User login
Second group B meningitis vaccine wins FDA nod of approval
A second serogroup B meningococcal vaccine has been approved by the Food and Drug Administration, based on an international study of over 2,500 adolescents and young adults, the agency announced on Jan. 23.
The vaccine is approved to prevent invasive meningococcal disease caused by Neisseria meningitidis serogroup B in people aged 10-25 years. The vaccine will be marketed as Bexsero, by Novartis Vaccines and Diagnostics, according to the FDA statement. The first serogroup B meningococcal vaccine was approved in October 2014; that vaccine is Trumenba, manufactured by Wyeth Pharmaceuticals. “The approval of these vaccines represents a major public health accomplishment toward preventing this life-threatening disease,” Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, said in the statement.
Before the two vaccines were approved, commercially available FDA-approved meningococcal vaccines covered four of the five main serogroups of N. meningitidis bacteria that cause meningococcal disease (A, C, Y, and W). In 2012, about 160 of the approximately 500 cases of meningococcal disease reported in the United States were caused by serogroup B, according to Centers for Disease Control and Prevention data cited in the FDA statement.
Approval of Bexsero was based on three studies of about 2,600 adolescents and young adults in Canada, Australia, Chile, and the United Kingdom. After two doses of the vaccine, 62%-88% of recipients “had antibodies in their blood that killed three different N. meningitidis serogroup B strains in tests carried out in a laboratory,” compared with 0-23% before they were vaccinated, according to the FDA statement. The most common adverse effects associated with the vaccine include pain and swelling at the injection site, headache, diarrhea, muscle pain, joint pain, fatigue, and chills. Safety information is from an evaluation of 5,000 people who received the vaccine in the United States and other countries, and in over 15,000 people who received the vaccine during two outbreaks of meningococcal disease at universities in the United States.
Almost 30,000 doses of the vaccine were provided to students and staff members during outbreaks of meningitis B at Princeton (New Jersey) University and the University of California, Santa Barbara, according to Novartis.
Because this was an accelerated approval, Novartis will conduct studies to determine if the vaccine is effective against other strains of N. meningitidis serogroup B. Bexsero was approved in people aged 2 months and older in Europe in January 2013.
More information on serogroup B meningococcal vaccine and the outbreaks is available on the CDC website at http://www.cdc.gov/meningococcal/outbreaks/vaccine-serogroupB.html
A second serogroup B meningococcal vaccine has been approved by the Food and Drug Administration, based on an international study of over 2,500 adolescents and young adults, the agency announced on Jan. 23.
The vaccine is approved to prevent invasive meningococcal disease caused by Neisseria meningitidis serogroup B in people aged 10-25 years. The vaccine will be marketed as Bexsero, by Novartis Vaccines and Diagnostics, according to the FDA statement. The first serogroup B meningococcal vaccine was approved in October 2014; that vaccine is Trumenba, manufactured by Wyeth Pharmaceuticals. “The approval of these vaccines represents a major public health accomplishment toward preventing this life-threatening disease,” Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, said in the statement.
Before the two vaccines were approved, commercially available FDA-approved meningococcal vaccines covered four of the five main serogroups of N. meningitidis bacteria that cause meningococcal disease (A, C, Y, and W). In 2012, about 160 of the approximately 500 cases of meningococcal disease reported in the United States were caused by serogroup B, according to Centers for Disease Control and Prevention data cited in the FDA statement.
Approval of Bexsero was based on three studies of about 2,600 adolescents and young adults in Canada, Australia, Chile, and the United Kingdom. After two doses of the vaccine, 62%-88% of recipients “had antibodies in their blood that killed three different N. meningitidis serogroup B strains in tests carried out in a laboratory,” compared with 0-23% before they were vaccinated, according to the FDA statement. The most common adverse effects associated with the vaccine include pain and swelling at the injection site, headache, diarrhea, muscle pain, joint pain, fatigue, and chills. Safety information is from an evaluation of 5,000 people who received the vaccine in the United States and other countries, and in over 15,000 people who received the vaccine during two outbreaks of meningococcal disease at universities in the United States.
Almost 30,000 doses of the vaccine were provided to students and staff members during outbreaks of meningitis B at Princeton (New Jersey) University and the University of California, Santa Barbara, according to Novartis.
Because this was an accelerated approval, Novartis will conduct studies to determine if the vaccine is effective against other strains of N. meningitidis serogroup B. Bexsero was approved in people aged 2 months and older in Europe in January 2013.
More information on serogroup B meningococcal vaccine and the outbreaks is available on the CDC website at http://www.cdc.gov/meningococcal/outbreaks/vaccine-serogroupB.html
A second serogroup B meningococcal vaccine has been approved by the Food and Drug Administration, based on an international study of over 2,500 adolescents and young adults, the agency announced on Jan. 23.
The vaccine is approved to prevent invasive meningococcal disease caused by Neisseria meningitidis serogroup B in people aged 10-25 years. The vaccine will be marketed as Bexsero, by Novartis Vaccines and Diagnostics, according to the FDA statement. The first serogroup B meningococcal vaccine was approved in October 2014; that vaccine is Trumenba, manufactured by Wyeth Pharmaceuticals. “The approval of these vaccines represents a major public health accomplishment toward preventing this life-threatening disease,” Dr. Karen Midthun, director of the FDA’s Center for Biologics Evaluation and Research, said in the statement.
Before the two vaccines were approved, commercially available FDA-approved meningococcal vaccines covered four of the five main serogroups of N. meningitidis bacteria that cause meningococcal disease (A, C, Y, and W). In 2012, about 160 of the approximately 500 cases of meningococcal disease reported in the United States were caused by serogroup B, according to Centers for Disease Control and Prevention data cited in the FDA statement.
Approval of Bexsero was based on three studies of about 2,600 adolescents and young adults in Canada, Australia, Chile, and the United Kingdom. After two doses of the vaccine, 62%-88% of recipients “had antibodies in their blood that killed three different N. meningitidis serogroup B strains in tests carried out in a laboratory,” compared with 0-23% before they were vaccinated, according to the FDA statement. The most common adverse effects associated with the vaccine include pain and swelling at the injection site, headache, diarrhea, muscle pain, joint pain, fatigue, and chills. Safety information is from an evaluation of 5,000 people who received the vaccine in the United States and other countries, and in over 15,000 people who received the vaccine during two outbreaks of meningococcal disease at universities in the United States.
Almost 30,000 doses of the vaccine were provided to students and staff members during outbreaks of meningitis B at Princeton (New Jersey) University and the University of California, Santa Barbara, according to Novartis.
Because this was an accelerated approval, Novartis will conduct studies to determine if the vaccine is effective against other strains of N. meningitidis serogroup B. Bexsero was approved in people aged 2 months and older in Europe in January 2013.
More information on serogroup B meningococcal vaccine and the outbreaks is available on the CDC website at http://www.cdc.gov/meningococcal/outbreaks/vaccine-serogroupB.html
FROM THE FDA
FDA panel backs antifungal for invasive aspergillosis, mucormycosis
SILVER SPRING, MD. – A novel treatment for invasive aspergillosis and invasive mucormycosis gained the support of a Food and Drug Administration advisory panel, although members were more ambivalent about the mucormycosis indication, based on the small amount of data available in those patients.
At a meeting on Jan. 22, the FDA’s Anti-Infective Drugs Advisory Committee voted 11-0 that there was “substantial evidence” that isavuconazonium, an antifungal prodrug, was safe and effective for the treatment of invasive aspergillosis, a life-threatening condition most commonly seen in immunocompromised patients. The drug was studied in a phase III study comparing isavuconazonium to voriconazole, the standard of care, in more than 500 patients.
The panel voted 8-2, with one abstention, that there was substantial evidence it was safe and effective for treating patients with invasive mucormycosis, with panelists voting on both sides of the question citing the study of only 37 patients that used historical controls as an issue. Those supporting approval for mucormycosis said that, if approved, the manufacturer, Astellas, should be required to conduct a phase IV trial further evaluating this treatment for patients with this infection.
Isavuconazonium is a prodrug of isavuconazole, a triazole antifungal, and would be available in an oral capsule formulation and a powder formulation that is reconstituted for intravenous administration (and needs to be administered through an in-line filter in case of particulate formation).
For aspergillosis, “I do believe that this drug provides a reasonable alternative to the current therapies that are available without additional toxicities,” said panelist Dr. Paige Waterman of the Global Emerging Infections Surveillance and Response System at the Walter Reed Army Medical Center, Silver Spring, Md. Other panelists agreed with her recommendations that labeling should make clear that it should not be used in people under age 18 years or in pregnant women, that a filter should be used when administered intravenously, and that labeling should include a statement about the increased risk of hepatotoxicity that also appears in the labeling of other drugs in the same class.
Because it has been associated with a shortened QT interval, she said that screening ECGs should be recommended for patients and that there should be some extra caution when it is prescribed to people who are Asian or of Asian descent, since drug concentrations were slightly higher among Asian patients who received the drug. Other recommendations from panelists included the need to conduct studies of the drug to provide information on therapeutic drug monitoring and in people under 18 years.
For treatment of invasive mucormycosis, an even rarer fungal infection which, in hospitals, is associated with the use of contaminated materials or organ transplantation, the panelists were more hesitant, but those voting in favor of approval cited the significance of the condition and the reasonable efficacy results in the study, stressing that postmarketing studies were critical. Panelists also noted that more clinical data are clearly needed in patients with this infection and the lack of data directly comparing it to amphotericin B – the only FDA-approved drug for this indication – was problematic.

“This drug really does fill an unmet need; I have high hopes that it is at least as good as amphotericin. But I do think we need more data to confirm that,” said Dr. Michael Neely, chair of antimicrobial stewardship at Children’s Hospital, Los Angeles, who was among those voting in favor of this indication. It appears to have a better safety profile and it would be available in both oral and intravenous formulations, he added. Amphotericin B is available only in an intravenous (IV) formulation.
If approved, isavuconazonium would provide an alternative to voriconazole for treating aspergillosis, and the IV formulation does not contain cyclodextrin, unlike the IV formulation of voriconazole, which limits its use in patients with moderate to severe renal dysfunction, according to Astellas. Safety concerns specific to isavuconazonium include QT-segment shortening and particulate formation in the IV formulation, according to the FDA.
The randomized, double-blind, international, noninferiority study compared treatment with isavuconazonium to voriconazole in 516 adults with invasive aspergillosis. In the randomized study, the mean age of patients was 51 years, 60% were men, most were white, 11% were in the United States and Canada, 20% had had an allogeneic bone marrow transplant, and 70% had an uncontrolled malignancy at baseline (Infect. Drug Resist. 2013;6:16374). The primary effectiveness endpoint, all-cause mortality through day 42, was 19% in those on isavuconazonium, compared with 20% in those on voriconazole. The study met the prespecified noninferiority margin. The rates of deaths and serious adverse events were similar to voriconazole, and there were fewer adverse events in those on isavuconazonium requiring discontinuation of the drug (14% vs. 23%).
Differences in adverse events included lower rates of hepatobiliary disorders, including hyperbilirubinemia, abnormal hepatic function, and jaundice (9% vs. 16%); skin and subcutaneous tissue disorders, including rash, erythema, and drug eruption (34% vs 43%) associated with isavuconazonium, compared with voriconazole. Common adverse events included nausea (28%), vomiting (25%), and diarrhea (24%). Decreases in the QT segment occurred in 7.5% of those on isavuconazonium, compared with 4.5% of those on voriconazole, but were not associated with clinical events.
The prospective, open-label, single-arm study evaluated isavuconazonium in 37 patients with proven or probable mucormycosis infections, whose mean age was 49 years; 59% had a hematologic malignancy, and about 40% were neutropenic at baseline. All-cause mortality at day 42 was almost 38%, which was similar to the mortality rate for amphotericin in the literature, according to Astellas.
There are about 12,000 cases of aspergillosis and about 500 cases of mucormycosis in the United States every year, the company said. In addition to voriconazole, other drugs approved for invasive aspergillosis include amphotericin formulations, itraconazole, and caspofungin. The FDA usually follows the recommendations of its advisory panels. The FDA is expected to make a decision by March 8, according to Astellas, which plans to market the drug as Cresemba if approved. It is also under review in Europe for the same indications.
Panelists were cleared of potential conflicts of interest related to the topic of the meeting. In some cases, a panelist may be given a waiver but not at this meeting.
Dr. Daniel Ouellette, FCCP, comments: Serious fungal infections are being increasingly recognized in patients who are immunocompromised or critically ill, but effective treatments with an acceptable side-effect profile have been sparse. Just a few years ago, the mainstay of treatment was amphotericin, an agent whose use has been associated with a variety of complications. However, novel agents have been developed that hold promise. The recent development of isavuconazonium, a pro-drug of isavuconazole, may lead to further augmentation of the clinician's armamentarium in fighting these illnesses.
Dr. Daniel Ouellette, FCCP, comments: Serious fungal infections are being increasingly recognized in patients who are immunocompromised or critically ill, but effective treatments with an acceptable side-effect profile have been sparse. Just a few years ago, the mainstay of treatment was amphotericin, an agent whose use has been associated with a variety of complications. However, novel agents have been developed that hold promise. The recent development of isavuconazonium, a pro-drug of isavuconazole, may lead to further augmentation of the clinician's armamentarium in fighting these illnesses.
Dr. Daniel Ouellette, FCCP, comments: Serious fungal infections are being increasingly recognized in patients who are immunocompromised or critically ill, but effective treatments with an acceptable side-effect profile have been sparse. Just a few years ago, the mainstay of treatment was amphotericin, an agent whose use has been associated with a variety of complications. However, novel agents have been developed that hold promise. The recent development of isavuconazonium, a pro-drug of isavuconazole, may lead to further augmentation of the clinician's armamentarium in fighting these illnesses.
SILVER SPRING, MD. – A novel treatment for invasive aspergillosis and invasive mucormycosis gained the support of a Food and Drug Administration advisory panel, although members were more ambivalent about the mucormycosis indication, based on the small amount of data available in those patients.
At a meeting on Jan. 22, the FDA’s Anti-Infective Drugs Advisory Committee voted 11-0 that there was “substantial evidence” that isavuconazonium, an antifungal prodrug, was safe and effective for the treatment of invasive aspergillosis, a life-threatening condition most commonly seen in immunocompromised patients. The drug was studied in a phase III study comparing isavuconazonium to voriconazole, the standard of care, in more than 500 patients.
The panel voted 8-2, with one abstention, that there was substantial evidence it was safe and effective for treating patients with invasive mucormycosis, with panelists voting on both sides of the question citing the study of only 37 patients that used historical controls as an issue. Those supporting approval for mucormycosis said that, if approved, the manufacturer, Astellas, should be required to conduct a phase IV trial further evaluating this treatment for patients with this infection.
Isavuconazonium is a prodrug of isavuconazole, a triazole antifungal, and would be available in an oral capsule formulation and a powder formulation that is reconstituted for intravenous administration (and needs to be administered through an in-line filter in case of particulate formation).
For aspergillosis, “I do believe that this drug provides a reasonable alternative to the current therapies that are available without additional toxicities,” said panelist Dr. Paige Waterman of the Global Emerging Infections Surveillance and Response System at the Walter Reed Army Medical Center, Silver Spring, Md. Other panelists agreed with her recommendations that labeling should make clear that it should not be used in people under age 18 years or in pregnant women, that a filter should be used when administered intravenously, and that labeling should include a statement about the increased risk of hepatotoxicity that also appears in the labeling of other drugs in the same class.
Because it has been associated with a shortened QT interval, she said that screening ECGs should be recommended for patients and that there should be some extra caution when it is prescribed to people who are Asian or of Asian descent, since drug concentrations were slightly higher among Asian patients who received the drug. Other recommendations from panelists included the need to conduct studies of the drug to provide information on therapeutic drug monitoring and in people under 18 years.
For treatment of invasive mucormycosis, an even rarer fungal infection which, in hospitals, is associated with the use of contaminated materials or organ transplantation, the panelists were more hesitant, but those voting in favor of approval cited the significance of the condition and the reasonable efficacy results in the study, stressing that postmarketing studies were critical. Panelists also noted that more clinical data are clearly needed in patients with this infection and the lack of data directly comparing it to amphotericin B – the only FDA-approved drug for this indication – was problematic.
