User login
Deutetrabenazine for tardive dyskinesia
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
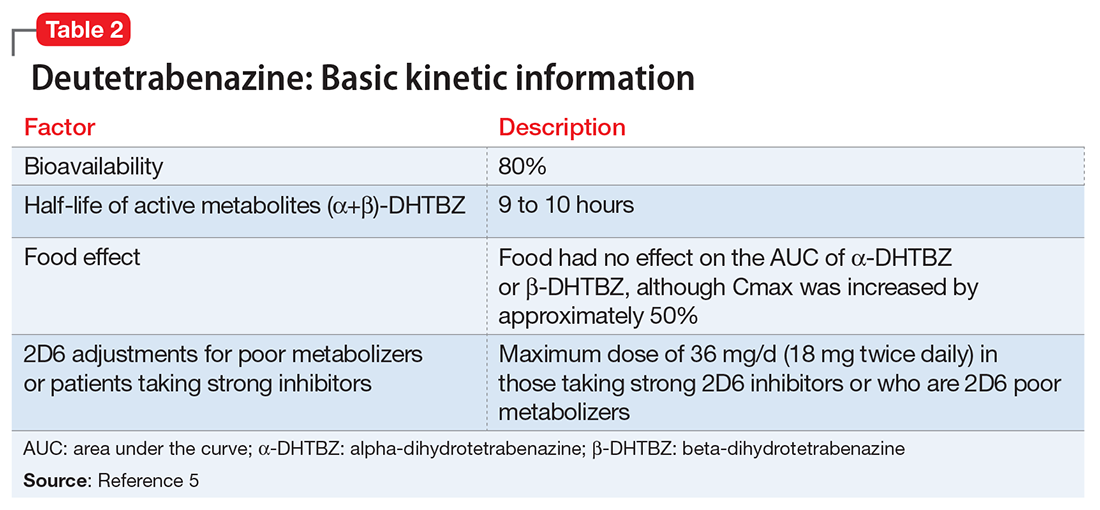
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
Self-disclosure as therapy: The benefits of expressive writing
As psychiatrists, we often provide our patients with a prescription in the hope that the medication will alleviate their symptoms. Perhaps we engage our patients in psychotherapy, encouraging them to reflect on their thoughts, behaviors, and emotions to alter their cognitions. We may remark that our goal is for the patient to “become their own therapist.” What if we encouraged our patients to express themselves in a less structured manner and become their own therapists through writing?
Benefits of expressive writing
Writing about an experienced traumatic event—specifically, to express emotions related to the event—has been associated with improved health outcomes.1,2 Many of these improvements are related to somatic health and basic function, including decreased use of health services, improved immune functioning, and a boost in grades or occupational performance.1 Patients who participate in expressive writing also have demonstrated improvements in distress, negative affect, depression, and posttraumatic stress disorder (PTSD) symptoms.1,2 Although improvement in PTSD symptoms with expressive writing has varied across studies, it appears that patients with PTSD who score high in trait negative emotion may receive the most benefit from the practice.3
Why does it work?
There are several theories regarding why expressive writing is an effective therapy. Originally, it was believed that the active inhibition of not talking about traumatic events was a form of physiological work and a long-term, low-lying stressor, and that writing about such events could reduce this stress. However, newer studies offer various explanations for its efficacy, including:
- repeat exposure to stressful or traumatic memories and consequent self-distancing
- creation of a narrative around the stressful event
- labeling of emotions
- self-affirmation and meaning-making related to the negative event.4
Rx writing
Encouraging your patients to use expressive writing is simple. You might ask a patient struggling with distress and negative affect following a traumatic experience to write about his (her) thoughts and feelings regarding the incident. For example:
Spend about 15 minutes writing your deepest thoughts and feelings about going through this traumatic experience. Discuss the ways it affected different areas of your life, including relationships with family and friends, school or work, or self-confidence and self-esteem. Don’t worry about spelling, grammar, or sentence structure.
Assure patients that you do not need to review their writing, but would like to hear about their experience writing. Many studies on expressive writing instructed participants to write for 3 to 5 consecutive days, 15 to 30 minutes each day.1,2 Patients may disclose a dramatic spectrum and intensity of experience and often are willing to do so.
Expressive writing is a simple, low-risk exercise that benefits many people. Perhaps by prescribing a course of writing, you will find your patients can benefit as well.
1. Baikie KA, Geerligs L, Wilhelm K. Expressive writing and positive writing for participants with mood disorders: an online randomized controlled trial. J Affect Disord. 2012;136(3):310-319.
2. Krpan KM, Kross E, Berman MG, et al. An everyday activity as a treatment for depression: the benefits of expressive writing for people diagnosed with major depressive disorder. J Affect Disord. 2013;150(3):1148-1151.
3. Hoyt T, Yeater EA. The effects of negative emotion and expressive writing on posttraumatic stress symptoms. J Soc Clin Psychol. 2011;30:549-569.
4. Niles AN, Byrne Haltom KE, Lieberman MD, et al. Writing content predicts benefit from written expressive disclosure: evidence for repeated exposure and self-affirmation. Cogn Emot. 2016;30(2):258-274.
As psychiatrists, we often provide our patients with a prescription in the hope that the medication will alleviate their symptoms. Perhaps we engage our patients in psychotherapy, encouraging them to reflect on their thoughts, behaviors, and emotions to alter their cognitions. We may remark that our goal is for the patient to “become their own therapist.” What if we encouraged our patients to express themselves in a less structured manner and become their own therapists through writing?
Benefits of expressive writing
Writing about an experienced traumatic event—specifically, to express emotions related to the event—has been associated with improved health outcomes.1,2 Many of these improvements are related to somatic health and basic function, including decreased use of health services, improved immune functioning, and a boost in grades or occupational performance.1 Patients who participate in expressive writing also have demonstrated improvements in distress, negative affect, depression, and posttraumatic stress disorder (PTSD) symptoms.1,2 Although improvement in PTSD symptoms with expressive writing has varied across studies, it appears that patients with PTSD who score high in trait negative emotion may receive the most benefit from the practice.3
Why does it work?
There are several theories regarding why expressive writing is an effective therapy. Originally, it was believed that the active inhibition of not talking about traumatic events was a form of physiological work and a long-term, low-lying stressor, and that writing about such events could reduce this stress. However, newer studies offer various explanations for its efficacy, including:
- repeat exposure to stressful or traumatic memories and consequent self-distancing
- creation of a narrative around the stressful event
- labeling of emotions
- self-affirmation and meaning-making related to the negative event.4
Rx writing
Encouraging your patients to use expressive writing is simple. You might ask a patient struggling with distress and negative affect following a traumatic experience to write about his (her) thoughts and feelings regarding the incident. For example:
Spend about 15 minutes writing your deepest thoughts and feelings about going through this traumatic experience. Discuss the ways it affected different areas of your life, including relationships with family and friends, school or work, or self-confidence and self-esteem. Don’t worry about spelling, grammar, or sentence structure.
Assure patients that you do not need to review their writing, but would like to hear about their experience writing. Many studies on expressive writing instructed participants to write for 3 to 5 consecutive days, 15 to 30 minutes each day.1,2 Patients may disclose a dramatic spectrum and intensity of experience and often are willing to do so.
Expressive writing is a simple, low-risk exercise that benefits many people. Perhaps by prescribing a course of writing, you will find your patients can benefit as well.
As psychiatrists, we often provide our patients with a prescription in the hope that the medication will alleviate their symptoms. Perhaps we engage our patients in psychotherapy, encouraging them to reflect on their thoughts, behaviors, and emotions to alter their cognitions. We may remark that our goal is for the patient to “become their own therapist.” What if we encouraged our patients to express themselves in a less structured manner and become their own therapists through writing?
Benefits of expressive writing
Writing about an experienced traumatic event—specifically, to express emotions related to the event—has been associated with improved health outcomes.1,2 Many of these improvements are related to somatic health and basic function, including decreased use of health services, improved immune functioning, and a boost in grades or occupational performance.1 Patients who participate in expressive writing also have demonstrated improvements in distress, negative affect, depression, and posttraumatic stress disorder (PTSD) symptoms.1,2 Although improvement in PTSD symptoms with expressive writing has varied across studies, it appears that patients with PTSD who score high in trait negative emotion may receive the most benefit from the practice.3
Why does it work?
There are several theories regarding why expressive writing is an effective therapy. Originally, it was believed that the active inhibition of not talking about traumatic events was a form of physiological work and a long-term, low-lying stressor, and that writing about such events could reduce this stress. However, newer studies offer various explanations for its efficacy, including:
- repeat exposure to stressful or traumatic memories and consequent self-distancing
- creation of a narrative around the stressful event
- labeling of emotions
- self-affirmation and meaning-making related to the negative event.4
Rx writing
Encouraging your patients to use expressive writing is simple. You might ask a patient struggling with distress and negative affect following a traumatic experience to write about his (her) thoughts and feelings regarding the incident. For example:
Spend about 15 minutes writing your deepest thoughts and feelings about going through this traumatic experience. Discuss the ways it affected different areas of your life, including relationships with family and friends, school or work, or self-confidence and self-esteem. Don’t worry about spelling, grammar, or sentence structure.
Assure patients that you do not need to review their writing, but would like to hear about their experience writing. Many studies on expressive writing instructed participants to write for 3 to 5 consecutive days, 15 to 30 minutes each day.1,2 Patients may disclose a dramatic spectrum and intensity of experience and often are willing to do so.
Expressive writing is a simple, low-risk exercise that benefits many people. Perhaps by prescribing a course of writing, you will find your patients can benefit as well.
1. Baikie KA, Geerligs L, Wilhelm K. Expressive writing and positive writing for participants with mood disorders: an online randomized controlled trial. J Affect Disord. 2012;136(3):310-319.
2. Krpan KM, Kross E, Berman MG, et al. An everyday activity as a treatment for depression: the benefits of expressive writing for people diagnosed with major depressive disorder. J Affect Disord. 2013;150(3):1148-1151.
3. Hoyt T, Yeater EA. The effects of negative emotion and expressive writing on posttraumatic stress symptoms. J Soc Clin Psychol. 2011;30:549-569.
4. Niles AN, Byrne Haltom KE, Lieberman MD, et al. Writing content predicts benefit from written expressive disclosure: evidence for repeated exposure and self-affirmation. Cogn Emot. 2016;30(2):258-274.
1. Baikie KA, Geerligs L, Wilhelm K. Expressive writing and positive writing for participants with mood disorders: an online randomized controlled trial. J Affect Disord. 2012;136(3):310-319.
2. Krpan KM, Kross E, Berman MG, et al. An everyday activity as a treatment for depression: the benefits of expressive writing for people diagnosed with major depressive disorder. J Affect Disord. 2013;150(3):1148-1151.
3. Hoyt T, Yeater EA. The effects of negative emotion and expressive writing on posttraumatic stress symptoms. J Soc Clin Psychol. 2011;30:549-569.
4. Niles AN, Byrne Haltom KE, Lieberman MD, et al. Writing content predicts benefit from written expressive disclosure: evidence for repeated exposure and self-affirmation. Cogn Emot. 2016;30(2):258-274.
Managing requests for gluten-, lactose-, and animal-free medications
Patients may ask their psychiatrist to prescribe gluten-, lactose-, or animal-free medications because of concerns about allergies, disease states, religious beliefs, or dietary preferences. Determining the source of non-active medication ingredients can be challenging and time-consuming, because ingredients vary across dosages and formulations of the same medication. We review how to address requests for gluten-, lactose-, and animal-free medications.
Gluten-free
Although the risk of a medication containing gluten is low,1 patients with Celiac disease must avoid gluten to prevent disease exacerbation. Therefore, physicians should thoroughly evaluate medication ingredients to prevent inadvertent gluten consumption.
Medication excipients that may contain gluten include:
- starch
- pregelatinized starch
- sodium starch glycolate.1
These starches can come from various sources, including corn, wheat, potato, and tapioca. Wheat-derived starch contains gluten and should be avoided by patients with Celiac disease. Advise patients to avoid any starch if its source cannot be determined.
Some sources may list sugar alcohols, such as mannitol and xylitol, as gluten–containing excipients because they may be extracted from starch sources, such as wheat; however, all gluten is removed during refinement and these products are safe.2
Lactose-free
How to respond to a patient’s request for lactose-free medication depends on whether the patient is lactose intolerant or has a milk allergy. The amount of lactose that patients with lactase deficiency can tolerate varies.3 Most medications are thought to contain minimal amounts of lactose. Case reports have described patients experiencing lactose intolerance symptoms after taking 1 or 2 medications, but this is rare.3 Therefore, it is reasonable to use lactose–containing products in patients with lactose intolerance. If such a patient develops symptoms after taking a medication that contains lactose, suggest that he (she):
- take the medication with food, if appropriate, to slow absorption and reduce symptoms
- take it with a lactase enzyme product
- substitute it with a medication that does not contain lactose (switch to a different product or formulation, as appropriate).
Compared with patients who are lactose intolerant, those with a milk allergy experience an immunoglobulin E–mediated reaction when they consume milk protein. Milk proteins typically are filtered out during manufacturing, but a small amount can remain. Although it has not been determined if oral medications containing milk protein can cause an allergic reaction, some researchers have hypothesized that these medications may be tolerated because acid and digestive enzymes break down the milk protein. However, because the respiratory tract lacks this protection, inhaled products that contain lactose may be more likely to cause an allergic reaction and should be avoided if possible. Because oral medications do not usually contain milk proteins, it may be reasonable to prescribe lactose–containing oral products to a patient with a milk allergy. If the patient experiences a reaction or wishes to avoid lactose, an alternative non-lactose–containing product or formulation may be prescribed.
Animal-free
Individuals who are members of certain religions, including Judaism, Islam, Orthodox Christianity, and the Seventh Day Adventist Church, typically avoid pork, and those who are Hindu or Buddhist may avoid beef products.4 Gelatin and stearic acid, which can be found in the gelatin shell of capsules and within extended-release (ER) tablets, frequently are derived from porcine or bovine sources. The source of gelatin and stearic acid may change from lot to lot, and manufacturers should be contacted to assist with identifying the source for a specific medication. Consider these options to reduce a patient’s exposure to animal-containing products:
- change from an ER to an immediate-release (IR) product (confirm that IR is gelatin- and stearic acid–free)
- use a non-capsule formulation
- remove the content of a capsule before ingestion, if appropriate
- try an alternative route of administration, such as transdermal.
How to best help patients
Before taking steps to accommodate a request for a gluten-, lactose-, or animal-free medication, which can be time-consuming, verify the reason for your patient’s request. It may be sufficient to explain to your patient that typically exposure to excipients within oral medications is small and does not cause problems for a patient with lactose intolerance or a milk allergy. The resources listed in the Table can help provide further education on these concerns; however, due to potential delays in updating a Web site, it may be necessary to contact the medication manufacturer directly to verify ingredients.
If your patient still has concerns about ingredients, consider the following steps:
- Use the National Library of Medicine’s Pillbox Web site (https://pillbox.nlm.nih.gov) to search for a medication, dose, or formulation that does not contain the concerning ingredients
- If the concerning ingredient is not listed, either prescribe the medication or contact the manufacturer for further information, depending on the patient’s reason for the request
- If the concerning ingredient is listed, work with the pharmacist to contact the manufacturer.
There are 2 additional points to consider regarding medication excipients. Be aware that generic medications are produced by multiple manufacturers, and each may use different excipients. Also, a manufacturer may not guarantee that a medication is gluten-free because of the potential for cross-contamination during manufacturing, although the risk is extremely low.
1. Plogsted S. Gluten in medication. Celiac Disease Foundation. https://celiac.org/live-gluten-free/glutenfreediet/gluten-medication. Accessed January 13, 2017.
2. Plogsted S. Gluten free drugs. http://www.glutenfreedrugs.com. Updated April 28, 2017. Accessed September 2, 2017.
3. Lactose in medications. Pharmacist’s Letter/Prescriber’s Letter. 2007;230779.
4. Sattar SP, Shakeel Ahmed M, Majeed F, et al. Inert medication ingredients causing nonadherence due to religious beliefs. Ann Pharmacother. 2004;38(4):621-624.
Patients may ask their psychiatrist to prescribe gluten-, lactose-, or animal-free medications because of concerns about allergies, disease states, religious beliefs, or dietary preferences. Determining the source of non-active medication ingredients can be challenging and time-consuming, because ingredients vary across dosages and formulations of the same medication. We review how to address requests for gluten-, lactose-, and animal-free medications.
Gluten-free
Although the risk of a medication containing gluten is low,1 patients with Celiac disease must avoid gluten to prevent disease exacerbation. Therefore, physicians should thoroughly evaluate medication ingredients to prevent inadvertent gluten consumption.
Medication excipients that may contain gluten include:
- starch
- pregelatinized starch
- sodium starch glycolate.1
These starches can come from various sources, including corn, wheat, potato, and tapioca. Wheat-derived starch contains gluten and should be avoided by patients with Celiac disease. Advise patients to avoid any starch if its source cannot be determined.
Some sources may list sugar alcohols, such as mannitol and xylitol, as gluten–containing excipients because they may be extracted from starch sources, such as wheat; however, all gluten is removed during refinement and these products are safe.2
Lactose-free
How to respond to a patient’s request for lactose-free medication depends on whether the patient is lactose intolerant or has a milk allergy. The amount of lactose that patients with lactase deficiency can tolerate varies.3 Most medications are thought to contain minimal amounts of lactose. Case reports have described patients experiencing lactose intolerance symptoms after taking 1 or 2 medications, but this is rare.3 Therefore, it is reasonable to use lactose–containing products in patients with lactose intolerance. If such a patient develops symptoms after taking a medication that contains lactose, suggest that he (she):
- take the medication with food, if appropriate, to slow absorption and reduce symptoms
- take it with a lactase enzyme product
- substitute it with a medication that does not contain lactose (switch to a different product or formulation, as appropriate).
Compared with patients who are lactose intolerant, those with a milk allergy experience an immunoglobulin E–mediated reaction when they consume milk protein. Milk proteins typically are filtered out during manufacturing, but a small amount can remain. Although it has not been determined if oral medications containing milk protein can cause an allergic reaction, some researchers have hypothesized that these medications may be tolerated because acid and digestive enzymes break down the milk protein. However, because the respiratory tract lacks this protection, inhaled products that contain lactose may be more likely to cause an allergic reaction and should be avoided if possible. Because oral medications do not usually contain milk proteins, it may be reasonable to prescribe lactose–containing oral products to a patient with a milk allergy. If the patient experiences a reaction or wishes to avoid lactose, an alternative non-lactose–containing product or formulation may be prescribed.
Animal-free
Individuals who are members of certain religions, including Judaism, Islam, Orthodox Christianity, and the Seventh Day Adventist Church, typically avoid pork, and those who are Hindu or Buddhist may avoid beef products.4 Gelatin and stearic acid, which can be found in the gelatin shell of capsules and within extended-release (ER) tablets, frequently are derived from porcine or bovine sources. The source of gelatin and stearic acid may change from lot to lot, and manufacturers should be contacted to assist with identifying the source for a specific medication. Consider these options to reduce a patient’s exposure to animal-containing products:
- change from an ER to an immediate-release (IR) product (confirm that IR is gelatin- and stearic acid–free)
- use a non-capsule formulation
- remove the content of a capsule before ingestion, if appropriate
- try an alternative route of administration, such as transdermal.
How to best help patients
Before taking steps to accommodate a request for a gluten-, lactose-, or animal-free medication, which can be time-consuming, verify the reason for your patient’s request. It may be sufficient to explain to your patient that typically exposure to excipients within oral medications is small and does not cause problems for a patient with lactose intolerance or a milk allergy. The resources listed in the Table can help provide further education on these concerns; however, due to potential delays in updating a Web site, it may be necessary to contact the medication manufacturer directly to verify ingredients.
If your patient still has concerns about ingredients, consider the following steps:
- Use the National Library of Medicine’s Pillbox Web site (https://pillbox.nlm.nih.gov) to search for a medication, dose, or formulation that does not contain the concerning ingredients
- If the concerning ingredient is not listed, either prescribe the medication or contact the manufacturer for further information, depending on the patient’s reason for the request
- If the concerning ingredient is listed, work with the pharmacist to contact the manufacturer.
There are 2 additional points to consider regarding medication excipients. Be aware that generic medications are produced by multiple manufacturers, and each may use different excipients. Also, a manufacturer may not guarantee that a medication is gluten-free because of the potential for cross-contamination during manufacturing, although the risk is extremely low.
Patients may ask their psychiatrist to prescribe gluten-, lactose-, or animal-free medications because of concerns about allergies, disease states, religious beliefs, or dietary preferences. Determining the source of non-active medication ingredients can be challenging and time-consuming, because ingredients vary across dosages and formulations of the same medication. We review how to address requests for gluten-, lactose-, and animal-free medications.
Gluten-free
Although the risk of a medication containing gluten is low,1 patients with Celiac disease must avoid gluten to prevent disease exacerbation. Therefore, physicians should thoroughly evaluate medication ingredients to prevent inadvertent gluten consumption.
Medication excipients that may contain gluten include:
- starch
- pregelatinized starch
- sodium starch glycolate.1
These starches can come from various sources, including corn, wheat, potato, and tapioca. Wheat-derived starch contains gluten and should be avoided by patients with Celiac disease. Advise patients to avoid any starch if its source cannot be determined.
Some sources may list sugar alcohols, such as mannitol and xylitol, as gluten–containing excipients because they may be extracted from starch sources, such as wheat; however, all gluten is removed during refinement and these products are safe.2
Lactose-free
How to respond to a patient’s request for lactose-free medication depends on whether the patient is lactose intolerant or has a milk allergy. The amount of lactose that patients with lactase deficiency can tolerate varies.3 Most medications are thought to contain minimal amounts of lactose. Case reports have described patients experiencing lactose intolerance symptoms after taking 1 or 2 medications, but this is rare.3 Therefore, it is reasonable to use lactose–containing products in patients with lactose intolerance. If such a patient develops symptoms after taking a medication that contains lactose, suggest that he (she):
- take the medication with food, if appropriate, to slow absorption and reduce symptoms
- take it with a lactase enzyme product
- substitute it with a medication that does not contain lactose (switch to a different product or formulation, as appropriate).
Compared with patients who are lactose intolerant, those with a milk allergy experience an immunoglobulin E–mediated reaction when they consume milk protein. Milk proteins typically are filtered out during manufacturing, but a small amount can remain. Although it has not been determined if oral medications containing milk protein can cause an allergic reaction, some researchers have hypothesized that these medications may be tolerated because acid and digestive enzymes break down the milk protein. However, because the respiratory tract lacks this protection, inhaled products that contain lactose may be more likely to cause an allergic reaction and should be avoided if possible. Because oral medications do not usually contain milk proteins, it may be reasonable to prescribe lactose–containing oral products to a patient with a milk allergy. If the patient experiences a reaction or wishes to avoid lactose, an alternative non-lactose–containing product or formulation may be prescribed.
Animal-free
Individuals who are members of certain religions, including Judaism, Islam, Orthodox Christianity, and the Seventh Day Adventist Church, typically avoid pork, and those who are Hindu or Buddhist may avoid beef products.4 Gelatin and stearic acid, which can be found in the gelatin shell of capsules and within extended-release (ER) tablets, frequently are derived from porcine or bovine sources. The source of gelatin and stearic acid may change from lot to lot, and manufacturers should be contacted to assist with identifying the source for a specific medication. Consider these options to reduce a patient’s exposure to animal-containing products:
- change from an ER to an immediate-release (IR) product (confirm that IR is gelatin- and stearic acid–free)
- use a non-capsule formulation
- remove the content of a capsule before ingestion, if appropriate
- try an alternative route of administration, such as transdermal.
How to best help patients
Before taking steps to accommodate a request for a gluten-, lactose-, or animal-free medication, which can be time-consuming, verify the reason for your patient’s request. It may be sufficient to explain to your patient that typically exposure to excipients within oral medications is small and does not cause problems for a patient with lactose intolerance or a milk allergy. The resources listed in the Table can help provide further education on these concerns; however, due to potential delays in updating a Web site, it may be necessary to contact the medication manufacturer directly to verify ingredients.
If your patient still has concerns about ingredients, consider the following steps:
- Use the National Library of Medicine’s Pillbox Web site (https://pillbox.nlm.nih.gov) to search for a medication, dose, or formulation that does not contain the concerning ingredients
- If the concerning ingredient is not listed, either prescribe the medication or contact the manufacturer for further information, depending on the patient’s reason for the request
- If the concerning ingredient is listed, work with the pharmacist to contact the manufacturer.
There are 2 additional points to consider regarding medication excipients. Be aware that generic medications are produced by multiple manufacturers, and each may use different excipients. Also, a manufacturer may not guarantee that a medication is gluten-free because of the potential for cross-contamination during manufacturing, although the risk is extremely low.
1. Plogsted S. Gluten in medication. Celiac Disease Foundation. https://celiac.org/live-gluten-free/glutenfreediet/gluten-medication. Accessed January 13, 2017.
2. Plogsted S. Gluten free drugs. http://www.glutenfreedrugs.com. Updated April 28, 2017. Accessed September 2, 2017.
3. Lactose in medications. Pharmacist’s Letter/Prescriber’s Letter. 2007;230779.
4. Sattar SP, Shakeel Ahmed M, Majeed F, et al. Inert medication ingredients causing nonadherence due to religious beliefs. Ann Pharmacother. 2004;38(4):621-624.
1. Plogsted S. Gluten in medication. Celiac Disease Foundation. https://celiac.org/live-gluten-free/glutenfreediet/gluten-medication. Accessed January 13, 2017.
2. Plogsted S. Gluten free drugs. http://www.glutenfreedrugs.com. Updated April 28, 2017. Accessed September 2, 2017.
3. Lactose in medications. Pharmacist’s Letter/Prescriber’s Letter. 2007;230779.
4. Sattar SP, Shakeel Ahmed M, Majeed F, et al. Inert medication ingredients causing nonadherence due to religious beliefs. Ann Pharmacother. 2004;38(4):621-624.
What to do after a patient assaults you
Physical assaults by patients are an occupational hazard of practicing medicine. Assaults can happen in any clinical setting, occur unexpectedly, and have a lasting impact on all involved. In an anonymous survey of 11,000 hospital workers, 18.8% reported being physically assaulted.1 Psychiatric clinicia
Remain calm. Although it may be difficult to do so immediately after being assaulted, remaining calm is essential. You may experience a myriad of emotions, such as anger, fear, vulnerability, shock, or guilt. Although these responses are normal, they can hinder your ability to accomplish subsequent tasks.
Recall the assault. Despite the unpleasantness of replaying the incident, recall as many details as you can and immediately write them down. Because of the copious amount of paperwork you may be required to file (eg, incident reports, employee health forms) and statements that you will likely repeat, having an accurate version of what happened is paramount to determining a course of action. You also may be required to give a statement to law enforcement officials.
Report the assault to your supervisor(s). Informing supervisors and colleagues of what happened could begin the implementation of corrective measures to decrease the risk of future assaults.
Talk about the incident with coworkers, supervisors, and friends to help process what happened, normalize what you are experiencing, and allow others to learn from you. Being assaulted can be traumatic and can result in experiencing post-assault symptoms, such as disruptions in sleep patterns, changes in appetite, and nightmares of the incident. These can be normal reactions to what is an abnormal situation. If necessary, seek medical assistance.
Evaluate the circumstances. Although you may not be at fault, consider if there may have been contributing factors:
- Were there signs of escalating aggressiveness in the patient’s behavior that you may have missed?
- Would the presence of a chaperone during interactions with the patient have reduced the risk of an assault?
- Did you maintain a safe distance from the patient?
- Were existing safety policies followed?
Examine your surroundings. Could the surroundings where the assault occurred have hindered your ability to escape? If so, can they be altered to increase your chance of escaping? Are there items that could be used as potential weapons and should be removed?Expect changes to processes and procedures as part of the reverberations after an assault. Your firsthand account of the assault can limit staff overreactions by analyzing whether existing policies were appropriately implemented, before deeming them ineffective and enacting new policies.
1. Pompeii LA, Schoenfisch AL, Lipscomb HJ, et al. Physical assault, physical threat, and verbal abuse perpetrated against hospital workers by patients or visitors in six U.S. hospitals. Am J Ind Med. 2015;58(11):1194-1204.
2. Phillips JP. Workplace violence against health care workers in the United States. N Engl J Med. 2016;374(17):1661-1669.
3. Privitera M, Weisman R, Cerulli C, et al. Violence toward mental health staff and safety in the work environment. Occup Med (Lond). 2005;55(6):480-486.
Physical assaults by patients are an occupational hazard of practicing medicine. Assaults can happen in any clinical setting, occur unexpectedly, and have a lasting impact on all involved. In an anonymous survey of 11,000 hospital workers, 18.8% reported being physically assaulted.1 Psychiatric clinicia
Remain calm. Although it may be difficult to do so immediately after being assaulted, remaining calm is essential. You may experience a myriad of emotions, such as anger, fear, vulnerability, shock, or guilt. Although these responses are normal, they can hinder your ability to accomplish subsequent tasks.
Recall the assault. Despite the unpleasantness of replaying the incident, recall as many details as you can and immediately write them down. Because of the copious amount of paperwork you may be required to file (eg, incident reports, employee health forms) and statements that you will likely repeat, having an accurate version of what happened is paramount to determining a course of action. You also may be required to give a statement to law enforcement officials.
Report the assault to your supervisor(s). Informing supervisors and colleagues of what happened could begin the implementation of corrective measures to decrease the risk of future assaults.
Talk about the incident with coworkers, supervisors, and friends to help process what happened, normalize what you are experiencing, and allow others to learn from you. Being assaulted can be traumatic and can result in experiencing post-assault symptoms, such as disruptions in sleep patterns, changes in appetite, and nightmares of the incident. These can be normal reactions to what is an abnormal situation. If necessary, seek medical assistance.
Evaluate the circumstances. Although you may not be at fault, consider if there may have been contributing factors:
- Were there signs of escalating aggressiveness in the patient’s behavior that you may have missed?
- Would the presence of a chaperone during interactions with the patient have reduced the risk of an assault?
- Did you maintain a safe distance from the patient?
- Were existing safety policies followed?
Examine your surroundings. Could the surroundings where the assault occurred have hindered your ability to escape? If so, can they be altered to increase your chance of escaping? Are there items that could be used as potential weapons and should be removed?Expect changes to processes and procedures as part of the reverberations after an assault. Your firsthand account of the assault can limit staff overreactions by analyzing whether existing policies were appropriately implemented, before deeming them ineffective and enacting new policies.
Physical assaults by patients are an occupational hazard of practicing medicine. Assaults can happen in any clinical setting, occur unexpectedly, and have a lasting impact on all involved. In an anonymous survey of 11,000 hospital workers, 18.8% reported being physically assaulted.1 Psychiatric clinicia
Remain calm. Although it may be difficult to do so immediately after being assaulted, remaining calm is essential. You may experience a myriad of emotions, such as anger, fear, vulnerability, shock, or guilt. Although these responses are normal, they can hinder your ability to accomplish subsequent tasks.
Recall the assault. Despite the unpleasantness of replaying the incident, recall as many details as you can and immediately write them down. Because of the copious amount of paperwork you may be required to file (eg, incident reports, employee health forms) and statements that you will likely repeat, having an accurate version of what happened is paramount to determining a course of action. You also may be required to give a statement to law enforcement officials.
Report the assault to your supervisor(s). Informing supervisors and colleagues of what happened could begin the implementation of corrective measures to decrease the risk of future assaults.
Talk about the incident with coworkers, supervisors, and friends to help process what happened, normalize what you are experiencing, and allow others to learn from you. Being assaulted can be traumatic and can result in experiencing post-assault symptoms, such as disruptions in sleep patterns, changes in appetite, and nightmares of the incident. These can be normal reactions to what is an abnormal situation. If necessary, seek medical assistance.
Evaluate the circumstances. Although you may not be at fault, consider if there may have been contributing factors:
- Were there signs of escalating aggressiveness in the patient’s behavior that you may have missed?
- Would the presence of a chaperone during interactions with the patient have reduced the risk of an assault?
- Did you maintain a safe distance from the patient?
- Were existing safety policies followed?
Examine your surroundings. Could the surroundings where the assault occurred have hindered your ability to escape? If so, can they be altered to increase your chance of escaping? Are there items that could be used as potential weapons and should be removed?Expect changes to processes and procedures as part of the reverberations after an assault. Your firsthand account of the assault can limit staff overreactions by analyzing whether existing policies were appropriately implemented, before deeming them ineffective and enacting new policies.
1. Pompeii LA, Schoenfisch AL, Lipscomb HJ, et al. Physical assault, physical threat, and verbal abuse perpetrated against hospital workers by patients or visitors in six U.S. hospitals. Am J Ind Med. 2015;58(11):1194-1204.
2. Phillips JP. Workplace violence against health care workers in the United States. N Engl J Med. 2016;374(17):1661-1669.
3. Privitera M, Weisman R, Cerulli C, et al. Violence toward mental health staff and safety in the work environment. Occup Med (Lond). 2005;55(6):480-486.
1. Pompeii LA, Schoenfisch AL, Lipscomb HJ, et al. Physical assault, physical threat, and verbal abuse perpetrated against hospital workers by patients or visitors in six U.S. hospitals. Am J Ind Med. 2015;58(11):1194-1204.
2. Phillips JP. Workplace violence against health care workers in the United States. N Engl J Med. 2016;374(17):1661-1669.
3. Privitera M, Weisman R, Cerulli C, et al. Violence toward mental health staff and safety in the work environment. Occup Med (Lond). 2005;55(6):480-486.
Advanced Stage and Relapsed/Refractory Hodgkin Lymphoma
INTRODUCTION
Hodgkin lymphoma, previously known as Hodgkin’s disease, is a B-cell lymphoproliferative disease characterized by a unique set of pathologic and epidemiologic features. The disease is characterized by the presence of multinucleate giant cells called Hodgkin Reed-Sternberg (HRS) cells.1 Hodgkin lymphoma is unique compared to other B-cell lymphomas because of the relative rarity of the malignant cells within affected tissues. The HRS cells, which usually account for only 0.1% to 10% of the cells, induce accumulation of nonmalignant lymphocytes, macrophages, granulocytes, eosinophils, plasma cells, and histiocytes, which then constitute the majority of tumor cellularity.2 Although the disease was first described by Sir Thomas Hodgkin in 1832, in part because of this unique histopathology, it was not until the 1990s that it was conclusively demonstrated that HRS cells are in fact monoclonal germinal center–derived B cells.
Due to the development of highly effective therapies for Hodgkin lymphoma, cure is a reasonable goal for most patients. Because of the high cure rate, late complications of therapy must be considered when selecting treatment. This article reviews the clinical features and treatment options for advanced stage and relapsed/refractory Hodgkin lymphoma. A previously published article reviewed the epidemiology, etiology/pathogenesis, pathologic classification, initial workup, and staging evaluation of Hodgkin lymphoma, as well as the prognostic stratification and treatment of patients with early-stage Hodgkin lymphoma.3
PRESENTATION, INITIAL EVALUATION, AND PROGNOSIS
Overall, classical Hodgkin lymphoma (cHL) usually presents with asymptomatic mediastinal or cervical lymphadenopathy. At least 50% of patients will have stage I or II disease.4 A mediastinal mass is seen in most patients with nodular sclerosis cHL, at times showing the characteristics of bulky (> 10 cm) disease. Constitutional, or B, symptoms (fever, night sweats, and weight loss) are present in approximately 25% of all patients with cHL, but 50% of advanced stage patients. Between 10% and 15% of patients will have extranodal disease, most commonly involving lung, bone, and liver. Lymphocyte-predominant Hodgkin lymphoma (LPHL) is a rare histological subtype of Hodgkin lymphoma that is differentiated from cHL by distinct clinicopathological features. The clinical course and treatment approach for LPHL are dependent upon the stage of disease. The clinicopathological features of LPHL are discussed in the early-stage Hodgkin lymphoma article.3
For the purposes of prognosis and selection of treatment, Hodgkin lymphoma is commonly classified as early stage favorable, early stage unfavorable, and advanced stage. For advanced stage Hodgkin lymphoma patients, prognosis can be defined using a tool commonly referred to as the International Prognostic Score (IPS). This index consists of 7 factors: male gender, age 45 years or older, stage IV disease, hemoglobin < 10.5 g/dL, white blood cell (WBC) count > 15,000/μL, lymphopenia (absolute lymphocyte count < 600 cells/μL or lymphocytes < 8% of WBC count), and serum albumin < 4 g/dL.5 In the original study by Hasenclever et al,5 the 5-year freedom from progression (FFP) ranged from 42% to 84% and the 5-year overall survival (OS) ranged from 56% to 90%, depending on the number of factors present. This scoring system, however, was developed using a patient population treated prior to 1992. Using a more recently treated patient population, the British Columbia Cancer Agency (BCCA) found that the IPS is still valid for prognostication, but outcomes have improved across all IPS groups, with 5-year FFP now ranging from 62% to 88% and 5-year OS ranging from 67% to 98%.6 This improvement is likely a reflection of improved therapy and supportive care. Table 1 shows the PFS and OS within each IPS group, comparing the data from the German Hodgkin Study Group (GHSG) and BCCA group.5,6
High expression of CD68 is associated with adverse outcomes, whereas high FOXP3 and CD20 expression on tumor cells are predictors of superior outcomes.8 A recent study found that CD68 expression was associated with OS. Five-year OS was 88% in those with less than 25% CD68 expression, versus 63% in those with greater than 25% CD68 expression.9
Roemer and colleagues evaluated 108 newly diagnosed cHL biopsy specimens and found that almost all cHL patients had concordant alteration of PD-L1 (programmed death ligand-1) and PD-L2 loci, with a spectrum of 9p24.1 alterations ranging from low level polysomy to near uniform 9p24.1 amplification. PD-L1/PD-L2 copy number alterations are therefore a defining pathobiological feature of cHL.10 PFS was significantly shorter for patients with 9p24.1 amplification, and those patients were likely to have advanced disease suggesting that 9p24.1 amplification is associated with less favorable prognosis.10 This may change with the increasing use of PD-1 inhibitors in the treatment of cHL.
High baseline metabolic tumor volume and total lesion glycolysis have also been associated with adverse outcomes in cHL. While not routinely assessed in practice currently, these tools may ultimately be used to assess prognosis and guide therapy in clinical practice.11
ADVANCED STAGE HODGKIN LYMPHOMA
FRONTLINE THERAPY
First-line Chemotherapy
Chemotherapy plays an essential role in the treatment of advanced stage Hodgkin lymphoma. In the 1960s, the MOPP regimen (nitrogen mustard, vincristine, procarbazine, prednisone) was developed, with a 10-year OS of 50% and a progression-free survival (PFS) of 52% reported in advanced stage patients. The complete remission (CR) rate was 81%, and 36% of patients who achieved CR relapsed later.12 This chemotherapy regimen is associated with a significant rate of myelosuppression and infertility as well as long-term risk of secondary myelodysplasia and acute leukemias.13,14 This led to the development of newer regimens such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine).15 In a randomized trial, ABVD showed improved failure-free survival (FFS) over MOPP (61% versus 50% at 5 years) but similar OS (66%–73%).16 In light of these findings, and considering the lower rate of infertility and myelotoxicity, ABVD became the standard of care for advanced stage cHL in the United States.
The Stanford V regimen was developed in an attempt to further minimize toxicity.17 Stanford V is a condensed, 12-week chemotherapy regimen that includes mechlorethamine, doxorubicin, vinblastine, etoposide, prednisone, vincristine, and bleomycin, followed by involved-field radiation therapy (IFRT). Subsequent trials compared the Stanford V and ABVD regimens and showed similar OS, freedom from treatment failure (FFTF), and response rates.18,19 The ABVD regimen was noted to have higher pulmonary toxicity, while other toxicities such as lymphopenia and neuropathy were higher with the Stanford V regimen. In addition, Stanford V requires patients to receive radiation therapy (RT) to original sites of disease larger than 5 cm in size and contiguous sites.
Another regimen which has been studied extensively for advanced stage Hodgkin lymphoma, and is considered a standard of care in some parts of the world, is escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone). In the HD9 study (n = 1196), the GHSG evaluated BEACOPP, escalated BEACOPP, and COPP/ABVD in advanced stage Hodgkin lymphoma.20 All arms of the study included 30 Gy RT to sites of bulky disease or residual disease. This study showed improved OS and FFTF with escalated BEACOPP, but at the cost of higher rates of toxicity. At 10 years, FFTF was 64%, 70%, and 82% with OS rates of 75%, 80%, and 86% for COPP/ABVD, baseline BEACOPP, and escalated BEACOPP, respectively (P < 0.001). The rate of secondary acute leukemia 10 years after treatment was 0.4% for COPP/ABVD, 1.5% for BEACOPP, and 3.0% for escalated BEACOPP. However, 3 subsequent randomized trials did not confirm a survival benefit with escalated BEACOPP relative to ABVD. In the HD 2000 trial (n = 295)21 and in a trial by Viviani and colleagues (n = 331),22 an improvement in OS was not demonstrated in favor of escalated BEACOPP. These studies also confirmed a higher rate of toxicities as well as secondary malignancies associated with the escalated BEACOPP regimen. In the EORTC20012 Intergroup trial (n = 549), 8 cycles of ABVD was compared with 4 cycles of escalated BEACOPP followed by 4 cycles of baseline BEACOPP, without radiation, in patients with clinical stage III or IV Hodgkin lymphoma with IPS score ≥ 3. Both regimens resulted in statistically similar FFS (63.7% in ABVD × 8 versus 69.3% in BEACOPP 4+4) and OS (86.7% in ABVD × 8 vs 90.3% in BEACOPP 4+4).23
In the United States, ABVD (6–8 cycles) is commonly used, although escalated BEACOPP (particularly for patients with an IPS of 4 or higher) and Stanford V are considered appropriate as well.24 In the North American Intergroup study comparing ABVD to Stanford V, and in the trial by Viviani et al, ABVD was associated with a 5- to 7-year FFS of 73% to 79% and OS of 84% to 92%.19,22 Given these excellent results, as well as the potential to cure patients with second-line therapy consisting of autologous hematopoietic cell transplantation (auto-HCT), the general consensus among most U.S. hematologists and oncologists is that ABVD remains the treatment of choice, and that the improved FFS/PFS with escalated BEACOPP is not outweighed by the additional toxicity associated with the regimen. There may, however, be a role for escalated BEACOPP in select patients who have a suboptimal response to ABVD as defined by interim positron emission tomography (iPET) scan (see below).
Brentuximab vedotin is an anti-CD30 antibody-drug conjugate (ADC) consisting of an anti-CD30 antibody linked to monomethyl auristatin E (MMAE), a potent antitubulin agent. CD30 is highly expressed on HRS cells and also in anaplastic large cell lymphoma. Upon binding to CD30, the ADC/CD30 complex is then internalized and directed to the lysosome, where the ADC is proteolytically cleaved, releasing MMAE from the antibody. MMAE then disrupts microtubule networks within the cell, leading to G2/M cycle arrest and apoptosis. CD30 is consistently expressed on HRS cells. In addition to being studied in the relapsed/refractory setting (described below), brentuximab has been studied in the first-line setting. In a phase 1 trial, brentuximab combined with ABVD was associated with increased pulmonary toxicity, while brentuximab + AVD had no significant pulmonary toxicity, with an excellent CR rate (96%), suggesting that substituting brentuximab for bleomycin may be an effective strategy. In addition to possibly being more efficacious, this strategy would also have the benefit of eliminating the risk of bleomycin pulmonary toxicity.25 Based on this data, a large international phase 3 study (the ECHELON-1 trial) comparing ABVD versus brentuximab + AVD in advanced stage cHL patients was recently completed. This study enrolled 1334 patients, and preliminary results were recently announced. With a median follow-up of 24 months, the brentuximab + AVD arm had a 4.9% absolute improvement in PFS relative to the ABVD arm (82.1% versus 77.2%). The brentuximab + AVD arm had an increased incidence of febrile neutropenia, managed with growth factors and peripheral neuropathy requiring dose adjustments, whereas the ABVD arm had an increased rate and severity of pulmonary toxicity.26 Further follow-up will be required to determine whether this will translate into a survival benefit. See Table 2 for a summary of recent large randomized prospective phase 3 trials in advanced stage Hodgkin lymphoma. 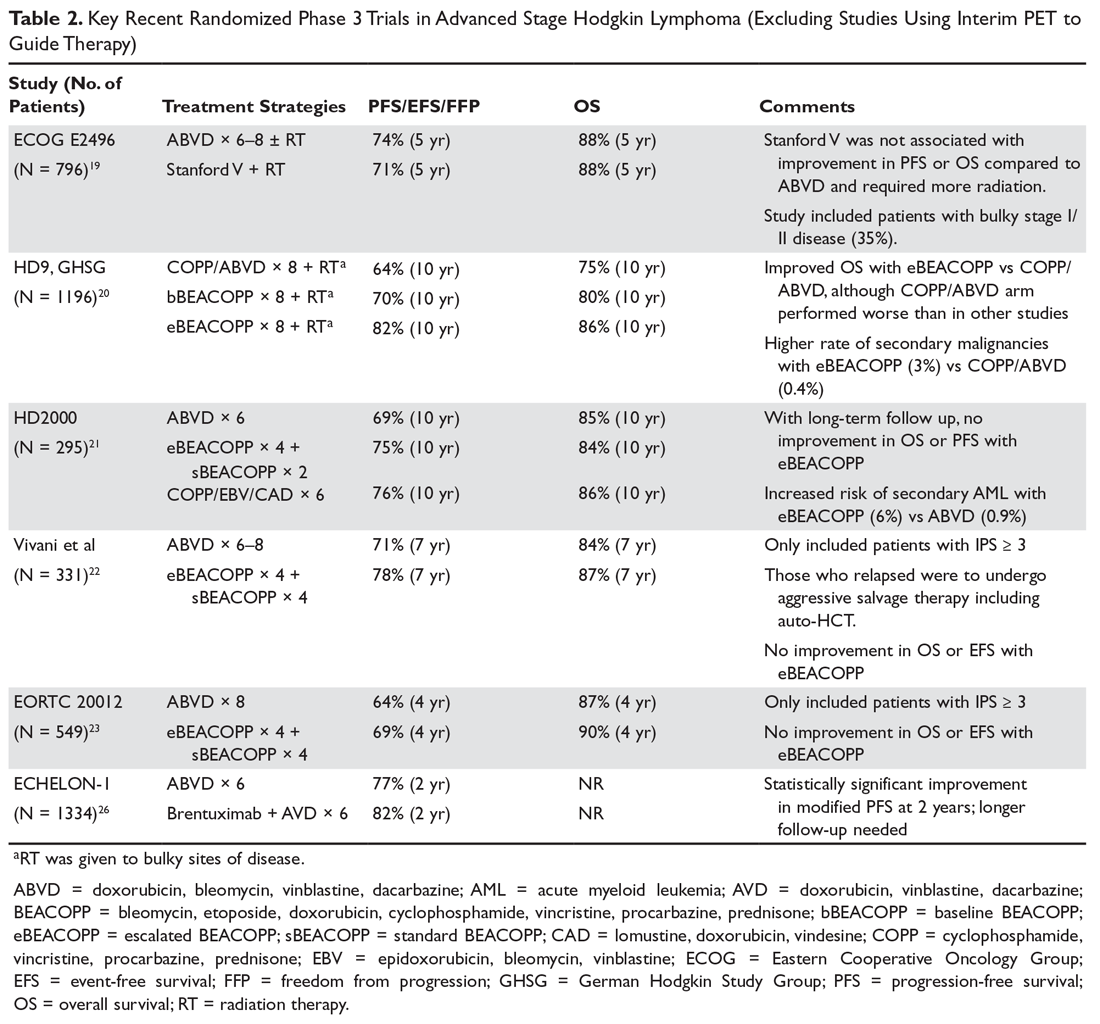
Alternative Regimens in Older Patients
Patients older than 60 years of age often have poor tolerance for ABVD and especially escalated BEACOPP. This results in increased treatment-related mortality and reduced overall dose intensity, with higher relapse rates and poor OS. In an attempt to improve on the results of treatment of elderly patients with Hodgkin lymphoma, alternative regimens have been explored. One example is PVAG (prednisone, vinblastine, doxorubicin, gemcitabine). With this regimen, the 3-year OS was 66% and PFS was 58%. One patient out of 59 died from treatment-related toxicity, which is much improved over the historical figures for elderly patients with Hodgkin lymphoma.27 Another commonly used approach in practice is to simply omit bleomycin from ABVD. In the early-stage setting (GHSG HD-13 trial), this regimen (referred to as AVD) led to 89.6% PFS at 5 years, compared to 93.5% with ABVD.28 It therefore stands to reason that this should be a reasonable option in older or more frail advanced stage cHL patients as well.
Brentuximab has been evaluated as a single-agent therapy for first-line therapy of elderly patients with Hodgkin lymphoma. In a phase 2 study, 27 patients (63% with advanced stage disease) were treated, with a 92% overall response rate and 73% CR rate. However the median duration of remission was disappointing at only 9.1 months.29 Based on this data, single-agent brentuximab appears to be a reasonable and well tolerated option for frail or elderly patients, although with the caveat that long-term disease control is relatively uncommon.
RESPONSE-ADAPTED FRONTLINE THERAPY USING INTERIM PET SCAN
In recent years, response-adapted treatment approaches have been extensively researched in cHL using iPET. The goal is to reduce toxicity by minimizing therapy in those who achieve negative iPET and/or to intensify treatment for patients with suboptimal response on iPET. Gallamini et al evaluated the prognostic role of an early iPET scan in advanced Hodgkin lymphoma patients (n = 190) treated with ABVD. This study found that patients with positive iPET had a 2-year PFS of 12.8% versus 95.0% in patients with negative iPET. This result was highly statistically significant (P < 0.0001). This study also showed that PET-2 (iPET after 2 cycles of ABVD) superseded the prognostic value of the IPS at diagnosis.30 As a result, numerous subsequent studies have been pursued using iPET for risk-adapted treatment in cHL.
A critical element to the conduct of iPET risk-adapted treatment for cHL is the interpretation of the iPET. In hopes of standardizing iPET interpretation in clinical trials, a scoring system called the Deauville score was developed. The Deauville score ranges from 1 to 5 (Table 3). 
The SWOG (Southwest Oncology Group) S0816 trial (n = 358) evaluated iPET-adapted treatment after 2 cycles of ABVD in stage III or IV Hodgkin lymphoma patients. Patients with positive iPET (Deauville score 4 to 5; n = 60) received escalated BEACOPP for 6 cycles, whereas iPET-negative (Deauville score 1 to 3; n = 271) patients continued to receive 4 more cycles of ABVD. The 2-year PFS was 64% for iPET-positive patients.33 This PFS was much higher than the expected 15% to 30% from prior studies such as Gallamini et al,30 suggesting that the treatment intensification may have been of benefit.
In the HD0801 study (n = 519), newly diagnosed advanced Hodgkin lymphoma patients with positive iPET after 2 cycles of ABVD (n = 103) received early ifosfamide-containing salvage therapy followed by high-dose therapy with autologous stem cell rescue. The 2-year PFS was 76% for PET-2–positive patients, comparable with PET-2–negative patients who had PFS of 81%.34 Again, this result for iPET-positive patients was much better than expected based on the historical control from Gallamini et al, suggesting that the treatment intensification may have been beneficial. It should be emphasized, however, that neither HD0801 nor S0816 were randomized prospective trials; rather, all iPET-positive patients were assigned to an intensified treatment approach.
In the HD18 trial (n = 1100), patients with advanced stage cHL started therapy with escalated BEACOPP and underwent an iPET after 2 cycles. For those with a positive iPET, rituximab was added to escalated BEACOPP in the experimental arm (n = 220) for cycles 3 through 8. The control group (n = 220) continued to receive 6 more cycles of escalated BEACOPP. In the 2 groups, the 3-year PFS was similar (91.4% in escalated BEACOPP, 93% in rituximab + escalated BEACOPP), suggesting no significant benefit with addition of rituximab.35 This study also calls into question whether iPET provides useful information for patients receiving intensive therapy such as escalated BEACOPP, and indicates that the historical control data for iPET-positive patients from Gallamini et al may not be consistently reproduced in other prospective trials. As a result, nonrandomized trials that implement an iPET risk-adapted approach should be interpreted with caution. See Table 4 for a summary of recent trials in advanced stage Hodgkin lymphoma using iPET scan to guide therapy. 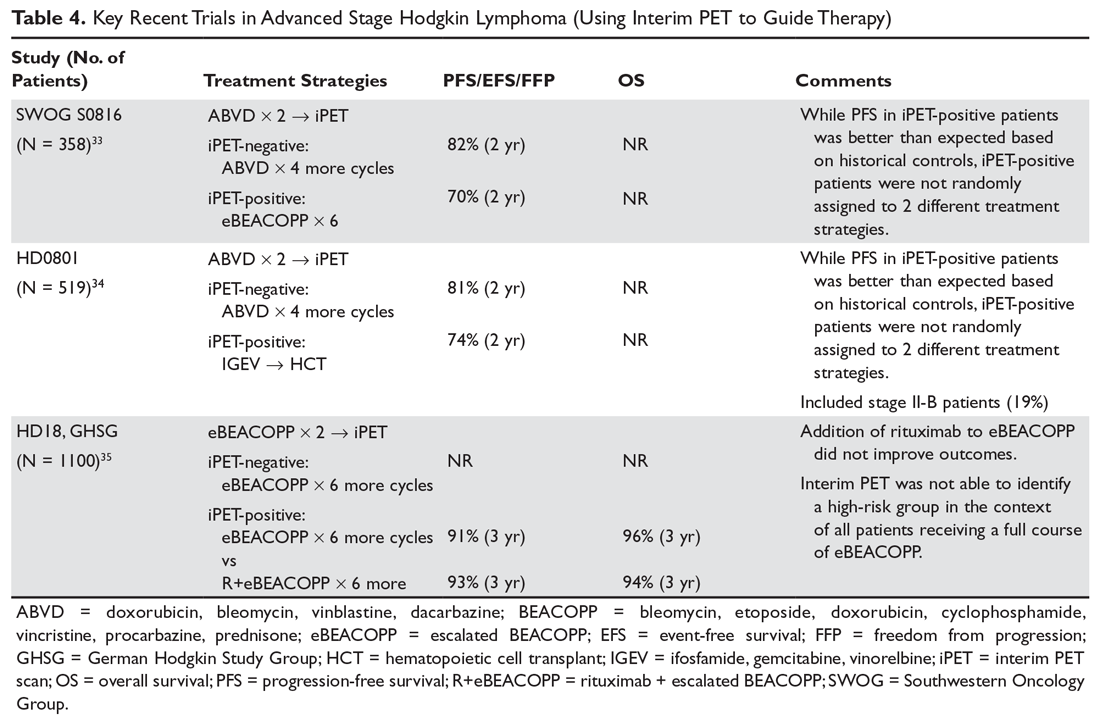
RADIATION THERAPY IN FRONTLINE TREATMENT
In patients with advanced stage Hodgkin lymphoma, IFRT to initial bulky sites of disease may be incorporated into frontline therapy to improve local control. However, whether this provides a survival benefit and which patients benefit most from consolidative RT remain unclear.
The European Organization for Research and Treatment of Cancer (EORTC) completed a randomized study in advanced stage Hodgkin lymphoma patients who achieved complete or partial remission after MOPP-ABV.36 Patients in CR were randomly assigned to receive no further treatment versus IFRT (24 Gy to all initially involved nodal areas and 16 to 24 Gy to all initially involved extranodal sites). Patients in partial remission (PR) were treated with 30 Gy to nodal areas and 18 to 24 Gy to extranodal sites. Among the CR patients, the 5-year event-free survival (EFS) was 79% to 84% and did not differ for those who received radiation versus those who did not. Five-year OS was 85% to 91% and also did not differ between the 2 groups. However, among the patients in PR after chemotherapy, the 5-year EFS was 79% and the 5-year OS was 87%, which is better than expected for PR patients, indicating a possible benefit to RT in patients with a partial response after chemotherapy. In the GHSG HD12 trial, patients with advanced stage Hodgkin lymphoma who had a residual lesion by computed tomography (CT) (but not analyzed by PET) had a very subtle improvement in FFTF (90% versus 87%) in favor of consolidation with IFRT, but again no survival benefit was seen.37
The EORTC and HD12 studies described above utilized CT scan for assigning remission status following chemotherapy, and it is now well known that many patients with residual masses (by CT) after chemotherapy may in fact be cured, as such residual radiographic abnormalities may simply be composed of fibrosis. PET scan is more accurate than CT in identifying patients who truly have residual active disease following chemotherapy. As a result, the EORTC study discussed above and the GHSG HD12 trial are of limited relevance in the modern era, in which patients routinely undergo PET scan at the end of therapy. Restricting IFRT to sites that remain PET-positive after completing chemotherapy may be a reasonable strategy that would allow for the avoidance of RT in many patients, and may obviate the need for aggressive second-line therapy (eg, high-dose therapy and autologous hematopoietic cell transplant [auto-HCT]). This approach was taken in the GHSG HD15 trial (n = 2182) in which advanced stage patients were treated with 3 variations on the BEACOPP regimen (8 cycles of escalated BEACOPP, 6 cycles of escalated BEACOPP, or 8 cycles of baseline BEACOPP, randomized in a 1:1:1 ratio). Patients with a residual mass of 2.5 cm or greater on CT scan then underwent a PET scan; if the lesion was PET positive, it was treated with 30 Gy of IFRT. This overall strategy was very effective, with 5-year FFTF rates of 84.4%, 89.3%, and 85.4%, respectively. The OS rates were 91.9%, 95.3%, and 94.5%, respectively. For patients with lesions that remained PET positive after chemotherapy, the PFS rate was 86.2% at 48 months, whereas patients in PR with persistent mass ≥ 2.5 cm but with negative PET had a PFS of 92.6%, similar to that of patients in CR.38 With this approach of BEACOPP followed by PET-guided radiation, the proportion of patients receiving RT was reduced from 71% (in the HD9 study) to only 11% in the HD15 study,38 with no apparent loss in overall efficacy when comparing the results of the 2 studies.
UPFRONT STEM CELL TRANSPLANTATION
To further improve outcomes of patients with advanced Hodgkin lymphoma with high-risk disease, high-dose therapy with auto-HCT has been explored as part of frontline therapy. While this has been shown to be feasible in such patients,39 randomized trials have not shown a clear benefit in terms of FFS or OS with upfront auto-HCT. 40,41 Therefore, auto-HCT is not considered a standard component of frontline therapy for cHL patients who achieve CR by PET/CT scan.
RELAPSED AND REFRACTORY HODGKIN LYMPHOMA
Depending on the stage, risk factors, and frontline regimen utilized, between 5% and 40% of patients with Hodgkin lymphoma can be expected to experience either primary induction failure or a relapse after attaining remission with frontline therapy.3 Primary refractory Hodgkin lymphoma, which occurs in up to 5% to 10% of patients, is defined as progression or no response during induction treatment or within 90 days of completing treatment. In cases where remission status is in question, an updated tissue biopsy is recommended. Biopsy is also recommended in cases in which new sites of disease have appeared or if relapse has occurred after a durable period of remission. Restaging is recommended at the time of relapse.
For younger patients with relapsed/refractory Hodgkin lymphoma, the standard of care in most cases is second-line (or salvage) chemotherapy followed by high-dose therapy and auto-HCT. For patients not felt to be candidates for auto-HCT, options include conventional second-line chemotherapy alone, salvage radiotherapy, novel agents such as brentuximab or immune checkpoint inhibitors, and/or participation in clinical trials.
CONVENTIONAL MULTI-AGENT CHEMOTHERAPY REGIMENS
Numerous conventional regimens have been shown in phase 2 studies to be active in relapsed and refractory Hodgkin lymphoma. These include platinum-based regimens, gemcitabine-based regimens, and alkylator-based regimens. No randomized trials in Hodgkin lymphoma have been conducted comparing these regimens. In general, regimens are chosen based on the patient’s age, performance status, comorbidities, and whether auto-HCT is being considered.
In the United States, platinum-based regimens such as ICE (ifosfamide, carboplatin, etoposide),42 DHAP (dexamethasone, cisplatin, high-dose cytarabine),43 ESHAP (etoposide, methylprednisolone, high-dose cytarabine, cisplatin),44 GDP (gemcitabine, cisplatin, dexamethasone),45 and GCD (gemcitabine, carboplatin, dexamethasone)46 are all considered appropriate second-line therapy options for patients being considered for auto-HCT, due to their high response rates and because autologous hematopoietic stem cell collection remains feasible after these regimens. Response rates range from 60% to 88%, with CR rates between 17% and 41%, and toxic death rates generally well below 5%.
Other gemcitabine-based regimens such as IGEV (ifosfamide, gemcitabine, vinorelbine) and GVD (gemcitabine, vinorelbine, liposomal doxorubicin) are also effective.47,48 GVD is an excellent choice since it is a generally well-tolerated outpatient regimen with a 60% response rate even in heavily pretreated patients.48 Stem cell collection remains feasible after both IGEV and GVD as well. ABVD can produce CR in approximately 20% to 50% of patients initially treated with MOPP.49–51 In practice, however, most patients today with relapsed or refractory Hodgkin lymphoma have already received ABVD as part of their first-line therapy, and retreatment with ABVD is not a good option because it would be associated with prohibitively high cumulative doses of doxorubicin.
These multi-agent chemotherapy regimens may not be tolerated well in patients over age 65 to 70 years or those with significant underlying comorbidities. In recent years, bendamustine has emerged as one of the most active conventional agents for cHL, with overall response rates of 53% to 58% in heavily pre-treated patients.52,53 Bendamustine can generally be tolerated even in elderly patients as well.
Some centers, particularly in Europe, investigated aggressive salvage regimens such as mini-BEAM (carmustine, etoposide, cytarabine, melphalan)54 or dexa-BEAM (BEAM plus dexamethasone).55 These regimens, however, are associated with significant hematologic toxicity and high (2%–5%) treatment-related mortality. As a result, these are rarely used in the United States.
For patients who have progressed after (or are not candidates for) platinum- and/or gemcitabine-based therapy, older alkylator-based regimens such as MOPP, C-MOPP, or ChlVPP (chlorambucil, vinblastine, procarbazine, prednisone) can be considered.56–58 However, these regimens are associated with significant bone marrow suppression, and autologous hematopoietic stem cell collection may no longer be feasible after such regimens. Therefore, these regimens should only be given to patients not felt to be auto-HCT candidates, or patients for whom autologous hematopoietic stem cell collection has already been completed. Weekly vinblastine or single-agent gemcitabine are palliative chemotherapy options, with response rates in the 60% to 80% range. Patients can sometimes be maintained on such low-intensity palliative regimens for 6 to 12 months or longer.59,60
BRENTUXIMAB VEDOTIN
Several trials are evaluating incorporation of brentuximab into second-line therapy in transplant-eligible patients. These approaches have used brentuximab prior to, concurrent with, or following platinum-based chemotherapy.61 While there is currently no consensus on the optimal way to incorporate brentuximab into salvage therapy, it is possible that the use of brentuximab or other novel agents in salvage therapy may allow for avoidance of conventional chemotherapy in some patients. In addition, this may translate into more patients proceeding to auto-HCT in a PET negative state. PET negativity prior to auto-HCT is a powerful predictor of long-term remission after auto-HCT, so any intervention that increases the rate of PET negativity prior to auto-HCT would be expected to improve outcomes with auto-HCT.62–65
For patients not being considered for autoHCT, or those for whom platinum-based salvage therapy was ineffective, single-agent brentuximab is an excellent option. In 2 phase 2 studies, an overall response rate (ORR) of 60% to 75% (including a CR rate of 22%–34%) was seen in relapsed and refractory Hodgkin lymphoma patients.66 The US Food and Drug Administration (FDA) approved brentuximab vedotin in August 2011 for treatment of relapsed and refractory Hodgkin lymphoma, after a failed auto-HCT, or in patients who are not auto-HCT candidates and who have received at least 2 prior chemotherapy regimens. With more extended follow-up, it has become clear that a proportion of patients who achieve CR to brentuximab may maintain remission long-term—58% at 3 years and 38% at 5 years.67 These patients may in fact be cured, in many cases without having undergone allogeneic HCT (allo-HCT) after brentuximab.
PD-1 (IMMUNE CHECKPOINT) INHIBITORS
As discussed earlier, PD-L1/PD-L2 copy number alterations represent a disease-defining feature of cHL. Alterations in chromosome 9p24.1 increase the expression of PD-1 ligands PD-L1 and PD-L2. Nivolumab and pembrolizumab are PD-1-blocking antibodies, which have recently been FDA approved for relapsed and refractory cHL. In a study with 23 patients, with 78% of them relapsing after auto-HCT and 78% relapsing after brentuximab, nivolumab produced an objective response in 87% of the patients, with 17% achieving CR and 70% achieving PR. The rate of PFS was 86% at 24 weeks.68 Pembrolizumab, another PD-1 antagonist, was also tested in relapsed and refractory Hodgkin lymphoma. In the KEYNOTE-087 study (n = 210), pembrolizumab produced an ORR of 64% to 70% in 3 different cohorts of relapsed and refractory cHL patients. Overall CR rate was 22%.69 In general, these agents are well tolerated, although patients must be monitored closely for
inflammatory/autoimmune-type toxicities including skin rash, diarrhea/colitis, transaminitis, endocrine abnormalities, and pneumonitis. Prompt recognition and initiation of corticosteroids is essential in managing these toxicities. Of note, PD-1 inhibitors should be given very cautiously to patients with a prior history of allo-HCT, since 30% to 55% of such patients will experience acute graft-versus-host disease (GVHD) in this setting. In 2 retrospective studies, the response rate was very high at 77% to 95%; however, 10% to 26% of all patients treated with PD-1 inhibitors post-allo-HCT died from GVHD induced by PD-1 inhibition.70,71 These risks and benefits therefore need to be carefully weighed in the post-allo-HCT setting. In another recent study, the outcomes were reported for 39 patients who underwent allo-HCT after prior therapy with a PD-1 inhibitor. Three patients (7.7%) developed lethal acute GVHD, suggesting there may be an increased risk of GVHD in patients undergoing allo-HCT after prior PD-1 inhibitor therapy.72
AUTOLOGOUS STEM CELL TRANSPLANTATION
Several studies have shown an improved disease-free survival (DFS) or FFS in patients with relapsed cHL treated by auto-HCT as compared to those receiving conventional chemotherapy alone.55,73,74 Overall, for relapsed disease, one can expect an approximately 50% to 60% chance for DFS at 5 years post-transplant. In a retrospective, matched-pair analysis, FFP was 62% for auto-HCT patients, compared to 32% for conventional chemotherapy patients. OS, however, was similar for the 2 groups (47%–54%). Patients failing induction therapy or relapsing within 1 year were seen to benefit the most from auto-HCT, including an OS benefit.74
A European prospective randomized trial was conducted comparing conventional salvage therapy to auto-HCT. In this study, 161 patients with relapsed Hodgkin lymphoma were treated with 2 cycles of dexa-BEAM. Those with chemo-sensitive disease were then randomized to either 2 more cycles of dexa-BEAM or high-dose BEAM with auto-HCT. Auto-HCT was associated with an approximately 55% FFTF at 3 years, versus 34% with conventional chemotherapy alone.55 This benefit again was most apparent for patients relapsing within 1 year of completion of primary therapy, although an OS benefit was not seen with auto-HCT. For patients with late relapse (>1 year after completion of primary therapy), auto-HCT was associated with an approximately 75% FFTF at 3 years, versus 50% with chemotherapy alone. One other small randomized trial of auto-HCT in relapsed and refractory Hodgkin lymphoma also showed an improved 3-year EFS in favor of auto-HCT (53% versus 10%), again with no difference in OS.73
The lack of OS benefit seen in these studies suggests that auto-HCT at first or second relapse provides comparable outcomes. Auto-HCT offers the benefit of avoiding the long-term toxicities associated with multiple salvage regimens and the anxiety associated with multiple relapses. In addition, the treatment-related mortality with auto-HCT is now in the 1% to 2% range in younger patients, at centers that perform the procedure routinely. For all of these reasons, auto-HCT is commonly recommended by physicians for Hodgkin lymphoma patients in first or second relapse. In most cases, transplant is favored in first relapse, since waiting until second relapse may be associated with a lower chance of achieving CR and difficulty collecting sufficient hematopoietic stem cells. For patients with early relapse or primary refractory disease, an even stronger case can be made for auto-HCT as the best option to achieve sustained control of the disease. For patients with late relapse, conventional salvage therapy alone may be a reasonable option, particularly in older or frail patients, or those with significant comorbid conditions.
The optimal conditioning regimen for autoHCT for relapsed and refractory Hodgkin lymphoma remains undefined. No randomized trials have been performed comparing conditioning regimens for relapsed and refractory Hodgkin lymphoma. One retrospective study compared 92 patients with Hodgkin lymphoma who underwent auto-HCT using a total-body irradiation (TBI) regimen versus a chemotherapy-alone regimen. No difference in 5-year OS or EFS was seen.75 Given the lack of benefit seen with TBI, along with reports of increased rates of secondary malignancies and myelodysplasia with TBI,76 chemotherapy-alone conditioning regimens are most widely employed. For example, in the United States, either the BEAM or CBV (cyclophosphamide, carmustine, etoposide) regimens are used in over 80% of cases.77 This practice was justified in a Center for International Blood and Marrow Transplant Research (CIBMTR) retrospective study comparing outcomes by conditioning regimens, in which no regimen performed better than BEAM or CBV.78
IFRT is often given as an adjunctive therapy to sites of initial and/or relapsed disease following auto-HCT. Although a relatively common practice, whether this truly enhances outcomes beyond that obtained with auto-HCT alone is unclear. Two retrospective studies have shown some benefit in terms of improvement in OS at 3 to 5 years in the group that received IFRT (70%–73% versus 40%–56%).79,80 Given the retrospective nature and small size of these studies, a prospective study would be needed to properly define the potential role for IFRT following auto-HCT in relapsed/refractory Hodgkin lymphoma. Another retrospective study (n = 73) that evaluated peri-transplant IFRT in Hodgkin lymphoma patients receiving auto transplant found no improvement in survival for patients who received peri-transplant IFRT. This study, however, did show a survival benefit in the subgroup of patients with limited stage disease.81
Prognostic Factors Associated with Outcome with Auto-HCT
The factor most consistently associated with improved outcome for patients with relapsed and refractory Hodgkin lymphoma who undergo auto-HCT is the disease status at transplant.63,77 Those in a second CR, versus a chemo-sensitive relapse (but not CR), versus a chemo-refractory relapse have DFS rates of 60% to 70%, 30% to 40%, and 10% to 20%, respectively.63 The duration between remission and relapse also has important prognostic significance. Late relapse (> 1 year after completion of frontline therapy) is associated with better outcomes as compared to early relapse.55 Other factors with prognostic significance at relapse include anemia, time to relapse and clinical stage, B symptoms, extranodal disease, number of prior chemotherapy regimens, and performance status.42,82 The prognostic impact of pretransplant disease status has been confirmed by studies using functional imaging (eg, FDG-PET or gallium scans). In a report by Moskowitz et al, patients with negative functional imaging following second-line therapy had a 77% EFS post-auto-HCT versus 33% in those whose functional imaging remained positive.62 Very similar findings have been reported by other groups.63–65
Post-Auto-HCT Brentuximab Maintenance
In the multicenter, randomized, double-blinded phase 3 AETHERA trial (n = 329), brentuximab (n = 165) was compared with placebo (n = 164) in patients with unfavorable risk relapsed or primary refractory cHL who had undergone autologous transplant. Eligible patients had at least 1 of the following risk factors for progression after auto-HCT: primary refractory Hodgkin lymphoma (failure to achieve complete remission), relapsed Hodgkin lymphoma with an initial remission duration of less than 12 months, or extranodal involvement at the start of pre-transplantation salvage chemotherapy. Patients were required to have CR, PR, or stable disease after pretransplant salvage chemotherapy with adequate kidney, liver, and bone marrow function. Patients who previously received brentuximab were excluded. Patients received 16 cycles of brentuximab or placebo once every 3 weeks starting 30 to 45 days after transplant. The PFS was significantly improved in the brentuximab group when compared to the placebo group (hazard ratio 0.57; P = 0.0013) after a median observation time of 30 months. Median PFS was 42.9 months in the brentuximab group versus 24.1 months in the placebo group; estimated 2-year PFS rates were 63% in the brentuximab group and 51% in the placebo group. OS was not significantly different between the study groups (~85%), presumably due to the fact that patients in the control group who relapsed likely went on to receive brentuximab as a subsequent therapy.83
PRIMARY REFRACTORY HODGKIN LYMPHOMA
Patients with primary refractory Hodgkin lymphoma have a poor outcome. Salvage therapy using conventional chemotherapy and/or RT results in long-term DFS in 10% or fewer of such patients.13,84 Given these poor outcomes with conventional salvage therapy, auto-HCT is considered to be the standard of care for this subset of patients. The GHSG retrospectively analyzed the prognostic factors and outcomes of patients with primary refractory Hodgkin lymphoma. The 5-year freedom-from-second-failure and the 5-year OS were reported to be 31% and 43%, respectively, for those patients treated with auto-HCT. Patients with poor functional status at time of transplant, age greater than 50 years, and failure to attain a temporary remission had a 0% 5-year OS, as compared to 55% in patients without any of these risk factors.85 A large retrospective European study showed that patients with chemo-resistant disease who underwent transplant had a 19% survival at 5 years.63 Hence, even patients with primary refractory Hodgkin lymphoma have some chance of achieving long-term survival following auto-HCT.
SALVAGE RADIOTHERAPY
The GHSG performed a retrospective analysis of the efficacy of salvage RT in patients with refractory or first-relapsed Hodgkin lymphoma. Five-year FFTF and OS rates were 28% and 51%, respectively. Patients with a limited-stage relapse and without B symptoms were more likely to benefit from salvage RT.86 Campbell et al reported on 81 patients undergoing salvage RT for persistent or recurrent Hodgkin lymphoma after chemotherapy. The 10-year FFTF and OS rates were 33% and 46%, respectively.87 Similarly, Wirth et al reported a 5-year FFS of 26% and 5-year OS of 57%. These figures were 36% and 75%, respectively, in patients whose relapse was limited to supradiaphragmatic nodal sites without B symptoms.88 RT therefore may be a useful strategy for a subset of patients who relapse following chemotherapy, particularly those with a limited-stage relapse, without B symptoms, and those with relapsed disease after a CR, as opposed to those with a partial response or lack of response to the prior chemotherapy regimen.
INVESTIGATIONAL AGENTS AND NOVEL COMBINATIONS
Several biological therapies are emerging as options for the treatment of refractory or relapsed disease. These therapies consist of monoclonal antibodies and ADCs that target cell surface antigens, or small molecules that inhibit key intracellular pathways within neoplastic cells.
Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody used widely in B-cell non-Hodgkin lymphomas. The CD20 molecule is typically highly expressed in nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL). Two studies (one in relapsed patients, the other in a mixture of relapsed and previously untreated patients) showed significant activity of rituximab in relapsed NLPHL, with ORRs ranging from 94% to 100%, CR rates ranging from 41% to 53%, and median duration of remission in the 10- to 33-month range.89,90 In cHL, CD20 is expressed in HRS cells in 20% to 30% of cases. In such cases, single-agent rituximab has also shown activity. There is also evidence that rituximab may be effective even in cases in which the HRS cells are CD20-negative, presumably by virtue of depleting reactive B lymphocytes from the microenvironment, which may enhance anti-tumor immunity, or by eliminating a putative CD20-expressing Hodgkin lymphoma stem cell.91,92
Lenalidomide
Lenalidomide is an immunomodulatory drug that has multiple modes of action, including direct induction of apoptosis in tumor cells, antiangiogenic effects, and the activation of immune cells, such as natural killer cells and T cells. Lenalidomide has been shown to modify many features of the microenvironment of HRS cells and has demonstrated activity in other B-cell neoplasms. As a result, lenalidomide has been evaluated in relapsed and refractory Hodgkin lymphoma patients. A multicenter phase 2 study by Fehniger et al included 35 patients, 87% of whom had previously undergone HCT and 55% of whom were refractory to the last therapy.93 All patients were given lenalidomide 25 mg/day from days 1 to 21 of a 28-day cycle until disease progression. One patient was noted to achieve CR, 6 achieved PR, and 5 had stable disease lasting more than 6 months, for an ORR of 19% and a “cytostatic overall response rate” of 33%. The median duration of CR/partial remission was 6 months, with the median time-to-treatment failure in responders (including those with stable disease > 6 months) being 15 months. Similarly, in another study, Böll et al evaluated 12 patients across 4 German centers with relapsed or refractory disease who were treated with oral lenalidomide for 21 days in a 28-day cycle. No radiological evidence of disease progression after 2 cycles of lenalidomide was seen in any of the enrolled patients. ORR was noted to be 50%, with 6 patients with stable disease and 5 patients achieving PR after 2 cycles.94
Novel Brentuximab Combination Therapies
Brentuximab plus bendamustine. The combination of brentuximab and bendamustine was tested as an outpatient regimen in a phase 1/2 study (n = 55) in primary refractory Hodgkin lymphoma or after first relapse. The CR rate of the combination was 74%, with an overall objective response (CR + PR) of 93%. The CR rates were 64% and 84%, respectively, for refractory and relapsed patients. The PFS at 12 months was 80%, establishing this combination therapy as an effective salvage regimen with durable response.95
Brentuximab plus nivolumab. Preliminary results have recently been presented from 2 studies96,97 evaluating the combination of brentuximab and nivolumab. While this combination would still be considered investigational, these studies showed very encouraging ORRs of 90% to 100% and a CR rate of 62% to 66%. Longer follow-up is needed to determine whether these responses are durable and to document the toxicity profile of this combination.
Mammalian Target of Rapamycin Inhibitors
Two mammalian target of rapamycin (mTOR) inhibitors, everolimus and temsirolimus, are currently available in the United States. While neither drug currently has FDA approval for Hodgkin lymphoma, everolimus was evaluated in a phase 2 trial in a heavily pretreated group of relapsed/refractory patients. An ORR of 47% was seen, with a median time to progression of 7.2 months.98
ALLOGENEIC STEM CELL TRANSPLANTATION
Historically, patients who relapse after having an auto-HCT generally had a poor outcome, with a median survival of 2 to 3 years after failure of auto-HCT.99 These patients may be offered palliative chemotherapy (see above), treatment with novel agents (see above), or enrollment in a clinical trial. Select patients may benefit from a second hematopoietic stem cell transplant, most commonly an allo-HCT. However, rare patients with late relapse after auto-HCT may be considered for a second auto-HCT, with a minority of such patients achieving a durable remission after the second auto-HCT.100,101 Because relapse or progressive disease occurs most commonly in the first several months following auto-HCT, patients are more often considered for allo-HCT than a second auto-HCT. In addition, a second auto-HCT may not be feasible due to impaired bone marrow reserve and/or concerns for development of secondary myelodysplasia or acute myeloid leukemia.
Several studies have evaluated allo-HCT in relapsed/ refractory Hodgkin lymphoma. Early studies evaluating myeloablative allo-HCT for Hodgkin lymphoma showed excessive treatment-related mortality (up to 50%) and disappointingly low rates of long-term survival (< 25%).102,103 This was likely related to the fact that, in that era, most of the patients with Hodgkin lymphoma evaluated for allo-HCT were heavily pretreated and therefore at a higher risk for toxicity as well as lymphoma progression.
More recently, several studies have focused on the use of reduced-intensity conditioning (RIC) allo-HCT for relapsed and refractory Hodgkin lymphoma. This approach relies more on a “graft-versus-lymphoma” effect, the existence of which has been debated in Hodgkin lymphoma. Three single-center studies of RIC allo-HCT in patients with multiply recurrent Hodgkin lymphoma showed improved rates of treatment-related mortality (8%–16%) but still relatively low rates of long-term PFS (23%–39% at 2 to 4 years).104–106 Interestingly, in one of these studies the outcomes were more favorable for patients who underwent haploidentical (versus matched sibling or matched unrelated donor) transplants.105
Two large registry studies have also reported on the outcomes of RIC allo-HCT in patients with relapsed and refractory Hodgkin lymphoma.107,108 These studies also confirmed a modest improvement in outcomes compared with those seen historically with myeloablative transplants. Treatment-related mortality at 1 to 2 years was 23% to 33%, depending on whether a matched sibling donor versus an unrelated donor was used. However, long-term PFS (18%–20% at 2 to 5 years) and OS (28%–37% at 2 to 5 years) remained poor, primarily due to high rates of progressive lymphoma post-transplant. In both of these studies, patients were heavily pretreated (84%–96% had received 3 or more prior lines of chemotherapy, and 62%–89% received a prior auto-HCT), with 47% to 55% of patients chemo-resistant prior to transplant. Of note, both of these registry studies reflect patients who underwent transplant prior to the widespread use of brentuximab and PD-1 inhibitors.
Based on the single-center and registry data above, a prospective multicenter European phase 2 trial was conducted to evaluate the benefit of RIC allo-HCT in Hodgkin lymphoma.109 Ninety-two patients (86% with prior auto-HCT, 90% with 3 or more prior lines of therapy) were enrolled and given salvage therapy. Those who had stable disease or better following salvage therapy remained on protocol (n = 78) and underwent RIC with fludarabine and melphalan, followed by allo-HCT (70% with matched sibling donors). Treatment-related mortality was 15% at 1 year. Relapse or progression occurred in 49% at 2 years (35% if chemo-sensitive prior to transplant). Chronic GVHD was associated with a decreased rate of relapse, supporting the existence of a graft-versus-lymphoma effect in Hodgkin lymphoma. Unfortunately, PFS among all allografted patients was still relatively poor (24% at 4 years). However, among patients in CR prior to allo-HCT, a 50% PFS was seen at 4 years. Therefore, even in a prospective multicenter study, RIC allo-HCT offered significant benefit with manageable toxicity in relapsed and refractory Hodgkin lymphoma patients with chemo-sensitive disease.
These studies suggest that outcomes with allo-HCT would improve further if implemented earlier in the course of disease and/or with a lower burden of disease at transplant. It has therefore been suggested that allo-HCT should be considered soon after failure of auto-HCT is documented. In a retrospective study by Sarina et al, 185 Hodgkin lymphoma patients who relapsed following auto-HCT were then immediately considered for reduced-intensity allo-HCT.110 Of these, 122 had a donor identified, and 104 (85%) actually underwent allo-HCT. These 104 patients were then compared to the other 81 patients who either had no donor identified or had a donor but did not receive the planned allo-HCT. Two-year PFS and OS were superior in the patients undergoing allo-HCT (39% versus 14% and 66% versus 42%, respectively, P < 0.001), with a median follow-up of 4 years. The presence of chronic GVHD again was associated with improved PFS and OS. Disease status prior to transplant remained highly predictive of PFS and OS by multivariate analysis. Two other smaller retrospective studies similarly found a survival benefit associated with allo-HCT compared with patients who underwent conventional salvage therapies alone.111,112 These studies, although subject to the usual limitations of retrospective analyses, suggest that the results with reduced-intensity allo-HCT are in fact enhanced if applied earlier in the disease course, and are superior to those with conventional therapy alone.
Currently, the exact role of allo-HSCT, including the optimal timing and optimal donor source (matched sibling versus haploidentical sibling versus matched unrelated donor), remain undefined for relapsed and refractory Hodgkin lymphoma. As discussed earlier, brentuximab is highly active in relapsed Hodgkin lymphoma patients, with a subset of patients still in CR at 5 years.67 For such patients, avoiding the risks of allo-HCT is a desirable goal.
For those who relapse or progress after auto-HCT, a reasonable strategy therefore is to treat initially with brentuximab, unless the patient is already known to have responded poorly to brentuximab, or already has significant neuropathy. Those who achieve a CR to brentuximab are then observed. A subset of those patients will remain in remission at 5 years without further therapy. For those who relapse, or who achieve less than a CR to brentuximab, additional treatment (with brentuximab re-treatment being one option) followed by a reduced-intensity allo-HCT is a reasonable consideration. This approach has the theoretical advantages of (1) avoiding the risk of allo-HCT in the subset potentially cured by brentuximab, (2) getting patients to allo-HCT with fewer comorbidities (due to a lower total exposure to conventional chemotherapy pre-transplant), and (3) applying allo-HCT in the setting of sensitive disease/lower disease burden (due to the high efficacy of brentuximab). The results of a small study suggest that brentuximab may in fact be a very effective “bridge” to allotransplant. Chen et al113 reported on 18 patients with relapsed/refractory Hodgkin lymphoma (17 of whom had previously undergone auto-HCT) who were treated on brentuximab vedotin clinical trials. The data were retrospectively evaluated to determine the efficacy and safety of subsequent reduced-intensity allo-HCT. Remarkably, at 1 year the OS was 100%, PFS was 92%, and nonrelapse mortality was 0% with a median follow-up of 14 months. Hence, brentuximab is safe for use prior to reduced-intensity allo-HCT in heavily pre-treated patients and appears to be associated with very favorable post-transplant outcomes, particularly in comparison to older studies of allo-HCT in the era prior to brentuximab.
SUMMARY
Currently, cure is possible for the majority of patients diagnosed with advanced stage Hodgkin lymphoma. The challenge to the clinician is to provide curative treatment with the lowest risk of serious toxicities. Which regimen will best provide this balance of risk and benefit needs to be assessed based on the relapse risk, age, frailty, and comorbidity profile for an individual patient. For many patients with relapsed or refractory Hodgkin lymphoma, cure remains possible using approaches based on hematopoietic stem cell transplantation, RT, and/or brentuximab. In addition, there are now numerous conventional chemotherapy agents, RT strategies, and exciting newer agents such as PD-1 inhibitors, that can provide significant clinical benefit even when cure is not feasible.
1. Kuppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6.
2. Kuppers R. The biology of Hodgkin’s lymphoma. Nature Rev Cancer 2009;9:15–27.
3. Narra RK, Pingali SR, Fenske TS. Early-stage Hodgkin’s lymphoma. Hospital Physician Hematology-Oncology Board Review Manual. 2017;12(Part 3).
4. Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993;71:2062–71.
5. Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med 1998;339:1506–14.
6. Moccia AA, Donaldson J, Chhanabhai M, et al. International Prognostic Score in advanced-stage Hodgkin’s lymphoma: altered utility in the modern era. J Clin Oncol 2012;30:3383–8.
7. Diefenbach CS, Li H, Hong F, et al. Evaluation of the International Prognostic Score (IPS-7) and a Simpler Prognostic Score (IPS-3) for advanced Hodgkin lymphoma in the modern era. Br J Haematol 2015;171:530–8.
8. Greaves P, Clear A, Coutinho R, et al. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of cassical hodgkin lymphoma is predictive of outcome. J Clin Oncol 2013;31:256–62.
9. Touati M, Delage-Corre M, Monteil J, et al. CD68-positive tumor-associated macrophages predict unfavorable treatment outcomes in classical Hodgkin lymphoma in correlation with interim fluorodeoxyglucose-positron emission tomography assessment. Leuk Lymphoma 2015;56:332–41.
10. Roemer MG, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7.
11. Pike LC, Kirkwood AA, Patrick P, et al. Can baseline PET-CT features predict outcomes in advanced hodgkin lymphoma? A prospective evaluation of UK patients in The RATHL Trial (CRUK/07/033). Hematological Oncology 2017;35(Supplement S2):37–8.
12. DeVita VT Jr, Simon RM, Hubbard SM, et al. Curability of advanced Hodgkin’s disease with chemotherapy. Long-term follow-up of MOPP-treated patients at the National Cancer Institute. Ann Intern Med 1980;92:587–95.
