User login
Canadian agency reports rare, severe skin reactions to cancer drug
A notice about the risk of severe skin reactions associated with the cancer drug capecitabine – including Stevens-Johnson syndrome – has been released by the Canadian health department.
"Very rare cases of severe cutaneous reactions such as Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), in some cases with fatal outcome, have been reported during treatment with" capecitabine, according to a letter to health care professionals, posted on the Health Canada website on Dec. 3. The letter states that treatment with capecitabine should be "immediately discontinued" if a patient develops signs and symptoms of SJS or TEN.
No such notice has been posted by the U.S. Food and Drug Administration. The FDA "constantly monitors and reviews reports from regulatory agencies, especially those concerning products that affect Americans’ health," and is reviewing related scientific information on this issue, an FDA spokesperson said in response to a query on the notice. The agency "will determine what, if any, actions to take," he added.
Capecitabine is a nucleoside metabolic inhibitor, and is marketed as Xeloda in Canada and the United States for treatment of colorectal cancer and breast cancer. In the United States, it was initially approved in 1998 for treating metastatic breast cancer, and is now also approved as a first-line treatment for metastatic colon cancer, as well as for adjuvant colon cancer in patients with Dukes’ C colon cancer. It is marketed by Roche, which the letter says is working with Health Canada to revise the product monograph in Canada. In the United States, a generic formulation is also available.
The U.S. label includes hand-and-foot syndrome (including grade 2 and 3 events), dermatitis, rash, and erythema among the skin and subcutaneous adverse events reported in clinical trials, but there are no reports of SJS or TEN listed.
Serious adverse events associated with capecitabine should be reported to MedWatch, the FDA Safety Information and Adverse Event Reporting Program, at www.fda.gov/Safety/MedWatch/default.htm.
A notice about the risk of severe skin reactions associated with the cancer drug capecitabine – including Stevens-Johnson syndrome – has been released by the Canadian health department.
"Very rare cases of severe cutaneous reactions such as Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), in some cases with fatal outcome, have been reported during treatment with" capecitabine, according to a letter to health care professionals, posted on the Health Canada website on Dec. 3. The letter states that treatment with capecitabine should be "immediately discontinued" if a patient develops signs and symptoms of SJS or TEN.
No such notice has been posted by the U.S. Food and Drug Administration. The FDA "constantly monitors and reviews reports from regulatory agencies, especially those concerning products that affect Americans’ health," and is reviewing related scientific information on this issue, an FDA spokesperson said in response to a query on the notice. The agency "will determine what, if any, actions to take," he added.
Capecitabine is a nucleoside metabolic inhibitor, and is marketed as Xeloda in Canada and the United States for treatment of colorectal cancer and breast cancer. In the United States, it was initially approved in 1998 for treating metastatic breast cancer, and is now also approved as a first-line treatment for metastatic colon cancer, as well as for adjuvant colon cancer in patients with Dukes’ C colon cancer. It is marketed by Roche, which the letter says is working with Health Canada to revise the product monograph in Canada. In the United States, a generic formulation is also available.
The U.S. label includes hand-and-foot syndrome (including grade 2 and 3 events), dermatitis, rash, and erythema among the skin and subcutaneous adverse events reported in clinical trials, but there are no reports of SJS or TEN listed.
Serious adverse events associated with capecitabine should be reported to MedWatch, the FDA Safety Information and Adverse Event Reporting Program, at www.fda.gov/Safety/MedWatch/default.htm.
A notice about the risk of severe skin reactions associated with the cancer drug capecitabine – including Stevens-Johnson syndrome – has been released by the Canadian health department.
"Very rare cases of severe cutaneous reactions such as Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), in some cases with fatal outcome, have been reported during treatment with" capecitabine, according to a letter to health care professionals, posted on the Health Canada website on Dec. 3. The letter states that treatment with capecitabine should be "immediately discontinued" if a patient develops signs and symptoms of SJS or TEN.
No such notice has been posted by the U.S. Food and Drug Administration. The FDA "constantly monitors and reviews reports from regulatory agencies, especially those concerning products that affect Americans’ health," and is reviewing related scientific information on this issue, an FDA spokesperson said in response to a query on the notice. The agency "will determine what, if any, actions to take," he added.
Capecitabine is a nucleoside metabolic inhibitor, and is marketed as Xeloda in Canada and the United States for treatment of colorectal cancer and breast cancer. In the United States, it was initially approved in 1998 for treating metastatic breast cancer, and is now also approved as a first-line treatment for metastatic colon cancer, as well as for adjuvant colon cancer in patients with Dukes’ C colon cancer. It is marketed by Roche, which the letter says is working with Health Canada to revise the product monograph in Canada. In the United States, a generic formulation is also available.
The U.S. label includes hand-and-foot syndrome (including grade 2 and 3 events), dermatitis, rash, and erythema among the skin and subcutaneous adverse events reported in clinical trials, but there are no reports of SJS or TEN listed.
Serious adverse events associated with capecitabine should be reported to MedWatch, the FDA Safety Information and Adverse Event Reporting Program, at www.fda.gov/Safety/MedWatch/default.htm.
FDA reviewing emergency contraception data in overweight women
Evidence that the levonorgestrel emergency contraceptive is less effective in overweight women is being reviewed by the Food and Drug Administration, which will determine whether this information should be added to the U.S. labeling of the product.
In November, a warning that Norlevo, which contains 1.5 mg of levonorgestrel, is less effective in women who weigh at least 75 kg (165 pounds) and is not effective in women who weigh more than 80 kg (176 pounds), was added to the label of the product in Europe. No such information is included in the labels of the levonorgestrel emergency contraceptives marketed in the United States. Norlevo, which is not available in the United States, contains the same dose as Plan B One-Step, a 1.5-mg levonorgestrel tablet, available over the counter for women aged 15 years and older in the United States.

In response, "the FDA is currently reviewing the available and related scientific information on this issue, including the publication upon which the Norlevo labeling change was based," an FDA spokesperson said. "The agency will then determine what, if any, labelling changes to approved emergency contraceptives are warranted." The FDA’s approval of Plan B did not include a weight-related assessment of efficacy.
The study, a meta-analysis of randomized studies comparing the efficacy of ulipristal acetate (marketed as ella), and levonorgestrel emergency contraceptives, conducted to identify factors associated with failure of the emergency contraceptives, was published more than 2 years ago. For all emergency contraceptives, the risk of pregnancy was more than three times higher (odds ratio, 3.06) among obese women compared with women with a normal body mass index (BMI). But when investigators looked at specific methods, the risk of pregnancy among obese women was four times higher among those who took levonorgestrel (OR, 4.41) compared with those who used ulipristal (Contraception 2011;84;363-7).
Ulipristal is taken as a single tablet and requires a prescription. Experts in the United States already recommend other emergency contraceptive options, when possible.
Dr. Carolyn Westhoff, professor of obstetrics and gynecology, and of epidemiology, New York–Presbyterian Hospital/Columbia University Medical Center, said she advises clinicians to inform women of any weight that the copper IUD is the most effective form of emergency contraception available. It can be inserted at any time during the cycle, provides ongoing contraceptive protection, and, if indicated, routine screening for chlamydia can be performed at the time of insertion, she pointed out.
After the copper IUD, the most effective emergency contraceptive is ulipristal acetate, which can be prescribed in advance to sexually active women of reproductive age, she added. "While the effectiveness of ella might diminish somewhat with greater weight, that effect is less pronounced," said Dr. Westhoff, who is also the medical adviser to Planned Parenthood Federation of America (PPFA).
"At this point in our understanding, it seems that Plan B should be last choice when providing EC. For women who do not have a relationship to a health care provider, it might be the only choice, but for those women who have a relationship with a clinician, the more effective methods are always preferable," she said.
Planned Parenthood does not recommend Plan B for women with a BMI over 30, according to Karen Shea, director of medical standards at PPFA.
A statement provided by PPFA says that over-the-counter forms of emergency contraception, such as Plan B One-Step, "may not be as effective among women with a BMI of 25 or higher," and points out that "regardless of a woman’s weight, after 3 days, over-the-counter forms diminish in effectiveness."
Generic two-pill formulations of levonorgestrel are also available in the United States.
Dr. Westhoff said she had no relevant financial disclosures. Teva Pharmaceuticals, which markets Plan B One-Step in the United States, did not respond to a request for a comment.
Evidence that the levonorgestrel emergency contraceptive is less effective in overweight women is being reviewed by the Food and Drug Administration, which will determine whether this information should be added to the U.S. labeling of the product.
In November, a warning that Norlevo, which contains 1.5 mg of levonorgestrel, is less effective in women who weigh at least 75 kg (165 pounds) and is not effective in women who weigh more than 80 kg (176 pounds), was added to the label of the product in Europe. No such information is included in the labels of the levonorgestrel emergency contraceptives marketed in the United States. Norlevo, which is not available in the United States, contains the same dose as Plan B One-Step, a 1.5-mg levonorgestrel tablet, available over the counter for women aged 15 years and older in the United States.
In response, "the FDA is currently reviewing the available and related scientific information on this issue, including the publication upon which the Norlevo labeling change was based," an FDA spokesperson said. "The agency will then determine what, if any, labelling changes to approved emergency contraceptives are warranted." The FDA’s approval of Plan B did not include a weight-related assessment of efficacy.
The study, a meta-analysis of randomized studies comparing the efficacy of ulipristal acetate (marketed as ella), and levonorgestrel emergency contraceptives, conducted to identify factors associated with failure of the emergency contraceptives, was published more than 2 years ago. For all emergency contraceptives, the risk of pregnancy was more than three times higher (odds ratio, 3.06) among obese women compared with women with a normal body mass index (BMI). But when investigators looked at specific methods, the risk of pregnancy among obese women was four times higher among those who took levonorgestrel (OR, 4.41) compared with those who used ulipristal (Contraception 2011;84;363-7).
Ulipristal is taken as a single tablet and requires a prescription. Experts in the United States already recommend other emergency contraceptive options, when possible.
Dr. Carolyn Westhoff, professor of obstetrics and gynecology, and of epidemiology, New York–Presbyterian Hospital/Columbia University Medical Center, said she advises clinicians to inform women of any weight that the copper IUD is the most effective form of emergency contraception available. It can be inserted at any time during the cycle, provides ongoing contraceptive protection, and, if indicated, routine screening for chlamydia can be performed at the time of insertion, she pointed out.
