User login
Lower BMD found in patients with severe hemophilia A
Men with severe hemophilia A showed reduced levels of bone mineral density, compared with controls representative of the general population, according to findings from a case-control study.
In addition, the decrease in bone mineral density (BMD) was correlated with reduced functional ability and body mass index (BMI), and vitamin D insufficiency or deficiency.
“We aimed to investigate the presence of low BMD in adult patients diagnosed with severe hemophilia A and to evaluate the potential risk factors associated with low BMD and musculoskeletal function levels,” wrote Omer Ekinci, MD, of Firat University in Elazig, Turkey, and colleagues in Haemophilia.
The study included 41 men with severe hemophilia A and 40 men without hemophilia who were matched for age. All patients with hemophilia A received regular prophylactic therapy, and one patient had a high titre (greater than 5 Bethesda units) inhibitor against FVIII.
The researchers performed several laboratory tests: BMD was measured using dual-energy x-ray absorptiometry; BMI was recorded; and laboratory tests were performed to ascertain levels of vitamin D, calcium, phosphorus, alkaline phosphatase, parathyroid hormone, and hepatitis C and HIV antibodies. The Functional Independence Score in Hemophilia (FISH) was used to measure functional-ability status only in the study group.
After analysis, the researchers found a significant difference between patients in the case and control groups for femoral neck and total hip BMD (P = .017 and P less than .001, respectively), but not for lumbar spine BMD (P = .071).
In patients with hemophilia aged younger than 50 years, 27.8% were found to have “low normal” BMD levels, and 19.4% showed “lower than expected” BMD levels with respect to age.
“Vitamin D insufficiency and deficiency were present in 63.4% of the patients with hemophilia, significantly higher than the control group [37.5%; P less than .001],” the researchers wrote.
There were also statistically significant positive correlations between FISH score and femoral neck BMD (P = .001, r = .530), femoral neck z score (P = .001, r = .514), femoral neck T score (P = .002, r = .524), and lumbar spine BMD (P = .033, r = .334). No correlation was found between dual-energy x-ray absorptiometry measurements and the other variables (age, calcium, phosphorus, and alkaline phosphatase levels), and no results were reported for hepatitis C or HIV because none of the participants tested positive for those measures.
The most frequently reported causes of reduced BMD levels was vitamin D deficiency, low BMI, and low functional movement ability, although none of these was a strong independent risk factor in multivariate analysis, the authors reported.
They acknowledged that the results may not be generalizable to all patients because the study was conducted at a single center in Turkey.
“The results of our study emphasize the importance of early detection of comorbid conditions that decrease bone mass in severe hemophilia A patients,” they concluded.
The study was funded by the Yüzüncü Yıl University Scientific Research Project Committee. The authors reported no conflicts of interest.
SOURCE: Ekinci O et al. Haemophilia. 2019 Aug 8. doi: 10.1111/hae.13836.
Men with severe hemophilia A showed reduced levels of bone mineral density, compared with controls representative of the general population, according to findings from a case-control study.
In addition, the decrease in bone mineral density (BMD) was correlated with reduced functional ability and body mass index (BMI), and vitamin D insufficiency or deficiency.
“We aimed to investigate the presence of low BMD in adult patients diagnosed with severe hemophilia A and to evaluate the potential risk factors associated with low BMD and musculoskeletal function levels,” wrote Omer Ekinci, MD, of Firat University in Elazig, Turkey, and colleagues in Haemophilia.
The study included 41 men with severe hemophilia A and 40 men without hemophilia who were matched for age. All patients with hemophilia A received regular prophylactic therapy, and one patient had a high titre (greater than 5 Bethesda units) inhibitor against FVIII.
The researchers performed several laboratory tests: BMD was measured using dual-energy x-ray absorptiometry; BMI was recorded; and laboratory tests were performed to ascertain levels of vitamin D, calcium, phosphorus, alkaline phosphatase, parathyroid hormone, and hepatitis C and HIV antibodies. The Functional Independence Score in Hemophilia (FISH) was used to measure functional-ability status only in the study group.
After analysis, the researchers found a significant difference between patients in the case and control groups for femoral neck and total hip BMD (P = .017 and P less than .001, respectively), but not for lumbar spine BMD (P = .071).
In patients with hemophilia aged younger than 50 years, 27.8% were found to have “low normal” BMD levels, and 19.4% showed “lower than expected” BMD levels with respect to age.
“Vitamin D insufficiency and deficiency were present in 63.4% of the patients with hemophilia, significantly higher than the control group [37.5%; P less than .001],” the researchers wrote.
There were also statistically significant positive correlations between FISH score and femoral neck BMD (P = .001, r = .530), femoral neck z score (P = .001, r = .514), femoral neck T score (P = .002, r = .524), and lumbar spine BMD (P = .033, r = .334). No correlation was found between dual-energy x-ray absorptiometry measurements and the other variables (age, calcium, phosphorus, and alkaline phosphatase levels), and no results were reported for hepatitis C or HIV because none of the participants tested positive for those measures.
The most frequently reported causes of reduced BMD levels was vitamin D deficiency, low BMI, and low functional movement ability, although none of these was a strong independent risk factor in multivariate analysis, the authors reported.
They acknowledged that the results may not be generalizable to all patients because the study was conducted at a single center in Turkey.
“The results of our study emphasize the importance of early detection of comorbid conditions that decrease bone mass in severe hemophilia A patients,” they concluded.
The study was funded by the Yüzüncü Yıl University Scientific Research Project Committee. The authors reported no conflicts of interest.
SOURCE: Ekinci O et al. Haemophilia. 2019 Aug 8. doi: 10.1111/hae.13836.
Men with severe hemophilia A showed reduced levels of bone mineral density, compared with controls representative of the general population, according to findings from a case-control study.
In addition, the decrease in bone mineral density (BMD) was correlated with reduced functional ability and body mass index (BMI), and vitamin D insufficiency or deficiency.
“We aimed to investigate the presence of low BMD in adult patients diagnosed with severe hemophilia A and to evaluate the potential risk factors associated with low BMD and musculoskeletal function levels,” wrote Omer Ekinci, MD, of Firat University in Elazig, Turkey, and colleagues in Haemophilia.
The study included 41 men with severe hemophilia A and 40 men without hemophilia who were matched for age. All patients with hemophilia A received regular prophylactic therapy, and one patient had a high titre (greater than 5 Bethesda units) inhibitor against FVIII.
The researchers performed several laboratory tests: BMD was measured using dual-energy x-ray absorptiometry; BMI was recorded; and laboratory tests were performed to ascertain levels of vitamin D, calcium, phosphorus, alkaline phosphatase, parathyroid hormone, and hepatitis C and HIV antibodies. The Functional Independence Score in Hemophilia (FISH) was used to measure functional-ability status only in the study group.
After analysis, the researchers found a significant difference between patients in the case and control groups for femoral neck and total hip BMD (P = .017 and P less than .001, respectively), but not for lumbar spine BMD (P = .071).
In patients with hemophilia aged younger than 50 years, 27.8% were found to have “low normal” BMD levels, and 19.4% showed “lower than expected” BMD levels with respect to age.
“Vitamin D insufficiency and deficiency were present in 63.4% of the patients with hemophilia, significantly higher than the control group [37.5%; P less than .001],” the researchers wrote.
There were also statistically significant positive correlations between FISH score and femoral neck BMD (P = .001, r = .530), femoral neck z score (P = .001, r = .514), femoral neck T score (P = .002, r = .524), and lumbar spine BMD (P = .033, r = .334). No correlation was found between dual-energy x-ray absorptiometry measurements and the other variables (age, calcium, phosphorus, and alkaline phosphatase levels), and no results were reported for hepatitis C or HIV because none of the participants tested positive for those measures.
The most frequently reported causes of reduced BMD levels was vitamin D deficiency, low BMI, and low functional movement ability, although none of these was a strong independent risk factor in multivariate analysis, the authors reported.
They acknowledged that the results may not be generalizable to all patients because the study was conducted at a single center in Turkey.
“The results of our study emphasize the importance of early detection of comorbid conditions that decrease bone mass in severe hemophilia A patients,” they concluded.
The study was funded by the Yüzüncü Yıl University Scientific Research Project Committee. The authors reported no conflicts of interest.
SOURCE: Ekinci O et al. Haemophilia. 2019 Aug 8. doi: 10.1111/hae.13836.
FROM HAEMOPHILIA
Fatal Drug-Resistant Invasive Pulmonary Aspergillus fumigatus in a 56-Year-Old Immunosuppressed Man (FULL)
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1). 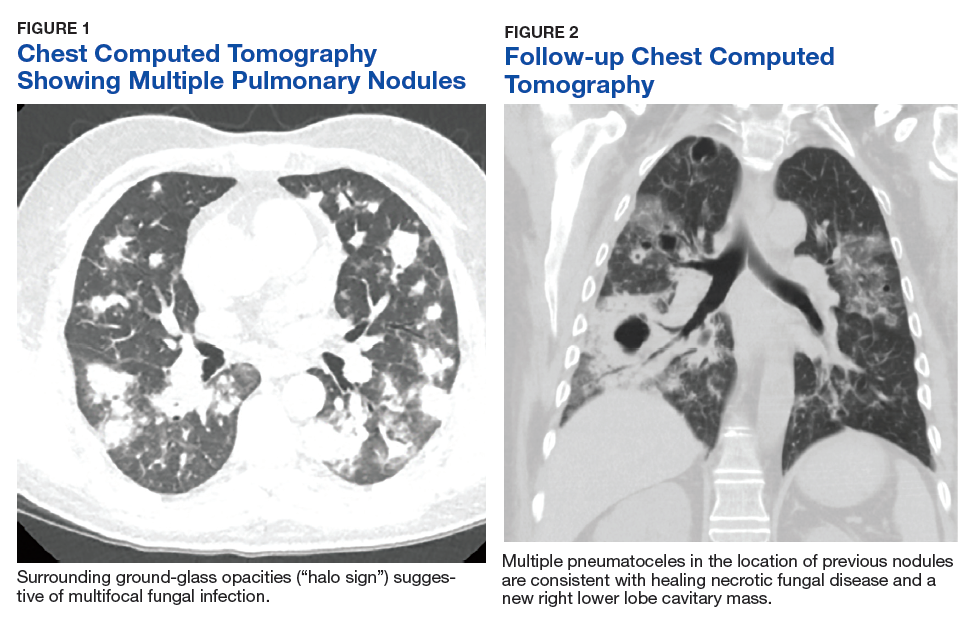
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4). 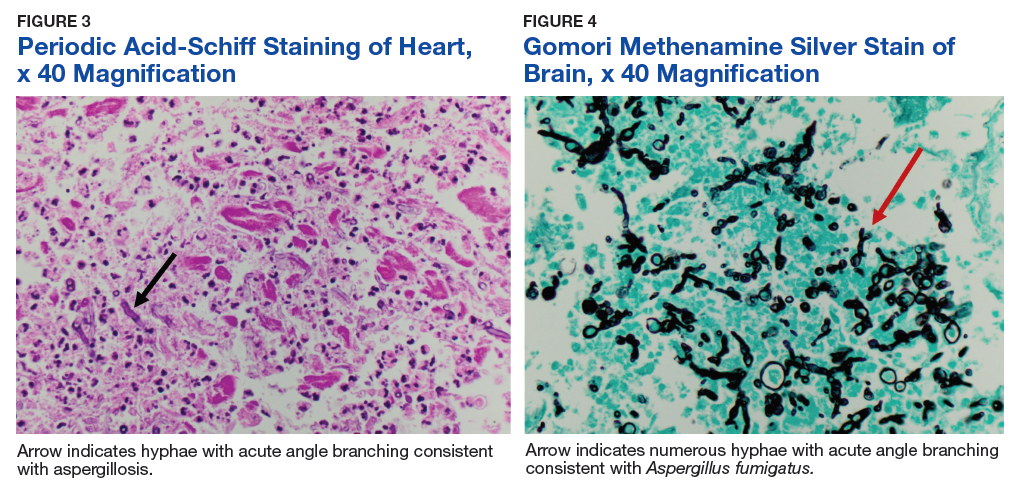
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Medicare’s CAR T-cell coverage decision draws praise, but cost issues linger
Physicians are praising the decision by officials at the Centers for Medicare & Medicaid Services to provide coverage of chimeric antigen receptor T-cell therapy, though they say the planned payment structure will still leave hospitals in the red when treatment is administered.

On Aug. 7, 2019, the agency issued a national coverage determination that outlines Medicare coverage of chimeric antigen receptor (CAR) T-cell therapies when they are provided in health care facilities enrolled in the Food and Drug Administration’s Risk Evaluation and Mitigation Strategies program. Medicare will cover treatments for both FDA-approved indications and off-label uses that are recommended in CMS-approved compendia.
“What you’ve seen in both the [Medicare Inpatient Prospective Payment System] rule, as well as this coverage determination, is recognition by CMS that, with CAR T cells, we are dealing with something different and something extraordinary,” Joseph Alvarnas, MD, vice president of government affairs at City of Hope, Duarte, Calif., said in an interview. “For a lot of patients who suffer with non-Hodgkin’s lymphoma, the consequences of being refractory to standard therapies mean that many patients have few great prospects for moving forward with curative treatments. CAR T cells represent a really innovative set of treatments for patients.”
A proposed national coverage determination, issued in February 2019, would have put in place a coverage with evidence development (CED) requirement for CAR T-cell therapy, covering treatment nationwide if it was offered through CMS-approved registries or clinical studies in which patients were monitored for 2 or more years following treatment.
Physicians applauded the decision not to restrict access by imposing the CED requirement.

“I think what CMS has put out is a good thing,” said Navneet Majhail, MD, director of the Cleveland Clinic’s blood and marrow transplant program, and president of the American Society for Transplantation and Cellular Therapy.
“Both at the Cleveland Clinic level and the society level, we have been asking CMS for something similar and we are really glad and excited that CMS did do this. The concern was that CMS might do this in the context of some other regulatory requirements like CED that they sometimes do. I am glad that CMS decided not to put that mechanism into place for the CAR T-cell therapies,” Dr. Majhail said.
Dr. Alvarnas, who also serves as chair of the American Society of Hematology Committee on Practice, agreed. “I see good. I don’t see bad. I have read through this and it strikes me as being written with fairly great clarity.”
Dr. Alvarnas added that he had been worried about potential restrictions, such as CED. “Once you put something under that whole rubric of coverage with evidence development, then what you do is you create a bottleneck around access to therapy because you have to have an accruing clinical trial for patients to, in fact, be able to participate in that form of therapy.”
By not imposing a CED requirement, it opens the door to better understanding the role CAR T cells play in treatment, Dr. Alvarnas noted.
“Over time, the number of patients for whom these therapeutics work, based upon real medical evidence, will escalate and grow at a pace that can far exceed the restrictions placed under a CED model,” he said, adding that the national coverage determination “gives us the license to deliver therapeutics to the right patients based upon medical evidence as it evolves, provided that these things get listed as part of the compendia. I think that is a fantastic recognition that new roles for drugs, agents, therapeutics ... are going to evolve at a pace far faster than what CMS can write rules about.”
While Medicare’s coverage determination garnered positive reviews, the agency’s Inpatient Prospective Payment System final rule – which outlines reimbursement for CAR T-cell therapy and other new technologies – got a more tepid response.
In the final rule, CMS raised the payment it makes to hospitals for administering CAR T-cell therapies through its new technology add-on payment. Payments will rise from 50% of the technology to 65%, an increase from $186,500 to $242,450 for CAR T-cell therapies, beginning on Oct. 1, 2019.
But even the bump up to 65% may not be enough.
“I see the move to 65% as a new technology add-on payment as an incremental step in the correct direction, but what we’ve done to some extent is that we’ve delayed getting to some sort of more wholly conceived system,” Dr. Alvarnas said, noting that a new system will be needed as the new technology add-on payment goes away in 2021.
Abhinav Deol, MD, of the Karmanos Cancer Institute in Detroit said it’s a challenge to cover costs for the treatment. “If you just look at the simple math, it is still going to be an economic challenge. The cells that are approved for lymphoma patients that will probably fall into the Medicare category, the list price of those cells is $373,000. Even with the 65% coverage, it’s about $235,000-$240,000 in reimbursement,” he said. “For a facility to be able to provide the care for patients, you have that delta that is still not covered. It is still going to be an economic challenge for many of the facilities to provide this care.”