“This drug really does fill an unmet need; I have high hopes that it is at least as good as amphotericin. But I do think we need more data to confirm that,” said Dr. Michael Neely, chair of antimicrobial stewardship at Children’s Hospital, Los Angeles, who was among those voting in favor of this indication. It appears to have a better safety profile and it would be available in both oral and intravenous formulations, he added. Amphotericin B is available only in an intravenous (IV) formulation.
If approved, isavuconazonium would provide an alternative to voriconazole for treating aspergillosis, and the IV formulation does not contain cyclodextrin, unlike the IV formulation of voriconazole, which limits its use in patients with moderate to severe renal dysfunction, according to Astellas. Safety concerns specific to isavuconazonium include QT-segment shortening and particulate formation in the IV formulation, according to the FDA.
The randomized, double-blind, international, noninferiority study compared treatment with isavuconazonium to voriconazole in 516 adults with invasive aspergillosis. In the randomized study, the mean age of patients was 51 years, 60% were men, most were white, 11% were in the United States and Canada, 20% had had an allogeneic bone marrow transplant, and 70% had an uncontrolled malignancy at baseline (Infect. Drug Resist. 2013;6:16374). The primary effectiveness endpoint, all-cause mortality through day 42, was 19% in those on isavuconazonium, compared with 20% in those on voriconazole. The study met the prespecified noninferiority margin. The rates of deaths and serious adverse events were similar to voriconazole, and there were fewer adverse events in those on isavuconazonium requiring discontinuation of the drug (14% vs. 23%).
Differences in adverse events included lower rates of hepatobiliary disorders, including hyperbilirubinemia, abnormal hepatic function, and jaundice (9% vs. 16%); skin and subcutaneous tissue disorders, including rash, erythema, and drug eruption (34% vs 43%) associated with isavuconazonium, compared with voriconazole. Common adverse events included nausea (28%), vomiting (25%), and diarrhea (24%). Decreases in the QT segment occurred in 7.5% of those on isavuconazonium, compared with 4.5% of those on voriconazole, but were not associated with clinical events.
The prospective, open-label, single-arm study evaluated isavuconazonium in 37 patients with proven or probable mucormycosis infections, whose mean age was 49 years; 59% had a hematologic malignancy, and about 40% were neutropenic at baseline. All-cause mortality at day 42 was almost 38%, which was similar to the mortality rate for amphotericin in the literature, according to Astellas.
There are about 12,000 cases of aspergillosis and about 500 cases of mucormycosis in the United States every year, the company said. In addition to voriconazole, other drugs approved for invasive aspergillosis include amphotericin formulations, itraconazole, and caspofungin. The FDA usually follows the recommendations of its advisory panels. The FDA is expected to make a decision by March 8, according to Astellas, which plans to market the drug as Cresemba if approved. It is also under review in Europe for the same indications.
Panelists were cleared of potential conflicts of interest related to the topic of the meeting. In some cases, a panelist may be given a waiver but not at this meeting.
SILVER SPRING, MD. – A novel treatment for invasive aspergillosis and invasive mucormycosis gained the support of a Food and Drug Administration advisory panel, although members were more ambivalent about the mucormycosis indication, based on the small amount of data available in those patients.
At a meeting on Jan. 22, the FDA’s Anti-Infective Drugs Advisory Committee voted 11-0 that there was “substantial evidence” that isavuconazonium, an antifungal prodrug, was safe and effective for the treatment of invasive aspergillosis, a life-threatening condition most commonly seen in immunocompromised patients. The drug was studied in a phase III study comparing isavuconazonium to voriconazole, the standard of care, in more than 500 patients.
The panel voted 8-2, with one abstention, that there was substantial evidence it was safe and effective for treating patients with invasive mucormycosis, with panelists voting on both sides of the question citing the study of only 37 patients that used historical controls as an issue. Those supporting approval for mucormycosis said that, if approved, the manufacturer, Astellas, should be required to conduct a phase IV trial further evaluating this treatment for patients with this infection.
Isavuconazonium is a prodrug of isavuconazole, a triazole antifungal, and would be available in an oral capsule formulation and a powder formulation that is reconstituted for intravenous administration (and needs to be administered through an in-line filter in case of particulate formation).
For aspergillosis, “I do believe that this drug provides a reasonable alternative to the current therapies that are available without additional toxicities,” said panelist Dr. Paige Waterman of the Global Emerging Infections Surveillance and Response System at the Walter Reed Army Medical Center, Silver Spring, Md. Other panelists agreed with her recommendations that labeling should make clear that it should not be used in people under age 18 years or in pregnant women, that a filter should be used when administered intravenously, and that labeling should include a statement about the increased risk of hepatotoxicity that also appears in the labeling of other drugs in the same class.
Because it has been associated with a shortened QT interval, she said that screening ECGs should be recommended for patients and that there should be some extra caution when it is prescribed to people who are Asian or of Asian descent, since drug concentrations were slightly higher among Asian patients who received the drug. Other recommendations from panelists included the need to conduct studies of the drug to provide information on therapeutic drug monitoring and in people under 18 years.
For treatment of invasive mucormycosis, an even rarer fungal infection which, in hospitals, is associated with the use of contaminated materials or organ transplantation, the panelists were more hesitant, but those voting in favor of approval cited the significance of the condition and the reasonable efficacy results in the study, stressing that postmarketing studies were critical. Panelists also noted that more clinical data are clearly needed in patients with this infection and the lack of data directly comparing it to amphotericin B – the only FDA-approved drug for this indication – was problematic.
“This drug really does fill an unmet need; I have high hopes that it is at least as good as amphotericin. But I do think we need more data to confirm that,” said Dr. Michael Neely, chair of antimicrobial stewardship at Children’s Hospital, Los Angeles, who was among those voting in favor of this indication. It appears to have a better safety profile and it would be available in both oral and intravenous formulations, he added. Amphotericin B is available only in an intravenous (IV) formulation.
If approved, isavuconazonium would provide an alternative to voriconazole for treating aspergillosis, and the IV formulation does not contain cyclodextrin, unlike the IV formulation of voriconazole, which limits its use in patients with moderate to severe renal dysfunction, according to Astellas. Safety concerns specific to isavuconazonium include QT-segment shortening and particulate formation in the IV formulation, according to the FDA.
The randomized, double-blind, international, noninferiority study compared treatment with isavuconazonium to voriconazole in 516 adults with invasive aspergillosis. In the randomized study, the mean age of patients was 51 years, 60% were men, most were white, 11% were in the United States and Canada, 20% had had an allogeneic bone marrow transplant, and 70% had an uncontrolled malignancy at baseline (Infect. Drug Resist. 2013;6:16374). The primary effectiveness endpoint, all-cause mortality through day 42, was 19% in those on isavuconazonium, compared with 20% in those on voriconazole. The study met the prespecified noninferiority margin. The rates of deaths and serious adverse events were similar to voriconazole, and there were fewer adverse events in those on isavuconazonium requiring discontinuation of the drug (14% vs. 23%).
Differences in adverse events included lower rates of hepatobiliary disorders, including hyperbilirubinemia, abnormal hepatic function, and jaundice (9% vs. 16%); skin and subcutaneous tissue disorders, including rash, erythema, and drug eruption (34% vs 43%) associated with isavuconazonium, compared with voriconazole. Common adverse events included nausea (28%), vomiting (25%), and diarrhea (24%). Decreases in the QT segment occurred in 7.5% of those on isavuconazonium, compared with 4.5% of those on voriconazole, but were not associated with clinical events.
The prospective, open-label, single-arm study evaluated isavuconazonium in 37 patients with proven or probable mucormycosis infections, whose mean age was 49 years; 59% had a hematologic malignancy, and about 40% were neutropenic at baseline. All-cause mortality at day 42 was almost 38%, which was similar to the mortality rate for amphotericin in the literature, according to Astellas.
There are about 12,000 cases of aspergillosis and about 500 cases of mucormycosis in the United States every year, the company said. In addition to voriconazole, other drugs approved for invasive aspergillosis include amphotericin formulations, itraconazole, and caspofungin. The FDA usually follows the recommendations of its advisory panels. The FDA is expected to make a decision by March 8, according to Astellas, which plans to market the drug as Cresemba if approved. It is also under review in Europe for the same indications.
Panelists were cleared of potential conflicts of interest related to the topic of the meeting. In some cases, a panelist may be given a waiver but not at this meeting.
AT AN FDA ADVISORY COMMITTEE MEETING
NIH report on long-term opioid treatment cites lack of data, research needs
The striking lack of data on the effectiveness and risks of long-term opioid treatment for the increasing number of people in the United States with chronic pain is reflected in the recommendations made by an expert panel convened by the National Institutes of Health.
To address the role of opioids in the treatment of chronic pain, the panelists met during an NIH Pathways to Prevention workshop in late September where more than 20 invited experts spoke on the topic. The panel produced a draft report shortly after the meeting concluded and received public comments, and the final report has been published on the NIH website. An abridged version of the panel’s final report, which highlights key issues surrounding the use of opioids and chronic pain treatment and provides recommendations on the types of research needed in this area, was published online Jan. 13 in the Annals of Internal Medicine (doi:10.7326/M14-2775).

“The overriding question posed to the panel is whether we as a nation are currently approaching chronic pain in the best possible manner that maximizes effectiveness and minimizes harm. The panel determined that the answer was an unequivocal no,” Dr. David Reuben, the panel chair and lead author of the report, said during a Jan. 16 telebriefing that was held to review the panel’s findings and recommendations. “We hope that the information presented in the panel report will shed light on the issues that need further attention and help facilitate research and better practice to improve outcomes for patients,” added Dr. Reuben, who is chief of geriatric medicine and professor of medicine at the University of California, Los Angeles.
The first recommendation in the panel’s position paper is that federal and non-federal agencies should fund research to identify what types of pain, diseases, and patients “are most likely to benefit and incur harm” from opioids. Agencies should also fund the development and evaluation of multi-disciplinary pain interventions, including cost-benefit analyses, and fund research to “develop and validate research measurement tools” that identify patient risk and outcomes, “related to long-term opioid use that can be adapted for clinical settings,” the panel recommended.

The one recommendation that directly pertains to current clinical practice states that “in the absence of definitive evidence, clinicians and health care systems should follow current guidelines by professional societies about which patients and which types of pain should be treated with opioids and about how best to monitor patients and mitigate risk for harm.” In addition, the report “identifies several key evidence gaps and research priorities that must be addressed so that physicians can recognize patients for whom opioids are most appropriate and use optimal regimens for these patients.”
Considering professional society guidelines is one of the main take home-messages of the report for clinicians, Dr. David Steffens, another panelist and author of the paper, said during the telebriefing. Dr. Steffens, professor and chair of the department of psychiatry, University of Connecticut, Farmington, observed that during the workshop, “the core part of what we heard was an astounding lack of data on efficacy of these drugs,” and noted that the majority of recommendations are “forward-looking in terms of a need to get more data.”
Among the main conclusions of the position paper is that while opioids are “clearly the best treatment for some patients with chronic pain ... there are probably more effective approaches” for many other patients. “The challenge is to identify the conditions in patients for which opioid use is most appropriate, the optimal regimens, the alternatives for those who are unlikely to benefit from opioids, and the best approach to ensuring that every patient’s needs are met by a patient-centered health care system,” the report concludes.
A systematic review of the effectiveness and risks of long-term opioids for chronic pain, prepared specifically for the workshop by the Pacific Northwest Evidence-based Practice Center (EPC) at Oregon Health & Science University, Portland, with funding by the Agency for Healthcare Research and Quality, was also published in the same issue of Annals of Internal Medicine (doi:10.7326/M14-2559).
The review, which evaluated evidence in the medical literature about the effectiveness and risks of long-term opioid therapy (more than 3 months) to treat chronic pain in adults, concluded that “reliable conclusions about the effectiveness of long-term opioid therapy for chronic pain are not possible due to the paucity of research to date.” Moreover, evidence indicating that long-term opioid therapy is associated with significant risks for overdoses, abuse, and other sequelae is increasing, according to the review, which defines chronic pain as pain “lasting longer than 3 months or past the normal time for tissue healing.” Dr. Roger Chou, director of the Pacific Northwest EPC and a physician in the OHSU internal medicine clinic, was the lead author of the review.

An estimated 100 million Americans have chronic pain, of whom about 25 million have moderate-to-severe pain that limits activities and adversely affects their quality of life, according to the position paper. Despite other available treatments, opioids are used for long-term management of chronic pain in an estimated 5-8 million Americans and prescriptions for opioids to treat pain increased from 76 million in 1991 to 219 million in 2011. This increase has been accompanied by a rise in opioid overdoses and treatment for addictions to prescription pain medications. The paper cites Centers for Disease Control and Prevention figures estimating that in 2011, there were more than 17,000 opioid-related overdose deaths.
The NIH Pathways to Prevention Workshop was sponsored by the NIH Office of Disease Prevention, the NIH Pain Consortium, the National Institute on Drug Abuse, and the National Institute of Neurological Disorders and Stroke.
None of the authors of the report had disclosures relevant to the topic. Dr. Chou’s disclosures included having received grants from the AHRQ during the study. He has also been a consultant for the U.S. Department of Health and Human Services and the Providers’ Clinical Support System for Opioid Therapies, which is funded by the Substance Abuse and Mental Health Services Administration.
The striking lack of data on the effectiveness and risks of long-term opioid treatment for the increasing number of people in the United States with chronic pain is reflected in the recommendations made by an expert panel convened by the National Institutes of Health.
To address the role of opioids in the treatment of chronic pain, the panelists met during an NIH Pathways to Prevention workshop in late September where more than 20 invited experts spoke on the topic. The panel produced a draft report shortly after the meeting concluded and received public comments, and the final report has been published on the NIH website. An abridged version of the panel’s final report, which highlights key issues surrounding the use of opioids and chronic pain treatment and provides recommendations on the types of research needed in this area, was published online Jan. 13 in the Annals of Internal Medicine (doi:10.7326/M14-2775).
“The overriding question posed to the panel is whether we as a nation are currently approaching chronic pain in the best possible manner that maximizes effectiveness and minimizes harm. The panel determined that the answer was an unequivocal no,” Dr. David Reuben, the panel chair and lead author of the report, said during a Jan. 16 telebriefing that was held to review the panel’s findings and recommendations. “We hope that the information presented in the panel report will shed light on the issues that need further attention and help facilitate research and better practice to improve outcomes for patients,” added Dr. Reuben, who is chief of geriatric medicine and professor of medicine at the University of California, Los Angeles.
The first recommendation in the panel’s position paper is that federal and non-federal agencies should fund research to identify what types of pain, diseases, and patients “are most likely to benefit and incur harm” from opioids. Agencies should also fund the development and evaluation of multi-disciplinary pain interventions, including cost-benefit analyses, and fund research to “develop and validate research measurement tools” that identify patient risk and outcomes, “related to long-term opioid use that can be adapted for clinical settings,” the panel recommended.
The one recommendation that directly pertains to current clinical practice states that “in the absence of definitive evidence, clinicians and health care systems should follow current guidelines by professional societies about which patients and which types of pain should be treated with opioids and about how best to monitor patients and mitigate risk for harm.” In addition, the report “identifies several key evidence gaps and research priorities that must be addressed so that physicians can recognize patients for whom opioids are most appropriate and use optimal regimens for these patients.”
Considering professional society guidelines is one of the main take home-messages of the report for clinicians, Dr. David Steffens, another panelist and author of the paper, said during the telebriefing. Dr. Steffens, professor and chair of the department of psychiatry, University of Connecticut, Farmington, observed that during the workshop, “the core part of what we heard was an astounding lack of data on efficacy of these drugs,” and noted that the majority of recommendations are “forward-looking in terms of a need to get more data.”
Among the main conclusions of the position paper is that while opioids are “clearly the best treatment for some patients with chronic pain ... there are probably more effective approaches” for many other patients. “The challenge is to identify the conditions in patients for which opioid use is most appropriate, the optimal regimens, the alternatives for those who are unlikely to benefit from opioids, and the best approach to ensuring that every patient’s needs are met by a patient-centered health care system,” the report concludes.
A systematic review of the effectiveness and risks of long-term opioids for chronic pain, prepared specifically for the workshop by the Pacific Northwest Evidence-based Practice Center (EPC) at Oregon Health & Science University, Portland, with funding by the Agency for Healthcare Research and Quality, was also published in the same issue of Annals of Internal Medicine (doi:10.7326/M14-2559).
