User login
Tumor-treating fields boost chemo for mesothelioma
GENEVA – For patients with malignant pleural mesothelioma, adding tumor-treating fields (TTFields) to standard pemetrexed plus platinum compound chemotherapy could boost median overall survival by about 6 months, according to final results from the phase 2 STELLAR trial.

The survival benefit of TTFields was greatest among patients with epithelioid mesothelioma, reported lead author Giovanni Luca Ceresoli, MD, of Humanitas Gavazzeni in Bergamo, Italy. According to Dr. Ceresoli, who presented findings at the at the European Lung Cancer Conference, TTFields offer a safe way to improve mesothelioma outcomes without increasing the risk of serious adverse events.
“TTFields are a locoregional treatment comprising low-intensity alternating electric fields delivered through a portable medical device,” Dr. Ceresoli explained at the meeting, presented by the European Society for Medical Oncology. “Their main mode of action is an anti-mitotic mechanism.” He noted that TTFields are already approved by the Food and Drug Administration for newly diagnosed glioblastoma.
The STELLAR trial involved 80 patients with mesothelioma who were treated with TTFields in combination with standard first-line chemotherapy, a combination of pemetrexed with cisplatin or carboplatin. Patients were instructed to self-administer continuous 150 kHz TTFields for at least 18 hours a day. Eligibility required an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1. Both ECOG status and cancer-related pain were followed with a visual analog scale until disease progression. Median overall survival (OS) was the primary endpoint.
The patient population was predominantly male (84%), with median age of 67 years. About 44% of the patients had an ECOG performance status of 1 and 66% had epithelioid histology. Median treatment time per day was 16.3 hours.
After a minimum follow-up of 1 year, patients treated with TTFields in combination with standard chemotherapy had a median overall survival of 18.2 months, compared with 12.1 months for standard chemotherapy alone, which Dr. Ceresoli cited as the historical benchmark. The survival benefit was 3 months longer among patients with epithelioid mesothelioma, who had a median overall survival of 21.2 months.
In addition to survival benefits, the investigators found that median time to decreased performance status was just over 1 year (13.1 months), and that pain did not increase to a clinically significant degree (33%) until an average of 8.4 months. Although no device-related serious adverse events occurred, 37 patients (46%) experienced TTFields-related dermatitis; 4 of these patients had grade 3 dermatitis. Dr. Ceresoli noted that dermatitis was typically “easily managed” with topical application of a corticosteroid, while patients with severe dermatitis took short treatment breaks.
“In conclusion, in the STELLAR trial, TTFields in combination with standard chemotherapy were effective and safe for first-line treatment of unresectable malignant pleural mesothelioma, and median overall survival was significantly longer as compared to historical controls,” Dr. Ceresoli said, pointing out better survival than in recent trials MAPS and LUME-Meso.
When asked by the invited discussant about future research, Dr. Ceresoli described a narrower focus for upcoming TTFields studies for mesothelioma. “As you well know, most patients have epithelioid histology, and in our hands, the patients with epithelioid histology had better prognoses,” he said. “So, in the future, I think we will focus on epithelioid tumors.”
Dr. Ceresoli disclosed travel funding from Novocure.
SOURCE: Ceresoli et al. ELCC 2019. Abstract 55O.
GENEVA – For patients with malignant pleural mesothelioma, adding tumor-treating fields (TTFields) to standard pemetrexed plus platinum compound chemotherapy could boost median overall survival by about 6 months, according to final results from the phase 2 STELLAR trial.
The survival benefit of TTFields was greatest among patients with epithelioid mesothelioma, reported lead author Giovanni Luca Ceresoli, MD, of Humanitas Gavazzeni in Bergamo, Italy. According to Dr. Ceresoli, who presented findings at the at the European Lung Cancer Conference, TTFields offer a safe way to improve mesothelioma outcomes without increasing the risk of serious adverse events.
“TTFields are a locoregional treatment comprising low-intensity alternating electric fields delivered through a portable medical device,” Dr. Ceresoli explained at the meeting, presented by the European Society for Medical Oncology. “Their main mode of action is an anti-mitotic mechanism.” He noted that TTFields are already approved by the Food and Drug Administration for newly diagnosed glioblastoma.
The STELLAR trial involved 80 patients with mesothelioma who were treated with TTFields in combination with standard first-line chemotherapy, a combination of pemetrexed with cisplatin or carboplatin. Patients were instructed to self-administer continuous 150 kHz TTFields for at least 18 hours a day. Eligibility required an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1. Both ECOG status and cancer-related pain were followed with a visual analog scale until disease progression. Median overall survival (OS) was the primary endpoint.
The patient population was predominantly male (84%), with median age of 67 years. About 44% of the patients had an ECOG performance status of 1 and 66% had epithelioid histology. Median treatment time per day was 16.3 hours.
After a minimum follow-up of 1 year, patients treated with TTFields in combination with standard chemotherapy had a median overall survival of 18.2 months, compared with 12.1 months for standard chemotherapy alone, which Dr. Ceresoli cited as the historical benchmark. The survival benefit was 3 months longer among patients with epithelioid mesothelioma, who had a median overall survival of 21.2 months.
In addition to survival benefits, the investigators found that median time to decreased performance status was just over 1 year (13.1 months), and that pain did not increase to a clinically significant degree (33%) until an average of 8.4 months. Although no device-related serious adverse events occurred, 37 patients (46%) experienced TTFields-related dermatitis; 4 of these patients had grade 3 dermatitis. Dr. Ceresoli noted that dermatitis was typically “easily managed” with topical application of a corticosteroid, while patients with severe dermatitis took short treatment breaks.
“In conclusion, in the STELLAR trial, TTFields in combination with standard chemotherapy were effective and safe for first-line treatment of unresectable malignant pleural mesothelioma, and median overall survival was significantly longer as compared to historical controls,” Dr. Ceresoli said, pointing out better survival than in recent trials MAPS and LUME-Meso.
When asked by the invited discussant about future research, Dr. Ceresoli described a narrower focus for upcoming TTFields studies for mesothelioma. “As you well know, most patients have epithelioid histology, and in our hands, the patients with epithelioid histology had better prognoses,” he said. “So, in the future, I think we will focus on epithelioid tumors.”
Dr. Ceresoli disclosed travel funding from Novocure.
SOURCE: Ceresoli et al. ELCC 2019. Abstract 55O.
GENEVA – For patients with malignant pleural mesothelioma, adding tumor-treating fields (TTFields) to standard pemetrexed plus platinum compound chemotherapy could boost median overall survival by about 6 months, according to final results from the phase 2 STELLAR trial.
The survival benefit of TTFields was greatest among patients with epithelioid mesothelioma, reported lead author Giovanni Luca Ceresoli, MD, of Humanitas Gavazzeni in Bergamo, Italy. According to Dr. Ceresoli, who presented findings at the at the European Lung Cancer Conference, TTFields offer a safe way to improve mesothelioma outcomes without increasing the risk of serious adverse events.
“TTFields are a locoregional treatment comprising low-intensity alternating electric fields delivered through a portable medical device,” Dr. Ceresoli explained at the meeting, presented by the European Society for Medical Oncology. “Their main mode of action is an anti-mitotic mechanism.” He noted that TTFields are already approved by the Food and Drug Administration for newly diagnosed glioblastoma.
The STELLAR trial involved 80 patients with mesothelioma who were treated with TTFields in combination with standard first-line chemotherapy, a combination of pemetrexed with cisplatin or carboplatin. Patients were instructed to self-administer continuous 150 kHz TTFields for at least 18 hours a day. Eligibility required an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1. Both ECOG status and cancer-related pain were followed with a visual analog scale until disease progression. Median overall survival (OS) was the primary endpoint.
The patient population was predominantly male (84%), with median age of 67 years. About 44% of the patients had an ECOG performance status of 1 and 66% had epithelioid histology. Median treatment time per day was 16.3 hours.
After a minimum follow-up of 1 year, patients treated with TTFields in combination with standard chemotherapy had a median overall survival of 18.2 months, compared with 12.1 months for standard chemotherapy alone, which Dr. Ceresoli cited as the historical benchmark. The survival benefit was 3 months longer among patients with epithelioid mesothelioma, who had a median overall survival of 21.2 months.
In addition to survival benefits, the investigators found that median time to decreased performance status was just over 1 year (13.1 months), and that pain did not increase to a clinically significant degree (33%) until an average of 8.4 months. Although no device-related serious adverse events occurred, 37 patients (46%) experienced TTFields-related dermatitis; 4 of these patients had grade 3 dermatitis. Dr. Ceresoli noted that dermatitis was typically “easily managed” with topical application of a corticosteroid, while patients with severe dermatitis took short treatment breaks.
“In conclusion, in the STELLAR trial, TTFields in combination with standard chemotherapy were effective and safe for first-line treatment of unresectable malignant pleural mesothelioma, and median overall survival was significantly longer as compared to historical controls,” Dr. Ceresoli said, pointing out better survival than in recent trials MAPS and LUME-Meso.
When asked by the invited discussant about future research, Dr. Ceresoli described a narrower focus for upcoming TTFields studies for mesothelioma. “As you well know, most patients have epithelioid histology, and in our hands, the patients with epithelioid histology had better prognoses,” he said. “So, in the future, I think we will focus on epithelioid tumors.”
Dr. Ceresoli disclosed travel funding from Novocure.
SOURCE: Ceresoli et al. ELCC 2019. Abstract 55O.
At ELCC 2019
Asboe-Hansen Sign in Toxic Epidermal Necrolysis
To the Editor:
A 25-year-old woman with no notable medical history was admitted to the hospital for suspected Stevens-Johnson syndrome (SJS). The patient was started on amoxicillin 7 days prior to the skin eruption for prophylaxis before removal of an intrauterine device. On the day of admission, she reported ocular discomfort, dysphagia, and dysuria. She developed erythema of the conjunctivae, face, chest, and proximal upper extremities, as well as erosions of the vermilion lips. She presented to the local emergency department and was transferred to our institution for urgent dermatologic consultation. On physical examination by the dermatology service, the patient had erythematous macules coalescing into patches with overlying flaccid bullae, some denuded, involving the face, chest, abdomen, back (Figure 1), bilateral upper extremities, bilateral thighs, and labia majora and minora. Additionally, she had conjunctivitis, superficial erosions of the vermilion lips, and tense bullae of the palms and soles. On palpation of the flaccid bullae, the Asboe-Hansen sign was elicited (Figure 2 and video). A shave biopsy of the newly elicited bullae was performed. Pathology showed a subepidermal bulla with confluent necrosis of the epidermis and minimal inflammatory infiltrate. An additional shave biopsy of perilesional skin was obtained for direct immunofluorescence, which was negative for IgG, C3, IgM, and IgA. Based on the clinical presentation involving more than 30% of the patient’s body surface area (BSA) and the pathology findings, a diagnosis of toxic epidermal necrolysis (TEN) was made. The patient remained in the intensive care unit with a multidisciplinary team consisting of dermatology, ophthalmology, gynecology, gastroenterology, and the general surgery burn group. Following treatment with intravenous immunoglobulin, systemic corticosteroids, and aggressive wound care, the patient made a full recovery.
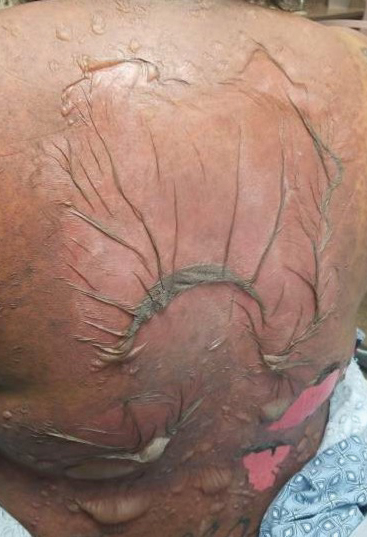
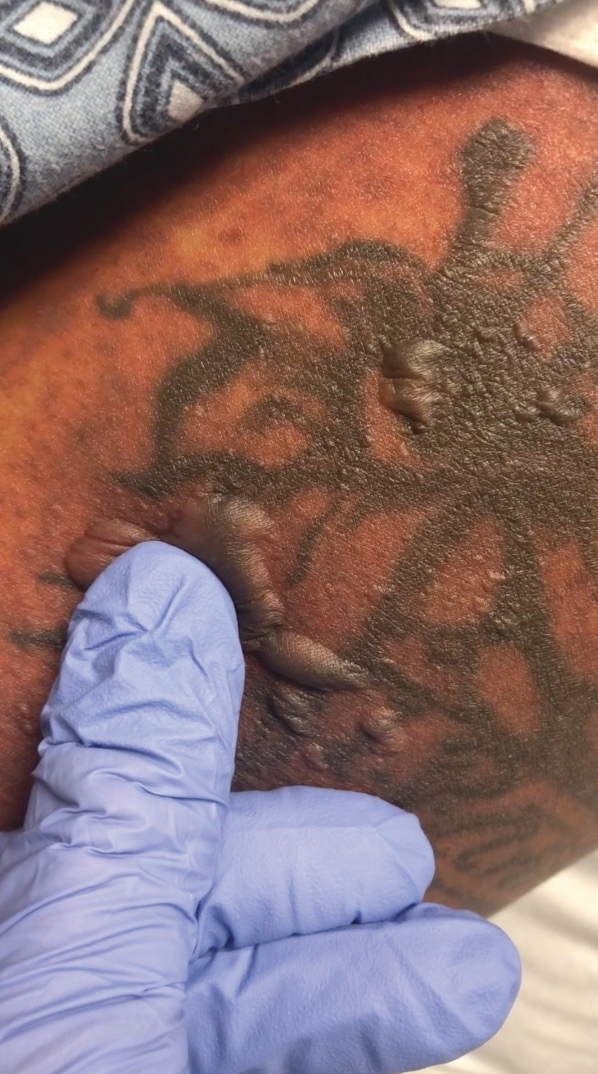
applied to an intact bulla.

Toxic epidermal necrolysis is a rare, acute, life-threatening mucocutaneous disease within a spectrum of adverse cutaneous drug reactions. The estimated worldwide incidence of TEN is 0.4 to 1.9 per million individuals annually.1 Toxic epidermal necrolysis is clinically characterized by diffuse exfoliation of the skin and mucosae with flaccid bullae. These clinical features are a consequence of extensive keratinocyte death, leading to dermoepidermal junction dissociation. Commonly, there is a prodrome of fever, pharyngitis, and painful skin preceding the diffuse erythema and sloughing of skin and mucous membranes. Lesions typically first appear on the trunk and then follow a centrifugal spread, often sparing the distal aspects of the arms and legs.
