD, PhD, from Houston Methodist Hospital summarized the way ADM mesh reached its current niche in routine, U.S. breast surgery.
About 20 years ago, plastic surgeons began using mesh during implant surgery to improve eventual breast cosmesis. Surgeons began to wrap the implant in mesh and then attached the mesh to the pectoral muscle so that the implant could go on top of the muscle and not beneath it. It greatly diminished capsular contraction around the implant over time, reduced the risk for implant movement, and allowed for more natural positioning of the breast with the implant inside, he said.
Another factor in the growing use of mesh was heavy promotion by manufacturers to a generation of plastic surgeons, Dr. Chevray said. But use of ADM may also lead to a slightly increased rate of seromas and infections.
“The benefit from mesh is hard to prove and is questionable” because it largely depends on a subjective assessment by a surgeon or patient, Dr. Chevray said. “The cost [of ADM] is substantial, but no data have shown that outcomes are better” with its use. Despite that, “nearly every surgeon uses mesh” these days, he noted.
A Food and Drug Administration advisory panel urged the agency to switch its recommended screening method for silent breast implant ruptures from MRI to ultrasound and to push the first screening examination back from the current 3 years post implant to 5 years.
Members of the FDA’s General and Plastic Surgery Advisory Panel also made suggestions to the FDA regarding how it might improve communication about the risks of breast implants to the public in general and to people considering implants in particular.
The panel also discussed the sort of safety and efficacy assessments the FDA should require for acellular dermal matrix (ADM), also known as mesh, to add the material’s label for use during breast reconstruction or implant augmentation. Surgeons have used mesh routinely as a surgical aid at other body sites, such as the abdomen. Although ADM is now also widely used during breast surgery, it has never undergone testing or labeling for use in that setting.
The FDA convened the advisory committee meeting largely to assess and discuss data and concerns about two recently appreciated complications of breast implant placement – breast implant–associated anaplastic large-cell lymphoma (BIA-ALCL) and a still poorly defined and described constellation of autoimmune and rheumatoid-like symptoms reported anecdotally by some breast implant recipients called Breast Implant Illness (BII). But agency officials asked the panel to also address these other issues related to the safety of breast implants and implant surgery.
The revised screening recommendations were primarily a response to a lack of compliance with current FDA recommendations to screen for breast implant rupture with MRI starting 3 years after placement and then every 2 years.
The problem is that a screening MRI costs about $1,500-$2,000 and is generally not covered by insurance when done for this purpose, although it is often covered when used to investigate a suspected rupture. The result is that less than 5% of implanted patients comply with the recommended screening schedule, noted committee chair Frank R. Lewis Jr., MD, executive director, emeritus, of the American Board of Surgery in Philadelphia.
“Effectively it’s a useless recommendation,” he said. “Ultrasound is far easier, quicker, and cheaper” and seems effective for screening.
The advisory panel recommended starting ultrasound screening 5 years after implantation, based on MRI screening data showing that virtually all ruptures don’t occur until after 5 years, and then following with ultrasound screening every 3 years after that. The panel recommended using MRI when the ultrasound result is equivocal or when the patient has symptoms suggesting rupture.
After hearing testimony during the sessions from several dozen women who told horror stories of the complications they experienced from breast implants, panel member Karen E. Burke, MD, PhD, spoke for many on the panel when she said “no doubt patients feel that the informed consent process failed them, that they were not aware of the risks.”
Dr. Burke suggested that patients must be informed so that they realize that breast implants are not static objects that will always sit unchanged in their body for the rest of their lives, that certain factors such as allergy or family history of tissue disease might predispose them to autoimmune-type reactions and that the diverse symptoms described for BII are possible sequelae.
A black box warning for the potential of developing anaplastic large-cell lymphoma should also go into the label, said Dr. Burke, a dermatologist who practices in New York City.
Dr. Lewis ridiculed the information booklets that implant manufacturers currently provide for patients as too long and dense. “They were not constructed to inform patients in the best way; they were constructed to provide legal protection.” He called for creating a two- or three-page list of potential adverse effects and points to consider.
Other panel members suggested public service advertisements similar to what is used to inform consumers about the risk from cigarettes. Dr. Burke recommended getting the word out about BII to other medical specialties that are more likely to see affected patients first, such as rheumatologists, immunologists, and dermatologists. She vowed to speak about these complications at an upcoming meeting of the American Academy of Dermatology. But other panel members noted that BII right now remains without any official medical definition nor clear causal link to breast implants.
The question of exactly what safety and efficacy data the FDA might require from manufacturers seeking a breast surgery indication for ADM was less clear.
Binita Ashar, MD, director of the FDA’s Division of Surgical Devices, highlighted the agency’s dilemma about considering data for a breast surgery indication. “The challenge for us is that we can’t expect a control arm because everyone today is using” mesh, she explained. “We’re looking for guidance on how to understand the risk-to-benefit profile” of ADM.
A plastic surgeon on the advisory panel, Pierre M. Chevray, MD, PhD, from Houston Methodist Hospital summarized the way ADM mesh reached its current niche in routine, U.S. breast surgery.
About 20 years ago, plastic surgeons began using mesh during implant surgery to improve eventual breast cosmesis. Surgeons began to wrap the implant in mesh and then attached the mesh to the pectoral muscle so that the implant could go on top of the muscle and not beneath it. It greatly diminished capsular contraction around the implant over time, reduced the risk for implant movement, and allowed for more natural positioning of the breast with the implant inside, he said.
Another factor in the growing use of mesh was heavy promotion by manufacturers to a generation of plastic surgeons, Dr. Chevray said. But use of ADM may also lead to a slightly increased rate of seromas and infections.
“The benefit from mesh is hard to prove and is questionable” because it largely depends on a subjective assessment by a surgeon or patient, Dr. Chevray said. “The cost [of ADM] is substantial, but no data have shown that outcomes are better” with its use. Despite that, “nearly every surgeon uses mesh” these days, he noted.
AT AN FDA ADVISORY PANEL MEETING
FDA approves siponimod for relapsing forms of MS
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.

Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.
Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
(MS), including clinically isolated syndrome, relapsing-remitting MS, and active secondary progressive MS.
