User login
Successful Treatment of Refractory Extensive Pityriasis Rubra Pilaris With Risankizumab and Acitretin
To the Editor:
Pityriasis rubra pilaris (PRP) is a rare papulosquamous condition with an unknown pathogenesis and limited efficacy data, which can make treatment challenging. Some cases of PRP spontaneously resolve in a few months, which is most common in the pediatric population.1 Pityriasis rubra pilaris in adults is likely to persist for years, and spontaneous resolution is unpredictable. Randomized clinical trials are difficult to perform due to the rarity of PRP.
Although there is no cure and no standard protocol for treating PRP, systemic retinoids historically are considered first-line therapy for moderate to severe cases.2 Additional management approaches include symptomatic control with moisturizers and psychological support. Alternative systemic treatments for moderate to severe cases include methotrexate, phototherapy, and cyclosporine.2
Pityriasis rubra pilaris demonstrates a favorable response to methotrexate treatment, especially in type I cases; however, patients on this alternative therapy should be monitored for severe adverse effects (eg, hepatotoxicity, pancytopenia, pneumonitis).2 Phototherapy should be approached with caution. Narrowband UVB, UVA1, and psoralen plus UVA therapy have successfully treated PRP; however, the response is variable. In some cases, the opposite effect can occur, in which the condition is photoaggravated. Phototherapy is a valid alternative form of treatment when used in combination with acitretin, and a phototest should be performed prior to starting this regimen. Cyclosporine is another immunosuppressant that can be considered for PRP treatment, though there are limited data demonstrating its efficacy.2
The introduction of biologic agents has changed the treatment approach for many dermatologic diseases, including PRP. Given the similar features between psoriasis and PRP, the biologics prescribed for psoriasis therapy also are used for patients with PRP that is challenging to treat, such as anti–tumor necrosis factor α inhibitors and IL inhibitors—specifically IL-17 and IL-23. Remission has been achieved with the use of biologics in combination with retinoid therapy.2
Biologic therapies used for PRP effectively inhibit cytokines and reduce the overall inflammatory processes involved in the development of the scaly patches and plaques seen in this condition. However, most reported clinical experiences are case studies, and more research in the form of randomized clinical trials is needed to understand the efficacy and long-term effects of this form of treatment in PRP. We present a case of a patient with refractory adult subtype I PRP that was successfully treated with the IL-23 inhibitor risankizumab.
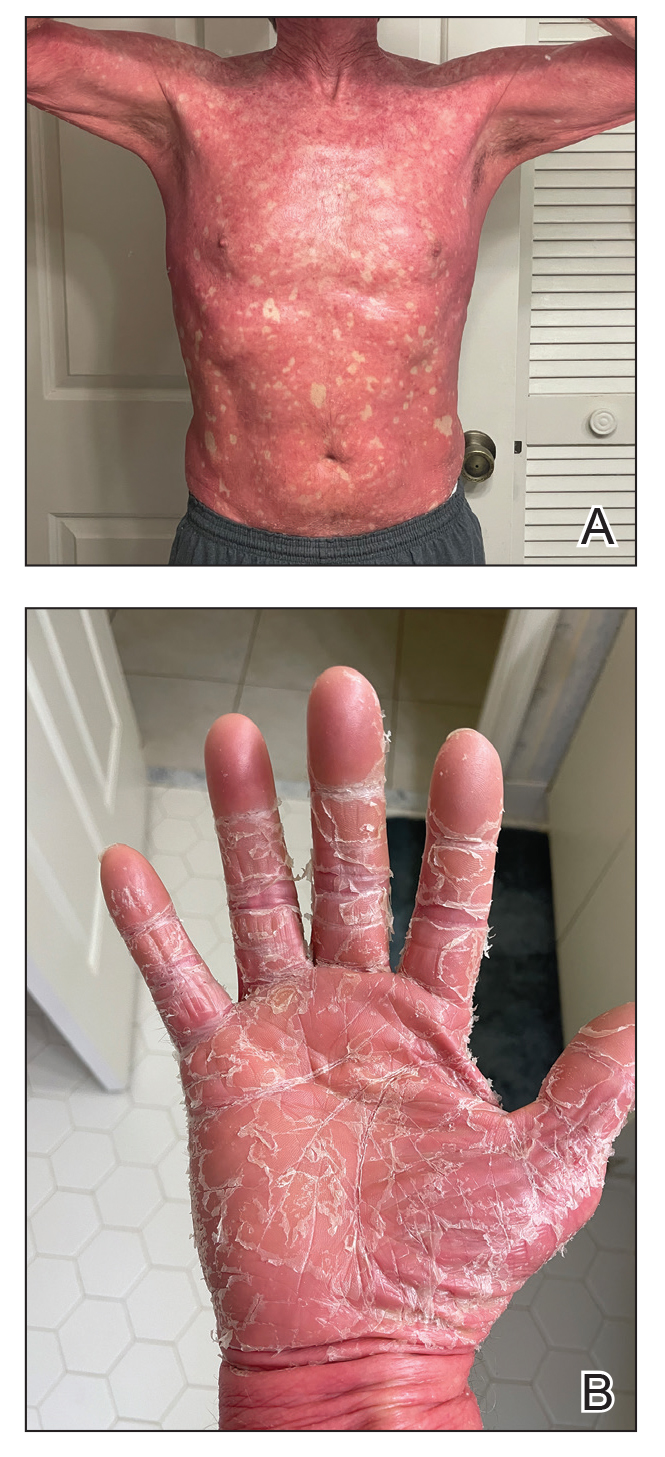
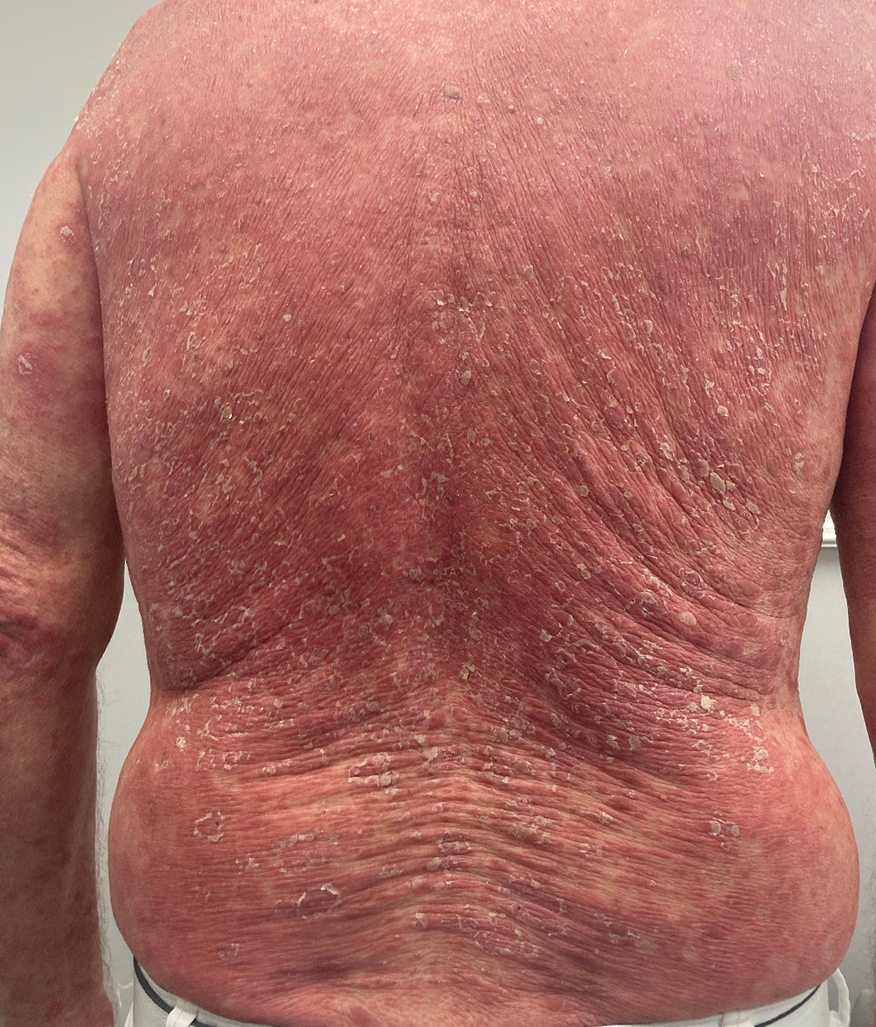
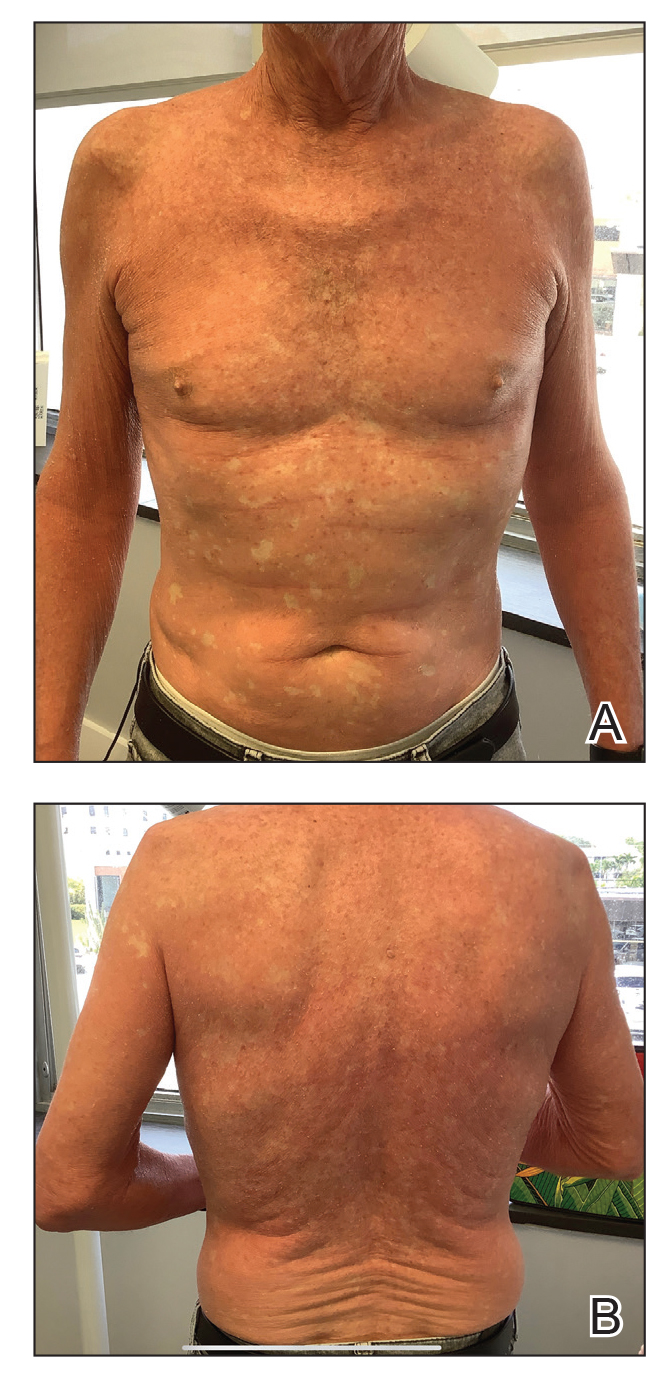
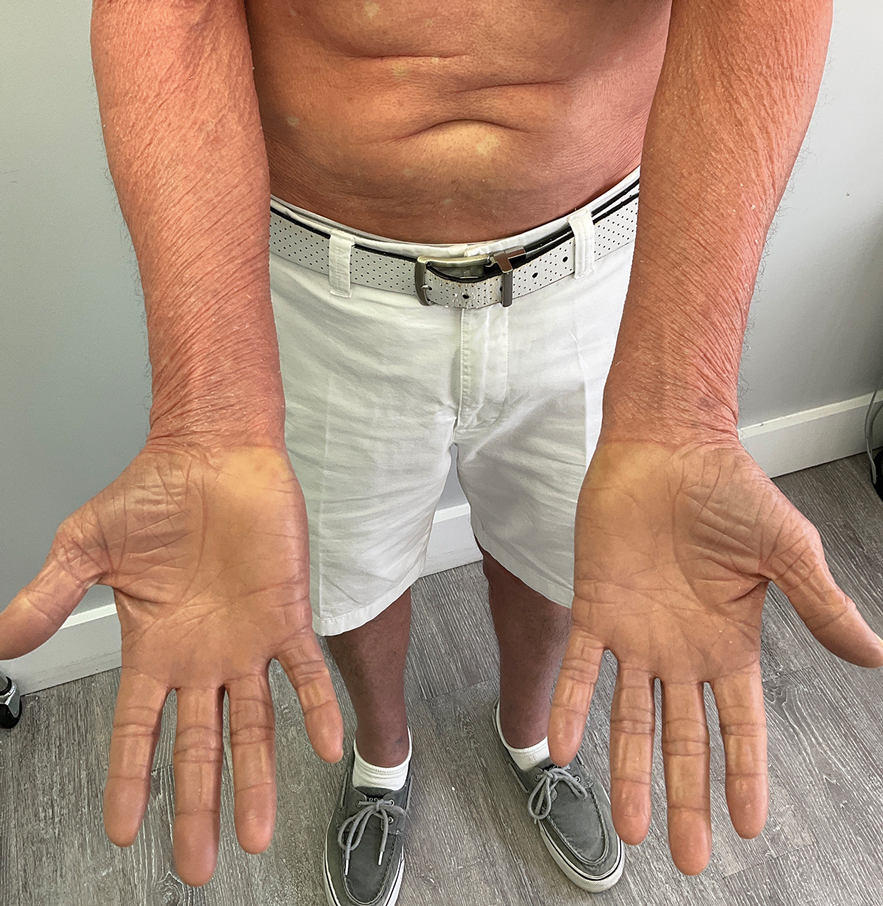
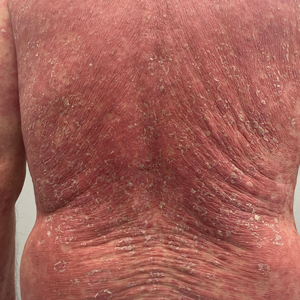
A 65-year-old man was referred to Florida Academic Dermatology Center (Coral Gables, Florida) with biopsy-proven PRP diagnosed 1 year prior. The patient reported experiencing a debilitating quality of life in the year since diagnosis (Figure 1). Treatment attempts with dupilumab, tralokinumab, intramuscular steroid injections, and topical corticosteroids had failed (Figure 2). Following evaluation at Florida Academic Dermatology Center, the patient was started on acitretin 25 mg every other day and received an initial subcutaneous injection of ixekizumab 160 mg (an IL-17 inhibitor) followed 2 weeks later by a second injection of 80 mg. After the 2 doses of ixekizumab, the patient’s condition worsened with the development of pinpoint hemorrhagic lesions. The medication was discontinued, and he was started on risankizumab 150 mg at the approved dosing regimen for plaque psoriasis in combination with the acitretin therapy. Prior to starting risankizumab, the affected body surface area (BSA) was 80%. At 1-month follow-up, he showed improvement with reduction in scaling and erythema and an affected BSA of 30% (Figure 3). At 4-month follow-up, he continued showing improvement with an affected BSA of 10% (Figure 4). Acitretin was discontinued, and the patient has been successfully maintained on risankizumab 150 mg/mL subcutaneous injections every 12 weeks since.
Oral retinoid therapy historically was considered first-line therapy for moderate to severe PRP. A systematic review (N=105) of retinoid therapies showed 83% of patients with PRP who were treated with acitretin plus biologic therapy had a favorable response, whereas only 36% of patients treated with acitretin as monotherapy had the same response, highlighting the importance of dual therapy.3 The use of ustekinumab, ixekizumab, and secukinumab (IL-17 inhibitors) for refractory PRP has been well documented, but a PubMed search of articles indexed for MEDLINE using the search terms risankizumab and pityriasis rubra pilaris yielded only 8 published cases of risankizumab for treatment of PRP.4-8 All patients were diagnosed with refractory PRP, and multiple treatment modalities failed.
Ustekinumab has been shown to create a rapid response and maintain it long term, especially in patients with type 1 PRP who did not respond to systemic therapies or anti–tumor necrosis factor α agents.2 An open-label, single-arm clinical trial found secukinumab was an effective therapy for PRP and demonstrated transcription heterogeneity of this dermatologic condition.9 The researchers proposed that some patients may respond to IL-17 inhibitors but others may not due to the differences in RNA molecules transcribed.9 Our patient demonstrated worsening of his condition with an IL-17 inhibitor but experienced remarkable improvement with risankizumab, an IL-23 inhibitor.
Risankizumab is indicated for the treatment of adults with moderate to severe plaque psoriasis. This humanized IgG1 monoclonal antibody targets the p19 subunit of IL-23, inhibiting its role in the pathogenic helper T cell (TH17) pathway. Research has shown that it is an efficacious and well-tolerated treatment modality for psoriatic conditions.10 It is well known that PRP and psoriasis have similar cytokine activations; therefore, we propose that combination therapy with risankizumab and acitretin may show promise for refractory PRP.
