User login
What Is Your Diagnosis? New World Cutaneous Leishmaniasis
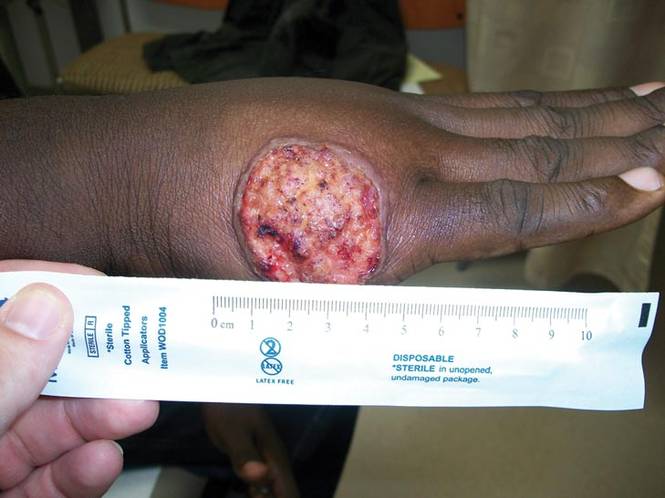
A 40-year-old man presented with a nonhealing ulcer on the right hand of 2 months’ duration. The lesion had started as a pruritic papule while he was visiting Guyana 2 months prior. The area had slowly enlarged with progressive ulceration. He denied any systemic signs including fever, chills, or weight loss, and his medical history was unremarkable. Physical examination revealed a 4-cm fungating ulceration with heaped-up borders on the dorsal aspect of the right hand.
The Diagnosis: New World Cutaneous Leishmaniasis
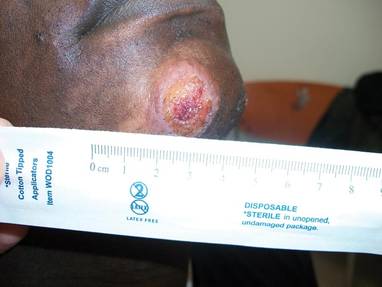
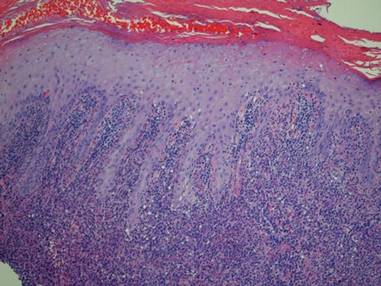
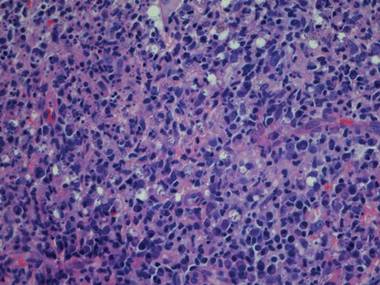
In addition to the ulceration on the right hand, a 2-cm ulcerated plaque also had developed on the right side of the chin a few days later (Figure 1). A biopsy was obtained from the lesion on the hand. Histopathologic examination revealed granulomatous inflammation with numerous histiocytes containing intracellular organisms (Figure 2). The microorganisms had pale pink nuclei with basophilic kinetoplasts (Figure 3). Tissue culture showed a mixed growth of gram-positive and gram-negative organisms but no predominant organism. Fungal and mycobacterial cultures were negative. A diagnosis of New World cutaneous leishmaniasis (CL) was made due to visualization of intracellular microorganisms containing basophilic kinetoplasts. Polymerase chain reaction on the tissue block confirmed the presence of Leishmania guyanensis.
|
Cutaneous leishmaniasis is caused by protozoa from the Leishmania species and is transmitted by the bite of the female sandfly. There are 2 classifications for the disease: Old World and New World. Old World CL is transmitted by the sandfly of the genus Phlebotomus, which is endemic in Asia, Africa, the Mediterranean, and the Middle East. New World CL is transmitted by the sandflies of the genus Lutzomyia, which are endemic in Mexico, Central America, and South America. There have been occasional cases of autochthonous transmission reported in Texas and Oklahoma,1 but there has been no known transmission of CL in Australia or the Pacific Islands.2 Human infection can be transmitted by 21 species of Leishmania and can be speciated by designated laboratories such as the US Centers for Disease Control and Prevention (CDC) and the Walter Reed Army Institute of Research (Silver Spring, Maryland) using tissue culture and polymerase chain reaction.1 The CDC can assist with the collection of specimens and supply of culture medium for cases occurring in the United States. Identification of the species is important because there are associated implications for treatment and prognosis.
There are 3 major forms of leishmaniasis: cutaneous, mucocutaneous, and visceral. Cutaneous leishmaniasis is the most common form. There are approximately 1.5 million new cases of CL each year worldwide,3 and more than 90% of these cases occur in Afghanistan, Algeria, Iran, Iraq, Saudi Arabia, Syria, Brazil, and Peru.1 New World CL is caused by 2 species complexes: Leishmania mexicana and Leishmania viannia, including the subspecies L guyanensis, which was seen in our patient.
Cutaneous leishmaniasis usually begins as a small, well-defined papule at the site of the insect bite that then enlarges and becomes a nodule or plaque. Next, the lesion becomes ulcerated with raised borders (Figure 1). The ulcer typically is painless unless there is secondary bacterial or fungal infection.3,4 The incubation period usually is 2 to 8 weeks and multiple lesions may be present, as seen in our patient.4
Old World CL can resolve without treatment, but New World CL is less likely to spontaneously resolve. Additionally, there is a greater risk for spread of the infection to mucous membranes or for systemic dissemination if New World CL is left untreated.2,5 Patients with multiple lesions (ie, ≥3); large lesions (ie, >2.5 cm); lesions on the face, hands, feet, or joints; and those who are immunocompromised should be treated promptly.2 Pentavalent antimonials (eg, meglumine antimoniate, sodium stibogluconate) are the treatment of choice for New World CL, except for infections caused by L guyanensis. The most common pentavalent antimonial agent used in the United States is sodium stibogluconate and is given at a standard intravenous dose of 20 mg antimony/kg daily for 20 days.2 The drug is only available through the CDC’s Drug Service under an Investigational New Drug protocol.1
Intramuscular pentamidine (3 mg/kg daily every other day for 4 doses) is the first-line treatment of CL caused by L guyanensis because systemic antimony usually is not effective.2,6,7 Intralesional injection with pentavalent antimonials usually is not recommended for treatment of New World CL because of the possibility of disseminated disease.2 Liposomal amphotericin B has mainly been used to treat visceral and mucosal leishmaniasis, but there have been some small studies and case reports that have showed it to be successful in treating CL.8-10 Larger controlled studies need to be performed. Oral antifungal drugs (eg, fluconazole, ketoconazole, itraconazole) also have been used to treat CL with variable results depending on the Leishmania species.11
There currently are no vaccines or drugs available to prevent against leishmaniasis. Preventive measures such as avoiding outdoor activities from dusk to dawn when sandflies are the most active, wearing protective clothing, and applying insect repellent that contains DEET (diethyltoluamide) can help reduce a traveler’s risk for becoming infected. Mosquito nets also should be treated with permethrin, which acts as an insect repellent, as sandflies are so small that they can penetrate mosquito nets.1,3,11
Acknowledgements—We would like to thank Francis Steurer, MS, and Barbara Herwaldt, MD, MPH, at the CDC in Atlanta, Georgia, for their help with the identification of the Leishmania species.
1. Herwaldt BL, Magill AJ. Infectious diseases related to travel: leishmaniasis, cutaneous. Centers for Disease Control and Prevention Web site. http://wwwnc.cdc.gov/travel/yellowbook/2010/chapter-5/cutaneous-leish maniasis.htm. Published August 1, 2013. Accessed March 5, 2015.
2. Mitropolos P, Konidas P, Durkin-Konidas M. New World cutaneous leishmaniasis: updated review of current and future diagnosis and treatment. J Am Acad Dermatol. 2010;63:309-322.
3. Hepburn NC. Cutaneous leishmaniasis: an overview. J Postgrad Med. 2003;49:50-54.
4. Markle WH, Makhoul K. Cutaneous leishmaniasis: recognition and treatment. Am Fam Physician. 2004;69:1455-1460.
5. Couppié P, Clyti E, Sainte Marie D, et al. Disseminated cutaneous leishmaniasis due to Leishmania guyanensis: case of a patient with 425 lesions. Am J Trop Med Hyg. 2004;71:558-560.
6. Nacher M, Carme B, Sainte Marie D, et al. Influence of clinical presentation on the efficacy of a short course of pentamidine in the treatment of cutaneous leishmaniasis in French Guiana. Ann Trop Med Parasitol. 2001;95:331-336.
7. Minodier P, Parola P. Cutaneous leishmaniasis treatment. Trav Med Inf Dis. 2007;5:150-158.
8. Solomon M, Baum S, Barzilai A, et al. Liposomal amphotericin B in comparison to sodium stibogluconate for cutaneous infection due to Leishmania braziliensis. J Am Acad Dermatol. 2007;56:612-616.
9. Konecny P, Stark DJ. An Australian case of New World cutaneous leishmaniasis. Med J Aust. 2007;186:315-317.
10. Brown M, Noursadeghi M, Boyle J, et al. Successful liposomal amphotericin B treatment of Leishmania braziliensis cutaneous leishmaniasis. Br J Dermatol. 2005;153:203-205.
11. Parasites: leishmaniasis. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/parasites/leishmaniasis/index.html. Updated January 10, 2013. Accessed March 5, 2015.
A 40-year-old man presented with a nonhealing ulcer on the right hand of 2 months’ duration. The lesion had started as a pruritic papule while he was visiting Guyana 2 months prior. The area had slowly enlarged with progressive ulceration. He denied any systemic signs including fever, chills, or weight loss, and his medical history was unremarkable. Physical examination revealed a 4-cm fungating ulceration with heaped-up borders on the dorsal aspect of the right hand.
The Diagnosis: New World Cutaneous Leishmaniasis
In addition to the ulceration on the right hand, a 2-cm ulcerated plaque also had developed on the right side of the chin a few days later (Figure 1). A biopsy was obtained from the lesion on the hand. Histopathologic examination revealed granulomatous inflammation with numerous histiocytes containing intracellular organisms (Figure 2). The microorganisms had pale pink nuclei with basophilic kinetoplasts (Figure 3). Tissue culture showed a mixed growth of gram-positive and gram-negative organisms but no predominant organism. Fungal and mycobacterial cultures were negative. A diagnosis of New World cutaneous leishmaniasis (CL) was made due to visualization of intracellular microorganisms containing basophilic kinetoplasts. Polymerase chain reaction on the tissue block confirmed the presence of Leishmania guyanensis.
|
Cutaneous leishmaniasis is caused by protozoa from the Leishmania species and is transmitted by the bite of the female sandfly. There are 2 classifications for the disease: Old World and New World. Old World CL is transmitted by the sandfly of the genus Phlebotomus, which is endemic in Asia, Africa, the Mediterranean, and the Middle East. New World CL is transmitted by the sandflies of the genus Lutzomyia, which are endemic in Mexico, Central America, and South America. There have been occasional cases of autochthonous transmission reported in Texas and Oklahoma,1 but there has been no known transmission of CL in Australia or the Pacific Islands.2 Human infection can be transmitted by 21 species of Leishmania and can be speciated by designated laboratories such as the US Centers for Disease Control and Prevention (CDC) and the Walter Reed Army Institute of Research (Silver Spring, Maryland) using tissue culture and polymerase chain reaction.1 The CDC can assist with the collection of specimens and supply of culture medium for cases occurring in the United States. Identification of the species is important because there are associated implications for treatment and prognosis.
There are 3 major forms of leishmaniasis: cutaneous, mucocutaneous, and visceral. Cutaneous leishmaniasis is the most common form. There are approximately 1.5 million new cases of CL each year worldwide,3 and more than 90% of these cases occur in Afghanistan, Algeria, Iran, Iraq, Saudi Arabia, Syria, Brazil, and Peru.1 New World CL is caused by 2 species complexes: Leishmania mexicana and Leishmania viannia, including the subspecies L guyanensis, which was seen in our patient.
Cutaneous leishmaniasis usually begins as a small, well-defined papule at the site of the insect bite that then enlarges and becomes a nodule or plaque. Next, the lesion becomes ulcerated with raised borders (Figure 1). The ulcer typically is painless unless there is secondary bacterial or fungal infection.3,4 The incubation period usually is 2 to 8 weeks and multiple lesions may be present, as seen in our patient.4
Old World CL can resolve without treatment, but New World CL is less likely to spontaneously resolve. Additionally, there is a greater risk for spread of the infection to mucous membranes or for systemic dissemination if New World CL is left untreated.2,5 Patients with multiple lesions (ie, ≥3); large lesions (ie, >2.5 cm); lesions on the face, hands, feet, or joints; and those who are immunocompromised should be treated promptly.2 Pentavalent antimonials (eg, meglumine antimoniate, sodium stibogluconate) are the treatment of choice for New World CL, except for infections caused by L guyanensis. The most common pentavalent antimonial agent used in the United States is sodium stibogluconate and is given at a standard intravenous dose of 20 mg antimony/kg daily for 20 days.2 The drug is only available through the CDC’s Drug Service under an Investigational New Drug protocol.1
Intramuscular pentamidine (3 mg/kg daily every other day for 4 doses) is the first-line treatment of CL caused by L guyanensis because systemic antimony usually is not effective.2,6,7 Intralesional injection with pentavalent antimonials usually is not recommended for treatment of New World CL because of the possibility of disseminated disease.2 Liposomal amphotericin B has mainly been used to treat visceral and mucosal leishmaniasis, but there have been some small studies and case reports that have showed it to be successful in treating CL.8-10 Larger controlled studies need to be performed. Oral antifungal drugs (eg, fluconazole, ketoconazole, itraconazole) also have been used to treat CL with variable results depending on the Leishmania species.11
There currently are no vaccines or drugs available to prevent against leishmaniasis. Preventive measures such as avoiding outdoor activities from dusk to dawn when sandflies are the most active, wearing protective clothing, and applying insect repellent that contains DEET (diethyltoluamide) can help reduce a traveler’s risk for becoming infected. Mosquito nets also should be treated with permethrin, which acts as an insect repellent, as sandflies are so small that they can penetrate mosquito nets.1,3,11
Acknowledgements—We would like to thank Francis Steurer, MS, and Barbara Herwaldt, MD, MPH, at the CDC in Atlanta, Georgia, for their help with the identification of the Leishmania species.
A 40-year-old man presented with a nonhealing ulcer on the right hand of 2 months’ duration. The lesion had started as a pruritic papule while he was visiting Guyana 2 months prior. The area had slowly enlarged with progressive ulceration. He denied any systemic signs including fever, chills, or weight loss, and his medical history was unremarkable. Physical examination revealed a 4-cm fungating ulceration with heaped-up borders on the dorsal aspect of the right hand.
The Diagnosis: New World Cutaneous Leishmaniasis
In addition to the ulceration on the right hand, a 2-cm ulcerated plaque also had developed on the right side of the chin a few days later (Figure 1). A biopsy was obtained from the lesion on the hand. Histopathologic examination revealed granulomatous inflammation with numerous histiocytes containing intracellular organisms (Figure 2). The microorganisms had pale pink nuclei with basophilic kinetoplasts (Figure 3). Tissue culture showed a mixed growth of gram-positive and gram-negative organisms but no predominant organism. Fungal and mycobacterial cultures were negative. A diagnosis of New World cutaneous leishmaniasis (CL) was made due to visualization of intracellular microorganisms containing basophilic kinetoplasts. Polymerase chain reaction on the tissue block confirmed the presence of Leishmania guyanensis.
|
Cutaneous leishmaniasis is caused by protozoa from the Leishmania species and is transmitted by the bite of the female sandfly. There are 2 classifications for the disease: Old World and New World. Old World CL is transmitted by the sandfly of the genus Phlebotomus, which is endemic in Asia, Africa, the Mediterranean, and the Middle East. New World CL is transmitted by the sandflies of the genus Lutzomyia, which are endemic in Mexico, Central America, and South America. There have been occasional cases of autochthonous transmission reported in Texas and Oklahoma,1 but there has been no known transmission of CL in Australia or the Pacific Islands.2 Human infection can be transmitted by 21 species of Leishmania and can be speciated by designated laboratories such as the US Centers for Disease Control and Prevention (CDC) and the Walter Reed Army Institute of Research (Silver Spring, Maryland) using tissue culture and polymerase chain reaction.1 The CDC can assist with the collection of specimens and supply of culture medium for cases occurring in the United States. Identification of the species is important because there are associated implications for treatment and prognosis.
There are 3 major forms of leishmaniasis: cutaneous, mucocutaneous, and visceral. Cutaneous leishmaniasis is the most common form. There are approximately 1.5 million new cases of CL each year worldwide,3 and more than 90% of these cases occur in Afghanistan, Algeria, Iran, Iraq, Saudi Arabia, Syria, Brazil, and Peru.1 New World CL is caused by 2 species complexes: Leishmania mexicana and Leishmania viannia, including the subspecies L guyanensis, which was seen in our patient.
Cutaneous leishmaniasis usually begins as a small, well-defined papule at the site of the insect bite that then enlarges and becomes a nodule or plaque. Next, the lesion becomes ulcerated with raised borders (Figure 1). The ulcer typically is painless unless there is secondary bacterial or fungal infection.3,4 The incubation period usually is 2 to 8 weeks and multiple lesions may be present, as seen in our patient.4
Old World CL can resolve without treatment, but New World CL is less likely to spontaneously resolve. Additionally, there is a greater risk for spread of the infection to mucous membranes or for systemic dissemination if New World CL is left untreated.2,5 Patients with multiple lesions (ie, ≥3); large lesions (ie, >2.5 cm); lesions on the face, hands, feet, or joints; and those who are immunocompromised should be treated promptly.2 Pentavalent antimonials (eg, meglumine antimoniate, sodium stibogluconate) are the treatment of choice for New World CL, except for infections caused by L guyanensis. The most common pentavalent antimonial agent used in the United States is sodium stibogluconate and is given at a standard intravenous dose of 20 mg antimony/kg daily for 20 days.2 The drug is only available through the CDC’s Drug Service under an Investigational New Drug protocol.1
Intramuscular pentamidine (3 mg/kg daily every other day for 4 doses) is the first-line treatment of CL caused by L guyanensis because systemic antimony usually is not effective.2,6,7 Intralesional injection with pentavalent antimonials usually is not recommended for treatment of New World CL because of the possibility of disseminated disease.2 Liposomal amphotericin B has mainly been used to treat visceral and mucosal leishmaniasis, but there have been some small studies and case reports that have showed it to be successful in treating CL.8-10 Larger controlled studies need to be performed. Oral antifungal drugs (eg, fluconazole, ketoconazole, itraconazole) also have been used to treat CL with variable results depending on the Leishmania species.11
There currently are no vaccines or drugs available to prevent against leishmaniasis. Preventive measures such as avoiding outdoor activities from dusk to dawn when sandflies are the most active, wearing protective clothing, and applying insect repellent that contains DEET (diethyltoluamide) can help reduce a traveler’s risk for becoming infected. Mosquito nets also should be treated with permethrin, which acts as an insect repellent, as sandflies are so small that they can penetrate mosquito nets.1,3,11
Acknowledgements—We would like to thank Francis Steurer, MS, and Barbara Herwaldt, MD, MPH, at the CDC in Atlanta, Georgia, for their help with the identification of the Leishmania species.
1. Herwaldt BL, Magill AJ. Infectious diseases related to travel: leishmaniasis, cutaneous. Centers for Disease Control and Prevention Web site. http://wwwnc.cdc.gov/travel/yellowbook/2010/chapter-5/cutaneous-leish maniasis.htm. Published August 1, 2013. Accessed March 5, 2015.
2. Mitropolos P, Konidas P, Durkin-Konidas M. New World cutaneous leishmaniasis: updated review of current and future diagnosis and treatment. J Am Acad Dermatol. 2010;63:309-322.
3. Hepburn NC. Cutaneous leishmaniasis: an overview. J Postgrad Med. 2003;49:50-54.
4. Markle WH, Makhoul K. Cutaneous leishmaniasis: recognition and treatment. Am Fam Physician. 2004;69:1455-1460.
5. Couppié P, Clyti E, Sainte Marie D, et al. Disseminated cutaneous leishmaniasis due to Leishmania guyanensis: case of a patient with 425 lesions. Am J Trop Med Hyg. 2004;71:558-560.
6. Nacher M, Carme B, Sainte Marie D, et al. Influence of clinical presentation on the efficacy of a short course of pentamidine in the treatment of cutaneous leishmaniasis in French Guiana. Ann Trop Med Parasitol. 2001;95:331-336.
7. Minodier P, Parola P. Cutaneous leishmaniasis treatment. Trav Med Inf Dis. 2007;5:150-158.
8. Solomon M, Baum S, Barzilai A, et al. Liposomal amphotericin B in comparison to sodium stibogluconate for cutaneous infection due to Leishmania braziliensis. J Am Acad Dermatol. 2007;56:612-616.
9. Konecny P, Stark DJ. An Australian case of New World cutaneous leishmaniasis. Med J Aust. 2007;186:315-317.
10. Brown M, Noursadeghi M, Boyle J, et al. Successful liposomal amphotericin B treatment of Leishmania braziliensis cutaneous leishmaniasis. Br J Dermatol. 2005;153:203-205.
11. Parasites: leishmaniasis. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/parasites/leishmaniasis/index.html. Updated January 10, 2013. Accessed March 5, 2015.
1. Herwaldt BL, Magill AJ. Infectious diseases related to travel: leishmaniasis, cutaneous. Centers for Disease Control and Prevention Web site. http://wwwnc.cdc.gov/travel/yellowbook/2010/chapter-5/cutaneous-leish maniasis.htm. Published August 1, 2013. Accessed March 5, 2015.
2. Mitropolos P, Konidas P, Durkin-Konidas M. New World cutaneous leishmaniasis: updated review of current and future diagnosis and treatment. J Am Acad Dermatol. 2010;63:309-322.
3. Hepburn NC. Cutaneous leishmaniasis: an overview. J Postgrad Med. 2003;49:50-54.
4. Markle WH, Makhoul K. Cutaneous leishmaniasis: recognition and treatment. Am Fam Physician. 2004;69:1455-1460.
5. Couppié P, Clyti E, Sainte Marie D, et al. Disseminated cutaneous leishmaniasis due to Leishmania guyanensis: case of a patient with 425 lesions. Am J Trop Med Hyg. 2004;71:558-560.
6. Nacher M, Carme B, Sainte Marie D, et al. Influence of clinical presentation on the efficacy of a short course of pentamidine in the treatment of cutaneous leishmaniasis in French Guiana. Ann Trop Med Parasitol. 2001;95:331-336.
7. Minodier P, Parola P. Cutaneous leishmaniasis treatment. Trav Med Inf Dis. 2007;5:150-158.
8. Solomon M, Baum S, Barzilai A, et al. Liposomal amphotericin B in comparison to sodium stibogluconate for cutaneous infection due to Leishmania braziliensis. J Am Acad Dermatol. 2007;56:612-616.
9. Konecny P, Stark DJ. An Australian case of New World cutaneous leishmaniasis. Med J Aust. 2007;186:315-317.
10. Brown M, Noursadeghi M, Boyle J, et al. Successful liposomal amphotericin B treatment of Leishmania braziliensis cutaneous leishmaniasis. Br J Dermatol. 2005;153:203-205.
11. Parasites: leishmaniasis. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/parasites/leishmaniasis/index.html. Updated January 10, 2013. Accessed March 5, 2015.
Fibrous Forehead Plaque
The Diagnosis: Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is an autosomal-dominant neurocutaneous syndrome characterized by multiple hamartomas distributed in multiple organs of the body, most commonly the skin, brain, eyes, heart, kidneys, liver, and lungs. In 1908, Vogt1 elucidated the classic diagnostic triad of seizures, mental retardation, and facial angiofibromas (formerly termed adenoma sebaceum). However, the full triad is evident in only 29% of cases; 6% of TSC patients have none of these 3 findings.2 The disease is caused by alterations in 2 TSC genes, TSC1 and TSC2, on chromosomes 9 and 16, respectively. Both are tumor suppressor genes and mutations in either can be responsible for all the complications seen in the disease.3 The gene products of the 2 genes, hamartin and tuberin, appear to act together in the regulation of cell growth by inhibiting a substance known as the mechanistic target of rapamycin.4,5 Up to two-thirds of cases may occur from spontaneous mutations.2,6 The disease is extremely variable in its manifestations, particularly the skin manifestations. This discussion will review the many cutaneous manifestations of TSC, most of them documented in our patient, and their frequency of occurrence.
