User login
Simplifying Allergic Contact Dermatitis Management with the Contact Allergen Management Program 2.0
Simplifying Allergic Contact Dermatitis Management with the Contact Allergen Management Program 2.0
While patch testing is the gold standard to diagnose type IV cutaneous hypersensitivity reactions, interpreting results can feel like trying to decipher a secret code, leaving patients feeling disempowered in avoiding their triggers. To truly manage allergic contact dermatitis (ACD), patients need comprehensive education on which allergens to avoid and ways to spot potential sources of exposure, including counseling, written guidelines, and lists of product alternatives.1 Patients who can recall and avoid their triggers experience greater improvement in clinical and quality-of-life scores.2 However, several studies have demonstrated that patients have difficulty recalling their allergens, even with longitudinal reminders.2-5 Quality-of-life and clinical outcomes also are not necessarily improved by successful allergen recall alone, as patients have reported limited success in actually avoiding allergens, highlighting the complexity of navigating exposures in daily life.2,6 To address these challenges, we examine common pitfalls patients encounter when avoiding allergens, highlight the benefits of utilizing safe lists and databases for allergen management, and introduce the updated Contact Allergen Management Program (CAMP) 2.0 as an optimal tool for long-term management of ACD.
Allergen Avoidance Pitfalls
Simply reading ingredient labels to avoid allergens is only marginally effective, as patients need to identify and interpret multiple chemical names as well as cross-reactors and related compounds to achieve success. Some allergens, such as fragrances or manufacturing impurities, are not explicitly identified on product labels. Even patients who can practice diligent label reading may struggle to find information on household or occupational products when full ingredient disclosure is not required.
Many of the allergens included in the American Contact Dermatitis Society (ACDS) Core 90 Series have alternative chemical aliases, and many have related compounds.6 For example, individuals with contact allergy to formaldehyde or a formaldehyde releaser usually need to avoid multiple other formaldehyde-releasing chemicals. Patients who test positive to amidoamine or dimethylaminopropylamine also must avoid the surfactant cocamidopropyl betaine—not because it is a cross-reactor, but because it is an impurity in the synthetic pathway.
Fragrance is one of the most common causes of ACD but can be challenging to avoid. Patients with allergies to fragrance or specific compounds (eg, limonene, linalool hydroperoxides) need to be savvy enough to navigate a broad spectrum of synthetic and botanical fragrance additives. Avoiding products that contain “fragrance” or “parfum” is simple enough, but patients also may need to recognize more than 3000 chemical names to identify individual fragrance ingredients that may be listed separately.7 Further, some fragrances are added for alternative purposes—preservative, medicinal, or emulsification—in which case products may deceptively tout themselves as being “fragrance free” yet still contain a fragrance allergen. This is made even more complex considering additional additives that commonly may cross-react with individual fragrance compounds; balsam of Peru, for example, is a botanical amalgam containing more than 250 compounds, including several fragrance components, making it an excellent indicator of fragrance allergy.8 While balsam of Peru and its fragrance constituents will almost never be listed on a product label, it cross-reacts with several benzyl derivatives commonly used in cosmetic formulations, such as benzyl alcohol, benzyl acetate, benzoic acid, benzyl benzoate, and benzyl cinnamate.9,10
Given that ACD is a common reason for patients to seek dermatologic care, it is crucial for clinicians to equip themselves with effective strategies to support patients after patch testing.11 This includes efficient translation of patch test results into practical advice while avoiding the oversimplified suggestion to read product labels; however, education alone cannot address the complexities of managing ACD, which is where contact allergen databases come into play.
An Essential Tool: Patient Allergen Databases and Safe Lists
Contact allergen databases are like a trusty sidekick for patients and clinicians, providing easily accessible information and tools to support allergen avoidance and improve ACD outcomes. While there are several existing resources, the ACDS launched its CAMP database in 2011 for ACDS members and their patients.12 The CAMP allows clinicians to easily generate personalized safe lists for household, medicament, and personal care products, facilitating seamless patient access both online and via a mobile application. The database also includes allergen-specific handouts to guide patient education.13 A key highlight of the CAMP is automated management of cross-reactors, which allows patients to choose products without having to memorize complex cross-reactor algorithms and helps avoid overly restrictive safe lists (Table).12-15
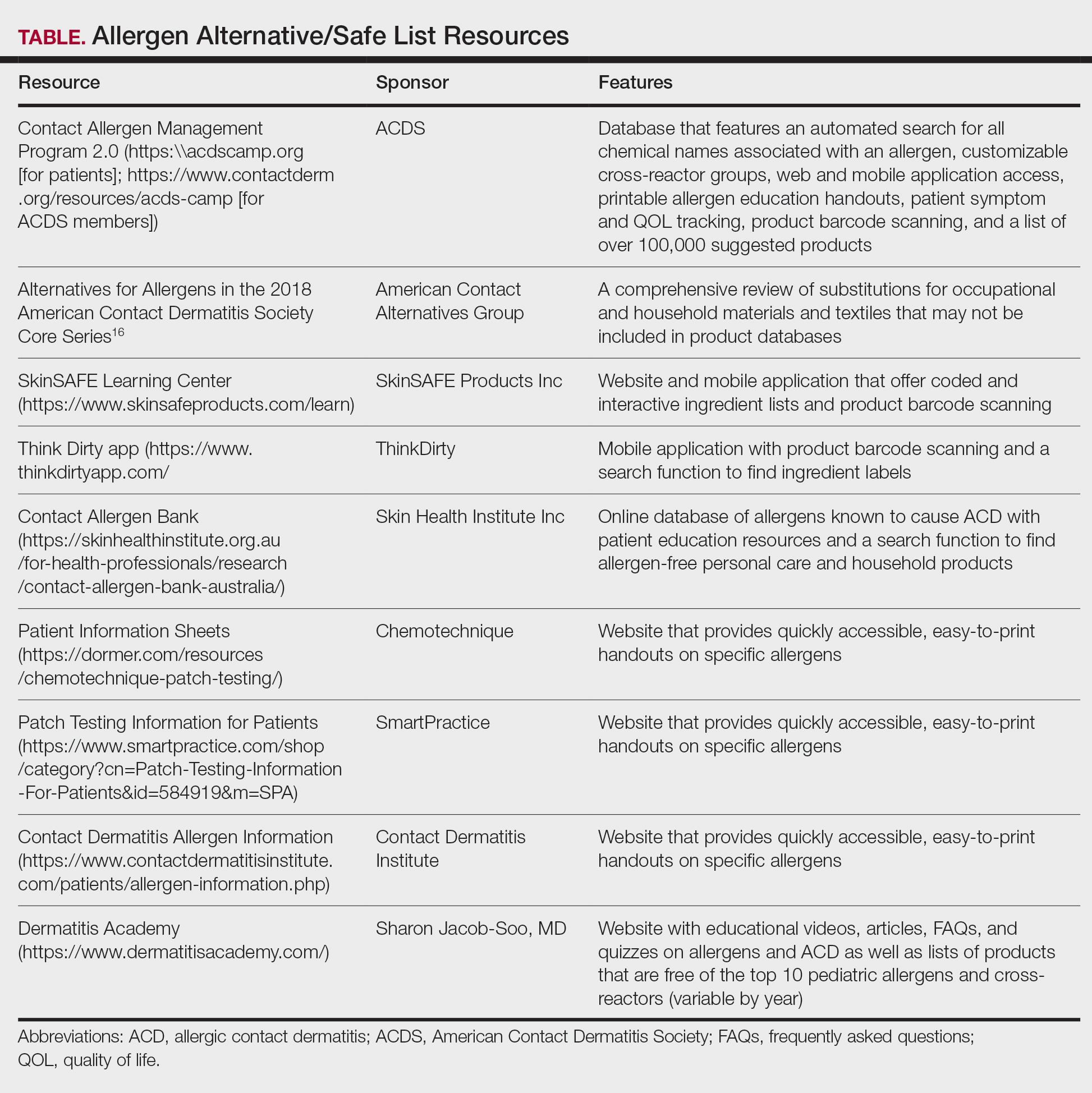
Other databases and online resources provide similar features, such as resources for patient education or finding safe products. The 2018 Alternatives for Allergens report is a vital adjunctive resource for guiding patients to suitable allergen-free products not included in commonly accessible product databases such as occupational materials, medical adhesives, shoes, or textiles.16
Introduction of CAMP 2.0
The latest version, CAMP 2.0, was launched in late 2024. The fully revamped database has a catalog of more than 100,000 products and comes packed with features that address many of the limitations found in the original CAMP. How does CAMP 2.0 work? The clinician inputs the patient’s allergens and makes choices about cross-reactor groups, and CAMP 2.0 outputs a list of allergen-free products that the patient can use when shopping for personal care products and the clinician can use for prescribing medicaments. The new user experience is intended to be more informative and engaging for all parties.
The CAMP 2.0 interface offers frequent product updates and streamlined database navigation, including enhanced search functions, barcode scanning, and a new mobile application for Apple and Android users. The mobile application also allows patients to track their symptoms and quality of life over time. With this additional functionality, there also is an extensive section for frequently asked questions and tutorials to help patients understand and utilize these features effectively.
Patients no longer have to wonder if a product that is not listed on their safe list is actually unsafe or just missing from the database. Several new features, including color-coded ingredient lists and organization of search codes into “safe” and “unsafe” product lists (Figure 1), help increase product transparency. These features can facilitate patient recognition of allergen names and cross-reactors in selected products. Future updates will include product purchasing through the mobile application and more educational handouts, including Spanish translations and dietary guidelines for systemic contact dermatitis.
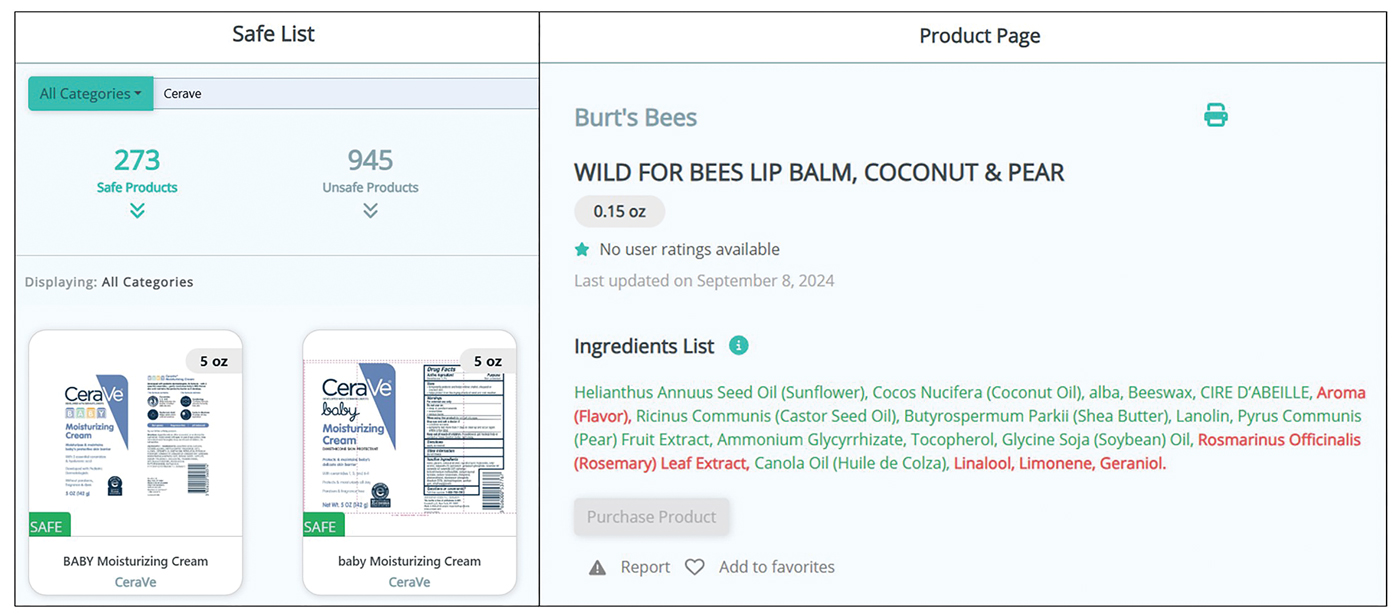
Patient Experience—Once patients complete patch testing with an ACDS member, they can access the CAMP 2.0 database for free via web-based or a mobile application. After setting up an account, patients gain immediate access to their allergen information, product database, and educational resources about ACD and CAMP 2.0. Patients can search for specific products using text or barcode scanning or browse through categorized lists of medical, household, and personal care items. Each product page contains the product name and brand along with a color-coded ingredient list to help patients identify safe and unsafe ingredients at a glance (Figure 1). Products not currently included in the database can be requested using the “Add Product” feature. Additional patient engagement features include options to mark favorite products, write reviews, and track quality of life over time.
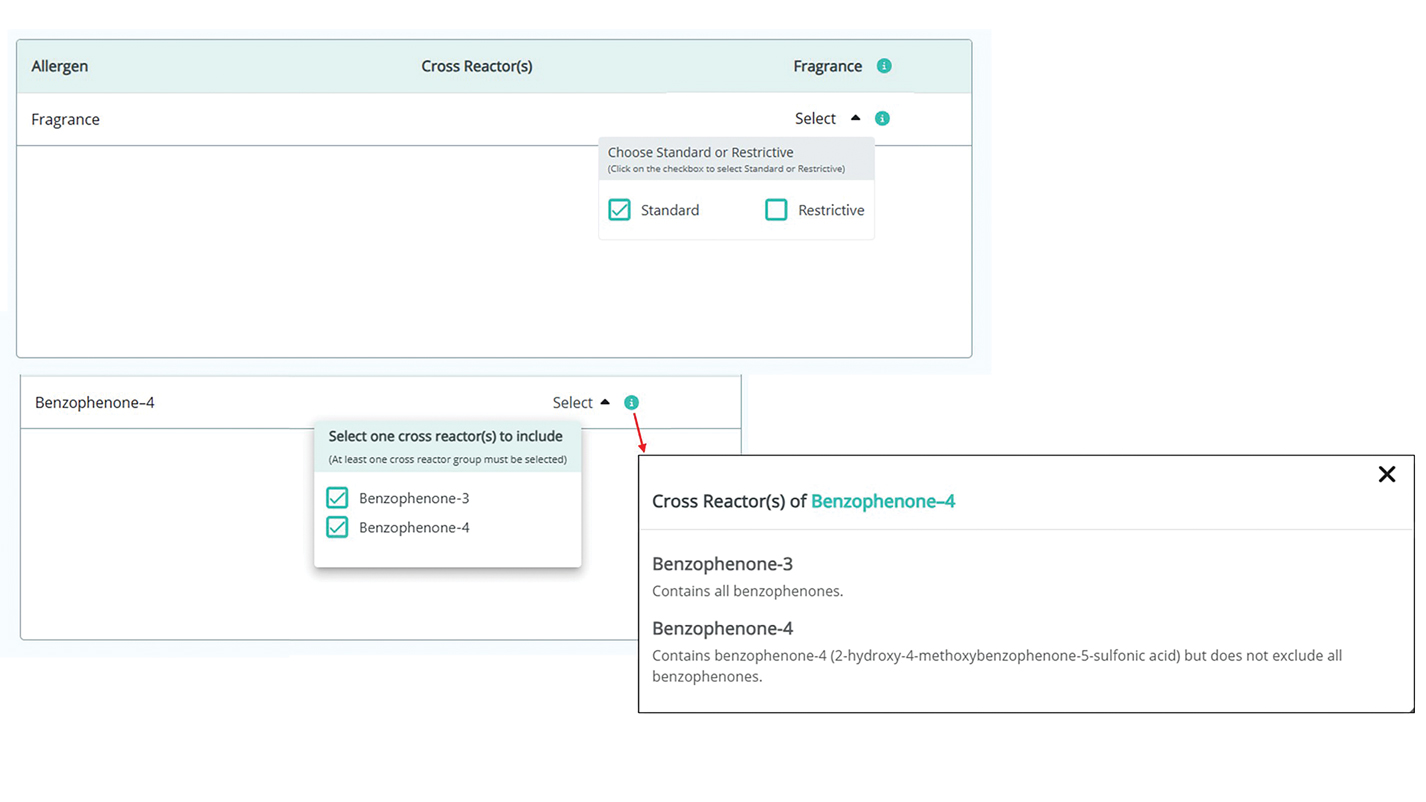
Physician Experience—The updated version includes several tutorials and frequently asked questions on how to improve ACD management and make the most of the new CAMP 2.0 tools and features. Generating patient allergen codes has been streamlined with an “Allergen Search” feature, allowing providers to quickly search and add or remove allergens to patients’ safe lists. Cross-reactor groups may be selectively added or removed for greater transparency and specificity in creating a patient safe list (Figure 2). Allergen codes now can be edited over time and are available for patient use via alphanumeric text or QR code format, which easily can be printed on a handout with instructions to help patients get acquainted with the system. For patient counseling, updated education handouts are available in the patient’s app and may be printed to provide supportive written educational material.
Approach to Long-Term Follow-up
When it comes to getting the most from patch testing, ongoing allergen avoidance is crucial. Patients may not see improvement unless they understand what ACD is and what needs to be done to improve it as well as become familiar with the names and common sources of their triggers.17 Clinicians can use CAMP 2.0 to facilitate patient improvement after patch testing, focusing on 3 key areas: continued patient education, patients’ ongoing progress in avoiding allergens, and monitored clinical improvement.
A solid understanding of ACD, such as its delayed (ie, 24-72 hours) onset after exposure, the need for allergen avoidance for at least 4 to 6 weeks before seeing improvement, and correlation of identified allergens with daily exposures, plays a major role in patient success. The CAMP 2.0 patch testing basics section is an excellent resource for patient-friendly explanations on patch testing and ACD. This resource, as well as allergen education handouts, may be reviewed at follow-up visits to continue to solidify patient learning.
Patients often have questions about allergen avoidance, such as occupational exposures, the suitability of specific products, or specific allergen names. These discussions are helpful for gauging how well patients are equipped to avoid their triggers as well as any hurdles they may be facing. If a patient still is experiencing flares after 6 to 8 weeks of safe-list adherence, it is important to take a thorough history of product use, daily exposures, and the patterns of distribution on the skin. Possible allergen exposures via topical medications also should be considered.18,19 Cross-checking products with a patient’s CAMP 2.0 safe list and correlating exposures with the continued ACD distribution are effective strategies to troubleshoot for unknown exposures to allergens.
Final Thoughts
Helping patients avoid allergens is essential to long-term management of ACD. The CAMP 2.0 safe list is an essential tool and a comprehensive reference for both patients and clinicians. With CAMP 2.0, allergen avoidance has never been more interactive or accessible.
- Tam I, Yu J. Allergic contact dermatitis in children: recommendations for patch testing. Curr Allergy Asthma Rep. 2020;20:41. doi:10.1007 /s11882-020-00939-z
- Dizdarevic A, Troensegaard W, Uldahl A, et al. Intervention study to evaluate the importance of information given to patients with contact allergy: a randomized, investigator-blinded clinical trial. Br J Dermatol. 2021;184:43-49. doi:10.1111/bjd.19119
- Jamil WN, Erikssohn I, Lindberg M. How well is the outcome of patch testing remembered by the patients? a 10-year follow-up of testing with the Swedish baseline series at the Department of Dermatology in Örebro, Sweden. Contact Dermatitis. 2012;66:215-220. doi:10.1111/j.1600-0536.2011.02039.x
- Scalf LA, Genebriera J, Davis MDP, et al. Patients’ perceptions of the usefulness and outcome of patch testing. J Am Acad Dermatol. 2007;56:928-932. doi:10.1016/j.jaad.2006.11.034
- Mossing K, Dizdarevic A, Svensson Å, et al. Impact on quality of life of an intervention providing additional information to patients with allergic contact dermatitis; a randomized clinical trial. J Eur Acad Dermatol Venereol. 2022;36:2166-2171. doi:10.1111/jdv.18412
- Schalock PC, Dunnick CA, Nedorost S, et al. American Contact Dermatitis Society Core Allergen Series: 2020 Update. Dermatitis. 2020;31:279-282. doi:10.1097/DER.0000000000000621
- Ingredient Breakdown: Fragrance. Think Dirty® Shop Clean. Accessed January 9, 2025. https://www.thinkdirtyapp.com/ingredient-breakdown-fragrance-3a8ef28f296a/
- Guarneri F, Corazza M, Stingeni L, et al. Myroxylon pereirae (balsam of Peru): still worth testing? Contact Dermatitis. 2021;85:269-273. doi:10.1111/cod.13839
- de Groot AC. Myroxylon pereirae resin (balsam of Peru)—a critical review of the literature and assessment of the significance of positive patch test reactions and the usefulness of restrictive diets. Contact Dermatitis. 2019;80:335-353. doi:10.1111/cod.13263
- Balsam of Peru: past and future. Allergic Contact Dermatitis Society; 2024. https://www.contactderm.org/UserFiles/members/Balsam_of_Peru___Past_and_Future.2.pdf
- Tramontana M, Hansel K, Bianchi L, et al. Advancing the understanding of allergic contact dermatitis: from pathophysiology to novel therapeutic approaches. Front Med. 2023;10. doi:10.3389 /fmed.2023.1184289
- McNamara D. ACDS launches Contact Allergen Management Program (CAMP). Internal Med News. March 7, 2011. Accessed December 31, 2024. https://www.mdedge.com/content/acds-launches-contact-allergen-management-program-camp-0
- Haque MZ, Rehman R, Guan L, et al. Recommendations to optimize patient education for allergic contact dermatitis: our approach. Contact Dermatitis. 2023;88:423-424. doi:10.1111/cod.14269
- Kist JM, el-Azhary RA, Hentz JG, et al. The Contact Allergen Replacement Database and treatment of allergic contact dermatitis. Arch Dermatol. 2004;140:1448-1450. doi:0.1001/archderm.140.12.1448
- El-Azhary RA, Yiannias JA. A new patient education approach in contact allergic dermatitis: the Contact Allergen Replacement Database (CARD). Int J Dermatol. 2004;43:278-280. doi:10.1111 /j.1365-4632.2004.01843.x
- Scheman A, Hylwa-Deufel S, Jacob SE, et al. Alternatives for allergens in the 2018 American Contact Dermatitis Society Core Series: report by the American Contact Alternatives Group. Dermatitis. 2019;30:87-105. doi:10.1097/DER.0000000000000453
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient management and education. J Am Acad Dermatol. 2016;74:1043-1054. doi:10.1016/j.jaad.2015.02.1144
- Ng A, Atwater AR, Reeder M. Contact allergy to topical medicaments, part 1: a double-edged sword. Cutis. 2021;108:271-275. doi:10.12788 /cutis.0390
- Nardelli A, D’Hooghe E, Drieghe J, et al. Allergic contact dermatitis from fragrance components in specific topical pharmaceutical products in Belgium. Contact Dermatitis. 2009;60:303-313. doi:10.1111 /j.1600-0536.2009.01542.x
While patch testing is the gold standard to diagnose type IV cutaneous hypersensitivity reactions, interpreting results can feel like trying to decipher a secret code, leaving patients feeling disempowered in avoiding their triggers. To truly manage allergic contact dermatitis (ACD), patients need comprehensive education on which allergens to avoid and ways to spot potential sources of exposure, including counseling, written guidelines, and lists of product alternatives.1 Patients who can recall and avoid their triggers experience greater improvement in clinical and quality-of-life scores.2 However, several studies have demonstrated that patients have difficulty recalling their allergens, even with longitudinal reminders.2-5 Quality-of-life and clinical outcomes also are not necessarily improved by successful allergen recall alone, as patients have reported limited success in actually avoiding allergens, highlighting the complexity of navigating exposures in daily life.2,6 To address these challenges, we examine common pitfalls patients encounter when avoiding allergens, highlight the benefits of utilizing safe lists and databases for allergen management, and introduce the updated Contact Allergen Management Program (CAMP) 2.0 as an optimal tool for long-term management of ACD.
Allergen Avoidance Pitfalls
Simply reading ingredient labels to avoid allergens is only marginally effective, as patients need to identify and interpret multiple chemical names as well as cross-reactors and related compounds to achieve success. Some allergens, such as fragrances or manufacturing impurities, are not explicitly identified on product labels. Even patients who can practice diligent label reading may struggle to find information on household or occupational products when full ingredient disclosure is not required.
Many of the allergens included in the American Contact Dermatitis Society (ACDS) Core 90 Series have alternative chemical aliases, and many have related compounds.6 For example, individuals with contact allergy to formaldehyde or a formaldehyde releaser usually need to avoid multiple other formaldehyde-releasing chemicals. Patients who test positive to amidoamine or dimethylaminopropylamine also must avoid the surfactant cocamidopropyl betaine—not because it is a cross-reactor, but because it is an impurity in the synthetic pathway.
Fragrance is one of the most common causes of ACD but can be challenging to avoid. Patients with allergies to fragrance or specific compounds (eg, limonene, linalool hydroperoxides) need to be savvy enough to navigate a broad spectrum of synthetic and botanical fragrance additives. Avoiding products that contain “fragrance” or “parfum” is simple enough, but patients also may need to recognize more than 3000 chemical names to identify individual fragrance ingredients that may be listed separately.7 Further, some fragrances are added for alternative purposes—preservative, medicinal, or emulsification—in which case products may deceptively tout themselves as being “fragrance free” yet still contain a fragrance allergen. This is made even more complex considering additional additives that commonly may cross-react with individual fragrance compounds; balsam of Peru, for example, is a botanical amalgam containing more than 250 compounds, including several fragrance components, making it an excellent indicator of fragrance allergy.8 While balsam of Peru and its fragrance constituents will almost never be listed on a product label, it cross-reacts with several benzyl derivatives commonly used in cosmetic formulations, such as benzyl alcohol, benzyl acetate, benzoic acid, benzyl benzoate, and benzyl cinnamate.9,10
Given that ACD is a common reason for patients to seek dermatologic care, it is crucial for clinicians to equip themselves with effective strategies to support patients after patch testing.11 This includes efficient translation of patch test results into practical advice while avoiding the oversimplified suggestion to read product labels; however, education alone cannot address the complexities of managing ACD, which is where contact allergen databases come into play.
An Essential Tool: Patient Allergen Databases and Safe Lists
Contact allergen databases are like a trusty sidekick for patients and clinicians, providing easily accessible information and tools to support allergen avoidance and improve ACD outcomes. While there are several existing resources, the ACDS launched its CAMP database in 2011 for ACDS members and their patients.12 The CAMP allows clinicians to easily generate personalized safe lists for household, medicament, and personal care products, facilitating seamless patient access both online and via a mobile application. The database also includes allergen-specific handouts to guide patient education.13 A key highlight of the CAMP is automated management of cross-reactors, which allows patients to choose products without having to memorize complex cross-reactor algorithms and helps avoid overly restrictive safe lists (Table).12-15
Other databases and online resources provide similar features, such as resources for patient education or finding safe products. The 2018 Alternatives for Allergens report is a vital adjunctive resource for guiding patients to suitable allergen-free products not included in commonly accessible product databases such as occupational materials, medical adhesives, shoes, or textiles.16
Introduction of CAMP 2.0
The latest version, CAMP 2.0, was launched in late 2024. The fully revamped database has a catalog of more than 100,000 products and comes packed with features that address many of the limitations found in the original CAMP. How does CAMP 2.0 work? The clinician inputs the patient’s allergens and makes choices about cross-reactor groups, and CAMP 2.0 outputs a list of allergen-free products that the patient can use when shopping for personal care products and the clinician can use for prescribing medicaments. The new user experience is intended to be more informative and engaging for all parties.
The CAMP 2.0 interface offers frequent product updates and streamlined database navigation, including enhanced search functions, barcode scanning, and a new mobile application for Apple and Android users. The mobile application also allows patients to track their symptoms and quality of life over time. With this additional functionality, there also is an extensive section for frequently asked questions and tutorials to help patients understand and utilize these features effectively.
Patients no longer have to wonder if a product that is not listed on their safe list is actually unsafe or just missing from the database. Several new features, including color-coded ingredient lists and organization of search codes into “safe” and “unsafe” product lists (Figure 1), help increase product transparency. These features can facilitate patient recognition of allergen names and cross-reactors in selected products. Future updates will include product purchasing through the mobile application and more educational handouts, including Spanish translations and dietary guidelines for systemic contact dermatitis.
Patient Experience—Once patients complete patch testing with an ACDS member, they can access the CAMP 2.0 database for free via web-based or a mobile application. After setting up an account, patients gain immediate access to their allergen information, product database, and educational resources about ACD and CAMP 2.0. Patients can search for specific products using text or barcode scanning or browse through categorized lists of medical, household, and personal care items. Each product page contains the product name and brand along with a color-coded ingredient list to help patients identify safe and unsafe ingredients at a glance (Figure 1). Products not currently included in the database can be requested using the “Add Product” feature. Additional patient engagement features include options to mark favorite products, write reviews, and track quality of life over time.
Physician Experience—The updated version includes several tutorials and frequently asked questions on how to improve ACD management and make the most of the new CAMP 2.0 tools and features. Generating patient allergen codes has been streamlined with an “Allergen Search” feature, allowing providers to quickly search and add or remove allergens to patients’ safe lists. Cross-reactor groups may be selectively added or removed for greater transparency and specificity in creating a patient safe list (Figure 2). Allergen codes now can be edited over time and are available for patient use via alphanumeric text or QR code format, which easily can be printed on a handout with instructions to help patients get acquainted with the system. For patient counseling, updated education handouts are available in the patient’s app and may be printed to provide supportive written educational material.
Approach to Long-Term Follow-up
When it comes to getting the most from patch testing, ongoing allergen avoidance is crucial. Patients may not see improvement unless they understand what ACD is and what needs to be done to improve it as well as become familiar with the names and common sources of their triggers.17 Clinicians can use CAMP 2.0 to facilitate patient improvement after patch testing, focusing on 3 key areas: continued patient education, patients’ ongoing progress in avoiding allergens, and monitored clinical improvement.
A solid understanding of ACD, such as its delayed (ie, 24-72 hours) onset after exposure, the need for allergen avoidance for at least 4 to 6 weeks before seeing improvement, and correlation of identified allergens with daily exposures, plays a major role in patient success. The CAMP 2.0 patch testing basics section is an excellent resource for patient-friendly explanations on patch testing and ACD. This resource, as well as allergen education handouts, may be reviewed at follow-up visits to continue to solidify patient learning.
Patients often have questions about allergen avoidance, such as occupational exposures, the suitability of specific products, or specific allergen names. These discussions are helpful for gauging how well patients are equipped to avoid their triggers as well as any hurdles they may be facing. If a patient still is experiencing flares after 6 to 8 weeks of safe-list adherence, it is important to take a thorough history of product use, daily exposures, and the patterns of distribution on the skin. Possible allergen exposures via topical medications also should be considered.18,19 Cross-checking products with a patient’s CAMP 2.0 safe list and correlating exposures with the continued ACD distribution are effective strategies to troubleshoot for unknown exposures to allergens.
Final Thoughts
Helping patients avoid allergens is essential to long-term management of ACD. The CAMP 2.0 safe list is an essential tool and a comprehensive reference for both patients and clinicians. With CAMP 2.0, allergen avoidance has never been more interactive or accessible.
While patch testing is the gold standard to diagnose type IV cutaneous hypersensitivity reactions, interpreting results can feel like trying to decipher a secret code, leaving patients feeling disempowered in avoiding their triggers. To truly manage allergic contact dermatitis (ACD), patients need comprehensive education on which allergens to avoid and ways to spot potential sources of exposure, including counseling, written guidelines, and lists of product alternatives.1 Patients who can recall and avoid their triggers experience greater improvement in clinical and quality-of-life scores.2 However, several studies have demonstrated that patients have difficulty recalling their allergens, even with longitudinal reminders.2-5 Quality-of-life and clinical outcomes also are not necessarily improved by successful allergen recall alone, as patients have reported limited success in actually avoiding allergens, highlighting the complexity of navigating exposures in daily life.2,6 To address these challenges, we examine common pitfalls patients encounter when avoiding allergens, highlight the benefits of utilizing safe lists and databases for allergen management, and introduce the updated Contact Allergen Management Program (CAMP) 2.0 as an optimal tool for long-term management of ACD.
Allergen Avoidance Pitfalls
Simply reading ingredient labels to avoid allergens is only marginally effective, as patients need to identify and interpret multiple chemical names as well as cross-reactors and related compounds to achieve success. Some allergens, such as fragrances or manufacturing impurities, are not explicitly identified on product labels. Even patients who can practice diligent label reading may struggle to find information on household or occupational products when full ingredient disclosure is not required.
Many of the allergens included in the American Contact Dermatitis Society (ACDS) Core 90 Series have alternative chemical aliases, and many have related compounds.6 For example, individuals with contact allergy to formaldehyde or a formaldehyde releaser usually need to avoid multiple other formaldehyde-releasing chemicals. Patients who test positive to amidoamine or dimethylaminopropylamine also must avoid the surfactant cocamidopropyl betaine—not because it is a cross-reactor, but because it is an impurity in the synthetic pathway.
Fragrance is one of the most common causes of ACD but can be challenging to avoid. Patients with allergies to fragrance or specific compounds (eg, limonene, linalool hydroperoxides) need to be savvy enough to navigate a broad spectrum of synthetic and botanical fragrance additives. Avoiding products that contain “fragrance” or “parfum” is simple enough, but patients also may need to recognize more than 3000 chemical names to identify individual fragrance ingredients that may be listed separately.7 Further, some fragrances are added for alternative purposes—preservative, medicinal, or emulsification—in which case products may deceptively tout themselves as being “fragrance free” yet still contain a fragrance allergen. This is made even more complex considering additional additives that commonly may cross-react with individual fragrance compounds; balsam of Peru, for example, is a botanical amalgam containing more than 250 compounds, including several fragrance components, making it an excellent indicator of fragrance allergy.8 While balsam of Peru and its fragrance constituents will almost never be listed on a product label, it cross-reacts with several benzyl derivatives commonly used in cosmetic formulations, such as benzyl alcohol, benzyl acetate, benzoic acid, benzyl benzoate, and benzyl cinnamate.9,10
Given that ACD is a common reason for patients to seek dermatologic care, it is crucial for clinicians to equip themselves with effective strategies to support patients after patch testing.11 This includes efficient translation of patch test results into practical advice while avoiding the oversimplified suggestion to read product labels; however, education alone cannot address the complexities of managing ACD, which is where contact allergen databases come into play.
An Essential Tool: Patient Allergen Databases and Safe Lists
Contact allergen databases are like a trusty sidekick for patients and clinicians, providing easily accessible information and tools to support allergen avoidance and improve ACD outcomes. While there are several existing resources, the ACDS launched its CAMP database in 2011 for ACDS members and their patients.12 The CAMP allows clinicians to easily generate personalized safe lists for household, medicament, and personal care products, facilitating seamless patient access both online and via a mobile application. The database also includes allergen-specific handouts to guide patient education.13 A key highlight of the CAMP is automated management of cross-reactors, which allows patients to choose products without having to memorize complex cross-reactor algorithms and helps avoid overly restrictive safe lists (Table).12-15
Other databases and online resources provide similar features, such as resources for patient education or finding safe products. The 2018 Alternatives for Allergens report is a vital adjunctive resource for guiding patients to suitable allergen-free products not included in commonly accessible product databases such as occupational materials, medical adhesives, shoes, or textiles.16
Introduction of CAMP 2.0
The latest version, CAMP 2.0, was launched in late 2024. The fully revamped database has a catalog of more than 100,000 products and comes packed with features that address many of the limitations found in the original CAMP. How does CAMP 2.0 work? The clinician inputs the patient’s allergens and makes choices about cross-reactor groups, and CAMP 2.0 outputs a list of allergen-free products that the patient can use when shopping for personal care products and the clinician can use for prescribing medicaments. The new user experience is intended to be more informative and engaging for all parties.
The CAMP 2.0 interface offers frequent product updates and streamlined database navigation, including enhanced search functions, barcode scanning, and a new mobile application for Apple and Android users. The mobile application also allows patients to track their symptoms and quality of life over time. With this additional functionality, there also is an extensive section for frequently asked questions and tutorials to help patients understand and utilize these features effectively.
Patients no longer have to wonder if a product that is not listed on their safe list is actually unsafe or just missing from the database. Several new features, including color-coded ingredient lists and organization of search codes into “safe” and “unsafe” product lists (Figure 1), help increase product transparency. These features can facilitate patient recognition of allergen names and cross-reactors in selected products. Future updates will include product purchasing through the mobile application and more educational handouts, including Spanish translations and dietary guidelines for systemic contact dermatitis.
Patient Experience—Once patients complete patch testing with an ACDS member, they can access the CAMP 2.0 database for free via web-based or a mobile application. After setting up an account, patients gain immediate access to their allergen information, product database, and educational resources about ACD and CAMP 2.0. Patients can search for specific products using text or barcode scanning or browse through categorized lists of medical, household, and personal care items. Each product page contains the product name and brand along with a color-coded ingredient list to help patients identify safe and unsafe ingredients at a glance (Figure 1). Products not currently included in the database can be requested using the “Add Product” feature. Additional patient engagement features include options to mark favorite products, write reviews, and track quality of life over time.
Physician Experience—The updated version includes several tutorials and frequently asked questions on how to improve ACD management and make the most of the new CAMP 2.0 tools and features. Generating patient allergen codes has been streamlined with an “Allergen Search” feature, allowing providers to quickly search and add or remove allergens to patients’ safe lists. Cross-reactor groups may be selectively added or removed for greater transparency and specificity in creating a patient safe list (Figure 2). Allergen codes now can be edited over time and are available for patient use via alphanumeric text or QR code format, which easily can be printed on a handout with instructions to help patients get acquainted with the system. For patient counseling, updated education handouts are available in the patient’s app and may be printed to provide supportive written educational material.