- Gelmetti C, Schiuma AA, Cerri D, et al. Pityriasis rubra pilaris in childhood: a long-term study of 29 cases. Pediatr Dermatol. 1986;3:446-451. doi:10.1111/j.1525-1470.1986.tb00648.x
- Moretta G, De Luca EV, Di Stefani A. Management of refractory pityriasis rubra pilaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2017;10:451-457. doi:10.2147/CCID.S124351
- Engelmann C, Elsner P, Miguel D. Treatment of pityriasis rubra pilaris type I: a systematic review. Eur J Dermatol. 2019;29:524-537. doi:10.1684/ejd.2019.3641
- Ricar J, Cetkovska P. Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab. Br J Dermatol. 2021;184:E148. doi:10.1111/bjd.19681
- Brocco E, Laffitte E. Risankizumab for pityriasis rubra pilaris. Clin Exp Dermatol. 2021;46:1322-1324. doi:10.1111/ced.14715
- Duarte B, Paiva Lopes MJ. Response to: ‘Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab.’ Br J Dermatol. 2021;185:235-236. doi:10.1111/bjd.20061
- Kromer C, Schön MP, Mössner R. Treatment of pityriasis rubra pilaris with risankizumab in two cases. J Dtsch Dermatol Ges. 2021;19:1207-1209. doi:10.1111/ddg.14504
- Kołt-Kamińska M, Osińska A, Kaznowska E, et al. Successful treatment of pityriasis rubra pilaris with risankizumab in children. Dermatol Ther (Heidelb). 2023;13:2431-2441. doi:10.1007/s13555-023-01005-y
- Boudreaux BW, Pincelli TP, Bhullar PK, et al. Secukinumab for the treatment of adult-onset pityriasis rubra pilaris: a single-arm clinical trial with transcriptomic analysis. Br J Dermatol. 2022;187:650-658. doi:10.1111/bjd.21708
- Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156:649-658. doi:10.1001/jamadermatol.2020.0723
To the Editor:
Pityriasis rubra pilaris (PRP) is a rare papulosquamous condition with an unknown pathogenesis and limited efficacy data, which can make treatment challenging. Some cases of PRP spontaneously resolve in a few months, which is most common in the pediatric population.1 Pityriasis rubra pilaris in adults is likely to persist for years, and spontaneous resolution is unpredictable. Randomized clinical trials are difficult to perform due to the rarity of PRP.
Although there is no cure and no standard protocol for treating PRP, systemic retinoids historically are considered first-line therapy for moderate to severe cases.2 Additional management approaches include symptomatic control with moisturizers and psychological support. Alternative systemic treatments for moderate to severe cases include methotrexate, phototherapy, and cyclosporine.2
Pityriasis rubra pilaris demonstrates a favorable response to methotrexate treatment, especially in type I cases; however, patients on this alternative therapy should be monitored for severe adverse effects (eg, hepatotoxicity, pancytopenia, pneumonitis).2 Phototherapy should be approached with caution. Narrowband UVB, UVA1, and psoralen plus UVA therapy have successfully treated PRP; however, the response is variable. In some cases, the opposite effect can occur, in which the condition is photoaggravated. Phototherapy is a valid alternative form of treatment when used in combination with acitretin, and a phototest should be performed prior to starting this regimen. Cyclosporine is another immunosuppressant that can be considered for PRP treatment, though there are limited data demonstrating its efficacy.2
The introduction of biologic agents has changed the treatment approach for many dermatologic diseases, including PRP. Given the similar features between psoriasis and PRP, the biologics prescribed for psoriasis therapy also are used for patients with PRP that is challenging to treat, such as anti–tumor necrosis factor α inhibitors and IL inhibitors—specifically IL-17 and IL-23. Remission has been achieved with the use of biologics in combination with retinoid therapy.2
Biologic therapies used for PRP effectively inhibit cytokines and reduce the overall inflammatory processes involved in the development of the scaly patches and plaques seen in this condition. However, most reported clinical experiences are case studies, and more research in the form of randomized clinical trials is needed to understand the efficacy and long-term effects of this form of treatment in PRP. We present a case of a patient with refractory adult subtype I PRP that was successfully treated with the IL-23 inhibitor risankizumab.
A 65-year-old man was referred to Florida Academic Dermatology Center (Coral Gables, Florida) with biopsy-proven PRP diagnosed 1 year prior. The patient reported experiencing a debilitating quality of life in the year since diagnosis (Figure 1). Treatment attempts with dupilumab, tralokinumab, intramuscular steroid injections, and topical corticosteroids had failed (Figure 2). Following evaluation at Florida Academic Dermatology Center, the patient was started on acitretin 25 mg every other day and received an initial subcutaneous injection of ixekizumab 160 mg (an IL-17 inhibitor) followed 2 weeks later by a second injection of 80 mg. After the 2 doses of ixekizumab, the patient’s condition worsened with the development of pinpoint hemorrhagic lesions. The medication was discontinued, and he was started on risankizumab 150 mg at the approved dosing regimen for plaque psoriasis in combination with the acitretin therapy. Prior to starting risankizumab, the affected body surface area (BSA) was 80%. At 1-month follow-up, he showed improvement with reduction in scaling and erythema and an affected BSA of 30% (Figure 3). At 4-month follow-up, he continued showing improvement with an affected BSA of 10% (Figure 4). Acitretin was discontinued, and the patient has been successfully maintained on risankizumab 150 mg/mL subcutaneous injections every 12 weeks since.
Oral retinoid therapy historically was considered first-line therapy for moderate to severe PRP. A systematic review (N=105) of retinoid therapies showed 83% of patients with PRP who were treated with acitretin plus biologic therapy had a favorable response, whereas only 36% of patients treated with acitretin as monotherapy had the same response, highlighting the importance of dual therapy.3 The use of ustekinumab, ixekizumab, and secukinumab (IL-17 inhibitors) for refractory PRP has been well documented, but a PubMed search of articles indexed for MEDLINE using the search terms risankizumab and pityriasis rubra pilaris yielded only 8 published cases of risankizumab for treatment of PRP.4-8 All patients were diagnosed with refractory PRP, and multiple treatment modalities failed.
Ustekinumab has been shown to create a rapid response and maintain it long term, especially in patients with type 1 PRP who did not respond to systemic therapies or anti–tumor necrosis factor α agents.2 An open-label, single-arm clinical trial found secukinumab was an effective therapy for PRP and demonstrated transcription heterogeneity of this dermatologic condition.9 The researchers proposed that some patients may respond to IL-17 inhibitors but others may not due to the differences in RNA molecules transcribed.9 Our patient demonstrated worsening of his condition with an IL-17 inhibitor but experienced remarkable improvement with risankizumab, an IL-23 inhibitor.
Risankizumab is indicated for the treatment of adults with moderate to severe plaque psoriasis. This humanized IgG1 monoclonal antibody targets the p19 subunit of IL-23, inhibiting its role in the pathogenic helper T cell (TH17) pathway. Research has shown that it is an efficacious and well-tolerated treatment modality for psoriatic conditions.10 It is well known that PRP and psoriasis have similar cytokine activations; therefore, we propose that combination therapy with risankizumab and acitretin may show promise for refractory PRP.
To the Editor:
Pityriasis rubra pilaris (PRP) is a rare papulosquamous condition with an unknown pathogenesis and limited efficacy data, which can make treatment challenging. Some cases of PRP spontaneously resolve in a few months, which is most common in the pediatric population.1 Pityriasis rubra pilaris in adults is likely to persist for years, and spontaneous resolution is unpredictable. Randomized clinical trials are difficult to perform due to the rarity of PRP.
Although there is no cure and no standard protocol for treating PRP, systemic retinoids historically are considered first-line therapy for moderate to severe cases.2 Additional management approaches include symptomatic control with moisturizers and psychological support. Alternative systemic treatments for moderate to severe cases include methotrexate, phototherapy, and cyclosporine.2
Pityriasis rubra pilaris demonstrates a favorable response to methotrexate treatment, especially in type I cases; however, patients on this alternative therapy should be monitored for severe adverse effects (eg, hepatotoxicity, pancytopenia, pneumonitis).2 Phototherapy should be approached with caution. Narrowband UVB, UVA1, and psoralen plus UVA therapy have successfully treated PRP; however, the response is variable. In some cases, the opposite effect can occur, in which the condition is photoaggravated. Phototherapy is a valid alternative form of treatment when used in combination with acitretin, and a phototest should be performed prior to starting this regimen. Cyclosporine is another immunosuppressant that can be considered for PRP treatment, though there are limited data demonstrating its efficacy.2
The introduction of biologic agents has changed the treatment approach for many dermatologic diseases, including PRP. Given the similar features between psoriasis and PRP, the biologics prescribed for psoriasis therapy also are used for patients with PRP that is challenging to treat, such as anti–tumor necrosis factor α inhibitors and IL inhibitors—specifically IL-17 and IL-23. Remission has been achieved with the use of biologics in combination with retinoid therapy.2
Biologic therapies used for PRP effectively inhibit cytokines and reduce the overall inflammatory processes involved in the development of the scaly patches and plaques seen in this condition. However, most reported clinical experiences are case studies, and more research in the form of randomized clinical trials is needed to understand the efficacy and long-term effects of this form of treatment in PRP. We present a case of a patient with refractory adult subtype I PRP that was successfully treated with the IL-23 inhibitor risankizumab.
A 65-year-old man was referred to Florida Academic Dermatology Center (Coral Gables, Florida) with biopsy-proven PRP diagnosed 1 year prior. The patient reported experiencing a debilitating quality of life in the year since diagnosis (Figure 1). Treatment attempts with dupilumab, tralokinumab, intramuscular steroid injections, and topical corticosteroids had failed (Figure 2). Following evaluation at Florida Academic Dermatology Center, the patient was started on acitretin 25 mg every other day and received an initial subcutaneous injection of ixekizumab 160 mg (an IL-17 inhibitor) followed 2 weeks later by a second injection of 80 mg. After the 2 doses of ixekizumab, the patient’s condition worsened with the development of pinpoint hemorrhagic lesions. The medication was discontinued, and he was started on risankizumab 150 mg at the approved dosing regimen for plaque psoriasis in combination with the acitretin therapy. Prior to starting risankizumab, the affected body surface area (BSA) was 80%. At 1-month follow-up, he showed improvement with reduction in scaling and erythema and an affected BSA of 30% (Figure 3). At 4-month follow-up, he continued showing improvement with an affected BSA of 10% (Figure 4). Acitretin was discontinued, and the patient has been successfully maintained on risankizumab 150 mg/mL subcutaneous injections every 12 weeks since.
Oral retinoid therapy historically was considered first-line therapy for moderate to severe PRP. A systematic review (N=105) of retinoid therapies showed 83% of patients with PRP who were treated with acitretin plus biologic therapy had a favorable response, whereas only 36% of patients treated with acitretin as monotherapy had the same response, highlighting the importance of dual therapy.3 The use of ustekinumab, ixekizumab, and secukinumab (IL-17 inhibitors) for refractory PRP has been well documented, but a PubMed search of articles indexed for MEDLINE using the search terms risankizumab and pityriasis rubra pilaris yielded only 8 published cases of risankizumab for treatment of PRP.4-8 All patients were diagnosed with refractory PRP, and multiple treatment modalities failed.
Ustekinumab has been shown to create a rapid response and maintain it long term, especially in patients with type 1 PRP who did not respond to systemic therapies or anti–tumor necrosis factor α agents.2 An open-label, single-arm clinical trial found secukinumab was an effective therapy for PRP and demonstrated transcription heterogeneity of this dermatologic condition.9 The researchers proposed that some patients may respond to IL-17 inhibitors but others may not due to the differences in RNA molecules transcribed.9 Our patient demonstrated worsening of his condition with an IL-17 inhibitor but experienced remarkable improvement with risankizumab, an IL-23 inhibitor.
Risankizumab is indicated for the treatment of adults with moderate to severe plaque psoriasis. This humanized IgG1 monoclonal antibody targets the p19 subunit of IL-23, inhibiting its role in the pathogenic helper T cell (TH17) pathway. Research has shown that it is an efficacious and well-tolerated treatment modality for psoriatic conditions.10 It is well known that PRP and psoriasis have similar cytokine activations; therefore, we propose that combination therapy with risankizumab and acitretin may show promise for refractory PRP.
- Gelmetti C, Schiuma AA, Cerri D, et al. Pityriasis rubra pilaris in childhood: a long-term study of 29 cases. Pediatr Dermatol. 1986;3:446-451. doi:10.1111/j.1525-1470.1986.tb00648.x
- Moretta G, De Luca EV, Di Stefani A. Management of refractory pityriasis rubra pilaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2017;10:451-457. doi:10.2147/CCID.S124351
- Engelmann C, Elsner P, Miguel D. Treatment of pityriasis rubra pilaris type I: a systematic review. Eur J Dermatol. 2019;29:524-537. doi:10.1684/ejd.2019.3641
- Ricar J, Cetkovska P. Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab. Br J Dermatol. 2021;184:E148. doi:10.1111/bjd.19681
- Brocco E, Laffitte E. Risankizumab for pityriasis rubra pilaris. Clin Exp Dermatol. 2021;46:1322-1324. doi:10.1111/ced.14715
- Duarte B, Paiva Lopes MJ. Response to: ‘Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab.’ Br J Dermatol. 2021;185:235-236. doi:10.1111/bjd.20061
- Kromer C, Schön MP, Mössner R. Treatment of pityriasis rubra pilaris with risankizumab in two cases. J Dtsch Dermatol Ges. 2021;19:1207-1209. doi:10.1111/ddg.14504
- Kołt-Kamińska M, Osińska A, Kaznowska E, et al. Successful treatment of pityriasis rubra pilaris with risankizumab in children. Dermatol Ther (Heidelb). 2023;13:2431-2441. doi:10.1007/s13555-023-01005-y
- Boudreaux BW, Pincelli TP, Bhullar PK, et al. Secukinumab for the treatment of adult-onset pityriasis rubra pilaris: a single-arm clinical trial with transcriptomic analysis. Br J Dermatol. 2022;187:650-658. doi:10.1111/bjd.21708
- Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156:649-658. doi:10.1001/jamadermatol.2020.0723
- Gelmetti C, Schiuma AA, Cerri D, et al. Pityriasis rubra pilaris in childhood: a long-term study of 29 cases. Pediatr Dermatol. 1986;3:446-451. doi:10.1111/j.1525-1470.1986.tb00648.x
- Moretta G, De Luca EV, Di Stefani A. Management of refractory pityriasis rubra pilaris: challenges and solutions. Clin Cosmet Investig Dermatol. 2017;10:451-457. doi:10.2147/CCID.S124351
- Engelmann C, Elsner P, Miguel D. Treatment of pityriasis rubra pilaris type I: a systematic review. Eur J Dermatol. 2019;29:524-537. doi:10.1684/ejd.2019.3641
- Ricar J, Cetkovska P. Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab. Br J Dermatol. 2021;184:E148. doi:10.1111/bjd.19681
- Brocco E, Laffitte E. Risankizumab for pityriasis rubra pilaris. Clin Exp Dermatol. 2021;46:1322-1324. doi:10.1111/ced.14715
- Duarte B, Paiva Lopes MJ. Response to: ‘Successful treatment of refractory extensive pityriasis rubra pilaris with risankizumab.’ Br J Dermatol. 2021;185:235-236. doi:10.1111/bjd.20061
- Kromer C, Schön MP, Mössner R. Treatment of pityriasis rubra pilaris with risankizumab in two cases. J Dtsch Dermatol Ges. 2021;19:1207-1209. doi:10.1111/ddg.14504
- Kołt-Kamińska M, Osińska A, Kaznowska E, et al. Successful treatment of pityriasis rubra pilaris with risankizumab in children. Dermatol Ther (Heidelb). 2023;13:2431-2441. doi:10.1007/s13555-023-01005-y
- Boudreaux BW, Pincelli TP, Bhullar PK, et al. Secukinumab for the treatment of adult-onset pityriasis rubra pilaris: a single-arm clinical trial with transcriptomic analysis. Br J Dermatol. 2022;187:650-658. doi:10.1111/bjd.21708
- Blauvelt A, Leonardi CL, Gooderham M, et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156:649-658. doi:10.1001/jamadermatol.2020.0723
Practice Points
- Pityriasis rubra pilaris (PRP) is a rare condition that is challenging to treat due to its unknown pathogenesis and limited efficacy data. Systemic retinoids historically were considered first-line therapy for moderate to severe cases of PRP.
- Biologics may be useful for refractory cases of PRP.
- Risankizumab is approved for moderate to severe plaque psoriasis and can be considered off-label for refractory PRP.
Second Treatment for Prurigo Nodularis Approved by FDA
On August 13, 2024, the
A first-in-class monoclonal antibody specifically designed to inhibit interleukin (IL)–31 signaling, nemolizumab, will be available in a prefilled pen for subcutaneous injection and will be marketed as Nemluvio. It is currently under FDA review for treating atopic dermatitis in adolescents and adults. 
Approval for PN is based on data from the phase 3 OLYMPIA clinical trial program, which evaluated the efficacy and safety of nemolizumab administered subcutaneously every 4 weeks in 560 patients with PN, according to a press release from Galderma, the manufacturer.
According to the press release, in OLYMPIA 1 and OLYMPIA 2, 58% and 56% of patients, respectively, achieved at least a 4-point reduction in itch intensity at week 16 as measured by the Peak Pruritus Numerical Rating Scale, compared with 16% in both placebo groups (P < .0001). At the same time, 26% and 38% of nemolizumab-treated patients reached clearance or almost-clearance of skin lesions on the Investigator Global Assessment score at week 16, compared with 7% and 11% in the placebo groups (P < .0001).
According to the company press release, the most common side effects of nemolizumab are headache and rashes in the form of eczema, atopic dermatitis, and nummular eczema.
“By inhibiting the signaling of IL-31, Nemluvio addresses a key driver of prurigo nodularis, safely and effectively improving itch as well as skin nodules,” Shawn G. Kwatra, MD, PhD, professor and chair of dermatology at the University of Maryland School of Medicine, Baltimore, and lead investigator of the OLYMPIA program, stated in the press release.
The regulatory submission of nemolizumab in atopic dermatitis is based on data from the phase 3 ARCADIA clinical trial program, which evaluated the efficacy and safety of nemolizumab administered subcutaneously every 4 weeks in adolescents and adults with moderate to severe atopic dermatitis. A decision on approval for this indication from the FDA is expected in December 2024.
In September 2022, dupilumab became the first FDA-approved treatment for PN in the United States.
A version of this article first appeared on Medscape.com.
On August 13, 2024, the
A first-in-class monoclonal antibody specifically designed to inhibit interleukin (IL)–31 signaling, nemolizumab, will be available in a prefilled pen for subcutaneous injection and will be marketed as Nemluvio. It is currently under FDA review for treating atopic dermatitis in adolescents and adults.
Approval for PN is based on data from the phase 3 OLYMPIA clinical trial program, which evaluated the efficacy and safety of nemolizumab administered subcutaneously every 4 weeks in 560 patients with PN, according to a press release from Galderma, the manufacturer.