Siponimod is a selective sphingosine 1-phosphate (S1P) receptor modulator that binds to S1P1 and S1P5 receptors. Its binding to the S1P1 receptor prevents lymphocytes from leaving the lymph nodes, which contributes to the treatment’s anti-inflammatory effects. Its binding to the S1P5 and S1P1 subreceptors on oligodendrocytes and astrocytes is intended to promote remyelination and prevent inflammation.
The treatment’s approval is based on the results of the phase 3 EXPAND study, according to the agency’s March 26 announcement. This randomized, double-blind study compared siponimod with placebo among 1,651 patients with secondary progressive MS. At baseline, the population’s mean age was 48 years, and mean disease duration was approximately 16 years. More than half the study population had a median Expanded Disability Status Scale score of 6.0 and relied on a walking aid.
Siponimod reduced the risk of 3-month confirmed disability progression (CDP) by 21%, compared with placebo (P = .013). Among participants with relapse activity in the 2 years prior to screening, siponimod reduced the risk of this outcome by 33%, compared with placebo (P = .0100). Siponimod delayed the risk of 6-month CDP by 26%, compared with placebo (P = .0058) and reduced the annualized relapse rate by 55%. In addition, the data suggested beneficial effects of siponimod on cognition, MRI disease activity, and brain volume loss. Siponimod did not provide significant improvements in patients with nonactive secondary progressive MS.
Common adverse events included headache, hypertension, and transaminase increase. The FDA requires siponimod to be dispensed with a medication guide that describes the treatment’s associated risks of infection, macular edema, decreased heart rate, and impaired lung function.
Novartis manufactures the drug. The company expects the drug to be available within 1 week, according to its press release.
United States now over 300 measles cases for the year
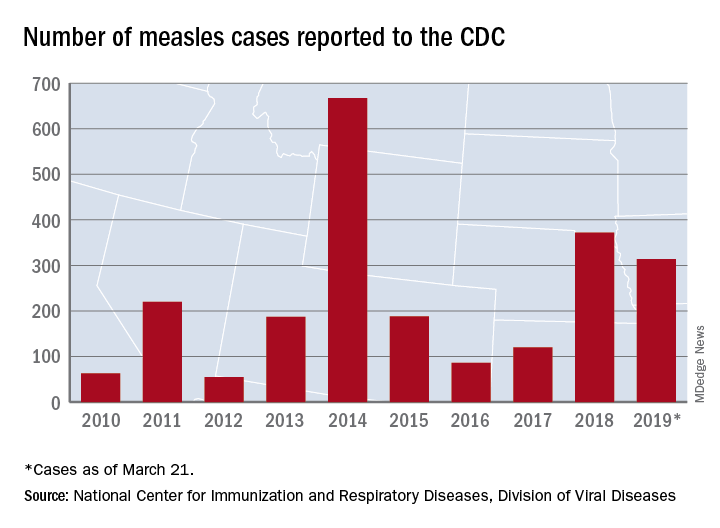
Despite those 46 new cases, the number of states with reported cases remains at 15, the CDC reported March 25.
For the fifth consecutive week the busiest outbreak was in Brooklyn, N.Y., which added 23 new cases. New York’s Rockland County, which is just north of New York City and has 46 confirmed cases for the year, is home to another of the six current outbreaks in the country, with the other four located in Washington (74 total cases for the state), Texas (14 cases), California (7 cases), and Illinois (6 cases). Other states with cases are Arizona, Colorado, Connecticut, Georgia, Kentucky, Missouri, New Hampshire, New Jersey, and Oregon, reported the CDC.
This year’s case total through less than 3 months is nearing the 372 that occurred in 2018, which was the second-worst year for measles in the last decade, but is still well off the 10-year high of 667 reported in 2014, the CDC said.
Despite those 46 new cases, the number of states with reported cases remains at 15, the CDC reported March 25.
For the fifth consecutive week the busiest outbreak was in Brooklyn, N.Y., which added 23 new cases. New York’s Rockland County, which is just north of New York City and has 46 confirmed cases for the year, is home to another of the six current outbreaks in the country, with the other four located in Washington (74 total cases for the state), Texas (14 cases), California (7 cases), and Illinois (6 cases). Other states with cases are Arizona, Colorado, Connecticut, Georgia, Kentucky, Missouri, New Hampshire, New Jersey, and Oregon, reported the CDC.
This year’s case total through less than 3 months is nearing the 372 that occurred in 2018, which was the second-worst year for measles in the last decade, but is still well off the 10-year high of 667 reported in 2014, the CDC said.
Despite those 46 new cases, the number of states with reported cases remains at 15, the CDC reported March 25.
For the fifth consecutive week the busiest outbreak was in Brooklyn, N.Y., which added 23 new cases. New York’s Rockland County, which is just north of New York City and has 46 confirmed cases for the year, is home to another of the six current outbreaks in the country, with the other four located in Washington (74 total cases for the state), Texas (14 cases), California (7 cases), and Illinois (6 cases). Other states with cases are Arizona, Colorado, Connecticut, Georgia, Kentucky, Missouri, New Hampshire, New Jersey, and Oregon, reported the CDC.
This year’s case total through less than 3 months is nearing the 372 that occurred in 2018, which was the second-worst year for measles in the last decade, but is still well off the 10-year high of 667 reported in 2014, the CDC said.
H3N2 putting a damper on flu season’s departure
The decline of influenza activity remains slow, largely “driven by a wave of H3N2 virus activity” in recent weeks, according to the Centers for Disease Control and Prevention.