Toxic epidermal necrolysis is part of a continuous spectrum with SJS. Less than 10% BSA involvement is considered SJS, 10% to 30% BSA involvement is SJS/TEN overlap, and more than 30% BSA detachment is TEN. Stevens-Johnson syndrome can progress to TEN. In TEN, the distribution of cutaneous lesions is more confluent, and mucosal involvement is more severe.2 The differential diagnosis may include staphylococcal scalded skin syndrome, drug-induced linear IgA bullous dermatosis, severe acute graft-vs-host disease, drug reaction with eosinophilia and systemic symptoms, and invasive fungal dermatitis. An accurate diagnosis of TEN is imperative, as the management and morbidity of these diseases are vastly different. Toxic epidermal necrolysis has an estimated mortality rate of 25% to 30%, with sepsis leading to multiorgan failure being the most common cause of death.3
Although the pathophysiology of TEN has yet to be fully elucidated, it is thought to be a T cell–mediated process with CD8+ cells acting as the primary means of keratinocyte death. An estimated 80% to 95% of cases are due to drug reactions.3 The medications that are most commonly associated with TEN include allopurinol, antibiotics, nonsteroidal anti-inflammatory drugs, and anticonvulsants. Symptoms typically begin 7 to 21 days after starting the drug. Less commonly, Mycoplasma pneumoniae, dengue virus, cytomegalovirus, and contrast medium have been reported as inciting factors for TEN.2
The diagnosis of TEN is established by correlating clinical features with a histopathologic examination obtained from a lesional skin biopsy. The classic cutaneous features of TEN begin as erythematous, flesh-colored, dusky to violaceous macules and/or morbilliform or targetoid lesions. These early lesions have the tendency to coalesce. The cutaneous findings will eventually progress into flaccid bullae, diffuse epidermal sloughing, and full-thickness skin necrosis.2,3 The evolution of skin lesions may be rapid or may take several days to develop. On palpation, the Nikolsky (lateral shearing of epidermis with minimal pressure) and Asboe-Hansen sign will be positive in patients with SJS/TEN, demonstrating that the associated blisters are flaccid and may be displaced peripherally.4 For an accurate diagnosis, the biopsy must contain full-thickness epidermis. It is imperative to choose a biopsy site from an acute blister, as old lesions of other diseases, such as erythema multiforme, will eventually become necrotic and mimic the histopathologic appearance of SJS/TEN, potentially leading to an incorrect diagnosis.4 Full-thickness epidermal necrosis has a high sensitivity but low specificity for TEN.3 The histologic features of TEN vary depending on the stage of the disease. Classic histologic findings include satellite necrosis of keratinocytes followed by full-thickness necrosis of keratinocytes and perivascular lymphoid infiltrates. The stratum corneum retains its original structure.4
The Asboe-Hansen sign, also known as the bulla spread sign, was originally described in 1960 as a diagnostic sign for pemphigus vulgaris.5 A positive Asboe-Hansen sign demonstrates the ability to enlarge a bulla in the lateral direction by applying perpendicular mechanical pressure to the roof of an intact bulla. The bulla is extended to adjacent nonblistered skin.6 A positive sign demonstrates decreased adhesion between keratinocytes or between the basal epidermal cells and the dermal connective tissue.5 In addition to pemphigus vulgaris, the Asboe-Hansen sign may be positive in TEN and SJS, as well as other diseases affecting the dermoepidermal junction including pemphigus foliaceus, pemphigus vegetans, and bullous pemphigoid. Asboe-Hansen5 made the argument that a fresh bulla should be biopsied if histopathologic diagnosis is necessary, as older bullae may exhibit epithelial cell regeneration and disturb an accurate diagnosis.
Accurate and early diagnosis of TEN is imperative, as prognosis is strongly correlated with the speed at which the offending drug is discontinued and appropriate medical treatment is initiated. Prompt withdrawal of the offending drug has been reported to reduce the risk for morbidity by 30% per day.7 Although classically associated with the pemphigus group of diseases, the Asboe-Hansen sign is of diagnostic value to the pathologist in diagnosing TEN by reproducing the same microscopic appearance of a fresh spontaneous blister. Due to the notable morbidity and mortality in SJS and TEN, the Asboe-Hansen sign should be attempted for the site of a lesional biopsy, as an accurate diagnosis relies on clinicopathologic correlation.
- Schwartz RA, McDonough PH, Lee BW, et al. Toxic epidermal necrolysis: part I. introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69:173.e1-173.e13.
- Frech LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:332-347.
- Schwartz RA, McDonough PH, Lee BW, et al. Toxic epidermal necrolysis: part II. prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69:187.e1–187.e16.
- Elston D, Stratman E, Miller S. Skin biopsy. J Am Acad Dermatol. 2016;74:1-16.
- Asboe-Hansen G. Blister-spread induced by finger-pressure, a diagnostic sign in pemphigus. J Invest Dermatol. 1960;34:5-9.
- Ganapati S. Eponymous dermatological signs in bullous dermatoses. Indian J Dermatol. 2014;59:21-23.
- Garcia-Doval I, Lecleach L, Bocquet H, et al. Toxic epidermal necrolysis and Stevens-Johnson syndrome: does early withdrawal of causative drugs decrease the risk of death? Arch Dermatol. 2000;136:323-327.
To the Editor:
A 25-year-old woman with no notable medical history was admitted to the hospital for suspected Stevens-Johnson syndrome (SJS). The patient was started on amoxicillin 7 days prior to the skin eruption for prophylaxis before removal of an intrauterine device. On the day of admission, she reported ocular discomfort, dysphagia, and dysuria. She developed erythema of the conjunctivae, face, chest, and proximal upper extremities, as well as erosions of the vermilion lips. She presented to the local emergency department and was transferred to our institution for urgent dermatologic consultation. On physical examination by the dermatology service, the patient had erythematous macules coalescing into patches with overlying flaccid bullae, some denuded, involving the face, chest, abdomen, back (Figure 1), bilateral upper extremities, bilateral thighs, and labia majora and minora. Additionally, she had conjunctivitis, superficial erosions of the vermilion lips, and tense bullae of the palms and soles. On palpation of the flaccid bullae, the Asboe-Hansen sign was elicited (Figure 2 and video). A shave biopsy of the newly elicited bullae was performed. Pathology showed a subepidermal bulla with confluent necrosis of the epidermis and minimal inflammatory infiltrate. An additional shave biopsy of perilesional skin was obtained for direct immunofluorescence, which was negative for IgG, C3, IgM, and IgA. Based on the clinical presentation involving more than 30% of the patient’s body surface area (BSA) and the pathology findings, a diagnosis of toxic epidermal necrolysis (TEN) was made. The patient remained in the intensive care unit with a multidisciplinary team consisting of dermatology, ophthalmology, gynecology, gastroenterology, and the general surgery burn group. Following treatment with intravenous immunoglobulin, systemic corticosteroids, and aggressive wound care, the patient made a full recovery.
applied to an intact bulla.
Toxic epidermal necrolysis is a rare, acute, life-threatening mucocutaneous disease within a spectrum of adverse cutaneous drug reactions. The estimated worldwide incidence of TEN is 0.4 to 1.9 per million individuals annually.1 Toxic epidermal necrolysis is clinically characterized by diffuse exfoliation of the skin and mucosae with flaccid bullae. These clinical features are a consequence of extensive keratinocyte death, leading to dermoepidermal junction dissociation. Commonly, there is a prodrome of fever, pharyngitis, and painful skin preceding the diffuse erythema and sloughing of skin and mucous membranes. Lesions typically first appear on the trunk and then follow a centrifugal spread, often sparing the distal aspects of the arms and legs.
Toxic epidermal necrolysis is part of a continuous spectrum with SJS. Less than 10% BSA involvement is considered SJS, 10% to 30% BSA involvement is SJS/TEN overlap, and more than 30% BSA detachment is TEN. Stevens-Johnson syndrome can progress to TEN. In TEN, the distribution of cutaneous lesions is more confluent, and mucosal involvement is more severe.2 The differential diagnosis may include staphylococcal scalded skin syndrome, drug-induced linear IgA bullous dermatosis, severe acute graft-vs-host disease, drug reaction with eosinophilia and systemic symptoms, and invasive fungal dermatitis. An accurate diagnosis of TEN is imperative, as the management and morbidity of these diseases are vastly different. Toxic epidermal necrolysis has an estimated mortality rate of 25% to 30%, with sepsis leading to multiorgan failure being the most common cause of death.3
Although the pathophysiology of TEN has yet to be fully elucidated, it is thought to be a T cell–mediated process with CD8+ cells acting as the primary means of keratinocyte death. An estimated 80% to 95% of cases are due to drug reactions.3 The medications that are most commonly associated with TEN include allopurinol, antibiotics, nonsteroidal anti-inflammatory drugs, and anticonvulsants. Symptoms typically begin 7 to 21 days after starting the drug. Less commonly, Mycoplasma pneumoniae, dengue virus, cytomegalovirus, and contrast medium have been reported as inciting factors for TEN.2
The diagnosis of TEN is established by correlating clinical features with a histopathologic examination obtained from a lesional skin biopsy. The classic cutaneous features of TEN begin as erythematous, flesh-colored, dusky to violaceous macules and/or morbilliform or targetoid lesions. These early lesions have the tendency to coalesce. The cutaneous findings will eventually progress into flaccid bullae, diffuse epidermal sloughing, and full-thickness skin necrosis.2,3 The evolution of skin lesions may be rapid or may take several days to develop. On palpation, the Nikolsky (lateral shearing of epidermis with minimal pressure) and Asboe-Hansen sign will be positive in patients with SJS/TEN, demonstrating that the associated blisters are flaccid and may be displaced peripherally.4 For an accurate diagnosis, the biopsy must contain full-thickness epidermis. It is imperative to choose a biopsy site from an acute blister, as old lesions of other diseases, such as erythema multiforme, will eventually become necrotic and mimic the histopathologic appearance of SJS/TEN, potentially leading to an incorrect diagnosis.4 Full-thickness epidermal necrosis has a high sensitivity but low specificity for TEN.3 The histologic features of TEN vary depending on the stage of the disease. Classic histologic findings include satellite necrosis of keratinocytes followed by full-thickness necrosis of keratinocytes and perivascular lymphoid infiltrates. The stratum corneum retains its original structure.4
The Asboe-Hansen sign, also known as the bulla spread sign, was originally described in 1960 as a diagnostic sign for pemphigus vulgaris.5 A positive Asboe-Hansen sign demonstrates the ability to enlarge a bulla in the lateral direction by applying perpendicular mechanical pressure to the roof of an intact bulla. The bulla is extended to adjacent nonblistered skin.6 A positive sign demonstrates decreased adhesion between keratinocytes or between the basal epidermal cells and the dermal connective tissue.5 In addition to pemphigus vulgaris, the Asboe-Hansen sign may be positive in TEN and SJS, as well as other diseases affecting the dermoepidermal junction including pemphigus foliaceus, pemphigus vegetans, and bullous pemphigoid. Asboe-Hansen5 made the argument that a fresh bulla should be biopsied if histopathologic diagnosis is necessary, as older bullae may exhibit epithelial cell regeneration and disturb an accurate diagnosis.
Accurate and early diagnosis of TEN is imperative, as prognosis is strongly correlated with the speed at which the offending drug is discontinued and appropriate medical treatment is initiated. Prompt withdrawal of the offending drug has been reported to reduce the risk for morbidity by 30% per day.7 Although classically associated with the pemphigus group of diseases, the Asboe-Hansen sign is of diagnostic value to the pathologist in diagnosing TEN by reproducing the same microscopic appearance of a fresh spontaneous blister. Due to the notable morbidity and mortality in SJS and TEN, the Asboe-Hansen sign should be attempted for the site of a lesional biopsy, as an accurate diagnosis relies on clinicopathologic correlation.
To the Editor:
A 25-year-old woman with no notable medical history was admitted to the hospital for suspected Stevens-Johnson syndrome (SJS). The patient was started on amoxicillin 7 days prior to the skin eruption for prophylaxis before removal of an intrauterine device. On the day of admission, she reported ocular discomfort, dysphagia, and dysuria. She developed erythema of the conjunctivae, face, chest, and proximal upper extremities, as well as erosions of the vermilion lips. She presented to the local emergency department and was transferred to our institution for urgent dermatologic consultation. On physical examination by the dermatology service, the patient had erythematous macules coalescing into patches with overlying flaccid bullae, some denuded, involving the face, chest, abdomen, back (Figure 1), bilateral upper extremities, bilateral thighs, and labia majora and minora. Additionally, she had conjunctivitis, superficial erosions of the vermilion lips, and tense bullae of the palms and soles. On palpation of the flaccid bullae, the Asboe-Hansen sign was elicited (Figure 2 and video). A shave biopsy of the newly elicited bullae was performed. Pathology showed a subepidermal bulla with confluent necrosis of the epidermis and minimal inflammatory infiltrate. An additional shave biopsy of perilesional skin was obtained for direct immunofluorescence, which was negative for IgG, C3, IgM, and IgA. Based on the clinical presentation involving more than 30% of the patient’s body surface area (BSA) and the pathology findings, a diagnosis of toxic epidermal necrolysis (TEN) was made. The patient remained in the intensive care unit with a multidisciplinary team consisting of dermatology, ophthalmology, gynecology, gastroenterology, and the general surgery burn group. Following treatment with intravenous immunoglobulin, systemic corticosteroids, and aggressive wound care, the patient made a full recovery.
applied to an intact bulla.
Toxic epidermal necrolysis is a rare, acute, life-threatening mucocutaneous disease within a spectrum of adverse cutaneous drug reactions. The estimated worldwide incidence of TEN is 0.4 to 1.9 per million individuals annually.1 Toxic epidermal necrolysis is clinically characterized by diffuse exfoliation of the skin and mucosae with flaccid bullae. These clinical features are a consequence of extensive keratinocyte death, leading to dermoepidermal junction dissociation. Commonly, there is a prodrome of fever, pharyngitis, and painful skin preceding the diffuse erythema and sloughing of skin and mucous membranes. Lesions typically first appear on the trunk and then follow a centrifugal spread, often sparing the distal aspects of the arms and legs.