The review, which evaluated evidence in the medical literature about the effectiveness and risks of long-term opioid therapy (more than 3 months) to treat chronic pain in adults, concluded that “reliable conclusions about the effectiveness of long-term opioid therapy for chronic pain are not possible due to the paucity of research to date.” Moreover, evidence indicating that long-term opioid therapy is associated with significant risks for overdoses, abuse, and other sequelae is increasing, according to the review, which defines chronic pain as pain “lasting longer than 3 months or past the normal time for tissue healing.” Dr. Roger Chou, director of the Pacific Northwest EPC and a physician in the OHSU internal medicine clinic, was the lead author of the review.
An estimated 100 million Americans have chronic pain, of whom about 25 million have moderate-to-severe pain that limits activities and adversely affects their quality of life, according to the position paper. Despite other available treatments, opioids are used for long-term management of chronic pain in an estimated 5-8 million Americans and prescriptions for opioids to treat pain increased from 76 million in 1991 to 219 million in 2011. This increase has been accompanied by a rise in opioid overdoses and treatment for addictions to prescription pain medications. The paper cites Centers for Disease Control and Prevention figures estimating that in 2011, there were more than 17,000 opioid-related overdose deaths.
The NIH Pathways to Prevention Workshop was sponsored by the NIH Office of Disease Prevention, the NIH Pain Consortium, the National Institute on Drug Abuse, and the National Institute of Neurological Disorders and Stroke.
None of the authors of the report had disclosures relevant to the topic. Dr. Chou’s disclosures included having received grants from the AHRQ during the study. He has also been a consultant for the U.S. Department of Health and Human Services and the Providers’ Clinical Support System for Opioid Therapies, which is funded by the Substance Abuse and Mental Health Services Administration.
The striking lack of data on the effectiveness and risks of long-term opioid treatment for the increasing number of people in the United States with chronic pain is reflected in the recommendations made by an expert panel convened by the National Institutes of Health.
To address the role of opioids in the treatment of chronic pain, the panelists met during an NIH Pathways to Prevention workshop in late September where more than 20 invited experts spoke on the topic. The panel produced a draft report shortly after the meeting concluded and received public comments, and the final report has been published on the NIH website. An abridged version of the panel’s final report, which highlights key issues surrounding the use of opioids and chronic pain treatment and provides recommendations on the types of research needed in this area, was published online Jan. 13 in the Annals of Internal Medicine (doi:10.7326/M14-2775).
“The overriding question posed to the panel is whether we as a nation are currently approaching chronic pain in the best possible manner that maximizes effectiveness and minimizes harm. The panel determined that the answer was an unequivocal no,” Dr. David Reuben, the panel chair and lead author of the report, said during a Jan. 16 telebriefing that was held to review the panel’s findings and recommendations. “We hope that the information presented in the panel report will shed light on the issues that need further attention and help facilitate research and better practice to improve outcomes for patients,” added Dr. Reuben, who is chief of geriatric medicine and professor of medicine at the University of California, Los Angeles.
The first recommendation in the panel’s position paper is that federal and non-federal agencies should fund research to identify what types of pain, diseases, and patients “are most likely to benefit and incur harm” from opioids. Agencies should also fund the development and evaluation of multi-disciplinary pain interventions, including cost-benefit analyses, and fund research to “develop and validate research measurement tools” that identify patient risk and outcomes, “related to long-term opioid use that can be adapted for clinical settings,” the panel recommended.
The one recommendation that directly pertains to current clinical practice states that “in the absence of definitive evidence, clinicians and health care systems should follow current guidelines by professional societies about which patients and which types of pain should be treated with opioids and about how best to monitor patients and mitigate risk for harm.” In addition, the report “identifies several key evidence gaps and research priorities that must be addressed so that physicians can recognize patients for whom opioids are most appropriate and use optimal regimens for these patients.”
Considering professional society guidelines is one of the main take home-messages of the report for clinicians, Dr. David Steffens, another panelist and author of the paper, said during the telebriefing. Dr. Steffens, professor and chair of the department of psychiatry, University of Connecticut, Farmington, observed that during the workshop, “the core part of what we heard was an astounding lack of data on efficacy of these drugs,” and noted that the majority of recommendations are “forward-looking in terms of a need to get more data.”
Among the main conclusions of the position paper is that while opioids are “clearly the best treatment for some patients with chronic pain ... there are probably more effective approaches” for many other patients. “The challenge is to identify the conditions in patients for which opioid use is most appropriate, the optimal regimens, the alternatives for those who are unlikely to benefit from opioids, and the best approach to ensuring that every patient’s needs are met by a patient-centered health care system,” the report concludes.
A systematic review of the effectiveness and risks of long-term opioids for chronic pain, prepared specifically for the workshop by the Pacific Northwest Evidence-based Practice Center (EPC) at Oregon Health & Science University, Portland, with funding by the Agency for Healthcare Research and Quality, was also published in the same issue of Annals of Internal Medicine (doi:10.7326/M14-2559).
The review, which evaluated evidence in the medical literature about the effectiveness and risks of long-term opioid therapy (more than 3 months) to treat chronic pain in adults, concluded that “reliable conclusions about the effectiveness of long-term opioid therapy for chronic pain are not possible due to the paucity of research to date.” Moreover, evidence indicating that long-term opioid therapy is associated with significant risks for overdoses, abuse, and other sequelae is increasing, according to the review, which defines chronic pain as pain “lasting longer than 3 months or past the normal time for tissue healing.” Dr. Roger Chou, director of the Pacific Northwest EPC and a physician in the OHSU internal medicine clinic, was the lead author of the review.
An estimated 100 million Americans have chronic pain, of whom about 25 million have moderate-to-severe pain that limits activities and adversely affects their quality of life, according to the position paper. Despite other available treatments, opioids are used for long-term management of chronic pain in an estimated 5-8 million Americans and prescriptions for opioids to treat pain increased from 76 million in 1991 to 219 million in 2011. This increase has been accompanied by a rise in opioid overdoses and treatment for addictions to prescription pain medications. The paper cites Centers for Disease Control and Prevention figures estimating that in 2011, there were more than 17,000 opioid-related overdose deaths.
The NIH Pathways to Prevention Workshop was sponsored by the NIH Office of Disease Prevention, the NIH Pain Consortium, the National Institute on Drug Abuse, and the National Institute of Neurological Disorders and Stroke.
None of the authors of the report had disclosures relevant to the topic. Dr. Chou’s disclosures included having received grants from the AHRQ during the study. He has also been a consultant for the U.S. Department of Health and Human Services and the Providers’ Clinical Support System for Opioid Therapies, which is funded by the Substance Abuse and Mental Health Services Administration.

FDA approves vagal blocking device for obesity
A novel implantable device that delivers electrical pulses to the intra-abdominal vagus nerve has been approved for treatment of obesity in adults, providing a less invasive alternative to bariatric surgery.
The Maestro Rechargeable System is “the first weight loss treatment device that targets the nerve pathway between the brain and the stomach that controls feelings of hunger and fullness,” according to the Food and Drug Administration statement released Jan. 14. The last device approved by the FDA for treatment of obesity was the Realize gastric band, in September 2007.
The device is approved for adults aged 18 and older with a body mass index of at least 40-45 kg/m2, or at least 35-39.9 kg/m2 with a related health condition such as high blood pressure or high cholesterol levels, who have tried to lose weight in a supervised weight management program within the past 5 years.
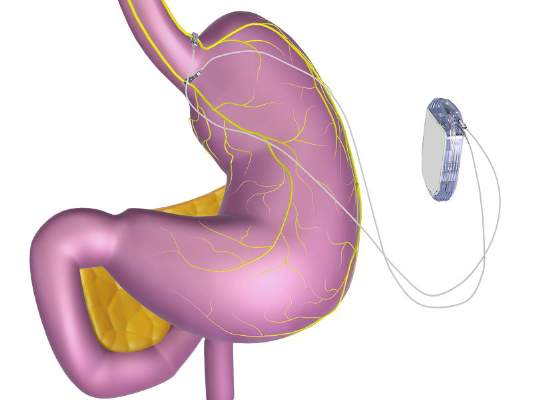
The system includes a rechargeable electrical pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure. “It works by sending intermittent electrical pulses to the trunks in the abdominal vagus nerve, which is involved in regulating stomach emptying and signaling to the brain that the stomach feels empty or full,” the FDA statement said, adding: “Although it is known that the electric stimulation blocks nerve activity between the brain and the stomach, the specific mechanisms for weight loss due to use of the device are unknown.”
The manufacturer, EnteroMedics, refers to the treatment as “VBLOC therapy,” delivered by the Maestro System. The company expects that the device will be available this year “on a limited basis” at select Bariatric Centers of Excellence in the United States, according to a statement issued by EnteroMedics on Jan. 14.
FDA approval was based on the results of the ReCharge study of 233 patients with a body mass index of at least 35 kg/m2; the device was activated in 157 patients, and the remaining patients had the device implanted but it was not activated and they served as controls.
After 12 months, those with the activated device lost 8.5% more excess weight than did the controls. Among those who had the device activated, almost 53% lost at least 20% of their excess weight and 38% lost at least 35% of their excess weight, according to the FDA.
The study did not meet the primary effectiveness endpoint, which was that those on active treatment would lose at least 10% more excess weight than would the controls. However, the majority of an FDA advisory panel that reviewed the data at a meeting in June 2014 supported approval, agreeing that the benefits outweighed the risks for the proposed indication. Panelists cited the fact that the study safety endpoint was met and that the device was effective in helping some people lose weight.
The FDA statement said the decision to approve the device was based on the panel’s recommendation, the study results, and an FDA survey of patient preferences for obesity devices, which found that “a group of patients would accept risks associated with this surgically implanted device for the amounts of weight loss expected to be provided by the device.”
As a condition for approval, EnteroMedics is required to conduct a 5-year postmarketing study that will collect safety and effectiveness data in at least 100 patients, including weight loss, adverse events, surgical revisions and explants and changes in obesity-related comorbidities, according to the FDA.
Serious adverse events in the ReCharge study were nausea, pain at the neuroregulator site, vomiting, and surgical complications; other adverse events were heartburn, problems swallowing, belching, mild nausea, and chest pain, the FDA noted.
The EnteroMedics statement says that contraindications for VBLOC therapy include liver cirrhosis, portal hypertension, esophageal varices or an uncorrectable, clinically significant hiatal hernia; patients for whom magnetic resonance imaging or diathermy use is planned; patients at high risk for surgical complications; and patients who have permanently implanted, electrically-powered medical devices or gastrointestinal devices or prostheses, such as pacemakers, implanted defibrillators, or neurostimulators.
A novel implantable device that delivers electrical pulses to the intra-abdominal vagus nerve has been approved for treatment of obesity in adults, providing a less invasive alternative to bariatric surgery.
The Maestro Rechargeable System is “the first weight loss treatment device that targets the nerve pathway between the brain and the stomach that controls feelings of hunger and fullness,” according to the Food and Drug Administration statement released Jan. 14. The last device approved by the FDA for treatment of obesity was the Realize gastric band, in September 2007.
The device is approved for adults aged 18 and older with a body mass index of at least 40-45 kg/m2, or at least 35-39.9 kg/m2 with a related health condition such as high blood pressure or high cholesterol levels, who have tried to lose weight in a supervised weight management program within the past 5 years.
The system includes a rechargeable electrical pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure. “It works by sending intermittent electrical pulses to the trunks in the abdominal vagus nerve, which is involved in regulating stomach emptying and signaling to the brain that the stomach feels empty or full,” the FDA statement said, adding: “Although it is known that the electric stimulation blocks nerve activity between the brain and the stomach, the specific mechanisms for weight loss due to use of the device are unknown.”
The manufacturer, EnteroMedics, refers to the treatment as “VBLOC therapy,” delivered by the Maestro System. The company expects that the device will be available this year “on a limited basis” at select Bariatric Centers of Excellence in the United States, according to a statement issued by EnteroMedics on Jan. 14.
FDA approval was based on the results of the ReCharge study of 233 patients with a body mass index of at least 35 kg/m2; the device was activated in 157 patients, and the remaining patients had the device implanted but it was not activated and they served as controls.
After 12 months, those with the activated device lost 8.5% more excess weight than did the controls. Among those who had the device activated, almost 53% lost at least 20% of their excess weight and 38% lost at least 35% of their excess weight, according to the FDA.
The study did not meet the primary effectiveness endpoint, which was that those on active treatment would lose at least 10% more excess weight than would the controls. However, the majority of an FDA advisory panel that reviewed the data at a meeting in June 2014 supported approval, agreeing that the benefits outweighed the risks for the proposed indication. Panelists cited the fact that the study safety endpoint was met and that the device was effective in helping some people lose weight.
The FDA statement said the decision to approve the device was based on the panel’s recommendation, the study results, and an FDA survey of patient preferences for obesity devices, which found that “a group of patients would accept risks associated with this surgically implanted device for the amounts of weight loss expected to be provided by the device.”
As a condition for approval, EnteroMedics is required to conduct a 5-year postmarketing study that will collect safety and effectiveness data in at least 100 patients, including weight loss, adverse events, surgical revisions and explants and changes in obesity-related comorbidities, according to the FDA.
Serious adverse events in the ReCharge study were nausea, pain at the neuroregulator site, vomiting, and surgical complications; other adverse events were heartburn, problems swallowing, belching, mild nausea, and chest pain, the FDA noted.
The EnteroMedics statement says that contraindications for VBLOC therapy include liver cirrhosis, portal hypertension, esophageal varices or an uncorrectable, clinically significant hiatal hernia; patients for whom magnetic resonance imaging or diathermy use is planned; patients at high risk for surgical complications; and patients who have permanently implanted, electrically-powered medical devices or gastrointestinal devices or prostheses, such as pacemakers, implanted defibrillators, or neurostimulators.
A novel implantable device that delivers electrical pulses to the intra-abdominal vagus nerve has been approved for treatment of obesity in adults, providing a less invasive alternative to bariatric surgery.
The Maestro Rechargeable System is “the first weight loss treatment device that targets the nerve pathway between the brain and the stomach that controls feelings of hunger and fullness,” according to the Food and Drug Administration statement released Jan. 14. The last device approved by the FDA for treatment of obesity was the Realize gastric band, in September 2007.
The device is approved for adults aged 18 and older with a body mass index of at least 40-45 kg/m2, or at least 35-39.9 kg/m2 with a related health condition such as high blood pressure or high cholesterol levels, who have tried to lose weight in a supervised weight management program within the past 5 years.
The system includes a rechargeable electrical pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure. “It works by sending intermittent electrical pulses to the trunks in the abdominal vagus nerve, which is involved in regulating stomach emptying and signaling to the brain that the stomach feels empty or full,” the FDA statement said, adding: “Although it is known that the electric stimulation blocks nerve activity between the brain and the stomach, the specific mechanisms for weight loss due to use of the device are unknown.”
The manufacturer, EnteroMedics, refers to the treatment as “VBLOC therapy,” delivered by the Maestro System. The company expects that the device will be available this year “on a limited basis” at select Bariatric Centers of Excellence in the United States, according to a statement issued by EnteroMedics on Jan. 14.
FDA approval was based on the results of the ReCharge study of 233 patients with a body mass index of at least 35 kg/m2; the device was activated in 157 patients, and the remaining patients had the device implanted but it was not activated and they served as controls.
After 12 months, those with the activated device lost 8.5% more excess weight than did the controls. Among those who had the device activated, almost 53% lost at least 20% of their excess weight and 38% lost at least 35% of their excess weight, according to the FDA.
The study did not meet the primary effectiveness endpoint, which was that those on active treatment would lose at least 10% more excess weight than would the controls. However, the majority of an FDA advisory panel that reviewed the data at a meeting in June 2014 supported approval, agreeing that the benefits outweighed the risks for the proposed indication. Panelists cited the fact that the study safety endpoint was met and that the device was effective in helping some people lose weight.
The FDA statement said the decision to approve the device was based on the panel’s recommendation, the study results, and an FDA survey of patient preferences for obesity devices, which found that “a group of patients would accept risks associated with this surgically implanted device for the amounts of weight loss expected to be provided by the device.”
As a condition for approval, EnteroMedics is required to conduct a 5-year postmarketing study that will collect safety and effectiveness data in at least 100 patients, including weight loss, adverse events, surgical revisions and explants and changes in obesity-related comorbidities, according to the FDA.