Early recognition of TSC is vital because prompt implementation of the recommended diagnostic evaluation (eg, neuroimaging studies, electroencephalogram, electrocardiography, renal ultrasonography, chest computed tomography) may prevent serious clinical consequences.7 Neurologic manifestations are the leading cause of morbidity and mortality in patients with TSC.2,8,9 Brain hamartomas in the form of cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas often are responsible for intractable seizures, most commonly as infantile spasms. Approximately 90% to 96% of TSC patients have seizures.6 Renal manifestations are strongly associated with TSC. Angiomyolipoma is the most common renal lesion found in TSC patients.4,9,10 Up to 80% of patients have renal angiomyolipomas. Their incidence increases with age.4,11,12 Tuberous sclerosis complex is known as a cause of epilepsy and mental retardation, but renal disease is a major cause of morbidity.13 Cardiovascular manifestations often are the earliest diagnostic findings in patients with TSC. Rhabdomyoma is the most common primary cardiac tumor in infants and children.11,13,14 The most common ocular findings in TSC are retinal hamartomas, appearing in 40% to 50% of patients.15
Cutaneous features frequently occur and are the most clinically apparent findings of TSC; if overlooked, diagnosis could be delayed, which ultimately increases mortality. The diagnosis of TSC continues to be based primarily on clinical grounds because most patients older than 5 years demonstrate multiple skin lesions.6 In 1992, specific diagnostic clinical criteria were created for TSC, which stratified the clinical features of TSC into 3 tiers—primary, secondary, and tertiary—based on their specificity.16 In subsequent years, some of the classic clinical signs once regarded as pathognomonic for TSC, such as single ungual fibroma or multiple renal angiomyolipomas, were questioned. New modifications to the original diagnostic criteria were necessary given the advances in both TSC clinical information and molecular genetics. In the summer of 1998, a consensus conference held in Annapolis, Maryland, was assembled by the Tuberous Sclerosis Alliance for the purpose of reevaluating and updating the clinical diagnostic criteria.7 The revised criteria were simplified into 2 main categories—major and minor features—based on the diagnostic importance and degree of specificity for TSC of each clinical and radiographic feature (Table 1). There are a number of considerations that merit close attention when applying these clinical criteria. Despite the large number of clinical features delineated, the diagnosis of TSC may be challenging early in life, particularly in patients younger than 2 years. The value of the clinical criteria for early diagnosis and prompt management of TSC is limited by the fact that many stigmata become apparent only in late childhood or adulthood. The lack of pathognomonic signs of TSC can add to the challenge of diagnosing subtle or atypical cases.6

A careful skin examination of individuals at risk for TSC is the best and the easiest method of establishing the diagnosis in most cases. According to Gomez,17 96% of patients with TSC have one or more main skin lesions of the disease, including facial angiofibromas, ungual fibromas, shagreen patches, or hypomelanotic macules. However, the diagnosis is more difficult in small children because many skin lesions become obvious with age; in fact, the full dermatological spectrum may never develop.17
Facial angiofibromas, also known as sebaceous adenomas, are the most visible and unsightly cutaneous manifestations of TSC, often resulting in stigmatization for both the affected individuals and their family members. These facial lesions often do not appear until late childhood or adulthood. In this case, the initial diagnosis often is made by the dermatologist who appreciates the significance of the white macules in a neonate. However, these hypomelanotic macules cannot be detected by the unaided eye, thus a physician would be wise to examine an infant with infantile spasms or mental retardation with a Wood lamp, which accentuates the lesions.2
Hypomelanotic macules were the overwhelmingly most common early findings in TSC. Infants with seizures or other possible stigmata of TSC should be carefully evaluated for these hypomelanotic macules as well as for other associated findings. Hypomelanotic macules can appear anywhere on the body, except the hands, feet, and genitalia; they are most commonly found on the lower extremities. Wood lamp examination reveals an accentuation of the hypopigmentation, which is a decrease in but not an absence of pigmentation. Lance ovate hypomelanotic macules are pathognomonic for TSC, thus the presence of 3 or more macules in an infant seriously suggests the diagnosis of TSC and echocardiography should be performed. The combination of white macules and seizures also increases the likelihood of TSC.18
Angiofibromas typically are found in the butterfly region of the face. Nico et al19 reported 2 patients with the complete syndrome of TSC who had multiple papules on the genital area in addition to classic facial lesions. Thus, it is important to examine the genital region of TSC patients and not misdiagnose these lesions as genital warts.
Jóźwiak et al18 screened 106 children with TSC and listed the prevalence of the most common cutaneous lesions. The results are presented in Table 2.
In a study by Webb et al,2 131 patients with TSC were examined and 126 (96%) exhibited skin signs. Although there was considerable variation in the age of expression of all the skin lesions, there was a trend toward the earlier expression of hypomelanotic macules and forehead fibrous plaques compared with facial angiofibromas and ungual fibromas. Shagreen patches usually were present by puberty. Ungual fibromas appeared for the first time as late as the fifth decade of life and were the only clinical feature in 3 patients. Ungual fibromas were present in 36% (47/131). Ten patients (8%) presented because of the skin manifestations and 21% (28/131) received treatment of symptomatic skin lesions. Two patients had large hamartomas at unusual sites, the occiput and forearm.2
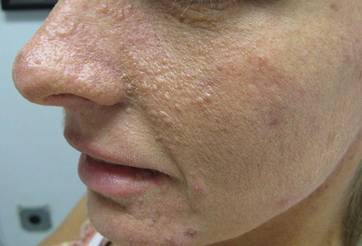
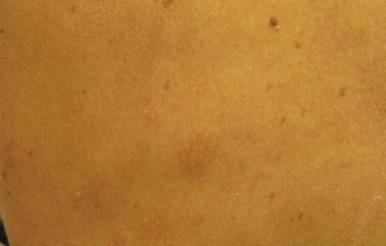
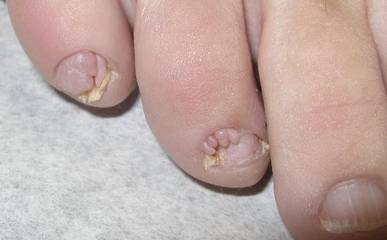
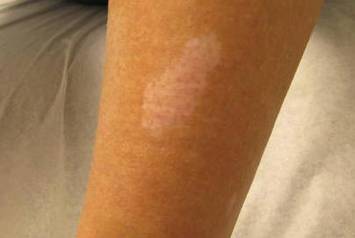
Our 30-year-old patient presented to our clinic accompanied by her grandmother who served as legal guardian and historian for the patient. The patient experienced her first seizure at 15 months of age. She was evaluated by the neurology department at that time and was diagnosed with epilepsy. In the months following, she developed strabismus of the eyes and was examined by the ophthalmology department. Retinal hamartomas were discovered and she was diagnosed with TSC. Her seizure disorder was managed by a neurologist with the use of topiramate. In her 20s, she had renal and cardiac workups, which were negative, and she continued to be free of any genitourinary or cardiac concerns. One year prior to presentation she underwent a brain computed tomography scan after a bicycle accident and was informed that she had “scarlike” lesions within the cerebral cortex. There was no known history of TSC in other family members. On physical examination, she was noted to be delayed in mental development and intelligence. She stated her first notable skin lesions appeared at 10 years of age and were skin tags of the upper back and neck and over the face. Soon after, the cheeks and nose developed numerous shiny brownish pink papules (Figure 1). Over the course of the next 20 years she developed other skin findings noted on our present skin examination. The superior aspect of the mid forehead revealed a 4×2-cm, raised, flesh-colored, firm tumorous plaque. The neck and upper back had multiple 2- to 5-mm, soft, pedunculated, polypoid, brownish pink papules. Her mid back revealed ill-defined, slightly elevated, rosy plaques and macules with a roughened speckled appearance (Figure 2). Several of the toenails and fingernails showed firm, fleshy, spiculelike papules around and under the nail bed (Figure 3). The lower anterior tibia revealed several oval-shaped, flat, well-demarcated, hypopigmented macules (Figure 4). Lesions on the face and mid back were biopsied and histopathology confirmed angiofibroma and shagreen patch, or connective tissue nevus, respectively. Our patient did not undergo treatment of any of these skin lesions, particularly the facial angiofibromas. Cosmetic treatment options such as laser or surgical removal were presented to her, but she declined.
| 
|
A brief review of the literature for treatment options of facial angiofibromas and the fibrous forehead plaque indicated that current treatments include but are not limited to cryotherapy, electrocautery, electrosurgery, shave excision, chemical peels, argon laser, CO2 laser, and radiofrequency equipment.20 For the best outcome, these treatments have to be repeated throughout childhood and teenage years. Rapamycin, also known as sirolimus, and its analogs are new therapeutic options for TSC. Rapamycin is an inhibitor of the mechanistic target of rapamycin and can normalize this unregulated pathway in a TSC patient. Various preclinical models have shown that rapamycin treatment reduces TSC-related tumors, including brain, skin, and kidney tumors.21 A pilot study by Foster et al22 revealed improvement in the facial angiofibromas of 4 children treated with 2 topical preparations of rapamycin. The study revealed that younger patients with smaller angiofibromas had the best response with near-complete clearance. Rapamycin topical preparations were more cost effective than pulsed dye laser under general anesthesia.22
| 
|
Tuberous sclerosis complex is not the only syndrome that can present with facial angiofibromas or numerous skin tag–type lesions such as those in our patient. It is important to also consider Birt-Hogg-Dubé syndrome and multiple endocrine neoplasia type 1 in the differential diagnosis of multiple facial angiofibromas, particularly with adult onset.23
Our case was presented to review the cutaneous manifestations of TSC and their presentation frequency, with the intent to enable the dermatologist to make a more accurate and earlier diagnosis of TSC. In doing so, morbidity of TSC patients could be decreased and quality of life increased.
1. Vogt H. Zur Diagnostik der tuberosen sklerose. Z Erforsch Behandl Jugeudl Schwachsinns. 1908;2:1-6.
2. Webb DW, Clarke A, Fryer A, et al. The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol. 1996;135:1-5.
3. van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805-808.
4. O’Callaghan F. Tuberous sclerosis complex: from basic science to clinical phenotypes. Arch Dis Child. 2004;89:985.
5. Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet. 2003;67:87-96.
6. Schwartz RA. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
7. Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13:624-628.
8. Tworetzky W, McElhinney DB, Margossian R, et al. Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol. 2003;92:487-489.
9. Casper KA, Donnelly LF, Chen B, et al. Tuberous sclerosis complex: renal imaging findings. Radiology. 2002;225:451-456.
10. El-Hashemite N, Zhang H, Henske EP, et al. Mutation in TSC2 and activation of mammalian target of rapamycin signaling pathway in renal angiomyolipoma. Lancet. 2003;361:1348-1349.
11. Jóźwiak S, Schwartz RA, Janniger CK, et al. Usefulness of diagnostic criteria of tuberous sclerosis complex in pediatric patients. J Child Neurol. 2000;15:652-659.
12. Ewalt DH, Sheffield E, Sparagana SP, et al. Renal lesion growth in children with tuberous sclerosis complex. J Urol. 1998;160:141-145.
13. Webb DW, Thomas RD, Osborne JP. Cardiac rhabdomyomas and their association with tuberous sclerosis. Arch Dis Child. 1993;68:367-370.
14. Holley DG, Martin GR, Brenner JI, et al. Diagnosis and management of fetal cardiac tumors: a multicenter experience and review of published reports. J Am Coll Cardiol. 1995;26:516-520.
15. Robertson DM. Ophthalmic manifestations of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:17-25.
16. Roach ES, Smith M, Huttenlocher P, et al. Diagnostic criteria: tuberous sclerosis complex. J Child Neurol. 1992;7:221-224.
17. Gomez MR. Tuberous sclerosis. In: Gomez MR, ed. Neurocutaneous Diseases. A Practical Approach. Boston, MA: Butterworth; 1987:30-52.
18. Jóźwiak S, Schwartz RA, Janniger CK, et al. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Int J Dermatol. 1998;37:911-917.
19. Nico MM, Ito LM, Valente NY. Genital angiofibromas in tuberous sclerosis: two cases. J Dermatol. 1999;26:111-114.
20. Gomes AA, Gomes YV, Lima FB, et al. Multiple facial angiofibromas treated with high frequency equipment. An Bras Dermatol. 2011;86:186-189.
21. Borkowska J, Schwartz RA, Kotulska K, et al. Tuberous sclerosis complex: tumors and tumorigenesis. Int J Dermatol. 2011;50:13-20.
22. Foster RS, Bint LJ, Halbert AR. Topical 0.1% rapamycin for angiofibromas in paediatric patients with tuberous sclerosis: a pilot study of four patients. Australas J Dermatol. 2012;53:52-56.
23. Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:108-111.
The Diagnosis: Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is an autosomal-dominant neurocutaneous syndrome characterized by multiple hamartomas distributed in multiple organs of the body, most commonly the skin, brain, eyes, heart, kidneys, liver, and lungs. In 1908, Vogt1 elucidated the classic diagnostic triad of seizures, mental retardation, and facial angiofibromas (formerly termed adenoma sebaceum). However, the full triad is evident in only 29% of cases; 6% of TSC patients have none of these 3 findings.2 The disease is caused by alterations in 2 TSC genes, TSC1 and TSC2, on chromosomes 9 and 16, respectively. Both are tumor suppressor genes and mutations in either can be responsible for all the complications seen in the disease.3 The gene products of the 2 genes, hamartin and tuberin, appear to act together in the regulation of cell growth by inhibiting a substance known as the mechanistic target of rapamycin.4,5 Up to two-thirds of cases may occur from spontaneous mutations.2,6 The disease is extremely variable in its manifestations, particularly the skin manifestations. This discussion will review the many cutaneous manifestations of TSC, most of them documented in our patient, and their frequency of occurrence.
Early recognition of TSC is vital because prompt implementation of the recommended diagnostic evaluation (eg, neuroimaging studies, electroencephalogram, electrocardiography, renal ultrasonography, chest computed tomography) may prevent serious clinical consequences.7 Neurologic manifestations are the leading cause of morbidity and mortality in patients with TSC.2,8,9 Brain hamartomas in the form of cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas often are responsible for intractable seizures, most commonly as infantile spasms. Approximately 90% to 96% of TSC patients have seizures.6 Renal manifestations are strongly associated with TSC. Angiomyolipoma is the most common renal lesion found in TSC patients.4,9,10 Up to 80% of patients have renal angiomyolipomas. Their incidence increases with age.4,11,12 Tuberous sclerosis complex is known as a cause of epilepsy and mental retardation, but renal disease is a major cause of morbidity.13 Cardiovascular manifestations often are the earliest diagnostic findings in patients with TSC. Rhabdomyoma is the most common primary cardiac tumor in infants and children.11,13,14 The most common ocular findings in TSC are retinal hamartomas, appearing in 40% to 50% of patients.15
Cutaneous features frequently occur and are the most clinically apparent findings of TSC; if overlooked, diagnosis could be delayed, which ultimately increases mortality. The diagnosis of TSC continues to be based primarily on clinical grounds because most patients older than 5 years demonstrate multiple skin lesions.6 In 1992, specific diagnostic clinical criteria were created for TSC, which stratified the clinical features of TSC into 3 tiers—primary, secondary, and tertiary—based on their specificity.16 In subsequent years, some of the classic clinical signs once regarded as pathognomonic for TSC, such as single ungual fibroma or multiple renal angiomyolipomas, were questioned. New modifications to the original diagnostic criteria were necessary given the advances in both TSC clinical information and molecular genetics. In the summer of 1998, a consensus conference held in Annapolis, Maryland, was assembled by the Tuberous Sclerosis Alliance for the purpose of reevaluating and updating the clinical diagnostic criteria.7 The revised criteria were simplified into 2 main categories—major and minor features—based on the diagnostic importance and degree of specificity for TSC of each clinical and radiographic feature (Table 1). There are a number of considerations that merit close attention when applying these clinical criteria. Despite the large number of clinical features delineated, the diagnosis of TSC may be challenging early in life, particularly in patients younger than 2 years. The value of the clinical criteria for early diagnosis and prompt management of TSC is limited by the fact that many stigmata become apparent only in late childhood or adulthood. The lack of pathognomonic signs of TSC can add to the challenge of diagnosing subtle or atypical cases.6
A careful skin examination of individuals at risk for TSC is the best and the easiest method of establishing the diagnosis in most cases. According to Gomez,17 96% of patients with TSC have one or more main skin lesions of the disease, including facial angiofibromas, ungual fibromas, shagreen patches, or hypomelanotic macules. However, the diagnosis is more difficult in small children because many skin lesions become obvious with age; in fact, the full dermatological spectrum may never develop.17
Facial angiofibromas, also known as sebaceous adenomas, are the most visible and unsightly cutaneous manifestations of TSC, often resulting in stigmatization for both the affected individuals and their family members. These facial lesions often do not appear until late childhood or adulthood. In this case, the initial diagnosis often is made by the dermatologist who appreciates the significance of the white macules in a neonate. However, these hypomelanotic macules cannot be detected by the unaided eye, thus a physician would be wise to examine an infant with infantile spasms or mental retardation with a Wood lamp, which accentuates the lesions.2
Hypomelanotic macules were the overwhelmingly most common early findings in TSC. Infants with seizures or other possible stigmata of TSC should be carefully evaluated for these hypomelanotic macules as well as for other associated findings. Hypomelanotic macules can appear anywhere on the body, except the hands, feet, and genitalia; they are most commonly found on the lower extremities. Wood lamp examination reveals an accentuation of the hypopigmentation, which is a decrease in but not an absence of pigmentation. Lance ovate hypomelanotic macules are pathognomonic for TSC, thus the presence of 3 or more macules in an infant seriously suggests the diagnosis of TSC and echocardiography should be performed. The combination of white macules and seizures also increases the likelihood of TSC.18
Angiofibromas typically are found in the butterfly region of the face. Nico et al19 reported 2 patients with the complete syndrome of TSC who had multiple papules on the genital area in addition to classic facial lesions. Thus, it is important to examine the genital region of TSC patients and not misdiagnose these lesions as genital warts.
Jóźwiak et al18 screened 106 children with TSC and listed the prevalence of the most common cutaneous lesions. The results are presented in Table 2.
In a study by Webb et al,2 131 patients with TSC were examined and 126 (96%) exhibited skin signs. Although there was considerable variation in the age of expression of all the skin lesions, there was a trend toward the earlier expression of hypomelanotic macules and forehead fibrous plaques compared with facial angiofibromas and ungual fibromas. Shagreen patches usually were present by puberty. Ungual fibromas appeared for the first time as late as the fifth decade of life and were the only clinical feature in 3 patients. Ungual fibromas were present in 36% (47/131). Ten patients (8%) presented because of the skin manifestations and 21% (28/131) received treatment of symptomatic skin lesions. Two patients had large hamartomas at unusual sites, the occiput and forearm.2
Our 30-year-old patient presented to our clinic accompanied by her grandmother who served as legal guardian and historian for the patient. The patient experienced her first seizure at 15 months of age. She was evaluated by the neurology department at that time and was diagnosed with epilepsy. In the months following, she developed strabismus of the eyes and was examined by the ophthalmology department. Retinal hamartomas were discovered and she was diagnosed with TSC. Her seizure disorder was managed by a neurologist with the use of topiramate. In her 20s, she had renal and cardiac workups, which were negative, and she continued to be free of any genitourinary or cardiac concerns. One year prior to presentation she underwent a brain computed tomography scan after a bicycle accident and was informed that she had “scarlike” lesions within the cerebral cortex. There was no known history of TSC in other family members. On physical examination, she was noted to be delayed in mental development and intelligence. She stated her first notable skin lesions appeared at 10 years of age and were skin tags of the upper back and neck and over the face. Soon after, the cheeks and nose developed numerous shiny brownish pink papules (Figure 1). Over the course of the next 20 years she developed other skin findings noted on our present skin examination. The superior aspect of the mid forehead revealed a 4×2-cm, raised, flesh-colored, firm tumorous plaque. The neck and upper back had multiple 2- to 5-mm, soft, pedunculated, polypoid, brownish pink papules. Her mid back revealed ill-defined, slightly elevated, rosy plaques and macules with a roughened speckled appearance (Figure 2). Several of the toenails and fingernails showed firm, fleshy, spiculelike papules around and under the nail bed (Figure 3). The lower anterior tibia revealed several oval-shaped, flat, well-demarcated, hypopigmented macules (Figure 4). Lesions on the face and mid back were biopsied and histopathology confirmed angiofibroma and shagreen patch, or connective tissue nevus, respectively. Our patient did not undergo treatment of any of these skin lesions, particularly the facial angiofibromas. Cosmetic treatment options such as laser or surgical removal were presented to her, but she declined.
|
|
|
A brief review of the literature for treatment options of facial angiofibromas and the fibrous forehead plaque indicated that current treatments include but are not limited to cryotherapy, electrocautery, electrosurgery, shave excision, chemical peels, argon laser, CO2 laser, and radiofrequency equipment.20 For the best outcome, these treatments have to be repeated throughout childhood and teenage years. Rapamycin, also known as sirolimus, and its analogs are new therapeutic options for TSC. Rapamycin is an inhibitor of the mechanistic target of rapamycin and can normalize this unregulated pathway in a TSC patient. Various preclinical models have shown that rapamycin treatment reduces TSC-related tumors, including brain, skin, and kidney tumors.21 A pilot study by Foster et al22 revealed improvement in the facial angiofibromas of 4 children treated with 2 topical preparations of rapamycin. The study revealed that younger patients with smaller angiofibromas had the best response with near-complete clearance. Rapamycin topical preparations were more cost effective than pulsed dye laser under general anesthesia.22
|
|
|
Tuberous sclerosis complex is not the only syndrome that can present with facial angiofibromas or numerous skin tag–type lesions such as those in our patient. It is important to also consider Birt-Hogg-Dubé syndrome and multiple endocrine neoplasia type 1 in the differential diagnosis of multiple facial angiofibromas, particularly with adult onset.23
Our case was presented to review the cutaneous manifestations of TSC and their presentation frequency, with the intent to enable the dermatologist to make a more accurate and earlier diagnosis of TSC. In doing so, morbidity of TSC patients could be decreased and quality of life increased.
The Diagnosis: Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is an autosomal-dominant neurocutaneous syndrome characterized by multiple hamartomas distributed in multiple organs of the body, most commonly the skin, brain, eyes, heart, kidneys, liver, and lungs. In 1908, Vogt1 elucidated the classic diagnostic triad of seizures, mental retardation, and facial angiofibromas (formerly termed adenoma sebaceum). However, the full triad is evident in only 29% of cases; 6% of TSC patients have none of these 3 findings.2 The disease is caused by alterations in 2 TSC genes, TSC1 and TSC2, on chromosomes 9 and 16, respectively. Both are tumor suppressor genes and mutations in either can be responsible for all the complications seen in the disease.3 The gene products of the 2 genes, hamartin and tuberin, appear to act together in the regulation of cell growth by inhibiting a substance known as the mechanistic target of rapamycin.4,5 Up to two-thirds of cases may occur from spontaneous mutations.2,6 The disease is extremely variable in its manifestations, particularly the skin manifestations. This discussion will review the many cutaneous manifestations of TSC, most of them documented in our patient, and their frequency of occurrence.
Early recognition of TSC is vital because prompt implementation of the recommended diagnostic evaluation (eg, neuroimaging studies, electroencephalogram, electrocardiography, renal ultrasonography, chest computed tomography) may prevent serious clinical consequences.7 Neurologic manifestations are the leading cause of morbidity and mortality in patients with TSC.2,8,9 Brain hamartomas in the form of cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas often are responsible for intractable seizures, most commonly as infantile spasms. Approximately 90% to 96% of TSC patients have seizures.6 Renal manifestations are strongly associated with TSC. Angiomyolipoma is the most common renal lesion found in TSC patients.4,9,10 Up to 80% of patients have renal angiomyolipomas. Their incidence increases with age.4,11,12 Tuberous sclerosis complex is known as a cause of epilepsy and mental retardation, but renal disease is a major cause of morbidity.13 Cardiovascular manifestations often are the earliest diagnostic findings in patients with TSC. Rhabdomyoma is the most common primary cardiac tumor in infants and children.11,13,14 The most common ocular findings in TSC are retinal hamartomas, appearing in 40% to 50% of patients.15
Cutaneous features frequently occur and are the most clinically apparent findings of TSC; if overlooked, diagnosis could be delayed, which ultimately increases mortality. The diagnosis of TSC continues to be based primarily on clinical grounds because most patients older than 5 years demonstrate multiple skin lesions.6 In 1992, specific diagnostic clinical criteria were created for TSC, which stratified the clinical features of TSC into 3 tiers—primary, secondary, and tertiary—based on their specificity.16 In subsequent years, some of the classic clinical signs once regarded as pathognomonic for TSC, such as single ungual fibroma or multiple renal angiomyolipomas, were questioned. New modifications to the original diagnostic criteria were necessary given the advances in both TSC clinical information and molecular genetics. In the summer of 1998, a consensus conference held in Annapolis, Maryland, was assembled by the Tuberous Sclerosis Alliance for the purpose of reevaluating and updating the clinical diagnostic criteria.7 The revised criteria were simplified into 2 main categories—major and minor features—based on the diagnostic importance and degree of specificity for TSC of each clinical and radiographic feature (Table 1). There are a number of considerations that merit close attention when applying these clinical criteria. Despite the large number of clinical features delineated, the diagnosis of TSC may be challenging early in life, particularly in patients younger than 2 years. The value of the clinical criteria for early diagnosis and prompt management of TSC is limited by the fact that many stigmata become apparent only in late childhood or adulthood. The lack of pathognomonic signs of TSC can add to the challenge of diagnosing subtle or atypical cases.6
A careful skin examination of individuals at risk for TSC is the best and the easiest method of establishing the diagnosis in most cases. According to Gomez,17 96% of patients with TSC have one or more main skin lesions of the disease, including facial angiofibromas, ungual fibromas, shagreen patches, or hypomelanotic macules. However, the diagnosis is more difficult in small children because many skin lesions become obvious with age; in fact, the full dermatological spectrum may never develop.17
Facial angiofibromas, also known as sebaceous adenomas, are the most visible and unsightly cutaneous manifestations of TSC, often resulting in stigmatization for both the affected individuals and their family members. These facial lesions often do not appear until late childhood or adulthood. In this case, the initial diagnosis often is made by the dermatologist who appreciates the significance of the white macules in a neonate. However, these hypomelanotic macules cannot be detected by the unaided eye, thus a physician would be wise to examine an infant with infantile spasms or mental retardation with a Wood lamp, which accentuates the lesions.2
Hypomelanotic macules were the overwhelmingly most common early findings in TSC. Infants with seizures or other possible stigmata of TSC should be carefully evaluated for these hypomelanotic macules as well as for other associated findings. Hypomelanotic macules can appear anywhere on the body, except the hands, feet, and genitalia; they are most commonly found on the lower extremities. Wood lamp examination reveals an accentuation of the hypopigmentation, which is a decrease in but not an absence of pigmentation. Lance ovate hypomelanotic macules are pathognomonic for TSC, thus the presence of 3 or more macules in an infant seriously suggests the diagnosis of TSC and echocardiography should be performed. The combination of white macules and seizures also increases the likelihood of TSC.18
Angiofibromas typically are found in the butterfly region of the face. Nico et al19 reported 2 patients with the complete syndrome of TSC who had multiple papules on the genital area in addition to classic facial lesions. Thus, it is important to examine the genital region of TSC patients and not misdiagnose these lesions as genital warts.
Jóźwiak et al18 screened 106 children with TSC and listed the prevalence of the most common cutaneous lesions. The results are presented in Table 2.