Approach to Long-Term Follow-up
When it comes to getting the most from patch testing, ongoing allergen avoidance is crucial. Patients may not see improvement unless they understand what ACD is and what needs to be done to improve it as well as become familiar with the names and common sources of their triggers.17 Clinicians can use CAMP 2.0 to facilitate patient improvement after patch testing, focusing on 3 key areas: continued patient education, patients’ ongoing progress in avoiding allergens, and monitored clinical improvement.
A solid understanding of ACD, such as its delayed (ie, 24-72 hours) onset after exposure, the need for allergen avoidance for at least 4 to 6 weeks before seeing improvement, and correlation of identified allergens with daily exposures, plays a major role in patient success. The CAMP 2.0 patch testing basics section is an excellent resource for patient-friendly explanations on patch testing and ACD. This resource, as well as allergen education handouts, may be reviewed at follow-up visits to continue to solidify patient learning.
Patients often have questions about allergen avoidance, such as occupational exposures, the suitability of specific products, or specific allergen names. These discussions are helpful for gauging how well patients are equipped to avoid their triggers as well as any hurdles they may be facing. If a patient still is experiencing flares after 6 to 8 weeks of safe-list adherence, it is important to take a thorough history of product use, daily exposures, and the patterns of distribution on the skin. Possible allergen exposures via topical medications also should be considered.18,19 Cross-checking products with a patient’s CAMP 2.0 safe list and correlating exposures with the continued ACD distribution are effective strategies to troubleshoot for unknown exposures to allergens.
Final Thoughts
Helping patients avoid allergens is essential to long-term management of ACD. The CAMP 2.0 safe list is an essential tool and a comprehensive reference for both patients and clinicians. With CAMP 2.0, allergen avoidance has never been more interactive or accessible.
- Tam I, Yu J. Allergic contact dermatitis in children: recommendations for patch testing. Curr Allergy Asthma Rep. 2020;20:41. doi:10.1007 /s11882-020-00939-z
- Dizdarevic A, Troensegaard W, Uldahl A, et al. Intervention study to evaluate the importance of information given to patients with contact allergy: a randomized, investigator-blinded clinical trial. Br J Dermatol. 2021;184:43-49. doi:10.1111/bjd.19119
- Jamil WN, Erikssohn I, Lindberg M. How well is the outcome of patch testing remembered by the patients? a 10-year follow-up of testing with the Swedish baseline series at the Department of Dermatology in Örebro, Sweden. Contact Dermatitis. 2012;66:215-220. doi:10.1111/j.1600-0536.2011.02039.x
- Scalf LA, Genebriera J, Davis MDP, et al. Patients’ perceptions of the usefulness and outcome of patch testing. J Am Acad Dermatol. 2007;56:928-932. doi:10.1016/j.jaad.2006.11.034
- Mossing K, Dizdarevic A, Svensson Å, et al. Impact on quality of life of an intervention providing additional information to patients with allergic contact dermatitis; a randomized clinical trial. J Eur Acad Dermatol Venereol. 2022;36:2166-2171. doi:10.1111/jdv.18412
- Schalock PC, Dunnick CA, Nedorost S, et al. American Contact Dermatitis Society Core Allergen Series: 2020 Update. Dermatitis. 2020;31:279-282. doi:10.1097/DER.0000000000000621
- Ingredient Breakdown: Fragrance. Think Dirty® Shop Clean. Accessed January 9, 2025. https://www.thinkdirtyapp.com/ingredient-breakdown-fragrance-3a8ef28f296a/
- Guarneri F, Corazza M, Stingeni L, et al. Myroxylon pereirae (balsam of Peru): still worth testing? Contact Dermatitis. 2021;85:269-273. doi:10.1111/cod.13839
- de Groot AC. Myroxylon pereirae resin (balsam of Peru)—a critical review of the literature and assessment of the significance of positive patch test reactions and the usefulness of restrictive diets. Contact Dermatitis. 2019;80:335-353. doi:10.1111/cod.13263
- Balsam of Peru: past and future. Allergic Contact Dermatitis Society; 2024. https://www.contactderm.org/UserFiles/members/Balsam_of_Peru___Past_and_Future.2.pdf
- Tramontana M, Hansel K, Bianchi L, et al. Advancing the understanding of allergic contact dermatitis: from pathophysiology to novel therapeutic approaches. Front Med. 2023;10. doi:10.3389 /fmed.2023.1184289
- McNamara D. ACDS launches Contact Allergen Management Program (CAMP). Internal Med News. March 7, 2011. Accessed December 31, 2024. https://www.mdedge.com/content/acds-launches-contact-allergen-management-program-camp-0
- Haque MZ, Rehman R, Guan L, et al. Recommendations to optimize patient education for allergic contact dermatitis: our approach. Contact Dermatitis. 2023;88:423-424. doi:10.1111/cod.14269
- Kist JM, el-Azhary RA, Hentz JG, et al. The Contact Allergen Replacement Database and treatment of allergic contact dermatitis. Arch Dermatol. 2004;140:1448-1450. doi:0.1001/archderm.140.12.1448
- El-Azhary RA, Yiannias JA. A new patient education approach in contact allergic dermatitis: the Contact Allergen Replacement Database (CARD). Int J Dermatol. 2004;43:278-280. doi:10.1111 /j.1365-4632.2004.01843.x
- Scheman A, Hylwa-Deufel S, Jacob SE, et al. Alternatives for allergens in the 2018 American Contact Dermatitis Society Core Series: report by the American Contact Alternatives Group. Dermatitis. 2019;30:87-105. doi:10.1097/DER.0000000000000453
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient management and education. J Am Acad Dermatol. 2016;74:1043-1054. doi:10.1016/j.jaad.2015.02.1144
- Ng A, Atwater AR, Reeder M. Contact allergy to topical medicaments, part 1: a double-edged sword. Cutis. 2021;108:271-275. doi:10.12788 /cutis.0390
- Nardelli A, D’Hooghe E, Drieghe J, et al. Allergic contact dermatitis from fragrance components in specific topical pharmaceutical products in Belgium. Contact Dermatitis. 2009;60:303-313. doi:10.1111 /j.1600-0536.2009.01542.x
- Tam I, Yu J. Allergic contact dermatitis in children: recommendations for patch testing. Curr Allergy Asthma Rep. 2020;20:41. doi:10.1007 /s11882-020-00939-z
- Dizdarevic A, Troensegaard W, Uldahl A, et al. Intervention study to evaluate the importance of information given to patients with contact allergy: a randomized, investigator-blinded clinical trial. Br J Dermatol. 2021;184:43-49. doi:10.1111/bjd.19119
- Jamil WN, Erikssohn I, Lindberg M. How well is the outcome of patch testing remembered by the patients? a 10-year follow-up of testing with the Swedish baseline series at the Department of Dermatology in Örebro, Sweden. Contact Dermatitis. 2012;66:215-220. doi:10.1111/j.1600-0536.2011.02039.x
- Scalf LA, Genebriera J, Davis MDP, et al. Patients’ perceptions of the usefulness and outcome of patch testing. J Am Acad Dermatol. 2007;56:928-932. doi:10.1016/j.jaad.2006.11.034
- Mossing K, Dizdarevic A, Svensson Å, et al. Impact on quality of life of an intervention providing additional information to patients with allergic contact dermatitis; a randomized clinical trial. J Eur Acad Dermatol Venereol. 2022;36:2166-2171. doi:10.1111/jdv.18412
- Schalock PC, Dunnick CA, Nedorost S, et al. American Contact Dermatitis Society Core Allergen Series: 2020 Update. Dermatitis. 2020;31:279-282. doi:10.1097/DER.0000000000000621
- Ingredient Breakdown: Fragrance. Think Dirty® Shop Clean. Accessed January 9, 2025. https://www.thinkdirtyapp.com/ingredient-breakdown-fragrance-3a8ef28f296a/
- Guarneri F, Corazza M, Stingeni L, et al. Myroxylon pereirae (balsam of Peru): still worth testing? Contact Dermatitis. 2021;85:269-273. doi:10.1111/cod.13839
- de Groot AC. Myroxylon pereirae resin (balsam of Peru)—a critical review of the literature and assessment of the significance of positive patch test reactions and the usefulness of restrictive diets. Contact Dermatitis. 2019;80:335-353. doi:10.1111/cod.13263
- Balsam of Peru: past and future. Allergic Contact Dermatitis Society; 2024. https://www.contactderm.org/UserFiles/members/Balsam_of_Peru___Past_and_Future.2.pdf
- Tramontana M, Hansel K, Bianchi L, et al. Advancing the understanding of allergic contact dermatitis: from pathophysiology to novel therapeutic approaches. Front Med. 2023;10. doi:10.3389 /fmed.2023.1184289
- McNamara D. ACDS launches Contact Allergen Management Program (CAMP). Internal Med News. March 7, 2011. Accessed December 31, 2024. https://www.mdedge.com/content/acds-launches-contact-allergen-management-program-camp-0
- Haque MZ, Rehman R, Guan L, et al. Recommendations to optimize patient education for allergic contact dermatitis: our approach. Contact Dermatitis. 2023;88:423-424. doi:10.1111/cod.14269
- Kist JM, el-Azhary RA, Hentz JG, et al. The Contact Allergen Replacement Database and treatment of allergic contact dermatitis. Arch Dermatol. 2004;140:1448-1450. doi:0.1001/archderm.140.12.1448
- El-Azhary RA, Yiannias JA. A new patient education approach in contact allergic dermatitis: the Contact Allergen Replacement Database (CARD). Int J Dermatol. 2004;43:278-280. doi:10.1111 /j.1365-4632.2004.01843.x
- Scheman A, Hylwa-Deufel S, Jacob SE, et al. Alternatives for allergens in the 2018 American Contact Dermatitis Society Core Series: report by the American Contact Alternatives Group. Dermatitis. 2019;30:87-105. doi:10.1097/DER.0000000000000453
- Mowad CM, Anderson B, Scheinman P, et al. Allergic contact dermatitis: patient management and education. J Am Acad Dermatol. 2016;74:1043-1054. doi:10.1016/j.jaad.2015.02.1144
- Ng A, Atwater AR, Reeder M. Contact allergy to topical medicaments, part 1: a double-edged sword. Cutis. 2021;108:271-275. doi:10.12788 /cutis.0390
- Nardelli A, D’Hooghe E, Drieghe J, et al. Allergic contact dermatitis from fragrance components in specific topical pharmaceutical products in Belgium. Contact Dermatitis. 2009;60:303-313. doi:10.1111 /j.1600-0536.2009.01542.x
Simplifying Allergic Contact Dermatitis Management with the Contact Allergen Management Program 2.0
Simplifying Allergic Contact Dermatitis Management with the Contact Allergen Management Program 2.0
PRACTICE POINTS
- Comprehensive patient education is critical for appropriate allergen avoidance after patch testing, and allergen databases and product safe lists are invaluable tools to complement clinical guidance.
- The updated Contact Allergen Management Program 2.0 offers an updated approach to patient guidance, including a database of more than 100,000 products and an easy-to-use platform to find safe, allergen-free products.
- Interactive learning resources, product pages, and quality-of-life tracking tools can help equip patients with information to encourage further autonomy in allergen avoidance.
Tattoo Granulomas With Uveitis Successfully Treated With CO2 Laser Ablation
Tattoo Granulomas With Uveitis Successfully Treated With CO2 Laser Ablation
To the Editor:
Uveitis associated with tattoos is common, yet the etiology and optimal treatment options for this phenomenon remain unclear. Possible causes include a delayed hypersensitivity reaction to tattoo ink antigen or systemic sarcoidosis localized to the skin.1 Long-term treatment options include topical, intralesional, and systemic corticosteroids or immunosuppressants.2 Short-term options often include direct surgical excision and laser treatment. However, laser removal of tattoo pigment typically involves multiple sessions over the course of years, and there is a risk for antigen dispersal that may lead to anaphylaxis. Determining the most effective and safe treatment for a patient with progressive and severe ocular symptoms can be challenging. We describe a patient with cutaneous blue ink tattoos who developed chronic bilateral glaucoma, iritis, uveitis, and ocular hypertension that was refractory to multiple systemic medications and ophthalmologic procedures but responded to CO2 laser ablation.
A 27-year-old man with an active smoking history presented to our laser surgery center with a rash of approximately 4 years’ duration in areas with blue tattoo ink on both forearms. He was referred by his ophthalmologist due to bilateral uveitis and iritis and subsequent ocular hypertension and glaucoma that developed approximately 5 years after tattoo placement on the bilateral forearms. When the rash first appeared, the skin in the areas of the blue tattoo ink had hyperpigmented pruritic plaques. The patient was treated by a dermatologist with topical steroids to help reduce the itching and inflammation. Around the same time, he also started having ocular symptoms—vitreous floaters, erythema, eye pain, and blurriness—and was diagnosed with iritis of unclear etiology by ophthalmology. Figure 1 documents the patient’s clinical course. Due to escalating intraocular pressure and symptoms, he was referred to a glaucoma specialist and a rheumatologist. Systemic and rheumatologic medical conditions were ruled out with negative results on a series of blood tests (eg, rheumatoid factor, HLA-B27, antinuclear antibody, lysozyme, interferon gamma release assay, erythrocyte sedimentation rate, C-reactive protein, hepatitis B/C virus, Treponema pallidum, HIV), and magnetic resonance imaging of the brain was negative, ruling out demyelinating disease. Laboratory workup for sarcoidosis also was performed. The angiotensin-converting enzyme level was 30 U/L (reference range, 9-67 U/L), and a chest radiograph and computed tomography with contrast indicated no evidence of cardiopulmonary involvement. Although sarcoidosis could not be definitively ruled out, no other cause could be determined, and the patient’s glaucoma specialist diagnosed him with tattoo-associated uveitis. The patient was started on brimonidine, latanoprost, prednisolone, and dorzolamidetimolol eye drops, as well as acetazolamide (500 mg twice daily) and oral prednisone (various doses). Over the next 3 years, the patient continued to have symptoms, and immunosuppressant medications—methotrexate 20-25 mg weekly and adalimumab 40 mg every 2 weeks—were added to his treatment regimen. The patient also underwent bilateral ophthalmologic procedures, including a Baerveldt glaucoma implant procedure in the left eye and circumferential trabeculectomy in the right eye.
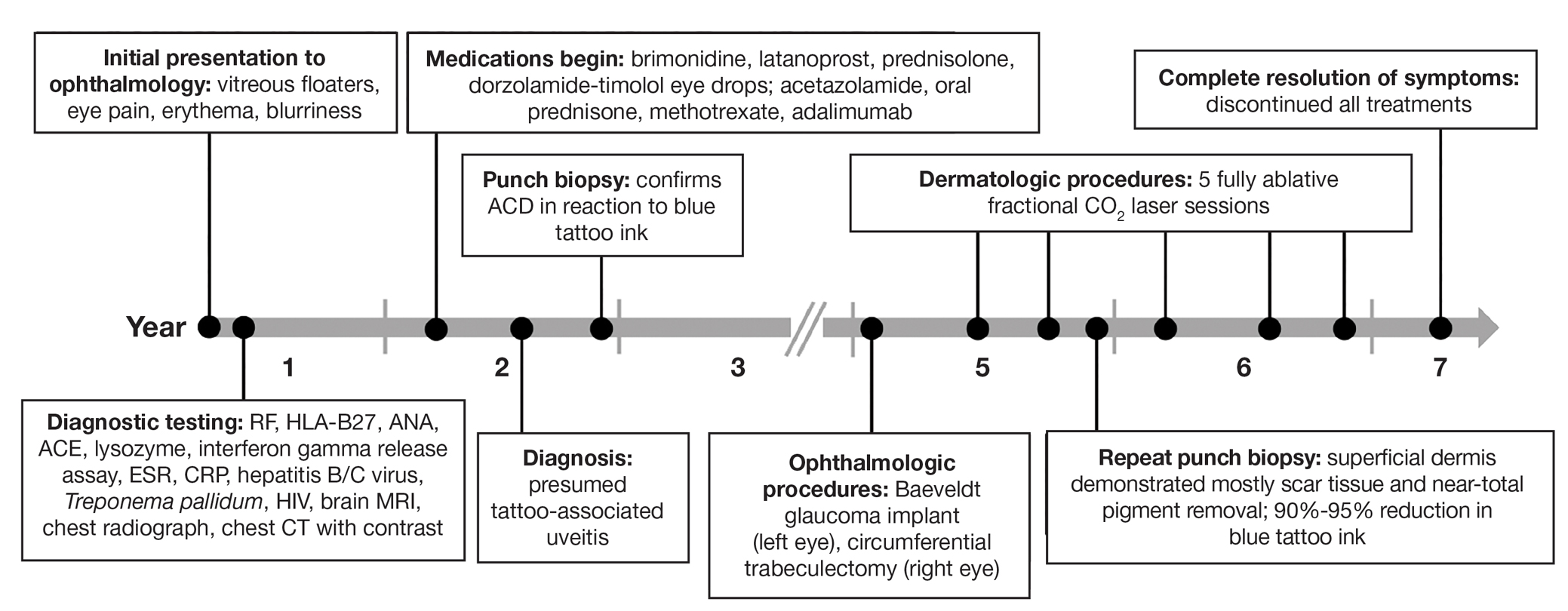
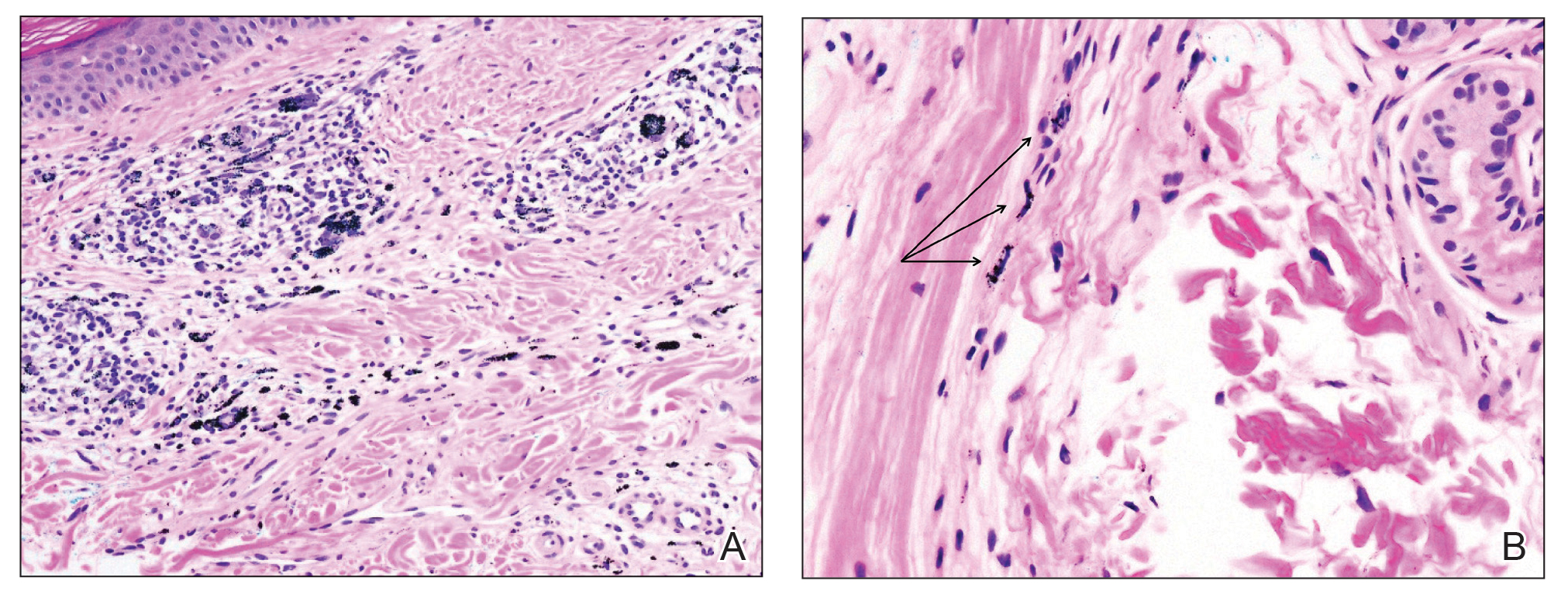
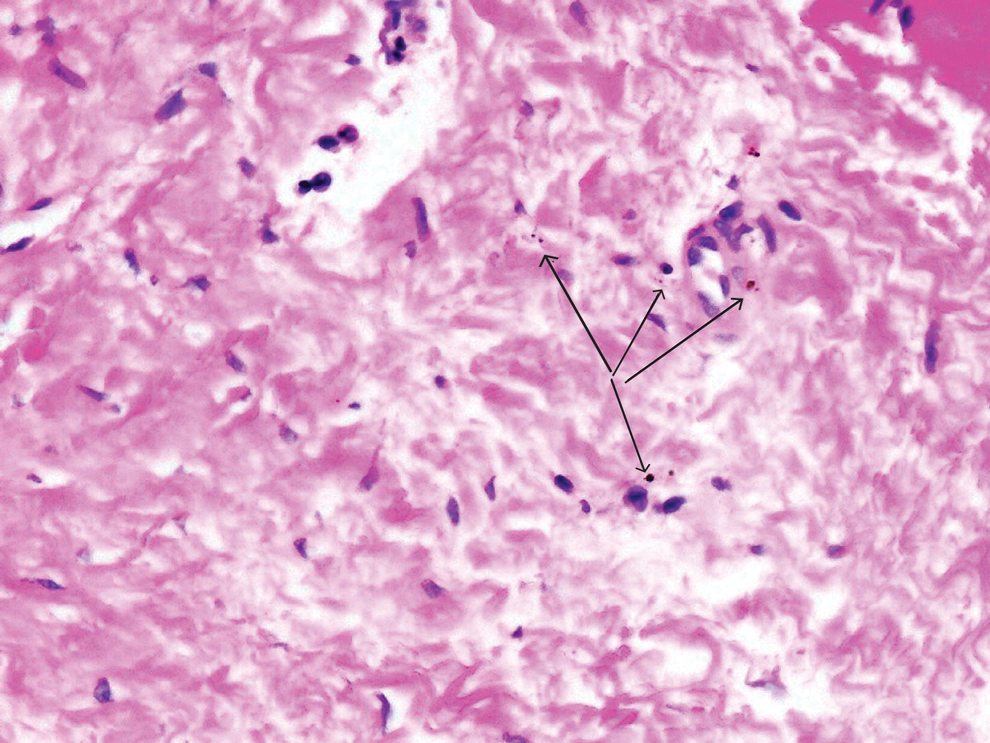
Despite these medications and procedures, the patient’s symptoms and intraocular pressure had not improved. At the current visit, punch biopsy of the tattooed skin and histologic examination showed dermal lymphoplasmacytic inflammation with scattered foreign-body giant cells associated with blue tattoo ink and overlying hyperkeratosis and spongiosis, consistent with allergic contact dermatitis (Figure 2). Because both immunosuppressant medications and ophthalmologic procedures had failed to control the progression of the ocular symptoms and the patient was at risk for permanent blindness, surgical excision and laser tattoo removal were considered as potential treatment options. Due to the large surface area and circumferential nature of the tattoos, there was a notable risk for disfiguring scars at both recipient and donor sites with surgical excision followed by graft placement. Thus, CO2 laser ablation was the preferred treatment option. However, this procedure was not without risk for anaphylaxis if the tattoo pigment were to be released into systemic circulation. Thus, at the first visit, ablation was performed on 3 test spots and the patient was prescribed cetirizine, diphenhydramine, and prophylactic prednisone for a few days. The patient then received a total of 5 fully ablative CO2 laser sessions (pulse energy: 200 mJ [15 J/cm2]; computerized pattern generator: 2-8-9 [85.2 J/cm2]; rate: 200 Hz [20 W], 3 passes) over 13 months to remove all visible blue ink in stages (Figure 3). Even with a shortened time course (as more time between laser sessions typically is preferred), the treatments were well tolerated with only mild hypertrophic scarring that responded to intralesional steroids (triamcinolone 10 mg/mL). On repeat skin biopsy during the treatment course, the superficial dermis demonstrated mostly scar tissue and near-total pigment removal—a 90% to 95% reduction in blue ink from prior biopsy—and minimal inflammation (Figure 4). Scant fine to coarse pigment deposition was seen in the deep dermis next to subcutaneous fat, which was unchanged from the previous biopsy. The patient’s ophthalmologic symptoms were tracked via improvement in intraocular pressure and stabilization of his vision, indicating rapid and complete resolution of the glaucoma after the last laser treatment. With resolution of his ocular symptoms, the patient was tapered off all immunosuppressant medications. The patient was lost to follow-up approximately 2 years after the final laser treatment.

Tattoo-associated uveitis initially was described in 1969 in 3 patients with light blue tattoos who developed tattoo granulomas and simultaneous uveitis. These cases were successfully treated with excision.3 Multiple cases have been reported since, often with bilateral uveitis and tattoos demonstrating noncaseating granulomatous inflammation that were treated with steroids.4 In 2018, a diagnosis of exclusion was proposed for uveitis associated with granulomatous tattoo reaction without sarcoidosis: tattoo granulomas with uveitis (TAGU).1
In this case, sarcoidosis initially was high on the differential diagnosis. Sarcoidosis is an immune-mediated systemic disease of unknown etiology characterized by the presence of widespread noncaseating epithelioid cell granulomas, primarily seen in the pulmonary and lymphatic systems. However, it often initially manifests with cutaneous involvement with noncaseating “naked” granulomas in the dermis and subcutaneous tissue. Although TAGU cases have demonstrated noncaseating granulomas in association with dermal tattoo pigment on histopathology,1,4 dermal lymphoplasmacytic inflammation with scattered foreign body giant cells was noted in our patient, which was more consistent with allergic contact dermatitis. Thus, it is important to consider that TAGU can be seen with varying histologic patterns. In patients with tattoos, sarcoidosis can manifest grossly as a papulonodular cutaneous reaction.5 Active smoking is associated with a decreased risk for sarcoidosis, and those who smoke are statistically more likely to have tattoos than the general population,6,7 so our patient’s smoking history may be relevant. However, sarcoidosis was an unlikely diagnosis due to the serum angiotensin-converting enzyme level; results of a chest radiograph (bilateral adenopathy and coarse reticular opacities) and computed tomography (hilar and mediastinal adenopathy); and nonsarcoidal histopathology.
An allergic reaction to tattoo ink is caused by a delayed-type hypersensitivity reaction to a pigment hapten that can develop abruptly months to years after tattoo placement—1 year after tattoo placement in our patient. This reaction was seen in our patient’s blue pigment tattoos, although it is more commonly seen in red pigment tattoos.8 Although the etiology of TAGU is poorly understood, it also is hypothesized to be a delayed-type hypersensitivity response to tattoo ink particles, suggested by the pattern of lymphocytes infiltrating the tattoo and atypical T-cell infiltrate on vitreous biopsy.9,10 Further research is required to elucidate the relationship between tattoos and uveitis.
Q-switched lasers (eg, 532-nm or 1064-nm Nd:YAG, alexandrite, or ruby lasers) are the standard treatment options for uncomplicated tattoo removal and employ a high-intensity, ultrashort pulse duration.11 However Q-switched lasers require multiple sessions and target pigment-containing cells, releasing the tattoo particles into systemic circulation, which can potentially induce a severe allergic response.12 In contrast, CO2 lasers use a different mechanism, emitting energy at a wavelength of 10,600 nm, which is absorbed by intracellular water and allows for the ablation of the superficial epidermis along with the embedded ink with subsequent re-epithelialization, as well as heat-mediated thermal injury to allow for dermal collagen remodeling.13 In a 2021 retrospective study of ablative laser therapy for allergic tattoo reactions, patients were treated with the 10,600-nm ablative CO2 laser and noted improvements in itching and burning with minimal adverse events.12 Although using a CO2 laser may not be considered a firstline treatment option for TAGU, the refractory clinical course and notable morbidity of surgical excision necessitated the use of ablative laser in our case.
Tattoo granulomas with uveitis is a rare diagnosis with the potential for serious permanent sequelae including blindness. Existing treatments such as topical and oral corticosteroids, immunosuppressants, surgical excision, and Q-switched lasers all are possible options, but in a patient with progressive ocular symptoms with other potential rheumatologic conditions and sarcoidosis ruled out, fully ablative CO2 laser may be an effective treatment option. Our case demonstrated the successful treatment of TAGU with CO2 laser ablation. Given the unclear etiology of TAGU and the limited evidence on treatment options and efficacy, our case contributes to the body of literature that can inform clinical management of this unusual and serious reaction.
- Kluger N. Tattoo-associated uveitis with or without systemic sarcoidosis: a comparative review of the literature. J Eur Acad Dermatol Venereol. 2018;32:1852-1861. doi:10.1111/jdv.15070
- Tiew S. Tattoo-associated panuveitis: a 10-year follow-up. Eur J Ophthalmol. 2019;29(1 suppl):18-21. doi:10.1177/1120672119846341
- Rorsman H, Brehmer-Andersson E, Dahlquist I, et al. Tattoo granuloma and uveitis. Lancet. 1969;2:27-28. doi:10.1016/s0140-6736(69)92600-2
- Ostheimer TA, Burkholder BM, Leung TG, et al. Tattoo-associated uveitis. Am J Ophthalmol. 2014;158:637-643.e1. doi:10.1016/j.ajo.2014.05.019
- Sepehri M, Hutton Carlsen K, Serup J. Papulo-nodular reactions in black tattoos as markers of sarcoidosis: study of 92 tattoo reactions from a hospital material. Dermatology. 2016;232:679-686. doi:10.1159/000453315
- Valeyre D, Prasse A, Nunes H, et al. Sarcoidosis. Lancet. 2014;383: 1155-1167. doi:10.1016/S0140-6736(13)60680-7
- Kluger N. Epidemiology of tattoos in industrialized countries. Curr Probl Dermatol. 2015;48:6-20. doi:10.1159/000369175
- Serup J, Hutton Carlsen K, Dommershausen N, et al. Identification of pigments related to allergic tattoo reactions in 104 human skin biopsies. Contact Dermatitis. 2020;82:73-82. doi:10.1111/cod.13423
- Mansour AM, Chan CC. Recurrent uveitis preceded by swelling of skin tattoos. Am J Ophthalmol. 1991;111:515-516. doi:10.1016/s0002-9394(14)72395-5
- Reddy AK, Shildkrot Y, Newman SA, et al. T-lymphocyte predominance and cellular atypia in tattoo-associated uveitis. JAMA Ophthalmol. 2015;133:1356-1357. doi:10.1001/jamaophthalmol.2015.3354
- Wenzel SM. Current concepts in laser tattoo removal. Skin Therapy Lett. 2010;15:3-5.
- van der Bent SAS, Huisman S, Rustemeyer T, et al. Ablative laser surgery for allergic tattoo reactions: a retrospective study. mLasers Med Sci. 2021;36:1241-1248. doi:10.1007/s10103-020-03164-2
- Yumeen S, Khan T. Laser carbon dioxide resurfacing. In: StatPearls. StatPearls Publishing; April 23, 2023. Accessed March 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK560544/
To the Editor:
Uveitis associated with tattoos is common, yet the etiology and optimal treatment options for this phenomenon remain unclear. Possible causes include a delayed hypersensitivity reaction to tattoo ink antigen or systemic sarcoidosis localized to the skin.1 Long-term treatment options include topical, intralesional, and systemic corticosteroids or immunosuppressants.2 Short-term options often include direct surgical excision and laser treatment. However, laser removal of tattoo pigment typically involves multiple sessions over the course of years, and there is a risk for antigen dispersal that may lead to anaphylaxis. Determining the most effective and safe treatment for a patient with progressive and severe ocular symptoms can be challenging. We describe a patient with cutaneous blue ink tattoos who developed chronic bilateral glaucoma, iritis, uveitis, and ocular hypertension that was refractory to multiple systemic medications and ophthalmologic procedures but responded to CO2 laser ablation.
A 27-year-old man with an active smoking history presented to our laser surgery center with a rash of approximately 4 years’ duration in areas with blue tattoo ink on both forearms. He was referred by his ophthalmologist due to bilateral uveitis and iritis and subsequent ocular hypertension and glaucoma that developed approximately 5 years after tattoo placement on the bilateral forearms. When the rash first appeared, the skin in the areas of the blue tattoo ink had hyperpigmented pruritic plaques. The patient was treated by a dermatologist with topical steroids to help reduce the itching and inflammation. Around the same time, he also started having ocular symptoms—vitreous floaters, erythema, eye pain, and blurriness—and was diagnosed with iritis of unclear etiology by ophthalmology. Figure 1 documents the patient’s clinical course. Due to escalating intraocular pressure and symptoms, he was referred to a glaucoma specialist and a rheumatologist. Systemic and rheumatologic medical conditions were ruled out with negative results on a series of blood tests (eg, rheumatoid factor, HLA-B27, antinuclear antibody, lysozyme, interferon gamma release assay, erythrocyte sedimentation rate, C-reactive protein, hepatitis B/C virus, Treponema pallidum, HIV), and magnetic resonance imaging of the brain was negative, ruling out demyelinating disease. Laboratory workup for sarcoidosis also was performed. The angiotensin-converting enzyme level was 30 U/L (reference range, 9-67 U/L), and a chest radiograph and computed tomography with contrast indicated no evidence of cardiopulmonary involvement. Although sarcoidosis could not be definitively ruled out, no other cause could be determined, and the patient’s glaucoma specialist diagnosed him with tattoo-associated uveitis. The patient was started on brimonidine, latanoprost, prednisolone, and dorzolamidetimolol eye drops, as well as acetazolamide (500 mg twice daily) and oral prednisone (various doses). Over the next 3 years, the patient continued to have symptoms, and immunosuppressant medications—methotrexate 20-25 mg weekly and adalimumab 40 mg every 2 weeks—were added to his treatment regimen. The patient also underwent bilateral ophthalmologic procedures, including a Baerveldt glaucoma implant procedure in the left eye and circumferential trabeculectomy in the right eye.
Despite these medications and procedures, the patient’s symptoms and intraocular pressure had not improved. At the current visit, punch biopsy of the tattooed skin and histologic examination showed dermal lymphoplasmacytic inflammation with scattered foreign-body giant cells associated with blue tattoo ink and overlying hyperkeratosis and spongiosis, consistent with allergic contact dermatitis (Figure 2). Because both immunosuppressant medications and ophthalmologic procedures had failed to control the progression of the ocular symptoms and the patient was at risk for permanent blindness, surgical excision and laser tattoo removal were considered as potential treatment options. Due to the large surface area and circumferential nature of the tattoos, there was a notable risk for disfiguring scars at both recipient and donor sites with surgical excision followed by graft placement. Thus, CO2 laser ablation was the preferred treatment option. However, this procedure was not without risk for anaphylaxis if the tattoo pigment were to be released into systemic circulation. Thus, at the first visit, ablation was performed on 3 test spots and the patient was prescribed cetirizine, diphenhydramine, and prophylactic prednisone for a few days. The patient then received a total of 5 fully ablative CO2 laser sessions (pulse energy: 200 mJ [15 J/cm2]; computerized pattern generator: 2-8-9 [85.2 J/cm2]; rate: 200 Hz [20 W], 3 passes) over 13 months to remove all visible blue ink in stages (Figure 3). Even with a shortened time course (as more time between laser sessions typically is preferred), the treatments were well tolerated with only mild hypertrophic scarring that responded to intralesional steroids (triamcinolone 10 mg/mL). On repeat skin biopsy during the treatment course, the superficial dermis demonstrated mostly scar tissue and near-total pigment removal—a 90% to 95% reduction in blue ink from prior biopsy—and minimal inflammation (Figure 4). Scant fine to coarse pigment deposition was seen in the deep dermis next to subcutaneous fat, which was unchanged from the previous biopsy. The patient’s ophthalmologic symptoms were tracked via improvement in intraocular pressure and stabilization of his vision, indicating rapid and complete resolution of the glaucoma after the last laser treatment. With resolution of his ocular symptoms, the patient was tapered off all immunosuppressant medications. The patient was lost to follow-up approximately 2 years after the final laser treatment.
Tattoo-associated uveitis initially was described in 1969 in 3 patients with light blue tattoos who developed tattoo granulomas and simultaneous uveitis. These cases were successfully treated with excision.3 Multiple cases have been reported since, often with bilateral uveitis and tattoos demonstrating noncaseating granulomatous inflammation that were treated with steroids.4 In 2018, a diagnosis of exclusion was proposed for uveitis associated with granulomatous tattoo reaction without sarcoidosis: tattoo granulomas with uveitis (TAGU).1
In this case, sarcoidosis initially was high on the differential diagnosis. Sarcoidosis is an immune-mediated systemic disease of unknown etiology characterized by the presence of widespread noncaseating epithelioid cell granulomas, primarily seen in the pulmonary and lymphatic systems. However, it often initially manifests with cutaneous involvement with noncaseating “naked” granulomas in the dermis and subcutaneous tissue. Although TAGU cases have demonstrated noncaseating granulomas in association with dermal tattoo pigment on histopathology,1,4 dermal lymphoplasmacytic inflammation with scattered foreign body giant cells was noted in our patient, which was more consistent with allergic contact dermatitis. Thus, it is important to consider that TAGU can be seen with varying histologic patterns. In patients with tattoos, sarcoidosis can manifest grossly as a papulonodular cutaneous reaction.5 Active smoking is associated with a decreased risk for sarcoidosis, and those who smoke are statistically more likely to have tattoos than the general population,6,7 so our patient’s smoking history may be relevant. However, sarcoidosis was an unlikely diagnosis due to the serum angiotensin-converting enzyme level; results of a chest radiograph (bilateral adenopathy and coarse reticular opacities) and computed tomography (hilar and mediastinal adenopathy); and nonsarcoidal histopathology.
