User login
Grouped Erythematous Papules and Plaques on the Trunk
The Diagnosis: Cutaneous B-Cell Lymphoma, Follicle Center Subtype
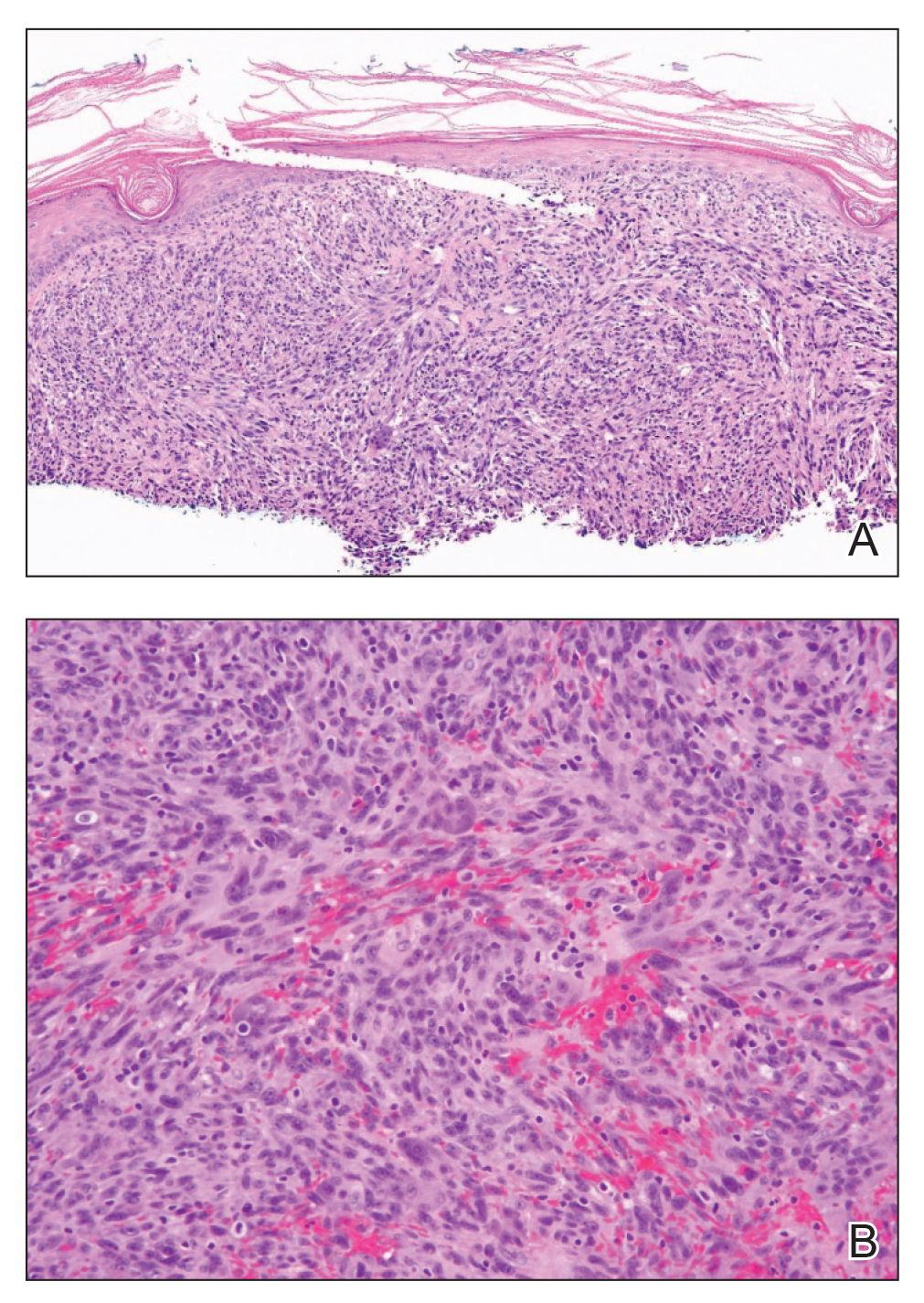
A 4-mm punch biopsy through the center of the largest lesion on the right posterior shoulder demonstrated a superficial and deep dermal atypical lymphoid infiltrate composed predominantly of small mature lymphocytes with interspersed intermediate-sized cells with irregular to cleaved nuclei, dispersed chromatin, one or more distinct nucleoli, occasional mitoses, and small amounts of cytoplasm (Figure, A). Immunoperoxidase studies showed the infiltrate to be a mixture of CD3+ T cells and CD20+ B cells (Figure, B). The B cells coexpressed B-cell lymphoma (Bcl) 6 protein (Figure, C) but were negative for multiple myeloma 1/interferon regulatory factor 4 and CD10; Bcl2 protein was positive in T cells but inconclusive for staining in B cells. Very few plasma cells were seen with CD138 stain. Fluorescence in situ hybridization studies were negative for IgH and BCL2 gene rearrangement. Molecular diagnostic studies for IgH and κ light chain gene rearrangement were positive for a clonal population. A clonal T-cell receptor γ chain gene rearrangement was not identified. The overall morphologic, immunophenotypic, and molecular findings were consistent with cutaneous involvement by a B-cell lymphoproliferative disorder, favoring primary cutaneous follicle center lymphoma (PCFCL).

The patient was referred to our cancer center for further workup consisting of a complete blood cell count with differential; comprehensive metabolic panel; lactate dehydrogenase; serum protein electrophoresis; peripheral blood flow cytometry; and computed tomography of the chest, abdomen, and pelvis. The analysis was unremarkable, supporting primary cutaneous disease. Additional studies suggested in the National Comprehensive Cancer Network (NCCN) Guidelines for primary cutaneous B-cell lymphomas include hepatitis B testing if the patient is being considered for immunotherapy and/or chemotherapy due to risk of reactivation, pregnancy testing in women of childbearing age, and human immunodeficiency virus testing.1 These tests were not performed in our patient because he did not have any risk factors for hepatitis B or human immunodeficiency virus.
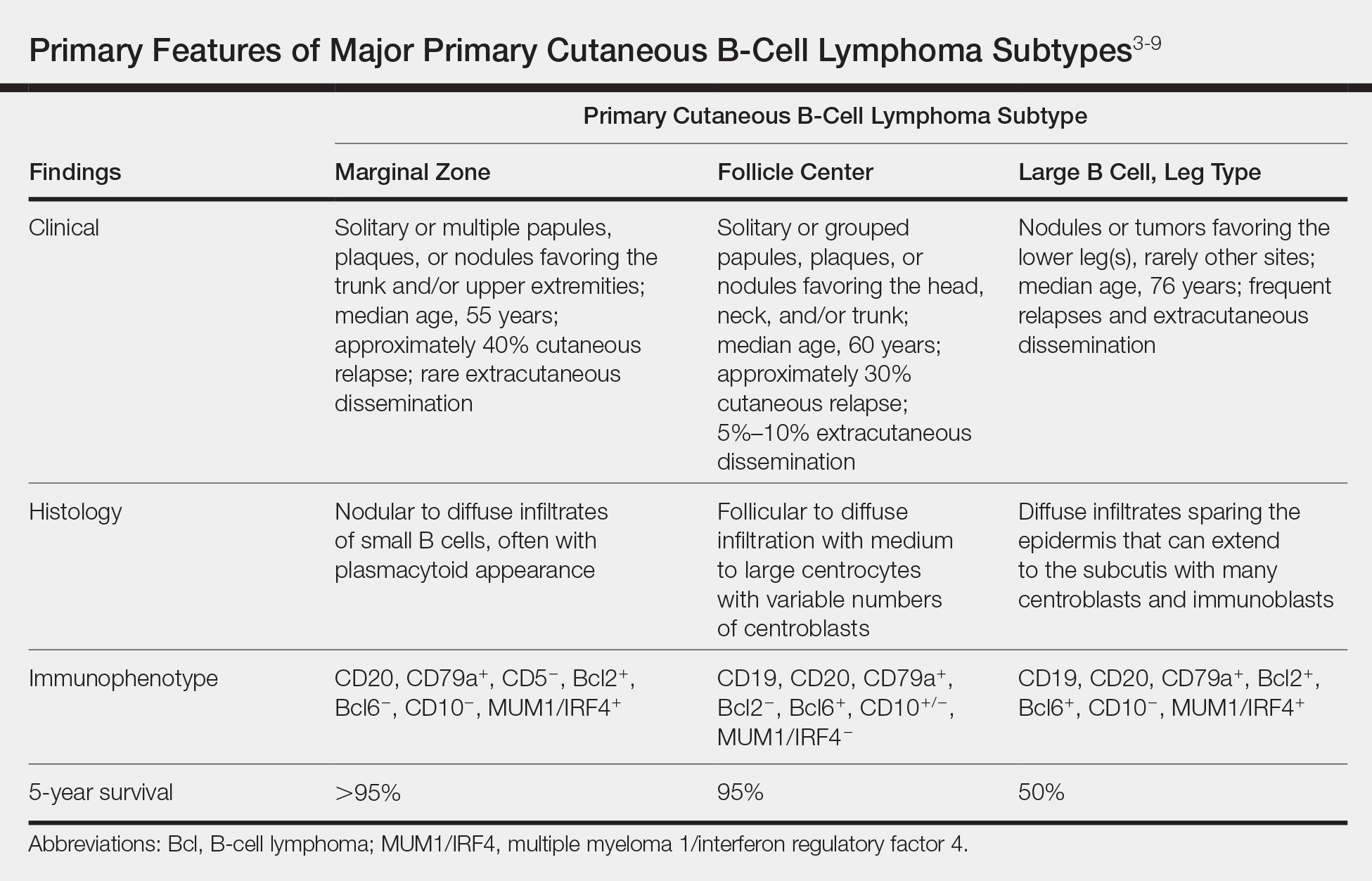
Primary cutaneous B-cell lymphomas originate in the skin without evidence of extracutaneous disease at presentation. They account for approximately 25% of primary cutaneous lymphomas in the United States, with primary cutaneous T-cell lymphoma being most common.2 The revised 2017 World Health Organization classification system defines 3 major subtypes of primary cutaneous B-cell lymphoma (Table).3-9 Primary cutaneous follicle center lymphoma is the most common subtype, accounting for approximately 60% of cases. In Europe, an association with Borrelia burgdorferi has been reported.10 The extent of skin involvement determines the T portion of TNM staging for PCFCL. It is based on the size and location of affected body regions that are delineated, such as the head and neck, chest, abdomen/genitalia, upper back, lower back/buttocks, each upper arm, each lower arm/hand, each upper leg, and each lower leg/foot. T1 is for solitary skin involvement in which the lesion is 5 cm or less in diameter (T1a) or greater than 5 cm (T1b). T2 is for regional skin involvement limited to 1 or 2 contiguous body regions, whereas T2a has all lesions confined to an area 15 cm or less in diameter, T2b has lesions confined to an area greater than 15 cm up to 30 cm in diameter, and the area for T2c is greater than 30 cm in diameter. Finally, T3 is generalized skin involvement, whereas T3a has multiple lesions in 2 noncontiguous body regions, and T3b has multiple lesions on 3 or more regions.11 At presentation, our patient was considered T2cN0M0, as his lesions were present on only 2 contiguous regions extending beyond 30 cm without any evidence of lymph node involvement or metastasis.
Treatment of PCFCL is tailored to each case, as there is a paucity of randomized data in this rare entity. It is guided by the number and location of cutaneous lesions, associated skin symptoms, age of the patient, and performance status. Local disease can be treated with intralesional corticosteroids, excision, or close monitoring if the patient is asymptomatic. Low-dose radiation therapy may be used as primary treatment or for local recurrence.12 Patients with more extensive skin lesions can relapse after clearing; those with refractory disease can be managed with single-agent rituximab.13 Our patient underwent low-dose radiation therapy with good response and has not experienced recurrence.
Lymphocytoma cutis, also known as benign reactive lymphoid hyperplasia, can be idiopathic or can arise after arthropod assault, penetrative skin trauma, drugs, or infections. In granuloma annulare, small dermal papules may present in isolation or coalesce to form annular plaques. It is a benign inflammatory disorder of unknown cause, can have mild pruritus, and usually is self-limited. Pyogenic granuloma is a benign vascular proliferation of unknown etiology. Sarcoidosis is an immune-mediated systemic disorder with granuloma formation that has a predilection for the lungs and the skin.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Primary Cutaneous B-Cell Lymphomas. Version 2.2018. https://oncolife.com.ua/doc/nccn/Primary_Cutaneous_B-Cell_Lymphomas.pdf. Published January 10, 2018. Accessed June 21, 2019.
- Dores GM, Anderson WF, Devesa SS. Cutaneous lymphomas reported to the National Cancer Institute's surveillance, epidemiology, and end results program: applying the new WHO-European Organisation for Research and Treatment of Cancer classification system. J Clin Oncol. 2005;23:7246-7248.
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC; 2017.
- Surveillance, Epidemiology, and End Results Program. National Cancer Institute website. https://seer.cancer.gov/. Accessed June 26, 2019.
- Cerroni L. B-cell lymphomas of the skin. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:2113-2126.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous follicle center lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-follicle-center-lymphoma. Updated February 7, 2018. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous marginal zone lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-marginal-zone-lymphoma. Updated March 6, 2019. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous large B cell lymphoma, leg type. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-large-b-cell-lymphoma-leg-type. Updated July 3, 2017. Accessed June 26, 2019.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 241-342.
- Goodlad JR, Davidson MM, Hollowood K, et al. Primary cutaneous B-cell lymphoma and Borrelia burgdorferi infection in patients from the Highlands of Scotand. Am J Surg Pathol. 2000;24:1279-1285.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Wilcon RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Morales AV, Advani R, Horwitz SM, et al. Indolent primary cutaneous B-cell lymphoma: experience using systemic rituximab. J Am Acad Dermatol. 2008;59:953-957.
The Diagnosis: Cutaneous B-Cell Lymphoma, Follicle Center Subtype
A 4-mm punch biopsy through the center of the largest lesion on the right posterior shoulder demonstrated a superficial and deep dermal atypical lymphoid infiltrate composed predominantly of small mature lymphocytes with interspersed intermediate-sized cells with irregular to cleaved nuclei, dispersed chromatin, one or more distinct nucleoli, occasional mitoses, and small amounts of cytoplasm (Figure, A). Immunoperoxidase studies showed the infiltrate to be a mixture of CD3+ T cells and CD20+ B cells (Figure, B). The B cells coexpressed B-cell lymphoma (Bcl) 6 protein (Figure, C) but were negative for multiple myeloma 1/interferon regulatory factor 4 and CD10; Bcl2 protein was positive in T cells but inconclusive for staining in B cells. Very few plasma cells were seen with CD138 stain. Fluorescence in situ hybridization studies were negative for IgH and BCL2 gene rearrangement. Molecular diagnostic studies for IgH and κ light chain gene rearrangement were positive for a clonal population. A clonal T-cell receptor γ chain gene rearrangement was not identified. The overall morphologic, immunophenotypic, and molecular findings were consistent with cutaneous involvement by a B-cell lymphoproliferative disorder, favoring primary cutaneous follicle center lymphoma (PCFCL).
The patient was referred to our cancer center for further workup consisting of a complete blood cell count with differential; comprehensive metabolic panel; lactate dehydrogenase; serum protein electrophoresis; peripheral blood flow cytometry; and computed tomography of the chest, abdomen, and pelvis. The analysis was unremarkable, supporting primary cutaneous disease. Additional studies suggested in the National Comprehensive Cancer Network (NCCN) Guidelines for primary cutaneous B-cell lymphomas include hepatitis B testing if the patient is being considered for immunotherapy and/or chemotherapy due to risk of reactivation, pregnancy testing in women of childbearing age, and human immunodeficiency virus testing.1 These tests were not performed in our patient because he did not have any risk factors for hepatitis B or human immunodeficiency virus.
Primary cutaneous B-cell lymphomas originate in the skin without evidence of extracutaneous disease at presentation. They account for approximately 25% of primary cutaneous lymphomas in the United States, with primary cutaneous T-cell lymphoma being most common.2 The revised 2017 World Health Organization classification system defines 3 major subtypes of primary cutaneous B-cell lymphoma (Table).3-9 Primary cutaneous follicle center lymphoma is the most common subtype, accounting for approximately 60% of cases. In Europe, an association with Borrelia burgdorferi has been reported.10 The extent of skin involvement determines the T portion of TNM staging for PCFCL. It is based on the size and location of affected body regions that are delineated, such as the head and neck, chest, abdomen/genitalia, upper back, lower back/buttocks, each upper arm, each lower arm/hand, each upper leg, and each lower leg/foot. T1 is for solitary skin involvement in which the lesion is 5 cm or less in diameter (T1a) or greater than 5 cm (T1b). T2 is for regional skin involvement limited to 1 or 2 contiguous body regions, whereas T2a has all lesions confined to an area 15 cm or less in diameter, T2b has lesions confined to an area greater than 15 cm up to 30 cm in diameter, and the area for T2c is greater than 30 cm in diameter. Finally, T3 is generalized skin involvement, whereas T3a has multiple lesions in 2 noncontiguous body regions, and T3b has multiple lesions on 3 or more regions.11 At presentation, our patient was considered T2cN0M0, as his lesions were present on only 2 contiguous regions extending beyond 30 cm without any evidence of lymph node involvement or metastasis.
Treatment of PCFCL is tailored to each case, as there is a paucity of randomized data in this rare entity. It is guided by the number and location of cutaneous lesions, associated skin symptoms, age of the patient, and performance status. Local disease can be treated with intralesional corticosteroids, excision, or close monitoring if the patient is asymptomatic. Low-dose radiation therapy may be used as primary treatment or for local recurrence.12 Patients with more extensive skin lesions can relapse after clearing; those with refractory disease can be managed with single-agent rituximab.13 Our patient underwent low-dose radiation therapy with good response and has not experienced recurrence.
Lymphocytoma cutis, also known as benign reactive lymphoid hyperplasia, can be idiopathic or can arise after arthropod assault, penetrative skin trauma, drugs, or infections. In granuloma annulare, small dermal papules may present in isolation or coalesce to form annular plaques. It is a benign inflammatory disorder of unknown cause, can have mild pruritus, and usually is self-limited. Pyogenic granuloma is a benign vascular proliferation of unknown etiology. Sarcoidosis is an immune-mediated systemic disorder with granuloma formation that has a predilection for the lungs and the skin.
The Diagnosis: Cutaneous B-Cell Lymphoma, Follicle Center Subtype
A 4-mm punch biopsy through the center of the largest lesion on the right posterior shoulder demonstrated a superficial and deep dermal atypical lymphoid infiltrate composed predominantly of small mature lymphocytes with interspersed intermediate-sized cells with irregular to cleaved nuclei, dispersed chromatin, one or more distinct nucleoli, occasional mitoses, and small amounts of cytoplasm (Figure, A). Immunoperoxidase studies showed the infiltrate to be a mixture of CD3+ T cells and CD20+ B cells (Figure, B). The B cells coexpressed B-cell lymphoma (Bcl) 6 protein (Figure, C) but were negative for multiple myeloma 1/interferon regulatory factor 4 and CD10; Bcl2 protein was positive in T cells but inconclusive for staining in B cells. Very few plasma cells were seen with CD138 stain. Fluorescence in situ hybridization studies were negative for IgH and BCL2 gene rearrangement. Molecular diagnostic studies for IgH and κ light chain gene rearrangement were positive for a clonal population. A clonal T-cell receptor γ chain gene rearrangement was not identified. The overall morphologic, immunophenotypic, and molecular findings were consistent with cutaneous involvement by a B-cell lymphoproliferative disorder, favoring primary cutaneous follicle center lymphoma (PCFCL).
The patient was referred to our cancer center for further workup consisting of a complete blood cell count with differential; comprehensive metabolic panel; lactate dehydrogenase; serum protein electrophoresis; peripheral blood flow cytometry; and computed tomography of the chest, abdomen, and pelvis. The analysis was unremarkable, supporting primary cutaneous disease. Additional studies suggested in the National Comprehensive Cancer Network (NCCN) Guidelines for primary cutaneous B-cell lymphomas include hepatitis B testing if the patient is being considered for immunotherapy and/or chemotherapy due to risk of reactivation, pregnancy testing in women of childbearing age, and human immunodeficiency virus testing.1 These tests were not performed in our patient because he did not have any risk factors for hepatitis B or human immunodeficiency virus.
Primary cutaneous B-cell lymphomas originate in the skin without evidence of extracutaneous disease at presentation. They account for approximately 25% of primary cutaneous lymphomas in the United States, with primary cutaneous T-cell lymphoma being most common.2 The revised 2017 World Health Organization classification system defines 3 major subtypes of primary cutaneous B-cell lymphoma (Table).3-9 Primary cutaneous follicle center lymphoma is the most common subtype, accounting for approximately 60% of cases. In Europe, an association with Borrelia burgdorferi has been reported.10 The extent of skin involvement determines the T portion of TNM staging for PCFCL. It is based on the size and location of affected body regions that are delineated, such as the head and neck, chest, abdomen/genitalia, upper back, lower back/buttocks, each upper arm, each lower arm/hand, each upper leg, and each lower leg/foot. T1 is for solitary skin involvement in which the lesion is 5 cm or less in diameter (T1a) or greater than 5 cm (T1b). T2 is for regional skin involvement limited to 1 or 2 contiguous body regions, whereas T2a has all lesions confined to an area 15 cm or less in diameter, T2b has lesions confined to an area greater than 15 cm up to 30 cm in diameter, and the area for T2c is greater than 30 cm in diameter. Finally, T3 is generalized skin involvement, whereas T3a has multiple lesions in 2 noncontiguous body regions, and T3b has multiple lesions on 3 or more regions.11 At presentation, our patient was considered T2cN0M0, as his lesions were present on only 2 contiguous regions extending beyond 30 cm without any evidence of lymph node involvement or metastasis.
Treatment of PCFCL is tailored to each case, as there is a paucity of randomized data in this rare entity. It is guided by the number and location of cutaneous lesions, associated skin symptoms, age of the patient, and performance status. Local disease can be treated with intralesional corticosteroids, excision, or close monitoring if the patient is asymptomatic. Low-dose radiation therapy may be used as primary treatment or for local recurrence.12 Patients with more extensive skin lesions can relapse after clearing; those with refractory disease can be managed with single-agent rituximab.13 Our patient underwent low-dose radiation therapy with good response and has not experienced recurrence.
Lymphocytoma cutis, also known as benign reactive lymphoid hyperplasia, can be idiopathic or can arise after arthropod assault, penetrative skin trauma, drugs, or infections. In granuloma annulare, small dermal papules may present in isolation or coalesce to form annular plaques. It is a benign inflammatory disorder of unknown cause, can have mild pruritus, and usually is self-limited. Pyogenic granuloma is a benign vascular proliferation of unknown etiology. Sarcoidosis is an immune-mediated systemic disorder with granuloma formation that has a predilection for the lungs and the skin.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Primary Cutaneous B-Cell Lymphomas. Version 2.2018. https://oncolife.com.ua/doc/nccn/Primary_Cutaneous_B-Cell_Lymphomas.pdf. Published January 10, 2018. Accessed June 21, 2019.
- Dores GM, Anderson WF, Devesa SS. Cutaneous lymphomas reported to the National Cancer Institute's surveillance, epidemiology, and end results program: applying the new WHO-European Organisation for Research and Treatment of Cancer classification system. J Clin Oncol. 2005;23:7246-7248.
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC; 2017.
- Surveillance, Epidemiology, and End Results Program. National Cancer Institute website. https://seer.cancer.gov/. Accessed June 26, 2019.
- Cerroni L. B-cell lymphomas of the skin. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:2113-2126.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous follicle center lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-follicle-center-lymphoma. Updated February 7, 2018. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous marginal zone lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-marginal-zone-lymphoma. Updated March 6, 2019. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous large B cell lymphoma, leg type. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-large-b-cell-lymphoma-leg-type. Updated July 3, 2017. Accessed June 26, 2019.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 241-342.
- Goodlad JR, Davidson MM, Hollowood K, et al. Primary cutaneous B-cell lymphoma and Borrelia burgdorferi infection in patients from the Highlands of Scotand. Am J Surg Pathol. 2000;24:1279-1285.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Wilcon RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Morales AV, Advani R, Horwitz SM, et al. Indolent primary cutaneous B-cell lymphoma: experience using systemic rituximab. J Am Acad Dermatol. 2008;59:953-957.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Primary Cutaneous B-Cell Lymphomas. Version 2.2018. https://oncolife.com.ua/doc/nccn/Primary_Cutaneous_B-Cell_Lymphomas.pdf. Published January 10, 2018. Accessed June 21, 2019.
- Dores GM, Anderson WF, Devesa SS. Cutaneous lymphomas reported to the National Cancer Institute's surveillance, epidemiology, and end results program: applying the new WHO-European Organisation for Research and Treatment of Cancer classification system. J Clin Oncol. 2005;23:7246-7248.
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC; 2017.
- Surveillance, Epidemiology, and End Results Program. National Cancer Institute website. https://seer.cancer.gov/. Accessed June 26, 2019.
- Cerroni L. B-cell lymphomas of the skin. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:2113-2126.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous follicle center lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-follicle-center-lymphoma. Updated February 7, 2018. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous marginal zone lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-marginal-zone-lymphoma. Updated March 6, 2019. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous large B cell lymphoma, leg type. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-large-b-cell-lymphoma-leg-type. Updated July 3, 2017. Accessed June 26, 2019.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 241-342.
- Goodlad JR, Davidson MM, Hollowood K, et al. Primary cutaneous B-cell lymphoma and Borrelia burgdorferi infection in patients from the Highlands of Scotand. Am J Surg Pathol. 2000;24:1279-1285.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Wilcon RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Morales AV, Advani R, Horwitz SM, et al. Indolent primary cutaneous B-cell lymphoma: experience using systemic rituximab. J Am Acad Dermatol. 2008;59:953-957.
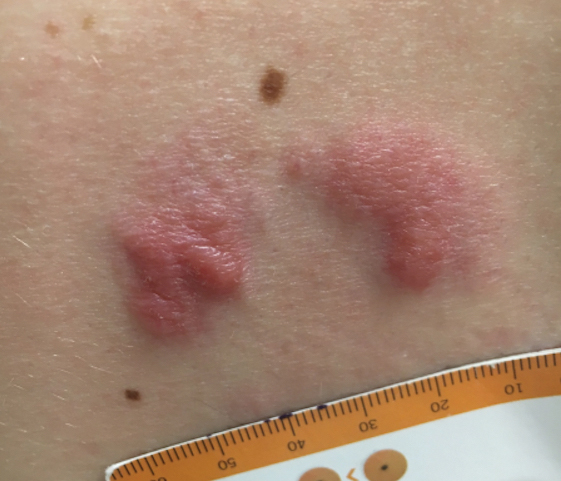
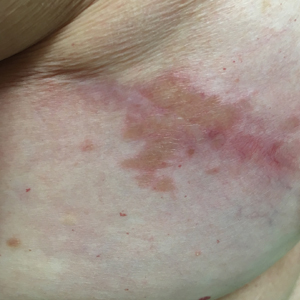
A 34-year-old man presented to the outpatient dermatology clinic with 3 groups of mildly pruritic, erythematous papules and plaques. The most prominent group appeared on the right posterior shoulder and had been slowly enlarging in size over the last 12 months (quiz image). A similar thinner group appeared on the left mid-back 6 months prior, and a third smaller group appeared over the left serratus anterior muscle 2 months prior. The patient reported having similar episodes dating back to his early 20s. In those instances, the lesions presented without an inciting incident, became more pronounced, and persisted for months to years before resolving. Previously affected areas included the upper and lateral back, flanks, and posterior upper arms. The patient used triamcinolone cream 0.1% up to 3 times daily on active lesions, which improved the pruritus and seemed to make the lesions resolve more quickly. He denied fever, chills, night sweats, anorexia, weight loss, fatigue, cough, and shortness of breath. His only medication was ranitidine 150 mg twice daily for gastroesophageal reflux disease. Physical examination revealed no palpable lymphadenopathy.