In a study by Webb et al,2 131 patients with TSC were examined and 126 (96%) exhibited skin signs. Although there was considerable variation in the age of expression of all the skin lesions, there was a trend toward the earlier expression of hypomelanotic macules and forehead fibrous plaques compared with facial angiofibromas and ungual fibromas. Shagreen patches usually were present by puberty. Ungual fibromas appeared for the first time as late as the fifth decade of life and were the only clinical feature in 3 patients. Ungual fibromas were present in 36% (47/131). Ten patients (8%) presented because of the skin manifestations and 21% (28/131) received treatment of symptomatic skin lesions. Two patients had large hamartomas at unusual sites, the occiput and forearm.2
Our 30-year-old patient presented to our clinic accompanied by her grandmother who served as legal guardian and historian for the patient. The patient experienced her first seizure at 15 months of age. She was evaluated by the neurology department at that time and was diagnosed with epilepsy. In the months following, she developed strabismus of the eyes and was examined by the ophthalmology department. Retinal hamartomas were discovered and she was diagnosed with TSC. Her seizure disorder was managed by a neurologist with the use of topiramate. In her 20s, she had renal and cardiac workups, which were negative, and she continued to be free of any genitourinary or cardiac concerns. One year prior to presentation she underwent a brain computed tomography scan after a bicycle accident and was informed that she had “scarlike” lesions within the cerebral cortex. There was no known history of TSC in other family members. On physical examination, she was noted to be delayed in mental development and intelligence. She stated her first notable skin lesions appeared at 10 years of age and were skin tags of the upper back and neck and over the face. Soon after, the cheeks and nose developed numerous shiny brownish pink papules (Figure 1). Over the course of the next 20 years she developed other skin findings noted on our present skin examination. The superior aspect of the mid forehead revealed a 4×2-cm, raised, flesh-colored, firm tumorous plaque. The neck and upper back had multiple 2- to 5-mm, soft, pedunculated, polypoid, brownish pink papules. Her mid back revealed ill-defined, slightly elevated, rosy plaques and macules with a roughened speckled appearance (Figure 2). Several of the toenails and fingernails showed firm, fleshy, spiculelike papules around and under the nail bed (Figure 3). The lower anterior tibia revealed several oval-shaped, flat, well-demarcated, hypopigmented macules (Figure 4). Lesions on the face and mid back were biopsied and histopathology confirmed angiofibroma and shagreen patch, or connective tissue nevus, respectively. Our patient did not undergo treatment of any of these skin lesions, particularly the facial angiofibromas. Cosmetic treatment options such as laser or surgical removal were presented to her, but she declined.
|
|
|
A brief review of the literature for treatment options of facial angiofibromas and the fibrous forehead plaque indicated that current treatments include but are not limited to cryotherapy, electrocautery, electrosurgery, shave excision, chemical peels, argon laser, CO2 laser, and radiofrequency equipment.20 For the best outcome, these treatments have to be repeated throughout childhood and teenage years. Rapamycin, also known as sirolimus, and its analogs are new therapeutic options for TSC. Rapamycin is an inhibitor of the mechanistic target of rapamycin and can normalize this unregulated pathway in a TSC patient. Various preclinical models have shown that rapamycin treatment reduces TSC-related tumors, including brain, skin, and kidney tumors.21 A pilot study by Foster et al22 revealed improvement in the facial angiofibromas of 4 children treated with 2 topical preparations of rapamycin. The study revealed that younger patients with smaller angiofibromas had the best response with near-complete clearance. Rapamycin topical preparations were more cost effective than pulsed dye laser under general anesthesia.22
|
|
|
Tuberous sclerosis complex is not the only syndrome that can present with facial angiofibromas or numerous skin tag–type lesions such as those in our patient. It is important to also consider Birt-Hogg-Dubé syndrome and multiple endocrine neoplasia type 1 in the differential diagnosis of multiple facial angiofibromas, particularly with adult onset.23
Our case was presented to review the cutaneous manifestations of TSC and their presentation frequency, with the intent to enable the dermatologist to make a more accurate and earlier diagnosis of TSC. In doing so, morbidity of TSC patients could be decreased and quality of life increased.
1. Vogt H. Zur Diagnostik der tuberosen sklerose. Z Erforsch Behandl Jugeudl Schwachsinns. 1908;2:1-6.
2. Webb DW, Clarke A, Fryer A, et al. The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol. 1996;135:1-5.
3. van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805-808.
4. O’Callaghan F. Tuberous sclerosis complex: from basic science to clinical phenotypes. Arch Dis Child. 2004;89:985.
5. Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet. 2003;67:87-96.
6. Schwartz RA. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
7. Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13:624-628.
8. Tworetzky W, McElhinney DB, Margossian R, et al. Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol. 2003;92:487-489.
9. Casper KA, Donnelly LF, Chen B, et al. Tuberous sclerosis complex: renal imaging findings. Radiology. 2002;225:451-456.
10. El-Hashemite N, Zhang H, Henske EP, et al. Mutation in TSC2 and activation of mammalian target of rapamycin signaling pathway in renal angiomyolipoma. Lancet. 2003;361:1348-1349.
11. Jóźwiak S, Schwartz RA, Janniger CK, et al. Usefulness of diagnostic criteria of tuberous sclerosis complex in pediatric patients. J Child Neurol. 2000;15:652-659.
12. Ewalt DH, Sheffield E, Sparagana SP, et al. Renal lesion growth in children with tuberous sclerosis complex. J Urol. 1998;160:141-145.
13. Webb DW, Thomas RD, Osborne JP. Cardiac rhabdomyomas and their association with tuberous sclerosis. Arch Dis Child. 1993;68:367-370.
14. Holley DG, Martin GR, Brenner JI, et al. Diagnosis and management of fetal cardiac tumors: a multicenter experience and review of published reports. J Am Coll Cardiol. 1995;26:516-520.
15. Robertson DM. Ophthalmic manifestations of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:17-25.
16. Roach ES, Smith M, Huttenlocher P, et al. Diagnostic criteria: tuberous sclerosis complex. J Child Neurol. 1992;7:221-224.
17. Gomez MR. Tuberous sclerosis. In: Gomez MR, ed. Neurocutaneous Diseases. A Practical Approach. Boston, MA: Butterworth; 1987:30-52.
18. Jóźwiak S, Schwartz RA, Janniger CK, et al. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Int J Dermatol. 1998;37:911-917.
19. Nico MM, Ito LM, Valente NY. Genital angiofibromas in tuberous sclerosis: two cases. J Dermatol. 1999;26:111-114.
20. Gomes AA, Gomes YV, Lima FB, et al. Multiple facial angiofibromas treated with high frequency equipment. An Bras Dermatol. 2011;86:186-189.
21. Borkowska J, Schwartz RA, Kotulska K, et al. Tuberous sclerosis complex: tumors and tumorigenesis. Int J Dermatol. 2011;50:13-20.
22. Foster RS, Bint LJ, Halbert AR. Topical 0.1% rapamycin for angiofibromas in paediatric patients with tuberous sclerosis: a pilot study of four patients. Australas J Dermatol. 2012;53:52-56.
23. Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:108-111.
1. Vogt H. Zur Diagnostik der tuberosen sklerose. Z Erforsch Behandl Jugeudl Schwachsinns. 1908;2:1-6.
2. Webb DW, Clarke A, Fryer A, et al. The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol. 1996;135:1-5.
3. van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805-808.
4. O’Callaghan F. Tuberous sclerosis complex: from basic science to clinical phenotypes. Arch Dis Child. 2004;89:985.
5. Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet. 2003;67:87-96.
6. Schwartz RA. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
7. Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13:624-628.
8. Tworetzky W, McElhinney DB, Margossian R, et al. Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol. 2003;92:487-489.
9. Casper KA, Donnelly LF, Chen B, et al. Tuberous sclerosis complex: renal imaging findings. Radiology. 2002;225:451-456.
10. El-Hashemite N, Zhang H, Henske EP, et al. Mutation in TSC2 and activation of mammalian target of rapamycin signaling pathway in renal angiomyolipoma. Lancet. 2003;361:1348-1349.
11. Jóźwiak S, Schwartz RA, Janniger CK, et al. Usefulness of diagnostic criteria of tuberous sclerosis complex in pediatric patients. J Child Neurol. 2000;15:652-659.
12. Ewalt DH, Sheffield E, Sparagana SP, et al. Renal lesion growth in children with tuberous sclerosis complex. J Urol. 1998;160:141-145.
13. Webb DW, Thomas RD, Osborne JP. Cardiac rhabdomyomas and their association with tuberous sclerosis. Arch Dis Child. 1993;68:367-370.
14. Holley DG, Martin GR, Brenner JI, et al. Diagnosis and management of fetal cardiac tumors: a multicenter experience and review of published reports. J Am Coll Cardiol. 1995;26:516-520.
15. Robertson DM. Ophthalmic manifestations of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:17-25.
16. Roach ES, Smith M, Huttenlocher P, et al. Diagnostic criteria: tuberous sclerosis complex. J Child Neurol. 1992;7:221-224.
17. Gomez MR. Tuberous sclerosis. In: Gomez MR, ed. Neurocutaneous Diseases. A Practical Approach. Boston, MA: Butterworth; 1987:30-52.
18. Jóźwiak S, Schwartz RA, Janniger CK, et al. Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Int J Dermatol. 1998;37:911-917.
19. Nico MM, Ito LM, Valente NY. Genital angiofibromas in tuberous sclerosis: two cases. J Dermatol. 1999;26:111-114.
20. Gomes AA, Gomes YV, Lima FB, et al. Multiple facial angiofibromas treated with high frequency equipment. An Bras Dermatol. 2011;86:186-189.
21. Borkowska J, Schwartz RA, Kotulska K, et al. Tuberous sclerosis complex: tumors and tumorigenesis. Int J Dermatol. 2011;50:13-20.
22. Foster RS, Bint LJ, Halbert AR. Topical 0.1% rapamycin for angiofibromas in paediatric patients with tuberous sclerosis: a pilot study of four patients. Australas J Dermatol. 2012;53:52-56.
23. Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:108-111.
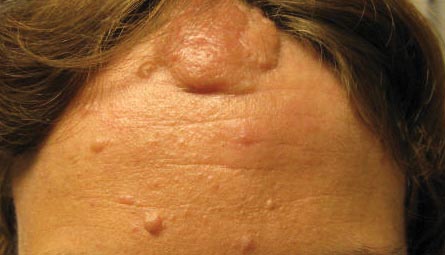
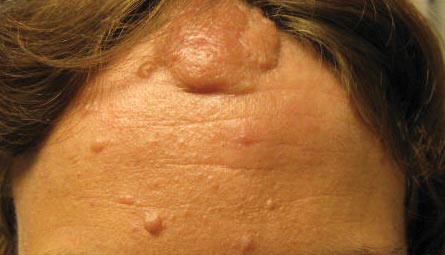
A 30-year-old woman presented to our dermatology clinic for evaluation and treatment options of a large 4×2-cm, firm, flesh-colored plaque on the superior aspect of the mid forehead. The plaque initially presented at 10 years of age and had grown since then. She reported no prior treatment of this nontender lesion and denied a family history of skin problems. She also had numerous skin tags all over the face, neck, and upper back that had increased in number for most of her adult life. In reviewing her medical history, she reported experiencing her first seizure at 15 months of age. She was evaluated by the neurology department at that time and was diagnosed with epilepsy.
Painful Purpura and Cutaneous Necrosis
The Diagnosis: Levamisole-Contaminated Cocaine
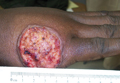
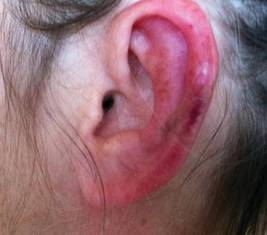
Physical examination revealed scattered palpable purpura including the nasal tip and large necrotizing skin lesions with bullae (Figure 1). Retiform purpura was noted on the patient’s trunk and legs. Purpuric plaques in various stages of necrosis were identified on the arms, trunk, breasts, and thighs, with an early lesion involving the left ear (Figure 2). Vitals revealed a blood pressure of 151/79 mm Hg and a temperature of 37.3°C. Although the patient initially denied illicit drug use, she later admitted to smoking crack cocaine prior to the skin eruption.
|
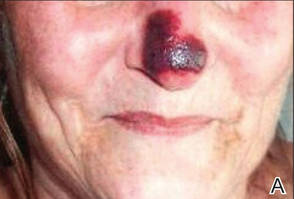
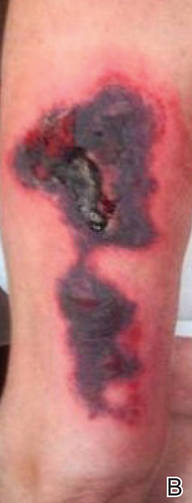
Levamisole, an agent used in the veterinary setting to deworm cattle and pigs, is a common additive found in nearly 77% of the seized cocaine supplies in the United States.1 Because of its immunomodulator effects, the agent was once used in humans to treat conditions such as colon cancer, rheumatoid arthritis, and nephritic syndrome. Therapeutic levamisole use has been associated with purpura on the external ears, cheeks, and nasal tip. The predilection for purpura on the ears was first described in children using levam-isole as treatment of nephritic syndrome.2 Antinuclear cytoplasmic antibodies (ANCA) and antiphospholipid antibodies also were associated with therapeutic use.3 Reported effects of levamisole in cocaine users include fever, agranulocytosis, and infection. The mechanism is currently unknown but is thought to be an immunological process as evidenced by the presence of positive autoantibodies.4 Characteristic purpura in a cocaine abuser should alert the physician to consider levamisole as the culprit.
The diagnosis is largely a clinical one and other serious etiologies must be ruled out. The differential should include purpura fulminans, a rare hemorrhagic condition caused by severe infection, such as meningococcemia or deficiency of the vitamin K–dependent anticoagulants protein C and protein S. Given our patient’s history of hepatitis, purpura fulminans could have been likely. Purpura fulminans is rapidly progressive and is usually accompanied by disseminated intravascular coagulation. Coagulation studies and evidence of a consumptive coagulopathy such as thrombocytopenia, prolonged partial thromboplastin time, and activated partial thromboplastin time would favor this diagnosis. These studies as well as protein C and protein S were normal in our patient. Mixed cryoglobulinemia also should be suspected in a patient with hepatitis C virus who presents with vasculitis and arthralgia. An undetectable viral load in addition to a negative assay for cryoglobulins and normal complement (C3 and C4) levels make this diagnosis unlikely. Further, the purpura in cryoglobulinemia is typically confined to the lower extremities.5 Warfarin-induced skin necrosis also should be considered in patients who are taking warfarin.
Laboratory tests can be used to confirm the presence of infection, agranulocytosis, and coagulopathy. Although urine drug screen can confirm exposure to cocaine, levamisole is not detected in routine toxicology screening. Its half-life is between 5 and 6 hours; therefore, detection in blood or urine should be done within 48 hours of exposure.3,4 Unfortunately, our patient presented 2 weeks after cocaine use, making detection of levamisole unfeasible. Rheumatologic screening also is appropriate in cases of suspected vasculitis. Our patient had a negative antinuclear antibody but tested positive for lupus anticoagulant and perinuclear ANCA. IgA and IgG anticardiolipin were negative with an indeterminate IgM anticardiolipin level. The literature supports these findings, as ANCA antibodies occurred in more than 90% of reported cases.3 Further, IgM anticardiolipin antibodies and lupus anticoagulant were positive in 65% and 51% of cases, respectively.3 Leukocyte and neutrophil count were normal in our patient.
Management of these patients is mainly supportive. Complete avoidance of the offending agent is absolutely essential for resolution and avoidance of recurrence. Provided that the patient abstains from cocaine, the skin findings typically resolve in 2 to 3 weeks.3,6 The antibodies, however, may be present for up to 14 months.2,3,6 Treatment with steroids and other immunosuppressants has been reported, but evidence is lacking on their role in the resolution of the lesions.3 In patients who develop a temporary antiphospholipid syndrome, aspirin may be recommended as a preventative measure.6 Pain management, treatment of secondary infection especially in situations with agranulocytosis, and surgical debridement of wounds are all potential problems that can arise.
Our patient had complete resolution of the purpura involving the nose and ear over the course of 3 weeks. She did, however, have to undergo surgical debridement of the nonhealing full-thickness necrotic lesions on her legs and arms.
The 2013 National Survey on Drug Use and Health estimates 1.5 million individuals aged 12 years or older were current (within the last month) users of cocaine.7 With levamisole becoming more prevalent in cocaine supplies, clinicians should be aware of this emerging condition. Rapid recognition can spare the patient unnecessary testing and inappropriate treatment regimens. Because testing for levamisole is difficult and not routinely performed, this case highlights the importance of clinical clues and an accurate social history in clinching the diagnosis.
1. National drug threat assessment: 2011. National Drug Intelligence Center Web site. http://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf. Published August 2011. Accessed March 9, 2015.
2. Rongioletti F, Gio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;40:948-951.
3. Gulati S, Donato AA. Lupus anticoagulant and ANCA associated thrombotic vasculopathy due to cocaine contaminated with levamisole: a case report and review of the literature. J Thromb Thrombolysis. 2012;34:7-10.
4. de la Hera I, Sanz V, Cullen D, et al. Necrosis of ears after cocaine probably adulterated with levamisole. Dermatology. 2011;223:25-28.
5. Seo P. Immune complex-mediated vasculitis. In: Klippel JH. Primer on the Rheumatic Diseases. 13th ed. New York, NY: Springer Science+Business Media, LLC; 2008:427-443.
6. Ching JA, Smith DS. Levamisole-induced necrosis of skin, soft tissue, and bone: case report and review of literature. J Burn Care Res. 2012;33:e1-e5.
7. Substance Abuse and Mental Health Services Administration. Results from the 2013 National Survey on Drug Use and Health: Summary of National Findings. NSDUH Series H-48, HHS Publication No. (SMA) 14-4863. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2014.
The Diagnosis: Levamisole-Contaminated Cocaine
Physical examination revealed scattered palpable purpura including the nasal tip and large necrotizing skin lesions with bullae (Figure 1). Retiform purpura was noted on the patient’s trunk and legs. Purpuric plaques in various stages of necrosis were identified on the arms, trunk, breasts, and thighs, with an early lesion involving the left ear (Figure 2). Vitals revealed a blood pressure of 151/79 mm Hg and a temperature of 37.3°C. Although the patient initially denied illicit drug use, she later admitted to smoking crack cocaine prior to the skin eruption.
|
Levamisole, an agent used in the veterinary setting to deworm cattle and pigs, is a common additive found in nearly 77% of the seized cocaine supplies in the United States.1 Because of its immunomodulator effects, the agent was once used in humans to treat conditions such as colon cancer, rheumatoid arthritis, and nephritic syndrome. Therapeutic levamisole use has been associated with purpura on the external ears, cheeks, and nasal tip. The predilection for purpura on the ears was first described in children using levam-isole as treatment of nephritic syndrome.2 Antinuclear cytoplasmic antibodies (ANCA) and antiphospholipid antibodies also were associated with therapeutic use.3 Reported effects of levamisole in cocaine users include fever, agranulocytosis, and infection. The mechanism is currently unknown but is thought to be an immunological process as evidenced by the presence of positive autoantibodies.4 Characteristic purpura in a cocaine abuser should alert the physician to consider levamisole as the culprit.
The diagnosis is largely a clinical one and other serious etiologies must be ruled out. The differential should include purpura fulminans, a rare hemorrhagic condition caused by severe infection, such as meningococcemia or deficiency of the vitamin K–dependent anticoagulants protein C and protein S. Given our patient’s history of hepatitis, purpura fulminans could have been likely. Purpura fulminans is rapidly progressive and is usually accompanied by disseminated intravascular coagulation. Coagulation studies and evidence of a consumptive coagulopathy such as thrombocytopenia, prolonged partial thromboplastin time, and activated partial thromboplastin time would favor this diagnosis. These studies as well as protein C and protein S were normal in our patient. Mixed cryoglobulinemia also should be suspected in a patient with hepatitis C virus who presents with vasculitis and arthralgia. An undetectable viral load in addition to a negative assay for cryoglobulins and normal complement (C3 and C4) levels make this diagnosis unlikely. Further, the purpura in cryoglobulinemia is typically confined to the lower extremities.5 Warfarin-induced skin necrosis also should be considered in patients who are taking warfarin.
Laboratory tests can be used to confirm the presence of infection, agranulocytosis, and coagulopathy. Although urine drug screen can confirm exposure to cocaine, levamisole is not detected in routine toxicology screening. Its half-life is between 5 and 6 hours; therefore, detection in blood or urine should be done within 48 hours of exposure.3,4 Unfortunately, our patient presented 2 weeks after cocaine use, making detection of levamisole unfeasible. Rheumatologic screening also is appropriate in cases of suspected vasculitis. Our patient had a negative antinuclear antibody but tested positive for lupus anticoagulant and perinuclear ANCA. IgA and IgG anticardiolipin were negative with an indeterminate IgM anticardiolipin level. The literature supports these findings, as ANCA antibodies occurred in more than 90% of reported cases.3 Further, IgM anticardiolipin antibodies and lupus anticoagulant were positive in 65% and 51% of cases, respectively.3 Leukocyte and neutrophil count were normal in our patient.
Management of these patients is mainly supportive. Complete avoidance of the offending agent is absolutely essential for resolution and avoidance of recurrence. Provided that the patient abstains from cocaine, the skin findings typically resolve in 2 to 3 weeks.3,6 The antibodies, however, may be present for up to 14 months.2,3,6 Treatment with steroids and other immunosuppressants has been reported, but evidence is lacking on their role in the resolution of the lesions.3 In patients who develop a temporary antiphospholipid syndrome, aspirin may be recommended as a preventative measure.6 Pain management, treatment of secondary infection especially in situations with agranulocytosis, and surgical debridement of wounds are all potential problems that can arise.
Our patient had complete resolution of the purpura involving the nose and ear over the course of 3 weeks. She did, however, have to undergo surgical debridement of the nonhealing full-thickness necrotic lesions on her legs and arms.
The 2013 National Survey on Drug Use and Health estimates 1.5 million individuals aged 12 years or older were current (within the last month) users of cocaine.7 With levamisole becoming more prevalent in cocaine supplies, clinicians should be aware of this emerging condition. Rapid recognition can spare the patient unnecessary testing and inappropriate treatment regimens. Because testing for levamisole is difficult and not routinely performed, this case highlights the importance of clinical clues and an accurate social history in clinching the diagnosis.
The Diagnosis: Levamisole-Contaminated Cocaine
Physical examination revealed scattered palpable purpura including the nasal tip and large necrotizing skin lesions with bullae (Figure 1). Retiform purpura was noted on the patient’s trunk and legs. Purpuric plaques in various stages of necrosis were identified on the arms, trunk, breasts, and thighs, with an early lesion involving the left ear (Figure 2). Vitals revealed a blood pressure of 151/79 mm Hg and a temperature of 37.3°C. Although the patient initially denied illicit drug use, she later admitted to smoking crack cocaine prior to the skin eruption.
|
Levamisole, an agent used in the veterinary setting to deworm cattle and pigs, is a common additive found in nearly 77% of the seized cocaine supplies in the United States.1 Because of its immunomodulator effects, the agent was once used in humans to treat conditions such as colon cancer, rheumatoid arthritis, and nephritic syndrome. Therapeutic levamisole use has been associated with purpura on the external ears, cheeks, and nasal tip. The predilection for purpura on the ears was first described in children using levam-isole as treatment of nephritic syndrome.2 Antinuclear cytoplasmic antibodies (ANCA) and antiphospholipid antibodies also were associated with therapeutic use.3 Reported effects of levamisole in cocaine users include fever, agranulocytosis, and infection. The mechanism is currently unknown but is thought to be an immunological process as evidenced by the presence of positive autoantibodies.4 Characteristic purpura in a cocaine abuser should alert the physician to consider levamisole as the culprit.
The diagnosis is largely a clinical one and other serious etiologies must be ruled out. The differential should include purpura fulminans, a rare hemorrhagic condition caused by severe infection, such as meningococcemia or deficiency of the vitamin K–dependent anticoagulants protein C and protein S. Given our patient’s history of hepatitis, purpura fulminans could have been likely. Purpura fulminans is rapidly progressive and is usually accompanied by disseminated intravascular coagulation. Coagulation studies and evidence of a consumptive coagulopathy such as thrombocytopenia, prolonged partial thromboplastin time, and activated partial thromboplastin time would favor this diagnosis. These studies as well as protein C and protein S were normal in our patient. Mixed cryoglobulinemia also should be suspected in a patient with hepatitis C virus who presents with vasculitis and arthralgia. An undetectable viral load in addition to a negative assay for cryoglobulins and normal complement (C3 and C4) levels make this diagnosis unlikely. Further, the purpura in cryoglobulinemia is typically confined to the lower extremities.5 Warfarin-induced skin necrosis also should be considered in patients who are taking warfarin.
Laboratory tests can be used to confirm the presence of infection, agranulocytosis, and coagulopathy. Although urine drug screen can confirm exposure to cocaine, levamisole is not detected in routine toxicology screening. Its half-life is between 5 and 6 hours; therefore, detection in blood or urine should be done within 48 hours of exposure.3,4 Unfortunately, our patient presented 2 weeks after cocaine use, making detection of levamisole unfeasible. Rheumatologic screening also is appropriate in cases of suspected vasculitis. Our patient had a negative antinuclear antibody but tested positive for lupus anticoagulant and perinuclear ANCA. IgA and IgG anticardiolipin were negative with an indeterminate IgM anticardiolipin level. The literature supports these findings, as ANCA antibodies occurred in more than 90% of reported cases.3 Further, IgM anticardiolipin antibodies and lupus anticoagulant were positive in 65% and 51% of cases, respectively.3 Leukocyte and neutrophil count were normal in our patient.
Management of these patients is mainly supportive. Complete avoidance of the offending agent is absolutely essential for resolution and avoidance of recurrence. Provided that the patient abstains from cocaine, the skin findings typically resolve in 2 to 3 weeks.3,6 The antibodies, however, may be present for up to 14 months.2,3,6 Treatment with steroids and other immunosuppressants has been reported, but evidence is lacking on their role in the resolution of the lesions.3 In patients who develop a temporary antiphospholipid syndrome, aspirin may be recommended as a preventative measure.6 Pain management, treatment of secondary infection especially in situations with agranulocytosis, and surgical debridement of wounds are all potential problems that can arise.
Our patient had complete resolution of the purpura involving the nose and ear over the course of 3 weeks. She did, however, have to undergo surgical debridement of the nonhealing full-thickness necrotic lesions on her legs and arms.
