User login
Aspirin plus a DOAC may do more harm than good in some
ORLANDO – in a large registry-based cohort.

The study, which involved a cohort of 2,045 patients who were followed at 6 anticoagulation clinics in Michigan during January 2009–June 2019, also found no apparent improvement in thrombosis incidence with the addition of aspirin, Jordan K. Schaefer, MD, reported during a press briefing at the annual meeting of the American Society of Hematology.
Of the cohort patients, 639 adults who received a DOAC plus aspirin after VTE or for NVAF without a clear indication were compared with 639 propensity-matched controls. The bleeding event rate per 100 patient years was 39.50 vs. 32.32 at an average of 15.2 months of follow-up in the combination therapy and DOAC monotherapy groups, respectively, said Dr. Schaefer of the division of hematology/oncology, department of internal medicine, University of Michigan, Ann Arbor.
“This result was statistically significant for clinically relevant non-major bleeding, with an 18.7 rate per 100 patient years, compared with 13.5 for DOAC monotherapy,” (P = .02), he said. “We also saw a significant increase in non-major bleeding with combination therapy, compared with direct oral anticoagulant monotherapy” (rate, 32.82 vs. 25.88; P =.04).
No significant difference was seen overall (P =.07) or for other specific types of bleeding, he noted.
The observed rates of thrombosis in the groups, respectively, were 2.35 and 2.23 per 100 patient years (P =.95), he said, noting that patients on combination therapy also had more emergency department visits and hospitalizations, but those differences were not statistically significant.
“Direct-acting oral anticoagulants, which include apixaban, dabigatran, edoxaban, and rivaroxaban, are increasingly used in clinical practice for indications that include the prevention of strokes for patients with nonvalvular atrial fibrillation, and the treatment and secondary prevention of venous thromboembolic disease,” Dr. Schaefer said.
Aspirin is commonly used in clinical practice for various indications, including primary prevention of heart attacks, strokes, and colorectal cancer, as well as for thromboprophylaxis in patients with certain blood disorders or with certain cardiac devices, he added.
“Aspirin is used for the secondary prevention of thrombosis for patients with known coronary artery disease, peripheral artery disease, or carotid artery disease,” he said. “And while adding aspirin to a DOAC is often appropriate after acute coronary syndromes or percutaneous coronary intervention, many patients receive the combination therapy without a clear indication, he said, noting that increasing evidence in recent years, largely from patients treated with warfarin and aspirin, suggest that the approach may do more harm than good for certain patients.
Specifically, there’s a question of whether aspirin is increasing the rates of bleeding without protecting patients from adverse thrombotic outcomes.
“This has specifically been a concern for patients who are on full-dose anticoagulation,” he said.
In the current study, patient demographics, comorbidities, and concurrent medications were well balanced in the treatment and control groups after propensity score matching, he said, noting that patients with a history of heart valve replacement, recent MI, or less than 3 months of follow-up were excluded.
“These findings need to be confirmed in larger studies, but until such data [are] available, clinicians and patients should continue to balance the relative risks and benefits of adding aspirin to their direct oral anticoagulant therapy,” Dr. Schaefer said. “Further research needs to evaluate key subgroups to see if any particular population may benefit from combination therapy compared to DOAC therapy alone.”
Dr. Schaefer reported having no disclosures.
SOURCE: Schaeffer J et al. ASH 2019. Abstract 787.
ORLANDO – in a large registry-based cohort.
The study, which involved a cohort of 2,045 patients who were followed at 6 anticoagulation clinics in Michigan during January 2009–June 2019, also found no apparent improvement in thrombosis incidence with the addition of aspirin, Jordan K. Schaefer, MD, reported during a press briefing at the annual meeting of the American Society of Hematology.
Of the cohort patients, 639 adults who received a DOAC plus aspirin after VTE or for NVAF without a clear indication were compared with 639 propensity-matched controls. The bleeding event rate per 100 patient years was 39.50 vs. 32.32 at an average of 15.2 months of follow-up in the combination therapy and DOAC monotherapy groups, respectively, said Dr. Schaefer of the division of hematology/oncology, department of internal medicine, University of Michigan, Ann Arbor.
“This result was statistically significant for clinically relevant non-major bleeding, with an 18.7 rate per 100 patient years, compared with 13.5 for DOAC monotherapy,” (P = .02), he said. “We also saw a significant increase in non-major bleeding with combination therapy, compared with direct oral anticoagulant monotherapy” (rate, 32.82 vs. 25.88; P =.04).
No significant difference was seen overall (P =.07) or for other specific types of bleeding, he noted.
The observed rates of thrombosis in the groups, respectively, were 2.35 and 2.23 per 100 patient years (P =.95), he said, noting that patients on combination therapy also had more emergency department visits and hospitalizations, but those differences were not statistically significant.
“Direct-acting oral anticoagulants, which include apixaban, dabigatran, edoxaban, and rivaroxaban, are increasingly used in clinical practice for indications that include the prevention of strokes for patients with nonvalvular atrial fibrillation, and the treatment and secondary prevention of venous thromboembolic disease,” Dr. Schaefer said.
Aspirin is commonly used in clinical practice for various indications, including primary prevention of heart attacks, strokes, and colorectal cancer, as well as for thromboprophylaxis in patients with certain blood disorders or with certain cardiac devices, he added.
“Aspirin is used for the secondary prevention of thrombosis for patients with known coronary artery disease, peripheral artery disease, or carotid artery disease,” he said. “And while adding aspirin to a DOAC is often appropriate after acute coronary syndromes or percutaneous coronary intervention, many patients receive the combination therapy without a clear indication, he said, noting that increasing evidence in recent years, largely from patients treated with warfarin and aspirin, suggest that the approach may do more harm than good for certain patients.
Specifically, there’s a question of whether aspirin is increasing the rates of bleeding without protecting patients from adverse thrombotic outcomes.
“This has specifically been a concern for patients who are on full-dose anticoagulation,” he said.
In the current study, patient demographics, comorbidities, and concurrent medications were well balanced in the treatment and control groups after propensity score matching, he said, noting that patients with a history of heart valve replacement, recent MI, or less than 3 months of follow-up were excluded.
“These findings need to be confirmed in larger studies, but until such data [are] available, clinicians and patients should continue to balance the relative risks and benefits of adding aspirin to their direct oral anticoagulant therapy,” Dr. Schaefer said. “Further research needs to evaluate key subgroups to see if any particular population may benefit from combination therapy compared to DOAC therapy alone.”
Dr. Schaefer reported having no disclosures.
SOURCE: Schaeffer J et al. ASH 2019. Abstract 787.
ORLANDO – in a large registry-based cohort.
The study, which involved a cohort of 2,045 patients who were followed at 6 anticoagulation clinics in Michigan during January 2009–June 2019, also found no apparent improvement in thrombosis incidence with the addition of aspirin, Jordan K. Schaefer, MD, reported during a press briefing at the annual meeting of the American Society of Hematology.
Of the cohort patients, 639 adults who received a DOAC plus aspirin after VTE or for NVAF without a clear indication were compared with 639 propensity-matched controls. The bleeding event rate per 100 patient years was 39.50 vs. 32.32 at an average of 15.2 months of follow-up in the combination therapy and DOAC monotherapy groups, respectively, said Dr. Schaefer of the division of hematology/oncology, department of internal medicine, University of Michigan, Ann Arbor.
“This result was statistically significant for clinically relevant non-major bleeding, with an 18.7 rate per 100 patient years, compared with 13.5 for DOAC monotherapy,” (P = .02), he said. “We also saw a significant increase in non-major bleeding with combination therapy, compared with direct oral anticoagulant monotherapy” (rate, 32.82 vs. 25.88; P =.04).
No significant difference was seen overall (P =.07) or for other specific types of bleeding, he noted.
The observed rates of thrombosis in the groups, respectively, were 2.35 and 2.23 per 100 patient years (P =.95), he said, noting that patients on combination therapy also had more emergency department visits and hospitalizations, but those differences were not statistically significant.
“Direct-acting oral anticoagulants, which include apixaban, dabigatran, edoxaban, and rivaroxaban, are increasingly used in clinical practice for indications that include the prevention of strokes for patients with nonvalvular atrial fibrillation, and the treatment and secondary prevention of venous thromboembolic disease,” Dr. Schaefer said.
Aspirin is commonly used in clinical practice for various indications, including primary prevention of heart attacks, strokes, and colorectal cancer, as well as for thromboprophylaxis in patients with certain blood disorders or with certain cardiac devices, he added.
“Aspirin is used for the secondary prevention of thrombosis for patients with known coronary artery disease, peripheral artery disease, or carotid artery disease,” he said. “And while adding aspirin to a DOAC is often appropriate after acute coronary syndromes or percutaneous coronary intervention, many patients receive the combination therapy without a clear indication, he said, noting that increasing evidence in recent years, largely from patients treated with warfarin and aspirin, suggest that the approach may do more harm than good for certain patients.
Specifically, there’s a question of whether aspirin is increasing the rates of bleeding without protecting patients from adverse thrombotic outcomes.
“This has specifically been a concern for patients who are on full-dose anticoagulation,” he said.
In the current study, patient demographics, comorbidities, and concurrent medications were well balanced in the treatment and control groups after propensity score matching, he said, noting that patients with a history of heart valve replacement, recent MI, or less than 3 months of follow-up were excluded.
“These findings need to be confirmed in larger studies, but until such data [are] available, clinicians and patients should continue to balance the relative risks and benefits of adding aspirin to their direct oral anticoagulant therapy,” Dr. Schaefer said. “Further research needs to evaluate key subgroups to see if any particular population may benefit from combination therapy compared to DOAC therapy alone.”
Dr. Schaefer reported having no disclosures.
SOURCE: Schaeffer J et al. ASH 2019. Abstract 787.
REPORTING FROM ASH 2019
Bispecific CAR T-cells yield high response rate in relapsed/refractory myeloma
ORLANDO – A dual-targeted chimeric antigen receptor (CAR) T-cell therapy has demonstrated a high overall response rate, a long response duration, and manageable safety in patients with relapsed or refractory multiple myeloma, according to an investigator in a phase 1 study.

The overall response rate exceeded 90%, and about three-quarters of patients remained progression-free at 9 months after treatment with the CAR T-cell therapy, which targets both B-cell maturation antigen (BCMA) and CD38, the study investigator reported.
Grade 3 or greater cytokine release syndrome (CRS) occurred in about one-quarter of the patients, and no neurotoxicity was observed, according to investigator Yu Hu, MD, of Tongji Medical College in Hubei, China.
“,” Dr. Hu said in a press conference.
Short-term relapse has been a “major challenge” with current CAR T-cell therapies currently under investigation for myeloma, most of which target BCMA, according to Dr. Hu.
He said the bispecific CAR T-cell therapy under investigation, known as BM38, was designed to target antigen loss and increase persistence of effector cells. According to the investigator, this was the first study to focus on an anti-BCMA and CD38 dual-targeted CAR T-cell therapy for patients with relapsed or refractory multiple myeloma.
Gary J. Schiller, MD, of UCLA Health, who moderated the press conference, said that while dual-targeting is a potentially “attractive” approach in these hard-to-treat patients, further follow-up is needed to see duration of response and to see if antigen escape re-emerges.
“Cellular therapy is costly, in terms of toxicity as well as financial costs, so you would like to see what the durability of responses is before engaging in that as a late-stage therapy, not to mention moving it up front,” Dr. Schiller said in an interview.
The median progression-free survival (PFS) duration had not been reached at the time of this report, though the 9-month PFS rate was 78.87%, according to the data presented by Dr. Hu.
In the phase 1 study, 22 patients received BM38 CAR T-cell infusions following a fludarabine and cyclophosphamide preconditioning regimen. The median patient age was 59 years, and 50% were male. Nearly three-quarters (72%) had a cytogenetic abnormality, and the median number of prior therapies approached four (range, two to nine prior therapies).
Twenty of the patients (90.9%) had a response: 12 who achieved stringent complete remission, 2 with very good partial response, 5 with partial responses, and 1 with a minimal response.
Of 9 patients with extramedullary disease, 8 achieved partial or complete elimination of tumors, Dr. Hu said in his presentation.
Cytokine release syndrome occurred in 20 patients (90.91%), 5 of whom experienced severe cases (22.73%), according to the reported data. There was no observed neurotoxicity, according to the report, while almost all had hematologic toxicities. Three experienced hepatotoxicity and one had nephrotoxicity, according to Dr. Hu.
The phase 1 study was supported by the National Natural Science Foundation of China, the Major Technological Innovation Special Project Fund of Hubei Province of China, and Cellyan Therapeutics. The senior author of the study was affiliated with Cellyan Therapeutics. Dr. Hu and coauthors reported that they had no relevant conflicts of interest to declare.
SOURCE: Li C et al. ASH 2019. Abstract 930.
ORLANDO – A dual-targeted chimeric antigen receptor (CAR) T-cell therapy has demonstrated a high overall response rate, a long response duration, and manageable safety in patients with relapsed or refractory multiple myeloma, according to an investigator in a phase 1 study.
The overall response rate exceeded 90%, and about three-quarters of patients remained progression-free at 9 months after treatment with the CAR T-cell therapy, which targets both B-cell maturation antigen (BCMA) and CD38, the study investigator reported.
Grade 3 or greater cytokine release syndrome (CRS) occurred in about one-quarter of the patients, and no neurotoxicity was observed, according to investigator Yu Hu, MD, of Tongji Medical College in Hubei, China.
“,” Dr. Hu said in a press conference.
Short-term relapse has been a “major challenge” with current CAR T-cell therapies currently under investigation for myeloma, most of which target BCMA, according to Dr. Hu.
He said the bispecific CAR T-cell therapy under investigation, known as BM38, was designed to target antigen loss and increase persistence of effector cells. According to the investigator, this was the first study to focus on an anti-BCMA and CD38 dual-targeted CAR T-cell therapy for patients with relapsed or refractory multiple myeloma.
Gary J. Schiller, MD, of UCLA Health, who moderated the press conference, said that while dual-targeting is a potentially “attractive” approach in these hard-to-treat patients, further follow-up is needed to see duration of response and to see if antigen escape re-emerges.
“Cellular therapy is costly, in terms of toxicity as well as financial costs, so you would like to see what the durability of responses is before engaging in that as a late-stage therapy, not to mention moving it up front,” Dr. Schiller said in an interview.
The median progression-free survival (PFS) duration had not been reached at the time of this report, though the 9-month PFS rate was 78.87%, according to the data presented by Dr. Hu.
In the phase 1 study, 22 patients received BM38 CAR T-cell infusions following a fludarabine and cyclophosphamide preconditioning regimen. The median patient age was 59 years, and 50% were male. Nearly three-quarters (72%) had a cytogenetic abnormality, and the median number of prior therapies approached four (range, two to nine prior therapies).
Twenty of the patients (90.9%) had a response: 12 who achieved stringent complete remission, 2 with very good partial response, 5 with partial responses, and 1 with a minimal response.
Of 9 patients with extramedullary disease, 8 achieved partial or complete elimination of tumors, Dr. Hu said in his presentation.
Cytokine release syndrome occurred in 20 patients (90.91%), 5 of whom experienced severe cases (22.73%), according to the reported data. There was no observed neurotoxicity, according to the report, while almost all had hematologic toxicities. Three experienced hepatotoxicity and one had nephrotoxicity, according to Dr. Hu.
The phase 1 study was supported by the National Natural Science Foundation of China, the Major Technological Innovation Special Project Fund of Hubei Province of China, and Cellyan Therapeutics. The senior author of the study was affiliated with Cellyan Therapeutics. Dr. Hu and coauthors reported that they had no relevant conflicts of interest to declare.
SOURCE: Li C et al. ASH 2019. Abstract 930.
ORLANDO – A dual-targeted chimeric antigen receptor (CAR) T-cell therapy has demonstrated a high overall response rate, a long response duration, and manageable safety in patients with relapsed or refractory multiple myeloma, according to an investigator in a phase 1 study.
The overall response rate exceeded 90%, and about three-quarters of patients remained progression-free at 9 months after treatment with the CAR T-cell therapy, which targets both B-cell maturation antigen (BCMA) and CD38, the study investigator reported.
Grade 3 or greater cytokine release syndrome (CRS) occurred in about one-quarter of the patients, and no neurotoxicity was observed, according to investigator Yu Hu, MD, of Tongji Medical College in Hubei, China.
“,” Dr. Hu said in a press conference.
Short-term relapse has been a “major challenge” with current CAR T-cell therapies currently under investigation for myeloma, most of which target BCMA, according to Dr. Hu.
He said the bispecific CAR T-cell therapy under investigation, known as BM38, was designed to target antigen loss and increase persistence of effector cells. According to the investigator, this was the first study to focus on an anti-BCMA and CD38 dual-targeted CAR T-cell therapy for patients with relapsed or refractory multiple myeloma.
Gary J. Schiller, MD, of UCLA Health, who moderated the press conference, said that while dual-targeting is a potentially “attractive” approach in these hard-to-treat patients, further follow-up is needed to see duration of response and to see if antigen escape re-emerges.
“Cellular therapy is costly, in terms of toxicity as well as financial costs, so you would like to see what the durability of responses is before engaging in that as a late-stage therapy, not to mention moving it up front,” Dr. Schiller said in an interview.
The median progression-free survival (PFS) duration had not been reached at the time of this report, though the 9-month PFS rate was 78.87%, according to the data presented by Dr. Hu.
In the phase 1 study, 22 patients received BM38 CAR T-cell infusions following a fludarabine and cyclophosphamide preconditioning regimen. The median patient age was 59 years, and 50% were male. Nearly three-quarters (72%) had a cytogenetic abnormality, and the median number of prior therapies approached four (range, two to nine prior therapies).
