User login
Symmetrical Pruriginous Nasal Rash
The Diagnosis: Irritant Contact Dermatitis
A slang term for volatile alkyl nitrites, poppers are inhaled for recreational purposes. They produce rapid-onset euphoria and sexual arousal, as well as relax anal and vaginal sphincters, facilitating sexual intercourse. Alkyl nitrites initially were developed to treat coronary disease and angina but were replaced by more potent drugs.1 Because of their psychoactive effects and smooth muscle relaxation properties, they are widely used by homosexual and bisexual men.1-3 The term poppers was originated by the sound generated when the glass vials are crushed; currently, they also may be found in other formats.1
Nausea, hypotension, and headache are mild common adverse effects of volatile alkyl nitrites1; cardiac arrhythmia, oxidative hemolysis,4 and poppers maculopathy5,6 with permanent eye damage also have been reported.7 On the skin, volatile alkyl nitrites induce irritant contact dermatitis that heals without scarring, characteristically involving the face and upper thoracic region, as they are volatile vapors.2 However, the reaction can occur elsewhere. There have been reports of contact dermatitis on other locations, such as the thigh or the ankle, due to vials broken while stored in pockets or on the cuff of the socks.1 There also is a report of irritant contact dermatitis manifesting as a penile ulcer.3 Albeit rare, allergic contact dermatitis to volatile alkyl nitrites and other nitrites also can occur.8
The abuse of alkyl nitrites may increase the risk for sexually transmitted infections (STIs), as they may decrease safer sexual practices and increase the propensity to engage in risky sexual behavior. It has been suggested to screen for STIs in patients with history of volatile alkyl nitrite use. In the past, volatile alkyl nitrites were believed to be a potential vector of human immunodeficiency virus.9 Other popular drugs used in social context or "club drugs," such as 3,4-methylenedioxymethamphetamine, gamma hydroxybutyrate, methamphetamine, and ketamine, do not produce irritant dermatitis as an adverse cutaneous reaction.10 The differential diagnosis in our patient included herpes simplex virus and contagious impetigo1 as well as bullous lupus erythematosus and periorificial dermatitis; however, the clinical picture, acute onset of the reaction, and the patient's medical history were critical in making the correct diagnosis.
The patient was treated with topical hydrocortisone and fusidic acid cream twice daily for 7 days with complete response. Sexually transmitted infection screening was unremarkable. We suggest performing an STI workup on patients with history of volatile alkyl nitrite use.
- Schauber J, Herzinger T. 'Poppers' dermatitis. Clin Exp Dermatol. 2012;37:587-588.
- Foroozan M, Studer M, Splingard B, et al. Facial dermatitis due to inhalation of poppers [in French]. Ann Dermatol Venereol. 2009;136:298-299.
- Latini A, Lora V, Zaccarelli M, et al. Unusual presentation of poppers dermatitis. JAMA Dermatol. 2017;153:233-234.
- Shortt J, Polizzotto MN, Opat SS, et al. Oxidative haemolysis due to 'poppers'. Br J Haematol. 2008;142:328.
- Davies AJ, Kelly SP, Naylor SG, et al. Adverse ophthalmic reaction in poppers users: case series of 'poppers maculopathy'. Eye (Lond). 2012;26:1479-1486.
- Davies AJ, Kelly SP, Bhatt PR. 'Poppers maculopathy'--an emerging ophthalmic reaction to recreational substance abuse. Eye (Lond). 2012;26:888.
- Vignal-Clermont C, Audo I, Sahel JA, et al. Poppers-associated retinal toxicity. N Engl J Med. 2010;363:1583-1585.
- Bos JD, Jansen FC, Timmer JG. Allergic contact dermatitis to amyl nitrite ('poppers'). Contact Dermatitis. 1985;12:109.
- Stratford M, Wilson PD. Agitation effects on microbial cell-cell interactions. Lett Appl Microbiol. 1990;11:1-6.
- Romanelli F, Smith KM, Thornton AC, et al. Poppers: epidemiology and clinical management of inhaled nitrite abuse. Pharmacotherapy. 2004;24:69-78.
The Diagnosis: Irritant Contact Dermatitis
A slang term for volatile alkyl nitrites, poppers are inhaled for recreational purposes. They produce rapid-onset euphoria and sexual arousal, as well as relax anal and vaginal sphincters, facilitating sexual intercourse. Alkyl nitrites initially were developed to treat coronary disease and angina but were replaced by more potent drugs.1 Because of their psychoactive effects and smooth muscle relaxation properties, they are widely used by homosexual and bisexual men.1-3 The term poppers was originated by the sound generated when the glass vials are crushed; currently, they also may be found in other formats.1
Nausea, hypotension, and headache are mild common adverse effects of volatile alkyl nitrites1; cardiac arrhythmia, oxidative hemolysis,4 and poppers maculopathy5,6 with permanent eye damage also have been reported.7 On the skin, volatile alkyl nitrites induce irritant contact dermatitis that heals without scarring, characteristically involving the face and upper thoracic region, as they are volatile vapors.2 However, the reaction can occur elsewhere. There have been reports of contact dermatitis on other locations, such as the thigh or the ankle, due to vials broken while stored in pockets or on the cuff of the socks.1 There also is a report of irritant contact dermatitis manifesting as a penile ulcer.3 Albeit rare, allergic contact dermatitis to volatile alkyl nitrites and other nitrites also can occur.8
The abuse of alkyl nitrites may increase the risk for sexually transmitted infections (STIs), as they may decrease safer sexual practices and increase the propensity to engage in risky sexual behavior. It has been suggested to screen for STIs in patients with history of volatile alkyl nitrite use. In the past, volatile alkyl nitrites were believed to be a potential vector of human immunodeficiency virus.9 Other popular drugs used in social context or "club drugs," such as 3,4-methylenedioxymethamphetamine, gamma hydroxybutyrate, methamphetamine, and ketamine, do not produce irritant dermatitis as an adverse cutaneous reaction.10 The differential diagnosis in our patient included herpes simplex virus and contagious impetigo1 as well as bullous lupus erythematosus and periorificial dermatitis; however, the clinical picture, acute onset of the reaction, and the patient's medical history were critical in making the correct diagnosis.
The patient was treated with topical hydrocortisone and fusidic acid cream twice daily for 7 days with complete response. Sexually transmitted infection screening was unremarkable. We suggest performing an STI workup on patients with history of volatile alkyl nitrite use.
The Diagnosis: Irritant Contact Dermatitis
A slang term for volatile alkyl nitrites, poppers are inhaled for recreational purposes. They produce rapid-onset euphoria and sexual arousal, as well as relax anal and vaginal sphincters, facilitating sexual intercourse. Alkyl nitrites initially were developed to treat coronary disease and angina but were replaced by more potent drugs.1 Because of their psychoactive effects and smooth muscle relaxation properties, they are widely used by homosexual and bisexual men.1-3 The term poppers was originated by the sound generated when the glass vials are crushed; currently, they also may be found in other formats.1
Nausea, hypotension, and headache are mild common adverse effects of volatile alkyl nitrites1; cardiac arrhythmia, oxidative hemolysis,4 and poppers maculopathy5,6 with permanent eye damage also have been reported.7 On the skin, volatile alkyl nitrites induce irritant contact dermatitis that heals without scarring, characteristically involving the face and upper thoracic region, as they are volatile vapors.2 However, the reaction can occur elsewhere. There have been reports of contact dermatitis on other locations, such as the thigh or the ankle, due to vials broken while stored in pockets or on the cuff of the socks.1 There also is a report of irritant contact dermatitis manifesting as a penile ulcer.3 Albeit rare, allergic contact dermatitis to volatile alkyl nitrites and other nitrites also can occur.8
The abuse of alkyl nitrites may increase the risk for sexually transmitted infections (STIs), as they may decrease safer sexual practices and increase the propensity to engage in risky sexual behavior. It has been suggested to screen for STIs in patients with history of volatile alkyl nitrite use. In the past, volatile alkyl nitrites were believed to be a potential vector of human immunodeficiency virus.9 Other popular drugs used in social context or "club drugs," such as 3,4-methylenedioxymethamphetamine, gamma hydroxybutyrate, methamphetamine, and ketamine, do not produce irritant dermatitis as an adverse cutaneous reaction.10 The differential diagnosis in our patient included herpes simplex virus and contagious impetigo1 as well as bullous lupus erythematosus and periorificial dermatitis; however, the clinical picture, acute onset of the reaction, and the patient's medical history were critical in making the correct diagnosis.
The patient was treated with topical hydrocortisone and fusidic acid cream twice daily for 7 days with complete response. Sexually transmitted infection screening was unremarkable. We suggest performing an STI workup on patients with history of volatile alkyl nitrite use.
- Schauber J, Herzinger T. 'Poppers' dermatitis. Clin Exp Dermatol. 2012;37:587-588.
- Foroozan M, Studer M, Splingard B, et al. Facial dermatitis due to inhalation of poppers [in French]. Ann Dermatol Venereol. 2009;136:298-299.
- Latini A, Lora V, Zaccarelli M, et al. Unusual presentation of poppers dermatitis. JAMA Dermatol. 2017;153:233-234.
- Shortt J, Polizzotto MN, Opat SS, et al. Oxidative haemolysis due to 'poppers'. Br J Haematol. 2008;142:328.
- Davies AJ, Kelly SP, Naylor SG, et al. Adverse ophthalmic reaction in poppers users: case series of 'poppers maculopathy'. Eye (Lond). 2012;26:1479-1486.
- Davies AJ, Kelly SP, Bhatt PR. 'Poppers maculopathy'--an emerging ophthalmic reaction to recreational substance abuse. Eye (Lond). 2012;26:888.
- Vignal-Clermont C, Audo I, Sahel JA, et al. Poppers-associated retinal toxicity. N Engl J Med. 2010;363:1583-1585.
- Bos JD, Jansen FC, Timmer JG. Allergic contact dermatitis to amyl nitrite ('poppers'). Contact Dermatitis. 1985;12:109.
- Stratford M, Wilson PD. Agitation effects on microbial cell-cell interactions. Lett Appl Microbiol. 1990;11:1-6.
- Romanelli F, Smith KM, Thornton AC, et al. Poppers: epidemiology and clinical management of inhaled nitrite abuse. Pharmacotherapy. 2004;24:69-78.
- Schauber J, Herzinger T. 'Poppers' dermatitis. Clin Exp Dermatol. 2012;37:587-588.
- Foroozan M, Studer M, Splingard B, et al. Facial dermatitis due to inhalation of poppers [in French]. Ann Dermatol Venereol. 2009;136:298-299.
- Latini A, Lora V, Zaccarelli M, et al. Unusual presentation of poppers dermatitis. JAMA Dermatol. 2017;153:233-234.
- Shortt J, Polizzotto MN, Opat SS, et al. Oxidative haemolysis due to 'poppers'. Br J Haematol. 2008;142:328.
- Davies AJ, Kelly SP, Naylor SG, et al. Adverse ophthalmic reaction in poppers users: case series of 'poppers maculopathy'. Eye (Lond). 2012;26:1479-1486.
- Davies AJ, Kelly SP, Bhatt PR. 'Poppers maculopathy'--an emerging ophthalmic reaction to recreational substance abuse. Eye (Lond). 2012;26:888.
- Vignal-Clermont C, Audo I, Sahel JA, et al. Poppers-associated retinal toxicity. N Engl J Med. 2010;363:1583-1585.
- Bos JD, Jansen FC, Timmer JG. Allergic contact dermatitis to amyl nitrite ('poppers'). Contact Dermatitis. 1985;12:109.
- Stratford M, Wilson PD. Agitation effects on microbial cell-cell interactions. Lett Appl Microbiol. 1990;11:1-6.
- Romanelli F, Smith KM, Thornton AC, et al. Poppers: epidemiology and clinical management of inhaled nitrite abuse. Pharmacotherapy. 2004;24:69-78.
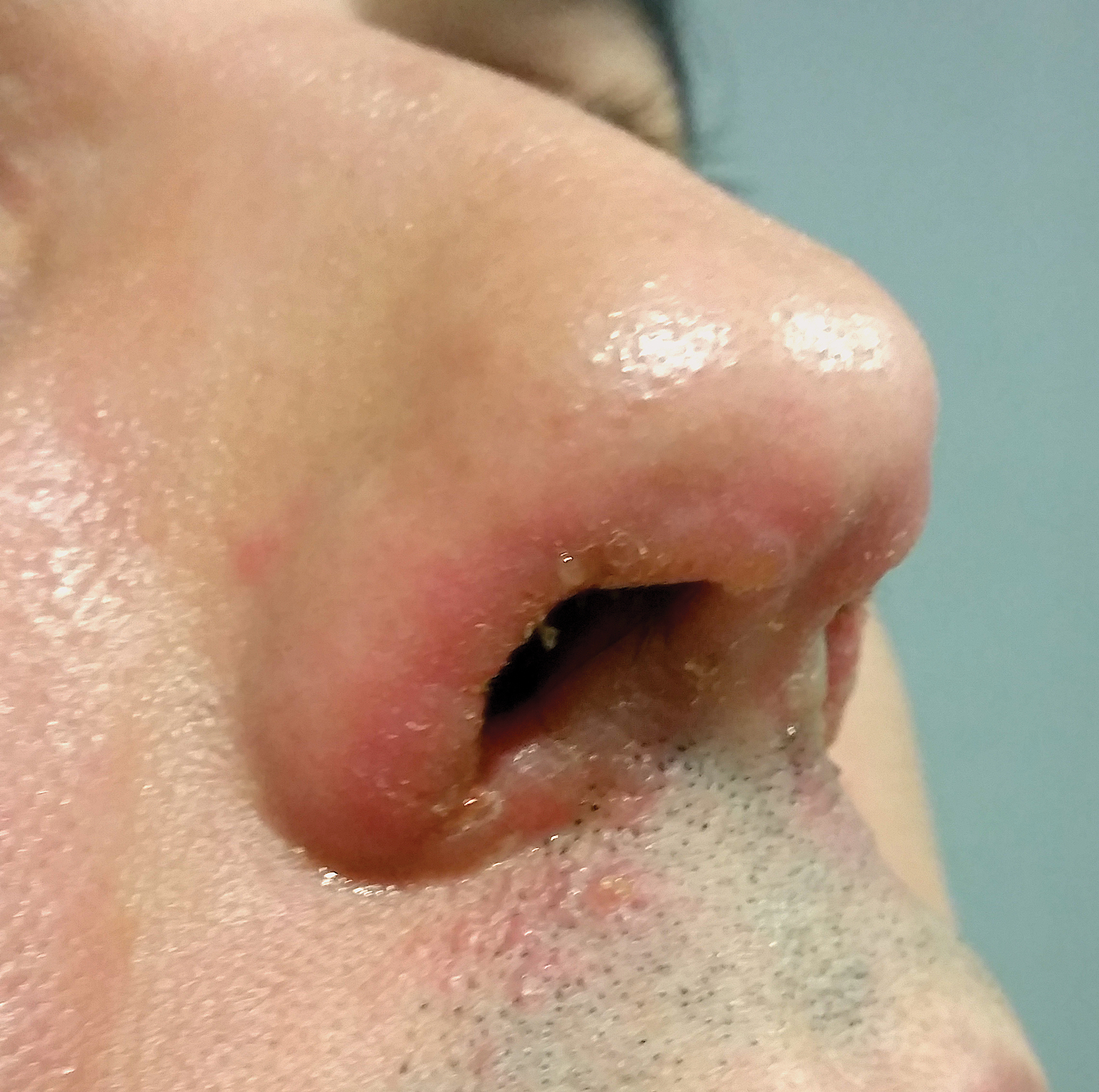
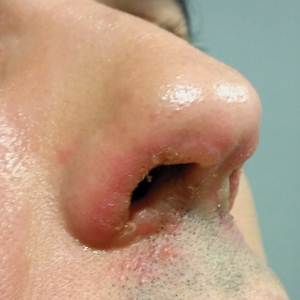
A 44-year-old man was referred to the department of dermatology for a pruriginous nasal rash. Physical examination revealed vesicles with clear content and crusts symmetrically in both nostrils and philtra. The remainder of the examination was otherwise unremarkable. The patient reported inhalation of poppers the prior night during a party. No history of connective tissue diseases was present. The patient was in overall good health with no fever or chills.
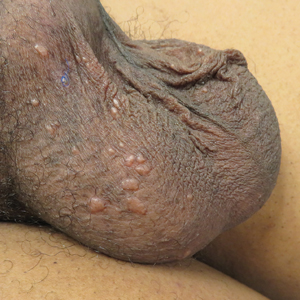
Flesh-Colored Papules on the Scrotum
The Diagnosis: Cutaneous Sarcoidosis
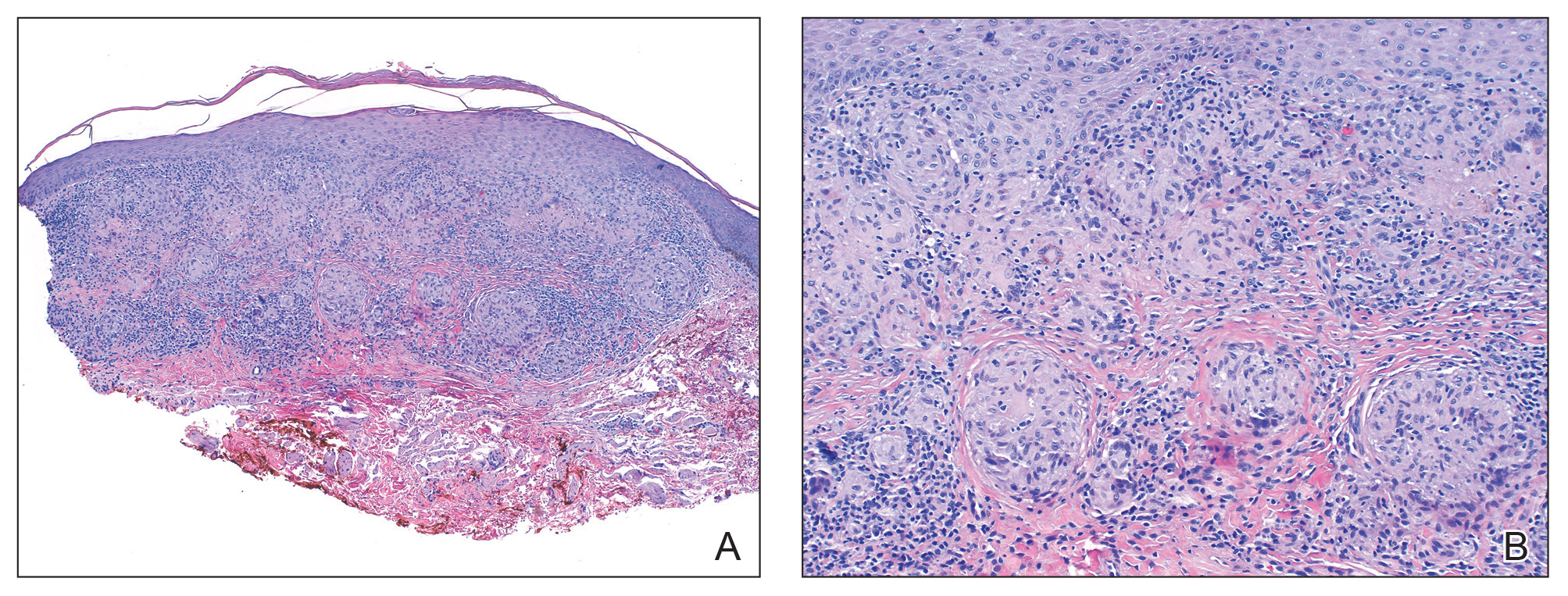
Histologic examination of the shave biopsy showed focal parakeratosis, irregular epidermal hyperplasia, and multiple noncaseating naked granulomas with occasional multinucleated giant cells in the dermis (Figure, A). The granulomas were surrounded by mild lymphocytic infiltration with rare eosinophils (Figure, B). Periodic acid-Schiff and Fite stains were negative for organisms, and polariscopic examination was negative; these findings confirmed the diagnosis of cutaneous sarcoidosis. Topical or intralesional steroids were recommended, but our patient declined treatment given that the lesions were asymptomatic.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects the skin in approximately 25% of patients.1 Cutaneous lesions manifest in 2 forms: specific and nonspecific. Noncaseating granulomas are considered specific. Nonspecific lesions include erythema nodosum, calcinosis cutis, Sweet syndrome, and nail clubbing. The most common sites of specific sarcoidosis lesions include the face, lips, neck, upper trunk, and extremities. Few cases have reported cutaneous sarcoidosis involving the genitalia; most reports describe vulvar cutaneous sarcoidosis.2-4 Although there have been reports of sarcoidosis involving the epididymis and testes, which presented as scrotal masses, cutaneous scrotal involvement with the skin as the primary site of involvement is rare.5,6 McLaughlin et al5 reported an extensive, pruritic, and eczematous eruption of the scrotum with associated edema and tenderness. Wei et al6 reported cutaneous sarcoidosis in the form of multiple indurated papules involving the penis and scrotum, similar to our case.
Comparing our patient to the case reported by Wei et al,6 both patients had Fitzpatrick skin type V or VI and systemic involvement including pulmonary disease. However, Wei et al6 did not clearly mention if the cutaneous manifestations preceded the diagnosis of systemic sarcoidosis or if they were present at the time of the diagnosis. Our patient developed cutaneous lesions 4 years after being diagnosed with systemic sarcoidosis from hilar lymphadenopathy. In addition to the scrotal lesions, he also had a lesion of lupus pernio presenting as a violaceous to brown plaque on the tip of the nose. Although both patients denied pruritus, the other patient's lesions were painful.6 Wei et al6 mentioned that treatment with topical, intralesional, and systemic steroids failed, and the patient's lesions continued to progress. Generally, topical and intralesional steroids are considered mainstay treatment of cutaneous sarcoidosis despite insufficient data to support their efficacy.7
The differential diagnosis of papules on the scrotum can be broad. Our provisional diagnoses for this particular morphology of small, flesh-colored, shiny, polygonal, flat-topped papules included condyloma acuminatum; lichen planus; idiopathic scrotal calcinosis; steatocystoma multiplex; and sarcoidosis (although uncommon for the site), given the history of pulmonary involvement. We considered a diagnosis of condyloma acuminatum, but the lesions were too shiny and smooth. On histology, condyloma acuminatum shows a hyperkeratotic and parakeratotic stratum corneum, an exophytic growth with marked acanthosis, and superficially located koilocytes. Morphologically, our patient's lesions resembled genital lichen planus. However, Wickham striae were absent, and our patient's lesions were asymptomatic while lesions of lichen planus usually are pruritic. Histologically, lichen planus is characterized by hyperkeratosis, hypergranulosis, sawtooth rete ridges, and lichenoid interface inflammation. Idiopathic scrotal calcinosis also could be included in the differential; however, the lesions would look whiter and firmer than those of our patient, and biopsy will clearly show calcium deposition. Steatocystoma multiplex is another condition that can affect the scrotum, along with the trunk, axillae, extremities, and neck. However, the lesions are expected to discharge oily material if squeezed and have a characteristic corrugated eosinophilic cuticle lining a cyst histologically.
Although it is undetermined if the risk for systemic involvement increases in patients with cutaneous sarcoidosis, evaluation for probable systemic involvement is necessary.1 Because cutaneous sarcoidosis generally can precede any systemic involvement, it would be reasonable to consider skin biopsies in patients who present with atypical wartlike lesions on the scrotum and penis to rule out sarcoidosis.
- Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis: relationship to severity and chronicity of disease. Clin Exp Dermatol. 2011;36:739.
- Vera C, Funaro D, Bouffard D. Vulvar sarcoidosis: case report and review of the literature. J Cutan Med Surg. 2013;17:287-290.
- Watkins S, Ismail A, McKay K, et al. Systemic sarcoidosis with unique vulvar involvement. JAMA Dermatol. 2014;150:666-667.
- Pereira IB, Khan A. Sarcoidosis rare cutaneous manifestations: vulval and perianal involvement. J Obstet Gynaecol. 2017;6:1-2.
- McLaughlin SS, Linquist AM, Burnett JW. Cutaneous sarcoidosis of the scrotum: a rare manifestation of systemic disease. Acta Derm Venereol. 2002;82:216-217.
- Wei H, Friedman KA, Rudikoff D. Multiple indurated papules on penis and scrotum. J Cutan Med Surg. 2000;4:202-204.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361.