Toxic epidermal necrolysis is part of a continuous spectrum with SJS. Less than 10% BSA involvement is considered SJS, 10% to 30% BSA involvement is SJS/TEN overlap, and more than 30% BSA detachment is TEN. Stevens-Johnson syndrome can progress to TEN. In TEN, the distribution of cutaneous lesions is more confluent, and mucosal involvement is more severe.2 The differential diagnosis may include staphylococcal scalded skin syndrome, drug-induced linear IgA bullous dermatosis, severe acute graft-vs-host disease, drug reaction with eosinophilia and systemic symptoms, and invasive fungal dermatitis. An accurate diagnosis of TEN is imperative, as the management and morbidity of these diseases are vastly different. Toxic epidermal necrolysis has an estimated mortality rate of 25% to 30%, with sepsis leading to multiorgan failure being the most common cause of death.3
Although the pathophysiology of TEN has yet to be fully elucidated, it is thought to be a T cell–mediated process with CD8+ cells acting as the primary means of keratinocyte death. An estimated 80% to 95% of cases are due to drug reactions.3 The medications that are most commonly associated with TEN include allopurinol, antibiotics, nonsteroidal anti-inflammatory drugs, and anticonvulsants. Symptoms typically begin 7 to 21 days after starting the drug. Less commonly, Mycoplasma pneumoniae, dengue virus, cytomegalovirus, and contrast medium have been reported as inciting factors for TEN.2
The diagnosis of TEN is established by correlating clinical features with a histopathologic examination obtained from a lesional skin biopsy. The classic cutaneous features of TEN begin as erythematous, flesh-colored, dusky to violaceous macules and/or morbilliform or targetoid lesions. These early lesions have the tendency to coalesce. The cutaneous findings will eventually progress into flaccid bullae, diffuse epidermal sloughing, and full-thickness skin necrosis.2,3 The evolution of skin lesions may be rapid or may take several days to develop. On palpation, the Nikolsky (lateral shearing of epidermis with minimal pressure) and Asboe-Hansen sign will be positive in patients with SJS/TEN, demonstrating that the associated blisters are flaccid and may be displaced peripherally.4 For an accurate diagnosis, the biopsy must contain full-thickness epidermis. It is imperative to choose a biopsy site from an acute blister, as old lesions of other diseases, such as erythema multiforme, will eventually become necrotic and mimic the histopathologic appearance of SJS/TEN, potentially leading to an incorrect diagnosis.4 Full-thickness epidermal necrosis has a high sensitivity but low specificity for TEN.3 The histologic features of TEN vary depending on the stage of the disease. Classic histologic findings include satellite necrosis of keratinocytes followed by full-thickness necrosis of keratinocytes and perivascular lymphoid infiltrates. The stratum corneum retains its original structure.4
The Asboe-Hansen sign, also known as the bulla spread sign, was originally described in 1960 as a diagnostic sign for pemphigus vulgaris.5 A positive Asboe-Hansen sign demonstrates the ability to enlarge a bulla in the lateral direction by applying perpendicular mechanical pressure to the roof of an intact bulla. The bulla is extended to adjacent nonblistered skin.6 A positive sign demonstrates decreased adhesion between keratinocytes or between the basal epidermal cells and the dermal connective tissue.5 In addition to pemphigus vulgaris, the Asboe-Hansen sign may be positive in TEN and SJS, as well as other diseases affecting the dermoepidermal junction including pemphigus foliaceus, pemphigus vegetans, and bullous pemphigoid. Asboe-Hansen5 made the argument that a fresh bulla should be biopsied if histopathologic diagnosis is necessary, as older bullae may exhibit epithelial cell regeneration and disturb an accurate diagnosis.
Accurate and early diagnosis of TEN is imperative, as prognosis is strongly correlated with the speed at which the offending drug is discontinued and appropriate medical treatment is initiated. Prompt withdrawal of the offending drug has been reported to reduce the risk for morbidity by 30% per day.7 Although classically associated with the pemphigus group of diseases, the Asboe-Hansen sign is of diagnostic value to the pathologist in diagnosing TEN by reproducing the same microscopic appearance of a fresh spontaneous blister. Due to the notable morbidity and mortality in SJS and TEN, the Asboe-Hansen sign should be attempted for the site of a lesional biopsy, as an accurate diagnosis relies on clinicopathologic correlation.
- Schwartz RA, McDonough PH, Lee BW, et al. Toxic epidermal necrolysis: part I. introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69:173.e1-173.e13.
- Frech LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:332-347.
- Schwartz RA, McDonough PH, Lee BW, et al. Toxic epidermal necrolysis: part II. prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69:187.e1–187.e16.
- Elston D, Stratman E, Miller S. Skin biopsy. J Am Acad Dermatol. 2016;74:1-16.
- Asboe-Hansen G. Blister-spread induced by finger-pressure, a diagnostic sign in pemphigus. J Invest Dermatol. 1960;34:5-9.
- Ganapati S. Eponymous dermatological signs in bullous dermatoses. Indian J Dermatol. 2014;59:21-23.
- Garcia-Doval I, Lecleach L, Bocquet H, et al. Toxic epidermal necrolysis and Stevens-Johnson syndrome: does early withdrawal of causative drugs decrease the risk of death? Arch Dermatol. 2000;136:323-327.
- Schwartz RA, McDonough PH, Lee BW, et al. Toxic epidermal necrolysis: part I. introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol. 2013;69:173.e1-173.e13.
- Frech LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:332-347.
- Schwartz RA, McDonough PH, Lee BW, et al. Toxic epidermal necrolysis: part II. prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69:187.e1–187.e16.
- Elston D, Stratman E, Miller S. Skin biopsy. J Am Acad Dermatol. 2016;74:1-16.
- Asboe-Hansen G. Blister-spread induced by finger-pressure, a diagnostic sign in pemphigus. J Invest Dermatol. 1960;34:5-9.
- Ganapati S. Eponymous dermatological signs in bullous dermatoses. Indian J Dermatol. 2014;59:21-23.
- Garcia-Doval I, Lecleach L, Bocquet H, et al. Toxic epidermal necrolysis and Stevens-Johnson syndrome: does early withdrawal of causative drugs decrease the risk of death? Arch Dermatol. 2000;136:323-327.
Practice Points
- Asboe-Hansen sign is a useful clinical tool for diagnosing toxic epidermal necrolysis (TEN).
- Asboe-Hansen sign can be employed to generate a fresh bulla for lesional skin biopsy in the evaluation of TEN.
Bendamustine/rituximab combo proves viable for comorbid CLL
A combination of bendamustine and rituximab generated an 88% overall response rate and 96% overall survival rate at 2 years among patients with chronic lymphocytic leukemia (CLL) in a study of 83 patients aged 53-83 years.
Although combined fludarabine, cyclophosphamide, and rituximab has demonstrated success in younger patients with CLL, this therapy is often considered too aggressive for the majority of CLL patients, who tend to be older and have multiple comorbidities, wrote Martin Špacek, MD, of Charles University and General University Hospital in Prague and his colleagues.
The alternative treatment combination of bendamustine and rituximab (BR) has not been well studied in patients with comorbidities, they said.
In a study published in Leukemia Research, the researchers enrolled 83 previously untreated adults with progressive CLL. The average age of the participants was 71 years, and 61% were men. The median creatinine clearance for the study population was 65 mL/min, and all patients had comorbidities, defined as scores greater than 6 on the Cumulative Illness Rating Scale (CIRS).
All patients were prescribed 90 mg/m2 bendamustine on days 1 and 2 combined with 375 mg/m2 rituximab on day 0 of the first course, and 500 mg/m2 rituximab on day 1 during subsequent courses every 28 days for a maximum of six cycles.
The overall response rate to BR was 88.0%, with a complete response rate of 20.5%. At 2 years, progression-free survival and overall survival rates were 69.9% and 96.2%, respectively.
A total of 51 patients (61.4%) experienced at least one grade 3 or 4 adverse event. The most common hematologic effects were neutropenia (40 patients), thrombocytopenia (14 patients), and anemia (8 patients). The most common nonhematologic effects were grade 3– or grade 4–level infections in 12 patients. Six patients developed severe skin rash.
Additionally, one patient developed sepsis during treatment and died after the first course of therapy.
“Age and CIRS failed to predict any severe toxicities or BR dose reduction,” the researchers noted.
The findings support data from previous studies and represent the largest study of CLL patients with significant comorbidities to be treated with BR, the researchers said.
More prospective research is needed, but the results demonstrate that “chemoimmunotherapy with BR is an effective therapeutic option with manageable toxicity for the initial treatment of CLL patients with significant comorbidities,” the investigators wrote.
The study was supported by the Ministry of Health, Czech Republic, the Charles University Progres program, and the Czech CLL Study Group. Researchers reported honoraria and travel grants from Mundipharma and Roche.
SOURCE: Spacek M et al. Leuk Res. 2019;79:17-21.
A combination of bendamustine and rituximab generated an 88% overall response rate and 96% overall survival rate at 2 years among patients with chronic lymphocytic leukemia (CLL) in a study of 83 patients aged 53-83 years.
Although combined fludarabine, cyclophosphamide, and rituximab has demonstrated success in younger patients with CLL, this therapy is often considered too aggressive for the majority of CLL patients, who tend to be older and have multiple comorbidities, wrote Martin Špacek, MD, of Charles University and General University Hospital in Prague and his colleagues.
The alternative treatment combination of bendamustine and rituximab (BR) has not been well studied in patients with comorbidities, they said.
In a study published in Leukemia Research, the researchers enrolled 83 previously untreated adults with progressive CLL. The average age of the participants was 71 years, and 61% were men. The median creatinine clearance for the study population was 65 mL/min, and all patients had comorbidities, defined as scores greater than 6 on the Cumulative Illness Rating Scale (CIRS).
All patients were prescribed 90 mg/m2 bendamustine on days 1 and 2 combined with 375 mg/m2 rituximab on day 0 of the first course, and 500 mg/m2 rituximab on day 1 during subsequent courses every 28 days for a maximum of six cycles.
The overall response rate to BR was 88.0%, with a complete response rate of 20.5%. At 2 years, progression-free survival and overall survival rates were 69.9% and 96.2%, respectively.
A total of 51 patients (61.4%) experienced at least one grade 3 or 4 adverse event. The most common hematologic effects were neutropenia (40 patients), thrombocytopenia (14 patients), and anemia (8 patients). The most common nonhematologic effects were grade 3– or grade 4–level infections in 12 patients. Six patients developed severe skin rash.
Additionally, one patient developed sepsis during treatment and died after the first course of therapy.
“Age and CIRS failed to predict any severe toxicities or BR dose reduction,” the researchers noted.
The findings support data from previous studies and represent the largest study of CLL patients with significant comorbidities to be treated with BR, the researchers said.
More prospective research is needed, but the results demonstrate that “chemoimmunotherapy with BR is an effective therapeutic option with manageable toxicity for the initial treatment of CLL patients with significant comorbidities,” the investigators wrote.
The study was supported by the Ministry of Health, Czech Republic, the Charles University Progres program, and the Czech CLL Study Group. Researchers reported honoraria and travel grants from Mundipharma and Roche.
SOURCE: Spacek M et al. Leuk Res. 2019;79:17-21.
A combination of bendamustine and rituximab generated an 88% overall response rate and 96% overall survival rate at 2 years among patients with chronic lymphocytic leukemia (CLL) in a study of 83 patients aged 53-83 years.
Although combined fludarabine, cyclophosphamide, and rituximab has demonstrated success in younger patients with CLL, this therapy is often considered too aggressive for the majority of CLL patients, who tend to be older and have multiple comorbidities, wrote Martin Špacek, MD, of Charles University and General University Hospital in Prague and his colleagues.
The alternative treatment combination of bendamustine and rituximab (BR) has not been well studied in patients with comorbidities, they said.
In a study published in Leukemia Research, the researchers enrolled 83 previously untreated adults with progressive CLL. The average age of the participants was 71 years, and 61% were men. The median creatinine clearance for the study population was 65 mL/min, and all patients had comorbidities, defined as scores greater than 6 on the Cumulative Illness Rating Scale (CIRS).
All patients were prescribed 90 mg/m2 bendamustine on days 1 and 2 combined with 375 mg/m2 rituximab on day 0 of the first course, and 500 mg/m2 rituximab on day 1 during subsequent courses every 28 days for a maximum of six cycles.
The overall response rate to BR was 88.0%, with a complete response rate of 20.5%. At 2 years, progression-free survival and overall survival rates were 69.9% and 96.2%, respectively.
A total of 51 patients (61.4%) experienced at least one grade 3 or 4 adverse event. The most common hematologic effects were neutropenia (40 patients), thrombocytopenia (14 patients), and anemia (8 patients). The most common nonhematologic effects were grade 3– or grade 4–level infections in 12 patients. Six patients developed severe skin rash.
Additionally, one patient developed sepsis during treatment and died after the first course of therapy.
“Age and CIRS failed to predict any severe toxicities or BR dose reduction,” the researchers noted.
The findings support data from previous studies and represent the largest study of CLL patients with significant comorbidities to be treated with BR, the researchers said.
More prospective research is needed, but the results demonstrate that “chemoimmunotherapy with BR is an effective therapeutic option with manageable toxicity for the initial treatment of CLL patients with significant comorbidities,” the investigators wrote.
The study was supported by the Ministry of Health, Czech Republic, the Charles University Progres program, and the Czech CLL Study Group. Researchers reported honoraria and travel grants from Mundipharma and Roche.
SOURCE: Spacek M et al. Leuk Res. 2019;79:17-21.
FROM LEUKEMIA RESEARCH
Key clinical point:
Major finding: The overall response rate for the combination therapy was 88.0%; complete response was 20.5%.
Study details: A prospective, observational study of 83 patients with chronic lymphocytic leukemia.
Disclosures: The study was supported by the Ministry of Health, Czech Republic, the Charles University Progres program, and the Czech CLL Study Group. Researchers reported honoraria and travel grants from Mundipharma and Roche.
Source: Spacek M et al. Leuk Res. 2019;79:17-21.
Don’t delay palliative care for IPF patients
and indicates that early, integrated palliative care should be a priority, according to the finding of a survey study.
“Patients with IPF suffer from exceptionally low [health-related quality of life] together with severe breathlessness and fatigue already two years before death. In addition, physical and emotional well-being further deteriorates near death concurrently with escalating overall symptom burden,” wrote Kaisa Rajala, MD, and her colleagues at Helsinki University Hospital.
They conducted a substudy of patients in the larger FinnishIPF study to assess health-related quality of life (HRQOL) and symptom burden in the period before death. Among 300 patients invited to participate, 247 agreed. Patient disease and sociodemographic data were collected from the FinnishIPF records and the study group completed questionnaires five times at 6 month intervals. The study began in April 2015 and continued until August 2017, by which time 92 (37%) of the patients had died (BMC Pulmonary Medicine 2018;18:172; doi: 0.1186/s12890-018-0738-x).
The investigators used self-reporting tools to look at HRQOL and symptom burden: RAND 36-item Health Survey (RAND-36), the Modified Medical Research and Council Dyspnea Scale (MMRC), the Modified Edmonton Symptom Assessment Scale (ESAS), and the Numeric Rating Scale (NRS).
About 35% of these patients were being treated with antifibrotic medication. Most of the patients had comorbidities, with cardiovascular disease being the most common.
The dimensions of HRQOL studied were physical function, general health, vitality, mental health, social function, and bodily pain. These patients experienced a gradual impairment in HRQOL similar to that of patients with chronic obstructive pulmonary disease, but with a pronounced, rapid deterioration beginning in the last 2 years of life.
The symptom burden also intensified in the last 2 years of life and ramped up significantly in the last 6 months before death. NRS scores are on a scale of 0-10, from no symptoms to worst symptoms. In most clinical situations, NRS scores equal to greater than 4 trigger more comprehensive symptom assessment. The scores for symptoms for these patients during the last 6 months were dyspnea, 7.1 (standard deviation 2.8); tiredness, 6.0 (SD 2.5), cough, 5.0 (SD 3.5), pain with movement, 3.9 (SD 3.1), insomnia, 3.9 (SD 2.9), anxiety, 3.9 (SD 2.9), and depression, 3.6 (SD 3.1).
