User login
Vitamin D and omega-3 supplements reduce autoimmune disease risk
For those of us who cannot sit in the sun and fish all day, the next best thing for preventing autoimmune diseases may be supplementation with vitamin D and fish oil-derived omega-3 fatty acids, results of a large prospective randomized trial suggest.

Among nearly 26,000 adults enrolled in a randomized trial designed primarily to study the effects of vitamin D and omega-3 supplementation on incident cancer and cardiovascular disease, 5, and 5 years of omega-3 fatty acid supplementation was associated with an 18% reduction in confirmed and probable incident autoimmune diseases, reported Karen H. Costenbader, MD, MPH, of Brigham & Women’s Hospital in Boston.
“The clinical importance of these results is very high, given that these are nontoxic, well-tolerated supplements, and that there are no other known effective therapies to reduce the incidence of autoimmune diseases,” she said during the virtual annual meeting of the American College of Rheumatology.
“People do have to take the supplements a long time to start to see the reduction in risk, especially for vitamin D, but they make biological sense, and autoimmune diseases develop slowly over time, so taking it today isn’t going to reduce risk of developing something tomorrow,” Dr. Costenbader said in an interview.
“These supplements have other health benefits. Obviously, fish oil is anti-inflammatory, and vitamin D is good for osteoporosis prevention, especially in our patients who take glucocorticoids. People who are otherwise healthy and have a family history of autoimmune disease might also consider starting to take these supplements,” she said.
After watching her presentation, session co-moderator Gregg Silverman, MD, from the NYU Langone School of Medicine in New York, who was not involved in the study, commented “I’m going to [nutrition store] GNC to get some vitamins.”
When asked for comment, the other session moderator, Tracy Frech, MD, of Vanderbilt University, Nashville, said, “I think Dr. Costenbader’s work is very important and her presentation excellent. My current practice is replacement of vitamin D in all autoimmune disease patients with low levels and per bone health guidelines. Additionally, I discuss omega-3 supplementation with Sjögren’s [syndrome] patients as a consideration.”
Evidence base
Dr. Costenbader noted that in a 2013 observational study from France, vitamin D derived through ultraviolet (UV) light exposure was associated with a lower risk for incident Crohn’s disease but not ulcerative colitis, and in two analyses of data in 2014 from the Nurses’ Health Study, both high plasma levels of 25-OH vitamin D and geographic residence in areas of high UV exposure were associated with a decreased incidence of rheumatoid arthritis (RA).

Other observational studies have supported omega-3 fatty acids for their anti-inflammatory properties, including a 2005 Danish prospective cohort study showing a lower risk for RA in participants who reported higher levels of fatty fish intake. In a separate study conducted in 2017, healthy volunteers with higher omega-3 fatty acid/total lipid proportions in red blood cell membranes had a lower prevalence of anti-cyclic citrullinated peptide (anti-CCP) antibodies and rheumatoid factor and a lower incidence of progression to inflammatory arthritis, she said.
Ancillary study
Despite the evidence, however, there have been no prospective randomized trials to test the effects of either vitamin D or omega-3 fatty acid supplementation on the incidence of autoimmune disease over time.
To rectify this, Dr. Costenbader and colleagues piggybacked an ancillary study onto the Vitamin D and Omega-3 Trial (VITAL), which had primary outcomes of cancer and cardiovascular disease incidence.
A total of 25,871 participants were enrolled, including 12,786 men aged 50 and older, and 13,085 women aged 55 and older.
The study had a 2 x 2 factorial design, with patients randomly assigned to vitamin D 2,000 IU/day or placebo, and then further randomized to either 1 g/day omega-3 fatty acids or placebo in both the vitamin D and placebo primary randomization arms.
At baseline 16,956 participants were assayed for 25-OH vitamin D and plasma omega 3 index, the ratio of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) to total fatty acids. Participants self-reported baseline and all incident autoimmune diseases annually, with the reports confirmed by medical record review and disease criteria whenever possible.
Results
At 5 years of follow-up, confirmed incident autoimmune diseases had occurred in 123 patients in the active vitamin D group, compared with 155 in the placebo vitamin D group, translating into a hazard ratio (HR) for vitamin D of 0.78 (P = .045).
In the active omega-3 arm, 130 participants developed an autoimmune disease, compared with 148 in the placebo omega-3 arm, which translated into a nonsignificant HR of 0.85.
There was no statistical interaction between the two supplements. The investigators did observe an interaction between vitamin D and body mass index, with the effect stronger among participants with low BMI (P = .02). There also was an interaction between omega-3 fatty acids with a family history of autoimmune disease (P = .03).
In multivariate analysis adjusted for age, sex, race, and other supplement arm, vitamin D alone was associated with an HR for incident autoimmune disease of 0.68 (P = .02), omega-3 alone was associated with a nonsignificant HR of 0.74, and the combination was associated with an HR of 0.69 (P = .03).
Dr. Costenbader and colleagues acknowledged that the study was limited by the lack of a high-risk or nutritionally-deficient population, where the effects of supplementation might be larger; the restriction of the sample to older adults; and to the difficulty of confirming incident autoimmune thyroid disease from patient reports.
Cheryl Koehn, an arthritis patient advocate from Vancouver, Canada, who was not involved in the study, commented in the “chat” section of the presentation that her rheumatologist “has recommended vitamin D for years now. Says basically everyone north of Boston is vitamin D deficient. I take 1,000 IU per day. Been taking it for years.” Ms. Koehn is the founder and president of Arthritis Consumer Experts, a website that provides education to those with arthritis.
“Agreed. I tell every patient to take vitamin D supplement,” commented Fatma Dedeoglu, MD, a rheumatologist at Boston Children’s Hospital.
A version of this article first appeared on Medscape.com.
For those of us who cannot sit in the sun and fish all day, the next best thing for preventing autoimmune diseases may be supplementation with vitamin D and fish oil-derived omega-3 fatty acids, results of a large prospective randomized trial suggest.
Among nearly 26,000 adults enrolled in a randomized trial designed primarily to study the effects of vitamin D and omega-3 supplementation on incident cancer and cardiovascular disease, 5, and 5 years of omega-3 fatty acid supplementation was associated with an 18% reduction in confirmed and probable incident autoimmune diseases, reported Karen H. Costenbader, MD, MPH, of Brigham & Women’s Hospital in Boston.
“The clinical importance of these results is very high, given that these are nontoxic, well-tolerated supplements, and that there are no other known effective therapies to reduce the incidence of autoimmune diseases,” she said during the virtual annual meeting of the American College of Rheumatology.
“People do have to take the supplements a long time to start to see the reduction in risk, especially for vitamin D, but they make biological sense, and autoimmune diseases develop slowly over time, so taking it today isn’t going to reduce risk of developing something tomorrow,” Dr. Costenbader said in an interview.
“These supplements have other health benefits. Obviously, fish oil is anti-inflammatory, and vitamin D is good for osteoporosis prevention, especially in our patients who take glucocorticoids. People who are otherwise healthy and have a family history of autoimmune disease might also consider starting to take these supplements,” she said.
After watching her presentation, session co-moderator Gregg Silverman, MD, from the NYU Langone School of Medicine in New York, who was not involved in the study, commented “I’m going to [nutrition store] GNC to get some vitamins.”
When asked for comment, the other session moderator, Tracy Frech, MD, of Vanderbilt University, Nashville, said, “I think Dr. Costenbader’s work is very important and her presentation excellent. My current practice is replacement of vitamin D in all autoimmune disease patients with low levels and per bone health guidelines. Additionally, I discuss omega-3 supplementation with Sjögren’s [syndrome] patients as a consideration.”
Evidence base
Dr. Costenbader noted that in a 2013 observational study from France, vitamin D derived through ultraviolet (UV) light exposure was associated with a lower risk for incident Crohn’s disease but not ulcerative colitis, and in two analyses of data in 2014 from the Nurses’ Health Study, both high plasma levels of 25-OH vitamin D and geographic residence in areas of high UV exposure were associated with a decreased incidence of rheumatoid arthritis (RA).
Other observational studies have supported omega-3 fatty acids for their anti-inflammatory properties, including a 2005 Danish prospective cohort study showing a lower risk for RA in participants who reported higher levels of fatty fish intake. In a separate study conducted in 2017, healthy volunteers with higher omega-3 fatty acid/total lipid proportions in red blood cell membranes had a lower prevalence of anti-cyclic citrullinated peptide (anti-CCP) antibodies and rheumatoid factor and a lower incidence of progression to inflammatory arthritis, she said.
Ancillary study
Despite the evidence, however, there have been no prospective randomized trials to test the effects of either vitamin D or omega-3 fatty acid supplementation on the incidence of autoimmune disease over time.
To rectify this, Dr. Costenbader and colleagues piggybacked an ancillary study onto the Vitamin D and Omega-3 Trial (VITAL), which had primary outcomes of cancer and cardiovascular disease incidence.
A total of 25,871 participants were enrolled, including 12,786 men aged 50 and older, and 13,085 women aged 55 and older.
The study had a 2 x 2 factorial design, with patients randomly assigned to vitamin D 2,000 IU/day or placebo, and then further randomized to either 1 g/day omega-3 fatty acids or placebo in both the vitamin D and placebo primary randomization arms.
At baseline 16,956 participants were assayed for 25-OH vitamin D and plasma omega 3 index, the ratio of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) to total fatty acids. Participants self-reported baseline and all incident autoimmune diseases annually, with the reports confirmed by medical record review and disease criteria whenever possible.
Results
At 5 years of follow-up, confirmed incident autoimmune diseases had occurred in 123 patients in the active vitamin D group, compared with 155 in the placebo vitamin D group, translating into a hazard ratio (HR) for vitamin D of 0.78 (P = .045).
In the active omega-3 arm, 130 participants developed an autoimmune disease, compared with 148 in the placebo omega-3 arm, which translated into a nonsignificant HR of 0.85.
There was no statistical interaction between the two supplements. The investigators did observe an interaction between vitamin D and body mass index, with the effect stronger among participants with low BMI (P = .02). There also was an interaction between omega-3 fatty acids with a family history of autoimmune disease (P = .03).
In multivariate analysis adjusted for age, sex, race, and other supplement arm, vitamin D alone was associated with an HR for incident autoimmune disease of 0.68 (P = .02), omega-3 alone was associated with a nonsignificant HR of 0.74, and the combination was associated with an HR of 0.69 (P = .03).
Dr. Costenbader and colleagues acknowledged that the study was limited by the lack of a high-risk or nutritionally-deficient population, where the effects of supplementation might be larger; the restriction of the sample to older adults; and to the difficulty of confirming incident autoimmune thyroid disease from patient reports.
Cheryl Koehn, an arthritis patient advocate from Vancouver, Canada, who was not involved in the study, commented in the “chat” section of the presentation that her rheumatologist “has recommended vitamin D for years now. Says basically everyone north of Boston is vitamin D deficient. I take 1,000 IU per day. Been taking it for years.” Ms. Koehn is the founder and president of Arthritis Consumer Experts, a website that provides education to those with arthritis.
“Agreed. I tell every patient to take vitamin D supplement,” commented Fatma Dedeoglu, MD, a rheumatologist at Boston Children’s Hospital.
A version of this article first appeared on Medscape.com.
For those of us who cannot sit in the sun and fish all day, the next best thing for preventing autoimmune diseases may be supplementation with vitamin D and fish oil-derived omega-3 fatty acids, results of a large prospective randomized trial suggest.
Among nearly 26,000 adults enrolled in a randomized trial designed primarily to study the effects of vitamin D and omega-3 supplementation on incident cancer and cardiovascular disease, 5, and 5 years of omega-3 fatty acid supplementation was associated with an 18% reduction in confirmed and probable incident autoimmune diseases, reported Karen H. Costenbader, MD, MPH, of Brigham & Women’s Hospital in Boston.
“The clinical importance of these results is very high, given that these are nontoxic, well-tolerated supplements, and that there are no other known effective therapies to reduce the incidence of autoimmune diseases,” she said during the virtual annual meeting of the American College of Rheumatology.
“People do have to take the supplements a long time to start to see the reduction in risk, especially for vitamin D, but they make biological sense, and autoimmune diseases develop slowly over time, so taking it today isn’t going to reduce risk of developing something tomorrow,” Dr. Costenbader said in an interview.
“These supplements have other health benefits. Obviously, fish oil is anti-inflammatory, and vitamin D is good for osteoporosis prevention, especially in our patients who take glucocorticoids. People who are otherwise healthy and have a family history of autoimmune disease might also consider starting to take these supplements,” she said.
After watching her presentation, session co-moderator Gregg Silverman, MD, from the NYU Langone School of Medicine in New York, who was not involved in the study, commented “I’m going to [nutrition store] GNC to get some vitamins.”
When asked for comment, the other session moderator, Tracy Frech, MD, of Vanderbilt University, Nashville, said, “I think Dr. Costenbader’s work is very important and her presentation excellent. My current practice is replacement of vitamin D in all autoimmune disease patients with low levels and per bone health guidelines. Additionally, I discuss omega-3 supplementation with Sjögren’s [syndrome] patients as a consideration.”
Evidence base
Dr. Costenbader noted that in a 2013 observational study from France, vitamin D derived through ultraviolet (UV) light exposure was associated with a lower risk for incident Crohn’s disease but not ulcerative colitis, and in two analyses of data in 2014 from the Nurses’ Health Study, both high plasma levels of 25-OH vitamin D and geographic residence in areas of high UV exposure were associated with a decreased incidence of rheumatoid arthritis (RA).
Other observational studies have supported omega-3 fatty acids for their anti-inflammatory properties, including a 2005 Danish prospective cohort study showing a lower risk for RA in participants who reported higher levels of fatty fish intake. In a separate study conducted in 2017, healthy volunteers with higher omega-3 fatty acid/total lipid proportions in red blood cell membranes had a lower prevalence of anti-cyclic citrullinated peptide (anti-CCP) antibodies and rheumatoid factor and a lower incidence of progression to inflammatory arthritis, she said.
Ancillary study
Despite the evidence, however, there have been no prospective randomized trials to test the effects of either vitamin D or omega-3 fatty acid supplementation on the incidence of autoimmune disease over time.
To rectify this, Dr. Costenbader and colleagues piggybacked an ancillary study onto the Vitamin D and Omega-3 Trial (VITAL), which had primary outcomes of cancer and cardiovascular disease incidence.
A total of 25,871 participants were enrolled, including 12,786 men aged 50 and older, and 13,085 women aged 55 and older.
The study had a 2 x 2 factorial design, with patients randomly assigned to vitamin D 2,000 IU/day or placebo, and then further randomized to either 1 g/day omega-3 fatty acids or placebo in both the vitamin D and placebo primary randomization arms.
At baseline 16,956 participants were assayed for 25-OH vitamin D and plasma omega 3 index, the ratio of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) to total fatty acids. Participants self-reported baseline and all incident autoimmune diseases annually, with the reports confirmed by medical record review and disease criteria whenever possible.
Results
At 5 years of follow-up, confirmed incident autoimmune diseases had occurred in 123 patients in the active vitamin D group, compared with 155 in the placebo vitamin D group, translating into a hazard ratio (HR) for vitamin D of 0.78 (P = .045).
In the active omega-3 arm, 130 participants developed an autoimmune disease, compared with 148 in the placebo omega-3 arm, which translated into a nonsignificant HR of 0.85.
There was no statistical interaction between the two supplements. The investigators did observe an interaction between vitamin D and body mass index, with the effect stronger among participants with low BMI (P = .02). There also was an interaction between omega-3 fatty acids with a family history of autoimmune disease (P = .03).
In multivariate analysis adjusted for age, sex, race, and other supplement arm, vitamin D alone was associated with an HR for incident autoimmune disease of 0.68 (P = .02), omega-3 alone was associated with a nonsignificant HR of 0.74, and the combination was associated with an HR of 0.69 (P = .03).
Dr. Costenbader and colleagues acknowledged that the study was limited by the lack of a high-risk or nutritionally-deficient population, where the effects of supplementation might be larger; the restriction of the sample to older adults; and to the difficulty of confirming incident autoimmune thyroid disease from patient reports.
Cheryl Koehn, an arthritis patient advocate from Vancouver, Canada, who was not involved in the study, commented in the “chat” section of the presentation that her rheumatologist “has recommended vitamin D for years now. Says basically everyone north of Boston is vitamin D deficient. I take 1,000 IU per day. Been taking it for years.” Ms. Koehn is the founder and president of Arthritis Consumer Experts, a website that provides education to those with arthritis.
“Agreed. I tell every patient to take vitamin D supplement,” commented Fatma Dedeoglu, MD, a rheumatologist at Boston Children’s Hospital.
A version of this article first appeared on Medscape.com.
FROM ACR 2021
‘Multimorbidity’ more commonly seen in people with lupus
People with systemic lupus erythematosus (SLE) have a threefold greater likelihood of having up to five or more comorbidities in comparison with people in the general population, according to the results of two separate U.S. population-based studies.
The higher rate of comorbidities seen included many of those commonly reported before, such as cardiovascular and renal disease, but also some that may be less frequently associated with SLE, notably chronic obstructive pulmonary disease (COPD) and cardiac arrhythmias.
“In the past, the characterization of SLE comorbidities has relied on individual comorbidity assessment,” Alí Duarte García, MD, said at the 14th International Congress on Systemic Lupus Erythematosus, held together will the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
“However, a patient-centric approach where a patient as a whole is seen and how many comorbidities they accrue has not been performed.” added Duarte García, who is a rheumatologist at the Mayo Clinic in Rochester, Minn.
Multiple conditions “overrepresented” in SLE patients
Dr. Duarte García reported the findings of one of the studies, both of which used data from the Rochester Epidemiology Project, a record-linkage system that collates clinical and hospital data from individuals who live in 19 counties in southeast Minnesota and eight counties in western Wisconsin; these patients have agreed to share their medical records for research.
The study population included 479 individuals diagnosed with SLE according to joint 2019 European Alliance of Associations for Rheumatology and American College of Rheumatology criteria. These were matched by age, sex, race, and county to 479 individuals without SLE.
The mean age of the study population was 53 years, 82% were women, and 86% were White.
“We defined multimorbidity as those patients who have two or more comorbidities and substantial multimorbidity as those patients who have five or more comorbidities,” Dr. Duarte García explained.