13. Longo DL, Duffey PL, Young RC, et al. Conventional-dose salvage combination chemotherapy in patients relapsing with Hodgkin’s disease after combination chemotherapy: the low probability for cure. J Clin Oncol 1992;10:210–8.
14. Kaldor JM, Day NE, Clarke EA, et al. Leukemia following Hodgkin’s disease. N Engl J Med 1990;322:7–13.
15. Bonadonna G, Zucali R, Monfardini S, et al. Combination chemotherapy of Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP. Cancer 1975;36:252–9.
16. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–84.
17. Horning SJ, Williams J, Bartlett NL, et al. Assessment of the stanford V regimen and consolidative radiotherapy for bulky and advanced Hodgkin’s disease: Eastern Cooperative Oncology Group pilot study E1492. J Clin Oncol 2000;18:972–80.
18. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the stanford V regimen and ABVD in the treatment of advanced Hodgkin’s Lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRCTN 64141244. J Clin Oncol 2009;27:5390–6.
19. Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013;31:684–91.
20. Engert A, Diehl V, Franklin J, et al. Escalated-dose BEACOPP in the treatment of patients with advanced-stage Hodgkin’s lymphoma: 10 years of follow-up of the GHSG HD9 study. J Clin Oncol 2009;27:4548–54.
21. Merli F, Luminari S, Gobbi PG, et al. Long-term results of the HD2000 trial comparing ABVD versus BEACOPP versus COPP-EBV-CAD in untreated patients with advanced Hodgkin lymphoma: a study by Fondazione Italiana Linfomi. J Clin Oncol 2016;34:1175–81.
22. Viviani S, Zinzani PL, Rambaldi A, et al. ABVD versus BEACOPP for Hodgkin’s lymphoma when high-dose salvage is planned. N Engl J Med 2011;365:203–12.
23. Carde P, Karrasch M, Fortpied C, et al. Eight cycles of ABVD versus four cycles of BEACOPPescalated plus four cycles of BEACOPPbaseline in stage III to IV, International Prognostic Score >/= 3, high-risk Hodgkin lymphoma: first results of the phase III EORTC 20012 intergroup trial. J Clin Oncol 2016;34:2028–36.
24. National Comprehensive Cancer Network I. NCCN Guidelines Version 1.2017 Hodgkin Lymphoma. Accessed July 20, 2017.
25. Younes A, Connors JM, Park SI, et al. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncology 2013;14:1348–56.
26. Takeda and Seattle Genetics announce positive results from phase 3 ECHELON-1 clinical trial evaluating ADCETRIS® (brentuximab vedotin) in frontline advanced Hodgkin lymphoma [press release]. Cambridge, MA: Takeda Oncology; June 26, 2017.
27. Boll B, Bredenfeld H, Gorgen H, et al. Phase 2 study of PVAG (prednisone, vinblastine, doxorubicin, gemcitabine) in elderly patients with early unfavorable or advanced stage Hodgkin lymphoma. Blood 2011;118:6292–8.
28. Behringer K, Goergen H, Hitz F, et al. Omission of dacarbazine or bleomycin, or both, from the ABVD regimen in treatment of early-stage favourable Hodgkin’s lymphoma (GHSG HD13): an open-label, randomised, non-inferiority trial. Lancet 2015;385:1418–27.
29. Forero-Torres A, Holkova B, Goldschmidt J, et al. Phase 2 study of frontline brentuximab vedotin monotherapy in Hodgkin lymphoma patients aged 60 years and older. Blood 2015;126:2798–804.
30. Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol 2007;25:3746–52.
31. Meignan M, Gallamini A, Meignan M, et al. Report on the First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymphoma 2009;50:1257–60.
32. Barrington SF, Mikhaeel NG, Kostakoglu L, et al. Role of Imaging in the Staging and Response Assessment of Lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 2014;32:3048–58.
33. Press OW, Li H, Schoder H, et al. US Intergroup trial of response-adapted therapy for stage III to IV Hodgkin lymphoma using early interim fluorodeoxyglucose-positron emission tomography imaging: Southwest Oncology Group S0816. J Clin Oncol 2016;34:2020–7.
34. Zinzani PL, Broccoli A, Gioia DM, et al. Interim positron emission tomography response-adapted therapy in advanced-stage Hodgkin lymphoma: final results of the pPhase II part of the HD0801 study. J Clin Oncol 2016;34:1376–85.
35. Borchmann P, Haverkamp H, Lohri A, et al. Progression-free survival of early interim PET-positive patients with advanced stage Hodgkin’s lymphoma treated with BEACOPPescalated alone or in combination with rituximab (HD18): an open-label, international, randomised phase 3 study by the German Hodgkin Study Group. Lancet Oncology 2017;18:454–63.
36. Aleman BM, Raemaekers JM, Tirelli U, et al. Involved-field radiotherapy for advanced Hodgkin’s lymphoma. N Engl J Med 2003;348:2396–406.
37. Borchmann P, Haverkamp H, Diehl V, et al. Eight cycles of escalated-dose BEACOPP compared with four cycles of escalated-dose BEACOPP followed by four cycles of baseline-dose BEACOPP with or without radiotherapy in patients with advanced-stage hodgkin’s lymphoma: final analysis of the HD12 trial of the German Hodgkin Study Group. J Clin Oncol 2011;29:4234–42.
38. Engert A, Haverkamp H, Kobe C, et al. Reduced-intensity chemotherapy and PET-guided radiotherapy in patients with advanced stage Hodgkin’s lymphoma (HD15 trial): a randomised, open-label, phase 3 non-inferiority trial. Lancet 2012;379:1791–9.
39. Nademanee A, Molina A, Fung H, et al. High-dose chemo/radiotherapy and autologous bone marrow or stem cell transplantation for poor-risk advanced-stage Hodgkin’s disease during first partial or complete remission. Biol Blood Marrow Transplant 1999;5:292–8.
40. Federico M, Bellei M, Brice P, et al. High-dose therapy and autologous stem-cell transplantation versus conventional therapy for patients with advanced Hodgkin’s lymphoma responding to front-line therapy. J Clin Oncol 2003;21:2320–5.
41. Arakelyan N, Berthou C, Desablens B, et al. Early versus late intensification for patients with high-risk Hodgkin lymphoma-3 cycles of intensive chemotherapy plus low-dose lymph node radiation therapy versus 4 cycles of combined doxorubicin, bleomycin, vinblastine, and dacarbazine plus myeloablative chemotherapy with autologous stem cell transplantation: five-year results of a randomized trial on behalf of the GOELAMS Group. Cancer 2008;113:3323–30.
42. Moskowitz CH, Nimer SD, Zelenetz AD, et al. A 2-step comprehensive high-dose chemoradiotherapy second-line program for relapsed and refractory Hodgkin disease: analysis by intent to treat and development of a prognostic model. Blood 2001;97:616–23.
43. Josting A, Rudolph C, Reiser M, et al. Time-intensified dexamethasone/cisplatin/cytarabine: an effective salvage therapy with low toxicity in patients with relapsed and refractory Hodgkin’s disease. Ann Oncol 2002;13:1628–35.
44. Aparicio J, Segura A, Garcera S, et al. ESHAP is an active regimen for relapsing Hodgkin’s disease. Ann Oncol 1999;10:593–5.
45. Baetz T, Belch A, Couban S, et al. Gemcitabine, dexamethasone and cisplatin is an active and non-toxic chemotherapy regimen in relapsed or refractory Hodgkin’s disease: a phase II study by the National Cancer Institute of Canada Clinical Trials Group. Ann Oncol 2003;14:1762–7.
46. Gopal AK, Press OW, Shustov AR, et al. Efficacy and safety of gemcitabine, carboplatin, dexamethasone, and rituximab in patients with relapsed/refractory lymphoma: a prospective multi-center phase II study by the Puget Sound Oncology Consortium. Leuk Lymphoma 2010;51:1523–9.
47. Santoro A, Magagnoli M, Spina M, et al. Ifosfamide, gemcitabine, and vinorelbine: a new induction regimen for refractory and relapsed Hodgkin’s lymphoma. Haematologica 2007;92:35–41.
48. Bartlett NL, Niedzwiecki D, Johnson JL, et al. Gemcitabine, vinorelbine, and pegylated liposomal doxorubicin (GVD), a salvage regimen in relapsed Hodgkin’s lymphoma: CALGB 59804. Ann Oncol 2007;18:1071–9.
49. Santoro A, Bonadonna G. Prolonged disease-free survival in MOPP-resistant Hodgkin’s disease after treatment with adriamycin, bleomycin, vinblastine and dacarbazine (ABVD). Cancer Chemother Pharmacol 1979;2:101–5.
50. Krikorian JG, Portlock CS, Rosenberg SA. Treatment of advanced Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide (ABVD) after failure of MOPP therapy. Cancer 1978;41:2107–11.
51. Piga A, Ambrosetti A, Todeschini G, et al. Doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) salvage of mechlorethamine, vincristine, prednisone, and procarbazine (MOPP)-resistant advanced Hodgkin’s disease. Cancer Treat Rep 1984;68:947–51.
52. Moskowitz AJ, Hamlin PA Jr, Perales MA, et al. Phase II study of bendamustine in relapsed and refractory Hodgkin lymphoma. J Clin Oncol 2013;31:456–60.
53. Anastasia A, Carlo-Stella C, Corradini P, et al. Bendamustine for relapsed/refractory classical Hodgkin lymphoma after high dose chemotherapy and or allogeneic transplant: a study of Fondazione Italiana Linfomi (FIL). Blood 2012;120:3652.
54. Martin A, Fernandez-Jimenez MC, Caballero MD, et al. Long-term follow-up in patients treated with Mini-BEAM as salvage therapy for relapsed or refractory Hodgkin’s disease. Br J Haematol 2001;113:161–71.
55. Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet 2002;359:2065–71.
56. Fisher RI, DeVita VT, Hubbard SP, et al. Prolonged disease-free survival in Hodgkin’s disease with MOPP reinduction after first relapse. Ann Intern Med 1979;90:761–3.
57. ChlVPP therapy for Hodgkin’s disease: experience of 960 patients. The International ChlVPP Treatment Group. Ann Oncol 1995;6:167–72.
58. Selby P, Patel P, Milan S, et al. ChlVPP combination chemotherapy for Hodgkin’s disease: long-term results. Br J Cancer 1990;62:279–85.
59. Santoro A, Bredenfeld H, Devizzi L, et al. Gemcitabine in the treatment of refractory Hodgkin’s disease: results of a multicenter phase II study. J Clin Oncol 2000;18:2615–9.
60. Little R, Wittes RE, Longo DL, Wilson WH. Vinblastine for recurrent Hodgkin’s disease following autologous bone marrow transplant. J Clin Oncol 1998;16:584–8.
61. Chen R, Palmer JM, Martin P, et al. Results of a multicenter phase II trial of brentuximab vedotin as second-line therapy before autologous transplantation in relapsed/refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2015;21:2136–40.
62. Moskowitz CH, Yahalom J, Zelenetz AD, et al. High-dose chemo-radiotherapy for relapsed or refractory Hodgkin lymphoma and the significance of pre-transplant functional imaging. Br J Haematol 2010;148:890–7.
63. Sureda A, Constans M, Iriondo A, et al. Prognostic factors affecting long-term outcome after stem cell transplantation in Hodgkin’s lymphoma autografted after a first relapse. Ann Oncol 2005;16:625–33.
64. Crocchiolo R, Canevari C, Assanelli A, et al. Pre-transplant 18FDG-PET predicts outcome in lymphoma patients treated with high-dose sequential chemotherapy followed by autologous stem cell transplantation. Leuk Lymphoma 2008;49:727–33.
65. Mocikova H, Pytlik R, Markova J, et al. Pre-transplant positron emission tomography in patients with relapsed Hodgkin lymphoma. Leuk Lymphoma 2011;52:1668–74.
66. Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol 2012;30:2183–9.
67. Gopal AK, Chen R, Smith SE, et al. Durable remissions in a pivotal phase 2 study of brentuximab vedotin in relapsed or refractory Hodgkin lymphoma. Blood 2015;125:1236–43.
68. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma.N Engl J Med 2015;372:311–9.
69. Chen R, Zinzani PL, Fanale MA, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 2017;35:2125–32.
70. Haverkos BM, Abbott D, Hamadani M, et al. PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood 2017;130:221–8.
71. Herbaux C, Gauthier J, Brice P, et al. Efficacy and tolerability of nivolumab after allogeneic transplantation for relapsed Hodgkin lymphoma. Blood 2017;129:2471–8.
72. Merryman RW, Kim HT, Zinzani PL et al. Safety and efficacy of allogeneic hematopoietic stem cell transplant after PD-1 blockage in relapsed/refractory lymphoma. Blood 2017;129:1380–8.
73. Linch DC, Winfield D, Goldstone AH, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet 1993;341:1051–4.
74. Yuen AR, Rosenberg SA, Hoppe RT, et al. Comparison between conventional salvage therapy and high-dose therapy with autografting for recurrent or refractory Hodgkin’s disease. Blood 1997;89:814–22.
75. Gutierrez-Delgado F, Holmberg L, Hooper H, et al. Autologous stem cell transplantation for Hodgkin’s disease: busulfan, melphalan and thiotepa compared to a radiation-based regimen. Bone Marrow Transplant 2003;32:279–85.
76. Hake CR, Graubert TA, Fenske TS. Does autologous transplantation directly increase the risk of secondary leukemia in lymphoma patients? Bone Marrow Transplant 2007;39:59–70.
77. Hahn T, McCarthy PL, Carreras J, et al. Comparison of prognostic models for autologous hematopoietic stem cell transplantation (AHCT) for relapsed Hodgkin lymphoma. Blood 2009;114:1215.
78. Chen Y-B, Lane AA, Logan BR, et al. Impact of conditioning regimen on outcomes for patients with lymphoma undergoing high-dose therapy with autologous hematopoietic cell transplantation. Biology Blood Marrow Transplant 2015;21:1046–53.
79. Wendland MM, Asch JD, Pulsipher MA, et al. The impact of involved field radiation therapy for patients receiving high-dose chemotherapy followed by hematopoietic progenitor cell transplant for the treatment of relapsed or refractory Hodgkin disease. Am J Clin Oncol 2006;29:189–95.
80. Biswas T, Culakova E, Friedberg JW, et al. Involved field radiation therapy following high dose chemotherapy and autologous stem cell transplant benefits local control and survival in refractory or recurrent Hodgkin lymphoma. Radiother Oncol 2012;103:367–72.
81. Levis M, Piva C, Filippi AR, et al. Potential benefit of involved-field radiotherapy for patients with Relapsed-refractory Hodgkin’s lymphoma with incomplete response before autologous stem cell transplantation. Clin Lymphoma Myeloma Leuk 2017;17:14–22.
82. Josting A, Franklin J, May M, et al. New prognostic score based on treatment outcome of patients with relapsed Hodgkin’s lymphoma registered in the database of the German Hodgkin’s lymphoma study group. J Clin Oncol 2002;20:221–30.
83. Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015;385:1853–62.
84. Bonfante V, Santoro A, Viviani S, et al. Outcome of patients with Hodgkin’s disease failing after primary MOPP-ABVD. J Clin Oncol 1997;15:528–34.
85. Josting A, Rueffer U, Franklin J, et al. Prognostic factors and treatment outcome in primary progressive Hodgkin lymphoma: a report from the German Hodgkin Lymphoma Study Group. Blood 2000;96:1280–6.
86. Josting A, Nogova L, Franklin J, et al. Salvage radiotherapy in patients with relapsed and refractory Hodgkin’s lymphoma: a retrospective analysis from the German Hodgkin Lymphoma Study Group. J Clin Oncol 2005;23:1522–9.
87. Campbell B WA, Milner A, Di Iulio J, et al. Long-term follow-up of salvage radiotherapy in Hodgkin’s lymphoma after chemotherapy failure. Int J Radiat Oncol Biol Phys 2005;63:1538–45.
88. Wirth A, Corry J, Laidlaw C, et al. Salvage radiotherapy for Hodgkin’s disease following chemotherapy failure. Int J Radiat Oncol Biol Phys 1997;39:599–607.
89. Schulz H, Rehwald U, Morschhauser F, et al. Rituximab in relapsed lymphocyte-predominant Hodgkin lymphoma: long-term results of a phase 2 trial by the German Hodgkin Lymphoma Study Group (GHSG). Blood 2008;111:109–11.
90. Ekstrand BC, Lucas JB, Horwitz SM, et al. Rituximab in lymphocyte-predominant Hodgkin disease: results of a phase 2 trial. Blood 2003;101:4285–9.
91. Younes A, Romaguera J, Hagemeister F, et al. A pilot study of rituximab in patients with recurrent, classic Hodgkin disease. Cancer 2003;98:310–4.
92. Rehwald U, Schulz H, Reiser M, et al. Treatment of relapsed CD20+ Hodgkin lymphoma with the monoclonal antibody rituximab is effective and well tolerated: results of a phase 2 trial of the German Hodgkin Lymphoma Study Group. Blood 2003;101:420–4.
93. Fehniger TA, Larson S, Trinkaus K, et al. A phase 2 multicenter study of lenalidomide in relapsed or refractory classical Hodgkin lymphoma. Blood 2011;118:5119–25.
94. Boll B, Borchmann P, Topp MS, et al. Lenalidomide in patients with refractory or multiple relapsed Hodgkin lymphoma. Br J Haematol 2010;148:480–2.
95. LaCasce AS, Bociek G, Sawas A, et al. Brentuximab vedotin plus bendamustine: a highly active salvage treatment regimen for patients with relapsed or refractory Hodgkin lymphoma. Blood 2015;126:3982.
96. Diefenbach CS, Hong F, David KA, et al. A phase I study with an expansion cohort of the combination of ipilimumab and nivolumab and brentuximab vedotin in patients with relapsed/refractory Hodgkin lymphoma: A trial of the ECOG-ACRIN Cancer Research Group (E4412 Arms D and E). Blood 2016;128:1106.
97. Herrera AF, Bartlett NL, Ramchandren R, et al. Preliminary results from a phase 1/2 study of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016;128:1105.
98. Johnston PB, Inwards DJ, Colgan JP, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol 2010;85:320–4.
99. Kewalramani T, Nimer SD, Zelenetz AD, et al. Progressive disease following autologous transplantation in patients with chemosensitive relapsed or primary refractory Hodgkin’s disease or aggressive non-Hodgkin’s lymphoma. Bone Marrow Transplant 2003;32:673–9.
100. Lin TS, Avalos BR, Penza SL, et al. Second autologous stem cell transplant for multiply relapsed Hodgkin’s disease. Bone Marrow Transplant 2002;29:763–7.
101. Smith SM, van Besien K, Carreras J, et al. Second autologous stem cell transplantation for relapsed lymphoma after a prior autologous transplant. Biol Blood Marrow Transplant 2008;14:904–12.
102. Gajewski JL, Phillips GL, Sobocinski KA, et al. Bone marrow transplants from HLA-identical siblings in advanced Hodgkin’s disease. J Clin Oncol 1996;14:572–8.
103. Peniket AJ, Ruiz de Elvira MC, Taghipour G, et al. An EBMT registry matched study of allogeneic stem cell transplants for lymphoma: allogeneic transplantation is associated with a lower relapse rate but a higher procedure-related mortality rate than autologous transplantation. Bone Marrow Transplant 2003;31:667–78.
104. Anderlini P, Saliba R, Acholonu S, et al. Fludarabine-melphalan as a preparative regimen for reduced-intensity conditioning allogeneic stem cell transplantation in relapsed and refractory Hodgkin’s lymphoma: the updated M.D. Anderson Cancer Center experience. Haematologica 2008;93:257–64.
105. Burroughs LM, O’Donnell PV, Sandmaier BM, et al. Comparison of outcomes of HLA-matched related, unrelated, or HLA-haploidentical related hematopoietic cell transplantation following nonmyeloablative conditioning for relapsed or refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2008;14:1279–87.
106. Peggs KS, Hunter A, Chopra R, et al. Clinical evidence of a graft-versus-Hodgkin’s-lymphoma effect after reduced-intensity allogeneic transplantation. Lancet 2005;365:1934–41.
107. Sureda A, Robinson S, Canals C, et al. Reduced-intensity conditioning compared with conventional allogeneic stem-cell transplantation in relapsed or refractory Hodgkin’s lymphoma: an analysis from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2008;26:455–62.
108. Devetten MP, Hari PN, Carreras J, et al. Unrelated donor reduced-intensity allogeneic hematopoietic stem cell transplantation for relapsed and refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:109–17.
109. Sureda A, Canals C, Arranz R, et al. Allogeneic stem cell transplantation after reduced intensity conditioning in patients with relapsed or refractory Hodgkin’s lymphoma. Results of the HDR-ALLO study - a prospective clinical trial by the Grupo Espanol de Linfomas/Trasplante de Medula Osea (GEL/TAMO) and the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. Haematologica 2012;97:310–7.
110. Sarina B, Castagna L, Farina L, et al. Allogeneic transplantation improves the overall and progression-free survival of Hodgkin lymphoma patients relapsing after autologous transplantation: a retrospective study based on the time of HLA typing and donor availability. Blood 2010;115:3671–7.
111. Castagna L, Sarina B, Todisco E, et al. Allogeneic stem cell transplantation compared with chemotherapy for poor-risk Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:432–8.
112. Thomson KJ, Peggs KS, Smith P, et al. Superiority of reduced-intensity allogeneic transplantation over conventional treatment for relapse of Hodgkin’s lymphoma following autologous stem cell transplantation. Bone Marrow Transplant 2008;41:765–70.
113. Chen R, Palmer JM, Thomas SH, et al. Brentuximab vedotin enables successful reduced-intensity allogeneic hematopoietic cell transplantation in patients with relapsed or refractory Hodgkin lymphoma. Blood 2012;119:6379–81.
INTRODUCTION
Hodgkin lymphoma, previously known as Hodgkin’s disease, is a B-cell lymphoproliferative disease characterized by a unique set of pathologic and epidemiologic features. The disease is characterized by the presence of multinucleate giant cells called Hodgkin Reed-Sternberg (HRS) cells.1 Hodgkin lymphoma is unique compared to other B-cell lymphomas because of the relative rarity of the malignant cells within affected tissues. The HRS cells, which usually account for only 0.1% to 10% of the cells, induce accumulation of nonmalignant lymphocytes, macrophages, granulocytes, eosinophils, plasma cells, and histiocytes, which then constitute the majority of tumor cellularity.2 Although the disease was first described by Sir Thomas Hodgkin in 1832, in part because of this unique histopathology, it was not until the 1990s that it was conclusively demonstrated that HRS cells are in fact monoclonal germinal center–derived B cells.
Due to the development of highly effective therapies for Hodgkin lymphoma, cure is a reasonable goal for most patients. Because of the high cure rate, late complications of therapy must be considered when selecting treatment. This article reviews the clinical features and treatment options for advanced stage and relapsed/refractory Hodgkin lymphoma. A previously published article reviewed the epidemiology, etiology/pathogenesis, pathologic classification, initial workup, and staging evaluation of Hodgkin lymphoma, as well as the prognostic stratification and treatment of patients with early-stage Hodgkin lymphoma.3
PRESENTATION, INITIAL EVALUATION, AND PROGNOSIS
Overall, classical Hodgkin lymphoma (cHL) usually presents with asymptomatic mediastinal or cervical lymphadenopathy. At least 50% of patients will have stage I or II disease.4 A mediastinal mass is seen in most patients with nodular sclerosis cHL, at times showing the characteristics of bulky (> 10 cm) disease. Constitutional, or B, symptoms (fever, night sweats, and weight loss) are present in approximately 25% of all patients with cHL, but 50% of advanced stage patients. Between 10% and 15% of patients will have extranodal disease, most commonly involving lung, bone, and liver. Lymphocyte-predominant Hodgkin lymphoma (LPHL) is a rare histological subtype of Hodgkin lymphoma that is differentiated from cHL by distinct clinicopathological features. The clinical course and treatment approach for LPHL are dependent upon the stage of disease. The clinicopathological features of LPHL are discussed in the early-stage Hodgkin lymphoma article.3
For the purposes of prognosis and selection of treatment, Hodgkin lymphoma is commonly classified as early stage favorable, early stage unfavorable, and advanced stage. For advanced stage Hodgkin lymphoma patients, prognosis can be defined using a tool commonly referred to as the International Prognostic Score (IPS). This index consists of 7 factors: male gender, age 45 years or older, stage IV disease, hemoglobin < 10.5 g/dL, white blood cell (WBC) count > 15,000/μL, lymphopenia (absolute lymphocyte count < 600 cells/μL or lymphocytes < 8% of WBC count), and serum albumin < 4 g/dL.5 In the original study by Hasenclever et al,5 the 5-year freedom from progression (FFP) ranged from 42% to 84% and the 5-year overall survival (OS) ranged from 56% to 90%, depending on the number of factors present. This scoring system, however, was developed using a patient population treated prior to 1992. Using a more recently treated patient population, the British Columbia Cancer Agency (BCCA) found that the IPS is still valid for prognostication, but outcomes have improved across all IPS groups, with 5-year FFP now ranging from 62% to 88% and 5-year OS ranging from 67% to 98%.6 This improvement is likely a reflection of improved therapy and supportive care. Table 1 shows the PFS and OS within each IPS group, comparing the data from the German Hodgkin Study Group (GHSG) and BCCA group.5,6
High expression of CD68 is associated with adverse outcomes, whereas high FOXP3 and CD20 expression on tumor cells are predictors of superior outcomes.8 A recent study found that CD68 expression was associated with OS. Five-year OS was 88% in those with less than 25% CD68 expression, versus 63% in those with greater than 25% CD68 expression.9
Roemer and colleagues evaluated 108 newly diagnosed cHL biopsy specimens and found that almost all cHL patients had concordant alteration of PD-L1 (programmed death ligand-1) and PD-L2 loci, with a spectrum of 9p24.1 alterations ranging from low level polysomy to near uniform 9p24.1 amplification. PD-L1/PD-L2 copy number alterations are therefore a defining pathobiological feature of cHL.10 PFS was significantly shorter for patients with 9p24.1 amplification, and those patients were likely to have advanced disease suggesting that 9p24.1 amplification is associated with less favorable prognosis.10 This may change with the increasing use of PD-1 inhibitors in the treatment of cHL.
High baseline metabolic tumor volume and total lesion glycolysis have also been associated with adverse outcomes in cHL. While not routinely assessed in practice currently, these tools may ultimately be used to assess prognosis and guide therapy in clinical practice.11
ADVANCED STAGE HODGKIN LYMPHOMA
FRONTLINE THERAPY
First-line Chemotherapy
Chemotherapy plays an essential role in the treatment of advanced stage Hodgkin lymphoma. In the 1960s, the MOPP regimen (nitrogen mustard, vincristine, procarbazine, prednisone) was developed, with a 10-year OS of 50% and a progression-free survival (PFS) of 52% reported in advanced stage patients. The complete remission (CR) rate was 81%, and 36% of patients who achieved CR relapsed later.12 This chemotherapy regimen is associated with a significant rate of myelosuppression and infertility as well as long-term risk of secondary myelodysplasia and acute leukemias.13,14 This led to the development of newer regimens such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine).15 In a randomized trial, ABVD showed improved failure-free survival (FFS) over MOPP (61% versus 50% at 5 years) but similar OS (66%–73%).16 In light of these findings, and considering the lower rate of infertility and myelotoxicity, ABVD became the standard of care for advanced stage cHL in the United States.
The Stanford V regimen was developed in an attempt to further minimize toxicity.17 Stanford V is a condensed, 12-week chemotherapy regimen that includes mechlorethamine, doxorubicin, vinblastine, etoposide, prednisone, vincristine, and bleomycin, followed by involved-field radiation therapy (IFRT). Subsequent trials compared the Stanford V and ABVD regimens and showed similar OS, freedom from treatment failure (FFTF), and response rates.18,19 The ABVD regimen was noted to have higher pulmonary toxicity, while other toxicities such as lymphopenia and neuropathy were higher with the Stanford V regimen. In addition, Stanford V requires patients to receive radiation therapy (RT) to original sites of disease larger than 5 cm in size and contiguous sites.
Another regimen which has been studied extensively for advanced stage Hodgkin lymphoma, and is considered a standard of care in some parts of the world, is escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone). In the HD9 study (n = 1196), the GHSG evaluated BEACOPP, escalated BEACOPP, and COPP/ABVD in advanced stage Hodgkin lymphoma.20 All arms of the study included 30 Gy RT to sites of bulky disease or residual disease. This study showed improved OS and FFTF with escalated BEACOPP, but at the cost of higher rates of toxicity. At 10 years, FFTF was 64%, 70%, and 82% with OS rates of 75%, 80%, and 86% for COPP/ABVD, baseline BEACOPP, and escalated BEACOPP, respectively (P < 0.001). The rate of secondary acute leukemia 10 years after treatment was 0.4% for COPP/ABVD, 1.5% for BEACOPP, and 3.0% for escalated BEACOPP. However, 3 subsequent randomized trials did not confirm a survival benefit with escalated BEACOPP relative to ABVD. In the HD 2000 trial (n = 295)21 and in a trial by Viviani and colleagues (n = 331),22 an improvement in OS was not demonstrated in favor of escalated BEACOPP. These studies also confirmed a higher rate of toxicities as well as secondary malignancies associated with the escalated BEACOPP regimen. In the EORTC20012 Intergroup trial (n = 549), 8 cycles of ABVD was compared with 4 cycles of escalated BEACOPP followed by 4 cycles of baseline BEACOPP, without radiation, in patients with clinical stage III or IV Hodgkin lymphoma with IPS score ≥ 3. Both regimens resulted in statistically similar FFS (63.7% in ABVD × 8 versus 69.3% in BEACOPP 4+4) and OS (86.7% in ABVD × 8 vs 90.3% in BEACOPP 4+4).23
In the United States, ABVD (6–8 cycles) is commonly used, although escalated BEACOPP (particularly for patients with an IPS of 4 or higher) and Stanford V are considered appropriate as well.24 In the North American Intergroup study comparing ABVD to Stanford V, and in the trial by Viviani et al, ABVD was associated with a 5- to 7-year FFS of 73% to 79% and OS of 84% to 92%.19,22 Given these excellent results, as well as the potential to cure patients with second-line therapy consisting of autologous hematopoietic cell transplantation (auto-HCT), the general consensus among most U.S. hematologists and oncologists is that ABVD remains the treatment of choice, and that the improved FFS/PFS with escalated BEACOPP is not outweighed by the additional toxicity associated with the regimen. There may, however, be a role for escalated BEACOPP in select patients who have a suboptimal response to ABVD as defined by interim positron emission tomography (iPET) scan (see below).
Brentuximab vedotin is an anti-CD30 antibody-drug conjugate (ADC) consisting of an anti-CD30 antibody linked to monomethyl auristatin E (MMAE), a potent antitubulin agent. CD30 is highly expressed on HRS cells and also in anaplastic large cell lymphoma. Upon binding to CD30, the ADC/CD30 complex is then internalized and directed to the lysosome, where the ADC is proteolytically cleaved, releasing MMAE from the antibody. MMAE then disrupts microtubule networks within the cell, leading to G2/M cycle arrest and apoptosis. CD30 is consistently expressed on HRS cells. In addition to being studied in the relapsed/refractory setting (described below), brentuximab has been studied in the first-line setting. In a phase 1 trial, brentuximab combined with ABVD was associated with increased pulmonary toxicity, while brentuximab + AVD had no significant pulmonary toxicity, with an excellent CR rate (96%), suggesting that substituting brentuximab for bleomycin may be an effective strategy. In addition to possibly being more efficacious, this strategy would also have the benefit of eliminating the risk of bleomycin pulmonary toxicity.25 Based on this data, a large international phase 3 study (the ECHELON-1 trial) comparing ABVD versus brentuximab + AVD in advanced stage cHL patients was recently completed. This study enrolled 1334 patients, and preliminary results were recently announced. With a median follow-up of 24 months, the brentuximab + AVD arm had a 4.9% absolute improvement in PFS relative to the ABVD arm (82.1% versus 77.2%). The brentuximab + AVD arm had an increased incidence of febrile neutropenia, managed with growth factors and peripheral neuropathy requiring dose adjustments, whereas the ABVD arm had an increased rate and severity of pulmonary toxicity.26 Further follow-up will be required to determine whether this will translate into a survival benefit. See Table 2 for a summary of recent large randomized prospective phase 3 trials in advanced stage Hodgkin lymphoma.
Alternative Regimens in Older Patients
Patients older than 60 years of age often have poor tolerance for ABVD and especially escalated BEACOPP. This results in increased treatment-related mortality and reduced overall dose intensity, with higher relapse rates and poor OS. In an attempt to improve on the results of treatment of elderly patients with Hodgkin lymphoma, alternative regimens have been explored. One example is PVAG (prednisone, vinblastine, doxorubicin, gemcitabine). With this regimen, the 3-year OS was 66% and PFS was 58%. One patient out of 59 died from treatment-related toxicity, which is much improved over the historical figures for elderly patients with Hodgkin lymphoma.27 Another commonly used approach in practice is to simply omit bleomycin from ABVD. In the early-stage setting (GHSG HD-13 trial), this regimen (referred to as AVD) led to 89.6% PFS at 5 years, compared to 93.5% with ABVD.28 It therefore stands to reason that this should be a reasonable option in older or more frail advanced stage cHL patients as well.
Brentuximab has been evaluated as a single-agent therapy for first-line therapy of elderly patients with Hodgkin lymphoma. In a phase 2 study, 27 patients (63% with advanced stage disease) were treated, with a 92% overall response rate and 73% CR rate. However the median duration of remission was disappointing at only 9.1 months.29 Based on this data, single-agent brentuximab appears to be a reasonable and well tolerated option for frail or elderly patients, although with the caveat that long-term disease control is relatively uncommon.
RESPONSE-ADAPTED FRONTLINE THERAPY USING INTERIM PET SCAN
In recent years, response-adapted treatment approaches have been extensively researched in cHL using iPET. The goal is to reduce toxicity by minimizing therapy in those who achieve negative iPET and/or to intensify treatment for patients with suboptimal response on iPET. Gallamini et al evaluated the prognostic role of an early iPET scan in advanced Hodgkin lymphoma patients (n = 190) treated with ABVD. This study found that patients with positive iPET had a 2-year PFS of 12.8% versus 95.0% in patients with negative iPET. This result was highly statistically significant (P < 0.0001). This study also showed that PET-2 (iPET after 2 cycles of ABVD) superseded the prognostic value of the IPS at diagnosis.30 As a result, numerous subsequent studies have been pursued using iPET for risk-adapted treatment in cHL.
A critical element to the conduct of iPET risk-adapted treatment for cHL is the interpretation of the iPET. In hopes of standardizing iPET interpretation in clinical trials, a scoring system called the Deauville score was developed. The Deauville score ranges from 1 to 5 (Table 3).
The SWOG (Southwest Oncology Group) S0816 trial (n = 358) evaluated iPET-adapted treatment after 2 cycles of ABVD in stage III or IV Hodgkin lymphoma patients. Patients with positive iPET (Deauville score 4 to 5; n = 60) received escalated BEACOPP for 6 cycles, whereas iPET-negative (Deauville score 1 to 3; n = 271) patients continued to receive 4 more cycles of ABVD. The 2-year PFS was 64% for iPET-positive patients.33 This PFS was much higher than the expected 15% to 30% from prior studies such as Gallamini et al,30 suggesting that the treatment intensification may have been of benefit.
In the HD0801 study (n = 519), newly diagnosed advanced Hodgkin lymphoma patients with positive iPET after 2 cycles of ABVD (n = 103) received early ifosfamide-containing salvage therapy followed by high-dose therapy with autologous stem cell rescue. The 2-year PFS was 76% for PET-2–positive patients, comparable with PET-2–negative patients who had PFS of 81%.34 Again, this result for iPET-positive patients was much better than expected based on the historical control from Gallamini et al, suggesting that the treatment intensification may have been beneficial. It should be emphasized, however, that neither HD0801 nor S0816 were randomized prospective trials; rather, all iPET-positive patients were assigned to an intensified treatment approach.
In the HD18 trial (n = 1100), patients with advanced stage cHL started therapy with escalated BEACOPP and underwent an iPET after 2 cycles. For those with a positive iPET, rituximab was added to escalated BEACOPP in the experimental arm (n = 220) for cycles 3 through 8. The control group (n = 220) continued to receive 6 more cycles of escalated BEACOPP. In the 2 groups, the 3-year PFS was similar (91.4% in escalated BEACOPP, 93% in rituximab + escalated BEACOPP), suggesting no significant benefit with addition of rituximab.35 This study also calls into question whether iPET provides useful information for patients receiving intensive therapy such as escalated BEACOPP, and indicates that the historical control data for iPET-positive patients from Gallamini et al may not be consistently reproduced in other prospective trials. As a result, nonrandomized trials that implement an iPET risk-adapted approach should be interpreted with caution. See Table 4 for a summary of recent trials in advanced stage Hodgkin lymphoma using iPET scan to guide therapy.
RADIATION THERAPY IN FRONTLINE TREATMENT
In patients with advanced stage Hodgkin lymphoma, IFRT to initial bulky sites of disease may be incorporated into frontline therapy to improve local control. However, whether this provides a survival benefit and which patients benefit most from consolidative RT remain unclear.
The European Organization for Research and Treatment of Cancer (EORTC) completed a randomized study in advanced stage Hodgkin lymphoma patients who achieved complete or partial remission after MOPP-ABV.36 Patients in CR were randomly assigned to receive no further treatment versus IFRT (24 Gy to all initially involved nodal areas and 16 to 24 Gy to all initially involved extranodal sites). Patients in partial remission (PR) were treated with 30 Gy to nodal areas and 18 to 24 Gy to extranodal sites. Among the CR patients, the 5-year event-free survival (EFS) was 79% to 84% and did not differ for those who received radiation versus those who did not. Five-year OS was 85% to 91% and also did not differ between the 2 groups. However, among the patients in PR after chemotherapy, the 5-year EFS was 79% and the 5-year OS was 87%, which is better than expected for PR patients, indicating a possible benefit to RT in patients with a partial response after chemotherapy. In the GHSG HD12 trial, patients with advanced stage Hodgkin lymphoma who had a residual lesion by computed tomography (CT) (but not analyzed by PET) had a very subtle improvement in FFTF (90% versus 87%) in favor of consolidation with IFRT, but again no survival benefit was seen.37
The EORTC and HD12 studies described above utilized CT scan for assigning remission status following chemotherapy, and it is now well known that many patients with residual masses (by CT) after chemotherapy may in fact be cured, as such residual radiographic abnormalities may simply be composed of fibrosis. PET scan is more accurate than CT in identifying patients who truly have residual active disease following chemotherapy. As a result, the EORTC study discussed above and the GHSG HD12 trial are of limited relevance in the modern era, in which patients routinely undergo PET scan at the end of therapy. Restricting IFRT to sites that remain PET-positive after completing chemotherapy may be a reasonable strategy that would allow for the avoidance of RT in many patients, and may obviate the need for aggressive second-line therapy (eg, high-dose therapy and autologous hematopoietic cell transplant [auto-HCT]). This approach was taken in the GHSG HD15 trial (n = 2182) in which advanced stage patients were treated with 3 variations on the BEACOPP regimen (8 cycles of escalated BEACOPP, 6 cycles of escalated BEACOPP, or 8 cycles of baseline BEACOPP, randomized in a 1:1:1 ratio). Patients with a residual mass of 2.5 cm or greater on CT scan then underwent a PET scan; if the lesion was PET positive, it was treated with 30 Gy of IFRT. This overall strategy was very effective, with 5-year FFTF rates of 84.4%, 89.3%, and 85.4%, respectively. The OS rates were 91.9%, 95.3%, and 94.5%, respectively. For patients with lesions that remained PET positive after chemotherapy, the PFS rate was 86.2% at 48 months, whereas patients in PR with persistent mass ≥ 2.5 cm but with negative PET had a PFS of 92.6%, similar to that of patients in CR.38 With this approach of BEACOPP followed by PET-guided radiation, the proportion of patients receiving RT was reduced from 71% (in the HD9 study) to only 11% in the HD15 study,38 with no apparent loss in overall efficacy when comparing the results of the 2 studies.
UPFRONT STEM CELL TRANSPLANTATION
To further improve outcomes of patients with advanced Hodgkin lymphoma with high-risk disease, high-dose therapy with auto-HCT has been explored as part of frontline therapy. While this has been shown to be feasible in such patients,39 randomized trials have not shown a clear benefit in terms of FFS or OS with upfront auto-HCT. 40,41 Therefore, auto-HCT is not considered a standard component of frontline therapy for cHL patients who achieve CR by PET/CT scan.
RELAPSED AND REFRACTORY HODGKIN LYMPHOMA
Depending on the stage, risk factors, and frontline regimen utilized, between 5% and 40% of patients with Hodgkin lymphoma can be expected to experience either primary induction failure or a relapse after attaining remission with frontline therapy.3 Primary refractory Hodgkin lymphoma, which occurs in up to 5% to 10% of patients, is defined as progression or no response during induction treatment or within 90 days of completing treatment. In cases where remission status is in question, an updated tissue biopsy is recommended. Biopsy is also recommended in cases in which new sites of disease have appeared or if relapse has occurred after a durable period of remission. Restaging is recommended at the time of relapse.
For younger patients with relapsed/refractory Hodgkin lymphoma, the standard of care in most cases is second-line (or salvage) chemotherapy followed by high-dose therapy and auto-HCT. For patients not felt to be candidates for auto-HCT, options include conventional second-line chemotherapy alone, salvage radiotherapy, novel agents such as brentuximab or immune checkpoint inhibitors, and/or participation in clinical trials.
CONVENTIONAL MULTI-AGENT CHEMOTHERAPY REGIMENS
Numerous conventional regimens have been shown in phase 2 studies to be active in relapsed and refractory Hodgkin lymphoma. These include platinum-based regimens, gemcitabine-based regimens, and alkylator-based regimens. No randomized trials in Hodgkin lymphoma have been conducted comparing these regimens. In general, regimens are chosen based on the patient’s age, performance status, comorbidities, and whether auto-HCT is being considered.
In the United States, platinum-based regimens such as ICE (ifosfamide, carboplatin, etoposide),42 DHAP (dexamethasone, cisplatin, high-dose cytarabine),43 ESHAP (etoposide, methylprednisolone, high-dose cytarabine, cisplatin),44 GDP (gemcitabine, cisplatin, dexamethasone),45 and GCD (gemcitabine, carboplatin, dexamethasone)46 are all considered appropriate second-line therapy options for patients being considered for auto-HCT, due to their high response rates and because autologous hematopoietic stem cell collection remains feasible after these regimens. Response rates range from 60% to 88%, with CR rates between 17% and 41%, and toxic death rates generally well below 5%.
Other gemcitabine-based regimens such as IGEV (ifosfamide, gemcitabine, vinorelbine) and GVD (gemcitabine, vinorelbine, liposomal doxorubicin) are also effective.47,48 GVD is an excellent choice since it is a generally well-tolerated outpatient regimen with a 60% response rate even in heavily pretreated patients.48 Stem cell collection remains feasible after both IGEV and GVD as well. ABVD can produce CR in approximately 20% to 50% of patients initially treated with MOPP.49–51 In practice, however, most patients today with relapsed or refractory Hodgkin lymphoma have already received ABVD as part of their first-line therapy, and retreatment with ABVD is not a good option because it would be associated with prohibitively high cumulative doses of doxorubicin.
These multi-agent chemotherapy regimens may not be tolerated well in patients over age 65 to 70 years or those with significant underlying comorbidities. In recent years, bendamustine has emerged as one of the most active conventional agents for cHL, with overall response rates of 53% to 58% in heavily pre-treated patients.52,53 Bendamustine can generally be tolerated even in elderly patients as well.
Some centers, particularly in Europe, investigated aggressive salvage regimens such as mini-BEAM (carmustine, etoposide, cytarabine, melphalan)54 or dexa-BEAM (BEAM plus dexamethasone).55 These regimens, however, are associated with significant hematologic toxicity and high (2%–5%) treatment-related mortality. As a result, these are rarely used in the United States.
For patients who have progressed after (or are not candidates for) platinum- and/or gemcitabine-based therapy, older alkylator-based regimens such as MOPP, C-MOPP, or ChlVPP (chlorambucil, vinblastine, procarbazine, prednisone) can be considered.56–58 However, these regimens are associated with significant bone marrow suppression, and autologous hematopoietic stem cell collection may no longer be feasible after such regimens. Therefore, these regimens should only be given to patients not felt to be auto-HCT candidates, or patients for whom autologous hematopoietic stem cell collection has already been completed. Weekly vinblastine or single-agent gemcitabine are palliative chemotherapy options, with response rates in the 60% to 80% range. Patients can sometimes be maintained on such low-intensity palliative regimens for 6 to 12 months or longer.59,60
BRENTUXIMAB VEDOTIN
Several trials are evaluating incorporation of brentuximab into second-line therapy in transplant-eligible patients. These approaches have used brentuximab prior to, concurrent with, or following platinum-based chemotherapy.61 While there is currently no consensus on the optimal way to incorporate brentuximab into salvage therapy, it is possible that the use of brentuximab or other novel agents in salvage therapy may allow for avoidance of conventional chemotherapy in some patients. In addition, this may translate into more patients proceeding to auto-HCT in a PET negative state. PET negativity prior to auto-HCT is a powerful predictor of long-term remission after auto-HCT, so any intervention that increases the rate of PET negativity prior to auto-HCT would be expected to improve outcomes with auto-HCT.62–65
For patients not being considered for autoHCT, or those for whom platinum-based salvage therapy was ineffective, single-agent brentuximab is an excellent option. In 2 phase 2 studies, an overall response rate (ORR) of 60% to 75% (including a CR rate of 22%–34%) was seen in relapsed and refractory Hodgkin lymphoma patients.66 The US Food and Drug Administration (FDA) approved brentuximab vedotin in August 2011 for treatment of relapsed and refractory Hodgkin lymphoma, after a failed auto-HCT, or in patients who are not auto-HCT candidates and who have received at least 2 prior chemotherapy regimens. With more extended follow-up, it has become clear that a proportion of patients who achieve CR to brentuximab may maintain remission long-term—58% at 3 years and 38% at 5 years.67 These patients may in fact be cured, in many cases without having undergone allogeneic HCT (allo-HCT) after brentuximab.
PD-1 (IMMUNE CHECKPOINT) INHIBITORS
As discussed earlier, PD-L1/PD-L2 copy number alterations represent a disease-defining feature of cHL. Alterations in chromosome 9p24.1 increase the expression of PD-1 ligands PD-L1 and PD-L2. Nivolumab and pembrolizumab are PD-1-blocking antibodies, which have recently been FDA approved for relapsed and refractory cHL. In a study with 23 patients, with 78% of them relapsing after auto-HCT and 78% relapsing after brentuximab, nivolumab produced an objective response in 87% of the patients, with 17% achieving CR and 70% achieving PR. The rate of PFS was 86% at 24 weeks.68 Pembrolizumab, another PD-1 antagonist, was also tested in relapsed and refractory Hodgkin lymphoma. In the KEYNOTE-087 study (n = 210), pembrolizumab produced an ORR of 64% to 70% in 3 different cohorts of relapsed and refractory cHL patients. Overall CR rate was 22%.69 In general, these agents are well tolerated, although patients must be monitored closely for
inflammatory/autoimmune-type toxicities including skin rash, diarrhea/colitis, transaminitis, endocrine abnormalities, and pneumonitis. Prompt recognition and initiation of corticosteroids is essential in managing these toxicities. Of note, PD-1 inhibitors should be given very cautiously to patients with a prior history of allo-HCT, since 30% to 55% of such patients will experience acute graft-versus-host disease (GVHD) in this setting. In 2 retrospective studies, the response rate was very high at 77% to 95%; however, 10% to 26% of all patients treated with PD-1 inhibitors post-allo-HCT died from GVHD induced by PD-1 inhibition.70,71 These risks and benefits therefore need to be carefully weighed in the post-allo-HCT setting. In another recent study, the outcomes were reported for 39 patients who underwent allo-HCT after prior therapy with a PD-1 inhibitor. Three patients (7.7%) developed lethal acute GVHD, suggesting there may be an increased risk of GVHD in patients undergoing allo-HCT after prior PD-1 inhibitor therapy.72
AUTOLOGOUS STEM CELL TRANSPLANTATION
Several studies have shown an improved disease-free survival (DFS) or FFS in patients with relapsed cHL treated by auto-HCT as compared to those receiving conventional chemotherapy alone.55,73,74 Overall, for relapsed disease, one can expect an approximately 50% to 60% chance for DFS at 5 years post-transplant. In a retrospective, matched-pair analysis, FFP was 62% for auto-HCT patients, compared to 32% for conventional chemotherapy patients. OS, however, was similar for the 2 groups (47%–54%). Patients failing induction therapy or relapsing within 1 year were seen to benefit the most from auto-HCT, including an OS benefit.74
A European prospective randomized trial was conducted comparing conventional salvage therapy to auto-HCT. In this study, 161 patients with relapsed Hodgkin lymphoma were treated with 2 cycles of dexa-BEAM. Those with chemo-sensitive disease were then randomized to either 2 more cycles of dexa-BEAM or high-dose BEAM with auto-HCT. Auto-HCT was associated with an approximately 55% FFTF at 3 years, versus 34% with conventional chemotherapy alone.55 This benefit again was most apparent for patients relapsing within 1 year of completion of primary therapy, although an OS benefit was not seen with auto-HCT. For patients with late relapse (>1 year after completion of primary therapy), auto-HCT was associated with an approximately 75% FFTF at 3 years, versus 50% with chemotherapy alone. One other small randomized trial of auto-HCT in relapsed and refractory Hodgkin lymphoma also showed an improved 3-year EFS in favor of auto-HCT (53% versus 10%), again with no difference in OS.73
The lack of OS benefit seen in these studies suggests that auto-HCT at first or second relapse provides comparable outcomes. Auto-HCT offers the benefit of avoiding the long-term toxicities associated with multiple salvage regimens and the anxiety associated with multiple relapses. In addition, the treatment-related mortality with auto-HCT is now in the 1% to 2% range in younger patients, at centers that perform the procedure routinely. For all of these reasons, auto-HCT is commonly recommended by physicians for Hodgkin lymphoma patients in first or second relapse. In most cases, transplant is favored in first relapse, since waiting until second relapse may be associated with a lower chance of achieving CR and difficulty collecting sufficient hematopoietic stem cells. For patients with early relapse or primary refractory disease, an even stronger case can be made for auto-HCT as the best option to achieve sustained control of the disease. For patients with late relapse, conventional salvage therapy alone may be a reasonable option, particularly in older or frail patients, or those with significant comorbid conditions.
The optimal conditioning regimen for autoHCT for relapsed and refractory Hodgkin lymphoma remains undefined. No randomized trials have been performed comparing conditioning regimens for relapsed and refractory Hodgkin lymphoma. One retrospective study compared 92 patients with Hodgkin lymphoma who underwent auto-HCT using a total-body irradiation (TBI) regimen versus a chemotherapy-alone regimen. No difference in 5-year OS or EFS was seen.75 Given the lack of benefit seen with TBI, along with reports of increased rates of secondary malignancies and myelodysplasia with TBI,76 chemotherapy-alone conditioning regimens are most widely employed. For example, in the United States, either the BEAM or CBV (cyclophosphamide, carmustine, etoposide) regimens are used in over 80% of cases.77 This practice was justified in a Center for International Blood and Marrow Transplant Research (CIBMTR) retrospective study comparing outcomes by conditioning regimens, in which no regimen performed better than BEAM or CBV.78
IFRT is often given as an adjunctive therapy to sites of initial and/or relapsed disease following auto-HCT. Although a relatively common practice, whether this truly enhances outcomes beyond that obtained with auto-HCT alone is unclear. Two retrospective studies have shown some benefit in terms of improvement in OS at 3 to 5 years in the group that received IFRT (70%–73% versus 40%–56%).79,80 Given the retrospective nature and small size of these studies, a prospective study would be needed to properly define the potential role for IFRT following auto-HCT in relapsed/refractory Hodgkin lymphoma. Another retrospective study (n = 73) that evaluated peri-transplant IFRT in Hodgkin lymphoma patients receiving auto transplant found no improvement in survival for patients who received peri-transplant IFRT. This study, however, did show a survival benefit in the subgroup of patients with limited stage disease.81
Prognostic Factors Associated with Outcome with Auto-HCT
The factor most consistently associated with improved outcome for patients with relapsed and refractory Hodgkin lymphoma who undergo auto-HCT is the disease status at transplant.63,77 Those in a second CR, versus a chemo-sensitive relapse (but not CR), versus a chemo-refractory relapse have DFS rates of 60% to 70%, 30% to 40%, and 10% to 20%, respectively.63 The duration between remission and relapse also has important prognostic significance. Late relapse (> 1 year after completion of frontline therapy) is associated with better outcomes as compared to early relapse.55 Other factors with prognostic significance at relapse include anemia, time to relapse and clinical stage, B symptoms, extranodal disease, number of prior chemotherapy regimens, and performance status.42,82 The prognostic impact of pretransplant disease status has been confirmed by studies using functional imaging (eg, FDG-PET or gallium scans). In a report by Moskowitz et al, patients with negative functional imaging following second-line therapy had a 77% EFS post-auto-HCT versus 33% in those whose functional imaging remained positive.62 Very similar findings have been reported by other groups.63–65
Post-Auto-HCT Brentuximab Maintenance
In the multicenter, randomized, double-blinded phase 3 AETHERA trial (n = 329), brentuximab (n = 165) was compared with placebo (n = 164) in patients with unfavorable risk relapsed or primary refractory cHL who had undergone autologous transplant. Eligible patients had at least 1 of the following risk factors for progression after auto-HCT: primary refractory Hodgkin lymphoma (failure to achieve complete remission), relapsed Hodgkin lymphoma with an initial remission duration of less than 12 months, or extranodal involvement at the start of pre-transplantation salvage chemotherapy. Patients were required to have CR, PR, or stable disease after pretransplant salvage chemotherapy with adequate kidney, liver, and bone marrow function. Patients who previously received brentuximab were excluded. Patients received 16 cycles of brentuximab or placebo once every 3 weeks starting 30 to 45 days after transplant. The PFS was significantly improved in the brentuximab group when compared to the placebo group (hazard ratio 0.57; P = 0.0013) after a median observation time of 30 months. Median PFS was 42.9 months in the brentuximab group versus 24.1 months in the placebo group; estimated 2-year PFS rates were 63% in the brentuximab group and 51% in the placebo group. OS was not significantly different between the study groups (~85%), presumably due to the fact that patients in the control group who relapsed likely went on to receive brentuximab as a subsequent therapy.83
PRIMARY REFRACTORY HODGKIN LYMPHOMA
Patients with primary refractory Hodgkin lymphoma have a poor outcome. Salvage therapy using conventional chemotherapy and/or RT results in long-term DFS in 10% or fewer of such patients.13,84 Given these poor outcomes with conventional salvage therapy, auto-HCT is considered to be the standard of care for this subset of patients. The GHSG retrospectively analyzed the prognostic factors and outcomes of patients with primary refractory Hodgkin lymphoma. The 5-year freedom-from-second-failure and the 5-year OS were reported to be 31% and 43%, respectively, for those patients treated with auto-HCT. Patients with poor functional status at time of transplant, age greater than 50 years, and failure to attain a temporary remission had a 0% 5-year OS, as compared to 55% in patients without any of these risk factors.85 A large retrospective European study showed that patients with chemo-resistant disease who underwent transplant had a 19% survival at 5 years.63 Hence, even patients with primary refractory Hodgkin lymphoma have some chance of achieving long-term survival following auto-HCT.
SALVAGE RADIOTHERAPY
The GHSG performed a retrospective analysis of the efficacy of salvage RT in patients with refractory or first-relapsed Hodgkin lymphoma. Five-year FFTF and OS rates were 28% and 51%, respectively. Patients with a limited-stage relapse and without B symptoms were more likely to benefit from salvage RT.86 Campbell et al reported on 81 patients undergoing salvage RT for persistent or recurrent Hodgkin lymphoma after chemotherapy. The 10-year FFTF and OS rates were 33% and 46%, respectively.87 Similarly, Wirth et al reported a 5-year FFS of 26% and 5-year OS of 57%. These figures were 36% and 75%, respectively, in patients whose relapse was limited to supradiaphragmatic nodal sites without B symptoms.88 RT therefore may be a useful strategy for a subset of patients who relapse following chemotherapy, particularly those with a limited-stage relapse, without B symptoms, and those with relapsed disease after a CR, as opposed to those with a partial response or lack of response to the prior chemotherapy regimen.
INVESTIGATIONAL AGENTS AND NOVEL COMBINATIONS
Several biological therapies are emerging as options for the treatment of refractory or relapsed disease. These therapies consist of monoclonal antibodies and ADCs that target cell surface antigens, or small molecules that inhibit key intracellular pathways within neoplastic cells.
Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody used widely in B-cell non-Hodgkin lymphomas. The CD20 molecule is typically highly expressed in nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL). Two studies (one in relapsed patients, the other in a mixture of relapsed and previously untreated patients) showed significant activity of rituximab in relapsed NLPHL, with ORRs ranging from 94% to 100%, CR rates ranging from 41% to 53%, and median duration of remission in the 10- to 33-month range.89,90 In cHL, CD20 is expressed in HRS cells in 20% to 30% of cases. In such cases, single-agent rituximab has also shown activity. There is also evidence that rituximab may be effective even in cases in which the HRS cells are CD20-negative, presumably by virtue of depleting reactive B lymphocytes from the microenvironment, which may enhance anti-tumor immunity, or by eliminating a putative CD20-expressing Hodgkin lymphoma stem cell.91,92
Lenalidomide
Lenalidomide is an immunomodulatory drug that has multiple modes of action, including direct induction of apoptosis in tumor cells, antiangiogenic effects, and the activation of immune cells, such as natural killer cells and T cells. Lenalidomide has been shown to modify many features of the microenvironment of HRS cells and has demonstrated activity in other B-cell neoplasms. As a result, lenalidomide has been evaluated in relapsed and refractory Hodgkin lymphoma patients. A multicenter phase 2 study by Fehniger et al included 35 patients, 87% of whom had previously undergone HCT and 55% of whom were refractory to the last therapy.93 All patients were given lenalidomide 25 mg/day from days 1 to 21 of a 28-day cycle until disease progression. One patient was noted to achieve CR, 6 achieved PR, and 5 had stable disease lasting more than 6 months, for an ORR of 19% and a “cytostatic overall response rate” of 33%. The median duration of CR/partial remission was 6 months, with the median time-to-treatment failure in responders (including those with stable disease > 6 months) being 15 months. Similarly, in another study, Böll et al evaluated 12 patients across 4 German centers with relapsed or refractory disease who were treated with oral lenalidomide for 21 days in a 28-day cycle. No radiological evidence of disease progression after 2 cycles of lenalidomide was seen in any of the enrolled patients. ORR was noted to be 50%, with 6 patients with stable disease and 5 patients achieving PR after 2 cycles.94
Novel Brentuximab Combination Therapies
Brentuximab plus bendamustine. The combination of brentuximab and bendamustine was tested as an outpatient regimen in a phase 1/2 study (n = 55) in primary refractory Hodgkin lymphoma or after first relapse. The CR rate of the combination was 74%, with an overall objective response (CR + PR) of 93%. The CR rates were 64% and 84%, respectively, for refractory and relapsed patients. The PFS at 12 months was 80%, establishing this combination therapy as an effective salvage regimen with durable response.95
Brentuximab plus nivolumab. Preliminary results have recently been presented from 2 studies96,97 evaluating the combination of brentuximab and nivolumab. While this combination would still be considered investigational, these studies showed very encouraging ORRs of 90% to 100% and a CR rate of 62% to 66%. Longer follow-up is needed to determine whether these responses are durable and to document the toxicity profile of this combination.
Mammalian Target of Rapamycin Inhibitors
Two mammalian target of rapamycin (mTOR) inhibitors, everolimus and temsirolimus, are currently available in the United States. While neither drug currently has FDA approval for Hodgkin lymphoma, everolimus was evaluated in a phase 2 trial in a heavily pretreated group of relapsed/refractory patients. An ORR of 47% was seen, with a median time to progression of 7.2 months.98
ALLOGENEIC STEM CELL TRANSPLANTATION
Historically, patients who relapse after having an auto-HCT generally had a poor outcome, with a median survival of 2 to 3 years after failure of auto-HCT.99 These patients may be offered palliative chemotherapy (see above), treatment with novel agents (see above), or enrollment in a clinical trial. Select patients may benefit from a second hematopoietic stem cell transplant, most commonly an allo-HCT. However, rare patients with late relapse after auto-HCT may be considered for a second auto-HCT, with a minority of such patients achieving a durable remission after the second auto-HCT.100,101 Because relapse or progressive disease occurs most commonly in the first several months following auto-HCT, patients are more often considered for allo-HCT than a second auto-HCT. In addition, a second auto-HCT may not be feasible due to impaired bone marrow reserve and/or concerns for development of secondary myelodysplasia or acute myeloid leukemia.
Several studies have evaluated allo-HCT in relapsed/ refractory Hodgkin lymphoma. Early studies evaluating myeloablative allo-HCT for Hodgkin lymphoma showed excessive treatment-related mortality (up to 50%) and disappointingly low rates of long-term survival (< 25%).102,103 This was likely related to the fact that, in that era, most of the patients with Hodgkin lymphoma evaluated for allo-HCT were heavily pretreated and therefore at a higher risk for toxicity as well as lymphoma progression.
More recently, several studies have focused on the use of reduced-intensity conditioning (RIC) allo-HCT for relapsed and refractory Hodgkin lymphoma. This approach relies more on a “graft-versus-lymphoma” effect, the existence of which has been debated in Hodgkin lymphoma. Three single-center studies of RIC allo-HCT in patients with multiply recurrent Hodgkin lymphoma showed improved rates of treatment-related mortality (8%–16%) but still relatively low rates of long-term PFS (23%–39% at 2 to 4 years).104–106 Interestingly, in one of these studies the outcomes were more favorable for patients who underwent haploidentical (versus matched sibling or matched unrelated donor) transplants.105
Two large registry studies have also reported on the outcomes of RIC allo-HCT in patients with relapsed and refractory Hodgkin lymphoma.107,108 These studies also confirmed a modest improvement in outcomes compared with those seen historically with myeloablative transplants. Treatment-related mortality at 1 to 2 years was 23% to 33%, depending on whether a matched sibling donor versus an unrelated donor was used. However, long-term PFS (18%–20% at 2 to 5 years) and OS (28%–37% at 2 to 5 years) remained poor, primarily due to high rates of progressive lymphoma post-transplant. In both of these studies, patients were heavily pretreated (84%–96% had received 3 or more prior lines of chemotherapy, and 62%–89% received a prior auto-HCT), with 47% to 55% of patients chemo-resistant prior to transplant. Of note, both of these registry studies reflect patients who underwent transplant prior to the widespread use of brentuximab and PD-1 inhibitors.
Based on the single-center and registry data above, a prospective multicenter European phase 2 trial was conducted to evaluate the benefit of RIC allo-HCT in Hodgkin lymphoma.109 Ninety-two patients (86% with prior auto-HCT, 90% with 3 or more prior lines of therapy) were enrolled and given salvage therapy. Those who had stable disease or better following salvage therapy remained on protocol (n = 78) and underwent RIC with fludarabine and melphalan, followed by allo-HCT (70% with matched sibling donors). Treatment-related mortality was 15% at 1 year. Relapse or progression occurred in 49% at 2 years (35% if chemo-sensitive prior to transplant). Chronic GVHD was associated with a decreased rate of relapse, supporting the existence of a graft-versus-lymphoma effect in Hodgkin lymphoma. Unfortunately, PFS among all allografted patients was still relatively poor (24% at 4 years). However, among patients in CR prior to allo-HCT, a 50% PFS was seen at 4 years. Therefore, even in a prospective multicenter study, RIC allo-HCT offered significant benefit with manageable toxicity in relapsed and refractory Hodgkin lymphoma patients with chemo-sensitive disease.
These studies suggest that outcomes with allo-HCT would improve further if implemented earlier in the course of disease and/or with a lower burden of disease at transplant. It has therefore been suggested that allo-HCT should be considered soon after failure of auto-HCT is documented. In a retrospective study by Sarina et al, 185 Hodgkin lymphoma patients who relapsed following auto-HCT were then immediately considered for reduced-intensity allo-HCT.110 Of these, 122 had a donor identified, and 104 (85%) actually underwent allo-HCT. These 104 patients were then compared to the other 81 patients who either had no donor identified or had a donor but did not receive the planned allo-HCT. Two-year PFS and OS were superior in the patients undergoing allo-HCT (39% versus 14% and 66% versus 42%, respectively, P < 0.001), with a median follow-up of 4 years. The presence of chronic GVHD again was associated with improved PFS and OS. Disease status prior to transplant remained highly predictive of PFS and OS by multivariate analysis. Two other smaller retrospective studies similarly found a survival benefit associated with allo-HCT compared with patients who underwent conventional salvage therapies alone.111,112 These studies, although subject to the usual limitations of retrospective analyses, suggest that the results with reduced-intensity allo-HCT are in fact enhanced if applied earlier in the disease course, and are superior to those with conventional therapy alone.
Currently, the exact role of allo-HSCT, including the optimal timing and optimal donor source (matched sibling versus haploidentical sibling versus matched unrelated donor), remain undefined for relapsed and refractory Hodgkin lymphoma. As discussed earlier, brentuximab is highly active in relapsed Hodgkin lymphoma patients, with a subset of patients still in CR at 5 years.67 For such patients, avoiding the risks of allo-HCT is a desirable goal.
For those who relapse or progress after auto-HCT, a reasonable strategy therefore is to treat initially with brentuximab, unless the patient is already known to have responded poorly to brentuximab, or already has significant neuropathy. Those who achieve a CR to brentuximab are then observed. A subset of those patients will remain in remission at 5 years without further therapy. For those who relapse, or who achieve less than a CR to brentuximab, additional treatment (with brentuximab re-treatment being one option) followed by a reduced-intensity allo-HCT is a reasonable consideration. This approach has the theoretical advantages of (1) avoiding the risk of allo-HCT in the subset potentially cured by brentuximab, (2) getting patients to allo-HCT with fewer comorbidities (due to a lower total exposure to conventional chemotherapy pre-transplant), and (3) applying allo-HCT in the setting of sensitive disease/lower disease burden (due to the high efficacy of brentuximab). The results of a small study suggest that brentuximab may in fact be a very effective “bridge” to allotransplant. Chen et al113 reported on 18 patients with relapsed/refractory Hodgkin lymphoma (17 of whom had previously undergone auto-HCT) who were treated on brentuximab vedotin clinical trials. The data were retrospectively evaluated to determine the efficacy and safety of subsequent reduced-intensity allo-HCT. Remarkably, at 1 year the OS was 100%, PFS was 92%, and nonrelapse mortality was 0% with a median follow-up of 14 months. Hence, brentuximab is safe for use prior to reduced-intensity allo-HCT in heavily pre-treated patients and appears to be associated with very favorable post-transplant outcomes, particularly in comparison to older studies of allo-HCT in the era prior to brentuximab.
SUMMARY
Currently, cure is possible for the majority of patients diagnosed with advanced stage Hodgkin lymphoma. The challenge to the clinician is to provide curative treatment with the lowest risk of serious toxicities. Which regimen will best provide this balance of risk and benefit needs to be assessed based on the relapse risk, age, frailty, and comorbidity profile for an individual patient. For many patients with relapsed or refractory Hodgkin lymphoma, cure remains possible using approaches based on hematopoietic stem cell transplantation, RT, and/or brentuximab. In addition, there are now numerous conventional chemotherapy agents, RT strategies, and exciting newer agents such as PD-1 inhibitors, that can provide significant clinical benefit even when cure is not feasible.
INTRODUCTION
Hodgkin lymphoma, previously known as Hodgkin’s disease, is a B-cell lymphoproliferative disease characterized by a unique set of pathologic and epidemiologic features. The disease is characterized by the presence of multinucleate giant cells called Hodgkin Reed-Sternberg (HRS) cells.1 Hodgkin lymphoma is unique compared to other B-cell lymphomas because of the relative rarity of the malignant cells within affected tissues. The HRS cells, which usually account for only 0.1% to 10% of the cells, induce accumulation of nonmalignant lymphocytes, macrophages, granulocytes, eosinophils, plasma cells, and histiocytes, which then constitute the majority of tumor cellularity.2 Although the disease was first described by Sir Thomas Hodgkin in 1832, in part because of this unique histopathology, it was not until the 1990s that it was conclusively demonstrated that HRS cells are in fact monoclonal germinal center–derived B cells.
Due to the development of highly effective therapies for Hodgkin lymphoma, cure is a reasonable goal for most patients. Because of the high cure rate, late complications of therapy must be considered when selecting treatment. This article reviews the clinical features and treatment options for advanced stage and relapsed/refractory Hodgkin lymphoma. A previously published article reviewed the epidemiology, etiology/pathogenesis, pathologic classification, initial workup, and staging evaluation of Hodgkin lymphoma, as well as the prognostic stratification and treatment of patients with early-stage Hodgkin lymphoma.3
PRESENTATION, INITIAL EVALUATION, AND PROGNOSIS
Overall, classical Hodgkin lymphoma (cHL) usually presents with asymptomatic mediastinal or cervical lymphadenopathy. At least 50% of patients will have stage I or II disease.4 A mediastinal mass is seen in most patients with nodular sclerosis cHL, at times showing the characteristics of bulky (> 10 cm) disease. Constitutional, or B, symptoms (fever, night sweats, and weight loss) are present in approximately 25% of all patients with cHL, but 50% of advanced stage patients. Between 10% and 15% of patients will have extranodal disease, most commonly involving lung, bone, and liver. Lymphocyte-predominant Hodgkin lymphoma (LPHL) is a rare histological subtype of Hodgkin lymphoma that is differentiated from cHL by distinct clinicopathological features. The clinical course and treatment approach for LPHL are dependent upon the stage of disease. The clinicopathological features of LPHL are discussed in the early-stage Hodgkin lymphoma article.3
For the purposes of prognosis and selection of treatment, Hodgkin lymphoma is commonly classified as early stage favorable, early stage unfavorable, and advanced stage. For advanced stage Hodgkin lymphoma patients, prognosis can be defined using a tool commonly referred to as the International Prognostic Score (IPS). This index consists of 7 factors: male gender, age 45 years or older, stage IV disease, hemoglobin < 10.5 g/dL, white blood cell (WBC) count > 15,000/μL, lymphopenia (absolute lymphocyte count < 600 cells/μL or lymphocytes < 8% of WBC count), and serum albumin < 4 g/dL.5 In the original study by Hasenclever et al,5 the 5-year freedom from progression (FFP) ranged from 42% to 84% and the 5-year overall survival (OS) ranged from 56% to 90%, depending on the number of factors present. This scoring system, however, was developed using a patient population treated prior to 1992. Using a more recently treated patient population, the British Columbia Cancer Agency (BCCA) found that the IPS is still valid for prognostication, but outcomes have improved across all IPS groups, with 5-year FFP now ranging from 62% to 88% and 5-year OS ranging from 67% to 98%.6 This improvement is likely a reflection of improved therapy and supportive care. Table 1 shows the PFS and OS within each IPS group, comparing the data from the German Hodgkin Study Group (GHSG) and BCCA group.5,6
High expression of CD68 is associated with adverse outcomes, whereas high FOXP3 and CD20 expression on tumor cells are predictors of superior outcomes.8 A recent study found that CD68 expression was associated with OS. Five-year OS was 88% in those with less than 25% CD68 expression, versus 63% in those with greater than 25% CD68 expression.9
Roemer and colleagues evaluated 108 newly diagnosed cHL biopsy specimens and found that almost all cHL patients had concordant alteration of PD-L1 (programmed death ligand-1) and PD-L2 loci, with a spectrum of 9p24.1 alterations ranging from low level polysomy to near uniform 9p24.1 amplification. PD-L1/PD-L2 copy number alterations are therefore a defining pathobiological feature of cHL.10 PFS was significantly shorter for patients with 9p24.1 amplification, and those patients were likely to have advanced disease suggesting that 9p24.1 amplification is associated with less favorable prognosis.10 This may change with the increasing use of PD-1 inhibitors in the treatment of cHL.
High baseline metabolic tumor volume and total lesion glycolysis have also been associated with adverse outcomes in cHL. While not routinely assessed in practice currently, these tools may ultimately be used to assess prognosis and guide therapy in clinical practice.11
ADVANCED STAGE HODGKIN LYMPHOMA
FRONTLINE THERAPY
First-line Chemotherapy
Chemotherapy plays an essential role in the treatment of advanced stage Hodgkin lymphoma. In the 1960s, the MOPP regimen (nitrogen mustard, vincristine, procarbazine, prednisone) was developed, with a 10-year OS of 50% and a progression-free survival (PFS) of 52% reported in advanced stage patients. The complete remission (CR) rate was 81%, and 36% of patients who achieved CR relapsed later.12 This chemotherapy regimen is associated with a significant rate of myelosuppression and infertility as well as long-term risk of secondary myelodysplasia and acute leukemias.13,14 This led to the development of newer regimens such as ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine).15 In a randomized trial, ABVD showed improved failure-free survival (FFS) over MOPP (61% versus 50% at 5 years) but similar OS (66%–73%).16 In light of these findings, and considering the lower rate of infertility and myelotoxicity, ABVD became the standard of care for advanced stage cHL in the United States.
The Stanford V regimen was developed in an attempt to further minimize toxicity.17 Stanford V is a condensed, 12-week chemotherapy regimen that includes mechlorethamine, doxorubicin, vinblastine, etoposide, prednisone, vincristine, and bleomycin, followed by involved-field radiation therapy (IFRT). Subsequent trials compared the Stanford V and ABVD regimens and showed similar OS, freedom from treatment failure (FFTF), and response rates.18,19 The ABVD regimen was noted to have higher pulmonary toxicity, while other toxicities such as lymphopenia and neuropathy were higher with the Stanford V regimen. In addition, Stanford V requires patients to receive radiation therapy (RT) to original sites of disease larger than 5 cm in size and contiguous sites.
Another regimen which has been studied extensively for advanced stage Hodgkin lymphoma, and is considered a standard of care in some parts of the world, is escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, prednisone). In the HD9 study (n = 1196), the GHSG evaluated BEACOPP, escalated BEACOPP, and COPP/ABVD in advanced stage Hodgkin lymphoma.20 All arms of the study included 30 Gy RT to sites of bulky disease or residual disease. This study showed improved OS and FFTF with escalated BEACOPP, but at the cost of higher rates of toxicity. At 10 years, FFTF was 64%, 70%, and 82% with OS rates of 75%, 80%, and 86% for COPP/ABVD, baseline BEACOPP, and escalated BEACOPP, respectively (P < 0.001). The rate of secondary acute leukemia 10 years after treatment was 0.4% for COPP/ABVD, 1.5% for BEACOPP, and 3.0% for escalated BEACOPP. However, 3 subsequent randomized trials did not confirm a survival benefit with escalated BEACOPP relative to ABVD. In the HD 2000 trial (n = 295)21 and in a trial by Viviani and colleagues (n = 331),22 an improvement in OS was not demonstrated in favor of escalated BEACOPP. These studies also confirmed a higher rate of toxicities as well as secondary malignancies associated with the escalated BEACOPP regimen. In the EORTC20012 Intergroup trial (n = 549), 8 cycles of ABVD was compared with 4 cycles of escalated BEACOPP followed by 4 cycles of baseline BEACOPP, without radiation, in patients with clinical stage III or IV Hodgkin lymphoma with IPS score ≥ 3. Both regimens resulted in statistically similar FFS (63.7% in ABVD × 8 versus 69.3% in BEACOPP 4+4) and OS (86.7% in ABVD × 8 vs 90.3% in BEACOPP 4+4).23
In the United States, ABVD (6–8 cycles) is commonly used, although escalated BEACOPP (particularly for patients with an IPS of 4 or higher) and Stanford V are considered appropriate as well.24 In the North American Intergroup study comparing ABVD to Stanford V, and in the trial by Viviani et al, ABVD was associated with a 5- to 7-year FFS of 73% to 79% and OS of 84% to 92%.19,22 Given these excellent results, as well as the potential to cure patients with second-line therapy consisting of autologous hematopoietic cell transplantation (auto-HCT), the general consensus among most U.S. hematologists and oncologists is that ABVD remains the treatment of choice, and that the improved FFS/PFS with escalated BEACOPP is not outweighed by the additional toxicity associated with the regimen. There may, however, be a role for escalated BEACOPP in select patients who have a suboptimal response to ABVD as defined by interim positron emission tomography (iPET) scan (see below).
Brentuximab vedotin is an anti-CD30 antibody-drug conjugate (ADC) consisting of an anti-CD30 antibody linked to monomethyl auristatin E (MMAE), a potent antitubulin agent. CD30 is highly expressed on HRS cells and also in anaplastic large cell lymphoma. Upon binding to CD30, the ADC/CD30 complex is then internalized and directed to the lysosome, where the ADC is proteolytically cleaved, releasing MMAE from the antibody. MMAE then disrupts microtubule networks within the cell, leading to G2/M cycle arrest and apoptosis. CD30 is consistently expressed on HRS cells. In addition to being studied in the relapsed/refractory setting (described below), brentuximab has been studied in the first-line setting. In a phase 1 trial, brentuximab combined with ABVD was associated with increased pulmonary toxicity, while brentuximab + AVD had no significant pulmonary toxicity, with an excellent CR rate (96%), suggesting that substituting brentuximab for bleomycin may be an effective strategy. In addition to possibly being more efficacious, this strategy would also have the benefit of eliminating the risk of bleomycin pulmonary toxicity.25 Based on this data, a large international phase 3 study (the ECHELON-1 trial) comparing ABVD versus brentuximab + AVD in advanced stage cHL patients was recently completed. This study enrolled 1334 patients, and preliminary results were recently announced. With a median follow-up of 24 months, the brentuximab + AVD arm had a 4.9% absolute improvement in PFS relative to the ABVD arm (82.1% versus 77.2%). The brentuximab + AVD arm had an increased incidence of febrile neutropenia, managed with growth factors and peripheral neuropathy requiring dose adjustments, whereas the ABVD arm had an increased rate and severity of pulmonary toxicity.26 Further follow-up will be required to determine whether this will translate into a survival benefit. See Table 2 for a summary of recent large randomized prospective phase 3 trials in advanced stage Hodgkin lymphoma.
Alternative Regimens in Older Patients
Patients older than 60 years of age often have poor tolerance for ABVD and especially escalated BEACOPP. This results in increased treatment-related mortality and reduced overall dose intensity, with higher relapse rates and poor OS. In an attempt to improve on the results of treatment of elderly patients with Hodgkin lymphoma, alternative regimens have been explored. One example is PVAG (prednisone, vinblastine, doxorubicin, gemcitabine). With this regimen, the 3-year OS was 66% and PFS was 58%. One patient out of 59 died from treatment-related toxicity, which is much improved over the historical figures for elderly patients with Hodgkin lymphoma.27 Another commonly used approach in practice is to simply omit bleomycin from ABVD. In the early-stage setting (GHSG HD-13 trial), this regimen (referred to as AVD) led to 89.6% PFS at 5 years, compared to 93.5% with ABVD.28 It therefore stands to reason that this should be a reasonable option in older or more frail advanced stage cHL patients as well.
Brentuximab has been evaluated as a single-agent therapy for first-line therapy of elderly patients with Hodgkin lymphoma. In a phase 2 study, 27 patients (63% with advanced stage disease) were treated, with a 92% overall response rate and 73% CR rate. However the median duration of remission was disappointing at only 9.1 months.29 Based on this data, single-agent brentuximab appears to be a reasonable and well tolerated option for frail or elderly patients, although with the caveat that long-term disease control is relatively uncommon.
RESPONSE-ADAPTED FRONTLINE THERAPY USING INTERIM PET SCAN
In recent years, response-adapted treatment approaches have been extensively researched in cHL using iPET. The goal is to reduce toxicity by minimizing therapy in those who achieve negative iPET and/or to intensify treatment for patients with suboptimal response on iPET. Gallamini et al evaluated the prognostic role of an early iPET scan in advanced Hodgkin lymphoma patients (n = 190) treated with ABVD. This study found that patients with positive iPET had a 2-year PFS of 12.8% versus 95.0% in patients with negative iPET. This result was highly statistically significant (P < 0.0001). This study also showed that PET-2 (iPET after 2 cycles of ABVD) superseded the prognostic value of the IPS at diagnosis.30 As a result, numerous subsequent studies have been pursued using iPET for risk-adapted treatment in cHL.
A critical element to the conduct of iPET risk-adapted treatment for cHL is the interpretation of the iPET. In hopes of standardizing iPET interpretation in clinical trials, a scoring system called the Deauville score was developed. The Deauville score ranges from 1 to 5 (Table 3).
The SWOG (Southwest Oncology Group) S0816 trial (n = 358) evaluated iPET-adapted treatment after 2 cycles of ABVD in stage III or IV Hodgkin lymphoma patients. Patients with positive iPET (Deauville score 4 to 5; n = 60) received escalated BEACOPP for 6 cycles, whereas iPET-negative (Deauville score 1 to 3; n = 271) patients continued to receive 4 more cycles of ABVD. The 2-year PFS was 64% for iPET-positive patients.33 This PFS was much higher than the expected 15% to 30% from prior studies such as Gallamini et al,30 suggesting that the treatment intensification may have been of benefit.
In the HD0801 study (n = 519), newly diagnosed advanced Hodgkin lymphoma patients with positive iPET after 2 cycles of ABVD (n = 103) received early ifosfamide-containing salvage therapy followed by high-dose therapy with autologous stem cell rescue. The 2-year PFS was 76% for PET-2–positive patients, comparable with PET-2–negative patients who had PFS of 81%.34 Again, this result for iPET-positive patients was much better than expected based on the historical control from Gallamini et al, suggesting that the treatment intensification may have been beneficial. It should be emphasized, however, that neither HD0801 nor S0816 were randomized prospective trials; rather, all iPET-positive patients were assigned to an intensified treatment approach.
In the HD18 trial (n = 1100), patients with advanced stage cHL started therapy with escalated BEACOPP and underwent an iPET after 2 cycles. For those with a positive iPET, rituximab was added to escalated BEACOPP in the experimental arm (n = 220) for cycles 3 through 8. The control group (n = 220) continued to receive 6 more cycles of escalated BEACOPP. In the 2 groups, the 3-year PFS was similar (91.4% in escalated BEACOPP, 93% in rituximab + escalated BEACOPP), suggesting no significant benefit with addition of rituximab.35 This study also calls into question whether iPET provides useful information for patients receiving intensive therapy such as escalated BEACOPP, and indicates that the historical control data for iPET-positive patients from Gallamini et al may not be consistently reproduced in other prospective trials. As a result, nonrandomized trials that implement an iPET risk-adapted approach should be interpreted with caution. See Table 4 for a summary of recent trials in advanced stage Hodgkin lymphoma using iPET scan to guide therapy.
RADIATION THERAPY IN FRONTLINE TREATMENT
In patients with advanced stage Hodgkin lymphoma, IFRT to initial bulky sites of disease may be incorporated into frontline therapy to improve local control. However, whether this provides a survival benefit and which patients benefit most from consolidative RT remain unclear.
The European Organization for Research and Treatment of Cancer (EORTC) completed a randomized study in advanced stage Hodgkin lymphoma patients who achieved complete or partial remission after MOPP-ABV.36 Patients in CR were randomly assigned to receive no further treatment versus IFRT (24 Gy to all initially involved nodal areas and 16 to 24 Gy to all initially involved extranodal sites). Patients in partial remission (PR) were treated with 30 Gy to nodal areas and 18 to 24 Gy to extranodal sites. Among the CR patients, the 5-year event-free survival (EFS) was 79% to 84% and did not differ for those who received radiation versus those who did not. Five-year OS was 85% to 91% and also did not differ between the 2 groups. However, among the patients in PR after chemotherapy, the 5-year EFS was 79% and the 5-year OS was 87%, which is better than expected for PR patients, indicating a possible benefit to RT in patients with a partial response after chemotherapy. In the GHSG HD12 trial, patients with advanced stage Hodgkin lymphoma who had a residual lesion by computed tomography (CT) (but not analyzed by PET) had a very subtle improvement in FFTF (90% versus 87%) in favor of consolidation with IFRT, but again no survival benefit was seen.37
The EORTC and HD12 studies described above utilized CT scan for assigning remission status following chemotherapy, and it is now well known that many patients with residual masses (by CT) after chemotherapy may in fact be cured, as such residual radiographic abnormalities may simply be composed of fibrosis. PET scan is more accurate than CT in identifying patients who truly have residual active disease following chemotherapy. As a result, the EORTC study discussed above and the GHSG HD12 trial are of limited relevance in the modern era, in which patients routinely undergo PET scan at the end of therapy. Restricting IFRT to sites that remain PET-positive after completing chemotherapy may be a reasonable strategy that would allow for the avoidance of RT in many patients, and may obviate the need for aggressive second-line therapy (eg, high-dose therapy and autologous hematopoietic cell transplant [auto-HCT]). This approach was taken in the GHSG HD15 trial (n = 2182) in which advanced stage patients were treated with 3 variations on the BEACOPP regimen (8 cycles of escalated BEACOPP, 6 cycles of escalated BEACOPP, or 8 cycles of baseline BEACOPP, randomized in a 1:1:1 ratio). Patients with a residual mass of 2.5 cm or greater on CT scan then underwent a PET scan; if the lesion was PET positive, it was treated with 30 Gy of IFRT. This overall strategy was very effective, with 5-year FFTF rates of 84.4%, 89.3%, and 85.4%, respectively. The OS rates were 91.9%, 95.3%, and 94.5%, respectively. For patients with lesions that remained PET positive after chemotherapy, the PFS rate was 86.2% at 48 months, whereas patients in PR with persistent mass ≥ 2.5 cm but with negative PET had a PFS of 92.6%, similar to that of patients in CR.38 With this approach of BEACOPP followed by PET-guided radiation, the proportion of patients receiving RT was reduced from 71% (in the HD9 study) to only 11% in the HD15 study,38 with no apparent loss in overall efficacy when comparing the results of the 2 studies.
UPFRONT STEM CELL TRANSPLANTATION
To further improve outcomes of patients with advanced Hodgkin lymphoma with high-risk disease, high-dose therapy with auto-HCT has been explored as part of frontline therapy. While this has been shown to be feasible in such patients,39 randomized trials have not shown a clear benefit in terms of FFS or OS with upfront auto-HCT. 40,41 Therefore, auto-HCT is not considered a standard component of frontline therapy for cHL patients who achieve CR by PET/CT scan.
RELAPSED AND REFRACTORY HODGKIN LYMPHOMA
Depending on the stage, risk factors, and frontline regimen utilized, between 5% and 40% of patients with Hodgkin lymphoma can be expected to experience either primary induction failure or a relapse after attaining remission with frontline therapy.3 Primary refractory Hodgkin lymphoma, which occurs in up to 5% to 10% of patients, is defined as progression or no response during induction treatment or within 90 days of completing treatment. In cases where remission status is in question, an updated tissue biopsy is recommended. Biopsy is also recommended in cases in which new sites of disease have appeared or if relapse has occurred after a durable period of remission. Restaging is recommended at the time of relapse.
For younger patients with relapsed/refractory Hodgkin lymphoma, the standard of care in most cases is second-line (or salvage) chemotherapy followed by high-dose therapy and auto-HCT. For patients not felt to be candidates for auto-HCT, options include conventional second-line chemotherapy alone, salvage radiotherapy, novel agents such as brentuximab or immune checkpoint inhibitors, and/or participation in clinical trials.