The 2013 National Survey on Drug Use and Health estimates 1.5 million individuals aged 12 years or older were current (within the last month) users of cocaine.7 With levamisole becoming more prevalent in cocaine supplies, clinicians should be aware of this emerging condition. Rapid recognition can spare the patient unnecessary testing and inappropriate treatment regimens. Because testing for levamisole is difficult and not routinely performed, this case highlights the importance of clinical clues and an accurate social history in clinching the diagnosis.
1. National drug threat assessment: 2011. National Drug Intelligence Center Web site. http://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf. Published August 2011. Accessed March 9, 2015.
2. Rongioletti F, Gio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;40:948-951.
3. Gulati S, Donato AA. Lupus anticoagulant and ANCA associated thrombotic vasculopathy due to cocaine contaminated with levamisole: a case report and review of the literature. J Thromb Thrombolysis. 2012;34:7-10.
4. de la Hera I, Sanz V, Cullen D, et al. Necrosis of ears after cocaine probably adulterated with levamisole. Dermatology. 2011;223:25-28.
5. Seo P. Immune complex-mediated vasculitis. In: Klippel JH. Primer on the Rheumatic Diseases. 13th ed. New York, NY: Springer Science+Business Media, LLC; 2008:427-443.
6. Ching JA, Smith DS. Levamisole-induced necrosis of skin, soft tissue, and bone: case report and review of literature. J Burn Care Res. 2012;33:e1-e5.
7. Substance Abuse and Mental Health Services Administration. Results from the 2013 National Survey on Drug Use and Health: Summary of National Findings. NSDUH Series H-48, HHS Publication No. (SMA) 14-4863. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2014.
1. National drug threat assessment: 2011. National Drug Intelligence Center Web site. http://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf. Published August 2011. Accessed March 9, 2015.
2. Rongioletti F, Gio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;40:948-951.
3. Gulati S, Donato AA. Lupus anticoagulant and ANCA associated thrombotic vasculopathy due to cocaine contaminated with levamisole: a case report and review of the literature. J Thromb Thrombolysis. 2012;34:7-10.
4. de la Hera I, Sanz V, Cullen D, et al. Necrosis of ears after cocaine probably adulterated with levamisole. Dermatology. 2011;223:25-28.
5. Seo P. Immune complex-mediated vasculitis. In: Klippel JH. Primer on the Rheumatic Diseases. 13th ed. New York, NY: Springer Science+Business Media, LLC; 2008:427-443.
6. Ching JA, Smith DS. Levamisole-induced necrosis of skin, soft tissue, and bone: case report and review of literature. J Burn Care Res. 2012;33:e1-e5.
7. Substance Abuse and Mental Health Services Administration. Results from the 2013 National Survey on Drug Use and Health: Summary of National Findings. NSDUH Series H-48, HHS Publication No. (SMA) 14-4863. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2014.
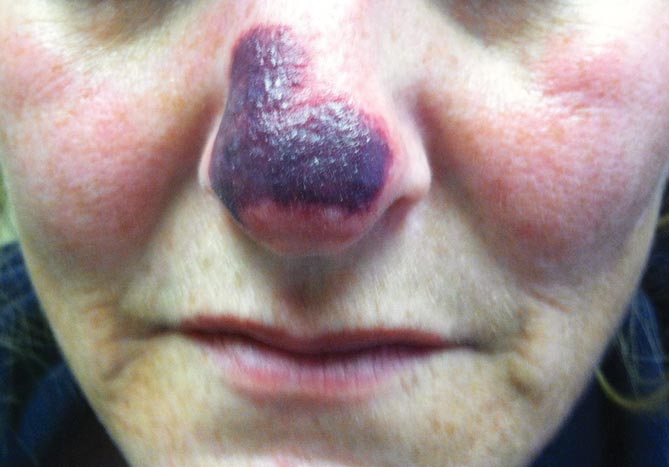
A 44-year-old woman with a history of hepatitis C virus and cocaine abuse presented with painful bruising and skin breakdown. Three weeks prior she was treated at an urgent care clinic with oral clindamycin for a recurrent Staphylococcus infection of her finger. One week later she started developing purpura and painful skin necrosis and ulceration. Most notable was the purpura on the tip of the nose. She also reported arthralgia and low-grade fever.
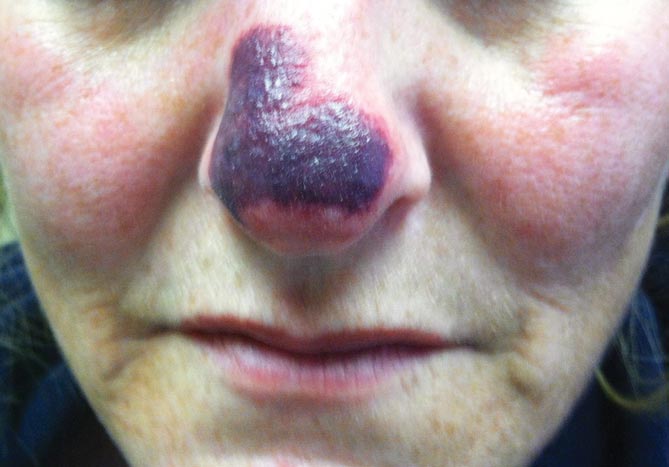
Friable Nodule on the Back
The Diagnosis: Spindle Cell Malignant Melanoma With Perineural Invasion
The incidence of melanoma has steadily increased in the United States since the 1930s when the incidence was reported at 1.0 per 100,000.1 In 1973 melanoma incidence was 6.8 per 100,000, and by 2007 the rate increased to 20.1 per 100,000.2 The American Cancer Society projects 73,870 new cases of melanoma in 2015, with melanoma as the fifth most common cancer in males and the seventh most common in females.3 Melanoma-related deaths are projected to be 9940. The lifetime probability of developing melanoma is 1 in 34 for males and 1 in 53 for females. Twice as many males are estimated to have melanoma-related deaths compared to females. The 5-year relative survival rate is 93% for white individuals and 75% for black individuals.3
Spindle cell melanoma is a rare variant of melanoma that was originally described by Conley et al4 in 1971. The lesion represents 2% to 4% of all melanomas and presents in older patients on sun-exposed skin as a pink or variably pigmented nodule measuring an average of 2 cm.5 Males are affected more than females, and prominent neural invasion is present in 30% to 100% of cases.6-10 Neural invasion can result in nerve palsies and/or dysesthesia. Because half of these lesions are amelanotic, they are often clinically misdiagnosed prior to biopsy.11
Histopathologically, these lesions can be quite challenging. Spindle cell melanoma histologically is an intradermal lesion composed of spindle cells distributed in bundles, fascicles, or nests, or singly between collagen fibers of the dermis. Other spindle tumors such as spindle cell squamous cell carcinoma, atypical fibroxanthoma, dermatofibrosarcoma protuberans, angiosarcoma, and leiomyosarcoma have a similar presentation with hematoxylin and eosin stain. The diagnostic process for spindle cell tumors is greatly aided by immunohistologic analysis. Spindle cell melanoma usually shows immunoreactivity with S-100. Human melanoma black 45, CD57, and neuron-specific enolase usually do not stain, and CD68 has been demonstrated in a minority of cases.
The initial biopsy specimen in our case displayed a dense dermal atypical spindle cell proliferation with hematoxylin and eosin stain. The differential diagnosis of the proliferation included desmoplastic melanoma, spindle cell squamous cell carcinoma, leiomyosarcoma, angiosarcoma, and atypical fibroxanthoma. Immunostains were used to further study the lesional biopsy. Cytokeratin 34bE12, CK5/6, cerium ammonium molybdate 5.2, CK7, epithelial membrane antigen, CK18, high-molecular-weight cytokeratin, S-100, Melan-A, human melanoma black 45, smooth muscle actin, desmin, CD68, CD34, CD10, and p63 were studied. The atypical dermal spindle cells were positive for S-100. S-100 and Melan-A highlighted an increased number of single melanocytes at the dermoepidermal junction. Other markers were negative.
A 1.0-cm wide excision to fascia was performed. Routine hematoxylin and eosin stain showed atypical dermal spindle cells with perineural invasion (Figure 1). The malignant spindle cells were S-100 positive (Figure 2). The spindle cells were negative for high-molecular-weight cytokeratin, CK5/6, p63, Melan-A, A103, microphthalmia, and tyrosinase. These findings confirm melanoma of the spindle cell type.
|
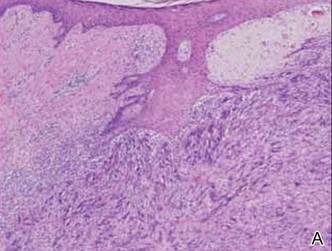
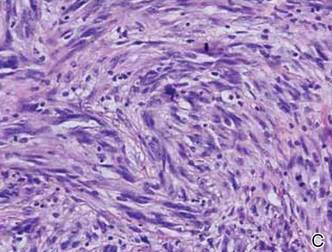
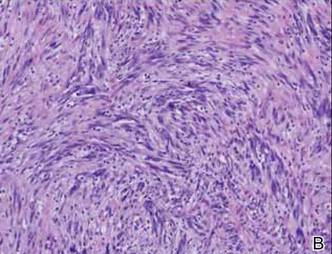
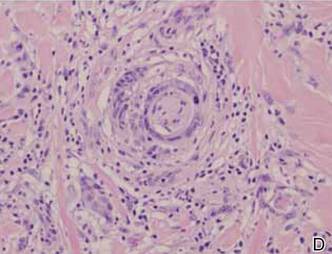
The lesion was a Clark level V melanoma with a Breslow thickness of at least 12 mm. Perineural invasion was noted and the mitotic index was 7 cells/mm2. Vascular and lymphatic invasion was negative and ulceration was present. Tumor-infiltrating lymphocytes were brisk, resulting in a pathologic staging of pT4NXMX.
On initial histologic study with hematoxylin and eosin stain, spindle cell melanoma lesions tend to be generally quite thick. Manganoni et al12 reported in their series a Breslow thickness ranging from 2.1 to 12 mm with a mean of 5.8 mm.
Treatment in our case involved a second wide incision to fascia with an additional 2-cm margin. The role of sentinel lymph node biopsy (SLNB) remains undefined. Patients with spindle cell melanoma have a lower frequency of positive sentinel lymph nodes than nondesmoplastic melanomas.13 As such, the need to perform SLNB has not been determined.13-15 Our patient had a history of breast cancer. The lesion appeared on the left side of the back and she pre-viously had a complete axillary lymphadenectomy on the left axillae. She declined SLNB. A systematic workup by the oncology service was negative. Continued follow-up has revealed no recurrent disease and recent workup was negative for metastatic disease.
Many studies report the increased incidence of local recurrence after excision for spindle cell melanoma as compared to non–spindle cell melanoma,16-18 which is likely related to perineural invasion as in our patient.
Spindle cell melanoma is a rare tumor that is often amelanotic and difficult to diagnose clinically. Routine hematoxylin and eosin staining shows a dermal spindle cell tumor. Immunohistochemical study is of great aid in defining the tumor. The clinician and pathologist must work together to correctly diagnose and treat this lesion.
1. Mikkilineni R, Weinstock MA. Epidemiology. In: Sober AJ, Haluska FG, eds. Atlas of Clinical Oncology: Skin Cancer. London, England: BC Decker; 2001:1-15.
2. Rigel D. Epidemiology of melanoma. Semin Cutan Med Surg. 2010;29:204-209.
3. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015. http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf. Accessed February 10, 2015.
4. Conley J, Lattes R, Orr W. Desmoplastic malignant melanoma (a rare variant of malignant melanoma. Cancer. 1971;28:914-936.
5. Repertinger SK, Teruya B, Sarma DP. Common spindle cell malignant neoplasms of the skin: differential diagnosis and review of the literature. Internet J Dermatol. 2009;7. https://ispub.com/IJD/7/2/11747. Accessed February 10, 2015.
6. Chang PC, Fischbein NJ, McCalmont TH, et al. Perineural spread of malignant melanoma of the head and neck: clinical and imaging features. AJNR Am J Neuroradial. 2004;25:5-11.
7. Cruz J, Reis-Filho JS, Lopes JM. Malignant peripheral nerve sheath tumor-like primary cutaneous melanoma. J Clin Pathol. 2004;57:218-220.
8. Tsao H, Sober AJ, Barnhill RL. Desmoplastic neurotropic melanoma. Semin Cutan Med Surg. 1997;16:45-47.
9. Kossard S, Doherty E, Murray E. Neurotropic melanoma. a variant of desmoplastic melanoma. Arch Dermatol. 1997;7:907-912.
10. Bruijn JA, Salasche S, Sober AJ, et al. Desmoplastic melanoma: clinicopathologic aspects of six cases. Dermatology. 1992;185:3-8.
11. Jesitus J. Desmoplastic melanoma. Dermatology Times. March 2009:1-2.
12. Manganoni AM, Farisoglio C, Bassissi S, et al. Desmoplastic melanoma: report of 5 cases. Dermatol Res Pract. 2009;2009:679010.
13. Gyorki DE, Busam K, Panageas K, et al. Sentinel lymph node biopsy for patients with desmoplastic melanoma. Ann Surg Oncol. 2003;10:403-407.
14. Livestro DP, Muzikansky A, Kaine EM, et al. of desmoplastic melanoma: a case-control comparison with other melanomas. J Clin Oncol. 2005;23:6739-6746.
15. Pawlik TM, Ross MI, Prieto VG, et al. Assessment of the role of sentinel lymph node biopsy for primary cutaneous desmoplastic melanoma. Cancer. 2006;106:900-906.
16. Smithers HM, McLeod GR, Little JH. Desmoplastic melanoma: patterns of recurrence. World J Surg. 1992;16:186-190.
17. McCarthy SW, Scolyer RA, Palmer AA. Desmoplastic melanoma: a diagnostic trap for the unwary. Pathology. 2004;36:445-451.
18. Bruijn JA, Mihm MC Jr, Barnhill RL. Desmoplastic melanoma. Histopathology. 1992;20:197-205.
The Diagnosis: Spindle Cell Malignant Melanoma With Perineural Invasion
The incidence of melanoma has steadily increased in the United States since the 1930s when the incidence was reported at 1.0 per 100,000.1 In 1973 melanoma incidence was 6.8 per 100,000, and by 2007 the rate increased to 20.1 per 100,000.2 The American Cancer Society projects 73,870 new cases of melanoma in 2015, with melanoma as the fifth most common cancer in males and the seventh most common in females.3 Melanoma-related deaths are projected to be 9940. The lifetime probability of developing melanoma is 1 in 34 for males and 1 in 53 for females. Twice as many males are estimated to have melanoma-related deaths compared to females. The 5-year relative survival rate is 93% for white individuals and 75% for black individuals.3
Spindle cell melanoma is a rare variant of melanoma that was originally described by Conley et al4 in 1971. The lesion represents 2% to 4% of all melanomas and presents in older patients on sun-exposed skin as a pink or variably pigmented nodule measuring an average of 2 cm.5 Males are affected more than females, and prominent neural invasion is present in 30% to 100% of cases.6-10 Neural invasion can result in nerve palsies and/or dysesthesia. Because half of these lesions are amelanotic, they are often clinically misdiagnosed prior to biopsy.11
Histopathologically, these lesions can be quite challenging. Spindle cell melanoma histologically is an intradermal lesion composed of spindle cells distributed in bundles, fascicles, or nests, or singly between collagen fibers of the dermis. Other spindle tumors such as spindle cell squamous cell carcinoma, atypical fibroxanthoma, dermatofibrosarcoma protuberans, angiosarcoma, and leiomyosarcoma have a similar presentation with hematoxylin and eosin stain. The diagnostic process for spindle cell tumors is greatly aided by immunohistologic analysis. Spindle cell melanoma usually shows immunoreactivity with S-100. Human melanoma black 45, CD57, and neuron-specific enolase usually do not stain, and CD68 has been demonstrated in a minority of cases.
The initial biopsy specimen in our case displayed a dense dermal atypical spindle cell proliferation with hematoxylin and eosin stain. The differential diagnosis of the proliferation included desmoplastic melanoma, spindle cell squamous cell carcinoma, leiomyosarcoma, angiosarcoma, and atypical fibroxanthoma. Immunostains were used to further study the lesional biopsy. Cytokeratin 34bE12, CK5/6, cerium ammonium molybdate 5.2, CK7, epithelial membrane antigen, CK18, high-molecular-weight cytokeratin, S-100, Melan-A, human melanoma black 45, smooth muscle actin, desmin, CD68, CD34, CD10, and p63 were studied. The atypical dermal spindle cells were positive for S-100. S-100 and Melan-A highlighted an increased number of single melanocytes at the dermoepidermal junction. Other markers were negative.
A 1.0-cm wide excision to fascia was performed. Routine hematoxylin and eosin stain showed atypical dermal spindle cells with perineural invasion (Figure 1). The malignant spindle cells were S-100 positive (Figure 2). The spindle cells were negative for high-molecular-weight cytokeratin, CK5/6, p63, Melan-A, A103, microphthalmia, and tyrosinase. These findings confirm melanoma of the spindle cell type.
|
The lesion was a Clark level V melanoma with a Breslow thickness of at least 12 mm. Perineural invasion was noted and the mitotic index was 7 cells/mm2. Vascular and lymphatic invasion was negative and ulceration was present. Tumor-infiltrating lymphocytes were brisk, resulting in a pathologic staging of pT4NXMX.
On initial histologic study with hematoxylin and eosin stain, spindle cell melanoma lesions tend to be generally quite thick. Manganoni et al12 reported in their series a Breslow thickness ranging from 2.1 to 12 mm with a mean of 5.8 mm.
Treatment in our case involved a second wide incision to fascia with an additional 2-cm margin. The role of sentinel lymph node biopsy (SLNB) remains undefined. Patients with spindle cell melanoma have a lower frequency of positive sentinel lymph nodes than nondesmoplastic melanomas.13 As such, the need to perform SLNB has not been determined.13-15 Our patient had a history of breast cancer. The lesion appeared on the left side of the back and she pre-viously had a complete axillary lymphadenectomy on the left axillae. She declined SLNB. A systematic workup by the oncology service was negative. Continued follow-up has revealed no recurrent disease and recent workup was negative for metastatic disease.
Many studies report the increased incidence of local recurrence after excision for spindle cell melanoma as compared to non–spindle cell melanoma,16-18 which is likely related to perineural invasion as in our patient.
Spindle cell melanoma is a rare tumor that is often amelanotic and difficult to diagnose clinically. Routine hematoxylin and eosin staining shows a dermal spindle cell tumor. Immunohistochemical study is of great aid in defining the tumor. The clinician and pathologist must work together to correctly diagnose and treat this lesion.
The Diagnosis: Spindle Cell Malignant Melanoma With Perineural Invasion
The incidence of melanoma has steadily increased in the United States since the 1930s when the incidence was reported at 1.0 per 100,000.1 In 1973 melanoma incidence was 6.8 per 100,000, and by 2007 the rate increased to 20.1 per 100,000.2 The American Cancer Society projects 73,870 new cases of melanoma in 2015, with melanoma as the fifth most common cancer in males and the seventh most common in females.3 Melanoma-related deaths are projected to be 9940. The lifetime probability of developing melanoma is 1 in 34 for males and 1 in 53 for females. Twice as many males are estimated to have melanoma-related deaths compared to females. The 5-year relative survival rate is 93% for white individuals and 75% for black individuals.3
Spindle cell melanoma is a rare variant of melanoma that was originally described by Conley et al4 in 1971. The lesion represents 2% to 4% of all melanomas and presents in older patients on sun-exposed skin as a pink or variably pigmented nodule measuring an average of 2 cm.5 Males are affected more than females, and prominent neural invasion is present in 30% to 100% of cases.6-10 Neural invasion can result in nerve palsies and/or dysesthesia. Because half of these lesions are amelanotic, they are often clinically misdiagnosed prior to biopsy.11
Histopathologically, these lesions can be quite challenging. Spindle cell melanoma histologically is an intradermal lesion composed of spindle cells distributed in bundles, fascicles, or nests, or singly between collagen fibers of the dermis. Other spindle tumors such as spindle cell squamous cell carcinoma, atypical fibroxanthoma, dermatofibrosarcoma protuberans, angiosarcoma, and leiomyosarcoma have a similar presentation with hematoxylin and eosin stain. The diagnostic process for spindle cell tumors is greatly aided by immunohistologic analysis. Spindle cell melanoma usually shows immunoreactivity with S-100. Human melanoma black 45, CD57, and neuron-specific enolase usually do not stain, and CD68 has been demonstrated in a minority of cases.
The initial biopsy specimen in our case displayed a dense dermal atypical spindle cell proliferation with hematoxylin and eosin stain. The differential diagnosis of the proliferation included desmoplastic melanoma, spindle cell squamous cell carcinoma, leiomyosarcoma, angiosarcoma, and atypical fibroxanthoma. Immunostains were used to further study the lesional biopsy. Cytokeratin 34bE12, CK5/6, cerium ammonium molybdate 5.2, CK7, epithelial membrane antigen, CK18, high-molecular-weight cytokeratin, S-100, Melan-A, human melanoma black 45, smooth muscle actin, desmin, CD68, CD34, CD10, and p63 were studied. The atypical dermal spindle cells were positive for S-100. S-100 and Melan-A highlighted an increased number of single melanocytes at the dermoepidermal junction. Other markers were negative.
A 1.0-cm wide excision to fascia was performed. Routine hematoxylin and eosin stain showed atypical dermal spindle cells with perineural invasion (Figure 1). The malignant spindle cells were S-100 positive (Figure 2). The spindle cells were negative for high-molecular-weight cytokeratin, CK5/6, p63, Melan-A, A103, microphthalmia, and tyrosinase. These findings confirm melanoma of the spindle cell type.
|
The lesion was a Clark level V melanoma with a Breslow thickness of at least 12 mm. Perineural invasion was noted and the mitotic index was 7 cells/mm2. Vascular and lymphatic invasion was negative and ulceration was present. Tumor-infiltrating lymphocytes were brisk, resulting in a pathologic staging of pT4NXMX.
On initial histologic study with hematoxylin and eosin stain, spindle cell melanoma lesions tend to be generally quite thick. Manganoni et al12 reported in their series a Breslow thickness ranging from 2.1 to 12 mm with a mean of 5.8 mm.
Treatment in our case involved a second wide incision to fascia with an additional 2-cm margin. The role of sentinel lymph node biopsy (SLNB) remains undefined. Patients with spindle cell melanoma have a lower frequency of positive sentinel lymph nodes than nondesmoplastic melanomas.13 As such, the need to perform SLNB has not been determined.13-15 Our patient had a history of breast cancer. The lesion appeared on the left side of the back and she pre-viously had a complete axillary lymphadenectomy on the left axillae. She declined SLNB. A systematic workup by the oncology service was negative. Continued follow-up has revealed no recurrent disease and recent workup was negative for metastatic disease.
Many studies report the increased incidence of local recurrence after excision for spindle cell melanoma as compared to non–spindle cell melanoma,16-18 which is likely related to perineural invasion as in our patient.
Spindle cell melanoma is a rare tumor that is often amelanotic and difficult to diagnose clinically. Routine hematoxylin and eosin staining shows a dermal spindle cell tumor. Immunohistochemical study is of great aid in defining the tumor. The clinician and pathologist must work together to correctly diagnose and treat this lesion.
1. Mikkilineni R, Weinstock MA. Epidemiology. In: Sober AJ, Haluska FG, eds. Atlas of Clinical Oncology: Skin Cancer. London, England: BC Decker; 2001:1-15.
2. Rigel D. Epidemiology of melanoma. Semin Cutan Med Surg. 2010;29:204-209.
3. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015. http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf. Accessed February 10, 2015.
4. Conley J, Lattes R, Orr W. Desmoplastic malignant melanoma (a rare variant of malignant melanoma. Cancer. 1971;28:914-936.
5. Repertinger SK, Teruya B, Sarma DP. Common spindle cell malignant neoplasms of the skin: differential diagnosis and review of the literature. Internet J Dermatol. 2009;7. https://ispub.com/IJD/7/2/11747. Accessed February 10, 2015.
6. Chang PC, Fischbein NJ, McCalmont TH, et al. Perineural spread of malignant melanoma of the head and neck: clinical and imaging features. AJNR Am J Neuroradial. 2004;25:5-11.
7. Cruz J, Reis-Filho JS, Lopes JM. Malignant peripheral nerve sheath tumor-like primary cutaneous melanoma. J Clin Pathol. 2004;57:218-220.
8. Tsao H, Sober AJ, Barnhill RL. Desmoplastic neurotropic melanoma. Semin Cutan Med Surg. 1997;16:45-47.
9. Kossard S, Doherty E, Murray E. Neurotropic melanoma. a variant of desmoplastic melanoma. Arch Dermatol. 1997;7:907-912.
10. Bruijn JA, Salasche S, Sober AJ, et al. Desmoplastic melanoma: clinicopathologic aspects of six cases. Dermatology. 1992;185:3-8.
11. Jesitus J. Desmoplastic melanoma. Dermatology Times. March 2009:1-2.
12. Manganoni AM, Farisoglio C, Bassissi S, et al. Desmoplastic melanoma: report of 5 cases. Dermatol Res Pract. 2009;2009:679010.
13. Gyorki DE, Busam K, Panageas K, et al. Sentinel lymph node biopsy for patients with desmoplastic melanoma. Ann Surg Oncol. 2003;10:403-407.
14. Livestro DP, Muzikansky A, Kaine EM, et al. of desmoplastic melanoma: a case-control comparison with other melanomas. J Clin Oncol. 2005;23:6739-6746.
15. Pawlik TM, Ross MI, Prieto VG, et al. Assessment of the role of sentinel lymph node biopsy for primary cutaneous desmoplastic melanoma. Cancer. 2006;106:900-906.
16. Smithers HM, McLeod GR, Little JH. Desmoplastic melanoma: patterns of recurrence. World J Surg. 1992;16:186-190.
17. McCarthy SW, Scolyer RA, Palmer AA. Desmoplastic melanoma: a diagnostic trap for the unwary. Pathology. 2004;36:445-451.
18. Bruijn JA, Mihm MC Jr, Barnhill RL. Desmoplastic melanoma. Histopathology. 1992;20:197-205.
1. Mikkilineni R, Weinstock MA. Epidemiology. In: Sober AJ, Haluska FG, eds. Atlas of Clinical Oncology: Skin Cancer. London, England: BC Decker; 2001:1-15.