After the copper IUD, the most effective emergency contraceptive is ulipristal acetate, which can be prescribed in advance to sexually active women of reproductive age, she added. "While the effectiveness of ella might diminish somewhat with greater weight, that effect is less pronounced," said Dr. Westhoff, who is also the medical adviser to Planned Parenthood Federation of America (PPFA).
"At this point in our understanding, it seems that Plan B should be last choice when providing EC. For women who do not have a relationship to a health care provider, it might be the only choice, but for those women who have a relationship with a clinician, the more effective methods are always preferable," she said.
Planned Parenthood does not recommend Plan B for women with a BMI over 30, according to Karen Shea, director of medical standards at PPFA.
A statement provided by PPFA says that over-the-counter forms of emergency contraception, such as Plan B One-Step, "may not be as effective among women with a BMI of 25 or higher," and points out that "regardless of a woman’s weight, after 3 days, over-the-counter forms diminish in effectiveness."
Generic two-pill formulations of levonorgestrel are also available in the United States.
Dr. Westhoff said she had no relevant financial disclosures. Teva Pharmaceuticals, which markets Plan B One-Step in the United States, did not respond to a request for a comment.
Evidence that the levonorgestrel emergency contraceptive is less effective in overweight women is being reviewed by the Food and Drug Administration, which will determine whether this information should be added to the U.S. labeling of the product.
In November, a warning that Norlevo, which contains 1.5 mg of levonorgestrel, is less effective in women who weigh at least 75 kg (165 pounds) and is not effective in women who weigh more than 80 kg (176 pounds), was added to the label of the product in Europe. No such information is included in the labels of the levonorgestrel emergency contraceptives marketed in the United States. Norlevo, which is not available in the United States, contains the same dose as Plan B One-Step, a 1.5-mg levonorgestrel tablet, available over the counter for women aged 15 years and older in the United States.
In response, "the FDA is currently reviewing the available and related scientific information on this issue, including the publication upon which the Norlevo labeling change was based," an FDA spokesperson said. "The agency will then determine what, if any, labelling changes to approved emergency contraceptives are warranted." The FDA’s approval of Plan B did not include a weight-related assessment of efficacy.
The study, a meta-analysis of randomized studies comparing the efficacy of ulipristal acetate (marketed as ella), and levonorgestrel emergency contraceptives, conducted to identify factors associated with failure of the emergency contraceptives, was published more than 2 years ago. For all emergency contraceptives, the risk of pregnancy was more than three times higher (odds ratio, 3.06) among obese women compared with women with a normal body mass index (BMI). But when investigators looked at specific methods, the risk of pregnancy among obese women was four times higher among those who took levonorgestrel (OR, 4.41) compared with those who used ulipristal (Contraception 2011;84;363-7).
Ulipristal is taken as a single tablet and requires a prescription. Experts in the United States already recommend other emergency contraceptive options, when possible.
Dr. Carolyn Westhoff, professor of obstetrics and gynecology, and of epidemiology, New York–Presbyterian Hospital/Columbia University Medical Center, said she advises clinicians to inform women of any weight that the copper IUD is the most effective form of emergency contraception available. It can be inserted at any time during the cycle, provides ongoing contraceptive protection, and, if indicated, routine screening for chlamydia can be performed at the time of insertion, she pointed out.
After the copper IUD, the most effective emergency contraceptive is ulipristal acetate, which can be prescribed in advance to sexually active women of reproductive age, she added. "While the effectiveness of ella might diminish somewhat with greater weight, that effect is less pronounced," said Dr. Westhoff, who is also the medical adviser to Planned Parenthood Federation of America (PPFA).
"At this point in our understanding, it seems that Plan B should be last choice when providing EC. For women who do not have a relationship to a health care provider, it might be the only choice, but for those women who have a relationship with a clinician, the more effective methods are always preferable," she said.
Planned Parenthood does not recommend Plan B for women with a BMI over 30, according to Karen Shea, director of medical standards at PPFA.
A statement provided by PPFA says that over-the-counter forms of emergency contraception, such as Plan B One-Step, "may not be as effective among women with a BMI of 25 or higher," and points out that "regardless of a woman’s weight, after 3 days, over-the-counter forms diminish in effectiveness."
Generic two-pill formulations of levonorgestrel are also available in the United States.
Dr. Westhoff said she had no relevant financial disclosures. Teva Pharmaceuticals, which markets Plan B One-Step in the United States, did not respond to a request for a comment.

Once-a-day HCV Protease Inhibitor Approved for Treating Hepatitis C
Simeprevir, a hepatitis C virus protease inhibitor, has been approved as a treatment for chronic hepatitis C in patients who have compensated liver disease, including cirrhosis, the Food and Drug Administration announced.
For patients with HCV genotype 1 infections, simeprevir’s approved dose is a 150-mg capsule taken once a day with food, in combination with peginterferon-alfa and ribavirin for 24-48 weeks. Before treatment, clinicians should screen patients with genotype 1a HCV infections for the presence of the NS3 Q80K polymorphism, which was associated with lower response rates in studies, "and to consider alternative therapy if the strain is detected," according to the FDA’s Nov. 22 statement announcing the approval.
Simeprevir is the third protease inhibitor to be approved by the FDA for treatment of hepatitis C. Boceprevir (Victrelis) and telaprevir (Incivek) were approved in 2011 and have been the standard of care, but they require taking 6-12 pills a day. Simeprevir is an NS3/4A protease inhibitor, which blocks the viral protease enzyme that enables HCV to replicate in host cells, according to Janssen Pharmaceuticals, which will market the drug as Olysio.
Simeprevir’s simpler dosing regimen, effectiveness, and favorable risk-benefit profile in clinical trials were cited by the FDA’s antiviral drug advisory committee in its unanimous recommendation to approve the drug at a meeting on Oct. 24.
Approval is based on five trials of 2,026 treatment-naive and treatment-experienced patients with chronic hepatitis C who were randomized to simeprevir plus peginterferon-alfa and ribavirin (PR) or to placebo plus PR. The primary endpoint was sustained virologic response rate at 12 weeks after treatment was planned to end (SVR12). SVR was defined as an HCV RNA level that was undetectable or detected only below a prespecified value.
Among the treatment-naive patients, the SVR12 rate was 80% for those treated with simeprevir plus PR and 50% in those who received placebo plus PR. In a study of treatment-experienced patients, 79% of those treated with the simeprevir regimen and 37% of those on peginterferon-alfa and ribavirin achieved SVR12.
In another study of treatment-experienced patients that included prior relapsers, partial responders, and null responders (those who did not respond to previous treatments at all), the addition of simeprevir to PR improved response rates when compared with treatment with PR alone, according to the FDA.
Rash, pruritus, and nausea were the most common adverse effects associated with treatment in the studies, the FDA said. There were cases of serious photosensitivity that required hospitalization, and the prescribing information recommends that patients use sun protection and limit sun exposure during treatment. The prescribing information notes that dosing recommendations cannot be made at this time for patients with moderate to severe hepatic impairment or those of East Asian descent. At the FDA panel meeting, company officials said that the appropriate dose in this population was being investigated.
Simeprevir was approved for treatment of genotype 1 HCV infection in September 2013 in Japan, where it is marketed as Sovriad (at a lower 100-mg dose), and in November in Canada, where it is marketed as Galexos, according to Janssen. Simeprevir is under review in Europe for the treatment of genotype 1 or 4 HCV.
Imminent approval of sofosbuvir, another HCV treatment with a different mechanism of action, is expected.
Simeprevir, a hepatitis C virus protease inhibitor, has been approved as a treatment for chronic hepatitis C in patients who have compensated liver disease, including cirrhosis, the Food and Drug Administration announced.
For patients with HCV genotype 1 infections, simeprevir’s approved dose is a 150-mg capsule taken once a day with food, in combination with peginterferon-alfa and ribavirin for 24-48 weeks. Before treatment, clinicians should screen patients with genotype 1a HCV infections for the presence of the NS3 Q80K polymorphism, which was associated with lower response rates in studies, "and to consider alternative therapy if the strain is detected," according to the FDA’s Nov. 22 statement announcing the approval.
Simeprevir is the third protease inhibitor to be approved by the FDA for treatment of hepatitis C. Boceprevir (Victrelis) and telaprevir (Incivek) were approved in 2011 and have been the standard of care, but they require taking 6-12 pills a day. Simeprevir is an NS3/4A protease inhibitor, which blocks the viral protease enzyme that enables HCV to replicate in host cells, according to Janssen Pharmaceuticals, which will market the drug as Olysio.
Simeprevir’s simpler dosing regimen, effectiveness, and favorable risk-benefit profile in clinical trials were cited by the FDA’s antiviral drug advisory committee in its unanimous recommendation to approve the drug at a meeting on Oct. 24.
Approval is based on five trials of 2,026 treatment-naive and treatment-experienced patients with chronic hepatitis C who were randomized to simeprevir plus peginterferon-alfa and ribavirin (PR) or to placebo plus PR. The primary endpoint was sustained virologic response rate at 12 weeks after treatment was planned to end (SVR12). SVR was defined as an HCV RNA level that was undetectable or detected only below a prespecified value.
Among the treatment-naive patients, the SVR12 rate was 80% for those treated with simeprevir plus PR and 50% in those who received placebo plus PR. In a study of treatment-experienced patients, 79% of those treated with the simeprevir regimen and 37% of those on peginterferon-alfa and ribavirin achieved SVR12.
In another study of treatment-experienced patients that included prior relapsers, partial responders, and null responders (those who did not respond to previous treatments at all), the addition of simeprevir to PR improved response rates when compared with treatment with PR alone, according to the FDA.
Rash, pruritus, and nausea were the most common adverse effects associated with treatment in the studies, the FDA said. There were cases of serious photosensitivity that required hospitalization, and the prescribing information recommends that patients use sun protection and limit sun exposure during treatment. The prescribing information notes that dosing recommendations cannot be made at this time for patients with moderate to severe hepatic impairment or those of East Asian descent. At the FDA panel meeting, company officials said that the appropriate dose in this population was being investigated.