CONVENTIONAL MULTI-AGENT CHEMOTHERAPY REGIMENS
Numerous conventional regimens have been shown in phase 2 studies to be active in relapsed and refractory Hodgkin lymphoma. These include platinum-based regimens, gemcitabine-based regimens, and alkylator-based regimens. No randomized trials in Hodgkin lymphoma have been conducted comparing these regimens. In general, regimens are chosen based on the patient’s age, performance status, comorbidities, and whether auto-HCT is being considered.
In the United States, platinum-based regimens such as ICE (ifosfamide, carboplatin, etoposide),42 DHAP (dexamethasone, cisplatin, high-dose cytarabine),43 ESHAP (etoposide, methylprednisolone, high-dose cytarabine, cisplatin),44 GDP (gemcitabine, cisplatin, dexamethasone),45 and GCD (gemcitabine, carboplatin, dexamethasone)46 are all considered appropriate second-line therapy options for patients being considered for auto-HCT, due to their high response rates and because autologous hematopoietic stem cell collection remains feasible after these regimens. Response rates range from 60% to 88%, with CR rates between 17% and 41%, and toxic death rates generally well below 5%.
Other gemcitabine-based regimens such as IGEV (ifosfamide, gemcitabine, vinorelbine) and GVD (gemcitabine, vinorelbine, liposomal doxorubicin) are also effective.47,48 GVD is an excellent choice since it is a generally well-tolerated outpatient regimen with a 60% response rate even in heavily pretreated patients.48 Stem cell collection remains feasible after both IGEV and GVD as well. ABVD can produce CR in approximately 20% to 50% of patients initially treated with MOPP.49–51 In practice, however, most patients today with relapsed or refractory Hodgkin lymphoma have already received ABVD as part of their first-line therapy, and retreatment with ABVD is not a good option because it would be associated with prohibitively high cumulative doses of doxorubicin.
These multi-agent chemotherapy regimens may not be tolerated well in patients over age 65 to 70 years or those with significant underlying comorbidities. In recent years, bendamustine has emerged as one of the most active conventional agents for cHL, with overall response rates of 53% to 58% in heavily pre-treated patients.52,53 Bendamustine can generally be tolerated even in elderly patients as well.
Some centers, particularly in Europe, investigated aggressive salvage regimens such as mini-BEAM (carmustine, etoposide, cytarabine, melphalan)54 or dexa-BEAM (BEAM plus dexamethasone).55 These regimens, however, are associated with significant hematologic toxicity and high (2%–5%) treatment-related mortality. As a result, these are rarely used in the United States.
For patients who have progressed after (or are not candidates for) platinum- and/or gemcitabine-based therapy, older alkylator-based regimens such as MOPP, C-MOPP, or ChlVPP (chlorambucil, vinblastine, procarbazine, prednisone) can be considered.56–58 However, these regimens are associated with significant bone marrow suppression, and autologous hematopoietic stem cell collection may no longer be feasible after such regimens. Therefore, these regimens should only be given to patients not felt to be auto-HCT candidates, or patients for whom autologous hematopoietic stem cell collection has already been completed. Weekly vinblastine or single-agent gemcitabine are palliative chemotherapy options, with response rates in the 60% to 80% range. Patients can sometimes be maintained on such low-intensity palliative regimens for 6 to 12 months or longer.59,60
BRENTUXIMAB VEDOTIN
Several trials are evaluating incorporation of brentuximab into second-line therapy in transplant-eligible patients. These approaches have used brentuximab prior to, concurrent with, or following platinum-based chemotherapy.61 While there is currently no consensus on the optimal way to incorporate brentuximab into salvage therapy, it is possible that the use of brentuximab or other novel agents in salvage therapy may allow for avoidance of conventional chemotherapy in some patients. In addition, this may translate into more patients proceeding to auto-HCT in a PET negative state. PET negativity prior to auto-HCT is a powerful predictor of long-term remission after auto-HCT, so any intervention that increases the rate of PET negativity prior to auto-HCT would be expected to improve outcomes with auto-HCT.62–65
For patients not being considered for autoHCT, or those for whom platinum-based salvage therapy was ineffective, single-agent brentuximab is an excellent option. In 2 phase 2 studies, an overall response rate (ORR) of 60% to 75% (including a CR rate of 22%–34%) was seen in relapsed and refractory Hodgkin lymphoma patients.66 The US Food and Drug Administration (FDA) approved brentuximab vedotin in August 2011 for treatment of relapsed and refractory Hodgkin lymphoma, after a failed auto-HCT, or in patients who are not auto-HCT candidates and who have received at least 2 prior chemotherapy regimens. With more extended follow-up, it has become clear that a proportion of patients who achieve CR to brentuximab may maintain remission long-term—58% at 3 years and 38% at 5 years.67 These patients may in fact be cured, in many cases without having undergone allogeneic HCT (allo-HCT) after brentuximab.
PD-1 (IMMUNE CHECKPOINT) INHIBITORS
As discussed earlier, PD-L1/PD-L2 copy number alterations represent a disease-defining feature of cHL. Alterations in chromosome 9p24.1 increase the expression of PD-1 ligands PD-L1 and PD-L2. Nivolumab and pembrolizumab are PD-1-blocking antibodies, which have recently been FDA approved for relapsed and refractory cHL. In a study with 23 patients, with 78% of them relapsing after auto-HCT and 78% relapsing after brentuximab, nivolumab produced an objective response in 87% of the patients, with 17% achieving CR and 70% achieving PR. The rate of PFS was 86% at 24 weeks.68 Pembrolizumab, another PD-1 antagonist, was also tested in relapsed and refractory Hodgkin lymphoma. In the KEYNOTE-087 study (n = 210), pembrolizumab produced an ORR of 64% to 70% in 3 different cohorts of relapsed and refractory cHL patients. Overall CR rate was 22%.69 In general, these agents are well tolerated, although patients must be monitored closely for
inflammatory/autoimmune-type toxicities including skin rash, diarrhea/colitis, transaminitis, endocrine abnormalities, and pneumonitis. Prompt recognition and initiation of corticosteroids is essential in managing these toxicities. Of note, PD-1 inhibitors should be given very cautiously to patients with a prior history of allo-HCT, since 30% to 55% of such patients will experience acute graft-versus-host disease (GVHD) in this setting. In 2 retrospective studies, the response rate was very high at 77% to 95%; however, 10% to 26% of all patients treated with PD-1 inhibitors post-allo-HCT died from GVHD induced by PD-1 inhibition.70,71 These risks and benefits therefore need to be carefully weighed in the post-allo-HCT setting. In another recent study, the outcomes were reported for 39 patients who underwent allo-HCT after prior therapy with a PD-1 inhibitor. Three patients (7.7%) developed lethal acute GVHD, suggesting there may be an increased risk of GVHD in patients undergoing allo-HCT after prior PD-1 inhibitor therapy.72
AUTOLOGOUS STEM CELL TRANSPLANTATION
Several studies have shown an improved disease-free survival (DFS) or FFS in patients with relapsed cHL treated by auto-HCT as compared to those receiving conventional chemotherapy alone.55,73,74 Overall, for relapsed disease, one can expect an approximately 50% to 60% chance for DFS at 5 years post-transplant. In a retrospective, matched-pair analysis, FFP was 62% for auto-HCT patients, compared to 32% for conventional chemotherapy patients. OS, however, was similar for the 2 groups (47%–54%). Patients failing induction therapy or relapsing within 1 year were seen to benefit the most from auto-HCT, including an OS benefit.74
A European prospective randomized trial was conducted comparing conventional salvage therapy to auto-HCT. In this study, 161 patients with relapsed Hodgkin lymphoma were treated with 2 cycles of dexa-BEAM. Those with chemo-sensitive disease were then randomized to either 2 more cycles of dexa-BEAM or high-dose BEAM with auto-HCT. Auto-HCT was associated with an approximately 55% FFTF at 3 years, versus 34% with conventional chemotherapy alone.55 This benefit again was most apparent for patients relapsing within 1 year of completion of primary therapy, although an OS benefit was not seen with auto-HCT. For patients with late relapse (>1 year after completion of primary therapy), auto-HCT was associated with an approximately 75% FFTF at 3 years, versus 50% with chemotherapy alone. One other small randomized trial of auto-HCT in relapsed and refractory Hodgkin lymphoma also showed an improved 3-year EFS in favor of auto-HCT (53% versus 10%), again with no difference in OS.73
The lack of OS benefit seen in these studies suggests that auto-HCT at first or second relapse provides comparable outcomes. Auto-HCT offers the benefit of avoiding the long-term toxicities associated with multiple salvage regimens and the anxiety associated with multiple relapses. In addition, the treatment-related mortality with auto-HCT is now in the 1% to 2% range in younger patients, at centers that perform the procedure routinely. For all of these reasons, auto-HCT is commonly recommended by physicians for Hodgkin lymphoma patients in first or second relapse. In most cases, transplant is favored in first relapse, since waiting until second relapse may be associated with a lower chance of achieving CR and difficulty collecting sufficient hematopoietic stem cells. For patients with early relapse or primary refractory disease, an even stronger case can be made for auto-HCT as the best option to achieve sustained control of the disease. For patients with late relapse, conventional salvage therapy alone may be a reasonable option, particularly in older or frail patients, or those with significant comorbid conditions.
The optimal conditioning regimen for autoHCT for relapsed and refractory Hodgkin lymphoma remains undefined. No randomized trials have been performed comparing conditioning regimens for relapsed and refractory Hodgkin lymphoma. One retrospective study compared 92 patients with Hodgkin lymphoma who underwent auto-HCT using a total-body irradiation (TBI) regimen versus a chemotherapy-alone regimen. No difference in 5-year OS or EFS was seen.75 Given the lack of benefit seen with TBI, along with reports of increased rates of secondary malignancies and myelodysplasia with TBI,76 chemotherapy-alone conditioning regimens are most widely employed. For example, in the United States, either the BEAM or CBV (cyclophosphamide, carmustine, etoposide) regimens are used in over 80% of cases.77 This practice was justified in a Center for International Blood and Marrow Transplant Research (CIBMTR) retrospective study comparing outcomes by conditioning regimens, in which no regimen performed better than BEAM or CBV.78
IFRT is often given as an adjunctive therapy to sites of initial and/or relapsed disease following auto-HCT. Although a relatively common practice, whether this truly enhances outcomes beyond that obtained with auto-HCT alone is unclear. Two retrospective studies have shown some benefit in terms of improvement in OS at 3 to 5 years in the group that received IFRT (70%–73% versus 40%–56%).79,80 Given the retrospective nature and small size of these studies, a prospective study would be needed to properly define the potential role for IFRT following auto-HCT in relapsed/refractory Hodgkin lymphoma. Another retrospective study (n = 73) that evaluated peri-transplant IFRT in Hodgkin lymphoma patients receiving auto transplant found no improvement in survival for patients who received peri-transplant IFRT. This study, however, did show a survival benefit in the subgroup of patients with limited stage disease.81
Prognostic Factors Associated with Outcome with Auto-HCT
The factor most consistently associated with improved outcome for patients with relapsed and refractory Hodgkin lymphoma who undergo auto-HCT is the disease status at transplant.63,77 Those in a second CR, versus a chemo-sensitive relapse (but not CR), versus a chemo-refractory relapse have DFS rates of 60% to 70%, 30% to 40%, and 10% to 20%, respectively.63 The duration between remission and relapse also has important prognostic significance. Late relapse (> 1 year after completion of frontline therapy) is associated with better outcomes as compared to early relapse.55 Other factors with prognostic significance at relapse include anemia, time to relapse and clinical stage, B symptoms, extranodal disease, number of prior chemotherapy regimens, and performance status.42,82 The prognostic impact of pretransplant disease status has been confirmed by studies using functional imaging (eg, FDG-PET or gallium scans). In a report by Moskowitz et al, patients with negative functional imaging following second-line therapy had a 77% EFS post-auto-HCT versus 33% in those whose functional imaging remained positive.62 Very similar findings have been reported by other groups.63–65
Post-Auto-HCT Brentuximab Maintenance
In the multicenter, randomized, double-blinded phase 3 AETHERA trial (n = 329), brentuximab (n = 165) was compared with placebo (n = 164) in patients with unfavorable risk relapsed or primary refractory cHL who had undergone autologous transplant. Eligible patients had at least 1 of the following risk factors for progression after auto-HCT: primary refractory Hodgkin lymphoma (failure to achieve complete remission), relapsed Hodgkin lymphoma with an initial remission duration of less than 12 months, or extranodal involvement at the start of pre-transplantation salvage chemotherapy. Patients were required to have CR, PR, or stable disease after pretransplant salvage chemotherapy with adequate kidney, liver, and bone marrow function. Patients who previously received brentuximab were excluded. Patients received 16 cycles of brentuximab or placebo once every 3 weeks starting 30 to 45 days after transplant. The PFS was significantly improved in the brentuximab group when compared to the placebo group (hazard ratio 0.57; P = 0.0013) after a median observation time of 30 months. Median PFS was 42.9 months in the brentuximab group versus 24.1 months in the placebo group; estimated 2-year PFS rates were 63% in the brentuximab group and 51% in the placebo group. OS was not significantly different between the study groups (~85%), presumably due to the fact that patients in the control group who relapsed likely went on to receive brentuximab as a subsequent therapy.83
PRIMARY REFRACTORY HODGKIN LYMPHOMA
Patients with primary refractory Hodgkin lymphoma have a poor outcome. Salvage therapy using conventional chemotherapy and/or RT results in long-term DFS in 10% or fewer of such patients.13,84 Given these poor outcomes with conventional salvage therapy, auto-HCT is considered to be the standard of care for this subset of patients. The GHSG retrospectively analyzed the prognostic factors and outcomes of patients with primary refractory Hodgkin lymphoma. The 5-year freedom-from-second-failure and the 5-year OS were reported to be 31% and 43%, respectively, for those patients treated with auto-HCT. Patients with poor functional status at time of transplant, age greater than 50 years, and failure to attain a temporary remission had a 0% 5-year OS, as compared to 55% in patients without any of these risk factors.85 A large retrospective European study showed that patients with chemo-resistant disease who underwent transplant had a 19% survival at 5 years.63 Hence, even patients with primary refractory Hodgkin lymphoma have some chance of achieving long-term survival following auto-HCT.
SALVAGE RADIOTHERAPY
The GHSG performed a retrospective analysis of the efficacy of salvage RT in patients with refractory or first-relapsed Hodgkin lymphoma. Five-year FFTF and OS rates were 28% and 51%, respectively. Patients with a limited-stage relapse and without B symptoms were more likely to benefit from salvage RT.86 Campbell et al reported on 81 patients undergoing salvage RT for persistent or recurrent Hodgkin lymphoma after chemotherapy. The 10-year FFTF and OS rates were 33% and 46%, respectively.87 Similarly, Wirth et al reported a 5-year FFS of 26% and 5-year OS of 57%. These figures were 36% and 75%, respectively, in patients whose relapse was limited to supradiaphragmatic nodal sites without B symptoms.88 RT therefore may be a useful strategy for a subset of patients who relapse following chemotherapy, particularly those with a limited-stage relapse, without B symptoms, and those with relapsed disease after a CR, as opposed to those with a partial response or lack of response to the prior chemotherapy regimen.
INVESTIGATIONAL AGENTS AND NOVEL COMBINATIONS
Several biological therapies are emerging as options for the treatment of refractory or relapsed disease. These therapies consist of monoclonal antibodies and ADCs that target cell surface antigens, or small molecules that inhibit key intracellular pathways within neoplastic cells.
Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody used widely in B-cell non-Hodgkin lymphomas. The CD20 molecule is typically highly expressed in nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL). Two studies (one in relapsed patients, the other in a mixture of relapsed and previously untreated patients) showed significant activity of rituximab in relapsed NLPHL, with ORRs ranging from 94% to 100%, CR rates ranging from 41% to 53%, and median duration of remission in the 10- to 33-month range.89,90 In cHL, CD20 is expressed in HRS cells in 20% to 30% of cases. In such cases, single-agent rituximab has also shown activity. There is also evidence that rituximab may be effective even in cases in which the HRS cells are CD20-negative, presumably by virtue of depleting reactive B lymphocytes from the microenvironment, which may enhance anti-tumor immunity, or by eliminating a putative CD20-expressing Hodgkin lymphoma stem cell.91,92
Lenalidomide
Lenalidomide is an immunomodulatory drug that has multiple modes of action, including direct induction of apoptosis in tumor cells, antiangiogenic effects, and the activation of immune cells, such as natural killer cells and T cells. Lenalidomide has been shown to modify many features of the microenvironment of HRS cells and has demonstrated activity in other B-cell neoplasms. As a result, lenalidomide has been evaluated in relapsed and refractory Hodgkin lymphoma patients. A multicenter phase 2 study by Fehniger et al included 35 patients, 87% of whom had previously undergone HCT and 55% of whom were refractory to the last therapy.93 All patients were given lenalidomide 25 mg/day from days 1 to 21 of a 28-day cycle until disease progression. One patient was noted to achieve CR, 6 achieved PR, and 5 had stable disease lasting more than 6 months, for an ORR of 19% and a “cytostatic overall response rate” of 33%. The median duration of CR/partial remission was 6 months, with the median time-to-treatment failure in responders (including those with stable disease > 6 months) being 15 months. Similarly, in another study, Böll et al evaluated 12 patients across 4 German centers with relapsed or refractory disease who were treated with oral lenalidomide for 21 days in a 28-day cycle. No radiological evidence of disease progression after 2 cycles of lenalidomide was seen in any of the enrolled patients. ORR was noted to be 50%, with 6 patients with stable disease and 5 patients achieving PR after 2 cycles.94
Novel Brentuximab Combination Therapies
Brentuximab plus bendamustine. The combination of brentuximab and bendamustine was tested as an outpatient regimen in a phase 1/2 study (n = 55) in primary refractory Hodgkin lymphoma or after first relapse. The CR rate of the combination was 74%, with an overall objective response (CR + PR) of 93%. The CR rates were 64% and 84%, respectively, for refractory and relapsed patients. The PFS at 12 months was 80%, establishing this combination therapy as an effective salvage regimen with durable response.95
Brentuximab plus nivolumab. Preliminary results have recently been presented from 2 studies96,97 evaluating the combination of brentuximab and nivolumab. While this combination would still be considered investigational, these studies showed very encouraging ORRs of 90% to 100% and a CR rate of 62% to 66%. Longer follow-up is needed to determine whether these responses are durable and to document the toxicity profile of this combination.
Mammalian Target of Rapamycin Inhibitors
Two mammalian target of rapamycin (mTOR) inhibitors, everolimus and temsirolimus, are currently available in the United States. While neither drug currently has FDA approval for Hodgkin lymphoma, everolimus was evaluated in a phase 2 trial in a heavily pretreated group of relapsed/refractory patients. An ORR of 47% was seen, with a median time to progression of 7.2 months.98
ALLOGENEIC STEM CELL TRANSPLANTATION
Historically, patients who relapse after having an auto-HCT generally had a poor outcome, with a median survival of 2 to 3 years after failure of auto-HCT.99 These patients may be offered palliative chemotherapy (see above), treatment with novel agents (see above), or enrollment in a clinical trial. Select patients may benefit from a second hematopoietic stem cell transplant, most commonly an allo-HCT. However, rare patients with late relapse after auto-HCT may be considered for a second auto-HCT, with a minority of such patients achieving a durable remission after the second auto-HCT.100,101 Because relapse or progressive disease occurs most commonly in the first several months following auto-HCT, patients are more often considered for allo-HCT than a second auto-HCT. In addition, a second auto-HCT may not be feasible due to impaired bone marrow reserve and/or concerns for development of secondary myelodysplasia or acute myeloid leukemia.
Several studies have evaluated allo-HCT in relapsed/ refractory Hodgkin lymphoma. Early studies evaluating myeloablative allo-HCT for Hodgkin lymphoma showed excessive treatment-related mortality (up to 50%) and disappointingly low rates of long-term survival (< 25%).102,103 This was likely related to the fact that, in that era, most of the patients with Hodgkin lymphoma evaluated for allo-HCT were heavily pretreated and therefore at a higher risk for toxicity as well as lymphoma progression.
More recently, several studies have focused on the use of reduced-intensity conditioning (RIC) allo-HCT for relapsed and refractory Hodgkin lymphoma. This approach relies more on a “graft-versus-lymphoma” effect, the existence of which has been debated in Hodgkin lymphoma. Three single-center studies of RIC allo-HCT in patients with multiply recurrent Hodgkin lymphoma showed improved rates of treatment-related mortality (8%–16%) but still relatively low rates of long-term PFS (23%–39% at 2 to 4 years).104–106 Interestingly, in one of these studies the outcomes were more favorable for patients who underwent haploidentical (versus matched sibling or matched unrelated donor) transplants.105
Two large registry studies have also reported on the outcomes of RIC allo-HCT in patients with relapsed and refractory Hodgkin lymphoma.107,108 These studies also confirmed a modest improvement in outcomes compared with those seen historically with myeloablative transplants. Treatment-related mortality at 1 to 2 years was 23% to 33%, depending on whether a matched sibling donor versus an unrelated donor was used. However, long-term PFS (18%–20% at 2 to 5 years) and OS (28%–37% at 2 to 5 years) remained poor, primarily due to high rates of progressive lymphoma post-transplant. In both of these studies, patients were heavily pretreated (84%–96% had received 3 or more prior lines of chemotherapy, and 62%–89% received a prior auto-HCT), with 47% to 55% of patients chemo-resistant prior to transplant. Of note, both of these registry studies reflect patients who underwent transplant prior to the widespread use of brentuximab and PD-1 inhibitors.
Based on the single-center and registry data above, a prospective multicenter European phase 2 trial was conducted to evaluate the benefit of RIC allo-HCT in Hodgkin lymphoma.109 Ninety-two patients (86% with prior auto-HCT, 90% with 3 or more prior lines of therapy) were enrolled and given salvage therapy. Those who had stable disease or better following salvage therapy remained on protocol (n = 78) and underwent RIC with fludarabine and melphalan, followed by allo-HCT (70% with matched sibling donors). Treatment-related mortality was 15% at 1 year. Relapse or progression occurred in 49% at 2 years (35% if chemo-sensitive prior to transplant). Chronic GVHD was associated with a decreased rate of relapse, supporting the existence of a graft-versus-lymphoma effect in Hodgkin lymphoma. Unfortunately, PFS among all allografted patients was still relatively poor (24% at 4 years). However, among patients in CR prior to allo-HCT, a 50% PFS was seen at 4 years. Therefore, even in a prospective multicenter study, RIC allo-HCT offered significant benefit with manageable toxicity in relapsed and refractory Hodgkin lymphoma patients with chemo-sensitive disease.
These studies suggest that outcomes with allo-HCT would improve further if implemented earlier in the course of disease and/or with a lower burden of disease at transplant. It has therefore been suggested that allo-HCT should be considered soon after failure of auto-HCT is documented. In a retrospective study by Sarina et al, 185 Hodgkin lymphoma patients who relapsed following auto-HCT were then immediately considered for reduced-intensity allo-HCT.110 Of these, 122 had a donor identified, and 104 (85%) actually underwent allo-HCT. These 104 patients were then compared to the other 81 patients who either had no donor identified or had a donor but did not receive the planned allo-HCT. Two-year PFS and OS were superior in the patients undergoing allo-HCT (39% versus 14% and 66% versus 42%, respectively, P < 0.001), with a median follow-up of 4 years. The presence of chronic GVHD again was associated with improved PFS and OS. Disease status prior to transplant remained highly predictive of PFS and OS by multivariate analysis. Two other smaller retrospective studies similarly found a survival benefit associated with allo-HCT compared with patients who underwent conventional salvage therapies alone.111,112 These studies, although subject to the usual limitations of retrospective analyses, suggest that the results with reduced-intensity allo-HCT are in fact enhanced if applied earlier in the disease course, and are superior to those with conventional therapy alone.
Currently, the exact role of allo-HSCT, including the optimal timing and optimal donor source (matched sibling versus haploidentical sibling versus matched unrelated donor), remain undefined for relapsed and refractory Hodgkin lymphoma. As discussed earlier, brentuximab is highly active in relapsed Hodgkin lymphoma patients, with a subset of patients still in CR at 5 years.67 For such patients, avoiding the risks of allo-HCT is a desirable goal.
For those who relapse or progress after auto-HCT, a reasonable strategy therefore is to treat initially with brentuximab, unless the patient is already known to have responded poorly to brentuximab, or already has significant neuropathy. Those who achieve a CR to brentuximab are then observed. A subset of those patients will remain in remission at 5 years without further therapy. For those who relapse, or who achieve less than a CR to brentuximab, additional treatment (with brentuximab re-treatment being one option) followed by a reduced-intensity allo-HCT is a reasonable consideration. This approach has the theoretical advantages of (1) avoiding the risk of allo-HCT in the subset potentially cured by brentuximab, (2) getting patients to allo-HCT with fewer comorbidities (due to a lower total exposure to conventional chemotherapy pre-transplant), and (3) applying allo-HCT in the setting of sensitive disease/lower disease burden (due to the high efficacy of brentuximab). The results of a small study suggest that brentuximab may in fact be a very effective “bridge” to allotransplant. Chen et al113 reported on 18 patients with relapsed/refractory Hodgkin lymphoma (17 of whom had previously undergone auto-HCT) who were treated on brentuximab vedotin clinical trials. The data were retrospectively evaluated to determine the efficacy and safety of subsequent reduced-intensity allo-HCT. Remarkably, at 1 year the OS was 100%, PFS was 92%, and nonrelapse mortality was 0% with a median follow-up of 14 months. Hence, brentuximab is safe for use prior to reduced-intensity allo-HCT in heavily pre-treated patients and appears to be associated with very favorable post-transplant outcomes, particularly in comparison to older studies of allo-HCT in the era prior to brentuximab.
SUMMARY
Currently, cure is possible for the majority of patients diagnosed with advanced stage Hodgkin lymphoma. The challenge to the clinician is to provide curative treatment with the lowest risk of serious toxicities. Which regimen will best provide this balance of risk and benefit needs to be assessed based on the relapse risk, age, frailty, and comorbidity profile for an individual patient. For many patients with relapsed or refractory Hodgkin lymphoma, cure remains possible using approaches based on hematopoietic stem cell transplantation, RT, and/or brentuximab. In addition, there are now numerous conventional chemotherapy agents, RT strategies, and exciting newer agents such as PD-1 inhibitors, that can provide significant clinical benefit even when cure is not feasible.
1. Kuppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6.
2. Kuppers R. The biology of Hodgkin’s lymphoma. Nature Rev Cancer 2009;9:15–27.
3. Narra RK, Pingali SR, Fenske TS. Early-stage Hodgkin’s lymphoma. Hospital Physician Hematology-Oncology Board Review Manual. 2017;12(Part 3).
4. Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993;71:2062–71.
5. Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med 1998;339:1506–14.
6. Moccia AA, Donaldson J, Chhanabhai M, et al. International Prognostic Score in advanced-stage Hodgkin’s lymphoma: altered utility in the modern era. J Clin Oncol 2012;30:3383–8.
7. Diefenbach CS, Li H, Hong F, et al. Evaluation of the International Prognostic Score (IPS-7) and a Simpler Prognostic Score (IPS-3) for advanced Hodgkin lymphoma in the modern era. Br J Haematol 2015;171:530–8.
8. Greaves P, Clear A, Coutinho R, et al. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of cassical hodgkin lymphoma is predictive of outcome. J Clin Oncol 2013;31:256–62.
9. Touati M, Delage-Corre M, Monteil J, et al. CD68-positive tumor-associated macrophages predict unfavorable treatment outcomes in classical Hodgkin lymphoma in correlation with interim fluorodeoxyglucose-positron emission tomography assessment. Leuk Lymphoma 2015;56:332–41.
10. Roemer MG, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7.
11. Pike LC, Kirkwood AA, Patrick P, et al. Can baseline PET-CT features predict outcomes in advanced hodgkin lymphoma? A prospective evaluation of UK patients in The RATHL Trial (CRUK/07/033). Hematological Oncology 2017;35(Supplement S2):37–8.
12. DeVita VT Jr, Simon RM, Hubbard SM, et al. Curability of advanced Hodgkin’s disease with chemotherapy. Long-term follow-up of MOPP-treated patients at the National Cancer Institute. Ann Intern Med 1980;92:587–95.
13. Longo DL, Duffey PL, Young RC, et al. Conventional-dose salvage combination chemotherapy in patients relapsing with Hodgkin’s disease after combination chemotherapy: the low probability for cure. J Clin Oncol 1992;10:210–8.
14. Kaldor JM, Day NE, Clarke EA, et al. Leukemia following Hodgkin’s disease. N Engl J Med 1990;322:7–13.
15. Bonadonna G, Zucali R, Monfardini S, et al. Combination chemotherapy of Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP. Cancer 1975;36:252–9.
16. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–84.
17. Horning SJ, Williams J, Bartlett NL, et al. Assessment of the stanford V regimen and consolidative radiotherapy for bulky and advanced Hodgkin’s disease: Eastern Cooperative Oncology Group pilot study E1492. J Clin Oncol 2000;18:972–80.
18. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the stanford V regimen and ABVD in the treatment of advanced Hodgkin’s Lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRCTN 64141244. J Clin Oncol 2009;27:5390–6.
19. Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013;31:684–91.
20. Engert A, Diehl V, Franklin J, et al. Escalated-dose BEACOPP in the treatment of patients with advanced-stage Hodgkin’s lymphoma: 10 years of follow-up of the GHSG HD9 study. J Clin Oncol 2009;27:4548–54.
21. Merli F, Luminari S, Gobbi PG, et al. Long-term results of the HD2000 trial comparing ABVD versus BEACOPP versus COPP-EBV-CAD in untreated patients with advanced Hodgkin lymphoma: a study by Fondazione Italiana Linfomi. J Clin Oncol 2016;34:1175–81.
22. Viviani S, Zinzani PL, Rambaldi A, et al. ABVD versus BEACOPP for Hodgkin’s lymphoma when high-dose salvage is planned. N Engl J Med 2011;365:203–12.
23. Carde P, Karrasch M, Fortpied C, et al. Eight cycles of ABVD versus four cycles of BEACOPPescalated plus four cycles of BEACOPPbaseline in stage III to IV, International Prognostic Score >/= 3, high-risk Hodgkin lymphoma: first results of the phase III EORTC 20012 intergroup trial. J Clin Oncol 2016;34:2028–36.
24. National Comprehensive Cancer Network I. NCCN Guidelines Version 1.2017 Hodgkin Lymphoma. Accessed July 20, 2017.
25. Younes A, Connors JM, Park SI, et al. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncology 2013;14:1348–56.
26. Takeda and Seattle Genetics announce positive results from phase 3 ECHELON-1 clinical trial evaluating ADCETRIS® (brentuximab vedotin) in frontline advanced Hodgkin lymphoma [press release]. Cambridge, MA: Takeda Oncology; June 26, 2017.
27. Boll B, Bredenfeld H, Gorgen H, et al. Phase 2 study of PVAG (prednisone, vinblastine, doxorubicin, gemcitabine) in elderly patients with early unfavorable or advanced stage Hodgkin lymphoma. Blood 2011;118:6292–8.
28. Behringer K, Goergen H, Hitz F, et al. Omission of dacarbazine or bleomycin, or both, from the ABVD regimen in treatment of early-stage favourable Hodgkin’s lymphoma (GHSG HD13): an open-label, randomised, non-inferiority trial. Lancet 2015;385:1418–27.
29. Forero-Torres A, Holkova B, Goldschmidt J, et al. Phase 2 study of frontline brentuximab vedotin monotherapy in Hodgkin lymphoma patients aged 60 years and older. Blood 2015;126:2798–804.
30. Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol 2007;25:3746–52.
31. Meignan M, Gallamini A, Meignan M, et al. Report on the First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymphoma 2009;50:1257–60.
32. Barrington SF, Mikhaeel NG, Kostakoglu L, et al. Role of Imaging in the Staging and Response Assessment of Lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 2014;32:3048–58.
33. Press OW, Li H, Schoder H, et al. US Intergroup trial of response-adapted therapy for stage III to IV Hodgkin lymphoma using early interim fluorodeoxyglucose-positron emission tomography imaging: Southwest Oncology Group S0816. J Clin Oncol 2016;34:2020–7.
34. Zinzani PL, Broccoli A, Gioia DM, et al. Interim positron emission tomography response-adapted therapy in advanced-stage Hodgkin lymphoma: final results of the pPhase II part of the HD0801 study. J Clin Oncol 2016;34:1376–85.
35. Borchmann P, Haverkamp H, Lohri A, et al. Progression-free survival of early interim PET-positive patients with advanced stage Hodgkin’s lymphoma treated with BEACOPPescalated alone or in combination with rituximab (HD18): an open-label, international, randomised phase 3 study by the German Hodgkin Study Group. Lancet Oncology 2017;18:454–63.
36. Aleman BM, Raemaekers JM, Tirelli U, et al. Involved-field radiotherapy for advanced Hodgkin’s lymphoma. N Engl J Med 2003;348:2396–406.
37. Borchmann P, Haverkamp H, Diehl V, et al. Eight cycles of escalated-dose BEACOPP compared with four cycles of escalated-dose BEACOPP followed by four cycles of baseline-dose BEACOPP with or without radiotherapy in patients with advanced-stage hodgkin’s lymphoma: final analysis of the HD12 trial of the German Hodgkin Study Group. J Clin Oncol 2011;29:4234–42.
38. Engert A, Haverkamp H, Kobe C, et al. Reduced-intensity chemotherapy and PET-guided radiotherapy in patients with advanced stage Hodgkin’s lymphoma (HD15 trial): a randomised, open-label, phase 3 non-inferiority trial. Lancet 2012;379:1791–9.
39. Nademanee A, Molina A, Fung H, et al. High-dose chemo/radiotherapy and autologous bone marrow or stem cell transplantation for poor-risk advanced-stage Hodgkin’s disease during first partial or complete remission. Biol Blood Marrow Transplant 1999;5:292–8.
40. Federico M, Bellei M, Brice P, et al. High-dose therapy and autologous stem-cell transplantation versus conventional therapy for patients with advanced Hodgkin’s lymphoma responding to front-line therapy. J Clin Oncol 2003;21:2320–5.
41. Arakelyan N, Berthou C, Desablens B, et al. Early versus late intensification for patients with high-risk Hodgkin lymphoma-3 cycles of intensive chemotherapy plus low-dose lymph node radiation therapy versus 4 cycles of combined doxorubicin, bleomycin, vinblastine, and dacarbazine plus myeloablative chemotherapy with autologous stem cell transplantation: five-year results of a randomized trial on behalf of the GOELAMS Group. Cancer 2008;113:3323–30.
42. Moskowitz CH, Nimer SD, Zelenetz AD, et al. A 2-step comprehensive high-dose chemoradiotherapy second-line program for relapsed and refractory Hodgkin disease: analysis by intent to treat and development of a prognostic model. Blood 2001;97:616–23.
43. Josting A, Rudolph C, Reiser M, et al. Time-intensified dexamethasone/cisplatin/cytarabine: an effective salvage therapy with low toxicity in patients with relapsed and refractory Hodgkin’s disease. Ann Oncol 2002;13:1628–35.
44. Aparicio J, Segura A, Garcera S, et al. ESHAP is an active regimen for relapsing Hodgkin’s disease. Ann Oncol 1999;10:593–5.
45. Baetz T, Belch A, Couban S, et al. Gemcitabine, dexamethasone and cisplatin is an active and non-toxic chemotherapy regimen in relapsed or refractory Hodgkin’s disease: a phase II study by the National Cancer Institute of Canada Clinical Trials Group. Ann Oncol 2003;14:1762–7.
46. Gopal AK, Press OW, Shustov AR, et al. Efficacy and safety of gemcitabine, carboplatin, dexamethasone, and rituximab in patients with relapsed/refractory lymphoma: a prospective multi-center phase II study by the Puget Sound Oncology Consortium. Leuk Lymphoma 2010;51:1523–9.
47. Santoro A, Magagnoli M, Spina M, et al. Ifosfamide, gemcitabine, and vinorelbine: a new induction regimen for refractory and relapsed Hodgkin’s lymphoma. Haematologica 2007;92:35–41.
48. Bartlett NL, Niedzwiecki D, Johnson JL, et al. Gemcitabine, vinorelbine, and pegylated liposomal doxorubicin (GVD), a salvage regimen in relapsed Hodgkin’s lymphoma: CALGB 59804. Ann Oncol 2007;18:1071–9.
49. Santoro A, Bonadonna G. Prolonged disease-free survival in MOPP-resistant Hodgkin’s disease after treatment with adriamycin, bleomycin, vinblastine and dacarbazine (ABVD). Cancer Chemother Pharmacol 1979;2:101–5.
50. Krikorian JG, Portlock CS, Rosenberg SA. Treatment of advanced Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide (ABVD) after failure of MOPP therapy. Cancer 1978;41:2107–11.
51. Piga A, Ambrosetti A, Todeschini G, et al. Doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) salvage of mechlorethamine, vincristine, prednisone, and procarbazine (MOPP)-resistant advanced Hodgkin’s disease. Cancer Treat Rep 1984;68:947–51.
52. Moskowitz AJ, Hamlin PA Jr, Perales MA, et al. Phase II study of bendamustine in relapsed and refractory Hodgkin lymphoma. J Clin Oncol 2013;31:456–60.
53. Anastasia A, Carlo-Stella C, Corradini P, et al. Bendamustine for relapsed/refractory classical Hodgkin lymphoma after high dose chemotherapy and or allogeneic transplant: a study of Fondazione Italiana Linfomi (FIL). Blood 2012;120:3652.
54. Martin A, Fernandez-Jimenez MC, Caballero MD, et al. Long-term follow-up in patients treated with Mini-BEAM as salvage therapy for relapsed or refractory Hodgkin’s disease. Br J Haematol 2001;113:161–71.
55. Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet 2002;359:2065–71.
56. Fisher RI, DeVita VT, Hubbard SP, et al. Prolonged disease-free survival in Hodgkin’s disease with MOPP reinduction after first relapse. Ann Intern Med 1979;90:761–3.
57. ChlVPP therapy for Hodgkin’s disease: experience of 960 patients. The International ChlVPP Treatment Group. Ann Oncol 1995;6:167–72.
58. Selby P, Patel P, Milan S, et al. ChlVPP combination chemotherapy for Hodgkin’s disease: long-term results. Br J Cancer 1990;62:279–85.
59. Santoro A, Bredenfeld H, Devizzi L, et al. Gemcitabine in the treatment of refractory Hodgkin’s disease: results of a multicenter phase II study. J Clin Oncol 2000;18:2615–9.
60. Little R, Wittes RE, Longo DL, Wilson WH. Vinblastine for recurrent Hodgkin’s disease following autologous bone marrow transplant. J Clin Oncol 1998;16:584–8.
61. Chen R, Palmer JM, Martin P, et al. Results of a multicenter phase II trial of brentuximab vedotin as second-line therapy before autologous transplantation in relapsed/refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2015;21:2136–40.
62. Moskowitz CH, Yahalom J, Zelenetz AD, et al. High-dose chemo-radiotherapy for relapsed or refractory Hodgkin lymphoma and the significance of pre-transplant functional imaging. Br J Haematol 2010;148:890–7.
63. Sureda A, Constans M, Iriondo A, et al. Prognostic factors affecting long-term outcome after stem cell transplantation in Hodgkin’s lymphoma autografted after a first relapse. Ann Oncol 2005;16:625–33.
64. Crocchiolo R, Canevari C, Assanelli A, et al. Pre-transplant 18FDG-PET predicts outcome in lymphoma patients treated with high-dose sequential chemotherapy followed by autologous stem cell transplantation. Leuk Lymphoma 2008;49:727–33.
65. Mocikova H, Pytlik R, Markova J, et al. Pre-transplant positron emission tomography in patients with relapsed Hodgkin lymphoma. Leuk Lymphoma 2011;52:1668–74.
66. Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol 2012;30:2183–9.
67. Gopal AK, Chen R, Smith SE, et al. Durable remissions in a pivotal phase 2 study of brentuximab vedotin in relapsed or refractory Hodgkin lymphoma. Blood 2015;125:1236–43.
68. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma.N Engl J Med 2015;372:311–9.
69. Chen R, Zinzani PL, Fanale MA, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 2017;35:2125–32.
70. Haverkos BM, Abbott D, Hamadani M, et al. PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood 2017;130:221–8.
71. Herbaux C, Gauthier J, Brice P, et al. Efficacy and tolerability of nivolumab after allogeneic transplantation for relapsed Hodgkin lymphoma. Blood 2017;129:2471–8.
72. Merryman RW, Kim HT, Zinzani PL et al. Safety and efficacy of allogeneic hematopoietic stem cell transplant after PD-1 blockage in relapsed/refractory lymphoma. Blood 2017;129:1380–8.
73. Linch DC, Winfield D, Goldstone AH, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet 1993;341:1051–4.
74. Yuen AR, Rosenberg SA, Hoppe RT, et al. Comparison between conventional salvage therapy and high-dose therapy with autografting for recurrent or refractory Hodgkin’s disease. Blood 1997;89:814–22.
75. Gutierrez-Delgado F, Holmberg L, Hooper H, et al. Autologous stem cell transplantation for Hodgkin’s disease: busulfan, melphalan and thiotepa compared to a radiation-based regimen. Bone Marrow Transplant 2003;32:279–85.
76. Hake CR, Graubert TA, Fenske TS. Does autologous transplantation directly increase the risk of secondary leukemia in lymphoma patients? Bone Marrow Transplant 2007;39:59–70.
77. Hahn T, McCarthy PL, Carreras J, et al. Comparison of prognostic models for autologous hematopoietic stem cell transplantation (AHCT) for relapsed Hodgkin lymphoma. Blood 2009;114:1215.
78. Chen Y-B, Lane AA, Logan BR, et al. Impact of conditioning regimen on outcomes for patients with lymphoma undergoing high-dose therapy with autologous hematopoietic cell transplantation. Biology Blood Marrow Transplant 2015;21:1046–53.
79. Wendland MM, Asch JD, Pulsipher MA, et al. The impact of involved field radiation therapy for patients receiving high-dose chemotherapy followed by hematopoietic progenitor cell transplant for the treatment of relapsed or refractory Hodgkin disease. Am J Clin Oncol 2006;29:189–95.
80. Biswas T, Culakova E, Friedberg JW, et al. Involved field radiation therapy following high dose chemotherapy and autologous stem cell transplant benefits local control and survival in refractory or recurrent Hodgkin lymphoma. Radiother Oncol 2012;103:367–72.
81. Levis M, Piva C, Filippi AR, et al. Potential benefit of involved-field radiotherapy for patients with Relapsed-refractory Hodgkin’s lymphoma with incomplete response before autologous stem cell transplantation. Clin Lymphoma Myeloma Leuk 2017;17:14–22.
82. Josting A, Franklin J, May M, et al. New prognostic score based on treatment outcome of patients with relapsed Hodgkin’s lymphoma registered in the database of the German Hodgkin’s lymphoma study group. J Clin Oncol 2002;20:221–30.
83. Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015;385:1853–62.
84. Bonfante V, Santoro A, Viviani S, et al. Outcome of patients with Hodgkin’s disease failing after primary MOPP-ABVD. J Clin Oncol 1997;15:528–34.
85. Josting A, Rueffer U, Franklin J, et al. Prognostic factors and treatment outcome in primary progressive Hodgkin lymphoma: a report from the German Hodgkin Lymphoma Study Group. Blood 2000;96:1280–6.
86. Josting A, Nogova L, Franklin J, et al. Salvage radiotherapy in patients with relapsed and refractory Hodgkin’s lymphoma: a retrospective analysis from the German Hodgkin Lymphoma Study Group. J Clin Oncol 2005;23:1522–9.
87. Campbell B WA, Milner A, Di Iulio J, et al. Long-term follow-up of salvage radiotherapy in Hodgkin’s lymphoma after chemotherapy failure. Int J Radiat Oncol Biol Phys 2005;63:1538–45.
88. Wirth A, Corry J, Laidlaw C, et al. Salvage radiotherapy for Hodgkin’s disease following chemotherapy failure. Int J Radiat Oncol Biol Phys 1997;39:599–607.
89. Schulz H, Rehwald U, Morschhauser F, et al. Rituximab in relapsed lymphocyte-predominant Hodgkin lymphoma: long-term results of a phase 2 trial by the German Hodgkin Lymphoma Study Group (GHSG). Blood 2008;111:109–11.
90. Ekstrand BC, Lucas JB, Horwitz SM, et al. Rituximab in lymphocyte-predominant Hodgkin disease: results of a phase 2 trial. Blood 2003;101:4285–9.
91. Younes A, Romaguera J, Hagemeister F, et al. A pilot study of rituximab in patients with recurrent, classic Hodgkin disease. Cancer 2003;98:310–4.
92. Rehwald U, Schulz H, Reiser M, et al. Treatment of relapsed CD20+ Hodgkin lymphoma with the monoclonal antibody rituximab is effective and well tolerated: results of a phase 2 trial of the German Hodgkin Lymphoma Study Group. Blood 2003;101:420–4.
93. Fehniger TA, Larson S, Trinkaus K, et al. A phase 2 multicenter study of lenalidomide in relapsed or refractory classical Hodgkin lymphoma. Blood 2011;118:5119–25.
94. Boll B, Borchmann P, Topp MS, et al. Lenalidomide in patients with refractory or multiple relapsed Hodgkin lymphoma. Br J Haematol 2010;148:480–2.
95. LaCasce AS, Bociek G, Sawas A, et al. Brentuximab vedotin plus bendamustine: a highly active salvage treatment regimen for patients with relapsed or refractory Hodgkin lymphoma. Blood 2015;126:3982.
96. Diefenbach CS, Hong F, David KA, et al. A phase I study with an expansion cohort of the combination of ipilimumab and nivolumab and brentuximab vedotin in patients with relapsed/refractory Hodgkin lymphoma: A trial of the ECOG-ACRIN Cancer Research Group (E4412 Arms D and E). Blood 2016;128:1106.
97. Herrera AF, Bartlett NL, Ramchandren R, et al. Preliminary results from a phase 1/2 study of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016;128:1105.
98. Johnston PB, Inwards DJ, Colgan JP, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol 2010;85:320–4.
99. Kewalramani T, Nimer SD, Zelenetz AD, et al. Progressive disease following autologous transplantation in patients with chemosensitive relapsed or primary refractory Hodgkin’s disease or aggressive non-Hodgkin’s lymphoma. Bone Marrow Transplant 2003;32:673–9.
100. Lin TS, Avalos BR, Penza SL, et al. Second autologous stem cell transplant for multiply relapsed Hodgkin’s disease. Bone Marrow Transplant 2002;29:763–7.
101. Smith SM, van Besien K, Carreras J, et al. Second autologous stem cell transplantation for relapsed lymphoma after a prior autologous transplant. Biol Blood Marrow Transplant 2008;14:904–12.
102. Gajewski JL, Phillips GL, Sobocinski KA, et al. Bone marrow transplants from HLA-identical siblings in advanced Hodgkin’s disease. J Clin Oncol 1996;14:572–8.
103. Peniket AJ, Ruiz de Elvira MC, Taghipour G, et al. An EBMT registry matched study of allogeneic stem cell transplants for lymphoma: allogeneic transplantation is associated with a lower relapse rate but a higher procedure-related mortality rate than autologous transplantation. Bone Marrow Transplant 2003;31:667–78.
104. Anderlini P, Saliba R, Acholonu S, et al. Fludarabine-melphalan as a preparative regimen for reduced-intensity conditioning allogeneic stem cell transplantation in relapsed and refractory Hodgkin’s lymphoma: the updated M.D. Anderson Cancer Center experience. Haematologica 2008;93:257–64.
105. Burroughs LM, O’Donnell PV, Sandmaier BM, et al. Comparison of outcomes of HLA-matched related, unrelated, or HLA-haploidentical related hematopoietic cell transplantation following nonmyeloablative conditioning for relapsed or refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2008;14:1279–87.
106. Peggs KS, Hunter A, Chopra R, et al. Clinical evidence of a graft-versus-Hodgkin’s-lymphoma effect after reduced-intensity allogeneic transplantation. Lancet 2005;365:1934–41.
107. Sureda A, Robinson S, Canals C, et al. Reduced-intensity conditioning compared with conventional allogeneic stem-cell transplantation in relapsed or refractory Hodgkin’s lymphoma: an analysis from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2008;26:455–62.
108. Devetten MP, Hari PN, Carreras J, et al. Unrelated donor reduced-intensity allogeneic hematopoietic stem cell transplantation for relapsed and refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:109–17.
109. Sureda A, Canals C, Arranz R, et al. Allogeneic stem cell transplantation after reduced intensity conditioning in patients with relapsed or refractory Hodgkin’s lymphoma. Results of the HDR-ALLO study - a prospective clinical trial by the Grupo Espanol de Linfomas/Trasplante de Medula Osea (GEL/TAMO) and the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. Haematologica 2012;97:310–7.
110. Sarina B, Castagna L, Farina L, et al. Allogeneic transplantation improves the overall and progression-free survival of Hodgkin lymphoma patients relapsing after autologous transplantation: a retrospective study based on the time of HLA typing and donor availability. Blood 2010;115:3671–7.
111. Castagna L, Sarina B, Todisco E, et al. Allogeneic stem cell transplantation compared with chemotherapy for poor-risk Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:432–8.
112. Thomson KJ, Peggs KS, Smith P, et al. Superiority of reduced-intensity allogeneic transplantation over conventional treatment for relapse of Hodgkin’s lymphoma following autologous stem cell transplantation. Bone Marrow Transplant 2008;41:765–70.
113. Chen R, Palmer JM, Thomas SH, et al. Brentuximab vedotin enables successful reduced-intensity allogeneic hematopoietic cell transplantation in patients with relapsed or refractory Hodgkin lymphoma. Blood 2012;119:6379–81.
1. Kuppers R, Rajewsky K, Zhao M, et al. Hodgkin disease: Hodgkin and Reed-Sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc Natl Acad Sci U S A 1994;91:10962–6.
2. Kuppers R. The biology of Hodgkin’s lymphoma. Nature Rev Cancer 2009;9:15–27.
3. Narra RK, Pingali SR, Fenske TS. Early-stage Hodgkin’s lymphoma. Hospital Physician Hematology-Oncology Board Review Manual. 2017;12(Part 3).
4. Mauch PM, Kalish LA, Kadin M, et al. Patterns of presentation of Hodgkin disease. Implications for etiology and pathogenesis. Cancer 1993;71:2062–71.
5. Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med 1998;339:1506–14.
6. Moccia AA, Donaldson J, Chhanabhai M, et al. International Prognostic Score in advanced-stage Hodgkin’s lymphoma: altered utility in the modern era. J Clin Oncol 2012;30:3383–8.
7. Diefenbach CS, Li H, Hong F, et al. Evaluation of the International Prognostic Score (IPS-7) and a Simpler Prognostic Score (IPS-3) for advanced Hodgkin lymphoma in the modern era. Br J Haematol 2015;171:530–8.
8. Greaves P, Clear A, Coutinho R, et al. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of cassical hodgkin lymphoma is predictive of outcome. J Clin Oncol 2013;31:256–62.
9. Touati M, Delage-Corre M, Monteil J, et al. CD68-positive tumor-associated macrophages predict unfavorable treatment outcomes in classical Hodgkin lymphoma in correlation with interim fluorodeoxyglucose-positron emission tomography assessment. Leuk Lymphoma 2015;56:332–41.
10. Roemer MG, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–7.
11. Pike LC, Kirkwood AA, Patrick P, et al. Can baseline PET-CT features predict outcomes in advanced hodgkin lymphoma? A prospective evaluation of UK patients in The RATHL Trial (CRUK/07/033). Hematological Oncology 2017;35(Supplement S2):37–8.
12. DeVita VT Jr, Simon RM, Hubbard SM, et al. Curability of advanced Hodgkin’s disease with chemotherapy. Long-term follow-up of MOPP-treated patients at the National Cancer Institute. Ann Intern Med 1980;92:587–95.
13. Longo DL, Duffey PL, Young RC, et al. Conventional-dose salvage combination chemotherapy in patients relapsing with Hodgkin’s disease after combination chemotherapy: the low probability for cure. J Clin Oncol 1992;10:210–8.
14. Kaldor JM, Day NE, Clarke EA, et al. Leukemia following Hodgkin’s disease. N Engl J Med 1990;322:7–13.
15. Bonadonna G, Zucali R, Monfardini S, et al. Combination chemotherapy of Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP. Cancer 1975;36:252–9.
16. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–84.
17. Horning SJ, Williams J, Bartlett NL, et al. Assessment of the stanford V regimen and consolidative radiotherapy for bulky and advanced Hodgkin’s disease: Eastern Cooperative Oncology Group pilot study E1492. J Clin Oncol 2000;18:972–80.
18. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the stanford V regimen and ABVD in the treatment of advanced Hodgkin’s Lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRCTN 64141244. J Clin Oncol 2009;27:5390–6.
19. Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496). J Clin Oncol 2013;31:684–91.
20. Engert A, Diehl V, Franklin J, et al. Escalated-dose BEACOPP in the treatment of patients with advanced-stage Hodgkin’s lymphoma: 10 years of follow-up of the GHSG HD9 study. J Clin Oncol 2009;27:4548–54.
21. Merli F, Luminari S, Gobbi PG, et al. Long-term results of the HD2000 trial comparing ABVD versus BEACOPP versus COPP-EBV-CAD in untreated patients with advanced Hodgkin lymphoma: a study by Fondazione Italiana Linfomi. J Clin Oncol 2016;34:1175–81.
22. Viviani S, Zinzani PL, Rambaldi A, et al. ABVD versus BEACOPP for Hodgkin’s lymphoma when high-dose salvage is planned. N Engl J Med 2011;365:203–12.
23. Carde P, Karrasch M, Fortpied C, et al. Eight cycles of ABVD versus four cycles of BEACOPPescalated plus four cycles of BEACOPPbaseline in stage III to IV, International Prognostic Score >/= 3, high-risk Hodgkin lymphoma: first results of the phase III EORTC 20012 intergroup trial. J Clin Oncol 2016;34:2028–36.
24. National Comprehensive Cancer Network I. NCCN Guidelines Version 1.2017 Hodgkin Lymphoma. Accessed July 20, 2017.
25. Younes A, Connors JM, Park SI, et al. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncology 2013;14:1348–56.
26. Takeda and Seattle Genetics announce positive results from phase 3 ECHELON-1 clinical trial evaluating ADCETRIS® (brentuximab vedotin) in frontline advanced Hodgkin lymphoma [press release]. Cambridge, MA: Takeda Oncology; June 26, 2017.
27. Boll B, Bredenfeld H, Gorgen H, et al. Phase 2 study of PVAG (prednisone, vinblastine, doxorubicin, gemcitabine) in elderly patients with early unfavorable or advanced stage Hodgkin lymphoma. Blood 2011;118:6292–8.
28. Behringer K, Goergen H, Hitz F, et al. Omission of dacarbazine or bleomycin, or both, from the ABVD regimen in treatment of early-stage favourable Hodgkin’s lymphoma (GHSG HD13): an open-label, randomised, non-inferiority trial. Lancet 2015;385:1418–27.
29. Forero-Torres A, Holkova B, Goldschmidt J, et al. Phase 2 study of frontline brentuximab vedotin monotherapy in Hodgkin lymphoma patients aged 60 years and older. Blood 2015;126:2798–804.
30. Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol 2007;25:3746–52.
31. Meignan M, Gallamini A, Meignan M, et al. Report on the First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymphoma 2009;50:1257–60.
32. Barrington SF, Mikhaeel NG, Kostakoglu L, et al. Role of Imaging in the Staging and Response Assessment of Lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 2014;32:3048–58.
33. Press OW, Li H, Schoder H, et al. US Intergroup trial of response-adapted therapy for stage III to IV Hodgkin lymphoma using early interim fluorodeoxyglucose-positron emission tomography imaging: Southwest Oncology Group S0816. J Clin Oncol 2016;34:2020–7.
34. Zinzani PL, Broccoli A, Gioia DM, et al. Interim positron emission tomography response-adapted therapy in advanced-stage Hodgkin lymphoma: final results of the pPhase II part of the HD0801 study. J Clin Oncol 2016;34:1376–85.
35. Borchmann P, Haverkamp H, Lohri A, et al. Progression-free survival of early interim PET-positive patients with advanced stage Hodgkin’s lymphoma treated with BEACOPPescalated alone or in combination with rituximab (HD18): an open-label, international, randomised phase 3 study by the German Hodgkin Study Group. Lancet Oncology 2017;18:454–63.
36. Aleman BM, Raemaekers JM, Tirelli U, et al. Involved-field radiotherapy for advanced Hodgkin’s lymphoma. N Engl J Med 2003;348:2396–406.
37. Borchmann P, Haverkamp H, Diehl V, et al. Eight cycles of escalated-dose BEACOPP compared with four cycles of escalated-dose BEACOPP followed by four cycles of baseline-dose BEACOPP with or without radiotherapy in patients with advanced-stage hodgkin’s lymphoma: final analysis of the HD12 trial of the German Hodgkin Study Group. J Clin Oncol 2011;29:4234–42.
38. Engert A, Haverkamp H, Kobe C, et al. Reduced-intensity chemotherapy and PET-guided radiotherapy in patients with advanced stage Hodgkin’s lymphoma (HD15 trial): a randomised, open-label, phase 3 non-inferiority trial. Lancet 2012;379:1791–9.
39. Nademanee A, Molina A, Fung H, et al. High-dose chemo/radiotherapy and autologous bone marrow or stem cell transplantation for poor-risk advanced-stage Hodgkin’s disease during first partial or complete remission. Biol Blood Marrow Transplant 1999;5:292–8.
40. Federico M, Bellei M, Brice P, et al. High-dose therapy and autologous stem-cell transplantation versus conventional therapy for patients with advanced Hodgkin’s lymphoma responding to front-line therapy. J Clin Oncol 2003;21:2320–5.
41. Arakelyan N, Berthou C, Desablens B, et al. Early versus late intensification for patients with high-risk Hodgkin lymphoma-3 cycles of intensive chemotherapy plus low-dose lymph node radiation therapy versus 4 cycles of combined doxorubicin, bleomycin, vinblastine, and dacarbazine plus myeloablative chemotherapy with autologous stem cell transplantation: five-year results of a randomized trial on behalf of the GOELAMS Group. Cancer 2008;113:3323–30.
42. Moskowitz CH, Nimer SD, Zelenetz AD, et al. A 2-step comprehensive high-dose chemoradiotherapy second-line program for relapsed and refractory Hodgkin disease: analysis by intent to treat and development of a prognostic model. Blood 2001;97:616–23.
43. Josting A, Rudolph C, Reiser M, et al. Time-intensified dexamethasone/cisplatin/cytarabine: an effective salvage therapy with low toxicity in patients with relapsed and refractory Hodgkin’s disease. Ann Oncol 2002;13:1628–35.
44. Aparicio J, Segura A, Garcera S, et al. ESHAP is an active regimen for relapsing Hodgkin’s disease. Ann Oncol 1999;10:593–5.
45. Baetz T, Belch A, Couban S, et al. Gemcitabine, dexamethasone and cisplatin is an active and non-toxic chemotherapy regimen in relapsed or refractory Hodgkin’s disease: a phase II study by the National Cancer Institute of Canada Clinical Trials Group. Ann Oncol 2003;14:1762–7.
46. Gopal AK, Press OW, Shustov AR, et al. Efficacy and safety of gemcitabine, carboplatin, dexamethasone, and rituximab in patients with relapsed/refractory lymphoma: a prospective multi-center phase II study by the Puget Sound Oncology Consortium. Leuk Lymphoma 2010;51:1523–9.
47. Santoro A, Magagnoli M, Spina M, et al. Ifosfamide, gemcitabine, and vinorelbine: a new induction regimen for refractory and relapsed Hodgkin’s lymphoma. Haematologica 2007;92:35–41.
48. Bartlett NL, Niedzwiecki D, Johnson JL, et al. Gemcitabine, vinorelbine, and pegylated liposomal doxorubicin (GVD), a salvage regimen in relapsed Hodgkin’s lymphoma: CALGB 59804. Ann Oncol 2007;18:1071–9.
49. Santoro A, Bonadonna G. Prolonged disease-free survival in MOPP-resistant Hodgkin’s disease after treatment with adriamycin, bleomycin, vinblastine and dacarbazine (ABVD). Cancer Chemother Pharmacol 1979;2:101–5.
50. Krikorian JG, Portlock CS, Rosenberg SA. Treatment of advanced Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide (ABVD) after failure of MOPP therapy. Cancer 1978;41:2107–11.
51. Piga A, Ambrosetti A, Todeschini G, et al. Doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) salvage of mechlorethamine, vincristine, prednisone, and procarbazine (MOPP)-resistant advanced Hodgkin’s disease. Cancer Treat Rep 1984;68:947–51.
52. Moskowitz AJ, Hamlin PA Jr, Perales MA, et al. Phase II study of bendamustine in relapsed and refractory Hodgkin lymphoma. J Clin Oncol 2013;31:456–60.
53. Anastasia A, Carlo-Stella C, Corradini P, et al. Bendamustine for relapsed/refractory classical Hodgkin lymphoma after high dose chemotherapy and or allogeneic transplant: a study of Fondazione Italiana Linfomi (FIL). Blood 2012;120:3652.
54. Martin A, Fernandez-Jimenez MC, Caballero MD, et al. Long-term follow-up in patients treated with Mini-BEAM as salvage therapy for relapsed or refractory Hodgkin’s disease. Br J Haematol 2001;113:161–71.
55. Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet 2002;359:2065–71.
56. Fisher RI, DeVita VT, Hubbard SP, et al. Prolonged disease-free survival in Hodgkin’s disease with MOPP reinduction after first relapse. Ann Intern Med 1979;90:761–3.
57. ChlVPP therapy for Hodgkin’s disease: experience of 960 patients. The International ChlVPP Treatment Group. Ann Oncol 1995;6:167–72.
58. Selby P, Patel P, Milan S, et al. ChlVPP combination chemotherapy for Hodgkin’s disease: long-term results. Br J Cancer 1990;62:279–85.
59. Santoro A, Bredenfeld H, Devizzi L, et al. Gemcitabine in the treatment of refractory Hodgkin’s disease: results of a multicenter phase II study. J Clin Oncol 2000;18:2615–9.
60. Little R, Wittes RE, Longo DL, Wilson WH. Vinblastine for recurrent Hodgkin’s disease following autologous bone marrow transplant. J Clin Oncol 1998;16:584–8.
61. Chen R, Palmer JM, Martin P, et al. Results of a multicenter phase II trial of brentuximab vedotin as second-line therapy before autologous transplantation in relapsed/refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2015;21:2136–40.
62. Moskowitz CH, Yahalom J, Zelenetz AD, et al. High-dose chemo-radiotherapy for relapsed or refractory Hodgkin lymphoma and the significance of pre-transplant functional imaging. Br J Haematol 2010;148:890–7.
63. Sureda A, Constans M, Iriondo A, et al. Prognostic factors affecting long-term outcome after stem cell transplantation in Hodgkin’s lymphoma autografted after a first relapse. Ann Oncol 2005;16:625–33.
64. Crocchiolo R, Canevari C, Assanelli A, et al. Pre-transplant 18FDG-PET predicts outcome in lymphoma patients treated with high-dose sequential chemotherapy followed by autologous stem cell transplantation. Leuk Lymphoma 2008;49:727–33.
65. Mocikova H, Pytlik R, Markova J, et al. Pre-transplant positron emission tomography in patients with relapsed Hodgkin lymphoma. Leuk Lymphoma 2011;52:1668–74.
66. Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol 2012;30:2183–9.
67. Gopal AK, Chen R, Smith SE, et al. Durable remissions in a pivotal phase 2 study of brentuximab vedotin in relapsed or refractory Hodgkin lymphoma. Blood 2015;125:1236–43.
68. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma.N Engl J Med 2015;372:311–9.
69. Chen R, Zinzani PL, Fanale MA, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol 2017;35:2125–32.
70. Haverkos BM, Abbott D, Hamadani M, et al. PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood 2017;130:221–8.
71. Herbaux C, Gauthier J, Brice P, et al. Efficacy and tolerability of nivolumab after allogeneic transplantation for relapsed Hodgkin lymphoma. Blood 2017;129:2471–8.
72. Merryman RW, Kim HT, Zinzani PL et al. Safety and efficacy of allogeneic hematopoietic stem cell transplant after PD-1 blockage in relapsed/refractory lymphoma. Blood 2017;129:1380–8.
73. Linch DC, Winfield D, Goldstone AH, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet 1993;341:1051–4.
74. Yuen AR, Rosenberg SA, Hoppe RT, et al. Comparison between conventional salvage therapy and high-dose therapy with autografting for recurrent or refractory Hodgkin’s disease. Blood 1997;89:814–22.
75. Gutierrez-Delgado F, Holmberg L, Hooper H, et al. Autologous stem cell transplantation for Hodgkin’s disease: busulfan, melphalan and thiotepa compared to a radiation-based regimen. Bone Marrow Transplant 2003;32:279–85.
76. Hake CR, Graubert TA, Fenske TS. Does autologous transplantation directly increase the risk of secondary leukemia in lymphoma patients? Bone Marrow Transplant 2007;39:59–70.
77. Hahn T, McCarthy PL, Carreras J, et al. Comparison of prognostic models for autologous hematopoietic stem cell transplantation (AHCT) for relapsed Hodgkin lymphoma. Blood 2009;114:1215.
78. Chen Y-B, Lane AA, Logan BR, et al. Impact of conditioning regimen on outcomes for patients with lymphoma undergoing high-dose therapy with autologous hematopoietic cell transplantation. Biology Blood Marrow Transplant 2015;21:1046–53.
79. Wendland MM, Asch JD, Pulsipher MA, et al. The impact of involved field radiation therapy for patients receiving high-dose chemotherapy followed by hematopoietic progenitor cell transplant for the treatment of relapsed or refractory Hodgkin disease. Am J Clin Oncol 2006;29:189–95.
80. Biswas T, Culakova E, Friedberg JW, et al. Involved field radiation therapy following high dose chemotherapy and autologous stem cell transplant benefits local control and survival in refractory or recurrent Hodgkin lymphoma. Radiother Oncol 2012;103:367–72.
81. Levis M, Piva C, Filippi AR, et al. Potential benefit of involved-field radiotherapy for patients with Relapsed-refractory Hodgkin’s lymphoma with incomplete response before autologous stem cell transplantation. Clin Lymphoma Myeloma Leuk 2017;17:14–22.
82. Josting A, Franklin J, May M, et al. New prognostic score based on treatment outcome of patients with relapsed Hodgkin’s lymphoma registered in the database of the German Hodgkin’s lymphoma study group. J Clin Oncol 2002;20:221–30.
83. Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015;385:1853–62.
84. Bonfante V, Santoro A, Viviani S, et al. Outcome of patients with Hodgkin’s disease failing after primary MOPP-ABVD. J Clin Oncol 1997;15:528–34.
85. Josting A, Rueffer U, Franklin J, et al. Prognostic factors and treatment outcome in primary progressive Hodgkin lymphoma: a report from the German Hodgkin Lymphoma Study Group. Blood 2000;96:1280–6.
86. Josting A, Nogova L, Franklin J, et al. Salvage radiotherapy in patients with relapsed and refractory Hodgkin’s lymphoma: a retrospective analysis from the German Hodgkin Lymphoma Study Group. J Clin Oncol 2005;23:1522–9.
87. Campbell B WA, Milner A, Di Iulio J, et al. Long-term follow-up of salvage radiotherapy in Hodgkin’s lymphoma after chemotherapy failure. Int J Radiat Oncol Biol Phys 2005;63:1538–45.
88. Wirth A, Corry J, Laidlaw C, et al. Salvage radiotherapy for Hodgkin’s disease following chemotherapy failure. Int J Radiat Oncol Biol Phys 1997;39:599–607.
89. Schulz H, Rehwald U, Morschhauser F, et al. Rituximab in relapsed lymphocyte-predominant Hodgkin lymphoma: long-term results of a phase 2 trial by the German Hodgkin Lymphoma Study Group (GHSG). Blood 2008;111:109–11.
90. Ekstrand BC, Lucas JB, Horwitz SM, et al. Rituximab in lymphocyte-predominant Hodgkin disease: results of a phase 2 trial. Blood 2003;101:4285–9.
91. Younes A, Romaguera J, Hagemeister F, et al. A pilot study of rituximab in patients with recurrent, classic Hodgkin disease. Cancer 2003;98:310–4.
92. Rehwald U, Schulz H, Reiser M, et al. Treatment of relapsed CD20+ Hodgkin lymphoma with the monoclonal antibody rituximab is effective and well tolerated: results of a phase 2 trial of the German Hodgkin Lymphoma Study Group. Blood 2003;101:420–4.
93. Fehniger TA, Larson S, Trinkaus K, et al. A phase 2 multicenter study of lenalidomide in relapsed or refractory classical Hodgkin lymphoma. Blood 2011;118:5119–25.
94. Boll B, Borchmann P, Topp MS, et al. Lenalidomide in patients with refractory or multiple relapsed Hodgkin lymphoma. Br J Haematol 2010;148:480–2.
95. LaCasce AS, Bociek G, Sawas A, et al. Brentuximab vedotin plus bendamustine: a highly active salvage treatment regimen for patients with relapsed or refractory Hodgkin lymphoma. Blood 2015;126:3982.
96. Diefenbach CS, Hong F, David KA, et al. A phase I study with an expansion cohort of the combination of ipilimumab and nivolumab and brentuximab vedotin in patients with relapsed/refractory Hodgkin lymphoma: A trial of the ECOG-ACRIN Cancer Research Group (E4412 Arms D and E). Blood 2016;128:1106.
97. Herrera AF, Bartlett NL, Ramchandren R, et al. Preliminary results from a phase 1/2 study of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016;128:1105.
98. Johnston PB, Inwards DJ, Colgan JP, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol 2010;85:320–4.
99. Kewalramani T, Nimer SD, Zelenetz AD, et al. Progressive disease following autologous transplantation in patients with chemosensitive relapsed or primary refractory Hodgkin’s disease or aggressive non-Hodgkin’s lymphoma. Bone Marrow Transplant 2003;32:673–9.
100. Lin TS, Avalos BR, Penza SL, et al. Second autologous stem cell transplant for multiply relapsed Hodgkin’s disease. Bone Marrow Transplant 2002;29:763–7.
101. Smith SM, van Besien K, Carreras J, et al. Second autologous stem cell transplantation for relapsed lymphoma after a prior autologous transplant. Biol Blood Marrow Transplant 2008;14:904–12.
102. Gajewski JL, Phillips GL, Sobocinski KA, et al. Bone marrow transplants from HLA-identical siblings in advanced Hodgkin’s disease. J Clin Oncol 1996;14:572–8.
103. Peniket AJ, Ruiz de Elvira MC, Taghipour G, et al. An EBMT registry matched study of allogeneic stem cell transplants for lymphoma: allogeneic transplantation is associated with a lower relapse rate but a higher procedure-related mortality rate than autologous transplantation. Bone Marrow Transplant 2003;31:667–78.
104. Anderlini P, Saliba R, Acholonu S, et al. Fludarabine-melphalan as a preparative regimen for reduced-intensity conditioning allogeneic stem cell transplantation in relapsed and refractory Hodgkin’s lymphoma: the updated M.D. Anderson Cancer Center experience. Haematologica 2008;93:257–64.
105. Burroughs LM, O’Donnell PV, Sandmaier BM, et al. Comparison of outcomes of HLA-matched related, unrelated, or HLA-haploidentical related hematopoietic cell transplantation following nonmyeloablative conditioning for relapsed or refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2008;14:1279–87.
106. Peggs KS, Hunter A, Chopra R, et al. Clinical evidence of a graft-versus-Hodgkin’s-lymphoma effect after reduced-intensity allogeneic transplantation. Lancet 2005;365:1934–41.
107. Sureda A, Robinson S, Canals C, et al. Reduced-intensity conditioning compared with conventional allogeneic stem-cell transplantation in relapsed or refractory Hodgkin’s lymphoma: an analysis from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2008;26:455–62.
108. Devetten MP, Hari PN, Carreras J, et al. Unrelated donor reduced-intensity allogeneic hematopoietic stem cell transplantation for relapsed and refractory Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:109–17.
109. Sureda A, Canals C, Arranz R, et al. Allogeneic stem cell transplantation after reduced intensity conditioning in patients with relapsed or refractory Hodgkin’s lymphoma. Results of the HDR-ALLO study - a prospective clinical trial by the Grupo Espanol de Linfomas/Trasplante de Medula Osea (GEL/TAMO) and the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. Haematologica 2012;97:310–7.
110. Sarina B, Castagna L, Farina L, et al. Allogeneic transplantation improves the overall and progression-free survival of Hodgkin lymphoma patients relapsing after autologous transplantation: a retrospective study based on the time of HLA typing and donor availability. Blood 2010;115:3671–7.
111. Castagna L, Sarina B, Todisco E, et al. Allogeneic stem cell transplantation compared with chemotherapy for poor-risk Hodgkin lymphoma. Biol Blood Marrow Transplant 2009;15:432–8.
112. Thomson KJ, Peggs KS, Smith P, et al. Superiority of reduced-intensity allogeneic transplantation over conventional treatment for relapse of Hodgkin’s lymphoma following autologous stem cell transplantation. Bone Marrow Transplant 2008;41:765–70.
113. Chen R, Palmer JM, Thomas SH, et al. Brentuximab vedotin enables successful reduced-intensity allogeneic hematopoietic cell transplantation in patients with relapsed or refractory Hodgkin lymphoma. Blood 2012;119:6379–81.
Tools, Clinical Prediction Rules, and Algorithms for the Insertion of Peripheral Intravenous Catheters in Adult Hospitalized Patients: A Systematic Scoping Review of Literature
Up to a billion peripheral intravenous catheters (PIVCs) are inserted annually; therefore, the importance of this invasive device in modern medicine cannot be argued.1 The insertion of a PIVC is a clinical procedure undertaken by a range of clinical staff and in a variety of patient populations and settings. In many clinical environments (for example, the emergency department [ED]), PIVCs are the predominant first-choice vascular access device (VAD).2,3 Researchers in one study estimated over 25 million PIVCs are used in French EDs each year,3 and intravenous therapy is the leading ED treatment in the United States.4
The purpose of this systematic scoping review was to investigate what PIVC decision-making approaches exist to facilitate FTIS of PIVCs in adult hospitalized patients. Our intention was to systematically synthesize the research on TRAs, to review significant associations identified with these TRAs, and to critique TRA validity and reliability.
METHODS
Scoping Review
We selected a scoping review method that, by definition, maps the evidence to identify gaps,13,14 set research agendas, and identify implications for decision making. This allowed a targeted approach to answering our 3 research questions:
- What published clinical TRAs exist to facilitate PIVC insertion in adults?
- What clinical, patient and/or product variables have been identified using TRAs as having significant associations with FTIS for PIVCs in adult patients?
- What is the reported reliability, validity, responsiveness, clinical feasibility, and utility of existing TRAs for PIVC insertion in adults?
Our aim was to identify the amount, variety and essential qualities of TRA literature rather than to critically appraise and evaluate the effectiveness of TRAs, a process reserved for systematic review and meta-analysis of interventional studies.13,14 We followed scoping review guidelines published by members and collaborators of the Joanna Briggs Institute, an internationally recognized leader in research synthesis, evidence use, and implementation. The guidance is based on 5 steps: (i) scoping review objective and question, (ii) background of the topic to support scoping review, (iii) study selection, (iv) charting the results, and (v) collating and summarizing results.15 Clinicometric assessment of a TRA or any clinical prediction rule requires 4 specific phases: (i) development (identification of predictors from data), (ii) validation (testing the rule in a separate population for reliability), (iii) impact analysis or responsiveness (How clinically useful is the rule in the clinical setting? Is it resource heavy or light? Is it cost effective?), and (iv) implementation and adoption (uptake into clinical practice).16
Search Strategy
We included studies that described the use or development of any TRA regarding PIVC insertion in the adult hospitalized population.
Inclusion Criteria
Studies were included if they were published in the English Language, included TRAs for PIVC insertion in adult hospital patients, and prospectively assessed a clinical category of patient for PIVC insertion using a traditional approach. We defined a traditional PIVC insertion approach as an assessment and/or insertion with touch and feel, therefore, without vessel-locating technology such as ultrasound and/or near infrared technology.
Exclusion Criteria
Exclusion criteria included pediatric studies, authors’ personal (nonresearch) experience of tools, TRAs focused on postinsertion assessment of the cannula (such as phlebitis, infiltration, and/or dressing failure), and papers with a focus on VADs other than PIVCs. We excluded studies using PIVC ultrasound and/or near infrared technology because these are not standard in all insertions and greatly change the information available for pre-insertion assessment as well as the likelihood of insertion success.
In June 2016, a systematic search of the Cochrane library, Ovid Medline® In-process & Other Non-Indexed Citations and Ovid MEDLINE(R) <1946 to Present>, EBSCO CINAHL databases, and Google Scholar with specific keywords to identify publications that identified or defined TRAs was undertaken. Medical subject headings were created with assistance from a research librarian using tailored functions within individual databases. With key search terms, we limited studies to those related to our inclusion criteria. See Appendix 1 for our search strategy for Medline and CINAHL.
We used Covidence, a web-based application specifically designed for systematic reviews to screen and evaluate eligible publications.17 Two authors (PJC and NSH) screened the initial retrieved searches based upon the predetermined inclusion and exclusion criteria.
Data Extraction
A paper template was developed and used by 2 reviewers (P.J.C. and N.S.H.). Data included the following: study sample, aim(s), design, setting and country in which the study took place, clinical and patient variables, and how the TRAs were developed and tested. Studies were categorized by TRA type. We also sought to identify if clinical trial registration (where appropriate) was evidenced, in addition to evidence of protocol publication and what standardized reporting guidelines were used (such as those outlined by the EQUATOR Network).18
Data Synthesis
Formal meta-analysis was beyond the scope and intention of this review. However, we provide the FTIS rate and the ranges of odds ratios (ORs) with 95% confidence intervals (CIs) for certain independent predictors.
RESULTS
Thirty-six references were imported for screening against title and abstract content, with 11 studies excluded and 25 studies assessed for full-text eligibility (see Figure, PRISMA Flowchart). We then excluded a further 12 studies (6 did not meet inclusion criteria, 2 were focused on the prehospital setting, 2 were personal correspondence and focused on another type of VAD, 1 was a protocol to establish a TRA, and 1 was a framework for all device types), leaving 13 studies included in the final review (see Figure). These studies presented data on 4 tools,19-22 4 predictive models3,23-25 (of which 3 present receiver operating characteristic/area under the curve scores),3,23,24 2 framed as risk factor studies,26,27 and 1 of each of the following: a scale,28 a score,29 and an estimation of the incidence report rate (Table 1).30 Seven studies had “difficult” or “difficulty” in their title as a term to use to describe insertion failure.3,19,24-27,30 One study was titled exclusively for the nursing profession,20 5 studies were reported in medical journals,3,24,26,29,30 and 6 were reported in nursing journals,19-22,25,27 with the remainder published in a vascular access journal.23,28
General Characteristics of Included Studies
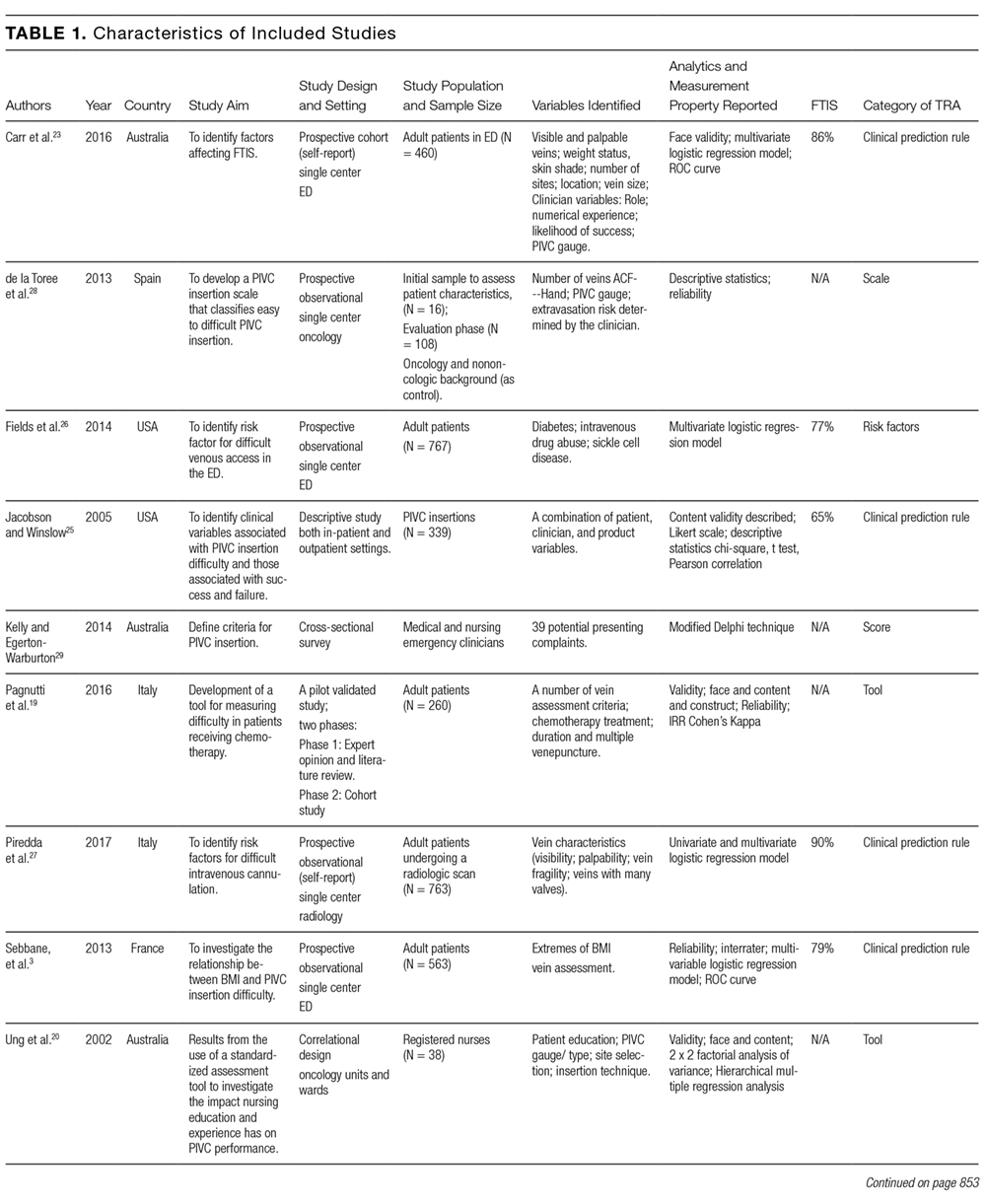
One TRA which was registered as a clinical trial24 involved a standardized reporting tool as is recommended by the EQUATOR Network.18
Nine of the 13 papers reported that TRA components were chosen based on identified predictors of successful insertion from observational data3,19,23-28,30, with 5 papers using multivariate logistic regression to identify independent predictors.3,23,24,26,2 At least 4330 insertion attempts on patients were reported. Seven papers reported FTIS, which ranged from 61%-90%.3,23-27,30
Two clinical settings accounted for 10 of the 13 included studies. We identified 5 papers from the ED setting3,23,26,29,30 and 5 studies specific to cancer settings.19-22,28 Two ED papers identified clinical predictors of insertion difficulty, with 1 identifying an existing medical diagnosis (such as sickle cell disease, diabetes, or intravenous drug abuse) and the other reporting a pragmatic patient self-report of difficulty.26,30 Three studies focused on patient-exclusive variables (such as vein characteristics)19,21,28 and some with a combined clinician and patient focus.3,23-25,27,30Relatively few studies reported interobserver measurements to describe the reliability of clinical assessments made.3,19,21,28 Webster et al. in Australia assessed interrater reliability of a vein assessment tool (VAT) and found high agreement (kappa 0.83 for medical imaging nurses and 0.93 for oncology nurses).21 Wells compared reliability with Altman’s K scores obtained from a different VAT when compared with the Deciding on Intravenous Access tool and found good agreement.22 Vein deterioration was proposed as a variable for inclusion when developing an assessment tool within an oncological context.31 In Spain, de la Torre and colleagues28 demonstrated good interrater agreement (with kappa, 0.77) for the Venous International Assessment (VIA) tool. The VIA offers a grading system scale to predict the patient’s declining vessel size while undergoing chemotherapy via peripheral veins with PIVCs. Grade I suggests little or no insertion failure, whereas a Grade V should predict insertion failure.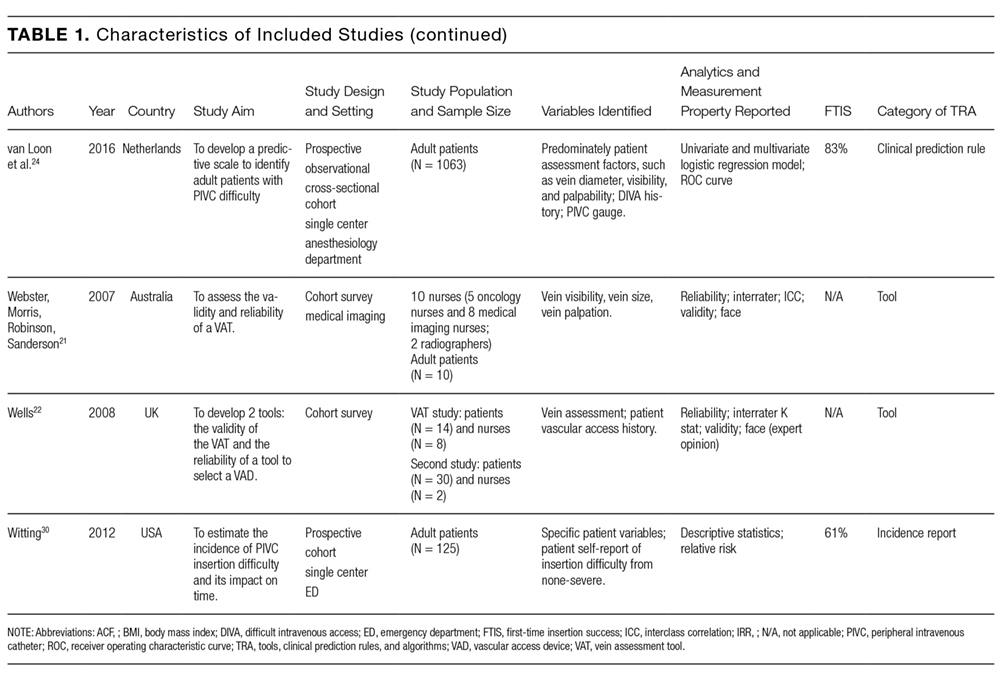
Patient Variables
Vein characteristics were significant independent factors associated with insertion success in a number of studies.3,19,23,24,27,28 These included the number of veins, descriptive quality (eg, small, medium, large), size, location, visible veins, and palpable veins. Other factors appear to be patient specific (such as chronic conditions), including diabetes (OR, 2.1 [adjusted to identify demographic risk factors]; 95% CI, 1.3-3.4), sickle cell disease (OR, 3.5; 95% CI, 1.4-4.8), and intravenous drug abuse (OR, 2.4; 95% CI, 1.1-5.3).26 It is unclear if a consistent relationship between weight classification and insertion outcomes exists. Despite a finding that BMI was not independently associated with insertion difficulty,26 one study reports that BMI was independently associated with insertion failure (BMI <18.5 [OR, 2.24; 95% CI, 1.07-4.67], BMI >30 [OR, 1.98; 95% CI, 1.9-3.60])3 and another reports emaciated patients were associated with greater failure when compared to normal weight patients (OR, 0.07; 95% CI, 0.02-0.34).23 Consequently, extremes of BMI appear to be associated with insertion outcomes despite 1 study reporting no significant association with BMI as an independent factor of insertion failure.26 A history of difficult intravenous access (DIVA) was reported in 1 study and independently associated with insertion failure (OR, 3.86; 95% CI, 2.39-6.25; see Table 2). DIVA appears to be the motivating factor in the title of 7 studies. When defined, the definitions of DIVA are heterogeneous and varied and include the following: >1 minute to insert a PIVC and requiring >1 attempt27; 2 failed attempts30; 3 or more PIVC attempts.26 In the remaining 4 studies, variables associated with difficulty are identified and, therefore, TRAs to target those in future with predicted difficulty prior to any attempts are proposed.3,19,24,25
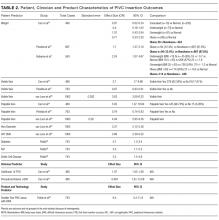
Clinician Variables

Specialist nurse certification, years of experience, and self-report skill level (P < 0.001) appear to be significantly associated with successful insertions.25 This is in part validated in another study reporting greater procedural inserting PIVCs as an independent predictor of success (OR, 4.404; 95% CI, 1.61-12-06; see Table 2).23 Two studies involved simple pragmatic percentage cut offs for PIVCs: likelihood of use29 and likelihood of insertion success.23 One paper using a cross-sectional design that surveyed ED clinicians suggested if the clinician’s predicted likelihood of the patient needing a PIVC was >80%, this was a reasonable trigger for PIVC insertion.29 The other, in a self-report cohort study, reported that a clinician’s likelihood estimation of PIVC FTIS prior to insertion is independently associated with FTIS (OR, 1.06; 95% CI, 1.04-1.07).23
Product Variables
In this review, higher failure rates were identified in smaller sizes (22-24 g).26 One study revealed gauge size was significantly associated with a failed first attempt in a univariate analysis (OR, 0.44; 95% CI, 0.34-0.58), but this was not retained in a multivariate model.24 Matching the PIVC size with vein assessment is considered in the VIA tool.28 It suggests a large PIVC (18 g) can be considered in patients with at least 6 vein options; smaller PIVCs of 22 to 24 g are recommended when 3 or fewer veins are found.28 One paper describes a greater proportion of success between PIVC brands.25
DISCUSSION
The published evidence for TRAs for PIVCs is limited, with few studies using 2 or more reliability, validity, responsiveness, clinical feasibility, or utility measurements in their development. There is a clear need to assess the clinical utility and clinical feasibility of these approaches so they can be externally validated prior to clinical adoption.16 For this reason, a validated TRA is likely required but must be appropriate for the capability of the healthcare services to use it. We suggest the consistent absence of all of these phases is owing to the variety of healthcare practitioners who are responsible for the insertion, the care and surveillance of peripheral cannulae, and the fragmentation of clinical approaches that exist.32
Previously, a comprehensive systematic review on the subject of PIVCs found that the presence of a visible and/or palpable vein is usually associated with FTIS.33 This current review found evidence of simple scores or cutoff percentage estimates in 2 TRA reports to predict either appropriate PIVC insertion or FTIS.23,29 If such methods are supported by future experimental trials, then such simple approaches could initiate huge clinical return, particularly given that idle or unused PIVCs are of substantial clinical concern.34-36 PIVCs transcend a variety of clinical environments with excessive use identified in the ED, where it may be performed for blood sampling alone and, hence, are labeled as “just in case” PIVCs and contribute to the term “idle PIVC.”23,34 Therefore, a clinical indication to perform PIVC insertion in the first instance must be embedded into any TRA; for example, clinical deterioration is likely and the risks are outweighed by benefit, intravenous fluids and/or medicines are required, and/or diagnostic or clinical procedures are requested (such as contrast scans or procedural sedation).