According to the press release, in OLYMPIA 1 and OLYMPIA 2, 58% and 56% of patients, respectively, achieved at least a 4-point reduction in itch intensity at week 16 as measured by the Peak Pruritus Numerical Rating Scale, compared with 16% in both placebo groups (P < .0001). At the same time, 26% and 38% of nemolizumab-treated patients reached clearance or almost-clearance of skin lesions on the Investigator Global Assessment score at week 16, compared with 7% and 11% in the placebo groups (P < .0001).
According to the company press release, the most common side effects of nemolizumab are headache and rashes in the form of eczema, atopic dermatitis, and nummular eczema.
“By inhibiting the signaling of IL-31, Nemluvio addresses a key driver of prurigo nodularis, safely and effectively improving itch as well as skin nodules,” Shawn G. Kwatra, MD, PhD, professor and chair of dermatology at the University of Maryland School of Medicine, Baltimore, and lead investigator of the OLYMPIA program, stated in the press release.
The regulatory submission of nemolizumab in atopic dermatitis is based on data from the phase 3 ARCADIA clinical trial program, which evaluated the efficacy and safety of nemolizumab administered subcutaneously every 4 weeks in adolescents and adults with moderate to severe atopic dermatitis. A decision on approval for this indication from the FDA is expected in December 2024.
In September 2022, dupilumab became the first FDA-approved treatment for PN in the United States.
A version of this article first appeared on Medscape.com.
On August 13, 2024, the
A first-in-class monoclonal antibody specifically designed to inhibit interleukin (IL)–31 signaling, nemolizumab, will be available in a prefilled pen for subcutaneous injection and will be marketed as Nemluvio. It is currently under FDA review for treating atopic dermatitis in adolescents and adults.
Approval for PN is based on data from the phase 3 OLYMPIA clinical trial program, which evaluated the efficacy and safety of nemolizumab administered subcutaneously every 4 weeks in 560 patients with PN, according to a press release from Galderma, the manufacturer.
According to the press release, in OLYMPIA 1 and OLYMPIA 2, 58% and 56% of patients, respectively, achieved at least a 4-point reduction in itch intensity at week 16 as measured by the Peak Pruritus Numerical Rating Scale, compared with 16% in both placebo groups (P < .0001). At the same time, 26% and 38% of nemolizumab-treated patients reached clearance or almost-clearance of skin lesions on the Investigator Global Assessment score at week 16, compared with 7% and 11% in the placebo groups (P < .0001).
According to the company press release, the most common side effects of nemolizumab are headache and rashes in the form of eczema, atopic dermatitis, and nummular eczema.
“By inhibiting the signaling of IL-31, Nemluvio addresses a key driver of prurigo nodularis, safely and effectively improving itch as well as skin nodules,” Shawn G. Kwatra, MD, PhD, professor and chair of dermatology at the University of Maryland School of Medicine, Baltimore, and lead investigator of the OLYMPIA program, stated in the press release.
The regulatory submission of nemolizumab in atopic dermatitis is based on data from the phase 3 ARCADIA clinical trial program, which evaluated the efficacy and safety of nemolizumab administered subcutaneously every 4 weeks in adolescents and adults with moderate to severe atopic dermatitis. A decision on approval for this indication from the FDA is expected in December 2024.
In September 2022, dupilumab became the first FDA-approved treatment for PN in the United States.
A version of this article first appeared on Medscape.com.
FDA Approves First Engineered Cell Therapy for a Solid Tumor
Afami-cel — the first engineered cell therapy for a solid tumor — is indicated specifically for adults with unresectable or metastatic synovial sarcoma who have received prior chemotherapy, are positive for several human leukocyte antigens (HLAs), and whose tumors express melanoma-associated antigen A4, as determined by FDA-authorized companion diagnostic devices.
The single-dose treatment targets solid tumors expressing melanoma-associated antigen A4, a protein highly expressed in synovial sarcoma.
Synovial sarcoma is a rare form of cancer, which affects about 1000 people in the US each year. Malignant cells develop and form a tumor in soft tissues, often in the extremities.
“Adults with metastatic synovial sarcoma, a life-threatening form of cancer, often face limited treatment options in addition to the risk of cancer spread or recurrence,” Nicole Verdun, MD, director of the Office of Therapeutic Products in the FDA’s Center for Biologics Evaluation and Research, said in the agency press release announcing the approval. “Today’s approval represents a significant milestone in the development of an innovative, safe and effective therapy for patients with this rare but potentially fatal disease.”
T-cell receptor therapy, like chimeric antigen receptor (CAR) T-cell (CAR-T) therapy, involves altering patient T cells to fight cancer. While CAR-T therapy inserts an artificial receptor to target a specific surface protein on cancer cells, the T-cell receptor therapy modifies existing receptors to recognize an array of antigens on the surface of cancer cells — a promising strategy for targeting solid tumors.
The accelerated approval of afami-cel was based on the phase 2 SPEARHEAD-1 trial in 44 patients with synovial sarcoma who received a single infusion of the therapy. The trial had enrolled 52 patients, but 8 did not receive afami-cel, including 3 who died and 1 who withdrew.
According to the FDA announcement, the overall response rate was 43.2%, with a median time to response of 4.9 weeks. The median duration of response was 6 months (95% CI, 4.6 months to not reached). Among patients who responded, 39% had a duration of response of 12 months or longer.
“These results suggest that a one-time treatment with afami-cel has the potential to extend life while allowing responders to go off chemotherapy,” said lead investigator Sandra D’Angelo, MD, a sarcoma specialist at Memorial Sloan Kettering Cancer Center in New York City, in a company press release.
The prescribing information includes a boxed warning for serious or fatal cytokine release syndrome.
The most common nonlaboratory adverse reactions, occurring in at least 20% of patients, included cytokine release syndrome, nausea, vomiting, fatigue, infections, pyrexia, constipation, dyspnea, tachycardia, hypotension, diarrhea, and edema. The most common grade 3 or 4 laboratory abnormalities, occurring in at least 20% of patients, included decreased lymphocyte count, neutrophil count, white cell blood count, red blood cell, and platelet count.
The recommended dose is between 2.68x109 to 10x109 MAGE-A4 T-cell receptor–positive T-cells. The FDA notice specifies not using a leukodepleting filter or prophylactic systemic corticosteroids.
The list price for the one-time therapy is $727,000, according to Fierce Pharma.
A version of this article first appeared on Medscape.com.
Afami-cel — the first engineered cell therapy for a solid tumor — is indicated specifically for adults with unresectable or metastatic synovial sarcoma who have received prior chemotherapy, are positive for several human leukocyte antigens (HLAs), and whose tumors express melanoma-associated antigen A4, as determined by FDA-authorized companion diagnostic devices.
The single-dose treatment targets solid tumors expressing melanoma-associated antigen A4, a protein highly expressed in synovial sarcoma.
Synovial sarcoma is a rare form of cancer, which affects about 1000 people in the US each year. Malignant cells develop and form a tumor in soft tissues, often in the extremities.
“Adults with metastatic synovial sarcoma, a life-threatening form of cancer, often face limited treatment options in addition to the risk of cancer spread or recurrence,” Nicole Verdun, MD, director of the Office of Therapeutic Products in the FDA’s Center for Biologics Evaluation and Research, said in the agency press release announcing the approval. “Today’s approval represents a significant milestone in the development of an innovative, safe and effective therapy for patients with this rare but potentially fatal disease.”
T-cell receptor therapy, like chimeric antigen receptor (CAR) T-cell (CAR-T) therapy, involves altering patient T cells to fight cancer. While CAR-T therapy inserts an artificial receptor to target a specific surface protein on cancer cells, the T-cell receptor therapy modifies existing receptors to recognize an array of antigens on the surface of cancer cells — a promising strategy for targeting solid tumors.
The accelerated approval of afami-cel was based on the phase 2 SPEARHEAD-1 trial in 44 patients with synovial sarcoma who received a single infusion of the therapy. The trial had enrolled 52 patients, but 8 did not receive afami-cel, including 3 who died and 1 who withdrew.
According to the FDA announcement, the overall response rate was 43.2%, with a median time to response of 4.9 weeks. The median duration of response was 6 months (95% CI, 4.6 months to not reached). Among patients who responded, 39% had a duration of response of 12 months or longer.
“These results suggest that a one-time treatment with afami-cel has the potential to extend life while allowing responders to go off chemotherapy,” said lead investigator Sandra D’Angelo, MD, a sarcoma specialist at Memorial Sloan Kettering Cancer Center in New York City, in a company press release.
The prescribing information includes a boxed warning for serious or fatal cytokine release syndrome.
The most common nonlaboratory adverse reactions, occurring in at least 20% of patients, included cytokine release syndrome, nausea, vomiting, fatigue, infections, pyrexia, constipation, dyspnea, tachycardia, hypotension, diarrhea, and edema. The most common grade 3 or 4 laboratory abnormalities, occurring in at least 20% of patients, included decreased lymphocyte count, neutrophil count, white cell blood count, red blood cell, and platelet count.
The recommended dose is between 2.68x109 to 10x109 MAGE-A4 T-cell receptor–positive T-cells. The FDA notice specifies not using a leukodepleting filter or prophylactic systemic corticosteroids.
The list price for the one-time therapy is $727,000, according to Fierce Pharma.
A version of this article first appeared on Medscape.com.
Afami-cel — the first engineered cell therapy for a solid tumor — is indicated specifically for adults with unresectable or metastatic synovial sarcoma who have received prior chemotherapy, are positive for several human leukocyte antigens (HLAs), and whose tumors express melanoma-associated antigen A4, as determined by FDA-authorized companion diagnostic devices.
The single-dose treatment targets solid tumors expressing melanoma-associated antigen A4, a protein highly expressed in synovial sarcoma.
Synovial sarcoma is a rare form of cancer, which affects about 1000 people in the US each year. Malignant cells develop and form a tumor in soft tissues, often in the extremities.
“Adults with metastatic synovial sarcoma, a life-threatening form of cancer, often face limited treatment options in addition to the risk of cancer spread or recurrence,” Nicole Verdun, MD, director of the Office of Therapeutic Products in the FDA’s Center for Biologics Evaluation and Research, said in the agency press release announcing the approval. “Today’s approval represents a significant milestone in the development of an innovative, safe and effective therapy for patients with this rare but potentially fatal disease.”
T-cell receptor therapy, like chimeric antigen receptor (CAR) T-cell (CAR-T) therapy, involves altering patient T cells to fight cancer. While CAR-T therapy inserts an artificial receptor to target a specific surface protein on cancer cells, the T-cell receptor therapy modifies existing receptors to recognize an array of antigens on the surface of cancer cells — a promising strategy for targeting solid tumors.
The accelerated approval of afami-cel was based on the phase 2 SPEARHEAD-1 trial in 44 patients with synovial sarcoma who received a single infusion of the therapy. The trial had enrolled 52 patients, but 8 did not receive afami-cel, including 3 who died and 1 who withdrew.
According to the FDA announcement, the overall response rate was 43.2%, with a median time to response of 4.9 weeks. The median duration of response was 6 months (95% CI, 4.6 months to not reached). Among patients who responded, 39% had a duration of response of 12 months or longer.
“These results suggest that a one-time treatment with afami-cel has the potential to extend life while allowing responders to go off chemotherapy,” said lead investigator Sandra D’Angelo, MD, a sarcoma specialist at Memorial Sloan Kettering Cancer Center in New York City, in a company press release.
The prescribing information includes a boxed warning for serious or fatal cytokine release syndrome.
The most common nonlaboratory adverse reactions, occurring in at least 20% of patients, included cytokine release syndrome, nausea, vomiting, fatigue, infections, pyrexia, constipation, dyspnea, tachycardia, hypotension, diarrhea, and edema. The most common grade 3 or 4 laboratory abnormalities, occurring in at least 20% of patients, included decreased lymphocyte count, neutrophil count, white cell blood count, red blood cell, and platelet count.
The recommended dose is between 2.68x109 to 10x109 MAGE-A4 T-cell receptor–positive T-cells. The FDA notice specifies not using a leukodepleting filter or prophylactic systemic corticosteroids.
The list price for the one-time therapy is $727,000, according to Fierce Pharma.
A version of this article first appeared on Medscape.com.
Lipedema: Current Diagnostic and Treatment Evidence
Lipedema affects about 11% of cisgender women, according to the Brazilian Society of Angiology and Vascular Surgery. Yet the condition remains wrapped in uncertainties. Despite significant advancements in understanding its physiology, diagnosis, and treatment, more clarity is needed as awareness and diagnoses increase.
At the latest International Congress on Obesity (ICO) in São Paulo, Brazil, Philipp Scherer, PhD, director of the Touchstone Diabetes Center, discussed the complexities of lipedema. “It is an extremely frustrating condition for someone like me, who has spent a lifetime studying functional and dysfunctional adipose tissue. We are trying to understand the physiology of this pathology, but it is challenging, and so far, we have not been able to find a concrete answer,” he noted.
Lipedema is characterized by the abnormal accumulation of subcutaneous adipose tissue, especially in the lower limbs, and almost exclusively affects cisgender women. The reason for this gender disparity is unclear. It could be an intrinsic characteristic of the disease or a result from clinicians’ lack of familiarity with lipedema, which often leads to misdiagnosis as obesity. This misdiagnosis results in fewer men seeking treatment.
Research has predominantly focused on women, and evidence suggests that hormones play a crucial role in the disease’s pathophysiology. Lipedema typically manifests during periods of hormonal changes, such as puberty, pregnancy, menopause, and hormone replacement therapies, reinforcing the idea that hormones significantly influence the condition’s development and progression.
Main Symptoms
Jonathan Kartt, CEO of the Lipedema Foundation, emphasized that intense pain in the areas of adipose tissue accumulation is a hallmark symptom of lipedema, setting it apart from obesity. Pain levels can vary widely among patients, ranging from moderate to severe, with unbearable peaks on certain days. Mr. Kartt stressed the importance of recognizing and addressing this often underestimated symptom.
Lipedema is characterized by a bilateral, symmetrical increase in mass compared with the rest of the body. This is commonly distinguished by the “cuff sign,” a separation between normal tissue in the feet and abnormal tissue from the ankle upward. Other frequent symptoms include a feeling of heaviness, discomfort, fatigue, frequent bruising, and tiredness. A notable sign is the presence of subcutaneous nodules with a texture similar to that of rice grains, which are crucial for differentiating lipedema from other conditions. Palpation during anamnesis is essential to identify these nodules and confirm the diagnosis.
“It is crucial to investigate the family history for genetic predisposition. Additionally, it is fundamental to ask whether, even with weight loss, the affected areas retain accumulated fat. Hormonal changes, pain symptoms, and impact on quality of life should also be carefully evaluated,” advised Mr. Kartt.
Diagnostic Tools
André Murad, MD, a clinical consultant at the Instituto Lipedema Brazil, has been exploring new diagnostic approaches for lipedema beyond traditional anamnesis. During his presentation at the ICO, he shared studies on the efficacy of imaging exams such as ultrasound, tomography, and MRI in diagnosing the characteristic lipedema-associated increase in subcutaneous tissue.
He also discussed lymphangiography and lymphoscintigraphy, highlighting the use of magnetic resonance lymphangiography to evaluate dilated lymphatic vessels often observed in patients with lipedema. “By injecting contrast into the feet, this technique allows the evaluation of vessels, which are usually dilated, indicating characteristic lymphatic system overload in lipedema. Lymphoscintigraphy is crucial for detecting associated lymphedema, revealing delayed lymphatic flow and asymmetry between limbs in cases of lipedema without lymphedema,” he explained.
Despite the various diagnostic options, Dr. Murad highlighted two highly effective studies. A Brazilian study used ultrasound to establish a cutoff point of 11.7 mm in the pretibial subcutaneous tissue thickness, achieving 96% specificity for diagnosis. Another study emphasized the value of dual-energy x-ray absorptiometry (DXA), which demonstrated 95% sensitivity. This method assesses fat distribution by correlating the amount present in the legs with the total body, providing a cost-effective and accessible option for specialists.
“DXA allows for a precise mathematical evaluation of fat distribution relative to the total body. A ratio of 0.38 in the leg-to-body relationship is a significant indicator of high suspicion of lipedema,” highlighted Dr. Murad. “In clinical practice, many patients self-diagnose with lipedema, but the clinical exam often reveals no disproportion, with the leg-to-body ratio below 0.38 being common in these cases,” he added.
Treatment Approaches
Treatments for lipedema are still evolving, with considerable debate about the best approach. While some specialists advocate exclusively for conservative treatment, others recommend combining these methods with surgical interventions, depending on the stage of the disease. The relative novelty of lipedema and the scarcity of robust, long-term studies contribute to the uncertainty around treatment efficacy.
Conservative treatment typically includes compression, lymphatic drainage techniques, and pressure therapy. An active lifestyle and a healthy diet are also recommended. Although these measures do not prevent the accumulation of adipose tissue, they help reduce inflammation and improve quality of life. “Even though the causes of lipedema are not fully known, lifestyle management is essential for controlling symptoms, starting with an anti-inflammatory diet,” emphasized Dr. Murad.
Because insulin promotes lipogenesis, a diet that avoids spikes in glycemic and insulin levels is advisable. Insulin resistance can exacerbate edema formation, so a Mediterranean diet may be beneficial. This diet limits fast-absorbing carbohydrates, such as added sugar, refined grains, and ultraprocessed foods, while promoting complex carbohydrates from whole grains and legumes.
Dr. Murad also presented a study evaluating the potential benefits of a low-carbohydrate, high-fat diet for patients with lipedema. The study demonstrated weight loss, reduced body fat, controlled leg volume, and, notably, pain relief.
For more advanced stages of lipedema, plastic surgery is often considered when conservative approaches do not yield satisfactory results. Some specialists advocate for surgery as an effective way to remove diseased adipose cells and reduce excess fat accumulation, which can improve physical appearance and associated pain. There is a growing consensus that surgical intervention should be performed early, ideally in stage I of IV, to maximize efficacy and prevent disease progression.
Fábio Masato Kamamoto, MD, a plastic surgeon and director of the Instituto Lipedema Brazil, shared insights into surgical treatments for lipedema. He discussed techniques from liposuction to advanced skin retraction and dermolipectomy, crucial for addressing more advanced stages of the condition. “It’s a complex process that demands precision to protect the lymphatic system, especially considering the characteristic nodules of lipedema,” he noted.
