User login
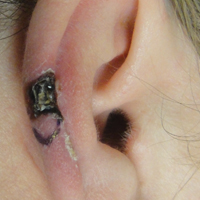
Hyperpigmented Papules and Plaques
The Diagnosis: Persistent Still Disease
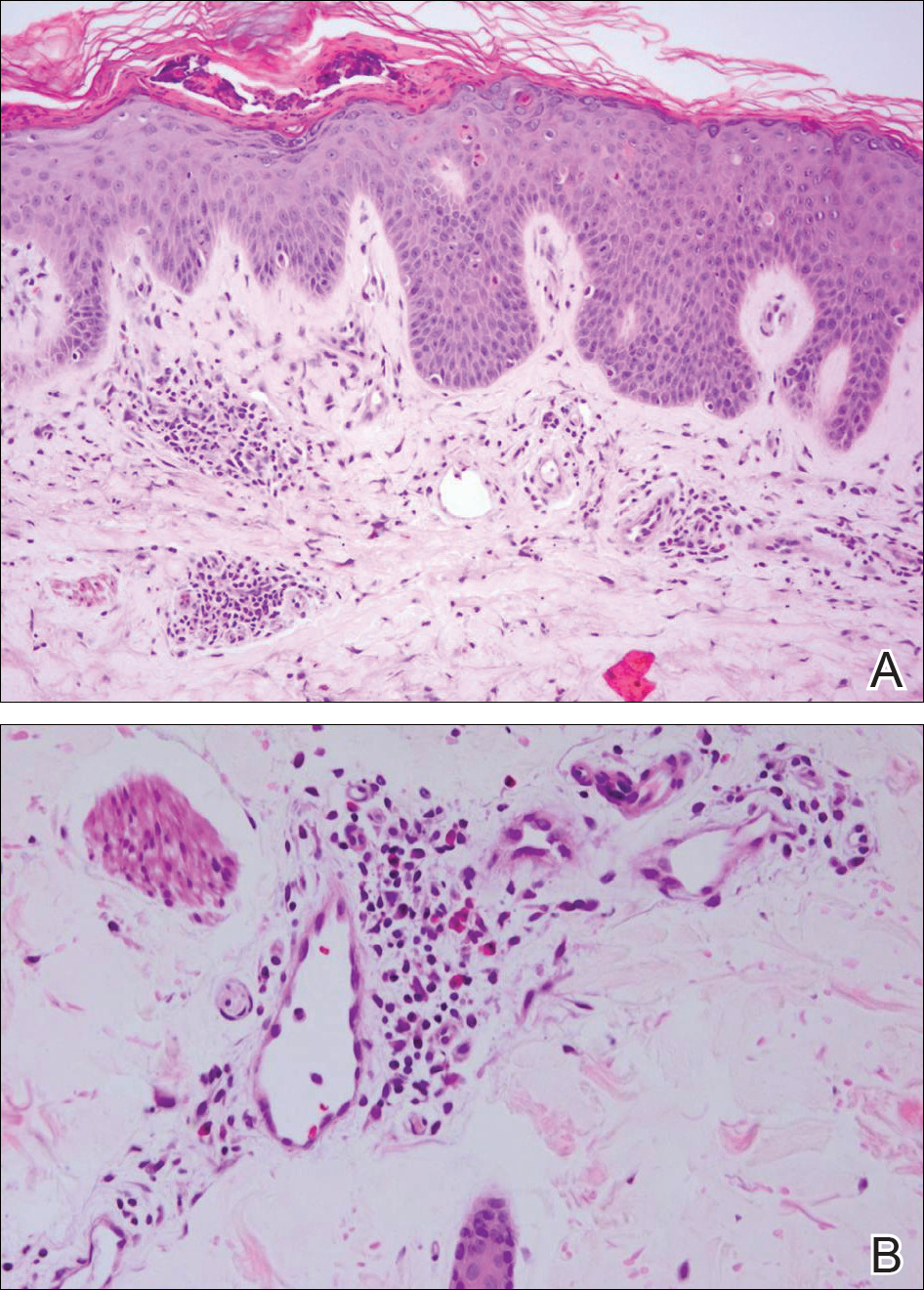
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
The Diagnosis: Persistent Still Disease
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
The Diagnosis: Persistent Still Disease
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
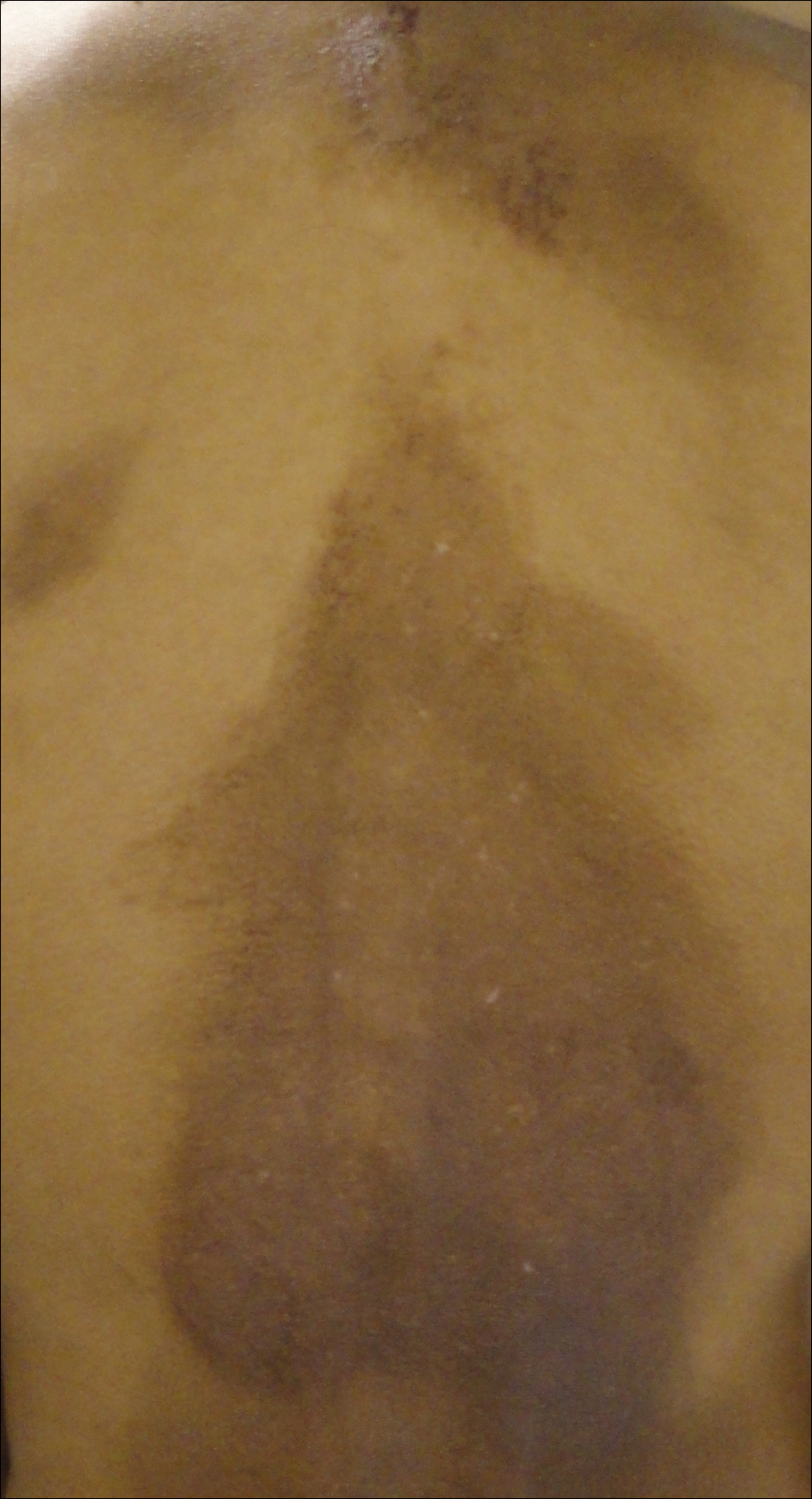
A 25-year-old Hispanic man with a history of juvenile idiopathic arthritis was admitted with a high-grade fever (temperature, >38.9°C) and diffuse nonlocalized abdominal pain of 2 days' duration. Physical examination revealed tachycardia, axillary lymphadenopathy, and hepatosplenomegaly. Cutaneous findings consisted of striking hyperpigmented patches on the chest and back, and hyperpigmented scaly lichenoid papules and plaques on the upper and lower extremities. The plaques on the lower extremities exhibited koebnerization. The patient reported that the eruption initially presented at 16 years of age as pruritic papules on the legs, which gradually spread to involve the arms, chest, and back. Prior treatments of juvenile idiopathic arthritis included prednisone, methotrexate, infliximab, and etanercept, though they were intermittent and temporary. Over time, the cutaneous eruption evolved into its current morphology and distribution, with periods of clearance observed while receiving systemic medications.
Bilateral Symmetric Onycholysis of Distal Fingernails
The Diagnosis: Allergic Contact Dermatitis
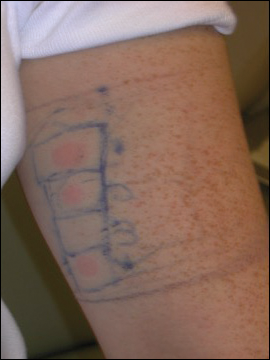
An allergic contact dermatitis (ACD) to acrylates was suspected and 4 patches were applied to the forearm (the North American Standard Series of the North American Contact Dermatitis Group). The patches were 2-hydroxyethyl methacrylate (2-HEMA) 2.0% permissible exposure limit (peL), ethyl acrylate 0.1% peL, tosylamide formaldehyde resin 10.0% peL, and methyl methacrylate 2.0% peL. A reading at 72 hours was performed and showed a positive reaction to hydroxyethyl methacrylate, ethyl acrylate, and methyl methacrylate, and a negative patch test to tosylamide formaldehyde resin (nail polish)(Figure). The patient was diagnosed with an allergic contact hypersensitivity to the aforementioned acrylates and instructed to avoid artificial nails and acrylate glues. She also was started on oral biotin supplements. On 6-month follow-up the patient had regrowth of all 10 fingernails without brittleness or splitting. She was able to use nail polishes but avoided all acrylic artificial nails and acrylate-containing personal care products.
Acrylate Allergy and Artificial Nails
Acrylates are plastic materials formed by polymerization of acrylic or methacrylic acid monomers and have been cited as a major cause of occupational and nonoccupational contact dermatitis. Contact dermatitis to acrylates in artificial nails was first reported in the 1950s.1,2 Products containing 100% methyl methacrylate monomers in acrylic nails were banned by the US Food and Drug Administration in the early 1970s after receiving a number of complaints.3 However, no regulation prohibits the use of methyl methacrylate monomer in cosmetic products, and various methacrylate and acrylate monomers remain widely used.4 With a growing popularity in artificial nails, it is expected the number of sensitized persons will increase.
Acrylate allergy from sculptured nails concern self-curing resins made from a polymer powder and a liquid monomer solution. Advantages of new UV-cured products include the lack of unpleasant smell and simplified modeling. They also do not require an irritant, such as methacrylic acid, as a bonding agent. Instead, 2-HEMA and 2-hydroxypropyl methacrylate are added. These photobonded nails colloquially are called gel nails (acid free) as opposed to acrylic nails (using methacrylic acid as a primer). It is important to note that the esters of acrylic acid but not the acid itself sensitize patients, and sensitization is not caused by the uncured gel or the monomer solution but by the remaining monomers in the cured plastic nail and the dust filings that are produced during the finishing process.
Clinical Presentation
Symptoms of an ACD to nail acrylates include pruritus and fingertip dermatitis along with nail plate dystrophy. There may be pruritus at the nail base, with subsequent dryness, thickening, and onycholysis. The brittle nails may become split, discolored, and develop paronychia. Inadvertent contact with glue monomers or other acrylate-containing substances may cause eczematous lesions at distant sites. Avoidance of the allergen often results in complete restoration of the normal nail and fingertip within months.
Sensitization
Acrylates and methacrylates are ubiquitous materials used for both industrial and commercial applications. Due to their widespread industrial use, contact allergies to acrylates including 2-HEMA, 2-hydroxypropyl methacrylate, and triethyleneglycol diacrylate (TREGDA) are common. Cross-reaction of these compounds has been observed and is postulated to be due to reaction of the (meth)acrylate carboxyethyl group with the receptors of antigen-presenting cells.5 As a result, an individual with an acrylate allergy sensitized to one allergen often is allergic to its similar compounds and cross-reactors and must avoid the assortment of compounds containing these ingredients, which is important for individuals with occupational sensitization to a particular acrylate who is subsequently susceptible to other acrylate-containing compounds triggering allergic reactions when reexposure occurs in different settings.
Allergens and Occupational Exposure
Acrylates in cosmetic nail products are a source of ACD for not only the customer but also the manicurist.6 The most frequently cited sources of ACD in beauticians are acrylate chemicals.7 However, acrylate compounds are an occupational hazard for a number of other specialists, including dentists and dental technicians, histology technicians, and individuals in the printing industry.8,9 Other individuals may be sensitized to acrylates through their inclusion in adhesives, dental bonding agents, hearing aids, electrocardiogram electrodes, artificial bone cement, and a myriad of other medical and nonmedical applications.4,10-12 For workers who cannot avoid occupational exposure to these allergens, polyvinyl alcohol and multilayer laminate gloves are recommended, as natural rubber latex gloves do not always provide adequate protection from many of these agents.10
Testing for Suspected Acrylate Allergy
Cross-reactivity among acrylates is widely considered in the literature but remains enigmatic and is an important consideration with regard to routine patch test screening.13 In the case of an acrylate allergy to nail products, using 2-HEMA and ethylene glycol dimethacrylate is effective in detecting sensitization by photobonded nails and in patients sensitized by powder liquid products.14 One study showed a patch test panel including 2-HEMA, ethylene glycol dimethacrylate, and TREGDA was effective in identifying the majority of individuals with an allergy to acrylates in nail products and nail technicians.15 Another study has shown the most commonly positive testing allergens to be HEMA, ethyl acrylate, and methyl methacrylate.16 If one is patch testing only one chemical, it appears 2-HEMA is preferred.17 However, broader panels of screening allergens are necessary to achieve an accurate diagnosis. Furthermore, different panels of test allergens have been shown to vary in their ability to detect an acrylate allergy in different occupational exposures.12
The time to patch test read also is important. A standard read at 72 hours is warranted; however, one study showed if only one read at day 3 was done without a subsequent day 7 read, then 25% of TREGDA and 50% of 2-HEMA allergies would have been missed in patients with occupational acrylate allergy.15 Other studies have reported late-appearing and long-lasting test reactions when testing for an acrylate allergy.18,19 Clinicians should be cognizant that an acrylate allergy may be present even if initial screening is negative but the history and clinical picture are suggestive.
- Canizares O. Contact dermatitis due to the acrylic materials used in artificial nails. AMA Arch Derm. 1956;74:141-143.
- Fisher AA, Franks A, Glick H. Allergic sensitization of the skin and nails to acrylic plastic nails. J Allergy. 1957;28:84-88.
- US Food and Drug Administration. Nail care products. http://www.fda.gov/Cosmetics/ProductsIngredients/Products/ucm127068.htm. Updated October 26, 2016. Accessed December 27, 2016.
- Haughton AM, Belsito DV. Acrylate allergy induced by acrylic nails resulting in prosthesis failure. J Am Acad Dermatol. 2008;59(5 suppl):S123-S124.
- Kanerva L. Cross-reactions of multifunctional methacrylates and acrylates. Acta Odontol Scand. 2001;59:320-329.
- Tammaro A, Narcisi A, Abruzzese C, et al. Fingertip dermatitis: occupational acrylate cross reaction. Allergol Int. 2014;63:609-610.
- Kwok C, Money A, Carder M, et al. Cases of occupational dermatitis and asthma in beauticians that were reported to The Health and Occupation Research (THOR) network from 1996 to 2011. Clin Exp Dermatol. 2014;39:590-595.
- Aalto-Korte K, Alanko K, Kuuliala O, et al. Methacrylate and acrylate allergy in dental personnel. Contact Dermatitis. 2007;57:324-330.
- Molina L, Amado A, Mattei PL 4th, et al. Contact dermatitis from acrylics in a histology laboratory assistant. Dermatitis. 2009;20:E11-E12.
- Prasad Hunasehally RY, Hughes TM, Stone NM. Atypical pattern of (meth)acrylate allergic contact dermatitis in dental professionals. Br Dent J. 2012;213:223-224.
- Stingeni L, Cerulli E, Spalletti A, et al. The role of acrylic acid impurity as a sensitizing component in electrocardiogram electrodes [published online January 27, 2015]. Contact Dermatitis. 2015;73:44-48.
- Sasseville D. Acrylates in contact dermatitis. Dermatitis. 2012;23:6-16.
- Fisher AA. Cross reactions between methyl methacrylate monomer and acrylic monomers presently used in acrylic nail preparations. Contact Dermatitis. 1980;6:345-347.
- Hemmer W, Focke M, Wantke F, et al. Allergic contact dermatitis to artificial fingernails prepared from UV light-cured acrylates. J Am Acad Dermatol. 1996;35(3, pt 1):377-380.
- Teik-Jin Goon A, Bruze M, Zimerson E, et al. Contact allergy to acrylates/methacrylates in the acrylate and nail acrylics series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2007;57:21-27.
- Drucker AM, Pratt MD. Acrylate contact allergy: patient characteristics and evaluation of screening allergens. Dermatitis. 2011;22:98-101.
- Ramos L, Cabral R, Goncalo M. Allergic contact dermatitis caused by acrylates and methacrylates--a 7-year study. Contact Dermatitis. 2014;71:102-107.
- Goon AT, Isaksson M, Zimerson E, et al. Contact allergy to (meth)acrylates in the dental series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2006;55:219-226.
- Isaksson M, Lindberg M, Sundberg K, et al. The development and course of patch-test reactions to 2-hydroxyethyl methacrylate and ethyleneglycol dimethacrylate. Contact Dermatitis. 2005;53:292-297.
The Diagnosis: Allergic Contact Dermatitis
An allergic contact dermatitis (ACD) to acrylates was suspected and 4 patches were applied to the forearm (the North American Standard Series of the North American Contact Dermatitis Group). The patches were 2-hydroxyethyl methacrylate (2-HEMA) 2.0% permissible exposure limit (peL), ethyl acrylate 0.1% peL, tosylamide formaldehyde resin 10.0% peL, and methyl methacrylate 2.0% peL. A reading at 72 hours was performed and showed a positive reaction to hydroxyethyl methacrylate, ethyl acrylate, and methyl methacrylate, and a negative patch test to tosylamide formaldehyde resin (nail polish)(Figure). The patient was diagnosed with an allergic contact hypersensitivity to the aforementioned acrylates and instructed to avoid artificial nails and acrylate glues. She also was started on oral biotin supplements. On 6-month follow-up the patient had regrowth of all 10 fingernails without brittleness or splitting. She was able to use nail polishes but avoided all acrylic artificial nails and acrylate-containing personal care products.
Acrylate Allergy and Artificial Nails
Acrylates are plastic materials formed by polymerization of acrylic or methacrylic acid monomers and have been cited as a major cause of occupational and nonoccupational contact dermatitis. Contact dermatitis to acrylates in artificial nails was first reported in the 1950s.1,2 Products containing 100% methyl methacrylate monomers in acrylic nails were banned by the US Food and Drug Administration in the early 1970s after receiving a number of complaints.3 However, no regulation prohibits the use of methyl methacrylate monomer in cosmetic products, and various methacrylate and acrylate monomers remain widely used.4 With a growing popularity in artificial nails, it is expected the number of sensitized persons will increase.