In the majority of papers reviewed, researchers described how to categorize patients into levels of anticipated and predicted difficulty, but none offered corresponding detailed recommendations for strategies to increase insertion success, such as insertion with ultrasound or vascular access expert. Hypothetically, adopting a TRA may assist with the early identification of difficult to cannulate patients who may require a more expert vascular access clinician. However, in this review, we identify that a uniform definition for DIVA is lacking. Both Webster et al.21 and Wells22 suggest that an expert inserter is required if difficult access is identified by their tools, but there is no clear description of the qualities of an expert inserter in the literature.37 Recently, consensus recommendations for the definition of vascular access specialist add to discussions about defining vascular access as an interdisciplinary specialist role.38 This is supported by other publications that highlight the association between PIVC procedural experience and increased insertion success.6,23,39-41With regards to products, PIVC gauge size may or may not be significantly associated with insertion success. For identifying a relationship of PIVC gauge with vein quality, both the vein diameter and description will help with the clinical interpretation of results. For example, it may be the case that bigger veins are easier to insert a PIVC and, thus, larger PIVCs are inserted. The opposite can occur when the veins are small and poorly visualized; hence, one may select a small gauge catheter. This argument is supported by Prottengeier et al.42 in a prehospital study that excluded PIVC size in a multivariate analysis because of confounding. However, gauge size is very likely to influence postinsertion complications. Prospective studies are contradictory and suggest 16 to 18 g PIVCs are more likely to contribute to superficial thrombus,43 phlebitis, and, thus, device failure, in contrast to others reporting more frequent dislodgement with smaller 22 g PIVCs.6,44Finally, the studies included did not assess survival times of the inserted PIVCs, given postinsertion failure in the hospitalized patient is prevalent45 and, importantly, modifiable.46 A TRA may yield initial insertion success, but if postinsertion the PIVC fails because of a modifiable reason that the TRA has not acknowledged, then it may be of negligible overall benefit. Therefore, TRAs for PIVC insertion need calibration, further development, and ongoing refinement prior to external validation testing.24 Future research should also examine the role of TRAs in settings where ultrasound or other insertion technology is routinely used.
CONCLUSION
This review identifies a clinically significant gap in vascular access science. The findings of this review support recent work on vessel health and preservation47-49 and appropriate device insertion.50 It also points to the need for further research on the development and testing of an appropriate clinical TRA to improve vascular access outcomes in clinical practice.
Acknowledgments
The authors thank Ms. Kylie Black and Mr. Simon Lewis, who are medical research librarians at The University of Western Australia.
Disclosure
Mr. Carr has received “speakers bureau” payment form CareFusion in 2013 and Becton Dickinson (BD) in 2014 for lectures on the subject of vascular access. He received a grant from CareFusion (facilitated by his institution at the time) to attend a scientific meeting on vascular access in the USA in 2012. Griffith University has received unrestricted investigator initiated research or educational grants on Marie Cooke’s behalf from product manufacturers: Baxter; Becton, Dickinson and Company; Centurion Medical Products and Entrotech Lifesciences. Griffith University has received unrestricted investigator initiated research or educational grants on Claire M. Rickard’s behalf from product manufacturers: 3M; Adhezion Biomedical, AngioDynamics; Bard, Baxter; B.Braun; Becton, Dickinson and Company; Centurion Medical Products; Cook Medical; Entrotech, Flomedical; ICU Medical; Medtronic; Smiths Medical, Teleflex. Griffith University has received consultancy payments on Claire M. Rickard’s behalf from product manufacturers: 3M, Bard; BBraun, BD, ResQDevices, Smiths Medical. Dr. Higgins and Dr. Rippey have nothing to disclose. All of the aforementioned have not biased or influenced this review.
All authors have made substantial contributions with this review. Each author has contributed to drafting and editing the manuscript and approves the final version for publishing.
1. Alexandrou E, Ray-Barruel G, Carr PJ, et al. International prevalence of the use of peripheral intravenous catheters. J Hosp Med. 2015;10(8):530-533. PubMed
2. Tiwari MM, Hermsen ED, Charlton ME, Anderson JR, Rupp ME. Inappropriate intravascular device use: a prospective study. J Hosp Infect. 2011;78(2):128-132. PubMed
3. Sebbane M, Claret PG, Lefebvre S, et al. Predicting peripheral venous access difficulty in the emergency department using body mass index and a clinical evaluation of venous accessibility. J Emerg Med. 2013;44(2):299-305. PubMed
4. Niska R, Bhuiya F, Xu J. National Hospital Ambulatory Medical Care Survey: 2007 emergency department summary. Natl Health Stat Report. 2010;(26):1-31. PubMed
5. Aulagnier J, Hoc C, Mathieu E, Dreyfus JF, Fischler M, Le Guen M. Efficacy of AccuVein to Facilitate Peripheral Intravenous Placement in Adults Presenting to an Emergency Department: A Randomized Clinical Trial. Acad Emerg Med. 2014;21(8):858-863. PubMed
6. Carr PJ, Glynn RW, Dineen B, Kropmans TJ. A pilot intravenous cannulation team: an Irish perspective. Br J Nurs. 2010;19(10):S19-S27. PubMed
7. Conaghan PG. Predicting outcomes in rheumatoid arthritis. Clin Rheumatol. 2011;30(Suppl 1):S41-S47. PubMed
8. Hendriksen JM, Geersing GJ, Moons KG, de Groot JA. Diagnostic and prognostic prediction models. J Thromb Haemost. 2013;11(Suppl 1):129-141. PubMed
9. Hodgson C, Needham D, Haines K, et al. Feasibility and inter-rater reliability of the ICU Mobility Scale. Heart Lung. 2014;43(1):19-24. PubMed
10. Pace NL, Eberhart LHJ, Kranke PR. Quantifying prognosis with risk predictions. Eur J Anaesthesiol. 2012;29(1):7-16. PubMed
11. Yen K, Riegert A, Gorelick MH. Derivation of the DIVA score: A clinical prediction rule for the identification of children with difficult intravenous access. Pediatr Emerg Care. 2008;24(3):143-147. PubMed
12. Manuel DG, Rosella LC, Hennessy D, Sanmartin C, Wilson K. Predictive risk algorithms in a population setting: An overview. J Epidemiol Community Health. 2012;66(10):859-865. PubMed
13. Tricco AC, Lillie E, Zarin W, et al. A scoping review on the conduct and reporting of scoping reviews. BMC Med Res Methodol. 2016;16:15. PubMed
14. Pham MT, Rajić A, Greig JD, Sargeant JM, Papadopoulos A, McEwen SA. A scoping review of scoping reviews: advancing the approach and enhancing the consistency. Res Synth Methods. 2014;5(4):371-385. PubMed
15. Peters MDJ, Godfrey CM, Khalil H, McInerney P, Parker D, Soares CB. Guidance for conducting systematic scoping reviews. Int J Evid Based Healthc. 2015;13(3):141-146. PubMed
16. Adams ST, Leveson SH. Clinical prediction rules. BMJ. 2012;344:d8312 PubMed
17. Babineau J. Product Review: Covidence (Systematic Review Software). J Can Health Libr Assoc. 2014;35(2):68-71.
18. Morris C. The EQUATOR network: Promoting the transparent and accurate reporting of research. Dev Med Child Neurol. 2008;50(10):723. PubMed
19. Pagnutti L, Bin A, Donato R, et al. Difficult intravenous access tool in patients receiving peripheral chemotherapy: A pilot-validation study. Eur J Oncol Nurs. 2016;20:58-63. PubMed
20. Ung L, Cook S, Edwards B, Hocking L, Osmond F, Buttergieg H. Peripheral intravenous cannulation in nursing: performance predictors. J Infus Nurs. 2002;25(3):189-195. PubMed
21. Webster J, Morris H-L, Robinson K, Sanderson U. Development and validation of a Vein Assessment Tool (VAT). Aust J Adv Nurs. 2007;24(4):5-7. PubMed
22. Wells S. Venous access in oncology and haematology patients: Part two. Nurs Stand. 2008;23(1):35–42. PubMed
23. Carr PJ, Rippey JA, Budgeon CA, Cooke ML, Higgins NS, Rickard Claire M. Insertion of peripheral intravenous cannulae in the Emergency Department: factors associated with first-time insertion success. J Vasc Access. 2016;17(2):182-190. PubMed
24. Loon FHJ van, Puijn LAPM, Houterman S, Bouwman ARA. Development of the A-DIVA Scale: A Clinical Predictive Scale to Identify Difficult Intravenous Access in Adult Patients Based on Clinical Observations. Medicine (Baltimore). 2016;95(16):e3428. PubMed
25. Jacobson AF, Winslow EH. Variables influencing intravenous catheter insertion difficulty and failure: an analysis of 339 intravenous catheter insertions. Heart Lung. 2005;34(5):345-359. PubMed
26. Fields MJ, Piela NE, Au AK, Ku BS. Risk factors associated with difficult venous access in adult ED patients. Am J Emerg Med. 2014;32(10):1179-1182 PubMed
27. Piredda M, Biagioli V, Barrella B, et al. Factors Affecting Difficult Peripheral Intravenous Cannulation in Adults: A Prospective Observational Study. J Clin Nurs. 2017;26(7-8):1074-1084 PubMed
28. de la Torre-Montero J-C, Montealegre-Sanz M, Faraldo-Cabana A, et al. Venous International Assessment, VIA scale, validated classification procedure for the peripheral venous system. J Vasc Access. 2014;15(1):45-50. PubMed
29. Kelly AM, Egerton-Warburton D. When is peripheral intravenous catheter insertion indicated in the emergency department? Emerg Med Australas. 2014;26(5):515–516. PubMed
30. Witting MD. IV access difficulty: Incidence and delays in an urban emergency department. J Emerg Med. 2012;42(4):483-487. PubMed
31. McGowan D, Wood S. Developing a venous assessment tool in IV chemotherapy administration. Br J Nurs. 2008;17(3):158-164. PubMed
32. Castro-Sánchez E, Charani E, Drumright LN, Sevdalis N, Shah N, Holmes AH. Fragmentation of Care Threatens Patient Safety in Peripheral Vascular Catheter Management in Acute Care–A Qualitative Study. PLoS One. 2014;9(1):e86167. PubMed
33. Sabri A, Szalas J, Holmes KS, Labib L, Mussivand T. Failed attempts and improvement strategies in peripheral intravenous catheterization. Biomed Mater Eng. 2013;23(1-2):93-108. PubMed
34. Limm EI, Fang X, Dendle C, Stuart RL, Egerton Warburton D. Half of All Peripheral Intravenous Lines in an Australian Tertiary Emergency Department Are Unused: Pain With No Gain? Ann Emerg Med. 2013;62(5):521-525 PubMed
35. Egerton-Warburton D, Ieraci S. First do no harm: In fact, first do nothing, at least not a cannula. Emerg Med Australas. 2013;25(4):289-290.
36. Becerra MB, Shirley D, Safdar N. Prevalence, risk factors, and outcomes of idle intravenous catheters: An integrative review. Am J Infect Control. 2016;44(10):e167-e172. PubMed
37. Carr PJ, Higgins NS, Cooke ML, Mihala G, Rickard CM. Vascular access specialist teams for device insertion and prevention of failure. Cochrane Library. John Wiley & Sons, Ltd; 2014.
38. Davis L, Owens AK, Thompson J. Defining the Specialty of Vascular Access through Consensus: Shaping the Future of Vascular Access. J Assoc Vasc Access. 2016;21(3):125-130.
39. Da Silva GA, Priebe S, Dias FN. Benefits of establishing an intravenous team and the standardization of peripheral intravenous catheters. J Infus Nurs. 2010;33(3):156-160. PubMed
40. Soifer NE, Borzak S, Edlin BR, Weinstein RA. Prevention of peripheral venous catheter complications with an intravenous therapy team: A randomized controlled trial. Arch Intern Med. 1998;158(5):473-477. PubMed
41. Cuper NJ, de Graaff JC, van Dijk AT, Verdaasdonk RM, van der Werff DB, Kalkman CJ. Predictive factors for difficult intravenous cannulation in pediatric patients at a tertiary pediatric hospital. Paediatr Anaesth. 2012;22(3):223-229. PubMed
42. Prottengeier J, Albermann M, Heinrich S, Birkholz T, Gall C, Schmidt J. The prehospital intravenous access assessment: a prospective study on intravenous access failure and access delay in prehospital emergency medicine. Eur J Emerg Med. 2016; 23(6)442-447. PubMed
43. Cicolini G, Bonghi AP, Di Labio L, Di Mascio R. Position of peripheral venous cannulae and the incidence of thrombophlebitis: an observational study. J Adv Nurs. 2009;65(6):1268-1273. PubMed
44. Wallis MC, McGrail M, Webster J, et al. Risk factors for peripheral intravenous catheter failure: a multivariate analysis of data from a randomized controlled trial. Infect Control Hosp Epidemiol. 2014;35(1):63-68. PubMed
45. Carr PJ, Rippey J, Moore T, et al. Reasons for Removal of Emergency Department-Inserted Peripheral Intravenous Cannulae in Admitted Patients: A Retrospective Medical Chart Audit in Australia. Infect Control Hosp Epidemiol. 2016;37(7):874-876. PubMed
46. Bugden S, Shean K, Scott M, et al. Skin Glue Reduces the Failure Rate of Emergency Department-Inserted Peripheral Intravenous Catheters: A Randomized Controlled Trial. Ann Emerg Med. 2016;68(2):196-201. PubMed
47. Moureau N, Trick N, Nifong T, Perry C, Kelley C, Carrico R, et al. Vessel health and preservation (Part 1): a new evidence-based approach to vascular access selection and management. J Vasc Access. 2012;13(3):351-356. PubMed
48. Jackson T, Hallam C, Corner T, Hill S. Right line, right patient, right time: Every choice matters. Br J Nurs. 2013;22(8):S24-S28. PubMed
49. Hallam C, Weston V, Denton A, et al. Development of the UK Vessel Health and Preservation (VHP) framework: a multi-organisational collaborative. J Infect Prev. 2016;17(2):65-72.
50. Chopra V, Flanders SA, Saint S, et al. The Michigan Appropriateness Guide for Intravenous Catheters (MAGIC): Results From a Multispecialty Panel Using the RAND/UCLA Appropriateness Method. Ann Intern Med. 2015;163(6 Suppl):S1-S40. PubMed
Up to a billion peripheral intravenous catheters (PIVCs) are inserted annually; therefore, the importance of this invasive device in modern medicine cannot be argued.1 The insertion of a PIVC is a clinical procedure undertaken by a range of clinical staff and in a variety of patient populations and settings. In many clinical environments (for example, the emergency department [ED]), PIVCs are the predominant first-choice vascular access device (VAD).2,3 Researchers in one study estimated over 25 million PIVCs are used in French EDs each year,3 and intravenous therapy is the leading ED treatment in the United States.4
The purpose of this systematic scoping review was to investigate what PIVC decision-making approaches exist to facilitate FTIS of PIVCs in adult hospitalized patients. Our intention was to systematically synthesize the research on TRAs, to review significant associations identified with these TRAs, and to critique TRA validity and reliability.
METHODS
Scoping Review
We selected a scoping review method that, by definition, maps the evidence to identify gaps,13,14 set research agendas, and identify implications for decision making. This allowed a targeted approach to answering our 3 research questions:
- What published clinical TRAs exist to facilitate PIVC insertion in adults?
- What clinical, patient and/or product variables have been identified using TRAs as having significant associations with FTIS for PIVCs in adult patients?
- What is the reported reliability, validity, responsiveness, clinical feasibility, and utility of existing TRAs for PIVC insertion in adults?
Our aim was to identify the amount, variety and essential qualities of TRA literature rather than to critically appraise and evaluate the effectiveness of TRAs, a process reserved for systematic review and meta-analysis of interventional studies.13,14 We followed scoping review guidelines published by members and collaborators of the Joanna Briggs Institute, an internationally recognized leader in research synthesis, evidence use, and implementation. The guidance is based on 5 steps: (i) scoping review objective and question, (ii) background of the topic to support scoping review, (iii) study selection, (iv) charting the results, and (v) collating and summarizing results.15 Clinicometric assessment of a TRA or any clinical prediction rule requires 4 specific phases: (i) development (identification of predictors from data), (ii) validation (testing the rule in a separate population for reliability), (iii) impact analysis or responsiveness (How clinically useful is the rule in the clinical setting? Is it resource heavy or light? Is it cost effective?), and (iv) implementation and adoption (uptake into clinical practice).16
Search Strategy
We included studies that described the use or development of any TRA regarding PIVC insertion in the adult hospitalized population.
Inclusion Criteria
Studies were included if they were published in the English Language, included TRAs for PIVC insertion in adult hospital patients, and prospectively assessed a clinical category of patient for PIVC insertion using a traditional approach. We defined a traditional PIVC insertion approach as an assessment and/or insertion with touch and feel, therefore, without vessel-locating technology such as ultrasound and/or near infrared technology.
Exclusion Criteria
Exclusion criteria included pediatric studies, authors’ personal (nonresearch) experience of tools, TRAs focused on postinsertion assessment of the cannula (such as phlebitis, infiltration, and/or dressing failure), and papers with a focus on VADs other than PIVCs. We excluded studies using PIVC ultrasound and/or near infrared technology because these are not standard in all insertions and greatly change the information available for pre-insertion assessment as well as the likelihood of insertion success.
In June 2016, a systematic search of the Cochrane library, Ovid Medline® In-process & Other Non-Indexed Citations and Ovid MEDLINE(R) <1946 to Present>, EBSCO CINAHL databases, and Google Scholar with specific keywords to identify publications that identified or defined TRAs was undertaken. Medical subject headings were created with assistance from a research librarian using tailored functions within individual databases. With key search terms, we limited studies to those related to our inclusion criteria. See Appendix 1 for our search strategy for Medline and CINAHL.
We used Covidence, a web-based application specifically designed for systematic reviews to screen and evaluate eligible publications.17 Two authors (PJC and NSH) screened the initial retrieved searches based upon the predetermined inclusion and exclusion criteria.
Data Extraction
A paper template was developed and used by 2 reviewers (P.J.C. and N.S.H.). Data included the following: study sample, aim(s), design, setting and country in which the study took place, clinical and patient variables, and how the TRAs were developed and tested. Studies were categorized by TRA type. We also sought to identify if clinical trial registration (where appropriate) was evidenced, in addition to evidence of protocol publication and what standardized reporting guidelines were used (such as those outlined by the EQUATOR Network).18
Data Synthesis
Formal meta-analysis was beyond the scope and intention of this review. However, we provide the FTIS rate and the ranges of odds ratios (ORs) with 95% confidence intervals (CIs) for certain independent predictors.
RESULTS
Thirty-six references were imported for screening against title and abstract content, with 11 studies excluded and 25 studies assessed for full-text eligibility (see Figure, PRISMA Flowchart). We then excluded a further 12 studies (6 did not meet inclusion criteria, 2 were focused on the prehospital setting, 2 were personal correspondence and focused on another type of VAD, 1 was a protocol to establish a TRA, and 1 was a framework for all device types), leaving 13 studies included in the final review (see Figure). These studies presented data on 4 tools,19-22 4 predictive models3,23-25 (of which 3 present receiver operating characteristic/area under the curve scores),3,23,24 2 framed as risk factor studies,26,27 and 1 of each of the following: a scale,28 a score,29 and an estimation of the incidence report rate (Table 1).30 Seven studies had “difficult” or “difficulty” in their title as a term to use to describe insertion failure.3,19,24-27,30 One study was titled exclusively for the nursing profession,20 5 studies were reported in medical journals,3,24,26,29,30 and 6 were reported in nursing journals,19-22,25,27 with the remainder published in a vascular access journal.23,28
General Characteristics of Included Studies
One TRA which was registered as a clinical trial24 involved a standardized reporting tool as is recommended by the EQUATOR Network.18
Nine of the 13 papers reported that TRA components were chosen based on identified predictors of successful insertion from observational data3,19,23-28,30, with 5 papers using multivariate logistic regression to identify independent predictors.3,23,24,26,2 At least 4330 insertion attempts on patients were reported. Seven papers reported FTIS, which ranged from 61%-90%.3,23-27,30
Two clinical settings accounted for 10 of the 13 included studies. We identified 5 papers from the ED setting3,23,26,29,30 and 5 studies specific to cancer settings.19-22,28 Two ED papers identified clinical predictors of insertion difficulty, with 1 identifying an existing medical diagnosis (such as sickle cell disease, diabetes, or intravenous drug abuse) and the other reporting a pragmatic patient self-report of difficulty.26,30 Three studies focused on patient-exclusive variables (such as vein characteristics)19,21,28 and some with a combined clinician and patient focus.3,23-25,27,30Relatively few studies reported interobserver measurements to describe the reliability of clinical assessments made.3,19,21,28 Webster et al. in Australia assessed interrater reliability of a vein assessment tool (VAT) and found high agreement (kappa 0.83 for medical imaging nurses and 0.93 for oncology nurses).21 Wells compared reliability with Altman’s K scores obtained from a different VAT when compared with the Deciding on Intravenous Access tool and found good agreement.22 Vein deterioration was proposed as a variable for inclusion when developing an assessment tool within an oncological context.31 In Spain, de la Torre and colleagues28 demonstrated good interrater agreement (with kappa, 0.77) for the Venous International Assessment (VIA) tool. The VIA offers a grading system scale to predict the patient’s declining vessel size while undergoing chemotherapy via peripheral veins with PIVCs. Grade I suggests little or no insertion failure, whereas a Grade V should predict insertion failure.
Patient Variables
Vein characteristics were significant independent factors associated with insertion success in a number of studies.3,19,23,24,27,28 These included the number of veins, descriptive quality (eg, small, medium, large), size, location, visible veins, and palpable veins. Other factors appear to be patient specific (such as chronic conditions), including diabetes (OR, 2.1 [adjusted to identify demographic risk factors]; 95% CI, 1.3-3.4), sickle cell disease (OR, 3.5; 95% CI, 1.4-4.8), and intravenous drug abuse (OR, 2.4; 95% CI, 1.1-5.3).26 It is unclear if a consistent relationship between weight classification and insertion outcomes exists. Despite a finding that BMI was not independently associated with insertion difficulty,26 one study reports that BMI was independently associated with insertion failure (BMI <18.5 [OR, 2.24; 95% CI, 1.07-4.67], BMI >30 [OR, 1.98; 95% CI, 1.9-3.60])3 and another reports emaciated patients were associated with greater failure when compared to normal weight patients (OR, 0.07; 95% CI, 0.02-0.34).23 Consequently, extremes of BMI appear to be associated with insertion outcomes despite 1 study reporting no significant association with BMI as an independent factor of insertion failure.26 A history of difficult intravenous access (DIVA) was reported in 1 study and independently associated with insertion failure (OR, 3.86; 95% CI, 2.39-6.25; see Table 2). DIVA appears to be the motivating factor in the title of 7 studies. When defined, the definitions of DIVA are heterogeneous and varied and include the following: >1 minute to insert a PIVC and requiring >1 attempt27; 2 failed attempts30; 3 or more PIVC attempts.26 In the remaining 4 studies, variables associated with difficulty are identified and, therefore, TRAs to target those in future with predicted difficulty prior to any attempts are proposed.3,19,24,25
Clinician Variables
Specialist nurse certification, years of experience, and self-report skill level (P < 0.001) appear to be significantly associated with successful insertions.25 This is in part validated in another study reporting greater procedural inserting PIVCs as an independent predictor of success (OR, 4.404; 95% CI, 1.61-12-06; see Table 2).23 Two studies involved simple pragmatic percentage cut offs for PIVCs: likelihood of use29 and likelihood of insertion success.23 One paper using a cross-sectional design that surveyed ED clinicians suggested if the clinician’s predicted likelihood of the patient needing a PIVC was >80%, this was a reasonable trigger for PIVC insertion.29 The other, in a self-report cohort study, reported that a clinician’s likelihood estimation of PIVC FTIS prior to insertion is independently associated with FTIS (OR, 1.06; 95% CI, 1.04-1.07).23
Product Variables
In this review, higher failure rates were identified in smaller sizes (22-24 g).26 One study revealed gauge size was significantly associated with a failed first attempt in a univariate analysis (OR, 0.44; 95% CI, 0.34-0.58), but this was not retained in a multivariate model.24 Matching the PIVC size with vein assessment is considered in the VIA tool.28 It suggests a large PIVC (18 g) can be considered in patients with at least 6 vein options; smaller PIVCs of 22 to 24 g are recommended when 3 or fewer veins are found.28 One paper describes a greater proportion of success between PIVC brands.25
DISCUSSION
The published evidence for TRAs for PIVCs is limited, with few studies using 2 or more reliability, validity, responsiveness, clinical feasibility, or utility measurements in their development. There is a clear need to assess the clinical utility and clinical feasibility of these approaches so they can be externally validated prior to clinical adoption.16 For this reason, a validated TRA is likely required but must be appropriate for the capability of the healthcare services to use it. We suggest the consistent absence of all of these phases is owing to the variety of healthcare practitioners who are responsible for the insertion, the care and surveillance of peripheral cannulae, and the fragmentation of clinical approaches that exist.32
Previously, a comprehensive systematic review on the subject of PIVCs found that the presence of a visible and/or palpable vein is usually associated with FTIS.33 This current review found evidence of simple scores or cutoff percentage estimates in 2 TRA reports to predict either appropriate PIVC insertion or FTIS.23,29 If such methods are supported by future experimental trials, then such simple approaches could initiate huge clinical return, particularly given that idle or unused PIVCs are of substantial clinical concern.34-36 PIVCs transcend a variety of clinical environments with excessive use identified in the ED, where it may be performed for blood sampling alone and, hence, are labeled as “just in case” PIVCs and contribute to the term “idle PIVC.”23,34 Therefore, a clinical indication to perform PIVC insertion in the first instance must be embedded into any TRA; for example, clinical deterioration is likely and the risks are outweighed by benefit, intravenous fluids and/or medicines are required, and/or diagnostic or clinical procedures are requested (such as contrast scans or procedural sedation).
In the majority of papers reviewed, researchers described how to categorize patients into levels of anticipated and predicted difficulty, but none offered corresponding detailed recommendations for strategies to increase insertion success, such as insertion with ultrasound or vascular access expert. Hypothetically, adopting a TRA may assist with the early identification of difficult to cannulate patients who may require a more expert vascular access clinician. However, in this review, we identify that a uniform definition for DIVA is lacking. Both Webster et al.21 and Wells22 suggest that an expert inserter is required if difficult access is identified by their tools, but there is no clear description of the qualities of an expert inserter in the literature.37 Recently, consensus recommendations for the definition of vascular access specialist add to discussions about defining vascular access as an interdisciplinary specialist role.38 This is supported by other publications that highlight the association between PIVC procedural experience and increased insertion success.6,23,39-41With regards to products, PIVC gauge size may or may not be significantly associated with insertion success. For identifying a relationship of PIVC gauge with vein quality, both the vein diameter and description will help with the clinical interpretation of results. For example, it may be the case that bigger veins are easier to insert a PIVC and, thus, larger PIVCs are inserted. The opposite can occur when the veins are small and poorly visualized; hence, one may select a small gauge catheter. This argument is supported by Prottengeier et al.42 in a prehospital study that excluded PIVC size in a multivariate analysis because of confounding. However, gauge size is very likely to influence postinsertion complications. Prospective studies are contradictory and suggest 16 to 18 g PIVCs are more likely to contribute to superficial thrombus,43 phlebitis, and, thus, device failure, in contrast to others reporting more frequent dislodgement with smaller 22 g PIVCs.6,44Finally, the studies included did not assess survival times of the inserted PIVCs, given postinsertion failure in the hospitalized patient is prevalent45 and, importantly, modifiable.46 A TRA may yield initial insertion success, but if postinsertion the PIVC fails because of a modifiable reason that the TRA has not acknowledged, then it may be of negligible overall benefit. Therefore, TRAs for PIVC insertion need calibration, further development, and ongoing refinement prior to external validation testing.24 Future research should also examine the role of TRAs in settings where ultrasound or other insertion technology is routinely used.
CONCLUSION
This review identifies a clinically significant gap in vascular access science. The findings of this review support recent work on vessel health and preservation47-49 and appropriate device insertion.50 It also points to the need for further research on the development and testing of an appropriate clinical TRA to improve vascular access outcomes in clinical practice.
Acknowledgments
The authors thank Ms. Kylie Black and Mr. Simon Lewis, who are medical research librarians at The University of Western Australia.
Disclosure
Mr. Carr has received “speakers bureau” payment form CareFusion in 2013 and Becton Dickinson (BD) in 2014 for lectures on the subject of vascular access. He received a grant from CareFusion (facilitated by his institution at the time) to attend a scientific meeting on vascular access in the USA in 2012. Griffith University has received unrestricted investigator initiated research or educational grants on Marie Cooke’s behalf from product manufacturers: Baxter; Becton, Dickinson and Company; Centurion Medical Products and Entrotech Lifesciences. Griffith University has received unrestricted investigator initiated research or educational grants on Claire M. Rickard’s behalf from product manufacturers: 3M; Adhezion Biomedical, AngioDynamics; Bard, Baxter; B.Braun; Becton, Dickinson and Company; Centurion Medical Products; Cook Medical; Entrotech, Flomedical; ICU Medical; Medtronic; Smiths Medical, Teleflex. Griffith University has received consultancy payments on Claire M. Rickard’s behalf from product manufacturers: 3M, Bard; BBraun, BD, ResQDevices, Smiths Medical. Dr. Higgins and Dr. Rippey have nothing to disclose. All of the aforementioned have not biased or influenced this review.
All authors have made substantial contributions with this review. Each author has contributed to drafting and editing the manuscript and approves the final version for publishing.
Up to a billion peripheral intravenous catheters (PIVCs) are inserted annually; therefore, the importance of this invasive device in modern medicine cannot be argued.1 The insertion of a PIVC is a clinical procedure undertaken by a range of clinical staff and in a variety of patient populations and settings. In many clinical environments (for example, the emergency department [ED]), PIVCs are the predominant first-choice vascular access device (VAD).2,3 Researchers in one study estimated over 25 million PIVCs are used in French EDs each year,3 and intravenous therapy is the leading ED treatment in the United States.4
The purpose of this systematic scoping review was to investigate what PIVC decision-making approaches exist to facilitate FTIS of PIVCs in adult hospitalized patients. Our intention was to systematically synthesize the research on TRAs, to review significant associations identified with these TRAs, and to critique TRA validity and reliability.
METHODS
Scoping Review
We selected a scoping review method that, by definition, maps the evidence to identify gaps,13,14 set research agendas, and identify implications for decision making. This allowed a targeted approach to answering our 3 research questions:
- What published clinical TRAs exist to facilitate PIVC insertion in adults?
- What clinical, patient and/or product variables have been identified using TRAs as having significant associations with FTIS for PIVCs in adult patients?
- What is the reported reliability, validity, responsiveness, clinical feasibility, and utility of existing TRAs for PIVC insertion in adults?
Our aim was to identify the amount, variety and essential qualities of TRA literature rather than to critically appraise and evaluate the effectiveness of TRAs, a process reserved for systematic review and meta-analysis of interventional studies.13,14 We followed scoping review guidelines published by members and collaborators of the Joanna Briggs Institute, an internationally recognized leader in research synthesis, evidence use, and implementation. The guidance is based on 5 steps: (i) scoping review objective and question, (ii) background of the topic to support scoping review, (iii) study selection, (iv) charting the results, and (v) collating and summarizing results.15 Clinicometric assessment of a TRA or any clinical prediction rule requires 4 specific phases: (i) development (identification of predictors from data), (ii) validation (testing the rule in a separate population for reliability), (iii) impact analysis or responsiveness (How clinically useful is the rule in the clinical setting? Is it resource heavy or light? Is it cost effective?), and (iv) implementation and adoption (uptake into clinical practice).16
Search Strategy
We included studies that described the use or development of any TRA regarding PIVC insertion in the adult hospitalized population.
Inclusion Criteria
Studies were included if they were published in the English Language, included TRAs for PIVC insertion in adult hospital patients, and prospectively assessed a clinical category of patient for PIVC insertion using a traditional approach. We defined a traditional PIVC insertion approach as an assessment and/or insertion with touch and feel, therefore, without vessel-locating technology such as ultrasound and/or near infrared technology.
Exclusion Criteria
Exclusion criteria included pediatric studies, authors’ personal (nonresearch) experience of tools, TRAs focused on postinsertion assessment of the cannula (such as phlebitis, infiltration, and/or dressing failure), and papers with a focus on VADs other than PIVCs. We excluded studies using PIVC ultrasound and/or near infrared technology because these are not standard in all insertions and greatly change the information available for pre-insertion assessment as well as the likelihood of insertion success.
In June 2016, a systematic search of the Cochrane library, Ovid Medline® In-process & Other Non-Indexed Citations and Ovid MEDLINE(R) <1946 to Present>, EBSCO CINAHL databases, and Google Scholar with specific keywords to identify publications that identified or defined TRAs was undertaken. Medical subject headings were created with assistance from a research librarian using tailored functions within individual databases. With key search terms, we limited studies to those related to our inclusion criteria. See Appendix 1 for our search strategy for Medline and CINAHL.
We used Covidence, a web-based application specifically designed for systematic reviews to screen and evaluate eligible publications.17 Two authors (PJC and NSH) screened the initial retrieved searches based upon the predetermined inclusion and exclusion criteria.
Data Extraction
A paper template was developed and used by 2 reviewers (P.J.C. and N.S.H.). Data included the following: study sample, aim(s), design, setting and country in which the study took place, clinical and patient variables, and how the TRAs were developed and tested. Studies were categorized by TRA type. We also sought to identify if clinical trial registration (where appropriate) was evidenced, in addition to evidence of protocol publication and what standardized reporting guidelines were used (such as those outlined by the EQUATOR Network).18
Data Synthesis
Formal meta-analysis was beyond the scope and intention of this review. However, we provide the FTIS rate and the ranges of odds ratios (ORs) with 95% confidence intervals (CIs) for certain independent predictors.
RESULTS
Thirty-six references were imported for screening against title and abstract content, with 11 studies excluded and 25 studies assessed for full-text eligibility (see Figure, PRISMA Flowchart). We then excluded a further 12 studies (6 did not meet inclusion criteria, 2 were focused on the prehospital setting, 2 were personal correspondence and focused on another type of VAD, 1 was a protocol to establish a TRA, and 1 was a framework for all device types), leaving 13 studies included in the final review (see Figure). These studies presented data on 4 tools,19-22 4 predictive models3,23-25 (of which 3 present receiver operating characteristic/area under the curve scores),3,23,24 2 framed as risk factor studies,26,27 and 1 of each of the following: a scale,28 a score,29 and an estimation of the incidence report rate (Table 1).30 Seven studies had “difficult” or “difficulty” in their title as a term to use to describe insertion failure.3,19,24-27,30 One study was titled exclusively for the nursing profession,20 5 studies were reported in medical journals,3,24,26,29,30 and 6 were reported in nursing journals,19-22,25,27 with the remainder published in a vascular access journal.23,28
General Characteristics of Included Studies
One TRA which was registered as a clinical trial24 involved a standardized reporting tool as is recommended by the EQUATOR Network.18
Nine of the 13 papers reported that TRA components were chosen based on identified predictors of successful insertion from observational data3,19,23-28,30, with 5 papers using multivariate logistic regression to identify independent predictors.3,23,24,26,2 At least 4330 insertion attempts on patients were reported. Seven papers reported FTIS, which ranged from 61%-90%.3,23-27,30
Two clinical settings accounted for 10 of the 13 included studies. We identified 5 papers from the ED setting3,23,26,29,30 and 5 studies specific to cancer settings.19-22,28 Two ED papers identified clinical predictors of insertion difficulty, with 1 identifying an existing medical diagnosis (such as sickle cell disease, diabetes, or intravenous drug abuse) and the other reporting a pragmatic patient self-report of difficulty.26,30 Three studies focused on patient-exclusive variables (such as vein characteristics)19,21,28 and some with a combined clinician and patient focus.3,23-25,27,30Relatively few studies reported interobserver measurements to describe the reliability of clinical assessments made.3,19,21,28 Webster et al. in Australia assessed interrater reliability of a vein assessment tool (VAT) and found high agreement (kappa 0.83 for medical imaging nurses and 0.93 for oncology nurses).21 Wells compared reliability with Altman’s K scores obtained from a different VAT when compared with the Deciding on Intravenous Access tool and found good agreement.22 Vein deterioration was proposed as a variable for inclusion when developing an assessment tool within an oncological context.31 In Spain, de la Torre and colleagues28 demonstrated good interrater agreement (with kappa, 0.77) for the Venous International Assessment (VIA) tool. The VIA offers a grading system scale to predict the patient’s declining vessel size while undergoing chemotherapy via peripheral veins with PIVCs. Grade I suggests little or no insertion failure, whereas a Grade V should predict insertion failure.
Patient Variables
Vein characteristics were significant independent factors associated with insertion success in a number of studies.3,19,23,24,27,28 These included the number of veins, descriptive quality (eg, small, medium, large), size, location, visible veins, and palpable veins. Other factors appear to be patient specific (such as chronic conditions), including diabetes (OR, 2.1 [adjusted to identify demographic risk factors]; 95% CI, 1.3-3.4), sickle cell disease (OR, 3.5; 95% CI, 1.4-4.8), and intravenous drug abuse (OR, 2.4; 95% CI, 1.1-5.3).26 It is unclear if a consistent relationship between weight classification and insertion outcomes exists. Despite a finding that BMI was not independently associated with insertion difficulty,26 one study reports that BMI was independently associated with insertion failure (BMI <18.5 [OR, 2.24; 95% CI, 1.07-4.67], BMI >30 [OR, 1.98; 95% CI, 1.9-3.60])3 and another reports emaciated patients were associated with greater failure when compared to normal weight patients (OR, 0.07; 95% CI, 0.02-0.34).23 Consequently, extremes of BMI appear to be associated with insertion outcomes despite 1 study reporting no significant association with BMI as an independent factor of insertion failure.26 A history of difficult intravenous access (DIVA) was reported in 1 study and independently associated with insertion failure (OR, 3.86; 95% CI, 2.39-6.25; see Table 2). DIVA appears to be the motivating factor in the title of 7 studies. When defined, the definitions of DIVA are heterogeneous and varied and include the following: >1 minute to insert a PIVC and requiring >1 attempt27; 2 failed attempts30; 3 or more PIVC attempts.26 In the remaining 4 studies, variables associated with difficulty are identified and, therefore, TRAs to target those in future with predicted difficulty prior to any attempts are proposed.3,19,24,25
Clinician Variables
Specialist nurse certification, years of experience, and self-report skill level (P < 0.001) appear to be significantly associated with successful insertions.25 This is in part validated in another study reporting greater procedural inserting PIVCs as an independent predictor of success (OR, 4.404; 95% CI, 1.61-12-06; see Table 2).23 Two studies involved simple pragmatic percentage cut offs for PIVCs: likelihood of use29 and likelihood of insertion success.23 One paper using a cross-sectional design that surveyed ED clinicians suggested if the clinician’s predicted likelihood of the patient needing a PIVC was >80%, this was a reasonable trigger for PIVC insertion.29 The other, in a self-report cohort study, reported that a clinician’s likelihood estimation of PIVC FTIS prior to insertion is independently associated with FTIS (OR, 1.06; 95% CI, 1.04-1.07).23
Product Variables
In this review, higher failure rates were identified in smaller sizes (22-24 g).26 One study revealed gauge size was significantly associated with a failed first attempt in a univariate analysis (OR, 0.44; 95% CI, 0.34-0.58), but this was not retained in a multivariate model.24 Matching the PIVC size with vein assessment is considered in the VIA tool.28 It suggests a large PIVC (18 g) can be considered in patients with at least 6 vein options; smaller PIVCs of 22 to 24 g are recommended when 3 or fewer veins are found.28 One paper describes a greater proportion of success between PIVC brands.25
DISCUSSION
The published evidence for TRAs for PIVCs is limited, with few studies using 2 or more reliability, validity, responsiveness, clinical feasibility, or utility measurements in their development. There is a clear need to assess the clinical utility and clinical feasibility of these approaches so they can be externally validated prior to clinical adoption.16 For this reason, a validated TRA is likely required but must be appropriate for the capability of the healthcare services to use it. We suggest the consistent absence of all of these phases is owing to the variety of healthcare practitioners who are responsible for the insertion, the care and surveillance of peripheral cannulae, and the fragmentation of clinical approaches that exist.32
Previously, a comprehensive systematic review on the subject of PIVCs found that the presence of a visible and/or palpable vein is usually associated with FTIS.33 This current review found evidence of simple scores or cutoff percentage estimates in 2 TRA reports to predict either appropriate PIVC insertion or FTIS.23,29 If such methods are supported by future experimental trials, then such simple approaches could initiate huge clinical return, particularly given that idle or unused PIVCs are of substantial clinical concern.34-36 PIVCs transcend a variety of clinical environments with excessive use identified in the ED, where it may be performed for blood sampling alone and, hence, are labeled as “just in case” PIVCs and contribute to the term “idle PIVC.”23,34 Therefore, a clinical indication to perform PIVC insertion in the first instance must be embedded into any TRA; for example, clinical deterioration is likely and the risks are outweighed by benefit, intravenous fluids and/or medicines are required, and/or diagnostic or clinical procedures are requested (such as contrast scans or procedural sedation).
In the majority of papers reviewed, researchers described how to categorize patients into levels of anticipated and predicted difficulty, but none offered corresponding detailed recommendations for strategies to increase insertion success, such as insertion with ultrasound or vascular access expert. Hypothetically, adopting a TRA may assist with the early identification of difficult to cannulate patients who may require a more expert vascular access clinician. However, in this review, we identify that a uniform definition for DIVA is lacking. Both Webster et al.21 and Wells22 suggest that an expert inserter is required if difficult access is identified by their tools, but there is no clear description of the qualities of an expert inserter in the literature.37 Recently, consensus recommendations for the definition of vascular access specialist add to discussions about defining vascular access as an interdisciplinary specialist role.38 This is supported by other publications that highlight the association between PIVC procedural experience and increased insertion success.6,23,39-41With regards to products, PIVC gauge size may or may not be significantly associated with insertion success. For identifying a relationship of PIVC gauge with vein quality, both the vein diameter and description will help with the clinical interpretation of results. For example, it may be the case that bigger veins are easier to insert a PIVC and, thus, larger PIVCs are inserted. The opposite can occur when the veins are small and poorly visualized; hence, one may select a small gauge catheter. This argument is supported by Prottengeier et al.42 in a prehospital study that excluded PIVC size in a multivariate analysis because of confounding. However, gauge size is very likely to influence postinsertion complications. Prospective studies are contradictory and suggest 16 to 18 g PIVCs are more likely to contribute to superficial thrombus,43 phlebitis, and, thus, device failure, in contrast to others reporting more frequent dislodgement with smaller 22 g PIVCs.6,44Finally, the studies included did not assess survival times of the inserted PIVCs, given postinsertion failure in the hospitalized patient is prevalent45 and, importantly, modifiable.46 A TRA may yield initial insertion success, but if postinsertion the PIVC fails because of a modifiable reason that the TRA has not acknowledged, then it may be of negligible overall benefit. Therefore, TRAs for PIVC insertion need calibration, further development, and ongoing refinement prior to external validation testing.24 Future research should also examine the role of TRAs in settings where ultrasound or other insertion technology is routinely used.
CONCLUSION
This review identifies a clinically significant gap in vascular access science. The findings of this review support recent work on vessel health and preservation47-49 and appropriate device insertion.50 It also points to the need for further research on the development and testing of an appropriate clinical TRA to improve vascular access outcomes in clinical practice.
Acknowledgments
The authors thank Ms. Kylie Black and Mr. Simon Lewis, who are medical research librarians at The University of Western Australia.
Disclosure
Mr. Carr has received “speakers bureau” payment form CareFusion in 2013 and Becton Dickinson (BD) in 2014 for lectures on the subject of vascular access. He received a grant from CareFusion (facilitated by his institution at the time) to attend a scientific meeting on vascular access in the USA in 2012. Griffith University has received unrestricted investigator initiated research or educational grants on Marie Cooke’s behalf from product manufacturers: Baxter; Becton, Dickinson and Company; Centurion Medical Products and Entrotech Lifesciences. Griffith University has received unrestricted investigator initiated research or educational grants on Claire M. Rickard’s behalf from product manufacturers: 3M; Adhezion Biomedical, AngioDynamics; Bard, Baxter; B.Braun; Becton, Dickinson and Company; Centurion Medical Products; Cook Medical; Entrotech, Flomedical; ICU Medical; Medtronic; Smiths Medical, Teleflex. Griffith University has received consultancy payments on Claire M. Rickard’s behalf from product manufacturers: 3M, Bard; BBraun, BD, ResQDevices, Smiths Medical. Dr. Higgins and Dr. Rippey have nothing to disclose. All of the aforementioned have not biased or influenced this review.
All authors have made substantial contributions with this review. Each author has contributed to drafting and editing the manuscript and approves the final version for publishing.
1. Alexandrou E, Ray-Barruel G, Carr PJ, et al. International prevalence of the use of peripheral intravenous catheters. J Hosp Med. 2015;10(8):530-533. PubMed
2. Tiwari MM, Hermsen ED, Charlton ME, Anderson JR, Rupp ME. Inappropriate intravascular device use: a prospective study. J Hosp Infect. 2011;78(2):128-132. PubMed
3. Sebbane M, Claret PG, Lefebvre S, et al. Predicting peripheral venous access difficulty in the emergency department using body mass index and a clinical evaluation of venous accessibility. J Emerg Med. 2013;44(2):299-305. PubMed
4. Niska R, Bhuiya F, Xu J. National Hospital Ambulatory Medical Care Survey: 2007 emergency department summary. Natl Health Stat Report. 2010;(26):1-31. PubMed
5. Aulagnier J, Hoc C, Mathieu E, Dreyfus JF, Fischler M, Le Guen M. Efficacy of AccuVein to Facilitate Peripheral Intravenous Placement in Adults Presenting to an Emergency Department: A Randomized Clinical Trial. Acad Emerg Med. 2014;21(8):858-863. PubMed
6. Carr PJ, Glynn RW, Dineen B, Kropmans TJ. A pilot intravenous cannulation team: an Irish perspective. Br J Nurs. 2010;19(10):S19-S27. PubMed
7. Conaghan PG. Predicting outcomes in rheumatoid arthritis. Clin Rheumatol. 2011;30(Suppl 1):S41-S47. PubMed
8. Hendriksen JM, Geersing GJ, Moons KG, de Groot JA. Diagnostic and prognostic prediction models. J Thromb Haemost. 2013;11(Suppl 1):129-141. PubMed
9. Hodgson C, Needham D, Haines K, et al. Feasibility and inter-rater reliability of the ICU Mobility Scale. Heart Lung. 2014;43(1):19-24. PubMed
10. Pace NL, Eberhart LHJ, Kranke PR. Quantifying prognosis with risk predictions. Eur J Anaesthesiol. 2012;29(1):7-16. PubMed
11. Yen K, Riegert A, Gorelick MH. Derivation of the DIVA score: A clinical prediction rule for the identification of children with difficult intravenous access. Pediatr Emerg Care. 2008;24(3):143-147. PubMed
12. Manuel DG, Rosella LC, Hennessy D, Sanmartin C, Wilson K. Predictive risk algorithms in a population setting: An overview. J Epidemiol Community Health. 2012;66(10):859-865. PubMed
13. Tricco AC, Lillie E, Zarin W, et al. A scoping review on the conduct and reporting of scoping reviews. BMC Med Res Methodol. 2016;16:15. PubMed
14. Pham MT, Rajić A, Greig JD, Sargeant JM, Papadopoulos A, McEwen SA. A scoping review of scoping reviews: advancing the approach and enhancing the consistency. Res Synth Methods. 2014;5(4):371-385. PubMed
15. Peters MDJ, Godfrey CM, Khalil H, McInerney P, Parker D, Soares CB. Guidance for conducting systematic scoping reviews. Int J Evid Based Healthc. 2015;13(3):141-146. PubMed
16. Adams ST, Leveson SH. Clinical prediction rules. BMJ. 2012;344:d8312 PubMed
17. Babineau J. Product Review: Covidence (Systematic Review Software). J Can Health Libr Assoc. 2014;35(2):68-71.
18. Morris C. The EQUATOR network: Promoting the transparent and accurate reporting of research. Dev Med Child Neurol. 2008;50(10):723. PubMed
19. Pagnutti L, Bin A, Donato R, et al. Difficult intravenous access tool in patients receiving peripheral chemotherapy: A pilot-validation study. Eur J Oncol Nurs. 2016;20:58-63. PubMed
20. Ung L, Cook S, Edwards B, Hocking L, Osmond F, Buttergieg H. Peripheral intravenous cannulation in nursing: performance predictors. J Infus Nurs. 2002;25(3):189-195. PubMed
21. Webster J, Morris H-L, Robinson K, Sanderson U. Development and validation of a Vein Assessment Tool (VAT). Aust J Adv Nurs. 2007;24(4):5-7. PubMed
22. Wells S. Venous access in oncology and haematology patients: Part two. Nurs Stand. 2008;23(1):35–42. PubMed
23. Carr PJ, Rippey JA, Budgeon CA, Cooke ML, Higgins NS, Rickard Claire M. Insertion of peripheral intravenous cannulae in the Emergency Department: factors associated with first-time insertion success. J Vasc Access. 2016;17(2):182-190. PubMed
24. Loon FHJ van, Puijn LAPM, Houterman S, Bouwman ARA. Development of the A-DIVA Scale: A Clinical Predictive Scale to Identify Difficult Intravenous Access in Adult Patients Based on Clinical Observations. Medicine (Baltimore). 2016;95(16):e3428. PubMed
25. Jacobson AF, Winslow EH. Variables influencing intravenous catheter insertion difficulty and failure: an analysis of 339 intravenous catheter insertions. Heart Lung. 2005;34(5):345-359. PubMed
26. Fields MJ, Piela NE, Au AK, Ku BS. Risk factors associated with difficult venous access in adult ED patients. Am J Emerg Med. 2014;32(10):1179-1182 PubMed
27. Piredda M, Biagioli V, Barrella B, et al. Factors Affecting Difficult Peripheral Intravenous Cannulation in Adults: A Prospective Observational Study. J Clin Nurs. 2017;26(7-8):1074-1084 PubMed
28. de la Torre-Montero J-C, Montealegre-Sanz M, Faraldo-Cabana A, et al. Venous International Assessment, VIA scale, validated classification procedure for the peripheral venous system. J Vasc Access. 2014;15(1):45-50. PubMed
29. Kelly AM, Egerton-Warburton D. When is peripheral intravenous catheter insertion indicated in the emergency department? Emerg Med Australas. 2014;26(5):515–516. PubMed
30. Witting MD. IV access difficulty: Incidence and delays in an urban emergency department. J Emerg Med. 2012;42(4):483-487. PubMed
31. McGowan D, Wood S. Developing a venous assessment tool in IV chemotherapy administration. Br J Nurs. 2008;17(3):158-164. PubMed
32. Castro-Sánchez E, Charani E, Drumright LN, Sevdalis N, Shah N, Holmes AH. Fragmentation of Care Threatens Patient Safety in Peripheral Vascular Catheter Management in Acute Care–A Qualitative Study. PLoS One. 2014;9(1):e86167. PubMed
33. Sabri A, Szalas J, Holmes KS, Labib L, Mussivand T. Failed attempts and improvement strategies in peripheral intravenous catheterization. Biomed Mater Eng. 2013;23(1-2):93-108. PubMed
34. Limm EI, Fang X, Dendle C, Stuart RL, Egerton Warburton D. Half of All Peripheral Intravenous Lines in an Australian Tertiary Emergency Department Are Unused: Pain With No Gain? Ann Emerg Med. 2013;62(5):521-525 PubMed
35. Egerton-Warburton D, Ieraci S. First do no harm: In fact, first do nothing, at least not a cannula. Emerg Med Australas. 2013;25(4):289-290.
36. Becerra MB, Shirley D, Safdar N. Prevalence, risk factors, and outcomes of idle intravenous catheters: An integrative review. Am J Infect Control. 2016;44(10):e167-e172. PubMed
37. Carr PJ, Higgins NS, Cooke ML, Mihala G, Rickard CM. Vascular access specialist teams for device insertion and prevention of failure. Cochrane Library. John Wiley & Sons, Ltd; 2014.
38. Davis L, Owens AK, Thompson J. Defining the Specialty of Vascular Access through Consensus: Shaping the Future of Vascular Access. J Assoc Vasc Access. 2016;21(3):125-130.
39. Da Silva GA, Priebe S, Dias FN. Benefits of establishing an intravenous team and the standardization of peripheral intravenous catheters. J Infus Nurs. 2010;33(3):156-160. PubMed
40. Soifer NE, Borzak S, Edlin BR, Weinstein RA. Prevention of peripheral venous catheter complications with an intravenous therapy team: A randomized controlled trial. Arch Intern Med. 1998;158(5):473-477. PubMed
41. Cuper NJ, de Graaff JC, van Dijk AT, Verdaasdonk RM, van der Werff DB, Kalkman CJ. Predictive factors for difficult intravenous cannulation in pediatric patients at a tertiary pediatric hospital. Paediatr Anaesth. 2012;22(3):223-229. PubMed
42. Prottengeier J, Albermann M, Heinrich S, Birkholz T, Gall C, Schmidt J. The prehospital intravenous access assessment: a prospective study on intravenous access failure and access delay in prehospital emergency medicine. Eur J Emerg Med. 2016; 23(6)442-447. PubMed
43. Cicolini G, Bonghi AP, Di Labio L, Di Mascio R. Position of peripheral venous cannulae and the incidence of thrombophlebitis: an observational study. J Adv Nurs. 2009;65(6):1268-1273. PubMed
44. Wallis MC, McGrail M, Webster J, et al. Risk factors for peripheral intravenous catheter failure: a multivariate analysis of data from a randomized controlled trial. Infect Control Hosp Epidemiol. 2014;35(1):63-68. PubMed
45. Carr PJ, Rippey J, Moore T, et al. Reasons for Removal of Emergency Department-Inserted Peripheral Intravenous Cannulae in Admitted Patients: A Retrospective Medical Chart Audit in Australia. Infect Control Hosp Epidemiol. 2016;37(7):874-876. PubMed
46. Bugden S, Shean K, Scott M, et al. Skin Glue Reduces the Failure Rate of Emergency Department-Inserted Peripheral Intravenous Catheters: A Randomized Controlled Trial. Ann Emerg Med. 2016;68(2):196-201. PubMed
47. Moureau N, Trick N, Nifong T, Perry C, Kelley C, Carrico R, et al. Vessel health and preservation (Part 1): a new evidence-based approach to vascular access selection and management. J Vasc Access. 2012;13(3):351-356. PubMed
48. Jackson T, Hallam C, Corner T, Hill S. Right line, right patient, right time: Every choice matters. Br J Nurs. 2013;22(8):S24-S28. PubMed
49. Hallam C, Weston V, Denton A, et al. Development of the UK Vessel Health and Preservation (VHP) framework: a multi-organisational collaborative. J Infect Prev. 2016;17(2):65-72.
50. Chopra V, Flanders SA, Saint S, et al. The Michigan Appropriateness Guide for Intravenous Catheters (MAGIC): Results From a Multispecialty Panel Using the RAND/UCLA Appropriateness Method. Ann Intern Med. 2015;163(6 Suppl):S1-S40. PubMed
1. Alexandrou E, Ray-Barruel G, Carr PJ, et al. International prevalence of the use of peripheral intravenous catheters. J Hosp Med. 2015;10(8):530-533. PubMed
2. Tiwari MM, Hermsen ED, Charlton ME, Anderson JR, Rupp ME. Inappropriate intravascular device use: a prospective study. J Hosp Infect. 2011;78(2):128-132. PubMed
3. Sebbane M, Claret PG, Lefebvre S, et al. Predicting peripheral venous access difficulty in the emergency department using body mass index and a clinical evaluation of venous accessibility. J Emerg Med. 2013;44(2):299-305. PubMed
4. Niska R, Bhuiya F, Xu J. National Hospital Ambulatory Medical Care Survey: 2007 emergency department summary. Natl Health Stat Report. 2010;(26):1-31. PubMed
5. Aulagnier J, Hoc C, Mathieu E, Dreyfus JF, Fischler M, Le Guen M. Efficacy of AccuVein to Facilitate Peripheral Intravenous Placement in Adults Presenting to an Emergency Department: A Randomized Clinical Trial. Acad Emerg Med. 2014;21(8):858-863. PubMed
6. Carr PJ, Glynn RW, Dineen B, Kropmans TJ. A pilot intravenous cannulation team: an Irish perspective. Br J Nurs. 2010;19(10):S19-S27. PubMed
7. Conaghan PG. Predicting outcomes in rheumatoid arthritis. Clin Rheumatol. 2011;30(Suppl 1):S41-S47. PubMed
8. Hendriksen JM, Geersing GJ, Moons KG, de Groot JA. Diagnostic and prognostic prediction models. J Thromb Haemost. 2013;11(Suppl 1):129-141. PubMed
9. Hodgson C, Needham D, Haines K, et al. Feasibility and inter-rater reliability of the ICU Mobility Scale. Heart Lung. 2014;43(1):19-24. PubMed
10. Pace NL, Eberhart LHJ, Kranke PR. Quantifying prognosis with risk predictions. Eur J Anaesthesiol. 2012;29(1):7-16. PubMed
11. Yen K, Riegert A, Gorelick MH. Derivation of the DIVA score: A clinical prediction rule for the identification of children with difficult intravenous access. Pediatr Emerg Care. 2008;24(3):143-147. PubMed
12. Manuel DG, Rosella LC, Hennessy D, Sanmartin C, Wilson K. Predictive risk algorithms in a population setting: An overview. J Epidemiol Community Health. 2012;66(10):859-865. PubMed
13. Tricco AC, Lillie E, Zarin W, et al. A scoping review on the conduct and reporting of scoping reviews. BMC Med Res Methodol. 2016;16:15. PubMed
14. Pham MT, Rajić A, Greig JD, Sargeant JM, Papadopoulos A, McEwen SA. A scoping review of scoping reviews: advancing the approach and enhancing the consistency. Res Synth Methods. 2014;5(4):371-385. PubMed
15. Peters MDJ, Godfrey CM, Khalil H, McInerney P, Parker D, Soares CB. Guidance for conducting systematic scoping reviews. Int J Evid Based Healthc. 2015;13(3):141-146. PubMed
16. Adams ST, Leveson SH. Clinical prediction rules. BMJ. 2012;344:d8312 PubMed
17. Babineau J. Product Review: Covidence (Systematic Review Software). J Can Health Libr Assoc. 2014;35(2):68-71.
18. Morris C. The EQUATOR network: Promoting the transparent and accurate reporting of research. Dev Med Child Neurol. 2008;50(10):723. PubMed
19. Pagnutti L, Bin A, Donato R, et al. Difficult intravenous access tool in patients receiving peripheral chemotherapy: A pilot-validation study. Eur J Oncol Nurs. 2016;20:58-63. PubMed
20. Ung L, Cook S, Edwards B, Hocking L, Osmond F, Buttergieg H. Peripheral intravenous cannulation in nursing: performance predictors. J Infus Nurs. 2002;25(3):189-195. PubMed
21. Webster J, Morris H-L, Robinson K, Sanderson U. Development and validation of a Vein Assessment Tool (VAT). Aust J Adv Nurs. 2007;24(4):5-7. PubMed
22. Wells S. Venous access in oncology and haematology patients: Part two. Nurs Stand. 2008;23(1):35–42. PubMed
23. Carr PJ, Rippey JA, Budgeon CA, Cooke ML, Higgins NS, Rickard Claire M. Insertion of peripheral intravenous cannulae in the Emergency Department: factors associated with first-time insertion success. J Vasc Access. 2016;17(2):182-190. PubMed
24. Loon FHJ van, Puijn LAPM, Houterman S, Bouwman ARA. Development of the A-DIVA Scale: A Clinical Predictive Scale to Identify Difficult Intravenous Access in Adult Patients Based on Clinical Observations. Medicine (Baltimore). 2016;95(16):e3428. PubMed
25. Jacobson AF, Winslow EH. Variables influencing intravenous catheter insertion difficulty and failure: an analysis of 339 intravenous catheter insertions. Heart Lung. 2005;34(5):345-359. PubMed
26. Fields MJ, Piela NE, Au AK, Ku BS. Risk factors associated with difficult venous access in adult ED patients. Am J Emerg Med. 2014;32(10):1179-1182 PubMed
27. Piredda M, Biagioli V, Barrella B, et al. Factors Affecting Difficult Peripheral Intravenous Cannulation in Adults: A Prospective Observational Study. J Clin Nurs. 2017;26(7-8):1074-1084 PubMed
28. de la Torre-Montero J-C, Montealegre-Sanz M, Faraldo-Cabana A, et al. Venous International Assessment, VIA scale, validated classification procedure for the peripheral venous system. J Vasc Access. 2014;15(1):45-50. PubMed
29. Kelly AM, Egerton-Warburton D. When is peripheral intravenous catheter insertion indicated in the emergency department? Emerg Med Australas. 2014;26(5):515–516. PubMed
30. Witting MD. IV access difficulty: Incidence and delays in an urban emergency department. J Emerg Med. 2012;42(4):483-487. PubMed
31. McGowan D, Wood S. Developing a venous assessment tool in IV chemotherapy administration. Br J Nurs. 2008;17(3):158-164. PubMed
32. Castro-Sánchez E, Charani E, Drumright LN, Sevdalis N, Shah N, Holmes AH. Fragmentation of Care Threatens Patient Safety in Peripheral Vascular Catheter Management in Acute Care–A Qualitative Study. PLoS One. 2014;9(1):e86167. PubMed
33. Sabri A, Szalas J, Holmes KS, Labib L, Mussivand T. Failed attempts and improvement strategies in peripheral intravenous catheterization. Biomed Mater Eng. 2013;23(1-2):93-108. PubMed
34. Limm EI, Fang X, Dendle C, Stuart RL, Egerton Warburton D. Half of All Peripheral Intravenous Lines in an Australian Tertiary Emergency Department Are Unused: Pain With No Gain? Ann Emerg Med. 2013;62(5):521-525 PubMed
35. Egerton-Warburton D, Ieraci S. First do no harm: In fact, first do nothing, at least not a cannula. Emerg Med Australas. 2013;25(4):289-290.
36. Becerra MB, Shirley D, Safdar N. Prevalence, risk factors, and outcomes of idle intravenous catheters: An integrative review. Am J Infect Control. 2016;44(10):e167-e172. PubMed
37. Carr PJ, Higgins NS, Cooke ML, Mihala G, Rickard CM. Vascular access specialist teams for device insertion and prevention of failure. Cochrane Library. John Wiley & Sons, Ltd; 2014.
38. Davis L, Owens AK, Thompson J. Defining the Specialty of Vascular Access through Consensus: Shaping the Future of Vascular Access. J Assoc Vasc Access. 2016;21(3):125-130.
39. Da Silva GA, Priebe S, Dias FN. Benefits of establishing an intravenous team and the standardization of peripheral intravenous catheters. J Infus Nurs. 2010;33(3):156-160. PubMed
40. Soifer NE, Borzak S, Edlin BR, Weinstein RA. Prevention of peripheral venous catheter complications with an intravenous therapy team: A randomized controlled trial. Arch Intern Med. 1998;158(5):473-477. PubMed
41. Cuper NJ, de Graaff JC, van Dijk AT, Verdaasdonk RM, van der Werff DB, Kalkman CJ. Predictive factors for difficult intravenous cannulation in pediatric patients at a tertiary pediatric hospital. Paediatr Anaesth. 2012;22(3):223-229. PubMed
42. Prottengeier J, Albermann M, Heinrich S, Birkholz T, Gall C, Schmidt J. The prehospital intravenous access assessment: a prospective study on intravenous access failure and access delay in prehospital emergency medicine. Eur J Emerg Med. 2016; 23(6)442-447. PubMed
43. Cicolini G, Bonghi AP, Di Labio L, Di Mascio R. Position of peripheral venous cannulae and the incidence of thrombophlebitis: an observational study. J Adv Nurs. 2009;65(6):1268-1273. PubMed
44. Wallis MC, McGrail M, Webster J, et al. Risk factors for peripheral intravenous catheter failure: a multivariate analysis of data from a randomized controlled trial. Infect Control Hosp Epidemiol. 2014;35(1):63-68. PubMed
45. Carr PJ, Rippey J, Moore T, et al. Reasons for Removal of Emergency Department-Inserted Peripheral Intravenous Cannulae in Admitted Patients: A Retrospective Medical Chart Audit in Australia. Infect Control Hosp Epidemiol. 2016;37(7):874-876. PubMed
46. Bugden S, Shean K, Scott M, et al. Skin Glue Reduces the Failure Rate of Emergency Department-Inserted Peripheral Intravenous Catheters: A Randomized Controlled Trial. Ann Emerg Med. 2016;68(2):196-201. PubMed
47. Moureau N, Trick N, Nifong T, Perry C, Kelley C, Carrico R, et al. Vessel health and preservation (Part 1): a new evidence-based approach to vascular access selection and management. J Vasc Access. 2012;13(3):351-356. PubMed
48. Jackson T, Hallam C, Corner T, Hill S. Right line, right patient, right time: Every choice matters. Br J Nurs. 2013;22(8):S24-S28. PubMed
49. Hallam C, Weston V, Denton A, et al. Development of the UK Vessel Health and Preservation (VHP) framework: a multi-organisational collaborative. J Infect Prev. 2016;17(2):65-72.
50. Chopra V, Flanders SA, Saint S, et al. The Michigan Appropriateness Guide for Intravenous Catheters (MAGIC): Results From a Multispecialty Panel Using the RAND/UCLA Appropriateness Method. Ann Intern Med. 2015;163(6 Suppl):S1-S40. PubMed
© 2017 Society of Hospital Medicine
Disseminated Intravascular Coagulation
INTRODUCTION
In the normal person, the process of coagulation is finely controlled at many levels to ensure the appropriate amount of hemostasis at the appropriate location. Broadly defined, disseminated intravascular coagulation (DIC) is the name given to any process that disrupts this fine tuning, leading to unregulated coagulation. Defined this way, DIC may be found in a variety of patients with a variety of disease states, and can present with a spectrum of findings ranging from asymptomatic abnormal laboratory results to florid bleeding or thrombosis. It is important to remember that DIC is always a consequence of an underlying pathological process and not a disease in and of itself. This article first reviews concepts common to all forms of DIC, and then reviews the more common disease states that lead to DIC.
PATHOGENESIS
At the most basic level, DIC is the clinical manifestation of inappropriate thrombin activation.1–5 Inappropriate thrombin activation can be due to underlying conditions such as sepsis and obstetric disasters. The activation of thrombin leads to (1) conversion of fibrinogen to fibrin, (2) activation of platelets (and their consumption), (3) activation of factors V and VIII, (4) activation of protein C (and degradation of factors Va and VIIIa), (5) activation of endothelial cells, and (6) activation of fibrinolysis (Table 1). 
1. Conversion of fibrinogen to fibrin, which leads to the formation of fibrin monomers and excessive thrombus formation. These thrombi are rapidly dissolved by excessive fibrinolysis in most patients. In certain clinical situations, especially cancer, excessive thrombosis will occur. In patients with cancer, this is most often a deep venous thrombosis. Rare patients, especially those with pancreatic cancer, may have severe DIC with multiple arterial and venous thromboses. Nonbacterial thrombotic endocarditis can also be seen in these patients, leading to widespread embolic complications.
2. Activation of platelets and their consumption. Thrombin is the most potent physiologic activator of platelets, so in DIC there is increased activation of platelets. These activated platelets are consumed, resulting in thrombocytopenia. Platelet dysfunction is also present. Platelets that have been activated and have released their contents but still circulate are known as “exhausted” platelets; these cells can no longer function to support coagulation. The fibrin degradation products (FDP) in DIC can also bind to GP IIb/IIIa and inhibit further platelet aggregation.
3. Activation of factors V, VIII, XI, and XIII. Activation of these factors can promote thrombosis, but they are then rapidly cleared by antithrombin (XI) or activated protein C (V and VIII) or by binding to the fibrin clot (XIII). This can lead to depletion of all the prothrombotic clotting factors and antithrombin, which in turn can lead to both thrombosis and bleeding.
4. Activation of protein C further promotes degradation of factors Va and VIIIa, enhances fibrinolysis, and decreases protein C levels.
5. Activation of endothelial cells, especially in the skin, may lead to thrombosis, and in certain patients, especially those with meningococcemia, purpura fulminans. Endothelial damage will down-regulate thrombomodulin, preventing activation of protein C and leading to further reductions in levels of activated protein C.56. Activation of fibrinolysis leads to the breakdown of fibrin monomers, formation of fibrin thrombi, and increased circulating fibrinogen. In most patients with DIC, the fibrinolytic response is brisk.6 This is why most patients with DIC present with bleeding and prolonged clotting times.
PATTERNS OF DIC
The clinical manifestations of DIC in a given patient depend on the balance of thrombin activation and secondary fibrinolysis plus the patient’s ability to compensate for the DIC. Patients with DIC can present in 1 of 4 patterns:1–3
1. Asymptomatic. Patients can present with laboratory evidence of DIC but no bleeding or thrombosis. This is often seen in patients with sepsis or cancer. However, with further progression of the underlying disease, these patients can rapidly become symptomatic.
2. Bleeding. The bleeding is due to a combination of factor depletion, platelet dysfunction, thrombocytopenia, and excessive fibrinolysis.1 These patients may present with diffuse bleeding from multiple sites (eg, intravenous sites, areas of instrumentation).
3. Thrombosis. Despite the general activation of the coagulation process, thrombosis is unusual in most patients with acute DIC. The exceptions include patients with cancer, trauma patients, and certain obstetrical patients. Most often the thrombosis is venous, but arterial thrombosis and nonbacterial thrombotic endocarditis have been reported.7
4. Purpura fulminans. This form of DIC is discussed in more detail later (see Specific DIC Syndromes section).
DIAGNOSIS
There is no one test that will diagnose DIC; one must match the test to the clinical situation (Table 2).8 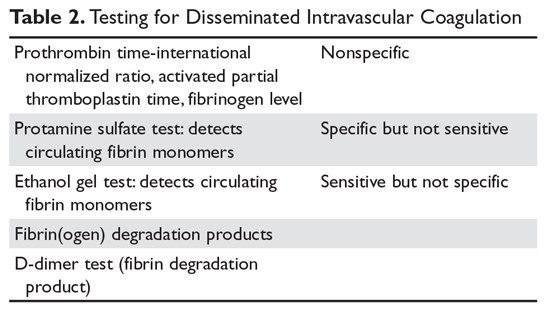
SCREENING TESTS
The prothrombin time-INR and activated thromboplastin time (aPPT) are usually elevated in severe DIC but may be normal or shortened in chronic forms.9 One may also see a shortened aPTT in severe acute DIC due to large amounts of activated thrombin and factor X “bypassing” the contact pathway. An aPTT as short as 10 seconds has been seen in acute DIC. The platelet count is usually reduced but may be normal in chronic DIC. Serum fibrinogen and platelets are decreased in acute DIC but again may be in the “normal” range in chronic DIC.10 The most sensitive screening test for DIC is a fall in the platelet count, with low counts seen in 98% of patients and counts under 50,000 cells/μL in 50%.9,11 The least specific test is fibrinogen, which tends to fall below normal only in severe acute DIC.9
SPECIFIC TESTS
This group of tests allows one to deduce that abnormally high concentrations of thrombin are present.
Ethanol Gel and Protamine Tests
Both of these older tests detected circulating fibrin monomers, whose appearance is an early sign of DIC. Circulating fibrin monomers are seen when thrombin acts on fibrinogen. Usually the monomer polymerizes with the fibrin clot, but when there is excess thrombin these monomers can circulate. Detection of circulating fibrin monomer means there is too much IIa and, ergo, DIC is present.
Fibrin(ogen) Degradation Products
Plasmin acts on the fibrin/fibrinogen molecule to cleave the molecule in specific places. The resulting degradation product levels will be elevated in situations of increased fibrin/fibrinogen destruction (DIC and fibrinolysis). The FDP are typically mildly elevated in renal and liver disease due to reduced clearance.
D-Dimers
When fibrin monomers bind to form a thrombus, factor XIII acts to bind their “D” domains together. This bond is resistant to plasmin and thus this degradation fragment is known as the “D-dimer.” High levels of D-dimer indicate that (1) IIa has acted on fibrinogen to form a fibrin monomer that bonded to another fibrin monomer, and (2) this thrombus was lysed by plasmin. Because D-dimers can be elevated (eg, with exercise, after surgery), an elevated D-dimer needs to be interpreted in the context of the clinical situation.11 Currently, this is the most common specific test for DIC performed.
Other Tests
Several other tests are sometimes helpful in diagnosing DIC.
Thrombin time. This test is performed by adding thrombin to plasma. Thrombin times are elevated in DIC (FDPs interfere with polymerization), in the presence of low fibrinogen levels, in dysfibrinogenemia, and in the presence of heparin (very sensitive).
Reptilase time is the same as thrombin time but is performed with a snake venom that is insensitive to heparin. Reptilase time is elevated in the same conditions as the thrombin time, with the exception of the presence of heparin. Thrombin time and reptilase time are most useful in evaluation of dysfibrinogenemia.
Prothrombin fragment 1.2 (F1.2). F1.2 is a small peptide cleaved off when prothrombin is activated to thrombin. Thus, high levels of F1.2 are found in DIC but can be seen in other thrombotic disorders. This test is still of limited clinical value.
DIC scoring system. A scoring system to both diagnose and quantify DIC has been proposed (Figure).11,12 
Thromboelastography (TEG). This is a point-of-care test that uses whole blood to determine specific coagulation parameters such as R time (time from start of test to clot formation), maximal amplitude (MA, maximum extent of thrombus), and LY30 (MA at 30 minutes, a measure of fibrinolysis).13 Studies have shown that TEG can identify DIC by demonstrating a shorter R time (excess thrombin generation) which prolongs as coagulation factors are consumed. The MA is decreased as fibrinogen is consumed and the LY30 shows excess fibrinolysis. TEG has been shown to be of particular value in the management of the complex coagulopathy of trauma.14
MIMICKERS OF DIC
It is important to recognize coagulation syndromes that are not DIC, especially those that have specific other therapies. The syndromes most frequently encountered are thrombotic thrombocytopenic purpura (TTP) and catastrophic antiphospholipid antibody syndrome (CAPS). One important clue to both of these syndromes is that, unlike DIC, there is no primary disorder (cancer, sepsis) that is driving the coagulation abnormalities.
TTP should be suspected when any patient presents with any combination of thrombocytopenia, microangiopathic hemolytic anemia (schistocytes and signs of hemolysis) plus end-organ damage.15–18 Patients with TTP most often present with intractable seizures, strokes, or sequelae of renal insufficiency. Many patients who present with TTP have been misdiagnosed as having sepsis, “lupus flare,” or vasculitis. The key diagnostic differentiator between TTP and DIC is the lack of activation of coagulation with TTP—fibrinogen is normal and D-dimers are minimally or not elevated. In TTP, lactate dehydrogenase is invariably elevated, often 2 to 3 times normal.19 The importance of identifying TTP is that untreated TTP is rapidly fatal. Mortality in the pre–plasma exchange era ranged from 95% to 100%. Today plasma exchange therapy is the foundation of TTP treatment and has reduced mortality to less than 20%.16,20–23Rarely patients with antiphospholipid antibody syndrome can present with fulminant multiorgan system failure.24–28 CAPS is caused by widespread microthrombi in multiple vascular fields. These patients will develop renal failure, encephalopathy, adult respiratory distress syndrome (often with pulmonary hemorrhage), cardiac failure, dramatic livedo reticularis, and worsening thrombocytopenia. Many of these patients have pre-existing autoimmune disorders and high-titer anticardiolipin antibodies. It appears that the best therapy for these patients is aggressive immunosuppression with steroids plus plasmapheresis, followed by rituximab or, if in the setting of lupus, intravenous cyclophosphamide monthly.27,29 Early recognition of CAPS can lead to quick therapy and resolution of the multiorgan system failure.
GENERAL THERAPY
The best way to treat DIC is to treat the underlying cause that is driving the thrombin generation.1,2,4,30,31 Fully addressing the underlying cause may not be possible or may take time, and in the meantime it is necessary to disrupt the cycle of thrombosis and/or hemorrhage. In the past, there was concern about using factor replacement due to fears of “feeding the fire,” or perpetuating the cycle of thrombosis. However, these concerns are not supported by evidence, and factors must be replaced if depletion occurs and bleeding ensues.32
Transfusion therapy of the patient with DIC is guided by the 5 laboratory tests that reflect the basic parameters essential for both hemostasis and blood volume status:33,34 hematocrit, platelet count, prothrombin time-INR, aPTT, and fibrinogen level. Decisions regarding replacement therapy are based on the results of these laboratory tests and the clinical situation of the patient (Table 3). 
The basic 5 laboratory tests should be repeated after administering the blood products. This allows one to ensure that adequate replacement therapy was given for the coagulation defects. Frequent checks of the coagulation tests also allow rapid identification and treatment of new coagulation defects in a timely fashion. A flow chart of the test and the blood products administered should also be maintained. This is important in acute situations such as trauma or obstetrical bleeding.
In theory, since DIC is the manifestation of exuberant thrombin production, blocking thrombin with heparin should decrease or shut down DIC. However, studies have shown that in most patients heparin administration has led to excessive bleeding. Currently, heparin therapy is reserved for patients who have thrombosis as a component of their DIC.2,41,42 Given the coagulopathy that is often present, specific heparin levels instead of the aPTT should be used to monitor anticoagulation.43,44
SPECIFIC DIC SYNDROMES
SEPSIS/INFECTIOUS DISEASE
Any overwhelming infection can lead to DIC.45 Classically, it was believed that gram-negative bacteria can lead tissue factor exposure via production of endotoxin, but recent studies indicate that DIC can be seen with any overwhelming infection.46,47 There are several potential avenues by which infections can lead to DIC. As mentioned, gram-negative bacteria produce endotoxin that can directly lead to tissue factor exposure, resulting in excess thrombin generation. In addition, any infection can lead to expression of inflammatory cytokines that induce tissue-factor expression by endothelium and monocytes. Some viruses and Rickettsia species can directly infect the vascular endothelium, converting it from an antithrombotic to a prothrombotic phenotype.48 When fighting infections, neutrophils can extrude their contents, including DNA, to help trap organisms. These neutrophil extracellular traps (NETS) may play an important role in promoting coagulopathy.49,50 The hypotension produced by sepsis leads to tissue hypoxia, which results in more DIC. The coagulopathy in sepsis can range from subtle abnormalities of testing to purpura fulminans. Thrombocytopenia is worsened by cytokine-induced hemophagocytic syndrome.
As with all forms of DIC, empiric therapy targeting the most likely source of infection and maintaining hemodynamic stability is the key to therapy. As discussed below, heparin and other forms of coagulation replacement appear to be of no benefit in therapy.
PURPURA FULMINANS
DIC in association with necrosis of the skin is seen in primary and secondary purpura fulminans.51,52 Primary purpura fulminans is most often seen after a viral infection.53 In these patients, the purpura fulminans starts with a painful red area on an extremity that rapidly progresses to a black ischemic area. In many patients, acquired deficiency of protein S is found.51,54,55 Secondary purpura fulminans is most often associated with meningococcemia infections but can be seen in any patient with overwhelming infection.56–58 Post-splenectomy sepsis syndrome patients and those with functional hyposplenism due to chronic liver diseases are also at risk.59 Patients present with signs of sepsis, and the skin lesions often involve the extremities and may lead to amputations. As opposed to primary purpura fulminans, those with the secondary form will have symmetrical ischemia distally (toes and fingers) that ascends as the process progresses. Rarely, adrenal infarction (Waterhouse-Friderichsen syndrome) occurs, which leads to severe hypotension.45
Recently, Warkenten has reported on limb gangrene in critically ill patients complicating sepsis or cardiogenic shock.60,61 These patients have DIC that is complicated by shock liver. Deep venous thrombosis with ischemic gangrene then develops, which can result in tissue loss and even amputation. The pathogenesis is hypothesized to be hepatic dysfunction leading to sudden drops in protein C and S plasma levels, which then leads to thrombophilia with widespread microvascular thrombosis. Therapy for purpura fulminans is controversial. Primary purpura fulminans, especially in those with postvaricella autoimmune protein S deficiency, has responded to plasma infusion titrated to keep the protein S level above 25%.51 Intravenous immunoglobulin has also been reported to help decrease the anti-protein S antibodies. Heparin has been reported to control the DIC and extent of necrosis.62 The starting dose in these patients is 5 to 8 units/kg/hr.2
Sick patients with secondary purpura fulminans have been treated with plasma drips, plasmapheresis, and continuous plasma ultrafiltration.62–66 Heparin therapy alone has not been shown to improve survival.66 Much attention has been given to replacement of natural anticoagulants such as protein C and antithrombin as therapy for purpura fulminans, but unfortunately randomized trials using antithrombin have shown mostly negative results.51,55,67–69 Trials using protein C concentrates have shown more promise in controlling the coagulopathy of purpura fulminans, but this is not widely available.63,70–72 Unfortunately, many patients will need debridement and amputation for their necrotic limbs, with one review showing approximately 66% of patients needing amputations.52
TRAUMA
Currently, the most common cause of acute DIC is trauma. The coagulation defects that occur in trauma patients are complex in origin and still controversial (including if even calling it DIC is appropriate!).73–76 The most common etiologies are
- Generation of excess activated protein C leading to increased consumption of factor V and VIII and increased fibrinolysis;
- Tissue damage leading to generation of excess thrombin generation;
- Dilution of hemostatic factors by blood or fluid resuscitation; and
- Activation of endothelial cells leading to generation of a prothrombotic surface and shedding of glycocalyx with antithrombotic properties.
Trauma patients are prone to hypothermia, and this can be the major complicating factor in their bleeding.77,78 Patients may be out “in the field” for a prolonged period of time and be hypothermic on arrival.79 Packed red cells are stored at 4°C, and the infusion of 1 unit can lower the body temperature by 0.16°C.80 Hypothermia has profound effects on the coagulation system that are associated with clinical bleeding.77,81,82 Even modest hypothermia can greatly augment bleeding and needs to be treated or prevented.
The initial management of the bleeding trauma patient is administration of red cells and plasma (FFP) in a 1:1 ratio. This has been shown by clinical studies to lessen the risk of exsanguination in the first 24 hours and to be associated with improved clinical outcomes.83,84 The basic set of coagulation tests should also be obtained to guide product replacement, especially as the bleeding is brought under control. Hypothermia can be prevented by several measures, including transfusing the blood through blood warmers. Devices are available that can warm 1 unit of blood per minute. An increasingly used technique is to perform “damage control” surgery. Patients are initially stabilized with control of damaged vessels and packing of oozing sites.85 Then the patient is taken to the intensive care unit to be warmed and have coagulation defects corrected.
For trauma patients at risk of serious bleeding, the use of tranexamic acid reduced all- cause mortality (relative risk 0.91), with death due to bleeding also being reduced (relative risk 0.85).86 There was no increase in thrombosis, but benefit was restricted to patients treated within 3 hours of the trauma. The dose of tranexamic acid was a 1-g bolus followed by a 1-g continuous infusion over 8 hours.
PREGNANCY-RELATED DIC SYNDROMES
Acute DIC of Pregnancy
Pregnancy can be associated with the rapid onset of severe DIC in 2 situations, abruption and amniotic fluid embolism.87,88 The separation of the placenta from the uterine wall creates a space for blood to occupy. Given the richness of the placenta in tissue factor, this leads to activation of coagulation both locally and systemically. Release of blood when this space reaches the vaginal opening can lead to rapid hemorrhage, further augmenting the coagulation abnormalities. Placental insufficiency can lead to fetal demise, which can also worsen the DIC. Management depends on the size of the abruption and the clinical status of both mother and fetus.87 For severe bleeding and DIC, blood product support is crucial to allow safe delivery. In pregnancy, the fibrinogen goal needs to be higher—200 mg/dL.89 For smaller abruption, close observation with early delivery is indicated.