Twenty of the patients (90.9%) had a response: 12 who achieved stringent complete remission, 2 with very good partial response, 5 with partial responses, and 1 with a minimal response.
Of 9 patients with extramedullary disease, 8 achieved partial or complete elimination of tumors, Dr. Hu said in his presentation.
Cytokine release syndrome occurred in 20 patients (90.91%), 5 of whom experienced severe cases (22.73%), according to the reported data. There was no observed neurotoxicity, according to the report, while almost all had hematologic toxicities. Three experienced hepatotoxicity and one had nephrotoxicity, according to Dr. Hu.
The phase 1 study was supported by the National Natural Science Foundation of China, the Major Technological Innovation Special Project Fund of Hubei Province of China, and Cellyan Therapeutics. The senior author of the study was affiliated with Cellyan Therapeutics. Dr. Hu and coauthors reported that they had no relevant conflicts of interest to declare.
SOURCE: Li C et al. ASH 2019. Abstract 930.
REPORTING FROM ASH 2019
High complete response rate seen with novel CAR-T for myeloma
ORLANDO – A novel chimeric antigen receptor T (CAR T) cell construct is associated with deep clinical responses in patients with multiple myeloma for whom prior lines of therapy – some numbering in the double digits – have failed.
Among 29 patients with multiple myeloma enrolled in a phase 1b/2 trial of JNJ-4528, the overall response rate (ORR) at 6 months median follow-up was 100%, including 69% complete responses, with 27 patients remaining free of disease progression at a median of 6 months, reported Deepu Madduri, MD, of Icahn School of Medicine at Mount Sinai, New York.

“These are very heavily pretreated patients, and so getting early and deep responses is quite amazing,” she said at a briefing prior to presentation of the data at the annual meeting of the American Society of Hematology.
JNJ-4528 is a second-generation CAR T containing two single-domain antibodies targeted against B-cell maturation protein (BCMA). As previously reported, an identical CAR T cell construct showed a high overall response with manageable toxicities in 74 patients with relapsed/refractory multiple myeloma. JNJ-4528 was granted a breakthrough therapy designation for relapsed/refractory multiple myeloma by the Food and Drug Administration on Dec. 6, 2019, and a priority medicines (PRIME) designation by the European Medicines Agency in April 2019.
BCMA was first described in myeloma in 2004 as a mechanism for the growth and survival of malignant plasma cells. Several research groups are currently investigating CAR T cells or monoclonal antibodies targeted to BCMA. The product closest to receiving FDA approval is likely BB2121.
At ASH 2019, Dr. Madduri presented results from the phase 1b portion of the CARTITUDE-1 trial. The investigators enrolled patients with multiple myeloma with measurable diseases as assessed by M-protein or serum free light chain levels who had experienced disease progression on at least 3 prior lines of therapy, or whose disease was refractory to at least two lines of therapy with a proteasome inhibitor, immunomodulatory drug (IMiD), and an anti-CD38 antibody.
Patients underwent apheresis for T-cell collection, with bridging therapy allowed until the expanded T cells could be delivered.
Following T-cell depletion with cyclophosphamide 300 mg/m2 and fludarabine 30 mg/m2 over 3 days, patients received a single weight-based infusion (compared with fixed-dose infusions used with other CAR T cell constructs).
The dose was targeted at 0.75x106 CAR-positive cells/kg, with a target range of 0.5–1.0x106, administered 5-7 days after the start of the conditioning regimen.
A total of 29 patients, median age 60, were evaluable for the safety and efficacy endpoints. One-fourth of the patients had a high-risk cytogenetic profile. The patients had received a median of 5 prior lines of therapy, with one patient receiving 18 prior lines. Of the 29 patients, 25 (86%) had previously undergone autologous transplantation.
As noted before, the ORR after a median follow-up of 6 months was 100%, with 69% completer responses, 17% very good partial responses, and 14% partial responses. The median time to complete response was 1 month (range 1 to 9 months). All but two patients remained free of disease progression at the median 6-month follow-up.
Nearly all patients (27) developed cytokine release syndrome (CRS), and one patient with prolonged grade 4 CRS died from related complications 99 days after infusion.
The median time to onset of CRS was 7 days with more than 90% of cases occurring between days 5 and 9.
Neurotoxicities, specifically immune effector cell–associated neurotoxicity syndrome (ICANS), were infrequent in CRS, and when they did occur were generally low grade, with only 1 grade 3 ICANS event.
Asked in an interview whether the impressive response rates seen with JNJ-4528 might persist over time, Dr. Madduri acknowledged that follow-up is still relatively short.
“This product is unique in that has a CD8 central memory phenotype preferentially, and we’re hoping that this would play a central role in the durability of response because they’re memory cells, but I think at this time we don’t know,” she said.
The CARTITUDE-1 trial is funded by Janssen Research & Development. Dr. Madduri disclosed serving as a consultant to Janssen and to Takeda, Foundation Medicine, AbbVie, and Celgene.
SOURCE: Madduri D et al. ASH 2019. Abstract 577.
ORLANDO – A novel chimeric antigen receptor T (CAR T) cell construct is associated with deep clinical responses in patients with multiple myeloma for whom prior lines of therapy – some numbering in the double digits – have failed.
Among 29 patients with multiple myeloma enrolled in a phase 1b/2 trial of JNJ-4528, the overall response rate (ORR) at 6 months median follow-up was 100%, including 69% complete responses, with 27 patients remaining free of disease progression at a median of 6 months, reported Deepu Madduri, MD, of Icahn School of Medicine at Mount Sinai, New York.
“These are very heavily pretreated patients, and so getting early and deep responses is quite amazing,” she said at a briefing prior to presentation of the data at the annual meeting of the American Society of Hematology.
JNJ-4528 is a second-generation CAR T containing two single-domain antibodies targeted against B-cell maturation protein (BCMA). As previously reported, an identical CAR T cell construct showed a high overall response with manageable toxicities in 74 patients with relapsed/refractory multiple myeloma. JNJ-4528 was granted a breakthrough therapy designation for relapsed/refractory multiple myeloma by the Food and Drug Administration on Dec. 6, 2019, and a priority medicines (PRIME) designation by the European Medicines Agency in April 2019.
BCMA was first described in myeloma in 2004 as a mechanism for the growth and survival of malignant plasma cells. Several research groups are currently investigating CAR T cells or monoclonal antibodies targeted to BCMA. The product closest to receiving FDA approval is likely BB2121.
At ASH 2019, Dr. Madduri presented results from the phase 1b portion of the CARTITUDE-1 trial. The investigators enrolled patients with multiple myeloma with measurable diseases as assessed by M-protein or serum free light chain levels who had experienced disease progression on at least 3 prior lines of therapy, or whose disease was refractory to at least two lines of therapy with a proteasome inhibitor, immunomodulatory drug (IMiD), and an anti-CD38 antibody.
Patients underwent apheresis for T-cell collection, with bridging therapy allowed until the expanded T cells could be delivered.
Following T-cell depletion with cyclophosphamide 300 mg/m2 and fludarabine 30 mg/m2 over 3 days, patients received a single weight-based infusion (compared with fixed-dose infusions used with other CAR T cell constructs).
The dose was targeted at 0.75x106 CAR-positive cells/kg, with a target range of 0.5–1.0x106, administered 5-7 days after the start of the conditioning regimen.
A total of 29 patients, median age 60, were evaluable for the safety and efficacy endpoints. One-fourth of the patients had a high-risk cytogenetic profile. The patients had received a median of 5 prior lines of therapy, with one patient receiving 18 prior lines. Of the 29 patients, 25 (86%) had previously undergone autologous transplantation.
As noted before, the ORR after a median follow-up of 6 months was 100%, with 69% completer responses, 17% very good partial responses, and 14% partial responses. The median time to complete response was 1 month (range 1 to 9 months). All but two patients remained free of disease progression at the median 6-month follow-up.
Nearly all patients (27) developed cytokine release syndrome (CRS), and one patient with prolonged grade 4 CRS died from related complications 99 days after infusion.
The median time to onset of CRS was 7 days with more than 90% of cases occurring between days 5 and 9.
Neurotoxicities, specifically immune effector cell–associated neurotoxicity syndrome (ICANS), were infrequent in CRS, and when they did occur were generally low grade, with only 1 grade 3 ICANS event.
Asked in an interview whether the impressive response rates seen with JNJ-4528 might persist over time, Dr. Madduri acknowledged that follow-up is still relatively short.
“This product is unique in that has a CD8 central memory phenotype preferentially, and we’re hoping that this would play a central role in the durability of response because they’re memory cells, but I think at this time we don’t know,” she said.
The CARTITUDE-1 trial is funded by Janssen Research & Development. Dr. Madduri disclosed serving as a consultant to Janssen and to Takeda, Foundation Medicine, AbbVie, and Celgene.
SOURCE: Madduri D et al. ASH 2019. Abstract 577.
ORLANDO – A novel chimeric antigen receptor T (CAR T) cell construct is associated with deep clinical responses in patients with multiple myeloma for whom prior lines of therapy – some numbering in the double digits – have failed.
Among 29 patients with multiple myeloma enrolled in a phase 1b/2 trial of JNJ-4528, the overall response rate (ORR) at 6 months median follow-up was 100%, including 69% complete responses, with 27 patients remaining free of disease progression at a median of 6 months, reported Deepu Madduri, MD, of Icahn School of Medicine at Mount Sinai, New York.
“These are very heavily pretreated patients, and so getting early and deep responses is quite amazing,” she said at a briefing prior to presentation of the data at the annual meeting of the American Society of Hematology.
JNJ-4528 is a second-generation CAR T containing two single-domain antibodies targeted against B-cell maturation protein (BCMA). As previously reported, an identical CAR T cell construct showed a high overall response with manageable toxicities in 74 patients with relapsed/refractory multiple myeloma. JNJ-4528 was granted a breakthrough therapy designation for relapsed/refractory multiple myeloma by the Food and Drug Administration on Dec. 6, 2019, and a priority medicines (PRIME) designation by the European Medicines Agency in April 2019.
BCMA was first described in myeloma in 2004 as a mechanism for the growth and survival of malignant plasma cells. Several research groups are currently investigating CAR T cells or monoclonal antibodies targeted to BCMA. The product closest to receiving FDA approval is likely BB2121.
At ASH 2019, Dr. Madduri presented results from the phase 1b portion of the CARTITUDE-1 trial. The investigators enrolled patients with multiple myeloma with measurable diseases as assessed by M-protein or serum free light chain levels who had experienced disease progression on at least 3 prior lines of therapy, or whose disease was refractory to at least two lines of therapy with a proteasome inhibitor, immunomodulatory drug (IMiD), and an anti-CD38 antibody.
Patients underwent apheresis for T-cell collection, with bridging therapy allowed until the expanded T cells could be delivered.
Following T-cell depletion with cyclophosphamide 300 mg/m2 and fludarabine 30 mg/m2 over 3 days, patients received a single weight-based infusion (compared with fixed-dose infusions used with other CAR T cell constructs).
The dose was targeted at 0.75x106 CAR-positive cells/kg, with a target range of 0.5–1.0x106, administered 5-7 days after the start of the conditioning regimen.
A total of 29 patients, median age 60, were evaluable for the safety and efficacy endpoints. One-fourth of the patients had a high-risk cytogenetic profile. The patients had received a median of 5 prior lines of therapy, with one patient receiving 18 prior lines. Of the 29 patients, 25 (86%) had previously undergone autologous transplantation.
As noted before, the ORR after a median follow-up of 6 months was 100%, with 69% completer responses, 17% very good partial responses, and 14% partial responses. The median time to complete response was 1 month (range 1 to 9 months). All but two patients remained free of disease progression at the median 6-month follow-up.
Nearly all patients (27) developed cytokine release syndrome (CRS), and one patient with prolonged grade 4 CRS died from related complications 99 days after infusion.
The median time to onset of CRS was 7 days with more than 90% of cases occurring between days 5 and 9.
Neurotoxicities, specifically immune effector cell–associated neurotoxicity syndrome (ICANS), were infrequent in CRS, and when they did occur were generally low grade, with only 1 grade 3 ICANS event.
Asked in an interview whether the impressive response rates seen with JNJ-4528 might persist over time, Dr. Madduri acknowledged that follow-up is still relatively short.
“This product is unique in that has a CD8 central memory phenotype preferentially, and we’re hoping that this would play a central role in the durability of response because they’re memory cells, but I think at this time we don’t know,” she said.
The CARTITUDE-1 trial is funded by Janssen Research & Development. Dr. Madduri disclosed serving as a consultant to Janssen and to Takeda, Foundation Medicine, AbbVie, and Celgene.
SOURCE: Madduri D et al. ASH 2019. Abstract 577.
REPORTING FROM ASH 2019
Patient-reported outcomes support first-line pembrolizumab for NSCLC
Patient-reported outcomes support pembrolizumab plus chemotherapy for first-line treatment of metastatic non–small cell lung cancer (NSCLC), based on results from the KEYNOTE-407 trial.
At week 18, patients given pembrolizumab more often reported clinically meaningful health-related quality of life improvements than those in the placebo group, according to lead author Julien Mazieres, MD, PhD, of Paul Sabatier University in Toulouse, France, and colleagues.
Writing in the Journal of Clinical Oncology, the investigators explained that these findings build upon previously published results from KEYNOTE-407, which showed that adding pembrolizumab to chemotherapy in the first line could extend both progression-free and overall survival among patients with NSCLC. The benefits to quality of life associated with pembrolizumab align with similar findings from the KEYNOTE-024 and KEYNOTE-189 trials, they added.
The present analysis involved 559 patients with treatment-naive metastatic NSCLC. Patients were randomized to receive 4 cycles of placebo or pembrolizumab once every 3 weeks with carboplatin-based chemotherapy, followed by pembrolizumab or placebo for an additional 31 cycles. Health-related quality of life was assessed by two measures: the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire–Core 30 (QLQ-C30) and QLQ–Lung Cancer Module 13 (QLQ-LC13).
Of the 559 patients enrolled, 554 completed at least one QLQ-C30 assessment and 553 completed at least one QLQ-LC13 assessment. These quality of life scores were compared temporally within treatment groups, from baseline to week 9 and week 18, and between groups. The investigators also analyzed median time to deterioration in chest pain, cough, and dyspnea.
Results showed that patients in the pembrolizumab group had statistically significant improvements in patient-reported outcomes over time and more frequently reported improvements than patients in the placebo group. Specifically, in the pembrolizumab group, least-squares mean score improved from baseline to week 9 (1.8) and week 18 (4.3); in comparison, least-squares mean score deteriorated in the placebo group from baseline to week 9 (–1.8) and week 18 (–0.57). Compared with placebo, treatment with pembrolizumab was associated with a least-squares mean change of 3.6 at week 9 (nominal P = .0337) and 4.9 at week 18 (nominal P = .0060). Stated differently, at week 18, compared with placebo, more patients in the pembrolizumab group reported clinically meaningful improvements in health-related quality of life (36.2% vs. 27.7%), and relatively fewer reported deterioration (22.8% vs. 31.3%). Median time to deterioration in symptoms was not reached in either treatment arm.
“These health-related quality of life findings, along with the improved efficacy (including overall survival benefit) of pembrolizumab plus carboplatin and paclitaxel/nab-paclitaxel, support its use as a first-line treatment of metastatic squamous NSCLC, regardless of programmed death–ligand 1 expression,” the investigators concluded.
The study was funded by Merck. The investigators reported additional relationships with Novartis, Genentech, Pfizer, and others.
SOURCE: Mazieres J et al. J Clin Oncol. 2019 Nov 21. doi: 10.1200/JCO.19.01348.
Patient-reported outcomes support pembrolizumab plus chemotherapy for first-line treatment of metastatic non–small cell lung cancer (NSCLC), based on results from the KEYNOTE-407 trial.
At week 18, patients given pembrolizumab more often reported clinically meaningful health-related quality of life improvements than those in the placebo group, according to lead author Julien Mazieres, MD, PhD, of Paul Sabatier University in Toulouse, France, and colleagues.
Writing in the Journal of Clinical Oncology, the investigators explained that these findings build upon previously published results from KEYNOTE-407, which showed that adding pembrolizumab to chemotherapy in the first line could extend both progression-free and overall survival among patients with NSCLC. The benefits to quality of life associated with pembrolizumab align with similar findings from the KEYNOTE-024 and KEYNOTE-189 trials, they added.
The present analysis involved 559 patients with treatment-naive metastatic NSCLC. Patients were randomized to receive 4 cycles of placebo or pembrolizumab once every 3 weeks with carboplatin-based chemotherapy, followed by pembrolizumab or placebo for an additional 31 cycles. Health-related quality of life was assessed by two measures: the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire–Core 30 (QLQ-C30) and QLQ–Lung Cancer Module 13 (QLQ-LC13).
Of the 559 patients enrolled, 554 completed at least one QLQ-C30 assessment and 553 completed at least one QLQ-LC13 assessment. These quality of life scores were compared temporally within treatment groups, from baseline to week 9 and week 18, and between groups. The investigators also analyzed median time to deterioration in chest pain, cough, and dyspnea.
Results showed that patients in the pembrolizumab group had statistically significant improvements in patient-reported outcomes over time and more frequently reported improvements than patients in the placebo group. Specifically, in the pembrolizumab group, least-squares mean score improved from baseline to week 9 (1.8) and week 18 (4.3); in comparison, least-squares mean score deteriorated in the placebo group from baseline to week 9 (–1.8) and week 18 (–0.57). Compared with placebo, treatment with pembrolizumab was associated with a least-squares mean change of 3.6 at week 9 (nominal P = .0337) and 4.9 at week 18 (nominal P = .0060). Stated differently, at week 18, compared with placebo, more patients in the pembrolizumab group reported clinically meaningful improvements in health-related quality of life (36.2% vs. 27.7%), and relatively fewer reported deterioration (22.8% vs. 31.3%). Median time to deterioration in symptoms was not reached in either treatment arm.
