User login
Enlarging Nodule on the Nipple
The Diagnosis: Nipple Adenoma (Florid Papillomatosis of the Nipple)
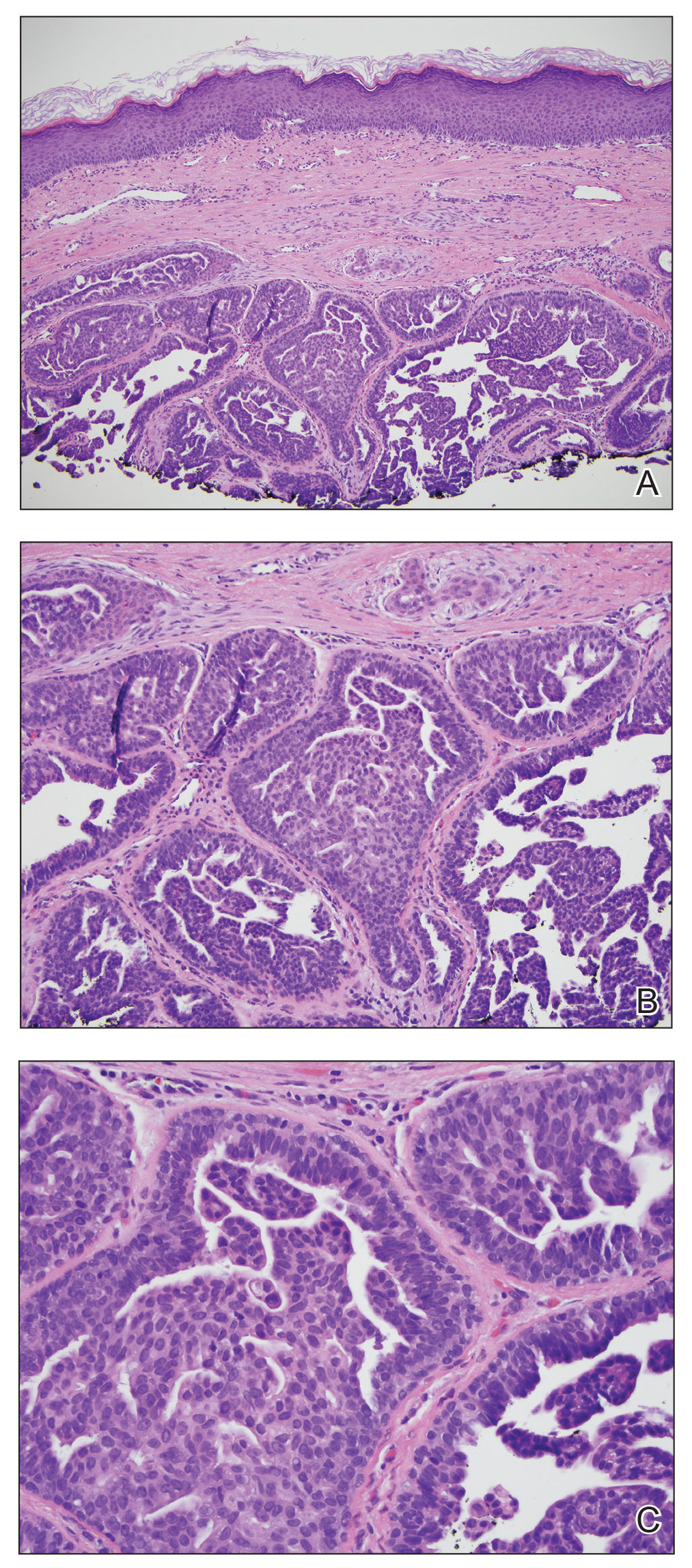
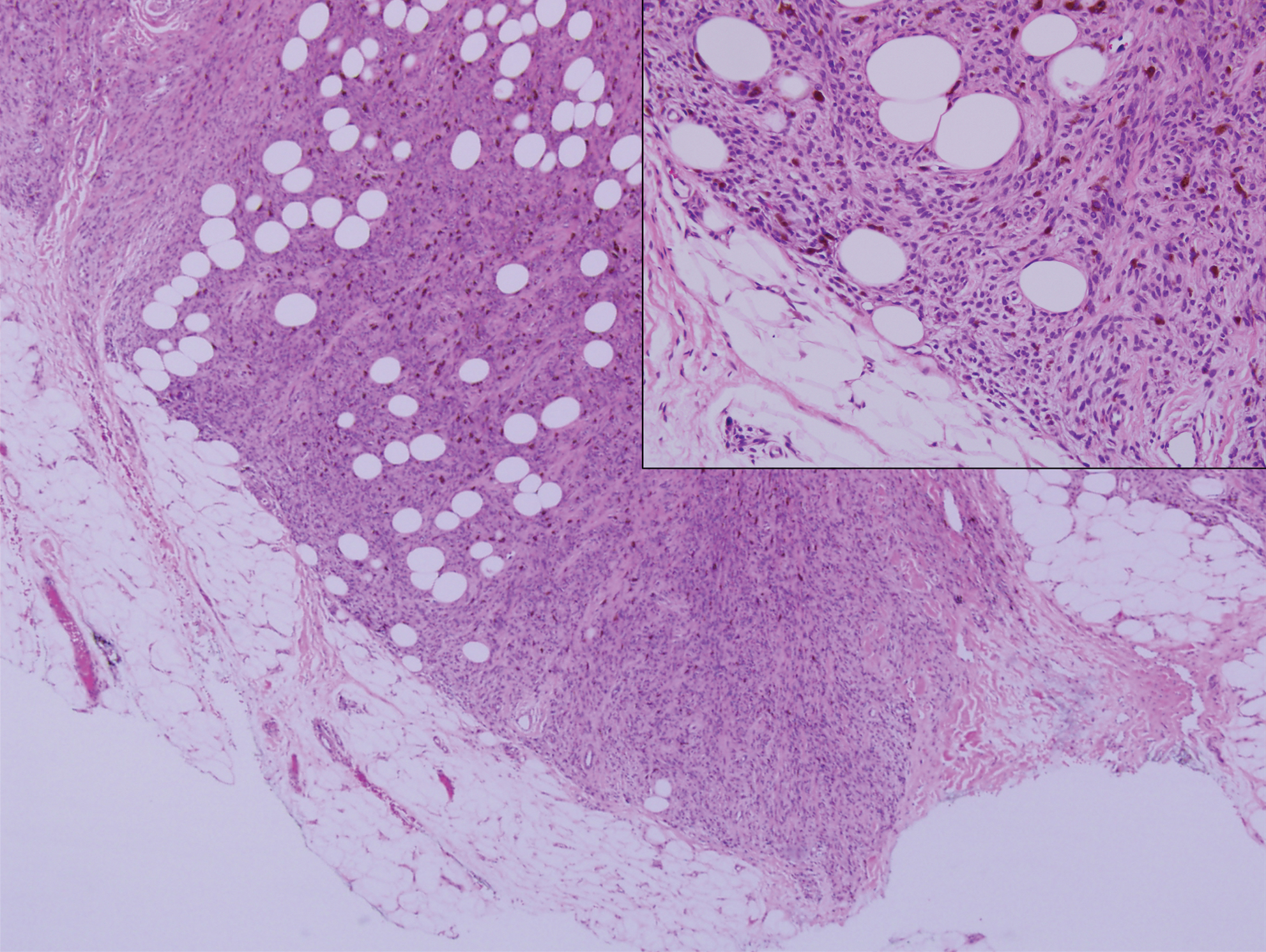
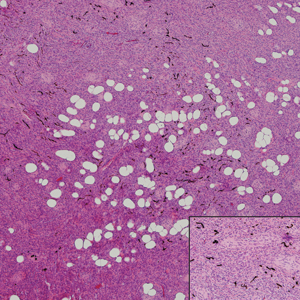
Biopsy of the nodule showed florid papillary hyperplasia of the ductal epithelium within the dermis that was sharply demarcated from the background stroma (Figure, A and B). Neither cytological nor architectural atypia were evident. There was no notable necrosis (Figure C). There was a background of fibrosis whereby the glandular ductal structures assumed a somewhat irregular growth pattern within the dermis with attendant hemorrhage. The patient underwent complete excision of the lesion. No evidence of carcinoma was seen on the final pathology, and the final margins were negative.
First described in 1923 and fully characterized in 1955, nipple adenoma (also known as florid papillomatosis of the nipple) is a benign proliferative neoplasm that originates in the lactiferous ducts of the nipple.1,2 It most commonly affects women aged 40 to 50 years (range, 0-89 years); less than 5% of cases are reported in men.3,4 It predominantly is unilateral, with only rare cases of bilateral papillomatosis reported. Patients often present with serous or serosanguineous discharge and an itching or burning sensation. Symptoms may worsen with the menstrual cycle.4 On physical examination, it presents as an ill-defined red nodule on the nipple with crusting, erosion, or erythema of the nipple surface. Although imaging generally is not used to confirm the diagnosis, mammography should be performed prior to biopsy to rule out underlying breast pathology. Dermoscopy may show linear cherry red structures or red serpiginous and annular structures.5,6 The differential diagnosis of nipple adenoma includes Paget disease of the breast, adenomyoepithelioma, subareolar subsclerosing duct hyperplasia, syringomatous adenoma, adenosis tumor, low-grade adenosquamous carcinoma, low-grade ductal carcinoma in situ, tubular carcinoma, and sweat gland tumors.3
Microscopic features of nipple adenoma have been categorized into 4 subtypes: sclerosing papillomatosis, papillomatosis, adenosis, and a mixed pattern.3,7 The tumors may have keratin cysts and focal necrosis but no atypia, and the myoepithelial cell layer is retained. Nipple adenomas show a glandular proliferation in the dermis that is relatively well circumscribed with glands that vary in appearance between a simple adenosislike pattern of growth to a papillary hyperplasia and/or usual ductal hyperplasia growth pattern. A pseudoinfiltrative pattern can occur when the glandular epithelium is entrapped within stromal fibrosis; however, the myoepithelial layer is retained. Occasionally, the glandular epithelium can grow in continuity with the surface squamous epithelium of the nipple, clinically simulating Paget disease of the breast.8 Immunohistochemical stains, specifically p63, p40, calponin 1, h-caldesmon, cytokeratin 5/6, CD10, and α; smooth muscle actin, highlight the myoepithelial cells, while cytokeratin 7 identifies the ductal epithelium, supporting the diagnosis.6 In addition to biopsy and microscopic tissue examination, touch preparation cytology, curettage cytology, and fine needle aspiration techniques have been used to perform cytologic examination of the lesions, aiding in identification of the benign or malignant nature of the neoplasm.6 Nipple adenoma also is referred to as florid papillomatosis of the nipple, papillary adenoma, erosive adenomatosis, and subareolar duct papillomatosis.7
Although nipple adenoma is a benign tumor, up to 16.5% of affected patients had an ipsilateral or contralateral mammary carcinoma.9 The majority arose coincidentally but separately in the same breast, and carcinoma arose directly from the nipple adenoma in 8 cases; 3 cases were carcinomas that arose in men.10 A definitive association or causal relationship between nipple adenoma and subsequent development of breast cancer has not been identified, and the incidence of nipple adenoma in patients with a positive family history of breast cancer has not been examined. Therefore, although various treatments including cryosurgery, nipple splitting enucleation, and Mohs micrographic surgery have been proposed, complete excision remains the gold standard of therapy. Regular breast examinations and digital mammography are necessary to screen for local recurrences.
- Miller E, Lewis D. The significance of serohemorrhagic or hemorrhagic discharge from the nipple. JAMA. 1923;81:1651-1657.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Rosen PP. Rosen's Breast Pathology. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:120-128.
- Brownstein MH, Phelps RG, Maqnin PH. Papillary adenoma of the nipple: analysis of fifteen new cases. J Am Acad Dermatol. 1985;12:707-715.
- Takashima S, Fujita Y, Miyauchi T, et al. Dermoscopic observation in adenoma of the nipple. J Dermatol. 2015;42:341-342.
- Spohn G, Trotter S, Tozbikian G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Shin SJ. Nipple adenoma (florid papillomatosis of the nipple). In: Dabbs DJ, ed. Breast Pathology. Philadelphia, PA: Elsevier Saunders; 2012:286-292.
- Schnitt SJ, Collins LC. Biopsy Interpretation of the Breast. 2nd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013.
- Salemis NS. Florid papillomatosis of the nipple: a rare presentation and review of the literature. Breast Dis. 2015;35:153-156.
- Di Bonito M, Cantile M, Collina F, et al. Adenoma of the nipple: a clinicopathological report of 13 cases. Oncol Lett. 2014;7:1839-1842.
The Diagnosis: Nipple Adenoma (Florid Papillomatosis of the Nipple)
Biopsy of the nodule showed florid papillary hyperplasia of the ductal epithelium within the dermis that was sharply demarcated from the background stroma (Figure, A and B). Neither cytological nor architectural atypia were evident. There was no notable necrosis (Figure C). There was a background of fibrosis whereby the glandular ductal structures assumed a somewhat irregular growth pattern within the dermis with attendant hemorrhage. The patient underwent complete excision of the lesion. No evidence of carcinoma was seen on the final pathology, and the final margins were negative.
First described in 1923 and fully characterized in 1955, nipple adenoma (also known as florid papillomatosis of the nipple) is a benign proliferative neoplasm that originates in the lactiferous ducts of the nipple.1,2 It most commonly affects women aged 40 to 50 years (range, 0-89 years); less than 5% of cases are reported in men.3,4 It predominantly is unilateral, with only rare cases of bilateral papillomatosis reported. Patients often present with serous or serosanguineous discharge and an itching or burning sensation. Symptoms may worsen with the menstrual cycle.4 On physical examination, it presents as an ill-defined red nodule on the nipple with crusting, erosion, or erythema of the nipple surface. Although imaging generally is not used to confirm the diagnosis, mammography should be performed prior to biopsy to rule out underlying breast pathology. Dermoscopy may show linear cherry red structures or red serpiginous and annular structures.5,6 The differential diagnosis of nipple adenoma includes Paget disease of the breast, adenomyoepithelioma, subareolar subsclerosing duct hyperplasia, syringomatous adenoma, adenosis tumor, low-grade adenosquamous carcinoma, low-grade ductal carcinoma in situ, tubular carcinoma, and sweat gland tumors.3
Microscopic features of nipple adenoma have been categorized into 4 subtypes: sclerosing papillomatosis, papillomatosis, adenosis, and a mixed pattern.3,7 The tumors may have keratin cysts and focal necrosis but no atypia, and the myoepithelial cell layer is retained. Nipple adenomas show a glandular proliferation in the dermis that is relatively well circumscribed with glands that vary in appearance between a simple adenosislike pattern of growth to a papillary hyperplasia and/or usual ductal hyperplasia growth pattern. A pseudoinfiltrative pattern can occur when the glandular epithelium is entrapped within stromal fibrosis; however, the myoepithelial layer is retained. Occasionally, the glandular epithelium can grow in continuity with the surface squamous epithelium of the nipple, clinically simulating Paget disease of the breast.8 Immunohistochemical stains, specifically p63, p40, calponin 1, h-caldesmon, cytokeratin 5/6, CD10, and α; smooth muscle actin, highlight the myoepithelial cells, while cytokeratin 7 identifies the ductal epithelium, supporting the diagnosis.6 In addition to biopsy and microscopic tissue examination, touch preparation cytology, curettage cytology, and fine needle aspiration techniques have been used to perform cytologic examination of the lesions, aiding in identification of the benign or malignant nature of the neoplasm.6 Nipple adenoma also is referred to as florid papillomatosis of the nipple, papillary adenoma, erosive adenomatosis, and subareolar duct papillomatosis.7
Although nipple adenoma is a benign tumor, up to 16.5% of affected patients had an ipsilateral or contralateral mammary carcinoma.9 The majority arose coincidentally but separately in the same breast, and carcinoma arose directly from the nipple adenoma in 8 cases; 3 cases were carcinomas that arose in men.10 A definitive association or causal relationship between nipple adenoma and subsequent development of breast cancer has not been identified, and the incidence of nipple adenoma in patients with a positive family history of breast cancer has not been examined. Therefore, although various treatments including cryosurgery, nipple splitting enucleation, and Mohs micrographic surgery have been proposed, complete excision remains the gold standard of therapy. Regular breast examinations and digital mammography are necessary to screen for local recurrences.
The Diagnosis: Nipple Adenoma (Florid Papillomatosis of the Nipple)
Biopsy of the nodule showed florid papillary hyperplasia of the ductal epithelium within the dermis that was sharply demarcated from the background stroma (Figure, A and B). Neither cytological nor architectural atypia were evident. There was no notable necrosis (Figure C). There was a background of fibrosis whereby the glandular ductal structures assumed a somewhat irregular growth pattern within the dermis with attendant hemorrhage. The patient underwent complete excision of the lesion. No evidence of carcinoma was seen on the final pathology, and the final margins were negative.
First described in 1923 and fully characterized in 1955, nipple adenoma (also known as florid papillomatosis of the nipple) is a benign proliferative neoplasm that originates in the lactiferous ducts of the nipple.1,2 It most commonly affects women aged 40 to 50 years (range, 0-89 years); less than 5% of cases are reported in men.3,4 It predominantly is unilateral, with only rare cases of bilateral papillomatosis reported. Patients often present with serous or serosanguineous discharge and an itching or burning sensation. Symptoms may worsen with the menstrual cycle.4 On physical examination, it presents as an ill-defined red nodule on the nipple with crusting, erosion, or erythema of the nipple surface. Although imaging generally is not used to confirm the diagnosis, mammography should be performed prior to biopsy to rule out underlying breast pathology. Dermoscopy may show linear cherry red structures or red serpiginous and annular structures.5,6 The differential diagnosis of nipple adenoma includes Paget disease of the breast, adenomyoepithelioma, subareolar subsclerosing duct hyperplasia, syringomatous adenoma, adenosis tumor, low-grade adenosquamous carcinoma, low-grade ductal carcinoma in situ, tubular carcinoma, and sweat gland tumors.3
Microscopic features of nipple adenoma have been categorized into 4 subtypes: sclerosing papillomatosis, papillomatosis, adenosis, and a mixed pattern.3,7 The tumors may have keratin cysts and focal necrosis but no atypia, and the myoepithelial cell layer is retained. Nipple adenomas show a glandular proliferation in the dermis that is relatively well circumscribed with glands that vary in appearance between a simple adenosislike pattern of growth to a papillary hyperplasia and/or usual ductal hyperplasia growth pattern. A pseudoinfiltrative pattern can occur when the glandular epithelium is entrapped within stromal fibrosis; however, the myoepithelial layer is retained. Occasionally, the glandular epithelium can grow in continuity with the surface squamous epithelium of the nipple, clinically simulating Paget disease of the breast.8 Immunohistochemical stains, specifically p63, p40, calponin 1, h-caldesmon, cytokeratin 5/6, CD10, and α; smooth muscle actin, highlight the myoepithelial cells, while cytokeratin 7 identifies the ductal epithelium, supporting the diagnosis.6 In addition to biopsy and microscopic tissue examination, touch preparation cytology, curettage cytology, and fine needle aspiration techniques have been used to perform cytologic examination of the lesions, aiding in identification of the benign or malignant nature of the neoplasm.6 Nipple adenoma also is referred to as florid papillomatosis of the nipple, papillary adenoma, erosive adenomatosis, and subareolar duct papillomatosis.7
Although nipple adenoma is a benign tumor, up to 16.5% of affected patients had an ipsilateral or contralateral mammary carcinoma.9 The majority arose coincidentally but separately in the same breast, and carcinoma arose directly from the nipple adenoma in 8 cases; 3 cases were carcinomas that arose in men.10 A definitive association or causal relationship between nipple adenoma and subsequent development of breast cancer has not been identified, and the incidence of nipple adenoma in patients with a positive family history of breast cancer has not been examined. Therefore, although various treatments including cryosurgery, nipple splitting enucleation, and Mohs micrographic surgery have been proposed, complete excision remains the gold standard of therapy. Regular breast examinations and digital mammography are necessary to screen for local recurrences.
- Miller E, Lewis D. The significance of serohemorrhagic or hemorrhagic discharge from the nipple. JAMA. 1923;81:1651-1657.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Rosen PP. Rosen's Breast Pathology. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:120-128.
- Brownstein MH, Phelps RG, Maqnin PH. Papillary adenoma of the nipple: analysis of fifteen new cases. J Am Acad Dermatol. 1985;12:707-715.
- Takashima S, Fujita Y, Miyauchi T, et al. Dermoscopic observation in adenoma of the nipple. J Dermatol. 2015;42:341-342.
- Spohn G, Trotter S, Tozbikian G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Shin SJ. Nipple adenoma (florid papillomatosis of the nipple). In: Dabbs DJ, ed. Breast Pathology. Philadelphia, PA: Elsevier Saunders; 2012:286-292.
- Schnitt SJ, Collins LC. Biopsy Interpretation of the Breast. 2nd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013.
- Salemis NS. Florid papillomatosis of the nipple: a rare presentation and review of the literature. Breast Dis. 2015;35:153-156.
- Di Bonito M, Cantile M, Collina F, et al. Adenoma of the nipple: a clinicopathological report of 13 cases. Oncol Lett. 2014;7:1839-1842.
- Miller E, Lewis D. The significance of serohemorrhagic or hemorrhagic discharge from the nipple. JAMA. 1923;81:1651-1657.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Rosen PP. Rosen's Breast Pathology. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:120-128.
- Brownstein MH, Phelps RG, Maqnin PH. Papillary adenoma of the nipple: analysis of fifteen new cases. J Am Acad Dermatol. 1985;12:707-715.
- Takashima S, Fujita Y, Miyauchi T, et al. Dermoscopic observation in adenoma of the nipple. J Dermatol. 2015;42:341-342.
- Spohn G, Trotter S, Tozbikian G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Shin SJ. Nipple adenoma (florid papillomatosis of the nipple). In: Dabbs DJ, ed. Breast Pathology. Philadelphia, PA: Elsevier Saunders; 2012:286-292.
- Schnitt SJ, Collins LC. Biopsy Interpretation of the Breast. 2nd ed. Philadelphia, PA: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013.
- Salemis NS. Florid papillomatosis of the nipple: a rare presentation and review of the literature. Breast Dis. 2015;35:153-156.
- Di Bonito M, Cantile M, Collina F, et al. Adenoma of the nipple: a clinicopathological report of 13 cases. Oncol Lett. 2014;7:1839-1842.
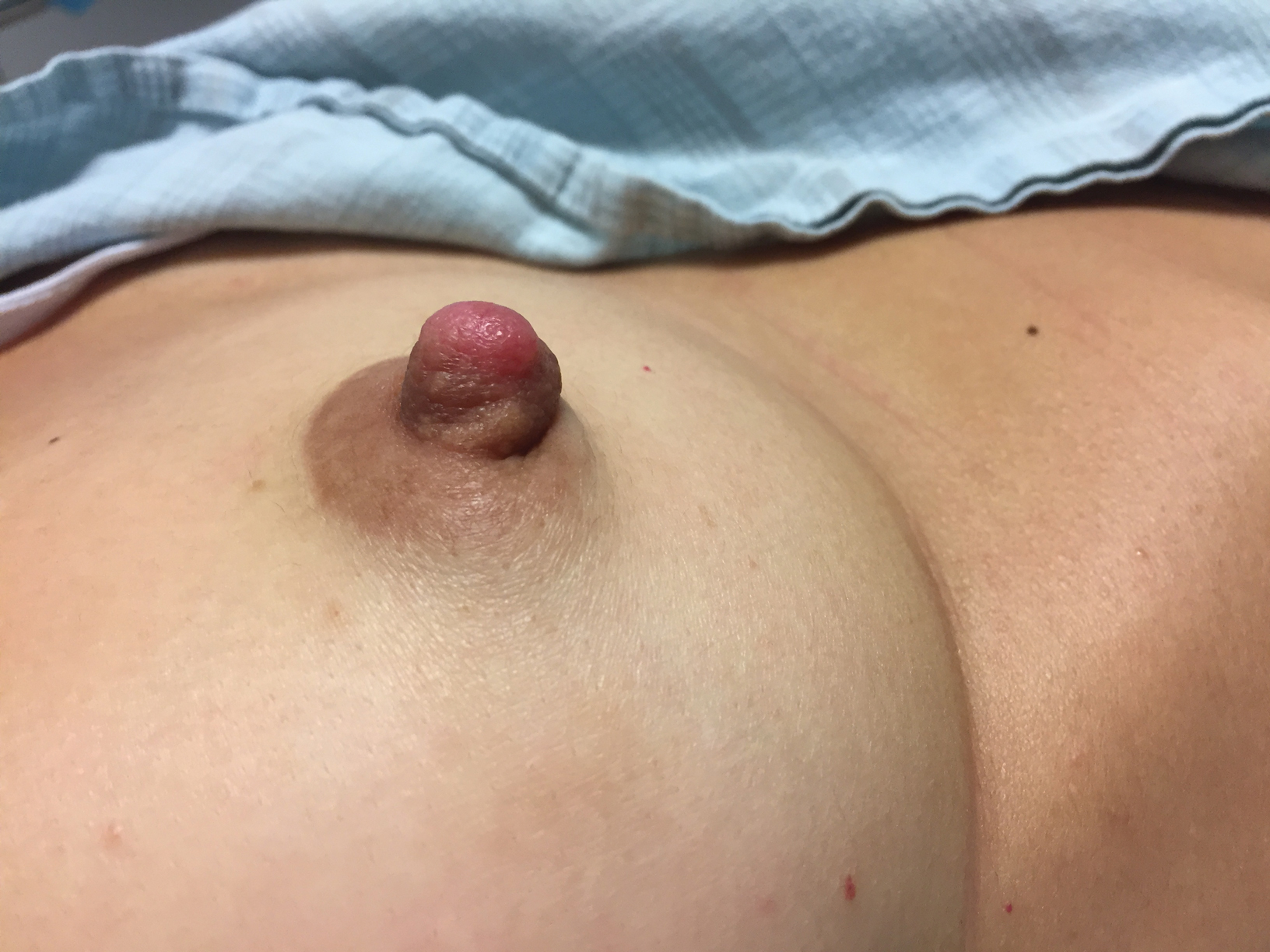
A healthy 48-year-old woman presented with a growth on the right nipple that had been slowly enlarging over the last few months. She initially noticed mild swelling in the area that persisted and formed a soft lump. She described mild pain with intermittent drainage but no bleeding. Her medical history was unremarkable, including a negative personal and family history of breast and skin cancer. She was taking no medications prior to development of the mass. She had no recent history of pregnancy or breastfeeding. A mammogram and breast ultrasound were not concerning for carcinoma. Physical examination showed a soft, exophytic, mildly tender, pink nodule on the right nipple that measured 12.2×7 mm; no drainage, bleeding, or ulceration was present. The surrounding skin of the areola and breast demonstrated no clinical changes. The contralateral breast, areola, and nipple were unaffected. The patient had no appreciable axillary or cervical lymphadenopathy. A deep shave biopsy of the nodule was performed and sent for histopathologic examination.
Pigmented Mass on the Shoulder
The Diagnosis: Pigmented Dermatofibrosarcoma Protuberans
Pigmented dermatofibrosarcoma protuberans (PDFSP), also known as Bednar tumor, is an uncommon variant of dermatofibrosarcoma protuberans (DFSP). Pigmented dermatofibrosarcoma protuberans constitutes 1% to 5% of all DFSP cases and most commonly is seen in nonwhite adults in the fourth decade of life, with occasional cases seen in pediatric patients, including some congenital cases. Typical sites of involvement include the shoulders, trunk, arms, legs, head, and neck.1,2 It also has been reported at sites of prior immunization, trauma, and insect bites.3
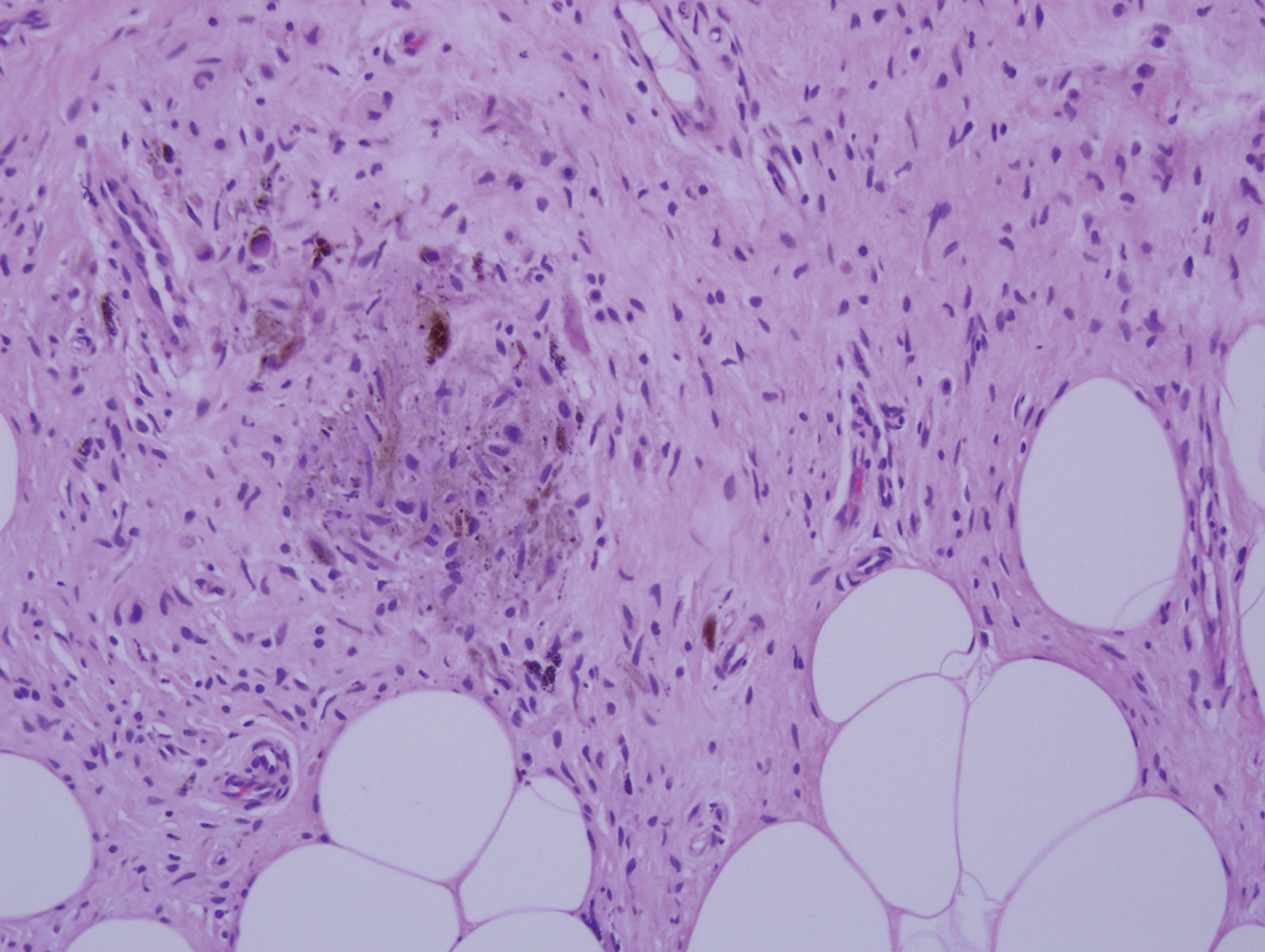
Histopathologic examination of our patient's shoulder nodule revealed an infiltrative neoplasm in the dermis and subcutaneous tissue composed of spindled cells with a storiform pattern and foci of scattered elongated dendritic pigmented cells. A narrow grenz zone separated the tumor from the epidermis, and characteristic honeycomb infiltration by tumor cells was noted in the subcutaneous fat. The nuclei were bland and monomorphous with areas of neuroid differentiation containing whorls and nerve cord-like structures (quiz image). The tumor cells were diffusely CD34 and vimentin positive, while S-100, SOX-10, neurofilament, smooth muscle actin, desmin, epithelial membrane antigen, and cytokeratins were negative. The immunophenotype excluded the possibility of neurogenic, pericytic, myofibroblastic, and myoid differentiation.