Acrylate allergy from sculptured nails concern self-curing resins made from a polymer powder and a liquid monomer solution. Advantages of new UV-cured products include the lack of unpleasant smell and simplified modeling. They also do not require an irritant, such as methacrylic acid, as a bonding agent. Instead, 2-HEMA and 2-hydroxypropyl methacrylate are added. These photobonded nails colloquially are called gel nails (acid free) as opposed to acrylic nails (using methacrylic acid as a primer). It is important to note that the esters of acrylic acid but not the acid itself sensitize patients, and sensitization is not caused by the uncured gel or the monomer solution but by the remaining monomers in the cured plastic nail and the dust filings that are produced during the finishing process.
Clinical Presentation
Symptoms of an ACD to nail acrylates include pruritus and fingertip dermatitis along with nail plate dystrophy. There may be pruritus at the nail base, with subsequent dryness, thickening, and onycholysis. The brittle nails may become split, discolored, and develop paronychia. Inadvertent contact with glue monomers or other acrylate-containing substances may cause eczematous lesions at distant sites. Avoidance of the allergen often results in complete restoration of the normal nail and fingertip within months.
Sensitization
Acrylates and methacrylates are ubiquitous materials used for both industrial and commercial applications. Due to their widespread industrial use, contact allergies to acrylates including 2-HEMA, 2-hydroxypropyl methacrylate, and triethyleneglycol diacrylate (TREGDA) are common. Cross-reaction of these compounds has been observed and is postulated to be due to reaction of the (meth)acrylate carboxyethyl group with the receptors of antigen-presenting cells.5 As a result, an individual with an acrylate allergy sensitized to one allergen often is allergic to its similar compounds and cross-reactors and must avoid the assortment of compounds containing these ingredients, which is important for individuals with occupational sensitization to a particular acrylate who is subsequently susceptible to other acrylate-containing compounds triggering allergic reactions when reexposure occurs in different settings.
Allergens and Occupational Exposure
Acrylates in cosmetic nail products are a source of ACD for not only the customer but also the manicurist.6 The most frequently cited sources of ACD in beauticians are acrylate chemicals.7 However, acrylate compounds are an occupational hazard for a number of other specialists, including dentists and dental technicians, histology technicians, and individuals in the printing industry.8,9 Other individuals may be sensitized to acrylates through their inclusion in adhesives, dental bonding agents, hearing aids, electrocardiogram electrodes, artificial bone cement, and a myriad of other medical and nonmedical applications.4,10-12 For workers who cannot avoid occupational exposure to these allergens, polyvinyl alcohol and multilayer laminate gloves are recommended, as natural rubber latex gloves do not always provide adequate protection from many of these agents.10
Testing for Suspected Acrylate Allergy
Cross-reactivity among acrylates is widely considered in the literature but remains enigmatic and is an important consideration with regard to routine patch test screening.13 In the case of an acrylate allergy to nail products, using 2-HEMA and ethylene glycol dimethacrylate is effective in detecting sensitization by photobonded nails and in patients sensitized by powder liquid products.14 One study showed a patch test panel including 2-HEMA, ethylene glycol dimethacrylate, and TREGDA was effective in identifying the majority of individuals with an allergy to acrylates in nail products and nail technicians.15 Another study has shown the most commonly positive testing allergens to be HEMA, ethyl acrylate, and methyl methacrylate.16 If one is patch testing only one chemical, it appears 2-HEMA is preferred.17 However, broader panels of screening allergens are necessary to achieve an accurate diagnosis. Furthermore, different panels of test allergens have been shown to vary in their ability to detect an acrylate allergy in different occupational exposures.12
The time to patch test read also is important. A standard read at 72 hours is warranted; however, one study showed if only one read at day 3 was done without a subsequent day 7 read, then 25% of TREGDA and 50% of 2-HEMA allergies would have been missed in patients with occupational acrylate allergy.15 Other studies have reported late-appearing and long-lasting test reactions when testing for an acrylate allergy.18,19 Clinicians should be cognizant that an acrylate allergy may be present even if initial screening is negative but the history and clinical picture are suggestive.
The Diagnosis: Allergic Contact Dermatitis
An allergic contact dermatitis (ACD) to acrylates was suspected and 4 patches were applied to the forearm (the North American Standard Series of the North American Contact Dermatitis Group). The patches were 2-hydroxyethyl methacrylate (2-HEMA) 2.0% permissible exposure limit (peL), ethyl acrylate 0.1% peL, tosylamide formaldehyde resin 10.0% peL, and methyl methacrylate 2.0% peL. A reading at 72 hours was performed and showed a positive reaction to hydroxyethyl methacrylate, ethyl acrylate, and methyl methacrylate, and a negative patch test to tosylamide formaldehyde resin (nail polish)(Figure). The patient was diagnosed with an allergic contact hypersensitivity to the aforementioned acrylates and instructed to avoid artificial nails and acrylate glues. She also was started on oral biotin supplements. On 6-month follow-up the patient had regrowth of all 10 fingernails without brittleness or splitting. She was able to use nail polishes but avoided all acrylic artificial nails and acrylate-containing personal care products.
Acrylate Allergy and Artificial Nails
Acrylates are plastic materials formed by polymerization of acrylic or methacrylic acid monomers and have been cited as a major cause of occupational and nonoccupational contact dermatitis. Contact dermatitis to acrylates in artificial nails was first reported in the 1950s.1,2 Products containing 100% methyl methacrylate monomers in acrylic nails were banned by the US Food and Drug Administration in the early 1970s after receiving a number of complaints.3 However, no regulation prohibits the use of methyl methacrylate monomer in cosmetic products, and various methacrylate and acrylate monomers remain widely used.4 With a growing popularity in artificial nails, it is expected the number of sensitized persons will increase.
Acrylate allergy from sculptured nails concern self-curing resins made from a polymer powder and a liquid monomer solution. Advantages of new UV-cured products include the lack of unpleasant smell and simplified modeling. They also do not require an irritant, such as methacrylic acid, as a bonding agent. Instead, 2-HEMA and 2-hydroxypropyl methacrylate are added. These photobonded nails colloquially are called gel nails (acid free) as opposed to acrylic nails (using methacrylic acid as a primer). It is important to note that the esters of acrylic acid but not the acid itself sensitize patients, and sensitization is not caused by the uncured gel or the monomer solution but by the remaining monomers in the cured plastic nail and the dust filings that are produced during the finishing process.
Clinical Presentation
Symptoms of an ACD to nail acrylates include pruritus and fingertip dermatitis along with nail plate dystrophy. There may be pruritus at the nail base, with subsequent dryness, thickening, and onycholysis. The brittle nails may become split, discolored, and develop paronychia. Inadvertent contact with glue monomers or other acrylate-containing substances may cause eczematous lesions at distant sites. Avoidance of the allergen often results in complete restoration of the normal nail and fingertip within months.
Sensitization
Acrylates and methacrylates are ubiquitous materials used for both industrial and commercial applications. Due to their widespread industrial use, contact allergies to acrylates including 2-HEMA, 2-hydroxypropyl methacrylate, and triethyleneglycol diacrylate (TREGDA) are common. Cross-reaction of these compounds has been observed and is postulated to be due to reaction of the (meth)acrylate carboxyethyl group with the receptors of antigen-presenting cells.5 As a result, an individual with an acrylate allergy sensitized to one allergen often is allergic to its similar compounds and cross-reactors and must avoid the assortment of compounds containing these ingredients, which is important for individuals with occupational sensitization to a particular acrylate who is subsequently susceptible to other acrylate-containing compounds triggering allergic reactions when reexposure occurs in different settings.
Allergens and Occupational Exposure
Acrylates in cosmetic nail products are a source of ACD for not only the customer but also the manicurist.6 The most frequently cited sources of ACD in beauticians are acrylate chemicals.7 However, acrylate compounds are an occupational hazard for a number of other specialists, including dentists and dental technicians, histology technicians, and individuals in the printing industry.8,9 Other individuals may be sensitized to acrylates through their inclusion in adhesives, dental bonding agents, hearing aids, electrocardiogram electrodes, artificial bone cement, and a myriad of other medical and nonmedical applications.4,10-12 For workers who cannot avoid occupational exposure to these allergens, polyvinyl alcohol and multilayer laminate gloves are recommended, as natural rubber latex gloves do not always provide adequate protection from many of these agents.10
Testing for Suspected Acrylate Allergy
Cross-reactivity among acrylates is widely considered in the literature but remains enigmatic and is an important consideration with regard to routine patch test screening.13 In the case of an acrylate allergy to nail products, using 2-HEMA and ethylene glycol dimethacrylate is effective in detecting sensitization by photobonded nails and in patients sensitized by powder liquid products.14 One study showed a patch test panel including 2-HEMA, ethylene glycol dimethacrylate, and TREGDA was effective in identifying the majority of individuals with an allergy to acrylates in nail products and nail technicians.15 Another study has shown the most commonly positive testing allergens to be HEMA, ethyl acrylate, and methyl methacrylate.16 If one is patch testing only one chemical, it appears 2-HEMA is preferred.17 However, broader panels of screening allergens are necessary to achieve an accurate diagnosis. Furthermore, different panels of test allergens have been shown to vary in their ability to detect an acrylate allergy in different occupational exposures.12
The time to patch test read also is important. A standard read at 72 hours is warranted; however, one study showed if only one read at day 3 was done without a subsequent day 7 read, then 25% of TREGDA and 50% of 2-HEMA allergies would have been missed in patients with occupational acrylate allergy.15 Other studies have reported late-appearing and long-lasting test reactions when testing for an acrylate allergy.18,19 Clinicians should be cognizant that an acrylate allergy may be present even if initial screening is negative but the history and clinical picture are suggestive.
- Canizares O. Contact dermatitis due to the acrylic materials used in artificial nails. AMA Arch Derm. 1956;74:141-143.
- Fisher AA, Franks A, Glick H. Allergic sensitization of the skin and nails to acrylic plastic nails. J Allergy. 1957;28:84-88.
- US Food and Drug Administration. Nail care products. http://www.fda.gov/Cosmetics/ProductsIngredients/Products/ucm127068.htm. Updated October 26, 2016. Accessed December 27, 2016.
- Haughton AM, Belsito DV. Acrylate allergy induced by acrylic nails resulting in prosthesis failure. J Am Acad Dermatol. 2008;59(5 suppl):S123-S124.
- Kanerva L. Cross-reactions of multifunctional methacrylates and acrylates. Acta Odontol Scand. 2001;59:320-329.
- Tammaro A, Narcisi A, Abruzzese C, et al. Fingertip dermatitis: occupational acrylate cross reaction. Allergol Int. 2014;63:609-610.
- Kwok C, Money A, Carder M, et al. Cases of occupational dermatitis and asthma in beauticians that were reported to The Health and Occupation Research (THOR) network from 1996 to 2011. Clin Exp Dermatol. 2014;39:590-595.
- Aalto-Korte K, Alanko K, Kuuliala O, et al. Methacrylate and acrylate allergy in dental personnel. Contact Dermatitis. 2007;57:324-330.
- Molina L, Amado A, Mattei PL 4th, et al. Contact dermatitis from acrylics in a histology laboratory assistant. Dermatitis. 2009;20:E11-E12.
- Prasad Hunasehally RY, Hughes TM, Stone NM. Atypical pattern of (meth)acrylate allergic contact dermatitis in dental professionals. Br Dent J. 2012;213:223-224.
- Stingeni L, Cerulli E, Spalletti A, et al. The role of acrylic acid impurity as a sensitizing component in electrocardiogram electrodes [published online January 27, 2015]. Contact Dermatitis. 2015;73:44-48.
- Sasseville D. Acrylates in contact dermatitis. Dermatitis. 2012;23:6-16.
- Fisher AA. Cross reactions between methyl methacrylate monomer and acrylic monomers presently used in acrylic nail preparations. Contact Dermatitis. 1980;6:345-347.
- Hemmer W, Focke M, Wantke F, et al. Allergic contact dermatitis to artificial fingernails prepared from UV light-cured acrylates. J Am Acad Dermatol. 1996;35(3, pt 1):377-380.
- Teik-Jin Goon A, Bruze M, Zimerson E, et al. Contact allergy to acrylates/methacrylates in the acrylate and nail acrylics series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2007;57:21-27.
- Drucker AM, Pratt MD. Acrylate contact allergy: patient characteristics and evaluation of screening allergens. Dermatitis. 2011;22:98-101.
- Ramos L, Cabral R, Goncalo M. Allergic contact dermatitis caused by acrylates and methacrylates--a 7-year study. Contact Dermatitis. 2014;71:102-107.
- Goon AT, Isaksson M, Zimerson E, et al. Contact allergy to (meth)acrylates in the dental series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2006;55:219-226.
- Isaksson M, Lindberg M, Sundberg K, et al. The development and course of patch-test reactions to 2-hydroxyethyl methacrylate and ethyleneglycol dimethacrylate. Contact Dermatitis. 2005;53:292-297.
- Canizares O. Contact dermatitis due to the acrylic materials used in artificial nails. AMA Arch Derm. 1956;74:141-143.
- Fisher AA, Franks A, Glick H. Allergic sensitization of the skin and nails to acrylic plastic nails. J Allergy. 1957;28:84-88.
- US Food and Drug Administration. Nail care products. http://www.fda.gov/Cosmetics/ProductsIngredients/Products/ucm127068.htm. Updated October 26, 2016. Accessed December 27, 2016.
- Haughton AM, Belsito DV. Acrylate allergy induced by acrylic nails resulting in prosthesis failure. J Am Acad Dermatol. 2008;59(5 suppl):S123-S124.
- Kanerva L. Cross-reactions of multifunctional methacrylates and acrylates. Acta Odontol Scand. 2001;59:320-329.
- Tammaro A, Narcisi A, Abruzzese C, et al. Fingertip dermatitis: occupational acrylate cross reaction. Allergol Int. 2014;63:609-610.
- Kwok C, Money A, Carder M, et al. Cases of occupational dermatitis and asthma in beauticians that were reported to The Health and Occupation Research (THOR) network from 1996 to 2011. Clin Exp Dermatol. 2014;39:590-595.
- Aalto-Korte K, Alanko K, Kuuliala O, et al. Methacrylate and acrylate allergy in dental personnel. Contact Dermatitis. 2007;57:324-330.
- Molina L, Amado A, Mattei PL 4th, et al. Contact dermatitis from acrylics in a histology laboratory assistant. Dermatitis. 2009;20:E11-E12.
- Prasad Hunasehally RY, Hughes TM, Stone NM. Atypical pattern of (meth)acrylate allergic contact dermatitis in dental professionals. Br Dent J. 2012;213:223-224.
- Stingeni L, Cerulli E, Spalletti A, et al. The role of acrylic acid impurity as a sensitizing component in electrocardiogram electrodes [published online January 27, 2015]. Contact Dermatitis. 2015;73:44-48.
- Sasseville D. Acrylates in contact dermatitis. Dermatitis. 2012;23:6-16.
- Fisher AA. Cross reactions between methyl methacrylate monomer and acrylic monomers presently used in acrylic nail preparations. Contact Dermatitis. 1980;6:345-347.
- Hemmer W, Focke M, Wantke F, et al. Allergic contact dermatitis to artificial fingernails prepared from UV light-cured acrylates. J Am Acad Dermatol. 1996;35(3, pt 1):377-380.
- Teik-Jin Goon A, Bruze M, Zimerson E, et al. Contact allergy to acrylates/methacrylates in the acrylate and nail acrylics series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2007;57:21-27.
- Drucker AM, Pratt MD. Acrylate contact allergy: patient characteristics and evaluation of screening allergens. Dermatitis. 2011;22:98-101.
- Ramos L, Cabral R, Goncalo M. Allergic contact dermatitis caused by acrylates and methacrylates--a 7-year study. Contact Dermatitis. 2014;71:102-107.
- Goon AT, Isaksson M, Zimerson E, et al. Contact allergy to (meth)acrylates in the dental series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2006;55:219-226.
- Isaksson M, Lindberg M, Sundberg K, et al. The development and course of patch-test reactions to 2-hydroxyethyl methacrylate and ethyleneglycol dimethacrylate. Contact Dermatitis. 2005;53:292-297.
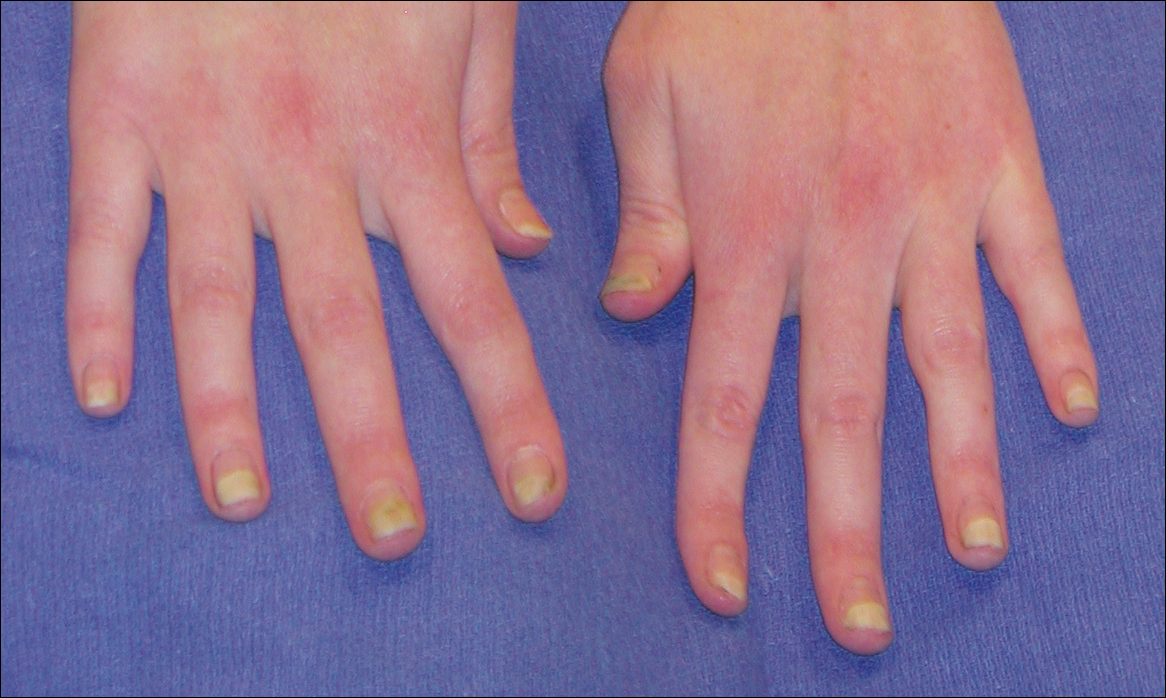
A 28-year-old woman presented with distal onycholysis of all 10 fingernails. The patient started to notice brittleness in the first, second, and third fingernails of the right hand 2 months prior. She had a 10-year history of wearing acrylic nails and reported a history of periungual eczema. On physical examination, all 10 fingernails had distal onycholysis and there was a green discoloration of the first fingernail on the left hand. On blood analysis, thyroid-stimulating hormone and free thyroxine were within reference range. A nail clipping showed onychodystrophy and a negative periodic acid-Schiff stain.
Painful Oral and Genital Ulcers
The Diagnosis: Pemphigus Vegetans
Pemphigus vegetans is a rare variant of pemphigus vulgaris. Clinically, pemphigus vegetans is characterized by vegetative lesions over the flexures, but any area of the skin may be involved. There have been case reports involving the scalp,1,2 mouth,3 and foot.4 There are 2 clinical subtypes: the Neumann type and the Hallopeau type.5 The Hallopeau type is relatively benign, requires lower doses of systemic corticosteroids, and has a prolonged remission, while the Neumann type necessitates higher doses of systemic corticosteroids and often presents with relapses and remissions.