Dr. Kamamoto discussed a former patient with stage III lipedema. In the initial stage, he performed liposuction, removing 8 L of fat and 3.4 kg of skin. After 6 months, a follow-up procedure resulted in a total removal of 15 kg. Complementary procedures, such as microneedling, were performed to stimulate collagen production and reduce skin sagging. In addition to cosmetic improvements, the procedure also removed the distinctive lipedema nodules, which Mr. Kartt described as feeling like “rice grains.” Removing these nodules significantly alleviates pain, according to Dr. Kamamoto.
The benefits of surgical treatment for lipedema can be long lasting. Dr. Kamamoto noted that fat tends not to reaccumulate in treated areas, with patients often experiencing lower weight, reduced edema, and decreased pain over time. “While we hope that patients do not regain weight, the benefits of surgery persist even if weight is regained. Therefore, combining conservative and surgical treatments remains a valid and effective approach,” he concluded.
Dr. Scherer highlighted that despite various approaches, there is still no definitive “magic signature” that fully explains lipedema. This lack of clarity directly affects the effectiveness of diagnoses and treatments. He expressed hope that future integration of data from different studies and approaches will lead to the identification of a clinically useful molecular signature. “The true cause of lipedema remains unknown, requiring more speculation, hypothesis formulation, and testing for significant discoveries. This situation is frustrating, as the disease affects many women who lack a clear diagnosis that differentiates them from patients with obesity, as well as evidence-based recommendations,” he concluded.
This story was translated from the Medscape Portuguese edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Lipedema affects about 11% of cisgender women, according to the Brazilian Society of Angiology and Vascular Surgery. Yet the condition remains wrapped in uncertainties. Despite significant advancements in understanding its physiology, diagnosis, and treatment, more clarity is needed as awareness and diagnoses increase.
At the latest International Congress on Obesity (ICO) in São Paulo, Brazil, Philipp Scherer, PhD, director of the Touchstone Diabetes Center, discussed the complexities of lipedema. “It is an extremely frustrating condition for someone like me, who has spent a lifetime studying functional and dysfunctional adipose tissue. We are trying to understand the physiology of this pathology, but it is challenging, and so far, we have not been able to find a concrete answer,” he noted.
Lipedema is characterized by the abnormal accumulation of subcutaneous adipose tissue, especially in the lower limbs, and almost exclusively affects cisgender women. The reason for this gender disparity is unclear. It could be an intrinsic characteristic of the disease or a result from clinicians’ lack of familiarity with lipedema, which often leads to misdiagnosis as obesity. This misdiagnosis results in fewer men seeking treatment.
Research has predominantly focused on women, and evidence suggests that hormones play a crucial role in the disease’s pathophysiology. Lipedema typically manifests during periods of hormonal changes, such as puberty, pregnancy, menopause, and hormone replacement therapies, reinforcing the idea that hormones significantly influence the condition’s development and progression.
Main Symptoms
Jonathan Kartt, CEO of the Lipedema Foundation, emphasized that intense pain in the areas of adipose tissue accumulation is a hallmark symptom of lipedema, setting it apart from obesity. Pain levels can vary widely among patients, ranging from moderate to severe, with unbearable peaks on certain days. Mr. Kartt stressed the importance of recognizing and addressing this often underestimated symptom.
Lipedema is characterized by a bilateral, symmetrical increase in mass compared with the rest of the body. This is commonly distinguished by the “cuff sign,” a separation between normal tissue in the feet and abnormal tissue from the ankle upward. Other frequent symptoms include a feeling of heaviness, discomfort, fatigue, frequent bruising, and tiredness. A notable sign is the presence of subcutaneous nodules with a texture similar to that of rice grains, which are crucial for differentiating lipedema from other conditions. Palpation during anamnesis is essential to identify these nodules and confirm the diagnosis.
“It is crucial to investigate the family history for genetic predisposition. Additionally, it is fundamental to ask whether, even with weight loss, the affected areas retain accumulated fat. Hormonal changes, pain symptoms, and impact on quality of life should also be carefully evaluated,” advised Mr. Kartt.
Diagnostic Tools
André Murad, MD, a clinical consultant at the Instituto Lipedema Brazil, has been exploring new diagnostic approaches for lipedema beyond traditional anamnesis. During his presentation at the ICO, he shared studies on the efficacy of imaging exams such as ultrasound, tomography, and MRI in diagnosing the characteristic lipedema-associated increase in subcutaneous tissue.
He also discussed lymphangiography and lymphoscintigraphy, highlighting the use of magnetic resonance lymphangiography to evaluate dilated lymphatic vessels often observed in patients with lipedema. “By injecting contrast into the feet, this technique allows the evaluation of vessels, which are usually dilated, indicating characteristic lymphatic system overload in lipedema. Lymphoscintigraphy is crucial for detecting associated lymphedema, revealing delayed lymphatic flow and asymmetry between limbs in cases of lipedema without lymphedema,” he explained.
Despite the various diagnostic options, Dr. Murad highlighted two highly effective studies. A Brazilian study used ultrasound to establish a cutoff point of 11.7 mm in the pretibial subcutaneous tissue thickness, achieving 96% specificity for diagnosis. Another study emphasized the value of dual-energy x-ray absorptiometry (DXA), which demonstrated 95% sensitivity. This method assesses fat distribution by correlating the amount present in the legs with the total body, providing a cost-effective and accessible option for specialists.
“DXA allows for a precise mathematical evaluation of fat distribution relative to the total body. A ratio of 0.38 in the leg-to-body relationship is a significant indicator of high suspicion of lipedema,” highlighted Dr. Murad. “In clinical practice, many patients self-diagnose with lipedema, but the clinical exam often reveals no disproportion, with the leg-to-body ratio below 0.38 being common in these cases,” he added.
Treatment Approaches
Treatments for lipedema are still evolving, with considerable debate about the best approach. While some specialists advocate exclusively for conservative treatment, others recommend combining these methods with surgical interventions, depending on the stage of the disease. The relative novelty of lipedema and the scarcity of robust, long-term studies contribute to the uncertainty around treatment efficacy.
Conservative treatment typically includes compression, lymphatic drainage techniques, and pressure therapy. An active lifestyle and a healthy diet are also recommended. Although these measures do not prevent the accumulation of adipose tissue, they help reduce inflammation and improve quality of life. “Even though the causes of lipedema are not fully known, lifestyle management is essential for controlling symptoms, starting with an anti-inflammatory diet,” emphasized Dr. Murad.
Because insulin promotes lipogenesis, a diet that avoids spikes in glycemic and insulin levels is advisable. Insulin resistance can exacerbate edema formation, so a Mediterranean diet may be beneficial. This diet limits fast-absorbing carbohydrates, such as added sugar, refined grains, and ultraprocessed foods, while promoting complex carbohydrates from whole grains and legumes.
Dr. Murad also presented a study evaluating the potential benefits of a low-carbohydrate, high-fat diet for patients with lipedema. The study demonstrated weight loss, reduced body fat, controlled leg volume, and, notably, pain relief.
For more advanced stages of lipedema, plastic surgery is often considered when conservative approaches do not yield satisfactory results. Some specialists advocate for surgery as an effective way to remove diseased adipose cells and reduce excess fat accumulation, which can improve physical appearance and associated pain. There is a growing consensus that surgical intervention should be performed early, ideally in stage I of IV, to maximize efficacy and prevent disease progression.
Fábio Masato Kamamoto, MD, a plastic surgeon and director of the Instituto Lipedema Brazil, shared insights into surgical treatments for lipedema. He discussed techniques from liposuction to advanced skin retraction and dermolipectomy, crucial for addressing more advanced stages of the condition. “It’s a complex process that demands precision to protect the lymphatic system, especially considering the characteristic nodules of lipedema,” he noted.
Dr. Kamamoto discussed a former patient with stage III lipedema. In the initial stage, he performed liposuction, removing 8 L of fat and 3.4 kg of skin. After 6 months, a follow-up procedure resulted in a total removal of 15 kg. Complementary procedures, such as microneedling, were performed to stimulate collagen production and reduce skin sagging. In addition to cosmetic improvements, the procedure also removed the distinctive lipedema nodules, which Mr. Kartt described as feeling like “rice grains.” Removing these nodules significantly alleviates pain, according to Dr. Kamamoto.
The benefits of surgical treatment for lipedema can be long lasting. Dr. Kamamoto noted that fat tends not to reaccumulate in treated areas, with patients often experiencing lower weight, reduced edema, and decreased pain over time. “While we hope that patients do not regain weight, the benefits of surgery persist even if weight is regained. Therefore, combining conservative and surgical treatments remains a valid and effective approach,” he concluded.
Dr. Scherer highlighted that despite various approaches, there is still no definitive “magic signature” that fully explains lipedema. This lack of clarity directly affects the effectiveness of diagnoses and treatments. He expressed hope that future integration of data from different studies and approaches will lead to the identification of a clinically useful molecular signature. “The true cause of lipedema remains unknown, requiring more speculation, hypothesis formulation, and testing for significant discoveries. This situation is frustrating, as the disease affects many women who lack a clear diagnosis that differentiates them from patients with obesity, as well as evidence-based recommendations,” he concluded.
This story was translated from the Medscape Portuguese edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Lipedema affects about 11% of cisgender women, according to the Brazilian Society of Angiology and Vascular Surgery. Yet the condition remains wrapped in uncertainties. Despite significant advancements in understanding its physiology, diagnosis, and treatment, more clarity is needed as awareness and diagnoses increase.
At the latest International Congress on Obesity (ICO) in São Paulo, Brazil, Philipp Scherer, PhD, director of the Touchstone Diabetes Center, discussed the complexities of lipedema. “It is an extremely frustrating condition for someone like me, who has spent a lifetime studying functional and dysfunctional adipose tissue. We are trying to understand the physiology of this pathology, but it is challenging, and so far, we have not been able to find a concrete answer,” he noted.
Lipedema is characterized by the abnormal accumulation of subcutaneous adipose tissue, especially in the lower limbs, and almost exclusively affects cisgender women. The reason for this gender disparity is unclear. It could be an intrinsic characteristic of the disease or a result from clinicians’ lack of familiarity with lipedema, which often leads to misdiagnosis as obesity. This misdiagnosis results in fewer men seeking treatment.
Research has predominantly focused on women, and evidence suggests that hormones play a crucial role in the disease’s pathophysiology. Lipedema typically manifests during periods of hormonal changes, such as puberty, pregnancy, menopause, and hormone replacement therapies, reinforcing the idea that hormones significantly influence the condition’s development and progression.
Main Symptoms
Jonathan Kartt, CEO of the Lipedema Foundation, emphasized that intense pain in the areas of adipose tissue accumulation is a hallmark symptom of lipedema, setting it apart from obesity. Pain levels can vary widely among patients, ranging from moderate to severe, with unbearable peaks on certain days. Mr. Kartt stressed the importance of recognizing and addressing this often underestimated symptom.
Lipedema is characterized by a bilateral, symmetrical increase in mass compared with the rest of the body. This is commonly distinguished by the “cuff sign,” a separation between normal tissue in the feet and abnormal tissue from the ankle upward. Other frequent symptoms include a feeling of heaviness, discomfort, fatigue, frequent bruising, and tiredness. A notable sign is the presence of subcutaneous nodules with a texture similar to that of rice grains, which are crucial for differentiating lipedema from other conditions. Palpation during anamnesis is essential to identify these nodules and confirm the diagnosis.
“It is crucial to investigate the family history for genetic predisposition. Additionally, it is fundamental to ask whether, even with weight loss, the affected areas retain accumulated fat. Hormonal changes, pain symptoms, and impact on quality of life should also be carefully evaluated,” advised Mr. Kartt.
Diagnostic Tools
André Murad, MD, a clinical consultant at the Instituto Lipedema Brazil, has been exploring new diagnostic approaches for lipedema beyond traditional anamnesis. During his presentation at the ICO, he shared studies on the efficacy of imaging exams such as ultrasound, tomography, and MRI in diagnosing the characteristic lipedema-associated increase in subcutaneous tissue.
He also discussed lymphangiography and lymphoscintigraphy, highlighting the use of magnetic resonance lymphangiography to evaluate dilated lymphatic vessels often observed in patients with lipedema. “By injecting contrast into the feet, this technique allows the evaluation of vessels, which are usually dilated, indicating characteristic lymphatic system overload in lipedema. Lymphoscintigraphy is crucial for detecting associated lymphedema, revealing delayed lymphatic flow and asymmetry between limbs in cases of lipedema without lymphedema,” he explained.
Despite the various diagnostic options, Dr. Murad highlighted two highly effective studies. A Brazilian study used ultrasound to establish a cutoff point of 11.7 mm in the pretibial subcutaneous tissue thickness, achieving 96% specificity for diagnosis. Another study emphasized the value of dual-energy x-ray absorptiometry (DXA), which demonstrated 95% sensitivity. This method assesses fat distribution by correlating the amount present in the legs with the total body, providing a cost-effective and accessible option for specialists.
“DXA allows for a precise mathematical evaluation of fat distribution relative to the total body. A ratio of 0.38 in the leg-to-body relationship is a significant indicator of high suspicion of lipedema,” highlighted Dr. Murad. “In clinical practice, many patients self-diagnose with lipedema, but the clinical exam often reveals no disproportion, with the leg-to-body ratio below 0.38 being common in these cases,” he added.
Treatment Approaches
Treatments for lipedema are still evolving, with considerable debate about the best approach. While some specialists advocate exclusively for conservative treatment, others recommend combining these methods with surgical interventions, depending on the stage of the disease. The relative novelty of lipedema and the scarcity of robust, long-term studies contribute to the uncertainty around treatment efficacy.
Conservative treatment typically includes compression, lymphatic drainage techniques, and pressure therapy. An active lifestyle and a healthy diet are also recommended. Although these measures do not prevent the accumulation of adipose tissue, they help reduce inflammation and improve quality of life. “Even though the causes of lipedema are not fully known, lifestyle management is essential for controlling symptoms, starting with an anti-inflammatory diet,” emphasized Dr. Murad.
Because insulin promotes lipogenesis, a diet that avoids spikes in glycemic and insulin levels is advisable. Insulin resistance can exacerbate edema formation, so a Mediterranean diet may be beneficial. This diet limits fast-absorbing carbohydrates, such as added sugar, refined grains, and ultraprocessed foods, while promoting complex carbohydrates from whole grains and legumes.
Dr. Murad also presented a study evaluating the potential benefits of a low-carbohydrate, high-fat diet for patients with lipedema. The study demonstrated weight loss, reduced body fat, controlled leg volume, and, notably, pain relief.
For more advanced stages of lipedema, plastic surgery is often considered when conservative approaches do not yield satisfactory results. Some specialists advocate for surgery as an effective way to remove diseased adipose cells and reduce excess fat accumulation, which can improve physical appearance and associated pain. There is a growing consensus that surgical intervention should be performed early, ideally in stage I of IV, to maximize efficacy and prevent disease progression.
Fábio Masato Kamamoto, MD, a plastic surgeon and director of the Instituto Lipedema Brazil, shared insights into surgical treatments for lipedema. He discussed techniques from liposuction to advanced skin retraction and dermolipectomy, crucial for addressing more advanced stages of the condition. “It’s a complex process that demands precision to protect the lymphatic system, especially considering the characteristic nodules of lipedema,” he noted.
Dr. Kamamoto discussed a former patient with stage III lipedema. In the initial stage, he performed liposuction, removing 8 L of fat and 3.4 kg of skin. After 6 months, a follow-up procedure resulted in a total removal of 15 kg. Complementary procedures, such as microneedling, were performed to stimulate collagen production and reduce skin sagging. In addition to cosmetic improvements, the procedure also removed the distinctive lipedema nodules, which Mr. Kartt described as feeling like “rice grains.” Removing these nodules significantly alleviates pain, according to Dr. Kamamoto.
The benefits of surgical treatment for lipedema can be long lasting. Dr. Kamamoto noted that fat tends not to reaccumulate in treated areas, with patients often experiencing lower weight, reduced edema, and decreased pain over time. “While we hope that patients do not regain weight, the benefits of surgery persist even if weight is regained. Therefore, combining conservative and surgical treatments remains a valid and effective approach,” he concluded.
Dr. Scherer highlighted that despite various approaches, there is still no definitive “magic signature” that fully explains lipedema. This lack of clarity directly affects the effectiveness of diagnoses and treatments. He expressed hope that future integration of data from different studies and approaches will lead to the identification of a clinically useful molecular signature. “The true cause of lipedema remains unknown, requiring more speculation, hypothesis formulation, and testing for significant discoveries. This situation is frustrating, as the disease affects many women who lack a clear diagnosis that differentiates them from patients with obesity, as well as evidence-based recommendations,” he concluded.
This story was translated from the Medscape Portuguese edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Dermatofibrosarcoma Protuberans More Common In Black Patients, Analysis Finds
TOPLINE:
that also found that larger tumor size and older age were associated with survival outcomes.
METHODOLOGY:
- Researchers used the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry from 2000 through 2018 to provide a comprehensive report on the incidence of DFSP, a rare, low-grade cutaneous soft tissue sarcoma, and factors associated with metastatic progression, overall survival (OS), and cancer-specific survival.
- A total of 7748 patients (mean age, 43.5 years; 53.3% women; 52% non-Hispanic White) were diagnosed with histologically confirmed DFSP of the skin and connective tissue and were included in the study.
- DFSP incidence was reported as cases per million person-years and age-adjusted to the 2000 US Standard Population, and factors influencing metastasis were assessed.
TAKEAWAY:
- The overall DFSP incidence rate was 6.25 cases per million person-years, with a higher incidence in Black individuals than in White individuals (8.74 vs 4.53).
- The 5-year OS rate was 95.8%. Older age (≥ 60 years; hazard ratio [HR], 6.66), male gender assigned at birth (HR, 1.79), and larger tumor size (≥ 3 cm; HR, 2.02) were associated with poorer OS (P < .001 for all).
- The 1-year and 5-year DFSP-specific survival rates were 99.9% and 99.2%, respectively. Older age (HR, 3.47; P < .001) and larger tumor size (≥ 3 cm; HR, 5.34; P = .002) were associated with significantly worse cancer-specific survival.