Wang and Yang4 previously reported a case of PDFSP with prominent meningothelial-like whorls focally resembling extracranial meningioma; however, the tumor cells were CD34 positive and epithelial membrane antigen negative, weighing against a diagnosis of meningioma. Most cases of PDFSP demonstrate the COL1A1-PDGFB (collagen type I α; 1/platelet-derived growth factor B-chain) fusion protein caused by the translocation t(17;22)(q22;q13), as in classic DFSP.5
Cellular blue nevus (CBN) is a benign melanocytic neoplasm that can present at any age and often occurs on the buttocks and in the sacrococcygeal region. Clinically, CBN presents as a firm, bluish black to bluish gray, dome-shaped nodule. The size varies from a few millimeters to several centimeters.6,7 Histologically, CBN is located completely in the dermis, extending along the adnexae into the subcutaneous tissue with a dumbbell-shaped outline (Figure 1).6-8 The tumor demonstrates oval epithelioid melanocytes with vesicular nuclei and prominent nucleoli. Immunohistochemically, tumor cells stain positively for melanocytic markers such as S-100, SOX-10, MART-1, and human melanoma black 45. CD34 expression rarely is reported in a subset of CBN.9
Pigmented neurofibroma is a rare variant of neurofibroma that produces melanin pigment and has a strong association with neurofibromatosis.10 It occurs most frequently in dark-skinned populations (Fitzpatrick skin types IV-VI). The most common location is the head and neck region.11,12 Histologically, pigmented neurofibroma resembles a diffuse neurofibroma admixed with melanin-producing cells (Figure 2).12 Immunostaining shows positivity for S-100 in both pigmented and Schwann cells; however, the pigmented cells stain positively for human melanoma black 45, Melan-A, and tyrosinase.10 CD34 can be fingerprint positive in neurofibroma, but a distinction from DFSP can be made by S-100 and SOX-10 immunostaining.13
Desmoplastic melanoma (DM) is an uncommon variant of malignant melanoma and has a higher tendency for persistent local growth and less frequent metastases than other variants of melanoma. It has a predilection for chronically sun-exposed areas such as the head and neck and occurs later in life. Clinically, DM appears as nonspecific, often amelanotic nodules or plaques or as scarlike lesions.14 Histologically, DM can be classified as mixed or pure based on the degree of desmoplasia and cellularity. A paucicellular proliferation of malignant spindled melanocytes within a densely fibrotic stroma with lymphoid nodules in the dermis is characteristic (Figure 3); perineural involvement is common.14,15 The most reliable confirmative stains are S-100 and SOX-10.16
Cutaneous meningioma is a rare tumor and could be subtyped into 3 groups. Type I is primary cutaneous meningioma and usually is present at birth on the scalp and paravertebral regions with a relatively good prognosis. Type II is ectopic soft-tissue meningioma that extends into the skin from around the sensory organs on the face. Type III is local invasion or true metastasis from a central nervous system meningioma. Types II and III develop later in life and the prognosis is poor.17,18 Clinically, lesions present as firm subcutaneous nodules or swellings. Cutaneous meningioma has several histopathologic variants. The classic presentation reveals concentric wrapping of tumor cells with round-oval nuclei containing delicate chromatin. Psammoma bodies are a common finding (Figure 4). Immunohistochemically, tumor cells are diffusely positive for epithelial membrane antigen and vimentin.18,19
- Amonkar GP, Rupani A, Shah A, et al. Bednar tumor: an uncommon entity. Dermatopathology (Basel). 2016;3:36-38.
- El Hachem M, Diociaiuti A, Latella E, et al. Congenital myxoid and pigmented dermatofibrosarcoma protuberans: a case report. Pediatr Dermatol. 2013;30:E74-E77.
- Anon-Requena MJ, Pico-Valimana M, Munoz-Arias G. Bednar tumor (pigmented dermatofibrosarcoma protuberans). Actas Dermosifiliogr. 2016;107:618-620.
- Wang J, Yang W. Pigmented dermatofibrosarcoma protuberans with prominent meningothelial-like whorls. J Cutan Pathol. 2008;35(suppl 1):65-69.
- Zardawi IM, Kattampallil J, Rode J. An unusual pigmented skin tumour. Bednar tumour, dorsum of left foot (pigmented dermatofibrosarcoma protuberans). Pathology. 2004;36:358-361.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and "malignant blue nevus": a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017;37:401-415.
- Zembowicz A, Granter SR, McKee PH, et al. Amelanotic cellular blue nevus: a hypopigmented variant of the cellular blue nevus: clinicopathologic analysis of 20 cases. Am J Surg Pathol. 2002;26:1493-1500.
- Smith K, Germain M, Williams J, et al. CD34-positive cellular blue nevi. J Cutan Pathol. 2001;28:145-150.
- Inaba M, Yamamoto T, Minami R, et al. Pigmented neurofibroma: report of two cases and literature review. Pathol Int. 2001;51:565-569.
- Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol. 2000;24:331-343.
- Motoi T, Ishida T, Kawato A, et al. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Hum Pathol. 2005;36:871-877.
- Yeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Schleich C, Ferringer T. Desmoplastic melanoma. Cutis. 2015;96:306, 313-314, 335.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas--a clinicopathologic study. Cancer. 1974;34:728-744.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol Lab Med. 2012;136:208-211.
- Bhanusali DG, Heath C, Gur D, et al. Metastatic meningioma of the scalp. Cutis. 2018;101:386-389.
The Diagnosis: Pigmented Dermatofibrosarcoma Protuberans
Pigmented dermatofibrosarcoma protuberans (PDFSP), also known as Bednar tumor, is an uncommon variant of dermatofibrosarcoma protuberans (DFSP). Pigmented dermatofibrosarcoma protuberans constitutes 1% to 5% of all DFSP cases and most commonly is seen in nonwhite adults in the fourth decade of life, with occasional cases seen in pediatric patients, including some congenital cases. Typical sites of involvement include the shoulders, trunk, arms, legs, head, and neck.1,2 It also has been reported at sites of prior immunization, trauma, and insect bites.3
Histopathologic examination of our patient's shoulder nodule revealed an infiltrative neoplasm in the dermis and subcutaneous tissue composed of spindled cells with a storiform pattern and foci of scattered elongated dendritic pigmented cells. A narrow grenz zone separated the tumor from the epidermis, and characteristic honeycomb infiltration by tumor cells was noted in the subcutaneous fat. The nuclei were bland and monomorphous with areas of neuroid differentiation containing whorls and nerve cord-like structures (quiz image). The tumor cells were diffusely CD34 and vimentin positive, while S-100, SOX-10, neurofilament, smooth muscle actin, desmin, epithelial membrane antigen, and cytokeratins were negative. The immunophenotype excluded the possibility of neurogenic, pericytic, myofibroblastic, and myoid differentiation.
Wang and Yang4 previously reported a case of PDFSP with prominent meningothelial-like whorls focally resembling extracranial meningioma; however, the tumor cells were CD34 positive and epithelial membrane antigen negative, weighing against a diagnosis of meningioma. Most cases of PDFSP demonstrate the COL1A1-PDGFB (collagen type I α; 1/platelet-derived growth factor B-chain) fusion protein caused by the translocation t(17;22)(q22;q13), as in classic DFSP.5
Cellular blue nevus (CBN) is a benign melanocytic neoplasm that can present at any age and often occurs on the buttocks and in the sacrococcygeal region. Clinically, CBN presents as a firm, bluish black to bluish gray, dome-shaped nodule. The size varies from a few millimeters to several centimeters.6,7 Histologically, CBN is located completely in the dermis, extending along the adnexae into the subcutaneous tissue with a dumbbell-shaped outline (Figure 1).6-8 The tumor demonstrates oval epithelioid melanocytes with vesicular nuclei and prominent nucleoli. Immunohistochemically, tumor cells stain positively for melanocytic markers such as S-100, SOX-10, MART-1, and human melanoma black 45. CD34 expression rarely is reported in a subset of CBN.9
Pigmented neurofibroma is a rare variant of neurofibroma that produces melanin pigment and has a strong association with neurofibromatosis.10 It occurs most frequently in dark-skinned populations (Fitzpatrick skin types IV-VI). The most common location is the head and neck region.11,12 Histologically, pigmented neurofibroma resembles a diffuse neurofibroma admixed with melanin-producing cells (Figure 2).12 Immunostaining shows positivity for S-100 in both pigmented and Schwann cells; however, the pigmented cells stain positively for human melanoma black 45, Melan-A, and tyrosinase.10 CD34 can be fingerprint positive in neurofibroma, but a distinction from DFSP can be made by S-100 and SOX-10 immunostaining.13
Desmoplastic melanoma (DM) is an uncommon variant of malignant melanoma and has a higher tendency for persistent local growth and less frequent metastases than other variants of melanoma. It has a predilection for chronically sun-exposed areas such as the head and neck and occurs later in life. Clinically, DM appears as nonspecific, often amelanotic nodules or plaques or as scarlike lesions.14 Histologically, DM can be classified as mixed or pure based on the degree of desmoplasia and cellularity. A paucicellular proliferation of malignant spindled melanocytes within a densely fibrotic stroma with lymphoid nodules in the dermis is characteristic (Figure 3); perineural involvement is common.14,15 The most reliable confirmative stains are S-100 and SOX-10.16
Cutaneous meningioma is a rare tumor and could be subtyped into 3 groups. Type I is primary cutaneous meningioma and usually is present at birth on the scalp and paravertebral regions with a relatively good prognosis. Type II is ectopic soft-tissue meningioma that extends into the skin from around the sensory organs on the face. Type III is local invasion or true metastasis from a central nervous system meningioma. Types II and III develop later in life and the prognosis is poor.17,18 Clinically, lesions present as firm subcutaneous nodules or swellings. Cutaneous meningioma has several histopathologic variants. The classic presentation reveals concentric wrapping of tumor cells with round-oval nuclei containing delicate chromatin. Psammoma bodies are a common finding (Figure 4). Immunohistochemically, tumor cells are diffusely positive for epithelial membrane antigen and vimentin.18,19
The Diagnosis: Pigmented Dermatofibrosarcoma Protuberans
Pigmented dermatofibrosarcoma protuberans (PDFSP), also known as Bednar tumor, is an uncommon variant of dermatofibrosarcoma protuberans (DFSP). Pigmented dermatofibrosarcoma protuberans constitutes 1% to 5% of all DFSP cases and most commonly is seen in nonwhite adults in the fourth decade of life, with occasional cases seen in pediatric patients, including some congenital cases. Typical sites of involvement include the shoulders, trunk, arms, legs, head, and neck.1,2 It also has been reported at sites of prior immunization, trauma, and insect bites.3
Histopathologic examination of our patient's shoulder nodule revealed an infiltrative neoplasm in the dermis and subcutaneous tissue composed of spindled cells with a storiform pattern and foci of scattered elongated dendritic pigmented cells. A narrow grenz zone separated the tumor from the epidermis, and characteristic honeycomb infiltration by tumor cells was noted in the subcutaneous fat. The nuclei were bland and monomorphous with areas of neuroid differentiation containing whorls and nerve cord-like structures (quiz image). The tumor cells were diffusely CD34 and vimentin positive, while S-100, SOX-10, neurofilament, smooth muscle actin, desmin, epithelial membrane antigen, and cytokeratins were negative. The immunophenotype excluded the possibility of neurogenic, pericytic, myofibroblastic, and myoid differentiation.
Wang and Yang4 previously reported a case of PDFSP with prominent meningothelial-like whorls focally resembling extracranial meningioma; however, the tumor cells were CD34 positive and epithelial membrane antigen negative, weighing against a diagnosis of meningioma. Most cases of PDFSP demonstrate the COL1A1-PDGFB (collagen type I α; 1/platelet-derived growth factor B-chain) fusion protein caused by the translocation t(17;22)(q22;q13), as in classic DFSP.5
Cellular blue nevus (CBN) is a benign melanocytic neoplasm that can present at any age and often occurs on the buttocks and in the sacrococcygeal region. Clinically, CBN presents as a firm, bluish black to bluish gray, dome-shaped nodule. The size varies from a few millimeters to several centimeters.6,7 Histologically, CBN is located completely in the dermis, extending along the adnexae into the subcutaneous tissue with a dumbbell-shaped outline (Figure 1).6-8 The tumor demonstrates oval epithelioid melanocytes with vesicular nuclei and prominent nucleoli. Immunohistochemically, tumor cells stain positively for melanocytic markers such as S-100, SOX-10, MART-1, and human melanoma black 45. CD34 expression rarely is reported in a subset of CBN.9
Pigmented neurofibroma is a rare variant of neurofibroma that produces melanin pigment and has a strong association with neurofibromatosis.10 It occurs most frequently in dark-skinned populations (Fitzpatrick skin types IV-VI). The most common location is the head and neck region.11,12 Histologically, pigmented neurofibroma resembles a diffuse neurofibroma admixed with melanin-producing cells (Figure 2).12 Immunostaining shows positivity for S-100 in both pigmented and Schwann cells; however, the pigmented cells stain positively for human melanoma black 45, Melan-A, and tyrosinase.10 CD34 can be fingerprint positive in neurofibroma, but a distinction from DFSP can be made by S-100 and SOX-10 immunostaining.13
Desmoplastic melanoma (DM) is an uncommon variant of malignant melanoma and has a higher tendency for persistent local growth and less frequent metastases than other variants of melanoma. It has a predilection for chronically sun-exposed areas such as the head and neck and occurs later in life. Clinically, DM appears as nonspecific, often amelanotic nodules or plaques or as scarlike lesions.14 Histologically, DM can be classified as mixed or pure based on the degree of desmoplasia and cellularity. A paucicellular proliferation of malignant spindled melanocytes within a densely fibrotic stroma with lymphoid nodules in the dermis is characteristic (Figure 3); perineural involvement is common.14,15 The most reliable confirmative stains are S-100 and SOX-10.16
Cutaneous meningioma is a rare tumor and could be subtyped into 3 groups. Type I is primary cutaneous meningioma and usually is present at birth on the scalp and paravertebral regions with a relatively good prognosis. Type II is ectopic soft-tissue meningioma that extends into the skin from around the sensory organs on the face. Type III is local invasion or true metastasis from a central nervous system meningioma. Types II and III develop later in life and the prognosis is poor.17,18 Clinically, lesions present as firm subcutaneous nodules or swellings. Cutaneous meningioma has several histopathologic variants. The classic presentation reveals concentric wrapping of tumor cells with round-oval nuclei containing delicate chromatin. Psammoma bodies are a common finding (Figure 4). Immunohistochemically, tumor cells are diffusely positive for epithelial membrane antigen and vimentin.18,19
- Amonkar GP, Rupani A, Shah A, et al. Bednar tumor: an uncommon entity. Dermatopathology (Basel). 2016;3:36-38.
- El Hachem M, Diociaiuti A, Latella E, et al. Congenital myxoid and pigmented dermatofibrosarcoma protuberans: a case report. Pediatr Dermatol. 2013;30:E74-E77.
- Anon-Requena MJ, Pico-Valimana M, Munoz-Arias G. Bednar tumor (pigmented dermatofibrosarcoma protuberans). Actas Dermosifiliogr. 2016;107:618-620.
- Wang J, Yang W. Pigmented dermatofibrosarcoma protuberans with prominent meningothelial-like whorls. J Cutan Pathol. 2008;35(suppl 1):65-69.
- Zardawi IM, Kattampallil J, Rode J. An unusual pigmented skin tumour. Bednar tumour, dorsum of left foot (pigmented dermatofibrosarcoma protuberans). Pathology. 2004;36:358-361.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and "malignant blue nevus": a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017;37:401-415.
- Zembowicz A, Granter SR, McKee PH, et al. Amelanotic cellular blue nevus: a hypopigmented variant of the cellular blue nevus: clinicopathologic analysis of 20 cases. Am J Surg Pathol. 2002;26:1493-1500.
- Smith K, Germain M, Williams J, et al. CD34-positive cellular blue nevi. J Cutan Pathol. 2001;28:145-150.
- Inaba M, Yamamoto T, Minami R, et al. Pigmented neurofibroma: report of two cases and literature review. Pathol Int. 2001;51:565-569.
- Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol. 2000;24:331-343.
- Motoi T, Ishida T, Kawato A, et al. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Hum Pathol. 2005;36:871-877.
- Yeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Schleich C, Ferringer T. Desmoplastic melanoma. Cutis. 2015;96:306, 313-314, 335.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas--a clinicopathologic study. Cancer. 1974;34:728-744.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol Lab Med. 2012;136:208-211.
- Bhanusali DG, Heath C, Gur D, et al. Metastatic meningioma of the scalp. Cutis. 2018;101:386-389.
- Amonkar GP, Rupani A, Shah A, et al. Bednar tumor: an uncommon entity. Dermatopathology (Basel). 2016;3:36-38.
- El Hachem M, Diociaiuti A, Latella E, et al. Congenital myxoid and pigmented dermatofibrosarcoma protuberans: a case report. Pediatr Dermatol. 2013;30:E74-E77.
- Anon-Requena MJ, Pico-Valimana M, Munoz-Arias G. Bednar tumor (pigmented dermatofibrosarcoma protuberans). Actas Dermosifiliogr. 2016;107:618-620.
- Wang J, Yang W. Pigmented dermatofibrosarcoma protuberans with prominent meningothelial-like whorls. J Cutan Pathol. 2008;35(suppl 1):65-69.
- Zardawi IM, Kattampallil J, Rode J. An unusual pigmented skin tumour. Bednar tumour, dorsum of left foot (pigmented dermatofibrosarcoma protuberans). Pathology. 2004;36:358-361.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and "malignant blue nevus": a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017;37:401-415.
- Zembowicz A, Granter SR, McKee PH, et al. Amelanotic cellular blue nevus: a hypopigmented variant of the cellular blue nevus: clinicopathologic analysis of 20 cases. Am J Surg Pathol. 2002;26:1493-1500.
- Smith K, Germain M, Williams J, et al. CD34-positive cellular blue nevi. J Cutan Pathol. 2001;28:145-150.
- Inaba M, Yamamoto T, Minami R, et al. Pigmented neurofibroma: report of two cases and literature review. Pathol Int. 2001;51:565-569.
- Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol. 2000;24:331-343.
- Motoi T, Ishida T, Kawato A, et al. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Hum Pathol. 2005;36:871-877.
- Yeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Schleich C, Ferringer T. Desmoplastic melanoma. Cutis. 2015;96:306, 313-314, 335.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas--a clinicopathologic study. Cancer. 1974;34:728-744.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol Lab Med. 2012;136:208-211.
- Bhanusali DG, Heath C, Gur D, et al. Metastatic meningioma of the scalp. Cutis. 2018;101:386-389.

A 37-year-old woman presented with an asymptomatic, indurated, pigmented, subcutaneous nodule on the right shoulder of more than 3 years' duration. The lesion had gradually increased in size with no associated symptoms. The patient had a history of endometrial adenocarcinoma and papillary thyroid carcinoma, which had been treated by hysterectomy-oophorectomy and right thyroidectomy, respectively. She had no other notable systemic abnormalities, and there was no family history of genetic disease or cancer. Physical examination demonstrated a 1.2×1.8-cm nontender, pigmented, subcutaneous nodule with a rough surface and indistinct borders. An excisional biopsy was performed.
Well-Circumscribed Tumor on the Hand
The Diagnosis: Nodular Kaposi Sarcoma
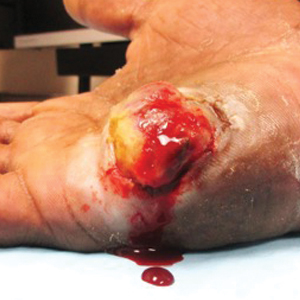
Epidemic Kaposi sarcoma (KS) primarily affects patients with human immunodeficiency virus (HIV) infection. Kaposi sarcoma can appear as brown, red, or blue-black macules, plaques, patches, nodules, or tumors, and it often is observed as multifocal cutaneous lesions located on the head, neck, and upper aspects of the trunk in a fulminant manner. Kaposi sarcoma portends a poor prognosis and is an AIDS-defining malignancy.1-3 Importantly, antiretroviral therapy does not preclude its consideration in those without AIDS-defining CD4 cell counts and undetectable HIV viremia presenting with cutaneous manifestations.2,3 A retrospective review by Daly et al4 reported KS lesions in patients with CD4 lymphocyte counts greater than 300 cells/µL, most of whom were antiretroviral therapy-naïve patients. Also, those with higher CD4 counts tended to have a solitary KS lesion at presentation, while those with CD4 counts less than 300 cells/µL tended to present with multiple foci.4 Epidemic KS lesions are clinically indistinguishable from other common cutaneous conditions in the differential diagnosis of KS, necessitating biopsy for histopathologic examination. Light microscopy findings help to delineate the diagnosis of KS. Immunohistochemical staining to the latent nuclear antigen 1 of human herpesvirus 8 (HHV-8) confirms the KS diagnosis.5,6 Our patient's presentation as a solitary acral lesion was atypical for KS.
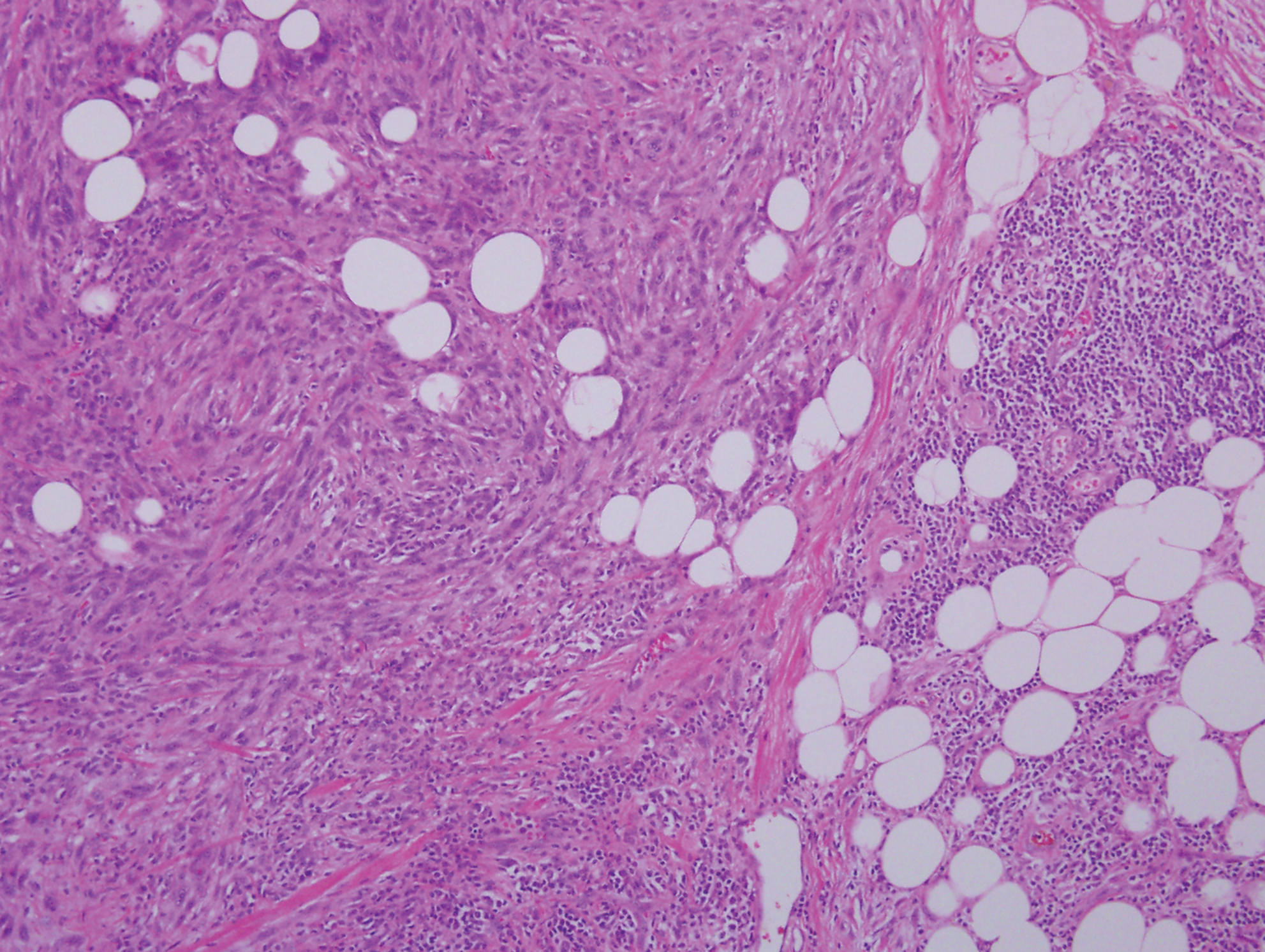
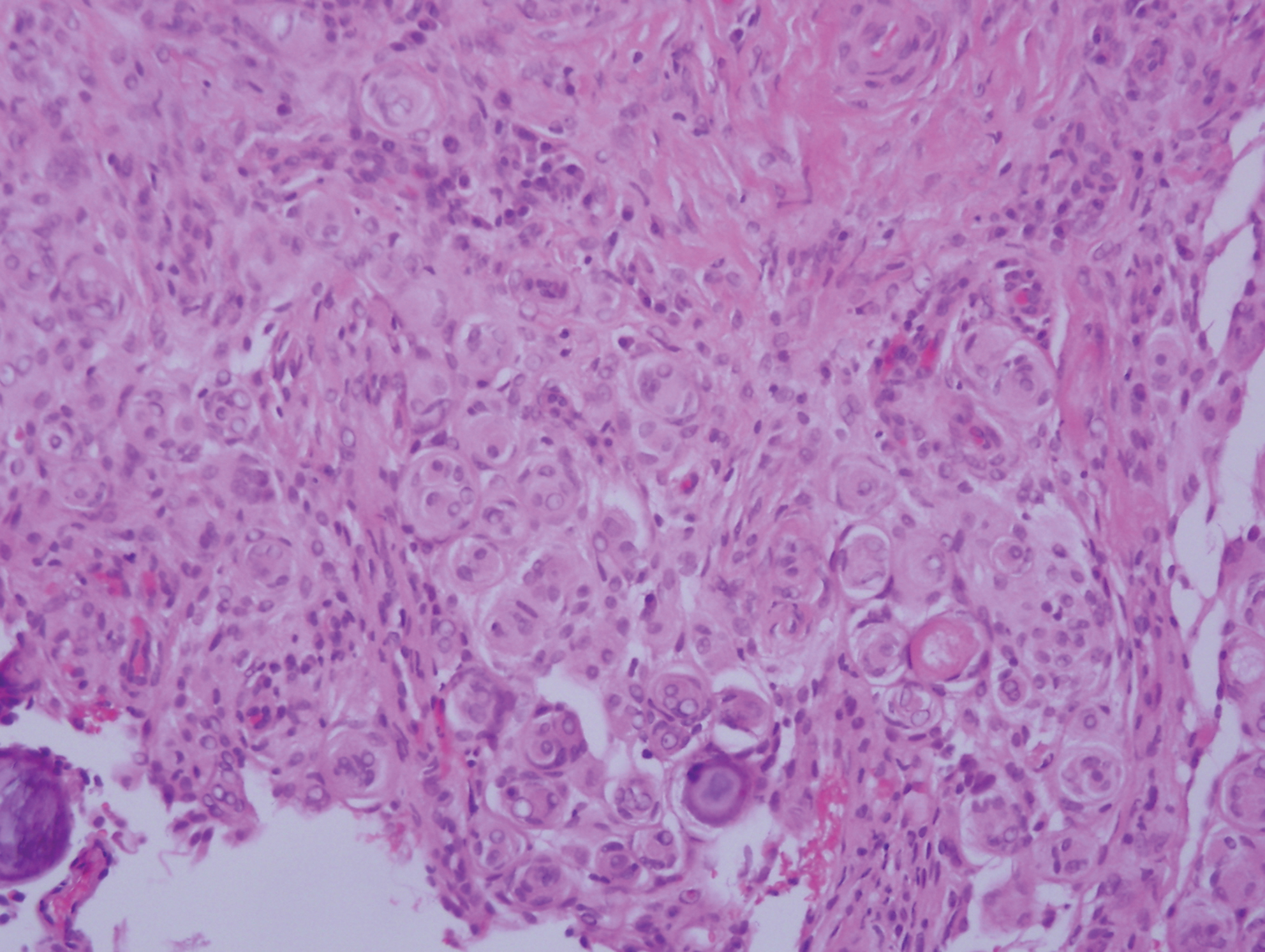
Light microscopy of our patient's biopsy demonstrated a large tumor on the acral surface of the right hand. Dermal collections of basophilic spindled cells clustered with small slitlike vascular spaces with abundant erythrocyte extravasation and numerous large ectatic vessels at the periphery were seen (Figure, A). At higher magnification, interlaced bundles of spindle cells with slitlike vessels with scattered lymphocytes and plasma cells were seen (Figure, B). An immunohistochemical stain for HHV-8 was positive and largely confined to spindle cells (Figure, C). These findings confirmed KS and met AIDS-defining criteria. Awareness of these histopathologic features is key in differentiating KS from other conditions in the differential diagnosis.