Rapidly Enlarging Neoplasm on the Face
The Diagnosis: Atypical Fibroxanthoma
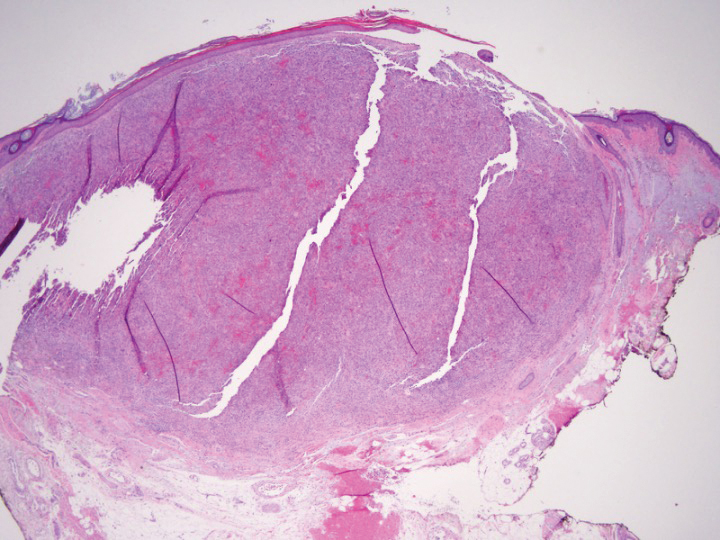
Shave biopsy showed the superficial aspect of a highly cellular tumor composed of pleomorphic spindle cells exhibiting storiform growth and increased mitotic activity (Figure 1). The tumor stained positive for factor XIIIa, CD163, CD68, and smooth muscle actin (mild), and negative for high-molecular-weight cytokeratin (HMW-CK), p63, S-100, and melan-A. Subsequent excision with 0.5-cm margins was performed, and histopathology showed a well-circumscribed tumor contained within the dermis with a histologic scar at the outer margin (Figure 2). There was no lymphovascular or perineural invasion by tumor cells. Re-excision with 0.3-cm margins demonstrated no residual scar or tumor, and external radiation was deferred due to clear surgical margins.
Atypical fibroxanthoma (AFX) belongs to a group of spindle cell neoplasms that can be diagnostically challenging, as they often lack specific morphologic features on examination or routine histology. These neoplasms--of which the differential includes malignant fibrous histiocytoma, spindle cell squamous cell carcinoma (SCC), desmoplastic melanoma, and leiomyosarcoma--may each appear as a rapidly enlarging solitary plaque or nodule on sun-damaged skin on the head and neck or less commonly on the trunk, arms, or legs. Histologically, the cells of AFX exhibit notable pleomorphism with frequent atypical mitotic figures and nonspecific surrounding dermal changes. Subcutaneous and lymphovascular or perineural invasion of tumor cells can point away from the diagnosis of AFX; however, these features are likely to be missed in small superficial shave biopsies.1,2 Therefore, immunohistochemistry (IHC) and adequate tumor sampling are essential in the accurate diagnosis of AFX and other spindle cell neoplasms.
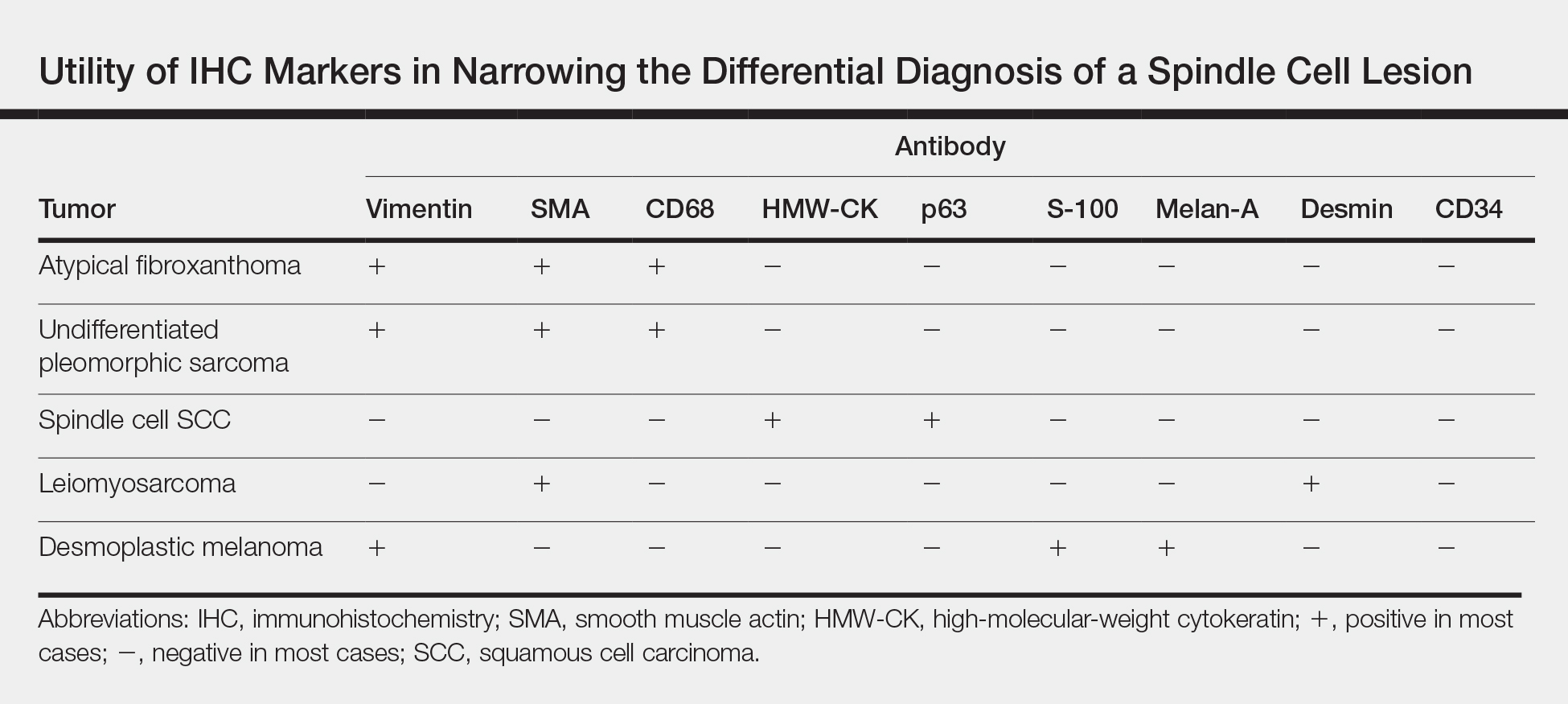
Several IHC markers have been employed in differentiating AFX from other spindle cell neoplasms.3-8 Positive stains for AFX include factor XIIIa (10%-25%), vimentin (>99%), CD10 (95%-100%), procollagen (87%), CD99 (35%-73%), CD163 (37%-79%), smooth muscle actin (50%), CD68 (>50%), and CD31 (43%). Other stains, such as HMW-CK, S-100, p63, desmin, CD34, and melan-A, typically are negative in AFX but are actively expressed in other pleomorphic spindle cell tumors. The Table summarizes the utility of these various markers in narrowing the differential diagnosis of a spindle cell lesion. Selection of an appropriate panel of IHC markers is critical for accurate diagnosis of AFX and exclusion of more aggressive, poorly differentiated spindle cell neoplasms. Key IHC markers include S-100 (negative in AFX; positive in desmoplastic melanoma), HMW-CK (negative in AFX; positive in spindle cell SCC), and p63 (negative in AFX; positive in spindle cell SCC). Benoit et al9 reported a case of a poorly differentiated spindle cell SCC misdiagnosed as AFX based on a limited IHC panel that was negative for pancytokeratin and S-100. Later, a more comprehensive IHC panel including HMW-CK and p63 confirmed spindle cell SCC, but by this time, a delay in therapy had allowed the tumor to metastasize, which ultimately proved fatal to the patient.9
In addition to incomplete IHC evaluation, accurate diagnosis of spindle cell tumors also may be obscured by inadequate tumor sampling. The cells of AFX tumors often are well circumscribed and dermally based, and an excisional biopsy is the preferred biopsy procedure for AFX. A tumor invading into subcutaneous tissue or into lymphovascular or perineural structures suggests a more aggressive, poorly differentiated spindle cell neoplasm.1,3 For example, the tumor cells of malignant fibrous histiocytoma, which belongs to the undifferentiated pleomorphic sarcoma group, may appear identical to those of AFX on histology, and the 2 tumors display similar IHC profiles.3 Malignant fibrous histiocytoma, however, extends into the subcutaneous space and portends a notably worse prognosis compared to AFX. Malignant fibrous histiocytoma tumors therefore require more aggressive treatment strategies such as external beam radiation therapy, whereas AFX can be safely treated with surgical removal alone. In our patient, complete visualization of tumor margins solidified the diagnosis of AFX and spared our patient from unnecessary radiation therapy. Overall, AFX has a good prognosis and metastasis is rare, particularly when good margin control is achieved.10
Our case highlights the importance of clinicopathologic correlation, including appropriate IHC analysis and adequate tumor sampling in the diagnostic workup of a pleomorphic spindle cell neoplasm. Although these tumors are well studied, their notable degree of clinical and histologic heterogeneity may pose a diagnostic challenge to even experienced dermatologists and require careful consideration of the potential pitfalls in diagnosis.
- Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Lopez L, Velez R. Atypical fibroxanthoma. Arch Pathol Lab Med. 2016;140:376-379.
- Hussein MR. Atypical fibroxanthoma: new insights. Expert Rev Anticancer Ther. 2014;14:1075-1088.
- Gleason BC, Calder KB, Cibull TL, et al. Utility of p63 in the differential diagnosis of atypical fibroxanthoma and spindle cell squamous cell carcinoma. J Cutan Pathol. 2009;36:543-547.
- Pouryazdanparast P, Yu L, Cutland JE, et al. Diagnostic value of CD163 in cutaneous spindle cell lesions. J Cutan Pathol. 2009;36:859-864.
- Beer TW. CD163 is not a sensitive marker for identification of atypical fibroxanthoma. J Cutan Pathol. 2012;39:29-32.
- Longacre TA, Smoller BR, Rouse RV. Atypical fibroxanthoma. multiple immunohistologic profiles. Am J Surg Pathol. 1993;17:1199-1209.
- Altman DA, Nickoloff BD, Fivenson DP. Differential expression of factor XIIa and CD34 in cutaneous mesenchymal tumors. J Cutan Pathol. 1993;20:154-158.
- Benoit A, Wisell J, Brown M. Cutaneous spindle cell carcinoma misdiagnosed as atypical fibroxanthoma based on immunohistochemical stains. JAAD Case Rep. 2015;1:392-394.
- New D, Bahrami S, Malone J, et al. Atypical fibroxanthoma with regional lymph node metastasis: report of a case and review of the literature. Arch Dermatol. 2010;146:1399-1404.
The Diagnosis: Atypical Fibroxanthoma
Shave biopsy showed the superficial aspect of a highly cellular tumor composed of pleomorphic spindle cells exhibiting storiform growth and increased mitotic activity (Figure 1). The tumor stained positive for factor XIIIa, CD163, CD68, and smooth muscle actin (mild), and negative for high-molecular-weight cytokeratin (HMW-CK), p63, S-100, and melan-A. Subsequent excision with 0.5-cm margins was performed, and histopathology showed a well-circumscribed tumor contained within the dermis with a histologic scar at the outer margin (Figure 2). There was no lymphovascular or perineural invasion by tumor cells. Re-excision with 0.3-cm margins demonstrated no residual scar or tumor, and external radiation was deferred due to clear surgical margins.
Atypical fibroxanthoma (AFX) belongs to a group of spindle cell neoplasms that can be diagnostically challenging, as they often lack specific morphologic features on examination or routine histology. These neoplasms--of which the differential includes malignant fibrous histiocytoma, spindle cell squamous cell carcinoma (SCC), desmoplastic melanoma, and leiomyosarcoma--may each appear as a rapidly enlarging solitary plaque or nodule on sun-damaged skin on the head and neck or less commonly on the trunk, arms, or legs. Histologically, the cells of AFX exhibit notable pleomorphism with frequent atypical mitotic figures and nonspecific surrounding dermal changes. Subcutaneous and lymphovascular or perineural invasion of tumor cells can point away from the diagnosis of AFX; however, these features are likely to be missed in small superficial shave biopsies.1,2 Therefore, immunohistochemistry (IHC) and adequate tumor sampling are essential in the accurate diagnosis of AFX and other spindle cell neoplasms.
Several IHC markers have been employed in differentiating AFX from other spindle cell neoplasms.3-8 Positive stains for AFX include factor XIIIa (10%-25%), vimentin (>99%), CD10 (95%-100%), procollagen (87%), CD99 (35%-73%), CD163 (37%-79%), smooth muscle actin (50%), CD68 (>50%), and CD31 (43%). Other stains, such as HMW-CK, S-100, p63, desmin, CD34, and melan-A, typically are negative in AFX but are actively expressed in other pleomorphic spindle cell tumors. The Table summarizes the utility of these various markers in narrowing the differential diagnosis of a spindle cell lesion. Selection of an appropriate panel of IHC markers is critical for accurate diagnosis of AFX and exclusion of more aggressive, poorly differentiated spindle cell neoplasms. Key IHC markers include S-100 (negative in AFX; positive in desmoplastic melanoma), HMW-CK (negative in AFX; positive in spindle cell SCC), and p63 (negative in AFX; positive in spindle cell SCC). Benoit et al9 reported a case of a poorly differentiated spindle cell SCC misdiagnosed as AFX based on a limited IHC panel that was negative for pancytokeratin and S-100. Later, a more comprehensive IHC panel including HMW-CK and p63 confirmed spindle cell SCC, but by this time, a delay in therapy had allowed the tumor to metastasize, which ultimately proved fatal to the patient.9
In addition to incomplete IHC evaluation, accurate diagnosis of spindle cell tumors also may be obscured by inadequate tumor sampling. The cells of AFX tumors often are well circumscribed and dermally based, and an excisional biopsy is the preferred biopsy procedure for AFX. A tumor invading into subcutaneous tissue or into lymphovascular or perineural structures suggests a more aggressive, poorly differentiated spindle cell neoplasm.1,3 For example, the tumor cells of malignant fibrous histiocytoma, which belongs to the undifferentiated pleomorphic sarcoma group, may appear identical to those of AFX on histology, and the 2 tumors display similar IHC profiles.3 Malignant fibrous histiocytoma, however, extends into the subcutaneous space and portends a notably worse prognosis compared to AFX. Malignant fibrous histiocytoma tumors therefore require more aggressive treatment strategies such as external beam radiation therapy, whereas AFX can be safely treated with surgical removal alone. In our patient, complete visualization of tumor margins solidified the diagnosis of AFX and spared our patient from unnecessary radiation therapy. Overall, AFX has a good prognosis and metastasis is rare, particularly when good margin control is achieved.10
Our case highlights the importance of clinicopathologic correlation, including appropriate IHC analysis and adequate tumor sampling in the diagnostic workup of a pleomorphic spindle cell neoplasm. Although these tumors are well studied, their notable degree of clinical and histologic heterogeneity may pose a diagnostic challenge to even experienced dermatologists and require careful consideration of the potential pitfalls in diagnosis.
The Diagnosis: Atypical Fibroxanthoma
Shave biopsy showed the superficial aspect of a highly cellular tumor composed of pleomorphic spindle cells exhibiting storiform growth and increased mitotic activity (Figure 1). The tumor stained positive for factor XIIIa, CD163, CD68, and smooth muscle actin (mild), and negative for high-molecular-weight cytokeratin (HMW-CK), p63, S-100, and melan-A. Subsequent excision with 0.5-cm margins was performed, and histopathology showed a well-circumscribed tumor contained within the dermis with a histologic scar at the outer margin (Figure 2). There was no lymphovascular or perineural invasion by tumor cells. Re-excision with 0.3-cm margins demonstrated no residual scar or tumor, and external radiation was deferred due to clear surgical margins.
Atypical fibroxanthoma (AFX) belongs to a group of spindle cell neoplasms that can be diagnostically challenging, as they often lack specific morphologic features on examination or routine histology. These neoplasms--of which the differential includes malignant fibrous histiocytoma, spindle cell squamous cell carcinoma (SCC), desmoplastic melanoma, and leiomyosarcoma--may each appear as a rapidly enlarging solitary plaque or nodule on sun-damaged skin on the head and neck or less commonly on the trunk, arms, or legs. Histologically, the cells of AFX exhibit notable pleomorphism with frequent atypical mitotic figures and nonspecific surrounding dermal changes. Subcutaneous and lymphovascular or perineural invasion of tumor cells can point away from the diagnosis of AFX; however, these features are likely to be missed in small superficial shave biopsies.1,2 Therefore, immunohistochemistry (IHC) and adequate tumor sampling are essential in the accurate diagnosis of AFX and other spindle cell neoplasms.
Several IHC markers have been employed in differentiating AFX from other spindle cell neoplasms.3-8 Positive stains for AFX include factor XIIIa (10%-25%), vimentin (>99%), CD10 (95%-100%), procollagen (87%), CD99 (35%-73%), CD163 (37%-79%), smooth muscle actin (50%), CD68 (>50%), and CD31 (43%). Other stains, such as HMW-CK, S-100, p63, desmin, CD34, and melan-A, typically are negative in AFX but are actively expressed in other pleomorphic spindle cell tumors. The Table summarizes the utility of these various markers in narrowing the differential diagnosis of a spindle cell lesion. Selection of an appropriate panel of IHC markers is critical for accurate diagnosis of AFX and exclusion of more aggressive, poorly differentiated spindle cell neoplasms. Key IHC markers include S-100 (negative in AFX; positive in desmoplastic melanoma), HMW-CK (negative in AFX; positive in spindle cell SCC), and p63 (negative in AFX; positive in spindle cell SCC). Benoit et al9 reported a case of a poorly differentiated spindle cell SCC misdiagnosed as AFX based on a limited IHC panel that was negative for pancytokeratin and S-100. Later, a more comprehensive IHC panel including HMW-CK and p63 confirmed spindle cell SCC, but by this time, a delay in therapy had allowed the tumor to metastasize, which ultimately proved fatal to the patient.9
In addition to incomplete IHC evaluation, accurate diagnosis of spindle cell tumors also may be obscured by inadequate tumor sampling. The cells of AFX tumors often are well circumscribed and dermally based, and an excisional biopsy is the preferred biopsy procedure for AFX. A tumor invading into subcutaneous tissue or into lymphovascular or perineural structures suggests a more aggressive, poorly differentiated spindle cell neoplasm.1,3 For example, the tumor cells of malignant fibrous histiocytoma, which belongs to the undifferentiated pleomorphic sarcoma group, may appear identical to those of AFX on histology, and the 2 tumors display similar IHC profiles.3 Malignant fibrous histiocytoma, however, extends into the subcutaneous space and portends a notably worse prognosis compared to AFX. Malignant fibrous histiocytoma tumors therefore require more aggressive treatment strategies such as external beam radiation therapy, whereas AFX can be safely treated with surgical removal alone. In our patient, complete visualization of tumor margins solidified the diagnosis of AFX and spared our patient from unnecessary radiation therapy. Overall, AFX has a good prognosis and metastasis is rare, particularly when good margin control is achieved.10
Our case highlights the importance of clinicopathologic correlation, including appropriate IHC analysis and adequate tumor sampling in the diagnostic workup of a pleomorphic spindle cell neoplasm. Although these tumors are well studied, their notable degree of clinical and histologic heterogeneity may pose a diagnostic challenge to even experienced dermatologists and require careful consideration of the potential pitfalls in diagnosis.
- Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Lopez L, Velez R. Atypical fibroxanthoma. Arch Pathol Lab Med. 2016;140:376-379.
- Hussein MR. Atypical fibroxanthoma: new insights. Expert Rev Anticancer Ther. 2014;14:1075-1088.
- Gleason BC, Calder KB, Cibull TL, et al. Utility of p63 in the differential diagnosis of atypical fibroxanthoma and spindle cell squamous cell carcinoma. J Cutan Pathol. 2009;36:543-547.
- Pouryazdanparast P, Yu L, Cutland JE, et al. Diagnostic value of CD163 in cutaneous spindle cell lesions. J Cutan Pathol. 2009;36:859-864.
- Beer TW. CD163 is not a sensitive marker for identification of atypical fibroxanthoma. J Cutan Pathol. 2012;39:29-32.
- Longacre TA, Smoller BR, Rouse RV. Atypical fibroxanthoma. multiple immunohistologic profiles. Am J Surg Pathol. 1993;17:1199-1209.
- Altman DA, Nickoloff BD, Fivenson DP. Differential expression of factor XIIa and CD34 in cutaneous mesenchymal tumors. J Cutan Pathol. 1993;20:154-158.
- Benoit A, Wisell J, Brown M. Cutaneous spindle cell carcinoma misdiagnosed as atypical fibroxanthoma based on immunohistochemical stains. JAAD Case Rep. 2015;1:392-394.
- New D, Bahrami S, Malone J, et al. Atypical fibroxanthoma with regional lymph node metastasis: report of a case and review of the literature. Arch Dermatol. 2010;146:1399-1404.
- Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Lopez L, Velez R. Atypical fibroxanthoma. Arch Pathol Lab Med. 2016;140:376-379.
- Hussein MR. Atypical fibroxanthoma: new insights. Expert Rev Anticancer Ther. 2014;14:1075-1088.
- Gleason BC, Calder KB, Cibull TL, et al. Utility of p63 in the differential diagnosis of atypical fibroxanthoma and spindle cell squamous cell carcinoma. J Cutan Pathol. 2009;36:543-547.
- Pouryazdanparast P, Yu L, Cutland JE, et al. Diagnostic value of CD163 in cutaneous spindle cell lesions. J Cutan Pathol. 2009;36:859-864.
- Beer TW. CD163 is not a sensitive marker for identification of atypical fibroxanthoma. J Cutan Pathol. 2012;39:29-32.
- Longacre TA, Smoller BR, Rouse RV. Atypical fibroxanthoma. multiple immunohistologic profiles. Am J Surg Pathol. 1993;17:1199-1209.
- Altman DA, Nickoloff BD, Fivenson DP. Differential expression of factor XIIa and CD34 in cutaneous mesenchymal tumors. J Cutan Pathol. 1993;20:154-158.
- Benoit A, Wisell J, Brown M. Cutaneous spindle cell carcinoma misdiagnosed as atypical fibroxanthoma based on immunohistochemical stains. JAAD Case Rep. 2015;1:392-394.
- New D, Bahrami S, Malone J, et al. Atypical fibroxanthoma with regional lymph node metastasis: report of a case and review of the literature. Arch Dermatol. 2010;146:1399-1404.
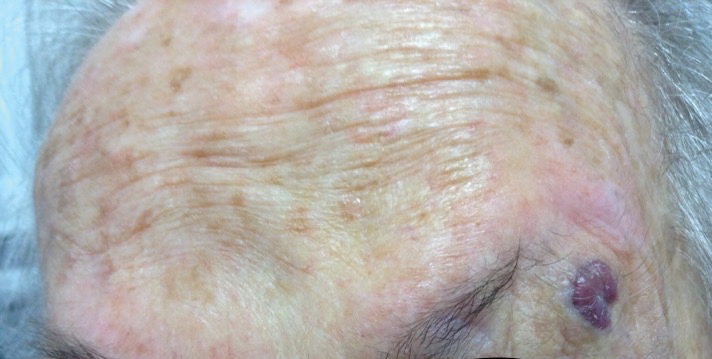
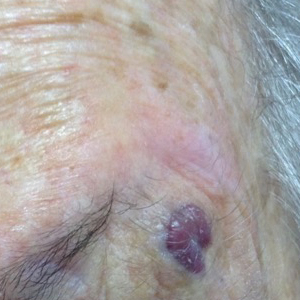
An 88-year-old woman presented for evaluation of an asymptomatic facial lesion that she first noticed 3 months prior, with rapid growth over the last month. Review of systems was negative, and she denied any history of connective tissue disease, skin cancer, or radiation to the head or neck area. Physical examination revealed a 1.5-cm, solitary, violaceous nodule on the left lateral eyebrow on a background of actinically damaged skin. The lesion was nontender and there were no similar lesions or palpable lymphadenopathy.
Recurrent Pruritic Multifocal Erythematous Rash
The Diagnosis: Wells Syndrome
Histopathologic examination of the biopsy demonstrated overlying acanthosis, focal spongiosis, and exocytosis. There also was proliferation and thickening of superficial capillaries and papillary fibrosis (Figure, A). There was a mixed interstitial and perivascular inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and eosinophils (Figure, A and B). Occasional flame figures were identified (Figure, C).

Wells syndrome, also known as eosinophilic cellulitis, was first described in 1971 by Wells1 as a recurrent granulomatous dermatitis with eosinophilia. Rarely reported worldwide, this chronic relapsing condition is characterized by a pronounced eosinophilic infiltrate of the dermis resembling urticaria or cellulitis.2 The exact etiology has not been elucidated; however, links to certain medications, vaccines, exaggerated arthropod reactions, infections, and malignancies have been documented.3
Wells syndrome is a diagnosis of exclusion and lacks a predictable dermatologic presentation, thereby mandating focused clinical follow-up as well as correlation with histopathology findings. Although the classic histologic hallmark of Wells syndrome is scattered flame figures, this finding is not specific and can be found in other hypereosinophilic conditions.2 Clinical manifestations most often consist of 2 distinct phases: an initial painful burning or pruritic sensation, followed by the development of erythematous and edematous dermal plaques that may heal with slight hyperpigmentation over 4 to 8 weeks. A case series of 19 patients demonstrated variants of Wells syndrome, with an annular granuloma-like appearance found primarily in adults and the signature plaque-type appearance predominating in children.4
Acute urticaria is characterized by pruritic erythematous wheals secondary to a histamine-mediated response brought on by a variety of triggers, typically allergic and self-resolving within 24 hours. When such lesions last longer than 24 hours, biopsy should be performed to exclude urticarial vasculitis, which is characterized by a burning or painful sensation rather than pruritis, in addition to dermal neutrophilia and perivascular infiltrate on histology. Erythema migrans of Lyme disease begins at the site of a tick bite, evolving from a red macule to an expanding targetoid lesion and typically is not pruritic. Infectious cellulitis presents with warm, tender, and poorly defined erythematous patches; can progress rapidly; and is accompanied by systemic symptoms such as fevers, malaise, and lymphadenopathy.