An allergic reaction to tattoo ink is caused by a delayed-type hypersensitivity reaction to a pigment hapten that can develop abruptly months to years after tattoo placement—1 year after tattoo placement in our patient. This reaction was seen in our patient’s blue pigment tattoos, although it is more commonly seen in red pigment tattoos.8 Although the etiology of TAGU is poorly understood, it also is hypothesized to be a delayed-type hypersensitivity response to tattoo ink particles, suggested by the pattern of lymphocytes infiltrating the tattoo and atypical T-cell infiltrate on vitreous biopsy.9,10 Further research is required to elucidate the relationship between tattoos and uveitis.
Q-switched lasers (eg, 532-nm or 1064-nm Nd:YAG, alexandrite, or ruby lasers) are the standard treatment options for uncomplicated tattoo removal and employ a high-intensity, ultrashort pulse duration.11 However Q-switched lasers require multiple sessions and target pigment-containing cells, releasing the tattoo particles into systemic circulation, which can potentially induce a severe allergic response.12 In contrast, CO2 lasers use a different mechanism, emitting energy at a wavelength of 10,600 nm, which is absorbed by intracellular water and allows for the ablation of the superficial epidermis along with the embedded ink with subsequent re-epithelialization, as well as heat-mediated thermal injury to allow for dermal collagen remodeling.13 In a 2021 retrospective study of ablative laser therapy for allergic tattoo reactions, patients were treated with the 10,600-nm ablative CO2 laser and noted improvements in itching and burning with minimal adverse events.12 Although using a CO2 laser may not be considered a firstline treatment option for TAGU, the refractory clinical course and notable morbidity of surgical excision necessitated the use of ablative laser in our case.
Tattoo granulomas with uveitis is a rare diagnosis with the potential for serious permanent sequelae including blindness. Existing treatments such as topical and oral corticosteroids, immunosuppressants, surgical excision, and Q-switched lasers all are possible options, but in a patient with progressive ocular symptoms with other potential rheumatologic conditions and sarcoidosis ruled out, fully ablative CO2 laser may be an effective treatment option. Our case demonstrated the successful treatment of TAGU with CO2 laser ablation. Given the unclear etiology of TAGU and the limited evidence on treatment options and efficacy, our case contributes to the body of literature that can inform clinical management of this unusual and serious reaction.
To the Editor:
Uveitis associated with tattoos is common, yet the etiology and optimal treatment options for this phenomenon remain unclear. Possible causes include a delayed hypersensitivity reaction to tattoo ink antigen or systemic sarcoidosis localized to the skin.1 Long-term treatment options include topical, intralesional, and systemic corticosteroids or immunosuppressants.2 Short-term options often include direct surgical excision and laser treatment. However, laser removal of tattoo pigment typically involves multiple sessions over the course of years, and there is a risk for antigen dispersal that may lead to anaphylaxis. Determining the most effective and safe treatment for a patient with progressive and severe ocular symptoms can be challenging. We describe a patient with cutaneous blue ink tattoos who developed chronic bilateral glaucoma, iritis, uveitis, and ocular hypertension that was refractory to multiple systemic medications and ophthalmologic procedures but responded to CO2 laser ablation.
A 27-year-old man with an active smoking history presented to our laser surgery center with a rash of approximately 4 years’ duration in areas with blue tattoo ink on both forearms. He was referred by his ophthalmologist due to bilateral uveitis and iritis and subsequent ocular hypertension and glaucoma that developed approximately 5 years after tattoo placement on the bilateral forearms. When the rash first appeared, the skin in the areas of the blue tattoo ink had hyperpigmented pruritic plaques. The patient was treated by a dermatologist with topical steroids to help reduce the itching and inflammation. Around the same time, he also started having ocular symptoms—vitreous floaters, erythema, eye pain, and blurriness—and was diagnosed with iritis of unclear etiology by ophthalmology. Figure 1 documents the patient’s clinical course. Due to escalating intraocular pressure and symptoms, he was referred to a glaucoma specialist and a rheumatologist. Systemic and rheumatologic medical conditions were ruled out with negative results on a series of blood tests (eg, rheumatoid factor, HLA-B27, antinuclear antibody, lysozyme, interferon gamma release assay, erythrocyte sedimentation rate, C-reactive protein, hepatitis B/C virus, Treponema pallidum, HIV), and magnetic resonance imaging of the brain was negative, ruling out demyelinating disease. Laboratory workup for sarcoidosis also was performed. The angiotensin-converting enzyme level was 30 U/L (reference range, 9-67 U/L), and a chest radiograph and computed tomography with contrast indicated no evidence of cardiopulmonary involvement. Although sarcoidosis could not be definitively ruled out, no other cause could be determined, and the patient’s glaucoma specialist diagnosed him with tattoo-associated uveitis. The patient was started on brimonidine, latanoprost, prednisolone, and dorzolamidetimolol eye drops, as well as acetazolamide (500 mg twice daily) and oral prednisone (various doses). Over the next 3 years, the patient continued to have symptoms, and immunosuppressant medications—methotrexate 20-25 mg weekly and adalimumab 40 mg every 2 weeks—were added to his treatment regimen. The patient also underwent bilateral ophthalmologic procedures, including a Baerveldt glaucoma implant procedure in the left eye and circumferential trabeculectomy in the right eye.
Despite these medications and procedures, the patient’s symptoms and intraocular pressure had not improved. At the current visit, punch biopsy of the tattooed skin and histologic examination showed dermal lymphoplasmacytic inflammation with scattered foreign-body giant cells associated with blue tattoo ink and overlying hyperkeratosis and spongiosis, consistent with allergic contact dermatitis (Figure 2). Because both immunosuppressant medications and ophthalmologic procedures had failed to control the progression of the ocular symptoms and the patient was at risk for permanent blindness, surgical excision and laser tattoo removal were considered as potential treatment options. Due to the large surface area and circumferential nature of the tattoos, there was a notable risk for disfiguring scars at both recipient and donor sites with surgical excision followed by graft placement. Thus, CO2 laser ablation was the preferred treatment option. However, this procedure was not without risk for anaphylaxis if the tattoo pigment were to be released into systemic circulation. Thus, at the first visit, ablation was performed on 3 test spots and the patient was prescribed cetirizine, diphenhydramine, and prophylactic prednisone for a few days. The patient then received a total of 5 fully ablative CO2 laser sessions (pulse energy: 200 mJ [15 J/cm2]; computerized pattern generator: 2-8-9 [85.2 J/cm2]; rate: 200 Hz [20 W], 3 passes) over 13 months to remove all visible blue ink in stages (Figure 3). Even with a shortened time course (as more time between laser sessions typically is preferred), the treatments were well tolerated with only mild hypertrophic scarring that responded to intralesional steroids (triamcinolone 10 mg/mL). On repeat skin biopsy during the treatment course, the superficial dermis demonstrated mostly scar tissue and near-total pigment removal—a 90% to 95% reduction in blue ink from prior biopsy—and minimal inflammation (Figure 4). Scant fine to coarse pigment deposition was seen in the deep dermis next to subcutaneous fat, which was unchanged from the previous biopsy. The patient’s ophthalmologic symptoms were tracked via improvement in intraocular pressure and stabilization of his vision, indicating rapid and complete resolution of the glaucoma after the last laser treatment. With resolution of his ocular symptoms, the patient was tapered off all immunosuppressant medications. The patient was lost to follow-up approximately 2 years after the final laser treatment.
Tattoo-associated uveitis initially was described in 1969 in 3 patients with light blue tattoos who developed tattoo granulomas and simultaneous uveitis. These cases were successfully treated with excision.3 Multiple cases have been reported since, often with bilateral uveitis and tattoos demonstrating noncaseating granulomatous inflammation that were treated with steroids.4 In 2018, a diagnosis of exclusion was proposed for uveitis associated with granulomatous tattoo reaction without sarcoidosis: tattoo granulomas with uveitis (TAGU).1
In this case, sarcoidosis initially was high on the differential diagnosis. Sarcoidosis is an immune-mediated systemic disease of unknown etiology characterized by the presence of widespread noncaseating epithelioid cell granulomas, primarily seen in the pulmonary and lymphatic systems. However, it often initially manifests with cutaneous involvement with noncaseating “naked” granulomas in the dermis and subcutaneous tissue. Although TAGU cases have demonstrated noncaseating granulomas in association with dermal tattoo pigment on histopathology,1,4 dermal lymphoplasmacytic inflammation with scattered foreign body giant cells was noted in our patient, which was more consistent with allergic contact dermatitis. Thus, it is important to consider that TAGU can be seen with varying histologic patterns. In patients with tattoos, sarcoidosis can manifest grossly as a papulonodular cutaneous reaction.5 Active smoking is associated with a decreased risk for sarcoidosis, and those who smoke are statistically more likely to have tattoos than the general population,6,7 so our patient’s smoking history may be relevant. However, sarcoidosis was an unlikely diagnosis due to the serum angiotensin-converting enzyme level; results of a chest radiograph (bilateral adenopathy and coarse reticular opacities) and computed tomography (hilar and mediastinal adenopathy); and nonsarcoidal histopathology.
An allergic reaction to tattoo ink is caused by a delayed-type hypersensitivity reaction to a pigment hapten that can develop abruptly months to years after tattoo placement—1 year after tattoo placement in our patient. This reaction was seen in our patient’s blue pigment tattoos, although it is more commonly seen in red pigment tattoos.8 Although the etiology of TAGU is poorly understood, it also is hypothesized to be a delayed-type hypersensitivity response to tattoo ink particles, suggested by the pattern of lymphocytes infiltrating the tattoo and atypical T-cell infiltrate on vitreous biopsy.9,10 Further research is required to elucidate the relationship between tattoos and uveitis.
Q-switched lasers (eg, 532-nm or 1064-nm Nd:YAG, alexandrite, or ruby lasers) are the standard treatment options for uncomplicated tattoo removal and employ a high-intensity, ultrashort pulse duration.11 However Q-switched lasers require multiple sessions and target pigment-containing cells, releasing the tattoo particles into systemic circulation, which can potentially induce a severe allergic response.12 In contrast, CO2 lasers use a different mechanism, emitting energy at a wavelength of 10,600 nm, which is absorbed by intracellular water and allows for the ablation of the superficial epidermis along with the embedded ink with subsequent re-epithelialization, as well as heat-mediated thermal injury to allow for dermal collagen remodeling.13 In a 2021 retrospective study of ablative laser therapy for allergic tattoo reactions, patients were treated with the 10,600-nm ablative CO2 laser and noted improvements in itching and burning with minimal adverse events.12 Although using a CO2 laser may not be considered a firstline treatment option for TAGU, the refractory clinical course and notable morbidity of surgical excision necessitated the use of ablative laser in our case.
Tattoo granulomas with uveitis is a rare diagnosis with the potential for serious permanent sequelae including blindness. Existing treatments such as topical and oral corticosteroids, immunosuppressants, surgical excision, and Q-switched lasers all are possible options, but in a patient with progressive ocular symptoms with other potential rheumatologic conditions and sarcoidosis ruled out, fully ablative CO2 laser may be an effective treatment option. Our case demonstrated the successful treatment of TAGU with CO2 laser ablation. Given the unclear etiology of TAGU and the limited evidence on treatment options and efficacy, our case contributes to the body of literature that can inform clinical management of this unusual and serious reaction.
- Kluger N. Tattoo-associated uveitis with or without systemic sarcoidosis: a comparative review of the literature. J Eur Acad Dermatol Venereol. 2018;32:1852-1861. doi:10.1111/jdv.15070
- Tiew S. Tattoo-associated panuveitis: a 10-year follow-up. Eur J Ophthalmol. 2019;29(1 suppl):18-21. doi:10.1177/1120672119846341
- Rorsman H, Brehmer-Andersson E, Dahlquist I, et al. Tattoo granuloma and uveitis. Lancet. 1969;2:27-28. doi:10.1016/s0140-6736(69)92600-2
- Ostheimer TA, Burkholder BM, Leung TG, et al. Tattoo-associated uveitis. Am J Ophthalmol. 2014;158:637-643.e1. doi:10.1016/j.ajo.2014.05.019
- Sepehri M, Hutton Carlsen K, Serup J. Papulo-nodular reactions in black tattoos as markers of sarcoidosis: study of 92 tattoo reactions from a hospital material. Dermatology. 2016;232:679-686. doi:10.1159/000453315
- Valeyre D, Prasse A, Nunes H, et al. Sarcoidosis. Lancet. 2014;383: 1155-1167. doi:10.1016/S0140-6736(13)60680-7
- Kluger N. Epidemiology of tattoos in industrialized countries. Curr Probl Dermatol. 2015;48:6-20. doi:10.1159/000369175
- Serup J, Hutton Carlsen K, Dommershausen N, et al. Identification of pigments related to allergic tattoo reactions in 104 human skin biopsies. Contact Dermatitis. 2020;82:73-82. doi:10.1111/cod.13423
- Mansour AM, Chan CC. Recurrent uveitis preceded by swelling of skin tattoos. Am J Ophthalmol. 1991;111:515-516. doi:10.1016/s0002-9394(14)72395-5
- Reddy AK, Shildkrot Y, Newman SA, et al. T-lymphocyte predominance and cellular atypia in tattoo-associated uveitis. JAMA Ophthalmol. 2015;133:1356-1357. doi:10.1001/jamaophthalmol.2015.3354
- Wenzel SM. Current concepts in laser tattoo removal. Skin Therapy Lett. 2010;15:3-5.
- van der Bent SAS, Huisman S, Rustemeyer T, et al. Ablative laser surgery for allergic tattoo reactions: a retrospective study. mLasers Med Sci. 2021;36:1241-1248. doi:10.1007/s10103-020-03164-2
- Yumeen S, Khan T. Laser carbon dioxide resurfacing. In: StatPearls. StatPearls Publishing; April 23, 2023. Accessed March 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK560544/
- Kluger N. Tattoo-associated uveitis with or without systemic sarcoidosis: a comparative review of the literature. J Eur Acad Dermatol Venereol. 2018;32:1852-1861. doi:10.1111/jdv.15070
- Tiew S. Tattoo-associated panuveitis: a 10-year follow-up. Eur J Ophthalmol. 2019;29(1 suppl):18-21. doi:10.1177/1120672119846341
- Rorsman H, Brehmer-Andersson E, Dahlquist I, et al. Tattoo granuloma and uveitis. Lancet. 1969;2:27-28. doi:10.1016/s0140-6736(69)92600-2
- Ostheimer TA, Burkholder BM, Leung TG, et al. Tattoo-associated uveitis. Am J Ophthalmol. 2014;158:637-643.e1. doi:10.1016/j.ajo.2014.05.019
- Sepehri M, Hutton Carlsen K, Serup J. Papulo-nodular reactions in black tattoos as markers of sarcoidosis: study of 92 tattoo reactions from a hospital material. Dermatology. 2016;232:679-686. doi:10.1159/000453315
- Valeyre D, Prasse A, Nunes H, et al. Sarcoidosis. Lancet. 2014;383: 1155-1167. doi:10.1016/S0140-6736(13)60680-7
- Kluger N. Epidemiology of tattoos in industrialized countries. Curr Probl Dermatol. 2015;48:6-20. doi:10.1159/000369175
- Serup J, Hutton Carlsen K, Dommershausen N, et al. Identification of pigments related to allergic tattoo reactions in 104 human skin biopsies. Contact Dermatitis. 2020;82:73-82. doi:10.1111/cod.13423
- Mansour AM, Chan CC. Recurrent uveitis preceded by swelling of skin tattoos. Am J Ophthalmol. 1991;111:515-516. doi:10.1016/s0002-9394(14)72395-5
- Reddy AK, Shildkrot Y, Newman SA, et al. T-lymphocyte predominance and cellular atypia in tattoo-associated uveitis. JAMA Ophthalmol. 2015;133:1356-1357. doi:10.1001/jamaophthalmol.2015.3354
- Wenzel SM. Current concepts in laser tattoo removal. Skin Therapy Lett. 2010;15:3-5.
- van der Bent SAS, Huisman S, Rustemeyer T, et al. Ablative laser surgery for allergic tattoo reactions: a retrospective study. mLasers Med Sci. 2021;36:1241-1248. doi:10.1007/s10103-020-03164-2
- Yumeen S, Khan T. Laser carbon dioxide resurfacing. In: StatPearls. StatPearls Publishing; April 23, 2023. Accessed March 13, 2025. https://www.ncbi.nlm.nih.gov/books/NBK560544/
Tattoo Granulomas With Uveitis Successfully Treated With CO2 Laser Ablation
Tattoo Granulomas With Uveitis Successfully Treated With CO2 Laser Ablation
PRACTICE POINTS
- Dermatologists should be aware that uveitis can develop as a delayed hypersensitivity reaction to tattoo ink, particularly in patients with blue ink tattoos.
- It is important to rule out systemic conditions such as sarcoidosis in patients presenting with uveitis and a history of tattoos.
- In a patient with progressive ocular symptoms, carbon dioxide laser ablation may be an effective treatment option if other potential rheumatologic conditions and sarcoidosis have been ruled out and other therapies have not resulted in improvement of symptoms.
- Continuous monitoring of ocular symptoms and intraocular pressure is vital to prevent complications such as glaucoma and potential blindness.

Managing Contact Dermatitis Related to Amputee Care
Managing Contact Dermatitis Related to Amputee Care
Amputees who use prosthetic devices are particularly susceptible to contact dermatitis due to moisture, irritation, and prolonged contact with components of the device. Contact dermatitis accounts for approximately one-third of the dermatoses encountered by amputees who wear a prosthesis.1 Diagnosing allergic contact dermatitis (ACD) and irritant contact dermatitis (ICD) is challenging due to errors of omission from the differential and the substantial clinical overlap with other eczematous dermatoses. Diagnosis relies on patient history, clinical examination, exposure assessment, diagnostic testing, and a high index of suspicion. Conventionally, ACD comprises approximately 20% of all contact dermatitis cases, whereas ICD accounts for 80%.2 Symptoms vary between the 2 conditions, with pruritus more common in ACD and burning and soreness more common in ICD.3 Onset of dermatitis relative to exposure is crucial, with ICD often manifesting more quickly and ACD requiring an initial sensitization phase.4 Additionally, the complexity of ICD as a condition with variable features adds to the diagnostic difficulty, especially when allergens also have irritant effects.
Understanding these 2 primary types of contact dermatitis is crucial for effective management and prevention strategies in amputees who use prosthetics. In this article, we describe common causes of ACD and ICD related to amputee prosthetics and propose a tailored patch testing panel in order to better diagnose ACD in this patient population.
Allergic Contact Dermatitis
Allergic contact dermatitis occurs when the skin comes into contact with a substance to which the individual is sensitized. In amputees who use prosthetics, the socket and sock liner materials are frequent culprits for triggering allergic reactions. Components such as rubber, metals (eg, nickel), adhesives, and various plastic monomers can induce ACD in susceptible individuals. Additionally, chronic friction and sweat augment hapten penetration, increasing the risk of developing ACD.5
Contact allergens (typically small molecules under 500 Da) penetrate the skin, engage dendritic cells, activate T lymphocytes, and trigger the immune response and memory.6 The skin contains a substantial population of memory T cells, with CD8+ T cells in the epidermis and CD4+ T cells in the dermis, expressing markers that facilitate skin reactivity. The balance between effector and regulatory T cells, which can produce suppressive cytokines such as IL-10, promotes clinical tolerance to allergens such as nickel.
Textile-driven ACD presents with a distinct clinical pattern, often manifesting as patchy generalized dermatitis that coincides with sites where garments fit most snugly. This presentation can mimic other forms of dermatitis, such as nummular or asteatotic dermatitis. The skin beneath undergarments such as underwear or prosthetic socks may be spared, as these act as shields from contact allergens. Notably, the face and hands typically are spared unless the patient has a cross-reaction to formaldehyde-based preservatives found in personal care products.4
Allergy to Components of the Prosthetic Socket and Sock Liner
A prosthesis consists of several key components, including a socket, sleeve, liner, and stump shrinker (eFigure 1). The prosthetic socket, custom-made to fit the residual limb, is the upper part of the prosthesis, while the lower part consists of prosthetic components such as joints and terminal devices ordered to meet individual needs. Prosthetic sleeves provide suspension by securely holding the prosthetic limb in place, while liners offer cushioning and protection to the residual limb, enhancing comfort and reducing friction. Stump shrinkers aid in reducing swelling and shaping the residual limb, facilitating a better fit for the prosthetic socket. Together, these components work in harmony to optimize stability, comfort, and functionality for the user, enabling them to navigate daily activities with greater ease and confidence. Common allergens found in components of the socket and sock liner include rubbers and other elastomers, metals, plastics, adhesives, and textiles.

Rubbers and Other Elastomers—Consumables, including liners, knee sleeves, and socks, are tailored to each client and utilize materials such as silicone and natural and synthetic rubbers for comfort and secure fit. Allergic reactions to natural rubber latex, more commonly used in earlier prosthetics, are associated with both type I and type IV hypersensitivity reactions.4 Proteins inherent to natural rubber are overwhelmingly associated with an immediate urticarial eruption, whereas chemical additives used to produce latex are mostly linked to delayed hypersensitivity reactions, manifesting as allergic reactions ranging from mild itching to severe skin blistering.4
Vulcanization is the process of using heat and other accelerators to manufacture rubber. Common rubber accelerators include thiurams (the most common allergen associated with rubbers and other elastomers), carbamates/carba mix, 1,3-diphenylguanidine, and mercaptobenzothiazole.4 Thiourea is an implicated cause of ACD to neoprene rubber.7 These sensitizing chemicals are all included in the North American 80 Comprehensive Series; only thiuram mix, carba mix, and mercaptobenzothiazole are available in the T.R.U.E. TEST (SmartPractice). Sensitization often occurs due to repeated exposure, particularly in individuals who have undergone multiple prosthetic fittings. Many modern prospective liners utilize a medical-grade silicone as an elastomer for its high flexibility; silicone is considered biologically nonreactive and generally is considered a rare cause of ACD.8
Metals—Nickel, a ubiquitous allergen found in metal alloys used in prosthetic hardware, can cause localized itching, redness, and even blistering upon contact with the skin. Other metals, such as cobalt and chromium, also may trigger allergic reactions in susceptible individuals. Though many elastic fitting prosthetic socks contain silver fibers to reduce odors and friction-causing blisters, pure silver used in clothing or jewelry rarely causes dermatitis.4
Plastics and Adhesives—Leg prosthesis sockets typically are finished with the application of varnish, plastics, and/or resins—all potential allergens—to improve the appearance of the device and protect it from external agents.9 Polyester plastics themselves can cause ICD, only rarely leading to ACD.4 Incomplete curing during their manufacture may result in inadvertent exposure to epoxy resins or other phenol- formaldehyde resins such as 4-tert-butylcatechol and 4-tert-butylphenol formaldehyde, demonstrated causes of ACD in amputees.10 Adhesives used in sock liners or tapes to secure prosthetic devices can contain ingredients such as acrylates (a well-known cause of nail allergens) and other formaldehyde resins.4 Additionally, benzophenone commonly is added to paints and rubbers as a UV light absorber, reducing UV degradation and enhancing the material’s durability under light exposure.11
Textiles—Cotton, a common component in prosthetic sock liners, is almost 100% cellulose and typically does not cause ACD; however, synthetic fibers such as polypropylene and elastane (spandex) can elicit allergic reactions.4 Allergy to textiles often is driven by the chemicals used in the manufacturing process, particularly textile finishes, dyes, and formaldehyde resins, which are commonly used as fabric treatments. Disperse dyes are another common cause of allergic reactions. Para-phenylenediamine, a dye found in permanent hair dye and other darkly colored fabrics, is a potent sensitizer that may cross-react with other compounds that also contain similar amine groups, such as ester anesthetics, sunscreens containing para-aminobenzoic acid, other para dyes, and sulfonamides.12 Sweat can exacerbate these reactions by causing allergens to leach out of textiles, increasing skin exposure. Additionally, prosthetics containing leather may trigger allergies to potassium dichromate and other chromium compounds used in the leather-tanning process.12
Allergy to Personal Care Products
Skin protectants and prosthetic cleansers are crucial in dermatologic care for amputees, working together to safeguard the skin and maintain prosthetic hygiene. Skin protectants form a barrier against irritation, friction, and moisture, protecting the residual limb from damage and enhancing comfort and mobility. Meanwhile, prosthetic cleansers remove sweat, oils, and bacteria from the prosthetic socket, reducing the risk of infections and odors and ensuring the longevity and optimal function of the prosthetic device. Together, they support skin health, comfort, and overall quality of life for amputees.
The socket should be cleaned with warm water prior to use, but more importantly, immediately after removing the prosthesis. If cleaning products are used at night, residual haptens may remain on the device, increasing the risk of sensitization. Common contact irritants found in personal care products utilized in amputee care include sulfates, surfactants, preservatives, and fragrances (eTable 1).4 Additionally, common household cleaners and disinfectants can damage the prosthesis, leading to breakdown and the release of the monomers, precipitating ACD.
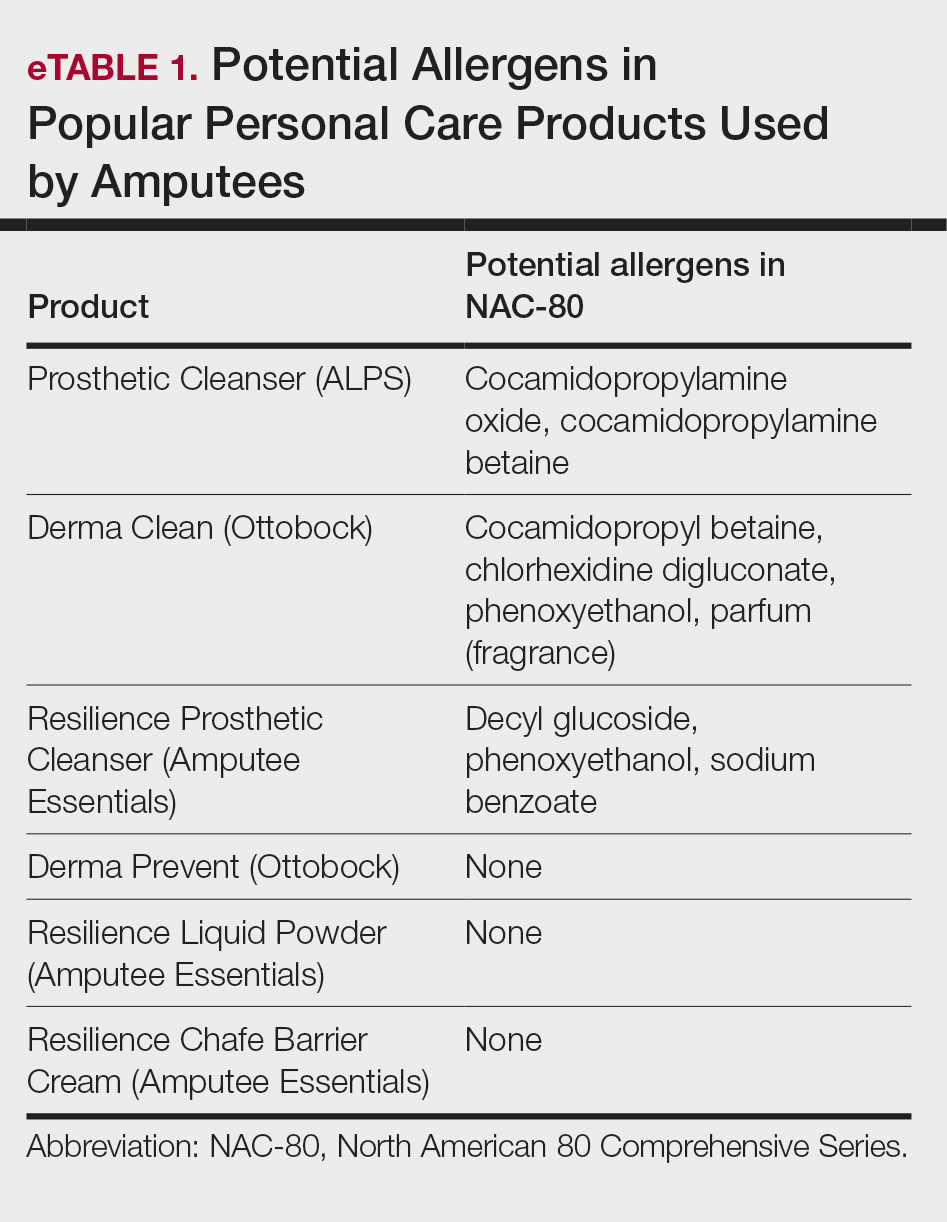
Patch Testing to Identify Causative Allergens
Patch testing is a valuable tool for identifying specific allergens responsible for ACD in amputees. This procedure involves applying small amounts of suspected allergens to the patient’s skin under occlusion and leaving the patches in place for 48 hours. After removal, the skin is assessed for reactions at 48 hours, with additional assessments conducted according to International Contact Dermatitis Research Group guidelines, typically at 72 and 96 hours, to identify delayed responses. This diagnostic approach helps pinpoint the substances to which the individual is allergic, enabling targeted avoidance strategies and treatment recommendations. Two widely used patch tests—the T.R.U.E. TEST, a preassembled patch test encompassing 35 allergens, and the North American 80 Comprehensive Series, which includes 80 allergens—demonstrate a sensitivity range between 70% and 80%.13,14 eTable 2 shows a recommended custom contact dermatitis panel to assess the most common causes of ACD related to amputee care.

Irritant Contact Dermatitis
Irritant contact dermatitis occurs when the skin’s protective barrier is damaged by repeated exposure to a particular irritant. In amputees, perspiration, friction, and pressure from prosthetic devices can exacerbate irritant reactions, leading to skin maceration, breakdown, and increased transepidermal penetration. Sweat accumulation within the prosthetic socket creates a moist environment conducive to ICD. The combination of sweat and friction can strip the skin of its natural oils, leading to dryness, chafing, and maceration. Continuous exposure to moisture also can exacerbate existing dermatitis and compromise skin integrity.4 Additionally, chronic irritation may increase transepidermal penetration of haptens, potentiating the development of ACD.15
Management of ICD in amputees involves a combination of treatments aimed at reducing friction, reducing sweating, and restoring barrier protection. Strategies to minimize mechanical trauma to the skin include ensuring proper socket fit, managing moisture, and protecting the skin. Using moisture-wicking sock liners and breathable prosthetic materials can help keep the skin dry. Topical antiperspirants containing aluminum chloride or similar compounds that help to block sweat glands often are the first line of treatment. Oral anticholinergics may be prescribed to reduce overall sweating, though they can have systemic side effects. Iontophoresis, a procedure where the affected area is exposed to a mild electrical current, can also be effective, especially for sweating of the hands and feet, though its application in amputees might be more limited.14
Recently, 2 treatments have emerged as options for managing excessive sweating (hyperhidrosis) in amputees: botulinum toxin injections and laser hair removal. By inhibiting the release of acetylcholine from sweat glands, botulinum toxin effectively reduces sweat production, thereby alleviating perspiration-induced skin irritation. Approximately 2 to 3 units of botulinum toxin at a dilution of 100 units in 1 mL of bacteriostatic saline 0.9% are injected transdermally at 1-cm intervals in a circumferential pattern on the skin covered by the prosthesis socket (typically a total of 300-500 units are utilized in the procedure)(eFigure 2).16 Laser hair removal can assist amputees with hyperhidrosis by reducing hair in the residual limb area, which decreases sweat retention and the potential for skin irritation due to friction.
Final Thoughts
In amputee dermatologic care, individuals with limb loss are particularly prone to contact dermatitis due to moisture, friction, and prolonged contact with prosthetic components. Diagnosing ACD and ICD is challenging due to overlapping symptoms and the potential for simultaneous occurrence. Distinguishing between these conditions is crucial for effective management. Understanding their causes, particularly in relation to prosthetic use, is essential for developing targeted prevention and treatment strategies, including the use of tailored patch testing panels to better diagnose ACD in amputees.
- Lyon CC, Kulkarni J, Zimersonc E, et al. Skin disorders in amputees. J Am Acad Dermatol. 2000;42:501-507.
- Bains SN, Nash P, Fonacier L. Irritant contact dermatitis. Clin Rev Allergy Immunol. 2018;56:99-109.
- Angelini G, Bonamonte D, Foti C, eds. Clinical Contact Dermatitis: A Practical Approach. Springer; 2021:57-92.
- Fisher AA, Rietschel RL, Fowler JF. Fisher’s Contact Dermatitis. BC Decker Inc; 2008.
- Johansen JD, Frosch PJ, Lepoittevin JP. Contact Dermatitis. Springer; 2010:43-90.
- Eisen HN, Orris L, Belman S. Elicitation of delayed allergic skin reactions with haptens: the dependence of elicitation on hapten combination with protein. J Exp Med. 1952;95:473-487.
- Johnson R. Wrist dermatitis: contact allergy to neoprene in a keyboard wrist rest. Am J Contact Dermat. 1997;8:172-174.
- Adams RM. Occupational Skin Disease. WB Saunders; 1999:501-551.
- Requena L, Vázquez F, Requena C, et al. Epoxy dermatitis of an amputation stump. Contact Dermatitis. 1986;14:320.
- Freeman S. Contact dermatitis of a limb stump caused by p-tertiary butyl catechol in the artificial limb. Contact Dermatitis. 1986;14:68-69.
- Heurung AR, Raju SI, Warshaw EM. Benzophenones. Dermatitis. 2014;25:3-10.
- Manneschi V, Palmerio B, Pauluzzi P, et al. Contact dermatitis from myoelectric prostheses. Contact Dermatitis. 1989;21:116-117.
- Heinrich D, Altmeyer P, Brasch J. “New” techniques for more sensitive patch testing? J Dtsch Dermatol Ges. 2011;9:889-896.
- James WD. Contact dermatitis update. Presented at: Walter Reed National Military Medical Center; April 18, 2024.
- Smith HR, Basketter DA, McFadden JP. Irritant dermatitis, irritancy and its role in allergic contact dermatitis. Clin Exp Dermatol. 2002;27:138-146.
- Lannan FM, Powell J, Kim GM, et al. Hyperhidrosis of the residual limb: a narrative review of the measurement and treatment of excess perspiration affecting individuals with amputation. Prosthet Orthot Int. 2021;45:477-486.
Amputees who use prosthetic devices are particularly susceptible to contact dermatitis due to moisture, irritation, and prolonged contact with components of the device. Contact dermatitis accounts for approximately one-third of the dermatoses encountered by amputees who wear a prosthesis.1 Diagnosing allergic contact dermatitis (ACD) and irritant contact dermatitis (ICD) is challenging due to errors of omission from the differential and the substantial clinical overlap with other eczematous dermatoses. Diagnosis relies on patient history, clinical examination, exposure assessment, diagnostic testing, and a high index of suspicion. Conventionally, ACD comprises approximately 20% of all contact dermatitis cases, whereas ICD accounts for 80%.2 Symptoms vary between the 2 conditions, with pruritus more common in ACD and burning and soreness more common in ICD.3 Onset of dermatitis relative to exposure is crucial, with ICD often manifesting more quickly and ACD requiring an initial sensitization phase.4 Additionally, the complexity of ICD as a condition with variable features adds to the diagnostic difficulty, especially when allergens also have irritant effects.
Understanding these 2 primary types of contact dermatitis is crucial for effective management and prevention strategies in amputees who use prosthetics. In this article, we describe common causes of ACD and ICD related to amputee prosthetics and propose a tailored patch testing panel in order to better diagnose ACD in this patient population.
Allergic Contact Dermatitis
Allergic contact dermatitis occurs when the skin comes into contact with a substance to which the individual is sensitized. In amputees who use prosthetics, the socket and sock liner materials are frequent culprits for triggering allergic reactions. Components such as rubber, metals (eg, nickel), adhesives, and various plastic monomers can induce ACD in susceptible individuals. Additionally, chronic friction and sweat augment hapten penetration, increasing the risk of developing ACD.5
Contact allergens (typically small molecules under 500 Da) penetrate the skin, engage dendritic cells, activate T lymphocytes, and trigger the immune response and memory.6 The skin contains a substantial population of memory T cells, with CD8+ T cells in the epidermis and CD4+ T cells in the dermis, expressing markers that facilitate skin reactivity. The balance between effector and regulatory T cells, which can produce suppressive cytokines such as IL-10, promotes clinical tolerance to allergens such as nickel.
Textile-driven ACD presents with a distinct clinical pattern, often manifesting as patchy generalized dermatitis that coincides with sites where garments fit most snugly. This presentation can mimic other forms of dermatitis, such as nummular or asteatotic dermatitis. The skin beneath undergarments such as underwear or prosthetic socks may be spared, as these act as shields from contact allergens. Notably, the face and hands typically are spared unless the patient has a cross-reaction to formaldehyde-based preservatives found in personal care products.4
Allergy to Components of the Prosthetic Socket and Sock Liner
A prosthesis consists of several key components, including a socket, sleeve, liner, and stump shrinker (eFigure 1). The prosthetic socket, custom-made to fit the residual limb, is the upper part of the prosthesis, while the lower part consists of prosthetic components such as joints and terminal devices ordered to meet individual needs. Prosthetic sleeves provide suspension by securely holding the prosthetic limb in place, while liners offer cushioning and protection to the residual limb, enhancing comfort and reducing friction. Stump shrinkers aid in reducing swelling and shaping the residual limb, facilitating a better fit for the prosthetic socket. Together, these components work in harmony to optimize stability, comfort, and functionality for the user, enabling them to navigate daily activities with greater ease and confidence. Common allergens found in components of the socket and sock liner include rubbers and other elastomers, metals, plastics, adhesives, and textiles.