Thomas LeBlanc, MD, an associate professor of medicine at Duke University, Durham, N.C., said that, while the coverage determination is a positive step, it’s not clear that it will provide meaningful access to CAR T-cell therapy because of the cost.
“These products are incredibly expensive, and the total cost of providing them is woefully underestimated in only focusing on the sticker price of the product,” he said. “Doing so ignores the significant hospital care, sometimes even critical care, as well as specialized knowledge and high touch supportive care, all of which is required to safely get patients through this revolutionary yet often risky treatment. So when CMS offers to pay just 65% of the sticker price, I suspect that many institutions will still lose six figures for each patient treated.”
Dr. LeBlanc predicted that many centers will decline to provide CAR T-cell therapy despite the increase in the new technology add-on payment, though he added that “I’d love to be wrong about this.”
Dr. Majhail agreed, noting that, even with the bump in the add-on payment, hospitals “won’t be whole in terms of providing care for these patients.”
“The reimbursement piece continues to be a challenge,” he said. “It is better than what it was, but there is still more work to be done. That is something we will have to keep working with the agency on.”
Physicians are praising the decision by officials at the Centers for Medicare & Medicaid Services to provide coverage of chimeric antigen receptor T-cell therapy, though they say the planned payment structure will still leave hospitals in the red when treatment is administered.
On Aug. 7, 2019, the agency issued a national coverage determination that outlines Medicare coverage of chimeric antigen receptor (CAR) T-cell therapies when they are provided in health care facilities enrolled in the Food and Drug Administration’s Risk Evaluation and Mitigation Strategies program. Medicare will cover treatments for both FDA-approved indications and off-label uses that are recommended in CMS-approved compendia.
“What you’ve seen in both the [Medicare Inpatient Prospective Payment System] rule, as well as this coverage determination, is recognition by CMS that, with CAR T cells, we are dealing with something different and something extraordinary,” Joseph Alvarnas, MD, vice president of government affairs at City of Hope, Duarte, Calif., said in an interview. “For a lot of patients who suffer with non-Hodgkin’s lymphoma, the consequences of being refractory to standard therapies mean that many patients have few great prospects for moving forward with curative treatments. CAR T cells represent a really innovative set of treatments for patients.”
A proposed national coverage determination, issued in February 2019, would have put in place a coverage with evidence development (CED) requirement for CAR T-cell therapy, covering treatment nationwide if it was offered through CMS-approved registries or clinical studies in which patients were monitored for 2 or more years following treatment.
Physicians applauded the decision not to restrict access by imposing the CED requirement.
“I think what CMS has put out is a good thing,” said Navneet Majhail, MD, director of the Cleveland Clinic’s blood and marrow transplant program, and president of the American Society for Transplantation and Cellular Therapy.
“Both at the Cleveland Clinic level and the society level, we have been asking CMS for something similar and we are really glad and excited that CMS did do this. The concern was that CMS might do this in the context of some other regulatory requirements like CED that they sometimes do. I am glad that CMS decided not to put that mechanism into place for the CAR T-cell therapies,” Dr. Majhail said.
Dr. Alvarnas, who also serves as chair of the American Society of Hematology Committee on Practice, agreed. “I see good. I don’t see bad. I have read through this and it strikes me as being written with fairly great clarity.”
Dr. Alvarnas added that he had been worried about potential restrictions, such as CED. “Once you put something under that whole rubric of coverage with evidence development, then what you do is you create a bottleneck around access to therapy because you have to have an accruing clinical trial for patients to, in fact, be able to participate in that form of therapy.”
By not imposing a CED requirement, it opens the door to better understanding the role CAR T cells play in treatment, Dr. Alvarnas noted.
“Over time, the number of patients for whom these therapeutics work, based upon real medical evidence, will escalate and grow at a pace that can far exceed the restrictions placed under a CED model,” he said, adding that the national coverage determination “gives us the license to deliver therapeutics to the right patients based upon medical evidence as it evolves, provided that these things get listed as part of the compendia. I think that is a fantastic recognition that new roles for drugs, agents, therapeutics ... are going to evolve at a pace far faster than what CMS can write rules about.”
While Medicare’s coverage determination garnered positive reviews, the agency’s Inpatient Prospective Payment System final rule – which outlines reimbursement for CAR T-cell therapy and other new technologies – got a more tepid response.
In the final rule, CMS raised the payment it makes to hospitals for administering CAR T-cell therapies through its new technology add-on payment. Payments will rise from 50% of the technology to 65%, an increase from $186,500 to $242,450 for CAR T-cell therapies, beginning on Oct. 1, 2019.
But even the bump up to 65% may not be enough.
“I see the move to 65% as a new technology add-on payment as an incremental step in the correct direction, but what we’ve done to some extent is that we’ve delayed getting to some sort of more wholly conceived system,” Dr. Alvarnas said, noting that a new system will be needed as the new technology add-on payment goes away in 2021.
Abhinav Deol, MD, of the Karmanos Cancer Institute in Detroit said it’s a challenge to cover costs for the treatment. “If you just look at the simple math, it is still going to be an economic challenge. The cells that are approved for lymphoma patients that will probably fall into the Medicare category, the list price of those cells is $373,000. Even with the 65% coverage, it’s about $235,000-$240,000 in reimbursement,” he said. “For a facility to be able to provide the care for patients, you have that delta that is still not covered. It is still going to be an economic challenge for many of the facilities to provide this care.”
Thomas LeBlanc, MD, an associate professor of medicine at Duke University, Durham, N.C., said that, while the coverage determination is a positive step, it’s not clear that it will provide meaningful access to CAR T-cell therapy because of the cost.
“These products are incredibly expensive, and the total cost of providing them is woefully underestimated in only focusing on the sticker price of the product,” he said. “Doing so ignores the significant hospital care, sometimes even critical care, as well as specialized knowledge and high touch supportive care, all of which is required to safely get patients through this revolutionary yet often risky treatment. So when CMS offers to pay just 65% of the sticker price, I suspect that many institutions will still lose six figures for each patient treated.”
Dr. LeBlanc predicted that many centers will decline to provide CAR T-cell therapy despite the increase in the new technology add-on payment, though he added that “I’d love to be wrong about this.”
Dr. Majhail agreed, noting that, even with the bump in the add-on payment, hospitals “won’t be whole in terms of providing care for these patients.”
“The reimbursement piece continues to be a challenge,” he said. “It is better than what it was, but there is still more work to be done. That is something we will have to keep working with the agency on.”
Physicians are praising the decision by officials at the Centers for Medicare & Medicaid Services to provide coverage of chimeric antigen receptor T-cell therapy, though they say the planned payment structure will still leave hospitals in the red when treatment is administered.
On Aug. 7, 2019, the agency issued a national coverage determination that outlines Medicare coverage of chimeric antigen receptor (CAR) T-cell therapies when they are provided in health care facilities enrolled in the Food and Drug Administration’s Risk Evaluation and Mitigation Strategies program. Medicare will cover treatments for both FDA-approved indications and off-label uses that are recommended in CMS-approved compendia.
“What you’ve seen in both the [Medicare Inpatient Prospective Payment System] rule, as well as this coverage determination, is recognition by CMS that, with CAR T cells, we are dealing with something different and something extraordinary,” Joseph Alvarnas, MD, vice president of government affairs at City of Hope, Duarte, Calif., said in an interview. “For a lot of patients who suffer with non-Hodgkin’s lymphoma, the consequences of being refractory to standard therapies mean that many patients have few great prospects for moving forward with curative treatments. CAR T cells represent a really innovative set of treatments for patients.”
A proposed national coverage determination, issued in February 2019, would have put in place a coverage with evidence development (CED) requirement for CAR T-cell therapy, covering treatment nationwide if it was offered through CMS-approved registries or clinical studies in which patients were monitored for 2 or more years following treatment.
Physicians applauded the decision not to restrict access by imposing the CED requirement.
“I think what CMS has put out is a good thing,” said Navneet Majhail, MD, director of the Cleveland Clinic’s blood and marrow transplant program, and president of the American Society for Transplantation and Cellular Therapy.
“Both at the Cleveland Clinic level and the society level, we have been asking CMS for something similar and we are really glad and excited that CMS did do this. The concern was that CMS might do this in the context of some other regulatory requirements like CED that they sometimes do. I am glad that CMS decided not to put that mechanism into place for the CAR T-cell therapies,” Dr. Majhail said.
Dr. Alvarnas, who also serves as chair of the American Society of Hematology Committee on Practice, agreed. “I see good. I don’t see bad. I have read through this and it strikes me as being written with fairly great clarity.”
Dr. Alvarnas added that he had been worried about potential restrictions, such as CED. “Once you put something under that whole rubric of coverage with evidence development, then what you do is you create a bottleneck around access to therapy because you have to have an accruing clinical trial for patients to, in fact, be able to participate in that form of therapy.”
By not imposing a CED requirement, it opens the door to better understanding the role CAR T cells play in treatment, Dr. Alvarnas noted.
“Over time, the number of patients for whom these therapeutics work, based upon real medical evidence, will escalate and grow at a pace that can far exceed the restrictions placed under a CED model,” he said, adding that the national coverage determination “gives us the license to deliver therapeutics to the right patients based upon medical evidence as it evolves, provided that these things get listed as part of the compendia. I think that is a fantastic recognition that new roles for drugs, agents, therapeutics ... are going to evolve at a pace far faster than what CMS can write rules about.”
While Medicare’s coverage determination garnered positive reviews, the agency’s Inpatient Prospective Payment System final rule – which outlines reimbursement for CAR T-cell therapy and other new technologies – got a more tepid response.
In the final rule, CMS raised the payment it makes to hospitals for administering CAR T-cell therapies through its new technology add-on payment. Payments will rise from 50% of the technology to 65%, an increase from $186,500 to $242,450 for CAR T-cell therapies, beginning on Oct. 1, 2019.
But even the bump up to 65% may not be enough.
“I see the move to 65% as a new technology add-on payment as an incremental step in the correct direction, but what we’ve done to some extent is that we’ve delayed getting to some sort of more wholly conceived system,” Dr. Alvarnas said, noting that a new system will be needed as the new technology add-on payment goes away in 2021.
Abhinav Deol, MD, of the Karmanos Cancer Institute in Detroit said it’s a challenge to cover costs for the treatment. “If you just look at the simple math, it is still going to be an economic challenge. The cells that are approved for lymphoma patients that will probably fall into the Medicare category, the list price of those cells is $373,000. Even with the 65% coverage, it’s about $235,000-$240,000 in reimbursement,” he said. “For a facility to be able to provide the care for patients, you have that delta that is still not covered. It is still going to be an economic challenge for many of the facilities to provide this care.”
Thomas LeBlanc, MD, an associate professor of medicine at Duke University, Durham, N.C., said that, while the coverage determination is a positive step, it’s not clear that it will provide meaningful access to CAR T-cell therapy because of the cost.
“These products are incredibly expensive, and the total cost of providing them is woefully underestimated in only focusing on the sticker price of the product,” he said. “Doing so ignores the significant hospital care, sometimes even critical care, as well as specialized knowledge and high touch supportive care, all of which is required to safely get patients through this revolutionary yet often risky treatment. So when CMS offers to pay just 65% of the sticker price, I suspect that many institutions will still lose six figures for each patient treated.”
Dr. LeBlanc predicted that many centers will decline to provide CAR T-cell therapy despite the increase in the new technology add-on payment, though he added that “I’d love to be wrong about this.”
Dr. Majhail agreed, noting that, even with the bump in the add-on payment, hospitals “won’t be whole in terms of providing care for these patients.”
“The reimbursement piece continues to be a challenge,” he said. “It is better than what it was, but there is still more work to be done. That is something we will have to keep working with the agency on.”
An unusual cause of bruising
A 61-year-old woman presented to our hematology clinic for evaluation of multiple episodes of bruising. The first episode occurred 8 months earlier, when she developed a large bruise after water skiing. Two months before coming to us, she went to her local emergency room because of new bruising and was found to have a prolonged activated partial thromboplastin time (aPTT) of 60 seconds (reference range 23.3–34.9), but she underwent no further testing at that time.
At presentation to our clinic, she reported having no fevers, night sweats, unintentional weight loss, swollen lymph nodes, joint pain, rashes, mouth sores, nosebleeds, or blood in the urine or stool. Her history was notable only for hypothyroidism, which was diagnosed in the previous year. Her medications included levothyroxine, vitamin D3, and vitamin C. She had been taking a baby aspirin daily for the past 10 years but had stopped 1 month earlier because of the bruising.

Ten years earlier she had been evaluated for a possible transient ischemic attack; laboratory results at that time included a normal aPTT of 25.1 seconds and a normal factor VIII level of 153% (reference range 50%–173%).


EVALUATION FOR AN ISOLATED PROLONGED aPTT
1. What is the appropriate next test to evaluate this patient’s prolonged aPTT?
- Lupus anticoagulant panel
- Coagulation factor levels
- Mixing studies
- Bethesda assay
Mixing studies
Once a prolonged aPTT is confirmed, the appropriate next step is a mixing study. This involves mixing the patient’s plasma with pooled normal plasma in a 1-to-1 ratio, then repeating the aPTT test immediately, and again after 1 hour of incubation at 37°C. If the patient does not have enough of one of the coagulation factors, the aPTT immediately returns to the normal range when plasma is mixed with the pooled plasma because the pooled plasma contains the factor that is lacking. If this happens, then factor assays should be performed to identify the deficient factor.1
Various antibodies that inhibit coagulation factors can also affect the aPTT. There are 2 general types: immediate-acting and delayed.
With an immediate-acting inhibitor, the aPTT does not correct into the normal range with initial mixing. Immediate-acting inhibitors are often seen together with lupus anticoagulants, which are nonspecific phospholipid antibodies. If an immediate-acting inhibitor is detected, further testing should focus on evaluation for lupus anticoagulant, including phospholipid-dependency studies.
With a delayed inhibitor, the aPTT initially comes down, but subsequently goes back up after incubation. Acquired factor VIII inhibitor is a classic delayed-type inhibitor and is also the most common factor inhibitor.1 If a delayed-acting inhibitor is found, specific intrinsic factor levels should be measured (factors VIII, IX, XI, and XII),2 and testing should also be done for lupus anticoagulant, as these inhibitors may occur together.
Bethesda assay

Case continued: Results of mixing and Bethesda studies

FACTOR VIII INHIBITOR EVALUATION
2. What is the most likely underlying condition associated with this patient’s factor VIII inhibitor?
- Autoimmune disease
- Malignancy
- A medication
- Unknown (idiopathic)
Acquired hemophilia A (AHA) is a rare disorder caused by autoantibodies against factor VIII. Its estimated incidence is about 1 person per million per year.4 It usually presents as unexplained bruising or bleeding and is only rarely diagnosed by an incidentally noted prolonged aPTT. The severity of bleeding is variable and can include subcutaneous, soft-tissue, retroperitoneal, gastrointestinal, and intracranial hemorrhage.5
AHA is considered idiopathic in more than half of cases. A study based on a European registry5 of 501 patients with AHA and a UK study6 of 172 patients found no underlying disease in 52% and 65% of patients, respectively. For patients with an identified cause, the most common causes were malignancy (12%5 and 15%6) and autoimmune disease (12%5 and 17%6).
Drugs have rarely been associated with factor VIII inhibitors. Such occurrences have been reported with interferon, blood thinners, antibiotics, and psychiatric medications, but no study yet has indicated causation. However, patients with congenital hemophilia A treated with factor VIII preparations have about a 15% chance of developing factor VIII inhibitors. In this setting, inhibitors develop in response to recombinant factor VIII exposure, unlike the autoimmune phenomena seen in AHA.
TREATMENT OF ACQUIRED HEMOPHILIA A
3. What is the most appropriate treatment for AHA?
- Desmopressin and prednisone
- Recombinant porcine factor VIII and prednisone plus cyclophosphamide
- Recombinant factor VIIa and rituximab
- Any of the above
Any of the above regimens can be used. In general, treatment of AHA has two purposes: to stop acute hemorrhage, and to reduce the level of factor VIII inhibitor. No standard treatment guidelines are available; evidence of the effectiveness of different drugs is based largely on data on congenital hemophilia A.3
Acute treatment to stop bleeding
Initial treatment of AHA often focuses on stopping an acute hemorrhage by either raising circulating levels of factor VIII or bypassing it in the coagulation cascade.
Desmopressin can temporarily raise factor VIII levels, but it is often ineffective in AHA unless the patient has very low inhibitor titers.3
Factor VIII concentrate (human or recombinant porcine factor VIII) may be effective in patients with low inhibitor titers (< 5 BU). Higher doses are often required than those used in congenital hemophilia A. Factor VIII concentrate is usually combined with immunosuppressive treatment to lower the factor VIII inhibitor level (described below).3
If these methods are ineffective or the patient has high inhibitor titers (> 5 BU), activated prothrombin complex concentrates, known as FEIBA (factor eight inhibitor bypassing activity), or recombinant factor VIIa is available. These agents bypass factor VIII in the clotting cascade.
Immunosuppression to reduce factor VIII inhibitor
Immunosuppressive agents are the mainstay of AHA treatment to lower the inhibitor level.
Regimens vary. A 2003 meta-analysis4 including 249 patients found that prednisone alone resulted in complete response in about 30% of patients, and the addition of cyclophosphamide increased the response rate to 60% to 100%. High-dose intravenous immunoglobulin led to conflicting results. Conclusions were limited by the variability of dosing and duration in treatment regimens among the 20 different studies included.
An analysis of 331 patients in the European Acquired Hemophilia Registry (EACH2)7 found that steroids alone produced remission in 48% of patients, while steroids combined with cyclophosphamide raised the rate to 70%. Rituximab-based regimens were successful in 59% but required twice as long to achieve remission as steroid or cyclophosphamide-based regimens. No benefit was noted from intravenous immunoglobulin.
Risks of disease and treatment
AHA is associated with significant risk of morbidity and death related to bleeding, complications of treatment, and underlying disease.
In EACH2, 16 of the 331 patients died of bleeding, 16 died of causes related to immunosuppression, and 45 died of causes related to the underlying condition.5 In the UK registry of 172 patients, 13 patients died of bleeding, and 12 died of sepsis related to immunosuppression.6
The factor VIII level and inhibitor titer are not necessarily useful in stratifying bleeding risk, as severe and fatal bleeding can occur at variable levels and patients remain at risk of bleeding as long as the inhibitor persists.6,7
CASE CONTINUED: TREATMENT, LYMPHOCYTOSIS
The patient was started on 60 mg daily of prednisone, resulting in a decrease in her aPTT, increase in factor VIII level, and lower Bethesda titer. On a return visit, her absolute lymphocyte count was 7.04 × 109/L (reference range 1.0–4.0). She reported no fevers, chills, or recent infections.
EVALUATING LYMPHOCYTOSIS
Lymphocytosis is defined in most laboratories as an absolute lymphocyte count greater than 4.0 × 109/L for adults. Normally, T cells (CD3+) make up 60% to 80% of lymphocytes, B cells (CD20+) 10% to 20%, and natural killer (NK) cells (CD3–, CD56+) 5% to 10%. Lymphocytosis is usually caused by infection, but it can have other causes, including malignancy.
Peripheral blood smear. If there is no clear cause of lymphocytosis, a peripheral blood smear can be used to assess lymphocyte morphology, providing clues to the underlying etiology. For example, atypical lymphocytes are often seen in infectious mononucleosis, while “smudge” lymphocytes are characteristic of chronic lymphocytic leukemia. If a peripheral smear shows abnormal morphology, further workup should include establishing whether the lymphocytes are polyclonal or clonal.8
CASE CONTINUED: LARGE GRANULAR LYMPHOCYTES