Serious adverse events in the ReCharge study were nausea, pain at the neuroregulator site, vomiting, and surgical complications; other adverse events were heartburn, problems swallowing, belching, mild nausea, and chest pain, the FDA noted.
The EnteroMedics statement says that contraindications for VBLOC therapy include liver cirrhosis, portal hypertension, esophageal varices or an uncorrectable, clinically significant hiatal hernia; patients for whom magnetic resonance imaging or diathermy use is planned; patients at high risk for surgical complications; and patients who have permanently implanted, electrically-powered medical devices or gastrointestinal devices or prostheses, such as pacemakers, implanted defibrillators, or neurostimulators.
FDA panel votes against approval of oral desmopressin for nocturia
HYATTSVILLE, MD. – The majority of a Food and Drug Administration advisory panel voted against approval of an orally disintegrating sublingual tablet formulation of desmopressin as a treatment for nocturia, citing uncertainties about the clinical benefits in the pivotal trials and concerns about the risk of hyponatremia.
At the meeting on Jan. 12, the FDA Endocrinologic and Metabolic Drugs Advisory Committee voted 10 to 5, with two abstentions, that the benefits of this desmopressin formulation in clinical trials did not outweigh the risks for the proposed indication, the treatment of nocturia due to nocturnal polyuria in adults who wake up two or more times each night to void. The indication proposed by Ferring Pharmaceuticals includes the statement that prior to treatment, “lifestyle changes and other treatable medical causes of nocturia should be addressed.”

Although the panel agreed that treatment was associated with statistically significant effects on the two primary endpoints in the two pivotal trials, those voting against approval said there were uncertainties about the clinical benefits, which several panelists said could be addressed in preapproval studies.
The panel generally agreed that the studies demonstrated a low risk of hyponatremia, the main safety concern associated with treatment. But they said that it was unclear whether the appropriate patients would be selected for treatment and would be properly monitored as recommended during treatment in real-world clinical situations. Examples included whether patients would remember to stop taking the medication when they developed an illness that could increase their risk of hyponatremia, or if they developed a disease or condition that affects sodium as they got older. There was also some uncertainty over the clinical significance of the small decreases in serum sodium in patients during the course of the 3-month studies, which evaluated two gender-specific doses that were lower than the higher doses previously studied that were associated with an increased risk of hyponatremia.
“The clinical benefit of the active medication relative to the substantial improvements seen in the placebo arm was relatively small,” said panelist Dr. Brendan Everett, director of the general cardiology inpatient service at Brigham and Women’s Hospital, Boston, who voted against approval. “The sponsor is on to something with respect to the time to the first nocturnal void being longer” among those on the drug, an exploratory endpoint that he recommended should be studied further. The company’s plan to address the outliers with substantial drops in sodium that are considered more dangerous was “reasonable,” he said, but added that he shared the concern of other committee members “that implementing that risk-mitigation strategy in the real world after approval would be difficult.”
The company developed lower doses to minimize the risk of hyponatremia and conducted two pivotal phase III confirmatory randomized, placebo-controlled studies of 268 women and almost 400 men with at least two voids per night (an average of 2.9 episodes), evaluating a 25-mcg dose in women and 50 mcg and 75 mcg in men over 3 months. Exclusion criteria included treatable causes of nocturia, such as diabetes insipidus and cardiac failure, and medical conditions that increased risk, such as hyponatremia and psychogenic polydipsia. (The company is not pursuing approval of the 75-mcg dose in men.)
Among the women, the number of voids per night over 3 months dropped by a mean of 1.46 among those on desmopressin vs. 1.24 among those on placebo, a 0.22 difference that was statistically significant, despite the large placebo effect. In the study of men, the number of voids per night dropped by a mean of 1.25 among those on the 50-mcg dose, vs. 0.88 among those on placebo, a difference of 0.37 that was also statistically significant.
In both studies, patients on desmopressin were almost twice as likely to achieve at least a 33% reduction in voids, the second primary endpoint. The large placebo effect in both studies, particularly among the women, is typically observed in studies of lower urinary tract dysfunction treatment, according to the company and several panelists.
An increase in the time to the first nocturnal void, a secondary endpoint, was increased by a mean of 49 minutes in women and by 39 minutes in men over placebo, which were both statistically significant differences.
There were two cases (1%) of hyponatremia among the women and three cases (3%) among the men on the 50-mcg dose; no patients on these two doses developed a serum sodium below 125 mmol/L (severe hyponatremia).
While the studies showed statistically significant decreases in the primary endpoints, the FDA reviewers questioned the magnitude of the clinical benefit. Other issues raised by FDA reviewers included the “urologically heterogeneous” population of patients in the two studies, which included, for example, patients with small-capacity bladders and those taking medications for overactive bladder or benign prostatic hypertrophy.
Currently, there is no FDA-approved treatment for nocturia or nocturnal polyuria. Desmopressin, in a tablet formulation, is approved in the United States for treating central diabetes insipidus and nocturnal enuresis in children and is occasionally used off-label for patients with nocturia. It is also approved for preventing bleeding in patients with hemophilia A or type 1 von Willebrand’s disease with a subcutaneous injection or nasal spray formulation.
The 25-mcg and the 50-mcg doses of the desmopressin orally disintegrating tablet are equivalent to the 42-mcg and 83-mcg doses of the desmopressin tablet, respectively, according to the FDA.
The orally disintegrating tablet formulation of desmopressin was approved in Canada in 2014 as a treatment for nocturia due to nocturnal polyuria at the same doses were proposed for approval in the United States. Higher doses have been approved in more than 65 countries).
The FDA usually follows the recommendations of its advisory panels.
In a statement issued after the meeting, Dr. Paul Korner, senior vice president for U.S. development at Ferring, said, “We look forward to working with the FDA to ensure it has the data needed to complete its evaluation of the safety and efficacy of this medication.”
The FDA is expected to make a decision during the first quarter of 2015, according to the company.
FDA panelists were cleared of conflicts.
HYATTSVILLE, MD. – The majority of a Food and Drug Administration advisory panel voted against approval of an orally disintegrating sublingual tablet formulation of desmopressin as a treatment for nocturia, citing uncertainties about the clinical benefits in the pivotal trials and concerns about the risk of hyponatremia.
At the meeting on Jan. 12, the FDA Endocrinologic and Metabolic Drugs Advisory Committee voted 10 to 5, with two abstentions, that the benefits of this desmopressin formulation in clinical trials did not outweigh the risks for the proposed indication, the treatment of nocturia due to nocturnal polyuria in adults who wake up two or more times each night to void. The indication proposed by Ferring Pharmaceuticals includes the statement that prior to treatment, “lifestyle changes and other treatable medical causes of nocturia should be addressed.”
Although the panel agreed that treatment was associated with statistically significant effects on the two primary endpoints in the two pivotal trials, those voting against approval said there were uncertainties about the clinical benefits, which several panelists said could be addressed in preapproval studies.
The panel generally agreed that the studies demonstrated a low risk of hyponatremia, the main safety concern associated with treatment. But they said that it was unclear whether the appropriate patients would be selected for treatment and would be properly monitored as recommended during treatment in real-world clinical situations. Examples included whether patients would remember to stop taking the medication when they developed an illness that could increase their risk of hyponatremia, or if they developed a disease or condition that affects sodium as they got older. There was also some uncertainty over the clinical significance of the small decreases in serum sodium in patients during the course of the 3-month studies, which evaluated two gender-specific doses that were lower than the higher doses previously studied that were associated with an increased risk of hyponatremia.
“The clinical benefit of the active medication relative to the substantial improvements seen in the placebo arm was relatively small,” said panelist Dr. Brendan Everett, director of the general cardiology inpatient service at Brigham and Women’s Hospital, Boston, who voted against approval. “The sponsor is on to something with respect to the time to the first nocturnal void being longer” among those on the drug, an exploratory endpoint that he recommended should be studied further. The company’s plan to address the outliers with substantial drops in sodium that are considered more dangerous was “reasonable,” he said, but added that he shared the concern of other committee members “that implementing that risk-mitigation strategy in the real world after approval would be difficult.”
The company developed lower doses to minimize the risk of hyponatremia and conducted two pivotal phase III confirmatory randomized, placebo-controlled studies of 268 women and almost 400 men with at least two voids per night (an average of 2.9 episodes), evaluating a 25-mcg dose in women and 50 mcg and 75 mcg in men over 3 months. Exclusion criteria included treatable causes of nocturia, such as diabetes insipidus and cardiac failure, and medical conditions that increased risk, such as hyponatremia and psychogenic polydipsia. (The company is not pursuing approval of the 75-mcg dose in men.)
Among the women, the number of voids per night over 3 months dropped by a mean of 1.46 among those on desmopressin vs. 1.24 among those on placebo, a 0.22 difference that was statistically significant, despite the large placebo effect. In the study of men, the number of voids per night dropped by a mean of 1.25 among those on the 50-mcg dose, vs. 0.88 among those on placebo, a difference of 0.37 that was also statistically significant.
In both studies, patients on desmopressin were almost twice as likely to achieve at least a 33% reduction in voids, the second primary endpoint. The large placebo effect in both studies, particularly among the women, is typically observed in studies of lower urinary tract dysfunction treatment, according to the company and several panelists.
An increase in the time to the first nocturnal void, a secondary endpoint, was increased by a mean of 49 minutes in women and by 39 minutes in men over placebo, which were both statistically significant differences.
There were two cases (1%) of hyponatremia among the women and three cases (3%) among the men on the 50-mcg dose; no patients on these two doses developed a serum sodium below 125 mmol/L (severe hyponatremia).
While the studies showed statistically significant decreases in the primary endpoints, the FDA reviewers questioned the magnitude of the clinical benefit. Other issues raised by FDA reviewers included the “urologically heterogeneous” population of patients in the two studies, which included, for example, patients with small-capacity bladders and those taking medications for overactive bladder or benign prostatic hypertrophy.
Currently, there is no FDA-approved treatment for nocturia or nocturnal polyuria. Desmopressin, in a tablet formulation, is approved in the United States for treating central diabetes insipidus and nocturnal enuresis in children and is occasionally used off-label for patients with nocturia. It is also approved for preventing bleeding in patients with hemophilia A or type 1 von Willebrand’s disease with a subcutaneous injection or nasal spray formulation.
The 25-mcg and the 50-mcg doses of the desmopressin orally disintegrating tablet are equivalent to the 42-mcg and 83-mcg doses of the desmopressin tablet, respectively, according to the FDA.
The orally disintegrating tablet formulation of desmopressin was approved in Canada in 2014 as a treatment for nocturia due to nocturnal polyuria at the same doses were proposed for approval in the United States. Higher doses have been approved in more than 65 countries).
The FDA usually follows the recommendations of its advisory panels.
In a statement issued after the meeting, Dr. Paul Korner, senior vice president for U.S. development at Ferring, said, “We look forward to working with the FDA to ensure it has the data needed to complete its evaluation of the safety and efficacy of this medication.”
The FDA is expected to make a decision during the first quarter of 2015, according to the company.
FDA panelists were cleared of conflicts.
HYATTSVILLE, MD. – The majority of a Food and Drug Administration advisory panel voted against approval of an orally disintegrating sublingual tablet formulation of desmopressin as a treatment for nocturia, citing uncertainties about the clinical benefits in the pivotal trials and concerns about the risk of hyponatremia.
At the meeting on Jan. 12, the FDA Endocrinologic and Metabolic Drugs Advisory Committee voted 10 to 5, with two abstentions, that the benefits of this desmopressin formulation in clinical trials did not outweigh the risks for the proposed indication, the treatment of nocturia due to nocturnal polyuria in adults who wake up two or more times each night to void. The indication proposed by Ferring Pharmaceuticals includes the statement that prior to treatment, “lifestyle changes and other treatable medical causes of nocturia should be addressed.”
Although the panel agreed that treatment was associated with statistically significant effects on the two primary endpoints in the two pivotal trials, those voting against approval said there were uncertainties about the clinical benefits, which several panelists said could be addressed in preapproval studies.
The panel generally agreed that the studies demonstrated a low risk of hyponatremia, the main safety concern associated with treatment. But they said that it was unclear whether the appropriate patients would be selected for treatment and would be properly monitored as recommended during treatment in real-world clinical situations. Examples included whether patients would remember to stop taking the medication when they developed an illness that could increase their risk of hyponatremia, or if they developed a disease or condition that affects sodium as they got older. There was also some uncertainty over the clinical significance of the small decreases in serum sodium in patients during the course of the 3-month studies, which evaluated two gender-specific doses that were lower than the higher doses previously studied that were associated with an increased risk of hyponatremia.
“The clinical benefit of the active medication relative to the substantial improvements seen in the placebo arm was relatively small,” said panelist Dr. Brendan Everett, director of the general cardiology inpatient service at Brigham and Women’s Hospital, Boston, who voted against approval. “The sponsor is on to something with respect to the time to the first nocturnal void being longer” among those on the drug, an exploratory endpoint that he recommended should be studied further. The company’s plan to address the outliers with substantial drops in sodium that are considered more dangerous was “reasonable,” he said, but added that he shared the concern of other committee members “that implementing that risk-mitigation strategy in the real world after approval would be difficult.”
The company developed lower doses to minimize the risk of hyponatremia and conducted two pivotal phase III confirmatory randomized, placebo-controlled studies of 268 women and almost 400 men with at least two voids per night (an average of 2.9 episodes), evaluating a 25-mcg dose in women and 50 mcg and 75 mcg in men over 3 months. Exclusion criteria included treatable causes of nocturia, such as diabetes insipidus and cardiac failure, and medical conditions that increased risk, such as hyponatremia and psychogenic polydipsia. (The company is not pursuing approval of the 75-mcg dose in men.)
Among the women, the number of voids per night over 3 months dropped by a mean of 1.46 among those on desmopressin vs. 1.24 among those on placebo, a 0.22 difference that was statistically significant, despite the large placebo effect. In the study of men, the number of voids per night dropped by a mean of 1.25 among those on the 50-mcg dose, vs. 0.88 among those on placebo, a difference of 0.37 that was also statistically significant.
In both studies, patients on desmopressin were almost twice as likely to achieve at least a 33% reduction in voids, the second primary endpoint. The large placebo effect in both studies, particularly among the women, is typically observed in studies of lower urinary tract dysfunction treatment, according to the company and several panelists.
An increase in the time to the first nocturnal void, a secondary endpoint, was increased by a mean of 49 minutes in women and by 39 minutes in men over placebo, which were both statistically significant differences.
There were two cases (1%) of hyponatremia among the women and three cases (3%) among the men on the 50-mcg dose; no patients on these two doses developed a serum sodium below 125 mmol/L (severe hyponatremia).
While the studies showed statistically significant decreases in the primary endpoints, the FDA reviewers questioned the magnitude of the clinical benefit. Other issues raised by FDA reviewers included the “urologically heterogeneous” population of patients in the two studies, which included, for example, patients with small-capacity bladders and those taking medications for overactive bladder or benign prostatic hypertrophy.
Currently, there is no FDA-approved treatment for nocturia or nocturnal polyuria. Desmopressin, in a tablet formulation, is approved in the United States for treating central diabetes insipidus and nocturnal enuresis in children and is occasionally used off-label for patients with nocturia. It is also approved for preventing bleeding in patients with hemophilia A or type 1 von Willebrand’s disease with a subcutaneous injection or nasal spray formulation.
The 25-mcg and the 50-mcg doses of the desmopressin orally disintegrating tablet are equivalent to the 42-mcg and 83-mcg doses of the desmopressin tablet, respectively, according to the FDA.
The orally disintegrating tablet formulation of desmopressin was approved in Canada in 2014 as a treatment for nocturia due to nocturnal polyuria at the same doses were proposed for approval in the United States. Higher doses have been approved in more than 65 countries).
The FDA usually follows the recommendations of its advisory panels.
In a statement issued after the meeting, Dr. Paul Korner, senior vice president for U.S. development at Ferring, said, “We look forward to working with the FDA to ensure it has the data needed to complete its evaluation of the safety and efficacy of this medication.”
The FDA is expected to make a decision during the first quarter of 2015, according to the company.
FDA panelists were cleared of conflicts.
AT AN FDA ADVISORY COMMITTEE MEETING

Making naloxone available to patients at risk for opioid overdoses
AVENTURA, FLA. – Clinicians interested in providing naloxone for their patients at risk for an opioid overdose should consider the pros and cons of the different formulations, addiction medicine specialists say.