- Large tumor size (odds ratio [OR], 2.24) and DFSP located on the head and neck (OR, 4.88), or genitalia (OR, 3.16) were significantly associated with increased metastasis risk. Higher socioeconomic status was linked to a lower risk for metastasis.
IN PRACTICE:
“Our findings highlight the increased incidence rates of DFSP among Black patients. We demonstrate the interplay between patient demographics and clinical factors in influencing DFSP metastasis, OS, and cancer-specific survival,” the authors wrote. The results, they added, “may be useful for further evaluation of proposed causes, which will ultimately lead to further understanding and prevention of this disease.”
SOURCE:
The study was led by Jalal Maghfour, MD, Department of Dermatology, Henry Ford Health, Detroit, and was published online on June 20 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
Details on specific cases in the SEER registry are limited. For 1752 patients, tumor size was not included, increasing the risk for misclassification bias. Because specific pathology reports were not available, the analysis did not address histologic grade.
DISCLOSURES:
The study did not receive any funding support. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
that also found that larger tumor size and older age were associated with survival outcomes.
METHODOLOGY:
- Researchers used the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry from 2000 through 2018 to provide a comprehensive report on the incidence of DFSP, a rare, low-grade cutaneous soft tissue sarcoma, and factors associated with metastatic progression, overall survival (OS), and cancer-specific survival.
- A total of 7748 patients (mean age, 43.5 years; 53.3% women; 52% non-Hispanic White) were diagnosed with histologically confirmed DFSP of the skin and connective tissue and were included in the study.
- DFSP incidence was reported as cases per million person-years and age-adjusted to the 2000 US Standard Population, and factors influencing metastasis were assessed.
TAKEAWAY:
- The overall DFSP incidence rate was 6.25 cases per million person-years, with a higher incidence in Black individuals than in White individuals (8.74 vs 4.53).
- The 5-year OS rate was 95.8%. Older age (≥ 60 years; hazard ratio [HR], 6.66), male gender assigned at birth (HR, 1.79), and larger tumor size (≥ 3 cm; HR, 2.02) were associated with poorer OS (P < .001 for all).
- The 1-year and 5-year DFSP-specific survival rates were 99.9% and 99.2%, respectively. Older age (HR, 3.47; P < .001) and larger tumor size (≥ 3 cm; HR, 5.34; P = .002) were associated with significantly worse cancer-specific survival.
- Large tumor size (odds ratio [OR], 2.24) and DFSP located on the head and neck (OR, 4.88), or genitalia (OR, 3.16) were significantly associated with increased metastasis risk. Higher socioeconomic status was linked to a lower risk for metastasis.
IN PRACTICE:
“Our findings highlight the increased incidence rates of DFSP among Black patients. We demonstrate the interplay between patient demographics and clinical factors in influencing DFSP metastasis, OS, and cancer-specific survival,” the authors wrote. The results, they added, “may be useful for further evaluation of proposed causes, which will ultimately lead to further understanding and prevention of this disease.”
SOURCE:
The study was led by Jalal Maghfour, MD, Department of Dermatology, Henry Ford Health, Detroit, and was published online on June 20 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
Details on specific cases in the SEER registry are limited. For 1752 patients, tumor size was not included, increasing the risk for misclassification bias. Because specific pathology reports were not available, the analysis did not address histologic grade.
DISCLOSURES:
The study did not receive any funding support. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
that also found that larger tumor size and older age were associated with survival outcomes.
METHODOLOGY:
- Researchers used the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) registry from 2000 through 2018 to provide a comprehensive report on the incidence of DFSP, a rare, low-grade cutaneous soft tissue sarcoma, and factors associated with metastatic progression, overall survival (OS), and cancer-specific survival.
- A total of 7748 patients (mean age, 43.5 years; 53.3% women; 52% non-Hispanic White) were diagnosed with histologically confirmed DFSP of the skin and connective tissue and were included in the study.
- DFSP incidence was reported as cases per million person-years and age-adjusted to the 2000 US Standard Population, and factors influencing metastasis were assessed.
TAKEAWAY:
- The overall DFSP incidence rate was 6.25 cases per million person-years, with a higher incidence in Black individuals than in White individuals (8.74 vs 4.53).
- The 5-year OS rate was 95.8%. Older age (≥ 60 years; hazard ratio [HR], 6.66), male gender assigned at birth (HR, 1.79), and larger tumor size (≥ 3 cm; HR, 2.02) were associated with poorer OS (P < .001 for all).
- The 1-year and 5-year DFSP-specific survival rates were 99.9% and 99.2%, respectively. Older age (HR, 3.47; P < .001) and larger tumor size (≥ 3 cm; HR, 5.34; P = .002) were associated with significantly worse cancer-specific survival.
- Large tumor size (odds ratio [OR], 2.24) and DFSP located on the head and neck (OR, 4.88), or genitalia (OR, 3.16) were significantly associated with increased metastasis risk. Higher socioeconomic status was linked to a lower risk for metastasis.
IN PRACTICE:
“Our findings highlight the increased incidence rates of DFSP among Black patients. We demonstrate the interplay between patient demographics and clinical factors in influencing DFSP metastasis, OS, and cancer-specific survival,” the authors wrote. The results, they added, “may be useful for further evaluation of proposed causes, which will ultimately lead to further understanding and prevention of this disease.”
SOURCE:
The study was led by Jalal Maghfour, MD, Department of Dermatology, Henry Ford Health, Detroit, and was published online on June 20 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
Details on specific cases in the SEER registry are limited. For 1752 patients, tumor size was not included, increasing the risk for misclassification bias. Because specific pathology reports were not available, the analysis did not address histologic grade.
DISCLOSURES:
The study did not receive any funding support. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Pyzchiva Receives FDA Approval as Third Ustekinumab Biosimilar
The Food and Drug Administration has approved ustekinumab-ttwe (Pyzchiva) as a biosimilar to ustekinumab (Stelara) for the treatment of multiple inflammatory conditions.
In addition, the agency “provisionally determined” that the medication would be interchangeable with the reference product but that designation would not take hold until the interchangeability exclusivity period for the first approved biosimilar ustekinumab-auub (Wezlana) expires, according to a press release. This designation would, depending on state law, allow a pharmacist to substitute the biosimilar for the reference product without involving the prescribing clinician. It’s unclear when ustekinumab-auub’s interchangeability exclusivity ends.
Ustekinumab-ttwe, a human interleukin (IL)-12 and IL-23 antagonist, is indicated for the treatment of:
- Moderate to severe plaque psoriasis in adults and pediatric patients aged 6 years or older who are candidates for phototherapy or systemic therapy
- Active psoriatic arthritis in adults and pediatric patients aged 6 years or older with moderately to severely active Crohn’s disease or ulcerative colitis
It is administered via subcutaneous injection in 45 mg/0.5 mL and 90 mg/mL prefilled syringes or via intravenous infusion in 130 mg/26 mL (5 mg/mL) single-dose vial.
Developed by Samsung Bioepis, ustekinumab-ttwe will be commercialized by Sandoz in the United States. Besides ustekinumab-auub, the other ustekinumab biosimilar is ustekinumab-aekn (Selarsdi).
Ustekinumab-ttwe is expected to launch in February 2025 “in accordance with the settlement and license agreement with Janssen Biotech,” which manufacturers the reference product, Sandoz said. The other approved ustekinumab biosimilars will launch within a similar time frame.
A version of this article appeared on Medscape.com.
The Food and Drug Administration has approved ustekinumab-ttwe (Pyzchiva) as a biosimilar to ustekinumab (Stelara) for the treatment of multiple inflammatory conditions.
In addition, the agency “provisionally determined” that the medication would be interchangeable with the reference product but that designation would not take hold until the interchangeability exclusivity period for the first approved biosimilar ustekinumab-auub (Wezlana) expires, according to a press release. This designation would, depending on state law, allow a pharmacist to substitute the biosimilar for the reference product without involving the prescribing clinician. It’s unclear when ustekinumab-auub’s interchangeability exclusivity ends.
Ustekinumab-ttwe, a human interleukin (IL)-12 and IL-23 antagonist, is indicated for the treatment of:
- Moderate to severe plaque psoriasis in adults and pediatric patients aged 6 years or older who are candidates for phototherapy or systemic therapy
- Active psoriatic arthritis in adults and pediatric patients aged 6 years or older with moderately to severely active Crohn’s disease or ulcerative colitis
It is administered via subcutaneous injection in 45 mg/0.5 mL and 90 mg/mL prefilled syringes or via intravenous infusion in 130 mg/26 mL (5 mg/mL) single-dose vial.
Developed by Samsung Bioepis, ustekinumab-ttwe will be commercialized by Sandoz in the United States. Besides ustekinumab-auub, the other ustekinumab biosimilar is ustekinumab-aekn (Selarsdi).
Ustekinumab-ttwe is expected to launch in February 2025 “in accordance with the settlement and license agreement with Janssen Biotech,” which manufacturers the reference product, Sandoz said. The other approved ustekinumab biosimilars will launch within a similar time frame.
A version of this article appeared on Medscape.com.
The Food and Drug Administration has approved ustekinumab-ttwe (Pyzchiva) as a biosimilar to ustekinumab (Stelara) for the treatment of multiple inflammatory conditions.
In addition, the agency “provisionally determined” that the medication would be interchangeable with the reference product but that designation would not take hold until the interchangeability exclusivity period for the first approved biosimilar ustekinumab-auub (Wezlana) expires, according to a press release. This designation would, depending on state law, allow a pharmacist to substitute the biosimilar for the reference product without involving the prescribing clinician. It’s unclear when ustekinumab-auub’s interchangeability exclusivity ends.
Ustekinumab-ttwe, a human interleukin (IL)-12 and IL-23 antagonist, is indicated for the treatment of:
- Moderate to severe plaque psoriasis in adults and pediatric patients aged 6 years or older who are candidates for phototherapy or systemic therapy
- Active psoriatic arthritis in adults and pediatric patients aged 6 years or older with moderately to severely active Crohn’s disease or ulcerative colitis
It is administered via subcutaneous injection in 45 mg/0.5 mL and 90 mg/mL prefilled syringes or via intravenous infusion in 130 mg/26 mL (5 mg/mL) single-dose vial.
Developed by Samsung Bioepis, ustekinumab-ttwe will be commercialized by Sandoz in the United States. Besides ustekinumab-auub, the other ustekinumab biosimilar is ustekinumab-aekn (Selarsdi).
Ustekinumab-ttwe is expected to launch in February 2025 “in accordance with the settlement and license agreement with Janssen Biotech,” which manufacturers the reference product, Sandoz said. The other approved ustekinumab biosimilars will launch within a similar time frame.
A version of this article appeared on Medscape.com.
‘Therapeutic Continuums’ Guide Systemic Sclerosis Treatment in Updated EULAR Recommendations
VIENNA – The use of immunosuppressive and antifibrotic drugs to treat skin and lung fibrosis leads updated recommendations from the European Alliance of Associations for Rheumatology (EULAR) for the treatment of systemic sclerosis.
“The most impactful new recommendation relates to the evidence for immunosuppressive agents and antifibrotics for the treatment of skin fibrosis and lung fibrosis,” said Francesco Del Galdo, MD, PhD, professor of experimental medicine, consultant rheumatologist, and scleroderma and connective tissue diseases specialist at Leeds Teaching Hospitals NHS Trust, Leeds, England. Dr. Del Galdo presented the update at the annual European Congress of Rheumatology.
“But there are also new recommendations, including a redefined target population for hematopoietic stem cell transplantation following cyclophosphamide, the upfront combination treatment at the time of diagnosis of pulmonary arterial hypertension [PAH], and a negative recommendation for the use of anticoagulants for pulmonary arterial hypertension,” noted Dr. Del Galdo, highlighting key updates in the 2024 recommendations.
Robert B.M. Landewé, MD, PhD, professor and rheumatologist at Amsterdam University Medical Center, Amsterdam, the Netherlands, and Zuyderland Medical Center, Heerlen, the Netherlands, co-moderated the session on EULAR recommendations. “The management of systemic sclerosis is a field in which a lot is happening,” he said. “The last update goes back to 2017, and in the meantime, many new approaches have seen the light, especially pertaining to skin fibrosis and interstitial lung disease. Six new recommendations have been coined, covering drugs like mycophenolate mofetil, nintedanib, rituximab, and tocilizumab. None of these therapies were present in the 2017 recommendations. It seems the field is now ready to further expand on targeted therapies for the management of musculoskeletal and gastrointestinal manifestations, calcinosis, and the local management of digital ulcers.”
‘Therapeutic Continuums’ Aid Disease Management
Dr. Del Galdo and his colleagues grouped the various interventions across what the recommendations label as evidence-backed “therapeutic continuums.” These span six of the eight different clinical manifestations of systemic sclerosis: Raynaud’s phenomenon, digital ulcers, pulmonary hypertension, musculoskeletal manifestations, skin fibrosis, interstitial lung disease (ILD), and gastrointestinal and renal crisis.
A slide showing the different strengths of evidence for various drugs across the eight manifestations illustrated the principle behind the therapeutic continuums. “These ‘therapeutic continuums’ suggest a common pathogenetic mechanism driving the various manifestations of disease,” said Dr. Del Galdo. For example, he noted, “If rituximab had a positive response in skin and in lung, it suggests that B cells play a role in the clinical manifestations of skin and lung in this disease.”
Dr. Del Galdo highlighted the new immunosuppression continuum and associated treatments for skin and lung fibrosis. “For skin involvement, the task force recommended mycophenolate, methotrexate, and rituximab, with tocilizumab having a lower level of evidence and lower recommendation strength; similarly, in interstitial lung disease, we have rituximab, mycophenolate, cyclophosphamide, and nintedanib, and these all have the highest strength of evidence. Tocilizumab is assigned one strength of evidence below the other drugs.”
He also cited the phosphodiesterase 5 inhibitor (PDE5i) drugs that are used across Raynaud’s phenomenon, digital ulcers, and pulmonary arterial hypertension, which together form a vascular therapeutic continuum.
The complexity of systemic sclerosis and multiple manifestations was a major determinant of the recommendations, Dr. Del Galdo pointed out. “The task force realized that since this is such a complex disease, we cannot recommend one treatment unconditionally. For example, with mycophenolate mofetil, what works for most patients for the skin and lung manifestations might not for someone who experiences severe diarrhea, in which mycophenolate is contraindicated. So, the highest degree of recommendation that the task force felt comfortable with was ‘should be considered.’ ”
Dr. Del Galdo stressed that the complex nature of systemic sclerosis means that “when thinking of treating one manifestation, you also always need to consider all the other clinical manifestations as experienced by the patient, and it is this multifaceted scenario that will ultimately lead to your final choice.”
Turning to new evidence around drug use, Dr. Del Galdo said that rituximab has the highest level of evidence across skin and lung manifestations, nintedanib is new in lung, and tocilizumab is new across both skin and lung.
To treat systemic sclerosis–pulmonary arterial hypertension (SSc-PAH), as long as there are no contraindications, the task force recommends using PDE5i and endothelin receptor antagonists (ERAs) at diagnosis. Data from phase 3 trials show a better outcome when the combination is established early.
The task force suggests avoiding the use of warfarin in PAH. “This is supported by a signal from two trials showing an increase in morbidity and mortality in these patients,” noted Dr. Del Galdo.
He also pointed out that selexipag and riociguat were new and important second-line additions for the treatment of PAH, and — consistent with the ERA approach — the EULAR recommendation supports frequent follow-up to establish a treat-to-target approach to maximizing clinical outcomes in SSc-PAH and SSc-ILD. “Specifically, for the first time, we recommend monitoring the effect of any chosen intervention selected within 3-6 months of starting. The evidence suggests there is a group of patients who respond and some who respond less well and who might benefit from a second-line intervention.”
For example, results of one trial support the approach of adding an antifibrotic agent to reduce progression in people with progressive lung fibrosis. “Similarly, for pulmonary hypertension, we recommend putting patients on dual treatment, and if this fails, place them on selexipag or switch the PDE5i to riociguat,” Dr. Del Galdo said.
Systemic Sclerosis Research Agenda and Recommendations Align
Dr. Del Galdo highlighted the value of therapeutic continuums in advancing disease understanding. “It is starting to teach us what we know and what we don’t and where do we need to build more evidence. Effectively, they determine where the gaps in therapy lie, and this starts to guide the research agenda.
“In fact, what is really interesting about this recommendation update — certainly from the perspective of disease understanding — is that we are starting to have a bird’s-eye view of the clinical manifestations of scleroderma that have so often been dealt with separately. Now we are starting to build a cumulative evidence map of this disease.”
In 2017, the research agenda largely advocated identifying immune-targeting drugs for skin and lung fibrosis, Dr. Del Galdo pointed out. “Now, we’ve done that — we’ve identified appropriate immunosuppressive drugs — and this is testimony to the importance of these recommendations because what prioritized the research agenda 10 years ago ended up informing the clinical trials and made it into the recommendations.”
“We definitely are one step forward compared to this 2017 recommendation and closer to what we would like to do,” he asserted.
Remission Elusive but Getting Closer
In some respects, according to Dr. Del Galdo, research and development is making relatively slow progress, especially compared with other rheumatologic diseases such as rheumatoid arthritis. “We cannot put patients with systemic sclerosis in remission yet. But I think we are one step ahead in that we’ve now established the treat-to-target approach to maximize the efficacy with which we can stall disease progression, but we cannot yet put these patients into remission,” he said. Systemic sclerosis has multiple manifestations, and fibrotic damage cannot be reversed. “Right now, the scar will remain there forever,” he noted.
Until remission is achievable, Dr. Del Galdo advises diagnosing and treating patients earlier to prevent fibrotic manifestations.
Dr. Del Galdo explained the three leading priorities on the systemic sclerosis research agenda. “There are three because it is such a complex disease. The first is considering the patient voice — this is the most important one, and the patients say they want a more holistic approach — so trialing and treating multiple manifestations together.”
Second, Dr. Del Galdo said, he would like to see a patient-reported measure developed that can capture the entire disease.
Third, from a physician’s point of view, Dr. Del Galdo said, “We want to send the patients into remission. We need to continue to further deconvolute the clinical manifestations and find the bottleneck at the beginning of the natural history of disease.