Simeprevir was approved for treatment of genotype 1 HCV infection in September 2013 in Japan, where it is marketed as Sovriad (at a lower 100-mg dose), and in November in Canada, where it is marketed as Galexos, according to Janssen. Simeprevir is under review in Europe for the treatment of genotype 1 or 4 HCV.
Imminent approval of sofosbuvir, another HCV treatment with a different mechanism of action, is expected.
Simeprevir, a hepatitis C virus protease inhibitor, has been approved as a treatment for chronic hepatitis C in patients who have compensated liver disease, including cirrhosis, the Food and Drug Administration announced.
For patients with HCV genotype 1 infections, simeprevir’s approved dose is a 150-mg capsule taken once a day with food, in combination with peginterferon-alfa and ribavirin for 24-48 weeks. Before treatment, clinicians should screen patients with genotype 1a HCV infections for the presence of the NS3 Q80K polymorphism, which was associated with lower response rates in studies, "and to consider alternative therapy if the strain is detected," according to the FDA’s Nov. 22 statement announcing the approval.
Simeprevir is the third protease inhibitor to be approved by the FDA for treatment of hepatitis C. Boceprevir (Victrelis) and telaprevir (Incivek) were approved in 2011 and have been the standard of care, but they require taking 6-12 pills a day. Simeprevir is an NS3/4A protease inhibitor, which blocks the viral protease enzyme that enables HCV to replicate in host cells, according to Janssen Pharmaceuticals, which will market the drug as Olysio.
Simeprevir’s simpler dosing regimen, effectiveness, and favorable risk-benefit profile in clinical trials were cited by the FDA’s antiviral drug advisory committee in its unanimous recommendation to approve the drug at a meeting on Oct. 24.
Approval is based on five trials of 2,026 treatment-naive and treatment-experienced patients with chronic hepatitis C who were randomized to simeprevir plus peginterferon-alfa and ribavirin (PR) or to placebo plus PR. The primary endpoint was sustained virologic response rate at 12 weeks after treatment was planned to end (SVR12). SVR was defined as an HCV RNA level that was undetectable or detected only below a prespecified value.
Among the treatment-naive patients, the SVR12 rate was 80% for those treated with simeprevir plus PR and 50% in those who received placebo plus PR. In a study of treatment-experienced patients, 79% of those treated with the simeprevir regimen and 37% of those on peginterferon-alfa and ribavirin achieved SVR12.
In another study of treatment-experienced patients that included prior relapsers, partial responders, and null responders (those who did not respond to previous treatments at all), the addition of simeprevir to PR improved response rates when compared with treatment with PR alone, according to the FDA.
Rash, pruritus, and nausea were the most common adverse effects associated with treatment in the studies, the FDA said. There were cases of serious photosensitivity that required hospitalization, and the prescribing information recommends that patients use sun protection and limit sun exposure during treatment. The prescribing information notes that dosing recommendations cannot be made at this time for patients with moderate to severe hepatic impairment or those of East Asian descent. At the FDA panel meeting, company officials said that the appropriate dose in this population was being investigated.
Simeprevir was approved for treatment of genotype 1 HCV infection in September 2013 in Japan, where it is marketed as Sovriad (at a lower 100-mg dose), and in November in Canada, where it is marketed as Galexos, according to Janssen. Simeprevir is under review in Europe for the treatment of genotype 1 or 4 HCV.
Imminent approval of sofosbuvir, another HCV treatment with a different mechanism of action, is expected.
Once-a-day HCV protease inhibitor approved for treating hepatitis C
Simeprevir, a hepatitis C virus protease inhibitor, has been approved as a treatment for chronic hepatitis C in patients who have compensated liver disease, including cirrhosis, the Food and Drug Administration announced.
For patients with HCV genotype 1 infections, simeprevir’s approved dose is a 150-mg capsule taken once a day with food, in combination with peginterferon-alfa and ribavirin for 24-48 weeks. Before treatment, clinicians should screen patients with genotype 1a HCV infections for the presence of the NS3 Q80K polymorphism, which was associated with lower response rates in studies, "and to consider alternative therapy if the strain is detected," according to the FDA’s Nov. 22 statement announcing the approval.
Simeprevir is the third protease inhibitor to be approved by the FDA for treatment of hepatitis C. Boceprevir (Victrelis) and telaprevir (Incivek) were approved in 2011 and have been the standard of care, but they require taking 6-12 pills a day. Simeprevir is an NS3/4A protease inhibitor, which blocks the viral protease enzyme that enables HCV to replicate in host cells, according to Janssen Pharmaceuticals, which will market the drug as Olysio.
Simeprevir’s simpler dosing regimen, effectiveness, and favorable risk-benefit profile in clinical trials were cited by the FDA’s antiviral drug advisory committee in its unanimous recommendation to approve the drug at a meeting on Oct. 24.
Approval is based on five trials of 2,026 treatment-naive and treatment-experienced patients with chronic hepatitis C who were randomized to simeprevir plus peginterferon-alfa and ribavirin (PR) or to placebo plus PR. The primary endpoint was sustained virologic response rate at 12 weeks after treatment was planned to end (SVR12). SVR was defined as an HCV RNA level that was undetectable or detected only below a prespecified value.
Among the treatment-naive patients, the SVR12 rate was 80% for those treated with simeprevir plus PR and 50% in those who received placebo plus PR. In a study of treatment-experienced patients, 79% of those treated with the simeprevir regimen and 37% of those on peginterferon-alfa and ribavirin achieved SVR12.
In another study of treatment-experienced patients that included prior relapsers, partial responders, and null responders (those who did not respond to previous treatments at all), the addition of simeprevir to PR improved response rates when compared with treatment with PR alone, according to the FDA.
Rash, pruritus, and nausea were the most common adverse effects associated with treatment in the studies, the FDA said. There were cases of serious photosensitivity that required hospitalization, and the prescribing information recommends that patients use sun protection and limit sun exposure during treatment. The prescribing information notes that dosing recommendations cannot be made at this time for patients with moderate to severe hepatic impairment or those of East Asian descent. At the FDA panel meeting, company officials said that the appropriate dose in this population was being investigated.
Simeprevir was approved for treatment of genotype 1 HCV infection in September 2013 in Japan, where it is marketed as Sovriad (at a lower 100-mg dose), and in November in Canada, where it is marketed as Galexos, according to Janssen. Simeprevir is under review in Europe for the treatment of genotype 1 or 4 HCV.
Imminent approval of sofosbuvir, another HCV treatment with a different mechanism of action, is expected.
Simeprevir, a hepatitis C virus protease inhibitor, has been approved as a treatment for chronic hepatitis C in patients who have compensated liver disease, including cirrhosis, the Food and Drug Administration announced.
For patients with HCV genotype 1 infections, simeprevir’s approved dose is a 150-mg capsule taken once a day with food, in combination with peginterferon-alfa and ribavirin for 24-48 weeks. Before treatment, clinicians should screen patients with genotype 1a HCV infections for the presence of the NS3 Q80K polymorphism, which was associated with lower response rates in studies, "and to consider alternative therapy if the strain is detected," according to the FDA’s Nov. 22 statement announcing the approval.
Simeprevir is the third protease inhibitor to be approved by the FDA for treatment of hepatitis C. Boceprevir (Victrelis) and telaprevir (Incivek) were approved in 2011 and have been the standard of care, but they require taking 6-12 pills a day. Simeprevir is an NS3/4A protease inhibitor, which blocks the viral protease enzyme that enables HCV to replicate in host cells, according to Janssen Pharmaceuticals, which will market the drug as Olysio.
Simeprevir’s simpler dosing regimen, effectiveness, and favorable risk-benefit profile in clinical trials were cited by the FDA’s antiviral drug advisory committee in its unanimous recommendation to approve the drug at a meeting on Oct. 24.
Approval is based on five trials of 2,026 treatment-naive and treatment-experienced patients with chronic hepatitis C who were randomized to simeprevir plus peginterferon-alfa and ribavirin (PR) or to placebo plus PR. The primary endpoint was sustained virologic response rate at 12 weeks after treatment was planned to end (SVR12). SVR was defined as an HCV RNA level that was undetectable or detected only below a prespecified value.
Among the treatment-naive patients, the SVR12 rate was 80% for those treated with simeprevir plus PR and 50% in those who received placebo plus PR. In a study of treatment-experienced patients, 79% of those treated with the simeprevir regimen and 37% of those on peginterferon-alfa and ribavirin achieved SVR12.
In another study of treatment-experienced patients that included prior relapsers, partial responders, and null responders (those who did not respond to previous treatments at all), the addition of simeprevir to PR improved response rates when compared with treatment with PR alone, according to the FDA.
Rash, pruritus, and nausea were the most common adverse effects associated with treatment in the studies, the FDA said. There were cases of serious photosensitivity that required hospitalization, and the prescribing information recommends that patients use sun protection and limit sun exposure during treatment. The prescribing information notes that dosing recommendations cannot be made at this time for patients with moderate to severe hepatic impairment or those of East Asian descent. At the FDA panel meeting, company officials said that the appropriate dose in this population was being investigated.
Simeprevir was approved for treatment of genotype 1 HCV infection in September 2013 in Japan, where it is marketed as Sovriad (at a lower 100-mg dose), and in November in Canada, where it is marketed as Galexos, according to Janssen. Simeprevir is under review in Europe for the treatment of genotype 1 or 4 HCV.
Imminent approval of sofosbuvir, another HCV treatment with a different mechanism of action, is expected.
Simeprevir, a hepatitis C virus protease inhibitor, has been approved as a treatment for chronic hepatitis C in patients who have compensated liver disease, including cirrhosis, the Food and Drug Administration announced.
For patients with HCV genotype 1 infections, simeprevir’s approved dose is a 150-mg capsule taken once a day with food, in combination with peginterferon-alfa and ribavirin for 24-48 weeks. Before treatment, clinicians should screen patients with genotype 1a HCV infections for the presence of the NS3 Q80K polymorphism, which was associated with lower response rates in studies, "and to consider alternative therapy if the strain is detected," according to the FDA’s Nov. 22 statement announcing the approval.