In addition, clinicians should become familiar with legislation affecting how naloxone can be prescribed to lay persons in their state, according to the specialists, who spoke at the annual meeting of the American Academy of Addiction Psychiatry.
During a workshop on expanding access to the opioid antagonist to patients at risk and their families to help prevent opioid overdoses, Dr. Eric D. Collins, physician-in-chief at Silver Hill Hospital, New Canaan, Conn., described naloxone as “an effective and generally safe intervention that requires minimal training” and can be administered by a layperson. “Any prescriber can provide naloxone to persons at risk of opioid overdose” and in some states, can provide naloxone to anyone at risk of witnessing an opioid overdose, he added.

Dr. Seddon R. Savage, director of the Dartmouth Center on Addiction Recovery and Education, Hanover, N.H., said that although providing naloxone is a late intervention, it saves lives. “So clearly, it is critical that we embrace that strategy,” she said.
Providing naloxone is among the current strategies used to address the misuse of prescription opioids and to reduce mortality and morbidity related to opioids. These strategies include the development of tamper-resistant products, public campaigns increasing awareness about the importance of locking up pain medications and disposing of them safely, drug take back programs, and pharmacy-based interventions.
Overdose death rates fall
To date, “good solid evidence for efficacy in reducing mortality and opioid overdoses only exists” for maintenance therapy of opioid dependence and naloxone distribution, said Dr. Savage, who also is medical director of the chronic pain and addiction program at Silver Hill Hospital. She cited a 2013 study that found a significant reduction in opioid overdose deaths rate after a program that provided education about overdoses and nasal naloxone kits to potential bystanders (users, families, and friends) was implemented in 19 communities in Massachusetts with high overdose rates. Training included 10-60 minutes of training in recognizing and intervening in an overdose (BMJ 2013; 346:f174).
For pain medicine specialists, Dr. Savage said, “it is reasonable to consider” providing naloxone to patients at risk for overuse tied to unmet pain needs, accidental overuse, or self-treatment of other symptoms; as well as those with history of recreational drug use or addiction. Primary care clinicians can target their patients being treated with opioids for pain and those with known or a suspected addiction to opioids. Addiction medicine specialists can consider prescribing naloxone to their patients with opioid addiction on or off opioid agonist therapy (OAT), such as buprenorphine.

However, challenges prevail in prescribing naloxone, which include wide variations in naloxone legislation and Good Samaritan laws in states, resources needed to train lay people to administer naloxone, and recent increases in the cost of intranasal naloxone. The latter is the most widely used form of the drug to treat overdoses, said Dr. Savage, who also is affiliated with Dartmouth Medical School in Hanover.
Familiarity with laws advised
Dr. Savage advised clinicians to become familiar with the policies regarding naloxone prescribing and Good Samaritan laws in their jurisdictions. Legislation that exists in some states but not others include protection from possession of controlled substance and paraphernalia (in 22 states and the District of Columbia [D.C.] as of August 2014), protection from criminal liability for lay administration of naloxone (23 states and D.C.), protection from civil liability for lay administration (20 states and D.C.) and protection of prescribers from criminal liability (13 states). In 24 states, third party prescribing is allowed.
Naloxone – which binds to opioid receptors and in patients on opioids, thereby reversing the effects of opioids, including respiratory depression – can precipitate opioid withdrawal symptoms in some patients, said Dr. Collins, who also is affiliated with Columbia University in New York. He referred to a 2004 study that found that the most common adverse events after naloxone was used to treated a suspected opioid overdose outside of the hospital were confusion ( 32%) and headache (22%), nausea and vomiting (9%), and aggressiveness (8%), but serious complications were rare (Eur. J. Emerg. Med. 2004;11:19-23). Seizures were reported in 4% and tachycardia was reported in 6%. Dr. Collins said he has never seen a case of allergy to naloxone, the only contraindication to the drug.
Since aggressiveness might be associated with withdrawal, Dr. Savage said training for naloxone providers often includes how to manage a combative person. The risk of tachycardia has been raised as a concern, particularly in situations where law enforcement personnel who are not medically trained are delivering naloxone, but those patients are transported to medical facilities, she pointed out.
Intranasal (IN) naloxone, which is not approved by the Food and Drug Administration, is the most widely used method of administering naloxone by police and other first responders. They use a standard dose of 0.4 contained in a syringe with an atomizer, delivering one 0.2 mg dose per nostril. Another option is the autoinjector that provides an injection of naloxone with a speaker that provides instructions guiding the user. This option was approved by the FDA in 2014.
Evidence that the intranasal form is effective dates back to 2005, with a study that found a dose of 2 mg/2 cc in a prefilled syringe delivered with an atomizer by Denver paramedics was effective in 43 of 52 (83%) people. IV naloxone was needed in nine individuals who did not respond to the IN dose, including five who had nasal pathology (J. Emerg. Med. 2005; 29:265-71). In a more recent study that randomized 100 patients admitted to an emergency department with an opioid overdose to a lower IN dose (0.4 mg) or IV naloxone, the IN route proved as effective as the IV route in reversing respiratory depression and central nervous system effects (Arch. Med. Sci. 2014;10:309-14).
Broad effectiveness found
Dr. Collins said the various options have advantages and drawbacks, and if only one option is available for whatever reason, “they all work.” IM and IV formulations have the fastest onset of action, are the least expensive, and are FDA-approved but carry the risk of a needlestick injury. IN naloxone is not FDA-approved, and might have a slightly slower and less predictable effect but has no risk of a needlestick injury. In addition, IN naloxone is inexpensive and easy to use. The recently FDA-approved Evzio autoinjector pre-filled with naloxone solution, administered IM or subcutaneously, has a faster onset with a predictable effect and no risk of a needle stick. It also includes audio instructions that walks the user through its use. But it costs about $400 per kit, which is a drawback, he said.
Dr. Savage pointed out that administering naloxone is only part of intervening in an opioid overdose and that training is needed when naloxone is dispensed or prescribed to patients or their families. Included in such training would be when to call for help, how to identify signs of an overdose, how to position patients for rescue, understanding rescue breathing, and knowing when the admininstration of naloxone is indicated. Since naloxone wears off in about half an hour, depending on the route and other variables, people should be told that they need to call for emergency support and transport for ongoing care because many opioids end up lasting longer than the effects of naloxone.
The FDA is considering making naloxone available over the counter, but such a move will take several years. It can be provided by pharmacies in states that have a collaborative practice agreement in place that supports distribution to people addicted to opioids, their families, and friends, she said. For example, in Rhode Island, a collaborative practice agreement exists allowing Walgreens pharmacies to dispense naloxone without a prescription.
Based on an informal poll taken during the workshop, none of those in attendance had started to prescribe naloxone for their patients, although they were interested in doing so or were figuring out how to make it available. During the discussion period, barriers they cited included the time and resources needed to train people to administer naloxone, the cost, and the lack of awareness among pharmacies about their ability to obtain intranasal kits. Suggestions among participants include having nurses or pain medicine fellows provide training and education, and using videos for training. A physician at the Atlanta Veterans Affairs Medical Center pointed out that naloxone is provided at no charge to all VA hospitals, although patients might have a co-pay.
Among the resources provided by Dr. Savage and Dr. Collins for clinicians interested in providing naloxone to patients at risk of an opioid overdose and to family members of others who might witness an overdose was an American Medical Association webinar on naloxone safety, information on how to start prescribing and dispensing naloxone rescue kits, and patient information videos. More information on how to start a program is available at http://www.naloxoneinfo.org.
Dr. Savage and Dr. Collins said they had no direct commercial interests related to the topic of the workshop.
AVENTURA, FLA. – Clinicians interested in providing naloxone for their patients at risk for an opioid overdose should consider the pros and cons of the different formulations, addiction medicine specialists say.
In addition, clinicians should become familiar with legislation affecting how naloxone can be prescribed to lay persons in their state, according to the specialists, who spoke at the annual meeting of the American Academy of Addiction Psychiatry.
During a workshop on expanding access to the opioid antagonist to patients at risk and their families to help prevent opioid overdoses, Dr. Eric D. Collins, physician-in-chief at Silver Hill Hospital, New Canaan, Conn., described naloxone as “an effective and generally safe intervention that requires minimal training” and can be administered by a layperson. “Any prescriber can provide naloxone to persons at risk of opioid overdose” and in some states, can provide naloxone to anyone at risk of witnessing an opioid overdose, he added.
Dr. Seddon R. Savage, director of the Dartmouth Center on Addiction Recovery and Education, Hanover, N.H., said that although providing naloxone is a late intervention, it saves lives. “So clearly, it is critical that we embrace that strategy,” she said.
Providing naloxone is among the current strategies used to address the misuse of prescription opioids and to reduce mortality and morbidity related to opioids. These strategies include the development of tamper-resistant products, public campaigns increasing awareness about the importance of locking up pain medications and disposing of them safely, drug take back programs, and pharmacy-based interventions.
Overdose death rates fall
To date, “good solid evidence for efficacy in reducing mortality and opioid overdoses only exists” for maintenance therapy of opioid dependence and naloxone distribution, said Dr. Savage, who also is medical director of the chronic pain and addiction program at Silver Hill Hospital. She cited a 2013 study that found a significant reduction in opioid overdose deaths rate after a program that provided education about overdoses and nasal naloxone kits to potential bystanders (users, families, and friends) was implemented in 19 communities in Massachusetts with high overdose rates. Training included 10-60 minutes of training in recognizing and intervening in an overdose (BMJ 2013; 346:f174).
For pain medicine specialists, Dr. Savage said, “it is reasonable to consider” providing naloxone to patients at risk for overuse tied to unmet pain needs, accidental overuse, or self-treatment of other symptoms; as well as those with history of recreational drug use or addiction. Primary care clinicians can target their patients being treated with opioids for pain and those with known or a suspected addiction to opioids. Addiction medicine specialists can consider prescribing naloxone to their patients with opioid addiction on or off opioid agonist therapy (OAT), such as buprenorphine.
However, challenges prevail in prescribing naloxone, which include wide variations in naloxone legislation and Good Samaritan laws in states, resources needed to train lay people to administer naloxone, and recent increases in the cost of intranasal naloxone. The latter is the most widely used form of the drug to treat overdoses, said Dr. Savage, who also is affiliated with Dartmouth Medical School in Hanover.
Familiarity with laws advised
Dr. Savage advised clinicians to become familiar with the policies regarding naloxone prescribing and Good Samaritan laws in their jurisdictions. Legislation that exists in some states but not others include protection from possession of controlled substance and paraphernalia (in 22 states and the District of Columbia [D.C.] as of August 2014), protection from criminal liability for lay administration of naloxone (23 states and D.C.), protection from civil liability for lay administration (20 states and D.C.) and protection of prescribers from criminal liability (13 states). In 24 states, third party prescribing is allowed.
Naloxone – which binds to opioid receptors and in patients on opioids, thereby reversing the effects of opioids, including respiratory depression – can precipitate opioid withdrawal symptoms in some patients, said Dr. Collins, who also is affiliated with Columbia University in New York. He referred to a 2004 study that found that the most common adverse events after naloxone was used to treated a suspected opioid overdose outside of the hospital were confusion ( 32%) and headache (22%), nausea and vomiting (9%), and aggressiveness (8%), but serious complications were rare (Eur. J. Emerg. Med. 2004;11:19-23). Seizures were reported in 4% and tachycardia was reported in 6%. Dr. Collins said he has never seen a case of allergy to naloxone, the only contraindication to the drug.
Since aggressiveness might be associated with withdrawal, Dr. Savage said training for naloxone providers often includes how to manage a combative person. The risk of tachycardia has been raised as a concern, particularly in situations where law enforcement personnel who are not medically trained are delivering naloxone, but those patients are transported to medical facilities, she pointed out.
Intranasal (IN) naloxone, which is not approved by the Food and Drug Administration, is the most widely used method of administering naloxone by police and other first responders. They use a standard dose of 0.4 contained in a syringe with an atomizer, delivering one 0.2 mg dose per nostril. Another option is the autoinjector that provides an injection of naloxone with a speaker that provides instructions guiding the user. This option was approved by the FDA in 2014.
Evidence that the intranasal form is effective dates back to 2005, with a study that found a dose of 2 mg/2 cc in a prefilled syringe delivered with an atomizer by Denver paramedics was effective in 43 of 52 (83%) people. IV naloxone was needed in nine individuals who did not respond to the IN dose, including five who had nasal pathology (J. Emerg. Med. 2005; 29:265-71). In a more recent study that randomized 100 patients admitted to an emergency department with an opioid overdose to a lower IN dose (0.4 mg) or IV naloxone, the IN route proved as effective as the IV route in reversing respiratory depression and central nervous system effects (Arch. Med. Sci. 2014;10:309-14).
Broad effectiveness found
Dr. Collins said the various options have advantages and drawbacks, and if only one option is available for whatever reason, “they all work.” IM and IV formulations have the fastest onset of action, are the least expensive, and are FDA-approved but carry the risk of a needlestick injury. IN naloxone is not FDA-approved, and might have a slightly slower and less predictable effect but has no risk of a needlestick injury. In addition, IN naloxone is inexpensive and easy to use. The recently FDA-approved Evzio autoinjector pre-filled with naloxone solution, administered IM or subcutaneously, has a faster onset with a predictable effect and no risk of a needle stick. It also includes audio instructions that walks the user through its use. But it costs about $400 per kit, which is a drawback, he said.
Dr. Savage pointed out that administering naloxone is only part of intervening in an opioid overdose and that training is needed when naloxone is dispensed or prescribed to patients or their families. Included in such training would be when to call for help, how to identify signs of an overdose, how to position patients for rescue, understanding rescue breathing, and knowing when the admininstration of naloxone is indicated. Since naloxone wears off in about half an hour, depending on the route and other variables, people should be told that they need to call for emergency support and transport for ongoing care because many opioids end up lasting longer than the effects of naloxone.
The FDA is considering making naloxone available over the counter, but such a move will take several years. It can be provided by pharmacies in states that have a collaborative practice agreement in place that supports distribution to people addicted to opioids, their families, and friends, she said. For example, in Rhode Island, a collaborative practice agreement exists allowing Walgreens pharmacies to dispense naloxone without a prescription.
Based on an informal poll taken during the workshop, none of those in attendance had started to prescribe naloxone for their patients, although they were interested in doing so or were figuring out how to make it available. During the discussion period, barriers they cited included the time and resources needed to train people to administer naloxone, the cost, and the lack of awareness among pharmacies about their ability to obtain intranasal kits. Suggestions among participants include having nurses or pain medicine fellows provide training and education, and using videos for training. A physician at the Atlanta Veterans Affairs Medical Center pointed out that naloxone is provided at no charge to all VA hospitals, although patients might have a co-pay.
Among the resources provided by Dr. Savage and Dr. Collins for clinicians interested in providing naloxone to patients at risk of an opioid overdose and to family members of others who might witness an overdose was an American Medical Association webinar on naloxone safety, information on how to start prescribing and dispensing naloxone rescue kits, and patient information videos. More information on how to start a program is available at http://www.naloxoneinfo.org.
Dr. Savage and Dr. Collins said they had no direct commercial interests related to the topic of the workshop.
AVENTURA, FLA. – Clinicians interested in providing naloxone for their patients at risk for an opioid overdose should consider the pros and cons of the different formulations, addiction medicine specialists say.
In addition, clinicians should become familiar with legislation affecting how naloxone can be prescribed to lay persons in their state, according to the specialists, who spoke at the annual meeting of the American Academy of Addiction Psychiatry.
During a workshop on expanding access to the opioid antagonist to patients at risk and their families to help prevent opioid overdoses, Dr. Eric D. Collins, physician-in-chief at Silver Hill Hospital, New Canaan, Conn., described naloxone as “an effective and generally safe intervention that requires minimal training” and can be administered by a layperson. “Any prescriber can provide naloxone to persons at risk of opioid overdose” and in some states, can provide naloxone to anyone at risk of witnessing an opioid overdose, he added.
Dr. Seddon R. Savage, director of the Dartmouth Center on Addiction Recovery and Education, Hanover, N.H., said that although providing naloxone is a late intervention, it saves lives. “So clearly, it is critical that we embrace that strategy,” she said.
