User login
Black Linear Streaks on the Face With Pruritic Plaques on the Trunk and Arms
The Diagnosis: Toxicodendron Dermatitis
Toxicodendron dermatitis is an allergic contact dermatitis that can occur after exposure to a plant from the Toxicodendron genus including poison ivy (Toxicodendron radicans), poison oak (Toxicodendron diversilobum), and poison sumac (Toxicodendron vernix). These plants produce urushiol in their oleoresinous sap, which causes intense pruritus, streaks of erythema, and edematous papules followed by vesicles and bullae. Previously sensitized individuals develop symptoms as quickly as 24 to 48 hours after exposure, with a range of 5 hours to 15 days.1-3 Rarely, black spots also can be found on the skin, most prominently after 72 hours of exposure.4
The color change of urushiol-containing sap from pale to black was first documented by Peter Kalm, a Swedish botanist who traveled to North America in the 1700s.5 The black-spot test can be used to identify Toxicodendron species because the sap will turn black when expressed on white paper after a few minutes.6 Manifestation of black lacquer streaks on the skin is rare because concentrated sap is necessary, which typically requires an unusually prolonged exposure with Toxicodendron plants.7
Without treatment, typical Toxicodendron dermatitis resolves in approximately 3 weeks, though it may take up to 6 weeks to clear.2 Early intervention is critical, as urushiol will fully absorb after 30 minutes.2 After contact, complete removal of the oleoresin by washing with mild soap and water within 10 minutes can prevent dermatitis. Early topical corticosteroid application can reduce erythema and pruritus. Extensive or severe involvement, which includes Toxicodendron dermatitis with black spots, is treated with systemic corticosteroids such as prednisone that is tapered over 2 to 3 weeks.1
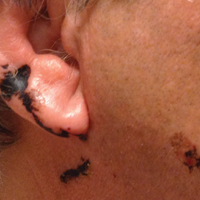
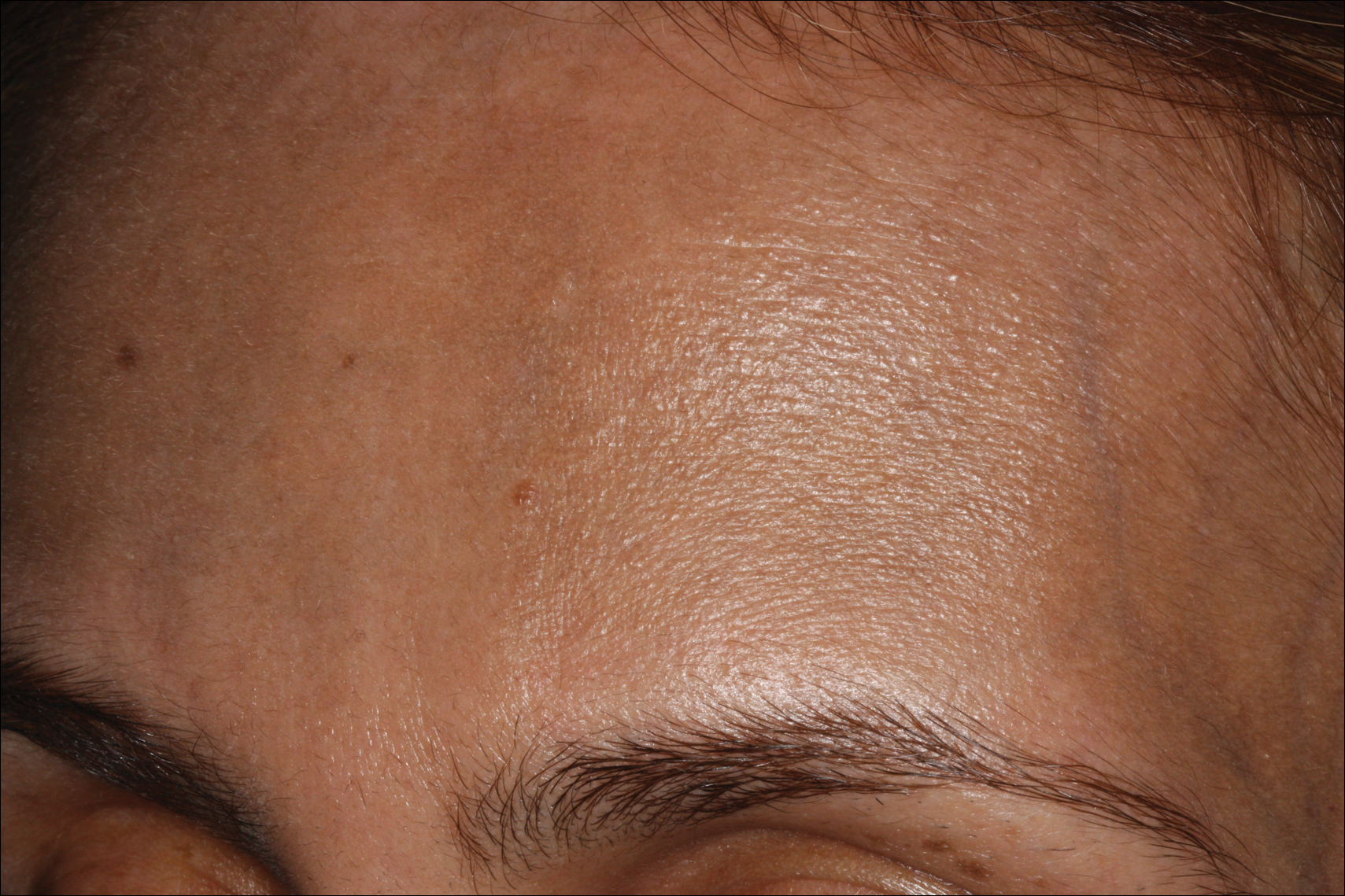
Our patient had classic findings of Toxicodendron dermatitis; however, initially there was concern for levamisole toxicity by the emergency department, as well-demarcated purpuric or dark skin lesions can be due to morbid conditions such as leukocytoclastic vasculitis or skin necrosis from drug toxicities or infectious etiologies. Dermatology was consulted and these concerns were alleviated on closer skin examination and further questioning. The patient reported that he spent several hours cutting brush that was known to be T vernix (poison sumac) 3 days prior to presentation. Interestingly, the patient did not have similar black streaks on the left side of the face, as he held the weed-trimming saw in his right hand and in effect protected the left side of the face from debris. Furthermore, he had pruritic erythematous plaques on both forearms. The facial black lacquer-like streaks were the result of urushiol oxidation in the setting of prolonged exposure to the poison sumac oleoresin sap during weed trimming. After dermatologic evaluation, the patient was discharged from the emergency department on a 15-day taper of oral prednisone, and he was instructed to wash involved areas and exposed clothing with soap and water, which led to complete resolution.
- Lee NP, Arriola ER. Poison ivy, oak, and sumac dermatitis. West J Med. 1999;171:354-355.
- Gladman AC. Toxicodendron dermatitis: poison ivy, oak, and sumac. Wilderness Environ Med. 2006;17:120-128.
- Gross M, Baer H, Fales H. Urushiols of poisonous anacardiaceae. Phytochemistry. 1975;14:2263-2266.
- Mallory SB, Miller OF 3dU, Tyler WB. Toxicodendron radicans dermatitis with black lacquer deposit on the skin. J Am Acad Dermatol. 1982;6:363-368.
- Benson AB. Peter Kalm's Travels in North America: The English Version of 1770. New York, NY: Dover Publications; 1937.
- Guin JD. The black spot test for recognizing poison ivy and related species. J Am Acad Dermatol. 1980;2:332-333.
- Kurlan JG, Lucky AW. Black spot poison ivy: a report of 5 cases and a review of the literature. J Am Acad Dermatol. 2001;45:246-249.
The Diagnosis: Toxicodendron Dermatitis
Toxicodendron dermatitis is an allergic contact dermatitis that can occur after exposure to a plant from the Toxicodendron genus including poison ivy (Toxicodendron radicans), poison oak (Toxicodendron diversilobum), and poison sumac (Toxicodendron vernix). These plants produce urushiol in their oleoresinous sap, which causes intense pruritus, streaks of erythema, and edematous papules followed by vesicles and bullae. Previously sensitized individuals develop symptoms as quickly as 24 to 48 hours after exposure, with a range of 5 hours to 15 days.1-3 Rarely, black spots also can be found on the skin, most prominently after 72 hours of exposure.4
The color change of urushiol-containing sap from pale to black was first documented by Peter Kalm, a Swedish botanist who traveled to North America in the 1700s.5 The black-spot test can be used to identify Toxicodendron species because the sap will turn black when expressed on white paper after a few minutes.6 Manifestation of black lacquer streaks on the skin is rare because concentrated sap is necessary, which typically requires an unusually prolonged exposure with Toxicodendron plants.7
Without treatment, typical Toxicodendron dermatitis resolves in approximately 3 weeks, though it may take up to 6 weeks to clear.2 Early intervention is critical, as urushiol will fully absorb after 30 minutes.2 After contact, complete removal of the oleoresin by washing with mild soap and water within 10 minutes can prevent dermatitis. Early topical corticosteroid application can reduce erythema and pruritus. Extensive or severe involvement, which includes Toxicodendron dermatitis with black spots, is treated with systemic corticosteroids such as prednisone that is tapered over 2 to 3 weeks.1
Our patient had classic findings of Toxicodendron dermatitis; however, initially there was concern for levamisole toxicity by the emergency department, as well-demarcated purpuric or dark skin lesions can be due to morbid conditions such as leukocytoclastic vasculitis or skin necrosis from drug toxicities or infectious etiologies. Dermatology was consulted and these concerns were alleviated on closer skin examination and further questioning. The patient reported that he spent several hours cutting brush that was known to be T vernix (poison sumac) 3 days prior to presentation. Interestingly, the patient did not have similar black streaks on the left side of the face, as he held the weed-trimming saw in his right hand and in effect protected the left side of the face from debris. Furthermore, he had pruritic erythematous plaques on both forearms. The facial black lacquer-like streaks were the result of urushiol oxidation in the setting of prolonged exposure to the poison sumac oleoresin sap during weed trimming. After dermatologic evaluation, the patient was discharged from the emergency department on a 15-day taper of oral prednisone, and he was instructed to wash involved areas and exposed clothing with soap and water, which led to complete resolution.
The Diagnosis: Toxicodendron Dermatitis
Toxicodendron dermatitis is an allergic contact dermatitis that can occur after exposure to a plant from the Toxicodendron genus including poison ivy (Toxicodendron radicans), poison oak (Toxicodendron diversilobum), and poison sumac (Toxicodendron vernix). These plants produce urushiol in their oleoresinous sap, which causes intense pruritus, streaks of erythema, and edematous papules followed by vesicles and bullae. Previously sensitized individuals develop symptoms as quickly as 24 to 48 hours after exposure, with a range of 5 hours to 15 days.1-3 Rarely, black spots also can be found on the skin, most prominently after 72 hours of exposure.4
The color change of urushiol-containing sap from pale to black was first documented by Peter Kalm, a Swedish botanist who traveled to North America in the 1700s.5 The black-spot test can be used to identify Toxicodendron species because the sap will turn black when expressed on white paper after a few minutes.6 Manifestation of black lacquer streaks on the skin is rare because concentrated sap is necessary, which typically requires an unusually prolonged exposure with Toxicodendron plants.7
Without treatment, typical Toxicodendron dermatitis resolves in approximately 3 weeks, though it may take up to 6 weeks to clear.2 Early intervention is critical, as urushiol will fully absorb after 30 minutes.2 After contact, complete removal of the oleoresin by washing with mild soap and water within 10 minutes can prevent dermatitis. Early topical corticosteroid application can reduce erythema and pruritus. Extensive or severe involvement, which includes Toxicodendron dermatitis with black spots, is treated with systemic corticosteroids such as prednisone that is tapered over 2 to 3 weeks.1
Our patient had classic findings of Toxicodendron dermatitis; however, initially there was concern for levamisole toxicity by the emergency department, as well-demarcated purpuric or dark skin lesions can be due to morbid conditions such as leukocytoclastic vasculitis or skin necrosis from drug toxicities or infectious etiologies. Dermatology was consulted and these concerns were alleviated on closer skin examination and further questioning. The patient reported that he spent several hours cutting brush that was known to be T vernix (poison sumac) 3 days prior to presentation. Interestingly, the patient did not have similar black streaks on the left side of the face, as he held the weed-trimming saw in his right hand and in effect protected the left side of the face from debris. Furthermore, he had pruritic erythematous plaques on both forearms. The facial black lacquer-like streaks were the result of urushiol oxidation in the setting of prolonged exposure to the poison sumac oleoresin sap during weed trimming. After dermatologic evaluation, the patient was discharged from the emergency department on a 15-day taper of oral prednisone, and he was instructed to wash involved areas and exposed clothing with soap and water, which led to complete resolution.
- Lee NP, Arriola ER. Poison ivy, oak, and sumac dermatitis. West J Med. 1999;171:354-355.
- Gladman AC. Toxicodendron dermatitis: poison ivy, oak, and sumac. Wilderness Environ Med. 2006;17:120-128.
- Gross M, Baer H, Fales H. Urushiols of poisonous anacardiaceae. Phytochemistry. 1975;14:2263-2266.
- Mallory SB, Miller OF 3dU, Tyler WB. Toxicodendron radicans dermatitis with black lacquer deposit on the skin. J Am Acad Dermatol. 1982;6:363-368.
- Benson AB. Peter Kalm's Travels in North America: The English Version of 1770. New York, NY: Dover Publications; 1937.
- Guin JD. The black spot test for recognizing poison ivy and related species. J Am Acad Dermatol. 1980;2:332-333.
- Kurlan JG, Lucky AW. Black spot poison ivy: a report of 5 cases and a review of the literature. J Am Acad Dermatol. 2001;45:246-249.
- Lee NP, Arriola ER. Poison ivy, oak, and sumac dermatitis. West J Med. 1999;171:354-355.
- Gladman AC. Toxicodendron dermatitis: poison ivy, oak, and sumac. Wilderness Environ Med. 2006;17:120-128.
- Gross M, Baer H, Fales H. Urushiols of poisonous anacardiaceae. Phytochemistry. 1975;14:2263-2266.
- Mallory SB, Miller OF 3dU, Tyler WB. Toxicodendron radicans dermatitis with black lacquer deposit on the skin. J Am Acad Dermatol. 1982;6:363-368.
- Benson AB. Peter Kalm's Travels in North America: The English Version of 1770. New York, NY: Dover Publications; 1937.
- Guin JD. The black spot test for recognizing poison ivy and related species. J Am Acad Dermatol. 1980;2:332-333.
- Kurlan JG, Lucky AW. Black spot poison ivy: a report of 5 cases and a review of the literature. J Am Acad Dermatol. 2001;45:246-249.
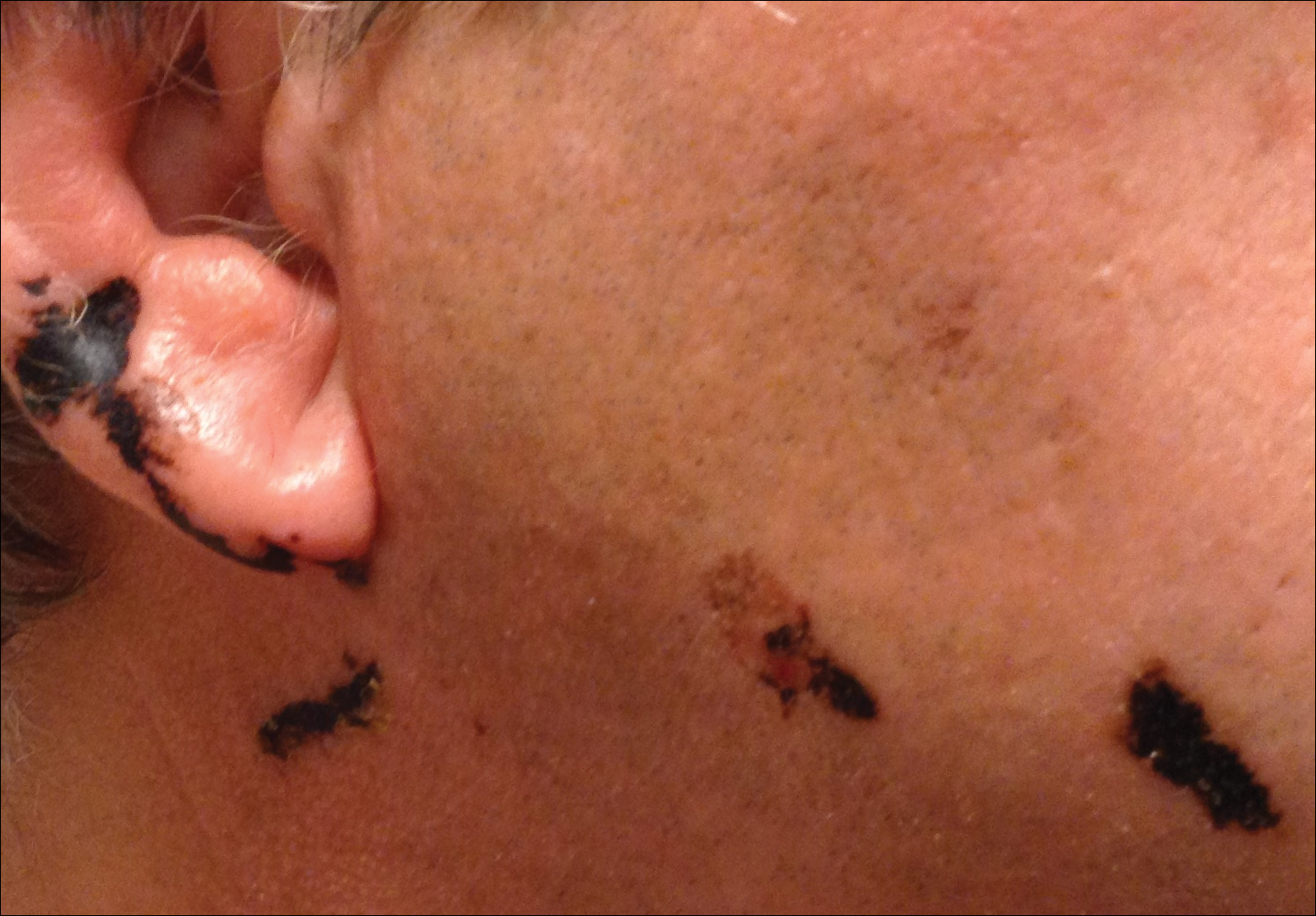
A 68-year-old man presented to the emergency department with pruritic, edematous, pink plaques on the trunk and arms, as well as black linear streaks on the face, prompting dermatology consultation for possible tissue necrosis. The patient reported working outdoors in his garden 3 days prior to presentation.