“These health-related quality of life findings, along with the improved efficacy (including overall survival benefit) of pembrolizumab plus carboplatin and paclitaxel/nab-paclitaxel, support its use as a first-line treatment of metastatic squamous NSCLC, regardless of programmed death–ligand 1 expression,” the investigators concluded.
The study was funded by Merck. The investigators reported additional relationships with Novartis, Genentech, Pfizer, and others.
SOURCE: Mazieres J et al. J Clin Oncol. 2019 Nov 21. doi: 10.1200/JCO.19.01348.
Patient-reported outcomes support pembrolizumab plus chemotherapy for first-line treatment of metastatic non–small cell lung cancer (NSCLC), based on results from the KEYNOTE-407 trial.
At week 18, patients given pembrolizumab more often reported clinically meaningful health-related quality of life improvements than those in the placebo group, according to lead author Julien Mazieres, MD, PhD, of Paul Sabatier University in Toulouse, France, and colleagues.
Writing in the Journal of Clinical Oncology, the investigators explained that these findings build upon previously published results from KEYNOTE-407, which showed that adding pembrolizumab to chemotherapy in the first line could extend both progression-free and overall survival among patients with NSCLC. The benefits to quality of life associated with pembrolizumab align with similar findings from the KEYNOTE-024 and KEYNOTE-189 trials, they added.
The present analysis involved 559 patients with treatment-naive metastatic NSCLC. Patients were randomized to receive 4 cycles of placebo or pembrolizumab once every 3 weeks with carboplatin-based chemotherapy, followed by pembrolizumab or placebo for an additional 31 cycles. Health-related quality of life was assessed by two measures: the European Organisation for the Research and Treatment of Cancer Quality of Life Questionnaire–Core 30 (QLQ-C30) and QLQ–Lung Cancer Module 13 (QLQ-LC13).
Of the 559 patients enrolled, 554 completed at least one QLQ-C30 assessment and 553 completed at least one QLQ-LC13 assessment. These quality of life scores were compared temporally within treatment groups, from baseline to week 9 and week 18, and between groups. The investigators also analyzed median time to deterioration in chest pain, cough, and dyspnea.
Results showed that patients in the pembrolizumab group had statistically significant improvements in patient-reported outcomes over time and more frequently reported improvements than patients in the placebo group. Specifically, in the pembrolizumab group, least-squares mean score improved from baseline to week 9 (1.8) and week 18 (4.3); in comparison, least-squares mean score deteriorated in the placebo group from baseline to week 9 (–1.8) and week 18 (–0.57). Compared with placebo, treatment with pembrolizumab was associated with a least-squares mean change of 3.6 at week 9 (nominal P = .0337) and 4.9 at week 18 (nominal P = .0060). Stated differently, at week 18, compared with placebo, more patients in the pembrolizumab group reported clinically meaningful improvements in health-related quality of life (36.2% vs. 27.7%), and relatively fewer reported deterioration (22.8% vs. 31.3%). Median time to deterioration in symptoms was not reached in either treatment arm.
“These health-related quality of life findings, along with the improved efficacy (including overall survival benefit) of pembrolizumab plus carboplatin and paclitaxel/nab-paclitaxel, support its use as a first-line treatment of metastatic squamous NSCLC, regardless of programmed death–ligand 1 expression,” the investigators concluded.
The study was funded by Merck. The investigators reported additional relationships with Novartis, Genentech, Pfizer, and others.
SOURCE: Mazieres J et al. J Clin Oncol. 2019 Nov 21. doi: 10.1200/JCO.19.01348.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Intensive BP control reduced dementia but increased brain atrophy and hurt cognition
SAN DIEGO – Intensive blood pressure control over 4 years reduced the overall risk of all-cause dementia by 17%, compared with standard care, but in subanalyses of the Systolic Blood Pressure Intervention Trial (SPRINT) it was also associated with significant decreases in cognitive function and total brain volume, researchers said at the Clinical Trials on Alzheimer’s Disease conference.

Whether these between-group differences were clinically meaningful was the topic of some debate, but they were enough to prompt Mary Sano, PhD, to strongly state her reservations.
“The cardiovascular effects of SPRINT were impressive, but I am concerned about minimizing the potentially negative effect on cognition,” said Dr. Sano, professor of psychiatry and director of the Alzheimer’s Disease Research Center at the Icahn School of Medicine at Mount Sinai, New York. “Do I really want to treat a healthy, nonimpaired patient like this if I have to warn them that their cognition might actually get worse? We just cannot minimize this risk. There is very strong evidence that [intensive treatment of blood pressure] might be a step backward in cognition. Would you lower your own blood pressure at a risk of losing some points on your cognition?”
The subanalyses were conducted as part of the SPRINT Memory and Cognition In Decreased Hypertension (SPRINT MIND) substudy, which looked at cardiovascular and mortality outcomes in 9,361 subjects whose hypertension was managed intensively or by standard care (target systolic blood pressure less than 120 mm Hg vs. less than 140 mm Hg). The trial was stopped early because of a 25% reduction in the primary composite cardiovascular disease endpoint and a 27% reduction in all-cause mortality in the intensive-treatment group.
SPRINT MIND examined the risks of incident probable dementia, mild cognitive impairment (MCI), and a composite outcome of both. Intensive control reduced the risk of MCI by 19% and the combined outcome by 15%.
At the conference, SPRINT MIND investigators presented three long-term subanalyses with a median intervention and follow-up time of about 4 years.
Sarah Gaussoin of Wake Forest University, Winston-Salem, N.C., presented unpublished data detailing the effects of intensive control on several dementia subtypes: nonamnestic single domain, nonamnestic multidomain, amnestic single domain, and amnestic multidomain. There were 640 subjects in this analysis.
After a median of 3.3 years of intervention and 5 years of follow-up, there were no differences in the rate of incident probable dementia between the single- and multidomain nonamnestic groups. “We did see a strong 22% decreased risk in single-domain versus multidomain amnestic MCI, however,” she said.
Nicholas Pajewski, PhD, also of Wake Forest University, discussed more detailed cognitive outcomes in SPRINT MIND among 2,900 subjects who had a full battery of cognitive testing at every assessment over 5 years. The outcomes included memory deficit and processing speed.
Dr. Pajewski reported finding no significant difference between the groups in the rates of memory decline in either outcome. But there was a greater rate of decline in processing speed in the intensively treated group, he added. The difference was small but statistically significant.
The difference was largely driven by results of a single cognitive test – the Trail Making Test Part A. “It corresponded to about a 1.25-second increase over 4 years,” in processing speed on this test, Dr. Pajewski said.
There were no between-group differences in any of the other domains explored, including language, executive function, global cognitive function, or the Montreal Cognitive Assessment.
“Obviously, these results are perplexing,” given the overall positive results of SPRINT MIND, he said. “Intensive blood pressure control is a beneficial thing, and we expected to see an effect on memory, or a blunting of decline, and instead we saw some small decrements going the other way. This led us to speculate about what’s going on.”
The trial relied on a narrow definition of MCI that might have affected the outcomes. There was also a very broad range of ages in the study, ranging from 53 to 86 years. More importantly, he said, the original SPRINT study didn’t collect cognitive data at baseline, so there was no way to know how many subjects already might have had MCI when they entered the trial.
Ilya Nasrallah, MD, PhD, of the University of Pennsylvania, Philadelphia, presented MRI data on white-matter lesions, hippocampal volume fractional anisotropy in the cingulum, and cerebral blood flow. The median time between scans was 4 years, with a median treatment time of 3.4 years.
The standard-care group showed a significantly greater increase in white-matter lesion volume at the follow-up scan than did the intensive-treatment group (1.45 cm3 vs. 0.92 cm3). But the intensively treated group had significantly more brain atrophy, losing a median of 30.6 cm3, compared with a loss of 26.9 cm3 in the standard-treatment group.
“It was a very small difference amounting to less than 1% of the total brain volume, but it was still statistically significant,” Dr. Nasrallah said.
Loss of gray-matter volume drove about two-thirds of the difference in the intensively treated group. There was a corresponding increase in cerebrospinal fluid volume that was driven by differences in the ventricles and the subarachnoid space.
However, there were no significant differences in right, left, or total hippocampal volume. There also were no differences in cingulate bundle anisotropy or cerebral blood flow.
SPRINT was funded by the National Institutes of Health. None of the investigators reported having financial conflicts of interest.
SAN DIEGO – Intensive blood pressure control over 4 years reduced the overall risk of all-cause dementia by 17%, compared with standard care, but in subanalyses of the Systolic Blood Pressure Intervention Trial (SPRINT) it was also associated with significant decreases in cognitive function and total brain volume, researchers said at the Clinical Trials on Alzheimer’s Disease conference.
Whether these between-group differences were clinically meaningful was the topic of some debate, but they were enough to prompt Mary Sano, PhD, to strongly state her reservations.
“The cardiovascular effects of SPRINT were impressive, but I am concerned about minimizing the potentially negative effect on cognition,” said Dr. Sano, professor of psychiatry and director of the Alzheimer’s Disease Research Center at the Icahn School of Medicine at Mount Sinai, New York. “Do I really want to treat a healthy, nonimpaired patient like this if I have to warn them that their cognition might actually get worse? We just cannot minimize this risk. There is very strong evidence that [intensive treatment of blood pressure] might be a step backward in cognition. Would you lower your own blood pressure at a risk of losing some points on your cognition?”
The subanalyses were conducted as part of the SPRINT Memory and Cognition In Decreased Hypertension (SPRINT MIND) substudy, which looked at cardiovascular and mortality outcomes in 9,361 subjects whose hypertension was managed intensively or by standard care (target systolic blood pressure less than 120 mm Hg vs. less than 140 mm Hg). The trial was stopped early because of a 25% reduction in the primary composite cardiovascular disease endpoint and a 27% reduction in all-cause mortality in the intensive-treatment group.
SPRINT MIND examined the risks of incident probable dementia, mild cognitive impairment (MCI), and a composite outcome of both. Intensive control reduced the risk of MCI by 19% and the combined outcome by 15%.
At the conference, SPRINT MIND investigators presented three long-term subanalyses with a median intervention and follow-up time of about 4 years.
Sarah Gaussoin of Wake Forest University, Winston-Salem, N.C., presented unpublished data detailing the effects of intensive control on several dementia subtypes: nonamnestic single domain, nonamnestic multidomain, amnestic single domain, and amnestic multidomain. There were 640 subjects in this analysis.
After a median of 3.3 years of intervention and 5 years of follow-up, there were no differences in the rate of incident probable dementia between the single- and multidomain nonamnestic groups. “We did see a strong 22% decreased risk in single-domain versus multidomain amnestic MCI, however,” she said.
Nicholas Pajewski, PhD, also of Wake Forest University, discussed more detailed cognitive outcomes in SPRINT MIND among 2,900 subjects who had a full battery of cognitive testing at every assessment over 5 years. The outcomes included memory deficit and processing speed.
Dr. Pajewski reported finding no significant difference between the groups in the rates of memory decline in either outcome. But there was a greater rate of decline in processing speed in the intensively treated group, he added. The difference was small but statistically significant.
The difference was largely driven by results of a single cognitive test – the Trail Making Test Part A. “It corresponded to about a 1.25-second increase over 4 years,” in processing speed on this test, Dr. Pajewski said.
There were no between-group differences in any of the other domains explored, including language, executive function, global cognitive function, or the Montreal Cognitive Assessment.
“Obviously, these results are perplexing,” given the overall positive results of SPRINT MIND, he said. “Intensive blood pressure control is a beneficial thing, and we expected to see an effect on memory, or a blunting of decline, and instead we saw some small decrements going the other way. This led us to speculate about what’s going on.”
The trial relied on a narrow definition of MCI that might have affected the outcomes. There was also a very broad range of ages in the study, ranging from 53 to 86 years. More importantly, he said, the original SPRINT study didn’t collect cognitive data at baseline, so there was no way to know how many subjects already might have had MCI when they entered the trial.
Ilya Nasrallah, MD, PhD, of the University of Pennsylvania, Philadelphia, presented MRI data on white-matter lesions, hippocampal volume fractional anisotropy in the cingulum, and cerebral blood flow. The median time between scans was 4 years, with a median treatment time of 3.4 years.
The standard-care group showed a significantly greater increase in white-matter lesion volume at the follow-up scan than did the intensive-treatment group (1.45 cm3 vs. 0.92 cm3). But the intensively treated group had significantly more brain atrophy, losing a median of 30.6 cm3, compared with a loss of 26.9 cm3 in the standard-treatment group.
“It was a very small difference amounting to less than 1% of the total brain volume, but it was still statistically significant,” Dr. Nasrallah said.
Loss of gray-matter volume drove about two-thirds of the difference in the intensively treated group. There was a corresponding increase in cerebrospinal fluid volume that was driven by differences in the ventricles and the subarachnoid space.
However, there were no significant differences in right, left, or total hippocampal volume. There also were no differences in cingulate bundle anisotropy or cerebral blood flow.
SPRINT was funded by the National Institutes of Health. None of the investigators reported having financial conflicts of interest.
SAN DIEGO – Intensive blood pressure control over 4 years reduced the overall risk of all-cause dementia by 17%, compared with standard care, but in subanalyses of the Systolic Blood Pressure Intervention Trial (SPRINT) it was also associated with significant decreases in cognitive function and total brain volume, researchers said at the Clinical Trials on Alzheimer’s Disease conference.
Whether these between-group differences were clinically meaningful was the topic of some debate, but they were enough to prompt Mary Sano, PhD, to strongly state her reservations.
“The cardiovascular effects of SPRINT were impressive, but I am concerned about minimizing the potentially negative effect on cognition,” said Dr. Sano, professor of psychiatry and director of the Alzheimer’s Disease Research Center at the Icahn School of Medicine at Mount Sinai, New York. “Do I really want to treat a healthy, nonimpaired patient like this if I have to warn them that their cognition might actually get worse? We just cannot minimize this risk. There is very strong evidence that [intensive treatment of blood pressure] might be a step backward in cognition. Would you lower your own blood pressure at a risk of losing some points on your cognition?”
The subanalyses were conducted as part of the SPRINT Memory and Cognition In Decreased Hypertension (SPRINT MIND) substudy, which looked at cardiovascular and mortality outcomes in 9,361 subjects whose hypertension was managed intensively or by standard care (target systolic blood pressure less than 120 mm Hg vs. less than 140 mm Hg). The trial was stopped early because of a 25% reduction in the primary composite cardiovascular disease endpoint and a 27% reduction in all-cause mortality in the intensive-treatment group.
SPRINT MIND examined the risks of incident probable dementia, mild cognitive impairment (MCI), and a composite outcome of both. Intensive control reduced the risk of MCI by 19% and the combined outcome by 15%.
At the conference, SPRINT MIND investigators presented three long-term subanalyses with a median intervention and follow-up time of about 4 years.
Sarah Gaussoin of Wake Forest University, Winston-Salem, N.C., presented unpublished data detailing the effects of intensive control on several dementia subtypes: nonamnestic single domain, nonamnestic multidomain, amnestic single domain, and amnestic multidomain. There were 640 subjects in this analysis.
After a median of 3.3 years of intervention and 5 years of follow-up, there were no differences in the rate of incident probable dementia between the single- and multidomain nonamnestic groups. “We did see a strong 22% decreased risk in single-domain versus multidomain amnestic MCI, however,” she said.
Nicholas Pajewski, PhD, also of Wake Forest University, discussed more detailed cognitive outcomes in SPRINT MIND among 2,900 subjects who had a full battery of cognitive testing at every assessment over 5 years. The outcomes included memory deficit and processing speed.
Dr. Pajewski reported finding no significant difference between the groups in the rates of memory decline in either outcome. But there was a greater rate of decline in processing speed in the intensively treated group, he added. The difference was small but statistically significant.
The difference was largely driven by results of a single cognitive test – the Trail Making Test Part A. “It corresponded to about a 1.25-second increase over 4 years,” in processing speed on this test, Dr. Pajewski said.
There were no between-group differences in any of the other domains explored, including language, executive function, global cognitive function, or the Montreal Cognitive Assessment.
“Obviously, these results are perplexing,” given the overall positive results of SPRINT MIND, he said. “Intensive blood pressure control is a beneficial thing, and we expected to see an effect on memory, or a blunting of decline, and instead we saw some small decrements going the other way. This led us to speculate about what’s going on.”
The trial relied on a narrow definition of MCI that might have affected the outcomes. There was also a very broad range of ages in the study, ranging from 53 to 86 years. More importantly, he said, the original SPRINT study didn’t collect cognitive data at baseline, so there was no way to know how many subjects already might have had MCI when they entered the trial.
Ilya Nasrallah, MD, PhD, of the University of Pennsylvania, Philadelphia, presented MRI data on white-matter lesions, hippocampal volume fractional anisotropy in the cingulum, and cerebral blood flow. The median time between scans was 4 years, with a median treatment time of 3.4 years.
The standard-care group showed a significantly greater increase in white-matter lesion volume at the follow-up scan than did the intensive-treatment group (1.45 cm3 vs. 0.92 cm3). But the intensively treated group had significantly more brain atrophy, losing a median of 30.6 cm3, compared with a loss of 26.9 cm3 in the standard-treatment group.
“It was a very small difference amounting to less than 1% of the total brain volume, but it was still statistically significant,” Dr. Nasrallah said.
Loss of gray-matter volume drove about two-thirds of the difference in the intensively treated group. There was a corresponding increase in cerebrospinal fluid volume that was driven by differences in the ventricles and the subarachnoid space.