Providing naloxone is among the current strategies used to address the misuse of prescription opioids and to reduce mortality and morbidity related to opioids. These strategies include the development of tamper-resistant products, public campaigns increasing awareness about the importance of locking up pain medications and disposing of them safely, drug take back programs, and pharmacy-based interventions.
Overdose death rates fall
To date, “good solid evidence for efficacy in reducing mortality and opioid overdoses only exists” for maintenance therapy of opioid dependence and naloxone distribution, said Dr. Savage, who also is medical director of the chronic pain and addiction program at Silver Hill Hospital. She cited a 2013 study that found a significant reduction in opioid overdose deaths rate after a program that provided education about overdoses and nasal naloxone kits to potential bystanders (users, families, and friends) was implemented in 19 communities in Massachusetts with high overdose rates. Training included 10-60 minutes of training in recognizing and intervening in an overdose (BMJ 2013; 346:f174).
For pain medicine specialists, Dr. Savage said, “it is reasonable to consider” providing naloxone to patients at risk for overuse tied to unmet pain needs, accidental overuse, or self-treatment of other symptoms; as well as those with history of recreational drug use or addiction. Primary care clinicians can target their patients being treated with opioids for pain and those with known or a suspected addiction to opioids. Addiction medicine specialists can consider prescribing naloxone to their patients with opioid addiction on or off opioid agonist therapy (OAT), such as buprenorphine.
However, challenges prevail in prescribing naloxone, which include wide variations in naloxone legislation and Good Samaritan laws in states, resources needed to train lay people to administer naloxone, and recent increases in the cost of intranasal naloxone. The latter is the most widely used form of the drug to treat overdoses, said Dr. Savage, who also is affiliated with Dartmouth Medical School in Hanover.
Familiarity with laws advised
Dr. Savage advised clinicians to become familiar with the policies regarding naloxone prescribing and Good Samaritan laws in their jurisdictions. Legislation that exists in some states but not others include protection from possession of controlled substance and paraphernalia (in 22 states and the District of Columbia [D.C.] as of August 2014), protection from criminal liability for lay administration of naloxone (23 states and D.C.), protection from civil liability for lay administration (20 states and D.C.) and protection of prescribers from criminal liability (13 states). In 24 states, third party prescribing is allowed.
Naloxone – which binds to opioid receptors and in patients on opioids, thereby reversing the effects of opioids, including respiratory depression – can precipitate opioid withdrawal symptoms in some patients, said Dr. Collins, who also is affiliated with Columbia University in New York. He referred to a 2004 study that found that the most common adverse events after naloxone was used to treated a suspected opioid overdose outside of the hospital were confusion ( 32%) and headache (22%), nausea and vomiting (9%), and aggressiveness (8%), but serious complications were rare (Eur. J. Emerg. Med. 2004;11:19-23). Seizures were reported in 4% and tachycardia was reported in 6%. Dr. Collins said he has never seen a case of allergy to naloxone, the only contraindication to the drug.
Since aggressiveness might be associated with withdrawal, Dr. Savage said training for naloxone providers often includes how to manage a combative person. The risk of tachycardia has been raised as a concern, particularly in situations where law enforcement personnel who are not medically trained are delivering naloxone, but those patients are transported to medical facilities, she pointed out.
Intranasal (IN) naloxone, which is not approved by the Food and Drug Administration, is the most widely used method of administering naloxone by police and other first responders. They use a standard dose of 0.4 contained in a syringe with an atomizer, delivering one 0.2 mg dose per nostril. Another option is the autoinjector that provides an injection of naloxone with a speaker that provides instructions guiding the user. This option was approved by the FDA in 2014.
Evidence that the intranasal form is effective dates back to 2005, with a study that found a dose of 2 mg/2 cc in a prefilled syringe delivered with an atomizer by Denver paramedics was effective in 43 of 52 (83%) people. IV naloxone was needed in nine individuals who did not respond to the IN dose, including five who had nasal pathology (J. Emerg. Med. 2005; 29:265-71). In a more recent study that randomized 100 patients admitted to an emergency department with an opioid overdose to a lower IN dose (0.4 mg) or IV naloxone, the IN route proved as effective as the IV route in reversing respiratory depression and central nervous system effects (Arch. Med. Sci. 2014;10:309-14).
Broad effectiveness found
Dr. Collins said the various options have advantages and drawbacks, and if only one option is available for whatever reason, “they all work.” IM and IV formulations have the fastest onset of action, are the least expensive, and are FDA-approved but carry the risk of a needlestick injury. IN naloxone is not FDA-approved, and might have a slightly slower and less predictable effect but has no risk of a needlestick injury. In addition, IN naloxone is inexpensive and easy to use. The recently FDA-approved Evzio autoinjector pre-filled with naloxone solution, administered IM or subcutaneously, has a faster onset with a predictable effect and no risk of a needle stick. It also includes audio instructions that walks the user through its use. But it costs about $400 per kit, which is a drawback, he said.
Dr. Savage pointed out that administering naloxone is only part of intervening in an opioid overdose and that training is needed when naloxone is dispensed or prescribed to patients or their families. Included in such training would be when to call for help, how to identify signs of an overdose, how to position patients for rescue, understanding rescue breathing, and knowing when the admininstration of naloxone is indicated. Since naloxone wears off in about half an hour, depending on the route and other variables, people should be told that they need to call for emergency support and transport for ongoing care because many opioids end up lasting longer than the effects of naloxone.
The FDA is considering making naloxone available over the counter, but such a move will take several years. It can be provided by pharmacies in states that have a collaborative practice agreement in place that supports distribution to people addicted to opioids, their families, and friends, she said. For example, in Rhode Island, a collaborative practice agreement exists allowing Walgreens pharmacies to dispense naloxone without a prescription.
Based on an informal poll taken during the workshop, none of those in attendance had started to prescribe naloxone for their patients, although they were interested in doing so or were figuring out how to make it available. During the discussion period, barriers they cited included the time and resources needed to train people to administer naloxone, the cost, and the lack of awareness among pharmacies about their ability to obtain intranasal kits. Suggestions among participants include having nurses or pain medicine fellows provide training and education, and using videos for training. A physician at the Atlanta Veterans Affairs Medical Center pointed out that naloxone is provided at no charge to all VA hospitals, although patients might have a co-pay.
Among the resources provided by Dr. Savage and Dr. Collins for clinicians interested in providing naloxone to patients at risk of an opioid overdose and to family members of others who might witness an overdose was an American Medical Association webinar on naloxone safety, information on how to start prescribing and dispensing naloxone rescue kits, and patient information videos. More information on how to start a program is available at http://www.naloxoneinfo.org.
Dr. Savage and Dr. Collins said they had no direct commercial interests related to the topic of the workshop.
EXPERT ANALYSIS FROM THE AAAP ANNUAL MEETING

Edoxaban approved for atrial fib, DVT, and PE indications
Edoxaban, a selective factor Xa-inhibitor, has been approved by the Food and Drug Administration for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, with a statement in the boxed warning that it should not be used in patients with normal renal function.
The warning reflects the results of a subgroup analysis in the pivotal trial, which found that the 60-mg dose was superior to warfarin in terms of reducing the stroke risk in mildly renally impaired patients, but was worse in patients with normal renal function. This was the main focus of a meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Panel meeting in October, in which the panel voted 9-1 to recommend approval of edoxaban for this indication, but had mixed opinions on whether approval should be limited to patients with mild to moderate renal impairment.
The approved prescribing information recommends that a patient’s creatinine clearance should be checked before edoxaban is prescribed. “Patients with creatinine clearance greater than 95 mL/min have an increased risk of stroke, compared to similar patients given warfarin,” and should be treated with another anticoagulant, the FDA said in the Jan. 9 statement announcing the approval. The recommended dose for those with a creatinine clearance between 50 mL/min and 95 mL/min is 60 mg once a day; for those with a creatinine clearance of 15-50 mL/min, the recommended dose is 30 mg once a day, according to the prescribing information.
Edoxaban, the fourth novel oral anticoagulant drug approved by the FDA, will be marketed as Savaysa by Daiichi Sankyo. It was also approved to treat deep vein thrombosis and pulmonary embolism following 5-10 days of initial therapy with a parenteral anticoagulant. The recommended dose for this indication is 60 mg once a day. For patients with a creatinine clearance of 15-50 mL/min, or who weigh up to 60 kg (about 132 pounds), or who are taking “certain P-glycoprotein inhibitors,” the 30-mg/day dose is recommended.
Approval for the nonvalvular AF indication was based on ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation) study, comparing once-daily edoxaban (60 mg and 30 mg) to warfarin in 21,015 patients with nonvalvular AF, at a moderate to high risk of thromboembolic events (N. Engl. J. Med. 2013;369:2093-104). Over a median of almost 3 years, both doses were noninferior to warfarin in the primary efficacy endpoint, the occurrence of first stroke or of a systemic embolic event. Overall, major bleeding events were significantly lower among those on the 60-mg and 30-mg doses, compared with those on warfarin. However, the rate of ischemic stroke was higher relative to warfarin in patients with a creatinine clearance over 95 mL/min.
About half of the edoxaban dose is eliminated by the kidneys, and patients with a creatinine clearance above 95 mL/min have lower plasma edoxaban levels, according to a statement in the clinical trials section of the prescribing information, which adds: “Given the clear relationship of dose and blood levels to effectiveness in the ENGAGE AF-TIMI 48 study, it could be anticipated that patients with better renal function would show a smaller effect of Savaysa, compared to warfarin than would patients with mildly impaired renal function, and this was in fact observed.”
Approval of the DVT and PE indication was based on the Hokusai-VTE study of about 8,200 people comparing edoxaban to warfarin, which found that the edoxaban 60 mg once a day was noninferior to warfarin in the rate of symptomatic venous thromboembolism (3.2% vs. 3.5% in those on warfarin). The rate of major or clinically relevant nonmajor bleeding events was 8.5% among those on edoxaban vs. 10.3% in those on warfarin (N. Engl. J. Med. 2013;369:1406-15).
Bleeding and anemia were the most common adverse events among patients with nonvalvular atrial fibrillation in clinical trials, and “as with other FDA-approved anticlotting drugs, bleeding, including life-threatening bleeding, is the most serious risk with Savaysa,” the FDA statement said. Among those treated for DVT and PE, the most common adverse events were bleeding, rash, abnormal liver function tests, and anemia.
Savaysa is the fourth novel oral anticoagulant to be cleared by the FDA, after dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis).
Serious adverse events associated with edoxaban should be reported to the FDA’s MedWatch program or at 800-332-1088.
A precedent does not come readily to mind where one restricts the use of a drug to patients with mild to moderately impaired renal function to optimize the benefit-risk balance. Typically, one avoids the drug or reduces the dose in such subgroups. For this reason, I think the drug is likely going to be a “nonstarter” for many clinicians.
During the FDA Advisory Committee panel meeting on edoxaban, the emphasis was on excluding patients with a creatinine clearance greater than 80 mL/min (representing about 37% of patients in the pivotal trial). The FDA approval used a cut-off of 95 mL/min (representing about 22% of patients in the pivotal trial). Even though the latter increases the eligible pool of patients for edoxaban, the major challenge is going to be clinical acceptability and marketability, especially given no unique advantage of this drug over other approved novel anticoagulants.
The CrCl cut off of 95mL/min applies to nonvalvular atrial fibrillation indication, and not to the venous thromboembolism (DVT/PE) indication. However, the latter indication requires 5-10 days of parenteral anticoagulant therapy, which puts it at a disadvantage compared with rivaroxaban or apixaban.
It is interesting to note that active pathological bleeding qualifies as a contraindication, but use in patients with normal renal function does not. There is a twofold increased risk of overall stroke or systemic embolism (primary endpoint) and ischemic stroke in patients with normal renal function (CrCl > 95 mL/min) along with an attenuation of the hemorrhagic risk advantage in this subgroup. Even though adjudicated major bleeding is still favorable for edoxaban in this subgroup, in my opinion the ischemic stroke risk outweighs the major bleeding advantage.
Sanjay Kaul, M.D., is professor of medicine at the University of California, Los Angeles. He was a member of the FDA’s Cardiovascular and Renal Drugs Advisory Committee that reviewed edoxaban at a meeting on Oct. 30, 2014.
A precedent does not come readily to mind where one restricts the use of a drug to patients with mild to moderately impaired renal function to optimize the benefit-risk balance. Typically, one avoids the drug or reduces the dose in such subgroups. For this reason, I think the drug is likely going to be a “nonstarter” for many clinicians.
During the FDA Advisory Committee panel meeting on edoxaban, the emphasis was on excluding patients with a creatinine clearance greater than 80 mL/min (representing about 37% of patients in the pivotal trial). The FDA approval used a cut-off of 95 mL/min (representing about 22% of patients in the pivotal trial). Even though the latter increases the eligible pool of patients for edoxaban, the major challenge is going to be clinical acceptability and marketability, especially given no unique advantage of this drug over other approved novel anticoagulants.
The CrCl cut off of 95mL/min applies to nonvalvular atrial fibrillation indication, and not to the venous thromboembolism (DVT/PE) indication. However, the latter indication requires 5-10 days of parenteral anticoagulant therapy, which puts it at a disadvantage compared with rivaroxaban or apixaban.
It is interesting to note that active pathological bleeding qualifies as a contraindication, but use in patients with normal renal function does not. There is a twofold increased risk of overall stroke or systemic embolism (primary endpoint) and ischemic stroke in patients with normal renal function (CrCl > 95 mL/min) along with an attenuation of the hemorrhagic risk advantage in this subgroup. Even though adjudicated major bleeding is still favorable for edoxaban in this subgroup, in my opinion the ischemic stroke risk outweighs the major bleeding advantage.
Sanjay Kaul, M.D., is professor of medicine at the University of California, Los Angeles. He was a member of the FDA’s Cardiovascular and Renal Drugs Advisory Committee that reviewed edoxaban at a meeting on Oct. 30, 2014.
A precedent does not come readily to mind where one restricts the use of a drug to patients with mild to moderately impaired renal function to optimize the benefit-risk balance. Typically, one avoids the drug or reduces the dose in such subgroups. For this reason, I think the drug is likely going to be a “nonstarter” for many clinicians.
During the FDA Advisory Committee panel meeting on edoxaban, the emphasis was on excluding patients with a creatinine clearance greater than 80 mL/min (representing about 37% of patients in the pivotal trial). The FDA approval used a cut-off of 95 mL/min (representing about 22% of patients in the pivotal trial). Even though the latter increases the eligible pool of patients for edoxaban, the major challenge is going to be clinical acceptability and marketability, especially given no unique advantage of this drug over other approved novel anticoagulants.
The CrCl cut off of 95mL/min applies to nonvalvular atrial fibrillation indication, and not to the venous thromboembolism (DVT/PE) indication. However, the latter indication requires 5-10 days of parenteral anticoagulant therapy, which puts it at a disadvantage compared with rivaroxaban or apixaban.
It is interesting to note that active pathological bleeding qualifies as a contraindication, but use in patients with normal renal function does not. There is a twofold increased risk of overall stroke or systemic embolism (primary endpoint) and ischemic stroke in patients with normal renal function (CrCl > 95 mL/min) along with an attenuation of the hemorrhagic risk advantage in this subgroup. Even though adjudicated major bleeding is still favorable for edoxaban in this subgroup, in my opinion the ischemic stroke risk outweighs the major bleeding advantage.
Sanjay Kaul, M.D., is professor of medicine at the University of California, Los Angeles. He was a member of the FDA’s Cardiovascular and Renal Drugs Advisory Committee that reviewed edoxaban at a meeting on Oct. 30, 2014.
Edoxaban, a selective factor Xa-inhibitor, has been approved by the Food and Drug Administration for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, with a statement in the boxed warning that it should not be used in patients with normal renal function.
The warning reflects the results of a subgroup analysis in the pivotal trial, which found that the 60-mg dose was superior to warfarin in terms of reducing the stroke risk in mildly renally impaired patients, but was worse in patients with normal renal function. This was the main focus of a meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Panel meeting in October, in which the panel voted 9-1 to recommend approval of edoxaban for this indication, but had mixed opinions on whether approval should be limited to patients with mild to moderate renal impairment.