The Diagnosis: Cutaneous Sarcoidosis
Histologic examination of the shave biopsy showed focal parakeratosis, irregular epidermal hyperplasia, and multiple noncaseating naked granulomas with occasional multinucleated giant cells in the dermis (Figure, A). The granulomas were surrounded by mild lymphocytic infiltration with rare eosinophils (Figure, B). Periodic acid-Schiff and Fite stains were negative for organisms, and polariscopic examination was negative; these findings confirmed the diagnosis of cutaneous sarcoidosis. Topical or intralesional steroids were recommended, but our patient declined treatment given that the lesions were asymptomatic.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects the skin in approximately 25% of patients.1 Cutaneous lesions manifest in 2 forms: specific and nonspecific. Noncaseating granulomas are considered specific. Nonspecific lesions include erythema nodosum, calcinosis cutis, Sweet syndrome, and nail clubbing. The most common sites of specific sarcoidosis lesions include the face, lips, neck, upper trunk, and extremities. Few cases have reported cutaneous sarcoidosis involving the genitalia; most reports describe vulvar cutaneous sarcoidosis.2-4 Although there have been reports of sarcoidosis involving the epididymis and testes, which presented as scrotal masses, cutaneous scrotal involvement with the skin as the primary site of involvement is rare.5,6 McLaughlin et al5 reported an extensive, pruritic, and eczematous eruption of the scrotum with associated edema and tenderness. Wei et al6 reported cutaneous sarcoidosis in the form of multiple indurated papules involving the penis and scrotum, similar to our case.
Comparing our patient to the case reported by Wei et al,6 both patients had Fitzpatrick skin type V or VI and systemic involvement including pulmonary disease. However, Wei et al6 did not clearly mention if the cutaneous manifestations preceded the diagnosis of systemic sarcoidosis or if they were present at the time of the diagnosis. Our patient developed cutaneous lesions 4 years after being diagnosed with systemic sarcoidosis from hilar lymphadenopathy. In addition to the scrotal lesions, he also had a lesion of lupus pernio presenting as a violaceous to brown plaque on the tip of the nose. Although both patients denied pruritus, the other patient's lesions were painful.6 Wei et al6 mentioned that treatment with topical, intralesional, and systemic steroids failed, and the patient's lesions continued to progress. Generally, topical and intralesional steroids are considered mainstay treatment of cutaneous sarcoidosis despite insufficient data to support their efficacy.7
The differential diagnosis of papules on the scrotum can be broad. Our provisional diagnoses for this particular morphology of small, flesh-colored, shiny, polygonal, flat-topped papules included condyloma acuminatum; lichen planus; idiopathic scrotal calcinosis; steatocystoma multiplex; and sarcoidosis (although uncommon for the site), given the history of pulmonary involvement. We considered a diagnosis of condyloma acuminatum, but the lesions were too shiny and smooth. On histology, condyloma acuminatum shows a hyperkeratotic and parakeratotic stratum corneum, an exophytic growth with marked acanthosis, and superficially located koilocytes. Morphologically, our patient's lesions resembled genital lichen planus. However, Wickham striae were absent, and our patient's lesions were asymptomatic while lesions of lichen planus usually are pruritic. Histologically, lichen planus is characterized by hyperkeratosis, hypergranulosis, sawtooth rete ridges, and lichenoid interface inflammation. Idiopathic scrotal calcinosis also could be included in the differential; however, the lesions would look whiter and firmer than those of our patient, and biopsy will clearly show calcium deposition. Steatocystoma multiplex is another condition that can affect the scrotum, along with the trunk, axillae, extremities, and neck. However, the lesions are expected to discharge oily material if squeezed and have a characteristic corrugated eosinophilic cuticle lining a cyst histologically.
Although it is undetermined if the risk for systemic involvement increases in patients with cutaneous sarcoidosis, evaluation for probable systemic involvement is necessary.1 Because cutaneous sarcoidosis generally can precede any systemic involvement, it would be reasonable to consider skin biopsies in patients who present with atypical wartlike lesions on the scrotum and penis to rule out sarcoidosis.
The Diagnosis: Cutaneous Sarcoidosis
Histologic examination of the shave biopsy showed focal parakeratosis, irregular epidermal hyperplasia, and multiple noncaseating naked granulomas with occasional multinucleated giant cells in the dermis (Figure, A). The granulomas were surrounded by mild lymphocytic infiltration with rare eosinophils (Figure, B). Periodic acid-Schiff and Fite stains were negative for organisms, and polariscopic examination was negative; these findings confirmed the diagnosis of cutaneous sarcoidosis. Topical or intralesional steroids were recommended, but our patient declined treatment given that the lesions were asymptomatic.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects the skin in approximately 25% of patients.1 Cutaneous lesions manifest in 2 forms: specific and nonspecific. Noncaseating granulomas are considered specific. Nonspecific lesions include erythema nodosum, calcinosis cutis, Sweet syndrome, and nail clubbing. The most common sites of specific sarcoidosis lesions include the face, lips, neck, upper trunk, and extremities. Few cases have reported cutaneous sarcoidosis involving the genitalia; most reports describe vulvar cutaneous sarcoidosis.2-4 Although there have been reports of sarcoidosis involving the epididymis and testes, which presented as scrotal masses, cutaneous scrotal involvement with the skin as the primary site of involvement is rare.5,6 McLaughlin et al5 reported an extensive, pruritic, and eczematous eruption of the scrotum with associated edema and tenderness. Wei et al6 reported cutaneous sarcoidosis in the form of multiple indurated papules involving the penis and scrotum, similar to our case.
Comparing our patient to the case reported by Wei et al,6 both patients had Fitzpatrick skin type V or VI and systemic involvement including pulmonary disease. However, Wei et al6 did not clearly mention if the cutaneous manifestations preceded the diagnosis of systemic sarcoidosis or if they were present at the time of the diagnosis. Our patient developed cutaneous lesions 4 years after being diagnosed with systemic sarcoidosis from hilar lymphadenopathy. In addition to the scrotal lesions, he also had a lesion of lupus pernio presenting as a violaceous to brown plaque on the tip of the nose. Although both patients denied pruritus, the other patient's lesions were painful.6 Wei et al6 mentioned that treatment with topical, intralesional, and systemic steroids failed, and the patient's lesions continued to progress. Generally, topical and intralesional steroids are considered mainstay treatment of cutaneous sarcoidosis despite insufficient data to support their efficacy.7
The differential diagnosis of papules on the scrotum can be broad. Our provisional diagnoses for this particular morphology of small, flesh-colored, shiny, polygonal, flat-topped papules included condyloma acuminatum; lichen planus; idiopathic scrotal calcinosis; steatocystoma multiplex; and sarcoidosis (although uncommon for the site), given the history of pulmonary involvement. We considered a diagnosis of condyloma acuminatum, but the lesions were too shiny and smooth. On histology, condyloma acuminatum shows a hyperkeratotic and parakeratotic stratum corneum, an exophytic growth with marked acanthosis, and superficially located koilocytes. Morphologically, our patient's lesions resembled genital lichen planus. However, Wickham striae were absent, and our patient's lesions were asymptomatic while lesions of lichen planus usually are pruritic. Histologically, lichen planus is characterized by hyperkeratosis, hypergranulosis, sawtooth rete ridges, and lichenoid interface inflammation. Idiopathic scrotal calcinosis also could be included in the differential; however, the lesions would look whiter and firmer than those of our patient, and biopsy will clearly show calcium deposition. Steatocystoma multiplex is another condition that can affect the scrotum, along with the trunk, axillae, extremities, and neck. However, the lesions are expected to discharge oily material if squeezed and have a characteristic corrugated eosinophilic cuticle lining a cyst histologically.
Although it is undetermined if the risk for systemic involvement increases in patients with cutaneous sarcoidosis, evaluation for probable systemic involvement is necessary.1 Because cutaneous sarcoidosis generally can precede any systemic involvement, it would be reasonable to consider skin biopsies in patients who present with atypical wartlike lesions on the scrotum and penis to rule out sarcoidosis.
- Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis: relationship to severity and chronicity of disease. Clin Exp Dermatol. 2011;36:739.
- Vera C, Funaro D, Bouffard D. Vulvar sarcoidosis: case report and review of the literature. J Cutan Med Surg. 2013;17:287-290.
- Watkins S, Ismail A, McKay K, et al. Systemic sarcoidosis with unique vulvar involvement. JAMA Dermatol. 2014;150:666-667.
- Pereira IB, Khan A. Sarcoidosis rare cutaneous manifestations: vulval and perianal involvement. J Obstet Gynaecol. 2017;6:1-2.
- McLaughlin SS, Linquist AM, Burnett JW. Cutaneous sarcoidosis of the scrotum: a rare manifestation of systemic disease. Acta Derm Venereol. 2002;82:216-217.
- Wei H, Friedman KA, Rudikoff D. Multiple indurated papules on penis and scrotum. J Cutan Med Surg. 2000;4:202-204.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361.
- Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis: relationship to severity and chronicity of disease. Clin Exp Dermatol. 2011;36:739.
- Vera C, Funaro D, Bouffard D. Vulvar sarcoidosis: case report and review of the literature. J Cutan Med Surg. 2013;17:287-290.
- Watkins S, Ismail A, McKay K, et al. Systemic sarcoidosis with unique vulvar involvement. JAMA Dermatol. 2014;150:666-667.
- Pereira IB, Khan A. Sarcoidosis rare cutaneous manifestations: vulval and perianal involvement. J Obstet Gynaecol. 2017;6:1-2.
- McLaughlin SS, Linquist AM, Burnett JW. Cutaneous sarcoidosis of the scrotum: a rare manifestation of systemic disease. Acta Derm Venereol. 2002;82:216-217.
- Wei H, Friedman KA, Rudikoff D. Multiple indurated papules on penis and scrotum. J Cutan Med Surg. 2000;4:202-204.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361.
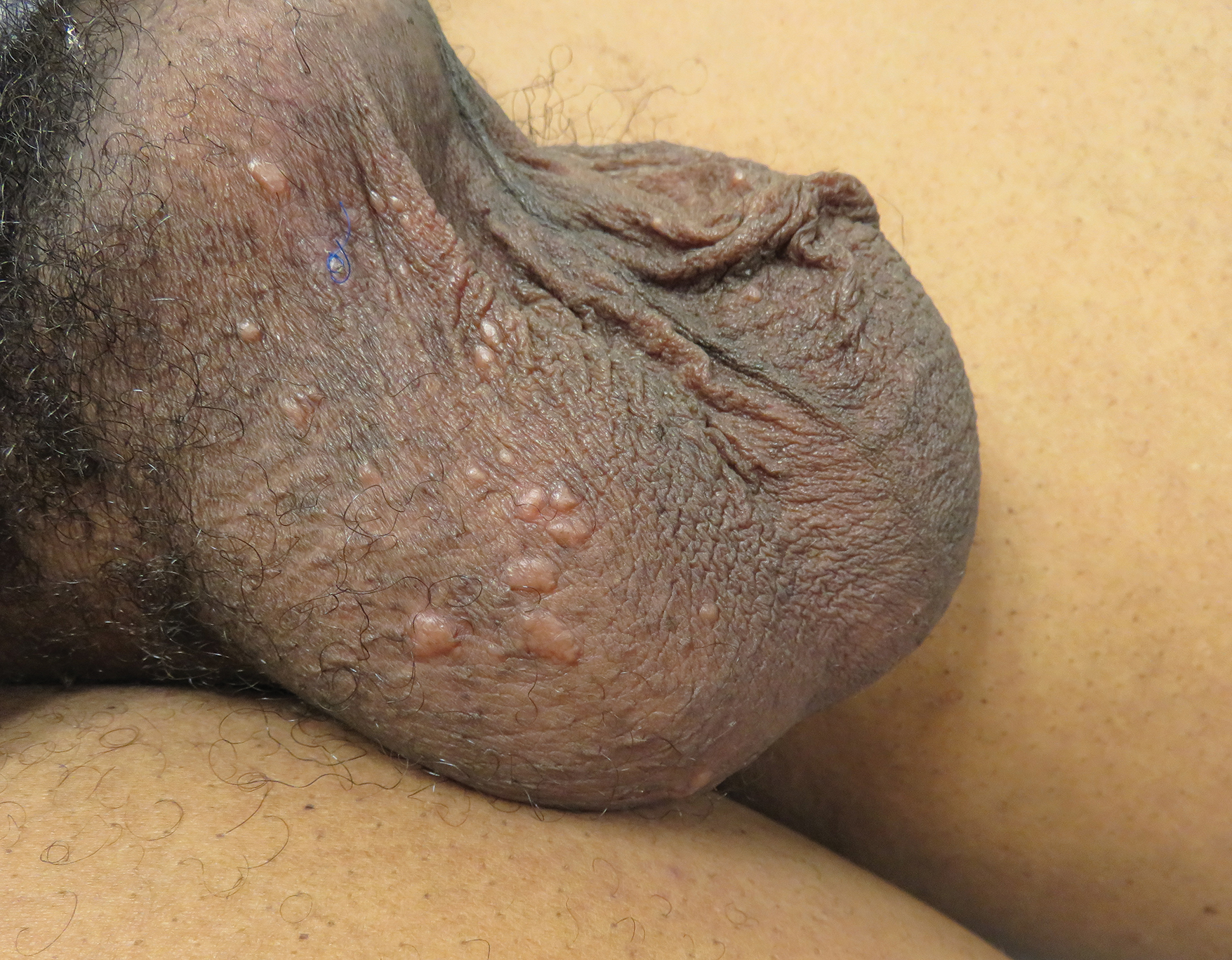
A 44-year-old black man presented with "bumps on the scrotum" of approximately 4 months' duration. They were asymptomatic and untreated. The patient denied extramarital sexual contacts or a history of any sexually transmitted infection. His medical history was notable for sarcoidosis diagnosed 4 years prior to presentation when hilar lymphadenopathy was incidentally found on routine screening. His condition was managed with regular follow-up without treatment. He also had a positive tuberculosis skin test in the past without radiologic evidence of active pulmonary disease. Physical examination revealed multiple 2- to 5-mm, flesh-colored, shiny, polygonal, flat-topped papules spread diffusely over the scrotum. A 1-cm, barely palpable, nonscaly, violaceous to brown plaque also was seen on the tip of the nose. A punch biopsy was taken from a lesion on the scrotum.
Painful and Pruritic Erosions on the Back
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
The Diagnosis: Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare blistering disease that affects patients with systemic lupus erythematosus (SLE). Our patient had a several-year history of SLE and was being managed by a rheumatologist. She was taking hydroxychloroquine at the time of the flare. Although BSLE tends to present in those with SLE that has already been diagnosed, BSLE has been reported as a possible initial manifestation of SLE.1
Bullous systemic lupus erythematosus is estimated to occur in less than 5% of patients with SLE and is more common in black women between the second and third decades of life,2 though it also can be seen in the pediatric population.3 The lesions of BSLE usually present as subepidermal blisters often located on the face, neck, and arms on an erythematous or possibly urticarial base. Although non-BSLE vesiculobullous eruptions may be seen in patients with SLE, BSLE is differentiated from these other eruptions by its appearance on sun-exposed and non-sun-exposed areas of the body, while other vesiculobullous eruptions associated with SLE typically are limited to sun-exposed sites.4
Due to its clinical presentation overlapping with several vesiculobullous conditions, a set of diagnostic criteria have been suggested for BSLE, including the following: (1) fulfillment of the American Rheumatism Association's criteria for SLE5; (2) a new-onset vesiculobullous eruption, primarily on sun-exposed skin; (3) histology showing a subepidermal blister with a predominantly neutrophilic infiltrate; (4) presence of IgG, IgA, IgM, and C3 at the basement membrane zone; (5) evidence of antibodies to type VII collagen; and (6) immunoelectron microscopy showing codistribution of immunoglobulin deposits with anchoring fibrils/type VII collagen. To meet the diagnosis of type I BSLE, all 6 criteria must be satisfied. To meet the diagnosis of type II BSLE, only criteria 1 to 4 need to be satisfied.6
Patients with BSLE may be presumed to have a different but clinically similar vesiculobullous condition (eg, bullous pemphigoid, cutaneous manifestations of SLE) and may be started on systemic corticosteroids. However, BSLE patients often do not show great improvement while on corticosteroids and may even flare shortly after beginning systemic corticosteroid treatment. The current treatment of choice for BSLE is dapsone, a sulfa drug that is thought to exhibit its anti-inflammatory properties via the inhibition of the alternative pathway of the complement system and through the inhibition of polymorphonuclear leukocyte functions.7 A response to dapsone helps differentiate BSLE from histopathologically and immunopathologically identical conditions such as epidermolysis bullosa acquisita.4 Bullous systemic lupus erythematosus can be differentiated from dermatitis herpetiformis with the presence of antigliadin and antitissue transglutaminase antibodies, which are found in the latter. Additionally, BSLE may show the presence of IgG and IgM deposition in addition to IgA deposition, as opposed to dermatitis herpetiformis where only IgA is found.8 The presence of these additional antibody depositions also help differentiate BSLE from linear IgA bullous dermatosis (LABD), as LABD will only have IgA depositions and often presents with an annular, crown of jewels-like appearance. Finally, there is a well-described phenomenon of LABD being drug induced, particularly after a course of vancomycin,9 and such an association with vancomycin has not been documented for BSLE.
Our patient was diagnosed with BSLE following the flare approximately 1.5 years prior to the current presentation. She had been started on dapsone 75 mg daily at that time and was taking 75 mg at the time of presentation. She was admitted and treated as an inpatient with high-dose (1 mg/kg) intravenous prednisone due to the extensive current flare.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Miziara ID, Mahmoud A, Chagury AA, et al. Bullous systemic lupus erythematosus: case report. Int Arch Otorhinolaryngol. 2013;17:344-346.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Grover C, Khurana A, Sharma S, et al. Bullous systemic lupus erythematosus. Indian J Dermatol. 2013;58:492.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of RheumatologyClassification Criteria for Systemic Lupus Erythematosus [published online August 6, 2019]. Arthritis Rheumatol. 2019;71:1400-1412.
- Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S.
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:6.
- Barbosa WS, Rodarte CM, Guerra JG, et al. Bullous systemic lupus erythematosus: differential diagnosis with dermatitis herpetiformis. An Bras Dermatol. 2011;86(4 suppl 1):S92-S95.
- Yordanova I, Valtchev V, Gospodinov D, et al. IgA linear bullous dermatosis in childhood. J IMAB. 2015;21:1012-1014.
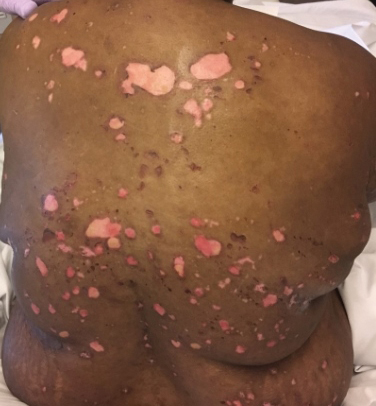
A 51-year-old black woman presented to the dermatology clinic with painful and pruritic erosions on the back, abdomen, neck, and arms of approximately 2 months' duration. The lesions started on the back and spread in a cephalocaudal manner. The patient denied any new changes in medication. Physical examination revealed large erosions with mild weeping of serosanguineous fluid on the back, abdomen, neck, and upper extremities. A few tense bullae were present on the dorsal aspect of the right hand. She had experienced a similar flare approximately 1.5 years prior to the current presentation. At that time, 2 shave biopsies from vesiculobullous lesions on the right side of the neck were sent for hematoxylin and eosin staining and direct immunofluorescence. Biopsy results showed a subepidermal blister that extended along the course of the hair follicle and was associated with an infiltrate of neutrophilic granulocytes that also extended along the course of the hair follicle. Direct immunofluorescence showed IgG and C3 deposition in the basement membrane zone extending along the floor of the blister where the epidermis was separated from the dermis.
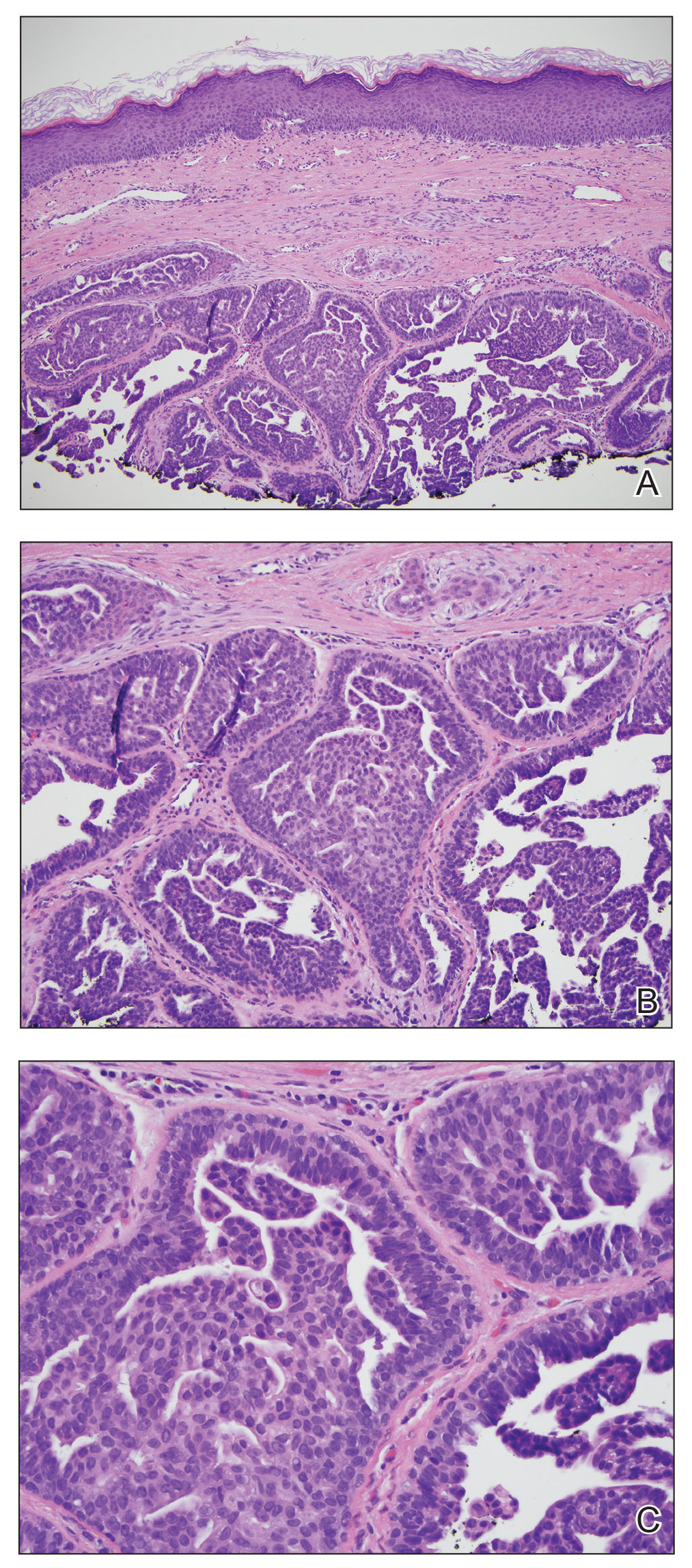
Enlarging Nodule on the Nipple
The Diagnosis: Nipple Adenoma (Florid Papillomatosis of the Nipple)
Biopsy of the nodule showed florid papillary hyperplasia of the ductal epithelium within the dermis that was sharply demarcated from the background stroma (Figure, A and B). Neither cytological nor architectural atypia were evident. There was no notable necrosis (Figure C). There was a background of fibrosis whereby the glandular ductal structures assumed a somewhat irregular growth pattern within the dermis with attendant hemorrhage. The patient underwent complete excision of the lesion. No evidence of carcinoma was seen on the final pathology, and the final margins were negative.