However, there were no significant differences in right, left, or total hippocampal volume. There also were no differences in cingulate bundle anisotropy or cerebral blood flow.
SPRINT was funded by the National Institutes of Health. None of the investigators reported having financial conflicts of interest.
REPORTING FROM CTAD 2019
Women experience more chemoradiotherapy toxicity in rectal cancer
Women are more likely to experience acute toxic effects from chemoradiotherapy for rectal cancer than men, but this does not appear to negatively impact treatment adherence or outcomes, research suggests.
In a research letter published in JAMA Oncology, Markus Diefenhardt, MD, from the University of Frankfurt and coauthors wrote that, while the risk of toxic chemotherapy effects was known to be greater in women for a number of cancers, this association was relatively unexplored for rectal cancer.
The researchers performed a pooled analysis of data from two phase 3, randomized clinical trials, involving 1,016 patients with rectal cancer – 28.6% of whom were female – treated with fluorouracil-based chemoradiotherapy followed by surgery and adjuvant fluorouracil.
They found that women experienced significantly higher rates of leukopenia and diarrhea than men. Grade 3-4 leukopenia was experienced by 28.6% of women, compared with 20.5% of men, and grades 3-4 diarrhea was experienced by 17.2% of women, compared with 8.1% of men.
Despite this, the study found similar rates of adherence to treatment between men and women both for neoadjuvant and adjuvant chemoradiotherapy. Women also had similar rates of disease-free survival and overall survival as men, and there were no significant differences in local recurrence or distant metastases.
“Although to our knowledge no data support using different chemotherapy regimens for men and women with rectal cancer, increased awareness of a higher risk of toxic effects among women may facilitate refinement of fluorouracil-based chemoradiotherapy and adjuvant chemotherapy, such as tailored patient education, closer monitoring of adverse effects, and earlier introduction of supportive measures,” the authors wrote.
The authors proposed several possible explanations for the higher rate of toxic effects in women. For example, women may have lower levels of the enzyme dihydropyridine dehydrogenase, which catabolizes fluorouracil, which could result in overdosing of fluorouracil. Similarly, sex-specific body fat composition could also contribute to fluorouracil overdosing in women.
The study also saw fewer postoperative complications in women, which the authors suggested could be related to the lower rate of abdominoperineal resections in women.
The two clinical trials included in the study were funded by German Cancer Aid. One author declared funding from German Cancer Aid, another declared a range of honoraria, research fees and institutional funding from the pharmaceutical sector. No other conflicts of interest were declared.
SOURCE: Diefendhardt M et al. JAMA Oncol. 2019 Dec 5. doi: 10.1001/jamaoncol.2019.5102.
Women are more likely to experience acute toxic effects from chemoradiotherapy for rectal cancer than men, but this does not appear to negatively impact treatment adherence or outcomes, research suggests.
In a research letter published in JAMA Oncology, Markus Diefenhardt, MD, from the University of Frankfurt and coauthors wrote that, while the risk of toxic chemotherapy effects was known to be greater in women for a number of cancers, this association was relatively unexplored for rectal cancer.
The researchers performed a pooled analysis of data from two phase 3, randomized clinical trials, involving 1,016 patients with rectal cancer – 28.6% of whom were female – treated with fluorouracil-based chemoradiotherapy followed by surgery and adjuvant fluorouracil.
They found that women experienced significantly higher rates of leukopenia and diarrhea than men. Grade 3-4 leukopenia was experienced by 28.6% of women, compared with 20.5% of men, and grades 3-4 diarrhea was experienced by 17.2% of women, compared with 8.1% of men.
Despite this, the study found similar rates of adherence to treatment between men and women both for neoadjuvant and adjuvant chemoradiotherapy. Women also had similar rates of disease-free survival and overall survival as men, and there were no significant differences in local recurrence or distant metastases.
“Although to our knowledge no data support using different chemotherapy regimens for men and women with rectal cancer, increased awareness of a higher risk of toxic effects among women may facilitate refinement of fluorouracil-based chemoradiotherapy and adjuvant chemotherapy, such as tailored patient education, closer monitoring of adverse effects, and earlier introduction of supportive measures,” the authors wrote.
The authors proposed several possible explanations for the higher rate of toxic effects in women. For example, women may have lower levels of the enzyme dihydropyridine dehydrogenase, which catabolizes fluorouracil, which could result in overdosing of fluorouracil. Similarly, sex-specific body fat composition could also contribute to fluorouracil overdosing in women.
The study also saw fewer postoperative complications in women, which the authors suggested could be related to the lower rate of abdominoperineal resections in women.
The two clinical trials included in the study were funded by German Cancer Aid. One author declared funding from German Cancer Aid, another declared a range of honoraria, research fees and institutional funding from the pharmaceutical sector. No other conflicts of interest were declared.
SOURCE: Diefendhardt M et al. JAMA Oncol. 2019 Dec 5. doi: 10.1001/jamaoncol.2019.5102.
Women are more likely to experience acute toxic effects from chemoradiotherapy for rectal cancer than men, but this does not appear to negatively impact treatment adherence or outcomes, research suggests.
In a research letter published in JAMA Oncology, Markus Diefenhardt, MD, from the University of Frankfurt and coauthors wrote that, while the risk of toxic chemotherapy effects was known to be greater in women for a number of cancers, this association was relatively unexplored for rectal cancer.
The researchers performed a pooled analysis of data from two phase 3, randomized clinical trials, involving 1,016 patients with rectal cancer – 28.6% of whom were female – treated with fluorouracil-based chemoradiotherapy followed by surgery and adjuvant fluorouracil.
They found that women experienced significantly higher rates of leukopenia and diarrhea than men. Grade 3-4 leukopenia was experienced by 28.6% of women, compared with 20.5% of men, and grades 3-4 diarrhea was experienced by 17.2% of women, compared with 8.1% of men.
Despite this, the study found similar rates of adherence to treatment between men and women both for neoadjuvant and adjuvant chemoradiotherapy. Women also had similar rates of disease-free survival and overall survival as men, and there were no significant differences in local recurrence or distant metastases.
“Although to our knowledge no data support using different chemotherapy regimens for men and women with rectal cancer, increased awareness of a higher risk of toxic effects among women may facilitate refinement of fluorouracil-based chemoradiotherapy and adjuvant chemotherapy, such as tailored patient education, closer monitoring of adverse effects, and earlier introduction of supportive measures,” the authors wrote.
The authors proposed several possible explanations for the higher rate of toxic effects in women. For example, women may have lower levels of the enzyme dihydropyridine dehydrogenase, which catabolizes fluorouracil, which could result in overdosing of fluorouracil. Similarly, sex-specific body fat composition could also contribute to fluorouracil overdosing in women.
The study also saw fewer postoperative complications in women, which the authors suggested could be related to the lower rate of abdominoperineal resections in women.
The two clinical trials included in the study were funded by German Cancer Aid. One author declared funding from German Cancer Aid, another declared a range of honoraria, research fees and institutional funding from the pharmaceutical sector. No other conflicts of interest were declared.
SOURCE: Diefendhardt M et al. JAMA Oncol. 2019 Dec 5. doi: 10.1001/jamaoncol.2019.5102.
FROM JAMA ONCOLOGY
Key clinical point: Women show significantly higher rates of toxic effects from rectal cancer chemoradiotherapy than men.
Major finding: Women experience significantly higher rates of leukopenia and diarrhea from rectal cancer chemoradiotherapy.
Study details: A pooled analysis of data from two phase 3, randomized, controlled trials in 1,016 patients.
Disclosures: The two clinical trials included in the study were funded by German Cancer Aid. One author declared funding from German Cancer Aid, another declared a range of honoraria, research fees and institutional funding from the pharmaceutical sector. No other conflicts of interest were declared.
Source: Diefendhardt M et al. JAMA Oncol. 2019 Dec 5. doi: 10.1001/jamaoncol.2019.5102.
Understanding Principles of High Reliability Organizations Through the Eyes of VIONE, A Clinical Program to Improve Patient Safety by Deprescribing Potentially Inappropriate Medications and Reducing Polypharmacy
High reliability organizations (HROs) incorporate continuous process improvement through leadership commitment to create a safety culture that works toward creating a zero-harm environment.1 The Veterans Health Administration (VHA) has set transformational goals for becoming an HRO. In this article, we describe VIONE, an expanding medication deprescribing clinical program, which exemplifies the translation of HRO principles into health care system models. Both VIONE and HRO are globally relevant.
Reducing medication errors and related adverse drug events are important for achieving zero harm. Preventable medical errors rank behind heart disease and cancer as the third leading cause of death in the US.2 The simultaneous use of multiple medications can lead to dangerous drug interactions, adverse outcomes, and challenges with adherence. When a person is taking multiple medicines, known as polypharmacy, it is more likely that some are potentially inappropriate medications (PIM). Current literature highlights the prevalence and dangers of polypharmacy, which ranks among the top 10 common causes of death in the US, as well as suggestions to address preventable adverse outcomes from polypharmacy and PIM.3-5
Deprescribing of PIM frequently results in better disease management with improved health outcomes and quality of life.4 Many health care settings lack standardized approaches or set expectations to proactively deprescribe PIM. There has been insufficient emphasis on how to make decisions for deprescribing medications when therapeutic benefits are not clear and/or when the adverse effects may outweigh the therapeutic benefits.5
It is imperative to provide practice guidance for deprescribing nonessential medications along with systems-based infrastructure to enable integrated and effective assessments during opportune moments in the health care continuum. Multimodal approaches that include education, risk stratification, population health management interventions, research and resource allocation can help transform organizational culture in health care facilities toward HRO models of care, aiming at zero harm to patients.
The practical lessons learned from VIONE implementation science experiences on various scales and under diverse circumstances, cumulative wisdom from hindsight, foresight and critical insights gathered during nationwide spread of VIONE over the past 3 years continues to propel us toward the desirable direction and core concepts of an HRO.
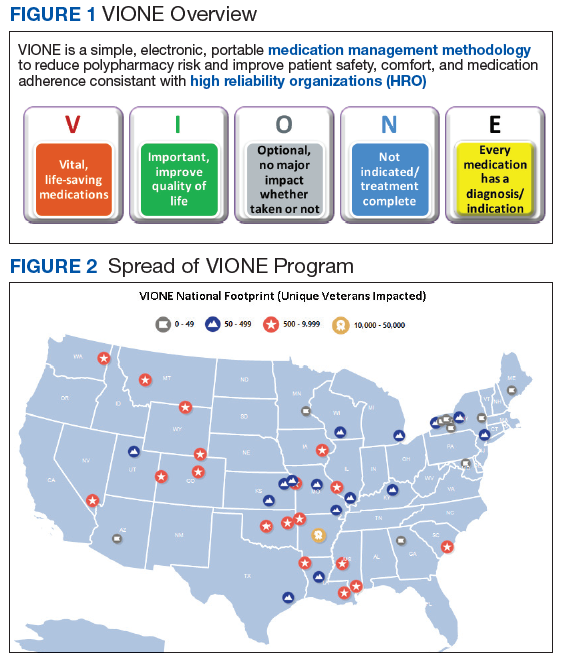
The VIONE program facilitates practical, real-time interventions that could be tailored to various health care settings, organizational needs, and available resources. VIONE implements an electronic Computerized Patient Record System (CPRS) tool to enable planned cessation of nonessential medications that are potentially harmful, inappropriate, not indicated, or not necessary. The VIONE tool supports systematic, individualized assessment and adjustment through 5 filters (Figure 1). It prompts providers to assign 1 of these filters intuitively and objectively. VIONE combines clinical evidence for best practices, an interprofessional team approach, patient engagement, adapted use of existing medical records systems, and HRO principles for effective implementation.
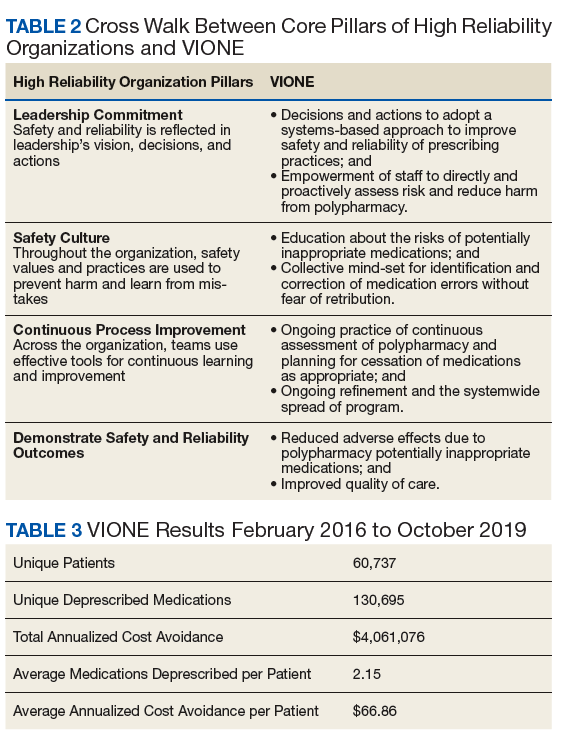
As a tool to support safer prescribing practices, VIONE aligns closely with HRO principles (Table 1) and core pillars (Table 2).6-8 A zero-harm safety culture necessitates that medications be used for correct reasons, over a correct duration of time, and following a correct schedule while monitoring for adverse outcomes. However, reality generally falls significantly short of this for a myriad of reasons, such as compromised health literacy, functional limitations, affordability, communication gaps, patients seen by multiple providers, and an accumulation of prescriptions due to comorbidities, symptom progression, and management of adverse effects. Through a sharpened focus on both precision medicine and competent prescription management, VIONE is a viable opportunity for investing in the zero-harm philosophy that is integral to an HRO.

Design and Implementation
Initially launched in 2016 in a 15-bed inpatient, subacute rehabilitation unit within a VHA tertiary care facility, VIONE has been sustained and gradually expanded to 38 other VHA facility programs (Figure 2). Recognizing the potential value if adopted into widespread use, VIONE was a Gold Status winner in the VHA Under Secretary for Health Shark Tank-style competition in 2017 and was selected by the VHA Diffusion of Excellence as an innovation worthy of scale and spread through national dissemination.9 A toolkit for VIONE implementation, patient and provider brochures, VIONE vignette, and National Dialog template also have been created.10
Implementing VIONE in a new facility requires an actively engaged core team committed to patient safety and reduction of polypharmacy and PIM, interest and availability to lead project implementation strategies, along with meaningful local organizational support. The current structure for VIONE spread is as follows:
- Interested VHA participants review information and contact [email protected].
- The VIONE team orients implementing champions, mainly pharmacists, physicians, nurse practitioners, and physician assistants at a facility program level, offering guidance and available resources.
- Clinical Application Coordinators at Central Arkansas VA Healthcare System and participating facilities collaborate to add deprescribing menu options in CPRS and install the VIONE Polypharmacy Reminder Dialog template.
- Through close and ongoing collaborations, medical providers and clinical pharmacists proceed with deprescribing, aiming at planned cessation of nonessential and PIM, using the mnemonic prompt of VIONE. Vital and Important medications are continued and consolidated while a methodical plan is developed to deprescribe any medications that could lead to more harm than benefit and qualify based on the filters of Optional, Not indicated, and Every medicine has a diagnosis/reason. They select the proper discontinuation reasons in the CPRS medication menu (Figure 3) and document the rationale in the progress notes. It is highly encouraged that the collaborating pharmacists and health care providers add each other as cosigners and communicate effectively. Clinical pharmacy specialists also use the VIONE Polypharmacy Reminder Dialog Template (RDT) to document complete medication reviews with veterans to include deprescribing rationale and document shared decision making.
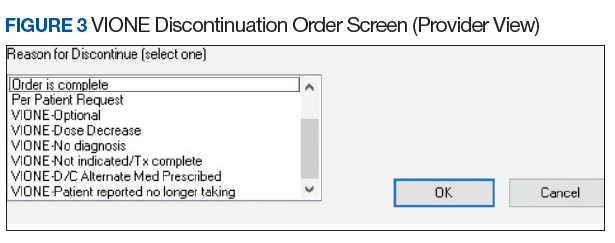
- A VIONE national dashboard captures deprescribing data in real time and automates reporting with daily updates that are readily accessible to all implementing facilities. Minimum data captured include the number of unique veterans impacted, number of medications deprescribed, cumulative cost avoidance to date, and number of prescriptions deprescribed per veteran. The dashboard facilitates real-time use of individual patient data and has also been designed to capture data from VHA administrative data portals and Corporate Data Warehouse.
Results
As of October 31, 2019, the assessment of polypharmacy using the VIONE tool across VHA sites has benefited > 60,000 unique veterans, of whom 49.2% were in urban areas, 47.7% in rural areas, and 3.1% in highly rural areas. Elderly male veterans comprised a clear majority. More than 128,000 medications have been deprescribed. The top classes of medications deprescribed are antihypertensives, over-the-counter medications, and antidiabetic medications. An annualized cost avoidance of > $4.0 million has been achieved. Cost avoidance is the cost of medications that otherwise would have continued to be filled and paid for by the VHA if they had not been deprescribed, projected for a maximum of 365 days. The calculation methodology can be summarized as follows:
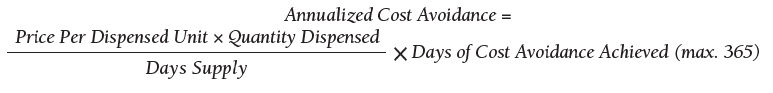
The calculations reported in Table 3 and Figure 4 are conservative and include only chronic outpatient prescriptions and do not account for medications deprescribed in inpatient units, nursing home, community living centers, or domiciliary populations. Data tracked separately from inpatient and community living center patient populations indicated an additional 25,536 deprescribed medications, across 28 VA facilities, impacting 7,076 veterans with an average 2.15 medications deprescribed per veteran. The additional achieved cost avoidance was $370,272 (based on $14.50 average cost per prescription). Medications restarted within 30 days of deprescribing are not included in these calculations.