A previously published list of 44 categories of comorbidities was used to classify the multimorbidity seen, and 27 of these were “overrepresented” in patients with SLE.
Patients with SLE averaged 5.3 comorbidities, whereas control study subjects had 2.9. Comparing SLE with non-SLE individuals, the odds ratio for having two or more comorbid conditions was 2.96, and for five or more comorbidities it was 3.06.
The highest odds ratio comparing SLE with non-SLE individuals was seen for pulmonary disorders (39.0).
Dr. Duarte García highlighted four comorbidities that occurred in SLE patients that were perhaps more unusual: congestive heart failure (OR, 13.3), valvular heart disease (OR, 4.2), cardiac arrhythmias (OR, 2.85), and COPD (OR, 2.7).
“Given the association of multimorbidity with poor outcomes, care delivery strategies to manage multimorbidity are needed in SLE,” Dr. Duarte García concluded.
Similar findings seen in cutaneous lupus
There is also an excess of comorbid conditions in people with cutaneous lupus erythematosus (CLE), Mehmet Hocaoglu, MD, said in reporting the findings of the second study.
Dr. Hocaoglu, an internal medicine resident at the University of Maryland Medical Center in Baltimore, and part of the same team of researchers as Dr. Duarte García, noted that in skin-related lupus the risk of multimorbidity was about doubled.
For this separate analysis, a total of 303 patients with cutaneous lupus had been matched to 303 controls from the general population. Odds ratios for having two or more or five or more comorbidities were a respective 2.27 and 1.65.
Among the comorbidities seen that were higher in those with cutaneous lupus than in the general population subjects were fibromyalgia, liver disease, hypertension, anemia, hypothyroidism, and COPD.
“Further research is definitely needed to identify if the driver of this multimorbidity in CLE patients is the disease itself or the treatments CLE patients are receiving or a multifactorial cause that is driving the disease association,” Dr. Hocaoglu said.
Comment and perspective
“Comorbidities that are not appropriate to the general population, compared to SLE,” seem to have been included in the overall SLE and the cutaneous lupus analyses, Raquel Faria, MD, suggested.
Dr. Faria, an internal medicine consultant at Unidade de Imunologia Clínica – Centro Hospitalar Universitário Porto (Portugal), chaired the poster discussion session in which the two studies had been presented.
She wondered if the researchers had analyzed the data while accounting for “the comorbidities that you knew are due to activity in lupus, like anemia?”
The number of patients with SLE who had pulmonary circulation disorders – 7.5% vs. 0.2% of the general population – also caught Dr. Faria’s attention.
That’s “a really huge number,” Dr. Faria pointed out, “I think it is pretty overrepresented.”
Dr. Duarte García acknowledged that they “took a very broad approach” in using a “very large comorbidity index.”
“What we were observing initially is precisely what you’re mentioning,” he responded to Dr. Faria.
“We were pulling patients who were having disease manifestation rather than a comorbidity,” Dr. Duarte-García said.
These are initial and very exploratory data, he stressed. “We have now moved on to modify the index.” Some of the changes that they have made were to incorporate the SLICC Damage Index Score and tighten up the list of ICD codes used.
No outside funding was received for either of the studies. Dr. Duarte García and Dr. Hocaoglu individually stated that they had no actual or potential conflicts of interest in relation to their presentations.
A version of this article first appeared on Medscape.com.
People with systemic lupus erythematosus (SLE) have a threefold greater likelihood of having up to five or more comorbidities in comparison with people in the general population, according to the results of two separate U.S. population-based studies.
The higher rate of comorbidities seen included many of those commonly reported before, such as cardiovascular and renal disease, but also some that may be less frequently associated with SLE, notably chronic obstructive pulmonary disease (COPD) and cardiac arrhythmias.
“In the past, the characterization of SLE comorbidities has relied on individual comorbidity assessment,” Alí Duarte García, MD, said at the 14th International Congress on Systemic Lupus Erythematosus, held together will the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
“However, a patient-centric approach where a patient as a whole is seen and how many comorbidities they accrue has not been performed.” added Duarte García, who is a rheumatologist at the Mayo Clinic in Rochester, Minn.
Multiple conditions “overrepresented” in SLE patients
Dr. Duarte García reported the findings of one of the studies, both of which used data from the Rochester Epidemiology Project, a record-linkage system that collates clinical and hospital data from individuals who live in 19 counties in southeast Minnesota and eight counties in western Wisconsin; these patients have agreed to share their medical records for research.
The study population included 479 individuals diagnosed with SLE according to joint 2019 European Alliance of Associations for Rheumatology and American College of Rheumatology criteria. These were matched by age, sex, race, and county to 479 individuals without SLE.
The mean age of the study population was 53 years, 82% were women, and 86% were White.
“We defined multimorbidity as those patients who have two or more comorbidities and substantial multimorbidity as those patients who have five or more comorbidities,” Dr. Duarte García explained.
A previously published list of 44 categories of comorbidities was used to classify the multimorbidity seen, and 27 of these were “overrepresented” in patients with SLE.
Patients with SLE averaged 5.3 comorbidities, whereas control study subjects had 2.9. Comparing SLE with non-SLE individuals, the odds ratio for having two or more comorbid conditions was 2.96, and for five or more comorbidities it was 3.06.
The highest odds ratio comparing SLE with non-SLE individuals was seen for pulmonary disorders (39.0).
Dr. Duarte García highlighted four comorbidities that occurred in SLE patients that were perhaps more unusual: congestive heart failure (OR, 13.3), valvular heart disease (OR, 4.2), cardiac arrhythmias (OR, 2.85), and COPD (OR, 2.7).
“Given the association of multimorbidity with poor outcomes, care delivery strategies to manage multimorbidity are needed in SLE,” Dr. Duarte García concluded.
Similar findings seen in cutaneous lupus
There is also an excess of comorbid conditions in people with cutaneous lupus erythematosus (CLE), Mehmet Hocaoglu, MD, said in reporting the findings of the second study.
Dr. Hocaoglu, an internal medicine resident at the University of Maryland Medical Center in Baltimore, and part of the same team of researchers as Dr. Duarte García, noted that in skin-related lupus the risk of multimorbidity was about doubled.
For this separate analysis, a total of 303 patients with cutaneous lupus had been matched to 303 controls from the general population. Odds ratios for having two or more or five or more comorbidities were a respective 2.27 and 1.65.
Among the comorbidities seen that were higher in those with cutaneous lupus than in the general population subjects were fibromyalgia, liver disease, hypertension, anemia, hypothyroidism, and COPD.
“Further research is definitely needed to identify if the driver of this multimorbidity in CLE patients is the disease itself or the treatments CLE patients are receiving or a multifactorial cause that is driving the disease association,” Dr. Hocaoglu said.
Comment and perspective
“Comorbidities that are not appropriate to the general population, compared to SLE,” seem to have been included in the overall SLE and the cutaneous lupus analyses, Raquel Faria, MD, suggested.
Dr. Faria, an internal medicine consultant at Unidade de Imunologia Clínica – Centro Hospitalar Universitário Porto (Portugal), chaired the poster discussion session in which the two studies had been presented.
She wondered if the researchers had analyzed the data while accounting for “the comorbidities that you knew are due to activity in lupus, like anemia?”
The number of patients with SLE who had pulmonary circulation disorders – 7.5% vs. 0.2% of the general population – also caught Dr. Faria’s attention.
That’s “a really huge number,” Dr. Faria pointed out, “I think it is pretty overrepresented.”
Dr. Duarte García acknowledged that they “took a very broad approach” in using a “very large comorbidity index.”
“What we were observing initially is precisely what you’re mentioning,” he responded to Dr. Faria.
“We were pulling patients who were having disease manifestation rather than a comorbidity,” Dr. Duarte-García said.
These are initial and very exploratory data, he stressed. “We have now moved on to modify the index.” Some of the changes that they have made were to incorporate the SLICC Damage Index Score and tighten up the list of ICD codes used.
No outside funding was received for either of the studies. Dr. Duarte García and Dr. Hocaoglu individually stated that they had no actual or potential conflicts of interest in relation to their presentations.
A version of this article first appeared on Medscape.com.
People with systemic lupus erythematosus (SLE) have a threefold greater likelihood of having up to five or more comorbidities in comparison with people in the general population, according to the results of two separate U.S. population-based studies.
The higher rate of comorbidities seen included many of those commonly reported before, such as cardiovascular and renal disease, but also some that may be less frequently associated with SLE, notably chronic obstructive pulmonary disease (COPD) and cardiac arrhythmias.
“In the past, the characterization of SLE comorbidities has relied on individual comorbidity assessment,” Alí Duarte García, MD, said at the 14th International Congress on Systemic Lupus Erythematosus, held together will the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
“However, a patient-centric approach where a patient as a whole is seen and how many comorbidities they accrue has not been performed.” added Duarte García, who is a rheumatologist at the Mayo Clinic in Rochester, Minn.
Multiple conditions “overrepresented” in SLE patients
Dr. Duarte García reported the findings of one of the studies, both of which used data from the Rochester Epidemiology Project, a record-linkage system that collates clinical and hospital data from individuals who live in 19 counties in southeast Minnesota and eight counties in western Wisconsin; these patients have agreed to share their medical records for research.
The study population included 479 individuals diagnosed with SLE according to joint 2019 European Alliance of Associations for Rheumatology and American College of Rheumatology criteria. These were matched by age, sex, race, and county to 479 individuals without SLE.
The mean age of the study population was 53 years, 82% were women, and 86% were White.
“We defined multimorbidity as those patients who have two or more comorbidities and substantial multimorbidity as those patients who have five or more comorbidities,” Dr. Duarte García explained.
A previously published list of 44 categories of comorbidities was used to classify the multimorbidity seen, and 27 of these were “overrepresented” in patients with SLE.
Patients with SLE averaged 5.3 comorbidities, whereas control study subjects had 2.9. Comparing SLE with non-SLE individuals, the odds ratio for having two or more comorbid conditions was 2.96, and for five or more comorbidities it was 3.06.
The highest odds ratio comparing SLE with non-SLE individuals was seen for pulmonary disorders (39.0).
Dr. Duarte García highlighted four comorbidities that occurred in SLE patients that were perhaps more unusual: congestive heart failure (OR, 13.3), valvular heart disease (OR, 4.2), cardiac arrhythmias (OR, 2.85), and COPD (OR, 2.7).
“Given the association of multimorbidity with poor outcomes, care delivery strategies to manage multimorbidity are needed in SLE,” Dr. Duarte García concluded.
Similar findings seen in cutaneous lupus
There is also an excess of comorbid conditions in people with cutaneous lupus erythematosus (CLE), Mehmet Hocaoglu, MD, said in reporting the findings of the second study.
Dr. Hocaoglu, an internal medicine resident at the University of Maryland Medical Center in Baltimore, and part of the same team of researchers as Dr. Duarte García, noted that in skin-related lupus the risk of multimorbidity was about doubled.
For this separate analysis, a total of 303 patients with cutaneous lupus had been matched to 303 controls from the general population. Odds ratios for having two or more or five or more comorbidities were a respective 2.27 and 1.65.
Among the comorbidities seen that were higher in those with cutaneous lupus than in the general population subjects were fibromyalgia, liver disease, hypertension, anemia, hypothyroidism, and COPD.
“Further research is definitely needed to identify if the driver of this multimorbidity in CLE patients is the disease itself or the treatments CLE patients are receiving or a multifactorial cause that is driving the disease association,” Dr. Hocaoglu said.
Comment and perspective
“Comorbidities that are not appropriate to the general population, compared to SLE,” seem to have been included in the overall SLE and the cutaneous lupus analyses, Raquel Faria, MD, suggested.
Dr. Faria, an internal medicine consultant at Unidade de Imunologia Clínica – Centro Hospitalar Universitário Porto (Portugal), chaired the poster discussion session in which the two studies had been presented.
She wondered if the researchers had analyzed the data while accounting for “the comorbidities that you knew are due to activity in lupus, like anemia?”
The number of patients with SLE who had pulmonary circulation disorders – 7.5% vs. 0.2% of the general population – also caught Dr. Faria’s attention.
That’s “a really huge number,” Dr. Faria pointed out, “I think it is pretty overrepresented.”
Dr. Duarte García acknowledged that they “took a very broad approach” in using a “very large comorbidity index.”
“What we were observing initially is precisely what you’re mentioning,” he responded to Dr. Faria.
“We were pulling patients who were having disease manifestation rather than a comorbidity,” Dr. Duarte-García said.
These are initial and very exploratory data, he stressed. “We have now moved on to modify the index.” Some of the changes that they have made were to incorporate the SLICC Damage Index Score and tighten up the list of ICD codes used.
No outside funding was received for either of the studies. Dr. Duarte García and Dr. Hocaoglu individually stated that they had no actual or potential conflicts of interest in relation to their presentations.
A version of this article first appeared on Medscape.com.
The tryptophan photoproduct FICZ and its effects on the skin
The melatonin precursor tryptophan, an amino acid essential in the human diet, has been shown to display antioxidant effects.1 FICZ (also known as 6-formylindolo[3,2-b]carbazole) is a photoproduct of tryptophan that is engendered by exposure to UVB.2 This column discusses the beneficial and detrimental influence of FICZ in skin health.

Antioxidant activity
In 2005, Trommer and Neubert devised a skin lipid model system to screen 47 various compounds (drugs, plant extracts, other plant constituents, and polysaccharides) for topical antioxidative activity in response to UV-induced lipid peroxidation. Among the drugs evaluated, they observed that tryptophan exerted antioxidant effects.3
Wound healing potential
A murine study by Bandeira et al. in 2015 revealed that tryptophan-induced mitigation of the inflammatory response and indoleamine 2, 3-dioxygenase expression may have enhanced skin wound healing in mice who were repeatedly stressed.4
Antifibrotic activity
In 2018, Murai et al. endeavored to clarify the role of FICZ in regulating the expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in normal human dermal fibroblasts. They found that FICZ assists in imparting UV-mediated antifibrotic effects through the AHR/MEK/ERK signal pathway in normal human dermal fibroblasts and, thus, shows promise as a therapeutic option for scleroderma.5
Cutaneous leishmaniasis
In 2019, Rodrigues et al. conducted a quantitative analysis of the relative expression of 170 genes involved in various biological processes in the skin biopsies from patients with cutaneous leishmaniasis caused by infection with either Leishmania major or L. tropica. They identified tryptophan-2,3-deoxygenase as a restriction factor for the disorder.6
Photosensitizing activity
Park et al. showed that FICZ, a tryptophan photoproduct and endogenous high-affinity aryl hydrocarbon receptor (AhR) agonist, exhibits nanomolar photodynamic activity as a UVA photosensitizer in epidermal keratinocytes and, thus, is possibly operative in human skin.7 Syed and Mukhtar add that FICZ is effective at nanomolar concentrations and that future research may elucidate its applicability against UV-induced adverse effects and inflammatory skin conditions.8
FICZ, oxidative stress, and cancer promotion
FICZ is known to display detrimental, as well as beneficial, influences in skin. The tryptophan photoproduct, comparable to UVB, ligates AhR, generates reactive oxygen species, and strongly photosensitizes for UVA. As Furue et al. note, FICZ upregulates the expression of terminal differentiation molecules (i.e., filaggrin and loricrin via AhR), and its application has been shown to suppress cutaneous inflammation in a psoriasis and dermatitis mouse model.2
In 2016, Reid et al. reported that the protein photodamage brought about by endogenous photosensitizers such as tryptophan tyrosine residues can contribute to the deleterious impact of UVA on human skin.9
In 2018, Tanaka et al. showed that FICZ imparts a cascade of events tantamount in some cases to UVB, as it promoted the synthesis of proinflammatory cytokines such as interleukin (IL)-1 alpha, IL-1 beta, and IL-6 and boosted reactive oxygen species generation in human HaCaT keratinocytes in an AhR-dependent fashion. They concluded that observing FICZ activity contributes to the understanding of how UVB damages organisms.10
That same year, Murai et al. assessed the effects of FICZ on TGF-beta-mediated ACTA2 and collagen I expression in normal human dermal fibroblasts. They determined that it may act as a key chromophore and one approach to mitigating the effects of photoaging may be to downregulate FICZ signaling.11
A year earlier, Brem et al. showed that the combined effect of FICZ and UVA engendered significant protein damage in HaCaT human keratinocytes, with the oxidation yielded from the combination of FICZ and UVA blocking the removal of potentially mutagenic UVB-induced DNA photolesions by nucleotide excision repair. The researchers concluded that the development of FICZ may raise the risk of incurring skin cancer resulting from sun exposure via the promotion of photochemical impairment of the nucleotide excision repair proteome, which in turn inhibits the removal of UVB-induced DNA lesions.12
Conclusion
However, the tryptophan photoproduct FICZ, which results from UVB exposure, presents as a complicated substance, conferring healthy and harmful effects. Much more research is necessary to determine how best to harness and direct the useful activities of tryptophan and FICZ without incurring damaging effects. Nanotechnology may be one useful avenue of investigation for this purpose.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann has written two textbooks and a New York Times Best Sellers book for consumers. Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Galderma, Revance, Evolus, and Burt’s Bees. She is the CEO of Skin Type Solutions Inc., a company that independently tests skin care products and makes recommendations to physicians on which skin care technologies are best. Write to her at [email protected].
References
1. Trommer H et al. J Pharm Pharmacol. 2003 Oct;55(10):1379-88.
2. Furue M et al. G Ital Dermatol Venereol. 2019 Feb;154(1):37-41.
3. Trommer H and Neubert RH. J Pharm Pharm Sci. 2005 Sep 15;8(3):494-506.
4. Bandeira LG et al. PLoS One. 2015 Jun 9:10(6):e0128439.
5. Murai M et al. J Dermatol Sci. 2018 Jul;91(1):97-103.
6. Rodrigues V et al. Front Cell Infect Microbiol. 2019 Oct 4;9:338. eCollection 2019.
7. Park SL et al. J Invest Dermatol. 2015 Jun;135(6):1649-58.
8. Syed DN and Mukhtar H. J Invest Dermatol. 2015 Jun;135(6):1478-81.
9. Reid LO et al. Biochemistry. 2016 Aug 30;55(34):4777-86.
10. Tanaka Y et al. Oxid Med Cell Longev. 2018 Nov 25;2018:9298052.
11. Murai M et al. J Dermatol Sci. 2018 Jan;89(1):19-26.
12. Brem R et al. Sci Rep. 2017 Jun 27;7(1):4310.
The melatonin precursor tryptophan, an amino acid essential in the human diet, has been shown to display antioxidant effects.1 FICZ (also known as 6-formylindolo[3,2-b]carbazole) is a photoproduct of tryptophan that is engendered by exposure to UVB.2 This column discusses the beneficial and detrimental influence of FICZ in skin health.
