User login
Isolated nail psoriasis may bring arthritis into play
for dermatologists to improve their diagnostic accuracy,” investigators said in a research letter.
Diagnosis of isolated NP was delayed by almost 3 years among the 87 cases recorded and “arthritis was most often diagnosed concurrently with NP,” at a major nail referral center between Jan. 1, 2001, and Dec. 21, 2022, Michelle J. Chang of Drexel University, Philadelphia, and associates reported.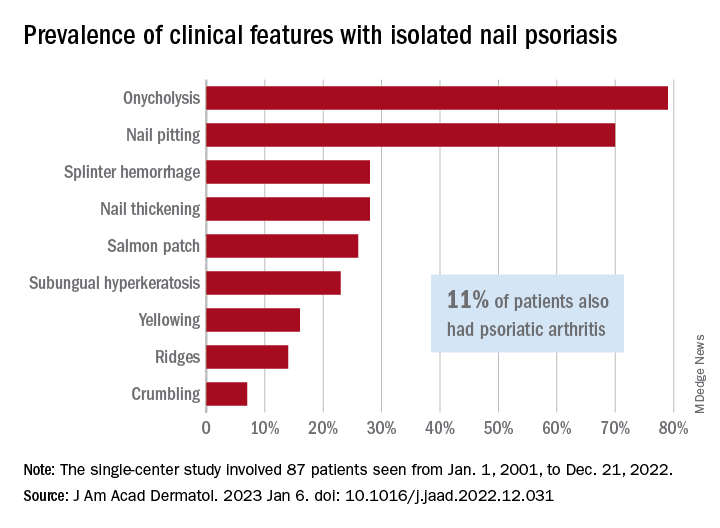
In what the authors say is, “the largest study documenting clinical and histologic features in patients with isolated NP,” the two most common clinical features were onycholysis and nail plate pitting, seen in 79% and 70% of cases, respectively. No other single feature had a prevalence higher than 28%.
The most frequent clinical dyad was onycholysis and pitting in 66% of patients, followed by onycholysis/nail thickening in 33% and onycholysis/splinter hemorrhage in 32%. The most common histologic features were parakeratosis in 79% and neutrophil infiltration in 48%, the investigators said.
Psoriatic arthritis (PsA), a focus of the study, occurred in 10 (11%) of the 87 individuals with isolated NP. Considering this finding, and “the close proximity between the nail apparatus and joint, we hypothesize a reciprocal relationship, with nail unit inflammation precipitating PsA,” Ms. Chang and associates wrote.
Senior author, Shari Lipner, MD, PhD, of the department of dermatology, Weill Cornell Medicine, New York, is a consultant for Ortho-Dermatologics, Hoth Therapeutics, and BelleTorus. Ms. Chang and the two other investigators had no conflicts of interest to declare.
for dermatologists to improve their diagnostic accuracy,” investigators said in a research letter.
Diagnosis of isolated NP was delayed by almost 3 years among the 87 cases recorded and “arthritis was most often diagnosed concurrently with NP,” at a major nail referral center between Jan. 1, 2001, and Dec. 21, 2022, Michelle J. Chang of Drexel University, Philadelphia, and associates reported.
In what the authors say is, “the largest study documenting clinical and histologic features in patients with isolated NP,” the two most common clinical features were onycholysis and nail plate pitting, seen in 79% and 70% of cases, respectively. No other single feature had a prevalence higher than 28%.
The most frequent clinical dyad was onycholysis and pitting in 66% of patients, followed by onycholysis/nail thickening in 33% and onycholysis/splinter hemorrhage in 32%. The most common histologic features were parakeratosis in 79% and neutrophil infiltration in 48%, the investigators said.
Psoriatic arthritis (PsA), a focus of the study, occurred in 10 (11%) of the 87 individuals with isolated NP. Considering this finding, and “the close proximity between the nail apparatus and joint, we hypothesize a reciprocal relationship, with nail unit inflammation precipitating PsA,” Ms. Chang and associates wrote.
Senior author, Shari Lipner, MD, PhD, of the department of dermatology, Weill Cornell Medicine, New York, is a consultant for Ortho-Dermatologics, Hoth Therapeutics, and BelleTorus. Ms. Chang and the two other investigators had no conflicts of interest to declare.
for dermatologists to improve their diagnostic accuracy,” investigators said in a research letter.
Diagnosis of isolated NP was delayed by almost 3 years among the 87 cases recorded and “arthritis was most often diagnosed concurrently with NP,” at a major nail referral center between Jan. 1, 2001, and Dec. 21, 2022, Michelle J. Chang of Drexel University, Philadelphia, and associates reported.
In what the authors say is, “the largest study documenting clinical and histologic features in patients with isolated NP,” the two most common clinical features were onycholysis and nail plate pitting, seen in 79% and 70% of cases, respectively. No other single feature had a prevalence higher than 28%.
The most frequent clinical dyad was onycholysis and pitting in 66% of patients, followed by onycholysis/nail thickening in 33% and onycholysis/splinter hemorrhage in 32%. The most common histologic features were parakeratosis in 79% and neutrophil infiltration in 48%, the investigators said.
Psoriatic arthritis (PsA), a focus of the study, occurred in 10 (11%) of the 87 individuals with isolated NP. Considering this finding, and “the close proximity between the nail apparatus and joint, we hypothesize a reciprocal relationship, with nail unit inflammation precipitating PsA,” Ms. Chang and associates wrote.
Senior author, Shari Lipner, MD, PhD, of the department of dermatology, Weill Cornell Medicine, New York, is a consultant for Ortho-Dermatologics, Hoth Therapeutics, and BelleTorus. Ms. Chang and the two other investigators had no conflicts of interest to declare.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Bacteremia, antibodies link periodontal disease to RA development and activity
a new longitudinal study suggests.
The results suggest that PD involves repeated oral mucosa breakdowns with the release of citrullinated oral bacteria into the bloodstream and that these bacteria activate inflammatory monocytes in the inflamed RA synovium and in the blood of patients with RA. The bacteria also activate anti-citrullinated protein antibody (ACPA)–positive B cells, promoting affinity maturation and epitope spreading to citrullinated human antigens, the authors wrote.
“Our study discovered frequent and repeated episodes of oral bacteria in the blood of patients with periodontal disease and rheumatoid arthritis,” senior study author Dana E. Orange, MD, associate professor of clinical investigation at Rockefeller University, New York, said in an interview. “We saw that these bacteria were triggering an inflammatory [monocyte] response that is similar to what we see in the inflamed joints of patients with RA.
“RA patients are less likely to benefit from RA treatment if they have concurrent periodontal disease,” she said.
“RA patients tend to harbor cyclic citrullinated peptide [CCP] autoantibodies. Many groups have noted that CCP antibodies are very highly mutated, a signature of a B-cell/antibody response that has been stimulated over and over again,” Dr. Orange explained. “We found that CCP antibodies also bind the same oral bacteria we detected in the bloodstream. That patients with PD experience frequent, repeated episodes of oral bacteria in the blood is consistent with the very high level of mutation burden of CCP antibodies.”
Periodontal disease is a large problem among older adults
Periodontal disease ranges from gingivitis with swollen, red gums that may bleed, to periodontitis with gums pulling away from teeth, bone loss, and loose or lost teeth. PD is very prevalent in the United States, with some form of it occurring in 47% of people 30 years of age and older and in 70% of those 65 years and older, according to the Centers for Disease Control and Prevention.
PD is more common in people with RA who have detectable ACPAs, and that link implicates oral mucosal inflammation in RA pathogenesis.
Investigating whether PD leads to RA
At Rockefeller University, Dr. Orange and her colleagues followed five female patients with RA who were seropositive for CCP over the course of 1-4 years. Two had severe PD, and three showed no signs of PD. Each week, participants provided finger-stick blood samples for RNA sequencing and B-cell repertoire sequencing, reported changes in their medication and dental work, and completed questionnaires. They also underwent monthly physical exams that evaluated tenderness and swelling in 28 joints, and they provided additional samples during self-reported flares.
At Stanford (Calif.) University, researchers isolated plasmablasts from blood samples collected from 12 donors with anti-CPP+ RA and healthy donors. They also analyzed synovial fluid samples from 65 RA and OA patients.
In addition, University of Colorado researchers collected plasma samples from patients with RA and healthy donors, and they classified the gingival health of participants.
The researchers found that the patients with PD also had repeated flare-ups of oral bacteria in the blood, mainly from the Streptococcaceae family, suggesting that the oral mucosa repeatedly broke down and introduced bacteria into the bloodstream. In the blood, inflammatory immune cells targeted the bacteria and released ACPAs, which have been linked with RA.
“Even without dental procedures, patients with RA and periodontal disease experience repeated episodes of bacteremia,” Dr. Orange said. “While dental procedures are known to be associated with dissemination of oral bacteria into the bloodstream, we didn’t expect to see this happening so frequently and independent of dental procedures. We also had thought that people with oral bacteria in their blood would have symptoms such as fever or malaise, but they were asymptomatic.”
Experts welcome the results
“It is well established that oral health impacts overall health. In particular, the link between periodontal disease and chronic systemic diseases has been explored in many robust studies,” Chi T. Viet, MD, DDS, PhD, oral and maxillofacial surgeon and associate professor at the Loma Linda (Calif.) University School of Dentistry, told this news organization.
“The study ... highlights the importance of early treatment and prevention of oral health issues, as they can have devastating systemic sequelae,” added Dr. Viet, who was not involved in the study.
Devon Charlton, MD, MPH, director of rheumatology at the University of Pittsburgh Medical Center Regional Orthopedics in New Castle, Pa., advised patients and providers to team up to manage RA and PD.
“If the patient and provider aggressively treat periodontal disease and dental health in general, the patient decreases their risk of infection and inflammation, decreases the potential development of RA, and increases their future quality of life,” said Dr. Charlton, also not involved in the study.
However, findings from the study population may be difficult to generalize to other populations, he added.
Dr. Orange and her colleagues suggest further related research, including studies on whether treating PD makes RA easier to treat and whether the treatment of PD in patients whose RA is in clinical remission raises the likelihood of being able to discontinue therapy safely.
The study was supported through grant funding from the National Institutes of Health, the National Science Foundation, the Robertson Foundation, Rockefeller University, the Bernard and Irene Schwartz Foundation, the Iris and Junming Le Foundation, and the Rheumatology Research Foundation. Dr. Orange reports no relevant financial relationships. Several coauthors report financial relationships with the pharmaceutical industry. Dr. Viet and Dr. Charlton report no relevant financial relationships. All experts commented by email.
A version of this article first appeared on Medscape.com.
a new longitudinal study suggests.
The results suggest that PD involves repeated oral mucosa breakdowns with the release of citrullinated oral bacteria into the bloodstream and that these bacteria activate inflammatory monocytes in the inflamed RA synovium and in the blood of patients with RA. The bacteria also activate anti-citrullinated protein antibody (ACPA)–positive B cells, promoting affinity maturation and epitope spreading to citrullinated human antigens, the authors wrote.
“Our study discovered frequent and repeated episodes of oral bacteria in the blood of patients with periodontal disease and rheumatoid arthritis,” senior study author Dana E. Orange, MD, associate professor of clinical investigation at Rockefeller University, New York, said in an interview. “We saw that these bacteria were triggering an inflammatory [monocyte] response that is similar to what we see in the inflamed joints of patients with RA.
“RA patients are less likely to benefit from RA treatment if they have concurrent periodontal disease,” she said.
“RA patients tend to harbor cyclic citrullinated peptide [CCP] autoantibodies. Many groups have noted that CCP antibodies are very highly mutated, a signature of a B-cell/antibody response that has been stimulated over and over again,” Dr. Orange explained. “We found that CCP antibodies also bind the same oral bacteria we detected in the bloodstream. That patients with PD experience frequent, repeated episodes of oral bacteria in the blood is consistent with the very high level of mutation burden of CCP antibodies.”
Periodontal disease is a large problem among older adults
Periodontal disease ranges from gingivitis with swollen, red gums that may bleed, to periodontitis with gums pulling away from teeth, bone loss, and loose or lost teeth. PD is very prevalent in the United States, with some form of it occurring in 47% of people 30 years of age and older and in 70% of those 65 years and older, according to the Centers for Disease Control and Prevention.
PD is more common in people with RA who have detectable ACPAs, and that link implicates oral mucosal inflammation in RA pathogenesis.
Investigating whether PD leads to RA
At Rockefeller University, Dr. Orange and her colleagues followed five female patients with RA who were seropositive for CCP over the course of 1-4 years. Two had severe PD, and three showed no signs of PD. Each week, participants provided finger-stick blood samples for RNA sequencing and B-cell repertoire sequencing, reported changes in their medication and dental work, and completed questionnaires. They also underwent monthly physical exams that evaluated tenderness and swelling in 28 joints, and they provided additional samples during self-reported flares.
At Stanford (Calif.) University, researchers isolated plasmablasts from blood samples collected from 12 donors with anti-CPP+ RA and healthy donors. They also analyzed synovial fluid samples from 65 RA and OA patients.
In addition, University of Colorado researchers collected plasma samples from patients with RA and healthy donors, and they classified the gingival health of participants.
The researchers found that the patients with PD also had repeated flare-ups of oral bacteria in the blood, mainly from the Streptococcaceae family, suggesting that the oral mucosa repeatedly broke down and introduced bacteria into the bloodstream. In the blood, inflammatory immune cells targeted the bacteria and released ACPAs, which have been linked with RA.
“Even without dental procedures, patients with RA and periodontal disease experience repeated episodes of bacteremia,” Dr. Orange said. “While dental procedures are known to be associated with dissemination of oral bacteria into the bloodstream, we didn’t expect to see this happening so frequently and independent of dental procedures. We also had thought that people with oral bacteria in their blood would have symptoms such as fever or malaise, but they were asymptomatic.”
Experts welcome the results
“It is well established that oral health impacts overall health. In particular, the link between periodontal disease and chronic systemic diseases has been explored in many robust studies,” Chi T. Viet, MD, DDS, PhD, oral and maxillofacial surgeon and associate professor at the Loma Linda (Calif.) University School of Dentistry, told this news organization.
“The study ... highlights the importance of early treatment and prevention of oral health issues, as they can have devastating systemic sequelae,” added Dr. Viet, who was not involved in the study.
Devon Charlton, MD, MPH, director of rheumatology at the University of Pittsburgh Medical Center Regional Orthopedics in New Castle, Pa., advised patients and providers to team up to manage RA and PD.
“If the patient and provider aggressively treat periodontal disease and dental health in general, the patient decreases their risk of infection and inflammation, decreases the potential development of RA, and increases their future quality of life,” said Dr. Charlton, also not involved in the study.
However, findings from the study population may be difficult to generalize to other populations, he added.
Dr. Orange and her colleagues suggest further related research, including studies on whether treating PD makes RA easier to treat and whether the treatment of PD in patients whose RA is in clinical remission raises the likelihood of being able to discontinue therapy safely.
The study was supported through grant funding from the National Institutes of Health, the National Science Foundation, the Robertson Foundation, Rockefeller University, the Bernard and Irene Schwartz Foundation, the Iris and Junming Le Foundation, and the Rheumatology Research Foundation. Dr. Orange reports no relevant financial relationships. Several coauthors report financial relationships with the pharmaceutical industry. Dr. Viet and Dr. Charlton report no relevant financial relationships. All experts commented by email.
A version of this article first appeared on Medscape.com.
a new longitudinal study suggests.
The results suggest that PD involves repeated oral mucosa breakdowns with the release of citrullinated oral bacteria into the bloodstream and that these bacteria activate inflammatory monocytes in the inflamed RA synovium and in the blood of patients with RA. The bacteria also activate anti-citrullinated protein antibody (ACPA)–positive B cells, promoting affinity maturation and epitope spreading to citrullinated human antigens, the authors wrote.
“Our study discovered frequent and repeated episodes of oral bacteria in the blood of patients with periodontal disease and rheumatoid arthritis,” senior study author Dana E. Orange, MD, associate professor of clinical investigation at Rockefeller University, New York, said in an interview. “We saw that these bacteria were triggering an inflammatory [monocyte] response that is similar to what we see in the inflamed joints of patients with RA.
“RA patients are less likely to benefit from RA treatment if they have concurrent periodontal disease,” she said.
“RA patients tend to harbor cyclic citrullinated peptide [CCP] autoantibodies. Many groups have noted that CCP antibodies are very highly mutated, a signature of a B-cell/antibody response that has been stimulated over and over again,” Dr. Orange explained. “We found that CCP antibodies also bind the same oral bacteria we detected in the bloodstream. That patients with PD experience frequent, repeated episodes of oral bacteria in the blood is consistent with the very high level of mutation burden of CCP antibodies.”
Periodontal disease is a large problem among older adults
Periodontal disease ranges from gingivitis with swollen, red gums that may bleed, to periodontitis with gums pulling away from teeth, bone loss, and loose or lost teeth. PD is very prevalent in the United States, with some form of it occurring in 47% of people 30 years of age and older and in 70% of those 65 years and older, according to the Centers for Disease Control and Prevention.
PD is more common in people with RA who have detectable ACPAs, and that link implicates oral mucosal inflammation in RA pathogenesis.
Investigating whether PD leads to RA
At Rockefeller University, Dr. Orange and her colleagues followed five female patients with RA who were seropositive for CCP over the course of 1-4 years. Two had severe PD, and three showed no signs of PD. Each week, participants provided finger-stick blood samples for RNA sequencing and B-cell repertoire sequencing, reported changes in their medication and dental work, and completed questionnaires. They also underwent monthly physical exams that evaluated tenderness and swelling in 28 joints, and they provided additional samples during self-reported flares.
At Stanford (Calif.) University, researchers isolated plasmablasts from blood samples collected from 12 donors with anti-CPP+ RA and healthy donors. They also analyzed synovial fluid samples from 65 RA and OA patients.
In addition, University of Colorado researchers collected plasma samples from patients with RA and healthy donors, and they classified the gingival health of participants.
The researchers found that the patients with PD also had repeated flare-ups of oral bacteria in the blood, mainly from the Streptococcaceae family, suggesting that the oral mucosa repeatedly broke down and introduced bacteria into the bloodstream. In the blood, inflammatory immune cells targeted the bacteria and released ACPAs, which have been linked with RA.
“Even without dental procedures, patients with RA and periodontal disease experience repeated episodes of bacteremia,” Dr. Orange said. “While dental procedures are known to be associated with dissemination of oral bacteria into the bloodstream, we didn’t expect to see this happening so frequently and independent of dental procedures. We also had thought that people with oral bacteria in their blood would have symptoms such as fever or malaise, but they were asymptomatic.”
Experts welcome the results
“It is well established that oral health impacts overall health. In particular, the link between periodontal disease and chronic systemic diseases has been explored in many robust studies,” Chi T. Viet, MD, DDS, PhD, oral and maxillofacial surgeon and associate professor at the Loma Linda (Calif.) University School of Dentistry, told this news organization.
“The study ... highlights the importance of early treatment and prevention of oral health issues, as they can have devastating systemic sequelae,” added Dr. Viet, who was not involved in the study.