First described in 1923 and fully characterized in 1955, nipple adenoma (also known as florid papillomatosis of the nipple) is a benign proliferative neoplasm that originates in the lactiferous ducts of the nipple.1,2 It most commonly affects women aged 40 to 50 years (range, 0-89 years); less than 5% of cases are reported in men.3,4 It predominantly is unilateral, with only rare cases of bilateral papillomatosis reported. Patients often present with serous or serosanguineous discharge and an itching or burning sensation. Symptoms may worsen with the menstrual cycle.4 On physical examination, it presents as an ill-defined red nodule on the nipple with crusting, erosion, or erythema of the nipple surface. Although imaging generally is not used to confirm the diagnosis, mammography should be performed prior to biopsy to rule out underlying breast pathology. Dermoscopy may show linear cherry red structures or red serpiginous and annular structures.5,6 The differential diagnosis of nipple adenoma includes Paget disease of the breast, adenomyoepithelioma, subareolar subsclerosing duct hyperplasia, syringomatous adenoma, adenosis tumor, low-grade adenosquamous carcinoma, low-grade ductal carcinoma in situ, tubular carcinoma, and sweat gland tumors.3
Microscopic features of nipple adenoma have been categorized into 4 subtypes: sclerosing papillomatosis, papillomatosis, adenosis, and a mixed pattern.3,7 The tumors may have keratin cysts and focal necrosis but no atypia, and the myoepithelial cell layer is retained. Nipple adenomas show a glandular proliferation in the dermis that is relatively well circumscribed with glands that vary in appearance between a simple adenosislike pattern of growth to a papillary hyperplasia and/or usual ductal hyperplasia growth pattern. A pseudoinfiltrative pattern can occur when the glandular epithelium is entrapped within stromal fibrosis; however, the myoepithelial layer is retained. Occasionally, the glandular epithelium can grow in continuity with the surface squamous epithelium of the nipple, clinically simulating Paget disease of the breast.8 Immunohistochemical stains, specifically p63, p40, calponin 1, h-caldesmon, cytokeratin 5/6, CD10, and α; smooth muscle actin, highlight the myoepithelial cells, while cytokeratin 7 identifies the ductal epithelium, supporting the diagnosis.6 In addition to biopsy and microscopic tissue examination, touch preparation cytology, curettage cytology, and fine needle aspiration techniques have been used to perform cytologic examination of the lesions, aiding in identification of the benign or malignant nature of the neoplasm.6 Nipple adenoma also is referred to as florid papillomatosis of the nipple, papillary adenoma, erosive adenomatosis, and subareolar duct papillomatosis.7
Although nipple adenoma is a benign tumor, up to 16.5% of affected patients had an ipsilateral or contralateral mammary carcinoma.9 The majority arose coincidentally but separately in the same breast, and carcinoma arose directly from the nipple adenoma in 8 cases; 3 cases were carcinomas that arose in men.10 A definitive association or causal relationship between nipple adenoma and subsequent development of breast cancer has not been identified, and the incidence of nipple adenoma in patients with a positive family history of breast cancer has not been examined. Therefore, although various treatments including cryosurgery, nipple splitting enucleation, and Mohs micrographic surgery have been proposed, complete excision remains the gold standard of therapy. Regular breast examinations and digital mammography are necessary to screen for local recurrences.
- Miller E, Lewis D. The significance of serohemorrhagic or hemorrhagic discharge from the nipple. JAMA. 1923;81:1651-1657.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Rosen PP. Rosen's Breast Pathology. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:120-128.
- Brownstein MH, Phelps RG, Maqnin PH. Papillary adenoma of the nipple: analysis of fifteen new cases. J Am Acad Dermatol. 1985;12:707-715.
- Takashima S, Fujita Y, Miyauchi T, et al. Dermoscopic observation in adenoma of the nipple. J Dermatol. 2015;42:341-342.
- Spohn G, Trotter S, Tozbikian G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Shin SJ. Nipple adenoma (florid papillomatosis of the nipple). In: Dabbs DJ, ed. Breast Pathology. Philadelphia, PA: Elsevier Saunders; 2012:286-292.
- Schnitt SJ, Collins LC. Biopsy Interpretation of the Breast. 2nd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013.
- Salemis NS. Florid papillomatosis of the nipple: a rare presentation and review of the literature. Breast Dis. 2015;35:153-156.
- Di Bonito M, Cantile M, Collina F, et al. Adenoma of the nipple: a clinicopathological report of 13 cases. Oncol Lett. 2014;7:1839-1842.
The Diagnosis: Nipple Adenoma (Florid Papillomatosis of the Nipple)
Biopsy of the nodule showed florid papillary hyperplasia of the ductal epithelium within the dermis that was sharply demarcated from the background stroma (Figure, A and B). Neither cytological nor architectural atypia were evident. There was no notable necrosis (Figure C). There was a background of fibrosis whereby the glandular ductal structures assumed a somewhat irregular growth pattern within the dermis with attendant hemorrhage. The patient underwent complete excision of the lesion. No evidence of carcinoma was seen on the final pathology, and the final margins were negative.
First described in 1923 and fully characterized in 1955, nipple adenoma (also known as florid papillomatosis of the nipple) is a benign proliferative neoplasm that originates in the lactiferous ducts of the nipple.1,2 It most commonly affects women aged 40 to 50 years (range, 0-89 years); less than 5% of cases are reported in men.3,4 It predominantly is unilateral, with only rare cases of bilateral papillomatosis reported. Patients often present with serous or serosanguineous discharge and an itching or burning sensation. Symptoms may worsen with the menstrual cycle.4 On physical examination, it presents as an ill-defined red nodule on the nipple with crusting, erosion, or erythema of the nipple surface. Although imaging generally is not used to confirm the diagnosis, mammography should be performed prior to biopsy to rule out underlying breast pathology. Dermoscopy may show linear cherry red structures or red serpiginous and annular structures.5,6 The differential diagnosis of nipple adenoma includes Paget disease of the breast, adenomyoepithelioma, subareolar subsclerosing duct hyperplasia, syringomatous adenoma, adenosis tumor, low-grade adenosquamous carcinoma, low-grade ductal carcinoma in situ, tubular carcinoma, and sweat gland tumors.3
Microscopic features of nipple adenoma have been categorized into 4 subtypes: sclerosing papillomatosis, papillomatosis, adenosis, and a mixed pattern.3,7 The tumors may have keratin cysts and focal necrosis but no atypia, and the myoepithelial cell layer is retained. Nipple adenomas show a glandular proliferation in the dermis that is relatively well circumscribed with glands that vary in appearance between a simple adenosislike pattern of growth to a papillary hyperplasia and/or usual ductal hyperplasia growth pattern. A pseudoinfiltrative pattern can occur when the glandular epithelium is entrapped within stromal fibrosis; however, the myoepithelial layer is retained. Occasionally, the glandular epithelium can grow in continuity with the surface squamous epithelium of the nipple, clinically simulating Paget disease of the breast.8 Immunohistochemical stains, specifically p63, p40, calponin 1, h-caldesmon, cytokeratin 5/6, CD10, and α; smooth muscle actin, highlight the myoepithelial cells, while cytokeratin 7 identifies the ductal epithelium, supporting the diagnosis.6 In addition to biopsy and microscopic tissue examination, touch preparation cytology, curettage cytology, and fine needle aspiration techniques have been used to perform cytologic examination of the lesions, aiding in identification of the benign or malignant nature of the neoplasm.6 Nipple adenoma also is referred to as florid papillomatosis of the nipple, papillary adenoma, erosive adenomatosis, and subareolar duct papillomatosis.7
Although nipple adenoma is a benign tumor, up to 16.5% of affected patients had an ipsilateral or contralateral mammary carcinoma.9 The majority arose coincidentally but separately in the same breast, and carcinoma arose directly from the nipple adenoma in 8 cases; 3 cases were carcinomas that arose in men.10 A definitive association or causal relationship between nipple adenoma and subsequent development of breast cancer has not been identified, and the incidence of nipple adenoma in patients with a positive family history of breast cancer has not been examined. Therefore, although various treatments including cryosurgery, nipple splitting enucleation, and Mohs micrographic surgery have been proposed, complete excision remains the gold standard of therapy. Regular breast examinations and digital mammography are necessary to screen for local recurrences.
The Diagnosis: Nipple Adenoma (Florid Papillomatosis of the Nipple)
Biopsy of the nodule showed florid papillary hyperplasia of the ductal epithelium within the dermis that was sharply demarcated from the background stroma (Figure, A and B). Neither cytological nor architectural atypia were evident. There was no notable necrosis (Figure C). There was a background of fibrosis whereby the glandular ductal structures assumed a somewhat irregular growth pattern within the dermis with attendant hemorrhage. The patient underwent complete excision of the lesion. No evidence of carcinoma was seen on the final pathology, and the final margins were negative.
First described in 1923 and fully characterized in 1955, nipple adenoma (also known as florid papillomatosis of the nipple) is a benign proliferative neoplasm that originates in the lactiferous ducts of the nipple.1,2 It most commonly affects women aged 40 to 50 years (range, 0-89 years); less than 5% of cases are reported in men.3,4 It predominantly is unilateral, with only rare cases of bilateral papillomatosis reported. Patients often present with serous or serosanguineous discharge and an itching or burning sensation. Symptoms may worsen with the menstrual cycle.4 On physical examination, it presents as an ill-defined red nodule on the nipple with crusting, erosion, or erythema of the nipple surface. Although imaging generally is not used to confirm the diagnosis, mammography should be performed prior to biopsy to rule out underlying breast pathology. Dermoscopy may show linear cherry red structures or red serpiginous and annular structures.5,6 The differential diagnosis of nipple adenoma includes Paget disease of the breast, adenomyoepithelioma, subareolar subsclerosing duct hyperplasia, syringomatous adenoma, adenosis tumor, low-grade adenosquamous carcinoma, low-grade ductal carcinoma in situ, tubular carcinoma, and sweat gland tumors.3
Microscopic features of nipple adenoma have been categorized into 4 subtypes: sclerosing papillomatosis, papillomatosis, adenosis, and a mixed pattern.3,7 The tumors may have keratin cysts and focal necrosis but no atypia, and the myoepithelial cell layer is retained. Nipple adenomas show a glandular proliferation in the dermis that is relatively well circumscribed with glands that vary in appearance between a simple adenosislike pattern of growth to a papillary hyperplasia and/or usual ductal hyperplasia growth pattern. A pseudoinfiltrative pattern can occur when the glandular epithelium is entrapped within stromal fibrosis; however, the myoepithelial layer is retained. Occasionally, the glandular epithelium can grow in continuity with the surface squamous epithelium of the nipple, clinically simulating Paget disease of the breast.8 Immunohistochemical stains, specifically p63, p40, calponin 1, h-caldesmon, cytokeratin 5/6, CD10, and α; smooth muscle actin, highlight the myoepithelial cells, while cytokeratin 7 identifies the ductal epithelium, supporting the diagnosis.6 In addition to biopsy and microscopic tissue examination, touch preparation cytology, curettage cytology, and fine needle aspiration techniques have been used to perform cytologic examination of the lesions, aiding in identification of the benign or malignant nature of the neoplasm.6 Nipple adenoma also is referred to as florid papillomatosis of the nipple, papillary adenoma, erosive adenomatosis, and subareolar duct papillomatosis.7
Although nipple adenoma is a benign tumor, up to 16.5% of affected patients had an ipsilateral or contralateral mammary carcinoma.9 The majority arose coincidentally but separately in the same breast, and carcinoma arose directly from the nipple adenoma in 8 cases; 3 cases were carcinomas that arose in men.10 A definitive association or causal relationship between nipple adenoma and subsequent development of breast cancer has not been identified, and the incidence of nipple adenoma in patients with a positive family history of breast cancer has not been examined. Therefore, although various treatments including cryosurgery, nipple splitting enucleation, and Mohs micrographic surgery have been proposed, complete excision remains the gold standard of therapy. Regular breast examinations and digital mammography are necessary to screen for local recurrences.
- Miller E, Lewis D. The significance of serohemorrhagic or hemorrhagic discharge from the nipple. JAMA. 1923;81:1651-1657.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Rosen PP. Rosen's Breast Pathology. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:120-128.
- Brownstein MH, Phelps RG, Maqnin PH. Papillary adenoma of the nipple: analysis of fifteen new cases. J Am Acad Dermatol. 1985;12:707-715.
- Takashima S, Fujita Y, Miyauchi T, et al. Dermoscopic observation in adenoma of the nipple. J Dermatol. 2015;42:341-342.
- Spohn G, Trotter S, Tozbikian G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Shin SJ. Nipple adenoma (florid papillomatosis of the nipple). In: Dabbs DJ, ed. Breast Pathology. Philadelphia, PA: Elsevier Saunders; 2012:286-292.
- Schnitt SJ, Collins LC. Biopsy Interpretation of the Breast. 2nd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013.
- Salemis NS. Florid papillomatosis of the nipple: a rare presentation and review of the literature. Breast Dis. 2015;35:153-156.
- Di Bonito M, Cantile M, Collina F, et al. Adenoma of the nipple: a clinicopathological report of 13 cases. Oncol Lett. 2014;7:1839-1842.
- Miller E, Lewis D. The significance of serohemorrhagic or hemorrhagic discharge from the nipple. JAMA. 1923;81:1651-1657.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Rosen PP. Rosen's Breast Pathology. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:120-128.
- Brownstein MH, Phelps RG, Maqnin PH. Papillary adenoma of the nipple: analysis of fifteen new cases. J Am Acad Dermatol. 1985;12:707-715.
- Takashima S, Fujita Y, Miyauchi T, et al. Dermoscopic observation in adenoma of the nipple. J Dermatol. 2015;42:341-342.
- Spohn G, Trotter S, Tozbikian G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Shin SJ. Nipple adenoma (florid papillomatosis of the nipple). In: Dabbs DJ, ed. Breast Pathology. Philadelphia, PA: Elsevier Saunders; 2012:286-292.
- Schnitt SJ, Collins LC. Biopsy Interpretation of the Breast. 2nd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013.
- Salemis NS. Florid papillomatosis of the nipple: a rare presentation and review of the literature. Breast Dis. 2015;35:153-156.
- Di Bonito M, Cantile M, Collina F, et al. Adenoma of the nipple: a clinicopathological report of 13 cases. Oncol Lett. 2014;7:1839-1842.
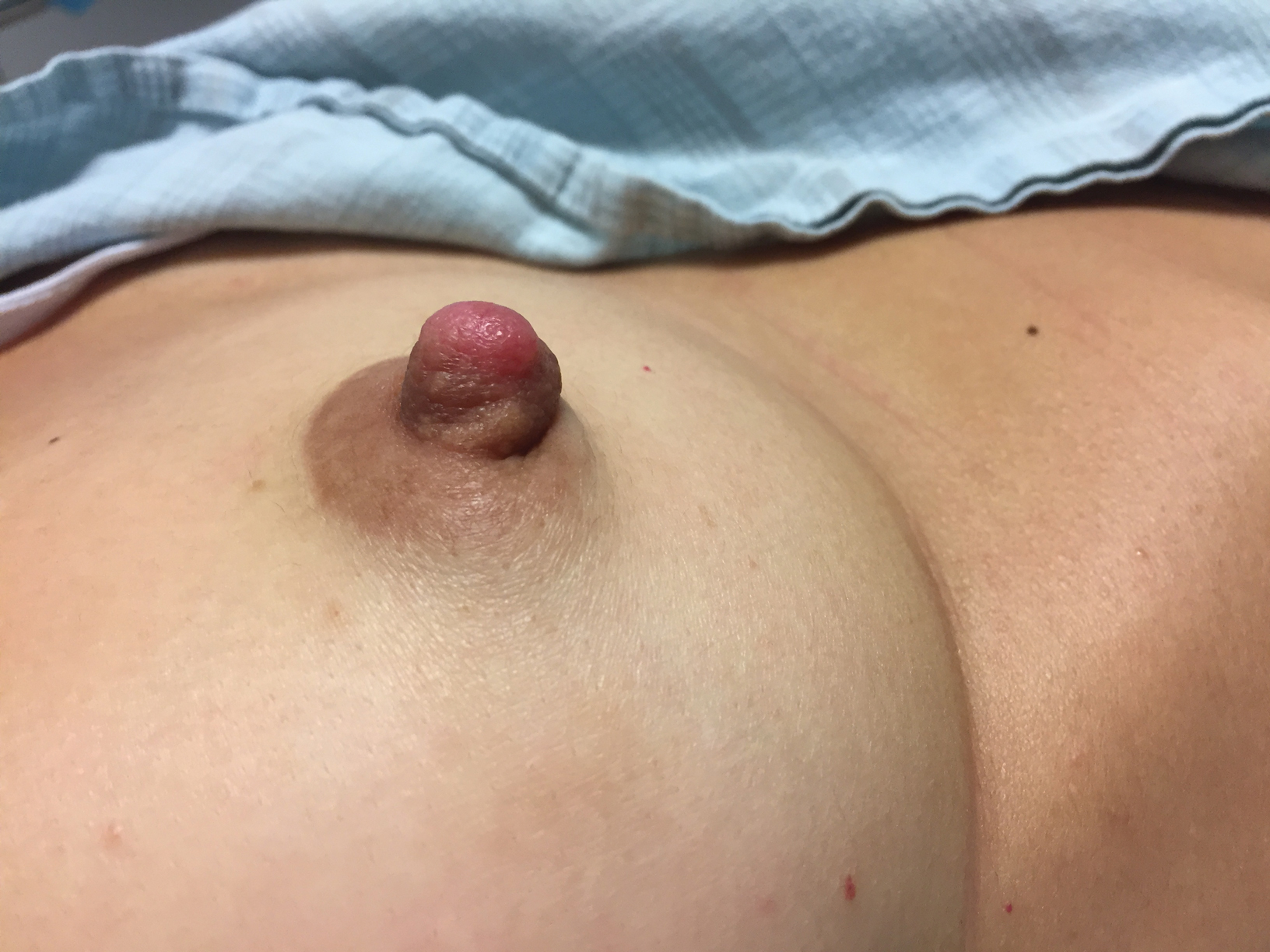
A healthy 48-year-old woman presented with a growth on the right nipple that had been slowly enlarging over the last few months. She initially noticed mild swelling in the area that persisted and formed a soft lump. She described mild pain with intermittent drainage but no bleeding. Her medical history was unremarkable, including a negative personal and family history of breast and skin cancer. She was taking no medications prior to development of the mass. She had no recent history of pregnancy or breastfeeding. A mammogram and breast ultrasound were not concerning for carcinoma. Physical examination showed a soft, exophytic, mildly tender, pink nodule on the right nipple that measured 12.2×7 mm; no drainage, bleeding, or ulceration was present. The surrounding skin of the areola and breast demonstrated no clinical changes. The contralateral breast, areola, and nipple were unaffected. The patient had no appreciable axillary or cervical lymphadenopathy. A deep shave biopsy of the nodule was performed and sent for histopathologic examination.
Violaceous Patches on the Arm
The Diagnosis: Phacomatosis Cesioflammea
Phacomatosis pigmentovascularis (PPV) encompasses a group of diseases that have a vascular nevus coupled with a pigmented nevus.1 It is divided into 5 types: Type I is defined by the presence of a vascular malformation and epidermal nevus; type II by a vascular malformation and dermal melanosis with or without nevus anemicus; type III by a vascular malformation and nevus spilus with or without nevus anemicus; type IV by a vascular malformation, dermal melanosis, and nevus spilus with or without nevus anemicus; and type V as cutis marmorata telangiectatica congenita and dermal melanosis.1
Happle2 proposed a descriptive classification system in 2005 that eliminated type I PPV because neither linear epidermal nevus nor Becker nevus are derived from pigmentary cells. An appended "a" denotes a subtype with isolated cutaneous findings, while "b" is associated with extracutaneous manifestations. Phacomatosis cesioflammea (type IIa/b) refers to blue-hued dermal melanocytosis and nevus flammeus. Phacomatosis spilorosea (type IIIa/b) refers to nevus spilus and rose-colored nevus flammeus. Phacomatosis cesiomarmorata (type Va/b) refers to dermal melanocytosis and cutis marmorata telangiectasia congenita. The last group (type IVa/b) is unclassifiable phacomatosis pigmentovascularis.2,3
Phacomatosis pigmentovascularis can be isolated to the skin or have associated extracutaneous findings, including ocular melanocytosis, seizures, or cognitive delay due to intracerebral vascular malformations. Patients also can develop limb and soft-tissue overgrowth.4 Phacomatosis pigmentovascularis has been found to be associated with mutations in the GNA11 and GNAQ genes. The theory behind PPV is twin spotting, resulting from a somatic mutation that leads to mosaic proliferation of 2 different cell lines.5 Phacomatosis pigmentovascularis can occur in isolation or can demonstrate the phenotype of Sturge-Weber syndrome or Klippel-Trenaunay syndrome. In Sturge-Weber syndrome, capillary malformations involve the face and underlying leptomeninges and cerebral cortex. Glaucoma and epilepsy also may be present. In Klippel-Trenaunay syndrome, capillary malformations involve the extremities (usually the legs) in association with varicose veins, soft-tissue hypertrophy, and skeletal overgrowth.6-9 Tuberous sclerosis is an autosomal-dominant neurocutaneous disease in which patients develop hamartomas throughout the body, including the brain, skin, eyes, kidneys, heart, and lungs. Cutaneous manifestations include facial angiofibromas, ungual fibromas, hypomelanotic macules (ash leaf spots, confetti-like lesions), shagreen patches or connective tissue hamartomas, and fibrous plaques on the forehead. Tuberous sclerosis does not include vascular malformations.10
Our patient was diagnosed with PPV type IIb, or phacomatosis cesioflammea. He had a large port-wine stain involving the right upper arm, back (Figure, A), and chest (Figure, B) with involvement of the bilateral conjunctivae (Figure, C). Our case is unique because our patient did not have dermal melanocytosis, only ocular melanocytosis.
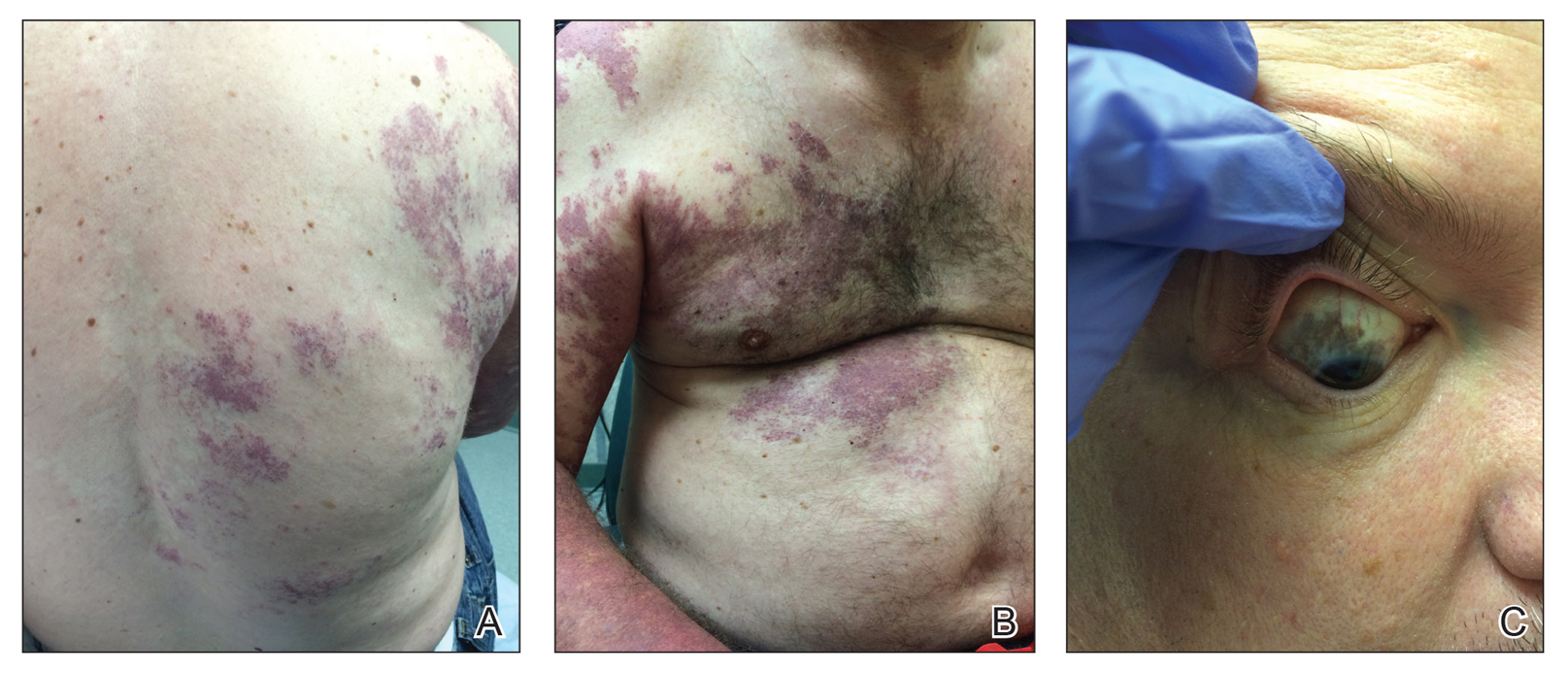
Once underlying neurologic and vascular anomalies have been ruled out, port-wine stains can be treated cosmetically. Pulsed dye laser is the gold standard therapy for capillary malformations, especially when instituted early. Follow-up with ophthalmology is advised to monitor ocular involvement. Shields et al11 suggested dilated fundoscopy for patients with port-wine stains because choroidal pigmentation often is the only ocular change seen. Ocular melanocytosis can progress to pigmented glaucoma or choroidal melanoma.