Rubbers and Other Elastomers—Consumables, including liners, knee sleeves, and socks, are tailored to each client and utilize materials such as silicone and natural and synthetic rubbers for comfort and secure fit. Allergic reactions to natural rubber latex, more commonly used in earlier prosthetics, are associated with both type I and type IV hypersensitivity reactions.4 Proteins inherent to natural rubber are overwhelmingly associated with an immediate urticarial eruption, whereas chemical additives used to produce latex are mostly linked to delayed hypersensitivity reactions, manifesting as allergic reactions ranging from mild itching to severe skin blistering.4
Vulcanization is the process of using heat and other accelerators to manufacture rubber. Common rubber accelerators include thiurams (the most common allergen associated with rubbers and other elastomers), carbamates/carba mix, 1,3-diphenylguanidine, and mercaptobenzothiazole.4 Thiourea is an implicated cause of ACD to neoprene rubber.7 These sensitizing chemicals are all included in the North American 80 Comprehensive Series; only thiuram mix, carba mix, and mercaptobenzothiazole are available in the T.R.U.E. TEST (SmartPractice). Sensitization often occurs due to repeated exposure, particularly in individuals who have undergone multiple prosthetic fittings. Many modern prospective liners utilize a medical-grade silicone as an elastomer for its high flexibility; silicone is considered biologically nonreactive and generally is considered a rare cause of ACD.8
Metals—Nickel, a ubiquitous allergen found in metal alloys used in prosthetic hardware, can cause localized itching, redness, and even blistering upon contact with the skin. Other metals, such as cobalt and chromium, also may trigger allergic reactions in susceptible individuals. Though many elastic fitting prosthetic socks contain silver fibers to reduce odors and friction-causing blisters, pure silver used in clothing or jewelry rarely causes dermatitis.4
Plastics and Adhesives—Leg prosthesis sockets typically are finished with the application of varnish, plastics, and/or resins—all potential allergens—to improve the appearance of the device and protect it from external agents.9 Polyester plastics themselves can cause ICD, only rarely leading to ACD.4 Incomplete curing during their manufacture may result in inadvertent exposure to epoxy resins or other phenol- formaldehyde resins such as 4-tert-butylcatechol and 4-tert-butylphenol formaldehyde, demonstrated causes of ACD in amputees.10 Adhesives used in sock liners or tapes to secure prosthetic devices can contain ingredients such as acrylates (a well-known cause of nail allergens) and other formaldehyde resins.4 Additionally, benzophenone commonly is added to paints and rubbers as a UV light absorber, reducing UV degradation and enhancing the material’s durability under light exposure.11
Textiles—Cotton, a common component in prosthetic sock liners, is almost 100% cellulose and typically does not cause ACD; however, synthetic fibers such as polypropylene and elastane (spandex) can elicit allergic reactions.4 Allergy to textiles often is driven by the chemicals used in the manufacturing process, particularly textile finishes, dyes, and formaldehyde resins, which are commonly used as fabric treatments. Disperse dyes are another common cause of allergic reactions. Para-phenylenediamine, a dye found in permanent hair dye and other darkly colored fabrics, is a potent sensitizer that may cross-react with other compounds that also contain similar amine groups, such as ester anesthetics, sunscreens containing para-aminobenzoic acid, other para dyes, and sulfonamides.12 Sweat can exacerbate these reactions by causing allergens to leach out of textiles, increasing skin exposure. Additionally, prosthetics containing leather may trigger allergies to potassium dichromate and other chromium compounds used in the leather-tanning process.12
Allergy to Personal Care Products
Skin protectants and prosthetic cleansers are crucial in dermatologic care for amputees, working together to safeguard the skin and maintain prosthetic hygiene. Skin protectants form a barrier against irritation, friction, and moisture, protecting the residual limb from damage and enhancing comfort and mobility. Meanwhile, prosthetic cleansers remove sweat, oils, and bacteria from the prosthetic socket, reducing the risk of infections and odors and ensuring the longevity and optimal function of the prosthetic device. Together, they support skin health, comfort, and overall quality of life for amputees.
The socket should be cleaned with warm water prior to use, but more importantly, immediately after removing the prosthesis. If cleaning products are used at night, residual haptens may remain on the device, increasing the risk of sensitization. Common contact irritants found in personal care products utilized in amputee care include sulfates, surfactants, preservatives, and fragrances (eTable 1).4 Additionally, common household cleaners and disinfectants can damage the prosthesis, leading to breakdown and the release of the monomers, precipitating ACD.
Patch Testing to Identify Causative Allergens
Patch testing is a valuable tool for identifying specific allergens responsible for ACD in amputees. This procedure involves applying small amounts of suspected allergens to the patient’s skin under occlusion and leaving the patches in place for 48 hours. After removal, the skin is assessed for reactions at 48 hours, with additional assessments conducted according to International Contact Dermatitis Research Group guidelines, typically at 72 and 96 hours, to identify delayed responses. This diagnostic approach helps pinpoint the substances to which the individual is allergic, enabling targeted avoidance strategies and treatment recommendations. Two widely used patch tests—the T.R.U.E. TEST, a preassembled patch test encompassing 35 allergens, and the North American 80 Comprehensive Series, which includes 80 allergens—demonstrate a sensitivity range between 70% and 80%.13,14 eTable 2 shows a recommended custom contact dermatitis panel to assess the most common causes of ACD related to amputee care.
Irritant Contact Dermatitis
Irritant contact dermatitis occurs when the skin’s protective barrier is damaged by repeated exposure to a particular irritant. In amputees, perspiration, friction, and pressure from prosthetic devices can exacerbate irritant reactions, leading to skin maceration, breakdown, and increased transepidermal penetration. Sweat accumulation within the prosthetic socket creates a moist environment conducive to ICD. The combination of sweat and friction can strip the skin of its natural oils, leading to dryness, chafing, and maceration. Continuous exposure to moisture also can exacerbate existing dermatitis and compromise skin integrity.4 Additionally, chronic irritation may increase transepidermal penetration of haptens, potentiating the development of ACD.15
Management of ICD in amputees involves a combination of treatments aimed at reducing friction, reducing sweating, and restoring barrier protection. Strategies to minimize mechanical trauma to the skin include ensuring proper socket fit, managing moisture, and protecting the skin. Using moisture-wicking sock liners and breathable prosthetic materials can help keep the skin dry. Topical antiperspirants containing aluminum chloride or similar compounds that help to block sweat glands often are the first line of treatment. Oral anticholinergics may be prescribed to reduce overall sweating, though they can have systemic side effects. Iontophoresis, a procedure where the affected area is exposed to a mild electrical current, can also be effective, especially for sweating of the hands and feet, though its application in amputees might be more limited.14
Recently, 2 treatments have emerged as options for managing excessive sweating (hyperhidrosis) in amputees: botulinum toxin injections and laser hair removal. By inhibiting the release of acetylcholine from sweat glands, botulinum toxin effectively reduces sweat production, thereby alleviating perspiration-induced skin irritation. Approximately 2 to 3 units of botulinum toxin at a dilution of 100 units in 1 mL of bacteriostatic saline 0.9% are injected transdermally at 1-cm intervals in a circumferential pattern on the skin covered by the prosthesis socket (typically a total of 300-500 units are utilized in the procedure)(eFigure 2).16 Laser hair removal can assist amputees with hyperhidrosis by reducing hair in the residual limb area, which decreases sweat retention and the potential for skin irritation due to friction.
Final Thoughts
In amputee dermatologic care, individuals with limb loss are particularly prone to contact dermatitis due to moisture, friction, and prolonged contact with prosthetic components. Diagnosing ACD and ICD is challenging due to overlapping symptoms and the potential for simultaneous occurrence. Distinguishing between these conditions is crucial for effective management. Understanding their causes, particularly in relation to prosthetic use, is essential for developing targeted prevention and treatment strategies, including the use of tailored patch testing panels to better diagnose ACD in amputees.
Amputees who use prosthetic devices are particularly susceptible to contact dermatitis due to moisture, irritation, and prolonged contact with components of the device. Contact dermatitis accounts for approximately one-third of the dermatoses encountered by amputees who wear a prosthesis.1 Diagnosing allergic contact dermatitis (ACD) and irritant contact dermatitis (ICD) is challenging due to errors of omission from the differential and the substantial clinical overlap with other eczematous dermatoses. Diagnosis relies on patient history, clinical examination, exposure assessment, diagnostic testing, and a high index of suspicion. Conventionally, ACD comprises approximately 20% of all contact dermatitis cases, whereas ICD accounts for 80%.2 Symptoms vary between the 2 conditions, with pruritus more common in ACD and burning and soreness more common in ICD.3 Onset of dermatitis relative to exposure is crucial, with ICD often manifesting more quickly and ACD requiring an initial sensitization phase.4 Additionally, the complexity of ICD as a condition with variable features adds to the diagnostic difficulty, especially when allergens also have irritant effects.
Understanding these 2 primary types of contact dermatitis is crucial for effective management and prevention strategies in amputees who use prosthetics. In this article, we describe common causes of ACD and ICD related to amputee prosthetics and propose a tailored patch testing panel in order to better diagnose ACD in this patient population.
Allergic Contact Dermatitis
Allergic contact dermatitis occurs when the skin comes into contact with a substance to which the individual is sensitized. In amputees who use prosthetics, the socket and sock liner materials are frequent culprits for triggering allergic reactions. Components such as rubber, metals (eg, nickel), adhesives, and various plastic monomers can induce ACD in susceptible individuals. Additionally, chronic friction and sweat augment hapten penetration, increasing the risk of developing ACD.5
Contact allergens (typically small molecules under 500 Da) penetrate the skin, engage dendritic cells, activate T lymphocytes, and trigger the immune response and memory.6 The skin contains a substantial population of memory T cells, with CD8+ T cells in the epidermis and CD4+ T cells in the dermis, expressing markers that facilitate skin reactivity. The balance between effector and regulatory T cells, which can produce suppressive cytokines such as IL-10, promotes clinical tolerance to allergens such as nickel.
Textile-driven ACD presents with a distinct clinical pattern, often manifesting as patchy generalized dermatitis that coincides with sites where garments fit most snugly. This presentation can mimic other forms of dermatitis, such as nummular or asteatotic dermatitis. The skin beneath undergarments such as underwear or prosthetic socks may be spared, as these act as shields from contact allergens. Notably, the face and hands typically are spared unless the patient has a cross-reaction to formaldehyde-based preservatives found in personal care products.4
Allergy to Components of the Prosthetic Socket and Sock Liner
A prosthesis consists of several key components, including a socket, sleeve, liner, and stump shrinker (eFigure 1). The prosthetic socket, custom-made to fit the residual limb, is the upper part of the prosthesis, while the lower part consists of prosthetic components such as joints and terminal devices ordered to meet individual needs. Prosthetic sleeves provide suspension by securely holding the prosthetic limb in place, while liners offer cushioning and protection to the residual limb, enhancing comfort and reducing friction. Stump shrinkers aid in reducing swelling and shaping the residual limb, facilitating a better fit for the prosthetic socket. Together, these components work in harmony to optimize stability, comfort, and functionality for the user, enabling them to navigate daily activities with greater ease and confidence. Common allergens found in components of the socket and sock liner include rubbers and other elastomers, metals, plastics, adhesives, and textiles.
Rubbers and Other Elastomers—Consumables, including liners, knee sleeves, and socks, are tailored to each client and utilize materials such as silicone and natural and synthetic rubbers for comfort and secure fit. Allergic reactions to natural rubber latex, more commonly used in earlier prosthetics, are associated with both type I and type IV hypersensitivity reactions.4 Proteins inherent to natural rubber are overwhelmingly associated with an immediate urticarial eruption, whereas chemical additives used to produce latex are mostly linked to delayed hypersensitivity reactions, manifesting as allergic reactions ranging from mild itching to severe skin blistering.4
Vulcanization is the process of using heat and other accelerators to manufacture rubber. Common rubber accelerators include thiurams (the most common allergen associated with rubbers and other elastomers), carbamates/carba mix, 1,3-diphenylguanidine, and mercaptobenzothiazole.4 Thiourea is an implicated cause of ACD to neoprene rubber.7 These sensitizing chemicals are all included in the North American 80 Comprehensive Series; only thiuram mix, carba mix, and mercaptobenzothiazole are available in the T.R.U.E. TEST (SmartPractice). Sensitization often occurs due to repeated exposure, particularly in individuals who have undergone multiple prosthetic fittings. Many modern prospective liners utilize a medical-grade silicone as an elastomer for its high flexibility; silicone is considered biologically nonreactive and generally is considered a rare cause of ACD.8
Metals—Nickel, a ubiquitous allergen found in metal alloys used in prosthetic hardware, can cause localized itching, redness, and even blistering upon contact with the skin. Other metals, such as cobalt and chromium, also may trigger allergic reactions in susceptible individuals. Though many elastic fitting prosthetic socks contain silver fibers to reduce odors and friction-causing blisters, pure silver used in clothing or jewelry rarely causes dermatitis.4
Plastics and Adhesives—Leg prosthesis sockets typically are finished with the application of varnish, plastics, and/or resins—all potential allergens—to improve the appearance of the device and protect it from external agents.9 Polyester plastics themselves can cause ICD, only rarely leading to ACD.4 Incomplete curing during their manufacture may result in inadvertent exposure to epoxy resins or other phenol- formaldehyde resins such as 4-tert-butylcatechol and 4-tert-butylphenol formaldehyde, demonstrated causes of ACD in amputees.10 Adhesives used in sock liners or tapes to secure prosthetic devices can contain ingredients such as acrylates (a well-known cause of nail allergens) and other formaldehyde resins.4 Additionally, benzophenone commonly is added to paints and rubbers as a UV light absorber, reducing UV degradation and enhancing the material’s durability under light exposure.11
Textiles—Cotton, a common component in prosthetic sock liners, is almost 100% cellulose and typically does not cause ACD; however, synthetic fibers such as polypropylene and elastane (spandex) can elicit allergic reactions.4 Allergy to textiles often is driven by the chemicals used in the manufacturing process, particularly textile finishes, dyes, and formaldehyde resins, which are commonly used as fabric treatments. Disperse dyes are another common cause of allergic reactions. Para-phenylenediamine, a dye found in permanent hair dye and other darkly colored fabrics, is a potent sensitizer that may cross-react with other compounds that also contain similar amine groups, such as ester anesthetics, sunscreens containing para-aminobenzoic acid, other para dyes, and sulfonamides.12 Sweat can exacerbate these reactions by causing allergens to leach out of textiles, increasing skin exposure. Additionally, prosthetics containing leather may trigger allergies to potassium dichromate and other chromium compounds used in the leather-tanning process.12
Allergy to Personal Care Products
Skin protectants and prosthetic cleansers are crucial in dermatologic care for amputees, working together to safeguard the skin and maintain prosthetic hygiene. Skin protectants form a barrier against irritation, friction, and moisture, protecting the residual limb from damage and enhancing comfort and mobility. Meanwhile, prosthetic cleansers remove sweat, oils, and bacteria from the prosthetic socket, reducing the risk of infections and odors and ensuring the longevity and optimal function of the prosthetic device. Together, they support skin health, comfort, and overall quality of life for amputees.
The socket should be cleaned with warm water prior to use, but more importantly, immediately after removing the prosthesis. If cleaning products are used at night, residual haptens may remain on the device, increasing the risk of sensitization. Common contact irritants found in personal care products utilized in amputee care include sulfates, surfactants, preservatives, and fragrances (eTable 1).4 Additionally, common household cleaners and disinfectants can damage the prosthesis, leading to breakdown and the release of the monomers, precipitating ACD.
Patch Testing to Identify Causative Allergens
Patch testing is a valuable tool for identifying specific allergens responsible for ACD in amputees. This procedure involves applying small amounts of suspected allergens to the patient’s skin under occlusion and leaving the patches in place for 48 hours. After removal, the skin is assessed for reactions at 48 hours, with additional assessments conducted according to International Contact Dermatitis Research Group guidelines, typically at 72 and 96 hours, to identify delayed responses. This diagnostic approach helps pinpoint the substances to which the individual is allergic, enabling targeted avoidance strategies and treatment recommendations. Two widely used patch tests—the T.R.U.E. TEST, a preassembled patch test encompassing 35 allergens, and the North American 80 Comprehensive Series, which includes 80 allergens—demonstrate a sensitivity range between 70% and 80%.13,14 eTable 2 shows a recommended custom contact dermatitis panel to assess the most common causes of ACD related to amputee care.
Irritant Contact Dermatitis
Irritant contact dermatitis occurs when the skin’s protective barrier is damaged by repeated exposure to a particular irritant. In amputees, perspiration, friction, and pressure from prosthetic devices can exacerbate irritant reactions, leading to skin maceration, breakdown, and increased transepidermal penetration. Sweat accumulation within the prosthetic socket creates a moist environment conducive to ICD. The combination of sweat and friction can strip the skin of its natural oils, leading to dryness, chafing, and maceration. Continuous exposure to moisture also can exacerbate existing dermatitis and compromise skin integrity.4 Additionally, chronic irritation may increase transepidermal penetration of haptens, potentiating the development of ACD.15
Management of ICD in amputees involves a combination of treatments aimed at reducing friction, reducing sweating, and restoring barrier protection. Strategies to minimize mechanical trauma to the skin include ensuring proper socket fit, managing moisture, and protecting the skin. Using moisture-wicking sock liners and breathable prosthetic materials can help keep the skin dry. Topical antiperspirants containing aluminum chloride or similar compounds that help to block sweat glands often are the first line of treatment. Oral anticholinergics may be prescribed to reduce overall sweating, though they can have systemic side effects. Iontophoresis, a procedure where the affected area is exposed to a mild electrical current, can also be effective, especially for sweating of the hands and feet, though its application in amputees might be more limited.14
Recently, 2 treatments have emerged as options for managing excessive sweating (hyperhidrosis) in amputees: botulinum toxin injections and laser hair removal. By inhibiting the release of acetylcholine from sweat glands, botulinum toxin effectively reduces sweat production, thereby alleviating perspiration-induced skin irritation. Approximately 2 to 3 units of botulinum toxin at a dilution of 100 units in 1 mL of bacteriostatic saline 0.9% are injected transdermally at 1-cm intervals in a circumferential pattern on the skin covered by the prosthesis socket (typically a total of 300-500 units are utilized in the procedure)(eFigure 2).16 Laser hair removal can assist amputees with hyperhidrosis by reducing hair in the residual limb area, which decreases sweat retention and the potential for skin irritation due to friction.
Final Thoughts
In amputee dermatologic care, individuals with limb loss are particularly prone to contact dermatitis due to moisture, friction, and prolonged contact with prosthetic components. Diagnosing ACD and ICD is challenging due to overlapping symptoms and the potential for simultaneous occurrence. Distinguishing between these conditions is crucial for effective management. Understanding their causes, particularly in relation to prosthetic use, is essential for developing targeted prevention and treatment strategies, including the use of tailored patch testing panels to better diagnose ACD in amputees.
- Lyon CC, Kulkarni J, Zimersonc E, et al. Skin disorders in amputees. J Am Acad Dermatol. 2000;42:501-507.
- Bains SN, Nash P, Fonacier L. Irritant contact dermatitis. Clin Rev Allergy Immunol. 2018;56:99-109.
- Angelini G, Bonamonte D, Foti C, eds. Clinical Contact Dermatitis: A Practical Approach. Springer; 2021:57-92.
- Fisher AA, Rietschel RL, Fowler JF. Fisher’s Contact Dermatitis. BC Decker Inc; 2008.
- Johansen JD, Frosch PJ, Lepoittevin JP. Contact Dermatitis. Springer; 2010:43-90.
- Eisen HN, Orris L, Belman S. Elicitation of delayed allergic skin reactions with haptens: the dependence of elicitation on hapten combination with protein. J Exp Med. 1952;95:473-487.
- Johnson R. Wrist dermatitis: contact allergy to neoprene in a keyboard wrist rest. Am J Contact Dermat. 1997;8:172-174.
- Adams RM. Occupational Skin Disease. WB Saunders; 1999:501-551.
- Requena L, Vázquez F, Requena C, et al. Epoxy dermatitis of an amputation stump. Contact Dermatitis. 1986;14:320.
- Freeman S. Contact dermatitis of a limb stump caused by p-tertiary butyl catechol in the artificial limb. Contact Dermatitis. 1986;14:68-69.
- Heurung AR, Raju SI, Warshaw EM. Benzophenones. Dermatitis. 2014;25:3-10.
- Manneschi V, Palmerio B, Pauluzzi P, et al. Contact dermatitis from myoelectric prostheses. Contact Dermatitis. 1989;21:116-117.
- Heinrich D, Altmeyer P, Brasch J. “New” techniques for more sensitive patch testing? J Dtsch Dermatol Ges. 2011;9:889-896.
- James WD. Contact dermatitis update. Presented at: Walter Reed National Military Medical Center; April 18, 2024.
- Smith HR, Basketter DA, McFadden JP. Irritant dermatitis, irritancy and its role in allergic contact dermatitis. Clin Exp Dermatol. 2002;27:138-146.
- Lannan FM, Powell J, Kim GM, et al. Hyperhidrosis of the residual limb: a narrative review of the measurement and treatment of excess perspiration affecting individuals with amputation. Prosthet Orthot Int. 2021;45:477-486.
- Lyon CC, Kulkarni J, Zimersonc E, et al. Skin disorders in amputees. J Am Acad Dermatol. 2000;42:501-507.
- Bains SN, Nash P, Fonacier L. Irritant contact dermatitis. Clin Rev Allergy Immunol. 2018;56:99-109.
- Angelini G, Bonamonte D, Foti C, eds. Clinical Contact Dermatitis: A Practical Approach. Springer; 2021:57-92.
- Fisher AA, Rietschel RL, Fowler JF. Fisher’s Contact Dermatitis. BC Decker Inc; 2008.
- Johansen JD, Frosch PJ, Lepoittevin JP. Contact Dermatitis. Springer; 2010:43-90.
- Eisen HN, Orris L, Belman S. Elicitation of delayed allergic skin reactions with haptens: the dependence of elicitation on hapten combination with protein. J Exp Med. 1952;95:473-487.
- Johnson R. Wrist dermatitis: contact allergy to neoprene in a keyboard wrist rest. Am J Contact Dermat. 1997;8:172-174.
- Adams RM. Occupational Skin Disease. WB Saunders; 1999:501-551.
- Requena L, Vázquez F, Requena C, et al. Epoxy dermatitis of an amputation stump. Contact Dermatitis. 1986;14:320.
- Freeman S. Contact dermatitis of a limb stump caused by p-tertiary butyl catechol in the artificial limb. Contact Dermatitis. 1986;14:68-69.
- Heurung AR, Raju SI, Warshaw EM. Benzophenones. Dermatitis. 2014;25:3-10.
- Manneschi V, Palmerio B, Pauluzzi P, et al. Contact dermatitis from myoelectric prostheses. Contact Dermatitis. 1989;21:116-117.
- Heinrich D, Altmeyer P, Brasch J. “New” techniques for more sensitive patch testing? J Dtsch Dermatol Ges. 2011;9:889-896.
- James WD. Contact dermatitis update. Presented at: Walter Reed National Military Medical Center; April 18, 2024.
- Smith HR, Basketter DA, McFadden JP. Irritant dermatitis, irritancy and its role in allergic contact dermatitis. Clin Exp Dermatol. 2002;27:138-146.
- Lannan FM, Powell J, Kim GM, et al. Hyperhidrosis of the residual limb: a narrative review of the measurement and treatment of excess perspiration affecting individuals with amputation. Prosthet Orthot Int. 2021;45:477-486.
Managing Contact Dermatitis Related to Amputee Care
Managing Contact Dermatitis Related to Amputee Care
PRACTICE POINTS
- Incorporating a tailored patch testing panel that includes common prosthetic-related allergens (eg, rubber, metals, adhesives) can greatly improve the diagnosis and treatment of allergic vs irritant contact dermatitis in amputees.
- Effective management of irritant contact dermatitis in amputees involves reducing moisture and friction in the prosthetic socket with moisture-wicking liners, ensuring proper fit, and utilizing treatments such as topical antiperspirants and botulinum toxin injections.

Pruritus: Diagnosing and Treating Older Adults
Chronic pruritus is a common problem among older individuals. During a session at the Dermatology Days of Paris 2024 conference dedicated to general practitioners, Juliette Delaunay, MD, a dermatologist and venereologist at Angers University Hospital Center in Angers, France, and Gabrielle Lisembard, MD, a general practitioner in the French town Grand-Fort-Philippe, discussed diagnostic approaches and key principles for the therapeutic management of pruritus.
Identifying Causes
“Pruritus in older people is most often linked to physiological changes in the skin caused by aging, leading to significant xerosis. However, before attributing it to aging, we need to rule out several causes,” Delaunay noted.
Beyond simple aging, one must consider autoimmune bullous dermatoses (bullous pemphigoid), drug-related causes, metabolic disorders (can occur at any age), cutaneous T-cell lymphomas, scabies, lice, and HIV infection.
Senile Pruritus
Aging-related xerosis can cause senile pruritus, often presenting as itching with scratch marks and dry skin. “This is a diagnosis of exclusion,” Delaunay insisted.
In older individuals with pruritus, initial examinations should include complete blood cell count (CBC), liver function tests, and thyroid-stimulating hormone levels. Syphilis serology, HIV testing, and beta-2 microglobulin levels are secondary evaluations. Renal function analysis may also be performed, and imaging may be required to investigate neoplasia.
“Annual etiological reassessment is essential if the initial evaluation is negative, as patients may later develop or report a neoplasia or hematological disorder,” Delaunay emphasized.
Paraneoplastic pruritus can occur, particularly those linked to hematological disorders (lymphomas, polycythemia, or myeloma).
Bullous Pemphigoid
Bullous pemphigoid often begins with pruritus, which can be severe and lead to insomnia. General practitioners should consider bullous pemphigoids when there is a bullous rash (tense blisters with citrine content) or an urticarial or chronic eczematous rash that does not heal spontaneously within a few days. The first-line biologic test to confirm the diagnosis is the CBC, which may reveal significant hypereosinophilia.
The diagnosis is confirmed by a skin biopsy showing a subepidermal blister with a preserved roof, unlike intraepidermal dermatoses, where the roof ruptures.
Direct immunofluorescence revealed deposits of immunoglobulin G antibodies along the dermoepidermal junction.
Approximately 40% of cases of bullous pemphigoid are associated with neurodegenerative diseases, such as stroke, parkinsonism, or dementia syndromes — occurring at a rate two to three times higher than in the general population.
It’s important to identify drugs that induce bullous pemphigoid, such as gliptins, anti-programmed cell death protein 1-programmed death-ligand 1 agents, loop diuretics (furosemide and bumetanide), anti-aldosterones (spironolactone), antiarrhythmics (amiodarone), and neuroleptics (phenothiazines).
“Stopping the medication is not mandatory if the bullous pemphigoid is well controlled by local or systemic treatments and the medication is essential. The decision to stop should be made on a case-by-case basis in consultation with the treating specialist,” Delaunay emphasized.
Treatment consists of very strong local corticosteroid therapy as the first-line treatment. If ineffective, systemic treatments based on methotrexate, oral corticosteroids, or immunomodulatory agents may be considered. Hospitalization is sometimes required.
Drug-Induced Pruritus
Drug-induced pruritus is common because older individuals often take multiple medications (antihypertensives, statins, oral hypoglycemics, psychotropic drugs, antiarrhythmics, etc.). “Sometimes, drug-induced pruritus can occur even if the medication was started several months or years ago,” Delaunay emphasized.
The lesions are generally nonspecific and scratching.
“This is a diagnosis of exclusion for other causes of pruritus. In the absence of specific lesions pointing to a dermatosis, eviction/reintroduction tests with treatments should be conducted one by one, which can be quite lengthy,” she explained.
Awareness for Scabies
Delaunay reminded attendees to consider scabies in older individuals when classic signs of pruritus flare up at night, with a rash affecting the face, scabs, or vesicles in the interdigital spaces of the hands, wrists, scrotal area, or the peri-mammary region.
“The incidence is increasing, particularly in nursing homes, where outbreaks pose a significant risk of rapid spread. Treatment involves three courses of topical and oral treatments administered on days 0, 7, and 14. All contact cases must also be treated. Sometimes, these thick lesions are stripped with 10% salicylated petroleum jelly. Environmental treatment with acaricides is essential, along with strict isolation measures,” Delaunay emphasized.
Adherent nits on the scalp or other hairy areas should raise suspicion of pediculosis.
Neurogenic and Psychogenic Origins
Neurogenic pruritus can occur during a stroke, presenting as contralateral pruritus, or in the presence of a brain tumor or following neurosurgery. Opioid-containing medications may also induce neurogenic pruritus.
The presence of unilateral painful or itchy sensations should prompt the investigation of shingles in older individuals.
Psychogenic pruritus is also common and can arise in the context of psychosis with parasitophobia or as part of anxiety-depression syndromes.
Supportive Measures
For managing pruritus, it is essential to:
- Keep nails trimmed short
- Wash with cold or lukewarm water
- Use lipid-rice soaps and syndets
- Avoid irritants, including antiseptics, cologne, no-rinse cleansers, and steroidal or nonsteroidal anti-inflammatory drugs
- Limit bathing frequency
- Avoid wearing nylon, wool, or tight clothing
- Minimize exposure to heat and excessive heating
“Alternatives to scratching, such as applying a moisturizing emollient, can be beneficial and may have a placebo effect,” explained the dermatologist. She further emphasized that local corticosteroids are effective only in the presence of inflammatory dermatosis and should not be applied to healthy skin. Similarly, antihistamines should only be prescribed if the pruritus is histamine-mediated.
Capsaicin may be useful in the treatment of localized neuropathic pruritus.
In cases of neurogenic pruritus, gabapentin and pregabalin may be prescribed, but tolerance can be problematic at this age. Other measures include acupuncture, cryotherapy, relaxation, hypnosis, psychotherapy, and music therapy. In cases of repeated therapeutic failure, patients may be treated with biotherapy (dupilumab) by a dermatologist.
This story was translated from Medscape’s French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Chronic pruritus is a common problem among older individuals. During a session at the Dermatology Days of Paris 2024 conference dedicated to general practitioners, Juliette Delaunay, MD, a dermatologist and venereologist at Angers University Hospital Center in Angers, France, and Gabrielle Lisembard, MD, a general practitioner in the French town Grand-Fort-Philippe, discussed diagnostic approaches and key principles for the therapeutic management of pruritus.
Identifying Causes
“Pruritus in older people is most often linked to physiological changes in the skin caused by aging, leading to significant xerosis. However, before attributing it to aging, we need to rule out several causes,” Delaunay noted.
Beyond simple aging, one must consider autoimmune bullous dermatoses (bullous pemphigoid), drug-related causes, metabolic disorders (can occur at any age), cutaneous T-cell lymphomas, scabies, lice, and HIV infection.
Senile Pruritus
Aging-related xerosis can cause senile pruritus, often presenting as itching with scratch marks and dry skin. “This is a diagnosis of exclusion,” Delaunay insisted.
In older individuals with pruritus, initial examinations should include complete blood cell count (CBC), liver function tests, and thyroid-stimulating hormone levels. Syphilis serology, HIV testing, and beta-2 microglobulin levels are secondary evaluations. Renal function analysis may also be performed, and imaging may be required to investigate neoplasia.
“Annual etiological reassessment is essential if the initial evaluation is negative, as patients may later develop or report a neoplasia or hematological disorder,” Delaunay emphasized.
Paraneoplastic pruritus can occur, particularly those linked to hematological disorders (lymphomas, polycythemia, or myeloma).
Bullous Pemphigoid
Bullous pemphigoid often begins with pruritus, which can be severe and lead to insomnia. General practitioners should consider bullous pemphigoids when there is a bullous rash (tense blisters with citrine content) or an urticarial or chronic eczematous rash that does not heal spontaneously within a few days. The first-line biologic test to confirm the diagnosis is the CBC, which may reveal significant hypereosinophilia.
The diagnosis is confirmed by a skin biopsy showing a subepidermal blister with a preserved roof, unlike intraepidermal dermatoses, where the roof ruptures.
Direct immunofluorescence revealed deposits of immunoglobulin G antibodies along the dermoepidermal junction.
Approximately 40% of cases of bullous pemphigoid are associated with neurodegenerative diseases, such as stroke, parkinsonism, or dementia syndromes — occurring at a rate two to three times higher than in the general population.
It’s important to identify drugs that induce bullous pemphigoid, such as gliptins, anti-programmed cell death protein 1-programmed death-ligand 1 agents, loop diuretics (furosemide and bumetanide), anti-aldosterones (spironolactone), antiarrhythmics (amiodarone), and neuroleptics (phenothiazines).
“Stopping the medication is not mandatory if the bullous pemphigoid is well controlled by local or systemic treatments and the medication is essential. The decision to stop should be made on a case-by-case basis in consultation with the treating specialist,” Delaunay emphasized.
Treatment consists of very strong local corticosteroid therapy as the first-line treatment. If ineffective, systemic treatments based on methotrexate, oral corticosteroids, or immunomodulatory agents may be considered. Hospitalization is sometimes required.
Drug-Induced Pruritus
Drug-induced pruritus is common because older individuals often take multiple medications (antihypertensives, statins, oral hypoglycemics, psychotropic drugs, antiarrhythmics, etc.). “Sometimes, drug-induced pruritus can occur even if the medication was started several months or years ago,” Delaunay emphasized.
The lesions are generally nonspecific and scratching.
“This is a diagnosis of exclusion for other causes of pruritus. In the absence of specific lesions pointing to a dermatosis, eviction/reintroduction tests with treatments should be conducted one by one, which can be quite lengthy,” she explained.
Awareness for Scabies
Delaunay reminded attendees to consider scabies in older individuals when classic signs of pruritus flare up at night, with a rash affecting the face, scabs, or vesicles in the interdigital spaces of the hands, wrists, scrotal area, or the peri-mammary region.
“The incidence is increasing, particularly in nursing homes, where outbreaks pose a significant risk of rapid spread. Treatment involves three courses of topical and oral treatments administered on days 0, 7, and 14. All contact cases must also be treated. Sometimes, these thick lesions are stripped with 10% salicylated petroleum jelly. Environmental treatment with acaricides is essential, along with strict isolation measures,” Delaunay emphasized.
Adherent nits on the scalp or other hairy areas should raise suspicion of pediculosis.
Neurogenic and Psychogenic Origins
Neurogenic pruritus can occur during a stroke, presenting as contralateral pruritus, or in the presence of a brain tumor or following neurosurgery. Opioid-containing medications may also induce neurogenic pruritus.
The presence of unilateral painful or itchy sensations should prompt the investigation of shingles in older individuals.
Psychogenic pruritus is also common and can arise in the context of psychosis with parasitophobia or as part of anxiety-depression syndromes.
Supportive Measures
For managing pruritus, it is essential to:
- Keep nails trimmed short
- Wash with cold or lukewarm water
- Use lipid-rice soaps and syndets
- Avoid irritants, including antiseptics, cologne, no-rinse cleansers, and steroidal or nonsteroidal anti-inflammatory drugs
- Limit bathing frequency
- Avoid wearing nylon, wool, or tight clothing
- Minimize exposure to heat and excessive heating
“Alternatives to scratching, such as applying a moisturizing emollient, can be beneficial and may have a placebo effect,” explained the dermatologist. She further emphasized that local corticosteroids are effective only in the presence of inflammatory dermatosis and should not be applied to healthy skin. Similarly, antihistamines should only be prescribed if the pruritus is histamine-mediated.
Capsaicin may be useful in the treatment of localized neuropathic pruritus.
In cases of neurogenic pruritus, gabapentin and pregabalin may be prescribed, but tolerance can be problematic at this age. Other measures include acupuncture, cryotherapy, relaxation, hypnosis, psychotherapy, and music therapy. In cases of repeated therapeutic failure, patients may be treated with biotherapy (dupilumab) by a dermatologist.
This story was translated from Medscape’s French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Chronic pruritus is a common problem among older individuals. During a session at the Dermatology Days of Paris 2024 conference dedicated to general practitioners, Juliette Delaunay, MD, a dermatologist and venereologist at Angers University Hospital Center in Angers, France, and Gabrielle Lisembard, MD, a general practitioner in the French town Grand-Fort-Philippe, discussed diagnostic approaches and key principles for the therapeutic management of pruritus.
Identifying Causes
“Pruritus in older people is most often linked to physiological changes in the skin caused by aging, leading to significant xerosis. However, before attributing it to aging, we need to rule out several causes,” Delaunay noted.
Beyond simple aging, one must consider autoimmune bullous dermatoses (bullous pemphigoid), drug-related causes, metabolic disorders (can occur at any age), cutaneous T-cell lymphomas, scabies, lice, and HIV infection.
Senile Pruritus
Aging-related xerosis can cause senile pruritus, often presenting as itching with scratch marks and dry skin. “This is a diagnosis of exclusion,” Delaunay insisted.
In older individuals with pruritus, initial examinations should include complete blood cell count (CBC), liver function tests, and thyroid-stimulating hormone levels. Syphilis serology, HIV testing, and beta-2 microglobulin levels are secondary evaluations. Renal function analysis may also be performed, and imaging may be required to investigate neoplasia.
“Annual etiological reassessment is essential if the initial evaluation is negative, as patients may later develop or report a neoplasia or hematological disorder,” Delaunay emphasized.
Paraneoplastic pruritus can occur, particularly those linked to hematological disorders (lymphomas, polycythemia, or myeloma).
Bullous Pemphigoid
Bullous pemphigoid often begins with pruritus, which can be severe and lead to insomnia. General practitioners should consider bullous pemphigoids when there is a bullous rash (tense blisters with citrine content) or an urticarial or chronic eczematous rash that does not heal spontaneously within a few days. The first-line biologic test to confirm the diagnosis is the CBC, which may reveal significant hypereosinophilia.