Devon Charlton, MD, MPH, director of rheumatology at the University of Pittsburgh Medical Center Regional Orthopedics in New Castle, Pa., advised patients and providers to team up to manage RA and PD.
“If the patient and provider aggressively treat periodontal disease and dental health in general, the patient decreases their risk of infection and inflammation, decreases the potential development of RA, and increases their future quality of life,” said Dr. Charlton, also not involved in the study.
However, findings from the study population may be difficult to generalize to other populations, he added.
Dr. Orange and her colleagues suggest further related research, including studies on whether treating PD makes RA easier to treat and whether the treatment of PD in patients whose RA is in clinical remission raises the likelihood of being able to discontinue therapy safely.
The study was supported through grant funding from the National Institutes of Health, the National Science Foundation, the Robertson Foundation, Rockefeller University, the Bernard and Irene Schwartz Foundation, the Iris and Junming Le Foundation, and the Rheumatology Research Foundation. Dr. Orange reports no relevant financial relationships. Several coauthors report financial relationships with the pharmaceutical industry. Dr. Viet and Dr. Charlton report no relevant financial relationships. All experts commented by email.
A version of this article first appeared on Medscape.com.
FROM SCIENCE TRANSLATIONAL MEDICINE
What’s holding back physicians from prescribing biosimilars? Four specialties weigh in
While most providers think that biosimilars will positively impact care, few feel that the economic benefits of biosimilars to date are enough to motivate switching.

In a new survey of over 350 dermatologists, gastroenterologists, ophthalmologists, and rheumatologists, clinicians shared their opinions on the rapidly evolving landscape of biosimilars, detailing top concerns about prescribing these medications and how they presently use biosimilars in clinical practice. Across all specialties, providers said they would be most likely to prescribe biosimilars to new patients or if a patient’s health plan mandated the switch. Most providers listed concerns about biosimilar efficacy and lack of economic benefit as the main barriers to adoption of biosimilars in clinical practice.
Cardinal Health, a health care services company based in Dublin, Ohio, conducted the surveys from July through October 2022.
Rheumatologists want cost-savings for patients
2023 is gearing up to be a big year for biosimilars for inflammatory diseases, with at least eight adalimumab biosimilars entering the market in the United States. Amjevita, manufactured by Amgen, was the first to become commercially available on Jan. 31. Out of 103 surveyed rheumatologists, 62% said they were very comfortable prescribing biosimilars to patients, and 32% said they were somewhat comfortable. Providers said they would be most likely to prescribe a biosimilar to new patients (40%) or if biosimilars were mandated by a patient’s health plan (41%). Nearly one-third (31%) of rheumatologists said that a discount of 21%-30% from a reference product would be necessary to consider switching a patient to a biosimilar.
There are several reasons why a rheumatologist might be wary of switching patients to biosimilars, said Marcus Snow, MD, chair of the American College of Rheumatology’s Committee on Rheumatologic Care. “Rheumatologists will always express concern about changing medications that work well for their patients. It is not ideal to ‘force switch’ to a different product, even if it is almost identical,” he told this news organization in an email. “Also, we must remember that a patient on a biologic has failed traditional medications, which speaks to the struggle a patient must endure to get their disease under control. Fail-first situations can cause a rheumatologist to be initially resistant or hesitant to any changes.”
The top concerns among rheumatologists about prescribing biosimilars were medication efficacy (36%), lack of economic benefit (24%), and evaluating when to prescribe a biosimilar versus a reference product (17%). For adalimumab biosimilars, rheumatologists said that interchangeability – a regulatory designation where a biosimilar can be automatically substituted for its reference product at the pharmacy – and citrate-free formulation were the most important product attributes. Sixty-four percent of providers also noted that patient out-of-pocket cost would be key when deciding to prescribe an adalimumab biosimilar.
“There needs to be a true reduction in price, to change providers’ opinions on the economic benefits of biosimilars – in the system generally and for the patient,” Dr. Snow said. “Things will get there eventually, but it is not there yet, based on the list prices we see for some biosimilars.”
Gastroenterologists emphasize patient education
Gastroenterology is another specialty to be affected by the influx of adalimumab biosimilars. Out of 72 surveyed gastroenterologists, 86% said they were very comfortable prescribing biosimilars. About half (49%) said they would be most likely to prescribe a biosimilar to patients with health plans mandating a biosimilar. More than 60% of surveyed gastroenterologists said that biosimilars would positively impact care; providers were divided on the current economic benefits of biosimilars, with 36% saying that the current discounts on biosimilars versus reference products were not favorable enough to motivate switching, and 35% stating that they were. A total of 40% of surveyed providers said that savings of 21%-30%, compared with savings of a reference product, would motivate them to switch patients to a biosimilar, with all other clinical factors being equal.
Gastroenterologists said that, along with the efficacy and cost savings of biosimilars, providing patient education (18%) was a top concern when prescribing biosimilars. Eighty-four percent of respondents said that educating patients about biosimilars as safe and effective treatment options was at least somewhat important. Nearly all participants (99%) cited device ease-of-use as at least somewhat important when considering prescribing adalimumab biosimilars, in addition to interchangeability (97%) and citrate-free formulation (93%).
“Despite general acceptance of biosimilars, there remains some uncertainty regarding their place in the current gastroenterology landscape,” wrote Vivek Kaul, MD, a professor of medicine at the University of Rochester (N.Y.) Medical Center, in the report. “This is likely because only half of the survey respondents believed that biosimilars will positively impact gastroenterology care, further highlighting the ongoing need for real-world data and incorporation of biosimilar use and interchangeability into clinical guidelines.”
Few dermatologists currently prescribe biosimilars
Eight out of ten dermatologists reported being at least somewhat comfortable prescribing biosimilars to patients, though fewer than 20% said they had prescribed a biosimilar in the past year. This indicates limited adoption of infliximab biosimilars, which were the only biosimilars with a dermatologic indication available in 2022, Alex Gross, MD, a dermatologist in Cumming, Ga., noted in his featured commentary in the report. Just 15% of respondents disagreed that biosimilars would have a positive impact on care, and 41% said they were excited about new biosimilars becoming available.
About half (47%) of dermatologists thought the economic benefits of biosimilars were not strong enough to motivate switching patients from reference products. Twenty-nine percent of respondents said that discounts of 21%-30% from a reference product would motivate them to switch patients to a biosimilar, with all other clinical factors being equal, while 20% said they were not likely to prescribe a biosimilar regardless of savings.
Dermatologists may be concerned that these cost savings may not be passed onto patients, said Alison Ehrlich, MD, a dermatologist in Washington, in an email to this news organization. Patient out-of-pocket cost savings would need to be “both significant and transparent” to begin to change providers’ minds, she noted.
Biosimilar efficacy was a top concern for 48% of dermatologists, while 13% said their main concern around prescribing biosimilars was lack of payer adoption. At least 95% of providers said that device ease-of-use and interchangeability were the most important attributes when considering adalimumab biosimilars. Nearly two-thirds (65%) reported that patient out-of-pocket cost would be key when deciding to prescribe an adalimumab biosimilar.
If both patients and providers are informed on biosimilar use and there are cost benefits, dermatologists’ opinions may become more favorable toward biosimilars, but that will take time, Dr. Ehrlich said. “We are very early in the game for biosimilar use in dermatology,” she added.
Ophthalmologists remain wary
Biosimilars have been relatively new to ophthalmology, with the first ranibizumab biosimilar becoming commercially available in July 2022. In the survey, 64 retina specialists were asked different questions than participants from other specialties to gauge ophthalmologists› familiarity with the biosimilars approval process and their overall comfort prescribing these medications. The primary concerns with prescribing biosimilars among respondents was payer coverage (52%), being uncomfortable with biosimilars from a clinical standpoint (48%), and administrative barriers (45%), such as prior authorization. Despite this lack of comfort with biosimilars, two-thirds of participants thought the U.S. Food and Drug Administration approval process for these medications was sufficient to evaluate their efficacy and safety. Still, fewer than half (48%) of providers said they do or would prescribe biosimilars.
George Williams, MD, a spokesperson for the American Academy of Ophthalmology, noted that the FDA approval process for biosimilars was not as rigorous as for the respective reference product, and fewer patients are followed over a shorter time period. “Since anti–[vascular endothelial growth factor (VEGF)] therapy for indications such as neovascular age-related macular degeneration continues indefinitely over years, ophthalmologists may have concerns about the long-term efficacy and safety when applied to larger real-world populations. Ophthalmologists are well aware of safety issues with VEGF inhibitors arising after FDA approval,” he told this news organization in an email.
When asked about the likelihood of using either aflibercept or ranibizumab biosimilars in their clinical practice once commercially available, 70% of ophthalmologists said they would be at least somewhat likely to prescribe aflibercept biosimilars, and 64% said they would be at least somewhat likely to prescribe ranibizumab biosimilars. About half of respondents said they would not likely switch a currently stable patient on either aflibercept or ranibizumab to the corresponding biosimilar. More than half of ophthalmologists (56%) said they would prescribe a biosimilar only if it had an interchangeability designation.
Out of all four specialties, ophthalmologists more frequently reported that higher discounts from a reference product would be necessary to consider switching a patient to a biosimilar. Currently, many ophthalmologists are comfortable with the off-label use of bevacizumab (Avastin) for treating wet age-related macular degeneration, which also offers more cost savings than any currently available biosimilar on the market, Dr. Williams said.
While the limited number of respondents makes it difficult to draw concrete conclusions, Dr. Williams emphasized that the AAO supported the use of biosimilars. “We believe that with clinical experience ophthalmic biosimilars will become useful therapeutic agents,” he noted.
A version of this article first appeared on Medscape.com.
While most providers think that biosimilars will positively impact care, few feel that the economic benefits of biosimilars to date are enough to motivate switching.
In a new survey of over 350 dermatologists, gastroenterologists, ophthalmologists, and rheumatologists, clinicians shared their opinions on the rapidly evolving landscape of biosimilars, detailing top concerns about prescribing these medications and how they presently use biosimilars in clinical practice. Across all specialties, providers said they would be most likely to prescribe biosimilars to new patients or if a patient’s health plan mandated the switch. Most providers listed concerns about biosimilar efficacy and lack of economic benefit as the main barriers to adoption of biosimilars in clinical practice.
Cardinal Health, a health care services company based in Dublin, Ohio, conducted the surveys from July through October 2022.
Rheumatologists want cost-savings for patients
2023 is gearing up to be a big year for biosimilars for inflammatory diseases, with at least eight adalimumab biosimilars entering the market in the United States. Amjevita, manufactured by Amgen, was the first to become commercially available on Jan. 31. Out of 103 surveyed rheumatologists, 62% said they were very comfortable prescribing biosimilars to patients, and 32% said they were somewhat comfortable. Providers said they would be most likely to prescribe a biosimilar to new patients (40%) or if biosimilars were mandated by a patient’s health plan (41%). Nearly one-third (31%) of rheumatologists said that a discount of 21%-30% from a reference product would be necessary to consider switching a patient to a biosimilar.
There are several reasons why a rheumatologist might be wary of switching patients to biosimilars, said Marcus Snow, MD, chair of the American College of Rheumatology’s Committee on Rheumatologic Care. “Rheumatologists will always express concern about changing medications that work well for their patients. It is not ideal to ‘force switch’ to a different product, even if it is almost identical,” he told this news organization in an email. “Also, we must remember that a patient on a biologic has failed traditional medications, which speaks to the struggle a patient must endure to get their disease under control. Fail-first situations can cause a rheumatologist to be initially resistant or hesitant to any changes.”
The top concerns among rheumatologists about prescribing biosimilars were medication efficacy (36%), lack of economic benefit (24%), and evaluating when to prescribe a biosimilar versus a reference product (17%). For adalimumab biosimilars, rheumatologists said that interchangeability – a regulatory designation where a biosimilar can be automatically substituted for its reference product at the pharmacy – and citrate-free formulation were the most important product attributes. Sixty-four percent of providers also noted that patient out-of-pocket cost would be key when deciding to prescribe an adalimumab biosimilar.
“There needs to be a true reduction in price, to change providers’ opinions on the economic benefits of biosimilars – in the system generally and for the patient,” Dr. Snow said. “Things will get there eventually, but it is not there yet, based on the list prices we see for some biosimilars.”
Gastroenterologists emphasize patient education
Gastroenterology is another specialty to be affected by the influx of adalimumab biosimilars. Out of 72 surveyed gastroenterologists, 86% said they were very comfortable prescribing biosimilars. About half (49%) said they would be most likely to prescribe a biosimilar to patients with health plans mandating a biosimilar. More than 60% of surveyed gastroenterologists said that biosimilars would positively impact care; providers were divided on the current economic benefits of biosimilars, with 36% saying that the current discounts on biosimilars versus reference products were not favorable enough to motivate switching, and 35% stating that they were. A total of 40% of surveyed providers said that savings of 21%-30%, compared with savings of a reference product, would motivate them to switch patients to a biosimilar, with all other clinical factors being equal.
Gastroenterologists said that, along with the efficacy and cost savings of biosimilars, providing patient education (18%) was a top concern when prescribing biosimilars. Eighty-four percent of respondents said that educating patients about biosimilars as safe and effective treatment options was at least somewhat important. Nearly all participants (99%) cited device ease-of-use as at least somewhat important when considering prescribing adalimumab biosimilars, in addition to interchangeability (97%) and citrate-free formulation (93%).
“Despite general acceptance of biosimilars, there remains some uncertainty regarding their place in the current gastroenterology landscape,” wrote Vivek Kaul, MD, a professor of medicine at the University of Rochester (N.Y.) Medical Center, in the report. “This is likely because only half of the survey respondents believed that biosimilars will positively impact gastroenterology care, further highlighting the ongoing need for real-world data and incorporation of biosimilar use and interchangeability into clinical guidelines.”
Few dermatologists currently prescribe biosimilars
Eight out of ten dermatologists reported being at least somewhat comfortable prescribing biosimilars to patients, though fewer than 20% said they had prescribed a biosimilar in the past year. This indicates limited adoption of infliximab biosimilars, which were the only biosimilars with a dermatologic indication available in 2022, Alex Gross, MD, a dermatologist in Cumming, Ga., noted in his featured commentary in the report. Just 15% of respondents disagreed that biosimilars would have a positive impact on care, and 41% said they were excited about new biosimilars becoming available.
About half (47%) of dermatologists thought the economic benefits of biosimilars were not strong enough to motivate switching patients from reference products. Twenty-nine percent of respondents said that discounts of 21%-30% from a reference product would motivate them to switch patients to a biosimilar, with all other clinical factors being equal, while 20% said they were not likely to prescribe a biosimilar regardless of savings.
Dermatologists may be concerned that these cost savings may not be passed onto patients, said Alison Ehrlich, MD, a dermatologist in Washington, in an email to this news organization. Patient out-of-pocket cost savings would need to be “both significant and transparent” to begin to change providers’ minds, she noted.
Biosimilar efficacy was a top concern for 48% of dermatologists, while 13% said their main concern around prescribing biosimilars was lack of payer adoption. At least 95% of providers said that device ease-of-use and interchangeability were the most important attributes when considering adalimumab biosimilars. Nearly two-thirds (65%) reported that patient out-of-pocket cost would be key when deciding to prescribe an adalimumab biosimilar.
If both patients and providers are informed on biosimilar use and there are cost benefits, dermatologists’ opinions may become more favorable toward biosimilars, but that will take time, Dr. Ehrlich said. “We are very early in the game for biosimilar use in dermatology,” she added.
Ophthalmologists remain wary
Biosimilars have been relatively new to ophthalmology, with the first ranibizumab biosimilar becoming commercially available in July 2022. In the survey, 64 retina specialists were asked different questions than participants from other specialties to gauge ophthalmologists› familiarity with the biosimilars approval process and their overall comfort prescribing these medications. The primary concerns with prescribing biosimilars among respondents was payer coverage (52%), being uncomfortable with biosimilars from a clinical standpoint (48%), and administrative barriers (45%), such as prior authorization. Despite this lack of comfort with biosimilars, two-thirds of participants thought the U.S. Food and Drug Administration approval process for these medications was sufficient to evaluate their efficacy and safety. Still, fewer than half (48%) of providers said they do or would prescribe biosimilars.
George Williams, MD, a spokesperson for the American Academy of Ophthalmology, noted that the FDA approval process for biosimilars was not as rigorous as for the respective reference product, and fewer patients are followed over a shorter time period. “Since anti–[vascular endothelial growth factor (VEGF)] therapy for indications such as neovascular age-related macular degeneration continues indefinitely over years, ophthalmologists may have concerns about the long-term efficacy and safety when applied to larger real-world populations. Ophthalmologists are well aware of safety issues with VEGF inhibitors arising after FDA approval,” he told this news organization in an email.
When asked about the likelihood of using either aflibercept or ranibizumab biosimilars in their clinical practice once commercially available, 70% of ophthalmologists said they would be at least somewhat likely to prescribe aflibercept biosimilars, and 64% said they would be at least somewhat likely to prescribe ranibizumab biosimilars. About half of respondents said they would not likely switch a currently stable patient on either aflibercept or ranibizumab to the corresponding biosimilar. More than half of ophthalmologists (56%) said they would prescribe a biosimilar only if it had an interchangeability designation.
Out of all four specialties, ophthalmologists more frequently reported that higher discounts from a reference product would be necessary to consider switching a patient to a biosimilar. Currently, many ophthalmologists are comfortable with the off-label use of bevacizumab (Avastin) for treating wet age-related macular degeneration, which also offers more cost savings than any currently available biosimilar on the market, Dr. Williams said.
While the limited number of respondents makes it difficult to draw concrete conclusions, Dr. Williams emphasized that the AAO supported the use of biosimilars. “We believe that with clinical experience ophthalmic biosimilars will become useful therapeutic agents,” he noted.
A version of this article first appeared on Medscape.com.
While most providers think that biosimilars will positively impact care, few feel that the economic benefits of biosimilars to date are enough to motivate switching.
In a new survey of over 350 dermatologists, gastroenterologists, ophthalmologists, and rheumatologists, clinicians shared their opinions on the rapidly evolving landscape of biosimilars, detailing top concerns about prescribing these medications and how they presently use biosimilars in clinical practice. Across all specialties, providers said they would be most likely to prescribe biosimilars to new patients or if a patient’s health plan mandated the switch. Most providers listed concerns about biosimilar efficacy and lack of economic benefit as the main barriers to adoption of biosimilars in clinical practice.
Cardinal Health, a health care services company based in Dublin, Ohio, conducted the surveys from July through October 2022.
Rheumatologists want cost-savings for patients
2023 is gearing up to be a big year for biosimilars for inflammatory diseases, with at least eight adalimumab biosimilars entering the market in the United States. Amjevita, manufactured by Amgen, was the first to become commercially available on Jan. 31. Out of 103 surveyed rheumatologists, 62% said they were very comfortable prescribing biosimilars to patients, and 32% said they were somewhat comfortable. Providers said they would be most likely to prescribe a biosimilar to new patients (40%) or if biosimilars were mandated by a patient’s health plan (41%). Nearly one-third (31%) of rheumatologists said that a discount of 21%-30% from a reference product would be necessary to consider switching a patient to a biosimilar.