Investigators noted the steep change in the proportion of patients with MMRC scores greater than or equal to 3 (needing to stop walking after approximately 100 m or a few minutes because of breathlessness) beginning in the last 2 years of life.
The study limitations are its relatively small size, the self-reported data, and the lack of lung function measurements in most patients in the last 6 months of life.
The findings point to the urgent need for early palliative care in IPF patients, the investigators concluded. They noted that the sharp decline in HRQOL is similar to that seen in lung cancer patients, in contrast to the more gradual trend seen in COPD patients.
But there are common benefits of an early palliative program for all of these patients, they stressed. “Early integrated palliative care for patients with lung cancer has shown substantial benefits, such as lower depression scores, higher HRQOL, better communication of end-of-life care preferences, less aggressive care at the end of life, and longer overall survival. Similarly, a randomized trial demonstrated better control of dyspnea and a survival benefit with integrated palliative care in patients with COPD and interstitial lung disease. In addition to cancer patients, early integrated palliative care may reduce end-of-life acute care utilization, and allow patients with IPF to die in their preferred locations. Integrated palliative care in IPF patients seems to lower respiratory-related emergency room visits and hospitalizations and may allow more patients to die at home.”
The study was funded by The Academy of Finland and various Finnish nonprofit organizations funded the study.
SOURCE: Rajala K et al. BMC Pulm Med. 2018;18:172. doi: 0.1186/s12890-018-0738-x.
and indicates that early, integrated palliative care should be a priority, according to the finding of a survey study.
“Patients with IPF suffer from exceptionally low [health-related quality of life] together with severe breathlessness and fatigue already two years before death. In addition, physical and emotional well-being further deteriorates near death concurrently with escalating overall symptom burden,” wrote Kaisa Rajala, MD, and her colleagues at Helsinki University Hospital.
They conducted a substudy of patients in the larger FinnishIPF study to assess health-related quality of life (HRQOL) and symptom burden in the period before death. Among 300 patients invited to participate, 247 agreed. Patient disease and sociodemographic data were collected from the FinnishIPF records and the study group completed questionnaires five times at 6 month intervals. The study began in April 2015 and continued until August 2017, by which time 92 (37%) of the patients had died (BMC Pulmonary Medicine 2018;18:172; doi: 0.1186/s12890-018-0738-x).
The investigators used self-reporting tools to look at HRQOL and symptom burden: RAND 36-item Health Survey (RAND-36), the Modified Medical Research and Council Dyspnea Scale (MMRC), the Modified Edmonton Symptom Assessment Scale (ESAS), and the Numeric Rating Scale (NRS).
About 35% of these patients were being treated with antifibrotic medication. Most of the patients had comorbidities, with cardiovascular disease being the most common.
The dimensions of HRQOL studied were physical function, general health, vitality, mental health, social function, and bodily pain. These patients experienced a gradual impairment in HRQOL similar to that of patients with chronic obstructive pulmonary disease, but with a pronounced, rapid deterioration beginning in the last 2 years of life.
The symptom burden also intensified in the last 2 years of life and ramped up significantly in the last 6 months before death. NRS scores are on a scale of 0-10, from no symptoms to worst symptoms. In most clinical situations, NRS scores equal to greater than 4 trigger more comprehensive symptom assessment. The scores for symptoms for these patients during the last 6 months were dyspnea, 7.1 (standard deviation 2.8); tiredness, 6.0 (SD 2.5), cough, 5.0 (SD 3.5), pain with movement, 3.9 (SD 3.1), insomnia, 3.9 (SD 2.9), anxiety, 3.9 (SD 2.9), and depression, 3.6 (SD 3.1).
Investigators noted the steep change in the proportion of patients with MMRC scores greater than or equal to 3 (needing to stop walking after approximately 100 m or a few minutes because of breathlessness) beginning in the last 2 years of life.
The study limitations are its relatively small size, the self-reported data, and the lack of lung function measurements in most patients in the last 6 months of life.
The findings point to the urgent need for early palliative care in IPF patients, the investigators concluded. They noted that the sharp decline in HRQOL is similar to that seen in lung cancer patients, in contrast to the more gradual trend seen in COPD patients.
But there are common benefits of an early palliative program for all of these patients, they stressed. “Early integrated palliative care for patients with lung cancer has shown substantial benefits, such as lower depression scores, higher HRQOL, better communication of end-of-life care preferences, less aggressive care at the end of life, and longer overall survival. Similarly, a randomized trial demonstrated better control of dyspnea and a survival benefit with integrated palliative care in patients with COPD and interstitial lung disease. In addition to cancer patients, early integrated palliative care may reduce end-of-life acute care utilization, and allow patients with IPF to die in their preferred locations. Integrated palliative care in IPF patients seems to lower respiratory-related emergency room visits and hospitalizations and may allow more patients to die at home.”
The study was funded by The Academy of Finland and various Finnish nonprofit organizations funded the study.
SOURCE: Rajala K et al. BMC Pulm Med. 2018;18:172. doi: 0.1186/s12890-018-0738-x.
and indicates that early, integrated palliative care should be a priority, according to the finding of a survey study.
“Patients with IPF suffer from exceptionally low [health-related quality of life] together with severe breathlessness and fatigue already two years before death. In addition, physical and emotional well-being further deteriorates near death concurrently with escalating overall symptom burden,” wrote Kaisa Rajala, MD, and her colleagues at Helsinki University Hospital.
They conducted a substudy of patients in the larger FinnishIPF study to assess health-related quality of life (HRQOL) and symptom burden in the period before death. Among 300 patients invited to participate, 247 agreed. Patient disease and sociodemographic data were collected from the FinnishIPF records and the study group completed questionnaires five times at 6 month intervals. The study began in April 2015 and continued until August 2017, by which time 92 (37%) of the patients had died (BMC Pulmonary Medicine 2018;18:172; doi: 0.1186/s12890-018-0738-x).
The investigators used self-reporting tools to look at HRQOL and symptom burden: RAND 36-item Health Survey (RAND-36), the Modified Medical Research and Council Dyspnea Scale (MMRC), the Modified Edmonton Symptom Assessment Scale (ESAS), and the Numeric Rating Scale (NRS).
About 35% of these patients were being treated with antifibrotic medication. Most of the patients had comorbidities, with cardiovascular disease being the most common.
The dimensions of HRQOL studied were physical function, general health, vitality, mental health, social function, and bodily pain. These patients experienced a gradual impairment in HRQOL similar to that of patients with chronic obstructive pulmonary disease, but with a pronounced, rapid deterioration beginning in the last 2 years of life.
The symptom burden also intensified in the last 2 years of life and ramped up significantly in the last 6 months before death. NRS scores are on a scale of 0-10, from no symptoms to worst symptoms. In most clinical situations, NRS scores equal to greater than 4 trigger more comprehensive symptom assessment. The scores for symptoms for these patients during the last 6 months were dyspnea, 7.1 (standard deviation 2.8); tiredness, 6.0 (SD 2.5), cough, 5.0 (SD 3.5), pain with movement, 3.9 (SD 3.1), insomnia, 3.9 (SD 2.9), anxiety, 3.9 (SD 2.9), and depression, 3.6 (SD 3.1).
Investigators noted the steep change in the proportion of patients with MMRC scores greater than or equal to 3 (needing to stop walking after approximately 100 m or a few minutes because of breathlessness) beginning in the last 2 years of life.
The study limitations are its relatively small size, the self-reported data, and the lack of lung function measurements in most patients in the last 6 months of life.
The findings point to the urgent need for early palliative care in IPF patients, the investigators concluded. They noted that the sharp decline in HRQOL is similar to that seen in lung cancer patients, in contrast to the more gradual trend seen in COPD patients.
But there are common benefits of an early palliative program for all of these patients, they stressed. “Early integrated palliative care for patients with lung cancer has shown substantial benefits, such as lower depression scores, higher HRQOL, better communication of end-of-life care preferences, less aggressive care at the end of life, and longer overall survival. Similarly, a randomized trial demonstrated better control of dyspnea and a survival benefit with integrated palliative care in patients with COPD and interstitial lung disease. In addition to cancer patients, early integrated palliative care may reduce end-of-life acute care utilization, and allow patients with IPF to die in their preferred locations. Integrated palliative care in IPF patients seems to lower respiratory-related emergency room visits and hospitalizations and may allow more patients to die at home.”
The study was funded by The Academy of Finland and various Finnish nonprofit organizations funded the study.
SOURCE: Rajala K et al. BMC Pulm Med. 2018;18:172. doi: 0.1186/s12890-018-0738-x.
FROM BMC PULMONARY MEDICINE
Polyglutamine diseases are rare, but not the mutations that cause them
Polyglutamine diseases are a group of hereditary neurodegenerative disorders caused by mutations in which a trinucleotide repeat expands pathologically on a disease-associated gene. The diseases are rare, with the most common among them – Huntington disease – affecting between 10 and 14 per 100,000 people in Western countries, where prevalence is highest.
In polyglutamine diseases, which include the spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy, and spinal bulbar muscular atrophy, higher CAG (cytosine-adenine-guanine) repeat numbers are associated with greater disease severity, faster progression, or earlier age at onset.
In research published April 1 in JAMA Neurology, investigators report that more than one-tenth of the European population carries CAG expansions that fall short of the repeats needed to cause any of 9 polyglutamine diseases – but are enough to put them at risk of having children who develop one. A smaller number of people – about 1% – carry enough CAG repeats to cause one of the diseases late in life.
For their research, Sarah L. Gardiner, MD, of Leiden (the Netherlands) University, and her colleagues looked at polyglutamine expansion variants for nine diseases in samples from 14,196 adults (56% of whom were women) from the Netherlands, Scotland, and Ireland. The samples were taken from five population-based cohort studies conducted between 1997 and 2012, and all subjects were without a history of polyglutamine disease or major depression.
Of these, 10.7% had a CAG repeat number on a disease-associated gene that was in the intermediate range, defined as a number of repeats that cannot cause disease but for which “expansion into the fully pathological range has been observed on intergenerational transmission,” Dr. Gardiner and her colleagues wrote. And some 1.3% of subjects were found to have CAG repeats within the disease-causing range, “mostly in the lower range associated with elderly onset.”
The investigators found no differences in sex, age, or body mass index between individuals with CAG repeat numbers within the pathological range and individuals with CAG repeat numbers within the normal or intermediate range.
Whether carriers of immediate or lower-range pathological CAG repeats went on to develop disease could not be measured, as follow-up data were not available. Another limitation of the study, the investigators acknowledged, was that the genotyping method used “did not allow us to determine the presence of trinucleotide interruptions,” which can affect disease penetrance.
“A late age at onset, a reduced penetrance, or the presence of interruptions could all explain the asymptomatic status of our carriers of intermediate and pathological polyglutamine disease–associated alleles at the time of assessment,” Dr. Gardiner and her colleagues wrote.
This study was funded by the European Union and Dutch government agencies; one of the population-based cohort studies from which the study sample was taken received some support from Bristol-Myers-Squibb. One of Dr. Gardiner’s coauthors, Raymund A. C. Roos, MD, PhD, disclosed being an adviser for UniQure, a gene-therapy firm, and no other conflicts of interest were reported.
SOURCE: Gardiner et al. JAMA Neurol. 2019 Apr 1. doi: 10.001/jamaneurol.2019.042.
Gardiner et al. describe the results of an appraisal of polyglutamine expansion variants in more than 14,000 individuals from the Netherlands, Scotland, and Ireland. Given the relative rarity of polyglutamine repeat disease, the first question that comes to mind is why were so many individuals identified with repeats in the pathogenic range? Based on our understanding of disease prevalence, it is unlikely that each of these individuals will become affected; therefore, this work suggests a reduced penetrance of these mutations. The findings are illustrative of a growing theme in human disease genetics: There are a very large number of apparently healthy individuals in the general population who carry mutations associated with various diseases. The phenomenon of reduced penetrance, where mutations cause disease in some but not all carriers, overlaps and arguably may be the same as that of variable expressivity, where the same mutation can lead to very different disease outcomes in different individuals. It is extremely likely that second-generation sequencing and population-scale screening will continue to reveal similar themes. We continue to appreciate the increasing complexity of the human genome and its relationship to disease, even those diseases we thought of previously as simple “single-gene” disorders.
Monia B. Hammer, PhD, and Andrew B. Singleton, PhD, are with the National Institute on Aging, National Institutes of Health, Bethesda, Md. Dr. Hammer and Dr. Singleton report no financial conflicts of interest related to their editorial.
Gardiner et al. describe the results of an appraisal of polyglutamine expansion variants in more than 14,000 individuals from the Netherlands, Scotland, and Ireland. Given the relative rarity of polyglutamine repeat disease, the first question that comes to mind is why were so many individuals identified with repeats in the pathogenic range? Based on our understanding of disease prevalence, it is unlikely that each of these individuals will become affected; therefore, this work suggests a reduced penetrance of these mutations. The findings are illustrative of a growing theme in human disease genetics: There are a very large number of apparently healthy individuals in the general population who carry mutations associated with various diseases. The phenomenon of reduced penetrance, where mutations cause disease in some but not all carriers, overlaps and arguably may be the same as that of variable expressivity, where the same mutation can lead to very different disease outcomes in different individuals. It is extremely likely that second-generation sequencing and population-scale screening will continue to reveal similar themes. We continue to appreciate the increasing complexity of the human genome and its relationship to disease, even those diseases we thought of previously as simple “single-gene” disorders.
Monia B. Hammer, PhD, and Andrew B. Singleton, PhD, are with the National Institute on Aging, National Institutes of Health, Bethesda, Md. Dr. Hammer and Dr. Singleton report no financial conflicts of interest related to their editorial.
Gardiner et al. describe the results of an appraisal of polyglutamine expansion variants in more than 14,000 individuals from the Netherlands, Scotland, and Ireland. Given the relative rarity of polyglutamine repeat disease, the first question that comes to mind is why were so many individuals identified with repeats in the pathogenic range? Based on our understanding of disease prevalence, it is unlikely that each of these individuals will become affected; therefore, this work suggests a reduced penetrance of these mutations. The findings are illustrative of a growing theme in human disease genetics: There are a very large number of apparently healthy individuals in the general population who carry mutations associated with various diseases. The phenomenon of reduced penetrance, where mutations cause disease in some but not all carriers, overlaps and arguably may be the same as that of variable expressivity, where the same mutation can lead to very different disease outcomes in different individuals. It is extremely likely that second-generation sequencing and population-scale screening will continue to reveal similar themes. We continue to appreciate the increasing complexity of the human genome and its relationship to disease, even those diseases we thought of previously as simple “single-gene” disorders.
Monia B. Hammer, PhD, and Andrew B. Singleton, PhD, are with the National Institute on Aging, National Institutes of Health, Bethesda, Md. Dr. Hammer and Dr. Singleton report no financial conflicts of interest related to their editorial.