The approved prescribing information recommends that a patient’s creatinine clearance should be checked before edoxaban is prescribed. “Patients with creatinine clearance greater than 95 mL/min have an increased risk of stroke, compared to similar patients given warfarin,” and should be treated with another anticoagulant, the FDA said in the Jan. 9 statement announcing the approval. The recommended dose for those with a creatinine clearance between 50 mL/min and 95 mL/min is 60 mg once a day; for those with a creatinine clearance of 15-50 mL/min, the recommended dose is 30 mg once a day, according to the prescribing information.
Edoxaban, the fourth novel oral anticoagulant drug approved by the FDA, will be marketed as Savaysa by Daiichi Sankyo. It was also approved to treat deep vein thrombosis and pulmonary embolism following 5-10 days of initial therapy with a parenteral anticoagulant. The recommended dose for this indication is 60 mg once a day. For patients with a creatinine clearance of 15-50 mL/min, or who weigh up to 60 kg (about 132 pounds), or who are taking “certain P-glycoprotein inhibitors,” the 30-mg/day dose is recommended.
Approval for the nonvalvular AF indication was based on ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation) study, comparing once-daily edoxaban (60 mg and 30 mg) to warfarin in 21,015 patients with nonvalvular AF, at a moderate to high risk of thromboembolic events (N. Engl. J. Med. 2013;369:2093-104). Over a median of almost 3 years, both doses were noninferior to warfarin in the primary efficacy endpoint, the occurrence of first stroke or of a systemic embolic event. Overall, major bleeding events were significantly lower among those on the 60-mg and 30-mg doses, compared with those on warfarin. However, the rate of ischemic stroke was higher relative to warfarin in patients with a creatinine clearance over 95 mL/min.
About half of the edoxaban dose is eliminated by the kidneys, and patients with a creatinine clearance above 95 mL/min have lower plasma edoxaban levels, according to a statement in the clinical trials section of the prescribing information, which adds: “Given the clear relationship of dose and blood levels to effectiveness in the ENGAGE AF-TIMI 48 study, it could be anticipated that patients with better renal function would show a smaller effect of Savaysa, compared to warfarin than would patients with mildly impaired renal function, and this was in fact observed.”
Approval of the DVT and PE indication was based on the Hokusai-VTE study of about 8,200 people comparing edoxaban to warfarin, which found that the edoxaban 60 mg once a day was noninferior to warfarin in the rate of symptomatic venous thromboembolism (3.2% vs. 3.5% in those on warfarin). The rate of major or clinically relevant nonmajor bleeding events was 8.5% among those on edoxaban vs. 10.3% in those on warfarin (N. Engl. J. Med. 2013;369:1406-15).
Bleeding and anemia were the most common adverse events among patients with nonvalvular atrial fibrillation in clinical trials, and “as with other FDA-approved anticlotting drugs, bleeding, including life-threatening bleeding, is the most serious risk with Savaysa,” the FDA statement said. Among those treated for DVT and PE, the most common adverse events were bleeding, rash, abnormal liver function tests, and anemia.
Savaysa is the fourth novel oral anticoagulant to be cleared by the FDA, after dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis).
Serious adverse events associated with edoxaban should be reported to the FDA’s MedWatch program or at 800-332-1088.
Edoxaban, a selective factor Xa-inhibitor, has been approved by the Food and Drug Administration for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, with a statement in the boxed warning that it should not be used in patients with normal renal function.
The warning reflects the results of a subgroup analysis in the pivotal trial, which found that the 60-mg dose was superior to warfarin in terms of reducing the stroke risk in mildly renally impaired patients, but was worse in patients with normal renal function. This was the main focus of a meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Panel meeting in October, in which the panel voted 9-1 to recommend approval of edoxaban for this indication, but had mixed opinions on whether approval should be limited to patients with mild to moderate renal impairment.
The approved prescribing information recommends that a patient’s creatinine clearance should be checked before edoxaban is prescribed. “Patients with creatinine clearance greater than 95 mL/min have an increased risk of stroke, compared to similar patients given warfarin,” and should be treated with another anticoagulant, the FDA said in the Jan. 9 statement announcing the approval. The recommended dose for those with a creatinine clearance between 50 mL/min and 95 mL/min is 60 mg once a day; for those with a creatinine clearance of 15-50 mL/min, the recommended dose is 30 mg once a day, according to the prescribing information.
Edoxaban, the fourth novel oral anticoagulant drug approved by the FDA, will be marketed as Savaysa by Daiichi Sankyo. It was also approved to treat deep vein thrombosis and pulmonary embolism following 5-10 days of initial therapy with a parenteral anticoagulant. The recommended dose for this indication is 60 mg once a day. For patients with a creatinine clearance of 15-50 mL/min, or who weigh up to 60 kg (about 132 pounds), or who are taking “certain P-glycoprotein inhibitors,” the 30-mg/day dose is recommended.
Approval for the nonvalvular AF indication was based on ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation) study, comparing once-daily edoxaban (60 mg and 30 mg) to warfarin in 21,015 patients with nonvalvular AF, at a moderate to high risk of thromboembolic events (N. Engl. J. Med. 2013;369:2093-104). Over a median of almost 3 years, both doses were noninferior to warfarin in the primary efficacy endpoint, the occurrence of first stroke or of a systemic embolic event. Overall, major bleeding events were significantly lower among those on the 60-mg and 30-mg doses, compared with those on warfarin. However, the rate of ischemic stroke was higher relative to warfarin in patients with a creatinine clearance over 95 mL/min.
About half of the edoxaban dose is eliminated by the kidneys, and patients with a creatinine clearance above 95 mL/min have lower plasma edoxaban levels, according to a statement in the clinical trials section of the prescribing information, which adds: “Given the clear relationship of dose and blood levels to effectiveness in the ENGAGE AF-TIMI 48 study, it could be anticipated that patients with better renal function would show a smaller effect of Savaysa, compared to warfarin than would patients with mildly impaired renal function, and this was in fact observed.”
Approval of the DVT and PE indication was based on the Hokusai-VTE study of about 8,200 people comparing edoxaban to warfarin, which found that the edoxaban 60 mg once a day was noninferior to warfarin in the rate of symptomatic venous thromboembolism (3.2% vs. 3.5% in those on warfarin). The rate of major or clinically relevant nonmajor bleeding events was 8.5% among those on edoxaban vs. 10.3% in those on warfarin (N. Engl. J. Med. 2013;369:1406-15).
Bleeding and anemia were the most common adverse events among patients with nonvalvular atrial fibrillation in clinical trials, and “as with other FDA-approved anticlotting drugs, bleeding, including life-threatening bleeding, is the most serious risk with Savaysa,” the FDA statement said. Among those treated for DVT and PE, the most common adverse events were bleeding, rash, abnormal liver function tests, and anemia.
Savaysa is the fourth novel oral anticoagulant to be cleared by the FDA, after dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis).
Serious adverse events associated with edoxaban should be reported to the FDA’s MedWatch program or at 800-332-1088.
Extended-release carbidopa-levodopa approved for Parkinson’s
An extended-release capsule formulation of carbidopa and levodopa in a 1:4 ratio was approved by the Food and Drug Administration for treating Parkinson’s disease, the manufacturer announced in a statement on Jan. 8.
The combination, which will be marketed as Rytary, also is approved for postencephalitic parkinsonism and parkinsonism that may occur after carbon monoxide intoxication or manganese intoxication, according to the company, Impax Pharmaceuticals.

The company studied the drug combination in patients with early to advanced Parkinson’s in the United States and Europe in the APEX-PD study and the ADVANCE-PD study.
The combination will be available in four strengths: 23.75 mg/95 mg (carbidopa/levodopa), 36.25 mg/145 mg, 48.75 mg/195 mg, and 61.25 mg/245 mg. The capsules, which can be swallowed whole, contain both immediate- and extended-release beads that can be sprinkled on applesauce for immediate consumption by patients who have trouble swallowing, the company said.
The company expects that the product will be available for distribution in February.
The prescribing information is available here.
An extended-release capsule formulation of carbidopa and levodopa in a 1:4 ratio was approved by the Food and Drug Administration for treating Parkinson’s disease, the manufacturer announced in a statement on Jan. 8.
The combination, which will be marketed as Rytary, also is approved for postencephalitic parkinsonism and parkinsonism that may occur after carbon monoxide intoxication or manganese intoxication, according to the company, Impax Pharmaceuticals.
The company studied the drug combination in patients with early to advanced Parkinson’s in the United States and Europe in the APEX-PD study and the ADVANCE-PD study.
The combination will be available in four strengths: 23.75 mg/95 mg (carbidopa/levodopa), 36.25 mg/145 mg, 48.75 mg/195 mg, and 61.25 mg/245 mg. The capsules, which can be swallowed whole, contain both immediate- and extended-release beads that can be sprinkled on applesauce for immediate consumption by patients who have trouble swallowing, the company said.
The company expects that the product will be available for distribution in February.
The prescribing information is available here.
An extended-release capsule formulation of carbidopa and levodopa in a 1:4 ratio was approved by the Food and Drug Administration for treating Parkinson’s disease, the manufacturer announced in a statement on Jan. 8.
The combination, which will be marketed as Rytary, also is approved for postencephalitic parkinsonism and parkinsonism that may occur after carbon monoxide intoxication or manganese intoxication, according to the company, Impax Pharmaceuticals.
The company studied the drug combination in patients with early to advanced Parkinson’s in the United States and Europe in the APEX-PD study and the ADVANCE-PD study.
The combination will be available in four strengths: 23.75 mg/95 mg (carbidopa/levodopa), 36.25 mg/145 mg, 48.75 mg/195 mg, and 61.25 mg/245 mg. The capsules, which can be swallowed whole, contain both immediate- and extended-release beads that can be sprinkled on applesauce for immediate consumption by patients who have trouble swallowing, the company said.
The company expects that the product will be available for distribution in February.
The prescribing information is available here.

FDA panel backs approval of filgrastim biosimilar
SILVER SPRING, MD. – With the unanimous support of a Food and Drug Administration advisory panel and a favorable review by the agency, a “biosimilar” version of filgrastim, the recombinant granulocyte colony-stimulating factor marketed as Neupogen, will likely become the first biosimilar product to be approved in the United States.
At a meeting on Jan. 7, the FDA’s Oncologic Drugs Advisory Committee voted 14-0 that the filgrastim biosimilar should be approved for the five indications approved for Neupogen in the United States, based on “the totality of the evidence,” which includes pharmacokinetic, pharmacodynamic, immunogenicity, and clinical data. With little debate, they agreed that other than minor differences in clinically inactive components, the biosimilar, currently referred to as “EP2006,” was “highly similar” to the reference product, and that there were “no clinically meaningful differences” between EP2006 and Neupogen – the components of the definition of biosimilarity.
The meeting was considered historic, since this is the first biosimilar to be reviewed by an FDA advisory panel, and if approved, will be the first biosimilar to become available as a result of the Biologics Price Competition and Innovation Act of 2009, which was passed as part of the Affordable Care Act, creating “an abbreviated licensure pathway for biological products shown to be ‘biosimilar’ or ‘interchangeable’ with an FDA-licensed biological product,” according to the FDA.
Neupogen was approved by the FDA in 1991 and is marketed by Amgen. If approved, EP2006 would be the first biosimilar product to be approved in the United States, and, like generic drugs, is expected to provide a cheaper version of Neupogen. Sandoz, the generic pharmaceuticals arm of Novartis, plans to market the biosimilar as Zarxio in the United States. The filgrastim biosimilar was approved in 2009 in the European Union, where it is marketed as Zarzio, and it is now approved in more than 60 countries, with more than 7.5 million days of exposure, according to Sandoz.
Between 2009 and 2012, the use of filgrastim increased by 30% in Europe, and the biosimilar is now the most commonly prescribed version of filgrastim in Europe. The cost of filgrastim dropped substantially after approval because of the competition, company officials said.
EP2006 will be available in prefilled syringes containing 300 mcg/0.5 mL or 480 mcg/0.8 mL administered subcutaneously or intravenously, the same strengths as U.S.-approved Neupogen. For approval, Sandoz submitted eight studies, including two studies comparing EP2006 to U.S.-approved Neupogen conducted specifically for the U.S. approval, one of 28 healthy volunteers and another of 218 women with breast cancer, treated with myelosuppressive chemotherapy, evaluating the pharmacokinetics (PK), pharmacodynamics (PD), safety, and immunogenicity (the breast cancer study also evaluated safety and efficacy). The remaining studies included studies in healthy volunteers and breast cancer patients, comparing the biosimilar to European-approved Neupogen, or single-arm studies.
At the meeting, Sigrid Balser, Ph.D., of the global clinical development department at Sandoz, said that in the U.S. study of healthy volunteers, there was a “high degree of similarity” between EP2006 and Neupogen in terms of the PD and PK results, and the absolute neutrophil count (ANC) and CD34+ cell responses. In the U.S. study of breast cancer patients, EP2006 and Neupogen were equivalent in terms of efficacy over the first chemotherapy cycle and had a similar safety profile over six chemotherapy cycles. There were no signs of immunogenicity associated with the biosimilar in the U.S. or European studies in patients with breast cancer or healthy volunteers.
Outside of the United States, more than 3,800 patients have been treated with Zarzio for various indications, including chemotherapy-induced neutropenia, hematopoietic stem cell mobilization, and severe chronic neutropenia, and have been monitored as part of postmarketing trials or routine pharmacovigilance. To date, there have been no signals of any differences in the safety profile of the filgrastim biosimilar, compared with Neupogen;no cases of immunogenicity; and safety and effectiveness has been “confirmed in clinical practice,” Dr. Balser added.
Based on the data, the FDA reviewers concluded that EP2006 is “highly similar to U.S.-licensed Neupogen, and that there are no clinically meaningful difference between the two products,” said Dr. Albert Deisseroth, medical officer and team leader in the division of hematology products, in the FDA’s office of hematology and oncology products.
“Clinically, they appear to really function pretty equally in terms of what you’re asking them to do,” said the panel chair, Dr. Deborah Armstrong, professor of oncology at Johns Hopkins University, Baltimore. The extensive use of the filgrastim biosimilar in other parts of the world provided evidence of “robust safety and efficacy,” which contributed to her support for approval, she added.
Cost is not allowed to be discussed during FDA panel meetings, and Sandoz did not specify any possible price of the biosimilar, but this issue was the “elephant in the room,” as one panelist pointed out. However, Dr. Louis Weiner, chairman of the department of oncology and director of the Lombardi Comprehensive Cancer Center at Georgetown University in Washington, who spoke on behalf of Sandoz at the meeting, said that the availability of the filgrastim biosimilar has “enormous promise in reducing cost and expanding access” to this treatment. Although it has “unquestioned clinical value,” he said that granulocyte colony-stimulating factor therapy is underused.
The five indications approved for Neupogen are to decrease the incidence of infection‚ as manifested by febrile neutropenia in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever; for reducing the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy treatment of adults with acute myeloid leukemia; to reduce the duration of neutropenia and neutropenia-related clinical sequelae; for the mobilization of hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis; and for chronic administration to reduce the incidence and duration of sequelae of neutropenia in symptomatic patients with congenital‚ cyclic‚ or idiopathic neutropenia.
The FDA usually follows the recommendations of its advisory panels. Panelists were cleared of potential conflicts of interest related to the topic of the meeting. In some cases, a panelist may be given a waiver but not at this meeting. Dr. Weiner’s disclosure included being a consultant to Sandoz.
SILVER SPRING, MD. – With the unanimous support of a Food and Drug Administration advisory panel and a favorable review by the agency, a “biosimilar” version of filgrastim, the recombinant granulocyte colony-stimulating factor marketed as Neupogen, will likely become the first biosimilar product to be approved in the United States.
At a meeting on Jan. 7, the FDA’s Oncologic Drugs Advisory Committee voted 14-0 that the filgrastim biosimilar should be approved for the five indications approved for Neupogen in the United States, based on “the totality of the evidence,” which includes pharmacokinetic, pharmacodynamic, immunogenicity, and clinical data. With little debate, they agreed that other than minor differences in clinically inactive components, the biosimilar, currently referred to as “EP2006,” was “highly similar” to the reference product, and that there were “no clinically meaningful differences” between EP2006 and Neupogen – the components of the definition of biosimilarity.
The meeting was considered historic, since this is the first biosimilar to be reviewed by an FDA advisory panel, and if approved, will be the first biosimilar to become available as a result of the Biologics Price Competition and Innovation Act of 2009, which was passed as part of the Affordable Care Act, creating “an abbreviated licensure pathway for biological products shown to be ‘biosimilar’ or ‘interchangeable’ with an FDA-licensed biological product,” according to the FDA.