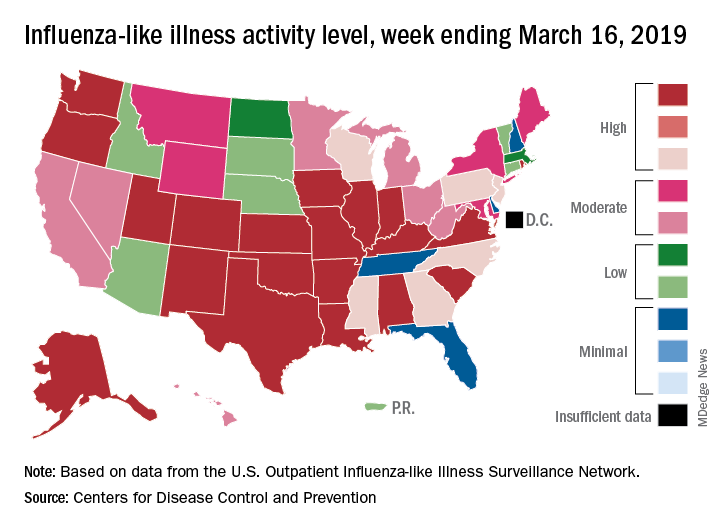
Fewer states reported the highest level of influenza-like illness (ILI) activity on the CDC’s 1-10 scale for the week ending March 16, but the national proportion of outpatient visits for ILI was 4.4% for the second consecutive week, the CDC’s influenza division reported March 22. The outpatient-visit figure for the week ending March 9 was originally reported as 4.5% last week, but it has been revised down to 4.4% this week.
Another measure of activity – the percentage of respiratory specimens testing positive for influenza viruses in clinical laboratories – actually increased slightly during the week ending March 16, the CDC noted.
For the current week, there were 26 states in the high (8-10) range of activity – 20 states were at level 10 and another 6 states were at level 8 – compared with the previous week, when 21 states were at level 10 and 30 states were in the high range, the CDC’s Outpatient ILI Surveillance Network reported.
There were eight ILI-related deaths in children reported during the week ending March 16, seven of which occurred in previous weeks. The total for the 2018-2019 season so far is 76, the CDC said.
New preliminary estimates on influenza’s burden nationally put the total number of deaths at 25,000-41,500 since the beginning of the season on Oct. 1, 2018. There also have been 375,000-454,000 flu-related hospitalizations, 13.2 million to 15.4 million medical visits, and 28.5 to 32.8 million individual illnesses, the CDC said.
Since the CDC “expects flu activity to remain elevated for a number of weeks,” it continues to recommend flu vaccination and the use of influenza antiviral drugs as “an important second line of defense that can be used to treat flu illness. H3N2 viruses are typically associated with more severe illness in older adults, and flu vaccine may protect less well against H3N2 illness in older adults, making prompt treatment with flu antivirals in this age group especially important during the current period of H3N2 predominance.”
The decline of influenza activity remains slow, largely “driven by a wave of H3N2 virus activity” in recent weeks, according to the Centers for Disease Control and Prevention.
Fewer states reported the highest level of influenza-like illness (ILI) activity on the CDC’s 1-10 scale for the week ending March 16, but the national proportion of outpatient visits for ILI was 4.4% for the second consecutive week, the CDC’s influenza division reported March 22. The outpatient-visit figure for the week ending March 9 was originally reported as 4.5% last week, but it has been revised down to 4.4% this week.
Another measure of activity – the percentage of respiratory specimens testing positive for influenza viruses in clinical laboratories – actually increased slightly during the week ending March 16, the CDC noted.
For the current week, there were 26 states in the high (8-10) range of activity – 20 states were at level 10 and another 6 states were at level 8 – compared with the previous week, when 21 states were at level 10 and 30 states were in the high range, the CDC’s Outpatient ILI Surveillance Network reported.
There were eight ILI-related deaths in children reported during the week ending March 16, seven of which occurred in previous weeks. The total for the 2018-2019 season so far is 76, the CDC said.
New preliminary estimates on influenza’s burden nationally put the total number of deaths at 25,000-41,500 since the beginning of the season on Oct. 1, 2018. There also have been 375,000-454,000 flu-related hospitalizations, 13.2 million to 15.4 million medical visits, and 28.5 to 32.8 million individual illnesses, the CDC said.
Since the CDC “expects flu activity to remain elevated for a number of weeks,” it continues to recommend flu vaccination and the use of influenza antiviral drugs as “an important second line of defense that can be used to treat flu illness. H3N2 viruses are typically associated with more severe illness in older adults, and flu vaccine may protect less well against H3N2 illness in older adults, making prompt treatment with flu antivirals in this age group especially important during the current period of H3N2 predominance.”
The decline of influenza activity remains slow, largely “driven by a wave of H3N2 virus activity” in recent weeks, according to the Centers for Disease Control and Prevention.
Fewer states reported the highest level of influenza-like illness (ILI) activity on the CDC’s 1-10 scale for the week ending March 16, but the national proportion of outpatient visits for ILI was 4.4% for the second consecutive week, the CDC’s influenza division reported March 22. The outpatient-visit figure for the week ending March 9 was originally reported as 4.5% last week, but it has been revised down to 4.4% this week.
Another measure of activity – the percentage of respiratory specimens testing positive for influenza viruses in clinical laboratories – actually increased slightly during the week ending March 16, the CDC noted.
For the current week, there were 26 states in the high (8-10) range of activity – 20 states were at level 10 and another 6 states were at level 8 – compared with the previous week, when 21 states were at level 10 and 30 states were in the high range, the CDC’s Outpatient ILI Surveillance Network reported.
There were eight ILI-related deaths in children reported during the week ending March 16, seven of which occurred in previous weeks. The total for the 2018-2019 season so far is 76, the CDC said.
New preliminary estimates on influenza’s burden nationally put the total number of deaths at 25,000-41,500 since the beginning of the season on Oct. 1, 2018. There also have been 375,000-454,000 flu-related hospitalizations, 13.2 million to 15.4 million medical visits, and 28.5 to 32.8 million individual illnesses, the CDC said.
Since the CDC “expects flu activity to remain elevated for a number of weeks,” it continues to recommend flu vaccination and the use of influenza antiviral drugs as “an important second line of defense that can be used to treat flu illness. H3N2 viruses are typically associated with more severe illness in older adults, and flu vaccine may protect less well against H3N2 illness in older adults, making prompt treatment with flu antivirals in this age group especially important during the current period of H3N2 predominance.”
FDA approves solriamfetol for daytime sleepiness treatment
The. It is the first dopamine and norepinephrine reuptake inhibitor approved to treat those conditions.
Approval was based on results from TONES (Treatment of Obstructive Sleep Apnea and Narcolepsy Excessive Sleepiness), a phase 3 study that combined four randomized, placebo-controlled trials assessing solriamfetol at various doses, compared with a placebo. After 12 weeks, 68%-74% of patients taking solriamfetol at 75 mg and 78%-90% of those taking solriamfetol at 150 mg reported improvement as assessed by the Patient Global Impression of Change scale.