Polyglutamine diseases are a group of hereditary neurodegenerative disorders caused by mutations in which a trinucleotide repeat expands pathologically on a disease-associated gene. The diseases are rare, with the most common among them – Huntington disease – affecting between 10 and 14 per 100,000 people in Western countries, where prevalence is highest.
In polyglutamine diseases, which include the spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy, and spinal bulbar muscular atrophy, higher CAG (cytosine-adenine-guanine) repeat numbers are associated with greater disease severity, faster progression, or earlier age at onset.
In research published April 1 in JAMA Neurology, investigators report that more than one-tenth of the European population carries CAG expansions that fall short of the repeats needed to cause any of 9 polyglutamine diseases – but are enough to put them at risk of having children who develop one. A smaller number of people – about 1% – carry enough CAG repeats to cause one of the diseases late in life.
For their research, Sarah L. Gardiner, MD, of Leiden (the Netherlands) University, and her colleagues looked at polyglutamine expansion variants for nine diseases in samples from 14,196 adults (56% of whom were women) from the Netherlands, Scotland, and Ireland. The samples were taken from five population-based cohort studies conducted between 1997 and 2012, and all subjects were without a history of polyglutamine disease or major depression.
Of these, 10.7% had a CAG repeat number on a disease-associated gene that was in the intermediate range, defined as a number of repeats that cannot cause disease but for which “expansion into the fully pathological range has been observed on intergenerational transmission,” Dr. Gardiner and her colleagues wrote. And some 1.3% of subjects were found to have CAG repeats within the disease-causing range, “mostly in the lower range associated with elderly onset.”
The investigators found no differences in sex, age, or body mass index between individuals with CAG repeat numbers within the pathological range and individuals with CAG repeat numbers within the normal or intermediate range.
Whether carriers of immediate or lower-range pathological CAG repeats went on to develop disease could not be measured, as follow-up data were not available. Another limitation of the study, the investigators acknowledged, was that the genotyping method used “did not allow us to determine the presence of trinucleotide interruptions,” which can affect disease penetrance.
“A late age at onset, a reduced penetrance, or the presence of interruptions could all explain the asymptomatic status of our carriers of intermediate and pathological polyglutamine disease–associated alleles at the time of assessment,” Dr. Gardiner and her colleagues wrote.
This study was funded by the European Union and Dutch government agencies; one of the population-based cohort studies from which the study sample was taken received some support from Bristol-Myers-Squibb. One of Dr. Gardiner’s coauthors, Raymund A. C. Roos, MD, PhD, disclosed being an adviser for UniQure, a gene-therapy firm, and no other conflicts of interest were reported.
SOURCE: Gardiner et al. JAMA Neurol. 2019 Apr 1. doi: 10.001/jamaneurol.2019.042.
Polyglutamine diseases are a group of hereditary neurodegenerative disorders caused by mutations in which a trinucleotide repeat expands pathologically on a disease-associated gene. The diseases are rare, with the most common among them – Huntington disease – affecting between 10 and 14 per 100,000 people in Western countries, where prevalence is highest.
In polyglutamine diseases, which include the spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy, and spinal bulbar muscular atrophy, higher CAG (cytosine-adenine-guanine) repeat numbers are associated with greater disease severity, faster progression, or earlier age at onset.
In research published April 1 in JAMA Neurology, investigators report that more than one-tenth of the European population carries CAG expansions that fall short of the repeats needed to cause any of 9 polyglutamine diseases – but are enough to put them at risk of having children who develop one. A smaller number of people – about 1% – carry enough CAG repeats to cause one of the diseases late in life.
For their research, Sarah L. Gardiner, MD, of Leiden (the Netherlands) University, and her colleagues looked at polyglutamine expansion variants for nine diseases in samples from 14,196 adults (56% of whom were women) from the Netherlands, Scotland, and Ireland. The samples were taken from five population-based cohort studies conducted between 1997 and 2012, and all subjects were without a history of polyglutamine disease or major depression.
Of these, 10.7% had a CAG repeat number on a disease-associated gene that was in the intermediate range, defined as a number of repeats that cannot cause disease but for which “expansion into the fully pathological range has been observed on intergenerational transmission,” Dr. Gardiner and her colleagues wrote. And some 1.3% of subjects were found to have CAG repeats within the disease-causing range, “mostly in the lower range associated with elderly onset.”
The investigators found no differences in sex, age, or body mass index between individuals with CAG repeat numbers within the pathological range and individuals with CAG repeat numbers within the normal or intermediate range.
Whether carriers of immediate or lower-range pathological CAG repeats went on to develop disease could not be measured, as follow-up data were not available. Another limitation of the study, the investigators acknowledged, was that the genotyping method used “did not allow us to determine the presence of trinucleotide interruptions,” which can affect disease penetrance.
“A late age at onset, a reduced penetrance, or the presence of interruptions could all explain the asymptomatic status of our carriers of intermediate and pathological polyglutamine disease–associated alleles at the time of assessment,” Dr. Gardiner and her colleagues wrote.
This study was funded by the European Union and Dutch government agencies; one of the population-based cohort studies from which the study sample was taken received some support from Bristol-Myers-Squibb. One of Dr. Gardiner’s coauthors, Raymund A. C. Roos, MD, PhD, disclosed being an adviser for UniQure, a gene-therapy firm, and no other conflicts of interest were reported.
SOURCE: Gardiner et al. JAMA Neurol. 2019 Apr 1. doi: 10.001/jamaneurol.2019.042.
FROM JAMA NEUROLOGY
Scurvy Masquerading as Reactive Arthritis
To the Editor:
A 28-year-old recently homeless white man with a history of heroin abuse was admitted with a worsening rash and left ankle pain of 1 week’s duration, as well as subjective fever after 3 weeks of a productive cough, sore throat, hoarse voice, and general malaise. Six days prior to presentation, he developed redness and swelling of the dorsal aspects of both hands with accompanying rash, and 2 days prior to presentation he developed a similar rash on the legs with associated left ankle pain, redness, and swelling. He also reported eye redness, pain, photophobia, crusty eye discharge, and a pins and needles sensation on the soles of both feet. Additionally, he had noted difficulty with urination over several days. He had been homeless for less than 1 month prior to admission.
On physical examination, the patient appeared to be well nourished. Skin examination was notable for scattered perifollicular hemorrhagic and hyperkeratotic papules ranging in size from 3 to 6 mm with associated nummular alopecia of the bilateral medial thighs (Figure); well-demarcated desquamated patches on the weight-bearing aspects of the plantar feet; and a 2.0-cm, well-demarcated, thinly raised erythematous patch of the inferolateral penile shaft. Oral examination was notable for multiple discrete areas of ulceration on the lateral aspects of the tongue. Ophthalmic examination revealed conjunctival injection and photophobia. The ankles were edematous and tender (the left ankle more than the right), and range of passive motion was limited by pain.

Laboratory values were remarkable for a hemoglobin count of 13.1 g/dL (reference range, 14.2–18 g/dL), erythrocyte sedimentation rate of 31 mm/h (reference range, 0–10 mm/h), and C-reactive protein level of 5.4 mg/dL (reference range, 0–0.8 mg/dL). Urinalysis was unremarkable, blood cultures were negative, and a chest radiograph was normal. Human immunodeficiency virus and rapid plasma reagin tests were negative, with normal levels of IgG, IgA, and IgM. IgE was elevated at 572 IU/mL (reference range, 0–100 IU/mL). Ultrasonography of the leg was negative for deep vein thrombosis, and a left ankle radiograph was negative for fracture. The patient previously was found to have antinuclear antibodies of 1:40 and negative antineutrophil cytoplasmic antibodies, anti–double-stranded DNA, anti–Sjögren syndrome antigens A and B, and cryoglobulins, as well as normal complement levels. The constellation of rash, arthritis, conjunctivitis, and difficulty with urination raised a high suspicion for reactive arthritis; however, the patient was found to be HLA-B27 negative with a negative urine chlamydia test.
The patient was mildly hypokalemic at 2.9 mmol/L (reference range, 3.5–5.0 mmol/L) and hypoalbuminemic at 3.6 g/dL (reference range, 3.9–5.0 g/dL). He had a slightly elevated international normalized ratio of 1.4 (reference range, 0.9–1.2). Further questioning revealed that his diet consisted mostly of soda and energy drinks; his vitamin C level was subsequently checked and found to be 0 mg/dL (reference range, 0.2–2.0 mg/dL). A diagnosis of scurvy was made, and his symptoms improved at the hospital while maintaining a diet with normal levels of vitamin C. His rash had markedly improved by hospital day 2, joint swelling decreased, and the conjunctival injection and eye pain had resolved. Upon outpatient follow-up, his rash and joint swelling continued to improve, and he had not experienced any further areas of hair loss.
Scurvy, a condition caused by vitamin C deficiency, is a disease of historical importance, as it ravaged ships full of sailors in days past; however, its incidence has decreased drastically since Lind1 first described its treatment using citrus fruits in 1753. Nonetheless, even with modern day access to foods rich in vitamin C, scurvy is far more common than expected in the developed world.
Vitamin C (ascorbic acid) plays a crucial role in human biochemistry. Although many plants and animals can synthesize ascorbic acid, humans and other animals such as guinea pigs lack the required enzyme, making vitamin C an essential nutrient required in dietary intake.2-4 Hypovitaminosis C leads to scurvy when collagen production becomes impaired due to lack of ascorbic acid as a required cofactor for its synthesis, which leads to tissue and capillary fragility, causing hemorrhage and perivascular edema.4 The diagnosis of scurvy is clinical and typically is based on signs such as perivascular hemorrhage, bleeding gums, anemia, impaired wound healing, and ecchymoses in the setting of vitamin C deficiency (<11 μmol/L or <0.2 mg/dL) with rapid resolution upon vitamin C supplementation.5
Important sources of vitamin C include citrus fruits, strawberries, broccoli, spinach, and potatoes. Recommended daily intake is 75 to 90 mg, with smokers requiring 110 to 125 mg daily because of increased oxidative stress.6-9 Although access to these foods in the modern United States is high, as many as 10% of males and 6.9% of females are vitamin C deficient, and in the subset of generally healthy middle-class Americans, as many as 6% are deficient.8,10 The highest risk groups tend to be smokers and individuals with low incomes.8 Although vitamin C deficiency does not automatically equate to scurvy, early studies on experimentally induced scurvy in prisoners showed that signs of scurvy may begin to develop in as few as 29 days of complete vitamin C deprivation, with overt scurvy developing after approximately 40 to 90 days.11,12
Patients with scurvy often pose a diagnostic dilemma for physicians because their presenting symptoms, such as fatigue, anemia, and rash, are nonspecific and can lead physicians down a laborious and costly road of unnecessary tests including vasculitic, infectious, and rheumatologic workups to determine the cause of the symptoms. Increased awareness of the current prevalence of hypovitaminosis C may help to decrease these unnecessary costs by putting scurvy higher on the differential for patients with this spectrum of symptoms.
Scurvy has been called the eternal masquerader because its nonspecific signs and symptoms have often led to misdiagnosis.13 Cases of scurvy mimicking diseases ranging from bone tumors14 to spondyloarthritis15 and vasculitis16 have been reported. The typical patient at risk for scurvy tends to fall in one of the following categories: psychiatric illness, gastrointestinal disorders, malnourishment, chronic alcoholism, drug use, elderly age, infants, restrictive dietary habits or food allergies, or those in developing countries.17-20 Our patient did not fit particularly well into any of the aforementioned high-risk categories; he had only recently become homeless and had a history of intravenous drug use but had not been using drugs in the months prior to the development of scurvy. Additionally, his salient symptoms were more consistent with reactive arthritis than with classic scurvy.
Although he had many symptoms consistent with scurvy such as generalized malaise, perifollicular hemorrhage and hyperkeratosis, spongy edema of the joints, and mild anemia on laboratory testing, he was missing several classic scurvy symptoms. Unlike many patients with scurvy, our patient did not describe any history of bruising easily or dental concerns, and examination was notably absent of ecchymoses as well as spongy or bleeding gums. He did, however, present with eye irritation and photophobia. These symptoms, consistent with keratoconjunctivitis sicca, are lesser known because ocular findings are rarely found in scurvy.21 Patients with scurvy can report eye burning and irritation, redness, blurry vision, and sensitivity to bright light secondary to increased dryness of the corneal surfaces. Horrobin et al22 postulated that this symptom may be mediated by regulation of prostaglandin E1 by vitamin C.
Another less common sign of scurvy found in our patient was patchy alopecia. Alopecia most often is seen in association with concomitant Sjögren syndrome.11,23 The etiology of the hair loss stems from the role of ascorbic acid in disulfide bonding during hair formation. The hair may fracture, coil into a corkscrew hair, or bend in several places, leading to a swan-neck deformity. Although a skin biopsy was not performed in our patient, results typically demonstrate a coiled hair in its follicle.24,25
We present the case of an otherwise generally healthy patient who developed vitamin C deficiency due to a diet consisting mostly of soda and energy drinks. His case presented a diagnostic dilemma, as his symptoms at first seemed most consistent with reactive arthritis and he was missing several of the risk factors and symptoms that would have led to an early diagnosis of scurvy. Vitamin C deficiency is not as uncommon as expected in the developed world; practitioners must be aware of the common as well as the unusual signs of scurvy.
- Lind J. A Treatise of the Scurvy. Edinburgh, Scotland: Sands, Murray, and Cochran; 1753.
- Levine M, Rumsey SC, Daruwala R, et al. Criteria and recommendations for vitamin C intake. JAMA. 1999;281:1415-1423.
- Jacob RA. Vitamin C. In: Shils ME, Olson JA, Shike M, et al, eds. Modern Nutrition in Health and Disease. Baltimore, MD: William & Wilkins; 1999:467-483.
- Levine M. New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314:892-902.
- Hirschman JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895-906.
- Bardnard ND, Weissinger R, Jaster BJ, et al, eds. Nutrition Guide for Clinicians. 2nd ed. Washington, DC: Physician’s Committee For Responsible Medicine; 2009:33.
- Institute of Medicine. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington DC: National Academic Press; 2000.
- Schleicher RL, Carroll MD, Ford ES, et al. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003-2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90:1252-1263.
- Schectman G, Byrd JC, Gruchow HW. The influence of smoking on vitamin C status in adults. Am J Public Health. 1989;79:158-162.
- Johnston CS, Thompson LL. Vitamin C status of an outpatient population. J Am Coll Nutr. 1998;17:366-370.
- Hodges RE, Baker EM, Hood J, et al. Experimental scurvy in man. Am J Clin Nutr. 1969;22:535-548.
- Hodges RE, Hood J, Canham JE, et al. Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24:432-443.
- Gupta P, Taneja K, Iyer PU, et al. Scurvy—the eternal masquerader. Ann Trop Paediatr. 1989;9:118-121.
- Haq RU, Dhammi IK, Jain AK, et al. Infantile scurvy masquerading as bone tumour. Ann Acad Med Singapore. 2013;42:363-365.