Antioxidant activity
In 2005, Trommer and Neubert devised a skin lipid model system to screen 47 various compounds (drugs, plant extracts, other plant constituents, and polysaccharides) for topical antioxidative activity in response to UV-induced lipid peroxidation. Among the drugs evaluated, they observed that tryptophan exerted antioxidant effects.3
Wound healing potential
A murine study by Bandeira et al. in 2015 revealed that tryptophan-induced mitigation of the inflammatory response and indoleamine 2, 3-dioxygenase expression may have enhanced skin wound healing in mice who were repeatedly stressed.4
Antifibrotic activity
In 2018, Murai et al. endeavored to clarify the role of FICZ in regulating the expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in normal human dermal fibroblasts. They found that FICZ assists in imparting UV-mediated antifibrotic effects through the AHR/MEK/ERK signal pathway in normal human dermal fibroblasts and, thus, shows promise as a therapeutic option for scleroderma.5
Cutaneous leishmaniasis
In 2019, Rodrigues et al. conducted a quantitative analysis of the relative expression of 170 genes involved in various biological processes in the skin biopsies from patients with cutaneous leishmaniasis caused by infection with either Leishmania major or L. tropica. They identified tryptophan-2,3-deoxygenase as a restriction factor for the disorder.6
Photosensitizing activity
Park et al. showed that FICZ, a tryptophan photoproduct and endogenous high-affinity aryl hydrocarbon receptor (AhR) agonist, exhibits nanomolar photodynamic activity as a UVA photosensitizer in epidermal keratinocytes and, thus, is possibly operative in human skin.7 Syed and Mukhtar add that FICZ is effective at nanomolar concentrations and that future research may elucidate its applicability against UV-induced adverse effects and inflammatory skin conditions.8
FICZ, oxidative stress, and cancer promotion
FICZ is known to display detrimental, as well as beneficial, influences in skin. The tryptophan photoproduct, comparable to UVB, ligates AhR, generates reactive oxygen species, and strongly photosensitizes for UVA. As Furue et al. note, FICZ upregulates the expression of terminal differentiation molecules (i.e., filaggrin and loricrin via AhR), and its application has been shown to suppress cutaneous inflammation in a psoriasis and dermatitis mouse model.2
In 2016, Reid et al. reported that the protein photodamage brought about by endogenous photosensitizers such as tryptophan tyrosine residues can contribute to the deleterious impact of UVA on human skin.9
In 2018, Tanaka et al. showed that FICZ imparts a cascade of events tantamount in some cases to UVB, as it promoted the synthesis of proinflammatory cytokines such as interleukin (IL)-1 alpha, IL-1 beta, and IL-6 and boosted reactive oxygen species generation in human HaCaT keratinocytes in an AhR-dependent fashion. They concluded that observing FICZ activity contributes to the understanding of how UVB damages organisms.10
That same year, Murai et al. assessed the effects of FICZ on TGF-beta-mediated ACTA2 and collagen I expression in normal human dermal fibroblasts. They determined that it may act as a key chromophore and one approach to mitigating the effects of photoaging may be to downregulate FICZ signaling.11
A year earlier, Brem et al. showed that the combined effect of FICZ and UVA engendered significant protein damage in HaCaT human keratinocytes, with the oxidation yielded from the combination of FICZ and UVA blocking the removal of potentially mutagenic UVB-induced DNA photolesions by nucleotide excision repair. The researchers concluded that the development of FICZ may raise the risk of incurring skin cancer resulting from sun exposure via the promotion of photochemical impairment of the nucleotide excision repair proteome, which in turn inhibits the removal of UVB-induced DNA lesions.12
Conclusion
However, the tryptophan photoproduct FICZ, which results from UVB exposure, presents as a complicated substance, conferring healthy and harmful effects. Much more research is necessary to determine how best to harness and direct the useful activities of tryptophan and FICZ without incurring damaging effects. Nanotechnology may be one useful avenue of investigation for this purpose.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann has written two textbooks and a New York Times Best Sellers book for consumers. Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Galderma, Revance, Evolus, and Burt’s Bees. She is the CEO of Skin Type Solutions Inc., a company that independently tests skin care products and makes recommendations to physicians on which skin care technologies are best. Write to her at [email protected].
References
1. Trommer H et al. J Pharm Pharmacol. 2003 Oct;55(10):1379-88.
2. Furue M et al. G Ital Dermatol Venereol. 2019 Feb;154(1):37-41.
3. Trommer H and Neubert RH. J Pharm Pharm Sci. 2005 Sep 15;8(3):494-506.
4. Bandeira LG et al. PLoS One. 2015 Jun 9:10(6):e0128439.
5. Murai M et al. J Dermatol Sci. 2018 Jul;91(1):97-103.
6. Rodrigues V et al. Front Cell Infect Microbiol. 2019 Oct 4;9:338. eCollection 2019.
7. Park SL et al. J Invest Dermatol. 2015 Jun;135(6):1649-58.
8. Syed DN and Mukhtar H. J Invest Dermatol. 2015 Jun;135(6):1478-81.
9. Reid LO et al. Biochemistry. 2016 Aug 30;55(34):4777-86.
10. Tanaka Y et al. Oxid Med Cell Longev. 2018 Nov 25;2018:9298052.
11. Murai M et al. J Dermatol Sci. 2018 Jan;89(1):19-26.
12. Brem R et al. Sci Rep. 2017 Jun 27;7(1):4310.
The melatonin precursor tryptophan, an amino acid essential in the human diet, has been shown to display antioxidant effects.1 FICZ (also known as 6-formylindolo[3,2-b]carbazole) is a photoproduct of tryptophan that is engendered by exposure to UVB.2 This column discusses the beneficial and detrimental influence of FICZ in skin health.
Antioxidant activity
In 2005, Trommer and Neubert devised a skin lipid model system to screen 47 various compounds (drugs, plant extracts, other plant constituents, and polysaccharides) for topical antioxidative activity in response to UV-induced lipid peroxidation. Among the drugs evaluated, they observed that tryptophan exerted antioxidant effects.3
Wound healing potential
A murine study by Bandeira et al. in 2015 revealed that tryptophan-induced mitigation of the inflammatory response and indoleamine 2, 3-dioxygenase expression may have enhanced skin wound healing in mice who were repeatedly stressed.4
Antifibrotic activity
In 2018, Murai et al. endeavored to clarify the role of FICZ in regulating the expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in normal human dermal fibroblasts. They found that FICZ assists in imparting UV-mediated antifibrotic effects through the AHR/MEK/ERK signal pathway in normal human dermal fibroblasts and, thus, shows promise as a therapeutic option for scleroderma.5
Cutaneous leishmaniasis
In 2019, Rodrigues et al. conducted a quantitative analysis of the relative expression of 170 genes involved in various biological processes in the skin biopsies from patients with cutaneous leishmaniasis caused by infection with either Leishmania major or L. tropica. They identified tryptophan-2,3-deoxygenase as a restriction factor for the disorder.6
Photosensitizing activity
Park et al. showed that FICZ, a tryptophan photoproduct and endogenous high-affinity aryl hydrocarbon receptor (AhR) agonist, exhibits nanomolar photodynamic activity as a UVA photosensitizer in epidermal keratinocytes and, thus, is possibly operative in human skin.7 Syed and Mukhtar add that FICZ is effective at nanomolar concentrations and that future research may elucidate its applicability against UV-induced adverse effects and inflammatory skin conditions.8
FICZ, oxidative stress, and cancer promotion
FICZ is known to display detrimental, as well as beneficial, influences in skin. The tryptophan photoproduct, comparable to UVB, ligates AhR, generates reactive oxygen species, and strongly photosensitizes for UVA. As Furue et al. note, FICZ upregulates the expression of terminal differentiation molecules (i.e., filaggrin and loricrin via AhR), and its application has been shown to suppress cutaneous inflammation in a psoriasis and dermatitis mouse model.2
In 2016, Reid et al. reported that the protein photodamage brought about by endogenous photosensitizers such as tryptophan tyrosine residues can contribute to the deleterious impact of UVA on human skin.9
In 2018, Tanaka et al. showed that FICZ imparts a cascade of events tantamount in some cases to UVB, as it promoted the synthesis of proinflammatory cytokines such as interleukin (IL)-1 alpha, IL-1 beta, and IL-6 and boosted reactive oxygen species generation in human HaCaT keratinocytes in an AhR-dependent fashion. They concluded that observing FICZ activity contributes to the understanding of how UVB damages organisms.10
That same year, Murai et al. assessed the effects of FICZ on TGF-beta-mediated ACTA2 and collagen I expression in normal human dermal fibroblasts. They determined that it may act as a key chromophore and one approach to mitigating the effects of photoaging may be to downregulate FICZ signaling.11
A year earlier, Brem et al. showed that the combined effect of FICZ and UVA engendered significant protein damage in HaCaT human keratinocytes, with the oxidation yielded from the combination of FICZ and UVA blocking the removal of potentially mutagenic UVB-induced DNA photolesions by nucleotide excision repair. The researchers concluded that the development of FICZ may raise the risk of incurring skin cancer resulting from sun exposure via the promotion of photochemical impairment of the nucleotide excision repair proteome, which in turn inhibits the removal of UVB-induced DNA lesions.12
Conclusion
However, the tryptophan photoproduct FICZ, which results from UVB exposure, presents as a complicated substance, conferring healthy and harmful effects. Much more research is necessary to determine how best to harness and direct the useful activities of tryptophan and FICZ without incurring damaging effects. Nanotechnology may be one useful avenue of investigation for this purpose.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann has written two textbooks and a New York Times Best Sellers book for consumers. Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Galderma, Revance, Evolus, and Burt’s Bees. She is the CEO of Skin Type Solutions Inc., a company that independently tests skin care products and makes recommendations to physicians on which skin care technologies are best. Write to her at [email protected].
References
1. Trommer H et al. J Pharm Pharmacol. 2003 Oct;55(10):1379-88.
2. Furue M et al. G Ital Dermatol Venereol. 2019 Feb;154(1):37-41.
3. Trommer H and Neubert RH. J Pharm Pharm Sci. 2005 Sep 15;8(3):494-506.
4. Bandeira LG et al. PLoS One. 2015 Jun 9:10(6):e0128439.
5. Murai M et al. J Dermatol Sci. 2018 Jul;91(1):97-103.
6. Rodrigues V et al. Front Cell Infect Microbiol. 2019 Oct 4;9:338. eCollection 2019.
7. Park SL et al. J Invest Dermatol. 2015 Jun;135(6):1649-58.
8. Syed DN and Mukhtar H. J Invest Dermatol. 2015 Jun;135(6):1478-81.
9. Reid LO et al. Biochemistry. 2016 Aug 30;55(34):4777-86.
10. Tanaka Y et al. Oxid Med Cell Longev. 2018 Nov 25;2018:9298052.
11. Murai M et al. J Dermatol Sci. 2018 Jan;89(1):19-26.
12. Brem R et al. Sci Rep. 2017 Jun 27;7(1):4310.
Lupus may confer higher risk of death from COVID-19
There is a significantly increased risk for acute respiratory distress syndrome (ARDS)–related death from COVID-19 among people with systemic lupus erythematous (SLE), compared with the general population, according to data collected in Brazil in 2020.
“Special care is therefore necessary for these patients, as well as reinforcement of the importance of preventive measures during a pandemic for this population,” said Eloisa Bonfá, MD, PhD, at the 14th International Congress on Systemic Lupus Erythematosus, which was held together with the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
“We know that lupus patients have an increased susceptibility to infections due to autoimmune dysregulation and use of immunosuppressive therapy,” explained Dr. Bonfá, who is clinical director of the largest tertiary referral center for autoimmune rheumatic diseases in Latin America, the University of São Paulo Faculty of Medicine Hospital Clinics.
“Our study demonstrates for the first time that lupus patients have an increased ARDS severity,” she added.
Prior to the meeting, the study was published in ACR Open Rheumatology.
Collating the evidence
Since the COVID-19 pandemic began, there have been more than 20 million confirmed cases of SARS-CoV-2 infection in Brazil and more than half a million deaths.
Dr. Bonfá presented the results of a cross-sectional study that was part of the country’s national Influenza Epidemiological Reporting Surveillance System. Data from 2020 were used, which included just over 252,000 individuals who had polymerase chain reaction–confirmed SARS-CoV-2 infection. Of these individuals, there were 319 consecutively recruited patients with SLE.
The aim was to look at the effect of being hospitalized for COVID-19–related ARDS on outcomes in people with SLE versus the general population.
ARDS was defined as a positive polymerase chain reaction test and accompanying flu-like symptoms with dyspnea, respiratory discomfort, persistent pressure in the chest, or desaturation less than 95% in room air or having a bluish tinge to the lips or face.
Other telling signs of a serious respiratory infection that were evaluated, but not mandatory for study eligibility, were loss of smell, impaired taste, typical CT findings, or having had contact with a confirmed COVID-19 case in the preceding 2 weeks.
Key findings
The risk for death from COVID-19–related ARDS was “more than double” in patients with SLE, compared with the general population, Dr. Bonfá reported. The relative risk in the fully adjusted, propensity-scored analysis was approximately 2.25.
That analysis did not account for other comorbidities but was fully adjusted for individuals’ age, sex, and region of Brazil where they lived. The latter was important, Dr. Bonfá said, because “we have a high disparity regarding health access and treatment among regions.”
Comorbidities considered as part of the analyses included arterial hypertension, diabetes, malignancies, neurologic disease, and diseases affecting the heart, lung, liver, and kidneys. Researchers also adjusted for smoking, alcohol intake, body weight, pregnancy, and transplantation.
SLE had a greater impact on individuals’ outcomes than all other comorbidities considered.
“We evaluated lupus as one comorbidity compared to all other comorbidities,” Dr. Bonfá explained.
SLE “more than doubled the chances” of dying from ARDS, she said. “This is [a] very impressive finding.”
They found that SLE was associated with an RR for death of 1.73, compared with non-SLE patients, when propensity-score matching without adjustment for comorbidities was used. The RR for death dropped to 1.40 but was still significant when researchers included comorbidities.
Dr. Bonfá and her team also looked at a combined endpoint of death, ICU admission, and need for mechanical ventilation. They found an increased risk in patients with SLE versus the general population in all their analyses, ranging from 1.70 if comorbidities were included in the model to 1.27 if they weren’t to 1.39 if propensity-score matching alone was used.
Got lupus? ‘Get vaccinated’
“The data we have are in nonvaccinated patients,” Dr. Bonfá said. “We didn’t have vaccines in 2020.”
Whether being vaccinated might make a different to the risks found in this study is an “interesting question,” and one that may be examined in the future.
Certainly, other work Dr. Bonfá has been involved in seems to point to a likely benefit of vaccination in patients with autoimmune diseases in terms of reducing mortality from COVID-19, even when rates of infection may be on the rise.
“There’s considerable vaccine hesitancy in SLE patients,” Chi-Chiu Mok, MD, of Tuen Mun Hospital in Hong Kong, observed in a separate presentation at the congress.
This may be for several reasons, such as worry that their disease may flare or the vaccine might compromise their drug treatment or result in uncommon complications.
However, “we should encourage our SLE patients to receive COVID-19 vaccination at a time of clinical remission or low disease activity state,” Dr. Mok advised.
“Physical distancing, protective masks, and personal hygiene [measures]” should also continue.
The bottom line for those with SLE is to get vaccinated, stressed Sandra Navarra, MD, of the University of Santo Tomas Hospital in Manila, the Philippines, during the discussion.
“There’s still so much out there that we do not know about,” she said. “Just get yourself vaccinated.”
The study had no outside funding. Dr. Bonfá, Dr. Mok, and Dr. Navarra reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
There is a significantly increased risk for acute respiratory distress syndrome (ARDS)–related death from COVID-19 among people with systemic lupus erythematous (SLE), compared with the general population, according to data collected in Brazil in 2020.
“Special care is therefore necessary for these patients, as well as reinforcement of the importance of preventive measures during a pandemic for this population,” said Eloisa Bonfá, MD, PhD, at the 14th International Congress on Systemic Lupus Erythematosus, which was held together with the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
“We know that lupus patients have an increased susceptibility to infections due to autoimmune dysregulation and use of immunosuppressive therapy,” explained Dr. Bonfá, who is clinical director of the largest tertiary referral center for autoimmune rheumatic diseases in Latin America, the University of São Paulo Faculty of Medicine Hospital Clinics.
“Our study demonstrates for the first time that lupus patients have an increased ARDS severity,” she added.
Prior to the meeting, the study was published in ACR Open Rheumatology.
Collating the evidence
Since the COVID-19 pandemic began, there have been more than 20 million confirmed cases of SARS-CoV-2 infection in Brazil and more than half a million deaths.
Dr. Bonfá presented the results of a cross-sectional study that was part of the country’s national Influenza Epidemiological Reporting Surveillance System. Data from 2020 were used, which included just over 252,000 individuals who had polymerase chain reaction–confirmed SARS-CoV-2 infection. Of these individuals, there were 319 consecutively recruited patients with SLE.
The aim was to look at the effect of being hospitalized for COVID-19–related ARDS on outcomes in people with SLE versus the general population.
ARDS was defined as a positive polymerase chain reaction test and accompanying flu-like symptoms with dyspnea, respiratory discomfort, persistent pressure in the chest, or desaturation less than 95% in room air or having a bluish tinge to the lips or face.
Other telling signs of a serious respiratory infection that were evaluated, but not mandatory for study eligibility, were loss of smell, impaired taste, typical CT findings, or having had contact with a confirmed COVID-19 case in the preceding 2 weeks.
Key findings
The risk for death from COVID-19–related ARDS was “more than double” in patients with SLE, compared with the general population, Dr. Bonfá reported. The relative risk in the fully adjusted, propensity-scored analysis was approximately 2.25.