Amniotic fluid embolism is sudden, with the vascular collapse of the woman soon after delivery. Due to the presence of procoagulant rich fluid in the circulatory system, there is often overwhelming DIC. Therapy is directed at both supporting blood volume and correcting hemostatic defects.
HELLP
The acronym HELLP (hemolysis, elevated liver tests, low platelets) describes a variant of preeclampsia.90 Classically, HELLP syndrome occurs after 28 weeks of gestation in a patient with preeclampsia, but can occur as early as 22 weeks in patients with antiphospholipid antibody syndrome.91–93 The preeclampsia need not be severe. The first sign of HELLP is a decrease in the platelet count followed by abnormal liver function tests. Signs of hemolysis are present with abundant schistocytes on the smear and a high lactate dehydrogenase level. HELLP can progress to liver failure, and deaths are also reported due to hepatic rupture. Unlike TTP, fetal involvement is present in the HELLP syndrome, with fetal thrombocytopenia reported in 30% of cases. In severe cases, elevated D-dimers consistent with DIC are also found. Delivery of the child will most often result in cessation of the HELLP syndrome, but refractory cases will require dexamethasone and plasma exchange.94 Patients should be closely observed for 1 to 2 days after delivery as the hematologic picture can transiently worsen before improving.95
Acute Fatty Liver of Pregnancy
Fatty liver of pregnancy also occurs late in pregnancy and is only associated with preeclampsia in 50% of cases.96,97 Patients first present with nonspecific symptoms of nausea and vomiting but can progress to fulminant liver failure. Patients develop thrombocytopenia early in the course, but in the later stages can develop DIC and very low fibrinogen levels. Mortality rates without therapy can be as high as 90%. Low blood glucose and high ammonia levels can help distinguish fatty liver from other pregnancy complications.98 Treatment consists of prompt delivery of the child and aggressive blood product support.
Retained Dead Fetus Syndrome
Becoming rarer in modern practices, the presence of a dead fetus for many weeks (usually ≥ 5) can result in a chronic DIC state with fibrinogen depletion and coagulopathy. In some women, this is worsened at delivery. In a stable patient, a short trial of heparin prior to planning delivery can control the DIC to allow the coagulopathy to stabilize.
DRUG-INDUCED HEMOLYTIC-DIC SYNDROMES
A severe variant of the drug-induced immune complex hemolysis associated with DIC has been recognized. Rare patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone) have developed this syndrome.99–104 The clinical syndrome starts 7 to 10 days after the drug is administered. Often the patient has only received the antibiotic for surgical prophylaxis. The patient will develop severe Coombs’-positive hemolysis with hypotension and DIC. The patients are often believed to have sepsis and in the management of the supposed sepsis often are re-exposed to the cephalosporin, resulting in worsening of the clinical picture. The outcome is often fatal due to massive hemolysis and thrombosis.101,105–107
Quinine is associated with a unique syndrome of drug-induced DIC.108–111 Approximately 24 to 96 hours after quinine exposure, the patient becomes acutely ill with nausea and vomiting. The patient then develops a microangiopathic hemolytic anemia, DIC, and renal failure. Some patients, besides having antiplatelet antibodies, also have antibodies binding to red cells and neutrophils, which may lead to the more severe syndrome. Despite therapy, patients with quinine-induced TTP have a high incidence of chronic renal failure.
Treatment of the drug-induced hemolytic-DIC syndrome is anecdotal. Patients have responded to aggressive therapy, including plasma exchange, dialysis, and prednisone. Early recognition of the hemolytic anemia and the suspicion it is drug related is important for early diagnosis so that the incriminated drug can be discontinued.
CANCER
Cancers, primarily adenocarcinomas, can result in DIC. The classic Trousseau syndrome referred to the association of migratory superficial thrombophlebitis with cancer112 but now refers to cancer associated with thrombotic DIC.113,114 Highly vascular tumor cells are known to express tissue factor.114,115 In addition, some tumor cells can express a direct activator of factor X (“cancer procoagulant”). Unlike many DIC states, cancer presents with thrombosis instead of bleeding. This may be due to the inflammatory state which accompanies cancer, or it may be a unique part of the chronic nature of cancer DIC biology that allows time for the body to compensate for loss of coagulation factors. In some patients, thrombosis is the first sign of an underlying cancer, sometimes predating the cancer diagnosis by months.115 Rarely, the DIC can result in nonthrombotic endocarditis with micro-emboli leading to widespread small-vessel thrombosis.113
Since effective antineoplastic therapy is lacking for many tumors associated with Trousseau syndrome, DIC therapy is aimed at suppressing thrombosis. An exception is prostate cancer, where hormonal therapy can markedly decrease the DIC.116 Due to the tumor directly activating coagulation factors, inhibition of active enzymes via heparin has been shown to reduce rates of recurrence compared with warfarin.114,115 Clinical trials have demonstrated that heparin therapy is associated with a lower thrombosis recurrence rate than warfarin.117,118 In some patients, the thrombotic process is so vigorous that new thrombosis can be seen within hours of stopping heparin.112
ACUTE PROMYELOCYTIC LEUKEMIA
There are multiple hemostatic defects in patients with acute promyelocytic leukemia (APL).119 Most, if not all, patients with APL have evidence of DIC at the time of diagnosis. Patients with APL have a higher risk of death during induction therapy as compared with patients with other forms of leukemia, with death most often due to bleeding. Once in remission, APL patients have a higher cure rate than most patients with leukemia. APL is also unique among leukemias in that biologic therapy with retinoic acid or arsenic is effective in inducing remission and cure in most patients. Although effective therapy is available, early death rates due to bleeding have not changed.119
APL patients can present with pancytopenia due to leukemic marrow replacement or with diffuse bleeding due to DIC and thrombocytopenia. Life-threatening bleeding such as intracranial hemorrhage may occur at any time until the leukemia is put into remission. The etiology of the hemostatic defects in APL is complex and is thought to be the result of DIC, fibrinolysis, and the release of prothrombotic extracellular chromatin and other procoagulant enzymes.119,120 The diagnosis of APL can be straightforward when the leukemic cells are promyelocytes with abundant Auer rods, although some patients have the microgranular form without obvious Auer rods. The precise diagnosis requires molecular methods, including obtaining FISH for detecting the t(15;17) in PML/RARA fusion. Upon diagnosis of APL, one should obtain a complete coagulation profile, including INR, aPTT, fibrinogen, platelet count, and D-dimers. Change in fibrinogen levels tends to be a good marker of progress in treating the coagulation defects.
Therapy of APL involves treating both the leukemia and the coagulopathy. Currently, the standard treatment for APL is trans-retinoic acid (ATRA) in combination with chemotherapy or arsenic.121,122 This approach will induce remission in more than 90% of patients, and a sizable majority of these patients will be cured of their APL. ATRA therapy will also lead to early correction of the coagulation defects, often within the first week of therapy.123 This is in stark contrast to the chemotherapy era when the coagulation defects would become worse with therapy. Given the marked beneficial effect of ATRA on the coagulopathy of APL and its low toxicity profile, it should be empirically started for any patients suspected of having APL while genetic testing is being performed. Rare reports of massive thrombosis complicating therapy with ATRA exist, but the relationship to either the APL or ATRA is unknown.
Therapy for the coagulation defects consists of aggressive transfusion therapy support and possible use of other pharmacologic agents to control DIC.124,125 The fibrinogen level should be maintained at over 150 mg/dL and the platelet count at over 50,000 cells/µL.126 Controversy still exists over the role of heparin in therapy of APL.104 Although attractive for its ability to quench thrombin, heparin use can lead to profound bleeding and its use in treating APL has fallen out of favor.
SNAKEBITES
Snake envenomation can lead to direct activation of multiple coagulation enzymes, including factors V, X, thrombin, and protein C, and lead to cleavage of fibrinogen.127,128 Envenomation can also activate coagulation and damage vascular endothelium. The DIC can be enhanced by widespread tissue necrosis and hypotension. The key to management of snake bites is administration of specific antivenom. The role of prophylactic factor replacement is controversial, but this therapy is indicated if there is clinical bleeding.129 One confounder is that some snake venoms, especially rattlesnake, can induce reversible platelet aggregation, which corrects with antivenom.
LOCAL VASCULAR ABNORMALITIES
Abnormal vascular structures, such as vascular tumors, vascular malformations, and aneurysms, can lead to localized areas of thrombin generation that can “spill-over” into the general circulation, leading to DIC. The diagnosis Kasabach-Merritt phenomenon should be reserved for children with vascular tumors such as angioma or hemangioendothelioma.130 Therapy depends on the lesion. Embolization to reduce blood flow of vascular malformations can either be definitive therapy or stabilize the patient for surgery. Aneurysms can be repaired by surgery or stenting. Rare patients with aneurysms with significant coagulopathy may require heparin to raise the fibrinogen level before surgery. Kasabach-Merritt disease can respond to steroids or therapy such as vincristine or interferon.130 Increasing data shows that use of the mTOR inhibitor sirolimus can shrink these vascular abnormalities leading to lessening of the coagulopathy.131
CONCLUSION
At the most basic level, DIC is the excess activity of thrombin. However, the clinical presentation and therapy can differ greatly depending on the primary cause. Both diagnosis and therapy involve close coordination of laboratory data and clinical assessment.
1. Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998;59:65–73.
2. De Jonge E, Levi M, Stoutenbeek CP, Van Deventer SJH. Current drug treatment strategies for disseminated intravascular coagulation. Drugs 1998;55:767–77.
3. Baker WF Jr. Clinical aspects of disseminated intravascular coagulation: a clinician’s point of view. Sem Thrombosis Hemostasis 1989;15:1–57.
4. Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999;341:586–92.
5. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037.
6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016;175:12–23.
7. Sharma S, Mayberry JC, DeLoughery TG, Mullins RJ. Fatal cerebroembolism from nonbacterial thrombotic endocarditis in a trauma patient: case report and review. Mil Med 2000;165:83–5.
8. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505–12.
9. Yu M, Nardella A, Pechet L. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med 2000;28:1777–80.
10. Mant MJ, King EG. Severe, acute disseminated intravascular coagulation. A reappraisal of its pathophysiology, clinical significance, and therapy based on 47 patients. Am J Med 1979;67:557–63.
11. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33.
12. Levi M. Disseminated intravascular coagulation. Crit Care Med 2007;35:2191–5.
13. Nogami K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br J Haematol 2016;174:503–14.
14. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin 2017;33:119–34.
15. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
16. George JN. How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 2000;96:1223–9.
17. Murrin RJ, Murray JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 2006;20:51–60.
18. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836–46.
19. Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol 1994;47:94–9.
20. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991;325:393–7.
21. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpurahemolytic uremic syndrome—clinical experience in 108 patients. N Engl J Med 1991;325:398–403.
22. Kaplan BS, Trachtman H. Improve survival with plasma exchange thrombotic thrombopenic purpura-hemolytic uremic syndrome. Am J Med 2001;110:156–7.
23. Kremer Hovinga JA, Coppo P, Lammle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 2017;3:17020.
24. Asherson RA. The catastrophic antiphospholipid syndrome [editorial]. J Rheumatol 1992;19:508–12.
25. Asherson RA, Piette JC. The catastrophic antiphospholipid syndrome 1996: acute multi-organ failure associated with antiphospholipid antibodies: a review of 31 patients. Lupus 1996;5:414–7.
26. Asherson RA, Cervera R. Castastrophic antiphospholipid syndrome. Curr Opinion Hematol 2000;5:325–9.
27. Merrill JT, Asherson RA. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rhuem 2006;2:81–9.
28. Rodriguez-Pinto I, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: The current management approach. Best Pract Res Clin Rheumatol 2016;30:239–9.
29. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 2016;28:218–27.
30. Hoffman JN, Faist E. Coagulation inhibitor replacement during sepsis: useless? Crit Care Med 2000;28(9 Suppl):S74–6.
31. Wada H, Asakura H, Okamoto K, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Thromb Res 2010;125:6–11.
32. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
33. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
34. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85:487–91.
35. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409–17.
36. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
37. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
38. Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794–801.
39. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
40. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
41. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
42. Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
43. Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993;119:104–9.
44. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782–8.
45. Yoshikawa T, Tanaka KR, Guze LB. Infection and disseminated intravascular coagulation. Medicine (Baltimore) 1971;50:237–58.
46. Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci 2004;328:196–204.
47. Lipinska-Gediga M. Coagulopathy in sepsis - a new look at an old problem. Anaesthesiol Intensive Ther 2016;48:352–9.
48. Van Gorp ECM, Suharti C, ten Cate H, et al. Review: Infections diseases and coagulation disorders. Journal of Infectious Diseases 1999;180:176–86.
49. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129:1357–67.
50. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61.
51. Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol 1998;15:169–83.
52. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol 2007;57:944–56.
53. Spicer TE, Rau JM. Purpura fulminans. Am J Med 1976;61:566–71.
54. Josephson C, Nuss R, Jacobson L, et al. The varicellaautoantibody syndrome. Pediatr Res 2001;50:345–52.
55. Smith OP, White B. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol 1999;104:202–7.
56. Gamper G, Oschatz E, Herkner H, et al. Sepsis-associated purpura fulminans in adults. Wien Klin Wochenschr 2001;113:107–12.
57. Ward KM, Celebi JT, Gmyrek R, Grossman ME. Acute infectious purpura fulminans associated with asplenism or hyposplenism. J Am Acad Dermatol 2002;47:493–6.
58. Childers BJ, Cobanov B. Acute infectious purpura fulminans: a 15-year retrospective review of 28 consecutive cases. Am Surg 2003;69:86–90.
59. Carpenter CT, Kaiser AB. Purpura fulminans in pneumococcal sepsis: case report and review. Scand J Infect Dis 1997;29:479–83.
60. Warkentin TE, Pai M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis: reply. J Thromb Haemost 2016;14:2317–9.
61. Warkentin TE. Ischemic limb gangrene with pulses. N Engl J Med 2015;373:642–55.
62. Duncan A. New therapies for severe meningococcal disease but better outcomes? Lancet 1997;350:1565–6.
63. Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet1997;350:1590–3.
64. Branson HE, Katz J. A structured approach to the management of purpura fulminans. J Natl Med Assoc 1983;75:821–5.
65. Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth 2001;86:581–6.
66. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. Heparin therapy and survival rate. Scand J Infect Dis 1971;3:127–33.
67. Giudici D, Baudo F, Palareti G, et al. Antithrombin replacement in patients with sepsis and septic shock. Haematologica 1999;84:452–60.
68. Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Crit Care Med 2000;28(9 Suppl):S38–43.
69. Levi M, De Jonge E, van der PT, ten Cate H. Novel approaches to the management of disseminated intravascular coagulation. Crit Care Med 2000;28(9 Suppl):S20–4.
70. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura fulminans in meningococcemia with protein C concentrate. J Pediatr 1995;126:646–52.
71. White B, Livingstone W, Murphy C, et al. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood 2000;96:3719–24.
72. Schellongowski P, Bauer E, Holzinger U, et al. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin). Vox Sang 2006;90:294–301.
73. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
74. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin 2017;33:101–18.
75. Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): a focused review on pathophysiology. Intern Emerg Med 2017 May 5.
76. Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 2016;128:1043–9.
77. Eddy VA, Morris JA Jr, Cullinane DC. Hypothermia, coagulopathy, and acidosis. Surg Clin North Am 2000;80:845–54.
78. Peng RY, Bongard FS. Hypothermia in trauma patients. J Am Coll Surg 1999;188:685–96.
79. Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma 1990;30:200–2.
80. Rajek A, Greif R, Sessler DI, et al. Core cooling by central venous infusion of ice-cold (4 degrees C and 20 degrees C) fluid: isolation of core and peripheral thermal compartments. Anesthesiol 2000;93:629–37.
81. Watts DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma 1998;44:846–54.
82. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg 1990;160:515–8.
83. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
84. Johansson PI, Stensballe J, Oliveri R, Wade CE, Ostrowski SR, Holcomb JB. How I treat patients with massive hemorrhage. Blood 2014;124:3052–8.
85. Stone HH, Strom PR, Mullins RJ. Management of the major coagulopathy with onset during laparotomy. Ann Surg 1983;197:532–5.
86. WOMAN Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
87. Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Semin Perinatol 2009;33:189–95.
88. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev 2009;23:167–76.
89. Collins P, Abdul-Kadir R, Thachil J, Subcommittees on Women’ s Health Issues in T, Haemostasis, on Disseminated Intravascular C. Management of coagulopathy associated with postpartum hemorrhage: guidance from the SSC of the ISTH. J Thromb Haemost 2016;14:205–10.
90. Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv 2004;59:838–45.
91. Egerman RS, Sibai BM. HELLP syndrome. Clin Obstetr Gynecol 1999;42:381–9.
92. Saphier CJ, Repke JT. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a review of diagnosis and management. Sem Perinatol 1998;22:118–33.
93. Le Thi TD, Tieulie N, Costedoat N, et al. The HELLP syndrome in the antiphospholipid syndrome: retrospective study of 16 cases in 15 women. Ann Rheum Dis 2005;64:273–8.
94. Martin JN Jr, Perry KG Jr, Blake PG, et al. Better maternal outcomes are achieved with dexamethasone therapy for postpartum HELLP (hemolysis, elevated liver enzymes, and thrombocytopenia) syndrome. Am J Obstet Gynecol 1997;177:1011–7.
95. Magann EF, Martin JN Jr. Twelve steps to optimal management of HELLP syndrome. Clinical Obstet Gynecol 1999;42:532–50.
96. Jwayyed SM, Blanda M, Kubina M. Acute fatty liver of pregnancy. J Emerg Medi 1999;17:673–7.
97. Bacq Y. Acute fatty liver of pregnancy. Sem Perinatol 1998;22:134–40.
98. Egerman RS, Sibai BM. Imitators of preeclampsia and eclampsia. Clin Obstet Gynecol 1999;42:551–62.
99. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Medi Rev 1993;7:255–67.
100. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis 1992;15:863–5.
101. Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion 1992;32:269–71.
102. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes:case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion 1999;39:306–9.
103. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion 1999;39:1239–46.
104. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol 2006;81:186–8.
105. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr 1995;126:813–5.
106. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1995;14:1116–7.
107. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone. J Pediatr 1995;126:816–7.
108. Gottschall JL, Elliot W, Lianos E, et al. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood 1991;77:306–10.
109. Gottschall JL, Neahring B, McFarland JG, et al. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol 1994;47:283–9.
110. Crum NF, Gable P. Quinine-induced hemolytic-uremic syndrome. South Med J 2000;93:726–8.
111. Vesely T, Vesely JN, George JN. Quinine-Induced thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): frequency, clinical features, and long-term outcomes. Blood 2000;96:629 [abstract].
112. Bell WR, Starksen NF, Tong S, Porterfield JK. Trousseau’s syndrome. Devastating coagulopathy in the absence of heparin. Am J Med 1985;79:423–30.
113. Sack GH, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinic, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
114. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007;110:1723–9.
115. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol 2005;6:401–10.
116. de la Fouchardiere C, Flechon A, Droz JP. Coagulopathy in prostate cancer. Neth J Med 2003;61:347–54.
117. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest 2008;133(6 Suppl):454S–545S.
118. Lee AY, Kamphuisen PW, Meyer G, et al. Tinzaparin vs warfarin for treatment of acute venous thromboembolism in patients with active cancer: a randomized clinical trial. JAMA 2015;314:677–86.
119. Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol 2012;87:596–603.
120. Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017;129:1855–64.
121. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
122. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–21.
123. Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated iwth acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2–9.
124. Falanga A, Rickles FR. Management of thrombohemorrhagic syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program 2007;2007:165–71
125. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
126. Sanz MA, Grimwade D, Tallman MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
127. Lu Q, Clemetson JM, Clemetson KJ. Snake venoms and hemostasis. J Thromb Haemost 2005;3:1791–9.
128. Berling I, Isbister GK. Hematologic effects and complications of snake envenoming. Transfus Med Rev 2015;29:82–9.
129. Isbister GK, Jayamanne S, Mohamed F, et al. A randomized controlled trial of fresh frozen plasma for coagulopathy in Russell’s viper (Daboia russelii) envenoming. J Thromb Haemost 2017;15:645–54.
130. Rodriguez V, Lee A, Witman PM, Anderson PA. Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 2009;31:522–6.
131. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 2017;27:86–90.
INTRODUCTION
In the normal person, the process of coagulation is finely controlled at many levels to ensure the appropriate amount of hemostasis at the appropriate location. Broadly defined, disseminated intravascular coagulation (DIC) is the name given to any process that disrupts this fine tuning, leading to unregulated coagulation. Defined this way, DIC may be found in a variety of patients with a variety of disease states, and can present with a spectrum of findings ranging from asymptomatic abnormal laboratory results to florid bleeding or thrombosis. It is important to remember that DIC is always a consequence of an underlying pathological process and not a disease in and of itself. This article first reviews concepts common to all forms of DIC, and then reviews the more common disease states that lead to DIC.
PATHOGENESIS
At the most basic level, DIC is the clinical manifestation of inappropriate thrombin activation.1–5 Inappropriate thrombin activation can be due to underlying conditions such as sepsis and obstetric disasters. The activation of thrombin leads to (1) conversion of fibrinogen to fibrin, (2) activation of platelets (and their consumption), (3) activation of factors V and VIII, (4) activation of protein C (and degradation of factors Va and VIIIa), (5) activation of endothelial cells, and (6) activation of fibrinolysis (Table 1).
1. Conversion of fibrinogen to fibrin, which leads to the formation of fibrin monomers and excessive thrombus formation. These thrombi are rapidly dissolved by excessive fibrinolysis in most patients. In certain clinical situations, especially cancer, excessive thrombosis will occur. In patients with cancer, this is most often a deep venous thrombosis. Rare patients, especially those with pancreatic cancer, may have severe DIC with multiple arterial and venous thromboses. Nonbacterial thrombotic endocarditis can also be seen in these patients, leading to widespread embolic complications.
2. Activation of platelets and their consumption. Thrombin is the most potent physiologic activator of platelets, so in DIC there is increased activation of platelets. These activated platelets are consumed, resulting in thrombocytopenia. Platelet dysfunction is also present. Platelets that have been activated and have released their contents but still circulate are known as “exhausted” platelets; these cells can no longer function to support coagulation. The fibrin degradation products (FDP) in DIC can also bind to GP IIb/IIIa and inhibit further platelet aggregation.
3. Activation of factors V, VIII, XI, and XIII. Activation of these factors can promote thrombosis, but they are then rapidly cleared by antithrombin (XI) or activated protein C (V and VIII) or by binding to the fibrin clot (XIII). This can lead to depletion of all the prothrombotic clotting factors and antithrombin, which in turn can lead to both thrombosis and bleeding.
4. Activation of protein C further promotes degradation of factors Va and VIIIa, enhances fibrinolysis, and decreases protein C levels.
5. Activation of endothelial cells, especially in the skin, may lead to thrombosis, and in certain patients, especially those with meningococcemia, purpura fulminans. Endothelial damage will down-regulate thrombomodulin, preventing activation of protein C and leading to further reductions in levels of activated protein C.56. Activation of fibrinolysis leads to the breakdown of fibrin monomers, formation of fibrin thrombi, and increased circulating fibrinogen. In most patients with DIC, the fibrinolytic response is brisk.6 This is why most patients with DIC present with bleeding and prolonged clotting times.
PATTERNS OF DIC
The clinical manifestations of DIC in a given patient depend on the balance of thrombin activation and secondary fibrinolysis plus the patient’s ability to compensate for the DIC. Patients with DIC can present in 1 of 4 patterns:1–3
1. Asymptomatic. Patients can present with laboratory evidence of DIC but no bleeding or thrombosis. This is often seen in patients with sepsis or cancer. However, with further progression of the underlying disease, these patients can rapidly become symptomatic.
2. Bleeding. The bleeding is due to a combination of factor depletion, platelet dysfunction, thrombocytopenia, and excessive fibrinolysis.1 These patients may present with diffuse bleeding from multiple sites (eg, intravenous sites, areas of instrumentation).
3. Thrombosis. Despite the general activation of the coagulation process, thrombosis is unusual in most patients with acute DIC. The exceptions include patients with cancer, trauma patients, and certain obstetrical patients. Most often the thrombosis is venous, but arterial thrombosis and nonbacterial thrombotic endocarditis have been reported.7
4. Purpura fulminans. This form of DIC is discussed in more detail later (see Specific DIC Syndromes section).
DIAGNOSIS
There is no one test that will diagnose DIC; one must match the test to the clinical situation (Table 2).8
SCREENING TESTS
The prothrombin time-INR and activated thromboplastin time (aPPT) are usually elevated in severe DIC but may be normal or shortened in chronic forms.9 One may also see a shortened aPTT in severe acute DIC due to large amounts of activated thrombin and factor X “bypassing” the contact pathway. An aPTT as short as 10 seconds has been seen in acute DIC. The platelet count is usually reduced but may be normal in chronic DIC. Serum fibrinogen and platelets are decreased in acute DIC but again may be in the “normal” range in chronic DIC.10 The most sensitive screening test for DIC is a fall in the platelet count, with low counts seen in 98% of patients and counts under 50,000 cells/μL in 50%.9,11 The least specific test is fibrinogen, which tends to fall below normal only in severe acute DIC.9
SPECIFIC TESTS
This group of tests allows one to deduce that abnormally high concentrations of thrombin are present.
Ethanol Gel and Protamine Tests
Both of these older tests detected circulating fibrin monomers, whose appearance is an early sign of DIC. Circulating fibrin monomers are seen when thrombin acts on fibrinogen. Usually the monomer polymerizes with the fibrin clot, but when there is excess thrombin these monomers can circulate. Detection of circulating fibrin monomer means there is too much IIa and, ergo, DIC is present.
Fibrin(ogen) Degradation Products
Plasmin acts on the fibrin/fibrinogen molecule to cleave the molecule in specific places. The resulting degradation product levels will be elevated in situations of increased fibrin/fibrinogen destruction (DIC and fibrinolysis). The FDP are typically mildly elevated in renal and liver disease due to reduced clearance.
D-Dimers
When fibrin monomers bind to form a thrombus, factor XIII acts to bind their “D” domains together. This bond is resistant to plasmin and thus this degradation fragment is known as the “D-dimer.” High levels of D-dimer indicate that (1) IIa has acted on fibrinogen to form a fibrin monomer that bonded to another fibrin monomer, and (2) this thrombus was lysed by plasmin. Because D-dimers can be elevated (eg, with exercise, after surgery), an elevated D-dimer needs to be interpreted in the context of the clinical situation.11 Currently, this is the most common specific test for DIC performed.
Other Tests
Several other tests are sometimes helpful in diagnosing DIC.
Thrombin time. This test is performed by adding thrombin to plasma. Thrombin times are elevated in DIC (FDPs interfere with polymerization), in the presence of low fibrinogen levels, in dysfibrinogenemia, and in the presence of heparin (very sensitive).
Reptilase time is the same as thrombin time but is performed with a snake venom that is insensitive to heparin. Reptilase time is elevated in the same conditions as the thrombin time, with the exception of the presence of heparin. Thrombin time and reptilase time are most useful in evaluation of dysfibrinogenemia.
Prothrombin fragment 1.2 (F1.2). F1.2 is a small peptide cleaved off when prothrombin is activated to thrombin. Thus, high levels of F1.2 are found in DIC but can be seen in other thrombotic disorders. This test is still of limited clinical value.
DIC scoring system. A scoring system to both diagnose and quantify DIC has been proposed (Figure).11,12
Thromboelastography (TEG). This is a point-of-care test that uses whole blood to determine specific coagulation parameters such as R time (time from start of test to clot formation), maximal amplitude (MA, maximum extent of thrombus), and LY30 (MA at 30 minutes, a measure of fibrinolysis).13 Studies have shown that TEG can identify DIC by demonstrating a shorter R time (excess thrombin generation) which prolongs as coagulation factors are consumed. The MA is decreased as fibrinogen is consumed and the LY30 shows excess fibrinolysis. TEG has been shown to be of particular value in the management of the complex coagulopathy of trauma.14
MIMICKERS OF DIC
It is important to recognize coagulation syndromes that are not DIC, especially those that have specific other therapies. The syndromes most frequently encountered are thrombotic thrombocytopenic purpura (TTP) and catastrophic antiphospholipid antibody syndrome (CAPS). One important clue to both of these syndromes is that, unlike DIC, there is no primary disorder (cancer, sepsis) that is driving the coagulation abnormalities.
TTP should be suspected when any patient presents with any combination of thrombocytopenia, microangiopathic hemolytic anemia (schistocytes and signs of hemolysis) plus end-organ damage.15–18 Patients with TTP most often present with intractable seizures, strokes, or sequelae of renal insufficiency. Many patients who present with TTP have been misdiagnosed as having sepsis, “lupus flare,” or vasculitis. The key diagnostic differentiator between TTP and DIC is the lack of activation of coagulation with TTP—fibrinogen is normal and D-dimers are minimally or not elevated. In TTP, lactate dehydrogenase is invariably elevated, often 2 to 3 times normal.19 The importance of identifying TTP is that untreated TTP is rapidly fatal. Mortality in the pre–plasma exchange era ranged from 95% to 100%. Today plasma exchange therapy is the foundation of TTP treatment and has reduced mortality to less than 20%.16,20–23Rarely patients with antiphospholipid antibody syndrome can present with fulminant multiorgan system failure.24–28 CAPS is caused by widespread microthrombi in multiple vascular fields. These patients will develop renal failure, encephalopathy, adult respiratory distress syndrome (often with pulmonary hemorrhage), cardiac failure, dramatic livedo reticularis, and worsening thrombocytopenia. Many of these patients have pre-existing autoimmune disorders and high-titer anticardiolipin antibodies. It appears that the best therapy for these patients is aggressive immunosuppression with steroids plus plasmapheresis, followed by rituximab or, if in the setting of lupus, intravenous cyclophosphamide monthly.27,29 Early recognition of CAPS can lead to quick therapy and resolution of the multiorgan system failure.
GENERAL THERAPY
The best way to treat DIC is to treat the underlying cause that is driving the thrombin generation.1,2,4,30,31 Fully addressing the underlying cause may not be possible or may take time, and in the meantime it is necessary to disrupt the cycle of thrombosis and/or hemorrhage. In the past, there was concern about using factor replacement due to fears of “feeding the fire,” or perpetuating the cycle of thrombosis. However, these concerns are not supported by evidence, and factors must be replaced if depletion occurs and bleeding ensues.32
Transfusion therapy of the patient with DIC is guided by the 5 laboratory tests that reflect the basic parameters essential for both hemostasis and blood volume status:33,34 hematocrit, platelet count, prothrombin time-INR, aPTT, and fibrinogen level. Decisions regarding replacement therapy are based on the results of these laboratory tests and the clinical situation of the patient (Table 3).
The basic 5 laboratory tests should be repeated after administering the blood products. This allows one to ensure that adequate replacement therapy was given for the coagulation defects. Frequent checks of the coagulation tests also allow rapid identification and treatment of new coagulation defects in a timely fashion. A flow chart of the test and the blood products administered should also be maintained. This is important in acute situations such as trauma or obstetrical bleeding.
In theory, since DIC is the manifestation of exuberant thrombin production, blocking thrombin with heparin should decrease or shut down DIC. However, studies have shown that in most patients heparin administration has led to excessive bleeding. Currently, heparin therapy is reserved for patients who have thrombosis as a component of their DIC.2,41,42 Given the coagulopathy that is often present, specific heparin levels instead of the aPTT should be used to monitor anticoagulation.43,44
SPECIFIC DIC SYNDROMES
SEPSIS/INFECTIOUS DISEASE
Any overwhelming infection can lead to DIC.45 Classically, it was believed that gram-negative bacteria can lead tissue factor exposure via production of endotoxin, but recent studies indicate that DIC can be seen with any overwhelming infection.46,47 There are several potential avenues by which infections can lead to DIC. As mentioned, gram-negative bacteria produce endotoxin that can directly lead to tissue factor exposure, resulting in excess thrombin generation. In addition, any infection can lead to expression of inflammatory cytokines that induce tissue-factor expression by endothelium and monocytes. Some viruses and Rickettsia species can directly infect the vascular endothelium, converting it from an antithrombotic to a prothrombotic phenotype.48 When fighting infections, neutrophils can extrude their contents, including DNA, to help trap organisms. These neutrophil extracellular traps (NETS) may play an important role in promoting coagulopathy.49,50 The hypotension produced by sepsis leads to tissue hypoxia, which results in more DIC. The coagulopathy in sepsis can range from subtle abnormalities of testing to purpura fulminans. Thrombocytopenia is worsened by cytokine-induced hemophagocytic syndrome.
As with all forms of DIC, empiric therapy targeting the most likely source of infection and maintaining hemodynamic stability is the key to therapy. As discussed below, heparin and other forms of coagulation replacement appear to be of no benefit in therapy.
PURPURA FULMINANS
DIC in association with necrosis of the skin is seen in primary and secondary purpura fulminans.51,52 Primary purpura fulminans is most often seen after a viral infection.53 In these patients, the purpura fulminans starts with a painful red area on an extremity that rapidly progresses to a black ischemic area. In many patients, acquired deficiency of protein S is found.51,54,55 Secondary purpura fulminans is most often associated with meningococcemia infections but can be seen in any patient with overwhelming infection.56–58 Post-splenectomy sepsis syndrome patients and those with functional hyposplenism due to chronic liver diseases are also at risk.59 Patients present with signs of sepsis, and the skin lesions often involve the extremities and may lead to amputations. As opposed to primary purpura fulminans, those with the secondary form will have symmetrical ischemia distally (toes and fingers) that ascends as the process progresses. Rarely, adrenal infarction (Waterhouse-Friderichsen syndrome) occurs, which leads to severe hypotension.45
Recently, Warkenten has reported on limb gangrene in critically ill patients complicating sepsis or cardiogenic shock.60,61 These patients have DIC that is complicated by shock liver. Deep venous thrombosis with ischemic gangrene then develops, which can result in tissue loss and even amputation. The pathogenesis is hypothesized to be hepatic dysfunction leading to sudden drops in protein C and S plasma levels, which then leads to thrombophilia with widespread microvascular thrombosis. Therapy for purpura fulminans is controversial. Primary purpura fulminans, especially in those with postvaricella autoimmune protein S deficiency, has responded to plasma infusion titrated to keep the protein S level above 25%.51 Intravenous immunoglobulin has also been reported to help decrease the anti-protein S antibodies. Heparin has been reported to control the DIC and extent of necrosis.62 The starting dose in these patients is 5 to 8 units/kg/hr.2
Sick patients with secondary purpura fulminans have been treated with plasma drips, plasmapheresis, and continuous plasma ultrafiltration.62–66 Heparin therapy alone has not been shown to improve survival.66 Much attention has been given to replacement of natural anticoagulants such as protein C and antithrombin as therapy for purpura fulminans, but unfortunately randomized trials using antithrombin have shown mostly negative results.51,55,67–69 Trials using protein C concentrates have shown more promise in controlling the coagulopathy of purpura fulminans, but this is not widely available.63,70–72 Unfortunately, many patients will need debridement and amputation for their necrotic limbs, with one review showing approximately 66% of patients needing amputations.52
TRAUMA
Currently, the most common cause of acute DIC is trauma. The coagulation defects that occur in trauma patients are complex in origin and still controversial (including if even calling it DIC is appropriate!).73–76 The most common etiologies are
- Generation of excess activated protein C leading to increased consumption of factor V and VIII and increased fibrinolysis;
- Tissue damage leading to generation of excess thrombin generation;
- Dilution of hemostatic factors by blood or fluid resuscitation; and
- Activation of endothelial cells leading to generation of a prothrombotic surface and shedding of glycocalyx with antithrombotic properties.
Trauma patients are prone to hypothermia, and this can be the major complicating factor in their bleeding.77,78 Patients may be out “in the field” for a prolonged period of time and be hypothermic on arrival.79 Packed red cells are stored at 4°C, and the infusion of 1 unit can lower the body temperature by 0.16°C.80 Hypothermia has profound effects on the coagulation system that are associated with clinical bleeding.77,81,82 Even modest hypothermia can greatly augment bleeding and needs to be treated or prevented.
The initial management of the bleeding trauma patient is administration of red cells and plasma (FFP) in a 1:1 ratio. This has been shown by clinical studies to lessen the risk of exsanguination in the first 24 hours and to be associated with improved clinical outcomes.83,84 The basic set of coagulation tests should also be obtained to guide product replacement, especially as the bleeding is brought under control. Hypothermia can be prevented by several measures, including transfusing the blood through blood warmers. Devices are available that can warm 1 unit of blood per minute. An increasingly used technique is to perform “damage control” surgery. Patients are initially stabilized with control of damaged vessels and packing of oozing sites.85 Then the patient is taken to the intensive care unit to be warmed and have coagulation defects corrected.
For trauma patients at risk of serious bleeding, the use of tranexamic acid reduced all- cause mortality (relative risk 0.91), with death due to bleeding also being reduced (relative risk 0.85).86 There was no increase in thrombosis, but benefit was restricted to patients treated within 3 hours of the trauma. The dose of tranexamic acid was a 1-g bolus followed by a 1-g continuous infusion over 8 hours.
PREGNANCY-RELATED DIC SYNDROMES
Acute DIC of Pregnancy
Pregnancy can be associated with the rapid onset of severe DIC in 2 situations, abruption and amniotic fluid embolism.87,88 The separation of the placenta from the uterine wall creates a space for blood to occupy. Given the richness of the placenta in tissue factor, this leads to activation of coagulation both locally and systemically. Release of blood when this space reaches the vaginal opening can lead to rapid hemorrhage, further augmenting the coagulation abnormalities. Placental insufficiency can lead to fetal demise, which can also worsen the DIC. Management depends on the size of the abruption and the clinical status of both mother and fetus.87 For severe bleeding and DIC, blood product support is crucial to allow safe delivery. In pregnancy, the fibrinogen goal needs to be higher—200 mg/dL.89 For smaller abruption, close observation with early delivery is indicated.
Amniotic fluid embolism is sudden, with the vascular collapse of the woman soon after delivery. Due to the presence of procoagulant rich fluid in the circulatory system, there is often overwhelming DIC. Therapy is directed at both supporting blood volume and correcting hemostatic defects.
HELLP
The acronym HELLP (hemolysis, elevated liver tests, low platelets) describes a variant of preeclampsia.90 Classically, HELLP syndrome occurs after 28 weeks of gestation in a patient with preeclampsia, but can occur as early as 22 weeks in patients with antiphospholipid antibody syndrome.91–93 The preeclampsia need not be severe. The first sign of HELLP is a decrease in the platelet count followed by abnormal liver function tests. Signs of hemolysis are present with abundant schistocytes on the smear and a high lactate dehydrogenase level. HELLP can progress to liver failure, and deaths are also reported due to hepatic rupture. Unlike TTP, fetal involvement is present in the HELLP syndrome, with fetal thrombocytopenia reported in 30% of cases. In severe cases, elevated D-dimers consistent with DIC are also found. Delivery of the child will most often result in cessation of the HELLP syndrome, but refractory cases will require dexamethasone and plasma exchange.94 Patients should be closely observed for 1 to 2 days after delivery as the hematologic picture can transiently worsen before improving.95
Acute Fatty Liver of Pregnancy
Fatty liver of pregnancy also occurs late in pregnancy and is only associated with preeclampsia in 50% of cases.96,97 Patients first present with nonspecific symptoms of nausea and vomiting but can progress to fulminant liver failure. Patients develop thrombocytopenia early in the course, but in the later stages can develop DIC and very low fibrinogen levels. Mortality rates without therapy can be as high as 90%. Low blood glucose and high ammonia levels can help distinguish fatty liver from other pregnancy complications.98 Treatment consists of prompt delivery of the child and aggressive blood product support.
Retained Dead Fetus Syndrome
Becoming rarer in modern practices, the presence of a dead fetus for many weeks (usually ≥ 5) can result in a chronic DIC state with fibrinogen depletion and coagulopathy. In some women, this is worsened at delivery. In a stable patient, a short trial of heparin prior to planning delivery can control the DIC to allow the coagulopathy to stabilize.
DRUG-INDUCED HEMOLYTIC-DIC SYNDROMES
A severe variant of the drug-induced immune complex hemolysis associated with DIC has been recognized. Rare patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone) have developed this syndrome.99–104 The clinical syndrome starts 7 to 10 days after the drug is administered. Often the patient has only received the antibiotic for surgical prophylaxis. The patient will develop severe Coombs’-positive hemolysis with hypotension and DIC. The patients are often believed to have sepsis and in the management of the supposed sepsis often are re-exposed to the cephalosporin, resulting in worsening of the clinical picture. The outcome is often fatal due to massive hemolysis and thrombosis.101,105–107
Quinine is associated with a unique syndrome of drug-induced DIC.108–111 Approximately 24 to 96 hours after quinine exposure, the patient becomes acutely ill with nausea and vomiting. The patient then develops a microangiopathic hemolytic anemia, DIC, and renal failure. Some patients, besides having antiplatelet antibodies, also have antibodies binding to red cells and neutrophils, which may lead to the more severe syndrome. Despite therapy, patients with quinine-induced TTP have a high incidence of chronic renal failure.
Treatment of the drug-induced hemolytic-DIC syndrome is anecdotal. Patients have responded to aggressive therapy, including plasma exchange, dialysis, and prednisone. Early recognition of the hemolytic anemia and the suspicion it is drug related is important for early diagnosis so that the incriminated drug can be discontinued.
CANCER
Cancers, primarily adenocarcinomas, can result in DIC. The classic Trousseau syndrome referred to the association of migratory superficial thrombophlebitis with cancer112 but now refers to cancer associated with thrombotic DIC.113,114 Highly vascular tumor cells are known to express tissue factor.114,115 In addition, some tumor cells can express a direct activator of factor X (“cancer procoagulant”). Unlike many DIC states, cancer presents with thrombosis instead of bleeding. This may be due to the inflammatory state which accompanies cancer, or it may be a unique part of the chronic nature of cancer DIC biology that allows time for the body to compensate for loss of coagulation factors. In some patients, thrombosis is the first sign of an underlying cancer, sometimes predating the cancer diagnosis by months.115 Rarely, the DIC can result in nonthrombotic endocarditis with micro-emboli leading to widespread small-vessel thrombosis.113
Since effective antineoplastic therapy is lacking for many tumors associated with Trousseau syndrome, DIC therapy is aimed at suppressing thrombosis. An exception is prostate cancer, where hormonal therapy can markedly decrease the DIC.116 Due to the tumor directly activating coagulation factors, inhibition of active enzymes via heparin has been shown to reduce rates of recurrence compared with warfarin.114,115 Clinical trials have demonstrated that heparin therapy is associated with a lower thrombosis recurrence rate than warfarin.117,118 In some patients, the thrombotic process is so vigorous that new thrombosis can be seen within hours of stopping heparin.112
ACUTE PROMYELOCYTIC LEUKEMIA
There are multiple hemostatic defects in patients with acute promyelocytic leukemia (APL).119 Most, if not all, patients with APL have evidence of DIC at the time of diagnosis. Patients with APL have a higher risk of death during induction therapy as compared with patients with other forms of leukemia, with death most often due to bleeding. Once in remission, APL patients have a higher cure rate than most patients with leukemia. APL is also unique among leukemias in that biologic therapy with retinoic acid or arsenic is effective in inducing remission and cure in most patients. Although effective therapy is available, early death rates due to bleeding have not changed.119
APL patients can present with pancytopenia due to leukemic marrow replacement or with diffuse bleeding due to DIC and thrombocytopenia. Life-threatening bleeding such as intracranial hemorrhage may occur at any time until the leukemia is put into remission. The etiology of the hemostatic defects in APL is complex and is thought to be the result of DIC, fibrinolysis, and the release of prothrombotic extracellular chromatin and other procoagulant enzymes.119,120 The diagnosis of APL can be straightforward when the leukemic cells are promyelocytes with abundant Auer rods, although some patients have the microgranular form without obvious Auer rods. The precise diagnosis requires molecular methods, including obtaining FISH for detecting the t(15;17) in PML/RARA fusion. Upon diagnosis of APL, one should obtain a complete coagulation profile, including INR, aPTT, fibrinogen, platelet count, and D-dimers. Change in fibrinogen levels tends to be a good marker of progress in treating the coagulation defects.
Therapy of APL involves treating both the leukemia and the coagulopathy. Currently, the standard treatment for APL is trans-retinoic acid (ATRA) in combination with chemotherapy or arsenic.121,122 This approach will induce remission in more than 90% of patients, and a sizable majority of these patients will be cured of their APL. ATRA therapy will also lead to early correction of the coagulation defects, often within the first week of therapy.123 This is in stark contrast to the chemotherapy era when the coagulation defects would become worse with therapy. Given the marked beneficial effect of ATRA on the coagulopathy of APL and its low toxicity profile, it should be empirically started for any patients suspected of having APL while genetic testing is being performed. Rare reports of massive thrombosis complicating therapy with ATRA exist, but the relationship to either the APL or ATRA is unknown.
Therapy for the coagulation defects consists of aggressive transfusion therapy support and possible use of other pharmacologic agents to control DIC.124,125 The fibrinogen level should be maintained at over 150 mg/dL and the platelet count at over 50,000 cells/µL.126 Controversy still exists over the role of heparin in therapy of APL.104 Although attractive for its ability to quench thrombin, heparin use can lead to profound bleeding and its use in treating APL has fallen out of favor.
SNAKEBITES
Snake envenomation can lead to direct activation of multiple coagulation enzymes, including factors V, X, thrombin, and protein C, and lead to cleavage of fibrinogen.127,128 Envenomation can also activate coagulation and damage vascular endothelium. The DIC can be enhanced by widespread tissue necrosis and hypotension. The key to management of snake bites is administration of specific antivenom. The role of prophylactic factor replacement is controversial, but this therapy is indicated if there is clinical bleeding.129 One confounder is that some snake venoms, especially rattlesnake, can induce reversible platelet aggregation, which corrects with antivenom.
LOCAL VASCULAR ABNORMALITIES
Abnormal vascular structures, such as vascular tumors, vascular malformations, and aneurysms, can lead to localized areas of thrombin generation that can “spill-over” into the general circulation, leading to DIC. The diagnosis Kasabach-Merritt phenomenon should be reserved for children with vascular tumors such as angioma or hemangioendothelioma.130 Therapy depends on the lesion. Embolization to reduce blood flow of vascular malformations can either be definitive therapy or stabilize the patient for surgery. Aneurysms can be repaired by surgery or stenting. Rare patients with aneurysms with significant coagulopathy may require heparin to raise the fibrinogen level before surgery. Kasabach-Merritt disease can respond to steroids or therapy such as vincristine or interferon.130 Increasing data shows that use of the mTOR inhibitor sirolimus can shrink these vascular abnormalities leading to lessening of the coagulopathy.131
CONCLUSION
At the most basic level, DIC is the excess activity of thrombin. However, the clinical presentation and therapy can differ greatly depending on the primary cause. Both diagnosis and therapy involve close coordination of laboratory data and clinical assessment.
INTRODUCTION
In the normal person, the process of coagulation is finely controlled at many levels to ensure the appropriate amount of hemostasis at the appropriate location. Broadly defined, disseminated intravascular coagulation (DIC) is the name given to any process that disrupts this fine tuning, leading to unregulated coagulation. Defined this way, DIC may be found in a variety of patients with a variety of disease states, and can present with a spectrum of findings ranging from asymptomatic abnormal laboratory results to florid bleeding or thrombosis. It is important to remember that DIC is always a consequence of an underlying pathological process and not a disease in and of itself. This article first reviews concepts common to all forms of DIC, and then reviews the more common disease states that lead to DIC.
PATHOGENESIS
At the most basic level, DIC is the clinical manifestation of inappropriate thrombin activation.1–5 Inappropriate thrombin activation can be due to underlying conditions such as sepsis and obstetric disasters. The activation of thrombin leads to (1) conversion of fibrinogen to fibrin, (2) activation of platelets (and their consumption), (3) activation of factors V and VIII, (4) activation of protein C (and degradation of factors Va and VIIIa), (5) activation of endothelial cells, and (6) activation of fibrinolysis (Table 1).
1. Conversion of fibrinogen to fibrin, which leads to the formation of fibrin monomers and excessive thrombus formation. These thrombi are rapidly dissolved by excessive fibrinolysis in most patients. In certain clinical situations, especially cancer, excessive thrombosis will occur. In patients with cancer, this is most often a deep venous thrombosis. Rare patients, especially those with pancreatic cancer, may have severe DIC with multiple arterial and venous thromboses. Nonbacterial thrombotic endocarditis can also be seen in these patients, leading to widespread embolic complications.
2. Activation of platelets and their consumption. Thrombin is the most potent physiologic activator of platelets, so in DIC there is increased activation of platelets. These activated platelets are consumed, resulting in thrombocytopenia. Platelet dysfunction is also present. Platelets that have been activated and have released their contents but still circulate are known as “exhausted” platelets; these cells can no longer function to support coagulation. The fibrin degradation products (FDP) in DIC can also bind to GP IIb/IIIa and inhibit further platelet aggregation.
3. Activation of factors V, VIII, XI, and XIII. Activation of these factors can promote thrombosis, but they are then rapidly cleared by antithrombin (XI) or activated protein C (V and VIII) or by binding to the fibrin clot (XIII). This can lead to depletion of all the prothrombotic clotting factors and antithrombin, which in turn can lead to both thrombosis and bleeding.
4. Activation of protein C further promotes degradation of factors Va and VIIIa, enhances fibrinolysis, and decreases protein C levels.
5. Activation of endothelial cells, especially in the skin, may lead to thrombosis, and in certain patients, especially those with meningococcemia, purpura fulminans. Endothelial damage will down-regulate thrombomodulin, preventing activation of protein C and leading to further reductions in levels of activated protein C.56. Activation of fibrinolysis leads to the breakdown of fibrin monomers, formation of fibrin thrombi, and increased circulating fibrinogen. In most patients with DIC, the fibrinolytic response is brisk.6 This is why most patients with DIC present with bleeding and prolonged clotting times.
PATTERNS OF DIC
The clinical manifestations of DIC in a given patient depend on the balance of thrombin activation and secondary fibrinolysis plus the patient’s ability to compensate for the DIC. Patients with DIC can present in 1 of 4 patterns:1–3
1. Asymptomatic. Patients can present with laboratory evidence of DIC but no bleeding or thrombosis. This is often seen in patients with sepsis or cancer. However, with further progression of the underlying disease, these patients can rapidly become symptomatic.
2. Bleeding. The bleeding is due to a combination of factor depletion, platelet dysfunction, thrombocytopenia, and excessive fibrinolysis.1 These patients may present with diffuse bleeding from multiple sites (eg, intravenous sites, areas of instrumentation).
3. Thrombosis. Despite the general activation of the coagulation process, thrombosis is unusual in most patients with acute DIC. The exceptions include patients with cancer, trauma patients, and certain obstetrical patients. Most often the thrombosis is venous, but arterial thrombosis and nonbacterial thrombotic endocarditis have been reported.7
4. Purpura fulminans. This form of DIC is discussed in more detail later (see Specific DIC Syndromes section).
DIAGNOSIS
There is no one test that will diagnose DIC; one must match the test to the clinical situation (Table 2).8
SCREENING TESTS
The prothrombin time-INR and activated thromboplastin time (aPPT) are usually elevated in severe DIC but may be normal or shortened in chronic forms.9 One may also see a shortened aPTT in severe acute DIC due to large amounts of activated thrombin and factor X “bypassing” the contact pathway. An aPTT as short as 10 seconds has been seen in acute DIC. The platelet count is usually reduced but may be normal in chronic DIC. Serum fibrinogen and platelets are decreased in acute DIC but again may be in the “normal” range in chronic DIC.10 The most sensitive screening test for DIC is a fall in the platelet count, with low counts seen in 98% of patients and counts under 50,000 cells/μL in 50%.9,11 The least specific test is fibrinogen, which tends to fall below normal only in severe acute DIC.9
SPECIFIC TESTS
This group of tests allows one to deduce that abnormally high concentrations of thrombin are present.
Ethanol Gel and Protamine Tests
Both of these older tests detected circulating fibrin monomers, whose appearance is an early sign of DIC. Circulating fibrin monomers are seen when thrombin acts on fibrinogen. Usually the monomer polymerizes with the fibrin clot, but when there is excess thrombin these monomers can circulate. Detection of circulating fibrin monomer means there is too much IIa and, ergo, DIC is present.
Fibrin(ogen) Degradation Products
Plasmin acts on the fibrin/fibrinogen molecule to cleave the molecule in specific places. The resulting degradation product levels will be elevated in situations of increased fibrin/fibrinogen destruction (DIC and fibrinolysis). The FDP are typically mildly elevated in renal and liver disease due to reduced clearance.
D-Dimers
When fibrin monomers bind to form a thrombus, factor XIII acts to bind their “D” domains together. This bond is resistant to plasmin and thus this degradation fragment is known as the “D-dimer.” High levels of D-dimer indicate that (1) IIa has acted on fibrinogen to form a fibrin monomer that bonded to another fibrin monomer, and (2) this thrombus was lysed by plasmin. Because D-dimers can be elevated (eg, with exercise, after surgery), an elevated D-dimer needs to be interpreted in the context of the clinical situation.11 Currently, this is the most common specific test for DIC performed.
Other Tests
Several other tests are sometimes helpful in diagnosing DIC.
Thrombin time. This test is performed by adding thrombin to plasma. Thrombin times are elevated in DIC (FDPs interfere with polymerization), in the presence of low fibrinogen levels, in dysfibrinogenemia, and in the presence of heparin (very sensitive).
Reptilase time is the same as thrombin time but is performed with a snake venom that is insensitive to heparin. Reptilase time is elevated in the same conditions as the thrombin time, with the exception of the presence of heparin. Thrombin time and reptilase time are most useful in evaluation of dysfibrinogenemia.
Prothrombin fragment 1.2 (F1.2). F1.2 is a small peptide cleaved off when prothrombin is activated to thrombin. Thus, high levels of F1.2 are found in DIC but can be seen in other thrombotic disorders. This test is still of limited clinical value.
DIC scoring system. A scoring system to both diagnose and quantify DIC has been proposed (Figure).11,12
Thromboelastography (TEG). This is a point-of-care test that uses whole blood to determine specific coagulation parameters such as R time (time from start of test to clot formation), maximal amplitude (MA, maximum extent of thrombus), and LY30 (MA at 30 minutes, a measure of fibrinolysis).13 Studies have shown that TEG can identify DIC by demonstrating a shorter R time (excess thrombin generation) which prolongs as coagulation factors are consumed. The MA is decreased as fibrinogen is consumed and the LY30 shows excess fibrinolysis. TEG has been shown to be of particular value in the management of the complex coagulopathy of trauma.14
MIMICKERS OF DIC
It is important to recognize coagulation syndromes that are not DIC, especially those that have specific other therapies. The syndromes most frequently encountered are thrombotic thrombocytopenic purpura (TTP) and catastrophic antiphospholipid antibody syndrome (CAPS). One important clue to both of these syndromes is that, unlike DIC, there is no primary disorder (cancer, sepsis) that is driving the coagulation abnormalities.
TTP should be suspected when any patient presents with any combination of thrombocytopenia, microangiopathic hemolytic anemia (schistocytes and signs of hemolysis) plus end-organ damage.15–18 Patients with TTP most often present with intractable seizures, strokes, or sequelae of renal insufficiency. Many patients who present with TTP have been misdiagnosed as having sepsis, “lupus flare,” or vasculitis. The key diagnostic differentiator between TTP and DIC is the lack of activation of coagulation with TTP—fibrinogen is normal and D-dimers are minimally or not elevated. In TTP, lactate dehydrogenase is invariably elevated, often 2 to 3 times normal.19 The importance of identifying TTP is that untreated TTP is rapidly fatal. Mortality in the pre–plasma exchange era ranged from 95% to 100%. Today plasma exchange therapy is the foundation of TTP treatment and has reduced mortality to less than 20%.16,20–23Rarely patients with antiphospholipid antibody syndrome can present with fulminant multiorgan system failure.24–28 CAPS is caused by widespread microthrombi in multiple vascular fields. These patients will develop renal failure, encephalopathy, adult respiratory distress syndrome (often with pulmonary hemorrhage), cardiac failure, dramatic livedo reticularis, and worsening thrombocytopenia. Many of these patients have pre-existing autoimmune disorders and high-titer anticardiolipin antibodies. It appears that the best therapy for these patients is aggressive immunosuppression with steroids plus plasmapheresis, followed by rituximab or, if in the setting of lupus, intravenous cyclophosphamide monthly.27,29 Early recognition of CAPS can lead to quick therapy and resolution of the multiorgan system failure.
GENERAL THERAPY
The best way to treat DIC is to treat the underlying cause that is driving the thrombin generation.1,2,4,30,31 Fully addressing the underlying cause may not be possible or may take time, and in the meantime it is necessary to disrupt the cycle of thrombosis and/or hemorrhage. In the past, there was concern about using factor replacement due to fears of “feeding the fire,” or perpetuating the cycle of thrombosis. However, these concerns are not supported by evidence, and factors must be replaced if depletion occurs and bleeding ensues.32
Transfusion therapy of the patient with DIC is guided by the 5 laboratory tests that reflect the basic parameters essential for both hemostasis and blood volume status:33,34 hematocrit, platelet count, prothrombin time-INR, aPTT, and fibrinogen level. Decisions regarding replacement therapy are based on the results of these laboratory tests and the clinical situation of the patient (Table 3).
The basic 5 laboratory tests should be repeated after administering the blood products. This allows one to ensure that adequate replacement therapy was given for the coagulation defects. Frequent checks of the coagulation tests also allow rapid identification and treatment of new coagulation defects in a timely fashion. A flow chart of the test and the blood products administered should also be maintained. This is important in acute situations such as trauma or obstetrical bleeding.
In theory, since DIC is the manifestation of exuberant thrombin production, blocking thrombin with heparin should decrease or shut down DIC. However, studies have shown that in most patients heparin administration has led to excessive bleeding. Currently, heparin therapy is reserved for patients who have thrombosis as a component of their DIC.2,41,42 Given the coagulopathy that is often present, specific heparin levels instead of the aPTT should be used to monitor anticoagulation.43,44
SPECIFIC DIC SYNDROMES
SEPSIS/INFECTIOUS DISEASE
Any overwhelming infection can lead to DIC.45 Classically, it was believed that gram-negative bacteria can lead tissue factor exposure via production of endotoxin, but recent studies indicate that DIC can be seen with any overwhelming infection.46,47 There are several potential avenues by which infections can lead to DIC. As mentioned, gram-negative bacteria produce endotoxin that can directly lead to tissue factor exposure, resulting in excess thrombin generation. In addition, any infection can lead to expression of inflammatory cytokines that induce tissue-factor expression by endothelium and monocytes. Some viruses and Rickettsia species can directly infect the vascular endothelium, converting it from an antithrombotic to a prothrombotic phenotype.48 When fighting infections, neutrophils can extrude their contents, including DNA, to help trap organisms. These neutrophil extracellular traps (NETS) may play an important role in promoting coagulopathy.49,50 The hypotension produced by sepsis leads to tissue hypoxia, which results in more DIC. The coagulopathy in sepsis can range from subtle abnormalities of testing to purpura fulminans. Thrombocytopenia is worsened by cytokine-induced hemophagocytic syndrome.
As with all forms of DIC, empiric therapy targeting the most likely source of infection and maintaining hemodynamic stability is the key to therapy. As discussed below, heparin and other forms of coagulation replacement appear to be of no benefit in therapy.
PURPURA FULMINANS
DIC in association with necrosis of the skin is seen in primary and secondary purpura fulminans.51,52 Primary purpura fulminans is most often seen after a viral infection.53 In these patients, the purpura fulminans starts with a painful red area on an extremity that rapidly progresses to a black ischemic area. In many patients, acquired deficiency of protein S is found.51,54,55 Secondary purpura fulminans is most often associated with meningococcemia infections but can be seen in any patient with overwhelming infection.56–58 Post-splenectomy sepsis syndrome patients and those with functional hyposplenism due to chronic liver diseases are also at risk.59 Patients present with signs of sepsis, and the skin lesions often involve the extremities and may lead to amputations. As opposed to primary purpura fulminans, those with the secondary form will have symmetrical ischemia distally (toes and fingers) that ascends as the process progresses. Rarely, adrenal infarction (Waterhouse-Friderichsen syndrome) occurs, which leads to severe hypotension.45
Recently, Warkenten has reported on limb gangrene in critically ill patients complicating sepsis or cardiogenic shock.60,61 These patients have DIC that is complicated by shock liver. Deep venous thrombosis with ischemic gangrene then develops, which can result in tissue loss and even amputation. The pathogenesis is hypothesized to be hepatic dysfunction leading to sudden drops in protein C and S plasma levels, which then leads to thrombophilia with widespread microvascular thrombosis. Therapy for purpura fulminans is controversial. Primary purpura fulminans, especially in those with postvaricella autoimmune protein S deficiency, has responded to plasma infusion titrated to keep the protein S level above 25%.51 Intravenous immunoglobulin has also been reported to help decrease the anti-protein S antibodies. Heparin has been reported to control the DIC and extent of necrosis.62 The starting dose in these patients is 5 to 8 units/kg/hr.2
Sick patients with secondary purpura fulminans have been treated with plasma drips, plasmapheresis, and continuous plasma ultrafiltration.62–66 Heparin therapy alone has not been shown to improve survival.66 Much attention has been given to replacement of natural anticoagulants such as protein C and antithrombin as therapy for purpura fulminans, but unfortunately randomized trials using antithrombin have shown mostly negative results.51,55,67–69 Trials using protein C concentrates have shown more promise in controlling the coagulopathy of purpura fulminans, but this is not widely available.63,70–72 Unfortunately, many patients will need debridement and amputation for their necrotic limbs, with one review showing approximately 66% of patients needing amputations.52
TRAUMA
Currently, the most common cause of acute DIC is trauma. The coagulation defects that occur in trauma patients are complex in origin and still controversial (including if even calling it DIC is appropriate!).73–76 The most common etiologies are
- Generation of excess activated protein C leading to increased consumption of factor V and VIII and increased fibrinolysis;
- Tissue damage leading to generation of excess thrombin generation;
- Dilution of hemostatic factors by blood or fluid resuscitation; and
- Activation of endothelial cells leading to generation of a prothrombotic surface and shedding of glycocalyx with antithrombotic properties.
Trauma patients are prone to hypothermia, and this can be the major complicating factor in their bleeding.77,78 Patients may be out “in the field” for a prolonged period of time and be hypothermic on arrival.79 Packed red cells are stored at 4°C, and the infusion of 1 unit can lower the body temperature by 0.16°C.80 Hypothermia has profound effects on the coagulation system that are associated with clinical bleeding.77,81,82 Even modest hypothermia can greatly augment bleeding and needs to be treated or prevented.
The initial management of the bleeding trauma patient is administration of red cells and plasma (FFP) in a 1:1 ratio. This has been shown by clinical studies to lessen the risk of exsanguination in the first 24 hours and to be associated with improved clinical outcomes.83,84 The basic set of coagulation tests should also be obtained to guide product replacement, especially as the bleeding is brought under control. Hypothermia can be prevented by several measures, including transfusing the blood through blood warmers. Devices are available that can warm 1 unit of blood per minute. An increasingly used technique is to perform “damage control” surgery. Patients are initially stabilized with control of damaged vessels and packing of oozing sites.85 Then the patient is taken to the intensive care unit to be warmed and have coagulation defects corrected.
For trauma patients at risk of serious bleeding, the use of tranexamic acid reduced all- cause mortality (relative risk 0.91), with death due to bleeding also being reduced (relative risk 0.85).86 There was no increase in thrombosis, but benefit was restricted to patients treated within 3 hours of the trauma. The dose of tranexamic acid was a 1-g bolus followed by a 1-g continuous infusion over 8 hours.
PREGNANCY-RELATED DIC SYNDROMES
Acute DIC of Pregnancy
Pregnancy can be associated with the rapid onset of severe DIC in 2 situations, abruption and amniotic fluid embolism.87,88 The separation of the placenta from the uterine wall creates a space for blood to occupy. Given the richness of the placenta in tissue factor, this leads to activation of coagulation both locally and systemically. Release of blood when this space reaches the vaginal opening can lead to rapid hemorrhage, further augmenting the coagulation abnormalities. Placental insufficiency can lead to fetal demise, which can also worsen the DIC. Management depends on the size of the abruption and the clinical status of both mother and fetus.87 For severe bleeding and DIC, blood product support is crucial to allow safe delivery. In pregnancy, the fibrinogen goal needs to be higher—200 mg/dL.89 For smaller abruption, close observation with early delivery is indicated.
Amniotic fluid embolism is sudden, with the vascular collapse of the woman soon after delivery. Due to the presence of procoagulant rich fluid in the circulatory system, there is often overwhelming DIC. Therapy is directed at both supporting blood volume and correcting hemostatic defects.
HELLP
The acronym HELLP (hemolysis, elevated liver tests, low platelets) describes a variant of preeclampsia.90 Classically, HELLP syndrome occurs after 28 weeks of gestation in a patient with preeclampsia, but can occur as early as 22 weeks in patients with antiphospholipid antibody syndrome.91–93 The preeclampsia need not be severe. The first sign of HELLP is a decrease in the platelet count followed by abnormal liver function tests. Signs of hemolysis are present with abundant schistocytes on the smear and a high lactate dehydrogenase level. HELLP can progress to liver failure, and deaths are also reported due to hepatic rupture. Unlike TTP, fetal involvement is present in the HELLP syndrome, with fetal thrombocytopenia reported in 30% of cases. In severe cases, elevated D-dimers consistent with DIC are also found. Delivery of the child will most often result in cessation of the HELLP syndrome, but refractory cases will require dexamethasone and plasma exchange.94 Patients should be closely observed for 1 to 2 days after delivery as the hematologic picture can transiently worsen before improving.95
Acute Fatty Liver of Pregnancy
Fatty liver of pregnancy also occurs late in pregnancy and is only associated with preeclampsia in 50% of cases.96,97 Patients first present with nonspecific symptoms of nausea and vomiting but can progress to fulminant liver failure. Patients develop thrombocytopenia early in the course, but in the later stages can develop DIC and very low fibrinogen levels. Mortality rates without therapy can be as high as 90%. Low blood glucose and high ammonia levels can help distinguish fatty liver from other pregnancy complications.98 Treatment consists of prompt delivery of the child and aggressive blood product support.
Retained Dead Fetus Syndrome
Becoming rarer in modern practices, the presence of a dead fetus for many weeks (usually ≥ 5) can result in a chronic DIC state with fibrinogen depletion and coagulopathy. In some women, this is worsened at delivery. In a stable patient, a short trial of heparin prior to planning delivery can control the DIC to allow the coagulopathy to stabilize.
DRUG-INDUCED HEMOLYTIC-DIC SYNDROMES
A severe variant of the drug-induced immune complex hemolysis associated with DIC has been recognized. Rare patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone) have developed this syndrome.99–104 The clinical syndrome starts 7 to 10 days after the drug is administered. Often the patient has only received the antibiotic for surgical prophylaxis. The patient will develop severe Coombs’-positive hemolysis with hypotension and DIC. The patients are often believed to have sepsis and in the management of the supposed sepsis often are re-exposed to the cephalosporin, resulting in worsening of the clinical picture. The outcome is often fatal due to massive hemolysis and thrombosis.101,105–107
Quinine is associated with a unique syndrome of drug-induced DIC.108–111 Approximately 24 to 96 hours after quinine exposure, the patient becomes acutely ill with nausea and vomiting. The patient then develops a microangiopathic hemolytic anemia, DIC, and renal failure. Some patients, besides having antiplatelet antibodies, also have antibodies binding to red cells and neutrophils, which may lead to the more severe syndrome. Despite therapy, patients with quinine-induced TTP have a high incidence of chronic renal failure.
Treatment of the drug-induced hemolytic-DIC syndrome is anecdotal. Patients have responded to aggressive therapy, including plasma exchange, dialysis, and prednisone. Early recognition of the hemolytic anemia and the suspicion it is drug related is important for early diagnosis so that the incriminated drug can be discontinued.
CANCER
Cancers, primarily adenocarcinomas, can result in DIC. The classic Trousseau syndrome referred to the association of migratory superficial thrombophlebitis with cancer112 but now refers to cancer associated with thrombotic DIC.113,114 Highly vascular tumor cells are known to express tissue factor.114,115 In addition, some tumor cells can express a direct activator of factor X (“cancer procoagulant”). Unlike many DIC states, cancer presents with thrombosis instead of bleeding. This may be due to the inflammatory state which accompanies cancer, or it may be a unique part of the chronic nature of cancer DIC biology that allows time for the body to compensate for loss of coagulation factors. In some patients, thrombosis is the first sign of an underlying cancer, sometimes predating the cancer diagnosis by months.115 Rarely, the DIC can result in nonthrombotic endocarditis with micro-emboli leading to widespread small-vessel thrombosis.113
Since effective antineoplastic therapy is lacking for many tumors associated with Trousseau syndrome, DIC therapy is aimed at suppressing thrombosis. An exception is prostate cancer, where hormonal therapy can markedly decrease the DIC.116 Due to the tumor directly activating coagulation factors, inhibition of active enzymes via heparin has been shown to reduce rates of recurrence compared with warfarin.114,115 Clinical trials have demonstrated that heparin therapy is associated with a lower thrombosis recurrence rate than warfarin.117,118 In some patients, the thrombotic process is so vigorous that new thrombosis can be seen within hours of stopping heparin.112
ACUTE PROMYELOCYTIC LEUKEMIA
There are multiple hemostatic defects in patients with acute promyelocytic leukemia (APL).119 Most, if not all, patients with APL have evidence of DIC at the time of diagnosis. Patients with APL have a higher risk of death during induction therapy as compared with patients with other forms of leukemia, with death most often due to bleeding. Once in remission, APL patients have a higher cure rate than most patients with leukemia. APL is also unique among leukemias in that biologic therapy with retinoic acid or arsenic is effective in inducing remission and cure in most patients. Although effective therapy is available, early death rates due to bleeding have not changed.119
APL patients can present with pancytopenia due to leukemic marrow replacement or with diffuse bleeding due to DIC and thrombocytopenia. Life-threatening bleeding such as intracranial hemorrhage may occur at any time until the leukemia is put into remission. The etiology of the hemostatic defects in APL is complex and is thought to be the result of DIC, fibrinolysis, and the release of prothrombotic extracellular chromatin and other procoagulant enzymes.119,120 The diagnosis of APL can be straightforward when the leukemic cells are promyelocytes with abundant Auer rods, although some patients have the microgranular form without obvious Auer rods. The precise diagnosis requires molecular methods, including obtaining FISH for detecting the t(15;17) in PML/RARA fusion. Upon diagnosis of APL, one should obtain a complete coagulation profile, including INR, aPTT, fibrinogen, platelet count, and D-dimers. Change in fibrinogen levels tends to be a good marker of progress in treating the coagulation defects.
Therapy of APL involves treating both the leukemia and the coagulopathy. Currently, the standard treatment for APL is trans-retinoic acid (ATRA) in combination with chemotherapy or arsenic.121,122 This approach will induce remission in more than 90% of patients, and a sizable majority of these patients will be cured of their APL. ATRA therapy will also lead to early correction of the coagulation defects, often within the first week of therapy.123 This is in stark contrast to the chemotherapy era when the coagulation defects would become worse with therapy. Given the marked beneficial effect of ATRA on the coagulopathy of APL and its low toxicity profile, it should be empirically started for any patients suspected of having APL while genetic testing is being performed. Rare reports of massive thrombosis complicating therapy with ATRA exist, but the relationship to either the APL or ATRA is unknown.
Therapy for the coagulation defects consists of aggressive transfusion therapy support and possible use of other pharmacologic agents to control DIC.124,125 The fibrinogen level should be maintained at over 150 mg/dL and the platelet count at over 50,000 cells/µL.126 Controversy still exists over the role of heparin in therapy of APL.104 Although attractive for its ability to quench thrombin, heparin use can lead to profound bleeding and its use in treating APL has fallen out of favor.
SNAKEBITES
Snake envenomation can lead to direct activation of multiple coagulation enzymes, including factors V, X, thrombin, and protein C, and lead to cleavage of fibrinogen.127,128 Envenomation can also activate coagulation and damage vascular endothelium. The DIC can be enhanced by widespread tissue necrosis and hypotension. The key to management of snake bites is administration of specific antivenom. The role of prophylactic factor replacement is controversial, but this therapy is indicated if there is clinical bleeding.129 One confounder is that some snake venoms, especially rattlesnake, can induce reversible platelet aggregation, which corrects with antivenom.
LOCAL VASCULAR ABNORMALITIES
Abnormal vascular structures, such as vascular tumors, vascular malformations, and aneurysms, can lead to localized areas of thrombin generation that can “spill-over” into the general circulation, leading to DIC. The diagnosis Kasabach-Merritt phenomenon should be reserved for children with vascular tumors such as angioma or hemangioendothelioma.130 Therapy depends on the lesion. Embolization to reduce blood flow of vascular malformations can either be definitive therapy or stabilize the patient for surgery. Aneurysms can be repaired by surgery or stenting. Rare patients with aneurysms with significant coagulopathy may require heparin to raise the fibrinogen level before surgery. Kasabach-Merritt disease can respond to steroids or therapy such as vincristine or interferon.130 Increasing data shows that use of the mTOR inhibitor sirolimus can shrink these vascular abnormalities leading to lessening of the coagulopathy.131
CONCLUSION
At the most basic level, DIC is the excess activity of thrombin. However, the clinical presentation and therapy can differ greatly depending on the primary cause. Both diagnosis and therapy involve close coordination of laboratory data and clinical assessment.
1. Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998;59:65–73.
2. De Jonge E, Levi M, Stoutenbeek CP, Van Deventer SJH. Current drug treatment strategies for disseminated intravascular coagulation. Drugs 1998;55:767–77.
3. Baker WF Jr. Clinical aspects of disseminated intravascular coagulation: a clinician’s point of view. Sem Thrombosis Hemostasis 1989;15:1–57.
4. Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999;341:586–92.
5. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037.
6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016;175:12–23.
7. Sharma S, Mayberry JC, DeLoughery TG, Mullins RJ. Fatal cerebroembolism from nonbacterial thrombotic endocarditis in a trauma patient: case report and review. Mil Med 2000;165:83–5.
8. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505–12.
9. Yu M, Nardella A, Pechet L. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med 2000;28:1777–80.
10. Mant MJ, King EG. Severe, acute disseminated intravascular coagulation. A reappraisal of its pathophysiology, clinical significance, and therapy based on 47 patients. Am J Med 1979;67:557–63.
11. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33.
12. Levi M. Disseminated intravascular coagulation. Crit Care Med 2007;35:2191–5.
13. Nogami K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br J Haematol 2016;174:503–14.
14. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin 2017;33:119–34.
15. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
16. George JN. How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 2000;96:1223–9.
17. Murrin RJ, Murray JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 2006;20:51–60.
18. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836–46.
19. Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol 1994;47:94–9.
20. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991;325:393–7.
21. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpurahemolytic uremic syndrome—clinical experience in 108 patients. N Engl J Med 1991;325:398–403.
22. Kaplan BS, Trachtman H. Improve survival with plasma exchange thrombotic thrombopenic purpura-hemolytic uremic syndrome. Am J Med 2001;110:156–7.
23. Kremer Hovinga JA, Coppo P, Lammle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 2017;3:17020.
24. Asherson RA. The catastrophic antiphospholipid syndrome [editorial]. J Rheumatol 1992;19:508–12.
25. Asherson RA, Piette JC. The catastrophic antiphospholipid syndrome 1996: acute multi-organ failure associated with antiphospholipid antibodies: a review of 31 patients. Lupus 1996;5:414–7.
26. Asherson RA, Cervera R. Castastrophic antiphospholipid syndrome. Curr Opinion Hematol 2000;5:325–9.
27. Merrill JT, Asherson RA. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rhuem 2006;2:81–9.
28. Rodriguez-Pinto I, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: The current management approach. Best Pract Res Clin Rheumatol 2016;30:239–9.
29. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 2016;28:218–27.
30. Hoffman JN, Faist E. Coagulation inhibitor replacement during sepsis: useless? Crit Care Med 2000;28(9 Suppl):S74–6.
31. Wada H, Asakura H, Okamoto K, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Thromb Res 2010;125:6–11.
32. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
33. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
34. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85:487–91.
35. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409–17.
36. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
37. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
38. Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794–801.
39. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
40. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
41. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
42. Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
43. Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993;119:104–9.
44. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782–8.
45. Yoshikawa T, Tanaka KR, Guze LB. Infection and disseminated intravascular coagulation. Medicine (Baltimore) 1971;50:237–58.
46. Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci 2004;328:196–204.
47. Lipinska-Gediga M. Coagulopathy in sepsis - a new look at an old problem. Anaesthesiol Intensive Ther 2016;48:352–9.
48. Van Gorp ECM, Suharti C, ten Cate H, et al. Review: Infections diseases and coagulation disorders. Journal of Infectious Diseases 1999;180:176–86.
49. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129:1357–67.
50. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61.
51. Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol 1998;15:169–83.
52. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol 2007;57:944–56.
53. Spicer TE, Rau JM. Purpura fulminans. Am J Med 1976;61:566–71.
54. Josephson C, Nuss R, Jacobson L, et al. The varicellaautoantibody syndrome. Pediatr Res 2001;50:345–52.
55. Smith OP, White B. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol 1999;104:202–7.
56. Gamper G, Oschatz E, Herkner H, et al. Sepsis-associated purpura fulminans in adults. Wien Klin Wochenschr 2001;113:107–12.
57. Ward KM, Celebi JT, Gmyrek R, Grossman ME. Acute infectious purpura fulminans associated with asplenism or hyposplenism. J Am Acad Dermatol 2002;47:493–6.
58. Childers BJ, Cobanov B. Acute infectious purpura fulminans: a 15-year retrospective review of 28 consecutive cases. Am Surg 2003;69:86–90.
59. Carpenter CT, Kaiser AB. Purpura fulminans in pneumococcal sepsis: case report and review. Scand J Infect Dis 1997;29:479–83.
60. Warkentin TE, Pai M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis: reply. J Thromb Haemost 2016;14:2317–9.
61. Warkentin TE. Ischemic limb gangrene with pulses. N Engl J Med 2015;373:642–55.
62. Duncan A. New therapies for severe meningococcal disease but better outcomes? Lancet 1997;350:1565–6.
63. Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet1997;350:1590–3.
64. Branson HE, Katz J. A structured approach to the management of purpura fulminans. J Natl Med Assoc 1983;75:821–5.
65. Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth 2001;86:581–6.
66. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. Heparin therapy and survival rate. Scand J Infect Dis 1971;3:127–33.
67. Giudici D, Baudo F, Palareti G, et al. Antithrombin replacement in patients with sepsis and septic shock. Haematologica 1999;84:452–60.
68. Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Crit Care Med 2000;28(9 Suppl):S38–43.
69. Levi M, De Jonge E, van der PT, ten Cate H. Novel approaches to the management of disseminated intravascular coagulation. Crit Care Med 2000;28(9 Suppl):S20–4.
70. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura fulminans in meningococcemia with protein C concentrate. J Pediatr 1995;126:646–52.
71. White B, Livingstone W, Murphy C, et al. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood 2000;96:3719–24.
72. Schellongowski P, Bauer E, Holzinger U, et al. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin). Vox Sang 2006;90:294–301.
73. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
74. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin 2017;33:101–18.
75. Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): a focused review on pathophysiology. Intern Emerg Med 2017 May 5.
76. Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 2016;128:1043–9.
77. Eddy VA, Morris JA Jr, Cullinane DC. Hypothermia, coagulopathy, and acidosis. Surg Clin North Am 2000;80:845–54.
78. Peng RY, Bongard FS. Hypothermia in trauma patients. J Am Coll Surg 1999;188:685–96.
79. Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma 1990;30:200–2.
80. Rajek A, Greif R, Sessler DI, et al. Core cooling by central venous infusion of ice-cold (4 degrees C and 20 degrees C) fluid: isolation of core and peripheral thermal compartments. Anesthesiol 2000;93:629–37.
81. Watts DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma 1998;44:846–54.
82. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg 1990;160:515–8.
83. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
84. Johansson PI, Stensballe J, Oliveri R, Wade CE, Ostrowski SR, Holcomb JB. How I treat patients with massive hemorrhage. Blood 2014;124:3052–8.
85. Stone HH, Strom PR, Mullins RJ. Management of the major coagulopathy with onset during laparotomy. Ann Surg 1983;197:532–5.
86. WOMAN Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
87. Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Semin Perinatol 2009;33:189–95.
88. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev 2009;23:167–76.
89. Collins P, Abdul-Kadir R, Thachil J, Subcommittees on Women’ s Health Issues in T, Haemostasis, on Disseminated Intravascular C. Management of coagulopathy associated with postpartum hemorrhage: guidance from the SSC of the ISTH. J Thromb Haemost 2016;14:205–10.
90. Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv 2004;59:838–45.
91. Egerman RS, Sibai BM. HELLP syndrome. Clin Obstetr Gynecol 1999;42:381–9.
92. Saphier CJ, Repke JT. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a review of diagnosis and management. Sem Perinatol 1998;22:118–33.
93. Le Thi TD, Tieulie N, Costedoat N, et al. The HELLP syndrome in the antiphospholipid syndrome: retrospective study of 16 cases in 15 women. Ann Rheum Dis 2005;64:273–8.
94. Martin JN Jr, Perry KG Jr, Blake PG, et al. Better maternal outcomes are achieved with dexamethasone therapy for postpartum HELLP (hemolysis, elevated liver enzymes, and thrombocytopenia) syndrome. Am J Obstet Gynecol 1997;177:1011–7.
95. Magann EF, Martin JN Jr. Twelve steps to optimal management of HELLP syndrome. Clinical Obstet Gynecol 1999;42:532–50.
96. Jwayyed SM, Blanda M, Kubina M. Acute fatty liver of pregnancy. J Emerg Medi 1999;17:673–7.
97. Bacq Y. Acute fatty liver of pregnancy. Sem Perinatol 1998;22:134–40.
98. Egerman RS, Sibai BM. Imitators of preeclampsia and eclampsia. Clin Obstet Gynecol 1999;42:551–62.
99. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Medi Rev 1993;7:255–67.
100. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis 1992;15:863–5.
101. Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion 1992;32:269–71.
102. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes:case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion 1999;39:306–9.
103. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion 1999;39:1239–46.
104. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol 2006;81:186–8.
105. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr 1995;126:813–5.
106. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1995;14:1116–7.
107. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone. J Pediatr 1995;126:816–7.
108. Gottschall JL, Elliot W, Lianos E, et al. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood 1991;77:306–10.
109. Gottschall JL, Neahring B, McFarland JG, et al. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol 1994;47:283–9.
110. Crum NF, Gable P. Quinine-induced hemolytic-uremic syndrome. South Med J 2000;93:726–8.
111. Vesely T, Vesely JN, George JN. Quinine-Induced thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): frequency, clinical features, and long-term outcomes. Blood 2000;96:629 [abstract].
112. Bell WR, Starksen NF, Tong S, Porterfield JK. Trousseau’s syndrome. Devastating coagulopathy in the absence of heparin. Am J Med 1985;79:423–30.
113. Sack GH, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinic, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
114. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007;110:1723–9.
115. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol 2005;6:401–10.
116. de la Fouchardiere C, Flechon A, Droz JP. Coagulopathy in prostate cancer. Neth J Med 2003;61:347–54.
117. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest 2008;133(6 Suppl):454S–545S.
118. Lee AY, Kamphuisen PW, Meyer G, et al. Tinzaparin vs warfarin for treatment of acute venous thromboembolism in patients with active cancer: a randomized clinical trial. JAMA 2015;314:677–86.
119. Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol 2012;87:596–603.
120. Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017;129:1855–64.
121. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
122. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–21.
123. Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated iwth acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2–9.
124. Falanga A, Rickles FR. Management of thrombohemorrhagic syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program 2007;2007:165–71
125. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
126. Sanz MA, Grimwade D, Tallman MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
127. Lu Q, Clemetson JM, Clemetson KJ. Snake venoms and hemostasis. J Thromb Haemost 2005;3:1791–9.
128. Berling I, Isbister GK. Hematologic effects and complications of snake envenoming. Transfus Med Rev 2015;29:82–9.
129. Isbister GK, Jayamanne S, Mohamed F, et al. A randomized controlled trial of fresh frozen plasma for coagulopathy in Russell’s viper (Daboia russelii) envenoming. J Thromb Haemost 2017;15:645–54.
130. Rodriguez V, Lee A, Witman PM, Anderson PA. Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 2009;31:522–6.
131. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 2017;27:86–90.
1. Carey MJ, Rodgers GM. Disseminated intravascular coagulation: clinical and laboratory aspects. Am J Hematol 1998;59:65–73.
2. De Jonge E, Levi M, Stoutenbeek CP, Van Deventer SJH. Current drug treatment strategies for disseminated intravascular coagulation. Drugs 1998;55:767–77.
3. Baker WF Jr. Clinical aspects of disseminated intravascular coagulation: a clinician’s point of view. Sem Thrombosis Hemostasis 1989;15:1–57.
4. Levi M, ten Cate H. Disseminated intravascular coagulation. N Engl J Med 1999;341:586–92.
5. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers 2016;2:16037.
6. Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol 2016;175:12–23.
7. Sharma S, Mayberry JC, DeLoughery TG, Mullins RJ. Fatal cerebroembolism from nonbacterial thrombotic endocarditis in a trauma patient: case report and review. Mil Med 2000;165:83–5.
8. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med 2016;36:505–12.
9. Yu M, Nardella A, Pechet L. Screening tests of disseminated intravascular coagulation: guidelines for rapid and specific laboratory diagnosis. Crit Care Med 2000;28:1777–80.
10. Mant MJ, King EG. Severe, acute disseminated intravascular coagulation. A reappraisal of its pathophysiology, clinical significance, and therapy based on 47 patients. Am J Med 1979;67:557–63.
11. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol 2009;145:24–33.
12. Levi M. Disseminated intravascular coagulation. Crit Care Med 2007;35:2191–5.
13. Nogami K. The utility of thromboelastography in inherited and acquired bleeding disorders. Br J Haematol 2016;174:503–14.
14. Gonzalez E, Moore EE, Moore HB. Management of trauma-induced coagulopathy with thrombelastography. Crit Care Clin 2017;33:119–34.
15. George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med 2006;354:1927–35.
16. George JN. How I treat patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Blood 2000;96:1223–9.
17. Murrin RJ, Murray JA. Thrombotic thrombocytopenic purpura: aetiology, pathophysiology and treatment. Blood Rev 2006;20:51–60.
18. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood 2017;129:2836–46.
19. Patton JF, Manning KR, Case D, Owen J. Serum lactate dehydrogenase and platelet count predict survival in thrombotic thrombocytopenic purpura. Am J Hematol 1994;47:94–9.
20. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991;325:393–7.
21. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpurahemolytic uremic syndrome—clinical experience in 108 patients. N Engl J Med 1991;325:398–403.
22. Kaplan BS, Trachtman H. Improve survival with plasma exchange thrombotic thrombopenic purpura-hemolytic uremic syndrome. Am J Med 2001;110:156–7.
23. Kremer Hovinga JA, Coppo P, Lammle B, et al. Thrombotic thrombocytopenic purpura. Nat Rev Dis Primers 2017;3:17020.
24. Asherson RA. The catastrophic antiphospholipid syndrome [editorial]. J Rheumatol 1992;19:508–12.
25. Asherson RA, Piette JC. The catastrophic antiphospholipid syndrome 1996: acute multi-organ failure associated with antiphospholipid antibodies: a review of 31 patients. Lupus 1996;5:414–7.