- Pazzola G, Possemato N, Germanò G, et al. Scurvy mimicking spondyloarthritis in a young man. Clin Exp Rheumatol. 2013;31:795.
- Friesgaard Christensen A, Clemmensen O, Junker P. Palpable purpura with an unexpected outcome. Case Rep Rheumatol. 2013;2013:678427.
- Des Roches A, Paradis L, Paradis J, et al. Food allergy as a new risk factor for scurvy. Allergy. 2006;61:1487-1488.
- Pimentel L. Scurvy: historical review and current diagnostic approach. Am J Emerg Med. 2003;21:328-332.
- Codreanu F, Jarlot S, Astier C, et al. An apple a day...chronic glossitis in a 4-year-old boy. Eur Ann Allergy Clin Immunol. 2012;44:86-88.
- Stephen R, Utecht T. Scurvy identified in the emergency department: a case report. J Emerg Med. 2001;21:235-237.
- Hood J, Hodges RE. Ocular lesions in scurvy. Am J Clin Nutr. 1969;22:559-567.
- Horrobin DF, Oka M, Manku MS. The regulation of prostaglandin E1 formation: a candidate for one of the fundamental mechanisms involved in the actions of vitamin C. Med Hypotheses. 1979;5:849-858.
- Hood J, Burns CA, Hodges RE. Sjogren’s syndrome in scurvy. N Engl J Med. 1970;282:1120-1124.
- Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130:177-179.
- Velandia B, Centor RM, McConnell V, et al. Scurvy is still present in developed countries. J Gen Intern Med. 2008;23:1281-1284.
To the Editor:
A 28-year-old recently homeless white man with a history of heroin abuse was admitted with a worsening rash and left ankle pain of 1 week’s duration, as well as subjective fever after 3 weeks of a productive cough, sore throat, hoarse voice, and general malaise. Six days prior to presentation, he developed redness and swelling of the dorsal aspects of both hands with accompanying rash, and 2 days prior to presentation he developed a similar rash on the legs with associated left ankle pain, redness, and swelling. He also reported eye redness, pain, photophobia, crusty eye discharge, and a pins and needles sensation on the soles of both feet. Additionally, he had noted difficulty with urination over several days. He had been homeless for less than 1 month prior to admission.
On physical examination, the patient appeared to be well nourished. Skin examination was notable for scattered perifollicular hemorrhagic and hyperkeratotic papules ranging in size from 3 to 6 mm with associated nummular alopecia of the bilateral medial thighs (Figure); well-demarcated desquamated patches on the weight-bearing aspects of the plantar feet; and a 2.0-cm, well-demarcated, thinly raised erythematous patch of the inferolateral penile shaft. Oral examination was notable for multiple discrete areas of ulceration on the lateral aspects of the tongue. Ophthalmic examination revealed conjunctival injection and photophobia. The ankles were edematous and tender (the left ankle more than the right), and range of passive motion was limited by pain.
Laboratory values were remarkable for a hemoglobin count of 13.1 g/dL (reference range, 14.2–18 g/dL), erythrocyte sedimentation rate of 31 mm/h (reference range, 0–10 mm/h), and C-reactive protein level of 5.4 mg/dL (reference range, 0–0.8 mg/dL). Urinalysis was unremarkable, blood cultures were negative, and a chest radiograph was normal. Human immunodeficiency virus and rapid plasma reagin tests were negative, with normal levels of IgG, IgA, and IgM. IgE was elevated at 572 IU/mL (reference range, 0–100 IU/mL). Ultrasonography of the leg was negative for deep vein thrombosis, and a left ankle radiograph was negative for fracture. The patient previously was found to have antinuclear antibodies of 1:40 and negative antineutrophil cytoplasmic antibodies, anti–double-stranded DNA, anti–Sjögren syndrome antigens A and B, and cryoglobulins, as well as normal complement levels. The constellation of rash, arthritis, conjunctivitis, and difficulty with urination raised a high suspicion for reactive arthritis; however, the patient was found to be HLA-B27 negative with a negative urine chlamydia test.
The patient was mildly hypokalemic at 2.9 mmol/L (reference range, 3.5–5.0 mmol/L) and hypoalbuminemic at 3.6 g/dL (reference range, 3.9–5.0 g/dL). He had a slightly elevated international normalized ratio of 1.4 (reference range, 0.9–1.2). Further questioning revealed that his diet consisted mostly of soda and energy drinks; his vitamin C level was subsequently checked and found to be 0 mg/dL (reference range, 0.2–2.0 mg/dL). A diagnosis of scurvy was made, and his symptoms improved at the hospital while maintaining a diet with normal levels of vitamin C. His rash had markedly improved by hospital day 2, joint swelling decreased, and the conjunctival injection and eye pain had resolved. Upon outpatient follow-up, his rash and joint swelling continued to improve, and he had not experienced any further areas of hair loss.
Scurvy, a condition caused by vitamin C deficiency, is a disease of historical importance, as it ravaged ships full of sailors in days past; however, its incidence has decreased drastically since Lind1 first described its treatment using citrus fruits in 1753. Nonetheless, even with modern day access to foods rich in vitamin C, scurvy is far more common than expected in the developed world.
Vitamin C (ascorbic acid) plays a crucial role in human biochemistry. Although many plants and animals can synthesize ascorbic acid, humans and other animals such as guinea pigs lack the required enzyme, making vitamin C an essential nutrient required in dietary intake.2-4 Hypovitaminosis C leads to scurvy when collagen production becomes impaired due to lack of ascorbic acid as a required cofactor for its synthesis, which leads to tissue and capillary fragility, causing hemorrhage and perivascular edema.4 The diagnosis of scurvy is clinical and typically is based on signs such as perivascular hemorrhage, bleeding gums, anemia, impaired wound healing, and ecchymoses in the setting of vitamin C deficiency (<11 μmol/L or <0.2 mg/dL) with rapid resolution upon vitamin C supplementation.5
Important sources of vitamin C include citrus fruits, strawberries, broccoli, spinach, and potatoes. Recommended daily intake is 75 to 90 mg, with smokers requiring 110 to 125 mg daily because of increased oxidative stress.6-9 Although access to these foods in the modern United States is high, as many as 10% of males and 6.9% of females are vitamin C deficient, and in the subset of generally healthy middle-class Americans, as many as 6% are deficient.8,10 The highest risk groups tend to be smokers and individuals with low incomes.8 Although vitamin C deficiency does not automatically equate to scurvy, early studies on experimentally induced scurvy in prisoners showed that signs of scurvy may begin to develop in as few as 29 days of complete vitamin C deprivation, with overt scurvy developing after approximately 40 to 90 days.11,12
Patients with scurvy often pose a diagnostic dilemma for physicians because their presenting symptoms, such as fatigue, anemia, and rash, are nonspecific and can lead physicians down a laborious and costly road of unnecessary tests including vasculitic, infectious, and rheumatologic workups to determine the cause of the symptoms. Increased awareness of the current prevalence of hypovitaminosis C may help to decrease these unnecessary costs by putting scurvy higher on the differential for patients with this spectrum of symptoms.
Scurvy has been called the eternal masquerader because its nonspecific signs and symptoms have often led to misdiagnosis.13 Cases of scurvy mimicking diseases ranging from bone tumors14 to spondyloarthritis15 and vasculitis16 have been reported. The typical patient at risk for scurvy tends to fall in one of the following categories: psychiatric illness, gastrointestinal disorders, malnourishment, chronic alcoholism, drug use, elderly age, infants, restrictive dietary habits or food allergies, or those in developing countries.17-20 Our patient did not fit particularly well into any of the aforementioned high-risk categories; he had only recently become homeless and had a history of intravenous drug use but had not been using drugs in the months prior to the development of scurvy. Additionally, his salient symptoms were more consistent with reactive arthritis than with classic scurvy.
Although he had many symptoms consistent with scurvy such as generalized malaise, perifollicular hemorrhage and hyperkeratosis, spongy edema of the joints, and mild anemia on laboratory testing, he was missing several classic scurvy symptoms. Unlike many patients with scurvy, our patient did not describe any history of bruising easily or dental concerns, and examination was notably absent of ecchymoses as well as spongy or bleeding gums. He did, however, present with eye irritation and photophobia. These symptoms, consistent with keratoconjunctivitis sicca, are lesser known because ocular findings are rarely found in scurvy.21 Patients with scurvy can report eye burning and irritation, redness, blurry vision, and sensitivity to bright light secondary to increased dryness of the corneal surfaces. Horrobin et al22 postulated that this symptom may be mediated by regulation of prostaglandin E1 by vitamin C.
Another less common sign of scurvy found in our patient was patchy alopecia. Alopecia most often is seen in association with concomitant Sjögren syndrome.11,23 The etiology of the hair loss stems from the role of ascorbic acid in disulfide bonding during hair formation. The hair may fracture, coil into a corkscrew hair, or bend in several places, leading to a swan-neck deformity. Although a skin biopsy was not performed in our patient, results typically demonstrate a coiled hair in its follicle.24,25
We present the case of an otherwise generally healthy patient who developed vitamin C deficiency due to a diet consisting mostly of soda and energy drinks. His case presented a diagnostic dilemma, as his symptoms at first seemed most consistent with reactive arthritis and he was missing several of the risk factors and symptoms that would have led to an early diagnosis of scurvy. Vitamin C deficiency is not as uncommon as expected in the developed world; practitioners must be aware of the common as well as the unusual signs of scurvy.
To the Editor:
A 28-year-old recently homeless white man with a history of heroin abuse was admitted with a worsening rash and left ankle pain of 1 week’s duration, as well as subjective fever after 3 weeks of a productive cough, sore throat, hoarse voice, and general malaise. Six days prior to presentation, he developed redness and swelling of the dorsal aspects of both hands with accompanying rash, and 2 days prior to presentation he developed a similar rash on the legs with associated left ankle pain, redness, and swelling. He also reported eye redness, pain, photophobia, crusty eye discharge, and a pins and needles sensation on the soles of both feet. Additionally, he had noted difficulty with urination over several days. He had been homeless for less than 1 month prior to admission.
On physical examination, the patient appeared to be well nourished. Skin examination was notable for scattered perifollicular hemorrhagic and hyperkeratotic papules ranging in size from 3 to 6 mm with associated nummular alopecia of the bilateral medial thighs (Figure); well-demarcated desquamated patches on the weight-bearing aspects of the plantar feet; and a 2.0-cm, well-demarcated, thinly raised erythematous patch of the inferolateral penile shaft. Oral examination was notable for multiple discrete areas of ulceration on the lateral aspects of the tongue. Ophthalmic examination revealed conjunctival injection and photophobia. The ankles were edematous and tender (the left ankle more than the right), and range of passive motion was limited by pain.
Laboratory values were remarkable for a hemoglobin count of 13.1 g/dL (reference range, 14.2–18 g/dL), erythrocyte sedimentation rate of 31 mm/h (reference range, 0–10 mm/h), and C-reactive protein level of 5.4 mg/dL (reference range, 0–0.8 mg/dL). Urinalysis was unremarkable, blood cultures were negative, and a chest radiograph was normal. Human immunodeficiency virus and rapid plasma reagin tests were negative, with normal levels of IgG, IgA, and IgM. IgE was elevated at 572 IU/mL (reference range, 0–100 IU/mL). Ultrasonography of the leg was negative for deep vein thrombosis, and a left ankle radiograph was negative for fracture. The patient previously was found to have antinuclear antibodies of 1:40 and negative antineutrophil cytoplasmic antibodies, anti–double-stranded DNA, anti–Sjögren syndrome antigens A and B, and cryoglobulins, as well as normal complement levels. The constellation of rash, arthritis, conjunctivitis, and difficulty with urination raised a high suspicion for reactive arthritis; however, the patient was found to be HLA-B27 negative with a negative urine chlamydia test.
The patient was mildly hypokalemic at 2.9 mmol/L (reference range, 3.5–5.0 mmol/L) and hypoalbuminemic at 3.6 g/dL (reference range, 3.9–5.0 g/dL). He had a slightly elevated international normalized ratio of 1.4 (reference range, 0.9–1.2). Further questioning revealed that his diet consisted mostly of soda and energy drinks; his vitamin C level was subsequently checked and found to be 0 mg/dL (reference range, 0.2–2.0 mg/dL). A diagnosis of scurvy was made, and his symptoms improved at the hospital while maintaining a diet with normal levels of vitamin C. His rash had markedly improved by hospital day 2, joint swelling decreased, and the conjunctival injection and eye pain had resolved. Upon outpatient follow-up, his rash and joint swelling continued to improve, and he had not experienced any further areas of hair loss.
Scurvy, a condition caused by vitamin C deficiency, is a disease of historical importance, as it ravaged ships full of sailors in days past; however, its incidence has decreased drastically since Lind1 first described its treatment using citrus fruits in 1753. Nonetheless, even with modern day access to foods rich in vitamin C, scurvy is far more common than expected in the developed world.
Vitamin C (ascorbic acid) plays a crucial role in human biochemistry. Although many plants and animals can synthesize ascorbic acid, humans and other animals such as guinea pigs lack the required enzyme, making vitamin C an essential nutrient required in dietary intake.2-4 Hypovitaminosis C leads to scurvy when collagen production becomes impaired due to lack of ascorbic acid as a required cofactor for its synthesis, which leads to tissue and capillary fragility, causing hemorrhage and perivascular edema.4 The diagnosis of scurvy is clinical and typically is based on signs such as perivascular hemorrhage, bleeding gums, anemia, impaired wound healing, and ecchymoses in the setting of vitamin C deficiency (<11 μmol/L or <0.2 mg/dL) with rapid resolution upon vitamin C supplementation.5
Important sources of vitamin C include citrus fruits, strawberries, broccoli, spinach, and potatoes. Recommended daily intake is 75 to 90 mg, with smokers requiring 110 to 125 mg daily because of increased oxidative stress.6-9 Although access to these foods in the modern United States is high, as many as 10% of males and 6.9% of females are vitamin C deficient, and in the subset of generally healthy middle-class Americans, as many as 6% are deficient.8,10 The highest risk groups tend to be smokers and individuals with low incomes.8 Although vitamin C deficiency does not automatically equate to scurvy, early studies on experimentally induced scurvy in prisoners showed that signs of scurvy may begin to develop in as few as 29 days of complete vitamin C deprivation, with overt scurvy developing after approximately 40 to 90 days.11,12
Patients with scurvy often pose a diagnostic dilemma for physicians because their presenting symptoms, such as fatigue, anemia, and rash, are nonspecific and can lead physicians down a laborious and costly road of unnecessary tests including vasculitic, infectious, and rheumatologic workups to determine the cause of the symptoms. Increased awareness of the current prevalence of hypovitaminosis C may help to decrease these unnecessary costs by putting scurvy higher on the differential for patients with this spectrum of symptoms.