There are several reasons why a rheumatologist might be wary of switching patients to biosimilars, said Marcus Snow, MD, chair of the American College of Rheumatology’s Committee on Rheumatologic Care. “Rheumatologists will always express concern about changing medications that work well for their patients. It is not ideal to ‘force switch’ to a different product, even if it is almost identical,” he told this news organization in an email. “Also, we must remember that a patient on a biologic has failed traditional medications, which speaks to the struggle a patient must endure to get their disease under control. Fail-first situations can cause a rheumatologist to be initially resistant or hesitant to any changes.”
The top concerns among rheumatologists about prescribing biosimilars were medication efficacy (36%), lack of economic benefit (24%), and evaluating when to prescribe a biosimilar versus a reference product (17%). For adalimumab biosimilars, rheumatologists said that interchangeability – a regulatory designation where a biosimilar can be automatically substituted for its reference product at the pharmacy – and citrate-free formulation were the most important product attributes. Sixty-four percent of providers also noted that patient out-of-pocket cost would be key when deciding to prescribe an adalimumab biosimilar.
“There needs to be a true reduction in price, to change providers’ opinions on the economic benefits of biosimilars – in the system generally and for the patient,” Dr. Snow said. “Things will get there eventually, but it is not there yet, based on the list prices we see for some biosimilars.”
Gastroenterologists emphasize patient education
Gastroenterology is another specialty to be affected by the influx of adalimumab biosimilars. Out of 72 surveyed gastroenterologists, 86% said they were very comfortable prescribing biosimilars. About half (49%) said they would be most likely to prescribe a biosimilar to patients with health plans mandating a biosimilar. More than 60% of surveyed gastroenterologists said that biosimilars would positively impact care; providers were divided on the current economic benefits of biosimilars, with 36% saying that the current discounts on biosimilars versus reference products were not favorable enough to motivate switching, and 35% stating that they were. A total of 40% of surveyed providers said that savings of 21%-30%, compared with savings of a reference product, would motivate them to switch patients to a biosimilar, with all other clinical factors being equal.
Gastroenterologists said that, along with the efficacy and cost savings of biosimilars, providing patient education (18%) was a top concern when prescribing biosimilars. Eighty-four percent of respondents said that educating patients about biosimilars as safe and effective treatment options was at least somewhat important. Nearly all participants (99%) cited device ease-of-use as at least somewhat important when considering prescribing adalimumab biosimilars, in addition to interchangeability (97%) and citrate-free formulation (93%).
“Despite general acceptance of biosimilars, there remains some uncertainty regarding their place in the current gastroenterology landscape,” wrote Vivek Kaul, MD, a professor of medicine at the University of Rochester (N.Y.) Medical Center, in the report. “This is likely because only half of the survey respondents believed that biosimilars will positively impact gastroenterology care, further highlighting the ongoing need for real-world data and incorporation of biosimilar use and interchangeability into clinical guidelines.”
Few dermatologists currently prescribe biosimilars
Eight out of ten dermatologists reported being at least somewhat comfortable prescribing biosimilars to patients, though fewer than 20% said they had prescribed a biosimilar in the past year. This indicates limited adoption of infliximab biosimilars, which were the only biosimilars with a dermatologic indication available in 2022, Alex Gross, MD, a dermatologist in Cumming, Ga., noted in his featured commentary in the report. Just 15% of respondents disagreed that biosimilars would have a positive impact on care, and 41% said they were excited about new biosimilars becoming available.
About half (47%) of dermatologists thought the economic benefits of biosimilars were not strong enough to motivate switching patients from reference products. Twenty-nine percent of respondents said that discounts of 21%-30% from a reference product would motivate them to switch patients to a biosimilar, with all other clinical factors being equal, while 20% said they were not likely to prescribe a biosimilar regardless of savings.
Dermatologists may be concerned that these cost savings may not be passed onto patients, said Alison Ehrlich, MD, a dermatologist in Washington, in an email to this news organization. Patient out-of-pocket cost savings would need to be “both significant and transparent” to begin to change providers’ minds, she noted.
Biosimilar efficacy was a top concern for 48% of dermatologists, while 13% said their main concern around prescribing biosimilars was lack of payer adoption. At least 95% of providers said that device ease-of-use and interchangeability were the most important attributes when considering adalimumab biosimilars. Nearly two-thirds (65%) reported that patient out-of-pocket cost would be key when deciding to prescribe an adalimumab biosimilar.
If both patients and providers are informed on biosimilar use and there are cost benefits, dermatologists’ opinions may become more favorable toward biosimilars, but that will take time, Dr. Ehrlich said. “We are very early in the game for biosimilar use in dermatology,” she added.
Ophthalmologists remain wary
Biosimilars have been relatively new to ophthalmology, with the first ranibizumab biosimilar becoming commercially available in July 2022. In the survey, 64 retina specialists were asked different questions than participants from other specialties to gauge ophthalmologists› familiarity with the biosimilars approval process and their overall comfort prescribing these medications. The primary concerns with prescribing biosimilars among respondents was payer coverage (52%), being uncomfortable with biosimilars from a clinical standpoint (48%), and administrative barriers (45%), such as prior authorization. Despite this lack of comfort with biosimilars, two-thirds of participants thought the U.S. Food and Drug Administration approval process for these medications was sufficient to evaluate their efficacy and safety. Still, fewer than half (48%) of providers said they do or would prescribe biosimilars.
George Williams, MD, a spokesperson for the American Academy of Ophthalmology, noted that the FDA approval process for biosimilars was not as rigorous as for the respective reference product, and fewer patients are followed over a shorter time period. “Since anti–[vascular endothelial growth factor (VEGF)] therapy for indications such as neovascular age-related macular degeneration continues indefinitely over years, ophthalmologists may have concerns about the long-term efficacy and safety when applied to larger real-world populations. Ophthalmologists are well aware of safety issues with VEGF inhibitors arising after FDA approval,” he told this news organization in an email.
When asked about the likelihood of using either aflibercept or ranibizumab biosimilars in their clinical practice once commercially available, 70% of ophthalmologists said they would be at least somewhat likely to prescribe aflibercept biosimilars, and 64% said they would be at least somewhat likely to prescribe ranibizumab biosimilars. About half of respondents said they would not likely switch a currently stable patient on either aflibercept or ranibizumab to the corresponding biosimilar. More than half of ophthalmologists (56%) said they would prescribe a biosimilar only if it had an interchangeability designation.
Out of all four specialties, ophthalmologists more frequently reported that higher discounts from a reference product would be necessary to consider switching a patient to a biosimilar. Currently, many ophthalmologists are comfortable with the off-label use of bevacizumab (Avastin) for treating wet age-related macular degeneration, which also offers more cost savings than any currently available biosimilar on the market, Dr. Williams said.
While the limited number of respondents makes it difficult to draw concrete conclusions, Dr. Williams emphasized that the AAO supported the use of biosimilars. “We believe that with clinical experience ophthalmic biosimilars will become useful therapeutic agents,” he noted.
A version of this article first appeared on Medscape.com.
Expert offers caveats to perioperative antirheumatic drug guideline
The latest guideline for perioperative management of antirheumatic medication in patients undergoing total hip (THA) and total knee arthroplasty (TKA) offers recommendations based on the latest evidence, but many of those recommendations are based on a low level of evidence, according to a speaker at the 2023 Rheumatology Winter Clinical Symposium.
Martin Bergman, MD, clinical professor of medicine at Drexel University, Philadelphia, said the development of the American College of Rheumatology/American Association of Hip and Knee Surgeons guideline was necessary because there was a lack of consensus on when to stop treatments prior to patients with rheumatologic disease undergoing THA and TKA, and when it was appropriate to restart those treatments.
“We all were having the same problem, and I think everybody recognized that just stopping medicines forever didn’t make sense, but maybe continuing medicines also didn’t make sense,” Dr. Bergman said.
While the 2017 ACR/AAHKS perioperative management guideline contained good recommendations, the “explosion” of new medications in rheumatology made it necessary to update the guideline with the latest data on new medications such as immunosuppressants.
2022 guideline recommendations
In the 2022 guideline, which covers disease-modifying treatments taken by patients with rheumatoid arthritis, spondyloarthritis, and psoriatic arthritis, the authors reaffirmed their recommendations to continue methotrexate, sulfasalazine, hydroxychloroquine, leflunomide, and apremilast through total joint arthroplasty.
Where the 2022 guideline differs from the 2017 guideline is in which biologics are covered and under what circumstances they should be withheld and restarted around surgery. The 2022 guideline includes recommendations for abatacept, adalimumab, anakinra, certolizumab pegol, etanercept, golimumab, guselkumab, infliximab, ixekizumab, rituximab, secukinumab, tocilizumab, and ustekinumab. Each biologic has its own recommended stop and restart times based around the dosing interval and respective method of administration. Dr. Bergman said a general rule with biologics under the new guideline is that the timing of surgery should occur approximately 1 week after the first missed dose of the medication. The only biologic that does not follow this pattern is rituximab, where surgery should be planned for 1 month after the last missed dose.
Dr. Bergman noted that how the guidelines handle interval dosing with infliximab may present a problem. The guideline provides recommendations for patients receiving infliximab every 4 weeks, every 6 weeks, and every 8 weeks. However, Dr. Bergman said this can create a scenario where a patient receiving infliximab at a dose of 3 mg/kg every 8 weeks has surgery at 9 weeks, a patient receiving 5 mg/kg every 6 weeks has surgery at 7 weeks, and a patient receiving 10 mg/kg every 4 weeks has surgery at 5 weeks. “There is some intellectual problem with it,” he said.
Another change from the 2017 guideline is how long to wait for surgery after stopping Janus kinase inhibitors. While the 2017 guideline recommended withholding JAK inhibitors 7 days before surgery, the 2022 guideline lowered that waiting period to 3 days, Dr. Bergman explained.
Concerning use of steroids around THA and TKA surgery, “the days of stress steroid dosing are done,” Dr. Bergman said. “You don’t have to stress dose them. You just follow them, and you keep them on their steroid dose.”
The new guideline recommends restarting therapy once the wound is healed and there is no physical evidence of infection at approximately 2 weeks. “There’s no data to support this,” he said, and his concern is that patients who have stopped a tumor necrosis factor inhibitor may flare if they don’t restart their medication.
While the guideline also covered recommendations for systemic lupus erythematosus, they are “very similar” to the recommendations for inflammatory arthritis, Dr. Bergman noted. “If you have somebody who is not very sick, you stop the medications,” he said, “but try to stop anything else about a week before the surgery. If they’re sick, you basically have to keep them on their medications.”
Caveats in guideline
The recommendations in the 2022 guideline come with a number of caveats, Dr. Bergman noted. For instance, the authors acknowledged limitations in the guideline regarding providing recommendations for only THA and TKA, the “paucity of evidence” around direct infection risk resulting from medications in the perioperative period for THA and TKA, the nonseparation of biologics when assessing infection risk, and the use of dosing interval as a metric for stopping the drug without considering the drug’s half-life.
A “crucial caveat,” Dr. Bergman said, was that the guideline focused on infection risk based on a statement from a panel of patients prior to the development of the 2017 guideline, which “stated very clearly any risk of infection, while rare, was more significant to them than the possibility of postoperative flares, despite flares being reported in over 60% of patients after surgery.
“For the patients, the paramount question was infection, infection, infection, infection. That’s all they cared about, and that is the basis behind a lot of the decision-making here,” Dr. Bergman said.
Another caveat came from a communication Dr. Bergman received from one of the panel members. The panel member noted there were no conclusions or recommendations provided in the guideline for how to manage perioperative flares, such as restarting a corticosteroid or biologic agent. “There was a lot of discussion about what to do with steroids if patients flare, or what to do with [other] medications if they flare, and they just couldn’t come to a consensus,” Dr. Bergman said. “It’s just not discussed.”
Dr. Bergman said he is “somewhat critical” of the ACR/AAHKS guideline, but noted it is an “ambitious project” given the lack of evidence for the recommendations. “The alternative was stop the medications forever and having people really flare, or at least try to get some semblance of rationality behind what we’re going to do,” he said.
Response from attendees
Jack Cush, MD, a rheumatologist based in Dallas and executive editor of RheumNow.com, took issue with the new recommendations surrounding stopping infliximab. When giving a patient infliximab every 8 weeks at 3 mg/kg, “you’re giving [it] at the nadir of the drug,” he said.
Rather than drug half-life, “it’s about inflammation,” he emphasized. “Inflammation is dominant in causing infection. It drives risk more than anything. The worst thing you can do is wash someone out.
“If you’re going beyond 8 weeks on infliximab, you’re getting closer to washing them out,” he pointed out. “I think it’s a really bad idea.”
Allan Gibofsky, MD, JD, professor of medicine at Weill Cornell Medicine and codirector of the Clinic for Inflammatory Arthritis and Biologic Therapy at Hospital for Special Surgery, both in New York, explained that the guideline is not standard of care, which would be subject to malpractice if not implemented properly.
“When you have guidelines, you follow them unless there are clinical situations which would necessitate another approach to the patient,” he said. “Professional institutions and associations will never put forth rules, they will put forth guidelines so you have the opportunity to deviate from them when the appropriate clinical situation dictates.”
Dr. Bergman reported being a speaker and consultant for AbbVie, Amgen, Bristol-Myers Squibb, GlaxoSmithKline, Novartis, Pfizer, and Regeneron; he holds stock in Johnson & Johnson and Merck.
The latest guideline for perioperative management of antirheumatic medication in patients undergoing total hip (THA) and total knee arthroplasty (TKA) offers recommendations based on the latest evidence, but many of those recommendations are based on a low level of evidence, according to a speaker at the 2023 Rheumatology Winter Clinical Symposium.
Martin Bergman, MD, clinical professor of medicine at Drexel University, Philadelphia, said the development of the American College of Rheumatology/American Association of Hip and Knee Surgeons guideline was necessary because there was a lack of consensus on when to stop treatments prior to patients with rheumatologic disease undergoing THA and TKA, and when it was appropriate to restart those treatments.
“We all were having the same problem, and I think everybody recognized that just stopping medicines forever didn’t make sense, but maybe continuing medicines also didn’t make sense,” Dr. Bergman said.
While the 2017 ACR/AAHKS perioperative management guideline contained good recommendations, the “explosion” of new medications in rheumatology made it necessary to update the guideline with the latest data on new medications such as immunosuppressants.
2022 guideline recommendations
In the 2022 guideline, which covers disease-modifying treatments taken by patients with rheumatoid arthritis, spondyloarthritis, and psoriatic arthritis, the authors reaffirmed their recommendations to continue methotrexate, sulfasalazine, hydroxychloroquine, leflunomide, and apremilast through total joint arthroplasty.
Where the 2022 guideline differs from the 2017 guideline is in which biologics are covered and under what circumstances they should be withheld and restarted around surgery. The 2022 guideline includes recommendations for abatacept, adalimumab, anakinra, certolizumab pegol, etanercept, golimumab, guselkumab, infliximab, ixekizumab, rituximab, secukinumab, tocilizumab, and ustekinumab. Each biologic has its own recommended stop and restart times based around the dosing interval and respective method of administration. Dr. Bergman said a general rule with biologics under the new guideline is that the timing of surgery should occur approximately 1 week after the first missed dose of the medication. The only biologic that does not follow this pattern is rituximab, where surgery should be planned for 1 month after the last missed dose.
Dr. Bergman noted that how the guidelines handle interval dosing with infliximab may present a problem. The guideline provides recommendations for patients receiving infliximab every 4 weeks, every 6 weeks, and every 8 weeks. However, Dr. Bergman said this can create a scenario where a patient receiving infliximab at a dose of 3 mg/kg every 8 weeks has surgery at 9 weeks, a patient receiving 5 mg/kg every 6 weeks has surgery at 7 weeks, and a patient receiving 10 mg/kg every 4 weeks has surgery at 5 weeks. “There is some intellectual problem with it,” he said.
Another change from the 2017 guideline is how long to wait for surgery after stopping Janus kinase inhibitors. While the 2017 guideline recommended withholding JAK inhibitors 7 days before surgery, the 2022 guideline lowered that waiting period to 3 days, Dr. Bergman explained.
Concerning use of steroids around THA and TKA surgery, “the days of stress steroid dosing are done,” Dr. Bergman said. “You don’t have to stress dose them. You just follow them, and you keep them on their steroid dose.”
The new guideline recommends restarting therapy once the wound is healed and there is no physical evidence of infection at approximately 2 weeks. “There’s no data to support this,” he said, and his concern is that patients who have stopped a tumor necrosis factor inhibitor may flare if they don’t restart their medication.
While the guideline also covered recommendations for systemic lupus erythematosus, they are “very similar” to the recommendations for inflammatory arthritis, Dr. Bergman noted. “If you have somebody who is not very sick, you stop the medications,” he said, “but try to stop anything else about a week before the surgery. If they’re sick, you basically have to keep them on their medications.”
Caveats in guideline
The recommendations in the 2022 guideline come with a number of caveats, Dr. Bergman noted. For instance, the authors acknowledged limitations in the guideline regarding providing recommendations for only THA and TKA, the “paucity of evidence” around direct infection risk resulting from medications in the perioperative period for THA and TKA, the nonseparation of biologics when assessing infection risk, and the use of dosing interval as a metric for stopping the drug without considering the drug’s half-life.
A “crucial caveat,” Dr. Bergman said, was that the guideline focused on infection risk based on a statement from a panel of patients prior to the development of the 2017 guideline, which “stated very clearly any risk of infection, while rare, was more significant to them than the possibility of postoperative flares, despite flares being reported in over 60% of patients after surgery.
“For the patients, the paramount question was infection, infection, infection, infection. That’s all they cared about, and that is the basis behind a lot of the decision-making here,” Dr. Bergman said.
Another caveat came from a communication Dr. Bergman received from one of the panel members. The panel member noted there were no conclusions or recommendations provided in the guideline for how to manage perioperative flares, such as restarting a corticosteroid or biologic agent. “There was a lot of discussion about what to do with steroids if patients flare, or what to do with [other] medications if they flare, and they just couldn’t come to a consensus,” Dr. Bergman said. “It’s just not discussed.”
Dr. Bergman said he is “somewhat critical” of the ACR/AAHKS guideline, but noted it is an “ambitious project” given the lack of evidence for the recommendations. “The alternative was stop the medications forever and having people really flare, or at least try to get some semblance of rationality behind what we’re going to do,” he said.
Response from attendees
Jack Cush, MD, a rheumatologist based in Dallas and executive editor of RheumNow.com, took issue with the new recommendations surrounding stopping infliximab. When giving a patient infliximab every 8 weeks at 3 mg/kg, “you’re giving [it] at the nadir of the drug,” he said.