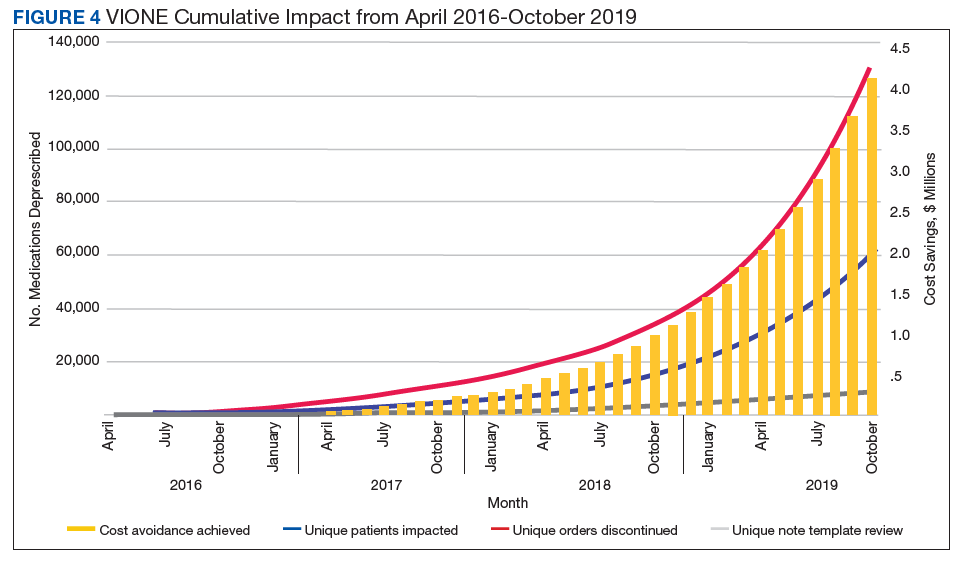
The cost avoidance calculation further excludes the effects of VIONE implementation on many other types of interventions. These interventions include, but are not limited to, changing from aggressive care to end of life, comfort care when strongly indicated; reduced emergency department visits or invasive diagnostic and therapeutic approaches, when not indicated; medical supplies, antimicrobial preparations; labor costs related to packaging, mailing, and administering prescriptions; reduced/prevented clinical waste; reduced decompensation of systemic illnesses and subsequent health care needs precipitated by iatrogenic disturbances and prolonged convalescence; and overall changes to prescribing practices through purposeful and targeted interactions with colleagues across various disciplines and various hierarchical levels.
Discussion
The VIONE clinical program exemplifies the translation of HRO principles into health care system practices. VIONE offers a systematic approach to improve medication management with an emphasis on deprescribing nonessential medications across various health care settings, facilitating VHA efforts toward zero harm. It demonstrates close alignment with the key building blocks of an HRO. Effective VIONE incorporation into an organizational culture reflects leadership commitment to safety and reliability in their vision and actions. By empowering staff to proactively reduce inappropriate medications and thereby prevent patient harm, VIONE contributes to enhancing an enterprise-wide culture of safety, with fewer errors and greater reliability. As a standardized decision support tool for the ongoing practice of assessment and planned cessation of potentially inappropriate medications, VIONE illustrates how continuous process improvement can be a part of staff-engaged, veteran-centered, highly reliable care. The standardization of the VIONE tool promotes achievement and sustainment of desired HRO principles and practices within health care delivery systems.
Conclusions
The VIONE program was launched not as a cost savings or research program but as a practical, real-time bedside or ambulatory care intervention to improve patient safety. Its value is reflected in the overwhelming response from scholarly and well-engaged colleagues expressing serious interests in expanding collaborations and tailoring efforts to add more depth and breadth to VIONE related efforts.
Acknowledgments
The authors express their gratitude to Central Arkansas VA Healthcare System leadership, Clinical Applications Coordinators, and colleagues for their unconditional support, to the Diffusion of Excellence programs at US Department of Veterans Affairs Central Office for their endorsement, and to the many VHA participants who renew our optimism and energy as we continue this exciting journey. We also thank Bridget B. Kelly for her assistance in writing and editing of the manuscript.
1. Chassin MR, Jerod ML. High-reliability health care: getting there from here. The Joint Commission. Milbank Q. 2013;91(3):459-490.
2. Makary MA, Daniel M. Medical error—the third leading cause of death in the US. BMJ. 2016;353:i2139.
3. Quinn KJ, Shah NH. A dataset quantifying polypharmacy in the United States. Sci Data. 2017;4:170167.
4. Scott IA, Hilmer SN, Reeve E, et al. Reducing inappropriate polypharmacy: the process of deprescribing. JAMA Intern Med. 2015;175(5):827-834.
5. Steinman MA. Polypharmacy—time to get beyond numbers. JAMA Intern Med. 2016;176(4):482-483.
6. US Department of Veterans Affairs. High reliability. https://dvagov.sharepoint.com/sites/OHT-PMO/high-reliability/Pages/default.aspx. [Nonpublic source, not verified.]
7. Gordon S, Mendenhall P, O’Connor BB. Beyond the Checklist: What Else Health Care Can Learn from Aviation Teamwork and Safety. Ithaca, NY: Cornell University Press; 2013.
8. Institute of Medicine (US) Committee on Quality of Health Care in America; Kohn LT, Corrigan JM, Donaldson MS, eds. To Err Is Human: Building a Safer Health System. Washington, DC: The National Academies Press; 2000.
9. US Department of Veterans Affairs. Diffusion of Excellence. https://www.va.gov/HEALTHCAREEXCELLENCE/diffusion-of-excellence/. Updated August 10, 2018. Accessed June 26, 2019.
10. US Department of Veterans Affairs. VIONE program toolkit. https://www.vapulse.net/docs/DOC-259375. [Nonpublic source, not verified.]
High reliability organizations (HROs) incorporate continuous process improvement through leadership commitment to create a safety culture that works toward creating a zero-harm environment.1 The Veterans Health Administration (VHA) has set transformational goals for becoming an HRO. In this article, we describe VIONE, an expanding medication deprescribing clinical program, which exemplifies the translation of HRO principles into health care system models. Both VIONE and HRO are globally relevant.
Reducing medication errors and related adverse drug events are important for achieving zero harm. Preventable medical errors rank behind heart disease and cancer as the third leading cause of death in the US.2 The simultaneous use of multiple medications can lead to dangerous drug interactions, adverse outcomes, and challenges with adherence. When a person is taking multiple medicines, known as polypharmacy, it is more likely that some are potentially inappropriate medications (PIM). Current literature highlights the prevalence and dangers of polypharmacy, which ranks among the top 10 common causes of death in the US, as well as suggestions to address preventable adverse outcomes from polypharmacy and PIM.3-5
Deprescribing of PIM frequently results in better disease management with improved health outcomes and quality of life.4 Many health care settings lack standardized approaches or set expectations to proactively deprescribe PIM. There has been insufficient emphasis on how to make decisions for deprescribing medications when therapeutic benefits are not clear and/or when the adverse effects may outweigh the therapeutic benefits.5
It is imperative to provide practice guidance for deprescribing nonessential medications along with systems-based infrastructure to enable integrated and effective assessments during opportune moments in the health care continuum. Multimodal approaches that include education, risk stratification, population health management interventions, research and resource allocation can help transform organizational culture in health care facilities toward HRO models of care, aiming at zero harm to patients.
The practical lessons learned from VIONE implementation science experiences on various scales and under diverse circumstances, cumulative wisdom from hindsight, foresight and critical insights gathered during nationwide spread of VIONE over the past 3 years continues to propel us toward the desirable direction and core concepts of an HRO.
The VIONE program facilitates practical, real-time interventions that could be tailored to various health care settings, organizational needs, and available resources. VIONE implements an electronic Computerized Patient Record System (CPRS) tool to enable planned cessation of nonessential medications that are potentially harmful, inappropriate, not indicated, or not necessary. The VIONE tool supports systematic, individualized assessment and adjustment through 5 filters (Figure 1). It prompts providers to assign 1 of these filters intuitively and objectively. VIONE combines clinical evidence for best practices, an interprofessional team approach, patient engagement, adapted use of existing medical records systems, and HRO principles for effective implementation.
As a tool to support safer prescribing practices, VIONE aligns closely with HRO principles (Table 1) and core pillars (Table 2).6-8 A zero-harm safety culture necessitates that medications be used for correct reasons, over a correct duration of time, and following a correct schedule while monitoring for adverse outcomes. However, reality generally falls significantly short of this for a myriad of reasons, such as compromised health literacy, functional limitations, affordability, communication gaps, patients seen by multiple providers, and an accumulation of prescriptions due to comorbidities, symptom progression, and management of adverse effects. Through a sharpened focus on both precision medicine and competent prescription management, VIONE is a viable opportunity for investing in the zero-harm philosophy that is integral to an HRO.
Design and Implementation
Initially launched in 2016 in a 15-bed inpatient, subacute rehabilitation unit within a VHA tertiary care facility, VIONE has been sustained and gradually expanded to 38 other VHA facility programs (Figure 2). Recognizing the potential value if adopted into widespread use, VIONE was a Gold Status winner in the VHA Under Secretary for Health Shark Tank-style competition in 2017 and was selected by the VHA Diffusion of Excellence as an innovation worthy of scale and spread through national dissemination.9 A toolkit for VIONE implementation, patient and provider brochures, VIONE vignette, and National Dialog template also have been created.10
Implementing VIONE in a new facility requires an actively engaged core team committed to patient safety and reduction of polypharmacy and PIM, interest and availability to lead project implementation strategies, along with meaningful local organizational support. The current structure for VIONE spread is as follows:
- Interested VHA participants review information and contact [email protected].
- The VIONE team orients implementing champions, mainly pharmacists, physicians, nurse practitioners, and physician assistants at a facility program level, offering guidance and available resources.
- Clinical Application Coordinators at Central Arkansas VA Healthcare System and participating facilities collaborate to add deprescribing menu options in CPRS and install the VIONE Polypharmacy Reminder Dialog template.
- Through close and ongoing collaborations, medical providers and clinical pharmacists proceed with deprescribing, aiming at planned cessation of nonessential and PIM, using the mnemonic prompt of VIONE. Vital and Important medications are continued and consolidated while a methodical plan is developed to deprescribe any medications that could lead to more harm than benefit and qualify based on the filters of Optional, Not indicated, and Every medicine has a diagnosis/reason. They select the proper discontinuation reasons in the CPRS medication menu (Figure 3) and document the rationale in the progress notes. It is highly encouraged that the collaborating pharmacists and health care providers add each other as cosigners and communicate effectively. Clinical pharmacy specialists also use the VIONE Polypharmacy Reminder Dialog Template (RDT) to document complete medication reviews with veterans to include deprescribing rationale and document shared decision making.
- A VIONE national dashboard captures deprescribing data in real time and automates reporting with daily updates that are readily accessible to all implementing facilities. Minimum data captured include the number of unique veterans impacted, number of medications deprescribed, cumulative cost avoidance to date, and number of prescriptions deprescribed per veteran. The dashboard facilitates real-time use of individual patient data and has also been designed to capture data from VHA administrative data portals and Corporate Data Warehouse.
Results
As of October 31, 2019, the assessment of polypharmacy using the VIONE tool across VHA sites has benefited > 60,000 unique veterans, of whom 49.2% were in urban areas, 47.7% in rural areas, and 3.1% in highly rural areas. Elderly male veterans comprised a clear majority. More than 128,000 medications have been deprescribed. The top classes of medications deprescribed are antihypertensives, over-the-counter medications, and antidiabetic medications. An annualized cost avoidance of > $4.0 million has been achieved. Cost avoidance is the cost of medications that otherwise would have continued to be filled and paid for by the VHA if they had not been deprescribed, projected for a maximum of 365 days. The calculation methodology can be summarized as follows:
The calculations reported in Table 3 and Figure 4 are conservative and include only chronic outpatient prescriptions and do not account for medications deprescribed in inpatient units, nursing home, community living centers, or domiciliary populations. Data tracked separately from inpatient and community living center patient populations indicated an additional 25,536 deprescribed medications, across 28 VA facilities, impacting 7,076 veterans with an average 2.15 medications deprescribed per veteran. The additional achieved cost avoidance was $370,272 (based on $14.50 average cost per prescription). Medications restarted within 30 days of deprescribing are not included in these calculations.
The cost avoidance calculation further excludes the effects of VIONE implementation on many other types of interventions. These interventions include, but are not limited to, changing from aggressive care to end of life, comfort care when strongly indicated; reduced emergency department visits or invasive diagnostic and therapeutic approaches, when not indicated; medical supplies, antimicrobial preparations; labor costs related to packaging, mailing, and administering prescriptions; reduced/prevented clinical waste; reduced decompensation of systemic illnesses and subsequent health care needs precipitated by iatrogenic disturbances and prolonged convalescence; and overall changes to prescribing practices through purposeful and targeted interactions with colleagues across various disciplines and various hierarchical levels.
Discussion
The VIONE clinical program exemplifies the translation of HRO principles into health care system practices. VIONE offers a systematic approach to improve medication management with an emphasis on deprescribing nonessential medications across various health care settings, facilitating VHA efforts toward zero harm. It demonstrates close alignment with the key building blocks of an HRO. Effective VIONE incorporation into an organizational culture reflects leadership commitment to safety and reliability in their vision and actions. By empowering staff to proactively reduce inappropriate medications and thereby prevent patient harm, VIONE contributes to enhancing an enterprise-wide culture of safety, with fewer errors and greater reliability. As a standardized decision support tool for the ongoing practice of assessment and planned cessation of potentially inappropriate medications, VIONE illustrates how continuous process improvement can be a part of staff-engaged, veteran-centered, highly reliable care. The standardization of the VIONE tool promotes achievement and sustainment of desired HRO principles and practices within health care delivery systems.
Conclusions
The VIONE program was launched not as a cost savings or research program but as a practical, real-time bedside or ambulatory care intervention to improve patient safety. Its value is reflected in the overwhelming response from scholarly and well-engaged colleagues expressing serious interests in expanding collaborations and tailoring efforts to add more depth and breadth to VIONE related efforts.
Acknowledgments
The authors express their gratitude to Central Arkansas VA Healthcare System leadership, Clinical Applications Coordinators, and colleagues for their unconditional support, to the Diffusion of Excellence programs at US Department of Veterans Affairs Central Office for their endorsement, and to the many VHA participants who renew our optimism and energy as we continue this exciting journey. We also thank Bridget B. Kelly for her assistance in writing and editing of the manuscript.
High reliability organizations (HROs) incorporate continuous process improvement through leadership commitment to create a safety culture that works toward creating a zero-harm environment.1 The Veterans Health Administration (VHA) has set transformational goals for becoming an HRO. In this article, we describe VIONE, an expanding medication deprescribing clinical program, which exemplifies the translation of HRO principles into health care system models. Both VIONE and HRO are globally relevant.
Reducing medication errors and related adverse drug events are important for achieving zero harm. Preventable medical errors rank behind heart disease and cancer as the third leading cause of death in the US.2 The simultaneous use of multiple medications can lead to dangerous drug interactions, adverse outcomes, and challenges with adherence. When a person is taking multiple medicines, known as polypharmacy, it is more likely that some are potentially inappropriate medications (PIM). Current literature highlights the prevalence and dangers of polypharmacy, which ranks among the top 10 common causes of death in the US, as well as suggestions to address preventable adverse outcomes from polypharmacy and PIM.3-5
Deprescribing of PIM frequently results in better disease management with improved health outcomes and quality of life.4 Many health care settings lack standardized approaches or set expectations to proactively deprescribe PIM. There has been insufficient emphasis on how to make decisions for deprescribing medications when therapeutic benefits are not clear and/or when the adverse effects may outweigh the therapeutic benefits.5
It is imperative to provide practice guidance for deprescribing nonessential medications along with systems-based infrastructure to enable integrated and effective assessments during opportune moments in the health care continuum. Multimodal approaches that include education, risk stratification, population health management interventions, research and resource allocation can help transform organizational culture in health care facilities toward HRO models of care, aiming at zero harm to patients.
The practical lessons learned from VIONE implementation science experiences on various scales and under diverse circumstances, cumulative wisdom from hindsight, foresight and critical insights gathered during nationwide spread of VIONE over the past 3 years continues to propel us toward the desirable direction and core concepts of an HRO.
The VIONE program facilitates practical, real-time interventions that could be tailored to various health care settings, organizational needs, and available resources. VIONE implements an electronic Computerized Patient Record System (CPRS) tool to enable planned cessation of nonessential medications that are potentially harmful, inappropriate, not indicated, or not necessary. The VIONE tool supports systematic, individualized assessment and adjustment through 5 filters (Figure 1). It prompts providers to assign 1 of these filters intuitively and objectively. VIONE combines clinical evidence for best practices, an interprofessional team approach, patient engagement, adapted use of existing medical records systems, and HRO principles for effective implementation.
As a tool to support safer prescribing practices, VIONE aligns closely with HRO principles (Table 1) and core pillars (Table 2).6-8 A zero-harm safety culture necessitates that medications be used for correct reasons, over a correct duration of time, and following a correct schedule while monitoring for adverse outcomes. However, reality generally falls significantly short of this for a myriad of reasons, such as compromised health literacy, functional limitations, affordability, communication gaps, patients seen by multiple providers, and an accumulation of prescriptions due to comorbidities, symptom progression, and management of adverse effects. Through a sharpened focus on both precision medicine and competent prescription management, VIONE is a viable opportunity for investing in the zero-harm philosophy that is integral to an HRO.