Scurvy has been called the eternal masquerader because its nonspecific signs and symptoms have often led to misdiagnosis.13 Cases of scurvy mimicking diseases ranging from bone tumors14 to spondyloarthritis15 and vasculitis16 have been reported. The typical patient at risk for scurvy tends to fall in one of the following categories: psychiatric illness, gastrointestinal disorders, malnourishment, chronic alcoholism, drug use, elderly age, infants, restrictive dietary habits or food allergies, or those in developing countries.17-20 Our patient did not fit particularly well into any of the aforementioned high-risk categories; he had only recently become homeless and had a history of intravenous drug use but had not been using drugs in the months prior to the development of scurvy. Additionally, his salient symptoms were more consistent with reactive arthritis than with classic scurvy.
Although he had many symptoms consistent with scurvy such as generalized malaise, perifollicular hemorrhage and hyperkeratosis, spongy edema of the joints, and mild anemia on laboratory testing, he was missing several classic scurvy symptoms. Unlike many patients with scurvy, our patient did not describe any history of bruising easily or dental concerns, and examination was notably absent of ecchymoses as well as spongy or bleeding gums. He did, however, present with eye irritation and photophobia. These symptoms, consistent with keratoconjunctivitis sicca, are lesser known because ocular findings are rarely found in scurvy.21 Patients with scurvy can report eye burning and irritation, redness, blurry vision, and sensitivity to bright light secondary to increased dryness of the corneal surfaces. Horrobin et al22 postulated that this symptom may be mediated by regulation of prostaglandin E1 by vitamin C.
Another less common sign of scurvy found in our patient was patchy alopecia. Alopecia most often is seen in association with concomitant Sjögren syndrome.11,23 The etiology of the hair loss stems from the role of ascorbic acid in disulfide bonding during hair formation. The hair may fracture, coil into a corkscrew hair, or bend in several places, leading to a swan-neck deformity. Although a skin biopsy was not performed in our patient, results typically demonstrate a coiled hair in its follicle.24,25
We present the case of an otherwise generally healthy patient who developed vitamin C deficiency due to a diet consisting mostly of soda and energy drinks. His case presented a diagnostic dilemma, as his symptoms at first seemed most consistent with reactive arthritis and he was missing several of the risk factors and symptoms that would have led to an early diagnosis of scurvy. Vitamin C deficiency is not as uncommon as expected in the developed world; practitioners must be aware of the common as well as the unusual signs of scurvy.
- Lind J. A Treatise of the Scurvy. Edinburgh, Scotland: Sands, Murray, and Cochran; 1753.
- Levine M, Rumsey SC, Daruwala R, et al. Criteria and recommendations for vitamin C intake. JAMA. 1999;281:1415-1423.
- Jacob RA. Vitamin C. In: Shils ME, Olson JA, Shike M, et al, eds. Modern Nutrition in Health and Disease. Baltimore, MD: William & Wilkins; 1999:467-483.
- Levine M. New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314:892-902.
- Hirschman JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895-906.
- Bardnard ND, Weissinger R, Jaster BJ, et al, eds. Nutrition Guide for Clinicians. 2nd ed. Washington, DC: Physician’s Committee For Responsible Medicine; 2009:33.
- Institute of Medicine. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington DC: National Academic Press; 2000.
- Schleicher RL, Carroll MD, Ford ES, et al. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003-2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90:1252-1263.
- Schectman G, Byrd JC, Gruchow HW. The influence of smoking on vitamin C status in adults. Am J Public Health. 1989;79:158-162.
- Johnston CS, Thompson LL. Vitamin C status of an outpatient population. J Am Coll Nutr. 1998;17:366-370.
- Hodges RE, Baker EM, Hood J, et al. Experimental scurvy in man. Am J Clin Nutr. 1969;22:535-548.
- Hodges RE, Hood J, Canham JE, et al. Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24:432-443.
- Gupta P, Taneja K, Iyer PU, et al. Scurvy—the eternal masquerader. Ann Trop Paediatr. 1989;9:118-121.
- Haq RU, Dhammi IK, Jain AK, et al. Infantile scurvy masquerading as bone tumour. Ann Acad Med Singapore. 2013;42:363-365.
- Pazzola G, Possemato N, Germanò G, et al. Scurvy mimicking spondyloarthritis in a young man. Clin Exp Rheumatol. 2013;31:795.
- Friesgaard Christensen A, Clemmensen O, Junker P. Palpable purpura with an unexpected outcome. Case Rep Rheumatol. 2013;2013:678427.
- Des Roches A, Paradis L, Paradis J, et al. Food allergy as a new risk factor for scurvy. Allergy. 2006;61:1487-1488.
- Pimentel L. Scurvy: historical review and current diagnostic approach. Am J Emerg Med. 2003;21:328-332.
- Codreanu F, Jarlot S, Astier C, et al. An apple a day...chronic glossitis in a 4-year-old boy. Eur Ann Allergy Clin Immunol. 2012;44:86-88.
- Stephen R, Utecht T. Scurvy identified in the emergency department: a case report. J Emerg Med. 2001;21:235-237.
- Hood J, Hodges RE. Ocular lesions in scurvy. Am J Clin Nutr. 1969;22:559-567.
- Horrobin DF, Oka M, Manku MS. The regulation of prostaglandin E1 formation: a candidate for one of the fundamental mechanisms involved in the actions of vitamin C. Med Hypotheses. 1979;5:849-858.
- Hood J, Burns CA, Hodges RE. Sjogren’s syndrome in scurvy. N Engl J Med. 1970;282:1120-1124.
- Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130:177-179.
- Velandia B, Centor RM, McConnell V, et al. Scurvy is still present in developed countries. J Gen Intern Med. 2008;23:1281-1284.
- Lind J. A Treatise of the Scurvy. Edinburgh, Scotland: Sands, Murray, and Cochran; 1753.
- Levine M, Rumsey SC, Daruwala R, et al. Criteria and recommendations for vitamin C intake. JAMA. 1999;281:1415-1423.
- Jacob RA. Vitamin C. In: Shils ME, Olson JA, Shike M, et al, eds. Modern Nutrition in Health and Disease. Baltimore, MD: William & Wilkins; 1999:467-483.
- Levine M. New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314:892-902.
- Hirschman JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895-906.
- Bardnard ND, Weissinger R, Jaster BJ, et al, eds. Nutrition Guide for Clinicians. 2nd ed. Washington, DC: Physician’s Committee For Responsible Medicine; 2009:33.
- Institute of Medicine. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington DC: National Academic Press; 2000.
- Schleicher RL, Carroll MD, Ford ES, et al. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003-2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90:1252-1263.
- Schectman G, Byrd JC, Gruchow HW. The influence of smoking on vitamin C status in adults. Am J Public Health. 1989;79:158-162.
- Johnston CS, Thompson LL. Vitamin C status of an outpatient population. J Am Coll Nutr. 1998;17:366-370.
- Hodges RE, Baker EM, Hood J, et al. Experimental scurvy in man. Am J Clin Nutr. 1969;22:535-548.
- Hodges RE, Hood J, Canham JE, et al. Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24:432-443.
- Gupta P, Taneja K, Iyer PU, et al. Scurvy—the eternal masquerader. Ann Trop Paediatr. 1989;9:118-121.
- Haq RU, Dhammi IK, Jain AK, et al. Infantile scurvy masquerading as bone tumour. Ann Acad Med Singapore. 2013;42:363-365.
- Pazzola G, Possemato N, Germanò G, et al. Scurvy mimicking spondyloarthritis in a young man. Clin Exp Rheumatol. 2013;31:795.
- Friesgaard Christensen A, Clemmensen O, Junker P. Palpable purpura with an unexpected outcome. Case Rep Rheumatol. 2013;2013:678427.
- Des Roches A, Paradis L, Paradis J, et al. Food allergy as a new risk factor for scurvy. Allergy. 2006;61:1487-1488.
- Pimentel L. Scurvy: historical review and current diagnostic approach. Am J Emerg Med. 2003;21:328-332.
- Codreanu F, Jarlot S, Astier C, et al. An apple a day...chronic glossitis in a 4-year-old boy. Eur Ann Allergy Clin Immunol. 2012;44:86-88.
- Stephen R, Utecht T. Scurvy identified in the emergency department: a case report. J Emerg Med. 2001;21:235-237.
- Hood J, Hodges RE. Ocular lesions in scurvy. Am J Clin Nutr. 1969;22:559-567.
- Horrobin DF, Oka M, Manku MS. The regulation of prostaglandin E1 formation: a candidate for one of the fundamental mechanisms involved in the actions of vitamin C. Med Hypotheses. 1979;5:849-858.
- Hood J, Burns CA, Hodges RE. Sjogren’s syndrome in scurvy. N Engl J Med. 1970;282:1120-1124.
- Walter JF. Scurvy resulting from a self-imposed diet. West J Med. 1979;130:177-179.
- Velandia B, Centor RM, McConnell V, et al. Scurvy is still present in developed countries. J Gen Intern Med. 2008;23:1281-1284.
Practice Points
- Patients with scurvy often pose a diagnostic dilemma because their presenting symptoms can lead physicians down a laborious and costly road of unnecessary tests including vasculitic, infectious, and rheumatologic workups.
- The diagnosis of scurvy is clinical and typically is based on signs such as perivascular hemorrhage, bleeding gums, anemia, impaired wound healing, and ecchymoses in the setting of vitamin C deficiency with rapid resolution upon vitamin C supplementation.
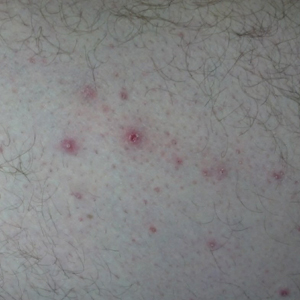
Genetic variant increases stroke risk in childhood cancer survivors
ATLANTA – Adult survivors of childhood cancers are at significantly greater risk than the general population for late-term complications related to therapy, including secondary cancers, cardiovascular disease, and cerebrovascular complications, including ischemic and hemorrhagic strokes.
In particular, childhood cancer survivors have an approximately eightfold higher risk for stroke, compared with their siblings, with a history of cranial irradiation being a strong, dose-dependent risk factor for stroke.
Researchers at St. Jude Children’s Research Hospital in Memphis, Tenn., are conducting a retrospective cohort study with prospective clinical follow-up and ongoing enrollment of childhood cancer survivors who are 5 or more years out of therapy.
The study includes publicly available, whole-genome sequencing data on 4,500 participants. Sifting through these data, Yadav Sapkota, PhD, a clinical research scientist at St. Jude, and his colleagues have identified a genetic variant strongly associated with stroke risk in survivors of European ancestry, and they have replicated the finding in survivors of African ancestry.
In a video interview at the annual meeting of the American Association for Cancer Research, Dr. Sapkota describes his group’s findings and potential research and clinical implications.
The study was sponsored by the National Cancer Institute and ALSAC, the fundraising and awareness organization of St. Jude. Dr. Sapkota declared no conflict of interest.
ATLANTA – Adult survivors of childhood cancers are at significantly greater risk than the general population for late-term complications related to therapy, including secondary cancers, cardiovascular disease, and cerebrovascular complications, including ischemic and hemorrhagic strokes.
In particular, childhood cancer survivors have an approximately eightfold higher risk for stroke, compared with their siblings, with a history of cranial irradiation being a strong, dose-dependent risk factor for stroke.
Researchers at St. Jude Children’s Research Hospital in Memphis, Tenn., are conducting a retrospective cohort study with prospective clinical follow-up and ongoing enrollment of childhood cancer survivors who are 5 or more years out of therapy.
The study includes publicly available, whole-genome sequencing data on 4,500 participants. Sifting through these data, Yadav Sapkota, PhD, a clinical research scientist at St. Jude, and his colleagues have identified a genetic variant strongly associated with stroke risk in survivors of European ancestry, and they have replicated the finding in survivors of African ancestry.
In a video interview at the annual meeting of the American Association for Cancer Research, Dr. Sapkota describes his group’s findings and potential research and clinical implications.
The study was sponsored by the National Cancer Institute and ALSAC, the fundraising and awareness organization of St. Jude. Dr. Sapkota declared no conflict of interest.
ATLANTA – Adult survivors of childhood cancers are at significantly greater risk than the general population for late-term complications related to therapy, including secondary cancers, cardiovascular disease, and cerebrovascular complications, including ischemic and hemorrhagic strokes.
In particular, childhood cancer survivors have an approximately eightfold higher risk for stroke, compared with their siblings, with a history of cranial irradiation being a strong, dose-dependent risk factor for stroke.
Researchers at St. Jude Children’s Research Hospital in Memphis, Tenn., are conducting a retrospective cohort study with prospective clinical follow-up and ongoing enrollment of childhood cancer survivors who are 5 or more years out of therapy.
The study includes publicly available, whole-genome sequencing data on 4,500 participants. Sifting through these data, Yadav Sapkota, PhD, a clinical research scientist at St. Jude, and his colleagues have identified a genetic variant strongly associated with stroke risk in survivors of European ancestry, and they have replicated the finding in survivors of African ancestry.
In a video interview at the annual meeting of the American Association for Cancer Research, Dr. Sapkota describes his group’s findings and potential research and clinical implications.
The study was sponsored by the National Cancer Institute and ALSAC, the fundraising and awareness organization of St. Jude. Dr. Sapkota declared no conflict of interest.
REPORTING FROM AACR 2019

CAR T cells target HER2 expression in advanced sarcomas
ATLANTA – Sarcomas of bone and soft tissues are considered to be “antigenically cold” tumors, with few identifiable mutations that may be susceptible to targeted therapies.
Some sarcoma subtypes such as osteosarcoma and rhabomyosarcoma, however, frequently express the human epidermal growth factor receptor 2 on tumor surfaces. Although HER2 expression in these tumors is at too low a level for HER2-targeted therapies such as trastuzumab (Herceptin), HER2 appears to be an opportunistic target for chimeric antigen receptor (CAR) T-cell therapy, according to Shoba Navai, MD, from Baylor College of Medicine, Houston.
In a video interview at the 2019 annual meeting of the American Association for Cancer Research, Dr. Navai describes her team’s early experience using a HER2-targeted CAR-T cell construct and preinfusion lymphodepletion in patients with advanced sarcomas.
Development of the CAR-T cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai reported having no disclosures.
ATLANTA – Sarcomas of bone and soft tissues are considered to be “antigenically cold” tumors, with few identifiable mutations that may be susceptible to targeted therapies.
Some sarcoma subtypes such as osteosarcoma and rhabomyosarcoma, however, frequently express the human epidermal growth factor receptor 2 on tumor surfaces. Although HER2 expression in these tumors is at too low a level for HER2-targeted therapies such as trastuzumab (Herceptin), HER2 appears to be an opportunistic target for chimeric antigen receptor (CAR) T-cell therapy, according to Shoba Navai, MD, from Baylor College of Medicine, Houston.
In a video interview at the 2019 annual meeting of the American Association for Cancer Research, Dr. Navai describes her team’s early experience using a HER2-targeted CAR-T cell construct and preinfusion lymphodepletion in patients with advanced sarcomas.
Development of the CAR-T cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai reported having no disclosures.
ATLANTA – Sarcomas of bone and soft tissues are considered to be “antigenically cold” tumors, with few identifiable mutations that may be susceptible to targeted therapies.
