User login
Post COVID-19, Long-term Risk for Autoimmune, Autoinflammatory Skin Disorders Increased, Study Finds
In addition, the authors reported that COVID-19 vaccination appears to reduce these risks.
The study was published in JAMA Dermatology.
‘Compelling Evidence’
“This well-executed study by Heo et al provides compelling evidence to support an association between COVID-19 infection and the development of subsequent autoimmune and autoinflammatory skin diseases,” wrote authors led by Lisa M. Arkin, MD, of the Department of Dermatology, University of Wisconsin School of Medicine and Public Health in Madison, in an accompanying editorial.
Using databases from Korea’s National Health Insurance Service and the Korea Disease Control and Prevention Agency, investigators led by Yeon-Woo Heo, MD, a dermatology resident at Yonsei University Wonju College of Medicine, Wonju, Republic of Korea, compared 3.1 million people who had COVID-19 with 3.8 million controls, all with at least 180 days’ follow-up through December 31, 2022.
At a mean follow-up of 287 days in both cohorts, authors found significantly elevated risks for AA and vitiligo (adjusted hazard ratio [aHR], 1.11 for both), AT (aHR, 1.24), Behçet disease (aHR, 1.45), and BP (aHR, 1.62) in the post–COVID-19 cohort. The infection also raised the risk for other conditions such as systemic lupus erythematosus (aHR, 1.14) and Crohn’s disease (aHR, 1.35).
In subgroup analyses, demographic factors were associated with diverse effects: COVID-19 infection was associated with significantly higher odds of developing AA (for both men and women), vitiligo (men), Behçet disease (men and women), Crohn’s disease (men), ulcerative colitis (men), rheumatoid arthritis (men and women), systemic lupus erythematosus (men), ankylosing spondylitis (men), AT (women), and BP (women) than controls.
Those aged under 40 years were more likely to develop AA, primary cicatricial alopecia, Behçet disease, and ulcerative colitis, while those aged 40 years or older were more likely to develop AA, AT, vitiligo, Behçet disease, Crohn’s disease, rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, ankylosing spondylitis, and BP.
Additionally, severe COVID-19 requiring intensive care unit admission was associated with a significantly increased risk for autoimmune diseases, including AA, psoriasis, BP, and sarcoidosis. By timeframe, risks for AA, AT, and psoriasis were significantly higher during the initial Delta-dominant period.
Vaccination Effect
Moreover, vaccinated individuals were less likely to develop AA, AT, psoriasis, Behçet disease, and various nondermatologic conditions than were those who were unvaccinated. This finding, wrote Heo and colleagues, “may provide evidence to support the hypothesis that COVID-19 vaccines can help prevent autoimmune diseases.”
“That’s the part we all need to take into our offices tomorrow,” said Brett King, MD, PhD, a Fairfield, Connecticut–based dermatologist in private practice. He was not involved with the study but was asked to comment.
Overall, King said, the study carries two main messages. “The first is that COVID-19 infection increases the likelihood of developing an autoimmune or autoinflammatory disease in a large population.” The second and very important message is that being vaccinated against COVID-19 provides protection against developing an autoimmune or autoinflammatory disease.
“My concern is that the popular media highlights the first part,” said King, “and everybody who develops alopecia areata, vitiligo, or sarcoidosis blames COVID-19. That’s not what this work says.”
The foregoing distinction is especially important during the fall and winter, he added, when people getting influenza vaccines are routinely offered COVID-19 vaccines. “Many patients have said, ‘I got the COVID vaccine and developed alopecia areata 6 months later.’ Nearly everybody who has developed a new or worsening health condition in the last almost 5 years has had the perfect fall guy — the COVID vaccine or infection.”
With virtually all patients asking if they should get an updated COVID-19 vaccine or booster, he added, many report having heard that such vaccines cause AA, vitiligo, or other diseases. “To anchor these conversations in real data and not just anecdotes from a blog or Facebook is very useful,” said King, “and now we have very good data saying that the COVID vaccine is protective against these disorders.”
George Han, MD, PhD, associate professor of dermatology at the Donald and Barbara Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York, applauds investigators’ use of a large, robust database but suggests interpreting results cautiously. He was not involved with the study but was asked to comment.
“You could do a large, well-done study,” Han said, “but it could still not necessarily be generalizable. These autoimmune conditions they’re looking at have clear ethnic and racial biases.” Heo and colleagues acknowledged shortcomings including their study population’s monomorphic nature.
Additional issues that limit the study’s impact, said Han, include the difficulty of conceptualizing a 10%-20% increase in conditions that at baseline are rare. And many of the findings reflected natural patterns, he said. For instance, BP more commonly affects older people, COVID-19 notwithstanding.
Han said that for him, the study’s main value going forward is helping to explain a rash of worsening inflammatory skin disease that many dermatologists saw early in the pandemic. “We would regularly see patients who were well controlled with, for example, psoriasis or eczema. But after COVID-19 infection or a vaccine (usually mRNA-type), in some cases they would come in flaring badly.” This happened at least a dozen times during the first year of post-shutdown appointments, he said.
“We’ve seen patients who have flared multiple times — they get the booster, then flare again,” Han added. Similar patterns occurred with pyoderma gangrenosum and other inflammatory skin diseases, he said.
Given the modest effect sizes of the associations reported in the Korean study, Arkin and colleagues wrote in their JAMA Dermatology editorial that surveillance for autoimmune disease is probably not warranted without new examination findings or symptoms. “For certain,” King said, “we should not go hunting for things that aren’t obviously there.”
Rather, Arkin and colleagues wrote, the higher autoimmunity rates seen among the unvaccinated, as well as during the Delta phase (when patients were sicker and hospitalizations were more likely) and in patients requiring intensive care, suggest that “interventions that reduce disease severity could also potentially reduce long-term risk of subsequent autoimmune sequelae.”
Future research addressing whether people with preexisting autoimmune conditions are at greater risk for flares or developing new autoimmune diseases following COVID-19 infection “would help to frame an evidence-based approach for patients with autoimmune disorders who develop COVID-19 infection, including the role for antiviral treatments,” they added.
The study was supported by grants from the Research Program of the Korea Medical Institute, the Korea Health Industry Development Institute, and the National Research Foundation of Korea. Han and King reported no relevant financial relationships. Arkin disclosed receiving research grants to her institution from Amgen and Eli Lilly, personal fees from Sanofi/Regeneron for consulting, and personal consulting fees from Merck outside the submitted work. Another author reported personal consulting fees from Dexcel Pharma and Honeydew outside the submitted work. No other disclosures were reported.
A version of this article appeared on Medscape.com.
In addition, the authors reported that COVID-19 vaccination appears to reduce these risks.
The study was published in JAMA Dermatology.
‘Compelling Evidence’
“This well-executed study by Heo et al provides compelling evidence to support an association between COVID-19 infection and the development of subsequent autoimmune and autoinflammatory skin diseases,” wrote authors led by Lisa M. Arkin, MD, of the Department of Dermatology, University of Wisconsin School of Medicine and Public Health in Madison, in an accompanying editorial.
Using databases from Korea’s National Health Insurance Service and the Korea Disease Control and Prevention Agency, investigators led by Yeon-Woo Heo, MD, a dermatology resident at Yonsei University Wonju College of Medicine, Wonju, Republic of Korea, compared 3.1 million people who had COVID-19 with 3.8 million controls, all with at least 180 days’ follow-up through December 31, 2022.
At a mean follow-up of 287 days in both cohorts, authors found significantly elevated risks for AA and vitiligo (adjusted hazard ratio [aHR], 1.11 for both), AT (aHR, 1.24), Behçet disease (aHR, 1.45), and BP (aHR, 1.62) in the post–COVID-19 cohort. The infection also raised the risk for other conditions such as systemic lupus erythematosus (aHR, 1.14) and Crohn’s disease (aHR, 1.35).
In subgroup analyses, demographic factors were associated with diverse effects: COVID-19 infection was associated with significantly higher odds of developing AA (for both men and women), vitiligo (men), Behçet disease (men and women), Crohn’s disease (men), ulcerative colitis (men), rheumatoid arthritis (men and women), systemic lupus erythematosus (men), ankylosing spondylitis (men), AT (women), and BP (women) than controls.
Those aged under 40 years were more likely to develop AA, primary cicatricial alopecia, Behçet disease, and ulcerative colitis, while those aged 40 years or older were more likely to develop AA, AT, vitiligo, Behçet disease, Crohn’s disease, rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, ankylosing spondylitis, and BP.
Additionally, severe COVID-19 requiring intensive care unit admission was associated with a significantly increased risk for autoimmune diseases, including AA, psoriasis, BP, and sarcoidosis. By timeframe, risks for AA, AT, and psoriasis were significantly higher during the initial Delta-dominant period.
Vaccination Effect
Moreover, vaccinated individuals were less likely to develop AA, AT, psoriasis, Behçet disease, and various nondermatologic conditions than were those who were unvaccinated. This finding, wrote Heo and colleagues, “may provide evidence to support the hypothesis that COVID-19 vaccines can help prevent autoimmune diseases.”
“That’s the part we all need to take into our offices tomorrow,” said Brett King, MD, PhD, a Fairfield, Connecticut–based dermatologist in private practice. He was not involved with the study but was asked to comment.
Overall, King said, the study carries two main messages. “The first is that COVID-19 infection increases the likelihood of developing an autoimmune or autoinflammatory disease in a large population.” The second and very important message is that being vaccinated against COVID-19 provides protection against developing an autoimmune or autoinflammatory disease.
“My concern is that the popular media highlights the first part,” said King, “and everybody who develops alopecia areata, vitiligo, or sarcoidosis blames COVID-19. That’s not what this work says.”
The foregoing distinction is especially important during the fall and winter, he added, when people getting influenza vaccines are routinely offered COVID-19 vaccines. “Many patients have said, ‘I got the COVID vaccine and developed alopecia areata 6 months later.’ Nearly everybody who has developed a new or worsening health condition in the last almost 5 years has had the perfect fall guy — the COVID vaccine or infection.”
With virtually all patients asking if they should get an updated COVID-19 vaccine or booster, he added, many report having heard that such vaccines cause AA, vitiligo, or other diseases. “To anchor these conversations in real data and not just anecdotes from a blog or Facebook is very useful,” said King, “and now we have very good data saying that the COVID vaccine is protective against these disorders.”
George Han, MD, PhD, associate professor of dermatology at the Donald and Barbara Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York, applauds investigators’ use of a large, robust database but suggests interpreting results cautiously. He was not involved with the study but was asked to comment.
“You could do a large, well-done study,” Han said, “but it could still not necessarily be generalizable. These autoimmune conditions they’re looking at have clear ethnic and racial biases.” Heo and colleagues acknowledged shortcomings including their study population’s monomorphic nature.
Additional issues that limit the study’s impact, said Han, include the difficulty of conceptualizing a 10%-20% increase in conditions that at baseline are rare. And many of the findings reflected natural patterns, he said. For instance, BP more commonly affects older people, COVID-19 notwithstanding.
Han said that for him, the study’s main value going forward is helping to explain a rash of worsening inflammatory skin disease that many dermatologists saw early in the pandemic. “We would regularly see patients who were well controlled with, for example, psoriasis or eczema. But after COVID-19 infection or a vaccine (usually mRNA-type), in some cases they would come in flaring badly.” This happened at least a dozen times during the first year of post-shutdown appointments, he said.
“We’ve seen patients who have flared multiple times — they get the booster, then flare again,” Han added. Similar patterns occurred with pyoderma gangrenosum and other inflammatory skin diseases, he said.
Given the modest effect sizes of the associations reported in the Korean study, Arkin and colleagues wrote in their JAMA Dermatology editorial that surveillance for autoimmune disease is probably not warranted without new examination findings or symptoms. “For certain,” King said, “we should not go hunting for things that aren’t obviously there.”
Rather, Arkin and colleagues wrote, the higher autoimmunity rates seen among the unvaccinated, as well as during the Delta phase (when patients were sicker and hospitalizations were more likely) and in patients requiring intensive care, suggest that “interventions that reduce disease severity could also potentially reduce long-term risk of subsequent autoimmune sequelae.”
Future research addressing whether people with preexisting autoimmune conditions are at greater risk for flares or developing new autoimmune diseases following COVID-19 infection “would help to frame an evidence-based approach for patients with autoimmune disorders who develop COVID-19 infection, including the role for antiviral treatments,” they added.
The study was supported by grants from the Research Program of the Korea Medical Institute, the Korea Health Industry Development Institute, and the National Research Foundation of Korea. Han and King reported no relevant financial relationships. Arkin disclosed receiving research grants to her institution from Amgen and Eli Lilly, personal fees from Sanofi/Regeneron for consulting, and personal consulting fees from Merck outside the submitted work. Another author reported personal consulting fees from Dexcel Pharma and Honeydew outside the submitted work. No other disclosures were reported.
A version of this article appeared on Medscape.com.
In addition, the authors reported that COVID-19 vaccination appears to reduce these risks.
The study was published in JAMA Dermatology.
‘Compelling Evidence’
“This well-executed study by Heo et al provides compelling evidence to support an association between COVID-19 infection and the development of subsequent autoimmune and autoinflammatory skin diseases,” wrote authors led by Lisa M. Arkin, MD, of the Department of Dermatology, University of Wisconsin School of Medicine and Public Health in Madison, in an accompanying editorial.
Using databases from Korea’s National Health Insurance Service and the Korea Disease Control and Prevention Agency, investigators led by Yeon-Woo Heo, MD, a dermatology resident at Yonsei University Wonju College of Medicine, Wonju, Republic of Korea, compared 3.1 million people who had COVID-19 with 3.8 million controls, all with at least 180 days’ follow-up through December 31, 2022.
At a mean follow-up of 287 days in both cohorts, authors found significantly elevated risks for AA and vitiligo (adjusted hazard ratio [aHR], 1.11 for both), AT (aHR, 1.24), Behçet disease (aHR, 1.45), and BP (aHR, 1.62) in the post–COVID-19 cohort. The infection also raised the risk for other conditions such as systemic lupus erythematosus (aHR, 1.14) and Crohn’s disease (aHR, 1.35).
In subgroup analyses, demographic factors were associated with diverse effects: COVID-19 infection was associated with significantly higher odds of developing AA (for both men and women), vitiligo (men), Behçet disease (men and women), Crohn’s disease (men), ulcerative colitis (men), rheumatoid arthritis (men and women), systemic lupus erythematosus (men), ankylosing spondylitis (men), AT (women), and BP (women) than controls.
Those aged under 40 years were more likely to develop AA, primary cicatricial alopecia, Behçet disease, and ulcerative colitis, while those aged 40 years or older were more likely to develop AA, AT, vitiligo, Behçet disease, Crohn’s disease, rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, ankylosing spondylitis, and BP.
Additionally, severe COVID-19 requiring intensive care unit admission was associated with a significantly increased risk for autoimmune diseases, including AA, psoriasis, BP, and sarcoidosis. By timeframe, risks for AA, AT, and psoriasis were significantly higher during the initial Delta-dominant period.
Vaccination Effect
Moreover, vaccinated individuals were less likely to develop AA, AT, psoriasis, Behçet disease, and various nondermatologic conditions than were those who were unvaccinated. This finding, wrote Heo and colleagues, “may provide evidence to support the hypothesis that COVID-19 vaccines can help prevent autoimmune diseases.”
“That’s the part we all need to take into our offices tomorrow,” said Brett King, MD, PhD, a Fairfield, Connecticut–based dermatologist in private practice. He was not involved with the study but was asked to comment.
Overall, King said, the study carries two main messages. “The first is that COVID-19 infection increases the likelihood of developing an autoimmune or autoinflammatory disease in a large population.” The second and very important message is that being vaccinated against COVID-19 provides protection against developing an autoimmune or autoinflammatory disease.
“My concern is that the popular media highlights the first part,” said King, “and everybody who develops alopecia areata, vitiligo, or sarcoidosis blames COVID-19. That’s not what this work says.”
The foregoing distinction is especially important during the fall and winter, he added, when people getting influenza vaccines are routinely offered COVID-19 vaccines. “Many patients have said, ‘I got the COVID vaccine and developed alopecia areata 6 months later.’ Nearly everybody who has developed a new or worsening health condition in the last almost 5 years has had the perfect fall guy — the COVID vaccine or infection.”
With virtually all patients asking if they should get an updated COVID-19 vaccine or booster, he added, many report having heard that such vaccines cause AA, vitiligo, or other diseases. “To anchor these conversations in real data and not just anecdotes from a blog or Facebook is very useful,” said King, “and now we have very good data saying that the COVID vaccine is protective against these disorders.”
George Han, MD, PhD, associate professor of dermatology at the Donald and Barbara Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York, applauds investigators’ use of a large, robust database but suggests interpreting results cautiously. He was not involved with the study but was asked to comment.
“You could do a large, well-done study,” Han said, “but it could still not necessarily be generalizable. These autoimmune conditions they’re looking at have clear ethnic and racial biases.” Heo and colleagues acknowledged shortcomings including their study population’s monomorphic nature.
Additional issues that limit the study’s impact, said Han, include the difficulty of conceptualizing a 10%-20% increase in conditions that at baseline are rare. And many of the findings reflected natural patterns, he said. For instance, BP more commonly affects older people, COVID-19 notwithstanding.
Han said that for him, the study’s main value going forward is helping to explain a rash of worsening inflammatory skin disease that many dermatologists saw early in the pandemic. “We would regularly see patients who were well controlled with, for example, psoriasis or eczema. But after COVID-19 infection or a vaccine (usually mRNA-type), in some cases they would come in flaring badly.” This happened at least a dozen times during the first year of post-shutdown appointments, he said.
“We’ve seen patients who have flared multiple times — they get the booster, then flare again,” Han added. Similar patterns occurred with pyoderma gangrenosum and other inflammatory skin diseases, he said.
Given the modest effect sizes of the associations reported in the Korean study, Arkin and colleagues wrote in their JAMA Dermatology editorial that surveillance for autoimmune disease is probably not warranted without new examination findings or symptoms. “For certain,” King said, “we should not go hunting for things that aren’t obviously there.”
Rather, Arkin and colleagues wrote, the higher autoimmunity rates seen among the unvaccinated, as well as during the Delta phase (when patients were sicker and hospitalizations were more likely) and in patients requiring intensive care, suggest that “interventions that reduce disease severity could also potentially reduce long-term risk of subsequent autoimmune sequelae.”
Future research addressing whether people with preexisting autoimmune conditions are at greater risk for flares or developing new autoimmune diseases following COVID-19 infection “would help to frame an evidence-based approach for patients with autoimmune disorders who develop COVID-19 infection, including the role for antiviral treatments,” they added.
The study was supported by grants from the Research Program of the Korea Medical Institute, the Korea Health Industry Development Institute, and the National Research Foundation of Korea. Han and King reported no relevant financial relationships. Arkin disclosed receiving research grants to her institution from Amgen and Eli Lilly, personal fees from Sanofi/Regeneron for consulting, and personal consulting fees from Merck outside the submitted work. Another author reported personal consulting fees from Dexcel Pharma and Honeydew outside the submitted work. No other disclosures were reported.
A version of this article appeared on Medscape.com.
FROM JAMA DERMATOLOGY
Home Spirometry Has Potential for Detecting Pulmonary Decline in Systemic Sclerosis
TOPLINE:
Home spirometry shows potential for early detection of pulmonary function decline in patients with systemic sclerosis–associated interstitial lung disease (SSc-ILD). It shows good cross-sectional correlation with hospital tests, along with 60% sensitivity and 87% specificity for detecting progressive ILD.
METHODOLOGY:
- Researchers conducted a prospective, observational study to examine the validity of home spirometry for detecting a decline in pulmonary function in patients with SSc-ILD.
- They included 43 patients aged 18 years or older with SSc-ILD from two tertiary referral centers in the Netherlands who received treatment with immunosuppressives for a maximum duration of 8 weeks prior to baseline.
- All participants were required to take weekly home spirometry measurements using a handheld spirometer for 1 year, with 35 completing 6 months of follow-up and 31 completing 12 months.
- Pulmonary function tests were conducted in the hospital at baseline and semiannual visits.
- The primary outcome was the κ (kappa statistic) agreement between home and hospital measurements after 1 year to detect a decline in forced vital capacity (FVC) of 5% or more; the sensitivity and specificity of home spirometry were also evaluated to detect an absolute decline in FVC%, using hospital tests as the gold standard.
TAKEAWAY:
- Home spirometry showed a fair agreement with the pulmonary function tests conducted at the hospital (κ, 0.40; 95% CI, 0.01-0.79).
- Home spirometry showed a sensitivity of 60% and specificity of 87% in detecting a decline in FVC% predicted of 5% or more.
- The intraclass correlation coefficient between home and hospital FVC measurements was moderate to high, with values of 0.85 at baseline, 0.84 at 6 months, and 0.72 at 12 months (P < .0001 for all).