4. What is the next step to evaluate the patient’s lymphocytosis?
- Bone marrow biopsy
- Karyotype analysis
- Flow cytometry
- Fluorescence in situ hybridization
Flow cytometry with V-beta analysis is the best first test to determine the cause of lymphocytosis after review of the peripheral smear. For persistent lymphocytosis, flow cytometry should be done even if a peripheral smear shows normal lymphocyte morphology.
Most T cells possess receptors composed of alpha and beta chains, each encoded by variable (V), diversity (D), joining (J), and constant (C) gene segments. The V, D, and J segments undergo rearrangement during T-cell development in the thymus based on antigen exposure, producing a diverse T-cell receptor population.
In a polyclonal population of lymphocytes, the T-cell receptors have a variety of gene segment arrangements, indicating normal T-cell development. But in a clonal population of lymphocytes, the T-cell receptors have a single identical gene segment arrangement, indicating they all originated from a single clone.9 Lymphocytosis in response to an infection is typically polyclonal, while malignant lymphocytosis is clonal.
Monoclonal antibodies against many of the variable regions of the beta chain (V-beta) of T-cell receptors have been developed, enabling flow cytometry to establish clonality.
T-cell receptor gene rearrangement studies can also be performed using polymerase chain reaction and Southern blot techniques.9
Karyotype analysis is usually not performed for the finding of LGLs, because most leukemias (eg, T-cell and NK-cell leukemias) have cells with a normal karyotype.
Bone marrow biopsy is invasive and usually not required to evaluate LGLs. It can be especially risky for a patient with a bleeding disorder such as a factor VIII inhibitor.10
Case continued: Flow cytometry confirms clonality
Subsequent flow cytometry found that more than 50% of the patient’s lymphocytes were LGLs that co-expressed CD3+, CD8+, CD56+, and CD57+, with aberrantly decreased CD7 expression. T-cell V-beta analysis demonstrated an expansion of the V-beta 17 family, and T-cell receptor gene analysis with polymerase chain reaction confirmed the presence of a clonal rearrangement.
LGL LEUKEMIA: CLASSIFICATION AND MANAGEMENT
LGLs normally account for 10% to 15% of peripheral mononuclear cells.11 LGL leukemia is caused by a clonal population of cytotoxic T cells or NK cells and involves an increased number of LGLs (usually > 2 × 109/L).10
LGL leukemia is divided into 3 categories according to the most recent World Health Organization classification10,12:
T-cell LGL leukemia (about 85% of cases) is considered indolent but can cause significant cytopenias and is often associated with autoimmune disease.13 Cells usually express a CD3+, CD8+, CD16+, and CD57+ phenotype. Survival is about 70% at 10 years.
Chronic NK-cell lymphocytosis (about 10%) also tends to have an indolent course with cytopenia and an autoimmune association, and with a similar prognosis to T-cell LGL leukemia. Cells express a CD3–, CD16+, and CD56+ phenotype.
Aggressive NK-cell LGL leukemia (about 5%) is associated with Epstein-Barr virus infection and occurs in younger patients. It is characterized by severe cytopenias, “B symptoms” (ie, fever, night sweats, weight loss), and has a very poor prognosis. Like chronic NK-cell lymphocytosis, cells express a CD3–, CD16+, and CD56+ phenotype. Fas (CD95) and Fas-ligand (CD178) are strongly expressed.10,13
Most cases of LGL leukemia can be diagnosed on the basis of classic morphology on peripheral blood smear and evidence of clonality on flow cytometry or gene rearrangement studies. T-cell receptor gene studies cannot be used to establish clonality in the NK subtypes, as NK cells do not express T-cell receptors.11
Case continued: Diagnosis, continued course
In our patient, T-cell LGL leukemia was diagnosed on the basis of the peripheral smear, flow cytometry results, and positive T-cell receptor gene studies for clonal rearrangement in the T-cell receptor beta region.
While her corticosteroid therapy was being tapered, her factor III inhibitor level increased, and she had a small episode of bleeding, prompting the start of cyclophosphamide 50 mg daily with lower doses of prednisone.