Best evidence favors the use of moderate- to high-dose corticosteroids as first-line treatment.5 The use of tumor necrosis factor blockers, various immunomodulating agents, and combination therapy with levocetirizine and hydroxyzine have demonstrated variable levels of efficacy, albeit often followed by high rates of relapse with drug discontinuation.6
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc. 1971;57:46-56.
- Aberer W, Konrad K, Wolff K. Wells' syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol. 1988;18:105-114.
- Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983-1984.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Ferreli C, Pinna AL, Atzori L, et al. Eosinophilic cellulitis (Well's syndrome): a new case description. J Eur Acad Dermatol Venereol. 1999;13:41-45.
- Cormerais M, Poizeau F, Darrieux L, et al. Wells' syndrome mimicking facial cellulitis: a report of two cases. Case Rep Dermatol. 2015;7:117-122.
The Diagnosis: Wells Syndrome
Histopathologic examination of the biopsy demonstrated overlying acanthosis, focal spongiosis, and exocytosis. There also was proliferation and thickening of superficial capillaries and papillary fibrosis (Figure, A). There was a mixed interstitial and perivascular inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and eosinophils (Figure, A and B). Occasional flame figures were identified (Figure, C).
Wells syndrome, also known as eosinophilic cellulitis, was first described in 1971 by Wells1 as a recurrent granulomatous dermatitis with eosinophilia. Rarely reported worldwide, this chronic relapsing condition is characterized by a pronounced eosinophilic infiltrate of the dermis resembling urticaria or cellulitis.2 The exact etiology has not been elucidated; however, links to certain medications, vaccines, exaggerated arthropod reactions, infections, and malignancies have been documented.3
Wells syndrome is a diagnosis of exclusion and lacks a predictable dermatologic presentation, thereby mandating focused clinical follow-up as well as correlation with histopathology findings. Although the classic histologic hallmark of Wells syndrome is scattered flame figures, this finding is not specific and can be found in other hypereosinophilic conditions.2 Clinical manifestations most often consist of 2 distinct phases: an initial painful burning or pruritic sensation, followed by the development of erythematous and edematous dermal plaques that may heal with slight hyperpigmentation over 4 to 8 weeks. A case series of 19 patients demonstrated variants of Wells syndrome, with an annular granuloma-like appearance found primarily in adults and the signature plaque-type appearance predominating in children.4
Acute urticaria is characterized by pruritic erythematous wheals secondary to a histamine-mediated response brought on by a variety of triggers, typically allergic and self-resolving within 24 hours. When such lesions last longer than 24 hours, biopsy should be performed to exclude urticarial vasculitis, which is characterized by a burning or painful sensation rather than pruritis, in addition to dermal neutrophilia and perivascular infiltrate on histology. Erythema migrans of Lyme disease begins at the site of a tick bite, evolving from a red macule to an expanding targetoid lesion and typically is not pruritic. Infectious cellulitis presents with warm, tender, and poorly defined erythematous patches; can progress rapidly; and is accompanied by systemic symptoms such as fevers, malaise, and lymphadenopathy.
Best evidence favors the use of moderate- to high-dose corticosteroids as first-line treatment.5 The use of tumor necrosis factor blockers, various immunomodulating agents, and combination therapy with levocetirizine and hydroxyzine have demonstrated variable levels of efficacy, albeit often followed by high rates of relapse with drug discontinuation.6
The Diagnosis: Wells Syndrome
Histopathologic examination of the biopsy demonstrated overlying acanthosis, focal spongiosis, and exocytosis. There also was proliferation and thickening of superficial capillaries and papillary fibrosis (Figure, A). There was a mixed interstitial and perivascular inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and eosinophils (Figure, A and B). Occasional flame figures were identified (Figure, C).
Wells syndrome, also known as eosinophilic cellulitis, was first described in 1971 by Wells1 as a recurrent granulomatous dermatitis with eosinophilia. Rarely reported worldwide, this chronic relapsing condition is characterized by a pronounced eosinophilic infiltrate of the dermis resembling urticaria or cellulitis.2 The exact etiology has not been elucidated; however, links to certain medications, vaccines, exaggerated arthropod reactions, infections, and malignancies have been documented.3
Wells syndrome is a diagnosis of exclusion and lacks a predictable dermatologic presentation, thereby mandating focused clinical follow-up as well as correlation with histopathology findings. Although the classic histologic hallmark of Wells syndrome is scattered flame figures, this finding is not specific and can be found in other hypereosinophilic conditions.2 Clinical manifestations most often consist of 2 distinct phases: an initial painful burning or pruritic sensation, followed by the development of erythematous and edematous dermal plaques that may heal with slight hyperpigmentation over 4 to 8 weeks. A case series of 19 patients demonstrated variants of Wells syndrome, with an annular granuloma-like appearance found primarily in adults and the signature plaque-type appearance predominating in children.4
Acute urticaria is characterized by pruritic erythematous wheals secondary to a histamine-mediated response brought on by a variety of triggers, typically allergic and self-resolving within 24 hours. When such lesions last longer than 24 hours, biopsy should be performed to exclude urticarial vasculitis, which is characterized by a burning or painful sensation rather than pruritis, in addition to dermal neutrophilia and perivascular infiltrate on histology. Erythema migrans of Lyme disease begins at the site of a tick bite, evolving from a red macule to an expanding targetoid lesion and typically is not pruritic. Infectious cellulitis presents with warm, tender, and poorly defined erythematous patches; can progress rapidly; and is accompanied by systemic symptoms such as fevers, malaise, and lymphadenopathy.
Best evidence favors the use of moderate- to high-dose corticosteroids as first-line treatment.5 The use of tumor necrosis factor blockers, various immunomodulating agents, and combination therapy with levocetirizine and hydroxyzine have demonstrated variable levels of efficacy, albeit often followed by high rates of relapse with drug discontinuation.6
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc. 1971;57:46-56.
- Aberer W, Konrad K, Wolff K. Wells' syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol. 1988;18:105-114.
- Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983-1984.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Ferreli C, Pinna AL, Atzori L, et al. Eosinophilic cellulitis (Well's syndrome): a new case description. J Eur Acad Dermatol Venereol. 1999;13:41-45.
- Cormerais M, Poizeau F, Darrieux L, et al. Wells' syndrome mimicking facial cellulitis: a report of two cases. Case Rep Dermatol. 2015;7:117-122.
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc. 1971;57:46-56.
- Aberer W, Konrad K, Wolff K. Wells' syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol. 1988;18:105-114.
- Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983-1984.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Ferreli C, Pinna AL, Atzori L, et al. Eosinophilic cellulitis (Well's syndrome): a new case description. J Eur Acad Dermatol Venereol. 1999;13:41-45.
- Cormerais M, Poizeau F, Darrieux L, et al. Wells' syndrome mimicking facial cellulitis: a report of two cases. Case Rep Dermatol. 2015;7:117-122.
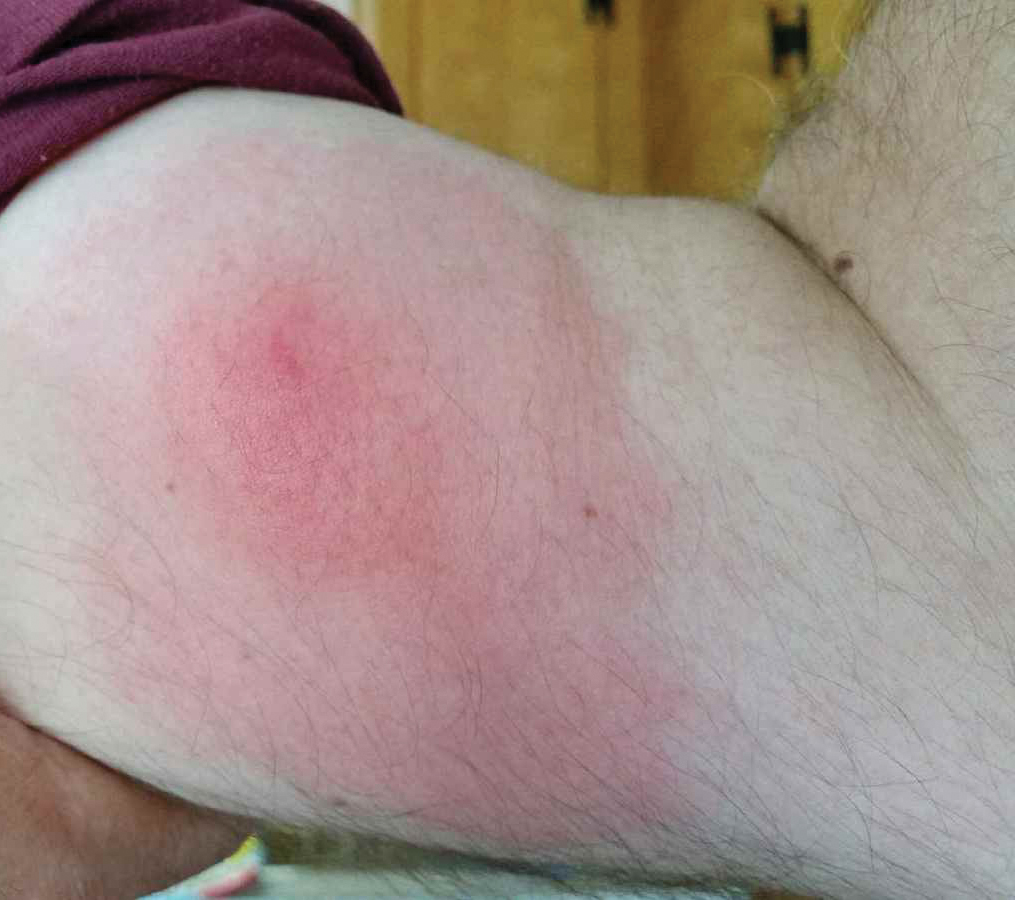
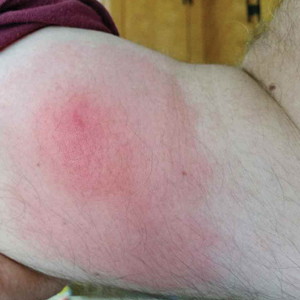
A 60-year-old man with a history of hyperlipidemia developed acute onset of an intensely pruritic and painful burning rash on the dorsal aspect of the left forearm of 8 days' duration. The patient described the rash as red and warm. It measured 2 cm at inception and peaked at 12 cm 6 months later when the patient presented. These symptoms resolved without therapeutic intervention.
Over the ensuing 6 months, he experienced 13 self-limited episodes of erythematous indurated cutaneous streaks, usually with proximal migration on the arms along with involvement of the posterior thorax and right leg. Five months prior to the onset of the initial rash, the patient had discontinued ezetimibe to treat hyperlipidemia due to swelling of the lips and tongue. He also reported that he regularly hunted in upstate Pennsylvania but reported no history of arthropod or animal bites. The patient did not take prescription or over-the-counter medications, and he denied the presence of fever, night sweats, fatigue, adenopathy, anorexia, weight loss, diarrhea, joint pain or swelling, or illicit drug use. Lyme titers, complete blood cell count, erythrocyte sedimentation rate, and comprehensive metabolic panel were within reference range. A punch biopsy was performed.
Rapidly Growing Cutaneous Nodules on the Scalp
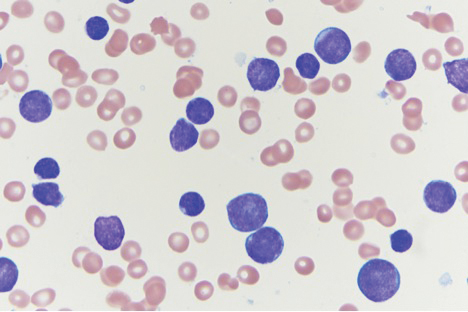
The Diagnosis: B-Cell Acute Lymphoblastic Leukemia
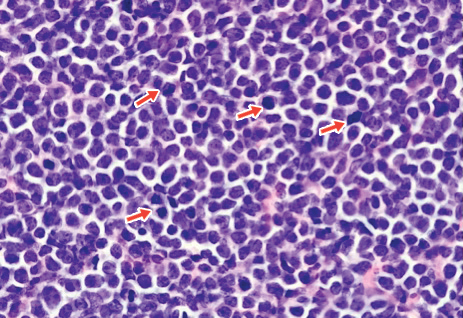
A 4-mm punch biopsy of one of the scalp lesions showed a diffuse infiltrate of intermediately sized cells with variably mature chromatin and irregular nuclear contours, consistent with a neoplastic process. Numerous mitotic figures were present, indicating a high proliferation rate (Figure 1). At that time there was no evidence of systemic involvement. A repeat biopsy with concurrent bone marrow biopsy was scheduled 10 days after the patient's initial presentation for further classification. Laboratory studies at that time revealed leukocytosis with elevated neutrophils and lymphocytes as well as a high absolute blast count.
On immunohistochemical staining, the neoplastic cells were positive for CD45, which indicated the neoplasm was hematopoietic, as well as CD10 and the B-cell antigens PAX-5 and CD79a. The cells were negative for CD20, which also is a B-cell marker, but this marker is only expressed in approximately half of pediatric acute lymphoblastic leukemia (ALL) cases with B-cell precursor origin.1 Markers that typically are expressed in B-cell acute lymphoblastic leukemia (B-ALL)--CD34 and terminal deoxynucleotidyl transferase--were both negative. These results were somewhat contradictory, and the differential remained open to both B-ALL and mature B-cell lymphoma. A bone marrow biopsy showed approximately 65% blasts or leukemic cells (Figure 2). Flow cytometry showed the cells were positive for CD10, CD19, weak CD79a, and variable lambda surface antigen expression. The cells were negative for expression of CD20, CD34, terminal deoxynucleotidyl transferase, myeloid antigens, and CD3. Ultimately, the morphology and immunophenotype were most consistent with a diagnosis of B-ALL. Fluorescence in situ hybridization revealed mixed lineage leukemia, MLL, gene rearrangements.
In general, when considering the differential diagnosis of superficial nodules, 5 elements are helpful to consider: the number of nodules (single vs multiple); the location; and the presence or absence of tenderness, pigmentation or erythema, and firmness.2 Our patient had multiple nodules on the scalp, which were erythematous to slightly purple and firm. The differential diagnosis can be categorized into malignant; infectious; and benign inflammatory, vascular, and fibrous tumors.
Potential oncologic processes include leukemia cutis, lymphoblastic leukemia/lymphoma, Langerhans cell histiocytosis, and rhabdomyosarcoma. Initial laboratory test results were reassuring. Infectious processes in the differential include deep fungal infections such as coccidioidomycosis and nontuberculous mycobacterial infections. Coccidioidomycosis was the most likely to cause skin lesions or masses in our patient; however, it was considered less likely because the patient's family had not traveled or been exposed to an endemic area.3
Benign tumors in the differential include deep hemangioma, which was deemed less likely in our patient because most hemangiomas reach 80% of their maximum size by 5 months of age.4 Another possible benign tumor is infantile myofibromatosis, which is rare but is the most common fibrous tumor of infancy.5
Early-onset childhood sarcoidosis also has been shown to produce multiple nontender firm nodules.2 This process was considered unlikely in our patient because not only is the disease relatively rare in the pediatric population, but most reported childhood cases have occurred in patients aged 13 to 15 years.6 Additionally, no uveitis or arthritis was observed in this case.
Ultimately, histopathology and bone marrow biopsy were necessary to determine the diagnosis of B-ALL. Although uncommon, cutaneous involvement can be an early sign of ALL in children.7 Thus, neoplastic etiologies should be considered in the workup of cutaneous nodules in children, especially when these nodules are hard, rapidly growing, ulcerated, fixed, and/or vascular.8 Once the diagnosis is established, initial workup of ALL in children should include complete blood cell count with manual differential, prothrombin time, partial thromboplastin time, electrolytes, uric acid, and renal and liver function tests. Often, baseline viral titers such as cytomegalovirus, Epstein-Barr virus, human immunodeficiency virus, hepatitis B virus, and varicella-zoster virus also are included. Patients are risk stratified to the appropriate level of treatment based on tumor immunophenotype, cytogenetic findings, patient age, white blood cell count at the time of diagnosis, and response to initial therapy. Treatment typically is comprised of a multidrug regimen divided into several phases--induction, consolidation, and maintenance--as well as therapy directed to the central nervous system. Treatment protocols usually take 2 to 3 years to complete.
Our patient was treated with 1 dose of intrathecal methotrexate before starting the Interfant-06 protocol with a 7-day methylprednisolone prophase. The patient's nodules shrank over time and were no longer present after 14 days of treatment.
- Dworzak MN, Schumich A, Printz D, et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed immunotherapy. Blood. 2008;112:3982-3988.
- Whelan JP, Zembowicz A. Case records of the Massachusetts General Hospital. case 19-2006. a 22-month-old boy with the rapid growth of subcutaneous nodules. N Engl J Med. 2006;354:2697-2704.
- Malo J, Luraschi-Monjagatta C, Wolk DM, et al. Update on the diagnosis of pulmonary coccidioidomycosis. Ann Am Thorac Soc. 2014;11:243-253.
- Chang LC, Haggstrom AN, Drolet BA, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360-367.
- Schurr P, Moulsdale W. Infantile myofibroma. Adv Neonatal Care. 2008;8:13-20.
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16.
- Millot F, Robert A, Bertrand Y, et al. Cutaneous involvement in children with acute lymphoblastic leukemia or lymphoblastic lymphoma. The Children's Leukemia Cooperative Group of the European Organization of Research and Treatment of Cancer (EORTC). Pediatrics. 1997;100:60-64.
- Fogelson S, Dohil M. Papular and nodular skin lesions in children. Semin Plast Surg. 2006;20:180-191.
The Diagnosis: B-Cell Acute Lymphoblastic Leukemia
A 4-mm punch biopsy of one of the scalp lesions showed a diffuse infiltrate of intermediately sized cells with variably mature chromatin and irregular nuclear contours, consistent with a neoplastic process. Numerous mitotic figures were present, indicating a high proliferation rate (Figure 1). At that time there was no evidence of systemic involvement. A repeat biopsy with concurrent bone marrow biopsy was scheduled 10 days after the patient's initial presentation for further classification. Laboratory studies at that time revealed leukocytosis with elevated neutrophils and lymphocytes as well as a high absolute blast count.
On immunohistochemical staining, the neoplastic cells were positive for CD45, which indicated the neoplasm was hematopoietic, as well as CD10 and the B-cell antigens PAX-5 and CD79a. The cells were negative for CD20, which also is a B-cell marker, but this marker is only expressed in approximately half of pediatric acute lymphoblastic leukemia (ALL) cases with B-cell precursor origin.1 Markers that typically are expressed in B-cell acute lymphoblastic leukemia (B-ALL)--CD34 and terminal deoxynucleotidyl transferase--were both negative. These results were somewhat contradictory, and the differential remained open to both B-ALL and mature B-cell lymphoma. A bone marrow biopsy showed approximately 65% blasts or leukemic cells (Figure 2). Flow cytometry showed the cells were positive for CD10, CD19, weak CD79a, and variable lambda surface antigen expression. The cells were negative for expression of CD20, CD34, terminal deoxynucleotidyl transferase, myeloid antigens, and CD3. Ultimately, the morphology and immunophenotype were most consistent with a diagnosis of B-ALL. Fluorescence in situ hybridization revealed mixed lineage leukemia, MLL, gene rearrangements.
In general, when considering the differential diagnosis of superficial nodules, 5 elements are helpful to consider: the number of nodules (single vs multiple); the location; and the presence or absence of tenderness, pigmentation or erythema, and firmness.2 Our patient had multiple nodules on the scalp, which were erythematous to slightly purple and firm. The differential diagnosis can be categorized into malignant; infectious; and benign inflammatory, vascular, and fibrous tumors.
Potential oncologic processes include leukemia cutis, lymphoblastic leukemia/lymphoma, Langerhans cell histiocytosis, and rhabdomyosarcoma. Initial laboratory test results were reassuring. Infectious processes in the differential include deep fungal infections such as coccidioidomycosis and nontuberculous mycobacterial infections. Coccidioidomycosis was the most likely to cause skin lesions or masses in our patient; however, it was considered less likely because the patient's family had not traveled or been exposed to an endemic area.3
Benign tumors in the differential include deep hemangioma, which was deemed less likely in our patient because most hemangiomas reach 80% of their maximum size by 5 months of age.4 Another possible benign tumor is infantile myofibromatosis, which is rare but is the most common fibrous tumor of infancy.5
Early-onset childhood sarcoidosis also has been shown to produce multiple nontender firm nodules.2 This process was considered unlikely in our patient because not only is the disease relatively rare in the pediatric population, but most reported childhood cases have occurred in patients aged 13 to 15 years.6 Additionally, no uveitis or arthritis was observed in this case.
Ultimately, histopathology and bone marrow biopsy were necessary to determine the diagnosis of B-ALL. Although uncommon, cutaneous involvement can be an early sign of ALL in children.7 Thus, neoplastic etiologies should be considered in the workup of cutaneous nodules in children, especially when these nodules are hard, rapidly growing, ulcerated, fixed, and/or vascular.8 Once the diagnosis is established, initial workup of ALL in children should include complete blood cell count with manual differential, prothrombin time, partial thromboplastin time, electrolytes, uric acid, and renal and liver function tests. Often, baseline viral titers such as cytomegalovirus, Epstein-Barr virus, human immunodeficiency virus, hepatitis B virus, and varicella-zoster virus also are included. Patients are risk stratified to the appropriate level of treatment based on tumor immunophenotype, cytogenetic findings, patient age, white blood cell count at the time of diagnosis, and response to initial therapy. Treatment typically is comprised of a multidrug regimen divided into several phases--induction, consolidation, and maintenance--as well as therapy directed to the central nervous system. Treatment protocols usually take 2 to 3 years to complete.
Our patient was treated with 1 dose of intrathecal methotrexate before starting the Interfant-06 protocol with a 7-day methylprednisolone prophase. The patient's nodules shrank over time and were no longer present after 14 days of treatment.
The Diagnosis: B-Cell Acute Lymphoblastic Leukemia
A 4-mm punch biopsy of one of the scalp lesions showed a diffuse infiltrate of intermediately sized cells with variably mature chromatin and irregular nuclear contours, consistent with a neoplastic process. Numerous mitotic figures were present, indicating a high proliferation rate (Figure 1). At that time there was no evidence of systemic involvement. A repeat biopsy with concurrent bone marrow biopsy was scheduled 10 days after the patient's initial presentation for further classification. Laboratory studies at that time revealed leukocytosis with elevated neutrophils and lymphocytes as well as a high absolute blast count.
On immunohistochemical staining, the neoplastic cells were positive for CD45, which indicated the neoplasm was hematopoietic, as well as CD10 and the B-cell antigens PAX-5 and CD79a. The cells were negative for CD20, which also is a B-cell marker, but this marker is only expressed in approximately half of pediatric acute lymphoblastic leukemia (ALL) cases with B-cell precursor origin.1 Markers that typically are expressed in B-cell acute lymphoblastic leukemia (B-ALL)--CD34 and terminal deoxynucleotidyl transferase--were both negative. These results were somewhat contradictory, and the differential remained open to both B-ALL and mature B-cell lymphoma. A bone marrow biopsy showed approximately 65% blasts or leukemic cells (Figure 2). Flow cytometry showed the cells were positive for CD10, CD19, weak CD79a, and variable lambda surface antigen expression. The cells were negative for expression of CD20, CD34, terminal deoxynucleotidyl transferase, myeloid antigens, and CD3. Ultimately, the morphology and immunophenotype were most consistent with a diagnosis of B-ALL. Fluorescence in situ hybridization revealed mixed lineage leukemia, MLL, gene rearrangements.
In general, when considering the differential diagnosis of superficial nodules, 5 elements are helpful to consider: the number of nodules (single vs multiple); the location; and the presence or absence of tenderness, pigmentation or erythema, and firmness.2 Our patient had multiple nodules on the scalp, which were erythematous to slightly purple and firm. The differential diagnosis can be categorized into malignant; infectious; and benign inflammatory, vascular, and fibrous tumors.
Potential oncologic processes include leukemia cutis, lymphoblastic leukemia/lymphoma, Langerhans cell histiocytosis, and rhabdomyosarcoma. Initial laboratory test results were reassuring. Infectious processes in the differential include deep fungal infections such as coccidioidomycosis and nontuberculous mycobacterial infections. Coccidioidomycosis was the most likely to cause skin lesions or masses in our patient; however, it was considered less likely because the patient's family had not traveled or been exposed to an endemic area.3
Benign tumors in the differential include deep hemangioma, which was deemed less likely in our patient because most hemangiomas reach 80% of their maximum size by 5 months of age.4 Another possible benign tumor is infantile myofibromatosis, which is rare but is the most common fibrous tumor of infancy.5
Early-onset childhood sarcoidosis also has been shown to produce multiple nontender firm nodules.2 This process was considered unlikely in our patient because not only is the disease relatively rare in the pediatric population, but most reported childhood cases have occurred in patients aged 13 to 15 years.6 Additionally, no uveitis or arthritis was observed in this case.
Ultimately, histopathology and bone marrow biopsy were necessary to determine the diagnosis of B-ALL. Although uncommon, cutaneous involvement can be an early sign of ALL in children.7 Thus, neoplastic etiologies should be considered in the workup of cutaneous nodules in children, especially when these nodules are hard, rapidly growing, ulcerated, fixed, and/or vascular.8 Once the diagnosis is established, initial workup of ALL in children should include complete blood cell count with manual differential, prothrombin time, partial thromboplastin time, electrolytes, uric acid, and renal and liver function tests. Often, baseline viral titers such as cytomegalovirus, Epstein-Barr virus, human immunodeficiency virus, hepatitis B virus, and varicella-zoster virus also are included. Patients are risk stratified to the appropriate level of treatment based on tumor immunophenotype, cytogenetic findings, patient age, white blood cell count at the time of diagnosis, and response to initial therapy. Treatment typically is comprised of a multidrug regimen divided into several phases--induction, consolidation, and maintenance--as well as therapy directed to the central nervous system. Treatment protocols usually take 2 to 3 years to complete.
Our patient was treated with 1 dose of intrathecal methotrexate before starting the Interfant-06 protocol with a 7-day methylprednisolone prophase. The patient's nodules shrank over time and were no longer present after 14 days of treatment.
- Dworzak MN, Schumich A, Printz D, et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed immunotherapy. Blood. 2008;112:3982-3988.
- Whelan JP, Zembowicz A. Case records of the Massachusetts General Hospital. case 19-2006. a 22-month-old boy with the rapid growth of subcutaneous nodules. N Engl J Med. 2006;354:2697-2704.
- Malo J, Luraschi-Monjagatta C, Wolk DM, et al. Update on the diagnosis of pulmonary coccidioidomycosis. Ann Am Thorac Soc. 2014;11:243-253.
- Chang LC, Haggstrom AN, Drolet BA, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360-367.
- Schurr P, Moulsdale W. Infantile myofibroma. Adv Neonatal Care. 2008;8:13-20.
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16.