- However, the longitudinal agreement between home and hospital measurements was lower with a correlation coefficient of 0.55.
IN PRACTICE:
“These findings suggest that home spirometry is both feasible and moderately accurate in patients with systemic sclerosis–associated ILD. However, where home spirometry fell short was the low sensitivity in detecting a decline in FVC% predicted,” experts wrote in an accompanying editorial.
“The results of this study support further evaluation of the implementation of home spirometry in addition to regular healthcare management but do not endorse relying solely on home monitoring to detect a decline in pulmonary function,” study authors wrote.
SOURCE:
The study was led by Arthiha Velauthapillai, MD, Department of Rheumatology, Radboud University Medical Center, Nijmegen, the Netherlands, and was published online November 8, 2024, in The Lancet Rheumatology.
LIMITATIONS:
The study might have been underpowered because of inaccuracies in initial assumptions, with a lower-than-anticipated prevalence of progressive ILD and a higher dropout rate. The study included only Dutch patients, which may have limited the generalizability of its findings to other settings with lower internet access or literacy rates.
DISCLOSURES:
This study was partly supported by grants from Galapagos and Boehringer Ingelheim. Some authors received grants or consulting or speaker fees from Boehringer Ingelheim, AstraZeneca, and other pharmaceutical companies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Home spirometry shows potential for early detection of pulmonary function decline in patients with systemic sclerosis–associated interstitial lung disease (SSc-ILD). It shows good cross-sectional correlation with hospital tests, along with 60% sensitivity and 87% specificity for detecting progressive ILD.
METHODOLOGY:
- Researchers conducted a prospective, observational study to examine the validity of home spirometry for detecting a decline in pulmonary function in patients with SSc-ILD.
- They included 43 patients aged 18 years or older with SSc-ILD from two tertiary referral centers in the Netherlands who received treatment with immunosuppressives for a maximum duration of 8 weeks prior to baseline.
- All participants were required to take weekly home spirometry measurements using a handheld spirometer for 1 year, with 35 completing 6 months of follow-up and 31 completing 12 months.
- Pulmonary function tests were conducted in the hospital at baseline and semiannual visits.
- The primary outcome was the κ (kappa statistic) agreement between home and hospital measurements after 1 year to detect a decline in forced vital capacity (FVC) of 5% or more; the sensitivity and specificity of home spirometry were also evaluated to detect an absolute decline in FVC%, using hospital tests as the gold standard.
TAKEAWAY:
- Home spirometry showed a fair agreement with the pulmonary function tests conducted at the hospital (κ, 0.40; 95% CI, 0.01-0.79).
- Home spirometry showed a sensitivity of 60% and specificity of 87% in detecting a decline in FVC% predicted of 5% or more.
- The intraclass correlation coefficient between home and hospital FVC measurements was moderate to high, with values of 0.85 at baseline, 0.84 at 6 months, and 0.72 at 12 months (P < .0001 for all).
- However, the longitudinal agreement between home and hospital measurements was lower with a correlation coefficient of 0.55.
IN PRACTICE:
“These findings suggest that home spirometry is both feasible and moderately accurate in patients with systemic sclerosis–associated ILD. However, where home spirometry fell short was the low sensitivity in detecting a decline in FVC% predicted,” experts wrote in an accompanying editorial.
“The results of this study support further evaluation of the implementation of home spirometry in addition to regular healthcare management but do not endorse relying solely on home monitoring to detect a decline in pulmonary function,” study authors wrote.
SOURCE:
The study was led by Arthiha Velauthapillai, MD, Department of Rheumatology, Radboud University Medical Center, Nijmegen, the Netherlands, and was published online November 8, 2024, in The Lancet Rheumatology.
LIMITATIONS:
The study might have been underpowered because of inaccuracies in initial assumptions, with a lower-than-anticipated prevalence of progressive ILD and a higher dropout rate. The study included only Dutch patients, which may have limited the generalizability of its findings to other settings with lower internet access or literacy rates.
DISCLOSURES:
This study was partly supported by grants from Galapagos and Boehringer Ingelheim. Some authors received grants or consulting or speaker fees from Boehringer Ingelheim, AstraZeneca, and other pharmaceutical companies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Home spirometry shows potential for early detection of pulmonary function decline in patients with systemic sclerosis–associated interstitial lung disease (SSc-ILD). It shows good cross-sectional correlation with hospital tests, along with 60% sensitivity and 87% specificity for detecting progressive ILD.
METHODOLOGY:
- Researchers conducted a prospective, observational study to examine the validity of home spirometry for detecting a decline in pulmonary function in patients with SSc-ILD.
- They included 43 patients aged 18 years or older with SSc-ILD from two tertiary referral centers in the Netherlands who received treatment with immunosuppressives for a maximum duration of 8 weeks prior to baseline.
- All participants were required to take weekly home spirometry measurements using a handheld spirometer for 1 year, with 35 completing 6 months of follow-up and 31 completing 12 months.
- Pulmonary function tests were conducted in the hospital at baseline and semiannual visits.
- The primary outcome was the κ (kappa statistic) agreement between home and hospital measurements after 1 year to detect a decline in forced vital capacity (FVC) of 5% or more; the sensitivity and specificity of home spirometry were also evaluated to detect an absolute decline in FVC%, using hospital tests as the gold standard.
TAKEAWAY:
- Home spirometry showed a fair agreement with the pulmonary function tests conducted at the hospital (κ, 0.40; 95% CI, 0.01-0.79).
- Home spirometry showed a sensitivity of 60% and specificity of 87% in detecting a decline in FVC% predicted of 5% or more.
- The intraclass correlation coefficient between home and hospital FVC measurements was moderate to high, with values of 0.85 at baseline, 0.84 at 6 months, and 0.72 at 12 months (P < .0001 for all).
- However, the longitudinal agreement between home and hospital measurements was lower with a correlation coefficient of 0.55.
IN PRACTICE:
“These findings suggest that home spirometry is both feasible and moderately accurate in patients with systemic sclerosis–associated ILD. However, where home spirometry fell short was the low sensitivity in detecting a decline in FVC% predicted,” experts wrote in an accompanying editorial.
“The results of this study support further evaluation of the implementation of home spirometry in addition to regular healthcare management but do not endorse relying solely on home monitoring to detect a decline in pulmonary function,” study authors wrote.
SOURCE:
The study was led by Arthiha Velauthapillai, MD, Department of Rheumatology, Radboud University Medical Center, Nijmegen, the Netherlands, and was published online November 8, 2024, in The Lancet Rheumatology.
LIMITATIONS:
The study might have been underpowered because of inaccuracies in initial assumptions, with a lower-than-anticipated prevalence of progressive ILD and a higher dropout rate. The study included only Dutch patients, which may have limited the generalizability of its findings to other settings with lower internet access or literacy rates.
DISCLOSURES:
This study was partly supported by grants from Galapagos and Boehringer Ingelheim. Some authors received grants or consulting or speaker fees from Boehringer Ingelheim, AstraZeneca, and other pharmaceutical companies.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Lichen Planus Responds to Treatment with Topical Ruxolitinib in Phase 2 Study
both when given twice daily and as needed, according to data from a phase 2 trial.
The research, presented at the European Academy of Dermatology and Venereology (EADV) 2024 Congress, involved 64 patients older than 18 years. Ruxolitinib cream (Opzelura) is a topical formulation of a Janus kinase (JAK)1/JAK2 inhibitor, approved by the Food and Drug Administration (FDA) for treating mild to moderate atopic dermatitis and for nonsegmental vitiligo in adults and children aged 12 years or older.

Ruxolitinib cream twice daily resulted in “significant improvements in cutaneous lichen planus disease severity vs vehicle” after 16 weeks of treatment, said the study presenter, Aaron R. Mangold, MD, a dermatologist at Mayo Clinic, Scottsdale, Arizona.
Further improvements were seen during another 16 weeks of additional open-label, as-needed application, he added, and the topical treatment was “generally well tolerated.”
Consequently, “ruxolitinib cream represents a promising potential treatment for cutaneous lichen planus,” Mangold concluded.
Asked to comment on the results, Adam Friedman, MD, Professor and Chair of Dermatology, George Washington University, Washington, DC, who was not involved with the study, said that in keeping with the characterization of lichen planus using the four Ps — purple, polygonal, pruritic, papules — it is “Pretty common, Predictably disabling and disfiguring, and Passed over again and again in the drug development world.”
He said in an interview that this chronic inflammatory skin condition, which affects roughly 2% of the population, also “lacks consensus on work-up and management, likely in part owing to the absence of sizable clinical trial data.”
A recent survey conducted at a meeting indicated that dermatologists “heavily lean on topical therapies for the management of all severity levels,” noted Friedman, one of the survey authors. “Therefore, the phase 2 data presented at EADV is a welcome addition to the mix.”
Phase 2 Study Results
At the meeting, Mangold said that a previous proof-of-concept single-arm study in 12 patients suggested that topical ruxolitinib was highly effective in treating cutaneous lichen planus.
The current phase 2 trial enrolled 64 patients with predominantly cutaneous disease who had an Investigator’s Global Assessment (IGA) score of 3 or 4 and an Itch Numeric Rating Scale (NRS) score of ≥ 4. Their median age was 57 years, and 71.9% were women. Nearly 63% were White, 28.1% were Black, and 6.3% were Asian. The median duration of disease was 4.9 years, and 90.6% had received prior treatment for their lichen planus.
They were randomized to receive 1.5% ruxolitinib cream or a vehicle cream twice daily for 16 weeks, and following a primary endpoint assessment, they were transferred to an open-label extension period, during which they used ruxolitinib cream as needed for another 16 weeks. There was an additional 30-day safety follow-up period.
At week 16, significantly more patients treated with the ruxolitinib cream (50.0%) vs vehicle cream (21.9%) achieved IGA treatment success (the primary endpoint), defined as an IGA score of 0 or 1 with ≥ 2-grade improvement from baseline (odds ratio, 4.04; P = .0129).
In the open-label extension, when all patients used the active cream as needed, the proportion achieving IGA treatment success increased to 60% among the patients originally treated with ruxolitinib cream and 60.9% among those who switched from the vehicle cream.
A similar pattern was seen with Itch NRS scores. At 16 weeks, 57.7% of those treated with the ruxolitinib cream and 19.2% of those given the vehicle cream achieved an Itch NRS score of ≥ 4 (P < .01), rising to 84.2% and 73.3%, respectively, during the open-label extension.
The time to achievement of an Itch NRS of ≥ 4 was also significantly shorter with the ruxolitinib cream than with the vehicle cream (median days, 17 vs 97; hazard ratio, 2.85; P = .0008).
In both treatment groups, Skin Pain NRS scores decreased by a mean of 3.0 with ruxolitinib cream and 1.3 with the vehicle cream at week 16. By the end of the open-label extension, scores dropped by 4.3 among those who continued on active treatment and by 3.5 among those who switched from vehicle to topical ruxolitinib.
There were few treatment-emergent adverse events, with just three ruxolitinib patients affected during the randomized phase of the trial. There was one grade ≥ 3 event considered unrelated to the study drug, and no serious treatment-emergent adverse events were reported.
The most common adverse events during the randomized period were nasopharyngitis, hypertension, and contusion, all experienced by fewer than 10% of patients, whereas sinusitis, increased blood cholesterol levels, and increased blood creatine phosphokinase were most common in the open-label extension, experienced by no more than 5% of patients.
In the interview, Friedman commented that “these data provide hope that one day soon, there will be an FDA-approved, effective, and well-tolerated approach for this condition, validating the patient and supporting the dermatologist with an evidence-based option.”
The study was funded by Incyte. Mangold declared relationships with Argenx, Boehringer Ingelheim, Bristol-Myers Squibb, Clarivate, Incyte Corporation, Janssen, Nuvig Therapeutics, Pfizer, Regeneron Pharmaceuticals, Soligenix, Tourmaline Bio, AbbVie, Corbus, Eli Lilly, Kyowa, Merck, miRagen Therapeutics, Palvella Therapeutics, Priovant Therapeutics, and Adelphi Values. Friedman declared a relationship with Incyte, but it is not related to this topic.
A version of this article first appeared on Medscape.com.
both when given twice daily and as needed, according to data from a phase 2 trial.
The research, presented at the European Academy of Dermatology and Venereology (EADV) 2024 Congress, involved 64 patients older than 18 years. Ruxolitinib cream (Opzelura) is a topical formulation of a Janus kinase (JAK)1/JAK2 inhibitor, approved by the Food and Drug Administration (FDA) for treating mild to moderate atopic dermatitis and for nonsegmental vitiligo in adults and children aged 12 years or older.
Ruxolitinib cream twice daily resulted in “significant improvements in cutaneous lichen planus disease severity vs vehicle” after 16 weeks of treatment, said the study presenter, Aaron R. Mangold, MD, a dermatologist at Mayo Clinic, Scottsdale, Arizona.
Further improvements were seen during another 16 weeks of additional open-label, as-needed application, he added, and the topical treatment was “generally well tolerated.”
Consequently, “ruxolitinib cream represents a promising potential treatment for cutaneous lichen planus,” Mangold concluded.
Asked to comment on the results, Adam Friedman, MD, Professor and Chair of Dermatology, George Washington University, Washington, DC, who was not involved with the study, said that in keeping with the characterization of lichen planus using the four Ps — purple, polygonal, pruritic, papules — it is “Pretty common, Predictably disabling and disfiguring, and Passed over again and again in the drug development world.”
He said in an interview that this chronic inflammatory skin condition, which affects roughly 2% of the population, also “lacks consensus on work-up and management, likely in part owing to the absence of sizable clinical trial data.”
A recent survey conducted at a meeting indicated that dermatologists “heavily lean on topical therapies for the management of all severity levels,” noted Friedman, one of the survey authors. “Therefore, the phase 2 data presented at EADV is a welcome addition to the mix.”
Phase 2 Study Results
At the meeting, Mangold said that a previous proof-of-concept single-arm study in 12 patients suggested that topical ruxolitinib was highly effective in treating cutaneous lichen planus.
The current phase 2 trial enrolled 64 patients with predominantly cutaneous disease who had an Investigator’s Global Assessment (IGA) score of 3 or 4 and an Itch Numeric Rating Scale (NRS) score of ≥ 4. Their median age was 57 years, and 71.9% were women. Nearly 63% were White, 28.1% were Black, and 6.3% were Asian. The median duration of disease was 4.9 years, and 90.6% had received prior treatment for their lichen planus.
They were randomized to receive 1.5% ruxolitinib cream or a vehicle cream twice daily for 16 weeks, and following a primary endpoint assessment, they were transferred to an open-label extension period, during which they used ruxolitinib cream as needed for another 16 weeks. There was an additional 30-day safety follow-up period.
At week 16, significantly more patients treated with the ruxolitinib cream (50.0%) vs vehicle cream (21.9%) achieved IGA treatment success (the primary endpoint), defined as an IGA score of 0 or 1 with ≥ 2-grade improvement from baseline (odds ratio, 4.04; P = .0129).
In the open-label extension, when all patients used the active cream as needed, the proportion achieving IGA treatment success increased to 60% among the patients originally treated with ruxolitinib cream and 60.9% among those who switched from the vehicle cream.
A similar pattern was seen with Itch NRS scores. At 16 weeks, 57.7% of those treated with the ruxolitinib cream and 19.2% of those given the vehicle cream achieved an Itch NRS score of ≥ 4 (P < .01), rising to 84.2% and 73.3%, respectively, during the open-label extension.
The time to achievement of an Itch NRS of ≥ 4 was also significantly shorter with the ruxolitinib cream than with the vehicle cream (median days, 17 vs 97; hazard ratio, 2.85; P = .0008).
In both treatment groups, Skin Pain NRS scores decreased by a mean of 3.0 with ruxolitinib cream and 1.3 with the vehicle cream at week 16. By the end of the open-label extension, scores dropped by 4.3 among those who continued on active treatment and by 3.5 among those who switched from vehicle to topical ruxolitinib.
There were few treatment-emergent adverse events, with just three ruxolitinib patients affected during the randomized phase of the trial. There was one grade ≥ 3 event considered unrelated to the study drug, and no serious treatment-emergent adverse events were reported.
The most common adverse events during the randomized period were nasopharyngitis, hypertension, and contusion, all experienced by fewer than 10% of patients, whereas sinusitis, increased blood cholesterol levels, and increased blood creatine phosphokinase were most common in the open-label extension, experienced by no more than 5% of patients.
In the interview, Friedman commented that “these data provide hope that one day soon, there will be an FDA-approved, effective, and well-tolerated approach for this condition, validating the patient and supporting the dermatologist with an evidence-based option.”
The study was funded by Incyte. Mangold declared relationships with Argenx, Boehringer Ingelheim, Bristol-Myers Squibb, Clarivate, Incyte Corporation, Janssen, Nuvig Therapeutics, Pfizer, Regeneron Pharmaceuticals, Soligenix, Tourmaline Bio, AbbVie, Corbus, Eli Lilly, Kyowa, Merck, miRagen Therapeutics, Palvella Therapeutics, Priovant Therapeutics, and Adelphi Values. Friedman declared a relationship with Incyte, but it is not related to this topic.
A version of this article first appeared on Medscape.com.
both when given twice daily and as needed, according to data from a phase 2 trial.
The research, presented at the European Academy of Dermatology and Venereology (EADV) 2024 Congress, involved 64 patients older than 18 years. Ruxolitinib cream (Opzelura) is a topical formulation of a Janus kinase (JAK)1/JAK2 inhibitor, approved by the Food and Drug Administration (FDA) for treating mild to moderate atopic dermatitis and for nonsegmental vitiligo in adults and children aged 12 years or older.
Ruxolitinib cream twice daily resulted in “significant improvements in cutaneous lichen planus disease severity vs vehicle” after 16 weeks of treatment, said the study presenter, Aaron R. Mangold, MD, a dermatologist at Mayo Clinic, Scottsdale, Arizona.
Further improvements were seen during another 16 weeks of additional open-label, as-needed application, he added, and the topical treatment was “generally well tolerated.”
Consequently, “ruxolitinib cream represents a promising potential treatment for cutaneous lichen planus,” Mangold concluded.
Asked to comment on the results, Adam Friedman, MD, Professor and Chair of Dermatology, George Washington University, Washington, DC, who was not involved with the study, said that in keeping with the characterization of lichen planus using the four Ps — purple, polygonal, pruritic, papules — it is “Pretty common, Predictably disabling and disfiguring, and Passed over again and again in the drug development world.”
He said in an interview that this chronic inflammatory skin condition, which affects roughly 2% of the population, also “lacks consensus on work-up and management, likely in part owing to the absence of sizable clinical trial data.”
A recent survey conducted at a meeting indicated that dermatologists “heavily lean on topical therapies for the management of all severity levels,” noted Friedman, one of the survey authors. “Therefore, the phase 2 data presented at EADV is a welcome addition to the mix.”
Phase 2 Study Results
At the meeting, Mangold said that a previous proof-of-concept single-arm study in 12 patients suggested that topical ruxolitinib was highly effective in treating cutaneous lichen planus.
The current phase 2 trial enrolled 64 patients with predominantly cutaneous disease who had an Investigator’s Global Assessment (IGA) score of 3 or 4 and an Itch Numeric Rating Scale (NRS) score of ≥ 4. Their median age was 57 years, and 71.9% were women. Nearly 63% were White, 28.1% were Black, and 6.3% were Asian. The median duration of disease was 4.9 years, and 90.6% had received prior treatment for their lichen planus.
They were randomized to receive 1.5% ruxolitinib cream or a vehicle cream twice daily for 16 weeks, and following a primary endpoint assessment, they were transferred to an open-label extension period, during which they used ruxolitinib cream as needed for another 16 weeks. There was an additional 30-day safety follow-up period.
At week 16, significantly more patients treated with the ruxolitinib cream (50.0%) vs vehicle cream (21.9%) achieved IGA treatment success (the primary endpoint), defined as an IGA score of 0 or 1 with ≥ 2-grade improvement from baseline (odds ratio, 4.04; P = .0129).
In the open-label extension, when all patients used the active cream as needed, the proportion achieving IGA treatment success increased to 60% among the patients originally treated with ruxolitinib cream and 60.9% among those who switched from the vehicle cream.
A similar pattern was seen with Itch NRS scores. At 16 weeks, 57.7% of those treated with the ruxolitinib cream and 19.2% of those given the vehicle cream achieved an Itch NRS score of ≥ 4 (P < .01), rising to 84.2% and 73.3%, respectively, during the open-label extension.
The time to achievement of an Itch NRS of ≥ 4 was also significantly shorter with the ruxolitinib cream than with the vehicle cream (median days, 17 vs 97; hazard ratio, 2.85; P = .0008).
In both treatment groups, Skin Pain NRS scores decreased by a mean of 3.0 with ruxolitinib cream and 1.3 with the vehicle cream at week 16. By the end of the open-label extension, scores dropped by 4.3 among those who continued on active treatment and by 3.5 among those who switched from vehicle to topical ruxolitinib.