The diagnosis of pemphigus vegetans is based on clinical suspicion and confirmed by histological examination and immunological findings. The diagnosis may be difficult, as its presentation varies and histopathological findings may resemble other conditions.
Systemic corticosteroids are the well-established drug of choice for treating pemphigus vegetans to induce remission and maintain healing before cautiously tapering down the dosage approximately 50% every 2 weeks.6 Adjuvant drugs used in conjunction with steroids for steroid-sparing purpose include azathioprine, cyclophosphamide, mycophenolate mofetil, methotrexate, and cyclosporine.6 Pulsed intravenous steroids,7 intravenous immunoglobulins,8 pulsed dexamethasone cyclophosphamide,9 and extracorporeal photopheresis10 are given for severe and recalcitrant disease.
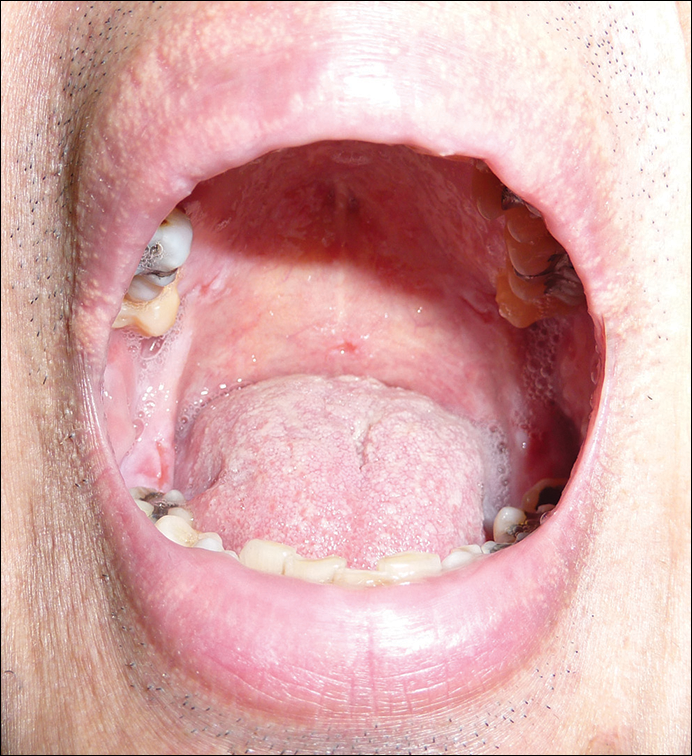
Laboratory investigations of our patient showed a normal complete blood cell count and a normal renal and liver profile. Herpes simplex virus serology was positive for type 1 and type 2 IgM and IgG. Urethral swab was dry and negative for gonorrhea. Serology for chlamydia, toxoplasma, amoebiasis, and leishmaniasis was negative. Human immunodeficiency virus serology, hepatitis screening, rapid plasma reagin, Treponema pallidum hemagglutination, rheumatoid factor, and antinuclear antibody all were negative. The patient was given a course of oral acyclovir 400 mg 3 times daily and empirical treatment with oral doxycycline 100 mg twice daily for a week with no clinical response.
Two biopsies from the perianal ulcers showed inflamed squamous papillomata with no Donovan bodies. A third biopsy from an intact blister showed acantholytic cells in the suprabasal bullae with eosinophilic and lymphocytic infiltrates at the upper dermis. Direct immunofluorescence demonstrated intercellular C3 and IgG deposits.
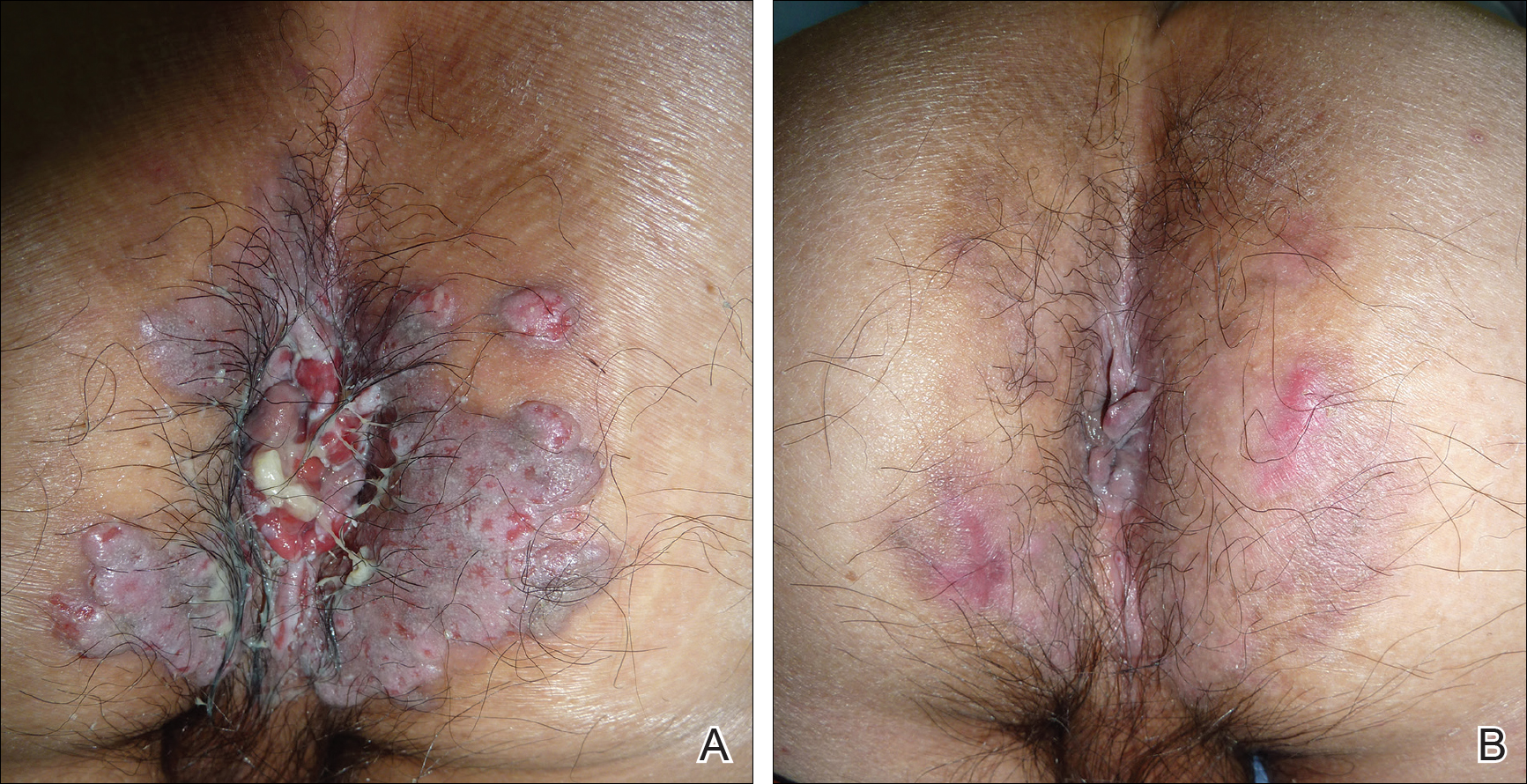
The patient was started on oral prednisolone at 1 mg/kg daily and oral azathioprine 50 mg daily with resolution of the perianal, penile, and oral ulcers (Figures 1 and 2). He achieved good suppression of further eruption. At the patient's most recent follow-up (2.5 years after the initial presentation), he was in remission and was currently taking oral azathioprine 100 mg once daily and no oral corticosteroids.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp [published online October 22,2014]. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Augusto de Oliveira M, Martins E Martins F, Lourenço S, et al. Oral pemphigus vegetans: a case report. Dermatol Online J. 2012;18:10.
- Ma DL, Fang K. Hallopeau type of pemphigus vegetans confined to the right foot: case report. Chin Med J (Engl). 2009;122:588-590.
- Ahmed AR, Blose DA. Pemphigus vegetans. Neumann type and Hallopeau type. Int J Dermatol. 1984;23:135-141.
- Harman KE, Albert S, Black MM, et al. Guidelines for the management of pemphigus vulgaris. Br J Dermatol. 2003;149:926-937.
- Chryssomallis F, Dimitriades A, Chaidemenos GC, et al. Steroid-pulse therapy in pemphigus vulgaris long term follow-up. Int J Dermatol. 1995;34:438-442.
- Ahmed AR. Intravenous immunoglobulin therapy in the treatment of patients with pemphigus vulgaris unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol. 2001;45:679-690.
- Pasricha JS, Khaitan BK, Raman RS, et al. Dexamethasone-cyclophosphamide pulse therapy for pemphigus. Int J Dermatol. 1995;34:875-882.
- Rook AH, Jegasothy BV, Heald P, et al. Extracorporeal photochemotherapy for drug-resistant pemphigus vulgaris. Ann Int Med. 1990;112:303-305.
The Diagnosis: Pemphigus Vegetans
Pemphigus vegetans is a rare variant of pemphigus vulgaris. Clinically, pemphigus vegetans is characterized by vegetative lesions over the flexures, but any area of the skin may be involved. There have been case reports involving the scalp,1,2 mouth,3 and foot.4 There are 2 clinical subtypes: the Neumann type and the Hallopeau type.5 The Hallopeau type is relatively benign, requires lower doses of systemic corticosteroids, and has a prolonged remission, while the Neumann type necessitates higher doses of systemic corticosteroids and often presents with relapses and remissions.
The diagnosis of pemphigus vegetans is based on clinical suspicion and confirmed by histological examination and immunological findings. The diagnosis may be difficult, as its presentation varies and histopathological findings may resemble other conditions.
Systemic corticosteroids are the well-established drug of choice for treating pemphigus vegetans to induce remission and maintain healing before cautiously tapering down the dosage approximately 50% every 2 weeks.6 Adjuvant drugs used in conjunction with steroids for steroid-sparing purpose include azathioprine, cyclophosphamide, mycophenolate mofetil, methotrexate, and cyclosporine.6 Pulsed intravenous steroids,7 intravenous immunoglobulins,8 pulsed dexamethasone cyclophosphamide,9 and extracorporeal photopheresis10 are given for severe and recalcitrant disease.
Laboratory investigations of our patient showed a normal complete blood cell count and a normal renal and liver profile. Herpes simplex virus serology was positive for type 1 and type 2 IgM and IgG. Urethral swab was dry and negative for gonorrhea. Serology for chlamydia, toxoplasma, amoebiasis, and leishmaniasis was negative. Human immunodeficiency virus serology, hepatitis screening, rapid plasma reagin, Treponema pallidum hemagglutination, rheumatoid factor, and antinuclear antibody all were negative. The patient was given a course of oral acyclovir 400 mg 3 times daily and empirical treatment with oral doxycycline 100 mg twice daily for a week with no clinical response.
Two biopsies from the perianal ulcers showed inflamed squamous papillomata with no Donovan bodies. A third biopsy from an intact blister showed acantholytic cells in the suprabasal bullae with eosinophilic and lymphocytic infiltrates at the upper dermis. Direct immunofluorescence demonstrated intercellular C3 and IgG deposits.
The patient was started on oral prednisolone at 1 mg/kg daily and oral azathioprine 50 mg daily with resolution of the perianal, penile, and oral ulcers (Figures 1 and 2). He achieved good suppression of further eruption. At the patient's most recent follow-up (2.5 years after the initial presentation), he was in remission and was currently taking oral azathioprine 100 mg once daily and no oral corticosteroids.
The Diagnosis: Pemphigus Vegetans
Pemphigus vegetans is a rare variant of pemphigus vulgaris. Clinically, pemphigus vegetans is characterized by vegetative lesions over the flexures, but any area of the skin may be involved. There have been case reports involving the scalp,1,2 mouth,3 and foot.4 There are 2 clinical subtypes: the Neumann type and the Hallopeau type.5 The Hallopeau type is relatively benign, requires lower doses of systemic corticosteroids, and has a prolonged remission, while the Neumann type necessitates higher doses of systemic corticosteroids and often presents with relapses and remissions.
The diagnosis of pemphigus vegetans is based on clinical suspicion and confirmed by histological examination and immunological findings. The diagnosis may be difficult, as its presentation varies and histopathological findings may resemble other conditions.
Systemic corticosteroids are the well-established drug of choice for treating pemphigus vegetans to induce remission and maintain healing before cautiously tapering down the dosage approximately 50% every 2 weeks.6 Adjuvant drugs used in conjunction with steroids for steroid-sparing purpose include azathioprine, cyclophosphamide, mycophenolate mofetil, methotrexate, and cyclosporine.6 Pulsed intravenous steroids,7 intravenous immunoglobulins,8 pulsed dexamethasone cyclophosphamide,9 and extracorporeal photopheresis10 are given for severe and recalcitrant disease.
Laboratory investigations of our patient showed a normal complete blood cell count and a normal renal and liver profile. Herpes simplex virus serology was positive for type 1 and type 2 IgM and IgG. Urethral swab was dry and negative for gonorrhea. Serology for chlamydia, toxoplasma, amoebiasis, and leishmaniasis was negative. Human immunodeficiency virus serology, hepatitis screening, rapid plasma reagin, Treponema pallidum hemagglutination, rheumatoid factor, and antinuclear antibody all were negative. The patient was given a course of oral acyclovir 400 mg 3 times daily and empirical treatment with oral doxycycline 100 mg twice daily for a week with no clinical response.
Two biopsies from the perianal ulcers showed inflamed squamous papillomata with no Donovan bodies. A third biopsy from an intact blister showed acantholytic cells in the suprabasal bullae with eosinophilic and lymphocytic infiltrates at the upper dermis. Direct immunofluorescence demonstrated intercellular C3 and IgG deposits.
The patient was started on oral prednisolone at 1 mg/kg daily and oral azathioprine 50 mg daily with resolution of the perianal, penile, and oral ulcers (Figures 1 and 2). He achieved good suppression of further eruption. At the patient's most recent follow-up (2.5 years after the initial presentation), he was in remission and was currently taking oral azathioprine 100 mg once daily and no oral corticosteroids.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp [published online October 22,2014]. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Augusto de Oliveira M, Martins E Martins F, Lourenço S, et al. Oral pemphigus vegetans: a case report. Dermatol Online J. 2012;18:10.
- Ma DL, Fang K. Hallopeau type of pemphigus vegetans confined to the right foot: case report. Chin Med J (Engl). 2009;122:588-590.
- Ahmed AR, Blose DA. Pemphigus vegetans. Neumann type and Hallopeau type. Int J Dermatol. 1984;23:135-141.
- Harman KE, Albert S, Black MM, et al. Guidelines for the management of pemphigus vulgaris. Br J Dermatol. 2003;149:926-937.
- Chryssomallis F, Dimitriades A, Chaidemenos GC, et al. Steroid-pulse therapy in pemphigus vulgaris long term follow-up. Int J Dermatol. 1995;34:438-442.
- Ahmed AR. Intravenous immunoglobulin therapy in the treatment of patients with pemphigus vulgaris unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol. 2001;45:679-690.
- Pasricha JS, Khaitan BK, Raman RS, et al. Dexamethasone-cyclophosphamide pulse therapy for pemphigus. Int J Dermatol. 1995;34:875-882.
- Rook AH, Jegasothy BV, Heald P, et al. Extracorporeal photochemotherapy for drug-resistant pemphigus vulgaris. Ann Int Med. 1990;112:303-305.
- Danopoulou I, Stavropoulos P, Stratigos A, et al. Pemphigus vegetans confined to the scalp. Int J Dermatol. 2006;45:1008-1009.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp [published online October 22,2014]. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Augusto de Oliveira M, Martins E Martins F, Lourenço S, et al. Oral pemphigus vegetans: a case report. Dermatol Online J. 2012;18:10.
- Ma DL, Fang K. Hallopeau type of pemphigus vegetans confined to the right foot: case report. Chin Med J (Engl). 2009;122:588-590.
- Ahmed AR, Blose DA. Pemphigus vegetans. Neumann type and Hallopeau type. Int J Dermatol. 1984;23:135-141.
- Harman KE, Albert S, Black MM, et al. Guidelines for the management of pemphigus vulgaris. Br J Dermatol. 2003;149:926-937.
- Chryssomallis F, Dimitriades A, Chaidemenos GC, et al. Steroid-pulse therapy in pemphigus vulgaris long term follow-up. Int J Dermatol. 1995;34:438-442.
- Ahmed AR. Intravenous immunoglobulin therapy in the treatment of patients with pemphigus vulgaris unresponsive to conventional immunosuppressive treatment. J Am Acad Dermatol. 2001;45:679-690.
- Pasricha JS, Khaitan BK, Raman RS, et al. Dexamethasone-cyclophosphamide pulse therapy for pemphigus. Int J Dermatol. 1995;34:875-882.
- Rook AH, Jegasothy BV, Heald P, et al. Extracorporeal photochemotherapy for drug-resistant pemphigus vulgaris. Ann Int Med. 1990;112:303-305.
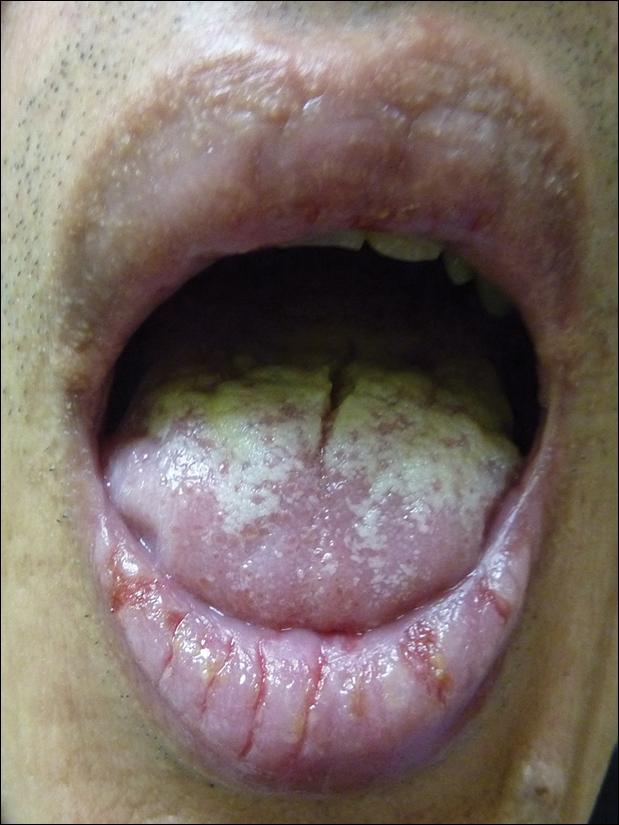
A 52-year-old man presented with persistent painful oral ulcers and penile and perianal erosions of 6 months' duration. He strongly denied engaging in high-risk sexual activities and had lost 10 kg over the last 6 months. He did not report taking any over-the-counter or alternative medications. On physical examination there were multiple fissures on the lower lip with erosive white plaques on the tongue and buccal mucosa. There were erosions over the foreskin and glans penis and a few erosive plaques on the perianal skin. Bilateral inguinal lymph nodes were enlarged.
Superficial Ulceration on the Vulva
Foscarnet-Induced Ulceration
Viral swabs were negative for herpes simplex virus. The diagnosis of foscarnet-induced ulceration was reached and the drug was discontinued. Symptomatic treatment with soap substitutes and lidocaine ointment was used.