- Fernandez-Guarino M, Boixeda P, De las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Villarreal DJ, Leal F. Phacomatosis pigmentovascularis of cesioflammea type. An Bras Dermatol. 2016;91(5 suppl 1):54-56.
- Thomas AC, Zeng Z, Riviere JB, et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. 2016;136:770-778.
- Krema H, Simpson R, McGowan H. Choroidal melanoma in phacomatosis pigmentovascularis cesioflammea. Can J Ophthalmol. 2013;48:E41-E42.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:301-310.
- Pradhan S, Patnaik S, Padhi T, et al. Phakomatosis pigmentovascularis type IIb, Sturge-Weber syndrome and cone shaped tongue: an unusual association. Indian J Dermatol Venereol Leprol. 2015;81:614-616.
- Turk BG, Turkmen M, Tuna A, et al. Phakomatosis pigmentovascularis type IIb associated with Klippel-Trenaunay syndrome and congenital triangular alopecia. J Am Acad Dermatol. 2011;65:E46-E49.
- Sen S, Bala S, Halder C, et al. Phakomatosis pigmentovascularis presenting with Sturge-Weber syndrome and Klippel-Trenaunay syndrome. Indian J Dermatol. 2015;60:77-79.
- Schwartz RA, Fernandez G, Kotulska K, et al. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
The Diagnosis: Phacomatosis Cesioflammea
Phacomatosis pigmentovascularis (PPV) encompasses a group of diseases that have a vascular nevus coupled with a pigmented nevus.1 It is divided into 5 types: Type I is defined by the presence of a vascular malformation and epidermal nevus; type II by a vascular malformation and dermal melanosis with or without nevus anemicus; type III by a vascular malformation and nevus spilus with or without nevus anemicus; type IV by a vascular malformation, dermal melanosis, and nevus spilus with or without nevus anemicus; and type V as cutis marmorata telangiectatica congenita and dermal melanosis.1
Happle2 proposed a descriptive classification system in 2005 that eliminated type I PPV because neither linear epidermal nevus nor Becker nevus are derived from pigmentary cells. An appended "a" denotes a subtype with isolated cutaneous findings, while "b" is associated with extracutaneous manifestations. Phacomatosis cesioflammea (type IIa/b) refers to blue-hued dermal melanocytosis and nevus flammeus. Phacomatosis spilorosea (type IIIa/b) refers to nevus spilus and rose-colored nevus flammeus. Phacomatosis cesiomarmorata (type Va/b) refers to dermal melanocytosis and cutis marmorata telangiectasia congenita. The last group (type IVa/b) is unclassifiable phacomatosis pigmentovascularis.2,3
Phacomatosis pigmentovascularis can be isolated to the skin or have associated extracutaneous findings, including ocular melanocytosis, seizures, or cognitive delay due to intracerebral vascular malformations. Patients also can develop limb and soft-tissue overgrowth.4 Phacomatosis pigmentovascularis has been found to be associated with mutations in the GNA11 and GNAQ genes. The theory behind PPV is twin spotting, resulting from a somatic mutation that leads to mosaic proliferation of 2 different cell lines.5 Phacomatosis pigmentovascularis can occur in isolation or can demonstrate the phenotype of Sturge-Weber syndrome or Klippel-Trenaunay syndrome. In Sturge-Weber syndrome, capillary malformations involve the face and underlying leptomeninges and cerebral cortex. Glaucoma and epilepsy also may be present. In Klippel-Trenaunay syndrome, capillary malformations involve the extremities (usually the legs) in association with varicose veins, soft-tissue hypertrophy, and skeletal overgrowth.6-9 Tuberous sclerosis is an autosomal-dominant neurocutaneous disease in which patients develop hamartomas throughout the body, including the brain, skin, eyes, kidneys, heart, and lungs. Cutaneous manifestations include facial angiofibromas, ungual fibromas, hypomelanotic macules (ash leaf spots, confetti-like lesions), shagreen patches or connective tissue hamartomas, and fibrous plaques on the forehead. Tuberous sclerosis does not include vascular malformations.10
Our patient was diagnosed with PPV type IIb, or phacomatosis cesioflammea. He had a large port-wine stain involving the right upper arm, back (Figure, A), and chest (Figure, B) with involvement of the bilateral conjunctivae (Figure, C). Our case is unique because our patient did not have dermal melanocytosis, only ocular melanocytosis.
Once underlying neurologic and vascular anomalies have been ruled out, port-wine stains can be treated cosmetically. Pulsed dye laser is the gold standard therapy for capillary malformations, especially when instituted early. Follow-up with ophthalmology is advised to monitor ocular involvement. Shields et al11 suggested dilated fundoscopy for patients with port-wine stains because choroidal pigmentation often is the only ocular change seen. Ocular melanocytosis can progress to pigmented glaucoma or choroidal melanoma.
The Diagnosis: Phacomatosis Cesioflammea
Phacomatosis pigmentovascularis (PPV) encompasses a group of diseases that have a vascular nevus coupled with a pigmented nevus.1 It is divided into 5 types: Type I is defined by the presence of a vascular malformation and epidermal nevus; type II by a vascular malformation and dermal melanosis with or without nevus anemicus; type III by a vascular malformation and nevus spilus with or without nevus anemicus; type IV by a vascular malformation, dermal melanosis, and nevus spilus with or without nevus anemicus; and type V as cutis marmorata telangiectatica congenita and dermal melanosis.1
Happle2 proposed a descriptive classification system in 2005 that eliminated type I PPV because neither linear epidermal nevus nor Becker nevus are derived from pigmentary cells. An appended "a" denotes a subtype with isolated cutaneous findings, while "b" is associated with extracutaneous manifestations. Phacomatosis cesioflammea (type IIa/b) refers to blue-hued dermal melanocytosis and nevus flammeus. Phacomatosis spilorosea (type IIIa/b) refers to nevus spilus and rose-colored nevus flammeus. Phacomatosis cesiomarmorata (type Va/b) refers to dermal melanocytosis and cutis marmorata telangiectasia congenita. The last group (type IVa/b) is unclassifiable phacomatosis pigmentovascularis.2,3
Phacomatosis pigmentovascularis can be isolated to the skin or have associated extracutaneous findings, including ocular melanocytosis, seizures, or cognitive delay due to intracerebral vascular malformations. Patients also can develop limb and soft-tissue overgrowth.4 Phacomatosis pigmentovascularis has been found to be associated with mutations in the GNA11 and GNAQ genes. The theory behind PPV is twin spotting, resulting from a somatic mutation that leads to mosaic proliferation of 2 different cell lines.5 Phacomatosis pigmentovascularis can occur in isolation or can demonstrate the phenotype of Sturge-Weber syndrome or Klippel-Trenaunay syndrome. In Sturge-Weber syndrome, capillary malformations involve the face and underlying leptomeninges and cerebral cortex. Glaucoma and epilepsy also may be present. In Klippel-Trenaunay syndrome, capillary malformations involve the extremities (usually the legs) in association with varicose veins, soft-tissue hypertrophy, and skeletal overgrowth.6-9 Tuberous sclerosis is an autosomal-dominant neurocutaneous disease in which patients develop hamartomas throughout the body, including the brain, skin, eyes, kidneys, heart, and lungs. Cutaneous manifestations include facial angiofibromas, ungual fibromas, hypomelanotic macules (ash leaf spots, confetti-like lesions), shagreen patches or connective tissue hamartomas, and fibrous plaques on the forehead. Tuberous sclerosis does not include vascular malformations.10
Our patient was diagnosed with PPV type IIb, or phacomatosis cesioflammea. He had a large port-wine stain involving the right upper arm, back (Figure, A), and chest (Figure, B) with involvement of the bilateral conjunctivae (Figure, C). Our case is unique because our patient did not have dermal melanocytosis, only ocular melanocytosis.
Once underlying neurologic and vascular anomalies have been ruled out, port-wine stains can be treated cosmetically. Pulsed dye laser is the gold standard therapy for capillary malformations, especially when instituted early. Follow-up with ophthalmology is advised to monitor ocular involvement. Shields et al11 suggested dilated fundoscopy for patients with port-wine stains because choroidal pigmentation often is the only ocular change seen. Ocular melanocytosis can progress to pigmented glaucoma or choroidal melanoma.
- Fernandez-Guarino M, Boixeda P, De las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Villarreal DJ, Leal F. Phacomatosis pigmentovascularis of cesioflammea type. An Bras Dermatol. 2016;91(5 suppl 1):54-56.
- Thomas AC, Zeng Z, Riviere JB, et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. 2016;136:770-778.
- Krema H, Simpson R, McGowan H. Choroidal melanoma in phacomatosis pigmentovascularis cesioflammea. Can J Ophthalmol. 2013;48:E41-E42.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:301-310.
- Pradhan S, Patnaik S, Padhi T, et al. Phakomatosis pigmentovascularis type IIb, Sturge-Weber syndrome and cone shaped tongue: an unusual association. Indian J Dermatol Venereol Leprol. 2015;81:614-616.
- Turk BG, Turkmen M, Tuna A, et al. Phakomatosis pigmentovascularis type IIb associated with Klippel-Trenaunay syndrome and congenital triangular alopecia. J Am Acad Dermatol. 2011;65:E46-E49.
- Sen S, Bala S, Halder C, et al. Phakomatosis pigmentovascularis presenting with Sturge-Weber syndrome and Klippel-Trenaunay syndrome. Indian J Dermatol. 2015;60:77-79.
- Schwartz RA, Fernandez G, Kotulska K, et al. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
- Fernandez-Guarino M, Boixeda P, De las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Villarreal DJ, Leal F. Phacomatosis pigmentovascularis of cesioflammea type. An Bras Dermatol. 2016;91(5 suppl 1):54-56.
- Thomas AC, Zeng Z, Riviere JB, et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. 2016;136:770-778.
- Krema H, Simpson R, McGowan H. Choroidal melanoma in phacomatosis pigmentovascularis cesioflammea. Can J Ophthalmol. 2013;48:E41-E42.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:301-310.
- Pradhan S, Patnaik S, Padhi T, et al. Phakomatosis pigmentovascularis type IIb, Sturge-Weber syndrome and cone shaped tongue: an unusual association. Indian J Dermatol Venereol Leprol. 2015;81:614-616.
- Turk BG, Turkmen M, Tuna A, et al. Phakomatosis pigmentovascularis type IIb associated with Klippel-Trenaunay syndrome and congenital triangular alopecia. J Am Acad Dermatol. 2011;65:E46-E49.
- Sen S, Bala S, Halder C, et al. Phakomatosis pigmentovascularis presenting with Sturge-Weber syndrome and Klippel-Trenaunay syndrome. Indian J Dermatol. 2015;60:77-79.
- Schwartz RA, Fernandez G, Kotulska K, et al. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
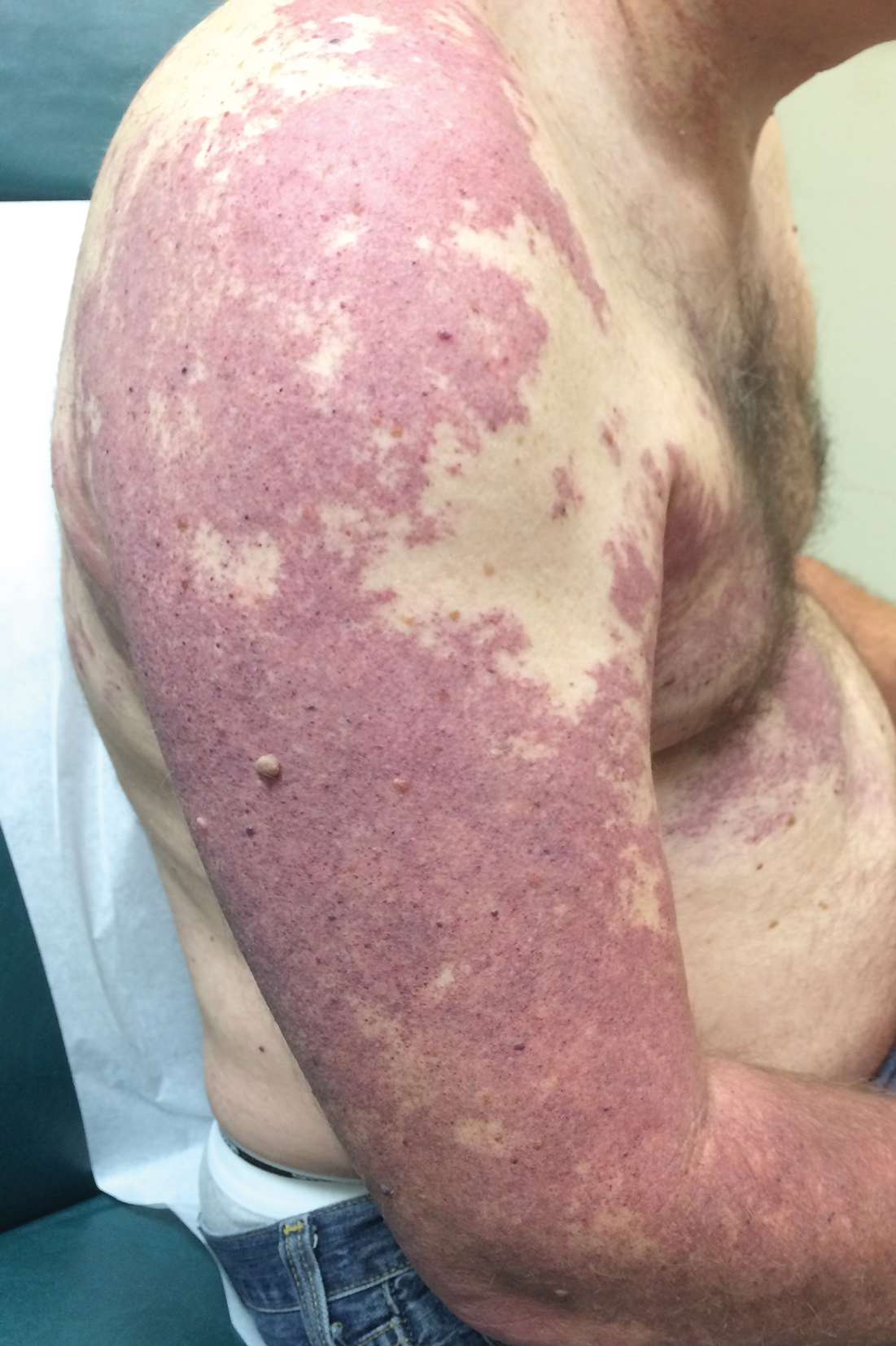
A 55-year-old man presented with red-violet patches on the right arm and chest that had been present since birth. The patches were asymptomatic and stable in size and shape. He denied any personal or family history of glaucoma or epilepsy. Physical examination demonstrated nonblanchable, violaceous to red patches on the right arm, back, and chest. No thrills or bruits were appreciable, and the right and left arms were of equal circumference and length. Further examination revealed hyperpigmented patches on the bilateral conjunctivae.
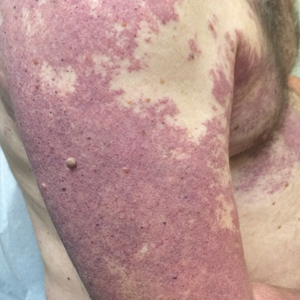
Erythematous Papules and Pustules on the Nose
The Diagnosis: Granulosis Rubra Nasi
A history of prominent nasal sweating was later elicited and the patient was subsequently diagnosed with granulosis rubra nasi. She was instructed to continue daily use of topical pimecrolimus with the addition of topical atropine, resulting in complete resolution of the eruption at 6-week follow-up (Figure, A). She was then maintained on topical atropine monotherapy, only noting recurrence with cessation of the atropine (Figure, B).
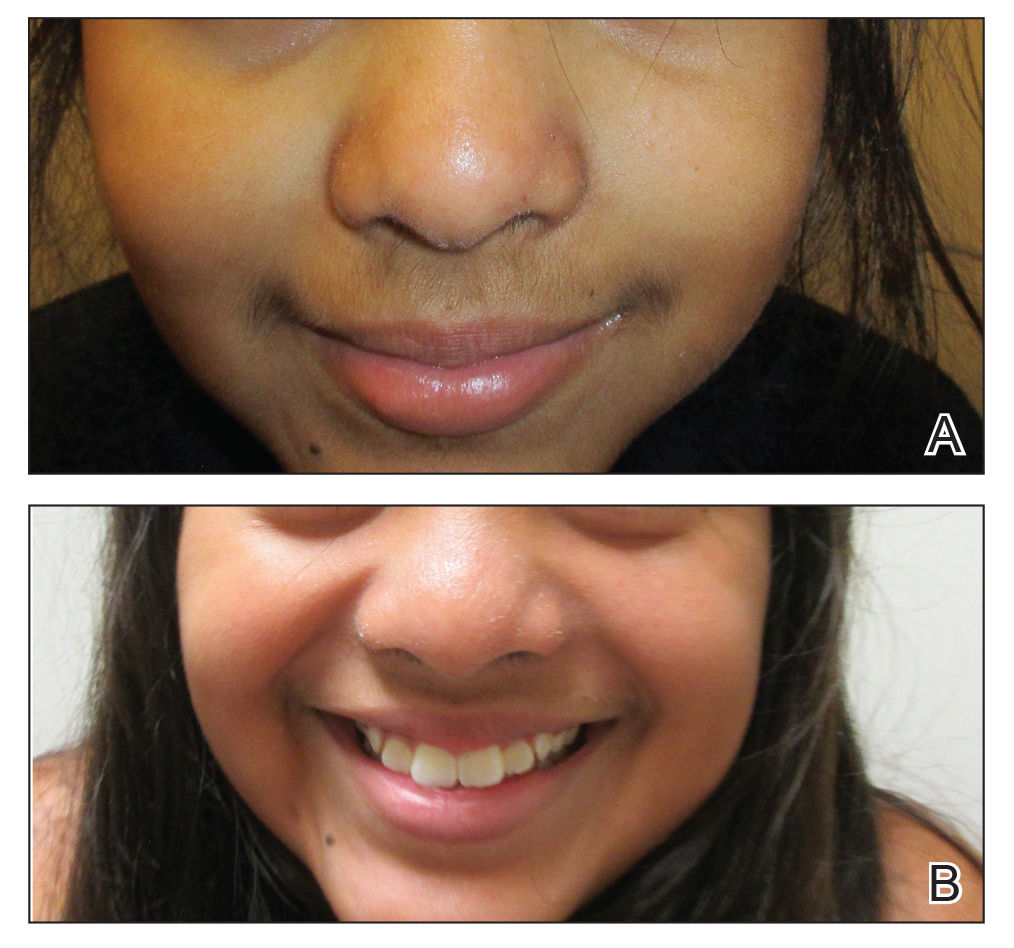
Other successful treatment regimens of granulosis rubra nasi include injection of botulinum toxin into the nose,1 monotherapy with topical tacrolimus,2 topical indomethacin, steroids, and cryotherapy, among other modalities.1 Topical atropine and pimecrolimus were selected as first-line agents for treating our pediatric patient due to tolerability and their anti-inflammatory and anticholinergic properties.
Granulosis rubra nasi is a form of focal hyperhidrosis that presents as erythematous papules, pustules, and vesicles of the midface, especially the nose.3 It is a fairly rare condition that can mimic many other common clinical entities, including comedonal acne, nevus comedonicus, periorificial dermatitis, and tinea faciei, but is resistant to treatments aimed at these disorders. It was first described as a "peculiar disease of the skin of the nose in children" in a case report by Jadassohn4 in 1901. It is most common in children aged 7 to 12 years and typically resolves at puberty; adults rarely are affected. Although the etiology has not yet been elucidated, autosomal-dominant transmission has been described, and the cutaneous changes are hypothesized to be secondary to hyperhidrosis.5 This postulation is further corroborated by a case report of a pheochromocytoma-associated granulosis rubra nasi that resolved with surgical excision of the pheochromocytoma.6 It is not uncommon for patients to have concomitant palmoplantar hyperhidrosis and acrocyanosis.5 Histopathologic examination is not necessary for diagnosis, but when performed, it discloses a mononuclear cellular infiltrate surrounding eccrine sweat ducts, blood vessels, and lymphatics without other abnormalities of the epidermis or pilosebaceous unit.1-3,7
- Grazziotin TC, Buffon RB, Da Silva Manzoni AP, et al. Treatment of granulosis rubra nasi with botulinum toxin. Dermatol Surg. 2009;35:1298-1299.
- Kumar P, Gosai A, Mondal AK, et al. Granulosis rubra nasi: a rare condition treated successfully with topical tacrolimus. Dermatol Reports. 2012;4:E5.
- Sargunam C, Thomas J, Ahmed NA. Granulosis rubra nasi. Indian Dermatol Online J. 2013;4:208-209.
- Jadassohn J. Ueber eine eigenartige erkrankung der nasenhaut bei kindern. Arch Derm Syph. 1901;58:145-158.
- Hellier FF. Granulosis rubra nasi in a mother and daughter. Br Med J. 1937;2:1068.
- Heid E, Samain F, Jelen G, et al. Granulosis rubra nasi and pheochromocytoma. Ann Dermatol Venereol. 1996;123:106-108.
- Akhdari N. Granulosis rubra nasi. Int J Dermatol. 2007;46:396.
The Diagnosis: Granulosis Rubra Nasi
A history of prominent nasal sweating was later elicited and the patient was subsequently diagnosed with granulosis rubra nasi. She was instructed to continue daily use of topical pimecrolimus with the addition of topical atropine, resulting in complete resolution of the eruption at 6-week follow-up (Figure, A). She was then maintained on topical atropine monotherapy, only noting recurrence with cessation of the atropine (Figure, B).
Other successful treatment regimens of granulosis rubra nasi include injection of botulinum toxin into the nose,1 monotherapy with topical tacrolimus,2 topical indomethacin, steroids, and cryotherapy, among other modalities.1 Topical atropine and pimecrolimus were selected as first-line agents for treating our pediatric patient due to tolerability and their anti-inflammatory and anticholinergic properties.
Granulosis rubra nasi is a form of focal hyperhidrosis that presents as erythematous papules, pustules, and vesicles of the midface, especially the nose.3 It is a fairly rare condition that can mimic many other common clinical entities, including comedonal acne, nevus comedonicus, periorificial dermatitis, and tinea faciei, but is resistant to treatments aimed at these disorders. It was first described as a "peculiar disease of the skin of the nose in children" in a case report by Jadassohn4 in 1901. It is most common in children aged 7 to 12 years and typically resolves at puberty; adults rarely are affected. Although the etiology has not yet been elucidated, autosomal-dominant transmission has been described, and the cutaneous changes are hypothesized to be secondary to hyperhidrosis.5 This postulation is further corroborated by a case report of a pheochromocytoma-associated granulosis rubra nasi that resolved with surgical excision of the pheochromocytoma.6 It is not uncommon for patients to have concomitant palmoplantar hyperhidrosis and acrocyanosis.5 Histopathologic examination is not necessary for diagnosis, but when performed, it discloses a mononuclear cellular infiltrate surrounding eccrine sweat ducts, blood vessels, and lymphatics without other abnormalities of the epidermis or pilosebaceous unit.1-3,7
The Diagnosis: Granulosis Rubra Nasi
A history of prominent nasal sweating was later elicited and the patient was subsequently diagnosed with granulosis rubra nasi. She was instructed to continue daily use of topical pimecrolimus with the addition of topical atropine, resulting in complete resolution of the eruption at 6-week follow-up (Figure, A). She was then maintained on topical atropine monotherapy, only noting recurrence with cessation of the atropine (Figure, B).
Other successful treatment regimens of granulosis rubra nasi include injection of botulinum toxin into the nose,1 monotherapy with topical tacrolimus,2 topical indomethacin, steroids, and cryotherapy, among other modalities.1 Topical atropine and pimecrolimus were selected as first-line agents for treating our pediatric patient due to tolerability and their anti-inflammatory and anticholinergic properties.