“If we can find a drug that is effective very early on, before the patients start getting the eight different manifestations with different levels of severity, then we will be on the right road, which we hope will end in remission.”
Dr. Del Galdo has served on the speakers bureau for AstraZeneca and Janssen; consulted for AstraZeneca, Boehringer Ingelheim, Capella, Chemomab, Janssen, and Mitsubishi-Tanabe; and received grant or research support from AbbVie, AstraZeneca, Boheringer Ingelheim, Capella, Chemomab, Kymab, Janssen, and Mitsubishi-Tanabe. Dr. Landewé had no relevant disclosures.
A version of this article first appeared on Medscape.com.
VIENNA – The use of immunosuppressive and antifibrotic drugs to treat skin and lung fibrosis leads updated recommendations from the European Alliance of Associations for Rheumatology (EULAR) for the treatment of systemic sclerosis.
“The most impactful new recommendation relates to the evidence for immunosuppressive agents and antifibrotics for the treatment of skin fibrosis and lung fibrosis,” said Francesco Del Galdo, MD, PhD, professor of experimental medicine, consultant rheumatologist, and scleroderma and connective tissue diseases specialist at Leeds Teaching Hospitals NHS Trust, Leeds, England. Dr. Del Galdo presented the update at the annual European Congress of Rheumatology.
“But there are also new recommendations, including a redefined target population for hematopoietic stem cell transplantation following cyclophosphamide, the upfront combination treatment at the time of diagnosis of pulmonary arterial hypertension [PAH], and a negative recommendation for the use of anticoagulants for pulmonary arterial hypertension,” noted Dr. Del Galdo, highlighting key updates in the 2024 recommendations.
Robert B.M. Landewé, MD, PhD, professor and rheumatologist at Amsterdam University Medical Center, Amsterdam, the Netherlands, and Zuyderland Medical Center, Heerlen, the Netherlands, co-moderated the session on EULAR recommendations. “The management of systemic sclerosis is a field in which a lot is happening,” he said. “The last update goes back to 2017, and in the meantime, many new approaches have seen the light, especially pertaining to skin fibrosis and interstitial lung disease. Six new recommendations have been coined, covering drugs like mycophenolate mofetil, nintedanib, rituximab, and tocilizumab. None of these therapies were present in the 2017 recommendations. It seems the field is now ready to further expand on targeted therapies for the management of musculoskeletal and gastrointestinal manifestations, calcinosis, and the local management of digital ulcers.”
‘Therapeutic Continuums’ Aid Disease Management
Dr. Del Galdo and his colleagues grouped the various interventions across what the recommendations label as evidence-backed “therapeutic continuums.” These span six of the eight different clinical manifestations of systemic sclerosis: Raynaud’s phenomenon, digital ulcers, pulmonary hypertension, musculoskeletal manifestations, skin fibrosis, interstitial lung disease (ILD), and gastrointestinal and renal crisis.
A slide showing the different strengths of evidence for various drugs across the eight manifestations illustrated the principle behind the therapeutic continuums. “These ‘therapeutic continuums’ suggest a common pathogenetic mechanism driving the various manifestations of disease,” said Dr. Del Galdo. For example, he noted, “If rituximab had a positive response in skin and in lung, it suggests that B cells play a role in the clinical manifestations of skin and lung in this disease.”
Dr. Del Galdo highlighted the new immunosuppression continuum and associated treatments for skin and lung fibrosis. “For skin involvement, the task force recommended mycophenolate, methotrexate, and rituximab, with tocilizumab having a lower level of evidence and lower recommendation strength; similarly, in interstitial lung disease, we have rituximab, mycophenolate, cyclophosphamide, and nintedanib, and these all have the highest strength of evidence. Tocilizumab is assigned one strength of evidence below the other drugs.”
He also cited the phosphodiesterase 5 inhibitor (PDE5i) drugs that are used across Raynaud’s phenomenon, digital ulcers, and pulmonary arterial hypertension, which together form a vascular therapeutic continuum.
The complexity of systemic sclerosis and multiple manifestations was a major determinant of the recommendations, Dr. Del Galdo pointed out. “The task force realized that since this is such a complex disease, we cannot recommend one treatment unconditionally. For example, with mycophenolate mofetil, what works for most patients for the skin and lung manifestations might not for someone who experiences severe diarrhea, in which mycophenolate is contraindicated. So, the highest degree of recommendation that the task force felt comfortable with was ‘should be considered.’ ”
Dr. Del Galdo stressed that the complex nature of systemic sclerosis means that “when thinking of treating one manifestation, you also always need to consider all the other clinical manifestations as experienced by the patient, and it is this multifaceted scenario that will ultimately lead to your final choice.”
Turning to new evidence around drug use, Dr. Del Galdo said that rituximab has the highest level of evidence across skin and lung manifestations, nintedanib is new in lung, and tocilizumab is new across both skin and lung.
To treat systemic sclerosis–pulmonary arterial hypertension (SSc-PAH), as long as there are no contraindications, the task force recommends using PDE5i and endothelin receptor antagonists (ERAs) at diagnosis. Data from phase 3 trials show a better outcome when the combination is established early.
The task force suggests avoiding the use of warfarin in PAH. “This is supported by a signal from two trials showing an increase in morbidity and mortality in these patients,” noted Dr. Del Galdo.
He also pointed out that selexipag and riociguat were new and important second-line additions for the treatment of PAH, and — consistent with the ERA approach — the EULAR recommendation supports frequent follow-up to establish a treat-to-target approach to maximizing clinical outcomes in SSc-PAH and SSc-ILD. “Specifically, for the first time, we recommend monitoring the effect of any chosen intervention selected within 3-6 months of starting. The evidence suggests there is a group of patients who respond and some who respond less well and who might benefit from a second-line intervention.”
For example, results of one trial support the approach of adding an antifibrotic agent to reduce progression in people with progressive lung fibrosis. “Similarly, for pulmonary hypertension, we recommend putting patients on dual treatment, and if this fails, place them on selexipag or switch the PDE5i to riociguat,” Dr. Del Galdo said.
Systemic Sclerosis Research Agenda and Recommendations Align
Dr. Del Galdo highlighted the value of therapeutic continuums in advancing disease understanding. “It is starting to teach us what we know and what we don’t and where do we need to build more evidence. Effectively, they determine where the gaps in therapy lie, and this starts to guide the research agenda.
“In fact, what is really interesting about this recommendation update — certainly from the perspective of disease understanding — is that we are starting to have a bird’s-eye view of the clinical manifestations of scleroderma that have so often been dealt with separately. Now we are starting to build a cumulative evidence map of this disease.”
In 2017, the research agenda largely advocated identifying immune-targeting drugs for skin and lung fibrosis, Dr. Del Galdo pointed out. “Now, we’ve done that — we’ve identified appropriate immunosuppressive drugs — and this is testimony to the importance of these recommendations because what prioritized the research agenda 10 years ago ended up informing the clinical trials and made it into the recommendations.”
“We definitely are one step forward compared to this 2017 recommendation and closer to what we would like to do,” he asserted.
Remission Elusive but Getting Closer
In some respects, according to Dr. Del Galdo, research and development is making relatively slow progress, especially compared with other rheumatologic diseases such as rheumatoid arthritis. “We cannot put patients with systemic sclerosis in remission yet. But I think we are one step ahead in that we’ve now established the treat-to-target approach to maximize the efficacy with which we can stall disease progression, but we cannot yet put these patients into remission,” he said. Systemic sclerosis has multiple manifestations, and fibrotic damage cannot be reversed. “Right now, the scar will remain there forever,” he noted.
Until remission is achievable, Dr. Del Galdo advises diagnosing and treating patients earlier to prevent fibrotic manifestations.
Dr. Del Galdo explained the three leading priorities on the systemic sclerosis research agenda. “There are three because it is such a complex disease. The first is considering the patient voice — this is the most important one, and the patients say they want a more holistic approach — so trialing and treating multiple manifestations together.”
Second, Dr. Del Galdo said, he would like to see a patient-reported measure developed that can capture the entire disease.
Third, from a physician’s point of view, Dr. Del Galdo said, “We want to send the patients into remission. We need to continue to further deconvolute the clinical manifestations and find the bottleneck at the beginning of the natural history of disease.
“If we can find a drug that is effective very early on, before the patients start getting the eight different manifestations with different levels of severity, then we will be on the right road, which we hope will end in remission.”
Dr. Del Galdo has served on the speakers bureau for AstraZeneca and Janssen; consulted for AstraZeneca, Boehringer Ingelheim, Capella, Chemomab, Janssen, and Mitsubishi-Tanabe; and received grant or research support from AbbVie, AstraZeneca, Boheringer Ingelheim, Capella, Chemomab, Kymab, Janssen, and Mitsubishi-Tanabe. Dr. Landewé had no relevant disclosures.
A version of this article first appeared on Medscape.com.
VIENNA – The use of immunosuppressive and antifibrotic drugs to treat skin and lung fibrosis leads updated recommendations from the European Alliance of Associations for Rheumatology (EULAR) for the treatment of systemic sclerosis.
“The most impactful new recommendation relates to the evidence for immunosuppressive agents and antifibrotics for the treatment of skin fibrosis and lung fibrosis,” said Francesco Del Galdo, MD, PhD, professor of experimental medicine, consultant rheumatologist, and scleroderma and connective tissue diseases specialist at Leeds Teaching Hospitals NHS Trust, Leeds, England. Dr. Del Galdo presented the update at the annual European Congress of Rheumatology.
“But there are also new recommendations, including a redefined target population for hematopoietic stem cell transplantation following cyclophosphamide, the upfront combination treatment at the time of diagnosis of pulmonary arterial hypertension [PAH], and a negative recommendation for the use of anticoagulants for pulmonary arterial hypertension,” noted Dr. Del Galdo, highlighting key updates in the 2024 recommendations.
Robert B.M. Landewé, MD, PhD, professor and rheumatologist at Amsterdam University Medical Center, Amsterdam, the Netherlands, and Zuyderland Medical Center, Heerlen, the Netherlands, co-moderated the session on EULAR recommendations. “The management of systemic sclerosis is a field in which a lot is happening,” he said. “The last update goes back to 2017, and in the meantime, many new approaches have seen the light, especially pertaining to skin fibrosis and interstitial lung disease. Six new recommendations have been coined, covering drugs like mycophenolate mofetil, nintedanib, rituximab, and tocilizumab. None of these therapies were present in the 2017 recommendations. It seems the field is now ready to further expand on targeted therapies for the management of musculoskeletal and gastrointestinal manifestations, calcinosis, and the local management of digital ulcers.”
‘Therapeutic Continuums’ Aid Disease Management
Dr. Del Galdo and his colleagues grouped the various interventions across what the recommendations label as evidence-backed “therapeutic continuums.” These span six of the eight different clinical manifestations of systemic sclerosis: Raynaud’s phenomenon, digital ulcers, pulmonary hypertension, musculoskeletal manifestations, skin fibrosis, interstitial lung disease (ILD), and gastrointestinal and renal crisis.
A slide showing the different strengths of evidence for various drugs across the eight manifestations illustrated the principle behind the therapeutic continuums. “These ‘therapeutic continuums’ suggest a common pathogenetic mechanism driving the various manifestations of disease,” said Dr. Del Galdo. For example, he noted, “If rituximab had a positive response in skin and in lung, it suggests that B cells play a role in the clinical manifestations of skin and lung in this disease.”
Dr. Del Galdo highlighted the new immunosuppression continuum and associated treatments for skin and lung fibrosis. “For skin involvement, the task force recommended mycophenolate, methotrexate, and rituximab, with tocilizumab having a lower level of evidence and lower recommendation strength; similarly, in interstitial lung disease, we have rituximab, mycophenolate, cyclophosphamide, and nintedanib, and these all have the highest strength of evidence. Tocilizumab is assigned one strength of evidence below the other drugs.”
He also cited the phosphodiesterase 5 inhibitor (PDE5i) drugs that are used across Raynaud’s phenomenon, digital ulcers, and pulmonary arterial hypertension, which together form a vascular therapeutic continuum.
The complexity of systemic sclerosis and multiple manifestations was a major determinant of the recommendations, Dr. Del Galdo pointed out. “The task force realized that since this is such a complex disease, we cannot recommend one treatment unconditionally. For example, with mycophenolate mofetil, what works for most patients for the skin and lung manifestations might not for someone who experiences severe diarrhea, in which mycophenolate is contraindicated. So, the highest degree of recommendation that the task force felt comfortable with was ‘should be considered.’ ”
Dr. Del Galdo stressed that the complex nature of systemic sclerosis means that “when thinking of treating one manifestation, you also always need to consider all the other clinical manifestations as experienced by the patient, and it is this multifaceted scenario that will ultimately lead to your final choice.”
Turning to new evidence around drug use, Dr. Del Galdo said that rituximab has the highest level of evidence across skin and lung manifestations, nintedanib is new in lung, and tocilizumab is new across both skin and lung.
To treat systemic sclerosis–pulmonary arterial hypertension (SSc-PAH), as long as there are no contraindications, the task force recommends using PDE5i and endothelin receptor antagonists (ERAs) at diagnosis. Data from phase 3 trials show a better outcome when the combination is established early.
The task force suggests avoiding the use of warfarin in PAH. “This is supported by a signal from two trials showing an increase in morbidity and mortality in these patients,” noted Dr. Del Galdo.
He also pointed out that selexipag and riociguat were new and important second-line additions for the treatment of PAH, and — consistent with the ERA approach — the EULAR recommendation supports frequent follow-up to establish a treat-to-target approach to maximizing clinical outcomes in SSc-PAH and SSc-ILD. “Specifically, for the first time, we recommend monitoring the effect of any chosen intervention selected within 3-6 months of starting. The evidence suggests there is a group of patients who respond and some who respond less well and who might benefit from a second-line intervention.”
For example, results of one trial support the approach of adding an antifibrotic agent to reduce progression in people with progressive lung fibrosis. “Similarly, for pulmonary hypertension, we recommend putting patients on dual treatment, and if this fails, place them on selexipag or switch the PDE5i to riociguat,” Dr. Del Galdo said.
Systemic Sclerosis Research Agenda and Recommendations Align
Dr. Del Galdo highlighted the value of therapeutic continuums in advancing disease understanding. “It is starting to teach us what we know and what we don’t and where do we need to build more evidence. Effectively, they determine where the gaps in therapy lie, and this starts to guide the research agenda.
“In fact, what is really interesting about this recommendation update — certainly from the perspective of disease understanding — is that we are starting to have a bird’s-eye view of the clinical manifestations of scleroderma that have so often been dealt with separately. Now we are starting to build a cumulative evidence map of this disease.”
In 2017, the research agenda largely advocated identifying immune-targeting drugs for skin and lung fibrosis, Dr. Del Galdo pointed out. “Now, we’ve done that — we’ve identified appropriate immunosuppressive drugs — and this is testimony to the importance of these recommendations because what prioritized the research agenda 10 years ago ended up informing the clinical trials and made it into the recommendations.”
“We definitely are one step forward compared to this 2017 recommendation and closer to what we would like to do,” he asserted.
Remission Elusive but Getting Closer
In some respects, according to Dr. Del Galdo, research and development is making relatively slow progress, especially compared with other rheumatologic diseases such as rheumatoid arthritis. “We cannot put patients with systemic sclerosis in remission yet. But I think we are one step ahead in that we’ve now established the treat-to-target approach to maximize the efficacy with which we can stall disease progression, but we cannot yet put these patients into remission,” he said. Systemic sclerosis has multiple manifestations, and fibrotic damage cannot be reversed. “Right now, the scar will remain there forever,” he noted.
Until remission is achievable, Dr. Del Galdo advises diagnosing and treating patients earlier to prevent fibrotic manifestations.
Dr. Del Galdo explained the three leading priorities on the systemic sclerosis research agenda. “There are three because it is such a complex disease. The first is considering the patient voice — this is the most important one, and the patients say they want a more holistic approach — so trialing and treating multiple manifestations together.”
Second, Dr. Del Galdo said, he would like to see a patient-reported measure developed that can capture the entire disease.
Third, from a physician’s point of view, Dr. Del Galdo said, “We want to send the patients into remission. We need to continue to further deconvolute the clinical manifestations and find the bottleneck at the beginning of the natural history of disease.
“If we can find a drug that is effective very early on, before the patients start getting the eight different manifestations with different levels of severity, then we will be on the right road, which we hope will end in remission.”
Dr. Del Galdo has served on the speakers bureau for AstraZeneca and Janssen; consulted for AstraZeneca, Boehringer Ingelheim, Capella, Chemomab, Janssen, and Mitsubishi-Tanabe; and received grant or research support from AbbVie, AstraZeneca, Boheringer Ingelheim, Capella, Chemomab, Kymab, Janssen, and Mitsubishi-Tanabe. Dr. Landewé had no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM EULAR 2024
First-line Canakinumab Without Steroids Shows Effectiveness for Systemic Juvenile Idiopathic Arthritis
VIENNA — The interleukin-1 receptor antagonist (IL-1RA) canakinumab provided control of systemic juvenile idiopathic arthritis (sJIA) without the use of glucocorticoids for up to a year in most study participants after three monthly injections.
In this study of 20 patients with newly diagnosed sJIA treated off glucocorticoids, fever was controlled after a single injection in all patients, and 16 patients reached the primary outcome of remission after three injections, said Gerd Horneff, MD, PhD, Asklepios Children’s Hospital, Sankt Augustin, Germany.
Results of this open-label study, called CANAKINUMAB FIRST, were presented as late-breaking findings at the European Alliance of Associations for Rheumatology (EULAR) 2024 Annual Meeting.
“Steroid-free, first-line treatment with canakinumab led to sustained responses in most patients, with a considerable number achieving remission,” said Dr. Horneff, adding that the observation in this group is ongoing.
Building on Earlier Data
The efficacy of canakinumab was previously reported in anecdotal experiences and one small patient series published 10 years ago. Dr. Horneff noted that he has offered this drug off label to patients with challenging cases.