The diagnosis is confirmed by a skin biopsy showing a subepidermal blister with a preserved roof, unlike intraepidermal dermatoses, where the roof ruptures.
Direct immunofluorescence revealed deposits of immunoglobulin G antibodies along the dermoepidermal junction.
Approximately 40% of cases of bullous pemphigoid are associated with neurodegenerative diseases, such as stroke, parkinsonism, or dementia syndromes — occurring at a rate two to three times higher than in the general population.
It’s important to identify drugs that induce bullous pemphigoid, such as gliptins, anti-programmed cell death protein 1-programmed death-ligand 1 agents, loop diuretics (furosemide and bumetanide), anti-aldosterones (spironolactone), antiarrhythmics (amiodarone), and neuroleptics (phenothiazines).
“Stopping the medication is not mandatory if the bullous pemphigoid is well controlled by local or systemic treatments and the medication is essential. The decision to stop should be made on a case-by-case basis in consultation with the treating specialist,” Delaunay emphasized.
Treatment consists of very strong local corticosteroid therapy as the first-line treatment. If ineffective, systemic treatments based on methotrexate, oral corticosteroids, or immunomodulatory agents may be considered. Hospitalization is sometimes required.
Drug-Induced Pruritus
Drug-induced pruritus is common because older individuals often take multiple medications (antihypertensives, statins, oral hypoglycemics, psychotropic drugs, antiarrhythmics, etc.). “Sometimes, drug-induced pruritus can occur even if the medication was started several months or years ago,” Delaunay emphasized.
The lesions are generally nonspecific and scratching.
“This is a diagnosis of exclusion for other causes of pruritus. In the absence of specific lesions pointing to a dermatosis, eviction/reintroduction tests with treatments should be conducted one by one, which can be quite lengthy,” she explained.
Awareness for Scabies
Delaunay reminded attendees to consider scabies in older individuals when classic signs of pruritus flare up at night, with a rash affecting the face, scabs, or vesicles in the interdigital spaces of the hands, wrists, scrotal area, or the peri-mammary region.
“The incidence is increasing, particularly in nursing homes, where outbreaks pose a significant risk of rapid spread. Treatment involves three courses of topical and oral treatments administered on days 0, 7, and 14. All contact cases must also be treated. Sometimes, these thick lesions are stripped with 10% salicylated petroleum jelly. Environmental treatment with acaricides is essential, along with strict isolation measures,” Delaunay emphasized.
Adherent nits on the scalp or other hairy areas should raise suspicion of pediculosis.
Neurogenic and Psychogenic Origins
Neurogenic pruritus can occur during a stroke, presenting as contralateral pruritus, or in the presence of a brain tumor or following neurosurgery. Opioid-containing medications may also induce neurogenic pruritus.
The presence of unilateral painful or itchy sensations should prompt the investigation of shingles in older individuals.
Psychogenic pruritus is also common and can arise in the context of psychosis with parasitophobia or as part of anxiety-depression syndromes.
Supportive Measures
For managing pruritus, it is essential to:
- Keep nails trimmed short
- Wash with cold or lukewarm water
- Use lipid-rice soaps and syndets
- Avoid irritants, including antiseptics, cologne, no-rinse cleansers, and steroidal or nonsteroidal anti-inflammatory drugs
- Limit bathing frequency
- Avoid wearing nylon, wool, or tight clothing
- Minimize exposure to heat and excessive heating
“Alternatives to scratching, such as applying a moisturizing emollient, can be beneficial and may have a placebo effect,” explained the dermatologist. She further emphasized that local corticosteroids are effective only in the presence of inflammatory dermatosis and should not be applied to healthy skin. Similarly, antihistamines should only be prescribed if the pruritus is histamine-mediated.
Capsaicin may be useful in the treatment of localized neuropathic pruritus.
In cases of neurogenic pruritus, gabapentin and pregabalin may be prescribed, but tolerance can be problematic at this age. Other measures include acupuncture, cryotherapy, relaxation, hypnosis, psychotherapy, and music therapy. In cases of repeated therapeutic failure, patients may be treated with biotherapy (dupilumab) by a dermatologist.
This story was translated from Medscape’s French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Central Line Skin Reactions in Children: Survey Addresses Treatment Protocols in Use
TOPLINE:
A and reported varying management approaches.
METHODOLOGY:
- Researchers developed and administered a 14-item Qualtrics survey to 107 dermatologists providing pediatric inpatient care through the Society for Pediatric Dermatology’s Inpatient Dermatology Section and Section Chief email lists.
- A total of 35 dermatologists (33%) from multiple institutions responded to the survey; most respondents (94%) specialized in pediatric dermatology.
- Researchers assessed management of CLD-associated adverse skin reactions.
TAKEAWAY:
- All respondents reported receiving CLD-related consults, but 66% indicated there was no personal or institutional standardized approach for managing CLD-associated skin reactions.
- Respondents said most reactions were in children aged 1-12 years (19 or 76% of 25 respondents) compared with those aged < 1 year (3 or 12% of 25 respondents).
- Management strategies included switching to alternative products, applying topical corticosteroids, and performing patch testing for allergies.
IN PRACTICE:
“Insights derived from this study, including variation in clinician familiarity with reaction patterns, underscore the necessity of a standardized protocol for classifying and managing cutaneous CLD reactions in pediatric patients,” the authors wrote. “Further investigation is needed to better characterize CLD-associated allergic CD [contact dermatitis], irritant CD, and skin infections, as well as at-risk populations, to better inform clinical approaches,” they added.
SOURCE:
The study was led by Carly Mulinda, Columbia University College of Physicians and Surgeons, New York, and was published online on December 16 in Pediatric Dermatology.
LIMITATIONS:
The authors noted variable respondent awareness of institutional CLD and potential recency bias as key limitations of the study.
DISCLOSURES:
Study funding source was not declared. The authors reported no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
A and reported varying management approaches.
METHODOLOGY:
- Researchers developed and administered a 14-item Qualtrics survey to 107 dermatologists providing pediatric inpatient care through the Society for Pediatric Dermatology’s Inpatient Dermatology Section and Section Chief email lists.
- A total of 35 dermatologists (33%) from multiple institutions responded to the survey; most respondents (94%) specialized in pediatric dermatology.
- Researchers assessed management of CLD-associated adverse skin reactions.
TAKEAWAY:
- All respondents reported receiving CLD-related consults, but 66% indicated there was no personal or institutional standardized approach for managing CLD-associated skin reactions.
- Respondents said most reactions were in children aged 1-12 years (19 or 76% of 25 respondents) compared with those aged < 1 year (3 or 12% of 25 respondents).
- Management strategies included switching to alternative products, applying topical corticosteroids, and performing patch testing for allergies.
IN PRACTICE:
“Insights derived from this study, including variation in clinician familiarity with reaction patterns, underscore the necessity of a standardized protocol for classifying and managing cutaneous CLD reactions in pediatric patients,” the authors wrote. “Further investigation is needed to better characterize CLD-associated allergic CD [contact dermatitis], irritant CD, and skin infections, as well as at-risk populations, to better inform clinical approaches,” they added.
SOURCE:
The study was led by Carly Mulinda, Columbia University College of Physicians and Surgeons, New York, and was published online on December 16 in Pediatric Dermatology.
LIMITATIONS:
The authors noted variable respondent awareness of institutional CLD and potential recency bias as key limitations of the study.
DISCLOSURES:
Study funding source was not declared. The authors reported no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
A and reported varying management approaches.
METHODOLOGY:
- Researchers developed and administered a 14-item Qualtrics survey to 107 dermatologists providing pediatric inpatient care through the Society for Pediatric Dermatology’s Inpatient Dermatology Section and Section Chief email lists.
- A total of 35 dermatologists (33%) from multiple institutions responded to the survey; most respondents (94%) specialized in pediatric dermatology.
- Researchers assessed management of CLD-associated adverse skin reactions.
TAKEAWAY:
- All respondents reported receiving CLD-related consults, but 66% indicated there was no personal or institutional standardized approach for managing CLD-associated skin reactions.
- Respondents said most reactions were in children aged 1-12 years (19 or 76% of 25 respondents) compared with those aged < 1 year (3 or 12% of 25 respondents).
- Management strategies included switching to alternative products, applying topical corticosteroids, and performing patch testing for allergies.
IN PRACTICE:
“Insights derived from this study, including variation in clinician familiarity with reaction patterns, underscore the necessity of a standardized protocol for classifying and managing cutaneous CLD reactions in pediatric patients,” the authors wrote. “Further investigation is needed to better characterize CLD-associated allergic CD [contact dermatitis], irritant CD, and skin infections, as well as at-risk populations, to better inform clinical approaches,” they added.
SOURCE:
The study was led by Carly Mulinda, Columbia University College of Physicians and Surgeons, New York, and was published online on December 16 in Pediatric Dermatology.
LIMITATIONS:
The authors noted variable respondent awareness of institutional CLD and potential recency bias as key limitations of the study.
DISCLOSURES:
Study funding source was not declared. The authors reported no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Allergic Contact Dermatitis: New Culprits
New allergens responsible for contact dermatitis emerge regularly. During the Dermatology Days of Paris 2024 conference, Angèle Soria, MD, PhD, a dermatologist at Tenon Hospital in Paris, France, outlined four major categories driving this trend. Among them are (meth)acrylates found in nail cosmetics used in salons or do-it-yourself false nail kits that can be bought online.
Isothiazolinones
a preservative used in many cosmetics; (meth)acrylates; essential oils; and epoxy resins used in industry and leisure activities.
Around 15 years ago, parabens, commonly used as preservatives in cosmetics, were identified as endocrine disruptors. In response, they were largely replaced by newer preservatives, notably MI. However, this led to a proliferation of allergic contact dermatitis in Europe between 2010 and 2013.
“About 10% of the population that we tested showed allergies to these preservatives, primarily found in cosmetics,” explained Soria. Since 2015, the use of MI in leave-on cosmetics has been prohibited in Europe and its concentration restricted in rinse-off products. However, cosmetics sold online from outside Europe may not comply with these regulations.
MI is also present in water-based paints to prevent mold. “A few years ago, we started seeing patients with facial angioedema, sometimes combined with asthma, caused by these isothiazolinone preservatives, including in patients who are not professional painters,” said Soria. More recently, attention has shifted to MI’s presence in household cleaning products. A 2020 Spanish study found MI in 76% of 34 analyzed cleaning products.
MI-based fungicides are also used to treat leather during transport, which can lead to contact allergies among professionals and consumers alike. Additionally, MI has been identified in children’s toys, including slime gels, and in florists’ gel cubes used to preserve flowers.
“We are therefore surrounded by these preservatives, which are no longer only in cosmetics,” warned the dermatologist.
(Meth)acrylates
Another major allergen category is (meth)acrylates, responsible for many cases of allergic contact dermatitis. Acrylates and their derivatives are widely used in everyday items. They are low–molecular weight monomers, sensitizing on contact with the skin. Their polymerized forms include materials like Plexiglas.
“We are currently witnessing an epidemic of contact dermatitis in the general population, mainly due to nail cosmetics, such as semipermanent nail polishes and at-home false nail kits,” reported Soria. Nail cosmetics account for 97% of new sensitization cases involving (meth)acrylates. These allergens often cause severe dermatitis, prompting the European Union to mandate labeling in 2020, warning that these products are “for professional use only” and can “cause allergic reactions.”
Beyond nail cosmetics, these allergens are also found in dental products (such as trays), ECG electrodes, prosthetics, glucose sensors, surgical adhesives, and some electronic devices like earbuds and phone screens. Notably, patients sensitized to acrylates via nail kits may experience reactions during dental treatments involving acrylates.
Investigating Essential Oil Use
Essential oils, distinct from vegetable oils like almond or argan, are another known allergen. Often considered risk-free due to their “natural” label, these products are widely used topically, orally, or via inhalation for various purposes, such as treating respiratory infections or creating relaxing atmospheres. However, essential oils contain fragrant molecules like terpenes, which can become highly allergenic over time, especially after repeated exposure.
Soria emphasized the importance of asking patients about their use of essential oils, especially tea tree and lavender oils, which are commonly used but rarely mentioned by patients unless prompted.
Epoxy Resins in Recreational Use
Epoxy resins are a growing cause of contact allergies, not just in professional settings such as aeronautics and construction work but also increasingly in recreational activities. Soria highlighted the case of a 12-year-old girl hospitalized for severe facial edema after engaging in resin crafts inspired by TikTok. For 6 months, she had been creating resin objects, such as bowls and cutting boards, using vinyl gloves and a Filtering FacePiece 2 mask under adult supervision.
“The growing popularity and online availability of epoxy resins mean that allergic reactions should now be considered even in nonprofessional contexts,” warned Soria.
Clinical Approach
When dermatologists suspect allergic contact dermatitis, the first step is to treat the condition with corticosteroid creams. This is followed by a detailed patient interview to identify suspected allergens in products they’ve used.
Patch testing is then conducted to confirm the allergen. Small chambers containing potential allergens are applied to the upper back for 48 hours without removal. Results are read 2-5 days later, with some cases requiring a 7-day follow-up.
The patient’s occupation is an important factor, as certain professions, such as hairdressing, healthcare, or beauty therapy, are known to trigger allergic contact dermatitis. Similarly, certain hobbies may also play a role.
A thorough approach ensures accurate diagnosis and targeted prevention strategies.
This story was translated from Medscape’s French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
New allergens responsible for contact dermatitis emerge regularly. During the Dermatology Days of Paris 2024 conference, Angèle Soria, MD, PhD, a dermatologist at Tenon Hospital in Paris, France, outlined four major categories driving this trend. Among them are (meth)acrylates found in nail cosmetics used in salons or do-it-yourself false nail kits that can be bought online.
Isothiazolinones
a preservative used in many cosmetics; (meth)acrylates; essential oils; and epoxy resins used in industry and leisure activities.
Around 15 years ago, parabens, commonly used as preservatives in cosmetics, were identified as endocrine disruptors. In response, they were largely replaced by newer preservatives, notably MI. However, this led to a proliferation of allergic contact dermatitis in Europe between 2010 and 2013.
“About 10% of the population that we tested showed allergies to these preservatives, primarily found in cosmetics,” explained Soria. Since 2015, the use of MI in leave-on cosmetics has been prohibited in Europe and its concentration restricted in rinse-off products. However, cosmetics sold online from outside Europe may not comply with these regulations.
MI is also present in water-based paints to prevent mold. “A few years ago, we started seeing patients with facial angioedema, sometimes combined with asthma, caused by these isothiazolinone preservatives, including in patients who are not professional painters,” said Soria. More recently, attention has shifted to MI’s presence in household cleaning products. A 2020 Spanish study found MI in 76% of 34 analyzed cleaning products.
MI-based fungicides are also used to treat leather during transport, which can lead to contact allergies among professionals and consumers alike. Additionally, MI has been identified in children’s toys, including slime gels, and in florists’ gel cubes used to preserve flowers.
“We are therefore surrounded by these preservatives, which are no longer only in cosmetics,” warned the dermatologist.
(Meth)acrylates
Another major allergen category is (meth)acrylates, responsible for many cases of allergic contact dermatitis. Acrylates and their derivatives are widely used in everyday items. They are low–molecular weight monomers, sensitizing on contact with the skin. Their polymerized forms include materials like Plexiglas.
“We are currently witnessing an epidemic of contact dermatitis in the general population, mainly due to nail cosmetics, such as semipermanent nail polishes and at-home false nail kits,” reported Soria. Nail cosmetics account for 97% of new sensitization cases involving (meth)acrylates. These allergens often cause severe dermatitis, prompting the European Union to mandate labeling in 2020, warning that these products are “for professional use only” and can “cause allergic reactions.”
Beyond nail cosmetics, these allergens are also found in dental products (such as trays), ECG electrodes, prosthetics, glucose sensors, surgical adhesives, and some electronic devices like earbuds and phone screens. Notably, patients sensitized to acrylates via nail kits may experience reactions during dental treatments involving acrylates.
Investigating Essential Oil Use
Essential oils, distinct from vegetable oils like almond or argan, are another known allergen. Often considered risk-free due to their “natural” label, these products are widely used topically, orally, or via inhalation for various purposes, such as treating respiratory infections or creating relaxing atmospheres. However, essential oils contain fragrant molecules like terpenes, which can become highly allergenic over time, especially after repeated exposure.
Soria emphasized the importance of asking patients about their use of essential oils, especially tea tree and lavender oils, which are commonly used but rarely mentioned by patients unless prompted.
Epoxy Resins in Recreational Use
Epoxy resins are a growing cause of contact allergies, not just in professional settings such as aeronautics and construction work but also increasingly in recreational activities. Soria highlighted the case of a 12-year-old girl hospitalized for severe facial edema after engaging in resin crafts inspired by TikTok. For 6 months, she had been creating resin objects, such as bowls and cutting boards, using vinyl gloves and a Filtering FacePiece 2 mask under adult supervision.
“The growing popularity and online availability of epoxy resins mean that allergic reactions should now be considered even in nonprofessional contexts,” warned Soria.
Clinical Approach
When dermatologists suspect allergic contact dermatitis, the first step is to treat the condition with corticosteroid creams. This is followed by a detailed patient interview to identify suspected allergens in products they’ve used.
Patch testing is then conducted to confirm the allergen. Small chambers containing potential allergens are applied to the upper back for 48 hours without removal. Results are read 2-5 days later, with some cases requiring a 7-day follow-up.
The patient’s occupation is an important factor, as certain professions, such as hairdressing, healthcare, or beauty therapy, are known to trigger allergic contact dermatitis. Similarly, certain hobbies may also play a role.
A thorough approach ensures accurate diagnosis and targeted prevention strategies.
This story was translated from Medscape’s French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
New allergens responsible for contact dermatitis emerge regularly. During the Dermatology Days of Paris 2024 conference, Angèle Soria, MD, PhD, a dermatologist at Tenon Hospital in Paris, France, outlined four major categories driving this trend. Among them are (meth)acrylates found in nail cosmetics used in salons or do-it-yourself false nail kits that can be bought online.
Isothiazolinones
a preservative used in many cosmetics; (meth)acrylates; essential oils; and epoxy resins used in industry and leisure activities.
Around 15 years ago, parabens, commonly used as preservatives in cosmetics, were identified as endocrine disruptors. In response, they were largely replaced by newer preservatives, notably MI. However, this led to a proliferation of allergic contact dermatitis in Europe between 2010 and 2013.
“About 10% of the population that we tested showed allergies to these preservatives, primarily found in cosmetics,” explained Soria. Since 2015, the use of MI in leave-on cosmetics has been prohibited in Europe and its concentration restricted in rinse-off products. However, cosmetics sold online from outside Europe may not comply with these regulations.
MI is also present in water-based paints to prevent mold. “A few years ago, we started seeing patients with facial angioedema, sometimes combined with asthma, caused by these isothiazolinone preservatives, including in patients who are not professional painters,” said Soria. More recently, attention has shifted to MI’s presence in household cleaning products. A 2020 Spanish study found MI in 76% of 34 analyzed cleaning products.
MI-based fungicides are also used to treat leather during transport, which can lead to contact allergies among professionals and consumers alike. Additionally, MI has been identified in children’s toys, including slime gels, and in florists’ gel cubes used to preserve flowers.
“We are therefore surrounded by these preservatives, which are no longer only in cosmetics,” warned the dermatologist.
(Meth)acrylates
Another major allergen category is (meth)acrylates, responsible for many cases of allergic contact dermatitis. Acrylates and their derivatives are widely used in everyday items. They are low–molecular weight monomers, sensitizing on contact with the skin. Their polymerized forms include materials like Plexiglas.
“We are currently witnessing an epidemic of contact dermatitis in the general population, mainly due to nail cosmetics, such as semipermanent nail polishes and at-home false nail kits,” reported Soria. Nail cosmetics account for 97% of new sensitization cases involving (meth)acrylates. These allergens often cause severe dermatitis, prompting the European Union to mandate labeling in 2020, warning that these products are “for professional use only” and can “cause allergic reactions.”
Beyond nail cosmetics, these allergens are also found in dental products (such as trays), ECG electrodes, prosthetics, glucose sensors, surgical adhesives, and some electronic devices like earbuds and phone screens. Notably, patients sensitized to acrylates via nail kits may experience reactions during dental treatments involving acrylates.
Investigating Essential Oil Use
Essential oils, distinct from vegetable oils like almond or argan, are another known allergen. Often considered risk-free due to their “natural” label, these products are widely used topically, orally, or via inhalation for various purposes, such as treating respiratory infections or creating relaxing atmospheres. However, essential oils contain fragrant molecules like terpenes, which can become highly allergenic over time, especially after repeated exposure.
Soria emphasized the importance of asking patients about their use of essential oils, especially tea tree and lavender oils, which are commonly used but rarely mentioned by patients unless prompted.
Epoxy Resins in Recreational Use
Epoxy resins are a growing cause of contact allergies, not just in professional settings such as aeronautics and construction work but also increasingly in recreational activities. Soria highlighted the case of a 12-year-old girl hospitalized for severe facial edema after engaging in resin crafts inspired by TikTok. For 6 months, she had been creating resin objects, such as bowls and cutting boards, using vinyl gloves and a Filtering FacePiece 2 mask under adult supervision.
“The growing popularity and online availability of epoxy resins mean that allergic reactions should now be considered even in nonprofessional contexts,” warned Soria.
Clinical Approach
When dermatologists suspect allergic contact dermatitis, the first step is to treat the condition with corticosteroid creams. This is followed by a detailed patient interview to identify suspected allergens in products they’ve used.
Patch testing is then conducted to confirm the allergen. Small chambers containing potential allergens are applied to the upper back for 48 hours without removal. Results are read 2-5 days later, with some cases requiring a 7-day follow-up.
The patient’s occupation is an important factor, as certain professions, such as hairdressing, healthcare, or beauty therapy, are known to trigger allergic contact dermatitis. Similarly, certain hobbies may also play a role.
A thorough approach ensures accurate diagnosis and targeted prevention strategies.
This story was translated from Medscape’s French edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Ob.Gyn. Says Collaboration with Dermatologists Essential for Managing Vulvar Dermatoses
— and she believes collaboration with dermatologists is essential, especially for complex cases in what she calls a neglected, data-poor area of medicine.
She also recommends that dermatologists have a good understanding of the vestibule, “one of the most important structures in vulvar medicine,” and that they become equipped to recognize generalized and localized causes of vulvar pain and/or itch.
“The problem is, we don’t talk about [vulvovaginal pain and itch] ... it’s taboo and we’re not taught about it in medical school,” Cigna, assistant professor of obstetrics and gynecology at The George Washington University (GWU), Washington, DC, said in a grand rounds lecture held recently at the GWU School of Medicine and Health Sciences Department of Dermatology.
“There are dermatologists who don’t have much training in vulvar dermatology, and a lot of gyns don’t get as much training” as they should, she said in an interview after the lecture. “So who’s looking at people’s vulvar skin and figuring out what’s going on and giving them effective treatments and evidence-based education?”
Cigna and dermatologist Emily Murphy, MD, will be co-directors of a joint ob.gyn-dermatology Vulvar Dermatology Clinic at GWU that will be launched in 2025, with monthly clinics for particularly challenging cases where the etiology is unclear or treatment is ineffective. “We want to collaborate in a more systematic way and put our heads together and think creatively about what will improve patient care,” Cigna said in the interview.
Dermatologists have valuable expertise in the immunology and genetic factors involved in skin disorders, Cigna said. Moreover, Murphy, assistant professor of dermatology and director of the Vulvar Health Program at GWU, said in an interview, dermatologists “are comfortable in going to off-label systemic medications that ob.gyns may not use that often” and bring to the table expertise in various types of procedures.
Murphy recently trained with Melissa Mauskar, MD, associate of dermatology and obstetrics and gynecology at the University of Texas Southwestern, Dallas, and founder and director of the Gynecologic Dermatology Clinic there. “It’s so important for dermatologists to be involved. It just takes some extra training that residents aren’t getting right now,” said Murphy, a member of the newly formed Vulvar Dermatoses Research Consortium.
In her grand rounds lecture, Cigna offered pearls to dermatologists for approaching a history and exam and covered highlights of the diagnosis and treatment of various problems, from vulvar Candida infections and lichen simplex chronicus to vulvar lichen sclerosus (LS), vulvar lichen planus (LP), vulvar Crohn’s disease, pudendal neuralgia, and pelvic floor muscle spasm, as well as the role of mast cell proliferation in vulvar issues.
Approaching the History and Exam
A comprehensive history covers the start, duration, and location of pain and/or itching as well as a detailed timeline (such as timing of potential causes, including injuries or births) and symptoms (such as burning, cutting, aching, and stinging). The question of whether pain “is on the outside, at the entrance, or deeper inside” is “crucial, especially for those in dermatology,” Cigna emphasized.
“And if you’re seeing a patient for a vulvar condition, please ask them about sex. Ask, is this affecting your sexual or intimate life with your partner because this can also give you a clue about what’s going on and how you can help them,” she told the audience of dermatologists.
Queries about trauma history (physical and emotional/verbal), competitive sports (such as daily cycling, equestrian, and heavy weight lifting), endometriosis/gynecologic surgery, connective tissue disorders (such as Ehler-Danlos syndrome), and irritable bowel syndrome are all potentially important to consider. It is important to ask about anxiety, depression, and obsessive-compulsive disorder, which do not cause — but are highly associated with — vulvar dermatoses, she said.
A surprisingly large number of people with vulvovaginal issues are being diagnosed with Ehler-Danlos syndrome, so “I’m always asking, are you hypermobile because this might be affecting the musculoskeletal system, which might be affecting the pelvis,” Cigna said. “Anything that affects the pelvis can affect the vulva as well.”
The pelvic examination should be “offered” rather than assumed to be part of the exam, as part of a trauma-informed approach that is crucial for earning trust, she advised. “Just saying, ‘we’re going to talk, and then I can offer you an exam if you like’…patients like it. It helps them feel safer and more open.”
Many diagnoses are differentiated by eliciting pain on the anterior vs the posterior half of the vulvar vestibule — the part of the vulva that lies between the labia minora and is composed of nonkeratinized tissue with embryonic origins in the endoderm. “If you touch on the keratinized skin (of the vulva) and they don’t have pain, but on the vestibule they do have pain, and there is no pain inside the vagina, this suggests there is a vestibular problem,” said Cigna.
Pain/tenderness isolated to the posterior half of the vestibule suggests a muscular cause, and pain in both the posterior and anterior parts of the vestibule suggests a cause that is more systemic or diffuse, which could be a result of a hormonal issue such as one related to oral contraceptives or decreased testosterone, or a nerve-related process.
Cigna uses gentle swipes of a Q-tip moistened with water or gel to examine the vulva rather than a poke or touch, with the exception being the posterior vestibule, which overlies muscle insertion sites. “Make sure to get a baseline in remote areas such as the inner thigh, and always distinguish between ‘scratchy/sensitive’ sensations and pain,” she said, noting the value of having the patient hold a mirror on her inner thigh.
Causes of Vulvar Itch: Infectious and Noninfectious
With vulvar candidiasis, a common infectious cause of vulvar itch, “you have to ask if they’re also itching on the inside because if you treat them with a topical and you don’t treat the vaginal yeast infection that may be co-occurring, they’ll keep reseeding their vulvar skin,” Cigna said, “and it will never be fully treated.”
Candida albicans is the most common cause of vulvar or vulvovaginal candidiasis, and resistance to antifungals has been rising. Non-albicans Candida “tends to have even higher resistance rates,” she said. Ordering a sensitivity panel along with the culture is helpful, but “comprehensive vaginal biome” panels are generally not useful. “It’s hard to correlate the information clinically,” she said, “and there’s not always a lot of information about susceptibilities, which is what I really like to know.”
Cigna’s treatments for vaginal infections include miconazole, terconazole, and fluconazole (and occasionally, itraconazole or voriconazole — a “decision we don’t take lightly”). Vulvar treatments include nystatin ointment, clotrimazole cream, and miconazole cream. Often, optimal treatment involves addressing “both inside and out,” she said, noting the importance of also killing yeast in undergarment fabric.
“In my experience, Diflucan [oral fluconazole] doesn’t treat persistent vulvar cutaneous skin yeast well, so while I might try Diflucan, I typically use something topical as well,” she said. “And with vaginal yeast, we do use boric acid from time to time, especially for non-albicans species because it tends to be a little more effective.”
Noninfectious causes of vulvar itch include allergic, neuropathic, and muscular causes, as well as autoimmune dermatoses and mast cell activation syndrome. Well known in dermatology are acute contact dermatitis and lichen simplex chronicus — both characterized by induration, thickening, and a “puffy” erythematous appearance, and worsening of pruritus at night. What may be less appreciated is the long list of implicated allergens , including Always menstrual pads made of a plastic-containing “dry weave” material, Cigna said. There are at least several cotton-only, low-preservative feminine products available on the market, she noted.
Common Autoimmune Vulvar Dermatoses: LS and LP
Vulvar LS has traditionally been thought to affect mainly prepubertal and postmenopausal women, but the autoimmune condition is now known to affect more reproductive-age people with vulvas than previously appreciated, Cigna said.
And notably, in an observational web-based study of premenopausal women (aged 18-50 years) with biopsy-confirmed vulvar LS, the leading symptom was not itch but dyspareunia and tearing with intercourse. This means “we’re missing people,” said Cigna, an author of the study. “We think the reason we’re not seeing itch as commonly in this population is that itch is likely mediated by the low estrogen state of pre- and postmenopausal people.”
Vulvar LS also occurs in pregnancy, with symptoms that are either stable or decrease during pregnancy and increase in the postpartum period, as demonstrated in a recently published online survey.
Patients with vulvar LS can present with hypopigmentation, lichenification, and scarring and architectural changes, the latter of which can involve clitoral phimosis, labial resorption, and narrowing of the introitus. (The vaginal mucosa is unaffected.) The presentation can be subtle, especially in premenopausal women, and differentiation between LS, vitiligo, and yeast is sometimes necessary.
A timely biopsy-driven definitive diagnosis is important because vulvar LS increases the risk for cancer if it’s not adequately treated and because long-term steroid use can affect the accuracy of pathology reports. “We really care about keeping this disease in remission as much as possible,” Cigna said. Experts in the field recommend long-term maintenance therapy with a mid-ultra-potent steroid one to three times/week or an alternative. “I’ve just started using ruxolitinib cream, a Janus kinase (JAK) inhibitor, and tacrolimus, a calcineurin inhibitor,” she said.
With vulvar LP, based on current evidence, the risk for malignant transformation is low, but “it crosses into the vagina and can cause vaginal adhesions, so if you’re diagnosing someone with lichen planus, you need to make sure you’re talking with them about dilators, and if you’re not comfortable, send them to [gyn],” she said.
The use of vulvoscopy is important for one’s ability to see the fine Wickham’s striae that often characterize vulvar LP, she noted. Medical treatments for vulvar LP include topical calcineurin inhibitors, high-potency steroids, and JAK inhibitors.
Surgical treatment of vulvar granuloma fissuratum caused by vulvar LS is possible (when the patient is in complete remission, to prevent koebnerization), with daily post-op application of clobetasol and retraction of tissues, noted Cigna, the author of a study on vulvar lysis of adhesions.
With both LS and LP, Cigna said, “don’t forget (consideration of) hormones” as an adjunctive treatment, especially in postmenopausal women. “Patients in a low hormone state will have more flares.”
Vulvar Crohn’s
“We all have to know how to look for this,” Cigna said. “Unilateral or asymmetric swelling is classic, but don’t rule out the diagnosis if you see symmetric swelling.” Patients also typically have linear “knife-like” fissures or ulcerations, the vulva “is very indurated,” and “swelling is so intense, the patients are miserable,” she said.
Vulvar Crohn’s disease may precede intestinal disease in 20%-30% of patients, so referral to a gastroenterologist — and ideally subsequent collaboration — is important, as vulvar manifestations are treated with systemic medications typical for Crohn’s.
A biopsy is required for diagnosis, and the pathologist should be advised to look for lichenified squamous mucosa with the Touton giant cell reaction. “Vulvar Crohn’s is a rare enough disorder that if you don’t have an experienced or informed pathologist looking at your specimen, they may miss it because they won’t be looking for it,” Cigna added in the interview. “You should be really clear about what you’re looking for.”
Neuropathic Itch, Pelvic Floor Muscle Spasm
Patients with pudendal neuralgia — caused by an injured, entrapped, or irritated pudendal nerve (originating from S2-S4) — typically present with chronic vulvar and pelvic pain that is often unprovoked and worsens with sitting. Itching upon touch is often another symptom, and some patients describe a foreign body sensation. The cause is often trauma (such as an accident or childbirth-related) as opposed to myofascial irritation, Cigna explained in her lecture.
“Your exam will be largely normal, with no skin findings, so patients will get sent away if you don’t know to look for pudendal neuralgia by pressing on the pudendal nerve or doing (or referring for) a diagnostic nerve block,” Cigna added in the interview.
Persistent genital arousal disorder (PGAD) is “more global” in that it can also originate not only from the pudendal nerve but also from nerve roots higher in the spine or even from the brain. “People feel a sense of arousal, but some describe it as an itch,” Cigna said in her lecture, referring to a 2021 consensus document on PGAD/genito-pelvic dysesthesia by the International Society for the Study of Women’s Sexual Health as a valuable resource for understanding and managing the condition.
Diagnosis and treatment usually start with a pudendal nerve block with a combination of steroid and anesthetic. If this does not relieve arousal/itching, the next step may be an MRI to look higher in the spine.
Pelvic Floor Muscle Spasm
Vulvar pain, skin itching, and irritation can be symptoms of pelvic floor muscle spasm. “Oftentimes people come to me and say, ‘I have a dermatologic problem,’” Cigna said. “The skin may look red and erythematous, but it’s probably more likely a muscle problem when you’re not finding anything, and no amount of steroid will help the itch go away when the problem lies underneath.”
Co-occurring symptoms can include vaginal dryness, clitoral pain, urethral discomfort, bladder pain/irritation, increased urgency, constipation, and anal fissures. The first-line treatment approach is pelvic floor therapy.
“Pelvic floor therapy is not just for incontinence. It’s also for pain and discomfort from muscles,” she said, noting that most patients with vulvar disorders are referred for pelvic floor therapy. “Almost all of them end up having pelvic floor dysfunction because the pelvic floor muscles spasm whenever there’s pain or inflammation.”
A Cautionary Word on Vulvodynia, and a Mast Cell Paradigm to Explore
Vulvodynia is defined as persistent pain of at least 3 months’ duration with no clear cause. “These are the patients with no skin findings,” Cigna said. But in most cases, she said, careful investigation identifies causes that are musculoskeletal, hormonal, or nerve-related.
“It’s a term that’s thrown around a lot — it’s kind of a catchall. Yet it should be a small minority of patients who truly have a diagnosis of vulvodynia,” she said.
In the early stages of investigation is the idea that mast cell proliferation and mast cell activation may play a role in some cases of vulvar and vestibular pain and itching. “We see that some patients with vulvodynia and vestibulodynia have mast cells that are increased in number in the epithelium and beneath the epithelium, and nerve staining shows an increased number of nerve endings traveling into the epithelium,” Cigna said.
“We do diagnose some people clinically” based on urticaria and other symptoms suggestive of mast cell proliferation/activation (such as flushing, abdominal cramping, diarrhea, hypotensive syncope or near syncope, and tachycardia), and “then we send them to the allergist for testing,” Cigna said.
Cigna and Murphy have no relevant financial disclosures.
A version of this article appeared on Medscape.com.
— and she believes collaboration with dermatologists is essential, especially for complex cases in what she calls a neglected, data-poor area of medicine.
She also recommends that dermatologists have a good understanding of the vestibule, “one of the most important structures in vulvar medicine,” and that they become equipped to recognize generalized and localized causes of vulvar pain and/or itch.
“The problem is, we don’t talk about [vulvovaginal pain and itch] ... it’s taboo and we’re not taught about it in medical school,” Cigna, assistant professor of obstetrics and gynecology at The George Washington University (GWU), Washington, DC, said in a grand rounds lecture held recently at the GWU School of Medicine and Health Sciences Department of Dermatology.
“There are dermatologists who don’t have much training in vulvar dermatology, and a lot of gyns don’t get as much training” as they should, she said in an interview after the lecture. “So who’s looking at people’s vulvar skin and figuring out what’s going on and giving them effective treatments and evidence-based education?”
Cigna and dermatologist Emily Murphy, MD, will be co-directors of a joint ob.gyn-dermatology Vulvar Dermatology Clinic at GWU that will be launched in 2025, with monthly clinics for particularly challenging cases where the etiology is unclear or treatment is ineffective. “We want to collaborate in a more systematic way and put our heads together and think creatively about what will improve patient care,” Cigna said in the interview.
Dermatologists have valuable expertise in the immunology and genetic factors involved in skin disorders, Cigna said. Moreover, Murphy, assistant professor of dermatology and director of the Vulvar Health Program at GWU, said in an interview, dermatologists “are comfortable in going to off-label systemic medications that ob.gyns may not use that often” and bring to the table expertise in various types of procedures.
Murphy recently trained with Melissa Mauskar, MD, associate of dermatology and obstetrics and gynecology at the University of Texas Southwestern, Dallas, and founder and director of the Gynecologic Dermatology Clinic there. “It’s so important for dermatologists to be involved. It just takes some extra training that residents aren’t getting right now,” said Murphy, a member of the newly formed Vulvar Dermatoses Research Consortium.
In her grand rounds lecture, Cigna offered pearls to dermatologists for approaching a history and exam and covered highlights of the diagnosis and treatment of various problems, from vulvar Candida infections and lichen simplex chronicus to vulvar lichen sclerosus (LS), vulvar lichen planus (LP), vulvar Crohn’s disease, pudendal neuralgia, and pelvic floor muscle spasm, as well as the role of mast cell proliferation in vulvar issues.