Granulosis rubra nasi is a form of focal hyperhidrosis that presents as erythematous papules, pustules, and vesicles of the midface, especially the nose.3 It is a fairly rare condition that can mimic many other common clinical entities, including comedonal acne, nevus comedonicus, periorificial dermatitis, and tinea faciei, but is resistant to treatments aimed at these disorders. It was first described as a "peculiar disease of the skin of the nose in children" in a case report by Jadassohn4 in 1901. It is most common in children aged 7 to 12 years and typically resolves at puberty; adults rarely are affected. Although the etiology has not yet been elucidated, autosomal-dominant transmission has been described, and the cutaneous changes are hypothesized to be secondary to hyperhidrosis.5 This postulation is further corroborated by a case report of a pheochromocytoma-associated granulosis rubra nasi that resolved with surgical excision of the pheochromocytoma.6 It is not uncommon for patients to have concomitant palmoplantar hyperhidrosis and acrocyanosis.5 Histopathologic examination is not necessary for diagnosis, but when performed, it discloses a mononuclear cellular infiltrate surrounding eccrine sweat ducts, blood vessels, and lymphatics without other abnormalities of the epidermis or pilosebaceous unit.1-3,7
- Grazziotin TC, Buffon RB, Da Silva Manzoni AP, et al. Treatment of granulosis rubra nasi with botulinum toxin. Dermatol Surg. 2009;35:1298-1299.
- Kumar P, Gosai A, Mondal AK, et al. Granulosis rubra nasi: a rare condition treated successfully with topical tacrolimus. Dermatol Reports. 2012;4:E5.
- Sargunam C, Thomas J, Ahmed NA. Granulosis rubra nasi. Indian Dermatol Online J. 2013;4:208-209.
- Jadassohn J. Ueber eine eigenartige erkrankung der nasenhaut bei kindern. Arch Derm Syph. 1901;58:145-158.
- Hellier FF. Granulosis rubra nasi in a mother and daughter. Br Med J. 1937;2:1068.
- Heid E, Samain F, Jelen G, et al. Granulosis rubra nasi and pheochromocytoma. Ann Dermatol Venereol. 1996;123:106-108.
- Akhdari N. Granulosis rubra nasi. Int J Dermatol. 2007;46:396.
- Grazziotin TC, Buffon RB, Da Silva Manzoni AP, et al. Treatment of granulosis rubra nasi with botulinum toxin. Dermatol Surg. 2009;35:1298-1299.
- Kumar P, Gosai A, Mondal AK, et al. Granulosis rubra nasi: a rare condition treated successfully with topical tacrolimus. Dermatol Reports. 2012;4:E5.
- Sargunam C, Thomas J, Ahmed NA. Granulosis rubra nasi. Indian Dermatol Online J. 2013;4:208-209.
- Jadassohn J. Ueber eine eigenartige erkrankung der nasenhaut bei kindern. Arch Derm Syph. 1901;58:145-158.
- Hellier FF. Granulosis rubra nasi in a mother and daughter. Br Med J. 1937;2:1068.
- Heid E, Samain F, Jelen G, et al. Granulosis rubra nasi and pheochromocytoma. Ann Dermatol Venereol. 1996;123:106-108.
- Akhdari N. Granulosis rubra nasi. Int J Dermatol. 2007;46:396.
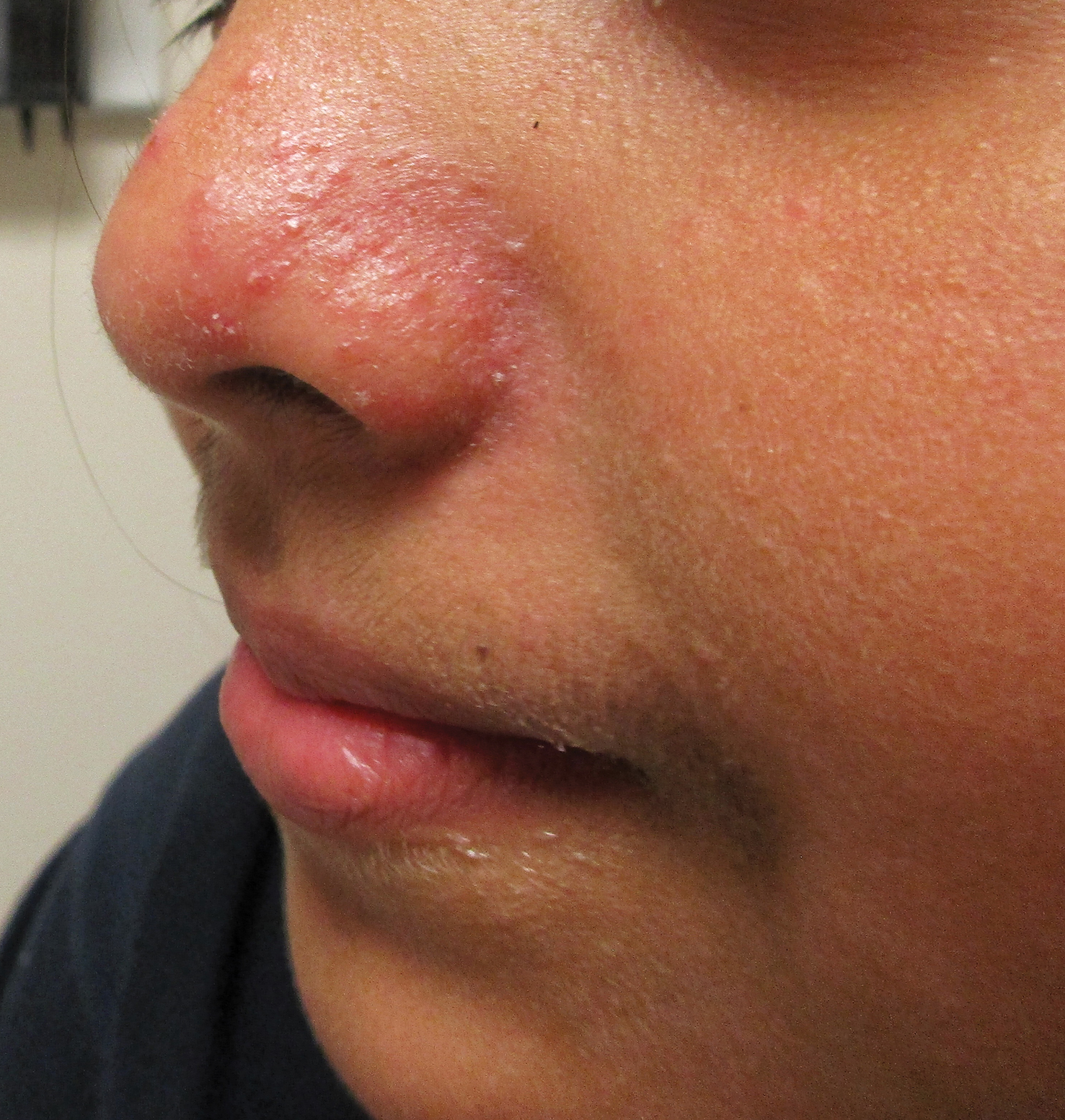
A healthy 9-year-old girl presented with a 2-year history of erythematous papules and pustules on the nose. There was no involvement of the rest of the face or body. At the time of presentation, she had been treated with several topical therapies including steroids, calcineurin inhibitors, antibiotics, and retinoids without improvement. A potassium hydroxide preparation from a pustule was performed and revealed only normal keratinocytes.
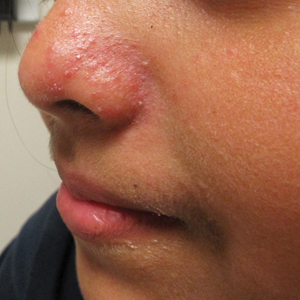
Well-Circumscribed Tumor on the Hand
The Diagnosis: Nodular Kaposi Sarcoma
Epidemic Kaposi sarcoma (KS) primarily affects patients with human immunodeficiency virus (HIV) infection. Kaposi sarcoma can appear as brown, red, or blue-black macules, plaques, patches, nodules, or tumors, and it often is observed as multifocal cutaneous lesions located on the head, neck, and upper aspects of the trunk in a fulminant manner. Kaposi sarcoma portends a poor prognosis and is an AIDS-defining malignancy.1-3 Importantly, antiretroviral therapy does not preclude its consideration in those without AIDS-defining CD4 cell counts and undetectable HIV viremia presenting with cutaneous manifestations.2,3 A retrospective review by Daly et al4 reported KS lesions in patients with CD4 lymphocyte counts greater than 300 cells/µL, most of whom were antiretroviral therapy-naïve patients. Also, those with higher CD4 counts tended to have a solitary KS lesion at presentation, while those with CD4 counts less than 300 cells/µL tended to present with multiple foci.4 Epidemic KS lesions are clinically indistinguishable from other common cutaneous conditions in the differential diagnosis of KS, necessitating biopsy for histopathologic examination. Light microscopy findings help to delineate the diagnosis of KS. Immunohistochemical staining to the latent nuclear antigen 1 of human herpesvirus 8 (HHV-8) confirms the KS diagnosis.5,6 Our patient's presentation as a solitary acral lesion was atypical for KS.
Light microscopy of our patient's biopsy demonstrated a large tumor on the acral surface of the right hand. Dermal collections of basophilic spindled cells clustered with small slitlike vascular spaces with abundant erythrocyte extravasation and numerous large ectatic vessels at the periphery were seen (Figure, A). At higher magnification, interlaced bundles of spindle cells with slitlike vessels with scattered lymphocytes and plasma cells were seen (Figure, B). An immunohistochemical stain for HHV-8 was positive and largely confined to spindle cells (Figure, C). These findings confirmed KS and met AIDS-defining criteria. Awareness of these histopathologic features is key in differentiating KS from other conditions in the differential diagnosis.

The patient's history of late latent syphilis coinfected with HIV and persistently elevated rapid plasma reagin that was recalcitrant to therapy placed an atypical nodular presentation within reason for the differential diagnosis. Deviations from the typical papulosquamous presentation with acral involvement in an immunocompromised patient mandates a consideration for syphilis with an atypical presentation. Atypical presentations include nodular, annular, pustular, lues maligna, frambesiform, corymbose, and photosensitive distributions.7,8 Notably, coinfection with HIV modifies the clinical presentation, serology, and efficacy of treatment.7-10 Atypical presentations are more common in coinfected HIV-positive patients, mandating a high degree of suspicion. Nodular secondary syphilis and the noduloulcerative form (lues maligna) often spare the palmar and plantar surfaces, and patients often have constitutional symptoms accompanying the cutaneous eruptions. In questionable cases, a biopsy lends clarification. Light microscopy on hematoxylin and eosin (H&E) staining may display acanthosis, superficial and deep perivascular swelling, plasma, histiocyte infiltrates, dermoepidermal junction changes, mixed patterns, epidermal hyperplasia, and dermal vascular thickening.7-9,11 Spirochetes may be observed on Warthin-Starry stain; however, artifact obscuration from melanin granules and reticular fibers or paucity of organisms can make identification difficult. Immunohistochemical staining may prove useful when H&E stains are atypical or have a paucity of organisms or plasma cells or when silver stains have artifactual obscuration.9 Our patient's solitary palmar lesion without constitutional symptoms made an atypical nodular secondary syphilis presentation less likely. Ultimately, the histopathologic findings were consistent with KS.
Bacillary angiomatosis (BA) is caused by Bartonella species and results in vascular proliferation with cutaneous manifestation. It frequently is observed in patients with HIV or other immunosuppressive conditions as well as patients with exposure to mammals or their vectors. Protean cutaneous manifestations and distributions of BA exist. The number of lesions can be singular to thousands. Solitary superficial pyogenic granuloma-like lesions can be clinically indistinguishable from both KS and pyogenic granuloma (PG). Superficial lesions often begin as red, violaceous, or flesh-colored papules that hemorrhage easily with trauma. The morphology of the papule can progress to be exophytic with dome-shaped or ulcerative surface features and is rubbery on palpation.12 Biopsy is required to differentiate BA from KS. Bacillary angiomatosis on light microscopy with H&E shows protuberant, lobulated, round vessels with plump endothelial cells with or without necrosis. A neutrophil infiltrate in close proximity to bacilli may be noted. Warthin-Starry stain demonstrates numerous bacilli juxtaposed to these endothelial cells. The lack of immunohistochemical staining for HHV-8 also differentiates BA from KS.12,13
Pyogenic granuloma is resultant from proliferation of endothelial cells with a lobular architecture. Pyogenic granulomas are benign, rapidly progressive, acquired lesions presenting in the skin and mucous membranes. Pyogenic granuloma often presents as a single painless papule or nodule with a glistening red-violaceous color that occasionally appears with a perilesional collarette. The lesions are friable and easily hemorrhage. Pyogenic granuloma has been associated with local skin trauma and estrogen hormones. Histopathologic examination of PG assists with differentiation from other nodular lesions. Light microscopy with standard H&E staining demonstrates a network of capillaries arranged into a lobule surrounded by a fibrous matrix. Endothelial cells appear round and protrude into the vascular lamina. Mitotic activity is increased. Lack of findings on Warthin-Starry stain assists with differentiating PG from BA, while the microscopy architecture and immunohistochemical staining differentiates PG from KS.6,13,14
Squamous cell carcinoma (SCC) is the primary malignant cancer of the hand. The dorsal aspect of the hand is the most common location; SCC less commonly is located on the palmar surface, fingers, nail bed, or intertriginous areas.15-17 Chakrabarti et al16 found that these lesions were invasive SCC when located on the palmar surface. Morphologically, SCC takes an exophytic papular, nodular, or scaly appearance with a red to flesh-colored appearance and poor demarcation of the borders. Progression to large ulcerated or secondarily infected lesions also can occur. The inflammatory reaction may cause tenderness to palpation and hemorrhage with trauma. Histopathologic examination of invasive SCC reveals atypical keratinocytes violating the basement membrane and abundant cytoplasm. Our patient's clinical presentation placed invasive SCC low on the differential diagnosis, and the histopathologic and immunohistochemical results eliminated SCC as the diagnosis.
- Antman K, Chang Y. Kaposi's sarcoma. N Engl J Med. 2000;342:1027-1038.
- Pipette WW. The incidence of second malignancies in subsets of Kaposi's sarcoma. J Am Acad Dermatol. 1987;16:855-861.
- Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12:6-11.
- Daly ML, Fogo A, McDonald C, et al. Kaposi sarcoma: no longer an AIDS-defining illness? a retrospective study of Kaposi sarcoma cases with CD4 counts above 300/mm³ at presentation. Clin Exp Dermatol. 2014;39:7-12.
- Broccolo F, Tassan Din C, Viganò MG, et al. HHV-8 DNA replication correlates with the clinical status in AIDS-related Kaposi's sarcoma. J Clin Virol. 2016;78:47-52.
- Pereira PF, Cuzzi T, Galhardo MC. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An Bras Dermatol. 2013;88:243-246.
- Gevorgyan O, Owen BD, Balavenkataraman A, et al. A nodular-ulcerative form of secondary syphilis in AIDS. Proc (Bayl Univ Med Cent). 2017;30:80-82.
- Balagula Y, Mattei PL, Wisco OJ, et al. The great imitator revisited: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;31:595-599.
- Yayli S, della Torre R, Hegyi I, et al. Late secondary syphilis with nodular lesions mimicking Kaposi sarcoma in a patient with human immunodeficiency virus. Int J Dermatol. 2014;53:E71-E73.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Cockerell CJ, LeBoit PE. Bacillary angiomatosis: a newly characterized, pseudoneoplastic, infectious, cutaneous vascular disorder. J Am Acad Dermatol. 1990;22:501-512.
- Forrestel AK, Naujokas A, Martin JN, et al. Bacillary angiomatosis masquerading as Kaposi's sarcoma in East Africa. J Int Assoc Provid AIDS Care. 2015;14:21-25.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma), localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol. 2007;29:408-411.
- Marks R. Squamous cell carcinoma. Lancet. 1996;347:735-738.
- Chakrabarti I, Watson JD, Dorrance H. Skin tumours of the hand. a 10-year review. J Hand Surg Br. 1993;18:484-486.
- Sobanko JF, Dagum AB, Davis IC, et al. Soft tissue tumors of the hand. 2. malignant. Dermatol Surg. 2007;33:771-785.
The Diagnosis: Nodular Kaposi Sarcoma
Epidemic Kaposi sarcoma (KS) primarily affects patients with human immunodeficiency virus (HIV) infection. Kaposi sarcoma can appear as brown, red, or blue-black macules, plaques, patches, nodules, or tumors, and it often is observed as multifocal cutaneous lesions located on the head, neck, and upper aspects of the trunk in a fulminant manner. Kaposi sarcoma portends a poor prognosis and is an AIDS-defining malignancy.1-3 Importantly, antiretroviral therapy does not preclude its consideration in those without AIDS-defining CD4 cell counts and undetectable HIV viremia presenting with cutaneous manifestations.2,3 A retrospective review by Daly et al4 reported KS lesions in patients with CD4 lymphocyte counts greater than 300 cells/µL, most of whom were antiretroviral therapy-naïve patients. Also, those with higher CD4 counts tended to have a solitary KS lesion at presentation, while those with CD4 counts less than 300 cells/µL tended to present with multiple foci.4 Epidemic KS lesions are clinically indistinguishable from other common cutaneous conditions in the differential diagnosis of KS, necessitating biopsy for histopathologic examination. Light microscopy findings help to delineate the diagnosis of KS. Immunohistochemical staining to the latent nuclear antigen 1 of human herpesvirus 8 (HHV-8) confirms the KS diagnosis.5,6 Our patient's presentation as a solitary acral lesion was atypical for KS.
Light microscopy of our patient's biopsy demonstrated a large tumor on the acral surface of the right hand. Dermal collections of basophilic spindled cells clustered with small slitlike vascular spaces with abundant erythrocyte extravasation and numerous large ectatic vessels at the periphery were seen (Figure, A). At higher magnification, interlaced bundles of spindle cells with slitlike vessels with scattered lymphocytes and plasma cells were seen (Figure, B). An immunohistochemical stain for HHV-8 was positive and largely confined to spindle cells (Figure, C). These findings confirmed KS and met AIDS-defining criteria. Awareness of these histopathologic features is key in differentiating KS from other conditions in the differential diagnosis.
The patient's history of late latent syphilis coinfected with HIV and persistently elevated rapid plasma reagin that was recalcitrant to therapy placed an atypical nodular presentation within reason for the differential diagnosis. Deviations from the typical papulosquamous presentation with acral involvement in an immunocompromised patient mandates a consideration for syphilis with an atypical presentation. Atypical presentations include nodular, annular, pustular, lues maligna, frambesiform, corymbose, and photosensitive distributions.7,8 Notably, coinfection with HIV modifies the clinical presentation, serology, and efficacy of treatment.7-10 Atypical presentations are more common in coinfected HIV-positive patients, mandating a high degree of suspicion. Nodular secondary syphilis and the noduloulcerative form (lues maligna) often spare the palmar and plantar surfaces, and patients often have constitutional symptoms accompanying the cutaneous eruptions. In questionable cases, a biopsy lends clarification. Light microscopy on hematoxylin and eosin (H&E) staining may display acanthosis, superficial and deep perivascular swelling, plasma, histiocyte infiltrates, dermoepidermal junction changes, mixed patterns, epidermal hyperplasia, and dermal vascular thickening.7-9,11 Spirochetes may be observed on Warthin-Starry stain; however, artifact obscuration from melanin granules and reticular fibers or paucity of organisms can make identification difficult. Immunohistochemical staining may prove useful when H&E stains are atypical or have a paucity of organisms or plasma cells or when silver stains have artifactual obscuration.9 Our patient's solitary palmar lesion without constitutional symptoms made an atypical nodular secondary syphilis presentation less likely. Ultimately, the histopathologic findings were consistent with KS.
Bacillary angiomatosis (BA) is caused by Bartonella species and results in vascular proliferation with cutaneous manifestation. It frequently is observed in patients with HIV or other immunosuppressive conditions as well as patients with exposure to mammals or their vectors. Protean cutaneous manifestations and distributions of BA exist. The number of lesions can be singular to thousands. Solitary superficial pyogenic granuloma-like lesions can be clinically indistinguishable from both KS and pyogenic granuloma (PG). Superficial lesions often begin as red, violaceous, or flesh-colored papules that hemorrhage easily with trauma. The morphology of the papule can progress to be exophytic with dome-shaped or ulcerative surface features and is rubbery on palpation.12 Biopsy is required to differentiate BA from KS. Bacillary angiomatosis on light microscopy with H&E shows protuberant, lobulated, round vessels with plump endothelial cells with or without necrosis. A neutrophil infiltrate in close proximity to bacilli may be noted. Warthin-Starry stain demonstrates numerous bacilli juxtaposed to these endothelial cells. The lack of immunohistochemical staining for HHV-8 also differentiates BA from KS.12,13
Pyogenic granuloma is resultant from proliferation of endothelial cells with a lobular architecture. Pyogenic granulomas are benign, rapidly progressive, acquired lesions presenting in the skin and mucous membranes. Pyogenic granuloma often presents as a single painless papule or nodule with a glistening red-violaceous color that occasionally appears with a perilesional collarette. The lesions are friable and easily hemorrhage. Pyogenic granuloma has been associated with local skin trauma and estrogen hormones. Histopathologic examination of PG assists with differentiation from other nodular lesions. Light microscopy with standard H&E staining demonstrates a network of capillaries arranged into a lobule surrounded by a fibrous matrix. Endothelial cells appear round and protrude into the vascular lamina. Mitotic activity is increased. Lack of findings on Warthin-Starry stain assists with differentiating PG from BA, while the microscopy architecture and immunohistochemical staining differentiates PG from KS.6,13,14
Squamous cell carcinoma (SCC) is the primary malignant cancer of the hand. The dorsal aspect of the hand is the most common location; SCC less commonly is located on the palmar surface, fingers, nail bed, or intertriginous areas.15-17 Chakrabarti et al16 found that these lesions were invasive SCC when located on the palmar surface. Morphologically, SCC takes an exophytic papular, nodular, or scaly appearance with a red to flesh-colored appearance and poor demarcation of the borders. Progression to large ulcerated or secondarily infected lesions also can occur. The inflammatory reaction may cause tenderness to palpation and hemorrhage with trauma. Histopathologic examination of invasive SCC reveals atypical keratinocytes violating the basement membrane and abundant cytoplasm. Our patient's clinical presentation placed invasive SCC low on the differential diagnosis, and the histopathologic and immunohistochemical results eliminated SCC as the diagnosis.
The Diagnosis: Nodular Kaposi Sarcoma
Epidemic Kaposi sarcoma (KS) primarily affects patients with human immunodeficiency virus (HIV) infection. Kaposi sarcoma can appear as brown, red, or blue-black macules, plaques, patches, nodules, or tumors, and it often is observed as multifocal cutaneous lesions located on the head, neck, and upper aspects of the trunk in a fulminant manner. Kaposi sarcoma portends a poor prognosis and is an AIDS-defining malignancy.1-3 Importantly, antiretroviral therapy does not preclude its consideration in those without AIDS-defining CD4 cell counts and undetectable HIV viremia presenting with cutaneous manifestations.2,3 A retrospective review by Daly et al4 reported KS lesions in patients with CD4 lymphocyte counts greater than 300 cells/µL, most of whom were antiretroviral therapy-naïve patients. Also, those with higher CD4 counts tended to have a solitary KS lesion at presentation, while those with CD4 counts less than 300 cells/µL tended to present with multiple foci.4 Epidemic KS lesions are clinically indistinguishable from other common cutaneous conditions in the differential diagnosis of KS, necessitating biopsy for histopathologic examination. Light microscopy findings help to delineate the diagnosis of KS. Immunohistochemical staining to the latent nuclear antigen 1 of human herpesvirus 8 (HHV-8) confirms the KS diagnosis.5,6 Our patient's presentation as a solitary acral lesion was atypical for KS.
Light microscopy of our patient's biopsy demonstrated a large tumor on the acral surface of the right hand. Dermal collections of basophilic spindled cells clustered with small slitlike vascular spaces with abundant erythrocyte extravasation and numerous large ectatic vessels at the periphery were seen (Figure, A). At higher magnification, interlaced bundles of spindle cells with slitlike vessels with scattered lymphocytes and plasma cells were seen (Figure, B). An immunohistochemical stain for HHV-8 was positive and largely confined to spindle cells (Figure, C). These findings confirmed KS and met AIDS-defining criteria. Awareness of these histopathologic features is key in differentiating KS from other conditions in the differential diagnosis.