2. Rigel D. Epidemiology of melanoma. Semin Cutan Med Surg. 2010;29:204-209.
3. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015. http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf. Accessed February 10, 2015.
4. Conley J, Lattes R, Orr W. Desmoplastic malignant melanoma (a rare variant of malignant melanoma. Cancer. 1971;28:914-936.
5. Repertinger SK, Teruya B, Sarma DP. Common spindle cell malignant neoplasms of the skin: differential diagnosis and review of the literature. Internet J Dermatol. 2009;7. https://ispub.com/IJD/7/2/11747. Accessed February 10, 2015.
6. Chang PC, Fischbein NJ, McCalmont TH, et al. Perineural spread of malignant melanoma of the head and neck: clinical and imaging features. AJNR Am J Neuroradial. 2004;25:5-11.
7. Cruz J, Reis-Filho JS, Lopes JM. Malignant peripheral nerve sheath tumor-like primary cutaneous melanoma. J Clin Pathol. 2004;57:218-220.
8. Tsao H, Sober AJ, Barnhill RL. Desmoplastic neurotropic melanoma. Semin Cutan Med Surg. 1997;16:45-47.
9. Kossard S, Doherty E, Murray E. Neurotropic melanoma. a variant of desmoplastic melanoma. Arch Dermatol. 1997;7:907-912.
10. Bruijn JA, Salasche S, Sober AJ, et al. Desmoplastic melanoma: clinicopathologic aspects of six cases. Dermatology. 1992;185:3-8.
11. Jesitus J. Desmoplastic melanoma. Dermatology Times. March 2009:1-2.
12. Manganoni AM, Farisoglio C, Bassissi S, et al. Desmoplastic melanoma: report of 5 cases. Dermatol Res Pract. 2009;2009:679010.
13. Gyorki DE, Busam K, Panageas K, et al. Sentinel lymph node biopsy for patients with desmoplastic melanoma. Ann Surg Oncol. 2003;10:403-407.
14. Livestro DP, Muzikansky A, Kaine EM, et al. of desmoplastic melanoma: a case-control comparison with other melanomas. J Clin Oncol. 2005;23:6739-6746.
15. Pawlik TM, Ross MI, Prieto VG, et al. Assessment of the role of sentinel lymph node biopsy for primary cutaneous desmoplastic melanoma. Cancer. 2006;106:900-906.
16. Smithers HM, McLeod GR, Little JH. Desmoplastic melanoma: patterns of recurrence. World J Surg. 1992;16:186-190.
17. McCarthy SW, Scolyer RA, Palmer AA. Desmoplastic melanoma: a diagnostic trap for the unwary. Pathology. 2004;36:445-451.
18. Bruijn JA, Mihm MC Jr, Barnhill RL. Desmoplastic melanoma. Histopathology. 1992;20:197-205.
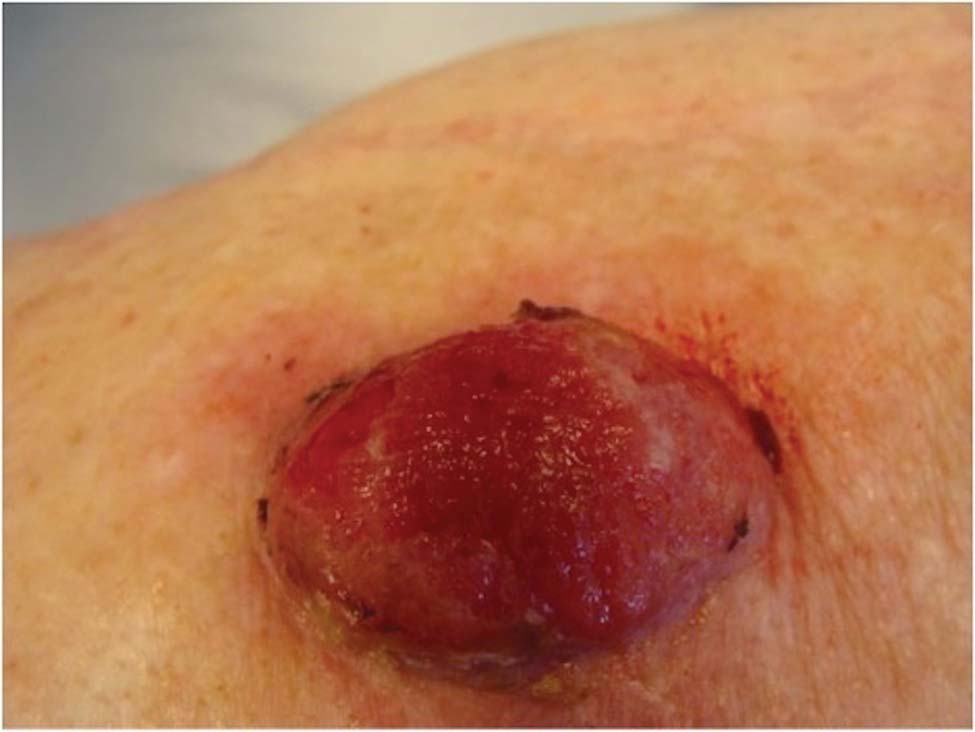
A 78-year-old woman presented with a large friable, sharply demarcated nodule of 3 months’ duration on the left side of the back. The lesion occasionally bled but was otherwise asymptomatic. There was no perilesional paresthesia. The patient’s medical history included hypertension, depression, chronic obstructive pulmonary disease, breast cancer, osteoporosis, and aortic valve disease.
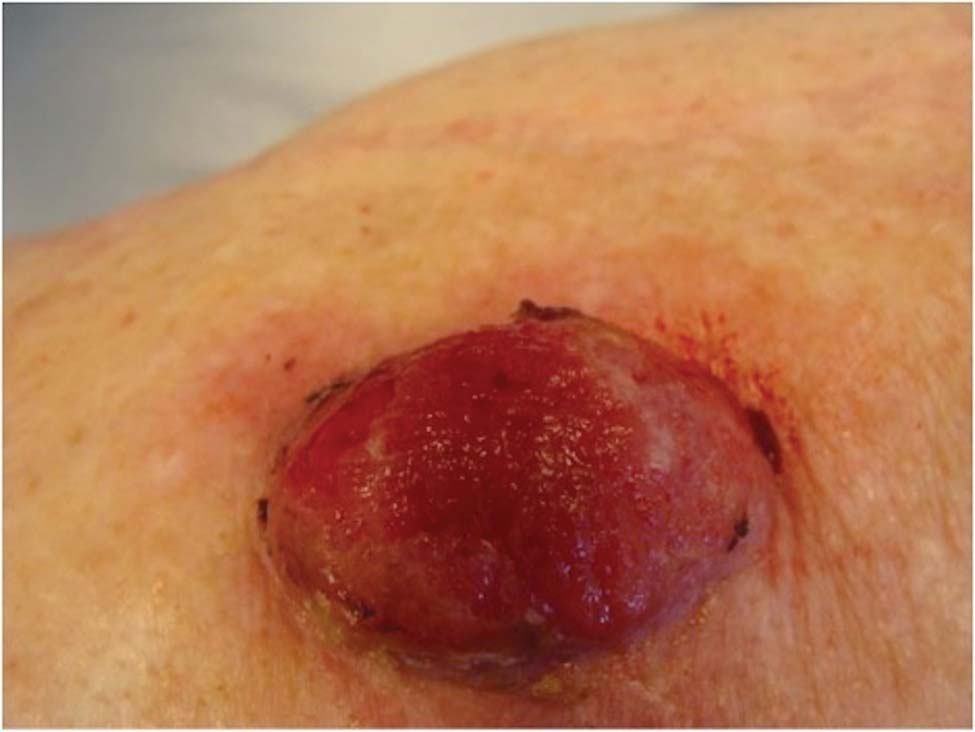
Telangiectases on the Cheeks and Nose
The Diagnosis: Hereditary Hemorrhagic Telangiectasia
Physical examination of our patient revealed multiple fine punctate telangiectases on the bilateral cheeks and the dorsum of the nose. Further examination revealed matlike telangiectases on the distal aspect of the tongue (Figure), buccal mucosa, palms, and fingers. Since diagnosis he has experienced several episodes of severe gastrointestinal tract bleeding. He underwent an esophagogastroduodenoscopy and was found to have bleeding gastric antral vascular ectasia that was treated with argon plasma coagulation. One year later he had a second esophagogastroduodenoscopy performed because of heme-positive stools and was found to have additional bleeding vascular ectasia that was treated again with argon plasma coagulation.
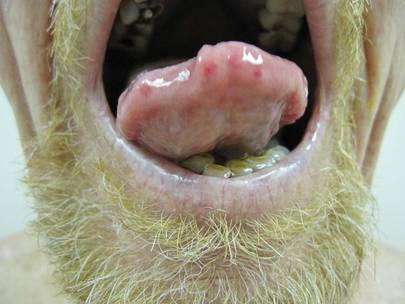
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal-dominant disorder that is characterized by mucocutaneous and visceral telangiectases, recurrent hemorrhages, and a familial occurrence.1 In North America, the overall incidence is approximately 1 per 5000 to 10,000 individuals per year.2 Although this disorder generally has an autosomal-dominant transmission, approximately 20% of cases do not have a familial component.2
Clinically these patients most commonly present with a recurrent episode of epistaxis that occurs within the first 2 decades of life. Shortly after the onset of these recurrent episodes, patients begin to develop punctate or splinterlike telangiectases on the lips, oral mucosa, upper extremities, nail beds, and trunk. These cutaneous telangiectases are a cosmetic problem and can sometimes cause hemorrhage, especially from the tongue and fingers.1 The serious complications of HHT arise from internal organ involvement. Gastrointestinal telangiectases and arteriovenous malformations (AVMs) can result in acute gastrointestinal hemorrhages or iron deficiency anemia in approximately 16% of patients. Central nervous system AVMs result in migraines, brain abscesses, paraparesis, ischemia, strokes, transient ischemic attacks, seizures, and both intracerebral and subarachnoid hemorrhage. Pulmonary AVMs can cause a right-to-left shunt, leading to hypoxemia and embolic events due to the bypass of the lungs, which act as a filtering capillary bed.3
Genetic linkage analysis has revealed 2 genes that are responsible for HHT. The first gene, ENG, encodes endoglin and is found on band 9q33-34.3 The second gene, ALK1, encodes activin receptorlike kinase 1 and is found on band 12q11-12. Both of these gene products are involved with vascular remodeling. They are integral membrane glycoproteins mainly expressed on vascular endothelial cells and act as surface receptors for transforming growth factor b. Mutations in ALK1 are associated with a generally more benign course, whereas mutations in ENG more commonly have pulmonary AVMs.3
The diagnosis of HHT is established if 3 of the following features are present: (1) epistaxis (ie, spontaneous recurrent nosebleeds); (2) telangiectases in characteristic sites (ie, oral cavity, lips, nose, fingers); (3) visceral lesions, such as gastrointestinal telangiectases (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, or spinal AVM; (4) family history (ie, a first-degree relative with HHT).3 Similar diseases with telangiectases should be considered in the differential diagnosis including CREST syndrome (characterized by calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia), generalized essential telangiectasia, and ataxia telangiectasia.2
Patients diagnosed with HHT who have a family history of the disease should be evaluated for pulmonary AVM through chest computed tomography and pulmonary angiography because they are at the highest risk. Magnetic resonance imaging is useful to rule out central nervous system involvement. Treatment of cutaneous telangiectases consists of electrocauterization; sclerotherapy; and a variety of lasers and light sources including the Nd:YAG laser, intense pulsed light, argon laser, or pulsed dye laser.1,2,4 An extremely useful resource for patients with HHT and family members is the HHT Foundation (http://curehht.org).
1. Fernández-Jorge B, Del Pozo Losada J, Paradela S, et al. Treatment of cutaneous and mucosal telangiectases in hereditary hemorrhagic telangiectasia: report of three cases. J Cosmet Laser Ther. 2007;9:29-33.
2. Lee HE, Sagong C, Yeo KY, et al. A case of hereditary hemorrhagic telangiectasia. Ann Dermatol. 2009;21:206-208.
3. Garzon MC, Huang JT, Enjolras O, et al. Vascular malformations. part II: associated syndromes. J Am Acad Dermatol. 2007;56:541-564.
4. Sato Y, Takayama T, Takahari D, et al. Successful treatment for gastro-intestinal bleeding of Osler-Weber-Rendu disease by argon plasma coagulation using double-balloon enteroscopy. Endoscopy. 2008;40(suppl 2):E228-E229.
The Diagnosis: Hereditary Hemorrhagic Telangiectasia
Physical examination of our patient revealed multiple fine punctate telangiectases on the bilateral cheeks and the dorsum of the nose. Further examination revealed matlike telangiectases on the distal aspect of the tongue (Figure), buccal mucosa, palms, and fingers. Since diagnosis he has experienced several episodes of severe gastrointestinal tract bleeding. He underwent an esophagogastroduodenoscopy and was found to have bleeding gastric antral vascular ectasia that was treated with argon plasma coagulation. One year later he had a second esophagogastroduodenoscopy performed because of heme-positive stools and was found to have additional bleeding vascular ectasia that was treated again with argon plasma coagulation.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal-dominant disorder that is characterized by mucocutaneous and visceral telangiectases, recurrent hemorrhages, and a familial occurrence.1 In North America, the overall incidence is approximately 1 per 5000 to 10,000 individuals per year.2 Although this disorder generally has an autosomal-dominant transmission, approximately 20% of cases do not have a familial component.2
Clinically these patients most commonly present with a recurrent episode of epistaxis that occurs within the first 2 decades of life. Shortly after the onset of these recurrent episodes, patients begin to develop punctate or splinterlike telangiectases on the lips, oral mucosa, upper extremities, nail beds, and trunk. These cutaneous telangiectases are a cosmetic problem and can sometimes cause hemorrhage, especially from the tongue and fingers.1 The serious complications of HHT arise from internal organ involvement. Gastrointestinal telangiectases and arteriovenous malformations (AVMs) can result in acute gastrointestinal hemorrhages or iron deficiency anemia in approximately 16% of patients. Central nervous system AVMs result in migraines, brain abscesses, paraparesis, ischemia, strokes, transient ischemic attacks, seizures, and both intracerebral and subarachnoid hemorrhage. Pulmonary AVMs can cause a right-to-left shunt, leading to hypoxemia and embolic events due to the bypass of the lungs, which act as a filtering capillary bed.3
Genetic linkage analysis has revealed 2 genes that are responsible for HHT. The first gene, ENG, encodes endoglin and is found on band 9q33-34.3 The second gene, ALK1, encodes activin receptorlike kinase 1 and is found on band 12q11-12. Both of these gene products are involved with vascular remodeling. They are integral membrane glycoproteins mainly expressed on vascular endothelial cells and act as surface receptors for transforming growth factor b. Mutations in ALK1 are associated with a generally more benign course, whereas mutations in ENG more commonly have pulmonary AVMs.3
The diagnosis of HHT is established if 3 of the following features are present: (1) epistaxis (ie, spontaneous recurrent nosebleeds); (2) telangiectases in characteristic sites (ie, oral cavity, lips, nose, fingers); (3) visceral lesions, such as gastrointestinal telangiectases (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, or spinal AVM; (4) family history (ie, a first-degree relative with HHT).3 Similar diseases with telangiectases should be considered in the differential diagnosis including CREST syndrome (characterized by calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia), generalized essential telangiectasia, and ataxia telangiectasia.2
Patients diagnosed with HHT who have a family history of the disease should be evaluated for pulmonary AVM through chest computed tomography and pulmonary angiography because they are at the highest risk. Magnetic resonance imaging is useful to rule out central nervous system involvement. Treatment of cutaneous telangiectases consists of electrocauterization; sclerotherapy; and a variety of lasers and light sources including the Nd:YAG laser, intense pulsed light, argon laser, or pulsed dye laser.1,2,4 An extremely useful resource for patients with HHT and family members is the HHT Foundation (http://curehht.org).
The Diagnosis: Hereditary Hemorrhagic Telangiectasia
Physical examination of our patient revealed multiple fine punctate telangiectases on the bilateral cheeks and the dorsum of the nose. Further examination revealed matlike telangiectases on the distal aspect of the tongue (Figure), buccal mucosa, palms, and fingers. Since diagnosis he has experienced several episodes of severe gastrointestinal tract bleeding. He underwent an esophagogastroduodenoscopy and was found to have bleeding gastric antral vascular ectasia that was treated with argon plasma coagulation. One year later he had a second esophagogastroduodenoscopy performed because of heme-positive stools and was found to have additional bleeding vascular ectasia that was treated again with argon plasma coagulation.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal-dominant disorder that is characterized by mucocutaneous and visceral telangiectases, recurrent hemorrhages, and a familial occurrence.1 In North America, the overall incidence is approximately 1 per 5000 to 10,000 individuals per year.2 Although this disorder generally has an autosomal-dominant transmission, approximately 20% of cases do not have a familial component.2
Clinically these patients most commonly present with a recurrent episode of epistaxis that occurs within the first 2 decades of life. Shortly after the onset of these recurrent episodes, patients begin to develop punctate or splinterlike telangiectases on the lips, oral mucosa, upper extremities, nail beds, and trunk. These cutaneous telangiectases are a cosmetic problem and can sometimes cause hemorrhage, especially from the tongue and fingers.1 The serious complications of HHT arise from internal organ involvement. Gastrointestinal telangiectases and arteriovenous malformations (AVMs) can result in acute gastrointestinal hemorrhages or iron deficiency anemia in approximately 16% of patients. Central nervous system AVMs result in migraines, brain abscesses, paraparesis, ischemia, strokes, transient ischemic attacks, seizures, and both intracerebral and subarachnoid hemorrhage. Pulmonary AVMs can cause a right-to-left shunt, leading to hypoxemia and embolic events due to the bypass of the lungs, which act as a filtering capillary bed.3
Genetic linkage analysis has revealed 2 genes that are responsible for HHT. The first gene, ENG, encodes endoglin and is found on band 9q33-34.3 The second gene, ALK1, encodes activin receptorlike kinase 1 and is found on band 12q11-12. Both of these gene products are involved with vascular remodeling. They are integral membrane glycoproteins mainly expressed on vascular endothelial cells and act as surface receptors for transforming growth factor b. Mutations in ALK1 are associated with a generally more benign course, whereas mutations in ENG more commonly have pulmonary AVMs.3
The diagnosis of HHT is established if 3 of the following features are present: (1) epistaxis (ie, spontaneous recurrent nosebleeds); (2) telangiectases in characteristic sites (ie, oral cavity, lips, nose, fingers); (3) visceral lesions, such as gastrointestinal telangiectases (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, or spinal AVM; (4) family history (ie, a first-degree relative with HHT).3 Similar diseases with telangiectases should be considered in the differential diagnosis including CREST syndrome (characterized by calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia), generalized essential telangiectasia, and ataxia telangiectasia.2
Patients diagnosed with HHT who have a family history of the disease should be evaluated for pulmonary AVM through chest computed tomography and pulmonary angiography because they are at the highest risk. Magnetic resonance imaging is useful to rule out central nervous system involvement. Treatment of cutaneous telangiectases consists of electrocauterization; sclerotherapy; and a variety of lasers and light sources including the Nd:YAG laser, intense pulsed light, argon laser, or pulsed dye laser.1,2,4 An extremely useful resource for patients with HHT and family members is the HHT Foundation (http://curehht.org).
1. Fernández-Jorge B, Del Pozo Losada J, Paradela S, et al. Treatment of cutaneous and mucosal telangiectases in hereditary hemorrhagic telangiectasia: report of three cases. J Cosmet Laser Ther. 2007;9:29-33.
2. Lee HE, Sagong C, Yeo KY, et al. A case of hereditary hemorrhagic telangiectasia. Ann Dermatol. 2009;21:206-208.
3. Garzon MC, Huang JT, Enjolras O, et al. Vascular malformations. part II: associated syndromes. J Am Acad Dermatol. 2007;56:541-564.
4. Sato Y, Takayama T, Takahari D, et al. Successful treatment for gastro-intestinal bleeding of Osler-Weber-Rendu disease by argon plasma coagulation using double-balloon enteroscopy. Endoscopy. 2008;40(suppl 2):E228-E229.
1. Fernández-Jorge B, Del Pozo Losada J, Paradela S, et al. Treatment of cutaneous and mucosal telangiectases in hereditary hemorrhagic telangiectasia: report of three cases. J Cosmet Laser Ther. 2007;9:29-33.
2. Lee HE, Sagong C, Yeo KY, et al. A case of hereditary hemorrhagic telangiectasia. Ann Dermatol. 2009;21:206-208.
3. Garzon MC, Huang JT, Enjolras O, et al. Vascular malformations. part II: associated syndromes. J Am Acad Dermatol. 2007;56:541-564.
4. Sato Y, Takayama T, Takahari D, et al. Successful treatment for gastro-intestinal bleeding of Osler-Weber-Rendu disease by argon plasma coagulation using double-balloon enteroscopy. Endoscopy. 2008;40(suppl 2):E228-E229.
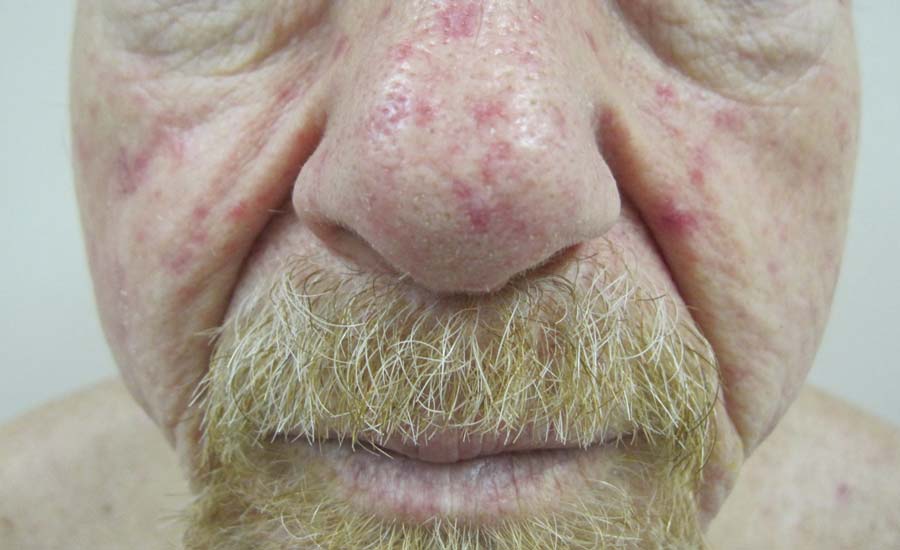
A 68-year-old man presented for evaluation of numerous telangiectases that developed on the cheeks, nose, lips, tongue, and fingers. He denied any history of gastrointestinal tract bleeding but admitted to numerous nosebleeds in the recent past. Family history indicated that his maternal grandmother died of an aneurysm, his mother died of a cerebral hemorrhage, 2 paternal cousins had hemochromatosis, and 2 brothers were healthy.
What Is Your Diagnosis? Extramammary Paget Disease

A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
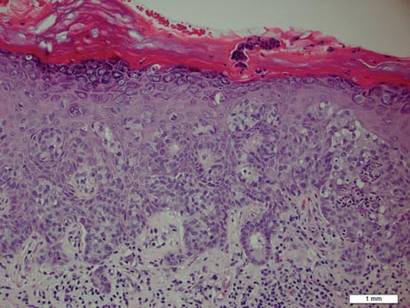
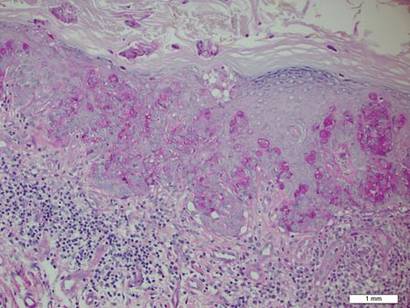
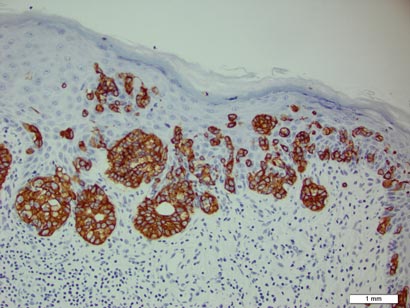
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
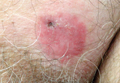
Bluish Red Verrucous Lesions on the Leg
The Diagnosis: Linear Verrucous Hemangioma
Verrucous hemangioma is a rare congenital vascular malformation of the cutaneous and subcutaneous tissues. Although almost invariably present at birth, it may appear later in childhood or even in adulthood. Lesions commonly are found on the legs and may be linear, multiple, and disseminated or sometimes confined to the digits. In the early phase of evolution, the lesions are nonkeratotic, soft, blue-red plaques, but they gradually become increasingly hyperkeratotic.1 Linear verrucous hemangioma is even more rare with few published reports.
In 1937, Halter2 first used the term verrucous hemangioma to describe a 16-year-old adolescent boy who presented with a linear purpuric cluster of plaques extending from the right buttock to the toes. Imperial and Helwig3 later described it as a distinct entity. Since then, similar lesions have been described using a variety of names such as angiokeratoma circumscriptum neviforme, angiokeratoma circumscriptum, angiokeratoma corporis neviforme, keratotic hemangioma, nevus vascularis unius lateralis, and nevus keratoangiomatosus.3 Therefore, the exact incidence is difficult to determine.
Lesions generally are noted at birth or in early childhood and are often located on the lower extremities. The early lesions are bluish red in color. Secondary infection is a frequent complication, resulting in reactive papillomatosis and hyperkeratosis; thus, the older lesions acquire a verrucous or warty surface.3 Clinically, they may resemble angiokeratoma, lym-phangioma circumscriptum, verrucous epidermal nevus, verrucous cancer, or even malignant melanoma. Lesions initially resemble port-wine stains and later may become soft, bluish red vascular swellings that tend to grow in size and become verrucous.1
The histologic appearance closely resembles angiokeratoma, as both lesions show vascular spaces beneath a papillomatous and hyperkeratotic epidermis.4 However, in contrast to angiokeratoma, the vascular spaces in verrucous hemangioma also involve the lower dermis and subcutaneous tissues.