Approaching the History and Exam
A comprehensive history covers the start, duration, and location of pain and/or itching as well as a detailed timeline (such as timing of potential causes, including injuries or births) and symptoms (such as burning, cutting, aching, and stinging). The question of whether pain “is on the outside, at the entrance, or deeper inside” is “crucial, especially for those in dermatology,” Cigna emphasized.
“And if you’re seeing a patient for a vulvar condition, please ask them about sex. Ask, is this affecting your sexual or intimate life with your partner because this can also give you a clue about what’s going on and how you can help them,” she told the audience of dermatologists.
Queries about trauma history (physical and emotional/verbal), competitive sports (such as daily cycling, equestrian, and heavy weight lifting), endometriosis/gynecologic surgery, connective tissue disorders (such as Ehler-Danlos syndrome), and irritable bowel syndrome are all potentially important to consider. It is important to ask about anxiety, depression, and obsessive-compulsive disorder, which do not cause — but are highly associated with — vulvar dermatoses, she said.
A surprisingly large number of people with vulvovaginal issues are being diagnosed with Ehler-Danlos syndrome, so “I’m always asking, are you hypermobile because this might be affecting the musculoskeletal system, which might be affecting the pelvis,” Cigna said. “Anything that affects the pelvis can affect the vulva as well.”
The pelvic examination should be “offered” rather than assumed to be part of the exam, as part of a trauma-informed approach that is crucial for earning trust, she advised. “Just saying, ‘we’re going to talk, and then I can offer you an exam if you like’…patients like it. It helps them feel safer and more open.”
Many diagnoses are differentiated by eliciting pain on the anterior vs the posterior half of the vulvar vestibule — the part of the vulva that lies between the labia minora and is composed of nonkeratinized tissue with embryonic origins in the endoderm. “If you touch on the keratinized skin (of the vulva) and they don’t have pain, but on the vestibule they do have pain, and there is no pain inside the vagina, this suggests there is a vestibular problem,” said Cigna.
Pain/tenderness isolated to the posterior half of the vestibule suggests a muscular cause, and pain in both the posterior and anterior parts of the vestibule suggests a cause that is more systemic or diffuse, which could be a result of a hormonal issue such as one related to oral contraceptives or decreased testosterone, or a nerve-related process.
Cigna uses gentle swipes of a Q-tip moistened with water or gel to examine the vulva rather than a poke or touch, with the exception being the posterior vestibule, which overlies muscle insertion sites. “Make sure to get a baseline in remote areas such as the inner thigh, and always distinguish between ‘scratchy/sensitive’ sensations and pain,” she said, noting the value of having the patient hold a mirror on her inner thigh.
Causes of Vulvar Itch: Infectious and Noninfectious
With vulvar candidiasis, a common infectious cause of vulvar itch, “you have to ask if they’re also itching on the inside because if you treat them with a topical and you don’t treat the vaginal yeast infection that may be co-occurring, they’ll keep reseeding their vulvar skin,” Cigna said, “and it will never be fully treated.”
Candida albicans is the most common cause of vulvar or vulvovaginal candidiasis, and resistance to antifungals has been rising. Non-albicans Candida “tends to have even higher resistance rates,” she said. Ordering a sensitivity panel along with the culture is helpful, but “comprehensive vaginal biome” panels are generally not useful. “It’s hard to correlate the information clinically,” she said, “and there’s not always a lot of information about susceptibilities, which is what I really like to know.”
Cigna’s treatments for vaginal infections include miconazole, terconazole, and fluconazole (and occasionally, itraconazole or voriconazole — a “decision we don’t take lightly”). Vulvar treatments include nystatin ointment, clotrimazole cream, and miconazole cream. Often, optimal treatment involves addressing “both inside and out,” she said, noting the importance of also killing yeast in undergarment fabric.
“In my experience, Diflucan [oral fluconazole] doesn’t treat persistent vulvar cutaneous skin yeast well, so while I might try Diflucan, I typically use something topical as well,” she said. “And with vaginal yeast, we do use boric acid from time to time, especially for non-albicans species because it tends to be a little more effective.”
Noninfectious causes of vulvar itch include allergic, neuropathic, and muscular causes, as well as autoimmune dermatoses and mast cell activation syndrome. Well known in dermatology are acute contact dermatitis and lichen simplex chronicus — both characterized by induration, thickening, and a “puffy” erythematous appearance, and worsening of pruritus at night. What may be less appreciated is the long list of implicated allergens , including Always menstrual pads made of a plastic-containing “dry weave” material, Cigna said. There are at least several cotton-only, low-preservative feminine products available on the market, she noted.
Common Autoimmune Vulvar Dermatoses: LS and LP
Vulvar LS has traditionally been thought to affect mainly prepubertal and postmenopausal women, but the autoimmune condition is now known to affect more reproductive-age people with vulvas than previously appreciated, Cigna said.
And notably, in an observational web-based study of premenopausal women (aged 18-50 years) with biopsy-confirmed vulvar LS, the leading symptom was not itch but dyspareunia and tearing with intercourse. This means “we’re missing people,” said Cigna, an author of the study. “We think the reason we’re not seeing itch as commonly in this population is that itch is likely mediated by the low estrogen state of pre- and postmenopausal people.”
Vulvar LS also occurs in pregnancy, with symptoms that are either stable or decrease during pregnancy and increase in the postpartum period, as demonstrated in a recently published online survey.
Patients with vulvar LS can present with hypopigmentation, lichenification, and scarring and architectural changes, the latter of which can involve clitoral phimosis, labial resorption, and narrowing of the introitus. (The vaginal mucosa is unaffected.) The presentation can be subtle, especially in premenopausal women, and differentiation between LS, vitiligo, and yeast is sometimes necessary.
A timely biopsy-driven definitive diagnosis is important because vulvar LS increases the risk for cancer if it’s not adequately treated and because long-term steroid use can affect the accuracy of pathology reports. “We really care about keeping this disease in remission as much as possible,” Cigna said. Experts in the field recommend long-term maintenance therapy with a mid-ultra-potent steroid one to three times/week or an alternative. “I’ve just started using ruxolitinib cream, a Janus kinase (JAK) inhibitor, and tacrolimus, a calcineurin inhibitor,” she said.
With vulvar LP, based on current evidence, the risk for malignant transformation is low, but “it crosses into the vagina and can cause vaginal adhesions, so if you’re diagnosing someone with lichen planus, you need to make sure you’re talking with them about dilators, and if you’re not comfortable, send them to [gyn],” she said.
The use of vulvoscopy is important for one’s ability to see the fine Wickham’s striae that often characterize vulvar LP, she noted. Medical treatments for vulvar LP include topical calcineurin inhibitors, high-potency steroids, and JAK inhibitors.
Surgical treatment of vulvar granuloma fissuratum caused by vulvar LS is possible (when the patient is in complete remission, to prevent koebnerization), with daily post-op application of clobetasol and retraction of tissues, noted Cigna, the author of a study on vulvar lysis of adhesions.
With both LS and LP, Cigna said, “don’t forget (consideration of) hormones” as an adjunctive treatment, especially in postmenopausal women. “Patients in a low hormone state will have more flares.”
Vulvar Crohn’s
“We all have to know how to look for this,” Cigna said. “Unilateral or asymmetric swelling is classic, but don’t rule out the diagnosis if you see symmetric swelling.” Patients also typically have linear “knife-like” fissures or ulcerations, the vulva “is very indurated,” and “swelling is so intense, the patients are miserable,” she said.
Vulvar Crohn’s disease may precede intestinal disease in 20%-30% of patients, so referral to a gastroenterologist — and ideally subsequent collaboration — is important, as vulvar manifestations are treated with systemic medications typical for Crohn’s.
A biopsy is required for diagnosis, and the pathologist should be advised to look for lichenified squamous mucosa with the Touton giant cell reaction. “Vulvar Crohn’s is a rare enough disorder that if you don’t have an experienced or informed pathologist looking at your specimen, they may miss it because they won’t be looking for it,” Cigna added in the interview. “You should be really clear about what you’re looking for.”
Neuropathic Itch, Pelvic Floor Muscle Spasm
Patients with pudendal neuralgia — caused by an injured, entrapped, or irritated pudendal nerve (originating from S2-S4) — typically present with chronic vulvar and pelvic pain that is often unprovoked and worsens with sitting. Itching upon touch is often another symptom, and some patients describe a foreign body sensation. The cause is often trauma (such as an accident or childbirth-related) as opposed to myofascial irritation, Cigna explained in her lecture.
“Your exam will be largely normal, with no skin findings, so patients will get sent away if you don’t know to look for pudendal neuralgia by pressing on the pudendal nerve or doing (or referring for) a diagnostic nerve block,” Cigna added in the interview.
Persistent genital arousal disorder (PGAD) is “more global” in that it can also originate not only from the pudendal nerve but also from nerve roots higher in the spine or even from the brain. “People feel a sense of arousal, but some describe it as an itch,” Cigna said in her lecture, referring to a 2021 consensus document on PGAD/genito-pelvic dysesthesia by the International Society for the Study of Women’s Sexual Health as a valuable resource for understanding and managing the condition.
Diagnosis and treatment usually start with a pudendal nerve block with a combination of steroid and anesthetic. If this does not relieve arousal/itching, the next step may be an MRI to look higher in the spine.
Pelvic Floor Muscle Spasm
Vulvar pain, skin itching, and irritation can be symptoms of pelvic floor muscle spasm. “Oftentimes people come to me and say, ‘I have a dermatologic problem,’” Cigna said. “The skin may look red and erythematous, but it’s probably more likely a muscle problem when you’re not finding anything, and no amount of steroid will help the itch go away when the problem lies underneath.”
Co-occurring symptoms can include vaginal dryness, clitoral pain, urethral discomfort, bladder pain/irritation, increased urgency, constipation, and anal fissures. The first-line treatment approach is pelvic floor therapy.
“Pelvic floor therapy is not just for incontinence. It’s also for pain and discomfort from muscles,” she said, noting that most patients with vulvar disorders are referred for pelvic floor therapy. “Almost all of them end up having pelvic floor dysfunction because the pelvic floor muscles spasm whenever there’s pain or inflammation.”
A Cautionary Word on Vulvodynia, and a Mast Cell Paradigm to Explore
Vulvodynia is defined as persistent pain of at least 3 months’ duration with no clear cause. “These are the patients with no skin findings,” Cigna said. But in most cases, she said, careful investigation identifies causes that are musculoskeletal, hormonal, or nerve-related.
“It’s a term that’s thrown around a lot — it’s kind of a catchall. Yet it should be a small minority of patients who truly have a diagnosis of vulvodynia,” she said.
In the early stages of investigation is the idea that mast cell proliferation and mast cell activation may play a role in some cases of vulvar and vestibular pain and itching. “We see that some patients with vulvodynia and vestibulodynia have mast cells that are increased in number in the epithelium and beneath the epithelium, and nerve staining shows an increased number of nerve endings traveling into the epithelium,” Cigna said.
“We do diagnose some people clinically” based on urticaria and other symptoms suggestive of mast cell proliferation/activation (such as flushing, abdominal cramping, diarrhea, hypotensive syncope or near syncope, and tachycardia), and “then we send them to the allergist for testing,” Cigna said.
Cigna and Murphy have no relevant financial disclosures.
A version of this article appeared on Medscape.com.
— and she believes collaboration with dermatologists is essential, especially for complex cases in what she calls a neglected, data-poor area of medicine.
She also recommends that dermatologists have a good understanding of the vestibule, “one of the most important structures in vulvar medicine,” and that they become equipped to recognize generalized and localized causes of vulvar pain and/or itch.
“The problem is, we don’t talk about [vulvovaginal pain and itch] ... it’s taboo and we’re not taught about it in medical school,” Cigna, assistant professor of obstetrics and gynecology at The George Washington University (GWU), Washington, DC, said in a grand rounds lecture held recently at the GWU School of Medicine and Health Sciences Department of Dermatology.
“There are dermatologists who don’t have much training in vulvar dermatology, and a lot of gyns don’t get as much training” as they should, she said in an interview after the lecture. “So who’s looking at people’s vulvar skin and figuring out what’s going on and giving them effective treatments and evidence-based education?”
Cigna and dermatologist Emily Murphy, MD, will be co-directors of a joint ob.gyn-dermatology Vulvar Dermatology Clinic at GWU that will be launched in 2025, with monthly clinics for particularly challenging cases where the etiology is unclear or treatment is ineffective. “We want to collaborate in a more systematic way and put our heads together and think creatively about what will improve patient care,” Cigna said in the interview.
Dermatologists have valuable expertise in the immunology and genetic factors involved in skin disorders, Cigna said. Moreover, Murphy, assistant professor of dermatology and director of the Vulvar Health Program at GWU, said in an interview, dermatologists “are comfortable in going to off-label systemic medications that ob.gyns may not use that often” and bring to the table expertise in various types of procedures.
Murphy recently trained with Melissa Mauskar, MD, associate of dermatology and obstetrics and gynecology at the University of Texas Southwestern, Dallas, and founder and director of the Gynecologic Dermatology Clinic there. “It’s so important for dermatologists to be involved. It just takes some extra training that residents aren’t getting right now,” said Murphy, a member of the newly formed Vulvar Dermatoses Research Consortium.
In her grand rounds lecture, Cigna offered pearls to dermatologists for approaching a history and exam and covered highlights of the diagnosis and treatment of various problems, from vulvar Candida infections and lichen simplex chronicus to vulvar lichen sclerosus (LS), vulvar lichen planus (LP), vulvar Crohn’s disease, pudendal neuralgia, and pelvic floor muscle spasm, as well as the role of mast cell proliferation in vulvar issues.
Approaching the History and Exam
A comprehensive history covers the start, duration, and location of pain and/or itching as well as a detailed timeline (such as timing of potential causes, including injuries or births) and symptoms (such as burning, cutting, aching, and stinging). The question of whether pain “is on the outside, at the entrance, or deeper inside” is “crucial, especially for those in dermatology,” Cigna emphasized.
“And if you’re seeing a patient for a vulvar condition, please ask them about sex. Ask, is this affecting your sexual or intimate life with your partner because this can also give you a clue about what’s going on and how you can help them,” she told the audience of dermatologists.
Queries about trauma history (physical and emotional/verbal), competitive sports (such as daily cycling, equestrian, and heavy weight lifting), endometriosis/gynecologic surgery, connective tissue disorders (such as Ehler-Danlos syndrome), and irritable bowel syndrome are all potentially important to consider. It is important to ask about anxiety, depression, and obsessive-compulsive disorder, which do not cause — but are highly associated with — vulvar dermatoses, she said.
A surprisingly large number of people with vulvovaginal issues are being diagnosed with Ehler-Danlos syndrome, so “I’m always asking, are you hypermobile because this might be affecting the musculoskeletal system, which might be affecting the pelvis,” Cigna said. “Anything that affects the pelvis can affect the vulva as well.”
The pelvic examination should be “offered” rather than assumed to be part of the exam, as part of a trauma-informed approach that is crucial for earning trust, she advised. “Just saying, ‘we’re going to talk, and then I can offer you an exam if you like’…patients like it. It helps them feel safer and more open.”
Many diagnoses are differentiated by eliciting pain on the anterior vs the posterior half of the vulvar vestibule — the part of the vulva that lies between the labia minora and is composed of nonkeratinized tissue with embryonic origins in the endoderm. “If you touch on the keratinized skin (of the vulva) and they don’t have pain, but on the vestibule they do have pain, and there is no pain inside the vagina, this suggests there is a vestibular problem,” said Cigna.
Pain/tenderness isolated to the posterior half of the vestibule suggests a muscular cause, and pain in both the posterior and anterior parts of the vestibule suggests a cause that is more systemic or diffuse, which could be a result of a hormonal issue such as one related to oral contraceptives or decreased testosterone, or a nerve-related process.
Cigna uses gentle swipes of a Q-tip moistened with water or gel to examine the vulva rather than a poke or touch, with the exception being the posterior vestibule, which overlies muscle insertion sites. “Make sure to get a baseline in remote areas such as the inner thigh, and always distinguish between ‘scratchy/sensitive’ sensations and pain,” she said, noting the value of having the patient hold a mirror on her inner thigh.
Causes of Vulvar Itch: Infectious and Noninfectious
With vulvar candidiasis, a common infectious cause of vulvar itch, “you have to ask if they’re also itching on the inside because if you treat them with a topical and you don’t treat the vaginal yeast infection that may be co-occurring, they’ll keep reseeding their vulvar skin,” Cigna said, “and it will never be fully treated.”
Candida albicans is the most common cause of vulvar or vulvovaginal candidiasis, and resistance to antifungals has been rising. Non-albicans Candida “tends to have even higher resistance rates,” she said. Ordering a sensitivity panel along with the culture is helpful, but “comprehensive vaginal biome” panels are generally not useful. “It’s hard to correlate the information clinically,” she said, “and there’s not always a lot of information about susceptibilities, which is what I really like to know.”
Cigna’s treatments for vaginal infections include miconazole, terconazole, and fluconazole (and occasionally, itraconazole or voriconazole — a “decision we don’t take lightly”). Vulvar treatments include nystatin ointment, clotrimazole cream, and miconazole cream. Often, optimal treatment involves addressing “both inside and out,” she said, noting the importance of also killing yeast in undergarment fabric.
“In my experience, Diflucan [oral fluconazole] doesn’t treat persistent vulvar cutaneous skin yeast well, so while I might try Diflucan, I typically use something topical as well,” she said. “And with vaginal yeast, we do use boric acid from time to time, especially for non-albicans species because it tends to be a little more effective.”
Noninfectious causes of vulvar itch include allergic, neuropathic, and muscular causes, as well as autoimmune dermatoses and mast cell activation syndrome. Well known in dermatology are acute contact dermatitis and lichen simplex chronicus — both characterized by induration, thickening, and a “puffy” erythematous appearance, and worsening of pruritus at night. What may be less appreciated is the long list of implicated allergens , including Always menstrual pads made of a plastic-containing “dry weave” material, Cigna said. There are at least several cotton-only, low-preservative feminine products available on the market, she noted.
Common Autoimmune Vulvar Dermatoses: LS and LP
Vulvar LS has traditionally been thought to affect mainly prepubertal and postmenopausal women, but the autoimmune condition is now known to affect more reproductive-age people with vulvas than previously appreciated, Cigna said.
And notably, in an observational web-based study of premenopausal women (aged 18-50 years) with biopsy-confirmed vulvar LS, the leading symptom was not itch but dyspareunia and tearing with intercourse. This means “we’re missing people,” said Cigna, an author of the study. “We think the reason we’re not seeing itch as commonly in this population is that itch is likely mediated by the low estrogen state of pre- and postmenopausal people.”
Vulvar LS also occurs in pregnancy, with symptoms that are either stable or decrease during pregnancy and increase in the postpartum period, as demonstrated in a recently published online survey.
Patients with vulvar LS can present with hypopigmentation, lichenification, and scarring and architectural changes, the latter of which can involve clitoral phimosis, labial resorption, and narrowing of the introitus. (The vaginal mucosa is unaffected.) The presentation can be subtle, especially in premenopausal women, and differentiation between LS, vitiligo, and yeast is sometimes necessary.
A timely biopsy-driven definitive diagnosis is important because vulvar LS increases the risk for cancer if it’s not adequately treated and because long-term steroid use can affect the accuracy of pathology reports. “We really care about keeping this disease in remission as much as possible,” Cigna said. Experts in the field recommend long-term maintenance therapy with a mid-ultra-potent steroid one to three times/week or an alternative. “I’ve just started using ruxolitinib cream, a Janus kinase (JAK) inhibitor, and tacrolimus, a calcineurin inhibitor,” she said.
With vulvar LP, based on current evidence, the risk for malignant transformation is low, but “it crosses into the vagina and can cause vaginal adhesions, so if you’re diagnosing someone with lichen planus, you need to make sure you’re talking with them about dilators, and if you’re not comfortable, send them to [gyn],” she said.
The use of vulvoscopy is important for one’s ability to see the fine Wickham’s striae that often characterize vulvar LP, she noted. Medical treatments for vulvar LP include topical calcineurin inhibitors, high-potency steroids, and JAK inhibitors.
Surgical treatment of vulvar granuloma fissuratum caused by vulvar LS is possible (when the patient is in complete remission, to prevent koebnerization), with daily post-op application of clobetasol and retraction of tissues, noted Cigna, the author of a study on vulvar lysis of adhesions.
With both LS and LP, Cigna said, “don’t forget (consideration of) hormones” as an adjunctive treatment, especially in postmenopausal women. “Patients in a low hormone state will have more flares.”
Vulvar Crohn’s
“We all have to know how to look for this,” Cigna said. “Unilateral or asymmetric swelling is classic, but don’t rule out the diagnosis if you see symmetric swelling.” Patients also typically have linear “knife-like” fissures or ulcerations, the vulva “is very indurated,” and “swelling is so intense, the patients are miserable,” she said.
Vulvar Crohn’s disease may precede intestinal disease in 20%-30% of patients, so referral to a gastroenterologist — and ideally subsequent collaboration — is important, as vulvar manifestations are treated with systemic medications typical for Crohn’s.
A biopsy is required for diagnosis, and the pathologist should be advised to look for lichenified squamous mucosa with the Touton giant cell reaction. “Vulvar Crohn’s is a rare enough disorder that if you don’t have an experienced or informed pathologist looking at your specimen, they may miss it because they won’t be looking for it,” Cigna added in the interview. “You should be really clear about what you’re looking for.”
Neuropathic Itch, Pelvic Floor Muscle Spasm
Patients with pudendal neuralgia — caused by an injured, entrapped, or irritated pudendal nerve (originating from S2-S4) — typically present with chronic vulvar and pelvic pain that is often unprovoked and worsens with sitting. Itching upon touch is often another symptom, and some patients describe a foreign body sensation. The cause is often trauma (such as an accident or childbirth-related) as opposed to myofascial irritation, Cigna explained in her lecture.
“Your exam will be largely normal, with no skin findings, so patients will get sent away if you don’t know to look for pudendal neuralgia by pressing on the pudendal nerve or doing (or referring for) a diagnostic nerve block,” Cigna added in the interview.
Persistent genital arousal disorder (PGAD) is “more global” in that it can also originate not only from the pudendal nerve but also from nerve roots higher in the spine or even from the brain. “People feel a sense of arousal, but some describe it as an itch,” Cigna said in her lecture, referring to a 2021 consensus document on PGAD/genito-pelvic dysesthesia by the International Society for the Study of Women’s Sexual Health as a valuable resource for understanding and managing the condition.
Diagnosis and treatment usually start with a pudendal nerve block with a combination of steroid and anesthetic. If this does not relieve arousal/itching, the next step may be an MRI to look higher in the spine.
Pelvic Floor Muscle Spasm
Vulvar pain, skin itching, and irritation can be symptoms of pelvic floor muscle spasm. “Oftentimes people come to me and say, ‘I have a dermatologic problem,’” Cigna said. “The skin may look red and erythematous, but it’s probably more likely a muscle problem when you’re not finding anything, and no amount of steroid will help the itch go away when the problem lies underneath.”
Co-occurring symptoms can include vaginal dryness, clitoral pain, urethral discomfort, bladder pain/irritation, increased urgency, constipation, and anal fissures. The first-line treatment approach is pelvic floor therapy.
“Pelvic floor therapy is not just for incontinence. It’s also for pain and discomfort from muscles,” she said, noting that most patients with vulvar disorders are referred for pelvic floor therapy. “Almost all of them end up having pelvic floor dysfunction because the pelvic floor muscles spasm whenever there’s pain or inflammation.”
A Cautionary Word on Vulvodynia, and a Mast Cell Paradigm to Explore
Vulvodynia is defined as persistent pain of at least 3 months’ duration with no clear cause. “These are the patients with no skin findings,” Cigna said. But in most cases, she said, careful investigation identifies causes that are musculoskeletal, hormonal, or nerve-related.
“It’s a term that’s thrown around a lot — it’s kind of a catchall. Yet it should be a small minority of patients who truly have a diagnosis of vulvodynia,” she said.
In the early stages of investigation is the idea that mast cell proliferation and mast cell activation may play a role in some cases of vulvar and vestibular pain and itching. “We see that some patients with vulvodynia and vestibulodynia have mast cells that are increased in number in the epithelium and beneath the epithelium, and nerve staining shows an increased number of nerve endings traveling into the epithelium,” Cigna said.
“We do diagnose some people clinically” based on urticaria and other symptoms suggestive of mast cell proliferation/activation (such as flushing, abdominal cramping, diarrhea, hypotensive syncope or near syncope, and tachycardia), and “then we send them to the allergist for testing,” Cigna said.
Cigna and Murphy have no relevant financial disclosures.
A version of this article appeared on Medscape.com.
Sulfites: The 2024 American Contact Dermatitis Society Allergen of the Year
The American Contact Dermatitis Society (ACDS) selected sulfites as the 2024 Allergen of the Year.1 Due to their preservative and antioxidant properties, sulfites are prevalent in a variety of foods, beverages, medications, and personal care products; however, sulfites also have been implicated as a potential contact allergen. In this article, we review common sources of sulfite exposure, clinical manifestations of allergic contact dermatitis (ACD) to sulfites, and patch testing considerations for this emerging allergen.
What Are Sulfites?
Sulfiting agents are compounds that contain the sulfite ion SO32-, including sulfur dioxide, sodium disulfite (sodium metabisulfite), and potassium metabisulfite.2 Sulfites occur naturally in the environment and commonly are used as preservatives, antibrowning agents, and antioxidants in various foods, beverages, medications, cosmetics, and skin care products. As antibrowning agents and antioxidants, sulfites help maintain the natural appearance of foods and other products and prevent premature spoiling by inactivating oxidative enzymes.3 It should be noted that sulfites and sulfates are distinct and unrelated compounds that do not cross-react.1
Common Sources of Sulfite Exposure
From a morning glass of juice to an evening shower, in the pharmacy and at the hair salon, sulfite exposure is ubiquitous in most daily routines. Sulfites are present in many foods and beverages, either as a byproduct of natural fermentation or as an additive to prevent spoiling and color change. The Table provides examples of foods with high sulfite content.1,4-6 In particular, dried fruit, bottled lemon juice, wine, grape juice, sauerkraut juice, and pickled onions have high sulfite content.

Topical medications and personal care products represent other potential sources of sulfite exposure. A number of reports have shown that sulfites may be included in topical steroids,7 antibiotics,8 antifungals,9 hemorrhoidal preparations,10 local anesthetics,11 and urinary catheterization gel,12 highlighting their many potential applications. In addition, a comprehensive ingredient analysis of 264 ophthalmic medications found that 3.8% of the products contained sodium disulfite.13 Sulfites may be found in personal care products, including facial and hand cleansers, shampoos, moisturizers, and toothpastes. Hair dyes also commonly contain sulfites,7 which are listed in as many as 90% of hair dye kits in the ACDS Contact Allergen Management Program database.1
Occupational exposures also are widespread, as sulfites are extensively utilized across diverse industries such as pharmaceuticals, health care, leather manufacturing, mineral extraction, food preparation, chemical manufacturing, textiles, alcohol brewing, and wine production.1
Sulfites also are used in the rubber industry—particularly in gloves—due to their anticoagulant and preservative properties.4 This is relevant to health care providers, who may use dozens of disposable gloves in a single day. In an experimental pilot study, researchers detected sulfites in 83% (5/6) of natural rubber latex gloves, 96% (23/24) of synthetic (nitrile) gloves, and 0% (0/5) of polyvinyl chloride gloves.14 While this study was limited to a small sample size, it demonstrates the common use of sulfites in certain rubber gloves and encourages future studies to determine whether there is a quantitative threshold to elicit allergic reactions.
Sulfite Allergy
In 1968, an early case report of ACD to sulfites was published involving a pharmaceutical worker who developed hand eczema after working at a factory for 3 months and had a positive patch test to potassium metabisulfite.15 There have been other cases published in the literature since then, including localized ACD as well as less common cases of systemic contact dermatitis following oral, injectable, and rectal sulfite exposures.16
The North American Contact Dermatitis Group found that, among 132 (2.7%) of 4885 patients with positive patch tests to sodium disulfite from 2017 to 2018, the most commonly involved body sites were the face (28.8%) and hands (20.5%) followed by a scattered/generalized distribution (13.6%). Involvement of the face and hands may correlate with the most frequent sources of exposure that were identified, including personal care products (particularly hair dyes)(18.9%), medications (9.1%), and foods (7.6%).17 A multicenter analysis of patch test results from Germany, Austria, and Switzerland from 1999 to 2013 showed that 357 (2.9%) of 12,156 patients had positive reactions to sodium disulfite, with the most commonly identified exposure sources being topical pharmaceutical agents (59.3%); cosmetics, creams, and sunscreens (13.6%); and systemic drugs (6.8%).18 However, it is not always possible to determine the clinical relevance of a positive patch test to sulfites.1
Other than the face and hands, there have been other unexpected anatomic locations for sulfite ACD (eg, the lower back), and systemic contact dermatitis has manifested with widespread rashes due to oral, rectal, and parenteral exposure.4,16,19 There is no definitive link between sulfite contact allergy and patient sex, but there seems to be a higher prevalence in patients older than 40 years, perhaps related to overall lifetime exposure.1
Immediate hypersensitivity reactions to sulfites also have been reported, including urticaria, angioedema, and anaphylaxis.4 Due to multiple cases of severe dermatologic and respiratory reactions to food products containing sulfites,20 the US Food and Drug Administration prohibited their use in fresh fruit and vegetables as antibrowning agents in 1986 and required labels on packaged foods that contained sulfites at more than 10 parts per million.21 However, food and drinks produced in restaurants, bakeries, and cafes as well as those that are distributed directly to consumers from the preparation site are exempt from these rules.17
In addition, consuming high amounts of dietary sulfites has been linked to headaches through unclear (ie, not necessarily allergic) mechanisms.4,22 One study found that wine with a higher sulfite concentration was associated with increased risk for headaches in participants who had a history of headaches related to wine consumption.22
Patch Testing to Sulfites
The North American Contact Dermatitis Group has tested sodium disulfite since 2017 and found an increased frequency of positive patch tests from 2.7% (N=4885) in 2017 and 201817 to 3.3% (N=4115) in 2019 and 202023 among patients referred for testing. Similarly, patch testing to sodium disulfite in nearly 40,000 patients in 9 European countries showed a pooled prevalence of reactions of 3.1%.17 However, this contact allergy may go unrecognized, as sulfites are not included in common patch test series, including the thin-layer rapid use epicutaneous test and the ACDS Core Allergen Series.24,25 The relatively high patch test positivity to sulfites along with the prevalence of daily exposures supports the addition of sulfites to more patch test screening series.
The recommended patch test concentration for sodium disulfite is 1% in petrolatum.5 Testing in aqueous solutions is not recommended because they can cause sulfites to break down, potentially producing false-positive or irritant patch test reactions.7,26,27
Recommendations for Patients With Sulfite Allergies
Individuals with contact allergies to sulfites should be counseled on exposure sources and should be given resources providing a list of safe products, such as the ACDS Contact Allergen Management Program (https://www.acdscamp.org/login) or SkinSAFE (https://www.skinsafeproducts.com/). Prescribers should be cognizant of sulfites that are present in prescription medications. Just because a patient has a positive patch test to sulfites does not automatically imply that they will need to modify their diet to avoid sulfite-containing foods; in the absence of cheilitis or a distribution suggestive of systemic contact dermatitis (eg, vesicular hand/foot dermatitis, intertriginous eruptions), this step may be unnecessary. On the other hand, individuals who have experienced immediate hypersensitivity reactions to sulfites should avoid sulfite-containing foods and carry an epinephrine autoinjector.
Final Interpretation
Sulfites are ubiquitous compounds found in various foods, beverages, medications, and personal care products in addition to a range of occupational exposures. The face and hands are the most common sites of sulfite ACD. Despite patch test positivity in as many as 3% of tested patients,17,23 sulfite allergy may be missed due to lack of routine testing on standard screening series.
- Ekstein SF, Warshaw EM. Sulfites: allergen of the year 2024. Dermatitis. 2024;35:6-12. doi:10.1089/derm.2023.0154
- Gunnison AF, Jacobsen DW. Sulfite hypersensitivity. a critical review. CRC Crit Rev Toxicol. 1987;17:185-214. doi:10.3109/10408448709071208
- Clough SR. Sodium sulfite. In: Wexler P, ed. Encyclopedia of Toxicology. 3rd ed. Academic Press; 2014: 341-343.
- Vally H, Misso NL, Madan V. Clinical effects of sulphite additives. Clin Exp Allergy. 2009;39:1643-1651. doi:10.1111/j.1365-2222.2009.03362.x
- Ralph N, Verma S, Merry S, et al. What is the relevance of contact allergy to sodium metabisulfite and which concentration of the allergen should we use? Dermatitis. 2015;26:162-165. doi:10.1097/der.0000000000000120
- Madan V, Walker SL, Beck MH. Sodium metabisulfite allergy is common but is it relevant? Contact Dermatitis. 2007;57:173-176. doi:10.1111/j.1600-0536.2007.01188.x
- García-Gavín J, Parente J, Goossens A. Allergic contact dermatitis caused by sodium metabisulfite: a challenging allergen. a case series and literature review. Contact Dermatitis. 2012;67:260-269. doi:10.1111/j.1600-0536.2012.02135.x
- Milpied B, van Wassenhove L, Larousse C, et al. Contact dermatitis from rifamycin. Contact Dermatitis. 1986;14:252-253. doi:10.1111/j.1600-0536.1986.tb01240.x
- Lodi A, Chiarelli G, Mancini LL, et al. Contact allergy to sodium sulfite contained in an antifungal preparation. Contact Dermatitis. 1993;29:97. doi:10.1111/j.1600-0536.1993.tb03493.x
- Sánchez-Pérez J, Abajo P, Córdoba S, et al. Allergic contact dermatitis from sodium metabisulfite in an antihemorrhoidal cream. Contact Dermatitis. 2000;42:176-177.
- Boyd AH, Warshaw EM. Sulfites: no longer a zebra? Dermatitis. 2017;28:364-366. doi:10.1097/der.0000000000000312
- Grosch E, Mahler V. Allergic contact dermatitis caused by a catheter system containing sodium metabisulfite. Contact Dermatitis. 2017;76:186-187. doi:10.1111/cod.12675
- Shaver RL, Warshaw EM. Contact allergens in prescription topical ophthalmic medications. Dermatitis. 2022;33:135-143. doi:10.1097/der.0000000000000751
- Dendooven E, Darrigade AS, Foubert K, et al. The presence of sulfites in ‘natural rubber latex’ and ‘synthetic’ rubber gloves: an experimental pilot study. Br J Dermatol. 2020;182:1054-1055. doi:10.1111/bjd.18608
- Nater JP. Allergic contact dermatitis caused by potassium metabisulfite. Dermatologica. 1968;136:477-478. doi:10.1159/000254143
- Borges AS, Valejo Coelho MM, Fernandes C, et al. Systemic allergic dermatitis caused by sodium metabisulfite in rectal enemas. Contact Dermatitis. 2018;78:429-430. doi:10.1111/cod.12971
- Warshaw EM, Buonomo M, DeKoven JG, et al. Patch testing with sodium disulfite: North American Contact Dermatitis Group experience, 2017 to 2018. Contact Dermatitis. 2021;85:285-296. doi:10.1111/cod.13860
- Häberle M, Geier J, Mahler V. Contact allergy to sulfites: clinical and occupational relevance—new data from the German Contact Dermatitis Research Group and the Information Network of Departments of Dermatology (IVDK). J Dtsch Dermatol Ges. 2016;14:938-941. doi:10.1111/ddg.13009
- Tan MG, Li HO, Pratt MD. Systemic allergic dermatitis to sodium metabisulfite in local anesthetic solution. Contact Dermatitis. 2022;86:120-121. doi:10.1111/cod.13978
- D’Amore T, Di Taranto A, Berardi G, et al. Sulfites in meat: occurrence, activity, toxicity, regulation, and detection. a comprehensive review. Compr Rev Food Sci Food Saf. 2020;19:2701-2720. doi:10.1111/1541-4337.12607
- Grotheer P, Marshall M, Simonne A. Sulfites: separating fact from fiction. May 11, 2022. UF IFAS Extension. University of Florida. Accessed October 4, 2024. https://edis.ifas.ufl.edu/publication/FY731
- Silva M, Gama J, Pinto N, et al. Sulfite concentration and the occurrence of headache in young adults: a prospective study. Eur J Clin Nutr. 2019;73:1316-1322. doi:10.1038/s41430-019-0420-2
- DeKoven JG, Warshaw EM, Reeder MJ, et al. North American Contact Dermatitis Group patch test results: 2019-2020. Dermatitis. 2023;34:90-104. doi:10.1089/derm.2022.29017.jdk
- T.R.U.E. Test. Thin-layer rapid use epicutaneous patch test. SmartPractice Dermatology Allergy. Accessed October 4, 2024. https://www.smartpractice.com/shop/category?id=581719&m=SPA
- Schalock PC, Dunnick CA, Nedorost, et al; American Contact Dermatitis Society Core Allergen Series Committee. American Contact Dermatitis Society Core Allergen Series: 2020 update. Dermatitis. 2020;31:279-282.
- Kaaman AC, Boman A, Wrangsjö K, et al. Contact allergy to sodium metabisulfite: an occupational problem. Contact Dermatitis. 2010;63:110-112. doi:10.1111/j.1600-0536.2010.01756.x
- Vena GA, Foti C, Angelini G. Sulfite contact allergy. Contact Dermatitis. 1994;31:172-175. doi:10.1111/j.1600-0536.1994.tb01959.x
The American Contact Dermatitis Society (ACDS) selected sulfites as the 2024 Allergen of the Year.1 Due to their preservative and antioxidant properties, sulfites are prevalent in a variety of foods, beverages, medications, and personal care products; however, sulfites also have been implicated as a potential contact allergen. In this article, we review common sources of sulfite exposure, clinical manifestations of allergic contact dermatitis (ACD) to sulfites, and patch testing considerations for this emerging allergen.