Rather than drug half-life, “it’s about inflammation,” he emphasized. “Inflammation is dominant in causing infection. It drives risk more than anything. The worst thing you can do is wash someone out.
“If you’re going beyond 8 weeks on infliximab, you’re getting closer to washing them out,” he pointed out. “I think it’s a really bad idea.”
Allan Gibofsky, MD, JD, professor of medicine at Weill Cornell Medicine and codirector of the Clinic for Inflammatory Arthritis and Biologic Therapy at Hospital for Special Surgery, both in New York, explained that the guideline is not standard of care, which would be subject to malpractice if not implemented properly.
“When you have guidelines, you follow them unless there are clinical situations which would necessitate another approach to the patient,” he said. “Professional institutions and associations will never put forth rules, they will put forth guidelines so you have the opportunity to deviate from them when the appropriate clinical situation dictates.”
Dr. Bergman reported being a speaker and consultant for AbbVie, Amgen, Bristol-Myers Squibb, GlaxoSmithKline, Novartis, Pfizer, and Regeneron; he holds stock in Johnson & Johnson and Merck.
The latest guideline for perioperative management of antirheumatic medication in patients undergoing total hip (THA) and total knee arthroplasty (TKA) offers recommendations based on the latest evidence, but many of those recommendations are based on a low level of evidence, according to a speaker at the 2023 Rheumatology Winter Clinical Symposium.
Martin Bergman, MD, clinical professor of medicine at Drexel University, Philadelphia, said the development of the American College of Rheumatology/American Association of Hip and Knee Surgeons guideline was necessary because there was a lack of consensus on when to stop treatments prior to patients with rheumatologic disease undergoing THA and TKA, and when it was appropriate to restart those treatments.
“We all were having the same problem, and I think everybody recognized that just stopping medicines forever didn’t make sense, but maybe continuing medicines also didn’t make sense,” Dr. Bergman said.
While the 2017 ACR/AAHKS perioperative management guideline contained good recommendations, the “explosion” of new medications in rheumatology made it necessary to update the guideline with the latest data on new medications such as immunosuppressants.
2022 guideline recommendations
In the 2022 guideline, which covers disease-modifying treatments taken by patients with rheumatoid arthritis, spondyloarthritis, and psoriatic arthritis, the authors reaffirmed their recommendations to continue methotrexate, sulfasalazine, hydroxychloroquine, leflunomide, and apremilast through total joint arthroplasty.
Where the 2022 guideline differs from the 2017 guideline is in which biologics are covered and under what circumstances they should be withheld and restarted around surgery. The 2022 guideline includes recommendations for abatacept, adalimumab, anakinra, certolizumab pegol, etanercept, golimumab, guselkumab, infliximab, ixekizumab, rituximab, secukinumab, tocilizumab, and ustekinumab. Each biologic has its own recommended stop and restart times based around the dosing interval and respective method of administration. Dr. Bergman said a general rule with biologics under the new guideline is that the timing of surgery should occur approximately 1 week after the first missed dose of the medication. The only biologic that does not follow this pattern is rituximab, where surgery should be planned for 1 month after the last missed dose.
Dr. Bergman noted that how the guidelines handle interval dosing with infliximab may present a problem. The guideline provides recommendations for patients receiving infliximab every 4 weeks, every 6 weeks, and every 8 weeks. However, Dr. Bergman said this can create a scenario where a patient receiving infliximab at a dose of 3 mg/kg every 8 weeks has surgery at 9 weeks, a patient receiving 5 mg/kg every 6 weeks has surgery at 7 weeks, and a patient receiving 10 mg/kg every 4 weeks has surgery at 5 weeks. “There is some intellectual problem with it,” he said.
Another change from the 2017 guideline is how long to wait for surgery after stopping Janus kinase inhibitors. While the 2017 guideline recommended withholding JAK inhibitors 7 days before surgery, the 2022 guideline lowered that waiting period to 3 days, Dr. Bergman explained.
Concerning use of steroids around THA and TKA surgery, “the days of stress steroid dosing are done,” Dr. Bergman said. “You don’t have to stress dose them. You just follow them, and you keep them on their steroid dose.”
The new guideline recommends restarting therapy once the wound is healed and there is no physical evidence of infection at approximately 2 weeks. “There’s no data to support this,” he said, and his concern is that patients who have stopped a tumor necrosis factor inhibitor may flare if they don’t restart their medication.
While the guideline also covered recommendations for systemic lupus erythematosus, they are “very similar” to the recommendations for inflammatory arthritis, Dr. Bergman noted. “If you have somebody who is not very sick, you stop the medications,” he said, “but try to stop anything else about a week before the surgery. If they’re sick, you basically have to keep them on their medications.”
Caveats in guideline
The recommendations in the 2022 guideline come with a number of caveats, Dr. Bergman noted. For instance, the authors acknowledged limitations in the guideline regarding providing recommendations for only THA and TKA, the “paucity of evidence” around direct infection risk resulting from medications in the perioperative period for THA and TKA, the nonseparation of biologics when assessing infection risk, and the use of dosing interval as a metric for stopping the drug without considering the drug’s half-life.
A “crucial caveat,” Dr. Bergman said, was that the guideline focused on infection risk based on a statement from a panel of patients prior to the development of the 2017 guideline, which “stated very clearly any risk of infection, while rare, was more significant to them than the possibility of postoperative flares, despite flares being reported in over 60% of patients after surgery.
“For the patients, the paramount question was infection, infection, infection, infection. That’s all they cared about, and that is the basis behind a lot of the decision-making here,” Dr. Bergman said.
Another caveat came from a communication Dr. Bergman received from one of the panel members. The panel member noted there were no conclusions or recommendations provided in the guideline for how to manage perioperative flares, such as restarting a corticosteroid or biologic agent. “There was a lot of discussion about what to do with steroids if patients flare, or what to do with [other] medications if they flare, and they just couldn’t come to a consensus,” Dr. Bergman said. “It’s just not discussed.”
Dr. Bergman said he is “somewhat critical” of the ACR/AAHKS guideline, but noted it is an “ambitious project” given the lack of evidence for the recommendations. “The alternative was stop the medications forever and having people really flare, or at least try to get some semblance of rationality behind what we’re going to do,” he said.
Response from attendees
Jack Cush, MD, a rheumatologist based in Dallas and executive editor of RheumNow.com, took issue with the new recommendations surrounding stopping infliximab. When giving a patient infliximab every 8 weeks at 3 mg/kg, “you’re giving [it] at the nadir of the drug,” he said.
Rather than drug half-life, “it’s about inflammation,” he emphasized. “Inflammation is dominant in causing infection. It drives risk more than anything. The worst thing you can do is wash someone out.
“If you’re going beyond 8 weeks on infliximab, you’re getting closer to washing them out,” he pointed out. “I think it’s a really bad idea.”
Allan Gibofsky, MD, JD, professor of medicine at Weill Cornell Medicine and codirector of the Clinic for Inflammatory Arthritis and Biologic Therapy at Hospital for Special Surgery, both in New York, explained that the guideline is not standard of care, which would be subject to malpractice if not implemented properly.
“When you have guidelines, you follow them unless there are clinical situations which would necessitate another approach to the patient,” he said. “Professional institutions and associations will never put forth rules, they will put forth guidelines so you have the opportunity to deviate from them when the appropriate clinical situation dictates.”
Dr. Bergman reported being a speaker and consultant for AbbVie, Amgen, Bristol-Myers Squibb, GlaxoSmithKline, Novartis, Pfizer, and Regeneron; he holds stock in Johnson & Johnson and Merck.
FROM RWCS 2023
How to recognize and treat hidden inflammation
“This then leads to inflammation, the healing of which the body is unable to keep under control,” explained Ulf Müller-Ladner, MD, PhD, chairperson of the German Society of Internal Medicine.
At the DGIM annual press conference, Dr. Müller-Ladner, who is also director of the department of rheumatology and clinical immunology at the Kerckhoff Clinic in Bad Nauheim, Germany, explained how IgG4 inflammation is triggered throughout the body and what therapeutic options are available.
Many manifestations
IgG4-associated inflammation can affect one or more organs or the surrounding connective tissue and cause fibrosis. As a result of fibrosis, the organ gradually loses function and is eventually transformed completely into scarred connective tissue.
“In the case of IgG4-associated inflammation, these fibroses have a histological structure, but extracting a sample is not possible from every affected organ,” said Dr. Müller-Ladner. Liver, bile ducts, blood vessels, skin, eyes, or even the central nervous system – practically every organ system can be affected by these inflammatory reactions.
IgG4-associated diseases have likely been around for some time, but it is only in the past 10 years that awareness has grown that, despite various manifestations, “they are all one and the same disease,” said Dr. Müller-Ladner.
IgG4-associated chronic, inflammatory, fibrosing diseases were only classified together as a single entity in the past few years. In terms of pathophysiology, B lymphocytes, IgG4-positive plasma cells, follicular T-helper cells, cytotoxic CD4-positive T cells, and macrophages work together and trigger an inflammatory reaction, which then encourages fibroblasts to overproduce connective tissue.
Beware inexplicable inflammation
It is estimated that 1 in 100,000 people suffer from the disease, but the number of incorrectly categorized patients may be significantly higher.
The diagnostic challenge lies in the fact that IgG4-associated inflammation occurs in almost every organ. It can cause different symptoms, depending on the organ affected.
Dr. Müller-Ladner provided the following take-home message: “Every inexplicable inflammation event and every organ dysfunction, especially if associated with an increase in connective tissue, could be an IgG4-associated disease. Keeping this in mind is the key to recovery.”
With most people, the inflammation persists for many years before any symptoms of the disease develop. Highly acute courses of progression are also possible.
Classic symptoms, such as fever, are not so characteristic of the latent inflammatory reaction, and according to classification criteria published by specialist rheumatology societies, they are an exclusion criterion. This is true with respect to the differential diagnosis for vasculitis, which also occurs throughout the body.
Histology is key
Blood levels of IgG4 and imaging are not always enough to confirm the diagnosis. In such cases, the histology is often a crucial factor in making a definitive diagnosis. Dominant organs in IgG4-associated diseases are the pancreas, the liver, the gallbladder, the intestines, the retroperitoneum, large blood vessels, the kidneys, the heart, the brain, saliva, tear ducts, as well all of the body’s connective tissue.
The kidneys play host to inflammation in the connective tissue and space-occupying masses in particular. “If the pancreas is affected, the signs can vary from diffuse swelling to the onset of diabetes mellitus. In contrast, if the aorta is affected, then the inflammation is characterized through a thickening of the vessel walls, aneurysms, and the corresponding circulation disorders,” said Dr. Müller-Ladner.
Because of the long period before the diagnosis is made, more than 50% of patients exhibit irreversible organ damage at the time of diagnosis, he added.
Glucocorticoids and immunosuppressants
Despite therapeutic intervention, the disease can have a fatal outcome, even if the patient is young, said Dr. Müller-Ladner. Glucocorticoids are the current therapy of choice. The dose is more than 0.5 mg of prednisolone equivalent per kg of body weight. “This usually leads to a rapid improvement in the inflammation. Subsequently, every organ is thoroughly diagnosed to assess the severity of the disease and to plan further treatment steps.”
In the long term, proven immunosuppressants, such as azathioprine, mycophenolate, leflunomide, and methotrexate, can be used, just as for many other chronic inflammatory diseases. Cyclophosphamide or cyclosporine is used more rarely, owing to their side effect profiles.
Because of the B-cell dominance, B-cell–depleting therapy with rituximab is currently a highly effective therapeutic option but one that must be applied for, because such use is off label. “If the body responds well to the medication, organ function often recovers,” said Dr. Müller-Ladner.
This article was translated from the Medscape German edition. A version appeared on Medscape.com.
“This then leads to inflammation, the healing of which the body is unable to keep under control,” explained Ulf Müller-Ladner, MD, PhD, chairperson of the German Society of Internal Medicine.
At the DGIM annual press conference, Dr. Müller-Ladner, who is also director of the department of rheumatology and clinical immunology at the Kerckhoff Clinic in Bad Nauheim, Germany, explained how IgG4 inflammation is triggered throughout the body and what therapeutic options are available.
Many manifestations
IgG4-associated inflammation can affect one or more organs or the surrounding connective tissue and cause fibrosis. As a result of fibrosis, the organ gradually loses function and is eventually transformed completely into scarred connective tissue.
“In the case of IgG4-associated inflammation, these fibroses have a histological structure, but extracting a sample is not possible from every affected organ,” said Dr. Müller-Ladner. Liver, bile ducts, blood vessels, skin, eyes, or even the central nervous system – practically every organ system can be affected by these inflammatory reactions.
IgG4-associated diseases have likely been around for some time, but it is only in the past 10 years that awareness has grown that, despite various manifestations, “they are all one and the same disease,” said Dr. Müller-Ladner.
IgG4-associated chronic, inflammatory, fibrosing diseases were only classified together as a single entity in the past few years. In terms of pathophysiology, B lymphocytes, IgG4-positive plasma cells, follicular T-helper cells, cytotoxic CD4-positive T cells, and macrophages work together and trigger an inflammatory reaction, which then encourages fibroblasts to overproduce connective tissue.
Beware inexplicable inflammation
It is estimated that 1 in 100,000 people suffer from the disease, but the number of incorrectly categorized patients may be significantly higher.
The diagnostic challenge lies in the fact that IgG4-associated inflammation occurs in almost every organ. It can cause different symptoms, depending on the organ affected.
Dr. Müller-Ladner provided the following take-home message: “Every inexplicable inflammation event and every organ dysfunction, especially if associated with an increase in connective tissue, could be an IgG4-associated disease. Keeping this in mind is the key to recovery.”
With most people, the inflammation persists for many years before any symptoms of the disease develop. Highly acute courses of progression are also possible.
Classic symptoms, such as fever, are not so characteristic of the latent inflammatory reaction, and according to classification criteria published by specialist rheumatology societies, they are an exclusion criterion. This is true with respect to the differential diagnosis for vasculitis, which also occurs throughout the body.
Histology is key
Blood levels of IgG4 and imaging are not always enough to confirm the diagnosis. In such cases, the histology is often a crucial factor in making a definitive diagnosis. Dominant organs in IgG4-associated diseases are the pancreas, the liver, the gallbladder, the intestines, the retroperitoneum, large blood vessels, the kidneys, the heart, the brain, saliva, tear ducts, as well all of the body’s connective tissue.
The kidneys play host to inflammation in the connective tissue and space-occupying masses in particular. “If the pancreas is affected, the signs can vary from diffuse swelling to the onset of diabetes mellitus. In contrast, if the aorta is affected, then the inflammation is characterized through a thickening of the vessel walls, aneurysms, and the corresponding circulation disorders,” said Dr. Müller-Ladner.
Because of the long period before the diagnosis is made, more than 50% of patients exhibit irreversible organ damage at the time of diagnosis, he added.
Glucocorticoids and immunosuppressants
Despite therapeutic intervention, the disease can have a fatal outcome, even if the patient is young, said Dr. Müller-Ladner. Glucocorticoids are the current therapy of choice. The dose is more than 0.5 mg of prednisolone equivalent per kg of body weight. “This usually leads to a rapid improvement in the inflammation. Subsequently, every organ is thoroughly diagnosed to assess the severity of the disease and to plan further treatment steps.”
In the long term, proven immunosuppressants, such as azathioprine, mycophenolate, leflunomide, and methotrexate, can be used, just as for many other chronic inflammatory diseases. Cyclophosphamide or cyclosporine is used more rarely, owing to their side effect profiles.
Because of the B-cell dominance, B-cell–depleting therapy with rituximab is currently a highly effective therapeutic option but one that must be applied for, because such use is off label. “If the body responds well to the medication, organ function often recovers,” said Dr. Müller-Ladner.
This article was translated from the Medscape German edition. A version appeared on Medscape.com.
“This then leads to inflammation, the healing of which the body is unable to keep under control,” explained Ulf Müller-Ladner, MD, PhD, chairperson of the German Society of Internal Medicine.
At the DGIM annual press conference, Dr. Müller-Ladner, who is also director of the department of rheumatology and clinical immunology at the Kerckhoff Clinic in Bad Nauheim, Germany, explained how IgG4 inflammation is triggered throughout the body and what therapeutic options are available.
Many manifestations
IgG4-associated inflammation can affect one or more organs or the surrounding connective tissue and cause fibrosis. As a result of fibrosis, the organ gradually loses function and is eventually transformed completely into scarred connective tissue.
“In the case of IgG4-associated inflammation, these fibroses have a histological structure, but extracting a sample is not possible from every affected organ,” said Dr. Müller-Ladner. Liver, bile ducts, blood vessels, skin, eyes, or even the central nervous system – practically every organ system can be affected by these inflammatory reactions.
IgG4-associated diseases have likely been around for some time, but it is only in the past 10 years that awareness has grown that, despite various manifestations, “they are all one and the same disease,” said Dr. Müller-Ladner.
IgG4-associated chronic, inflammatory, fibrosing diseases were only classified together as a single entity in the past few years. In terms of pathophysiology, B lymphocytes, IgG4-positive plasma cells, follicular T-helper cells, cytotoxic CD4-positive T cells, and macrophages work together and trigger an inflammatory reaction, which then encourages fibroblasts to overproduce connective tissue.
Beware inexplicable inflammation
It is estimated that 1 in 100,000 people suffer from the disease, but the number of incorrectly categorized patients may be significantly higher.
The diagnostic challenge lies in the fact that IgG4-associated inflammation occurs in almost every organ. It can cause different symptoms, depending on the organ affected.
Dr. Müller-Ladner provided the following take-home message: “Every inexplicable inflammation event and every organ dysfunction, especially if associated with an increase in connective tissue, could be an IgG4-associated disease. Keeping this in mind is the key to recovery.”
With most people, the inflammation persists for many years before any symptoms of the disease develop. Highly acute courses of progression are also possible.
Classic symptoms, such as fever, are not so characteristic of the latent inflammatory reaction, and according to classification criteria published by specialist rheumatology societies, they are an exclusion criterion. This is true with respect to the differential diagnosis for vasculitis, which also occurs throughout the body.
Histology is key
Blood levels of IgG4 and imaging are not always enough to confirm the diagnosis. In such cases, the histology is often a crucial factor in making a definitive diagnosis. Dominant organs in IgG4-associated diseases are the pancreas, the liver, the gallbladder, the intestines, the retroperitoneum, large blood vessels, the kidneys, the heart, the brain, saliva, tear ducts, as well all of the body’s connective tissue.
The kidneys play host to inflammation in the connective tissue and space-occupying masses in particular. “If the pancreas is affected, the signs can vary from diffuse swelling to the onset of diabetes mellitus. In contrast, if the aorta is affected, then the inflammation is characterized through a thickening of the vessel walls, aneurysms, and the corresponding circulation disorders,” said Dr. Müller-Ladner.
Because of the long period before the diagnosis is made, more than 50% of patients exhibit irreversible organ damage at the time of diagnosis, he added.