Foscarnet is an antiviral agent used when resistance develops to first-line therapies.1 It is a pyrophosphate analogue that inhibits viral DNA polymerase, thereby preventing viral replication. It is used in treating cytomegalovirus and herpes simplex virus, which are resistant to first-line therapies, or patients who develop hematologic toxicity from antivirals. The main side effects of foscarnet include nephrotoxicity, alteration of calcium homeostasis, and malaise. Genital ulceration is a known side effect of therapy, though it is rare and more commonly seen in uncircumcised males. Approximately 94% of the drug is excreted unchanged in the urine, which causes an irritant dermatitis that is more pronounced in males as the urine stays in the subpreputial area.1
Vulval ulceration2 and penile ulceration3 has been reported in AIDS patients treated with foscarnet. In these patients, the onset of ulceration is temporally related to foscarnet therapy, occurring at approximately day 7 to 24 of treatment and resolving after discontinuation of therapy.
- Wagstaff AJ, Bryson HM. Foscarnet. a reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs. 1994;48:199-226.
- Lacey HB, Ness A, Mandal BK. Vulval ulceration associated with foscarnet. Genitourin Med. 1992;68:182.
- Moyle G, Barton S, Gazzard BG. Penile ulceration with foscarnet therapy. AIDS. 1993;7:140-141.
Foscarnet-Induced Ulceration
Viral swabs were negative for herpes simplex virus. The diagnosis of foscarnet-induced ulceration was reached and the drug was discontinued. Symptomatic treatment with soap substitutes and lidocaine ointment was used.
Foscarnet is an antiviral agent used when resistance develops to first-line therapies.1 It is a pyrophosphate analogue that inhibits viral DNA polymerase, thereby preventing viral replication. It is used in treating cytomegalovirus and herpes simplex virus, which are resistant to first-line therapies, or patients who develop hematologic toxicity from antivirals. The main side effects of foscarnet include nephrotoxicity, alteration of calcium homeostasis, and malaise. Genital ulceration is a known side effect of therapy, though it is rare and more commonly seen in uncircumcised males. Approximately 94% of the drug is excreted unchanged in the urine, which causes an irritant dermatitis that is more pronounced in males as the urine stays in the subpreputial area.1
Vulval ulceration2 and penile ulceration3 has been reported in AIDS patients treated with foscarnet. In these patients, the onset of ulceration is temporally related to foscarnet therapy, occurring at approximately day 7 to 24 of treatment and resolving after discontinuation of therapy.
Foscarnet-Induced Ulceration
Viral swabs were negative for herpes simplex virus. The diagnosis of foscarnet-induced ulceration was reached and the drug was discontinued. Symptomatic treatment with soap substitutes and lidocaine ointment was used.
Foscarnet is an antiviral agent used when resistance develops to first-line therapies.1 It is a pyrophosphate analogue that inhibits viral DNA polymerase, thereby preventing viral replication. It is used in treating cytomegalovirus and herpes simplex virus, which are resistant to first-line therapies, or patients who develop hematologic toxicity from antivirals. The main side effects of foscarnet include nephrotoxicity, alteration of calcium homeostasis, and malaise. Genital ulceration is a known side effect of therapy, though it is rare and more commonly seen in uncircumcised males. Approximately 94% of the drug is excreted unchanged in the urine, which causes an irritant dermatitis that is more pronounced in males as the urine stays in the subpreputial area.1
Vulval ulceration2 and penile ulceration3 has been reported in AIDS patients treated with foscarnet. In these patients, the onset of ulceration is temporally related to foscarnet therapy, occurring at approximately day 7 to 24 of treatment and resolving after discontinuation of therapy.
- Wagstaff AJ, Bryson HM. Foscarnet. a reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs. 1994;48:199-226.
- Lacey HB, Ness A, Mandal BK. Vulval ulceration associated with foscarnet. Genitourin Med. 1992;68:182.
- Moyle G, Barton S, Gazzard BG. Penile ulceration with foscarnet therapy. AIDS. 1993;7:140-141.
- Wagstaff AJ, Bryson HM. Foscarnet. a reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs. 1994;48:199-226.
- Lacey HB, Ness A, Mandal BK. Vulval ulceration associated with foscarnet. Genitourin Med. 1992;68:182.
- Moyle G, Barton S, Gazzard BG. Penile ulceration with foscarnet therapy. AIDS. 1993;7:140-141.
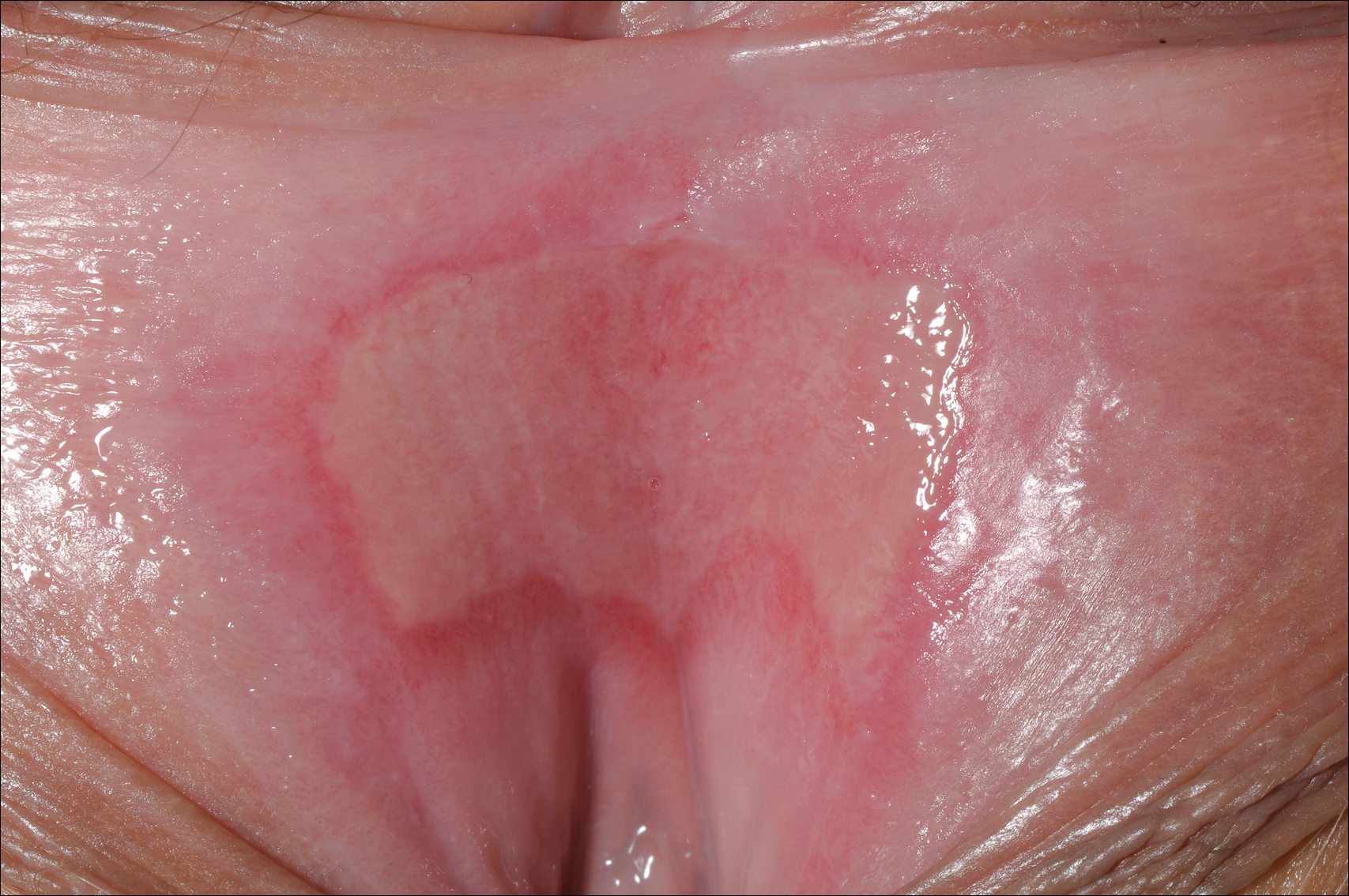
A 23-year-old woman who was immunosuppressed secondary to cyclophosphamide and prednisolone treatment of autoimmune panniculitis was admitted to intensive care with dyspnea. Cytomegalovirus and Pneumocystis jiroveci pneumonia were diagnosed on bronchoscopy and bronchial washings. Management with valganciclovir was started but worsened the patient's pancytopenia. She was started on intravenous foscarnet. After a week of therapy, the patient reported vulval soreness and painful micturition. On examination there was superficial ulceration of the labia minora. The affected area was symmetrical, and there was some extension into the vestibule. There were no vesicles or lesions on the cutaneous skin.
Purple Curvilinear Papules on the Back
The Diagnosis: Blaschkoid Graft-vs-host Disease
The patient had a history of myelodysplastic syndrome and underwent a bone marrow transplant 1 year prior to presentation. She had acute graft-vs-host disease (GVHD) 6 weeks following the transplant, which resolved with high-dose prednisone followed by UVB phototherapy. Skin biopsy demonstrated lichenoid dermatitis with vacuolar degeneration, dyskeratosis, and prominent pigment incontinence (Figure). Based on these findings and her clinical presentation, a diagnosis of blaschkoid GVHD was made.

Although acute GVHD is the result of immunocompetent donor T cells recognizing host tissues as foreign and initiating an immune response, the pathophysiology of chronic GVHD is not well understood.1,2 Theories for disease pathogenesis in chronic GVHD suggest an underlying autoimmune and/or alloreactive process.2-5 The skin often is the first organ affected in acute GVHD, and patients generally present with a pruritic morbilliform eruption that begins on the trunk and spreads to the rest of the body.1,2 Cutaneous manifestations of chronic GVHD may be protean. Lesions can resemble systemic sclerosis or morphea, lichen planus, psoriasis, ichthyosis, and many other conditions.2
The differential diagnosis of linear dermatoses includes herpes zoster, contact dermatitis, lichen striatus (blaschkitis), nevus unius lateris, inflammatory linear verrucous epidermal nevus, and incontinentia pigmenti.6,7 Lichen planus-like chronic GVHD occurring in a linear distribution has been described.6-14 Distinction between dermatomal and blaschkoid processes is diagnostically important. In the case of GVHD, dermatomal distribution may suggest an association between GVHD and prior herpes simplex virus or varicella-zoster virus infection.6,8 Herpesvirus may alter surface antigens of keratinocytes, rendering them targets of donor lymphocytes, and antibodies to viral particles may cross-react with host keratinocyte HLA antigens. It also is possible that dermatomal GVHD may simply be a type of isomorphic response (Köbner phenomenon).8
When cutaneous GVHD follows Blaschko lines, other mechanisms appear to be at play.9-14 It is plausible that these patients have an underlying genetic mosaicism, perhaps the result of a postzygotic mutation, that results in a daughter cell population that expresses surface antigens different from those of the primary cell population found elsewhere in the skin. Donor lymphocytes may selectively react to this mosaic population, leading to the clinical picture of chronic GVHD oriented along Blaschko lines.10,11,13,14
In conclusion, lichenoid linear GVHD following Blaschko lines is an uncommon presentation of chronic GVHD that highlights the heterogeneity of this disease and should be considered in the appropriate clinical setting.
- Ferrara JL, Levine JE, Reddy P, et al. Graft-versus-host disease. Lancet. 2009;373:1550-1561.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-515.e18; quiz 533-534.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
- Shimada M, Onizuka M, Machida S, et al. Association of autoimmune disease-related gene polymorphisms with chronic graft-versus-host disease. Br J Haematol. 2007;139:458-463.
- Zhang C, Todorov I, Zhang Z, et al. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood. 2006;107:2993-3001.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Kikuchi A, Okamoto S, Takahashi S, et al. Linear chronic cutaneous graft-versus-host disease. J Am Acad Dermatol. 1997;37:1004-1006.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Kennedy FE, Hilari H, Ferrer B, et al. Lichenoid chronic graft-vs-host disease following Blaschko lines. ActasDermosifiliogr. 2014;105:89-92.
- Lee SW, Kim YC, Lee E, et al. Linear lichenoid graft versus host disease: an unusual configuration following Blaschko's lines. J Dermatol. 2006;33:583-584.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson B, Lockman D. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130(9):1206-1208.
- Reisfeld PL. Lichenoid chronic graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Vassallo C, Derlino F, Ripamonti F, et al. Lichenoid cutaneous chronic GvHD following Blaschko lines. Int J Dermatol. 2014;53:473-475.
The Diagnosis: Blaschkoid Graft-vs-host Disease
The patient had a history of myelodysplastic syndrome and underwent a bone marrow transplant 1 year prior to presentation. She had acute graft-vs-host disease (GVHD) 6 weeks following the transplant, which resolved with high-dose prednisone followed by UVB phototherapy. Skin biopsy demonstrated lichenoid dermatitis with vacuolar degeneration, dyskeratosis, and prominent pigment incontinence (Figure). Based on these findings and her clinical presentation, a diagnosis of blaschkoid GVHD was made.
Although acute GVHD is the result of immunocompetent donor T cells recognizing host tissues as foreign and initiating an immune response, the pathophysiology of chronic GVHD is not well understood.1,2 Theories for disease pathogenesis in chronic GVHD suggest an underlying autoimmune and/or alloreactive process.2-5 The skin often is the first organ affected in acute GVHD, and patients generally present with a pruritic morbilliform eruption that begins on the trunk and spreads to the rest of the body.1,2 Cutaneous manifestations of chronic GVHD may be protean. Lesions can resemble systemic sclerosis or morphea, lichen planus, psoriasis, ichthyosis, and many other conditions.2
The differential diagnosis of linear dermatoses includes herpes zoster, contact dermatitis, lichen striatus (blaschkitis), nevus unius lateris, inflammatory linear verrucous epidermal nevus, and incontinentia pigmenti.6,7 Lichen planus-like chronic GVHD occurring in a linear distribution has been described.6-14 Distinction between dermatomal and blaschkoid processes is diagnostically important. In the case of GVHD, dermatomal distribution may suggest an association between GVHD and prior herpes simplex virus or varicella-zoster virus infection.6,8 Herpesvirus may alter surface antigens of keratinocytes, rendering them targets of donor lymphocytes, and antibodies to viral particles may cross-react with host keratinocyte HLA antigens. It also is possible that dermatomal GVHD may simply be a type of isomorphic response (Köbner phenomenon).8
When cutaneous GVHD follows Blaschko lines, other mechanisms appear to be at play.9-14 It is plausible that these patients have an underlying genetic mosaicism, perhaps the result of a postzygotic mutation, that results in a daughter cell population that expresses surface antigens different from those of the primary cell population found elsewhere in the skin. Donor lymphocytes may selectively react to this mosaic population, leading to the clinical picture of chronic GVHD oriented along Blaschko lines.10,11,13,14
In conclusion, lichenoid linear GVHD following Blaschko lines is an uncommon presentation of chronic GVHD that highlights the heterogeneity of this disease and should be considered in the appropriate clinical setting.
The Diagnosis: Blaschkoid Graft-vs-host Disease
The patient had a history of myelodysplastic syndrome and underwent a bone marrow transplant 1 year prior to presentation. She had acute graft-vs-host disease (GVHD) 6 weeks following the transplant, which resolved with high-dose prednisone followed by UVB phototherapy. Skin biopsy demonstrated lichenoid dermatitis with vacuolar degeneration, dyskeratosis, and prominent pigment incontinence (Figure). Based on these findings and her clinical presentation, a diagnosis of blaschkoid GVHD was made.
Although acute GVHD is the result of immunocompetent donor T cells recognizing host tissues as foreign and initiating an immune response, the pathophysiology of chronic GVHD is not well understood.1,2 Theories for disease pathogenesis in chronic GVHD suggest an underlying autoimmune and/or alloreactive process.2-5 The skin often is the first organ affected in acute GVHD, and patients generally present with a pruritic morbilliform eruption that begins on the trunk and spreads to the rest of the body.1,2 Cutaneous manifestations of chronic GVHD may be protean. Lesions can resemble systemic sclerosis or morphea, lichen planus, psoriasis, ichthyosis, and many other conditions.2
The differential diagnosis of linear dermatoses includes herpes zoster, contact dermatitis, lichen striatus (blaschkitis), nevus unius lateris, inflammatory linear verrucous epidermal nevus, and incontinentia pigmenti.6,7 Lichen planus-like chronic GVHD occurring in a linear distribution has been described.6-14 Distinction between dermatomal and blaschkoid processes is diagnostically important. In the case of GVHD, dermatomal distribution may suggest an association between GVHD and prior herpes simplex virus or varicella-zoster virus infection.6,8 Herpesvirus may alter surface antigens of keratinocytes, rendering them targets of donor lymphocytes, and antibodies to viral particles may cross-react with host keratinocyte HLA antigens. It also is possible that dermatomal GVHD may simply be a type of isomorphic response (Köbner phenomenon).8
When cutaneous GVHD follows Blaschko lines, other mechanisms appear to be at play.9-14 It is plausible that these patients have an underlying genetic mosaicism, perhaps the result of a postzygotic mutation, that results in a daughter cell population that expresses surface antigens different from those of the primary cell population found elsewhere in the skin. Donor lymphocytes may selectively react to this mosaic population, leading to the clinical picture of chronic GVHD oriented along Blaschko lines.10,11,13,14
In conclusion, lichenoid linear GVHD following Blaschko lines is an uncommon presentation of chronic GVHD that highlights the heterogeneity of this disease and should be considered in the appropriate clinical setting.
- Ferrara JL, Levine JE, Reddy P, et al. Graft-versus-host disease. Lancet. 2009;373:1550-1561.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-515.e18; quiz 533-534.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
- Shimada M, Onizuka M, Machida S, et al. Association of autoimmune disease-related gene polymorphisms with chronic graft-versus-host disease. Br J Haematol. 2007;139:458-463.
- Zhang C, Todorov I, Zhang Z, et al. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood. 2006;107:2993-3001.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Kikuchi A, Okamoto S, Takahashi S, et al. Linear chronic cutaneous graft-versus-host disease. J Am Acad Dermatol. 1997;37:1004-1006.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Kennedy FE, Hilari H, Ferrer B, et al. Lichenoid chronic graft-vs-host disease following Blaschko lines. ActasDermosifiliogr. 2014;105:89-92.
- Lee SW, Kim YC, Lee E, et al. Linear lichenoid graft versus host disease: an unusual configuration following Blaschko's lines. J Dermatol. 2006;33:583-584.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson B, Lockman D. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130(9):1206-1208.
- Reisfeld PL. Lichenoid chronic graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Vassallo C, Derlino F, Ripamonti F, et al. Lichenoid cutaneous chronic GvHD following Blaschko lines. Int J Dermatol. 2014;53:473-475.
- Ferrara JL, Levine JE, Reddy P, et al. Graft-versus-host disease. Lancet. 2009;373:1550-1561.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-515.e18; quiz 533-534.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
- Shimada M, Onizuka M, Machida S, et al. Association of autoimmune disease-related gene polymorphisms with chronic graft-versus-host disease. Br J Haematol. 2007;139:458-463.
- Zhang C, Todorov I, Zhang Z, et al. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood. 2006;107:2993-3001.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Kikuchi A, Okamoto S, Takahashi S, et al. Linear chronic cutaneous graft-versus-host disease. J Am Acad Dermatol. 1997;37:1004-1006.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Kennedy FE, Hilari H, Ferrer B, et al. Lichenoid chronic graft-vs-host disease following Blaschko lines. ActasDermosifiliogr. 2014;105:89-92.
- Lee SW, Kim YC, Lee E, et al. Linear lichenoid graft versus host disease: an unusual configuration following Blaschko's lines. J Dermatol. 2006;33:583-584.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson B, Lockman D. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130(9):1206-1208.