Simeprevir is the third protease inhibitor to be approved by the FDA for treatment of hepatitis C. Boceprevir (Victrelis) and telaprevir (Incivek) were approved in 2011 and have been the standard of care, but they require taking 6-12 pills a day. Simeprevir is an NS3/4A protease inhibitor, which blocks the viral protease enzyme that enables HCV to replicate in host cells, according to Janssen Pharmaceuticals, which will market the drug as Olysio.
Simeprevir’s simpler dosing regimen, effectiveness, and favorable risk-benefit profile in clinical trials were cited by the FDA’s antiviral drug advisory committee in its unanimous recommendation to approve the drug at a meeting on Oct. 24.
Approval is based on five trials of 2,026 treatment-naive and treatment-experienced patients with chronic hepatitis C who were randomized to simeprevir plus peginterferon-alfa and ribavirin (PR) or to placebo plus PR. The primary endpoint was sustained virologic response rate at 12 weeks after treatment was planned to end (SVR12). SVR was defined as an HCV RNA level that was undetectable or detected only below a prespecified value.
Among the treatment-naive patients, the SVR12 rate was 80% for those treated with simeprevir plus PR and 50% in those who received placebo plus PR. In a study of treatment-experienced patients, 79% of those treated with the simeprevir regimen and 37% of those on peginterferon-alfa and ribavirin achieved SVR12.
In another study of treatment-experienced patients that included prior relapsers, partial responders, and null responders (those who did not respond to previous treatments at all), the addition of simeprevir to PR improved response rates when compared with treatment with PR alone, according to the FDA.
Rash, pruritus, and nausea were the most common adverse effects associated with treatment in the studies, the FDA said. There were cases of serious photosensitivity that required hospitalization, and the prescribing information recommends that patients use sun protection and limit sun exposure during treatment. The prescribing information notes that dosing recommendations cannot be made at this time for patients with moderate to severe hepatic impairment or those of East Asian descent. At the FDA panel meeting, company officials said that the appropriate dose in this population was being investigated.
Simeprevir was approved for treatment of genotype 1 HCV infection in September 2013 in Japan, where it is marketed as Sovriad (at a lower 100-mg dose), and in November in Canada, where it is marketed as Galexos, according to Janssen. Simeprevir is under review in Europe for the treatment of genotype 1 or 4 HCV.
Imminent approval of sofosbuvir, another HCV treatment with a different mechanism of action, is expected.
FDA Lifts Rosiglitazone Prescribing Restrictions
Limitations on prescribing the drug rosiglitazone to a small group of people with type 2 diabetes, and other restrictions that have been in place since 2010, are being removed, the Food and Drug Administration announced.
Concurring with the recommendations of two expert advisory panels in June, the FDA is modifying the cardiovascular safety statement in the labels of rosiglitazone products, changing the rosiglitazone Risk Evaluation and Mitigation Strategy (REMS), and releasing the manufacturer from a requirement to conduct a postmarketing safety study, according to the announcement.
The FDA is also lifting the requirement that physicians, pharmacies, and patients enroll in the REMS program, to prescribe, dispense, or receive any of the available rosiglitazone products.
Rosiglitazone, a thiazolidinedione, is marketed as Avandia, and as Avandamet (combined with metformin), and Avandaryl (combined with glimepiride); generic formulations are also available.
"Although some scientific uncertainty about the cardiovascular safety of rosiglitazone medicines still remains, in light of the new reevaluation of the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial, FDA’s concern is substantially reduced and the rosiglitazone REMS program requirements will be modified," the statement said.
The drug was restricted in 2010, based on a recommendations of FDA advisory panels. The panel members reviewed data that included a meta-analysis suggesting rosiglitazone was associated with an increased cardiovascular risk (N. Engl. J. Med. 2007;356:2457-71). Under the REMS, rosiglitazone products can be prescribed only to patients after the prescriber has discussed the potential increased risk of MI associated with the drug – and only in patients who are already taking rosiglitazone or are unable to achieve glycemic control on other medications.
But at the joint meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee in June, 20 of the 26 panel members voted to modify (13) or remove (6) the required REMS, based on the readjudicated results of the RECORD trial, conducted independently by the Duke Clinical Research Institute in Durham, N.C. The reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all-cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
In the Nov. 25 statement, Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research, said the FDA’s actions "reflect the most current scientific knowledge about the risks and benefits" of rosiglitazone. "Given these new results, our level of concern is considerably reduced; thus, we are requiring the removal of certain prescribing restrictions."
The remaining requirement under the REMS is that manufacturers of rosiglitazone products need to ensure that health care professionals "who are likely to prescribe rosiglitazone medicines" receive training "based on the current state of knowledge concerning the cardiovascular risk of rosiglitazone medicines."
The FDA’s action on rosiglitazone is a little like putting the milk back in the refrigerator after it has spoiled and seeing if it will become good again. Unless there are clinical champions for thiazolidinediones, of which there are precious few these days, the field of diabetes has moved on. That said, rosiglitazone and pioglitazone were very important and helpful medications for the vast majority of patients of took them.
Dr. Daniel Einhorn, FACP, FACE, is medical director of Scripps Whittier Diabetes Institute and clinical professor of medicine at the University of California, San Diego. He had no relevant disclosures.
The FDA’s action on rosiglitazone is a little like putting the milk back in the refrigerator after it has spoiled and seeing if it will become good again. Unless there are clinical champions for thiazolidinediones, of which there are precious few these days, the field of diabetes has moved on. That said, rosiglitazone and pioglitazone were very important and helpful medications for the vast majority of patients of took them.
Dr. Daniel Einhorn, FACP, FACE, is medical director of Scripps Whittier Diabetes Institute and clinical professor of medicine at the University of California, San Diego. He had no relevant disclosures.
The FDA’s action on rosiglitazone is a little like putting the milk back in the refrigerator after it has spoiled and seeing if it will become good again. Unless there are clinical champions for thiazolidinediones, of which there are precious few these days, the field of diabetes has moved on. That said, rosiglitazone and pioglitazone were very important and helpful medications for the vast majority of patients of took them.
Dr. Daniel Einhorn, FACP, FACE, is medical director of Scripps Whittier Diabetes Institute and clinical professor of medicine at the University of California, San Diego. He had no relevant disclosures.
Limitations on prescribing the drug rosiglitazone to a small group of people with type 2 diabetes, and other restrictions that have been in place since 2010, are being removed, the Food and Drug Administration announced.
Concurring with the recommendations of two expert advisory panels in June, the FDA is modifying the cardiovascular safety statement in the labels of rosiglitazone products, changing the rosiglitazone Risk Evaluation and Mitigation Strategy (REMS), and releasing the manufacturer from a requirement to conduct a postmarketing safety study, according to the announcement.
The FDA is also lifting the requirement that physicians, pharmacies, and patients enroll in the REMS program, to prescribe, dispense, or receive any of the available rosiglitazone products.
Rosiglitazone, a thiazolidinedione, is marketed as Avandia, and as Avandamet (combined with metformin), and Avandaryl (combined with glimepiride); generic formulations are also available.
"Although some scientific uncertainty about the cardiovascular safety of rosiglitazone medicines still remains, in light of the new reevaluation of the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial, FDA’s concern is substantially reduced and the rosiglitazone REMS program requirements will be modified," the statement said.
The drug was restricted in 2010, based on a recommendations of FDA advisory panels. The panel members reviewed data that included a meta-analysis suggesting rosiglitazone was associated with an increased cardiovascular risk (N. Engl. J. Med. 2007;356:2457-71). Under the REMS, rosiglitazone products can be prescribed only to patients after the prescriber has discussed the potential increased risk of MI associated with the drug – and only in patients who are already taking rosiglitazone or are unable to achieve glycemic control on other medications.
But at the joint meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee in June, 20 of the 26 panel members voted to modify (13) or remove (6) the required REMS, based on the readjudicated results of the RECORD trial, conducted independently by the Duke Clinical Research Institute in Durham, N.C. The reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all-cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
In the Nov. 25 statement, Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research, said the FDA’s actions "reflect the most current scientific knowledge about the risks and benefits" of rosiglitazone. "Given these new results, our level of concern is considerably reduced; thus, we are requiring the removal of certain prescribing restrictions."
The remaining requirement under the REMS is that manufacturers of rosiglitazone products need to ensure that health care professionals "who are likely to prescribe rosiglitazone medicines" receive training "based on the current state of knowledge concerning the cardiovascular risk of rosiglitazone medicines."
Limitations on prescribing the drug rosiglitazone to a small group of people with type 2 diabetes, and other restrictions that have been in place since 2010, are being removed, the Food and Drug Administration announced.
Concurring with the recommendations of two expert advisory panels in June, the FDA is modifying the cardiovascular safety statement in the labels of rosiglitazone products, changing the rosiglitazone Risk Evaluation and Mitigation Strategy (REMS), and releasing the manufacturer from a requirement to conduct a postmarketing safety study, according to the announcement.
The FDA is also lifting the requirement that physicians, pharmacies, and patients enroll in the REMS program, to prescribe, dispense, or receive any of the available rosiglitazone products.
Rosiglitazone, a thiazolidinedione, is marketed as Avandia, and as Avandamet (combined with metformin), and Avandaryl (combined with glimepiride); generic formulations are also available.
"Although some scientific uncertainty about the cardiovascular safety of rosiglitazone medicines still remains, in light of the new reevaluation of the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial, FDA’s concern is substantially reduced and the rosiglitazone REMS program requirements will be modified," the statement said.
The drug was restricted in 2010, based on a recommendations of FDA advisory panels. The panel members reviewed data that included a meta-analysis suggesting rosiglitazone was associated with an increased cardiovascular risk (N. Engl. J. Med. 2007;356:2457-71). Under the REMS, rosiglitazone products can be prescribed only to patients after the prescriber has discussed the potential increased risk of MI associated with the drug – and only in patients who are already taking rosiglitazone or are unable to achieve glycemic control on other medications.
But at the joint meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee in June, 20 of the 26 panel members voted to modify (13) or remove (6) the required REMS, based on the readjudicated results of the RECORD trial, conducted independently by the Duke Clinical Research Institute in Durham, N.C. The reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all-cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
In the Nov. 25 statement, Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research, said the FDA’s actions "reflect the most current scientific knowledge about the risks and benefits" of rosiglitazone. "Given these new results, our level of concern is considerably reduced; thus, we are requiring the removal of certain prescribing restrictions."