Recalcitrant Solitary Erythematous Scaly Patch on the Foot
The Diagnosis: Pagetoid Reticulosis
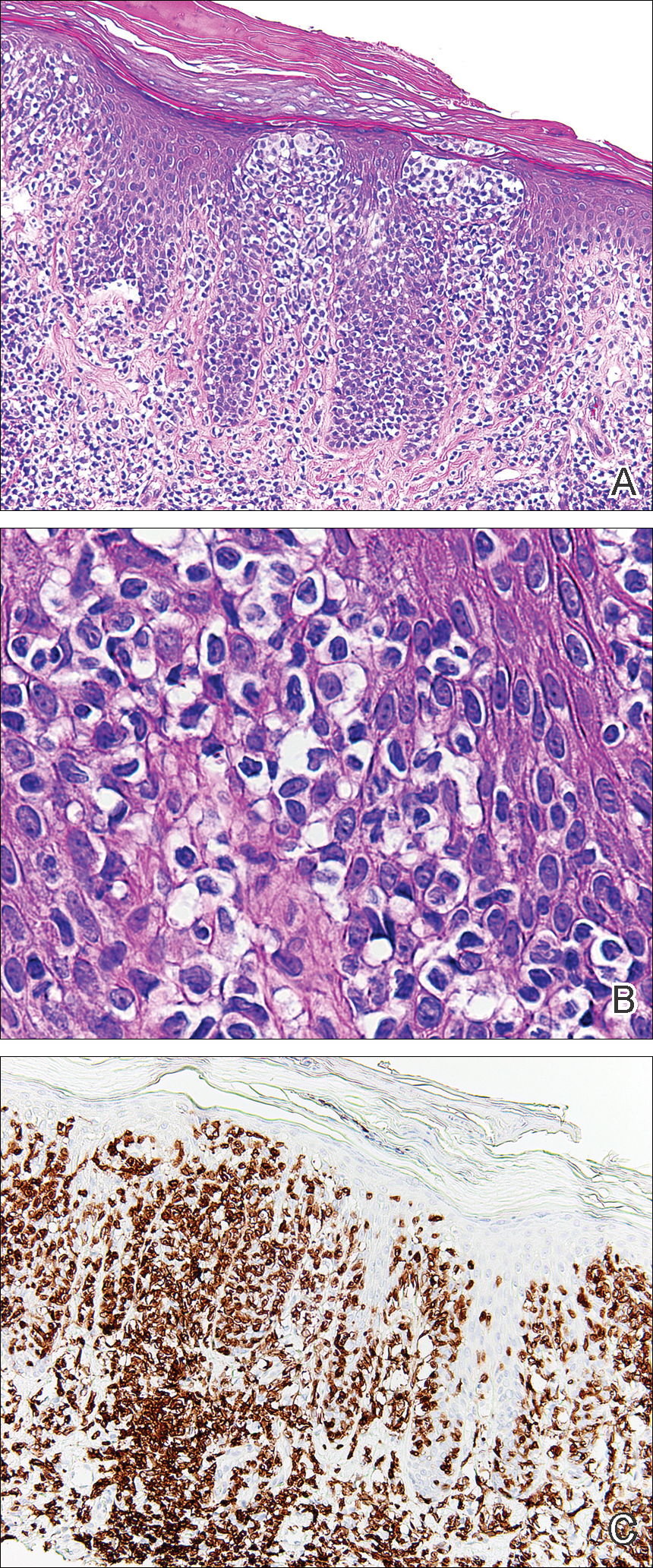
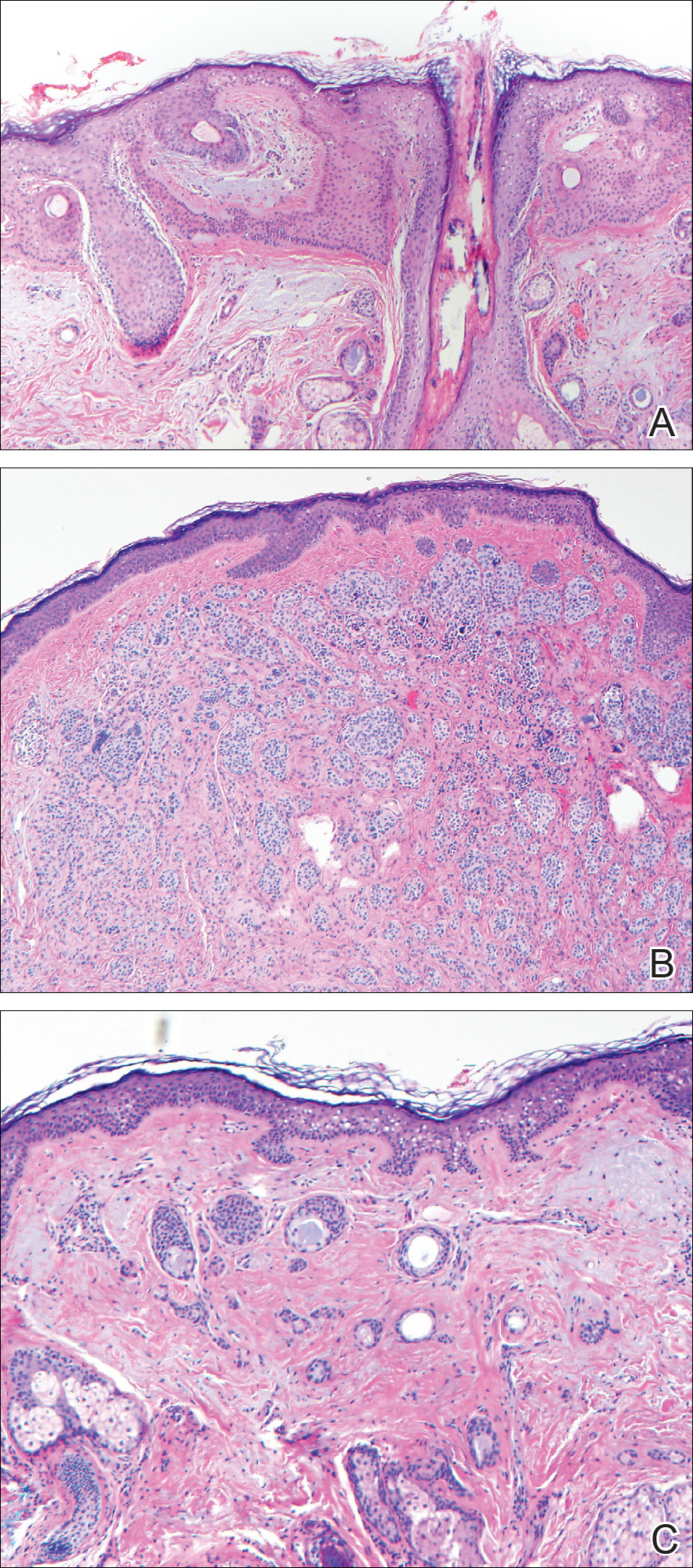
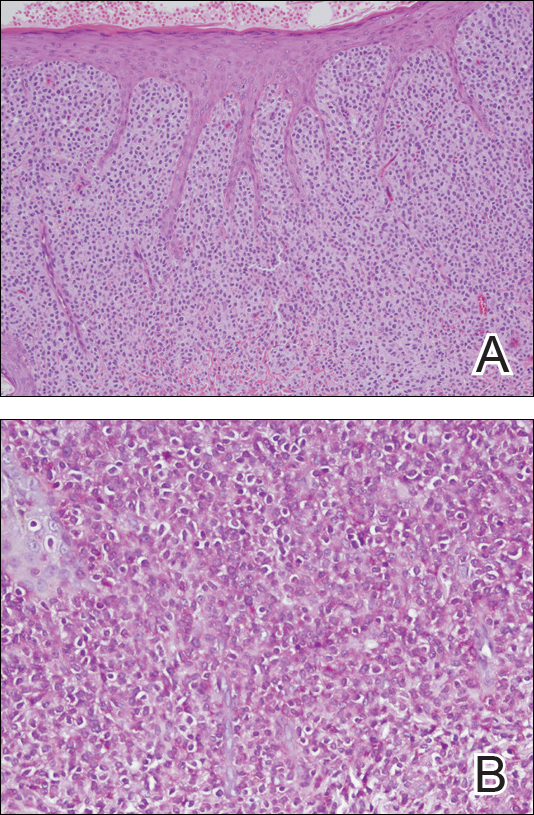
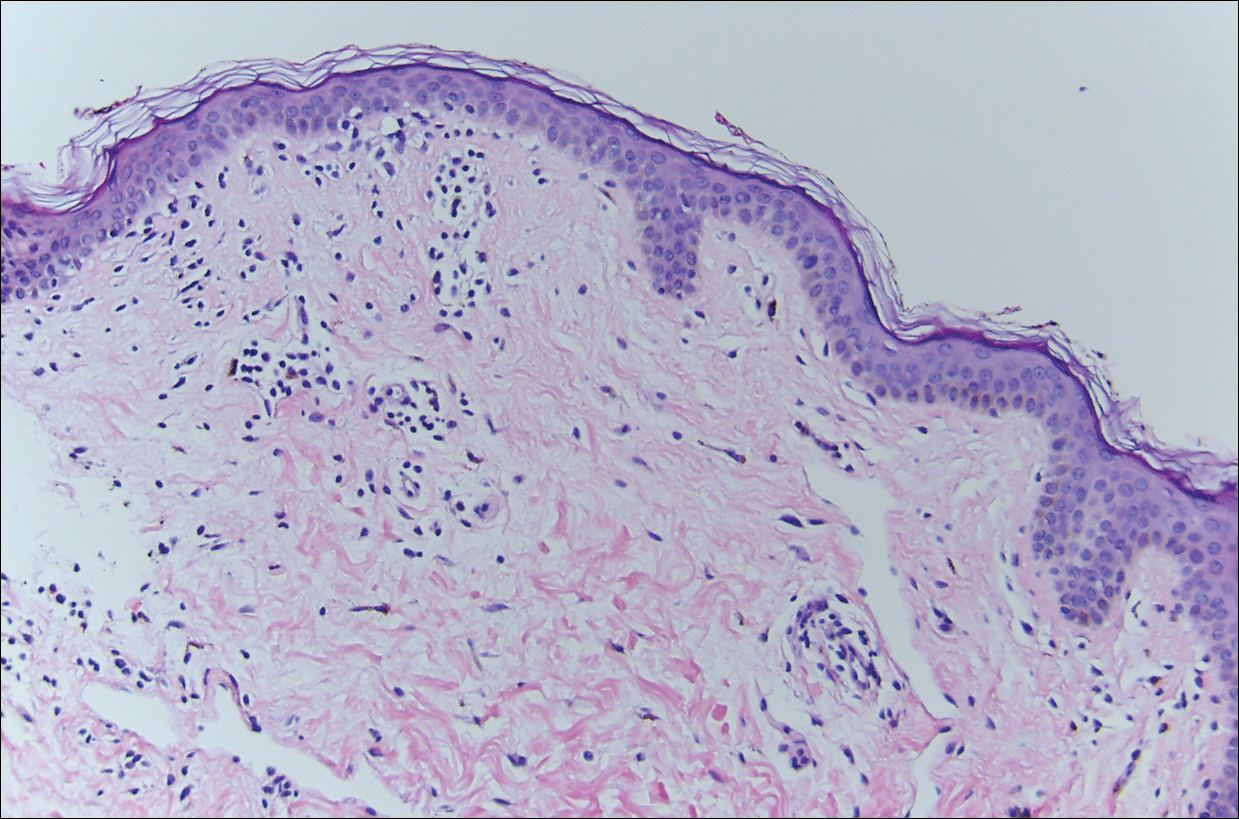
Histopathologic examination demonstrated a dense infiltrate and psoriasiform pattern epidermal hyperplasia (Figure, A). There was conspicuous epidermotropism of moderately enlarged, hyperchromatic lymphocytes. Intraepidermal lymphocytes were slightly larger, darker, and more convoluted than those in the subjacent dermis (Figure, B). These cells exhibited CD3+ T-cell differentiation with an abnormal CD4-CD7-CD8- phenotype (Figure, C). The histopathologic finding of atypical epidermotropic T-cell infiltrate was compatible with a rare variant of mycosis fungoides known as pagetoid reticulosis (PR). After discussing the diagnosis and treatment options, the patient elected to begin with a conservative approach to therapy. We prescribed fluocinonide ointment 0.05% twice daily under occlusion. At 1 month follow-up, the patient experienced marked improvement of the erythema and scaling of the lesion.
Pagetoid reticulosis is a primary cutaneous T-cell lymphoma that has been categorized as an indolent localized variant of mycosis fungoides. This rare skin disorder was originally described by Woringer and Kolopp in 19391 and was further renamed in 1973 by Braun-Falco et al.2 At that time the term pagetoid reticulosis was introduced due to similarities in histopathologic findings seen in Paget disease of the nipple. Two variants of the disease have been described since then: the localized type and the disseminated type. The localized type, also known as Woringer-Kolopp disease (WKD), typically presents as a persistent, sharply localized, scaly patch that slowly expands over several years. The lesion is classically located on the extensor surface of the hand or foot and often is asymptomatic. Due to the benign presentation, WKD can easily be confused with much more common diseases, such as psoriasis or fungal infections, resulting in a substantial delay in the diagnosis. The patient will often report a medical history notable for frequent office visits and numerous failed therapies. Even though it is exceedingly uncommon, these findings should prompt the practitioner to add WKD to their differential. The disseminated type of PR (also known as Ketron-Goodman disease) is characterized by diffuse cutaneous involvement, carries a much more progressive course, and often leads to a poor outcome.3 The histopathologic features of WKD and Ketron-Goodman disease are identical, and the 2 types are distinguished on clinical grounds alone.
Histopathologic features of PR are unique and often distinct in comparison to mycosis fungoides. Pagetoid reticulosis often is described as epidermal hyperplasia with parakeratosis, prominent acanthosis, and excessive epidermotropism of atypical lymphocytes scattered throughout the epidermis.3 The distinct pattern of epidermotropism seen in PR is the characteristic finding. Review of immunocytochemistry from reported cases has shown that CD marker expression of neoplastic T cells in PR can be variable in nature.4 Although it is known that immunophenotyping can be useful in diagnosing and distinguishing PR from other types of primary cutaneous T-cell lymphoma, the clinical significance of the observed phenotypic variation remains a mystery. As of now, it appears to be prognostically irrelevant.5
There are numerous therapeutic options available for PR. Depending on the size and extent of the disease, surgical excision and radiotherapy may be an option and are the most effective.6 For patients who are not good candidates or opt out of these options, there are various pharmacotherapies that also have proven to work. Traditional therapies include topical corticosteroids, corticosteroid injections, and phototherapy. However, more recent trials with retinoids, such as alitretinoin or bexarotene, appear to offer a promising therapeutic approach.7
Pagetoid reticulosis is a true malignant lymphoma of T-cell lineage, but it typically carries an excellent prognosis. Rare cases have been reported to progress to disseminated lymphoma.8 Therefore, long-term follow-up for a patient diagnosed with PR is recommended.
- Woringer FR, Kolopp P. Lésion érythémato-squameuse polycyclique de l'avant-bras évoluantdepuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol. 1939;10:945-948.
- Braun-Falco O, Marghescu S, Wolff HH. Pagetoid reticulosis--Woringer-Kolopp's disease [in German]. Hautarzt. 1973;24:11-21.
- Haghighi B, Smoller BR, Leboit PE, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502-510.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Mourtzinos N, Puri PK, Wang G, et al. CD4/CD8 double negative pagetoid reticulosis: a case report and literature review. J Cutan Pathol. 2010;37:491-496.
- Lee J, Viakhireva N, Cesca C, et al. Clinicopathologic features and treatment outcomes in Woringer-Kolopp disease. J Am Acad Dermatol. 2008;59:706-712.
- Schmitz L, Bierhoff E, Dirschka T. Alitretinoin: an effective treatment option for pagetoid reticulosis. J Dtsch Dermatol Ges. 2013;11:1194-1195.
- Ioannides G, Engel MF, Rywlin AM. Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol. 1983;5:153-158.
The Diagnosis: Pagetoid Reticulosis
Histopathologic examination demonstrated a dense infiltrate and psoriasiform pattern epidermal hyperplasia (Figure, A). There was conspicuous epidermotropism of moderately enlarged, hyperchromatic lymphocytes. Intraepidermal lymphocytes were slightly larger, darker, and more convoluted than those in the subjacent dermis (Figure, B). These cells exhibited CD3+ T-cell differentiation with an abnormal CD4-CD7-CD8- phenotype (Figure, C). The histopathologic finding of atypical epidermotropic T-cell infiltrate was compatible with a rare variant of mycosis fungoides known as pagetoid reticulosis (PR). After discussing the diagnosis and treatment options, the patient elected to begin with a conservative approach to therapy. We prescribed fluocinonide ointment 0.05% twice daily under occlusion. At 1 month follow-up, the patient experienced marked improvement of the erythema and scaling of the lesion.
Pagetoid reticulosis is a primary cutaneous T-cell lymphoma that has been categorized as an indolent localized variant of mycosis fungoides. This rare skin disorder was originally described by Woringer and Kolopp in 19391 and was further renamed in 1973 by Braun-Falco et al.2 At that time the term pagetoid reticulosis was introduced due to similarities in histopathologic findings seen in Paget disease of the nipple. Two variants of the disease have been described since then: the localized type and the disseminated type. The localized type, also known as Woringer-Kolopp disease (WKD), typically presents as a persistent, sharply localized, scaly patch that slowly expands over several years. The lesion is classically located on the extensor surface of the hand or foot and often is asymptomatic. Due to the benign presentation, WKD can easily be confused with much more common diseases, such as psoriasis or fungal infections, resulting in a substantial delay in the diagnosis. The patient will often report a medical history notable for frequent office visits and numerous failed therapies. Even though it is exceedingly uncommon, these findings should prompt the practitioner to add WKD to their differential. The disseminated type of PR (also known as Ketron-Goodman disease) is characterized by diffuse cutaneous involvement, carries a much more progressive course, and often leads to a poor outcome.3 The histopathologic features of WKD and Ketron-Goodman disease are identical, and the 2 types are distinguished on clinical grounds alone.
Histopathologic features of PR are unique and often distinct in comparison to mycosis fungoides. Pagetoid reticulosis often is described as epidermal hyperplasia with parakeratosis, prominent acanthosis, and excessive epidermotropism of atypical lymphocytes scattered throughout the epidermis.3 The distinct pattern of epidermotropism seen in PR is the characteristic finding. Review of immunocytochemistry from reported cases has shown that CD marker expression of neoplastic T cells in PR can be variable in nature.4 Although it is known that immunophenotyping can be useful in diagnosing and distinguishing PR from other types of primary cutaneous T-cell lymphoma, the clinical significance of the observed phenotypic variation remains a mystery. As of now, it appears to be prognostically irrelevant.5
There are numerous therapeutic options available for PR. Depending on the size and extent of the disease, surgical excision and radiotherapy may be an option and are the most effective.6 For patients who are not good candidates or opt out of these options, there are various pharmacotherapies that also have proven to work. Traditional therapies include topical corticosteroids, corticosteroid injections, and phototherapy. However, more recent trials with retinoids, such as alitretinoin or bexarotene, appear to offer a promising therapeutic approach.7
Pagetoid reticulosis is a true malignant lymphoma of T-cell lineage, but it typically carries an excellent prognosis. Rare cases have been reported to progress to disseminated lymphoma.8 Therefore, long-term follow-up for a patient diagnosed with PR is recommended.
The Diagnosis: Pagetoid Reticulosis
Histopathologic examination demonstrated a dense infiltrate and psoriasiform pattern epidermal hyperplasia (Figure, A). There was conspicuous epidermotropism of moderately enlarged, hyperchromatic lymphocytes. Intraepidermal lymphocytes were slightly larger, darker, and more convoluted than those in the subjacent dermis (Figure, B). These cells exhibited CD3+ T-cell differentiation with an abnormal CD4-CD7-CD8- phenotype (Figure, C). The histopathologic finding of atypical epidermotropic T-cell infiltrate was compatible with a rare variant of mycosis fungoides known as pagetoid reticulosis (PR). After discussing the diagnosis and treatment options, the patient elected to begin with a conservative approach to therapy. We prescribed fluocinonide ointment 0.05% twice daily under occlusion. At 1 month follow-up, the patient experienced marked improvement of the erythema and scaling of the lesion.
Pagetoid reticulosis is a primary cutaneous T-cell lymphoma that has been categorized as an indolent localized variant of mycosis fungoides. This rare skin disorder was originally described by Woringer and Kolopp in 19391 and was further renamed in 1973 by Braun-Falco et al.2 At that time the term pagetoid reticulosis was introduced due to similarities in histopathologic findings seen in Paget disease of the nipple. Two variants of the disease have been described since then: the localized type and the disseminated type. The localized type, also known as Woringer-Kolopp disease (WKD), typically presents as a persistent, sharply localized, scaly patch that slowly expands over several years. The lesion is classically located on the extensor surface of the hand or foot and often is asymptomatic. Due to the benign presentation, WKD can easily be confused with much more common diseases, such as psoriasis or fungal infections, resulting in a substantial delay in the diagnosis. The patient will often report a medical history notable for frequent office visits and numerous failed therapies. Even though it is exceedingly uncommon, these findings should prompt the practitioner to add WKD to their differential. The disseminated type of PR (also known as Ketron-Goodman disease) is characterized by diffuse cutaneous involvement, carries a much more progressive course, and often leads to a poor outcome.3 The histopathologic features of WKD and Ketron-Goodman disease are identical, and the 2 types are distinguished on clinical grounds alone.
Histopathologic features of PR are unique and often distinct in comparison to mycosis fungoides. Pagetoid reticulosis often is described as epidermal hyperplasia with parakeratosis, prominent acanthosis, and excessive epidermotropism of atypical lymphocytes scattered throughout the epidermis.3 The distinct pattern of epidermotropism seen in PR is the characteristic finding. Review of immunocytochemistry from reported cases has shown that CD marker expression of neoplastic T cells in PR can be variable in nature.4 Although it is known that immunophenotyping can be useful in diagnosing and distinguishing PR from other types of primary cutaneous T-cell lymphoma, the clinical significance of the observed phenotypic variation remains a mystery. As of now, it appears to be prognostically irrelevant.5
There are numerous therapeutic options available for PR. Depending on the size and extent of the disease, surgical excision and radiotherapy may be an option and are the most effective.6 For patients who are not good candidates or opt out of these options, there are various pharmacotherapies that also have proven to work. Traditional therapies include topical corticosteroids, corticosteroid injections, and phototherapy. However, more recent trials with retinoids, such as alitretinoin or bexarotene, appear to offer a promising therapeutic approach.7
Pagetoid reticulosis is a true malignant lymphoma of T-cell lineage, but it typically carries an excellent prognosis. Rare cases have been reported to progress to disseminated lymphoma.8 Therefore, long-term follow-up for a patient diagnosed with PR is recommended.
- Woringer FR, Kolopp P. Lésion érythémato-squameuse polycyclique de l'avant-bras évoluantdepuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol. 1939;10:945-948.
- Braun-Falco O, Marghescu S, Wolff HH. Pagetoid reticulosis--Woringer-Kolopp's disease [in German]. Hautarzt. 1973;24:11-21.
- Haghighi B, Smoller BR, Leboit PE, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502-510.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Mourtzinos N, Puri PK, Wang G, et al. CD4/CD8 double negative pagetoid reticulosis: a case report and literature review. J Cutan Pathol. 2010;37:491-496.
- Lee J, Viakhireva N, Cesca C, et al. Clinicopathologic features and treatment outcomes in Woringer-Kolopp disease. J Am Acad Dermatol. 2008;59:706-712.
- Schmitz L, Bierhoff E, Dirschka T. Alitretinoin: an effective treatment option for pagetoid reticulosis. J Dtsch Dermatol Ges. 2013;11:1194-1195.
- Ioannides G, Engel MF, Rywlin AM. Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol. 1983;5:153-158.
- Woringer FR, Kolopp P. Lésion érythémato-squameuse polycyclique de l'avant-bras évoluantdepuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol. 1939;10:945-948.
- Braun-Falco O, Marghescu S, Wolff HH. Pagetoid reticulosis--Woringer-Kolopp's disease [in German]. Hautarzt. 1973;24:11-21.
- Haghighi B, Smoller BR, Leboit PE, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502-510.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Mourtzinos N, Puri PK, Wang G, et al. CD4/CD8 double negative pagetoid reticulosis: a case report and literature review. J Cutan Pathol. 2010;37:491-496.
- Lee J, Viakhireva N, Cesca C, et al. Clinicopathologic features and treatment outcomes in Woringer-Kolopp disease. J Am Acad Dermatol. 2008;59:706-712.
- Schmitz L, Bierhoff E, Dirschka T. Alitretinoin: an effective treatment option for pagetoid reticulosis. J Dtsch Dermatol Ges. 2013;11:1194-1195.
- Ioannides G, Engel MF, Rywlin AM. Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol. 1983;5:153-158.
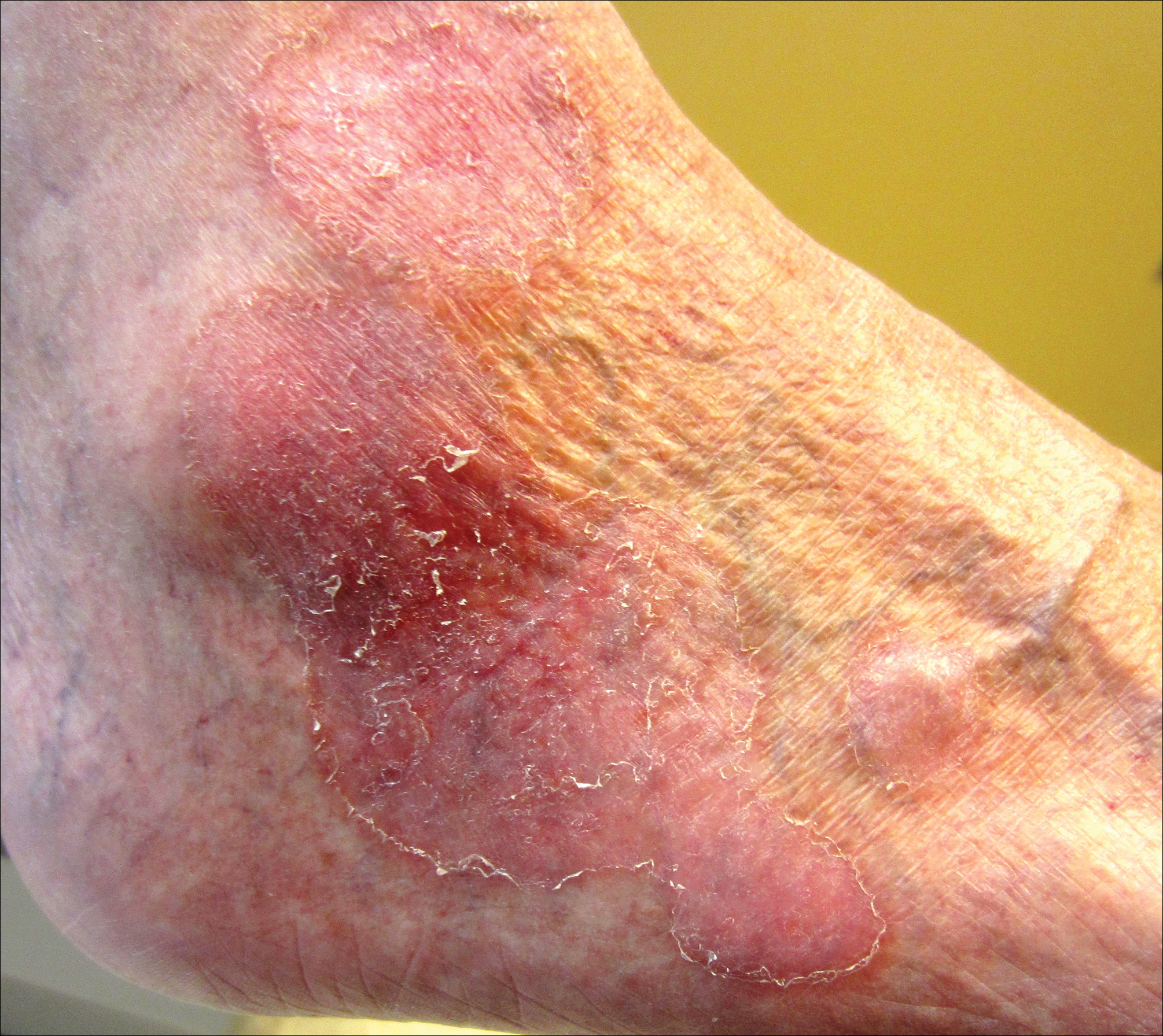
An 80-year-old man with a history of malignant melanoma and squamous cell carcinoma presented to the dermatology clinic with a chronic rash of 20 years' duration on the right ankle that extended to the instep of the right foot. His medical history was notable for hypertension and hyperlipidemia. Family history was unremarkable. The patient described the rash as red and scaly but denied associated pain or pruritus. Over the last 2 to 3 years he had tried treating the affected area with petroleum jelly, topical and oral antifungals, and mild topical steroids with minimal improvement. Complete review of systems was performed and was negative other than some mild constipation. Physical examination revealed an erythematous scaly patch on the dorsal aspect of the right ankle. Potassium hydroxide preparation and fungal culture swab yielded negative results, and a shave biopsy was performed.
Flesh-Colored Nodule With Underlying Sclerotic Plaque
The Diagnosis: Collision Tumor
Excisional biopsy and histopathological examination demonstrated a collision tumor composed of a benign intradermal melanocytic nevus, tumor of follicular infundibulum, and an underlying sclerosing epithelial neoplasm, with a differential diagnosis of desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma (Figure).