Solriamfetol is approved at 75 mg and 150 mg for patients with narcolepsy and at 37.5 mg, 75 mg, and 150 mg for patients with obstructive sleep apnea. The most common adverse events associated with solriamfetol are headache, nausea, decreased appetite, and anxiety.
“Excessive daytime sleepiness can negatively impact the daily lives of people living with narcolepsy or obstructive sleep apnea at work, at home, or in daily activities. With this approval, a new, daytime medicine that can provide sustained wakefulness throughout the day will be available for patients,” Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals, said in the press release.
Find the full press release on the Jazz Pharmaceuticals website.
The. It is the first dopamine and norepinephrine reuptake inhibitor approved to treat those conditions.
Approval was based on results from TONES (Treatment of Obstructive Sleep Apnea and Narcolepsy Excessive Sleepiness), a phase 3 study that combined four randomized, placebo-controlled trials assessing solriamfetol at various doses, compared with a placebo. After 12 weeks, 68%-74% of patients taking solriamfetol at 75 mg and 78%-90% of those taking solriamfetol at 150 mg reported improvement as assessed by the Patient Global Impression of Change scale.
Solriamfetol is approved at 75 mg and 150 mg for patients with narcolepsy and at 37.5 mg, 75 mg, and 150 mg for patients with obstructive sleep apnea. The most common adverse events associated with solriamfetol are headache, nausea, decreased appetite, and anxiety.
“Excessive daytime sleepiness can negatively impact the daily lives of people living with narcolepsy or obstructive sleep apnea at work, at home, or in daily activities. With this approval, a new, daytime medicine that can provide sustained wakefulness throughout the day will be available for patients,” Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals, said in the press release.
Find the full press release on the Jazz Pharmaceuticals website.
The. It is the first dopamine and norepinephrine reuptake inhibitor approved to treat those conditions.
Approval was based on results from TONES (Treatment of Obstructive Sleep Apnea and Narcolepsy Excessive Sleepiness), a phase 3 study that combined four randomized, placebo-controlled trials assessing solriamfetol at various doses, compared with a placebo. After 12 weeks, 68%-74% of patients taking solriamfetol at 75 mg and 78%-90% of those taking solriamfetol at 150 mg reported improvement as assessed by the Patient Global Impression of Change scale.
Solriamfetol is approved at 75 mg and 150 mg for patients with narcolepsy and at 37.5 mg, 75 mg, and 150 mg for patients with obstructive sleep apnea. The most common adverse events associated with solriamfetol are headache, nausea, decreased appetite, and anxiety.
“Excessive daytime sleepiness can negatively impact the daily lives of people living with narcolepsy or obstructive sleep apnea at work, at home, or in daily activities. With this approval, a new, daytime medicine that can provide sustained wakefulness throughout the day will be available for patients,” Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals, said in the press release.
Find the full press release on the Jazz Pharmaceuticals website.
FDA examines changing donation policies for men who have sex with men
The
At a meeting of the FDA’s Blood Products Advisory Committee, the agency shared the content of the 5-item questionnaire and reviewed the proposed study design with committee members, who were asked to comment – but not vote – on the best path forward for MSM donation policies.
The FDA is “committed to ongoing evaluation of the MSM deferral policy” and remains open to adjusting the policy based on the best available scientific evidence, said Barbee Whitaker, PhD, a lead scientist in the agency’s Office of Emerging and Transfusion Transmitted Disease
After recruiting 2,000 men who have had sex with men at least once during the past 3 months, the study will aim to identify individuals who have very recently become HIV infected, in order to assess the discriminant function of the set of behavioral questions that are proposed in the questionnaire.
The crux of the problem currently, noted Dr. Whitaker, is identifying those individuals who are very recently infected with HIV. Nucleic acid testing has tightened the window of undetectability considerably, but the current 12-month deferral window after men have had sexual contact with other men is designed to ensure safety of the blood supply.
Social justice concerns have been raised about the blanket deferral, said Dr. Whitaker; the behavioral questions in the pilot study will ask about the number of different sexual partners men have had within the past 1, 3, and 12 months and ask about the type of sexual contact (oral sex, or anal penetrative or receptive intercourse). The questionnaire also asks about sex with a partner known to be HIV positive, condom use, and use of pre-exposure prophylaxis (PrEP).
The FDA will ask for proposals to conduct the study with an eye to having sites in such cities as Washington, Atlanta, and Miami, which have high incidences of HIV, to improve chances of early detection.
The behavioral questionnaire is not seen as an immediate replacement for the 12-month deferral policy, the FDA made clear in its briefing documents and in discussion with the committee. Instead, its utility will be in the information gleaned from the pilot study and a follow-on that may include several hundred thousand individuals. These data should provide “population-based evidence upon which to base regulatory decisions to ensure blood safety,” she said.
Donation policies outside the United States
Whether a change in blood donation deferral policies for MSM would be a shortened window or a move toward a behavioral questionnaire is currently not known. Globally, a variety of practices are used for blood screening, said Mindy Goldman, MD, medical director of Canadian Blood Services, who reviewed international perspectives on blood donation for MSM.
“There’s no general consensus on donation deferrals internationally,” she said. Factors influencing policy can include epidemiology, risk analysis, modeling, and history of response to threats in the past.
However, “there’s basically a couple of main approaches” to handling deferrals for MSM, Dr. Goldman said. One is time-based deferral – the strategy used in the United States, as well as Canada, the United Kingdom, Japan, and Australia.
Japan and the U.K. have recently moved to 3-month deferral periods, a figure arrived at by doubling the window period for nucleic acid testing for HIV, roughly, Dr. Goldman said. Early data from the U.K. experience has not shown an increase in HIV rates among donors, or an increase in NAT-only positive donors, she said. An application to move from a 12-month to a 3-month deferral period is pending in Canada.
A strong advantage of time-based deferral as a risk management strategy, Dr. Goldman said, is standardization. “For us, standardization is close to godliness.”