Design and Implementation
Initially launched in 2016 in a 15-bed inpatient, subacute rehabilitation unit within a VHA tertiary care facility, VIONE has been sustained and gradually expanded to 38 other VHA facility programs (Figure 2). Recognizing the potential value if adopted into widespread use, VIONE was a Gold Status winner in the VHA Under Secretary for Health Shark Tank-style competition in 2017 and was selected by the VHA Diffusion of Excellence as an innovation worthy of scale and spread through national dissemination.9 A toolkit for VIONE implementation, patient and provider brochures, VIONE vignette, and National Dialog template also have been created.10
Implementing VIONE in a new facility requires an actively engaged core team committed to patient safety and reduction of polypharmacy and PIM, interest and availability to lead project implementation strategies, along with meaningful local organizational support. The current structure for VIONE spread is as follows:
- Interested VHA participants review information and contact [email protected].
- The VIONE team orients implementing champions, mainly pharmacists, physicians, nurse practitioners, and physician assistants at a facility program level, offering guidance and available resources.
- Clinical Application Coordinators at Central Arkansas VA Healthcare System and participating facilities collaborate to add deprescribing menu options in CPRS and install the VIONE Polypharmacy Reminder Dialog template.
- Through close and ongoing collaborations, medical providers and clinical pharmacists proceed with deprescribing, aiming at planned cessation of nonessential and PIM, using the mnemonic prompt of VIONE. Vital and Important medications are continued and consolidated while a methodical plan is developed to deprescribe any medications that could lead to more harm than benefit and qualify based on the filters of Optional, Not indicated, and Every medicine has a diagnosis/reason. They select the proper discontinuation reasons in the CPRS medication menu (Figure 3) and document the rationale in the progress notes. It is highly encouraged that the collaborating pharmacists and health care providers add each other as cosigners and communicate effectively. Clinical pharmacy specialists also use the VIONE Polypharmacy Reminder Dialog Template (RDT) to document complete medication reviews with veterans to include deprescribing rationale and document shared decision making.
- A VIONE national dashboard captures deprescribing data in real time and automates reporting with daily updates that are readily accessible to all implementing facilities. Minimum data captured include the number of unique veterans impacted, number of medications deprescribed, cumulative cost avoidance to date, and number of prescriptions deprescribed per veteran. The dashboard facilitates real-time use of individual patient data and has also been designed to capture data from VHA administrative data portals and Corporate Data Warehouse.
Results
As of October 31, 2019, the assessment of polypharmacy using the VIONE tool across VHA sites has benefited > 60,000 unique veterans, of whom 49.2% were in urban areas, 47.7% in rural areas, and 3.1% in highly rural areas. Elderly male veterans comprised a clear majority. More than 128,000 medications have been deprescribed. The top classes of medications deprescribed are antihypertensives, over-the-counter medications, and antidiabetic medications. An annualized cost avoidance of > $4.0 million has been achieved. Cost avoidance is the cost of medications that otherwise would have continued to be filled and paid for by the VHA if they had not been deprescribed, projected for a maximum of 365 days. The calculation methodology can be summarized as follows:
The calculations reported in Table 3 and Figure 4 are conservative and include only chronic outpatient prescriptions and do not account for medications deprescribed in inpatient units, nursing home, community living centers, or domiciliary populations. Data tracked separately from inpatient and community living center patient populations indicated an additional 25,536 deprescribed medications, across 28 VA facilities, impacting 7,076 veterans with an average 2.15 medications deprescribed per veteran. The additional achieved cost avoidance was $370,272 (based on $14.50 average cost per prescription). Medications restarted within 30 days of deprescribing are not included in these calculations.
The cost avoidance calculation further excludes the effects of VIONE implementation on many other types of interventions. These interventions include, but are not limited to, changing from aggressive care to end of life, comfort care when strongly indicated; reduced emergency department visits or invasive diagnostic and therapeutic approaches, when not indicated; medical supplies, antimicrobial preparations; labor costs related to packaging, mailing, and administering prescriptions; reduced/prevented clinical waste; reduced decompensation of systemic illnesses and subsequent health care needs precipitated by iatrogenic disturbances and prolonged convalescence; and overall changes to prescribing practices through purposeful and targeted interactions with colleagues across various disciplines and various hierarchical levels.
Discussion
The VIONE clinical program exemplifies the translation of HRO principles into health care system practices. VIONE offers a systematic approach to improve medication management with an emphasis on deprescribing nonessential medications across various health care settings, facilitating VHA efforts toward zero harm. It demonstrates close alignment with the key building blocks of an HRO. Effective VIONE incorporation into an organizational culture reflects leadership commitment to safety and reliability in their vision and actions. By empowering staff to proactively reduce inappropriate medications and thereby prevent patient harm, VIONE contributes to enhancing an enterprise-wide culture of safety, with fewer errors and greater reliability. As a standardized decision support tool for the ongoing practice of assessment and planned cessation of potentially inappropriate medications, VIONE illustrates how continuous process improvement can be a part of staff-engaged, veteran-centered, highly reliable care. The standardization of the VIONE tool promotes achievement and sustainment of desired HRO principles and practices within health care delivery systems.
Conclusions
The VIONE program was launched not as a cost savings or research program but as a practical, real-time bedside or ambulatory care intervention to improve patient safety. Its value is reflected in the overwhelming response from scholarly and well-engaged colleagues expressing serious interests in expanding collaborations and tailoring efforts to add more depth and breadth to VIONE related efforts.
Acknowledgments
The authors express their gratitude to Central Arkansas VA Healthcare System leadership, Clinical Applications Coordinators, and colleagues for their unconditional support, to the Diffusion of Excellence programs at US Department of Veterans Affairs Central Office for their endorsement, and to the many VHA participants who renew our optimism and energy as we continue this exciting journey. We also thank Bridget B. Kelly for her assistance in writing and editing of the manuscript.
1. Chassin MR, Jerod ML. High-reliability health care: getting there from here. The Joint Commission. Milbank Q. 2013;91(3):459-490.
2. Makary MA, Daniel M. Medical error—the third leading cause of death in the US. BMJ. 2016;353:i2139.
3. Quinn KJ, Shah NH. A dataset quantifying polypharmacy in the United States. Sci Data. 2017;4:170167.
4. Scott IA, Hilmer SN, Reeve E, et al. Reducing inappropriate polypharmacy: the process of deprescribing. JAMA Intern Med. 2015;175(5):827-834.
5. Steinman MA. Polypharmacy—time to get beyond numbers. JAMA Intern Med. 2016;176(4):482-483.
6. US Department of Veterans Affairs. High reliability. https://dvagov.sharepoint.com/sites/OHT-PMO/high-reliability/Pages/default.aspx. [Nonpublic source, not verified.]
7. Gordon S, Mendenhall P, O’Connor BB. Beyond the Checklist: What Else Health Care Can Learn from Aviation Teamwork and Safety. Ithaca, NY: Cornell University Press; 2013.
8. Institute of Medicine (US) Committee on Quality of Health Care in America; Kohn LT, Corrigan JM, Donaldson MS, eds. To Err Is Human: Building a Safer Health System. Washington, DC: The National Academies Press; 2000.
9. US Department of Veterans Affairs. Diffusion of Excellence. https://www.va.gov/HEALTHCAREEXCELLENCE/diffusion-of-excellence/. Updated August 10, 2018. Accessed June 26, 2019.
10. US Department of Veterans Affairs. VIONE program toolkit. https://www.vapulse.net/docs/DOC-259375. [Nonpublic source, not verified.]
1. Chassin MR, Jerod ML. High-reliability health care: getting there from here. The Joint Commission. Milbank Q. 2013;91(3):459-490.
2. Makary MA, Daniel M. Medical error—the third leading cause of death in the US. BMJ. 2016;353:i2139.
3. Quinn KJ, Shah NH. A dataset quantifying polypharmacy in the United States. Sci Data. 2017;4:170167.
4. Scott IA, Hilmer SN, Reeve E, et al. Reducing inappropriate polypharmacy: the process of deprescribing. JAMA Intern Med. 2015;175(5):827-834.
5. Steinman MA. Polypharmacy—time to get beyond numbers. JAMA Intern Med. 2016;176(4):482-483.
6. US Department of Veterans Affairs. High reliability. https://dvagov.sharepoint.com/sites/OHT-PMO/high-reliability/Pages/default.aspx. [Nonpublic source, not verified.]
7. Gordon S, Mendenhall P, O’Connor BB. Beyond the Checklist: What Else Health Care Can Learn from Aviation Teamwork and Safety. Ithaca, NY: Cornell University Press; 2013.
8. Institute of Medicine (US) Committee on Quality of Health Care in America; Kohn LT, Corrigan JM, Donaldson MS, eds. To Err Is Human: Building a Safer Health System. Washington, DC: The National Academies Press; 2000.
9. US Department of Veterans Affairs. Diffusion of Excellence. https://www.va.gov/HEALTHCAREEXCELLENCE/diffusion-of-excellence/. Updated August 10, 2018. Accessed June 26, 2019.
10. US Department of Veterans Affairs. VIONE program toolkit. https://www.vapulse.net/docs/DOC-259375. [Nonpublic source, not verified.]
Sequential CRT, immunotherapy nets high PFS in node-positive cervical cancer
Sequential chemoradiotherapy (CRT) and immunotherapy is safe, well tolerated, and efficacious among patients with locally advanced cervical cancer being treated with curative intent, a multicenter phase 1 trial suggests.
Less than 10% of patients treated with this sequence experienced a grade 3 toxicity. Meanwhile, more than 80% were alive and free of disease progression at 1 year.
“Despite standard CRT, most women with lymph node–positive cervical cancer experience disease recurrence,” note the investigators, led by Jyoti S. Mayadev, MD, associate professor in the department of radiation medicine and applied sciences, University of California, San Diego, in La Jolla. “Our study is potentially transformative in the standard treatment schema of locally advanced cervical cancer, with the prospect for immuno-oncology to add durable survival in patients with node-positive disease, a current unmet oncologic need.”
The investigators enrolled in the trial 32 women from Gynecology Oncology Cooperative Group member institutions who had stage IB2 to IVA cervical cancer with positive pelvic and/or para-aortic lymph nodes. Treatment consisted of six weekly doses of cisplatin, 40 mg/m2, concurrent with extended-field, 3-dimensional conformal radiotherapy, followed by the immune checkpoint inhibitor ipilimumab (Yervoy) every 21 days for four cycles.
Results reported in JAMA Oncology showed that all 32 patients completed CRT and 21 patients went on to receive ipilimumab. Among the latter, 86% completed all four planned cycles and the rest completed two cycles.
In the group receiving sequential CRT and ipilimumab, 9.5% experienced grade 3 toxicity (lipase increase in one case and dermatitis in another case). Both toxicities were self-limited.
With a 14.8-month median follow-up, the patients treated with CRT-ipilimumab had a 12-month overall survival rate of 90%, and a 12-month progression-free survival rate of 81% (median durations were not reached). Neither human papillomavirus genotype nor HLA subtype was associated with these outcomes.
Translational analyses showed that patients experienced an increase in peripheral blood T cells expressing programmed cell death 1 (PD-1) after CRT that was then sustained with ipilimumab therapy. “[T]he use of an immune checkpoint inhibitor could stimulate the antitumor activity of tumor-specific cytotoxic T cells and augment radiation-induced neoantigen load,” the investigators proposed.
“To our knowledge, this phase 1 study is the first to show tolerability with a signal of efficacy of an immune check-point inhibitor ... as a part of the definitive treatment of locally advanced cervical cancer,” they concluded. “Our findings show promise for the use of immunotherapy in the definitive setting of locally advanced, node-positive cervical cancer; patients with this cancer historically have a poor prognosis with standard therapy alone.”
Dr. Mayadev disclosed receiving a grant from the National Cancer Institute during the conduct of the study, personal fees from AstraZeneca, grants from NRG Oncology, and personal fees and nonfinancial support from the Gynecology Oncology Group Foundation outside the submitted work; receiving compensation for serving on the advisory board of Varian Medical Systems in 2018; and being a speaker for Samsung Medical Systems in 2017. The study was supported by the National Cancer Institute and by institutional funds.
SOURCE: Mayadev JS et al. JAMA Oncol. 2019 Nov 27. doi: 10.1001/jamaoncol.2019.3857.
Sequential chemoradiotherapy (CRT) and immunotherapy is safe, well tolerated, and efficacious among patients with locally advanced cervical cancer being treated with curative intent, a multicenter phase 1 trial suggests.
Less than 10% of patients treated with this sequence experienced a grade 3 toxicity. Meanwhile, more than 80% were alive and free of disease progression at 1 year.
“Despite standard CRT, most women with lymph node–positive cervical cancer experience disease recurrence,” note the investigators, led by Jyoti S. Mayadev, MD, associate professor in the department of radiation medicine and applied sciences, University of California, San Diego, in La Jolla. “Our study is potentially transformative in the standard treatment schema of locally advanced cervical cancer, with the prospect for immuno-oncology to add durable survival in patients with node-positive disease, a current unmet oncologic need.”
The investigators enrolled in the trial 32 women from Gynecology Oncology Cooperative Group member institutions who had stage IB2 to IVA cervical cancer with positive pelvic and/or para-aortic lymph nodes. Treatment consisted of six weekly doses of cisplatin, 40 mg/m2, concurrent with extended-field, 3-dimensional conformal radiotherapy, followed by the immune checkpoint inhibitor ipilimumab (Yervoy) every 21 days for four cycles.
Results reported in JAMA Oncology showed that all 32 patients completed CRT and 21 patients went on to receive ipilimumab. Among the latter, 86% completed all four planned cycles and the rest completed two cycles.
In the group receiving sequential CRT and ipilimumab, 9.5% experienced grade 3 toxicity (lipase increase in one case and dermatitis in another case). Both toxicities were self-limited.
With a 14.8-month median follow-up, the patients treated with CRT-ipilimumab had a 12-month overall survival rate of 90%, and a 12-month progression-free survival rate of 81% (median durations were not reached). Neither human papillomavirus genotype nor HLA subtype was associated with these outcomes.
Translational analyses showed that patients experienced an increase in peripheral blood T cells expressing programmed cell death 1 (PD-1) after CRT that was then sustained with ipilimumab therapy. “[T]he use of an immune checkpoint inhibitor could stimulate the antitumor activity of tumor-specific cytotoxic T cells and augment radiation-induced neoantigen load,” the investigators proposed.
“To our knowledge, this phase 1 study is the first to show tolerability with a signal of efficacy of an immune check-point inhibitor ... as a part of the definitive treatment of locally advanced cervical cancer,” they concluded. “Our findings show promise for the use of immunotherapy in the definitive setting of locally advanced, node-positive cervical cancer; patients with this cancer historically have a poor prognosis with standard therapy alone.”
Dr. Mayadev disclosed receiving a grant from the National Cancer Institute during the conduct of the study, personal fees from AstraZeneca, grants from NRG Oncology, and personal fees and nonfinancial support from the Gynecology Oncology Group Foundation outside the submitted work; receiving compensation for serving on the advisory board of Varian Medical Systems in 2018; and being a speaker for Samsung Medical Systems in 2017. The study was supported by the National Cancer Institute and by institutional funds.
SOURCE: Mayadev JS et al. JAMA Oncol. 2019 Nov 27. doi: 10.1001/jamaoncol.2019.3857.
Sequential chemoradiotherapy (CRT) and immunotherapy is safe, well tolerated, and efficacious among patients with locally advanced cervical cancer being treated with curative intent, a multicenter phase 1 trial suggests.
Less than 10% of patients treated with this sequence experienced a grade 3 toxicity. Meanwhile, more than 80% were alive and free of disease progression at 1 year.
“Despite standard CRT, most women with lymph node–positive cervical cancer experience disease recurrence,” note the investigators, led by Jyoti S. Mayadev, MD, associate professor in the department of radiation medicine and applied sciences, University of California, San Diego, in La Jolla. “Our study is potentially transformative in the standard treatment schema of locally advanced cervical cancer, with the prospect for immuno-oncology to add durable survival in patients with node-positive disease, a current unmet oncologic need.”
The investigators enrolled in the trial 32 women from Gynecology Oncology Cooperative Group member institutions who had stage IB2 to IVA cervical cancer with positive pelvic and/or para-aortic lymph nodes. Treatment consisted of six weekly doses of cisplatin, 40 mg/m2, concurrent with extended-field, 3-dimensional conformal radiotherapy, followed by the immune checkpoint inhibitor ipilimumab (Yervoy) every 21 days for four cycles.
Results reported in JAMA Oncology showed that all 32 patients completed CRT and 21 patients went on to receive ipilimumab. Among the latter, 86% completed all four planned cycles and the rest completed two cycles.
In the group receiving sequential CRT and ipilimumab, 9.5% experienced grade 3 toxicity (lipase increase in one case and dermatitis in another case). Both toxicities were self-limited.
With a 14.8-month median follow-up, the patients treated with CRT-ipilimumab had a 12-month overall survival rate of 90%, and a 12-month progression-free survival rate of 81% (median durations were not reached). Neither human papillomavirus genotype nor HLA subtype was associated with these outcomes.
Translational analyses showed that patients experienced an increase in peripheral blood T cells expressing programmed cell death 1 (PD-1) after CRT that was then sustained with ipilimumab therapy. “[T]he use of an immune checkpoint inhibitor could stimulate the antitumor activity of tumor-specific cytotoxic T cells and augment radiation-induced neoantigen load,” the investigators proposed.