Common acquired melanocytic nevus presents clinically as a macule, papule, or nodule with smooth regular borders. The pigmented variant displays an evenly distributed pigment on the lesion. Intradermal melanocytic nevus often presents as a flesh-colored nodule, as in our case. Histopathologically, benign intradermal nevus typically is composed of a proliferation of melanocytes that exhibit dispersion as they go deeper in the dermis and maturation that manifests as melanocytes becoming smaller and more spindled in the deeper portions of the lesion.1 These 2 characteristics plus the bland cytology seen in the present case confirm the benign characteristic of this lesion (Figure, B).
In addition to the benign intradermal melanocytic nevus, an adjacent tumor of follicular infundibulum was noted. Tumor of follicular infundibulum is a rare adnexal tumor. It occurs frequently on the head and neck and shows some female predominance.2,3 Multiple lesions and eruptive lesions are rare forms that also have been reported.4 Histopathologically, the tumor demonstrates an epithelial plate that is present in the papillary dermis and is connected to the epidermis at multiple points with attachment to the follicular outer root sheath. Peripheral palisading is characteristically present above an eosinophilic basement membrane (Figure, A). Rare reports have documented sebaceous and eccrine differentiation.5,6
Tumor of follicular infundibulum has been reported to be associated with other tumors. Organoid nevus (nevus sebaceous), trichilemmal tumor, and fibroma have been reported to occur as a collision tumor with tumor of follicular infundibulum. An association with Cowden disease also has been described.7 Biopsies that represent partial samples should be interpreted cautiously, as step sections can reveal basal cell carcinoma.
The term sclerosing epithelial neoplasm describes tumors that share a paisley tielike epithelial pattern and sclerotic stroma. Small specimens often require clinicopathologic correlation (Figure, C). The differential diagnosis includes morpheaform basal cell carcinoma, desmoplastic trichoepithelioma, and microcystic adnexal carcinoma. A panel of stains using Ber-EP4, PHLDA1, cytokeratin 15, and cytokeratin 19 has been proposed to help differentiate these entities.8 CD34 and cytokeratin 20 also have been used with varying success in small specimens.9,10
- Ferringer T, Peckham S, Ko CJ, et al. Melanocytic neoplasms. In: Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:105-109.
- Headington JT. Tumors of the hair follicle. Am J Pathol. 1976;85:480-505.
- Davis DA, Cohen PR. Hair follicle nevus: case report and review of the literature. Pediatr Dermatol. 1996;13:135-138.
- Ikeda S, Kawada J, Yaguchi H, et al. A case of unilateral, systematized linear hair follicle nevi associated with epidermal nevus-like lesions. Dermatology. 2003;206:172-174.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Mahalingam M, Bhawan J, Finn R, et al. Tumor of the follicular infundibulum with sebaceous differentiation. J Cutan Pathol. 2001;28:314-317.
- Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
- Sellheyer K, Nelson P, Kutzner H, et al. The immunohistochemical differential diagnosis of microcystic adnexal carcinoma, desmoplastic trichoepithelioma and morpheaform basal cell carcinoma using BerEP4 and stem cell markers. J Cutan Pathol. 2013;40:363-370.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
The Diagnosis: Collision Tumor
Excisional biopsy and histopathological examination demonstrated a collision tumor composed of a benign intradermal melanocytic nevus, tumor of follicular infundibulum, and an underlying sclerosing epithelial neoplasm, with a differential diagnosis of desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma (Figure).
Common acquired melanocytic nevus presents clinically as a macule, papule, or nodule with smooth regular borders. The pigmented variant displays an evenly distributed pigment on the lesion. Intradermal melanocytic nevus often presents as a flesh-colored nodule, as in our case. Histopathologically, benign intradermal nevus typically is composed of a proliferation of melanocytes that exhibit dispersion as they go deeper in the dermis and maturation that manifests as melanocytes becoming smaller and more spindled in the deeper portions of the lesion.1 These 2 characteristics plus the bland cytology seen in the present case confirm the benign characteristic of this lesion (Figure, B).
In addition to the benign intradermal melanocytic nevus, an adjacent tumor of follicular infundibulum was noted. Tumor of follicular infundibulum is a rare adnexal tumor. It occurs frequently on the head and neck and shows some female predominance.2,3 Multiple lesions and eruptive lesions are rare forms that also have been reported.4 Histopathologically, the tumor demonstrates an epithelial plate that is present in the papillary dermis and is connected to the epidermis at multiple points with attachment to the follicular outer root sheath. Peripheral palisading is characteristically present above an eosinophilic basement membrane (Figure, A). Rare reports have documented sebaceous and eccrine differentiation.5,6
Tumor of follicular infundibulum has been reported to be associated with other tumors. Organoid nevus (nevus sebaceous), trichilemmal tumor, and fibroma have been reported to occur as a collision tumor with tumor of follicular infundibulum. An association with Cowden disease also has been described.7 Biopsies that represent partial samples should be interpreted cautiously, as step sections can reveal basal cell carcinoma.
The term sclerosing epithelial neoplasm describes tumors that share a paisley tielike epithelial pattern and sclerotic stroma. Small specimens often require clinicopathologic correlation (Figure, C). The differential diagnosis includes morpheaform basal cell carcinoma, desmoplastic trichoepithelioma, and microcystic adnexal carcinoma. A panel of stains using Ber-EP4, PHLDA1, cytokeratin 15, and cytokeratin 19 has been proposed to help differentiate these entities.8 CD34 and cytokeratin 20 also have been used with varying success in small specimens.9,10
The Diagnosis: Collision Tumor
Excisional biopsy and histopathological examination demonstrated a collision tumor composed of a benign intradermal melanocytic nevus, tumor of follicular infundibulum, and an underlying sclerosing epithelial neoplasm, with a differential diagnosis of desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma (Figure).
Common acquired melanocytic nevus presents clinically as a macule, papule, or nodule with smooth regular borders. The pigmented variant displays an evenly distributed pigment on the lesion. Intradermal melanocytic nevus often presents as a flesh-colored nodule, as in our case. Histopathologically, benign intradermal nevus typically is composed of a proliferation of melanocytes that exhibit dispersion as they go deeper in the dermis and maturation that manifests as melanocytes becoming smaller and more spindled in the deeper portions of the lesion.1 These 2 characteristics plus the bland cytology seen in the present case confirm the benign characteristic of this lesion (Figure, B).
In addition to the benign intradermal melanocytic nevus, an adjacent tumor of follicular infundibulum was noted. Tumor of follicular infundibulum is a rare adnexal tumor. It occurs frequently on the head and neck and shows some female predominance.2,3 Multiple lesions and eruptive lesions are rare forms that also have been reported.4 Histopathologically, the tumor demonstrates an epithelial plate that is present in the papillary dermis and is connected to the epidermis at multiple points with attachment to the follicular outer root sheath. Peripheral palisading is characteristically present above an eosinophilic basement membrane (Figure, A). Rare reports have documented sebaceous and eccrine differentiation.5,6
Tumor of follicular infundibulum has been reported to be associated with other tumors. Organoid nevus (nevus sebaceous), trichilemmal tumor, and fibroma have been reported to occur as a collision tumor with tumor of follicular infundibulum. An association with Cowden disease also has been described.7 Biopsies that represent partial samples should be interpreted cautiously, as step sections can reveal basal cell carcinoma.
The term sclerosing epithelial neoplasm describes tumors that share a paisley tielike epithelial pattern and sclerotic stroma. Small specimens often require clinicopathologic correlation (Figure, C). The differential diagnosis includes morpheaform basal cell carcinoma, desmoplastic trichoepithelioma, and microcystic adnexal carcinoma. A panel of stains using Ber-EP4, PHLDA1, cytokeratin 15, and cytokeratin 19 has been proposed to help differentiate these entities.8 CD34 and cytokeratin 20 also have been used with varying success in small specimens.9,10
- Ferringer T, Peckham S, Ko CJ, et al. Melanocytic neoplasms. In: Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:105-109.
- Headington JT. Tumors of the hair follicle. Am J Pathol. 1976;85:480-505.
- Davis DA, Cohen PR. Hair follicle nevus: case report and review of the literature. Pediatr Dermatol. 1996;13:135-138.
- Ikeda S, Kawada J, Yaguchi H, et al. A case of unilateral, systematized linear hair follicle nevi associated with epidermal nevus-like lesions. Dermatology. 2003;206:172-174.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Mahalingam M, Bhawan J, Finn R, et al. Tumor of the follicular infundibulum with sebaceous differentiation. J Cutan Pathol. 2001;28:314-317.
- Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
- Sellheyer K, Nelson P, Kutzner H, et al. The immunohistochemical differential diagnosis of microcystic adnexal carcinoma, desmoplastic trichoepithelioma and morpheaform basal cell carcinoma using BerEP4 and stem cell markers. J Cutan Pathol. 2013;40:363-370.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
- Ferringer T, Peckham S, Ko CJ, et al. Melanocytic neoplasms. In: Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:105-109.
- Headington JT. Tumors of the hair follicle. Am J Pathol. 1976;85:480-505.
- Davis DA, Cohen PR. Hair follicle nevus: case report and review of the literature. Pediatr Dermatol. 1996;13:135-138.
- Ikeda S, Kawada J, Yaguchi H, et al. A case of unilateral, systematized linear hair follicle nevi associated with epidermal nevus-like lesions. Dermatology. 2003;206:172-174.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Mahalingam M, Bhawan J, Finn R, et al. Tumor of the follicular infundibulum with sebaceous differentiation. J Cutan Pathol. 2001;28:314-317.
- Cribier B, Grosshans E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33:979-984.
- Sellheyer K, Nelson P, Kutzner H, et al. The immunohistochemical differential diagnosis of microcystic adnexal carcinoma, desmoplastic trichoepithelioma and morpheaform basal cell carcinoma using BerEP4 and stem cell markers. J Cutan Pathol. 2013;40:363-370.
- Abesamis-Cubillan E, El-Shabrawi-Caelen L, LeBoit PE. Merkel cells and sclerosing epithelial neoplasms. Am J Dermatopathol. 2000;22:311-315.
- Smith KJ, Williams J, Corbett D, et al. Microcystic adnexal carcinoma: an immunohistochemical study including markers of proliferation and apoptosis. Am J Surg Pathol. 2001;25:464-471.
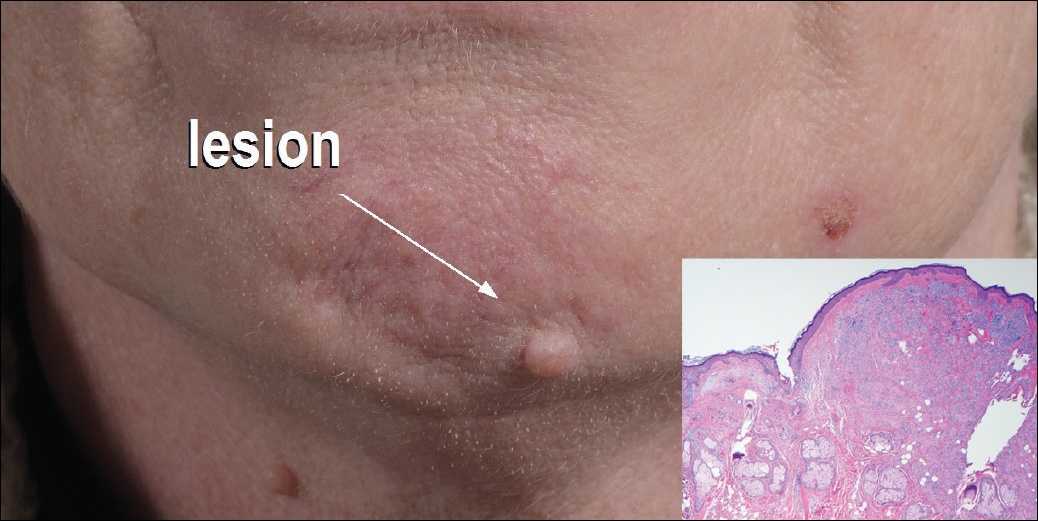
A 54-year-old man presented with a flesh-colored lesion on the chin. The nodule measured 0.6 cm in diameter. There was an underlying sclerotic plaque with indistinct borders.
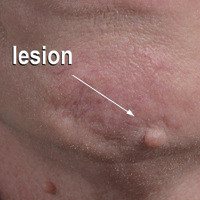
Redness and Painful Ulcerations in the Perineal Area
The Diagnosis: PELVIS Syndrome
Infantile hemangiomas (IHs) are present in up to 10% of infants by 1 year of age and are most commonly located on the face and upper extremities. Less than 10% of IHs develop in the perineum.1 Perineal IHs are benign tumors of the vascular endothelium that present as plaques and commonly are accompanied by painful ulcerations. Ulceration is more common in the diaper area secondary to irritation from urine, stool, and friction.2 Although most IHs are benign isolated findings, facial IHs have been associated with several syndromes including Sturge-Weber and PHACE (posterior fossa brain malformations, hemangiomas, arterial anomalies, cardiac anomalies and coarctation of the aorta, and eye and endocrine abnormalities) syndromes.3 Researchers also have identified an association between lumbosacral IHs and spinal dysraphism (tethered spinal cord).4
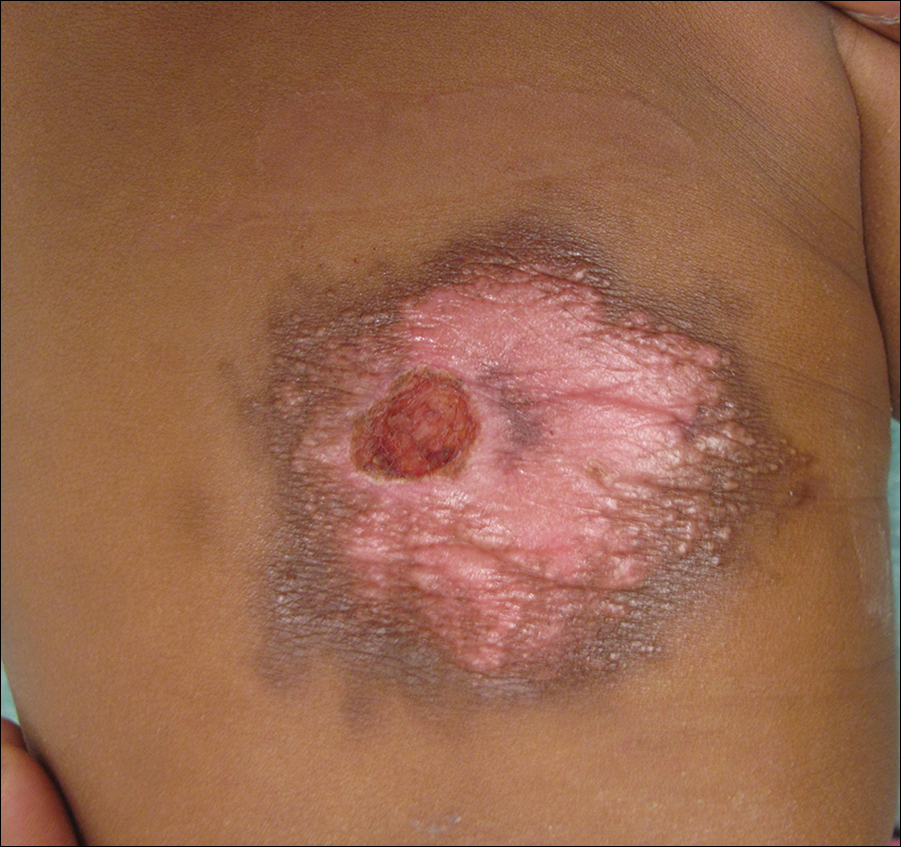
A smaller number of studies have investigated congenital anomalies related to perineal IH,1,5 specifically PELVIS syndrome. The acronym PELVIS has been used to describe a syndrome of congenital malformations including perineal hemangioma, external genital malformations, lipomyelomeningocele, vesicorenal abnormalities, imperforate anus, and skin tag.1 An alternative description of similar findings is LUMBAR (lower body hemangioma and other cutaneous defects; urogenital anomalies, ulceration; myelopathy; bony deformities; anorectal malformations, arterial anomalies; and renal anomalies).5 Researchers have suggested that both of these acronyms describe the same syndrome, and it is common for the syndrome to be incomplete.6 One study (N=11) found that perineal hemangiomas are most commonly associated with anal malformations (8 patients), followed by urinary tract abnormalities (7 patients) and malformation of the external genitalia (7 patients). A skin tag was present in 5 patients.1 The pathogenesis of PELVIS syndrome is unknown.
When an infant presents with a perineal hemangioma and physical examination suggests PELVIS syndrome, imaging should be performed to evaluate for other anomalies. Before 4 months of age, ultrasound should be utilized to investigate the presence of reno-genitourinary or spinal malformations. Magnetic resonance imaging is the preferred imaging modality in children older than 4 months.7 Management of PELVIS syndrome requires a multidisciplinary approach and early recognition of the full extent of congenital malformations. Pediatric dermatologists, urologists, endocrinologists, and neonatologists have a role in its diagnosis and treatment.
- Girard C, Bigorre M, Guillot B, et al. PELVIS syndrome. Arch Dermatol. 2006;142:884-888.
- Bruckner AL, Frieden IJ. Hemangiomas of infancy. J Am Acad Dermatol. 2003;48:477-496.
- Frieden IJ, Reese V, Cohen D. PHACE syndrome: the association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132:307-311.
- Albright AL, Gartner JC, Wiener ES. Lumbar cutaneous hemangiomas as indicators of tethered spinal cords. Pediatrics. 1989;83:977-980.
- Iacobas I, Burrows PE, Frieden IJ, et al. LUMBAR: association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies. J Pediatr. 2010;157:795-801.
- Frade FN, Kadlub V, Soupre S, et al. PELVIS or LUMBAR syndrome: the same entity. two case reports. Arch Pediatr. 2012;19:55-58.
- Berk DR, Bayliss SJ, Merritt DF. Management quandary: extensive perineal infantile hemangioma with associated congenital anomalies: an example of the PELVIS syndrome. J Pediatr Adolesc Gynecol. 2007;20:105-108.
The Diagnosis: PELVIS Syndrome
Infantile hemangiomas (IHs) are present in up to 10% of infants by 1 year of age and are most commonly located on the face and upper extremities. Less than 10% of IHs develop in the perineum.1 Perineal IHs are benign tumors of the vascular endothelium that present as plaques and commonly are accompanied by painful ulcerations. Ulceration is more common in the diaper area secondary to irritation from urine, stool, and friction.2 Although most IHs are benign isolated findings, facial IHs have been associated with several syndromes including Sturge-Weber and PHACE (posterior fossa brain malformations, hemangiomas, arterial anomalies, cardiac anomalies and coarctation of the aorta, and eye and endocrine abnormalities) syndromes.3 Researchers also have identified an association between lumbosacral IHs and spinal dysraphism (tethered spinal cord).4
A smaller number of studies have investigated congenital anomalies related to perineal IH,1,5 specifically PELVIS syndrome. The acronym PELVIS has been used to describe a syndrome of congenital malformations including perineal hemangioma, external genital malformations, lipomyelomeningocele, vesicorenal abnormalities, imperforate anus, and skin tag.1 An alternative description of similar findings is LUMBAR (lower body hemangioma and other cutaneous defects; urogenital anomalies, ulceration; myelopathy; bony deformities; anorectal malformations, arterial anomalies; and renal anomalies).5 Researchers have suggested that both of these acronyms describe the same syndrome, and it is common for the syndrome to be incomplete.6 One study (N=11) found that perineal hemangiomas are most commonly associated with anal malformations (8 patients), followed by urinary tract abnormalities (7 patients) and malformation of the external genitalia (7 patients). A skin tag was present in 5 patients.1 The pathogenesis of PELVIS syndrome is unknown.
When an infant presents with a perineal hemangioma and physical examination suggests PELVIS syndrome, imaging should be performed to evaluate for other anomalies. Before 4 months of age, ultrasound should be utilized to investigate the presence of reno-genitourinary or spinal malformations. Magnetic resonance imaging is the preferred imaging modality in children older than 4 months.7 Management of PELVIS syndrome requires a multidisciplinary approach and early recognition of the full extent of congenital malformations. Pediatric dermatologists, urologists, endocrinologists, and neonatologists have a role in its diagnosis and treatment.
The Diagnosis: PELVIS Syndrome
Infantile hemangiomas (IHs) are present in up to 10% of infants by 1 year of age and are most commonly located on the face and upper extremities. Less than 10% of IHs develop in the perineum.1 Perineal IHs are benign tumors of the vascular endothelium that present as plaques and commonly are accompanied by painful ulcerations. Ulceration is more common in the diaper area secondary to irritation from urine, stool, and friction.2 Although most IHs are benign isolated findings, facial IHs have been associated with several syndromes including Sturge-Weber and PHACE (posterior fossa brain malformations, hemangiomas, arterial anomalies, cardiac anomalies and coarctation of the aorta, and eye and endocrine abnormalities) syndromes.3 Researchers also have identified an association between lumbosacral IHs and spinal dysraphism (tethered spinal cord).4
A smaller number of studies have investigated congenital anomalies related to perineal IH,1,5 specifically PELVIS syndrome. The acronym PELVIS has been used to describe a syndrome of congenital malformations including perineal hemangioma, external genital malformations, lipomyelomeningocele, vesicorenal abnormalities, imperforate anus, and skin tag.1 An alternative description of similar findings is LUMBAR (lower body hemangioma and other cutaneous defects; urogenital anomalies, ulceration; myelopathy; bony deformities; anorectal malformations, arterial anomalies; and renal anomalies).5 Researchers have suggested that both of these acronyms describe the same syndrome, and it is common for the syndrome to be incomplete.6 One study (N=11) found that perineal hemangiomas are most commonly associated with anal malformations (8 patients), followed by urinary tract abnormalities (7 patients) and malformation of the external genitalia (7 patients). A skin tag was present in 5 patients.1 The pathogenesis of PELVIS syndrome is unknown.
When an infant presents with a perineal hemangioma and physical examination suggests PELVIS syndrome, imaging should be performed to evaluate for other anomalies. Before 4 months of age, ultrasound should be utilized to investigate the presence of reno-genitourinary or spinal malformations. Magnetic resonance imaging is the preferred imaging modality in children older than 4 months.7 Management of PELVIS syndrome requires a multidisciplinary approach and early recognition of the full extent of congenital malformations. Pediatric dermatologists, urologists, endocrinologists, and neonatologists have a role in its diagnosis and treatment.
- Girard C, Bigorre M, Guillot B, et al. PELVIS syndrome. Arch Dermatol. 2006;142:884-888.
- Bruckner AL, Frieden IJ. Hemangiomas of infancy. J Am Acad Dermatol. 2003;48:477-496.
- Frieden IJ, Reese V, Cohen D. PHACE syndrome: the association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132:307-311.
- Albright AL, Gartner JC, Wiener ES. Lumbar cutaneous hemangiomas as indicators of tethered spinal cords. Pediatrics. 1989;83:977-980.
- Iacobas I, Burrows PE, Frieden IJ, et al. LUMBAR: association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies. J Pediatr. 2010;157:795-801.
- Frade FN, Kadlub V, Soupre S, et al. PELVIS or LUMBAR syndrome: the same entity. two case reports. Arch Pediatr. 2012;19:55-58.
- Berk DR, Bayliss SJ, Merritt DF. Management quandary: extensive perineal infantile hemangioma with associated congenital anomalies: an example of the PELVIS syndrome. J Pediatr Adolesc Gynecol. 2007;20:105-108.
- Girard C, Bigorre M, Guillot B, et al. PELVIS syndrome. Arch Dermatol. 2006;142:884-888.
- Bruckner AL, Frieden IJ. Hemangiomas of infancy. J Am Acad Dermatol. 2003;48:477-496.
- Frieden IJ, Reese V, Cohen D. PHACE syndrome: the association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132:307-311.
- Albright AL, Gartner JC, Wiener ES. Lumbar cutaneous hemangiomas as indicators of tethered spinal cords. Pediatrics. 1989;83:977-980.
- Iacobas I, Burrows PE, Frieden IJ, et al. LUMBAR: association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies. J Pediatr. 2010;157:795-801.