The remaining requirement under the REMS is that manufacturers of rosiglitazone products need to ensure that health care professionals "who are likely to prescribe rosiglitazone medicines" receive training "based on the current state of knowledge concerning the cardiovascular risk of rosiglitazone medicines."
FDA lifts rosiglitazone prescribing restrictions
Limitations on prescribing the drug rosiglitazone to a small group of people with type 2 diabetes, and other restrictions that have been in place since 2010, are being removed, the Food and Drug Administration announced on Nov. 25.
Concurring with the recommendations of two expert advisory panels in June, the FDA is modifying the cardiovascular safety statement in the labels of rosiglitazone products, changing the rosiglitazone Risk Evaluation and Mitigation Strategy (REMS), and releasing the manufacturer from a requirement to conduct a postmarketing safety study, according to the announcement.
The FDA is also lifting the requirement that physicians, pharmacies, and patients enroll in the REMS program, to prescribe, dispense, or receive any of the available rosiglitazone products.
Rosiglitazone, a thiazolidinedione, is marketed as Avandia, and as Avandamet (combined with metformin), and Avandaryl (combined with glimepiride); generic formulations are also available.
"Although some scientific uncertainty about the cardiovascular safety of rosiglitazone medicines still remains, in light of the new reevaluation of the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial, FDA’s concern is substantially reduced and the rosiglitazone REMS program requirements will be modified," the statement said.
The drug was restricted in 2010, based on a recommendations of FDA advisory panels. The panel members reviewed data that included a meta-analysis suggesting rosiglitazone was associated with an increased cardiovascular risk (N. Engl. J. Med. 2007;356:2457-71). Under the REMS, rosiglitazone products can be prescribed only to patients after the prescriber has discussed the potential increased risk of MI associated with the drug – and only in patients who are already taking rosiglitazone or are unable to achieve glycemic control on other medications.
But at the joint meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee in June, 20 of the 26 panel members voted to modify (13) or remove (6) the required REMS, based on the readjudicated results of the RECORD trial, conducted independently by the Duke Clinical Research Institute in Durham, N.C. The reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all-cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
In the Nov. 25 statement, Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research, said the FDA’s actions "reflect the most current scientific knowledge about the risks and benefits" of rosiglitazone. "Given these new results, our level of concern is considerably reduced; thus, we are requiring the removal of certain prescribing restrictions."
The remaining requirement under the REMS is that manufacturers of rosiglitazone products need to ensure that health care professionals "who are likely to prescribe rosiglitazone medicines" receive training "based on the current state of knowledge concerning the cardiovascular risk of rosiglitazone medicines."
The FDA’s action on rosiglitazone is a little like putting the milk back in the refrigerator after it has spoiled and seeing if it will become good again. Unless there are clinical champions for thiazolidinediones, of which there are precious few these days, the field of diabetes has moved on. That said, rosiglitazone and pioglitazone were very important and helpful medications for the vast majority of patients of took them.
Dr. Daniel Einhorn, FACP, FACE, is medical director of Scripps Whittier Diabetes Institute and clinical professor of medicine at the University of California, San Diego. He had no relevant disclosures.
The FDA’s action on rosiglitazone is a little like putting the milk back in the refrigerator after it has spoiled and seeing if it will become good again. Unless there are clinical champions for thiazolidinediones, of which there are precious few these days, the field of diabetes has moved on. That said, rosiglitazone and pioglitazone were very important and helpful medications for the vast majority of patients of took them.
Dr. Daniel Einhorn, FACP, FACE, is medical director of Scripps Whittier Diabetes Institute and clinical professor of medicine at the University of California, San Diego. He had no relevant disclosures.
The FDA’s action on rosiglitazone is a little like putting the milk back in the refrigerator after it has spoiled and seeing if it will become good again. Unless there are clinical champions for thiazolidinediones, of which there are precious few these days, the field of diabetes has moved on. That said, rosiglitazone and pioglitazone were very important and helpful medications for the vast majority of patients of took them.
Dr. Daniel Einhorn, FACP, FACE, is medical director of Scripps Whittier Diabetes Institute and clinical professor of medicine at the University of California, San Diego. He had no relevant disclosures.
Limitations on prescribing the drug rosiglitazone to a small group of people with type 2 diabetes, and other restrictions that have been in place since 2010, are being removed, the Food and Drug Administration announced on Nov. 25.
Concurring with the recommendations of two expert advisory panels in June, the FDA is modifying the cardiovascular safety statement in the labels of rosiglitazone products, changing the rosiglitazone Risk Evaluation and Mitigation Strategy (REMS), and releasing the manufacturer from a requirement to conduct a postmarketing safety study, according to the announcement.
The FDA is also lifting the requirement that physicians, pharmacies, and patients enroll in the REMS program, to prescribe, dispense, or receive any of the available rosiglitazone products.
Rosiglitazone, a thiazolidinedione, is marketed as Avandia, and as Avandamet (combined with metformin), and Avandaryl (combined with glimepiride); generic formulations are also available.
"Although some scientific uncertainty about the cardiovascular safety of rosiglitazone medicines still remains, in light of the new reevaluation of the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial, FDA’s concern is substantially reduced and the rosiglitazone REMS program requirements will be modified," the statement said.
The drug was restricted in 2010, based on a recommendations of FDA advisory panels. The panel members reviewed data that included a meta-analysis suggesting rosiglitazone was associated with an increased cardiovascular risk (N. Engl. J. Med. 2007;356:2457-71). Under the REMS, rosiglitazone products can be prescribed only to patients after the prescriber has discussed the potential increased risk of MI associated with the drug – and only in patients who are already taking rosiglitazone or are unable to achieve glycemic control on other medications.
But at the joint meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee in June, 20 of the 26 panel members voted to modify (13) or remove (6) the required REMS, based on the readjudicated results of the RECORD trial, conducted independently by the Duke Clinical Research Institute in Durham, N.C. The reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all-cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
In the Nov. 25 statement, Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research, said the FDA’s actions "reflect the most current scientific knowledge about the risks and benefits" of rosiglitazone. "Given these new results, our level of concern is considerably reduced; thus, we are requiring the removal of certain prescribing restrictions."
The remaining requirement under the REMS is that manufacturers of rosiglitazone products need to ensure that health care professionals "who are likely to prescribe rosiglitazone medicines" receive training "based on the current state of knowledge concerning the cardiovascular risk of rosiglitazone medicines."
Limitations on prescribing the drug rosiglitazone to a small group of people with type 2 diabetes, and other restrictions that have been in place since 2010, are being removed, the Food and Drug Administration announced on Nov. 25.
Concurring with the recommendations of two expert advisory panels in June, the FDA is modifying the cardiovascular safety statement in the labels of rosiglitazone products, changing the rosiglitazone Risk Evaluation and Mitigation Strategy (REMS), and releasing the manufacturer from a requirement to conduct a postmarketing safety study, according to the announcement.
The FDA is also lifting the requirement that physicians, pharmacies, and patients enroll in the REMS program, to prescribe, dispense, or receive any of the available rosiglitazone products.
Rosiglitazone, a thiazolidinedione, is marketed as Avandia, and as Avandamet (combined with metformin), and Avandaryl (combined with glimepiride); generic formulations are also available.
"Although some scientific uncertainty about the cardiovascular safety of rosiglitazone medicines still remains, in light of the new reevaluation of the Rosiglitazone Evaluated for Cardiovascular Outcomes and Regulation of Glycemia in Diabetes (RECORD) trial, FDA’s concern is substantially reduced and the rosiglitazone REMS program requirements will be modified," the statement said.
The drug was restricted in 2010, based on a recommendations of FDA advisory panels. The panel members reviewed data that included a meta-analysis suggesting rosiglitazone was associated with an increased cardiovascular risk (N. Engl. J. Med. 2007;356:2457-71). Under the REMS, rosiglitazone products can be prescribed only to patients after the prescriber has discussed the potential increased risk of MI associated with the drug – and only in patients who are already taking rosiglitazone or are unable to achieve glycemic control on other medications.
But at the joint meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee in June, 20 of the 26 panel members voted to modify (13) or remove (6) the required REMS, based on the readjudicated results of the RECORD trial, conducted independently by the Duke Clinical Research Institute in Durham, N.C. The reanalysis found no significant differences in the composite of CV death, MI, or stroke; numerically fewer CV deaths and numerically fewer strokes in the rosiglitazone arm; and a lower risk of all-cause death. There were more MIs in the rosiglitazone arm, but the difference was not significant.
In the Nov. 25 statement, Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research, said the FDA’s actions "reflect the most current scientific knowledge about the risks and benefits" of rosiglitazone. "Given these new results, our level of concern is considerably reduced; thus, we are requiring the removal of certain prescribing restrictions."
The remaining requirement under the REMS is that manufacturers of rosiglitazone products need to ensure that health care professionals "who are likely to prescribe rosiglitazone medicines" receive training "based on the current state of knowledge concerning the cardiovascular risk of rosiglitazone medicines."
Sorafenib approval now includes late-stage thyroid cancer
Sorafenib’s approval has been expanded to include late-stage differentiated thyroid cancer, the Food and Drug Administration announced on Nov. 22.
The kinase inhibitor was approved for the treatment of "locally recurrent or metastatic, progressive, differentiated thyroid carcinoma refractory to radioactive iodine treatment," according to the prescribing information.
The expedited approval –completed in 6 months – was based on a study of 417 people with locally recurrent or metastatic, progressive differentiated thyroid cancer that had not responded to radioactive iodine treatment. Subjects were randomized to 400 mg of sorafenib twice a day or placebo. The median progression-free survival was 10.8 months among those on sorafenib and 5.8 months among those on placebo, a statistically significant 41% difference.
Diarrhea, fatigue, infection, alopecia, hand-foot skin reaction, rash, weight loss, decreased appetite, nausea, gastrointestinal and abdominal pains, and hypertension were among the most common adverse effects associated with treatment, according to the FDA. In addition, thyroid stimulating hormone, which can promote thyroid cancer, "is more likely to become elevated while on treatment with Nexavar, requiring adjustment of thyroid hormone replacement therapy," the statement said.
Sorafenib, marketed as Nexavar by Bayer HealthCare Pharmaceuticals was approved for treating advanced renal cell carcinoma in 2005 and for unresectable hepatocellular carcinoma in 2007.