26. Asherson RA, Cervera R. Castastrophic antiphospholipid syndrome. Curr Opinion Hematol 2000;5:325–9.
27. Merrill JT, Asherson RA. Catastrophic antiphospholipid syndrome. Nat Clin Pract Rhuem 2006;2:81–9.
28. Rodriguez-Pinto I, Espinosa G, Cervera R. Catastrophic antiphospholipid syndrome: The current management approach. Best Pract Res Clin Rheumatol 2016;30:239–9.
29. Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol 2016;28:218–27.
30. Hoffman JN, Faist E. Coagulation inhibitor replacement during sepsis: useless? Crit Care Med 2000;28(9 Suppl):S74–6.
31. Wada H, Asakura H, Okamoto K, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Thromb Res 2010;125:6–11.
32. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
33. Counts RB, Haisch C, Simon TL, et al. Hemostasis in massively transfused trauma patients. Ann Surg 1979;190:91–9.
34. Stainsby D, MacLennan S, Hamilton PJ. Management of massive blood loss: a template guideline. Br J Anaesth 2000;85:487–91.
35. Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med 1999;340:409–17.
36. Blair SD, Janvrin SB, McCollum CN, Greenhalgh RM. Effect of early blood transfusion on gastrointestinal haemorrhage. Br J Surg 1986;73:783–5.
37. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013;368:11–21.
38. Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794–801.
39. Ciavarella D, Reed RL, Counts RB, et al. Clotting factor levels and the risk of diffuse microvascular bleeding in the massively transfused patient. Br J Haematol 1987;67:365–8.
40. Chowdhury P, Saayman AG, Paulus U, et al. Efficacy of standard dose and 30 ml/kg fresh frozen plasma in correcting laboratory parameters of haemostasis in critically ill patients. Br J Haematol 2004;125:69–73.
41. Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood 1982;60:284–7.
42. Callander N, Rapaport SI. Trousseau’s syndrome. West J Med 1993;158:364–71.
43. Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993;119:104–9.
44. Olson JD, Arkin CF, Brandt JT, et al. College of American Pathologists Conference XXXI on laboratory monitoring of anticoagulant therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998;122:782–8.
45. Yoshikawa T, Tanaka KR, Guze LB. Infection and disseminated intravascular coagulation. Medicine (Baltimore) 1971;50:237–58.
46. Jagneaux T, Taylor DE, Kantrow SP. Coagulation in sepsis. Am J Med Sci 2004;328:196–204.
47. Lipinska-Gediga M. Coagulopathy in sepsis - a new look at an old problem. Anaesthesiol Intensive Ther 2016;48:352–9.
48. Van Gorp ECM, Suharti C, ten Cate H, et al. Review: Infections diseases and coagulation disorders. Journal of Infectious Diseases 1999;180:176–86.
49. McDonald B, Davis RP, Kim SJ, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017;129:1357–67.
50. Semeraro F, Ammollo CT, Morrissey JH, et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–61.
51. Darmstadt GL. Acute infectious purpura fulminans: pathogenesis and medical management. Pediatr Dermatol 1998;15:169–83.
52. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol 2007;57:944–56.
53. Spicer TE, Rau JM. Purpura fulminans. Am J Med 1976;61:566–71.
54. Josephson C, Nuss R, Jacobson L, et al. The varicellaautoantibody syndrome. Pediatr Res 2001;50:345–52.
55. Smith OP, White B. Infectious purpura fulminans: diagnosis and treatment. Br J Haematol 1999;104:202–7.
56. Gamper G, Oschatz E, Herkner H, et al. Sepsis-associated purpura fulminans in adults. Wien Klin Wochenschr 2001;113:107–12.
57. Ward KM, Celebi JT, Gmyrek R, Grossman ME. Acute infectious purpura fulminans associated with asplenism or hyposplenism. J Am Acad Dermatol 2002;47:493–6.
58. Childers BJ, Cobanov B. Acute infectious purpura fulminans: a 15-year retrospective review of 28 consecutive cases. Am Surg 2003;69:86–90.
59. Carpenter CT, Kaiser AB. Purpura fulminans in pneumococcal sepsis: case report and review. Scand J Infect Dis 1997;29:479–83.
60. Warkentin TE, Pai M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis: reply. J Thromb Haemost 2016;14:2317–9.
61. Warkentin TE. Ischemic limb gangrene with pulses. N Engl J Med 2015;373:642–55.
62. Duncan A. New therapies for severe meningococcal disease but better outcomes? Lancet 1997;350:1565–6.
63. Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet1997;350:1590–3.
64. Branson HE, Katz J. A structured approach to the management of purpura fulminans. J Natl Med Assoc 1983;75:821–5.
65. Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth 2001;86:581–6.
66. Manios SG, Kanakoudi F, Maniati E. Fulminant meningococcemia. Heparin therapy and survival rate. Scand J Infect Dis 1971;3:127–33.
67. Giudici D, Baudo F, Palareti G, et al. Antithrombin replacement in patients with sepsis and septic shock. Haematologica 1999;84:452–60.
68. Fourrier F, Jourdain M, Tournoys A. Clinical trial results with antithrombin III in sepsis. Crit Care Med 2000;28(9 Suppl):S38–43.
69. Levi M, De Jonge E, van der PT, ten Cate H. Novel approaches to the management of disseminated intravascular coagulation. Crit Care Med 2000;28(9 Suppl):S20–4.
70. Rivard GE, David M, Farrell C, Schwarz HP. Treatment of purpura fulminans in meningococcemia with protein C concentrate. J Pediatr 1995;126:646–52.
71. White B, Livingstone W, Murphy C, et al. An open-label study of the role of adjuvant hemostatic support with protein C replacement therapy in purpura fulminans-associated meningococcemia. Blood 2000;96:3719–24.
72. Schellongowski P, Bauer E, Holzinger U, et al. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin). Vox Sang 2006;90:294–301.
73. DeLoughery TG. Coagulation defects in trauma patients: etiology, recognition, and therapy. Crit Care Clin 2004;20:13–24.
74. Cohen MJ, Christie SA. Coagulopathy of trauma. Crit Care Clin 2017;33:101–18.
75. Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): a focused review on pathophysiology. Intern Emerg Med 2017 May 5.
76. Chang R, Cardenas JC, Wade CE, Holcomb JB. Advances in the understanding of trauma-induced coagulopathy. Blood 2016;128:1043–9.
77. Eddy VA, Morris JA Jr, Cullinane DC. Hypothermia, coagulopathy, and acidosis. Surg Clin North Am 2000;80:845–54.
78. Peng RY, Bongard FS. Hypothermia in trauma patients. J Am Coll Surg 1999;188:685–96.
79. Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma 1990;30:200–2.
80. Rajek A, Greif R, Sessler DI, et al. Core cooling by central venous infusion of ice-cold (4 degrees C and 20 degrees C) fluid: isolation of core and peripheral thermal compartments. Anesthesiol 2000;93:629–37.
81. Watts DD, Trask A, Soeken K, et al. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma 1998;44:846–54.
82. Ferrara A, MacArthur JD, Wright HK, et al. Hypothermia and acidosis worsen coagulopathy in the patient requiring massive transfusion. Am J Surg 1990;160:515–8.
83. Holcomb JB, Tilley BC, Baraniuk S, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015;313:471–82.
84. Johansson PI, Stensballe J, Oliveri R, Wade CE, Ostrowski SR, Holcomb JB. How I treat patients with massive hemorrhage. Blood 2014;124:3052–8.
85. Stone HH, Strom PR, Mullins RJ. Management of the major coagulopathy with onset during laparotomy. Ann Surg 1983;197:532–5.
86. WOMAN Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23–32.
87. Hall DR. Abruptio placentae and disseminated intravascular coagulopathy. Semin Perinatol 2009;33:189–95.
88. Thachil J, Toh CH. Disseminated intravascular coagulation in obstetric disorders and its acute haematological management. Blood Rev 2009;23:167–76.
89. Collins P, Abdul-Kadir R, Thachil J, Subcommittees on Women’ s Health Issues in T, Haemostasis, on Disseminated Intravascular C. Management of coagulopathy associated with postpartum hemorrhage: guidance from the SSC of the ISTH. J Thromb Haemost 2016;14:205–10.
90. Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv 2004;59:838–45.
91. Egerman RS, Sibai BM. HELLP syndrome. Clin Obstetr Gynecol 1999;42:381–9.
92. Saphier CJ, Repke JT. Hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a review of diagnosis and management. Sem Perinatol 1998;22:118–33.
93. Le Thi TD, Tieulie N, Costedoat N, et al. The HELLP syndrome in the antiphospholipid syndrome: retrospective study of 16 cases in 15 women. Ann Rheum Dis 2005;64:273–8.
94. Martin JN Jr, Perry KG Jr, Blake PG, et al. Better maternal outcomes are achieved with dexamethasone therapy for postpartum HELLP (hemolysis, elevated liver enzymes, and thrombocytopenia) syndrome. Am J Obstet Gynecol 1997;177:1011–7.
95. Magann EF, Martin JN Jr. Twelve steps to optimal management of HELLP syndrome. Clinical Obstet Gynecol 1999;42:532–50.
96. Jwayyed SM, Blanda M, Kubina M. Acute fatty liver of pregnancy. J Emerg Medi 1999;17:673–7.
97. Bacq Y. Acute fatty liver of pregnancy. Sem Perinatol 1998;22:134–40.
98. Egerman RS, Sibai BM. Imitators of preeclampsia and eclampsia. Clin Obstet Gynecol 1999;42:551–62.
99. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Medi Rev 1993;7:255–67.
100. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis 1992;15:863–5.
101. Garratty G, Nance S, Lloyd M, Domen R. Fatal immune hemolytic anemia due to cefotetan. Transfusion 1992;32:269–71.
102. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes:case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion 1999;39:306–9.
103. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion 1999;39:1239–46.
104. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol 2006;81:186–8.
105. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr 1995;126:813–5.
106. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J 1995;14:1116–7.
107. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone. J Pediatr 1995;126:816–7.
108. Gottschall JL, Elliot W, Lianos E, et al. Quinine-induced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood 1991;77:306–10.
109. Gottschall JL, Neahring B, McFarland JG, et al. Quinine-induced immune thrombocytopenia with hemolytic uremic syndrome: clinical and serological findings in nine patients and review of literature. Am J Hematol 1994;47:283–9.
110. Crum NF, Gable P. Quinine-induced hemolytic-uremic syndrome. South Med J 2000;93:726–8.
111. Vesely T, Vesely JN, George JN. Quinine-Induced thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS): frequency, clinical features, and long-term outcomes. Blood 2000;96:629 [abstract].
112. Bell WR, Starksen NF, Tong S, Porterfield JK. Trousseau’s syndrome. Devastating coagulopathy in the absence of heparin. Am J Med 1985;79:423–30.
113. Sack GH, Levin J, Bell WR. Trousseau’s syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinic, pathophysiologic, and therapeutic features. Medicine 1977;56:1–37.
114. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007;110:1723–9.
115. Prandoni P, Falanga A, Piccioli A. Cancer and venous thromboembolism. Lancet Oncol 2005;6:401–10.
116. de la Fouchardiere C, Flechon A, Droz JP. Coagulopathy in prostate cancer. Neth J Med 2003;61:347–54.
117. Kearon C, Kahn SR, Agnelli G, et al. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. 8th ed. Chest 2008;133(6 Suppl):454S–545S.
118. Lee AY, Kamphuisen PW, Meyer G, et al. Tinzaparin vs warfarin for treatment of acute venous thromboembolism in patients with active cancer: a randomized clinical trial. JAMA 2015;314:677–86.
119. Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol 2012;87:596–603.
120. Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood 2017;129:1855–64.
121. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008;111:2505–15.
122. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013;369:111–21.
123. Dombret H, Scrobohaci ML, Ghorra P, et al. Coagulation disorders associated iwth acute promyelocytic leukemia: Corrective effect of all-trans retinoic acid treatment. Leukemia 1993;7:2–9.
124. Falanga A, Rickles FR. Management of thrombohemorrhagic syndromes (THS) in hematologic malignancies. Hematology Am Soc Hematol Educ Program 2007;2007:165–71
125. Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood 2009;114:5126–35.
126. Sanz MA, Grimwade D, Tallman MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009;113:1875–91.
127. Lu Q, Clemetson JM, Clemetson KJ. Snake venoms and hemostasis. J Thromb Haemost 2005;3:1791–9.
128. Berling I, Isbister GK. Hematologic effects and complications of snake envenoming. Transfus Med Rev 2015;29:82–9.
129. Isbister GK, Jayamanne S, Mohamed F, et al. A randomized controlled trial of fresh frozen plasma for coagulopathy in Russell’s viper (Daboia russelii) envenoming. J Thromb Haemost 2017;15:645–54.
130. Rodriguez V, Lee A, Witman PM, Anderson PA. Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 2009;31:522–6.
131. Triana P, Dore M, Cerezo VN, et al. Sirolimus in the treatment of vascular anomalies. Eur J Pediatr Surg 2017;27:86–90.
Cancer-Related Fatigue: Approach to Assessment and Management
INTRODUCTION
Fatigue is a common distressing effect of cancer.1 It impairs the quality of life of patients undergoing active cancer treatment and of post-treatment survivors alike. The National Comprehensive Cancer Network (NCCN) defines cancer-related fatigue (CRF) as “a distressing, persistent, subjective sense of physical, emotional and/or cognitive tiredness related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning.”2 CRF differs from fatigue reported by individuals without cancer in that CRF is more severe and is not relieved by rest. The prevalence of CRF in cancer patients and survivors is highly variable, with estimates ranging between 25% and 99%.2,3 The methods used for screening patients for fatigue and the characteristics of the patient groups may account for this variability.
PATHOPHYSIOLOGY
The specific pathophysiologic mechanism underlying CRF is unknown, making targeted treatment a challenge. The multidimensional and subjective nature of CRF has limited the development of research methodologies to explain this condition. However, research has been done in both human and animal models, and several theories have been proposed to explain the pathophysiology of CRF. While pro-inflammatory cytokines remain the central factor playing a significant role at multiple levels in CRF, there may be a complex interplay of multiple mechanisms contributing to fatigue in an individual patient.
CENTRAL NERVOUS SYSTEM DISTURBANCES
The basal ganglia are known to influence motivation. Lack of motivation and drive may cause failure to complete physical and mental tasks, even with preserved cognitive ability and motor function. In a study of melanoma patients receiving interferon, increased activity of the basal ganglia and the cerebellum resulted in higher fatigue scores.4 Increased levels of cytokines may alter blood flow to the cerebellum and lead to the perception of fatigue. In a study of 12 patients and matched controls, when patients were asked to perform sustained elbow flexion until they perceived exhaustion, CRF patients perceived physical exhaustion sooner than controls. In CRF patients in this study, muscle fatigue measured by electromyogram was less than that in healthy individuals at the time of exhaustion, suggesting the role of the central nervous system in CRF.5 However, there is not enough evidence at this time to support central nervous system disturbance as the main factor contributing to fatigue in cancer patients.
CIRCADIAN RHYTHM DYSREGULATION
Circadian rhythm is regulated by the suprachiasmatic nucleus in the hypothalamus through cortisol and melatonin. Sleep disturbances occur with disruption of the circadian rhythm. Tumor-related peptides such as epidermal growth factor or alterations in serotonin and cortisol can influence the suprachiasmatic nucleus and the complex signaling pathways.2 Positive feedback loops that are activated by cortisol under the influence of cytokines may lead to continuous cytokine production and altered circadian rhythm. Bower et al showed that changes in the cortisol curve influence fatigue in breast cancer survivors.6 These patients had a late evening peak in cortisol levels, compared with an early morning peak in individuals without cancer.
INHIBITION OF HYPOTHALAMIC-PITUITARY-ADRENAL AXIS
The hypothalamic–pituitary–adrenal (HPA) axis regulates the release of the stress hormone cortisol. One of several hypotheses advanced to explain the effect of serotonin and the HPA axis on CRF suggests that lower serotonin levels cause decreased activation of 5-hydroxytrytophan 1-a (5-HT1-a) receptors in the hypothalamus, leading to decreased activity of the HPA axis.6 Inhibition of the HPA axis may occur with higher levels of serotonin as well.7 The 5-HT1-a receptors are also triggered by cytokines. However, the correction of serotonin levels by antidepressants was not shown to improve fatigue.8 Inhibition of the HPA axis can also lead to lower testosterone, progesterone, or estrogen levels, which may indirectly contribute to fatigue.2
SKELETAL MUSCLE EFFECT
Chemotherapy- and tumor-related cachexia have a direct effect on the metabolism of skeletal muscles. This effect may lead to impaired adenosine triphosphate (ATP) generation during muscle contraction.9 ATP infusion improved muscle strength in 1 trial, but this was not confirmed in another trial.10,11 Muscle contraction studies showed no differences in the contractile properties of muscles in fatigued patients who failed earlier in motor tasks and healthy controls.12 This finding suggests that there could be a failure of skeletal muscle activation by the central nervous system or inhibition of skeletal muscle activity. Cytokines and other neurotransmitters activate vagal efferent nerve fibers, which may lead to reflex inhibition in skeletal muscles.13,14
PRO-INFLAMMATORY CYTOKINES
Tumors or treatment of them may cause tissue injury, which triggers immune cells to release cytokines, signaling the brain to manifest the symptom of fatigue. Inflammatory pathways are influenced by psychological, behavioral, and biological factors, which play a role as risk factors in CRF. Levels of interleukin 6 (IL-6), interleukin-1 receptor antagonist, interleukin-1, and tumor necrosis factor (TNF) have been shown to be elevated in fatigued patients being treated for leukemia and non-Hodgkin lymphoma.15 IL-6 was also associated with increased fatigue in breast cancer survivors.16 Similar findings were reported in patients undergoing stem cell transplantation and high-dose chemotherapy.17 Elevated levels of IL-6 and C-reactive protein were also linked to fatigue in terminally ill cancer patients.18,19 Furthermore, TNF-α signaling was associated with post-chemotherapy fatigue in breast cancer patients.20 Leukocytes in breast cancer survivors with fatigue also have increased gene expression of pro-inflammatory cytokines, emphasizing the role of cytokines and inflammation in the pathogenesis of CRF.21
OTHER HYPOTHESES
Several other hypotheses for CRF pathogenesis have been proposed. Activation of latent viruses such as Epstein-Barr virus, lack of social support,22 genetic alterations in the immune pathway,23 epigenetic changes,24 accumulation of neurotoxic metabolites and depletion of serotonin by indoleamine 2,3-dioxygenase pathway activation,25 elevated vascular endothelial growth factor levels,26 and hypoxia-related organ dysfunction due to anemia or hemoglobin dysfunction13 all have been postulated to cause CRF.
EVALUATION AND TREATMENT
Fours steps are involved in the evaluation and treatment of CRF (Figure).
SCREENING
Because patients and health care professionals may be unaware of the treatment options available for CRF, patients may not report fatigue levels to their clinicians, and clinicians may not understand the impact of fatigue on their patients’ quality of life. This leads to under-recognition of the problem. The NCCN recommends screening every cancer patient and post-treatment survivor for fatigue.2 Patients should be screened at their first visit and then at periodic intervals during and after cancer treatment.
Many scales are available to screen patients for CRF in clinical practice and clinical trials.27 A single item that asks patients to rate their fatigue on a scale from 0 to 10—in which 0 indicates no fatigue, 1 to 3 indicates mild fatigue, 4 to 6 indicates moderate fatigue, 7 to 9 indicates severe fatigue, and 10 indicates the worst fatigue imaginable—is commonly used to screen for CRF.2 This scale was adapted from the MD Anderson Symptom Inventory scale and is based on a large nationwide study of cancer patients and survivors.28 The statistically derived cutoff points in this study are consistent with other scales such as the Brief Fatigue Inventory (BFI) and support the cutoff points (4–6 for moderate and ≥ 7 for severe fatigue) used in various fatigue management guidelines. Furthermore, studies of fatigue in cancer patients have revealed a marked decrease in physical function at levels of 7 or higher, suggesting 7 as an optimal cutoff to identify severe fatigue.29,30 The Visual Analog Scale is another simple-to-use tool that helps in understanding variations in fatigue throughout the course of the day.31 The 9-item BFI is often used in clinical trials.29 It measures the severity of fatigue over the previous 24 hours and has been validated in patients who do not speak English.32
CRF affects not only the somatic domain, but also the cognitive, behavioral, and affective domains; therefore, multidimensional scales have been developed for screening. One such tool is the Multidimensional Fatigue Inventory, which assesses 5 dimensions of fatigue—general fatigue, physical fatigue, reduced motivation, reduced activity, and mental fatigue—and compares the patient’s results with those of individuals without cancer.33,34 The Functional Assessment of Cancer Therapy for Fatigue (FACT-F) is a 13-item questionnaire that has been used to measure CRF in clinical trials as well as in patients receiving various treatments.35
Although many scales are available, the validity of self-reporting on simple fatigue-rating scales is equal to or better than most complex, lengthy scales.36 Therefore, unidimensional tools such as the numeric rating scale of 0–10 are commonly used in clinical practice. Mild fatigue (0–3) requires periodic reevaluation, and moderate and severe fatigue need further evaluation and management.37
PRIMARY EVALUATION
This phase involves a focused history and physical examination and assessment of concurrent symptoms and contributing factors.
History and Physical Examination
A detailed history of the patient’s malignancy and type of previous and current treatment may help reveal the cause of fatigue. New-onset fatigue or increase in fatigue may be related to the progression of disease in patients with active malignancy or recurrence of cancer in survivors. These patients may require appropriate testing to assess the underlying disease pattern. A detailed review of systems may help identify some of the contributing factors, which are discussed below. A detailed history regarding medications, including over-the-counter drugs, complementary agents, and past and prior cancer therapies, is helpful as medications can contribute to fatigue. For example, opioids may cause drowsiness and fatigue, which could be improved by dose adjustments. A focused history of fatigue should be obtained in all patients with moderate to severe CRF, which includes the onset, pattern, duration, associated or alleviating factors, and interference with functioning, including activities of daily living.37 Physical examination should focus on identifying signs of organ dysfunction and features of substance or alcohol abuse, which may cause poor sleep and fatigue.
Assessment of Contributing Factors
The management of fatigue should be multifactorial, with a comprehensive assessment and treatment plan to address all modifiable fatigue etiologies. The Table lists potential contributing factors to fatigue that should be considered when evaluating patients for CRF; several common conditions are discussed below. 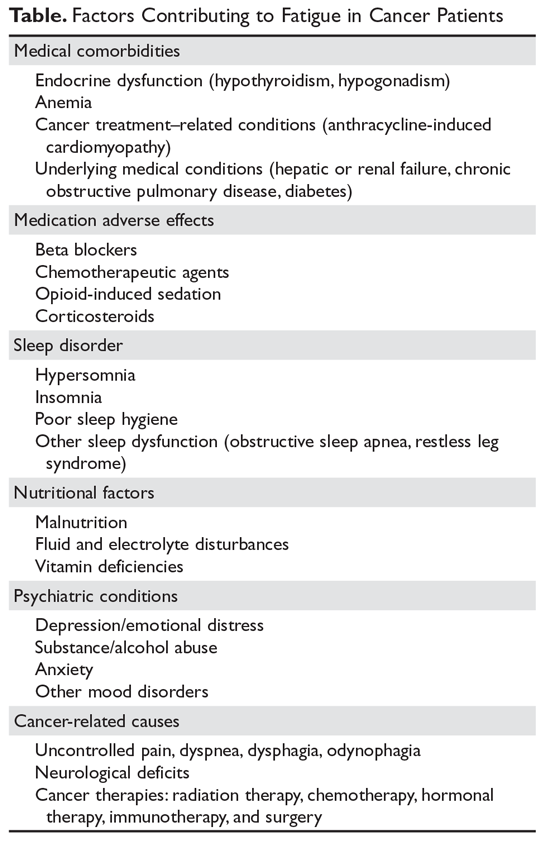
Anemia. Anemia has been correlated with fatigue and quality of life. In a study of 4382 cancer patients receiving chemotherapy, quality-of-life measures using FACT-Anemia scores improved with increased hemoglobin levels.38 Cancer patients may have anemia due to marrow-suppressing effects of chemotherapy and may also have iron deficiency anemia due to blood loss or auto-immune hemolytic anemia. Therefore, a detailed work-up is required to identify the etiology of anemia. Patients with CRF whose anemia is related to chemotherapy or anemia of chronic disease may benefit from red blood cell transfusion or erythropoiesis-stimulating agents (ESAs). ESAs have been studied extensively; however, their use is controversial because of concerns about thromboembolic side effects leading to adverse outcomes.39 Also, ESA therapy is not recommended in patients with hematologic malignancies. ESA use should be restricted to patients with chemotherapy-related anemia with hemoglobin below 10 mg/dL and should be discontinued in 6 to 8 weeks if patients do not respond.40 Other patients may benefit from blood transfusions, which were shown to help in patients with hemoglobin levels between 7.5 and 8.5 g/dL.41
Sleep disturbance. Poor sleep is common in fatigued cancer survivors.42 Pro-inflammatory cytokines can disrupt the sleep–wake cycle, causing changes in the HPA axis and neuroendocrine system, which in turn may lead to increasing fatigue. Heckler et al showed that improvement in nighttime sleep leads to improvement of fatigue.43 Cognitive behavioral therapy and sleep hygiene are important in caring for patients with CRF and poor sleep.44 Taking a warm bath and/or drinking a glass of milk before bedtime, avoiding caffeinated drinks, and avoiding frequent napping in the day might help. Some patients may require medications such as benzodiazepines or non-benzodiazepine hypnotics (eg, zolpidem) to promote sleep.45 Melatonin agonists are approved for insomnia in the United States, but not in Europe.46
Malnutrition. Patients with advanced-stage cancer and with cancers affecting the gastrointestinal tract frequently develop mechanical bowel obstructions, especially at the end of their life, which cause malnutrition. Chemotherapy-related nausea and vomiting may also cause poor oral intake and malnutrition, causing fatigue from muscle weakness. Dehydration and electrolyte imbalances frequently occur as a result of poor oral intake, which might worsen fatigue. In our experience, modifying dietary intake with appropriate caloric exchanges with the help of a nutrition expert has lessened fatigue in some patients. However, terminally ill patients are advised to eat based on their comfort.
Medical comorbidities. Congestive heart failure from anthracycline chemotherapy, hypothyroidism after radiation therapy for head and neck cancers, renal failure, or hepatic failure from chemotherapy may indirectly lead to fatigue. Chemotherapy, opioids, and steroids may cause hypogonadism, which can contribute to fatigue in men.47
Assessment of Concurrent Symptoms
Depression is common in cancer patients and coexists with pain, insomnia, fatigue, and anxiety as a symptom cluster.48 A symptom cluster is defined as 2 or more concurrent and interrelated symptoms occurring together; treating one of these symptoms without addressing other symptoms is not effective.49 Therefore, screening for and management of depression, anxiety, and insomnia play an important role in the management of CRF.
Physical symptoms due to the tumor or to therapy— such as pain, dyspnea, nausea, and decreased physical activity—may also contribute to fatigue both directly and indirectly. Patients with lung cancer may have hypoxemia, which can contribute to dyspnea with activity and a perception of fatigue. Optimal management of pain and other physical symptoms in patients with cancer may significantly alleviate fatigue.50
MANAGEMENT
Management of CRF is individualized based on the patient’s clinical status: active cancer treatment, survivor, or end of life. Education and counselling of patients and their caregivers play an important role in CRF. NCCN guidelines recommend focusing on pain control, distress management, energy conservation, physical activity, nutrition, and sleep hygiene.
Nonpharmacologic Interventions
Energy conservation. Energy conservation strategies, in which patients are advised to set priorities and realistic expectations, are highly recommended. Some energy-conserving strategies are to pace oneself, delegate and schedule activities at times of peak energy, postpone nonessential activities, attend to 1 activity at a time, structure daily routines, and maintain a diary to identify their peak energy period and structure activities around that time.51,52 When patients approach the end of life, increasing fatigue may limit their activity level, and they may depend on caregivers for assistance with activities of daily living, monitoring treatment-related adverse effects, and taking medications, especially elderly patients.53
Cognitive behavioral therapy. Cognitive behavioral therapy (CBT) has been shown to improve CRF during active treatment, and the benefits persist for a minimum of 2 years after therapy.54 CBT interventions that optimize sleep quality may improve fatigue.55 More studies are needed to understand whether referral to a psychologist for formal CBT is required. Randomized clinical trials showed patient fatigue education, learned self-care, coping techniques, and balancing rest and activity benefit patients with CRF.56
Exercise. Physical activity is highly encouraged in patients with CRF. Exercise increases muscle protein synthesis, improves cytokine response, and decreases the rate of sarcopenia in healthy populations.57 Studies have shown that exercise helps CRF at all phases of the cancer journey, including radiation therapy, chemotherapy, and survivorship.58 Some patients may feel less motivated to exercise and may not believe that exercise is possible or could potentially help them. Counselling is needed for such patients.
Older cancer survivors have a decline in their functional capacity and reduced muscle mass. Exercise can improve their cardiorespiratory fitness, muscle strength, and body composition.57 Exercise not only helps at the cellular level but also has psychosocial benefits from improved self-esteem. Therefore, exercise may be recommended for younger patients as well as for the older population, who may have comorbidities and less motivation than younger patients.
In a meta-analysis of 56 randomized controlled trials involving 4068 participants, aerobic exercise was found to have beneficial effects on CRF for patients during and after chemotherapy, specifically for patients with solid tumors.59 In another meta-analysis of breast and prostate cancer survivors, a combination of aerobic exercise with resistance training (3–6 metabolic equivalents, 60%–80% range of motion) was shown to reduce CRF more than aerobic exercise alone.60 This effect was also shown in a randomized controlled trial of 160 patients with stage 0 to III breast cancer undergoing radiation therapy.61 The control group in this study had a group-based non-exercise intervention/relaxation; therefore, the study showed that the effect of resistance training extends beyond the psychosocial benefits of group-based interventions. The intervention comprised 8 progressive machine-based resistance exercises (3 sets, 8–12 repetitions at 60%–80% of 1 repetition maximum) for 60 minutes twice weekly for 12 weeks. However, fatigue assessment questionnaire scores showed benefits only in the physical fatigue components, but not in the affective and cognitive components.
The American Society of Clinical Oncology’s guidelines for cancer survivors with fatigue recommends 150 minutes of moderate aerobic exercise (eg, fast walking, cycling, or swimming) per week, with 2 or 3 sessions of strength training per week.62 An individualized approach to exercise is recommended, as patients’ ability to perform certain types of exercises may be limited by thrombocytopenia, neutropenia, or lytic bone metastasis. Routine use of pre-exercise cardiovascular testing is not recommended but may be considered in high-risk populations, especially patients with risk factors for coronary heart disease and diabetes.63 Patients with comorbidities, substantial deconditioning, functional and anatomic defects, or recent major surgery may benefit from referral to physical therapy.37 Patients near end of life may also benefit from an exercise program, as demonstrated in several studies that showed benefit in CRF and quality of life.64,65 We recommend that physicians use their best clinical judgement in suggesting the type and intensity of exercise program, as it may not be feasible in some patients.
Mind-body interventions. Mindfulness-based stress reduction (MBSR) has shown promise in breast cancer survivors, who reported immediate improvements in fatigue severity that continued up to 6 weeks after cessation of the training.66 Prior studies had similar findings, suggesting that MBSR modestly decreases fatigue and sleep disturbances and has a greater effect on the degree to which symptoms interfere with many facets of life.67
Yoga. A study of a yoga intervention showed a benefit in older cancer survivors.68 In breast cancer patients undergoing chemotherapy, yoga was shown to benefit both physical and cognitive fatigue.69 DVD-based yoga had benefits similar to strengthening exercises in a study of 34 early-stage breast cancer survivors with CRF.70 More studies are needed in men and patients and survivors of other cancers, as most studies of yoga were conducted in women with breast cancer.
Tai chi/qigong. Like yoga, tai chi and qigong are practices of meditative movement. These practices use postures or movements with a focus on breath and a meditative state to bring about deep states of relaxation. Qigong is a series of simple, repeated practices including body posture/movement, breath practice, and meditation performed in synchrony. Tai chi easy (TCE) is a simplified set of common, repetitive tai chi movements. In a trial, qigong/TCE was compared with sham qigong, which had physical movements but no breathing or meditative practice. Breast cancer survivors in the qigong/TCE group had improved fatigue scores, and the effect persisted for 3 months.71 Additional research is needed in this area.
Acupuncture. A randomized controlled trial in breast cancer patients with CRF showed an improvement in the mean general fatigue score (per the Multidimensional Fatigue Inventory) in patients who received acupuncture versus those who did not (−3.11 [95% confidence interval −3.97 to −2.25]; P < 0.001) at 6 weeks. Improvements were seen in both the mental and physical aspects of fatigue.72 However, Deng et al noted that true acupuncture was no more effective than sham acupuncture for reducing post-chemotherapy chronic fatigue.73 Presently, there is not sufficient evidence to evaluate the benefits of acupuncture in CRF.
Other modalities. Massage therapy, music therapy, hypnosis, therapeutic touch, biofield therapies, relaxation, and reiki are other therapies for which few studies have been done; of the studies that have been done, the results are mixed, and additional research is needed.74 Currently, there are not sufficient data to recommend any of these modalities.
Pharmacologic Interventions
Psychostimulants. Methylphenidate and modafinil are psychostimulants or wakefulness-promoting agents. Pilot studies showed benefit from methylphenidate and modafinil in CRF,75–77 but randomized controlled trials have yielded mixed results. Therefore, in patients with severe fatigue during cancer therapy, the initial management strategy involves evaluation and treatment of medical conditions such as anemia and a trial of nonpharmacological strategies as discussed above. If symptoms persist, then a therapeutic trial of a psychostimulant may be considered per NCCN guidelines for patients undergoing active cancer treatment.37
Methylphenidate directly stimulates adrenergic receptors and indirectly releases dopamine and norepinephrine from presynaptic terminals, which may explain why the drug benefits patients receiving opioid-induced sedation. It is a commonly studied psychostimulant, though its mechanism of action in CRF is unclear. Randomized controlled trials of methylphenidate have resulted in a wide range of findings due to the heterogeneity of study populations and variations in the dosage of methylphenidate. A meta-analysis of 7 studies indicates that methylphenidate benefitted the subgroup of patients with CRF.78 Likewise, in an analysis of 5 randomized controlled trials, Minton et al showed a benefit of psychostimulants in fatigue compared with placebo.79 However, another study of methylphenidate in patients with CRF showed a benefit only in patients with severe fatigue or advanced disease.80 Methylphenidate was found to benefit cancer patients receiving opioid-induced sedation, as methylphenidate promotes wakefulness, though fatigue was not studied specifically.81 In a trial with 30 hospice patients in which the methylphenidate dose was titrated based on response and adverse effects, Kerr at al found that the drug improved fatigue in a dose-dependent manner.82 However, a study in patients with CRF at the University of Texas MD Anderson Cancer Center found no significant difference in BFI scores between patients receiving methylphenidate and those receiving placebo at the end of 2 weeks of treatment.83 Also, other randomized controlled trials in patients undergoing adjuvant chemotherapy for breast cancer84 and patients receiving radiation therapy for brain tumors85 failed to demonstrate the efficacy of methylphenidate in CRF. It should be used cautiously after ruling out other causes of fatigue. The drug is overall well tolerated and side effects include headache and nausea.
Modafinil is a non-amphetamine psychostimulant that has been approved for the treatment of narcolepsy. In a trial studying the effect of modafinil on patients receiving docetaxel-based chemotherapy for metastatic breast or prostate cancer, there was a modest but not statistically significant improvement in fatigue scores on the MD Anderson Symptom Inventory compared with placebo. Nausea and vomiting were higher in the modafinil arm than in the placebo arm.86 Similarly, modafinil was not superior to placebo for CRF in 208 patients with non-squamous cell lung cancer not undergoing chemotherapy or radiation.87 A placebo effect was also noted in patients with multiple myeloma88 and patients with primary brain tumors.89 In a phase 3, multicenter, randomized, placebo-controlled, double-blind clinical trial of modafinil for CRF in 867 patients undergoing chemotherapy, there was a reduction in fatigue only for patients with severe baseline fatigue, with no significant effect on mild to moderate fatigue.90 In another recent study, modafinil was shown to reduce depressive symptoms only in patients with severe fatigue (BFI item 3 score ≥ 7).91 This finding is consistent with previous studies showing benefit in patients with high baseline fatigue, but additional randomized controlled trials are needed to provide clarity. NCCN guidelines do not recommend the use of modafinil to treat CRF.37
Other pharmacologic interventions. Corticosteroids are often used for symptom control in cancer patients. These drugs have anti-inflammatory effects through their modulation of pro-inflammatory cytokines.92 In a randomized controlled trial evaluating the efficacy of corticosteroids, patients receiving dexamethasone (4 mg twice daily) experienced significant improvement in their FACT-F scores compared with patients receiving placebo.93 A similar benefit in fatigue was demonstrated in another study of methylprednisolone (32 mg daily) versus placebo.94 Despite the benefits of steroids, their adverse effects, such as mood swings, gastritis, hyperglycemia, and immune suppression, limit their long-term use. Therefore, the use of steroids should be restricted to terminally ill fatigued patients with other symptoms such as anorexia, brain metastasis, or pain related to bone metastasis.37
Testosterone replacement has been shown to diminish fatigue in non-cancer patients. Many men with advanced cancer have hypogonadism leading to low serum testosterone, which may cause fatigue. In a small trial in which cancer patients with hypogonadism received intramuscular testosterone every 14 days or placebo, the group receiving testosterone showed improvement in FACT-F scores, but there was no significant difference in FACT-F scores between the 2 groups.95
Antidepressants have failed to demonstrate benefit in CRF without depression.8 However, if a patient has both fatigue and depression, antidepressants may help.96 A selective serotonin receptor inhibitor is recommended as a first-line antidepressant.97 Patients with cancer are often receiving multiple medications, and medication interactions should be considered to prevent adverse events such as serotonin syndrome.
Complementary and Alternative Supplements
Studies using vitamin supplementation have been inconclusive in patients with CRF.74 The use of other dietary supplements has yielded mixed results, and coenzyme Q has shown no benefit for patients with CRF.98
The benefit of ginseng was studied in a RCT involving 364 patients with CRF. There was an improvement in Multidimensional Fatigue Symptom Inventory-short form (MFSI-SF) scores at 8 weeks in patients receiving 2000 mg of Wisconsin ginseng compared with patients receiving placebo.99 Patients on active treatment had greater improvement as compared to the post-treatment group in this trial. In another study of high-dose panax ginseng (ginseng root) at 800 mg daily for 29 days, patients had improvement of CRF as well as overall quality of life, appetite, and sleep at night. It was also well tolerated with few adverse effects.100 Interaction with warfarin, calcium channel blockers, antiplatelet agents, thrombolytic agents, imatinib, and other agents may occur; therefore, ginseng must be used with careful monitoring in selected patients. There is not enough evidence at this time to support the routine use of ginseng in CRF.
The seed extract of the guarana plant (Paullinia cupana) traditionally has been used as a stimulant. An improvement in fatigue scores was seen with the use of oral guarana (100 mg daily) at the end of 21 days in breast cancer patients receiving chemotherapy.101 Further studies are needed for these results to be generalized and to understand the adverse effects and interaction profile of guarana.
Reevaluation
Patients who have completed cancer treatment must be monitored for fatigue over the long term, as fatigue may exist beyond the period of active treatment. Many studies have shown fatigue in breast cancer survivors, and fatigue has been demonstrated in survivors of colorectal, lung, and prostate cancers as well as myeloproliferative neoplasms.28 Therefore, it is important to screen patients for fatigue during follow-up visits. There are currently no studies evaluating the long-term treatment of fatigue. In our experience, the timing of follow-up depends on the level of fatigue and interventions prescribed. Once fatigue is stabilized to a level with which the patient is able to cope, the time interval for follow-up may be lengthened. Annual visits may suffice in patients with mild fatigue. Follow-up of patients with moderate to severe fatigue depends on the level of fatigue, the ability to cope, choice of treatment, and presence of contributing factors.
CONCLUSION
CRF is a complex condition that places a significant burden on patients and caregivers, resulting in emotional distress, poor functioning, and suffering. Fatigue can occur before, during, and long after cancer treatment. The approach to CRF begins with screening for and educating patients and their caregivers about the symptoms. Many screening scales are available that may be used to follow patients’ progress over time. The evaluation and management of contributing conditions may help improve fatigue. If the fatigue persists, an individualized approach with a combination of nonpharmacologic and pharmacologic approaches should be considered. More research is needed to understand brain signaling pathways, cytokine changes, and genomic changes in cancer patients with fatigue. Though many hypotheses have been proposed, to date there is no biological marker to assess this condition. Biomarker research needs to be advanced to help to identify patients at risk for fatigue. As cytokines have a major role in CRF, targeted therapy to block cytokine pathways may also be explored in the future.
Acknowledgment: The authors thank Bryan Tutt for providing editorial assistance during the writing of this article.
1. Scherber RM, Kosiorek HE, Senyak Z, et al. Comprehensively understanding fatigue in patients with myeloproliferative neoplasms. Cancer 2016;122:477–85.
2. Neefjes EC, van der Vorst MJ, Blauwhoff-Buskermolen S, Verheul HM. Aiming for a better understanding and management of cancer-related fatigue. Oncologist 2013;18:1135–43.
3. Radbruch L, Strasser F, Elsner F, et al. Fatigue in palliative care patients—an EAPC approach. Palliat Med 2008;22:13–32.
4. Capuron L, Pagnoni G, Demetrashvili MF, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 2007;32:2384–92.
5. Kisiel-Sajewicz K, Siemionow V, Seyidova-Khoshknabi D, et al. Myoelectrical manifestation of fatigue less prominent in patients with cancer related fatigue. PLoS One 2013;8:e83636.
6. Bower JE, Ganz PA, Aziz N. Altered cortisol response to psychologic stress in breast cancer survivors with persistent fatigue. Psychosom Med 2005;67:277–80.
7. Barsevick A, Frost M, Zwinderman A, et al. I’m so tired: biological and genetic mechanisms of cancer-related fatigue. Qual Life Res 2010;19:1419–27.
8. Morrow GR, Hickok JT, Roscoe JA, et al. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol 2003;21:4635–41.
9. Fontes-Oliveira CC, Busquets S, Toledo M, et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 2013;1830:2770–8.
10. Agteresch HJ, Dagnelie PC, van der Gaast A, et al. Randomized clinical trial of adenosine 5’-triphosphate in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst 2000;92:321–8.
11. Beijer S, Hupperets PS, van den Borne BE, et al. Randomized clinical trial on the effects of adenosine 5’-triphosphate infusions on quality of life, functional status, and fatigue in preterminal cancer patients. J Pain Symptom Manage 2010;40:520–30.
12. Kisiel-Sajewicz K, Davis MP, Siemionow V, et al. Lack of muscle contractile property changes at the time of perceived physical exhaustion suggests central mechanisms contributing to early motor task failure in patients with cancer-related fatigue. J Pain Symptom Manage 2012;44:351–61.
13. Ryan JL, Carroll JK, Ryan EP, et al. Mechanisms of cancer-related fatigue. Oncologist 2007;12 Suppl 1:22–34.
14. Seruga B, Zhang H, Bernstein LJ, Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer 2008;8:887–99.
15. Wang XS, Giralt SA, Mendoza TR, et al. Clinical factors associated with cancer-related fatigue in patients being treated for leukemia and non-Hodgkin’s lymphoma. J Clin Oncol 2002;20:1319–28.
16. Collado-Hidalgo A, Bower JE, Ganz PA, et al. Inflammatory biomarkers for persistent fatigue in breast cancer survivors. Clin Cancer Res 2006;12:2759–66.
17. Wang XS, Shi Q, Williams LA, et al. Serum interleukin-6 predicts the development of multiple symptoms at nadir of allogeneic hematopoietic stem cell transplantation. Cancer 2008;113:2102–9.
18. Inagaki M, Isono M, Okuyama T, et al. Plasma interleukin-6 and fatigue in terminally ill cancer patients. J Pain Symptom Manage 2008;35:153–61.
19. Laird BJ, McMillan DC, Fayers P, et al. The systemic inflammatory response and its relationship to pain and other symptoms in advanced cancer. Oncologist 2013;18:1050–5.
20. Bower JE, Ganz PA, Irwin MR, et al. Inflammation and behavioral symptoms after breast cancer treatment: do fatigue, depression, and sleep disturbance share a common underlying mechanism? J Clin Oncol 2011;29:3517–22.
21. Whistler T, Taylor R, Craddock RC, et al. Gene expression correlates of unexplained fatigue. Pharmacogenomics 2006;7:395–405.
22. Fagundes CP, Bennett JM, Alfano CM, et al. Social support and socioeconomic status interact to predict Epstein-Barr virus latency in women awaiting diagnosis or newly diagnosed with breast cancer. Health Psychol 2012;31:11–19.
23. Landmark-Hoyvik H, Reinertsen KV, Loge JH, et al. Alterations of gene expression in blood cells associated with chronic fatigue in breast cancer survivors. Pharmacogenomics J 2009;9:333–40.
24. Smith AK, Conneely KN, Pace TW, et al. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain Behav Immun 2014;38:227–36.
25. Kim S, Miller BJ, Stefanek ME, Miller AH. Inflammation-induced activation of the indoleamine 2,3-dioxygenase pathway: Relevance to cancer-related fatigue. Cancer 2015;121:2129–36.
26. Mills PJ, Parker B, Dimsdale JE, et al. The relationship between fatigue and quality of life and inflammation during anthracycline-based chemotherapy in breast cancer. Biol Psychol 2005;69:85–96.
27. Jean-Pierre P, Figueroa-Moseley CD, Kohli S, et al. Assessment of cancer-related fatigue: implications for clinical diagnosis and treatment. Oncologist 2007;12 Suppl 1:11–21.
28. Wang XS, Zhao F, Fisch MJ, et al. Prevalence and characteristics of moderate to severe fatigue: a multicenter study in cancer patients and survivors. Cancer 2014;120:425–32.
29. Mendoza TR, Wang XS, Cleeland CS, et al. The rapid assessment of fatigue severity in cancer patients: use of the Brief Fatigue Inventory. Cancer 1999;85:1186–96.
30. Mendoza ME, Capafons A, Gralow JR, et al. Randomized controlled trial of the Valencia model of waking hypnosis plus CBT for pain, fatigue, and sleep management in patients with cancer and cancer survivors. Psychooncology 2016 Jul 28.
31. Glaus A. Assessment of fatigue in cancer and non-cancer patients and in healthy individuals. Support Care Cancer 1993;1:305–15.
32. Seyidova-Khoshknabi D, Davis MP, Walsh D. A systematic review of cancer-related fatigue measurement questionnaires. Am J Hosp Palliat Care 2011;28:119–29.
33. Holzner B, Kemmler G, Greil R, et al. The impact of hemoglobin levels on fatigue and quality of life in cancer patients. Ann Oncol 2002;13:965–73.
34. Stein KD, Jacobsen PB, Blanchard CM, Thors C. Further validation of the multidimensional fatigue symptom inventory-short form. J Pain Symptom Manage 2004;27:14–23.
35. Hwang SS, Chang VT, Rue M, Kasimis B. Multidimensional independent predictors of cancer-related fatigue. J Pain Symptom Manage 2003;26:604–14.
36. Peterson DR. Scope and generality of verbally defined personality factors. Psychol Rev 1965;72:48–59.
37. Berger AM, Abernethy AP, Atkinson A, et al. NCCN Clinical Practice Guidelines Cancer-related fatigue. J Natl Compr Canc Netw 2010;8:904–31.
38. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer 2002;95:888–95.
39. Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012;12:CD003407.
40. Rizzo JD, Brouwers M, Hurley P, et al. American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update on the use of epoetin and darbepoetin in adult patients with cancer. Blood 2010;116:4045–59.
41. Preston NJ, Hurlow A, Brine J, Bennett MI. Blood transfusions for anaemia in patients with advanced cancer. Cochrane Database Syst Rev 2012(2):CD009007.
42. Minton O, Stone PC. A comparison of cognitive function, sleep and activity levels in disease-free breast cancer patients with or without cancer-related fatigue syndrome. BMJ Support Palliat Care 2012;2:231–8.
43. Heckler CE, Garland SN, Peoples AR, et al. Cognitive behavioral therapy for insomnia, but not armodafinil, improves fatigue in cancer survivors with insomnia: a randomized placebo-controlled trial. Support Care Cancer 2016;24:2059–66.
44. Howell D, Oliver TK, Keller-Olaman S, et al. Sleep disturbance in adults with cancer: a systematic review of evidence for best practices in assessment and management for clinical practice. Ann Oncol 2014;25:791–800.
45. Wilt TJ, MacDonald R, Brasure M, et al. Pharmacologic treatment of insomnia disorder: an evidence report for a clinical practice guideline by the American College of Physicians. Ann Intern Med 2016;165:103–12.
46. Kuriyama A, Honda M, Hayashino Y. Ramelteon for the treatment of insomnia in adults: a systematic review and meta-analysis. Sleep Med 2014;15:385–92.
47. Strasser F, Palmer JL, Schover LR, et al. The impact of hypogonadism and autonomic dysfunction on fatigue, emotional function, and sexual desire in male patients with advanced cancer: a pilot study. Cancer 2006;107:2949–57.
48. Agasi-Idenburg SC, Thong MS, Punt CJ, et al. Comparison of symptom clusters associated with fatigue in older and younger survivors of colorectal cancer. Support Care Cancer 2017;25:625–32.
49. Miaskowski C, Aouizerat BE. Is there a biological basis for the clustering of symptoms? Semin Oncol Nurs 2007;23:99–105.
50. de Raaf PJ, de Klerk C, Timman R, et al. Systematic monitoring and treatment of physical symptoms to alleviate fatigue in patients with advanced cancer: a randomized controlled trial. J Clin Oncol 2013;31:716–23.
51. Barsevick AM, Whitmer K, Sweeney C, Nail LM. A pilot study examining energy conservation for cancer treatment-related fatigue. Cancer Nurs 2002;25:333–41.
52. Barsevick AM, Dudley W, Beck S, et a;. A randomized clinical trial of energy conservation for patients with cancer-related fatigue. Cancer 2004;100:1302–10.
53. Luciani A, Jacobsen PB, Extermann M, et al. Fatigue and functional dependence in older cancer patients. Am J Clin Oncol 2008;31:424–30.
54. Abrahams HJ, Gielissen MF, Goedendorp MM, et al. A randomized controlled trial of web-based cognitive behavioral therapy for severely fatigued breast cancer survivors (CHANGE-study): study protocol. BMC Cancer 2015;15:765.
55. Quesnel C, Savard J, Simard S, et al. Efficacy of cognitive-behavioral therapy for insomnia in women treated for nonmetastatic breast cancer. J Consult Clin Psychol 2003;71:189–200.
56. Goedendorp MM, Gielissen MF, Verhagen CA, Bleijenberg G. Psychosocial interventions for reducing fatigue during cancer treatment in adults. Cochrane Database Syst Rev 2009(1):CD006953.
57. Greiwe JS, Cheng B, Rubin DC, et al. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J 2001;15:475–82.
58. Furmaniak AC, Menig M, Markes MH. Exercise for women receiving adjuvant therapy for breast cancer. Cochrane Database Syst Rev 2016;(9):CD005001.
59. Cramp F, Byron-Daniel J. Exercise for the management of cancer-related fatigue in adults. Cochrane Database Syst Rev 2012;(11):CD006145.
60. Brown JC, Huedo-Medina TB, Pescatello LS, et al. Efficacy of exercise interventions in modulating cancer-related fatigue among adult cancer survivors: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2011;20:123–33.
61. Steindorf K, Schmidt ME, Klassen O, et al. Randomized, controlled trial of resistance training in breast cancer patients receiving adjuvant radiotherapy: results on cancer-related fatigue and quality of life. Ann Oncol 2014;25:2237–43.
62. Bower JE, Bak K, Berger A, et al. Screening, assessment, and management of fatigue in adult survivors of cancer: an American Society of Clinical oncology clinical practice guideline adaptation. J Clin Oncol 2014;32:1840–50.
63. Kenjale AA, Hornsby WE, Crowgey T, et al. Pre-exercise participation cardiovascular screening in a heterogeneous cohort of adult cancer patients. Oncologist 2014;19:999–1005.
64. Oldervoll LM, Loge JH, Paltiel H, et al. The effect of a physical exercise program in palliative care: A phase II study. J Pain Symptom Manage 2006;31:421–30.
65. Porock D, Kristjanson LJ, Tinnelly K, et al. An exercise intervention for advanced cancer patients experiencing fatigue: a pilot study. J Palliat Care 2000;16:30–6.
66. Lengacher CA, Kip KE, Reich RR, et al. A cost-effective mindfulness stress reduction program: a randomized control trial for breast cancer survivors. Nursing Econ 2015;33:210–8, 32.
67. Lengacher CA, Reich RR, Post-White J, et al. Mindfulness based stress reduction in post-treatment breast cancer patients: an examination of symptoms and symptom clusters. J Behav Med 2012;35:86–94.
68. Sprod LK, Fernandez ID, Janelsins MC, et al. Effects of yoga on cancer-related fatigue and global side-effect burden in older cancer survivors. J Geriatr Oncol 2015;6:8–14.
69. Wang G, Wang S, Jiang P, Zeng C. [Effect of Yoga on cancer related fatigue in breast cancer patients with chemotherapy]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2014;39:1077–82.
70. Stan DL, Croghan KA, Croghan IT, et al. Randomized pilot trial of yoga versus strengthening exercises in breast cancer survivors with cancer-related fatigue. Support Care Cancer 2016;24:4005–15.
71. Larkey LK, Roe DJ, Weihs KL, et al. Randomized controlled trial of Qigong/Tai Chi Easy on cancer-related fatigue in breast cancer survivors. Ann Behav Med 2015;49:165–76.
72. Molassiotis A, Bardy J, Finnegan-John J, et al. Acupuncture for cancer-related fatigue in patients with breast cancer: a pragmatic randomized controlled trial. J Clin Oncol 2012;30:4470–6.
73. Deng G, Chan Y, Sjoberg D, et al. Acupuncture for the treatment of post-chemotherapy chronic fatigue: a randomized, blinded, sham-controlled trial. Support Care Cancer 2013;21:1735–41.
74. Finnegan-John J, Molassiotis A, Richardson A, Ream E. A systematic review of complementary and alternative medicine interventions for the management of cancer-related fatigue. Integr Cancer Ther 2013;12:276–90.
75. Schwartz AL, Thompson JA, Masood N. Interferon-induced fatigue in patients with melanoma: a pilot study of exercise and methylphenidate. Oncol Nurs Forum 2002;29:E85–90.
76. Spathis A, Dhillan R, Booden D, et al. Modafinil for the treatment of fatigue in lung cancer: a pilot study. Palliat Med 2009;23:325–31.
77. Blackhall L, Petroni G, Shu J, et al. A pilot study evaluating the safety and efficacy of modafinal for cancer-related fatigue. J Palliat Med 2009;12:433–9.
78. Qu D, Zhang Z, Yu X, et al. Psychotropic drugs for the management of cancer-related fatigue: a systematic review and meta-analysis. Eur J Cancer Care (Engl) 2015;25:970–9.
79. Minton O, Richardson A, Sharpe M, et al. Drug therapy for the management of cancer-related fatigue. Cochrane Database Syst Rev 2010(7):CD006704.
80. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol 2010;28:3673–9.
81. Bruera E, Driver L, Barnes EA, et al. Patient-controlled methylphenidate for the management of fatigue in patients with advanced cancer: a preliminary report. J Clin Oncol 2003;21:4439–43.
82. Kerr CW, Drake J, Milch RA, et al. Effects of methylphenidate on fatigue and depression: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage 2012;43:68–77.
83. Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J 2014;20:8–14.
84. Mar Fan HG, Clemons M, Xu W, et al. A randomised, placebo-controlled, double-blind trial of the effects of d-methylphenidate on fatigue and cognitive dysfunction in women undergoing adjuvant chemotherapy for breast cancer. Support Care Cancer 2008;16:577–83.
85. Butler JM Jr, Case LD, Atkins J, et al. A phase III, double-blind, placebo-controlled prospective randomized clinical trial of d-threo-methylphenidate HCl in brain tumor patients receiving radiation therapy. Int J Radiat Oncol Biol Phys 2007;69:1496–501.
86. Hovey E, de Souza P, Marx G, et al. Phase III, randomized, double-blind, placebo-controlled study of modafinil for fatigue in patients treated with docetaxel-based chemotherapy. Support Care Cancer 2014;22:1233–42.
87. Spathis A, Fife K, Blackhall F, et al. Modafinil for the treatment of fatigue in lung cancer: results of a placebo-controlled, double-blind, randomized trial. J Clin Oncol 2014;32:1882–8.
88. Berenson JR, Yellin O, Shamasunder HK, et al. A phase 3 trial of armodafinil for the treatment of cancer-related fatigue for patients with multiple myeloma. Support Care Cancer 2015; 23:1503–12.
89. Boele FW, Douw L, de Groot M, et al. The effect of modafinil on fatigue, cognitive functioning, and mood in primary brain tumor patients: a multicenter randomized controlled trial. Neuro Oncol 2013;15:1420–8.
90. Jean-Pierre P, Morrow GR, Roscoe JA, et al. A phase 3 randomized, placebo-controlled, double-blind, clinical trial of the effect of modafinil on cancer-related fatigue among 631 patients receiving chemotherapy: a University of Rochester Cancer Center Community Clinical Oncology Program Research base study. Cancer 2010;116:3513–20.
91. Conley CC, Kamen CS, Heckler CE, et al. Modafinil moderates the relationship between cancer-related fatigue and depression in 541 patients receiving chemotherapy. J Clin Psychopharmacol 2016;36:82–5.
92. Brattsand R, Linden M. Cytokine modulation by glucocorticoids: mechanisms and actions in cellular studies. Aliment Pharmacol Ther 1996;10 Suppl 2:81–90.
93. Yennurajalingam S, Frisbee-Hume S, Palmer JL, et al. Reduction of cancer-related fatigue with dexamethasone: a double-blind, randomized, placebo-controlled trial in patients with advanced cancer. J Clin Oncol 2013;31:3076–82.
94. Bruera E, Roca E, Cedaro L, et al. Action of oral methylprednisolone in terminal cancer patients: a prospective randomized double-blind study. Cancer Treat Rep 1985;69:751–4.
95. Pulivarthi K, Dev R, Garcia J, et al. Testosterone replacement for fatigue in male hypogonadic patients with advanced cancer: A preliminary double-blind placebo-controlled trial. J Clin Oncol 2012;30 (suppl). Abstract e19643.
96. Palesh OG, Mustian KM, Peppone LJ, et al. Impact of paroxetine on sleep problems in 426 cancer patients receiving chemotherapy: a trial from the University of Rochester Cancer Center Community Clinical Oncology Program. Sleep Med 2012;13:1184–90.
97. Thekdi SM, Trinidad A, Roth A. Psychopharmacology in Cancer. Curr Psychiatry Rep 2014;17:529.
98. Lesser GJ. Case D, Stark N, et al. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol 2013;11:31–42.
99. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst 2013;105:1230–8.
100. Yennurajalingam S, Reddy A, Tannir NM, et al. High-dose Asian ginseng (panax ginseng) for cancer-related fatigue: a preliminary report. Integr Cancer Ther 2015;14:419–27.
101. Howell D, Keller-Olaman S, Oliver TK, et al. A pan-Canadian practice guideline and algorithm: screening, assessment, and supportive care of adults with cancer-related fatigue. Curr Oncol 2013;20:e233–46.
INTRODUCTION
Fatigue is a common distressing effect of cancer.1 It impairs the quality of life of patients undergoing active cancer treatment and of post-treatment survivors alike. The National Comprehensive Cancer Network (NCCN) defines cancer-related fatigue (CRF) as “a distressing, persistent, subjective sense of physical, emotional and/or cognitive tiredness related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning.”2 CRF differs from fatigue reported by individuals without cancer in that CRF is more severe and is not relieved by rest. The prevalence of CRF in cancer patients and survivors is highly variable, with estimates ranging between 25% and 99%.2,3 The methods used for screening patients for fatigue and the characteristics of the patient groups may account for this variability.
PATHOPHYSIOLOGY
The specific pathophysiologic mechanism underlying CRF is unknown, making targeted treatment a challenge. The multidimensional and subjective nature of CRF has limited the development of research methodologies to explain this condition. However, research has been done in both human and animal models, and several theories have been proposed to explain the pathophysiology of CRF. While pro-inflammatory cytokines remain the central factor playing a significant role at multiple levels in CRF, there may be a complex interplay of multiple mechanisms contributing to fatigue in an individual patient.
CENTRAL NERVOUS SYSTEM DISTURBANCES
The basal ganglia are known to influence motivation. Lack of motivation and drive may cause failure to complete physical and mental tasks, even with preserved cognitive ability and motor function. In a study of melanoma patients receiving interferon, increased activity of the basal ganglia and the cerebellum resulted in higher fatigue scores.4 Increased levels of cytokines may alter blood flow to the cerebellum and lead to the perception of fatigue. In a study of 12 patients and matched controls, when patients were asked to perform sustained elbow flexion until they perceived exhaustion, CRF patients perceived physical exhaustion sooner than controls. In CRF patients in this study, muscle fatigue measured by electromyogram was less than that in healthy individuals at the time of exhaustion, suggesting the role of the central nervous system in CRF.5 However, there is not enough evidence at this time to support central nervous system disturbance as the main factor contributing to fatigue in cancer patients.
CIRCADIAN RHYTHM DYSREGULATION
Circadian rhythm is regulated by the suprachiasmatic nucleus in the hypothalamus through cortisol and melatonin. Sleep disturbances occur with disruption of the circadian rhythm. Tumor-related peptides such as epidermal growth factor or alterations in serotonin and cortisol can influence the suprachiasmatic nucleus and the complex signaling pathways.2 Positive feedback loops that are activated by cortisol under the influence of cytokines may lead to continuous cytokine production and altered circadian rhythm. Bower et al showed that changes in the cortisol curve influence fatigue in breast cancer survivors.6 These patients had a late evening peak in cortisol levels, compared with an early morning peak in individuals without cancer.
INHIBITION OF HYPOTHALAMIC-PITUITARY-ADRENAL AXIS
The hypothalamic–pituitary–adrenal (HPA) axis regulates the release of the stress hormone cortisol. One of several hypotheses advanced to explain the effect of serotonin and the HPA axis on CRF suggests that lower serotonin levels cause decreased activation of 5-hydroxytrytophan 1-a (5-HT1-a) receptors in the hypothalamus, leading to decreased activity of the HPA axis.6 Inhibition of the HPA axis may occur with higher levels of serotonin as well.7 The 5-HT1-a receptors are also triggered by cytokines. However, the correction of serotonin levels by antidepressants was not shown to improve fatigue.8 Inhibition of the HPA axis can also lead to lower testosterone, progesterone, or estrogen levels, which may indirectly contribute to fatigue.2
SKELETAL MUSCLE EFFECT
Chemotherapy- and tumor-related cachexia have a direct effect on the metabolism of skeletal muscles. This effect may lead to impaired adenosine triphosphate (ATP) generation during muscle contraction.9 ATP infusion improved muscle strength in 1 trial, but this was not confirmed in another trial.10,11 Muscle contraction studies showed no differences in the contractile properties of muscles in fatigued patients who failed earlier in motor tasks and healthy controls.12 This finding suggests that there could be a failure of skeletal muscle activation by the central nervous system or inhibition of skeletal muscle activity. Cytokines and other neurotransmitters activate vagal efferent nerve fibers, which may lead to reflex inhibition in skeletal muscles.13,14
PRO-INFLAMMATORY CYTOKINES
Tumors or treatment of them may cause tissue injury, which triggers immune cells to release cytokines, signaling the brain to manifest the symptom of fatigue. Inflammatory pathways are influenced by psychological, behavioral, and biological factors, which play a role as risk factors in CRF. Levels of interleukin 6 (IL-6), interleukin-1 receptor antagonist, interleukin-1, and tumor necrosis factor (TNF) have been shown to be elevated in fatigued patients being treated for leukemia and non-Hodgkin lymphoma.15 IL-6 was also associated with increased fatigue in breast cancer survivors.16 Similar findings were reported in patients undergoing stem cell transplantation and high-dose chemotherapy.17 Elevated levels of IL-6 and C-reactive protein were also linked to fatigue in terminally ill cancer patients.18,19 Furthermore, TNF-α signaling was associated with post-chemotherapy fatigue in breast cancer patients.20 Leukocytes in breast cancer survivors with fatigue also have increased gene expression of pro-inflammatory cytokines, emphasizing the role of cytokines and inflammation in the pathogenesis of CRF.21
OTHER HYPOTHESES
Several other hypotheses for CRF pathogenesis have been proposed. Activation of latent viruses such as Epstein-Barr virus, lack of social support,22 genetic alterations in the immune pathway,23 epigenetic changes,24 accumulation of neurotoxic metabolites and depletion of serotonin by indoleamine 2,3-dioxygenase pathway activation,25 elevated vascular endothelial growth factor levels,26 and hypoxia-related organ dysfunction due to anemia or hemoglobin dysfunction13 all have been postulated to cause CRF.
EVALUATION AND TREATMENT
Fours steps are involved in the evaluation and treatment of CRF (Figure).
SCREENING
Because patients and health care professionals may be unaware of the treatment options available for CRF, patients may not report fatigue levels to their clinicians, and clinicians may not understand the impact of fatigue on their patients’ quality of life. This leads to under-recognition of the problem. The NCCN recommends screening every cancer patient and post-treatment survivor for fatigue.2 Patients should be screened at their first visit and then at periodic intervals during and after cancer treatment.
Many scales are available to screen patients for CRF in clinical practice and clinical trials.27 A single item that asks patients to rate their fatigue on a scale from 0 to 10—in which 0 indicates no fatigue, 1 to 3 indicates mild fatigue, 4 to 6 indicates moderate fatigue, 7 to 9 indicates severe fatigue, and 10 indicates the worst fatigue imaginable—is commonly used to screen for CRF.2 This scale was adapted from the MD Anderson Symptom Inventory scale and is based on a large nationwide study of cancer patients and survivors.28 The statistically derived cutoff points in this study are consistent with other scales such as the Brief Fatigue Inventory (BFI) and support the cutoff points (4–6 for moderate and ≥ 7 for severe fatigue) used in various fatigue management guidelines. Furthermore, studies of fatigue in cancer patients have revealed a marked decrease in physical function at levels of 7 or higher, suggesting 7 as an optimal cutoff to identify severe fatigue.29,30 The Visual Analog Scale is another simple-to-use tool that helps in understanding variations in fatigue throughout the course of the day.31 The 9-item BFI is often used in clinical trials.29 It measures the severity of fatigue over the previous 24 hours and has been validated in patients who do not speak English.32
CRF affects not only the somatic domain, but also the cognitive, behavioral, and affective domains; therefore, multidimensional scales have been developed for screening. One such tool is the Multidimensional Fatigue Inventory, which assesses 5 dimensions of fatigue—general fatigue, physical fatigue, reduced motivation, reduced activity, and mental fatigue—and compares the patient’s results with those of individuals without cancer.33,34 The Functional Assessment of Cancer Therapy for Fatigue (FACT-F) is a 13-item questionnaire that has been used to measure CRF in clinical trials as well as in patients receiving various treatments.35
Although many scales are available, the validity of self-reporting on simple fatigue-rating scales is equal to or better than most complex, lengthy scales.36 Therefore, unidimensional tools such as the numeric rating scale of 0–10 are commonly used in clinical practice. Mild fatigue (0–3) requires periodic reevaluation, and moderate and severe fatigue need further evaluation and management.37
PRIMARY EVALUATION
This phase involves a focused history and physical examination and assessment of concurrent symptoms and contributing factors.
History and Physical Examination
A detailed history of the patient’s malignancy and type of previous and current treatment may help reveal the cause of fatigue. New-onset fatigue or increase in fatigue may be related to the progression of disease in patients with active malignancy or recurrence of cancer in survivors. These patients may require appropriate testing to assess the underlying disease pattern. A detailed review of systems may help identify some of the contributing factors, which are discussed below. A detailed history regarding medications, including over-the-counter drugs, complementary agents, and past and prior cancer therapies, is helpful as medications can contribute to fatigue. For example, opioids may cause drowsiness and fatigue, which could be improved by dose adjustments. A focused history of fatigue should be obtained in all patients with moderate to severe CRF, which includes the onset, pattern, duration, associated or alleviating factors, and interference with functioning, including activities of daily living.37 Physical examination should focus on identifying signs of organ dysfunction and features of substance or alcohol abuse, which may cause poor sleep and fatigue.
Assessment of Contributing Factors
The management of fatigue should be multifactorial, with a comprehensive assessment and treatment plan to address all modifiable fatigue etiologies. The Table lists potential contributing factors to fatigue that should be considered when evaluating patients for CRF; several common conditions are discussed below.
Anemia. Anemia has been correlated with fatigue and quality of life. In a study of 4382 cancer patients receiving chemotherapy, quality-of-life measures using FACT-Anemia scores improved with increased hemoglobin levels.38 Cancer patients may have anemia due to marrow-suppressing effects of chemotherapy and may also have iron deficiency anemia due to blood loss or auto-immune hemolytic anemia. Therefore, a detailed work-up is required to identify the etiology of anemia. Patients with CRF whose anemia is related to chemotherapy or anemia of chronic disease may benefit from red blood cell transfusion or erythropoiesis-stimulating agents (ESAs). ESAs have been studied extensively; however, their use is controversial because of concerns about thromboembolic side effects leading to adverse outcomes.39 Also, ESA therapy is not recommended in patients with hematologic malignancies. ESA use should be restricted to patients with chemotherapy-related anemia with hemoglobin below 10 mg/dL and should be discontinued in 6 to 8 weeks if patients do not respond.40 Other patients may benefit from blood transfusions, which were shown to help in patients with hemoglobin levels between 7.5 and 8.5 g/dL.41
Sleep disturbance. Poor sleep is common in fatigued cancer survivors.42 Pro-inflammatory cytokines can disrupt the sleep–wake cycle, causing changes in the HPA axis and neuroendocrine system, which in turn may lead to increasing fatigue. Heckler et al showed that improvement in nighttime sleep leads to improvement of fatigue.43 Cognitive behavioral therapy and sleep hygiene are important in caring for patients with CRF and poor sleep.44 Taking a warm bath and/or drinking a glass of milk before bedtime, avoiding caffeinated drinks, and avoiding frequent napping in the day might help. Some patients may require medications such as benzodiazepines or non-benzodiazepine hypnotics (eg, zolpidem) to promote sleep.45 Melatonin agonists are approved for insomnia in the United States, but not in Europe.46
Malnutrition. Patients with advanced-stage cancer and with cancers affecting the gastrointestinal tract frequently develop mechanical bowel obstructions, especially at the end of their life, which cause malnutrition. Chemotherapy-related nausea and vomiting may also cause poor oral intake and malnutrition, causing fatigue from muscle weakness. Dehydration and electrolyte imbalances frequently occur as a result of poor oral intake, which might worsen fatigue. In our experience, modifying dietary intake with appropriate caloric exchanges with the help of a nutrition expert has lessened fatigue in some patients. However, terminally ill patients are advised to eat based on their comfort.
Medical comorbidities. Congestive heart failure from anthracycline chemotherapy, hypothyroidism after radiation therapy for head and neck cancers, renal failure, or hepatic failure from chemotherapy may indirectly lead to fatigue. Chemotherapy, opioids, and steroids may cause hypogonadism, which can contribute to fatigue in men.47
Assessment of Concurrent Symptoms
Depression is common in cancer patients and coexists with pain, insomnia, fatigue, and anxiety as a symptom cluster.48 A symptom cluster is defined as 2 or more concurrent and interrelated symptoms occurring together; treating one of these symptoms without addressing other symptoms is not effective.49 Therefore, screening for and management of depression, anxiety, and insomnia play an important role in the management of CRF.
Physical symptoms due to the tumor or to therapy— such as pain, dyspnea, nausea, and decreased physical activity—may also contribute to fatigue both directly and indirectly. Patients with lung cancer may have hypoxemia, which can contribute to dyspnea with activity and a perception of fatigue. Optimal management of pain and other physical symptoms in patients with cancer may significantly alleviate fatigue.50
MANAGEMENT
Management of CRF is individualized based on the patient’s clinical status: active cancer treatment, survivor, or end of life. Education and counselling of patients and their caregivers play an important role in CRF. NCCN guidelines recommend focusing on pain control, distress management, energy conservation, physical activity, nutrition, and sleep hygiene.
Nonpharmacologic Interventions
Energy conservation. Energy conservation strategies, in which patients are advised to set priorities and realistic expectations, are highly recommended. Some energy-conserving strategies are to pace oneself, delegate and schedule activities at times of peak energy, postpone nonessential activities, attend to 1 activity at a time, structure daily routines, and maintain a diary to identify their peak energy period and structure activities around that time.51,52 When patients approach the end of life, increasing fatigue may limit their activity level, and they may depend on caregivers for assistance with activities of daily living, monitoring treatment-related adverse effects, and taking medications, especially elderly patients.53
Cognitive behavioral therapy. Cognitive behavioral therapy (CBT) has been shown to improve CRF during active treatment, and the benefits persist for a minimum of 2 years after therapy.54 CBT interventions that optimize sleep quality may improve fatigue.55 More studies are needed to understand whether referral to a psychologist for formal CBT is required. Randomized clinical trials showed patient fatigue education, learned self-care, coping techniques, and balancing rest and activity benefit patients with CRF.56
Exercise. Physical activity is highly encouraged in patients with CRF. Exercise increases muscle protein synthesis, improves cytokine response, and decreases the rate of sarcopenia in healthy populations.57 Studies have shown that exercise helps CRF at all phases of the cancer journey, including radiation therapy, chemotherapy, and survivorship.58 Some patients may feel less motivated to exercise and may not believe that exercise is possible or could potentially help them. Counselling is needed for such patients.
Older cancer survivors have a decline in their functional capacity and reduced muscle mass. Exercise can improve their cardiorespiratory fitness, muscle strength, and body composition.57 Exercise not only helps at the cellular level but also has psychosocial benefits from improved self-esteem. Therefore, exercise may be recommended for younger patients as well as for the older population, who may have comorbidities and less motivation than younger patients.
In a meta-analysis of 56 randomized controlled trials involving 4068 participants, aerobic exercise was found to have beneficial effects on CRF for patients during and after chemotherapy, specifically for patients with solid tumors.59 In another meta-analysis of breast and prostate cancer survivors, a combination of aerobic exercise with resistance training (3–6 metabolic equivalents, 60%–80% range of motion) was shown to reduce CRF more than aerobic exercise alone.60 This effect was also shown in a randomized controlled trial of 160 patients with stage 0 to III breast cancer undergoing radiation therapy.61 The control group in this study had a group-based non-exercise intervention/relaxation; therefore, the study showed that the effect of resistance training extends beyond the psychosocial benefits of group-based interventions. The intervention comprised 8 progressive machine-based resistance exercises (3 sets, 8–12 repetitions at 60%–80% of 1 repetition maximum) for 60 minutes twice weekly for 12 weeks. However, fatigue assessment questionnaire scores showed benefits only in the physical fatigue components, but not in the affective and cognitive components.
The American Society of Clinical Oncology’s guidelines for cancer survivors with fatigue recommends 150 minutes of moderate aerobic exercise (eg, fast walking, cycling, or swimming) per week, with 2 or 3 sessions of strength training per week.62 An individualized approach to exercise is recommended, as patients’ ability to perform certain types of exercises may be limited by thrombocytopenia, neutropenia, or lytic bone metastasis. Routine use of pre-exercise cardiovascular testing is not recommended but may be considered in high-risk populations, especially patients with risk factors for coronary heart disease and diabetes.63 Patients with comorbidities, substantial deconditioning, functional and anatomic defects, or recent major surgery may benefit from referral to physical therapy.37 Patients near end of life may also benefit from an exercise program, as demonstrated in several studies that showed benefit in CRF and quality of life.64,65 We recommend that physicians use their best clinical judgement in suggesting the type and intensity of exercise program, as it may not be feasible in some patients.
Mind-body interventions. Mindfulness-based stress reduction (MBSR) has shown promise in breast cancer survivors, who reported immediate improvements in fatigue severity that continued up to 6 weeks after cessation of the training.66 Prior studies had similar findings, suggesting that MBSR modestly decreases fatigue and sleep disturbances and has a greater effect on the degree to which symptoms interfere with many facets of life.67
Yoga. A study of a yoga intervention showed a benefit in older cancer survivors.68 In breast cancer patients undergoing chemotherapy, yoga was shown to benefit both physical and cognitive fatigue.69 DVD-based yoga had benefits similar to strengthening exercises in a study of 34 early-stage breast cancer survivors with CRF.70 More studies are needed in men and patients and survivors of other cancers, as most studies of yoga were conducted in women with breast cancer.
Tai chi/qigong. Like yoga, tai chi and qigong are practices of meditative movement. These practices use postures or movements with a focus on breath and a meditative state to bring about deep states of relaxation. Qigong is a series of simple, repeated practices including body posture/movement, breath practice, and meditation performed in synchrony. Tai chi easy (TCE) is a simplified set of common, repetitive tai chi movements. In a trial, qigong/TCE was compared with sham qigong, which had physical movements but no breathing or meditative practice. Breast cancer survivors in the qigong/TCE group had improved fatigue scores, and the effect persisted for 3 months.71 Additional research is needed in this area.
Acupuncture. A randomized controlled trial in breast cancer patients with CRF showed an improvement in the mean general fatigue score (per the Multidimensional Fatigue Inventory) in patients who received acupuncture versus those who did not (−3.11 [95% confidence interval −3.97 to −2.25]; P < 0.001) at 6 weeks. Improvements were seen in both the mental and physical aspects of fatigue.72 However, Deng et al noted that true acupuncture was no more effective than sham acupuncture for reducing post-chemotherapy chronic fatigue.73 Presently, there is not sufficient evidence to evaluate the benefits of acupuncture in CRF.
Other modalities. Massage therapy, music therapy, hypnosis, therapeutic touch, biofield therapies, relaxation, and reiki are other therapies for which few studies have been done; of the studies that have been done, the results are mixed, and additional research is needed.74 Currently, there are not sufficient data to recommend any of these modalities.
Pharmacologic Interventions
Psychostimulants. Methylphenidate and modafinil are psychostimulants or wakefulness-promoting agents. Pilot studies showed benefit from methylphenidate and modafinil in CRF,75–77 but randomized controlled trials have yielded mixed results. Therefore, in patients with severe fatigue during cancer therapy, the initial management strategy involves evaluation and treatment of medical conditions such as anemia and a trial of nonpharmacological strategies as discussed above. If symptoms persist, then a therapeutic trial of a psychostimulant may be considered per NCCN guidelines for patients undergoing active cancer treatment.37
Methylphenidate directly stimulates adrenergic receptors and indirectly releases dopamine and norepinephrine from presynaptic terminals, which may explain why the drug benefits patients receiving opioid-induced sedation. It is a commonly studied psychostimulant, though its mechanism of action in CRF is unclear. Randomized controlled trials of methylphenidate have resulted in a wide range of findings due to the heterogeneity of study populations and variations in the dosage of methylphenidate. A meta-analysis of 7 studies indicates that methylphenidate benefitted the subgroup of patients with CRF.78 Likewise, in an analysis of 5 randomized controlled trials, Minton et al showed a benefit of psychostimulants in fatigue compared with placebo.79 However, another study of methylphenidate in patients with CRF showed a benefit only in patients with severe fatigue or advanced disease.80 Methylphenidate was found to benefit cancer patients receiving opioid-induced sedation, as methylphenidate promotes wakefulness, though fatigue was not studied specifically.81 In a trial with 30 hospice patients in which the methylphenidate dose was titrated based on response and adverse effects, Kerr at al found that the drug improved fatigue in a dose-dependent manner.82 However, a study in patients with CRF at the University of Texas MD Anderson Cancer Center found no significant difference in BFI scores between patients receiving methylphenidate and those receiving placebo at the end of 2 weeks of treatment.83 Also, other randomized controlled trials in patients undergoing adjuvant chemotherapy for breast cancer84 and patients receiving radiation therapy for brain tumors85 failed to demonstrate the efficacy of methylphenidate in CRF. It should be used cautiously after ruling out other causes of fatigue. The drug is overall well tolerated and side effects include headache and nausea.
Modafinil is a non-amphetamine psychostimulant that has been approved for the treatment of narcolepsy. In a trial studying the effect of modafinil on patients receiving docetaxel-based chemotherapy for metastatic breast or prostate cancer, there was a modest but not statistically significant improvement in fatigue scores on the MD Anderson Symptom Inventory compared with placebo. Nausea and vomiting were higher in the modafinil arm than in the placebo arm.86 Similarly, modafinil was not superior to placebo for CRF in 208 patients with non-squamous cell lung cancer not undergoing chemotherapy or radiation.87 A placebo effect was also noted in patients with multiple myeloma88 and patients with primary brain tumors.89 In a phase 3, multicenter, randomized, placebo-controlled, double-blind clinical trial of modafinil for CRF in 867 patients undergoing chemotherapy, there was a reduction in fatigue only for patients with severe baseline fatigue, with no significant effect on mild to moderate fatigue.90 In another recent study, modafinil was shown to reduce depressive symptoms only in patients with severe fatigue (BFI item 3 score ≥ 7).91 This finding is consistent with previous studies showing benefit in patients with high baseline fatigue, but additional randomized controlled trials are needed to provide clarity. NCCN guidelines do not recommend the use of modafinil to treat CRF.37
Other pharmacologic interventions. Corticosteroids are often used for symptom control in cancer patients. These drugs have anti-inflammatory effects through their modulation of pro-inflammatory cytokines.92 In a randomized controlled trial evaluating the efficacy of corticosteroids, patients receiving dexamethasone (4 mg twice daily) experienced significant improvement in their FACT-F scores compared with patients receiving placebo.93 A similar benefit in fatigue was demonstrated in another study of methylprednisolone (32 mg daily) versus placebo.94 Despite the benefits of steroids, their adverse effects, such as mood swings, gastritis, hyperglycemia, and immune suppression, limit their long-term use. Therefore, the use of steroids should be restricted to terminally ill fatigued patients with other symptoms such as anorexia, brain metastasis, or pain related to bone metastasis.37
Testosterone replacement has been shown to diminish fatigue in non-cancer patients. Many men with advanced cancer have hypogonadism leading to low serum testosterone, which may cause fatigue. In a small trial in which cancer patients with hypogonadism received intramuscular testosterone every 14 days or placebo, the group receiving testosterone showed improvement in FACT-F scores, but there was no significant difference in FACT-F scores between the 2 groups.95
Antidepressants have failed to demonstrate benefit in CRF without depression.8 However, if a patient has both fatigue and depression, antidepressants may help.96 A selective serotonin receptor inhibitor is recommended as a first-line antidepressant.97 Patients with cancer are often receiving multiple medications, and medication interactions should be considered to prevent adverse events such as serotonin syndrome.
Complementary and Alternative Supplements
Studies using vitamin supplementation have been inconclusive in patients with CRF.74 The use of other dietary supplements has yielded mixed results, and coenzyme Q has shown no benefit for patients with CRF.98
The benefit of ginseng was studied in a RCT involving 364 patients with CRF. There was an improvement in Multidimensional Fatigue Symptom Inventory-short form (MFSI-SF) scores at 8 weeks in patients receiving 2000 mg of Wisconsin ginseng compared with patients receiving placebo.99 Patients on active treatment had greater improvement as compared to the post-treatment group in this trial. In another study of high-dose panax ginseng (ginseng root) at 800 mg daily for 29 days, patients had improvement of CRF as well as overall quality of life, appetite, and sleep at night. It was also well tolerated with few adverse effects.100 Interaction with warfarin, calcium channel blockers, antiplatelet agents, thrombolytic agents, imatinib, and other agents may occur; therefore, ginseng must be used with careful monitoring in selected patients. There is not enough evidence at this time to support the routine use of ginseng in CRF.
The seed extract of the guarana plant (Paullinia cupana) traditionally has been used as a stimulant. An improvement in fatigue scores was seen with the use of oral guarana (100 mg daily) at the end of 21 days in breast cancer patients receiving chemotherapy.101 Further studies are needed for these results to be generalized and to understand the adverse effects and interaction profile of guarana.
Reevaluation
Patients who have completed cancer treatment must be monitored for fatigue over the long term, as fatigue may exist beyond the period of active treatment. Many studies have shown fatigue in breast cancer survivors, and fatigue has been demonstrated in survivors of colorectal, lung, and prostate cancers as well as myeloproliferative neoplasms.28 Therefore, it is important to screen patients for fatigue during follow-up visits. There are currently no studies evaluating the long-term treatment of fatigue. In our experience, the timing of follow-up depends on the level of fatigue and interventions prescribed. Once fatigue is stabilized to a level with which the patient is able to cope, the time interval for follow-up may be lengthened. Annual visits may suffice in patients with mild fatigue. Follow-up of patients with moderate to severe fatigue depends on the level of fatigue, the ability to cope, choice of treatment, and presence of contributing factors.
CONCLUSION
CRF is a complex condition that places a significant burden on patients and caregivers, resulting in emotional distress, poor functioning, and suffering. Fatigue can occur before, during, and long after cancer treatment. The approach to CRF begins with screening for and educating patients and their caregivers about the symptoms. Many screening scales are available that may be used to follow patients’ progress over time. The evaluation and management of contributing conditions may help improve fatigue. If the fatigue persists, an individualized approach with a combination of nonpharmacologic and pharmacologic approaches should be considered. More research is needed to understand brain signaling pathways, cytokine changes, and genomic changes in cancer patients with fatigue. Though many hypotheses have been proposed, to date there is no biological marker to assess this condition. Biomarker research needs to be advanced to help to identify patients at risk for fatigue. As cytokines have a major role in CRF, targeted therapy to block cytokine pathways may also be explored in the future.
Acknowledgment: The authors thank Bryan Tutt for providing editorial assistance during the writing of this article.
INTRODUCTION
Fatigue is a common distressing effect of cancer.1 It impairs the quality of life of patients undergoing active cancer treatment and of post-treatment survivors alike. The National Comprehensive Cancer Network (NCCN) defines cancer-related fatigue (CRF) as “a distressing, persistent, subjective sense of physical, emotional and/or cognitive tiredness related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning.”2 CRF differs from fatigue reported by individuals without cancer in that CRF is more severe and is not relieved by rest. The prevalence of CRF in cancer patients and survivors is highly variable, with estimates ranging between 25% and 99%.2,3 The methods used for screening patients for fatigue and the characteristics of the patient groups may account for this variability.
PATHOPHYSIOLOGY
The specific pathophysiologic mechanism underlying CRF is unknown, making targeted treatment a challenge. The multidimensional and subjective nature of CRF has limited the development of research methodologies to explain this condition. However, research has been done in both human and animal models, and several theories have been proposed to explain the pathophysiology of CRF. While pro-inflammatory cytokines remain the central factor playing a significant role at multiple levels in CRF, there may be a complex interplay of multiple mechanisms contributing to fatigue in an individual patient.