Glucocorticoids and immunosuppressants
Despite therapeutic intervention, the disease can have a fatal outcome, even if the patient is young, said Dr. Müller-Ladner. Glucocorticoids are the current therapy of choice. The dose is more than 0.5 mg of prednisolone equivalent per kg of body weight. “This usually leads to a rapid improvement in the inflammation. Subsequently, every organ is thoroughly diagnosed to assess the severity of the disease and to plan further treatment steps.”
In the long term, proven immunosuppressants, such as azathioprine, mycophenolate, leflunomide, and methotrexate, can be used, just as for many other chronic inflammatory diseases. Cyclophosphamide or cyclosporine is used more rarely, owing to their side effect profiles.
Because of the B-cell dominance, B-cell–depleting therapy with rituximab is currently a highly effective therapeutic option but one that must be applied for, because such use is off label. “If the body responds well to the medication, organ function often recovers,” said Dr. Müller-Ladner.
This article was translated from the Medscape German edition. A version appeared on Medscape.com.
Are repeat radiographs necessary in rheumatoid and psoriatic arthritis?
Follow-up radiographs after an initial baseline reading in patients with rheumatoid arthritis or psoriatic arthritis may still show radiographic progression despite treatment with current therapies, but it’s unclear if they will affect treatment decisions between patients and doctors that may take place regardless of the radiographic information, according to arguments made for and against their usefulness in a point-counterpoint session at the 2023 Rheumatology Winter Clinical Symposium.
Alvin Wells, MD, PhD, director of the department of rheumatology at Advocate Aurora Health in Franklin, Wisc., said that x-rays “reflect the history of joint pathology” and can get worse over time, correlating with disease activity and severity.
While RA does not yet have the “holy grail” of complete or true remission, Dr. Wells argued, the combination of clinical remission, laboratory remission, and imaging remission gets patients with RA close to the ideal when measured over time. “You need to continue to monitor these patients as you follow them along,” he said.
The BARFOT study, which evaluated 1,938 patients with early RA in two cohorts during 1992-1999 and again between 2000 and 2006, showed that more active treatments in the 2000s did not result in improvements in Health Assessment Questionnaire (HAQ) and pain scores, compared with patients treated in the 1990s. “You can see in some of those patients those scores do increase, and that even despite aggressive therapies that we had in 2006, you can still see some of those patients still have progression of the disease,” Dr. Wells explained. “How did they know? Because they looked.”

He also cited a study from researchers at the Mayo Clinic who examined 586 patients with RA that showed a higher prevalence of functional disability in patients with RA who also had radiographic changes, compared with patients without RA. “Radiographic changes correlate with disease severity and functional disability as well,” Dr. Wells said.
Just as prostate-specific antigen levels are used in prostate cancer screening and hemoglobin A1c is measured in diabetes management, radiographs should be used to track progression of disease in RA and PsA, Dr. Wells argued. “[I]f you don’t know, you can’t treat,” he said.
Some patients near remission may have radiographic progression even though disease activity measurements such as C-reactive protein (CRP) values do not show presence of active disease. In a study analyzing 1,184 patients with RA in the ASPIRE, ERA, Leflunomide, PREMIER and TEMPO trials, swollen joint count (SJC) was a better predictor of radiographic progression than CRP in patients near remission.
“[E]ven where you don’t see smoke, there still could be fire,” Dr. Wells said. “Some of these patients still progress and these are outliers, and the way they saw that [was] because they followed those patients along. If you don’t look, you don’t know.”
Radiographic progression can also be seen among nonswollen joints in patients with RA and PsA. In a study of 1,207 joints in 55 patients with RA and 352 joints in 18 patients with PsA, researchers in Austria found tenderness in nonswollen joints was associated with radiographic progression.
Despite having effective treatments in RA and PsA, “none of our therapies show that they’re able to prevent progression,” Dr. Wells said.
When it comes to hitting the treatment target in RA, some rheumatologists may think they can accomplish it without use of repeated radiographs. “I have a different perspective on that – that you really do indeed need to do the x-rays today and follow those x-rays along, especially if it’s going to change your treatment paradigm and what your treatment decision would be for the patient,” he said.
Counterpoint: Repeat radiographs aren’t helpful
Almost all rheumatologists would likely order an initial radiograph for their patients with RA or PsA, Roy M. Fleischmann, MD, clinical professor of medicine at the University of Texas and codirector of the Metroplex Clinical Research Center, both in Dallas, said in his presentation.
“If you see erosions when you start, chances are you’re going to be much more aggressive,” Dr. Fleischmann said. “So it is justification for early, more aggressive treatment of disease.”
In recent decades, radiographic progression in RA has decreased as more effective antirheumatic treatments have come into use, Dr. Fleischmann argued.

“We had x-ray progression in virtually everybody, and it was consistent no matter what we treated with, which was gold or penicillamine or any of the NSAIDs or sulfasalazine,” he said. “With methotrexate ... about 60% of patients actually have no x-ray progression, and that was a major change, and that’s one of the reasons why methotrexate has become the keystone of therapy. But even with methotrexate, [we] still had many patients who progressed.”
After the introduction of tumor necrosis factor inhibitors and other mechanisms in the late 1990s, “all of a sudden, you don’t see x-ray progression – mean x-ray progression – in a group of patients,” he noted.
Many rheumatologists now use a treat-to-target strategy, and if the patient achieves true clinical remission or sustained very low disease activity as measured by Boolean remission, Simple Disease Activity Index, or Clinical Disease Activity Index, they have “very little chance of radiographic progression and functional decline,” he said.
“If a patient doesn’t achieve remission or very low disease activity, obtaining a radiograph doesn’t change what you do because the patient’s not where they want to be, where you want them to be; you’re going to make a change anyway,” Dr. Fleischmann explained. “The radiograph isn’t going to help you do that.”
If a patient is in sustained remission but a radiograph is ordered and shows disease progression, he questioned what the rheumatologist would do in that situation.
“Now the patient’s in, let’s say, a Boolean remission. They have no tender joints. They have no swollen joints ... their pain assessment is zero, their CRP is zero, and they do have some x-ray progression. Where are you going to change?” Dr. Fleischmann asked. “There’s no data that anything else would work. I don’t know what you would do. So, in conclusion, I would say you really don’t need to repeat an x-ray.”
AI reading x-rays?
Commenting on the point-counterpoint session, Arthur Kavanaugh, MD, professor of medicine at the University of California, San Diego, and director of RWCS, asked Dr. Fleischmann and Dr. Wells how they address the issue of how many radiologists seem to be unfamiliar with reading hand radiographs and RA progression.
Dr. Fleischmann said he was trained in how to read hand radiographs in medical school, but that training no longer appears to be occurring. “If you have a good bone radiologist, of which there are not a lot, you’re great. But if you don’t have a really good bone radiologist, it’s difficult,” he said.
Dr. Kavanaugh alluded to the advancement of artificial intelligence (AI) in radiology and posed the question of how both rheumatologists felt about AI reading and interpreting their radiographs. “If you could reliably submit x-rays and they would say what the Sharp score was and where the differences were, would that change anything?” he asked.
“I think having artificial intelligence read the x-ray or an MRI is really, really good. It’ll be better than the radiologists,” Dr. Fleischmann responded. “But I don’t think that you really need to repeat the x-ray. I mean, I really don’t think you need to repeat it. You need to treat the patient.”
Dr. Wells reported having financial relationships with numerous pharmaceutical companies. Dr. Fleischmann reported no relevant financial relationships.
Follow-up radiographs after an initial baseline reading in patients with rheumatoid arthritis or psoriatic arthritis may still show radiographic progression despite treatment with current therapies, but it’s unclear if they will affect treatment decisions between patients and doctors that may take place regardless of the radiographic information, according to arguments made for and against their usefulness in a point-counterpoint session at the 2023 Rheumatology Winter Clinical Symposium.
Alvin Wells, MD, PhD, director of the department of rheumatology at Advocate Aurora Health in Franklin, Wisc., said that x-rays “reflect the history of joint pathology” and can get worse over time, correlating with disease activity and severity.
While RA does not yet have the “holy grail” of complete or true remission, Dr. Wells argued, the combination of clinical remission, laboratory remission, and imaging remission gets patients with RA close to the ideal when measured over time. “You need to continue to monitor these patients as you follow them along,” he said.
The BARFOT study, which evaluated 1,938 patients with early RA in two cohorts during 1992-1999 and again between 2000 and 2006, showed that more active treatments in the 2000s did not result in improvements in Health Assessment Questionnaire (HAQ) and pain scores, compared with patients treated in the 1990s. “You can see in some of those patients those scores do increase, and that even despite aggressive therapies that we had in 2006, you can still see some of those patients still have progression of the disease,” Dr. Wells explained. “How did they know? Because they looked.”
He also cited a study from researchers at the Mayo Clinic who examined 586 patients with RA that showed a higher prevalence of functional disability in patients with RA who also had radiographic changes, compared with patients without RA. “Radiographic changes correlate with disease severity and functional disability as well,” Dr. Wells said.
Just as prostate-specific antigen levels are used in prostate cancer screening and hemoglobin A1c is measured in diabetes management, radiographs should be used to track progression of disease in RA and PsA, Dr. Wells argued. “[I]f you don’t know, you can’t treat,” he said.
Some patients near remission may have radiographic progression even though disease activity measurements such as C-reactive protein (CRP) values do not show presence of active disease. In a study analyzing 1,184 patients with RA in the ASPIRE, ERA, Leflunomide, PREMIER and TEMPO trials, swollen joint count (SJC) was a better predictor of radiographic progression than CRP in patients near remission.
“[E]ven where you don’t see smoke, there still could be fire,” Dr. Wells said. “Some of these patients still progress and these are outliers, and the way they saw that [was] because they followed those patients along. If you don’t look, you don’t know.”
Radiographic progression can also be seen among nonswollen joints in patients with RA and PsA. In a study of 1,207 joints in 55 patients with RA and 352 joints in 18 patients with PsA, researchers in Austria found tenderness in nonswollen joints was associated with radiographic progression.
Despite having effective treatments in RA and PsA, “none of our therapies show that they’re able to prevent progression,” Dr. Wells said.
When it comes to hitting the treatment target in RA, some rheumatologists may think they can accomplish it without use of repeated radiographs. “I have a different perspective on that – that you really do indeed need to do the x-rays today and follow those x-rays along, especially if it’s going to change your treatment paradigm and what your treatment decision would be for the patient,” he said.
Counterpoint: Repeat radiographs aren’t helpful
Almost all rheumatologists would likely order an initial radiograph for their patients with RA or PsA, Roy M. Fleischmann, MD, clinical professor of medicine at the University of Texas and codirector of the Metroplex Clinical Research Center, both in Dallas, said in his presentation.
“If you see erosions when you start, chances are you’re going to be much more aggressive,” Dr. Fleischmann said. “So it is justification for early, more aggressive treatment of disease.”
In recent decades, radiographic progression in RA has decreased as more effective antirheumatic treatments have come into use, Dr. Fleischmann argued.
“We had x-ray progression in virtually everybody, and it was consistent no matter what we treated with, which was gold or penicillamine or any of the NSAIDs or sulfasalazine,” he said. “With methotrexate ... about 60% of patients actually have no x-ray progression, and that was a major change, and that’s one of the reasons why methotrexate has become the keystone of therapy. But even with methotrexate, [we] still had many patients who progressed.”
After the introduction of tumor necrosis factor inhibitors and other mechanisms in the late 1990s, “all of a sudden, you don’t see x-ray progression – mean x-ray progression – in a group of patients,” he noted.
Many rheumatologists now use a treat-to-target strategy, and if the patient achieves true clinical remission or sustained very low disease activity as measured by Boolean remission, Simple Disease Activity Index, or Clinical Disease Activity Index, they have “very little chance of radiographic progression and functional decline,” he said.
“If a patient doesn’t achieve remission or very low disease activity, obtaining a radiograph doesn’t change what you do because the patient’s not where they want to be, where you want them to be; you’re going to make a change anyway,” Dr. Fleischmann explained. “The radiograph isn’t going to help you do that.”
If a patient is in sustained remission but a radiograph is ordered and shows disease progression, he questioned what the rheumatologist would do in that situation.
“Now the patient’s in, let’s say, a Boolean remission. They have no tender joints. They have no swollen joints ... their pain assessment is zero, their CRP is zero, and they do have some x-ray progression. Where are you going to change?” Dr. Fleischmann asked. “There’s no data that anything else would work. I don’t know what you would do. So, in conclusion, I would say you really don’t need to repeat an x-ray.”
AI reading x-rays?
Commenting on the point-counterpoint session, Arthur Kavanaugh, MD, professor of medicine at the University of California, San Diego, and director of RWCS, asked Dr. Fleischmann and Dr. Wells how they address the issue of how many radiologists seem to be unfamiliar with reading hand radiographs and RA progression.
Dr. Fleischmann said he was trained in how to read hand radiographs in medical school, but that training no longer appears to be occurring. “If you have a good bone radiologist, of which there are not a lot, you’re great. But if you don’t have a really good bone radiologist, it’s difficult,” he said.
Dr. Kavanaugh alluded to the advancement of artificial intelligence (AI) in radiology and posed the question of how both rheumatologists felt about AI reading and interpreting their radiographs. “If you could reliably submit x-rays and they would say what the Sharp score was and where the differences were, would that change anything?” he asked.
“I think having artificial intelligence read the x-ray or an MRI is really, really good. It’ll be better than the radiologists,” Dr. Fleischmann responded. “But I don’t think that you really need to repeat the x-ray. I mean, I really don’t think you need to repeat it. You need to treat the patient.”
Dr. Wells reported having financial relationships with numerous pharmaceutical companies. Dr. Fleischmann reported no relevant financial relationships.
Follow-up radiographs after an initial baseline reading in patients with rheumatoid arthritis or psoriatic arthritis may still show radiographic progression despite treatment with current therapies, but it’s unclear if they will affect treatment decisions between patients and doctors that may take place regardless of the radiographic information, according to arguments made for and against their usefulness in a point-counterpoint session at the 2023 Rheumatology Winter Clinical Symposium.
Alvin Wells, MD, PhD, director of the department of rheumatology at Advocate Aurora Health in Franklin, Wisc., said that x-rays “reflect the history of joint pathology” and can get worse over time, correlating with disease activity and severity.
While RA does not yet have the “holy grail” of complete or true remission, Dr. Wells argued, the combination of clinical remission, laboratory remission, and imaging remission gets patients with RA close to the ideal when measured over time. “You need to continue to monitor these patients as you follow them along,” he said.
The BARFOT study, which evaluated 1,938 patients with early RA in two cohorts during 1992-1999 and again between 2000 and 2006, showed that more active treatments in the 2000s did not result in improvements in Health Assessment Questionnaire (HAQ) and pain scores, compared with patients treated in the 1990s. “You can see in some of those patients those scores do increase, and that even despite aggressive therapies that we had in 2006, you can still see some of those patients still have progression of the disease,” Dr. Wells explained. “How did they know? Because they looked.”
He also cited a study from researchers at the Mayo Clinic who examined 586 patients with RA that showed a higher prevalence of functional disability in patients with RA who also had radiographic changes, compared with patients without RA. “Radiographic changes correlate with disease severity and functional disability as well,” Dr. Wells said.
Just as prostate-specific antigen levels are used in prostate cancer screening and hemoglobin A1c is measured in diabetes management, radiographs should be used to track progression of disease in RA and PsA, Dr. Wells argued. “[I]f you don’t know, you can’t treat,” he said.
Some patients near remission may have radiographic progression even though disease activity measurements such as C-reactive protein (CRP) values do not show presence of active disease. In a study analyzing 1,184 patients with RA in the ASPIRE, ERA, Leflunomide, PREMIER and TEMPO trials, swollen joint count (SJC) was a better predictor of radiographic progression than CRP in patients near remission.
“[E]ven where you don’t see smoke, there still could be fire,” Dr. Wells said. “Some of these patients still progress and these are outliers, and the way they saw that [was] because they followed those patients along. If you don’t look, you don’t know.”
Radiographic progression can also be seen among nonswollen joints in patients with RA and PsA. In a study of 1,207 joints in 55 patients with RA and 352 joints in 18 patients with PsA, researchers in Austria found tenderness in nonswollen joints was associated with radiographic progression.
Despite having effective treatments in RA and PsA, “none of our therapies show that they’re able to prevent progression,” Dr. Wells said.
When it comes to hitting the treatment target in RA, some rheumatologists may think they can accomplish it without use of repeated radiographs. “I have a different perspective on that – that you really do indeed need to do the x-rays today and follow those x-rays along, especially if it’s going to change your treatment paradigm and what your treatment decision would be for the patient,” he said.
Counterpoint: Repeat radiographs aren’t helpful
Almost all rheumatologists would likely order an initial radiograph for their patients with RA or PsA, Roy M. Fleischmann, MD, clinical professor of medicine at the University of Texas and codirector of the Metroplex Clinical Research Center, both in Dallas, said in his presentation.
“If you see erosions when you start, chances are you’re going to be much more aggressive,” Dr. Fleischmann said. “So it is justification for early, more aggressive treatment of disease.”
In recent decades, radiographic progression in RA has decreased as more effective antirheumatic treatments have come into use, Dr. Fleischmann argued.
“We had x-ray progression in virtually everybody, and it was consistent no matter what we treated with, which was gold or penicillamine or any of the NSAIDs or sulfasalazine,” he said. “With methotrexate ... about 60% of patients actually have no x-ray progression, and that was a major change, and that’s one of the reasons why methotrexate has become the keystone of therapy. But even with methotrexate, [we] still had many patients who progressed.”
After the introduction of tumor necrosis factor inhibitors and other mechanisms in the late 1990s, “all of a sudden, you don’t see x-ray progression – mean x-ray progression – in a group of patients,” he noted.
Many rheumatologists now use a treat-to-target strategy, and if the patient achieves true clinical remission or sustained very low disease activity as measured by Boolean remission, Simple Disease Activity Index, or Clinical Disease Activity Index, they have “very little chance of radiographic progression and functional decline,” he said.
“If a patient doesn’t achieve remission or very low disease activity, obtaining a radiograph doesn’t change what you do because the patient’s not where they want to be, where you want them to be; you’re going to make a change anyway,” Dr. Fleischmann explained. “The radiograph isn’t going to help you do that.”
If a patient is in sustained remission but a radiograph is ordered and shows disease progression, he questioned what the rheumatologist would do in that situation.
“Now the patient’s in, let’s say, a Boolean remission. They have no tender joints. They have no swollen joints ... their pain assessment is zero, their CRP is zero, and they do have some x-ray progression. Where are you going to change?” Dr. Fleischmann asked. “There’s no data that anything else would work. I don’t know what you would do. So, in conclusion, I would say you really don’t need to repeat an x-ray.”
AI reading x-rays?
Commenting on the point-counterpoint session, Arthur Kavanaugh, MD, professor of medicine at the University of California, San Diego, and director of RWCS, asked Dr. Fleischmann and Dr. Wells how they address the issue of how many radiologists seem to be unfamiliar with reading hand radiographs and RA progression.