- Millot F, Robert A, Bertrand Y, et al. Cutaneous involvement in children with acute lymphoblastic leukemia or lymphoblastic lymphoma. The Children's Leukemia Cooperative Group of the European Organization of Research and Treatment of Cancer (EORTC). Pediatrics. 1997;100:60-64.
- Fogelson S, Dohil M. Papular and nodular skin lesions in children. Semin Plast Surg. 2006;20:180-191.
- Dworzak MN, Schumich A, Printz D, et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed immunotherapy. Blood. 2008;112:3982-3988.
- Whelan JP, Zembowicz A. Case records of the Massachusetts General Hospital. case 19-2006. a 22-month-old boy with the rapid growth of subcutaneous nodules. N Engl J Med. 2006;354:2697-2704.
- Malo J, Luraschi-Monjagatta C, Wolk DM, et al. Update on the diagnosis of pulmonary coccidioidomycosis. Ann Am Thorac Soc. 2014;11:243-253.
- Chang LC, Haggstrom AN, Drolet BA, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360-367.
- Schurr P, Moulsdale W. Infantile myofibroma. Adv Neonatal Care. 2008;8:13-20.
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16.
- Millot F, Robert A, Bertrand Y, et al. Cutaneous involvement in children with acute lymphoblastic leukemia or lymphoblastic lymphoma. The Children's Leukemia Cooperative Group of the European Organization of Research and Treatment of Cancer (EORTC). Pediatrics. 1997;100:60-64.
- Fogelson S, Dohil M. Papular and nodular skin lesions in children. Semin Plast Surg. 2006;20:180-191.
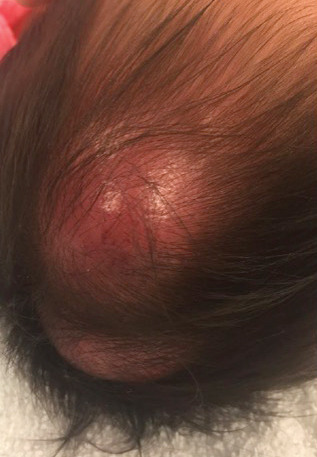
An 8-month-old infant girl presented with rapidly growing cutaneous nodules on the scalp of 1 month's duration. Her parents reported that she disliked lying flat but was otherwise growing and developing normally. Nondiagnostic ultrasonography of the head and brain had been performed as well as a skull radiograph, which found no evidence of lytic lesions. On physical examination, 3 erythematous to violaceous, subcutaneous, firm, fixed nodules were observed on the scalp. Notable cervical lymphadenopathy with several distinct, fixed, firm, subcutaneous nodules in the postauricular lymph chains also were noted. The patient had no pertinent medical history and was born via normal spontaneous vaginal delivery to healthy parents. The remainder of the physical examination and review of systems was negative.
Painless Nodule on the Leg
The Diagnosis: Plasmablastic Lymphoma
Histopathologic examination revealed a diffuse dense proliferation of large, atypical, and pleomorphic mononuclear cells with prominent nucleoli and many mitotic figures representing plasmacytoid cells in the dermis (Figure). Immunostaining was positive for MUM-1 (marker of late-stage plasma cells and activated T cells) and BCL-2 (antiapoptotic marker). Fluorescent polymerase chain reaction was positive for clonal IgH gene arrangement, and fluorescence in situ hybridization was positive for C-MYC rearrangement in 94% of cells. Epstein-Barr encoding region in situ hybridization also was positive. Rare cells stained positive for T-cell markers. CD20, BCL-6, and CD30 immunostains were negative, suggesting that these cells were not B or T cells, though terminally differentiated B cells also can lack these markers. Bone marrow biopsy showed a similar staining pattern to the skin with 10% atypical plasmacytoid cells. Computed tomography of the left leg showed an enlargement of the semimembranosus muscle with internal areas of high density and heterogeneous enhancement. The patient underwent decompression of the left peroneal nerve. Biopsy showed a staining pattern similar to the right skin nodule and bone marrow, consistent with lymphoma.
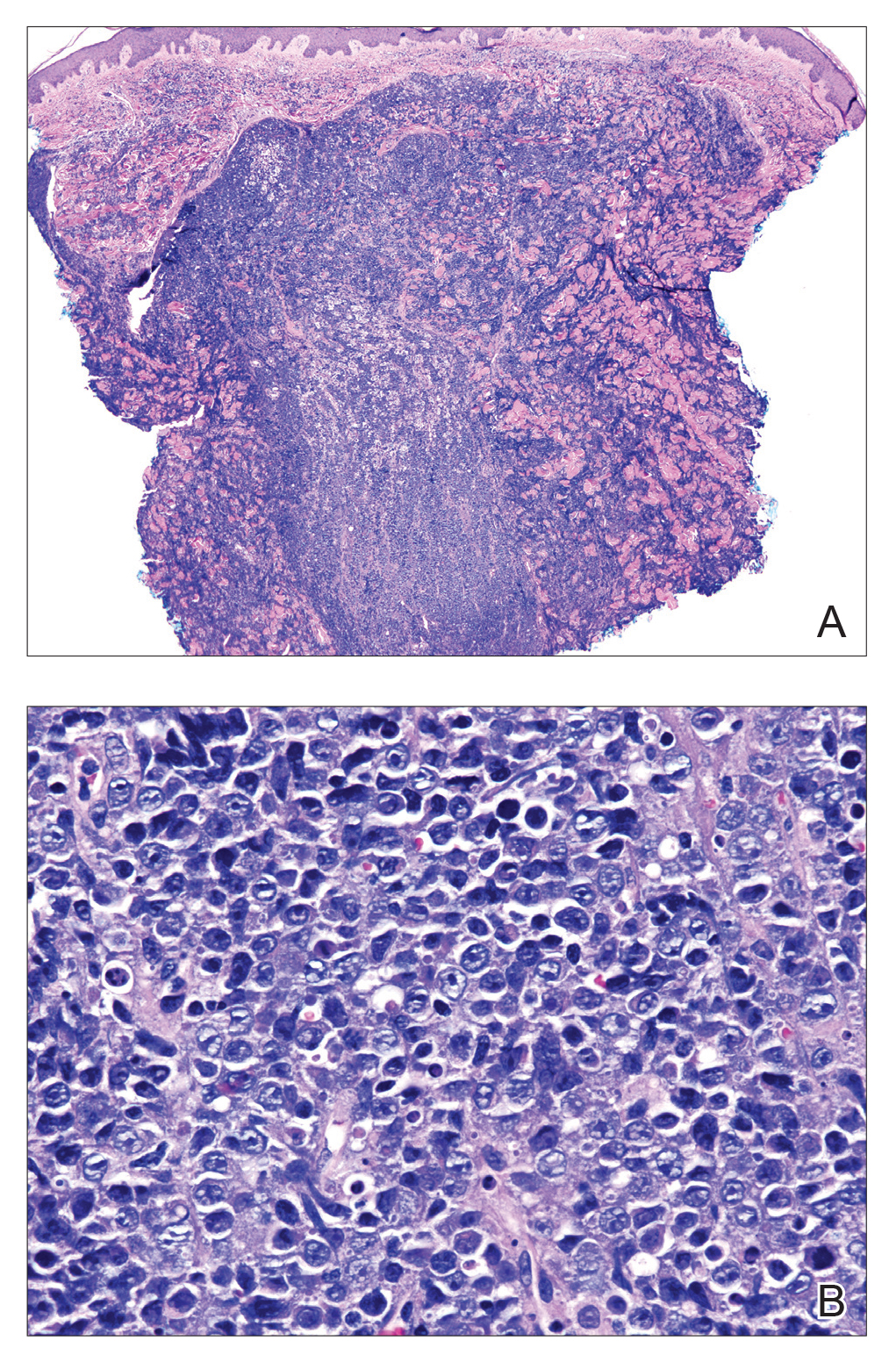
He was diagnosed with stage IV human immunodeficiency virus (HIV)-associated plasmablastic lymphoma (PBL) and received 6 cycles of R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) without vincristine with intrathecal methotrexate, followed by 3 cycles of DHAP (dexamethasone, high dose Ara C, cisplatin) with bortezomib and daratumumab after relapse. Ultimately, he underwent autologous stem cell transplantation and was alive 13 months after diagnosis.
Plasmablastic lymphoma is a rare subtype of non-Hodgkin lymphoma that most commonly arises in the oral cavity of individuals with HIV.1 In addition to HIV infection, PBL also is seen in patients with other causes of immunodeficiency such as iatrogenic immunosuppression following solid organ transplantation.1 The typical disease presentation is an expanding mass in the oral cavity; however, 34% (52/151) of reported cases arose at extraoral primary sites, with a minority of cases confined to cutaneous sites with no systemic involvement.2 Cutaneous PBL presentations may include flesh-colored or purple, grouped or solitary nodules; an erythematous infiltrated plaque; or purple-red ulcerated nodules. The lesions usually are asymptomatic and located on the arms and legs.3
On histologic examination, PBL is characterized by a diffuse monomorphic lymphoid infiltrate that sometimes invades the surrounding soft tissue.4-6 The neoplastic cells have eccentric round nucleoli. Plasmablastic lymphoma characteristically displays a high proliferation index with many mitotic figures and signs of apoptosis.4-6 Definitive diagnosis requires immunohistochemical staining. Typical B-cell antigens (CD20) as well as CD45 are negative, while plasma cell markers such as CD38 are positive. Other B- and T-cell markers usually are negative.5,7 The pathogenesis of PBL is thought to be related to Epstein-Barr virus or human herpesvirus 8 infection. In a series of PBL cases, Epstein-Barr virus and human herpesvirus 8 was positive in 75% (97/129) and 17% (13/75) of tested cases, respectively.1
The prognosis for PBL is poor, with a median overall survival of 15 months and a 3-year survival rate of 25% in HIV-infected individuals.8 However, cutaneous PBL without systemic involvement has a considerably better prognosis, with only 1 of 12 cases resulting in death.2,3,9 Treatment of PBL depends on the extent of the disease. Cutaneous PBL can be treated with surgery and adjuvant radiation.3 Chemotherapy is required for patients with multiple lesions or systemic involvement. Current treatment regimens are similar to those used for other aggressive lymphomas such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone).1 Transplant recipients should have their immunosuppression reduced, and HIV-infected patients should have their highly active antiretroviral therapy regimens optimized. Patients presenting with PBL without HIV should be tested for HIV, as PBL has previously been reported to be the presenting manifestation of HIV infection.10
The differential diagnosis for a rapidly expanding, vascular-appearing, red mass on the legs in an immunosuppressed individual includes abscess, malignancy, Kaposi sarcoma, Sweet syndrome, and tertiary syphilis.
Acknowledgment
We thank Sameera Husain, MD (New York, New York), for her assistance with histopathologic photographs and interpretation.
- Riedel DJ, Gonzalez-Cuyar LF, Zhao XF, et al. Plasmablastic lymphoma of the oral cavity: a rapidly progressive lymphoma associated with HIV infection. Lancet Infect Dis. 2008;8:261-267.
- Heiser D, Müller H, Kempf W, et al. Primary cutaneous plasmablastic lymphoma of the lower leg in an HIV-negative patient. J Am Acad Dermatol. 2012;67:E202-E205.
- Jambusaria A, Shafer D, Wu H, et al. Cutaneous plasmablastic lymphoma. J Am Acad Dermatol. 2008;58:676-678.
- Delecluse HJ, Anagnostopoulos I, Dallenbach F, et al. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413-1420.
- Gaidano G, Cerri M, Capello D, et al. Molecular histogenesis of plasmablastic lymphoma of the oral cavity. Br J Haematol. 2002;119:622-628.
- Folk GS, Abbondanzo SL, Childers EL, et al. Plasmablastic lymphoma: a clinicopathologic correlation. Ann Diagn Pathol. 2006;10:8-12.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Castillo J, Pantanowitz L, Dezube BJ. HIV-associated plasmablastic lymphoma: lessons learned from 112 published cases. Am J Hematol. 2008;83:804-809.
- Horna P, Hamill JR, Sokol L, et al. Primary cutaneous plasmablastic lymphoma in an immunocompetent patient. J Am Acad Dermatol. 2013;69:E274-E276.
- Desai RS, Vanaki SS, Puranik RS, et al. Plasmablastic lymphoma presenting as a gingival growth in a previously undiagnosed HIV-positive patient: a case report. J Oral Maxillofac Surg. 2007;65:1358-1361.
The Diagnosis: Plasmablastic Lymphoma
Histopathologic examination revealed a diffuse dense proliferation of large, atypical, and pleomorphic mononuclear cells with prominent nucleoli and many mitotic figures representing plasmacytoid cells in the dermis (Figure). Immunostaining was positive for MUM-1 (marker of late-stage plasma cells and activated T cells) and BCL-2 (antiapoptotic marker). Fluorescent polymerase chain reaction was positive for clonal IgH gene arrangement, and fluorescence in situ hybridization was positive for C-MYC rearrangement in 94% of cells. Epstein-Barr encoding region in situ hybridization also was positive. Rare cells stained positive for T-cell markers. CD20, BCL-6, and CD30 immunostains were negative, suggesting that these cells were not B or T cells, though terminally differentiated B cells also can lack these markers. Bone marrow biopsy showed a similar staining pattern to the skin with 10% atypical plasmacytoid cells. Computed tomography of the left leg showed an enlargement of the semimembranosus muscle with internal areas of high density and heterogeneous enhancement. The patient underwent decompression of the left peroneal nerve. Biopsy showed a staining pattern similar to the right skin nodule and bone marrow, consistent with lymphoma.
He was diagnosed with stage IV human immunodeficiency virus (HIV)-associated plasmablastic lymphoma (PBL) and received 6 cycles of R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) without vincristine with intrathecal methotrexate, followed by 3 cycles of DHAP (dexamethasone, high dose Ara C, cisplatin) with bortezomib and daratumumab after relapse. Ultimately, he underwent autologous stem cell transplantation and was alive 13 months after diagnosis.
Plasmablastic lymphoma is a rare subtype of non-Hodgkin lymphoma that most commonly arises in the oral cavity of individuals with HIV.1 In addition to HIV infection, PBL also is seen in patients with other causes of immunodeficiency such as iatrogenic immunosuppression following solid organ transplantation.1 The typical disease presentation is an expanding mass in the oral cavity; however, 34% (52/151) of reported cases arose at extraoral primary sites, with a minority of cases confined to cutaneous sites with no systemic involvement.2 Cutaneous PBL presentations may include flesh-colored or purple, grouped or solitary nodules; an erythematous infiltrated plaque; or purple-red ulcerated nodules. The lesions usually are asymptomatic and located on the arms and legs.3
On histologic examination, PBL is characterized by a diffuse monomorphic lymphoid infiltrate that sometimes invades the surrounding soft tissue.4-6 The neoplastic cells have eccentric round nucleoli. Plasmablastic lymphoma characteristically displays a high proliferation index with many mitotic figures and signs of apoptosis.4-6 Definitive diagnosis requires immunohistochemical staining. Typical B-cell antigens (CD20) as well as CD45 are negative, while plasma cell markers such as CD38 are positive. Other B- and T-cell markers usually are negative.5,7 The pathogenesis of PBL is thought to be related to Epstein-Barr virus or human herpesvirus 8 infection. In a series of PBL cases, Epstein-Barr virus and human herpesvirus 8 was positive in 75% (97/129) and 17% (13/75) of tested cases, respectively.1
The prognosis for PBL is poor, with a median overall survival of 15 months and a 3-year survival rate of 25% in HIV-infected individuals.8 However, cutaneous PBL without systemic involvement has a considerably better prognosis, with only 1 of 12 cases resulting in death.2,3,9 Treatment of PBL depends on the extent of the disease. Cutaneous PBL can be treated with surgery and adjuvant radiation.3 Chemotherapy is required for patients with multiple lesions or systemic involvement. Current treatment regimens are similar to those used for other aggressive lymphomas such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone).1 Transplant recipients should have their immunosuppression reduced, and HIV-infected patients should have their highly active antiretroviral therapy regimens optimized. Patients presenting with PBL without HIV should be tested for HIV, as PBL has previously been reported to be the presenting manifestation of HIV infection.10
The differential diagnosis for a rapidly expanding, vascular-appearing, red mass on the legs in an immunosuppressed individual includes abscess, malignancy, Kaposi sarcoma, Sweet syndrome, and tertiary syphilis.
Acknowledgment
We thank Sameera Husain, MD (New York, New York), for her assistance with histopathologic photographs and interpretation.
The Diagnosis: Plasmablastic Lymphoma
Histopathologic examination revealed a diffuse dense proliferation of large, atypical, and pleomorphic mononuclear cells with prominent nucleoli and many mitotic figures representing plasmacytoid cells in the dermis (Figure). Immunostaining was positive for MUM-1 (marker of late-stage plasma cells and activated T cells) and BCL-2 (antiapoptotic marker). Fluorescent polymerase chain reaction was positive for clonal IgH gene arrangement, and fluorescence in situ hybridization was positive for C-MYC rearrangement in 94% of cells. Epstein-Barr encoding region in situ hybridization also was positive. Rare cells stained positive for T-cell markers. CD20, BCL-6, and CD30 immunostains were negative, suggesting that these cells were not B or T cells, though terminally differentiated B cells also can lack these markers. Bone marrow biopsy showed a similar staining pattern to the skin with 10% atypical plasmacytoid cells. Computed tomography of the left leg showed an enlargement of the semimembranosus muscle with internal areas of high density and heterogeneous enhancement. The patient underwent decompression of the left peroneal nerve. Biopsy showed a staining pattern similar to the right skin nodule and bone marrow, consistent with lymphoma.
He was diagnosed with stage IV human immunodeficiency virus (HIV)-associated plasmablastic lymphoma (PBL) and received 6 cycles of R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) without vincristine with intrathecal methotrexate, followed by 3 cycles of DHAP (dexamethasone, high dose Ara C, cisplatin) with bortezomib and daratumumab after relapse. Ultimately, he underwent autologous stem cell transplantation and was alive 13 months after diagnosis.
Plasmablastic lymphoma is a rare subtype of non-Hodgkin lymphoma that most commonly arises in the oral cavity of individuals with HIV.1 In addition to HIV infection, PBL also is seen in patients with other causes of immunodeficiency such as iatrogenic immunosuppression following solid organ transplantation.1 The typical disease presentation is an expanding mass in the oral cavity; however, 34% (52/151) of reported cases arose at extraoral primary sites, with a minority of cases confined to cutaneous sites with no systemic involvement.2 Cutaneous PBL presentations may include flesh-colored or purple, grouped or solitary nodules; an erythematous infiltrated plaque; or purple-red ulcerated nodules. The lesions usually are asymptomatic and located on the arms and legs.3
On histologic examination, PBL is characterized by a diffuse monomorphic lymphoid infiltrate that sometimes invades the surrounding soft tissue.4-6 The neoplastic cells have eccentric round nucleoli. Plasmablastic lymphoma characteristically displays a high proliferation index with many mitotic figures and signs of apoptosis.4-6 Definitive diagnosis requires immunohistochemical staining. Typical B-cell antigens (CD20) as well as CD45 are negative, while plasma cell markers such as CD38 are positive. Other B- and T-cell markers usually are negative.5,7 The pathogenesis of PBL is thought to be related to Epstein-Barr virus or human herpesvirus 8 infection. In a series of PBL cases, Epstein-Barr virus and human herpesvirus 8 was positive in 75% (97/129) and 17% (13/75) of tested cases, respectively.1
The prognosis for PBL is poor, with a median overall survival of 15 months and a 3-year survival rate of 25% in HIV-infected individuals.8 However, cutaneous PBL without systemic involvement has a considerably better prognosis, with only 1 of 12 cases resulting in death.2,3,9 Treatment of PBL depends on the extent of the disease. Cutaneous PBL can be treated with surgery and adjuvant radiation.3 Chemotherapy is required for patients with multiple lesions or systemic involvement. Current treatment regimens are similar to those used for other aggressive lymphomas such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone).1 Transplant recipients should have their immunosuppression reduced, and HIV-infected patients should have their highly active antiretroviral therapy regimens optimized. Patients presenting with PBL without HIV should be tested for HIV, as PBL has previously been reported to be the presenting manifestation of HIV infection.10
The differential diagnosis for a rapidly expanding, vascular-appearing, red mass on the legs in an immunosuppressed individual includes abscess, malignancy, Kaposi sarcoma, Sweet syndrome, and tertiary syphilis.
Acknowledgment
We thank Sameera Husain, MD (New York, New York), for her assistance with histopathologic photographs and interpretation.
- Riedel DJ, Gonzalez-Cuyar LF, Zhao XF, et al. Plasmablastic lymphoma of the oral cavity: a rapidly progressive lymphoma associated with HIV infection. Lancet Infect Dis. 2008;8:261-267.
- Heiser D, Müller H, Kempf W, et al. Primary cutaneous plasmablastic lymphoma of the lower leg in an HIV-negative patient. J Am Acad Dermatol. 2012;67:E202-E205.
- Jambusaria A, Shafer D, Wu H, et al. Cutaneous plasmablastic lymphoma. J Am Acad Dermatol. 2008;58:676-678.
- Delecluse HJ, Anagnostopoulos I, Dallenbach F, et al. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413-1420.
- Gaidano G, Cerri M, Capello D, et al. Molecular histogenesis of plasmablastic lymphoma of the oral cavity. Br J Haematol. 2002;119:622-628.
- Folk GS, Abbondanzo SL, Childers EL, et al. Plasmablastic lymphoma: a clinicopathologic correlation. Ann Diagn Pathol. 2006;10:8-12.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Castillo J, Pantanowitz L, Dezube BJ. HIV-associated plasmablastic lymphoma: lessons learned from 112 published cases. Am J Hematol. 2008;83:804-809.
- Horna P, Hamill JR, Sokol L, et al. Primary cutaneous plasmablastic lymphoma in an immunocompetent patient. J Am Acad Dermatol. 2013;69:E274-E276.
- Desai RS, Vanaki SS, Puranik RS, et al. Plasmablastic lymphoma presenting as a gingival growth in a previously undiagnosed HIV-positive patient: a case report. J Oral Maxillofac Surg. 2007;65:1358-1361.
- Riedel DJ, Gonzalez-Cuyar LF, Zhao XF, et al. Plasmablastic lymphoma of the oral cavity: a rapidly progressive lymphoma associated with HIV infection. Lancet Infect Dis. 2008;8:261-267.
- Heiser D, Müller H, Kempf W, et al. Primary cutaneous plasmablastic lymphoma of the lower leg in an HIV-negative patient. J Am Acad Dermatol. 2012;67:E202-E205.
- Jambusaria A, Shafer D, Wu H, et al. Cutaneous plasmablastic lymphoma. J Am Acad Dermatol. 2008;58:676-678.
- Delecluse HJ, Anagnostopoulos I, Dallenbach F, et al. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413-1420.
- Gaidano G, Cerri M, Capello D, et al. Molecular histogenesis of plasmablastic lymphoma of the oral cavity. Br J Haematol. 2002;119:622-628.
- Folk GS, Abbondanzo SL, Childers EL, et al. Plasmablastic lymphoma: a clinicopathologic correlation. Ann Diagn Pathol. 2006;10:8-12.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Castillo J, Pantanowitz L, Dezube BJ. HIV-associated plasmablastic lymphoma: lessons learned from 112 published cases. Am J Hematol. 2008;83:804-809.
- Horna P, Hamill JR, Sokol L, et al. Primary cutaneous plasmablastic lymphoma in an immunocompetent patient. J Am Acad Dermatol. 2013;69:E274-E276.
- Desai RS, Vanaki SS, Puranik RS, et al. Plasmablastic lymphoma presenting as a gingival growth in a previously undiagnosed HIV-positive patient: a case report. J Oral Maxillofac Surg. 2007;65:1358-1361.
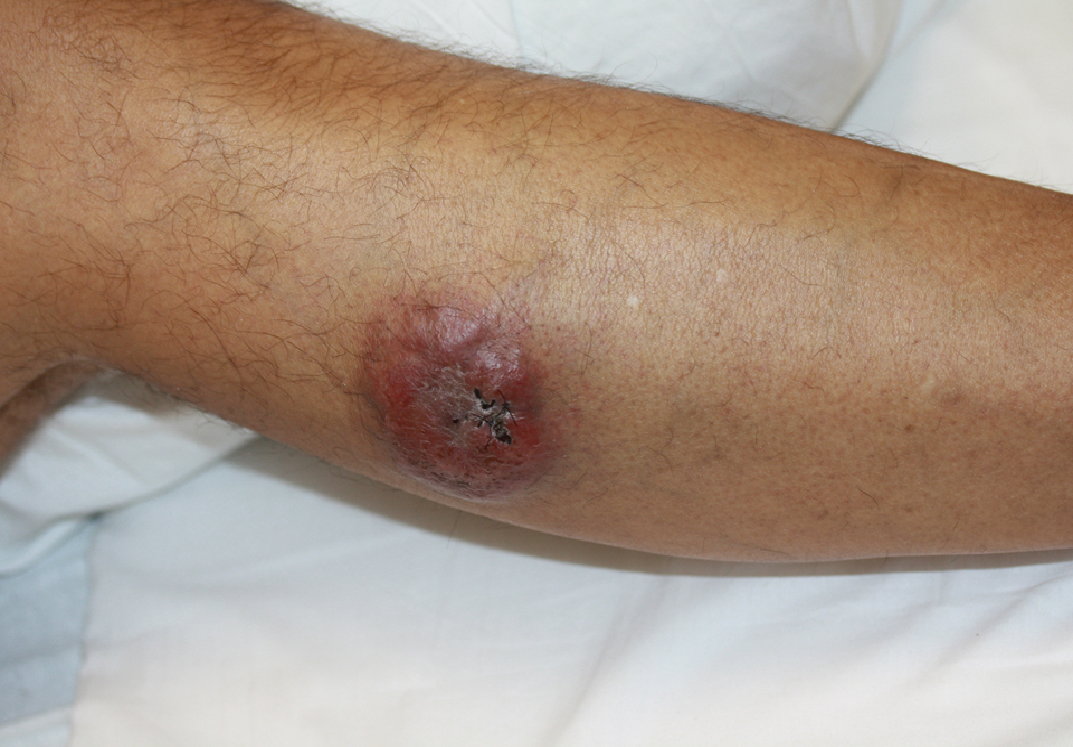
A 44-year-old man presented with numbness and a burning sensation of the left lateral leg and dorsal foot of 3 days' duration as well as a left foot drop of 1 day's duration. A painless red nodule on the right shin also developed over a 10-day period. He had been diagnosed with human immunodeficiency virus a year prior and reported compliance with antiretroviral therapy. There was a newly identified, well-demarcated, 6-cm, round, red-purple, flat-topped, nodular tumor with central depression on the right lateral shin. Ultrasonography of the nodule revealed a heterogeneous septate structure with increased vascularity. There was no regional or generalized lymphadenopathy. Laboratory values were notable for microcytic anemia. The white blood cell count was within reference range. Human immunodeficiency virus RNA viral load was elevated (3183 viral copies/mL [reference range, <20 viral copies/mL]). Two punch biopsies of the nodule were performed.