The patient's history of late latent syphilis coinfected with HIV and persistently elevated rapid plasma reagin that was recalcitrant to therapy placed an atypical nodular presentation within reason for the differential diagnosis. Deviations from the typical papulosquamous presentation with acral involvement in an immunocompromised patient mandates a consideration for syphilis with an atypical presentation. Atypical presentations include nodular, annular, pustular, lues maligna, frambesiform, corymbose, and photosensitive distributions.7,8 Notably, coinfection with HIV modifies the clinical presentation, serology, and efficacy of treatment.7-10 Atypical presentations are more common in coinfected HIV-positive patients, mandating a high degree of suspicion. Nodular secondary syphilis and the noduloulcerative form (lues maligna) often spare the palmar and plantar surfaces, and patients often have constitutional symptoms accompanying the cutaneous eruptions. In questionable cases, a biopsy lends clarification. Light microscopy on hematoxylin and eosin (H&E) staining may display acanthosis, superficial and deep perivascular swelling, plasma, histiocyte infiltrates, dermoepidermal junction changes, mixed patterns, epidermal hyperplasia, and dermal vascular thickening.7-9,11 Spirochetes may be observed on Warthin-Starry stain; however, artifact obscuration from melanin granules and reticular fibers or paucity of organisms can make identification difficult. Immunohistochemical staining may prove useful when H&E stains are atypical or have a paucity of organisms or plasma cells or when silver stains have artifactual obscuration.9 Our patient's solitary palmar lesion without constitutional symptoms made an atypical nodular secondary syphilis presentation less likely. Ultimately, the histopathologic findings were consistent with KS.
Bacillary angiomatosis (BA) is caused by Bartonella species and results in vascular proliferation with cutaneous manifestation. It frequently is observed in patients with HIV or other immunosuppressive conditions as well as patients with exposure to mammals or their vectors. Protean cutaneous manifestations and distributions of BA exist. The number of lesions can be singular to thousands. Solitary superficial pyogenic granuloma-like lesions can be clinically indistinguishable from both KS and pyogenic granuloma (PG). Superficial lesions often begin as red, violaceous, or flesh-colored papules that hemorrhage easily with trauma. The morphology of the papule can progress to be exophytic with dome-shaped or ulcerative surface features and is rubbery on palpation.12 Biopsy is required to differentiate BA from KS. Bacillary angiomatosis on light microscopy with H&E shows protuberant, lobulated, round vessels with plump endothelial cells with or without necrosis. A neutrophil infiltrate in close proximity to bacilli may be noted. Warthin-Starry stain demonstrates numerous bacilli juxtaposed to these endothelial cells. The lack of immunohistochemical staining for HHV-8 also differentiates BA from KS.12,13
Pyogenic granuloma is resultant from proliferation of endothelial cells with a lobular architecture. Pyogenic granulomas are benign, rapidly progressive, acquired lesions presenting in the skin and mucous membranes. Pyogenic granuloma often presents as a single painless papule or nodule with a glistening red-violaceous color that occasionally appears with a perilesional collarette. The lesions are friable and easily hemorrhage. Pyogenic granuloma has been associated with local skin trauma and estrogen hormones. Histopathologic examination of PG assists with differentiation from other nodular lesions. Light microscopy with standard H&E staining demonstrates a network of capillaries arranged into a lobule surrounded by a fibrous matrix. Endothelial cells appear round and protrude into the vascular lamina. Mitotic activity is increased. Lack of findings on Warthin-Starry stain assists with differentiating PG from BA, while the microscopy architecture and immunohistochemical staining differentiates PG from KS.6,13,14
Squamous cell carcinoma (SCC) is the primary malignant cancer of the hand. The dorsal aspect of the hand is the most common location; SCC less commonly is located on the palmar surface, fingers, nail bed, or intertriginous areas.15-17 Chakrabarti et al16 found that these lesions were invasive SCC when located on the palmar surface. Morphologically, SCC takes an exophytic papular, nodular, or scaly appearance with a red to flesh-colored appearance and poor demarcation of the borders. Progression to large ulcerated or secondarily infected lesions also can occur. The inflammatory reaction may cause tenderness to palpation and hemorrhage with trauma. Histopathologic examination of invasive SCC reveals atypical keratinocytes violating the basement membrane and abundant cytoplasm. Our patient's clinical presentation placed invasive SCC low on the differential diagnosis, and the histopathologic and immunohistochemical results eliminated SCC as the diagnosis.
- Antman K, Chang Y. Kaposi's sarcoma. N Engl J Med. 2000;342:1027-1038.
- Pipette WW. The incidence of second malignancies in subsets of Kaposi's sarcoma. J Am Acad Dermatol. 1987;16:855-861.
- Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12:6-11.
- Daly ML, Fogo A, McDonald C, et al. Kaposi sarcoma: no longer an AIDS-defining illness? a retrospective study of Kaposi sarcoma cases with CD4 counts above 300/mm³ at presentation. Clin Exp Dermatol. 2014;39:7-12.
- Broccolo F, Tassan Din C, Viganò MG, et al. HHV-8 DNA replication correlates with the clinical status in AIDS-related Kaposi's sarcoma. J Clin Virol. 2016;78:47-52.
- Pereira PF, Cuzzi T, Galhardo MC. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An Bras Dermatol. 2013;88:243-246.
- Gevorgyan O, Owen BD, Balavenkataraman A, et al. A nodular-ulcerative form of secondary syphilis in AIDS. Proc (Bayl Univ Med Cent). 2017;30:80-82.
- Balagula Y, Mattei PL, Wisco OJ, et al. The great imitator revisited: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;31:595-599.
- Yayli S, della Torre R, Hegyi I, et al. Late secondary syphilis with nodular lesions mimicking Kaposi sarcoma in a patient with human immunodeficiency virus. Int J Dermatol. 2014;53:E71-E73.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Cockerell CJ, LeBoit PE. Bacillary angiomatosis: a newly characterized, pseudoneoplastic, infectious, cutaneous vascular disorder. J Am Acad Dermatol. 1990;22:501-512.
- Forrestel AK, Naujokas A, Martin JN, et al. Bacillary angiomatosis masquerading as Kaposi's sarcoma in East Africa. J Int Assoc Provid AIDS Care. 2015;14:21-25.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma), localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol. 2007;29:408-411.
- Marks R. Squamous cell carcinoma. Lancet. 1996;347:735-738.
- Chakrabarti I, Watson JD, Dorrance H. Skin tumours of the hand. a 10-year review. J Hand Surg Br. 1993;18:484-486.
- Sobanko JF, Dagum AB, Davis IC, et al. Soft tissue tumors of the hand. 2. malignant. Dermatol Surg. 2007;33:771-785.
The Diagnosis: Nodular Kaposi Sarcoma
Epidemic Kaposi sarcoma (KS) primarily affects patients with human immunodeficiency virus (HIV) infection. Kaposi sarcoma can appear as brown, red, or blue-black macules, plaques, patches, nodules, or tumors, and it often is observed as multifocal cutaneous lesions located on the head, neck, and upper aspects of the trunk in a fulminant manner. Kaposi sarcoma portends a poor prognosis and is an AIDS-defining malignancy.1-3 Importantly, antiretroviral therapy does not preclude its consideration in those without AIDS-defining CD4 cell counts and undetectable HIV viremia presenting with cutaneous manifestations.2,3 A retrospective review by Daly et al4 reported KS lesions in patients with CD4 lymphocyte counts greater than 300 cells/µL, most of whom were antiretroviral therapy-naïve patients. Also, those with higher CD4 counts tended to have a solitary KS lesion at presentation, while those with CD4 counts less than 300 cells/µL tended to present with multiple foci.4 Epidemic KS lesions are clinically indistinguishable from other common cutaneous conditions in the differential diagnosis of KS, necessitating biopsy for histopathologic examination. Light microscopy findings help to delineate the diagnosis of KS. Immunohistochemical staining to the latent nuclear antigen 1 of human herpesvirus 8 (HHV-8) confirms the KS diagnosis.5,6 Our patient's presentation as a solitary acral lesion was atypical for KS.
Light microscopy of our patient's biopsy demonstrated a large tumor on the acral surface of the right hand. Dermal collections of basophilic spindled cells clustered with small slitlike vascular spaces with abundant erythrocyte extravasation and numerous large ectatic vessels at the periphery were seen (Figure, A). At higher magnification, interlaced bundles of spindle cells with slitlike vessels with scattered lymphocytes and plasma cells were seen (Figure, B). An immunohistochemical stain for HHV-8 was positive and largely confined to spindle cells (Figure, C). These findings confirmed KS and met AIDS-defining criteria. Awareness of these histopathologic features is key in differentiating KS from other conditions in the differential diagnosis.
The patient's history of late latent syphilis coinfected with HIV and persistently elevated rapid plasma reagin that was recalcitrant to therapy placed an atypical nodular presentation within reason for the differential diagnosis. Deviations from the typical papulosquamous presentation with acral involvement in an immunocompromised patient mandates a consideration for syphilis with an atypical presentation. Atypical presentations include nodular, annular, pustular, lues maligna, frambesiform, corymbose, and photosensitive distributions.7,8 Notably, coinfection with HIV modifies the clinical presentation, serology, and efficacy of treatment.7-10 Atypical presentations are more common in coinfected HIV-positive patients, mandating a high degree of suspicion. Nodular secondary syphilis and the noduloulcerative form (lues maligna) often spare the palmar and plantar surfaces, and patients often have constitutional symptoms accompanying the cutaneous eruptions. In questionable cases, a biopsy lends clarification. Light microscopy on hematoxylin and eosin (H&E) staining may display acanthosis, superficial and deep perivascular swelling, plasma, histiocyte infiltrates, dermoepidermal junction changes, mixed patterns, epidermal hyperplasia, and dermal vascular thickening.7-9,11 Spirochetes may be observed on Warthin-Starry stain; however, artifact obscuration from melanin granules and reticular fibers or paucity of organisms can make identification difficult. Immunohistochemical staining may prove useful when H&E stains are atypical or have a paucity of organisms or plasma cells or when silver stains have artifactual obscuration.9 Our patient's solitary palmar lesion without constitutional symptoms made an atypical nodular secondary syphilis presentation less likely. Ultimately, the histopathologic findings were consistent with KS.
Bacillary angiomatosis (BA) is caused by Bartonella species and results in vascular proliferation with cutaneous manifestation. It frequently is observed in patients with HIV or other immunosuppressive conditions as well as patients with exposure to mammals or their vectors. Protean cutaneous manifestations and distributions of BA exist. The number of lesions can be singular to thousands. Solitary superficial pyogenic granuloma-like lesions can be clinically indistinguishable from both KS and pyogenic granuloma (PG). Superficial lesions often begin as red, violaceous, or flesh-colored papules that hemorrhage easily with trauma. The morphology of the papule can progress to be exophytic with dome-shaped or ulcerative surface features and is rubbery on palpation.12 Biopsy is required to differentiate BA from KS. Bacillary angiomatosis on light microscopy with H&E shows protuberant, lobulated, round vessels with plump endothelial cells with or without necrosis. A neutrophil infiltrate in close proximity to bacilli may be noted. Warthin-Starry stain demonstrates numerous bacilli juxtaposed to these endothelial cells. The lack of immunohistochemical staining for HHV-8 also differentiates BA from KS.12,13
Pyogenic granuloma is resultant from proliferation of endothelial cells with a lobular architecture. Pyogenic granulomas are benign, rapidly progressive, acquired lesions presenting in the skin and mucous membranes. Pyogenic granuloma often presents as a single painless papule or nodule with a glistening red-violaceous color that occasionally appears with a perilesional collarette. The lesions are friable and easily hemorrhage. Pyogenic granuloma has been associated with local skin trauma and estrogen hormones. Histopathologic examination of PG assists with differentiation from other nodular lesions. Light microscopy with standard H&E staining demonstrates a network of capillaries arranged into a lobule surrounded by a fibrous matrix. Endothelial cells appear round and protrude into the vascular lamina. Mitotic activity is increased. Lack of findings on Warthin-Starry stain assists with differentiating PG from BA, while the microscopy architecture and immunohistochemical staining differentiates PG from KS.6,13,14
Squamous cell carcinoma (SCC) is the primary malignant cancer of the hand. The dorsal aspect of the hand is the most common location; SCC less commonly is located on the palmar surface, fingers, nail bed, or intertriginous areas.15-17 Chakrabarti et al16 found that these lesions were invasive SCC when located on the palmar surface. Morphologically, SCC takes an exophytic papular, nodular, or scaly appearance with a red to flesh-colored appearance and poor demarcation of the borders. Progression to large ulcerated or secondarily infected lesions also can occur. The inflammatory reaction may cause tenderness to palpation and hemorrhage with trauma. Histopathologic examination of invasive SCC reveals atypical keratinocytes violating the basement membrane and abundant cytoplasm. Our patient's clinical presentation placed invasive SCC low on the differential diagnosis, and the histopathologic and immunohistochemical results eliminated SCC as the diagnosis.
The Diagnosis: Nodular Kaposi Sarcoma
Epidemic Kaposi sarcoma (KS) primarily affects patients with human immunodeficiency virus (HIV) infection. Kaposi sarcoma can appear as brown, red, or blue-black macules, plaques, patches, nodules, or tumors, and it often is observed as multifocal cutaneous lesions located on the head, neck, and upper aspects of the trunk in a fulminant manner. Kaposi sarcoma portends a poor prognosis and is an AIDS-defining malignancy.1-3 Importantly, antiretroviral therapy does not preclude its consideration in those without AIDS-defining CD4 cell counts and undetectable HIV viremia presenting with cutaneous manifestations.2,3 A retrospective review by Daly et al4 reported KS lesions in patients with CD4 lymphocyte counts greater than 300 cells/µL, most of whom were antiretroviral therapy-naïve patients. Also, those with higher CD4 counts tended to have a solitary KS lesion at presentation, while those with CD4 counts less than 300 cells/µL tended to present with multiple foci.4 Epidemic KS lesions are clinically indistinguishable from other common cutaneous conditions in the differential diagnosis of KS, necessitating biopsy for histopathologic examination. Light microscopy findings help to delineate the diagnosis of KS. Immunohistochemical staining to the latent nuclear antigen 1 of human herpesvirus 8 (HHV-8) confirms the KS diagnosis.5,6 Our patient's presentation as a solitary acral lesion was atypical for KS.
Light microscopy of our patient's biopsy demonstrated a large tumor on the acral surface of the right hand. Dermal collections of basophilic spindled cells clustered with small slitlike vascular spaces with abundant erythrocyte extravasation and numerous large ectatic vessels at the periphery were seen (Figure, A). At higher magnification, interlaced bundles of spindle cells with slitlike vessels with scattered lymphocytes and plasma cells were seen (Figure, B). An immunohistochemical stain for HHV-8 was positive and largely confined to spindle cells (Figure, C). These findings confirmed KS and met AIDS-defining criteria. Awareness of these histopathologic features is key in differentiating KS from other conditions in the differential diagnosis.
The patient's history of late latent syphilis coinfected with HIV and persistently elevated rapid plasma reagin that was recalcitrant to therapy placed an atypical nodular presentation within reason for the differential diagnosis. Deviations from the typical papulosquamous presentation with acral involvement in an immunocompromised patient mandates a consideration for syphilis with an atypical presentation. Atypical presentations include nodular, annular, pustular, lues maligna, frambesiform, corymbose, and photosensitive distributions.7,8 Notably, coinfection with HIV modifies the clinical presentation, serology, and efficacy of treatment.7-10 Atypical presentations are more common in coinfected HIV-positive patients, mandating a high degree of suspicion. Nodular secondary syphilis and the noduloulcerative form (lues maligna) often spare the palmar and plantar surfaces, and patients often have constitutional symptoms accompanying the cutaneous eruptions. In questionable cases, a biopsy lends clarification. Light microscopy on hematoxylin and eosin (H&E) staining may display acanthosis, superficial and deep perivascular swelling, plasma, histiocyte infiltrates, dermoepidermal junction changes, mixed patterns, epidermal hyperplasia, and dermal vascular thickening.7-9,11 Spirochetes may be observed on Warthin-Starry stain; however, artifact obscuration from melanin granules and reticular fibers or paucity of organisms can make identification difficult. Immunohistochemical staining may prove useful when H&E stains are atypical or have a paucity of organisms or plasma cells or when silver stains have artifactual obscuration.9 Our patient's solitary palmar lesion without constitutional symptoms made an atypical nodular secondary syphilis presentation less likely. Ultimately, the histopathologic findings were consistent with KS.
Bacillary angiomatosis (BA) is caused by Bartonella species and results in vascular proliferation with cutaneous manifestation. It frequently is observed in patients with HIV or other immunosuppressive conditions as well as patients with exposure to mammals or their vectors. Protean cutaneous manifestations and distributions of BA exist. The number of lesions can be singular to thousands. Solitary superficial pyogenic granuloma-like lesions can be clinically indistinguishable from both KS and pyogenic granuloma (PG). Superficial lesions often begin as red, violaceous, or flesh-colored papules that hemorrhage easily with trauma. The morphology of the papule can progress to be exophytic with dome-shaped or ulcerative surface features and is rubbery on palpation.12 Biopsy is required to differentiate BA from KS. Bacillary angiomatosis on light microscopy with H&E shows protuberant, lobulated, round vessels with plump endothelial cells with or without necrosis. A neutrophil infiltrate in close proximity to bacilli may be noted. Warthin-Starry stain demonstrates numerous bacilli juxtaposed to these endothelial cells. The lack of immunohistochemical staining for HHV-8 also differentiates BA from KS.12,13
Pyogenic granuloma is resultant from proliferation of endothelial cells with a lobular architecture. Pyogenic granulomas are benign, rapidly progressive, acquired lesions presenting in the skin and mucous membranes. Pyogenic granuloma often presents as a single painless papule or nodule with a glistening red-violaceous color that occasionally appears with a perilesional collarette. The lesions are friable and easily hemorrhage. Pyogenic granuloma has been associated with local skin trauma and estrogen hormones. Histopathologic examination of PG assists with differentiation from other nodular lesions. Light microscopy with standard H&E staining demonstrates a network of capillaries arranged into a lobule surrounded by a fibrous matrix. Endothelial cells appear round and protrude into the vascular lamina. Mitotic activity is increased. Lack of findings on Warthin-Starry stain assists with differentiating PG from BA, while the microscopy architecture and immunohistochemical staining differentiates PG from KS.6,13,14
Squamous cell carcinoma (SCC) is the primary malignant cancer of the hand. The dorsal aspect of the hand is the most common location; SCC less commonly is located on the palmar surface, fingers, nail bed, or intertriginous areas.15-17 Chakrabarti et al16 found that these lesions were invasive SCC when located on the palmar surface. Morphologically, SCC takes an exophytic papular, nodular, or scaly appearance with a red to flesh-colored appearance and poor demarcation of the borders. Progression to large ulcerated or secondarily infected lesions also can occur. The inflammatory reaction may cause tenderness to palpation and hemorrhage with trauma. Histopathologic examination of invasive SCC reveals atypical keratinocytes violating the basement membrane and abundant cytoplasm. Our patient's clinical presentation placed invasive SCC low on the differential diagnosis, and the histopathologic and immunohistochemical results eliminated SCC as the diagnosis.
- Antman K, Chang Y. Kaposi's sarcoma. N Engl J Med. 2000;342:1027-1038.
- Pipette WW. The incidence of second malignancies in subsets of Kaposi's sarcoma. J Am Acad Dermatol. 1987;16:855-861.
- Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12:6-11.
- Daly ML, Fogo A, McDonald C, et al. Kaposi sarcoma: no longer an AIDS-defining illness? a retrospective study of Kaposi sarcoma cases with CD4 counts above 300/mm³ at presentation. Clin Exp Dermatol. 2014;39:7-12.
- Broccolo F, Tassan Din C, Viganò MG, et al. HHV-8 DNA replication correlates with the clinical status in AIDS-related Kaposi's sarcoma. J Clin Virol. 2016;78:47-52.
- Pereira PF, Cuzzi T, Galhardo MC. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An Bras Dermatol. 2013;88:243-246.
- Gevorgyan O, Owen BD, Balavenkataraman A, et al. A nodular-ulcerative form of secondary syphilis in AIDS. Proc (Bayl Univ Med Cent). 2017;30:80-82.
- Balagula Y, Mattei PL, Wisco OJ, et al. The great imitator revisited: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;31:595-599.
- Yayli S, della Torre R, Hegyi I, et al. Late secondary syphilis with nodular lesions mimicking Kaposi sarcoma in a patient with human immunodeficiency virus. Int J Dermatol. 2014;53:E71-E73.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Cockerell CJ, LeBoit PE. Bacillary angiomatosis: a newly characterized, pseudoneoplastic, infectious, cutaneous vascular disorder. J Am Acad Dermatol. 1990;22:501-512.
- Forrestel AK, Naujokas A, Martin JN, et al. Bacillary angiomatosis masquerading as Kaposi's sarcoma in East Africa. J Int Assoc Provid AIDS Care. 2015;14:21-25.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma), localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol. 2007;29:408-411.
- Marks R. Squamous cell carcinoma. Lancet. 1996;347:735-738.
- Chakrabarti I, Watson JD, Dorrance H. Skin tumours of the hand. a 10-year review. J Hand Surg Br. 1993;18:484-486.
- Sobanko JF, Dagum AB, Davis IC, et al. Soft tissue tumors of the hand. 2. malignant. Dermatol Surg. 2007;33:771-785.
- Antman K, Chang Y. Kaposi's sarcoma. N Engl J Med. 2000;342:1027-1038.
- Pipette WW. The incidence of second malignancies in subsets of Kaposi's sarcoma. J Am Acad Dermatol. 1987;16:855-861.
- Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12:6-11.
- Daly ML, Fogo A, McDonald C, et al. Kaposi sarcoma: no longer an AIDS-defining illness? a retrospective study of Kaposi sarcoma cases with CD4 counts above 300/mm³ at presentation. Clin Exp Dermatol. 2014;39:7-12.
- Broccolo F, Tassan Din C, Viganò MG, et al. HHV-8 DNA replication correlates with the clinical status in AIDS-related Kaposi's sarcoma. J Clin Virol. 2016;78:47-52.
- Pereira PF, Cuzzi T, Galhardo MC. Immunohistochemical detection of the latent nuclear antigen-1 of the human herpesvirus type 8 to differentiate cutaneous epidemic Kaposi sarcoma and its histological simulators. An Bras Dermatol. 2013;88:243-246.
- Gevorgyan O, Owen BD, Balavenkataraman A, et al. A nodular-ulcerative form of secondary syphilis in AIDS. Proc (Bayl Univ Med Cent). 2017;30:80-82.
- Balagula Y, Mattei PL, Wisco OJ, et al. The great imitator revisited: the spectrum of atypical cutaneous manifestations of secondary syphilis. Int J Dermatol. 2014;53:1434-1441.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;31:595-599.
- Yayli S, della Torre R, Hegyi I, et al. Late secondary syphilis with nodular lesions mimicking Kaposi sarcoma in a patient with human immunodeficiency virus. Int J Dermatol. 2014;53:E71-E73.
- Jeerapaet P, Ackerman AB. Histologic patterns of secondary syphilis. Arch Dermatol. 1973;107:373-377.
- Cockerell CJ, LeBoit PE. Bacillary angiomatosis: a newly characterized, pseudoneoplastic, infectious, cutaneous vascular disorder. J Am Acad Dermatol. 1990;22:501-512.
- Forrestel AK, Naujokas A, Martin JN, et al. Bacillary angiomatosis masquerading as Kaposi's sarcoma in East Africa. J Int Assoc Provid AIDS Care. 2015;14:21-25.
- Fortna RR, Junkins-Hopkins JM. A case of lobular capillary hemangioma (pyogenic granuloma), localized to the subcutaneous tissue, and a review of the literature. Am J Dermatopathol. 2007;29:408-411.
- Marks R. Squamous cell carcinoma. Lancet. 1996;347:735-738.
- Chakrabarti I, Watson JD, Dorrance H. Skin tumours of the hand. a 10-year review. J Hand Surg Br. 1993;18:484-486.
- Sobanko JF, Dagum AB, Davis IC, et al. Soft tissue tumors of the hand. 2. malignant. Dermatol Surg. 2007;33:771-785.

A 52-year-old man presented to the dermatology clinic with a 2×3-cm, fungating, dome-shaped, ulcerative, moist, well-circumscribed tumor with peripheral maceration on the volar aspect of the right hand of 3 months’ duration. The tumor was malodorous, painful, and hemorrhaged easily with minimal trauma. The patient’s medical history was notable for human immunodeficiency virus and latent syphilis, with elevated rapid plasma reagin titers and a positive Treponema palladium antibody on chemiluminescent immunoassay, that was refractory to 3 treatments with penicillin. The patient was not on antiretroviral therapy. He had a CD4+ lymphocyte count of 980 cells/µL (reference range, 359–1519 cells/µL) and a viral load of 8560 copies/mL (reference range, <200 copies/mL). No other skin or systemic concerns were noted, and the patient denied any recent travel, exposure to animals, or constitutional symptoms. A deep shave biopsy of the lesion was performed.
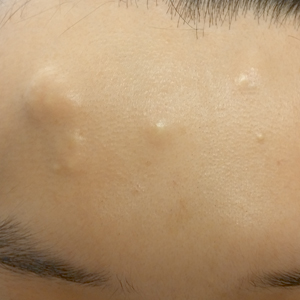
Rapidly Growing Retroauricular Tumor
The Diagnosis: Milia En Plaque
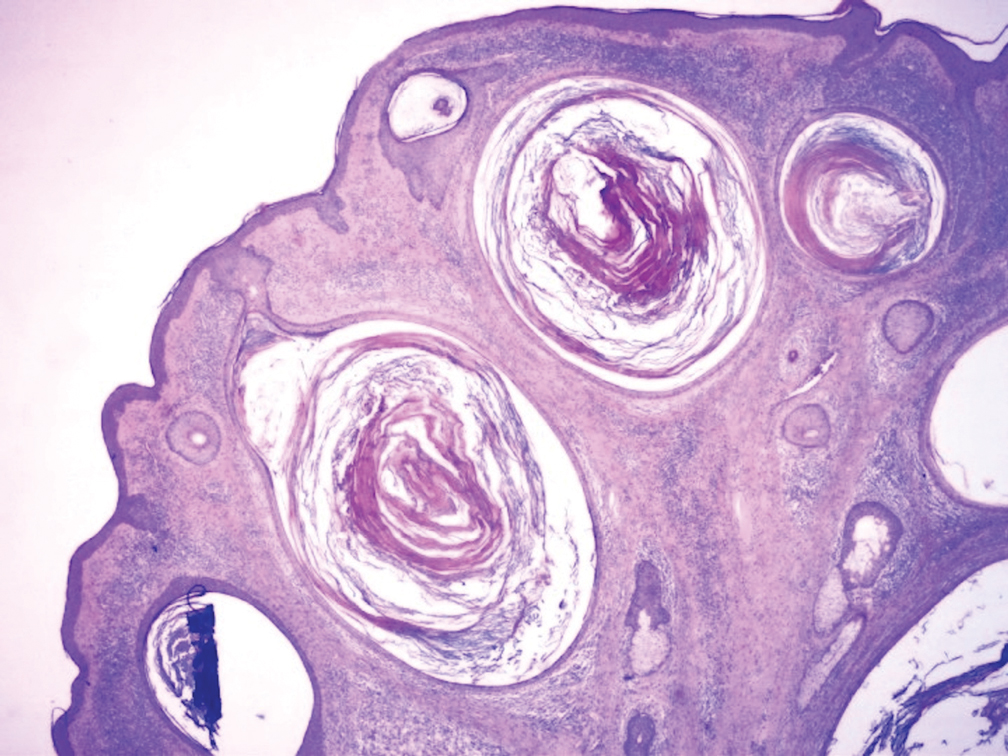
Biopsy results revealed a normal epidermis; the dermis showed multiple small cystic structures lined by a stratified squamous epithelium containing eosinophilic keratin surrounded by a mononuclear cell infiltrate and some melanophages (Figure).
Milia en plaque was first described in 1903 by Balzer and Fouquet.1 In 1978, Hubler et al2 presented 2 cases with an asymptomatic, erythematous, and edematous plaque and white milialike lesions. On histopathology, they showed multiple cystic structures characterized by central laminated keratin and an intense polymorphic inflammatory reaction surrounding the cyst and epidermal appendages. Both patients were treated with topical tretinoin with complete response at 3 months. The authors suggested the term milia en plaque to describe this clinical entity.2
Milia en plaque is described as an infrequent condition that more often presents on the head, neck, and trunk, as well as the periocular, periauricular, and perinasal areas. It has been reported to occur at any age3 but appears more frequently in middle-aged adults and females. A congenital case also has been reported.4 It has been associated with pseudoxanthoma elasticum, lichen planus, trauma, kidney transplant, and cyclosporine use, but it also can present in healthy individuals,3 as in our patient. No clear cause has been identified.
Pathology is characteristic, with multiple cysts filled with keratin and surrounded by 2 or 3 layers of epithelial cells, associated with a mononuclear, nonlichenoid, mononuclear infiltrate.5 Structures similar to follicular infundibular tumors have been described, suggesting a common origin of follicular lesions as milia en plaque.6
Treatment includes surgical excision, cryosurgery, dermabrasion, electrodesiccation, trichloroacetic acid, photodynamic therapy, CO2 and erbium lasers, topical retinoids, minocycline, and etretinate.7 We performed a complete surgical excision in our patient.
In acneform reactions, erythematous papules and pustules can be found on the cheeks and forehead. Nevus comedonicus appears during childhood and presents with multiple open comedones. Postinflammatory milia is present in chronic inflammatory pathologies such as porphyria cutanea tarda. Histopathologic findings in adnexal tumors show a benign proliferation of any cellular type of a cutaneous annex.
Milia en plaque is an unusual but benign condition that is distinguished clinically by its characteristic presentation.
- Balzer F, Fouquet C. Milium confluent retroauricularies bilateral. Bull Soc Fr Dermatol Syphiligr. 1903;14:361.
- Hubler WR, Rudolph AH, Kelleher RM. Milia en plaque. Cutis. 1978;22:67-70.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Wang AR, Bercovitch L. Congenital milia en plaque. Pediatr Dermatol. 2016;33:258-259.
- Muñoz-Martínez R, Santamarina-Albertos A, Sanz-Muñoz C, et al. Milia en plaque. Actas Dermosifiliogr. 2013;104:638-640.
- Terui H, Hashimoto A, Yamasaki K, et al. Milia en plaque as a distinct follicular hamartoma with cystic trichoepitheliomatous features. Am J Dermatopathol. 2016;38:212-217.