There were few treatment-emergent adverse events, with just three ruxolitinib patients affected during the randomized phase of the trial. There was one grade ≥ 3 event considered unrelated to the study drug, and no serious treatment-emergent adverse events were reported.
The most common adverse events during the randomized period were nasopharyngitis, hypertension, and contusion, all experienced by fewer than 10% of patients, whereas sinusitis, increased blood cholesterol levels, and increased blood creatine phosphokinase were most common in the open-label extension, experienced by no more than 5% of patients.
In the interview, Friedman commented that “these data provide hope that one day soon, there will be an FDA-approved, effective, and well-tolerated approach for this condition, validating the patient and supporting the dermatologist with an evidence-based option.”
The study was funded by Incyte. Mangold declared relationships with Argenx, Boehringer Ingelheim, Bristol-Myers Squibb, Clarivate, Incyte Corporation, Janssen, Nuvig Therapeutics, Pfizer, Regeneron Pharmaceuticals, Soligenix, Tourmaline Bio, AbbVie, Corbus, Eli Lilly, Kyowa, Merck, miRagen Therapeutics, Palvella Therapeutics, Priovant Therapeutics, and Adelphi Values. Friedman declared a relationship with Incyte, but it is not related to this topic.
A version of this article first appeared on Medscape.com.
FROM EADV 2024
Pemphigus, Bullous Pemphigoid Risk Increased After COVID-19 Infection
TOPLINE:
according to a study that also found that vaccination against COVID-19 is associated with a reduced risk for these conditions.
METHODOLOGY:
- Researchers conducted a population-based retrospective cohort study using data from the TriNetX Analytics Network, encompassing over 112 million electronic health records in the United States.
- The study compared the risk for AIBD within 3 months among individuals who had COVID-19 infection and no COVID-19 vaccination 6 months prior to the infection (n = 4,787,106), individuals who had COVID-19 vaccination but did not have COVID-19 infection (n = 3,466,536), and individuals who did not have COVID-19 infection or vaccination (n = 5,609,197).
- The mean age of the three groups was 44.9, 52.3, and 49.3 years, respectively.
- Propensity score matching included 4,408,748 individuals each for the comparison between COVID-19 infection and controls, 3,465,420 for COVID-19 vaccination and controls, and 3,362,850 for COVID-19 infection and vaccination. The mean follow-up ranged from 72.2 to 76.3 days.
TAKEAWAY:
- Individuals with COVID-19 infection showed a 50.8% increased risk for AIBD within 3 months (P < .001) compared with those without infection or vaccination. The risk was more pronounced for pemphigus (hazard ratio [HR], 2.432; P < .001) than bullous pemphigoid (HR, 1.376; P = .036).
- On the contrary, individuals who had the COVID-19 vaccination showed almost half the risk for AIBD (HR, 0.514; P < .001). The risk reduction was significant for pemphigus (HR, 0.477; P = .030), but not for bullous pemphigoid (HR, 0.846).
- When the infection and vaccination groups were compared, COVID-19 infection increased AIBD risk by more than threefold (HR, 3.130; P < .001), with a particularly high risk for pemphigus (HR, 5.508; P < .001). A significant risk was also seen for bullous pemphigoid (HR, 1.587; P = .008).
IN PRACTICE:
“The findings underscore the importance of vaccination not only in preventing severe COVID-19 outcomes but also in potentially protecting against autoimmune complications,” the authors wrote, adding that “this potential dual benefit of vaccination should be a key message in public health campaigns and clinical practice to enhance vaccine uptake and ultimately improve health outcomes.”
SOURCE:
The study was led by Philip Curman, MD, PhD, of the Dermato-Venereology Clinic at Karolinska University Hospital, Stockholm, Sweden, and was published online on November 7 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The retrospective design has inherent biases, there is potential underreporting of COVID-19 cases and vaccinations, and there is misallocation of individuals. Unmeasured confounding factors may be present.
DISCLOSURES:
This study was funded by grant from the State of Schleswig-Holstein. Two authors were employees of TriNetX. Some authors received financial support and travel grants from various sources, including TriNetX. Additional disclosures are noted in the article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
according to a study that also found that vaccination against COVID-19 is associated with a reduced risk for these conditions.
METHODOLOGY:
- Researchers conducted a population-based retrospective cohort study using data from the TriNetX Analytics Network, encompassing over 112 million electronic health records in the United States.
- The study compared the risk for AIBD within 3 months among individuals who had COVID-19 infection and no COVID-19 vaccination 6 months prior to the infection (n = 4,787,106), individuals who had COVID-19 vaccination but did not have COVID-19 infection (n = 3,466,536), and individuals who did not have COVID-19 infection or vaccination (n = 5,609,197).
- The mean age of the three groups was 44.9, 52.3, and 49.3 years, respectively.
- Propensity score matching included 4,408,748 individuals each for the comparison between COVID-19 infection and controls, 3,465,420 for COVID-19 vaccination and controls, and 3,362,850 for COVID-19 infection and vaccination. The mean follow-up ranged from 72.2 to 76.3 days.
TAKEAWAY:
- Individuals with COVID-19 infection showed a 50.8% increased risk for AIBD within 3 months (P < .001) compared with those without infection or vaccination. The risk was more pronounced for pemphigus (hazard ratio [HR], 2.432; P < .001) than bullous pemphigoid (HR, 1.376; P = .036).
- On the contrary, individuals who had the COVID-19 vaccination showed almost half the risk for AIBD (HR, 0.514; P < .001). The risk reduction was significant for pemphigus (HR, 0.477; P = .030), but not for bullous pemphigoid (HR, 0.846).
- When the infection and vaccination groups were compared, COVID-19 infection increased AIBD risk by more than threefold (HR, 3.130; P < .001), with a particularly high risk for pemphigus (HR, 5.508; P < .001). A significant risk was also seen for bullous pemphigoid (HR, 1.587; P = .008).
IN PRACTICE:
“The findings underscore the importance of vaccination not only in preventing severe COVID-19 outcomes but also in potentially protecting against autoimmune complications,” the authors wrote, adding that “this potential dual benefit of vaccination should be a key message in public health campaigns and clinical practice to enhance vaccine uptake and ultimately improve health outcomes.”
SOURCE:
The study was led by Philip Curman, MD, PhD, of the Dermato-Venereology Clinic at Karolinska University Hospital, Stockholm, Sweden, and was published online on November 7 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The retrospective design has inherent biases, there is potential underreporting of COVID-19 cases and vaccinations, and there is misallocation of individuals. Unmeasured confounding factors may be present.
DISCLOSURES:
This study was funded by grant from the State of Schleswig-Holstein. Two authors were employees of TriNetX. Some authors received financial support and travel grants from various sources, including TriNetX. Additional disclosures are noted in the article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
according to a study that also found that vaccination against COVID-19 is associated with a reduced risk for these conditions.
METHODOLOGY:
- Researchers conducted a population-based retrospective cohort study using data from the TriNetX Analytics Network, encompassing over 112 million electronic health records in the United States.
- The study compared the risk for AIBD within 3 months among individuals who had COVID-19 infection and no COVID-19 vaccination 6 months prior to the infection (n = 4,787,106), individuals who had COVID-19 vaccination but did not have COVID-19 infection (n = 3,466,536), and individuals who did not have COVID-19 infection or vaccination (n = 5,609,197).
- The mean age of the three groups was 44.9, 52.3, and 49.3 years, respectively.
- Propensity score matching included 4,408,748 individuals each for the comparison between COVID-19 infection and controls, 3,465,420 for COVID-19 vaccination and controls, and 3,362,850 for COVID-19 infection and vaccination. The mean follow-up ranged from 72.2 to 76.3 days.
TAKEAWAY:
- Individuals with COVID-19 infection showed a 50.8% increased risk for AIBD within 3 months (P < .001) compared with those without infection or vaccination. The risk was more pronounced for pemphigus (hazard ratio [HR], 2.432; P < .001) than bullous pemphigoid (HR, 1.376; P = .036).
- On the contrary, individuals who had the COVID-19 vaccination showed almost half the risk for AIBD (HR, 0.514; P < .001). The risk reduction was significant for pemphigus (HR, 0.477; P = .030), but not for bullous pemphigoid (HR, 0.846).
- When the infection and vaccination groups were compared, COVID-19 infection increased AIBD risk by more than threefold (HR, 3.130; P < .001), with a particularly high risk for pemphigus (HR, 5.508; P < .001). A significant risk was also seen for bullous pemphigoid (HR, 1.587; P = .008).
IN PRACTICE:
“The findings underscore the importance of vaccination not only in preventing severe COVID-19 outcomes but also in potentially protecting against autoimmune complications,” the authors wrote, adding that “this potential dual benefit of vaccination should be a key message in public health campaigns and clinical practice to enhance vaccine uptake and ultimately improve health outcomes.”
SOURCE:
The study was led by Philip Curman, MD, PhD, of the Dermato-Venereology Clinic at Karolinska University Hospital, Stockholm, Sweden, and was published online on November 7 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The retrospective design has inherent biases, there is potential underreporting of COVID-19 cases and vaccinations, and there is misallocation of individuals. Unmeasured confounding factors may be present.
DISCLOSURES:
This study was funded by grant from the State of Schleswig-Holstein. Two authors were employees of TriNetX. Some authors received financial support and travel grants from various sources, including TriNetX. Additional disclosures are noted in the article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Botulinum Toxin Injection for Treatment of Scleroderma-Related Anterior Neck Sclerosis
To the Editor:
Scleroderma is a chronic autoimmune connective tissue disease that results in excessive collagen deposition in the skin and other organs throughout the body. On its own or in the setting of mixed connective tissue disease, scleroderma can result in systemic or localized symptoms that can limit patients’ functional capabilities, cause pain and discomfort, and reduce self-esteem—all negatively impacting patients’ quality of life.1,2 Neck sclerosis is a common manifestation of scleroderma. There is no curative treatment for scleroderma; thus, therapy is focused on slowing disease progression and improving quality of life. We present a case of neck sclerosis in a 44-year-old woman with scleroderma that was successfully treated with botulinum toxin (BTX) type A injection, resulting in improved skin laxity and appearance with high patient satisfaction. Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region.
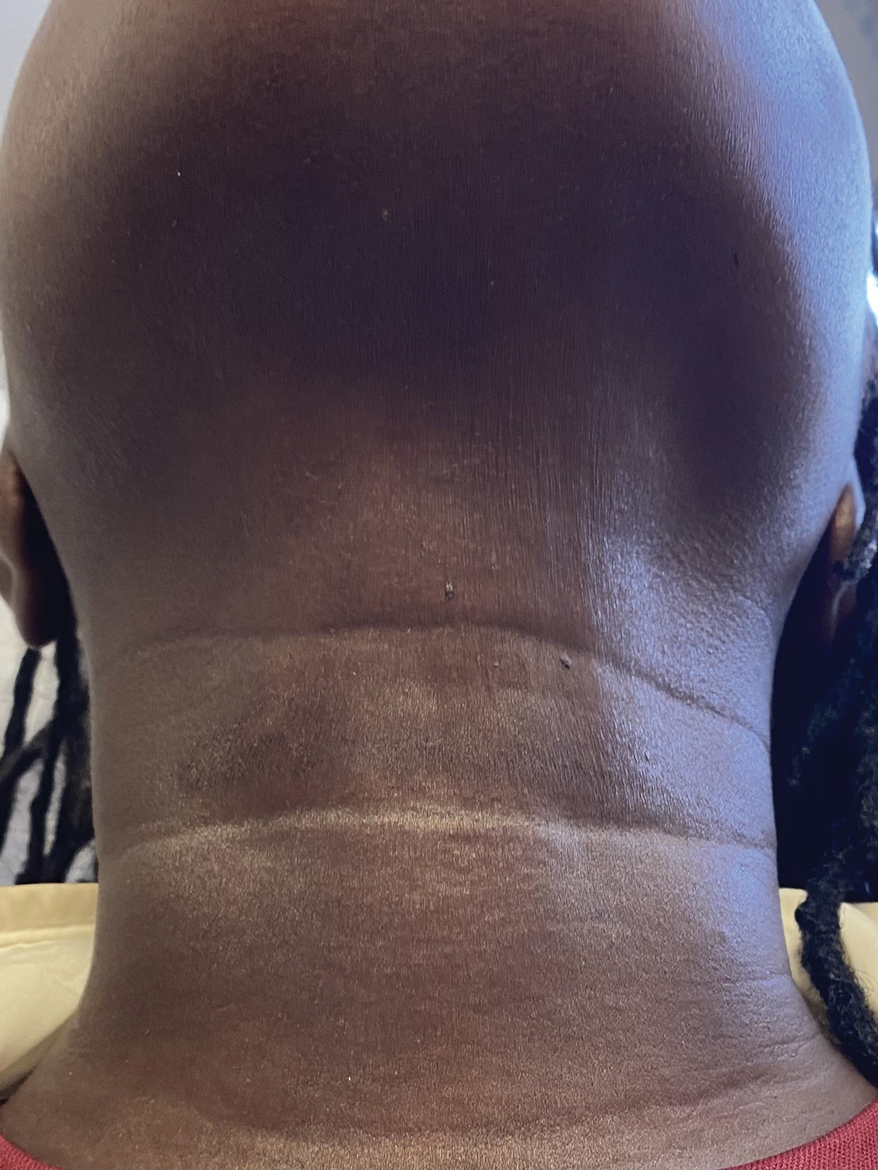
A 44-year-old woman presented to the dermatology clinic for treatment of thickened neck skin with stiffness and tightness that had been present for months to years. She had a history of mixed connective tissue disease (MCTD)(positive anti-ribonucleoprotein, anti–Sjögren syndrome–related antigen, and anti-Smith antibodies) with features of scleroderma and polyarthritis. The patient currently was taking sulfasalazine for the polyarthritis; she previously had taken hydroxychloroquine but discontinued treatment due to ineffectiveness. She was not taking any topical or systemic medications for scleroderma. On physical examination, the skin on the anterior neck appeared thickened with shiny patches (Figure 1). Pinching the skin in the affected area demonstrated sclerosis with high tension.
The dermatologist (J.J.) discussed potential treatment options to help relax the tension in the skin of the anterior neck, including BTX injections. After receiving counsel on adverse effects, alternative treatments, and postprocedural care, the patient decided to proceed with the procedure. The anterior neck was cleansed with an alcohol swab and 37 units (range, 25–50 units) of incobotulinumtoxinA (reconstituted using 2.5-mL bacteriostatic normal saline per 100 units) was injected transdermally using a 9-point injection technique, with each injection placed approximately 1 cm apart. The approximate treatment area included the space between the sternocleidomastoid anterior edges and below the hyoid bone up to the cricothyroid membrane (anatomic zone II).
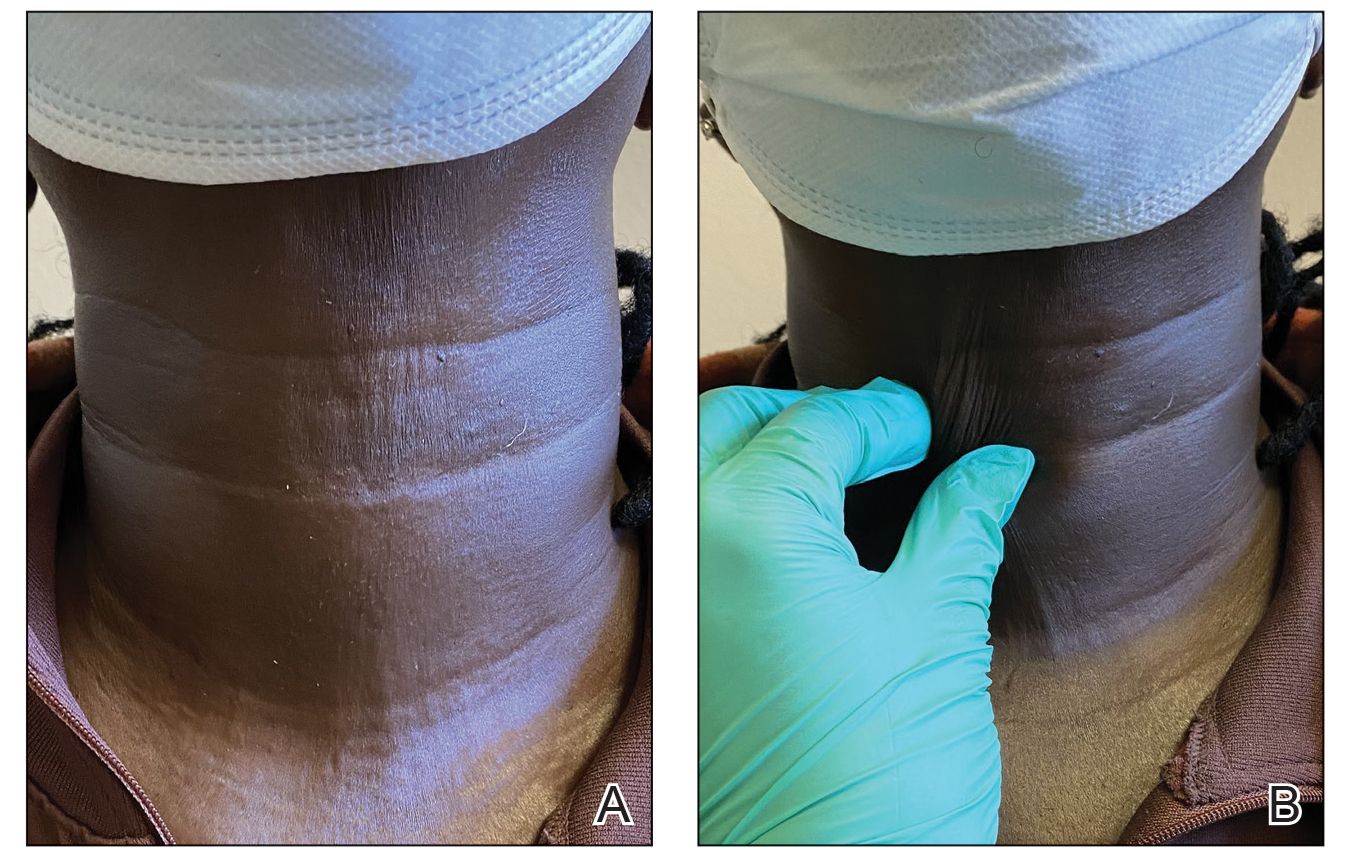
When the patient returned for follow-up 3 weeks later, she reported considerable improvement in the stiffness and appearance of the skin on the anterior neck. On physical examination, the skin of the neck appeared softened, and improved laxity was seen on pinching the skin compared to the initial presentation (Figure 2). The patient expressed satisfaction with the results and denied any adverse events following the procedure.
Mixed connective tissue disease manifests with a combination of features from various disorders—mainly lupus, scleroderma, polymyositis, and rheumatoid arthritis. It is most prevalent in females and often is diagnosed in the third decade of life.3 It is associated with positive antinuclear antibodies and human leukocyte antigen (HLA) II alleles (HLA-DR4, HLA-DR1, and HLA-DR2). Raynaud phenomenon (RP), one of the most common skin manifestations in both scleroderma and MCTD, is present in 75% to 90% of patients with MCTD.3
Scleroderma is a chronic connective tissue disorder that results in excessive collagen deposition in the skin and other organs throughout the body.4 Although the etiology is unknown, scleroderma develops when overactivation of the immune system leads to CD4+ T-lymphocyte infiltration in the skin, along with the release of profibrotic interleukins and growth factors, resulting in fibrosis.4 Subtypes include localized scleroderma (morphea), limited cutaneous systemic sclerosis (formerly known as CREST [calcinosis, RP, esophageal dysmotility, sclerodactyly, and telangiectasia] syndrome), diffuse cutaneous systemic sclerosis, and systemic sclerosis sine scleroderma.5 Scleroderma is associated with positive antinuclear antibodies and HLA II alleles (HLA-DR2 and HLA-DR5).
On its own or in the setting of MCTD, scleroderma can result in systemic or localized symptoms. Overall, the most common symptom is RP.5 Localized scleroderma and limited cutaneous systemic sclerosis manifest with symptoms of the skin and underlying tissues. Diffuse cutaneous systemic sclerosis involves cutaneous and visceral symptoms, including lung, esophageal, and vascular involvement.6 Similar to MCTD, scleroderma is most prevalent in middle-aged females,7 though it occurs at a higher rate and with a more severe disease course in Black patients.8
A highly sensitive and specific test for scleroderma that can aid in diagnosis is the neck sign—tightening of the skin of the neck when the head extends.9,10 In one study, the neck sign was positive in more than 90% of patients with scleroderma and negative for control patients and those with primary RP.9 Thus, neck sclerosis is a common manifestation of scleroderma for which patients may seek treatment.