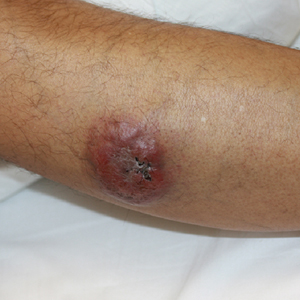
Linear Vulvar Lesions
The Diagnosis: Vestibular Papillomatosis
Vestibular papillomatosis (VP), the female equivalent of pearly penile papules, is characterized by multiple papules in a linear array on the labia minora and is considered a normal anatomic variant. It typically presents as monomorphous, soft, flesh-colored, filiform papules that are distributed in a symmetric fashion. In women, the papules present as linear arrays on the inner aspects of the labia minora, whereas in men, they present in a circumferential array along the sulcus of the glans penis.1 Lesions often are asymptomatic but may cause itching, burning, and dyspareunia.2 Previously believed to be associated with human papillomavirus infection,3 VP is now considered a noninfectious condition. Biopsy reveals parakeratosis and perinuclear vacuolization in the absence of true koilocytes.4,5 Dermoscopy and reflectance confocal microscopy have been used to differentiate VP from clinically similar lesions (eg, condyloma acuminatum).6,7 The prevalence of this condition is not well established; however, one study found VP in 1% of women attending genitourinary medicine clinics.3
Condyloma acuminatum, known colloquially as genital warts, is a human papillomavirus infection. Lesions tend to be painless and firm and are distributed asymmetrically with a cauliflowerlike appearance.1 Condyloma latum, found in secondary syphilis, is characterized by papules that are pale, smooth, flat topped, and moist.8 Molluscum contagiosum is an infection caused by a poxvirus presenting with flesh-colored, dome-shaped papules with central umbilication.9 The lesions of papulosquamous lichen planus are violaceous polygonal papules that affect the clitoral hood and labia minora and may cause pruritus. The cause of lichen planus is unknown; however, clinically similar lesions may occur in a lichenoid drug eruption due to certain medications.
Vestibular papillomatosis typically does not require treatment, except in symptomatic cases. To date, limited studies have reported variable treatment success utilizing destructive techniques such as CO2 laser or topical application of 5-fluorouracil or trichloroacetic acid.10
The lesions on our patient's left medial labia minora were successfully treated with low-voltage (3.0 V) electrodesiccation. Following local anesthesia with 1% lidocaine, each papule was gently electrodesiccated utilizing a standard hyfrecation electrode tip to a light gray discoloration. Postprocedural care involved only twice-daily cleansing with a gentle soap and application of petrolatum. The patient tolerated the procedure well and was satisfied with the cosmetic and functional results. She subsequently underwent treatment of the lesions on the right labia minora with equivalent treatment success.
- Moyal-Barracco M, Leibowitch M, Orth G. Vestibular papillae of the vulva. lack of evidence for human papillomavirus etiology. Arch Dermatol. 1990;126:1594-1598.
- Strand A, Wilander E, Zehbe I, et al. Vulvar papillomatosis, aceto-white lesions, and normal-looking vulvar mucosa evaluated by microscopy and human papillomavirus analysis. Gynecol Obstet Invest. 1995;40:265-270.
- Welch JM, Nayagam M, Parry G, et al. What is vestibular papillomatosis? a study of its prevalence, aetiology and natural history. Br J Obstet Gynaecol. 1993;100:939-942.
- Wilkinson EJ, Guerrero E, Daniel R, et al. Vulvar vestibulitis is rarely associated with human papillomavirus infection types 6, 11, 16, or 18. Int J Gynecol Pathol. 1993;12:344-349.
- Beznos G, Coates V, Focchi J, et al. Biomolecular study of the correlation between papillomatosis of the vulvar vestibule in adolescents and human papillomavirus. ScientificWorldJournal. 2006;6:628-636.
- Kim SH, Seo SH, Ko HC, et al. The use of dermatoscopy to differentiate vestibular papillae, a normal variant of the female external genitalia, from condyloma acuminata. J Am Acad Dermatol. 2009;60:353-355.
- Ozkur E, Falay T, Turgut Erdemir AV, et al. Vestibular papillomatosis: an important differential diagnosis of vulvar papillomas. Dermatol Online J. 2016;22. pii:13030/qt7933q377
- Chang GJ, Welton ML. Human papillomavirus, condylomata acuminata, and anal neoplasia. Clin Colon Rectal Surg. 2004;17:221-230.
- Lynch PJ, Moyal-Barracco M, Bogliatto F, et al. 2006 ISSVD classification of vulvar dermatoses: pathologic subsets and their clinical correlates. J Reprod Med. 2007;52:3-9.
- Bergeron C, Ferenczy A, Richart RM, et al. Micropapillomatosis labialis appears unrelated to human papillomavirus. Obstet Gynecol. 1990;76:281-286.
The Diagnosis: Vestibular Papillomatosis
Vestibular papillomatosis (VP), the female equivalent of pearly penile papules, is characterized by multiple papules in a linear array on the labia minora and is considered a normal anatomic variant. It typically presents as monomorphous, soft, flesh-colored, filiform papules that are distributed in a symmetric fashion. In women, the papules present as linear arrays on the inner aspects of the labia minora, whereas in men, they present in a circumferential array along the sulcus of the glans penis.1 Lesions often are asymptomatic but may cause itching, burning, and dyspareunia.2 Previously believed to be associated with human papillomavirus infection,3 VP is now considered a noninfectious condition. Biopsy reveals parakeratosis and perinuclear vacuolization in the absence of true koilocytes.4,5 Dermoscopy and reflectance confocal microscopy have been used to differentiate VP from clinically similar lesions (eg, condyloma acuminatum).6,7 The prevalence of this condition is not well established; however, one study found VP in 1% of women attending genitourinary medicine clinics.3
Condyloma acuminatum, known colloquially as genital warts, is a human papillomavirus infection. Lesions tend to be painless and firm and are distributed asymmetrically with a cauliflowerlike appearance.1 Condyloma latum, found in secondary syphilis, is characterized by papules that are pale, smooth, flat topped, and moist.8 Molluscum contagiosum is an infection caused by a poxvirus presenting with flesh-colored, dome-shaped papules with central umbilication.9 The lesions of papulosquamous lichen planus are violaceous polygonal papules that affect the clitoral hood and labia minora and may cause pruritus. The cause of lichen planus is unknown; however, clinically similar lesions may occur in a lichenoid drug eruption due to certain medications.
Vestibular papillomatosis typically does not require treatment, except in symptomatic cases. To date, limited studies have reported variable treatment success utilizing destructive techniques such as CO2 laser or topical application of 5-fluorouracil or trichloroacetic acid.10
The lesions on our patient's left medial labia minora were successfully treated with low-voltage (3.0 V) electrodesiccation. Following local anesthesia with 1% lidocaine, each papule was gently electrodesiccated utilizing a standard hyfrecation electrode tip to a light gray discoloration. Postprocedural care involved only twice-daily cleansing with a gentle soap and application of petrolatum. The patient tolerated the procedure well and was satisfied with the cosmetic and functional results. She subsequently underwent treatment of the lesions on the right labia minora with equivalent treatment success.
The Diagnosis: Vestibular Papillomatosis
Vestibular papillomatosis (VP), the female equivalent of pearly penile papules, is characterized by multiple papules in a linear array on the labia minora and is considered a normal anatomic variant. It typically presents as monomorphous, soft, flesh-colored, filiform papules that are distributed in a symmetric fashion. In women, the papules present as linear arrays on the inner aspects of the labia minora, whereas in men, they present in a circumferential array along the sulcus of the glans penis.1 Lesions often are asymptomatic but may cause itching, burning, and dyspareunia.2 Previously believed to be associated with human papillomavirus infection,3 VP is now considered a noninfectious condition. Biopsy reveals parakeratosis and perinuclear vacuolization in the absence of true koilocytes.4,5 Dermoscopy and reflectance confocal microscopy have been used to differentiate VP from clinically similar lesions (eg, condyloma acuminatum).6,7 The prevalence of this condition is not well established; however, one study found VP in 1% of women attending genitourinary medicine clinics.3
Condyloma acuminatum, known colloquially as genital warts, is a human papillomavirus infection. Lesions tend to be painless and firm and are distributed asymmetrically with a cauliflowerlike appearance.1 Condyloma latum, found in secondary syphilis, is characterized by papules that are pale, smooth, flat topped, and moist.8 Molluscum contagiosum is an infection caused by a poxvirus presenting with flesh-colored, dome-shaped papules with central umbilication.9 The lesions of papulosquamous lichen planus are violaceous polygonal papules that affect the clitoral hood and labia minora and may cause pruritus. The cause of lichen planus is unknown; however, clinically similar lesions may occur in a lichenoid drug eruption due to certain medications.
Vestibular papillomatosis typically does not require treatment, except in symptomatic cases. To date, limited studies have reported variable treatment success utilizing destructive techniques such as CO2 laser or topical application of 5-fluorouracil or trichloroacetic acid.10
The lesions on our patient's left medial labia minora were successfully treated with low-voltage (3.0 V) electrodesiccation. Following local anesthesia with 1% lidocaine, each papule was gently electrodesiccated utilizing a standard hyfrecation electrode tip to a light gray discoloration. Postprocedural care involved only twice-daily cleansing with a gentle soap and application of petrolatum. The patient tolerated the procedure well and was satisfied with the cosmetic and functional results. She subsequently underwent treatment of the lesions on the right labia minora with equivalent treatment success.
- Moyal-Barracco M, Leibowitch M, Orth G. Vestibular papillae of the vulva. lack of evidence for human papillomavirus etiology. Arch Dermatol. 1990;126:1594-1598.
- Strand A, Wilander E, Zehbe I, et al. Vulvar papillomatosis, aceto-white lesions, and normal-looking vulvar mucosa evaluated by microscopy and human papillomavirus analysis. Gynecol Obstet Invest. 1995;40:265-270.
- Welch JM, Nayagam M, Parry G, et al. What is vestibular papillomatosis? a study of its prevalence, aetiology and natural history. Br J Obstet Gynaecol. 1993;100:939-942.
- Wilkinson EJ, Guerrero E, Daniel R, et al. Vulvar vestibulitis is rarely associated with human papillomavirus infection types 6, 11, 16, or 18. Int J Gynecol Pathol. 1993;12:344-349.
- Beznos G, Coates V, Focchi J, et al. Biomolecular study of the correlation between papillomatosis of the vulvar vestibule in adolescents and human papillomavirus. ScientificWorldJournal. 2006;6:628-636.
- Kim SH, Seo SH, Ko HC, et al. The use of dermatoscopy to differentiate vestibular papillae, a normal variant of the female external genitalia, from condyloma acuminata. J Am Acad Dermatol. 2009;60:353-355.
- Ozkur E, Falay T, Turgut Erdemir AV, et al. Vestibular papillomatosis: an important differential diagnosis of vulvar papillomas. Dermatol Online J. 2016;22. pii:13030/qt7933q377
- Chang GJ, Welton ML. Human papillomavirus, condylomata acuminata, and anal neoplasia. Clin Colon Rectal Surg. 2004;17:221-230.
- Lynch PJ, Moyal-Barracco M, Bogliatto F, et al. 2006 ISSVD classification of vulvar dermatoses: pathologic subsets and their clinical correlates. J Reprod Med. 2007;52:3-9.
- Bergeron C, Ferenczy A, Richart RM, et al. Micropapillomatosis labialis appears unrelated to human papillomavirus. Obstet Gynecol. 1990;76:281-286.
- Moyal-Barracco M, Leibowitch M, Orth G. Vestibular papillae of the vulva. lack of evidence for human papillomavirus etiology. Arch Dermatol. 1990;126:1594-1598.
- Strand A, Wilander E, Zehbe I, et al. Vulvar papillomatosis, aceto-white lesions, and normal-looking vulvar mucosa evaluated by microscopy and human papillomavirus analysis. Gynecol Obstet Invest. 1995;40:265-270.
- Welch JM, Nayagam M, Parry G, et al. What is vestibular papillomatosis? a study of its prevalence, aetiology and natural history. Br J Obstet Gynaecol. 1993;100:939-942.
- Wilkinson EJ, Guerrero E, Daniel R, et al. Vulvar vestibulitis is rarely associated with human papillomavirus infection types 6, 11, 16, or 18. Int J Gynecol Pathol. 1993;12:344-349.
- Beznos G, Coates V, Focchi J, et al. Biomolecular study of the correlation between papillomatosis of the vulvar vestibule in adolescents and human papillomavirus. ScientificWorldJournal. 2006;6:628-636.
- Kim SH, Seo SH, Ko HC, et al. The use of dermatoscopy to differentiate vestibular papillae, a normal variant of the female external genitalia, from condyloma acuminata. J Am Acad Dermatol. 2009;60:353-355.
- Ozkur E, Falay T, Turgut Erdemir AV, et al. Vestibular papillomatosis: an important differential diagnosis of vulvar papillomas. Dermatol Online J. 2016;22. pii:13030/qt7933q377
- Chang GJ, Welton ML. Human papillomavirus, condylomata acuminata, and anal neoplasia. Clin Colon Rectal Surg. 2004;17:221-230.
- Lynch PJ, Moyal-Barracco M, Bogliatto F, et al. 2006 ISSVD classification of vulvar dermatoses: pathologic subsets and their clinical correlates. J Reprod Med. 2007;52:3-9.
- Bergeron C, Ferenczy A, Richart RM, et al. Micropapillomatosis labialis appears unrelated to human papillomavirus. Obstet Gynecol. 1990;76:281-286.
A 30-year-old woman with congenital absence of the uterus presented to dermatology for a second opinion of vulvar lesions that were first noted during adolescence. The patient reported that the lesions had not changed and were painful during sexual intercourse. The lesions were otherwise asymptomatic, and she had no additional relevant medical history or family history of similar lesions. She denied any history of sexually transmitted infections. Physical examination revealed multiple, soft, flesh-colored, 1- to 2-mm, discrete and coalescing, filiform papules distributed symmetrically in a linear array on the inner aspect of the bilateral medial labia minora. The rest of the mucocutaneous examination was normal.
The lesions on the left medial labia minora were treated with low-voltage (3.0 V) electrodesiccation following local anesthesia with 1% lidocaine (red arrow), while the lesions on the right medial labia minora were left untreated (black arrow). The clinical image shows the left labia minora approximately 1 month after treatment; the papules on the right labia minora were unchanged from the prior examination.
Asymptomatic, Slowly Enlarging Papule on the Nipple
The Diagnosis: Erosive Adenomatosis of the Nipple
Biopsy of the lesion revealed proliferative sections of glandular epithelium demonstrating apocrine differentiation, connecting to the epidermis and traversing throughout the entire dermis of the specimen (Figure). There were papillary projections of dilated ducts with a retained layer of myoepithelial cells surrounding the epithelial layers. Cytologic atypia was not appreciated. The patient was diagnosed with erosive adenomatosis of the nipple (EAN), also known as nipple adenoma. The lesion subsequently was treated and cleared with Mohs micrographic surgery (MMS). At 8-month follow-up there was no clinical recurrence of the lesion, and the patient was satisfied with the overall cosmetic appearance and conservation of the areola. The patient was followed clinically with annual breast examinations and mammography to monitor future recurrence.

Erosive adenomatosis of the nipple is an uncommon benign proliferative process of the lactiferous ducts of the nipple. Recognizing EAN is important because it resembles malignant breast diseases such as Paget disease of the nipple and invasive breast carcinoma. Due to these similarities, early cases of EAN have resulted in unnecessary mastectomies before the benignity of the condition was established.1 Accurate diagnosis is important to both the patient and the clinician for treatment planning as well as psychosocial consequences associated with the potential removal of this anatomically and cosmetically sensitive area.
Reviewing the literature on EAN is complicated by the variety of terms used to describe this condition, including but not limited to nipple adenoma, nipple duct adenoma, papillary adenoma of the nipple, and florid papillomatosis of the nipple. In 1955, Jones2 described EAN using the term florid papillomatosis of the nipple ducts. In 1962, Handley and Thackray1 argued that adenoma of the nipple was a more descriptive term because it more closely described the appearance of a sweat gland adenoma. They reasoned that adenoma of the nipple is a separate process from ductal papilloma due to the adenomatous proliferation into the nipple stroma rather than the lumen of the nipple ducts.1 The term adenoma of the nipple was further supported in 1965 by Taylor and Robertson.3 In 1959, Le Gal et al4 used the term erosive adenomatosis of the nipple to describe the erosive nature of nipple adenoma. The term nipple adenoma was published in the 2012 WHO Classification of Tumors of the Breast with 4 common histologic subtypes.5,6
Erosive adenomatosis of the nipple is clinically indistinguishable from Paget disease of the nipple, thus biopsy is essential for accurate diagnosis. In contrast to Paget disease, EAN tends to present in younger patients and progresses more slowly, and symptoms may be exacerbated around menstruation.1 Case reports demonstrate that patients may wait years before seeking medical attention for EAN.1,3,7,8 Presenting symptoms may include inflammation, crusting, nipple skin erosion, itching, and pain. Serous or sanguineous discharge from the lesions also is commonly reported. Palpation may reveal a small, hard, or elastic nodule within or underlying the nipple. In addition to Paget disease, EAN may resemble squamous cell carcinoma of the nipple, eczema, psoriasis, or a skin infection.6 Axillary lymphadenopathy is not present in the absence of a concomitant breast malignancy.8 On biopsy, nipple adenoma represents ductal proliferation of glandular structures within the stroma of the nipple that is well circumscribed but without borders. The erosive appearance of the lesion is produced by extensions of the glandular epithelium on the surface of the nipple.1,6 Specific to EAN is the presence of 2 cell types: an inner columnar epithelium and an outer cuboidal myoepithelium. These 2 cell types are present in normal lactiferous ducts; however, normal ducts are highly organized compared to EAN.9
After confirmation of EAN by nipple biopsy, complete surgical excision has been the gold standard for treatment, followed by reconstructive surgery.6 Handley and Thackray1 advocated for total excision of the nipple and areola with an underlying wedge of breast tissue to facilitate wound closure. More recently, successful alternative forms of treatment have been utilized to minimize disfiguring surgery. Alternative treatments include MMS,8 cryotherapy,10 and nipple splitting enucleation.6 Treatment with MMS has resulted in nipple sparing with the least amount of surface area sacrificed (1.1 cm2).9 Our case and prior case reports demonstrate that the tissue sparing potential of MMS is appropriate for the treatment of EAN, though traditionally it has been reserved for more malignant tumors. Preserving this sensitive area is both cosmetically and psychologically advantageous for the patient and thus should be considered when reviewing treatment options for EAN.
- Handley RS, Thackray AC. Adenoma of nipple. Br J Cancer. 1962;16:187-194.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Taylor HB, Robertson AG. Adenomas of the nipple. Cancer. 1965:18:995-1002.
- Le Gal Y, Gros CM, Bader P. Erosive adenomatosis of the nipple [in French]. Ann Anat Pathol (Paris). 1959;4:292-304.
- Eusebi V, Lester S. Tumours of the nipple. In: Lakhani SR, Ellis IO, Schnitt SJ, et al, eds. WHO Classification of Tumours of the Breast. Lyon, France: IARC; 2012.
- Spohn GP, Trotter SC, Tozbician G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Kowal R, Miller CJ, Elenitsas R. Eroded patch on the nipple of a 57-year-old woman. Arch Dermatol. 2008;144:933-938.
- Van Mierlo PL, Geelen GM, Neumann HA. Mohs micrographic surgery for an erosive adenomatosis of the nipple. Dermatol Surg. 1998;24:681-683.
- Brankov N, Nino T, Hsiang D, et al. Utilizing Mohs surgery for tissue preservation in erosive adenomatosis of the nipple. Dermatol Surg. 2016;42:684-686.
- Kuflik EG. Erosive adenomatosis of the nipple treated with cryosurgery. J Am Acad Dermatol. 1998;38:270-271.
The Diagnosis: Erosive Adenomatosis of the Nipple
Biopsy of the lesion revealed proliferative sections of glandular epithelium demonstrating apocrine differentiation, connecting to the epidermis and traversing throughout the entire dermis of the specimen (Figure). There were papillary projections of dilated ducts with a retained layer of myoepithelial cells surrounding the epithelial layers. Cytologic atypia was not appreciated. The patient was diagnosed with erosive adenomatosis of the nipple (EAN), also known as nipple adenoma. The lesion subsequently was treated and cleared with Mohs micrographic surgery (MMS). At 8-month follow-up there was no clinical recurrence of the lesion, and the patient was satisfied with the overall cosmetic appearance and conservation of the areola. The patient was followed clinically with annual breast examinations and mammography to monitor future recurrence.
Erosive adenomatosis of the nipple is an uncommon benign proliferative process of the lactiferous ducts of the nipple. Recognizing EAN is important because it resembles malignant breast diseases such as Paget disease of the nipple and invasive breast carcinoma. Due to these similarities, early cases of EAN have resulted in unnecessary mastectomies before the benignity of the condition was established.1 Accurate diagnosis is important to both the patient and the clinician for treatment planning as well as psychosocial consequences associated with the potential removal of this anatomically and cosmetically sensitive area.
Reviewing the literature on EAN is complicated by the variety of terms used to describe this condition, including but not limited to nipple adenoma, nipple duct adenoma, papillary adenoma of the nipple, and florid papillomatosis of the nipple. In 1955, Jones2 described EAN using the term florid papillomatosis of the nipple ducts. In 1962, Handley and Thackray1 argued that adenoma of the nipple was a more descriptive term because it more closely described the appearance of a sweat gland adenoma. They reasoned that adenoma of the nipple is a separate process from ductal papilloma due to the adenomatous proliferation into the nipple stroma rather than the lumen of the nipple ducts.1 The term adenoma of the nipple was further supported in 1965 by Taylor and Robertson.3 In 1959, Le Gal et al4 used the term erosive adenomatosis of the nipple to describe the erosive nature of nipple adenoma. The term nipple adenoma was published in the 2012 WHO Classification of Tumors of the Breast with 4 common histologic subtypes.5,6
Erosive adenomatosis of the nipple is clinically indistinguishable from Paget disease of the nipple, thus biopsy is essential for accurate diagnosis. In contrast to Paget disease, EAN tends to present in younger patients and progresses more slowly, and symptoms may be exacerbated around menstruation.1 Case reports demonstrate that patients may wait years before seeking medical attention for EAN.1,3,7,8 Presenting symptoms may include inflammation, crusting, nipple skin erosion, itching, and pain. Serous or sanguineous discharge from the lesions also is commonly reported. Palpation may reveal a small, hard, or elastic nodule within or underlying the nipple. In addition to Paget disease, EAN may resemble squamous cell carcinoma of the nipple, eczema, psoriasis, or a skin infection.6 Axillary lymphadenopathy is not present in the absence of a concomitant breast malignancy.8 On biopsy, nipple adenoma represents ductal proliferation of glandular structures within the stroma of the nipple that is well circumscribed but without borders. The erosive appearance of the lesion is produced by extensions of the glandular epithelium on the surface of the nipple.1,6 Specific to EAN is the presence of 2 cell types: an inner columnar epithelium and an outer cuboidal myoepithelium. These 2 cell types are present in normal lactiferous ducts; however, normal ducts are highly organized compared to EAN.9
After confirmation of EAN by nipple biopsy, complete surgical excision has been the gold standard for treatment, followed by reconstructive surgery.6 Handley and Thackray1 advocated for total excision of the nipple and areola with an underlying wedge of breast tissue to facilitate wound closure. More recently, successful alternative forms of treatment have been utilized to minimize disfiguring surgery. Alternative treatments include MMS,8 cryotherapy,10 and nipple splitting enucleation.6 Treatment with MMS has resulted in nipple sparing with the least amount of surface area sacrificed (1.1 cm2).9 Our case and prior case reports demonstrate that the tissue sparing potential of MMS is appropriate for the treatment of EAN, though traditionally it has been reserved for more malignant tumors. Preserving this sensitive area is both cosmetically and psychologically advantageous for the patient and thus should be considered when reviewing treatment options for EAN.
The Diagnosis: Erosive Adenomatosis of the Nipple
Biopsy of the lesion revealed proliferative sections of glandular epithelium demonstrating apocrine differentiation, connecting to the epidermis and traversing throughout the entire dermis of the specimen (Figure). There were papillary projections of dilated ducts with a retained layer of myoepithelial cells surrounding the epithelial layers. Cytologic atypia was not appreciated. The patient was diagnosed with erosive adenomatosis of the nipple (EAN), also known as nipple adenoma. The lesion subsequently was treated and cleared with Mohs micrographic surgery (MMS). At 8-month follow-up there was no clinical recurrence of the lesion, and the patient was satisfied with the overall cosmetic appearance and conservation of the areola. The patient was followed clinically with annual breast examinations and mammography to monitor future recurrence.
Erosive adenomatosis of the nipple is an uncommon benign proliferative process of the lactiferous ducts of the nipple. Recognizing EAN is important because it resembles malignant breast diseases such as Paget disease of the nipple and invasive breast carcinoma. Due to these similarities, early cases of EAN have resulted in unnecessary mastectomies before the benignity of the condition was established.1 Accurate diagnosis is important to both the patient and the clinician for treatment planning as well as psychosocial consequences associated with the potential removal of this anatomically and cosmetically sensitive area.
Reviewing the literature on EAN is complicated by the variety of terms used to describe this condition, including but not limited to nipple adenoma, nipple duct adenoma, papillary adenoma of the nipple, and florid papillomatosis of the nipple. In 1955, Jones2 described EAN using the term florid papillomatosis of the nipple ducts. In 1962, Handley and Thackray1 argued that adenoma of the nipple was a more descriptive term because it more closely described the appearance of a sweat gland adenoma. They reasoned that adenoma of the nipple is a separate process from ductal papilloma due to the adenomatous proliferation into the nipple stroma rather than the lumen of the nipple ducts.1 The term adenoma of the nipple was further supported in 1965 by Taylor and Robertson.3 In 1959, Le Gal et al4 used the term erosive adenomatosis of the nipple to describe the erosive nature of nipple adenoma. The term nipple adenoma was published in the 2012 WHO Classification of Tumors of the Breast with 4 common histologic subtypes.5,6
Erosive adenomatosis of the nipple is clinically indistinguishable from Paget disease of the nipple, thus biopsy is essential for accurate diagnosis. In contrast to Paget disease, EAN tends to present in younger patients and progresses more slowly, and symptoms may be exacerbated around menstruation.1 Case reports demonstrate that patients may wait years before seeking medical attention for EAN.1,3,7,8 Presenting symptoms may include inflammation, crusting, nipple skin erosion, itching, and pain. Serous or sanguineous discharge from the lesions also is commonly reported. Palpation may reveal a small, hard, or elastic nodule within or underlying the nipple. In addition to Paget disease, EAN may resemble squamous cell carcinoma of the nipple, eczema, psoriasis, or a skin infection.6 Axillary lymphadenopathy is not present in the absence of a concomitant breast malignancy.8 On biopsy, nipple adenoma represents ductal proliferation of glandular structures within the stroma of the nipple that is well circumscribed but without borders. The erosive appearance of the lesion is produced by extensions of the glandular epithelium on the surface of the nipple.1,6 Specific to EAN is the presence of 2 cell types: an inner columnar epithelium and an outer cuboidal myoepithelium. These 2 cell types are present in normal lactiferous ducts; however, normal ducts are highly organized compared to EAN.9
After confirmation of EAN by nipple biopsy, complete surgical excision has been the gold standard for treatment, followed by reconstructive surgery.6 Handley and Thackray1 advocated for total excision of the nipple and areola with an underlying wedge of breast tissue to facilitate wound closure. More recently, successful alternative forms of treatment have been utilized to minimize disfiguring surgery. Alternative treatments include MMS,8 cryotherapy,10 and nipple splitting enucleation.6 Treatment with MMS has resulted in nipple sparing with the least amount of surface area sacrificed (1.1 cm2).9 Our case and prior case reports demonstrate that the tissue sparing potential of MMS is appropriate for the treatment of EAN, though traditionally it has been reserved for more malignant tumors. Preserving this sensitive area is both cosmetically and psychologically advantageous for the patient and thus should be considered when reviewing treatment options for EAN.