That analysis did not account for other comorbidities but was fully adjusted for individuals’ age, sex, and region of Brazil where they lived. The latter was important, Dr. Bonfá said, because “we have a high disparity regarding health access and treatment among regions.”
Comorbidities considered as part of the analyses included arterial hypertension, diabetes, malignancies, neurologic disease, and diseases affecting the heart, lung, liver, and kidneys. Researchers also adjusted for smoking, alcohol intake, body weight, pregnancy, and transplantation.
SLE had a greater impact on individuals’ outcomes than all other comorbidities considered.
“We evaluated lupus as one comorbidity compared to all other comorbidities,” Dr. Bonfá explained.
SLE “more than doubled the chances” of dying from ARDS, she said. “This is [a] very impressive finding.”
They found that SLE was associated with an RR for death of 1.73, compared with non-SLE patients, when propensity-score matching without adjustment for comorbidities was used. The RR for death dropped to 1.40 but was still significant when researchers included comorbidities.
Dr. Bonfá and her team also looked at a combined endpoint of death, ICU admission, and need for mechanical ventilation. They found an increased risk in patients with SLE versus the general population in all their analyses, ranging from 1.70 if comorbidities were included in the model to 1.27 if they weren’t to 1.39 if propensity-score matching alone was used.
Got lupus? ‘Get vaccinated’
“The data we have are in nonvaccinated patients,” Dr. Bonfá said. “We didn’t have vaccines in 2020.”
Whether being vaccinated might make a different to the risks found in this study is an “interesting question,” and one that may be examined in the future.
Certainly, other work Dr. Bonfá has been involved in seems to point to a likely benefit of vaccination in patients with autoimmune diseases in terms of reducing mortality from COVID-19, even when rates of infection may be on the rise.
“There’s considerable vaccine hesitancy in SLE patients,” Chi-Chiu Mok, MD, of Tuen Mun Hospital in Hong Kong, observed in a separate presentation at the congress.
This may be for several reasons, such as worry that their disease may flare or the vaccine might compromise their drug treatment or result in uncommon complications.
However, “we should encourage our SLE patients to receive COVID-19 vaccination at a time of clinical remission or low disease activity state,” Dr. Mok advised.
“Physical distancing, protective masks, and personal hygiene [measures]” should also continue.
The bottom line for those with SLE is to get vaccinated, stressed Sandra Navarra, MD, of the University of Santo Tomas Hospital in Manila, the Philippines, during the discussion.
“There’s still so much out there that we do not know about,” she said. “Just get yourself vaccinated.”
The study had no outside funding. Dr. Bonfá, Dr. Mok, and Dr. Navarra reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
There is a significantly increased risk for acute respiratory distress syndrome (ARDS)–related death from COVID-19 among people with systemic lupus erythematous (SLE), compared with the general population, according to data collected in Brazil in 2020.
“Special care is therefore necessary for these patients, as well as reinforcement of the importance of preventive measures during a pandemic for this population,” said Eloisa Bonfá, MD, PhD, at the 14th International Congress on Systemic Lupus Erythematosus, which was held together with the 6th International Congress on Controversies in Rheumatology and Autoimmunity.
“We know that lupus patients have an increased susceptibility to infections due to autoimmune dysregulation and use of immunosuppressive therapy,” explained Dr. Bonfá, who is clinical director of the largest tertiary referral center for autoimmune rheumatic diseases in Latin America, the University of São Paulo Faculty of Medicine Hospital Clinics.
“Our study demonstrates for the first time that lupus patients have an increased ARDS severity,” she added.
Prior to the meeting, the study was published in ACR Open Rheumatology.
Collating the evidence
Since the COVID-19 pandemic began, there have been more than 20 million confirmed cases of SARS-CoV-2 infection in Brazil and more than half a million deaths.
Dr. Bonfá presented the results of a cross-sectional study that was part of the country’s national Influenza Epidemiological Reporting Surveillance System. Data from 2020 were used, which included just over 252,000 individuals who had polymerase chain reaction–confirmed SARS-CoV-2 infection. Of these individuals, there were 319 consecutively recruited patients with SLE.
The aim was to look at the effect of being hospitalized for COVID-19–related ARDS on outcomes in people with SLE versus the general population.
ARDS was defined as a positive polymerase chain reaction test and accompanying flu-like symptoms with dyspnea, respiratory discomfort, persistent pressure in the chest, or desaturation less than 95% in room air or having a bluish tinge to the lips or face.
Other telling signs of a serious respiratory infection that were evaluated, but not mandatory for study eligibility, were loss of smell, impaired taste, typical CT findings, or having had contact with a confirmed COVID-19 case in the preceding 2 weeks.
Key findings
The risk for death from COVID-19–related ARDS was “more than double” in patients with SLE, compared with the general population, Dr. Bonfá reported. The relative risk in the fully adjusted, propensity-scored analysis was approximately 2.25.
That analysis did not account for other comorbidities but was fully adjusted for individuals’ age, sex, and region of Brazil where they lived. The latter was important, Dr. Bonfá said, because “we have a high disparity regarding health access and treatment among regions.”
Comorbidities considered as part of the analyses included arterial hypertension, diabetes, malignancies, neurologic disease, and diseases affecting the heart, lung, liver, and kidneys. Researchers also adjusted for smoking, alcohol intake, body weight, pregnancy, and transplantation.
SLE had a greater impact on individuals’ outcomes than all other comorbidities considered.
“We evaluated lupus as one comorbidity compared to all other comorbidities,” Dr. Bonfá explained.
SLE “more than doubled the chances” of dying from ARDS, she said. “This is [a] very impressive finding.”
They found that SLE was associated with an RR for death of 1.73, compared with non-SLE patients, when propensity-score matching without adjustment for comorbidities was used. The RR for death dropped to 1.40 but was still significant when researchers included comorbidities.
Dr. Bonfá and her team also looked at a combined endpoint of death, ICU admission, and need for mechanical ventilation. They found an increased risk in patients with SLE versus the general population in all their analyses, ranging from 1.70 if comorbidities were included in the model to 1.27 if they weren’t to 1.39 if propensity-score matching alone was used.
Got lupus? ‘Get vaccinated’
“The data we have are in nonvaccinated patients,” Dr. Bonfá said. “We didn’t have vaccines in 2020.”
Whether being vaccinated might make a different to the risks found in this study is an “interesting question,” and one that may be examined in the future.
Certainly, other work Dr. Bonfá has been involved in seems to point to a likely benefit of vaccination in patients with autoimmune diseases in terms of reducing mortality from COVID-19, even when rates of infection may be on the rise.
“There’s considerable vaccine hesitancy in SLE patients,” Chi-Chiu Mok, MD, of Tuen Mun Hospital in Hong Kong, observed in a separate presentation at the congress.
This may be for several reasons, such as worry that their disease may flare or the vaccine might compromise their drug treatment or result in uncommon complications.
However, “we should encourage our SLE patients to receive COVID-19 vaccination at a time of clinical remission or low disease activity state,” Dr. Mok advised.
“Physical distancing, protective masks, and personal hygiene [measures]” should also continue.
The bottom line for those with SLE is to get vaccinated, stressed Sandra Navarra, MD, of the University of Santo Tomas Hospital in Manila, the Philippines, during the discussion.
“There’s still so much out there that we do not know about,” she said. “Just get yourself vaccinated.”
The study had no outside funding. Dr. Bonfá, Dr. Mok, and Dr. Navarra reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Duration of Adalimumab Therapy in Hidradenitis Suppurativa With and Without Oral Immunosuppressants
To the Editor:
The tumor necrosis factor α inhibitor adalimumab is the only US Food and Drug Administration–approved treatment of hidradenitis suppurativa (HS). Although 50.6% of patients fulfilled Hidradenitis Suppurativa Clinical Response criteria with adalimumab at 12 weeks, many responders were not satisfied with their disease control, and secondary loss of Hidradenitis Suppurativa Clinical Response fulfillment occurred in 15.9% of patients within approximately 3 years.1 Without other US Food and Drug Administration–approved HS treatments, some dermatologists have combined adalimumab with methotrexate (MTX) and/or mycophenolate mofetil (MMF) to attempt to increase the duration of satisfactory disease control while on adalimumab. Combining tumor necrosis factor α inhibitors with oral immunosuppressants is a well-established approach in psoriasis, psoriatic arthritis, and inflammatory bowel disease; however, to the best of our knowledge, this approach has not been studied for HS.2,3
To assess whether there is a role for combining adalimumab with MTX and/or MMF in the treatment of HS, we performed a single-institution retrospective chart review at the University of Connecticut Department of Dermatology to determine whether patients receiving combination therapy stayed on adalimumab longer than those who received adalimumab monotherapy. All patients receiving adalimumab for the treatment of HS with at least 1 follow-up visit 3 or more months after treatment initiation were included. Duration of treatment with adalimumab was defined as the length of time between initiation and termination of adalimumab, regardless of flares, adverse events, or addition of adjuvant therapy that occurred during this time span. Because standardized rating scales measuring the severity of HS at this time are not recorded routinely at our institution, treatment duration with adalimumab was used as a surrogate for measuring therapeutic success. Additionally, treatment duration is a meaningful end point, as patients with HS may require indefinite treatment. Patients were eligible for inclusion if they were receiving adalimumab for the treatment of HS. Patients were excluded if they were lost to follow-up or had received adalimumab for less than 6 months, as data suggest that biologics do not reach peak effect for up to 6 months in HS.4
We identified 116 eligible patients with HS, 32 of whom received combination therapy. Five patients received 40 mg of adalimumab every other week, and 111 patients received 40 mg of adalimumab each week. Patients receiving oral immunosuppressants were more likely to be male and as likely to be biologic naïve compared to patients on monotherapy (Table). The average weekly dose of MTX was 14.63 mg, and the average daily dose of MMF was 1000 mg. The average number of days between starting adalimumab and starting an oral immunosuppressant was 114.5 (SD, 217; median, 0) days. Reasons for discontinuation of adalimumab included insufficient response, noncompliance, dislike of injections, adverse events, fear of adverse events, other medical issues unrelated to HS, and insurance coverage issues. Patients who ended treatment with adalimumab owing to insurance coverage issues were still included in our study because insurance coverage remains a major determinant of treatment choice in HS and is relevant to the dynamics of medical decision-making.
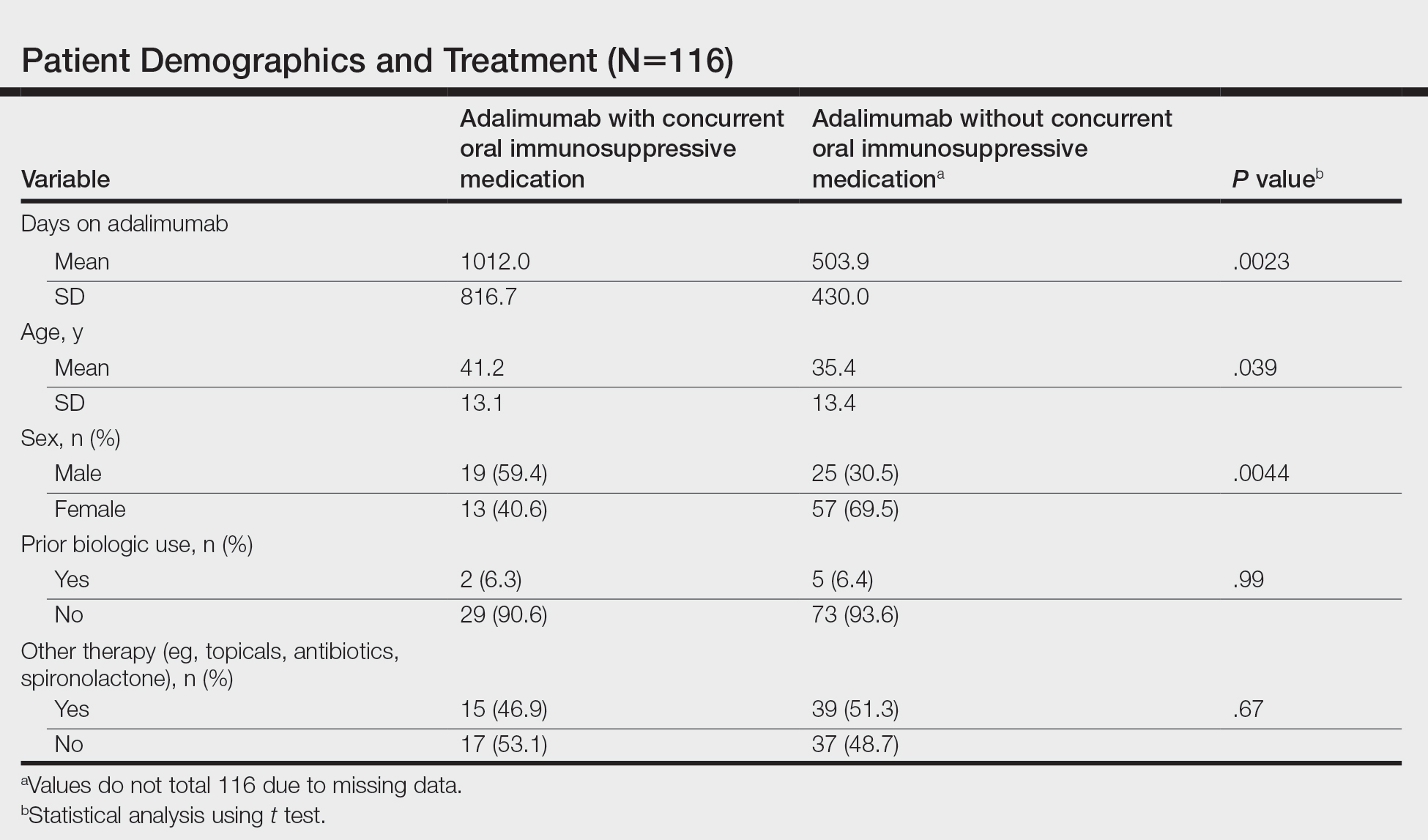
Statistical analysis was conducted on all patients inclusive of any reason for discontinuation to avoid bias in the calculation of treatment duration. Cox regression analysis was conducted for all independent variables and was noncontributory. Kaplan-Meier methodology was used to assess the duration of treatment of adalimumab with and without concomitant oral immunosuppressants, and quartile survival times were calculated. Quartile survival time is the time point after adalimumab initiation at which 25% of patients have discontinued adalimumab. We chose quartile survival time instead of average treatment duration to adequately power this study, given our small patient pool.
Although patients receiving adalimumab with oral immunosuppressants had a longer quartile treatment duration (450 days; 95% CI, 185-1800) than the group without oral immunosuppressants (360 days; 95% CI, 200-700), neither MTX nor MMF was shown to significantly prolong duration of therapy with adalimumab (log-rank test: P=.12). Additionally, patients receiving combination therapy were just as likely to discontinue adalimumab as those on monotherapy (χ2 test: P=.93). Patients who took both MTX and MMF at different times did show a statistically significant increase in adalimumab quartile treatment duration (1710 days; 95% CI, 1620 [upper limit not calculable]), but this is likely because these patients were kept on adalimumab while trialing adjunctive medications.
The results of our study indicate that MTX and MMF do not prolong duration of adalimumab therapy, which suggests that adalimumab combination therapy with MTX and MMF may not improve HS more than adalimumab alone, and/or partial responders to adalimumab monotherapy are unlikely to be converted to satisfactory responders with the addition of oral immunosuppressants. Limitations of our study include that it was retrospective, used treatment duration as a surrogate for objective efficacy measures, and relied on a single-institution data source. Additionally, owing to our small sample size, we were unable to account for certain potential confounders, including patient weight and insurance status. Future controlled prospective studies using objective end points are needed to further elucidate whether oral immunosuppressants have a role as an adjunct in the treatment of HS.
- Zouboulis CC, Okun MM, Prens EP, et al. Long-term adalimumab efficacy in patients with moderate-to-severe hidradenitis suppurativa/acne inversa: 3-year results of a phase 3 open-label extension study. J Am Acad Dermatol. 2019;80:60-69.e2. doi:10.1016/j.jaad.2018.05.040
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80:1029-1072. doi:10.1016/j.jaad.2018.11.057
- Sultan KS, Berkowitz JC, Khan S. Combination therapy for inflammatory bowel disease. World J Gastrointest Pharmacol Ther. 2017;8:103-113. doi:10.4292/wjgpt.v8.i2.103
- Prussick L, Rothstein B, Joshipura D, et al. Open-label, investigator-initiated, single-site exploratory trial evaluating secukinumab, an anti-interleukin-17A monoclonal antibody, for patients with moderate-to-severe hidradenitis suppurativa. Br J Dermatol. 2019;181:609-611.
To the Editor:
The tumor necrosis factor α inhibitor adalimumab is the only US Food and Drug Administration–approved treatment of hidradenitis suppurativa (HS). Although 50.6% of patients fulfilled Hidradenitis Suppurativa Clinical Response criteria with adalimumab at 12 weeks, many responders were not satisfied with their disease control, and secondary loss of Hidradenitis Suppurativa Clinical Response fulfillment occurred in 15.9% of patients within approximately 3 years.1 Without other US Food and Drug Administration–approved HS treatments, some dermatologists have combined adalimumab with methotrexate (MTX) and/or mycophenolate mofetil (MMF) to attempt to increase the duration of satisfactory disease control while on adalimumab. Combining tumor necrosis factor α inhibitors with oral immunosuppressants is a well-established approach in psoriasis, psoriatic arthritis, and inflammatory bowel disease; however, to the best of our knowledge, this approach has not been studied for HS.2,3
To assess whether there is a role for combining adalimumab with MTX and/or MMF in the treatment of HS, we performed a single-institution retrospective chart review at the University of Connecticut Department of Dermatology to determine whether patients receiving combination therapy stayed on adalimumab longer than those who received adalimumab monotherapy. All patients receiving adalimumab for the treatment of HS with at least 1 follow-up visit 3 or more months after treatment initiation were included. Duration of treatment with adalimumab was defined as the length of time between initiation and termination of adalimumab, regardless of flares, adverse events, or addition of adjuvant therapy that occurred during this time span. Because standardized rating scales measuring the severity of HS at this time are not recorded routinely at our institution, treatment duration with adalimumab was used as a surrogate for measuring therapeutic success. Additionally, treatment duration is a meaningful end point, as patients with HS may require indefinite treatment. Patients were eligible for inclusion if they were receiving adalimumab for the treatment of HS. Patients were excluded if they were lost to follow-up or had received adalimumab for less than 6 months, as data suggest that biologics do not reach peak effect for up to 6 months in HS.4
We identified 116 eligible patients with HS, 32 of whom received combination therapy. Five patients received 40 mg of adalimumab every other week, and 111 patients received 40 mg of adalimumab each week. Patients receiving oral immunosuppressants were more likely to be male and as likely to be biologic naïve compared to patients on monotherapy (Table). The average weekly dose of MTX was 14.63 mg, and the average daily dose of MMF was 1000 mg. The average number of days between starting adalimumab and starting an oral immunosuppressant was 114.5 (SD, 217; median, 0) days. Reasons for discontinuation of adalimumab included insufficient response, noncompliance, dislike of injections, adverse events, fear of adverse events, other medical issues unrelated to HS, and insurance coverage issues. Patients who ended treatment with adalimumab owing to insurance coverage issues were still included in our study because insurance coverage remains a major determinant of treatment choice in HS and is relevant to the dynamics of medical decision-making.