LGL LEUKEMIA AND AUTOIMMUNE DISEASE
Patients with LGL leukemia commonly have or develop autoimmune conditions. Immune-mediated cytopenias including pure red cell aplasia, aplastic anemia, and autoimmune hemolytic anemias can occur. Neutropenia, the most common cytopenia in LGL leukemia, is thought to be at least partly autoimmune, as the degree of neutropenia is often worse than would be expected solely from bone-marrow infiltration of LGL cells.10,14,15
Rheumatoid arthritis is the most common autoimmune condition associated with LGL leukemia, with a reported incidence between 11% and 36%.13–15
Felty syndrome (rheumatoid arthritis, splenomegaly, and neutropenia) is often associated with LGL leukemia and is thought by some to be part of the same disease process.15
Treat with immunosuppressives if needed
Indications for treating LGL leukemia include the development of cytopenias and associated autoimmune diseases. Immunosuppressive agents, such as methotrexate, cyclophosphamide, and cyclosporine, are commonly used.10,11,14 Most evidence of treatment efficacy is from retrospective studies and case reports, with widely variable response rates that overall are around 50%.10
ACQUIRED HEMOPHILIA A AND HEMATOLOGIC MALIGNANCY
A systematic review found 30 cases of AHA associated with hematologic malignancies.16 The largest case series17 in this analysis had 8 patients, and included diagnoses of chronic lymphocytic leukemia, erythroleukemia, myelofibrosis, multiple myeloma, and myelodysplastic syndrome. In 3 of these patients, the appearance of the inhibitor preceded the diagnosis of the underlying malignancy by an average of 3.5 months. In 1 patient with erythroleukemia and another with multiple myeloma, the activity of the inhibitor could be clearly correlated with the underlying malignancy. In the other 6 patients, no association between the two could be made.
In the same series, complete resolution of the inhibitor was related only to the level of Bethesda titer present at diagnosis, with those who achieved resolution having lower mean Bethesda titers.17 Similarly, in EACH2, lower inhibitor Bethesda titers and higher factor VIII levels at presentation were associated with faster inhibitor eradication and normalization of factor VIII levels.7
Murphy et al18 described a 62-year-old woman with Felty syndrome who developed a factor VIII inhibitor and was subsequently given a diagnosis of LGL leukemia. Treatment with immunosuppressive agents, including cyclophosphamide, azathioprine, and rituximab, successfully eradicated her factor VIII inhibitor, although the LGL leukemia persisted.
Case conclusion: Eradication of factor VIII inhibitor
Our patient, similar to the patient described by Murphy et al18 above, had eradication of the factor VIII inhibitor despite persistence of LGL leukemia. Between the time of diagnosis at our clinic, when she had 54% LGLs, and eradication of the inhibitor 3 months later, the LGL percentage ranged from 45% to 89%. No clear direct correlation between LGL and factor VIII inhibitor levels could be detected.
Given the strong association of LGL leukemia with autoimmune disease, it is tempting to believe that her factor VIII inhibitor was somehow related to her malignancy, although the exact mechanism remained unclear. The average age at diagnosis is 60 for LGL leukemia11 and over 70 for AHA,5,6 so advanced age may be the common denominator. Whether or not our patient will have recurrence of her factor VIII inhibitor or the development of other autoimmune diseases with the persistence of her LGL leukemia remains to be seen.
At last follow-up, our patient was off all therapy and continued to have normal aPTT and factor VIII levels. Repeat flow cytometry after treatment of her factor VIII inhibitor showed persistence of a clonal T-cell population, although reduced from 72% to 60%. It may be that the 2 entities were unrelated, and the clonal T-cell population was simply fluctuating over time. This can be determined only with further observation. As the patient had no symptoms from her LGL leukemia, she continued to be observed without treatment.
TAKE-HOME POINTS
- The coagulation assay is key to initially assessing a bleeding abnormality; whether the prothrombin time and aPTT are normal or prolonged narrows the differential diagnosis and determines next steps in evaluation.
- Mixing studies can help pinpoint the responsible deficient factor.
- Acquired factor VIII deficiency, also known as AHA, may be caused by autoimmune disease, malignancy, or medications, but it is usually idiopathic.
- AHA treatment is focused on achieving hemostasis and reducing factor VIII inhibitor.
- Lymphocytosis should be evaluated with a peripheral blood smear and flow cytometry to determine if the population is polyclonal (associated with infection) or clonal (associated with malignancy).
- LGL leukemia is usually a chronic, indolent disease, although an uncommon subtype has an aggressive course.
- The association between AHA and LGL leukemia is unclear, and both conditions must be monitored and managed.
- Kamal AH, Tefferi A, Pruthi RK. How to interpret and pursue an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time in adults. Mayo Clin Proc 2007; 82(7):864–873. doi:10.4065/82.7.864
- Tcherniantchouk O, Laposata M, Marques MB. The isolated prolonged PTT. Am J Hematol 2013; 88(1):82–85. doi:10.1002/ajh.23285
- Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program 2006:432–437. doi:10.1182/asheducation-2006.1.432
- Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol 2003; 121(1):21–35. pmid:12670328
- Knoebl P, Marco P, Baudo F, et al; EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10(4):622–631. doi:10.1111/j.1538-7836.2012.04654.x
- Collins PW, Hirsch S, Baglin TP, et al; UK Haemophilia Centre Doctors’ Organisation. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007; 109(5):1870–1877. doi:10.1182/blood-2006-06-029850
- Collins P, Baudo F, Knoebl P, et al; EACH2 Registry Collaborators. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood 2012; 120(1):47–55. doi:10.1182/blood-2012-02-409185
- George TI. Malignant or benign leukocytosis. Hematology Am Soc Hematol Educ Program 2012; 2012:475–484. doi:10.1182/asheducation-2012.1.475
- Watters RJ, Liu X, Loughran TP Jr. T-cell and natural killer-cell large granular lymphocyte leukemia neoplasias. Leuk Lymphoma 2011; 52(12):2217–2225. doi:10.3109/10428194.2011.593276
- Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood 2017; 129(9):1082–1094. doi:10.1182/blood-2016-08-692590
- Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program 2012; 2012:652–659. doi:10.1182/asheducation-2012.1.652
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127(20):2375–2390. doi:10.1182/blood-2016-01-643569
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist 2004; 9(3):247–258. pmid:15169980
- Bockorny B, Dasanu CA. Autoimmune manifestations in large granular lymphocyte leukemia. Clin Lymphoma Myeloma Leuk 2012; 12(6):400–405. doi:10.1016/j.clml.2012.06.006
- Liu X, Loughran TP Jr. The spectrum of large granular lymphocyte leukemia and Felty’s syndrome. Curr Opin Hematol 2011; 18(4):254–259. doi:10.1097/MOH.0b013e32834760fb
- Franchini M, Lippi G. Acquired factor V inhibitors: a systematic review. J Thromb Thrombolysis 2011; 31(4):449–457. doi:10.1007/s11239-010-0529-6
- Sallah S, Nguyen NP, Abdallah JM, Hanrahan LR. Acquired hemophilia in patients with hematologic malignancies. Arch Pathol Lab Med 2000; 124(5):730–734.
- Murphy PW, Brett LK, Verla-Tebit E, Macik BG, Loughran TP Jr. Acquired inhibitors to factor VIII and fibrinogen in the setting of T-cell large granular lymphocyte leukemia: a case report and review of the literature. Blood Coagul Fibrinolysis 2015; 26(2):211–213. doi:10.1097/MBC.0000000000000209
A 61-year-old woman presented to our hematology clinic for evaluation of multiple episodes of bruising. The first episode occurred 8 months earlier, when she developed a large bruise after water skiing. Two months before coming to us, she went to her local emergency room because of new bruising and was found to have a prolonged activated partial thromboplastin time (aPTT) of 60 seconds (reference range 23.3–34.9), but she underwent no further testing at that time.
At presentation to our clinic, she reported having no fevers, night sweats, unintentional weight loss, swollen lymph nodes, joint pain, rashes, mouth sores, nosebleeds, or blood in the urine or stool. Her history was notable only for hypothyroidism, which was diagnosed in the previous year. Her medications included levothyroxine, vitamin D3, and vitamin C. She had been taking a baby aspirin daily for the past 10 years but had stopped 1 month earlier because of the bruising.
Ten years earlier she had been evaluated for a possible transient ischemic attack; laboratory results at that time included a normal aPTT of 25.1 seconds and a normal factor VIII level of 153% (reference range 50%–173%).
EVALUATION FOR AN ISOLATED PROLONGED aPTT
1. What is the appropriate next test to evaluate this patient’s prolonged aPTT?
- Lupus anticoagulant panel
- Coagulation factor levels
- Mixing studies
- Bethesda assay
Mixing studies
Once a prolonged aPTT is confirmed, the appropriate next step is a mixing study. This involves mixing the patient’s plasma with pooled normal plasma in a 1-to-1 ratio, then repeating the aPTT test immediately, and again after 1 hour of incubation at 37°C. If the patient does not have enough of one of the coagulation factors, the aPTT immediately returns to the normal range when plasma is mixed with the pooled plasma because the pooled plasma contains the factor that is lacking. If this happens, then factor assays should be performed to identify the deficient factor.1
Various antibodies that inhibit coagulation factors can also affect the aPTT. There are 2 general types: immediate-acting and delayed.
With an immediate-acting inhibitor, the aPTT does not correct into the normal range with initial mixing. Immediate-acting inhibitors are often seen together with lupus anticoagulants, which are nonspecific phospholipid antibodies. If an immediate-acting inhibitor is detected, further testing should focus on evaluation for lupus anticoagulant, including phospholipid-dependency studies.
With a delayed inhibitor, the aPTT initially comes down, but subsequently goes back up after incubation. Acquired factor VIII inhibitor is a classic delayed-type inhibitor and is also the most common factor inhibitor.1 If a delayed-acting inhibitor is found, specific intrinsic factor levels should be measured (factors VIII, IX, XI, and XII),2 and testing should also be done for lupus anticoagulant, as these inhibitors may occur together.
Bethesda assay
Case continued: Results of mixing and Bethesda studies
FACTOR VIII INHIBITOR EVALUATION
2. What is the most likely underlying condition associated with this patient’s factor VIII inhibitor?
- Autoimmune disease
- Malignancy
- A medication
- Unknown (idiopathic)
Acquired hemophilia A (AHA) is a rare disorder caused by autoantibodies against factor VIII. Its estimated incidence is about 1 person per million per year.4 It usually presents as unexplained bruising or bleeding and is only rarely diagnosed by an incidentally noted prolonged aPTT. The severity of bleeding is variable and can include subcutaneous, soft-tissue, retroperitoneal, gastrointestinal, and intracranial hemorrhage.5
AHA is considered idiopathic in more than half of cases. A study based on a European registry5 of 501 patients with AHA and a UK study6 of 172 patients found no underlying disease in 52% and 65% of patients, respectively. For patients with an identified cause, the most common causes were malignancy (12%5 and 15%6) and autoimmune disease (12%5 and 17%6).
Drugs have rarely been associated with factor VIII inhibitors. Such occurrences have been reported with interferon, blood thinners, antibiotics, and psychiatric medications, but no study yet has indicated causation. However, patients with congenital hemophilia A treated with factor VIII preparations have about a 15% chance of developing factor VIII inhibitors. In this setting, inhibitors develop in response to recombinant factor VIII exposure, unlike the autoimmune phenomena seen in AHA.
TREATMENT OF ACQUIRED HEMOPHILIA A
3. What is the most appropriate treatment for AHA?
- Desmopressin and prednisone
- Recombinant porcine factor VIII and prednisone plus cyclophosphamide
- Recombinant factor VIIa and rituximab
- Any of the above
Any of the above regimens can be used. In general, treatment of AHA has two purposes: to stop acute hemorrhage, and to reduce the level of factor VIII inhibitor. No standard treatment guidelines are available; evidence of the effectiveness of different drugs is based largely on data on congenital hemophilia A.3
Acute treatment to stop bleeding
Initial treatment of AHA often focuses on stopping an acute hemorrhage by either raising circulating levels of factor VIII or bypassing it in the coagulation cascade.
Desmopressin can temporarily raise factor VIII levels, but it is often ineffective in AHA unless the patient has very low inhibitor titers.3
Factor VIII concentrate (human or recombinant porcine factor VIII) may be effective in patients with low inhibitor titers (< 5 BU). Higher doses are often required than those used in congenital hemophilia A. Factor VIII concentrate is usually combined with immunosuppressive treatment to lower the factor VIII inhibitor level (described below).3
If these methods are ineffective or the patient has high inhibitor titers (> 5 BU), activated prothrombin complex concentrates, known as FEIBA (factor eight inhibitor bypassing activity), or recombinant factor VIIa is available. These agents bypass factor VIII in the clotting cascade.
Immunosuppression to reduce factor VIII inhibitor
Immunosuppressive agents are the mainstay of AHA treatment to lower the inhibitor level.
Regimens vary. A 2003 meta-analysis4 including 249 patients found that prednisone alone resulted in complete response in about 30% of patients, and the addition of cyclophosphamide increased the response rate to 60% to 100%. High-dose intravenous immunoglobulin led to conflicting results. Conclusions were limited by the variability of dosing and duration in treatment regimens among the 20 different studies included.
An analysis of 331 patients in the European Acquired Hemophilia Registry (EACH2)7 found that steroids alone produced remission in 48% of patients, while steroids combined with cyclophosphamide raised the rate to 70%. Rituximab-based regimens were successful in 59% but required twice as long to achieve remission as steroid or cyclophosphamide-based regimens. No benefit was noted from intravenous immunoglobulin.
Risks of disease and treatment
AHA is associated with significant risk of morbidity and death related to bleeding, complications of treatment, and underlying disease.
In EACH2, 16 of the 331 patients died of bleeding, 16 died of causes related to immunosuppression, and 45 died of causes related to the underlying condition.5 In the UK registry of 172 patients, 13 patients died of bleeding, and 12 died of sepsis related to immunosuppression.6
The factor VIII level and inhibitor titer are not necessarily useful in stratifying bleeding risk, as severe and fatal bleeding can occur at variable levels and patients remain at risk of bleeding as long as the inhibitor persists.6,7
CASE CONTINUED: TREATMENT, LYMPHOCYTOSIS
The patient was started on 60 mg daily of prednisone, resulting in a decrease in her aPTT, increase in factor VIII level, and lower Bethesda titer. On a return visit, her absolute lymphocyte count was 7.04 × 109/L (reference range 1.0–4.0). She reported no fevers, chills, or recent infections.
EVALUATING LYMPHOCYTOSIS
Lymphocytosis is defined in most laboratories as an absolute lymphocyte count greater than 4.0 × 109/L for adults. Normally, T cells (CD3+) make up 60% to 80% of lymphocytes, B cells (CD20+) 10% to 20%, and natural killer (NK) cells (CD3–, CD56+) 5% to 10%. Lymphocytosis is usually caused by infection, but it can have other causes, including malignancy.
Peripheral blood smear. If there is no clear cause of lymphocytosis, a peripheral blood smear can be used to assess lymphocyte morphology, providing clues to the underlying etiology. For example, atypical lymphocytes are often seen in infectious mononucleosis, while “smudge” lymphocytes are characteristic of chronic lymphocytic leukemia. If a peripheral smear shows abnormal morphology, further workup should include establishing whether the lymphocytes are polyclonal or clonal.8
CASE CONTINUED: LARGE GRANULAR LYMPHOCYTES
4. What is the next step to evaluate the patient’s lymphocytosis?
- Bone marrow biopsy
- Karyotype analysis
- Flow cytometry
- Fluorescence in situ hybridization
Flow cytometry with V-beta analysis is the best first test to determine the cause of lymphocytosis after review of the peripheral smear. For persistent lymphocytosis, flow cytometry should be done even if a peripheral smear shows normal lymphocyte morphology.
Most T cells possess receptors composed of alpha and beta chains, each encoded by variable (V), diversity (D), joining (J), and constant (C) gene segments. The V, D, and J segments undergo rearrangement during T-cell development in the thymus based on antigen exposure, producing a diverse T-cell receptor population.
In a polyclonal population of lymphocytes, the T-cell receptors have a variety of gene segment arrangements, indicating normal T-cell development. But in a clonal population of lymphocytes, the T-cell receptors have a single identical gene segment arrangement, indicating they all originated from a single clone.9 Lymphocytosis in response to an infection is typically polyclonal, while malignant lymphocytosis is clonal.
Monoclonal antibodies against many of the variable regions of the beta chain (V-beta) of T-cell receptors have been developed, enabling flow cytometry to establish clonality.
T-cell receptor gene rearrangement studies can also be performed using polymerase chain reaction and Southern blot techniques.9
Karyotype analysis is usually not performed for the finding of LGLs, because most leukemias (eg, T-cell and NK-cell leukemias) have cells with a normal karyotype.
Bone marrow biopsy is invasive and usually not required to evaluate LGLs. It can be especially risky for a patient with a bleeding disorder such as a factor VIII inhibitor.10
Case continued: Flow cytometry confirms clonality
Subsequent flow cytometry found that more than 50% of the patient’s lymphocytes were LGLs that co-expressed CD3+, CD8+, CD56+, and CD57+, with aberrantly decreased CD7 expression. T-cell V-beta analysis demonstrated an expansion of the V-beta 17 family, and T-cell receptor gene analysis with polymerase chain reaction confirmed the presence of a clonal rearrangement.
LGL LEUKEMIA: CLASSIFICATION AND MANAGEMENT
LGLs normally account for 10% to 15% of peripheral mononuclear cells.11 LGL leukemia is caused by a clonal population of cytotoxic T cells or NK cells and involves an increased number of LGLs (usually > 2 × 109/L).10
LGL leukemia is divided into 3 categories according to the most recent World Health Organization classification10,12:
T-cell LGL leukemia (about 85% of cases) is considered indolent but can cause significant cytopenias and is often associated with autoimmune disease.13 Cells usually express a CD3+, CD8+, CD16+, and CD57+ phenotype. Survival is about 70% at 10 years.
Chronic NK-cell lymphocytosis (about 10%) also tends to have an indolent course with cytopenia and an autoimmune association, and with a similar prognosis to T-cell LGL leukemia. Cells express a CD3–, CD16+, and CD56+ phenotype.
Aggressive NK-cell LGL leukemia (about 5%) is associated with Epstein-Barr virus infection and occurs in younger patients. It is characterized by severe cytopenias, “B symptoms” (ie, fever, night sweats, weight loss), and has a very poor prognosis. Like chronic NK-cell lymphocytosis, cells express a CD3–, CD16+, and CD56+ phenotype. Fas (CD95) and Fas-ligand (CD178) are strongly expressed.10,13
Most cases of LGL leukemia can be diagnosed on the basis of classic morphology on peripheral blood smear and evidence of clonality on flow cytometry or gene rearrangement studies. T-cell receptor gene studies cannot be used to establish clonality in the NK subtypes, as NK cells do not express T-cell receptors.11
Case continued: Diagnosis, continued course
In our patient, T-cell LGL leukemia was diagnosed on the basis of the peripheral smear, flow cytometry results, and positive T-cell receptor gene studies for clonal rearrangement in the T-cell receptor beta region.
While her corticosteroid therapy was being tapered, her factor III inhibitor level increased, and she had a small episode of bleeding, prompting the start of cyclophosphamide 50 mg daily with lower doses of prednisone.
LGL LEUKEMIA AND AUTOIMMUNE DISEASE
Patients with LGL leukemia commonly have or develop autoimmune conditions. Immune-mediated cytopenias including pure red cell aplasia, aplastic anemia, and autoimmune hemolytic anemias can occur. Neutropenia, the most common cytopenia in LGL leukemia, is thought to be at least partly autoimmune, as the degree of neutropenia is often worse than would be expected solely from bone-marrow infiltration of LGL cells.10,14,15
Rheumatoid arthritis is the most common autoimmune condition associated with LGL leukemia, with a reported incidence between 11% and 36%.13–15
Felty syndrome (rheumatoid arthritis, splenomegaly, and neutropenia) is often associated with LGL leukemia and is thought by some to be part of the same disease process.15
Treat with immunosuppressives if needed
Indications for treating LGL leukemia include the development of cytopenias and associated autoimmune diseases. Immunosuppressive agents, such as methotrexate, cyclophosphamide, and cyclosporine, are commonly used.10,11,14 Most evidence of treatment efficacy is from retrospective studies and case reports, with widely variable response rates that overall are around 50%.10
ACQUIRED HEMOPHILIA A AND HEMATOLOGIC MALIGNANCY
A systematic review found 30 cases of AHA associated with hematologic malignancies.16 The largest case series17 in this analysis had 8 patients, and included diagnoses of chronic lymphocytic leukemia, erythroleukemia, myelofibrosis, multiple myeloma, and myelodysplastic syndrome. In 3 of these patients, the appearance of the inhibitor preceded the diagnosis of the underlying malignancy by an average of 3.5 months. In 1 patient with erythroleukemia and another with multiple myeloma, the activity of the inhibitor could be clearly correlated with the underlying malignancy. In the other 6 patients, no association between the two could be made.
In the same series, complete resolution of the inhibitor was related only to the level of Bethesda titer present at diagnosis, with those who achieved resolution having lower mean Bethesda titers.17 Similarly, in EACH2, lower inhibitor Bethesda titers and higher factor VIII levels at presentation were associated with faster inhibitor eradication and normalization of factor VIII levels.7
Murphy et al18 described a 62-year-old woman with Felty syndrome who developed a factor VIII inhibitor and was subsequently given a diagnosis of LGL leukemia. Treatment with immunosuppressive agents, including cyclophosphamide, azathioprine, and rituximab, successfully eradicated her factor VIII inhibitor, although the LGL leukemia persisted.
Case conclusion: Eradication of factor VIII inhibitor
Our patient, similar to the patient described by Murphy et al18 above, had eradication of the factor VIII inhibitor despite persistence of LGL leukemia. Between the time of diagnosis at our clinic, when she had 54% LGLs, and eradication of the inhibitor 3 months later, the LGL percentage ranged from 45% to 89%. No clear direct correlation between LGL and factor VIII inhibitor levels could be detected.
Given the strong association of LGL leukemia with autoimmune disease, it is tempting to believe that her factor VIII inhibitor was somehow related to her malignancy, although the exact mechanism remained unclear. The average age at diagnosis is 60 for LGL leukemia11 and over 70 for AHA,5,6 so advanced age may be the common denominator. Whether or not our patient will have recurrence of her factor VIII inhibitor or the development of other autoimmune diseases with the persistence of her LGL leukemia remains to be seen.
At last follow-up, our patient was off all therapy and continued to have normal aPTT and factor VIII levels. Repeat flow cytometry after treatment of her factor VIII inhibitor showed persistence of a clonal T-cell population, although reduced from 72% to 60%. It may be that the 2 entities were unrelated, and the clonal T-cell population was simply fluctuating over time. This can be determined only with further observation. As the patient had no symptoms from her LGL leukemia, she continued to be observed without treatment.