- Reisfeld PL. Lichenoid chronic graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Vassallo C, Derlino F, Ripamonti F, et al. Lichenoid cutaneous chronic GvHD following Blaschko lines. Int J Dermatol. 2014;53:473-475.
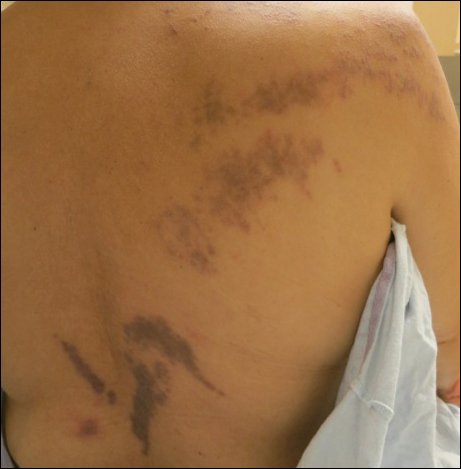
A 56-year-old woman with a history of bone marrow transplant presented for evaluation of a nonpruritic rash of 3 months' duration. Physical examination revealed confluent purple-colored and hyperpigmented papules localized to the back and right arm in a curvilinear pattern. Laboratory results were notable for mildly elevated aspartate transaminase and alanine transaminase levels.
Pruritic and Painful Nodules on the Tongue
The Diagnosis: Chronic Hyperplastic Candidiasis (Nodular Form)
Chronic hyperplastic candidiasis (CHC) is a rare form of oropharyngeal candidiasis. The most frequent clinical presentation is a white plaque that cannot be detached (also known as candidal leukoplakia). It usually involves the anterior buccal mucosa, mainly the commissural area, though the palate and tongue also can be affected. The nodular type of CHC is even less common. Our patient exhibited the typical clinical presentation of the nodular type of CHC.1-3 The differential diagnosis includes leukoplakia, premalignant and malignant epithelial lesions, granular cell tumor, and florid oral papillomatosis.1,3 A biopsy usually is required for diagnostic confirmation. Histologically, CHC is characterized by parakeratosis and a hyperplastic epithelium invaded by Candida hyphae.4 Because Candida species are commensal in up to 50% of the healthy population, superficial colonization of tissues is not enough to indicate notable disease.1 In our patient, the histopathology revealed a hyperplastic mucosa without atypia and numerous hyphae (Figure). Both lingual swab and tissue cultures revealed high growth of Candida albicans.
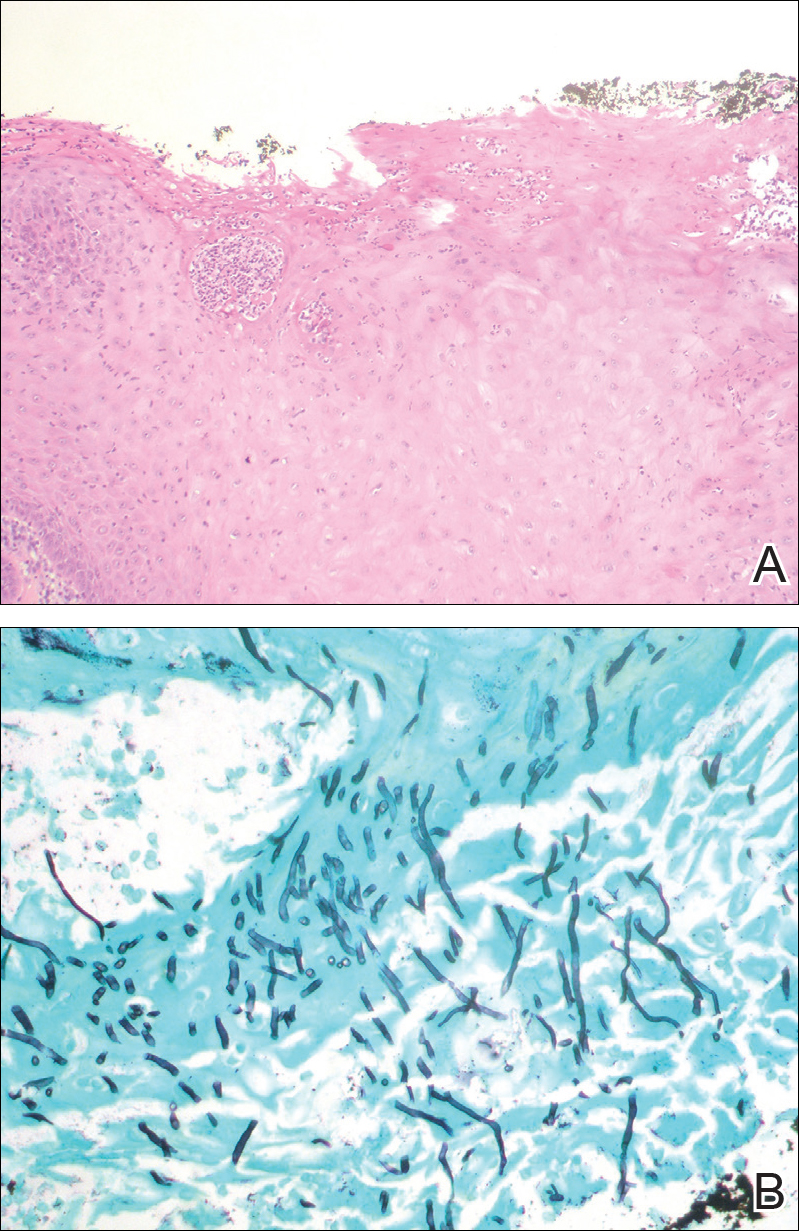
Infection by C albicans depends on pathogen virulence and host factors such as wearing dentures, reduced salivary production, smoking habit, or immunosuppression.1,4 Apart from wearing dentures, our patient did not present with other predisposing factors. It is possible that the immunosuppressive status related to old age and associated oral changes contributed to Candida infection in this case.
Topical or systemic antifungal agents together with the elimination of predisposing factors are usual first-line treatments. Because of the relationship with atypia and the possibility of evolving into carcinoma in untreated or persistent lesions, follow-up is necessary to verify complete resolution after treatment.1,3,4 In the case reported herein, the lesions disappeared after 15 days of oral fluconazole treatment.
- Shibata T, Yamashita D, Hasegawa S, et al. Oral candidiasis mimicking tongue cancer [published online January 12, 2011]. Auris Nasus Larynx. 2011;38:418-420.
- Scardina GA, Ruggieri A, Messina P. Chronic hyperplastic candidosis: a pilot study of the efficacy of 0.18% isotretinoin. J Oral Sci. 2009;51:407-410.
- Sitheeque MA, Samaranayake LP. Chronic hyperplastic candidosis/candidiasis (candidal leukoplakia). Crit Rev Oral Biol Med. 2003;14:253-267.
- Williams DW, Bartie KL, Potts AJ, et al. Strain persistence of invasive Candida albicans in chronic hyperplastic candidosis that underwent malignant change. Gerodontology. 2001;18:73-78.
The Diagnosis: Chronic Hyperplastic Candidiasis (Nodular Form)
Chronic hyperplastic candidiasis (CHC) is a rare form of oropharyngeal candidiasis. The most frequent clinical presentation is a white plaque that cannot be detached (also known as candidal leukoplakia). It usually involves the anterior buccal mucosa, mainly the commissural area, though the palate and tongue also can be affected. The nodular type of CHC is even less common. Our patient exhibited the typical clinical presentation of the nodular type of CHC.1-3 The differential diagnosis includes leukoplakia, premalignant and malignant epithelial lesions, granular cell tumor, and florid oral papillomatosis.1,3 A biopsy usually is required for diagnostic confirmation. Histologically, CHC is characterized by parakeratosis and a hyperplastic epithelium invaded by Candida hyphae.4 Because Candida species are commensal in up to 50% of the healthy population, superficial colonization of tissues is not enough to indicate notable disease.1 In our patient, the histopathology revealed a hyperplastic mucosa without atypia and numerous hyphae (Figure). Both lingual swab and tissue cultures revealed high growth of Candida albicans.
Infection by C albicans depends on pathogen virulence and host factors such as wearing dentures, reduced salivary production, smoking habit, or immunosuppression.1,4 Apart from wearing dentures, our patient did not present with other predisposing factors. It is possible that the immunosuppressive status related to old age and associated oral changes contributed to Candida infection in this case.
Topical or systemic antifungal agents together with the elimination of predisposing factors are usual first-line treatments. Because of the relationship with atypia and the possibility of evolving into carcinoma in untreated or persistent lesions, follow-up is necessary to verify complete resolution after treatment.1,3,4 In the case reported herein, the lesions disappeared after 15 days of oral fluconazole treatment.
The Diagnosis: Chronic Hyperplastic Candidiasis (Nodular Form)
Chronic hyperplastic candidiasis (CHC) is a rare form of oropharyngeal candidiasis. The most frequent clinical presentation is a white plaque that cannot be detached (also known as candidal leukoplakia). It usually involves the anterior buccal mucosa, mainly the commissural area, though the palate and tongue also can be affected. The nodular type of CHC is even less common. Our patient exhibited the typical clinical presentation of the nodular type of CHC.1-3 The differential diagnosis includes leukoplakia, premalignant and malignant epithelial lesions, granular cell tumor, and florid oral papillomatosis.1,3 A biopsy usually is required for diagnostic confirmation. Histologically, CHC is characterized by parakeratosis and a hyperplastic epithelium invaded by Candida hyphae.4 Because Candida species are commensal in up to 50% of the healthy population, superficial colonization of tissues is not enough to indicate notable disease.1 In our patient, the histopathology revealed a hyperplastic mucosa without atypia and numerous hyphae (Figure). Both lingual swab and tissue cultures revealed high growth of Candida albicans.
Infection by C albicans depends on pathogen virulence and host factors such as wearing dentures, reduced salivary production, smoking habit, or immunosuppression.1,4 Apart from wearing dentures, our patient did not present with other predisposing factors. It is possible that the immunosuppressive status related to old age and associated oral changes contributed to Candida infection in this case.
Topical or systemic antifungal agents together with the elimination of predisposing factors are usual first-line treatments. Because of the relationship with atypia and the possibility of evolving into carcinoma in untreated or persistent lesions, follow-up is necessary to verify complete resolution after treatment.1,3,4 In the case reported herein, the lesions disappeared after 15 days of oral fluconazole treatment.
- Shibata T, Yamashita D, Hasegawa S, et al. Oral candidiasis mimicking tongue cancer [published online January 12, 2011]. Auris Nasus Larynx. 2011;38:418-420.
- Scardina GA, Ruggieri A, Messina P. Chronic hyperplastic candidosis: a pilot study of the efficacy of 0.18% isotretinoin. J Oral Sci. 2009;51:407-410.
- Sitheeque MA, Samaranayake LP. Chronic hyperplastic candidosis/candidiasis (candidal leukoplakia). Crit Rev Oral Biol Med. 2003;14:253-267.
- Williams DW, Bartie KL, Potts AJ, et al. Strain persistence of invasive Candida albicans in chronic hyperplastic candidosis that underwent malignant change. Gerodontology. 2001;18:73-78.
- Shibata T, Yamashita D, Hasegawa S, et al. Oral candidiasis mimicking tongue cancer [published online January 12, 2011]. Auris Nasus Larynx. 2011;38:418-420.
- Scardina GA, Ruggieri A, Messina P. Chronic hyperplastic candidosis: a pilot study of the efficacy of 0.18% isotretinoin. J Oral Sci. 2009;51:407-410.
- Sitheeque MA, Samaranayake LP. Chronic hyperplastic candidosis/candidiasis (candidal leukoplakia). Crit Rev Oral Biol Med. 2003;14:253-267.
- Williams DW, Bartie KL, Potts AJ, et al. Strain persistence of invasive Candida albicans in chronic hyperplastic candidosis that underwent malignant change. Gerodontology. 2001;18:73-78.

An 82-year-old woman with atrial fibrillation and chronic obstructive pulmonary disease presented with pruritic and painful lesions on the tongue of 10 years' duration. She had not undergone treatment with systemic or inhaled corticosteroids during the course of the pulmonary disease. On physical examination, several fleshy and well-defined erythematous papules speckled with whitish areas were observed on the dorsal aspect and anterior border of the tongue. Superficial whitish areas could not be removed by scraping.
Firm Pink Nodule on the Scalp
The Diagnosis: Metastatic Renal Cell Carcinoma
A shave biopsy of the right occipital scalp lesion demonstrated large clear cells arranged in oval nests with mildly atypical central nuclei (Figure). The findings were consistent with the clear cell type of metastatic renal cell carcinoma (mRCC) with tumor involvement in the deep margin. PAX8 immunostain was positive in the tumor, further supporting the diagnosis of mRCC.
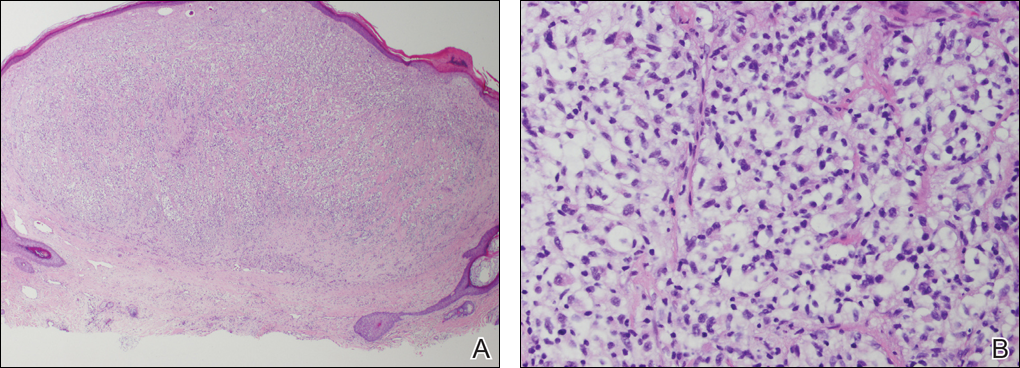
The most common patient demographic diagnosed with renal cell carcinoma (RCC) is men in the sixth and seventh decades of life.1,2 The classic presentation of RCC includes the triad of flank pain, hematuria, and a palpable abdominal mass. Other symptoms may include fatigue, weight loss, and anemia; however, small localized tumors rarely are symptomatic, and less than 10% of patients with RCC present with this classic triad.3 Many RCCs are diagnosed from incidental findings on abdominal imaging or from the presentation of metastatic disease.1,2 The lungs, bones, and liver are the most common sites of metastases, while skin metastases are rare.2 One study found only 3.3% (10/306) of RCC cases had cutaneous metastases and the scalp was the most common location. In half of these patients, the skin metastases were present at initial RCC diagnosis.4
The most common presentation of a cutaneous metastasis of RCC entails the rapid development of a reddish blue nodule on the face or scalp.4,5 The differential diagnosis may include basal cell carcinoma, hemangioma, cutaneous angiosarcoma, pyogenic granuloma, and atypical fibroxanthoma. Careful elicitation of a medical history indicating any of the classic or systemic signs associated with RCC or a personal history of RCC should raise the suspicion for mRCC and prompt a biopsy.
Clear cell RCC is the most common type of primary RCC and 81% of mRCCs were found to be of the clear cell type. Clear cell RCC classically exhibits clear ballooned cytoplasm with distinct cell borders.6 Immunohistochemistry stains with PAX2 and PAX8 are helpful in diagnosing the renal origin of the metastatic tissue, with PAX8 being especially helpful (89% sensitivity).7
The von Hippel-Lindau gene, VHL, is involved in up to 60% of sporadic clear cell RCC cases, in addition to its involvement in VHL syndrome.1 von Hippel-Lindau syndrome is associated with clear cell RCC, retinal angiomas, hemangioblastomas of the central nervous system, and pheochromocytomas.1 The mutated VHL gene is associated with an increased expression of vascular endothelial growth factor (VEGF), which promotes angiogenesis, endothelial mutagenesis, and vascular permeability.8 These physiologic responses are likely responsible for the proliferation and dissemination of neoplastic cells in clear cell RCC.9 The understanding of this pathogenic mechanism has led to the development of targeted effective treatments in RCC.
Systemic treatments for mRCC are rapidly evolving and improving.9 Vascular endothelial growth factor tyrosine kinase inhibitors such as sorafenib, sunitinib, and pazopanib, as well as the VEGF monoclonal antibody bevacizumab, have all demonstrated notable efficacy in the treatment of RCC. Phase 3 clinical trials for the treatment of mRCC have demonstrated that the new, more biochemically potent VEGF tyrosine kinase inhibitor axitinib has superior efficacy to sorafenib, the prior standard of care.9 Our patient noted that the cutaneous mRCC lesion seemed to be improving after starting treatment with axitinib 2 months prior to presentation. In addition to systemic chemotherapy, the surgical removal of lesions often is indicated for the treatment of cutaneous mRCC.5,10 Our patient has continued axitinib and is doing well.
- Cohen HT, McGovern FJ. Medical progress: renal-cell carcinoma. N Engl J Med. 2005;353:2477-2490.
- Schlesinger-Raab A, Treiber U, Zaak D, et al. Metastatic renal cell carcinoma: results of a population-based study with 25 years follow-up. Eur J Cancer. 2008;44:2485-2495.
- Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. N Engl J Med. 1996;335:865-875.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Arrabal-Polo MA, Arias-Santiago SA, Aneiros-Fernandez J, et al. Cutaneous metastases in renal cell carcinoma: a case report. Cases J. 2009;2:7948.
- Mai KT, Alhalouly T, Lamba M, et al. Distribution of subtypes of metastatic renal-cell carcinoma: correlating findings of fine-needle aspiration biopsy and surgical pathology. Diagn Cytopathol. 2003;28:66-70.
- Ozcan A, Roza F, Ro JY, et al. PAX2 and PAX8 expression in primary and metastatic renal tumors: a comprehensive comparison. Arch Pathol Lab Med. 2012;136:1541-1551.
- Albiges L, Salem M, Rini B, et al. Vascular endothelial growth factor-targeted therapies in advanced renal cell carcinoma. Hematol Oncol Clin North Am. 2011;25:813-833.
- Mittal K, Wood LS, Rini BI. Axitinib in metastatic renal cell carcinoma. Biol Ther. 2012;2:5.
- Kassam K, Tiong E, Nigar E, et al. Exophytic parietal skin metastases of renal cell carcinoma [published online December 26, 2013]. Case Rep Dermatol Med. 2013;2013:196016.
The Diagnosis: Metastatic Renal Cell Carcinoma
A shave biopsy of the right occipital scalp lesion demonstrated large clear cells arranged in oval nests with mildly atypical central nuclei (Figure). The findings were consistent with the clear cell type of metastatic renal cell carcinoma (mRCC) with tumor involvement in the deep margin. PAX8 immunostain was positive in the tumor, further supporting the diagnosis of mRCC.
The most common patient demographic diagnosed with renal cell carcinoma (RCC) is men in the sixth and seventh decades of life.1,2 The classic presentation of RCC includes the triad of flank pain, hematuria, and a palpable abdominal mass. Other symptoms may include fatigue, weight loss, and anemia; however, small localized tumors rarely are symptomatic, and less than 10% of patients with RCC present with this classic triad.3 Many RCCs are diagnosed from incidental findings on abdominal imaging or from the presentation of metastatic disease.1,2 The lungs, bones, and liver are the most common sites of metastases, while skin metastases are rare.2 One study found only 3.3% (10/306) of RCC cases had cutaneous metastases and the scalp was the most common location. In half of these patients, the skin metastases were present at initial RCC diagnosis.4
The most common presentation of a cutaneous metastasis of RCC entails the rapid development of a reddish blue nodule on the face or scalp.4,5 The differential diagnosis may include basal cell carcinoma, hemangioma, cutaneous angiosarcoma, pyogenic granuloma, and atypical fibroxanthoma. Careful elicitation of a medical history indicating any of the classic or systemic signs associated with RCC or a personal history of RCC should raise the suspicion for mRCC and prompt a biopsy.