While there is no curative treatment for scleroderma, skin manifestations can be treated with mycophenolate mofetil or methotrexate.5 Systemic treatments may be recommended if the patient has additional symptoms, such as azathioprine for myositis/arthritis and cyclophosphamide for interstitial lung disease.5 However, it is important to note that these medications are associated with risk for gastrointestinal upset, mouth sores, fatigue, or other complications.
Botulinum toxin is a bacterial protein toxin and neuromodulator that inhibits neurotransmitter release by cleaving SNARE proteins at peripheral nerve terminal junctions.11 It has been used in a variety of dermatologic and nondermatologic conditions, including migraines, hyperhidrosis, contractures, scars, and overactive bladder. It also has been used in aesthetics for facial rejuvenation and minimization of wrinkle appearance. Dermatologists and rheumatologists have successfully used BTX to treat primary and secondary RP—the most common symptom of scleroderma—due to its vasodilatation properties.12 Although our patient did not have RP, use of BTX to treat other features of scleroderma, including en coup de sabre, thoracic outlet syndrome, dyspareunia, gastroparesis, pterygium inversum unguis, and dysphagia has been documented.13-18 An in vivo mouse study that examined the possible mechanism for BTX as a treatment in scleroderma found that BTX injections significantly decreased dermal thickness and inflammation in fibrosis (P<.05). An analysis of oxidative stress and mRNA expression showed that BTX may treat fibrosis by suppressing oxidative stress and inflammatory cells, resulting in decreased apoptosis and oxidant-induced intracellular accumulation of reactive oxygen species.19 Another animal study demonstrated the positive effects of BTX treatment for fibrosis of the bladder in rats.20 In one case report, a female patient with scleroderma and facial fibrosis received perioral BTX injections for cosmetic purposes but also observed improvement in mouth constriction, demonstrating the potential efficacy of BTX for facial fibrosis.21
Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region. We recommend assessing the efficacy of the initial BTX treatment after 2 to 3 weeks, with additional injections as needed to achieve the patient’s desired level of comfort and appearance at approximately 3-month intervals (aligning with the expected duration of efficacy of BTX).22 Our patient experienced considerable relief and high satisfaction with BTX treatment. Given the limitations of sclerosis treatments and the unwanted adverse-effect profile of systemic treatments, BTX injections may be a preferrable treatment option for cutaneous manifestations of scleroderma among patients. Future studies with larger patient populations and a control group are warranted to further explore the use of BTX for the dermatologic treatment of scleroderma.
- Lis-S´wie¸ty A, Skrzypek-Salamon A, Ranosz-Janicka I, et al. Health-related quality of life and its influencing factors in adult patients with localized scleroderma—a cross-sectional study. Health Qual Life Outcomes. 2020;18:133. doi:10.1186/s12955-020-01386-0
- Almeida C, Almeida I, Vasconcelos C. Quality of life in systemic sclerosis. Autoimmun Rev. 2015;14:1087-1096. doi:10.1016/j.autrev.2015.07.012
- Ortega-Hernandez OD, Shoenfeld Y. Mixed connective tissue disease: an overview of clinical manifestations, diagnosis and treatment. Best Pract Res Clin Rheumatol. 2012;26:61-72. doi:10.1016/j.berh.2012.01.009
- Rongioletti F, Ferreli C, Atzori L, et al. Scleroderma with an update about clinico-pathological correlation. G Ital Dermatol Venereol. 2018;153:208-215. doi:10.23736/S0392-0488.18.05922-9
- Fett N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437. doi:10.1016/j.clindermatol.2013.01.010
- Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90:62-73. doi:10.1590/abd1806-4841.20152890
- Calderon LM, Pope JE. Scleroderma epidemiology update. Curr Opin Rheumatol. 2021;33:122-127. doi:10.1097/BOR.0000000000000785
- Morgan ND, Gelber AC. African Americans and scleroderma: examining the root cause of the association. Arthritis Care Res (Hoboken). 2019;71:1151-1153. doi:10.1002/acr.23860
- Barnett AJ. The “neck sign” in scleroderma. Arthritis Rheum. 1989;32:209-211. doi:10.1002/anr.1780320215
- Barnett AJ, Miller M, Littlejohn GO. The diagnosis and classification of scleroderma (systemic sclerosis). Postgrad Med J. 1988;64:121-125. doi:10.1136/pgmj.64.748.121
- Rossetto O, Pirazzini M, Fabris F, et al. Botulinum neurotoxins: mechanism of action. Handb Exp Pharmacol. 2021;263:35-47.doi:10.1007/164_2020_355
- Ennis D, Ahmad Z, Anderson MA, et al. Botulinum toxin in the management of primary and secondary Raynaud’s phenomenon. Best Pract Res Clin Rheumatol. 2021;35:101684. doi:10.1016/j.berh.2021.101684
- Turkmani MG, Alnomair N. Enhancement of the aesthetic outcome of scleroderma en coup de sabre with botulinum toxin injection. JAAD Case Rep. 2018;4:579-581. doi:10.1016/j.jdcr.2018.03.023
- Le EN, Freischlag JA, Christo PJ, et al. Thoracic outlet syndrome secondary to localized scleroderma treated with botulinum toxin injection. Arthritis Care Res (Hoboken). 2010;62:430-433. doi:10.1002/acr.20099
- Mousty E, Rathat G, Rouleau C, et al. Botulinum toxin type A for treatment of dyspareunia caused by localized scleroderma. Acta Obstet Gynecol Scand. 2011;90:926-927. doi:10.1111/j.1600-0412.2011.01183.x
- Tang DM, Friedenberg FK. Gastroparesis: approach, diagnostic evaluation, and management. Dis Mon. 2011;57:74-101. doi:10.1016/j.disamonth.2010.12.007
- Katschinski M. [Diagnosis and treatment of esophageal motility disorders]. Ther Umsch. 2001;58:128-133. doi:10.1024/0040-5930.58.3.128
- Kim DJ, Odell ID. Improvement of pterygium inversum unguis and Raynaud phenomenon with interdigital botulinum toxin injections. JAAD Case Rep. 2022;26:79-81. doi:10.1016/j.jdcr.2022.06.009
- Baral H, Sekiguchi A, Uchiyama A, et al. Inhibition of skin fibrosis in systemic sclerosis by botulinum toxin B via the suppression of oxidative stress. J Dermatol. 2021;48:1052-1061. doi:10.1111/1346-8138.15888
- Jia C, Xing T, Shang Z, et al. Botulinum toxin A improves neurogenic bladder fibrosis by suppressing transforming growth factor β1 expression in rats. Transl Androl Urol. 2021;10:2000-2007. doi:10.21037/tau-21-62
- Hoverson K, Love T, Lam TK, et al. A novel treatment for limited mouth opening due to facial fibrosis: a case series. J Am Acad Dermatol. 2018;78:190-192. doi:10.1016/j.jaad.2017.07.006
- Kollewe K, Mohammadi B, Köhler S, et al. Blepharospasm: long-term treatment with either Botox®, Xeomin® or Dysport®. J Neural Transm (Vienna). 2015;122:427-431. doi:10.1007/s00702-014-1278-z
To the Editor:
Scleroderma is a chronic autoimmune connective tissue disease that results in excessive collagen deposition in the skin and other organs throughout the body. On its own or in the setting of mixed connective tissue disease, scleroderma can result in systemic or localized symptoms that can limit patients’ functional capabilities, cause pain and discomfort, and reduce self-esteem—all negatively impacting patients’ quality of life.1,2 Neck sclerosis is a common manifestation of scleroderma. There is no curative treatment for scleroderma; thus, therapy is focused on slowing disease progression and improving quality of life. We present a case of neck sclerosis in a 44-year-old woman with scleroderma that was successfully treated with botulinum toxin (BTX) type A injection, resulting in improved skin laxity and appearance with high patient satisfaction. Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region.
A 44-year-old woman presented to the dermatology clinic for treatment of thickened neck skin with stiffness and tightness that had been present for months to years. She had a history of mixed connective tissue disease (MCTD)(positive anti-ribonucleoprotein, anti–Sjögren syndrome–related antigen, and anti-Smith antibodies) with features of scleroderma and polyarthritis. The patient currently was taking sulfasalazine for the polyarthritis; she previously had taken hydroxychloroquine but discontinued treatment due to ineffectiveness. She was not taking any topical or systemic medications for scleroderma. On physical examination, the skin on the anterior neck appeared thickened with shiny patches (Figure 1). Pinching the skin in the affected area demonstrated sclerosis with high tension.
The dermatologist (J.J.) discussed potential treatment options to help relax the tension in the skin of the anterior neck, including BTX injections. After receiving counsel on adverse effects, alternative treatments, and postprocedural care, the patient decided to proceed with the procedure. The anterior neck was cleansed with an alcohol swab and 37 units (range, 25–50 units) of incobotulinumtoxinA (reconstituted using 2.5-mL bacteriostatic normal saline per 100 units) was injected transdermally using a 9-point injection technique, with each injection placed approximately 1 cm apart. The approximate treatment area included the space between the sternocleidomastoid anterior edges and below the hyoid bone up to the cricothyroid membrane (anatomic zone II).
When the patient returned for follow-up 3 weeks later, she reported considerable improvement in the stiffness and appearance of the skin on the anterior neck. On physical examination, the skin of the neck appeared softened, and improved laxity was seen on pinching the skin compared to the initial presentation (Figure 2). The patient expressed satisfaction with the results and denied any adverse events following the procedure.
Mixed connective tissue disease manifests with a combination of features from various disorders—mainly lupus, scleroderma, polymyositis, and rheumatoid arthritis. It is most prevalent in females and often is diagnosed in the third decade of life.3 It is associated with positive antinuclear antibodies and human leukocyte antigen (HLA) II alleles (HLA-DR4, HLA-DR1, and HLA-DR2). Raynaud phenomenon (RP), one of the most common skin manifestations in both scleroderma and MCTD, is present in 75% to 90% of patients with MCTD.3
Scleroderma is a chronic connective tissue disorder that results in excessive collagen deposition in the skin and other organs throughout the body.4 Although the etiology is unknown, scleroderma develops when overactivation of the immune system leads to CD4+ T-lymphocyte infiltration in the skin, along with the release of profibrotic interleukins and growth factors, resulting in fibrosis.4 Subtypes include localized scleroderma (morphea), limited cutaneous systemic sclerosis (formerly known as CREST [calcinosis, RP, esophageal dysmotility, sclerodactyly, and telangiectasia] syndrome), diffuse cutaneous systemic sclerosis, and systemic sclerosis sine scleroderma.5 Scleroderma is associated with positive antinuclear antibodies and HLA II alleles (HLA-DR2 and HLA-DR5).
On its own or in the setting of MCTD, scleroderma can result in systemic or localized symptoms. Overall, the most common symptom is RP.5 Localized scleroderma and limited cutaneous systemic sclerosis manifest with symptoms of the skin and underlying tissues. Diffuse cutaneous systemic sclerosis involves cutaneous and visceral symptoms, including lung, esophageal, and vascular involvement.6 Similar to MCTD, scleroderma is most prevalent in middle-aged females,7 though it occurs at a higher rate and with a more severe disease course in Black patients.8
A highly sensitive and specific test for scleroderma that can aid in diagnosis is the neck sign—tightening of the skin of the neck when the head extends.9,10 In one study, the neck sign was positive in more than 90% of patients with scleroderma and negative for control patients and those with primary RP.9 Thus, neck sclerosis is a common manifestation of scleroderma for which patients may seek treatment.
While there is no curative treatment for scleroderma, skin manifestations can be treated with mycophenolate mofetil or methotrexate.5 Systemic treatments may be recommended if the patient has additional symptoms, such as azathioprine for myositis/arthritis and cyclophosphamide for interstitial lung disease.5 However, it is important to note that these medications are associated with risk for gastrointestinal upset, mouth sores, fatigue, or other complications.
Botulinum toxin is a bacterial protein toxin and neuromodulator that inhibits neurotransmitter release by cleaving SNARE proteins at peripheral nerve terminal junctions.11 It has been used in a variety of dermatologic and nondermatologic conditions, including migraines, hyperhidrosis, contractures, scars, and overactive bladder. It also has been used in aesthetics for facial rejuvenation and minimization of wrinkle appearance. Dermatologists and rheumatologists have successfully used BTX to treat primary and secondary RP—the most common symptom of scleroderma—due to its vasodilatation properties.12 Although our patient did not have RP, use of BTX to treat other features of scleroderma, including en coup de sabre, thoracic outlet syndrome, dyspareunia, gastroparesis, pterygium inversum unguis, and dysphagia has been documented.13-18 An in vivo mouse study that examined the possible mechanism for BTX as a treatment in scleroderma found that BTX injections significantly decreased dermal thickness and inflammation in fibrosis (P<.05). An analysis of oxidative stress and mRNA expression showed that BTX may treat fibrosis by suppressing oxidative stress and inflammatory cells, resulting in decreased apoptosis and oxidant-induced intracellular accumulation of reactive oxygen species.19 Another animal study demonstrated the positive effects of BTX treatment for fibrosis of the bladder in rats.20 In one case report, a female patient with scleroderma and facial fibrosis received perioral BTX injections for cosmetic purposes but also observed improvement in mouth constriction, demonstrating the potential efficacy of BTX for facial fibrosis.21
Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region. We recommend assessing the efficacy of the initial BTX treatment after 2 to 3 weeks, with additional injections as needed to achieve the patient’s desired level of comfort and appearance at approximately 3-month intervals (aligning with the expected duration of efficacy of BTX).22 Our patient experienced considerable relief and high satisfaction with BTX treatment. Given the limitations of sclerosis treatments and the unwanted adverse-effect profile of systemic treatments, BTX injections may be a preferrable treatment option for cutaneous manifestations of scleroderma among patients. Future studies with larger patient populations and a control group are warranted to further explore the use of BTX for the dermatologic treatment of scleroderma.
To the Editor:
Scleroderma is a chronic autoimmune connective tissue disease that results in excessive collagen deposition in the skin and other organs throughout the body. On its own or in the setting of mixed connective tissue disease, scleroderma can result in systemic or localized symptoms that can limit patients’ functional capabilities, cause pain and discomfort, and reduce self-esteem—all negatively impacting patients’ quality of life.1,2 Neck sclerosis is a common manifestation of scleroderma. There is no curative treatment for scleroderma; thus, therapy is focused on slowing disease progression and improving quality of life. We present a case of neck sclerosis in a 44-year-old woman with scleroderma that was successfully treated with botulinum toxin (BTX) type A injection, resulting in improved skin laxity and appearance with high patient satisfaction. Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region.
A 44-year-old woman presented to the dermatology clinic for treatment of thickened neck skin with stiffness and tightness that had been present for months to years. She had a history of mixed connective tissue disease (MCTD)(positive anti-ribonucleoprotein, anti–Sjögren syndrome–related antigen, and anti-Smith antibodies) with features of scleroderma and polyarthritis. The patient currently was taking sulfasalazine for the polyarthritis; she previously had taken hydroxychloroquine but discontinued treatment due to ineffectiveness. She was not taking any topical or systemic medications for scleroderma. On physical examination, the skin on the anterior neck appeared thickened with shiny patches (Figure 1). Pinching the skin in the affected area demonstrated sclerosis with high tension.
The dermatologist (J.J.) discussed potential treatment options to help relax the tension in the skin of the anterior neck, including BTX injections. After receiving counsel on adverse effects, alternative treatments, and postprocedural care, the patient decided to proceed with the procedure. The anterior neck was cleansed with an alcohol swab and 37 units (range, 25–50 units) of incobotulinumtoxinA (reconstituted using 2.5-mL bacteriostatic normal saline per 100 units) was injected transdermally using a 9-point injection technique, with each injection placed approximately 1 cm apart. The approximate treatment area included the space between the sternocleidomastoid anterior edges and below the hyoid bone up to the cricothyroid membrane (anatomic zone II).
When the patient returned for follow-up 3 weeks later, she reported considerable improvement in the stiffness and appearance of the skin on the anterior neck. On physical examination, the skin of the neck appeared softened, and improved laxity was seen on pinching the skin compared to the initial presentation (Figure 2). The patient expressed satisfaction with the results and denied any adverse events following the procedure.
Mixed connective tissue disease manifests with a combination of features from various disorders—mainly lupus, scleroderma, polymyositis, and rheumatoid arthritis. It is most prevalent in females and often is diagnosed in the third decade of life.3 It is associated with positive antinuclear antibodies and human leukocyte antigen (HLA) II alleles (HLA-DR4, HLA-DR1, and HLA-DR2). Raynaud phenomenon (RP), one of the most common skin manifestations in both scleroderma and MCTD, is present in 75% to 90% of patients with MCTD.3
Scleroderma is a chronic connective tissue disorder that results in excessive collagen deposition in the skin and other organs throughout the body.4 Although the etiology is unknown, scleroderma develops when overactivation of the immune system leads to CD4+ T-lymphocyte infiltration in the skin, along with the release of profibrotic interleukins and growth factors, resulting in fibrosis.4 Subtypes include localized scleroderma (morphea), limited cutaneous systemic sclerosis (formerly known as CREST [calcinosis, RP, esophageal dysmotility, sclerodactyly, and telangiectasia] syndrome), diffuse cutaneous systemic sclerosis, and systemic sclerosis sine scleroderma.5 Scleroderma is associated with positive antinuclear antibodies and HLA II alleles (HLA-DR2 and HLA-DR5).
On its own or in the setting of MCTD, scleroderma can result in systemic or localized symptoms. Overall, the most common symptom is RP.5 Localized scleroderma and limited cutaneous systemic sclerosis manifest with symptoms of the skin and underlying tissues. Diffuse cutaneous systemic sclerosis involves cutaneous and visceral symptoms, including lung, esophageal, and vascular involvement.6 Similar to MCTD, scleroderma is most prevalent in middle-aged females,7 though it occurs at a higher rate and with a more severe disease course in Black patients.8
A highly sensitive and specific test for scleroderma that can aid in diagnosis is the neck sign—tightening of the skin of the neck when the head extends.9,10 In one study, the neck sign was positive in more than 90% of patients with scleroderma and negative for control patients and those with primary RP.9 Thus, neck sclerosis is a common manifestation of scleroderma for which patients may seek treatment.
While there is no curative treatment for scleroderma, skin manifestations can be treated with mycophenolate mofetil or methotrexate.5 Systemic treatments may be recommended if the patient has additional symptoms, such as azathioprine for myositis/arthritis and cyclophosphamide for interstitial lung disease.5 However, it is important to note that these medications are associated with risk for gastrointestinal upset, mouth sores, fatigue, or other complications.
Botulinum toxin is a bacterial protein toxin and neuromodulator that inhibits neurotransmitter release by cleaving SNARE proteins at peripheral nerve terminal junctions.11 It has been used in a variety of dermatologic and nondermatologic conditions, including migraines, hyperhidrosis, contractures, scars, and overactive bladder. It also has been used in aesthetics for facial rejuvenation and minimization of wrinkle appearance. Dermatologists and rheumatologists have successfully used BTX to treat primary and secondary RP—the most common symptom of scleroderma—due to its vasodilatation properties.12 Although our patient did not have RP, use of BTX to treat other features of scleroderma, including en coup de sabre, thoracic outlet syndrome, dyspareunia, gastroparesis, pterygium inversum unguis, and dysphagia has been documented.13-18 An in vivo mouse study that examined the possible mechanism for BTX as a treatment in scleroderma found that BTX injections significantly decreased dermal thickness and inflammation in fibrosis (P<.05). An analysis of oxidative stress and mRNA expression showed that BTX may treat fibrosis by suppressing oxidative stress and inflammatory cells, resulting in decreased apoptosis and oxidant-induced intracellular accumulation of reactive oxygen species.19 Another animal study demonstrated the positive effects of BTX treatment for fibrosis of the bladder in rats.20 In one case report, a female patient with scleroderma and facial fibrosis received perioral BTX injections for cosmetic purposes but also observed improvement in mouth constriction, demonstrating the potential efficacy of BTX for facial fibrosis.21
Our case demonstrates the potential positive effects of BTX treatment in patients with features of sclerosis or fibrosis, particularly in the neck region. We recommend assessing the efficacy of the initial BTX treatment after 2 to 3 weeks, with additional injections as needed to achieve the patient’s desired level of comfort and appearance at approximately 3-month intervals (aligning with the expected duration of efficacy of BTX).22 Our patient experienced considerable relief and high satisfaction with BTX treatment. Given the limitations of sclerosis treatments and the unwanted adverse-effect profile of systemic treatments, BTX injections may be a preferrable treatment option for cutaneous manifestations of scleroderma among patients. Future studies with larger patient populations and a control group are warranted to further explore the use of BTX for the dermatologic treatment of scleroderma.