The objective was to evaluate canakinumab as a first-line monotherapy administered in the absence of glucocorticoids. The study was open to children aged 2-18 years with active sJIA/juvenile Still disease confirmed with published criteria. All were naive to biologic or nonbiologic disease-modifying antirheumatic drugs as well as steroids.
The median age of the children was 8.4 years. A total of 60% were men. The median disease duration at the time of entry was 1.2 months. Most had fever (95%) and rash (80%) with high levels of inflammatory markers at baseline. The mean number of painful joints was 3.1, and the mean number of systemic manifestations was 2.8. No patient was without any systemic involvement, but four of the patients did not have any painful joints.
At enrollment, patients were scheduled to receive three injections of canakinumab at monthly intervals during an active treatment phase, after which they entered an observation phase lasting 40 weeks. In the event of nonresponse or flares in either phase, they were transitioned to usual care.
Symptoms Resolve After Single Injection
After the first injection, active joint disease and all systemic manifestations resolved in 16 (80%) of the 20 patients. Joint activity and systemic manifestations also remained controlled after the second and third injections in 16 of the 20 patients.
One patient in this series achieved inactive disease after a single injection but developed what appeared to be a treatment-related allergic reaction. He received no further treatment and was excluded from the study, although he is being followed separately.
“According to sJADAS [systemic JIA Disease Activity Score] criteria at month 3, 14 had inactive disease, three had minimal disease activity, and one patient had moderate disease activity,” Dr. Horneff said.
At week 24, or 3 months after the last injection, there was still no joint activity in 16 patients. Systemic manifestations remained controlled in 13 patients, but 1 patient by this point had a flare. Another flare occurred after this point, and other patients have not yet completed the 52-week observation period.
“Of the 10 patients who remained in the study and have completed the 52-week observation period, eight have had a drug-free remission,” Dr. Horneff said.
MAS Event Observed in One Patient
In addition to the allergic skin reaction, which was considered probably related to the study drug, there were three flares, one of which was a macrophage activation syndrome (MAS) event. The MAS occurred 8 weeks after the last injection, but it was managed successfully.
Of 30 infections that developed during the observation period, 18 involved the upper airway. All were treated successfully. There were also two injection-site reactions and one case of cytopenia.
Among the studies planned for follow-up, investigators will examine genomic and gene activation in relation to disease activity and the effect of canakinumab.
Comoderator of the abstract session and chair of the EULAR 2024 Abstract Selection Committee, Christian Dejaco, MD, PhD, a consultant rheumatologist and associate professor at the Medical University of Graz in Graz, Austria, suggested that these are highly encouraging data for a disease that does not currently have any approved therapies. Clearly, larger studies with a longer follow-up period are needed, but he pointed out that phase 3 trials in a rare disease like sJIA are challenging.
Because of the limited number of cases, “it will be difficult to conduct a placebo-controlled trial,” he pointed out. However, he hopes this study will provide the basis for larger studies and sufficient data to lead to an indication for this therapy.
In the meantime, he also believes that these data are likely to support empirical use in a difficult disease, even in advance of formal regulatory approval.
“We heard that canakinumab is already being used off label in JIA, and these data might encourage more of that,” he said.
Dr. Horneff reported financial relationships with AbbVie, Boehringer Ingelheim, Celgene, Chugai, GlaxoSmithKline, Janssen, Merck Sharpe & Dohme, Novartis, Pfizer, Roche, Sanofi, and Sobe. Dr. Dejaco reported no potential conflicts of interest.
A version of this article first appeared on Medscape.com.
VIENNA — The interleukin-1 receptor antagonist (IL-1RA) canakinumab provided control of systemic juvenile idiopathic arthritis (sJIA) without the use of glucocorticoids for up to a year in most study participants after three monthly injections.
In this study of 20 patients with newly diagnosed sJIA treated off glucocorticoids, fever was controlled after a single injection in all patients, and 16 patients reached the primary outcome of remission after three injections, said Gerd Horneff, MD, PhD, Asklepios Children’s Hospital, Sankt Augustin, Germany.
Results of this open-label study, called CANAKINUMAB FIRST, were presented as late-breaking findings at the European Alliance of Associations for Rheumatology (EULAR) 2024 Annual Meeting.
“Steroid-free, first-line treatment with canakinumab led to sustained responses in most patients, with a considerable number achieving remission,” said Dr. Horneff, adding that the observation in this group is ongoing.
Building on Earlier Data
The efficacy of canakinumab was previously reported in anecdotal experiences and one small patient series published 10 years ago. Dr. Horneff noted that he has offered this drug off label to patients with challenging cases.
The objective was to evaluate canakinumab as a first-line monotherapy administered in the absence of glucocorticoids. The study was open to children aged 2-18 years with active sJIA/juvenile Still disease confirmed with published criteria. All were naive to biologic or nonbiologic disease-modifying antirheumatic drugs as well as steroids.
The median age of the children was 8.4 years. A total of 60% were men. The median disease duration at the time of entry was 1.2 months. Most had fever (95%) and rash (80%) with high levels of inflammatory markers at baseline. The mean number of painful joints was 3.1, and the mean number of systemic manifestations was 2.8. No patient was without any systemic involvement, but four of the patients did not have any painful joints.
At enrollment, patients were scheduled to receive three injections of canakinumab at monthly intervals during an active treatment phase, after which they entered an observation phase lasting 40 weeks. In the event of nonresponse or flares in either phase, they were transitioned to usual care.
Symptoms Resolve After Single Injection
After the first injection, active joint disease and all systemic manifestations resolved in 16 (80%) of the 20 patients. Joint activity and systemic manifestations also remained controlled after the second and third injections in 16 of the 20 patients.
One patient in this series achieved inactive disease after a single injection but developed what appeared to be a treatment-related allergic reaction. He received no further treatment and was excluded from the study, although he is being followed separately.
“According to sJADAS [systemic JIA Disease Activity Score] criteria at month 3, 14 had inactive disease, three had minimal disease activity, and one patient had moderate disease activity,” Dr. Horneff said.
At week 24, or 3 months after the last injection, there was still no joint activity in 16 patients. Systemic manifestations remained controlled in 13 patients, but 1 patient by this point had a flare. Another flare occurred after this point, and other patients have not yet completed the 52-week observation period.
“Of the 10 patients who remained in the study and have completed the 52-week observation period, eight have had a drug-free remission,” Dr. Horneff said.
MAS Event Observed in One Patient
In addition to the allergic skin reaction, which was considered probably related to the study drug, there were three flares, one of which was a macrophage activation syndrome (MAS) event. The MAS occurred 8 weeks after the last injection, but it was managed successfully.
Of 30 infections that developed during the observation period, 18 involved the upper airway. All were treated successfully. There were also two injection-site reactions and one case of cytopenia.
Among the studies planned for follow-up, investigators will examine genomic and gene activation in relation to disease activity and the effect of canakinumab.
Comoderator of the abstract session and chair of the EULAR 2024 Abstract Selection Committee, Christian Dejaco, MD, PhD, a consultant rheumatologist and associate professor at the Medical University of Graz in Graz, Austria, suggested that these are highly encouraging data for a disease that does not currently have any approved therapies. Clearly, larger studies with a longer follow-up period are needed, but he pointed out that phase 3 trials in a rare disease like sJIA are challenging.
Because of the limited number of cases, “it will be difficult to conduct a placebo-controlled trial,” he pointed out. However, he hopes this study will provide the basis for larger studies and sufficient data to lead to an indication for this therapy.
In the meantime, he also believes that these data are likely to support empirical use in a difficult disease, even in advance of formal regulatory approval.
“We heard that canakinumab is already being used off label in JIA, and these data might encourage more of that,” he said.
Dr. Horneff reported financial relationships with AbbVie, Boehringer Ingelheim, Celgene, Chugai, GlaxoSmithKline, Janssen, Merck Sharpe & Dohme, Novartis, Pfizer, Roche, Sanofi, and Sobe. Dr. Dejaco reported no potential conflicts of interest.
A version of this article first appeared on Medscape.com.
VIENNA — The interleukin-1 receptor antagonist (IL-1RA) canakinumab provided control of systemic juvenile idiopathic arthritis (sJIA) without the use of glucocorticoids for up to a year in most study participants after three monthly injections.
In this study of 20 patients with newly diagnosed sJIA treated off glucocorticoids, fever was controlled after a single injection in all patients, and 16 patients reached the primary outcome of remission after three injections, said Gerd Horneff, MD, PhD, Asklepios Children’s Hospital, Sankt Augustin, Germany.
Results of this open-label study, called CANAKINUMAB FIRST, were presented as late-breaking findings at the European Alliance of Associations for Rheumatology (EULAR) 2024 Annual Meeting.
“Steroid-free, first-line treatment with canakinumab led to sustained responses in most patients, with a considerable number achieving remission,” said Dr. Horneff, adding that the observation in this group is ongoing.
Building on Earlier Data
The efficacy of canakinumab was previously reported in anecdotal experiences and one small patient series published 10 years ago. Dr. Horneff noted that he has offered this drug off label to patients with challenging cases.
The objective was to evaluate canakinumab as a first-line monotherapy administered in the absence of glucocorticoids. The study was open to children aged 2-18 years with active sJIA/juvenile Still disease confirmed with published criteria. All were naive to biologic or nonbiologic disease-modifying antirheumatic drugs as well as steroids.
The median age of the children was 8.4 years. A total of 60% were men. The median disease duration at the time of entry was 1.2 months. Most had fever (95%) and rash (80%) with high levels of inflammatory markers at baseline. The mean number of painful joints was 3.1, and the mean number of systemic manifestations was 2.8. No patient was without any systemic involvement, but four of the patients did not have any painful joints.
At enrollment, patients were scheduled to receive three injections of canakinumab at monthly intervals during an active treatment phase, after which they entered an observation phase lasting 40 weeks. In the event of nonresponse or flares in either phase, they were transitioned to usual care.
Symptoms Resolve After Single Injection
After the first injection, active joint disease and all systemic manifestations resolved in 16 (80%) of the 20 patients. Joint activity and systemic manifestations also remained controlled after the second and third injections in 16 of the 20 patients.
One patient in this series achieved inactive disease after a single injection but developed what appeared to be a treatment-related allergic reaction. He received no further treatment and was excluded from the study, although he is being followed separately.
“According to sJADAS [systemic JIA Disease Activity Score] criteria at month 3, 14 had inactive disease, three had minimal disease activity, and one patient had moderate disease activity,” Dr. Horneff said.
At week 24, or 3 months after the last injection, there was still no joint activity in 16 patients. Systemic manifestations remained controlled in 13 patients, but 1 patient by this point had a flare. Another flare occurred after this point, and other patients have not yet completed the 52-week observation period.
“Of the 10 patients who remained in the study and have completed the 52-week observation period, eight have had a drug-free remission,” Dr. Horneff said.
MAS Event Observed in One Patient
In addition to the allergic skin reaction, which was considered probably related to the study drug, there were three flares, one of which was a macrophage activation syndrome (MAS) event. The MAS occurred 8 weeks after the last injection, but it was managed successfully.
Of 30 infections that developed during the observation period, 18 involved the upper airway. All were treated successfully. There were also two injection-site reactions and one case of cytopenia.
Among the studies planned for follow-up, investigators will examine genomic and gene activation in relation to disease activity and the effect of canakinumab.
Comoderator of the abstract session and chair of the EULAR 2024 Abstract Selection Committee, Christian Dejaco, MD, PhD, a consultant rheumatologist and associate professor at the Medical University of Graz in Graz, Austria, suggested that these are highly encouraging data for a disease that does not currently have any approved therapies. Clearly, larger studies with a longer follow-up period are needed, but he pointed out that phase 3 trials in a rare disease like sJIA are challenging.
Because of the limited number of cases, “it will be difficult to conduct a placebo-controlled trial,” he pointed out. However, he hopes this study will provide the basis for larger studies and sufficient data to lead to an indication for this therapy.
In the meantime, he also believes that these data are likely to support empirical use in a difficult disease, even in advance of formal regulatory approval.
“We heard that canakinumab is already being used off label in JIA, and these data might encourage more of that,” he said.
Dr. Horneff reported financial relationships with AbbVie, Boehringer Ingelheim, Celgene, Chugai, GlaxoSmithKline, Janssen, Merck Sharpe & Dohme, Novartis, Pfizer, Roche, Sanofi, and Sobe. Dr. Dejaco reported no potential conflicts of interest.
A version of this article first appeared on Medscape.com.
FROM EULAR 2024
Sex-Related Differences Found in IgG4-Related Disease Epidemiology
TOPLINE:
Men with immunoglobulin G4 (IgG4)-related disease exhibit significantly lower serum lipase levels and a greater likelihood of organ involvement than women, highlighting significant sex-dependent differences in disease manifestations.
METHODOLOGY:
- Researchers conducted a retrospective study of 328 patients (69% men) diagnosed with IgG4-related disease at the Massachusetts General Hospital – Rheumatology Clinic, Boston, who met the American College of Rheumatology–European Alliance of Associations for Rheumatology (ACR-EULAR) classification criteria between January 2008 and May 2023.
- Among the 328 patients, 69% were men and 31% were women, with a significant male-to-female ratio of 2.2:1.0. Men were typically older at diagnosis (median age, 63.7 vs 58.2 years).
- Data on serum lipase levels, renal involvement, and other clinical and laboratory parameters were collected.
TAKEAWAY:
- Men had higher baseline ACR-EULAR scores, indicating more severe disease (median score of 35.0 vs 29.5; P = .0010).
- Male patients demonstrated a median baseline serum lipase concentration of 24.5 U/L, significantly lower than the 33.5 U/L observed in women.
- Pancreatic (50% vs 26%) or renal (36% vs 18%) involvement was more common in men.
- Men exhibited higher IgG4 levels (P = .0050) and active B-cell responses in the blood (P = .0095).
IN PRACTICE:
According to the authors, this work confirms “the impression of an important sex disparity among patients with IgG4-related disease, with most patients being male, and male patients demonstrating strong tendencies toward more severe disease than female patients.”
SOURCE:
The study was led by Isha Jha, MD, Massachusetts General Hospital, Boston. It was published online on May 30, 2024, in The Lancet Rheumatology.
LIMITATIONS:
The study’s retrospective design may limit the ability to establish causality between sex differences and IgG4-related disease manifestations. A relatively small percentage of patients were assessed before receiving any immunosuppressive treatment, potentially influencing the observed clinical parameters.
DISCLOSURES:
This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases, the Rheumatology Research Foundation, and the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Some authors declared financial ties outside this work.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Men with immunoglobulin G4 (IgG4)-related disease exhibit significantly lower serum lipase levels and a greater likelihood of organ involvement than women, highlighting significant sex-dependent differences in disease manifestations.
METHODOLOGY:
- Researchers conducted a retrospective study of 328 patients (69% men) diagnosed with IgG4-related disease at the Massachusetts General Hospital – Rheumatology Clinic, Boston, who met the American College of Rheumatology–European Alliance of Associations for Rheumatology (ACR-EULAR) classification criteria between January 2008 and May 2023.
- Among the 328 patients, 69% were men and 31% were women, with a significant male-to-female ratio of 2.2:1.0. Men were typically older at diagnosis (median age, 63.7 vs 58.2 years).
- Data on serum lipase levels, renal involvement, and other clinical and laboratory parameters were collected.
TAKEAWAY:
- Men had higher baseline ACR-EULAR scores, indicating more severe disease (median score of 35.0 vs 29.5; P = .0010).
- Male patients demonstrated a median baseline serum lipase concentration of 24.5 U/L, significantly lower than the 33.5 U/L observed in women.
- Pancreatic (50% vs 26%) or renal (36% vs 18%) involvement was more common in men.
- Men exhibited higher IgG4 levels (P = .0050) and active B-cell responses in the blood (P = .0095).
IN PRACTICE:
According to the authors, this work confirms “the impression of an important sex disparity among patients with IgG4-related disease, with most patients being male, and male patients demonstrating strong tendencies toward more severe disease than female patients.”
SOURCE:
The study was led by Isha Jha, MD, Massachusetts General Hospital, Boston. It was published online on May 30, 2024, in The Lancet Rheumatology.
LIMITATIONS:
The study’s retrospective design may limit the ability to establish causality between sex differences and IgG4-related disease manifestations. A relatively small percentage of patients were assessed before receiving any immunosuppressive treatment, potentially influencing the observed clinical parameters.
DISCLOSURES:
This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases, the Rheumatology Research Foundation, and the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Some authors declared financial ties outside this work.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Men with immunoglobulin G4 (IgG4)-related disease exhibit significantly lower serum lipase levels and a greater likelihood of organ involvement than women, highlighting significant sex-dependent differences in disease manifestations.
METHODOLOGY:
- Researchers conducted a retrospective study of 328 patients (69% men) diagnosed with IgG4-related disease at the Massachusetts General Hospital – Rheumatology Clinic, Boston, who met the American College of Rheumatology–European Alliance of Associations for Rheumatology (ACR-EULAR) classification criteria between January 2008 and May 2023.
- Among the 328 patients, 69% were men and 31% were women, with a significant male-to-female ratio of 2.2:1.0. Men were typically older at diagnosis (median age, 63.7 vs 58.2 years).
- Data on serum lipase levels, renal involvement, and other clinical and laboratory parameters were collected.
TAKEAWAY:
- Men had higher baseline ACR-EULAR scores, indicating more severe disease (median score of 35.0 vs 29.5; P = .0010).
- Male patients demonstrated a median baseline serum lipase concentration of 24.5 U/L, significantly lower than the 33.5 U/L observed in women.
- Pancreatic (50% vs 26%) or renal (36% vs 18%) involvement was more common in men.
- Men exhibited higher IgG4 levels (P = .0050) and active B-cell responses in the blood (P = .0095).
IN PRACTICE:
According to the authors, this work confirms “the impression of an important sex disparity among patients with IgG4-related disease, with most patients being male, and male patients demonstrating strong tendencies toward more severe disease than female patients.”
SOURCE:
The study was led by Isha Jha, MD, Massachusetts General Hospital, Boston. It was published online on May 30, 2024, in The Lancet Rheumatology.
LIMITATIONS:
The study’s retrospective design may limit the ability to establish causality between sex differences and IgG4-related disease manifestations. A relatively small percentage of patients were assessed before receiving any immunosuppressive treatment, potentially influencing the observed clinical parameters.