Statistical analysis was conducted on all patients inclusive of any reason for discontinuation to avoid bias in the calculation of treatment duration. Cox regression analysis was conducted for all independent variables and was noncontributory. Kaplan-Meier methodology was used to assess the duration of treatment of adalimumab with and without concomitant oral immunosuppressants, and quartile survival times were calculated. Quartile survival time is the time point after adalimumab initiation at which 25% of patients have discontinued adalimumab. We chose quartile survival time instead of average treatment duration to adequately power this study, given our small patient pool.
Although patients receiving adalimumab with oral immunosuppressants had a longer quartile treatment duration (450 days; 95% CI, 185-1800) than the group without oral immunosuppressants (360 days; 95% CI, 200-700), neither MTX nor MMF was shown to significantly prolong duration of therapy with adalimumab (log-rank test: P=.12). Additionally, patients receiving combination therapy were just as likely to discontinue adalimumab as those on monotherapy (χ2 test: P=.93). Patients who took both MTX and MMF at different times did show a statistically significant increase in adalimumab quartile treatment duration (1710 days; 95% CI, 1620 [upper limit not calculable]), but this is likely because these patients were kept on adalimumab while trialing adjunctive medications.
The results of our study indicate that MTX and MMF do not prolong duration of adalimumab therapy, which suggests that adalimumab combination therapy with MTX and MMF may not improve HS more than adalimumab alone, and/or partial responders to adalimumab monotherapy are unlikely to be converted to satisfactory responders with the addition of oral immunosuppressants. Limitations of our study include that it was retrospective, used treatment duration as a surrogate for objective efficacy measures, and relied on a single-institution data source. Additionally, owing to our small sample size, we were unable to account for certain potential confounders, including patient weight and insurance status. Future controlled prospective studies using objective end points are needed to further elucidate whether oral immunosuppressants have a role as an adjunct in the treatment of HS.
To the Editor:
The tumor necrosis factor α inhibitor adalimumab is the only US Food and Drug Administration–approved treatment of hidradenitis suppurativa (HS). Although 50.6% of patients fulfilled Hidradenitis Suppurativa Clinical Response criteria with adalimumab at 12 weeks, many responders were not satisfied with their disease control, and secondary loss of Hidradenitis Suppurativa Clinical Response fulfillment occurred in 15.9% of patients within approximately 3 years.1 Without other US Food and Drug Administration–approved HS treatments, some dermatologists have combined adalimumab with methotrexate (MTX) and/or mycophenolate mofetil (MMF) to attempt to increase the duration of satisfactory disease control while on adalimumab. Combining tumor necrosis factor α inhibitors with oral immunosuppressants is a well-established approach in psoriasis, psoriatic arthritis, and inflammatory bowel disease; however, to the best of our knowledge, this approach has not been studied for HS.2,3
To assess whether there is a role for combining adalimumab with MTX and/or MMF in the treatment of HS, we performed a single-institution retrospective chart review at the University of Connecticut Department of Dermatology to determine whether patients receiving combination therapy stayed on adalimumab longer than those who received adalimumab monotherapy. All patients receiving adalimumab for the treatment of HS with at least 1 follow-up visit 3 or more months after treatment initiation were included. Duration of treatment with adalimumab was defined as the length of time between initiation and termination of adalimumab, regardless of flares, adverse events, or addition of adjuvant therapy that occurred during this time span. Because standardized rating scales measuring the severity of HS at this time are not recorded routinely at our institution, treatment duration with adalimumab was used as a surrogate for measuring therapeutic success. Additionally, treatment duration is a meaningful end point, as patients with HS may require indefinite treatment. Patients were eligible for inclusion if they were receiving adalimumab for the treatment of HS. Patients were excluded if they were lost to follow-up or had received adalimumab for less than 6 months, as data suggest that biologics do not reach peak effect for up to 6 months in HS.4
We identified 116 eligible patients with HS, 32 of whom received combination therapy. Five patients received 40 mg of adalimumab every other week, and 111 patients received 40 mg of adalimumab each week. Patients receiving oral immunosuppressants were more likely to be male and as likely to be biologic naïve compared to patients on monotherapy (Table). The average weekly dose of MTX was 14.63 mg, and the average daily dose of MMF was 1000 mg. The average number of days between starting adalimumab and starting an oral immunosuppressant was 114.5 (SD, 217; median, 0) days. Reasons for discontinuation of adalimumab included insufficient response, noncompliance, dislike of injections, adverse events, fear of adverse events, other medical issues unrelated to HS, and insurance coverage issues. Patients who ended treatment with adalimumab owing to insurance coverage issues were still included in our study because insurance coverage remains a major determinant of treatment choice in HS and is relevant to the dynamics of medical decision-making.
Statistical analysis was conducted on all patients inclusive of any reason for discontinuation to avoid bias in the calculation of treatment duration. Cox regression analysis was conducted for all independent variables and was noncontributory. Kaplan-Meier methodology was used to assess the duration of treatment of adalimumab with and without concomitant oral immunosuppressants, and quartile survival times were calculated. Quartile survival time is the time point after adalimumab initiation at which 25% of patients have discontinued adalimumab. We chose quartile survival time instead of average treatment duration to adequately power this study, given our small patient pool.
Although patients receiving adalimumab with oral immunosuppressants had a longer quartile treatment duration (450 days; 95% CI, 185-1800) than the group without oral immunosuppressants (360 days; 95% CI, 200-700), neither MTX nor MMF was shown to significantly prolong duration of therapy with adalimumab (log-rank test: P=.12). Additionally, patients receiving combination therapy were just as likely to discontinue adalimumab as those on monotherapy (χ2 test: P=.93). Patients who took both MTX and MMF at different times did show a statistically significant increase in adalimumab quartile treatment duration (1710 days; 95% CI, 1620 [upper limit not calculable]), but this is likely because these patients were kept on adalimumab while trialing adjunctive medications.
The results of our study indicate that MTX and MMF do not prolong duration of adalimumab therapy, which suggests that adalimumab combination therapy with MTX and MMF may not improve HS more than adalimumab alone, and/or partial responders to adalimumab monotherapy are unlikely to be converted to satisfactory responders with the addition of oral immunosuppressants. Limitations of our study include that it was retrospective, used treatment duration as a surrogate for objective efficacy measures, and relied on a single-institution data source. Additionally, owing to our small sample size, we were unable to account for certain potential confounders, including patient weight and insurance status. Future controlled prospective studies using objective end points are needed to further elucidate whether oral immunosuppressants have a role as an adjunct in the treatment of HS.
- Zouboulis CC, Okun MM, Prens EP, et al. Long-term adalimumab efficacy in patients with moderate-to-severe hidradenitis suppurativa/acne inversa: 3-year results of a phase 3 open-label extension study. J Am Acad Dermatol. 2019;80:60-69.e2. doi:10.1016/j.jaad.2018.05.040
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80:1029-1072. doi:10.1016/j.jaad.2018.11.057
- Sultan KS, Berkowitz JC, Khan S. Combination therapy for inflammatory bowel disease. World J Gastrointest Pharmacol Ther. 2017;8:103-113. doi:10.4292/wjgpt.v8.i2.103
- Prussick L, Rothstein B, Joshipura D, et al. Open-label, investigator-initiated, single-site exploratory trial evaluating secukinumab, an anti-interleukin-17A monoclonal antibody, for patients with moderate-to-severe hidradenitis suppurativa. Br J Dermatol. 2019;181:609-611.
- Zouboulis CC, Okun MM, Prens EP, et al. Long-term adalimumab efficacy in patients with moderate-to-severe hidradenitis suppurativa/acne inversa: 3-year results of a phase 3 open-label extension study. J Am Acad Dermatol. 2019;80:60-69.e2. doi:10.1016/j.jaad.2018.05.040
- Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80:1029-1072. doi:10.1016/j.jaad.2018.11.057
- Sultan KS, Berkowitz JC, Khan S. Combination therapy for inflammatory bowel disease. World J Gastrointest Pharmacol Ther. 2017;8:103-113. doi:10.4292/wjgpt.v8.i2.103
- Prussick L, Rothstein B, Joshipura D, et al. Open-label, investigator-initiated, single-site exploratory trial evaluating secukinumab, an anti-interleukin-17A monoclonal antibody, for patients with moderate-to-severe hidradenitis suppurativa. Br J Dermatol. 2019;181:609-611.
Practice Points
- Adalimumab is the only medication approved by the US Food and Drug Administration for treatment of hidradenitis suppurativa (HS), yet many patients on adalimumab do not achieve satisfactory results. New treatment options are in demand for patients affected by HS.
- Although combining tumor necrosis factor α inhibitors with oral immunosuppressants such as methotrexate and mycophenolate mofetil appears to be beneficial in treating other conditions such as psoriasis, these treatments may not have as great a benefit for patients with HS.
FDA approves avacopan for rare ANCA autoimmune disease
U.S. regulators approved avacopan (Tavneos) for a rare immune disorder after receiving additional information to address concerns raised about the drug that were previously discussed at a public meeting in May.

ChemoCentryx, the drug’s manufacturer, today announced that the U.S. Food and Drug Administration approved the drug as an adjunctive treatment for severe active antineutrophil cytoplasmic autoantibody–associated vasculitis (also known as ANCA-associated vasculitis or ANCA vasculitis).
This systemic disease results from overactivation of the complement system, leading to inflammation and eventual destruction of small blood vessels. This can lead to organ damage and failure, with the kidney as the major target, said the company in a statement.
The avacopan approval was based in large part on the results of the ADVOCATE trial, which were highlighted in a February 2021 editorial in the New England Journal of Medicine , titled “Avacopan – Time to replace glucocorticoids?” But the FDA-approved indication for avacopan is as an adjunctive treatment of adult patients with severe active ANCA-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. “Tavneos does not eliminate glucocorticoid use,” the label states.
The ADVOCATE trial was a global, randomized, double-blind, active-controlled, double-dummy phase 3 trial of 330 patients with ANCA-associated vasculitis conducted in 20 countries, ChemoCentryx said. Participants were randomly assigned to receive either rituximab or cyclophosphamide (followed by azathioprine/mycophenolate) and either avacopan or study-supplied oral prednisone.
Subjects in both treatment groups could also receive nonprotocol glucocorticoids as needed. The study met its primary endpoints of disease remission at 26 weeks and sustained remission at 52 weeks, as assessed by the Birmingham Vasculitis Activity Score (BVAS), ChemoCentryx said. Common adverse reactions among study participants included nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increase, and paresthesia.
In the ChemoCentryx statement, Peter A. Merkel, MD, MPH, a consultant to the company and the chief of rheumatology at the University of Pennsylvania, Philadelphia, called the avacopan clearance a “first-in-a-decade approval of a medicine for ANCA-associated vasculitis.”
“Patients will now have access to a new class of medication that provides beneficial effects for the treatment of ANCA-associated vasculitis,” Dr. Merkel said.
In reviewing the avacopan application, the FDA noted that the medicine is intended to treat “a rare and serious disease associated with high morbidity and increased mortality.”
“It is also a disease with high unmet need for new therapies,” the FDA staff said in a review of the ChemoCentryx application for approval of avacopan, which was posted online ahead of a meeting this past May.
Previous FDA concerns
In that review, FDA staff made public various concerns about the evidence used in seeking approval of the medicine. The FDA staff said there were “substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.”
Members of the FDA’s Arthritis Advisory Committee voted 10-8 on May 6 on a question of whether the risk-benefit profile of avacopan is adequate to support approval. The panel also voted 9-9 on whether the efficacy data support approval of avacopan, and 10-8 that the safety profile of avacopan is adequate to support approval.
ChemoCentryx in July said it filed an amendment to its new drug application (NDA) for avacopan. This appears to have answered regulators’ questions about the drug.
On a call with analysts Friday, ChemoCentryx officials outlined a marketing strategy for avacopan, with efforts focused on reaching influential rheumatologists and nephrologists. The company will set a U.S. wholesale acquisition cost for the drug of about $150,000-$200,000 a patient, in keeping with the range of prices often seen for orphan drugs. ChemoCentryx said it intends to offer financial support programs for the medicine.
ChemoCentryx said avacopan is also approved for the treatment of microscopic polyangiitis and granulomatosis with polyangiitis (the two main forms of ANCA-associated vasculitis) in Japan. The regulatory decision in Europe is expected by the end of this year.
A version of this article first appeared on Medscape.com.
U.S. regulators approved avacopan (Tavneos) for a rare immune disorder after receiving additional information to address concerns raised about the drug that were previously discussed at a public meeting in May.
ChemoCentryx, the drug’s manufacturer, today announced that the U.S. Food and Drug Administration approved the drug as an adjunctive treatment for severe active antineutrophil cytoplasmic autoantibody–associated vasculitis (also known as ANCA-associated vasculitis or ANCA vasculitis).
This systemic disease results from overactivation of the complement system, leading to inflammation and eventual destruction of small blood vessels. This can lead to organ damage and failure, with the kidney as the major target, said the company in a statement.
The avacopan approval was based in large part on the results of the ADVOCATE trial, which were highlighted in a February 2021 editorial in the New England Journal of Medicine , titled “Avacopan – Time to replace glucocorticoids?” But the FDA-approved indication for avacopan is as an adjunctive treatment of adult patients with severe active ANCA-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. “Tavneos does not eliminate glucocorticoid use,” the label states.
The ADVOCATE trial was a global, randomized, double-blind, active-controlled, double-dummy phase 3 trial of 330 patients with ANCA-associated vasculitis conducted in 20 countries, ChemoCentryx said. Participants were randomly assigned to receive either rituximab or cyclophosphamide (followed by azathioprine/mycophenolate) and either avacopan or study-supplied oral prednisone.
Subjects in both treatment groups could also receive nonprotocol glucocorticoids as needed. The study met its primary endpoints of disease remission at 26 weeks and sustained remission at 52 weeks, as assessed by the Birmingham Vasculitis Activity Score (BVAS), ChemoCentryx said. Common adverse reactions among study participants included nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increase, and paresthesia.
In the ChemoCentryx statement, Peter A. Merkel, MD, MPH, a consultant to the company and the chief of rheumatology at the University of Pennsylvania, Philadelphia, called the avacopan clearance a “first-in-a-decade approval of a medicine for ANCA-associated vasculitis.”
“Patients will now have access to a new class of medication that provides beneficial effects for the treatment of ANCA-associated vasculitis,” Dr. Merkel said.
In reviewing the avacopan application, the FDA noted that the medicine is intended to treat “a rare and serious disease associated with high morbidity and increased mortality.”
“It is also a disease with high unmet need for new therapies,” the FDA staff said in a review of the ChemoCentryx application for approval of avacopan, which was posted online ahead of a meeting this past May.
Previous FDA concerns
In that review, FDA staff made public various concerns about the evidence used in seeking approval of the medicine. The FDA staff said there were “substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.”
Members of the FDA’s Arthritis Advisory Committee voted 10-8 on May 6 on a question of whether the risk-benefit profile of avacopan is adequate to support approval. The panel also voted 9-9 on whether the efficacy data support approval of avacopan, and 10-8 that the safety profile of avacopan is adequate to support approval.
ChemoCentryx in July said it filed an amendment to its new drug application (NDA) for avacopan. This appears to have answered regulators’ questions about the drug.
On a call with analysts Friday, ChemoCentryx officials outlined a marketing strategy for avacopan, with efforts focused on reaching influential rheumatologists and nephrologists. The company will set a U.S. wholesale acquisition cost for the drug of about $150,000-$200,000 a patient, in keeping with the range of prices often seen for orphan drugs. ChemoCentryx said it intends to offer financial support programs for the medicine.
ChemoCentryx said avacopan is also approved for the treatment of microscopic polyangiitis and granulomatosis with polyangiitis (the two main forms of ANCA-associated vasculitis) in Japan. The regulatory decision in Europe is expected by the end of this year.
A version of this article first appeared on Medscape.com.
U.S. regulators approved avacopan (Tavneos) for a rare immune disorder after receiving additional information to address concerns raised about the drug that were previously discussed at a public meeting in May.
ChemoCentryx, the drug’s manufacturer, today announced that the U.S. Food and Drug Administration approved the drug as an adjunctive treatment for severe active antineutrophil cytoplasmic autoantibody–associated vasculitis (also known as ANCA-associated vasculitis or ANCA vasculitis).
This systemic disease results from overactivation of the complement system, leading to inflammation and eventual destruction of small blood vessels. This can lead to organ damage and failure, with the kidney as the major target, said the company in a statement.
The avacopan approval was based in large part on the results of the ADVOCATE trial, which were highlighted in a February 2021 editorial in the New England Journal of Medicine , titled “Avacopan – Time to replace glucocorticoids?” But the FDA-approved indication for avacopan is as an adjunctive treatment of adult patients with severe active ANCA-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. “Tavneos does not eliminate glucocorticoid use,” the label states.
The ADVOCATE trial was a global, randomized, double-blind, active-controlled, double-dummy phase 3 trial of 330 patients with ANCA-associated vasculitis conducted in 20 countries, ChemoCentryx said. Participants were randomly assigned to receive either rituximab or cyclophosphamide (followed by azathioprine/mycophenolate) and either avacopan or study-supplied oral prednisone.