- Frade FN, Kadlub V, Soupre S, et al. PELVIS or LUMBAR syndrome: the same entity. two case reports. Arch Pediatr. 2012;19:55-58.
- Berk DR, Bayliss SJ, Merritt DF. Management quandary: extensive perineal infantile hemangioma with associated congenital anomalies: an example of the PELVIS syndrome. J Pediatr Adolesc Gynecol. 2007;20:105-108.
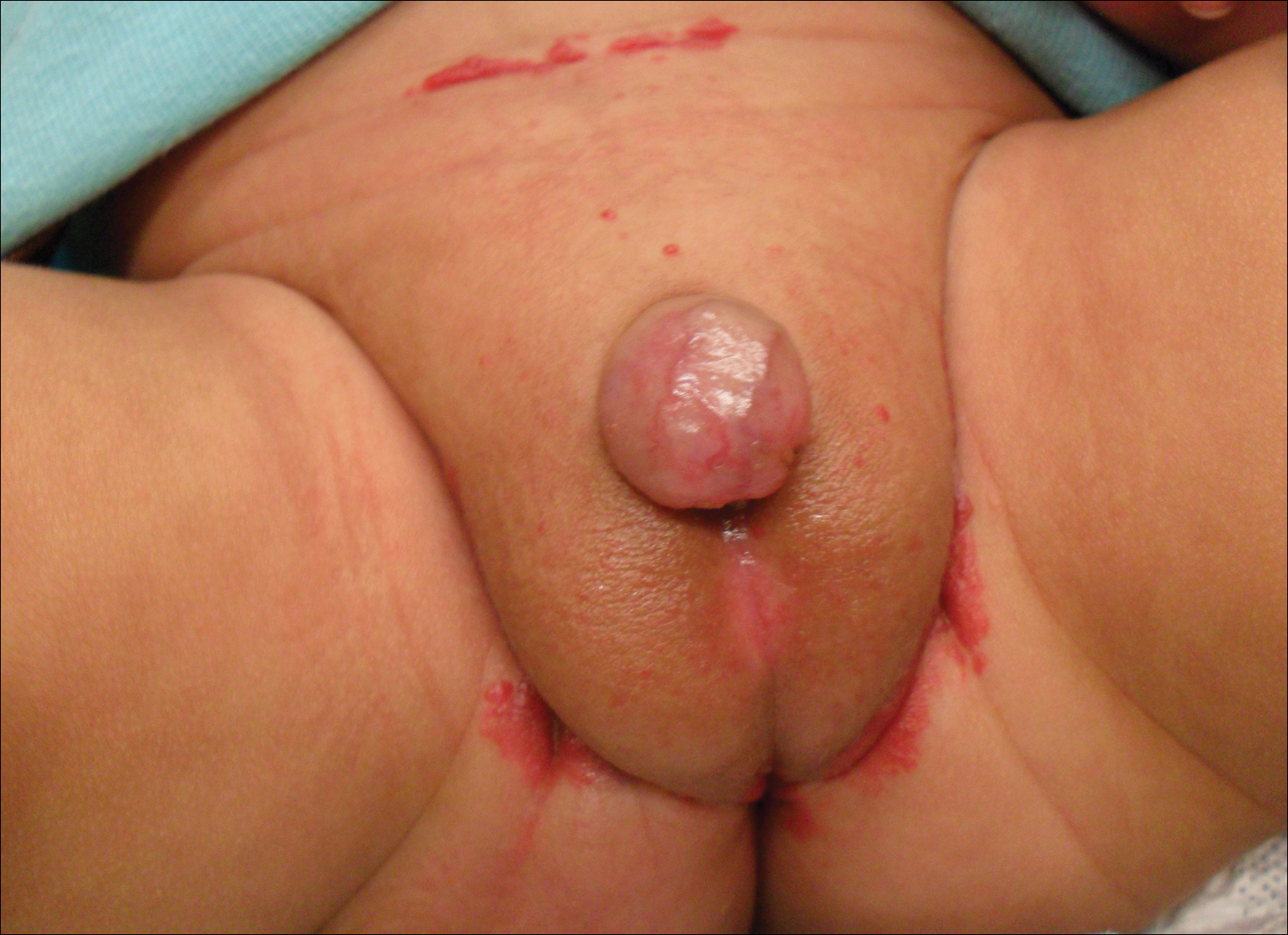
A 7-week-old boy with ambiguous genitalia presented for evaluation of what the parents described as progressively worsening diaper rash. The patient was born at full-term after an uncomplicated gestation via normal spontaneous vaginal delivery. Examination of the external genitalia revealed microphallus with phimosis and a bifid scrotum. Two weeks after birth, the patient developed redness and painful ulcerations in the diaper area. At the time of presentation, the patient had bright red plaques along the suprapubic lines, inguinal creases, and in the perineal region. Physical examination also was notable for tender ulcerations of the inguinal creases and perineum and a perineal skin tag.
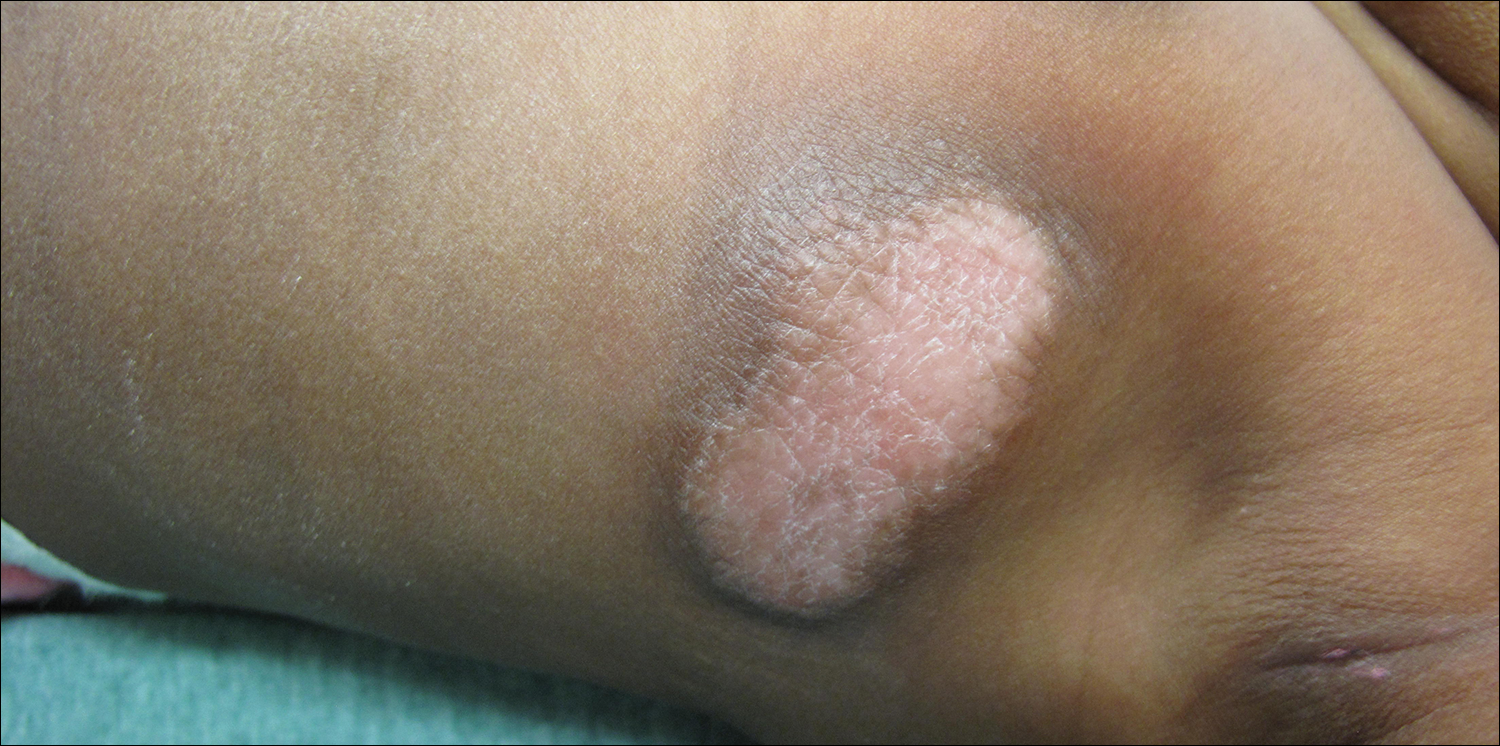
Large Hyperpigmented Plaques on the Trunk of a Newborn
The Diagnosis: Cutaneous Mastocytoma
Physical examination revealed a 58×51-mm hyperpigmented plaque with central pink coloration and scale on the right side of the back as well as a 39×33-mm pink plaque with a hyperpigmented border on the left side of the flank (Figure 1). At follow-up 2 weeks later, the patient's parents reported that blisters formed within both of the plaques. The blisters ruptured a few hours after forming and drained clear fluid with scant blood. Both plaques contained erosions from the ruptured bullae but remained the same size with no surrounding erythema or warmth. A 4-mm punch biopsy was performed of intact skin from the back lesion (Figure 2A). Histologic examination revealed a cellular infiltrate of monotonous bland cells that completely filled the dermis without epidermal involvement, along with occasional intermixed eosinophils. The morphology of these infiltrating cells was compatible with mast cells confirmed by strongly positive Leder staining (Figure 2B).
Mastocytosis encompasses a rare group of disorders characterized by abnormal mast cell accumulation or mast cell mediator release in various tissues. These disorders can be classified as either systemic mastocytosis with mast cell infiltration into bone marrow or other extracutaneous organs, or cutaneous mastocytosis with disease limited to the skin.1 Mutations involving activation of the c-Kit receptor in stimulating mast cell growth and development have been implicated in both systemic and cutaneous forms of the disease.2,3
Cutaneous mastocytosis is most often diagnosed in childhood and typically is characterized by spontaneous regression before puberty in a majority of cases.1,4 Under the World Health Organization classification system, cutaneous mastocytosis can be further subdivided into 3 disorders (listed in order of most to least common): urticaria pigmentosa (also known as maculopapular cutaneous mastocytosis) with typical, plaque, and nodular forms; cutaneous mastocytoma (as seen in this patient); and diffuse cutaneous mastocytosis.5 Compared to the widespread distribution of small macules and papules in urticaria pigmentosa, the cutaneous mastocytoma subtype presents with 1 to 6 brown to orange-yellow plaques or nodules measuring more than 1 cm in diameter. Cutaneous mastocytoma typically presents in infancy and is located most commonly on the trunk and extremities, though it may be found on the face or scalp. The plaques of mastocytoma often have well-defined margins, and these lesions may become bullous or demonstrate Darier sign of urtication and erythema on physical stimulation. Patients most commonly experience pruritus from mast cell degranulation and rarely exhibit systemic symptoms of mast cell mediator release; however, generalized flushing, hypotension, headaches, and gastrointestinal symptoms may occur, particularly if the lesion is vigorously rubbed.6,7 Conditions in the differential include aplasia cutis congenita, connective tissue nevus, epidermal nevus, and epidermolysis bullosa. They should not elicit a blister if rubbed, except for epidermolysis bullosa, which can easily be differentiated based on histology.
The workup for cutaneous mastocytosis in the pediatric population may include a biopsy of lesional skin, though in many cases the characteristic cutaneous manifestations are sufficient to make a diagnosis. Histologically, biopsy results often reveal abundant diffuse dermal infiltration of mast cells, which are characterized by their large pink granular cytoplasm and round dense central nuclei. In pediatric patients, mast cells typically are restricted to the dermis, and there is a low risk for hematologic abnormalities, thereby precluding the need for bone marrow examination in the absence of organomegaly or notable peripheral blood abnormalities such as severe cytopenia.5,6
Management of cutaneous mastocytosis consists of avoidance of mast cell degranulation triggers and symptomatic treatment of histamine release. Triggers include certain medications (eg, narcotic analgesics, aspirin, nonsteroidal anti-inflammatory drugs, iodinated contrast agents, antibiotics, muscle relaxants), mechanical irritation, insect stings, spicy foods, stress, or extreme temperature changes.8 Symptomatic treatment can be achieved through topical corticosteroid or oral antihistamine use. Along with decreasing pruritus, topical corticosteroids also may be helpful in decreasing time to spontaneous resolution and healing.7 The patient in this case was treated with desonide ointment 0.05% daily to both lesions as well as mupirocin ointment 2% as needed for erosions. These treatments helped reduce the patient's symptoms, but her lesions persisted over a follow-up period of 4 months.
- Valent P, Sperr WR, Schwartz LB, et al. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114:3-11.
- Bibi S, Langenfeld F, Jeanningros S, et al. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol Allergy Clin North Am. 2014;34:239-262.
- Yavuz AS, Lipsky PE, Yavuz S, et al. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood. 2002;100:661-665.
- Méni C, Bruneau J, Georgin-Lavialle S, et al. Paediatric mastocytosis: a systematic review of 1747 cases. Br J Dermatol. 2015;172:642-651.
- Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25:603-625.
- Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res. 2001;25:519-528.
- Patrizi A, Tabanelli M, Neri I, et al. Topical corticosteroids versus "wait and see" in the management of solitary mastocytoma in pediatric patients: a long-term follow-up. Dermatol Ther. 2015;28:57-61.
- Bonadonna P, Lombardo C. Drug allergy in mastocytosis. Immunol Allergy Clin North Am. 2014;34:397-405.
The Diagnosis: Cutaneous Mastocytoma
Physical examination revealed a 58×51-mm hyperpigmented plaque with central pink coloration and scale on the right side of the back as well as a 39×33-mm pink plaque with a hyperpigmented border on the left side of the flank (Figure 1). At follow-up 2 weeks later, the patient's parents reported that blisters formed within both of the plaques. The blisters ruptured a few hours after forming and drained clear fluid with scant blood. Both plaques contained erosions from the ruptured bullae but remained the same size with no surrounding erythema or warmth. A 4-mm punch biopsy was performed of intact skin from the back lesion (Figure 2A). Histologic examination revealed a cellular infiltrate of monotonous bland cells that completely filled the dermis without epidermal involvement, along with occasional intermixed eosinophils. The morphology of these infiltrating cells was compatible with mast cells confirmed by strongly positive Leder staining (Figure 2B).
Mastocytosis encompasses a rare group of disorders characterized by abnormal mast cell accumulation or mast cell mediator release in various tissues. These disorders can be classified as either systemic mastocytosis with mast cell infiltration into bone marrow or other extracutaneous organs, or cutaneous mastocytosis with disease limited to the skin.1 Mutations involving activation of the c-Kit receptor in stimulating mast cell growth and development have been implicated in both systemic and cutaneous forms of the disease.2,3
Cutaneous mastocytosis is most often diagnosed in childhood and typically is characterized by spontaneous regression before puberty in a majority of cases.1,4 Under the World Health Organization classification system, cutaneous mastocytosis can be further subdivided into 3 disorders (listed in order of most to least common): urticaria pigmentosa (also known as maculopapular cutaneous mastocytosis) with typical, plaque, and nodular forms; cutaneous mastocytoma (as seen in this patient); and diffuse cutaneous mastocytosis.5 Compared to the widespread distribution of small macules and papules in urticaria pigmentosa, the cutaneous mastocytoma subtype presents with 1 to 6 brown to orange-yellow plaques or nodules measuring more than 1 cm in diameter. Cutaneous mastocytoma typically presents in infancy and is located most commonly on the trunk and extremities, though it may be found on the face or scalp. The plaques of mastocytoma often have well-defined margins, and these lesions may become bullous or demonstrate Darier sign of urtication and erythema on physical stimulation. Patients most commonly experience pruritus from mast cell degranulation and rarely exhibit systemic symptoms of mast cell mediator release; however, generalized flushing, hypotension, headaches, and gastrointestinal symptoms may occur, particularly if the lesion is vigorously rubbed.6,7 Conditions in the differential include aplasia cutis congenita, connective tissue nevus, epidermal nevus, and epidermolysis bullosa. They should not elicit a blister if rubbed, except for epidermolysis bullosa, which can easily be differentiated based on histology.
The workup for cutaneous mastocytosis in the pediatric population may include a biopsy of lesional skin, though in many cases the characteristic cutaneous manifestations are sufficient to make a diagnosis. Histologically, biopsy results often reveal abundant diffuse dermal infiltration of mast cells, which are characterized by their large pink granular cytoplasm and round dense central nuclei. In pediatric patients, mast cells typically are restricted to the dermis, and there is a low risk for hematologic abnormalities, thereby precluding the need for bone marrow examination in the absence of organomegaly or notable peripheral blood abnormalities such as severe cytopenia.5,6
Management of cutaneous mastocytosis consists of avoidance of mast cell degranulation triggers and symptomatic treatment of histamine release. Triggers include certain medications (eg, narcotic analgesics, aspirin, nonsteroidal anti-inflammatory drugs, iodinated contrast agents, antibiotics, muscle relaxants), mechanical irritation, insect stings, spicy foods, stress, or extreme temperature changes.8 Symptomatic treatment can be achieved through topical corticosteroid or oral antihistamine use. Along with decreasing pruritus, topical corticosteroids also may be helpful in decreasing time to spontaneous resolution and healing.7 The patient in this case was treated with desonide ointment 0.05% daily to both lesions as well as mupirocin ointment 2% as needed for erosions. These treatments helped reduce the patient's symptoms, but her lesions persisted over a follow-up period of 4 months.
The Diagnosis: Cutaneous Mastocytoma
Physical examination revealed a 58×51-mm hyperpigmented plaque with central pink coloration and scale on the right side of the back as well as a 39×33-mm pink plaque with a hyperpigmented border on the left side of the flank (Figure 1). At follow-up 2 weeks later, the patient's parents reported that blisters formed within both of the plaques. The blisters ruptured a few hours after forming and drained clear fluid with scant blood. Both plaques contained erosions from the ruptured bullae but remained the same size with no surrounding erythema or warmth. A 4-mm punch biopsy was performed of intact skin from the back lesion (Figure 2A). Histologic examination revealed a cellular infiltrate of monotonous bland cells that completely filled the dermis without epidermal involvement, along with occasional intermixed eosinophils. The morphology of these infiltrating cells was compatible with mast cells confirmed by strongly positive Leder staining (Figure 2B).
Mastocytosis encompasses a rare group of disorders characterized by abnormal mast cell accumulation or mast cell mediator release in various tissues. These disorders can be classified as either systemic mastocytosis with mast cell infiltration into bone marrow or other extracutaneous organs, or cutaneous mastocytosis with disease limited to the skin.1 Mutations involving activation of the c-Kit receptor in stimulating mast cell growth and development have been implicated in both systemic and cutaneous forms of the disease.2,3
Cutaneous mastocytosis is most often diagnosed in childhood and typically is characterized by spontaneous regression before puberty in a majority of cases.1,4 Under the World Health Organization classification system, cutaneous mastocytosis can be further subdivided into 3 disorders (listed in order of most to least common): urticaria pigmentosa (also known as maculopapular cutaneous mastocytosis) with typical, plaque, and nodular forms; cutaneous mastocytoma (as seen in this patient); and diffuse cutaneous mastocytosis.5 Compared to the widespread distribution of small macules and papules in urticaria pigmentosa, the cutaneous mastocytoma subtype presents with 1 to 6 brown to orange-yellow plaques or nodules measuring more than 1 cm in diameter. Cutaneous mastocytoma typically presents in infancy and is located most commonly on the trunk and extremities, though it may be found on the face or scalp. The plaques of mastocytoma often have well-defined margins, and these lesions may become bullous or demonstrate Darier sign of urtication and erythema on physical stimulation. Patients most commonly experience pruritus from mast cell degranulation and rarely exhibit systemic symptoms of mast cell mediator release; however, generalized flushing, hypotension, headaches, and gastrointestinal symptoms may occur, particularly if the lesion is vigorously rubbed.6,7 Conditions in the differential include aplasia cutis congenita, connective tissue nevus, epidermal nevus, and epidermolysis bullosa. They should not elicit a blister if rubbed, except for epidermolysis bullosa, which can easily be differentiated based on histology.
The workup for cutaneous mastocytosis in the pediatric population may include a biopsy of lesional skin, though in many cases the characteristic cutaneous manifestations are sufficient to make a diagnosis. Histologically, biopsy results often reveal abundant diffuse dermal infiltration of mast cells, which are characterized by their large pink granular cytoplasm and round dense central nuclei. In pediatric patients, mast cells typically are restricted to the dermis, and there is a low risk for hematologic abnormalities, thereby precluding the need for bone marrow examination in the absence of organomegaly or notable peripheral blood abnormalities such as severe cytopenia.5,6
Management of cutaneous mastocytosis consists of avoidance of mast cell degranulation triggers and symptomatic treatment of histamine release. Triggers include certain medications (eg, narcotic analgesics, aspirin, nonsteroidal anti-inflammatory drugs, iodinated contrast agents, antibiotics, muscle relaxants), mechanical irritation, insect stings, spicy foods, stress, or extreme temperature changes.8 Symptomatic treatment can be achieved through topical corticosteroid or oral antihistamine use. Along with decreasing pruritus, topical corticosteroids also may be helpful in decreasing time to spontaneous resolution and healing.7 The patient in this case was treated with desonide ointment 0.05% daily to both lesions as well as mupirocin ointment 2% as needed for erosions. These treatments helped reduce the patient's symptoms, but her lesions persisted over a follow-up period of 4 months.
- Valent P, Sperr WR, Schwartz LB, et al. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114:3-11.
- Bibi S, Langenfeld F, Jeanningros S, et al. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol Allergy Clin North Am. 2014;34:239-262.
- Yavuz AS, Lipsky PE, Yavuz S, et al. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood. 2002;100:661-665.
- Méni C, Bruneau J, Georgin-Lavialle S, et al. Paediatric mastocytosis: a systematic review of 1747 cases. Br J Dermatol. 2015;172:642-651.
- Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25:603-625.
- Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res. 2001;25:519-528.
- Patrizi A, Tabanelli M, Neri I, et al. Topical corticosteroids versus "wait and see" in the management of solitary mastocytoma in pediatric patients: a long-term follow-up. Dermatol Ther. 2015;28:57-61.
- Bonadonna P, Lombardo C. Drug allergy in mastocytosis. Immunol Allergy Clin North Am. 2014;34:397-405.
- Valent P, Sperr WR, Schwartz LB, et al. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114:3-11.
- Bibi S, Langenfeld F, Jeanningros S, et al. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol Allergy Clin North Am. 2014;34:239-262.
- Yavuz AS, Lipsky PE, Yavuz S, et al. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood. 2002;100:661-665.
- Méni C, Bruneau J, Georgin-Lavialle S, et al. Paediatric mastocytosis: a systematic review of 1747 cases. Br J Dermatol. 2015;172:642-651.
- Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25:603-625.
- Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res. 2001;25:519-528.
- Patrizi A, Tabanelli M, Neri I, et al. Topical corticosteroids versus "wait and see" in the management of solitary mastocytoma in pediatric patients: a long-term follow-up. Dermatol Ther. 2015;28:57-61.
- Bonadonna P, Lombardo C. Drug allergy in mastocytosis. Immunol Allergy Clin North Am. 2014;34:397-405.
A 4-day-old girl with no notable medical history presented with 2 pink lesions on the right side of the back and left side of the flank. Both lesions were present at birth and had not changed in size, shape, or color in the first 4 days of life. She had no constitutional symptoms. The child was a full-term newborn, and her mother experienced no pregnancy or delivery complications. She had no family history of similar skin findings.
Bluish Gray Hyperpigmentation on the Face and Neck
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7
- Ramirez CO. Los cenicientos: problema clinica. In: Memoria del Primer Congresso Centroamericano de Dermatologica, December 5-8, 1957. San Salvador, El Salvador; 1957:122-130.
- Lee SJ, Chung KY. Erythema dyschromicum perstans in early childhood. J Dermatol. 1999;26:119-121.
- Homez-Chacin, Barroso C. On the etiopathogenic of the erythema dyschromicum perstans: possibility of a melanosis neurocutaneous. Dermatol Venez. 1996;4:149-151.
- Correa MC, Memije EV, Vargas-Alarcon G, et al. HLA-DR association with the genetic susceptibility to develop ashy dermatosis in Mexican Mestizo patients [published online November 20, 2006]. J Am Acad Dermatol. 2007;56:617-620.