Although cases of linear verrucous hemangioma have been reported, its distribution along the lines of Blaschko is rare.5,6 It has been proposed that these lesions may actually be following dermatomal patterns or that the linear arrangement represents genetic mosaicism.5 In our case, the lesion started as a small plaque in childhood and gradually spread linearly to the buttock (Figure 1). A biopsy was taken from the lesion on the right buttock, which showed numerous dilated capillaries in the dermis (Figure 2).
|
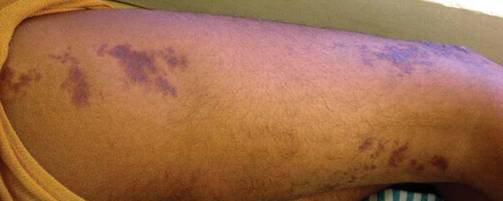
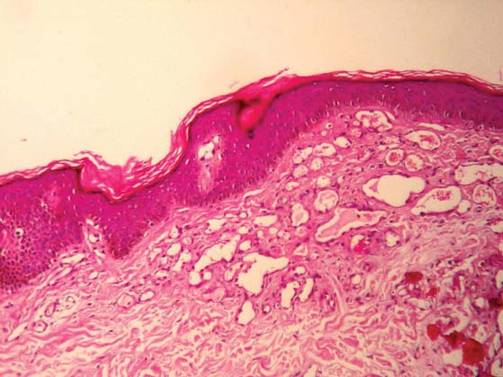
Verrucous hemangiomas are best treated by excision. Larger lesions will need grafting. There is a tendency for recurrence to occur unless excision is complete.1 Yang and Ohara7 reported 14 patients with small localized lesions that were cured by 1 session of surgery without recurrence; 9 patients with wider and more extensive lesions required combination therapy in several stages for optimal results.
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. London, England: Blackwell Scientific Publications; 2004:15-60.
2. Halter K. Haemangioma verrucosum mit Osteoatrophie. Dermatol Z. 1937;75:271-279.
3. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathological study of 21 cases. Arch Dermatol. 1967;96:247-253.
4. Calduch L, Ortega C, Navarro V, et al. Verrucous hemangioma: Report of two cases and review of the literature. Pediatr Dermatol. 2000;17:213-217.
5. Wentscher U, Happle R. Linear verrucous hemangioma. J Am Acad Dermatol. 2000;42:516-518.
6. Jain VK, Aggarwal K, Jain S. Linear verrucous hemangioma on the leg. Indian J Dermatol Venereol Leprol. 2008;74:656-658.
7. Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919.
The Diagnosis: Linear Verrucous Hemangioma
Verrucous hemangioma is a rare congenital vascular malformation of the cutaneous and subcutaneous tissues. Although almost invariably present at birth, it may appear later in childhood or even in adulthood. Lesions commonly are found on the legs and may be linear, multiple, and disseminated or sometimes confined to the digits. In the early phase of evolution, the lesions are nonkeratotic, soft, blue-red plaques, but they gradually become increasingly hyperkeratotic.1 Linear verrucous hemangioma is even more rare with few published reports.
In 1937, Halter2 first used the term verrucous hemangioma to describe a 16-year-old adolescent boy who presented with a linear purpuric cluster of plaques extending from the right buttock to the toes. Imperial and Helwig3 later described it as a distinct entity. Since then, similar lesions have been described using a variety of names such as angiokeratoma circumscriptum neviforme, angiokeratoma circumscriptum, angiokeratoma corporis neviforme, keratotic hemangioma, nevus vascularis unius lateralis, and nevus keratoangiomatosus.3 Therefore, the exact incidence is difficult to determine.
Lesions generally are noted at birth or in early childhood and are often located on the lower extremities. The early lesions are bluish red in color. Secondary infection is a frequent complication, resulting in reactive papillomatosis and hyperkeratosis; thus, the older lesions acquire a verrucous or warty surface.3 Clinically, they may resemble angiokeratoma, lym-phangioma circumscriptum, verrucous epidermal nevus, verrucous cancer, or even malignant melanoma. Lesions initially resemble port-wine stains and later may become soft, bluish red vascular swellings that tend to grow in size and become verrucous.1
The histologic appearance closely resembles angiokeratoma, as both lesions show vascular spaces beneath a papillomatous and hyperkeratotic epidermis.4 However, in contrast to angiokeratoma, the vascular spaces in verrucous hemangioma also involve the lower dermis and subcutaneous tissues.
Although cases of linear verrucous hemangioma have been reported, its distribution along the lines of Blaschko is rare.5,6 It has been proposed that these lesions may actually be following dermatomal patterns or that the linear arrangement represents genetic mosaicism.5 In our case, the lesion started as a small plaque in childhood and gradually spread linearly to the buttock (Figure 1). A biopsy was taken from the lesion on the right buttock, which showed numerous dilated capillaries in the dermis (Figure 2).
|
Verrucous hemangiomas are best treated by excision. Larger lesions will need grafting. There is a tendency for recurrence to occur unless excision is complete.1 Yang and Ohara7 reported 14 patients with small localized lesions that were cured by 1 session of surgery without recurrence; 9 patients with wider and more extensive lesions required combination therapy in several stages for optimal results.
The Diagnosis: Linear Verrucous Hemangioma
Verrucous hemangioma is a rare congenital vascular malformation of the cutaneous and subcutaneous tissues. Although almost invariably present at birth, it may appear later in childhood or even in adulthood. Lesions commonly are found on the legs and may be linear, multiple, and disseminated or sometimes confined to the digits. In the early phase of evolution, the lesions are nonkeratotic, soft, blue-red plaques, but they gradually become increasingly hyperkeratotic.1 Linear verrucous hemangioma is even more rare with few published reports.
In 1937, Halter2 first used the term verrucous hemangioma to describe a 16-year-old adolescent boy who presented with a linear purpuric cluster of plaques extending from the right buttock to the toes. Imperial and Helwig3 later described it as a distinct entity. Since then, similar lesions have been described using a variety of names such as angiokeratoma circumscriptum neviforme, angiokeratoma circumscriptum, angiokeratoma corporis neviforme, keratotic hemangioma, nevus vascularis unius lateralis, and nevus keratoangiomatosus.3 Therefore, the exact incidence is difficult to determine.
Lesions generally are noted at birth or in early childhood and are often located on the lower extremities. The early lesions are bluish red in color. Secondary infection is a frequent complication, resulting in reactive papillomatosis and hyperkeratosis; thus, the older lesions acquire a verrucous or warty surface.3 Clinically, they may resemble angiokeratoma, lym-phangioma circumscriptum, verrucous epidermal nevus, verrucous cancer, or even malignant melanoma. Lesions initially resemble port-wine stains and later may become soft, bluish red vascular swellings that tend to grow in size and become verrucous.1
The histologic appearance closely resembles angiokeratoma, as both lesions show vascular spaces beneath a papillomatous and hyperkeratotic epidermis.4 However, in contrast to angiokeratoma, the vascular spaces in verrucous hemangioma also involve the lower dermis and subcutaneous tissues.
Although cases of linear verrucous hemangioma have been reported, its distribution along the lines of Blaschko is rare.5,6 It has been proposed that these lesions may actually be following dermatomal patterns or that the linear arrangement represents genetic mosaicism.5 In our case, the lesion started as a small plaque in childhood and gradually spread linearly to the buttock (Figure 1). A biopsy was taken from the lesion on the right buttock, which showed numerous dilated capillaries in the dermis (Figure 2).
|
Verrucous hemangiomas are best treated by excision. Larger lesions will need grafting. There is a tendency for recurrence to occur unless excision is complete.1 Yang and Ohara7 reported 14 patients with small localized lesions that were cured by 1 session of surgery without recurrence; 9 patients with wider and more extensive lesions required combination therapy in several stages for optimal results.
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. London, England: Blackwell Scientific Publications; 2004:15-60.
2. Halter K. Haemangioma verrucosum mit Osteoatrophie. Dermatol Z. 1937;75:271-279.
3. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathological study of 21 cases. Arch Dermatol. 1967;96:247-253.
4. Calduch L, Ortega C, Navarro V, et al. Verrucous hemangioma: Report of two cases and review of the literature. Pediatr Dermatol. 2000;17:213-217.
5. Wentscher U, Happle R. Linear verrucous hemangioma. J Am Acad Dermatol. 2000;42:516-518.
6. Jain VK, Aggarwal K, Jain S. Linear verrucous hemangioma on the leg. Indian J Dermatol Venereol Leprol. 2008;74:656-658.
7. Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919.
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. London, England: Blackwell Scientific Publications; 2004:15-60.
2. Halter K. Haemangioma verrucosum mit Osteoatrophie. Dermatol Z. 1937;75:271-279.
3. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathological study of 21 cases. Arch Dermatol. 1967;96:247-253.
4. Calduch L, Ortega C, Navarro V, et al. Verrucous hemangioma: Report of two cases and review of the literature. Pediatr Dermatol. 2000;17:213-217.
5. Wentscher U, Happle R. Linear verrucous hemangioma. J Am Acad Dermatol. 2000;42:516-518.
6. Jain VK, Aggarwal K, Jain S. Linear verrucous hemangioma on the leg. Indian J Dermatol Venereol Leprol. 2008;74:656-658.
7. Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919.
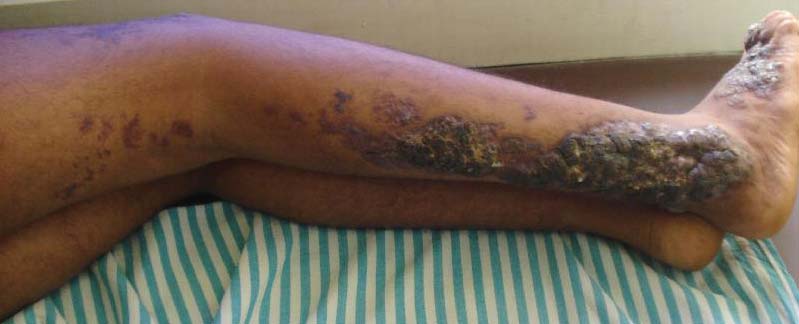
An 18-year-old man presented with a history of multiple bluish verrucous lesions over the right leg. A small nodule on the lateral aspect of the right ankle was present at birth; it increased in size and number and gradually extended to the right buttock. He had recurrent bleeding and infection over the lesions. No other remarkable comorbidities were noted. Dermatologic examination revealed multiple well-circumscribed, bluish red, verrucous lesions distributed linearly along the lateral aspect of the leg. The surface of the lesions was verruciform and showed crusting at places. The second and third toes on the right foot were involved. On the buttock, multiple well-defined, bluish red plaques were present. Both limbs were of equal length.
Waxy Indurated Plaques on the Eyelids
The Diagnosis: Primary (Myeloma-Associated) Systemic Amyloidosis
Amyloidosis is a broad term describing the abnormal autoaggregation of normally soluble proteins as extracellular deposits. More than 2 dozen protein precursors to these amyloid fibrils have been characterized; however, all amyloid deposits invariably act to disrupt normal tissue structure and function. The term amyloid dates back to the 19th century when it was used by Rudolph Virchow to describe the lardaceous appearance of affected tissue. In the 1920s amyloid was found to avidly stain with Congo red, and shortly thereafter the classical apple green birefringence was described when these plaques were exposed to polarized light1 (Figure 1). Currently, amyloidosis is classified by the World Health Organization based on the type of protein deposited; there are more than 20 types of low-molecular-weight proteins that form amyloid fibrils. Generally, amyloidosis can be viewed as primary (AL), secondary (AA), or hereditary in origin. Hereditary amyloidosis usually arises in the setting of familial mutations that potentiate the aggregation of plasma proteins, while secondary amyloidosis develops in the setting of chronic inflammatory diseases (eg, rheumatoid arthritis, lupus). Primary systemic amyloidosis, which presented in our patient, is caused by the overproduction and deposition of clonal immunoglobulin light chain fragments.
Prevalence
Primary systemic amyloidosis (AL amyloidosis) is the most common cause of amyloidosis in the United States, with an annual incidence of 6 to 7 cases per million. The median age at diagnosis is 73.5 years and approximately 60% of patients are male.2 Due to the rare nature of this disease, few large-scale studies have been conducted looking at the pathogenesis, presentation, and best treatment options for AL amyloidosis. However, the following trends were compiled based on the available literature and serve as a useful clinical guide. Primary systemic AL amyloidosis is characterized by an underlying plasma cell dyscrasia, such as multiple myeloma or monoclonal gammopathy of undetermined significance.3 Although AL amyloidosis frequently is associated with multiple myeloma, only 10% of these patients have coexisting multiple myeloma. Conversely, 12% to 15% of multiple myeloma patients are eventually diagnosed with AL amyloi-dosis. The plasma cells in AL amyloidosis are clonal and produce immunoglobulin κ and λ light chains. Interestingly, the ratio of λ to κ light chains in AL amyloidosis is 3 to 1, which is the reverse of the ratio usually seen in multiple myeloma.4
Clinical Presentation
Because AL amyloidosis can affect virtually every organ system except the brain parenchyma, the clinical presentation is extremely variable and initial symptoms are nonspecific, including fatigue and weight loss. Therefore, the diagnosis is frequently delayed until end-organ dysfunction becomes evident.5 At presentation, three-quarters of patients have involvement of 2 or more organs. In a study of 445 patients with AL amyloidosis, the most commonly involved organs included the kidneys (40%–70%), heart (30%–60%), liver (10%–25%), gastrointestinal tract (10%–20%), peripheral nervous system (5%–20%), and soft tissue infiltration (15%), with macroglossia being a pathognomonic finding. Macroglossia arises from the direct deposition of monoclonal immunoglobulin light chains within the tongue and is observed in up to 40% of cases.6 Dental indentations can be observed on the lateral aspects of the tongue. Macroglossia can lead to dysphagia as well as respiratory complications.
Diagnosis
Distinct cutaneous manifestations of amyloidosis are evident in 29% to 40% of cases and can prompt the astute dermatologist to test for underlying disease. Purpura and ecchymotic lesions occur in 15% of patients and are usually preceded by minor trauma (pinch purpura) or maneuvers that increase intravascular pressure (eg, Valsalva maneuver, vomiting).7 Periorbital distribution is the most classic finding, as was evident in our patient; however, all flexural regions including the neck, axillae, and anogenital regions can be involved. Our patient also had purpura present on the thumb (Figure 2) and shoulder (Figure 3), demonstrating the variable presentation of amyloidosis on the skin. Mechanistically, AL amyloidosis is thought to contribute to purpura by infiltrating blood vessels, thus making them more fragile. Furthermore, AL amyloidosis can potentiate a bleeding diathesis by depleting the body of factor X secondary to the binding of vitamin K–dependent factors to amyloid deposits. Additional cutaneous findings in AL amyloidosis may include smooth, shiny, waxy papules that may coalesce to form nodules and plaques of various sizes. Lesions typically are distributed in the flexural areas, including the mouth, eyes, neck, inguinal folds, and axillae. Lesions occurring in the perianal and vulvar skin may resemble giant condylomata or xanthomas.8 Although rare, nail dystrophy characterized by brittle nail plates and longitudinal ridging also can occur in patients with systemic AL amyloidosis. Another rare but extremely important cutaneous manifestation is hemorrhagic or clear subepidermal bullae. Histopathology can aid in the diagnosis of bullous amyloidosis.9 Diagnosis is usually accomplished by tissue biopsy of an affected organ or surrogate site as well as screening of the serum and urine for monoclonal immunoglobulins (detected in >90% of cases). The choice of a suitable biopsy site is at times complicated by accessibility and morbidity of the procedure as well as the varying yields. The biopsy sites with the highest yield in AL amyloidosis are the kidneys and liver (90%), followed by abdominal fat-pad (60%–84%), rectum (50%–80%), bone marrow (50%–55%), and skin (50%).10 Given the invasive nature of kidney and liver biopsies, the abdominal fat-pad frequently is sampled first, followed by more invasive procedures if the initial biopsy is negative and clinical suspicion remains high. In the presence of clinically evident mucocutaneous lesions, skin biopsy also can be used for initial diagnosis; however, the yield drops precipitously when skin involvement is absent. Amyloid has an eosinophilic amorphous appearance on hematoxylin and eosin stains and is definitively diagnosed by Congo red staining (apple green birefringence under polarized light). Because Congo red staining only confirms the presence of amyloid and not the precipitated protein, typing of the amyloid deposits should be done either by immunohistochemical staining or electron microscopy. Because of the association of AL amyloidosis with plasma cell dyscrasia, a bone marrow biopsy is warranted to grade the degree of plasma cell infiltration of the marrow. If no plasma cell dyscrasia is identified in the setting of a biopsy suggestive of amyloid, other causes of amyloidosis should be considered including familial (10% of cases) and secondary variants.11 End-organ involvement also should be investigated with echocardiography, electrocardiography peripheral nerve studies, skeletal survey, and renal and liver function testing. Prognosis is poor with expected survival of 12 to 14 months and is heavily dependent on the degree and type of end-organ involvement at the time of diagnosis, with median survival ranging from 4 to 6 months in patients with congestive heart failure to as long as 2 years in patients with primarily renal involvement.5,10,12,13 Poor prognosticating factors in AL amyloidosis involve dominant cardiac involvement, coexisting multiple myeloma, and increased peripheral plasma cell counts.14
|
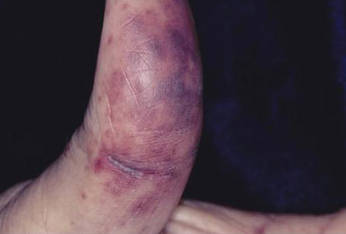
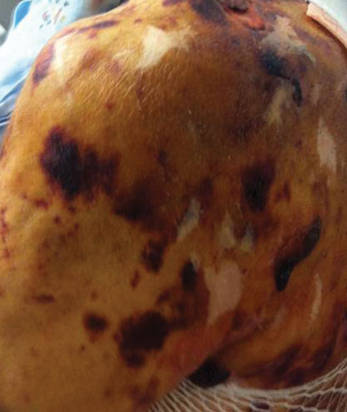
Treatment
Treatment regimens for AL amyloidosis have historically been adapted from those used to treat multiple myeloma, with the primary focus being the reduction of the amyloidogenic clone. The first effective therapy for AL amyloidosis was melphalan and prednisone. Although this regimen extended survival from half a year (6–8 months) to a year (12–18 months), it also was plagued by severe side effects and delayed response times. Increased response rates and fewer side effects have been achieved by replacing prednisone with dexamethasone in the aforementioned regimen.15 High-dose melphalan conditioning followed by autologous stem cell transplant has shown tremendous promise, with complete remission rates approaching 40% and median survival exceeding 4 years in some studies.16 However, the treatment-related mortality of stem cell transplant regimens is 10% to 20%, thus limiting this approach to younger and healthier patients. Exciting early work is emerging to support the efficacy of novel agents in the treatment of AL amyloidosis, including thalidomide, lenalidomide, and bortezomib. Because these agents do not have many of the traditional side effects associated with chemotherapy, they are increasingly being used in patients who are not candidates for stem cell transplant or melphalan-dexamethasone. They also can be used as salvage therapy in patients with relapsed disease.
Conclusion
Primary AL amyloidosis is a rare disorder that carries a poor prognosis. One of the major challenges for clinicians is diagnosis, as nearly any organ system can be affected, resulting in variable presentations. Although cutaneous manifestations are highly characteristic of AL amyloidosis and may help aid in its diagnosis, only a limited number of patients have involvement of the skin. As such, further research is needed to facilitate earlier diagnosis of primary AL amyloidosis as well as to develop novel therapies for this devastating disease.
1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-538.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
3. Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150-1151.
4. Solomon AB. Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. 1982;70:453-460.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
6. Thibault I, Vallieres I. Macroglossia due to systemic amyloidosis: is there a role for radiotherapy? Case Rep Oncol. 2011;4:392-399.
7. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1-16.
8. Chapman RS, Neville EA, Lawson JW. Xanthoma-like skin lesions as a presenting feature in primary systemic amyloidosis. Br J Clin Pract. 1973;27:271-273.
9. Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271-299.
11. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-1791.
12. Kyle RA, Greipp PR. Amyloidosis (AL). clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-683.
13. Madan SA, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232-238.
14. Pardanani A, Witzig TE, Schroeder G, et al. Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis. Blood. 2003;101:827-830.
15. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936-2938.
16. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
The Diagnosis: Primary (Myeloma-Associated) Systemic Amyloidosis
Amyloidosis is a broad term describing the abnormal autoaggregation of normally soluble proteins as extracellular deposits. More than 2 dozen protein precursors to these amyloid fibrils have been characterized; however, all amyloid deposits invariably act to disrupt normal tissue structure and function. The term amyloid dates back to the 19th century when it was used by Rudolph Virchow to describe the lardaceous appearance of affected tissue. In the 1920s amyloid was found to avidly stain with Congo red, and shortly thereafter the classical apple green birefringence was described when these plaques were exposed to polarized light1 (Figure 1). Currently, amyloidosis is classified by the World Health Organization based on the type of protein deposited; there are more than 20 types of low-molecular-weight proteins that form amyloid fibrils. Generally, amyloidosis can be viewed as primary (AL), secondary (AA), or hereditary in origin. Hereditary amyloidosis usually arises in the setting of familial mutations that potentiate the aggregation of plasma proteins, while secondary amyloidosis develops in the setting of chronic inflammatory diseases (eg, rheumatoid arthritis, lupus). Primary systemic amyloidosis, which presented in our patient, is caused by the overproduction and deposition of clonal immunoglobulin light chain fragments.
Prevalence
Primary systemic amyloidosis (AL amyloidosis) is the most common cause of amyloidosis in the United States, with an annual incidence of 6 to 7 cases per million. The median age at diagnosis is 73.5 years and approximately 60% of patients are male.2 Due to the rare nature of this disease, few large-scale studies have been conducted looking at the pathogenesis, presentation, and best treatment options for AL amyloidosis. However, the following trends were compiled based on the available literature and serve as a useful clinical guide. Primary systemic AL amyloidosis is characterized by an underlying plasma cell dyscrasia, such as multiple myeloma or monoclonal gammopathy of undetermined significance.3 Although AL amyloidosis frequently is associated with multiple myeloma, only 10% of these patients have coexisting multiple myeloma. Conversely, 12% to 15% of multiple myeloma patients are eventually diagnosed with AL amyloi-dosis. The plasma cells in AL amyloidosis are clonal and produce immunoglobulin κ and λ light chains. Interestingly, the ratio of λ to κ light chains in AL amyloidosis is 3 to 1, which is the reverse of the ratio usually seen in multiple myeloma.4
Clinical Presentation
Because AL amyloidosis can affect virtually every organ system except the brain parenchyma, the clinical presentation is extremely variable and initial symptoms are nonspecific, including fatigue and weight loss. Therefore, the diagnosis is frequently delayed until end-organ dysfunction becomes evident.5 At presentation, three-quarters of patients have involvement of 2 or more organs. In a study of 445 patients with AL amyloidosis, the most commonly involved organs included the kidneys (40%–70%), heart (30%–60%), liver (10%–25%), gastrointestinal tract (10%–20%), peripheral nervous system (5%–20%), and soft tissue infiltration (15%), with macroglossia being a pathognomonic finding. Macroglossia arises from the direct deposition of monoclonal immunoglobulin light chains within the tongue and is observed in up to 40% of cases.6 Dental indentations can be observed on the lateral aspects of the tongue. Macroglossia can lead to dysphagia as well as respiratory complications.
Diagnosis
Distinct cutaneous manifestations of amyloidosis are evident in 29% to 40% of cases and can prompt the astute dermatologist to test for underlying disease. Purpura and ecchymotic lesions occur in 15% of patients and are usually preceded by minor trauma (pinch purpura) or maneuvers that increase intravascular pressure (eg, Valsalva maneuver, vomiting).7 Periorbital distribution is the most classic finding, as was evident in our patient; however, all flexural regions including the neck, axillae, and anogenital regions can be involved. Our patient also had purpura present on the thumb (Figure 2) and shoulder (Figure 3), demonstrating the variable presentation of amyloidosis on the skin. Mechanistically, AL amyloidosis is thought to contribute to purpura by infiltrating blood vessels, thus making them more fragile. Furthermore, AL amyloidosis can potentiate a bleeding diathesis by depleting the body of factor X secondary to the binding of vitamin K–dependent factors to amyloid deposits. Additional cutaneous findings in AL amyloidosis may include smooth, shiny, waxy papules that may coalesce to form nodules and plaques of various sizes. Lesions typically are distributed in the flexural areas, including the mouth, eyes, neck, inguinal folds, and axillae. Lesions occurring in the perianal and vulvar skin may resemble giant condylomata or xanthomas.8 Although rare, nail dystrophy characterized by brittle nail plates and longitudinal ridging also can occur in patients with systemic AL amyloidosis. Another rare but extremely important cutaneous manifestation is hemorrhagic or clear subepidermal bullae. Histopathology can aid in the diagnosis of bullous amyloidosis.9 Diagnosis is usually accomplished by tissue biopsy of an affected organ or surrogate site as well as screening of the serum and urine for monoclonal immunoglobulins (detected in >90% of cases). The choice of a suitable biopsy site is at times complicated by accessibility and morbidity of the procedure as well as the varying yields. The biopsy sites with the highest yield in AL amyloidosis are the kidneys and liver (90%), followed by abdominal fat-pad (60%–84%), rectum (50%–80%), bone marrow (50%–55%), and skin (50%).10 Given the invasive nature of kidney and liver biopsies, the abdominal fat-pad frequently is sampled first, followed by more invasive procedures if the initial biopsy is negative and clinical suspicion remains high. In the presence of clinically evident mucocutaneous lesions, skin biopsy also can be used for initial diagnosis; however, the yield drops precipitously when skin involvement is absent. Amyloid has an eosinophilic amorphous appearance on hematoxylin and eosin stains and is definitively diagnosed by Congo red staining (apple green birefringence under polarized light). Because Congo red staining only confirms the presence of amyloid and not the precipitated protein, typing of the amyloid deposits should be done either by immunohistochemical staining or electron microscopy. Because of the association of AL amyloidosis with plasma cell dyscrasia, a bone marrow biopsy is warranted to grade the degree of plasma cell infiltration of the marrow. If no plasma cell dyscrasia is identified in the setting of a biopsy suggestive of amyloid, other causes of amyloidosis should be considered including familial (10% of cases) and secondary variants.11 End-organ involvement also should be investigated with echocardiography, electrocardiography peripheral nerve studies, skeletal survey, and renal and liver function testing. Prognosis is poor with expected survival of 12 to 14 months and is heavily dependent on the degree and type of end-organ involvement at the time of diagnosis, with median survival ranging from 4 to 6 months in patients with congestive heart failure to as long as 2 years in patients with primarily renal involvement.5,10,12,13 Poor prognosticating factors in AL amyloidosis involve dominant cardiac involvement, coexisting multiple myeloma, and increased peripheral plasma cell counts.14
|
Treatment
Treatment regimens for AL amyloidosis have historically been adapted from those used to treat multiple myeloma, with the primary focus being the reduction of the amyloidogenic clone. The first effective therapy for AL amyloidosis was melphalan and prednisone. Although this regimen extended survival from half a year (6–8 months) to a year (12–18 months), it also was plagued by severe side effects and delayed response times. Increased response rates and fewer side effects have been achieved by replacing prednisone with dexamethasone in the aforementioned regimen.15 High-dose melphalan conditioning followed by autologous stem cell transplant has shown tremendous promise, with complete remission rates approaching 40% and median survival exceeding 4 years in some studies.16 However, the treatment-related mortality of stem cell transplant regimens is 10% to 20%, thus limiting this approach to younger and healthier patients. Exciting early work is emerging to support the efficacy of novel agents in the treatment of AL amyloidosis, including thalidomide, lenalidomide, and bortezomib. Because these agents do not have many of the traditional side effects associated with chemotherapy, they are increasingly being used in patients who are not candidates for stem cell transplant or melphalan-dexamethasone. They also can be used as salvage therapy in patients with relapsed disease.