Sorafenib’s approval has been expanded to include late-stage differentiated thyroid cancer, the Food and Drug Administration announced on Nov. 22.
The kinase inhibitor was approved for the treatment of "locally recurrent or metastatic, progressive, differentiated thyroid carcinoma refractory to radioactive iodine treatment," according to the prescribing information.
The expedited approval –completed in 6 months – was based on a study of 417 people with locally recurrent or metastatic, progressive differentiated thyroid cancer that had not responded to radioactive iodine treatment. Subjects were randomized to 400 mg of sorafenib twice a day or placebo. The median progression-free survival was 10.8 months among those on sorafenib and 5.8 months among those on placebo, a statistically significant 41% difference.
Diarrhea, fatigue, infection, alopecia, hand-foot skin reaction, rash, weight loss, decreased appetite, nausea, gastrointestinal and abdominal pains, and hypertension were among the most common adverse effects associated with treatment, according to the FDA. In addition, thyroid stimulating hormone, which can promote thyroid cancer, "is more likely to become elevated while on treatment with Nexavar, requiring adjustment of thyroid hormone replacement therapy," the statement said.
Sorafenib, marketed as Nexavar by Bayer HealthCare Pharmaceuticals was approved for treating advanced renal cell carcinoma in 2005 and for unresectable hepatocellular carcinoma in 2007.
Sorafenib’s approval has been expanded to include late-stage differentiated thyroid cancer, the Food and Drug Administration announced on Nov. 22.
The kinase inhibitor was approved for the treatment of "locally recurrent or metastatic, progressive, differentiated thyroid carcinoma refractory to radioactive iodine treatment," according to the prescribing information.
The expedited approval –completed in 6 months – was based on a study of 417 people with locally recurrent or metastatic, progressive differentiated thyroid cancer that had not responded to radioactive iodine treatment. Subjects were randomized to 400 mg of sorafenib twice a day or placebo. The median progression-free survival was 10.8 months among those on sorafenib and 5.8 months among those on placebo, a statistically significant 41% difference.
Diarrhea, fatigue, infection, alopecia, hand-foot skin reaction, rash, weight loss, decreased appetite, nausea, gastrointestinal and abdominal pains, and hypertension were among the most common adverse effects associated with treatment, according to the FDA. In addition, thyroid stimulating hormone, which can promote thyroid cancer, "is more likely to become elevated while on treatment with Nexavar, requiring adjustment of thyroid hormone replacement therapy," the statement said.
Sorafenib, marketed as Nexavar by Bayer HealthCare Pharmaceuticals was approved for treating advanced renal cell carcinoma in 2005 and for unresectable hepatocellular carcinoma in 2007.
FDA grants full approval to crizotinib for NSCLC indication
The kinase inhibitor crizotinib has received full approval for the treatment of metastatic non–small cell lung cancer, based on a study that found treatment with the drug was associated with superior progression-free survival and overall response rates compared with chemotherapy.
The Food and Drug Administration announced the approval Nov. 21 in a written statement.
In August 2011, crizotinib received accelerated approval for the treatment of metastatic non–small cell lung cancer (NSCLC) in patients whose tumors are anaplastic lymphoma kinase (ALK) positive, as detected by a test approved by the FDA at the same time. The drug approval was based on objective response rates of 50% and 61% in two single-arm open-label trials; full approval was contingent on providing evidence that confirmed the clinical benefits.
This evidence was provided by an open-label, multinational randomized study of 347 patients with ALK-positive metastatic NSCLC, who had progressed after treatment with platinum-based chemotherapy. The patients were randomized to treatment with crizotinib or chemotherapy, according to the statement. Those who received chemotherapy received pemetrexed or docetaxel if they previously had been treated with pemetrexed.
The median progression-free survival was 7.7 months among those on crizotinib, compared with 3.0 months among those on chemotherapy, a statistically significant difference that represented a reduced risk of 51% (hazard ratio = 0.49). The objective response rate was 65% among those on crizotinib, compared with 20% of those on chemotherapy, also a significant difference. The median response durations were 7.4 months and 5.6 months, respectively. In a planned interim analysis, however, overall survival was the same in both groups, according to the FDA.
Common adverse events associated with crizotinib, affecting at least 25% of treated patients, included nausea, diarrhea, vomiting, visual disorders, constipation, edema, elevated transaminase levels, and fatigue.
In a safety evaluation of 172 patients treated with crizotinib in the study, 37% had serious adverse events, the FDA said. Pneumonia, pulmonary embolism, dyspnea, and interstitial lung disease were the most common. Adverse events, which included acute respiratory distress syndrome, arrhythmia, dyspnea, pneumonia, pneumonitis, pulmonary embolism, interstitial lung disease, respiratory failure, and sepsis, were fatal in nine patients.
Crizotinib is marketed as Xalkori, by Pfizer. It comes in a capsule formulation and is administered at a dose of 250 mg, twice a day, with or without food.
The prescribing information is available here.
Serious adverse events associated with crizotinib should be reported to the FDA’s MedWatch program at 800-332-1088 or here.
The kinase inhibitor crizotinib has received full approval for the treatment of metastatic non–small cell lung cancer, based on a study that found treatment with the drug was associated with superior progression-free survival and overall response rates compared with chemotherapy.
The Food and Drug Administration announced the approval Nov. 21 in a written statement.
In August 2011, crizotinib received accelerated approval for the treatment of metastatic non–small cell lung cancer (NSCLC) in patients whose tumors are anaplastic lymphoma kinase (ALK) positive, as detected by a test approved by the FDA at the same time. The drug approval was based on objective response rates of 50% and 61% in two single-arm open-label trials; full approval was contingent on providing evidence that confirmed the clinical benefits.
This evidence was provided by an open-label, multinational randomized study of 347 patients with ALK-positive metastatic NSCLC, who had progressed after treatment with platinum-based chemotherapy. The patients were randomized to treatment with crizotinib or chemotherapy, according to the statement. Those who received chemotherapy received pemetrexed or docetaxel if they previously had been treated with pemetrexed.
The median progression-free survival was 7.7 months among those on crizotinib, compared with 3.0 months among those on chemotherapy, a statistically significant difference that represented a reduced risk of 51% (hazard ratio = 0.49). The objective response rate was 65% among those on crizotinib, compared with 20% of those on chemotherapy, also a significant difference. The median response durations were 7.4 months and 5.6 months, respectively. In a planned interim analysis, however, overall survival was the same in both groups, according to the FDA.
Common adverse events associated with crizotinib, affecting at least 25% of treated patients, included nausea, diarrhea, vomiting, visual disorders, constipation, edema, elevated transaminase levels, and fatigue.
In a safety evaluation of 172 patients treated with crizotinib in the study, 37% had serious adverse events, the FDA said. Pneumonia, pulmonary embolism, dyspnea, and interstitial lung disease were the most common. Adverse events, which included acute respiratory distress syndrome, arrhythmia, dyspnea, pneumonia, pneumonitis, pulmonary embolism, interstitial lung disease, respiratory failure, and sepsis, were fatal in nine patients.
Crizotinib is marketed as Xalkori, by Pfizer. It comes in a capsule formulation and is administered at a dose of 250 mg, twice a day, with or without food.
The prescribing information is available here.
Serious adverse events associated with crizotinib should be reported to the FDA’s MedWatch program at 800-332-1088 or here.
The kinase inhibitor crizotinib has received full approval for the treatment of metastatic non–small cell lung cancer, based on a study that found treatment with the drug was associated with superior progression-free survival and overall response rates compared with chemotherapy.
The Food and Drug Administration announced the approval Nov. 21 in a written statement.
In August 2011, crizotinib received accelerated approval for the treatment of metastatic non–small cell lung cancer (NSCLC) in patients whose tumors are anaplastic lymphoma kinase (ALK) positive, as detected by a test approved by the FDA at the same time. The drug approval was based on objective response rates of 50% and 61% in two single-arm open-label trials; full approval was contingent on providing evidence that confirmed the clinical benefits.
This evidence was provided by an open-label, multinational randomized study of 347 patients with ALK-positive metastatic NSCLC, who had progressed after treatment with platinum-based chemotherapy. The patients were randomized to treatment with crizotinib or chemotherapy, according to the statement. Those who received chemotherapy received pemetrexed or docetaxel if they previously had been treated with pemetrexed.
The median progression-free survival was 7.7 months among those on crizotinib, compared with 3.0 months among those on chemotherapy, a statistically significant difference that represented a reduced risk of 51% (hazard ratio = 0.49). The objective response rate was 65% among those on crizotinib, compared with 20% of those on chemotherapy, also a significant difference. The median response durations were 7.4 months and 5.6 months, respectively. In a planned interim analysis, however, overall survival was the same in both groups, according to the FDA.
Common adverse events associated with crizotinib, affecting at least 25% of treated patients, included nausea, diarrhea, vomiting, visual disorders, constipation, edema, elevated transaminase levels, and fatigue.
In a safety evaluation of 172 patients treated with crizotinib in the study, 37% had serious adverse events, the FDA said. Pneumonia, pulmonary embolism, dyspnea, and interstitial lung disease were the most common. Adverse events, which included acute respiratory distress syndrome, arrhythmia, dyspnea, pneumonia, pneumonitis, pulmonary embolism, interstitial lung disease, respiratory failure, and sepsis, were fatal in nine patients.
Crizotinib is marketed as Xalkori, by Pfizer. It comes in a capsule formulation and is administered at a dose of 250 mg, twice a day, with or without food.
The prescribing information is available here.
Serious adverse events associated with crizotinib should be reported to the FDA’s MedWatch program at 800-332-1088 or here.
FDA approves neurostimulator device for treatment-resistant epilepsy
An implantable neurostimulator has been approved as a treatment for people with medically intractable epilepsy, based on a 3-month study of 191 patients, the Food and Drug Administration announced on Nov. 14.
The RNS stimulator, manufactured by Neuropace, is implanted in the cranium and is connected to 1-2 leads implanted near the seizure focus. It is designed "to detect abnormal electrical activity in the brain and respond by delivering imperceptible levels of electrical stimulation to normalize brain activity before an individual experiences seizures," according to Neuropace.