Clear cell RCC is the most common type of primary RCC and 81% of mRCCs were found to be of the clear cell type. Clear cell RCC classically exhibits clear ballooned cytoplasm with distinct cell borders.6 Immunohistochemistry stains with PAX2 and PAX8 are helpful in diagnosing the renal origin of the metastatic tissue, with PAX8 being especially helpful (89% sensitivity).7
The von Hippel-Lindau gene, VHL, is involved in up to 60% of sporadic clear cell RCC cases, in addition to its involvement in VHL syndrome.1 von Hippel-Lindau syndrome is associated with clear cell RCC, retinal angiomas, hemangioblastomas of the central nervous system, and pheochromocytomas.1 The mutated VHL gene is associated with an increased expression of vascular endothelial growth factor (VEGF), which promotes angiogenesis, endothelial mutagenesis, and vascular permeability.8 These physiologic responses are likely responsible for the proliferation and dissemination of neoplastic cells in clear cell RCC.9 The understanding of this pathogenic mechanism has led to the development of targeted effective treatments in RCC.
Systemic treatments for mRCC are rapidly evolving and improving.9 Vascular endothelial growth factor tyrosine kinase inhibitors such as sorafenib, sunitinib, and pazopanib, as well as the VEGF monoclonal antibody bevacizumab, have all demonstrated notable efficacy in the treatment of RCC. Phase 3 clinical trials for the treatment of mRCC have demonstrated that the new, more biochemically potent VEGF tyrosine kinase inhibitor axitinib has superior efficacy to sorafenib, the prior standard of care.9 Our patient noted that the cutaneous mRCC lesion seemed to be improving after starting treatment with axitinib 2 months prior to presentation. In addition to systemic chemotherapy, the surgical removal of lesions often is indicated for the treatment of cutaneous mRCC.5,10 Our patient has continued axitinib and is doing well.
The Diagnosis: Metastatic Renal Cell Carcinoma
A shave biopsy of the right occipital scalp lesion demonstrated large clear cells arranged in oval nests with mildly atypical central nuclei (Figure). The findings were consistent with the clear cell type of metastatic renal cell carcinoma (mRCC) with tumor involvement in the deep margin. PAX8 immunostain was positive in the tumor, further supporting the diagnosis of mRCC.
The most common patient demographic diagnosed with renal cell carcinoma (RCC) is men in the sixth and seventh decades of life.1,2 The classic presentation of RCC includes the triad of flank pain, hematuria, and a palpable abdominal mass. Other symptoms may include fatigue, weight loss, and anemia; however, small localized tumors rarely are symptomatic, and less than 10% of patients with RCC present with this classic triad.3 Many RCCs are diagnosed from incidental findings on abdominal imaging or from the presentation of metastatic disease.1,2 The lungs, bones, and liver are the most common sites of metastases, while skin metastases are rare.2 One study found only 3.3% (10/306) of RCC cases had cutaneous metastases and the scalp was the most common location. In half of these patients, the skin metastases were present at initial RCC diagnosis.4
The most common presentation of a cutaneous metastasis of RCC entails the rapid development of a reddish blue nodule on the face or scalp.4,5 The differential diagnosis may include basal cell carcinoma, hemangioma, cutaneous angiosarcoma, pyogenic granuloma, and atypical fibroxanthoma. Careful elicitation of a medical history indicating any of the classic or systemic signs associated with RCC or a personal history of RCC should raise the suspicion for mRCC and prompt a biopsy.
Clear cell RCC is the most common type of primary RCC and 81% of mRCCs were found to be of the clear cell type. Clear cell RCC classically exhibits clear ballooned cytoplasm with distinct cell borders.6 Immunohistochemistry stains with PAX2 and PAX8 are helpful in diagnosing the renal origin of the metastatic tissue, with PAX8 being especially helpful (89% sensitivity).7
The von Hippel-Lindau gene, VHL, is involved in up to 60% of sporadic clear cell RCC cases, in addition to its involvement in VHL syndrome.1 von Hippel-Lindau syndrome is associated with clear cell RCC, retinal angiomas, hemangioblastomas of the central nervous system, and pheochromocytomas.1 The mutated VHL gene is associated with an increased expression of vascular endothelial growth factor (VEGF), which promotes angiogenesis, endothelial mutagenesis, and vascular permeability.8 These physiologic responses are likely responsible for the proliferation and dissemination of neoplastic cells in clear cell RCC.9 The understanding of this pathogenic mechanism has led to the development of targeted effective treatments in RCC.
Systemic treatments for mRCC are rapidly evolving and improving.9 Vascular endothelial growth factor tyrosine kinase inhibitors such as sorafenib, sunitinib, and pazopanib, as well as the VEGF monoclonal antibody bevacizumab, have all demonstrated notable efficacy in the treatment of RCC. Phase 3 clinical trials for the treatment of mRCC have demonstrated that the new, more biochemically potent VEGF tyrosine kinase inhibitor axitinib has superior efficacy to sorafenib, the prior standard of care.9 Our patient noted that the cutaneous mRCC lesion seemed to be improving after starting treatment with axitinib 2 months prior to presentation. In addition to systemic chemotherapy, the surgical removal of lesions often is indicated for the treatment of cutaneous mRCC.5,10 Our patient has continued axitinib and is doing well.
- Cohen HT, McGovern FJ. Medical progress: renal-cell carcinoma. N Engl J Med. 2005;353:2477-2490.
- Schlesinger-Raab A, Treiber U, Zaak D, et al. Metastatic renal cell carcinoma: results of a population-based study with 25 years follow-up. Eur J Cancer. 2008;44:2485-2495.
- Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. N Engl J Med. 1996;335:865-875.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Arrabal-Polo MA, Arias-Santiago SA, Aneiros-Fernandez J, et al. Cutaneous metastases in renal cell carcinoma: a case report. Cases J. 2009;2:7948.
- Mai KT, Alhalouly T, Lamba M, et al. Distribution of subtypes of metastatic renal-cell carcinoma: correlating findings of fine-needle aspiration biopsy and surgical pathology. Diagn Cytopathol. 2003;28:66-70.
- Ozcan A, Roza F, Ro JY, et al. PAX2 and PAX8 expression in primary and metastatic renal tumors: a comprehensive comparison. Arch Pathol Lab Med. 2012;136:1541-1551.
- Albiges L, Salem M, Rini B, et al. Vascular endothelial growth factor-targeted therapies in advanced renal cell carcinoma. Hematol Oncol Clin North Am. 2011;25:813-833.
- Mittal K, Wood LS, Rini BI. Axitinib in metastatic renal cell carcinoma. Biol Ther. 2012;2:5.
- Kassam K, Tiong E, Nigar E, et al. Exophytic parietal skin metastases of renal cell carcinoma [published online December 26, 2013]. Case Rep Dermatol Med. 2013;2013:196016.
- Cohen HT, McGovern FJ. Medical progress: renal-cell carcinoma. N Engl J Med. 2005;353:2477-2490.
- Schlesinger-Raab A, Treiber U, Zaak D, et al. Metastatic renal cell carcinoma: results of a population-based study with 25 years follow-up. Eur J Cancer. 2008;44:2485-2495.
- Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. N Engl J Med. 1996;335:865-875.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Arrabal-Polo MA, Arias-Santiago SA, Aneiros-Fernandez J, et al. Cutaneous metastases in renal cell carcinoma: a case report. Cases J. 2009;2:7948.
- Mai KT, Alhalouly T, Lamba M, et al. Distribution of subtypes of metastatic renal-cell carcinoma: correlating findings of fine-needle aspiration biopsy and surgical pathology. Diagn Cytopathol. 2003;28:66-70.
- Ozcan A, Roza F, Ro JY, et al. PAX2 and PAX8 expression in primary and metastatic renal tumors: a comprehensive comparison. Arch Pathol Lab Med. 2012;136:1541-1551.
- Albiges L, Salem M, Rini B, et al. Vascular endothelial growth factor-targeted therapies in advanced renal cell carcinoma. Hematol Oncol Clin North Am. 2011;25:813-833.
- Mittal K, Wood LS, Rini BI. Axitinib in metastatic renal cell carcinoma. Biol Ther. 2012;2:5.
- Kassam K, Tiong E, Nigar E, et al. Exophytic parietal skin metastases of renal cell carcinoma [published online December 26, 2013]. Case Rep Dermatol Med. 2013;2013:196016.
A 69-year-old man with stage IV renal cell carcinoma (RCC) but no history of skin cancer presented with a nodule on the right side of the posterior scalp of 2 months' duration. He noted that the lesion bled intermittently and crusted over. Three years prior, the patient had a left-sided nephrectomy, which showed a 7-cm tumor consistent with clear cell RCC (Fuhrman nuclear grade 2). Over the last 6 months, RCC metastases to the right kidney, lungs, and right cervical lymph nodes were found. His current chemotherapy treatment was axitinib; he was previously on pazopanib and everolimus. Physical examination revealed a 10×10-mm, pink-yellow, firm nodule with overlying telangiectases on the right side of the posterior scalp.
Diffuse Rash With Associated Ulceration
The Diagnosis: Epidermotropic CD8+ T-Cell Lymphoma
Epidermotropic CD8+ T-cell lymphoma is a rare aggressive form of cutaneous T-cell lymphoma (CTCL), accounting for less than 1% of all cases.1 Since this subtype of CTCL was first described in 1999 by Berti et al,2 approximately 45 cases have been reported in the literature.1 It typically is found in elderly men and presents as disseminated or localized papules, patches, plaques, nodules, and tumors, often with central necrosis, ulceration, crusting, and hemorrhage (Figure 1).1,3 These lesions rapidly progress and can affect any skin site, but acral accentuation and mucosal involvement are common.4 Due to the rapidly progressive nature of this disease, patients typically present with widespread plaque- and tumor-stage disease.3 Frequency of systemic spread is high, with metastasis to the central nervous system, lungs, and testes being most common. Lymph nodes typically are spared, helping to differentiate this form of CTCL from classic mycosis fungoides.
Diagnosis of epidermotropic CD8+ T-cell lymphoma is based on a combination of clinical, histopathologic, and immunohistochemical features. Histopathologic components include epidermotropism, particularly in the basal cell layer, in a pagetoid or linear pattern. A second feature is a dermal infiltrate consisting of a nodular or diffuse pattern of atypical lymphocytes that extend to the subcutaneous fat (Figure 2). All cases of epidermotropic CD8+ T-cell lymphoma express the CD8+ phenotype and most have a high Ki-67 proliferation index and are CD3, CD45RA, and/or T-cell intracellular antigen 1 positive.1
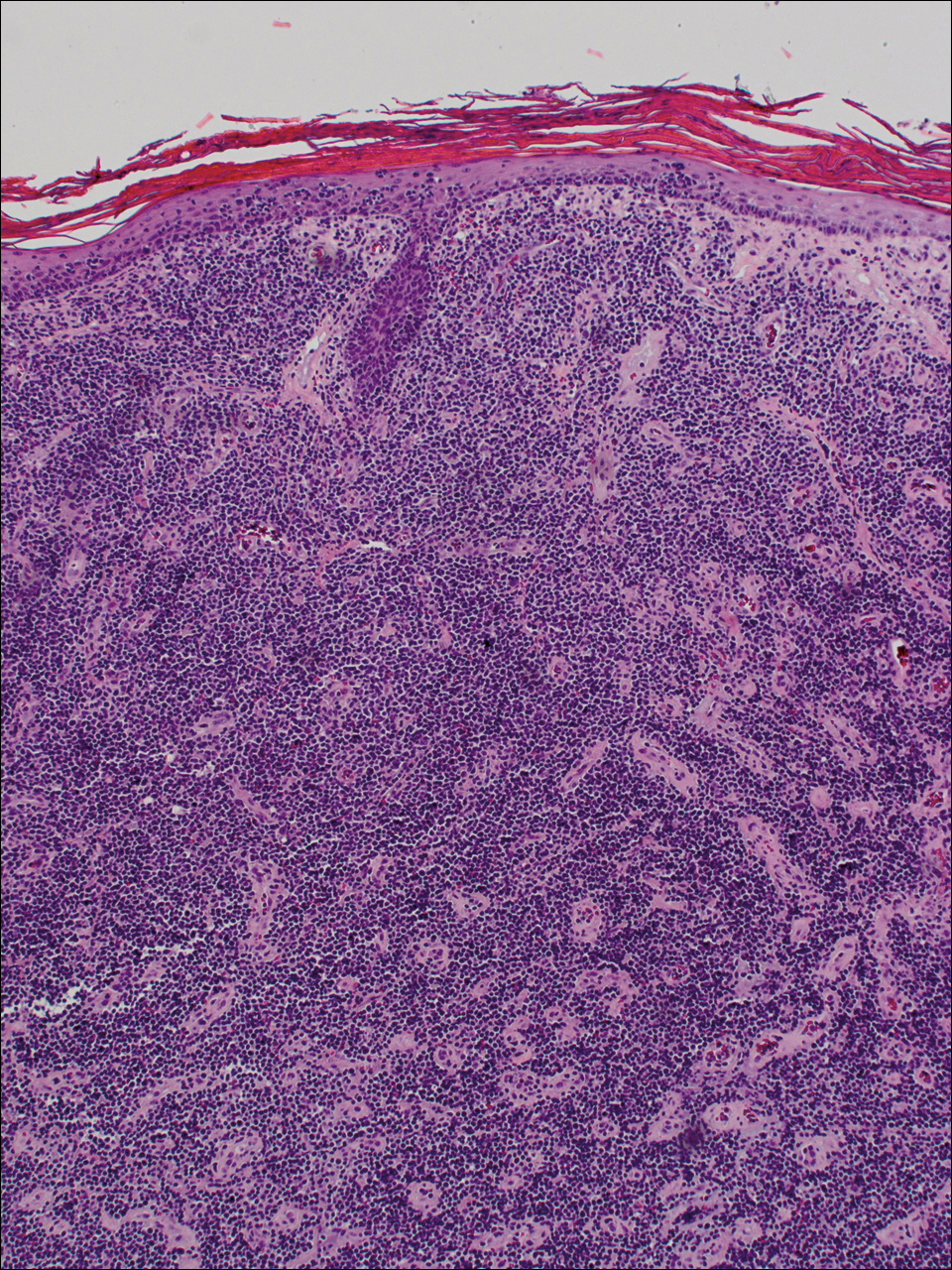
Due to its aggressive nature, epidermotropic CD8+ T-cell lymphoma has a poor prognosis, with an average 5-year survival rate of 18% and median survival of 22.5 months.3 Treatment proves difficult as conventional therapies for CD4+ CTCL have proven ineffective for epidermotropic CD8+ T-cell lymphoma. Partial response has been seen with bexarotene alone and with total skin electron beam therapy combined with oral retinoids.1
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: proposed diagnostic criteria and therapeutic evaluation. J Am Acad Dermatol. 2012;67:748-759.
- Berti E, Tomasini D, Vermeer MH, et al. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas. a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483-492.
- Gormley RH, Hess SD, Anand D, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma. J Am Acad Dermatol. 2010;62:300-307.
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T cell lymphoma: a diagnostic and therapeutic challenge. Int J Dermatol. 2014;53:76-81.
The Diagnosis: Epidermotropic CD8+ T-Cell Lymphoma
Epidermotropic CD8+ T-cell lymphoma is a rare aggressive form of cutaneous T-cell lymphoma (CTCL), accounting for less than 1% of all cases.1 Since this subtype of CTCL was first described in 1999 by Berti et al,2 approximately 45 cases have been reported in the literature.1 It typically is found in elderly men and presents as disseminated or localized papules, patches, plaques, nodules, and tumors, often with central necrosis, ulceration, crusting, and hemorrhage (Figure 1).1,3 These lesions rapidly progress and can affect any skin site, but acral accentuation and mucosal involvement are common.4 Due to the rapidly progressive nature of this disease, patients typically present with widespread plaque- and tumor-stage disease.3 Frequency of systemic spread is high, with metastasis to the central nervous system, lungs, and testes being most common. Lymph nodes typically are spared, helping to differentiate this form of CTCL from classic mycosis fungoides.
Diagnosis of epidermotropic CD8+ T-cell lymphoma is based on a combination of clinical, histopathologic, and immunohistochemical features. Histopathologic components include epidermotropism, particularly in the basal cell layer, in a pagetoid or linear pattern. A second feature is a dermal infiltrate consisting of a nodular or diffuse pattern of atypical lymphocytes that extend to the subcutaneous fat (Figure 2). All cases of epidermotropic CD8+ T-cell lymphoma express the CD8+ phenotype and most have a high Ki-67 proliferation index and are CD3, CD45RA, and/or T-cell intracellular antigen 1 positive.1
Due to its aggressive nature, epidermotropic CD8+ T-cell lymphoma has a poor prognosis, with an average 5-year survival rate of 18% and median survival of 22.5 months.3 Treatment proves difficult as conventional therapies for CD4+ CTCL have proven ineffective for epidermotropic CD8+ T-cell lymphoma. Partial response has been seen with bexarotene alone and with total skin electron beam therapy combined with oral retinoids.1
The Diagnosis: Epidermotropic CD8+ T-Cell Lymphoma
Epidermotropic CD8+ T-cell lymphoma is a rare aggressive form of cutaneous T-cell lymphoma (CTCL), accounting for less than 1% of all cases.1 Since this subtype of CTCL was first described in 1999 by Berti et al,2 approximately 45 cases have been reported in the literature.1 It typically is found in elderly men and presents as disseminated or localized papules, patches, plaques, nodules, and tumors, often with central necrosis, ulceration, crusting, and hemorrhage (Figure 1).1,3 These lesions rapidly progress and can affect any skin site, but acral accentuation and mucosal involvement are common.4 Due to the rapidly progressive nature of this disease, patients typically present with widespread plaque- and tumor-stage disease.3 Frequency of systemic spread is high, with metastasis to the central nervous system, lungs, and testes being most common. Lymph nodes typically are spared, helping to differentiate this form of CTCL from classic mycosis fungoides.
Diagnosis of epidermotropic CD8+ T-cell lymphoma is based on a combination of clinical, histopathologic, and immunohistochemical features. Histopathologic components include epidermotropism, particularly in the basal cell layer, in a pagetoid or linear pattern. A second feature is a dermal infiltrate consisting of a nodular or diffuse pattern of atypical lymphocytes that extend to the subcutaneous fat (Figure 2). All cases of epidermotropic CD8+ T-cell lymphoma express the CD8+ phenotype and most have a high Ki-67 proliferation index and are CD3, CD45RA, and/or T-cell intracellular antigen 1 positive.1
Due to its aggressive nature, epidermotropic CD8+ T-cell lymphoma has a poor prognosis, with an average 5-year survival rate of 18% and median survival of 22.5 months.3 Treatment proves difficult as conventional therapies for CD4+ CTCL have proven ineffective for epidermotropic CD8+ T-cell lymphoma. Partial response has been seen with bexarotene alone and with total skin electron beam therapy combined with oral retinoids.1
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: proposed diagnostic criteria and therapeutic evaluation. J Am Acad Dermatol. 2012;67:748-759.
- Berti E, Tomasini D, Vermeer MH, et al. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas. a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483-492.
- Gormley RH, Hess SD, Anand D, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma. J Am Acad Dermatol. 2010;62:300-307.
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T cell lymphoma: a diagnostic and therapeutic challenge. Int J Dermatol. 2014;53:76-81.
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: proposed diagnostic criteria and therapeutic evaluation. J Am Acad Dermatol. 2012;67:748-759.