Some sarcoma subtypes such as osteosarcoma and rhabomyosarcoma, however, frequently express the human epidermal growth factor receptor 2 on tumor surfaces. Although HER2 expression in these tumors is at too low a level for HER2-targeted therapies such as trastuzumab (Herceptin), HER2 appears to be an opportunistic target for chimeric antigen receptor (CAR) T-cell therapy, according to Shoba Navai, MD, from Baylor College of Medicine, Houston.
In a video interview at the 2019 annual meeting of the American Association for Cancer Research, Dr. Navai describes her team’s early experience using a HER2-targeted CAR-T cell construct and preinfusion lymphodepletion in patients with advanced sarcomas.
Development of the CAR-T cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai reported having no disclosures.
REPORTING FROM AACR 2019

Trofinetide may benefit patients with Rett syndrome
Furthermore, the treatment is safe and well tolerated, according to results published online ahead of print March 27 in Neurology.

“These are very promising data for the Rett community that is currently without any [Food and Drug Administration]–approved treatment option,” Daniel Glaze, MD, said in a press release from Acadia Pharmaceuticals, which is developing trofinetide.
In 2017, a phase 2 study indicated that the drug was safe and tolerable when administered in doses of 70 mg/kg b.i.d. to adolescent and adult females with Rett syndrome. The study also provided initial evidence of the drug’s efficacy. Dr. Glaze, professor of pediatrics and neurology at Baylor College of Medicine in Houston, and his colleagues decided to conduct a larger phase 2 study that examined higher doses and a longer treatment duration.
The researchers first enrolled 62 participants in the study, all of whom received placebo b.i.d. for 14 days. They next randomized participants in equal groups to placebo or one of three twice-daily doses of trofinetide (i.e., 50 mg/kg, 100 mg/kg, and 200 mg/kg). After a blinded review of safety and tolerability data, Dr. Glaze and his colleagues enrolled 20 more participants and randomized them in equal groups to placebo or 200 mg/kg b.i.d. of trofinetide. This modification in study design was intended to increase the likelihood of detecting a clinical benefit. Randomized, double-blind treatment lasted for 42 days. Participants presented for a final visit at approximately 10 days after the treatment period ended.
A total of 82 girls aged 5-15 years participated in the study. They all met the 2010 diagnostic criteria for classic Rett syndrome, had a documented pathogenic MECP2 variant, were in the postregression stage of the syndrome, and had been stable on pharmacologic and behavioral treatments for at least 4 weeks. The sample’s mean age was 9.7 years, 94% were white, and mean weight was 26.1 kg. The treatment groups’ demographic characteristics were balanced.
All three doses of trofinetide were safe and tolerable. One participant in the 200-mg/kg b.i.d. group was withdrawn from the study at her parents’ request. Serious adverse events occurred in one control participant, one receiving the 100-mg/kg b.i.d. dose, and one receiving the 200-mg/kg b.i.d. dose. These serious adverse events were considered unrelated to study medication and resolved by the study’s end. The most common adverse events were diarrhea, vomiting, upper respiratory tract infection, and pyrexia. Most were mild or moderate and considered unrelated to trofinetide.
The 200-mg/kg b.i.d. dose of trofinetide significantly improved outcomes on the Rett Syndrome Behavior Questionnaire (RSBQ), Clinical Global Impression Scale–Improvement (CGI-I), and Rett Syndrome Domain Specific Concerns (RTT-DSC), compared with placebo. The change of the median RTT-DSC score was 15%, and the change of the mean RSBQ score was 16%. More than 20% of participants receiving 200 mg/kg b.i.d. of trofinetide were rated much improved on the CGI-I, compared with less than 5% of controls.
“Overall, the observed clinical improvement in the present pediatric trial was more manifest than in the previous trial, with younger age (i.e., greater neuroplasticity), higher doses (i.e., higher drug exposure), and longer drug treatment duration (i.e., 28 days in Rett-001 vs. 42 days in Rett-002) as potential contributors,” said Dr. Glaze and his colleagues. “Future studies aiming at replicating the results of this pediatric trial would benefit from including the RSBQ as a primary endpoint and should also consider evaluating RSBQ subscales such as the General Mood.”
Neuren Pharmaceuticals and Rettsyndrome.org funded the study. Dr. Glaze is a consultant to Neuren Pharmaceuticals.
SOURCE: Glaze DG et al. Neurology. 2019 Mar 27. doi: 10.1212/WNL.0000000000007316.
Furthermore, the treatment is safe and well tolerated, according to results published online ahead of print March 27 in Neurology.
“These are very promising data for the Rett community that is currently without any [Food and Drug Administration]–approved treatment option,” Daniel Glaze, MD, said in a press release from Acadia Pharmaceuticals, which is developing trofinetide.
In 2017, a phase 2 study indicated that the drug was safe and tolerable when administered in doses of 70 mg/kg b.i.d. to adolescent and adult females with Rett syndrome. The study also provided initial evidence of the drug’s efficacy. Dr. Glaze, professor of pediatrics and neurology at Baylor College of Medicine in Houston, and his colleagues decided to conduct a larger phase 2 study that examined higher doses and a longer treatment duration.
The researchers first enrolled 62 participants in the study, all of whom received placebo b.i.d. for 14 days. They next randomized participants in equal groups to placebo or one of three twice-daily doses of trofinetide (i.e., 50 mg/kg, 100 mg/kg, and 200 mg/kg). After a blinded review of safety and tolerability data, Dr. Glaze and his colleagues enrolled 20 more participants and randomized them in equal groups to placebo or 200 mg/kg b.i.d. of trofinetide. This modification in study design was intended to increase the likelihood of detecting a clinical benefit. Randomized, double-blind treatment lasted for 42 days. Participants presented for a final visit at approximately 10 days after the treatment period ended.
A total of 82 girls aged 5-15 years participated in the study. They all met the 2010 diagnostic criteria for classic Rett syndrome, had a documented pathogenic MECP2 variant, were in the postregression stage of the syndrome, and had been stable on pharmacologic and behavioral treatments for at least 4 weeks. The sample’s mean age was 9.7 years, 94% were white, and mean weight was 26.1 kg. The treatment groups’ demographic characteristics were balanced.
All three doses of trofinetide were safe and tolerable. One participant in the 200-mg/kg b.i.d. group was withdrawn from the study at her parents’ request. Serious adverse events occurred in one control participant, one receiving the 100-mg/kg b.i.d. dose, and one receiving the 200-mg/kg b.i.d. dose. These serious adverse events were considered unrelated to study medication and resolved by the study’s end. The most common adverse events were diarrhea, vomiting, upper respiratory tract infection, and pyrexia. Most were mild or moderate and considered unrelated to trofinetide.
The 200-mg/kg b.i.d. dose of trofinetide significantly improved outcomes on the Rett Syndrome Behavior Questionnaire (RSBQ), Clinical Global Impression Scale–Improvement (CGI-I), and Rett Syndrome Domain Specific Concerns (RTT-DSC), compared with placebo. The change of the median RTT-DSC score was 15%, and the change of the mean RSBQ score was 16%. More than 20% of participants receiving 200 mg/kg b.i.d. of trofinetide were rated much improved on the CGI-I, compared with less than 5% of controls.
“Overall, the observed clinical improvement in the present pediatric trial was more manifest than in the previous trial, with younger age (i.e., greater neuroplasticity), higher doses (i.e., higher drug exposure), and longer drug treatment duration (i.e., 28 days in Rett-001 vs. 42 days in Rett-002) as potential contributors,” said Dr. Glaze and his colleagues. “Future studies aiming at replicating the results of this pediatric trial would benefit from including the RSBQ as a primary endpoint and should also consider evaluating RSBQ subscales such as the General Mood.”
Neuren Pharmaceuticals and Rettsyndrome.org funded the study. Dr. Glaze is a consultant to Neuren Pharmaceuticals.
SOURCE: Glaze DG et al. Neurology. 2019 Mar 27. doi: 10.1212/WNL.0000000000007316.
Furthermore, the treatment is safe and well tolerated, according to results published online ahead of print March 27 in Neurology.
“These are very promising data for the Rett community that is currently without any [Food and Drug Administration]–approved treatment option,” Daniel Glaze, MD, said in a press release from Acadia Pharmaceuticals, which is developing trofinetide.
In 2017, a phase 2 study indicated that the drug was safe and tolerable when administered in doses of 70 mg/kg b.i.d. to adolescent and adult females with Rett syndrome. The study also provided initial evidence of the drug’s efficacy. Dr. Glaze, professor of pediatrics and neurology at Baylor College of Medicine in Houston, and his colleagues decided to conduct a larger phase 2 study that examined higher doses and a longer treatment duration.
The researchers first enrolled 62 participants in the study, all of whom received placebo b.i.d. for 14 days. They next randomized participants in equal groups to placebo or one of three twice-daily doses of trofinetide (i.e., 50 mg/kg, 100 mg/kg, and 200 mg/kg). After a blinded review of safety and tolerability data, Dr. Glaze and his colleagues enrolled 20 more participants and randomized them in equal groups to placebo or 200 mg/kg b.i.d. of trofinetide. This modification in study design was intended to increase the likelihood of detecting a clinical benefit. Randomized, double-blind treatment lasted for 42 days. Participants presented for a final visit at approximately 10 days after the treatment period ended.
A total of 82 girls aged 5-15 years participated in the study. They all met the 2010 diagnostic criteria for classic Rett syndrome, had a documented pathogenic MECP2 variant, were in the postregression stage of the syndrome, and had been stable on pharmacologic and behavioral treatments for at least 4 weeks. The sample’s mean age was 9.7 years, 94% were white, and mean weight was 26.1 kg. The treatment groups’ demographic characteristics were balanced.
All three doses of trofinetide were safe and tolerable. One participant in the 200-mg/kg b.i.d. group was withdrawn from the study at her parents’ request. Serious adverse events occurred in one control participant, one receiving the 100-mg/kg b.i.d. dose, and one receiving the 200-mg/kg b.i.d. dose. These serious adverse events were considered unrelated to study medication and resolved by the study’s end. The most common adverse events were diarrhea, vomiting, upper respiratory tract infection, and pyrexia. Most were mild or moderate and considered unrelated to trofinetide.
The 200-mg/kg b.i.d. dose of trofinetide significantly improved outcomes on the Rett Syndrome Behavior Questionnaire (RSBQ), Clinical Global Impression Scale–Improvement (CGI-I), and Rett Syndrome Domain Specific Concerns (RTT-DSC), compared with placebo. The change of the median RTT-DSC score was 15%, and the change of the mean RSBQ score was 16%. More than 20% of participants receiving 200 mg/kg b.i.d. of trofinetide were rated much improved on the CGI-I, compared with less than 5% of controls.
“Overall, the observed clinical improvement in the present pediatric trial was more manifest than in the previous trial, with younger age (i.e., greater neuroplasticity), higher doses (i.e., higher drug exposure), and longer drug treatment duration (i.e., 28 days in Rett-001 vs. 42 days in Rett-002) as potential contributors,” said Dr. Glaze and his colleagues. “Future studies aiming at replicating the results of this pediatric trial would benefit from including the RSBQ as a primary endpoint and should also consider evaluating RSBQ subscales such as the General Mood.”
Neuren Pharmaceuticals and Rettsyndrome.org funded the study. Dr. Glaze is a consultant to Neuren Pharmaceuticals.
SOURCE: Glaze DG et al. Neurology. 2019 Mar 27. doi: 10.1212/WNL.0000000000007316.
FROM NEUROLOGY
Key clinical point: Trofinetide provides clinically meaningful improvements in core symptoms of Rett syndrome.
Major finding: The 200 mg/kg b.i.d. dose of trofinetide improved outcomes on the Rett Syndrome Behavior Questionnaire by 16%.
Study details: A phase 2, double-blind, placebo-controlled study of 82 children and adolescents with Rett syndrome.
Disclosures: Neuren Pharmaceuticals and Rettsyndrome.org funded the study.
Source: Glaze DG et al. Neurology. 2019 Mar 27. doi: 10.1212/WNL.0000000000007316.
Promising New Approach for Treating Cystic Fibrosis
In cystic fibrosis (CF), a defective gene makes a defective protein that causes acidic and sticky mucus that clogs the lungs and puts patients at risk for bacterial infections. Because different people have different protein mutations, and 10% of patients with CF make no protein at all, treatments are limited. But amphotericin apparently has the potential to work regardless of the mutation and even when the protein is missing. The researchers liken it to a “molecular prosthetic,” because it restores function much as a prosthetic device replaces a limb.
In the study, supported in part by the National Heart, Lung, and Blood Institute, researchers used lung tissue from patients with CF as well as animal models. Instead of trying to correct the protein or do gene therapy, which the researchers say is not yet effective in the lung, they used a small molecule protein that can perform the channel function of the missing or defective protein.
They found that amphotericin restored pH levels, improved viscosity, and increased antibacterial activity.
Amphotericin also can be delivered directly to the lungs to avoid common adverse effects, the researchers say. Although more studies are needed, according to the National Institute of Health, “experts are hopeful.”
In cystic fibrosis (CF), a defective gene makes a defective protein that causes acidic and sticky mucus that clogs the lungs and puts patients at risk for bacterial infections. Because different people have different protein mutations, and 10% of patients with CF make no protein at all, treatments are limited. But amphotericin apparently has the potential to work regardless of the mutation and even when the protein is missing. The researchers liken it to a “molecular prosthetic,” because it restores function much as a prosthetic device replaces a limb.
In the study, supported in part by the National Heart, Lung, and Blood Institute, researchers used lung tissue from patients with CF as well as animal models. Instead of trying to correct the protein or do gene therapy, which the researchers say is not yet effective in the lung, they used a small molecule protein that can perform the channel function of the missing or defective protein.
They found that amphotericin restored pH levels, improved viscosity, and increased antibacterial activity.
Amphotericin also can be delivered directly to the lungs to avoid common adverse effects, the researchers say. Although more studies are needed, according to the National Institute of Health, “experts are hopeful.”
In cystic fibrosis (CF), a defective gene makes a defective protein that causes acidic and sticky mucus that clogs the lungs and puts patients at risk for bacterial infections. Because different people have different protein mutations, and 10% of patients with CF make no protein at all, treatments are limited. But amphotericin apparently has the potential to work regardless of the mutation and even when the protein is missing. The researchers liken it to a “molecular prosthetic,” because it restores function much as a prosthetic device replaces a limb.
In the study, supported in part by the National Heart, Lung, and Blood Institute, researchers used lung tissue from patients with CF as well as animal models. Instead of trying to correct the protein or do gene therapy, which the researchers say is not yet effective in the lung, they used a small molecule protein that can perform the channel function of the missing or defective protein.
They found that amphotericin restored pH levels, improved viscosity, and increased antibacterial activity.
Amphotericin also can be delivered directly to the lungs to avoid common adverse effects, the researchers say. Although more studies are needed, according to the National Institute of Health, “experts are hopeful.”