Subjects in both treatment groups could also receive nonprotocol glucocorticoids as needed. The study met its primary endpoints of disease remission at 26 weeks and sustained remission at 52 weeks, as assessed by the Birmingham Vasculitis Activity Score (BVAS), ChemoCentryx said. Common adverse reactions among study participants included nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increase, and paresthesia.
In the ChemoCentryx statement, Peter A. Merkel, MD, MPH, a consultant to the company and the chief of rheumatology at the University of Pennsylvania, Philadelphia, called the avacopan clearance a “first-in-a-decade approval of a medicine for ANCA-associated vasculitis.”
“Patients will now have access to a new class of medication that provides beneficial effects for the treatment of ANCA-associated vasculitis,” Dr. Merkel said.
In reviewing the avacopan application, the FDA noted that the medicine is intended to treat “a rare and serious disease associated with high morbidity and increased mortality.”
“It is also a disease with high unmet need for new therapies,” the FDA staff said in a review of the ChemoCentryx application for approval of avacopan, which was posted online ahead of a meeting this past May.
Previous FDA concerns
In that review, FDA staff made public various concerns about the evidence used in seeking approval of the medicine. The FDA staff said there were “substantial uncertainties around the phase 3 study design and results, raising questions about the adequacy of this single trial to inform the benefit-risk assessment.”
Members of the FDA’s Arthritis Advisory Committee voted 10-8 on May 6 on a question of whether the risk-benefit profile of avacopan is adequate to support approval. The panel also voted 9-9 on whether the efficacy data support approval of avacopan, and 10-8 that the safety profile of avacopan is adequate to support approval.
ChemoCentryx in July said it filed an amendment to its new drug application (NDA) for avacopan. This appears to have answered regulators’ questions about the drug.
On a call with analysts Friday, ChemoCentryx officials outlined a marketing strategy for avacopan, with efforts focused on reaching influential rheumatologists and nephrologists. The company will set a U.S. wholesale acquisition cost for the drug of about $150,000-$200,000 a patient, in keeping with the range of prices often seen for orphan drugs. ChemoCentryx said it intends to offer financial support programs for the medicine.
ChemoCentryx said avacopan is also approved for the treatment of microscopic polyangiitis and granulomatosis with polyangiitis (the two main forms of ANCA-associated vasculitis) in Japan. The regulatory decision in Europe is expected by the end of this year.
A version of this article first appeared on Medscape.com.
NIAMS director reflects on her mentors, spotlights research projects underway
After many years at the University of California, San Francisco, Lindsey A. Criswell, MD, MPH, DSc, began a new chapter in February 2021 as the director of the National Institute of Arthritis and Musculoskeletal and Skin Disease, part of the National Institutes of Health. NIH Director Francis S. Collins, MD, PhD, selected her for the post.

“Dr. Criswell has rich experience as a clinician, researcher, and administrator,” Dr. Collins said in a prepared statement. “Her ability to oversee the research program of one of the country’s top research-intensive medical schools, and her expertise in autoimmune diseases, including rheumatoid arthritis and lupus, make her well positioned to direct NIAMS.” Dr. Criswell, a rheumatologist, was named a full professor of medicine at UCSF in 2007 and had served as vice chancellor of research at the university since 2017. She has authored more than 250 peer-reviewed scientific papers, and her efforts have contributed to the identification of more than 30 genes linked to autoimmune disorders. In her first media interview, Dr. Criswell opens up about her mentors, operational challenges posed by the COVID-19 pandemic, and highlights many NIAMS research projects underway.
Who inspired you most early in your career as a physician scientist? I have had great opportunities to work with fabulous mentors. Wallace (Wally) Epstein, MD, was my mentor when I was a rheumatology fellow and junior faculty member at UCSF. He was broadly admired for the breadth of his experience as a clinician and a researcher, and he was noteworthy at that time for his strong support for women and students of color. One of the many things I appreciated about him was his diverse range of interests outside of work, which included cello playing and woodworking.
Another mentor was Ephraim (Eph) Engleman, MD, the first academic rheumatologist in California. Eph continued to see patients beyond the age of 100. Perhaps his most important contributions were his efforts towards advocacy for funding for research and education in rheumatology. A prodigy violinist, he too had a broad range of personal interests.
What research into the genetics and epidemiology of human autoimmune disease that you have been a part of has most surprised you, in term of its ultimate clinical impact? Some of my most rewarding and impactful work has focused on the shared genetic basis of autoimmune diseases. We’ve identified dozens of genes that contribute to the risk and outcome of rheumatoid arthritis, lupus, and other autoimmune disorders. These discoveries regarding shared genes and pathways among such a diverse set of conditions have helped to inform optimal therapeutic target and treatment strategies across multiple diseases. For example, exploration of RA genes and pathways has revealed that approved agents for other conditions, such as cancer, may be appropriately repurposed for the treatment of RA. These are critical observations that have the potential to dramatically accelerate progress in developing new therapies for autoimmune diseases, such as RA.
Did you have much interaction with Stephen I. Katz, MD, PhD, your longtime predecessor who passed away unexpectedly in 2018? If so, what do you remember most about him? I regret that I had very little interaction with Steve, but I am well aware of the impact he had on NIAMS, NIH, and the research enterprise overall. He inspired so many people in a personal way, and I am energized by the legacy that he left behind.
What are your goals for the early part of your tenure as the new director of NIAMS? An important goal is getting to know the NIAMS community and expanding my knowledge of the Institute’s musculoskeletal and skin portfolios. I am also conducting outreach to Institute/Center directors and other NIH leadership to increase opportunities for input and advice. In doing this, I am identifying shared research interests, best practices, and potential partners for possible future collaborations. Another important goal is to increase NIAMS’ visibility within and beyond NIH. Ultimately, I want to contribute to the great work of the Institute and improve the lives of people with rheumatic, musculoskeletal, and skin diseases.
How would you characterize your management style? I like to lead with a flat hierarchy and work collectively to address opportunities and challenges. I value team building and tend to tap a variety of perspectives and expertise at all levels to achieve consensus, where possible.
The Accelerating Medicines Partnership (AMP) program was launched in 2014, with projects in three disease areas including the autoimmune disorders RA and lupus. What are some recent highlights from this program with respect to RA and lupus? AMP RA/SLE was dedicated to identifying promising therapeutic targets for RA and systemic lupus erythematosus. AMP-funded researchers have applied cutting-edge technologies to study cells from the synovial tissues of the joints of people with RA, and from the kidneys of people with lupus nephritis. In 2014, studying tissues in patients where the disease is active was a novel approach, since most research was conducted in mouse models or human blood samples.
The AMP RA/SLE Network developed a rich dataset that is available to the research community. Investigators are now using the data to facilitate RA and lupus research. For example, using AMP data, NIAMS-supported researchers identified potential biomarkers that could help predict an imminent RA flare. Work from another NIAMS-supported group suggests that targeting the regulatory transcription factor HIF-1, which drives inflammation and tissue damage, might be an effective approach for treating renal injury in lupus.
The data generated are accessible to the scientific community through two NIH websites: the database of Genotypes and Phenotypes (dbGaP) and the Immunology Database and Analysis Portal (IMMPORT).
Given the success of AMP RA/SLE, NIH plans to launch an “AMP 2.0” later in 2021. The AMP Autoimmune and Immune-Mediated Diseases (AMP AIM) program will provide an opportunity to leverage the accomplishments of AMP RA/SLE to new conditions, including psoriatic spectrum diseases and Sjögren’s syndrome.
What are some recent highlights from NIAMS-supported research in skin diseases? NIAMS-supported investigators continue to make significant strides in our understanding of skin biology and disease. For example, researchers recently demonstrated that imiquimod, a drug used to treat precancerous skin lesions, can help mouse ear wounds heal without scarring.
Another team addressed the safety and potential benefit of Staphylococcus hominis A9, a bacterium isolated from healthy human skin, as a topical therapy for atopic dermatitis.
Moving forward, AMP AIM will refine and extend the single-cell analysis of tissues to additional diseases, including psoriasis, setting the stage for the discovery of new therapeutic targets for the disease.
How has the COVID-19 pandemic changed the landscape of research, at least for the short term? This is a once-in-a-century pandemic that none of us were fully prepared for. We understand that it has been particularly challenging for women scientists, scientists with young children, and trainees and junior faculty who are at critically important and vulnerable stages of their careers. There isn’t a lab or clinical setting that hasn’t been negatively impacted in some way.
During the pandemic, the NIH instituted administrative flexibilities to support the grantee community, including extensions in time. In addition, the agency has issued several funding opportunities specific to COVID-19, some of which involve NIAMS participation.
What is NIAMS doing to help early/young investigators as well as female investigators and those from minority groups? Structural racism in biomedical research is a heightened concern. Earlier this year, Dr. Collins established the UNITE initiative to address structural racism and promote racial equity and inclusion at the NIH and within the larger biomedical community that we support. NIAMS is fully committed to this effort. One example is the Diversity Supplement Program, which is designed to attract and encourage eligible individuals from underrepresented populations to research careers.
Early-stage investigators are another top priority. In a tribute to the beloved former NIAMS director, NIH recently established the Stephen I. Katz Early Stage Investigator Research Grant Program. The R01 award provides support for a project unrelated to an early investigator’s area of postdoctoral study. (No preliminary data are allowed.) This award mechanism is a unique opportunity for early-stage investigators to take their research in a completely new direction.
Managing work and family life is an important concern, particularly for female investigators. Many NIH grant awards allow for reimbursement of actual, allowable costs incurred for childcare and parental leave. The NIH is exploring initiatives to promote research continuity and retention of eligible investigators facing major life events, such as pregnancy, childbirth, and adoption, at vulnerable career stages.
Who inspires you most in your work today? I am inspired by the ongoing struggles of our patients, junior investigators, and by the committed staff members on my team.
After many years at the University of California, San Francisco, Lindsey A. Criswell, MD, MPH, DSc, began a new chapter in February 2021 as the director of the National Institute of Arthritis and Musculoskeletal and Skin Disease, part of the National Institutes of Health. NIH Director Francis S. Collins, MD, PhD, selected her for the post.
“Dr. Criswell has rich experience as a clinician, researcher, and administrator,” Dr. Collins said in a prepared statement. “Her ability to oversee the research program of one of the country’s top research-intensive medical schools, and her expertise in autoimmune diseases, including rheumatoid arthritis and lupus, make her well positioned to direct NIAMS.” Dr. Criswell, a rheumatologist, was named a full professor of medicine at UCSF in 2007 and had served as vice chancellor of research at the university since 2017. She has authored more than 250 peer-reviewed scientific papers, and her efforts have contributed to the identification of more than 30 genes linked to autoimmune disorders. In her first media interview, Dr. Criswell opens up about her mentors, operational challenges posed by the COVID-19 pandemic, and highlights many NIAMS research projects underway.
Who inspired you most early in your career as a physician scientist? I have had great opportunities to work with fabulous mentors. Wallace (Wally) Epstein, MD, was my mentor when I was a rheumatology fellow and junior faculty member at UCSF. He was broadly admired for the breadth of his experience as a clinician and a researcher, and he was noteworthy at that time for his strong support for women and students of color. One of the many things I appreciated about him was his diverse range of interests outside of work, which included cello playing and woodworking.
Another mentor was Ephraim (Eph) Engleman, MD, the first academic rheumatologist in California. Eph continued to see patients beyond the age of 100. Perhaps his most important contributions were his efforts towards advocacy for funding for research and education in rheumatology. A prodigy violinist, he too had a broad range of personal interests.
What research into the genetics and epidemiology of human autoimmune disease that you have been a part of has most surprised you, in term of its ultimate clinical impact? Some of my most rewarding and impactful work has focused on the shared genetic basis of autoimmune diseases. We’ve identified dozens of genes that contribute to the risk and outcome of rheumatoid arthritis, lupus, and other autoimmune disorders. These discoveries regarding shared genes and pathways among such a diverse set of conditions have helped to inform optimal therapeutic target and treatment strategies across multiple diseases. For example, exploration of RA genes and pathways has revealed that approved agents for other conditions, such as cancer, may be appropriately repurposed for the treatment of RA. These are critical observations that have the potential to dramatically accelerate progress in developing new therapies for autoimmune diseases, such as RA.
Did you have much interaction with Stephen I. Katz, MD, PhD, your longtime predecessor who passed away unexpectedly in 2018? If so, what do you remember most about him? I regret that I had very little interaction with Steve, but I am well aware of the impact he had on NIAMS, NIH, and the research enterprise overall. He inspired so many people in a personal way, and I am energized by the legacy that he left behind.
What are your goals for the early part of your tenure as the new director of NIAMS? An important goal is getting to know the NIAMS community and expanding my knowledge of the Institute’s musculoskeletal and skin portfolios. I am also conducting outreach to Institute/Center directors and other NIH leadership to increase opportunities for input and advice. In doing this, I am identifying shared research interests, best practices, and potential partners for possible future collaborations. Another important goal is to increase NIAMS’ visibility within and beyond NIH. Ultimately, I want to contribute to the great work of the Institute and improve the lives of people with rheumatic, musculoskeletal, and skin diseases.
How would you characterize your management style? I like to lead with a flat hierarchy and work collectively to address opportunities and challenges. I value team building and tend to tap a variety of perspectives and expertise at all levels to achieve consensus, where possible.
The Accelerating Medicines Partnership (AMP) program was launched in 2014, with projects in three disease areas including the autoimmune disorders RA and lupus. What are some recent highlights from this program with respect to RA and lupus? AMP RA/SLE was dedicated to identifying promising therapeutic targets for RA and systemic lupus erythematosus. AMP-funded researchers have applied cutting-edge technologies to study cells from the synovial tissues of the joints of people with RA, and from the kidneys of people with lupus nephritis. In 2014, studying tissues in patients where the disease is active was a novel approach, since most research was conducted in mouse models or human blood samples.
The AMP RA/SLE Network developed a rich dataset that is available to the research community. Investigators are now using the data to facilitate RA and lupus research. For example, using AMP data, NIAMS-supported researchers identified potential biomarkers that could help predict an imminent RA flare. Work from another NIAMS-supported group suggests that targeting the regulatory transcription factor HIF-1, which drives inflammation and tissue damage, might be an effective approach for treating renal injury in lupus.
The data generated are accessible to the scientific community through two NIH websites: the database of Genotypes and Phenotypes (dbGaP) and the Immunology Database and Analysis Portal (IMMPORT).
Given the success of AMP RA/SLE, NIH plans to launch an “AMP 2.0” later in 2021. The AMP Autoimmune and Immune-Mediated Diseases (AMP AIM) program will provide an opportunity to leverage the accomplishments of AMP RA/SLE to new conditions, including psoriatic spectrum diseases and Sjögren’s syndrome.
What are some recent highlights from NIAMS-supported research in skin diseases? NIAMS-supported investigators continue to make significant strides in our understanding of skin biology and disease. For example, researchers recently demonstrated that imiquimod, a drug used to treat precancerous skin lesions, can help mouse ear wounds heal without scarring.
Another team addressed the safety and potential benefit of Staphylococcus hominis A9, a bacterium isolated from healthy human skin, as a topical therapy for atopic dermatitis.
Moving forward, AMP AIM will refine and extend the single-cell analysis of tissues to additional diseases, including psoriasis, setting the stage for the discovery of new therapeutic targets for the disease.
How has the COVID-19 pandemic changed the landscape of research, at least for the short term? This is a once-in-a-century pandemic that none of us were fully prepared for. We understand that it has been particularly challenging for women scientists, scientists with young children, and trainees and junior faculty who are at critically important and vulnerable stages of their careers. There isn’t a lab or clinical setting that hasn’t been negatively impacted in some way.
During the pandemic, the NIH instituted administrative flexibilities to support the grantee community, including extensions in time. In addition, the agency has issued several funding opportunities specific to COVID-19, some of which involve NIAMS participation.
What is NIAMS doing to help early/young investigators as well as female investigators and those from minority groups? Structural racism in biomedical research is a heightened concern. Earlier this year, Dr. Collins established the UNITE initiative to address structural racism and promote racial equity and inclusion at the NIH and within the larger biomedical community that we support. NIAMS is fully committed to this effort. One example is the Diversity Supplement Program, which is designed to attract and encourage eligible individuals from underrepresented populations to research careers.
Early-stage investigators are another top priority. In a tribute to the beloved former NIAMS director, NIH recently established the Stephen I. Katz Early Stage Investigator Research Grant Program. The R01 award provides support for a project unrelated to an early investigator’s area of postdoctoral study. (No preliminary data are allowed.) This award mechanism is a unique opportunity for early-stage investigators to take their research in a completely new direction.
Managing work and family life is an important concern, particularly for female investigators. Many NIH grant awards allow for reimbursement of actual, allowable costs incurred for childcare and parental leave. The NIH is exploring initiatives to promote research continuity and retention of eligible investigators facing major life events, such as pregnancy, childbirth, and adoption, at vulnerable career stages.
Who inspires you most in your work today? I am inspired by the ongoing struggles of our patients, junior investigators, and by the committed staff members on my team.
After many years at the University of California, San Francisco, Lindsey A. Criswell, MD, MPH, DSc, began a new chapter in February 2021 as the director of the National Institute of Arthritis and Musculoskeletal and Skin Disease, part of the National Institutes of Health. NIH Director Francis S. Collins, MD, PhD, selected her for the post.
“Dr. Criswell has rich experience as a clinician, researcher, and administrator,” Dr. Collins said in a prepared statement. “Her ability to oversee the research program of one of the country’s top research-intensive medical schools, and her expertise in autoimmune diseases, including rheumatoid arthritis and lupus, make her well positioned to direct NIAMS.” Dr. Criswell, a rheumatologist, was named a full professor of medicine at UCSF in 2007 and had served as vice chancellor of research at the university since 2017. She has authored more than 250 peer-reviewed scientific papers, and her efforts have contributed to the identification of more than 30 genes linked to autoimmune disorders. In her first media interview, Dr. Criswell opens up about her mentors, operational challenges posed by the COVID-19 pandemic, and highlights many NIAMS research projects underway.
Who inspired you most early in your career as a physician scientist? I have had great opportunities to work with fabulous mentors. Wallace (Wally) Epstein, MD, was my mentor when I was a rheumatology fellow and junior faculty member at UCSF. He was broadly admired for the breadth of his experience as a clinician and a researcher, and he was noteworthy at that time for his strong support for women and students of color. One of the many things I appreciated about him was his diverse range of interests outside of work, which included cello playing and woodworking.