- Handley RS, Thackray AC. Adenoma of nipple. Br J Cancer. 1962;16:187-194.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Taylor HB, Robertson AG. Adenomas of the nipple. Cancer. 1965:18:995-1002.
- Le Gal Y, Gros CM, Bader P. Erosive adenomatosis of the nipple [in French]. Ann Anat Pathol (Paris). 1959;4:292-304.
- Eusebi V, Lester S. Tumours of the nipple. In: Lakhani SR, Ellis IO, Schnitt SJ, et al, eds. WHO Classification of Tumours of the Breast. Lyon, France: IARC; 2012.
- Spohn GP, Trotter SC, Tozbician G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Kowal R, Miller CJ, Elenitsas R. Eroded patch on the nipple of a 57-year-old woman. Arch Dermatol. 2008;144:933-938.
- Van Mierlo PL, Geelen GM, Neumann HA. Mohs micrographic surgery for an erosive adenomatosis of the nipple. Dermatol Surg. 1998;24:681-683.
- Brankov N, Nino T, Hsiang D, et al. Utilizing Mohs surgery for tissue preservation in erosive adenomatosis of the nipple. Dermatol Surg. 2016;42:684-686.
- Kuflik EG. Erosive adenomatosis of the nipple treated with cryosurgery. J Am Acad Dermatol. 1998;38:270-271.
- Handley RS, Thackray AC. Adenoma of nipple. Br J Cancer. 1962;16:187-194.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Taylor HB, Robertson AG. Adenomas of the nipple. Cancer. 1965:18:995-1002.
- Le Gal Y, Gros CM, Bader P. Erosive adenomatosis of the nipple [in French]. Ann Anat Pathol (Paris). 1959;4:292-304.
- Eusebi V, Lester S. Tumours of the nipple. In: Lakhani SR, Ellis IO, Schnitt SJ, et al, eds. WHO Classification of Tumours of the Breast. Lyon, France: IARC; 2012.
- Spohn GP, Trotter SC, Tozbician G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Kowal R, Miller CJ, Elenitsas R. Eroded patch on the nipple of a 57-year-old woman. Arch Dermatol. 2008;144:933-938.
- Van Mierlo PL, Geelen GM, Neumann HA. Mohs micrographic surgery for an erosive adenomatosis of the nipple. Dermatol Surg. 1998;24:681-683.
- Brankov N, Nino T, Hsiang D, et al. Utilizing Mohs surgery for tissue preservation in erosive adenomatosis of the nipple. Dermatol Surg. 2016;42:684-686.
- Kuflik EG. Erosive adenomatosis of the nipple treated with cryosurgery. J Am Acad Dermatol. 1998;38:270-271.
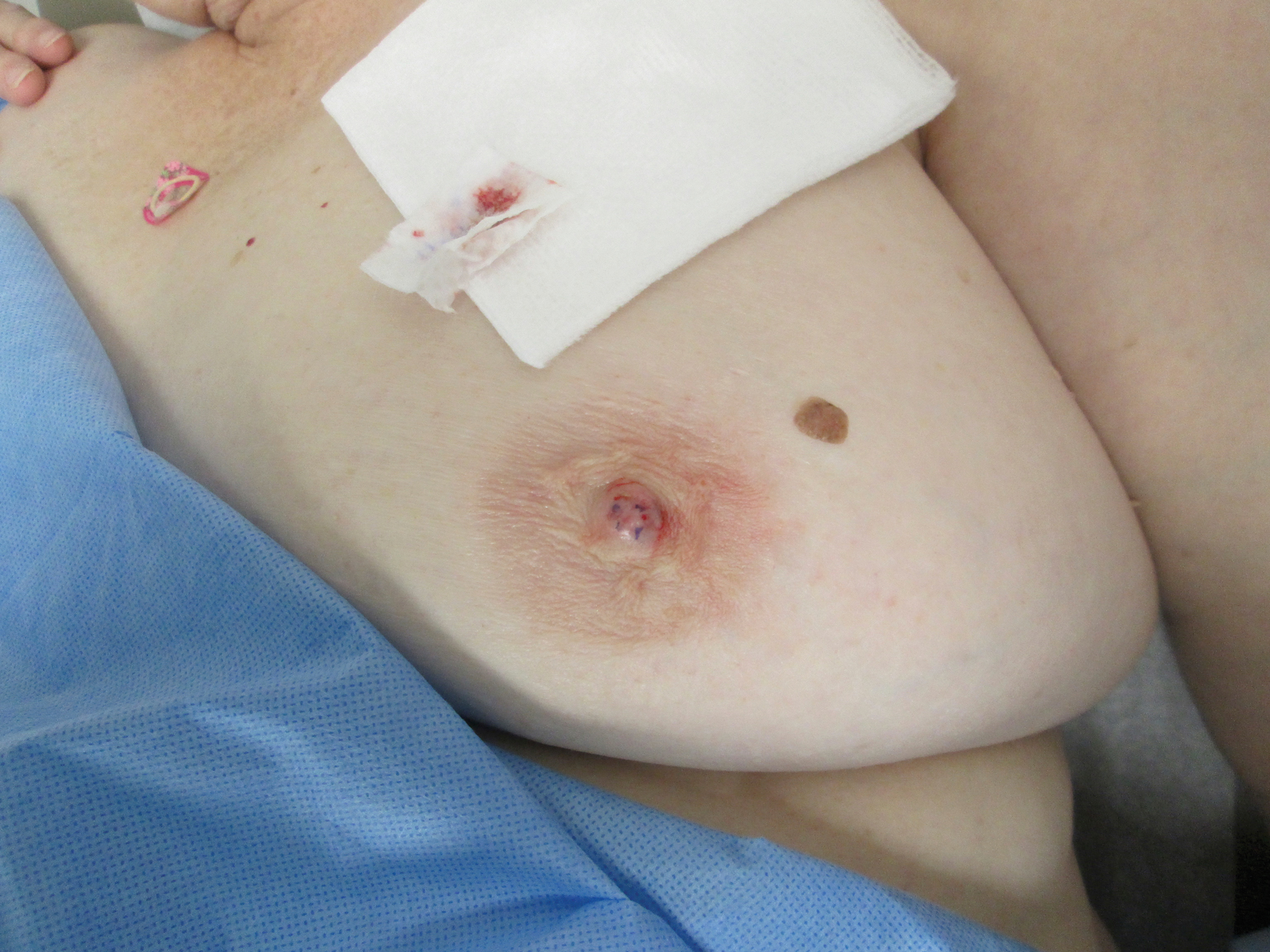
A 61-year-old woman presented with an asymptomatic, slowly enlarging, 9-mm, firm, red papule on the left nipple of 2 years' duration. She had no notable medical history, including a BI-RADS (Breast Imaging Reporting and Data System) mammogram score of 2 that was suggestive of benign findings 2 years prior. A repeat mammogram ordered by radiology and completed before presenting to dermatology had a BI-RADS score of 4, noting a concerning feature in the area of the lesion and prompting a biopsy.
Violaceous Nodules on the Hard Palate
The Diagnosis: Kaposi Sarcoma
A 4-mm punch biopsy from the border of an ulcerated nodular lesion on the hard palate demonstrated diffusely distributed spindle cells, cleftlike microvascularity with extravasated erythrocytes, and widespread human herpesvirus 8 immunoreactivity on histopathology (Figure 1). Serologic tests were positive for human immunodeficiency virus (HIV) infection; HIV RNA was 14,584 IU/mL and the CD4 count was 254/mm3. The patient was diagnosed with Kaposi sarcoma (KS) and referred to the infectious disease department for initiation of antiviral therapy. Marked regression was detected after 6 months of highly active antiretroviral therapy (HAART) without any additional treatment (Figure 2).
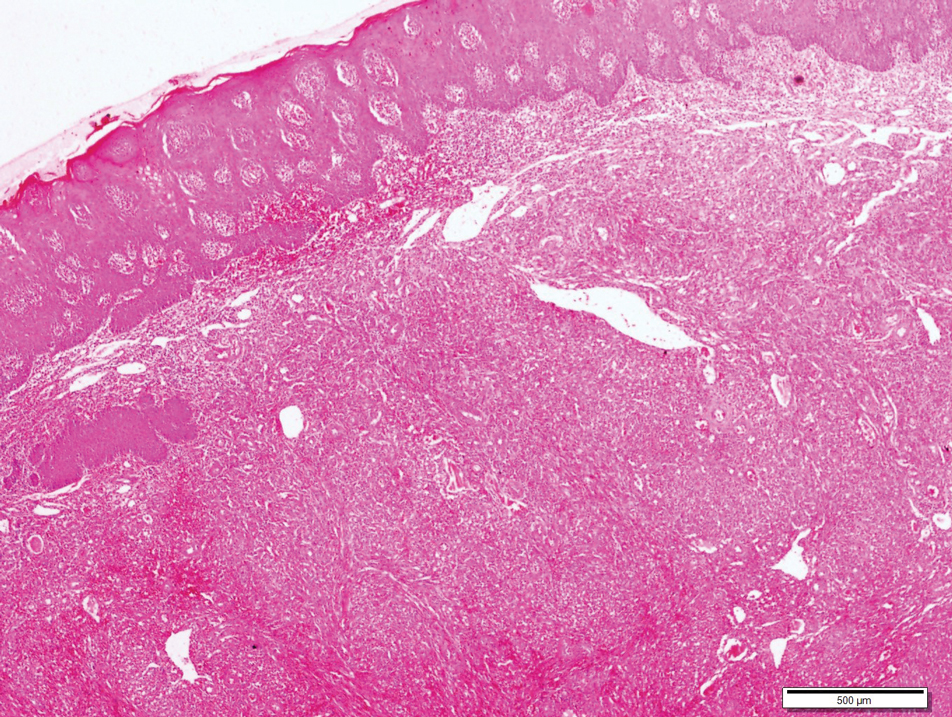
widespread human herpesvirus 8 immunoreactivity (H&E, original magnification ×4).
Kaposi sarcoma is a human herpesvirus 8-associated angioproliferative disorder with low-grade malignant potential. There are 4 well-known clinical types: classic, endemic, iatrogenic, and AIDS associated.1 Involvement of the oral cavity may be seen in all types but mostly is associated with the AIDS-associated type, which also could be a signal for undiagnosed asymptomatic HIV infection.2 Oral KS most often affects the hard and soft palate, gingiva, and dorsal tongue, with plaques or tumors ranging from nonpigmented to brownish red or violaceous. AIDS-associated KS is known to be related to cytokine expression, which is induced by HIV infection causing immune dysregulation by altering the expression of cytokines, including IL-1, tumor necrosis factor α, and IL-6.1 An in vitro study showed that cytokines secrete a number of angiogenic growth factors that, along with HIV proteins, induce and proliferate cells to become sarcoma cells. Integrins and the apoptosis process also are important in proliferation and neovascularization of KS tumor cells.3
Bacillary angiomatosis (BA) is a rare manifestation of infection caused by Bartonella species, which leads to vasoproliferative lesions of the skin and other organs. Bacillary angiomatosis affects individuals with advanced HIV or other immunocompromised individuals and may clinically mimic KS, which is similarly characterized by red-purple papules, nodules, or plaques. Differentiating BA from KS largely depends on histopathologic examination, with BA demonstrating protuberant endothelial cells surrounded by clumps of bacilli that are visible on Warthin-Starry silver stain.
Lymphangioma is a benign hamartomatous hyperplasia of the lymphatic vessels. The majority of lymphangiomas are superficial, but a few may extend deeply into the connective tissue. Intraoral lymphangiomas occur more frequently on the dorsum of the tongue, followed by the palate, buccal mucosa, gingiva, and lips. They may be differentiated with their soft quality, pebblelike surface, and translucent vesicles.
Malignant tumors of the oral cavity are rare, representing only 5% of tumors occurring in the body.4 Among malignant tumors of the oral cavity, squamous cell carcinomas are the most frequent type (90%-98%), and lymphomas and melanoma are the most outstanding among the remaining 2% to 10%. Both for lymphoma and mucosal melanoma, the most common sites of involvement are the soft tissues of the oral cavity, palatal mucosa, gingiva, tongue, cheeks, floor of the mouth, and lips.4 Although mucosal melanoma lesions usually are characterized by pigmented and ulcerated lesions, amelanotic variants also should be kept in mind. Histopathologic examination is mandatory for diagnosis.
Intralesional chemotherapy with vinblastine or bleomycin, radiotherapy, electrochemotherapy, systemic antiretroviral therapy (ie, HAART), and chemotherapy with daunorubicin and pegylated liposomal doxorubicin are the main treatment options.5,6 The immune system activator role of HAART leads to an increased CD4 count and reduces HIV proteins, which helps induction of the proliferation and neovascularization of KS tumor cells.3 This effect may help resolution of KS with localized involvement and allows physicians to utilize HAART without any other additional local and systemic chemotherapy treatment.
- Fatahzadeh M, Schwartz RA. Oral Kaposi's sarcoma: a review and update. Int J Dermatol. 2013;52:666-672.
- Martorano LM, Cannella JD, Lloyd JR. Mucocutaneous presentation of Kaposi sarcoma in an asymptomatic human immunodeficiency virus-positive man. Cutis. 2015;95:E19-E22.
- Stebbing J, Portsmouth S, Gazzard B. How does HAART lead to the resolution of Kaposi's sarcoma? J Antimicrobial Chemother. 2003;51:1095-1098.
- Guevara-Canales JO, Morales-Vadillo R, Sacsaquispe-Contreras SJ, et al. Malignant lymphoma of the oral cavity and the maxillofacial region: overall survivalprognostic factors. Med Oral Patol Oral Cir Bucal. 2013;18:E619-E626.
- Donato V, Guarnaccia R, Dognini J, et al. Radiation therapy in the treatment of HIV-related Kaposi's sarcoma. Anticancer Res. 2013;33:2153-2157.
- Gbabe OF, Okwundu CI, Dedicoat M, et al. Treatment of severe or progressive Kaposi's sarcoma in HIV-infected adults. Cochrane Database Syst Rev. 2014:CD003256.
The Diagnosis: Kaposi Sarcoma
A 4-mm punch biopsy from the border of an ulcerated nodular lesion on the hard palate demonstrated diffusely distributed spindle cells, cleftlike microvascularity with extravasated erythrocytes, and widespread human herpesvirus 8 immunoreactivity on histopathology (Figure 1). Serologic tests were positive for human immunodeficiency virus (HIV) infection; HIV RNA was 14,584 IU/mL and the CD4 count was 254/mm3. The patient was diagnosed with Kaposi sarcoma (KS) and referred to the infectious disease department for initiation of antiviral therapy. Marked regression was detected after 6 months of highly active antiretroviral therapy (HAART) without any additional treatment (Figure 2).
widespread human herpesvirus 8 immunoreactivity (H&E, original magnification ×4).
Kaposi sarcoma is a human herpesvirus 8-associated angioproliferative disorder with low-grade malignant potential. There are 4 well-known clinical types: classic, endemic, iatrogenic, and AIDS associated.1 Involvement of the oral cavity may be seen in all types but mostly is associated with the AIDS-associated type, which also could be a signal for undiagnosed asymptomatic HIV infection.2 Oral KS most often affects the hard and soft palate, gingiva, and dorsal tongue, with plaques or tumors ranging from nonpigmented to brownish red or violaceous. AIDS-associated KS is known to be related to cytokine expression, which is induced by HIV infection causing immune dysregulation by altering the expression of cytokines, including IL-1, tumor necrosis factor α, and IL-6.1 An in vitro study showed that cytokines secrete a number of angiogenic growth factors that, along with HIV proteins, induce and proliferate cells to become sarcoma cells. Integrins and the apoptosis process also are important in proliferation and neovascularization of KS tumor cells.3
Bacillary angiomatosis (BA) is a rare manifestation of infection caused by Bartonella species, which leads to vasoproliferative lesions of the skin and other organs. Bacillary angiomatosis affects individuals with advanced HIV or other immunocompromised individuals and may clinically mimic KS, which is similarly characterized by red-purple papules, nodules, or plaques. Differentiating BA from KS largely depends on histopathologic examination, with BA demonstrating protuberant endothelial cells surrounded by clumps of bacilli that are visible on Warthin-Starry silver stain.
Lymphangioma is a benign hamartomatous hyperplasia of the lymphatic vessels. The majority of lymphangiomas are superficial, but a few may extend deeply into the connective tissue. Intraoral lymphangiomas occur more frequently on the dorsum of the tongue, followed by the palate, buccal mucosa, gingiva, and lips. They may be differentiated with their soft quality, pebblelike surface, and translucent vesicles.
Malignant tumors of the oral cavity are rare, representing only 5% of tumors occurring in the body.4 Among malignant tumors of the oral cavity, squamous cell carcinomas are the most frequent type (90%-98%), and lymphomas and melanoma are the most outstanding among the remaining 2% to 10%. Both for lymphoma and mucosal melanoma, the most common sites of involvement are the soft tissues of the oral cavity, palatal mucosa, gingiva, tongue, cheeks, floor of the mouth, and lips.4 Although mucosal melanoma lesions usually are characterized by pigmented and ulcerated lesions, amelanotic variants also should be kept in mind. Histopathologic examination is mandatory for diagnosis.
Intralesional chemotherapy with vinblastine or bleomycin, radiotherapy, electrochemotherapy, systemic antiretroviral therapy (ie, HAART), and chemotherapy with daunorubicin and pegylated liposomal doxorubicin are the main treatment options.5,6 The immune system activator role of HAART leads to an increased CD4 count and reduces HIV proteins, which helps induction of the proliferation and neovascularization of KS tumor cells.3 This effect may help resolution of KS with localized involvement and allows physicians to utilize HAART without any other additional local and systemic chemotherapy treatment.
The Diagnosis: Kaposi Sarcoma
A 4-mm punch biopsy from the border of an ulcerated nodular lesion on the hard palate demonstrated diffusely distributed spindle cells, cleftlike microvascularity with extravasated erythrocytes, and widespread human herpesvirus 8 immunoreactivity on histopathology (Figure 1). Serologic tests were positive for human immunodeficiency virus (HIV) infection; HIV RNA was 14,584 IU/mL and the CD4 count was 254/mm3. The patient was diagnosed with Kaposi sarcoma (KS) and referred to the infectious disease department for initiation of antiviral therapy. Marked regression was detected after 6 months of highly active antiretroviral therapy (HAART) without any additional treatment (Figure 2).
widespread human herpesvirus 8 immunoreactivity (H&E, original magnification ×4).
Kaposi sarcoma is a human herpesvirus 8-associated angioproliferative disorder with low-grade malignant potential. There are 4 well-known clinical types: classic, endemic, iatrogenic, and AIDS associated.1 Involvement of the oral cavity may be seen in all types but mostly is associated with the AIDS-associated type, which also could be a signal for undiagnosed asymptomatic HIV infection.2 Oral KS most often affects the hard and soft palate, gingiva, and dorsal tongue, with plaques or tumors ranging from nonpigmented to brownish red or violaceous. AIDS-associated KS is known to be related to cytokine expression, which is induced by HIV infection causing immune dysregulation by altering the expression of cytokines, including IL-1, tumor necrosis factor α, and IL-6.1 An in vitro study showed that cytokines secrete a number of angiogenic growth factors that, along with HIV proteins, induce and proliferate cells to become sarcoma cells. Integrins and the apoptosis process also are important in proliferation and neovascularization of KS tumor cells.3
Bacillary angiomatosis (BA) is a rare manifestation of infection caused by Bartonella species, which leads to vasoproliferative lesions of the skin and other organs. Bacillary angiomatosis affects individuals with advanced HIV or other immunocompromised individuals and may clinically mimic KS, which is similarly characterized by red-purple papules, nodules, or plaques. Differentiating BA from KS largely depends on histopathologic examination, with BA demonstrating protuberant endothelial cells surrounded by clumps of bacilli that are visible on Warthin-Starry silver stain.
Lymphangioma is a benign hamartomatous hyperplasia of the lymphatic vessels. The majority of lymphangiomas are superficial, but a few may extend deeply into the connective tissue. Intraoral lymphangiomas occur more frequently on the dorsum of the tongue, followed by the palate, buccal mucosa, gingiva, and lips. They may be differentiated with their soft quality, pebblelike surface, and translucent vesicles.
Malignant tumors of the oral cavity are rare, representing only 5% of tumors occurring in the body.4 Among malignant tumors of the oral cavity, squamous cell carcinomas are the most frequent type (90%-98%), and lymphomas and melanoma are the most outstanding among the remaining 2% to 10%. Both for lymphoma and mucosal melanoma, the most common sites of involvement are the soft tissues of the oral cavity, palatal mucosa, gingiva, tongue, cheeks, floor of the mouth, and lips.4 Although mucosal melanoma lesions usually are characterized by pigmented and ulcerated lesions, amelanotic variants also should be kept in mind. Histopathologic examination is mandatory for diagnosis.
Intralesional chemotherapy with vinblastine or bleomycin, radiotherapy, electrochemotherapy, systemic antiretroviral therapy (ie, HAART), and chemotherapy with daunorubicin and pegylated liposomal doxorubicin are the main treatment options.5,6 The immune system activator role of HAART leads to an increased CD4 count and reduces HIV proteins, which helps induction of the proliferation and neovascularization of KS tumor cells.3 This effect may help resolution of KS with localized involvement and allows physicians to utilize HAART without any other additional local and systemic chemotherapy treatment.
- Fatahzadeh M, Schwartz RA. Oral Kaposi's sarcoma: a review and update. Int J Dermatol. 2013;52:666-672.
- Martorano LM, Cannella JD, Lloyd JR. Mucocutaneous presentation of Kaposi sarcoma in an asymptomatic human immunodeficiency virus-positive man. Cutis. 2015;95:E19-E22.
- Stebbing J, Portsmouth S, Gazzard B. How does HAART lead to the resolution of Kaposi's sarcoma? J Antimicrobial Chemother. 2003;51:1095-1098.
- Guevara-Canales JO, Morales-Vadillo R, Sacsaquispe-Contreras SJ, et al. Malignant lymphoma of the oral cavity and the maxillofacial region: overall survivalprognostic factors. Med Oral Patol Oral Cir Bucal. 2013;18:E619-E626.
- Donato V, Guarnaccia R, Dognini J, et al. Radiation therapy in the treatment of HIV-related Kaposi's sarcoma. Anticancer Res. 2013;33:2153-2157.
- Gbabe OF, Okwundu CI, Dedicoat M, et al. Treatment of severe or progressive Kaposi's sarcoma in HIV-infected adults. Cochrane Database Syst Rev. 2014:CD003256.
- Fatahzadeh M, Schwartz RA. Oral Kaposi's sarcoma: a review and update. Int J Dermatol. 2013;52:666-672.
- Martorano LM, Cannella JD, Lloyd JR. Mucocutaneous presentation of Kaposi sarcoma in an asymptomatic human immunodeficiency virus-positive man. Cutis. 2015;95:E19-E22.
- Stebbing J, Portsmouth S, Gazzard B. How does HAART lead to the resolution of Kaposi's sarcoma? J Antimicrobial Chemother. 2003;51:1095-1098.
- Guevara-Canales JO, Morales-Vadillo R, Sacsaquispe-Contreras SJ, et al. Malignant lymphoma of the oral cavity and the maxillofacial region: overall survivalprognostic factors. Med Oral Patol Oral Cir Bucal. 2013;18:E619-E626.
- Donato V, Guarnaccia R, Dognini J, et al. Radiation therapy in the treatment of HIV-related Kaposi's sarcoma. Anticancer Res. 2013;33:2153-2157.
- Gbabe OF, Okwundu CI, Dedicoat M, et al. Treatment of severe or progressive Kaposi's sarcoma in HIV-infected adults. Cochrane Database Syst Rev. 2014:CD003256.
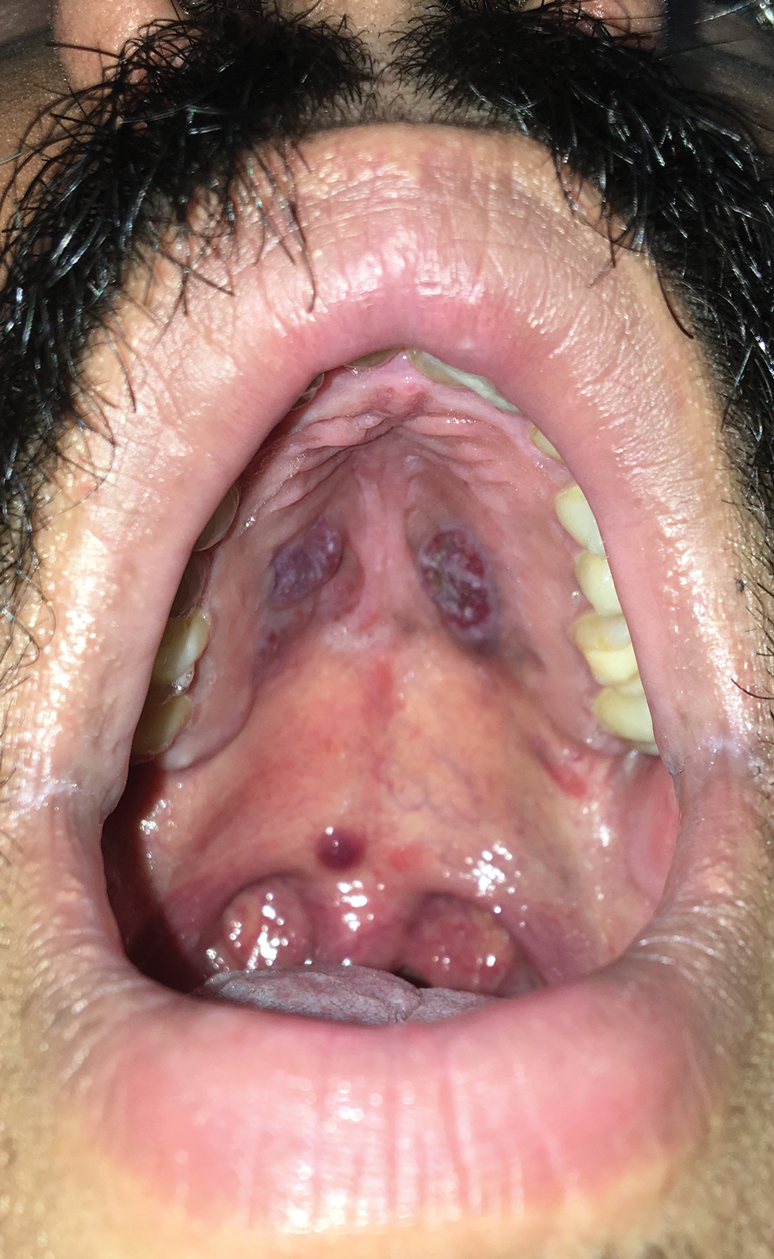
A 30-year-old man presented to our outpatient clinic with rapidly growing, ulcerated, violaceous lesions on the hard palate of 4 months' duration. Physical examination revealed approximately 2.0×1.5-cm, centrally ulcerated, violaceous, nodular lesions on the hard palate, as well as a 4-mm pinkish papular lesion on the soft palate.