- Tenna S, Filoni A, Pagliarello C, et al. Eyelid milia en plaque: a treatment challenge with a new CO2 fractional laser. Dermatol Ther. 2014;27:65-67.
The Diagnosis: Milia En Plaque
Biopsy results revealed a normal epidermis; the dermis showed multiple small cystic structures lined by a stratified squamous epithelium containing eosinophilic keratin surrounded by a mononuclear cell infiltrate and some melanophages (Figure).
Milia en plaque was first described in 1903 by Balzer and Fouquet.1 In 1978, Hubler et al2 presented 2 cases with an asymptomatic, erythematous, and edematous plaque and white milialike lesions. On histopathology, they showed multiple cystic structures characterized by central laminated keratin and an intense polymorphic inflammatory reaction surrounding the cyst and epidermal appendages. Both patients were treated with topical tretinoin with complete response at 3 months. The authors suggested the term milia en plaque to describe this clinical entity.2
Milia en plaque is described as an infrequent condition that more often presents on the head, neck, and trunk, as well as the periocular, periauricular, and perinasal areas. It has been reported to occur at any age3 but appears more frequently in middle-aged adults and females. A congenital case also has been reported.4 It has been associated with pseudoxanthoma elasticum, lichen planus, trauma, kidney transplant, and cyclosporine use, but it also can present in healthy individuals,3 as in our patient. No clear cause has been identified.
Pathology is characteristic, with multiple cysts filled with keratin and surrounded by 2 or 3 layers of epithelial cells, associated with a mononuclear, nonlichenoid, mononuclear infiltrate.5 Structures similar to follicular infundibular tumors have been described, suggesting a common origin of follicular lesions as milia en plaque.6
Treatment includes surgical excision, cryosurgery, dermabrasion, electrodesiccation, trichloroacetic acid, photodynamic therapy, CO2 and erbium lasers, topical retinoids, minocycline, and etretinate.7 We performed a complete surgical excision in our patient.
In acneform reactions, erythematous papules and pustules can be found on the cheeks and forehead. Nevus comedonicus appears during childhood and presents with multiple open comedones. Postinflammatory milia is present in chronic inflammatory pathologies such as porphyria cutanea tarda. Histopathologic findings in adnexal tumors show a benign proliferation of any cellular type of a cutaneous annex.
Milia en plaque is an unusual but benign condition that is distinguished clinically by its characteristic presentation.
The Diagnosis: Milia En Plaque
Biopsy results revealed a normal epidermis; the dermis showed multiple small cystic structures lined by a stratified squamous epithelium containing eosinophilic keratin surrounded by a mononuclear cell infiltrate and some melanophages (Figure).
Milia en plaque was first described in 1903 by Balzer and Fouquet.1 In 1978, Hubler et al2 presented 2 cases with an asymptomatic, erythematous, and edematous plaque and white milialike lesions. On histopathology, they showed multiple cystic structures characterized by central laminated keratin and an intense polymorphic inflammatory reaction surrounding the cyst and epidermal appendages. Both patients were treated with topical tretinoin with complete response at 3 months. The authors suggested the term milia en plaque to describe this clinical entity.2
Milia en plaque is described as an infrequent condition that more often presents on the head, neck, and trunk, as well as the periocular, periauricular, and perinasal areas. It has been reported to occur at any age3 but appears more frequently in middle-aged adults and females. A congenital case also has been reported.4 It has been associated with pseudoxanthoma elasticum, lichen planus, trauma, kidney transplant, and cyclosporine use, but it also can present in healthy individuals,3 as in our patient. No clear cause has been identified.
Pathology is characteristic, with multiple cysts filled with keratin and surrounded by 2 or 3 layers of epithelial cells, associated with a mononuclear, nonlichenoid, mononuclear infiltrate.5 Structures similar to follicular infundibular tumors have been described, suggesting a common origin of follicular lesions as milia en plaque.6
Treatment includes surgical excision, cryosurgery, dermabrasion, electrodesiccation, trichloroacetic acid, photodynamic therapy, CO2 and erbium lasers, topical retinoids, minocycline, and etretinate.7 We performed a complete surgical excision in our patient.
In acneform reactions, erythematous papules and pustules can be found on the cheeks and forehead. Nevus comedonicus appears during childhood and presents with multiple open comedones. Postinflammatory milia is present in chronic inflammatory pathologies such as porphyria cutanea tarda. Histopathologic findings in adnexal tumors show a benign proliferation of any cellular type of a cutaneous annex.
Milia en plaque is an unusual but benign condition that is distinguished clinically by its characteristic presentation.
- Balzer F, Fouquet C. Milium confluent retroauricularies bilateral. Bull Soc Fr Dermatol Syphiligr. 1903;14:361.
- Hubler WR, Rudolph AH, Kelleher RM. Milia en plaque. Cutis. 1978;22:67-70.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Wang AR, Bercovitch L. Congenital milia en plaque. Pediatr Dermatol. 2016;33:258-259.
- Muñoz-Martínez R, Santamarina-Albertos A, Sanz-Muñoz C, et al. Milia en plaque. Actas Dermosifiliogr. 2013;104:638-640.
- Terui H, Hashimoto A, Yamasaki K, et al. Milia en plaque as a distinct follicular hamartoma with cystic trichoepitheliomatous features. Am J Dermatopathol. 2016;38:212-217.
- Tenna S, Filoni A, Pagliarello C, et al. Eyelid milia en plaque: a treatment challenge with a new CO2 fractional laser. Dermatol Ther. 2014;27:65-67.
- Balzer F, Fouquet C. Milium confluent retroauricularies bilateral. Bull Soc Fr Dermatol Syphiligr. 1903;14:361.
- Hubler WR, Rudolph AH, Kelleher RM. Milia en plaque. Cutis. 1978;22:67-70.
- Berk DR, Bayliss SJ. Milia: a review and classification. J Am Acad Dermatol. 2008;59:1050-1063.
- Wang AR, Bercovitch L. Congenital milia en plaque. Pediatr Dermatol. 2016;33:258-259.
- Muñoz-Martínez R, Santamarina-Albertos A, Sanz-Muñoz C, et al. Milia en plaque. Actas Dermosifiliogr. 2013;104:638-640.
- Terui H, Hashimoto A, Yamasaki K, et al. Milia en plaque as a distinct follicular hamartoma with cystic trichoepitheliomatous features. Am J Dermatopathol. 2016;38:212-217.
- Tenna S, Filoni A, Pagliarello C, et al. Eyelid milia en plaque: a treatment challenge with a new CO2 fractional laser. Dermatol Ther. 2014;27:65-67.
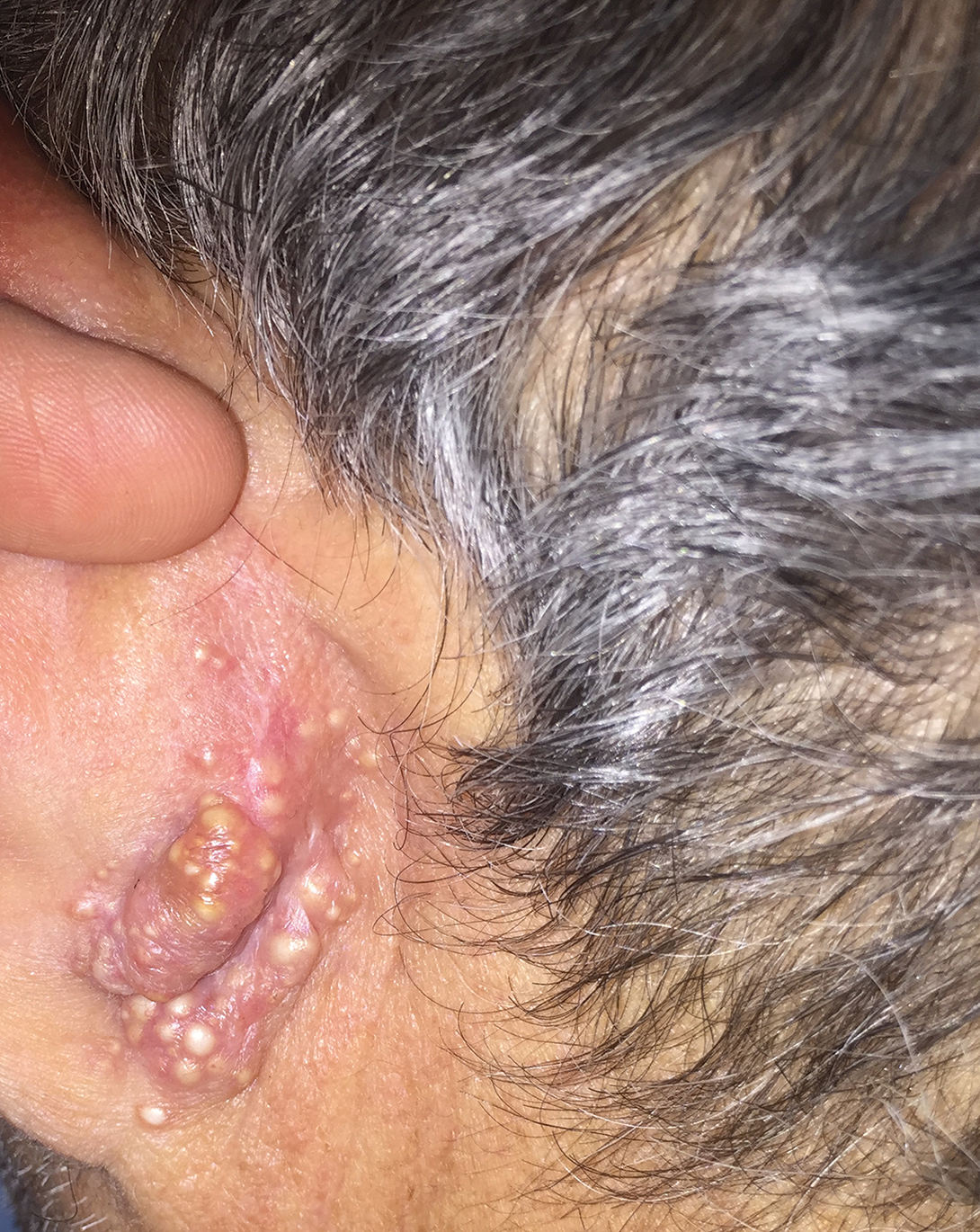
A 72-year-old man with a history of hypertension presented with a rapidly growing left retroauricular tumor of 3 months' duration. When manipulated, whitish material with a foul-smelling odor was expressed from the lesion. Physical examination showed an erythematous 3.2 ×1-cm tumor on the left posterior ear with multiple 1- to 2-mm white-yellow papules on its surface. A biopsy of the lesion was performed.
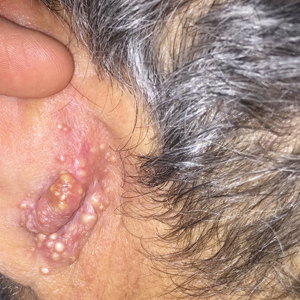
Ill-Defined Macule on the Abdomen
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
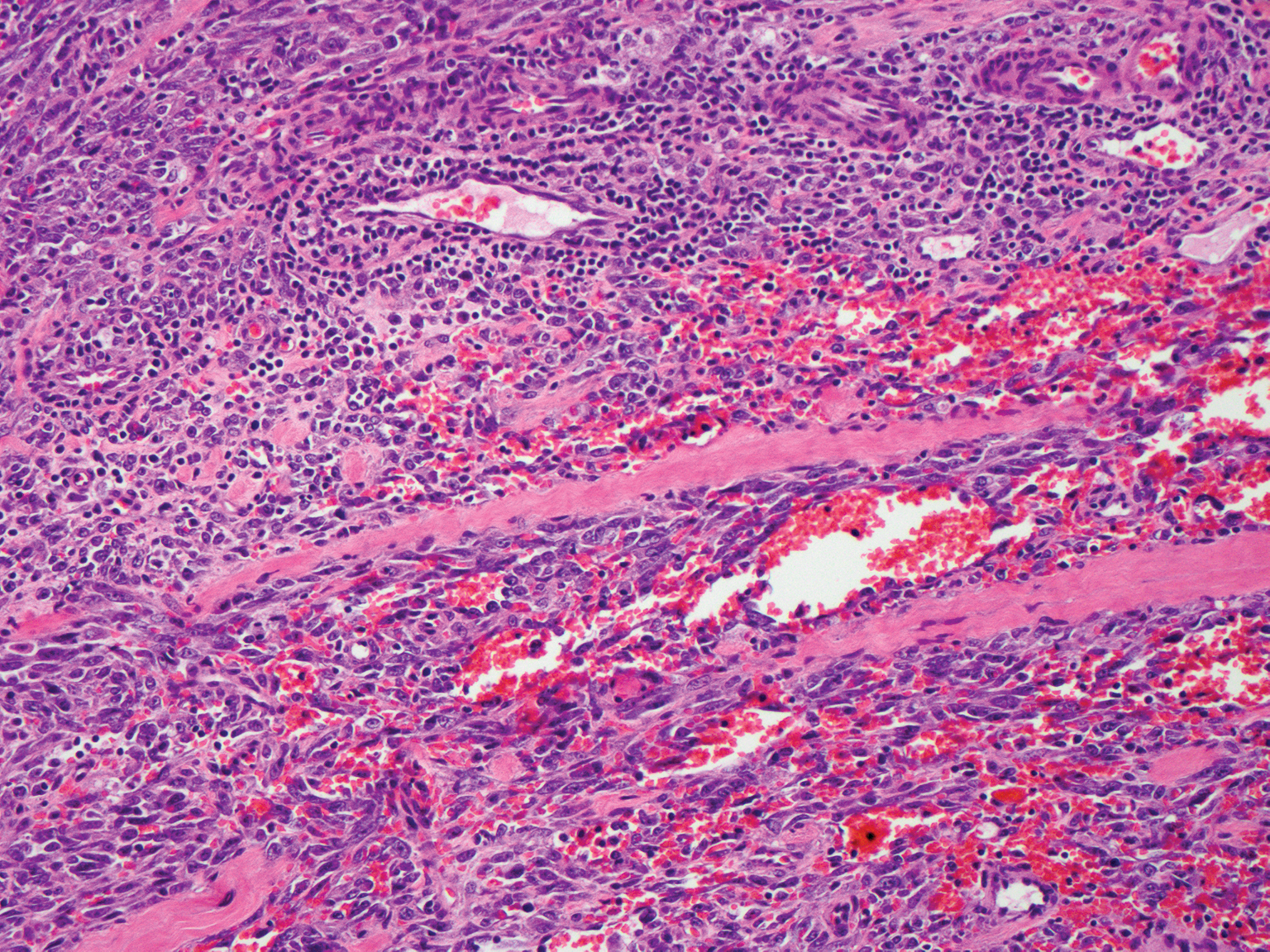
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
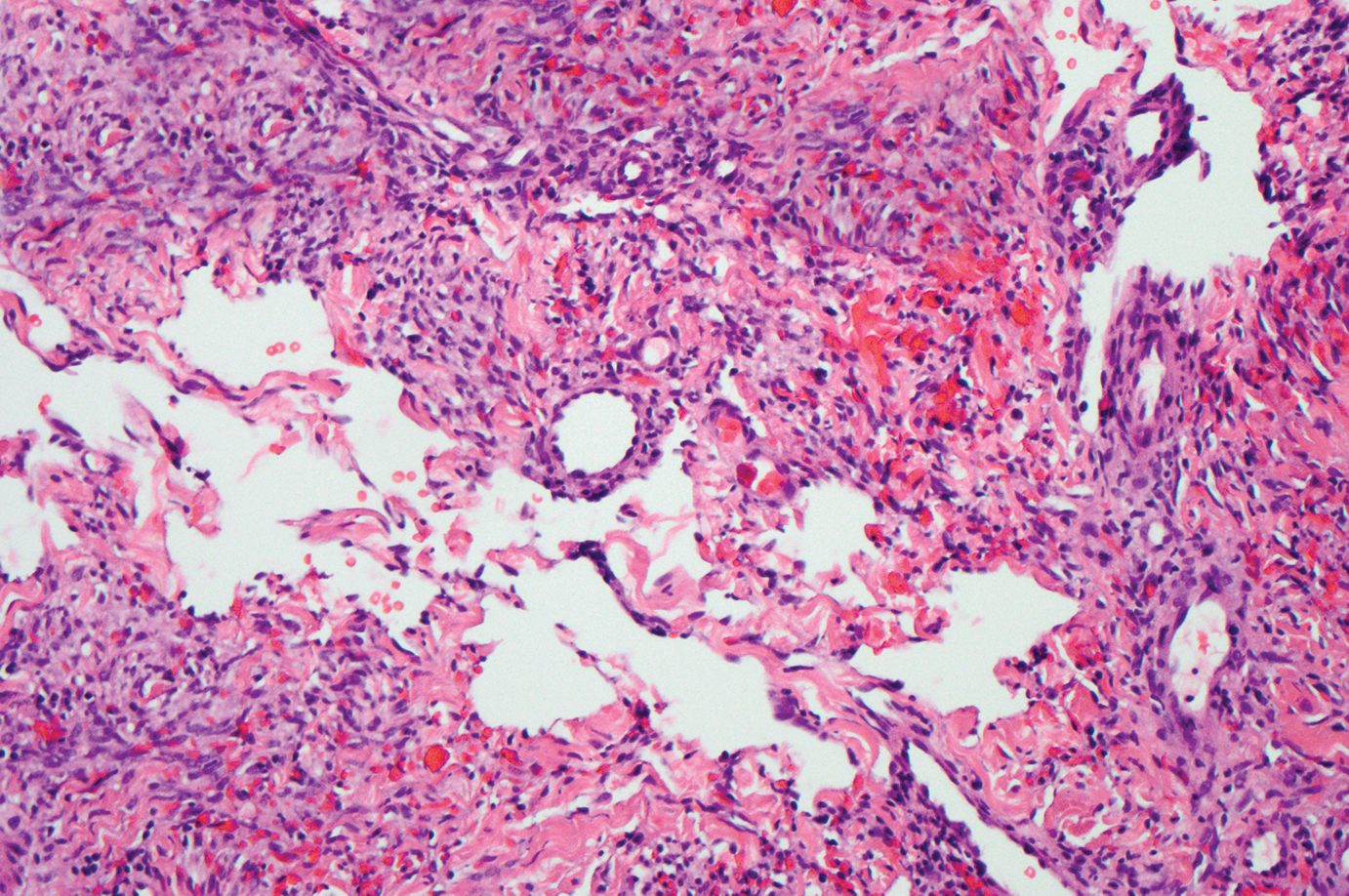
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
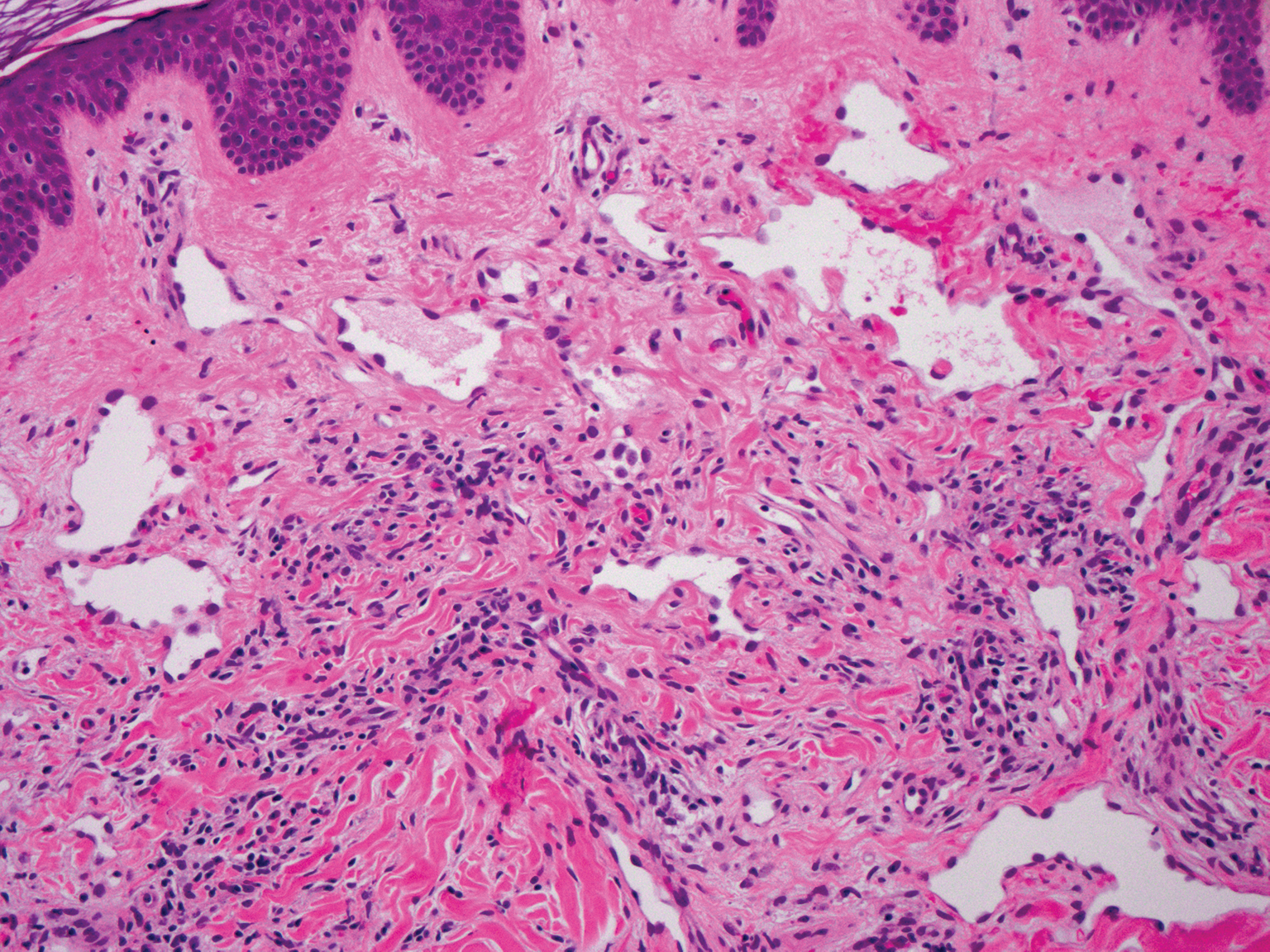
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15

- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15
- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
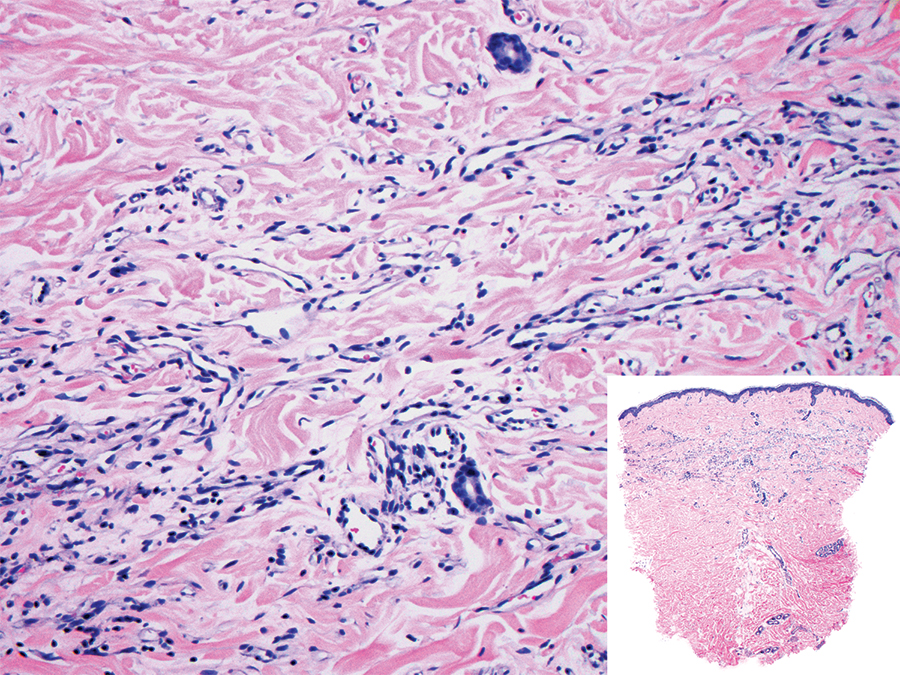
A 38-year-old woman presented with an asymptomatic lesion on the abdomen. On physical examination, there was a 5×2-mm, solitary, ill-defined pink macule on the right side of the abdomen. The patient denied recent change in size or color of the lesion, prior trauma, or a personal or family history of similar lesions. Due to the uncertain diagnostic appearance, a punch biopsy was performed.
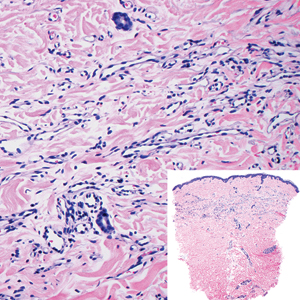
Grouped Erythematous Papules and Plaques on the Trunk
The Diagnosis: Cutaneous B-Cell Lymphoma, Follicle Center Subtype
A 4-mm punch biopsy through the center of the largest lesion on the right posterior shoulder demonstrated a superficial and deep dermal atypical lymphoid infiltrate composed predominantly of small mature lymphocytes with interspersed intermediate-sized cells with irregular to cleaved nuclei, dispersed chromatin, one or more distinct nucleoli, occasional mitoses, and small amounts of cytoplasm (Figure, A). Immunoperoxidase studies showed the infiltrate to be a mixture of CD3+ T cells and CD20+ B cells (Figure, B). The B cells coexpressed B-cell lymphoma (Bcl) 6 protein (Figure, C) but were negative for multiple myeloma 1/interferon regulatory factor 4 and CD10; Bcl2 protein was positive in T cells but inconclusive for staining in B cells. Very few plasma cells were seen with CD138 stain. Fluorescence in situ hybridization studies were negative for IgH and BCL2 gene rearrangement. Molecular diagnostic studies for IgH and κ light chain gene rearrangement were positive for a clonal population. A clonal T-cell receptor γ chain gene rearrangement was not identified. The overall morphologic, immunophenotypic, and molecular findings were consistent with cutaneous involvement by a B-cell lymphoproliferative disorder, favoring primary cutaneous follicle center lymphoma (PCFCL).

The patient was referred to our cancer center for further workup consisting of a complete blood cell count with differential; comprehensive metabolic panel; lactate dehydrogenase; serum protein electrophoresis; peripheral blood flow cytometry; and computed tomography of the chest, abdomen, and pelvis. The analysis was unremarkable, supporting primary cutaneous disease. Additional studies suggested in the National Comprehensive Cancer Network (NCCN) Guidelines for primary cutaneous B-cell lymphomas include hepatitis B testing if the patient is being considered for immunotherapy and/or chemotherapy due to risk of reactivation, pregnancy testing in women of childbearing age, and human immunodeficiency virus testing.1 These tests were not performed in our patient because he did not have any risk factors for hepatitis B or human immunodeficiency virus.
Primary cutaneous B-cell lymphomas originate in the skin without evidence of extracutaneous disease at presentation. They account for approximately 25% of primary cutaneous lymphomas in the United States, with primary cutaneous T-cell lymphoma being most common.2 The revised 2017 World Health Organization classification system defines 3 major subtypes of primary cutaneous B-cell lymphoma (Table).3-9 Primary cutaneous follicle center lymphoma is the most common subtype, accounting for approximately 60% of cases. In Europe, an association with Borrelia burgdorferi has been reported.10 The extent of skin involvement determines the T portion of TNM staging for PCFCL. It is based on the size and location of affected body regions that are delineated, such as the head and neck, chest, abdomen/genitalia, upper back, lower back/buttocks, each upper arm, each lower arm/hand, each upper leg, and each lower leg/foot. T1 is for solitary skin involvement in which the lesion is 5 cm or less in diameter (T1a) or greater than 5 cm (T1b). T2 is for regional skin involvement limited to 1 or 2 contiguous body regions, whereas T2a has all lesions confined to an area 15 cm or less in diameter, T2b has lesions confined to an area greater than 15 cm up to 30 cm in diameter, and the area for T2c is greater than 30 cm in diameter. Finally, T3 is generalized skin involvement, whereas T3a has multiple lesions in 2 noncontiguous body regions, and T3b has multiple lesions on 3 or more regions.11 At presentation, our patient was considered T2cN0M0, as his lesions were present on only 2 contiguous regions extending beyond 30 cm without any evidence of lymph node involvement or metastasis.
Treatment of PCFCL is tailored to each case, as there is a paucity of randomized data in this rare entity. It is guided by the number and location of cutaneous lesions, associated skin symptoms, age of the patient, and performance status. Local disease can be treated with intralesional corticosteroids, excision, or close monitoring if the patient is asymptomatic. Low-dose radiation therapy may be used as primary treatment or for local recurrence.12 Patients with more extensive skin lesions can relapse after clearing; those with refractory disease can be managed with single-agent rituximab.13 Our patient underwent low-dose radiation therapy with good response and has not experienced recurrence.
Lymphocytoma cutis, also known as benign reactive lymphoid hyperplasia, can be idiopathic or can arise after arthropod assault, penetrative skin trauma, drugs, or infections. In granuloma annulare, small dermal papules may present in isolation or coalesce to form annular plaques. It is a benign inflammatory disorder of unknown cause, can have mild pruritus, and usually is self-limited. Pyogenic granuloma is a benign vascular proliferation of unknown etiology. Sarcoidosis is an immune-mediated systemic disorder with granuloma formation that has a predilection for the lungs and the skin.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Primary Cutaneous B-Cell Lymphomas. Version 2.2018. https://oncolife.com.ua/doc/nccn/Primary_Cutaneous_B-Cell_Lymphomas.pdf. Published January 10, 2018. Accessed June 21, 2019.
- Dores GM, Anderson WF, Devesa SS. Cutaneous lymphomas reported to the National Cancer Institute's surveillance, epidemiology, and end results program: applying the new WHO-European Organisation for Research and Treatment of Cancer classification system. J Clin Oncol. 2005;23:7246-7248.
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC; 2017.
- Surveillance, Epidemiology, and End Results Program. National Cancer Institute website. https://seer.cancer.gov/. Accessed June 26, 2019.
- Cerroni L. B-cell lymphomas of the skin. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:2113-2126.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous follicle center lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-follicle-center-lymphoma. Updated February 7, 2018. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous marginal zone lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-marginal-zone-lymphoma. Updated March 6, 2019. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous large B cell lymphoma, leg type. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-large-b-cell-lymphoma-leg-type. Updated July 3, 2017. Accessed June 26, 2019.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 241-342.