- Berti E, Tomasini D, Vermeer MH, et al. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas. a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483-492.
- Gormley RH, Hess SD, Anand D, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma. J Am Acad Dermatol. 2010;62:300-307.
- Nofal A, Abdel-Mawla MY, Assaf M, et al. Primary cutaneous aggressive epidermotropic CD8+ T cell lymphoma: a diagnostic and therapeutic challenge. Int J Dermatol. 2014;53:76-81.
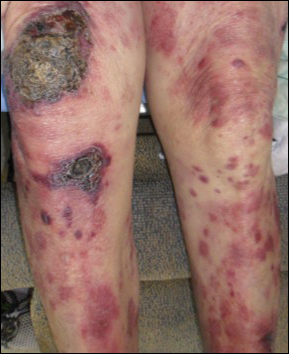
A 72-year-old woman who was admitted for pneumonia and acute hypoxic respiratory failure was seen for an inpatient consultation for a diffuse rash with associated ulceration. She reported a rash of 20 months' duration that began on the legs and then spread to the trunk, arms, head, and neck with minimal pruritus and no pain or photosensitivity. She had been treated with hydroxychloroquine, mycophenolate mofetil, and prednisone without improvement. The patient noted recent ulceration on the rash. Physical examination revealed violaceous patches, plaques, nodules, and tumors with rare ulceration involving the face, trunk, and extremities. Biopsy showed a diffuse infiltration of the dermis with medium-sized atypical lymphocytes with scant cytoplasm and round to irregular hyperchromatic nuclei with clumped chromatin. Epidermotropism with small collections of atypical lymphocytes also was present within the epidermis.
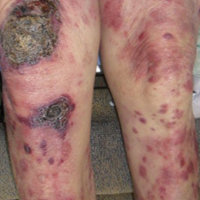
Discharging Nodule on the Jaw
The Diagnosis: Dental Sinus Secondary to Osteonecrosis of the Jaw
Cone beam computed tomography revealed an area of lucency measuring 40×20 mm in the body of the right mandible (Figure). The patient subsequently underwent curettage of the wound with sequestrectomy of the involved area.
Osteonecrosis of the jaw is a form of avascular necrosis. It is an uncommon but potentially serious side effect of bisphosphonate use.1 Bisphosphonates commonly are used as first-line therapy for osteoporosis, with proven efficacy to reduce fracture risk by exerting an antiresorptive effect on bones.2 Bisphosphonate-related osteonecrosis of the jaw (BRONJ) is defined by the American Association of Oral and Maxillofacial Surgeons as the presence of exposed necrotic bone in the maxillofacial region that has persisted for more than 8 weeks, with current or prior treatment with a bisphosphonate and absence of prior radiation therapy to the jaw.3 Bisphosphonate-related osteonecrosis of the jaw can be associated with infections, pathologic fractures, extraoral fistulae, or osteolysis extending to the inferior border.
Our patient had a dental sinus that resulted from the underlying BRONJ. The jawbones, unlike the long bones, are in a special environment in that both acute and chronic infections occur often within the bone, and surgical procedures as well as masticatory trauma expose the bone to a bacteria-laden environment.4 Infection around the root apex of a tooth results in a dental abscess and a sinus tract can develop from the abscess, draining either intraorally or extraorally.5 Facial sinus tracts can be either odontogenic or nonodontogenic, and sometimes the lesions of dental origin may be confused with dermatological lesions.
Bisphosphonates inhibit osteoclasts, which are responsible for bone resorption. Antiangiogenetic effects also have been reported in bisphosphonates, resulting in devitalized bone.6 The potent and prolonged inhibition of bone remodeling likely plays an important role in BRONJ. The more frequently occurring microdamage inflicted on the lower jawbone with mastication also may represent a contributory factor.7
Bisphosphonate-related osteonecrosis of the jaw more often is associated with the use of high-dose intravenous (IV) bisphosphonate in cancer-related hypercalcemia and less so with oral bisphosphonates, which are generally used to treat osteoporosis.3 In a Swedish study conducted from 2003 to 2010, 55 cases of BRONJ were documented in a population of 1.2 million individuals. The prevalence of BRONJ in patients on oral bisphosphonates and IV bisphosphonates was estimated to be 0.024% and 2.8%, respectively.8
Bisphosphonates are widely used worldwide as the main treatment of osteoporosis. The association between osteonecrosis of the jaw and oral bisphosphonates is contentious among the osteoporosis population, as most studies focus on IV bisphosphonate use in cancer patients.9 Bisphosphonate-related osteonecrosis of the jaw adversely affects the patient's quality of life, producing notable morbidity in afflicted patients. Thus, a complete dental assessment and treatment is recommended before the initiation of bisphosphonate treatment. The risk for developing BRONJ associated with oral bisphosphonates increases when the duration of therapy exceeds 3 years.3 It has been reported that antifracture efficacy would persist for 1 to 2 years following discontinuation of alendronate or risedronate that had been taken for 3 to 5 years, but patients with low bone mineral density at the femoral neck (T-score below -2.5) after 3 to 5 years of treatment of bisphosphonates are at the highest risk for vertebral fractures and therefore appear to benefit most from continuation of therapy.10 For dental procedures, the American Association of Oral and Maxillofacial Surgeons suggests that if systemic conditions persist, the clinician might consider discontinuation of oral bisphosphonates for a 3-month period before and after elective invasive dental surgery to lower the risk for BRONJ.3 When possible, invasive dentoalveolar procedures such as extractions should be avoided; conservative endodontic treatment is preferable.
Bisphosphonate-related osteonecrosis of the jaw is a devastating condition that is difficult to treat and manage, thus the focus should be on prevention through dental clearance prior to starting bisphosphonates. It also is crucial to have a high index of suspicion for BRONJ in patients presenting with orofacial lesions so that they can be treated expediently.
- Edwards BJ, Gounder M, McKoy JM, et al. Pharmacovigilance and reporting oversight in US FDA fast-track process: bisphosphonates and osteonecrosis of the jaw. Lancet Oncol. 2008;9:1166-1172.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med. 2013;126:13-20.
- Ruggiero SL, Dodson TB, Assael LA, et al; American Association of Oral and Maxillofacial Surgeons. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws--2009 update. J Oral Maxillofac Surg. 2009;67(5 suppl):2-12.
- Mawardi H, Treister N, Richardson P, et al. Sinus tracts--an early sign of bisphosphonate-associated osteonecrosis of the jaws? J Oral Maxillofac Surg. 2009;67:593-601.
- Sammut S, Malden N, Lopes V. Facial cutaneous sinuses of dental origin--a diagnostic challenge. Br Dent J. 2013;215:555-558.
- Wood J, Bonjean K, Ruetz S, et al. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055-1061.
- Hoefert S, Schmitz I, Tannapfel A, et al. Importance of microcracks in etiology of bisphosphonate-related osteonecrosis of the jaw: a possible pathogenetic model of symptomatic and non-symptomatic osteonecrosis of the jaw based on scanning electron microscopy findings. Clin Oral Investig. 2010;14:271-284.
- Hallmer F, Bjørnland T, Nicklasson A, et al. Osteonecrosis of the jaw in patients treated with oral and intravenous bisphosphonates: experience in Sweden. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:202-208.
- Lin TC, Yang CY, Kao Yang YH, et al. Incidence and risk of osteonecrosis of the jaw among the Taiwan osteoporosis population [published online February 11, 2014]. Osteoporos Int. 2014;25:1503-1511.
- Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab. 2010;95:1555-1565.
The Diagnosis: Dental Sinus Secondary to Osteonecrosis of the Jaw
Cone beam computed tomography revealed an area of lucency measuring 40×20 mm in the body of the right mandible (Figure). The patient subsequently underwent curettage of the wound with sequestrectomy of the involved area.
Osteonecrosis of the jaw is a form of avascular necrosis. It is an uncommon but potentially serious side effect of bisphosphonate use.1 Bisphosphonates commonly are used as first-line therapy for osteoporosis, with proven efficacy to reduce fracture risk by exerting an antiresorptive effect on bones.2 Bisphosphonate-related osteonecrosis of the jaw (BRONJ) is defined by the American Association of Oral and Maxillofacial Surgeons as the presence of exposed necrotic bone in the maxillofacial region that has persisted for more than 8 weeks, with current or prior treatment with a bisphosphonate and absence of prior radiation therapy to the jaw.3 Bisphosphonate-related osteonecrosis of the jaw can be associated with infections, pathologic fractures, extraoral fistulae, or osteolysis extending to the inferior border.
Our patient had a dental sinus that resulted from the underlying BRONJ. The jawbones, unlike the long bones, are in a special environment in that both acute and chronic infections occur often within the bone, and surgical procedures as well as masticatory trauma expose the bone to a bacteria-laden environment.4 Infection around the root apex of a tooth results in a dental abscess and a sinus tract can develop from the abscess, draining either intraorally or extraorally.5 Facial sinus tracts can be either odontogenic or nonodontogenic, and sometimes the lesions of dental origin may be confused with dermatological lesions.
Bisphosphonates inhibit osteoclasts, which are responsible for bone resorption. Antiangiogenetic effects also have been reported in bisphosphonates, resulting in devitalized bone.6 The potent and prolonged inhibition of bone remodeling likely plays an important role in BRONJ. The more frequently occurring microdamage inflicted on the lower jawbone with mastication also may represent a contributory factor.7
Bisphosphonate-related osteonecrosis of the jaw more often is associated with the use of high-dose intravenous (IV) bisphosphonate in cancer-related hypercalcemia and less so with oral bisphosphonates, which are generally used to treat osteoporosis.3 In a Swedish study conducted from 2003 to 2010, 55 cases of BRONJ were documented in a population of 1.2 million individuals. The prevalence of BRONJ in patients on oral bisphosphonates and IV bisphosphonates was estimated to be 0.024% and 2.8%, respectively.8
Bisphosphonates are widely used worldwide as the main treatment of osteoporosis. The association between osteonecrosis of the jaw and oral bisphosphonates is contentious among the osteoporosis population, as most studies focus on IV bisphosphonate use in cancer patients.9 Bisphosphonate-related osteonecrosis of the jaw adversely affects the patient's quality of life, producing notable morbidity in afflicted patients. Thus, a complete dental assessment and treatment is recommended before the initiation of bisphosphonate treatment. The risk for developing BRONJ associated with oral bisphosphonates increases when the duration of therapy exceeds 3 years.3 It has been reported that antifracture efficacy would persist for 1 to 2 years following discontinuation of alendronate or risedronate that had been taken for 3 to 5 years, but patients with low bone mineral density at the femoral neck (T-score below -2.5) after 3 to 5 years of treatment of bisphosphonates are at the highest risk for vertebral fractures and therefore appear to benefit most from continuation of therapy.10 For dental procedures, the American Association of Oral and Maxillofacial Surgeons suggests that if systemic conditions persist, the clinician might consider discontinuation of oral bisphosphonates for a 3-month period before and after elective invasive dental surgery to lower the risk for BRONJ.3 When possible, invasive dentoalveolar procedures such as extractions should be avoided; conservative endodontic treatment is preferable.
Bisphosphonate-related osteonecrosis of the jaw is a devastating condition that is difficult to treat and manage, thus the focus should be on prevention through dental clearance prior to starting bisphosphonates. It also is crucial to have a high index of suspicion for BRONJ in patients presenting with orofacial lesions so that they can be treated expediently.
The Diagnosis: Dental Sinus Secondary to Osteonecrosis of the Jaw
Cone beam computed tomography revealed an area of lucency measuring 40×20 mm in the body of the right mandible (Figure). The patient subsequently underwent curettage of the wound with sequestrectomy of the involved area.
Osteonecrosis of the jaw is a form of avascular necrosis. It is an uncommon but potentially serious side effect of bisphosphonate use.1 Bisphosphonates commonly are used as first-line therapy for osteoporosis, with proven efficacy to reduce fracture risk by exerting an antiresorptive effect on bones.2 Bisphosphonate-related osteonecrosis of the jaw (BRONJ) is defined by the American Association of Oral and Maxillofacial Surgeons as the presence of exposed necrotic bone in the maxillofacial region that has persisted for more than 8 weeks, with current or prior treatment with a bisphosphonate and absence of prior radiation therapy to the jaw.3 Bisphosphonate-related osteonecrosis of the jaw can be associated with infections, pathologic fractures, extraoral fistulae, or osteolysis extending to the inferior border.
Our patient had a dental sinus that resulted from the underlying BRONJ. The jawbones, unlike the long bones, are in a special environment in that both acute and chronic infections occur often within the bone, and surgical procedures as well as masticatory trauma expose the bone to a bacteria-laden environment.4 Infection around the root apex of a tooth results in a dental abscess and a sinus tract can develop from the abscess, draining either intraorally or extraorally.5 Facial sinus tracts can be either odontogenic or nonodontogenic, and sometimes the lesions of dental origin may be confused with dermatological lesions.
Bisphosphonates inhibit osteoclasts, which are responsible for bone resorption. Antiangiogenetic effects also have been reported in bisphosphonates, resulting in devitalized bone.6 The potent and prolonged inhibition of bone remodeling likely plays an important role in BRONJ. The more frequently occurring microdamage inflicted on the lower jawbone with mastication also may represent a contributory factor.7
Bisphosphonate-related osteonecrosis of the jaw more often is associated with the use of high-dose intravenous (IV) bisphosphonate in cancer-related hypercalcemia and less so with oral bisphosphonates, which are generally used to treat osteoporosis.3 In a Swedish study conducted from 2003 to 2010, 55 cases of BRONJ were documented in a population of 1.2 million individuals. The prevalence of BRONJ in patients on oral bisphosphonates and IV bisphosphonates was estimated to be 0.024% and 2.8%, respectively.8
Bisphosphonates are widely used worldwide as the main treatment of osteoporosis. The association between osteonecrosis of the jaw and oral bisphosphonates is contentious among the osteoporosis population, as most studies focus on IV bisphosphonate use in cancer patients.9 Bisphosphonate-related osteonecrosis of the jaw adversely affects the patient's quality of life, producing notable morbidity in afflicted patients. Thus, a complete dental assessment and treatment is recommended before the initiation of bisphosphonate treatment. The risk for developing BRONJ associated with oral bisphosphonates increases when the duration of therapy exceeds 3 years.3 It has been reported that antifracture efficacy would persist for 1 to 2 years following discontinuation of alendronate or risedronate that had been taken for 3 to 5 years, but patients with low bone mineral density at the femoral neck (T-score below -2.5) after 3 to 5 years of treatment of bisphosphonates are at the highest risk for vertebral fractures and therefore appear to benefit most from continuation of therapy.10 For dental procedures, the American Association of Oral and Maxillofacial Surgeons suggests that if systemic conditions persist, the clinician might consider discontinuation of oral bisphosphonates for a 3-month period before and after elective invasive dental surgery to lower the risk for BRONJ.3 When possible, invasive dentoalveolar procedures such as extractions should be avoided; conservative endodontic treatment is preferable.
Bisphosphonate-related osteonecrosis of the jaw is a devastating condition that is difficult to treat and manage, thus the focus should be on prevention through dental clearance prior to starting bisphosphonates. It also is crucial to have a high index of suspicion for BRONJ in patients presenting with orofacial lesions so that they can be treated expediently.
- Edwards BJ, Gounder M, McKoy JM, et al. Pharmacovigilance and reporting oversight in US FDA fast-track process: bisphosphonates and osteonecrosis of the jaw. Lancet Oncol. 2008;9:1166-1172.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med. 2013;126:13-20.
- Ruggiero SL, Dodson TB, Assael LA, et al; American Association of Oral and Maxillofacial Surgeons. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws--2009 update. J Oral Maxillofac Surg. 2009;67(5 suppl):2-12.
- Mawardi H, Treister N, Richardson P, et al. Sinus tracts--an early sign of bisphosphonate-associated osteonecrosis of the jaws? J Oral Maxillofac Surg. 2009;67:593-601.
- Sammut S, Malden N, Lopes V. Facial cutaneous sinuses of dental origin--a diagnostic challenge. Br Dent J. 2013;215:555-558.
- Wood J, Bonjean K, Ruetz S, et al. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055-1061.
- Hoefert S, Schmitz I, Tannapfel A, et al. Importance of microcracks in etiology of bisphosphonate-related osteonecrosis of the jaw: a possible pathogenetic model of symptomatic and non-symptomatic osteonecrosis of the jaw based on scanning electron microscopy findings. Clin Oral Investig. 2010;14:271-284.
- Hallmer F, Bjørnland T, Nicklasson A, et al. Osteonecrosis of the jaw in patients treated with oral and intravenous bisphosphonates: experience in Sweden. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:202-208.
- Lin TC, Yang CY, Kao Yang YH, et al. Incidence and risk of osteonecrosis of the jaw among the Taiwan osteoporosis population [published online February 11, 2014]. Osteoporos Int. 2014;25:1503-1511.
- Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab. 2010;95:1555-1565.
- Edwards BJ, Gounder M, McKoy JM, et al. Pharmacovigilance and reporting oversight in US FDA fast-track process: bisphosphonates and osteonecrosis of the jaw. Lancet Oncol. 2008;9:1166-1172.
- McClung M, Harris ST, Miller PD, et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med. 2013;126:13-20.
- Ruggiero SL, Dodson TB, Assael LA, et al; American Association of Oral and Maxillofacial Surgeons. American Association of Oral and Maxillofacial Surgeons position paper on bisphosphonate-related osteonecrosis of the jaws--2009 update. J Oral Maxillofac Surg. 2009;67(5 suppl):2-12.
- Mawardi H, Treister N, Richardson P, et al. Sinus tracts--an early sign of bisphosphonate-associated osteonecrosis of the jaws? J Oral Maxillofac Surg. 2009;67:593-601.
- Sammut S, Malden N, Lopes V. Facial cutaneous sinuses of dental origin--a diagnostic challenge. Br Dent J. 2013;215:555-558.
- Wood J, Bonjean K, Ruetz S, et al. Novel antiangiogenic effects of the bisphosphonate compound zoledronic acid. J Pharmacol Exp Ther. 2002;302:1055-1061.
- Hoefert S, Schmitz I, Tannapfel A, et al. Importance of microcracks in etiology of bisphosphonate-related osteonecrosis of the jaw: a possible pathogenetic model of symptomatic and non-symptomatic osteonecrosis of the jaw based on scanning electron microscopy findings. Clin Oral Investig. 2010;14:271-284.
- Hallmer F, Bjørnland T, Nicklasson A, et al. Osteonecrosis of the jaw in patients treated with oral and intravenous bisphosphonates: experience in Sweden. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:202-208.
- Lin TC, Yang CY, Kao Yang YH, et al. Incidence and risk of osteonecrosis of the jaw among the Taiwan osteoporosis population [published online February 11, 2014]. Osteoporos Int. 2014;25:1503-1511.
- Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab. 2010;95:1555-1565.