- Jablonska S. Ingestion of ammonium nitrate as a possible cause of erythema dyschromicum perstans (ashy dermatosis). Dermatologica. 1975;150:287-291.
- Stevenson JR, Miura M. Erythema dyschromicum perstans (ashy dermatosis). Arch Dermatol. 1966;94:196-199.
- Baranda L, Torres-Alvarez B, Cortes-Franco R, et al. Involvement of cell adhesion and activation molecules in the pathogenesis of erythema dyschromicum perstans (ashy dermatitis). the effect of clofazimine therapy. Arch Dermatol. 1997;133:325-329.
- Vasquez-Ochoa LA, Isaza-Guzman DM, Orozco-Mora B, et al. Immunopathologic study of erythema dyschromicum perstans (ashy dermatosis). Int J Dermatol. 2006;45:937-941.
- Convit J, Kerdel-Vegas F, Roderiguez G. Erythema dyschromicum perstans: a hiltherto undescribed skin disease. J Invest Dermatol. 1961;36:457-462.
- Ono S, Miyachi Y, Kabashima K. Ashy dermatosis with prior pruritic and scaling skin lesions. J Dermatol. 2012;39:1103-1104.
- Sanchez NP, Pathak MA, Sato SS, et al. Circumscribed dermal melaninoses: classification, light, histochemical, and electron microscopic studies on three patients with the erythema dyschromicum perstans type. Int J Dermatol. 1982;21:25-32.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Ahmed SV, Sajjan R. Chrysiasis: a gold "curse!" [published online May 21, 2009]. BMJ Case Rep. 2009;2009.
- Fiscus V, Hankinson A, Alweis R. Minocycline-induced hyperpigmentation. J Community Hosp Intern Med Perspect. 2014;4. doi:10.3402/jchimp.v4.24063.
- Cox AJ, Marich KW. Gold in the dermis following gold therapy for rheumatoid arthritis. Arch Dermatol. 1973;108:655-657.
- al-Talib RK, Wright DH, Theaker JM. Orange-red birefringence of gold particles in paraffin wax embedded sections: an aid to the diagnosis of chrysiasis. Histopathology. 1994;24:176-178.
- Meyer AJ, Nahass GT. Hyperpigmented patches on the dorsa of the feet. minocycline pigmentation. Arch Dermatol. 1995;131:1447-1450.
- Bayne-Poorman M, Shubrook J. Bluish pigmentation of face and sclera. J Fam Pract. 2010;59:519-522.
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7
- Ramirez CO. Los cenicientos: problema clinica. In: Memoria del Primer Congresso Centroamericano de Dermatologica, December 5-8, 1957. San Salvador, El Salvador; 1957:122-130.
- Lee SJ, Chung KY. Erythema dyschromicum perstans in early childhood. J Dermatol. 1999;26:119-121.
- Homez-Chacin, Barroso C. On the etiopathogenic of the erythema dyschromicum perstans: possibility of a melanosis neurocutaneous. Dermatol Venez. 1996;4:149-151.
- Correa MC, Memije EV, Vargas-Alarcon G, et al. HLA-DR association with the genetic susceptibility to develop ashy dermatosis in Mexican Mestizo patients [published online November 20, 2006]. J Am Acad Dermatol. 2007;56:617-620.
- Jablonska S. Ingestion of ammonium nitrate as a possible cause of erythema dyschromicum perstans (ashy dermatosis). Dermatologica. 1975;150:287-291.
- Stevenson JR, Miura M. Erythema dyschromicum perstans (ashy dermatosis). Arch Dermatol. 1966;94:196-199.
- Baranda L, Torres-Alvarez B, Cortes-Franco R, et al. Involvement of cell adhesion and activation molecules in the pathogenesis of erythema dyschromicum perstans (ashy dermatitis). the effect of clofazimine therapy. Arch Dermatol. 1997;133:325-329.
- Vasquez-Ochoa LA, Isaza-Guzman DM, Orozco-Mora B, et al. Immunopathologic study of erythema dyschromicum perstans (ashy dermatosis). Int J Dermatol. 2006;45:937-941.
- Convit J, Kerdel-Vegas F, Roderiguez G. Erythema dyschromicum perstans: a hiltherto undescribed skin disease. J Invest Dermatol. 1961;36:457-462.
- Ono S, Miyachi Y, Kabashima K. Ashy dermatosis with prior pruritic and scaling skin lesions. J Dermatol. 2012;39:1103-1104.
- Sanchez NP, Pathak MA, Sato SS, et al. Circumscribed dermal melaninoses: classification, light, histochemical, and electron microscopic studies on three patients with the erythema dyschromicum perstans type. Int J Dermatol. 1982;21:25-32.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Ahmed SV, Sajjan R. Chrysiasis: a gold "curse!" [published online May 21, 2009]. BMJ Case Rep. 2009;2009.
- Fiscus V, Hankinson A, Alweis R. Minocycline-induced hyperpigmentation. J Community Hosp Intern Med Perspect. 2014;4. doi:10.3402/jchimp.v4.24063.
- Cox AJ, Marich KW. Gold in the dermis following gold therapy for rheumatoid arthritis. Arch Dermatol. 1973;108:655-657.
- al-Talib RK, Wright DH, Theaker JM. Orange-red birefringence of gold particles in paraffin wax embedded sections: an aid to the diagnosis of chrysiasis. Histopathology. 1994;24:176-178.
- Meyer AJ, Nahass GT. Hyperpigmented patches on the dorsa of the feet. minocycline pigmentation. Arch Dermatol. 1995;131:1447-1450.
- Bayne-Poorman M, Shubrook J. Bluish pigmentation of face and sclera. J Fam Pract. 2010;59:519-522.
- Ramirez CO. Los cenicientos: problema clinica. In: Memoria del Primer Congresso Centroamericano de Dermatologica, December 5-8, 1957. San Salvador, El Salvador; 1957:122-130.
- Lee SJ, Chung KY. Erythema dyschromicum perstans in early childhood. J Dermatol. 1999;26:119-121.
- Homez-Chacin, Barroso C. On the etiopathogenic of the erythema dyschromicum perstans: possibility of a melanosis neurocutaneous. Dermatol Venez. 1996;4:149-151.
- Correa MC, Memije EV, Vargas-Alarcon G, et al. HLA-DR association with the genetic susceptibility to develop ashy dermatosis in Mexican Mestizo patients [published online November 20, 2006]. J Am Acad Dermatol. 2007;56:617-620.
- Jablonska S. Ingestion of ammonium nitrate as a possible cause of erythema dyschromicum perstans (ashy dermatosis). Dermatologica. 1975;150:287-291.
- Stevenson JR, Miura M. Erythema dyschromicum perstans (ashy dermatosis). Arch Dermatol. 1966;94:196-199.
- Baranda L, Torres-Alvarez B, Cortes-Franco R, et al. Involvement of cell adhesion and activation molecules in the pathogenesis of erythema dyschromicum perstans (ashy dermatitis). the effect of clofazimine therapy. Arch Dermatol. 1997;133:325-329.
- Vasquez-Ochoa LA, Isaza-Guzman DM, Orozco-Mora B, et al. Immunopathologic study of erythema dyschromicum perstans (ashy dermatosis). Int J Dermatol. 2006;45:937-941.
- Convit J, Kerdel-Vegas F, Roderiguez G. Erythema dyschromicum perstans: a hiltherto undescribed skin disease. J Invest Dermatol. 1961;36:457-462.
- Ono S, Miyachi Y, Kabashima K. Ashy dermatosis with prior pruritic and scaling skin lesions. J Dermatol. 2012;39:1103-1104.
- Sanchez NP, Pathak MA, Sato SS, et al. Circumscribed dermal melaninoses: classification, light, histochemical, and electron microscopic studies on three patients with the erythema dyschromicum perstans type. Int J Dermatol. 1982;21:25-32.
- Vega ME, Waxtein L, Arenas R, et al. Ashy dermatosis and lichen planus pigmentosus: a clinicopathologic study of 31 cases. Int J Dermatol. 1992;31:90-94.
- Ahmed SV, Sajjan R. Chrysiasis: a gold "curse!" [published online May 21, 2009]. BMJ Case Rep. 2009;2009.
- Fiscus V, Hankinson A, Alweis R. Minocycline-induced hyperpigmentation. J Community Hosp Intern Med Perspect. 2014;4. doi:10.3402/jchimp.v4.24063.
- Cox AJ, Marich KW. Gold in the dermis following gold therapy for rheumatoid arthritis. Arch Dermatol. 1973;108:655-657.
- al-Talib RK, Wright DH, Theaker JM. Orange-red birefringence of gold particles in paraffin wax embedded sections: an aid to the diagnosis of chrysiasis. Histopathology. 1994;24:176-178.
- Meyer AJ, Nahass GT. Hyperpigmented patches on the dorsa of the feet. minocycline pigmentation. Arch Dermatol. 1995;131:1447-1450.
- Bayne-Poorman M, Shubrook J. Bluish pigmentation of face and sclera. J Fam Pract. 2010;59:519-522.
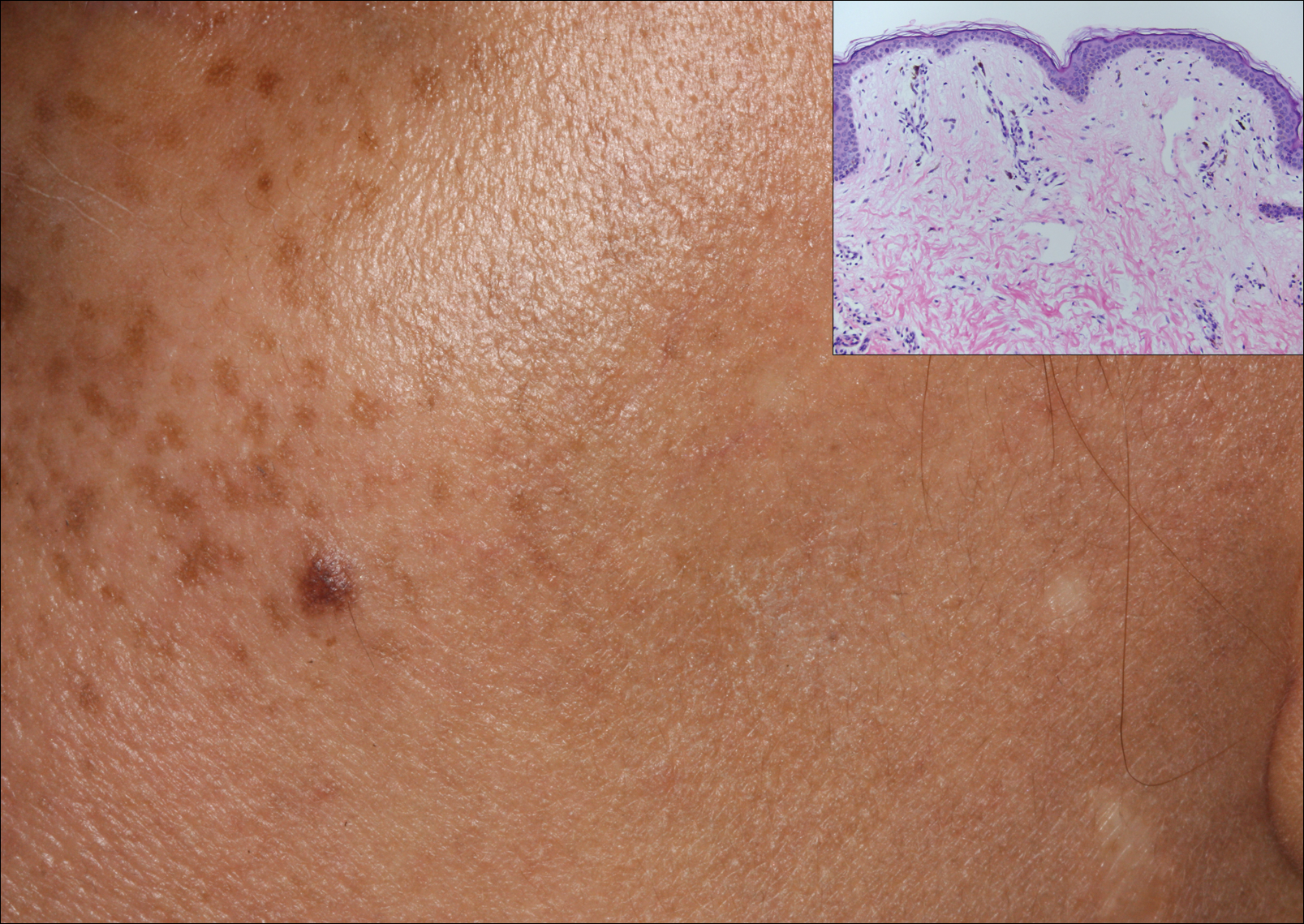
A middle-aged woman with Fitzpatrick skin type IV was evaluated for progressive hyperpigmentation of several months' duration involving the neck, jawline, both sides of the face, and forehead. The lesions were mildly pruritic. She denied contact with any new substance and there was no history of an eruption preceding the hyperpigmentation. Medical history included chronic anemia that was managed with iron supplementation. On physical examination, blue-gray nonscaly macules and patches were observed distributed symmetrically on the neck, jawline, sides of the face, and forehead. Microscopic examination of 2 shave biopsies revealed subtle vacuolar interface dermatitis with mild perivascular lymphocytic infiltrate and dermal melanophages (inset).
Recalcitrant Hyperkeratotic Plaques
The Diagnosis: Hypertrophic Lupus Erythematosus
Physical examination at initial presentation revealed well-demarcated, 2- to 3-cm plaques with scale distributed most extensively on the elbows and shins with lesser involvement of the chest and abdomen. After treatment with topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, new annular, erythematous, and edematous lesions began to appear on the chest and abdomen (Figure 1). These new lesions appeared less hyperkeratotic than the older ones.
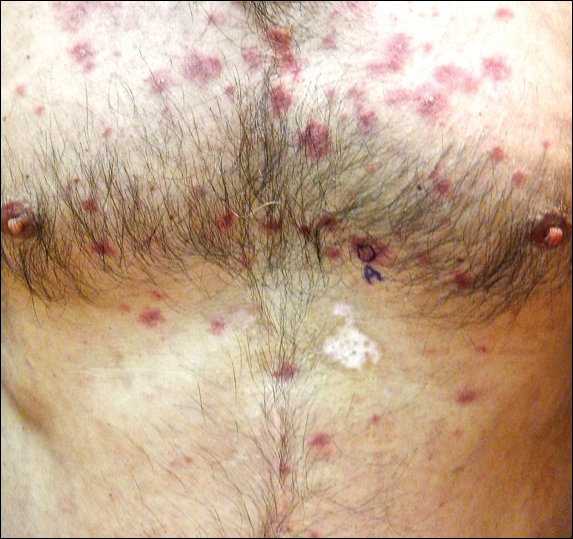
Biopsy of a hyperkeratotic lesion from the patient's arm revealed marked hyperkeratosis, parakeratosis, epidermal hyperplasia, focal vacuolar change, solar elastosis, and transepidermal elastotic elimination (Figure 2A). A second biopsy performed on a newer chest lesion revealed interface changes, degeneration of the basal layer, follicular plugging, and dermal mucin (Figure 2B). Serology revealed an antinuclear antibody (ANA) titer of 1:1280 (reference range, <1:40 dilution) and hemoglobin of 11.5 g/dL (reference range, 14.0-17.5 g/dL). On the basis of clinical, histologic, and serologic findings, hypertrophic lupus erythematosus (LE) was diagnosed. The patient was treated with oral prednisone, which resulted in rapid improvement.
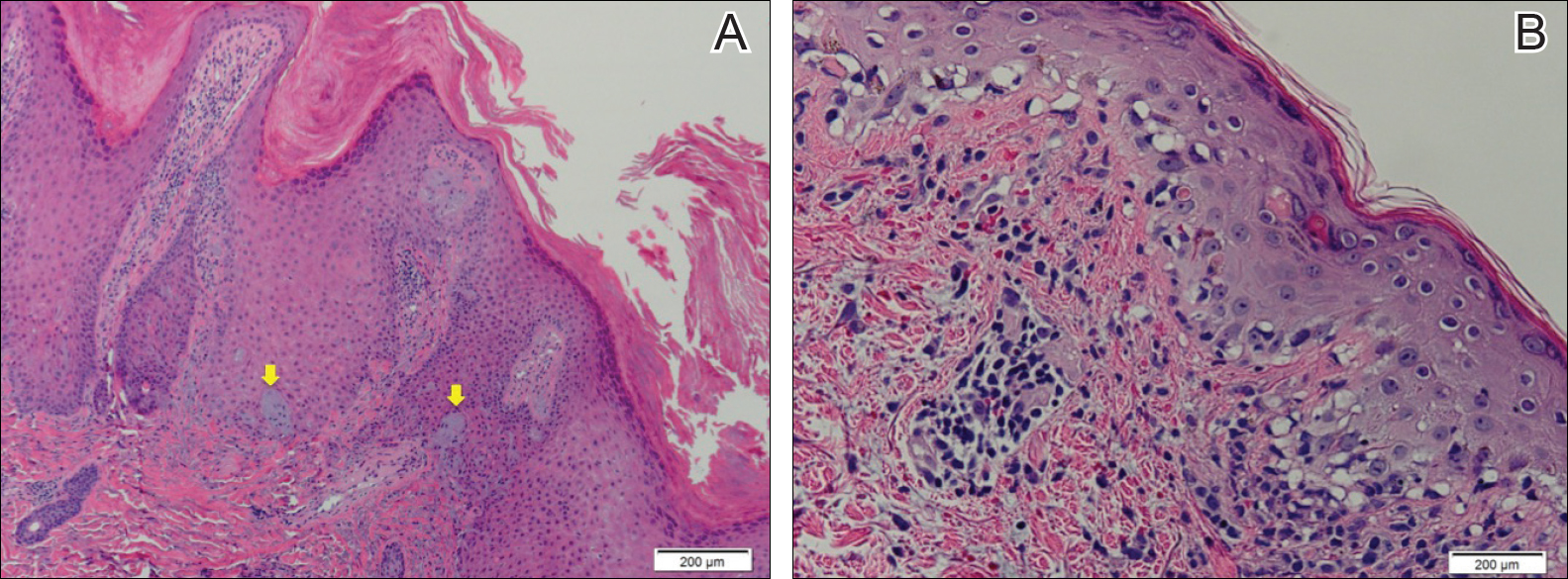
Hypertrophic LE is a rare subset of chronic cutaneous lupus first described by Behcet1 in 1942. Lesions are identified as verrucous keratotic plaques with a characteristic erythematous indurated border.2 Patients predominantly are middle-aged women with lesions distributed on sun-exposed areas. Most often, hypertrophic LE is seen in association with the classic lesions of discoid LE; however, patients may present exclusively with the cutaneous manifestations of hypertrophic LE. More rarely, as seen in this case, hypertrophic LE may present in conjunction with systemic features.3 The diagnosis of systemic LE requires 4 of the following criteria be fulfilled: malar rash; discoid rash; photosensitivity; oral ulcers; arthritis; cardiopulmonary serositis; renal involvement; positive ANA titer; and neurologic, hematologic, or immunologic disorders.4 Our patient qualified for discoid rash, photosensitivity, cardiopulmonary involvement with mitral valve defects and pulmonary pleuritis, hematologic disorder (anemia), and a positive ANA titer. Furthermore, in patients with only cutaneous discoid LE, serology generally reveals negative or low-titer ANA and negative anti-Ro antibodies.5
Hypertrophic LE is characterized histologically by irregular epidermal hyperplasia in association with features of classic cutaneous LE. Distinctive features of cutaneous LE include interface changes, follicular plugging, dermal mucin, and angiocentric lymphocytic inflammation.6 Notably, additional biopsies of the less hyperkeratotic lesions on our patient's chest and abdomen were performed, which revealed classic cutaneous LE features (Figure 2B).
Hypertrophic LE has 2 histological variants: lichen planus-like and keratoacanthoma (KA)-like patterns. Most cases are described as lichen planus-like, with a dense bandlike infiltrate in association with irregular epidermal hyperplasia, vacuolar interface changes, and reactive squamous atypia.5 In contrast, the less common KA-like lesions consist of a keratinous center with vigorous squamous epithelial proliferation.6
Clinically, hypertrophic LE may resemble hypertrophic psoriasis, lichen planus, KA, or squamous cell carcinoma (SCC). Due to the presence of pseudocarcinomatous hyperplasia, the histopathologic differential includes hypertrophic lichen planus, SCC, KA, and deep fungal infections. However, these other diseases lack the classic features of cutaneous LE, which include interface changes, follicular plugging, dermal mucin, and perivascular lymphocytic inflammation. Additionally, transepidermal elastotic elimination (Figure 2A) helps distinguish hypertrophic LE from other diagnoses.7 One of the most important tasks is distinguishing hypertrophic LE from SCC. Hypertrophic LE does not typically display eosinophil infiltrates, which differentiates it from SCC and KA. Additionally, studies report that CD123 positivity can be useful.6 Positive plasmacytoid dendritic cells are abundant at the dermoepidermal junction in hypertrophic LE, while only single or rare clusters of CD123+ cells are seen in SCC.8 Also, SCC has been found to arise in long-standing cutaneous LE lesions including both discoid and hypertrophic LE. Therefore, clinical and sometimes histological follow-up is required.
Hypertrophic LE often is challenging to treat and frequently is resistant to antimalarial drugs. The primary goals of treatment involve reducing inflammatory infiltrate and minimizing hyperkeratinization. Topical corticosteroids and calcineurin inhibitors often are inadequate as monotherapy due to reduced penetrance through the thick lesions; however, intralesional corticosteroids may be beneficial in patients with localized disease.9 Unfortunately, topical or intralesional treatments are impractical in patients with extensive lesions, as seen in our patient, in which case systemic corticosteroids can be beneficial.
Topical retinoids also have been found to be highly effective.10 Specifically, retinoids such as acitretin and isotretinoin, in some cases combined with antimalarial drugs, are effective in reducing the keratinization of these lesions. Successful treatment also has been reported with ustekinumab, thalidomide, mycophenolate mofetil, and pulsed dye laser.11 As in other types of cutaneous LE, hyperkeratotic LE is photosensitive; avoidance of prolonged sun exposure should be advised.8
- Bechet PE. Lupus erythematosus hypertrophicus et profundus. Arch Derm Syphilol. 1942;45:33-39.
- Bernardi M, Bahrami S, Callen JP. Hypertrophic lupus erythematous complicating long-standing systemic lupus erythematous. Lupus. 2011;20:549-550.
- Spann CR, Callen JP, Klein JB, et al. Clinical, serologic and immunogenetic studies in patients with chronic cutaneous (discoid) lupus erythematosus who have verrucous and/or hypertrophic skin lesions. J Rheumatol. 1988;15:256-261.
- Yu C, Gershwin E, Chang C. Diagnostic criteria for systemic lupus erythematosus: a critical review [published online January 21, 2014]. J Autoimmun. 2014;48-49:10-13.
- Provost TT. The relationship between discoid and systemic lupus erythematous. Arch Dermatol. 1994;130:1308-1310.
- Arps DP, Patel RM. Cutaneous hypertrophic lupus erythematous: a challenging histopathologic diagnosis in the absence of clinical information. Arch Pathol Lab Med. 2013;137:1205-1210.
- Daldon PE, De Souza EM, Cintra ML. Hypertrophic lupus erythematous: a clinicopathological study of 14 cases. J Cutan Pathol. 2003;30:443-448.
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophiclupus erythematous: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10:366-381.
- Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32:482-486.
- Winchester D, Duffin KC, Hansen C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic cutaneous lupus. Lupus. 2012;12:1007-1010.
The Diagnosis: Hypertrophic Lupus Erythematosus
Physical examination at initial presentation revealed well-demarcated, 2- to 3-cm plaques with scale distributed most extensively on the elbows and shins with lesser involvement of the chest and abdomen. After treatment with topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, new annular, erythematous, and edematous lesions began to appear on the chest and abdomen (Figure 1). These new lesions appeared less hyperkeratotic than the older ones.