However, she added, “another major limitation is that you’re still deferring all sexually active MSM, including those who are in a stable monogamous relationship from donating. From a justice perspective for the lowest risk population of MSM – they are still being deferred using this type of approach.”
Some nations, such as Spain and Italy, use individual risk assessment via physician-led interviews. These approaches are often not standardized. “There’s no national uniform questionnaire, so there’s less standardization, and more variability between blood centers,” Dr. Goldman said. “So you wind up trying to compare apples with oranges.”
This means the results are harder to evaluate on a national level. However, there appears to be higher residual risk, with HIV rates among first-time donors approaching those of the general population, Dr. Goldman said.
Another strategy, used in France, is a test-retest model, where blood from first-time MSM that initially tests negative for HIV is held until the individual returns for re-testing or an additional donation, with a second negative test. This approach increases operational complexity and cost, noted Dr. Goldman, and because of the short shelf life of platelets, it’s not practical for this blood component.
In general questioning and discussion after this and other background presentations, the committee could agree on one point: this isn’t an easy question.
“I’m increasingly struck by how difficult this problem is,” said committee member Roger Lewis, MD, PhD, professor at the University of California, Los Angeles, and chair of the department of emergency medicine at Harbor-UCLA Medical Center. Regarding just the problem of completing the pilot study, Dr. Lewis commented, “It sounds like it’s going to be impossible to get the data that directly answers the questions.”
Peter Marx, MD, PhD, who directs the FDA’s Center for Biologics Evaluation and Research (CBER), which oversees blood products safety, joined the discussion to acknowledge the difficulty, but underscore the social importance of a careful examination of the current MSM donation policy.
“We understand the issues here…. With all due respect to our European colleagues, there’s not enough data. That’s the point of this study; we also know that the U.S. has a very different epidemiology of HIV than the U.K. and a lot of other places,” Dr. Marx said. “The pilot study is a way to get some data where we might be able to get away from a time-based deferral. The LGBT community finds any time-based deferral discriminatory.”
Pathogen reduction technology
The committee heard a proposal for a completely different strategy during its afternoon session: pathogen reduction technology (PRT) holds promise to achieve virtual elimination of HIV and other pathogens from donated blood products.
The FDA is reviewing a variance request from the nonprofit blood donation organization Bloodworks Northwest organization to use PRT for apheresis platelet donations from MSM who would otherwise be deferred because of sexual activity within the 12-month deferral window.
James AuBuchon, MD, president of Bloodworks Northwest, explained that his organization takes in about 225,000 donations annually. The variance sought would use the FDA-approved INTERCEPT device to achieve pathogen reduction for donations that meet all requirements except the MSM deferral, and that would still undergo all relevant transfusion transmitted infection testing.
The INTERCEPT device uses amotosalen, which intercalates with DNA and RNA, inactivating it after exposure to ultraviolet A light. Amotosalen is then removed from the blood product before administration. The pathogen reduction activity doesn’t interfere with platelets or plasma, and is active against a wide range of viruses, bacteria, and fungal pathogens, explained Dr. AuBuchon, who is also a professor of hematology at the University of Washington, Seattle.
Dr. AuBuchon walked the committee through procedures designed to flag donors for PRT platelet apheresis, and to ensure these donations receive the intended PRT treatment. Platelets were chosen for this variance request, he explained, because demand outstrips supply. “We are all spending additional time and resources in recruiting a new framework and demographic, and it is exceedingly difficult to keep enough donors coming through the door,” he said. “Our platelet utilization climbs continually – it’s up 15% in the last 4 years.”
Committee members circled around the idea that all risk can’t be eliminated, even with the highly effective PRT technology. But the risk is exceedingly low, said committee chair Richard Kaufman, MD, medical director of the adult transfusion service at Brigham and Women’s Hospital, Boston. “It’s not possible to get rid of the window. We can kind of hammer down the risk by shrinking down the window by using incredibly sensitive tests. But that risk continues to exist. Pathogen reduction can take care of that residual risk…. So what’s left is really quite a low risk,” Dr. Kaufman said.
Susan Stramer, PhD, vice president of scientific affairs for the American Red Cross, concurred, noting that pathogen reduction techniques are already in use for many other blood products, particularly within the plasma industry.
Wrapping up, Dr. Kaufman asked individual committee members to summarize their position on the variance request, though the FDA had not placed a voting question before the committee. Consensus in the room was that this real-world examination of PRT could point to a path to expanding the donor pool while maintaining patient safety – a concern all agreed was paramount.
The FDA usually follows the recommendations of its committees.
The
At a meeting of the FDA’s Blood Products Advisory Committee, the agency shared the content of the 5-item questionnaire and reviewed the proposed study design with committee members, who were asked to comment – but not vote – on the best path forward for MSM donation policies.
The FDA is “committed to ongoing evaluation of the MSM deferral policy” and remains open to adjusting the policy based on the best available scientific evidence, said Barbee Whitaker, PhD, a lead scientist in the agency’s Office of Emerging and Transfusion Transmitted Disease
After recruiting 2,000 men who have had sex with men at least once during the past 3 months, the study will aim to identify individuals who have very recently become HIV infected, in order to assess the discriminant function of the set of behavioral questions that are proposed in the questionnaire.
The crux of the problem currently, noted Dr. Whitaker, is identifying those individuals who are very recently infected with HIV. Nucleic acid testing has tightened the window of undetectability considerably, but the current 12-month deferral window after men have had sexual contact with other men is designed to ensure safety of the blood supply.
Social justice concerns have been raised about the blanket deferral, said Dr. Whitaker; the behavioral questions in the pilot study will ask about the number of different sexual partners men have had within the past 1, 3, and 12 months and ask about the type of sexual contact (oral sex, or anal penetrative or receptive intercourse). The questionnaire also asks about sex with a partner known to be HIV positive, condom use, and use of pre-exposure prophylaxis (PrEP).
The FDA will ask for proposals to conduct the study with an eye to having sites in such cities as Washington, Atlanta, and Miami, which have high incidences of HIV, to improve chances of early detection.
The behavioral questionnaire is not seen as an immediate replacement for the 12-month deferral policy, the FDA made clear in its briefing documents and in discussion with the committee. Instead, its utility will be in the information gleaned from the pilot study and a follow-on that may include several hundred thousand individuals. These data should provide “population-based evidence upon which to base regulatory decisions to ensure blood safety,” she said.