Conclusion
Primary AL amyloidosis is a rare disorder that carries a poor prognosis. One of the major challenges for clinicians is diagnosis, as nearly any organ system can be affected, resulting in variable presentations. Although cutaneous manifestations are highly characteristic of AL amyloidosis and may help aid in its diagnosis, only a limited number of patients have involvement of the skin. As such, further research is needed to facilitate earlier diagnosis of primary AL amyloidosis as well as to develop novel therapies for this devastating disease.
The Diagnosis: Primary (Myeloma-Associated) Systemic Amyloidosis
Amyloidosis is a broad term describing the abnormal autoaggregation of normally soluble proteins as extracellular deposits. More than 2 dozen protein precursors to these amyloid fibrils have been characterized; however, all amyloid deposits invariably act to disrupt normal tissue structure and function. The term amyloid dates back to the 19th century when it was used by Rudolph Virchow to describe the lardaceous appearance of affected tissue. In the 1920s amyloid was found to avidly stain with Congo red, and shortly thereafter the classical apple green birefringence was described when these plaques were exposed to polarized light1 (Figure 1). Currently, amyloidosis is classified by the World Health Organization based on the type of protein deposited; there are more than 20 types of low-molecular-weight proteins that form amyloid fibrils. Generally, amyloidosis can be viewed as primary (AL), secondary (AA), or hereditary in origin. Hereditary amyloidosis usually arises in the setting of familial mutations that potentiate the aggregation of plasma proteins, while secondary amyloidosis develops in the setting of chronic inflammatory diseases (eg, rheumatoid arthritis, lupus). Primary systemic amyloidosis, which presented in our patient, is caused by the overproduction and deposition of clonal immunoglobulin light chain fragments.
Prevalence
Primary systemic amyloidosis (AL amyloidosis) is the most common cause of amyloidosis in the United States, with an annual incidence of 6 to 7 cases per million. The median age at diagnosis is 73.5 years and approximately 60% of patients are male.2 Due to the rare nature of this disease, few large-scale studies have been conducted looking at the pathogenesis, presentation, and best treatment options for AL amyloidosis. However, the following trends were compiled based on the available literature and serve as a useful clinical guide. Primary systemic AL amyloidosis is characterized by an underlying plasma cell dyscrasia, such as multiple myeloma or monoclonal gammopathy of undetermined significance.3 Although AL amyloidosis frequently is associated with multiple myeloma, only 10% of these patients have coexisting multiple myeloma. Conversely, 12% to 15% of multiple myeloma patients are eventually diagnosed with AL amyloi-dosis. The plasma cells in AL amyloidosis are clonal and produce immunoglobulin κ and λ light chains. Interestingly, the ratio of λ to κ light chains in AL amyloidosis is 3 to 1, which is the reverse of the ratio usually seen in multiple myeloma.4
Clinical Presentation
Because AL amyloidosis can affect virtually every organ system except the brain parenchyma, the clinical presentation is extremely variable and initial symptoms are nonspecific, including fatigue and weight loss. Therefore, the diagnosis is frequently delayed until end-organ dysfunction becomes evident.5 At presentation, three-quarters of patients have involvement of 2 or more organs. In a study of 445 patients with AL amyloidosis, the most commonly involved organs included the kidneys (40%–70%), heart (30%–60%), liver (10%–25%), gastrointestinal tract (10%–20%), peripheral nervous system (5%–20%), and soft tissue infiltration (15%), with macroglossia being a pathognomonic finding. Macroglossia arises from the direct deposition of monoclonal immunoglobulin light chains within the tongue and is observed in up to 40% of cases.6 Dental indentations can be observed on the lateral aspects of the tongue. Macroglossia can lead to dysphagia as well as respiratory complications.
Diagnosis
Distinct cutaneous manifestations of amyloidosis are evident in 29% to 40% of cases and can prompt the astute dermatologist to test for underlying disease. Purpura and ecchymotic lesions occur in 15% of patients and are usually preceded by minor trauma (pinch purpura) or maneuvers that increase intravascular pressure (eg, Valsalva maneuver, vomiting).7 Periorbital distribution is the most classic finding, as was evident in our patient; however, all flexural regions including the neck, axillae, and anogenital regions can be involved. Our patient also had purpura present on the thumb (Figure 2) and shoulder (Figure 3), demonstrating the variable presentation of amyloidosis on the skin. Mechanistically, AL amyloidosis is thought to contribute to purpura by infiltrating blood vessels, thus making them more fragile. Furthermore, AL amyloidosis can potentiate a bleeding diathesis by depleting the body of factor X secondary to the binding of vitamin K–dependent factors to amyloid deposits. Additional cutaneous findings in AL amyloidosis may include smooth, shiny, waxy papules that may coalesce to form nodules and plaques of various sizes. Lesions typically are distributed in the flexural areas, including the mouth, eyes, neck, inguinal folds, and axillae. Lesions occurring in the perianal and vulvar skin may resemble giant condylomata or xanthomas.8 Although rare, nail dystrophy characterized by brittle nail plates and longitudinal ridging also can occur in patients with systemic AL amyloidosis. Another rare but extremely important cutaneous manifestation is hemorrhagic or clear subepidermal bullae. Histopathology can aid in the diagnosis of bullous amyloidosis.9 Diagnosis is usually accomplished by tissue biopsy of an affected organ or surrogate site as well as screening of the serum and urine for monoclonal immunoglobulins (detected in >90% of cases). The choice of a suitable biopsy site is at times complicated by accessibility and morbidity of the procedure as well as the varying yields. The biopsy sites with the highest yield in AL amyloidosis are the kidneys and liver (90%), followed by abdominal fat-pad (60%–84%), rectum (50%–80%), bone marrow (50%–55%), and skin (50%).10 Given the invasive nature of kidney and liver biopsies, the abdominal fat-pad frequently is sampled first, followed by more invasive procedures if the initial biopsy is negative and clinical suspicion remains high. In the presence of clinically evident mucocutaneous lesions, skin biopsy also can be used for initial diagnosis; however, the yield drops precipitously when skin involvement is absent. Amyloid has an eosinophilic amorphous appearance on hematoxylin and eosin stains and is definitively diagnosed by Congo red staining (apple green birefringence under polarized light). Because Congo red staining only confirms the presence of amyloid and not the precipitated protein, typing of the amyloid deposits should be done either by immunohistochemical staining or electron microscopy. Because of the association of AL amyloidosis with plasma cell dyscrasia, a bone marrow biopsy is warranted to grade the degree of plasma cell infiltration of the marrow. If no plasma cell dyscrasia is identified in the setting of a biopsy suggestive of amyloid, other causes of amyloidosis should be considered including familial (10% of cases) and secondary variants.11 End-organ involvement also should be investigated with echocardiography, electrocardiography peripheral nerve studies, skeletal survey, and renal and liver function testing. Prognosis is poor with expected survival of 12 to 14 months and is heavily dependent on the degree and type of end-organ involvement at the time of diagnosis, with median survival ranging from 4 to 6 months in patients with congestive heart failure to as long as 2 years in patients with primarily renal involvement.5,10,12,13 Poor prognosticating factors in AL amyloidosis involve dominant cardiac involvement, coexisting multiple myeloma, and increased peripheral plasma cell counts.14
|
Treatment
Treatment regimens for AL amyloidosis have historically been adapted from those used to treat multiple myeloma, with the primary focus being the reduction of the amyloidogenic clone. The first effective therapy for AL amyloidosis was melphalan and prednisone. Although this regimen extended survival from half a year (6–8 months) to a year (12–18 months), it also was plagued by severe side effects and delayed response times. Increased response rates and fewer side effects have been achieved by replacing prednisone with dexamethasone in the aforementioned regimen.15 High-dose melphalan conditioning followed by autologous stem cell transplant has shown tremendous promise, with complete remission rates approaching 40% and median survival exceeding 4 years in some studies.16 However, the treatment-related mortality of stem cell transplant regimens is 10% to 20%, thus limiting this approach to younger and healthier patients. Exciting early work is emerging to support the efficacy of novel agents in the treatment of AL amyloidosis, including thalidomide, lenalidomide, and bortezomib. Because these agents do not have many of the traditional side effects associated with chemotherapy, they are increasingly being used in patients who are not candidates for stem cell transplant or melphalan-dexamethasone. They also can be used as salvage therapy in patients with relapsed disease.
Conclusion
Primary AL amyloidosis is a rare disorder that carries a poor prognosis. One of the major challenges for clinicians is diagnosis, as nearly any organ system can be affected, resulting in variable presentations. Although cutaneous manifestations are highly characteristic of AL amyloidosis and may help aid in its diagnosis, only a limited number of patients have involvement of the skin. As such, further research is needed to facilitate earlier diagnosis of primary AL amyloidosis as well as to develop novel therapies for this devastating disease.
1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-538.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
3. Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150-1151.
4. Solomon AB. Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. 1982;70:453-460.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
6. Thibault I, Vallieres I. Macroglossia due to systemic amyloidosis: is there a role for radiotherapy? Case Rep Oncol. 2011;4:392-399.
7. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1-16.
8. Chapman RS, Neville EA, Lawson JW. Xanthoma-like skin lesions as a presenting feature in primary systemic amyloidosis. Br J Clin Pract. 1973;27:271-273.
9. Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271-299.
11. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-1791.
12. Kyle RA, Greipp PR. Amyloidosis (AL). clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-683.
13. Madan SA, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232-238.
14. Pardanani A, Witzig TE, Schroeder G, et al. Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis. Blood. 2003;101:827-830.
15. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936-2938.
16. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
1. Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-538.
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817-1822.
3. Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150-1151.
4. Solomon AB. Frangione B, Franklin EC. Bence Jones proteins and light chains of immunoglobulins. preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda). J Clin Invest. 1982;70:453-460.
5. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
6. Thibault I, Vallieres I. Macroglossia due to systemic amyloidosis: is there a role for radiotherapy? Case Rep Oncol. 2011;4:392-399.
7. Breathnach SM. Amyloid and amyloidosis. J Am Acad Dermatol. 1988;18:1-16.
8. Chapman RS, Neville EA, Lawson JW. Xanthoma-like skin lesions as a presenting feature in primary systemic amyloidosis. Br J Clin Pract. 1973;27:271-273.
9. Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
10. Kyle RA, Bayrd ED. Amyloidosis: review of 236 cases. Medicine. 1975;54:271-299.
11. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-1791.
12. Kyle RA, Greipp PR. Amyloidosis (AL). clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-683.
13. Madan SA, Dispenzieri A, Lacy MQ, et al. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc. 2010;85:232-238.
14. Pardanani A, Witzig TE, Schroeder G, et al. Circulating peripheral blood plasma cells as a prognostic indicator in patients with primary systemic amyloidosis. Blood. 2003;101:827-830.
15. Palladini G, Perfetti V, Obici L, et al. Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation. Blood. 2004;103:2936-2938.
16. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140:85-93.
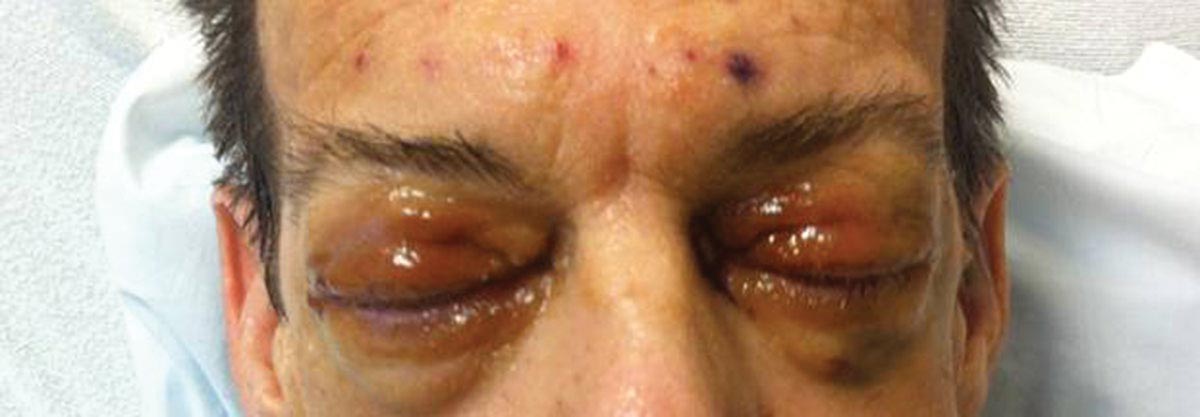
A 57-year-old man who underwent an orthotopic liver transplant 10 months prior for autoimmune hepatitis developed a rash on the trunk, arms, and legs. A dermatology consultation was requested. On evaluation, the patient was noted to have purpuric patches on the trunk and extremities, as well as prominent, bilateral, waxy and indurated periorbital plaques. The patient’s medical history revealed anemia, osteopenia, multiple bone fractures, and renal impairment.
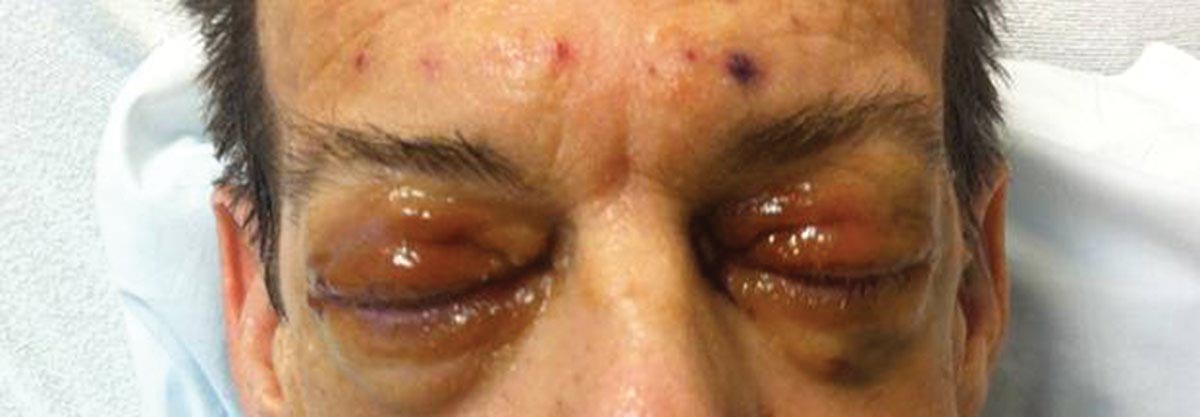
Multiple Papules on the Eyelid Margin
The Diagnosis: Molluscum Contagiosum
Dermoscopy showed multiple whitish amorphous structures with peripheral blood vessels (Figure 1). A skin biopsy specimen from the lower eyelid revealed loculated and endophytic epidermal hyperplasia. The keratinocytes contained large eosinophilic intracytoplasmic inclusion bodies, and the diagnosis of molluscum contagiosum (MC) was confirmed (Figure 2). Laboratory results were positive for human immunodeficiency virus (HIV) infection with a CD4 lymphocyte count of 18 cells/mm3 and viral load of 199,686 copies/mL. The patient was treated with CO2 laser therapy for eyelid lesions. A regimen of highly active antiretroviral therapy (HAART) was later started using a combination of lamivudine-zidovudine (150 mg and 300 mg) as well as lopinavir-ritonavir (400 mg and 100 mg), both twice daily. There was no recurrence at 3-month follow-up.
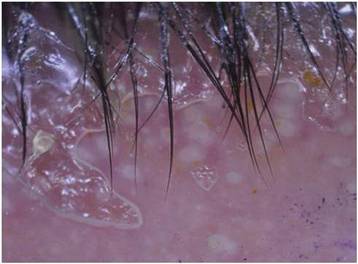
|
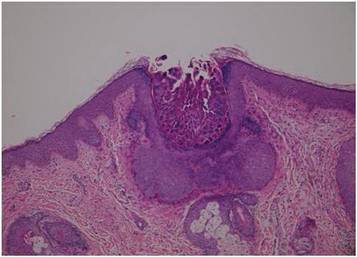
|
Molluscum contagiosum is a common cutaneous infection that is caused by a double-stranded DNA poxvirus. The clinical manifestations of MC are solitary or multiple, tiny, dome-shaped, pale, waxy or flesh-colored papules with central umbilication. The skin lesions can be located anywhere on the body. It occurs mostly in children, but adults also may be affected. In patients with atopic dermatitis or immunocompromised status such as AIDS, acute lymphoblastic leukemia, multiple myeloma, hyperimmunoglobulin E syndrome, or treatment with prednisone and methotrexate, the cutaneous lesions may be more extensive with an atypical presentation.1-6
The diagnosis of MC is mainly made by clinical inspection. However, Giemsa staining, Papanicolaou tests, and histopathology are useful for diagnosis of atypical MC.7 Dermoscopy is a noninvasive and fast diagnostic tool for MC.8,9 In dermoscopy, MC is characterized by multiple spherical, whitish, amorphous structures with a crown of blood vessels surrounding the periphery, termed red corona.8,9 The whitish amorphous structures and red corona correlate with inclusion bodies and dermal dilated blood vessels, respectively.
Approximately 13% of HIV patients have cutaneous MC, and the lesions tend to be more diffuse and refractory to treatment.10 A giant variant and abscess formation also have been described.11,12 Molluscum contagiosum of the eyelids often occurs in advanced HIV infection with a CD4 count less than 80 cells/mm3.1 These patients often have been diagnosed with HIV before developing eyelid MC. The severity of MC in immunocompromised patients may be related to the deficits of cell-mediated immunity, especially the T helper 1 (TH1) cytokine pathway. One case report also showed the clinical remission of MC after restoration of CD4 count with HAART.13 Our patient was not previously diagnosed with HIV and the MC of the eyelid margin was the early presentation of AIDS.
Molluscum contagiosum of the eyelids may cause chronic keratoconjunctivitis or even vascular infiltration and scarring of the peripheral cornea.14 These manifestations may be attributed to a hypersensitivity reaction to viral protein in tear film. Therefore, individuals with eyelid MC should accept thorough examination of the conjunctiva and cornea.
Treatment options include surgical excision, CO2 laser, curettage, and hyperfocal cryotherapy.15 Several reports also have demonstrated effectiveness of cidofovir for treatment of extensive MC lesions.16 Highly active antiretroviral therapy may play a role in the treatment of patients with AIDS by restoring the CD4 count.13 However, a few patients may develop immune reconstitution inflammatory syndrome, an intensive inflammatory reaction to pathogens after HAART, leading to paradoxical worsening of existing infection. Spontaneous corneal perforation due to immune reconstitution inflammatory syndrome in a case with eyelid and conjunctival MC has been reported.17 Therefore, physicians should perform MC therapy before HAART and mucocutaneous lesions should be followed regularly to prevent possible morbidity.
In summary, we report a case of AIDS with the initial presentation of MC on the eyelid margin. Physicians should test for HIV infection in patients with an atypical presentation of MC. The ocular mucosa also should be examined in patients with MC of the eyelid to prevent possible complications.
1. Pérez-Blázquez E, Villafruela I, Madero S. Eyelid molluscum contagiosum in patients with human immunodeficiency virus infection. Orbit. 1999;18:75-81.
2. Ozyürek E, Sentürk N, Kefeli M, et al. Ulcerating molluscum contagiosum in a boy with relapsed acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:e114-e116.
3. Moradi P, Bhogal M, Thaung C, et al. Epibulbar molluscum contagiosum lesions in multiple myeloma. Cornea. 2011;30:910-911.
4. Rosenberg EW, Yusk JW. Molluscum contagiosum. eruption following treatment with prednisone and methotrexate. Arch Dermatol. 1970;101:439-441.
5. Yang CH, Lee WI, Hsu TS. Disseminated white papules. Arch Dermatol. 2006;142:775-780.
6. Fotiadou C, Lazaridou E, Lekkas D, et al. Disseminated, eruptive molluscum contagiosum lesions in a psoriasis patient under treatment with methotrexate and cyclosporine. Eur J Dermatol. 2012;22:147-148.
7. Kumar N, Okiro P, Wasike R. Cytological diagnosis of molluscum contagiosum with an unusual clinical presentation at an unusual site. J Dermatol Case Rep. 2010;4:63-65.
8. Micali G, Lacarrubba F. Augmented diagnostic capability using videodermatoscopy on selected infectious and non-infectious penile growths. Int J Dermatol. 2011;50:1501-1505.
9. Micali G, Lacarrubba F, Massimino D, et al. Dermatoscopy: alternative uses in daily clinical practice. J Am Acad Dermatol. 2011;64:1135-1146.
10. Tzung TY, Yang CY, Chao SC, et al. Cutaneous manifestations of human immunodeficiency virus infection in Taiwan. Kaohsiung J Med Sci. 2004;20:216-224.
11. Chang W-Y, Chang C-P, Yang S-A, et al. Giant molluscum contagiosum with concurrence of molluscum dermatitis. Dermatol Sinica. 2005;23:81-85.
12. Bates CM, Carey PB, Dhar J, et al. Molluscum contagiosum—a novel presentation. Int J STD AIDS. 2001;12:614-615.
13. Schulz D, Sarra GM, Koerner UB, et al. Evolution of HIV-1-related conjunctival molluscum contagiosum under HAART: report of a bilaterally manifesting case and literature review. Graefes Arch Clin Exp Ophthalmol. 2004;242:951-955.
14. Redmond RM. Molluscum contagiosum is not always benign. BMJ. 2004;329:403.
15. Bardenstein DS, Elmets C. Hyperfocal cryotherapy of multiple molluscum contagiosum lesions in patients with the acquired immune deficiency syndrome. Ophthalmology. 1995;102:131-134.
16. Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous cidofovir in the treatment of giant molluscum contagiosum in a patient with human immunodeficiency virus. Arch Dermatol. 2011;147:652-654.
17. Williamson W, Dorot N, Mortemousque B, et al. Spontaneous corneal perforation and conjunctival molluscum contagiosum in a AIDS patient [in French]. J Fr Ophtalmol. 1995;18:703-707.
The Diagnosis: Molluscum Contagiosum
Dermoscopy showed multiple whitish amorphous structures with peripheral blood vessels (Figure 1). A skin biopsy specimen from the lower eyelid revealed loculated and endophytic epidermal hyperplasia. The keratinocytes contained large eosinophilic intracytoplasmic inclusion bodies, and the diagnosis of molluscum contagiosum (MC) was confirmed (Figure 2). Laboratory results were positive for human immunodeficiency virus (HIV) infection with a CD4 lymphocyte count of 18 cells/mm3 and viral load of 199,686 copies/mL. The patient was treated with CO2 laser therapy for eyelid lesions. A regimen of highly active antiretroviral therapy (HAART) was later started using a combination of lamivudine-zidovudine (150 mg and 300 mg) as well as lopinavir-ritonavir (400 mg and 100 mg), both twice daily. There was no recurrence at 3-month follow-up.
|
|
|
|
Molluscum contagiosum is a common cutaneous infection that is caused by a double-stranded DNA poxvirus. The clinical manifestations of MC are solitary or multiple, tiny, dome-shaped, pale, waxy or flesh-colored papules with central umbilication. The skin lesions can be located anywhere on the body. It occurs mostly in children, but adults also may be affected. In patients with atopic dermatitis or immunocompromised status such as AIDS, acute lymphoblastic leukemia, multiple myeloma, hyperimmunoglobulin E syndrome, or treatment with prednisone and methotrexate, the cutaneous lesions may be more extensive with an atypical presentation.1-6
The diagnosis of MC is mainly made by clinical inspection. However, Giemsa staining, Papanicolaou tests, and histopathology are useful for diagnosis of atypical MC.7 Dermoscopy is a noninvasive and fast diagnostic tool for MC.8,9 In dermoscopy, MC is characterized by multiple spherical, whitish, amorphous structures with a crown of blood vessels surrounding the periphery, termed red corona.8,9 The whitish amorphous structures and red corona correlate with inclusion bodies and dermal dilated blood vessels, respectively.
Approximately 13% of HIV patients have cutaneous MC, and the lesions tend to be more diffuse and refractory to treatment.10 A giant variant and abscess formation also have been described.11,12 Molluscum contagiosum of the eyelids often occurs in advanced HIV infection with a CD4 count less than 80 cells/mm3.1 These patients often have been diagnosed with HIV before developing eyelid MC. The severity of MC in immunocompromised patients may be related to the deficits of cell-mediated immunity, especially the T helper 1 (TH1) cytokine pathway. One case report also showed the clinical remission of MC after restoration of CD4 count with HAART.13 Our patient was not previously diagnosed with HIV and the MC of the eyelid margin was the early presentation of AIDS.
Molluscum contagiosum of the eyelids may cause chronic keratoconjunctivitis or even vascular infiltration and scarring of the peripheral cornea.14 These manifestations may be attributed to a hypersensitivity reaction to viral protein in tear film. Therefore, individuals with eyelid MC should accept thorough examination of the conjunctiva and cornea.