Biopsy of a hyperkeratotic lesion from the patient's arm revealed marked hyperkeratosis, parakeratosis, epidermal hyperplasia, focal vacuolar change, solar elastosis, and transepidermal elastotic elimination (Figure 2A). A second biopsy performed on a newer chest lesion revealed interface changes, degeneration of the basal layer, follicular plugging, and dermal mucin (Figure 2B). Serology revealed an antinuclear antibody (ANA) titer of 1:1280 (reference range, <1:40 dilution) and hemoglobin of 11.5 g/dL (reference range, 14.0-17.5 g/dL). On the basis of clinical, histologic, and serologic findings, hypertrophic lupus erythematosus (LE) was diagnosed. The patient was treated with oral prednisone, which resulted in rapid improvement.
Hypertrophic LE is a rare subset of chronic cutaneous lupus first described by Behcet1 in 1942. Lesions are identified as verrucous keratotic plaques with a characteristic erythematous indurated border.2 Patients predominantly are middle-aged women with lesions distributed on sun-exposed areas. Most often, hypertrophic LE is seen in association with the classic lesions of discoid LE; however, patients may present exclusively with the cutaneous manifestations of hypertrophic LE. More rarely, as seen in this case, hypertrophic LE may present in conjunction with systemic features.3 The diagnosis of systemic LE requires 4 of the following criteria be fulfilled: malar rash; discoid rash; photosensitivity; oral ulcers; arthritis; cardiopulmonary serositis; renal involvement; positive ANA titer; and neurologic, hematologic, or immunologic disorders.4 Our patient qualified for discoid rash, photosensitivity, cardiopulmonary involvement with mitral valve defects and pulmonary pleuritis, hematologic disorder (anemia), and a positive ANA titer. Furthermore, in patients with only cutaneous discoid LE, serology generally reveals negative or low-titer ANA and negative anti-Ro antibodies.5
Hypertrophic LE is characterized histologically by irregular epidermal hyperplasia in association with features of classic cutaneous LE. Distinctive features of cutaneous LE include interface changes, follicular plugging, dermal mucin, and angiocentric lymphocytic inflammation.6 Notably, additional biopsies of the less hyperkeratotic lesions on our patient's chest and abdomen were performed, which revealed classic cutaneous LE features (Figure 2B).
Hypertrophic LE has 2 histological variants: lichen planus-like and keratoacanthoma (KA)-like patterns. Most cases are described as lichen planus-like, with a dense bandlike infiltrate in association with irregular epidermal hyperplasia, vacuolar interface changes, and reactive squamous atypia.5 In contrast, the less common KA-like lesions consist of a keratinous center with vigorous squamous epithelial proliferation.6
Clinically, hypertrophic LE may resemble hypertrophic psoriasis, lichen planus, KA, or squamous cell carcinoma (SCC). Due to the presence of pseudocarcinomatous hyperplasia, the histopathologic differential includes hypertrophic lichen planus, SCC, KA, and deep fungal infections. However, these other diseases lack the classic features of cutaneous LE, which include interface changes, follicular plugging, dermal mucin, and perivascular lymphocytic inflammation. Additionally, transepidermal elastotic elimination (Figure 2A) helps distinguish hypertrophic LE from other diagnoses.7 One of the most important tasks is distinguishing hypertrophic LE from SCC. Hypertrophic LE does not typically display eosinophil infiltrates, which differentiates it from SCC and KA. Additionally, studies report that CD123 positivity can be useful.6 Positive plasmacytoid dendritic cells are abundant at the dermoepidermal junction in hypertrophic LE, while only single or rare clusters of CD123+ cells are seen in SCC.8 Also, SCC has been found to arise in long-standing cutaneous LE lesions including both discoid and hypertrophic LE. Therefore, clinical and sometimes histological follow-up is required.
Hypertrophic LE often is challenging to treat and frequently is resistant to antimalarial drugs. The primary goals of treatment involve reducing inflammatory infiltrate and minimizing hyperkeratinization. Topical corticosteroids and calcineurin inhibitors often are inadequate as monotherapy due to reduced penetrance through the thick lesions; however, intralesional corticosteroids may be beneficial in patients with localized disease.9 Unfortunately, topical or intralesional treatments are impractical in patients with extensive lesions, as seen in our patient, in which case systemic corticosteroids can be beneficial.
Topical retinoids also have been found to be highly effective.10 Specifically, retinoids such as acitretin and isotretinoin, in some cases combined with antimalarial drugs, are effective in reducing the keratinization of these lesions. Successful treatment also has been reported with ustekinumab, thalidomide, mycophenolate mofetil, and pulsed dye laser.11 As in other types of cutaneous LE, hyperkeratotic LE is photosensitive; avoidance of prolonged sun exposure should be advised.8
The Diagnosis: Hypertrophic Lupus Erythematosus
Physical examination at initial presentation revealed well-demarcated, 2- to 3-cm plaques with scale distributed most extensively on the elbows and shins with lesser involvement of the chest and abdomen. After treatment with topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, new annular, erythematous, and edematous lesions began to appear on the chest and abdomen (Figure 1). These new lesions appeared less hyperkeratotic than the older ones.
Biopsy of a hyperkeratotic lesion from the patient's arm revealed marked hyperkeratosis, parakeratosis, epidermal hyperplasia, focal vacuolar change, solar elastosis, and transepidermal elastotic elimination (Figure 2A). A second biopsy performed on a newer chest lesion revealed interface changes, degeneration of the basal layer, follicular plugging, and dermal mucin (Figure 2B). Serology revealed an antinuclear antibody (ANA) titer of 1:1280 (reference range, <1:40 dilution) and hemoglobin of 11.5 g/dL (reference range, 14.0-17.5 g/dL). On the basis of clinical, histologic, and serologic findings, hypertrophic lupus erythematosus (LE) was diagnosed. The patient was treated with oral prednisone, which resulted in rapid improvement.
Hypertrophic LE is a rare subset of chronic cutaneous lupus first described by Behcet1 in 1942. Lesions are identified as verrucous keratotic plaques with a characteristic erythematous indurated border.2 Patients predominantly are middle-aged women with lesions distributed on sun-exposed areas. Most often, hypertrophic LE is seen in association with the classic lesions of discoid LE; however, patients may present exclusively with the cutaneous manifestations of hypertrophic LE. More rarely, as seen in this case, hypertrophic LE may present in conjunction with systemic features.3 The diagnosis of systemic LE requires 4 of the following criteria be fulfilled: malar rash; discoid rash; photosensitivity; oral ulcers; arthritis; cardiopulmonary serositis; renal involvement; positive ANA titer; and neurologic, hematologic, or immunologic disorders.4 Our patient qualified for discoid rash, photosensitivity, cardiopulmonary involvement with mitral valve defects and pulmonary pleuritis, hematologic disorder (anemia), and a positive ANA titer. Furthermore, in patients with only cutaneous discoid LE, serology generally reveals negative or low-titer ANA and negative anti-Ro antibodies.5
Hypertrophic LE is characterized histologically by irregular epidermal hyperplasia in association with features of classic cutaneous LE. Distinctive features of cutaneous LE include interface changes, follicular plugging, dermal mucin, and angiocentric lymphocytic inflammation.6 Notably, additional biopsies of the less hyperkeratotic lesions on our patient's chest and abdomen were performed, which revealed classic cutaneous LE features (Figure 2B).
Hypertrophic LE has 2 histological variants: lichen planus-like and keratoacanthoma (KA)-like patterns. Most cases are described as lichen planus-like, with a dense bandlike infiltrate in association with irregular epidermal hyperplasia, vacuolar interface changes, and reactive squamous atypia.5 In contrast, the less common KA-like lesions consist of a keratinous center with vigorous squamous epithelial proliferation.6
Clinically, hypertrophic LE may resemble hypertrophic psoriasis, lichen planus, KA, or squamous cell carcinoma (SCC). Due to the presence of pseudocarcinomatous hyperplasia, the histopathologic differential includes hypertrophic lichen planus, SCC, KA, and deep fungal infections. However, these other diseases lack the classic features of cutaneous LE, which include interface changes, follicular plugging, dermal mucin, and perivascular lymphocytic inflammation. Additionally, transepidermal elastotic elimination (Figure 2A) helps distinguish hypertrophic LE from other diagnoses.7 One of the most important tasks is distinguishing hypertrophic LE from SCC. Hypertrophic LE does not typically display eosinophil infiltrates, which differentiates it from SCC and KA. Additionally, studies report that CD123 positivity can be useful.6 Positive plasmacytoid dendritic cells are abundant at the dermoepidermal junction in hypertrophic LE, while only single or rare clusters of CD123+ cells are seen in SCC.8 Also, SCC has been found to arise in long-standing cutaneous LE lesions including both discoid and hypertrophic LE. Therefore, clinical and sometimes histological follow-up is required.
Hypertrophic LE often is challenging to treat and frequently is resistant to antimalarial drugs. The primary goals of treatment involve reducing inflammatory infiltrate and minimizing hyperkeratinization. Topical corticosteroids and calcineurin inhibitors often are inadequate as monotherapy due to reduced penetrance through the thick lesions; however, intralesional corticosteroids may be beneficial in patients with localized disease.9 Unfortunately, topical or intralesional treatments are impractical in patients with extensive lesions, as seen in our patient, in which case systemic corticosteroids can be beneficial.
Topical retinoids also have been found to be highly effective.10 Specifically, retinoids such as acitretin and isotretinoin, in some cases combined with antimalarial drugs, are effective in reducing the keratinization of these lesions. Successful treatment also has been reported with ustekinumab, thalidomide, mycophenolate mofetil, and pulsed dye laser.11 As in other types of cutaneous LE, hyperkeratotic LE is photosensitive; avoidance of prolonged sun exposure should be advised.8
- Bechet PE. Lupus erythematosus hypertrophicus et profundus. Arch Derm Syphilol. 1942;45:33-39.
- Bernardi M, Bahrami S, Callen JP. Hypertrophic lupus erythematous complicating long-standing systemic lupus erythematous. Lupus. 2011;20:549-550.
- Spann CR, Callen JP, Klein JB, et al. Clinical, serologic and immunogenetic studies in patients with chronic cutaneous (discoid) lupus erythematosus who have verrucous and/or hypertrophic skin lesions. J Rheumatol. 1988;15:256-261.
- Yu C, Gershwin E, Chang C. Diagnostic criteria for systemic lupus erythematosus: a critical review [published online January 21, 2014]. J Autoimmun. 2014;48-49:10-13.
- Provost TT. The relationship between discoid and systemic lupus erythematous. Arch Dermatol. 1994;130:1308-1310.
- Arps DP, Patel RM. Cutaneous hypertrophic lupus erythematous: a challenging histopathologic diagnosis in the absence of clinical information. Arch Pathol Lab Med. 2013;137:1205-1210.
- Daldon PE, De Souza EM, Cintra ML. Hypertrophic lupus erythematous: a clinicopathological study of 14 cases. J Cutan Pathol. 2003;30:443-448.
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophiclupus erythematous: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10:366-381.
- Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32:482-486.
- Winchester D, Duffin KC, Hansen C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic cutaneous lupus. Lupus. 2012;12:1007-1010.
- Bechet PE. Lupus erythematosus hypertrophicus et profundus. Arch Derm Syphilol. 1942;45:33-39.
- Bernardi M, Bahrami S, Callen JP. Hypertrophic lupus erythematous complicating long-standing systemic lupus erythematous. Lupus. 2011;20:549-550.
- Spann CR, Callen JP, Klein JB, et al. Clinical, serologic and immunogenetic studies in patients with chronic cutaneous (discoid) lupus erythematosus who have verrucous and/or hypertrophic skin lesions. J Rheumatol. 1988;15:256-261.
- Yu C, Gershwin E, Chang C. Diagnostic criteria for systemic lupus erythematosus: a critical review [published online January 21, 2014]. J Autoimmun. 2014;48-49:10-13.
- Provost TT. The relationship between discoid and systemic lupus erythematous. Arch Dermatol. 1994;130:1308-1310.
- Arps DP, Patel RM. Cutaneous hypertrophic lupus erythematous: a challenging histopathologic diagnosis in the absence of clinical information. Arch Pathol Lab Med. 2013;137:1205-1210.
- Daldon PE, De Souza EM, Cintra ML. Hypertrophic lupus erythematous: a clinicopathological study of 14 cases. J Cutan Pathol. 2003;30:443-448.
- Ko CJ, Srivastava B, Braverman I, et al. Hypertrophiclupus erythematous: the diagnostic utility of CD123 staining. J Cutan Pathol. 2011;38:889-892.
- Walling HW, Sontheimer RD. Cutaneous lupus erythematosus. issues in diagnosis and treatment. Am J Clin Dermatol. 2009;10:366-381.
- Al-Mutairi N, Rijhwani M, Nour-Eldin O. Hypertrophic lupus erythematosus treated successfully with acitretin as monotherapy. J Dermatol. 2005;32:482-486.
- Winchester D, Duffin KC, Hansen C. Response to ustekinumab in a patient with both severe psoriasis and hypertrophic cutaneous lupus. Lupus. 2012;12:1007-1010.
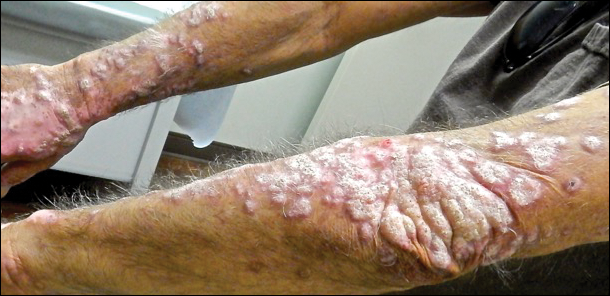
A 53-year-old man presented with a persistent, hyperkeratotic, pruritic rash on the arms, chest, and abdomen. The patient was treated for presumed psoriasis for 9 months by a primary care physician. However, despite an extensive treatment history, which included topical steroids, adalimumab, methotrexate, and narrowband UVB phototherapy, his condition worsened, and new erythematous and edematous lesions with no scale appeared on the back and chest. The patient's history also was notable for splenic rupture and mitral valve defects for which he was maintained on warfarin. In addition, he was evaluated by an allergist for new-onset dyspnea and treated with prednisone, which subsequently resulted in partial resolution of the skin lesions.
Friable Warty Plaque on the Heel
The Diagnosis: Verrucous Hemangioma
Verrucous hemangioma (VH) is a rare vascular anomaly that has not been definitively delineated as a malformation or a tumor, as it has features of both. Verrucous hemangioma presents at birth as a compressible soft mass with a red violaceous hue favoring the legs.1,2 Over time VH will develop a warty, friable, and keratotic surface that can begin to evolve as early as 6 months or as late as 34 years of age.3 Verrucous hemangioma does not involute and tends to grow proportionally with the patient. Thus, VH classically has been considered a vascular malformation.
On histopathology VH shows collections of uniform, thin-walled vessels with a multilamellated basement membrane throughout the dermis, similar to an infantile hemangioma (IH). These lesions extend deep into the subcutaneous tissue and often involve the underlying fascia. The papillary dermis has large ectatic vessels, while the epidermis displays verrucous hyperkeratosis, papillomatosis, and irregular acanthosis without viral change (Figure).4,5 The superficial component can resemble an angiokeratoma; however, VH is differentiated by a deeper component that is often larger in size and has a more protracted clinical course.
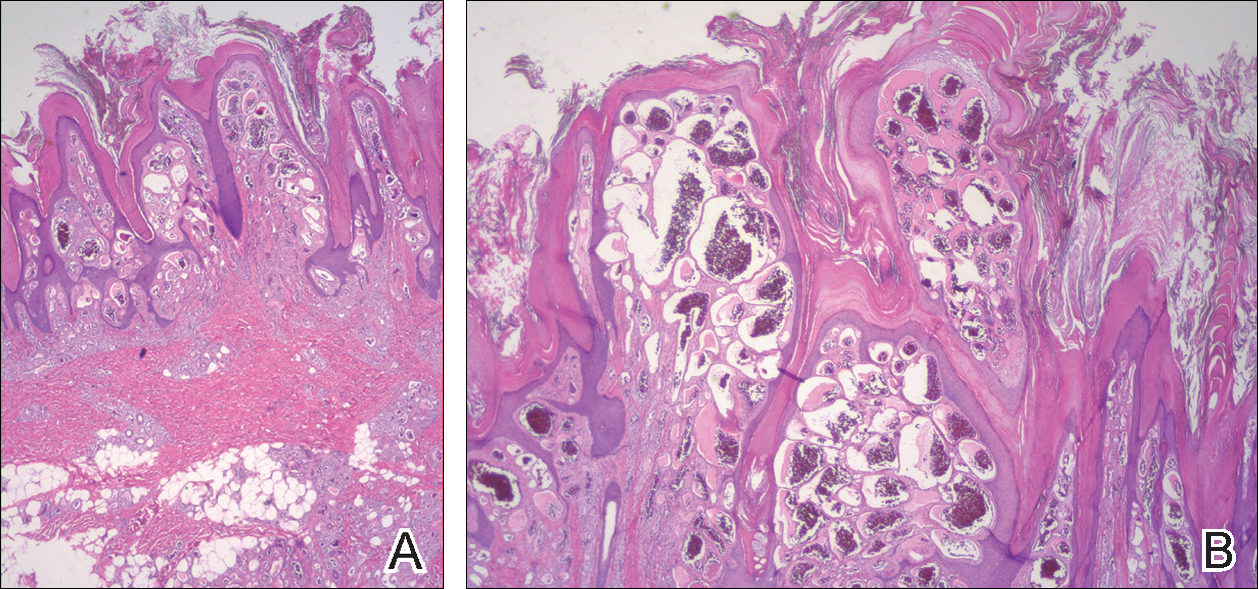
Similar to IH, immunohistochemical studies have shown that VH expresses Wilms tumor 1 and glucose transporter 1 but is negative for D2-40.4 These findings suggest that VH is a vascular tumor rather than a vascular malformation, as was previously reported.6 Additional research has shown that the immunohistochemical staining profile of VH is nearly identical to IH, which has led to postulation that VH may be of placental mesodermal origin, as has been hypothesized for IH.5
Due to its deep infiltration and tendency for recurrence, VH is most effectively treated with wide local excision.3,6-8 Preoperative planning with magnetic resonance imaging may be indicated. Although laser monotherapy and other local destructive therapies have been largely unsuccessful, postsurgical laser therapy with CO2 lasers as well as dual pulsed dye laser and Nd:YAG laser have shown promise in preventing recurrence.3
- Tennant LB, Mulliken JB, Perez-Atayde AR, et al. Verrucous hemangioma revisited. Pediatr Dermatol. 2006;23:208-215.
- Koc M, Kavala M, Kocatür E, et al. An unusual vascular tumor: verrucous hemangioma. Dermatol Online J. 2009;15:7.
- Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919; discussion 920.
- Trindade F, Torrelo A, Requena L, et al. An immunohistochemical study of verrucous hemangiomas. J Cutan Pathol. 2013;40:472-476.
- Laing EL, Brasch HD, Steel R, et al. Verrucous hemangioma expresses primitive markers. J Cutan Pathol. 2013;40:391-396.
- Mankani MH, Dufresne CR. Verrucous malformations: their presentation and management. Ann Plast Surg. 2000;45:31-36.
- Clairwood MQ, Bruckner AL, Dadras SS. Verrucous hemangioma: a report of two cases and review of the literature. J Cutan Pathol. 2011;38:740-746.
- Segura Palacios JM, Boixeda P, Rocha J, et al. Laser treatment for verrucous hemangioma. Laser Med Sci. 2012;27:681-684.
The Diagnosis: Verrucous Hemangioma
Verrucous hemangioma (VH) is a rare vascular anomaly that has not been definitively delineated as a malformation or a tumor, as it has features of both. Verrucous hemangioma presents at birth as a compressible soft mass with a red violaceous hue favoring the legs.1,2 Over time VH will develop a warty, friable, and keratotic surface that can begin to evolve as early as 6 months or as late as 34 years of age.3 Verrucous hemangioma does not involute and tends to grow proportionally with the patient. Thus, VH classically has been considered a vascular malformation.
On histopathology VH shows collections of uniform, thin-walled vessels with a multilamellated basement membrane throughout the dermis, similar to an infantile hemangioma (IH). These lesions extend deep into the subcutaneous tissue and often involve the underlying fascia. The papillary dermis has large ectatic vessels, while the epidermis displays verrucous hyperkeratosis, papillomatosis, and irregular acanthosis without viral change (Figure).4,5 The superficial component can resemble an angiokeratoma; however, VH is differentiated by a deeper component that is often larger in size and has a more protracted clinical course.
Similar to IH, immunohistochemical studies have shown that VH expresses Wilms tumor 1 and glucose transporter 1 but is negative for D2-40.4 These findings suggest that VH is a vascular tumor rather than a vascular malformation, as was previously reported.6 Additional research has shown that the immunohistochemical staining profile of VH is nearly identical to IH, which has led to postulation that VH may be of placental mesodermal origin, as has been hypothesized for IH.5
Due to its deep infiltration and tendency for recurrence, VH is most effectively treated with wide local excision.3,6-8 Preoperative planning with magnetic resonance imaging may be indicated. Although laser monotherapy and other local destructive therapies have been largely unsuccessful, postsurgical laser therapy with CO2 lasers as well as dual pulsed dye laser and Nd:YAG laser have shown promise in preventing recurrence.3
The Diagnosis: Verrucous Hemangioma
Verrucous hemangioma (VH) is a rare vascular anomaly that has not been definitively delineated as a malformation or a tumor, as it has features of both. Verrucous hemangioma presents at birth as a compressible soft mass with a red violaceous hue favoring the legs.1,2 Over time VH will develop a warty, friable, and keratotic surface that can begin to evolve as early as 6 months or as late as 34 years of age.3 Verrucous hemangioma does not involute and tends to grow proportionally with the patient. Thus, VH classically has been considered a vascular malformation.
On histopathology VH shows collections of uniform, thin-walled vessels with a multilamellated basement membrane throughout the dermis, similar to an infantile hemangioma (IH). These lesions extend deep into the subcutaneous tissue and often involve the underlying fascia. The papillary dermis has large ectatic vessels, while the epidermis displays verrucous hyperkeratosis, papillomatosis, and irregular acanthosis without viral change (Figure).4,5 The superficial component can resemble an angiokeratoma; however, VH is differentiated by a deeper component that is often larger in size and has a more protracted clinical course.
Similar to IH, immunohistochemical studies have shown that VH expresses Wilms tumor 1 and glucose transporter 1 but is negative for D2-40.4 These findings suggest that VH is a vascular tumor rather than a vascular malformation, as was previously reported.6 Additional research has shown that the immunohistochemical staining profile of VH is nearly identical to IH, which has led to postulation that VH may be of placental mesodermal origin, as has been hypothesized for IH.5
Due to its deep infiltration and tendency for recurrence, VH is most effectively treated with wide local excision.3,6-8 Preoperative planning with magnetic resonance imaging may be indicated. Although laser monotherapy and other local destructive therapies have been largely unsuccessful, postsurgical laser therapy with CO2 lasers as well as dual pulsed dye laser and Nd:YAG laser have shown promise in preventing recurrence.3
- Tennant LB, Mulliken JB, Perez-Atayde AR, et al. Verrucous hemangioma revisited. Pediatr Dermatol. 2006;23:208-215.
- Koc M, Kavala M, Kocatür E, et al. An unusual vascular tumor: verrucous hemangioma. Dermatol Online J. 2009;15:7.
- Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919; discussion 920.
- Trindade F, Torrelo A, Requena L, et al. An immunohistochemical study of verrucous hemangiomas. J Cutan Pathol. 2013;40:472-476.
- Laing EL, Brasch HD, Steel R, et al. Verrucous hemangioma expresses primitive markers. J Cutan Pathol. 2013;40:391-396.
- Mankani MH, Dufresne CR. Verrucous malformations: their presentation and management. Ann Plast Surg. 2000;45:31-36.
- Clairwood MQ, Bruckner AL, Dadras SS. Verrucous hemangioma: a report of two cases and review of the literature. J Cutan Pathol. 2011;38:740-746.
- Segura Palacios JM, Boixeda P, Rocha J, et al. Laser treatment for verrucous hemangioma. Laser Med Sci. 2012;27:681-684.