Donation policies outside the United States
Whether a change in blood donation deferral policies for MSM would be a shortened window or a move toward a behavioral questionnaire is currently not known. Globally, a variety of practices are used for blood screening, said Mindy Goldman, MD, medical director of Canadian Blood Services, who reviewed international perspectives on blood donation for MSM.
“There’s no general consensus on donation deferrals internationally,” she said. Factors influencing policy can include epidemiology, risk analysis, modeling, and history of response to threats in the past.
However, “there’s basically a couple of main approaches” to handling deferrals for MSM, Dr. Goldman said. One is time-based deferral – the strategy used in the United States, as well as Canada, the United Kingdom, Japan, and Australia.
Japan and the U.K. have recently moved to 3-month deferral periods, a figure arrived at by doubling the window period for nucleic acid testing for HIV, roughly, Dr. Goldman said. Early data from the U.K. experience has not shown an increase in HIV rates among donors, or an increase in NAT-only positive donors, she said. An application to move from a 12-month to a 3-month deferral period is pending in Canada.
A strong advantage of time-based deferral as a risk management strategy, Dr. Goldman said, is standardization. “For us, standardization is close to godliness.”
However, she added, “another major limitation is that you’re still deferring all sexually active MSM, including those who are in a stable monogamous relationship from donating. From a justice perspective for the lowest risk population of MSM – they are still being deferred using this type of approach.”
Some nations, such as Spain and Italy, use individual risk assessment via physician-led interviews. These approaches are often not standardized. “There’s no national uniform questionnaire, so there’s less standardization, and more variability between blood centers,” Dr. Goldman said. “So you wind up trying to compare apples with oranges.”
This means the results are harder to evaluate on a national level. However, there appears to be higher residual risk, with HIV rates among first-time donors approaching those of the general population, Dr. Goldman said.
Another strategy, used in France, is a test-retest model, where blood from first-time MSM that initially tests negative for HIV is held until the individual returns for re-testing or an additional donation, with a second negative test. This approach increases operational complexity and cost, noted Dr. Goldman, and because of the short shelf life of platelets, it’s not practical for this blood component.
In general questioning and discussion after this and other background presentations, the committee could agree on one point: this isn’t an easy question.
“I’m increasingly struck by how difficult this problem is,” said committee member Roger Lewis, MD, PhD, professor at the University of California, Los Angeles, and chair of the department of emergency medicine at Harbor-UCLA Medical Center. Regarding just the problem of completing the pilot study, Dr. Lewis commented, “It sounds like it’s going to be impossible to get the data that directly answers the questions.”
Peter Marx, MD, PhD, who directs the FDA’s Center for Biologics Evaluation and Research (CBER), which oversees blood products safety, joined the discussion to acknowledge the difficulty, but underscore the social importance of a careful examination of the current MSM donation policy.
“We understand the issues here…. With all due respect to our European colleagues, there’s not enough data. That’s the point of this study; we also know that the U.S. has a very different epidemiology of HIV than the U.K. and a lot of other places,” Dr. Marx said. “The pilot study is a way to get some data where we might be able to get away from a time-based deferral. The LGBT community finds any time-based deferral discriminatory.”
Pathogen reduction technology
The committee heard a proposal for a completely different strategy during its afternoon session: pathogen reduction technology (PRT) holds promise to achieve virtual elimination of HIV and other pathogens from donated blood products.
The FDA is reviewing a variance request from the nonprofit blood donation organization Bloodworks Northwest organization to use PRT for apheresis platelet donations from MSM who would otherwise be deferred because of sexual activity within the 12-month deferral window.
James AuBuchon, MD, president of Bloodworks Northwest, explained that his organization takes in about 225,000 donations annually. The variance sought would use the FDA-approved INTERCEPT device to achieve pathogen reduction for donations that meet all requirements except the MSM deferral, and that would still undergo all relevant transfusion transmitted infection testing.
The INTERCEPT device uses amotosalen, which intercalates with DNA and RNA, inactivating it after exposure to ultraviolet A light. Amotosalen is then removed from the blood product before administration. The pathogen reduction activity doesn’t interfere with platelets or plasma, and is active against a wide range of viruses, bacteria, and fungal pathogens, explained Dr. AuBuchon, who is also a professor of hematology at the University of Washington, Seattle.
Dr. AuBuchon walked the committee through procedures designed to flag donors for PRT platelet apheresis, and to ensure these donations receive the intended PRT treatment. Platelets were chosen for this variance request, he explained, because demand outstrips supply. “We are all spending additional time and resources in recruiting a new framework and demographic, and it is exceedingly difficult to keep enough donors coming through the door,” he said. “Our platelet utilization climbs continually – it’s up 15% in the last 4 years.”
Committee members circled around the idea that all risk can’t be eliminated, even with the highly effective PRT technology. But the risk is exceedingly low, said committee chair Richard Kaufman, MD, medical director of the adult transfusion service at Brigham and Women’s Hospital, Boston. “It’s not possible to get rid of the window. We can kind of hammer down the risk by shrinking down the window by using incredibly sensitive tests. But that risk continues to exist. Pathogen reduction can take care of that residual risk…. So what’s left is really quite a low risk,” Dr. Kaufman said.
Susan Stramer, PhD, vice president of scientific affairs for the American Red Cross, concurred, noting that pathogen reduction techniques are already in use for many other blood products, particularly within the plasma industry.
Wrapping up, Dr. Kaufman asked individual committee members to summarize their position on the variance request, though the FDA had not placed a voting question before the committee. Consensus in the room was that this real-world examination of PRT could point to a path to expanding the donor pool while maintaining patient safety – a concern all agreed was paramount.
The FDA usually follows the recommendations of its committees.
The
At a meeting of the FDA’s Blood Products Advisory Committee, the agency shared the content of the 5-item questionnaire and reviewed the proposed study design with committee members, who were asked to comment – but not vote – on the best path forward for MSM donation policies.