Neupogen was approved by the FDA in 1991 and is marketed by Amgen. If approved, EP2006 would be the first biosimilar product to be approved in the United States, and, like generic drugs, is expected to provide a cheaper version of Neupogen. Sandoz, the generic pharmaceuticals arm of Novartis, plans to market the biosimilar as Zarxio in the United States. The filgrastim biosimilar was approved in 2009 in the European Union, where it is marketed as Zarzio, and it is now approved in more than 60 countries, with more than 7.5 million days of exposure, according to Sandoz.
Between 2009 and 2012, the use of filgrastim increased by 30% in Europe, and the biosimilar is now the most commonly prescribed version of filgrastim in Europe. The cost of filgrastim dropped substantially after approval because of the competition, company officials said.
EP2006 will be available in prefilled syringes containing 300 mcg/0.5 mL or 480 mcg/0.8 mL administered subcutaneously or intravenously, the same strengths as U.S.-approved Neupogen. For approval, Sandoz submitted eight studies, including two studies comparing EP2006 to U.S.-approved Neupogen conducted specifically for the U.S. approval, one of 28 healthy volunteers and another of 218 women with breast cancer, treated with myelosuppressive chemotherapy, evaluating the pharmacokinetics (PK), pharmacodynamics (PD), safety, and immunogenicity (the breast cancer study also evaluated safety and efficacy). The remaining studies included studies in healthy volunteers and breast cancer patients, comparing the biosimilar to European-approved Neupogen, or single-arm studies.
At the meeting, Sigrid Balser, Ph.D., of the global clinical development department at Sandoz, said that in the U.S. study of healthy volunteers, there was a “high degree of similarity” between EP2006 and Neupogen in terms of the PD and PK results, and the absolute neutrophil count (ANC) and CD34+ cell responses. In the U.S. study of breast cancer patients, EP2006 and Neupogen were equivalent in terms of efficacy over the first chemotherapy cycle and had a similar safety profile over six chemotherapy cycles. There were no signs of immunogenicity associated with the biosimilar in the U.S. or European studies in patients with breast cancer or healthy volunteers.
Outside of the United States, more than 3,800 patients have been treated with Zarzio for various indications, including chemotherapy-induced neutropenia, hematopoietic stem cell mobilization, and severe chronic neutropenia, and have been monitored as part of postmarketing trials or routine pharmacovigilance. To date, there have been no signals of any differences in the safety profile of the filgrastim biosimilar, compared with Neupogen;no cases of immunogenicity; and safety and effectiveness has been “confirmed in clinical practice,” Dr. Balser added.
Based on the data, the FDA reviewers concluded that EP2006 is “highly similar to U.S.-licensed Neupogen, and that there are no clinically meaningful difference between the two products,” said Dr. Albert Deisseroth, medical officer and team leader in the division of hematology products, in the FDA’s office of hematology and oncology products.
“Clinically, they appear to really function pretty equally in terms of what you’re asking them to do,” said the panel chair, Dr. Deborah Armstrong, professor of oncology at Johns Hopkins University, Baltimore. The extensive use of the filgrastim biosimilar in other parts of the world provided evidence of “robust safety and efficacy,” which contributed to her support for approval, she added.
Cost is not allowed to be discussed during FDA panel meetings, and Sandoz did not specify any possible price of the biosimilar, but this issue was the “elephant in the room,” as one panelist pointed out. However, Dr. Louis Weiner, chairman of the department of oncology and director of the Lombardi Comprehensive Cancer Center at Georgetown University in Washington, who spoke on behalf of Sandoz at the meeting, said that the availability of the filgrastim biosimilar has “enormous promise in reducing cost and expanding access” to this treatment. Although it has “unquestioned clinical value,” he said that granulocyte colony-stimulating factor therapy is underused.
The five indications approved for Neupogen are to decrease the incidence of infection‚ as manifested by febrile neutropenia in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever; for reducing the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy treatment of adults with acute myeloid leukemia; to reduce the duration of neutropenia and neutropenia-related clinical sequelae; for the mobilization of hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis; and for chronic administration to reduce the incidence and duration of sequelae of neutropenia in symptomatic patients with congenital‚ cyclic‚ or idiopathic neutropenia.
The FDA usually follows the recommendations of its advisory panels. Panelists were cleared of potential conflicts of interest related to the topic of the meeting. In some cases, a panelist may be given a waiver but not at this meeting. Dr. Weiner’s disclosure included being a consultant to Sandoz.
SILVER SPRING, MD. – With the unanimous support of a Food and Drug Administration advisory panel and a favorable review by the agency, a “biosimilar” version of filgrastim, the recombinant granulocyte colony-stimulating factor marketed as Neupogen, will likely become the first biosimilar product to be approved in the United States.
At a meeting on Jan. 7, the FDA’s Oncologic Drugs Advisory Committee voted 14-0 that the filgrastim biosimilar should be approved for the five indications approved for Neupogen in the United States, based on “the totality of the evidence,” which includes pharmacokinetic, pharmacodynamic, immunogenicity, and clinical data. With little debate, they agreed that other than minor differences in clinically inactive components, the biosimilar, currently referred to as “EP2006,” was “highly similar” to the reference product, and that there were “no clinically meaningful differences” between EP2006 and Neupogen – the components of the definition of biosimilarity.
The meeting was considered historic, since this is the first biosimilar to be reviewed by an FDA advisory panel, and if approved, will be the first biosimilar to become available as a result of the Biologics Price Competition and Innovation Act of 2009, which was passed as part of the Affordable Care Act, creating “an abbreviated licensure pathway for biological products shown to be ‘biosimilar’ or ‘interchangeable’ with an FDA-licensed biological product,” according to the FDA.
Neupogen was approved by the FDA in 1991 and is marketed by Amgen. If approved, EP2006 would be the first biosimilar product to be approved in the United States, and, like generic drugs, is expected to provide a cheaper version of Neupogen. Sandoz, the generic pharmaceuticals arm of Novartis, plans to market the biosimilar as Zarxio in the United States. The filgrastim biosimilar was approved in 2009 in the European Union, where it is marketed as Zarzio, and it is now approved in more than 60 countries, with more than 7.5 million days of exposure, according to Sandoz.
Between 2009 and 2012, the use of filgrastim increased by 30% in Europe, and the biosimilar is now the most commonly prescribed version of filgrastim in Europe. The cost of filgrastim dropped substantially after approval because of the competition, company officials said.
EP2006 will be available in prefilled syringes containing 300 mcg/0.5 mL or 480 mcg/0.8 mL administered subcutaneously or intravenously, the same strengths as U.S.-approved Neupogen. For approval, Sandoz submitted eight studies, including two studies comparing EP2006 to U.S.-approved Neupogen conducted specifically for the U.S. approval, one of 28 healthy volunteers and another of 218 women with breast cancer, treated with myelosuppressive chemotherapy, evaluating the pharmacokinetics (PK), pharmacodynamics (PD), safety, and immunogenicity (the breast cancer study also evaluated safety and efficacy). The remaining studies included studies in healthy volunteers and breast cancer patients, comparing the biosimilar to European-approved Neupogen, or single-arm studies.
At the meeting, Sigrid Balser, Ph.D., of the global clinical development department at Sandoz, said that in the U.S. study of healthy volunteers, there was a “high degree of similarity” between EP2006 and Neupogen in terms of the PD and PK results, and the absolute neutrophil count (ANC) and CD34+ cell responses. In the U.S. study of breast cancer patients, EP2006 and Neupogen were equivalent in terms of efficacy over the first chemotherapy cycle and had a similar safety profile over six chemotherapy cycles. There were no signs of immunogenicity associated with the biosimilar in the U.S. or European studies in patients with breast cancer or healthy volunteers.
Outside of the United States, more than 3,800 patients have been treated with Zarzio for various indications, including chemotherapy-induced neutropenia, hematopoietic stem cell mobilization, and severe chronic neutropenia, and have been monitored as part of postmarketing trials or routine pharmacovigilance. To date, there have been no signals of any differences in the safety profile of the filgrastim biosimilar, compared with Neupogen;no cases of immunogenicity; and safety and effectiveness has been “confirmed in clinical practice,” Dr. Balser added.
Based on the data, the FDA reviewers concluded that EP2006 is “highly similar to U.S.-licensed Neupogen, and that there are no clinically meaningful difference between the two products,” said Dr. Albert Deisseroth, medical officer and team leader in the division of hematology products, in the FDA’s office of hematology and oncology products.
“Clinically, they appear to really function pretty equally in terms of what you’re asking them to do,” said the panel chair, Dr. Deborah Armstrong, professor of oncology at Johns Hopkins University, Baltimore. The extensive use of the filgrastim biosimilar in other parts of the world provided evidence of “robust safety and efficacy,” which contributed to her support for approval, she added.
Cost is not allowed to be discussed during FDA panel meetings, and Sandoz did not specify any possible price of the biosimilar, but this issue was the “elephant in the room,” as one panelist pointed out. However, Dr. Louis Weiner, chairman of the department of oncology and director of the Lombardi Comprehensive Cancer Center at Georgetown University in Washington, who spoke on behalf of Sandoz at the meeting, said that the availability of the filgrastim biosimilar has “enormous promise in reducing cost and expanding access” to this treatment. Although it has “unquestioned clinical value,” he said that granulocyte colony-stimulating factor therapy is underused.
The five indications approved for Neupogen are to decrease the incidence of infection‚ as manifested by febrile neutropenia in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever; for reducing the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy treatment of adults with acute myeloid leukemia; to reduce the duration of neutropenia and neutropenia-related clinical sequelae; for the mobilization of hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis; and for chronic administration to reduce the incidence and duration of sequelae of neutropenia in symptomatic patients with congenital‚ cyclic‚ or idiopathic neutropenia.
The FDA usually follows the recommendations of its advisory panels. Panelists were cleared of potential conflicts of interest related to the topic of the meeting. In some cases, a panelist may be given a waiver but not at this meeting. Dr. Weiner’s disclosure included being a consultant to Sandoz.
AT AN FDA ADVISORY COMMITTEE MEETING
FDA approves IV antibacterial for complicated UTIs, abdominal infections
A combination of a cephalosporin and a beta-lactamase inhibitor in an intravenous formulation has been approved for treating complicated intra-abdominal infections and complicated urinary tract infections in adults, the Food and Drug Administration announced on Dec. 19.
The cephalosporin is ceftolozane and the beta-lactamase inhibitor is tazobactam; it will be marketed as Zerbaxa by Cubist Pharmaceuticals.
This is the fourth antibacterial drug product approved by the FDA in 2014 and, like the other three, it was designated as a Qualified Infectious Disease Product (QIDP) and was given priority review, according to the FDA statement. Zerbaxa was granted a QIDP designation under the Generating Antibiotic Incentives Now (GAIN) Act of the FDA Safety and Innovation Act, “because it is an antibacterial or antifungal human drug intended to treat a serious or life-threatening infection,” the statement said.
As part of the QIDP program, the manufacturer has also been granted an extra 5 years of “exclusivity” – exclusive marketing rights – by the FDA.
Ceftolozane-tazobactam was approved for treating complicated intra-abdominal infections in combination with metronidazole; approval for this indication was based on a study of 979 adults, randomized to the combination or to meropenem.
Approval for complicated urinary tract infections, including pyelonephritis, was based on a study of 1,068 adults, randomized to treatment with ceftolozane-tazobactam or levofloxacin.
The prescribing information includes a warning about decreased efficacy in patients with renal impairment (a baseline creatinine clearance of 30-50 mL/min or less, and the recommendation to monitor creatinine clearance “at least daily in patients with changing renal function,” and to adjust dose accordingly. Nausea, diarrhea, headache, and fever were the most common adverse events in studies, according to the FDA statement.
The other antibacterials approved by the FDA in 2014 were approved for treating acute bacterial skin and skin structure infections caused by certain susceptible bacteria. They were dalbavancin (Dalvance), approved in May; tedizolid (Sivextro), approved in June; and oritavancin (Orbactiv), approved in August.
Serious adverse events associated with Zerbaxa should be reported to the FDA’s MedWatch program at 800-332-1088 or http://www.fda.gov/Safety/MedWatch/HowToReport/default.htm.
A combination of a cephalosporin and a beta-lactamase inhibitor in an intravenous formulation has been approved for treating complicated intra-abdominal infections and complicated urinary tract infections in adults, the Food and Drug Administration announced on Dec. 19.
The cephalosporin is ceftolozane and the beta-lactamase inhibitor is tazobactam; it will be marketed as Zerbaxa by Cubist Pharmaceuticals.
This is the fourth antibacterial drug product approved by the FDA in 2014 and, like the other three, it was designated as a Qualified Infectious Disease Product (QIDP) and was given priority review, according to the FDA statement. Zerbaxa was granted a QIDP designation under the Generating Antibiotic Incentives Now (GAIN) Act of the FDA Safety and Innovation Act, “because it is an antibacterial or antifungal human drug intended to treat a serious or life-threatening infection,” the statement said.
As part of the QIDP program, the manufacturer has also been granted an extra 5 years of “exclusivity” – exclusive marketing rights – by the FDA.
Ceftolozane-tazobactam was approved for treating complicated intra-abdominal infections in combination with metronidazole; approval for this indication was based on a study of 979 adults, randomized to the combination or to meropenem.
Approval for complicated urinary tract infections, including pyelonephritis, was based on a study of 1,068 adults, randomized to treatment with ceftolozane-tazobactam or levofloxacin.
The prescribing information includes a warning about decreased efficacy in patients with renal impairment (a baseline creatinine clearance of 30-50 mL/min or less, and the recommendation to monitor creatinine clearance “at least daily in patients with changing renal function,” and to adjust dose accordingly. Nausea, diarrhea, headache, and fever were the most common adverse events in studies, according to the FDA statement.
The other antibacterials approved by the FDA in 2014 were approved for treating acute bacterial skin and skin structure infections caused by certain susceptible bacteria. They were dalbavancin (Dalvance), approved in May; tedizolid (Sivextro), approved in June; and oritavancin (Orbactiv), approved in August.
Serious adverse events associated with Zerbaxa should be reported to the FDA’s MedWatch program at 800-332-1088 or http://www.fda.gov/Safety/MedWatch/HowToReport/default.htm.
A combination of a cephalosporin and a beta-lactamase inhibitor in an intravenous formulation has been approved for treating complicated intra-abdominal infections and complicated urinary tract infections in adults, the Food and Drug Administration announced on Dec. 19.
The cephalosporin is ceftolozane and the beta-lactamase inhibitor is tazobactam; it will be marketed as Zerbaxa by Cubist Pharmaceuticals.
This is the fourth antibacterial drug product approved by the FDA in 2014 and, like the other three, it was designated as a Qualified Infectious Disease Product (QIDP) and was given priority review, according to the FDA statement. Zerbaxa was granted a QIDP designation under the Generating Antibiotic Incentives Now (GAIN) Act of the FDA Safety and Innovation Act, “because it is an antibacterial or antifungal human drug intended to treat a serious or life-threatening infection,” the statement said.
As part of the QIDP program, the manufacturer has also been granted an extra 5 years of “exclusivity” – exclusive marketing rights – by the FDA.
Ceftolozane-tazobactam was approved for treating complicated intra-abdominal infections in combination with metronidazole; approval for this indication was based on a study of 979 adults, randomized to the combination or to meropenem.
Approval for complicated urinary tract infections, including pyelonephritis, was based on a study of 1,068 adults, randomized to treatment with ceftolozane-tazobactam or levofloxacin.
The prescribing information includes a warning about decreased efficacy in patients with renal impairment (a baseline creatinine clearance of 30-50 mL/min or less, and the recommendation to monitor creatinine clearance “at least daily in patients with changing renal function,” and to adjust dose accordingly. Nausea, diarrhea, headache, and fever were the most common adverse events in studies, according to the FDA statement.
The other antibacterials approved by the FDA in 2014 were approved for treating acute bacterial skin and skin structure infections caused by certain susceptible bacteria. They were dalbavancin (Dalvance), approved in May; tedizolid (Sivextro), approved in June; and oritavancin (Orbactiv), approved in August.
Serious adverse events associated with Zerbaxa should be reported to the FDA’s MedWatch program at 800-332-1088 or http://www.fda.gov/Safety/MedWatch/HowToReport/default.htm.