- Goodlad JR, Davidson MM, Hollowood K, et al. Primary cutaneous B-cell lymphoma and Borrelia burgdorferi infection in patients from the Highlands of Scotand. Am J Surg Pathol. 2000;24:1279-1285.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Wilcon RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Morales AV, Advani R, Horwitz SM, et al. Indolent primary cutaneous B-cell lymphoma: experience using systemic rituximab. J Am Acad Dermatol. 2008;59:953-957.
The Diagnosis: Cutaneous B-Cell Lymphoma, Follicle Center Subtype
A 4-mm punch biopsy through the center of the largest lesion on the right posterior shoulder demonstrated a superficial and deep dermal atypical lymphoid infiltrate composed predominantly of small mature lymphocytes with interspersed intermediate-sized cells with irregular to cleaved nuclei, dispersed chromatin, one or more distinct nucleoli, occasional mitoses, and small amounts of cytoplasm (Figure, A). Immunoperoxidase studies showed the infiltrate to be a mixture of CD3+ T cells and CD20+ B cells (Figure, B). The B cells coexpressed B-cell lymphoma (Bcl) 6 protein (Figure, C) but were negative for multiple myeloma 1/interferon regulatory factor 4 and CD10; Bcl2 protein was positive in T cells but inconclusive for staining in B cells. Very few plasma cells were seen with CD138 stain. Fluorescence in situ hybridization studies were negative for IgH and BCL2 gene rearrangement. Molecular diagnostic studies for IgH and κ light chain gene rearrangement were positive for a clonal population. A clonal T-cell receptor γ chain gene rearrangement was not identified. The overall morphologic, immunophenotypic, and molecular findings were consistent with cutaneous involvement by a B-cell lymphoproliferative disorder, favoring primary cutaneous follicle center lymphoma (PCFCL).
The patient was referred to our cancer center for further workup consisting of a complete blood cell count with differential; comprehensive metabolic panel; lactate dehydrogenase; serum protein electrophoresis; peripheral blood flow cytometry; and computed tomography of the chest, abdomen, and pelvis. The analysis was unremarkable, supporting primary cutaneous disease. Additional studies suggested in the National Comprehensive Cancer Network (NCCN) Guidelines for primary cutaneous B-cell lymphomas include hepatitis B testing if the patient is being considered for immunotherapy and/or chemotherapy due to risk of reactivation, pregnancy testing in women of childbearing age, and human immunodeficiency virus testing.1 These tests were not performed in our patient because he did not have any risk factors for hepatitis B or human immunodeficiency virus.
Primary cutaneous B-cell lymphomas originate in the skin without evidence of extracutaneous disease at presentation. They account for approximately 25% of primary cutaneous lymphomas in the United States, with primary cutaneous T-cell lymphoma being most common.2 The revised 2017 World Health Organization classification system defines 3 major subtypes of primary cutaneous B-cell lymphoma (Table).3-9 Primary cutaneous follicle center lymphoma is the most common subtype, accounting for approximately 60% of cases. In Europe, an association with Borrelia burgdorferi has been reported.10 The extent of skin involvement determines the T portion of TNM staging for PCFCL. It is based on the size and location of affected body regions that are delineated, such as the head and neck, chest, abdomen/genitalia, upper back, lower back/buttocks, each upper arm, each lower arm/hand, each upper leg, and each lower leg/foot. T1 is for solitary skin involvement in which the lesion is 5 cm or less in diameter (T1a) or greater than 5 cm (T1b). T2 is for regional skin involvement limited to 1 or 2 contiguous body regions, whereas T2a has all lesions confined to an area 15 cm or less in diameter, T2b has lesions confined to an area greater than 15 cm up to 30 cm in diameter, and the area for T2c is greater than 30 cm in diameter. Finally, T3 is generalized skin involvement, whereas T3a has multiple lesions in 2 noncontiguous body regions, and T3b has multiple lesions on 3 or more regions.11 At presentation, our patient was considered T2cN0M0, as his lesions were present on only 2 contiguous regions extending beyond 30 cm without any evidence of lymph node involvement or metastasis.
Treatment of PCFCL is tailored to each case, as there is a paucity of randomized data in this rare entity. It is guided by the number and location of cutaneous lesions, associated skin symptoms, age of the patient, and performance status. Local disease can be treated with intralesional corticosteroids, excision, or close monitoring if the patient is asymptomatic. Low-dose radiation therapy may be used as primary treatment or for local recurrence.12 Patients with more extensive skin lesions can relapse after clearing; those with refractory disease can be managed with single-agent rituximab.13 Our patient underwent low-dose radiation therapy with good response and has not experienced recurrence.
Lymphocytoma cutis, also known as benign reactive lymphoid hyperplasia, can be idiopathic or can arise after arthropod assault, penetrative skin trauma, drugs, or infections. In granuloma annulare, small dermal papules may present in isolation or coalesce to form annular plaques. It is a benign inflammatory disorder of unknown cause, can have mild pruritus, and usually is self-limited. Pyogenic granuloma is a benign vascular proliferation of unknown etiology. Sarcoidosis is an immune-mediated systemic disorder with granuloma formation that has a predilection for the lungs and the skin.
The Diagnosis: Cutaneous B-Cell Lymphoma, Follicle Center Subtype
A 4-mm punch biopsy through the center of the largest lesion on the right posterior shoulder demonstrated a superficial and deep dermal atypical lymphoid infiltrate composed predominantly of small mature lymphocytes with interspersed intermediate-sized cells with irregular to cleaved nuclei, dispersed chromatin, one or more distinct nucleoli, occasional mitoses, and small amounts of cytoplasm (Figure, A). Immunoperoxidase studies showed the infiltrate to be a mixture of CD3+ T cells and CD20+ B cells (Figure, B). The B cells coexpressed B-cell lymphoma (Bcl) 6 protein (Figure, C) but were negative for multiple myeloma 1/interferon regulatory factor 4 and CD10; Bcl2 protein was positive in T cells but inconclusive for staining in B cells. Very few plasma cells were seen with CD138 stain. Fluorescence in situ hybridization studies were negative for IgH and BCL2 gene rearrangement. Molecular diagnostic studies for IgH and κ light chain gene rearrangement were positive for a clonal population. A clonal T-cell receptor γ chain gene rearrangement was not identified. The overall morphologic, immunophenotypic, and molecular findings were consistent with cutaneous involvement by a B-cell lymphoproliferative disorder, favoring primary cutaneous follicle center lymphoma (PCFCL).
The patient was referred to our cancer center for further workup consisting of a complete blood cell count with differential; comprehensive metabolic panel; lactate dehydrogenase; serum protein electrophoresis; peripheral blood flow cytometry; and computed tomography of the chest, abdomen, and pelvis. The analysis was unremarkable, supporting primary cutaneous disease. Additional studies suggested in the National Comprehensive Cancer Network (NCCN) Guidelines for primary cutaneous B-cell lymphomas include hepatitis B testing if the patient is being considered for immunotherapy and/or chemotherapy due to risk of reactivation, pregnancy testing in women of childbearing age, and human immunodeficiency virus testing.1 These tests were not performed in our patient because he did not have any risk factors for hepatitis B or human immunodeficiency virus.
Primary cutaneous B-cell lymphomas originate in the skin without evidence of extracutaneous disease at presentation. They account for approximately 25% of primary cutaneous lymphomas in the United States, with primary cutaneous T-cell lymphoma being most common.2 The revised 2017 World Health Organization classification system defines 3 major subtypes of primary cutaneous B-cell lymphoma (Table).3-9 Primary cutaneous follicle center lymphoma is the most common subtype, accounting for approximately 60% of cases. In Europe, an association with Borrelia burgdorferi has been reported.10 The extent of skin involvement determines the T portion of TNM staging for PCFCL. It is based on the size and location of affected body regions that are delineated, such as the head and neck, chest, abdomen/genitalia, upper back, lower back/buttocks, each upper arm, each lower arm/hand, each upper leg, and each lower leg/foot. T1 is for solitary skin involvement in which the lesion is 5 cm or less in diameter (T1a) or greater than 5 cm (T1b). T2 is for regional skin involvement limited to 1 or 2 contiguous body regions, whereas T2a has all lesions confined to an area 15 cm or less in diameter, T2b has lesions confined to an area greater than 15 cm up to 30 cm in diameter, and the area for T2c is greater than 30 cm in diameter. Finally, T3 is generalized skin involvement, whereas T3a has multiple lesions in 2 noncontiguous body regions, and T3b has multiple lesions on 3 or more regions.11 At presentation, our patient was considered T2cN0M0, as his lesions were present on only 2 contiguous regions extending beyond 30 cm without any evidence of lymph node involvement or metastasis.
Treatment of PCFCL is tailored to each case, as there is a paucity of randomized data in this rare entity. It is guided by the number and location of cutaneous lesions, associated skin symptoms, age of the patient, and performance status. Local disease can be treated with intralesional corticosteroids, excision, or close monitoring if the patient is asymptomatic. Low-dose radiation therapy may be used as primary treatment or for local recurrence.12 Patients with more extensive skin lesions can relapse after clearing; those with refractory disease can be managed with single-agent rituximab.13 Our patient underwent low-dose radiation therapy with good response and has not experienced recurrence.
Lymphocytoma cutis, also known as benign reactive lymphoid hyperplasia, can be idiopathic or can arise after arthropod assault, penetrative skin trauma, drugs, or infections. In granuloma annulare, small dermal papules may present in isolation or coalesce to form annular plaques. It is a benign inflammatory disorder of unknown cause, can have mild pruritus, and usually is self-limited. Pyogenic granuloma is a benign vascular proliferation of unknown etiology. Sarcoidosis is an immune-mediated systemic disorder with granuloma formation that has a predilection for the lungs and the skin.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Primary Cutaneous B-Cell Lymphomas. Version 2.2018. https://oncolife.com.ua/doc/nccn/Primary_Cutaneous_B-Cell_Lymphomas.pdf. Published January 10, 2018. Accessed June 21, 2019.
- Dores GM, Anderson WF, Devesa SS. Cutaneous lymphomas reported to the National Cancer Institute's surveillance, epidemiology, and end results program: applying the new WHO-European Organisation for Research and Treatment of Cancer classification system. J Clin Oncol. 2005;23:7246-7248.
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC; 2017.
- Surveillance, Epidemiology, and End Results Program. National Cancer Institute website. https://seer.cancer.gov/. Accessed June 26, 2019.
- Cerroni L. B-cell lymphomas of the skin. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:2113-2126.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous follicle center lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-follicle-center-lymphoma. Updated February 7, 2018. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous marginal zone lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-marginal-zone-lymphoma. Updated March 6, 2019. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous large B cell lymphoma, leg type. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-large-b-cell-lymphoma-leg-type. Updated July 3, 2017. Accessed June 26, 2019.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 241-342.
- Goodlad JR, Davidson MM, Hollowood K, et al. Primary cutaneous B-cell lymphoma and Borrelia burgdorferi infection in patients from the Highlands of Scotand. Am J Surg Pathol. 2000;24:1279-1285.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Wilcon RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Morales AV, Advani R, Horwitz SM, et al. Indolent primary cutaneous B-cell lymphoma: experience using systemic rituximab. J Am Acad Dermatol. 2008;59:953-957.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Primary Cutaneous B-Cell Lymphomas. Version 2.2018. https://oncolife.com.ua/doc/nccn/Primary_Cutaneous_B-Cell_Lymphomas.pdf. Published January 10, 2018. Accessed June 21, 2019.
- Dores GM, Anderson WF, Devesa SS. Cutaneous lymphomas reported to the National Cancer Institute's surveillance, epidemiology, and end results program: applying the new WHO-European Organisation for Research and Treatment of Cancer classification system. J Clin Oncol. 2005;23:7246-7248.
- Swerdlow SH, Campo E, Harris NL, et al, eds. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC; 2017.
- Surveillance, Epidemiology, and End Results Program. National Cancer Institute website. https://seer.cancer.gov/. Accessed June 26, 2019.
- Cerroni L. B-cell lymphomas of the skin. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:2113-2126.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous follicle center lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-follicle-center-lymphoma. Updated February 7, 2018. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous marginal zone lymphoma. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-marginal-zone-lymphoma. Updated March 6, 2019. Accessed June 26, 2019.
- Jacobsen E, Freedman AS, Willemze R. Primary cutaneous large B cell lymphoma, leg type. UpToDate website. https://www.uptodate.com/contents/primary-cutaneous-large-b-cell-lymphoma-leg-type. Updated July 3, 2017. Accessed June 26, 2019.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-13; quiz 241-342.
- Goodlad JR, Davidson MM, Hollowood K, et al. Primary cutaneous B-cell lymphoma and Borrelia burgdorferi infection in patients from the Highlands of Scotand. Am J Surg Pathol. 2000;24:1279-1285.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Wilcon RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Morales AV, Advani R, Horwitz SM, et al. Indolent primary cutaneous B-cell lymphoma: experience using systemic rituximab. J Am Acad Dermatol. 2008;59:953-957.
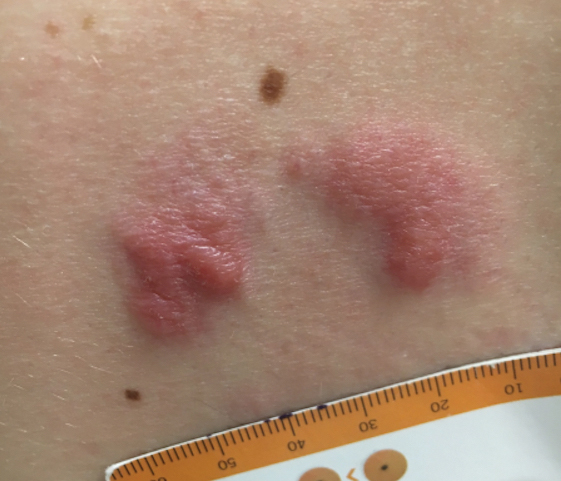
A 34-year-old man presented to the outpatient dermatology clinic with 3 groups of mildly pruritic, erythematous papules and plaques. The most prominent group appeared on the right posterior shoulder and had been slowly enlarging in size over the last 12 months (quiz image). A similar thinner group appeared on the left mid-back 6 months prior, and a third smaller group appeared over the left serratus anterior muscle 2 months prior. The patient reported having similar episodes dating back to his early 20s. In those instances, the lesions presented without an inciting incident, became more pronounced, and persisted for months to years before resolving. Previously affected areas included the upper and lateral back, flanks, and posterior upper arms. The patient used triamcinolone cream 0.1% up to 3 times daily on active lesions, which improved the pruritus and seemed to make the lesions resolve more quickly. He denied fever, chills, night sweats, anorexia, weight loss, fatigue, cough, and shortness of breath. His only medication was ranitidine 150 mg twice daily for gastroesophageal reflux disease. Physical examination revealed no palpable lymphadenopathy.
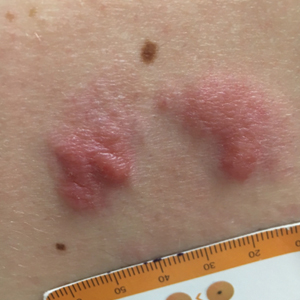
Rapidly Enlarging Neoplasm on the Face
The Diagnosis: Atypical Fibroxanthoma
Shave biopsy showed the superficial aspect of a highly cellular tumor composed of pleomorphic spindle cells exhibiting storiform growth and increased mitotic activity (Figure 1). The tumor stained positive for factor XIIIa, CD163, CD68, and smooth muscle actin (mild), and negative for high-molecular-weight cytokeratin (HMW-CK), p63, S-100, and melan-A. Subsequent excision with 0.5-cm margins was performed, and histopathology showed a well-circumscribed tumor contained within the dermis with a histologic scar at the outer margin (Figure 2). There was no lymphovascular or perineural invasion by tumor cells. Re-excision with 0.3-cm margins demonstrated no residual scar or tumor, and external radiation was deferred due to clear surgical margins.
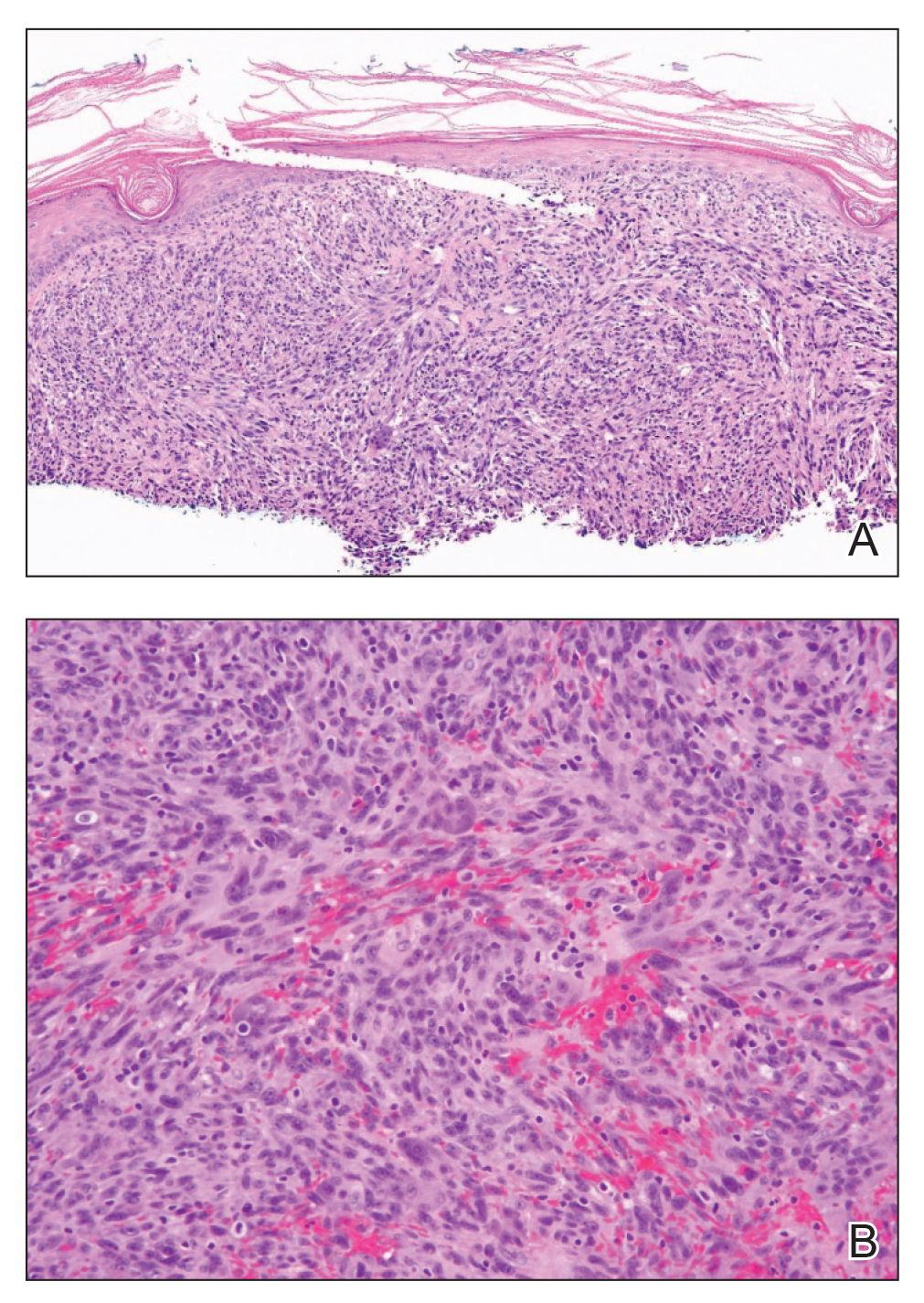
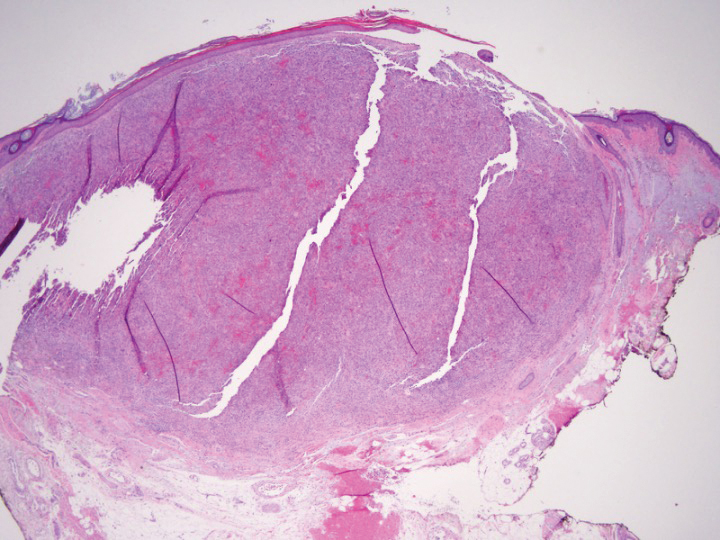
Atypical fibroxanthoma (AFX) belongs to a group of spindle cell neoplasms that can be diagnostically challenging, as they often lack specific morphologic features on examination or routine histology. These neoplasms--of which the differential includes malignant fibrous histiocytoma, spindle cell squamous cell carcinoma (SCC), desmoplastic melanoma, and leiomyosarcoma--may each appear as a rapidly enlarging solitary plaque or nodule on sun-damaged skin on the head and neck or less commonly on the trunk, arms, or legs. Histologically, the cells of AFX exhibit notable pleomorphism with frequent atypical mitotic figures and nonspecific surrounding dermal changes. Subcutaneous and lymphovascular or perineural invasion of tumor cells can point away from the diagnosis of AFX; however, these features are likely to be missed in small superficial shave biopsies.1,2 Therefore, immunohistochemistry (IHC) and adequate tumor sampling are essential in the accurate diagnosis of AFX and other spindle cell neoplasms.
Several IHC markers have been employed in differentiating AFX from other spindle cell neoplasms.3-8 Positive stains for AFX include factor XIIIa (10%-25%), vimentin (>99%), CD10 (95%-100%), procollagen (87%), CD99 (35%-73%), CD163 (37%-79%), smooth muscle actin (50%), CD68 (>50%), and CD31 (43%). Other stains, such as HMW-CK, S-100, p63, desmin, CD34, and melan-A, typically are negative in AFX but are actively expressed in other pleomorphic spindle cell tumors. The Table summarizes the utility of these various markers in narrowing the differential diagnosis of a spindle cell lesion. Selection of an appropriate panel of IHC markers is critical for accurate diagnosis of AFX and exclusion of more aggressive, poorly differentiated spindle cell neoplasms. Key IHC markers include S-100 (negative in AFX; positive in desmoplastic melanoma), HMW-CK (negative in AFX; positive in spindle cell SCC), and p63 (negative in AFX; positive in spindle cell SCC). Benoit et al9 reported a case of a poorly differentiated spindle cell SCC misdiagnosed as AFX based on a limited IHC panel that was negative for pancytokeratin and S-100. Later, a more comprehensive IHC panel including HMW-CK and p63 confirmed spindle cell SCC, but by this time, a delay in therapy had allowed the tumor to metastasize, which ultimately proved fatal to the patient.9
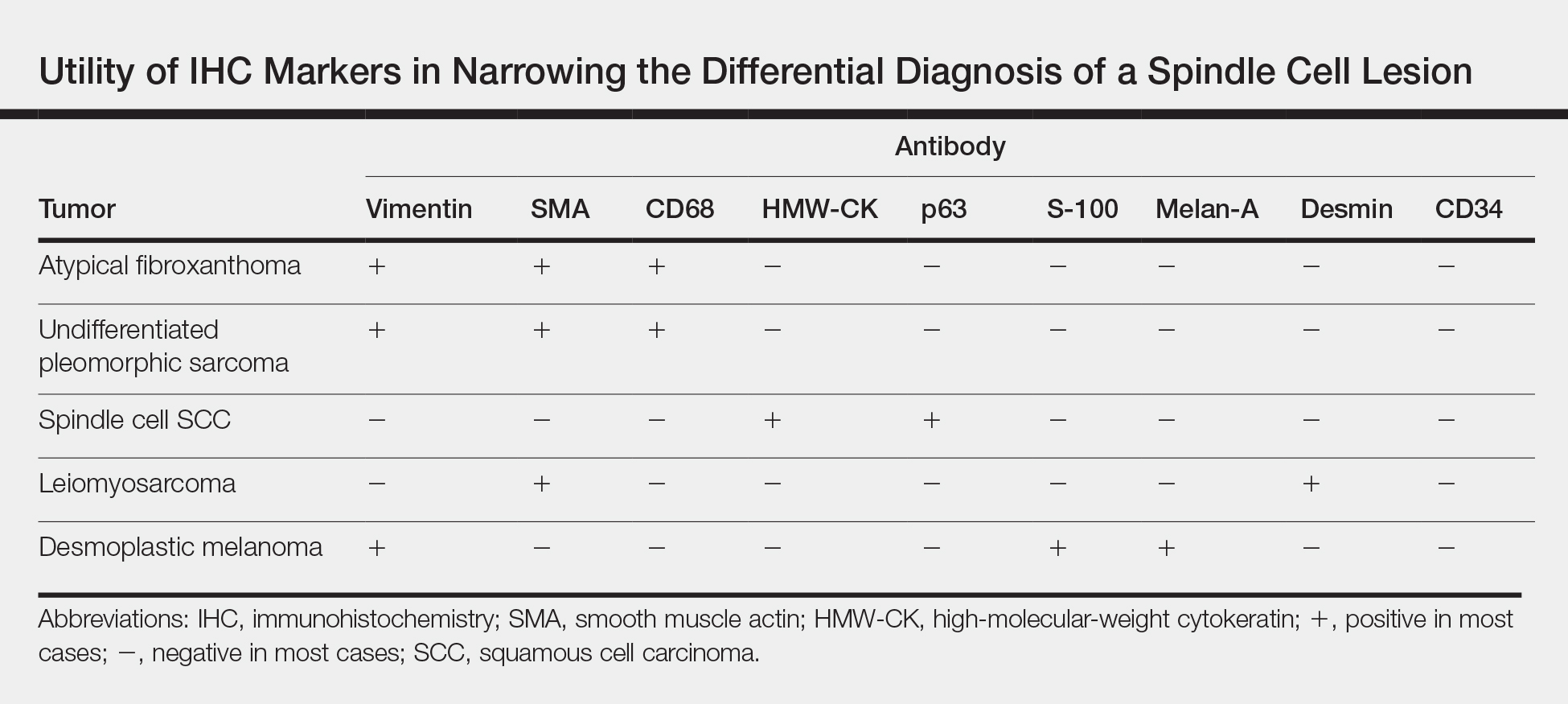
In addition to incomplete IHC evaluation, accurate diagnosis of spindle cell tumors also may be obscured by inadequate tumor sampling. The cells of AFX tumors often are well circumscribed and dermally based, and an excisional biopsy is the preferred biopsy procedure for AFX. A tumor invading into subcutaneous tissue or into lymphovascular or perineural structures suggests a more aggressive, poorly differentiated spindle cell neoplasm.1,3 For example, the tumor cells of malignant fibrous histiocytoma, which belongs to the undifferentiated pleomorphic sarcoma group, may appear identical to those of AFX on histology, and the 2 tumors display similar IHC profiles.3 Malignant fibrous histiocytoma, however, extends into the subcutaneous space and portends a notably worse prognosis compared to AFX. Malignant fibrous histiocytoma tumors therefore require more aggressive treatment strategies such as external beam radiation therapy, whereas AFX can be safely treated with surgical removal alone. In our patient, complete visualization of tumor margins solidified the diagnosis of AFX and spared our patient from unnecessary radiation therapy. Overall, AFX has a good prognosis and metastasis is rare, particularly when good margin control is achieved.10
Our case highlights the importance of clinicopathologic correlation, including appropriate IHC analysis and adequate tumor sampling in the diagnostic workup of a pleomorphic spindle cell neoplasm. Although these tumors are well studied, their notable degree of clinical and histologic heterogeneity may pose a diagnostic challenge to even experienced dermatologists and require careful consideration of the potential pitfalls in diagnosis.
- Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Lopez L, Velez R. Atypical fibroxanthoma. Arch Pathol Lab Med. 2016;140:376-379.
- Hussein MR. Atypical fibroxanthoma: new insights. Expert Rev Anticancer Ther. 2014;14:1075-1088.
- Gleason BC, Calder KB, Cibull TL, et al. Utility of p63 in the differential diagnosis of atypical fibroxanthoma and spindle cell squamous cell carcinoma. J Cutan Pathol. 2009;36:543-547.
- Pouryazdanparast P, Yu L, Cutland JE, et al. Diagnostic value of CD163 in cutaneous spindle cell lesions. J Cutan Pathol. 2009;36:859-864.
- Beer TW. CD163 is not a sensitive marker for identification of atypical fibroxanthoma. J Cutan Pathol. 2012;39:29-32.
- Longacre TA, Smoller BR, Rouse RV. Atypical fibroxanthoma. multiple immunohistologic profiles. Am J Surg Pathol. 1993;17:1199-1209.
- Altman DA, Nickoloff BD, Fivenson DP. Differential expression of factor XIIa and CD34 in cutaneous mesenchymal tumors. J Cutan Pathol. 1993;20:154-158.
- Benoit A, Wisell J, Brown M. Cutaneous spindle cell carcinoma misdiagnosed as atypical fibroxanthoma based on immunohistochemical stains. JAAD Case Rep. 2015;1:392-394.
- New D, Bahrami S, Malone J, et al. Atypical fibroxanthoma with regional lymph node metastasis: report of a case and review of the literature. Arch Dermatol. 2010;146:1399-1404.
The Diagnosis: Atypical Fibroxanthoma
Shave biopsy showed the superficial aspect of a highly cellular tumor composed of pleomorphic spindle cells exhibiting storiform growth and increased mitotic activity (Figure 1). The tumor stained positive for factor XIIIa, CD163, CD68, and smooth muscle actin (mild), and negative for high-molecular-weight cytokeratin (HMW-CK), p63, S-100, and melan-A. Subsequent excision with 0.5-cm margins was performed, and histopathology showed a well-circumscribed tumor contained within the dermis with a histologic scar at the outer margin (Figure 2). There was no lymphovascular or perineural invasion by tumor cells. Re-excision with 0.3-cm margins demonstrated no residual scar or tumor, and external radiation was deferred due to clear surgical margins.