Another mentor was Ephraim (Eph) Engleman, MD, the first academic rheumatologist in California. Eph continued to see patients beyond the age of 100. Perhaps his most important contributions were his efforts towards advocacy for funding for research and education in rheumatology. A prodigy violinist, he too had a broad range of personal interests.
What research into the genetics and epidemiology of human autoimmune disease that you have been a part of has most surprised you, in term of its ultimate clinical impact? Some of my most rewarding and impactful work has focused on the shared genetic basis of autoimmune diseases. We’ve identified dozens of genes that contribute to the risk and outcome of rheumatoid arthritis, lupus, and other autoimmune disorders. These discoveries regarding shared genes and pathways among such a diverse set of conditions have helped to inform optimal therapeutic target and treatment strategies across multiple diseases. For example, exploration of RA genes and pathways has revealed that approved agents for other conditions, such as cancer, may be appropriately repurposed for the treatment of RA. These are critical observations that have the potential to dramatically accelerate progress in developing new therapies for autoimmune diseases, such as RA.
Did you have much interaction with Stephen I. Katz, MD, PhD, your longtime predecessor who passed away unexpectedly in 2018? If so, what do you remember most about him? I regret that I had very little interaction with Steve, but I am well aware of the impact he had on NIAMS, NIH, and the research enterprise overall. He inspired so many people in a personal way, and I am energized by the legacy that he left behind.
What are your goals for the early part of your tenure as the new director of NIAMS? An important goal is getting to know the NIAMS community and expanding my knowledge of the Institute’s musculoskeletal and skin portfolios. I am also conducting outreach to Institute/Center directors and other NIH leadership to increase opportunities for input and advice. In doing this, I am identifying shared research interests, best practices, and potential partners for possible future collaborations. Another important goal is to increase NIAMS’ visibility within and beyond NIH. Ultimately, I want to contribute to the great work of the Institute and improve the lives of people with rheumatic, musculoskeletal, and skin diseases.
How would you characterize your management style? I like to lead with a flat hierarchy and work collectively to address opportunities and challenges. I value team building and tend to tap a variety of perspectives and expertise at all levels to achieve consensus, where possible.
The Accelerating Medicines Partnership (AMP) program was launched in 2014, with projects in three disease areas including the autoimmune disorders RA and lupus. What are some recent highlights from this program with respect to RA and lupus? AMP RA/SLE was dedicated to identifying promising therapeutic targets for RA and systemic lupus erythematosus. AMP-funded researchers have applied cutting-edge technologies to study cells from the synovial tissues of the joints of people with RA, and from the kidneys of people with lupus nephritis. In 2014, studying tissues in patients where the disease is active was a novel approach, since most research was conducted in mouse models or human blood samples.
The AMP RA/SLE Network developed a rich dataset that is available to the research community. Investigators are now using the data to facilitate RA and lupus research. For example, using AMP data, NIAMS-supported researchers identified potential biomarkers that could help predict an imminent RA flare. Work from another NIAMS-supported group suggests that targeting the regulatory transcription factor HIF-1, which drives inflammation and tissue damage, might be an effective approach for treating renal injury in lupus.
The data generated are accessible to the scientific community through two NIH websites: the database of Genotypes and Phenotypes (dbGaP) and the Immunology Database and Analysis Portal (IMMPORT).
Given the success of AMP RA/SLE, NIH plans to launch an “AMP 2.0” later in 2021. The AMP Autoimmune and Immune-Mediated Diseases (AMP AIM) program will provide an opportunity to leverage the accomplishments of AMP RA/SLE to new conditions, including psoriatic spectrum diseases and Sjögren’s syndrome.
What are some recent highlights from NIAMS-supported research in skin diseases? NIAMS-supported investigators continue to make significant strides in our understanding of skin biology and disease. For example, researchers recently demonstrated that imiquimod, a drug used to treat precancerous skin lesions, can help mouse ear wounds heal without scarring.
Another team addressed the safety and potential benefit of Staphylococcus hominis A9, a bacterium isolated from healthy human skin, as a topical therapy for atopic dermatitis.
Moving forward, AMP AIM will refine and extend the single-cell analysis of tissues to additional diseases, including psoriasis, setting the stage for the discovery of new therapeutic targets for the disease.
How has the COVID-19 pandemic changed the landscape of research, at least for the short term? This is a once-in-a-century pandemic that none of us were fully prepared for. We understand that it has been particularly challenging for women scientists, scientists with young children, and trainees and junior faculty who are at critically important and vulnerable stages of their careers. There isn’t a lab or clinical setting that hasn’t been negatively impacted in some way.
During the pandemic, the NIH instituted administrative flexibilities to support the grantee community, including extensions in time. In addition, the agency has issued several funding opportunities specific to COVID-19, some of which involve NIAMS participation.
What is NIAMS doing to help early/young investigators as well as female investigators and those from minority groups? Structural racism in biomedical research is a heightened concern. Earlier this year, Dr. Collins established the UNITE initiative to address structural racism and promote racial equity and inclusion at the NIH and within the larger biomedical community that we support. NIAMS is fully committed to this effort. One example is the Diversity Supplement Program, which is designed to attract and encourage eligible individuals from underrepresented populations to research careers.
Early-stage investigators are another top priority. In a tribute to the beloved former NIAMS director, NIH recently established the Stephen I. Katz Early Stage Investigator Research Grant Program. The R01 award provides support for a project unrelated to an early investigator’s area of postdoctoral study. (No preliminary data are allowed.) This award mechanism is a unique opportunity for early-stage investigators to take their research in a completely new direction.
Managing work and family life is an important concern, particularly for female investigators. Many NIH grant awards allow for reimbursement of actual, allowable costs incurred for childcare and parental leave. The NIH is exploring initiatives to promote research continuity and retention of eligible investigators facing major life events, such as pregnancy, childbirth, and adoption, at vulnerable career stages.
Who inspires you most in your work today? I am inspired by the ongoing struggles of our patients, junior investigators, and by the committed staff members on my team.
Temporary hold of mycophenolate helps immune response to SARS-CoV-2 vaccination
Withholding mycophenolate around the time of vaccination against SARS-CoV-2 proved safe and augmented the humoral response to vaccination among a group of patients at one center who were taking the immunosuppressive drug for a variety of rheumatic and musculoskeletal diseases (RMDs).
Previous studies have shown that use of mycophenolate attenuates the humoral response to SARS-CoV-2 vaccination, and the most up-to-date recommendations from the American College of Rheumatology on SARS-CoV-2 vaccination in patients with RMDs advise that mycophenolate should be withheld for a week after receiving the vaccine.
To understand better how withholding mycophenolate would affect immune response to SARS-CoV-2 vaccination, rheumatology fellow Caoilfhionn M. Connolly, MD, and coauthors at Johns Hopkins University, Baltimore, described in their report – published online Sept. 23, 2021, in Annals of the Rheumatic Diseases – how they compared the immune responses to vaccination in 24 patients who withheld mycophenolate and 171 patients who did not stop taking it. All but 1 of the 24 patients who withheld mycophenolate were female, with a median age of 51 years, and they had mostly systemic lupus erythematosus (6 patients), myositis (5), scleroderma (4), or overlap connective tissue disease (4). Three patients received the Janssen/Johnson & Johnson vaccine; all others received either the two-dose Moderna or Pfizer/BioNTech mRNA series.
At a median of 32 days after vaccination, all but two of the patients (92%) who withheld mycophenolate had detectable antibodies against the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, compared with 65% of those who continued the drug (P = .01). This calculated to patients who withheld the drug as having nearly sixfold higher odds for a positive antibody response (odds ratio, 5.8; 95% CI, 1.3-25.5; P = .02). The association remained statistically significant in an logistic regression analysis that was adjusted for age, sex, race, vaccine type, and use of rituximab and glucocorticoids.
The withholding group also had significantly higher median anti-RBD immunoglobulin titers than did the group that continued therapy (125 vs. 7 U/L; P = .004).
Two patients who reported a flare of their underlying disease during the perivaccination period were treated with topical and oral glucocorticoids.
The patients who withdrew mycophenolate had taken it with twice daily dosing at a median total daily dose of 2,000 mg. They ended up withholding a median of 20 doses around the time of vaccination, with 54% withholding before, 38% both before and after, and 8% only after vaccination.
The researchers said that the conclusions that can be drawn from the study were limited by its small sample size, which “did not allow for evaluation of optimal duration of withholding therapy,” and also its “nonrandomized design, lack of data on cellular response, and limited information on dosing of other immunosuppressive agents.”
Three of the authors disclosed receiving consulting and speaking honoraria from Sanofi, Novartis, CSL Behring, Jazz Pharmaceuticals, Veloxis, Mallincrodt, and Thermo Fisher Scientific. A fourth author has received consulting fees from Janssen, Boehringer Ingelheim, Mallinckrodt, EMD Serono, Allogene, and ArgenX.
Withholding mycophenolate around the time of vaccination against SARS-CoV-2 proved safe and augmented the humoral response to vaccination among a group of patients at one center who were taking the immunosuppressive drug for a variety of rheumatic and musculoskeletal diseases (RMDs).
Previous studies have shown that use of mycophenolate attenuates the humoral response to SARS-CoV-2 vaccination, and the most up-to-date recommendations from the American College of Rheumatology on SARS-CoV-2 vaccination in patients with RMDs advise that mycophenolate should be withheld for a week after receiving the vaccine.
To understand better how withholding mycophenolate would affect immune response to SARS-CoV-2 vaccination, rheumatology fellow Caoilfhionn M. Connolly, MD, and coauthors at Johns Hopkins University, Baltimore, described in their report – published online Sept. 23, 2021, in Annals of the Rheumatic Diseases – how they compared the immune responses to vaccination in 24 patients who withheld mycophenolate and 171 patients who did not stop taking it. All but 1 of the 24 patients who withheld mycophenolate were female, with a median age of 51 years, and they had mostly systemic lupus erythematosus (6 patients), myositis (5), scleroderma (4), or overlap connective tissue disease (4). Three patients received the Janssen/Johnson & Johnson vaccine; all others received either the two-dose Moderna or Pfizer/BioNTech mRNA series.
At a median of 32 days after vaccination, all but two of the patients (92%) who withheld mycophenolate had detectable antibodies against the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, compared with 65% of those who continued the drug (P = .01). This calculated to patients who withheld the drug as having nearly sixfold higher odds for a positive antibody response (odds ratio, 5.8; 95% CI, 1.3-25.5; P = .02). The association remained statistically significant in an logistic regression analysis that was adjusted for age, sex, race, vaccine type, and use of rituximab and glucocorticoids.
The withholding group also had significantly higher median anti-RBD immunoglobulin titers than did the group that continued therapy (125 vs. 7 U/L; P = .004).
Two patients who reported a flare of their underlying disease during the perivaccination period were treated with topical and oral glucocorticoids.
The patients who withdrew mycophenolate had taken it with twice daily dosing at a median total daily dose of 2,000 mg. They ended up withholding a median of 20 doses around the time of vaccination, with 54% withholding before, 38% both before and after, and 8% only after vaccination.
The researchers said that the conclusions that can be drawn from the study were limited by its small sample size, which “did not allow for evaluation of optimal duration of withholding therapy,” and also its “nonrandomized design, lack of data on cellular response, and limited information on dosing of other immunosuppressive agents.”
Three of the authors disclosed receiving consulting and speaking honoraria from Sanofi, Novartis, CSL Behring, Jazz Pharmaceuticals, Veloxis, Mallincrodt, and Thermo Fisher Scientific. A fourth author has received consulting fees from Janssen, Boehringer Ingelheim, Mallinckrodt, EMD Serono, Allogene, and ArgenX.
Withholding mycophenolate around the time of vaccination against SARS-CoV-2 proved safe and augmented the humoral response to vaccination among a group of patients at one center who were taking the immunosuppressive drug for a variety of rheumatic and musculoskeletal diseases (RMDs).
Previous studies have shown that use of mycophenolate attenuates the humoral response to SARS-CoV-2 vaccination, and the most up-to-date recommendations from the American College of Rheumatology on SARS-CoV-2 vaccination in patients with RMDs advise that mycophenolate should be withheld for a week after receiving the vaccine.
To understand better how withholding mycophenolate would affect immune response to SARS-CoV-2 vaccination, rheumatology fellow Caoilfhionn M. Connolly, MD, and coauthors at Johns Hopkins University, Baltimore, described in their report – published online Sept. 23, 2021, in Annals of the Rheumatic Diseases – how they compared the immune responses to vaccination in 24 patients who withheld mycophenolate and 171 patients who did not stop taking it. All but 1 of the 24 patients who withheld mycophenolate were female, with a median age of 51 years, and they had mostly systemic lupus erythematosus (6 patients), myositis (5), scleroderma (4), or overlap connective tissue disease (4). Three patients received the Janssen/Johnson & Johnson vaccine; all others received either the two-dose Moderna or Pfizer/BioNTech mRNA series.
At a median of 32 days after vaccination, all but two of the patients (92%) who withheld mycophenolate had detectable antibodies against the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, compared with 65% of those who continued the drug (P = .01). This calculated to patients who withheld the drug as having nearly sixfold higher odds for a positive antibody response (odds ratio, 5.8; 95% CI, 1.3-25.5; P = .02). The association remained statistically significant in an logistic regression analysis that was adjusted for age, sex, race, vaccine type, and use of rituximab and glucocorticoids.
The withholding group also had significantly higher median anti-RBD immunoglobulin titers than did the group that continued therapy (125 vs. 7 U/L; P = .004).
Two patients who reported a flare of their underlying disease during the perivaccination period were treated with topical and oral glucocorticoids.
The patients who withdrew mycophenolate had taken it with twice daily dosing at a median total daily dose of 2,000 mg. They ended up withholding a median of 20 doses around the time of vaccination, with 54% withholding before, 38% both before and after, and 8% only after vaccination.
The researchers said that the conclusions that can be drawn from the study were limited by its small sample size, which “did not allow for evaluation of optimal duration of withholding therapy,” and also its “nonrandomized design, lack of data on cellular response, and limited information on dosing of other immunosuppressive agents.”
Three of the authors disclosed receiving consulting and speaking honoraria from Sanofi, Novartis, CSL Behring, Jazz Pharmaceuticals, Veloxis, Mallincrodt, and Thermo Fisher Scientific. A fourth author has received consulting fees from Janssen, Boehringer Ingelheim, Mallinckrodt, EMD Serono, Allogene, and ArgenX.
FROM ANNALS OF THE RHEUMATIC DISEASES
NIH to study COVID vaccine booster in people with autoimmune disease
In the wake of the Centers for Disease Control and Prevention’s recommendation for a third COVID-19 mRNA vaccine dose for immunocompromised people and the Food and Drug Administration’s authorization of the third dose, the according to an announcement.
The investigators of the trial, called COVID‐19 Booster Vaccine in Autoimmune Disease Non‐Responders, also want to determine if pausing immunosuppressive therapy for autoimmune disease improves the antibody response to an extra dose of a COVID-19 vaccine.
The trial will specifically look at the effects of mycophenolate mofetil (MMF) or mycophenolic acid (MPA), and methotrexate (MTX), or receipt of B cell–depletion therapy such as rituximab within the past 12 months on immune response to a booster dose in people with systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, systemic sclerosis, or pemphigus. They have to have either no serologic response to their initial COVID-19 vaccine regimen or a suboptimal response, defined as a Roche Elecsys Anti-SARS-CoV-2 S (RBD) result greater than or equal to 50 U/mL.
The results of studies conducted in solid-organ transplant recipients who take immunosuppressants showed that an extra dose of vaccine could improve the immune response to the vaccine in many of the individuals, which suggests that the same approach might work in people with autoimmune disease who need treatment with immunosuppressive drugs. Improving the immune response of people with autoimmune disease to COVID-19 vaccines is important because higher rates of severe COVID-19 and death have been reported in this group of patients than in the general population, and it is unclear whether this is attributable to the autoimmune disease, the immunosuppressive medications taken to treat it, or both.
The open-label trial, conducted by the NIAID-funded Autoimmunity Centers of Excellence, aims to enroll 600 people aged 18 years and older with those conditions at 15-20 sites in the United States.
Because medications commonly taken by people with these conditions have been associated with poorer immune responses to vaccines, the trial will randomize the following two cohorts to stop or continue taking their immunosuppressive medication(s) or stop them before and after the booster according to protocol:
- Cohort 1 includes people who are taking MMF or MPA, without additional B cell–depleting medications or MTX.
- Cohort 2 includes people who are taking MTX without additional B cell–depleting medications or MMF/MPA.
A third, nonrandomized cohort consists of people who have received B cell–depletion therapy within the past 12 months regardless of whether they are also taking MMF/MPA or MTX.
Besides the cohort-specific exclusions, other rheumatic disease medications, including biologics, are allowed in the groups.
The primary outcome of the trial is the proportion of participants who have a protective antibody response at week 4. Secondary outcomes will examine various antibody responses at intervals, changes in disease activity across autoimmune diseases, adverse events, and SARS-CoV-2 infections out to 48 weeks.
Study participants will be followed for a total of 13 months. Preliminary results are expected in November 2021, according to the National Institutes of Health.
The trial is being led by Judith James, MD, PhD; Meggan Mackay, MD, MS; Dinesh Khanna, MBBS, MSc; and Amit Bar-Or, MD.
In the wake of the Centers for Disease Control and Prevention’s recommendation for a third COVID-19 mRNA vaccine dose for immunocompromised people and the Food and Drug Administration’s authorization of the third dose, the according to an announcement.
The investigators of the trial, called COVID‐19 Booster Vaccine in Autoimmune Disease Non‐Responders, also want to determine if pausing immunosuppressive therapy for autoimmune disease improves the antibody response to an extra dose of a COVID-19 vaccine.
The trial will specifically look at the effects of mycophenolate mofetil (MMF) or mycophenolic acid (MPA), and methotrexate (MTX), or receipt of B cell–depletion therapy such as rituximab within the past 12 months on immune response to a booster dose in people with systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, systemic sclerosis, or pemphigus. They have to have either no serologic response to their initial COVID-19 vaccine regimen or a suboptimal response, defined as a Roche Elecsys Anti-SARS-CoV-2 S (RBD) result greater than or equal to 50 U/mL.