- Tennant LB, Mulliken JB, Perez-Atayde AR, et al. Verrucous hemangioma revisited. Pediatr Dermatol. 2006;23:208-215.
- Koc M, Kavala M, Kocatür E, et al. An unusual vascular tumor: verrucous hemangioma. Dermatol Online J. 2009;15:7.
- Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919; discussion 920.
- Trindade F, Torrelo A, Requena L, et al. An immunohistochemical study of verrucous hemangiomas. J Cutan Pathol. 2013;40:472-476.
- Laing EL, Brasch HD, Steel R, et al. Verrucous hemangioma expresses primitive markers. J Cutan Pathol. 2013;40:391-396.
- Mankani MH, Dufresne CR. Verrucous malformations: their presentation and management. Ann Plast Surg. 2000;45:31-36.
- Clairwood MQ, Bruckner AL, Dadras SS. Verrucous hemangioma: a report of two cases and review of the literature. J Cutan Pathol. 2011;38:740-746.
- Segura Palacios JM, Boixeda P, Rocha J, et al. Laser treatment for verrucous hemangioma. Laser Med Sci. 2012;27:681-684.
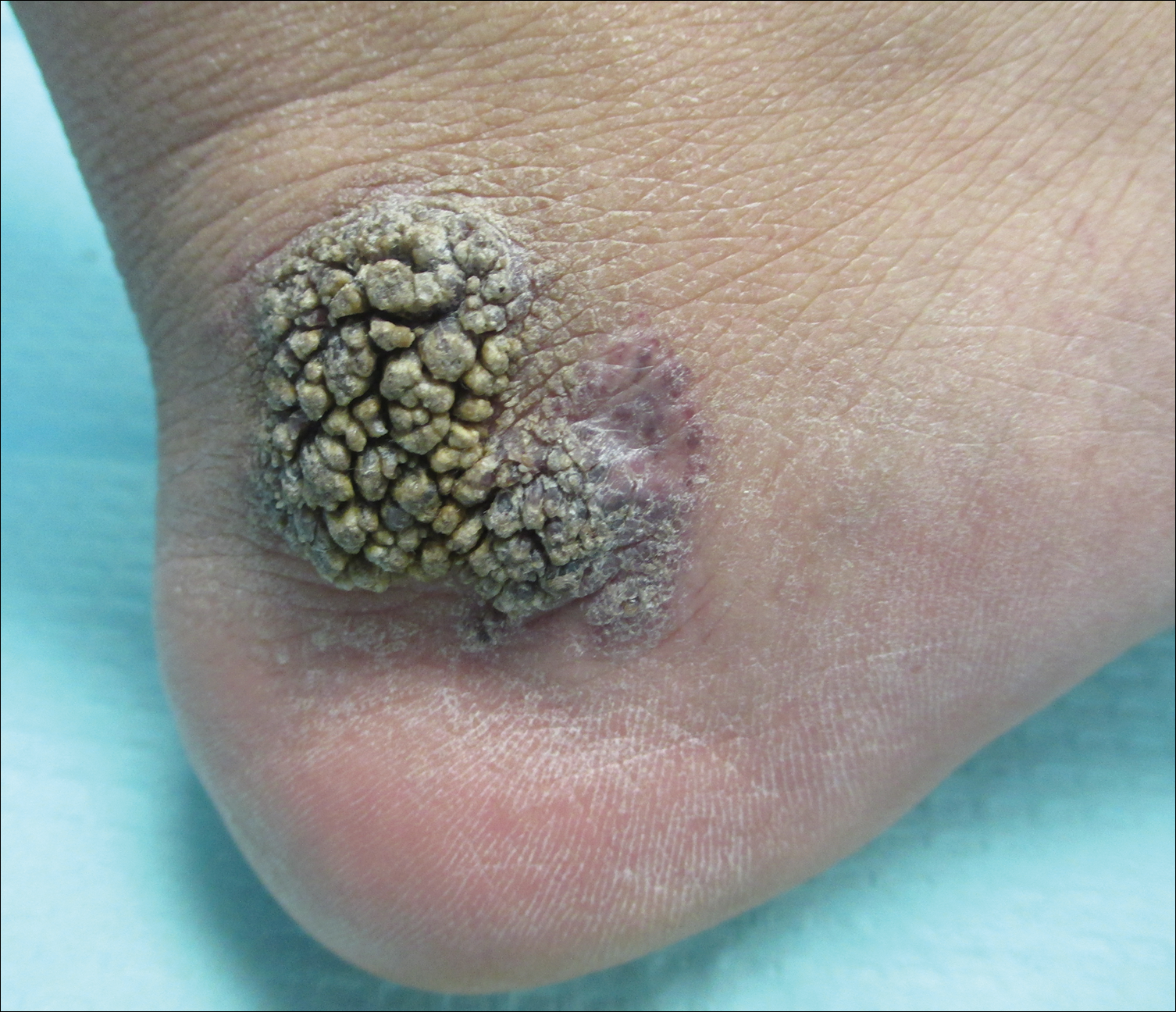
A 31-year-old man presented with a large friable and warty plaque on the left heel. He recalled that the lesion had been present since birth as a flat red birthmark that grew proportionally with him. Throughout his adolescence its surface became increasingly rough and bumpy. The patient described receiving laser treatment twice in his early 20s without notable improvement. He wanted the lesion removed because it was easily traumatized, resulting in bleeding, pain, and infection. The patient reported being otherwise healthy.
Expanding Pruritic Plaque on the Forearm
The Diagnosis: Cutaneous Protothecosis
A 4-mm punch biopsy of the plaque on the right forearm was performed. The biopsy showed chronic inflammation with prominent histiocytes, foreign body giant cells, plasma cells, and abundant eosinophils (Figure 1). Grocott-Gomori methenamine-silver stain demonstrated abundant soccer ball-like or floretlike sporangia that were 3 to 11 μm, consistent with a diagnosis of protothecosis (Figure 2).
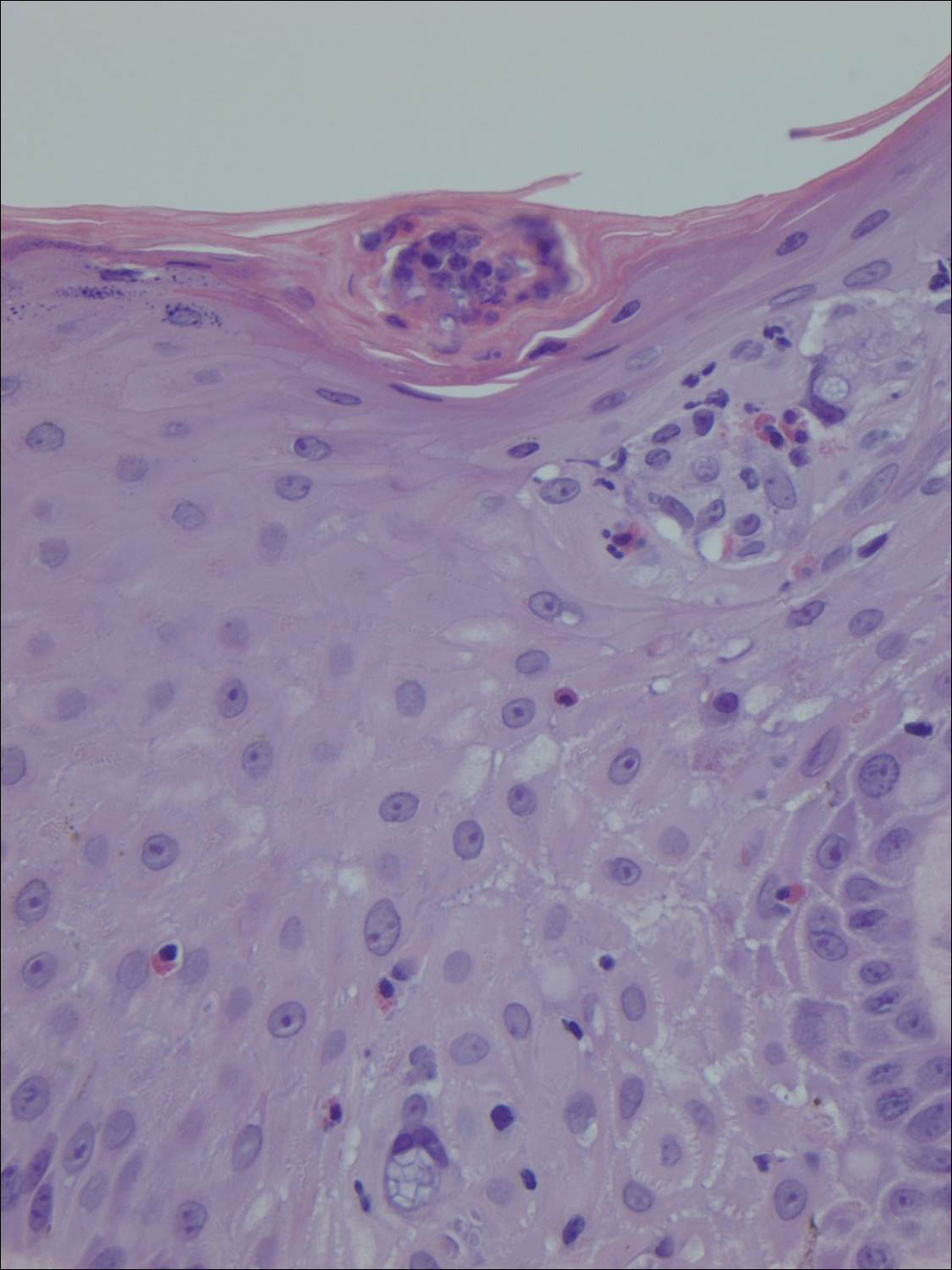

Cutaneous protothecosis is an infection caused by chlorophyll-lacking algae of the genus Prototheca.1 It is ubiquitous in nature and can be isolated from various reservoirs such as trees, grass, water, and food sources.2 Protothecosis is present worldwide and in the United States; it is most prevalent in the Southeast. Prototheca species are rare but often endemic in cattle and can cause bovine mastitis and enteritis.3 However, they are rare opportunistic infections in humans.
The pathogenesis of cutaneous protothecosis is largely unknown.4 However, most infections are thought to be caused by traumatic inoculation into subcutaneous tissues.1,2 The majority of cases occur in patients older than 30 years. To date, approximately 160 cases have been reported in the literature worldwide.5 There are 3 main species of Prototheca, but almost all human infections are caused by Prototheca wickerhamii.2 Clinically, most patients with protothecosis present with cutaneous findings, but olecranon bursitis and systemic forms also have been reported.1
Risk factors for protothecosis include immunosuppression, most often due to steroids, in addition to malignancies, diabetes mellitus, and certain occupations.1 The presentation can be variable from papules and plaques to even herpetiform appearances.4 Protothecosis usually affects the skin and soft tissues of exposed areas such as the extremities or the face.6 Diagnosis largely is made on detection of characteristic floretlike sporangia with a prominent cell wall on histopathological examination. Prototheca wickerhamii specifically produces a morula form of sporangia with endospores arranged symmetrically, giving it a characteristic soccer ball appearance.2
Treatment of protothecosis is difficult and remains controversial.1 There are no established protothecosis treatment protocols or guidelines due to the small number of cases.7 In vitro studies have demonstrated sensitivity to amphotericin B and various azoles as well as a wide range of antibiotics.1 Olecranon bursitis and small skin lesions can be treated by surgical excision. All other Prototheca infections require systemic treatment with azoles or intravenous amphotericin B for immunocompromised patients or those with disseminated disease.5 However, failure to respond to medical management often occurs, requiring surgical excision.1,6
Our patient was treated with a 3-month course of voriconazole but therapy failed and the plaque continued to expand. The patient underwent a wide excision that was repaired with a partial-thickness skin graft. Rebiopsy of the papule adjacent to the skin graft showed no further recurrence.
In conclusion, protothecosis generally is not clinically suspected and patients are subjected to various treatments without adequate results. A definitive diagnosis easily can be established with a skin biopsy, which can direct timely and appropriate treatment.
- Lass-Flörl C, Mary A. Human protothecosis. Clin Microbiol Rev. 2007;20:230-242.
- Mayorga J, Barba-Gómez JF, Verduzco-Martínez AP, et al. Protothecosis. Clin Dermatol. 2012;30:432-436.
- Jensen HE, Aalbaek B, Bloch B, et al. Bovine mammary protothecosis due to Prototheca zopfii. Med Mycol. 1998;36:89-95.
- Boyd AS, Langley M, King LE Jr. Cutaneous manifestations of Prototheca infections. J Am Acad Dermatol. 1995;32:758-764.
- Todd JR, King JW, Oberle A, et al. Protothecosis: report of a case with 20-year follow-up, and review of previously published cases. Med Mycol. 2012;50:673-689.
- Hightower KD, Messina JL. Cutaneous protothecosis: a case report and review of the literature. Cutis. 2007;80:129-131.
- Yamada N, Yoshida Y, Ohsawa T, et al. A case of cutaneous protothecosis successfully treated with local thermal therapy as an adjunct to itraconazole therapy in an immunocompromised host. Med Mycol. 2010;48:643-646.
The Diagnosis: Cutaneous Protothecosis
A 4-mm punch biopsy of the plaque on the right forearm was performed. The biopsy showed chronic inflammation with prominent histiocytes, foreign body giant cells, plasma cells, and abundant eosinophils (Figure 1). Grocott-Gomori methenamine-silver stain demonstrated abundant soccer ball-like or floretlike sporangia that were 3 to 11 μm, consistent with a diagnosis of protothecosis (Figure 2).
Cutaneous protothecosis is an infection caused by chlorophyll-lacking algae of the genus Prototheca.1 It is ubiquitous in nature and can be isolated from various reservoirs such as trees, grass, water, and food sources.2 Protothecosis is present worldwide and in the United States; it is most prevalent in the Southeast. Prototheca species are rare but often endemic in cattle and can cause bovine mastitis and enteritis.3 However, they are rare opportunistic infections in humans.
The pathogenesis of cutaneous protothecosis is largely unknown.4 However, most infections are thought to be caused by traumatic inoculation into subcutaneous tissues.1,2 The majority of cases occur in patients older than 30 years. To date, approximately 160 cases have been reported in the literature worldwide.5 There are 3 main species of Prototheca, but almost all human infections are caused by Prototheca wickerhamii.2 Clinically, most patients with protothecosis present with cutaneous findings, but olecranon bursitis and systemic forms also have been reported.1
Risk factors for protothecosis include immunosuppression, most often due to steroids, in addition to malignancies, diabetes mellitus, and certain occupations.1 The presentation can be variable from papules and plaques to even herpetiform appearances.4 Protothecosis usually affects the skin and soft tissues of exposed areas such as the extremities or the face.6 Diagnosis largely is made on detection of characteristic floretlike sporangia with a prominent cell wall on histopathological examination. Prototheca wickerhamii specifically produces a morula form of sporangia with endospores arranged symmetrically, giving it a characteristic soccer ball appearance.2
Treatment of protothecosis is difficult and remains controversial.1 There are no established protothecosis treatment protocols or guidelines due to the small number of cases.7 In vitro studies have demonstrated sensitivity to amphotericin B and various azoles as well as a wide range of antibiotics.1 Olecranon bursitis and small skin lesions can be treated by surgical excision. All other Prototheca infections require systemic treatment with azoles or intravenous amphotericin B for immunocompromised patients or those with disseminated disease.5 However, failure to respond to medical management often occurs, requiring surgical excision.1,6
Our patient was treated with a 3-month course of voriconazole but therapy failed and the plaque continued to expand. The patient underwent a wide excision that was repaired with a partial-thickness skin graft. Rebiopsy of the papule adjacent to the skin graft showed no further recurrence.
In conclusion, protothecosis generally is not clinically suspected and patients are subjected to various treatments without adequate results. A definitive diagnosis easily can be established with a skin biopsy, which can direct timely and appropriate treatment.
The Diagnosis: Cutaneous Protothecosis
A 4-mm punch biopsy of the plaque on the right forearm was performed. The biopsy showed chronic inflammation with prominent histiocytes, foreign body giant cells, plasma cells, and abundant eosinophils (Figure 1). Grocott-Gomori methenamine-silver stain demonstrated abundant soccer ball-like or floretlike sporangia that were 3 to 11 μm, consistent with a diagnosis of protothecosis (Figure 2).
Cutaneous protothecosis is an infection caused by chlorophyll-lacking algae of the genus Prototheca.1 It is ubiquitous in nature and can be isolated from various reservoirs such as trees, grass, water, and food sources.2 Protothecosis is present worldwide and in the United States; it is most prevalent in the Southeast. Prototheca species are rare but often endemic in cattle and can cause bovine mastitis and enteritis.3 However, they are rare opportunistic infections in humans.
The pathogenesis of cutaneous protothecosis is largely unknown.4 However, most infections are thought to be caused by traumatic inoculation into subcutaneous tissues.1,2 The majority of cases occur in patients older than 30 years. To date, approximately 160 cases have been reported in the literature worldwide.5 There are 3 main species of Prototheca, but almost all human infections are caused by Prototheca wickerhamii.2 Clinically, most patients with protothecosis present with cutaneous findings, but olecranon bursitis and systemic forms also have been reported.1
Risk factors for protothecosis include immunosuppression, most often due to steroids, in addition to malignancies, diabetes mellitus, and certain occupations.1 The presentation can be variable from papules and plaques to even herpetiform appearances.4 Protothecosis usually affects the skin and soft tissues of exposed areas such as the extremities or the face.6 Diagnosis largely is made on detection of characteristic floretlike sporangia with a prominent cell wall on histopathological examination. Prototheca wickerhamii specifically produces a morula form of sporangia with endospores arranged symmetrically, giving it a characteristic soccer ball appearance.2
Treatment of protothecosis is difficult and remains controversial.1 There are no established protothecosis treatment protocols or guidelines due to the small number of cases.7 In vitro studies have demonstrated sensitivity to amphotericin B and various azoles as well as a wide range of antibiotics.1 Olecranon bursitis and small skin lesions can be treated by surgical excision. All other Prototheca infections require systemic treatment with azoles or intravenous amphotericin B for immunocompromised patients or those with disseminated disease.5 However, failure to respond to medical management often occurs, requiring surgical excision.1,6
Our patient was treated with a 3-month course of voriconazole but therapy failed and the plaque continued to expand. The patient underwent a wide excision that was repaired with a partial-thickness skin graft. Rebiopsy of the papule adjacent to the skin graft showed no further recurrence.
In conclusion, protothecosis generally is not clinically suspected and patients are subjected to various treatments without adequate results. A definitive diagnosis easily can be established with a skin biopsy, which can direct timely and appropriate treatment.
- Lass-Flörl C, Mary A. Human protothecosis. Clin Microbiol Rev. 2007;20:230-242.
- Mayorga J, Barba-Gómez JF, Verduzco-Martínez AP, et al. Protothecosis. Clin Dermatol. 2012;30:432-436.
- Jensen HE, Aalbaek B, Bloch B, et al. Bovine mammary protothecosis due to Prototheca zopfii. Med Mycol. 1998;36:89-95.
- Boyd AS, Langley M, King LE Jr. Cutaneous manifestations of Prototheca infections. J Am Acad Dermatol. 1995;32:758-764.
- Todd JR, King JW, Oberle A, et al. Protothecosis: report of a case with 20-year follow-up, and review of previously published cases. Med Mycol. 2012;50:673-689.
- Hightower KD, Messina JL. Cutaneous protothecosis: a case report and review of the literature. Cutis. 2007;80:129-131.
- Yamada N, Yoshida Y, Ohsawa T, et al. A case of cutaneous protothecosis successfully treated with local thermal therapy as an adjunct to itraconazole therapy in an immunocompromised host. Med Mycol. 2010;48:643-646.
- Lass-Flörl C, Mary A. Human protothecosis. Clin Microbiol Rev. 2007;20:230-242.
- Mayorga J, Barba-Gómez JF, Verduzco-Martínez AP, et al. Protothecosis. Clin Dermatol. 2012;30:432-436.
- Jensen HE, Aalbaek B, Bloch B, et al. Bovine mammary protothecosis due to Prototheca zopfii. Med Mycol. 1998;36:89-95.
- Boyd AS, Langley M, King LE Jr. Cutaneous manifestations of Prototheca infections. J Am Acad Dermatol. 1995;32:758-764.
- Todd JR, King JW, Oberle A, et al. Protothecosis: report of a case with 20-year follow-up, and review of previously published cases. Med Mycol. 2012;50:673-689.
- Hightower KD, Messina JL. Cutaneous protothecosis: a case report and review of the literature. Cutis. 2007;80:129-131.
- Yamada N, Yoshida Y, Ohsawa T, et al. A case of cutaneous protothecosis successfully treated with local thermal therapy as an adjunct to itraconazole therapy in an immunocompromised host. Med Mycol. 2010;48:643-646.
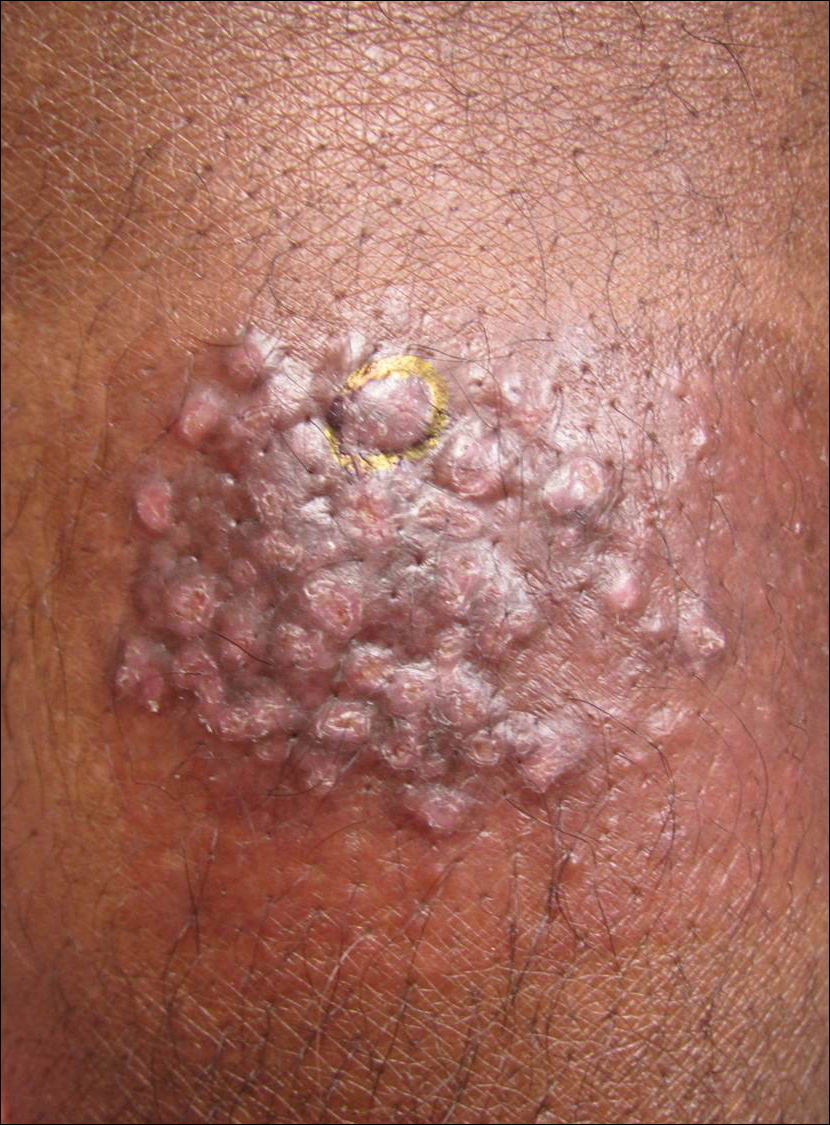
A 66-year-old male firefighter initially presented to the emergency department with an expanding pruritic plaque on the dorsal aspect of the right forearm. The patient recalled the appearance of a single 3-mm papule shortly after doing yardwork in Biloxi, Mississippi. He remembered getting wet grass on the arms, which he later washed off without any notable trauma. The single papule grew into a larger plaque over the next month. In the emergency department he was treated with sulfamethoxazole-trimethoprim, mupirocin, and clotrimazole without response. He was referred to the dermatology department 6 months later and was noted to have multiple 3- to 4-mm papules that coalesced into a 4-cm lichenified plaque with surrounding erythema on the right forearm. His medical history was notable for type 2 diabetes mellitus, hypertension, and hyperlipidemia. The remainder of the physical examination and review of systems was negative.