Atypical fibroxanthoma (AFX) belongs to a group of spindle cell neoplasms that can be diagnostically challenging, as they often lack specific morphologic features on examination or routine histology. These neoplasms--of which the differential includes malignant fibrous histiocytoma, spindle cell squamous cell carcinoma (SCC), desmoplastic melanoma, and leiomyosarcoma--may each appear as a rapidly enlarging solitary plaque or nodule on sun-damaged skin on the head and neck or less commonly on the trunk, arms, or legs. Histologically, the cells of AFX exhibit notable pleomorphism with frequent atypical mitotic figures and nonspecific surrounding dermal changes. Subcutaneous and lymphovascular or perineural invasion of tumor cells can point away from the diagnosis of AFX; however, these features are likely to be missed in small superficial shave biopsies.1,2 Therefore, immunohistochemistry (IHC) and adequate tumor sampling are essential in the accurate diagnosis of AFX and other spindle cell neoplasms.
Several IHC markers have been employed in differentiating AFX from other spindle cell neoplasms.3-8 Positive stains for AFX include factor XIIIa (10%-25%), vimentin (>99%), CD10 (95%-100%), procollagen (87%), CD99 (35%-73%), CD163 (37%-79%), smooth muscle actin (50%), CD68 (>50%), and CD31 (43%). Other stains, such as HMW-CK, S-100, p63, desmin, CD34, and melan-A, typically are negative in AFX but are actively expressed in other pleomorphic spindle cell tumors. The Table summarizes the utility of these various markers in narrowing the differential diagnosis of a spindle cell lesion. Selection of an appropriate panel of IHC markers is critical for accurate diagnosis of AFX and exclusion of more aggressive, poorly differentiated spindle cell neoplasms. Key IHC markers include S-100 (negative in AFX; positive in desmoplastic melanoma), HMW-CK (negative in AFX; positive in spindle cell SCC), and p63 (negative in AFX; positive in spindle cell SCC). Benoit et al9 reported a case of a poorly differentiated spindle cell SCC misdiagnosed as AFX based on a limited IHC panel that was negative for pancytokeratin and S-100. Later, a more comprehensive IHC panel including HMW-CK and p63 confirmed spindle cell SCC, but by this time, a delay in therapy had allowed the tumor to metastasize, which ultimately proved fatal to the patient.9
In addition to incomplete IHC evaluation, accurate diagnosis of spindle cell tumors also may be obscured by inadequate tumor sampling. The cells of AFX tumors often are well circumscribed and dermally based, and an excisional biopsy is the preferred biopsy procedure for AFX. A tumor invading into subcutaneous tissue or into lymphovascular or perineural structures suggests a more aggressive, poorly differentiated spindle cell neoplasm.1,3 For example, the tumor cells of malignant fibrous histiocytoma, which belongs to the undifferentiated pleomorphic sarcoma group, may appear identical to those of AFX on histology, and the 2 tumors display similar IHC profiles.3 Malignant fibrous histiocytoma, however, extends into the subcutaneous space and portends a notably worse prognosis compared to AFX. Malignant fibrous histiocytoma tumors therefore require more aggressive treatment strategies such as external beam radiation therapy, whereas AFX can be safely treated with surgical removal alone. In our patient, complete visualization of tumor margins solidified the diagnosis of AFX and spared our patient from unnecessary radiation therapy. Overall, AFX has a good prognosis and metastasis is rare, particularly when good margin control is achieved.10
Our case highlights the importance of clinicopathologic correlation, including appropriate IHC analysis and adequate tumor sampling in the diagnostic workup of a pleomorphic spindle cell neoplasm. Although these tumors are well studied, their notable degree of clinical and histologic heterogeneity may pose a diagnostic challenge to even experienced dermatologists and require careful consideration of the potential pitfalls in diagnosis.
The Diagnosis: Atypical Fibroxanthoma
Shave biopsy showed the superficial aspect of a highly cellular tumor composed of pleomorphic spindle cells exhibiting storiform growth and increased mitotic activity (Figure 1). The tumor stained positive for factor XIIIa, CD163, CD68, and smooth muscle actin (mild), and negative for high-molecular-weight cytokeratin (HMW-CK), p63, S-100, and melan-A. Subsequent excision with 0.5-cm margins was performed, and histopathology showed a well-circumscribed tumor contained within the dermis with a histologic scar at the outer margin (Figure 2). There was no lymphovascular or perineural invasion by tumor cells. Re-excision with 0.3-cm margins demonstrated no residual scar or tumor, and external radiation was deferred due to clear surgical margins.
Atypical fibroxanthoma (AFX) belongs to a group of spindle cell neoplasms that can be diagnostically challenging, as they often lack specific morphologic features on examination or routine histology. These neoplasms--of which the differential includes malignant fibrous histiocytoma, spindle cell squamous cell carcinoma (SCC), desmoplastic melanoma, and leiomyosarcoma--may each appear as a rapidly enlarging solitary plaque or nodule on sun-damaged skin on the head and neck or less commonly on the trunk, arms, or legs. Histologically, the cells of AFX exhibit notable pleomorphism with frequent atypical mitotic figures and nonspecific surrounding dermal changes. Subcutaneous and lymphovascular or perineural invasion of tumor cells can point away from the diagnosis of AFX; however, these features are likely to be missed in small superficial shave biopsies.1,2 Therefore, immunohistochemistry (IHC) and adequate tumor sampling are essential in the accurate diagnosis of AFX and other spindle cell neoplasms.
Several IHC markers have been employed in differentiating AFX from other spindle cell neoplasms.3-8 Positive stains for AFX include factor XIIIa (10%-25%), vimentin (>99%), CD10 (95%-100%), procollagen (87%), CD99 (35%-73%), CD163 (37%-79%), smooth muscle actin (50%), CD68 (>50%), and CD31 (43%). Other stains, such as HMW-CK, S-100, p63, desmin, CD34, and melan-A, typically are negative in AFX but are actively expressed in other pleomorphic spindle cell tumors. The Table summarizes the utility of these various markers in narrowing the differential diagnosis of a spindle cell lesion. Selection of an appropriate panel of IHC markers is critical for accurate diagnosis of AFX and exclusion of more aggressive, poorly differentiated spindle cell neoplasms. Key IHC markers include S-100 (negative in AFX; positive in desmoplastic melanoma), HMW-CK (negative in AFX; positive in spindle cell SCC), and p63 (negative in AFX; positive in spindle cell SCC). Benoit et al9 reported a case of a poorly differentiated spindle cell SCC misdiagnosed as AFX based on a limited IHC panel that was negative for pancytokeratin and S-100. Later, a more comprehensive IHC panel including HMW-CK and p63 confirmed spindle cell SCC, but by this time, a delay in therapy had allowed the tumor to metastasize, which ultimately proved fatal to the patient.9
In addition to incomplete IHC evaluation, accurate diagnosis of spindle cell tumors also may be obscured by inadequate tumor sampling. The cells of AFX tumors often are well circumscribed and dermally based, and an excisional biopsy is the preferred biopsy procedure for AFX. A tumor invading into subcutaneous tissue or into lymphovascular or perineural structures suggests a more aggressive, poorly differentiated spindle cell neoplasm.1,3 For example, the tumor cells of malignant fibrous histiocytoma, which belongs to the undifferentiated pleomorphic sarcoma group, may appear identical to those of AFX on histology, and the 2 tumors display similar IHC profiles.3 Malignant fibrous histiocytoma, however, extends into the subcutaneous space and portends a notably worse prognosis compared to AFX. Malignant fibrous histiocytoma tumors therefore require more aggressive treatment strategies such as external beam radiation therapy, whereas AFX can be safely treated with surgical removal alone. In our patient, complete visualization of tumor margins solidified the diagnosis of AFX and spared our patient from unnecessary radiation therapy. Overall, AFX has a good prognosis and metastasis is rare, particularly when good margin control is achieved.10
Our case highlights the importance of clinicopathologic correlation, including appropriate IHC analysis and adequate tumor sampling in the diagnostic workup of a pleomorphic spindle cell neoplasm. Although these tumors are well studied, their notable degree of clinical and histologic heterogeneity may pose a diagnostic challenge to even experienced dermatologists and require careful consideration of the potential pitfalls in diagnosis.
- Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Lopez L, Velez R. Atypical fibroxanthoma. Arch Pathol Lab Med. 2016;140:376-379.
- Hussein MR. Atypical fibroxanthoma: new insights. Expert Rev Anticancer Ther. 2014;14:1075-1088.
- Gleason BC, Calder KB, Cibull TL, et al. Utility of p63 in the differential diagnosis of atypical fibroxanthoma and spindle cell squamous cell carcinoma. J Cutan Pathol. 2009;36:543-547.
- Pouryazdanparast P, Yu L, Cutland JE, et al. Diagnostic value of CD163 in cutaneous spindle cell lesions. J Cutan Pathol. 2009;36:859-864.
- Beer TW. CD163 is not a sensitive marker for identification of atypical fibroxanthoma. J Cutan Pathol. 2012;39:29-32.
- Longacre TA, Smoller BR, Rouse RV. Atypical fibroxanthoma. multiple immunohistologic profiles. Am J Surg Pathol. 1993;17:1199-1209.
- Altman DA, Nickoloff BD, Fivenson DP. Differential expression of factor XIIa and CD34 in cutaneous mesenchymal tumors. J Cutan Pathol. 1993;20:154-158.
- Benoit A, Wisell J, Brown M. Cutaneous spindle cell carcinoma misdiagnosed as atypical fibroxanthoma based on immunohistochemical stains. JAAD Case Rep. 2015;1:392-394.
- New D, Bahrami S, Malone J, et al. Atypical fibroxanthoma with regional lymph node metastasis: report of a case and review of the literature. Arch Dermatol. 2010;146:1399-1404.
- Iorizzo LJ, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Lopez L, Velez R. Atypical fibroxanthoma. Arch Pathol Lab Med. 2016;140:376-379.
- Hussein MR. Atypical fibroxanthoma: new insights. Expert Rev Anticancer Ther. 2014;14:1075-1088.
- Gleason BC, Calder KB, Cibull TL, et al. Utility of p63 in the differential diagnosis of atypical fibroxanthoma and spindle cell squamous cell carcinoma. J Cutan Pathol. 2009;36:543-547.
- Pouryazdanparast P, Yu L, Cutland JE, et al. Diagnostic value of CD163 in cutaneous spindle cell lesions. J Cutan Pathol. 2009;36:859-864.
- Beer TW. CD163 is not a sensitive marker for identification of atypical fibroxanthoma. J Cutan Pathol. 2012;39:29-32.
- Longacre TA, Smoller BR, Rouse RV. Atypical fibroxanthoma. multiple immunohistologic profiles. Am J Surg Pathol. 1993;17:1199-1209.
- Altman DA, Nickoloff BD, Fivenson DP. Differential expression of factor XIIa and CD34 in cutaneous mesenchymal tumors. J Cutan Pathol. 1993;20:154-158.
- Benoit A, Wisell J, Brown M. Cutaneous spindle cell carcinoma misdiagnosed as atypical fibroxanthoma based on immunohistochemical stains. JAAD Case Rep. 2015;1:392-394.
- New D, Bahrami S, Malone J, et al. Atypical fibroxanthoma with regional lymph node metastasis: report of a case and review of the literature. Arch Dermatol. 2010;146:1399-1404.
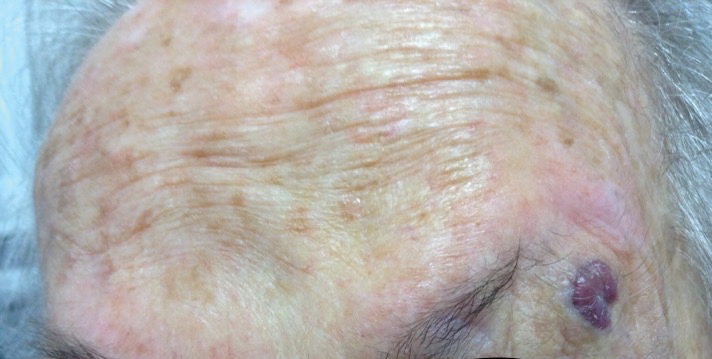
An 88-year-old woman presented for evaluation of an asymptomatic facial lesion that she first noticed 3 months prior, with rapid growth over the last month. Review of systems was negative, and she denied any history of connective tissue disease, skin cancer, or radiation to the head or neck area. Physical examination revealed a 1.5-cm, solitary, violaceous nodule on the left lateral eyebrow on a background of actinically damaged skin. The lesion was nontender and there were no similar lesions or palpable lymphadenopathy.
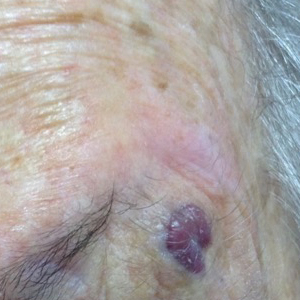
Recurrent Pruritic Multifocal Erythematous Rash
The Diagnosis: Wells Syndrome
Histopathologic examination of the biopsy demonstrated overlying acanthosis, focal spongiosis, and exocytosis. There also was proliferation and thickening of superficial capillaries and papillary fibrosis (Figure, A). There was a mixed interstitial and perivascular inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and eosinophils (Figure, A and B). Occasional flame figures were identified (Figure, C).

Wells syndrome, also known as eosinophilic cellulitis, was first described in 1971 by Wells1 as a recurrent granulomatous dermatitis with eosinophilia. Rarely reported worldwide, this chronic relapsing condition is characterized by a pronounced eosinophilic infiltrate of the dermis resembling urticaria or cellulitis.2 The exact etiology has not been elucidated; however, links to certain medications, vaccines, exaggerated arthropod reactions, infections, and malignancies have been documented.3
Wells syndrome is a diagnosis of exclusion and lacks a predictable dermatologic presentation, thereby mandating focused clinical follow-up as well as correlation with histopathology findings. Although the classic histologic hallmark of Wells syndrome is scattered flame figures, this finding is not specific and can be found in other hypereosinophilic conditions.2 Clinical manifestations most often consist of 2 distinct phases: an initial painful burning or pruritic sensation, followed by the development of erythematous and edematous dermal plaques that may heal with slight hyperpigmentation over 4 to 8 weeks. A case series of 19 patients demonstrated variants of Wells syndrome, with an annular granuloma-like appearance found primarily in adults and the signature plaque-type appearance predominating in children.4
Acute urticaria is characterized by pruritic erythematous wheals secondary to a histamine-mediated response brought on by a variety of triggers, typically allergic and self-resolving within 24 hours. When such lesions last longer than 24 hours, biopsy should be performed to exclude urticarial vasculitis, which is characterized by a burning or painful sensation rather than pruritis, in addition to dermal neutrophilia and perivascular infiltrate on histology. Erythema migrans of Lyme disease begins at the site of a tick bite, evolving from a red macule to an expanding targetoid lesion and typically is not pruritic. Infectious cellulitis presents with warm, tender, and poorly defined erythematous patches; can progress rapidly; and is accompanied by systemic symptoms such as fevers, malaise, and lymphadenopathy.
Best evidence favors the use of moderate- to high-dose corticosteroids as first-line treatment.5 The use of tumor necrosis factor blockers, various immunomodulating agents, and combination therapy with levocetirizine and hydroxyzine have demonstrated variable levels of efficacy, albeit often followed by high rates of relapse with drug discontinuation.6
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc. 1971;57:46-56.
- Aberer W, Konrad K, Wolff K. Wells' syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol. 1988;18:105-114.
- Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983-1984.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Ferreli C, Pinna AL, Atzori L, et al. Eosinophilic cellulitis (Well's syndrome): a new case description. J Eur Acad Dermatol Venereol. 1999;13:41-45.
- Cormerais M, Poizeau F, Darrieux L, et al. Wells' syndrome mimicking facial cellulitis: a report of two cases. Case Rep Dermatol. 2015;7:117-122.
The Diagnosis: Wells Syndrome
Histopathologic examination of the biopsy demonstrated overlying acanthosis, focal spongiosis, and exocytosis. There also was proliferation and thickening of superficial capillaries and papillary fibrosis (Figure, A). There was a mixed interstitial and perivascular inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and eosinophils (Figure, A and B). Occasional flame figures were identified (Figure, C).
Wells syndrome, also known as eosinophilic cellulitis, was first described in 1971 by Wells1 as a recurrent granulomatous dermatitis with eosinophilia. Rarely reported worldwide, this chronic relapsing condition is characterized by a pronounced eosinophilic infiltrate of the dermis resembling urticaria or cellulitis.2 The exact etiology has not been elucidated; however, links to certain medications, vaccines, exaggerated arthropod reactions, infections, and malignancies have been documented.3
Wells syndrome is a diagnosis of exclusion and lacks a predictable dermatologic presentation, thereby mandating focused clinical follow-up as well as correlation with histopathology findings. Although the classic histologic hallmark of Wells syndrome is scattered flame figures, this finding is not specific and can be found in other hypereosinophilic conditions.2 Clinical manifestations most often consist of 2 distinct phases: an initial painful burning or pruritic sensation, followed by the development of erythematous and edematous dermal plaques that may heal with slight hyperpigmentation over 4 to 8 weeks. A case series of 19 patients demonstrated variants of Wells syndrome, with an annular granuloma-like appearance found primarily in adults and the signature plaque-type appearance predominating in children.4
Acute urticaria is characterized by pruritic erythematous wheals secondary to a histamine-mediated response brought on by a variety of triggers, typically allergic and self-resolving within 24 hours. When such lesions last longer than 24 hours, biopsy should be performed to exclude urticarial vasculitis, which is characterized by a burning or painful sensation rather than pruritis, in addition to dermal neutrophilia and perivascular infiltrate on histology. Erythema migrans of Lyme disease begins at the site of a tick bite, evolving from a red macule to an expanding targetoid lesion and typically is not pruritic. Infectious cellulitis presents with warm, tender, and poorly defined erythematous patches; can progress rapidly; and is accompanied by systemic symptoms such as fevers, malaise, and lymphadenopathy.
Best evidence favors the use of moderate- to high-dose corticosteroids as first-line treatment.5 The use of tumor necrosis factor blockers, various immunomodulating agents, and combination therapy with levocetirizine and hydroxyzine have demonstrated variable levels of efficacy, albeit often followed by high rates of relapse with drug discontinuation.6
The Diagnosis: Wells Syndrome
Histopathologic examination of the biopsy demonstrated overlying acanthosis, focal spongiosis, and exocytosis. There also was proliferation and thickening of superficial capillaries and papillary fibrosis (Figure, A). There was a mixed interstitial and perivascular inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and eosinophils (Figure, A and B). Occasional flame figures were identified (Figure, C).
Wells syndrome, also known as eosinophilic cellulitis, was first described in 1971 by Wells1 as a recurrent granulomatous dermatitis with eosinophilia. Rarely reported worldwide, this chronic relapsing condition is characterized by a pronounced eosinophilic infiltrate of the dermis resembling urticaria or cellulitis.2 The exact etiology has not been elucidated; however, links to certain medications, vaccines, exaggerated arthropod reactions, infections, and malignancies have been documented.3
Wells syndrome is a diagnosis of exclusion and lacks a predictable dermatologic presentation, thereby mandating focused clinical follow-up as well as correlation with histopathology findings. Although the classic histologic hallmark of Wells syndrome is scattered flame figures, this finding is not specific and can be found in other hypereosinophilic conditions.2 Clinical manifestations most often consist of 2 distinct phases: an initial painful burning or pruritic sensation, followed by the development of erythematous and edematous dermal plaques that may heal with slight hyperpigmentation over 4 to 8 weeks. A case series of 19 patients demonstrated variants of Wells syndrome, with an annular granuloma-like appearance found primarily in adults and the signature plaque-type appearance predominating in children.4
Acute urticaria is characterized by pruritic erythematous wheals secondary to a histamine-mediated response brought on by a variety of triggers, typically allergic and self-resolving within 24 hours. When such lesions last longer than 24 hours, biopsy should be performed to exclude urticarial vasculitis, which is characterized by a burning or painful sensation rather than pruritis, in addition to dermal neutrophilia and perivascular infiltrate on histology. Erythema migrans of Lyme disease begins at the site of a tick bite, evolving from a red macule to an expanding targetoid lesion and typically is not pruritic. Infectious cellulitis presents with warm, tender, and poorly defined erythematous patches; can progress rapidly; and is accompanied by systemic symptoms such as fevers, malaise, and lymphadenopathy.
Best evidence favors the use of moderate- to high-dose corticosteroids as first-line treatment.5 The use of tumor necrosis factor blockers, various immunomodulating agents, and combination therapy with levocetirizine and hydroxyzine have demonstrated variable levels of efficacy, albeit often followed by high rates of relapse with drug discontinuation.6
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc. 1971;57:46-56.
- Aberer W, Konrad K, Wolff K. Wells' syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol. 1988;18:105-114.
- Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983-1984.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Ferreli C, Pinna AL, Atzori L, et al. Eosinophilic cellulitis (Well's syndrome): a new case description. J Eur Acad Dermatol Venereol. 1999;13:41-45.
- Cormerais M, Poizeau F, Darrieux L, et al. Wells' syndrome mimicking facial cellulitis: a report of two cases. Case Rep Dermatol. 2015;7:117-122.
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc. 1971;57:46-56.
- Aberer W, Konrad K, Wolff K. Wells' syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol. 1988;18:105-114.
- Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983-1984.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Ferreli C, Pinna AL, Atzori L, et al. Eosinophilic cellulitis (Well's syndrome): a new case description. J Eur Acad Dermatol Venereol. 1999;13:41-45.
- Cormerais M, Poizeau F, Darrieux L, et al. Wells' syndrome mimicking facial cellulitis: a report of two cases. Case Rep Dermatol. 2015;7:117-122.
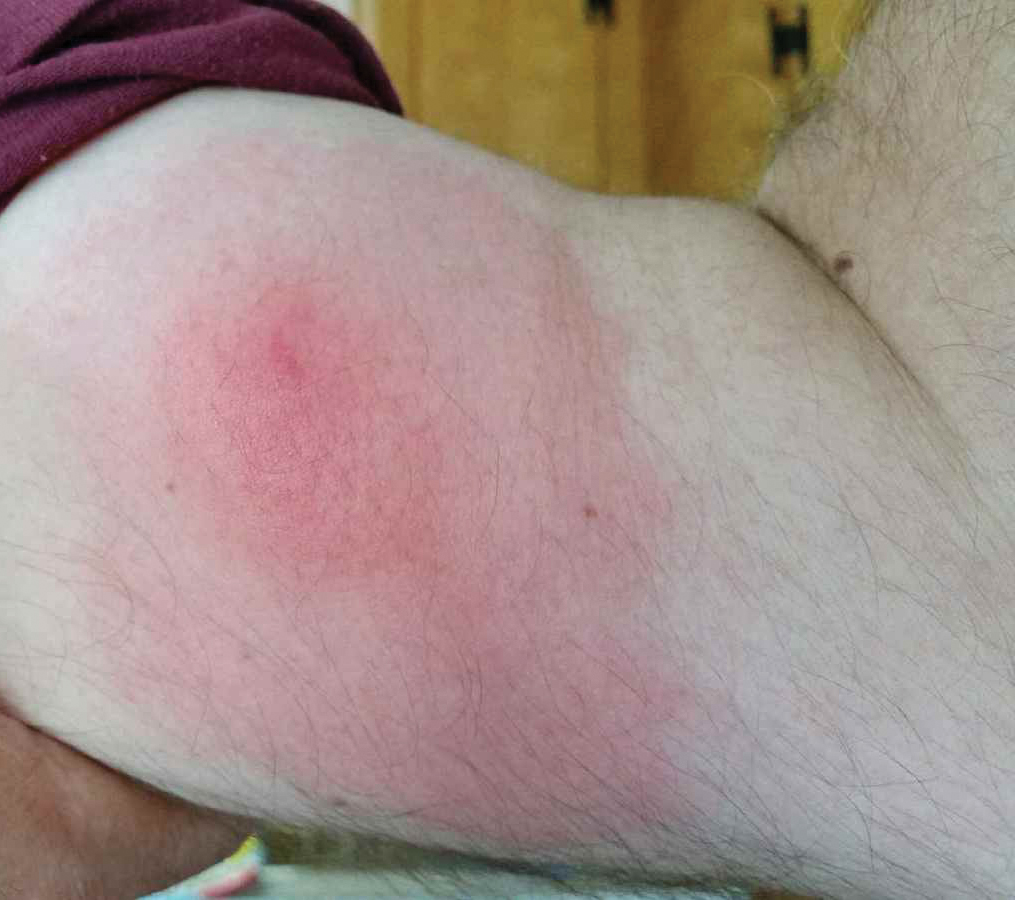
A 60-year-old man with a history of hyperlipidemia developed acute onset of an intensely pruritic and painful burning rash on the dorsal aspect of the left forearm of 8 days' duration. The patient described the rash as red and warm. It measured 2 cm at inception and peaked at 12 cm 6 months later when the patient presented. These symptoms resolved without therapeutic intervention.
Over the ensuing 6 months, he experienced 13 self-limited episodes of erythematous indurated cutaneous streaks, usually with proximal migration on the arms along with involvement of the posterior thorax and right leg. Five months prior to the onset of the initial rash, the patient had discontinued ezetimibe to treat hyperlipidemia due to swelling of the lips and tongue. He also reported that he regularly hunted in upstate Pennsylvania but reported no history of arthropod or animal bites. The patient did not take prescription or over-the-counter medications, and he denied the presence of fever, night sweats, fatigue, adenopathy, anorexia, weight loss, diarrhea, joint pain or swelling, or illicit drug use. Lyme titers, complete blood cell count, erythrocyte sedimentation rate, and comprehensive metabolic panel were within reference range. A punch biopsy was performed.
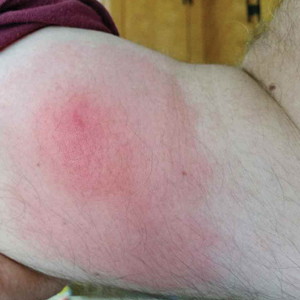
Erythema Gyratum Repens–like Eruption in Sézary Syndrome: Evidence for the Role of a Dermatophyte
Case Report
A 65-year-old woman presented with stage IVA2 mycosis fungoides (MF)(T4N3M0B2)/Sézary syndrome (SS). A peripheral blood count contained 6000 Sézary cells with cerebriform nuclei, a CD2+/−CD3+CD4+CD5+/−CD7+CD8−CD26−immunophenotype, and a highly abnormal CD4 to CD8 ratio (70:1). Positron emission tomography and computed tomography demonstrated hypermetabolic subcutaneous nodules in the base of the neck and generalized lymphadenopathy. Lymph node biopsy showed involvement by T-cell lymphoma and dominant T-cell receptor γ clonality by polymerase chain reaction.
On initial presentation to the Cutaneous Lymphoma Clinic at the University of Wisconsin-Madison, the patient was erythrodermic. She also was noted to have undulating wavy bands and concentric annular, ringlike, thin, erythematous plaques with trailing scale, giving a wood grain, zebra hide–like appearance involving the buttocks, abdomen, and lower extremities (Figure 1). Lesions were markedly pruritic and were advancing rapidly. A diagnosis of erythema gyratum repens (EGR)–like eruption was made.
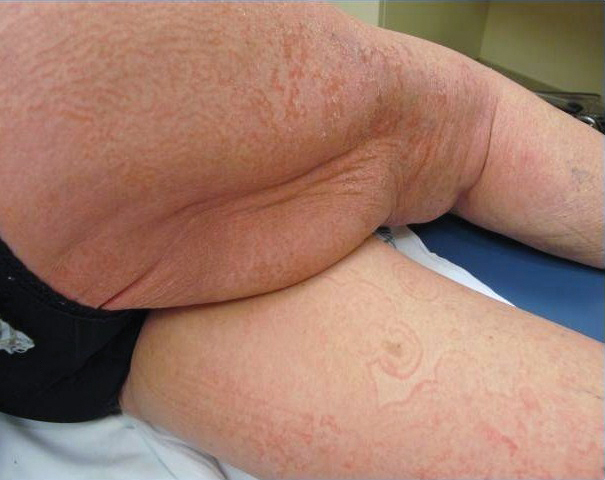
Biopsy of an EGR-like area on the leg showed a superficial perivascular and somewhat lichenoid lymphoid infiltrate (Figure 2). Lymphocytes were lined up along the basal layer, occasionally forming nests within the epidermis. Nearly all mononuclear cells in the epidermis and dermis exhibited positive CD3 and CD4 staining, with only scattered CD8 cells. These features were compatible with cutaneous involvement in SS. A concurrent biopsy from diffusely erythrodermic forearm skin, which lacked EGR-like morphology, showed similar histopathologic and immunophenotypic features.
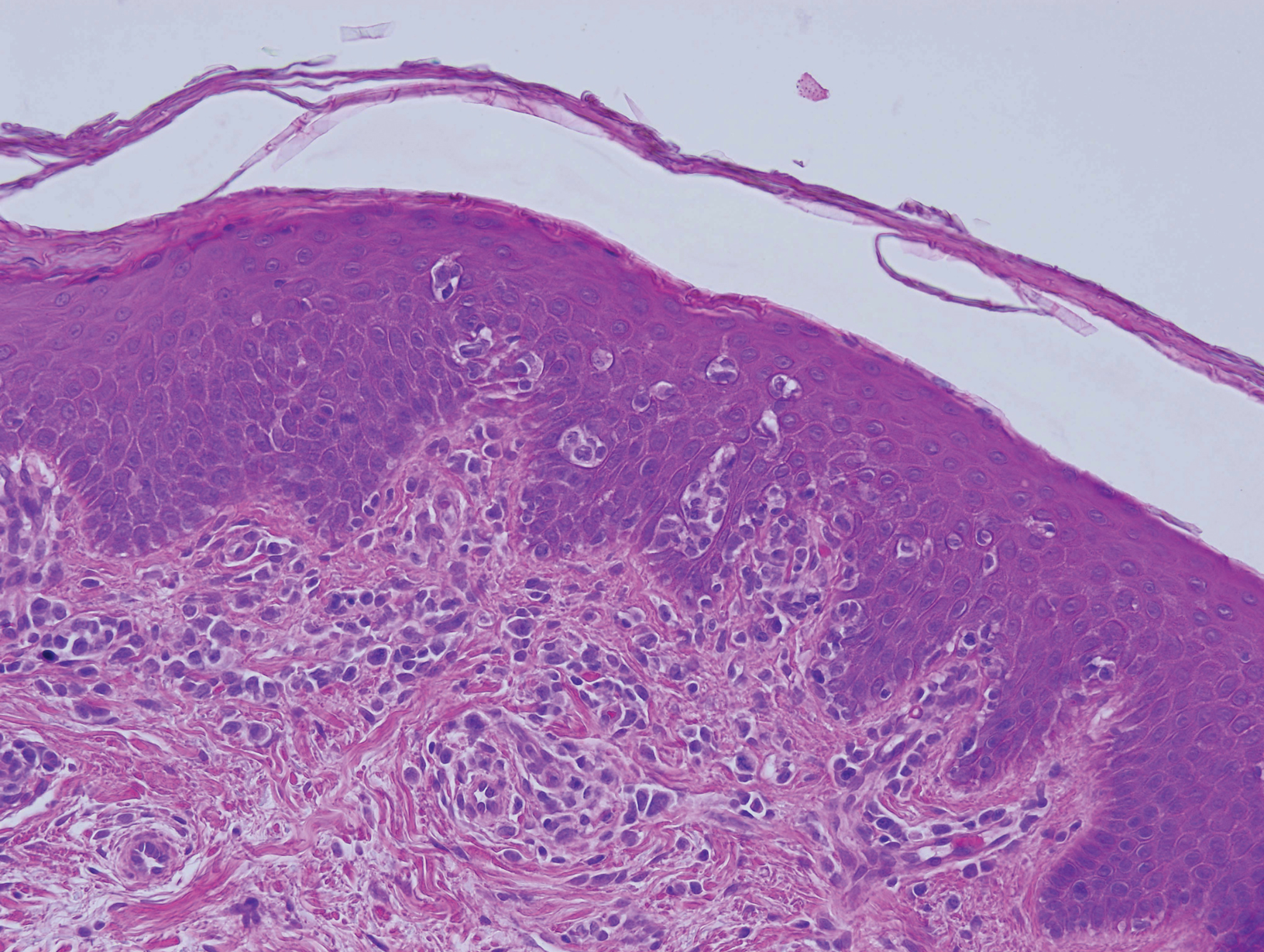
Periodic acid–Schiff (PAS) with diastase stain revealed numerous septate hyphae within the stratum corneum in both skin biopsy specimens (Figure 3). Fungal culture of EGR-like lesions was positive for a nonsporulating filamentous fungus, identified as Trichophyton rubrum by DNA sequencing.