The FDA is “committed to ongoing evaluation of the MSM deferral policy” and remains open to adjusting the policy based on the best available scientific evidence, said Barbee Whitaker, PhD, a lead scientist in the agency’s Office of Emerging and Transfusion Transmitted Disease
After recruiting 2,000 men who have had sex with men at least once during the past 3 months, the study will aim to identify individuals who have very recently become HIV infected, in order to assess the discriminant function of the set of behavioral questions that are proposed in the questionnaire.
The crux of the problem currently, noted Dr. Whitaker, is identifying those individuals who are very recently infected with HIV. Nucleic acid testing has tightened the window of undetectability considerably, but the current 12-month deferral window after men have had sexual contact with other men is designed to ensure safety of the blood supply.
Social justice concerns have been raised about the blanket deferral, said Dr. Whitaker; the behavioral questions in the pilot study will ask about the number of different sexual partners men have had within the past 1, 3, and 12 months and ask about the type of sexual contact (oral sex, or anal penetrative or receptive intercourse). The questionnaire also asks about sex with a partner known to be HIV positive, condom use, and use of pre-exposure prophylaxis (PrEP).
The FDA will ask for proposals to conduct the study with an eye to having sites in such cities as Washington, Atlanta, and Miami, which have high incidences of HIV, to improve chances of early detection.
The behavioral questionnaire is not seen as an immediate replacement for the 12-month deferral policy, the FDA made clear in its briefing documents and in discussion with the committee. Instead, its utility will be in the information gleaned from the pilot study and a follow-on that may include several hundred thousand individuals. These data should provide “population-based evidence upon which to base regulatory decisions to ensure blood safety,” she said.
Donation policies outside the United States
Whether a change in blood donation deferral policies for MSM would be a shortened window or a move toward a behavioral questionnaire is currently not known. Globally, a variety of practices are used for blood screening, said Mindy Goldman, MD, medical director of Canadian Blood Services, who reviewed international perspectives on blood donation for MSM.
“There’s no general consensus on donation deferrals internationally,” she said. Factors influencing policy can include epidemiology, risk analysis, modeling, and history of response to threats in the past.
However, “there’s basically a couple of main approaches” to handling deferrals for MSM, Dr. Goldman said. One is time-based deferral – the strategy used in the United States, as well as Canada, the United Kingdom, Japan, and Australia.
Japan and the U.K. have recently moved to 3-month deferral periods, a figure arrived at by doubling the window period for nucleic acid testing for HIV, roughly, Dr. Goldman said. Early data from the U.K. experience has not shown an increase in HIV rates among donors, or an increase in NAT-only positive donors, she said. An application to move from a 12-month to a 3-month deferral period is pending in Canada.
A strong advantage of time-based deferral as a risk management strategy, Dr. Goldman said, is standardization. “For us, standardization is close to godliness.”
However, she added, “another major limitation is that you’re still deferring all sexually active MSM, including those who are in a stable monogamous relationship from donating. From a justice perspective for the lowest risk population of MSM – they are still being deferred using this type of approach.”
Some nations, such as Spain and Italy, use individual risk assessment via physician-led interviews. These approaches are often not standardized. “There’s no national uniform questionnaire, so there’s less standardization, and more variability between blood centers,” Dr. Goldman said. “So you wind up trying to compare apples with oranges.”
This means the results are harder to evaluate on a national level. However, there appears to be higher residual risk, with HIV rates among first-time donors approaching those of the general population, Dr. Goldman said.
Another strategy, used in France, is a test-retest model, where blood from first-time MSM that initially tests negative for HIV is held until the individual returns for re-testing or an additional donation, with a second negative test. This approach increases operational complexity and cost, noted Dr. Goldman, and because of the short shelf life of platelets, it’s not practical for this blood component.
In general questioning and discussion after this and other background presentations, the committee could agree on one point: this isn’t an easy question.
“I’m increasingly struck by how difficult this problem is,” said committee member Roger Lewis, MD, PhD, professor at the University of California, Los Angeles, and chair of the department of emergency medicine at Harbor-UCLA Medical Center. Regarding just the problem of completing the pilot study, Dr. Lewis commented, “It sounds like it’s going to be impossible to get the data that directly answers the questions.”
Peter Marx, MD, PhD, who directs the FDA’s Center for Biologics Evaluation and Research (CBER), which oversees blood products safety, joined the discussion to acknowledge the difficulty, but underscore the social importance of a careful examination of the current MSM donation policy.
“We understand the issues here…. With all due respect to our European colleagues, there’s not enough data. That’s the point of this study; we also know that the U.S. has a very different epidemiology of HIV than the U.K. and a lot of other places,” Dr. Marx said. “The pilot study is a way to get some data where we might be able to get away from a time-based deferral. The LGBT community finds any time-based deferral discriminatory.”
Pathogen reduction technology
The committee heard a proposal for a completely different strategy during its afternoon session: pathogen reduction technology (PRT) holds promise to achieve virtual elimination of HIV and other pathogens from donated blood products.
The FDA is reviewing a variance request from the nonprofit blood donation organization Bloodworks Northwest organization to use PRT for apheresis platelet donations from MSM who would otherwise be deferred because of sexual activity within the 12-month deferral window.
James AuBuchon, MD, president of Bloodworks Northwest, explained that his organization takes in about 225,000 donations annually. The variance sought would use the FDA-approved INTERCEPT device to achieve pathogen reduction for donations that meet all requirements except the MSM deferral, and that would still undergo all relevant transfusion transmitted infection testing.
The INTERCEPT device uses amotosalen, which intercalates with DNA and RNA, inactivating it after exposure to ultraviolet A light. Amotosalen is then removed from the blood product before administration. The pathogen reduction activity doesn’t interfere with platelets or plasma, and is active against a wide range of viruses, bacteria, and fungal pathogens, explained Dr. AuBuchon, who is also a professor of hematology at the University of Washington, Seattle.
Dr. AuBuchon walked the committee through procedures designed to flag donors for PRT platelet apheresis, and to ensure these donations receive the intended PRT treatment. Platelets were chosen for this variance request, he explained, because demand outstrips supply. “We are all spending additional time and resources in recruiting a new framework and demographic, and it is exceedingly difficult to keep enough donors coming through the door,” he said. “Our platelet utilization climbs continually – it’s up 15% in the last 4 years.”