- Lis-S´wie¸ty A, Skrzypek-Salamon A, Ranosz-Janicka I, et al. Health-related quality of life and its influencing factors in adult patients with localized scleroderma—a cross-sectional study. Health Qual Life Outcomes. 2020;18:133. doi:10.1186/s12955-020-01386-0
- Almeida C, Almeida I, Vasconcelos C. Quality of life in systemic sclerosis. Autoimmun Rev. 2015;14:1087-1096. doi:10.1016/j.autrev.2015.07.012
- Ortega-Hernandez OD, Shoenfeld Y. Mixed connective tissue disease: an overview of clinical manifestations, diagnosis and treatment. Best Pract Res Clin Rheumatol. 2012;26:61-72. doi:10.1016/j.berh.2012.01.009
- Rongioletti F, Ferreli C, Atzori L, et al. Scleroderma with an update about clinico-pathological correlation. G Ital Dermatol Venereol. 2018;153:208-215. doi:10.23736/S0392-0488.18.05922-9
- Fett N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437. doi:10.1016/j.clindermatol.2013.01.010
- Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90:62-73. doi:10.1590/abd1806-4841.20152890
- Calderon LM, Pope JE. Scleroderma epidemiology update. Curr Opin Rheumatol. 2021;33:122-127. doi:10.1097/BOR.0000000000000785
- Morgan ND, Gelber AC. African Americans and scleroderma: examining the root cause of the association. Arthritis Care Res (Hoboken). 2019;71:1151-1153. doi:10.1002/acr.23860
- Barnett AJ. The “neck sign” in scleroderma. Arthritis Rheum. 1989;32:209-211. doi:10.1002/anr.1780320215
- Barnett AJ, Miller M, Littlejohn GO. The diagnosis and classification of scleroderma (systemic sclerosis). Postgrad Med J. 1988;64:121-125. doi:10.1136/pgmj.64.748.121
- Rossetto O, Pirazzini M, Fabris F, et al. Botulinum neurotoxins: mechanism of action. Handb Exp Pharmacol. 2021;263:35-47.doi:10.1007/164_2020_355
- Ennis D, Ahmad Z, Anderson MA, et al. Botulinum toxin in the management of primary and secondary Raynaud’s phenomenon. Best Pract Res Clin Rheumatol. 2021;35:101684. doi:10.1016/j.berh.2021.101684
- Turkmani MG, Alnomair N. Enhancement of the aesthetic outcome of scleroderma en coup de sabre with botulinum toxin injection. JAAD Case Rep. 2018;4:579-581. doi:10.1016/j.jdcr.2018.03.023
- Le EN, Freischlag JA, Christo PJ, et al. Thoracic outlet syndrome secondary to localized scleroderma treated with botulinum toxin injection. Arthritis Care Res (Hoboken). 2010;62:430-433. doi:10.1002/acr.20099
- Mousty E, Rathat G, Rouleau C, et al. Botulinum toxin type A for treatment of dyspareunia caused by localized scleroderma. Acta Obstet Gynecol Scand. 2011;90:926-927. doi:10.1111/j.1600-0412.2011.01183.x
- Tang DM, Friedenberg FK. Gastroparesis: approach, diagnostic evaluation, and management. Dis Mon. 2011;57:74-101. doi:10.1016/j.disamonth.2010.12.007
- Katschinski M. [Diagnosis and treatment of esophageal motility disorders]. Ther Umsch. 2001;58:128-133. doi:10.1024/0040-5930.58.3.128
- Kim DJ, Odell ID. Improvement of pterygium inversum unguis and Raynaud phenomenon with interdigital botulinum toxin injections. JAAD Case Rep. 2022;26:79-81. doi:10.1016/j.jdcr.2022.06.009
- Baral H, Sekiguchi A, Uchiyama A, et al. Inhibition of skin fibrosis in systemic sclerosis by botulinum toxin B via the suppression of oxidative stress. J Dermatol. 2021;48:1052-1061. doi:10.1111/1346-8138.15888
- Jia C, Xing T, Shang Z, et al. Botulinum toxin A improves neurogenic bladder fibrosis by suppressing transforming growth factor β1 expression in rats. Transl Androl Urol. 2021;10:2000-2007. doi:10.21037/tau-21-62
- Hoverson K, Love T, Lam TK, et al. A novel treatment for limited mouth opening due to facial fibrosis: a case series. J Am Acad Dermatol. 2018;78:190-192. doi:10.1016/j.jaad.2017.07.006
- Kollewe K, Mohammadi B, Köhler S, et al. Blepharospasm: long-term treatment with either Botox®, Xeomin® or Dysport®. J Neural Transm (Vienna). 2015;122:427-431. doi:10.1007/s00702-014-1278-z
- Lis-S´wie¸ty A, Skrzypek-Salamon A, Ranosz-Janicka I, et al. Health-related quality of life and its influencing factors in adult patients with localized scleroderma—a cross-sectional study. Health Qual Life Outcomes. 2020;18:133. doi:10.1186/s12955-020-01386-0
- Almeida C, Almeida I, Vasconcelos C. Quality of life in systemic sclerosis. Autoimmun Rev. 2015;14:1087-1096. doi:10.1016/j.autrev.2015.07.012
- Ortega-Hernandez OD, Shoenfeld Y. Mixed connective tissue disease: an overview of clinical manifestations, diagnosis and treatment. Best Pract Res Clin Rheumatol. 2012;26:61-72. doi:10.1016/j.berh.2012.01.009
- Rongioletti F, Ferreli C, Atzori L, et al. Scleroderma with an update about clinico-pathological correlation. G Ital Dermatol Venereol. 2018;153:208-215. doi:10.23736/S0392-0488.18.05922-9
- Fett N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437. doi:10.1016/j.clindermatol.2013.01.010
- Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90:62-73. doi:10.1590/abd1806-4841.20152890
- Calderon LM, Pope JE. Scleroderma epidemiology update. Curr Opin Rheumatol. 2021;33:122-127. doi:10.1097/BOR.0000000000000785
- Morgan ND, Gelber AC. African Americans and scleroderma: examining the root cause of the association. Arthritis Care Res (Hoboken). 2019;71:1151-1153. doi:10.1002/acr.23860
- Barnett AJ. The “neck sign” in scleroderma. Arthritis Rheum. 1989;32:209-211. doi:10.1002/anr.1780320215
- Barnett AJ, Miller M, Littlejohn GO. The diagnosis and classification of scleroderma (systemic sclerosis). Postgrad Med J. 1988;64:121-125. doi:10.1136/pgmj.64.748.121
- Rossetto O, Pirazzini M, Fabris F, et al. Botulinum neurotoxins: mechanism of action. Handb Exp Pharmacol. 2021;263:35-47.doi:10.1007/164_2020_355
- Ennis D, Ahmad Z, Anderson MA, et al. Botulinum toxin in the management of primary and secondary Raynaud’s phenomenon. Best Pract Res Clin Rheumatol. 2021;35:101684. doi:10.1016/j.berh.2021.101684
- Turkmani MG, Alnomair N. Enhancement of the aesthetic outcome of scleroderma en coup de sabre with botulinum toxin injection. JAAD Case Rep. 2018;4:579-581. doi:10.1016/j.jdcr.2018.03.023
- Le EN, Freischlag JA, Christo PJ, et al. Thoracic outlet syndrome secondary to localized scleroderma treated with botulinum toxin injection. Arthritis Care Res (Hoboken). 2010;62:430-433. doi:10.1002/acr.20099
- Mousty E, Rathat G, Rouleau C, et al. Botulinum toxin type A for treatment of dyspareunia caused by localized scleroderma. Acta Obstet Gynecol Scand. 2011;90:926-927. doi:10.1111/j.1600-0412.2011.01183.x
- Tang DM, Friedenberg FK. Gastroparesis: approach, diagnostic evaluation, and management. Dis Mon. 2011;57:74-101. doi:10.1016/j.disamonth.2010.12.007
- Katschinski M. [Diagnosis and treatment of esophageal motility disorders]. Ther Umsch. 2001;58:128-133. doi:10.1024/0040-5930.58.3.128
- Kim DJ, Odell ID. Improvement of pterygium inversum unguis and Raynaud phenomenon with interdigital botulinum toxin injections. JAAD Case Rep. 2022;26:79-81. doi:10.1016/j.jdcr.2022.06.009
- Baral H, Sekiguchi A, Uchiyama A, et al. Inhibition of skin fibrosis in systemic sclerosis by botulinum toxin B via the suppression of oxidative stress. J Dermatol. 2021;48:1052-1061. doi:10.1111/1346-8138.15888
- Jia C, Xing T, Shang Z, et al. Botulinum toxin A improves neurogenic bladder fibrosis by suppressing transforming growth factor β1 expression in rats. Transl Androl Urol. 2021;10:2000-2007. doi:10.21037/tau-21-62
- Hoverson K, Love T, Lam TK, et al. A novel treatment for limited mouth opening due to facial fibrosis: a case series. J Am Acad Dermatol. 2018;78:190-192. doi:10.1016/j.jaad.2017.07.006
- Kollewe K, Mohammadi B, Köhler S, et al. Blepharospasm: long-term treatment with either Botox®, Xeomin® or Dysport®. J Neural Transm (Vienna). 2015;122:427-431. doi:10.1007/s00702-014-1278-z
Practice Points
- Scleroderma is a chronic autoimmune connective tissue disease that results in excessive collagen deposition in the skin and other organs throughout the body.
- Although there is no curative treatment for scleroderma, there are options to slow disease progression and improve quality of life.
- Botulinum toxin injection may be a preferred treatment option in patients with features of sclerosis or fibrosis related to scleroderma, particularly in the neck region.
Novel Treatment Promising for Cutaneous Lupus in Phase 2 Trial
TOPLINE:
particularly in subacute and chronic cases.
METHODOLOGY:
- Researchers conducted a randomized phase 2 trial to evaluate the efficacy and safety of iberdomide in 288 patients with CLE (mean age, 45 years; 97% women). Iberdomide is a cereblon modulator, which results in degradation of two transcription factors of immune cell development and homeostasis — Ikaros and Aiolos — that have been implicated in the genetic predisposition of systemic lupus.
- CLE Disease Area and Severity Index Activity (CLASI-A) endpoints included mean percent change from baseline and ≥ 50% reduction from baseline (CLASI-50), which were evaluated in all patients with baseline CLASI-A scores ≥ 8 and by CLE subtypes (acute, subacute, and chronic).
- At baseline, 56% of patients had acute CLE, 29% had chronic CLE, and 16% had subacute CLE; 28% of patients had a baseline CLASI-A score ≥ 8.
- Patients were randomly assigned to receive oral iberdomide (0.45 mg, 0.30 mg, 0.15 mg, or placebo daily) for 24 weeks while continuing standard lupus medications. At week 24, patients on placebo were rerandomized to iberdomide 0.45 mg or 0.30 mg once a day, while those on iberdomide continued their assigned dose through week 52.
TAKEAWAY:
- Among patients with baseline CLASI-A ≥ 8, the mean change in CLASI-A score from baseline at week 24 was −66.7% for those on iberdomide 0.45 mg vs −54.2% for placebo (P = .295).
- At week 24, patients with subacute CLE showed a significantly greater mean percent change from baseline in CLASI-A with iberdomide 0.45 mg vs placebo (−90.5% vs −51.2%; P = .007), while no significant differences were observed with the 0.45-mg dose vs placebo in patients with chronic or acute CLE.
- Overall, CLASI-50 responses were not significantly different among those on 0.45 mg vs placebo (55.6% vs 44.6%). The proportions of patients achieving CLASI-50 at week 24 were significantly greater for iberdomide 0.45 mg vs placebo for those with subacute CLE (91.7% vs 52.9%; P = .035) and chronic CLE (62.1% vs 27.8%; P = .029), but not for those with baseline CLASI-A ≥ 8 (66.7% vs 50%).
- More than 80% of patients had treatment-emergent adverse events (TEAEs), which were mostly mild to moderate. Over 2 years, the most common were urinary tract infections, upper respiratory tract infections, neutropenia, and nasopharyngitis. TEAEs leading to iberdomide discontinuation in one or more patients were neutropenia (n = 7), rash (n = 7), increased hepatic enzymes (n = 4), and deep vein thrombosis (n = 3).
IN PRACTICE:
“Data from this phase 2 trial of iberdomide in patients with SLE suggest that a greater proportion of patients with subacute or chronic CLE who received the higher dose of 0.45 mg iberdomide achieved CLASI-50 vs placebo. For the overall population, CLASI-50 response was not significantly different between treatment groups at week 24, partly due to a high placebo response that may have been driven by patients with acute CLE,” the authors wrote.
SOURCE:
The study was led by Victoria P. Werth, MD, of the University of Pennsylvania and the Veteran’s Administration Medical Center, both in Philadelphia, and was published online in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The study included small patient subgroups for different CLE subtypes, which may affect the generalizability of the findings. CLE subtype was determined by the investigator without additional photographic adjudication. Additionally, the use of background lupus medications could have influenced the placebo group’s response, limiting the ability to observe the treatment effect of iberdomide monotherapy.
DISCLOSURES:
The study was funded by Bristol-Myers Squibb. Six authors reported being employed by Bristol-Myers Squibb, and several others reported consultancy and research support from various sources including Bristol-Myers Squibb.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
particularly in subacute and chronic cases.
METHODOLOGY:
- Researchers conducted a randomized phase 2 trial to evaluate the efficacy and safety of iberdomide in 288 patients with CLE (mean age, 45 years; 97% women). Iberdomide is a cereblon modulator, which results in degradation of two transcription factors of immune cell development and homeostasis — Ikaros and Aiolos — that have been implicated in the genetic predisposition of systemic lupus.
- CLE Disease Area and Severity Index Activity (CLASI-A) endpoints included mean percent change from baseline and ≥ 50% reduction from baseline (CLASI-50), which were evaluated in all patients with baseline CLASI-A scores ≥ 8 and by CLE subtypes (acute, subacute, and chronic).
- At baseline, 56% of patients had acute CLE, 29% had chronic CLE, and 16% had subacute CLE; 28% of patients had a baseline CLASI-A score ≥ 8.
- Patients were randomly assigned to receive oral iberdomide (0.45 mg, 0.30 mg, 0.15 mg, or placebo daily) for 24 weeks while continuing standard lupus medications. At week 24, patients on placebo were rerandomized to iberdomide 0.45 mg or 0.30 mg once a day, while those on iberdomide continued their assigned dose through week 52.
TAKEAWAY:
- Among patients with baseline CLASI-A ≥ 8, the mean change in CLASI-A score from baseline at week 24 was −66.7% for those on iberdomide 0.45 mg vs −54.2% for placebo (P = .295).
- At week 24, patients with subacute CLE showed a significantly greater mean percent change from baseline in CLASI-A with iberdomide 0.45 mg vs placebo (−90.5% vs −51.2%; P = .007), while no significant differences were observed with the 0.45-mg dose vs placebo in patients with chronic or acute CLE.
- Overall, CLASI-50 responses were not significantly different among those on 0.45 mg vs placebo (55.6% vs 44.6%). The proportions of patients achieving CLASI-50 at week 24 were significantly greater for iberdomide 0.45 mg vs placebo for those with subacute CLE (91.7% vs 52.9%; P = .035) and chronic CLE (62.1% vs 27.8%; P = .029), but not for those with baseline CLASI-A ≥ 8 (66.7% vs 50%).
- More than 80% of patients had treatment-emergent adverse events (TEAEs), which were mostly mild to moderate. Over 2 years, the most common were urinary tract infections, upper respiratory tract infections, neutropenia, and nasopharyngitis. TEAEs leading to iberdomide discontinuation in one or more patients were neutropenia (n = 7), rash (n = 7), increased hepatic enzymes (n = 4), and deep vein thrombosis (n = 3).
IN PRACTICE:
“Data from this phase 2 trial of iberdomide in patients with SLE suggest that a greater proportion of patients with subacute or chronic CLE who received the higher dose of 0.45 mg iberdomide achieved CLASI-50 vs placebo. For the overall population, CLASI-50 response was not significantly different between treatment groups at week 24, partly due to a high placebo response that may have been driven by patients with acute CLE,” the authors wrote.
SOURCE:
The study was led by Victoria P. Werth, MD, of the University of Pennsylvania and the Veteran’s Administration Medical Center, both in Philadelphia, and was published online in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The study included small patient subgroups for different CLE subtypes, which may affect the generalizability of the findings. CLE subtype was determined by the investigator without additional photographic adjudication. Additionally, the use of background lupus medications could have influenced the placebo group’s response, limiting the ability to observe the treatment effect of iberdomide monotherapy.
DISCLOSURES:
The study was funded by Bristol-Myers Squibb. Six authors reported being employed by Bristol-Myers Squibb, and several others reported consultancy and research support from various sources including Bristol-Myers Squibb.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
particularly in subacute and chronic cases.
METHODOLOGY:
- Researchers conducted a randomized phase 2 trial to evaluate the efficacy and safety of iberdomide in 288 patients with CLE (mean age, 45 years; 97% women). Iberdomide is a cereblon modulator, which results in degradation of two transcription factors of immune cell development and homeostasis — Ikaros and Aiolos — that have been implicated in the genetic predisposition of systemic lupus.
- CLE Disease Area and Severity Index Activity (CLASI-A) endpoints included mean percent change from baseline and ≥ 50% reduction from baseline (CLASI-50), which were evaluated in all patients with baseline CLASI-A scores ≥ 8 and by CLE subtypes (acute, subacute, and chronic).
- At baseline, 56% of patients had acute CLE, 29% had chronic CLE, and 16% had subacute CLE; 28% of patients had a baseline CLASI-A score ≥ 8.
- Patients were randomly assigned to receive oral iberdomide (0.45 mg, 0.30 mg, 0.15 mg, or placebo daily) for 24 weeks while continuing standard lupus medications. At week 24, patients on placebo were rerandomized to iberdomide 0.45 mg or 0.30 mg once a day, while those on iberdomide continued their assigned dose through week 52.
TAKEAWAY:
- Among patients with baseline CLASI-A ≥ 8, the mean change in CLASI-A score from baseline at week 24 was −66.7% for those on iberdomide 0.45 mg vs −54.2% for placebo (P = .295).
- At week 24, patients with subacute CLE showed a significantly greater mean percent change from baseline in CLASI-A with iberdomide 0.45 mg vs placebo (−90.5% vs −51.2%; P = .007), while no significant differences were observed with the 0.45-mg dose vs placebo in patients with chronic or acute CLE.
- Overall, CLASI-50 responses were not significantly different among those on 0.45 mg vs placebo (55.6% vs 44.6%). The proportions of patients achieving CLASI-50 at week 24 were significantly greater for iberdomide 0.45 mg vs placebo for those with subacute CLE (91.7% vs 52.9%; P = .035) and chronic CLE (62.1% vs 27.8%; P = .029), but not for those with baseline CLASI-A ≥ 8 (66.7% vs 50%).
- More than 80% of patients had treatment-emergent adverse events (TEAEs), which were mostly mild to moderate. Over 2 years, the most common were urinary tract infections, upper respiratory tract infections, neutropenia, and nasopharyngitis. TEAEs leading to iberdomide discontinuation in one or more patients were neutropenia (n = 7), rash (n = 7), increased hepatic enzymes (n = 4), and deep vein thrombosis (n = 3).
IN PRACTICE:
“Data from this phase 2 trial of iberdomide in patients with SLE suggest that a greater proportion of patients with subacute or chronic CLE who received the higher dose of 0.45 mg iberdomide achieved CLASI-50 vs placebo. For the overall population, CLASI-50 response was not significantly different between treatment groups at week 24, partly due to a high placebo response that may have been driven by patients with acute CLE,” the authors wrote.
SOURCE:
The study was led by Victoria P. Werth, MD, of the University of Pennsylvania and the Veteran’s Administration Medical Center, both in Philadelphia, and was published online in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The study included small patient subgroups for different CLE subtypes, which may affect the generalizability of the findings. CLE subtype was determined by the investigator without additional photographic adjudication. Additionally, the use of background lupus medications could have influenced the placebo group’s response, limiting the ability to observe the treatment effect of iberdomide monotherapy.
DISCLOSURES:
The study was funded by Bristol-Myers Squibb. Six authors reported being employed by Bristol-Myers Squibb, and several others reported consultancy and research support from various sources including Bristol-Myers Squibb.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Small Bowel Dysmotility Brings Challenges to Patients With Systemic Sclerosis
TOPLINE:
Patients with systemic sclerosis (SSc) who exhibit abnormal small bowel transit are more likely to be men, experience more severe cardiac involvement, have a higher mortality risk, and show fewer sicca symptoms.
METHODOLOGY:
- Researchers enrolled 130 patients with SSc having gastrointestinal (GI) symptoms (mean age at symptom onset, 56.8 years; 90% women; 81% White) seen at the Johns Hopkins Scleroderma Center, Baltimore, from October 2014 to May 2022.