TAKE-HOME POINTS
- The coagulation assay is key to initially assessing a bleeding abnormality; whether the prothrombin time and aPTT are normal or prolonged narrows the differential diagnosis and determines next steps in evaluation.
- Mixing studies can help pinpoint the responsible deficient factor.
- Acquired factor VIII deficiency, also known as AHA, may be caused by autoimmune disease, malignancy, or medications, but it is usually idiopathic.
- AHA treatment is focused on achieving hemostasis and reducing factor VIII inhibitor.
- Lymphocytosis should be evaluated with a peripheral blood smear and flow cytometry to determine if the population is polyclonal (associated with infection) or clonal (associated with malignancy).
- LGL leukemia is usually a chronic, indolent disease, although an uncommon subtype has an aggressive course.
- The association between AHA and LGL leukemia is unclear, and both conditions must be monitored and managed.
A 61-year-old woman presented to our hematology clinic for evaluation of multiple episodes of bruising. The first episode occurred 8 months earlier, when she developed a large bruise after water skiing. Two months before coming to us, she went to her local emergency room because of new bruising and was found to have a prolonged activated partial thromboplastin time (aPTT) of 60 seconds (reference range 23.3–34.9), but she underwent no further testing at that time.
At presentation to our clinic, she reported having no fevers, night sweats, unintentional weight loss, swollen lymph nodes, joint pain, rashes, mouth sores, nosebleeds, or blood in the urine or stool. Her history was notable only for hypothyroidism, which was diagnosed in the previous year. Her medications included levothyroxine, vitamin D3, and vitamin C. She had been taking a baby aspirin daily for the past 10 years but had stopped 1 month earlier because of the bruising.
Ten years earlier she had been evaluated for a possible transient ischemic attack; laboratory results at that time included a normal aPTT of 25.1 seconds and a normal factor VIII level of 153% (reference range 50%–173%).
EVALUATION FOR AN ISOLATED PROLONGED aPTT
1. What is the appropriate next test to evaluate this patient’s prolonged aPTT?
- Lupus anticoagulant panel
- Coagulation factor levels
- Mixing studies
- Bethesda assay
Mixing studies
Once a prolonged aPTT is confirmed, the appropriate next step is a mixing study. This involves mixing the patient’s plasma with pooled normal plasma in a 1-to-1 ratio, then repeating the aPTT test immediately, and again after 1 hour of incubation at 37°C. If the patient does not have enough of one of the coagulation factors, the aPTT immediately returns to the normal range when plasma is mixed with the pooled plasma because the pooled plasma contains the factor that is lacking. If this happens, then factor assays should be performed to identify the deficient factor.1
Various antibodies that inhibit coagulation factors can also affect the aPTT. There are 2 general types: immediate-acting and delayed.
With an immediate-acting inhibitor, the aPTT does not correct into the normal range with initial mixing. Immediate-acting inhibitors are often seen together with lupus anticoagulants, which are nonspecific phospholipid antibodies. If an immediate-acting inhibitor is detected, further testing should focus on evaluation for lupus anticoagulant, including phospholipid-dependency studies.
With a delayed inhibitor, the aPTT initially comes down, but subsequently goes back up after incubation. Acquired factor VIII inhibitor is a classic delayed-type inhibitor and is also the most common factor inhibitor.1 If a delayed-acting inhibitor is found, specific intrinsic factor levels should be measured (factors VIII, IX, XI, and XII),2 and testing should also be done for lupus anticoagulant, as these inhibitors may occur together.
Bethesda assay
Case continued: Results of mixing and Bethesda studies
FACTOR VIII INHIBITOR EVALUATION
2. What is the most likely underlying condition associated with this patient’s factor VIII inhibitor?
- Autoimmune disease
- Malignancy
- A medication
- Unknown (idiopathic)
Acquired hemophilia A (AHA) is a rare disorder caused by autoantibodies against factor VIII. Its estimated incidence is about 1 person per million per year.4 It usually presents as unexplained bruising or bleeding and is only rarely diagnosed by an incidentally noted prolonged aPTT. The severity of bleeding is variable and can include subcutaneous, soft-tissue, retroperitoneal, gastrointestinal, and intracranial hemorrhage.5
AHA is considered idiopathic in more than half of cases. A study based on a European registry5 of 501 patients with AHA and a UK study6 of 172 patients found no underlying disease in 52% and 65% of patients, respectively. For patients with an identified cause, the most common causes were malignancy (12%5 and 15%6) and autoimmune disease (12%5 and 17%6).
Drugs have rarely been associated with factor VIII inhibitors. Such occurrences have been reported with interferon, blood thinners, antibiotics, and psychiatric medications, but no study yet has indicated causation. However, patients with congenital hemophilia A treated with factor VIII preparations have about a 15% chance of developing factor VIII inhibitors. In this setting, inhibitors develop in response to recombinant factor VIII exposure, unlike the autoimmune phenomena seen in AHA.
TREATMENT OF ACQUIRED HEMOPHILIA A
3. What is the most appropriate treatment for AHA?
- Desmopressin and prednisone
- Recombinant porcine factor VIII and prednisone plus cyclophosphamide
- Recombinant factor VIIa and rituximab
- Any of the above
Any of the above regimens can be used. In general, treatment of AHA has two purposes: to stop acute hemorrhage, and to reduce the level of factor VIII inhibitor. No standard treatment guidelines are available; evidence of the effectiveness of different drugs is based largely on data on congenital hemophilia A.3
Acute treatment to stop bleeding
Initial treatment of AHA often focuses on stopping an acute hemorrhage by either raising circulating levels of factor VIII or bypassing it in the coagulation cascade.
Desmopressin can temporarily raise factor VIII levels, but it is often ineffective in AHA unless the patient has very low inhibitor titers.3
Factor VIII concentrate (human or recombinant porcine factor VIII) may be effective in patients with low inhibitor titers (< 5 BU). Higher doses are often required than those used in congenital hemophilia A. Factor VIII concentrate is usually combined with immunosuppressive treatment to lower the factor VIII inhibitor level (described below).3
If these methods are ineffective or the patient has high inhibitor titers (> 5 BU), activated prothrombin complex concentrates, known as FEIBA (factor eight inhibitor bypassing activity), or recombinant factor VIIa is available. These agents bypass factor VIII in the clotting cascade.
Immunosuppression to reduce factor VIII inhibitor
Immunosuppressive agents are the mainstay of AHA treatment to lower the inhibitor level.
Regimens vary. A 2003 meta-analysis4 including 249 patients found that prednisone alone resulted in complete response in about 30% of patients, and the addition of cyclophosphamide increased the response rate to 60% to 100%. High-dose intravenous immunoglobulin led to conflicting results. Conclusions were limited by the variability of dosing and duration in treatment regimens among the 20 different studies included.
An analysis of 331 patients in the European Acquired Hemophilia Registry (EACH2)7 found that steroids alone produced remission in 48% of patients, while steroids combined with cyclophosphamide raised the rate to 70%. Rituximab-based regimens were successful in 59% but required twice as long to achieve remission as steroid or cyclophosphamide-based regimens. No benefit was noted from intravenous immunoglobulin.
Risks of disease and treatment
AHA is associated with significant risk of morbidity and death related to bleeding, complications of treatment, and underlying disease.
In EACH2, 16 of the 331 patients died of bleeding, 16 died of causes related to immunosuppression, and 45 died of causes related to the underlying condition.5 In the UK registry of 172 patients, 13 patients died of bleeding, and 12 died of sepsis related to immunosuppression.6
The factor VIII level and inhibitor titer are not necessarily useful in stratifying bleeding risk, as severe and fatal bleeding can occur at variable levels and patients remain at risk of bleeding as long as the inhibitor persists.6,7
CASE CONTINUED: TREATMENT, LYMPHOCYTOSIS
The patient was started on 60 mg daily of prednisone, resulting in a decrease in her aPTT, increase in factor VIII level, and lower Bethesda titer. On a return visit, her absolute lymphocyte count was 7.04 × 109/L (reference range 1.0–4.0). She reported no fevers, chills, or recent infections.
EVALUATING LYMPHOCYTOSIS
Lymphocytosis is defined in most laboratories as an absolute lymphocyte count greater than 4.0 × 109/L for adults. Normally, T cells (CD3+) make up 60% to 80% of lymphocytes, B cells (CD20+) 10% to 20%, and natural killer (NK) cells (CD3–, CD56+) 5% to 10%. Lymphocytosis is usually caused by infection, but it can have other causes, including malignancy.
Peripheral blood smear. If there is no clear cause of lymphocytosis, a peripheral blood smear can be used to assess lymphocyte morphology, providing clues to the underlying etiology. For example, atypical lymphocytes are often seen in infectious mononucleosis, while “smudge” lymphocytes are characteristic of chronic lymphocytic leukemia. If a peripheral smear shows abnormal morphology, further workup should include establishing whether the lymphocytes are polyclonal or clonal.8
CASE CONTINUED: LARGE GRANULAR LYMPHOCYTES
4. What is the next step to evaluate the patient’s lymphocytosis?
- Bone marrow biopsy
- Karyotype analysis
- Flow cytometry
- Fluorescence in situ hybridization
Flow cytometry with V-beta analysis is the best first test to determine the cause of lymphocytosis after review of the peripheral smear. For persistent lymphocytosis, flow cytometry should be done even if a peripheral smear shows normal lymphocyte morphology.
Most T cells possess receptors composed of alpha and beta chains, each encoded by variable (V), diversity (D), joining (J), and constant (C) gene segments. The V, D, and J segments undergo rearrangement during T-cell development in the thymus based on antigen exposure, producing a diverse T-cell receptor population.
In a polyclonal population of lymphocytes, the T-cell receptors have a variety of gene segment arrangements, indicating normal T-cell development. But in a clonal population of lymphocytes, the T-cell receptors have a single identical gene segment arrangement, indicating they all originated from a single clone.9 Lymphocytosis in response to an infection is typically polyclonal, while malignant lymphocytosis is clonal.
Monoclonal antibodies against many of the variable regions of the beta chain (V-beta) of T-cell receptors have been developed, enabling flow cytometry to establish clonality.
T-cell receptor gene rearrangement studies can also be performed using polymerase chain reaction and Southern blot techniques.9
Karyotype analysis is usually not performed for the finding of LGLs, because most leukemias (eg, T-cell and NK-cell leukemias) have cells with a normal karyotype.
Bone marrow biopsy is invasive and usually not required to evaluate LGLs. It can be especially risky for a patient with a bleeding disorder such as a factor VIII inhibitor.10
Case continued: Flow cytometry confirms clonality
Subsequent flow cytometry found that more than 50% of the patient’s lymphocytes were LGLs that co-expressed CD3+, CD8+, CD56+, and CD57+, with aberrantly decreased CD7 expression. T-cell V-beta analysis demonstrated an expansion of the V-beta 17 family, and T-cell receptor gene analysis with polymerase chain reaction confirmed the presence of a clonal rearrangement.
LGL LEUKEMIA: CLASSIFICATION AND MANAGEMENT
LGLs normally account for 10% to 15% of peripheral mononuclear cells.11 LGL leukemia is caused by a clonal population of cytotoxic T cells or NK cells and involves an increased number of LGLs (usually > 2 × 109/L).10
LGL leukemia is divided into 3 categories according to the most recent World Health Organization classification10,12:
T-cell LGL leukemia (about 85% of cases) is considered indolent but can cause significant cytopenias and is often associated with autoimmune disease.13 Cells usually express a CD3+, CD8+, CD16+, and CD57+ phenotype. Survival is about 70% at 10 years.
Chronic NK-cell lymphocytosis (about 10%) also tends to have an indolent course with cytopenia and an autoimmune association, and with a similar prognosis to T-cell LGL leukemia. Cells express a CD3–, CD16+, and CD56+ phenotype.
Aggressive NK-cell LGL leukemia (about 5%) is associated with Epstein-Barr virus infection and occurs in younger patients. It is characterized by severe cytopenias, “B symptoms” (ie, fever, night sweats, weight loss), and has a very poor prognosis. Like chronic NK-cell lymphocytosis, cells express a CD3–, CD16+, and CD56+ phenotype. Fas (CD95) and Fas-ligand (CD178) are strongly expressed.10,13
Most cases of LGL leukemia can be diagnosed on the basis of classic morphology on peripheral blood smear and evidence of clonality on flow cytometry or gene rearrangement studies. T-cell receptor gene studies cannot be used to establish clonality in the NK subtypes, as NK cells do not express T-cell receptors.11
Case continued: Diagnosis, continued course
In our patient, T-cell LGL leukemia was diagnosed on the basis of the peripheral smear, flow cytometry results, and positive T-cell receptor gene studies for clonal rearrangement in the T-cell receptor beta region.
While her corticosteroid therapy was being tapered, her factor III inhibitor level increased, and she had a small episode of bleeding, prompting the start of cyclophosphamide 50 mg daily with lower doses of prednisone.
LGL LEUKEMIA AND AUTOIMMUNE DISEASE
Patients with LGL leukemia commonly have or develop autoimmune conditions. Immune-mediated cytopenias including pure red cell aplasia, aplastic anemia, and autoimmune hemolytic anemias can occur. Neutropenia, the most common cytopenia in LGL leukemia, is thought to be at least partly autoimmune, as the degree of neutropenia is often worse than would be expected solely from bone-marrow infiltration of LGL cells.10,14,15
Rheumatoid arthritis is the most common autoimmune condition associated with LGL leukemia, with a reported incidence between 11% and 36%.13–15
Felty syndrome (rheumatoid arthritis, splenomegaly, and neutropenia) is often associated with LGL leukemia and is thought by some to be part of the same disease process.15
Treat with immunosuppressives if needed
Indications for treating LGL leukemia include the development of cytopenias and associated autoimmune diseases. Immunosuppressive agents, such as methotrexate, cyclophosphamide, and cyclosporine, are commonly used.10,11,14 Most evidence of treatment efficacy is from retrospective studies and case reports, with widely variable response rates that overall are around 50%.10
ACQUIRED HEMOPHILIA A AND HEMATOLOGIC MALIGNANCY
A systematic review found 30 cases of AHA associated with hematologic malignancies.16 The largest case series17 in this analysis had 8 patients, and included diagnoses of chronic lymphocytic leukemia, erythroleukemia, myelofibrosis, multiple myeloma, and myelodysplastic syndrome. In 3 of these patients, the appearance of the inhibitor preceded the diagnosis of the underlying malignancy by an average of 3.5 months. In 1 patient with erythroleukemia and another with multiple myeloma, the activity of the inhibitor could be clearly correlated with the underlying malignancy. In the other 6 patients, no association between the two could be made.
In the same series, complete resolution of the inhibitor was related only to the level of Bethesda titer present at diagnosis, with those who achieved resolution having lower mean Bethesda titers.17 Similarly, in EACH2, lower inhibitor Bethesda titers and higher factor VIII levels at presentation were associated with faster inhibitor eradication and normalization of factor VIII levels.7
Murphy et al18 described a 62-year-old woman with Felty syndrome who developed a factor VIII inhibitor and was subsequently given a diagnosis of LGL leukemia. Treatment with immunosuppressive agents, including cyclophosphamide, azathioprine, and rituximab, successfully eradicated her factor VIII inhibitor, although the LGL leukemia persisted.
Case conclusion: Eradication of factor VIII inhibitor
Our patient, similar to the patient described by Murphy et al18 above, had eradication of the factor VIII inhibitor despite persistence of LGL leukemia. Between the time of diagnosis at our clinic, when she had 54% LGLs, and eradication of the inhibitor 3 months later, the LGL percentage ranged from 45% to 89%. No clear direct correlation between LGL and factor VIII inhibitor levels could be detected.
Given the strong association of LGL leukemia with autoimmune disease, it is tempting to believe that her factor VIII inhibitor was somehow related to her malignancy, although the exact mechanism remained unclear. The average age at diagnosis is 60 for LGL leukemia11 and over 70 for AHA,5,6 so advanced age may be the common denominator. Whether or not our patient will have recurrence of her factor VIII inhibitor or the development of other autoimmune diseases with the persistence of her LGL leukemia remains to be seen.
At last follow-up, our patient was off all therapy and continued to have normal aPTT and factor VIII levels. Repeat flow cytometry after treatment of her factor VIII inhibitor showed persistence of a clonal T-cell population, although reduced from 72% to 60%. It may be that the 2 entities were unrelated, and the clonal T-cell population was simply fluctuating over time. This can be determined only with further observation. As the patient had no symptoms from her LGL leukemia, she continued to be observed without treatment.
TAKE-HOME POINTS
- The coagulation assay is key to initially assessing a bleeding abnormality; whether the prothrombin time and aPTT are normal or prolonged narrows the differential diagnosis and determines next steps in evaluation.
- Mixing studies can help pinpoint the responsible deficient factor.
- Acquired factor VIII deficiency, also known as AHA, may be caused by autoimmune disease, malignancy, or medications, but it is usually idiopathic.
- AHA treatment is focused on achieving hemostasis and reducing factor VIII inhibitor.
- Lymphocytosis should be evaluated with a peripheral blood smear and flow cytometry to determine if the population is polyclonal (associated with infection) or clonal (associated with malignancy).
- LGL leukemia is usually a chronic, indolent disease, although an uncommon subtype has an aggressive course.
- The association between AHA and LGL leukemia is unclear, and both conditions must be monitored and managed.
- Kamal AH, Tefferi A, Pruthi RK. How to interpret and pursue an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time in adults. Mayo Clin Proc 2007; 82(7):864–873. doi:10.4065/82.7.864
- Tcherniantchouk O, Laposata M, Marques MB. The isolated prolonged PTT. Am J Hematol 2013; 88(1):82–85. doi:10.1002/ajh.23285
- Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program 2006:432–437. doi:10.1182/asheducation-2006.1.432
- Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol 2003; 121(1):21–35. pmid:12670328
- Knoebl P, Marco P, Baudo F, et al; EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10(4):622–631. doi:10.1111/j.1538-7836.2012.04654.x
- Collins PW, Hirsch S, Baglin TP, et al; UK Haemophilia Centre Doctors’ Organisation. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007; 109(5):1870–1877. doi:10.1182/blood-2006-06-029850
- Collins P, Baudo F, Knoebl P, et al; EACH2 Registry Collaborators. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood 2012; 120(1):47–55. doi:10.1182/blood-2012-02-409185
- George TI. Malignant or benign leukocytosis. Hematology Am Soc Hematol Educ Program 2012; 2012:475–484. doi:10.1182/asheducation-2012.1.475
- Watters RJ, Liu X, Loughran TP Jr. T-cell and natural killer-cell large granular lymphocyte leukemia neoplasias. Leuk Lymphoma 2011; 52(12):2217–2225. doi:10.3109/10428194.2011.593276
- Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood 2017; 129(9):1082–1094. doi:10.1182/blood-2016-08-692590
- Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program 2012; 2012:652–659. doi:10.1182/asheducation-2012.1.652
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127(20):2375–2390. doi:10.1182/blood-2016-01-643569
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist 2004; 9(3):247–258. pmid:15169980
- Bockorny B, Dasanu CA. Autoimmune manifestations in large granular lymphocyte leukemia. Clin Lymphoma Myeloma Leuk 2012; 12(6):400–405. doi:10.1016/j.clml.2012.06.006
- Liu X, Loughran TP Jr. The spectrum of large granular lymphocyte leukemia and Felty’s syndrome. Curr Opin Hematol 2011; 18(4):254–259. doi:10.1097/MOH.0b013e32834760fb
- Franchini M, Lippi G. Acquired factor V inhibitors: a systematic review. J Thromb Thrombolysis 2011; 31(4):449–457. doi:10.1007/s11239-010-0529-6
- Sallah S, Nguyen NP, Abdallah JM, Hanrahan LR. Acquired hemophilia in patients with hematologic malignancies. Arch Pathol Lab Med 2000; 124(5):730–734.
- Murphy PW, Brett LK, Verla-Tebit E, Macik BG, Loughran TP Jr. Acquired inhibitors to factor VIII and fibrinogen in the setting of T-cell large granular lymphocyte leukemia: a case report and review of the literature. Blood Coagul Fibrinolysis 2015; 26(2):211–213. doi:10.1097/MBC.0000000000000209
- Kamal AH, Tefferi A, Pruthi RK. How to interpret and pursue an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time in adults. Mayo Clin Proc 2007; 82(7):864–873. doi:10.4065/82.7.864
- Tcherniantchouk O, Laposata M, Marques MB. The isolated prolonged PTT. Am J Hematol 2013; 88(1):82–85. doi:10.1002/ajh.23285
- Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program 2006:432–437. doi:10.1182/asheducation-2006.1.432
- Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol 2003; 121(1):21–35. pmid:12670328
- Knoebl P, Marco P, Baudo F, et al; EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10(4):622–631. doi:10.1111/j.1538-7836.2012.04654.x
- Collins PW, Hirsch S, Baglin TP, et al; UK Haemophilia Centre Doctors’ Organisation. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007; 109(5):1870–1877. doi:10.1182/blood-2006-06-029850
- Collins P, Baudo F, Knoebl P, et al; EACH2 Registry Collaborators. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood 2012; 120(1):47–55. doi:10.1182/blood-2012-02-409185
- George TI. Malignant or benign leukocytosis. Hematology Am Soc Hematol Educ Program 2012; 2012:475–484. doi:10.1182/asheducation-2012.1.475
- Watters RJ, Liu X, Loughran TP Jr. T-cell and natural killer-cell large granular lymphocyte leukemia neoplasias. Leuk Lymphoma 2011; 52(12):2217–2225. doi:10.3109/10428194.2011.593276
- Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood 2017; 129(9):1082–1094. doi:10.1182/blood-2016-08-692590
- Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program 2012; 2012:652–659. doi:10.1182/asheducation-2012.1.652
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127(20):2375–2390. doi:10.1182/blood-2016-01-643569
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist 2004; 9(3):247–258. pmid:15169980
- Bockorny B, Dasanu CA. Autoimmune manifestations in large granular lymphocyte leukemia. Clin Lymphoma Myeloma Leuk 2012; 12(6):400–405. doi:10.1016/j.clml.2012.06.006
- Liu X, Loughran TP Jr. The spectrum of large granular lymphocyte leukemia and Felty’s syndrome. Curr Opin Hematol 2011; 18(4):254–259. doi:10.1097/MOH.0b013e32834760fb
- Franchini M, Lippi G. Acquired factor V inhibitors: a systematic review. J Thromb Thrombolysis 2011; 31(4):449–457. doi:10.1007/s11239-010-0529-6
- Sallah S, Nguyen NP, Abdallah JM, Hanrahan LR. Acquired hemophilia in patients with hematologic malignancies. Arch Pathol Lab Med 2000; 124(5):730–734.
- Murphy PW, Brett LK, Verla-Tebit E, Macik BG, Loughran TP Jr. Acquired inhibitors to factor VIII and fibrinogen in the setting of T-cell large granular lymphocyte leukemia: a case report and review of the literature. Blood Coagul Fibrinolysis 2015; 26(2):211–213. doi:10.1097/MBC.0000000000000209