“To our knowledge, this phase 1 study is the first to show tolerability with a signal of efficacy of an immune check-point inhibitor ... as a part of the definitive treatment of locally advanced cervical cancer,” they concluded. “Our findings show promise for the use of immunotherapy in the definitive setting of locally advanced, node-positive cervical cancer; patients with this cancer historically have a poor prognosis with standard therapy alone.”
Dr. Mayadev disclosed receiving a grant from the National Cancer Institute during the conduct of the study, personal fees from AstraZeneca, grants from NRG Oncology, and personal fees and nonfinancial support from the Gynecology Oncology Group Foundation outside the submitted work; receiving compensation for serving on the advisory board of Varian Medical Systems in 2018; and being a speaker for Samsung Medical Systems in 2017. The study was supported by the National Cancer Institute and by institutional funds.
SOURCE: Mayadev JS et al. JAMA Oncol. 2019 Nov 27. doi: 10.1001/jamaoncol.2019.3857.
FROM JAMA ONCOLOGY
Icosapent ethyl cost effective in REDUCE-IT analysis
PHILADELPHIA – The overall costs of icosapent ethyl were less than placebo, and the medication reduced cardiovascular events by 30% at a cost that fits well within acceptable quality-adjusted life-year (QALY) parameters, according to a cost-effectiveness analysis of the REDUCE-IT trial.

Days before the presentation of the analysis at the American Heart Association scientific sessions, a Food and Drug Administration advisory panel unanimously recommended approval of icosapent ethyl (Vascepa) for a new indication for reducing CV event risk. Icosapent ethyl, a highly purified form of the ethyl ester of eicosapentaenoic acid derived from fish oil, received FDA approval in 2012 for treatment of triglyceride levels of at least 500 mg/dL.
“What we found here is that icosapent ethyl is a dominant strategy,” said William S. Weintraub, MD, director of outcomes research at MedStar Heart & Vascular Institute in Washington, in reporting preliminary cost-analysis findings from REDUCE-IT (Reduction of Cardiovascular Events With Icosapent Ethyl – Intervention Trial). “It’s offering better outcomes at a lower cost.”
The dominant strategy was demonstrated by cost savings in 70% of simulations the cost-effectiveness analysis ran, Dr. Weintraub said.
“These are very impressive results,” said session moderator Seth S. Martin, MD, an internist and cardiologist at Johns Hopkins University, Baltimore. “We don’t often see dominant strategies for new drugs. This is very exciting.”
“Almost never,” Dr. Weintraub responded.
REDUCE-IT randomized 8,179 patients with a diagnosis of CVD or with diabetes and other risk factors who had been on statins and had triglycerides of 135-499 mg/dL to either 4 g of icosapent ethyl daily or placebo (N Engl J Med. 2019;380:11-22). Trial results showed the treatment group had an absolute risk reduction of 4.8% and a relative risk reduction of 25% of first CV events and a 30% relative risk reduction for total events, Dr. Weintraub said.
The analysis determined that the QALYs for icosapent ethyl versus those for placebo were 3.34 and 3.27, respectively, during the trial period and 11.61 and 11.35 over a lifetime. The mean costs for the two treatments were $27,576 and $28,205 during the trial period and $235,352 and $236,636 lifetime, respectively, Dr. Weintraub said.
An analysis of cost effectiveness showed that almost all of the estimates fell below the willingness-to-pay (WTP) threshold of $50,000 per QALY gained, Dr. Weintraub said. “In fact, some 70% plus are in what’s called quadrant two; that is, decreased cost and increased efficacy.”
The analysis also calculated the value of icosapent ethyl at three different WTP thresholds: up to $6 a day at a WTP of $50,000, up to $12 a day at $100,000, and up to $18 a day at $150,000. The analysis used the actual net pricing of $4.16 a day, Dr. Weintraub said. “That’s why we showed we have the dominant strategy,” he said.
Further cost-effectiveness analyses of the REDUCE-IT data will focus on subgroups, such as U.S. and non–U.S. patients and people with diabetes. He also emphasized the data he reported were preliminary. “We have a lot more work to do,” Dr. Weintraub said.
Dr. Weintraub reported having financial relationships with Amarin Pharma, which markets Vascepa, and AstraZeneca.
SOURCE: Weintraub WS. AHA 2019, Session FS.AOS.F1.
PHILADELPHIA – The overall costs of icosapent ethyl were less than placebo, and the medication reduced cardiovascular events by 30% at a cost that fits well within acceptable quality-adjusted life-year (QALY) parameters, according to a cost-effectiveness analysis of the REDUCE-IT trial.
Days before the presentation of the analysis at the American Heart Association scientific sessions, a Food and Drug Administration advisory panel unanimously recommended approval of icosapent ethyl (Vascepa) for a new indication for reducing CV event risk. Icosapent ethyl, a highly purified form of the ethyl ester of eicosapentaenoic acid derived from fish oil, received FDA approval in 2012 for treatment of triglyceride levels of at least 500 mg/dL.
“What we found here is that icosapent ethyl is a dominant strategy,” said William S. Weintraub, MD, director of outcomes research at MedStar Heart & Vascular Institute in Washington, in reporting preliminary cost-analysis findings from REDUCE-IT (Reduction of Cardiovascular Events With Icosapent Ethyl – Intervention Trial). “It’s offering better outcomes at a lower cost.”
The dominant strategy was demonstrated by cost savings in 70% of simulations the cost-effectiveness analysis ran, Dr. Weintraub said.
“These are very impressive results,” said session moderator Seth S. Martin, MD, an internist and cardiologist at Johns Hopkins University, Baltimore. “We don’t often see dominant strategies for new drugs. This is very exciting.”
“Almost never,” Dr. Weintraub responded.
REDUCE-IT randomized 8,179 patients with a diagnosis of CVD or with diabetes and other risk factors who had been on statins and had triglycerides of 135-499 mg/dL to either 4 g of icosapent ethyl daily or placebo (N Engl J Med. 2019;380:11-22). Trial results showed the treatment group had an absolute risk reduction of 4.8% and a relative risk reduction of 25% of first CV events and a 30% relative risk reduction for total events, Dr. Weintraub said.
The analysis determined that the QALYs for icosapent ethyl versus those for placebo were 3.34 and 3.27, respectively, during the trial period and 11.61 and 11.35 over a lifetime. The mean costs for the two treatments were $27,576 and $28,205 during the trial period and $235,352 and $236,636 lifetime, respectively, Dr. Weintraub said.
An analysis of cost effectiveness showed that almost all of the estimates fell below the willingness-to-pay (WTP) threshold of $50,000 per QALY gained, Dr. Weintraub said. “In fact, some 70% plus are in what’s called quadrant two; that is, decreased cost and increased efficacy.”
The analysis also calculated the value of icosapent ethyl at three different WTP thresholds: up to $6 a day at a WTP of $50,000, up to $12 a day at $100,000, and up to $18 a day at $150,000. The analysis used the actual net pricing of $4.16 a day, Dr. Weintraub said. “That’s why we showed we have the dominant strategy,” he said.
Further cost-effectiveness analyses of the REDUCE-IT data will focus on subgroups, such as U.S. and non–U.S. patients and people with diabetes. He also emphasized the data he reported were preliminary. “We have a lot more work to do,” Dr. Weintraub said.
Dr. Weintraub reported having financial relationships with Amarin Pharma, which markets Vascepa, and AstraZeneca.
SOURCE: Weintraub WS. AHA 2019, Session FS.AOS.F1.
PHILADELPHIA – The overall costs of icosapent ethyl were less than placebo, and the medication reduced cardiovascular events by 30% at a cost that fits well within acceptable quality-adjusted life-year (QALY) parameters, according to a cost-effectiveness analysis of the REDUCE-IT trial.
Days before the presentation of the analysis at the American Heart Association scientific sessions, a Food and Drug Administration advisory panel unanimously recommended approval of icosapent ethyl (Vascepa) for a new indication for reducing CV event risk. Icosapent ethyl, a highly purified form of the ethyl ester of eicosapentaenoic acid derived from fish oil, received FDA approval in 2012 for treatment of triglyceride levels of at least 500 mg/dL.
“What we found here is that icosapent ethyl is a dominant strategy,” said William S. Weintraub, MD, director of outcomes research at MedStar Heart & Vascular Institute in Washington, in reporting preliminary cost-analysis findings from REDUCE-IT (Reduction of Cardiovascular Events With Icosapent Ethyl – Intervention Trial). “It’s offering better outcomes at a lower cost.”
The dominant strategy was demonstrated by cost savings in 70% of simulations the cost-effectiveness analysis ran, Dr. Weintraub said.
“These are very impressive results,” said session moderator Seth S. Martin, MD, an internist and cardiologist at Johns Hopkins University, Baltimore. “We don’t often see dominant strategies for new drugs. This is very exciting.”
“Almost never,” Dr. Weintraub responded.
REDUCE-IT randomized 8,179 patients with a diagnosis of CVD or with diabetes and other risk factors who had been on statins and had triglycerides of 135-499 mg/dL to either 4 g of icosapent ethyl daily or placebo (N Engl J Med. 2019;380:11-22). Trial results showed the treatment group had an absolute risk reduction of 4.8% and a relative risk reduction of 25% of first CV events and a 30% relative risk reduction for total events, Dr. Weintraub said.
The analysis determined that the QALYs for icosapent ethyl versus those for placebo were 3.34 and 3.27, respectively, during the trial period and 11.61 and 11.35 over a lifetime. The mean costs for the two treatments were $27,576 and $28,205 during the trial period and $235,352 and $236,636 lifetime, respectively, Dr. Weintraub said.
An analysis of cost effectiveness showed that almost all of the estimates fell below the willingness-to-pay (WTP) threshold of $50,000 per QALY gained, Dr. Weintraub said. “In fact, some 70% plus are in what’s called quadrant two; that is, decreased cost and increased efficacy.”
The analysis also calculated the value of icosapent ethyl at three different WTP thresholds: up to $6 a day at a WTP of $50,000, up to $12 a day at $100,000, and up to $18 a day at $150,000. The analysis used the actual net pricing of $4.16 a day, Dr. Weintraub said. “That’s why we showed we have the dominant strategy,” he said.
Further cost-effectiveness analyses of the REDUCE-IT data will focus on subgroups, such as U.S. and non–U.S. patients and people with diabetes. He also emphasized the data he reported were preliminary. “We have a lot more work to do,” Dr. Weintraub said.
Dr. Weintraub reported having financial relationships with Amarin Pharma, which markets Vascepa, and AstraZeneca.
SOURCE: Weintraub WS. AHA 2019, Session FS.AOS.F1.
REPORTING FROM AHA 2019
DAPA-HF: Dapagliflozin benefits regardless of age, HF severity
PHILADELPHIA – The substantial benefits from adding dapagliflozin to guideline-directed medical therapy for patients with heart failure with reduced ejection fraction enrolled in the DAPA-HF trial applied to patients regardless of their age or baseline health status, a pair of new post hoc analyses suggest.
These findings emerged a day after a report that more fully delineated dapagliflozin’s consistent safety and efficacy in patients with heart failure with reduced ejection fraction (HFrEF) regardless of whether they also had type 2 diabetes. One of the new, post hoc analyses reported at the American Heart Association scientific sessions suggested that even the most elderly enrolled patients, 75 years and older, had a similar cut in mortality and acute heart failure exacerbations, compared with younger patients. A second post hoc analysis indicated that patients with severe heart failure symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with mild baseline symptoms, measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ).
The primary results from the DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial, first reported in August 2019, showed that among more than 4,700 patients with HFrEF randomized to receive the sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (Farxiga) on top of standard HFrEF medications or placebo, those who received dapagliflozin had a statistically significant, 26% decrease in their incidence of the primary study endpoint over a median 18 months, regardless of diabetes status (N Engl J Med. 2019 Nov 21;381[21]:1995-2008).
“These benefits were entirely consistent across the range of ages studied,” extending from patients younger than 55 years to those older than 75 years, John McMurray, MD, said at the meeting. “In many parts of the world, particularly North America and Western Europe, we have an increasingly elderly population. Many patients with heart failure are much older than in clinical trials,” he said.

“The thing of concern is whether elderly patients get as much benefit and tolerate treatment as well as younger patients,” said Dr. McMurray, professor of medical cardiology at the University of Glasgow.
“Dapagliflozin worked across all ages, including some very elderly patients enrolled in the trial,” said Mary Norine Walsh, MD, medical director of the heart failure and transplant program at St. Vincent Heart Center of Indiana in Indianapolis. “Many trials have not looked at age like this. I hope this is a new way to analyze trials to produce more information that can help patients,” she said in an interview.

Quality-of-life outcomes
The other new, post hoc analysis showed that patients with severe HF symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with milder baseline symptoms and less impaired function, measured by the KCCQ. Dapagliflozin treatment “improved cardiovascular death and worsening heart failure to a similar extent across the entire range of KCCQ at baseline,” Mikhail N. Kosiborod, MD, said in a separate talk at the meeting. In addition, dapagliflozin treatment increased the rate of small, moderate, and large clinically meaningful improvements in patients’ KCCQ scores across all key domains of the metric, which scores symptom frequency and severity, physical and social limitations, and quality of life, said Dr. Kosiborod, a cardiologist and professor of medicine at the University of Missouri–Kansas City.

After the first 8 months of treatment in the DAPA-HF trial, 58% of the 2,373 patients who received dapagliflozin had a clinically meaningful improvement in their total KCCQ symptom score of at least 5 points, compared with a 51% rate in the 2,371 patients in the control arm, a statistically significant difference. This meant that the number needed to treat with dapagliflozin was 14 patients to produce one additional patient with at least a 5-point KCCQ improvement compared with controls, a “very small” number needed to treat, Dr. Kosiborod said in an interview.
Addition of the KCCQ to the panel of assessments that patients underwent during DAPA-HF reflected an evolved approach to measuring efficacy outcomes in clinical trials by including patient-reported outcomes. Earlier in 2019, the Food and Drug Administration released draft guidance for heart failure drug development that explicitly called for efficacy endpoints in pivotal studies that measure how patients feel and function, and stating that these endpoints can be the basis for new drug approvals.
“To many patients, how they feel matters as much if not more than how long they live,” Dr. Kosiborod noted. The goals of heart failure treatments are not only to extend survival and reduced hospitalizations, but also to improve symptoms, function, and quality of life, he said.
“There is a lot of interest now in having outcomes in heart failure trials that are more meaningful to patients, like feeling better and being able to do more,” noted Dr. Walsh.
The DAPA-HF results also showed that patients had similar rates of reduction in death, heart failure hospitalization, or urgent clinical visits, regardless of how severely they were affected by their heart failure when they began dapagliflozin treatment. The researchers ran an analysis that divided the entire trial population into tertiles based on their KCCQ score on entering the study. Patients in the most severely-affected tertile had a 30% cut in their rate of death or acute heart failure exacerbation on dapagliflozin compared with placebo, while patients in the tertile with the mildest symptoms at baseline had a 38% reduction in their primary outcome incidence compared with controls who received placebo. Concurrently with Dr. Kosiborod’s report, the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044138).
Outcomes by age
Not surprisingly in DAPA-HF, the older patients were, the sicker, Dr. McMurray observed. Of the study’s 1,149 patients (24% of the study cohort) who were at least 75 years old, 62% had chronic kidney disease, compared with a 14% prevalence among the 636 patients younger than age 55. The 75-and-older group showed a steeper, 32% decline in incidence of the primary endpoint – a composite of cardiovascular (CV) death, HF hospitalization, or urgent HF visit requiring intravenous therapy – than in the other studied age groups: a 24% decline in those 65-74 years old, a 29% cut in those 55-64 years old, and a 13% drop in patients younger than 55 years old.
In addition, patients aged 75 years or greater were just as likely as the overall group to show at least a 5-point improvement in their KCCQ Total Symptom Score on dapagliflozin, as well as about the same reduced rate of deterioration compared with placebo as tracked with the KCCQ.
Patients “got as much benefit in terms of symptoms as well as morbidity and mortality,” Dr. McMurray concluded. Concurrently with the meeting report the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044133).
“These data are of critical importance, as improving patient-reported outcomes in heart failure, especially in highly symptomatic patients, is an important goal in drug development,” G. Michael Felker, MD, wrote in an editorial accompanying the two published analyses (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044578). These new analyses also highlight another attractive feature of dapagliflozin and, apparently, the entire class of SGLT2 inhibitors: They “ ‘play well with others’ when it comes to overlapping intolerances that often limit (either in reality or in perception) optimization of GDMT [guideline-directed medical therapy]. Although SGLT2 inhibitor therapy may lead to volume depletion and require adjustment of diuretics, the SGLT2 inhibitors generally lack some of the other dose-limiting adverse effects (such as renal dysfunction, hyperkalemia, and hypotension) that can make aggressive up-titration of GDMT problematic, particularly in older patients or those with more advanced disease,“ wrote Dr. Felker, professor of medicine at Duke University in Durham, N.C. “We stand at the beginning of a new era of ‘quadruple therapy’ for HFrEF with beta-blockers, an angiotensin receptor neprilysin inhibitor, mineralocorticoid receptor antagonists, and SGLT2 inhibitors,” he concluded.
A version of this article also appears on Medscape.com
In DAPA-HF, treatment with dapagliflozin met the three critical goals of heart failure management. When used on top of current guideline-directed medical therapy, the treatment reduced mortality, cut hospitalizations, and improved heart failure–related health status – all to a similar extent regardless of patients’ age or symptom severity at entry. These new, post hoc findings provide important, additional data supporting inhibition of sodium-glucose cotransporter (SGLT) 2 with dapagliflozin as the newest foundational pillar of treatment for heart failure with reduced ejection fraction (HFrEF).

The results of the analysis by baseline symptoms severity as measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ) showed similar treatment effects from dapagliflozin regardless of a patient’s baseline KCCQ score, suggesting that the prior report of a blunted effect of dapagliflozin in patients classified at baseline as being in New York Heart Association functional class III or IV compared with class I and II patients was likely a chance finding.