The patient's history of late latent syphilis coinfected with HIV and persistently elevated rapid plasma reagin that was recalcitrant to therapy placed an atypical nodular presentation within reason for the differential diagnosis. Deviations from the typical papulosquamous presentation with acral involvement in an immunocompromised patient mandates a consideration for syphilis with an atypical presentation. Atypical presentations include nodular, annular, pustular, lues maligna, frambesiform, corymbose, and photosensitive distributions.7,8 Notably, coinfection with HIV modifies the clinical presentation, serology, and efficacy of treatment.7-10 Atypical presentations are more common in coinfected HIV-positive patients, mandating a high degree of suspicion. Nodular secondary syphilis and the noduloulcerative form (lues maligna) often spare the palmar and plantar surfaces, and patients often have constitutional symptoms accompanying the cutaneous eruptions. In questionable cases, a biopsy lends clarification. Light microscopy on hematoxylin and eosin (H&E) staining may display acanthosis, superficial and deep perivascular swelling, plasma, histiocyte infiltrates, dermoepidermal junction changes, mixed patterns, epidermal hyperplasia, and dermal vascular thickening.7-9,11 Spirochetes may be observed on Warthin-Starry stain; however, artifact obscuration from melanin granules and reticular fibers or paucity of organisms can make identification difficult. Immunohistochemical staining may prove useful when H&E stains are atypical or have a paucity of organisms or plasma cells or when silver stains have artifactual obscuration.9 Our patient's solitary palmar lesion without constitutional symptoms made an atypical nodular secondary syphilis presentation less likely. Ultimately, the histopathologic findings were consistent with KS.
Bacillary angiomatosis (BA) is caused by Bartonella species and results in vascular proliferation with cutaneous manifestation. It frequently is observed in patients with HIV or other immunosuppressive conditions as well as patients with exposure to mammals or their vectors. Protean cutaneous manifestations and distributions of BA exist. The number of lesions can be singular to thousands. Solitary superficial pyogenic granuloma-like lesions can be clinically indistinguishable from both KS and pyogenic granuloma (PG). Superficial lesions often begin as red, violaceous, or flesh-colored papules that hemorrhage easily with trauma. The morphology of the papule can progress to be exophytic with dome-shaped or ulcerative surface features and is rubbery on palpation.12 Biopsy is required to differentiate BA from KS. Bacillary angiomatosis on light microscopy with H&E shows protuberant, lobulated, round vessels with plump endothelial cells with or without necrosis. A neutrophil infiltrate in close proximity to bacilli may be noted. Warthin-Starry stain demonstrates numerous bacilli juxtaposed to these endothelial cells. The lack of immunohistochemical staining for HHV-8 also differentiates BA from KS.12,13
Pyogenic granuloma is resultant from proliferation of endothelial cells with a lobular architecture. Pyogenic granulomas are benign, rapidly progressive, acquired lesions presenting in the skin and mucous membranes. Pyogenic granuloma often presents as a single painless papule or nodule with a glistening red-violaceous color that occasionally appears with a perilesional collarette. The lesions are friable and easily hemorrhage. Pyogenic granuloma has been associated with local skin trauma and estrogen hormones. Histopathologic examination of PG assists with differentiation from other nodular lesions. Light microscopy with standard H&E staining demonstrates a network of capillaries arranged into a lobule surrounded by a fibrous matrix. Endothelial cells appear round and protrude into the vascular lamina. Mitotic activity is increased. Lack of findings on Warthin-Starry stain assists with differentiating PG from BA, while the microscopy architecture and immunohistochemical staining differentiates PG from KS.6,13,14
Squamous cell carcinoma (SCC) is the primary malignant cancer of the hand. The dorsal aspect of the hand is the most common location; SCC less commonly is located on the palmar surface, fingers, nail bed, or intertriginous areas.15-17 Chakrabarti et al16 found that these lesions were invasive SCC when located on the palmar surface. Morphologically, SCC takes an exophytic papular, nodular, or scaly appearance with a red to flesh-colored appearance and poor demarcation of the borders. Progression to large ulcerated or secondarily infected lesions also can occur. The inflammatory reaction may cause tenderness to palpation and hemorrhage with trauma. Histopathologic examination of invasive SCC reveals atypical keratinocytes violating the basement membrane and abundant cytoplasm. Our patient's clinical presentation placed invasive SCC low on the differential diagnosis, and the histopathologic and immunohistochemical results eliminated SCC as the diagnosis.
- Antman K, Chang Y. Kaposi's sarcoma. N Engl J Med. 2000;342:1027-1038.
- Pipette WW. The incidence of second malignancies in subsets of Kaposi's sarcoma. J Am Acad Dermatol. 1987;16:855-861.
- Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12:6-11.
- Daly ML, Fogo A, McDonald C, et al. Kaposi sarcoma: no longer an AIDS-defining illness? a retrospective study of Kaposi sarcoma cases with CD4 counts above 300/mm³ at presentation. Clin Exp Dermatol. 2014;39:7-12.
- Broccolo F, Tassan Din C, Viganò MG, et al. HHV-8 DNA replication correlates with the clinical status in AIDS-related Kaposi's sarcoma. J Clin Virol. 2016;78:47-52.
- Pereira PF, Cuzzi T, Galhardo MC. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An Bras Dermatol. 2013;88:243-246.
- Gevorgyan O, Owen BD, Balavenkataraman A, et al. A nodular-ulcerative form of secondary syphilis in AIDS. Proc (Bayl Univ Med Cent). 2017;30:80-82.
- Balagula Y, Mattei PL, Wisco OJ, et al. The great imitator revisited: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;31:595-599.
- Yayli S, della Torre R, Hegyi I, et al. Late secondary syphilis with nodular lesions mimicking Kaposi sarcoma in a patient with human immunodeficiency virus. Int J Dermatol. 2014;53:E71-E73.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Cockerell CJ, LeBoit PE. Bacillary angiomatosis: a newly characterized, pseudoneoplastic, infectious, cutaneous vascular disorder. J Am Acad Dermatol. 1990;22:501-512.
- Forrestel AK, Naujokas A, Martin JN, et al. Bacillary angiomatosis masquerading as Kaposi's sarcoma in East Africa. J Int Assoc Provid AIDS Care. 2015;14:21-25.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma), localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol. 2007;29:408-411.
- Marks R. Squamous cell carcinoma. Lancet. 1996;347:735-738.
- Chakrabarti I, Watson JD, Dorrance H. Skin tumours of the hand. a 10-year review. J Hand Surg Br. 1993;18:484-486.
- Sobanko JF, Dagum AB, Davis IC, et al. Soft tissue tumors of the hand. 2. malignant. Dermatol Surg. 2007;33:771-785.
- Antman K, Chang Y. Kaposi's sarcoma. N Engl J Med. 2000;342:1027-1038.
- Pipette WW. The incidence of second malignancies in subsets of Kaposi's sarcoma. J Am Acad Dermatol. 1987;16:855-861.
- Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12:6-11.
- Daly ML, Fogo A, McDonald C, et al. Kaposi sarcoma: no longer an AIDS-defining illness? a retrospective study of Kaposi sarcoma cases with CD4 counts above 300/mm³ at presentation. Clin Exp Dermatol. 2014;39:7-12.
- Broccolo F, Tassan Din C, Viganò MG, et al. HHV-8 DNA replication correlates with the clinical status in AIDS-related Kaposi's sarcoma. J Clin Virol. 2016;78:47-52.
- Pereira PF, Cuzzi T, Galhardo MC. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An Bras Dermatol. 2013;88:243-246.
- Gevorgyan O, Owen BD, Balavenkataraman A, et al. A nodular-ulcerative form of secondary syphilis in AIDS. Proc (Bayl Univ Med Cent). 2017;30:80-82.
- Balagula Y, Mattei PL, Wisco OJ, et al. The great imitator revisited: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;31:595-599.
- Yayli S, della Torre R, Hegyi I, et al. Late secondary syphilis with nodular lesions mimicking Kaposi sarcoma in a patient with human immunodeficiency virus. Int J Dermatol. 2014;53:E71-E73.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Cockerell CJ, LeBoit PE. Bacillary angiomatosis: a newly characterized, pseudoneoplastic, infectious, cutaneous vascular disorder. J Am Acad Dermatol. 1990;22:501-512.
- Forrestel AK, Naujokas A, Martin JN, et al. Bacillary angiomatosis masquerading as Kaposi's sarcoma in East Africa. J Int Assoc Provid AIDS Care. 2015;14:21-25.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma), localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol. 2007;29:408-411.
- Marks R. Squamous cell carcinoma. Lancet. 1996;347:735-738.
- Chakrabarti I, Watson JD, Dorrance H. Skin tumours of the hand. a 10-year review. J Hand Surg Br. 1993;18:484-486.
- Sobanko JF, Dagum AB, Davis IC, et al. Soft tissue tumors of the hand. 2. malignant. Dermatol Surg. 2007;33:771-785.

A 52-year-old man presented to the dermatology clinic with a 2×3-cm, fungating, dome-shaped, ulcerative, moist, well-circumscribed tumor with peripheral maceration on the volar aspect of the right hand of 3 months’ duration. The tumor was malodorous, painful, and hemorrhaged easily with minimal trauma. The patient’s medical history was notable for human immunodeficiency virus and latent syphilis, with elevated rapid plasma reagin titers and a positive Treponema palladium antibody on chemiluminescent immunoassay, that was refractory to 3 treatments with penicillin. The patient was not on antiretroviral therapy. He had a CD4+ lymphocyte count of 980 cells/µL (reference range, 359–1519 cells/µL) and a viral load of 8560 copies/mL (reference range, <200 copies/mL). No other skin or systemic concerns were noted, and the patient denied any recent travel, exposure to animals, or constitutional symptoms. A deep shave biopsy of the lesion was performed.
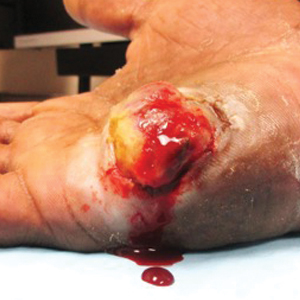
Rubbery Nodule on the Face of an Infant
The Diagnosis: Juvenile Xanthogranuloma
Juvenile xanthogranuloma (JXG) was first described in 1905 by Adamson1 as solitary or multiple plaquiform or nodular lesions that are yellow to yellowish brown. In 1954, Helwig and Hackney2 coined the term juvenile xanthogranuloma to define this histiocytic cutaneous granulomatous tumor.
Juvenile xanthogranuloma is a rare dermatologic disorder that may be present at birth and primarily affects infants and young children. The benign lesions generally occur in the first 4 years of life, with a median age of onset of 2 years.3 Lesions range in size from millimeters to several centimeters in diameter.4 The skin of the head and neck is the most commonly involved site in JXG. The most frequent noncutaneous site of JXG involvement is the eye, particularly the iris, accounting for 0.4% of cases.5,6 Extracutaneous sites such as the heart, liver, lungs, spleen, oral cavity, and brain also may be involved.4
Most children with JXG are asymptomatic. Skin lesions present as well-demarcated, rubbery, tan-orange papules or nodules. They usually are solitary, and multiple nodules can increase the risk for extracutaneous involvement.4 A case series of patients with neurofibromatosis type 1 showed 14 of 77 (18%) patients examined in the first year of life presented with JXG or other non–Langerhans cell histiocytosis.7 The adult form of cutaneous xanthogranuloma often presents with severe bronchial asthma.8
Histopathologic examination of a biopsy of the lesion typically demonstrates well-circumscribed nodules with dense infiltrates of polyhedral histiocytes with vaculoles.3-7 In 85% of cases, Touton giant cells are present.4,9 A prominent vascular network often is present, which was observed in our patient’s biopsy (Figure 1). Immunohistochemistry typically is positive for CD14, CD68, CD163, fascin, and factor XIIIa.4,10 Classically, the cells are negative for S-100 and CD1a, which differentiates these lesions from Langerhans cell histiocytosis.4-7,10 Our patient demonstrated scattered S-100–positive cells representing background dendritic cells and macrophages (Figure 2). The remainder of the clinical, morphologic, and immunophenotypic findings were consistent with non–Langerhans cell histiocytosis, specifically JXG. 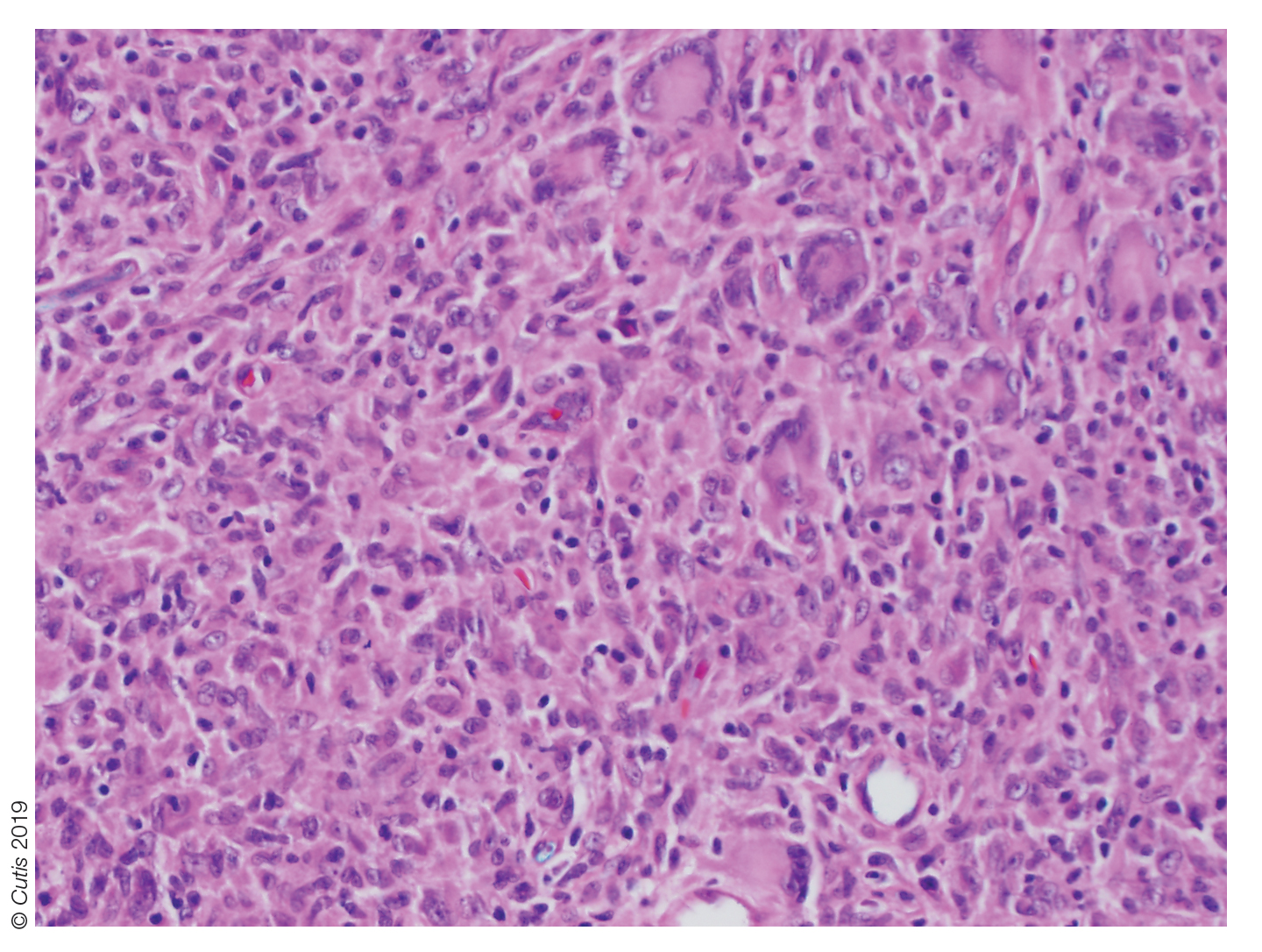
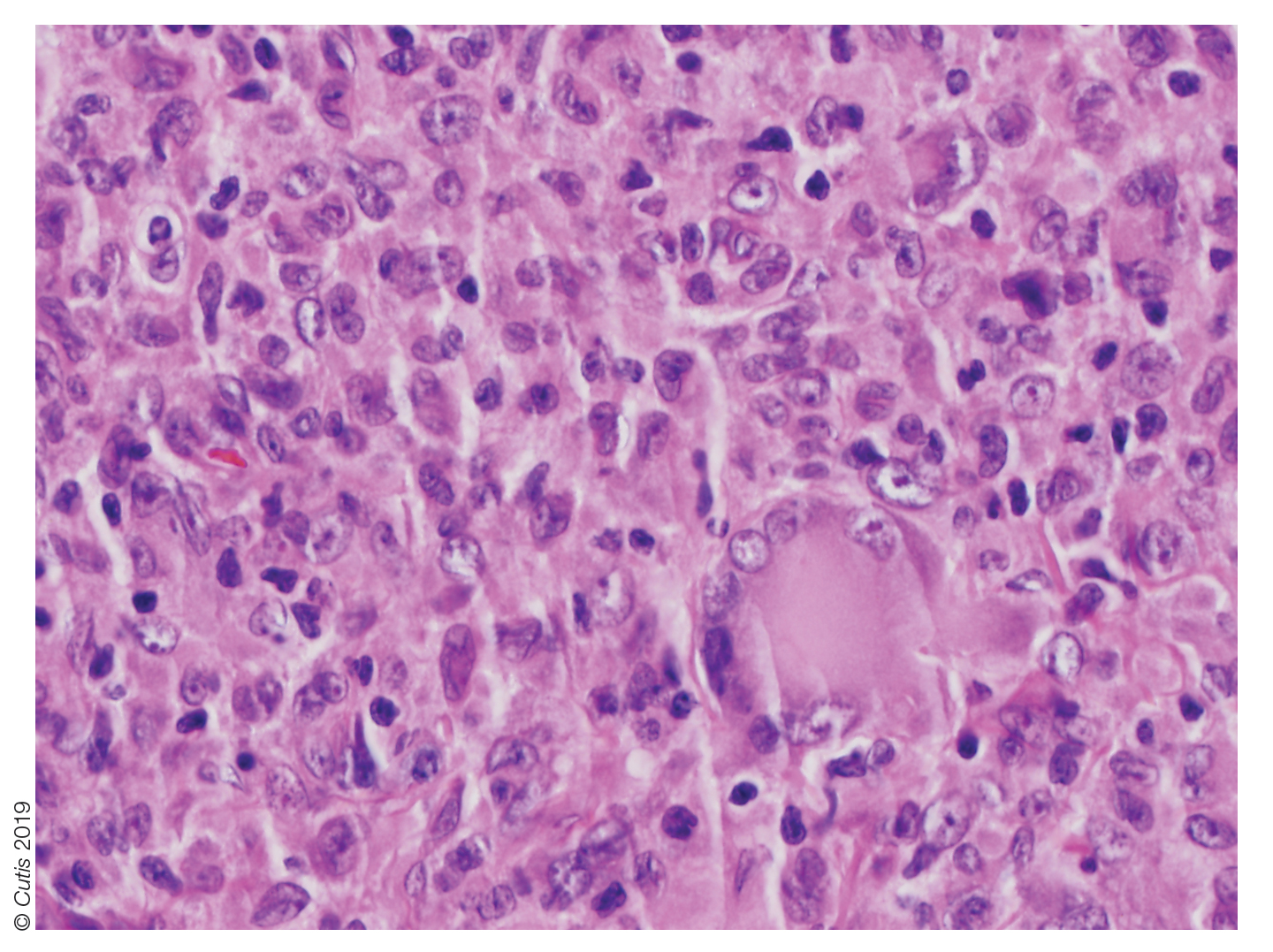
Biopsy should be performed in all cases, and basic laboratory test results such as a complete blood cell count and basic metabolic panel also are appropriate. Routine referral of all patients with cutaneous JXG for ophthalmologic evaluation is not recommended.11 Most patients with ocular involvement present with acute ocular concerns; asymptomatic eye involvement is rare. It is reasonable to consider referral to ophthalmology for patients younger than 2 years who present with multiple lesions, as they may have a higher risk for ocular involvement.11
Juvenile xanthogranuloma usually is a benign disorder with management involving observation, as the lesions typically involute spontaneously.3-7,9,10,12 Systemic or intralesional corticosteroids may be used for treatment in lesions that do not resolve. Ocular JXG refractory to steroid treatment has been managed in several cases with intravitreal bevacizumab.13 Additionally, surgical excision can be considered if malignancy is suspected, the lesion does not resolve with observation or steroid treatment, or the lesion is located near vital structures.4-7,9-13
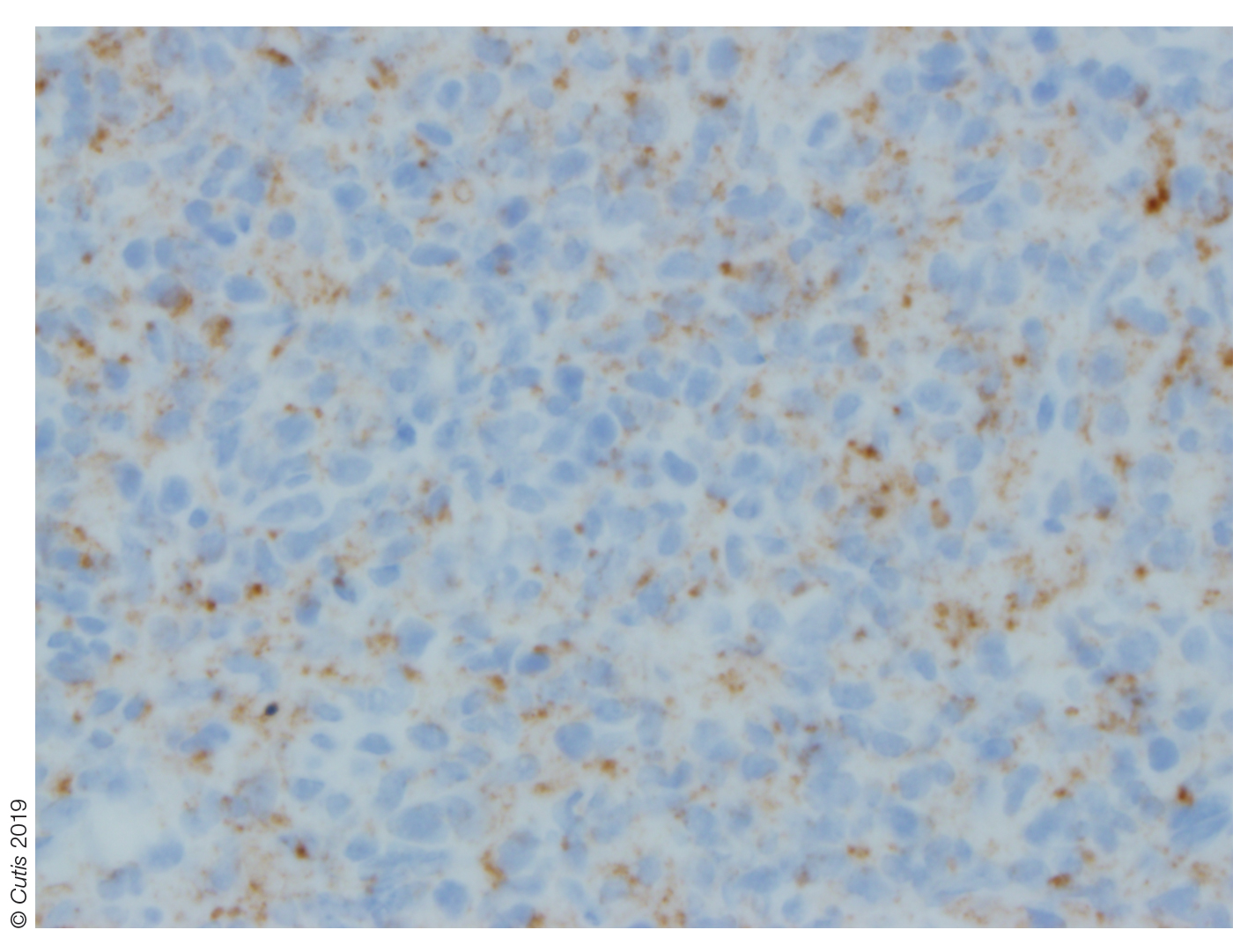
Spitz nevus presents as a single dome-shaped papule, but histology shows a symmetrical proliferation of spindle and epithelioid cells. Trachoma can present in and around the eye as a follicular hypertrophy but most commonly is seen in the conjunctiva. Dermoid cysts present as solitary subcutaneous cystic lesions; histology demonstrates a lining of keratinizing squamous epithelium with the presence of pilosebaceous structures. Dermatofibroma appears as a tan to reddish-brown papule in an area of prior minor trauma; pathology demonstrates an acanthotic epidermis with an underlying zone of normal papillary dermis and unencapsulated lesions with spindle cells overlapping in fascicles and whorls at the periphery.