Dr. Fleischmann said he was trained in how to read hand radiographs in medical school, but that training no longer appears to be occurring. “If you have a good bone radiologist, of which there are not a lot, you’re great. But if you don’t have a really good bone radiologist, it’s difficult,” he said.
Dr. Kavanaugh alluded to the advancement of artificial intelligence (AI) in radiology and posed the question of how both rheumatologists felt about AI reading and interpreting their radiographs. “If you could reliably submit x-rays and they would say what the Sharp score was and where the differences were, would that change anything?” he asked.
“I think having artificial intelligence read the x-ray or an MRI is really, really good. It’ll be better than the radiologists,” Dr. Fleischmann responded. “But I don’t think that you really need to repeat the x-ray. I mean, I really don’t think you need to repeat it. You need to treat the patient.”
Dr. Wells reported having financial relationships with numerous pharmaceutical companies. Dr. Fleischmann reported no relevant financial relationships.
FROM RWCS 2023
New tool better estimates cardiovascular risk in people with lupus
Current risk estimators are inaccurate
A tool that incorporates lupus-related variables with traditional risk factors provides a much more accurate assessment of cardiovascular (CV) risk in patients with systemic lupus erythematosus (SLE), according to data presented at the annual meeting of the Canadian Rheumatology Association.
In the initial clinical assessment of this tool, called the SLECRISK, “it identified high-risk lupus patients who would otherwise be missed by traditional methods of CV risk assessment,” reported May Y. Choi, MD, associate director of translational research at the University of Calgary’s (Alta.) Lupus Centre of Excellence.
It is well known that patients with SLE face an increased risk of CV events starting at an age long before risk begins climbing in the general population, according to Dr. Choi. She cited one study that showed women aged 35-44 years have a 50-fold greater risk of myocardial infarction than healthy individuals.
All major guidelines recognize this increased risk and recommend CV risk assessment in patients with SLE, even though Dr. Choi pointed out that traditional tools, such as the American College of Cardiology atherosclerotic cardiovascular disease (ASCVD) risk calculator or the Framingham Risk Score (FRS) have a limited ability to detect the patients with SLE who are most likely to have an event.
In SLE, current tools are inadequate
“These risk assessment tools perform poorly in SLE patients because they do not capture SLE-related inflammation,” Dr. Choi said. Of several examples, Dr. Choi cited a study showing “seven times more MIs and strokes observed than expected in SLE patients on the basis of the FRS.”
The disparity between expected and observed MIs and strokes is worse with increasing severity of SLE. In a study she presented 3 years ago, rates of CV events were 12 times higher in those with inactive or mild SLE, rising to a 16-fold increase among those with moderate disease and jumping to a 32-fold increase in those with severe SLE.
The SLECRISK tool was developed from the Brigham and Women’s Hospital SLE Registry, which was initiated in 1992. Patients without a history of CV disease were evaluated for traditional CV risk factors and for SLE-specific characteristics such as disease activity, levels of the complement proteins C3 and C4, kidney function, the presence of nephritis, and SLE duration. The value of these characteristics as predictors of CV events were then assessed over a 10-year follow-up period before being assembled into the SLECRISK tool.
In an example of the risk equation, Dr. Choi described a 50-year-old patient with SLE and a 5% 10-year ASCVD risk score, which is low. After adjustment for SLE risks, which included 10 years disease duration, high disease activity, elevated creatinine, and positive anti–double stranded DNA status, the 10-year CV risk score climbed to 16.2%, which is moderate.
The performance of the SLECRISK was evaluated in 1,243 patients providing 8,946.51 person-years of follow-up. During this period, there were 90 major adverse cardiac events (MACE), of which 82% were adjudicated by cardiologists, and 211 secondary events.
Relative to the ASCVD risk score, the SLECRISK identified about twice as many patients with SLE as having moderate risk and 3.5-fold more patients as having high risk. Among patients who experienced CV events, traditional CV risk factors were more common but so were SLE-specific risk factors, including greater disease severity, a greater likelihood of lupus nephritis, increased complement levels, and greater exposure to glucocorticoids, according to Dr. Choi.
Specificities for CV events higher on SLECRISK
In predicting CV events, the differences in specificities were in the same general range, although somewhat higher for the ASCVD risk score in regard to predicting MACE (83% vs. 72%) and MACE plus secondary events (90% vs. 79%). However, the sensitivities were much higher for SLECRISK relative to the ASCVD risk score for MACE alone (64% vs. 41%) and for MACE plus secondary events (58% vs. 35%).
When comparing those who had an MI or stroke, the ASCVD risk score identified 8 (7%) patients missed by SLECRISK, whereas SLECRISK identified 89 (73%) missed by the ASCVD risk score. The remaining 25 patients (20%) were identified by both. The advantage of SLECRISK was similar for MACE plus secondary outcomes.
Dr. Choi noted that all of the SLE-specific variables in SLECRISK are readily obtained and often already available in patient charts. She said that there is a plan to validate the tool in larger groups, but with a goal of creating a tool available online for clinicians and their patients to use. There is also an even more ambitious plan for the future.
“We have funding to look at machine learning to evaluate predictive variables in SLE patients,” Dr. Choi said. Rather than adding SLE-specific variables to traditional risks, the plan is to “start from scratch,” letting artificial intelligence assemble predictors without prejudice to what might or might not be relevant.
A SLE-specific tool for evaluating CV risk is an important “unmet need,” according to Karen H. Costenbader, MD, professor in the division of rheumatology, inflammation, and immunity at Brigham and Women’s Hospital and Harvard Medical School, both in Boston. In an interview, she reiterated that measuring CV risk in SLE is already guideline recommended, but conventional tools have been shown to be inaccurate.
“I can envision it being used in clinical encounters to help guide shared decision-making with patients,” explained Dr. Costenbader, who was not involved in the presentation at the CRA meeting but worked with Dr. Choi in developing SLECRISK. “It would give us more precise estimates, allowing us to risk stratify our patients and informing us as to which modifiable SLE-specific and nonspecific factors are contributing most to CV risk.’
The problem of using conventional risk assessments in SLE has been well recognized. Of those who have written on this subject, Maureen McMahon, MD, site director of the Lupus Clinical Trials Network at the University of California, Los Angeles, said: “There is a critical need for the development of SLE-specific risk assessment tools like SLECRISK.”
Author of several studies looking at alternatives for CV risk assessment in SLE, including a study looking at a panel of biomarkers that was published in ACR Open Rheumatology, Dr. McMahon said in an interview that CV risk in SLE is high but conventional risk assessments are flawed.
“Multiple previous studies have demonstrated that these currently available calculators are not adequate for identifying risk in the lupus patient population,” she said. According to Dr. McMahon, the fact that rheumatologists remain “dependent upon [these conventional] cardiovascular risk calculators” is a well-recognized problem that needs resolution.
Dr. Choi has financial relationships with AstraZeneca, GlaxoSmithKline, Mallinckrodt. MitogenDx, Organon, and Werfen International. Dr. Costenbader reports no potential conflicts of interest. Dr. McMahon has financial relationships with AstraZeneca, Aurinia Pharmaceuticals, Eli Lilly, and GlaxoSmithKline.
Current risk estimators are inaccurate
Current risk estimators are inaccurate
A tool that incorporates lupus-related variables with traditional risk factors provides a much more accurate assessment of cardiovascular (CV) risk in patients with systemic lupus erythematosus (SLE), according to data presented at the annual meeting of the Canadian Rheumatology Association.
In the initial clinical assessment of this tool, called the SLECRISK, “it identified high-risk lupus patients who would otherwise be missed by traditional methods of CV risk assessment,” reported May Y. Choi, MD, associate director of translational research at the University of Calgary’s (Alta.) Lupus Centre of Excellence.
It is well known that patients with SLE face an increased risk of CV events starting at an age long before risk begins climbing in the general population, according to Dr. Choi. She cited one study that showed women aged 35-44 years have a 50-fold greater risk of myocardial infarction than healthy individuals.
All major guidelines recognize this increased risk and recommend CV risk assessment in patients with SLE, even though Dr. Choi pointed out that traditional tools, such as the American College of Cardiology atherosclerotic cardiovascular disease (ASCVD) risk calculator or the Framingham Risk Score (FRS) have a limited ability to detect the patients with SLE who are most likely to have an event.
In SLE, current tools are inadequate
“These risk assessment tools perform poorly in SLE patients because they do not capture SLE-related inflammation,” Dr. Choi said. Of several examples, Dr. Choi cited a study showing “seven times more MIs and strokes observed than expected in SLE patients on the basis of the FRS.”
The disparity between expected and observed MIs and strokes is worse with increasing severity of SLE. In a study she presented 3 years ago, rates of CV events were 12 times higher in those with inactive or mild SLE, rising to a 16-fold increase among those with moderate disease and jumping to a 32-fold increase in those with severe SLE.
The SLECRISK tool was developed from the Brigham and Women’s Hospital SLE Registry, which was initiated in 1992. Patients without a history of CV disease were evaluated for traditional CV risk factors and for SLE-specific characteristics such as disease activity, levels of the complement proteins C3 and C4, kidney function, the presence of nephritis, and SLE duration. The value of these characteristics as predictors of CV events were then assessed over a 10-year follow-up period before being assembled into the SLECRISK tool.
In an example of the risk equation, Dr. Choi described a 50-year-old patient with SLE and a 5% 10-year ASCVD risk score, which is low. After adjustment for SLE risks, which included 10 years disease duration, high disease activity, elevated creatinine, and positive anti–double stranded DNA status, the 10-year CV risk score climbed to 16.2%, which is moderate.
The performance of the SLECRISK was evaluated in 1,243 patients providing 8,946.51 person-years of follow-up. During this period, there were 90 major adverse cardiac events (MACE), of which 82% were adjudicated by cardiologists, and 211 secondary events.
Relative to the ASCVD risk score, the SLECRISK identified about twice as many patients with SLE as having moderate risk and 3.5-fold more patients as having high risk. Among patients who experienced CV events, traditional CV risk factors were more common but so were SLE-specific risk factors, including greater disease severity, a greater likelihood of lupus nephritis, increased complement levels, and greater exposure to glucocorticoids, according to Dr. Choi.
Specificities for CV events higher on SLECRISK
In predicting CV events, the differences in specificities were in the same general range, although somewhat higher for the ASCVD risk score in regard to predicting MACE (83% vs. 72%) and MACE plus secondary events (90% vs. 79%). However, the sensitivities were much higher for SLECRISK relative to the ASCVD risk score for MACE alone (64% vs. 41%) and for MACE plus secondary events (58% vs. 35%).
When comparing those who had an MI or stroke, the ASCVD risk score identified 8 (7%) patients missed by SLECRISK, whereas SLECRISK identified 89 (73%) missed by the ASCVD risk score. The remaining 25 patients (20%) were identified by both. The advantage of SLECRISK was similar for MACE plus secondary outcomes.
Dr. Choi noted that all of the SLE-specific variables in SLECRISK are readily obtained and often already available in patient charts. She said that there is a plan to validate the tool in larger groups, but with a goal of creating a tool available online for clinicians and their patients to use. There is also an even more ambitious plan for the future.
“We have funding to look at machine learning to evaluate predictive variables in SLE patients,” Dr. Choi said. Rather than adding SLE-specific variables to traditional risks, the plan is to “start from scratch,” letting artificial intelligence assemble predictors without prejudice to what might or might not be relevant.
A SLE-specific tool for evaluating CV risk is an important “unmet need,” according to Karen H. Costenbader, MD, professor in the division of rheumatology, inflammation, and immunity at Brigham and Women’s Hospital and Harvard Medical School, both in Boston. In an interview, she reiterated that measuring CV risk in SLE is already guideline recommended, but conventional tools have been shown to be inaccurate.
“I can envision it being used in clinical encounters to help guide shared decision-making with patients,” explained Dr. Costenbader, who was not involved in the presentation at the CRA meeting but worked with Dr. Choi in developing SLECRISK. “It would give us more precise estimates, allowing us to risk stratify our patients and informing us as to which modifiable SLE-specific and nonspecific factors are contributing most to CV risk.’
The problem of using conventional risk assessments in SLE has been well recognized. Of those who have written on this subject, Maureen McMahon, MD, site director of the Lupus Clinical Trials Network at the University of California, Los Angeles, said: “There is a critical need for the development of SLE-specific risk assessment tools like SLECRISK.”
Author of several studies looking at alternatives for CV risk assessment in SLE, including a study looking at a panel of biomarkers that was published in ACR Open Rheumatology, Dr. McMahon said in an interview that CV risk in SLE is high but conventional risk assessments are flawed.
“Multiple previous studies have demonstrated that these currently available calculators are not adequate for identifying risk in the lupus patient population,” she said. According to Dr. McMahon, the fact that rheumatologists remain “dependent upon [these conventional] cardiovascular risk calculators” is a well-recognized problem that needs resolution.
Dr. Choi has financial relationships with AstraZeneca, GlaxoSmithKline, Mallinckrodt. MitogenDx, Organon, and Werfen International. Dr. Costenbader reports no potential conflicts of interest. Dr. McMahon has financial relationships with AstraZeneca, Aurinia Pharmaceuticals, Eli Lilly, and GlaxoSmithKline.
A tool that incorporates lupus-related variables with traditional risk factors provides a much more accurate assessment of cardiovascular (CV) risk in patients with systemic lupus erythematosus (SLE), according to data presented at the annual meeting of the Canadian Rheumatology Association.
In the initial clinical assessment of this tool, called the SLECRISK, “it identified high-risk lupus patients who would otherwise be missed by traditional methods of CV risk assessment,” reported May Y. Choi, MD, associate director of translational research at the University of Calgary’s (Alta.) Lupus Centre of Excellence.
It is well known that patients with SLE face an increased risk of CV events starting at an age long before risk begins climbing in the general population, according to Dr. Choi. She cited one study that showed women aged 35-44 years have a 50-fold greater risk of myocardial infarction than healthy individuals.
All major guidelines recognize this increased risk and recommend CV risk assessment in patients with SLE, even though Dr. Choi pointed out that traditional tools, such as the American College of Cardiology atherosclerotic cardiovascular disease (ASCVD) risk calculator or the Framingham Risk Score (FRS) have a limited ability to detect the patients with SLE who are most likely to have an event.
In SLE, current tools are inadequate
“These risk assessment tools perform poorly in SLE patients because they do not capture SLE-related inflammation,” Dr. Choi said. Of several examples, Dr. Choi cited a study showing “seven times more MIs and strokes observed than expected in SLE patients on the basis of the FRS.”
The disparity between expected and observed MIs and strokes is worse with increasing severity of SLE. In a study she presented 3 years ago, rates of CV events were 12 times higher in those with inactive or mild SLE, rising to a 16-fold increase among those with moderate disease and jumping to a 32-fold increase in those with severe SLE.
The SLECRISK tool was developed from the Brigham and Women’s Hospital SLE Registry, which was initiated in 1992. Patients without a history of CV disease were evaluated for traditional CV risk factors and for SLE-specific characteristics such as disease activity, levels of the complement proteins C3 and C4, kidney function, the presence of nephritis, and SLE duration. The value of these characteristics as predictors of CV events were then assessed over a 10-year follow-up period before being assembled into the SLECRISK tool.
In an example of the risk equation, Dr. Choi described a 50-year-old patient with SLE and a 5% 10-year ASCVD risk score, which is low. After adjustment for SLE risks, which included 10 years disease duration, high disease activity, elevated creatinine, and positive anti–double stranded DNA status, the 10-year CV risk score climbed to 16.2%, which is moderate.
The performance of the SLECRISK was evaluated in 1,243 patients providing 8,946.51 person-years of follow-up. During this period, there were 90 major adverse cardiac events (MACE), of which 82% were adjudicated by cardiologists, and 211 secondary events.
Relative to the ASCVD risk score, the SLECRISK identified about twice as many patients with SLE as having moderate risk and 3.5-fold more patients as having high risk. Among patients who experienced CV events, traditional CV risk factors were more common but so were SLE-specific risk factors, including greater disease severity, a greater likelihood of lupus nephritis, increased complement levels, and greater exposure to glucocorticoids, according to Dr. Choi.
Specificities for CV events higher on SLECRISK
In predicting CV events, the differences in specificities were in the same general range, although somewhat higher for the ASCVD risk score in regard to predicting MACE (83% vs. 72%) and MACE plus secondary events (90% vs. 79%). However, the sensitivities were much higher for SLECRISK relative to the ASCVD risk score for MACE alone (64% vs. 41%) and for MACE plus secondary events (58% vs. 35%).
When comparing those who had an MI or stroke, the ASCVD risk score identified 8 (7%) patients missed by SLECRISK, whereas SLECRISK identified 89 (73%) missed by the ASCVD risk score. The remaining 25 patients (20%) were identified by both. The advantage of SLECRISK was similar for MACE plus secondary outcomes.
Dr. Choi noted that all of the SLE-specific variables in SLECRISK are readily obtained and often already available in patient charts. She said that there is a plan to validate the tool in larger groups, but with a goal of creating a tool available online for clinicians and their patients to use. There is also an even more ambitious plan for the future.
“We have funding to look at machine learning to evaluate predictive variables in SLE patients,” Dr. Choi said. Rather than adding SLE-specific variables to traditional risks, the plan is to “start from scratch,” letting artificial intelligence assemble predictors without prejudice to what might or might not be relevant.
A SLE-specific tool for evaluating CV risk is an important “unmet need,” according to Karen H. Costenbader, MD, professor in the division of rheumatology, inflammation, and immunity at Brigham and Women’s Hospital and Harvard Medical School, both in Boston. In an interview, she reiterated that measuring CV risk in SLE is already guideline recommended, but conventional tools have been shown to be inaccurate.
“I can envision it being used in clinical encounters to help guide shared decision-making with patients,” explained Dr. Costenbader, who was not involved in the presentation at the CRA meeting but worked with Dr. Choi in developing SLECRISK. “It would give us more precise estimates, allowing us to risk stratify our patients and informing us as to which modifiable SLE-specific and nonspecific factors are contributing most to CV risk.’
The problem of using conventional risk assessments in SLE has been well recognized. Of those who have written on this subject, Maureen McMahon, MD, site director of the Lupus Clinical Trials Network at the University of California, Los Angeles, said: “There is a critical need for the development of SLE-specific risk assessment tools like SLECRISK.”
Author of several studies looking at alternatives for CV risk assessment in SLE, including a study looking at a panel of biomarkers that was published in ACR Open Rheumatology, Dr. McMahon said in an interview that CV risk in SLE is high but conventional risk assessments are flawed.
“Multiple previous studies have demonstrated that these currently available calculators are not adequate for identifying risk in the lupus patient population,” she said. According to Dr. McMahon, the fact that rheumatologists remain “dependent upon [these conventional] cardiovascular risk calculators” is a well-recognized problem that needs resolution.
Dr. Choi has financial relationships with AstraZeneca, GlaxoSmithKline, Mallinckrodt. MitogenDx, Organon, and Werfen International. Dr. Costenbader reports no potential conflicts of interest. Dr. McMahon has financial relationships with AstraZeneca, Aurinia Pharmaceuticals, Eli Lilly, and GlaxoSmithKline.