DISCLOSURES:
This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases, the Rheumatology Research Foundation, and the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Some authors declared financial ties outside this work.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
VEXAS Syndrome: Study Highlights Cutaneous Symptoms
Additionally, the most common histologic findings include leukocytoclastic vasculitis, neutrophilic dermatosis, and perivascular dermatitis; different variants in the UBA1 gene are associated with specific skin manifestations.
Those are key findings from a cohort study of 112 patients with VEXAS published online in JAMA Dermatology. The study, conducted by researchers at the National Institutes of Health (NIH) and several other institutions, aimed to define the spectrum of cutaneous manifestations in VEXAS in association with genetic, histologic, and other clinical findings.

First described in 2020, VEXAS syndrome is an adult-onset multisystem disease that can pose a diagnostic challenge to clinicians, the study’s corresponding author, Edward W. Cowen, MD, MHSc, of the dermatology branch at the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), said in an interview. The disease is caused by pathogenic variants in the UBA1 gene, located on the X chromosome. Affected individuals exhibit a wide range of manifestations, including cytopenia/myelodysplasia, multiorgan systemic inflammation, and cutaneous involvement.
“Patients may present to a variety of disease specialists depending on their symptoms and providers may not immediately consider a genetic etiology in an older individual,” Dr. Cowen said in an interview. “Although skin involvement occurs in more than 80% of patients, it is pleomorphic and may resemble a variety of other conditions such as vasculitis and Sweet syndrome.”
To better understand the cutaneous manifestations of VEXAS syndrome, the researchers evaluated data from 112 patients with VEXAS-defining genetic variants in the UBA1 gene between 2019 and 2023. Of the 112 patients, 73 underwent medical record review only, and 39 were prospectively evaluated at NIH. All but one of the patients were men, 94% were White individuals, and their mean age was 64 years. Skin involvement occurred in 83% of cases and was the most common presenting feature of VEXAS in 61% of cases.
Of the 64 histopathologic reports available from 60 patients, the main skin histopathologic findings were leukocytoclastic vasculitis in 23 patients (36%), neutrophilic dermatosis in 22 patients (34%), and perivascular dermatitis in 19 patients (30%). According to Dr. Cowen, one key histologic finding was a distinct pattern of “histiocytoid” dermal neutrophilic inflammation, which was present in 13 of 15 specimens (86%) that underwent central re-review. “This pattern can occasionally also be seen in patients with Sweet syndrome, unrelated to VEXAS, but was a hallmark feature found in the majority of skin biopsies of patients with VEXAS,” he said.
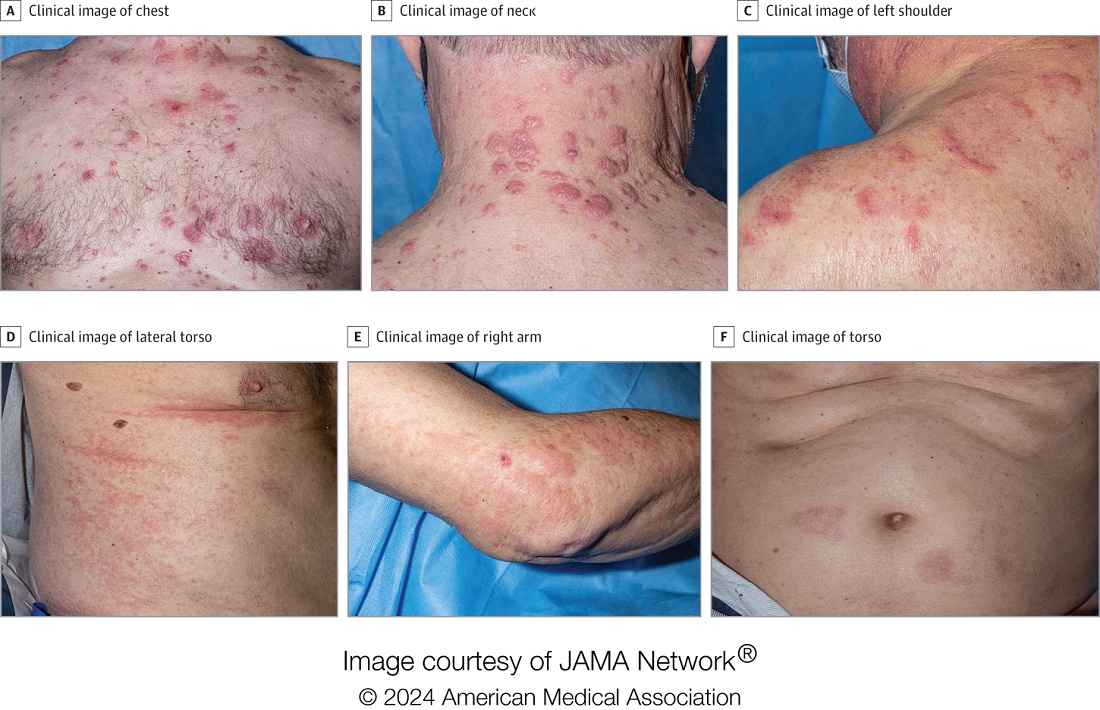
“Together with another pathologic finding, leukocytoclasia, these features can be useful clues to alert the pathologist to a potential diagnosis of VEXAS. This myeloid predominant pattern of skin inflammation was also most strongly associated with the leucine pathogenic variant of the UBA1 gene.” In contrast, cutaneous vasculitis was most strongly associated with the valine pathogenic variant of UBA1. “This is important because the valine variant has been previously independently linked to decreased survival,” he said.
In findings related to pathogenic genetic variants, the researchers observed that the p.Met41Leu variant was most frequently associated with neutrophilic dermal infiltrates in 14 of 17 patients (82%) with this variant and often resembled histiocytoid Sweet syndrome. In addition, the p.Met41Val variant was associated with vasculitic lesions in 11 of 20 patients (55%) with this variant and with a mixed leukocytic infiltrate in 17 of these 20 patients (85%).
Treatment Outcomes
In the realm of therapies, skin manifestations improved in 67 of 73 patients (92%) treated with oral prednisone, while treatment with the interleukin-1 receptor antagonist anakinra improved cutaneous disease in 9 of the 16 (56%) who received it. However, 12 (75%) of those who received anakinra developed severe injection-site reactions, including ulceration in two patients and abscess formation in one patient.
Dr. Cowen noted that VEXAS is associated with high mortality (22% in this cohort), and a high degree of suspicion is required to diagnose patients with VEXAS before significant end organ damage has occurred. “This diagnosis should be considered in all older male patients who present with neutrophilic dermatosis — particularly histiocytoid Sweet syndrome, vasculitis, or leukocytoclasia without vasculitis. Patients who appear to have isolated skin involvement may have cytopenias and acute phase reactants. Therefore, complete blood count with differential and ESR and CRP should be considered to investigate for macrocytosis, cytopenias, and systemic inflammation.”
He acknowledged certain limitations of the study, including the fact that many patients were first evaluated at the NIH after having disease symptoms for many months or years. “It is possible that patients with VEXAS referred to the NIH, either for genetic testing or in person evaluation, represent a population with more aggressive disease.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Connecticut, who was asked to comment on the study, emphasized the importance of the UBA1 mutation in the diagnosis of this complex syndrome. “Dermatologists should be aware of VEXAS syndrome as the majority of patients present with skin lesions, which can range from urticarial to Sweet syndrome–like to palpable purpura,” Dr. Ko said.
“Chondritis and periorbital edema, sometimes unilateral, are also associated. Histopathologic clues include a predominantly histiocytoid infiltrate,” she noted. In addition, “the prominent myxoid stroma around blood vessels and adnexal structures as a clue to VEXAS syndrome surprised me; I had not read that before.”
The study was supported by the Intramural Research Program of NIAMS. One of the study authors reported personal fees from Alexion, Novartis, and Sobi outside of the submitted work. No other disclosures were reported. Dr. Ko reported having no disclosures.
A version of this article appeared on Medscape.com .
Additionally, the most common histologic findings include leukocytoclastic vasculitis, neutrophilic dermatosis, and perivascular dermatitis; different variants in the UBA1 gene are associated with specific skin manifestations.
Those are key findings from a cohort study of 112 patients with VEXAS published online in JAMA Dermatology. The study, conducted by researchers at the National Institutes of Health (NIH) and several other institutions, aimed to define the spectrum of cutaneous manifestations in VEXAS in association with genetic, histologic, and other clinical findings.
First described in 2020, VEXAS syndrome is an adult-onset multisystem disease that can pose a diagnostic challenge to clinicians, the study’s corresponding author, Edward W. Cowen, MD, MHSc, of the dermatology branch at the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), said in an interview. The disease is caused by pathogenic variants in the UBA1 gene, located on the X chromosome. Affected individuals exhibit a wide range of manifestations, including cytopenia/myelodysplasia, multiorgan systemic inflammation, and cutaneous involvement.
“Patients may present to a variety of disease specialists depending on their symptoms and providers may not immediately consider a genetic etiology in an older individual,” Dr. Cowen said in an interview. “Although skin involvement occurs in more than 80% of patients, it is pleomorphic and may resemble a variety of other conditions such as vasculitis and Sweet syndrome.”
To better understand the cutaneous manifestations of VEXAS syndrome, the researchers evaluated data from 112 patients with VEXAS-defining genetic variants in the UBA1 gene between 2019 and 2023. Of the 112 patients, 73 underwent medical record review only, and 39 were prospectively evaluated at NIH. All but one of the patients were men, 94% were White individuals, and their mean age was 64 years. Skin involvement occurred in 83% of cases and was the most common presenting feature of VEXAS in 61% of cases.
Of the 64 histopathologic reports available from 60 patients, the main skin histopathologic findings were leukocytoclastic vasculitis in 23 patients (36%), neutrophilic dermatosis in 22 patients (34%), and perivascular dermatitis in 19 patients (30%). According to Dr. Cowen, one key histologic finding was a distinct pattern of “histiocytoid” dermal neutrophilic inflammation, which was present in 13 of 15 specimens (86%) that underwent central re-review. “This pattern can occasionally also be seen in patients with Sweet syndrome, unrelated to VEXAS, but was a hallmark feature found in the majority of skin biopsies of patients with VEXAS,” he said.
“Together with another pathologic finding, leukocytoclasia, these features can be useful clues to alert the pathologist to a potential diagnosis of VEXAS. This myeloid predominant pattern of skin inflammation was also most strongly associated with the leucine pathogenic variant of the UBA1 gene.” In contrast, cutaneous vasculitis was most strongly associated with the valine pathogenic variant of UBA1. “This is important because the valine variant has been previously independently linked to decreased survival,” he said.
In findings related to pathogenic genetic variants, the researchers observed that the p.Met41Leu variant was most frequently associated with neutrophilic dermal infiltrates in 14 of 17 patients (82%) with this variant and often resembled histiocytoid Sweet syndrome. In addition, the p.Met41Val variant was associated with vasculitic lesions in 11 of 20 patients (55%) with this variant and with a mixed leukocytic infiltrate in 17 of these 20 patients (85%).
Treatment Outcomes
In the realm of therapies, skin manifestations improved in 67 of 73 patients (92%) treated with oral prednisone, while treatment with the interleukin-1 receptor antagonist anakinra improved cutaneous disease in 9 of the 16 (56%) who received it. However, 12 (75%) of those who received anakinra developed severe injection-site reactions, including ulceration in two patients and abscess formation in one patient.
Dr. Cowen noted that VEXAS is associated with high mortality (22% in this cohort), and a high degree of suspicion is required to diagnose patients with VEXAS before significant end organ damage has occurred. “This diagnosis should be considered in all older male patients who present with neutrophilic dermatosis — particularly histiocytoid Sweet syndrome, vasculitis, or leukocytoclasia without vasculitis. Patients who appear to have isolated skin involvement may have cytopenias and acute phase reactants. Therefore, complete blood count with differential and ESR and CRP should be considered to investigate for macrocytosis, cytopenias, and systemic inflammation.”
He acknowledged certain limitations of the study, including the fact that many patients were first evaluated at the NIH after having disease symptoms for many months or years. “It is possible that patients with VEXAS referred to the NIH, either for genetic testing or in person evaluation, represent a population with more aggressive disease.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Connecticut, who was asked to comment on the study, emphasized the importance of the UBA1 mutation in the diagnosis of this complex syndrome. “Dermatologists should be aware of VEXAS syndrome as the majority of patients present with skin lesions, which can range from urticarial to Sweet syndrome–like to palpable purpura,” Dr. Ko said.
“Chondritis and periorbital edema, sometimes unilateral, are also associated. Histopathologic clues include a predominantly histiocytoid infiltrate,” she noted. In addition, “the prominent myxoid stroma around blood vessels and adnexal structures as a clue to VEXAS syndrome surprised me; I had not read that before.”
The study was supported by the Intramural Research Program of NIAMS. One of the study authors reported personal fees from Alexion, Novartis, and Sobi outside of the submitted work. No other disclosures were reported. Dr. Ko reported having no disclosures.
A version of this article appeared on Medscape.com .
Additionally, the most common histologic findings include leukocytoclastic vasculitis, neutrophilic dermatosis, and perivascular dermatitis; different variants in the UBA1 gene are associated with specific skin manifestations.
Those are key findings from a cohort study of 112 patients with VEXAS published online in JAMA Dermatology. The study, conducted by researchers at the National Institutes of Health (NIH) and several other institutions, aimed to define the spectrum of cutaneous manifestations in VEXAS in association with genetic, histologic, and other clinical findings.
First described in 2020, VEXAS syndrome is an adult-onset multisystem disease that can pose a diagnostic challenge to clinicians, the study’s corresponding author, Edward W. Cowen, MD, MHSc, of the dermatology branch at the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), said in an interview. The disease is caused by pathogenic variants in the UBA1 gene, located on the X chromosome. Affected individuals exhibit a wide range of manifestations, including cytopenia/myelodysplasia, multiorgan systemic inflammation, and cutaneous involvement.
“Patients may present to a variety of disease specialists depending on their symptoms and providers may not immediately consider a genetic etiology in an older individual,” Dr. Cowen said in an interview. “Although skin involvement occurs in more than 80% of patients, it is pleomorphic and may resemble a variety of other conditions such as vasculitis and Sweet syndrome.”
To better understand the cutaneous manifestations of VEXAS syndrome, the researchers evaluated data from 112 patients with VEXAS-defining genetic variants in the UBA1 gene between 2019 and 2023. Of the 112 patients, 73 underwent medical record review only, and 39 were prospectively evaluated at NIH. All but one of the patients were men, 94% were White individuals, and their mean age was 64 years. Skin involvement occurred in 83% of cases and was the most common presenting feature of VEXAS in 61% of cases.
Of the 64 histopathologic reports available from 60 patients, the main skin histopathologic findings were leukocytoclastic vasculitis in 23 patients (36%), neutrophilic dermatosis in 22 patients (34%), and perivascular dermatitis in 19 patients (30%). According to Dr. Cowen, one key histologic finding was a distinct pattern of “histiocytoid” dermal neutrophilic inflammation, which was present in 13 of 15 specimens (86%) that underwent central re-review. “This pattern can occasionally also be seen in patients with Sweet syndrome, unrelated to VEXAS, but was a hallmark feature found in the majority of skin biopsies of patients with VEXAS,” he said.
“Together with another pathologic finding, leukocytoclasia, these features can be useful clues to alert the pathologist to a potential diagnosis of VEXAS. This myeloid predominant pattern of skin inflammation was also most strongly associated with the leucine pathogenic variant of the UBA1 gene.” In contrast, cutaneous vasculitis was most strongly associated with the valine pathogenic variant of UBA1. “This is important because the valine variant has been previously independently linked to decreased survival,” he said.
In findings related to pathogenic genetic variants, the researchers observed that the p.Met41Leu variant was most frequently associated with neutrophilic dermal infiltrates in 14 of 17 patients (82%) with this variant and often resembled histiocytoid Sweet syndrome. In addition, the p.Met41Val variant was associated with vasculitic lesions in 11 of 20 patients (55%) with this variant and with a mixed leukocytic infiltrate in 17 of these 20 patients (85%).
Treatment Outcomes
In the realm of therapies, skin manifestations improved in 67 of 73 patients (92%) treated with oral prednisone, while treatment with the interleukin-1 receptor antagonist anakinra improved cutaneous disease in 9 of the 16 (56%) who received it. However, 12 (75%) of those who received anakinra developed severe injection-site reactions, including ulceration in two patients and abscess formation in one patient.
Dr. Cowen noted that VEXAS is associated with high mortality (22% in this cohort), and a high degree of suspicion is required to diagnose patients with VEXAS before significant end organ damage has occurred. “This diagnosis should be considered in all older male patients who present with neutrophilic dermatosis — particularly histiocytoid Sweet syndrome, vasculitis, or leukocytoclasia without vasculitis. Patients who appear to have isolated skin involvement may have cytopenias and acute phase reactants. Therefore, complete blood count with differential and ESR and CRP should be considered to investigate for macrocytosis, cytopenias, and systemic inflammation.”
He acknowledged certain limitations of the study, including the fact that many patients were first evaluated at the NIH after having disease symptoms for many months or years. “It is possible that patients with VEXAS referred to the NIH, either for genetic testing or in person evaluation, represent a population with more aggressive disease.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Connecticut, who was asked to comment on the study, emphasized the importance of the UBA1 mutation in the diagnosis of this complex syndrome. “Dermatologists should be aware of VEXAS syndrome as the majority of patients present with skin lesions, which can range from urticarial to Sweet syndrome–like to palpable purpura,” Dr. Ko said.
“Chondritis and periorbital edema, sometimes unilateral, are also associated. Histopathologic clues include a predominantly histiocytoid infiltrate,” she noted. In addition, “the prominent myxoid stroma around blood vessels and adnexal structures as a clue to VEXAS syndrome surprised me; I had not read that before.”
The study was supported by the Intramural Research Program of NIAMS. One of the study authors reported personal fees from Alexion, Novartis, and Sobi outside of the submitted work. No other disclosures were reported. Dr. Ko reported having no disclosures.
A version of this article appeared on Medscape.com .
FROM JAMA DERMATOLOGY