1. Adamson HG. Society intelligence: the Dermatological Society of
London. Br J Dermatol. 1905;17:222-223.
2. Helwig E, Hackney VC. Juvenile xanthogranuloma (nevoxanthoendothelioma).
Am J Pathol. 1954;30:625-626.
3. Farrugia EJ, Stephen AP, Raza SA. Juvenile xanthogranuloma of
temporal bone—a case report. J Laryngol Otol. 1997;111:63-65.
4. Cypel TK, Zuker RM. Juvenile xanthogranuloma: case report and review
of the literature. Can J Plast Surg. 2008;16:175-177.
5. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445-449.
6. Zimmerman LE. Ocular lesions of juvenile xanthogranuloma.
nevoxanthoendothelioma. Am J Ophthalmol. 1965;60:1011-1035.
7. Cambiaghi S, Restano L, Caputo R. Juvenile xanthogranuloma associated
with neurofibromatosis 1: 14 patients without evidence of hematologic
malignancies. Pediatr Dermatol. 2004;21:97-101.
8. Stover DG, Alapati S, Regueira O, et al. Treatment of juvenile xanthogranuloma.
Pediatr Blood Cancer. 2008;51:130-133.
9. Dehner LP. Juvenile xanthogranulomas in the first two decades of life. a
clinicopathologic study of 174 cases with cutaneous and extracutaneous
manifestations. Am J Surg Pathol. 2003;27:579-593.
10. Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-
Langerhans cell histiocytoses. Pediatr Blood Cancer. 2005;45:256-264.
11. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445.
12. Eggli KD, Caro P, Quioque T, et al. Juvenile xanthogranuloma: non-X
histiocytosis with systemic involvement. Pediatr Radiol. 1992;22:374-376.
13. Ashkenazy N, Henry CR, Abbey AM, et al. Successful treatment of juvenile
xanthogranuloma using bevacizumab. J AAPOS. 2014;18:295-297.
The Diagnosis: Juvenile Xanthogranuloma
Juvenile xanthogranuloma (JXG) was first described in 1905 by Adamson1 as solitary or multiple plaquiform or nodular lesions that are yellow to yellowish brown. In 1954, Helwig and Hackney2 coined the term juvenile xanthogranuloma to define this histiocytic cutaneous granulomatous tumor.
Juvenile xanthogranuloma is a rare dermatologic disorder that may be present at birth and primarily affects infants and young children. The benign lesions generally occur in the first 4 years of life, with a median age of onset of 2 years.3 Lesions range in size from millimeters to several centimeters in diameter.4 The skin of the head and neck is the most commonly involved site in JXG. The most frequent noncutaneous site of JXG involvement is the eye, particularly the iris, accounting for 0.4% of cases.5,6 Extracutaneous sites such as the heart, liver, lungs, spleen, oral cavity, and brain also may be involved.4
Most children with JXG are asymptomatic. Skin lesions present as well-demarcated, rubbery, tan-orange papules or nodules. They usually are solitary, and multiple nodules can increase the risk for extracutaneous involvement.4 A case series of patients with neurofibromatosis type 1 showed 14 of 77 (18%) patients examined in the first year of life presented with JXG or other non–Langerhans cell histiocytosis.7 The adult form of cutaneous xanthogranuloma often presents with severe bronchial asthma.8
Histopathologic examination of a biopsy of the lesion typically demonstrates well-circumscribed nodules with dense infiltrates of polyhedral histiocytes with vaculoles.3-7 In 85% of cases, Touton giant cells are present.4,9 A prominent vascular network often is present, which was observed in our patient’s biopsy (Figure 1). Immunohistochemistry typically is positive for CD14, CD68, CD163, fascin, and factor XIIIa.4,10 Classically, the cells are negative for S-100 and CD1a, which differentiates these lesions from Langerhans cell histiocytosis.4-7,10 Our patient demonstrated scattered S-100–positive cells representing background dendritic cells and macrophages (Figure 2). The remainder of the clinical, morphologic, and immunophenotypic findings were consistent with non–Langerhans cell histiocytosis, specifically JXG.
Biopsy should be performed in all cases, and basic laboratory test results such as a complete blood cell count and basic metabolic panel also are appropriate. Routine referral of all patients with cutaneous JXG for ophthalmologic evaluation is not recommended.11 Most patients with ocular involvement present with acute ocular concerns; asymptomatic eye involvement is rare. It is reasonable to consider referral to ophthalmology for patients younger than 2 years who present with multiple lesions, as they may have a higher risk for ocular involvement.11
Juvenile xanthogranuloma usually is a benign disorder with management involving observation, as the lesions typically involute spontaneously.3-7,9,10,12 Systemic or intralesional corticosteroids may be used for treatment in lesions that do not resolve. Ocular JXG refractory to steroid treatment has been managed in several cases with intravitreal bevacizumab.13 Additionally, surgical excision can be considered if malignancy is suspected, the lesion does not resolve with observation or steroid treatment, or the lesion is located near vital structures.4-7,9-13
Spitz nevus presents as a single dome-shaped papule, but histology shows a symmetrical proliferation of spindle and epithelioid cells. Trachoma can present in and around the eye as a follicular hypertrophy but most commonly is seen in the conjunctiva. Dermoid cysts present as solitary subcutaneous cystic lesions; histology demonstrates a lining of keratinizing squamous epithelium with the presence of pilosebaceous structures. Dermatofibroma appears as a tan to reddish-brown papule in an area of prior minor trauma; pathology demonstrates an acanthotic epidermis with an underlying zone of normal papillary dermis and unencapsulated lesions with spindle cells overlapping in fascicles and whorls at the periphery.
The Diagnosis: Juvenile Xanthogranuloma
Juvenile xanthogranuloma (JXG) was first described in 1905 by Adamson1 as solitary or multiple plaquiform or nodular lesions that are yellow to yellowish brown. In 1954, Helwig and Hackney2 coined the term juvenile xanthogranuloma to define this histiocytic cutaneous granulomatous tumor.
Juvenile xanthogranuloma is a rare dermatologic disorder that may be present at birth and primarily affects infants and young children. The benign lesions generally occur in the first 4 years of life, with a median age of onset of 2 years.3 Lesions range in size from millimeters to several centimeters in diameter.4 The skin of the head and neck is the most commonly involved site in JXG. The most frequent noncutaneous site of JXG involvement is the eye, particularly the iris, accounting for 0.4% of cases.5,6 Extracutaneous sites such as the heart, liver, lungs, spleen, oral cavity, and brain also may be involved.4
Most children with JXG are asymptomatic. Skin lesions present as well-demarcated, rubbery, tan-orange papules or nodules. They usually are solitary, and multiple nodules can increase the risk for extracutaneous involvement.4 A case series of patients with neurofibromatosis type 1 showed 14 of 77 (18%) patients examined in the first year of life presented with JXG or other non–Langerhans cell histiocytosis.7 The adult form of cutaneous xanthogranuloma often presents with severe bronchial asthma.8
Histopathologic examination of a biopsy of the lesion typically demonstrates well-circumscribed nodules with dense infiltrates of polyhedral histiocytes with vaculoles.3-7 In 85% of cases, Touton giant cells are present.4,9 A prominent vascular network often is present, which was observed in our patient’s biopsy (Figure 1). Immunohistochemistry typically is positive for CD14, CD68, CD163, fascin, and factor XIIIa.4,10 Classically, the cells are negative for S-100 and CD1a, which differentiates these lesions from Langerhans cell histiocytosis.4-7,10 Our patient demonstrated scattered S-100–positive cells representing background dendritic cells and macrophages (Figure 2). The remainder of the clinical, morphologic, and immunophenotypic findings were consistent with non–Langerhans cell histiocytosis, specifically JXG.
Biopsy should be performed in all cases, and basic laboratory test results such as a complete blood cell count and basic metabolic panel also are appropriate. Routine referral of all patients with cutaneous JXG for ophthalmologic evaluation is not recommended.11 Most patients with ocular involvement present with acute ocular concerns; asymptomatic eye involvement is rare. It is reasonable to consider referral to ophthalmology for patients younger than 2 years who present with multiple lesions, as they may have a higher risk for ocular involvement.11
Juvenile xanthogranuloma usually is a benign disorder with management involving observation, as the lesions typically involute spontaneously.3-7,9,10,12 Systemic or intralesional corticosteroids may be used for treatment in lesions that do not resolve. Ocular JXG refractory to steroid treatment has been managed in several cases with intravitreal bevacizumab.13 Additionally, surgical excision can be considered if malignancy is suspected, the lesion does not resolve with observation or steroid treatment, or the lesion is located near vital structures.4-7,9-13
Spitz nevus presents as a single dome-shaped papule, but histology shows a symmetrical proliferation of spindle and epithelioid cells. Trachoma can present in and around the eye as a follicular hypertrophy but most commonly is seen in the conjunctiva. Dermoid cysts present as solitary subcutaneous cystic lesions; histology demonstrates a lining of keratinizing squamous epithelium with the presence of pilosebaceous structures. Dermatofibroma appears as a tan to reddish-brown papule in an area of prior minor trauma; pathology demonstrates an acanthotic epidermis with an underlying zone of normal papillary dermis and unencapsulated lesions with spindle cells overlapping in fascicles and whorls at the periphery.
1. Adamson HG. Society intelligence: the Dermatological Society of
London. Br J Dermatol. 1905;17:222-223.
2. Helwig E, Hackney VC. Juvenile xanthogranuloma (nevoxanthoendothelioma).
Am J Pathol. 1954;30:625-626.
3. Farrugia EJ, Stephen AP, Raza SA. Juvenile xanthogranuloma of
temporal bone—a case report. J Laryngol Otol. 1997;111:63-65.
4. Cypel TK, Zuker RM. Juvenile xanthogranuloma: case report and review
of the literature. Can J Plast Surg. 2008;16:175-177.
5. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445-449.
6. Zimmerman LE. Ocular lesions of juvenile xanthogranuloma.
nevoxanthoendothelioma. Am J Ophthalmol. 1965;60:1011-1035.
7. Cambiaghi S, Restano L, Caputo R. Juvenile xanthogranuloma associated
with neurofibromatosis 1: 14 patients without evidence of hematologic
malignancies. Pediatr Dermatol. 2004;21:97-101.
8. Stover DG, Alapati S, Regueira O, et al. Treatment of juvenile xanthogranuloma.
Pediatr Blood Cancer. 2008;51:130-133.
9. Dehner LP. Juvenile xanthogranulomas in the first two decades of life. a
clinicopathologic study of 174 cases with cutaneous and extracutaneous
manifestations. Am J Surg Pathol. 2003;27:579-593.
10. Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-
Langerhans cell histiocytoses. Pediatr Blood Cancer. 2005;45:256-264.
11. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445.
12. Eggli KD, Caro P, Quioque T, et al. Juvenile xanthogranuloma: non-X
histiocytosis with systemic involvement. Pediatr Radiol. 1992;22:374-376.
13. Ashkenazy N, Henry CR, Abbey AM, et al. Successful treatment of juvenile
xanthogranuloma using bevacizumab. J AAPOS. 2014;18:295-297.
1. Adamson HG. Society intelligence: the Dermatological Society of
London. Br J Dermatol. 1905;17:222-223.
2. Helwig E, Hackney VC. Juvenile xanthogranuloma (nevoxanthoendothelioma).
Am J Pathol. 1954;30:625-626.
3. Farrugia EJ, Stephen AP, Raza SA. Juvenile xanthogranuloma of
temporal bone—a case report. J Laryngol Otol. 1997;111:63-65.
4. Cypel TK, Zuker RM. Juvenile xanthogranuloma: case report and review
of the literature. Can J Plast Surg. 2008;16:175-177.
5. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445-449.
6. Zimmerman LE. Ocular lesions of juvenile xanthogranuloma.
nevoxanthoendothelioma. Am J Ophthalmol. 1965;60:1011-1035.
7. Cambiaghi S, Restano L, Caputo R. Juvenile xanthogranuloma associated
with neurofibromatosis 1: 14 patients without evidence of hematologic
malignancies. Pediatr Dermatol. 2004;21:97-101.
8. Stover DG, Alapati S, Regueira O, et al. Treatment of juvenile xanthogranuloma.
Pediatr Blood Cancer. 2008;51:130-133.
9. Dehner LP. Juvenile xanthogranulomas in the first two decades of life. a
clinicopathologic study of 174 cases with cutaneous and extracutaneous
manifestations. Am J Surg Pathol. 2003;27:579-593.
10. Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-
Langerhans cell histiocytoses. Pediatr Blood Cancer. 2005;45:256-264.
11. Chang MW, Frieden IJ, Good W. The risk of intraocular juvenile
xanthogranuloma: survey of current practices and assessment of risk.
J Am Acad Dermatol. 1996;34:445.
12. Eggli KD, Caro P, Quioque T, et al. Juvenile xanthogranuloma: non-X
histiocytosis with systemic involvement. Pediatr Radiol. 1992;22:374-376.
13. Ashkenazy N, Henry CR, Abbey AM, et al. Successful treatment of juvenile
xanthogranuloma using bevacizumab. J AAPOS. 2014;18:295-297.
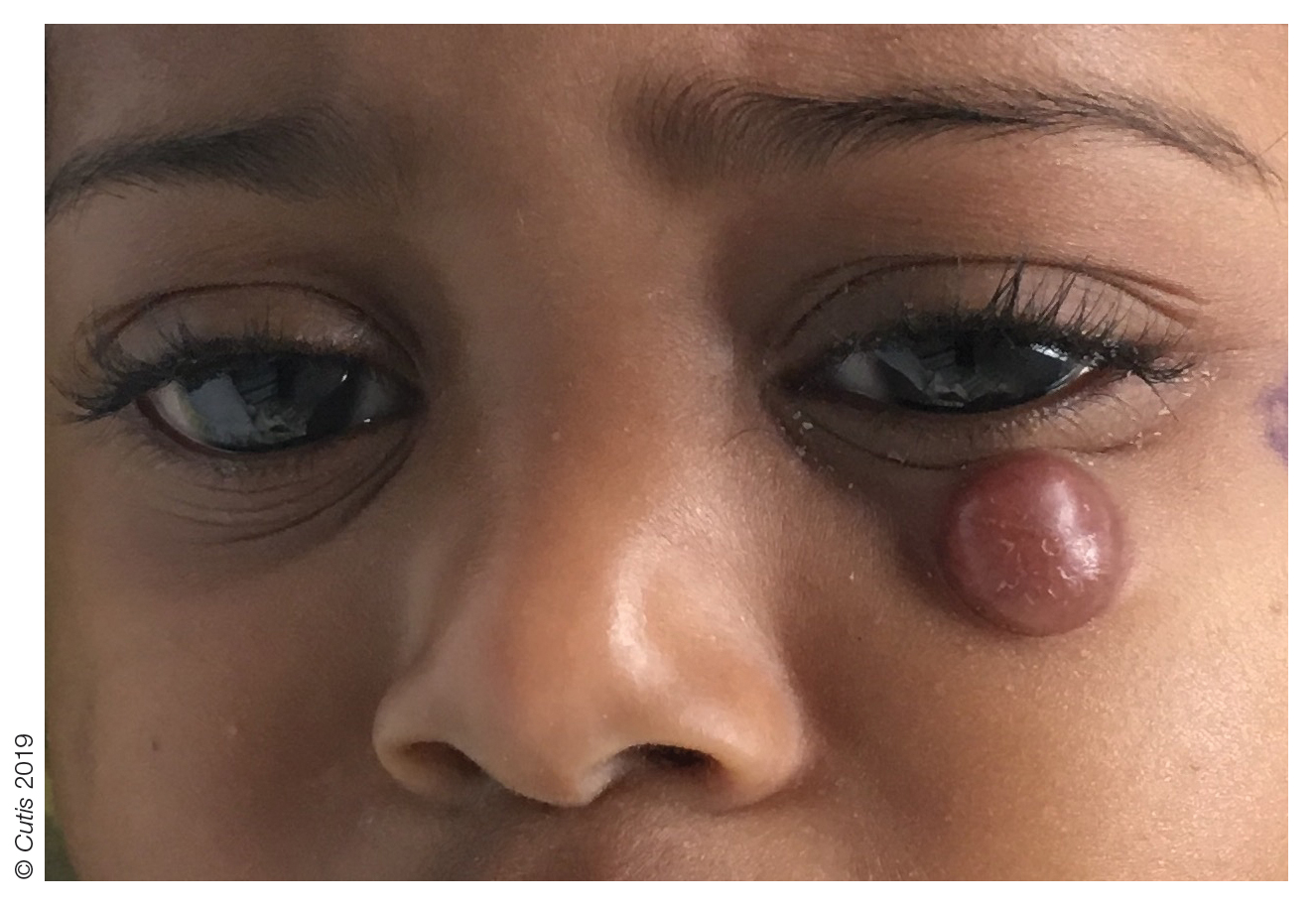
A 10-month-old girl presented with a facial nodule of 7 months’ duration that started as a small lesion. On physical examination, a single 10×10-mm, nontender, well-circumscribed, firm, freely mobile nodule was observed in the left infraorbital area. The patient was born full term at 37 weeks’ gestation via spontaneous vaginal delivery and had no other notable findings on physical examination. Excision was performed by an oculoplastic surgeon. Pathology revealed a relatively well-circumscribed, diffuse, dermal infiltrate of cells arranged in short fascicles and a storiform pattern. The cells had abundant clear to amphophilic cytoplasm, ovoid to reniform nuclei with vesicular chromatin and focal grooves, and diffuse CD68+ immunoreactivity, as well as scattered S-100–positive cells. The patient did well with the excision and no new lesions have developed.
Rapidly Growing Retroauricular Tumor
The Diagnosis: Milia En Plaque
Biopsy results revealed a normal epidermis; the dermis showed multiple small cystic structures lined by a stratified squamous epithelium containing eosinophilic keratin surrounded by a mononuclear cell infiltrate and some melanophages (Figure).
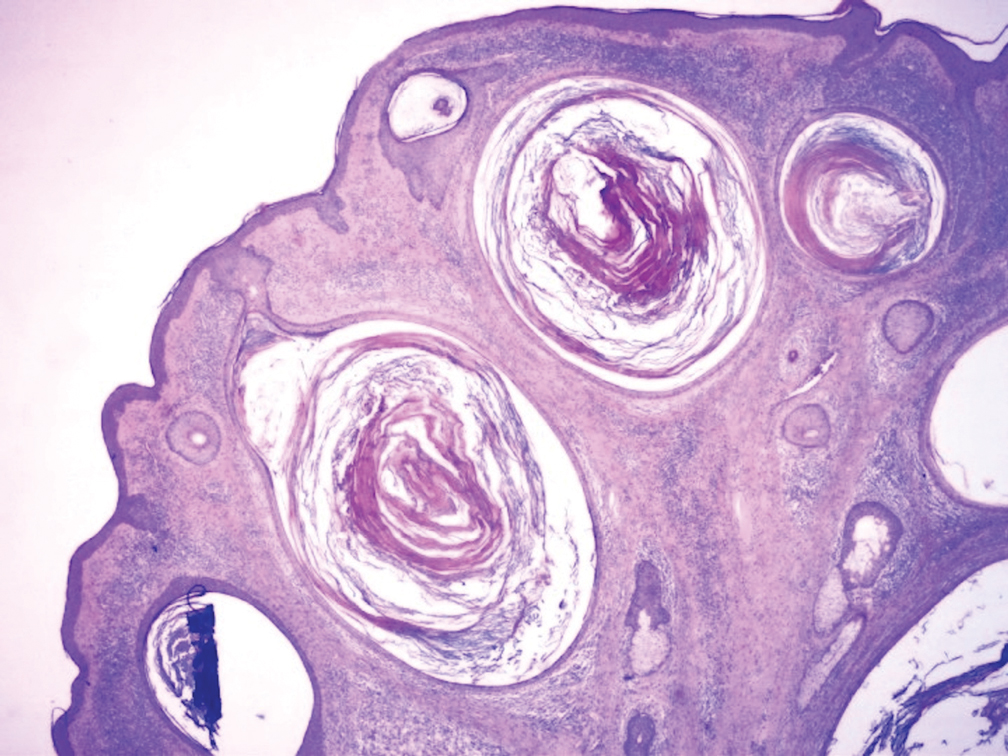
Milia en plaque was first described in 1903 by Balzer and Fouquet.1 In 1978, Hubler et al2 presented 2 cases with an asymptomatic, erythematous, and edematous plaque and white milialike lesions. On histopathology, they showed multiple cystic structures characterized by central laminated keratin and an intense polymorphic inflammatory reaction surrounding the cyst and epidermal appendages. Both patients were treated with topical tretinoin with complete response at 3 months. The authors suggested the term milia en plaque to describe this clinical entity.2
Milia en plaque is described as an infrequent condition that more often presents on the head, neck, and trunk, as well as the periocular, periauricular, and perinasal areas. It has been reported to occur at any age3 but appears more frequently in middle-aged adults and females. A congenital case also has been reported.4 It has been associated with pseudoxanthoma elasticum, lichen planus, trauma, kidney transplant, and cyclosporine use, but it also can present in healthy individuals,3 as in our patient. No clear cause has been identified.
Pathology is characteristic, with multiple cysts filled with keratin and surrounded by 2 or 3 layers of epithelial cells, associated with a mononuclear, nonlichenoid, mononuclear infiltrate.5 Structures similar to follicular infundibular tumors have been described, suggesting a common origin of follicular lesions as milia en plaque.6
Treatment includes surgical excision, cryosurgery, dermabrasion, electrodesiccation, trichloroacetic acid, photodynamic therapy, CO2 and erbium lasers, topical retinoids, minocycline, and etretinate.7 We performed a complete surgical excision in our patient.
In acneform reactions, erythematous papules and pustules can be found on the cheeks and forehead. Nevus comedonicus appears during childhood and presents with multiple open comedones. Postinflammatory milia is present in chronic inflammatory pathologies such as porphyria cutanea tarda. Histopathologic findings in adnexal tumors show a benign proliferation of any cellular type of a cutaneous annex.
Milia en plaque is an unusual but benign condition that is distinguished clinically by its characteristic presentation.
- Balzer F, Fouquet C. Milium confluent retroauricularies bilateral. Bull Soc Fr Dermatol Syphiligr. 1903;14:361.
- Hubler WR, Rudolph AH, Kelleher RM. Milia en plaque. Cutis. 1978;22:67-70.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Wang AR, Bercovitch L. Congenital milia en plaque. Pediatr Dermatol. 2016;33:258-259.
- Muñoz-Martínez R, Santamarina-Albertos A, Sanz-Muñoz C, et al. Milia en plaque. Actas Dermosifiliogr. 2013;104:638-640.
- Terui H, Hashimoto A, Yamasaki K, et al. Milia en plaque as a distinct follicular hamartoma with cystic trichoepitheliomatous features. Am J Dermatopathol. 2016;38:212-217.
- Tenna S, Filoni A, Pagliarello C, et al. Eyelid milia en plaque: a treatment challenge with a new CO2 fractional laser. Dermatol Ther. 2014;27:65-67.
The Diagnosis: Milia En Plaque
Biopsy results revealed a normal epidermis; the dermis showed multiple small cystic structures lined by a stratified squamous epithelium containing eosinophilic keratin surrounded by a mononuclear cell infiltrate and some melanophages (Figure).
Milia en plaque was first described in 1903 by Balzer and Fouquet.1 In 1978, Hubler et al2 presented 2 cases with an asymptomatic, erythematous, and edematous plaque and white milialike lesions. On histopathology, they showed multiple cystic structures characterized by central laminated keratin and an intense polymorphic inflammatory reaction surrounding the cyst and epidermal appendages. Both patients were treated with topical tretinoin with complete response at 3 months. The authors suggested the term milia en plaque to describe this clinical entity.2
Milia en plaque is described as an infrequent condition that more often presents on the head, neck, and trunk, as well as the periocular, periauricular, and perinasal areas. It has been reported to occur at any age3 but appears more frequently in middle-aged adults and females. A congenital case also has been reported.4 It has been associated with pseudoxanthoma elasticum, lichen planus, trauma, kidney transplant, and cyclosporine use, but it also can present in healthy individuals,3 as in our patient. No clear cause has been identified.