FROM CRA 2023
PsA prediction tool approaches clinical utility
Easily collected variables establish risk
A new tool for predicting which patients with psoriasis will develop psoriatic arthritis (PsA) is showing promise for such clinical applications as early treatment in those at risk or trials to prevent PsA, according to a summary of progress at the annual meeting of the Canadian Rheumatology Association.
Based on current levels of sensitivity and specificity, psoriasis “can be predicted with reasonable accuracy,” reported Lihi Eder, MD, PhD, director of research in the rheumatology division at the University of Toronto.

The predictive method, called PRESTO (Prediction of Psoriatic Arthritis Tool), is based on variables readily available in clinical practice, according to Dr. Eder. Once values are assigned to the risk factors, the risk of PsA over a 1-year or 5-year time frame can be estimated with a calculator.
She called PRESTO the “first clinical tool for predicting PsA among psoriasis patients.”
The work on this tool began in 2006 when the International Psoriasis and Arthritis Research Team (IPART) initiated a prospectively collected cohort of psoriasis patients. To be enrolled, patients had to be free of signs and symptoms of arthritis upon examination by a rheumatologist. They were then invited to return annually for follow-up that again included screening for joint involvement by a rheumatologist.
At baseline and at follow-up evaluations, 13 predictors were evaluated. These involved psoriasis characteristics, such as nail pitting; symptoms, such as stiffness; comorbidities, such as additional inflammatory diseases; and laboratory values, such as upregulated markers of inflammation.
Symptoms and signs used to predict PsA
Dr. Eder and her colleagues applied regression models to select an optimal combination of variables weighted for predictive value. Variables offering predictive value included higher PASI (Psoriasis Area and Severity Index), greater fatigue score as measured by FACIT (Functional Assessment of Chronic Illness Therapy) score, greater morning stiffness, and greater pain.
When applied to 635 patients in the IPART cohort, in which there were 51 incident PsA cases over 1 year and 75 incident cases over 5 years, the area under the curve (AUC) for PRESTO at the cutoffs studied was 72% for the 1-year time window and 75% for the 5-year time window.
These levels are associated with adequate accuracy, according to Dr. Eder, who explained that “an AUC greater than 70% is considered reasonable” for clinical applicability.
Moreover, the cutoffs can be adjusted for the specific purpose of the predictive tool. For example, to screen patients for risk, lower cutoffs could be employed to increase sensitivity. In order to select patients for a clinical trial to prevent PsA, higher cutoffs could be employed to increase specificity.
But sensitivities and specificities move in opposite directions when cutoffs are adjusted. Showing data from the 5-year prediction model, Dr. Eder reported that specificities climbed from about 58% to 97% as cutoffs were increased. The sensitivities with these adjustments fell from 79% to 14%.
In general, Dr. Eder said there was “excellent calibration” for the cutoffs employed when they compared the predicted and observed rates of PsA according to quintile of predictive probability. The differences were particularly minor over a 1-year time period. Over the 5-year period, observed rates were somewhat higher than predicted in the fourth and fifth quintile, but, again, this discrepancy could be modified for specific applications with cutoff adjustments.
Validation studies are planned
Even though psoriasis patients in IPART represents one of the largest cohorts of prospectively collected psoriasis patients, Dr. Eder acknowledged that the sample size would be considered “moderate” for developing a predictive model. However, the fact that the data were collected prospectively using standardized methodology strengthens the findings and provides the basis for the next step.
“Validation studies are planned with external cohorts,” said Dr. Eder, who indicated that a viable tool for identifying psoriasis patients at risk for PsA is likely. Even if it is not employed routinely in its current form at the level of individual patient care, she predicted that it will have value at a research level for understanding the relationship of psoriasis to PsA.

Christopher T. Ritchlin, MD, a professor and researcher at the University of Rochester (N.Y.), agreed that PRESTO has important potential as a clinical tool. Dr. Ritchlin has been involved in the development of PRESTO but was not involved in the presentation made at the CRA annual meeting.
“The PRESTO tool has the ability to predict the 2- and 5-year risk of developing psoriatic arthritis, which is an important advance if confirmed,” he said in an interview. He pointed out that approximately 25%-30% who develop psoriasis will go on to develop PsA but until now there has been no way to identify them.
“This tool may provide a pathway to early intervention,” he said.
Dr. Eder has financial relationships with AbbVie, Eli Lilly, Fresenius Kabi, Janssen, Novartis, Pfizer, Sandoz, and UCB. Dr. Ritchlin has financial relationships with many of the same companies.
Easily collected variables establish risk
Easily collected variables establish risk
A new tool for predicting which patients with psoriasis will develop psoriatic arthritis (PsA) is showing promise for such clinical applications as early treatment in those at risk or trials to prevent PsA, according to a summary of progress at the annual meeting of the Canadian Rheumatology Association.
Based on current levels of sensitivity and specificity, psoriasis “can be predicted with reasonable accuracy,” reported Lihi Eder, MD, PhD, director of research in the rheumatology division at the University of Toronto.
The predictive method, called PRESTO (Prediction of Psoriatic Arthritis Tool), is based on variables readily available in clinical practice, according to Dr. Eder. Once values are assigned to the risk factors, the risk of PsA over a 1-year or 5-year time frame can be estimated with a calculator.
She called PRESTO the “first clinical tool for predicting PsA among psoriasis patients.”
The work on this tool began in 2006 when the International Psoriasis and Arthritis Research Team (IPART) initiated a prospectively collected cohort of psoriasis patients. To be enrolled, patients had to be free of signs and symptoms of arthritis upon examination by a rheumatologist. They were then invited to return annually for follow-up that again included screening for joint involvement by a rheumatologist.
At baseline and at follow-up evaluations, 13 predictors were evaluated. These involved psoriasis characteristics, such as nail pitting; symptoms, such as stiffness; comorbidities, such as additional inflammatory diseases; and laboratory values, such as upregulated markers of inflammation.
Symptoms and signs used to predict PsA
Dr. Eder and her colleagues applied regression models to select an optimal combination of variables weighted for predictive value. Variables offering predictive value included higher PASI (Psoriasis Area and Severity Index), greater fatigue score as measured by FACIT (Functional Assessment of Chronic Illness Therapy) score, greater morning stiffness, and greater pain.
When applied to 635 patients in the IPART cohort, in which there were 51 incident PsA cases over 1 year and 75 incident cases over 5 years, the area under the curve (AUC) for PRESTO at the cutoffs studied was 72% for the 1-year time window and 75% for the 5-year time window.
These levels are associated with adequate accuracy, according to Dr. Eder, who explained that “an AUC greater than 70% is considered reasonable” for clinical applicability.
Moreover, the cutoffs can be adjusted for the specific purpose of the predictive tool. For example, to screen patients for risk, lower cutoffs could be employed to increase sensitivity. In order to select patients for a clinical trial to prevent PsA, higher cutoffs could be employed to increase specificity.
But sensitivities and specificities move in opposite directions when cutoffs are adjusted. Showing data from the 5-year prediction model, Dr. Eder reported that specificities climbed from about 58% to 97% as cutoffs were increased. The sensitivities with these adjustments fell from 79% to 14%.
In general, Dr. Eder said there was “excellent calibration” for the cutoffs employed when they compared the predicted and observed rates of PsA according to quintile of predictive probability. The differences were particularly minor over a 1-year time period. Over the 5-year period, observed rates were somewhat higher than predicted in the fourth and fifth quintile, but, again, this discrepancy could be modified for specific applications with cutoff adjustments.
Validation studies are planned
Even though psoriasis patients in IPART represents one of the largest cohorts of prospectively collected psoriasis patients, Dr. Eder acknowledged that the sample size would be considered “moderate” for developing a predictive model. However, the fact that the data were collected prospectively using standardized methodology strengthens the findings and provides the basis for the next step.
“Validation studies are planned with external cohorts,” said Dr. Eder, who indicated that a viable tool for identifying psoriasis patients at risk for PsA is likely. Even if it is not employed routinely in its current form at the level of individual patient care, she predicted that it will have value at a research level for understanding the relationship of psoriasis to PsA.
Christopher T. Ritchlin, MD, a professor and researcher at the University of Rochester (N.Y.), agreed that PRESTO has important potential as a clinical tool. Dr. Ritchlin has been involved in the development of PRESTO but was not involved in the presentation made at the CRA annual meeting.
“The PRESTO tool has the ability to predict the 2- and 5-year risk of developing psoriatic arthritis, which is an important advance if confirmed,” he said in an interview. He pointed out that approximately 25%-30% who develop psoriasis will go on to develop PsA but until now there has been no way to identify them.
“This tool may provide a pathway to early intervention,” he said.
Dr. Eder has financial relationships with AbbVie, Eli Lilly, Fresenius Kabi, Janssen, Novartis, Pfizer, Sandoz, and UCB. Dr. Ritchlin has financial relationships with many of the same companies.
A new tool for predicting which patients with psoriasis will develop psoriatic arthritis (PsA) is showing promise for such clinical applications as early treatment in those at risk or trials to prevent PsA, according to a summary of progress at the annual meeting of the Canadian Rheumatology Association.
Based on current levels of sensitivity and specificity, psoriasis “can be predicted with reasonable accuracy,” reported Lihi Eder, MD, PhD, director of research in the rheumatology division at the University of Toronto.
The predictive method, called PRESTO (Prediction of Psoriatic Arthritis Tool), is based on variables readily available in clinical practice, according to Dr. Eder. Once values are assigned to the risk factors, the risk of PsA over a 1-year or 5-year time frame can be estimated with a calculator.
She called PRESTO the “first clinical tool for predicting PsA among psoriasis patients.”
The work on this tool began in 2006 when the International Psoriasis and Arthritis Research Team (IPART) initiated a prospectively collected cohort of psoriasis patients. To be enrolled, patients had to be free of signs and symptoms of arthritis upon examination by a rheumatologist. They were then invited to return annually for follow-up that again included screening for joint involvement by a rheumatologist.
At baseline and at follow-up evaluations, 13 predictors were evaluated. These involved psoriasis characteristics, such as nail pitting; symptoms, such as stiffness; comorbidities, such as additional inflammatory diseases; and laboratory values, such as upregulated markers of inflammation.
Symptoms and signs used to predict PsA
Dr. Eder and her colleagues applied regression models to select an optimal combination of variables weighted for predictive value. Variables offering predictive value included higher PASI (Psoriasis Area and Severity Index), greater fatigue score as measured by FACIT (Functional Assessment of Chronic Illness Therapy) score, greater morning stiffness, and greater pain.
When applied to 635 patients in the IPART cohort, in which there were 51 incident PsA cases over 1 year and 75 incident cases over 5 years, the area under the curve (AUC) for PRESTO at the cutoffs studied was 72% for the 1-year time window and 75% for the 5-year time window.
These levels are associated with adequate accuracy, according to Dr. Eder, who explained that “an AUC greater than 70% is considered reasonable” for clinical applicability.
Moreover, the cutoffs can be adjusted for the specific purpose of the predictive tool. For example, to screen patients for risk, lower cutoffs could be employed to increase sensitivity. In order to select patients for a clinical trial to prevent PsA, higher cutoffs could be employed to increase specificity.
But sensitivities and specificities move in opposite directions when cutoffs are adjusted. Showing data from the 5-year prediction model, Dr. Eder reported that specificities climbed from about 58% to 97% as cutoffs were increased. The sensitivities with these adjustments fell from 79% to 14%.
In general, Dr. Eder said there was “excellent calibration” for the cutoffs employed when they compared the predicted and observed rates of PsA according to quintile of predictive probability. The differences were particularly minor over a 1-year time period. Over the 5-year period, observed rates were somewhat higher than predicted in the fourth and fifth quintile, but, again, this discrepancy could be modified for specific applications with cutoff adjustments.
Validation studies are planned
Even though psoriasis patients in IPART represents one of the largest cohorts of prospectively collected psoriasis patients, Dr. Eder acknowledged that the sample size would be considered “moderate” for developing a predictive model. However, the fact that the data were collected prospectively using standardized methodology strengthens the findings and provides the basis for the next step.
“Validation studies are planned with external cohorts,” said Dr. Eder, who indicated that a viable tool for identifying psoriasis patients at risk for PsA is likely. Even if it is not employed routinely in its current form at the level of individual patient care, she predicted that it will have value at a research level for understanding the relationship of psoriasis to PsA.
Christopher T. Ritchlin, MD, a professor and researcher at the University of Rochester (N.Y.), agreed that PRESTO has important potential as a clinical tool. Dr. Ritchlin has been involved in the development of PRESTO but was not involved in the presentation made at the CRA annual meeting.
“The PRESTO tool has the ability to predict the 2- and 5-year risk of developing psoriatic arthritis, which is an important advance if confirmed,” he said in an interview. He pointed out that approximately 25%-30% who develop psoriasis will go on to develop PsA but until now there has been no way to identify them.
“This tool may provide a pathway to early intervention,” he said.
Dr. Eder has financial relationships with AbbVie, Eli Lilly, Fresenius Kabi, Janssen, Novartis, Pfizer, Sandoz, and UCB. Dr. Ritchlin has financial relationships with many of the same companies.
FROM CRA 2023
Health plans get very poor scores for access to autoimmune drugs
Both public and private health plans score poorly when it comes to providing access to autoimmune medication, according to a report commissioned by the Autoimmune Association and Let My Doctors Decide, a national partnership of health care professionals. The analysis, published Jan. 26, found that 75% of insurers in the United States have policies that can limit coverage for Food and Drug Administration–approved medications for Crohn’s disease, lupus nephritis, multiple sclerosis, psoriasis, psoriatic arthritis, rheumatoid arthritis, and ulcerative colitis.
“Choice among health plans is a hallmark of the American health insurance system, yet this analysis shows that people living with autoimmune conditions have few, if any, coverage choices that do not involve significant to severe access restrictions,” the authors wrote.
The study looked at three common utilization management policies by health plans that can limit coverage of certain medications: step therapy, formulary/tier placement, and prior authorization. To compare health plans, researchers weighted these policies using a point system. Each medication indicated for each condition was given a score of 0-4 based on access restrictions in a health plan. If a plan used step therapy, it received one point, and requiring prior authorization added an additional point. They also added points based on where a drug appeared on a plan’s formulary. A lower total score meant fewer access barriers. The numbers were then added, and each health plan received a grade of A, B, C, or F based on their average score. The datasets and analysis were provided and performed by the data analytics firm MMIT.
Nearly 9 in 10 Medicare plans received a C or worse for coverage of medication received via mail order or the pharmacy. In commercial plans, the majority of plans scored Cs or Fs for six of the seven conditions, excluding lupus nephritis, where 67% of all commercial health plans scored a B for access to these medications.
Physician-administered medications tended to receive poorer coverage than drugs received via pharmacy. Across all conditions, 65% of Medicare Advantage plans scored an F for physician-administered medication access. For both psoriasis and multiple sclerosis, at least 80% of Medicare plans earned failing scores because of these restrictions. Coverage was poorer on both commercial and health exchange plans, where across all conditions, 83% achieved failing scores. Two exceptions were the Southern and Northern California PPO plans by the Kaiser Foundation Health Plan. Out of the largest 25 health plans in the United States, these two plans earned As in coverage for physician-administered medications across all seven autoimmune conditions.
The report shows “a growing disconnect between science and health insurance benefit designs that were developed in the 1960s and 1970s,” Kenneth Thorpe, PhD, of Emory University, Atlanta, said in an interview. Insurers originally designed these benefits to prevent excessive utilization in a population of mostly acutely ill patients, he said, whereas now, 90% of healthcare spending is linked to chronic conditions. For these patients, research shows that incentivizing patients to adhere to medications results in fewer hospitalizations and, therefore, more cost savings, Thorpe noted. These plans also do not consider that there is no average patient, he said, and healthcare providers should be able to match each patient to the best treatment option for them rather than trying out other less expensive medications first. “To the extent that physicians can have the flexibility to provide medications and treatments to patients that are going to have the best clinical response, that’s better outcomes at lower cost,” Dr. Thorpe said. While research shows heterogeneity in patient outcomes with different medication, “benefit designs from the past just don’t recognize that.”
Neither America’s Health Insurance Plans nor Pharmaceutical Care Management Association responded to a request for comment.
Quardricos Driskell, executive director of Let My Doctors Decide and vice president of government relations and public policy at the Autoimmune Association, hopes the study will spur action by policy makers and health plans to improve access to medications for the people who need them. Another larger point of the report is to “uphold the sanctity of protecting the doctor and patient relationship,” he said in an interview, adding “that decisions fundamentally need to be made not by insurance plans or middleman pharmacy benefit managers, but by the provider and patient.”
Mr. Driskell and Dr. Thorpe reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Both public and private health plans score poorly when it comes to providing access to autoimmune medication, according to a report commissioned by the Autoimmune Association and Let My Doctors Decide, a national partnership of health care professionals. The analysis, published Jan. 26, found that 75% of insurers in the United States have policies that can limit coverage for Food and Drug Administration–approved medications for Crohn’s disease, lupus nephritis, multiple sclerosis, psoriasis, psoriatic arthritis, rheumatoid arthritis, and ulcerative colitis.
“Choice among health plans is a hallmark of the American health insurance system, yet this analysis shows that people living with autoimmune conditions have few, if any, coverage choices that do not involve significant to severe access restrictions,” the authors wrote.
The study looked at three common utilization management policies by health plans that can limit coverage of certain medications: step therapy, formulary/tier placement, and prior authorization. To compare health plans, researchers weighted these policies using a point system. Each medication indicated for each condition was given a score of 0-4 based on access restrictions in a health plan. If a plan used step therapy, it received one point, and requiring prior authorization added an additional point. They also added points based on where a drug appeared on a plan’s formulary. A lower total score meant fewer access barriers. The numbers were then added, and each health plan received a grade of A, B, C, or F based on their average score. The datasets and analysis were provided and performed by the data analytics firm MMIT.
Nearly 9 in 10 Medicare plans received a C or worse for coverage of medication received via mail order or the pharmacy. In commercial plans, the majority of plans scored Cs or Fs for six of the seven conditions, excluding lupus nephritis, where 67% of all commercial health plans scored a B for access to these medications.
Physician-administered medications tended to receive poorer coverage than drugs received via pharmacy. Across all conditions, 65% of Medicare Advantage plans scored an F for physician-administered medication access. For both psoriasis and multiple sclerosis, at least 80% of Medicare plans earned failing scores because of these restrictions. Coverage was poorer on both commercial and health exchange plans, where across all conditions, 83% achieved failing scores. Two exceptions were the Southern and Northern California PPO plans by the Kaiser Foundation Health Plan. Out of the largest 25 health plans in the United States, these two plans earned As in coverage for physician-administered medications across all seven autoimmune conditions.