Timing, volume of transfusion may not matter in children with severe anemia
Trial results suggest African children with uncomplicated, severe anemia may not require immediate blood transfusion, and the volume of transfusion may only matter in the context of fever.

The TRACT trial showed no significant differences in 28-day mortality or other clinical outcomes between children who received immediate transfusions and those who did not.
Similarly, there was no significant difference in 28-day mortality among children who received transfusions of 20 mL/kg and those who received transfusions of 30 mL/kg. There was evidence to suggest a higher transfusion volume may benefit children without fevers, but this was an exploratory endpoint. The findings were published in the New England Journal of Medicine.
These results suggest “there is no credible reason to transfuse immediately or to transfuse a higher volume of blood, at least in pediatric populations in regions such as these two sub-Saharan countries [Uganda and Malawi],” Julie R. Ingelfinger, MD, of Massachusetts General Hospital in Boston, wrote in an accompanying editorial, also published in the New England Journal of Medicine (2019;381:475-6).
“The possible effect of higher volume transfusion in patients with fever may trigger additional and potentially useful studies,” she added.
Immediate transfusion
One goal of the TRACT trial was to determine if blood transfusion is the best treatment for children with severe anemia. With this in mind, Kathryn Maitland, MD, PhD, of Imperial College London and colleagues evaluated 1,565 Ugandan and Malawian children with uncomplicated, severe anemia. The patients’ median age was 26 months, and 984 (62.9%) had malaria.
The children were randomized to immediate transfusion (n = 778) or no immediate transfusion (n = 787). Children who did not have an immediate transfusion (control group) could receive a transfusion if they exhibited new signs of clinical severity or had their hemoglobin decrease to below 4 g/dL.
All children in the immediate-transfusion group received a transfusion, as did 386 (49.0%) in the control group. The median time to transfusion was 1.3 hours in the immediate group and 24.9 hours in the control group. The mean total blood volume transfused per child was 314 plus or minus 228 mL and 142 plus or minus 224, respectively. The follow-up period was 180 days, and 4.5% of patients (n = 71) were lost to follow-up.
The researchers found no significant difference between the treatment groups with regard to mortality, other clinical outcomes, or the cost of care.
The 28-day mortality rate was 0.9% in the immediate-transfusion group and 1.7% in the control group (hazard ratio, 0.54; 95% confidence interval, 0.22-1.36; P = .19). The 180-day mortality was 4.5% and 6.0%, respectively (HR, 0.75; 95% CI, 0.48-1.15).
Transfusion volume
To assess the effects of transfusion volume, Dr. Maitland and colleagues evaluated 3,196 Ugandan and Malawian children with severe anemia. The median age of the children was 37 months, and 2,050 (64.1%) had malaria.
The children received a transfusion of 30 mL/kg (n = 1,592) or 20 mL/kg (n = 1,596) at a median of 1.2 hours after randomization. Some children – 197 in the 30-mL/kg group and 300 in the 20-mL/kg group – received additional transfusions. The mean volume of total blood transfused per child was 475 plus or minus 385 mL, and 353 plus or minus 348 mL, respectively.
Overall, there was no significant between-group difference with regard to mortality. The 28-day mortality rate was 3.4% in the 30 mL/kg group and 4.5% in the 20 mL/kg group (HR = 0.76; 95% CI, 0.54 to 1.08; P = .12).
However, the 28-day mortality rate did differ according to the presence of fever at screening. The mortality rate was lower in the 30 mL/kg group for children without fevers (HR = 0.43; 95% CI, 0.27 to 0.69) but higher in the 30 mL/kg group for febrile children (HR = 1.91; 95% CI, 1.04 to 3.49).
For other outcomes, including readmissions and serious adverse events, the researchers found no significant between-group differences.
This trial was supported by a grant from the United Kingdom Medical Research Council through a concordat with the Department for International Development. One researcher has a Wellcome Senior Research Fellowship, and another is a National Institute for Health Research Senior Investigator. Dr. Ingelfinger is a deputy editor at the New England Journal of Medicine. No other relevant conflicts of interest were reported.
SOURCES: Maitland K et al. N Engl J Med. 2019;381:407-19. Maitland K et al. N Engl J Med. 2019;381:420-31.
Trial results suggest African children with uncomplicated, severe anemia may not require immediate blood transfusion, and the volume of transfusion may only matter in the context of fever.
The TRACT trial showed no significant differences in 28-day mortality or other clinical outcomes between children who received immediate transfusions and those who did not.
Similarly, there was no significant difference in 28-day mortality among children who received transfusions of 20 mL/kg and those who received transfusions of 30 mL/kg. There was evidence to suggest a higher transfusion volume may benefit children without fevers, but this was an exploratory endpoint. The findings were published in the New England Journal of Medicine.
These results suggest “there is no credible reason to transfuse immediately or to transfuse a higher volume of blood, at least in pediatric populations in regions such as these two sub-Saharan countries [Uganda and Malawi],” Julie R. Ingelfinger, MD, of Massachusetts General Hospital in Boston, wrote in an accompanying editorial, also published in the New England Journal of Medicine (2019;381:475-6).
“The possible effect of higher volume transfusion in patients with fever may trigger additional and potentially useful studies,” she added.
Immediate transfusion
One goal of the TRACT trial was to determine if blood transfusion is the best treatment for children with severe anemia. With this in mind, Kathryn Maitland, MD, PhD, of Imperial College London and colleagues evaluated 1,565 Ugandan and Malawian children with uncomplicated, severe anemia. The patients’ median age was 26 months, and 984 (62.9%) had malaria.
The children were randomized to immediate transfusion (n = 778) or no immediate transfusion (n = 787). Children who did not have an immediate transfusion (control group) could receive a transfusion if they exhibited new signs of clinical severity or had their hemoglobin decrease to below 4 g/dL.
All children in the immediate-transfusion group received a transfusion, as did 386 (49.0%) in the control group. The median time to transfusion was 1.3 hours in the immediate group and 24.9 hours in the control group. The mean total blood volume transfused per child was 314 plus or minus 228 mL and 142 plus or minus 224, respectively. The follow-up period was 180 days, and 4.5% of patients (n = 71) were lost to follow-up.
The researchers found no significant difference between the treatment groups with regard to mortality, other clinical outcomes, or the cost of care.
The 28-day mortality rate was 0.9% in the immediate-transfusion group and 1.7% in the control group (hazard ratio, 0.54; 95% confidence interval, 0.22-1.36; P = .19). The 180-day mortality was 4.5% and 6.0%, respectively (HR, 0.75; 95% CI, 0.48-1.15).
Transfusion volume
To assess the effects of transfusion volume, Dr. Maitland and colleagues evaluated 3,196 Ugandan and Malawian children with severe anemia. The median age of the children was 37 months, and 2,050 (64.1%) had malaria.
The children received a transfusion of 30 mL/kg (n = 1,592) or 20 mL/kg (n = 1,596) at a median of 1.2 hours after randomization. Some children – 197 in the 30-mL/kg group and 300 in the 20-mL/kg group – received additional transfusions. The mean volume of total blood transfused per child was 475 plus or minus 385 mL, and 353 plus or minus 348 mL, respectively.
Overall, there was no significant between-group difference with regard to mortality. The 28-day mortality rate was 3.4% in the 30 mL/kg group and 4.5% in the 20 mL/kg group (HR = 0.76; 95% CI, 0.54 to 1.08; P = .12).
However, the 28-day mortality rate did differ according to the presence of fever at screening. The mortality rate was lower in the 30 mL/kg group for children without fevers (HR = 0.43; 95% CI, 0.27 to 0.69) but higher in the 30 mL/kg group for febrile children (HR = 1.91; 95% CI, 1.04 to 3.49).
For other outcomes, including readmissions and serious adverse events, the researchers found no significant between-group differences.
This trial was supported by a grant from the United Kingdom Medical Research Council through a concordat with the Department for International Development. One researcher has a Wellcome Senior Research Fellowship, and another is a National Institute for Health Research Senior Investigator. Dr. Ingelfinger is a deputy editor at the New England Journal of Medicine. No other relevant conflicts of interest were reported.
SOURCES: Maitland K et al. N Engl J Med. 2019;381:407-19. Maitland K et al. N Engl J Med. 2019;381:420-31.
Trial results suggest African children with uncomplicated, severe anemia may not require immediate blood transfusion, and the volume of transfusion may only matter in the context of fever.
The TRACT trial showed no significant differences in 28-day mortality or other clinical outcomes between children who received immediate transfusions and those who did not.
Similarly, there was no significant difference in 28-day mortality among children who received transfusions of 20 mL/kg and those who received transfusions of 30 mL/kg. There was evidence to suggest a higher transfusion volume may benefit children without fevers, but this was an exploratory endpoint. The findings were published in the New England Journal of Medicine.
These results suggest “there is no credible reason to transfuse immediately or to transfuse a higher volume of blood, at least in pediatric populations in regions such as these two sub-Saharan countries [Uganda and Malawi],” Julie R. Ingelfinger, MD, of Massachusetts General Hospital in Boston, wrote in an accompanying editorial, also published in the New England Journal of Medicine (2019;381:475-6).
“The possible effect of higher volume transfusion in patients with fever may trigger additional and potentially useful studies,” she added.
Immediate transfusion
One goal of the TRACT trial was to determine if blood transfusion is the best treatment for children with severe anemia. With this in mind, Kathryn Maitland, MD, PhD, of Imperial College London and colleagues evaluated 1,565 Ugandan and Malawian children with uncomplicated, severe anemia. The patients’ median age was 26 months, and 984 (62.9%) had malaria.
The children were randomized to immediate transfusion (n = 778) or no immediate transfusion (n = 787). Children who did not have an immediate transfusion (control group) could receive a transfusion if they exhibited new signs of clinical severity or had their hemoglobin decrease to below 4 g/dL.
All children in the immediate-transfusion group received a transfusion, as did 386 (49.0%) in the control group. The median time to transfusion was 1.3 hours in the immediate group and 24.9 hours in the control group. The mean total blood volume transfused per child was 314 plus or minus 228 mL and 142 plus or minus 224, respectively. The follow-up period was 180 days, and 4.5% of patients (n = 71) were lost to follow-up.
The researchers found no significant difference between the treatment groups with regard to mortality, other clinical outcomes, or the cost of care.
The 28-day mortality rate was 0.9% in the immediate-transfusion group and 1.7% in the control group (hazard ratio, 0.54; 95% confidence interval, 0.22-1.36; P = .19). The 180-day mortality was 4.5% and 6.0%, respectively (HR, 0.75; 95% CI, 0.48-1.15).
Transfusion volume
To assess the effects of transfusion volume, Dr. Maitland and colleagues evaluated 3,196 Ugandan and Malawian children with severe anemia. The median age of the children was 37 months, and 2,050 (64.1%) had malaria.
The children received a transfusion of 30 mL/kg (n = 1,592) or 20 mL/kg (n = 1,596) at a median of 1.2 hours after randomization. Some children – 197 in the 30-mL/kg group and 300 in the 20-mL/kg group – received additional transfusions. The mean volume of total blood transfused per child was 475 plus or minus 385 mL, and 353 plus or minus 348 mL, respectively.
Overall, there was no significant between-group difference with regard to mortality. The 28-day mortality rate was 3.4% in the 30 mL/kg group and 4.5% in the 20 mL/kg group (HR = 0.76; 95% CI, 0.54 to 1.08; P = .12).
However, the 28-day mortality rate did differ according to the presence of fever at screening. The mortality rate was lower in the 30 mL/kg group for children without fevers (HR = 0.43; 95% CI, 0.27 to 0.69) but higher in the 30 mL/kg group for febrile children (HR = 1.91; 95% CI, 1.04 to 3.49).
For other outcomes, including readmissions and serious adverse events, the researchers found no significant between-group differences.
This trial was supported by a grant from the United Kingdom Medical Research Council through a concordat with the Department for International Development. One researcher has a Wellcome Senior Research Fellowship, and another is a National Institute for Health Research Senior Investigator. Dr. Ingelfinger is a deputy editor at the New England Journal of Medicine. No other relevant conflicts of interest were reported.
SOURCES: Maitland K et al. N Engl J Med. 2019;381:407-19. Maitland K et al. N Engl J Med. 2019;381:420-31.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: The 28-day mortality was 0.9% in patients who had immediate transfusions and 1.7% in those who did not (hazard ratio, 0.54; P = .19). The 28-day mortality rate was 3.4% in patients who received transfusions of 30 mL/kg and 4.5% in those who received transfusions of 20 mL/kg (HR, 0.76; P = .12). However, the mortality rate was lower in the 30-mL/kg group for children without fevers (HR, 0.43) and higher in the 30-mL/kg group for febrile children (HR, 1.91).
Study details: A phase 3 trial of African children with severe anemia who were randomized to immediate transfusion (n = 778) or no immediate transfusion (n = 787) and transfusions of 30 mL/kg (n = 1,592) or 20 mL/kg (n = 1,596)
Disclosures: The trial was supported by a grant from the United Kingdom Medical Research Council through a concordat with the Department for International Development. One researcher has a Wellcome Senior Research Fellowship, and another is a National Institute for Health Research Senior Investigator.
Sources: Maitland K et al. N Engl J Med. 2019;381:407-19. Maitland K et al. N Engl J Med. 2019;381:420-31.
RNA interference drug fitusiran looks effective in both hemophilia A and B
MELBOURNE – An investigational RNA interference therapeutic that suppresses the production of antithrombin has shown significant reductions in bleeding rates with no major safety events, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Fitusiran is a once-monthly, fixed-dose subcutaneous therapy that uses RNA interference to silence the gene for the endogenous anticoagulant antithrombin.