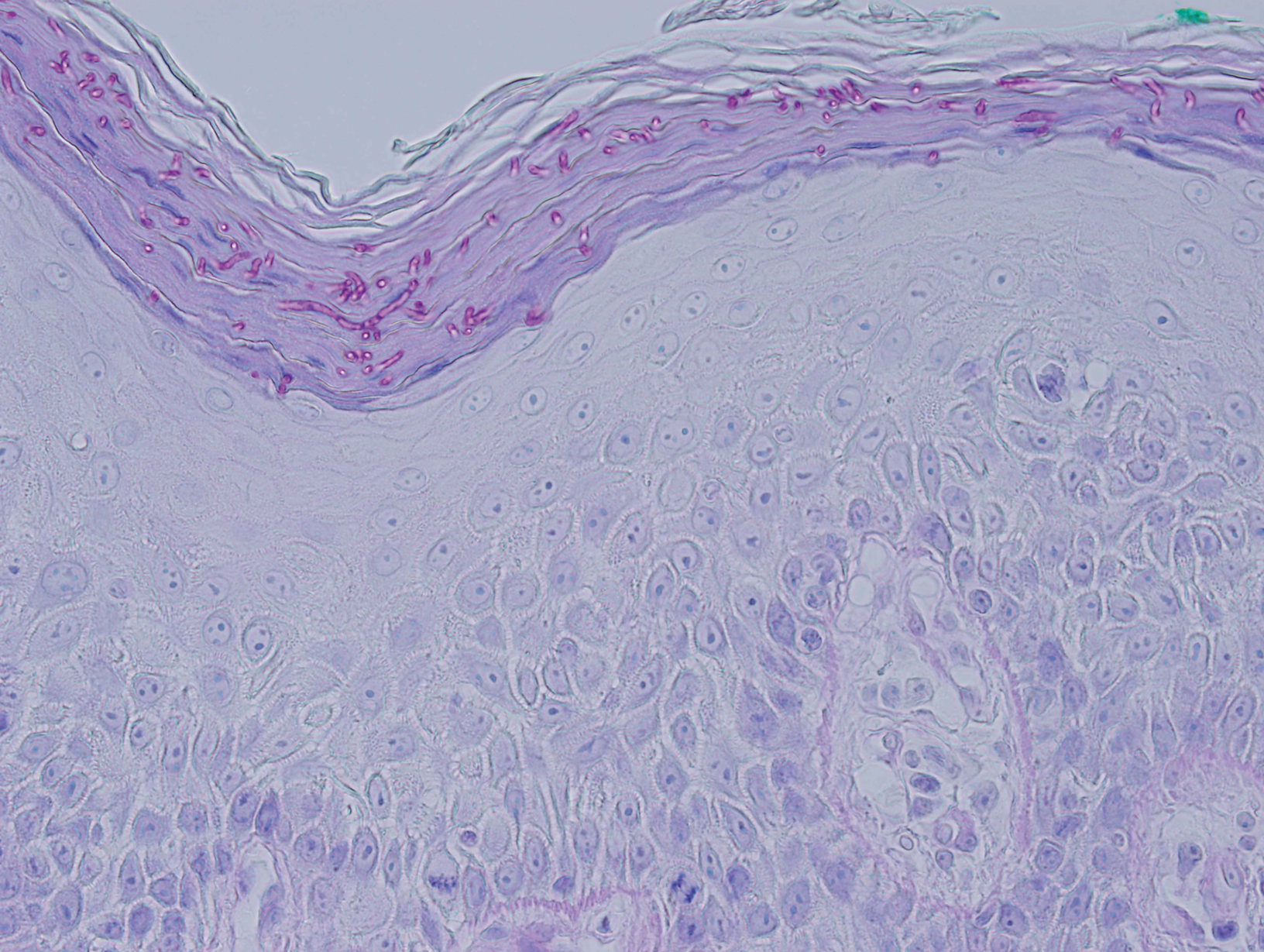
A diagnosis of EGR-like eruption secondary to tinea corporis in SS was made. The possibility of tinea incognito also was considered to explain the presence of dermatophytes in the biopsy from skin that exhibited only erythroderma clinically; however, the patient did not have a history of corticosteroid use.
Interferon alfa-2b and methotrexate therapy was initiated. Additionally, oral terbinafine (250 mg/d) was initiated for 14 days, resulting in complete resolution of the EGR-like eruption; nevertheless, diffuse erythema remained. Subsequently, within 3 months of treatment, the cutaneous T-cell lymphoma (CTCL) improved with continued interferon alfa-2b and methotrexate. Erythroderma became minimal; the circulating Sézary cell count decreased by 50%. The patient ultimately had multiple relapses in erythroderma and progression of SS. Erythema gyratum repens–like lesions recurred on multiple occasions, with a temporary response to repeat courses of oral terbinafine.
Comment
Defining True EGR vs EGR-like Eruption
Sézary syndrome represents the leukemic stage of CTCL, which is defined by the triad of erythroderma; generalized lymphadenopathy; and neoplastic T cells in the skin, lymph nodes, and peripheral blood. It is well known that CTCL can mimic multiple benign and malignant dermatoses. One rare presentation of CTCL is an EGR-like eruption.
Erythema gyratum repens presents as rapidly advancing, erythematous, concentric bands that can be figurate, gyrate, or annular, with a fine trailing edge of scale (wood grain pattern). The diagnosis is based on the characteristic clinical pattern of EGR and by ruling out other mimicking conditions with biopsy.1 Patients with the characteristic clinical pattern but with an alternate underlying dermatosis are described as having an EGR-like eruption rather than true EGR.
True EGR is most often but not always associated with underlying malignancy. Biopsy of true EGR eruptions show nonspecific histopathologic features, with perivascular superficial mononuclear dermatitis, occasional mild spongiosis, and focal parakeratosis; specific features of an alternate dermatosis are lacking.2 In addition to CTCL, EGR-like eruptions have been described in a number of diseases, including systemic lupus erythematosus, erythema annulare centrifugum, bullous dermatosis, erythrokeratodermia variabilis, urticarial vasculitis, leukocytoclastic vasculitis, and neutrophilic dermatoses.
Prior Reports of EGR-like Eruption in Association With MF
According to a PubMed search of articles indexed for MEDLINE using the terms erythema gyratum repens in mycosis fungoides, mycosis fungoides with tinea, and concentric wood grain erythema, there have been 6 other cases of an EGR-like eruption in association with MF (Table). Poonawalla et al3 first described an EGR-like eruption (utilizing the term tinea pseudoimbricata) in a 55-year-old man with stage IB MF (T2N0M0B0). The patient had a preceding history of tinea pedis and tinea corporis that preceded the diagnosis of MF. At the time of MF diagnosis, the patient presented with extensive concentric, gyrate, wood grain, annular lesions. His MF was resistant to topical mechlorethamine, psoralen plus UVA, and oral bexarotene. The body surface area involvement decreased from 60% to less than 1% after institution of oral and topical antifungal therapy. It was postulated that the widespread dermatophytosis that preceded the development of MF may have been the persistent antigen leading to his disease. Preceding the diagnosis of MF, skin scrapings were floridly positive for dermatophyte hyphae. Fungal cultures from the affected areas of skin grew T rubrum.3
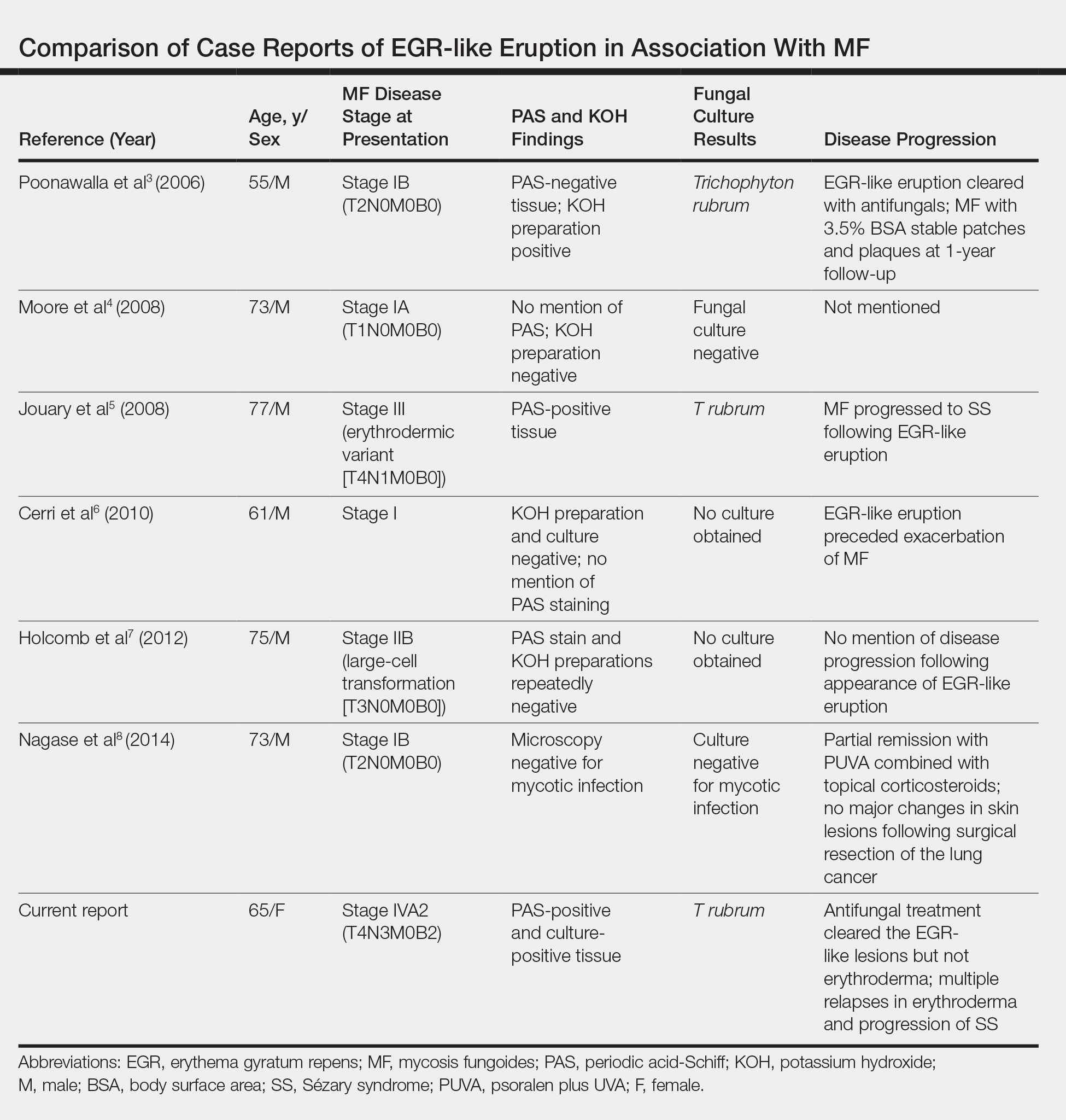
Moore et al4 described an EGR-like eruption on the trunk of a 73-year-old man with stage IA MF (T1N0M0B0). Biopsy was consistent with MF, but no fungal organisms were seen. Potassium hydroxide preparation and fungal cultures of the lesions also were negative for organisms. The patient was successfully treated with topical betamethasone.4Jouary et al5 described an EGR-like eruption in a 77-year-old man with stage III erythrodermic MF (T4N1M0B0). Biopsy showed mycelia on PAS stain. Subsequent culture isolated T rubrum. Terbinafine (250 mg/d) and ketoconazole cream 2% daily were initiated and the patient’s EGR-like rash quickly cleared, while MF progressed to SS.5
Cerri et al6 later described a case of EGR-like eruption in a 61-year-old man with stage I MF and an EGR-like eruption. Microscopic examination of potassium hydroxide (KOH) preparations and fungal culture of the lesions failed to demonstrate mycotic infection. There was no mention of PAS stain of skin biopsy specimens. In this case, the authors mentioned that EGR-like lesions preceded exacerbation of MF and questioned the prognostic significance of the EGR-like eruption in relation to MF.6
Holcomb et al7 reported the next case of a 75-year-old man with stage IIB MF (T3N0M0B0) with CD25+ and CD30+ large cell transformation who presented with an EGR-like eruption. In this case, PAS stain and KOH preparations were repeatedly negative for mycotic infection. Disease progression was not mentioned following the appearance of the EGR-like eruption.7
Nagase et al8 most recently described a case of a 73-year-old Japanese man with stage IB (T2N0M0B0) CD4−CD8− MF and lung cancer who developed a cutaneous eruption mimicking EGR. Microscopy and culture excluded the presence of a mycotic infection. The patient achieved partial remission with photochemotherapy (psoralen plus UVA) combined with topical corticosteroids. No major changes in the patient’s skin lesions were noted following surgical resection of the lung cancer.8
Dermatophyte Infection
It is known that conventional tinea corporis can occur in the setting of CTCL. However, EGR-like eruptions in CTCL can be distinguished from standard tinea corporis by the classic morphology of EGR and clinical history of rapid migration of these characteristic lesions.
Tinea imbricata is known to have a clinical appearance that is similar to EGR, but the infection is caused by Tinea concentricum, which is limited to southwest Polynesia, Melanesia, Southeast Asia, India, and Central America. Although T rubrum was the dermatophyte isolated by Poonawalla et al,3 Jouary et al,5 and in our case, whether T rubrum infection in the setting of CTCL has any impact on prognosis needs further study.
Our case of an EGR-like eruption presented in a patient with SS and tinea corporis. Biopsy specimens showed CTCL and concomitant dermatophytic infection that was confirmed with PAS stain and identified as T rubrum. Interestingly, our patient’s EGR-like eruption cleared with oral terbinafine therapy, consistent with findings described by Poonawalla et al3 and Jouary et al5 in which treatment of the dermatophytic infection led to resolution of the EGR-like eruption, suggesting a causative role.
However, testing for dermatophytes was negative in the other reported cases of EGR-like eruptions in patients with MF, despite screening for the presence of fungal microorganisms using KOH preparation, PAS staining, or fungal culture, or a combination of these methods,3-8 which raises the question: Do the cases reported without dermatophytic infection represent false-negative test results, or can the distinct clinical appearance of EGR indeed be seen in patients with CTCL who lack superimposed dermatophytosis? In 3 prior reported cases of EGR-like eruptions in MF, the eruption was preceded by immunosuppressive therapy.5-7
Further investigation is needed to correlate the role of dermatophytic infection in EGR-like eruptions. Our case and the Jouary et al5 case reported dermatophyte-positive EGR-like eruptions in MF and SS detected with histopathologic analysis and PAS stain. This low-cost screening method should be considered in future cases. If the test result is dermatophyte positive, a 14-day course of oral terbinafine (250 mg/d) might induce resolution of the EGR-like eruption.
Conclusion
The role of dermatophyte-induced EGR or EGR-like eruptions in other settings also warrants further investigation to shed light on this poorly understood yet striking dermatologic condition. Our patient showed both MF and dermatophytes in skin biopsy results, regardless of whether those sites showed erythroderma or EGR-like features clinically. On 3 occasions, antifungal treatment cleared the EGR-like lesions and associated pruritus but not erythroderma. Therefore, it appears that the mere presence of dermatophytes was necessary but not sufficient to produce the EGR-like lesions observed in our case.
- Rongioletti F, Fausti V, Parodi A. Erythema gyratum repens is not an obligate paraneoplastic disease: a systematic review of the literature and personal experience. J Eur Acad Dermatol Venereol. 2012;28:112-115.
- Albers SE, Fenske NA, Glass LF. Erythema gyratum repens: direct immunofluorescence microscopic findings. J Am Acad Dermatol. 1993;29:493-494.
- Poonawalla T, Chen W, Duvic M. Mycosis fungoides with tinea pseudoimbricata owing to Trichophyton rubrum infection. J Cutan Med Surg. 2006;10:52-56.
- Moore E, McFarlane R, Olerud J. Concentric wood grain erythema on the trunk. Arch Dermatol. 2008;144:673-678.
- Jouary T, Lalanne N, Stanislas S, et al. Erythema gyratum repens-like eruption in mycosis fungoides: is dermatophyte superinfection underdiagnosed in cutaneous T-cell lymphomas? J Eur Acad Dermatol Venereol. 2008;22:1276-1278.
- Cerri A, Vezzoli P, Serini SM, et al. Mycosis fungoides mimicking erythema gyratum repens: an additional variant? Eur J Dermatol. 2010;20:540-541.
- Holcomb M, Duvic M, Cutlan J. Erythema gyratum repens-like eruptions with large cell transformation in a patient with mycosis fungoides. Int J Dermatol. 2012;51:1231-1233.
- Nagase K, Shirai R, Okawa T, et al. CD4/CD8 double-negative mycosis fungoides mimicking erythema gyratum repens in a patient with underlying lung cancer. Acta Derm Venereol. 2014;94:89-90.
Case Report
A 65-year-old woman presented with stage IVA2 mycosis fungoides (MF)(T4N3M0B2)/Sézary syndrome (SS). A peripheral blood count contained 6000 Sézary cells with cerebriform nuclei, a CD2+/−CD3+CD4+CD5+/−CD7+CD8−CD26−immunophenotype, and a highly abnormal CD4 to CD8 ratio (70:1). Positron emission tomography and computed tomography demonstrated hypermetabolic subcutaneous nodules in the base of the neck and generalized lymphadenopathy. Lymph node biopsy showed involvement by T-cell lymphoma and dominant T-cell receptor γ clonality by polymerase chain reaction.
On initial presentation to the Cutaneous Lymphoma Clinic at the University of Wisconsin-Madison, the patient was erythrodermic. She also was noted to have undulating wavy bands and concentric annular, ringlike, thin, erythematous plaques with trailing scale, giving a wood grain, zebra hide–like appearance involving the buttocks, abdomen, and lower extremities (Figure 1). Lesions were markedly pruritic and were advancing rapidly. A diagnosis of erythema gyratum repens (EGR)–like eruption was made.
Biopsy of an EGR-like area on the leg showed a superficial perivascular and somewhat lichenoid lymphoid infiltrate (Figure 2). Lymphocytes were lined up along the basal layer, occasionally forming nests within the epidermis. Nearly all mononuclear cells in the epidermis and dermis exhibited positive CD3 and CD4 staining, with only scattered CD8 cells. These features were compatible with cutaneous involvement in SS. A concurrent biopsy from diffusely erythrodermic forearm skin, which lacked EGR-like morphology, showed similar histopathologic and immunophenotypic features.
Periodic acid–Schiff (PAS) with diastase stain revealed numerous septate hyphae within the stratum corneum in both skin biopsy specimens (Figure 3). Fungal culture of EGR-like lesions was positive for a nonsporulating filamentous fungus, identified as Trichophyton rubrum by DNA sequencing.
A diagnosis of EGR-like eruption secondary to tinea corporis in SS was made. The possibility of tinea incognito also was considered to explain the presence of dermatophytes in the biopsy from skin that exhibited only erythroderma clinically; however, the patient did not have a history of corticosteroid use.
Interferon alfa-2b and methotrexate therapy was initiated. Additionally, oral terbinafine (250 mg/d) was initiated for 14 days, resulting in complete resolution of the EGR-like eruption; nevertheless, diffuse erythema remained. Subsequently, within 3 months of treatment, the cutaneous T-cell lymphoma (CTCL) improved with continued interferon alfa-2b and methotrexate. Erythroderma became minimal; the circulating Sézary cell count decreased by 50%. The patient ultimately had multiple relapses in erythroderma and progression of SS. Erythema gyratum repens–like lesions recurred on multiple occasions, with a temporary response to repeat courses of oral terbinafine.
Comment
Defining True EGR vs EGR-like Eruption
Sézary syndrome represents the leukemic stage of CTCL, which is defined by the triad of erythroderma; generalized lymphadenopathy; and neoplastic T cells in the skin, lymph nodes, and peripheral blood. It is well known that CTCL can mimic multiple benign and malignant dermatoses. One rare presentation of CTCL is an EGR-like eruption.
Erythema gyratum repens presents as rapidly advancing, erythematous, concentric bands that can be figurate, gyrate, or annular, with a fine trailing edge of scale (wood grain pattern). The diagnosis is based on the characteristic clinical pattern of EGR and by ruling out other mimicking conditions with biopsy.1 Patients with the characteristic clinical pattern but with an alternate underlying dermatosis are described as having an EGR-like eruption rather than true EGR.
True EGR is most often but not always associated with underlying malignancy. Biopsy of true EGR eruptions show nonspecific histopathologic features, with perivascular superficial mononuclear dermatitis, occasional mild spongiosis, and focal parakeratosis; specific features of an alternate dermatosis are lacking.2 In addition to CTCL, EGR-like eruptions have been described in a number of diseases, including systemic lupus erythematosus, erythema annulare centrifugum, bullous dermatosis, erythrokeratodermia variabilis, urticarial vasculitis, leukocytoclastic vasculitis, and neutrophilic dermatoses.
Prior Reports of EGR-like Eruption in Association With MF
According to a PubMed search of articles indexed for MEDLINE using the terms erythema gyratum repens in mycosis fungoides, mycosis fungoides with tinea, and concentric wood grain erythema, there have been 6 other cases of an EGR-like eruption in association with MF (Table). Poonawalla et al3 first described an EGR-like eruption (utilizing the term tinea pseudoimbricata) in a 55-year-old man with stage IB MF (T2N0M0B0). The patient had a preceding history of tinea pedis and tinea corporis that preceded the diagnosis of MF. At the time of MF diagnosis, the patient presented with extensive concentric, gyrate, wood grain, annular lesions. His MF was resistant to topical mechlorethamine, psoralen plus UVA, and oral bexarotene. The body surface area involvement decreased from 60% to less than 1% after institution of oral and topical antifungal therapy. It was postulated that the widespread dermatophytosis that preceded the development of MF may have been the persistent antigen leading to his disease. Preceding the diagnosis of MF, skin scrapings were floridly positive for dermatophyte hyphae. Fungal cultures from the affected areas of skin grew T rubrum.3
Moore et al4 described an EGR-like eruption on the trunk of a 73-year-old man with stage IA MF (T1N0M0B0). Biopsy was consistent with MF, but no fungal organisms were seen. Potassium hydroxide preparation and fungal cultures of the lesions also were negative for organisms. The patient was successfully treated with topical betamethasone.4Jouary et al5 described an EGR-like eruption in a 77-year-old man with stage III erythrodermic MF (T4N1M0B0). Biopsy showed mycelia on PAS stain. Subsequent culture isolated T rubrum. Terbinafine (250 mg/d) and ketoconazole cream 2% daily were initiated and the patient’s EGR-like rash quickly cleared, while MF progressed to SS.5
Cerri et al6 later described a case of EGR-like eruption in a 61-year-old man with stage I MF and an EGR-like eruption. Microscopic examination of potassium hydroxide (KOH) preparations and fungal culture of the lesions failed to demonstrate mycotic infection. There was no mention of PAS stain of skin biopsy specimens. In this case, the authors mentioned that EGR-like lesions preceded exacerbation of MF and questioned the prognostic significance of the EGR-like eruption in relation to MF.6
Holcomb et al7 reported the next case of a 75-year-old man with stage IIB MF (T3N0M0B0) with CD25+ and CD30+ large cell transformation who presented with an EGR-like eruption. In this case, PAS stain and KOH preparations were repeatedly negative for mycotic infection. Disease progression was not mentioned following the appearance of the EGR-like eruption.7
Nagase et al8 most recently described a case of a 73-year-old Japanese man with stage IB (T2N0M0B0) CD4−CD8− MF and lung cancer who developed a cutaneous eruption mimicking EGR. Microscopy and culture excluded the presence of a mycotic infection. The patient achieved partial remission with photochemotherapy (psoralen plus UVA) combined with topical corticosteroids. No major changes in the patient’s skin lesions were noted following surgical resection of the lung cancer.8
Dermatophyte Infection
It is known that conventional tinea corporis can occur in the setting of CTCL. However, EGR-like eruptions in CTCL can be distinguished from standard tinea corporis by the classic morphology of EGR and clinical history of rapid migration of these characteristic lesions.
Tinea imbricata is known to have a clinical appearance that is similar to EGR, but the infection is caused by Tinea concentricum, which is limited to southwest Polynesia, Melanesia, Southeast Asia, India, and Central America. Although T rubrum was the dermatophyte isolated by Poonawalla et al,3 Jouary et al,5 and in our case, whether T rubrum infection in the setting of CTCL has any impact on prognosis needs further study.
Our case of an EGR-like eruption presented in a patient with SS and tinea corporis. Biopsy specimens showed CTCL and concomitant dermatophytic infection that was confirmed with PAS stain and identified as T rubrum. Interestingly, our patient’s EGR-like eruption cleared with oral terbinafine therapy, consistent with findings described by Poonawalla et al3 and Jouary et al5 in which treatment of the dermatophytic infection led to resolution of the EGR-like eruption, suggesting a causative role.
However, testing for dermatophytes was negative in the other reported cases of EGR-like eruptions in patients with MF, despite screening for the presence of fungal microorganisms using KOH preparation, PAS staining, or fungal culture, or a combination of these methods,3-8 which raises the question: Do the cases reported without dermatophytic infection represent false-negative test results, or can the distinct clinical appearance of EGR indeed be seen in patients with CTCL who lack superimposed dermatophytosis? In 3 prior reported cases of EGR-like eruptions in MF, the eruption was preceded by immunosuppressive therapy.5-7
Further investigation is needed to correlate the role of dermatophytic infection in EGR-like eruptions. Our case and the Jouary et al5 case reported dermatophyte-positive EGR-like eruptions in MF and SS detected with histopathologic analysis and PAS stain. This low-cost screening method should be considered in future cases. If the test result is dermatophyte positive, a 14-day course of oral terbinafine (250 mg/d) might induce resolution of the EGR-like eruption.
Conclusion
The role of dermatophyte-induced EGR or EGR-like eruptions in other settings also warrants further investigation to shed light on this poorly understood yet striking dermatologic condition. Our patient showed both MF and dermatophytes in skin biopsy results, regardless of whether those sites showed erythroderma or EGR-like features clinically. On 3 occasions, antifungal treatment cleared the EGR-like lesions and associated pruritus but not erythroderma. Therefore, it appears that the mere presence of dermatophytes was necessary but not sufficient to produce the EGR-like lesions observed in our case.
Case Report
A 65-year-old woman presented with stage IVA2 mycosis fungoides (MF)(T4N3M0B2)/Sézary syndrome (SS). A peripheral blood count contained 6000 Sézary cells with cerebriform nuclei, a CD2+/−CD3+CD4+CD5+/−CD7+CD8−CD26−immunophenotype, and a highly abnormal CD4 to CD8 ratio (70:1). Positron emission tomography and computed tomography demonstrated hypermetabolic subcutaneous nodules in the base of the neck and generalized lymphadenopathy. Lymph node biopsy showed involvement by T-cell lymphoma and dominant T-cell receptor γ clonality by polymerase chain reaction.
On initial presentation to the Cutaneous Lymphoma Clinic at the University of Wisconsin-Madison, the patient was erythrodermic. She also was noted to have undulating wavy bands and concentric annular, ringlike, thin, erythematous plaques with trailing scale, giving a wood grain, zebra hide–like appearance involving the buttocks, abdomen, and lower extremities (Figure 1). Lesions were markedly pruritic and were advancing rapidly. A diagnosis of erythema gyratum repens (EGR)–like eruption was made.
Biopsy of an EGR-like area on the leg showed a superficial perivascular and somewhat lichenoid lymphoid infiltrate (Figure 2). Lymphocytes were lined up along the basal layer, occasionally forming nests within the epidermis. Nearly all mononuclear cells in the epidermis and dermis exhibited positive CD3 and CD4 staining, with only scattered CD8 cells. These features were compatible with cutaneous involvement in SS. A concurrent biopsy from diffusely erythrodermic forearm skin, which lacked EGR-like morphology, showed similar histopathologic and immunophenotypic features.
Periodic acid–Schiff (PAS) with diastase stain revealed numerous septate hyphae within the stratum corneum in both skin biopsy specimens (Figure 3). Fungal culture of EGR-like lesions was positive for a nonsporulating filamentous fungus, identified as Trichophyton rubrum by DNA sequencing.
A diagnosis of EGR-like eruption secondary to tinea corporis in SS was made. The possibility of tinea incognito also was considered to explain the presence of dermatophytes in the biopsy from skin that exhibited only erythroderma clinically; however, the patient did not have a history of corticosteroid use.
Interferon alfa-2b and methotrexate therapy was initiated. Additionally, oral terbinafine (250 mg/d) was initiated for 14 days, resulting in complete resolution of the EGR-like eruption; nevertheless, diffuse erythema remained. Subsequently, within 3 months of treatment, the cutaneous T-cell lymphoma (CTCL) improved with continued interferon alfa-2b and methotrexate. Erythroderma became minimal; the circulating Sézary cell count decreased by 50%. The patient ultimately had multiple relapses in erythroderma and progression of SS. Erythema gyratum repens–like lesions recurred on multiple occasions, with a temporary response to repeat courses of oral terbinafine.