What Are Sulfites?
Sulfiting agents are compounds that contain the sulfite ion SO32-, including sulfur dioxide, sodium disulfite (sodium metabisulfite), and potassium metabisulfite.2 Sulfites occur naturally in the environment and commonly are used as preservatives, antibrowning agents, and antioxidants in various foods, beverages, medications, cosmetics, and skin care products. As antibrowning agents and antioxidants, sulfites help maintain the natural appearance of foods and other products and prevent premature spoiling by inactivating oxidative enzymes.3 It should be noted that sulfites and sulfates are distinct and unrelated compounds that do not cross-react.1
Common Sources of Sulfite Exposure
From a morning glass of juice to an evening shower, in the pharmacy and at the hair salon, sulfite exposure is ubiquitous in most daily routines. Sulfites are present in many foods and beverages, either as a byproduct of natural fermentation or as an additive to prevent spoiling and color change. The Table provides examples of foods with high sulfite content.1,4-6 In particular, dried fruit, bottled lemon juice, wine, grape juice, sauerkraut juice, and pickled onions have high sulfite content.
Topical medications and personal care products represent other potential sources of sulfite exposure. A number of reports have shown that sulfites may be included in topical steroids,7 antibiotics,8 antifungals,9 hemorrhoidal preparations,10 local anesthetics,11 and urinary catheterization gel,12 highlighting their many potential applications. In addition, a comprehensive ingredient analysis of 264 ophthalmic medications found that 3.8% of the products contained sodium disulfite.13 Sulfites may be found in personal care products, including facial and hand cleansers, shampoos, moisturizers, and toothpastes. Hair dyes also commonly contain sulfites,7 which are listed in as many as 90% of hair dye kits in the ACDS Contact Allergen Management Program database.1
Occupational exposures also are widespread, as sulfites are extensively utilized across diverse industries such as pharmaceuticals, health care, leather manufacturing, mineral extraction, food preparation, chemical manufacturing, textiles, alcohol brewing, and wine production.1
Sulfites also are used in the rubber industry—particularly in gloves—due to their anticoagulant and preservative properties.4 This is relevant to health care providers, who may use dozens of disposable gloves in a single day. In an experimental pilot study, researchers detected sulfites in 83% (5/6) of natural rubber latex gloves, 96% (23/24) of synthetic (nitrile) gloves, and 0% (0/5) of polyvinyl chloride gloves.14 While this study was limited to a small sample size, it demonstrates the common use of sulfites in certain rubber gloves and encourages future studies to determine whether there is a quantitative threshold to elicit allergic reactions.
Sulfite Allergy
In 1968, an early case report of ACD to sulfites was published involving a pharmaceutical worker who developed hand eczema after working at a factory for 3 months and had a positive patch test to potassium metabisulfite.15 There have been other cases published in the literature since then, including localized ACD as well as less common cases of systemic contact dermatitis following oral, injectable, and rectal sulfite exposures.16
The North American Contact Dermatitis Group found that, among 132 (2.7%) of 4885 patients with positive patch tests to sodium disulfite from 2017 to 2018, the most commonly involved body sites were the face (28.8%) and hands (20.5%) followed by a scattered/generalized distribution (13.6%). Involvement of the face and hands may correlate with the most frequent sources of exposure that were identified, including personal care products (particularly hair dyes)(18.9%), medications (9.1%), and foods (7.6%).17 A multicenter analysis of patch test results from Germany, Austria, and Switzerland from 1999 to 2013 showed that 357 (2.9%) of 12,156 patients had positive reactions to sodium disulfite, with the most commonly identified exposure sources being topical pharmaceutical agents (59.3%); cosmetics, creams, and sunscreens (13.6%); and systemic drugs (6.8%).18 However, it is not always possible to determine the clinical relevance of a positive patch test to sulfites.1
Other than the face and hands, there have been other unexpected anatomic locations for sulfite ACD (eg, the lower back), and systemic contact dermatitis has manifested with widespread rashes due to oral, rectal, and parenteral exposure.4,16,19 There is no definitive link between sulfite contact allergy and patient sex, but there seems to be a higher prevalence in patients older than 40 years, perhaps related to overall lifetime exposure.1
Immediate hypersensitivity reactions to sulfites also have been reported, including urticaria, angioedema, and anaphylaxis.4 Due to multiple cases of severe dermatologic and respiratory reactions to food products containing sulfites,20 the US Food and Drug Administration prohibited their use in fresh fruit and vegetables as antibrowning agents in 1986 and required labels on packaged foods that contained sulfites at more than 10 parts per million.21 However, food and drinks produced in restaurants, bakeries, and cafes as well as those that are distributed directly to consumers from the preparation site are exempt from these rules.17
In addition, consuming high amounts of dietary sulfites has been linked to headaches through unclear (ie, not necessarily allergic) mechanisms.4,22 One study found that wine with a higher sulfite concentration was associated with increased risk for headaches in participants who had a history of headaches related to wine consumption.22
Patch Testing to Sulfites
The North American Contact Dermatitis Group has tested sodium disulfite since 2017 and found an increased frequency of positive patch tests from 2.7% (N=4885) in 2017 and 201817 to 3.3% (N=4115) in 2019 and 202023 among patients referred for testing. Similarly, patch testing to sodium disulfite in nearly 40,000 patients in 9 European countries showed a pooled prevalence of reactions of 3.1%.17 However, this contact allergy may go unrecognized, as sulfites are not included in common patch test series, including the thin-layer rapid use epicutaneous test and the ACDS Core Allergen Series.24,25 The relatively high patch test positivity to sulfites along with the prevalence of daily exposures supports the addition of sulfites to more patch test screening series.
The recommended patch test concentration for sodium disulfite is 1% in petrolatum.5 Testing in aqueous solutions is not recommended because they can cause sulfites to break down, potentially producing false-positive or irritant patch test reactions.7,26,27
Recommendations for Patients With Sulfite Allergies
Individuals with contact allergies to sulfites should be counseled on exposure sources and should be given resources providing a list of safe products, such as the ACDS Contact Allergen Management Program (https://www.acdscamp.org/login) or SkinSAFE (https://www.skinsafeproducts.com/). Prescribers should be cognizant of sulfites that are present in prescription medications. Just because a patient has a positive patch test to sulfites does not automatically imply that they will need to modify their diet to avoid sulfite-containing foods; in the absence of cheilitis or a distribution suggestive of systemic contact dermatitis (eg, vesicular hand/foot dermatitis, intertriginous eruptions), this step may be unnecessary. On the other hand, individuals who have experienced immediate hypersensitivity reactions to sulfites should avoid sulfite-containing foods and carry an epinephrine autoinjector.
Final Interpretation
Sulfites are ubiquitous compounds found in various foods, beverages, medications, and personal care products in addition to a range of occupational exposures. The face and hands are the most common sites of sulfite ACD. Despite patch test positivity in as many as 3% of tested patients,17,23 sulfite allergy may be missed due to lack of routine testing on standard screening series.
The American Contact Dermatitis Society (ACDS) selected sulfites as the 2024 Allergen of the Year.1 Due to their preservative and antioxidant properties, sulfites are prevalent in a variety of foods, beverages, medications, and personal care products; however, sulfites also have been implicated as a potential contact allergen. In this article, we review common sources of sulfite exposure, clinical manifestations of allergic contact dermatitis (ACD) to sulfites, and patch testing considerations for this emerging allergen.
What Are Sulfites?
Sulfiting agents are compounds that contain the sulfite ion SO32-, including sulfur dioxide, sodium disulfite (sodium metabisulfite), and potassium metabisulfite.2 Sulfites occur naturally in the environment and commonly are used as preservatives, antibrowning agents, and antioxidants in various foods, beverages, medications, cosmetics, and skin care products. As antibrowning agents and antioxidants, sulfites help maintain the natural appearance of foods and other products and prevent premature spoiling by inactivating oxidative enzymes.3 It should be noted that sulfites and sulfates are distinct and unrelated compounds that do not cross-react.1
Common Sources of Sulfite Exposure
From a morning glass of juice to an evening shower, in the pharmacy and at the hair salon, sulfite exposure is ubiquitous in most daily routines. Sulfites are present in many foods and beverages, either as a byproduct of natural fermentation or as an additive to prevent spoiling and color change. The Table provides examples of foods with high sulfite content.1,4-6 In particular, dried fruit, bottled lemon juice, wine, grape juice, sauerkraut juice, and pickled onions have high sulfite content.
Topical medications and personal care products represent other potential sources of sulfite exposure. A number of reports have shown that sulfites may be included in topical steroids,7 antibiotics,8 antifungals,9 hemorrhoidal preparations,10 local anesthetics,11 and urinary catheterization gel,12 highlighting their many potential applications. In addition, a comprehensive ingredient analysis of 264 ophthalmic medications found that 3.8% of the products contained sodium disulfite.13 Sulfites may be found in personal care products, including facial and hand cleansers, shampoos, moisturizers, and toothpastes. Hair dyes also commonly contain sulfites,7 which are listed in as many as 90% of hair dye kits in the ACDS Contact Allergen Management Program database.1
Occupational exposures also are widespread, as sulfites are extensively utilized across diverse industries such as pharmaceuticals, health care, leather manufacturing, mineral extraction, food preparation, chemical manufacturing, textiles, alcohol brewing, and wine production.1
Sulfites also are used in the rubber industry—particularly in gloves—due to their anticoagulant and preservative properties.4 This is relevant to health care providers, who may use dozens of disposable gloves in a single day. In an experimental pilot study, researchers detected sulfites in 83% (5/6) of natural rubber latex gloves, 96% (23/24) of synthetic (nitrile) gloves, and 0% (0/5) of polyvinyl chloride gloves.14 While this study was limited to a small sample size, it demonstrates the common use of sulfites in certain rubber gloves and encourages future studies to determine whether there is a quantitative threshold to elicit allergic reactions.
Sulfite Allergy
In 1968, an early case report of ACD to sulfites was published involving a pharmaceutical worker who developed hand eczema after working at a factory for 3 months and had a positive patch test to potassium metabisulfite.15 There have been other cases published in the literature since then, including localized ACD as well as less common cases of systemic contact dermatitis following oral, injectable, and rectal sulfite exposures.16
The North American Contact Dermatitis Group found that, among 132 (2.7%) of 4885 patients with positive patch tests to sodium disulfite from 2017 to 2018, the most commonly involved body sites were the face (28.8%) and hands (20.5%) followed by a scattered/generalized distribution (13.6%). Involvement of the face and hands may correlate with the most frequent sources of exposure that were identified, including personal care products (particularly hair dyes)(18.9%), medications (9.1%), and foods (7.6%).17 A multicenter analysis of patch test results from Germany, Austria, and Switzerland from 1999 to 2013 showed that 357 (2.9%) of 12,156 patients had positive reactions to sodium disulfite, with the most commonly identified exposure sources being topical pharmaceutical agents (59.3%); cosmetics, creams, and sunscreens (13.6%); and systemic drugs (6.8%).18 However, it is not always possible to determine the clinical relevance of a positive patch test to sulfites.1
Other than the face and hands, there have been other unexpected anatomic locations for sulfite ACD (eg, the lower back), and systemic contact dermatitis has manifested with widespread rashes due to oral, rectal, and parenteral exposure.4,16,19 There is no definitive link between sulfite contact allergy and patient sex, but there seems to be a higher prevalence in patients older than 40 years, perhaps related to overall lifetime exposure.1
Immediate hypersensitivity reactions to sulfites also have been reported, including urticaria, angioedema, and anaphylaxis.4 Due to multiple cases of severe dermatologic and respiratory reactions to food products containing sulfites,20 the US Food and Drug Administration prohibited their use in fresh fruit and vegetables as antibrowning agents in 1986 and required labels on packaged foods that contained sulfites at more than 10 parts per million.21 However, food and drinks produced in restaurants, bakeries, and cafes as well as those that are distributed directly to consumers from the preparation site are exempt from these rules.17
In addition, consuming high amounts of dietary sulfites has been linked to headaches through unclear (ie, not necessarily allergic) mechanisms.4,22 One study found that wine with a higher sulfite concentration was associated with increased risk for headaches in participants who had a history of headaches related to wine consumption.22
Patch Testing to Sulfites
The North American Contact Dermatitis Group has tested sodium disulfite since 2017 and found an increased frequency of positive patch tests from 2.7% (N=4885) in 2017 and 201817 to 3.3% (N=4115) in 2019 and 202023 among patients referred for testing. Similarly, patch testing to sodium disulfite in nearly 40,000 patients in 9 European countries showed a pooled prevalence of reactions of 3.1%.17 However, this contact allergy may go unrecognized, as sulfites are not included in common patch test series, including the thin-layer rapid use epicutaneous test and the ACDS Core Allergen Series.24,25 The relatively high patch test positivity to sulfites along with the prevalence of daily exposures supports the addition of sulfites to more patch test screening series.
The recommended patch test concentration for sodium disulfite is 1% in petrolatum.5 Testing in aqueous solutions is not recommended because they can cause sulfites to break down, potentially producing false-positive or irritant patch test reactions.7,26,27
Recommendations for Patients With Sulfite Allergies
Individuals with contact allergies to sulfites should be counseled on exposure sources and should be given resources providing a list of safe products, such as the ACDS Contact Allergen Management Program (https://www.acdscamp.org/login) or SkinSAFE (https://www.skinsafeproducts.com/). Prescribers should be cognizant of sulfites that are present in prescription medications. Just because a patient has a positive patch test to sulfites does not automatically imply that they will need to modify their diet to avoid sulfite-containing foods; in the absence of cheilitis or a distribution suggestive of systemic contact dermatitis (eg, vesicular hand/foot dermatitis, intertriginous eruptions), this step may be unnecessary. On the other hand, individuals who have experienced immediate hypersensitivity reactions to sulfites should avoid sulfite-containing foods and carry an epinephrine autoinjector.
Final Interpretation
Sulfites are ubiquitous compounds found in various foods, beverages, medications, and personal care products in addition to a range of occupational exposures. The face and hands are the most common sites of sulfite ACD. Despite patch test positivity in as many as 3% of tested patients,17,23 sulfite allergy may be missed due to lack of routine testing on standard screening series.
- Ekstein SF, Warshaw EM. Sulfites: allergen of the year 2024. Dermatitis. 2024;35:6-12. doi:10.1089/derm.2023.0154
- Gunnison AF, Jacobsen DW. Sulfite hypersensitivity. a critical review. CRC Crit Rev Toxicol. 1987;17:185-214. doi:10.3109/10408448709071208
- Clough SR. Sodium sulfite. In: Wexler P, ed. Encyclopedia of Toxicology. 3rd ed. Academic Press; 2014: 341-343.
- Vally H, Misso NL, Madan V. Clinical effects of sulphite additives. Clin Exp Allergy. 2009;39:1643-1651. doi:10.1111/j.1365-2222.2009.03362.x
- Ralph N, Verma S, Merry S, et al. What is the relevance of contact allergy to sodium metabisulfite and which concentration of the allergen should we use? Dermatitis. 2015;26:162-165. doi:10.1097/der.0000000000000120
- Madan V, Walker SL, Beck MH. Sodium metabisulfite allergy is common but is it relevant? Contact Dermatitis. 2007;57:173-176. doi:10.1111/j.1600-0536.2007.01188.x
- García-Gavín J, Parente J, Goossens A. Allergic contact dermatitis caused by sodium metabisulfite: a challenging allergen. a case series and literature review. Contact Dermatitis. 2012;67:260-269. doi:10.1111/j.1600-0536.2012.02135.x
- Milpied B, van Wassenhove L, Larousse C, et al. Contact dermatitis from rifamycin. Contact Dermatitis. 1986;14:252-253. doi:10.1111/j.1600-0536.1986.tb01240.x
- Lodi A, Chiarelli G, Mancini LL, et al. Contact allergy to sodium sulfite contained in an antifungal preparation. Contact Dermatitis. 1993;29:97. doi:10.1111/j.1600-0536.1993.tb03493.x
- Sánchez-Pérez J, Abajo P, Córdoba S, et al. Allergic contact dermatitis from sodium metabisulfite in an antihemorrhoidal cream. Contact Dermatitis. 2000;42:176-177.
- Boyd AH, Warshaw EM. Sulfites: no longer a zebra? Dermatitis. 2017;28:364-366. doi:10.1097/der.0000000000000312
- Grosch E, Mahler V. Allergic contact dermatitis caused by a catheter system containing sodium metabisulfite. Contact Dermatitis. 2017;76:186-187. doi:10.1111/cod.12675
- Shaver RL, Warshaw EM. Contact allergens in prescription topical ophthalmic medications. Dermatitis. 2022;33:135-143. doi:10.1097/der.0000000000000751
- Dendooven E, Darrigade AS, Foubert K, et al. The presence of sulfites in ‘natural rubber latex’ and ‘synthetic’ rubber gloves: an experimental pilot study. Br J Dermatol. 2020;182:1054-1055. doi:10.1111/bjd.18608
- Nater JP. Allergic contact dermatitis caused by potassium metabisulfite. Dermatologica. 1968;136:477-478. doi:10.1159/000254143
- Borges AS, Valejo Coelho MM, Fernandes C, et al. Systemic allergic dermatitis caused by sodium metabisulfite in rectal enemas. Contact Dermatitis. 2018;78:429-430. doi:10.1111/cod.12971
- Warshaw EM, Buonomo M, DeKoven JG, et al. Patch testing with sodium disulfite: North American Contact Dermatitis Group experience, 2017 to 2018. Contact Dermatitis. 2021;85:285-296. doi:10.1111/cod.13860
- Häberle M, Geier J, Mahler V. Contact allergy to sulfites: clinical and occupational relevance—new data from the German Contact Dermatitis Research Group and the Information Network of Departments of Dermatology (IVDK). J Dtsch Dermatol Ges. 2016;14:938-941. doi:10.1111/ddg.13009
- Tan MG, Li HO, Pratt MD. Systemic allergic dermatitis to sodium metabisulfite in local anesthetic solution. Contact Dermatitis. 2022;86:120-121. doi:10.1111/cod.13978
- D’Amore T, Di Taranto A, Berardi G, et al. Sulfites in meat: occurrence, activity, toxicity, regulation, and detection. a comprehensive review. Compr Rev Food Sci Food Saf. 2020;19:2701-2720. doi:10.1111/1541-4337.12607
- Grotheer P, Marshall M, Simonne A. Sulfites: separating fact from fiction. May 11, 2022. UF IFAS Extension. University of Florida. Accessed October 4, 2024. https://edis.ifas.ufl.edu/publication/FY731
- Silva M, Gama J, Pinto N, et al. Sulfite concentration and the occurrence of headache in young adults: a prospective study. Eur J Clin Nutr. 2019;73:1316-1322. doi:10.1038/s41430-019-0420-2
- DeKoven JG, Warshaw EM, Reeder MJ, et al. North American Contact Dermatitis Group patch test results: 2019-2020. Dermatitis. 2023;34:90-104. doi:10.1089/derm.2022.29017.jdk
- T.R.U.E. Test. Thin-layer rapid use epicutaneous patch test. SmartPractice Dermatology Allergy. Accessed October 4, 2024. https://www.smartpractice.com/shop/category?id=581719&m=SPA
- Schalock PC, Dunnick CA, Nedorost, et al; American Contact Dermatitis Society Core Allergen Series Committee. American Contact Dermatitis Society Core Allergen Series: 2020 update. Dermatitis. 2020;31:279-282.
- Kaaman AC, Boman A, Wrangsjö K, et al. Contact allergy to sodium metabisulfite: an occupational problem. Contact Dermatitis. 2010;63:110-112. doi:10.1111/j.1600-0536.2010.01756.x
- Vena GA, Foti C, Angelini G. Sulfite contact allergy. Contact Dermatitis. 1994;31:172-175. doi:10.1111/j.1600-0536.1994.tb01959.x
- Ekstein SF, Warshaw EM. Sulfites: allergen of the year 2024. Dermatitis. 2024;35:6-12. doi:10.1089/derm.2023.0154
- Gunnison AF, Jacobsen DW. Sulfite hypersensitivity. a critical review. CRC Crit Rev Toxicol. 1987;17:185-214. doi:10.3109/10408448709071208
- Clough SR. Sodium sulfite. In: Wexler P, ed. Encyclopedia of Toxicology. 3rd ed. Academic Press; 2014: 341-343.
- Vally H, Misso NL, Madan V. Clinical effects of sulphite additives. Clin Exp Allergy. 2009;39:1643-1651. doi:10.1111/j.1365-2222.2009.03362.x
- Ralph N, Verma S, Merry S, et al. What is the relevance of contact allergy to sodium metabisulfite and which concentration of the allergen should we use? Dermatitis. 2015;26:162-165. doi:10.1097/der.0000000000000120
- Madan V, Walker SL, Beck MH. Sodium metabisulfite allergy is common but is it relevant? Contact Dermatitis. 2007;57:173-176. doi:10.1111/j.1600-0536.2007.01188.x
- García-Gavín J, Parente J, Goossens A. Allergic contact dermatitis caused by sodium metabisulfite: a challenging allergen. a case series and literature review. Contact Dermatitis. 2012;67:260-269. doi:10.1111/j.1600-0536.2012.02135.x
- Milpied B, van Wassenhove L, Larousse C, et al. Contact dermatitis from rifamycin. Contact Dermatitis. 1986;14:252-253. doi:10.1111/j.1600-0536.1986.tb01240.x
- Lodi A, Chiarelli G, Mancini LL, et al. Contact allergy to sodium sulfite contained in an antifungal preparation. Contact Dermatitis. 1993;29:97. doi:10.1111/j.1600-0536.1993.tb03493.x
- Sánchez-Pérez J, Abajo P, Córdoba S, et al. Allergic contact dermatitis from sodium metabisulfite in an antihemorrhoidal cream. Contact Dermatitis. 2000;42:176-177.
- Boyd AH, Warshaw EM. Sulfites: no longer a zebra? Dermatitis. 2017;28:364-366. doi:10.1097/der.0000000000000312
- Grosch E, Mahler V. Allergic contact dermatitis caused by a catheter system containing sodium metabisulfite. Contact Dermatitis. 2017;76:186-187. doi:10.1111/cod.12675
- Shaver RL, Warshaw EM. Contact allergens in prescription topical ophthalmic medications. Dermatitis. 2022;33:135-143. doi:10.1097/der.0000000000000751
- Dendooven E, Darrigade AS, Foubert K, et al. The presence of sulfites in ‘natural rubber latex’ and ‘synthetic’ rubber gloves: an experimental pilot study. Br J Dermatol. 2020;182:1054-1055. doi:10.1111/bjd.18608
- Nater JP. Allergic contact dermatitis caused by potassium metabisulfite. Dermatologica. 1968;136:477-478. doi:10.1159/000254143
- Borges AS, Valejo Coelho MM, Fernandes C, et al. Systemic allergic dermatitis caused by sodium metabisulfite in rectal enemas. Contact Dermatitis. 2018;78:429-430. doi:10.1111/cod.12971
- Warshaw EM, Buonomo M, DeKoven JG, et al. Patch testing with sodium disulfite: North American Contact Dermatitis Group experience, 2017 to 2018. Contact Dermatitis. 2021;85:285-296. doi:10.1111/cod.13860
- Häberle M, Geier J, Mahler V. Contact allergy to sulfites: clinical and occupational relevance—new data from the German Contact Dermatitis Research Group and the Information Network of Departments of Dermatology (IVDK). J Dtsch Dermatol Ges. 2016;14:938-941. doi:10.1111/ddg.13009
- Tan MG, Li HO, Pratt MD. Systemic allergic dermatitis to sodium metabisulfite in local anesthetic solution. Contact Dermatitis. 2022;86:120-121. doi:10.1111/cod.13978
- D’Amore T, Di Taranto A, Berardi G, et al. Sulfites in meat: occurrence, activity, toxicity, regulation, and detection. a comprehensive review. Compr Rev Food Sci Food Saf. 2020;19:2701-2720. doi:10.1111/1541-4337.12607
- Grotheer P, Marshall M, Simonne A. Sulfites: separating fact from fiction. May 11, 2022. UF IFAS Extension. University of Florida. Accessed October 4, 2024. https://edis.ifas.ufl.edu/publication/FY731
- Silva M, Gama J, Pinto N, et al. Sulfite concentration and the occurrence of headache in young adults: a prospective study. Eur J Clin Nutr. 2019;73:1316-1322. doi:10.1038/s41430-019-0420-2
- DeKoven JG, Warshaw EM, Reeder MJ, et al. North American Contact Dermatitis Group patch test results: 2019-2020. Dermatitis. 2023;34:90-104. doi:10.1089/derm.2022.29017.jdk
- T.R.U.E. Test. Thin-layer rapid use epicutaneous patch test. SmartPractice Dermatology Allergy. Accessed October 4, 2024. https://www.smartpractice.com/shop/category?id=581719&m=SPA
- Schalock PC, Dunnick CA, Nedorost, et al; American Contact Dermatitis Society Core Allergen Series Committee. American Contact Dermatitis Society Core Allergen Series: 2020 update. Dermatitis. 2020;31:279-282.
- Kaaman AC, Boman A, Wrangsjö K, et al. Contact allergy to sodium metabisulfite: an occupational problem. Contact Dermatitis. 2010;63:110-112. doi:10.1111/j.1600-0536.2010.01756.x
- Vena GA, Foti C, Angelini G. Sulfite contact allergy. Contact Dermatitis. 1994;31:172-175. doi:10.1111/j.1600-0536.1994.tb01959.x
Practice Points
- Sulfites are ubiquitous compounds that serve as preservatives and antioxidants in various foods, beverages, medications, and personal care products.
- Allergic contact dermatitis to sulfites most commonly affects the face and hands.
- Because sulfites are not included in most patch test screening series, contact allergy to sulfites may be missed unless expanded testing is performed.

Cosmetic Dermatology Product Recalls Still Common, Analysis Finds
TOPLINE:
Between 2011 and 2023, the US Food and Drug Administration (FDA) reported recalls of 334 cosmetic dermatology products in the United States, affecting over 77 million units, predominantly due to bacterial contamination.
METHODOLOGY:
- Researchers conducted a cross-sectional analysis of the FDA Enforcement Report database for cosmetic dermatology products from 2011 to 2023.
- Cosmetic products are any article “intended for body cleaning or beauty enhancement,” as defined by the FDA.
- Recalls were categorized by product type, reason for the recall, microbial contaminant, inorganic contaminant, distribution, and risk classification.
TAKEAWAY:
- During the study period, 334 voluntary and manufacturer-initiated recalls of cosmetic products were reported, affecting 77,135,700 units.
- A total of 297 recalls (88.9%) were categorized as Class II, indicating that they caused “medically reversible health consequences.” The median recall duration was 307 days.
- Hygiene and cleaning products accounted for most of the recalls (51.5%). Makeup gels, soaps, shampoos, tattoo ink, wipes, and lotions were the most recalled product categories. Nearly 51% of the products were distributed internationally.
- Microbial and inorganic contamination accounted for 76.8% and 10.2% of the recalls (the two most common reasons for the recall), respectively, with bacteria (80%) the most common contaminating pathogen (primarily Pseudomonas and Burkholderia species).
IN PRACTICE:
With 77 million units recalled by the FDA over 12 years, cosmetic recalls have remained common, the authors concluded, adding that “dermatologists should be key voices in pharmacovigilance given scientific expertise and frontline experience managing products and associated concerns.” Dermatologists, they added, “should also be aware of FDA enforcement reports for recall updates given that average recall termination took approximately 1 year.”
SOURCE:
The study was led by Kaushik P. Venkatesh, MBA, MPH, Harvard Medical School, Boston, and was published online on October 29 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The study’s limitations include the potential underreporting of Class III recalls (products that are unlikely to cause any adverse health reaction but violate FDA labeling or manufacturing laws) and lack of complete information on contaminants.
DISCLOSURES:
No information on funding was provided in the study. No conflicts of interest were reported.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Between 2011 and 2023, the US Food and Drug Administration (FDA) reported recalls of 334 cosmetic dermatology products in the United States, affecting over 77 million units, predominantly due to bacterial contamination.
METHODOLOGY:
- Researchers conducted a cross-sectional analysis of the FDA Enforcement Report database for cosmetic dermatology products from 2011 to 2023.
- Cosmetic products are any article “intended for body cleaning or beauty enhancement,” as defined by the FDA.
- Recalls were categorized by product type, reason for the recall, microbial contaminant, inorganic contaminant, distribution, and risk classification.
TAKEAWAY:
- During the study period, 334 voluntary and manufacturer-initiated recalls of cosmetic products were reported, affecting 77,135,700 units.
- A total of 297 recalls (88.9%) were categorized as Class II, indicating that they caused “medically reversible health consequences.” The median recall duration was 307 days.
- Hygiene and cleaning products accounted for most of the recalls (51.5%). Makeup gels, soaps, shampoos, tattoo ink, wipes, and lotions were the most recalled product categories. Nearly 51% of the products were distributed internationally.
- Microbial and inorganic contamination accounted for 76.8% and 10.2% of the recalls (the two most common reasons for the recall), respectively, with bacteria (80%) the most common contaminating pathogen (primarily Pseudomonas and Burkholderia species).
IN PRACTICE:
With 77 million units recalled by the FDA over 12 years, cosmetic recalls have remained common, the authors concluded, adding that “dermatologists should be key voices in pharmacovigilance given scientific expertise and frontline experience managing products and associated concerns.” Dermatologists, they added, “should also be aware of FDA enforcement reports for recall updates given that average recall termination took approximately 1 year.”
SOURCE:
The study was led by Kaushik P. Venkatesh, MBA, MPH, Harvard Medical School, Boston, and was published online on October 29 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The study’s limitations include the potential underreporting of Class III recalls (products that are unlikely to cause any adverse health reaction but violate FDA labeling or manufacturing laws) and lack of complete information on contaminants.
DISCLOSURES:
No information on funding was provided in the study. No conflicts of interest were reported.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
Between 2011 and 2023, the US Food and Drug Administration (FDA) reported recalls of 334 cosmetic dermatology products in the United States, affecting over 77 million units, predominantly due to bacterial contamination.
METHODOLOGY:
- Researchers conducted a cross-sectional analysis of the FDA Enforcement Report database for cosmetic dermatology products from 2011 to 2023.
- Cosmetic products are any article “intended for body cleaning or beauty enhancement,” as defined by the FDA.
- Recalls were categorized by product type, reason for the recall, microbial contaminant, inorganic contaminant, distribution, and risk classification.
TAKEAWAY:
- During the study period, 334 voluntary and manufacturer-initiated recalls of cosmetic products were reported, affecting 77,135,700 units.
- A total of 297 recalls (88.9%) were categorized as Class II, indicating that they caused “medically reversible health consequences.” The median recall duration was 307 days.
- Hygiene and cleaning products accounted for most of the recalls (51.5%). Makeup gels, soaps, shampoos, tattoo ink, wipes, and lotions were the most recalled product categories. Nearly 51% of the products were distributed internationally.
- Microbial and inorganic contamination accounted for 76.8% and 10.2% of the recalls (the two most common reasons for the recall), respectively, with bacteria (80%) the most common contaminating pathogen (primarily Pseudomonas and Burkholderia species).
IN PRACTICE:
With 77 million units recalled by the FDA over 12 years, cosmetic recalls have remained common, the authors concluded, adding that “dermatologists should be key voices in pharmacovigilance given scientific expertise and frontline experience managing products and associated concerns.” Dermatologists, they added, “should also be aware of FDA enforcement reports for recall updates given that average recall termination took approximately 1 year.”
SOURCE:
The study was led by Kaushik P. Venkatesh, MBA, MPH, Harvard Medical School, Boston, and was published online on October 29 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The study’s limitations include the potential underreporting of Class III recalls (products that are unlikely to cause any adverse health reaction but violate FDA labeling or manufacturing laws) and lack of complete information on contaminants.
DISCLOSURES:
No information on funding was provided in the study. No conflicts of interest were reported.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Asteraceae Dermatitis: Everyday Plants With Allergenic Potential
The Asteraceae (formerly Compositae) family of plants is derived from the ancient Greek word aster, meaning “star,” referring to the starlike arrangement of flower petals around a central disc known as a capitulum. What initially appears as a single flower is actually a composite of several smaller flowers, hence the former name Compositae.1 Well-known members of the Asteraceae family include ornamental annuals (eg, sunflowers, marigolds, cosmos), herbaceous perennials (eg, chrysanthemums, dandelions), vegetables (eg, lettuce, chicory, artichokes), herbs (eg, chamomile, tarragon), and weeds (eg, ragweed, horseweed, capeweed)(Figure 1).2

There are more than 25,000 species of Asteraceae plants that thrive in a wide range of climates worldwide. Cases of Asteraceae-induced skin reactions have been reported in North America, Europe, Asia, and Australia.3 Members of the Asteraceae family are ubiquitous in gardens, along roadsides, and in the wilderness. Occupational exposure commonly affects gardeners, florists, farmers, and forestry workers through either direct contact with plants or via airborne pollen. Furthermore, plants of the Asteraceae family are used in various products, including pediculicides (eg, insect repellents), cosmetics (eg, eye creams, body washes), and food products (eg, cooking oils, sweetening agents, coffee substitutes, herbal teas).4-6 These plants have substantial allergic potential, resulting in numerous cutaneous reactions.
Allergic Potential
Asteraceae plants can elicit both immediate and delayed hypersensitivity reactions (HSRs); for instance, exposure to ragweed pollen may cause an IgE-mediated type 1 HSR manifesting as allergic rhinitis or a type IV HSR manifesting as airborne allergic contact dermatitis.7,8 The main contact allergens present in Asteraceae plants are sesquiterpene lactones, which are found in the leaves, stems, flowers, and pollen.9-11 Sesquiterpene lactones consist of an α-methyl group attached to a lactone ring combined with a sesquiterpene.12 Patch testing can be used to diagnose Asteraceae allergy; however, the results are not consistently reliable because there is no perfect screening allergen. Patch test preparations commonly used to detect Asteraceae allergy include Compositae mix (consisting of Anthemis nobilis extract, Chamomilla recutita extract, Achillea millefolium extract, Tanacetum vulgare extract, Arnica montana extract, and parthenolide) and sesquiterpene lactone mix (consisting of alantolactone, dehydrocostus lactone, and costunolide). In North America, the prevalence of positive patch tests to Compositae mix and sesquiterpene lactone mix is approximately 2% and 0.5%, respectively.13 When patch testing is performed, both Compositae mix and sesquiterpene lactone mix should be utilized to minimize the risk of missing Asteraceae allergy, as sesquiterpene lactone mix alone does not detect all Compositae-sensitized patients. Additionally, it may be necessary to test supplemental Asteraceae allergens, including preparations from specific plants to which the patient has been exposed. Exposure to Asteraceae-containing cosmetic products may lead to dermatitis, though this is highly dependent on the particular plant species involved. For instance, the prevalence of sensitization is high in arnica (tincture) and elecampane but low with more commonly used species such as German chamomile.14
Cutaneous Manifestations
Asteraceae dermatitis, which also is known as Australian bush dermatitis, weed dermatitis, and chrysanthemum dermatitis,2 can manifest on any area of the body that directly contacts the plant or is exposed to the pollen. Asteraceae dermatitis historically was reported in older adults with a recent history of plant exposure.6,15 However, recent data have shown a female preponderance and a younger mean age of onset (46–49 years).16
There are multiple distinct clinical manifestations of Asteraceae dermatitis. The most common cutaneous finding is localized vesicular or eczematous patches on the hands or wrists. Other variations include eczematous rashes on the exposed skin of the hands, arms, face, and neck; generalized eczema; and isolated facial eczema.16,17 These variations can be attributed to contact dermatitis caused by airborne pollen, which may mimic photodermatitis. However, airborne Asteraceae dermatitis can be distinguished clinically from photodermatitis by the involvement of sun-protected areas such as the skinfolds of the eyelids, retroauricular sulci, and nasolabial folds (Figure 2).2,9 In rare cases, systemic allergic contact dermatitis can occur if the Asteraceae allergen is ingested.2,18
Other diagnostic clues include dermatitis that flares during the summer, at the peak of the growing season, with remission in the cooler months. Potential risk factors include a childhood history of atopic dermatitis and allergic rhinitis.16 With prolonged exposure, patients may develop chronic actinic dermatitis, an immunologically mediated photodermatosis characterized by lichenified and pruritic eczematous plaques located predominantly on sun-exposed areas with notable sparing of the skin folds.19 The association between Asteraceae dermatitis and chronic actinic dermatitis is highly variable, with some studies reporting a 25% correlation and others finding a stronger association of up to 80%.2,15,20 Asteraceae allergy appears to be a relatively uncommon cause of photoallergy in North America. In one recent study, 16% (3/19) of patients with chronic actinic dermatitis had positive patch or photopatch tests to sesquiterpene lactone mix, but in another large study of photopatch testing it was reported to be a rare photoallergen.21,22
Parthenium dermatitis is an allergic contact dermatitis caused by exposure to Parthenium hysterophorus, a weed of the Asteraceae family that is responsible for 30% of cases of contact dermatitis in India.23,24 Unlike the more classic manifestation of Asteraceae dermatitis, which primarily affects the upper extremities in cases from North America and Europe, Parthenium dermatitis typically occurs in an airborne pattern distribution.24
Management
While complete avoidance of Asteraceae plants is ideal, it often is unrealistic due to their abundance in nature. Therefore, minimizing exposure to the causative plants is recommended. Primary preventive measures such as wearing protective gloves and clothing and applying bentonite clay prior to exposure should be taken when working outdoors. Promptly showering after contact with plants also can reduce the risk for Asteraceae dermatitis.