- Clinical data and serum samples were longitudinally collected from all actively followed patients at the time of enrollment and every 6 months thereafter (median disease duration, 8.4 years).
- Participants underwent whole gut transit scintigraphy for the assessment of small bowel motility.
- A cross-sectional analysis compared the clinical features of patients with (n = 22; mean age at symptom onset, 61.4 years) and without (n = 108; mean age at symptom onset, 55.8 years) abnormal small bowel transit.
TAKEAWAY:
- Men with SSc (odds ratio [OR], 3.70; P = .038) and those with severe cardiac involvement (OR, 3.98; P = .035) were more likely to have abnormal small bowel transit.
- Sicca symptoms were negatively associated with abnormal small bowel transit in patients with SSc (adjusted OR, 0.28; P = .043).
- Patients with abnormal small bowel transit reported significantly worse (P = .028) and social functioning (P = .015) than those having a normal transit.
- A multivariate analysis showed that patients with abnormal small bowel transit had higher mortality than those with a normal transit (adjusted hazard ratio, 5.03; P = .005).
IN PRACTICE:
“Our findings improve our understanding of risk factors associated with abnormal small bowel transit in SSc patients and shed light on the lived experience of patients with this GI [gastrointestinal] complication,” the authors wrote. “Overall, these findings are important for patient risk stratification and monitoring and will help to identify a more homogeneous group of patients for future clinical and translational studies,” they added.
SOURCE:
The study was led by Jenice X. Cheah, MD, University of California, Los Angeles. It was published online on October 7, 2024, in Rheumatology.
LIMITATIONS:
The study may be subject to referral bias as it was conducted at a tertiary referral center, potentially including patients with a more severe disease status. Furthermore, this study was retrospective in nature, and whole gut transit studies were not conducted in all the patients seen at the referral center. Additionally, the cross-sectional design limited the ability to establish causality between the clinical features and abnormal small bowel transit.
DISCLOSURES:
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Patients with systemic sclerosis (SSc) who exhibit abnormal small bowel transit are more likely to be men, experience more severe cardiac involvement, have a higher mortality risk, and show fewer sicca symptoms.
METHODOLOGY:
- Researchers enrolled 130 patients with SSc having gastrointestinal (GI) symptoms (mean age at symptom onset, 56.8 years; 90% women; 81% White) seen at the Johns Hopkins Scleroderma Center, Baltimore, from October 2014 to May 2022.
- Clinical data and serum samples were longitudinally collected from all actively followed patients at the time of enrollment and every 6 months thereafter (median disease duration, 8.4 years).
- Participants underwent whole gut transit scintigraphy for the assessment of small bowel motility.
- A cross-sectional analysis compared the clinical features of patients with (n = 22; mean age at symptom onset, 61.4 years) and without (n = 108; mean age at symptom onset, 55.8 years) abnormal small bowel transit.
TAKEAWAY:
- Men with SSc (odds ratio [OR], 3.70; P = .038) and those with severe cardiac involvement (OR, 3.98; P = .035) were more likely to have abnormal small bowel transit.
- Sicca symptoms were negatively associated with abnormal small bowel transit in patients with SSc (adjusted OR, 0.28; P = .043).
- Patients with abnormal small bowel transit reported significantly worse (P = .028) and social functioning (P = .015) than those having a normal transit.
- A multivariate analysis showed that patients with abnormal small bowel transit had higher mortality than those with a normal transit (adjusted hazard ratio, 5.03; P = .005).
IN PRACTICE:
“Our findings improve our understanding of risk factors associated with abnormal small bowel transit in SSc patients and shed light on the lived experience of patients with this GI [gastrointestinal] complication,” the authors wrote. “Overall, these findings are important for patient risk stratification and monitoring and will help to identify a more homogeneous group of patients for future clinical and translational studies,” they added.
SOURCE:
The study was led by Jenice X. Cheah, MD, University of California, Los Angeles. It was published online on October 7, 2024, in Rheumatology.
LIMITATIONS:
The study may be subject to referral bias as it was conducted at a tertiary referral center, potentially including patients with a more severe disease status. Furthermore, this study was retrospective in nature, and whole gut transit studies were not conducted in all the patients seen at the referral center. Additionally, the cross-sectional design limited the ability to establish causality between the clinical features and abnormal small bowel transit.
DISCLOSURES:
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Patients with systemic sclerosis (SSc) who exhibit abnormal small bowel transit are more likely to be men, experience more severe cardiac involvement, have a higher mortality risk, and show fewer sicca symptoms.
METHODOLOGY:
- Researchers enrolled 130 patients with SSc having gastrointestinal (GI) symptoms (mean age at symptom onset, 56.8 years; 90% women; 81% White) seen at the Johns Hopkins Scleroderma Center, Baltimore, from October 2014 to May 2022.
- Clinical data and serum samples were longitudinally collected from all actively followed patients at the time of enrollment and every 6 months thereafter (median disease duration, 8.4 years).
- Participants underwent whole gut transit scintigraphy for the assessment of small bowel motility.
- A cross-sectional analysis compared the clinical features of patients with (n = 22; mean age at symptom onset, 61.4 years) and without (n = 108; mean age at symptom onset, 55.8 years) abnormal small bowel transit.
TAKEAWAY:
- Men with SSc (odds ratio [OR], 3.70; P = .038) and those with severe cardiac involvement (OR, 3.98; P = .035) were more likely to have abnormal small bowel transit.
- Sicca symptoms were negatively associated with abnormal small bowel transit in patients with SSc (adjusted OR, 0.28; P = .043).
- Patients with abnormal small bowel transit reported significantly worse (P = .028) and social functioning (P = .015) than those having a normal transit.
- A multivariate analysis showed that patients with abnormal small bowel transit had higher mortality than those with a normal transit (adjusted hazard ratio, 5.03; P = .005).
IN PRACTICE:
“Our findings improve our understanding of risk factors associated with abnormal small bowel transit in SSc patients and shed light on the lived experience of patients with this GI [gastrointestinal] complication,” the authors wrote. “Overall, these findings are important for patient risk stratification and monitoring and will help to identify a more homogeneous group of patients for future clinical and translational studies,” they added.
SOURCE:
The study was led by Jenice X. Cheah, MD, University of California, Los Angeles. It was published online on October 7, 2024, in Rheumatology.
LIMITATIONS:
The study may be subject to referral bias as it was conducted at a tertiary referral center, potentially including patients with a more severe disease status. Furthermore, this study was retrospective in nature, and whole gut transit studies were not conducted in all the patients seen at the referral center. Additionally, the cross-sectional design limited the ability to establish causality between the clinical features and abnormal small bowel transit.
DISCLOSURES:
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Considerations for the Use of Biologics in Pregnancy
Biologics have revolutionized dermatologic treatment, offering substantial relief from chronic and debilitating skin conditions such as psoriasis,
Biologics for Cutaneous Conditions
Biologics—tumor necrosis factor (TNF) α inhibitors; IL-17, IL-23, IL-12, and IL-36 inhibitors; and agents such as omalizumab and dupilumab—have shown remarkable efficacy in controlling severe or recalcitrant dermatologic conditions and typically are more effective than traditional systemic therapies.1 For instance, randomized clinical trials (RCTs) and real-world data have shown that patients with psoriasis can achieve considerable skin clearance with biologics, greatly enhancing QOL.2 Adalimumab and secukinumab, which have been approved for use in moderate to severe cases of hidradenitis suppurativa, reduce the frequency of painful nodules and abscesses, thereby decreasing pain and improving QOL. Dupilumab, an IL-4/13 receptor antagonist, has revolutionized the treatment of AD by drastically reducing itch and skin lesions and improving QOL.3 For chronic urticaria, the anti-IgE antibody omalizumab has effectively reduced the incidence of hives and itching, providing pronounced symptom relief when traditional antihistamines fail.4 Use of rituximab, an anti-CD20 monoclonal antibody, has led to remission in severe cases of pemphigus vulgaris and bullous pemphigoid.5
Impact of Untreated Cutaneous Conditions in Pregnancy
When treating patients who are pregnant, dermatologists must consider the health of both the expectant mother and the developing fetus. This dual focus complicates decision-making, particularly with the use of biologics. Untreated cutaneous conditions can profoundly impact a pregnant patient’s health and QOL as well as lead to pregnancy complications affecting the fetus, such as preterm birth or low birth weight. In some studies, moderate to severe psoriasis has been associated with increased risk for complications during pregnancy, including preeclampsia and intrauterine growth restriction.6 Although specific data on hidradenitis suppurativa are lacking, the highly inflammatory nature of the condition suggests similar adverse effects on pregnancy.7 Atopic dermatitis can be exacerbated during pregnancy due to a shift in the immune system to become more allergic dominant.8 Generalized pustular psoriasis manifests with widespread pustules, fever, and systemic inflammation, posing serious risks to both the mother and the fetus if left untreated9; in such a life-threatening scenario, the use of potent treatments such as spesolimab, an IL-36 receptor antagonist, may be warranted. Therefore, managing these conditions effectively is crucial not only for the mother’s health but also for fetal well-being.
Which Biologics Can Dermatologists Safely Prescribe?
Despite the benefits, many dermatologists are hesitant to prescribe biologics to pregnant patients due to the lack of understanding and definitive safety data.10,11 Although there are no RCTs that involve pregnant patients, current evidence suggests that several biologics are not teratogenic and do not cause fetal malformations. Extensive postexposure data support the safety of TNF-α inhibitors during pregnancy.12 Research has shown that children exposed to these agents in utero have normal development, infection rates, and vaccination outcomes comparable to nonexposed children. For example, a systematic review and meta-analysis found no significant increase in the risk for major congenital malformations, spontaneous abortions, or preterm births among patients exposed to anti–TNF-α agents during pregnancy.2 The Organization of Teratology Information Specialists Autoimmune Diseases in Pregnancy Project has provided valuable real-world data indicating that the use of TNF-α inhibitors in pregnancy, particularly during the first trimester, does not substantially elevate the risk for adverse outcomes.13 These findings have been corroborated by several other registry studies and RCTs, providing a robust safety profile for these agents during pregnancy.14
Similarly, postexposure data on IL-17 and IL-12/23 inhibitors indicate a favorable safety profile, though the sample sizes are smaller than those for anti–TNF-α agents.12,14 Studies of drugs such as secukinumab (IL-17 inhibitor), guselkumab (IL-23 inhibitor), or ustekinumab (IL-12/23 inhibitor) have shown no association with teratogenic effects or increased risk for miscarriage.14 However, agents such as spesolimab (IL-36 inhibitor) are relatively new, and ongoing studies are expected to provide more comprehensive safety data.15 Similarly, omalizumab and dupilumab have not been associated with increased risk for fetal malformations or adverse pregnancy outcomes. Omalizumab, indicated for chronic urticaria, has a good safety profile in pregnancy, with no significant increase in adverse outcomes reported in studies and registries.16 Dupilumab, used for AD, has demonstrated safety in pregnancy, with ongoing studies continuing to monitor outcomes.17
Conversely, rituximab (an anti-CD20 antibody for autoimmune bullous diseases) has shown evidence of adverse pregnancy outcomes, including fetal harm.18 Its use generally is discouraged unless deemed absolutely necessary, and no safer alternatives are available. Rituximab can cross the placenta, especially in the second and third trimesters, and has been associated with B-cell depletion in the fetus, leading to potential immunosuppression and increased risk for infections.5
Although the data on the safety of biologics in pregnancy are largely reassuring, it is essential to recognize that potential risks have not been ruled out entirely. There are extensive safety data for anti–TNF-α inhibitors, which provides a level of confidence; although newer agents such as IL-17 and IL-23 inhibitors have shown promising early results, further research is required to solidify their safety profiles during pregnancy.
Dermatologists must balance the risks and benefits of using biologics in pregnant patients. This decision-making process involves careful consideration of the severity of the mother’s condition, the potential risks to the fetus, and the availability of alternative treatments. For many severe dermatologic conditions, the benefits of biologics in controlling disease activity and improving QOL may outweigh the potential risks, especially when other treatments have failed or are not suitable.
Final Thoughts
The increasing use of biologics in dermatology has undoubtedly improved the management of severe skin conditions, substantially enhancing patients’ QOL. As more data become available and clinical guidelines evolve, health care providers will be better equipped to make informed decisions about the use of biologics, particularly in pregnant patients. Collaborative efforts between dermatologists, obstetricians, and researchers will help refine treatment guidelines and ensure that pregnant patients with severe dermatologic conditions receive the best possible care.
For now, although the current evidence supports the safety of many biologics during pregnancy,10,11 individualized care and informed decision-making remain paramount. Careful management and adherence to current guidelines make it possible to navigate the complexities of treating severe dermatologic conditions in pregnant patients, ensuring the best outcomes for both mother and child.
- Sehgal VN, Pandhi D, Khurana A. Biologics in dermatology: an integrated review. Indian J Dermatol. 2014; 59:425-441. doi:10.4103/0019-5154.139859
- Mahadevan U, Wolf DC, Dubinsky M, et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11:286-292. doi:10.1016/j.cgh.2012.11.011
- Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Saini SS, Bindslev-Jensen C, Maurer M, et al. Efficacy and safety of omalizumab in patients with chronic idiopathic/spontaneous urticaria who remain symptomatic on H1 antihistamines: a randomized, placebo-controlled study. J Invest Dermatol. 2015;135:67-75. doi:10.1038/jid.2014.306
- Mariette X, Forger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77:228-233. doi:10.1136/annrheumdis-2017-212196
- Yang Y-W, Chen C-S, Chen Y-H, et al. Psoriasis and pregnancy outcomes: a nationwide population-based study. J Am Acad Dermatol. 2011;64:71-77.
- Zouboulis CC, Del Marmol V, Mrowietz U, et al. Hidradenitis suppurativa/acne inversa: criteria for diagnosis, severity assessment, classification and disease evaluation. Dermatology. 2015;231:184-190.
- Balakirski G, Novak N. Atopic dermatitis and pregnancy. J Allergy Clin Immunol. 2022;149:1185-1194. doi:10.1016/j.jaci.2022.01.010
- Bachelez H, Choon S-E, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380:981-983.
- McMullan P, Yaghi M, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part I: pregnancy. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.072
- Yaghi M, McMullan P, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part II: lactation. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.071
- Owczarek W, Walecka I, Lesiak A, et al. The use of biological drugs in psoriasis patients prior to pregnancy, during pregnancy and lactation: a review of current clinical guidelines. Postepy Dermatol Alergol. 2020;37:821-830. doi:10.5114/ada.2020.102089
- Organization of Teratology Information Services (OTIS) Autoimmune Diseases in Pregnancy Project. ClinicalTrials.gov identifier: NCT00116272. Updated October 6, 2023. Accessed August 29, 2024. https://clinicaltrials.gov/study/NCT00116272
- Sanchez-Garcia V, Hernandez-Quiles R, de-Miguel-Balsa E, et al. Exposure to biologic therapy before and during pregnancy in patients with psoriasis: systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2023;37:1971-1990. doi:10.1111/jdv.19238
- Silverberg JI, Boguniewicz M, Hanifin J, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis is efficacious regardless of age of disease onset: a post hoc analysis of two phase 3 clinical trials. Dermatol Ther (Heidelb). 2022;12:2731-2746. doi:10.1007/s13555-022-00822-x
- Levi-Schaffer F, Mankuta D. Omalizumab safety in pregnancy. J Allergy Clin Immunol. 2020;145:481-483. doi:10.1016/j.jaci.2019.11.018
- Thaci D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387:40-52.
- Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506. doi:10.1182/blood-2010-07-295444
Biologics have revolutionized dermatologic treatment, offering substantial relief from chronic and debilitating skin conditions such as psoriasis,
Biologics for Cutaneous Conditions
Biologics—tumor necrosis factor (TNF) α inhibitors; IL-17, IL-23, IL-12, and IL-36 inhibitors; and agents such as omalizumab and dupilumab—have shown remarkable efficacy in controlling severe or recalcitrant dermatologic conditions and typically are more effective than traditional systemic therapies.1 For instance, randomized clinical trials (RCTs) and real-world data have shown that patients with psoriasis can achieve considerable skin clearance with biologics, greatly enhancing QOL.2 Adalimumab and secukinumab, which have been approved for use in moderate to severe cases of hidradenitis suppurativa, reduce the frequency of painful nodules and abscesses, thereby decreasing pain and improving QOL. Dupilumab, an IL-4/13 receptor antagonist, has revolutionized the treatment of AD by drastically reducing itch and skin lesions and improving QOL.3 For chronic urticaria, the anti-IgE antibody omalizumab has effectively reduced the incidence of hives and itching, providing pronounced symptom relief when traditional antihistamines fail.4 Use of rituximab, an anti-CD20 monoclonal antibody, has led to remission in severe cases of pemphigus vulgaris and bullous pemphigoid.5
Impact of Untreated Cutaneous Conditions in Pregnancy
When treating patients who are pregnant, dermatologists must consider the health of both the expectant mother and the developing fetus. This dual focus complicates decision-making, particularly with the use of biologics. Untreated cutaneous conditions can profoundly impact a pregnant patient’s health and QOL as well as lead to pregnancy complications affecting the fetus, such as preterm birth or low birth weight. In some studies, moderate to severe psoriasis has been associated with increased risk for complications during pregnancy, including preeclampsia and intrauterine growth restriction.6 Although specific data on hidradenitis suppurativa are lacking, the highly inflammatory nature of the condition suggests similar adverse effects on pregnancy.7 Atopic dermatitis can be exacerbated during pregnancy due to a shift in the immune system to become more allergic dominant.8 Generalized pustular psoriasis manifests with widespread pustules, fever, and systemic inflammation, posing serious risks to both the mother and the fetus if left untreated9; in such a life-threatening scenario, the use of potent treatments such as spesolimab, an IL-36 receptor antagonist, may be warranted. Therefore, managing these conditions effectively is crucial not only for the mother’s health but also for fetal well-being.
Which Biologics Can Dermatologists Safely Prescribe?
Despite the benefits, many dermatologists are hesitant to prescribe biologics to pregnant patients due to the lack of understanding and definitive safety data.10,11 Although there are no RCTs that involve pregnant patients, current evidence suggests that several biologics are not teratogenic and do not cause fetal malformations. Extensive postexposure data support the safety of TNF-α inhibitors during pregnancy.12 Research has shown that children exposed to these agents in utero have normal development, infection rates, and vaccination outcomes comparable to nonexposed children. For example, a systematic review and meta-analysis found no significant increase in the risk for major congenital malformations, spontaneous abortions, or preterm births among patients exposed to anti–TNF-α agents during pregnancy.2 The Organization of Teratology Information Specialists Autoimmune Diseases in Pregnancy Project has provided valuable real-world data indicating that the use of TNF-α inhibitors in pregnancy, particularly during the first trimester, does not substantially elevate the risk for adverse outcomes.13 These findings have been corroborated by several other registry studies and RCTs, providing a robust safety profile for these agents during pregnancy.14
Similarly, postexposure data on IL-17 and IL-12/23 inhibitors indicate a favorable safety profile, though the sample sizes are smaller than those for anti–TNF-α agents.12,14 Studies of drugs such as secukinumab (IL-17 inhibitor), guselkumab (IL-23 inhibitor), or ustekinumab (IL-12/23 inhibitor) have shown no association with teratogenic effects or increased risk for miscarriage.14 However, agents such as spesolimab (IL-36 inhibitor) are relatively new, and ongoing studies are expected to provide more comprehensive safety data.15 Similarly, omalizumab and dupilumab have not been associated with increased risk for fetal malformations or adverse pregnancy outcomes. Omalizumab, indicated for chronic urticaria, has a good safety profile in pregnancy, with no significant increase in adverse outcomes reported in studies and registries.16 Dupilumab, used for AD, has demonstrated safety in pregnancy, with ongoing studies continuing to monitor outcomes.17
Conversely, rituximab (an anti-CD20 antibody for autoimmune bullous diseases) has shown evidence of adverse pregnancy outcomes, including fetal harm.18 Its use generally is discouraged unless deemed absolutely necessary, and no safer alternatives are available. Rituximab can cross the placenta, especially in the second and third trimesters, and has been associated with B-cell depletion in the fetus, leading to potential immunosuppression and increased risk for infections.5
Although the data on the safety of biologics in pregnancy are largely reassuring, it is essential to recognize that potential risks have not been ruled out entirely. There are extensive safety data for anti–TNF-α inhibitors, which provides a level of confidence; although newer agents such as IL-17 and IL-23 inhibitors have shown promising early results, further research is required to solidify their safety profiles during pregnancy.
Dermatologists must balance the risks and benefits of using biologics in pregnant patients. This decision-making process involves careful consideration of the severity of the mother’s condition, the potential risks to the fetus, and the availability of alternative treatments. For many severe dermatologic conditions, the benefits of biologics in controlling disease activity and improving QOL may outweigh the potential risks, especially when other treatments have failed or are not suitable.