“The therapeutic hypothesis is based on the fact that hemophilia A and B are essentially thrombin-deficiency disorders, so if we lack factor VIII or factor IX, we can’t generate enough thrombin and we can’t produce a significant and substantial blood clot,” John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust. “If we, however, administer fitusiran, which will suppress antithrombin production, we can rebalance coagulation, generate more thrombin and form a much more substantial clot.”
Dr. Pasi presented results of an interim analysis of safety and efficacy data from an open-label, phase 2 extension study in 34 individuals with hemophilia A or B, with or without inhibitors, who were treated either with 50-mg or 80-mg doses of fitusiran for a median of at least 2 years.
Researchers saw significant declines in annualized bleeding rates in patients with hemophilia A and B, with and without inhibitors. Among those without inhibitors, the median annualized bleeding rate declined from 2.00 in patients already on hemophilia prophylaxis and 12.00 in those using on-demand treatment to 1.08 overall. In patients with inhibitors, the median annualized bleeding rate dropped from 42.00 to 1.04.
The treatment was also associated with substantial reductions in antithrombin production and increases in thrombin generation.
One patient in the phase 1 study experienced a fatal cerebral venous sinus thrombosis, which subsequently led to introduction of a bleed management protocol.
“Following that last case, we revised and reviewed the bleed management guidelines in view of the fact that there might potentially be an interaction between the amount of replacement therapy and thrombin generation,” Dr. Pasi said. Since introduction of that protocol, there have been no related thrombotic events.
The majority of adverse events reported were mild and deemed not related to the study drug, Dr. Pasi said. These included headache, injection site erythema, and arthralgia. A total of 14 subjects – all of whom were positive for hepatitis C at baseline – experienced rises in ALT levels but these were asymptomatic and resolved spontaneously.
One patient with chronic active hepatitis C infection also showed significant ALT/AST elevation which led to discontinuation of treatment.
In an interview, Dr. Pasi said one of the biggest advantages of fitusiran was that it could be used in patients with hemophilia A and B. “You’ve got patients with hemophilia B who’ve got no options at the moment. That would be an obvious specific group that would gain from this.”
Another advantage was fitusiran’s stability and dosing, he said, pointing out that the treatment was fixed dosing and stable at room temperature. Fitusiran is now undergoing phase 3 trials.
The study was funded by Sanofi Genzyme and Alnylam Pharmaceuticals, and six authors were employees of Sanofi Genzyme. Dr. Pasi reported financial relationships with pharmaceutical companies, including Alnylam.
SOURCE: Pasi J et al. 2019 ISTH Congress, Abstract OC 11.3.
MELBOURNE – An investigational RNA interference therapeutic that suppresses the production of antithrombin has shown significant reductions in bleeding rates with no major safety events, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Fitusiran is a once-monthly, fixed-dose subcutaneous therapy that uses RNA interference to silence the gene for the endogenous anticoagulant antithrombin.
“The therapeutic hypothesis is based on the fact that hemophilia A and B are essentially thrombin-deficiency disorders, so if we lack factor VIII or factor IX, we can’t generate enough thrombin and we can’t produce a significant and substantial blood clot,” John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust. “If we, however, administer fitusiran, which will suppress antithrombin production, we can rebalance coagulation, generate more thrombin and form a much more substantial clot.”
Dr. Pasi presented results of an interim analysis of safety and efficacy data from an open-label, phase 2 extension study in 34 individuals with hemophilia A or B, with or without inhibitors, who were treated either with 50-mg or 80-mg doses of fitusiran for a median of at least 2 years.
Researchers saw significant declines in annualized bleeding rates in patients with hemophilia A and B, with and without inhibitors. Among those without inhibitors, the median annualized bleeding rate declined from 2.00 in patients already on hemophilia prophylaxis and 12.00 in those using on-demand treatment to 1.08 overall. In patients with inhibitors, the median annualized bleeding rate dropped from 42.00 to 1.04.
The treatment was also associated with substantial reductions in antithrombin production and increases in thrombin generation.
One patient in the phase 1 study experienced a fatal cerebral venous sinus thrombosis, which subsequently led to introduction of a bleed management protocol.
“Following that last case, we revised and reviewed the bleed management guidelines in view of the fact that there might potentially be an interaction between the amount of replacement therapy and thrombin generation,” Dr. Pasi said. Since introduction of that protocol, there have been no related thrombotic events.
The majority of adverse events reported were mild and deemed not related to the study drug, Dr. Pasi said. These included headache, injection site erythema, and arthralgia. A total of 14 subjects – all of whom were positive for hepatitis C at baseline – experienced rises in ALT levels but these were asymptomatic and resolved spontaneously.
One patient with chronic active hepatitis C infection also showed significant ALT/AST elevation which led to discontinuation of treatment.
In an interview, Dr. Pasi said one of the biggest advantages of fitusiran was that it could be used in patients with hemophilia A and B. “You’ve got patients with hemophilia B who’ve got no options at the moment. That would be an obvious specific group that would gain from this.”
Another advantage was fitusiran’s stability and dosing, he said, pointing out that the treatment was fixed dosing and stable at room temperature. Fitusiran is now undergoing phase 3 trials.
The study was funded by Sanofi Genzyme and Alnylam Pharmaceuticals, and six authors were employees of Sanofi Genzyme. Dr. Pasi reported financial relationships with pharmaceutical companies, including Alnylam.
SOURCE: Pasi J et al. 2019 ISTH Congress, Abstract OC 11.3.
MELBOURNE – An investigational RNA interference therapeutic that suppresses the production of antithrombin has shown significant reductions in bleeding rates with no major safety events, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Fitusiran is a once-monthly, fixed-dose subcutaneous therapy that uses RNA interference to silence the gene for the endogenous anticoagulant antithrombin.
“The therapeutic hypothesis is based on the fact that hemophilia A and B are essentially thrombin-deficiency disorders, so if we lack factor VIII or factor IX, we can’t generate enough thrombin and we can’t produce a significant and substantial blood clot,” John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust. “If we, however, administer fitusiran, which will suppress antithrombin production, we can rebalance coagulation, generate more thrombin and form a much more substantial clot.”
Dr. Pasi presented results of an interim analysis of safety and efficacy data from an open-label, phase 2 extension study in 34 individuals with hemophilia A or B, with or without inhibitors, who were treated either with 50-mg or 80-mg doses of fitusiran for a median of at least 2 years.
Researchers saw significant declines in annualized bleeding rates in patients with hemophilia A and B, with and without inhibitors. Among those without inhibitors, the median annualized bleeding rate declined from 2.00 in patients already on hemophilia prophylaxis and 12.00 in those using on-demand treatment to 1.08 overall. In patients with inhibitors, the median annualized bleeding rate dropped from 42.00 to 1.04.
The treatment was also associated with substantial reductions in antithrombin production and increases in thrombin generation.
One patient in the phase 1 study experienced a fatal cerebral venous sinus thrombosis, which subsequently led to introduction of a bleed management protocol.
“Following that last case, we revised and reviewed the bleed management guidelines in view of the fact that there might potentially be an interaction between the amount of replacement therapy and thrombin generation,” Dr. Pasi said. Since introduction of that protocol, there have been no related thrombotic events.
The majority of adverse events reported were mild and deemed not related to the study drug, Dr. Pasi said. These included headache, injection site erythema, and arthralgia. A total of 14 subjects – all of whom were positive for hepatitis C at baseline – experienced rises in ALT levels but these were asymptomatic and resolved spontaneously.
One patient with chronic active hepatitis C infection also showed significant ALT/AST elevation which led to discontinuation of treatment.
In an interview, Dr. Pasi said one of the biggest advantages of fitusiran was that it could be used in patients with hemophilia A and B. “You’ve got patients with hemophilia B who’ve got no options at the moment. That would be an obvious specific group that would gain from this.”
Another advantage was fitusiran’s stability and dosing, he said, pointing out that the treatment was fixed dosing and stable at room temperature. Fitusiran is now undergoing phase 3 trials.
The study was funded by Sanofi Genzyme and Alnylam Pharmaceuticals, and six authors were employees of Sanofi Genzyme. Dr. Pasi reported financial relationships with pharmaceutical companies, including Alnylam.
SOURCE: Pasi J et al. 2019 ISTH Congress, Abstract OC 11.3.
REPORTING FROM 2019 ISTH CONGRESS
Early phase trial shows durable responses to gene therapy for hemophilia A
MELBOURNE – A gene therapy treatment for hemophilia A has shown sustained reductions in bleeding rates 3 years after treatment, with no major safety issues, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Valoctocogene roxaparvovec is an investigational gene therapy that involves using an adenovirus-associated virus to deliver the gene for clotting factor VIII.

John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust, presented the 3-year efficacy and safety results from the phase 1/2 trial of the therapy, involving 15 men with hemophilia A without inhibitors who received a single intravenous dose – either 4 x 1013 vector genomes (vg) per kg or 6 x 1013 vg/kg – of the therapy.
Participants’ mean annualized bleeding rate at baseline ranged from 6.5 among men who had been receiving prophylactic therapy to 25 among those who had been historically been treated on demand.
The treatment was associated with a substantial, significant reduction in mean annualized bleed rates; a 96% reduction in the 6 x 1013 vg/kg group by year 3, and 92% reduction in the 4 x 1013 vg/kg group by year 2.
By year 3, 86% of patients in the higher dose group had not experienced a bleed in the prior 12 months, all patients were off prophylaxis, and all had experienced resolution of target joints.
Mean factor VIII usage also decreased significantly, with a 96% reduction by year 3 in the higher dose cohort, and a 97% reduction by year 2 in the lower dose cohort.
The study also showed significant improvements in quality of life across all domains, Dr. Pasi reported.
There were no significant safety concerns raised during the study. Several patients experienced mild to moderate, transient rises in alanine aminotransferase levels at around 8-16 weeks after treatment, but there was no significant impact on liver function or on corticosteroid use. Two patients reported mild infusion reactions, which resolved within 48 hours with altering treatment.
The researchers also examined durability of factor VIII activity levels following the gene therapy, which was monitored using chromogenic assays. This revealed that after the initial increase following therapy, the factor VIII levels plateaued between years 2 and 3.
“We’ve got what we feel is really good clinical evidence of a persistent effect and we think this is dramatic,” Dr. Pasi said. A phase 3 trial is now underway.
A commenter from the audience, who remarked that the data were incredible and would make a huge difference for patients, asked about whether this represented a possible cure for the disease.
It’s premature to talk about a cure, Dr. Pasi said.
“It’s like watching paint dry; it’s going to take years before we know where we are,” he said in an interview.
However, this could represent massive and transformational change in the management of hemophilia A, he added.
On the question of whether this approach might also work in patients with inhibitors, Dr. Pasi said there were animal data suggesting that gene therapy could work in individuals with inhibitors, but the focus for the moment was on patients without inhibitors.
“But for patients that previously had a history of inhibitors and are now tolerant, that’s quite a significant group of patients that we were going to have to think about how we deal with that in due course,” he said.
The study was sponsored by manufacturer BioMarin Pharmaceutical. Dr. Pasi reported financial relationships with the study sponsor and other companies.
SOURCE: Pasi KJ et al. 2019 ISTH Congress, Abstract LB 01.2.
MELBOURNE – A gene therapy treatment for hemophilia A has shown sustained reductions in bleeding rates 3 years after treatment, with no major safety issues, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Valoctocogene roxaparvovec is an investigational gene therapy that involves using an adenovirus-associated virus to deliver the gene for clotting factor VIII.
John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust, presented the 3-year efficacy and safety results from the phase 1/2 trial of the therapy, involving 15 men with hemophilia A without inhibitors who received a single intravenous dose – either 4 x 1013 vector genomes (vg) per kg or 6 x 1013 vg/kg – of the therapy.
Participants’ mean annualized bleeding rate at baseline ranged from 6.5 among men who had been receiving prophylactic therapy to 25 among those who had been historically been treated on demand.
The treatment was associated with a substantial, significant reduction in mean annualized bleed rates; a 96% reduction in the 6 x 1013 vg/kg group by year 3, and 92% reduction in the 4 x 1013 vg/kg group by year 2.
By year 3, 86% of patients in the higher dose group had not experienced a bleed in the prior 12 months, all patients were off prophylaxis, and all had experienced resolution of target joints.
Mean factor VIII usage also decreased significantly, with a 96% reduction by year 3 in the higher dose cohort, and a 97% reduction by year 2 in the lower dose cohort.
The study also showed significant improvements in quality of life across all domains, Dr. Pasi reported.
There were no significant safety concerns raised during the study. Several patients experienced mild to moderate, transient rises in alanine aminotransferase levels at around 8-16 weeks after treatment, but there was no significant impact on liver function or on corticosteroid use. Two patients reported mild infusion reactions, which resolved within 48 hours with altering treatment.
The researchers also examined durability of factor VIII activity levels following the gene therapy, which was monitored using chromogenic assays. This revealed that after the initial increase following therapy, the factor VIII levels plateaued between years 2 and 3.
“We’ve got what we feel is really good clinical evidence of a persistent effect and we think this is dramatic,” Dr. Pasi said. A phase 3 trial is now underway.
A commenter from the audience, who remarked that the data were incredible and would make a huge difference for patients, asked about whether this represented a possible cure for the disease.
It’s premature to talk about a cure, Dr. Pasi said.
“It’s like watching paint dry; it’s going to take years before we know where we are,” he said in an interview.
However, this could represent massive and transformational change in the management of hemophilia A, he added.
On the question of whether this approach might also work in patients with inhibitors, Dr. Pasi said there were animal data suggesting that gene therapy could work in individuals with inhibitors, but the focus for the moment was on patients without inhibitors.
“But for patients that previously had a history of inhibitors and are now tolerant, that’s quite a significant group of patients that we were going to have to think about how we deal with that in due course,” he said.
The study was sponsored by manufacturer BioMarin Pharmaceutical. Dr. Pasi reported financial relationships with the study sponsor and other companies.
SOURCE: Pasi KJ et al. 2019 ISTH Congress, Abstract LB 01.2.
MELBOURNE – A gene therapy treatment for hemophilia A has shown sustained reductions in bleeding rates 3 years after treatment, with no major safety issues, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Valoctocogene roxaparvovec is an investigational gene therapy that involves using an adenovirus-associated virus to deliver the gene for clotting factor VIII.
John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust, presented the 3-year efficacy and safety results from the phase 1/2 trial of the therapy, involving 15 men with hemophilia A without inhibitors who received a single intravenous dose – either 4 x 1013 vector genomes (vg) per kg or 6 x 1013 vg/kg – of the therapy.
Participants’ mean annualized bleeding rate at baseline ranged from 6.5 among men who had been receiving prophylactic therapy to 25 among those who had been historically been treated on demand.
The treatment was associated with a substantial, significant reduction in mean annualized bleed rates; a 96% reduction in the 6 x 1013 vg/kg group by year 3, and 92% reduction in the 4 x 1013 vg/kg group by year 2.
By year 3, 86% of patients in the higher dose group had not experienced a bleed in the prior 12 months, all patients were off prophylaxis, and all had experienced resolution of target joints.
Mean factor VIII usage also decreased significantly, with a 96% reduction by year 3 in the higher dose cohort, and a 97% reduction by year 2 in the lower dose cohort.
The study also showed significant improvements in quality of life across all domains, Dr. Pasi reported.
There were no significant safety concerns raised during the study. Several patients experienced mild to moderate, transient rises in alanine aminotransferase levels at around 8-16 weeks after treatment, but there was no significant impact on liver function or on corticosteroid use. Two patients reported mild infusion reactions, which resolved within 48 hours with altering treatment.
The researchers also examined durability of factor VIII activity levels following the gene therapy, which was monitored using chromogenic assays. This revealed that after the initial increase following therapy, the factor VIII levels plateaued between years 2 and 3.
“We’ve got what we feel is really good clinical evidence of a persistent effect and we think this is dramatic,” Dr. Pasi said. A phase 3 trial is now underway.
A commenter from the audience, who remarked that the data were incredible and would make a huge difference for patients, asked about whether this represented a possible cure for the disease.
It’s premature to talk about a cure, Dr. Pasi said.
“It’s like watching paint dry; it’s going to take years before we know where we are,” he said in an interview.
However, this could represent massive and transformational change in the management of hemophilia A, he added.
On the question of whether this approach might also work in patients with inhibitors, Dr. Pasi said there were animal data suggesting that gene therapy could work in individuals with inhibitors, but the focus for the moment was on patients without inhibitors.
“But for patients that previously had a history of inhibitors and are now tolerant, that’s quite a significant group of patients that we were going to have to think about how we deal with that in due course,” he said.
The study was sponsored by manufacturer BioMarin Pharmaceutical. Dr. Pasi reported financial relationships with the study sponsor and other companies.
SOURCE: Pasi KJ et al. 2019 ISTH Congress, Abstract LB 01.2.
REPORTING FROM 2019 ISTH CONGRESS
BRCA2 mutations linked to childhood NHL
Pediatric non-Hodgkin lymphomas should be added to the list of cancers associated with BRCA2 mutations, and survivors of childhood NHL should be considered for genetic counseling, investigators suggest.
Among 1,380 survivors of childhood lymphomas, those who were retrospectively found to be carriers of BRCA2 mutations had a fivefold higher risk for non-Hodgkin lymphoma than controls without cancer, reported Zhaoming Wang, PhD, and colleagues from St. Jude Children’s Research Hospital in Memphis.
“Genetic counseling and the option of BRCA2 genetic testing should be offered to survivors of pediatric or adolescent non–Hodgkin lymphoma, particularly those with a family history of BRCA2-associated cancers,” they wrote in JAMA Oncology.
The investigators had previously reported that BRCA2 was the third-most frequently mutated gene among 3006 survivors of childhood cancers, with the highest number of mutations seen in lymphoma survivors. In that study, 7 of 586 survivors of Hodgkin and non-Hodgkin lymphoma (1.2%) were found to carry BRCA2 mutations.
In the current study, the investigators performed germline whole-genome sequencing on samples from 815 survivors of childhood Hodgkin lymphoma and 748 survivors of non-Hodgkin lymphoma from the St. Jude Lifetime Cohort and Childhood Cancer Survivor studies and compared the data with those of controls without cancer from the Genome Aggregation Database.
They identified mutations in five Hodgkin lymphoma survivors (0.6%) and eight non-Hodgkin lymphoma survivors.
A comparison of cancer risk among lymphoma survivors and controls found that non-Hodgkin lymphoma survivors and BRCA2 carriers had an odds ratio for cancer of 5.0, compared with controls who were not BRCA2 carriers (P less than .001). Among Hodgkin lymphoma survivors the OR for carriers vs. controls was 2.1, but was not statistically significant.
Available family histories for seven of the eight non-Hodgkin lymphoma BRCA2 mutation carriers showed histories of BRCA2-linked cancers, including breast, prostate, and pancreas tumors and malignant melanoma.
“Survivors whose test results are positive for mutation should be offered surveillance for BRCA2-associated cancers, such as breast and ovarian, and counseled about cancer risk–reducing strategies. Currently, it remains unclear whether surveillance for non–Hodgkin lymphoma is associated with early detection of lymphomas or with other medical advantages,” the investigators wrote.
“This study was funded by a grant to St Jude Children’s Research Hospital from the American Lebanese Syrian Associated Charities and by grants to St Jude Children’s Research Hospital from the National Institutes of Health. The authors reported having no conflicts of interest.
SOURCE: Wang Z et al. JAMA Oncology. 2019 Jul 25. doi: 10.1001/jamaoncol.2019.2203.
Pediatric non-Hodgkin lymphomas should be added to the list of cancers associated with BRCA2 mutations, and survivors of childhood NHL should be considered for genetic counseling, investigators suggest.
Among 1,380 survivors of childhood lymphomas, those who were retrospectively found to be carriers of BRCA2 mutations had a fivefold higher risk for non-Hodgkin lymphoma than controls without cancer, reported Zhaoming Wang, PhD, and colleagues from St. Jude Children’s Research Hospital in Memphis.
“Genetic counseling and the option of BRCA2 genetic testing should be offered to survivors of pediatric or adolescent non–Hodgkin lymphoma, particularly those with a family history of BRCA2-associated cancers,” they wrote in JAMA Oncology.
The investigators had previously reported that BRCA2 was the third-most frequently mutated gene among 3006 survivors of childhood cancers, with the highest number of mutations seen in lymphoma survivors. In that study, 7 of 586 survivors of Hodgkin and non-Hodgkin lymphoma (1.2%) were found to carry BRCA2 mutations.
In the current study, the investigators performed germline whole-genome sequencing on samples from 815 survivors of childhood Hodgkin lymphoma and 748 survivors of non-Hodgkin lymphoma from the St. Jude Lifetime Cohort and Childhood Cancer Survivor studies and compared the data with those of controls without cancer from the Genome Aggregation Database.
They identified mutations in five Hodgkin lymphoma survivors (0.6%) and eight non-Hodgkin lymphoma survivors.
A comparison of cancer risk among lymphoma survivors and controls found that non-Hodgkin lymphoma survivors and BRCA2 carriers had an odds ratio for cancer of 5.0, compared with controls who were not BRCA2 carriers (P less than .001). Among Hodgkin lymphoma survivors the OR for carriers vs. controls was 2.1, but was not statistically significant.
Available family histories for seven of the eight non-Hodgkin lymphoma BRCA2 mutation carriers showed histories of BRCA2-linked cancers, including breast, prostate, and pancreas tumors and malignant melanoma.
“Survivors whose test results are positive for mutation should be offered surveillance for BRCA2-associated cancers, such as breast and ovarian, and counseled about cancer risk–reducing strategies. Currently, it remains unclear whether surveillance for non–Hodgkin lymphoma is associated with early detection of lymphomas or with other medical advantages,” the investigators wrote.
“This study was funded by a grant to St Jude Children’s Research Hospital from the American Lebanese Syrian Associated Charities and by grants to St Jude Children’s Research Hospital from the National Institutes of Health. The authors reported having no conflicts of interest.
SOURCE: Wang Z et al. JAMA Oncology. 2019 Jul 25. doi: 10.1001/jamaoncol.2019.2203.
Pediatric non-Hodgkin lymphomas should be added to the list of cancers associated with BRCA2 mutations, and survivors of childhood NHL should be considered for genetic counseling, investigators suggest.
Among 1,380 survivors of childhood lymphomas, those who were retrospectively found to be carriers of BRCA2 mutations had a fivefold higher risk for non-Hodgkin lymphoma than controls without cancer, reported Zhaoming Wang, PhD, and colleagues from St. Jude Children’s Research Hospital in Memphis.
“Genetic counseling and the option of BRCA2 genetic testing should be offered to survivors of pediatric or adolescent non–Hodgkin lymphoma, particularly those with a family history of BRCA2-associated cancers,” they wrote in JAMA Oncology.
The investigators had previously reported that BRCA2 was the third-most frequently mutated gene among 3006 survivors of childhood cancers, with the highest number of mutations seen in lymphoma survivors. In that study, 7 of 586 survivors of Hodgkin and non-Hodgkin lymphoma (1.2%) were found to carry BRCA2 mutations.
In the current study, the investigators performed germline whole-genome sequencing on samples from 815 survivors of childhood Hodgkin lymphoma and 748 survivors of non-Hodgkin lymphoma from the St. Jude Lifetime Cohort and Childhood Cancer Survivor studies and compared the data with those of controls without cancer from the Genome Aggregation Database.
They identified mutations in five Hodgkin lymphoma survivors (0.6%) and eight non-Hodgkin lymphoma survivors.
A comparison of cancer risk among lymphoma survivors and controls found that non-Hodgkin lymphoma survivors and BRCA2 carriers had an odds ratio for cancer of 5.0, compared with controls who were not BRCA2 carriers (P less than .001). Among Hodgkin lymphoma survivors the OR for carriers vs. controls was 2.1, but was not statistically significant.
Available family histories for seven of the eight non-Hodgkin lymphoma BRCA2 mutation carriers showed histories of BRCA2-linked cancers, including breast, prostate, and pancreas tumors and malignant melanoma.
“Survivors whose test results are positive for mutation should be offered surveillance for BRCA2-associated cancers, such as breast and ovarian, and counseled about cancer risk–reducing strategies. Currently, it remains unclear whether surveillance for non–Hodgkin lymphoma is associated with early detection of lymphomas or with other medical advantages,” the investigators wrote.
“This study was funded by a grant to St Jude Children’s Research Hospital from the American Lebanese Syrian Associated Charities and by grants to St Jude Children’s Research Hospital from the National Institutes of Health. The authors reported having no conflicts of interest.
SOURCE: Wang Z et al. JAMA Oncology. 2019 Jul 25. doi: 10.1001/jamaoncol.2019.2203.
FROM JAMA ONCOLOGY
Sickle cell unit running 24/7 reduces readmissions, emergency visits
FORT LAUDERDALE, FLA. — A dedicated, 24-hour, 7-day-a-week sickle cell inpatient observation unit staffed by a multidisciplinary care team significantly reduced inpatient admissions and emergency department visits per patient, according to an analysis of a Philadelphia program.