CENTRAL NERVOUS SYSTEM DISTURBANCES
The basal ganglia are known to influence motivation. Lack of motivation and drive may cause failure to complete physical and mental tasks, even with preserved cognitive ability and motor function. In a study of melanoma patients receiving interferon, increased activity of the basal ganglia and the cerebellum resulted in higher fatigue scores.4 Increased levels of cytokines may alter blood flow to the cerebellum and lead to the perception of fatigue. In a study of 12 patients and matched controls, when patients were asked to perform sustained elbow flexion until they perceived exhaustion, CRF patients perceived physical exhaustion sooner than controls. In CRF patients in this study, muscle fatigue measured by electromyogram was less than that in healthy individuals at the time of exhaustion, suggesting the role of the central nervous system in CRF.5 However, there is not enough evidence at this time to support central nervous system disturbance as the main factor contributing to fatigue in cancer patients.
CIRCADIAN RHYTHM DYSREGULATION
Circadian rhythm is regulated by the suprachiasmatic nucleus in the hypothalamus through cortisol and melatonin. Sleep disturbances occur with disruption of the circadian rhythm. Tumor-related peptides such as epidermal growth factor or alterations in serotonin and cortisol can influence the suprachiasmatic nucleus and the complex signaling pathways.2 Positive feedback loops that are activated by cortisol under the influence of cytokines may lead to continuous cytokine production and altered circadian rhythm. Bower et al showed that changes in the cortisol curve influence fatigue in breast cancer survivors.6 These patients had a late evening peak in cortisol levels, compared with an early morning peak in individuals without cancer.
INHIBITION OF HYPOTHALAMIC-PITUITARY-ADRENAL AXIS
The hypothalamic–pituitary–adrenal (HPA) axis regulates the release of the stress hormone cortisol. One of several hypotheses advanced to explain the effect of serotonin and the HPA axis on CRF suggests that lower serotonin levels cause decreased activation of 5-hydroxytrytophan 1-a (5-HT1-a) receptors in the hypothalamus, leading to decreased activity of the HPA axis.6 Inhibition of the HPA axis may occur with higher levels of serotonin as well.7 The 5-HT1-a receptors are also triggered by cytokines. However, the correction of serotonin levels by antidepressants was not shown to improve fatigue.8 Inhibition of the HPA axis can also lead to lower testosterone, progesterone, or estrogen levels, which may indirectly contribute to fatigue.2
SKELETAL MUSCLE EFFECT
Chemotherapy- and tumor-related cachexia have a direct effect on the metabolism of skeletal muscles. This effect may lead to impaired adenosine triphosphate (ATP) generation during muscle contraction.9 ATP infusion improved muscle strength in 1 trial, but this was not confirmed in another trial.10,11 Muscle contraction studies showed no differences in the contractile properties of muscles in fatigued patients who failed earlier in motor tasks and healthy controls.12 This finding suggests that there could be a failure of skeletal muscle activation by the central nervous system or inhibition of skeletal muscle activity. Cytokines and other neurotransmitters activate vagal efferent nerve fibers, which may lead to reflex inhibition in skeletal muscles.13,14
PRO-INFLAMMATORY CYTOKINES
Tumors or treatment of them may cause tissue injury, which triggers immune cells to release cytokines, signaling the brain to manifest the symptom of fatigue. Inflammatory pathways are influenced by psychological, behavioral, and biological factors, which play a role as risk factors in CRF. Levels of interleukin 6 (IL-6), interleukin-1 receptor antagonist, interleukin-1, and tumor necrosis factor (TNF) have been shown to be elevated in fatigued patients being treated for leukemia and non-Hodgkin lymphoma.15 IL-6 was also associated with increased fatigue in breast cancer survivors.16 Similar findings were reported in patients undergoing stem cell transplantation and high-dose chemotherapy.17 Elevated levels of IL-6 and C-reactive protein were also linked to fatigue in terminally ill cancer patients.18,19 Furthermore, TNF-α signaling was associated with post-chemotherapy fatigue in breast cancer patients.20 Leukocytes in breast cancer survivors with fatigue also have increased gene expression of pro-inflammatory cytokines, emphasizing the role of cytokines and inflammation in the pathogenesis of CRF.21
OTHER HYPOTHESES
Several other hypotheses for CRF pathogenesis have been proposed. Activation of latent viruses such as Epstein-Barr virus, lack of social support,22 genetic alterations in the immune pathway,23 epigenetic changes,24 accumulation of neurotoxic metabolites and depletion of serotonin by indoleamine 2,3-dioxygenase pathway activation,25 elevated vascular endothelial growth factor levels,26 and hypoxia-related organ dysfunction due to anemia or hemoglobin dysfunction13 all have been postulated to cause CRF.
EVALUATION AND TREATMENT
Fours steps are involved in the evaluation and treatment of CRF (Figure).
SCREENING
Because patients and health care professionals may be unaware of the treatment options available for CRF, patients may not report fatigue levels to their clinicians, and clinicians may not understand the impact of fatigue on their patients’ quality of life. This leads to under-recognition of the problem. The NCCN recommends screening every cancer patient and post-treatment survivor for fatigue.2 Patients should be screened at their first visit and then at periodic intervals during and after cancer treatment.
Many scales are available to screen patients for CRF in clinical practice and clinical trials.27 A single item that asks patients to rate their fatigue on a scale from 0 to 10—in which 0 indicates no fatigue, 1 to 3 indicates mild fatigue, 4 to 6 indicates moderate fatigue, 7 to 9 indicates severe fatigue, and 10 indicates the worst fatigue imaginable—is commonly used to screen for CRF.2 This scale was adapted from the MD Anderson Symptom Inventory scale and is based on a large nationwide study of cancer patients and survivors.28 The statistically derived cutoff points in this study are consistent with other scales such as the Brief Fatigue Inventory (BFI) and support the cutoff points (4–6 for moderate and ≥ 7 for severe fatigue) used in various fatigue management guidelines. Furthermore, studies of fatigue in cancer patients have revealed a marked decrease in physical function at levels of 7 or higher, suggesting 7 as an optimal cutoff to identify severe fatigue.29,30 The Visual Analog Scale is another simple-to-use tool that helps in understanding variations in fatigue throughout the course of the day.31 The 9-item BFI is often used in clinical trials.29 It measures the severity of fatigue over the previous 24 hours and has been validated in patients who do not speak English.32
CRF affects not only the somatic domain, but also the cognitive, behavioral, and affective domains; therefore, multidimensional scales have been developed for screening. One such tool is the Multidimensional Fatigue Inventory, which assesses 5 dimensions of fatigue—general fatigue, physical fatigue, reduced motivation, reduced activity, and mental fatigue—and compares the patient’s results with those of individuals without cancer.33,34 The Functional Assessment of Cancer Therapy for Fatigue (FACT-F) is a 13-item questionnaire that has been used to measure CRF in clinical trials as well as in patients receiving various treatments.35
Although many scales are available, the validity of self-reporting on simple fatigue-rating scales is equal to or better than most complex, lengthy scales.36 Therefore, unidimensional tools such as the numeric rating scale of 0–10 are commonly used in clinical practice. Mild fatigue (0–3) requires periodic reevaluation, and moderate and severe fatigue need further evaluation and management.37
PRIMARY EVALUATION
This phase involves a focused history and physical examination and assessment of concurrent symptoms and contributing factors.
History and Physical Examination
A detailed history of the patient’s malignancy and type of previous and current treatment may help reveal the cause of fatigue. New-onset fatigue or increase in fatigue may be related to the progression of disease in patients with active malignancy or recurrence of cancer in survivors. These patients may require appropriate testing to assess the underlying disease pattern. A detailed review of systems may help identify some of the contributing factors, which are discussed below. A detailed history regarding medications, including over-the-counter drugs, complementary agents, and past and prior cancer therapies, is helpful as medications can contribute to fatigue. For example, opioids may cause drowsiness and fatigue, which could be improved by dose adjustments. A focused history of fatigue should be obtained in all patients with moderate to severe CRF, which includes the onset, pattern, duration, associated or alleviating factors, and interference with functioning, including activities of daily living.37 Physical examination should focus on identifying signs of organ dysfunction and features of substance or alcohol abuse, which may cause poor sleep and fatigue.
Assessment of Contributing Factors
The management of fatigue should be multifactorial, with a comprehensive assessment and treatment plan to address all modifiable fatigue etiologies. The Table lists potential contributing factors to fatigue that should be considered when evaluating patients for CRF; several common conditions are discussed below.
Anemia. Anemia has been correlated with fatigue and quality of life. In a study of 4382 cancer patients receiving chemotherapy, quality-of-life measures using FACT-Anemia scores improved with increased hemoglobin levels.38 Cancer patients may have anemia due to marrow-suppressing effects of chemotherapy and may also have iron deficiency anemia due to blood loss or auto-immune hemolytic anemia. Therefore, a detailed work-up is required to identify the etiology of anemia. Patients with CRF whose anemia is related to chemotherapy or anemia of chronic disease may benefit from red blood cell transfusion or erythropoiesis-stimulating agents (ESAs). ESAs have been studied extensively; however, their use is controversial because of concerns about thromboembolic side effects leading to adverse outcomes.39 Also, ESA therapy is not recommended in patients with hematologic malignancies. ESA use should be restricted to patients with chemotherapy-related anemia with hemoglobin below 10 mg/dL and should be discontinued in 6 to 8 weeks if patients do not respond.40 Other patients may benefit from blood transfusions, which were shown to help in patients with hemoglobin levels between 7.5 and 8.5 g/dL.41
Sleep disturbance. Poor sleep is common in fatigued cancer survivors.42 Pro-inflammatory cytokines can disrupt the sleep–wake cycle, causing changes in the HPA axis and neuroendocrine system, which in turn may lead to increasing fatigue. Heckler et al showed that improvement in nighttime sleep leads to improvement of fatigue.43 Cognitive behavioral therapy and sleep hygiene are important in caring for patients with CRF and poor sleep.44 Taking a warm bath and/or drinking a glass of milk before bedtime, avoiding caffeinated drinks, and avoiding frequent napping in the day might help. Some patients may require medications such as benzodiazepines or non-benzodiazepine hypnotics (eg, zolpidem) to promote sleep.45 Melatonin agonists are approved for insomnia in the United States, but not in Europe.46
Malnutrition. Patients with advanced-stage cancer and with cancers affecting the gastrointestinal tract frequently develop mechanical bowel obstructions, especially at the end of their life, which cause malnutrition. Chemotherapy-related nausea and vomiting may also cause poor oral intake and malnutrition, causing fatigue from muscle weakness. Dehydration and electrolyte imbalances frequently occur as a result of poor oral intake, which might worsen fatigue. In our experience, modifying dietary intake with appropriate caloric exchanges with the help of a nutrition expert has lessened fatigue in some patients. However, terminally ill patients are advised to eat based on their comfort.
Medical comorbidities. Congestive heart failure from anthracycline chemotherapy, hypothyroidism after radiation therapy for head and neck cancers, renal failure, or hepatic failure from chemotherapy may indirectly lead to fatigue. Chemotherapy, opioids, and steroids may cause hypogonadism, which can contribute to fatigue in men.47
Assessment of Concurrent Symptoms
Depression is common in cancer patients and coexists with pain, insomnia, fatigue, and anxiety as a symptom cluster.48 A symptom cluster is defined as 2 or more concurrent and interrelated symptoms occurring together; treating one of these symptoms without addressing other symptoms is not effective.49 Therefore, screening for and management of depression, anxiety, and insomnia play an important role in the management of CRF.
Physical symptoms due to the tumor or to therapy— such as pain, dyspnea, nausea, and decreased physical activity—may also contribute to fatigue both directly and indirectly. Patients with lung cancer may have hypoxemia, which can contribute to dyspnea with activity and a perception of fatigue. Optimal management of pain and other physical symptoms in patients with cancer may significantly alleviate fatigue.50
MANAGEMENT
Management of CRF is individualized based on the patient’s clinical status: active cancer treatment, survivor, or end of life. Education and counselling of patients and their caregivers play an important role in CRF. NCCN guidelines recommend focusing on pain control, distress management, energy conservation, physical activity, nutrition, and sleep hygiene.
Nonpharmacologic Interventions
Energy conservation. Energy conservation strategies, in which patients are advised to set priorities and realistic expectations, are highly recommended. Some energy-conserving strategies are to pace oneself, delegate and schedule activities at times of peak energy, postpone nonessential activities, attend to 1 activity at a time, structure daily routines, and maintain a diary to identify their peak energy period and structure activities around that time.51,52 When patients approach the end of life, increasing fatigue may limit their activity level, and they may depend on caregivers for assistance with activities of daily living, monitoring treatment-related adverse effects, and taking medications, especially elderly patients.53
Cognitive behavioral therapy. Cognitive behavioral therapy (CBT) has been shown to improve CRF during active treatment, and the benefits persist for a minimum of 2 years after therapy.54 CBT interventions that optimize sleep quality may improve fatigue.55 More studies are needed to understand whether referral to a psychologist for formal CBT is required. Randomized clinical trials showed patient fatigue education, learned self-care, coping techniques, and balancing rest and activity benefit patients with CRF.56
Exercise. Physical activity is highly encouraged in patients with CRF. Exercise increases muscle protein synthesis, improves cytokine response, and decreases the rate of sarcopenia in healthy populations.57 Studies have shown that exercise helps CRF at all phases of the cancer journey, including radiation therapy, chemotherapy, and survivorship.58 Some patients may feel less motivated to exercise and may not believe that exercise is possible or could potentially help them. Counselling is needed for such patients.
Older cancer survivors have a decline in their functional capacity and reduced muscle mass. Exercise can improve their cardiorespiratory fitness, muscle strength, and body composition.57 Exercise not only helps at the cellular level but also has psychosocial benefits from improved self-esteem. Therefore, exercise may be recommended for younger patients as well as for the older population, who may have comorbidities and less motivation than younger patients.
In a meta-analysis of 56 randomized controlled trials involving 4068 participants, aerobic exercise was found to have beneficial effects on CRF for patients during and after chemotherapy, specifically for patients with solid tumors.59 In another meta-analysis of breast and prostate cancer survivors, a combination of aerobic exercise with resistance training (3–6 metabolic equivalents, 60%–80% range of motion) was shown to reduce CRF more than aerobic exercise alone.60 This effect was also shown in a randomized controlled trial of 160 patients with stage 0 to III breast cancer undergoing radiation therapy.61 The control group in this study had a group-based non-exercise intervention/relaxation; therefore, the study showed that the effect of resistance training extends beyond the psychosocial benefits of group-based interventions. The intervention comprised 8 progressive machine-based resistance exercises (3 sets, 8–12 repetitions at 60%–80% of 1 repetition maximum) for 60 minutes twice weekly for 12 weeks. However, fatigue assessment questionnaire scores showed benefits only in the physical fatigue components, but not in the affective and cognitive components.
The American Society of Clinical Oncology’s guidelines for cancer survivors with fatigue recommends 150 minutes of moderate aerobic exercise (eg, fast walking, cycling, or swimming) per week, with 2 or 3 sessions of strength training per week.62 An individualized approach to exercise is recommended, as patients’ ability to perform certain types of exercises may be limited by thrombocytopenia, neutropenia, or lytic bone metastasis. Routine use of pre-exercise cardiovascular testing is not recommended but may be considered in high-risk populations, especially patients with risk factors for coronary heart disease and diabetes.63 Patients with comorbidities, substantial deconditioning, functional and anatomic defects, or recent major surgery may benefit from referral to physical therapy.37 Patients near end of life may also benefit from an exercise program, as demonstrated in several studies that showed benefit in CRF and quality of life.64,65 We recommend that physicians use their best clinical judgement in suggesting the type and intensity of exercise program, as it may not be feasible in some patients.
Mind-body interventions. Mindfulness-based stress reduction (MBSR) has shown promise in breast cancer survivors, who reported immediate improvements in fatigue severity that continued up to 6 weeks after cessation of the training.66 Prior studies had similar findings, suggesting that MBSR modestly decreases fatigue and sleep disturbances and has a greater effect on the degree to which symptoms interfere with many facets of life.67
Yoga. A study of a yoga intervention showed a benefit in older cancer survivors.68 In breast cancer patients undergoing chemotherapy, yoga was shown to benefit both physical and cognitive fatigue.69 DVD-based yoga had benefits similar to strengthening exercises in a study of 34 early-stage breast cancer survivors with CRF.70 More studies are needed in men and patients and survivors of other cancers, as most studies of yoga were conducted in women with breast cancer.
Tai chi/qigong. Like yoga, tai chi and qigong are practices of meditative movement. These practices use postures or movements with a focus on breath and a meditative state to bring about deep states of relaxation. Qigong is a series of simple, repeated practices including body posture/movement, breath practice, and meditation performed in synchrony. Tai chi easy (TCE) is a simplified set of common, repetitive tai chi movements. In a trial, qigong/TCE was compared with sham qigong, which had physical movements but no breathing or meditative practice. Breast cancer survivors in the qigong/TCE group had improved fatigue scores, and the effect persisted for 3 months.71 Additional research is needed in this area.
Acupuncture. A randomized controlled trial in breast cancer patients with CRF showed an improvement in the mean general fatigue score (per the Multidimensional Fatigue Inventory) in patients who received acupuncture versus those who did not (−3.11 [95% confidence interval −3.97 to −2.25]; P < 0.001) at 6 weeks. Improvements were seen in both the mental and physical aspects of fatigue.72 However, Deng et al noted that true acupuncture was no more effective than sham acupuncture for reducing post-chemotherapy chronic fatigue.73 Presently, there is not sufficient evidence to evaluate the benefits of acupuncture in CRF.
Other modalities. Massage therapy, music therapy, hypnosis, therapeutic touch, biofield therapies, relaxation, and reiki are other therapies for which few studies have been done; of the studies that have been done, the results are mixed, and additional research is needed.74 Currently, there are not sufficient data to recommend any of these modalities.
Pharmacologic Interventions
Psychostimulants. Methylphenidate and modafinil are psychostimulants or wakefulness-promoting agents. Pilot studies showed benefit from methylphenidate and modafinil in CRF,75–77 but randomized controlled trials have yielded mixed results. Therefore, in patients with severe fatigue during cancer therapy, the initial management strategy involves evaluation and treatment of medical conditions such as anemia and a trial of nonpharmacological strategies as discussed above. If symptoms persist, then a therapeutic trial of a psychostimulant may be considered per NCCN guidelines for patients undergoing active cancer treatment.37
Methylphenidate directly stimulates adrenergic receptors and indirectly releases dopamine and norepinephrine from presynaptic terminals, which may explain why the drug benefits patients receiving opioid-induced sedation. It is a commonly studied psychostimulant, though its mechanism of action in CRF is unclear. Randomized controlled trials of methylphenidate have resulted in a wide range of findings due to the heterogeneity of study populations and variations in the dosage of methylphenidate. A meta-analysis of 7 studies indicates that methylphenidate benefitted the subgroup of patients with CRF.78 Likewise, in an analysis of 5 randomized controlled trials, Minton et al showed a benefit of psychostimulants in fatigue compared with placebo.79 However, another study of methylphenidate in patients with CRF showed a benefit only in patients with severe fatigue or advanced disease.80 Methylphenidate was found to benefit cancer patients receiving opioid-induced sedation, as methylphenidate promotes wakefulness, though fatigue was not studied specifically.81 In a trial with 30 hospice patients in which the methylphenidate dose was titrated based on response and adverse effects, Kerr at al found that the drug improved fatigue in a dose-dependent manner.82 However, a study in patients with CRF at the University of Texas MD Anderson Cancer Center found no significant difference in BFI scores between patients receiving methylphenidate and those receiving placebo at the end of 2 weeks of treatment.83 Also, other randomized controlled trials in patients undergoing adjuvant chemotherapy for breast cancer84 and patients receiving radiation therapy for brain tumors85 failed to demonstrate the efficacy of methylphenidate in CRF. It should be used cautiously after ruling out other causes of fatigue. The drug is overall well tolerated and side effects include headache and nausea.
Modafinil is a non-amphetamine psychostimulant that has been approved for the treatment of narcolepsy. In a trial studying the effect of modafinil on patients receiving docetaxel-based chemotherapy for metastatic breast or prostate cancer, there was a modest but not statistically significant improvement in fatigue scores on the MD Anderson Symptom Inventory compared with placebo. Nausea and vomiting were higher in the modafinil arm than in the placebo arm.86 Similarly, modafinil was not superior to placebo for CRF in 208 patients with non-squamous cell lung cancer not undergoing chemotherapy or radiation.87 A placebo effect was also noted in patients with multiple myeloma88 and patients with primary brain tumors.89 In a phase 3, multicenter, randomized, placebo-controlled, double-blind clinical trial of modafinil for CRF in 867 patients undergoing chemotherapy, there was a reduction in fatigue only for patients with severe baseline fatigue, with no significant effect on mild to moderate fatigue.90 In another recent study, modafinil was shown to reduce depressive symptoms only in patients with severe fatigue (BFI item 3 score ≥ 7).91 This finding is consistent with previous studies showing benefit in patients with high baseline fatigue, but additional randomized controlled trials are needed to provide clarity. NCCN guidelines do not recommend the use of modafinil to treat CRF.37
Other pharmacologic interventions. Corticosteroids are often used for symptom control in cancer patients. These drugs have anti-inflammatory effects through their modulation of pro-inflammatory cytokines.92 In a randomized controlled trial evaluating the efficacy of corticosteroids, patients receiving dexamethasone (4 mg twice daily) experienced significant improvement in their FACT-F scores compared with patients receiving placebo.93 A similar benefit in fatigue was demonstrated in another study of methylprednisolone (32 mg daily) versus placebo.94 Despite the benefits of steroids, their adverse effects, such as mood swings, gastritis, hyperglycemia, and immune suppression, limit their long-term use. Therefore, the use of steroids should be restricted to terminally ill fatigued patients with other symptoms such as anorexia, brain metastasis, or pain related to bone metastasis.37
Testosterone replacement has been shown to diminish fatigue in non-cancer patients. Many men with advanced cancer have hypogonadism leading to low serum testosterone, which may cause fatigue. In a small trial in which cancer patients with hypogonadism received intramuscular testosterone every 14 days or placebo, the group receiving testosterone showed improvement in FACT-F scores, but there was no significant difference in FACT-F scores between the 2 groups.95
Antidepressants have failed to demonstrate benefit in CRF without depression.8 However, if a patient has both fatigue and depression, antidepressants may help.96 A selective serotonin receptor inhibitor is recommended as a first-line antidepressant.97 Patients with cancer are often receiving multiple medications, and medication interactions should be considered to prevent adverse events such as serotonin syndrome.
Complementary and Alternative Supplements
Studies using vitamin supplementation have been inconclusive in patients with CRF.74 The use of other dietary supplements has yielded mixed results, and coenzyme Q has shown no benefit for patients with CRF.98
The benefit of ginseng was studied in a RCT involving 364 patients with CRF. There was an improvement in Multidimensional Fatigue Symptom Inventory-short form (MFSI-SF) scores at 8 weeks in patients receiving 2000 mg of Wisconsin ginseng compared with patients receiving placebo.99 Patients on active treatment had greater improvement as compared to the post-treatment group in this trial. In another study of high-dose panax ginseng (ginseng root) at 800 mg daily for 29 days, patients had improvement of CRF as well as overall quality of life, appetite, and sleep at night. It was also well tolerated with few adverse effects.100 Interaction with warfarin, calcium channel blockers, antiplatelet agents, thrombolytic agents, imatinib, and other agents may occur; therefore, ginseng must be used with careful monitoring in selected patients. There is not enough evidence at this time to support the routine use of ginseng in CRF.
The seed extract of the guarana plant (Paullinia cupana) traditionally has been used as a stimulant. An improvement in fatigue scores was seen with the use of oral guarana (100 mg daily) at the end of 21 days in breast cancer patients receiving chemotherapy.101 Further studies are needed for these results to be generalized and to understand the adverse effects and interaction profile of guarana.
Reevaluation
Patients who have completed cancer treatment must be monitored for fatigue over the long term, as fatigue may exist beyond the period of active treatment. Many studies have shown fatigue in breast cancer survivors, and fatigue has been demonstrated in survivors of colorectal, lung, and prostate cancers as well as myeloproliferative neoplasms.28 Therefore, it is important to screen patients for fatigue during follow-up visits. There are currently no studies evaluating the long-term treatment of fatigue. In our experience, the timing of follow-up depends on the level of fatigue and interventions prescribed. Once fatigue is stabilized to a level with which the patient is able to cope, the time interval for follow-up may be lengthened. Annual visits may suffice in patients with mild fatigue. Follow-up of patients with moderate to severe fatigue depends on the level of fatigue, the ability to cope, choice of treatment, and presence of contributing factors.
CONCLUSION
CRF is a complex condition that places a significant burden on patients and caregivers, resulting in emotional distress, poor functioning, and suffering. Fatigue can occur before, during, and long after cancer treatment. The approach to CRF begins with screening for and educating patients and their caregivers about the symptoms. Many screening scales are available that may be used to follow patients’ progress over time. The evaluation and management of contributing conditions may help improve fatigue. If the fatigue persists, an individualized approach with a combination of nonpharmacologic and pharmacologic approaches should be considered. More research is needed to understand brain signaling pathways, cytokine changes, and genomic changes in cancer patients with fatigue. Though many hypotheses have been proposed, to date there is no biological marker to assess this condition. Biomarker research needs to be advanced to help to identify patients at risk for fatigue. As cytokines have a major role in CRF, targeted therapy to block cytokine pathways may also be explored in the future.
Acknowledgment: The authors thank Bryan Tutt for providing editorial assistance during the writing of this article.
1. Scherber RM, Kosiorek HE, Senyak Z, et al. Comprehensively understanding fatigue in patients with myeloproliferative neoplasms. Cancer 2016;122:477–85.
2. Neefjes EC, van der Vorst MJ, Blauwhoff-Buskermolen S, Verheul HM. Aiming for a better understanding and management of cancer-related fatigue. Oncologist 2013;18:1135–43.
3. Radbruch L, Strasser F, Elsner F, et al. Fatigue in palliative care patients—an EAPC approach. Palliat Med 2008;22:13–32.
4. Capuron L, Pagnoni G, Demetrashvili MF, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 2007;32:2384–92.
5. Kisiel-Sajewicz K, Siemionow V, Seyidova-Khoshknabi D, et al. Myoelectrical manifestation of fatigue less prominent in patients with cancer related fatigue. PLoS One 2013;8:e83636.
6. Bower JE, Ganz PA, Aziz N. Altered cortisol response to psychologic stress in breast cancer survivors with persistent fatigue. Psychosom Med 2005;67:277–80.
7. Barsevick A, Frost M, Zwinderman A, et al. I’m so tired: biological and genetic mechanisms of cancer-related fatigue. Qual Life Res 2010;19:1419–27.
8. Morrow GR, Hickok JT, Roscoe JA, et al. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol 2003;21:4635–41.
9. Fontes-Oliveira CC, Busquets S, Toledo M, et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 2013;1830:2770–8.
10. Agteresch HJ, Dagnelie PC, van der Gaast A, et al. Randomized clinical trial of adenosine 5’-triphosphate in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst 2000;92:321–8.
11. Beijer S, Hupperets PS, van den Borne BE, et al. Randomized clinical trial on the effects of adenosine 5’-triphosphate infusions on quality of life, functional status, and fatigue in preterminal cancer patients. J Pain Symptom Manage 2010;40:520–30.
12. Kisiel-Sajewicz K, Davis MP, Siemionow V, et al. Lack of muscle contractile property changes at the time of perceived physical exhaustion suggests central mechanisms contributing to early motor task failure in patients with cancer-related fatigue. J Pain Symptom Manage 2012;44:351–61.
13. Ryan JL, Carroll JK, Ryan EP, et al. Mechanisms of cancer-related fatigue. Oncologist 2007;12 Suppl 1:22–34.
14. Seruga B, Zhang H, Bernstein LJ, Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer 2008;8:887–99.
15. Wang XS, Giralt SA, Mendoza TR, et al. Clinical factors associated with cancer-related fatigue in patients being treated for leukemia and non-Hodgkin’s lymphoma. J Clin Oncol 2002;20:1319–28.
16. Collado-Hidalgo A, Bower JE, Ganz PA, et al. Inflammatory biomarkers for persistent fatigue in breast cancer survivors. Clin Cancer Res 2006;12:2759–66.
17. Wang XS, Shi Q, Williams LA, et al. Serum interleukin-6 predicts the development of multiple symptoms at nadir of allogeneic hematopoietic stem cell transplantation. Cancer 2008;113:2102–9.
18. Inagaki M, Isono M, Okuyama T, et al. Plasma interleukin-6 and fatigue in terminally ill cancer patients. J Pain Symptom Manage 2008;35:153–61.
19. Laird BJ, McMillan DC, Fayers P, et al. The systemic inflammatory response and its relationship to pain and other symptoms in advanced cancer. Oncologist 2013;18:1050–5.
20. Bower JE, Ganz PA, Irwin MR, et al. Inflammation and behavioral symptoms after breast cancer treatment: do fatigue, depression, and sleep disturbance share a common underlying mechanism? J Clin Oncol 2011;29:3517–22.
21. Whistler T, Taylor R, Craddock RC, et al. Gene expression correlates of unexplained fatigue. Pharmacogenomics 2006;7:395–405.
22. Fagundes CP, Bennett JM, Alfano CM, et al. Social support and socioeconomic status interact to predict Epstein-Barr virus latency in women awaiting diagnosis or newly diagnosed with breast cancer. Health Psychol 2012;31:11–19.
23. Landmark-Hoyvik H, Reinertsen KV, Loge JH, et al. Alterations of gene expression in blood cells associated with chronic fatigue in breast cancer survivors. Pharmacogenomics J 2009;9:333–40.
24. Smith AK, Conneely KN, Pace TW, et al. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain Behav Immun 2014;38:227–36.
25. Kim S, Miller BJ, Stefanek ME, Miller AH. Inflammation-induced activation of the indoleamine 2,3-dioxygenase pathway: Relevance to cancer-related fatigue. Cancer 2015;121:2129–36.
26. Mills PJ, Parker B, Dimsdale JE, et al. The relationship between fatigue and quality of life and inflammation during anthracycline-based chemotherapy in breast cancer. Biol Psychol 2005;69:85–96.
27. Jean-Pierre P, Figueroa-Moseley CD, Kohli S, et al. Assessment of cancer-related fatigue: implications for clinical diagnosis and treatment. Oncologist 2007;12 Suppl 1:11–21.
28. Wang XS, Zhao F, Fisch MJ, et al. Prevalence and characteristics of moderate to severe fatigue: a multicenter study in cancer patients and survivors. Cancer 2014;120:425–32.
29. Mendoza TR, Wang XS, Cleeland CS, et al. The rapid assessment of fatigue severity in cancer patients: use of the Brief Fatigue Inventory. Cancer 1999;85:1186–96.
30. Mendoza ME, Capafons A, Gralow JR, et al. Randomized controlled trial of the Valencia model of waking hypnosis plus CBT for pain, fatigue, and sleep management in patients with cancer and cancer survivors. Psychooncology 2016 Jul 28.
31. Glaus A. Assessment of fatigue in cancer and non-cancer patients and in healthy individuals. Support Care Cancer 1993;1:305–15.
32. Seyidova-Khoshknabi D, Davis MP, Walsh D. A systematic review of cancer-related fatigue measurement questionnaires. Am J Hosp Palliat Care 2011;28:119–29.
33. Holzner B, Kemmler G, Greil R, et al. The impact of hemoglobin levels on fatigue and quality of life in cancer patients. Ann Oncol 2002;13:965–73.
34. Stein KD, Jacobsen PB, Blanchard CM, Thors C. Further validation of the multidimensional fatigue symptom inventory-short form. J Pain Symptom Manage 2004;27:14–23.
35. Hwang SS, Chang VT, Rue M, Kasimis B. Multidimensional independent predictors of cancer-related fatigue. J Pain Symptom Manage 2003;26:604–14.
36. Peterson DR. Scope and generality of verbally defined personality factors. Psychol Rev 1965;72:48–59.
37. Berger AM, Abernethy AP, Atkinson A, et al. NCCN Clinical Practice Guidelines Cancer-related fatigue. J Natl Compr Canc Netw 2010;8:904–31.
38. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer 2002;95:888–95.
39. Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012;12:CD003407.
40. Rizzo JD, Brouwers M, Hurley P, et al. American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update on the use of epoetin and darbepoetin in adult patients with cancer. Blood 2010;116:4045–59.
41. Preston NJ, Hurlow A, Brine J, Bennett MI. Blood transfusions for anaemia in patients with advanced cancer. Cochrane Database Syst Rev 2012(2):CD009007.
42. Minton O, Stone PC. A comparison of cognitive function, sleep and activity levels in disease-free breast cancer patients with or without cancer-related fatigue syndrome. BMJ Support Palliat Care 2012;2:231–8.
43. Heckler CE, Garland SN, Peoples AR, et al. Cognitive behavioral therapy for insomnia, but not armodafinil, improves fatigue in cancer survivors with insomnia: a randomized placebo-controlled trial. Support Care Cancer 2016;24:2059–66.
44. Howell D, Oliver TK, Keller-Olaman S, et al. Sleep disturbance in adults with cancer: a systematic review of evidence for best practices in assessment and management for clinical practice. Ann Oncol 2014;25:791–800.
45. Wilt TJ, MacDonald R, Brasure M, et al. Pharmacologic treatment of insomnia disorder: an evidence report for a clinical practice guideline by the American College of Physicians. Ann Intern Med 2016;165:103–12.
46. Kuriyama A, Honda M, Hayashino Y. Ramelteon for the treatment of insomnia in adults: a systematic review and meta-analysis. Sleep Med 2014;15:385–92.
47. Strasser F, Palmer JL, Schover LR, et al. The impact of hypogonadism and autonomic dysfunction on fatigue, emotional function, and sexual desire in male patients with advanced cancer: a pilot study. Cancer 2006;107:2949–57.
48. Agasi-Idenburg SC, Thong MS, Punt CJ, et al. Comparison of symptom clusters associated with fatigue in older and younger survivors of colorectal cancer. Support Care Cancer 2017;25:625–32.
49. Miaskowski C, Aouizerat BE. Is there a biological basis for the clustering of symptoms? Semin Oncol Nurs 2007;23:99–105.
50. de Raaf PJ, de Klerk C, Timman R, et al. Systematic monitoring and treatment of physical symptoms to alleviate fatigue in patients with advanced cancer: a randomized controlled trial. J Clin Oncol 2013;31:716–23.
51. Barsevick AM, Whitmer K, Sweeney C, Nail LM. A pilot study examining energy conservation for cancer treatment-related fatigue. Cancer Nurs 2002;25:333–41.
52. Barsevick AM, Dudley W, Beck S, et a;. A randomized clinical trial of energy conservation for patients with cancer-related fatigue. Cancer 2004;100:1302–10.
53. Luciani A, Jacobsen PB, Extermann M, et al. Fatigue and functional dependence in older cancer patients. Am J Clin Oncol 2008;31:424–30.
54. Abrahams HJ, Gielissen MF, Goedendorp MM, et al. A randomized controlled trial of web-based cognitive behavioral therapy for severely fatigued breast cancer survivors (CHANGE-study): study protocol. BMC Cancer 2015;15:765.
55. Quesnel C, Savard J, Simard S, et al. Efficacy of cognitive-behavioral therapy for insomnia in women treated for nonmetastatic breast cancer. J Consult Clin Psychol 2003;71:189–200.
56. Goedendorp MM, Gielissen MF, Verhagen CA, Bleijenberg G. Psychosocial interventions for reducing fatigue during cancer treatment in adults. Cochrane Database Syst Rev 2009(1):CD006953.
57. Greiwe JS, Cheng B, Rubin DC, et al. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J 2001;15:475–82.
58. Furmaniak AC, Menig M, Markes MH. Exercise for women receiving adjuvant therapy for breast cancer. Cochrane Database Syst Rev 2016;(9):CD005001.
59. Cramp F, Byron-Daniel J. Exercise for the management of cancer-related fatigue in adults. Cochrane Database Syst Rev 2012;(11):CD006145.
60. Brown JC, Huedo-Medina TB, Pescatello LS, et al. Efficacy of exercise interventions in modulating cancer-related fatigue among adult cancer survivors: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2011;20:123–33.
61. Steindorf K, Schmidt ME, Klassen O, et al. Randomized, controlled trial of resistance training in breast cancer patients receiving adjuvant radiotherapy: results on cancer-related fatigue and quality of life. Ann Oncol 2014;25:2237–43.
62. Bower JE, Bak K, Berger A, et al. Screening, assessment, and management of fatigue in adult survivors of cancer: an American Society of Clinical oncology clinical practice guideline adaptation. J Clin Oncol 2014;32:1840–50.
63. Kenjale AA, Hornsby WE, Crowgey T, et al. Pre-exercise participation cardiovascular screening in a heterogeneous cohort of adult cancer patients. Oncologist 2014;19:999–1005.
64. Oldervoll LM, Loge JH, Paltiel H, et al. The effect of a physical exercise program in palliative care: A phase II study. J Pain Symptom Manage 2006;31:421–30.
65. Porock D, Kristjanson LJ, Tinnelly K, et al. An exercise intervention for advanced cancer patients experiencing fatigue: a pilot study. J Palliat Care 2000;16:30–6.
66. Lengacher CA, Kip KE, Reich RR, et al. A cost-effective mindfulness stress reduction program: a randomized control trial for breast cancer survivors. Nursing Econ 2015;33:210–8, 32.
67. Lengacher CA, Reich RR, Post-White J, et al. Mindfulness based stress reduction in post-treatment breast cancer patients: an examination of symptoms and symptom clusters. J Behav Med 2012;35:86–94.
68. Sprod LK, Fernandez ID, Janelsins MC, et al. Effects of yoga on cancer-related fatigue and global side-effect burden in older cancer survivors. J Geriatr Oncol 2015;6:8–14.
69. Wang G, Wang S, Jiang P, Zeng C. [Effect of Yoga on cancer related fatigue in breast cancer patients with chemotherapy]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2014;39:1077–82.
70. Stan DL, Croghan KA, Croghan IT, et al. Randomized pilot trial of yoga versus strengthening exercises in breast cancer survivors with cancer-related fatigue. Support Care Cancer 2016;24:4005–15.
71. Larkey LK, Roe DJ, Weihs KL, et al. Randomized controlled trial of Qigong/Tai Chi Easy on cancer-related fatigue in breast cancer survivors. Ann Behav Med 2015;49:165–76.
72. Molassiotis A, Bardy J, Finnegan-John J, et al. Acupuncture for cancer-related fatigue in patients with breast cancer: a pragmatic randomized controlled trial. J Clin Oncol 2012;30:4470–6.
73. Deng G, Chan Y, Sjoberg D, et al. Acupuncture for the treatment of post-chemotherapy chronic fatigue: a randomized, blinded, sham-controlled trial. Support Care Cancer 2013;21:1735–41.
74. Finnegan-John J, Molassiotis A, Richardson A, Ream E. A systematic review of complementary and alternative medicine interventions for the management of cancer-related fatigue. Integr Cancer Ther 2013;12:276–90.
75. Schwartz AL, Thompson JA, Masood N. Interferon-induced fatigue in patients with melanoma: a pilot study of exercise and methylphenidate. Oncol Nurs Forum 2002;29:E85–90.
76. Spathis A, Dhillan R, Booden D, et al. Modafinil for the treatment of fatigue in lung cancer: a pilot study. Palliat Med 2009;23:325–31.
77. Blackhall L, Petroni G, Shu J, et al. A pilot study evaluating the safety and efficacy of modafinal for cancer-related fatigue. J Palliat Med 2009;12:433–9.
78. Qu D, Zhang Z, Yu X, et al. Psychotropic drugs for the management of cancer-related fatigue: a systematic review and meta-analysis. Eur J Cancer Care (Engl) 2015;25:970–9.
79. Minton O, Richardson A, Sharpe M, et al. Drug therapy for the management of cancer-related fatigue. Cochrane Database Syst Rev 2010(7):CD006704.
80. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol 2010;28:3673–9.
81. Bruera E, Driver L, Barnes EA, et al. Patient-controlled methylphenidate for the management of fatigue in patients with advanced cancer: a preliminary report. J Clin Oncol 2003;21:4439–43.
82. Kerr CW, Drake J, Milch RA, et al. Effects of methylphenidate on fatigue and depression: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage 2012;43:68–77.
83. Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J 2014;20:8–14.
84. Mar Fan HG, Clemons M, Xu W, et al. A randomised, placebo-controlled, double-blind trial of the effects of d-methylphenidate on fatigue and cognitive dysfunction in women undergoing adjuvant chemotherapy for breast cancer. Support Care Cancer 2008;16:577–83.
85. Butler JM Jr, Case LD, Atkins J, et al. A phase III, double-blind, placebo-controlled prospective randomized clinical trial of d-threo-methylphenidate HCl in brain tumor patients receiving radiation therapy. Int J Radiat Oncol Biol Phys 2007;69:1496–501.
86. Hovey E, de Souza P, Marx G, et al. Phase III, randomized, double-blind, placebo-controlled study of modafinil for fatigue in patients treated with docetaxel-based chemotherapy. Support Care Cancer 2014;22:1233–42.
87. Spathis A, Fife K, Blackhall F, et al. Modafinil for the treatment of fatigue in lung cancer: results of a placebo-controlled, double-blind, randomized trial. J Clin Oncol 2014;32:1882–8.
88. Berenson JR, Yellin O, Shamasunder HK, et al. A phase 3 trial of armodafinil for the treatment of cancer-related fatigue for patients with multiple myeloma. Support Care Cancer 2015; 23:1503–12.
89. Boele FW, Douw L, de Groot M, et al. The effect of modafinil on fatigue, cognitive functioning, and mood in primary brain tumor patients: a multicenter randomized controlled trial. Neuro Oncol 2013;15:1420–8.
90. Jean-Pierre P, Morrow GR, Roscoe JA, et al. A phase 3 randomized, placebo-controlled, double-blind, clinical trial of the effect of modafinil on cancer-related fatigue among 631 patients receiving chemotherapy: a University of Rochester Cancer Center Community Clinical Oncology Program Research base study. Cancer 2010;116:3513–20.
91. Conley CC, Kamen CS, Heckler CE, et al. Modafinil moderates the relationship between cancer-related fatigue and depression in 541 patients receiving chemotherapy. J Clin Psychopharmacol 2016;36:82–5.
92. Brattsand R, Linden M. Cytokine modulation by glucocorticoids: mechanisms and actions in cellular studies. Aliment Pharmacol Ther 1996;10 Suppl 2:81–90.
93. Yennurajalingam S, Frisbee-Hume S, Palmer JL, et al. Reduction of cancer-related fatigue with dexamethasone: a double-blind, randomized, placebo-controlled trial in patients with advanced cancer. J Clin Oncol 2013;31:3076–82.
94. Bruera E, Roca E, Cedaro L, et al. Action of oral methylprednisolone in terminal cancer patients: a prospective randomized double-blind study. Cancer Treat Rep 1985;69:751–4.
95. Pulivarthi K, Dev R, Garcia J, et al. Testosterone replacement for fatigue in male hypogonadic patients with advanced cancer: A preliminary double-blind placebo-controlled trial. J Clin Oncol 2012;30 (suppl). Abstract e19643.
96. Palesh OG, Mustian KM, Peppone LJ, et al. Impact of paroxetine on sleep problems in 426 cancer patients receiving chemotherapy: a trial from the University of Rochester Cancer Center Community Clinical Oncology Program. Sleep Med 2012;13:1184–90.
97. Thekdi SM, Trinidad A, Roth A. Psychopharmacology in Cancer. Curr Psychiatry Rep 2014;17:529.
98. Lesser GJ. Case D, Stark N, et al. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol 2013;11:31–42.
99. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst 2013;105:1230–8.
100. Yennurajalingam S, Reddy A, Tannir NM, et al. High-dose Asian ginseng (panax ginseng) for cancer-related fatigue: a preliminary report. Integr Cancer Ther 2015;14:419–27.
101. Howell D, Keller-Olaman S, Oliver TK, et al. A pan-Canadian practice guideline and algorithm: screening, assessment, and supportive care of adults with cancer-related fatigue. Curr Oncol 2013;20:e233–46.
1. Scherber RM, Kosiorek HE, Senyak Z, et al. Comprehensively understanding fatigue in patients with myeloproliferative neoplasms. Cancer 2016;122:477–85.
2. Neefjes EC, van der Vorst MJ, Blauwhoff-Buskermolen S, Verheul HM. Aiming for a better understanding and management of cancer-related fatigue. Oncologist 2013;18:1135–43.
3. Radbruch L, Strasser F, Elsner F, et al. Fatigue in palliative care patients—an EAPC approach. Palliat Med 2008;22:13–32.
4. Capuron L, Pagnoni G, Demetrashvili MF, et al. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 2007;32:2384–92.
5. Kisiel-Sajewicz K, Siemionow V, Seyidova-Khoshknabi D, et al. Myoelectrical manifestation of fatigue less prominent in patients with cancer related fatigue. PLoS One 2013;8:e83636.
6. Bower JE, Ganz PA, Aziz N. Altered cortisol response to psychologic stress in breast cancer survivors with persistent fatigue. Psychosom Med 2005;67:277–80.
7. Barsevick A, Frost M, Zwinderman A, et al. I’m so tired: biological and genetic mechanisms of cancer-related fatigue. Qual Life Res 2010;19:1419–27.
8. Morrow GR, Hickok JT, Roscoe JA, et al. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J Clin Oncol 2003;21:4635–41.
9. Fontes-Oliveira CC, Busquets S, Toledo M, et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 2013;1830:2770–8.
10. Agteresch HJ, Dagnelie PC, van der Gaast A, et al. Randomized clinical trial of adenosine 5’-triphosphate in patients with advanced non-small-cell lung cancer. J Natl Cancer Inst 2000;92:321–8.
11. Beijer S, Hupperets PS, van den Borne BE, et al. Randomized clinical trial on the effects of adenosine 5’-triphosphate infusions on quality of life, functional status, and fatigue in preterminal cancer patients. J Pain Symptom Manage 2010;40:520–30.
12. Kisiel-Sajewicz K, Davis MP, Siemionow V, et al. Lack of muscle contractile property changes at the time of perceived physical exhaustion suggests central mechanisms contributing to early motor task failure in patients with cancer-related fatigue. J Pain Symptom Manage 2012;44:351–61.
13. Ryan JL, Carroll JK, Ryan EP, et al. Mechanisms of cancer-related fatigue. Oncologist 2007;12 Suppl 1:22–34.
14. Seruga B, Zhang H, Bernstein LJ, Tannock IF. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer 2008;8:887–99.
15. Wang XS, Giralt SA, Mendoza TR, et al. Clinical factors associated with cancer-related fatigue in patients being treated for leukemia and non-Hodgkin’s lymphoma. J Clin Oncol 2002;20:1319–28.
16. Collado-Hidalgo A, Bower JE, Ganz PA, et al. Inflammatory biomarkers for persistent fatigue in breast cancer survivors. Clin Cancer Res 2006;12:2759–66.
17. Wang XS, Shi Q, Williams LA, et al. Serum interleukin-6 predicts the development of multiple symptoms at nadir of allogeneic hematopoietic stem cell transplantation. Cancer 2008;113:2102–9.
18. Inagaki M, Isono M, Okuyama T, et al. Plasma interleukin-6 and fatigue in terminally ill cancer patients. J Pain Symptom Manage 2008;35:153–61.
19. Laird BJ, McMillan DC, Fayers P, et al. The systemic inflammatory response and its relationship to pain and other symptoms in advanced cancer. Oncologist 2013;18:1050–5.
20. Bower JE, Ganz PA, Irwin MR, et al. Inflammation and behavioral symptoms after breast cancer treatment: do fatigue, depression, and sleep disturbance share a common underlying mechanism? J Clin Oncol 2011;29:3517–22.
21. Whistler T, Taylor R, Craddock RC, et al. Gene expression correlates of unexplained fatigue. Pharmacogenomics 2006;7:395–405.
22. Fagundes CP, Bennett JM, Alfano CM, et al. Social support and socioeconomic status interact to predict Epstein-Barr virus latency in women awaiting diagnosis or newly diagnosed with breast cancer. Health Psychol 2012;31:11–19.
23. Landmark-Hoyvik H, Reinertsen KV, Loge JH, et al. Alterations of gene expression in blood cells associated with chronic fatigue in breast cancer survivors. Pharmacogenomics J 2009;9:333–40.
24. Smith AK, Conneely KN, Pace TW, et al. Epigenetic changes associated with inflammation in breast cancer patients treated with chemotherapy. Brain Behav Immun 2014;38:227–36.
25. Kim S, Miller BJ, Stefanek ME, Miller AH. Inflammation-induced activation of the indoleamine 2,3-dioxygenase pathway: Relevance to cancer-related fatigue. Cancer 2015;121:2129–36.
26. Mills PJ, Parker B, Dimsdale JE, et al. The relationship between fatigue and quality of life and inflammation during anthracycline-based chemotherapy in breast cancer. Biol Psychol 2005;69:85–96.
27. Jean-Pierre P, Figueroa-Moseley CD, Kohli S, et al. Assessment of cancer-related fatigue: implications for clinical diagnosis and treatment. Oncologist 2007;12 Suppl 1:11–21.
28. Wang XS, Zhao F, Fisch MJ, et al. Prevalence and characteristics of moderate to severe fatigue: a multicenter study in cancer patients and survivors. Cancer 2014;120:425–32.
29. Mendoza TR, Wang XS, Cleeland CS, et al. The rapid assessment of fatigue severity in cancer patients: use of the Brief Fatigue Inventory. Cancer 1999;85:1186–96.
30. Mendoza ME, Capafons A, Gralow JR, et al. Randomized controlled trial of the Valencia model of waking hypnosis plus CBT for pain, fatigue, and sleep management in patients with cancer and cancer survivors. Psychooncology 2016 Jul 28.
31. Glaus A. Assessment of fatigue in cancer and non-cancer patients and in healthy individuals. Support Care Cancer 1993;1:305–15.
32. Seyidova-Khoshknabi D, Davis MP, Walsh D. A systematic review of cancer-related fatigue measurement questionnaires. Am J Hosp Palliat Care 2011;28:119–29.
33. Holzner B, Kemmler G, Greil R, et al. The impact of hemoglobin levels on fatigue and quality of life in cancer patients. Ann Oncol 2002;13:965–73.
34. Stein KD, Jacobsen PB, Blanchard CM, Thors C. Further validation of the multidimensional fatigue symptom inventory-short form. J Pain Symptom Manage 2004;27:14–23.
35. Hwang SS, Chang VT, Rue M, Kasimis B. Multidimensional independent predictors of cancer-related fatigue. J Pain Symptom Manage 2003;26:604–14.
36. Peterson DR. Scope and generality of verbally defined personality factors. Psychol Rev 1965;72:48–59.
37. Berger AM, Abernethy AP, Atkinson A, et al. NCCN Clinical Practice Guidelines Cancer-related fatigue. J Natl Compr Canc Netw 2010;8:904–31.
38. Crawford J, Cella D, Cleeland CS, et al. Relationship between changes in hemoglobin level and quality of life during chemotherapy in anemic cancer patients receiving epoetin alfa therapy. Cancer 2002;95:888–95.
39. Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012;12:CD003407.
40. Rizzo JD, Brouwers M, Hurley P, et al. American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update on the use of epoetin and darbepoetin in adult patients with cancer. Blood 2010;116:4045–59.
41. Preston NJ, Hurlow A, Brine J, Bennett MI. Blood transfusions for anaemia in patients with advanced cancer. Cochrane Database Syst Rev 2012(2):CD009007.
42. Minton O, Stone PC. A comparison of cognitive function, sleep and activity levels in disease-free breast cancer patients with or without cancer-related fatigue syndrome. BMJ Support Palliat Care 2012;2:231–8.
43. Heckler CE, Garland SN, Peoples AR, et al. Cognitive behavioral therapy for insomnia, but not armodafinil, improves fatigue in cancer survivors with insomnia: a randomized placebo-controlled trial. Support Care Cancer 2016;24:2059–66.
44. Howell D, Oliver TK, Keller-Olaman S, et al. Sleep disturbance in adults with cancer: a systematic review of evidence for best practices in assessment and management for clinical practice. Ann Oncol 2014;25:791–800.
45. Wilt TJ, MacDonald R, Brasure M, et al. Pharmacologic treatment of insomnia disorder: an evidence report for a clinical practice guideline by the American College of Physicians. Ann Intern Med 2016;165:103–12.
46. Kuriyama A, Honda M, Hayashino Y. Ramelteon for the treatment of insomnia in adults: a systematic review and meta-analysis. Sleep Med 2014;15:385–92.
47. Strasser F, Palmer JL, Schover LR, et al. The impact of hypogonadism and autonomic dysfunction on fatigue, emotional function, and sexual desire in male patients with advanced cancer: a pilot study. Cancer 2006;107:2949–57.
48. Agasi-Idenburg SC, Thong MS, Punt CJ, et al. Comparison of symptom clusters associated with fatigue in older and younger survivors of colorectal cancer. Support Care Cancer 2017;25:625–32.
49. Miaskowski C, Aouizerat BE. Is there a biological basis for the clustering of symptoms? Semin Oncol Nurs 2007;23:99–105.
50. de Raaf PJ, de Klerk C, Timman R, et al. Systematic monitoring and treatment of physical symptoms to alleviate fatigue in patients with advanced cancer: a randomized controlled trial. J Clin Oncol 2013;31:716–23.
51. Barsevick AM, Whitmer K, Sweeney C, Nail LM. A pilot study examining energy conservation for cancer treatment-related fatigue. Cancer Nurs 2002;25:333–41.
52. Barsevick AM, Dudley W, Beck S, et a;. A randomized clinical trial of energy conservation for patients with cancer-related fatigue. Cancer 2004;100:1302–10.
53. Luciani A, Jacobsen PB, Extermann M, et al. Fatigue and functional dependence in older cancer patients. Am J Clin Oncol 2008;31:424–30.
54. Abrahams HJ, Gielissen MF, Goedendorp MM, et al. A randomized controlled trial of web-based cognitive behavioral therapy for severely fatigued breast cancer survivors (CHANGE-study): study protocol. BMC Cancer 2015;15:765.
55. Quesnel C, Savard J, Simard S, et al. Efficacy of cognitive-behavioral therapy for insomnia in women treated for nonmetastatic breast cancer. J Consult Clin Psychol 2003;71:189–200.
56. Goedendorp MM, Gielissen MF, Verhagen CA, Bleijenberg G. Psychosocial interventions for reducing fatigue during cancer treatment in adults. Cochrane Database Syst Rev 2009(1):CD006953.
57. Greiwe JS, Cheng B, Rubin DC, et al. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J 2001;15:475–82.
58. Furmaniak AC, Menig M, Markes MH. Exercise for women receiving adjuvant therapy for breast cancer. Cochrane Database Syst Rev 2016;(9):CD005001.
59. Cramp F, Byron-Daniel J. Exercise for the management of cancer-related fatigue in adults. Cochrane Database Syst Rev 2012;(11):CD006145.
60. Brown JC, Huedo-Medina TB, Pescatello LS, et al. Efficacy of exercise interventions in modulating cancer-related fatigue among adult cancer survivors: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2011;20:123–33.
61. Steindorf K, Schmidt ME, Klassen O, et al. Randomized, controlled trial of resistance training in breast cancer patients receiving adjuvant radiotherapy: results on cancer-related fatigue and quality of life. Ann Oncol 2014;25:2237–43.
62. Bower JE, Bak K, Berger A, et al. Screening, assessment, and management of fatigue in adult survivors of cancer: an American Society of Clinical oncology clinical practice guideline adaptation. J Clin Oncol 2014;32:1840–50.
63. Kenjale AA, Hornsby WE, Crowgey T, et al. Pre-exercise participation cardiovascular screening in a heterogeneous cohort of adult cancer patients. Oncologist 2014;19:999–1005.
64. Oldervoll LM, Loge JH, Paltiel H, et al. The effect of a physical exercise program in palliative care: A phase II study. J Pain Symptom Manage 2006;31:421–30.
65. Porock D, Kristjanson LJ, Tinnelly K, et al. An exercise intervention for advanced cancer patients experiencing fatigue: a pilot study. J Palliat Care 2000;16:30–6.
66. Lengacher CA, Kip KE, Reich RR, et al. A cost-effective mindfulness stress reduction program: a randomized control trial for breast cancer survivors. Nursing Econ 2015;33:210–8, 32.
67. Lengacher CA, Reich RR, Post-White J, et al. Mindfulness based stress reduction in post-treatment breast cancer patients: an examination of symptoms and symptom clusters. J Behav Med 2012;35:86–94.
68. Sprod LK, Fernandez ID, Janelsins MC, et al. Effects of yoga on cancer-related fatigue and global side-effect burden in older cancer survivors. J Geriatr Oncol 2015;6:8–14.
69. Wang G, Wang S, Jiang P, Zeng C. [Effect of Yoga on cancer related fatigue in breast cancer patients with chemotherapy]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2014;39:1077–82.
70. Stan DL, Croghan KA, Croghan IT, et al. Randomized pilot trial of yoga versus strengthening exercises in breast cancer survivors with cancer-related fatigue. Support Care Cancer 2016;24:4005–15.
71. Larkey LK, Roe DJ, Weihs KL, et al. Randomized controlled trial of Qigong/Tai Chi Easy on cancer-related fatigue in breast cancer survivors. Ann Behav Med 2015;49:165–76.
72. Molassiotis A, Bardy J, Finnegan-John J, et al. Acupuncture for cancer-related fatigue in patients with breast cancer: a pragmatic randomized controlled trial. J Clin Oncol 2012;30:4470–6.
73. Deng G, Chan Y, Sjoberg D, et al. Acupuncture for the treatment of post-chemotherapy chronic fatigue: a randomized, blinded, sham-controlled trial. Support Care Cancer 2013;21:1735–41.
74. Finnegan-John J, Molassiotis A, Richardson A, Ream E. A systematic review of complementary and alternative medicine interventions for the management of cancer-related fatigue. Integr Cancer Ther 2013;12:276–90.
75. Schwartz AL, Thompson JA, Masood N. Interferon-induced fatigue in patients with melanoma: a pilot study of exercise and methylphenidate. Oncol Nurs Forum 2002;29:E85–90.
76. Spathis A, Dhillan R, Booden D, et al. Modafinil for the treatment of fatigue in lung cancer: a pilot study. Palliat Med 2009;23:325–31.
77. Blackhall L, Petroni G, Shu J, et al. A pilot study evaluating the safety and efficacy of modafinal for cancer-related fatigue. J Palliat Med 2009;12:433–9.
78. Qu D, Zhang Z, Yu X, et al. Psychotropic drugs for the management of cancer-related fatigue: a systematic review and meta-analysis. Eur J Cancer Care (Engl) 2015;25:970–9.
79. Minton O, Richardson A, Sharpe M, et al. Drug therapy for the management of cancer-related fatigue. Cochrane Database Syst Rev 2010(7):CD006704.
80. Moraska AR, Sood A, Dakhil SR, et al. Phase III, randomized, double-blind, placebo-controlled study of long-acting methylphenidate for cancer-related fatigue: North Central Cancer Treatment Group NCCTG-N05C7 trial. J Clin Oncol 2010;28:3673–9.
81. Bruera E, Driver L, Barnes EA, et al. Patient-controlled methylphenidate for the management of fatigue in patients with advanced cancer: a preliminary report. J Clin Oncol 2003;21:4439–43.
82. Kerr CW, Drake J, Milch RA, et al. Effects of methylphenidate on fatigue and depression: a randomized, double-blind, placebo-controlled trial. J Pain Symptom Manage 2012;43:68–77.
83. Escalante CP, Meyers C, Reuben JM, et al. A randomized, double-blind, 2-period, placebo-controlled crossover trial of a sustained-release methylphenidate in the treatment of fatigue in cancer patients. Cancer J 2014;20:8–14.
84. Mar Fan HG, Clemons M, Xu W, et al. A randomised, placebo-controlled, double-blind trial of the effects of d-methylphenidate on fatigue and cognitive dysfunction in women undergoing adjuvant chemotherapy for breast cancer. Support Care Cancer 2008;16:577–83.
85. Butler JM Jr, Case LD, Atkins J, et al. A phase III, double-blind, placebo-controlled prospective randomized clinical trial of d-threo-methylphenidate HCl in brain tumor patients receiving radiation therapy. Int J Radiat Oncol Biol Phys 2007;69:1496–501.
86. Hovey E, de Souza P, Marx G, et al. Phase III, randomized, double-blind, placebo-controlled study of modafinil for fatigue in patients treated with docetaxel-based chemotherapy. Support Care Cancer 2014;22:1233–42.
87. Spathis A, Fife K, Blackhall F, et al. Modafinil for the treatment of fatigue in lung cancer: results of a placebo-controlled, double-blind, randomized trial. J Clin Oncol 2014;32:1882–8.
88. Berenson JR, Yellin O, Shamasunder HK, et al. A phase 3 trial of armodafinil for the treatment of cancer-related fatigue for patients with multiple myeloma. Support Care Cancer 2015; 23:1503–12.
89. Boele FW, Douw L, de Groot M, et al. The effect of modafinil on fatigue, cognitive functioning, and mood in primary brain tumor patients: a multicenter randomized controlled trial. Neuro Oncol 2013;15:1420–8.
90. Jean-Pierre P, Morrow GR, Roscoe JA, et al. A phase 3 randomized, placebo-controlled, double-blind, clinical trial of the effect of modafinil on cancer-related fatigue among 631 patients receiving chemotherapy: a University of Rochester Cancer Center Community Clinical Oncology Program Research base study. Cancer 2010;116:3513–20.
91. Conley CC, Kamen CS, Heckler CE, et al. Modafinil moderates the relationship between cancer-related fatigue and depression in 541 patients receiving chemotherapy. J Clin Psychopharmacol 2016;36:82–5.
92. Brattsand R, Linden M. Cytokine modulation by glucocorticoids: mechanisms and actions in cellular studies. Aliment Pharmacol Ther 1996;10 Suppl 2:81–90.
93. Yennurajalingam S, Frisbee-Hume S, Palmer JL, et al. Reduction of cancer-related fatigue with dexamethasone: a double-blind, randomized, placebo-controlled trial in patients with advanced cancer. J Clin Oncol 2013;31:3076–82.
94. Bruera E, Roca E, Cedaro L, et al. Action of oral methylprednisolone in terminal cancer patients: a prospective randomized double-blind study. Cancer Treat Rep 1985;69:751–4.
95. Pulivarthi K, Dev R, Garcia J, et al. Testosterone replacement for fatigue in male hypogonadic patients with advanced cancer: A preliminary double-blind placebo-controlled trial. J Clin Oncol 2012;30 (suppl). Abstract e19643.
96. Palesh OG, Mustian KM, Peppone LJ, et al. Impact of paroxetine on sleep problems in 426 cancer patients receiving chemotherapy: a trial from the University of Rochester Cancer Center Community Clinical Oncology Program. Sleep Med 2012;13:1184–90.
97. Thekdi SM, Trinidad A, Roth A. Psychopharmacology in Cancer. Curr Psychiatry Rep 2014;17:529.
98. Lesser GJ. Case D, Stark N, et al. A randomized, double-blind, placebo-controlled study of oral coenzyme Q10 to relieve self-reported treatment-related fatigue in newly diagnosed patients with breast cancer. J Support Oncol 2013;11:31–42.
99. Barton DL, Liu H, Dakhil SR, et al. Wisconsin Ginseng (Panax quinquefolius) to improve cancer-related fatigue: a randomized, double-blind trial, N07C2. J Natl Cancer Inst 2013;105:1230–8.
100. Yennurajalingam S, Reddy A, Tannir NM, et al. High-dose Asian ginseng (panax ginseng) for cancer-related fatigue: a preliminary report. Integr Cancer Ther 2015;14:419–27.
101. Howell D, Keller-Olaman S, Oliver TK, et al. A pan-Canadian practice guideline and algorithm: screening, assessment, and supportive care of adults with cancer-related fatigue. Curr Oncol 2013;20:e233–46.
ICD-10-CM code changes: What's new for 2018
The list of new and revised International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) codes has been published, with changes becoming effective on October 1, 2017. This year, you can look forward to minimal code changes for gynecologic conditions. The biggest change in this category is the addition of codes that describe a lump in the breast according to which breast and the location on the breast, as well as the addition of a code for prophylactic removal of the fallopian tubes. The codes related to obstetrics will have more extensive changes. You will find revisions to the codes for ectopic pregnancy, a new set of codes for addressing an abnormal fetal heart rate during the antepartum period, and, most welcome of all, an expansion of the single code for antenatal testing to 17 very specific codes.
Gynecologic code changes
The single code N63, Unspecified lump in breast, has been expanded to include codes for both the right and the left breast. Code N63 therefore will be considered invalid after October 1, 2018. Expanded codes include:
- N63.0, Unspecified lump in unspecified breast
- N63.1, Unspecified lump in the right breast
- N63.10, Unspecified lump in the right breast, unspecified quadrant
- N63.11, Unspecified lump in the right breast, upper outer quadrant
- N63.12, Unspecified lump in the right breast, upper inner quadrant
- N63.13, Unspecified lump in the right breast, lower outer quadrant
- N63.14, Unspecified lump in the right breast, lower inner quadrant
- N63.2, Unspecified lump in the left breast
- N63.20, Unspecified lump in the left breast, unspecified quadrant
- N63.21, Unspecified lump in the left breast, upper outer quadrant
- N63.22, Unspecified lump in the left breast, upper inner quadrant
- N63.23, Unspecified lump in the left breast, lower outer quadrant
- N63.24, Unspecified lump in the left breast, lower inner quadrant
- N63.3, Unspecified lump in axillary tail
- N63.31, Unspecified lump in axillary tail of the right breast
- N63.32, Unspecified lump in axillary tail of the left breast
- N63.4, Unspecified lump in breast, subareolar
- N63.41, Unspecified lump in right breast, subareolar
- N63.42, Unspecified lump in left breast, subareolar
Other changes to gynecologic codes
There has been a minor change to the description for code Z31.5, Encounter for genetic counseling. It now is described as "Encounter for procreative genetic counseling."
The code Z40.02, Encounter for prophylactic removal of ovary(s), now refers to one or more ovaries without distinction of right or left, and a new code has been added for removal of the fallopian tube(s), Z40.03, Encounter for prophylactic removal of fallopian tube(s).
The inclusion term "endometriosis of the thorax" has been added to code N80.8, Other endometriosis.
The inclusion term "premenstrual dysphoric disorder" has been deleted from code N94.3, Premenstrual tension syndrome. If the patient has been diagnosed with this condition, the code F32.81, Premenstrual dysphoric disorder, should be reported, as this is considered a mental health issue.
The "excludes 1" note under code Z87.41, Personal history of dysplasia of the female genital tract, has been revised. The codes for personal history of intraepithelial neoplasia III that may not be reported with this code are Z86.001 and Z86.008.
Obstetric code changes
The ectopic pregnancy codes have been revised to include references to the right or left structure affected, which means the codes change from 5 digits to 6 digits in length, and the codes O00.10, O00.11, O00.20, and O00.21 will no longer be valid.
- O00.1, Tubal pregnancy
- O00.10, Tubal pregnancy without intrauterine pregnancy
- O00.101, Right tubal pregnancy without intrauterine pregnancy
- O00.102, Left tubal pregnancy without intrauterine pregnancy
- O00.109, Unspecified tubal pregnancy without intrauterine pregnancy
- O00.11, Tubal pregnancy with intrauterine pregnancy
- O00.111, Right tubal pregnancy with intrauterine pregnancy
- O00.112, Left tubal pregnancy with intrauterine pregnancy
- O00.119, Unspecified tubal pregnancy with intrauterine pregnancy
- O00.10, Tubal pregnancy without intrauterine pregnancy
- O00.2, Ovarian pregnancy
- O00.20, Ovarian pregnancy without intrauterine pregnancy
- O00.201, Right ovarian pregnancy without intrauterine pregnancy
- O00.202, Left ovarian pregnancy without intrauterine pregnancy
- O00.209, Unspecified ovarian pregnancy without intrauterine pregnancy
- O00.21, Ovarian pregnancy with intrauterine pregnancy
- O00.211, Right ovarian pregnancy with intrauterine pregnancy
- O00.212, Left ovarian pregnancy with intrauterine pregnancy
- O00.219, Unspecified ovarian pregnancy with intrauterine pregnancy
- O00.20, Ovarian pregnancy without intrauterine pregnancy
New codes for fetal heart rate abnormalities
New codes have been added to report a fetal heart rate or rhythm abnormality during the antepartum period. Until now, there only has been a code that addresses this issue during labor and delivery, O76, Abnormality in fetal heart rate and rhythm complicating labor and delivery.
- O36.83, Maternal care for abnormalities of the fetal heart rate or rhythm
- O36.831, Maternal care for abnormalities of the fetal heart rate or rhythm, first trimester
- O36.832, Maternal care for abnormalities of the fetal heart rate or rhythm, second trimester
- O36.833, Maternal care for abnormalities of the fetal heart rate or rhythm, third trimester
- O36.839, Maternal care for abnormalities of the fetal heart rate or rhythm, unspecified trimester
Several codes redefined
ICD-10 has corrected an "excludes" note error for the code O99.1, Other diseases of the blood and blood-forming organs and certain disorders involving the immune mechanism complicating pregnancy, childbirth, and the puerperium. In 2017, any hemorrhage with coagulation defects defined in code category O45.- or codes O46.0-, O67.0, or O72.3 could be reported with O99.1. This set of codes has now been redefined as an "excludes 1" note, which means that they may not be reported with O99.1 since they are considered inclusive.
More specific codes added for antenatal screening
Great news for those awaiting a more specific code for antenatal screening: The code Z36 has been expanded to more closely match the codes that were available in ICD-9-CM, but it goes beyond the basic list in that codes have been added for things like nonvisualization of anatomic structures on a previous scan and screening for cervical length and fetal lung maturity. Be sure to pay attention to the excludes notes and other inclusive terms.
- Z36, Encounter for antenatal screening of mother (Screening is the testing for disease or disease precursors in asymptomatic individuals so that early detection and treatment can be provided for those who test positive for the disease.)
Includes: Encounter for placental sample (taken vaginally)
Excludes 2: O28.-, Abnormal findings on antenatal screening of mother
- Z36.5, Encounter for antenatal screening for isoimmunization
- Z36.4, Encounter for antenatal screening for fetal growth retardation
Intrauterine growth restriction (IUGR)/small-for-dates - Z36.3, Encounter for antenatal screening for malformations
Screening for a suspected anomaly - Z36.2, Encounter for other antenatal screening follow-up
Nonvisualized anatomy on a previous scan - Z36.1, Encounter for antenatal screening for raised alpha-fetoprotein level
Encounter for antenatal screening for elevated maternal serum alpha-fetoprotein level - Z36.0, Encounter for antenatal screening for chromosomal anomalies
- Z36.81, Encounter for antenatal screening for hydrops fetalis
- Z36.8A, Encounter for antenatal screening for other genetic defects
- Z36.89, Encounter for other specified antenatal screening
- Z36.88, Encounter for antenatal screening for fetal macrosomia
Screening for large-for-dates - Z36.87, Encounter for antenatal screening for uncertain dates
- Z36.86, Encounter for antenatal screening for cervical length
Screening for risk of preterm labor - Z36.85, Encounter for antenatal screening for Streptococcus B
- Z36.84, Encounter for antenatal screening for fetal lung maturity
- Z36.83, Encounter for fetal screening for congenital cardiac abnormalities
- Z36.82, Encounter for antenatal screening for nuchal translucency
- Z36.81, Encounter for antenatal screening for hydrops fetalis
- Z36.9, Encounter for antenatal screening, unspecified
Code changes for abortion and complications
The code range for use with Z3A, weeks of gestation, has changed from O00-O9A to O09-O9A to reflect the guideline change last year to remove the requirement to use this code with any code that describes pregnancy with an abortive outcome (codes O00-O08).
In addition, if a patient has retained products of conception (POC) after either a spontaneous or elective abortion, report the "without complication" code for the retained POC (O03.4, Incomplete spontaneous abortion without complication, or O07.4, Failed attempted termination of pregnancy without complication). If any other complication occurred in addition to the retained POC, use the code for that particular complication and not O03.4 or O07.4.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.

Ms. Witt is an independent coding and documentation consultant and former program manager, department of coding and nomenclature, American Congress of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Ms. Witt is an independent coding and documentation consultant and former program manager, department of coding and nomenclature, American Congress of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
Ms. Witt is an independent coding and documentation consultant and former program manager, department of coding and nomenclature, American Congress of Obstetricians and Gynecologists.
The author reports no financial relationships relevant to this article.
The list of new and revised International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) codes has been published, with changes becoming effective on October 1, 2017. This year, you can look forward to minimal code changes for gynecologic conditions. The biggest change in this category is the addition of codes that describe a lump in the breast according to which breast and the location on the breast, as well as the addition of a code for prophylactic removal of the fallopian tubes. The codes related to obstetrics will have more extensive changes. You will find revisions to the codes for ectopic pregnancy, a new set of codes for addressing an abnormal fetal heart rate during the antepartum period, and, most welcome of all, an expansion of the single code for antenatal testing to 17 very specific codes.
Gynecologic code changes
The single code N63, Unspecified lump in breast, has been expanded to include codes for both the right and the left breast. Code N63 therefore will be considered invalid after October 1, 2018. Expanded codes include:
- N63.0, Unspecified lump in unspecified breast
- N63.1, Unspecified lump in the right breast
- N63.10, Unspecified lump in the right breast, unspecified quadrant
- N63.11, Unspecified lump in the right breast, upper outer quadrant
- N63.12, Unspecified lump in the right breast, upper inner quadrant
- N63.13, Unspecified lump in the right breast, lower outer quadrant
- N63.14, Unspecified lump in the right breast, lower inner quadrant
- N63.2, Unspecified lump in the left breast
- N63.20, Unspecified lump in the left breast, unspecified quadrant
- N63.21, Unspecified lump in the left breast, upper outer quadrant
- N63.22, Unspecified lump in the left breast, upper inner quadrant
- N63.23, Unspecified lump in the left breast, lower outer quadrant
- N63.24, Unspecified lump in the left breast, lower inner quadrant
- N63.3, Unspecified lump in axillary tail
- N63.31, Unspecified lump in axillary tail of the right breast
- N63.32, Unspecified lump in axillary tail of the left breast
- N63.4, Unspecified lump in breast, subareolar
- N63.41, Unspecified lump in right breast, subareolar
- N63.42, Unspecified lump in left breast, subareolar
Other changes to gynecologic codes
There has been a minor change to the description for code Z31.5, Encounter for genetic counseling. It now is described as "Encounter for procreative genetic counseling."
The code Z40.02, Encounter for prophylactic removal of ovary(s), now refers to one or more ovaries without distinction of right or left, and a new code has been added for removal of the fallopian tube(s), Z40.03, Encounter for prophylactic removal of fallopian tube(s).
The inclusion term "endometriosis of the thorax" has been added to code N80.8, Other endometriosis.
The inclusion term "premenstrual dysphoric disorder" has been deleted from code N94.3, Premenstrual tension syndrome. If the patient has been diagnosed with this condition, the code F32.81, Premenstrual dysphoric disorder, should be reported, as this is considered a mental health issue.
The "excludes 1" note under code Z87.41, Personal history of dysplasia of the female genital tract, has been revised. The codes for personal history of intraepithelial neoplasia III that may not be reported with this code are Z86.001 and Z86.008.
Obstetric code changes
The ectopic pregnancy codes have been revised to include references to the right or left structure affected, which means the codes change from 5 digits to 6 digits in length, and the codes O00.10, O00.11, O00.20, and O00.21 will no longer be valid.
- O00.1, Tubal pregnancy
- O00.10, Tubal pregnancy without intrauterine pregnancy
- O00.101, Right tubal pregnancy without intrauterine pregnancy
- O00.102, Left tubal pregnancy without intrauterine pregnancy
- O00.109, Unspecified tubal pregnancy without intrauterine pregnancy
- O00.11, Tubal pregnancy with intrauterine pregnancy
- O00.111, Right tubal pregnancy with intrauterine pregnancy
- O00.112, Left tubal pregnancy with intrauterine pregnancy
- O00.119, Unspecified tubal pregnancy with intrauterine pregnancy
- O00.10, Tubal pregnancy without intrauterine pregnancy
- O00.2, Ovarian pregnancy
- O00.20, Ovarian pregnancy without intrauterine pregnancy
- O00.201, Right ovarian pregnancy without intrauterine pregnancy
- O00.202, Left ovarian pregnancy without intrauterine pregnancy
- O00.209, Unspecified ovarian pregnancy without intrauterine pregnancy
- O00.21, Ovarian pregnancy with intrauterine pregnancy
- O00.211, Right ovarian pregnancy with intrauterine pregnancy
- O00.212, Left ovarian pregnancy with intrauterine pregnancy
- O00.219, Unspecified ovarian pregnancy with intrauterine pregnancy
- O00.20, Ovarian pregnancy without intrauterine pregnancy
New codes for fetal heart rate abnormalities
New codes have been added to report a fetal heart rate or rhythm abnormality during the antepartum period. Until now, there only has been a code that addresses this issue during labor and delivery, O76, Abnormality in fetal heart rate and rhythm complicating labor and delivery.
- O36.83, Maternal care for abnormalities of the fetal heart rate or rhythm
- O36.831, Maternal care for abnormalities of the fetal heart rate or rhythm, first trimester
- O36.832, Maternal care for abnormalities of the fetal heart rate or rhythm, second trimester
- O36.833, Maternal care for abnormalities of the fetal heart rate or rhythm, third trimester
- O36.839, Maternal care for abnormalities of the fetal heart rate or rhythm, unspecified trimester
Several codes redefined
ICD-10 has corrected an "excludes" note error for the code O99.1, Other diseases of the blood and blood-forming organs and certain disorders involving the immune mechanism complicating pregnancy, childbirth, and the puerperium. In 2017, any hemorrhage with coagulation defects defined in code category O45.- or codes O46.0-, O67.0, or O72.3 could be reported with O99.1. This set of codes has now been redefined as an "excludes 1" note, which means that they may not be reported with O99.1 since they are considered inclusive.
More specific codes added for antenatal screening
Great news for those awaiting a more specific code for antenatal screening: The code Z36 has been expanded to more closely match the codes that were available in ICD-9-CM, but it goes beyond the basic list in that codes have been added for things like nonvisualization of anatomic structures on a previous scan and screening for cervical length and fetal lung maturity. Be sure to pay attention to the excludes notes and other inclusive terms.
- Z36, Encounter for antenatal screening of mother (Screening is the testing for disease or disease precursors in asymptomatic individuals so that early detection and treatment can be provided for those who test positive for the disease.)
Includes: Encounter for placental sample (taken vaginally)
Excludes 2: O28.-, Abnormal findings on antenatal screening of mother
- Z36.5, Encounter for antenatal screening for isoimmunization
- Z36.4, Encounter for antenatal screening for fetal growth retardation
Intrauterine growth restriction (IUGR)/small-for-dates - Z36.3, Encounter for antenatal screening for malformations
Screening for a suspected anomaly - Z36.2, Encounter for other antenatal screening follow-up
Nonvisualized anatomy on a previous scan - Z36.1, Encounter for antenatal screening for raised alpha-fetoprotein level
Encounter for antenatal screening for elevated maternal serum alpha-fetoprotein level - Z36.0, Encounter for antenatal screening for chromosomal anomalies
- Z36.81, Encounter for antenatal screening for hydrops fetalis
- Z36.8A, Encounter for antenatal screening for other genetic defects
- Z36.89, Encounter for other specified antenatal screening
- Z36.88, Encounter for antenatal screening for fetal macrosomia
Screening for large-for-dates - Z36.87, Encounter for antenatal screening for uncertain dates
- Z36.86, Encounter for antenatal screening for cervical length
Screening for risk of preterm labor - Z36.85, Encounter for antenatal screening for Streptococcus B
- Z36.84, Encounter for antenatal screening for fetal lung maturity
- Z36.83, Encounter for fetal screening for congenital cardiac abnormalities
- Z36.82, Encounter for antenatal screening for nuchal translucency
- Z36.81, Encounter for antenatal screening for hydrops fetalis
- Z36.9, Encounter for antenatal screening, unspecified
Code changes for abortion and complications
The code range for use with Z3A, weeks of gestation, has changed from O00-O9A to O09-O9A to reflect the guideline change last year to remove the requirement to use this code with any code that describes pregnancy with an abortive outcome (codes O00-O08).
In addition, if a patient has retained products of conception (POC) after either a spontaneous or elective abortion, report the "without complication" code for the retained POC (O03.4, Incomplete spontaneous abortion without complication, or O07.4, Failed attempted termination of pregnancy without complication). If any other complication occurred in addition to the retained POC, use the code for that particular complication and not O03.4 or O07.4.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
The list of new and revised International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM) codes has been published, with changes becoming effective on October 1, 2017. This year, you can look forward to minimal code changes for gynecologic conditions. The biggest change in this category is the addition of codes that describe a lump in the breast according to which breast and the location on the breast, as well as the addition of a code for prophylactic removal of the fallopian tubes. The codes related to obstetrics will have more extensive changes. You will find revisions to the codes for ectopic pregnancy, a new set of codes for addressing an abnormal fetal heart rate during the antepartum period, and, most welcome of all, an expansion of the single code for antenatal testing to 17 very specific codes.
Gynecologic code changes
The single code N63, Unspecified lump in breast, has been expanded to include codes for both the right and the left breast. Code N63 therefore will be considered invalid after October 1, 2018. Expanded codes include:
- N63.0, Unspecified lump in unspecified breast
- N63.1, Unspecified lump in the right breast
- N63.10, Unspecified lump in the right breast, unspecified quadrant
- N63.11, Unspecified lump in the right breast, upper outer quadrant
- N63.12, Unspecified lump in the right breast, upper inner quadrant
- N63.13, Unspecified lump in the right breast, lower outer quadrant
- N63.14, Unspecified lump in the right breast, lower inner quadrant
- N63.2, Unspecified lump in the left breast
- N63.20, Unspecified lump in the left breast, unspecified quadrant
- N63.21, Unspecified lump in the left breast, upper outer quadrant
- N63.22, Unspecified lump in the left breast, upper inner quadrant
- N63.23, Unspecified lump in the left breast, lower outer quadrant
- N63.24, Unspecified lump in the left breast, lower inner quadrant
- N63.3, Unspecified lump in axillary tail
- N63.31, Unspecified lump in axillary tail of the right breast
- N63.32, Unspecified lump in axillary tail of the left breast
- N63.4, Unspecified lump in breast, subareolar
- N63.41, Unspecified lump in right breast, subareolar
- N63.42, Unspecified lump in left breast, subareolar
Other changes to gynecologic codes
There has been a minor change to the description for code Z31.5, Encounter for genetic counseling. It now is described as "Encounter for procreative genetic counseling."
The code Z40.02, Encounter for prophylactic removal of ovary(s), now refers to one or more ovaries without distinction of right or left, and a new code has been added for removal of the fallopian tube(s), Z40.03, Encounter for prophylactic removal of fallopian tube(s).
The inclusion term "endometriosis of the thorax" has been added to code N80.8, Other endometriosis.
The inclusion term "premenstrual dysphoric disorder" has been deleted from code N94.3, Premenstrual tension syndrome. If the patient has been diagnosed with this condition, the code F32.81, Premenstrual dysphoric disorder, should be reported, as this is considered a mental health issue.
The "excludes 1" note under code Z87.41, Personal history of dysplasia of the female genital tract, has been revised. The codes for personal history of intraepithelial neoplasia III that may not be reported with this code are Z86.001 and Z86.008.
Obstetric code changes
The ectopic pregnancy codes have been revised to include references to the right or left structure affected, which means the codes change from 5 digits to 6 digits in length, and the codes O00.10, O00.11, O00.20, and O00.21 will no longer be valid.
- O00.1, Tubal pregnancy
- O00.10, Tubal pregnancy without intrauterine pregnancy
- O00.101, Right tubal pregnancy without intrauterine pregnancy
- O00.102, Left tubal pregnancy without intrauterine pregnancy
- O00.109, Unspecified tubal pregnancy without intrauterine pregnancy
- O00.11, Tubal pregnancy with intrauterine pregnancy
- O00.111, Right tubal pregnancy with intrauterine pregnancy
- O00.112, Left tubal pregnancy with intrauterine pregnancy
- O00.119, Unspecified tubal pregnancy with intrauterine pregnancy
- O00.10, Tubal pregnancy without intrauterine pregnancy
- O00.2, Ovarian pregnancy
- O00.20, Ovarian pregnancy without intrauterine pregnancy
- O00.201, Right ovarian pregnancy without intrauterine pregnancy
- O00.202, Left ovarian pregnancy without intrauterine pregnancy
- O00.209, Unspecified ovarian pregnancy without intrauterine pregnancy
- O00.21, Ovarian pregnancy with intrauterine pregnancy
- O00.211, Right ovarian pregnancy with intrauterine pregnancy
- O00.212, Left ovarian pregnancy with intrauterine pregnancy
- O00.219, Unspecified ovarian pregnancy with intrauterine pregnancy
- O00.20, Ovarian pregnancy without intrauterine pregnancy
New codes for fetal heart rate abnormalities
New codes have been added to report a fetal heart rate or rhythm abnormality during the antepartum period. Until now, there only has been a code that addresses this issue during labor and delivery, O76, Abnormality in fetal heart rate and rhythm complicating labor and delivery.
- O36.83, Maternal care for abnormalities of the fetal heart rate or rhythm
- O36.831, Maternal care for abnormalities of the fetal heart rate or rhythm, first trimester
- O36.832, Maternal care for abnormalities of the fetal heart rate or rhythm, second trimester
- O36.833, Maternal care for abnormalities of the fetal heart rate or rhythm, third trimester
- O36.839, Maternal care for abnormalities of the fetal heart rate or rhythm, unspecified trimester
Several codes redefined
ICD-10 has corrected an "excludes" note error for the code O99.1, Other diseases of the blood and blood-forming organs and certain disorders involving the immune mechanism complicating pregnancy, childbirth, and the puerperium. In 2017, any hemorrhage with coagulation defects defined in code category O45.- or codes O46.0-, O67.0, or O72.3 could be reported with O99.1. This set of codes has now been redefined as an "excludes 1" note, which means that they may not be reported with O99.1 since they are considered inclusive.
More specific codes added for antenatal screening
Great news for those awaiting a more specific code for antenatal screening: The code Z36 has been expanded to more closely match the codes that were available in ICD-9-CM, but it goes beyond the basic list in that codes have been added for things like nonvisualization of anatomic structures on a previous scan and screening for cervical length and fetal lung maturity. Be sure to pay attention to the excludes notes and other inclusive terms.
- Z36, Encounter for antenatal screening of mother (Screening is the testing for disease or disease precursors in asymptomatic individuals so that early detection and treatment can be provided for those who test positive for the disease.)
Includes: Encounter for placental sample (taken vaginally)
Excludes 2: O28.-, Abnormal findings on antenatal screening of mother
- Z36.5, Encounter for antenatal screening for isoimmunization
- Z36.4, Encounter for antenatal screening for fetal growth retardation
Intrauterine growth restriction (IUGR)/small-for-dates - Z36.3, Encounter for antenatal screening for malformations
Screening for a suspected anomaly - Z36.2, Encounter for other antenatal screening follow-up
Nonvisualized anatomy on a previous scan - Z36.1, Encounter for antenatal screening for raised alpha-fetoprotein level
Encounter for antenatal screening for elevated maternal serum alpha-fetoprotein level - Z36.0, Encounter for antenatal screening for chromosomal anomalies
- Z36.81, Encounter for antenatal screening for hydrops fetalis
- Z36.8A, Encounter for antenatal screening for other genetic defects
- Z36.89, Encounter for other specified antenatal screening
- Z36.88, Encounter for antenatal screening for fetal macrosomia
Screening for large-for-dates - Z36.87, Encounter for antenatal screening for uncertain dates
- Z36.86, Encounter for antenatal screening for cervical length
Screening for risk of preterm labor - Z36.85, Encounter for antenatal screening for Streptococcus B
- Z36.84, Encounter for antenatal screening for fetal lung maturity
- Z36.83, Encounter for fetal screening for congenital cardiac abnormalities
- Z36.82, Encounter for antenatal screening for nuchal translucency
- Z36.81, Encounter for antenatal screening for hydrops fetalis
- Z36.9, Encounter for antenatal screening, unspecified
Code changes for abortion and complications
The code range for use with Z3A, weeks of gestation, has changed from O00-O9A to O09-O9A to reflect the guideline change last year to remove the requirement to use this code with any code that describes pregnancy with an abortive outcome (codes O00-O08).