Final Thoughts
The increasing use of biologics in dermatology has undoubtedly improved the management of severe skin conditions, substantially enhancing patients’ QOL. As more data become available and clinical guidelines evolve, health care providers will be better equipped to make informed decisions about the use of biologics, particularly in pregnant patients. Collaborative efforts between dermatologists, obstetricians, and researchers will help refine treatment guidelines and ensure that pregnant patients with severe dermatologic conditions receive the best possible care.
For now, although the current evidence supports the safety of many biologics during pregnancy,10,11 individualized care and informed decision-making remain paramount. Careful management and adherence to current guidelines make it possible to navigate the complexities of treating severe dermatologic conditions in pregnant patients, ensuring the best outcomes for both mother and child.
Biologics have revolutionized dermatologic treatment, offering substantial relief from chronic and debilitating skin conditions such as psoriasis,
Biologics for Cutaneous Conditions
Biologics—tumor necrosis factor (TNF) α inhibitors; IL-17, IL-23, IL-12, and IL-36 inhibitors; and agents such as omalizumab and dupilumab—have shown remarkable efficacy in controlling severe or recalcitrant dermatologic conditions and typically are more effective than traditional systemic therapies.1 For instance, randomized clinical trials (RCTs) and real-world data have shown that patients with psoriasis can achieve considerable skin clearance with biologics, greatly enhancing QOL.2 Adalimumab and secukinumab, which have been approved for use in moderate to severe cases of hidradenitis suppurativa, reduce the frequency of painful nodules and abscesses, thereby decreasing pain and improving QOL. Dupilumab, an IL-4/13 receptor antagonist, has revolutionized the treatment of AD by drastically reducing itch and skin lesions and improving QOL.3 For chronic urticaria, the anti-IgE antibody omalizumab has effectively reduced the incidence of hives and itching, providing pronounced symptom relief when traditional antihistamines fail.4 Use of rituximab, an anti-CD20 monoclonal antibody, has led to remission in severe cases of pemphigus vulgaris and bullous pemphigoid.5
Impact of Untreated Cutaneous Conditions in Pregnancy
When treating patients who are pregnant, dermatologists must consider the health of both the expectant mother and the developing fetus. This dual focus complicates decision-making, particularly with the use of biologics. Untreated cutaneous conditions can profoundly impact a pregnant patient’s health and QOL as well as lead to pregnancy complications affecting the fetus, such as preterm birth or low birth weight. In some studies, moderate to severe psoriasis has been associated with increased risk for complications during pregnancy, including preeclampsia and intrauterine growth restriction.6 Although specific data on hidradenitis suppurativa are lacking, the highly inflammatory nature of the condition suggests similar adverse effects on pregnancy.7 Atopic dermatitis can be exacerbated during pregnancy due to a shift in the immune system to become more allergic dominant.8 Generalized pustular psoriasis manifests with widespread pustules, fever, and systemic inflammation, posing serious risks to both the mother and the fetus if left untreated9; in such a life-threatening scenario, the use of potent treatments such as spesolimab, an IL-36 receptor antagonist, may be warranted. Therefore, managing these conditions effectively is crucial not only for the mother’s health but also for fetal well-being.
Which Biologics Can Dermatologists Safely Prescribe?
Despite the benefits, many dermatologists are hesitant to prescribe biologics to pregnant patients due to the lack of understanding and definitive safety data.10,11 Although there are no RCTs that involve pregnant patients, current evidence suggests that several biologics are not teratogenic and do not cause fetal malformations. Extensive postexposure data support the safety of TNF-α inhibitors during pregnancy.12 Research has shown that children exposed to these agents in utero have normal development, infection rates, and vaccination outcomes comparable to nonexposed children. For example, a systematic review and meta-analysis found no significant increase in the risk for major congenital malformations, spontaneous abortions, or preterm births among patients exposed to anti–TNF-α agents during pregnancy.2 The Organization of Teratology Information Specialists Autoimmune Diseases in Pregnancy Project has provided valuable real-world data indicating that the use of TNF-α inhibitors in pregnancy, particularly during the first trimester, does not substantially elevate the risk for adverse outcomes.13 These findings have been corroborated by several other registry studies and RCTs, providing a robust safety profile for these agents during pregnancy.14
Similarly, postexposure data on IL-17 and IL-12/23 inhibitors indicate a favorable safety profile, though the sample sizes are smaller than those for anti–TNF-α agents.12,14 Studies of drugs such as secukinumab (IL-17 inhibitor), guselkumab (IL-23 inhibitor), or ustekinumab (IL-12/23 inhibitor) have shown no association with teratogenic effects or increased risk for miscarriage.14 However, agents such as spesolimab (IL-36 inhibitor) are relatively new, and ongoing studies are expected to provide more comprehensive safety data.15 Similarly, omalizumab and dupilumab have not been associated with increased risk for fetal malformations or adverse pregnancy outcomes. Omalizumab, indicated for chronic urticaria, has a good safety profile in pregnancy, with no significant increase in adverse outcomes reported in studies and registries.16 Dupilumab, used for AD, has demonstrated safety in pregnancy, with ongoing studies continuing to monitor outcomes.17
Conversely, rituximab (an anti-CD20 antibody for autoimmune bullous diseases) has shown evidence of adverse pregnancy outcomes, including fetal harm.18 Its use generally is discouraged unless deemed absolutely necessary, and no safer alternatives are available. Rituximab can cross the placenta, especially in the second and third trimesters, and has been associated with B-cell depletion in the fetus, leading to potential immunosuppression and increased risk for infections.5
Although the data on the safety of biologics in pregnancy are largely reassuring, it is essential to recognize that potential risks have not been ruled out entirely. There are extensive safety data for anti–TNF-α inhibitors, which provides a level of confidence; although newer agents such as IL-17 and IL-23 inhibitors have shown promising early results, further research is required to solidify their safety profiles during pregnancy.
Dermatologists must balance the risks and benefits of using biologics in pregnant patients. This decision-making process involves careful consideration of the severity of the mother’s condition, the potential risks to the fetus, and the availability of alternative treatments. For many severe dermatologic conditions, the benefits of biologics in controlling disease activity and improving QOL may outweigh the potential risks, especially when other treatments have failed or are not suitable.
Final Thoughts
The increasing use of biologics in dermatology has undoubtedly improved the management of severe skin conditions, substantially enhancing patients’ QOL. As more data become available and clinical guidelines evolve, health care providers will be better equipped to make informed decisions about the use of biologics, particularly in pregnant patients. Collaborative efforts between dermatologists, obstetricians, and researchers will help refine treatment guidelines and ensure that pregnant patients with severe dermatologic conditions receive the best possible care.
For now, although the current evidence supports the safety of many biologics during pregnancy,10,11 individualized care and informed decision-making remain paramount. Careful management and adherence to current guidelines make it possible to navigate the complexities of treating severe dermatologic conditions in pregnant patients, ensuring the best outcomes for both mother and child.
- Sehgal VN, Pandhi D, Khurana A. Biologics in dermatology: an integrated review. Indian J Dermatol. 2014; 59:425-441. doi:10.4103/0019-5154.139859
- Mahadevan U, Wolf DC, Dubinsky M, et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11:286-292. doi:10.1016/j.cgh.2012.11.011
- Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Saini SS, Bindslev-Jensen C, Maurer M, et al. Efficacy and safety of omalizumab in patients with chronic idiopathic/spontaneous urticaria who remain symptomatic on H1 antihistamines: a randomized, placebo-controlled study. J Invest Dermatol. 2015;135:67-75. doi:10.1038/jid.2014.306
- Mariette X, Forger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77:228-233. doi:10.1136/annrheumdis-2017-212196
- Yang Y-W, Chen C-S, Chen Y-H, et al. Psoriasis and pregnancy outcomes: a nationwide population-based study. J Am Acad Dermatol. 2011;64:71-77.
- Zouboulis CC, Del Marmol V, Mrowietz U, et al. Hidradenitis suppurativa/acne inversa: criteria for diagnosis, severity assessment, classification and disease evaluation. Dermatology. 2015;231:184-190.
- Balakirski G, Novak N. Atopic dermatitis and pregnancy. J Allergy Clin Immunol. 2022;149:1185-1194. doi:10.1016/j.jaci.2022.01.010
- Bachelez H, Choon S-E, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380:981-983.
- McMullan P, Yaghi M, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part I: pregnancy. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.072
- Yaghi M, McMullan P, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part II: lactation. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.071
- Owczarek W, Walecka I, Lesiak A, et al. The use of biological drugs in psoriasis patients prior to pregnancy, during pregnancy and lactation: a review of current clinical guidelines. Postepy Dermatol Alergol. 2020;37:821-830. doi:10.5114/ada.2020.102089
- Organization of Teratology Information Services (OTIS) Autoimmune Diseases in Pregnancy Project. ClinicalTrials.gov identifier: NCT00116272. Updated October 6, 2023. Accessed August 29, 2024. https://clinicaltrials.gov/study/NCT00116272
- Sanchez-Garcia V, Hernandez-Quiles R, de-Miguel-Balsa E, et al. Exposure to biologic therapy before and during pregnancy in patients with psoriasis: systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2023;37:1971-1990. doi:10.1111/jdv.19238
- Silverberg JI, Boguniewicz M, Hanifin J, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis is efficacious regardless of age of disease onset: a post hoc analysis of two phase 3 clinical trials. Dermatol Ther (Heidelb). 2022;12:2731-2746. doi:10.1007/s13555-022-00822-x
- Levi-Schaffer F, Mankuta D. Omalizumab safety in pregnancy. J Allergy Clin Immunol. 2020;145:481-483. doi:10.1016/j.jaci.2019.11.018
- Thaci D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387:40-52.
- Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506. doi:10.1182/blood-2010-07-295444
- Sehgal VN, Pandhi D, Khurana A. Biologics in dermatology: an integrated review. Indian J Dermatol. 2014; 59:425-441. doi:10.4103/0019-5154.139859
- Mahadevan U, Wolf DC, Dubinsky M, et al. Placental transfer of anti-tumor necrosis factor agents in pregnant patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11:286-292. doi:10.1016/j.cgh.2012.11.011
- Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Saini SS, Bindslev-Jensen C, Maurer M, et al. Efficacy and safety of omalizumab in patients with chronic idiopathic/spontaneous urticaria who remain symptomatic on H1 antihistamines: a randomized, placebo-controlled study. J Invest Dermatol. 2015;135:67-75. doi:10.1038/jid.2014.306
- Mariette X, Forger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77:228-233. doi:10.1136/annrheumdis-2017-212196
- Yang Y-W, Chen C-S, Chen Y-H, et al. Psoriasis and pregnancy outcomes: a nationwide population-based study. J Am Acad Dermatol. 2011;64:71-77.
- Zouboulis CC, Del Marmol V, Mrowietz U, et al. Hidradenitis suppurativa/acne inversa: criteria for diagnosis, severity assessment, classification and disease evaluation. Dermatology. 2015;231:184-190.
- Balakirski G, Novak N. Atopic dermatitis and pregnancy. J Allergy Clin Immunol. 2022;149:1185-1194. doi:10.1016/j.jaci.2022.01.010
- Bachelez H, Choon S-E, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380:981-983.
- McMullan P, Yaghi M, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part I: pregnancy. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.072
- Yaghi M, McMullan P, Truong TM, et al. Safety of dermatologic medications in pregnancy and lactation: an update—part II: lactation. J Am Acad Dermatol. Published online January 25, 2024. doi:10.1016/j.jaad.2023.10.071
- Owczarek W, Walecka I, Lesiak A, et al. The use of biological drugs in psoriasis patients prior to pregnancy, during pregnancy and lactation: a review of current clinical guidelines. Postepy Dermatol Alergol. 2020;37:821-830. doi:10.5114/ada.2020.102089
- Organization of Teratology Information Services (OTIS) Autoimmune Diseases in Pregnancy Project. ClinicalTrials.gov identifier: NCT00116272. Updated October 6, 2023. Accessed August 29, 2024. https://clinicaltrials.gov/study/NCT00116272
- Sanchez-Garcia V, Hernandez-Quiles R, de-Miguel-Balsa E, et al. Exposure to biologic therapy before and during pregnancy in patients with psoriasis: systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2023;37:1971-1990. doi:10.1111/jdv.19238
- Silverberg JI, Boguniewicz M, Hanifin J, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis is efficacious regardless of age of disease onset: a post hoc analysis of two phase 3 clinical trials. Dermatol Ther (Heidelb). 2022;12:2731-2746. doi:10.1007/s13555-022-00822-x
- Levi-Schaffer F, Mankuta D. Omalizumab safety in pregnancy. J Allergy Clin Immunol. 2020;145:481-483. doi:10.1016/j.jaci.2019.11.018
- Thaci D, Simpson EL, Beck LA, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387:40-52.
- Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506. doi:10.1182/blood-2010-07-295444
Dermatomyositis Cancer Screening Guidelines Get Real-World Validation
Newly issued guidelines for cancer screening in patients with dermatomyositis had 100% sensitivity in a single institution’s cohort, though most of the cancers found would have been detected with standard cancer screenings recommended for the general population, according to a research letter published in JAMA Dermatology.
“These early results emphasize the continued need to refine risk assessment and cancer screening for patients with dermatomyositis while balancing resource use and outcomes,” concluded Caroline J. Stone and her colleagues at the Department of Dermatology, Perelman School of Medicine, University of Pennsylvania, Philadelphia.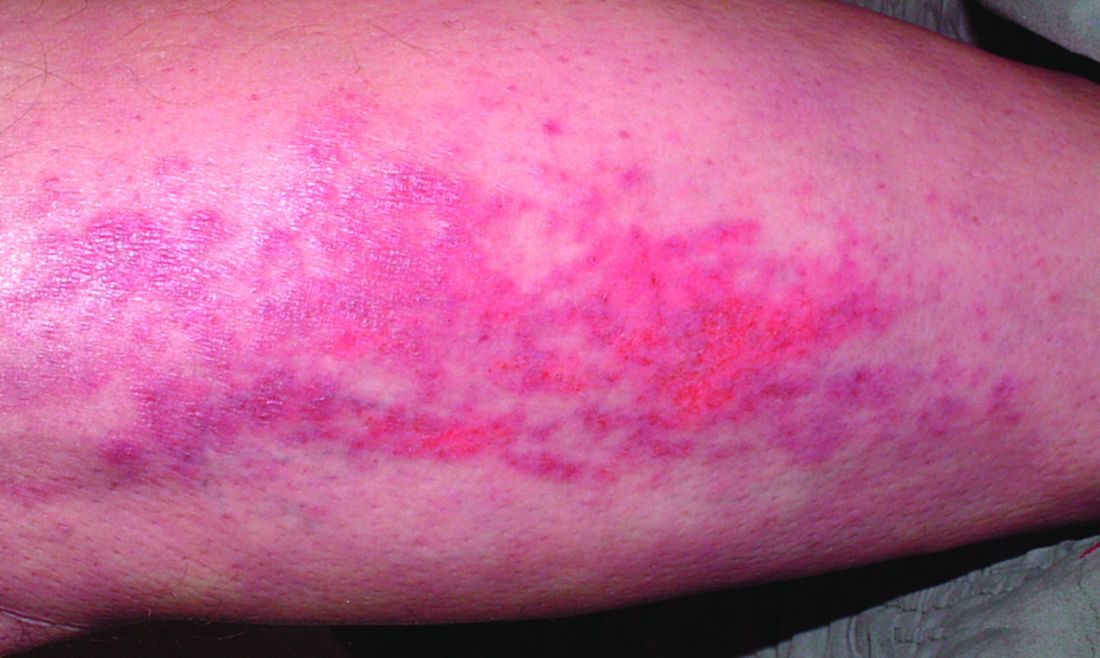
Patients with dermatomyositis have approximately a 4.7 times greater risk for cancer than those without it, according to a 2016 meta-analysis. Despite the well-established link between cancer and dermatomyositis, cancer in people with idiopathic inflammatory myopathies is commonly diagnosed at a later stage and is the leading cause of death in people with these conditions.
Guidelines First Presented in 2022 and Published in 2023
A wide variability in screening practices eventually led the International Myositis Assessment & Clinical Studies Group (IMACS) to present the first evidence-based and consensus-based guidelines for cancer screening of patients with idiopathic inflammatory myopathies, including those with dermatomyositis, at the 2022 annual meeting of the American College of Rheumatology and publish them in 2023 in Nature Reviews Rheumatology. The guidelines advise low-risk patients to undergo basic cancer screening with routine blood and urine studies, liver function tests, plain chest radiography, and age- and sex-appropriate cancer screening.
Intermediate- and high-risk patients are recommended to undergo enhanced screening that can include mammography, Pap tests, endoscopy/colonoscopy, pelvic and transvaginal ultrasonography, prostate-specific antigen or cancer antigen 125 blood tests, fecal occult blood tests, and CT of the neck, thorax, abdomen, and pelvis.
But because the guidelines are new, little evidence exists regarding their validation in real-world cohorts. Researchers, therefore, assessed the IMACS guidelines in 370 patients, aged 18-80 years, who visited the University of Pennsylvania rheumatology-dermatology specialty clinic between July 2008 and January 2024. All participants had dermatomyositis and at least 3 years of follow-up and were an average 48 years old. The vast majority were women (87%) and White participants (89%).
Most (68.6%) had myositis-specific autoantibody test results, one of the factors included in the guidelines for determining whether the patient should be classified as low, intermediate, or high risk. Other factors for risk stratification included myositis subtype, age at disease onset, and clinical features. About half (49.2%) had classic dermatomyositis, 42.4% had amyopathic dermatomyositis, 3.8% had juvenile dermatomyositis, 3.2% had hypomyopathic dermatomyositis, 0.8% had antisynthetase syndrome, and 0.5% had immune-mediated necrotizing myopathy.
Just over half the patients (54%) were classified as high risk, while 37.3% were classified as intermediate risk and 8.9% as low risk using the guidelines. Among the 18 patients (4.9%) with paraneoplastic dermatomyositis, 15 were classified as high risk and 3 as intermediate risk.
Of the patients diagnosed with cancer, 55% of cases were diagnosed about a year before their dermatomyositis diagnosis. In three patients, symptoms “suggestive of cancer at the time of dermatomyositis diagnosis, including lymphadenopathy and unexplained weight loss,” led to diagnostic testing that found an underlying cancer.
In the eight patients diagnosed with cancer after their dermatomyositis diagnosis, 75% of the cancers were identified during the first year of follow-up and 25% in the second year. Five were identified based on basic cancer screening and three on enhanced screening.
A total of 11 patients (3%) developed intravenous contrast allergies, and no other adverse events were reported to be associated with cancer screening, but the study was not designed to capture other types of adverse screening effects, such as cost, quality of life, or risk from radiation exposure.
The most common neoplasm identified was breast cancer, found in nine (50%) of the patients using mammography. Two patients had lung cancer identified with chest radiography and two had ovarian cancer identified with abdominal radiography and CT. The remaining five patients included one each with bladder cancer, papillary thyroid cancer, renal cell carcinoma, non-Hodgkin lymphoma, and adenocarcinoma with unknown primary.
The sensitivity of the guidelines in detecting cancer related to dermatomyositis was 100%, though the authors noted that the “IMACS risk-stratification scheme may overestimate cancer risk and encourage enhanced screening protocols of unclear benefit.” Most of the cancers found after dermatomyositis diagnosis were detected with routine age- and sex-related screening that already falls under basic cancer screening recommendations for the general population. Nonetheless, 90% of the participants fell into the intermediate- and high-risk groups, warranting a more comprehensive and costly enhanced screening protocol.
Will the Guidelines Lead to Overscreening?
The 4.9% cancer prevalence is considerably lower than the typical 15%-25% prevalence among patients with dermatomyositis, but the findings, regardless, suggest the guidelines will lead to overscreening, wrote Andrea D. Maderal, MD, University of Miami Miller School of Medicine in Florida, and Alisa Femia, MD, New York University Grossman School of Medicine, New York City, in an accompanying editorial. Given that the median age in patients with cancer in the study was 58 years — 18 years older than the age cutoff for high-risk criteria — one way to refine the guidelines may be to increase the age for the high-risk category, they suggested.
“While these guidelines led to many ultimately unnecessary screening tests based on currently recommended designations of intermediate-risk and high-risk patients, these guidelines reflect a more conservative approach to screening than was previously performed,” Dr. Maderal and Dr. Femia wrote.
Jeff Gehlhausen, MD, PhD, an assistant professor of dermatology at Yale School of Medicine, New Haven, Connecticut, said he is not concerned about overscreening in patients, however, and is “very enthusiastic” about the findings.
“Patients are very anxious for good reason,” given the typical cancer prevalence of 25% in this population, he said in an interview. “I think therein lies the challenge — with that risk, what is ‘enough’ screening?” Yet this “incredibly impressive” study “provides real insights into the applicability of the IMACS screenings to our dermatomyositis management,” including relevance to his own patients. “Their findings are instructive for how to better evaluate these patients in a more mindful fashion,” he said, and they are particularly welcome, given how widely variable practice has historically been before the guidelines were issued.