The results of studies conducted in solid-organ transplant recipients who take immunosuppressants showed that an extra dose of vaccine could improve the immune response to the vaccine in many of the individuals, which suggests that the same approach might work in people with autoimmune disease who need treatment with immunosuppressive drugs. Improving the immune response of people with autoimmune disease to COVID-19 vaccines is important because higher rates of severe COVID-19 and death have been reported in this group of patients than in the general population, and it is unclear whether this is attributable to the autoimmune disease, the immunosuppressive medications taken to treat it, or both.
The open-label trial, conducted by the NIAID-funded Autoimmunity Centers of Excellence, aims to enroll 600 people aged 18 years and older with those conditions at 15-20 sites in the United States.
Because medications commonly taken by people with these conditions have been associated with poorer immune responses to vaccines, the trial will randomize the following two cohorts to stop or continue taking their immunosuppressive medication(s) or stop them before and after the booster according to protocol:
- Cohort 1 includes people who are taking MMF or MPA, without additional B cell–depleting medications or MTX.
- Cohort 2 includes people who are taking MTX without additional B cell–depleting medications or MMF/MPA.
A third, nonrandomized cohort consists of people who have received B cell–depletion therapy within the past 12 months regardless of whether they are also taking MMF/MPA or MTX.
Besides the cohort-specific exclusions, other rheumatic disease medications, including biologics, are allowed in the groups.
The primary outcome of the trial is the proportion of participants who have a protective antibody response at week 4. Secondary outcomes will examine various antibody responses at intervals, changes in disease activity across autoimmune diseases, adverse events, and SARS-CoV-2 infections out to 48 weeks.
Study participants will be followed for a total of 13 months. Preliminary results are expected in November 2021, according to the National Institutes of Health.
The trial is being led by Judith James, MD, PhD; Meggan Mackay, MD, MS; Dinesh Khanna, MBBS, MSc; and Amit Bar-Or, MD.
In the wake of the Centers for Disease Control and Prevention’s recommendation for a third COVID-19 mRNA vaccine dose for immunocompromised people and the Food and Drug Administration’s authorization of the third dose, the according to an announcement.
The investigators of the trial, called COVID‐19 Booster Vaccine in Autoimmune Disease Non‐Responders, also want to determine if pausing immunosuppressive therapy for autoimmune disease improves the antibody response to an extra dose of a COVID-19 vaccine.
The trial will specifically look at the effects of mycophenolate mofetil (MMF) or mycophenolic acid (MPA), and methotrexate (MTX), or receipt of B cell–depletion therapy such as rituximab within the past 12 months on immune response to a booster dose in people with systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, systemic sclerosis, or pemphigus. They have to have either no serologic response to their initial COVID-19 vaccine regimen or a suboptimal response, defined as a Roche Elecsys Anti-SARS-CoV-2 S (RBD) result greater than or equal to 50 U/mL.
The results of studies conducted in solid-organ transplant recipients who take immunosuppressants showed that an extra dose of vaccine could improve the immune response to the vaccine in many of the individuals, which suggests that the same approach might work in people with autoimmune disease who need treatment with immunosuppressive drugs. Improving the immune response of people with autoimmune disease to COVID-19 vaccines is important because higher rates of severe COVID-19 and death have been reported in this group of patients than in the general population, and it is unclear whether this is attributable to the autoimmune disease, the immunosuppressive medications taken to treat it, or both.
The open-label trial, conducted by the NIAID-funded Autoimmunity Centers of Excellence, aims to enroll 600 people aged 18 years and older with those conditions at 15-20 sites in the United States.
Because medications commonly taken by people with these conditions have been associated with poorer immune responses to vaccines, the trial will randomize the following two cohorts to stop or continue taking their immunosuppressive medication(s) or stop them before and after the booster according to protocol:
- Cohort 1 includes people who are taking MMF or MPA, without additional B cell–depleting medications or MTX.
- Cohort 2 includes people who are taking MTX without additional B cell–depleting medications or MMF/MPA.
A third, nonrandomized cohort consists of people who have received B cell–depletion therapy within the past 12 months regardless of whether they are also taking MMF/MPA or MTX.
Besides the cohort-specific exclusions, other rheumatic disease medications, including biologics, are allowed in the groups.
The primary outcome of the trial is the proportion of participants who have a protective antibody response at week 4. Secondary outcomes will examine various antibody responses at intervals, changes in disease activity across autoimmune diseases, adverse events, and SARS-CoV-2 infections out to 48 weeks.
Study participants will be followed for a total of 13 months. Preliminary results are expected in November 2021, according to the National Institutes of Health.
The trial is being led by Judith James, MD, PhD; Meggan Mackay, MD, MS; Dinesh Khanna, MBBS, MSc; and Amit Bar-Or, MD.
Hep B vaccine response varied among youth with inflammatory, autoimmune disorders

“Hepatitis B is a common viral infection with 2 billion people worldwide having evidence of prior or current infection, and it can present as an acute or chronic infection,” or with chronic sequelae, including cirrhosis and hepatocellular carcinoma, Alexandra Ritter said during the annual meeting of the Society for Pediatric Dermatology. A three-dose vaccination series is recommended beginning at birth, and in 2016, the Centers for Disease Control and Prevention reported that 90.5% of U.S. children aged 19-35 months had completed the series.
While the vaccine series provides protection in healthy individuals more than 95% of the time, a decreased response has been noted in specific pediatric populations, including those with inflammatory and autoimmune diseases. “This is important to note and investigate further because a decreased vaccine response increases the risk for this high-risk population, and the use of boosters is currently debated,” said Ms. Ritter, who is a fourth-year student at the Medical University of South Carolina, Charleston.
To determine the percent of pediatric patients with inflammatory or autoimmune disease who lack evidence of immunity following the hepatitis B vaccine series, Ms. Ritter and colleagues Abigail Truitt and pediatric dermatologist Lara Wine Lee, MD, PhD, of MUSC, retrospectively reviewed the charts of 160 patients between the ages of 6 months and 21 years, who were diagnosed with an autoimmune or autoinflammatory disease, or inflammatory bowel disease (IBD), and had documented evidence of vaccination and serologic testing prior to the start of immunosuppressive therapy.
Of the 160 patients, 100 (63%) had IBD, 34 (21%) had an autoimmune disease, 26 (16%) had an autoinflammatory disease, 89 (56%) were female, and their mean age was 15 years.
The researchers observed variation in the testing ordered between the three patient groups. Specifically, 88.2% of autoimmune patients had hepatitis B surface antigen (HBsAg) testing, compared with 96.15% of patients with an autoinflammatory disease and 67% of patients with IBD, while 76.47% of patients with an autoimmune disease had hepatitis B core antibody (anti-HBc) testing, compared with 88.46% of patients with an autoinflammatory disease and 31% of patients with IBD.
In addition, 82.35% of patients with an autoimmune disease had HBsAg testing, compared with 100% of patients with an autoinflammatory disease and 94% of patients with IBD.
Of the 148 patients who had HBsAg testing ordered and completed prior to starting an immunosuppressive drug, there was no statistically significant difference in the percent of patients showing evidence of an immune response to the hepatitis B vaccine (32.14% among patients with an autoimmune disease, 34.62% among patients with an autoinflammatory disease, and 31.91% among patients with IBD). Combined, 67.57% of tested negative for the hepatitis B surface antibody.
“Our study showed that the majority of these patients did not show serologic evidence of immunity despite being fully vaccinated,” Ms. Ritter said. “There was also variation in the testing ordered and a more standardized approach is needed in this high-risk population.” She acknowledged certain limitations of the study, including its retrospective design and lack of a control group.
“This brings us to our next question of whether this indicates a failure of the vaccine, or the way immunity is tested,” she continued. “The CDC and the European Consensus Group on Hepatitis B Immunity recommend a cutoff of greater than 10 mIU/mL. Those that achieve immunity are protected for up to 20 years due to immune memory, even if their antibody levels later drop. There have been rare cases of immunocompetent individuals having evidence of transient asymptomatic infections when antibody levels drop. The chronic disease has only been documented in infants born to positive mothers. In hemodialysis patients, however, clinically significant infections have been documented when antibody levels drop.”
The CDC only recommends postvaccination testing to infants born to positive mothers, health care workers at high risk, hemodialysis patients, people with HIV and other immunocompromised people, and needle-sharing partners of chronically infected people. This is completed 1-2 months following the third vaccine dose, and those with antibody levels less than 10 mIU/mL should be revaccinated. “As some groups do not respond to the vaccine series, alternative dosing and the intradermal vaccine have been studied and shown to be effective in certain groups,” she said.
When it comes to monitoring immunocompromised individuals and giving booster shots, however, there are conflicting recommendations. The CDC recommends yearly testing and booster shots when levels drop below 10 mIU/mL only in hemodialysis patients, while the European Consensus Group recommends testing every 6-12 months for immunocompromised individuals and boosters when their levels drop below 10 mIU/mL.
“The CDC has not yet determined if other immunocompromised individuals should receive a booster, with more research required, but studies have shown it to be effective,” Ms. Ritter said. In a similar study looking at evidence of immunity in children with connective tissue disease who were on immunosuppressive treatment, 50% had no evidence of protective antibodies, compared with 96% in the control group. “In that study, a booster shot was given, and protective antibody concentrations were found at follow-up,” she said.
The researchers reported having no financial disclosures.
[email protected]
“Hepatitis B is a common viral infection with 2 billion people worldwide having evidence of prior or current infection, and it can present as an acute or chronic infection,” or with chronic sequelae, including cirrhosis and hepatocellular carcinoma, Alexandra Ritter said during the annual meeting of the Society for Pediatric Dermatology. A three-dose vaccination series is recommended beginning at birth, and in 2016, the Centers for Disease Control and Prevention reported that 90.5% of U.S. children aged 19-35 months had completed the series.
While the vaccine series provides protection in healthy individuals more than 95% of the time, a decreased response has been noted in specific pediatric populations, including those with inflammatory and autoimmune diseases. “This is important to note and investigate further because a decreased vaccine response increases the risk for this high-risk population, and the use of boosters is currently debated,” said Ms. Ritter, who is a fourth-year student at the Medical University of South Carolina, Charleston.
To determine the percent of pediatric patients with inflammatory or autoimmune disease who lack evidence of immunity following the hepatitis B vaccine series, Ms. Ritter and colleagues Abigail Truitt and pediatric dermatologist Lara Wine Lee, MD, PhD, of MUSC, retrospectively reviewed the charts of 160 patients between the ages of 6 months and 21 years, who were diagnosed with an autoimmune or autoinflammatory disease, or inflammatory bowel disease (IBD), and had documented evidence of vaccination and serologic testing prior to the start of immunosuppressive therapy.
Of the 160 patients, 100 (63%) had IBD, 34 (21%) had an autoimmune disease, 26 (16%) had an autoinflammatory disease, 89 (56%) were female, and their mean age was 15 years.
The researchers observed variation in the testing ordered between the three patient groups. Specifically, 88.2% of autoimmune patients had hepatitis B surface antigen (HBsAg) testing, compared with 96.15% of patients with an autoinflammatory disease and 67% of patients with IBD, while 76.47% of patients with an autoimmune disease had hepatitis B core antibody (anti-HBc) testing, compared with 88.46% of patients with an autoinflammatory disease and 31% of patients with IBD.
In addition, 82.35% of patients with an autoimmune disease had HBsAg testing, compared with 100% of patients with an autoinflammatory disease and 94% of patients with IBD.
Of the 148 patients who had HBsAg testing ordered and completed prior to starting an immunosuppressive drug, there was no statistically significant difference in the percent of patients showing evidence of an immune response to the hepatitis B vaccine (32.14% among patients with an autoimmune disease, 34.62% among patients with an autoinflammatory disease, and 31.91% among patients with IBD). Combined, 67.57% of tested negative for the hepatitis B surface antibody.
“Our study showed that the majority of these patients did not show serologic evidence of immunity despite being fully vaccinated,” Ms. Ritter said. “There was also variation in the testing ordered and a more standardized approach is needed in this high-risk population.” She acknowledged certain limitations of the study, including its retrospective design and lack of a control group.
“This brings us to our next question of whether this indicates a failure of the vaccine, or the way immunity is tested,” she continued. “The CDC and the European Consensus Group on Hepatitis B Immunity recommend a cutoff of greater than 10 mIU/mL. Those that achieve immunity are protected for up to 20 years due to immune memory, even if their antibody levels later drop. There have been rare cases of immunocompetent individuals having evidence of transient asymptomatic infections when antibody levels drop. The chronic disease has only been documented in infants born to positive mothers. In hemodialysis patients, however, clinically significant infections have been documented when antibody levels drop.”
The CDC only recommends postvaccination testing to infants born to positive mothers, health care workers at high risk, hemodialysis patients, people with HIV and other immunocompromised people, and needle-sharing partners of chronically infected people. This is completed 1-2 months following the third vaccine dose, and those with antibody levels less than 10 mIU/mL should be revaccinated. “As some groups do not respond to the vaccine series, alternative dosing and the intradermal vaccine have been studied and shown to be effective in certain groups,” she said.
When it comes to monitoring immunocompromised individuals and giving booster shots, however, there are conflicting recommendations. The CDC recommends yearly testing and booster shots when levels drop below 10 mIU/mL only in hemodialysis patients, while the European Consensus Group recommends testing every 6-12 months for immunocompromised individuals and boosters when their levels drop below 10 mIU/mL.
“The CDC has not yet determined if other immunocompromised individuals should receive a booster, with more research required, but studies have shown it to be effective,” Ms. Ritter said. In a similar study looking at evidence of immunity in children with connective tissue disease who were on immunosuppressive treatment, 50% had no evidence of protective antibodies, compared with 96% in the control group. “In that study, a booster shot was given, and protective antibody concentrations were found at follow-up,” she said.
The researchers reported having no financial disclosures.
[email protected]
“Hepatitis B is a common viral infection with 2 billion people worldwide having evidence of prior or current infection, and it can present as an acute or chronic infection,” or with chronic sequelae, including cirrhosis and hepatocellular carcinoma, Alexandra Ritter said during the annual meeting of the Society for Pediatric Dermatology. A three-dose vaccination series is recommended beginning at birth, and in 2016, the Centers for Disease Control and Prevention reported that 90.5% of U.S. children aged 19-35 months had completed the series.
While the vaccine series provides protection in healthy individuals more than 95% of the time, a decreased response has been noted in specific pediatric populations, including those with inflammatory and autoimmune diseases. “This is important to note and investigate further because a decreased vaccine response increases the risk for this high-risk population, and the use of boosters is currently debated,” said Ms. Ritter, who is a fourth-year student at the Medical University of South Carolina, Charleston.
To determine the percent of pediatric patients with inflammatory or autoimmune disease who lack evidence of immunity following the hepatitis B vaccine series, Ms. Ritter and colleagues Abigail Truitt and pediatric dermatologist Lara Wine Lee, MD, PhD, of MUSC, retrospectively reviewed the charts of 160 patients between the ages of 6 months and 21 years, who were diagnosed with an autoimmune or autoinflammatory disease, or inflammatory bowel disease (IBD), and had documented evidence of vaccination and serologic testing prior to the start of immunosuppressive therapy.
Of the 160 patients, 100 (63%) had IBD, 34 (21%) had an autoimmune disease, 26 (16%) had an autoinflammatory disease, 89 (56%) were female, and their mean age was 15 years.
The researchers observed variation in the testing ordered between the three patient groups. Specifically, 88.2% of autoimmune patients had hepatitis B surface antigen (HBsAg) testing, compared with 96.15% of patients with an autoinflammatory disease and 67% of patients with IBD, while 76.47% of patients with an autoimmune disease had hepatitis B core antibody (anti-HBc) testing, compared with 88.46% of patients with an autoinflammatory disease and 31% of patients with IBD.
In addition, 82.35% of patients with an autoimmune disease had HBsAg testing, compared with 100% of patients with an autoinflammatory disease and 94% of patients with IBD.
Of the 148 patients who had HBsAg testing ordered and completed prior to starting an immunosuppressive drug, there was no statistically significant difference in the percent of patients showing evidence of an immune response to the hepatitis B vaccine (32.14% among patients with an autoimmune disease, 34.62% among patients with an autoinflammatory disease, and 31.91% among patients with IBD). Combined, 67.57% of tested negative for the hepatitis B surface antibody.
“Our study showed that the majority of these patients did not show serologic evidence of immunity despite being fully vaccinated,” Ms. Ritter said. “There was also variation in the testing ordered and a more standardized approach is needed in this high-risk population.” She acknowledged certain limitations of the study, including its retrospective design and lack of a control group.
“This brings us to our next question of whether this indicates a failure of the vaccine, or the way immunity is tested,” she continued. “The CDC and the European Consensus Group on Hepatitis B Immunity recommend a cutoff of greater than 10 mIU/mL. Those that achieve immunity are protected for up to 20 years due to immune memory, even if their antibody levels later drop. There have been rare cases of immunocompetent individuals having evidence of transient asymptomatic infections when antibody levels drop. The chronic disease has only been documented in infants born to positive mothers. In hemodialysis patients, however, clinically significant infections have been documented when antibody levels drop.”
The CDC only recommends postvaccination testing to infants born to positive mothers, health care workers at high risk, hemodialysis patients, people with HIV and other immunocompromised people, and needle-sharing partners of chronically infected people. This is completed 1-2 months following the third vaccine dose, and those with antibody levels less than 10 mIU/mL should be revaccinated. “As some groups do not respond to the vaccine series, alternative dosing and the intradermal vaccine have been studied and shown to be effective in certain groups,” she said.
When it comes to monitoring immunocompromised individuals and giving booster shots, however, there are conflicting recommendations. The CDC recommends yearly testing and booster shots when levels drop below 10 mIU/mL only in hemodialysis patients, while the European Consensus Group recommends testing every 6-12 months for immunocompromised individuals and boosters when their levels drop below 10 mIU/mL.
“The CDC has not yet determined if other immunocompromised individuals should receive a booster, with more research required, but studies have shown it to be effective,” Ms. Ritter said. In a similar study looking at evidence of immunity in children with connective tissue disease who were on immunosuppressive treatment, 50% had no evidence of protective antibodies, compared with 96% in the control group. “In that study, a booster shot was given, and protective antibody concentrations were found at follow-up,” she said.
The researchers reported having no financial disclosures.
[email protected]
FROM SPD 2021