In addition, if a patient has retained products of conception (POC) after either a spontaneous or elective abortion, report the "without complication" code for the retained POC (O03.4, Incomplete spontaneous abortion without complication, or O07.4, Failed attempted termination of pregnancy without complication). If any other complication occurred in addition to the retained POC, use the code for that particular complication and not O03.4 or O07.4.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Management of polycythemia vera in the community oncology setting
Polycythemia vera, classified as a myeloproliferative neoplasm (MPN) and characterized by uncontrolled, clonal, myeloid expansion with predominant erythrocytosis,1 affects about 100,000 individuals in the United States.2 It is a chronic and burdensome disease associated with shortened survival.3 Patients are at an increased risk of cardiovascular events, solid tumors, and transformation to myelofibrosis (MF) and/or acute myeloid leukemia (AML).4,5 Furthermore, patients generally have a reduced quality of life (QoL) stemming from prevalent and occasionally severe polycythemia vera–related signs and symptoms, including fatigue, pruritus, and splenomegaly.6 In general, the classical Philadelphia chromosome-negative MPNs are associated with driver mutations in the following three genes: Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia virus oncogene (MPL).7 Almost all patients with polycythemia vera have an activating mutation in the cytoplasmic signal transduction protein JAK2.4 Patients with essential thrombocythemia (ET) or MF can have mutations in JAK2, CALR, or MPL. However, CALR and MPL mutations are absent or exceedingly rare in patients with polycythemia vera.7 Diagnosis can be challenging and is currently based on 2016 World Health Organization (WHO) diagnostic criteria.1
Management strategies include the use of aspirin, phlebotomy, and cytoreductive therapy. Ruxolitinib is a newer treatment option available for patients with polycythemia vera who are either resistant to or intolerant of hydroxyurea8,9— a population that previously had few treatment options. It is important for community oncologists and other treating clinicians to understand current diagnostic strategy and management options based on established guidelines, recent clinical evidence, and regulatory updates.
Search and selection process for research sources
In September 2016, we searched PubMed for articles published since 2006 with polycythemia vera included in the abstract or title. The initial 1,730 publications were screened by eye to select 46 key articles that guide current management of polycythemia vera. Four studies published before 2006 were also included based on their continued relevance.
Epidemiology and pathophysiology
Based on a meta-analysis of patients from Europe and the United States, the annual incidence of polycythemia vera estimated to be between 0.7 and 2.6 per 100,000 people.10 The age-adjusted prevalence of polycythemia vera in the United States is about 45-57 per 100,000 people,2 however, the true prevalence might be considerably greater.
Patients with polycythemia vera are at increased risk of cardiovascular events, thrombosis, and death.3-5 Risk is highest among patients older than 60 years or with a history of thrombosis.11 Uncontrolled myeloproliferation has also been identified as a risk factor for cardiovascular mortality and thrombosis. This was demonstrated in the prospective Cytoreductive Therapy in Polycythemia Vera (CYTO-PV) trial, which reported more cardiovascular events in patients with hematocrit levels of 45%-50%, compared with those whose hematocrit levels were <45%.12 In addition, retrospective data suggest leukocytosis is a potential risk factor for thromboembolic events and poor outcomes.13
Dysregulated JAK2 signaling is the principal driver of polycythemia vera pathophysiology. About 95% of patients with polycythemia vera will have an identifiable JAK2 V617F exon 14 mutation, with an additional 3%-5% demonstrating a JAK2 exon 12 mutation.4,14 Under physiologic conditions, JAK2 interacts with the STAT family of signal transduction proteins and serves as an important regulator of normal hematopoiesis.15 Mutated, constitutively activated JAK2 signaling promotes the various polycythemia vera disease manifestations, including excessive myeloproliferation, splenomegaly,15 and constitutional symptoms.14,16,17
Burden of disease for the individual
Mortality
Patients with polycythemia vera have an increased risk of mortality compared with an unaffected, age- and gender-matched cohort of the general population.3 A retrospective study of Medicare patients with polycythemia vera (mean age at diagnosis, 76.1 years) reported a median survival of 5.4 years, compared with 8.7 years for a matched cohort.3 A second retrospective study reported a median survival of 13.5 years (median age at diagnosis, 64 years; median follow-up time, 11.8 years).18
Leading causes of death for patients with polycythemia vera include cardiovascular and thrombotic events, the development of secondary solid tumors, and disease transformation to MF and/or AML. In the prospective European Collaboration on Low-Dose Aspirin in Polycythemia Vera (ECLAP) study of 1,638 patients, 45% of deaths (74/164) resulted from cardiovascular causes (1.7 per 100 patient-years).5 Thirteen percent of deaths were related to either leukemic or myelofibrotic transformation, and 20% of deaths were attributed to secondary solid tumors.5 In a retrospective analysis of 1,545 patients with polycythemia vera followed for a median of 6.9 years after diagnosis, 347 had died, primarily from acute leukemia (10%), secondary malignancies (10%), and thrombotic events (9%).4 Arterial and venous thrombotic events occurred in 12% and 9% of patients, respectively, with disease transformation to MF and AML occurring in 9% and 3% of patients. Further support of an increased risk of secondary malignancies comes from a retrospective analysis of a large Swedish cancer registry (1958–2006) that found an increased risk of secondary endocrine, renal, and skin malignancies; MF; and leukemia among patients with polycythemia vera.19
Symptoms and quality of life
Symptoms of polycythemia vera vary in severity, and patients often fail to attribute symptoms to the disease.20 Moreover, clinicians may underestimate a patient’s true disease burden or the effect it has on QoL.20 Point-of-care metrics, such as the MPN Symptom Assessment Form (MPN-SAF), were developed to aid in identifying and grading symptom burden. Studies using this metric have reported fatigue as the most common and most severe symptom (incidence, 73%-92%), with a variety of other symptoms also affecting a majority of patients (Figure 1).6,21-24 Although fatigue, pruritus, and a higher MPN-SAF total symptom score are significantly correlated with reduced QoL,22,25 the recent MPN Landmark survey suggests that even patients with low symptom severity scores have a reduction in their QoL.6 This study also highlighted that polycythemia vera can adversely affect multiple aspects of daily living: 48% of patients reported disease interfering with daily activities; 63% with family or social life; and 37% with employment, feeling compelled to work reduced hours.6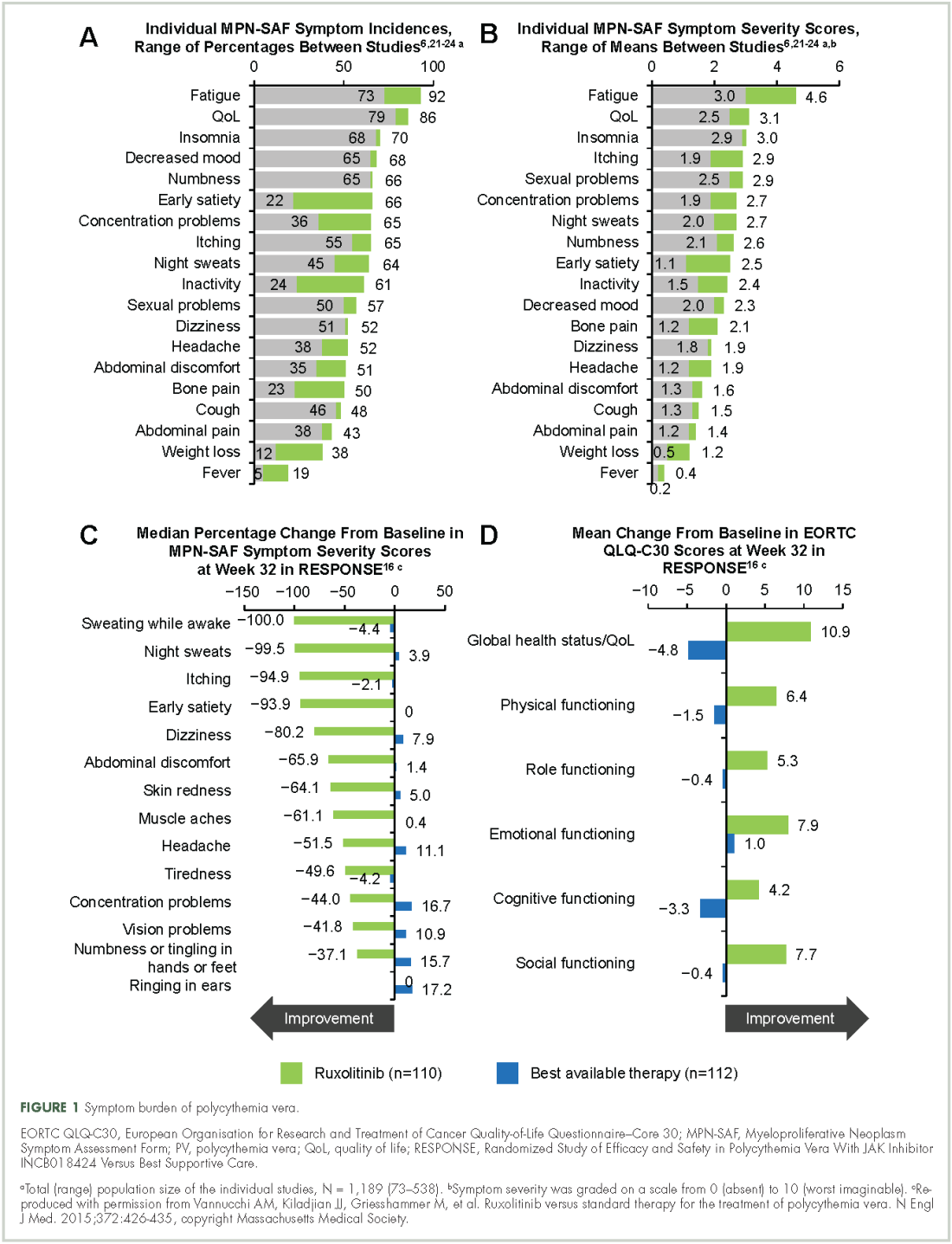
Splenomegaly is a common feature of polycythemia vera, affecting an estimated one in three patients, which may result in discomfort and early satiety.4
Identification and diagnosis
Most patients diagnosed with polycythemia vera are between the ages of 60 and 76 years,3-5 although about 25% are diagnosed before age 50.4 Tefferi and colleagues reported in a retrospective study that common features at presentation include JAK2 mutations (98%), elevated hemoglobin (73%), endogenous erythroid colony growth (73%), white blood cell count of >10.5 × 109/L (49%), and platelet count of ≥450 × 109/L (53%).4 In that same study, about a third of patients presented with a palpable spleen or polycythemia vera–related symptoms, including pruritus and vasomotor symptoms. However, many patients were asymptomatic at presentation, diagnosed incidentally by abnormal laboratory values.4 Patients can present with vascular thrombosis, occasionally involving atypical sites (eg, Budd-Chiari syndrome, other abdominal blood clots),26 thus, a heightened awareness and testing for JAK2 mutations may be appropriate in the evaluation of such individuals.
Evidence suggests many clinicians may not rigidly apply the WHO diagnostic criteria to establish a diagnosis.1,27,28 A recent retrospective claims analysis showed that only 40% of 121 patients diagnosed with polycythemia vera met the 2008 WHO diagnostic criteria, and for some patients, the diagnosis was based solely on the presence of the JAK2 V617F mutation.29 One should be aware of individuals with “masked” polycythemia vera, who may present with characteristic polycythemia vera features but have hemoglobin levels below those established by the WHO in 2008, typically owing to iron deficiency and/or a disproportionate expansion of plasma volume.30 To improve polycythemia vera diagnosis, the WHO diagnostic criteria were updated in 2016 with reduced hemoglobin diagnostic thresholds (Figure 2).1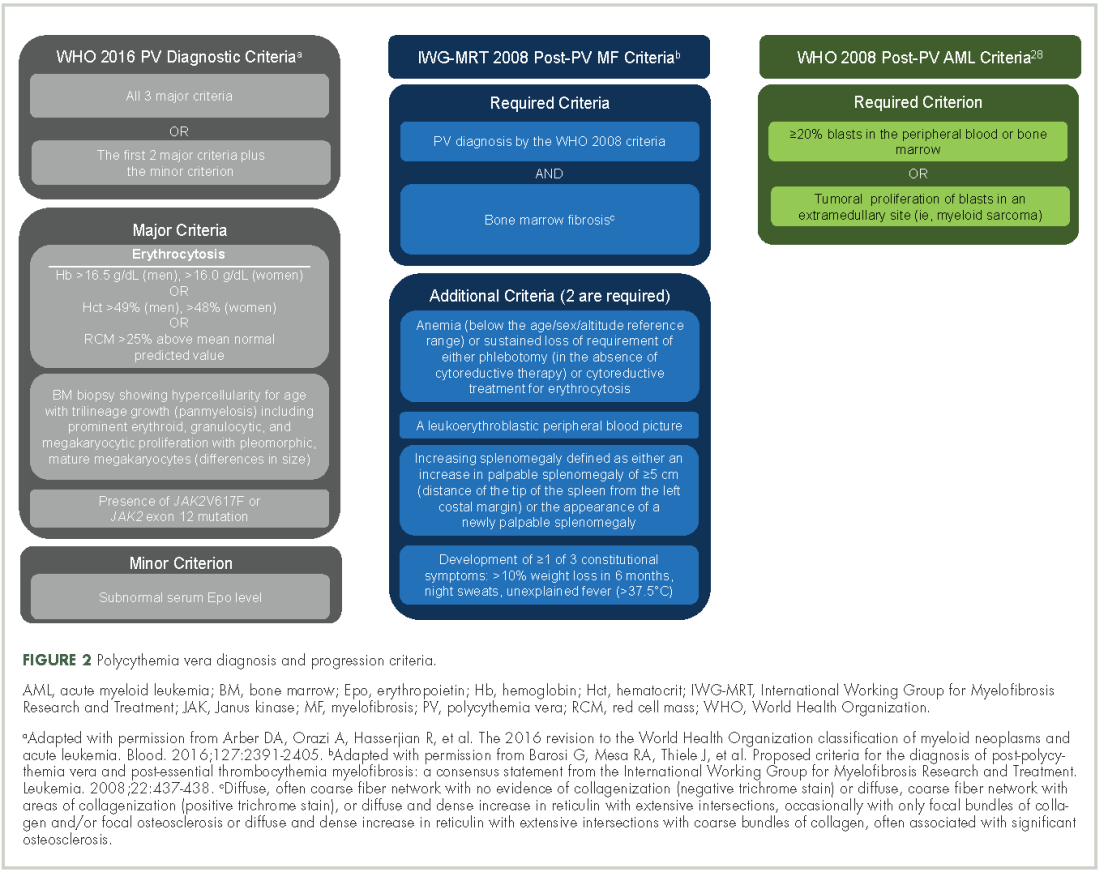
Management strategy
Treatment goals
The primary polycythemia vera–treatment goals are to reduce the risk of cardiovascular, thrombotic, and hemorrhagic events; reduce the risk of fibrotic and/or leukemic transformation; and alleviate polycythemia vera–related symptoms.11,31
Traditional treatment options
Aspirin. To reduce the risk of death from cardiovascular events, patients with polycythemia vera should receive low-dose aspirin32 and undergo phlebotomy to maintain a target hematocrit <45%, as established by the ECLAP and CYTO-PV trials (Figure 3).4,5,12,13,16,32-36 Higher doses of aspirin (ie, 325 mg 2 or more times a week) are associated with a dose-dependent increased risk of gastrointestinal bleeding.37 Low-dose aspirin is generally well tolerated; however, patients with extreme thrombocytosis may develop bleeding as a consequence of a well-described, thrombocytosis-associated acquired von Willebrand disease.11,38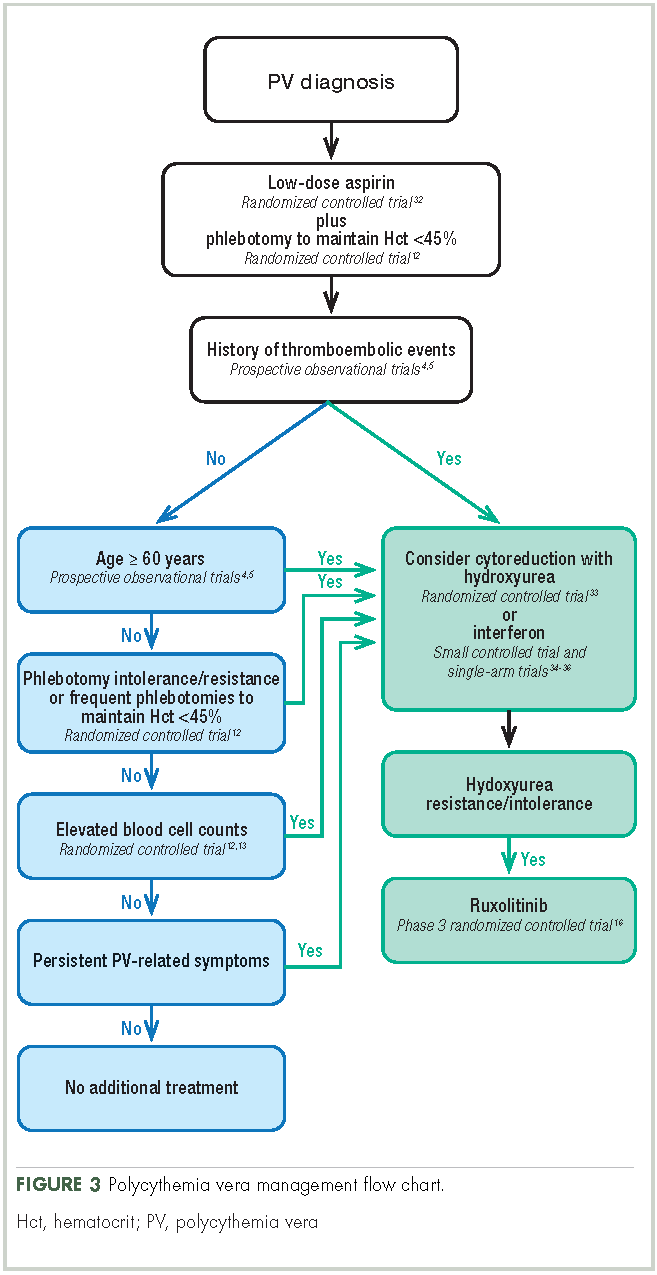
Phlebotomy. This procedure is generally tolerated by most patients, although it can occasionally engender extreme anxiety in some patients39 and may promote clinical manifestations of iron deficiency, including restless leg syndrome,40 impaired cognition, and worsening of fatigue.41 In the CYTO-PV study, 28% of patients with a target hematocrit 45%-50% discontinued phlebotomy treatment, although the percentage that discontinued because of poor tolerance was not reported.12 To avoid potential complications in patients with underlying cardiovascular disease, smaller-volume phlebotomies are often pursued.42
Cytoreductive therapy. Cytoreductive therapy with hydroxyurea or interferon (IFN) is recommended for high-risk patients (ie, those with a history of thrombosis or older than 60 years) as well as those with intolerable symptoms, progressive splenomegaly, or a burdensome phlebotomy requirement.11,31 Hydroxyurea is the typical first-line cytoreductive therapy11 based on clinical benefit,33,43 low cost, and feasibility of long-term treatment.33
Most patients benefit from long-term treatment with hydroxyurea; however, 25% develop resistance to or intolerance of hydroxyurea therapy.44 Intolerance typically manifests as leg ulcers or other mucocutaneous toxicity, gastrointestinal side effects, or fever.44 Resistance to hydroxyurea is defined as failure to achieve phlebotomy independence, persistent leukothrombocytosis or splenomegaly despite adequate doses of hydroxyurea, or inability to deliver the drug owing to dose-limiting cytopenias. The European LeukemiaNet (ELN) formally codified and published a definition of hydroxyurea resistance/intolerance45 (Table), which can be used to identify patients at high risk of poor outcomes.44 In a retrospective chart review of 261 patients with polycythemia vera, those meeting the ELN definition of hydroxyurea resistance had a 5.6-fold greater risk of mortality and a 6.8-fold increased risk of fibrotic and/or leukemic disease transformation.44
The use of IFN-α and pegylated variants are associated with clinical benefit, including normalization of blood counts, reduction of splenomegaly, symptom mitigation, and reduction in JAK2 V617F allele burden.46 However, poor tolerance46 and an inconvenient route of administration often preclude the long-term use of these agents. Adverse events associated with IFN-α include chills, depression, diarrhea, fatigue, fever, headache, musculoskeletal pain, myalgia, nausea, and weight loss.46 In clinical trials, recombinant IFN-α discontinuation rates within the first year of administration were as high as 29% and may have been dose dependent.46
Traditional treatment options may not effectively alleviate polycythemia vera–related symptoms.23,47 Two prospective studies failed to show an improvement in patient-reported MPN-SAF scores after treatment with hydroxyurea, aspirin, phlebotomy, IFN-α, busulfan, or radiophosphorus,23,47 and symptoms may worsen with the use of IFN-α.47
Allogenic transplantation. Although allogeneic transplantation is a potentially curative treatment option, it has been reserved primarily for younger patients with MPNs (age <60 years31). Furthermore, a recent systematic review concluded that overall survival was worse following allogeneic transplantation compared with a nontransplant approach (ie, phlebotomy and aspirin).48
Ruxolitinib. The oral JAK1/JAK2 inhibitor ruxolitinib has been approved by the US Food and Drug Administration (FDA) for the treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea,8 and by the European Medicines Agency (EMA) for adult patients with polycythemia vera who are resistant to or intolerant of hydroxyurea.9 Ruxolitinib is also approved by the FDA for patients with intermediate- or high-risk MF, including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF,8 and for similar patient populations by the EMA.9
Approval of ruxolitinib for the treatment of patients with polycythemia vera was based on the phase 3 randomized, open-label, multicenter RESPONSE trial in which 222 patients with polycythemia vera who met the modified ELN criteria for hydroxyurea resistance or intolerance (Table)16,31 were randomized to ruxolitinib or best available therapy (BAT). Compared with BAT, a greater proportion of patients treated with ruxolitinib achieved the primary composite endpoint of hematocrit control without the need for phlebotomy and ≥35% reduction in spleen volume by week 32 (22.7% vs 0.9%; P < .001).16,49 When looked at individually, hematocrit control and reduction in spleen size favored ruxolitinib over BAT (hematocrit control, 60.0% vs 18.8%, ruxolitinib and BAT, respectively; ≥35% reduction in spleen volume, 40.0% vs 0.9%). Furthermore, more patients receiving ruxolitinib achieved the key secondary endpoint of complete hematologic remission than did those receiving BAT (ie, normalization of blood counts; 23.6% vs 8.0%; P = .0016).16,49 Of note is that most patients who achieved primary treatment responses maintained disease control for ≥80 weeks.49
Results from RESPONSE indicate that ruxolitinib may substantially improve polycythemia vera–related symptoms. Treatment with ruxolitinib was associated with a greater improvement in nearly all symptoms evaluated by the MPN-SAF as well as greater improvements in QoL and functional measures with the EORTC QLQ-C30 trial metric compared with BAT (Figure 1).16 In addition, a post hoc exploratory analysis of RESPONSE indicated that patients receiving ruxolitinib showed a rapid normalization of abnormal iron indices at baseline, compared with those receiving BAT.50
Treatment safety and tolerability are particularly important considerations for patients with polycythemia vera, given the long natural history of the disease. In a preplanned analysis of RESPONSE at 80 weeks, 83% of patients randomized to receive treatment with ruxolitinib remained on treatment (median exposure, 111 weeks).49 Most adverse events reported in both treatment arms were grade 1/2.16,49 The most frequent nonhematologic adverse events (per 100 patient-years of exposure) in the ruxolitinib arm were headache (10.5%), diarrhea (9.7%), pruritus (9.7%), and fatigue (8.3%). The most common grade 3/4 nonhematologic adverse events (occurring at a rate of ≥0.9 per 100 patient-years of exposure) were limited to dyspnea (1.3%), abdominal pain (0.9%), headache (0.9%), and herpes zoster (0.9%).49 Hematologic adverse event rates in the ruxolitinib and BAT arms included anemia (any grade, 27.2% vs 47.6%, respectively; grade 3/4, 0.9% vs 0%), lymphopenia (27.2% vs 78.8%; 9.7% vs 27.2%), and thrombocytopenia (14.9% vs 29.9%; 2.6% vs 5.4%).49 Herpes zoster infections occurred more frequently in the ruxolitinib arm (any grade, 5.3%; grade 3/4, 0.9%) compared with the BAT arm (no herpes zoster events).49 There was a higher rate of nonmelanoma skin cancer (NMSC) in the ruxolitinib arm (4.4%), compared with the BAT arm (2.7%),49 most of which occurred in patients with a history of NMSC or precancerous skin lesions.16 Grade 1 or 2 elevations in serum lipids and cholesterol were observed with ruxolitinib but not BAT; however, subsequent effects on patient outcomes have not been determined.8,16 The rates of MF and AML transformations were 1.3% and 0.4%, respectively, in patients randomized to receive ruxolitinib,49 similar to previously published reports for patients with polycythemia vera.44
Additional insight regarding the effect of ruxolitinib on polycythemia vera–related symptoms is available from the RELIEF trial, a randomized, multicenter, double-blind, double-dummy, phase 3b clinical trial. In RELIEF, 110 patients were randomized to receive ruxolitinib or a stable dose of hydroxyurea and were then asked to record disease-related symptoms.51 Although the study failed to meet its primary endpoint (a ≥50% improvement by week 16 in MPN-SAF total symptom score for the cytokine symptom cluster [sum of individual scores for tiredness, itching, muscle aches, night sweats, and sweats while awake]), a numerically greater proportion of patients receiving ruxolitinib achieved the primary endpoint compared with those receiving hydroxyurea (43.4% and 29.6%, respectively; P = .139; odds ratio, 1.82; 95% confidence interval, 0.82-4.04). Similarly, the proportion of patients reporting a ≥50% improvement in pruritus and fatigue favored ruxolitinib over hydroxyurea (itching, 40.0% vs 26.4%; tiredness, 54.2% vs 32.0%). The safety profile for ruxolitinib was similar to that reported in the RESPONSE trial.
Possible future treatment options
Other possible treatment options for patients with polycythemia vera that are currently in clinical development include three pegylated IFN-α (PEG-IFN-α) variants and the telomerase inhibitor, imetelstat.
Pegylated interferon-α. PEG–IFN-α has the advantage of a longer plasma half-life compared with conventional IFN-α, permitting administration once per week or less often.46 Currently, three variants are under active investigation in phase 3 clinical trials: PEG–IFN-α2a (NCT01259856 and NCT01387763), PEG–IFN-α2b (NCT01387763), and AOP2014/P1101 (NCT02218047, NCT02523638, and NCT01949805).
Imetelstat. The telomerase inhibitor imetelstat is in clinical development for patients with MPNs. Clinical benefit was previously observed in patients with primary MF as well as post-polycythemia vera and post-ET MF.52 Imetelstat was evaluated in a phase 2 trial in patients with polycythemia vera or ET who required cytoreductive therapy and were resistant to or intolerant of ≥1 previous line of therapy or who refused standard therapy (NCT01243073). Results in patients with ET were published,53 however, findings from the polycythemia vera cohort have not been reported.
Community oncologist role in managing disease burden
Most patients with polycythemia vera are managed in the community setting. Consequently, the community oncologist plays a critical role in the initial diagnosis, risk stratification, patient education, and disease management.
Early disease recognition allows prompt therapeutic intervention with low-dose aspirin and phlebotomy, interventions shown to reduce the risk of cardiovascular events based on the ECLAP32 and CYTO-PV trials,12 respectively. The diagnosis of polycythemia vera is facilitated by applying the WHO diagnostic criteria (Figure 2);1 however, one should be aware of atypical presentations, including “masked” polycythemia vera,30 as well as the development of thrombosis at atypical sites.26
Optimal management strategies must include the frequent assessment of symptom burden and its effect on a patient’s QoL, with a keen awareness of the nonspecific nature of polycythemia vera–related symptoms, and the potential for patients and clinicians to minimize that effect.20 Patient-reported symptom severity and QoL should be assessed at each office visit with validated instruments, such as the MPN-SAF 10-item questionnaire.22It is important for the community oncologist to define treatment goals and implement a plan that reduces disease-associated morbidity and mortality. A critical treatment goal is to maintain hematocrit <45% by the appropriate use of phlebotomy12 and/or cytoreductive agents.12,16,46 Continued reassessment is important to identify patients with progressive disease and those who fail to achieve stated treatment goals or require an adjustment in cytoreductive therapy. Oncologists should be familiar with the concept of hydroxyurea resistance/intolerance as defined by the ELN (Table)31,45 to allow early identification of those patients who are most likely to benefit from a treatment change for continued optimal outcome.
Conclusions
Polycythemia vera is a clonal myeloproliferative neoplasm associated with significant disease-related morbidity and mortality. Appropriate management includes early diagnosis and implementation of appropriate therapy according to patient risk and therapeutic tolerance. Patients should initially receive aspirin32 and phlebotomy,12 with the goal of maintaining hematocrit <45%. Higher-risk patients and those who had inadequate disease control with phlebotomy alone require cytoreduction, typically with hydroxyurea. Although most patients will achieve adequate disease control with hydroxyurea,33,43 one in four patients will develop drug resistance or intolerance.44 Ruxolitinib is approved by the FDA and the EMA for the treatment of patients with polycythemia vera who are resistant to or intolerant of hydroxyurea. Compared with BAT, ruxolitinib is associated with improved hematocrit control, reductions in spleen size, a greater probability of blood count normalization, and improvement in polycythemia vera–related symptoms.16
Acknowledgments
Writing assistance was provided by Cory Pfeiffenberger, PhD, of Complete Healthcare Communications LLC, and was funded by Incyte Corporation, the maker of ruxolitinib.
1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405.
2. Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55:595-600.
3. Price GL, Davis KL, Karve S, Pohl G, Walgren RA. Survival patterns in United States (US) Medicare enrollees with non-CML myeloproliferative neoplasms (MPN). PLoS ONE. 2014;9:e90299.
4. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27:1874-1881.
5. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23:2224-2232.
6. Mesa R, Miller CB, Thyne M, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer. 2016;16:167.
7. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379-2390.
8. JAKAFI (ruxolitinib). Full Prescribing Information, Incyte Corporation, Wilmington, DE, USA, 2016.
9. JAKAVI (ruxolitinib). Summary of Product Characteristics, Novartis Pharma GmbH, Nuremberg, Germany, 2015.
10. Johansson P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 2006;32:171-173.
11. Vannucchi AM. How I treat polycythemia vera. Blood. 2014;124:3212-3220.
12. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368:22-33.
13. Barbui T, Masciulli A, Marfisi MR, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. 2015;126:560-561.
14. Passamonti F, Rumi E, Pietra D, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. 2010;24:1574-1579.
15. Quintás-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov. 2011;10:127-140.
16. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372:426-435.
17. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia. 2007;21:1952-1959.
18. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly-annotated essential thrombocythemia, polycythemia vera and myelofibrosis. Blood. 2014;124:2507-2513.
19. Fallah M, Kharazmi E, Sundquist J, Hemminki K. Higher risk of primary cancers after polycythaemia vera and vice versa. Br J Haematol. 2011;153:283-285.
20. Mesa R, Miller C, Thyne M, et al. Differences in treatment goals and perception of symptom burden between patients with MPNs and hematologists/oncologists in the United States: findings from the MPN landmark survey. Cancer. 2017;123:449-458.
21. Abelsson J, Andreasson B, Samuelsson J, et al. Patients with polycythemia vera have the worst impairment of quality of life among patients with newly diagnosed myeloproliferative neoplasms. Leuk Lymphoma. 2013;54:2226-2230.
22. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative Neoplasm (MPN) Symptom Assessment Form Total Symptom Score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30:4098-4103.
23. Johansson P, Mesa R, Scherber R, et al. Association between quality of life and clinical parameters in patients with myeloproliferative neoplasms. Leuk Lymphoma. 2012;53:441-444.
24. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011;118:401-408.
25. Siegel FP, Tauscher J, Petrides PE. Aquagenic pruritus in polycythemia vera: characteristics and influence on quality of life in 441 patients. Am J Hematol. 2013;88:665-669.
26. Smalberg JH, Arends LR, Valla DC, Kiladjian JJ, Janssen HL, Leebeek FW. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood. 2012;120:4921-4928.
27. Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008;22:437-438.
28. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937-951.
29. Roda P, Ferrari A, Tang X, et al. Determination of accuracy of polycythemia vera diagnoses and use of the JAK2V617F test in the diagnostic scheme. Ann Hematol. 2014;93:1467-1472.
30. Barbui T, Thiele J, Gisslinger H, et al. Masked polycythemia vera (mPV): results of an international study. Am J Hematol. 2014;89:52-54.
31. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29:761-770.
32. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350:114-124.
33. Kiladjian JJ, Chevret S, Dosquet C, Chomienne C, Rain JD. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29:3907-3913.
34. Quintás-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27:5418-5424.
35. Sacchi S, Leoni P, Liberati M, et al. A prospective comparison between treatment with phlebotomy alone and with interferon-alpha in patients with polycythemia vera. Ann Hematol. 1994;68:247-250.
36. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer. 2006;107:451-458.
37. Huang ES, Strate LL, Ho WW, Lee SS, Chan AT. Long-term use of aspirin and the risk of gastrointestinal bleeding. Am J Med. 2011;124:426-433.
38. Shetty S, Kasatkar P, Ghosh K. Pathophysiology of acquired von Willebrand disease: a concise review. Eur J Haematol. 2011;87:99-106.
39. Deacon B, Abramowitz J. Fear of needles and vasovagal reactions among phlebotomy patients. J Anxiety Disord. 2006;20:946-960.
40. Tobiasson M, Alyass B, Soderlund S, Birgegard G. High prevalence of restless legs syndrome among patients with polycytemia vera treated with venesectio. Med Oncol. 2010;27:105-107.
41. Greig AJ, Patterson AJ, Collins CE, Chalmers KA. Iron deficiency, cognition, mental health and fatigue in women of childbearing age: a systematic review. J Nutr Sci. 2013;2:e14.
42. Passamonti F. How I treat polycythemia vera. Blood. 2012;120:275-284.
43. Fruchtman SM, Mack K, Kaplan ME, Peterson P, Berk PD, Wasserman LR. From efficacy to safety: a Polycythemia Vera Study Group report on hydroxyurea in patients with polycythemia vera. Semin Hematol. 1997;34:17-23.
44. Alvarez-Lárran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119:1363-1369.
45. Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol. 2010;148:961-963.
46. Hasselbalch HC. A new era for IFN-α in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Expert Rev Hematol. 2011;4:637-655.
47. Emanuel R, Dueck AC, Kiladjian JJ, et al. Conventional therapeutic options have limited impact on MPN symptoms: insights from a prospective analysis of the MPN-SAF [abstract 366]. Presented at: European Hematology Association, June 14-17, 2012; Amsterdam, Netherlands.
48. Lacevic J, Reljic T, El Jurdi N, et al. Conservative management vs. allogeneic hematopoietic cell transplantation for polycythemia vera: a systematic review and decision-analysis. Blood. 2013;122:abstract 5372.
49. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80 week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
50. Verstovsek S, Harrison CN, Kiladjian J-J, et al. Effect of ruxolitinib on markers of iron deficiency: an analysis of the RESPONSE trial. Haematologica (EHA Annual Meeting Abstracts). 2015;100:abstract P672.
51. Mesa R, Vannucchi AM, Yacoub A, et al. The efficacy and safety of continued hydroxyurea therapy versus switching to ruxolitinib in patients with polycythemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Blood (ASH Annual Meeting Abstracts). 2014;124:abstract 3168.
52. Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373:908-919.
53. Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015;373:920-928.
Polycythemia vera, classified as a myeloproliferative neoplasm (MPN) and characterized by uncontrolled, clonal, myeloid expansion with predominant erythrocytosis,1 affects about 100,000 individuals in the United States.2 It is a chronic and burdensome disease associated with shortened survival.3 Patients are at an increased risk of cardiovascular events, solid tumors, and transformation to myelofibrosis (MF) and/or acute myeloid leukemia (AML).4,5 Furthermore, patients generally have a reduced quality of life (QoL) stemming from prevalent and occasionally severe polycythemia vera–related signs and symptoms, including fatigue, pruritus, and splenomegaly.6 In general, the classical Philadelphia chromosome-negative MPNs are associated with driver mutations in the following three genes: Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia virus oncogene (MPL).7 Almost all patients with polycythemia vera have an activating mutation in the cytoplasmic signal transduction protein JAK2.4 Patients with essential thrombocythemia (ET) or MF can have mutations in JAK2, CALR, or MPL. However, CALR and MPL mutations are absent or exceedingly rare in patients with polycythemia vera.7 Diagnosis can be challenging and is currently based on 2016 World Health Organization (WHO) diagnostic criteria.1
Management strategies include the use of aspirin, phlebotomy, and cytoreductive therapy. Ruxolitinib is a newer treatment option available for patients with polycythemia vera who are either resistant to or intolerant of hydroxyurea8,9— a population that previously had few treatment options. It is important for community oncologists and other treating clinicians to understand current diagnostic strategy and management options based on established guidelines, recent clinical evidence, and regulatory updates.
Search and selection process for research sources
In September 2016, we searched PubMed for articles published since 2006 with polycythemia vera included in the abstract or title. The initial 1,730 publications were screened by eye to select 46 key articles that guide current management of polycythemia vera. Four studies published before 2006 were also included based on their continued relevance.
Epidemiology and pathophysiology
Based on a meta-analysis of patients from Europe and the United States, the annual incidence of polycythemia vera estimated to be between 0.7 and 2.6 per 100,000 people.10 The age-adjusted prevalence of polycythemia vera in the United States is about 45-57 per 100,000 people,2 however, the true prevalence might be considerably greater.
Patients with polycythemia vera are at increased risk of cardiovascular events, thrombosis, and death.3-5 Risk is highest among patients older than 60 years or with a history of thrombosis.11 Uncontrolled myeloproliferation has also been identified as a risk factor for cardiovascular mortality and thrombosis. This was demonstrated in the prospective Cytoreductive Therapy in Polycythemia Vera (CYTO-PV) trial, which reported more cardiovascular events in patients with hematocrit levels of 45%-50%, compared with those whose hematocrit levels were <45%.12 In addition, retrospective data suggest leukocytosis is a potential risk factor for thromboembolic events and poor outcomes.13
Dysregulated JAK2 signaling is the principal driver of polycythemia vera pathophysiology. About 95% of patients with polycythemia vera will have an identifiable JAK2 V617F exon 14 mutation, with an additional 3%-5% demonstrating a JAK2 exon 12 mutation.4,14 Under physiologic conditions, JAK2 interacts with the STAT family of signal transduction proteins and serves as an important regulator of normal hematopoiesis.15 Mutated, constitutively activated JAK2 signaling promotes the various polycythemia vera disease manifestations, including excessive myeloproliferation, splenomegaly,15 and constitutional symptoms.14,16,17
Burden of disease for the individual
Mortality
Patients with polycythemia vera have an increased risk of mortality compared with an unaffected, age- and gender-matched cohort of the general population.3 A retrospective study of Medicare patients with polycythemia vera (mean age at diagnosis, 76.1 years) reported a median survival of 5.4 years, compared with 8.7 years for a matched cohort.3 A second retrospective study reported a median survival of 13.5 years (median age at diagnosis, 64 years; median follow-up time, 11.8 years).18
Leading causes of death for patients with polycythemia vera include cardiovascular and thrombotic events, the development of secondary solid tumors, and disease transformation to MF and/or AML. In the prospective European Collaboration on Low-Dose Aspirin in Polycythemia Vera (ECLAP) study of 1,638 patients, 45% of deaths (74/164) resulted from cardiovascular causes (1.7 per 100 patient-years).5 Thirteen percent of deaths were related to either leukemic or myelofibrotic transformation, and 20% of deaths were attributed to secondary solid tumors.5 In a retrospective analysis of 1,545 patients with polycythemia vera followed for a median of 6.9 years after diagnosis, 347 had died, primarily from acute leukemia (10%), secondary malignancies (10%), and thrombotic events (9%).4 Arterial and venous thrombotic events occurred in 12% and 9% of patients, respectively, with disease transformation to MF and AML occurring in 9% and 3% of patients. Further support of an increased risk of secondary malignancies comes from a retrospective analysis of a large Swedish cancer registry (1958–2006) that found an increased risk of secondary endocrine, renal, and skin malignancies; MF; and leukemia among patients with polycythemia vera.19
Symptoms and quality of life
Symptoms of polycythemia vera vary in severity, and patients often fail to attribute symptoms to the disease.20 Moreover, clinicians may underestimate a patient’s true disease burden or the effect it has on QoL.20 Point-of-care metrics, such as the MPN Symptom Assessment Form (MPN-SAF), were developed to aid in identifying and grading symptom burden. Studies using this metric have reported fatigue as the most common and most severe symptom (incidence, 73%-92%), with a variety of other symptoms also affecting a majority of patients (Figure 1).6,21-24 Although fatigue, pruritus, and a higher MPN-SAF total symptom score are significantly correlated with reduced QoL,22,25 the recent MPN Landmark survey suggests that even patients with low symptom severity scores have a reduction in their QoL.6 This study also highlighted that polycythemia vera can adversely affect multiple aspects of daily living: 48% of patients reported disease interfering with daily activities; 63% with family or social life; and 37% with employment, feeling compelled to work reduced hours.6
Splenomegaly is a common feature of polycythemia vera, affecting an estimated one in three patients, which may result in discomfort and early satiety.4
Identification and diagnosis
Most patients diagnosed with polycythemia vera are between the ages of 60 and 76 years,3-5 although about 25% are diagnosed before age 50.4 Tefferi and colleagues reported in a retrospective study that common features at presentation include JAK2 mutations (98%), elevated hemoglobin (73%), endogenous erythroid colony growth (73%), white blood cell count of >10.5 × 109/L (49%), and platelet count of ≥450 × 109/L (53%).4 In that same study, about a third of patients presented with a palpable spleen or polycythemia vera–related symptoms, including pruritus and vasomotor symptoms. However, many patients were asymptomatic at presentation, diagnosed incidentally by abnormal laboratory values.4 Patients can present with vascular thrombosis, occasionally involving atypical sites (eg, Budd-Chiari syndrome, other abdominal blood clots),26 thus, a heightened awareness and testing for JAK2 mutations may be appropriate in the evaluation of such individuals.
Evidence suggests many clinicians may not rigidly apply the WHO diagnostic criteria to establish a diagnosis.1,27,28 A recent retrospective claims analysis showed that only 40% of 121 patients diagnosed with polycythemia vera met the 2008 WHO diagnostic criteria, and for some patients, the diagnosis was based solely on the presence of the JAK2 V617F mutation.29 One should be aware of individuals with “masked” polycythemia vera, who may present with characteristic polycythemia vera features but have hemoglobin levels below those established by the WHO in 2008, typically owing to iron deficiency and/or a disproportionate expansion of plasma volume.30 To improve polycythemia vera diagnosis, the WHO diagnostic criteria were updated in 2016 with reduced hemoglobin diagnostic thresholds (Figure 2).1
Management strategy
Treatment goals
The primary polycythemia vera–treatment goals are to reduce the risk of cardiovascular, thrombotic, and hemorrhagic events; reduce the risk of fibrotic and/or leukemic transformation; and alleviate polycythemia vera–related symptoms.11,31
Traditional treatment options
Aspirin. To reduce the risk of death from cardiovascular events, patients with polycythemia vera should receive low-dose aspirin32 and undergo phlebotomy to maintain a target hematocrit <45%, as established by the ECLAP and CYTO-PV trials (Figure 3).4,5,12,13,16,32-36 Higher doses of aspirin (ie, 325 mg 2 or more times a week) are associated with a dose-dependent increased risk of gastrointestinal bleeding.37 Low-dose aspirin is generally well tolerated; however, patients with extreme thrombocytosis may develop bleeding as a consequence of a well-described, thrombocytosis-associated acquired von Willebrand disease.11,38
Phlebotomy. This procedure is generally tolerated by most patients, although it can occasionally engender extreme anxiety in some patients39 and may promote clinical manifestations of iron deficiency, including restless leg syndrome,40 impaired cognition, and worsening of fatigue.41 In the CYTO-PV study, 28% of patients with a target hematocrit 45%-50% discontinued phlebotomy treatment, although the percentage that discontinued because of poor tolerance was not reported.12 To avoid potential complications in patients with underlying cardiovascular disease, smaller-volume phlebotomies are often pursued.42
Cytoreductive therapy. Cytoreductive therapy with hydroxyurea or interferon (IFN) is recommended for high-risk patients (ie, those with a history of thrombosis or older than 60 years) as well as those with intolerable symptoms, progressive splenomegaly, or a burdensome phlebotomy requirement.11,31 Hydroxyurea is the typical first-line cytoreductive therapy11 based on clinical benefit,33,43 low cost, and feasibility of long-term treatment.33
Most patients benefit from long-term treatment with hydroxyurea; however, 25% develop resistance to or intolerance of hydroxyurea therapy.44 Intolerance typically manifests as leg ulcers or other mucocutaneous toxicity, gastrointestinal side effects, or fever.44 Resistance to hydroxyurea is defined as failure to achieve phlebotomy independence, persistent leukothrombocytosis or splenomegaly despite adequate doses of hydroxyurea, or inability to deliver the drug owing to dose-limiting cytopenias. The European LeukemiaNet (ELN) formally codified and published a definition of hydroxyurea resistance/intolerance45 (Table), which can be used to identify patients at high risk of poor outcomes.44 In a retrospective chart review of 261 patients with polycythemia vera, those meeting the ELN definition of hydroxyurea resistance had a 5.6-fold greater risk of mortality and a 6.8-fold increased risk of fibrotic and/or leukemic disease transformation.44
The use of IFN-α and pegylated variants are associated with clinical benefit, including normalization of blood counts, reduction of splenomegaly, symptom mitigation, and reduction in JAK2 V617F allele burden.46 However, poor tolerance46 and an inconvenient route of administration often preclude the long-term use of these agents. Adverse events associated with IFN-α include chills, depression, diarrhea, fatigue, fever, headache, musculoskeletal pain, myalgia, nausea, and weight loss.46 In clinical trials, recombinant IFN-α discontinuation rates within the first year of administration were as high as 29% and may have been dose dependent.46
Traditional treatment options may not effectively alleviate polycythemia vera–related symptoms.23,47 Two prospective studies failed to show an improvement in patient-reported MPN-SAF scores after treatment with hydroxyurea, aspirin, phlebotomy, IFN-α, busulfan, or radiophosphorus,23,47 and symptoms may worsen with the use of IFN-α.47
Allogenic transplantation. Although allogeneic transplantation is a potentially curative treatment option, it has been reserved primarily for younger patients with MPNs (age <60 years31). Furthermore, a recent systematic review concluded that overall survival was worse following allogeneic transplantation compared with a nontransplant approach (ie, phlebotomy and aspirin).48
Ruxolitinib. The oral JAK1/JAK2 inhibitor ruxolitinib has been approved by the US Food and Drug Administration (FDA) for the treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea,8 and by the European Medicines Agency (EMA) for adult patients with polycythemia vera who are resistant to or intolerant of hydroxyurea.9 Ruxolitinib is also approved by the FDA for patients with intermediate- or high-risk MF, including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF,8 and for similar patient populations by the EMA.9
Approval of ruxolitinib for the treatment of patients with polycythemia vera was based on the phase 3 randomized, open-label, multicenter RESPONSE trial in which 222 patients with polycythemia vera who met the modified ELN criteria for hydroxyurea resistance or intolerance (Table)16,31 were randomized to ruxolitinib or best available therapy (BAT). Compared with BAT, a greater proportion of patients treated with ruxolitinib achieved the primary composite endpoint of hematocrit control without the need for phlebotomy and ≥35% reduction in spleen volume by week 32 (22.7% vs 0.9%; P < .001).16,49 When looked at individually, hematocrit control and reduction in spleen size favored ruxolitinib over BAT (hematocrit control, 60.0% vs 18.8%, ruxolitinib and BAT, respectively; ≥35% reduction in spleen volume, 40.0% vs 0.9%). Furthermore, more patients receiving ruxolitinib achieved the key secondary endpoint of complete hematologic remission than did those receiving BAT (ie, normalization of blood counts; 23.6% vs 8.0%; P = .0016).16,49 Of note is that most patients who achieved primary treatment responses maintained disease control for ≥80 weeks.49
Results from RESPONSE indicate that ruxolitinib may substantially improve polycythemia vera–related symptoms. Treatment with ruxolitinib was associated with a greater improvement in nearly all symptoms evaluated by the MPN-SAF as well as greater improvements in QoL and functional measures with the EORTC QLQ-C30 trial metric compared with BAT (Figure 1).16 In addition, a post hoc exploratory analysis of RESPONSE indicated that patients receiving ruxolitinib showed a rapid normalization of abnormal iron indices at baseline, compared with those receiving BAT.50
Treatment safety and tolerability are particularly important considerations for patients with polycythemia vera, given the long natural history of the disease. In a preplanned analysis of RESPONSE at 80 weeks, 83% of patients randomized to receive treatment with ruxolitinib remained on treatment (median exposure, 111 weeks).49 Most adverse events reported in both treatment arms were grade 1/2.16,49 The most frequent nonhematologic adverse events (per 100 patient-years of exposure) in the ruxolitinib arm were headache (10.5%), diarrhea (9.7%), pruritus (9.7%), and fatigue (8.3%). The most common grade 3/4 nonhematologic adverse events (occurring at a rate of ≥0.9 per 100 patient-years of exposure) were limited to dyspnea (1.3%), abdominal pain (0.9%), headache (0.9%), and herpes zoster (0.9%).49 Hematologic adverse event rates in the ruxolitinib and BAT arms included anemia (any grade, 27.2% vs 47.6%, respectively; grade 3/4, 0.9% vs 0%), lymphopenia (27.2% vs 78.8%; 9.7% vs 27.2%), and thrombocytopenia (14.9% vs 29.9%; 2.6% vs 5.4%).49 Herpes zoster infections occurred more frequently in the ruxolitinib arm (any grade, 5.3%; grade 3/4, 0.9%) compared with the BAT arm (no herpes zoster events).49 There was a higher rate of nonmelanoma skin cancer (NMSC) in the ruxolitinib arm (4.4%), compared with the BAT arm (2.7%),49 most of which occurred in patients with a history of NMSC or precancerous skin lesions.16 Grade 1 or 2 elevations in serum lipids and cholesterol were observed with ruxolitinib but not BAT; however, subsequent effects on patient outcomes have not been determined.8,16 The rates of MF and AML transformations were 1.3% and 0.4%, respectively, in patients randomized to receive ruxolitinib,49 similar to previously published reports for patients with polycythemia vera.44
Additional insight regarding the effect of ruxolitinib on polycythemia vera–related symptoms is available from the RELIEF trial, a randomized, multicenter, double-blind, double-dummy, phase 3b clinical trial. In RELIEF, 110 patients were randomized to receive ruxolitinib or a stable dose of hydroxyurea and were then asked to record disease-related symptoms.51 Although the study failed to meet its primary endpoint (a ≥50% improvement by week 16 in MPN-SAF total symptom score for the cytokine symptom cluster [sum of individual scores for tiredness, itching, muscle aches, night sweats, and sweats while awake]), a numerically greater proportion of patients receiving ruxolitinib achieved the primary endpoint compared with those receiving hydroxyurea (43.4% and 29.6%, respectively; P = .139; odds ratio, 1.82; 95% confidence interval, 0.82-4.04). Similarly, the proportion of patients reporting a ≥50% improvement in pruritus and fatigue favored ruxolitinib over hydroxyurea (itching, 40.0% vs 26.4%; tiredness, 54.2% vs 32.0%). The safety profile for ruxolitinib was similar to that reported in the RESPONSE trial.
Possible future treatment options
Other possible treatment options for patients with polycythemia vera that are currently in clinical development include three pegylated IFN-α (PEG-IFN-α) variants and the telomerase inhibitor, imetelstat.
Pegylated interferon-α. PEG–IFN-α has the advantage of a longer plasma half-life compared with conventional IFN-α, permitting administration once per week or less often.46 Currently, three variants are under active investigation in phase 3 clinical trials: PEG–IFN-α2a (NCT01259856 and NCT01387763), PEG–IFN-α2b (NCT01387763), and AOP2014/P1101 (NCT02218047, NCT02523638, and NCT01949805).
Imetelstat. The telomerase inhibitor imetelstat is in clinical development for patients with MPNs. Clinical benefit was previously observed in patients with primary MF as well as post-polycythemia vera and post-ET MF.52 Imetelstat was evaluated in a phase 2 trial in patients with polycythemia vera or ET who required cytoreductive therapy and were resistant to or intolerant of ≥1 previous line of therapy or who refused standard therapy (NCT01243073). Results in patients with ET were published,53 however, findings from the polycythemia vera cohort have not been reported.
Community oncologist role in managing disease burden
Most patients with polycythemia vera are managed in the community setting. Consequently, the community oncologist plays a critical role in the initial diagnosis, risk stratification, patient education, and disease management.
Early disease recognition allows prompt therapeutic intervention with low-dose aspirin and phlebotomy, interventions shown to reduce the risk of cardiovascular events based on the ECLAP32 and CYTO-PV trials,12 respectively. The diagnosis of polycythemia vera is facilitated by applying the WHO diagnostic criteria (Figure 2);1 however, one should be aware of atypical presentations, including “masked” polycythemia vera,30 as well as the development of thrombosis at atypical sites.26
Optimal management strategies must include the frequent assessment of symptom burden and its effect on a patient’s QoL, with a keen awareness of the nonspecific nature of polycythemia vera–related symptoms, and the potential for patients and clinicians to minimize that effect.20 Patient-reported symptom severity and QoL should be assessed at each office visit with validated instruments, such as the MPN-SAF 10-item questionnaire.22It is important for the community oncologist to define treatment goals and implement a plan that reduces disease-associated morbidity and mortality. A critical treatment goal is to maintain hematocrit <45% by the appropriate use of phlebotomy12 and/or cytoreductive agents.12,16,46 Continued reassessment is important to identify patients with progressive disease and those who fail to achieve stated treatment goals or require an adjustment in cytoreductive therapy. Oncologists should be familiar with the concept of hydroxyurea resistance/intolerance as defined by the ELN (Table)31,45 to allow early identification of those patients who are most likely to benefit from a treatment change for continued optimal outcome.
Conclusions
Polycythemia vera is a clonal myeloproliferative neoplasm associated with significant disease-related morbidity and mortality. Appropriate management includes early diagnosis and implementation of appropriate therapy according to patient risk and therapeutic tolerance. Patients should initially receive aspirin32 and phlebotomy,12 with the goal of maintaining hematocrit <45%. Higher-risk patients and those who had inadequate disease control with phlebotomy alone require cytoreduction, typically with hydroxyurea. Although most patients will achieve adequate disease control with hydroxyurea,33,43 one in four patients will develop drug resistance or intolerance.44 Ruxolitinib is approved by the FDA and the EMA for the treatment of patients with polycythemia vera who are resistant to or intolerant of hydroxyurea. Compared with BAT, ruxolitinib is associated with improved hematocrit control, reductions in spleen size, a greater probability of blood count normalization, and improvement in polycythemia vera–related symptoms.16
Acknowledgments
Writing assistance was provided by Cory Pfeiffenberger, PhD, of Complete Healthcare Communications LLC, and was funded by Incyte Corporation, the maker of ruxolitinib.
Polycythemia vera, classified as a myeloproliferative neoplasm (MPN) and characterized by uncontrolled, clonal, myeloid expansion with predominant erythrocytosis,1 affects about 100,000 individuals in the United States.2 It is a chronic and burdensome disease associated with shortened survival.3 Patients are at an increased risk of cardiovascular events, solid tumors, and transformation to myelofibrosis (MF) and/or acute myeloid leukemia (AML).4,5 Furthermore, patients generally have a reduced quality of life (QoL) stemming from prevalent and occasionally severe polycythemia vera–related signs and symptoms, including fatigue, pruritus, and splenomegaly.6 In general, the classical Philadelphia chromosome-negative MPNs are associated with driver mutations in the following three genes: Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia virus oncogene (MPL).7 Almost all patients with polycythemia vera have an activating mutation in the cytoplasmic signal transduction protein JAK2.4 Patients with essential thrombocythemia (ET) or MF can have mutations in JAK2, CALR, or MPL. However, CALR and MPL mutations are absent or exceedingly rare in patients with polycythemia vera.7 Diagnosis can be challenging and is currently based on 2016 World Health Organization (WHO) diagnostic criteria.1
Management strategies include the use of aspirin, phlebotomy, and cytoreductive therapy. Ruxolitinib is a newer treatment option available for patients with polycythemia vera who are either resistant to or intolerant of hydroxyurea8,9— a population that previously had few treatment options. It is important for community oncologists and other treating clinicians to understand current diagnostic strategy and management options based on established guidelines, recent clinical evidence, and regulatory updates.
Search and selection process for research sources
In September 2016, we searched PubMed for articles published since 2006 with polycythemia vera included in the abstract or title. The initial 1,730 publications were screened by eye to select 46 key articles that guide current management of polycythemia vera. Four studies published before 2006 were also included based on their continued relevance.
Epidemiology and pathophysiology
Based on a meta-analysis of patients from Europe and the United States, the annual incidence of polycythemia vera estimated to be between 0.7 and 2.6 per 100,000 people.10 The age-adjusted prevalence of polycythemia vera in the United States is about 45-57 per 100,000 people,2 however, the true prevalence might be considerably greater.
Patients with polycythemia vera are at increased risk of cardiovascular events, thrombosis, and death.3-5 Risk is highest among patients older than 60 years or with a history of thrombosis.11 Uncontrolled myeloproliferation has also been identified as a risk factor for cardiovascular mortality and thrombosis. This was demonstrated in the prospective Cytoreductive Therapy in Polycythemia Vera (CYTO-PV) trial, which reported more cardiovascular events in patients with hematocrit levels of 45%-50%, compared with those whose hematocrit levels were <45%.12 In addition, retrospective data suggest leukocytosis is a potential risk factor for thromboembolic events and poor outcomes.13
Dysregulated JAK2 signaling is the principal driver of polycythemia vera pathophysiology. About 95% of patients with polycythemia vera will have an identifiable JAK2 V617F exon 14 mutation, with an additional 3%-5% demonstrating a JAK2 exon 12 mutation.4,14 Under physiologic conditions, JAK2 interacts with the STAT family of signal transduction proteins and serves as an important regulator of normal hematopoiesis.15 Mutated, constitutively activated JAK2 signaling promotes the various polycythemia vera disease manifestations, including excessive myeloproliferation, splenomegaly,15 and constitutional symptoms.14,16,17
Burden of disease for the individual
Mortality
Patients with polycythemia vera have an increased risk of mortality compared with an unaffected, age- and gender-matched cohort of the general population.3 A retrospective study of Medicare patients with polycythemia vera (mean age at diagnosis, 76.1 years) reported a median survival of 5.4 years, compared with 8.7 years for a matched cohort.3 A second retrospective study reported a median survival of 13.5 years (median age at diagnosis, 64 years; median follow-up time, 11.8 years).18
Leading causes of death for patients with polycythemia vera include cardiovascular and thrombotic events, the development of secondary solid tumors, and disease transformation to MF and/or AML. In the prospective European Collaboration on Low-Dose Aspirin in Polycythemia Vera (ECLAP) study of 1,638 patients, 45% of deaths (74/164) resulted from cardiovascular causes (1.7 per 100 patient-years).5 Thirteen percent of deaths were related to either leukemic or myelofibrotic transformation, and 20% of deaths were attributed to secondary solid tumors.5 In a retrospective analysis of 1,545 patients with polycythemia vera followed for a median of 6.9 years after diagnosis, 347 had died, primarily from acute leukemia (10%), secondary malignancies (10%), and thrombotic events (9%).4 Arterial and venous thrombotic events occurred in 12% and 9% of patients, respectively, with disease transformation to MF and AML occurring in 9% and 3% of patients. Further support of an increased risk of secondary malignancies comes from a retrospective analysis of a large Swedish cancer registry (1958–2006) that found an increased risk of secondary endocrine, renal, and skin malignancies; MF; and leukemia among patients with polycythemia vera.19
Symptoms and quality of life
Symptoms of polycythemia vera vary in severity, and patients often fail to attribute symptoms to the disease.20 Moreover, clinicians may underestimate a patient’s true disease burden or the effect it has on QoL.20 Point-of-care metrics, such as the MPN Symptom Assessment Form (MPN-SAF), were developed to aid in identifying and grading symptom burden. Studies using this metric have reported fatigue as the most common and most severe symptom (incidence, 73%-92%), with a variety of other symptoms also affecting a majority of patients (Figure 1).6,21-24 Although fatigue, pruritus, and a higher MPN-SAF total symptom score are significantly correlated with reduced QoL,22,25 the recent MPN Landmark survey suggests that even patients with low symptom severity scores have a reduction in their QoL.6 This study also highlighted that polycythemia vera can adversely affect multiple aspects of daily living: 48% of patients reported disease interfering with daily activities; 63% with family or social life; and 37% with employment, feeling compelled to work reduced hours.6
Splenomegaly is a common feature of polycythemia vera, affecting an estimated one in three patients, which may result in discomfort and early satiety.4
Identification and diagnosis
Most patients diagnosed with polycythemia vera are between the ages of 60 and 76 years,3-5 although about 25% are diagnosed before age 50.4 Tefferi and colleagues reported in a retrospective study that common features at presentation include JAK2 mutations (98%), elevated hemoglobin (73%), endogenous erythroid colony growth (73%), white blood cell count of >10.5 × 109/L (49%), and platelet count of ≥450 × 109/L (53%).4 In that same study, about a third of patients presented with a palpable spleen or polycythemia vera–related symptoms, including pruritus and vasomotor symptoms. However, many patients were asymptomatic at presentation, diagnosed incidentally by abnormal laboratory values.4 Patients can present with vascular thrombosis, occasionally involving atypical sites (eg, Budd-Chiari syndrome, other abdominal blood clots),26 thus, a heightened awareness and testing for JAK2 mutations may be appropriate in the evaluation of such individuals.
Evidence suggests many clinicians may not rigidly apply the WHO diagnostic criteria to establish a diagnosis.1,27,28 A recent retrospective claims analysis showed that only 40% of 121 patients diagnosed with polycythemia vera met the 2008 WHO diagnostic criteria, and for some patients, the diagnosis was based solely on the presence of the JAK2 V617F mutation.29 One should be aware of individuals with “masked” polycythemia vera, who may present with characteristic polycythemia vera features but have hemoglobin levels below those established by the WHO in 2008, typically owing to iron deficiency and/or a disproportionate expansion of plasma volume.30 To improve polycythemia vera diagnosis, the WHO diagnostic criteria were updated in 2016 with reduced hemoglobin diagnostic thresholds (Figure 2).1
Management strategy
Treatment goals
The primary polycythemia vera–treatment goals are to reduce the risk of cardiovascular, thrombotic, and hemorrhagic events; reduce the risk of fibrotic and/or leukemic transformation; and alleviate polycythemia vera–related symptoms.11,31
Traditional treatment options
Aspirin. To reduce the risk of death from cardiovascular events, patients with polycythemia vera should receive low-dose aspirin32 and undergo phlebotomy to maintain a target hematocrit <45%, as established by the ECLAP and CYTO-PV trials (Figure 3).4,5,12,13,16,32-36 Higher doses of aspirin (ie, 325 mg 2 or more times a week) are associated with a dose-dependent increased risk of gastrointestinal bleeding.37 Low-dose aspirin is generally well tolerated; however, patients with extreme thrombocytosis may develop bleeding as a consequence of a well-described, thrombocytosis-associated acquired von Willebrand disease.11,38
Phlebotomy. This procedure is generally tolerated by most patients, although it can occasionally engender extreme anxiety in some patients39 and may promote clinical manifestations of iron deficiency, including restless leg syndrome,40 impaired cognition, and worsening of fatigue.41 In the CYTO-PV study, 28% of patients with a target hematocrit 45%-50% discontinued phlebotomy treatment, although the percentage that discontinued because of poor tolerance was not reported.12 To avoid potential complications in patients with underlying cardiovascular disease, smaller-volume phlebotomies are often pursued.42
Cytoreductive therapy. Cytoreductive therapy with hydroxyurea or interferon (IFN) is recommended for high-risk patients (ie, those with a history of thrombosis or older than 60 years) as well as those with intolerable symptoms, progressive splenomegaly, or a burdensome phlebotomy requirement.11,31 Hydroxyurea is the typical first-line cytoreductive therapy11 based on clinical benefit,33,43 low cost, and feasibility of long-term treatment.33
Most patients benefit from long-term treatment with hydroxyurea; however, 25% develop resistance to or intolerance of hydroxyurea therapy.44 Intolerance typically manifests as leg ulcers or other mucocutaneous toxicity, gastrointestinal side effects, or fever.44 Resistance to hydroxyurea is defined as failure to achieve phlebotomy independence, persistent leukothrombocytosis or splenomegaly despite adequate doses of hydroxyurea, or inability to deliver the drug owing to dose-limiting cytopenias. The European LeukemiaNet (ELN) formally codified and published a definition of hydroxyurea resistance/intolerance45 (Table), which can be used to identify patients at high risk of poor outcomes.44 In a retrospective chart review of 261 patients with polycythemia vera, those meeting the ELN definition of hydroxyurea resistance had a 5.6-fold greater risk of mortality and a 6.8-fold increased risk of fibrotic and/or leukemic disease transformation.44
The use of IFN-α and pegylated variants are associated with clinical benefit, including normalization of blood counts, reduction of splenomegaly, symptom mitigation, and reduction in JAK2 V617F allele burden.46 However, poor tolerance46 and an inconvenient route of administration often preclude the long-term use of these agents. Adverse events associated with IFN-α include chills, depression, diarrhea, fatigue, fever, headache, musculoskeletal pain, myalgia, nausea, and weight loss.46 In clinical trials, recombinant IFN-α discontinuation rates within the first year of administration were as high as 29% and may have been dose dependent.46
Traditional treatment options may not effectively alleviate polycythemia vera–related symptoms.23,47 Two prospective studies failed to show an improvement in patient-reported MPN-SAF scores after treatment with hydroxyurea, aspirin, phlebotomy, IFN-α, busulfan, or radiophosphorus,23,47 and symptoms may worsen with the use of IFN-α.47
Allogenic transplantation. Although allogeneic transplantation is a potentially curative treatment option, it has been reserved primarily for younger patients with MPNs (age <60 years31). Furthermore, a recent systematic review concluded that overall survival was worse following allogeneic transplantation compared with a nontransplant approach (ie, phlebotomy and aspirin).48
Ruxolitinib. The oral JAK1/JAK2 inhibitor ruxolitinib has been approved by the US Food and Drug Administration (FDA) for the treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea,8 and by the European Medicines Agency (EMA) for adult patients with polycythemia vera who are resistant to or intolerant of hydroxyurea.9 Ruxolitinib is also approved by the FDA for patients with intermediate- or high-risk MF, including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF,8 and for similar patient populations by the EMA.9
Approval of ruxolitinib for the treatment of patients with polycythemia vera was based on the phase 3 randomized, open-label, multicenter RESPONSE trial in which 222 patients with polycythemia vera who met the modified ELN criteria for hydroxyurea resistance or intolerance (Table)16,31 were randomized to ruxolitinib or best available therapy (BAT). Compared with BAT, a greater proportion of patients treated with ruxolitinib achieved the primary composite endpoint of hematocrit control without the need for phlebotomy and ≥35% reduction in spleen volume by week 32 (22.7% vs 0.9%; P < .001).16,49 When looked at individually, hematocrit control and reduction in spleen size favored ruxolitinib over BAT (hematocrit control, 60.0% vs 18.8%, ruxolitinib and BAT, respectively; ≥35% reduction in spleen volume, 40.0% vs 0.9%). Furthermore, more patients receiving ruxolitinib achieved the key secondary endpoint of complete hematologic remission than did those receiving BAT (ie, normalization of blood counts; 23.6% vs 8.0%; P = .0016).16,49 Of note is that most patients who achieved primary treatment responses maintained disease control for ≥80 weeks.49
Results from RESPONSE indicate that ruxolitinib may substantially improve polycythemia vera–related symptoms. Treatment with ruxolitinib was associated with a greater improvement in nearly all symptoms evaluated by the MPN-SAF as well as greater improvements in QoL and functional measures with the EORTC QLQ-C30 trial metric compared with BAT (Figure 1).16 In addition, a post hoc exploratory analysis of RESPONSE indicated that patients receiving ruxolitinib showed a rapid normalization of abnormal iron indices at baseline, compared with those receiving BAT.50
Treatment safety and tolerability are particularly important considerations for patients with polycythemia vera, given the long natural history of the disease. In a preplanned analysis of RESPONSE at 80 weeks, 83% of patients randomized to receive treatment with ruxolitinib remained on treatment (median exposure, 111 weeks).49 Most adverse events reported in both treatment arms were grade 1/2.16,49 The most frequent nonhematologic adverse events (per 100 patient-years of exposure) in the ruxolitinib arm were headache (10.5%), diarrhea (9.7%), pruritus (9.7%), and fatigue (8.3%). The most common grade 3/4 nonhematologic adverse events (occurring at a rate of ≥0.9 per 100 patient-years of exposure) were limited to dyspnea (1.3%), abdominal pain (0.9%), headache (0.9%), and herpes zoster (0.9%).49 Hematologic adverse event rates in the ruxolitinib and BAT arms included anemia (any grade, 27.2% vs 47.6%, respectively; grade 3/4, 0.9% vs 0%), lymphopenia (27.2% vs 78.8%; 9.7% vs 27.2%), and thrombocytopenia (14.9% vs 29.9%; 2.6% vs 5.4%).49 Herpes zoster infections occurred more frequently in the ruxolitinib arm (any grade, 5.3%; grade 3/4, 0.9%) compared with the BAT arm (no herpes zoster events).49 There was a higher rate of nonmelanoma skin cancer (NMSC) in the ruxolitinib arm (4.4%), compared with the BAT arm (2.7%),49 most of which occurred in patients with a history of NMSC or precancerous skin lesions.16 Grade 1 or 2 elevations in serum lipids and cholesterol were observed with ruxolitinib but not BAT; however, subsequent effects on patient outcomes have not been determined.8,16 The rates of MF and AML transformations were 1.3% and 0.4%, respectively, in patients randomized to receive ruxolitinib,49 similar to previously published reports for patients with polycythemia vera.44
Additional insight regarding the effect of ruxolitinib on polycythemia vera–related symptoms is available from the RELIEF trial, a randomized, multicenter, double-blind, double-dummy, phase 3b clinical trial. In RELIEF, 110 patients were randomized to receive ruxolitinib or a stable dose of hydroxyurea and were then asked to record disease-related symptoms.51 Although the study failed to meet its primary endpoint (a ≥50% improvement by week 16 in MPN-SAF total symptom score for the cytokine symptom cluster [sum of individual scores for tiredness, itching, muscle aches, night sweats, and sweats while awake]), a numerically greater proportion of patients receiving ruxolitinib achieved the primary endpoint compared with those receiving hydroxyurea (43.4% and 29.6%, respectively; P = .139; odds ratio, 1.82; 95% confidence interval, 0.82-4.04). Similarly, the proportion of patients reporting a ≥50% improvement in pruritus and fatigue favored ruxolitinib over hydroxyurea (itching, 40.0% vs 26.4%; tiredness, 54.2% vs 32.0%). The safety profile for ruxolitinib was similar to that reported in the RESPONSE trial.
Possible future treatment options
Other possible treatment options for patients with polycythemia vera that are currently in clinical development include three pegylated IFN-α (PEG-IFN-α) variants and the telomerase inhibitor, imetelstat.
Pegylated interferon-α. PEG–IFN-α has the advantage of a longer plasma half-life compared with conventional IFN-α, permitting administration once per week or less often.46 Currently, three variants are under active investigation in phase 3 clinical trials: PEG–IFN-α2a (NCT01259856 and NCT01387763), PEG–IFN-α2b (NCT01387763), and AOP2014/P1101 (NCT02218047, NCT02523638, and NCT01949805).
Imetelstat. The telomerase inhibitor imetelstat is in clinical development for patients with MPNs. Clinical benefit was previously observed in patients with primary MF as well as post-polycythemia vera and post-ET MF.52 Imetelstat was evaluated in a phase 2 trial in patients with polycythemia vera or ET who required cytoreductive therapy and were resistant to or intolerant of ≥1 previous line of therapy or who refused standard therapy (NCT01243073). Results in patients with ET were published,53 however, findings from the polycythemia vera cohort have not been reported.
Community oncologist role in managing disease burden
Most patients with polycythemia vera are managed in the community setting. Consequently, the community oncologist plays a critical role in the initial diagnosis, risk stratification, patient education, and disease management.
Early disease recognition allows prompt therapeutic intervention with low-dose aspirin and phlebotomy, interventions shown to reduce the risk of cardiovascular events based on the ECLAP32 and CYTO-PV trials,12 respectively. The diagnosis of polycythemia vera is facilitated by applying the WHO diagnostic criteria (Figure 2);1 however, one should be aware of atypical presentations, including “masked” polycythemia vera,30 as well as the development of thrombosis at atypical sites.26
Optimal management strategies must include the frequent assessment of symptom burden and its effect on a patient’s QoL, with a keen awareness of the nonspecific nature of polycythemia vera–related symptoms, and the potential for patients and clinicians to minimize that effect.20 Patient-reported symptom severity and QoL should be assessed at each office visit with validated instruments, such as the MPN-SAF 10-item questionnaire.22It is important for the community oncologist to define treatment goals and implement a plan that reduces disease-associated morbidity and mortality. A critical treatment goal is to maintain hematocrit <45% by the appropriate use of phlebotomy12 and/or cytoreductive agents.12,16,46 Continued reassessment is important to identify patients with progressive disease and those who fail to achieve stated treatment goals or require an adjustment in cytoreductive therapy. Oncologists should be familiar with the concept of hydroxyurea resistance/intolerance as defined by the ELN (Table)31,45 to allow early identification of those patients who are most likely to benefit from a treatment change for continued optimal outcome.
Conclusions
Polycythemia vera is a clonal myeloproliferative neoplasm associated with significant disease-related morbidity and mortality. Appropriate management includes early diagnosis and implementation of appropriate therapy according to patient risk and therapeutic tolerance. Patients should initially receive aspirin32 and phlebotomy,12 with the goal of maintaining hematocrit <45%. Higher-risk patients and those who had inadequate disease control with phlebotomy alone require cytoreduction, typically with hydroxyurea. Although most patients will achieve adequate disease control with hydroxyurea,33,43 one in four patients will develop drug resistance or intolerance.44 Ruxolitinib is approved by the FDA and the EMA for the treatment of patients with polycythemia vera who are resistant to or intolerant of hydroxyurea. Compared with BAT, ruxolitinib is associated with improved hematocrit control, reductions in spleen size, a greater probability of blood count normalization, and improvement in polycythemia vera–related symptoms.16
Acknowledgments
Writing assistance was provided by Cory Pfeiffenberger, PhD, of Complete Healthcare Communications LLC, and was funded by Incyte Corporation, the maker of ruxolitinib.
1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405.
2. Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55:595-600.
3. Price GL, Davis KL, Karve S, Pohl G, Walgren RA. Survival patterns in United States (US) Medicare enrollees with non-CML myeloproliferative neoplasms (MPN). PLoS ONE. 2014;9:e90299.
4. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27:1874-1881.
5. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23:2224-2232.
6. Mesa R, Miller CB, Thyne M, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer. 2016;16:167.
7. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379-2390.
8. JAKAFI (ruxolitinib). Full Prescribing Information, Incyte Corporation, Wilmington, DE, USA, 2016.
9. JAKAVI (ruxolitinib). Summary of Product Characteristics, Novartis Pharma GmbH, Nuremberg, Germany, 2015.
10. Johansson P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 2006;32:171-173.
11. Vannucchi AM. How I treat polycythemia vera. Blood. 2014;124:3212-3220.
12. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368:22-33.
13. Barbui T, Masciulli A, Marfisi MR, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. 2015;126:560-561.
14. Passamonti F, Rumi E, Pietra D, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. 2010;24:1574-1579.
15. Quintás-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov. 2011;10:127-140.
16. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372:426-435.
17. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia. 2007;21:1952-1959.
18. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly-annotated essential thrombocythemia, polycythemia vera and myelofibrosis. Blood. 2014;124:2507-2513.
19. Fallah M, Kharazmi E, Sundquist J, Hemminki K. Higher risk of primary cancers after polycythaemia vera and vice versa. Br J Haematol. 2011;153:283-285.
20. Mesa R, Miller C, Thyne M, et al. Differences in treatment goals and perception of symptom burden between patients with MPNs and hematologists/oncologists in the United States: findings from the MPN landmark survey. Cancer. 2017;123:449-458.
21. Abelsson J, Andreasson B, Samuelsson J, et al. Patients with polycythemia vera have the worst impairment of quality of life among patients with newly diagnosed myeloproliferative neoplasms. Leuk Lymphoma. 2013;54:2226-2230.
22. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative Neoplasm (MPN) Symptom Assessment Form Total Symptom Score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30:4098-4103.
23. Johansson P, Mesa R, Scherber R, et al. Association between quality of life and clinical parameters in patients with myeloproliferative neoplasms. Leuk Lymphoma. 2012;53:441-444.
24. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011;118:401-408.
25. Siegel FP, Tauscher J, Petrides PE. Aquagenic pruritus in polycythemia vera: characteristics and influence on quality of life in 441 patients. Am J Hematol. 2013;88:665-669.
26. Smalberg JH, Arends LR, Valla DC, Kiladjian JJ, Janssen HL, Leebeek FW. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood. 2012;120:4921-4928.
27. Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008;22:437-438.
28. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937-951.
29. Roda P, Ferrari A, Tang X, et al. Determination of accuracy of polycythemia vera diagnoses and use of the JAK2V617F test in the diagnostic scheme. Ann Hematol. 2014;93:1467-1472.
30. Barbui T, Thiele J, Gisslinger H, et al. Masked polycythemia vera (mPV): results of an international study. Am J Hematol. 2014;89:52-54.
31. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29:761-770.
32. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350:114-124.
33. Kiladjian JJ, Chevret S, Dosquet C, Chomienne C, Rain JD. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29:3907-3913.
34. Quintás-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27:5418-5424.
35. Sacchi S, Leoni P, Liberati M, et al. A prospective comparison between treatment with phlebotomy alone and with interferon-alpha in patients with polycythemia vera. Ann Hematol. 1994;68:247-250.
36. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer. 2006;107:451-458.
37. Huang ES, Strate LL, Ho WW, Lee SS, Chan AT. Long-term use of aspirin and the risk of gastrointestinal bleeding. Am J Med. 2011;124:426-433.
38. Shetty S, Kasatkar P, Ghosh K. Pathophysiology of acquired von Willebrand disease: a concise review. Eur J Haematol. 2011;87:99-106.
39. Deacon B, Abramowitz J. Fear of needles and vasovagal reactions among phlebotomy patients. J Anxiety Disord. 2006;20:946-960.
40. Tobiasson M, Alyass B, Soderlund S, Birgegard G. High prevalence of restless legs syndrome among patients with polycytemia vera treated with venesectio. Med Oncol. 2010;27:105-107.
41. Greig AJ, Patterson AJ, Collins CE, Chalmers KA. Iron deficiency, cognition, mental health and fatigue in women of childbearing age: a systematic review. J Nutr Sci. 2013;2:e14.
42. Passamonti F. How I treat polycythemia vera. Blood. 2012;120:275-284.
43. Fruchtman SM, Mack K, Kaplan ME, Peterson P, Berk PD, Wasserman LR. From efficacy to safety: a Polycythemia Vera Study Group report on hydroxyurea in patients with polycythemia vera. Semin Hematol. 1997;34:17-23.
44. Alvarez-Lárran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119:1363-1369.
45. Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol. 2010;148:961-963.
46. Hasselbalch HC. A new era for IFN-α in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Expert Rev Hematol. 2011;4:637-655.
47. Emanuel R, Dueck AC, Kiladjian JJ, et al. Conventional therapeutic options have limited impact on MPN symptoms: insights from a prospective analysis of the MPN-SAF [abstract 366]. Presented at: European Hematology Association, June 14-17, 2012; Amsterdam, Netherlands.
48. Lacevic J, Reljic T, El Jurdi N, et al. Conservative management vs. allogeneic hematopoietic cell transplantation for polycythemia vera: a systematic review and decision-analysis. Blood. 2013;122:abstract 5372.
49. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80 week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
50. Verstovsek S, Harrison CN, Kiladjian J-J, et al. Effect of ruxolitinib on markers of iron deficiency: an analysis of the RESPONSE trial. Haematologica (EHA Annual Meeting Abstracts). 2015;100:abstract P672.
51. Mesa R, Vannucchi AM, Yacoub A, et al. The efficacy and safety of continued hydroxyurea therapy versus switching to ruxolitinib in patients with polycythemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Blood (ASH Annual Meeting Abstracts). 2014;124:abstract 3168.
52. Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373:908-919.
53. Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015;373:920-928.
1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405.
2. Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55:595-600.
3. Price GL, Davis KL, Karve S, Pohl G, Walgren RA. Survival patterns in United States (US) Medicare enrollees with non-CML myeloproliferative neoplasms (MPN). PLoS ONE. 2014;9:e90299.
4. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27:1874-1881.
5. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23:2224-2232.
6. Mesa R, Miller CB, Thyne M, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer. 2016;16:167.
7. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379-2390.
8. JAKAFI (ruxolitinib). Full Prescribing Information, Incyte Corporation, Wilmington, DE, USA, 2016.
9. JAKAVI (ruxolitinib). Summary of Product Characteristics, Novartis Pharma GmbH, Nuremberg, Germany, 2015.
10. Johansson P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 2006;32:171-173.
11. Vannucchi AM. How I treat polycythemia vera. Blood. 2014;124:3212-3220.
12. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368:22-33.
13. Barbui T, Masciulli A, Marfisi MR, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. 2015;126:560-561.
14. Passamonti F, Rumi E, Pietra D, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. 2010;24:1574-1579.
15. Quintás-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov. 2011;10:127-140.
16. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372:426-435.
17. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia. 2007;21:1952-1959.
18. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly-annotated essential thrombocythemia, polycythemia vera and myelofibrosis. Blood. 2014;124:2507-2513.
19. Fallah M, Kharazmi E, Sundquist J, Hemminki K. Higher risk of primary cancers after polycythaemia vera and vice versa. Br J Haematol. 2011;153:283-285.
20. Mesa R, Miller C, Thyne M, et al. Differences in treatment goals and perception of symptom burden between patients with MPNs and hematologists/oncologists in the United States: findings from the MPN landmark survey. Cancer. 2017;123:449-458.
21. Abelsson J, Andreasson B, Samuelsson J, et al. Patients with polycythemia vera have the worst impairment of quality of life among patients with newly diagnosed myeloproliferative neoplasms. Leuk Lymphoma. 2013;54:2226-2230.
22. Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative Neoplasm (MPN) Symptom Assessment Form Total Symptom Score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30:4098-4103.
23. Johansson P, Mesa R, Scherber R, et al. Association between quality of life and clinical parameters in patients with myeloproliferative neoplasms. Leuk Lymphoma. 2012;53:441-444.
24. Scherber R, Dueck AC, Johansson P, et al. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011;118:401-408.
25. Siegel FP, Tauscher J, Petrides PE. Aquagenic pruritus in polycythemia vera: characteristics and influence on quality of life in 441 patients. Am J Hematol. 2013;88:665-669.
26. Smalberg JH, Arends LR, Valla DC, Kiladjian JJ, Janssen HL, Leebeek FW. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood. 2012;120:4921-4928.
27. Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008;22:437-438.
28. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937-951.
29. Roda P, Ferrari A, Tang X, et al. Determination of accuracy of polycythemia vera diagnoses and use of the JAK2V617F test in the diagnostic scheme. Ann Hematol. 2014;93:1467-1472.
30. Barbui T, Thiele J, Gisslinger H, et al. Masked polycythemia vera (mPV): results of an international study. Am J Hematol. 2014;89:52-54.
31. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29:761-770.
32. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350:114-124.
33. Kiladjian JJ, Chevret S, Dosquet C, Chomienne C, Rain JD. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29:3907-3913.
34. Quintás-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27:5418-5424.
35. Sacchi S, Leoni P, Liberati M, et al. A prospective comparison between treatment with phlebotomy alone and with interferon-alpha in patients with polycythemia vera. Ann Hematol. 1994;68:247-250.
36. Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer. 2006;107:451-458.
37. Huang ES, Strate LL, Ho WW, Lee SS, Chan AT. Long-term use of aspirin and the risk of gastrointestinal bleeding. Am J Med. 2011;124:426-433.
38. Shetty S, Kasatkar P, Ghosh K. Pathophysiology of acquired von Willebrand disease: a concise review. Eur J Haematol. 2011;87:99-106.
39. Deacon B, Abramowitz J. Fear of needles and vasovagal reactions among phlebotomy patients. J Anxiety Disord. 2006;20:946-960.
40. Tobiasson M, Alyass B, Soderlund S, Birgegard G. High prevalence of restless legs syndrome among patients with polycytemia vera treated with venesectio. Med Oncol. 2010;27:105-107.
41. Greig AJ, Patterson AJ, Collins CE, Chalmers KA. Iron deficiency, cognition, mental health and fatigue in women of childbearing age: a systematic review. J Nutr Sci. 2013;2:e14.
42. Passamonti F. How I treat polycythemia vera. Blood. 2012;120:275-284.
43. Fruchtman SM, Mack K, Kaplan ME, Peterson P, Berk PD, Wasserman LR. From efficacy to safety: a Polycythemia Vera Study Group report on hydroxyurea in patients with polycythemia vera. Semin Hematol. 1997;34:17-23.
44. Alvarez-Lárran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119:1363-1369.
45. Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol. 2010;148:961-963.
46. Hasselbalch HC. A new era for IFN-α in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Expert Rev Hematol. 2011;4:637-655.
47. Emanuel R, Dueck AC, Kiladjian JJ, et al. Conventional therapeutic options have limited impact on MPN symptoms: insights from a prospective analysis of the MPN-SAF [abstract 366]. Presented at: European Hematology Association, June 14-17, 2012; Amsterdam, Netherlands.
48. Lacevic J, Reljic T, El Jurdi N, et al. Conservative management vs. allogeneic hematopoietic cell transplantation for polycythemia vera: a systematic review and decision-analysis. Blood. 2013;122:abstract 5372.
49. Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80 week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
50. Verstovsek S, Harrison CN, Kiladjian J-J, et al. Effect of ruxolitinib on markers of iron deficiency: an analysis of the RESPONSE trial. Haematologica (EHA Annual Meeting Abstracts). 2015;100:abstract P672.
51. Mesa R, Vannucchi AM, Yacoub A, et al. The efficacy and safety of continued hydroxyurea therapy versus switching to ruxolitinib in patients with polycythemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Blood (ASH Annual Meeting Abstracts). 2014;124:abstract 3168.
52. Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373:908-919.
53. Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015;373:920-928.