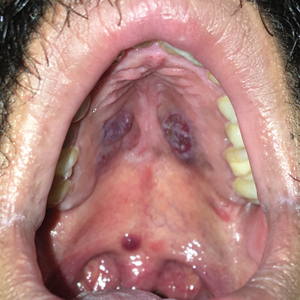
Cystic Scalp Lesion
The Diagnosis: Merkel Cell Carcinoma
An excisional biopsy revealed that the dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma (Figure 1). The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli. There was a tendency for smudgy artifacts at the periphery of the infiltrate, and the cells had relatively scant cytoplasm with slight streaming. Occasional apoptotic forms were present. Immunohistochemistry showed strong dotlike staining with cytokeratin 20 and moderate positivity with synaptophysin and chromogranin A (Figure 2). Unusually, there also was weak staining in a few tumor cells with thyroid transcription factor 1, a marker usually indicative of small cell carcinoma of the lungs that typically is negative in Merkel cell carcinoma (MCC). A second thyroid transcription factor 1 monoclonal antibody used in a double immunostain for lung adenocarcinomas was completely negative. This second antibody is more specific but less sensitive than the stand-alone version. The skin biopsy results confirmed the diagnosis of MCC. Given the patient's frailty and comorbidities, wide local excision was not performed and the patient was referred to radiation oncology. He died several months later from metastatic MCC.
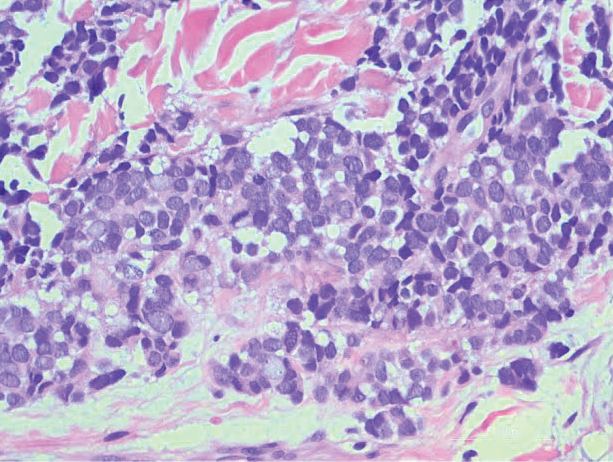
dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma. The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli (H&E, original magnification ×200).
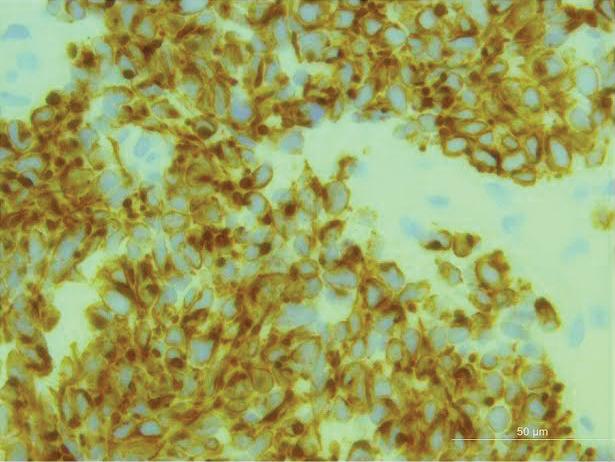
Merkel cell carcinoma (original magnification ×200).
Merkel cell carcinoma is an uncommon skin malignancy that can be easily mistaken for other conditions if the clinician is not familiar with its typical presentation. It most commonly is found on the head and neck in elderly individuals, most often aged 60 to 80 years,1 with a notable history of sun exposure and/or immunosuppression. It is an aggressive skin cancer that originally was thought to be due to pathogenic changes of Merkel cells,2 which are specialized touch receptors located at the dermoepidermal junction of the skin; however, newer evidence has suggested that MCC arises from malignant changes to skin stem cells.3 It shares more characteristics with extracutaneous neuroendocrine tumors and is more aptly labeled by pathologists as a primary neuroendocrine carcinoma of the skin.4
The frequency of MCC is highest in Australia, likely due to intense sun exposure, where the age-adjusted incidence rate reported in Queensland was 1.6 per 100,000 individuals from 2006 to 2010.5 The lowest incidence rates were reported in Finland (0.11 and 0.12 per 100,000 males and females, respectively)6 and Denmark (2.2 cases per million person-years).7 The clinical features of MCC are summarized by the mnemonic AEIOU: asymptomatic/lack of tenderness, expanding rapidly, immune suppression, older than 50 years, UV-exposed site on a person with fair skin.8 In a 2008 study of 195 patients, 89% of primary MCC lesions met 3 or more criteria, 32% met 4 or more criteria, and 7% met all 5 criteria.8
The classic presentation of MCC is a pink-red to violaceous nodule on the head or neck in an elderly patient, but there is a need to maintain suspicion of malignancy when examining a presumed infected cystic lesion, especially when a round of antibiotics has not ameliorated the symptoms. According to Heath et al,8 of 106 patients treated for MCC, 56% of first clinical impressions were benign. A PubMed and Scopus search was performed with the MeSH headings Merkel cell carcinoma +/- presentation to uncover similar unusual presentations between 1970 and the present day. Merkel cell carcinoma has been misdiagnosed as seemingly benign lesions including lipoma,9 allergic contact dermatitis,10 and atheroma.11 The differential diagnosis of MCC also includes cysts, amelanotic melanoma, basal cell carcinoma, dermatofibrosarcoma protuberans, squamous cell carcinoma, fungal kerion, leiomyosarcoma, neurothekeoma, abscesses, and cutaneous lymphoma.
Merkel cell polyomavirus has been implicated in the malignant transformation of MCC. It is a small, human, nonenveloped, double-stranded DNA virus1 and is found in approximately 70% to 80% of MCC cases.12 Merkel cell polyomavirus is a respiratory tract pathogen that is acquired by immunocompetent infants; it integrates itself into the host's genome and then enters a long latency period to later reactivate in immunocompromised adults.13
Wide local excision down to fascia is the mainstay of treatment of MCC, with recommended margins of 1 to 2 cm.14 Mohs micrographic surgery also can be considered.15 Similar to other neuroendocrine tumors, MCC is considered a radiosensitive tumor; radiation likely improves local control and is recommended in early-stage disease.16,17 It also has been described as the sole treatment modality in patients who are not candidates for surgery. The role of chemotherapy is more controversial, as responses do not appear to be long-lasting but should be considered in patients with advanced disease.14,18 There have been major advances in immunotherapy with the recent approvals of avelumab, an anti-PD-L1 inhibitor,19 and pembrolizumab,20 an anti-PD-1 inhibitor, for metastatic MCC. Clinical trials for MCC using kinase inhibitors and somatostatin analogues currently are ongoing.21
Several studies have demonstrated high rates of occult nodal disease in clinically node-negative patients, which has led to widespread use of sentinel lymph node biopsies.22,23 A sentinel lymph node biopsy is recommended at the time of surgery to aid with treatment decisions and prognosis.24
Merkel cell carcinoma is highly aggressive, and more than one-third of patients die from their disease, making it twice as lethal as melanoma. Overall survival rates remain low (5-year overall survival, 0%-18%) for advanced disease.5 Unfortunately, progression to metastasis is common and most often occurs within 2 years of diagnosis.17,25 Follow-up after treatment of MCC is crucial, with the 2019 National Comprehensive Cancer Network (NCCN) guidelines suggesting a physical examination with complete skin and complete lymph node examination every 3 to 6 months for 3 years and every 6 to 12 months thereafter.15
This case is an important reminder to include MCC in the differential diagnosis of presumed infected cysts, particularly on sun-exposed sites in elderly patients, as our patient was treated with antibiotics twice without improvement. An infected cyst with a lack of response to antibiotics should alert clinicians to the potential of malignancy.
- Sourvinos G, Mammas IN, Spandidos GA. 2015 Merkel cell polyoma virus infections in childhood. Arch Virol. 2015;160:887-892.
- Sibley RK, Rosai J, Foucar E, et al. Neuroendocrine (Merkel cell) carcinoma of the skin. a histologic and ultrastructural study of two cases. Am J Surg Pathol. 1980;4:211-221.
- Tilling T, Moll I. Which are the cells of origin in Merkel cell carcinoma? J Skin Cancer. 2012;2012:1-7.
- Succaria F, Radfar A, Bhawan J. Merkel cell carcinoma (primary neuroendocrine carcinoma of skin) mimicking basal cell carcinoma with review of different histopathologic features. Am J Dermatopathol. 2014;36:160-166.
- Youlden DR, Soyer HP, Youl PH, et al. Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993-2010. JAMA Dermatol. 2014;150:864-872.
- Kukko H, Böhling T, Koljonen V, et al. Merkel cell carcinoma--a population-based epidemiological study in Finland with a clinical series of 181 cases. Eur J Cancer. 2012;48:737-742.
- Kaae J, Hansen AV, Biggar RJ, et al. Merkel cell carcinoma: incidence, mortality, and risk of other cancers. J Natl Cancer Inst. 2010;102:793-801.
- Heath M, Jaimes N, Lamos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis of 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;59:375-381.
- Sarma DP, Heagley DE, Chalupa J, et al. An unusual clinical presentation of Merkel cell carcinoma: a case report. Case Rep Med. 2010;2010:905414.
- Craven E, Alexandroff A, Liu JK, et al. Merkel cell carcinoma mistaken for allergic contact dermatitis. BMJ. 2015;351:h4635.
- Kinoshita A, Hoashi T, Okazaki S, et al. Atypical case of Merkel cell carcinoma difficult to diagnose clinically. J Dermatol. 2017;44:E158-E159.
- Donepudi S, DeConti LC, Samlowski WE. Recent advances in the understanding of the genetics, etiology, and treatment of Merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
- Abedi Kiasari B, Vallely PJ, Klapper PE. Merkel cell polyoma virus DNA in immunocompetent and immunocompromised patients with respiratory disease. J Med Virol. 2011;83:2220-2224.
- Tai P. A practical update of surgical management of Merkel cell carcinoma of the skin. ISRN Surg. 2013;2013:850797.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Merkel Cell Carcinoma. Version 2.2019. Fort Washington, PA: National Comprehensive Cancer Network; 2019.
- Jabbour J. Merkel cell carcinoma: assessing the effect of wide local excision, lymph node dissection, and radiotherapy on recurrence and survival in early-stage disease--results from a review of 82 consecutive cases diagnosed between 1992 and 2004. Ann Surg Oncol. 2007;14:1943-1952.
- Medina-Franco H, Urist MM, Fiveash J, et al. Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol. 2001;8:204-208.
- Akhtar S, Oza KK, Wright J. Merkel cell carcinoma: report of 10 cases and review of the literature. J Am Acad Dermatol 2000;43:755-767.
- Palla AR, Doll D. Immunotherapy in Merkel cell carcinoma: role of avelumab. Immunotargets Ther. 2018;7:15-19.
- FDA approves pembrolizumab for Merkel cell carcinoma. US Food & Drug Administration website. http://www.fda.gov/Drugs/Information OnDrugs/ApprovedDrugs/ucm628867.htm. Published December 19, 2018. Accessed April 23, 2019.
- Schadendorff D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: epidemiology, prognosis, therapy, and unmet medical needs. Eur J Cancer. 2017;71:53-69.
- Schwartz JL, Griffith KA, Lowe L, et al. Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol. 2011;29:1036-1041.
- Kachare SD, Wong JH, Vohra NA, et al. Sentinel lymph node biopsy is associated with improved survival in Merkel cell carcinoma. Ann Surg Oncol. 2014;21:1624-1630.
- Gupta SG, Wang LC, Penas LC, et al. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: the Dana-Farber experience and meta-analysis of the literature. Arch Dermatol. 2006;142:685-690.
- Bajetta E, Celio L, Platania M, et al. Single-institution series of early-stage Merkel cell carcinoma: long-term outcomes in 95 patients managed with surgery alone. Ann Surg Oncol. 2009;16:2985-2993.
The Diagnosis: Merkel Cell Carcinoma
An excisional biopsy revealed that the dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma (Figure 1). The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli. There was a tendency for smudgy artifacts at the periphery of the infiltrate, and the cells had relatively scant cytoplasm with slight streaming. Occasional apoptotic forms were present. Immunohistochemistry showed strong dotlike staining with cytokeratin 20 and moderate positivity with synaptophysin and chromogranin A (Figure 2). Unusually, there also was weak staining in a few tumor cells with thyroid transcription factor 1, a marker usually indicative of small cell carcinoma of the lungs that typically is negative in Merkel cell carcinoma (MCC). A second thyroid transcription factor 1 monoclonal antibody used in a double immunostain for lung adenocarcinomas was completely negative. This second antibody is more specific but less sensitive than the stand-alone version. The skin biopsy results confirmed the diagnosis of MCC. Given the patient's frailty and comorbidities, wide local excision was not performed and the patient was referred to radiation oncology. He died several months later from metastatic MCC.
dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma. The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli (H&E, original magnification ×200).
Merkel cell carcinoma (original magnification ×200).
Merkel cell carcinoma is an uncommon skin malignancy that can be easily mistaken for other conditions if the clinician is not familiar with its typical presentation. It most commonly is found on the head and neck in elderly individuals, most often aged 60 to 80 years,1 with a notable history of sun exposure and/or immunosuppression. It is an aggressive skin cancer that originally was thought to be due to pathogenic changes of Merkel cells,2 which are specialized touch receptors located at the dermoepidermal junction of the skin; however, newer evidence has suggested that MCC arises from malignant changes to skin stem cells.3 It shares more characteristics with extracutaneous neuroendocrine tumors and is more aptly labeled by pathologists as a primary neuroendocrine carcinoma of the skin.4
The frequency of MCC is highest in Australia, likely due to intense sun exposure, where the age-adjusted incidence rate reported in Queensland was 1.6 per 100,000 individuals from 2006 to 2010.5 The lowest incidence rates were reported in Finland (0.11 and 0.12 per 100,000 males and females, respectively)6 and Denmark (2.2 cases per million person-years).7 The clinical features of MCC are summarized by the mnemonic AEIOU: asymptomatic/lack of tenderness, expanding rapidly, immune suppression, older than 50 years, UV-exposed site on a person with fair skin.8 In a 2008 study of 195 patients, 89% of primary MCC lesions met 3 or more criteria, 32% met 4 or more criteria, and 7% met all 5 criteria.8
The classic presentation of MCC is a pink-red to violaceous nodule on the head or neck in an elderly patient, but there is a need to maintain suspicion of malignancy when examining a presumed infected cystic lesion, especially when a round of antibiotics has not ameliorated the symptoms. According to Heath et al,8 of 106 patients treated for MCC, 56% of first clinical impressions were benign. A PubMed and Scopus search was performed with the MeSH headings Merkel cell carcinoma +/- presentation to uncover similar unusual presentations between 1970 and the present day. Merkel cell carcinoma has been misdiagnosed as seemingly benign lesions including lipoma,9 allergic contact dermatitis,10 and atheroma.11 The differential diagnosis of MCC also includes cysts, amelanotic melanoma, basal cell carcinoma, dermatofibrosarcoma protuberans, squamous cell carcinoma, fungal kerion, leiomyosarcoma, neurothekeoma, abscesses, and cutaneous lymphoma.
Merkel cell polyomavirus has been implicated in the malignant transformation of MCC. It is a small, human, nonenveloped, double-stranded DNA virus1 and is found in approximately 70% to 80% of MCC cases.12 Merkel cell polyomavirus is a respiratory tract pathogen that is acquired by immunocompetent infants; it integrates itself into the host's genome and then enters a long latency period to later reactivate in immunocompromised adults.13
Wide local excision down to fascia is the mainstay of treatment of MCC, with recommended margins of 1 to 2 cm.14 Mohs micrographic surgery also can be considered.15 Similar to other neuroendocrine tumors, MCC is considered a radiosensitive tumor; radiation likely improves local control and is recommended in early-stage disease.16,17 It also has been described as the sole treatment modality in patients who are not candidates for surgery. The role of chemotherapy is more controversial, as responses do not appear to be long-lasting but should be considered in patients with advanced disease.14,18 There have been major advances in immunotherapy with the recent approvals of avelumab, an anti-PD-L1 inhibitor,19 and pembrolizumab,20 an anti-PD-1 inhibitor, for metastatic MCC. Clinical trials for MCC using kinase inhibitors and somatostatin analogues currently are ongoing.21
Several studies have demonstrated high rates of occult nodal disease in clinically node-negative patients, which has led to widespread use of sentinel lymph node biopsies.22,23 A sentinel lymph node biopsy is recommended at the time of surgery to aid with treatment decisions and prognosis.24
Merkel cell carcinoma is highly aggressive, and more than one-third of patients die from their disease, making it twice as lethal as melanoma. Overall survival rates remain low (5-year overall survival, 0%-18%) for advanced disease.5 Unfortunately, progression to metastasis is common and most often occurs within 2 years of diagnosis.17,25 Follow-up after treatment of MCC is crucial, with the 2019 National Comprehensive Cancer Network (NCCN) guidelines suggesting a physical examination with complete skin and complete lymph node examination every 3 to 6 months for 3 years and every 6 to 12 months thereafter.15
This case is an important reminder to include MCC in the differential diagnosis of presumed infected cysts, particularly on sun-exposed sites in elderly patients, as our patient was treated with antibiotics twice without improvement. An infected cyst with a lack of response to antibiotics should alert clinicians to the potential of malignancy.
The Diagnosis: Merkel Cell Carcinoma
An excisional biopsy revealed that the dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma (Figure 1). The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli. There was a tendency for smudgy artifacts at the periphery of the infiltrate, and the cells had relatively scant cytoplasm with slight streaming. Occasional apoptotic forms were present. Immunohistochemistry showed strong dotlike staining with cytokeratin 20 and moderate positivity with synaptophysin and chromogranin A (Figure 2). Unusually, there also was weak staining in a few tumor cells with thyroid transcription factor 1, a marker usually indicative of small cell carcinoma of the lungs that typically is negative in Merkel cell carcinoma (MCC). A second thyroid transcription factor 1 monoclonal antibody used in a double immunostain for lung adenocarcinomas was completely negative. This second antibody is more specific but less sensitive than the stand-alone version. The skin biopsy results confirmed the diagnosis of MCC. Given the patient's frailty and comorbidities, wide local excision was not performed and the patient was referred to radiation oncology. He died several months later from metastatic MCC.
dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma. The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli (H&E, original magnification ×200).
Merkel cell carcinoma (original magnification ×200).
Merkel cell carcinoma is an uncommon skin malignancy that can be easily mistaken for other conditions if the clinician is not familiar with its typical presentation. It most commonly is found on the head and neck in elderly individuals, most often aged 60 to 80 years,1 with a notable history of sun exposure and/or immunosuppression. It is an aggressive skin cancer that originally was thought to be due to pathogenic changes of Merkel cells,2 which are specialized touch receptors located at the dermoepidermal junction of the skin; however, newer evidence has suggested that MCC arises from malignant changes to skin stem cells.3 It shares more characteristics with extracutaneous neuroendocrine tumors and is more aptly labeled by pathologists as a primary neuroendocrine carcinoma of the skin.4
The frequency of MCC is highest in Australia, likely due to intense sun exposure, where the age-adjusted incidence rate reported in Queensland was 1.6 per 100,000 individuals from 2006 to 2010.5 The lowest incidence rates were reported in Finland (0.11 and 0.12 per 100,000 males and females, respectively)6 and Denmark (2.2 cases per million person-years).7 The clinical features of MCC are summarized by the mnemonic AEIOU: asymptomatic/lack of tenderness, expanding rapidly, immune suppression, older than 50 years, UV-exposed site on a person with fair skin.8 In a 2008 study of 195 patients, 89% of primary MCC lesions met 3 or more criteria, 32% met 4 or more criteria, and 7% met all 5 criteria.8
The classic presentation of MCC is a pink-red to violaceous nodule on the head or neck in an elderly patient, but there is a need to maintain suspicion of malignancy when examining a presumed infected cystic lesion, especially when a round of antibiotics has not ameliorated the symptoms. According to Heath et al,8 of 106 patients treated for MCC, 56% of first clinical impressions were benign. A PubMed and Scopus search was performed with the MeSH headings Merkel cell carcinoma +/- presentation to uncover similar unusual presentations between 1970 and the present day. Merkel cell carcinoma has been misdiagnosed as seemingly benign lesions including lipoma,9 allergic contact dermatitis,10 and atheroma.11 The differential diagnosis of MCC also includes cysts, amelanotic melanoma, basal cell carcinoma, dermatofibrosarcoma protuberans, squamous cell carcinoma, fungal kerion, leiomyosarcoma, neurothekeoma, abscesses, and cutaneous lymphoma.
Merkel cell polyomavirus has been implicated in the malignant transformation of MCC. It is a small, human, nonenveloped, double-stranded DNA virus1 and is found in approximately 70% to 80% of MCC cases.12 Merkel cell polyomavirus is a respiratory tract pathogen that is acquired by immunocompetent infants; it integrates itself into the host's genome and then enters a long latency period to later reactivate in immunocompromised adults.13
Wide local excision down to fascia is the mainstay of treatment of MCC, with recommended margins of 1 to 2 cm.14 Mohs micrographic surgery also can be considered.15 Similar to other neuroendocrine tumors, MCC is considered a radiosensitive tumor; radiation likely improves local control and is recommended in early-stage disease.16,17 It also has been described as the sole treatment modality in patients who are not candidates for surgery. The role of chemotherapy is more controversial, as responses do not appear to be long-lasting but should be considered in patients with advanced disease.14,18 There have been major advances in immunotherapy with the recent approvals of avelumab, an anti-PD-L1 inhibitor,19 and pembrolizumab,20 an anti-PD-1 inhibitor, for metastatic MCC. Clinical trials for MCC using kinase inhibitors and somatostatin analogues currently are ongoing.21
Several studies have demonstrated high rates of occult nodal disease in clinically node-negative patients, which has led to widespread use of sentinel lymph node biopsies.22,23 A sentinel lymph node biopsy is recommended at the time of surgery to aid with treatment decisions and prognosis.24
Merkel cell carcinoma is highly aggressive, and more than one-third of patients die from their disease, making it twice as lethal as melanoma. Overall survival rates remain low (5-year overall survival, 0%-18%) for advanced disease.5 Unfortunately, progression to metastasis is common and most often occurs within 2 years of diagnosis.17,25 Follow-up after treatment of MCC is crucial, with the 2019 National Comprehensive Cancer Network (NCCN) guidelines suggesting a physical examination with complete skin and complete lymph node examination every 3 to 6 months for 3 years and every 6 to 12 months thereafter.15
This case is an important reminder to include MCC in the differential diagnosis of presumed infected cysts, particularly on sun-exposed sites in elderly patients, as our patient was treated with antibiotics twice without improvement. An infected cyst with a lack of response to antibiotics should alert clinicians to the potential of malignancy.
- Sourvinos G, Mammas IN, Spandidos GA. 2015 Merkel cell polyoma virus infections in childhood. Arch Virol. 2015;160:887-892.
- Sibley RK, Rosai J, Foucar E, et al. Neuroendocrine (Merkel cell) carcinoma of the skin. a histologic and ultrastructural study of two cases. Am J Surg Pathol. 1980;4:211-221.
- Tilling T, Moll I. Which are the cells of origin in Merkel cell carcinoma? J Skin Cancer. 2012;2012:1-7.
- Succaria F, Radfar A, Bhawan J. Merkel cell carcinoma (primary neuroendocrine carcinoma of skin) mimicking basal cell carcinoma with review of different histopathologic features. Am J Dermatopathol. 2014;36:160-166.
- Youlden DR, Soyer HP, Youl PH, et al. Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993-2010. JAMA Dermatol. 2014;150:864-872.
- Kukko H, Böhling T, Koljonen V, et al. Merkel cell carcinoma--a population-based epidemiological study in Finland with a clinical series of 181 cases. Eur J Cancer. 2012;48:737-742.
- Kaae J, Hansen AV, Biggar RJ, et al. Merkel cell carcinoma: incidence, mortality, and risk of other cancers. J Natl Cancer Inst. 2010;102:793-801.
- Heath M, Jaimes N, Lamos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis of 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;59:375-381.
- Sarma DP, Heagley DE, Chalupa J, et al. An unusual clinical presentation of Merkel cell carcinoma: a case report. Case Rep Med. 2010;2010:905414.
- Craven E, Alexandroff A, Liu JK, et al. Merkel cell carcinoma mistaken for allergic contact dermatitis. BMJ. 2015;351:h4635.
- Kinoshita A, Hoashi T, Okazaki S, et al. Atypical case of Merkel cell carcinoma difficult to diagnose clinically. J Dermatol. 2017;44:E158-E159.
- Donepudi S, DeConti LC, Samlowski WE. Recent advances in the understanding of the genetics, etiology, and treatment of Merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
- Abedi Kiasari B, Vallely PJ, Klapper PE. Merkel cell polyoma virus DNA in immunocompetent and immunocompromised patients with respiratory disease. J Med Virol. 2011;83:2220-2224.
- Tai P. A practical update of surgical management of Merkel cell carcinoma of the skin. ISRN Surg. 2013;2013:850797.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Merkel Cell Carcinoma. Version 2.2019. Fort Washington, PA: National Comprehensive Cancer Network; 2019.
- Jabbour J. Merkel cell carcinoma: assessing the effect of wide local excision, lymph node dissection, and radiotherapy on recurrence and survival in early-stage disease--results from a review of 82 consecutive cases diagnosed between 1992 and 2004. Ann Surg Oncol. 2007;14:1943-1952.
- Medina-Franco H, Urist MM, Fiveash J, et al. Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol. 2001;8:204-208.
- Akhtar S, Oza KK, Wright J. Merkel cell carcinoma: report of 10 cases and review of the literature. J Am Acad Dermatol 2000;43:755-767.
- Palla AR, Doll D. Immunotherapy in Merkel cell carcinoma: role of avelumab. Immunotargets Ther. 2018;7:15-19.
- FDA approves pembrolizumab for Merkel cell carcinoma. US Food & Drug Administration website. http://www.fda.gov/Drugs/Information OnDrugs/ApprovedDrugs/ucm628867.htm. Published December 19, 2018. Accessed April 23, 2019.
- Schadendorff D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: epidemiology, prognosis, therapy, and unmet medical needs. Eur J Cancer. 2017;71:53-69.
- Schwartz JL, Griffith KA, Lowe L, et al. Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol. 2011;29:1036-1041.