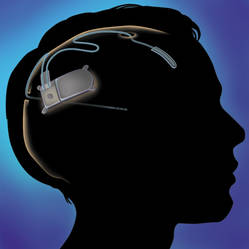
In the study of 191 patients, the number of seizures per month dropped by an average of 38% when the device was turned on, compared with an average 17% drop when the device was turned off, the FDA said in the statement announcing the approval. At the end of 3 months, seizures had been reduced by a median of 34% when the device was turned on, compared with a median 19% reduction when it was turned off. During a 2-year follow-up unblinded phase of the study, "data demonstrated a persistent reduction in seizure frequency," according to the FDA announcement.
The most common adverse effects of treatment were infections at the implant site and premature battery depletion, the agency said.
Patients who have RNS stimulators should not undergo magnetic resonance imaging, diathermy procedures, electroconvulsive therapy, or transcranial magnetic stimulation, which can cause permanent brain damage, even when the device is turned off, the FDA statement said.
An implantable neurostimulator has been approved as a treatment for people with medically intractable epilepsy, based on a 3-month study of 191 patients, the Food and Drug Administration announced on Nov. 14.
The RNS stimulator, manufactured by Neuropace, is implanted in the cranium and is connected to 1-2 leads implanted near the seizure focus. It is designed "to detect abnormal electrical activity in the brain and respond by delivering imperceptible levels of electrical stimulation to normalize brain activity before an individual experiences seizures," according to Neuropace.
In the study of 191 patients, the number of seizures per month dropped by an average of 38% when the device was turned on, compared with an average 17% drop when the device was turned off, the FDA said in the statement announcing the approval. At the end of 3 months, seizures had been reduced by a median of 34% when the device was turned on, compared with a median 19% reduction when it was turned off. During a 2-year follow-up unblinded phase of the study, "data demonstrated a persistent reduction in seizure frequency," according to the FDA announcement.
The most common adverse effects of treatment were infections at the implant site and premature battery depletion, the agency said.
Patients who have RNS stimulators should not undergo magnetic resonance imaging, diathermy procedures, electroconvulsive therapy, or transcranial magnetic stimulation, which can cause permanent brain damage, even when the device is turned off, the FDA statement said.
An implantable neurostimulator has been approved as a treatment for people with medically intractable epilepsy, based on a 3-month study of 191 patients, the Food and Drug Administration announced on Nov. 14.
The RNS stimulator, manufactured by Neuropace, is implanted in the cranium and is connected to 1-2 leads implanted near the seizure focus. It is designed "to detect abnormal electrical activity in the brain and respond by delivering imperceptible levels of electrical stimulation to normalize brain activity before an individual experiences seizures," according to Neuropace.
In the study of 191 patients, the number of seizures per month dropped by an average of 38% when the device was turned on, compared with an average 17% drop when the device was turned off, the FDA said in the statement announcing the approval. At the end of 3 months, seizures had been reduced by a median of 34% when the device was turned on, compared with a median 19% reduction when it was turned off. During a 2-year follow-up unblinded phase of the study, "data demonstrated a persistent reduction in seizure frequency," according to the FDA announcement.
The most common adverse effects of treatment were infections at the implant site and premature battery depletion, the agency said.
Patients who have RNS stimulators should not undergo magnetic resonance imaging, diathermy procedures, electroconvulsive therapy, or transcranial magnetic stimulation, which can cause permanent brain damage, even when the device is turned off, the FDA statement said.
FDA panel has mixed votes on alemtuzumab for multiple sclerosis
SILVER SPRING, MD. – A Food and Drug Administration advisory panel agreed that phase III clinical trials of alemtuzumab for multiple sclerosis were unblinded and likely biased but still voted 12-6 that the manufacturer had provided substantial evidence that the immunomodulating drug was effective as a treatment for patients with relapsing forms of multiple sclerosis.
At a Nov. 13 meeting of the FDA’s Peripheral and Central Nervous System Advisory committee, the six panelists who voted no on this question did so because the studies were not well controlled. But the panelists who voted yes said that the drug clearly had some activity against multiple sclerosis (MS), had not been shown to be worse than the active comparator, and in treated patients had beneficial effects that were seen on MRIs, a more objective measure.
Despite an increased risk of serious side effects of treatment that included immune thrombocytopenia, the panel voted 17-0, with 1 abstention, that safety concerns did not preclude approval of the drug, when the potential benefits of the drug were considered. To address the serious risks of alemtuzumab, the manufacturer Genzyme has proposed a risk evaluation and mitigation strategy (REMS) that includes restricted distribution of the drug to physicians trained and certified in prescribing the drug, administration only in certified infusion centers, and close monitoring of patients for adverse events.
The panel was not asked specifically to recommend approval of the drug, which Genzyme has proposed be approved for treatment of patients with relapsing forms of multiple sclerosis. But panelists indicated in their comments that the drug could benefit some patients with MS – although not as a first-line treatment at this point.
Like other panelists who voted that the safety issues did not preclude approval, the panel chair, Dr. Nathan Fountain, professor of neurology at the University of Virginia, Charlottesville, said he would not want to deny the drug to patients severely affected by MS, who are well informed about the risks, with a stringent risk-management program in place. But, he added, "I don’t think this is appropriate at all for drug-naive, simple, uncomplicated relapsing-remitting patients after their first exacerbation."
Alemtuzumab is a monoclonal antibody that binds to the CD52 antigen present at high levels on the surface of T and B lymphocytes. Its exact mechanism on disease activity in patients with MS is not entirely clear, but is thought to be related to the depletion and repopulation of lymphocytes that occur after the drug is administered, according to Genzyme. The proposed dose is 12 mg/day in an intravenous infusion for 5 consecutive days, followed by the same dose for 3 consecutive days 1 year later.
In 2001, alemtuzumab was approved for treating B-cell chronic lymphocytic leukemia in the United States; it was approved in September 2013 for MS in the European Union. It is marketed as Campath in the United States, but if approved for MS, it will be marketed as Lemtrada.
In two phase III studies of patients with MS, who were treatment-naive or had failed a previous treatment, alemtuzumab (12 mg/day) administered in a series of IV infusions a year apart was compared with treatment with a subcutaneous injection of 44 mcg of interferon beta-1a (Rebif), approved in 2002 for treatment of relapsing forms of MS.
The rate of relapse (new neurologic symptoms or worsening of previous neurologic symptoms) over 2 years and sustained accumulation of disability over 6 months were the primary efficacy endpoints.
In the study of 581 recently diagnosed, untreated patients, the relapse rate over 2 years was 18% among those treated with alemtuzumab, compared with 39% among those on interferon beta-1a – a 55% reduction in the relapse rate that was statistically significant, according to the manufacturer. At the end of 2 years, 8% of those treated with alemtuzumab had experienced sustained disability, compared with 11% of those on interferon beta-1a, which was not statistically significant.
In the study of 840 MS patients who had experienced disease activity on another treatment, the relapse rate over 2 years was 26% among those on alemtuzumab, compared with 52% of those on interferon beta-1a, a 49% reduction in the relapse rate that was statistically significant. The risk of sustained disability over 2 years was almost 13% among those on alemtuzumab, compared with 21% among those on interferon beta-1a, a 42% reduction in risk that was statistically significant, according to Genzyme.
Major concerns raised by the FDA were elevated rates of serious adverse events in about 1,200 patients treated with alemtuzumab, which include serious thyroid-related adverse events (2.1%), immune thrombocytopenia (1.6%), autoimmune hemolytic anemia (0.2%), nephropathies (0.5%), acquired hemophilia A (0.1%), type 1 diabetes (0.2%), pneumonitis (0.5%), thyroid cancer (0.4%), melanoma (0.2%), and serious infusion reactions (2.6%). These adverse events cannot be prevented with close monitoring of patients or with preventive measures and can also occur years after the last dose is administered, the FDA reviewers pointed out.
Besides safety, the other major issue at the meeting was the disagreement between the FDA and Genzyme over the design of the clinical trials, which were open label, with clinicians and patients unblinded to the treatment. Dr. Billy Dunn, acting deputy director of the FDA’s division of neurology products, said that the FDA reviewers believed that bias was "pervasive" in these studies but that the impact the bias had on efficacy results was unclear.
There was a large difference in dropout rates between the treatment groups after randomization before the initiation of treatment between the two groups – almost 13% among those assigned to interferon beta-1a vs. 2% among those assigned to alemtuzumab in one of the studies. This could be explained by patients assigned to interferon beta-1a wanting the newer drug and was among the evidence FDA reviewers said illustrated the bias that was introduced into the studies because of unblinding.
In other questions posed by the FDA, most of the panel voted that the company had not provided substantial evidence that alemtuzumab had a beneficial on disability (14-2 with 2 abstentions) and that the two phase III studies were not adequate and well controlled (11-6 with 1 abstention).
Genzyme, a Sanofi company, is conducting an extension study of alemtuzumab in about 1,300 patients (including those in the three studies) that is evaluating safety and efficacy for up to 5 years, which includes patients now being treated in Europe.
A decision on approval is expected in December. The FDA usually follows the recommendations of its advisory panels. Members have been cleared of potential conflicts; occasionally, a panelist is given a waiver for a potential conflict, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel agreed that phase III clinical trials of alemtuzumab for multiple sclerosis were unblinded and likely biased but still voted 12-6 that the manufacturer had provided substantial evidence that the immunomodulating drug was effective as a treatment for patients with relapsing forms of multiple sclerosis.
At a Nov. 13 meeting of the FDA’s Peripheral and Central Nervous System Advisory committee, the six panelists who voted no on this question did so because the studies were not well controlled. But the panelists who voted yes said that the drug clearly had some activity against multiple sclerosis (MS), had not been shown to be worse than the active comparator, and in treated patients had beneficial effects that were seen on MRIs, a more objective measure.
Despite an increased risk of serious side effects of treatment that included immune thrombocytopenia, the panel voted 17-0, with 1 abstention, that safety concerns did not preclude approval of the drug, when the potential benefits of the drug were considered. To address the serious risks of alemtuzumab, the manufacturer Genzyme has proposed a risk evaluation and mitigation strategy (REMS) that includes restricted distribution of the drug to physicians trained and certified in prescribing the drug, administration only in certified infusion centers, and close monitoring of patients for adverse events.