“These findings confirm the need for an individualized approach to treatment,” Sanaa Rizk, MD, director of the hereditary anemia program at Thomas Jefferson University Hospital, Philadelphia, said at the annual meeting of the Foundation for Sickle Cell Disease Research. “The potential strength of a multidisciplinary approach and personalized interventions toward high-utilizing subpopulations may offer the greatest impact.”
The study evaluated what Dr. Rizk called “the second clinical transformation” in care of sickle cell disease patients, which Thomas Jefferson University implemented in 2015. The comprehensive sickle cell center first opened in 2003, and the first transformation in November 2013 consisted of opening a four-bed sickle cell day unit to treat uncomplicated sickle cell vaso-occlusive crises with personalized pain treatment protocols (including IV fluids and opioids). It was staffed by a nurse practitioner, medical assistant, and two registered nurses from 8 a.m. to 5 p.m.
The second transformation transferred care to the inpatient observation unit on the hospital floor with access to 12 patient beds. A sickle cell nurse practitioner sees patients for same-day appointments, conducts sick visits, performs outreach, and handles follow-up with patients. The rest of the multidisciplinary team includes hospitalists, hematologists, internists, and a social worker who performs weekly inpatient rounds and meets monthly with ED leaders and pharmacists.
With the first transformation, ED visits per patient fell from 3.67 to 2.14 a year (P less than .001), and inpatient admissions per patient fell from 1.33 to 0.63 (P less than .0001), Dr. Rizk reported.
The second transformation reduced those per-patient rates even further, to 0.47 ED visits (P less than .01) and 0.29 inpatient admissions (P less than .001), she said.
“The expansion of the service reduced admissions and ED use significantly,” Dr. Rizk said.
She added that a subanalysis of the high-utilizer subgroup showed a decrease in average total medical charges by approximately $100,000/patient per year.
Dr. Rizk reported having no relevant financial disclosures.
SOURCE: Rizk S et al. FSCDR 2019, Abstract JSCDH-D-19-00049.
FORT LAUDERDALE, FLA. — A dedicated, 24-hour, 7-day-a-week sickle cell inpatient observation unit staffed by a multidisciplinary care team significantly reduced inpatient admissions and emergency department visits per patient, according to an analysis of a Philadelphia program.
“These findings confirm the need for an individualized approach to treatment,” Sanaa Rizk, MD, director of the hereditary anemia program at Thomas Jefferson University Hospital, Philadelphia, said at the annual meeting of the Foundation for Sickle Cell Disease Research. “The potential strength of a multidisciplinary approach and personalized interventions toward high-utilizing subpopulations may offer the greatest impact.”
The study evaluated what Dr. Rizk called “the second clinical transformation” in care of sickle cell disease patients, which Thomas Jefferson University implemented in 2015. The comprehensive sickle cell center first opened in 2003, and the first transformation in November 2013 consisted of opening a four-bed sickle cell day unit to treat uncomplicated sickle cell vaso-occlusive crises with personalized pain treatment protocols (including IV fluids and opioids). It was staffed by a nurse practitioner, medical assistant, and two registered nurses from 8 a.m. to 5 p.m.
The second transformation transferred care to the inpatient observation unit on the hospital floor with access to 12 patient beds. A sickle cell nurse practitioner sees patients for same-day appointments, conducts sick visits, performs outreach, and handles follow-up with patients. The rest of the multidisciplinary team includes hospitalists, hematologists, internists, and a social worker who performs weekly inpatient rounds and meets monthly with ED leaders and pharmacists.
With the first transformation, ED visits per patient fell from 3.67 to 2.14 a year (P less than .001), and inpatient admissions per patient fell from 1.33 to 0.63 (P less than .0001), Dr. Rizk reported.
The second transformation reduced those per-patient rates even further, to 0.47 ED visits (P less than .01) and 0.29 inpatient admissions (P less than .001), she said.
“The expansion of the service reduced admissions and ED use significantly,” Dr. Rizk said.
She added that a subanalysis of the high-utilizer subgroup showed a decrease in average total medical charges by approximately $100,000/patient per year.
Dr. Rizk reported having no relevant financial disclosures.
SOURCE: Rizk S et al. FSCDR 2019, Abstract JSCDH-D-19-00049.
FORT LAUDERDALE, FLA. — A dedicated, 24-hour, 7-day-a-week sickle cell inpatient observation unit staffed by a multidisciplinary care team significantly reduced inpatient admissions and emergency department visits per patient, according to an analysis of a Philadelphia program.
“These findings confirm the need for an individualized approach to treatment,” Sanaa Rizk, MD, director of the hereditary anemia program at Thomas Jefferson University Hospital, Philadelphia, said at the annual meeting of the Foundation for Sickle Cell Disease Research. “The potential strength of a multidisciplinary approach and personalized interventions toward high-utilizing subpopulations may offer the greatest impact.”
The study evaluated what Dr. Rizk called “the second clinical transformation” in care of sickle cell disease patients, which Thomas Jefferson University implemented in 2015. The comprehensive sickle cell center first opened in 2003, and the first transformation in November 2013 consisted of opening a four-bed sickle cell day unit to treat uncomplicated sickle cell vaso-occlusive crises with personalized pain treatment protocols (including IV fluids and opioids). It was staffed by a nurse practitioner, medical assistant, and two registered nurses from 8 a.m. to 5 p.m.
The second transformation transferred care to the inpatient observation unit on the hospital floor with access to 12 patient beds. A sickle cell nurse practitioner sees patients for same-day appointments, conducts sick visits, performs outreach, and handles follow-up with patients. The rest of the multidisciplinary team includes hospitalists, hematologists, internists, and a social worker who performs weekly inpatient rounds and meets monthly with ED leaders and pharmacists.
With the first transformation, ED visits per patient fell from 3.67 to 2.14 a year (P less than .001), and inpatient admissions per patient fell from 1.33 to 0.63 (P less than .0001), Dr. Rizk reported.
The second transformation reduced those per-patient rates even further, to 0.47 ED visits (P less than .01) and 0.29 inpatient admissions (P less than .001), she said.
“The expansion of the service reduced admissions and ED use significantly,” Dr. Rizk said.
She added that a subanalysis of the high-utilizer subgroup showed a decrease in average total medical charges by approximately $100,000/patient per year.
Dr. Rizk reported having no relevant financial disclosures.
SOURCE: Rizk S et al. FSCDR 2019, Abstract JSCDH-D-19-00049.
REPORTING FROM FSCDR 2019
Ibrutinib-venetoclax found highly active in hard-to-treat CLL
The strategy of simultaneously inhibiting proliferation and reactivating apoptosis can eradicate chronic lymphocytic leukemia (CLL) in a large share of patients, suggest results from the phase 2 CLARITY trial.

“Both ibrutinib and venetoclax are active in CLL with improved survival; however, as monotherapies, both currently are given until disease progression,” wrote Peter Hillmen, MBChB, PhD, St. James’s University Hospital, Leeds, England, and his colleagues.
In the single-arm, open-label trial, the investigators treated 53 patients with relapsed or refractory CLL with combination ibrutinib (Imbruvica), a small-molecule inhibitor of Bruton’s tyrosine kinase, and venetoclax (Venclexta), a small molecule inhibitor of the anti-apoptotic protein Bcl-2. The primary endpoint was MRD negativity, defined as presence of fewer than one CLL cell in 10,000 leukocytes, after 12 months of combination therapy.
Results reported in the Journal of Clinical Oncology showed that the combination was highly active, with 53% of patients achieving MRD negativity in the blood and 36% achieving MRD negativity in the marrow.
Most patients, 89%, had a treatment response, and slightly more than half, 51%, achieved a complete remission. With a median 21.1-month follow-up, only a single patient experienced progression and all were still alive.
Adverse effects were generally manageable. Grade 3-4 adverse events of special interest included 34 cases of neutropenia and 1 case of biochemical tumor lysis syndrome that was managed by delaying venetoclax.
“We have demonstrated promising efficacy that indicates potent synergy between ibrutinib and venetoclax for inducing MRD-negative responses with manageable adverse effects,” the investigators wrote. “The observation that a significant proportion of patients experience MRD-negative remission indicates that this combination can be given for a limited period and then stopped after patients achieve a deep remission.”
Whether the combination leads to permanent disease eradication in certain patients is still unclear, the investigators added.
The trial was supported by Bloodwise under the Trials Acceleration Programme, by the National Institute for Health Research Leeds Clinical Research Facility, and by an unrestricted educational grant from Janssen-Cilag and AbbVie. Ibrutinib was provided free of charge by Janssen-Cilag, and venetoclax was provided free of charge by AbbVie. Dr. Hillman reported financial relationships with Janssen, AbbVie, Roche, Pharmacyclics, and Gilead Sciences.
SOURCE: Hillmen P et al. J Clin Oncol. 2019 Jul 11. doi: 10.1200/JCO.19.00894.
The strategy of simultaneously inhibiting proliferation and reactivating apoptosis can eradicate chronic lymphocytic leukemia (CLL) in a large share of patients, suggest results from the phase 2 CLARITY trial.
“Both ibrutinib and venetoclax are active in CLL with improved survival; however, as monotherapies, both currently are given until disease progression,” wrote Peter Hillmen, MBChB, PhD, St. James’s University Hospital, Leeds, England, and his colleagues.
In the single-arm, open-label trial, the investigators treated 53 patients with relapsed or refractory CLL with combination ibrutinib (Imbruvica), a small-molecule inhibitor of Bruton’s tyrosine kinase, and venetoclax (Venclexta), a small molecule inhibitor of the anti-apoptotic protein Bcl-2. The primary endpoint was MRD negativity, defined as presence of fewer than one CLL cell in 10,000 leukocytes, after 12 months of combination therapy.
Results reported in the Journal of Clinical Oncology showed that the combination was highly active, with 53% of patients achieving MRD negativity in the blood and 36% achieving MRD negativity in the marrow.
Most patients, 89%, had a treatment response, and slightly more than half, 51%, achieved a complete remission. With a median 21.1-month follow-up, only a single patient experienced progression and all were still alive.
Adverse effects were generally manageable. Grade 3-4 adverse events of special interest included 34 cases of neutropenia and 1 case of biochemical tumor lysis syndrome that was managed by delaying venetoclax.
“We have demonstrated promising efficacy that indicates potent synergy between ibrutinib and venetoclax for inducing MRD-negative responses with manageable adverse effects,” the investigators wrote. “The observation that a significant proportion of patients experience MRD-negative remission indicates that this combination can be given for a limited period and then stopped after patients achieve a deep remission.”
Whether the combination leads to permanent disease eradication in certain patients is still unclear, the investigators added.
The trial was supported by Bloodwise under the Trials Acceleration Programme, by the National Institute for Health Research Leeds Clinical Research Facility, and by an unrestricted educational grant from Janssen-Cilag and AbbVie. Ibrutinib was provided free of charge by Janssen-Cilag, and venetoclax was provided free of charge by AbbVie. Dr. Hillman reported financial relationships with Janssen, AbbVie, Roche, Pharmacyclics, and Gilead Sciences.
SOURCE: Hillmen P et al. J Clin Oncol. 2019 Jul 11. doi: 10.1200/JCO.19.00894.
The strategy of simultaneously inhibiting proliferation and reactivating apoptosis can eradicate chronic lymphocytic leukemia (CLL) in a large share of patients, suggest results from the phase 2 CLARITY trial.
“Both ibrutinib and venetoclax are active in CLL with improved survival; however, as monotherapies, both currently are given until disease progression,” wrote Peter Hillmen, MBChB, PhD, St. James’s University Hospital, Leeds, England, and his colleagues.
In the single-arm, open-label trial, the investigators treated 53 patients with relapsed or refractory CLL with combination ibrutinib (Imbruvica), a small-molecule inhibitor of Bruton’s tyrosine kinase, and venetoclax (Venclexta), a small molecule inhibitor of the anti-apoptotic protein Bcl-2. The primary endpoint was MRD negativity, defined as presence of fewer than one CLL cell in 10,000 leukocytes, after 12 months of combination therapy.
Results reported in the Journal of Clinical Oncology showed that the combination was highly active, with 53% of patients achieving MRD negativity in the blood and 36% achieving MRD negativity in the marrow.
Most patients, 89%, had a treatment response, and slightly more than half, 51%, achieved a complete remission. With a median 21.1-month follow-up, only a single patient experienced progression and all were still alive.
Adverse effects were generally manageable. Grade 3-4 adverse events of special interest included 34 cases of neutropenia and 1 case of biochemical tumor lysis syndrome that was managed by delaying venetoclax.
“We have demonstrated promising efficacy that indicates potent synergy between ibrutinib and venetoclax for inducing MRD-negative responses with manageable adverse effects,” the investigators wrote. “The observation that a significant proportion of patients experience MRD-negative remission indicates that this combination can be given for a limited period and then stopped after patients achieve a deep remission.”
Whether the combination leads to permanent disease eradication in certain patients is still unclear, the investigators added.
The trial was supported by Bloodwise under the Trials Acceleration Programme, by the National Institute for Health Research Leeds Clinical Research Facility, and by an unrestricted educational grant from Janssen-Cilag and AbbVie. Ibrutinib was provided free of charge by Janssen-Cilag, and venetoclax was provided free of charge by AbbVie. Dr. Hillman reported financial relationships with Janssen, AbbVie, Roche, Pharmacyclics, and Gilead Sciences.
SOURCE: Hillmen P et al. J Clin Oncol. 2019 Jul 11. doi: 10.1200/JCO.19.00894.
FROM THE JOURNAL OF CLINICAL ONCOLOGY