Committee members circled around the idea that all risk can’t be eliminated, even with the highly effective PRT technology. But the risk is exceedingly low, said committee chair Richard Kaufman, MD, medical director of the adult transfusion service at Brigham and Women’s Hospital, Boston. “It’s not possible to get rid of the window. We can kind of hammer down the risk by shrinking down the window by using incredibly sensitive tests. But that risk continues to exist. Pathogen reduction can take care of that residual risk…. So what’s left is really quite a low risk,” Dr. Kaufman said.
Susan Stramer, PhD, vice president of scientific affairs for the American Red Cross, concurred, noting that pathogen reduction techniques are already in use for many other blood products, particularly within the plasma industry.
Wrapping up, Dr. Kaufman asked individual committee members to summarize their position on the variance request, though the FDA had not placed a voting question before the committee. Consensus in the room was that this real-world examination of PRT could point to a path to expanding the donor pool while maintaining patient safety – a concern all agreed was paramount.
The FDA usually follows the recommendations of its committees.
FROM AN FDA ADVISORY COMMITTEE MEETING
Measles cases confirmed in three more states
Forty more cases and three more states were added to the measles count in the last week, bringing the U.S. total to 268 cases in 15 states so far in 2019, according to the Centers for Disease Control and Prevention.
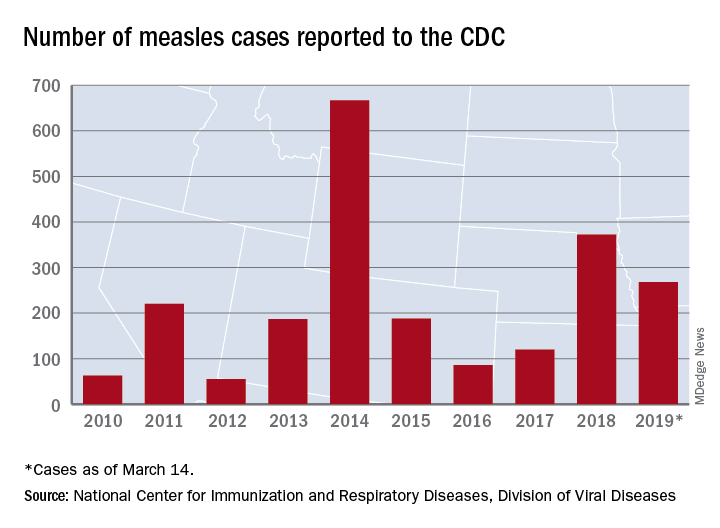
Arizona, Michigan, and Missouri reported their first confirmed cases of the year, joining California (one outbreak), Colorado, Connecticut, Georgia, Illinois (one outbreak), Kentucky, New Hampshire, New Jersey, New York (two outbreaks), Oregon, Texas (one outbreak), and Washington (one outbreak), the CDC reported March 18.
Brooklyn, N.Y., has become the epicenter of measles activity since mid-February, and with 25 of the 40 new cases occurring there, the borough has now led the nation for four consecutive weeks. There have been 157 confirmed cases in Brooklyn and one in Queens since the outbreak began in October of 2018, the New York City Department of Health and Mental Hygiene reported.
Michigan’s first case of measles is travel related and involved an individual visiting from Israel following a stay in New York, the Michigan Department of Health and Human Services and Oakland County Health Division announced March 13.
In Arizona, the state department of health services and the Pima County Public Health Department announced that a 12-month-old infant from Pima County has been diagnosed with measles after traveling to Asia.
A single case of measles, contracted while the person was traveling out of state, has been reported in Jefferson County, Mo., and is being managed by the county health department, according to St. Louis Public Radio.
Forty more cases and three more states were added to the measles count in the last week, bringing the U.S. total to 268 cases in 15 states so far in 2019, according to the Centers for Disease Control and Prevention.
Arizona, Michigan, and Missouri reported their first confirmed cases of the year, joining California (one outbreak), Colorado, Connecticut, Georgia, Illinois (one outbreak), Kentucky, New Hampshire, New Jersey, New York (two outbreaks), Oregon, Texas (one outbreak), and Washington (one outbreak), the CDC reported March 18.
Brooklyn, N.Y., has become the epicenter of measles activity since mid-February, and with 25 of the 40 new cases occurring there, the borough has now led the nation for four consecutive weeks. There have been 157 confirmed cases in Brooklyn and one in Queens since the outbreak began in October of 2018, the New York City Department of Health and Mental Hygiene reported.
Michigan’s first case of measles is travel related and involved an individual visiting from Israel following a stay in New York, the Michigan Department of Health and Human Services and Oakland County Health Division announced March 13.
In Arizona, the state department of health services and the Pima County Public Health Department announced that a 12-month-old infant from Pima County has been diagnosed with measles after traveling to Asia.
A single case of measles, contracted while the person was traveling out of state, has been reported in Jefferson County, Mo., and is being managed by the county health department, according to St. Louis Public Radio.
Forty more cases and three more states were added to the measles count in the last week, bringing the U.S. total to 268 cases in 15 states so far in 2019, according to the Centers for Disease Control and Prevention.
Arizona, Michigan, and Missouri reported their first confirmed cases of the year, joining California (one outbreak), Colorado, Connecticut, Georgia, Illinois (one outbreak), Kentucky, New Hampshire, New Jersey, New York (two outbreaks), Oregon, Texas (one outbreak), and Washington (one outbreak), the CDC reported March 18.
Brooklyn, N.Y., has become the epicenter of measles activity since mid-February, and with 25 of the 40 new cases occurring there, the borough has now led the nation for four consecutive weeks. There have been 157 confirmed cases in Brooklyn and one in Queens since the outbreak began in October of 2018, the New York City Department of Health and Mental Hygiene reported.
Michigan’s first case of measles is travel related and involved an individual visiting from Israel following a stay in New York, the Michigan Department of Health and Human Services and Oakland County Health Division announced March 13.
In Arizona, the state department of health services and the Pima County Public Health Department announced that a 12-month-old infant from Pima County has been diagnosed with measles after traveling to Asia.
A single case of measles, contracted while the person was traveling out of state, has been reported in Jefferson County, Mo., and is being managed by the county health department, according to St. Louis Public Radio.