“This question has been an outstanding one for decades, and nearly every doctor has a different answer,” Dr. Gehlhausen said. “The introduction of the guidelines alone are now much more actionable with this study, and that’s why it’s such an important one for our community.”
Benedict Wu, DO, PhD, director of Inpatient Dermatology and an assistant professor at Montefiore Einstein and a member of the Montefiore Einstein Comprehensive Cancer Center in New York City, similarly regarded the findings as reassuring, though he was surprised at the low prevalence of cancer in the patients.
“The most reassuring finding was that the detection of most malignancies was possible by using routine age- and sex-related screening combined with basic cancer screening,” Wu said in an interview. “Basic cancer screening can reduce costs while keeping patients safe.”
He also found it reassuring that all the paraneoplastic dermatomyositis was in intermediate- or high-risk patients, and while he does not see the IMACS guidelines as overestimating cancer risk, he does think “the risk stratification and recommended screening tests could be revised to be less ‘aggressive.’ ”
The overall low rate of cancer in the group “calls into question the need for stringent and annual cancer screening,” he said. “In this large cohort of patients, the fact that malignancy was detected within 2 years of dermatomyositis diagnosis will help guide us with long-term screening recommendations.”
Despite the study’s small size and single-center design, the demographics of the patients nearly represents exactly what is found in the United States more broadly, Wu noted. He also drew attention to how many patients lacked the myositis antibody profile performed, and he agreed with the authors that more extensive and prospective studies need to be conducted. He also emphasized the need to keep in mind that “the primary goal of dermatomyositis management should focus on controlling/reducing the disease burden.”
The research was funded by the National Institutes of Health and the US Department of Veterans Affairs. The authors had no disclosures. Dr. Maderal reported personal fees from argenx. No disclosures were noted for Dr. Gehlhausen and Dr. Wu.
A version of this article appeared on Medscape.com.
Newly issued guidelines for cancer screening in patients with dermatomyositis had 100% sensitivity in a single institution’s cohort, though most of the cancers found would have been detected with standard cancer screenings recommended for the general population, according to a research letter published in JAMA Dermatology.
“These early results emphasize the continued need to refine risk assessment and cancer screening for patients with dermatomyositis while balancing resource use and outcomes,” concluded Caroline J. Stone and her colleagues at the Department of Dermatology, Perelman School of Medicine, University of Pennsylvania, Philadelphia.
Patients with dermatomyositis have approximately a 4.7 times greater risk for cancer than those without it, according to a 2016 meta-analysis. Despite the well-established link between cancer and dermatomyositis, cancer in people with idiopathic inflammatory myopathies is commonly diagnosed at a later stage and is the leading cause of death in people with these conditions.
Guidelines First Presented in 2022 and Published in 2023
A wide variability in screening practices eventually led the International Myositis Assessment & Clinical Studies Group (IMACS) to present the first evidence-based and consensus-based guidelines for cancer screening of patients with idiopathic inflammatory myopathies, including those with dermatomyositis, at the 2022 annual meeting of the American College of Rheumatology and publish them in 2023 in Nature Reviews Rheumatology. The guidelines advise low-risk patients to undergo basic cancer screening with routine blood and urine studies, liver function tests, plain chest radiography, and age- and sex-appropriate cancer screening.
Intermediate- and high-risk patients are recommended to undergo enhanced screening that can include mammography, Pap tests, endoscopy/colonoscopy, pelvic and transvaginal ultrasonography, prostate-specific antigen or cancer antigen 125 blood tests, fecal occult blood tests, and CT of the neck, thorax, abdomen, and pelvis.
But because the guidelines are new, little evidence exists regarding their validation in real-world cohorts. Researchers, therefore, assessed the IMACS guidelines in 370 patients, aged 18-80 years, who visited the University of Pennsylvania rheumatology-dermatology specialty clinic between July 2008 and January 2024. All participants had dermatomyositis and at least 3 years of follow-up and were an average 48 years old. The vast majority were women (87%) and White participants (89%).
Most (68.6%) had myositis-specific autoantibody test results, one of the factors included in the guidelines for determining whether the patient should be classified as low, intermediate, or high risk. Other factors for risk stratification included myositis subtype, age at disease onset, and clinical features. About half (49.2%) had classic dermatomyositis, 42.4% had amyopathic dermatomyositis, 3.8% had juvenile dermatomyositis, 3.2% had hypomyopathic dermatomyositis, 0.8% had antisynthetase syndrome, and 0.5% had immune-mediated necrotizing myopathy.
Just over half the patients (54%) were classified as high risk, while 37.3% were classified as intermediate risk and 8.9% as low risk using the guidelines. Among the 18 patients (4.9%) with paraneoplastic dermatomyositis, 15 were classified as high risk and 3 as intermediate risk.
Of the patients diagnosed with cancer, 55% of cases were diagnosed about a year before their dermatomyositis diagnosis. In three patients, symptoms “suggestive of cancer at the time of dermatomyositis diagnosis, including lymphadenopathy and unexplained weight loss,” led to diagnostic testing that found an underlying cancer.
In the eight patients diagnosed with cancer after their dermatomyositis diagnosis, 75% of the cancers were identified during the first year of follow-up and 25% in the second year. Five were identified based on basic cancer screening and three on enhanced screening.
A total of 11 patients (3%) developed intravenous contrast allergies, and no other adverse events were reported to be associated with cancer screening, but the study was not designed to capture other types of adverse screening effects, such as cost, quality of life, or risk from radiation exposure.
The most common neoplasm identified was breast cancer, found in nine (50%) of the patients using mammography. Two patients had lung cancer identified with chest radiography and two had ovarian cancer identified with abdominal radiography and CT. The remaining five patients included one each with bladder cancer, papillary thyroid cancer, renal cell carcinoma, non-Hodgkin lymphoma, and adenocarcinoma with unknown primary.
The sensitivity of the guidelines in detecting cancer related to dermatomyositis was 100%, though the authors noted that the “IMACS risk-stratification scheme may overestimate cancer risk and encourage enhanced screening protocols of unclear benefit.” Most of the cancers found after dermatomyositis diagnosis were detected with routine age- and sex-related screening that already falls under basic cancer screening recommendations for the general population. Nonetheless, 90% of the participants fell into the intermediate- and high-risk groups, warranting a more comprehensive and costly enhanced screening protocol.
Will the Guidelines Lead to Overscreening?
The 4.9% cancer prevalence is considerably lower than the typical 15%-25% prevalence among patients with dermatomyositis, but the findings, regardless, suggest the guidelines will lead to overscreening, wrote Andrea D. Maderal, MD, University of Miami Miller School of Medicine in Florida, and Alisa Femia, MD, New York University Grossman School of Medicine, New York City, in an accompanying editorial. Given that the median age in patients with cancer in the study was 58 years — 18 years older than the age cutoff for high-risk criteria — one way to refine the guidelines may be to increase the age for the high-risk category, they suggested.
“While these guidelines led to many ultimately unnecessary screening tests based on currently recommended designations of intermediate-risk and high-risk patients, these guidelines reflect a more conservative approach to screening than was previously performed,” Dr. Maderal and Dr. Femia wrote.
Jeff Gehlhausen, MD, PhD, an assistant professor of dermatology at Yale School of Medicine, New Haven, Connecticut, said he is not concerned about overscreening in patients, however, and is “very enthusiastic” about the findings.
“Patients are very anxious for good reason,” given the typical cancer prevalence of 25% in this population, he said in an interview. “I think therein lies the challenge — with that risk, what is ‘enough’ screening?” Yet this “incredibly impressive” study “provides real insights into the applicability of the IMACS screenings to our dermatomyositis management,” including relevance to his own patients. “Their findings are instructive for how to better evaluate these patients in a more mindful fashion,” he said, and they are particularly welcome, given how widely variable practice has historically been before the guidelines were issued.
“This question has been an outstanding one for decades, and nearly every doctor has a different answer,” Dr. Gehlhausen said. “The introduction of the guidelines alone are now much more actionable with this study, and that’s why it’s such an important one for our community.”
Benedict Wu, DO, PhD, director of Inpatient Dermatology and an assistant professor at Montefiore Einstein and a member of the Montefiore Einstein Comprehensive Cancer Center in New York City, similarly regarded the findings as reassuring, though he was surprised at the low prevalence of cancer in the patients.
“The most reassuring finding was that the detection of most malignancies was possible by using routine age- and sex-related screening combined with basic cancer screening,” Wu said in an interview. “Basic cancer screening can reduce costs while keeping patients safe.”
He also found it reassuring that all the paraneoplastic dermatomyositis was in intermediate- or high-risk patients, and while he does not see the IMACS guidelines as overestimating cancer risk, he does think “the risk stratification and recommended screening tests could be revised to be less ‘aggressive.’ ”
The overall low rate of cancer in the group “calls into question the need for stringent and annual cancer screening,” he said. “In this large cohort of patients, the fact that malignancy was detected within 2 years of dermatomyositis diagnosis will help guide us with long-term screening recommendations.”
Despite the study’s small size and single-center design, the demographics of the patients nearly represents exactly what is found in the United States more broadly, Wu noted. He also drew attention to how many patients lacked the myositis antibody profile performed, and he agreed with the authors that more extensive and prospective studies need to be conducted. He also emphasized the need to keep in mind that “the primary goal of dermatomyositis management should focus on controlling/reducing the disease burden.”
The research was funded by the National Institutes of Health and the US Department of Veterans Affairs. The authors had no disclosures. Dr. Maderal reported personal fees from argenx. No disclosures were noted for Dr. Gehlhausen and Dr. Wu.
A version of this article appeared on Medscape.com.
Newly issued guidelines for cancer screening in patients with dermatomyositis had 100% sensitivity in a single institution’s cohort, though most of the cancers found would have been detected with standard cancer screenings recommended for the general population, according to a research letter published in JAMA Dermatology.
“These early results emphasize the continued need to refine risk assessment and cancer screening for patients with dermatomyositis while balancing resource use and outcomes,” concluded Caroline J. Stone and her colleagues at the Department of Dermatology, Perelman School of Medicine, University of Pennsylvania, Philadelphia.
Patients with dermatomyositis have approximately a 4.7 times greater risk for cancer than those without it, according to a 2016 meta-analysis. Despite the well-established link between cancer and dermatomyositis, cancer in people with idiopathic inflammatory myopathies is commonly diagnosed at a later stage and is the leading cause of death in people with these conditions.
Guidelines First Presented in 2022 and Published in 2023
A wide variability in screening practices eventually led the International Myositis Assessment & Clinical Studies Group (IMACS) to present the first evidence-based and consensus-based guidelines for cancer screening of patients with idiopathic inflammatory myopathies, including those with dermatomyositis, at the 2022 annual meeting of the American College of Rheumatology and publish them in 2023 in Nature Reviews Rheumatology. The guidelines advise low-risk patients to undergo basic cancer screening with routine blood and urine studies, liver function tests, plain chest radiography, and age- and sex-appropriate cancer screening.
Intermediate- and high-risk patients are recommended to undergo enhanced screening that can include mammography, Pap tests, endoscopy/colonoscopy, pelvic and transvaginal ultrasonography, prostate-specific antigen or cancer antigen 125 blood tests, fecal occult blood tests, and CT of the neck, thorax, abdomen, and pelvis.
But because the guidelines are new, little evidence exists regarding their validation in real-world cohorts. Researchers, therefore, assessed the IMACS guidelines in 370 patients, aged 18-80 years, who visited the University of Pennsylvania rheumatology-dermatology specialty clinic between July 2008 and January 2024. All participants had dermatomyositis and at least 3 years of follow-up and were an average 48 years old. The vast majority were women (87%) and White participants (89%).
Most (68.6%) had myositis-specific autoantibody test results, one of the factors included in the guidelines for determining whether the patient should be classified as low, intermediate, or high risk. Other factors for risk stratification included myositis subtype, age at disease onset, and clinical features. About half (49.2%) had classic dermatomyositis, 42.4% had amyopathic dermatomyositis, 3.8% had juvenile dermatomyositis, 3.2% had hypomyopathic dermatomyositis, 0.8% had antisynthetase syndrome, and 0.5% had immune-mediated necrotizing myopathy.
Just over half the patients (54%) were classified as high risk, while 37.3% were classified as intermediate risk and 8.9% as low risk using the guidelines. Among the 18 patients (4.9%) with paraneoplastic dermatomyositis, 15 were classified as high risk and 3 as intermediate risk.
Of the patients diagnosed with cancer, 55% of cases were diagnosed about a year before their dermatomyositis diagnosis. In three patients, symptoms “suggestive of cancer at the time of dermatomyositis diagnosis, including lymphadenopathy and unexplained weight loss,” led to diagnostic testing that found an underlying cancer.
In the eight patients diagnosed with cancer after their dermatomyositis diagnosis, 75% of the cancers were identified during the first year of follow-up and 25% in the second year. Five were identified based on basic cancer screening and three on enhanced screening.
A total of 11 patients (3%) developed intravenous contrast allergies, and no other adverse events were reported to be associated with cancer screening, but the study was not designed to capture other types of adverse screening effects, such as cost, quality of life, or risk from radiation exposure.
The most common neoplasm identified was breast cancer, found in nine (50%) of the patients using mammography. Two patients had lung cancer identified with chest radiography and two had ovarian cancer identified with abdominal radiography and CT. The remaining five patients included one each with bladder cancer, papillary thyroid cancer, renal cell carcinoma, non-Hodgkin lymphoma, and adenocarcinoma with unknown primary.
The sensitivity of the guidelines in detecting cancer related to dermatomyositis was 100%, though the authors noted that the “IMACS risk-stratification scheme may overestimate cancer risk and encourage enhanced screening protocols of unclear benefit.” Most of the cancers found after dermatomyositis diagnosis were detected with routine age- and sex-related screening that already falls under basic cancer screening recommendations for the general population. Nonetheless, 90% of the participants fell into the intermediate- and high-risk groups, warranting a more comprehensive and costly enhanced screening protocol.
Will the Guidelines Lead to Overscreening?
The 4.9% cancer prevalence is considerably lower than the typical 15%-25% prevalence among patients with dermatomyositis, but the findings, regardless, suggest the guidelines will lead to overscreening, wrote Andrea D. Maderal, MD, University of Miami Miller School of Medicine in Florida, and Alisa Femia, MD, New York University Grossman School of Medicine, New York City, in an accompanying editorial. Given that the median age in patients with cancer in the study was 58 years — 18 years older than the age cutoff for high-risk criteria — one way to refine the guidelines may be to increase the age for the high-risk category, they suggested.
“While these guidelines led to many ultimately unnecessary screening tests based on currently recommended designations of intermediate-risk and high-risk patients, these guidelines reflect a more conservative approach to screening than was previously performed,” Dr. Maderal and Dr. Femia wrote.
Jeff Gehlhausen, MD, PhD, an assistant professor of dermatology at Yale School of Medicine, New Haven, Connecticut, said he is not concerned about overscreening in patients, however, and is “very enthusiastic” about the findings.
“Patients are very anxious for good reason,” given the typical cancer prevalence of 25% in this population, he said in an interview. “I think therein lies the challenge — with that risk, what is ‘enough’ screening?” Yet this “incredibly impressive” study “provides real insights into the applicability of the IMACS screenings to our dermatomyositis management,” including relevance to his own patients. “Their findings are instructive for how to better evaluate these patients in a more mindful fashion,” he said, and they are particularly welcome, given how widely variable practice has historically been before the guidelines were issued.
“This question has been an outstanding one for decades, and nearly every doctor has a different answer,” Dr. Gehlhausen said. “The introduction of the guidelines alone are now much more actionable with this study, and that’s why it’s such an important one for our community.”
Benedict Wu, DO, PhD, director of Inpatient Dermatology and an assistant professor at Montefiore Einstein and a member of the Montefiore Einstein Comprehensive Cancer Center in New York City, similarly regarded the findings as reassuring, though he was surprised at the low prevalence of cancer in the patients.
“The most reassuring finding was that the detection of most malignancies was possible by using routine age- and sex-related screening combined with basic cancer screening,” Wu said in an interview. “Basic cancer screening can reduce costs while keeping patients safe.”
He also found it reassuring that all the paraneoplastic dermatomyositis was in intermediate- or high-risk patients, and while he does not see the IMACS guidelines as overestimating cancer risk, he does think “the risk stratification and recommended screening tests could be revised to be less ‘aggressive.’ ”
The overall low rate of cancer in the group “calls into question the need for stringent and annual cancer screening,” he said. “In this large cohort of patients, the fact that malignancy was detected within 2 years of dermatomyositis diagnosis will help guide us with long-term screening recommendations.”
Despite the study’s small size and single-center design, the demographics of the patients nearly represents exactly what is found in the United States more broadly, Wu noted. He also drew attention to how many patients lacked the myositis antibody profile performed, and he agreed with the authors that more extensive and prospective studies need to be conducted. He also emphasized the need to keep in mind that “the primary goal of dermatomyositis management should focus on controlling/reducing the disease burden.”
The research was funded by the National Institutes of Health and the US Department of Veterans Affairs. The authors had no disclosures. Dr. Maderal reported personal fees from argenx. No disclosures were noted for Dr. Gehlhausen and Dr. Wu.
A version of this article appeared on Medscape.com.
FROM JAMA DERMATOLOGY
A 71-year-old White female developed erosions after hip replacement surgery 2 months prior to presentation
The patient had been diagnosed with pemphigus vulgaris (PV) 1 year prior to presentation with erosions on the axilla. Biopsy at that time revealed intraepithelial acantholytic blistering with areas of suprabasilar and subcorneal clefting. Direct immunofluorescence was positive for linear/granular IgG deposition throughout the epithelial cell surfaces, as well as linear/granular C3 deposits of the lower two thirds of the epithelial strata, consistent for pemphigus vulgaris.
There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, PV presents with flaccid blistering lesions that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions can involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
There are numerous reports in the literature of PV occurring in previous surgical scars, and areas of friction or trauma. This so-called Koebner’s phenomenon is seen more commonly in several dermatologic conditions, such as psoriasis, lichen planus, verruca vulgaris, and vitiligo.

Treatment for PV is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid sparing agent such as mycophenolate mofetil. Other steroid sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Cerottini JP et al. Eur J Dermatol. 2000 Oct-Nov;10(7):546-7.
Reichert-Penetrat S et al. Eur J Dermatol. 1998 Jan-Feb;8(1):60-2.
Saini P et al. Skinmed. 2020 Aug 1;18(4):252-253.
The patient had been diagnosed with pemphigus vulgaris (PV) 1 year prior to presentation with erosions on the axilla. Biopsy at that time revealed intraepithelial acantholytic blistering with areas of suprabasilar and subcorneal clefting. Direct immunofluorescence was positive for linear/granular IgG deposition throughout the epithelial cell surfaces, as well as linear/granular C3 deposits of the lower two thirds of the epithelial strata, consistent for pemphigus vulgaris.
There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, PV presents with flaccid blistering lesions that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions can involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
There are numerous reports in the literature of PV occurring in previous surgical scars, and areas of friction or trauma. This so-called Koebner’s phenomenon is seen more commonly in several dermatologic conditions, such as psoriasis, lichen planus, verruca vulgaris, and vitiligo.
Treatment for PV is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid sparing agent such as mycophenolate mofetil. Other steroid sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Cerottini JP et al. Eur J Dermatol. 2000 Oct-Nov;10(7):546-7.
Reichert-Penetrat S et al. Eur J Dermatol. 1998 Jan-Feb;8(1):60-2.
Saini P et al. Skinmed. 2020 Aug 1;18(4):252-253.
The patient had been diagnosed with pemphigus vulgaris (PV) 1 year prior to presentation with erosions on the axilla. Biopsy at that time revealed intraepithelial acantholytic blistering with areas of suprabasilar and subcorneal clefting. Direct immunofluorescence was positive for linear/granular IgG deposition throughout the epithelial cell surfaces, as well as linear/granular C3 deposits of the lower two thirds of the epithelial strata, consistent for pemphigus vulgaris.
There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, PV presents with flaccid blistering lesions that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions can involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
There are numerous reports in the literature of PV occurring in previous surgical scars, and areas of friction or trauma. This so-called Koebner’s phenomenon is seen more commonly in several dermatologic conditions, such as psoriasis, lichen planus, verruca vulgaris, and vitiligo.
Treatment for PV is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid sparing agent such as mycophenolate mofetil. Other steroid sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Cerottini JP et al. Eur J Dermatol. 2000 Oct-Nov;10(7):546-7.
Reichert-Penetrat S et al. Eur J Dermatol. 1998 Jan-Feb;8(1):60-2.
Saini P et al. Skinmed. 2020 Aug 1;18(4):252-253.