Progressive Papular Eruption on the Face and Groin
The Diagnosis: Xanthoma Disseminatum
Genital examination revealed approximately 1.5×3-cm soft, yellow-pink plaques extending from the bilateral inguinal folds to the proximal medial thighs (Figure 1). There was no mucosal, axillary, extensor extremity, or palmoplantar involvement. Histopathologic examination of a biopsy from a plaque on the left side of the lower abdomen revealed sheets of foamy histiocytes distributed throughout a fibrotic dermis. Both mononucleated and multinucleated histiocytes were present, including many Touton giant cells (Figure 2). A patchy infiltrate of lymphocytes and rare eosinophils also was noted. The histiocytes labeled with factor XIIIa but not with S-100. Laboratory tests were performed with the following pertinent findings: low-density lipoprotein, 150 mg/dL (reference range, <130 mg/dL); high-density lipoprotein, 30 mg/dL (reference range, >40 mg/dL). Total cholesterol and triglyceride levels were within reference range, and complete blood cell count and basic metabolic panel were normal.
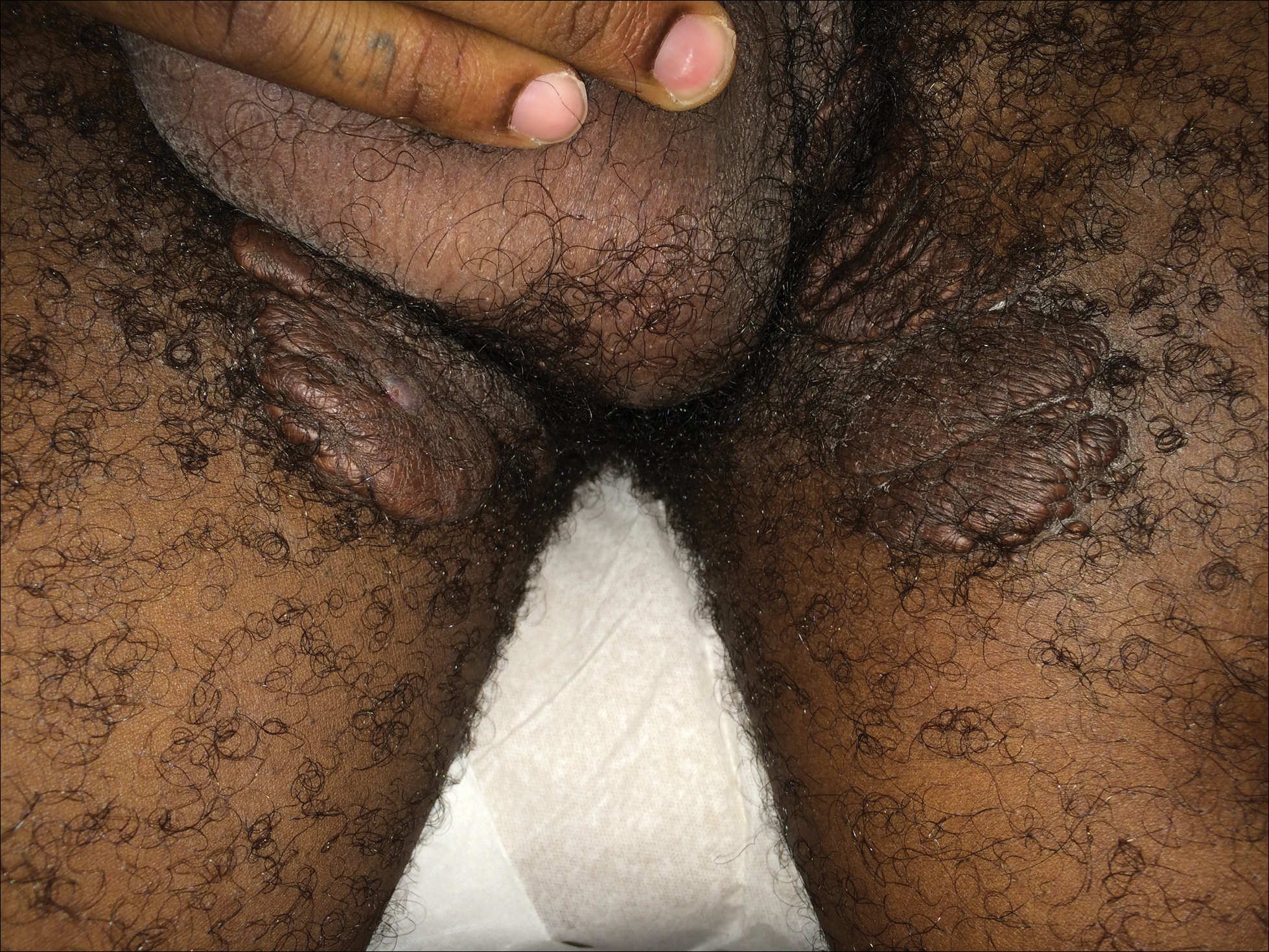
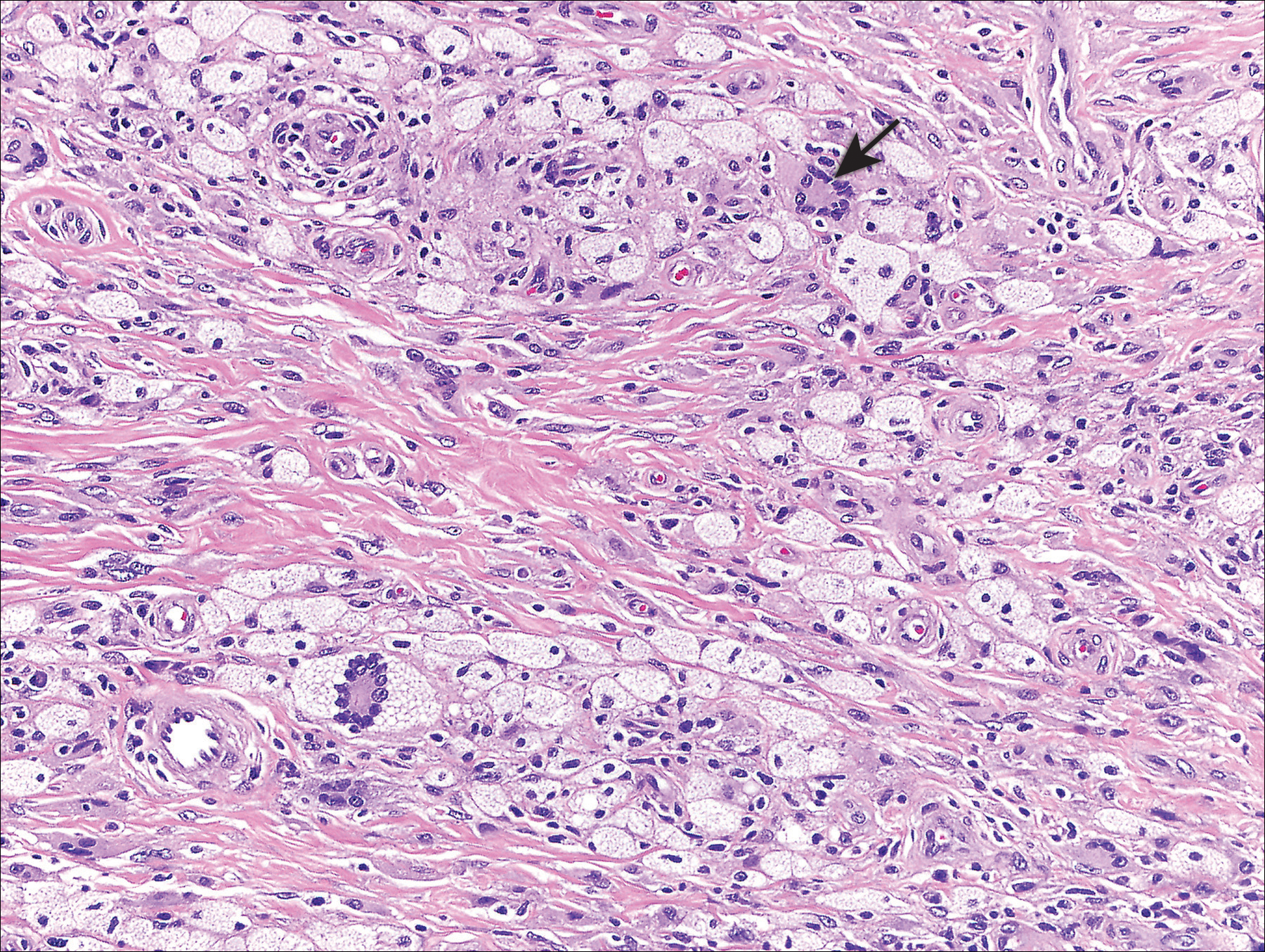
Xanthoma disseminatum (XD)(also known as Montgomery syndrome) is a rare, nonfamilial, normolipemic non-Langerhans cell histiocytosis characterized by extensive lipid deposition in the skin, mucous membranes, and internal organs. The pathogenesis of XD is poorly understood, but it may represent a macrophage-mediated reactive process triggered by superantigens.1
Xanthoma disseminatum most commonly affects males aged 5 to 25 years.2 Clinically, it is characterized by red-brown to yellowish papules and plaques symmetrically distributed over the eyelids, trunk, face, and proximal extremities. There is a predilection for involvement of flexural and intertriginous surfaces and tendency for extension along Langer lines. Extracutaneous involvement can be a notable cause of morbidity and mortality, underscoring the importance of distinguishing XD from other clinically similar xanthomatoses. Mucous membrane involvement occurs in 40% to 60% of patients.3 The oropharynx, larynx, and corneal and conjunctival membranes are most commonly affected, resulting in dysphagia, dysphonia or dyspnea, and visual impairment, respectively. Symptoms of internal organ involvement can be manifold, including pain or limited mobility secondary to osteolytic bone lesions or muscle or synovial membrane involvement, as well as seizures, strabismus, and cerebellar ataxia due to central nervous system lesions.2-4 Approximately 40% of patients develop diabetes insipidus secondary to involvement of the pituitary meninges.3
The differential diagnosis of XD includes juvenile xanthogranuloma, papular xanthomas, eruptive xanthomas, generalized eruptive histiocytosis, progressive nodular histiocytosis, multicentric reticulohistiocytosis, eruptive syringomas, sarcoidosis, and Langerhans cell histiocytosis; the latter should be considered, especially when there is concomitant diabetes insipidus.5 Laboratory studies typically are unremarkable. Although the majority of patients are normolipemic, rates of hyperlipemia within this group are comparable to the general population, occasionally rendering it difficult for the clinician to distinguish XD from hyperlipemic xanthomatoses. As such, diagnosis and differentiation from other xanthomatous processes rests on clinicopathological correlation. Histopathology reveals dermal collections of histiocytes, some with foamy cytoplasm, that range in appearance from spindled to scalloped to Touton-like. Early histopathology demonstrates scalloped macrophages with few foamy cells; a mixture of foamy cells, scalloped cells, inflammatory cells, and Touton and foreign body giant cells is characteristic of late lesions. Immunohistochemistry stains positive for non-Langerhans cell surface markers CD68 and factor XIIIa. Electron microscopy demonstrates dense and myeloid bodies, cholesterol crystals, and lipid vacuoles.5
Three subtypes of XD have been described based on the distinct clinical courses that have been observed in patients: a common, persistent, cutaneous form; a self-limited form with spontaneous resolution; and a progressive subtype with internal organ involvement. No consistently efficacious therapies have been identified, but isolated case reports attest to the efficacy of various agents, including azathioprine, clofibrate, cyclophosphamide, glucocorticoids, chlorambucil, and combination or monotherapy with lipid-lowering agents.3,5,6 Surgical resection, cryotherapy, radiotherapy, and CO2 laser therapy may offer some temporary benefit but do not alter the typically relapsing course of the disease.7,8 Remission and long-term control of lesions was reported with use of 2-chlorodeoxyadenosine, a purine nucleoside analogue, for 5 of 8 patients in a case series.3
- Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207-1212.
- Mahajan V, Sharma A, Chauhan P, et al. Xanthoma disseminatum: a red herring xanthomatosis. Indian J Dermatol Venereol Leprol. 2013;79:253-254.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol. 2011;147:459-464.
- Weiss N, Keller C. Xanthoma disseminatum: a rare normolipemic xanthomatosis. Clin Investig. 1993;71:233-238.
- Park HY, Cho DH, Joe DH, et al. A case of xanthoma disseminatum with spontaneous resolution over 10 years: review of the literature on long-term follow-up [published online May 26, 2011]. Dermatology. 2011;222:236-243.
- Kim SM, Waters P, Vincent A, et al. Sjogren's syndrome myelopathy: spinal cord involvement in Sjogren's syndrome might be a manifestation of neuromyelitis optica. Mult Scler. 2009;15:1062-1068.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol. 2008;58(2 suppl):S47-S49.
- Kim JY, Jung HD, Choe YS, et al. A case of xanthoma disseminatum accentuating over the eyelids. Ann Dermatol. 2010;22:353-357.
The Diagnosis: Xanthoma Disseminatum
Genital examination revealed approximately 1.5×3-cm soft, yellow-pink plaques extending from the bilateral inguinal folds to the proximal medial thighs (Figure 1). There was no mucosal, axillary, extensor extremity, or palmoplantar involvement. Histopathologic examination of a biopsy from a plaque on the left side of the lower abdomen revealed sheets of foamy histiocytes distributed throughout a fibrotic dermis. Both mononucleated and multinucleated histiocytes were present, including many Touton giant cells (Figure 2). A patchy infiltrate of lymphocytes and rare eosinophils also was noted. The histiocytes labeled with factor XIIIa but not with S-100. Laboratory tests were performed with the following pertinent findings: low-density lipoprotein, 150 mg/dL (reference range, <130 mg/dL); high-density lipoprotein, 30 mg/dL (reference range, >40 mg/dL). Total cholesterol and triglyceride levels were within reference range, and complete blood cell count and basic metabolic panel were normal.
Xanthoma disseminatum (XD)(also known as Montgomery syndrome) is a rare, nonfamilial, normolipemic non-Langerhans cell histiocytosis characterized by extensive lipid deposition in the skin, mucous membranes, and internal organs. The pathogenesis of XD is poorly understood, but it may represent a macrophage-mediated reactive process triggered by superantigens.1
Xanthoma disseminatum most commonly affects males aged 5 to 25 years.2 Clinically, it is characterized by red-brown to yellowish papules and plaques symmetrically distributed over the eyelids, trunk, face, and proximal extremities. There is a predilection for involvement of flexural and intertriginous surfaces and tendency for extension along Langer lines. Extracutaneous involvement can be a notable cause of morbidity and mortality, underscoring the importance of distinguishing XD from other clinically similar xanthomatoses. Mucous membrane involvement occurs in 40% to 60% of patients.3 The oropharynx, larynx, and corneal and conjunctival membranes are most commonly affected, resulting in dysphagia, dysphonia or dyspnea, and visual impairment, respectively. Symptoms of internal organ involvement can be manifold, including pain or limited mobility secondary to osteolytic bone lesions or muscle or synovial membrane involvement, as well as seizures, strabismus, and cerebellar ataxia due to central nervous system lesions.2-4 Approximately 40% of patients develop diabetes insipidus secondary to involvement of the pituitary meninges.3
The differential diagnosis of XD includes juvenile xanthogranuloma, papular xanthomas, eruptive xanthomas, generalized eruptive histiocytosis, progressive nodular histiocytosis, multicentric reticulohistiocytosis, eruptive syringomas, sarcoidosis, and Langerhans cell histiocytosis; the latter should be considered, especially when there is concomitant diabetes insipidus.5 Laboratory studies typically are unremarkable. Although the majority of patients are normolipemic, rates of hyperlipemia within this group are comparable to the general population, occasionally rendering it difficult for the clinician to distinguish XD from hyperlipemic xanthomatoses. As such, diagnosis and differentiation from other xanthomatous processes rests on clinicopathological correlation. Histopathology reveals dermal collections of histiocytes, some with foamy cytoplasm, that range in appearance from spindled to scalloped to Touton-like. Early histopathology demonstrates scalloped macrophages with few foamy cells; a mixture of foamy cells, scalloped cells, inflammatory cells, and Touton and foreign body giant cells is characteristic of late lesions. Immunohistochemistry stains positive for non-Langerhans cell surface markers CD68 and factor XIIIa. Electron microscopy demonstrates dense and myeloid bodies, cholesterol crystals, and lipid vacuoles.5
Three subtypes of XD have been described based on the distinct clinical courses that have been observed in patients: a common, persistent, cutaneous form; a self-limited form with spontaneous resolution; and a progressive subtype with internal organ involvement. No consistently efficacious therapies have been identified, but isolated case reports attest to the efficacy of various agents, including azathioprine, clofibrate, cyclophosphamide, glucocorticoids, chlorambucil, and combination or monotherapy with lipid-lowering agents.3,5,6 Surgical resection, cryotherapy, radiotherapy, and CO2 laser therapy may offer some temporary benefit but do not alter the typically relapsing course of the disease.7,8 Remission and long-term control of lesions was reported with use of 2-chlorodeoxyadenosine, a purine nucleoside analogue, for 5 of 8 patients in a case series.3
The Diagnosis: Xanthoma Disseminatum
Genital examination revealed approximately 1.5×3-cm soft, yellow-pink plaques extending from the bilateral inguinal folds to the proximal medial thighs (Figure 1). There was no mucosal, axillary, extensor extremity, or palmoplantar involvement. Histopathologic examination of a biopsy from a plaque on the left side of the lower abdomen revealed sheets of foamy histiocytes distributed throughout a fibrotic dermis. Both mononucleated and multinucleated histiocytes were present, including many Touton giant cells (Figure 2). A patchy infiltrate of lymphocytes and rare eosinophils also was noted. The histiocytes labeled with factor XIIIa but not with S-100. Laboratory tests were performed with the following pertinent findings: low-density lipoprotein, 150 mg/dL (reference range, <130 mg/dL); high-density lipoprotein, 30 mg/dL (reference range, >40 mg/dL). Total cholesterol and triglyceride levels were within reference range, and complete blood cell count and basic metabolic panel were normal.
Xanthoma disseminatum (XD)(also known as Montgomery syndrome) is a rare, nonfamilial, normolipemic non-Langerhans cell histiocytosis characterized by extensive lipid deposition in the skin, mucous membranes, and internal organs. The pathogenesis of XD is poorly understood, but it may represent a macrophage-mediated reactive process triggered by superantigens.1
Xanthoma disseminatum most commonly affects males aged 5 to 25 years.2 Clinically, it is characterized by red-brown to yellowish papules and plaques symmetrically distributed over the eyelids, trunk, face, and proximal extremities. There is a predilection for involvement of flexural and intertriginous surfaces and tendency for extension along Langer lines. Extracutaneous involvement can be a notable cause of morbidity and mortality, underscoring the importance of distinguishing XD from other clinically similar xanthomatoses. Mucous membrane involvement occurs in 40% to 60% of patients.3 The oropharynx, larynx, and corneal and conjunctival membranes are most commonly affected, resulting in dysphagia, dysphonia or dyspnea, and visual impairment, respectively. Symptoms of internal organ involvement can be manifold, including pain or limited mobility secondary to osteolytic bone lesions or muscle or synovial membrane involvement, as well as seizures, strabismus, and cerebellar ataxia due to central nervous system lesions.2-4 Approximately 40% of patients develop diabetes insipidus secondary to involvement of the pituitary meninges.3
The differential diagnosis of XD includes juvenile xanthogranuloma, papular xanthomas, eruptive xanthomas, generalized eruptive histiocytosis, progressive nodular histiocytosis, multicentric reticulohistiocytosis, eruptive syringomas, sarcoidosis, and Langerhans cell histiocytosis; the latter should be considered, especially when there is concomitant diabetes insipidus.5 Laboratory studies typically are unremarkable. Although the majority of patients are normolipemic, rates of hyperlipemia within this group are comparable to the general population, occasionally rendering it difficult for the clinician to distinguish XD from hyperlipemic xanthomatoses. As such, diagnosis and differentiation from other xanthomatous processes rests on clinicopathological correlation. Histopathology reveals dermal collections of histiocytes, some with foamy cytoplasm, that range in appearance from spindled to scalloped to Touton-like. Early histopathology demonstrates scalloped macrophages with few foamy cells; a mixture of foamy cells, scalloped cells, inflammatory cells, and Touton and foreign body giant cells is characteristic of late lesions. Immunohistochemistry stains positive for non-Langerhans cell surface markers CD68 and factor XIIIa. Electron microscopy demonstrates dense and myeloid bodies, cholesterol crystals, and lipid vacuoles.5
Three subtypes of XD have been described based on the distinct clinical courses that have been observed in patients: a common, persistent, cutaneous form; a self-limited form with spontaneous resolution; and a progressive subtype with internal organ involvement. No consistently efficacious therapies have been identified, but isolated case reports attest to the efficacy of various agents, including azathioprine, clofibrate, cyclophosphamide, glucocorticoids, chlorambucil, and combination or monotherapy with lipid-lowering agents.3,5,6 Surgical resection, cryotherapy, radiotherapy, and CO2 laser therapy may offer some temporary benefit but do not alter the typically relapsing course of the disease.7,8 Remission and long-term control of lesions was reported with use of 2-chlorodeoxyadenosine, a purine nucleoside analogue, for 5 of 8 patients in a case series.3
- Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207-1212.
- Mahajan V, Sharma A, Chauhan P, et al. Xanthoma disseminatum: a red herring xanthomatosis. Indian J Dermatol Venereol Leprol. 2013;79:253-254.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol. 2011;147:459-464.
- Weiss N, Keller C. Xanthoma disseminatum: a rare normolipemic xanthomatosis. Clin Investig. 1993;71:233-238.
- Park HY, Cho DH, Joe DH, et al. A case of xanthoma disseminatum with spontaneous resolution over 10 years: review of the literature on long-term follow-up [published online May 26, 2011]. Dermatology. 2011;222:236-243.
- Kim SM, Waters P, Vincent A, et al. Sjogren's syndrome myelopathy: spinal cord involvement in Sjogren's syndrome might be a manifestation of neuromyelitis optica. Mult Scler. 2009;15:1062-1068.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol. 2008;58(2 suppl):S47-S49.
- Kim JY, Jung HD, Choe YS, et al. A case of xanthoma disseminatum accentuating over the eyelids. Ann Dermatol. 2010;22:353-357.
- Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207-1212.
- Mahajan V, Sharma A, Chauhan P, et al. Xanthoma disseminatum: a red herring xanthomatosis. Indian J Dermatol Venereol Leprol. 2013;79:253-254.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol. 2011;147:459-464.
- Weiss N, Keller C. Xanthoma disseminatum: a rare normolipemic xanthomatosis. Clin Investig. 1993;71:233-238.
- Park HY, Cho DH, Joe DH, et al. A case of xanthoma disseminatum with spontaneous resolution over 10 years: review of the literature on long-term follow-up [published online May 26, 2011]. Dermatology. 2011;222:236-243.
- Kim SM, Waters P, Vincent A, et al. Sjogren's syndrome myelopathy: spinal cord involvement in Sjogren's syndrome might be a manifestation of neuromyelitis optica. Mult Scler. 2009;15:1062-1068.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol. 2008;58(2 suppl):S47-S49.
- Kim JY, Jung HD, Choe YS, et al. A case of xanthoma disseminatum accentuating over the eyelids. Ann Dermatol. 2010;22:353-357.

A 28-year-old man presented for evaluation of numerous papules on the face and groin that first appeared in adolescence and had been increasing in size and number over the last several years. The lesions occasionally were pruritic. Review of systems was noncontributory. His medical history was notable for asthma, and there were no affected family members. Physical examination revealed numerous symmetrically distributed, soft, yellow-pink, 1- to 5-mm papules coalescing into plaques on the bilateral malar cheeks extending to the medial canthi and the maxillary, mandibular, zygomatic, and submental regions, as well as the bilateral external auditory meatus.