Both the analyses by age and by KCCQ scores were limited by their post hoc status using data collected in a single study. No evidence addresses whether these are class effects for all drugs in the SGLT2-inhibitor class, whether these findings from DAPA-HF are generalizable to real world practice, or whether treatment with dapagliflozin would have similar effects on outcomes if it had been used more often in combination with sacubitril/valsartan. In DAPA-HF, 11% of patients also received sacubitril/valsartan even though existing management guidelines recommend sacubitril/valsartan as the preferred agent for inhibiting the renin-angiotensin system.
It’s also unclear whether patient-reported outcomes such as those measured by the KCCQ will help in sequencing the introduction of drugs for HFrEF patients, or drug selection by patients, providers, payers, and in guidelines.
Carolyn S.P. Lam, MD, is professor of medicine at Duke-National University of Singapore. She has been a consultant to and has received research funding from AstraZeneca and several other companies. She made these comments as designated discussant for the two reports.
In DAPA-HF, treatment with dapagliflozin met the three critical goals of heart failure management. When used on top of current guideline-directed medical therapy, the treatment reduced mortality, cut hospitalizations, and improved heart failure–related health status – all to a similar extent regardless of patients’ age or symptom severity at entry. These new, post hoc findings provide important, additional data supporting inhibition of sodium-glucose cotransporter (SGLT) 2 with dapagliflozin as the newest foundational pillar of treatment for heart failure with reduced ejection fraction (HFrEF).
The results of the analysis by baseline symptoms severity as measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ) showed similar treatment effects from dapagliflozin regardless of a patient’s baseline KCCQ score, suggesting that the prior report of a blunted effect of dapagliflozin in patients classified at baseline as being in New York Heart Association functional class III or IV compared with class I and II patients was likely a chance finding.
Both the analyses by age and by KCCQ scores were limited by their post hoc status using data collected in a single study. No evidence addresses whether these are class effects for all drugs in the SGLT2-inhibitor class, whether these findings from DAPA-HF are generalizable to real world practice, or whether treatment with dapagliflozin would have similar effects on outcomes if it had been used more often in combination with sacubitril/valsartan. In DAPA-HF, 11% of patients also received sacubitril/valsartan even though existing management guidelines recommend sacubitril/valsartan as the preferred agent for inhibiting the renin-angiotensin system.
It’s also unclear whether patient-reported outcomes such as those measured by the KCCQ will help in sequencing the introduction of drugs for HFrEF patients, or drug selection by patients, providers, payers, and in guidelines.
Carolyn S.P. Lam, MD, is professor of medicine at Duke-National University of Singapore. She has been a consultant to and has received research funding from AstraZeneca and several other companies. She made these comments as designated discussant for the two reports.
In DAPA-HF, treatment with dapagliflozin met the three critical goals of heart failure management. When used on top of current guideline-directed medical therapy, the treatment reduced mortality, cut hospitalizations, and improved heart failure–related health status – all to a similar extent regardless of patients’ age or symptom severity at entry. These new, post hoc findings provide important, additional data supporting inhibition of sodium-glucose cotransporter (SGLT) 2 with dapagliflozin as the newest foundational pillar of treatment for heart failure with reduced ejection fraction (HFrEF).
The results of the analysis by baseline symptoms severity as measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ) showed similar treatment effects from dapagliflozin regardless of a patient’s baseline KCCQ score, suggesting that the prior report of a blunted effect of dapagliflozin in patients classified at baseline as being in New York Heart Association functional class III or IV compared with class I and II patients was likely a chance finding.
Both the analyses by age and by KCCQ scores were limited by their post hoc status using data collected in a single study. No evidence addresses whether these are class effects for all drugs in the SGLT2-inhibitor class, whether these findings from DAPA-HF are generalizable to real world practice, or whether treatment with dapagliflozin would have similar effects on outcomes if it had been used more often in combination with sacubitril/valsartan. In DAPA-HF, 11% of patients also received sacubitril/valsartan even though existing management guidelines recommend sacubitril/valsartan as the preferred agent for inhibiting the renin-angiotensin system.
It’s also unclear whether patient-reported outcomes such as those measured by the KCCQ will help in sequencing the introduction of drugs for HFrEF patients, or drug selection by patients, providers, payers, and in guidelines.
Carolyn S.P. Lam, MD, is professor of medicine at Duke-National University of Singapore. She has been a consultant to and has received research funding from AstraZeneca and several other companies. She made these comments as designated discussant for the two reports.
PHILADELPHIA – The substantial benefits from adding dapagliflozin to guideline-directed medical therapy for patients with heart failure with reduced ejection fraction enrolled in the DAPA-HF trial applied to patients regardless of their age or baseline health status, a pair of new post hoc analyses suggest.
These findings emerged a day after a report that more fully delineated dapagliflozin’s consistent safety and efficacy in patients with heart failure with reduced ejection fraction (HFrEF) regardless of whether they also had type 2 diabetes. One of the new, post hoc analyses reported at the American Heart Association scientific sessions suggested that even the most elderly enrolled patients, 75 years and older, had a similar cut in mortality and acute heart failure exacerbations, compared with younger patients. A second post hoc analysis indicated that patients with severe heart failure symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with mild baseline symptoms, measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ).
The primary results from the DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial, first reported in August 2019, showed that among more than 4,700 patients with HFrEF randomized to receive the sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (Farxiga) on top of standard HFrEF medications or placebo, those who received dapagliflozin had a statistically significant, 26% decrease in their incidence of the primary study endpoint over a median 18 months, regardless of diabetes status (N Engl J Med. 2019 Nov 21;381[21]:1995-2008).
“These benefits were entirely consistent across the range of ages studied,” extending from patients younger than 55 years to those older than 75 years, John McMurray, MD, said at the meeting. “In many parts of the world, particularly North America and Western Europe, we have an increasingly elderly population. Many patients with heart failure are much older than in clinical trials,” he said.
“The thing of concern is whether elderly patients get as much benefit and tolerate treatment as well as younger patients,” said Dr. McMurray, professor of medical cardiology at the University of Glasgow.
“Dapagliflozin worked across all ages, including some very elderly patients enrolled in the trial,” said Mary Norine Walsh, MD, medical director of the heart failure and transplant program at St. Vincent Heart Center of Indiana in Indianapolis. “Many trials have not looked at age like this. I hope this is a new way to analyze trials to produce more information that can help patients,” she said in an interview.
Quality-of-life outcomes
The other new, post hoc analysis showed that patients with severe HF symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with milder baseline symptoms and less impaired function, measured by the KCCQ. Dapagliflozin treatment “improved cardiovascular death and worsening heart failure to a similar extent across the entire range of KCCQ at baseline,” Mikhail N. Kosiborod, MD, said in a separate talk at the meeting. In addition, dapagliflozin treatment increased the rate of small, moderate, and large clinically meaningful improvements in patients’ KCCQ scores across all key domains of the metric, which scores symptom frequency and severity, physical and social limitations, and quality of life, said Dr. Kosiborod, a cardiologist and professor of medicine at the University of Missouri–Kansas City.
After the first 8 months of treatment in the DAPA-HF trial, 58% of the 2,373 patients who received dapagliflozin had a clinically meaningful improvement in their total KCCQ symptom score of at least 5 points, compared with a 51% rate in the 2,371 patients in the control arm, a statistically significant difference. This meant that the number needed to treat with dapagliflozin was 14 patients to produce one additional patient with at least a 5-point KCCQ improvement compared with controls, a “very small” number needed to treat, Dr. Kosiborod said in an interview.
Addition of the KCCQ to the panel of assessments that patients underwent during DAPA-HF reflected an evolved approach to measuring efficacy outcomes in clinical trials by including patient-reported outcomes. Earlier in 2019, the Food and Drug Administration released draft guidance for heart failure drug development that explicitly called for efficacy endpoints in pivotal studies that measure how patients feel and function, and stating that these endpoints can be the basis for new drug approvals.
“To many patients, how they feel matters as much if not more than how long they live,” Dr. Kosiborod noted. The goals of heart failure treatments are not only to extend survival and reduced hospitalizations, but also to improve symptoms, function, and quality of life, he said.
“There is a lot of interest now in having outcomes in heart failure trials that are more meaningful to patients, like feeling better and being able to do more,” noted Dr. Walsh.
The DAPA-HF results also showed that patients had similar rates of reduction in death, heart failure hospitalization, or urgent clinical visits, regardless of how severely they were affected by their heart failure when they began dapagliflozin treatment. The researchers ran an analysis that divided the entire trial population into tertiles based on their KCCQ score on entering the study. Patients in the most severely-affected tertile had a 30% cut in their rate of death or acute heart failure exacerbation on dapagliflozin compared with placebo, while patients in the tertile with the mildest symptoms at baseline had a 38% reduction in their primary outcome incidence compared with controls who received placebo. Concurrently with Dr. Kosiborod’s report, the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044138).
Outcomes by age
Not surprisingly in DAPA-HF, the older patients were, the sicker, Dr. McMurray observed. Of the study’s 1,149 patients (24% of the study cohort) who were at least 75 years old, 62% had chronic kidney disease, compared with a 14% prevalence among the 636 patients younger than age 55. The 75-and-older group showed a steeper, 32% decline in incidence of the primary endpoint – a composite of cardiovascular (CV) death, HF hospitalization, or urgent HF visit requiring intravenous therapy – than in the other studied age groups: a 24% decline in those 65-74 years old, a 29% cut in those 55-64 years old, and a 13% drop in patients younger than 55 years old.
In addition, patients aged 75 years or greater were just as likely as the overall group to show at least a 5-point improvement in their KCCQ Total Symptom Score on dapagliflozin, as well as about the same reduced rate of deterioration compared with placebo as tracked with the KCCQ.
Patients “got as much benefit in terms of symptoms as well as morbidity and mortality,” Dr. McMurray concluded. Concurrently with the meeting report the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044133).
“These data are of critical importance, as improving patient-reported outcomes in heart failure, especially in highly symptomatic patients, is an important goal in drug development,” G. Michael Felker, MD, wrote in an editorial accompanying the two published analyses (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044578). These new analyses also highlight another attractive feature of dapagliflozin and, apparently, the entire class of SGLT2 inhibitors: They “ ‘play well with others’ when it comes to overlapping intolerances that often limit (either in reality or in perception) optimization of GDMT [guideline-directed medical therapy]. Although SGLT2 inhibitor therapy may lead to volume depletion and require adjustment of diuretics, the SGLT2 inhibitors generally lack some of the other dose-limiting adverse effects (such as renal dysfunction, hyperkalemia, and hypotension) that can make aggressive up-titration of GDMT problematic, particularly in older patients or those with more advanced disease,“ wrote Dr. Felker, professor of medicine at Duke University in Durham, N.C. “We stand at the beginning of a new era of ‘quadruple therapy’ for HFrEF with beta-blockers, an angiotensin receptor neprilysin inhibitor, mineralocorticoid receptor antagonists, and SGLT2 inhibitors,” he concluded.
A version of this article also appears on Medscape.com
PHILADELPHIA – The substantial benefits from adding dapagliflozin to guideline-directed medical therapy for patients with heart failure with reduced ejection fraction enrolled in the DAPA-HF trial applied to patients regardless of their age or baseline health status, a pair of new post hoc analyses suggest.
These findings emerged a day after a report that more fully delineated dapagliflozin’s consistent safety and efficacy in patients with heart failure with reduced ejection fraction (HFrEF) regardless of whether they also had type 2 diabetes. One of the new, post hoc analyses reported at the American Heart Association scientific sessions suggested that even the most elderly enrolled patients, 75 years and older, had a similar cut in mortality and acute heart failure exacerbations, compared with younger patients. A second post hoc analysis indicated that patients with severe heart failure symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with mild baseline symptoms, measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ).
The primary results from the DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial, first reported in August 2019, showed that among more than 4,700 patients with HFrEF randomized to receive the sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (Farxiga) on top of standard HFrEF medications or placebo, those who received dapagliflozin had a statistically significant, 26% decrease in their incidence of the primary study endpoint over a median 18 months, regardless of diabetes status (N Engl J Med. 2019 Nov 21;381[21]:1995-2008).
“These benefits were entirely consistent across the range of ages studied,” extending from patients younger than 55 years to those older than 75 years, John McMurray, MD, said at the meeting. “In many parts of the world, particularly North America and Western Europe, we have an increasingly elderly population. Many patients with heart failure are much older than in clinical trials,” he said.
“The thing of concern is whether elderly patients get as much benefit and tolerate treatment as well as younger patients,” said Dr. McMurray, professor of medical cardiology at the University of Glasgow.
“Dapagliflozin worked across all ages, including some very elderly patients enrolled in the trial,” said Mary Norine Walsh, MD, medical director of the heart failure and transplant program at St. Vincent Heart Center of Indiana in Indianapolis. “Many trials have not looked at age like this. I hope this is a new way to analyze trials to produce more information that can help patients,” she said in an interview.
Quality-of-life outcomes
The other new, post hoc analysis showed that patients with severe HF symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with milder baseline symptoms and less impaired function, measured by the KCCQ. Dapagliflozin treatment “improved cardiovascular death and worsening heart failure to a similar extent across the entire range of KCCQ at baseline,” Mikhail N. Kosiborod, MD, said in a separate talk at the meeting. In addition, dapagliflozin treatment increased the rate of small, moderate, and large clinically meaningful improvements in patients’ KCCQ scores across all key domains of the metric, which scores symptom frequency and severity, physical and social limitations, and quality of life, said Dr. Kosiborod, a cardiologist and professor of medicine at the University of Missouri–Kansas City.
After the first 8 months of treatment in the DAPA-HF trial, 58% of the 2,373 patients who received dapagliflozin had a clinically meaningful improvement in their total KCCQ symptom score of at least 5 points, compared with a 51% rate in the 2,371 patients in the control arm, a statistically significant difference. This meant that the number needed to treat with dapagliflozin was 14 patients to produce one additional patient with at least a 5-point KCCQ improvement compared with controls, a “very small” number needed to treat, Dr. Kosiborod said in an interview.
Addition of the KCCQ to the panel of assessments that patients underwent during DAPA-HF reflected an evolved approach to measuring efficacy outcomes in clinical trials by including patient-reported outcomes. Earlier in 2019, the Food and Drug Administration released draft guidance for heart failure drug development that explicitly called for efficacy endpoints in pivotal studies that measure how patients feel and function, and stating that these endpoints can be the basis for new drug approvals.
“To many patients, how they feel matters as much if not more than how long they live,” Dr. Kosiborod noted. The goals of heart failure treatments are not only to extend survival and reduced hospitalizations, but also to improve symptoms, function, and quality of life, he said.
“There is a lot of interest now in having outcomes in heart failure trials that are more meaningful to patients, like feeling better and being able to do more,” noted Dr. Walsh.
The DAPA-HF results also showed that patients had similar rates of reduction in death, heart failure hospitalization, or urgent clinical visits, regardless of how severely they were affected by their heart failure when they began dapagliflozin treatment. The researchers ran an analysis that divided the entire trial population into tertiles based on their KCCQ score on entering the study. Patients in the most severely-affected tertile had a 30% cut in their rate of death or acute heart failure exacerbation on dapagliflozin compared with placebo, while patients in the tertile with the mildest symptoms at baseline had a 38% reduction in their primary outcome incidence compared with controls who received placebo. Concurrently with Dr. Kosiborod’s report, the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044138).
Outcomes by age
Not surprisingly in DAPA-HF, the older patients were, the sicker, Dr. McMurray observed. Of the study’s 1,149 patients (24% of the study cohort) who were at least 75 years old, 62% had chronic kidney disease, compared with a 14% prevalence among the 636 patients younger than age 55. The 75-and-older group showed a steeper, 32% decline in incidence of the primary endpoint – a composite of cardiovascular (CV) death, HF hospitalization, or urgent HF visit requiring intravenous therapy – than in the other studied age groups: a 24% decline in those 65-74 years old, a 29% cut in those 55-64 years old, and a 13% drop in patients younger than 55 years old.
In addition, patients aged 75 years or greater were just as likely as the overall group to show at least a 5-point improvement in their KCCQ Total Symptom Score on dapagliflozin, as well as about the same reduced rate of deterioration compared with placebo as tracked with the KCCQ.
Patients “got as much benefit in terms of symptoms as well as morbidity and mortality,” Dr. McMurray concluded. Concurrently with the meeting report the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044133).
“These data are of critical importance, as improving patient-reported outcomes in heart failure, especially in highly symptomatic patients, is an important goal in drug development,” G. Michael Felker, MD, wrote in an editorial accompanying the two published analyses (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044578). These new analyses also highlight another attractive feature of dapagliflozin and, apparently, the entire class of SGLT2 inhibitors: They “ ‘play well with others’ when it comes to overlapping intolerances that often limit (either in reality or in perception) optimization of GDMT [guideline-directed medical therapy]. Although SGLT2 inhibitor therapy may lead to volume depletion and require adjustment of diuretics, the SGLT2 inhibitors generally lack some of the other dose-limiting adverse effects (such as renal dysfunction, hyperkalemia, and hypotension) that can make aggressive up-titration of GDMT problematic, particularly in older patients or those with more advanced disease,“ wrote Dr. Felker, professor of medicine at Duke University in Durham, N.C. “We stand at the beginning of a new era of ‘quadruple therapy’ for HFrEF with beta-blockers, an angiotensin receptor neprilysin inhibitor, mineralocorticoid receptor antagonists, and SGLT2 inhibitors,” he concluded.
A version of this article also appears on Medscape.com
REPORTING FROM THE AHA SCIENTIFIC SESSIONS