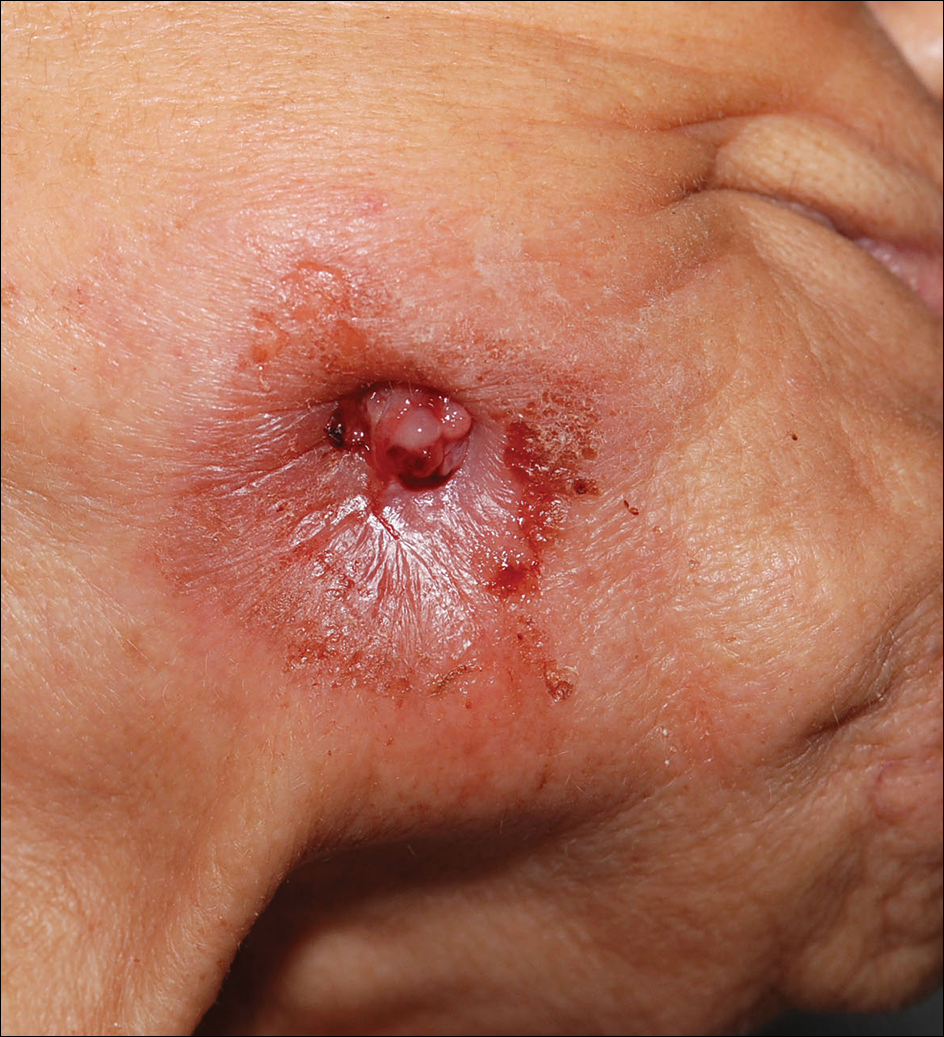
An 83-year-old woman presented with a painless, discharging, swollen nodule on the right side of the jaw of 6 months' duration. She had a history of osteoporosis diagnosed 3 years prior for which she was taking alendronate and cholecalciferol. Bone mineral density test scores were -3.93 (spine) and -2.81 (hip)(reference range, -2 and above). She also had hypertension that was treated with amlodipine. On examination there was fetor oris and a discharging sinus with purulent discharge at the jaw. The lower jaw was edentulous. A 5-mm area of red beefy granulation tissue was attached to underlying bone. An exposed sequestrum was seen intraorally with a 3-cm opening at the mandible. There also was submandibular lymphadenopathy.
Necrotic Lesion of the Ear
The Diagnosis: Chondrodermatitis Nodularis Chronica Helicis
Histopathologic examination revealed focal epidermal erosion and ulceration directly overlying the hyaline cartilage with degenerative changes (Figure). The dermis was relatively noninflamed with fibroplasia of the vasculature. The blood vessels indirectly beneath the ulceration were found to be unremarkable with no indications of fibrinoid necrosis, vasculitis, or the presence of thrombi. The patient was informed of the diagnosis, at which point she reported that she slept on the right side. The excisional biopsy site healed well without recurrence of chondrodermatitis nodularis chronica helicis (CNH).
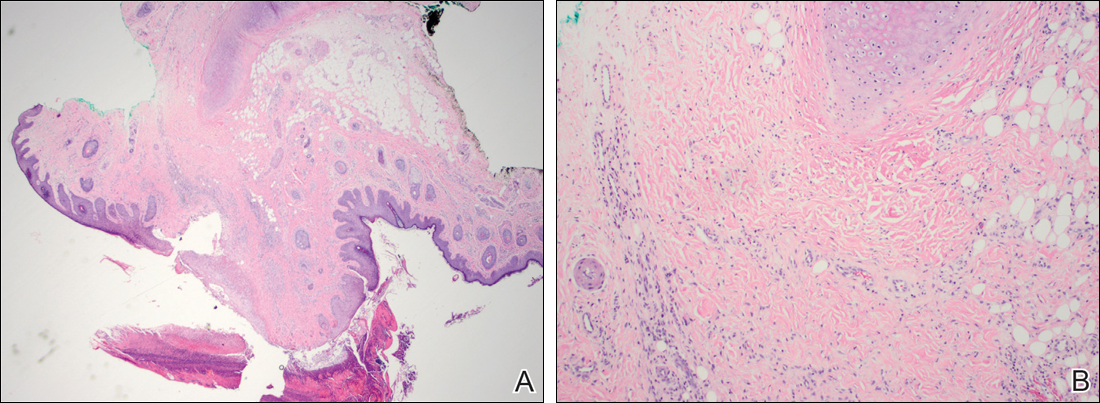
Chondrodermatitis nodularis chronica helicis, also known as clavus helicis, is a benign, usually solitary, painful lesion. Historically, it was first described in 1915 by Winkler1 and in the 1960s the most common documented cases were attributed to the headpieces of telephone operators and nuns.2 In the early 2000s, cell phones were determined to be a growing cause.3 Chondrodermatitis nodularis chronica helicis is most commonly found on the helix with the antihelix being affected less often.4 The condition is more common in men, with a male to female ratio being reported as high as 10:1. Possible causes of this disorder stem from damage to cartilage associated with pressure, sun exposure, cold temperatures, and microvascular disease. Additionally, some researchers have hypothesized that the cartilaginous damage resulting from solar elastosis and minor trauma leaves a susceptibility to CNH. This disorder usually presents as a small, exquisitely tender nodule that may ulcerate and crust.4 Chondrodermatitis nodularis chronica helicis may be mistaken for basal cell carcinoma, squamous cell carcinoma, actinic keratosis, and weathering nodules, though CNH tends to be more painful.
The diagnosis of CNH often is clinical but may require a skin biopsy. Histopathology of CNH shows a benign inflammatory lesion with an acanthotic hyperkeratotic epidermis that may be ulcerated. A primarily lymphocytic infiltrate usually is observed with variable presence of histiocytes and neutrophils. Cartilaginous changes range from simple perichondral thickening to notable areas of degeneration with calcification and ossification.4
Although the diagnosis of CNH often is straightforward, the remarkable necrosis present in our case made for an interesting differential diagnosis. Pernio, cryoglobulinemia, and levamisole-induced vasculopathy were all considered. Pernio, caused by cold-induced vasoconstriction and hypoxemia, classically presents as erythematous lesions with a symmetrical distribution on acral sites.5 Cryoglobulinemia involves proteins that precipitate at cold temperatures causing damage via an occlusive vasculopathy or an immune complex-mediated vasculitis. The presence of cryoglobulinemia is strongly associated with concomitant hepatitis C virus infection.6 Ulcerated and purpuric lesions of cryoglobulinemia may become necrotic. Levamisole is a veterinary antihelminthic drug and common cocaine contaminant, often added to cocaine as a cutting agent. Levamisole-induced vasculopathy favors acral sites and often is noted on the ears as purpuric patches, sometimes with necrosis.7
Several therapies for CNH have been reported with variable effectiveness.8 First-line treatments are the use of pressure-relieving devices including a doughnut-shaped pillow during sleep and intralesional corticosteroids.9 Surgical treatments including cryotherapy, simple excision, electrodesiccation and curettage, wedge resection with helical rim advancement flap, punch and graft technique, and CO2 laser have been tried.8 Photodynamic therapy and topical nitroglycerine also have shown to be of benefit.8,9
Our case of CNH is unique because of the remarkable degree of necrosis present on clinical examination. Chondrodermatitis nodularis chronica helicis with such an impressive necrotic presentation is rare. We speculate that the patient's underlying hypercoagulable state may have contributed to the dramatic presentation. It is important to keep CNH in mind when evaluating any necrotic lesion on the ear.
- Winkler M. Knötcehnformige Erkrankung am helix. chondrodermatitis nodularis chronic helicis. Arch für Dermatologie und Syphilis. 1915;121:278-285.
- Barker L, Young AW, Sachs W. Chondrodermatitis of the ears: a differential study of nodules of the helix and antihelix. Arch Dermatol. 1960;81:15-25.
- Elgart M. Cell phone chondrodermatitis. Arch Dermatol. 2000;136:1568.
- Cribier B, Scrivener Y, Peltre B. Neural hyperplasia in chondrodermatitis nodularis chronica helicis. J Am Acad Dermatol. 2006;55:844-848.
- King JM, Plotner AN, Adams BB. Perniosis induced by a cold-therapy system. Arch Dermatol. 2012;148:1101-1102.
- Berk DR, Mallory SB, Keeffe EB, et al. Dermatologic disorders associated with chronic hepatitis C: effect of interferon therapy. Clin Gastroenterol Hepatol. 2007;5:142-151.
- Hennings C, Miller J. Illicit drugs: what dermatologists need to know. J Am Acad Dermatol. 2013;69:135-142.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;64:531-536.
- Gilaberte Y, Frias M, Pérez-Lorenz J. Chondrodermatitis nodularis helicis successfully treated with photodynamic therapy. Arch Dermatol. 2010;146:1080-1082.
The Diagnosis: Chondrodermatitis Nodularis Chronica Helicis
Histopathologic examination revealed focal epidermal erosion and ulceration directly overlying the hyaline cartilage with degenerative changes (Figure). The dermis was relatively noninflamed with fibroplasia of the vasculature. The blood vessels indirectly beneath the ulceration were found to be unremarkable with no indications of fibrinoid necrosis, vasculitis, or the presence of thrombi. The patient was informed of the diagnosis, at which point she reported that she slept on the right side. The excisional biopsy site healed well without recurrence of chondrodermatitis nodularis chronica helicis (CNH).
Chondrodermatitis nodularis chronica helicis, also known as clavus helicis, is a benign, usually solitary, painful lesion. Historically, it was first described in 1915 by Winkler1 and in the 1960s the most common documented cases were attributed to the headpieces of telephone operators and nuns.2 In the early 2000s, cell phones were determined to be a growing cause.3 Chondrodermatitis nodularis chronica helicis is most commonly found on the helix with the antihelix being affected less often.4 The condition is more common in men, with a male to female ratio being reported as high as 10:1. Possible causes of this disorder stem from damage to cartilage associated with pressure, sun exposure, cold temperatures, and microvascular disease. Additionally, some researchers have hypothesized that the cartilaginous damage resulting from solar elastosis and minor trauma leaves a susceptibility to CNH. This disorder usually presents as a small, exquisitely tender nodule that may ulcerate and crust.4 Chondrodermatitis nodularis chronica helicis may be mistaken for basal cell carcinoma, squamous cell carcinoma, actinic keratosis, and weathering nodules, though CNH tends to be more painful.
The diagnosis of CNH often is clinical but may require a skin biopsy. Histopathology of CNH shows a benign inflammatory lesion with an acanthotic hyperkeratotic epidermis that may be ulcerated. A primarily lymphocytic infiltrate usually is observed with variable presence of histiocytes and neutrophils. Cartilaginous changes range from simple perichondral thickening to notable areas of degeneration with calcification and ossification.4
Although the diagnosis of CNH often is straightforward, the remarkable necrosis present in our case made for an interesting differential diagnosis. Pernio, cryoglobulinemia, and levamisole-induced vasculopathy were all considered. Pernio, caused by cold-induced vasoconstriction and hypoxemia, classically presents as erythematous lesions with a symmetrical distribution on acral sites.5 Cryoglobulinemia involves proteins that precipitate at cold temperatures causing damage via an occlusive vasculopathy or an immune complex-mediated vasculitis. The presence of cryoglobulinemia is strongly associated with concomitant hepatitis C virus infection.6 Ulcerated and purpuric lesions of cryoglobulinemia may become necrotic. Levamisole is a veterinary antihelminthic drug and common cocaine contaminant, often added to cocaine as a cutting agent. Levamisole-induced vasculopathy favors acral sites and often is noted on the ears as purpuric patches, sometimes with necrosis.7
Several therapies for CNH have been reported with variable effectiveness.8 First-line treatments are the use of pressure-relieving devices including a doughnut-shaped pillow during sleep and intralesional corticosteroids.9 Surgical treatments including cryotherapy, simple excision, electrodesiccation and curettage, wedge resection with helical rim advancement flap, punch and graft technique, and CO2 laser have been tried.8 Photodynamic therapy and topical nitroglycerine also have shown to be of benefit.8,9
Our case of CNH is unique because of the remarkable degree of necrosis present on clinical examination. Chondrodermatitis nodularis chronica helicis with such an impressive necrotic presentation is rare. We speculate that the patient's underlying hypercoagulable state may have contributed to the dramatic presentation. It is important to keep CNH in mind when evaluating any necrotic lesion on the ear.
The Diagnosis: Chondrodermatitis Nodularis Chronica Helicis
Histopathologic examination revealed focal epidermal erosion and ulceration directly overlying the hyaline cartilage with degenerative changes (Figure). The dermis was relatively noninflamed with fibroplasia of the vasculature. The blood vessels indirectly beneath the ulceration were found to be unremarkable with no indications of fibrinoid necrosis, vasculitis, or the presence of thrombi. The patient was informed of the diagnosis, at which point she reported that she slept on the right side. The excisional biopsy site healed well without recurrence of chondrodermatitis nodularis chronica helicis (CNH).
Chondrodermatitis nodularis chronica helicis, also known as clavus helicis, is a benign, usually solitary, painful lesion. Historically, it was first described in 1915 by Winkler1 and in the 1960s the most common documented cases were attributed to the headpieces of telephone operators and nuns.2 In the early 2000s, cell phones were determined to be a growing cause.3 Chondrodermatitis nodularis chronica helicis is most commonly found on the helix with the antihelix being affected less often.4 The condition is more common in men, with a male to female ratio being reported as high as 10:1. Possible causes of this disorder stem from damage to cartilage associated with pressure, sun exposure, cold temperatures, and microvascular disease. Additionally, some researchers have hypothesized that the cartilaginous damage resulting from solar elastosis and minor trauma leaves a susceptibility to CNH. This disorder usually presents as a small, exquisitely tender nodule that may ulcerate and crust.4 Chondrodermatitis nodularis chronica helicis may be mistaken for basal cell carcinoma, squamous cell carcinoma, actinic keratosis, and weathering nodules, though CNH tends to be more painful.
The diagnosis of CNH often is clinical but may require a skin biopsy. Histopathology of CNH shows a benign inflammatory lesion with an acanthotic hyperkeratotic epidermis that may be ulcerated. A primarily lymphocytic infiltrate usually is observed with variable presence of histiocytes and neutrophils. Cartilaginous changes range from simple perichondral thickening to notable areas of degeneration with calcification and ossification.4
Although the diagnosis of CNH often is straightforward, the remarkable necrosis present in our case made for an interesting differential diagnosis. Pernio, cryoglobulinemia, and levamisole-induced vasculopathy were all considered. Pernio, caused by cold-induced vasoconstriction and hypoxemia, classically presents as erythematous lesions with a symmetrical distribution on acral sites.5 Cryoglobulinemia involves proteins that precipitate at cold temperatures causing damage via an occlusive vasculopathy or an immune complex-mediated vasculitis. The presence of cryoglobulinemia is strongly associated with concomitant hepatitis C virus infection.6 Ulcerated and purpuric lesions of cryoglobulinemia may become necrotic. Levamisole is a veterinary antihelminthic drug and common cocaine contaminant, often added to cocaine as a cutting agent. Levamisole-induced vasculopathy favors acral sites and often is noted on the ears as purpuric patches, sometimes with necrosis.7
Several therapies for CNH have been reported with variable effectiveness.8 First-line treatments are the use of pressure-relieving devices including a doughnut-shaped pillow during sleep and intralesional corticosteroids.9 Surgical treatments including cryotherapy, simple excision, electrodesiccation and curettage, wedge resection with helical rim advancement flap, punch and graft technique, and CO2 laser have been tried.8 Photodynamic therapy and topical nitroglycerine also have shown to be of benefit.8,9
Our case of CNH is unique because of the remarkable degree of necrosis present on clinical examination. Chondrodermatitis nodularis chronica helicis with such an impressive necrotic presentation is rare. We speculate that the patient's underlying hypercoagulable state may have contributed to the dramatic presentation. It is important to keep CNH in mind when evaluating any necrotic lesion on the ear.
- Winkler M. Knötcehnformige Erkrankung am helix. chondrodermatitis nodularis chronic helicis. Arch für Dermatologie und Syphilis. 1915;121:278-285.
- Barker L, Young AW, Sachs W. Chondrodermatitis of the ears: a differential study of nodules of the helix and antihelix. Arch Dermatol. 1960;81:15-25.
- Elgart M. Cell phone chondrodermatitis. Arch Dermatol. 2000;136:1568.
- Cribier B, Scrivener Y, Peltre B. Neural hyperplasia in chondrodermatitis nodularis chronica helicis. J Am Acad Dermatol. 2006;55:844-848.
- King JM, Plotner AN, Adams BB. Perniosis induced by a cold-therapy system. Arch Dermatol. 2012;148:1101-1102.
- Berk DR, Mallory SB, Keeffe EB, et al. Dermatologic disorders associated with chronic hepatitis C: effect of interferon therapy. Clin Gastroenterol Hepatol. 2007;5:142-151.
- Hennings C, Miller J. Illicit drugs: what dermatologists need to know. J Am Acad Dermatol. 2013;69:135-142.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;64:531-536.
- Gilaberte Y, Frias M, Pérez-Lorenz J. Chondrodermatitis nodularis helicis successfully treated with photodynamic therapy. Arch Dermatol. 2010;146:1080-1082.
- Winkler M. Knötcehnformige Erkrankung am helix. chondrodermatitis nodularis chronic helicis. Arch für Dermatologie und Syphilis. 1915;121:278-285.
- Barker L, Young AW, Sachs W. Chondrodermatitis of the ears: a differential study of nodules of the helix and antihelix. Arch Dermatol. 1960;81:15-25.
- Elgart M. Cell phone chondrodermatitis. Arch Dermatol. 2000;136:1568.
- Cribier B, Scrivener Y, Peltre B. Neural hyperplasia in chondrodermatitis nodularis chronica helicis. J Am Acad Dermatol. 2006;55:844-848.
- King JM, Plotner AN, Adams BB. Perniosis induced by a cold-therapy system. Arch Dermatol. 2012;148:1101-1102.
- Berk DR, Mallory SB, Keeffe EB, et al. Dermatologic disorders associated with chronic hepatitis C: effect of interferon therapy. Clin Gastroenterol Hepatol. 2007;5:142-151.
- Hennings C, Miller J. Illicit drugs: what dermatologists need to know. J Am Acad Dermatol. 2013;69:135-142.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;64:531-536.
- Gilaberte Y, Frias M, Pérez-Lorenz J. Chondrodermatitis nodularis helicis successfully treated with photodynamic therapy. Arch Dermatol. 2010;146:1080-1082.
A 43-year-old woman presented with a painful necrotic lesion on the right ear of 1 month's duration. She denied trauma to the ear and had no other skin lesions elsewhere on the body. A course of doxycycline prior to presentation did not result in improvement. Her medical history was remarkable for diabetes mellitus, deep vein thrombosis, depression, and gastroesophageal reflux disease. She had been taking warfarin regularly for years. She denied using recreational drugs. On physical examination, the right ear demonstrated a 6-mm necrotic area with surrounding tender erythema. Examinations of the left ear, face, and legs were normal. An excisional biopsy was performed.