Treatment options include surgical excision, CO2 laser, curettage, and hyperfocal cryotherapy.15 Several reports also have demonstrated effectiveness of cidofovir for treatment of extensive MC lesions.16 Highly active antiretroviral therapy may play a role in the treatment of patients with AIDS by restoring the CD4 count.13 However, a few patients may develop immune reconstitution inflammatory syndrome, an intensive inflammatory reaction to pathogens after HAART, leading to paradoxical worsening of existing infection. Spontaneous corneal perforation due to immune reconstitution inflammatory syndrome in a case with eyelid and conjunctival MC has been reported.17 Therefore, physicians should perform MC therapy before HAART and mucocutaneous lesions should be followed regularly to prevent possible morbidity.
In summary, we report a case of AIDS with the initial presentation of MC on the eyelid margin. Physicians should test for HIV infection in patients with an atypical presentation of MC. The ocular mucosa also should be examined in patients with MC of the eyelid to prevent possible complications.
The Diagnosis: Molluscum Contagiosum
Dermoscopy showed multiple whitish amorphous structures with peripheral blood vessels (Figure 1). A skin biopsy specimen from the lower eyelid revealed loculated and endophytic epidermal hyperplasia. The keratinocytes contained large eosinophilic intracytoplasmic inclusion bodies, and the diagnosis of molluscum contagiosum (MC) was confirmed (Figure 2). Laboratory results were positive for human immunodeficiency virus (HIV) infection with a CD4 lymphocyte count of 18 cells/mm3 and viral load of 199,686 copies/mL. The patient was treated with CO2 laser therapy for eyelid lesions. A regimen of highly active antiretroviral therapy (HAART) was later started using a combination of lamivudine-zidovudine (150 mg and 300 mg) as well as lopinavir-ritonavir (400 mg and 100 mg), both twice daily. There was no recurrence at 3-month follow-up.
|
|
|
|
Molluscum contagiosum is a common cutaneous infection that is caused by a double-stranded DNA poxvirus. The clinical manifestations of MC are solitary or multiple, tiny, dome-shaped, pale, waxy or flesh-colored papules with central umbilication. The skin lesions can be located anywhere on the body. It occurs mostly in children, but adults also may be affected. In patients with atopic dermatitis or immunocompromised status such as AIDS, acute lymphoblastic leukemia, multiple myeloma, hyperimmunoglobulin E syndrome, or treatment with prednisone and methotrexate, the cutaneous lesions may be more extensive with an atypical presentation.1-6
The diagnosis of MC is mainly made by clinical inspection. However, Giemsa staining, Papanicolaou tests, and histopathology are useful for diagnosis of atypical MC.7 Dermoscopy is a noninvasive and fast diagnostic tool for MC.8,9 In dermoscopy, MC is characterized by multiple spherical, whitish, amorphous structures with a crown of blood vessels surrounding the periphery, termed red corona.8,9 The whitish amorphous structures and red corona correlate with inclusion bodies and dermal dilated blood vessels, respectively.
Approximately 13% of HIV patients have cutaneous MC, and the lesions tend to be more diffuse and refractory to treatment.10 A giant variant and abscess formation also have been described.11,12 Molluscum contagiosum of the eyelids often occurs in advanced HIV infection with a CD4 count less than 80 cells/mm3.1 These patients often have been diagnosed with HIV before developing eyelid MC. The severity of MC in immunocompromised patients may be related to the deficits of cell-mediated immunity, especially the T helper 1 (TH1) cytokine pathway. One case report also showed the clinical remission of MC after restoration of CD4 count with HAART.13 Our patient was not previously diagnosed with HIV and the MC of the eyelid margin was the early presentation of AIDS.
Molluscum contagiosum of the eyelids may cause chronic keratoconjunctivitis or even vascular infiltration and scarring of the peripheral cornea.14 These manifestations may be attributed to a hypersensitivity reaction to viral protein in tear film. Therefore, individuals with eyelid MC should accept thorough examination of the conjunctiva and cornea.
Treatment options include surgical excision, CO2 laser, curettage, and hyperfocal cryotherapy.15 Several reports also have demonstrated effectiveness of cidofovir for treatment of extensive MC lesions.16 Highly active antiretroviral therapy may play a role in the treatment of patients with AIDS by restoring the CD4 count.13 However, a few patients may develop immune reconstitution inflammatory syndrome, an intensive inflammatory reaction to pathogens after HAART, leading to paradoxical worsening of existing infection. Spontaneous corneal perforation due to immune reconstitution inflammatory syndrome in a case with eyelid and conjunctival MC has been reported.17 Therefore, physicians should perform MC therapy before HAART and mucocutaneous lesions should be followed regularly to prevent possible morbidity.
In summary, we report a case of AIDS with the initial presentation of MC on the eyelid margin. Physicians should test for HIV infection in patients with an atypical presentation of MC. The ocular mucosa also should be examined in patients with MC of the eyelid to prevent possible complications.
1. Pérez-Blázquez E, Villafruela I, Madero S. Eyelid molluscum contagiosum in patients with human immunodeficiency virus infection. Orbit. 1999;18:75-81.
2. Ozyürek E, Sentürk N, Kefeli M, et al. Ulcerating molluscum contagiosum in a boy with relapsed acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:e114-e116.
3. Moradi P, Bhogal M, Thaung C, et al. Epibulbar molluscum contagiosum lesions in multiple myeloma. Cornea. 2011;30:910-911.
4. Rosenberg EW, Yusk JW. Molluscum contagiosum. eruption following treatment with prednisone and methotrexate. Arch Dermatol. 1970;101:439-441.
5. Yang CH, Lee WI, Hsu TS. Disseminated white papules. Arch Dermatol. 2006;142:775-780.
6. Fotiadou C, Lazaridou E, Lekkas D, et al. Disseminated, eruptive molluscum contagiosum lesions in a psoriasis patient under treatment with methotrexate and cyclosporine. Eur J Dermatol. 2012;22:147-148.
7. Kumar N, Okiro P, Wasike R. Cytological diagnosis of molluscum contagiosum with an unusual clinical presentation at an unusual site. J Dermatol Case Rep. 2010;4:63-65.
8. Micali G, Lacarrubba F. Augmented diagnostic capability using videodermatoscopy on selected infectious and non-infectious penile growths. Int J Dermatol. 2011;50:1501-1505.
9. Micali G, Lacarrubba F, Massimino D, et al. Dermatoscopy: alternative uses in daily clinical practice. J Am Acad Dermatol. 2011;64:1135-1146.
10. Tzung TY, Yang CY, Chao SC, et al. Cutaneous manifestations of human immunodeficiency virus infection in Taiwan. Kaohsiung J Med Sci. 2004;20:216-224.
11. Chang W-Y, Chang C-P, Yang S-A, et al. Giant molluscum contagiosum with concurrence of molluscum dermatitis. Dermatol Sinica. 2005;23:81-85.
12. Bates CM, Carey PB, Dhar J, et al. Molluscum contagiosum—a novel presentation. Int J STD AIDS. 2001;12:614-615.
13. Schulz D, Sarra GM, Koerner UB, et al. Evolution of HIV-1-related conjunctival molluscum contagiosum under HAART: report of a bilaterally manifesting case and literature review. Graefes Arch Clin Exp Ophthalmol. 2004;242:951-955.
14. Redmond RM. Molluscum contagiosum is not always benign. BMJ. 2004;329:403.
15. Bardenstein DS, Elmets C. Hyperfocal cryotherapy of multiple molluscum contagiosum lesions in patients with the acquired immune deficiency syndrome. Ophthalmology. 1995;102:131-134.
16. Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous cidofovir in the treatment of giant molluscum contagiosum in a patient with human immunodeficiency virus. Arch Dermatol. 2011;147:652-654.
17. Williamson W, Dorot N, Mortemousque B, et al. Spontaneous corneal perforation and conjunctival molluscum contagiosum in a AIDS patient [in French]. J Fr Ophtalmol. 1995;18:703-707.
1. Pérez-Blázquez E, Villafruela I, Madero S. Eyelid molluscum contagiosum in patients with human immunodeficiency virus infection. Orbit. 1999;18:75-81.
2. Ozyürek E, Sentürk N, Kefeli M, et al. Ulcerating molluscum contagiosum in a boy with relapsed acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:e114-e116.
3. Moradi P, Bhogal M, Thaung C, et al. Epibulbar molluscum contagiosum lesions in multiple myeloma. Cornea. 2011;30:910-911.
4. Rosenberg EW, Yusk JW. Molluscum contagiosum. eruption following treatment with prednisone and methotrexate. Arch Dermatol. 1970;101:439-441.
5. Yang CH, Lee WI, Hsu TS. Disseminated white papules. Arch Dermatol. 2006;142:775-780.
6. Fotiadou C, Lazaridou E, Lekkas D, et al. Disseminated, eruptive molluscum contagiosum lesions in a psoriasis patient under treatment with methotrexate and cyclosporine. Eur J Dermatol. 2012;22:147-148.
7. Kumar N, Okiro P, Wasike R. Cytological diagnosis of molluscum contagiosum with an unusual clinical presentation at an unusual site. J Dermatol Case Rep. 2010;4:63-65.
8. Micali G, Lacarrubba F. Augmented diagnostic capability using videodermatoscopy on selected infectious and non-infectious penile growths. Int J Dermatol. 2011;50:1501-1505.
9. Micali G, Lacarrubba F, Massimino D, et al. Dermatoscopy: alternative uses in daily clinical practice. J Am Acad Dermatol. 2011;64:1135-1146.
10. Tzung TY, Yang CY, Chao SC, et al. Cutaneous manifestations of human immunodeficiency virus infection in Taiwan. Kaohsiung J Med Sci. 2004;20:216-224.
11. Chang W-Y, Chang C-P, Yang S-A, et al. Giant molluscum contagiosum with concurrence of molluscum dermatitis. Dermatol Sinica. 2005;23:81-85.
12. Bates CM, Carey PB, Dhar J, et al. Molluscum contagiosum—a novel presentation. Int J STD AIDS. 2001;12:614-615.
13. Schulz D, Sarra GM, Koerner UB, et al. Evolution of HIV-1-related conjunctival molluscum contagiosum under HAART: report of a bilaterally manifesting case and literature review. Graefes Arch Clin Exp Ophthalmol. 2004;242:951-955.
14. Redmond RM. Molluscum contagiosum is not always benign. BMJ. 2004;329:403.
15. Bardenstein DS, Elmets C. Hyperfocal cryotherapy of multiple molluscum contagiosum lesions in patients with the acquired immune deficiency syndrome. Ophthalmology. 1995;102:131-134.
16. Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous cidofovir in the treatment of giant molluscum contagiosum in a patient with human immunodeficiency virus. Arch Dermatol. 2011;147:652-654.
17. Williamson W, Dorot N, Mortemousque B, et al. Spontaneous corneal perforation and conjunctival molluscum contagiosum in a AIDS patient [in French]. J Fr Ophtalmol. 1995;18:703-707.
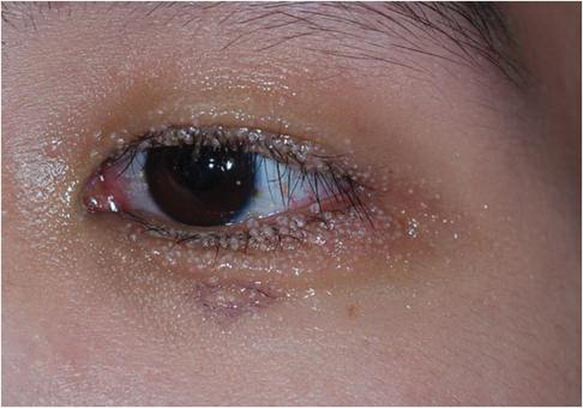
A 24-year-old man presented with multiple tiny papules over the left eyelid margin of 2 to 3 months’ duration. There were a couple of papules on the left upper eyelid initially, but they progressed to the upper and lower eyelid margin after scratching. On physical examination, multiple whitish to flesh-colored pearly papules measuring 1 to 2 mm were located on the left eyelid margin. Palpebral follicular conjunctivitis also was noted.
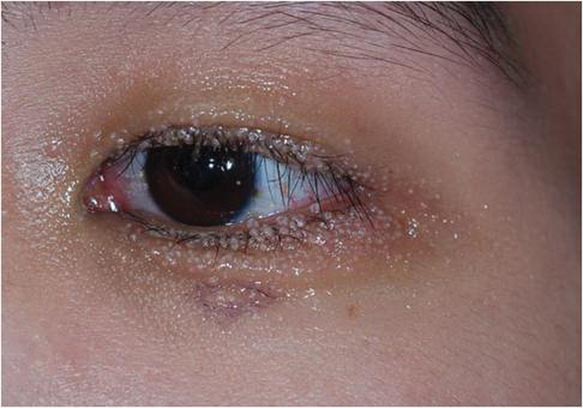
What Is Your Diagnosis? Acquired Lymphangiectasia
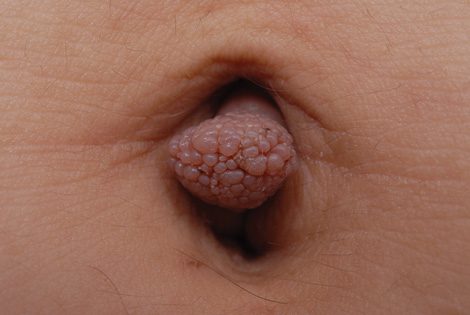
A 19-year-old woman presented with an umbilical mass of 5 months’ duration that had grown in size. Physical examination revealed a 1×1-cm brownish, pedunculated, cauliflower-shaped lesion on the umbilicus. There were no other signs or symptoms of disease. The patient’s personal and family disease history were unremarkable. An excisional biopsy was performed.
The Diagnosis: Acquired Lymphangiectasia
On histopathology numerous dilated channels lined by a single flat layer of endothelial cells were noted within the dermis. The overlying epidermis was papillomatous and acanthotic (Figure 1). The endothelial cells lining the dilated channels were D2-40 positive (Figure 2). Furthermore, the channels contained a pinkish amorphous material and a few red blood cells. The surrounding stroma showed scattered lymphocyte infiltration. These findings were consistent with lymphangiectasia. The lesion has not recurred 4 years following total excision.
|
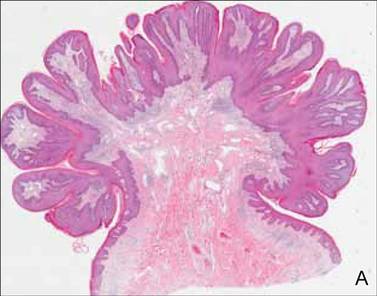

Acquired lymphangiectasia is known by various names, including lymphangioma, acquired lymphangioma, and acquired lymphangioma circumscriptum, which has led to confusion.1 Acquired lymphangiectasia, which is characterized by dilated superficial lymphatics, develops following damage to previously normal lymphatic channels, leading to a buildup of lymph pressure and backflow.2 Acquired lymphangiectasia has been reported as clinically and histologically indistinguishable from lymphangioma circumscriptum2; however, unlike in lymphangiectasia, the suffix -oma denotes a tumor. Our case matched more closely with the typical concept of lymphangiectasia rather than lymphangioma.
Clinical findings of acquired lymphangiectasia usually include translucent, flat or slightly raised, 2- to 5-mm, flesh-colored papules and vesicles.3,4 Acquired lymphangiectasia has been described with lesions that have verrucous surfaces mimicking warts, condyloma acuminata, or molluscum contagiosum.5,6 Our case suggests that acquired lymphangiectasia also can present with a pedunculated cauliflowerlike appearance. In general, it develops secondary to certain conditions such as recovery from trauma or surgery, postsurgical fibrosis, and irradiation. Lymphangiectasia often is seen on the arms, axillae, chest wall, and genital area in women and the scrotum, penis, thighs, and pubic region in men, both who have undergone radical surgery and irradiation for treatment of breast and prostate cancer, respectively.3 Our patient did not report any history of trauma to the umbilicus.
On histopathology acquired lymphangiectasia typically shows edematous polypoid nodules with dilated lymphatics. The overlying epidermis usually shows a spectrum of proliferation ranging from mild acanthosis to florid pseudoepitheliomatous hyperplasia with marked hyperkeratosis and parakeratosis. The distinctive finding of lymphangiectasia is the presence of dilated lymphatic spaces within the dermis. The dilated channels are filled with lymphatic fluid and often red and white blood cells. The single layer of flattened endothelial cells generally exhibits immunoreactivity to D2-40 and CD31.1
Treatment of lymphangiectasia is focused on reducing the pressure within the lymph vessels and managing consequent lymphedema with compression dressings. Simple surgical excision of lesions on sites such as the vulva or legs often is effective.3 If surgical intervention is not an option, cryotherapy, sclerotherapy, cauterization, and treatment with CO2 lasers also have been utilized with good outcomes.7 In the current case, total surgical excision was performed, which provided good results.
1. Stewart CJ, Chan T, Platten M. Acquired lymphangiectasia (‘lymphangioma circumscriptum’) of the vulva: a report of eight cases. Pathology. 2009;41:448-453.
2. Celis AV, Gaughf CN, Sangueza OP, et al. Acquired lymphangiectasis. South Med J. 1999;92:69-72.
3. Verma SB. Lymphangiectasias of the skin: victims of confusing nomenclature. Clin Exp Dermatol. 2009;34:566-569.
4. Mortimer PS. Disorder of lymphatic vessels. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 3. 8th ed. Hoboken, NJ: Wiley-Blackwell; 2010:48.28-48.29.
5. Sharma R, Tomar S, Chandra M. Acquired vulval lymphangiectases mimicking genital warts. Indian J Dermatol Venereol Leprol. 2002;68:166-167.
6. Horn LC, Kühndel K, Pawlowitsch T, et al. Acquired lymphangioma circumscriptum of the vulva mimicking genital warts. Eur J Obstet Gynecol Reprod Biol. 2005;123:118-120.
7. Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
A 19-year-old woman presented with an umbilical mass of 5 months’ duration that had grown in size. Physical examination revealed a 1×1-cm brownish, pedunculated, cauliflower-shaped lesion on the umbilicus. There were no other signs or symptoms of disease. The patient’s personal and family disease history were unremarkable. An excisional biopsy was performed.
The Diagnosis: Acquired Lymphangiectasia
On histopathology numerous dilated channels lined by a single flat layer of endothelial cells were noted within the dermis. The overlying epidermis was papillomatous and acanthotic (Figure 1). The endothelial cells lining the dilated channels were D2-40 positive (Figure 2). Furthermore, the channels contained a pinkish amorphous material and a few red blood cells. The surrounding stroma showed scattered lymphocyte infiltration. These findings were consistent with lymphangiectasia. The lesion has not recurred 4 years following total excision.
|
Acquired lymphangiectasia is known by various names, including lymphangioma, acquired lymphangioma, and acquired lymphangioma circumscriptum, which has led to confusion.1 Acquired lymphangiectasia, which is characterized by dilated superficial lymphatics, develops following damage to previously normal lymphatic channels, leading to a buildup of lymph pressure and backflow.2 Acquired lymphangiectasia has been reported as clinically and histologically indistinguishable from lymphangioma circumscriptum2; however, unlike in lymphangiectasia, the suffix -oma denotes a tumor. Our case matched more closely with the typical concept of lymphangiectasia rather than lymphangioma.
Clinical findings of acquired lymphangiectasia usually include translucent, flat or slightly raised, 2- to 5-mm, flesh-colored papules and vesicles.3,4 Acquired lymphangiectasia has been described with lesions that have verrucous surfaces mimicking warts, condyloma acuminata, or molluscum contagiosum.5,6 Our case suggests that acquired lymphangiectasia also can present with a pedunculated cauliflowerlike appearance. In general, it develops secondary to certain conditions such as recovery from trauma or surgery, postsurgical fibrosis, and irradiation. Lymphangiectasia often is seen on the arms, axillae, chest wall, and genital area in women and the scrotum, penis, thighs, and pubic region in men, both who have undergone radical surgery and irradiation for treatment of breast and prostate cancer, respectively.3 Our patient did not report any history of trauma to the umbilicus.
On histopathology acquired lymphangiectasia typically shows edematous polypoid nodules with dilated lymphatics. The overlying epidermis usually shows a spectrum of proliferation ranging from mild acanthosis to florid pseudoepitheliomatous hyperplasia with marked hyperkeratosis and parakeratosis. The distinctive finding of lymphangiectasia is the presence of dilated lymphatic spaces within the dermis. The dilated channels are filled with lymphatic fluid and often red and white blood cells. The single layer of flattened endothelial cells generally exhibits immunoreactivity to D2-40 and CD31.1
Treatment of lymphangiectasia is focused on reducing the pressure within the lymph vessels and managing consequent lymphedema with compression dressings. Simple surgical excision of lesions on sites such as the vulva or legs often is effective.3 If surgical intervention is not an option, cryotherapy, sclerotherapy, cauterization, and treatment with CO2 lasers also have been utilized with good outcomes.7 In the current case, total surgical excision was performed, which provided good results.
A 19-year-old woman presented with an umbilical mass of 5 months’ duration that had grown in size. Physical examination revealed a 1×1-cm brownish, pedunculated, cauliflower-shaped lesion on the umbilicus. There were no other signs or symptoms of disease. The patient’s personal and family disease history were unremarkable. An excisional biopsy was performed.
The Diagnosis: Acquired Lymphangiectasia
On histopathology numerous dilated channels lined by a single flat layer of endothelial cells were noted within the dermis. The overlying epidermis was papillomatous and acanthotic (Figure 1). The endothelial cells lining the dilated channels were D2-40 positive (Figure 2). Furthermore, the channels contained a pinkish amorphous material and a few red blood cells. The surrounding stroma showed scattered lymphocyte infiltration. These findings were consistent with lymphangiectasia. The lesion has not recurred 4 years following total excision.
|
Acquired lymphangiectasia is known by various names, including lymphangioma, acquired lymphangioma, and acquired lymphangioma circumscriptum, which has led to confusion.1 Acquired lymphangiectasia, which is characterized by dilated superficial lymphatics, develops following damage to previously normal lymphatic channels, leading to a buildup of lymph pressure and backflow.2 Acquired lymphangiectasia has been reported as clinically and histologically indistinguishable from lymphangioma circumscriptum2; however, unlike in lymphangiectasia, the suffix -oma denotes a tumor. Our case matched more closely with the typical concept of lymphangiectasia rather than lymphangioma.
Clinical findings of acquired lymphangiectasia usually include translucent, flat or slightly raised, 2- to 5-mm, flesh-colored papules and vesicles.3,4 Acquired lymphangiectasia has been described with lesions that have verrucous surfaces mimicking warts, condyloma acuminata, or molluscum contagiosum.5,6 Our case suggests that acquired lymphangiectasia also can present with a pedunculated cauliflowerlike appearance. In general, it develops secondary to certain conditions such as recovery from trauma or surgery, postsurgical fibrosis, and irradiation. Lymphangiectasia often is seen on the arms, axillae, chest wall, and genital area in women and the scrotum, penis, thighs, and pubic region in men, both who have undergone radical surgery and irradiation for treatment of breast and prostate cancer, respectively.3 Our patient did not report any history of trauma to the umbilicus.
On histopathology acquired lymphangiectasia typically shows edematous polypoid nodules with dilated lymphatics. The overlying epidermis usually shows a spectrum of proliferation ranging from mild acanthosis to florid pseudoepitheliomatous hyperplasia with marked hyperkeratosis and parakeratosis. The distinctive finding of lymphangiectasia is the presence of dilated lymphatic spaces within the dermis. The dilated channels are filled with lymphatic fluid and often red and white blood cells. The single layer of flattened endothelial cells generally exhibits immunoreactivity to D2-40 and CD31.1
Treatment of lymphangiectasia is focused on reducing the pressure within the lymph vessels and managing consequent lymphedema with compression dressings. Simple surgical excision of lesions on sites such as the vulva or legs often is effective.3 If surgical intervention is not an option, cryotherapy, sclerotherapy, cauterization, and treatment with CO2 lasers also have been utilized with good outcomes.7 In the current case, total surgical excision was performed, which provided good results.
1. Stewart CJ, Chan T, Platten M. Acquired lymphangiectasia (‘lymphangioma circumscriptum’) of the vulva: a report of eight cases. Pathology. 2009;41:448-453.
2. Celis AV, Gaughf CN, Sangueza OP, et al. Acquired lymphangiectasis. South Med J. 1999;92:69-72.
3. Verma SB. Lymphangiectasias of the skin: victims of confusing nomenclature. Clin Exp Dermatol. 2009;34:566-569.
4. Mortimer PS. Disorder of lymphatic vessels. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 3. 8th ed. Hoboken, NJ: Wiley-Blackwell; 2010:48.28-48.29.
5. Sharma R, Tomar S, Chandra M. Acquired vulval lymphangiectases mimicking genital warts. Indian J Dermatol Venereol Leprol. 2002;68:166-167.
6. Horn LC, Kühndel K, Pawlowitsch T, et al. Acquired lymphangioma circumscriptum of the vulva mimicking genital warts. Eur J Obstet Gynecol Reprod Biol. 2005;123:118-120.
7. Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
1. Stewart CJ, Chan T, Platten M. Acquired lymphangiectasia (‘lymphangioma circumscriptum’) of the vulva: a report of eight cases. Pathology. 2009;41:448-453.
2. Celis AV, Gaughf CN, Sangueza OP, et al. Acquired lymphangiectasis. South Med J. 1999;92:69-72.
3. Verma SB. Lymphangiectasias of the skin: victims of confusing nomenclature. Clin Exp Dermatol. 2009;34:566-569.
4. Mortimer PS. Disorder of lymphatic vessels. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. Vol 3. 8th ed. Hoboken, NJ: Wiley-Blackwell; 2010:48.28-48.29.
5. Sharma R, Tomar S, Chandra M. Acquired vulval lymphangiectases mimicking genital warts. Indian J Dermatol Venereol Leprol. 2002;68:166-167.
6. Horn LC, Kühndel K, Pawlowitsch T, et al. Acquired lymphangioma circumscriptum of the vulva mimicking genital warts. Eur J Obstet Gynecol Reprod Biol. 2005;123:118-120.
7. Patel GA, Schwartz RA. Cutaneous lymphangioma circumscriptum: frog spawn on the skin. Int J Dermatol. 2009;48:1290-1295.