The panel was not asked specifically to recommend approval of the drug, which Genzyme has proposed be approved for treatment of patients with relapsing forms of multiple sclerosis. But panelists indicated in their comments that the drug could benefit some patients with MS – although not as a first-line treatment at this point.
Like other panelists who voted that the safety issues did not preclude approval, the panel chair, Dr. Nathan Fountain, professor of neurology at the University of Virginia, Charlottesville, said he would not want to deny the drug to patients severely affected by MS, who are well informed about the risks, with a stringent risk-management program in place. But, he added, "I don’t think this is appropriate at all for drug-naive, simple, uncomplicated relapsing-remitting patients after their first exacerbation."
Alemtuzumab is a monoclonal antibody that binds to the CD52 antigen present at high levels on the surface of T and B lymphocytes. Its exact mechanism on disease activity in patients with MS is not entirely clear, but is thought to be related to the depletion and repopulation of lymphocytes that occur after the drug is administered, according to Genzyme. The proposed dose is 12 mg/day in an intravenous infusion for 5 consecutive days, followed by the same dose for 3 consecutive days 1 year later.
In 2001, alemtuzumab was approved for treating B-cell chronic lymphocytic leukemia in the United States; it was approved in September 2013 for MS in the European Union. It is marketed as Campath in the United States, but if approved for MS, it will be marketed as Lemtrada.
In two phase III studies of patients with MS, who were treatment-naive or had failed a previous treatment, alemtuzumab (12 mg/day) administered in a series of IV infusions a year apart was compared with treatment with a subcutaneous injection of 44 mcg of interferon beta-1a (Rebif), approved in 2002 for treatment of relapsing forms of MS.
The rate of relapse (new neurologic symptoms or worsening of previous neurologic symptoms) over 2 years and sustained accumulation of disability over 6 months were the primary efficacy endpoints.
In the study of 581 recently diagnosed, untreated patients, the relapse rate over 2 years was 18% among those treated with alemtuzumab, compared with 39% among those on interferon beta-1a – a 55% reduction in the relapse rate that was statistically significant, according to the manufacturer. At the end of 2 years, 8% of those treated with alemtuzumab had experienced sustained disability, compared with 11% of those on interferon beta-1a, which was not statistically significant.
In the study of 840 MS patients who had experienced disease activity on another treatment, the relapse rate over 2 years was 26% among those on alemtuzumab, compared with 52% of those on interferon beta-1a, a 49% reduction in the relapse rate that was statistically significant. The risk of sustained disability over 2 years was almost 13% among those on alemtuzumab, compared with 21% among those on interferon beta-1a, a 42% reduction in risk that was statistically significant, according to Genzyme.
Major concerns raised by the FDA were elevated rates of serious adverse events in about 1,200 patients treated with alemtuzumab, which include serious thyroid-related adverse events (2.1%), immune thrombocytopenia (1.6%), autoimmune hemolytic anemia (0.2%), nephropathies (0.5%), acquired hemophilia A (0.1%), type 1 diabetes (0.2%), pneumonitis (0.5%), thyroid cancer (0.4%), melanoma (0.2%), and serious infusion reactions (2.6%). These adverse events cannot be prevented with close monitoring of patients or with preventive measures and can also occur years after the last dose is administered, the FDA reviewers pointed out.
Besides safety, the other major issue at the meeting was the disagreement between the FDA and Genzyme over the design of the clinical trials, which were open label, with clinicians and patients unblinded to the treatment. Dr. Billy Dunn, acting deputy director of the FDA’s division of neurology products, said that the FDA reviewers believed that bias was "pervasive" in these studies but that the impact the bias had on efficacy results was unclear.
There was a large difference in dropout rates between the treatment groups after randomization before the initiation of treatment between the two groups – almost 13% among those assigned to interferon beta-1a vs. 2% among those assigned to alemtuzumab in one of the studies. This could be explained by patients assigned to interferon beta-1a wanting the newer drug and was among the evidence FDA reviewers said illustrated the bias that was introduced into the studies because of unblinding.
In other questions posed by the FDA, most of the panel voted that the company had not provided substantial evidence that alemtuzumab had a beneficial on disability (14-2 with 2 abstentions) and that the two phase III studies were not adequate and well controlled (11-6 with 1 abstention).
Genzyme, a Sanofi company, is conducting an extension study of alemtuzumab in about 1,300 patients (including those in the three studies) that is evaluating safety and efficacy for up to 5 years, which includes patients now being treated in Europe.
A decision on approval is expected in December. The FDA usually follows the recommendations of its advisory panels. Members have been cleared of potential conflicts; occasionally, a panelist is given a waiver for a potential conflict, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel agreed that phase III clinical trials of alemtuzumab for multiple sclerosis were unblinded and likely biased but still voted 12-6 that the manufacturer had provided substantial evidence that the immunomodulating drug was effective as a treatment for patients with relapsing forms of multiple sclerosis.
At a Nov. 13 meeting of the FDA’s Peripheral and Central Nervous System Advisory committee, the six panelists who voted no on this question did so because the studies were not well controlled. But the panelists who voted yes said that the drug clearly had some activity against multiple sclerosis (MS), had not been shown to be worse than the active comparator, and in treated patients had beneficial effects that were seen on MRIs, a more objective measure.
Despite an increased risk of serious side effects of treatment that included immune thrombocytopenia, the panel voted 17-0, with 1 abstention, that safety concerns did not preclude approval of the drug, when the potential benefits of the drug were considered. To address the serious risks of alemtuzumab, the manufacturer Genzyme has proposed a risk evaluation and mitigation strategy (REMS) that includes restricted distribution of the drug to physicians trained and certified in prescribing the drug, administration only in certified infusion centers, and close monitoring of patients for adverse events.
The panel was not asked specifically to recommend approval of the drug, which Genzyme has proposed be approved for treatment of patients with relapsing forms of multiple sclerosis. But panelists indicated in their comments that the drug could benefit some patients with MS – although not as a first-line treatment at this point.
Like other panelists who voted that the safety issues did not preclude approval, the panel chair, Dr. Nathan Fountain, professor of neurology at the University of Virginia, Charlottesville, said he would not want to deny the drug to patients severely affected by MS, who are well informed about the risks, with a stringent risk-management program in place. But, he added, "I don’t think this is appropriate at all for drug-naive, simple, uncomplicated relapsing-remitting patients after their first exacerbation."
Alemtuzumab is a monoclonal antibody that binds to the CD52 antigen present at high levels on the surface of T and B lymphocytes. Its exact mechanism on disease activity in patients with MS is not entirely clear, but is thought to be related to the depletion and repopulation of lymphocytes that occur after the drug is administered, according to Genzyme. The proposed dose is 12 mg/day in an intravenous infusion for 5 consecutive days, followed by the same dose for 3 consecutive days 1 year later.
In 2001, alemtuzumab was approved for treating B-cell chronic lymphocytic leukemia in the United States; it was approved in September 2013 for MS in the European Union. It is marketed as Campath in the United States, but if approved for MS, it will be marketed as Lemtrada.
In two phase III studies of patients with MS, who were treatment-naive or had failed a previous treatment, alemtuzumab (12 mg/day) administered in a series of IV infusions a year apart was compared with treatment with a subcutaneous injection of 44 mcg of interferon beta-1a (Rebif), approved in 2002 for treatment of relapsing forms of MS.
The rate of relapse (new neurologic symptoms or worsening of previous neurologic symptoms) over 2 years and sustained accumulation of disability over 6 months were the primary efficacy endpoints.
In the study of 581 recently diagnosed, untreated patients, the relapse rate over 2 years was 18% among those treated with alemtuzumab, compared with 39% among those on interferon beta-1a – a 55% reduction in the relapse rate that was statistically significant, according to the manufacturer. At the end of 2 years, 8% of those treated with alemtuzumab had experienced sustained disability, compared with 11% of those on interferon beta-1a, which was not statistically significant.
In the study of 840 MS patients who had experienced disease activity on another treatment, the relapse rate over 2 years was 26% among those on alemtuzumab, compared with 52% of those on interferon beta-1a, a 49% reduction in the relapse rate that was statistically significant. The risk of sustained disability over 2 years was almost 13% among those on alemtuzumab, compared with 21% among those on interferon beta-1a, a 42% reduction in risk that was statistically significant, according to Genzyme.
Major concerns raised by the FDA were elevated rates of serious adverse events in about 1,200 patients treated with alemtuzumab, which include serious thyroid-related adverse events (2.1%), immune thrombocytopenia (1.6%), autoimmune hemolytic anemia (0.2%), nephropathies (0.5%), acquired hemophilia A (0.1%), type 1 diabetes (0.2%), pneumonitis (0.5%), thyroid cancer (0.4%), melanoma (0.2%), and serious infusion reactions (2.6%). These adverse events cannot be prevented with close monitoring of patients or with preventive measures and can also occur years after the last dose is administered, the FDA reviewers pointed out.
Besides safety, the other major issue at the meeting was the disagreement between the FDA and Genzyme over the design of the clinical trials, which were open label, with clinicians and patients unblinded to the treatment. Dr. Billy Dunn, acting deputy director of the FDA’s division of neurology products, said that the FDA reviewers believed that bias was "pervasive" in these studies but that the impact the bias had on efficacy results was unclear.
There was a large difference in dropout rates between the treatment groups after randomization before the initiation of treatment between the two groups – almost 13% among those assigned to interferon beta-1a vs. 2% among those assigned to alemtuzumab in one of the studies. This could be explained by patients assigned to interferon beta-1a wanting the newer drug and was among the evidence FDA reviewers said illustrated the bias that was introduced into the studies because of unblinding.
In other questions posed by the FDA, most of the panel voted that the company had not provided substantial evidence that alemtuzumab had a beneficial on disability (14-2 with 2 abstentions) and that the two phase III studies were not adequate and well controlled (11-6 with 1 abstention).
Genzyme, a Sanofi company, is conducting an extension study of alemtuzumab in about 1,300 patients (including those in the three studies) that is evaluating safety and efficacy for up to 5 years, which includes patients now being treated in Europe.
A decision on approval is expected in December. The FDA usually follows the recommendations of its advisory panels. Members have been cleared of potential conflicts; occasionally, a panelist is given a waiver for a potential conflict, but not at this meeting.
AT AN FDA ADVISORY PANEL MEETING