The report shows “a growing disconnect between science and health insurance benefit designs that were developed in the 1960s and 1970s,” Kenneth Thorpe, PhD, of Emory University, Atlanta, said in an interview. Insurers originally designed these benefits to prevent excessive utilization in a population of mostly acutely ill patients, he said, whereas now, 90% of healthcare spending is linked to chronic conditions. For these patients, research shows that incentivizing patients to adhere to medications results in fewer hospitalizations and, therefore, more cost savings, Thorpe noted. These plans also do not consider that there is no average patient, he said, and healthcare providers should be able to match each patient to the best treatment option for them rather than trying out other less expensive medications first. “To the extent that physicians can have the flexibility to provide medications and treatments to patients that are going to have the best clinical response, that’s better outcomes at lower cost,” Dr. Thorpe said. While research shows heterogeneity in patient outcomes with different medication, “benefit designs from the past just don’t recognize that.”
Neither America’s Health Insurance Plans nor Pharmaceutical Care Management Association responded to a request for comment.
Quardricos Driskell, executive director of Let My Doctors Decide and vice president of government relations and public policy at the Autoimmune Association, hopes the study will spur action by policy makers and health plans to improve access to medications for the people who need them. Another larger point of the report is to “uphold the sanctity of protecting the doctor and patient relationship,” he said in an interview, adding “that decisions fundamentally need to be made not by insurance plans or middleman pharmacy benefit managers, but by the provider and patient.”
Mr. Driskell and Dr. Thorpe reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Both public and private health plans score poorly when it comes to providing access to autoimmune medication, according to a report commissioned by the Autoimmune Association and Let My Doctors Decide, a national partnership of health care professionals. The analysis, published Jan. 26, found that 75% of insurers in the United States have policies that can limit coverage for Food and Drug Administration–approved medications for Crohn’s disease, lupus nephritis, multiple sclerosis, psoriasis, psoriatic arthritis, rheumatoid arthritis, and ulcerative colitis.
“Choice among health plans is a hallmark of the American health insurance system, yet this analysis shows that people living with autoimmune conditions have few, if any, coverage choices that do not involve significant to severe access restrictions,” the authors wrote.
The study looked at three common utilization management policies by health plans that can limit coverage of certain medications: step therapy, formulary/tier placement, and prior authorization. To compare health plans, researchers weighted these policies using a point system. Each medication indicated for each condition was given a score of 0-4 based on access restrictions in a health plan. If a plan used step therapy, it received one point, and requiring prior authorization added an additional point. They also added points based on where a drug appeared on a plan’s formulary. A lower total score meant fewer access barriers. The numbers were then added, and each health plan received a grade of A, B, C, or F based on their average score. The datasets and analysis were provided and performed by the data analytics firm MMIT.
Nearly 9 in 10 Medicare plans received a C or worse for coverage of medication received via mail order or the pharmacy. In commercial plans, the majority of plans scored Cs or Fs for six of the seven conditions, excluding lupus nephritis, where 67% of all commercial health plans scored a B for access to these medications.
Physician-administered medications tended to receive poorer coverage than drugs received via pharmacy. Across all conditions, 65% of Medicare Advantage plans scored an F for physician-administered medication access. For both psoriasis and multiple sclerosis, at least 80% of Medicare plans earned failing scores because of these restrictions. Coverage was poorer on both commercial and health exchange plans, where across all conditions, 83% achieved failing scores. Two exceptions were the Southern and Northern California PPO plans by the Kaiser Foundation Health Plan. Out of the largest 25 health plans in the United States, these two plans earned As in coverage for physician-administered medications across all seven autoimmune conditions.
The report shows “a growing disconnect between science and health insurance benefit designs that were developed in the 1960s and 1970s,” Kenneth Thorpe, PhD, of Emory University, Atlanta, said in an interview. Insurers originally designed these benefits to prevent excessive utilization in a population of mostly acutely ill patients, he said, whereas now, 90% of healthcare spending is linked to chronic conditions. For these patients, research shows that incentivizing patients to adhere to medications results in fewer hospitalizations and, therefore, more cost savings, Thorpe noted. These plans also do not consider that there is no average patient, he said, and healthcare providers should be able to match each patient to the best treatment option for them rather than trying out other less expensive medications first. “To the extent that physicians can have the flexibility to provide medications and treatments to patients that are going to have the best clinical response, that’s better outcomes at lower cost,” Dr. Thorpe said. While research shows heterogeneity in patient outcomes with different medication, “benefit designs from the past just don’t recognize that.”
Neither America’s Health Insurance Plans nor Pharmaceutical Care Management Association responded to a request for comment.
Quardricos Driskell, executive director of Let My Doctors Decide and vice president of government relations and public policy at the Autoimmune Association, hopes the study will spur action by policy makers and health plans to improve access to medications for the people who need them. Another larger point of the report is to “uphold the sanctity of protecting the doctor and patient relationship,” he said in an interview, adding “that decisions fundamentally need to be made not by insurance plans or middleman pharmacy benefit managers, but by the provider and patient.”
Mr. Driskell and Dr. Thorpe reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Two biomarkers identify high-risk lupus nephritis
Levels at the time of flare predict outcomes
At the time of a nephritis flare in patients with systemic lupus erythematosus (SLE), elevated levels of two neutrophil extracellular trap (NET) protein complexes, elastase-DNA and HMGB1-DNA, predict declining renal function, poor response to therapy, and adverse renal outcomes, according to work presented at the annual meeting of the Canadian Rheumatology Association.
“These proteins are not only predominantly elevated in patients with proliferative lupus nephritis, but they correlate with adverse renal outcomes when patients are followed over 24 months,” reported Laura P. Whittall-Garcia, MD, a clinical fellow in rheumatology at the University of Toronto.
Lupus nephritis is common in SLE, developing in about 50% of patients, according to Dr. Whittall-Garcia. Of these, up to 20% will not respond to standard therapies, typically resulting in end-stage renal disease. Up until now, there has been no reliable method of predicting this adverse clinical course.
Proteins identified in NETs
The series of studies conducted by Dr. Whittall-Garcia and coinvestigators were focused on NETs, a network of strings of DNA that typically bind pathogenic microbes to prevent infection but can participate in the pathology of immune-mediated conditions. As Dr. Whittall-Garcia explained, DNA extruded from NETs has been a source of autoantigens.
Based on earlier work, they pursued the hypothesis that high mobility group box 1 (HMGB1) and elastase, which are both NET components, mediate NETosis, the immune response that protects against microbes in healthy individuals but contributes to tissue damage in patients with immune-related disorders. The first aim of this work was to confirm that elevations of elastase-DNA and HMGB1-DNA correlate with active lupus nephritis. The second aim was to determine if levels of these proteins at the time of lupus nephritis flare predicted renal outcomes at 12 and 24 months.
To pursue the first hypothesis, 49 patients with active SLE (18 of whom had active lupus nephritis) were evaluated along with 23 patients with inactive SLE and 20 healthy controls.
Highest levels seen in proliferative nephritis
Relative to healthy controls, patients with active SLE have highly significantly increased levels of both proteins (P < .0001). And relative to those with inactive SLE, the levels of active patients were higher but fell short of statistical significance. However, when the researchers compared those with active lupus nephritis with those who had active SLE but no nephritis, both proteins were significantly higher (P < .04), and the levels in patients with proliferative relative to nonproliferative lupus nephritis were higher still (P < .009).
To pursue the second aim of the study, the researchers retrospectively evaluated 109 patients with SLE. All had active lupus nephritis, a baseline estimated glomerular filtration rate (eGFR) greater than 30 mL/min prior to the flare, and at least 2 years of follow-up. They evaluated complete response at 12 and 24 months, percent decline in eGFR, and severe renal impairment (eGFR ≤ 30 mL/min) in the context of levels of elastase and HMGB1.
With elevations in either NET remnant, the odds ratio of failing to achieve a complete response at 24 months were approximately doubled for elastase-DNA (OR, 1.96; P = .01) and for HMGB1 (OR, 2.61; P = .02). For the endpoint of severe renal impairment 24 months after a lupus nephritis flare, there was also a positive association with both elastase-DNA (OR, 1.55; P = .005) and HMBG1-DNA (OR, 1.91; P = .01).
“For every 100-unit increase in elastase-DNA complexes, there is a 4.8% decrease in eGFR,” reported Dr. Whittall-Garcia, who noted this relationship was highly statistically significant (P < .0001). For HMGB1-DNA, each 100-unit increase was associated with a 5.3% decrease in eGFR (P = .0006).
No other biomarkers compare for prognosis
“After adjusting for multiple variables, these protein levels at the time of the flare outperformed all conventional biomarkers, including proteinuria and complement levels,” Dr. Whittall-Garcia said.
Larger validating studies are needed, but Dr. Whittall-Garcia is optimistic that measuring these NET remnant levels will prove useful for monitoring patients at the time of the lupus nephritis flare and over time for the purposes of predicting adverse outcomes and response to therapy.
Although more work is needed, Adegbenga A. Bankole, MD, associate professor of medicine at Virginia Tech University and chief of the rheumatology division at the Carilion Clinic, both in Roanoke, agreed that this is a promising research direction. He reported that NETs have been attracting interest at several research centers for their potential in helping to understand the pathogenesis of lupus nephritis.
“It is unlikely that any one test will ever be a panacea in the diagnosis or predictor of outcomes in lupus nephritis,” Dr. Bankole said in an interview, but “given the importance of NETosis in the development of this disease, studies like this will form the basis of the multistep process through which we will improve patient care.”
With further progress in this area, Dr. Bankole predicted that these studies will lead to clinical applications.
“Dr. Whittall-Garcia and her team will help in the development of diagnostic and/or predictive algorithms that may go on to help improve survival of future patients with SLE,” he said.Dr. Whittall-Garcia and Dr. Bankole report they have no relevant financial relationships.
Levels at the time of flare predict outcomes
Levels at the time of flare predict outcomes
At the time of a nephritis flare in patients with systemic lupus erythematosus (SLE), elevated levels of two neutrophil extracellular trap (NET) protein complexes, elastase-DNA and HMGB1-DNA, predict declining renal function, poor response to therapy, and adverse renal outcomes, according to work presented at the annual meeting of the Canadian Rheumatology Association.
“These proteins are not only predominantly elevated in patients with proliferative lupus nephritis, but they correlate with adverse renal outcomes when patients are followed over 24 months,” reported Laura P. Whittall-Garcia, MD, a clinical fellow in rheumatology at the University of Toronto.
Lupus nephritis is common in SLE, developing in about 50% of patients, according to Dr. Whittall-Garcia. Of these, up to 20% will not respond to standard therapies, typically resulting in end-stage renal disease. Up until now, there has been no reliable method of predicting this adverse clinical course.
Proteins identified in NETs
The series of studies conducted by Dr. Whittall-Garcia and coinvestigators were focused on NETs, a network of strings of DNA that typically bind pathogenic microbes to prevent infection but can participate in the pathology of immune-mediated conditions. As Dr. Whittall-Garcia explained, DNA extruded from NETs has been a source of autoantigens.
Based on earlier work, they pursued the hypothesis that high mobility group box 1 (HMGB1) and elastase, which are both NET components, mediate NETosis, the immune response that protects against microbes in healthy individuals but contributes to tissue damage in patients with immune-related disorders. The first aim of this work was to confirm that elevations of elastase-DNA and HMGB1-DNA correlate with active lupus nephritis. The second aim was to determine if levels of these proteins at the time of lupus nephritis flare predicted renal outcomes at 12 and 24 months.
To pursue the first hypothesis, 49 patients with active SLE (18 of whom had active lupus nephritis) were evaluated along with 23 patients with inactive SLE and 20 healthy controls.
Highest levels seen in proliferative nephritis
Relative to healthy controls, patients with active SLE have highly significantly increased levels of both proteins (P < .0001). And relative to those with inactive SLE, the levels of active patients were higher but fell short of statistical significance. However, when the researchers compared those with active lupus nephritis with those who had active SLE but no nephritis, both proteins were significantly higher (P < .04), and the levels in patients with proliferative relative to nonproliferative lupus nephritis were higher still (P < .009).
To pursue the second aim of the study, the researchers retrospectively evaluated 109 patients with SLE. All had active lupus nephritis, a baseline estimated glomerular filtration rate (eGFR) greater than 30 mL/min prior to the flare, and at least 2 years of follow-up. They evaluated complete response at 12 and 24 months, percent decline in eGFR, and severe renal impairment (eGFR ≤ 30 mL/min) in the context of levels of elastase and HMGB1.
With elevations in either NET remnant, the odds ratio of failing to achieve a complete response at 24 months were approximately doubled for elastase-DNA (OR, 1.96; P = .01) and for HMGB1 (OR, 2.61; P = .02). For the endpoint of severe renal impairment 24 months after a lupus nephritis flare, there was also a positive association with both elastase-DNA (OR, 1.55; P = .005) and HMBG1-DNA (OR, 1.91; P = .01).
“For every 100-unit increase in elastase-DNA complexes, there is a 4.8% decrease in eGFR,” reported Dr. Whittall-Garcia, who noted this relationship was highly statistically significant (P < .0001). For HMGB1-DNA, each 100-unit increase was associated with a 5.3% decrease in eGFR (P = .0006).
No other biomarkers compare for prognosis
“After adjusting for multiple variables, these protein levels at the time of the flare outperformed all conventional biomarkers, including proteinuria and complement levels,” Dr. Whittall-Garcia said.
Larger validating studies are needed, but Dr. Whittall-Garcia is optimistic that measuring these NET remnant levels will prove useful for monitoring patients at the time of the lupus nephritis flare and over time for the purposes of predicting adverse outcomes and response to therapy.
Although more work is needed, Adegbenga A. Bankole, MD, associate professor of medicine at Virginia Tech University and chief of the rheumatology division at the Carilion Clinic, both in Roanoke, agreed that this is a promising research direction. He reported that NETs have been attracting interest at several research centers for their potential in helping to understand the pathogenesis of lupus nephritis.
“It is unlikely that any one test will ever be a panacea in the diagnosis or predictor of outcomes in lupus nephritis,” Dr. Bankole said in an interview, but “given the importance of NETosis in the development of this disease, studies like this will form the basis of the multistep process through which we will improve patient care.”
With further progress in this area, Dr. Bankole predicted that these studies will lead to clinical applications.
“Dr. Whittall-Garcia and her team will help in the development of diagnostic and/or predictive algorithms that may go on to help improve survival of future patients with SLE,” he said.Dr. Whittall-Garcia and Dr. Bankole report they have no relevant financial relationships.
At the time of a nephritis flare in patients with systemic lupus erythematosus (SLE), elevated levels of two neutrophil extracellular trap (NET) protein complexes, elastase-DNA and HMGB1-DNA, predict declining renal function, poor response to therapy, and adverse renal outcomes, according to work presented at the annual meeting of the Canadian Rheumatology Association.
“These proteins are not only predominantly elevated in patients with proliferative lupus nephritis, but they correlate with adverse renal outcomes when patients are followed over 24 months,” reported Laura P. Whittall-Garcia, MD, a clinical fellow in rheumatology at the University of Toronto.
Lupus nephritis is common in SLE, developing in about 50% of patients, according to Dr. Whittall-Garcia. Of these, up to 20% will not respond to standard therapies, typically resulting in end-stage renal disease. Up until now, there has been no reliable method of predicting this adverse clinical course.
Proteins identified in NETs
The series of studies conducted by Dr. Whittall-Garcia and coinvestigators were focused on NETs, a network of strings of DNA that typically bind pathogenic microbes to prevent infection but can participate in the pathology of immune-mediated conditions. As Dr. Whittall-Garcia explained, DNA extruded from NETs has been a source of autoantigens.
Based on earlier work, they pursued the hypothesis that high mobility group box 1 (HMGB1) and elastase, which are both NET components, mediate NETosis, the immune response that protects against microbes in healthy individuals but contributes to tissue damage in patients with immune-related disorders. The first aim of this work was to confirm that elevations of elastase-DNA and HMGB1-DNA correlate with active lupus nephritis. The second aim was to determine if levels of these proteins at the time of lupus nephritis flare predicted renal outcomes at 12 and 24 months.
To pursue the first hypothesis, 49 patients with active SLE (18 of whom had active lupus nephritis) were evaluated along with 23 patients with inactive SLE and 20 healthy controls.
Highest levels seen in proliferative nephritis
Relative to healthy controls, patients with active SLE have highly significantly increased levels of both proteins (P < .0001). And relative to those with inactive SLE, the levels of active patients were higher but fell short of statistical significance. However, when the researchers compared those with active lupus nephritis with those who had active SLE but no nephritis, both proteins were significantly higher (P < .04), and the levels in patients with proliferative relative to nonproliferative lupus nephritis were higher still (P < .009).
To pursue the second aim of the study, the researchers retrospectively evaluated 109 patients with SLE. All had active lupus nephritis, a baseline estimated glomerular filtration rate (eGFR) greater than 30 mL/min prior to the flare, and at least 2 years of follow-up. They evaluated complete response at 12 and 24 months, percent decline in eGFR, and severe renal impairment (eGFR ≤ 30 mL/min) in the context of levels of elastase and HMGB1.
With elevations in either NET remnant, the odds ratio of failing to achieve a complete response at 24 months were approximately doubled for elastase-DNA (OR, 1.96; P = .01) and for HMGB1 (OR, 2.61; P = .02). For the endpoint of severe renal impairment 24 months after a lupus nephritis flare, there was also a positive association with both elastase-DNA (OR, 1.55; P = .005) and HMBG1-DNA (OR, 1.91; P = .01).
“For every 100-unit increase in elastase-DNA complexes, there is a 4.8% decrease in eGFR,” reported Dr. Whittall-Garcia, who noted this relationship was highly statistically significant (P < .0001). For HMGB1-DNA, each 100-unit increase was associated with a 5.3% decrease in eGFR (P = .0006).
No other biomarkers compare for prognosis
“After adjusting for multiple variables, these protein levels at the time of the flare outperformed all conventional biomarkers, including proteinuria and complement levels,” Dr. Whittall-Garcia said.
Larger validating studies are needed, but Dr. Whittall-Garcia is optimistic that measuring these NET remnant levels will prove useful for monitoring patients at the time of the lupus nephritis flare and over time for the purposes of predicting adverse outcomes and response to therapy.
Although more work is needed, Adegbenga A. Bankole, MD, associate professor of medicine at Virginia Tech University and chief of the rheumatology division at the Carilion Clinic, both in Roanoke, agreed that this is a promising research direction. He reported that NETs have been attracting interest at several research centers for their potential in helping to understand the pathogenesis of lupus nephritis.
“It is unlikely that any one test will ever be a panacea in the diagnosis or predictor of outcomes in lupus nephritis,” Dr. Bankole said in an interview, but “given the importance of NETosis in the development of this disease, studies like this will form the basis of the multistep process through which we will improve patient care.”
With further progress in this area, Dr. Bankole predicted that these studies will lead to clinical applications.
“Dr. Whittall-Garcia and her team will help in the development of diagnostic and/or predictive algorithms that may go on to help improve survival of future patients with SLE,” he said.Dr. Whittall-Garcia and Dr. Bankole report they have no relevant financial relationships.
FROM CRA 2023