- Kachare SD, Wong JH, Vohra NA, et al. Sentinel lymph node biopsy is associated with improved survival in Merkel cell carcinoma. Ann Surg Oncol. 2014;21:1624-1630.
- Gupta SG, Wang LC, Penas LC, et al. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: the Dana-Farber experience and meta-analysis of the literature. Arch Dermatol. 2006;142:685-690.
- Bajetta E, Celio L, Platania M, et al. Single-institution series of early-stage Merkel cell carcinoma: long-term outcomes in 95 patients managed with surgery alone. Ann Surg Oncol. 2009;16:2985-2993.
- Sourvinos G, Mammas IN, Spandidos GA. 2015 Merkel cell polyoma virus infections in childhood. Arch Virol. 2015;160:887-892.
- Sibley RK, Rosai J, Foucar E, et al. Neuroendocrine (Merkel cell) carcinoma of the skin. a histologic and ultrastructural study of two cases. Am J Surg Pathol. 1980;4:211-221.
- Tilling T, Moll I. Which are the cells of origin in Merkel cell carcinoma? J Skin Cancer. 2012;2012:1-7.
- Succaria F, Radfar A, Bhawan J. Merkel cell carcinoma (primary neuroendocrine carcinoma of skin) mimicking basal cell carcinoma with review of different histopathologic features. Am J Dermatopathol. 2014;36:160-166.
- Youlden DR, Soyer HP, Youl PH, et al. Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993-2010. JAMA Dermatol. 2014;150:864-872.
- Kukko H, Böhling T, Koljonen V, et al. Merkel cell carcinoma--a population-based epidemiological study in Finland with a clinical series of 181 cases. Eur J Cancer. 2012;48:737-742.
- Kaae J, Hansen AV, Biggar RJ, et al. Merkel cell carcinoma: incidence, mortality, and risk of other cancers. J Natl Cancer Inst. 2010;102:793-801.
- Heath M, Jaimes N, Lamos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis of 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;59:375-381.
- Sarma DP, Heagley DE, Chalupa J, et al. An unusual clinical presentation of Merkel cell carcinoma: a case report. Case Rep Med. 2010;2010:905414.
- Craven E, Alexandroff A, Liu JK, et al. Merkel cell carcinoma mistaken for allergic contact dermatitis. BMJ. 2015;351:h4635.
- Kinoshita A, Hoashi T, Okazaki S, et al. Atypical case of Merkel cell carcinoma difficult to diagnose clinically. J Dermatol. 2017;44:E158-E159.
- Donepudi S, DeConti LC, Samlowski WE. Recent advances in the understanding of the genetics, etiology, and treatment of Merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
- Abedi Kiasari B, Vallely PJ, Klapper PE. Merkel cell polyoma virus DNA in immunocompetent and immunocompromised patients with respiratory disease. J Med Virol. 2011;83:2220-2224.
- Tai P. A practical update of surgical management of Merkel cell carcinoma of the skin. ISRN Surg. 2013;2013:850797.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Merkel Cell Carcinoma. Version 2.2019. Fort Washington, PA: National Comprehensive Cancer Network; 2019.
- Jabbour J. Merkel cell carcinoma: assessing the effect of wide local excision, lymph node dissection, and radiotherapy on recurrence and survival in early-stage disease--results from a review of 82 consecutive cases diagnosed between 1992 and 2004. Ann Surg Oncol. 2007;14:1943-1952.
- Medina-Franco H, Urist MM, Fiveash J, et al. Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol. 2001;8:204-208.
- Akhtar S, Oza KK, Wright J. Merkel cell carcinoma: report of 10 cases and review of the literature. J Am Acad Dermatol 2000;43:755-767.
- Palla AR, Doll D. Immunotherapy in Merkel cell carcinoma: role of avelumab. Immunotargets Ther. 2018;7:15-19.
- FDA approves pembrolizumab for Merkel cell carcinoma. US Food & Drug Administration website. http://www.fda.gov/Drugs/Information OnDrugs/ApprovedDrugs/ucm628867.htm. Published December 19, 2018. Accessed April 23, 2019.
- Schadendorff D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: epidemiology, prognosis, therapy, and unmet medical needs. Eur J Cancer. 2017;71:53-69.
- Schwartz JL, Griffith KA, Lowe L, et al. Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol. 2011;29:1036-1041.
- Kachare SD, Wong JH, Vohra NA, et al. Sentinel lymph node biopsy is associated with improved survival in Merkel cell carcinoma. Ann Surg Oncol. 2014;21:1624-1630.
- Gupta SG, Wang LC, Penas LC, et al. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: the Dana-Farber experience and meta-analysis of the literature. Arch Dermatol. 2006;142:685-690.
- Bajetta E, Celio L, Platania M, et al. Single-institution series of early-stage Merkel cell carcinoma: long-term outcomes in 95 patients managed with surgery alone. Ann Surg Oncol. 2009;16:2985-2993.

A frail 85-year-old man presented to the emergency department for treatment of a 4.0.2 ×2.5-cm, erythematous, tender nodule on the scalp. The area was increasingly painful with persistent throbbing, which led to sleep disruption. The nodule did not express any material and was not aspirated or surgically treated. The lesion had been present for 1 to 2 years and was small and stable in size until it grew rapidly in the 6 weeks prior to presentation. The patient initially presented to his general practitioner during this period of rapid growth and was diagnosed with an infected sebaceous cyst that was treated with a course of oral cephalexin without improvement. Bacterial or fungal cultures were not performed. No other similar lesions were present, but there was 1 palpable lymph node in the right posterior cervical chain. At the time of presentation to the emergency department, the patient felt well and denied weight loss, night sweats, or fevers. He was given a dose of intravenous cefazolin by the emergency physician and then was referred to surgery for management of an infected sebaceous cyst.
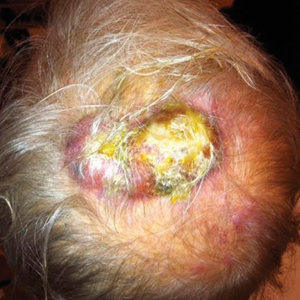
Coalescing Papules on Bilateral Mastectomy Scars
The Diagnosis: Scar Sarcoidosis
Although scars on both breasts were involved, the decision was made to biopsy the right breast because the patient reported more pain on the left breast. Biopsy showed noncaseating granulomas consistent with scar sarcoidosis (Figure). Additional screening tests were performed to evaluate for any systemic involvement of sarcoidosis, including a complete blood cell count, comprehensive metabolic panel, angiotensin-converting enzyme level, tuberculosis serology screening, electrocardiogram, chest radiograph, and pulmonary function tests. She also was referred to rheumatology and ophthalmology for consultation. The results of all screenings were within reference range, and no sign of systemic sarcoidosis was found. She was treated with hydrocortisone ointment 2.5% for several weeks without notable improvement. She elected not to pursue any additional treatment and to monitor the symptoms with close follow-up only. One year after the initial visit, the skin lesions spontaneously and notably improved.
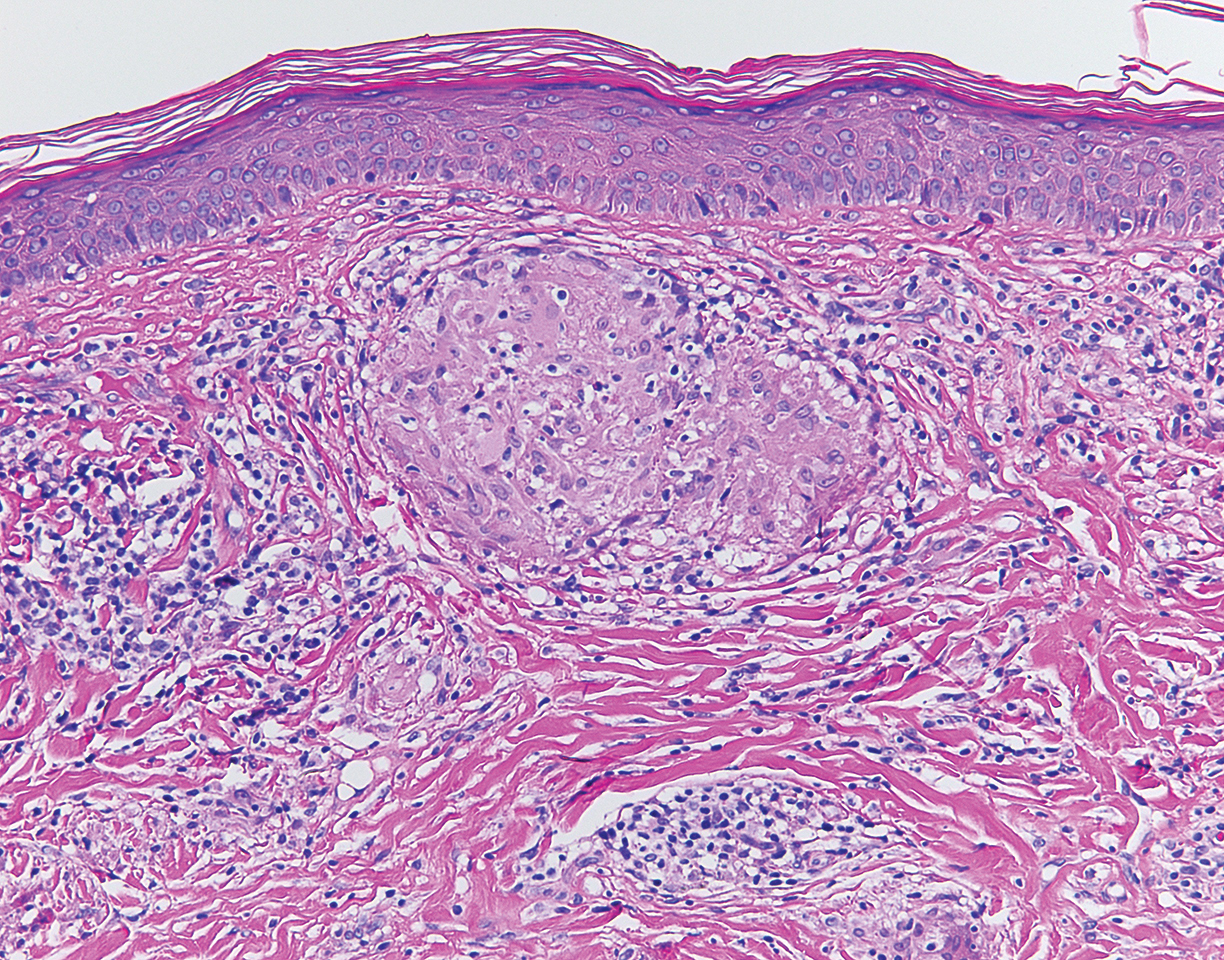
Sarcoidosis is a systemic granulomatous disorder of unknown etiology that most commonly affects the lungs. It also can involve the lymph nodes, liver, spleen, bones, gastrointestinal tract, eyes, and skin. Cutaneous sarcoidosis has been documented in the literature since the late 1800s and occurs in up to one-third of sarcoid patients.1 Cutaneous lesions developing within a preexisting scar is a well-known variant, occurring in 29% of patients with cutaneous sarcoidosis in one clinical study (N=818).2 There have been many reports describing scar sarcoidosis, with its development at prior sites of surgery, trauma, acne, or venipuncture.3 Other case reports have described variants of scar sarcoidosis developing at sites of hyaluronic acid injection, laser surgery, ritual scarification, tattoos, and desensitization injections, as well as prior herpes zoster infections.4-9
Cutaneous sarcoidosis has a wide range of clinical presentations. Lesions can be described as specific or nonspecific. Specific lesions demonstrate the typical sarcoid granuloma on histology and more often are seen in chronic disease, while nonspecific lesions more often are seen in acute disease.3,10 Scar sarcoidosis is an example of a specific lesion in which old scars become infiltrated with noncaseating granulomas. The granulomas typically are in the superficial dermis but may involve the full thickness of the dermis, extending into the subcutaneous tissue.11 The cause of granulomas developing in scars is unknown. Prior contamination of the scar with foreign material, possibly at the time of the trauma, is a possible underlying cause.12
Typical scar sarcoidosis presents as swollen, erythematous, indurated lesions with a purple-red hue that may become brown.3,12 Tenderness or pruritus also may be present.13 Interestingly, our patient's scar sarcoidosis presented with a yellow hue at both mastectomy sites.
Diagnosing scar sarcoidosis can be challenging. Patients are diagnosed with sarcoidosis when a compatible clinical or radiologic picture is present along with histologic evidence of a noncaseating granuloma and other potential causes are excluded.11 The differential includes an infectious etiology, other types of granulomatous dermatitis, hypertrophic scar, keloid, or foreign body granuloma.
Scar sarcoidosis can be isolated in occurrence. It also can precede or occur concomitantly or during a relapse of systemic sarcoidosis.10 Most commonly, patients with scar sarcoidosis also have systemic manifestations of sarcoidosis, and changing scars may be an indicator of disease exacerbation or relapse.10 For patients who only demonstrate specific skin lesions of cutaneous sarcoidosis, approximately 30% develop systemic involvement later in life.3 For this reason, close monitoring and regular follow-up are necessary.
Treatment of scar sarcoidosis is dependent on the extent of the disease and presence of systemic sarcoidosis. Topical and systemic corticosteroids, hydroxychloroquine, chloroquine phosphate, and methotrexate all have been shown to be helpful in treating cutaneous sarcoidosis.3 For scar sarcoidosis that is limited to only the scar site, as seen in our case, monitoring and close follow-up is acceptable. Topical steroids can be prescribed for symptomatic relief. Scar sarcoidosis can resolve slowly and spontaneously over time.10 Our patient notably improved 1 year after the initial presentation without treatment.
Scar sarcoidosis is a well-documented variant of cutaneous sarcoidosis that can have important implications for diagnosing systemic sarcoidosis. Although there are typical lesions that represent scar sarcoidosis, it is important to have a high degree of suspicion with any changing scar. Once diagnosed through biopsy, a thorough investigation for systemic signs of sarcoidosis needs to be performed to guide treatment.
- Bolognia JL, Jorizzo JL, Shaffer JV, eds. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med. 1983;52:525-533.
- Mañá J, Marcoval J, Graells J, et al. Cutaneous involvement in sarcoidosis: relationship to systemic disease. Arch Dermatol. 1997;133:882-888.
- Dal Sacco D, Cozzani E, Parodi A, et al. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol. 2005;44:411-412.
- Kormeili T, Neel V, Moy RL. Cutaneous sarcoidosis at sites of previous laser surgery. Cutis. 2004;73:53-55.
- Nayar M. Sarcoidosis on ritual scarification. Int J Dermatol. 1993;32:116-118.
- James WD, Elston DM, Berger TG, et al. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Healsmith MF, Hutchinson PE. The development of scar sarcoidosis at the site of desensitization injections. Clin Exp Dermatol. 1992;17:369-370.
- Singal A, Vij A, Pandhi D. Post herpes-zoster scar sarcoidosis with pulmonary involvement. Indian Dermatol Online J. 2014;5:77-79.
- Chudomirova K, Velichkova L, Anavi B, et al. Recurrent sarcoidosis in skin scars accompanying systemic sarcoidosis. J Eur Acad Dermatol Venereol. 2003;17:360-361.
- Selim A, Ehrsam E, Atassi MB, et al. Scar sarcoidosis: a case report and brief review. Cutis. 2006;78:418-422.
- Singal A, Thami GP, Goraya JS. Scar sarcoidosis in childhood: case report and review of the literature. Clin Exp Dermatol. 2005;30:244-246.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
The Diagnosis: Scar Sarcoidosis
Although scars on both breasts were involved, the decision was made to biopsy the right breast because the patient reported more pain on the left breast. Biopsy showed noncaseating granulomas consistent with scar sarcoidosis (Figure). Additional screening tests were performed to evaluate for any systemic involvement of sarcoidosis, including a complete blood cell count, comprehensive metabolic panel, angiotensin-converting enzyme level, tuberculosis serology screening, electrocardiogram, chest radiograph, and pulmonary function tests. She also was referred to rheumatology and ophthalmology for consultation. The results of all screenings were within reference range, and no sign of systemic sarcoidosis was found. She was treated with hydrocortisone ointment 2.5% for several weeks without notable improvement. She elected not to pursue any additional treatment and to monitor the symptoms with close follow-up only. One year after the initial visit, the skin lesions spontaneously and notably improved.
Sarcoidosis is a systemic granulomatous disorder of unknown etiology that most commonly affects the lungs. It also can involve the lymph nodes, liver, spleen, bones, gastrointestinal tract, eyes, and skin. Cutaneous sarcoidosis has been documented in the literature since the late 1800s and occurs in up to one-third of sarcoid patients.1 Cutaneous lesions developing within a preexisting scar is a well-known variant, occurring in 29% of patients with cutaneous sarcoidosis in one clinical study (N=818).2 There have been many reports describing scar sarcoidosis, with its development at prior sites of surgery, trauma, acne, or venipuncture.3 Other case reports have described variants of scar sarcoidosis developing at sites of hyaluronic acid injection, laser surgery, ritual scarification, tattoos, and desensitization injections, as well as prior herpes zoster infections.4-9
Cutaneous sarcoidosis has a wide range of clinical presentations. Lesions can be described as specific or nonspecific. Specific lesions demonstrate the typical sarcoid granuloma on histology and more often are seen in chronic disease, while nonspecific lesions more often are seen in acute disease.3,10 Scar sarcoidosis is an example of a specific lesion in which old scars become infiltrated with noncaseating granulomas. The granulomas typically are in the superficial dermis but may involve the full thickness of the dermis, extending into the subcutaneous tissue.11 The cause of granulomas developing in scars is unknown. Prior contamination of the scar with foreign material, possibly at the time of the trauma, is a possible underlying cause.12
Typical scar sarcoidosis presents as swollen, erythematous, indurated lesions with a purple-red hue that may become brown.3,12 Tenderness or pruritus also may be present.13 Interestingly, our patient's scar sarcoidosis presented with a yellow hue at both mastectomy sites.
Diagnosing scar sarcoidosis can be challenging. Patients are diagnosed with sarcoidosis when a compatible clinical or radiologic picture is present along with histologic evidence of a noncaseating granuloma and other potential causes are excluded.11 The differential includes an infectious etiology, other types of granulomatous dermatitis, hypertrophic scar, keloid, or foreign body granuloma.
Scar sarcoidosis can be isolated in occurrence. It also can precede or occur concomitantly or during a relapse of systemic sarcoidosis.10 Most commonly, patients with scar sarcoidosis also have systemic manifestations of sarcoidosis, and changing scars may be an indicator of disease exacerbation or relapse.10 For patients who only demonstrate specific skin lesions of cutaneous sarcoidosis, approximately 30% develop systemic involvement later in life.3 For this reason, close monitoring and regular follow-up are necessary.
Treatment of scar sarcoidosis is dependent on the extent of the disease and presence of systemic sarcoidosis. Topical and systemic corticosteroids, hydroxychloroquine, chloroquine phosphate, and methotrexate all have been shown to be helpful in treating cutaneous sarcoidosis.3 For scar sarcoidosis that is limited to only the scar site, as seen in our case, monitoring and close follow-up is acceptable. Topical steroids can be prescribed for symptomatic relief. Scar sarcoidosis can resolve slowly and spontaneously over time.10 Our patient notably improved 1 year after the initial presentation without treatment.
Scar sarcoidosis is a well-documented variant of cutaneous sarcoidosis that can have important implications for diagnosing systemic sarcoidosis. Although there are typical lesions that represent scar sarcoidosis, it is important to have a high degree of suspicion with any changing scar. Once diagnosed through biopsy, a thorough investigation for systemic signs of sarcoidosis needs to be performed to guide treatment.
The Diagnosis: Scar Sarcoidosis
Although scars on both breasts were involved, the decision was made to biopsy the right breast because the patient reported more pain on the left breast. Biopsy showed noncaseating granulomas consistent with scar sarcoidosis (Figure). Additional screening tests were performed to evaluate for any systemic involvement of sarcoidosis, including a complete blood cell count, comprehensive metabolic panel, angiotensin-converting enzyme level, tuberculosis serology screening, electrocardiogram, chest radiograph, and pulmonary function tests. She also was referred to rheumatology and ophthalmology for consultation. The results of all screenings were within reference range, and no sign of systemic sarcoidosis was found. She was treated with hydrocortisone ointment 2.5% for several weeks without notable improvement. She elected not to pursue any additional treatment and to monitor the symptoms with close follow-up only. One year after the initial visit, the skin lesions spontaneously and notably improved.
Sarcoidosis is a systemic granulomatous disorder of unknown etiology that most commonly affects the lungs. It also can involve the lymph nodes, liver, spleen, bones, gastrointestinal tract, eyes, and skin. Cutaneous sarcoidosis has been documented in the literature since the late 1800s and occurs in up to one-third of sarcoid patients.1 Cutaneous lesions developing within a preexisting scar is a well-known variant, occurring in 29% of patients with cutaneous sarcoidosis in one clinical study (N=818).2 There have been many reports describing scar sarcoidosis, with its development at prior sites of surgery, trauma, acne, or venipuncture.3 Other case reports have described variants of scar sarcoidosis developing at sites of hyaluronic acid injection, laser surgery, ritual scarification, tattoos, and desensitization injections, as well as prior herpes zoster infections.4-9
Cutaneous sarcoidosis has a wide range of clinical presentations. Lesions can be described as specific or nonspecific. Specific lesions demonstrate the typical sarcoid granuloma on histology and more often are seen in chronic disease, while nonspecific lesions more often are seen in acute disease.3,10 Scar sarcoidosis is an example of a specific lesion in which old scars become infiltrated with noncaseating granulomas. The granulomas typically are in the superficial dermis but may involve the full thickness of the dermis, extending into the subcutaneous tissue.11 The cause of granulomas developing in scars is unknown. Prior contamination of the scar with foreign material, possibly at the time of the trauma, is a possible underlying cause.12
Typical scar sarcoidosis presents as swollen, erythematous, indurated lesions with a purple-red hue that may become brown.3,12 Tenderness or pruritus also may be present.13 Interestingly, our patient's scar sarcoidosis presented with a yellow hue at both mastectomy sites.
Diagnosing scar sarcoidosis can be challenging. Patients are diagnosed with sarcoidosis when a compatible clinical or radiologic picture is present along with histologic evidence of a noncaseating granuloma and other potential causes are excluded.11 The differential includes an infectious etiology, other types of granulomatous dermatitis, hypertrophic scar, keloid, or foreign body granuloma.
Scar sarcoidosis can be isolated in occurrence. It also can precede or occur concomitantly or during a relapse of systemic sarcoidosis.10 Most commonly, patients with scar sarcoidosis also have systemic manifestations of sarcoidosis, and changing scars may be an indicator of disease exacerbation or relapse.10 For patients who only demonstrate specific skin lesions of cutaneous sarcoidosis, approximately 30% develop systemic involvement later in life.3 For this reason, close monitoring and regular follow-up are necessary.
Treatment of scar sarcoidosis is dependent on the extent of the disease and presence of systemic sarcoidosis. Topical and systemic corticosteroids, hydroxychloroquine, chloroquine phosphate, and methotrexate all have been shown to be helpful in treating cutaneous sarcoidosis.3 For scar sarcoidosis that is limited to only the scar site, as seen in our case, monitoring and close follow-up is acceptable. Topical steroids can be prescribed for symptomatic relief. Scar sarcoidosis can resolve slowly and spontaneously over time.10 Our patient notably improved 1 year after the initial presentation without treatment.
Scar sarcoidosis is a well-documented variant of cutaneous sarcoidosis that can have important implications for diagnosing systemic sarcoidosis. Although there are typical lesions that represent scar sarcoidosis, it is important to have a high degree of suspicion with any changing scar. Once diagnosed through biopsy, a thorough investigation for systemic signs of sarcoidosis needs to be performed to guide treatment.
- Bolognia JL, Jorizzo JL, Shaffer JV, eds. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med. 1983;52:525-533.
- Mañá J, Marcoval J, Graells J, et al. Cutaneous involvement in sarcoidosis: relationship to systemic disease. Arch Dermatol. 1997;133:882-888.
- Dal Sacco D, Cozzani E, Parodi A, et al. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol. 2005;44:411-412.
- Kormeili T, Neel V, Moy RL. Cutaneous sarcoidosis at sites of previous laser surgery. Cutis. 2004;73:53-55.
- Nayar M. Sarcoidosis on ritual scarification. Int J Dermatol. 1993;32:116-118.
- James WD, Elston DM, Berger TG, et al. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Healsmith MF, Hutchinson PE. The development of scar sarcoidosis at the site of desensitization injections. Clin Exp Dermatol. 1992;17:369-370.
- Singal A, Vij A, Pandhi D. Post herpes-zoster scar sarcoidosis with pulmonary involvement. Indian Dermatol Online J. 2014;5:77-79.
- Chudomirova K, Velichkova L, Anavi B, et al. Recurrent sarcoidosis in skin scars accompanying systemic sarcoidosis. J Eur Acad Dermatol Venereol. 2003;17:360-361.
- Selim A, Ehrsam E, Atassi MB, et al. Scar sarcoidosis: a case report and brief review. Cutis. 2006;78:418-422.
- Singal A, Thami GP, Goraya JS. Scar sarcoidosis in childhood: case report and review of the literature. Clin Exp Dermatol. 2005;30:244-246.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
- Bolognia JL, Jorizzo JL, Shaffer JV, eds. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med. 1983;52:525-533.
- Mañá J, Marcoval J, Graells J, et al. Cutaneous involvement in sarcoidosis: relationship to systemic disease. Arch Dermatol. 1997;133:882-888.
- Dal Sacco D, Cozzani E, Parodi A, et al. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol. 2005;44:411-412.
- Kormeili T, Neel V, Moy RL. Cutaneous sarcoidosis at sites of previous laser surgery. Cutis. 2004;73:53-55.
- Nayar M. Sarcoidosis on ritual scarification. Int J Dermatol. 1993;32:116-118.
- James WD, Elston DM, Berger TG, et al. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Healsmith MF, Hutchinson PE. The development of scar sarcoidosis at the site of desensitization injections. Clin Exp Dermatol. 1992;17:369-370.
- Singal A, Vij A, Pandhi D. Post herpes-zoster scar sarcoidosis with pulmonary involvement. Indian Dermatol Online J. 2014;5:77-79.
- Chudomirova K, Velichkova L, Anavi B, et al. Recurrent sarcoidosis in skin scars accompanying systemic sarcoidosis. J Eur Acad Dermatol Venereol. 2003;17:360-361.
- Selim A, Ehrsam E, Atassi MB, et al. Scar sarcoidosis: a case report and brief review. Cutis. 2006;78:418-422.
- Singal A, Thami GP, Goraya JS. Scar sarcoidosis in childhood: case report and review of the literature. Clin Exp Dermatol. 2005;30:244-246.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
A 57-year-old woman with triple-negative ductal breast cancer presented with a mildly pruritic rash on bilateral mastectomy scars of 3 to 4 months' duration. More than a year prior to presentation, she was diagnosed with breast cancer and treated with a bilateral mastectomy and chemotherapy. On physical examination, faintly yellow, slightly indurated, coalescing papules with red rims were present on the bilateral mastectomy scars, with the scar on the left side appearing worse than the right. She previously had not sought treatment.