Pathology is characteristic, with multiple cysts filled with keratin and surrounded by 2 or 3 layers of epithelial cells, associated with a mononuclear, nonlichenoid, mononuclear infiltrate.5 Structures similar to follicular infundibular tumors have been described, suggesting a common origin of follicular lesions as milia en plaque.6
Treatment includes surgical excision, cryosurgery, dermabrasion, electrodesiccation, trichloroacetic acid, photodynamic therapy, CO2 and erbium lasers, topical retinoids, minocycline, and etretinate.7 We performed a complete surgical excision in our patient.
In acneform reactions, erythematous papules and pustules can be found on the cheeks and forehead. Nevus comedonicus appears during childhood and presents with multiple open comedones. Postinflammatory milia is present in chronic inflammatory pathologies such as porphyria cutanea tarda. Histopathologic findings in adnexal tumors show a benign proliferation of any cellular type of a cutaneous annex.
Milia en plaque is an unusual but benign condition that is distinguished clinically by its characteristic presentation.
The Diagnosis: Milia En Plaque
Biopsy results revealed a normal epidermis; the dermis showed multiple small cystic structures lined by a stratified squamous epithelium containing eosinophilic keratin surrounded by a mononuclear cell infiltrate and some melanophages (Figure).
Milia en plaque was first described in 1903 by Balzer and Fouquet.1 In 1978, Hubler et al2 presented 2 cases with an asymptomatic, erythematous, and edematous plaque and white milialike lesions. On histopathology, they showed multiple cystic structures characterized by central laminated keratin and an intense polymorphic inflammatory reaction surrounding the cyst and epidermal appendages. Both patients were treated with topical tretinoin with complete response at 3 months. The authors suggested the term milia en plaque to describe this clinical entity.2
Milia en plaque is described as an infrequent condition that more often presents on the head, neck, and trunk, as well as the periocular, periauricular, and perinasal areas. It has been reported to occur at any age3 but appears more frequently in middle-aged adults and females. A congenital case also has been reported.4 It has been associated with pseudoxanthoma elasticum, lichen planus, trauma, kidney transplant, and cyclosporine use, but it also can present in healthy individuals,3 as in our patient. No clear cause has been identified.
Pathology is characteristic, with multiple cysts filled with keratin and surrounded by 2 or 3 layers of epithelial cells, associated with a mononuclear, nonlichenoid, mononuclear infiltrate.5 Structures similar to follicular infundibular tumors have been described, suggesting a common origin of follicular lesions as milia en plaque.6
Treatment includes surgical excision, cryosurgery, dermabrasion, electrodesiccation, trichloroacetic acid, photodynamic therapy, CO2 and erbium lasers, topical retinoids, minocycline, and etretinate.7 We performed a complete surgical excision in our patient.
In acneform reactions, erythematous papules and pustules can be found on the cheeks and forehead. Nevus comedonicus appears during childhood and presents with multiple open comedones. Postinflammatory milia is present in chronic inflammatory pathologies such as porphyria cutanea tarda. Histopathologic findings in adnexal tumors show a benign proliferation of any cellular type of a cutaneous annex.
Milia en plaque is an unusual but benign condition that is distinguished clinically by its characteristic presentation.
- Balzer F, Fouquet C. Milium confluent retroauricularies bilateral. Bull Soc Fr Dermatol Syphiligr. 1903;14:361.
- Hubler WR, Rudolph AH, Kelleher RM. Milia en plaque. Cutis. 1978;22:67-70.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Wang AR, Bercovitch L. Congenital milia en plaque. Pediatr Dermatol. 2016;33:258-259.
- Muñoz-Martínez R, Santamarina-Albertos A, Sanz-Muñoz C, et al. Milia en plaque. Actas Dermosifiliogr. 2013;104:638-640.
- Terui H, Hashimoto A, Yamasaki K, et al. Milia en plaque as a distinct follicular hamartoma with cystic trichoepitheliomatous features. Am J Dermatopathol. 2016;38:212-217.
- Tenna S, Filoni A, Pagliarello C, et al. Eyelid milia en plaque: a treatment challenge with a new CO2 fractional laser. Dermatol Ther. 2014;27:65-67.
- Balzer F, Fouquet C. Milium confluent retroauricularies bilateral. Bull Soc Fr Dermatol Syphiligr. 1903;14:361.
- Hubler WR, Rudolph AH, Kelleher RM. Milia en plaque. Cutis. 1978;22:67-70.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Wang AR, Bercovitch L. Congenital milia en plaque. Pediatr Dermatol. 2016;33:258-259.
- Muñoz-Martínez R, Santamarina-Albertos A, Sanz-Muñoz C, et al. Milia en plaque. Actas Dermosifiliogr. 2013;104:638-640.
- Terui H, Hashimoto A, Yamasaki K, et al. Milia en plaque as a distinct follicular hamartoma with cystic trichoepitheliomatous features. Am J Dermatopathol. 2016;38:212-217.
- Tenna S, Filoni A, Pagliarello C, et al. Eyelid milia en plaque: a treatment challenge with a new CO2 fractional laser. Dermatol Ther. 2014;27:65-67.
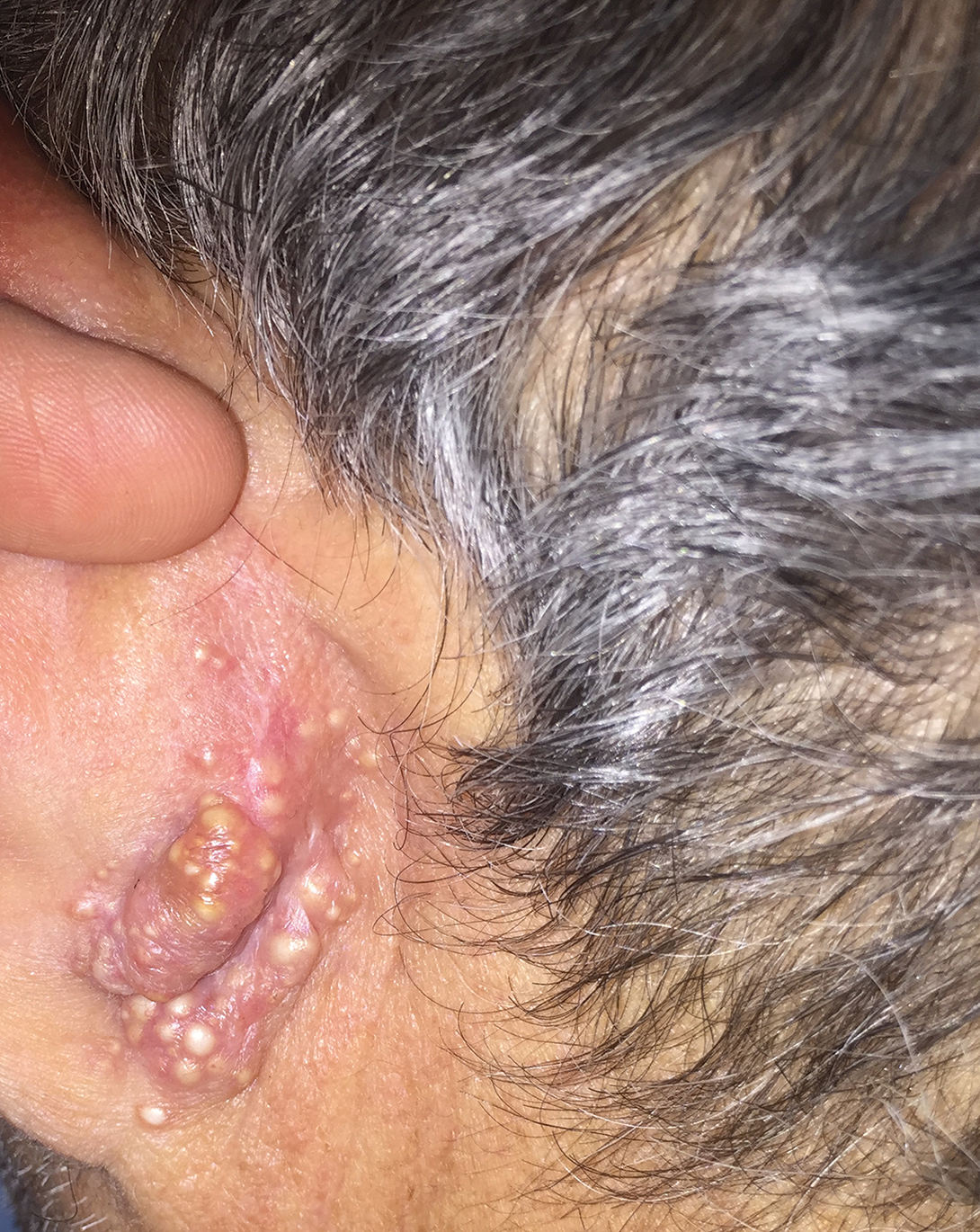
A 72-year-old man with a history of hypertension presented with a rapidly growing left retroauricular tumor of 3 months' duration. When manipulated, whitish material with a foul-smelling odor was expressed from the lesion. Physical examination showed an erythematous 3.2 ×1-cm tumor on the left posterior ear with multiple 1- to 2-mm white-yellow papules on its surface. A biopsy of the lesion was performed.
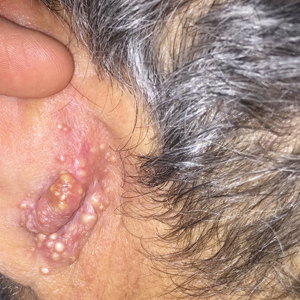
Painless Purple Streaks on the Arms and Chest
The Diagnosis: Factitial Purpura
Factitial dermatologic disorders are characterized by skin findings triggered by deliberate manipulation of the skin with objects to create lesions and feign signs of a dermatologic condition to seek emotional and psychological benefit.1 The etiology of the lesions is unclear, and the patient's history of the injury is hollow.2 Most often, there is sudden onset of the lesions without any warning or symptoms. When giving the history, the patient may appear unemotional, does not report pain, and denies self-infliction.1
In factitial purpura, the purple patches are clearly demarcated from uninvolved skin and have an unusual angular or geometric shape. The pattern typically takes the shape of the object used to create the purpura and lacks the features of recognizable dermatoses.2 In our patient and those with similar linear purpuric streaks, we use the term penny purpura to indicate that the lesions resulted from rubbing with a penny or other blunt object, similar to coining. The lesions occur in areas that are easily accessible and visible such as the arms, chest, or chin. It is suggested that the child unconsciously wants the lesions to be seen. Histologic findings in factitial purpura include disruption of collagen fiber bundles and extravasated red blood cells in the dermis.3 Unfortunately, evolving lesions may give nonspecific histologic findings; when the clinical lesions are typical, skin biopsy usually is unnecessary and may be misleading. Laboratory test results such as complete blood cell count, prothrombin time, and partial thromboplastin time usually are within reference range, as in our patient.
When evaluating these patients, confrontation is not recommended. More than two-thirds of affected patients have a history of trauma such as sexual/physical abuse or neglect, and the lesions typically arise during times of stress.1,3 Thus, treatment includes nonaccusatory measures and referral for psychologic evaluation. The purpura will rapidly heal when covered with an occlusive dressing.2
The differential diagnosis for penny purpura includes lesions that evolve from cupping and coining. Cupping is a type of complementary and alternative medicine that acts by correcting imbalances in the internal biofield and restoring the flow of qi, which determines the state of one's health and life span.4 Cupping is performed by placing a glass cup over a painful body part. A partial vacuum is created by flaming, mechanical withdrawal, or thermal cooling of the entrapped air under the cup. When the flame exhausts the supply of oxygen, the skin is sucked into the mouth of the glass, and the skin is bruised painlessly.4
The differential also includes child maltreatment syndrome and other disorders that would potentiate bruising. Intravascular etiologies include idiopathic thrombocytopenic purpura, leukemia, coagulation disorders, and other causes of thrombocytopenia or platelet dysfunction.3 Extravascular etiologies include hereditary collagen vascular disease (eg, Ehlers-Danlos syndrome), malnutrition, and other disorders associated with a decrease in collagen and other tissues that support cutaneous vessels. Vascular etiologies include infectious (eg, Rocky Mountain spotted fever, meningococcemia) and noninfectious vasculitis (eg, Henoch-Schönlein purpura), leaky capillary syndrome, drug reactions, and other disorders associated with a loss of vascular integrity.3
It is important to be able to differentiate self-inflicted lesions in a person who repeatedly acts as if he/she has a physical disorder from those that are created during the practices of cupping or any other cultural healing practice. Vascular disorders, malnutrition, and child abuse also should be excluded.3
For our patient with factitial purpura, we gently encouraged the family to work with the child's pediatrician and a pediatric psychologist to deal with stress related to the recurrent rash and asked them to think of the rash as a result of an external cause; however, we were careful not to blame anyone for the rash.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372; quiz 373.
- Al Hawsawi K, Pope E. Pediatric psychocutaneous disorders: a review of primary psychiatric disorders with dermatologic manifestations. Am J Clin Dermatol. 2011;12:247-257.
- Ring HC, Miller IM, Benfeldt E, et al. Artefactual skin lesions in children and adolescents: review of the literature and two cases of factitious purpura. Int J Dermatol. 2015;54:E27-E32.
- Mehta P, Dhapte V. Cupping therapy: a prudent remedy for a plethora of medical ailments. J Tradit Complement Med. 2015;5:127-134.
The Diagnosis: Factitial Purpura
Factitial dermatologic disorders are characterized by skin findings triggered by deliberate manipulation of the skin with objects to create lesions and feign signs of a dermatologic condition to seek emotional and psychological benefit.1 The etiology of the lesions is unclear, and the patient's history of the injury is hollow.2 Most often, there is sudden onset of the lesions without any warning or symptoms. When giving the history, the patient may appear unemotional, does not report pain, and denies self-infliction.1
In factitial purpura, the purple patches are clearly demarcated from uninvolved skin and have an unusual angular or geometric shape. The pattern typically takes the shape of the object used to create the purpura and lacks the features of recognizable dermatoses.2 In our patient and those with similar linear purpuric streaks, we use the term penny purpura to indicate that the lesions resulted from rubbing with a penny or other blunt object, similar to coining. The lesions occur in areas that are easily accessible and visible such as the arms, chest, or chin. It is suggested that the child unconsciously wants the lesions to be seen. Histologic findings in factitial purpura include disruption of collagen fiber bundles and extravasated red blood cells in the dermis.3 Unfortunately, evolving lesions may give nonspecific histologic findings; when the clinical lesions are typical, skin biopsy usually is unnecessary and may be misleading. Laboratory test results such as complete blood cell count, prothrombin time, and partial thromboplastin time usually are within reference range, as in our patient.
When evaluating these patients, confrontation is not recommended. More than two-thirds of affected patients have a history of trauma such as sexual/physical abuse or neglect, and the lesions typically arise during times of stress.1,3 Thus, treatment includes nonaccusatory measures and referral for psychologic evaluation. The purpura will rapidly heal when covered with an occlusive dressing.2
The differential diagnosis for penny purpura includes lesions that evolve from cupping and coining. Cupping is a type of complementary and alternative medicine that acts by correcting imbalances in the internal biofield and restoring the flow of qi, which determines the state of one's health and life span.4 Cupping is performed by placing a glass cup over a painful body part. A partial vacuum is created by flaming, mechanical withdrawal, or thermal cooling of the entrapped air under the cup. When the flame exhausts the supply of oxygen, the skin is sucked into the mouth of the glass, and the skin is bruised painlessly.4
The differential also includes child maltreatment syndrome and other disorders that would potentiate bruising. Intravascular etiologies include idiopathic thrombocytopenic purpura, leukemia, coagulation disorders, and other causes of thrombocytopenia or platelet dysfunction.3 Extravascular etiologies include hereditary collagen vascular disease (eg, Ehlers-Danlos syndrome), malnutrition, and other disorders associated with a decrease in collagen and other tissues that support cutaneous vessels. Vascular etiologies include infectious (eg, Rocky Mountain spotted fever, meningococcemia) and noninfectious vasculitis (eg, Henoch-Schönlein purpura), leaky capillary syndrome, drug reactions, and other disorders associated with a loss of vascular integrity.3
It is important to be able to differentiate self-inflicted lesions in a person who repeatedly acts as if he/she has a physical disorder from those that are created during the practices of cupping or any other cultural healing practice. Vascular disorders, malnutrition, and child abuse also should be excluded.3
For our patient with factitial purpura, we gently encouraged the family to work with the child's pediatrician and a pediatric psychologist to deal with stress related to the recurrent rash and asked them to think of the rash as a result of an external cause; however, we were careful not to blame anyone for the rash.
The Diagnosis: Factitial Purpura
Factitial dermatologic disorders are characterized by skin findings triggered by deliberate manipulation of the skin with objects to create lesions and feign signs of a dermatologic condition to seek emotional and psychological benefit.1 The etiology of the lesions is unclear, and the patient's history of the injury is hollow.2 Most often, there is sudden onset of the lesions without any warning or symptoms. When giving the history, the patient may appear unemotional, does not report pain, and denies self-infliction.1
In factitial purpura, the purple patches are clearly demarcated from uninvolved skin and have an unusual angular or geometric shape. The pattern typically takes the shape of the object used to create the purpura and lacks the features of recognizable dermatoses.2 In our patient and those with similar linear purpuric streaks, we use the term penny purpura to indicate that the lesions resulted from rubbing with a penny or other blunt object, similar to coining. The lesions occur in areas that are easily accessible and visible such as the arms, chest, or chin. It is suggested that the child unconsciously wants the lesions to be seen. Histologic findings in factitial purpura include disruption of collagen fiber bundles and extravasated red blood cells in the dermis.3 Unfortunately, evolving lesions may give nonspecific histologic findings; when the clinical lesions are typical, skin biopsy usually is unnecessary and may be misleading. Laboratory test results such as complete blood cell count, prothrombin time, and partial thromboplastin time usually are within reference range, as in our patient.
When evaluating these patients, confrontation is not recommended. More than two-thirds of affected patients have a history of trauma such as sexual/physical abuse or neglect, and the lesions typically arise during times of stress.1,3 Thus, treatment includes nonaccusatory measures and referral for psychologic evaluation. The purpura will rapidly heal when covered with an occlusive dressing.2
The differential diagnosis for penny purpura includes lesions that evolve from cupping and coining. Cupping is a type of complementary and alternative medicine that acts by correcting imbalances in the internal biofield and restoring the flow of qi, which determines the state of one's health and life span.4 Cupping is performed by placing a glass cup over a painful body part. A partial vacuum is created by flaming, mechanical withdrawal, or thermal cooling of the entrapped air under the cup. When the flame exhausts the supply of oxygen, the skin is sucked into the mouth of the glass, and the skin is bruised painlessly.4
The differential also includes child maltreatment syndrome and other disorders that would potentiate bruising. Intravascular etiologies include idiopathic thrombocytopenic purpura, leukemia, coagulation disorders, and other causes of thrombocytopenia or platelet dysfunction.3 Extravascular etiologies include hereditary collagen vascular disease (eg, Ehlers-Danlos syndrome), malnutrition, and other disorders associated with a decrease in collagen and other tissues that support cutaneous vessels. Vascular etiologies include infectious (eg, Rocky Mountain spotted fever, meningococcemia) and noninfectious vasculitis (eg, Henoch-Schönlein purpura), leaky capillary syndrome, drug reactions, and other disorders associated with a loss of vascular integrity.3
It is important to be able to differentiate self-inflicted lesions in a person who repeatedly acts as if he/she has a physical disorder from those that are created during the practices of cupping or any other cultural healing practice. Vascular disorders, malnutrition, and child abuse also should be excluded.3
For our patient with factitial purpura, we gently encouraged the family to work with the child's pediatrician and a pediatric psychologist to deal with stress related to the recurrent rash and asked them to think of the rash as a result of an external cause; however, we were careful not to blame anyone for the rash.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372; quiz 373.
- Al Hawsawi K, Pope E. Pediatric psychocutaneous disorders: a review of primary psychiatric disorders with dermatologic manifestations. Am J Clin Dermatol. 2011;12:247-257.
- Ring HC, Miller IM, Benfeldt E, et al. Artefactual skin lesions in children and adolescents: review of the literature and two cases of factitious purpura. Int J Dermatol. 2015;54:E27-E32.
- Mehta P, Dhapte V. Cupping therapy: a prudent remedy for a plethora of medical ailments. J Tradit Complement Med. 2015;5:127-134.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372; quiz 373.
- Al Hawsawi K, Pope E. Pediatric psychocutaneous disorders: a review of primary psychiatric disorders with dermatologic manifestations. Am J Clin Dermatol. 2011;12:247-257.
- Ring HC, Miller IM, Benfeldt E, et al. Artefactual skin lesions in children and adolescents: review of the literature and two cases of factitious purpura. Int J Dermatol. 2015;54:E27-E32.
- Mehta P, Dhapte V. Cupping therapy: a prudent remedy for a plethora of medical ailments. J Tradit Complement Med. 2015;5:127-134.
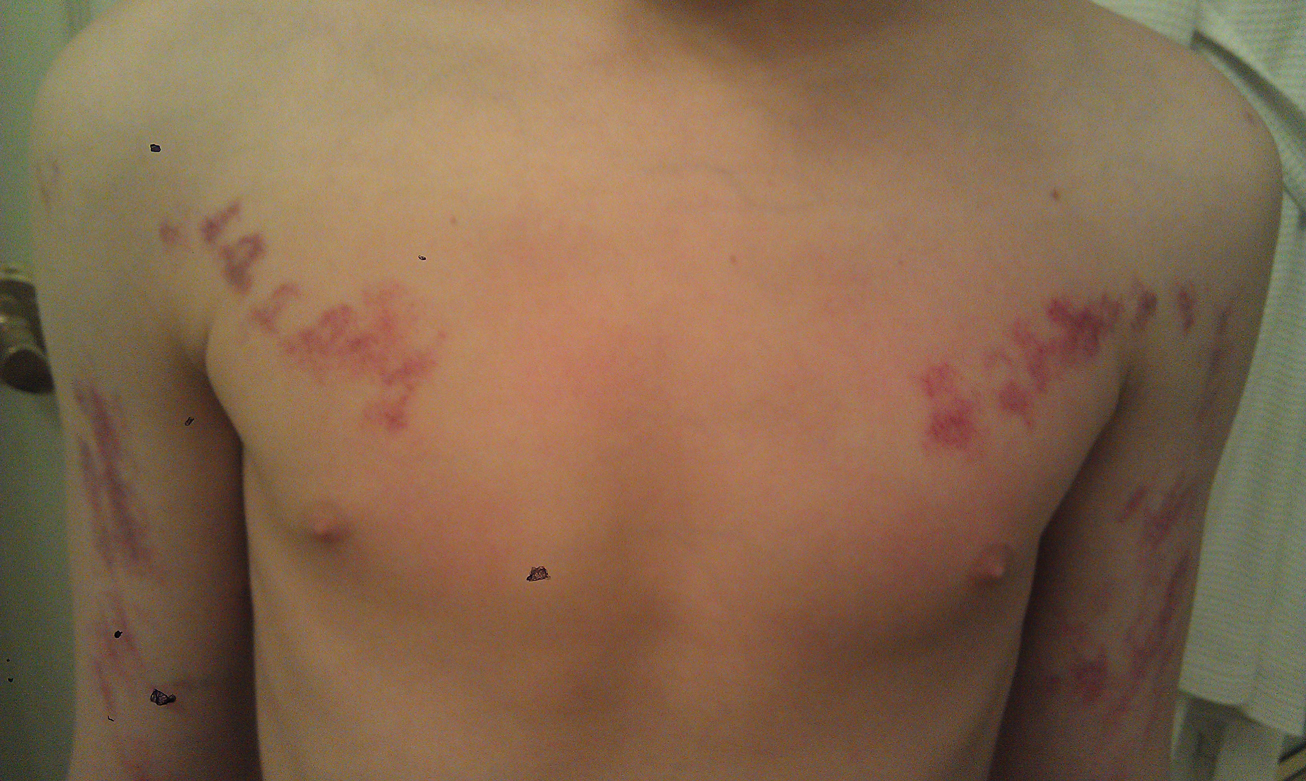
A 10-year-old boy presented with painless purple streaks on the arms and chest of 2 months' duration. The rash recurred several times per month and cleared without treatment in 3 to 5 days. There was no history of trauma or medication exposure, and he was growing and developing normally.