Symptomatic treatment is appropriate for mild cases and includes topical corticosteroids and calcineurin inhibitors. For severe cases, systemic corticosteroids may be needed for acute flares, with azathioprine, mycophenolate, cyclosporine, or methotrexate available for recalcitrant disease. Verma et al25 found that treatment with azathioprine for 6 months resulted in greater than 60% clearance in all 12 patients, with a majority achieving 80% to 100% clearance. Methotrexate has been used at doses of 15 mg once weekly.26 Narrowband UVB and psoralen plus UVA have been effective in extensive cases; however, care should be exercised in patients with photosensitive dermatitis, who instead should practice strict photoprotection.27-29 Lakshmi et al30 reported the use of cyclosporine during the acute phase of Asteraceae dermatitis at a dose of 2.5 mg/kg daily for 4 to 8 weeks. There have been several case reports of dupilumab treating allergic contact dermatitis; however, there have been 3 cases of patients with atopic dermatitis developing Asteraceae dermatitis while taking dupilumab.31,32 Recently, oral Janus kinase inhibitors have shown success in treating refractory cases of airborne Asteraceae dermatitis.33,34 Further research is needed to determine the safety and efficacy of dupilumab and Janus kinase inhibitors for treatment of Asteraceae dermatitis.
Final Thoughts
The Asteraceae plant family is vast and diverse, with more than 200 species reported to cause allergic contact dermatitis.12 Common modes of contact include gardening, occupational exposure, airborne pollen, and use of pediculicides and cosmetics that contain components of Asteraceae plants. Educating patients on how to minimize contact with Asteraceae plants is the most effective management strategy; topical agents and oral immunosuppressives can be used for symptomatic treatment.
- Morhardt S, Morhardt E. California Desert Flowers: An Introduction to Families, Genera, and Species. University of California Press; 2004.
- Gordon LA. Compositae dermatitis. Australas J Dermatol. 1999;40:123-130. doi:10.1046/j.1440-0960.1999.00341.x
- Denisow-Pietrzyk M, Pietrzyk Ł, Denisow B. Asteraceae species as potential environmental factors of allergy. Environ Sci Pollut Res Int. 2019;26:6290-6300. doi:10.1007/s11356-019-04146-w
- Paulsen E, Chistensen LP, Andersen KE. Cosmetics and herbal remedies with Compositae plant extracts—are they tolerated by Compositae-allergic patients? Contact Dermatitis. 2008;58:15-23. doi:10.1111/j.1600-0536.2007.01250.x
- Burry JN, Reid JG, Kirk J. Australian bush dermatitis. Contact Dermatitis. 1975;1:263-264. doi:10.1111/j.1600-0536.1975.tb05422.x
- Punchihewa N, Palmer A, Nixon R. Allergic contact dermatitis to Compositae: an Australian case series. Contact Dermatitis. 2022;87:356-362. doi:10.1111/cod.14162
- Chen KW, Marusciac L, Tamas PT, et al. Ragweed pollen allergy: burden, characteristics, and management of an imported allergen source in Europe. Int Arch Allergy Immunol. 2018;176:163-180. doi:10.1159/000487997
- Schloemer JA, Zirwas MJ, Burkhart CG. Airborne contact dermatitis: common causes in the USA. Int J Dermatol. 2015;54:271-274. doi:10.1111/ijd.12692
- Arlette J, Mitchell JC. Compositae dermatitis. current aspects. Contact Dermatitis. 1981;7:129-136. doi:10.1111/j.1600-0536.1981.tb04584.x
- Mitchell JC, Dupuis G. Allergic contact dermatitis from sesquiterpenoids of the Compositae family of plants. Br J Dermatol. 1971;84:139-150. doi:10.1111/j.1365-2133.1971.tb06857.x
- Salapovic H, Geier J, Reznicek G. Quantification of Sesquiterpene lactones in Asteraceae plant extracts: evaluation of their allergenic potential. Sci Pharm. 2013;81:807-818. doi:10.3797/scipharm.1306-17
- Paulsen E. Compositae dermatitis: a survey. Contact Dermatitis. 1992;26:76-86. doi:10.1111/j.1600-0536.1992.tb00888.x. Published correction appears in Contact Dermatitis. 1992;27:208.
- DeKoven JG, Silverberg JI, Warshaw EM, et al. North American Contact Dermatitis Group patch test results: 2017-2018. Dermatitis. 2021;32:111-123. doi:10.1097/DER.0000000000000729
- Paulsen E. Contact sensitization from Compositae-containing herbal remedies and cosmetics. Contact Dermatitis. 2002;47:189-198. doi:10.1034/j.1600-0536.2002.470401.x
- Frain-Bell W, Johnson BE. Contact allergic sensitivity to plants and the photosensitivity dermatitis and actinic reticuloid syndrome. Br J Dermatol. 1979;101:503-512.
- Paulsen E, Andersen KE. Clinical patterns of Compositae dermatitis in Danish monosensitized patients. Contact Dermatitis. 2018;78:185-193. doi:10.1111/cod.12916
- Jovanovic´ M, Poljacki M. Compositae dermatitis. Med Pregl. 2003;56:43-49. doi:10.2298/mpns0302043j
- Krook G. Occupational dermatitis from Lactuca sativa (lettuce) and Cichorium (endive). simultaneous occurrence of immediate and delayed allergy as a cause of contact dermatitis. Contact Dermatitis. 1977;3:27-36. doi:10.1111/j.1600-0536.1977.tb03583.x
- Paek SY, Lim HW. Chronic actinic dermatitis. Dermatol Clin. 2014;32:355-361, viii-ix. doi:10.1016/j.det.2014.03.007
- du P Menagé H, Hawk JL, White IR. Sesquiterpene lactone mix contact sensitivity and its relationship to chronic actinic dermatitis: a follow-up study. Contact Dermatitis. 1998;39:119-122. doi:10.1111/j.1600-0536.1998.tb05859.x
- Wang CX, Belsito DV. Chronic actinic dermatitis revisited. Dermatitis. 2020;31:68-74. doi:10.1097/DER.0000000000000531
- DeLeo VA, Adler BL, Warshaw EM, et al. Photopatch test results of the North American contact dermatitis group, 1999-2009. Photodermatol Photoimmunol Photomed. 2022;38:288-291. doi:10.1111/phpp.12742
- McGovern TW, LaWarre S. Botanical briefs: the scourge of India—Parthenium hysterophorus L. Cutis. 2001;67:27-34. Published correction appears in Cutis. 2001;67:154.
- Sharma VK, Verma P, Maharaja K. Parthenium dermatitis. Photochem Photobiol Sci. 2013;12:85-94. doi:10.1039/c2pp25186h
- Verma KK, Bansal A, Sethuraman G. Parthenium dermatitis treated with azathioprine weekly pulse doses. Indian J Dermatol Venereol Leprol. 2006;72:24-27. doi:10.4103/0378-6323.19713
- Sharma VK, Bhat R, Sethuraman G, et al. Treatment of Parthenium dermatitis with methotrexate. Contact Dermatitis. 2007;57:118-119. doi:10.1111/j.1600-0536.2006.00950.x
- Burke DA, Corey G, Storrs FJ. Psoralen plus UVA protocol for Compositae photosensitivity. Am J Contact Dermat. 1996;7:171-176.
- Lovell CR. Allergic contact dermatitis due to plants. In: Plants and the Skin. Blackwell Scientific Publications; 1993:96-254.
- Dogra S, Parsad D, Handa S. Narrowband ultraviolet B in airborne contact dermatitis: a ray of hope! Br J Dermatol. 2004;150:373-374. doi:10.1111/j.1365-2133.2004.05724.x
- Lakshmi C, Srinivas CR, Jayaraman A. Ciclosporin in Parthenium dermatitis—a report of 2 cases. Contact Dermatitis. 2008;59:245-248. doi:10.1111/j.1600-0536.2007.01208.x
- Hendricks AJ, Yosipovitch G, Shi VY. Dupilumab use in dermatologic conditions beyond atopic dermatitis—a systematic review. J Dermatolog Treat. 2021;32:19-28. doi:10.1080/09546634.2019.1689227
- Napolitano M, Fabbrocini G, Patruno C. Allergic contact dermatitis to Compositae: a possible cause of dupilumab-associated facial and neck dermatitis in atopic dermatitis patients? Contact Dermatitis. 2021;85:473-474. doi:10.1111/cod.13898
- Muddebihal A, Sardana K, Sinha S, et al. Tofacitinib in refractory Parthenium-induced airborne allergic contact dermatitis. Contact Dermatitis. 2023;88:150-152. doi:10.1111/cod.14234
- Baltazar D, Shinamoto SR, Hamann CP, et al. Occupational airborne allergic contact dermatitis to invasive Compositae species treated with abrocitinib: a case report. Contact Dermatitis. 2022;87:542-544. doi:10.1111/cod.14204
The Asteraceae (formerly Compositae) family of plants is derived from the ancient Greek word aster, meaning “star,” referring to the starlike arrangement of flower petals around a central disc known as a capitulum. What initially appears as a single flower is actually a composite of several smaller flowers, hence the former name Compositae.1 Well-known members of the Asteraceae family include ornamental annuals (eg, sunflowers, marigolds, cosmos), herbaceous perennials (eg, chrysanthemums, dandelions), vegetables (eg, lettuce, chicory, artichokes), herbs (eg, chamomile, tarragon), and weeds (eg, ragweed, horseweed, capeweed)(Figure 1).2
There are more than 25,000 species of Asteraceae plants that thrive in a wide range of climates worldwide. Cases of Asteraceae-induced skin reactions have been reported in North America, Europe, Asia, and Australia.3 Members of the Asteraceae family are ubiquitous in gardens, along roadsides, and in the wilderness. Occupational exposure commonly affects gardeners, florists, farmers, and forestry workers through either direct contact with plants or via airborne pollen. Furthermore, plants of the Asteraceae family are used in various products, including pediculicides (eg, insect repellents), cosmetics (eg, eye creams, body washes), and food products (eg, cooking oils, sweetening agents, coffee substitutes, herbal teas).4-6 These plants have substantial allergic potential, resulting in numerous cutaneous reactions.
Allergic Potential
Asteraceae plants can elicit both immediate and delayed hypersensitivity reactions (HSRs); for instance, exposure to ragweed pollen may cause an IgE-mediated type 1 HSR manifesting as allergic rhinitis or a type IV HSR manifesting as airborne allergic contact dermatitis.7,8 The main contact allergens present in Asteraceae plants are sesquiterpene lactones, which are found in the leaves, stems, flowers, and pollen.9-11 Sesquiterpene lactones consist of an α-methyl group attached to a lactone ring combined with a sesquiterpene.12 Patch testing can be used to diagnose Asteraceae allergy; however, the results are not consistently reliable because there is no perfect screening allergen. Patch test preparations commonly used to detect Asteraceae allergy include Compositae mix (consisting of Anthemis nobilis extract, Chamomilla recutita extract, Achillea millefolium extract, Tanacetum vulgare extract, Arnica montana extract, and parthenolide) and sesquiterpene lactone mix (consisting of alantolactone, dehydrocostus lactone, and costunolide). In North America, the prevalence of positive patch tests to Compositae mix and sesquiterpene lactone mix is approximately 2% and 0.5%, respectively.13 When patch testing is performed, both Compositae mix and sesquiterpene lactone mix should be utilized to minimize the risk of missing Asteraceae allergy, as sesquiterpene lactone mix alone does not detect all Compositae-sensitized patients. Additionally, it may be necessary to test supplemental Asteraceae allergens, including preparations from specific plants to which the patient has been exposed. Exposure to Asteraceae-containing cosmetic products may lead to dermatitis, though this is highly dependent on the particular plant species involved. For instance, the prevalence of sensitization is high in arnica (tincture) and elecampane but low with more commonly used species such as German chamomile.14
Cutaneous Manifestations
Asteraceae dermatitis, which also is known as Australian bush dermatitis, weed dermatitis, and chrysanthemum dermatitis,2 can manifest on any area of the body that directly contacts the plant or is exposed to the pollen. Asteraceae dermatitis historically was reported in older adults with a recent history of plant exposure.6,15 However, recent data have shown a female preponderance and a younger mean age of onset (46–49 years).16
There are multiple distinct clinical manifestations of Asteraceae dermatitis. The most common cutaneous finding is localized vesicular or eczematous patches on the hands or wrists. Other variations include eczematous rashes on the exposed skin of the hands, arms, face, and neck; generalized eczema; and isolated facial eczema.16,17 These variations can be attributed to contact dermatitis caused by airborne pollen, which may mimic photodermatitis. However, airborne Asteraceae dermatitis can be distinguished clinically from photodermatitis by the involvement of sun-protected areas such as the skinfolds of the eyelids, retroauricular sulci, and nasolabial folds (Figure 2).2,9 In rare cases, systemic allergic contact dermatitis can occur if the Asteraceae allergen is ingested.2,18
Other diagnostic clues include dermatitis that flares during the summer, at the peak of the growing season, with remission in the cooler months. Potential risk factors include a childhood history of atopic dermatitis and allergic rhinitis.16 With prolonged exposure, patients may develop chronic actinic dermatitis, an immunologically mediated photodermatosis characterized by lichenified and pruritic eczematous plaques located predominantly on sun-exposed areas with notable sparing of the skin folds.19 The association between Asteraceae dermatitis and chronic actinic dermatitis is highly variable, with some studies reporting a 25% correlation and others finding a stronger association of up to 80%.2,15,20 Asteraceae allergy appears to be a relatively uncommon cause of photoallergy in North America. In one recent study, 16% (3/19) of patients with chronic actinic dermatitis had positive patch or photopatch tests to sesquiterpene lactone mix, but in another large study of photopatch testing it was reported to be a rare photoallergen.21,22
Parthenium dermatitis is an allergic contact dermatitis caused by exposure to Parthenium hysterophorus, a weed of the Asteraceae family that is responsible for 30% of cases of contact dermatitis in India.23,24 Unlike the more classic manifestation of Asteraceae dermatitis, which primarily affects the upper extremities in cases from North America and Europe, Parthenium dermatitis typically occurs in an airborne pattern distribution.24
Management
While complete avoidance of Asteraceae plants is ideal, it often is unrealistic due to their abundance in nature. Therefore, minimizing exposure to the causative plants is recommended. Primary preventive measures such as wearing protective gloves and clothing and applying bentonite clay prior to exposure should be taken when working outdoors. Promptly showering after contact with plants also can reduce the risk for Asteraceae dermatitis.
Symptomatic treatment is appropriate for mild cases and includes topical corticosteroids and calcineurin inhibitors. For severe cases, systemic corticosteroids may be needed for acute flares, with azathioprine, mycophenolate, cyclosporine, or methotrexate available for recalcitrant disease. Verma et al25 found that treatment with azathioprine for 6 months resulted in greater than 60% clearance in all 12 patients, with a majority achieving 80% to 100% clearance. Methotrexate has been used at doses of 15 mg once weekly.26 Narrowband UVB and psoralen plus UVA have been effective in extensive cases; however, care should be exercised in patients with photosensitive dermatitis, who instead should practice strict photoprotection.27-29 Lakshmi et al30 reported the use of cyclosporine during the acute phase of Asteraceae dermatitis at a dose of 2.5 mg/kg daily for 4 to 8 weeks. There have been several case reports of dupilumab treating allergic contact dermatitis; however, there have been 3 cases of patients with atopic dermatitis developing Asteraceae dermatitis while taking dupilumab.31,32 Recently, oral Janus kinase inhibitors have shown success in treating refractory cases of airborne Asteraceae dermatitis.33,34 Further research is needed to determine the safety and efficacy of dupilumab and Janus kinase inhibitors for treatment of Asteraceae dermatitis.
Final Thoughts
The Asteraceae plant family is vast and diverse, with more than 200 species reported to cause allergic contact dermatitis.12 Common modes of contact include gardening, occupational exposure, airborne pollen, and use of pediculicides and cosmetics that contain components of Asteraceae plants. Educating patients on how to minimize contact with Asteraceae plants is the most effective management strategy; topical agents and oral immunosuppressives can be used for symptomatic treatment.
The Asteraceae (formerly Compositae) family of plants is derived from the ancient Greek word aster, meaning “star,” referring to the starlike arrangement of flower petals around a central disc known as a capitulum. What initially appears as a single flower is actually a composite of several smaller flowers, hence the former name Compositae.1 Well-known members of the Asteraceae family include ornamental annuals (eg, sunflowers, marigolds, cosmos), herbaceous perennials (eg, chrysanthemums, dandelions), vegetables (eg, lettuce, chicory, artichokes), herbs (eg, chamomile, tarragon), and weeds (eg, ragweed, horseweed, capeweed)(Figure 1).2
There are more than 25,000 species of Asteraceae plants that thrive in a wide range of climates worldwide. Cases of Asteraceae-induced skin reactions have been reported in North America, Europe, Asia, and Australia.3 Members of the Asteraceae family are ubiquitous in gardens, along roadsides, and in the wilderness. Occupational exposure commonly affects gardeners, florists, farmers, and forestry workers through either direct contact with plants or via airborne pollen. Furthermore, plants of the Asteraceae family are used in various products, including pediculicides (eg, insect repellents), cosmetics (eg, eye creams, body washes), and food products (eg, cooking oils, sweetening agents, coffee substitutes, herbal teas).4-6 These plants have substantial allergic potential, resulting in numerous cutaneous reactions.
Allergic Potential
Asteraceae plants can elicit both immediate and delayed hypersensitivity reactions (HSRs); for instance, exposure to ragweed pollen may cause an IgE-mediated type 1 HSR manifesting as allergic rhinitis or a type IV HSR manifesting as airborne allergic contact dermatitis.7,8 The main contact allergens present in Asteraceae plants are sesquiterpene lactones, which are found in the leaves, stems, flowers, and pollen.9-11 Sesquiterpene lactones consist of an α-methyl group attached to a lactone ring combined with a sesquiterpene.12 Patch testing can be used to diagnose Asteraceae allergy; however, the results are not consistently reliable because there is no perfect screening allergen. Patch test preparations commonly used to detect Asteraceae allergy include Compositae mix (consisting of Anthemis nobilis extract, Chamomilla recutita extract, Achillea millefolium extract, Tanacetum vulgare extract, Arnica montana extract, and parthenolide) and sesquiterpene lactone mix (consisting of alantolactone, dehydrocostus lactone, and costunolide). In North America, the prevalence of positive patch tests to Compositae mix and sesquiterpene lactone mix is approximately 2% and 0.5%, respectively.13 When patch testing is performed, both Compositae mix and sesquiterpene lactone mix should be utilized to minimize the risk of missing Asteraceae allergy, as sesquiterpene lactone mix alone does not detect all Compositae-sensitized patients. Additionally, it may be necessary to test supplemental Asteraceae allergens, including preparations from specific plants to which the patient has been exposed. Exposure to Asteraceae-containing cosmetic products may lead to dermatitis, though this is highly dependent on the particular plant species involved. For instance, the prevalence of sensitization is high in arnica (tincture) and elecampane but low with more commonly used species such as German chamomile.14
Cutaneous Manifestations
Asteraceae dermatitis, which also is known as Australian bush dermatitis, weed dermatitis, and chrysanthemum dermatitis,2 can manifest on any area of the body that directly contacts the plant or is exposed to the pollen. Asteraceae dermatitis historically was reported in older adults with a recent history of plant exposure.6,15 However, recent data have shown a female preponderance and a younger mean age of onset (46–49 years).16
There are multiple distinct clinical manifestations of Asteraceae dermatitis. The most common cutaneous finding is localized vesicular or eczematous patches on the hands or wrists. Other variations include eczematous rashes on the exposed skin of the hands, arms, face, and neck; generalized eczema; and isolated facial eczema.16,17 These variations can be attributed to contact dermatitis caused by airborne pollen, which may mimic photodermatitis. However, airborne Asteraceae dermatitis can be distinguished clinically from photodermatitis by the involvement of sun-protected areas such as the skinfolds of the eyelids, retroauricular sulci, and nasolabial folds (Figure 2).2,9 In rare cases, systemic allergic contact dermatitis can occur if the Asteraceae allergen is ingested.2,18
Other diagnostic clues include dermatitis that flares during the summer, at the peak of the growing season, with remission in the cooler months. Potential risk factors include a childhood history of atopic dermatitis and allergic rhinitis.16 With prolonged exposure, patients may develop chronic actinic dermatitis, an immunologically mediated photodermatosis characterized by lichenified and pruritic eczematous plaques located predominantly on sun-exposed areas with notable sparing of the skin folds.19 The association between Asteraceae dermatitis and chronic actinic dermatitis is highly variable, with some studies reporting a 25% correlation and others finding a stronger association of up to 80%.2,15,20 Asteraceae allergy appears to be a relatively uncommon cause of photoallergy in North America. In one recent study, 16% (3/19) of patients with chronic actinic dermatitis had positive patch or photopatch tests to sesquiterpene lactone mix, but in another large study of photopatch testing it was reported to be a rare photoallergen.21,22
Parthenium dermatitis is an allergic contact dermatitis caused by exposure to Parthenium hysterophorus, a weed of the Asteraceae family that is responsible for 30% of cases of contact dermatitis in India.23,24 Unlike the more classic manifestation of Asteraceae dermatitis, which primarily affects the upper extremities in cases from North America and Europe, Parthenium dermatitis typically occurs in an airborne pattern distribution.24
Management
While complete avoidance of Asteraceae plants is ideal, it often is unrealistic due to their abundance in nature. Therefore, minimizing exposure to the causative plants is recommended. Primary preventive measures such as wearing protective gloves and clothing and applying bentonite clay prior to exposure should be taken when working outdoors. Promptly showering after contact with plants also can reduce the risk for Asteraceae dermatitis.
Symptomatic treatment is appropriate for mild cases and includes topical corticosteroids and calcineurin inhibitors. For severe cases, systemic corticosteroids may be needed for acute flares, with azathioprine, mycophenolate, cyclosporine, or methotrexate available for recalcitrant disease. Verma et al25 found that treatment with azathioprine for 6 months resulted in greater than 60% clearance in all 12 patients, with a majority achieving 80% to 100% clearance. Methotrexate has been used at doses of 15 mg once weekly.26 Narrowband UVB and psoralen plus UVA have been effective in extensive cases; however, care should be exercised in patients with photosensitive dermatitis, who instead should practice strict photoprotection.27-29 Lakshmi et al30 reported the use of cyclosporine during the acute phase of Asteraceae dermatitis at a dose of 2.5 mg/kg daily for 4 to 8 weeks. There have been several case reports of dupilumab treating allergic contact dermatitis; however, there have been 3 cases of patients with atopic dermatitis developing Asteraceae dermatitis while taking dupilumab.31,32 Recently, oral Janus kinase inhibitors have shown success in treating refractory cases of airborne Asteraceae dermatitis.33,34 Further research is needed to determine the safety and efficacy of dupilumab and Janus kinase inhibitors for treatment of Asteraceae dermatitis.
Final Thoughts
The Asteraceae plant family is vast and diverse, with more than 200 species reported to cause allergic contact dermatitis.12 Common modes of contact include gardening, occupational exposure, airborne pollen, and use of pediculicides and cosmetics that contain components of Asteraceae plants. Educating patients on how to minimize contact with Asteraceae plants is the most effective management strategy; topical agents and oral immunosuppressives can be used for symptomatic treatment.
- Morhardt S, Morhardt E. California Desert Flowers: An Introduction to Families, Genera, and Species. University of California Press; 2004.
- Gordon LA. Compositae dermatitis. Australas J Dermatol. 1999;40:123-130. doi:10.1046/j.1440-0960.1999.00341.x
- Denisow-Pietrzyk M, Pietrzyk Ł, Denisow B. Asteraceae species as potential environmental factors of allergy. Environ Sci Pollut Res Int. 2019;26:6290-6300. doi:10.1007/s11356-019-04146-w
- Paulsen E, Chistensen LP, Andersen KE. Cosmetics and herbal remedies with Compositae plant extracts—are they tolerated by Compositae-allergic patients? Contact Dermatitis. 2008;58:15-23. doi:10.1111/j.1600-0536.2007.01250.x
- Burry JN, Reid JG, Kirk J. Australian bush dermatitis. Contact Dermatitis. 1975;1:263-264. doi:10.1111/j.1600-0536.1975.tb05422.x
- Punchihewa N, Palmer A, Nixon R. Allergic contact dermatitis to Compositae: an Australian case series. Contact Dermatitis. 2022;87:356-362. doi:10.1111/cod.14162
- Chen KW, Marusciac L, Tamas PT, et al. Ragweed pollen allergy: burden, characteristics, and management of an imported allergen source in Europe. Int Arch Allergy Immunol. 2018;176:163-180. doi:10.1159/000487997
- Schloemer JA, Zirwas MJ, Burkhart CG. Airborne contact dermatitis: common causes in the USA. Int J Dermatol. 2015;54:271-274. doi:10.1111/ijd.12692
- Arlette J, Mitchell JC. Compositae dermatitis. current aspects. Contact Dermatitis. 1981;7:129-136. doi:10.1111/j.1600-0536.1981.tb04584.x
- Mitchell JC, Dupuis G. Allergic contact dermatitis from sesquiterpenoids of the Compositae family of plants. Br J Dermatol. 1971;84:139-150. doi:10.1111/j.1365-2133.1971.tb06857.x
- Salapovic H, Geier J, Reznicek G. Quantification of Sesquiterpene lactones in Asteraceae plant extracts: evaluation of their allergenic potential. Sci Pharm. 2013;81:807-818. doi:10.3797/scipharm.1306-17
- Paulsen E. Compositae dermatitis: a survey. Contact Dermatitis. 1992;26:76-86. doi:10.1111/j.1600-0536.1992.tb00888.x. Published correction appears in Contact Dermatitis. 1992;27:208.
- DeKoven JG, Silverberg JI, Warshaw EM, et al. North American Contact Dermatitis Group patch test results: 2017-2018. Dermatitis. 2021;32:111-123. doi:10.1097/DER.0000000000000729
- Paulsen E. Contact sensitization from Compositae-containing herbal remedies and cosmetics. Contact Dermatitis. 2002;47:189-198. doi:10.1034/j.1600-0536.2002.470401.x
- Frain-Bell W, Johnson BE. Contact allergic sensitivity to plants and the photosensitivity dermatitis and actinic reticuloid syndrome. Br J Dermatol. 1979;101:503-512.
- Paulsen E, Andersen KE. Clinical patterns of Compositae dermatitis in Danish monosensitized patients. Contact Dermatitis. 2018;78:185-193. doi:10.1111/cod.12916
- Jovanovic´ M, Poljacki M. Compositae dermatitis. Med Pregl. 2003;56:43-49. doi:10.2298/mpns0302043j
- Krook G. Occupational dermatitis from Lactuca sativa (lettuce) and Cichorium (endive). simultaneous occurrence of immediate and delayed allergy as a cause of contact dermatitis. Contact Dermatitis. 1977;3:27-36. doi:10.1111/j.1600-0536.1977.tb03583.x
- Paek SY, Lim HW. Chronic actinic dermatitis. Dermatol Clin. 2014;32:355-361, viii-ix. doi:10.1016/j.det.2014.03.007
- du P Menagé H, Hawk JL, White IR. Sesquiterpene lactone mix contact sensitivity and its relationship to chronic actinic dermatitis: a follow-up study. Contact Dermatitis. 1998;39:119-122. doi:10.1111/j.1600-0536.1998.tb05859.x
- Wang CX, Belsito DV. Chronic actinic dermatitis revisited. Dermatitis. 2020;31:68-74. doi:10.1097/DER.0000000000000531
- DeLeo VA, Adler BL, Warshaw EM, et al. Photopatch test results of the North American contact dermatitis group, 1999-2009. Photodermatol Photoimmunol Photomed. 2022;38:288-291. doi:10.1111/phpp.12742
- McGovern TW, LaWarre S. Botanical briefs: the scourge of India—Parthenium hysterophorus L. Cutis. 2001;67:27-34. Published correction appears in Cutis. 2001;67:154.
- Sharma VK, Verma P, Maharaja K. Parthenium dermatitis. Photochem Photobiol Sci. 2013;12:85-94. doi:10.1039/c2pp25186h
- Verma KK, Bansal A, Sethuraman G. Parthenium dermatitis treated with azathioprine weekly pulse doses. Indian J Dermatol Venereol Leprol. 2006;72:24-27. doi:10.4103/0378-6323.19713
- Sharma VK, Bhat R, Sethuraman G, et al. Treatment of Parthenium dermatitis with methotrexate. Contact Dermatitis. 2007;57:118-119. doi:10.1111/j.1600-0536.2006.00950.x
- Burke DA, Corey G, Storrs FJ. Psoralen plus UVA protocol for Compositae photosensitivity. Am J Contact Dermat. 1996;7:171-176.
- Lovell CR. Allergic contact dermatitis due to plants. In: Plants and the Skin. Blackwell Scientific Publications; 1993:96-254.
- Dogra S, Parsad D, Handa S. Narrowband ultraviolet B in airborne contact dermatitis: a ray of hope! Br J Dermatol. 2004;150:373-374. doi:10.1111/j.1365-2133.2004.05724.x
- Lakshmi C, Srinivas CR, Jayaraman A. Ciclosporin in Parthenium dermatitis—a report of 2 cases. Contact Dermatitis. 2008;59:245-248. doi:10.1111/j.1600-0536.2007.01208.x
- Hendricks AJ, Yosipovitch G, Shi VY. Dupilumab use in dermatologic conditions beyond atopic dermatitis—a systematic review. J Dermatolog Treat. 2021;32:19-28. doi:10.1080/09546634.2019.1689227
- Napolitano M, Fabbrocini G, Patruno C. Allergic contact dermatitis to Compositae: a possible cause of dupilumab-associated facial and neck dermatitis in atopic dermatitis patients? Contact Dermatitis. 2021;85:473-474. doi:10.1111/cod.13898
- Muddebihal A, Sardana K, Sinha S, et al. Tofacitinib in refractory Parthenium-induced airborne allergic contact dermatitis. Contact Dermatitis. 2023;88:150-152. doi:10.1111/cod.14234
- Baltazar D, Shinamoto SR, Hamann CP, et al. Occupational airborne allergic contact dermatitis to invasive Compositae species treated with abrocitinib: a case report. Contact Dermatitis. 2022;87:542-544. doi:10.1111/cod.14204
- Morhardt S, Morhardt E. California Desert Flowers: An Introduction to Families, Genera, and Species. University of California Press; 2004.
- Gordon LA. Compositae dermatitis. Australas J Dermatol. 1999;40:123-130. doi:10.1046/j.1440-0960.1999.00341.x
- Denisow-Pietrzyk M, Pietrzyk Ł, Denisow B. Asteraceae species as potential environmental factors of allergy. Environ Sci Pollut Res Int. 2019;26:6290-6300. doi:10.1007/s11356-019-04146-w
- Paulsen E, Chistensen LP, Andersen KE. Cosmetics and herbal remedies with Compositae plant extracts—are they tolerated by Compositae-allergic patients? Contact Dermatitis. 2008;58:15-23. doi:10.1111/j.1600-0536.2007.01250.x
- Burry JN, Reid JG, Kirk J. Australian bush dermatitis. Contact Dermatitis. 1975;1:263-264. doi:10.1111/j.1600-0536.1975.tb05422.x
- Punchihewa N, Palmer A, Nixon R. Allergic contact dermatitis to Compositae: an Australian case series. Contact Dermatitis. 2022;87:356-362. doi:10.1111/cod.14162
- Chen KW, Marusciac L, Tamas PT, et al. Ragweed pollen allergy: burden, characteristics, and management of an imported allergen source in Europe. Int Arch Allergy Immunol. 2018;176:163-180. doi:10.1159/000487997
- Schloemer JA, Zirwas MJ, Burkhart CG. Airborne contact dermatitis: common causes in the USA. Int J Dermatol. 2015;54:271-274. doi:10.1111/ijd.12692
- Arlette J, Mitchell JC. Compositae dermatitis. current aspects. Contact Dermatitis. 1981;7:129-136. doi:10.1111/j.1600-0536.1981.tb04584.x
- Mitchell JC, Dupuis G. Allergic contact dermatitis from sesquiterpenoids of the Compositae family of plants. Br J Dermatol. 1971;84:139-150. doi:10.1111/j.1365-2133.1971.tb06857.x
- Salapovic H, Geier J, Reznicek G. Quantification of Sesquiterpene lactones in Asteraceae plant extracts: evaluation of their allergenic potential. Sci Pharm. 2013;81:807-818. doi:10.3797/scipharm.1306-17
- Paulsen E. Compositae dermatitis: a survey. Contact Dermatitis. 1992;26:76-86. doi:10.1111/j.1600-0536.1992.tb00888.x. Published correction appears in Contact Dermatitis. 1992;27:208.
- DeKoven JG, Silverberg JI, Warshaw EM, et al. North American Contact Dermatitis Group patch test results: 2017-2018. Dermatitis. 2021;32:111-123. doi:10.1097/DER.0000000000000729
- Paulsen E. Contact sensitization from Compositae-containing herbal remedies and cosmetics. Contact Dermatitis. 2002;47:189-198. doi:10.1034/j.1600-0536.2002.470401.x
- Frain-Bell W, Johnson BE. Contact allergic sensitivity to plants and the photosensitivity dermatitis and actinic reticuloid syndrome. Br J Dermatol. 1979;101:503-512.
- Paulsen E, Andersen KE. Clinical patterns of Compositae dermatitis in Danish monosensitized patients. Contact Dermatitis. 2018;78:185-193. doi:10.1111/cod.12916
- Jovanovic´ M, Poljacki M. Compositae dermatitis. Med Pregl. 2003;56:43-49. doi:10.2298/mpns0302043j
- Krook G. Occupational dermatitis from Lactuca sativa (lettuce) and Cichorium (endive). simultaneous occurrence of immediate and delayed allergy as a cause of contact dermatitis. Contact Dermatitis. 1977;3:27-36. doi:10.1111/j.1600-0536.1977.tb03583.x
- Paek SY, Lim HW. Chronic actinic dermatitis. Dermatol Clin. 2014;32:355-361, viii-ix. doi:10.1016/j.det.2014.03.007
- du P Menagé H, Hawk JL, White IR. Sesquiterpene lactone mix contact sensitivity and its relationship to chronic actinic dermatitis: a follow-up study. Contact Dermatitis. 1998;39:119-122. doi:10.1111/j.1600-0536.1998.tb05859.x
- Wang CX, Belsito DV. Chronic actinic dermatitis revisited. Dermatitis. 2020;31:68-74. doi:10.1097/DER.0000000000000531
- DeLeo VA, Adler BL, Warshaw EM, et al. Photopatch test results of the North American contact dermatitis group, 1999-2009. Photodermatol Photoimmunol Photomed. 2022;38:288-291. doi:10.1111/phpp.12742
- McGovern TW, LaWarre S. Botanical briefs: the scourge of India—Parthenium hysterophorus L. Cutis. 2001;67:27-34. Published correction appears in Cutis. 2001;67:154.
- Sharma VK, Verma P, Maharaja K. Parthenium dermatitis. Photochem Photobiol Sci. 2013;12:85-94. doi:10.1039/c2pp25186h
- Verma KK, Bansal A, Sethuraman G. Parthenium dermatitis treated with azathioprine weekly pulse doses. Indian J Dermatol Venereol Leprol. 2006;72:24-27. doi:10.4103/0378-6323.19713
- Sharma VK, Bhat R, Sethuraman G, et al. Treatment of Parthenium dermatitis with methotrexate. Contact Dermatitis. 2007;57:118-119. doi:10.1111/j.1600-0536.2006.00950.x
- Burke DA, Corey G, Storrs FJ. Psoralen plus UVA protocol for Compositae photosensitivity. Am J Contact Dermat. 1996;7:171-176.
- Lovell CR. Allergic contact dermatitis due to plants. In: Plants and the Skin. Blackwell Scientific Publications; 1993:96-254.
- Dogra S, Parsad D, Handa S. Narrowband ultraviolet B in airborne contact dermatitis: a ray of hope! Br J Dermatol. 2004;150:373-374. doi:10.1111/j.1365-2133.2004.05724.x
- Lakshmi C, Srinivas CR, Jayaraman A. Ciclosporin in Parthenium dermatitis—a report of 2 cases. Contact Dermatitis. 2008;59:245-248. doi:10.1111/j.1600-0536.2007.01208.x
- Hendricks AJ, Yosipovitch G, Shi VY. Dupilumab use in dermatologic conditions beyond atopic dermatitis—a systematic review. J Dermatolog Treat. 2021;32:19-28. doi:10.1080/09546634.2019.1689227
- Napolitano M, Fabbrocini G, Patruno C. Allergic contact dermatitis to Compositae: a possible cause of dupilumab-associated facial and neck dermatitis in atopic dermatitis patients? Contact Dermatitis. 2021;85:473-474. doi:10.1111/cod.13898
- Muddebihal A, Sardana K, Sinha S, et al. Tofacitinib in refractory Parthenium-induced airborne allergic contact dermatitis. Contact Dermatitis. 2023;88:150-152. doi:10.1111/cod.14234
- Baltazar D, Shinamoto SR, Hamann CP, et al. Occupational airborne allergic contact dermatitis to invasive Compositae species treated with abrocitinib: a case report. Contact Dermatitis. 2022;87:542-544. doi:10.1111/cod.14204
Practice Points
- Asteraceae dermatitis can occur from direct contact with plants of the Asteraceae family; through airborne pollen; or from exposure to topical medications, cooking products, and cosmetics.
- Patient education on primary prevention, especially protective clothing, is crucial, as these plants are ubiquitous outdoors and have diverse phenotypes.
- Management of mild Asteraceae dermatitis consists primarily of topical corticosteroids and calcineurin inhibitors, while systemic corticosteroids and other immunosuppressive agents are utilized for severe or recalcitrant cases.