Comment
Defining True EGR vs EGR-like Eruption
Sézary syndrome represents the leukemic stage of CTCL, which is defined by the triad of erythroderma; generalized lymphadenopathy; and neoplastic T cells in the skin, lymph nodes, and peripheral blood. It is well known that CTCL can mimic multiple benign and malignant dermatoses. One rare presentation of CTCL is an EGR-like eruption.
Erythema gyratum repens presents as rapidly advancing, erythematous, concentric bands that can be figurate, gyrate, or annular, with a fine trailing edge of scale (wood grain pattern). The diagnosis is based on the characteristic clinical pattern of EGR and by ruling out other mimicking conditions with biopsy.1 Patients with the characteristic clinical pattern but with an alternate underlying dermatosis are described as having an EGR-like eruption rather than true EGR.
True EGR is most often but not always associated with underlying malignancy. Biopsy of true EGR eruptions show nonspecific histopathologic features, with perivascular superficial mononuclear dermatitis, occasional mild spongiosis, and focal parakeratosis; specific features of an alternate dermatosis are lacking.2 In addition to CTCL, EGR-like eruptions have been described in a number of diseases, including systemic lupus erythematosus, erythema annulare centrifugum, bullous dermatosis, erythrokeratodermia variabilis, urticarial vasculitis, leukocytoclastic vasculitis, and neutrophilic dermatoses.
Prior Reports of EGR-like Eruption in Association With MF
According to a PubMed search of articles indexed for MEDLINE using the terms erythema gyratum repens in mycosis fungoides, mycosis fungoides with tinea, and concentric wood grain erythema, there have been 6 other cases of an EGR-like eruption in association with MF (Table). Poonawalla et al3 first described an EGR-like eruption (utilizing the term tinea pseudoimbricata) in a 55-year-old man with stage IB MF (T2N0M0B0). The patient had a preceding history of tinea pedis and tinea corporis that preceded the diagnosis of MF. At the time of MF diagnosis, the patient presented with extensive concentric, gyrate, wood grain, annular lesions. His MF was resistant to topical mechlorethamine, psoralen plus UVA, and oral bexarotene. The body surface area involvement decreased from 60% to less than 1% after institution of oral and topical antifungal therapy. It was postulated that the widespread dermatophytosis that preceded the development of MF may have been the persistent antigen leading to his disease. Preceding the diagnosis of MF, skin scrapings were floridly positive for dermatophyte hyphae. Fungal cultures from the affected areas of skin grew T rubrum.3
Moore et al4 described an EGR-like eruption on the trunk of a 73-year-old man with stage IA MF (T1N0M0B0). Biopsy was consistent with MF, but no fungal organisms were seen. Potassium hydroxide preparation and fungal cultures of the lesions also were negative for organisms. The patient was successfully treated with topical betamethasone.4Jouary et al5 described an EGR-like eruption in a 77-year-old man with stage III erythrodermic MF (T4N1M0B0). Biopsy showed mycelia on PAS stain. Subsequent culture isolated T rubrum. Terbinafine (250 mg/d) and ketoconazole cream 2% daily were initiated and the patient’s EGR-like rash quickly cleared, while MF progressed to SS.5
Cerri et al6 later described a case of EGR-like eruption in a 61-year-old man with stage I MF and an EGR-like eruption. Microscopic examination of potassium hydroxide (KOH) preparations and fungal culture of the lesions failed to demonstrate mycotic infection. There was no mention of PAS stain of skin biopsy specimens. In this case, the authors mentioned that EGR-like lesions preceded exacerbation of MF and questioned the prognostic significance of the EGR-like eruption in relation to MF.6
Holcomb et al7 reported the next case of a 75-year-old man with stage IIB MF (T3N0M0B0) with CD25+ and CD30+ large cell transformation who presented with an EGR-like eruption. In this case, PAS stain and KOH preparations were repeatedly negative for mycotic infection. Disease progression was not mentioned following the appearance of the EGR-like eruption.7
Nagase et al8 most recently described a case of a 73-year-old Japanese man with stage IB (T2N0M0B0) CD4−CD8− MF and lung cancer who developed a cutaneous eruption mimicking EGR. Microscopy and culture excluded the presence of a mycotic infection. The patient achieved partial remission with photochemotherapy (psoralen plus UVA) combined with topical corticosteroids. No major changes in the patient’s skin lesions were noted following surgical resection of the lung cancer.8
Dermatophyte Infection
It is known that conventional tinea corporis can occur in the setting of CTCL. However, EGR-like eruptions in CTCL can be distinguished from standard tinea corporis by the classic morphology of EGR and clinical history of rapid migration of these characteristic lesions.
Tinea imbricata is known to have a clinical appearance that is similar to EGR, but the infection is caused by Tinea concentricum, which is limited to southwest Polynesia, Melanesia, Southeast Asia, India, and Central America. Although T rubrum was the dermatophyte isolated by Poonawalla et al,3 Jouary et al,5 and in our case, whether T rubrum infection in the setting of CTCL has any impact on prognosis needs further study.
Our case of an EGR-like eruption presented in a patient with SS and tinea corporis. Biopsy specimens showed CTCL and concomitant dermatophytic infection that was confirmed with PAS stain and identified as T rubrum. Interestingly, our patient’s EGR-like eruption cleared with oral terbinafine therapy, consistent with findings described by Poonawalla et al3 and Jouary et al5 in which treatment of the dermatophytic infection led to resolution of the EGR-like eruption, suggesting a causative role.
However, testing for dermatophytes was negative in the other reported cases of EGR-like eruptions in patients with MF, despite screening for the presence of fungal microorganisms using KOH preparation, PAS staining, or fungal culture, or a combination of these methods,3-8 which raises the question: Do the cases reported without dermatophytic infection represent false-negative test results, or can the distinct clinical appearance of EGR indeed be seen in patients with CTCL who lack superimposed dermatophytosis? In 3 prior reported cases of EGR-like eruptions in MF, the eruption was preceded by immunosuppressive therapy.5-7
Further investigation is needed to correlate the role of dermatophytic infection in EGR-like eruptions. Our case and the Jouary et al5 case reported dermatophyte-positive EGR-like eruptions in MF and SS detected with histopathologic analysis and PAS stain. This low-cost screening method should be considered in future cases. If the test result is dermatophyte positive, a 14-day course of oral terbinafine (250 mg/d) might induce resolution of the EGR-like eruption.
Conclusion
The role of dermatophyte-induced EGR or EGR-like eruptions in other settings also warrants further investigation to shed light on this poorly understood yet striking dermatologic condition. Our patient showed both MF and dermatophytes in skin biopsy results, regardless of whether those sites showed erythroderma or EGR-like features clinically. On 3 occasions, antifungal treatment cleared the EGR-like lesions and associated pruritus but not erythroderma. Therefore, it appears that the mere presence of dermatophytes was necessary but not sufficient to produce the EGR-like lesions observed in our case.
- Rongioletti F, Fausti V, Parodi A. Erythema gyratum repens is not an obligate paraneoplastic disease: a systematic review of the literature and personal experience. J Eur Acad Dermatol Venereol. 2012;28:112-115.
- Albers SE, Fenske NA, Glass LF. Erythema gyratum repens: direct immunofluorescence microscopic findings. J Am Acad Dermatol. 1993;29:493-494.
- Poonawalla T, Chen W, Duvic M. Mycosis fungoides with tinea pseudoimbricata owing to Trichophyton rubrum infection. J Cutan Med Surg. 2006;10:52-56.
- Moore E, McFarlane R, Olerud J. Concentric wood grain erythema on the trunk. Arch Dermatol. 2008;144:673-678.
- Jouary T, Lalanne N, Stanislas S, et al. Erythema gyratum repens-like eruption in mycosis fungoides: is dermatophyte superinfection underdiagnosed in cutaneous T-cell lymphomas? J Eur Acad Dermatol Venereol. 2008;22:1276-1278.
- Cerri A, Vezzoli P, Serini SM, et al. Mycosis fungoides mimicking erythema gyratum repens: an additional variant? Eur J Dermatol. 2010;20:540-541.
- Holcomb M, Duvic M, Cutlan J. Erythema gyratum repens-like eruptions with large cell transformation in a patient with mycosis fungoides. Int J Dermatol. 2012;51:1231-1233.
- Nagase K, Shirai R, Okawa T, et al. CD4/CD8 double-negative mycosis fungoides mimicking erythema gyratum repens in a patient with underlying lung cancer. Acta Derm Venereol. 2014;94:89-90.
- Rongioletti F, Fausti V, Parodi A. Erythema gyratum repens is not an obligate paraneoplastic disease: a systematic review of the literature and personal experience. J Eur Acad Dermatol Venereol. 2012;28:112-115.
- Albers SE, Fenske NA, Glass LF. Erythema gyratum repens: direct immunofluorescence microscopic findings. J Am Acad Dermatol. 1993;29:493-494.
- Poonawalla T, Chen W, Duvic M. Mycosis fungoides with tinea pseudoimbricata owing to Trichophyton rubrum infection. J Cutan Med Surg. 2006;10:52-56.
- Moore E, McFarlane R, Olerud J. Concentric wood grain erythema on the trunk. Arch Dermatol. 2008;144:673-678.
- Jouary T, Lalanne N, Stanislas S, et al. Erythema gyratum repens-like eruption in mycosis fungoides: is dermatophyte superinfection underdiagnosed in cutaneous T-cell lymphomas? J Eur Acad Dermatol Venereol. 2008;22:1276-1278.
- Cerri A, Vezzoli P, Serini SM, et al. Mycosis fungoides mimicking erythema gyratum repens: an additional variant? Eur J Dermatol. 2010;20:540-541.
- Holcomb M, Duvic M, Cutlan J. Erythema gyratum repens-like eruptions with large cell transformation in a patient with mycosis fungoides. Int J Dermatol. 2012;51:1231-1233.
- Nagase K, Shirai R, Okawa T, et al. CD4/CD8 double-negative mycosis fungoides mimicking erythema gyratum repens in a patient with underlying lung cancer. Acta Derm Venereol. 2014;94:89-90.
Practice Points
- Erythema gyratum repens (EGR) presents as rapidly advancing, erythematous, concentric bands that can be figurate, gyrate, or annular, with fine trailing scale.
- Although EGR typically is associated with underlying malignancy, it is not an obligate paraneoplastic syndrome. There are numerous cases that are not associated with underlying neoplasms.
- An EGR-like eruption may be observed in Sézary syndrome, and an overlying superficial dermatophyte infection may play a role.
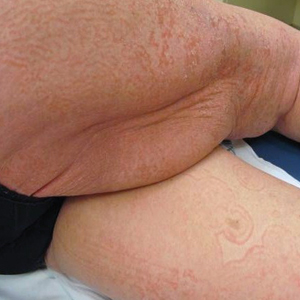
Multiple Subcutaneous Dermoid Cysts
To the Editor:
A 30-year-old man with no notable medical history presented to the dermatology clinic with multiple subcutaneous nodules on the forehead of 5 years’ duration. He reported no history of forehead trauma or manipulation of the lesions, and there was no accompanying pruritis, pain, erythema, or purulent discharge. There was no family history of skin or gastrointestinal tract tumors. On physical examination, the patient had 5 firm, flesh-colored to yellow nodules measuring approximately 0.2 to 1.5 cm in diameter without central punctae scattered over the central forehead (Figure 1). Due to cosmetic concerns, the patient elected to pursue surgical excision of the lesions, which occurred over several office visits. During surgical excision, the lesions were found to be smooth, encapsulated, and mobile, and they were excised without surgical complication. Histopathologic examination showed subcutaneous cysts lined by squamous epithelium with associated sebaceous glands (Figure 2A) and hair follicles in the cyst lumen (Figure 2B). These findings confirmed the diagnosis of multiple subcutaneous dermoid cysts.
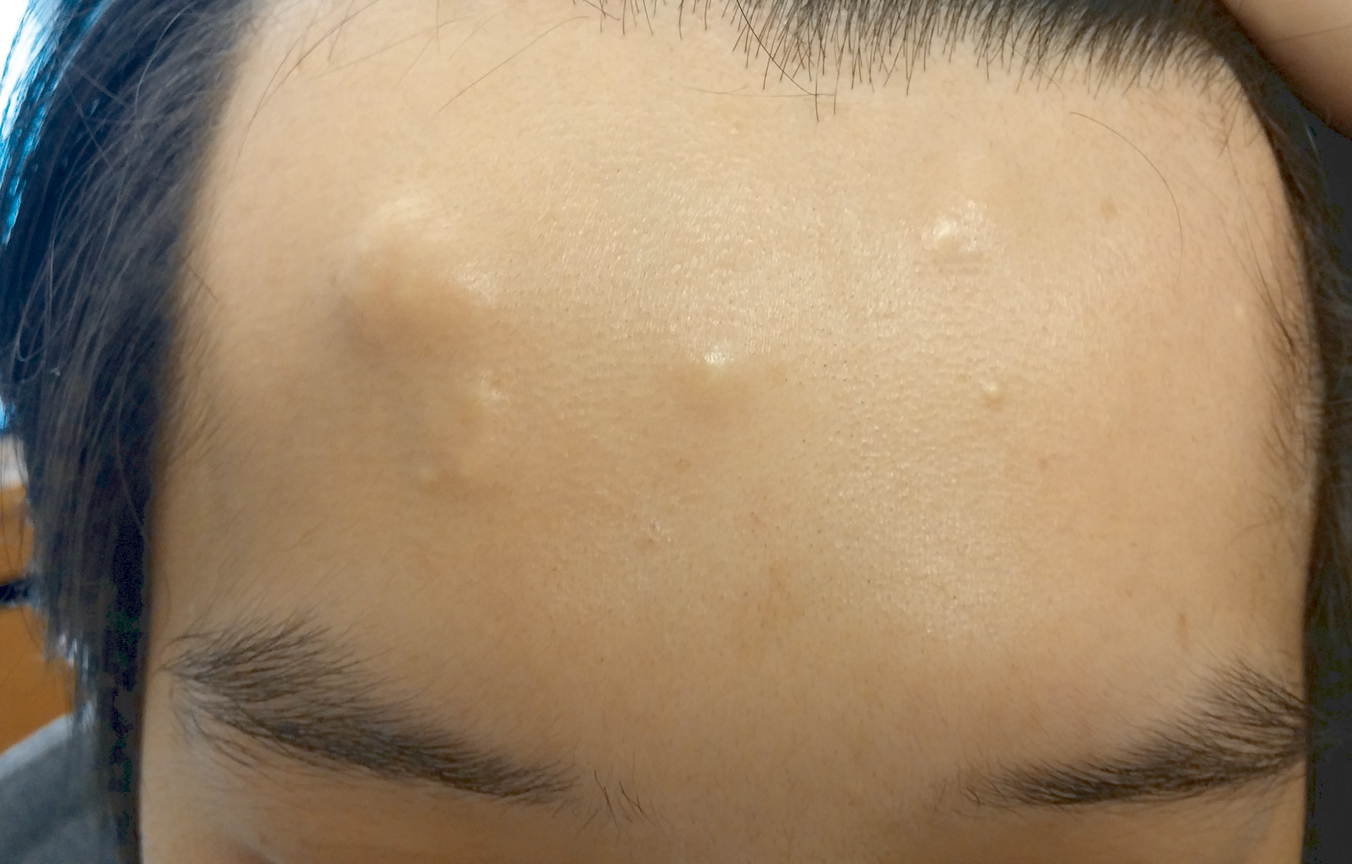
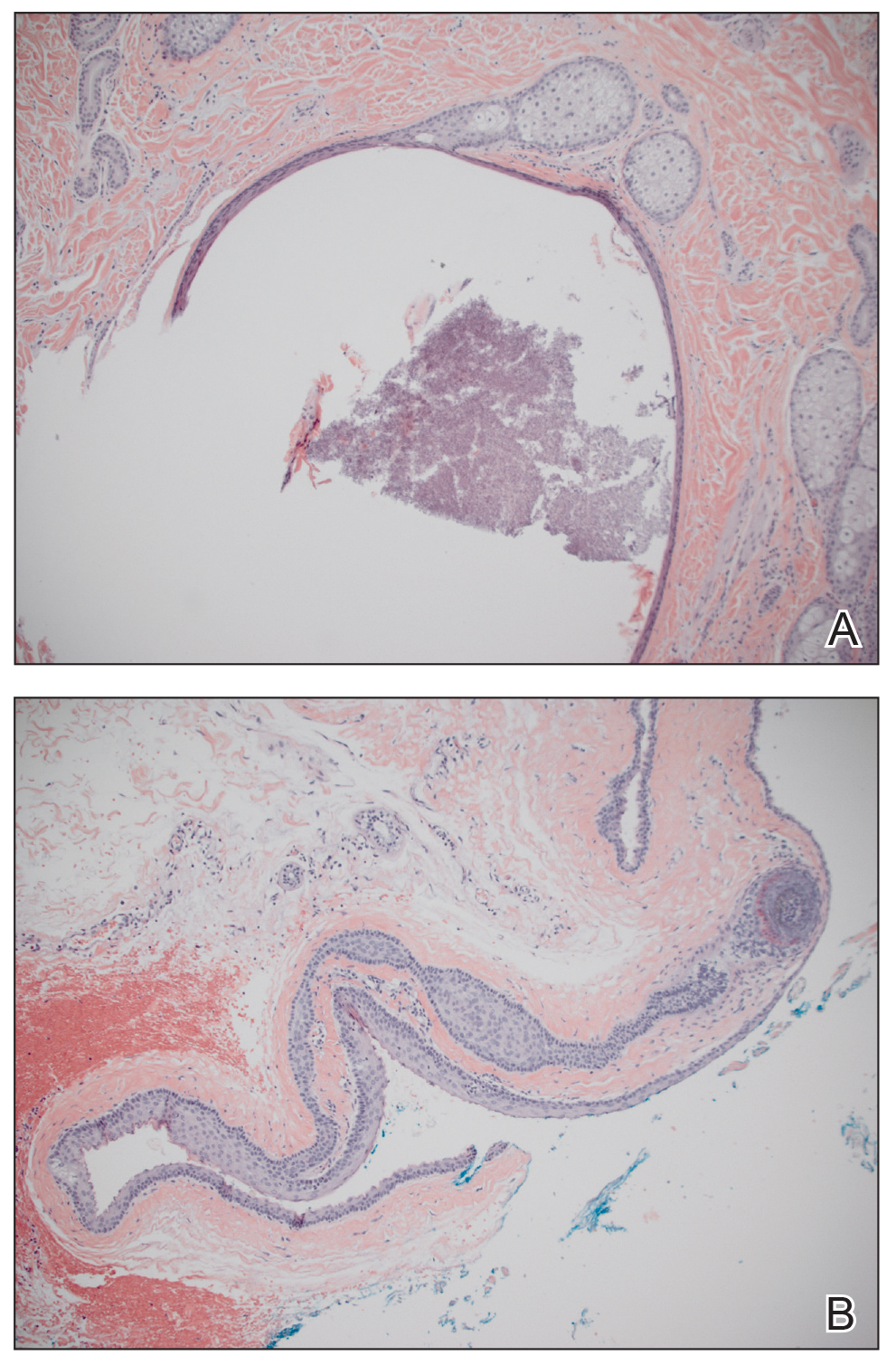
Dermoid cysts are relatively uncommon, benign tumors consisting of tissue derived from ectodermal and mesodermal germ cell layers. Dermoid cysts may be distinguished from teratomas, which may contain tissues derived from all 3 germ cell layers and typically consist of types of tissues foreign to the site of origin, such as dental, thyroid, gastrointestinal, or neural tissue.1,2 The majority of dermoid cysts are congenitally developed along the lines of embryologic fusion due to an error in the division of the ectoderm and mesoderm3,4; however, some dermoid cysts may be acquired from epidermal elements being traumatically implanted into the dermis.5
Our patient’s presentation with multiple dermoid cysts was atypical, as dermoid cysts are almost always solitary tumors. A similar case was reported in a 41-year-old man who developed multiple dermoid cysts on the forehead over a 20-year period.This patient also was otherwise healthy, denied prior trauma to the forehead, and reported no family history of skin or gastrointestinal tract tumors.5
Another unusual feature in our case was the location of the dermoid cysts on the central forehead. The most common location for dermoid cysts is the lateral third of the eyebrows (47%–70% of cases).1,4,6-10 These cysts occur because of sequestration of the surface ectoderm during fusion along the naso-optic groove.2 Dermoid cysts also have been noted in other anatomical areas such as the scalp, nose, anterior neck, and trunk.6
Dermoid cysts tend to be small, round, smooth, and slowly growing until sudden enlargement prompts surgical evaluation.4,6 During surgical excision, they often are fixed to the underlying bone but also may be freely mobile, as in our patient.6 Histopathologic examination reveals a stratified squamous epithelium with associated adnexal structures such as sebaceous glands or hair follicles.1 Smooth muscle fibers, prominent vascular stroma, small nerves, and collagen and elastic fibers also may be found within the lumen of dermoid cysts.2
In some cases, dermoid cysts may be invasive and carry the risk of bony erosion, intracranial extension, osteomyelitis, meningitis, or cerebral abscess. Imaging studies sometimes are needed to rule out intracranial or intraspinal extension, particularly for midline dermoid cysts.6 The standard of treatment for dermoid cysts is surgical excision and complete enucleation without disruption of the cyst wall; however, invasive dermoid cysts may require endoscopic excision, orbitotomy, or craniotomy.4,6
- Brownstein MH, Helwig EB. Subcutaneous dermoid cysts. Arch Dermatol. 1973;107:237-239.
- Smirniotopoulos JG, Chiechi MV. Teratomas, dermoids, and epidermoids of the head and neck. Radiographics. 1995;15:1437-1455.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Yamaki T, Higuchi R, Sasaki K, et al. Multiple dermoid cysts on the forehead. case report. Scand J Plast Reconstr Surg Hand Surg. 1996;30:321-324.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52-57.
- McAvoy JM, Zuckerbraun L. Dermoid cysts of the head and neck in children. Arch Otolaryngol. 1976;102:529-531.
- Taylor BW, Erich JB, Dockerty MB. Dermoids of the head and neck. Minnesota Med. 1966;49:1535-1540.
- Golden BA, Zide MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613-1619.
To the Editor:
A 30-year-old man with no notable medical history presented to the dermatology clinic with multiple subcutaneous nodules on the forehead of 5 years’ duration. He reported no history of forehead trauma or manipulation of the lesions, and there was no accompanying pruritis, pain, erythema, or purulent discharge. There was no family history of skin or gastrointestinal tract tumors. On physical examination, the patient had 5 firm, flesh-colored to yellow nodules measuring approximately 0.2 to 1.5 cm in diameter without central punctae scattered over the central forehead (Figure 1). Due to cosmetic concerns, the patient elected to pursue surgical excision of the lesions, which occurred over several office visits. During surgical excision, the lesions were found to be smooth, encapsulated, and mobile, and they were excised without surgical complication. Histopathologic examination showed subcutaneous cysts lined by squamous epithelium with associated sebaceous glands (Figure 2A) and hair follicles in the cyst lumen (Figure 2B). These findings confirmed the diagnosis of multiple subcutaneous dermoid cysts.
Dermoid cysts are relatively uncommon, benign tumors consisting of tissue derived from ectodermal and mesodermal germ cell layers. Dermoid cysts may be distinguished from teratomas, which may contain tissues derived from all 3 germ cell layers and typically consist of types of tissues foreign to the site of origin, such as dental, thyroid, gastrointestinal, or neural tissue.1,2 The majority of dermoid cysts are congenitally developed along the lines of embryologic fusion due to an error in the division of the ectoderm and mesoderm3,4; however, some dermoid cysts may be acquired from epidermal elements being traumatically implanted into the dermis.5
Our patient’s presentation with multiple dermoid cysts was atypical, as dermoid cysts are almost always solitary tumors. A similar case was reported in a 41-year-old man who developed multiple dermoid cysts on the forehead over a 20-year period.This patient also was otherwise healthy, denied prior trauma to the forehead, and reported no family history of skin or gastrointestinal tract tumors.5
Another unusual feature in our case was the location of the dermoid cysts on the central forehead. The most common location for dermoid cysts is the lateral third of the eyebrows (47%–70% of cases).1,4,6-10 These cysts occur because of sequestration of the surface ectoderm during fusion along the naso-optic groove.2 Dermoid cysts also have been noted in other anatomical areas such as the scalp, nose, anterior neck, and trunk.6
Dermoid cysts tend to be small, round, smooth, and slowly growing until sudden enlargement prompts surgical evaluation.4,6 During surgical excision, they often are fixed to the underlying bone but also may be freely mobile, as in our patient.6 Histopathologic examination reveals a stratified squamous epithelium with associated adnexal structures such as sebaceous glands or hair follicles.1 Smooth muscle fibers, prominent vascular stroma, small nerves, and collagen and elastic fibers also may be found within the lumen of dermoid cysts.2
In some cases, dermoid cysts may be invasive and carry the risk of bony erosion, intracranial extension, osteomyelitis, meningitis, or cerebral abscess. Imaging studies sometimes are needed to rule out intracranial or intraspinal extension, particularly for midline dermoid cysts.6 The standard of treatment for dermoid cysts is surgical excision and complete enucleation without disruption of the cyst wall; however, invasive dermoid cysts may require endoscopic excision, orbitotomy, or craniotomy.4,6
To the Editor:
A 30-year-old man with no notable medical history presented to the dermatology clinic with multiple subcutaneous nodules on the forehead of 5 years’ duration. He reported no history of forehead trauma or manipulation of the lesions, and there was no accompanying pruritis, pain, erythema, or purulent discharge. There was no family history of skin or gastrointestinal tract tumors. On physical examination, the patient had 5 firm, flesh-colored to yellow nodules measuring approximately 0.2 to 1.5 cm in diameter without central punctae scattered over the central forehead (Figure 1). Due to cosmetic concerns, the patient elected to pursue surgical excision of the lesions, which occurred over several office visits. During surgical excision, the lesions were found to be smooth, encapsulated, and mobile, and they were excised without surgical complication. Histopathologic examination showed subcutaneous cysts lined by squamous epithelium with associated sebaceous glands (Figure 2A) and hair follicles in the cyst lumen (Figure 2B). These findings confirmed the diagnosis of multiple subcutaneous dermoid cysts.
Dermoid cysts are relatively uncommon, benign tumors consisting of tissue derived from ectodermal and mesodermal germ cell layers. Dermoid cysts may be distinguished from teratomas, which may contain tissues derived from all 3 germ cell layers and typically consist of types of tissues foreign to the site of origin, such as dental, thyroid, gastrointestinal, or neural tissue.1,2 The majority of dermoid cysts are congenitally developed along the lines of embryologic fusion due to an error in the division of the ectoderm and mesoderm3,4; however, some dermoid cysts may be acquired from epidermal elements being traumatically implanted into the dermis.5
Our patient’s presentation with multiple dermoid cysts was atypical, as dermoid cysts are almost always solitary tumors. A similar case was reported in a 41-year-old man who developed multiple dermoid cysts on the forehead over a 20-year period.This patient also was otherwise healthy, denied prior trauma to the forehead, and reported no family history of skin or gastrointestinal tract tumors.5
Another unusual feature in our case was the location of the dermoid cysts on the central forehead. The most common location for dermoid cysts is the lateral third of the eyebrows (47%–70% of cases).1,4,6-10 These cysts occur because of sequestration of the surface ectoderm during fusion along the naso-optic groove.2 Dermoid cysts also have been noted in other anatomical areas such as the scalp, nose, anterior neck, and trunk.6
Dermoid cysts tend to be small, round, smooth, and slowly growing until sudden enlargement prompts surgical evaluation.4,6 During surgical excision, they often are fixed to the underlying bone but also may be freely mobile, as in our patient.6 Histopathologic examination reveals a stratified squamous epithelium with associated adnexal structures such as sebaceous glands or hair follicles.1 Smooth muscle fibers, prominent vascular stroma, small nerves, and collagen and elastic fibers also may be found within the lumen of dermoid cysts.2
In some cases, dermoid cysts may be invasive and carry the risk of bony erosion, intracranial extension, osteomyelitis, meningitis, or cerebral abscess. Imaging studies sometimes are needed to rule out intracranial or intraspinal extension, particularly for midline dermoid cysts.6 The standard of treatment for dermoid cysts is surgical excision and complete enucleation without disruption of the cyst wall; however, invasive dermoid cysts may require endoscopic excision, orbitotomy, or craniotomy.4,6
- Brownstein MH, Helwig EB. Subcutaneous dermoid cysts. Arch Dermatol. 1973;107:237-239.
- Smirniotopoulos JG, Chiechi MV. Teratomas, dermoids, and epidermoids of the head and neck. Radiographics. 1995;15:1437-1455.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Yamaki T, Higuchi R, Sasaki K, et al. Multiple dermoid cysts on the forehead. case report. Scand J Plast Reconstr Surg Hand Surg. 1996;30:321-324.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52-57.
- McAvoy JM, Zuckerbraun L. Dermoid cysts of the head and neck in children. Arch Otolaryngol. 1976;102:529-531.
- Taylor BW, Erich JB, Dockerty MB. Dermoids of the head and neck. Minnesota Med. 1966;49:1535-1540.
- Golden BA, Zide MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613-1619.
- Brownstein MH, Helwig EB. Subcutaneous dermoid cysts. Arch Dermatol. 1973;107:237-239.
- Smirniotopoulos JG, Chiechi MV. Teratomas, dermoids, and epidermoids of the head and neck. Radiographics. 1995;15:1437-1455.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Yamaki T, Higuchi R, Sasaki K, et al. Multiple dermoid cysts on the forehead. case report. Scand J Plast Reconstr Surg Hand Surg. 1996;30:321-324.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52-57.
- McAvoy JM, Zuckerbraun L. Dermoid cysts of the head and neck in children. Arch Otolaryngol. 1976;102:529-531.
- Taylor BW, Erich JB, Dockerty MB. Dermoids of the head and neck. Minnesota Med. 1966;49:1535-1540.
- Golden BA, Zide MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613-1619.
Practice Points
- The majority of dermoid cysts are congenital; however, they may be acquired from traumatic implantation of epidermal elements into the dermis.
- The most common location for dermoid cysts is the lateral third of the eyebrows; however, they also may occur on the mid forehead, scalp, nose, anterior neck, and trunk.
- Imaging studies may be needed to rule out intracranial or intraspinal extension of dermoid cysts, particularly for those presenting in the midline.