User login
FDA approves two-drug combination for advanced melanoma
A combination of two targeted, orally administered drug therapies – trametinib and dabrafenib – has been approved by the Food and Drug Administration for treating patients with unresectable melanoma or metastatic melanoma with BRAF V600E or V600K mutations, manufacturer GlaxoSmithKline announced on Jan. 9.
An FDA-approved test is used to detect the mutations, according to the company statement.
Trametinib, marketed as Mekinist, and dabrafenib, marketed as Tafinlar, were each approved in May 2013 as separate treatments for metastatic or unresectable melanomas that express the mutations – along with a companion test to detect the mutations. Both are kinase inhibitors.
The GSK statement noted that the accelerated approval of the combination therapy is based on the response rate and median duration of response results in a phase I/II study of 108 patients, which compared the combination (150 mg of dabrafenib twice a day and 2 mg of trametinib once a day) to treatment with dabrafenib alone. The overall response rates, as assessed by the investigators, were 76% among those on the combination and 54% for those on dabrafenib alone. The median duration of response was 10.5 months among those on the combination, vs. 5.6 months among those on dabrafenib alone.
The most common adverse events associated with the combination treatment included fever in 71% of patients, chills in 58%, fatigue in 52%, rash in 45%, nausea in 44%, vomiting in 40%, and diarrhea in 36%. Renal failure, pyrexia, back pain, and hemorrhage were among the most common grade-3 or -4 adverse events among those on the combination.
Serious adverse effects associated with treatment, including some potentially fatal effects, are new primary cutaneous skin cancers, tumor promotion in wild-type BRAF melanoma, and hemorrhagic events, according to GSK.
The FDA approval occurred through an accelerated process based on clinical evidence that the treatment has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit to patients. As a condition of accelerated approval, manufacturers are required to provide more clinical data confirming benefit, and if the follow-up studies fail to confirm benefit, the FDA can withdraw approval.
Full approval is dependent on the results of an ongoing phase III study, GSK said.
The updated trametinib label states that "improvement in disease-related symptoms or overall survival has not been demonstrated" for the combination treatment.
The prescribing information for Mekinist is available at http://us.gsk.com/products/assets/us_mekinist.pdf. The prescribing information for Tafinlar (which does not yet include the updated information on the combination therapy approval) is available at http://us.gsk.com/products/assets/us_tafinlar.pdf.
A combination of two targeted, orally administered drug therapies – trametinib and dabrafenib – has been approved by the Food and Drug Administration for treating patients with unresectable melanoma or metastatic melanoma with BRAF V600E or V600K mutations, manufacturer GlaxoSmithKline announced on Jan. 9.
An FDA-approved test is used to detect the mutations, according to the company statement.
Trametinib, marketed as Mekinist, and dabrafenib, marketed as Tafinlar, were each approved in May 2013 as separate treatments for metastatic or unresectable melanomas that express the mutations – along with a companion test to detect the mutations. Both are kinase inhibitors.
The GSK statement noted that the accelerated approval of the combination therapy is based on the response rate and median duration of response results in a phase I/II study of 108 patients, which compared the combination (150 mg of dabrafenib twice a day and 2 mg of trametinib once a day) to treatment with dabrafenib alone. The overall response rates, as assessed by the investigators, were 76% among those on the combination and 54% for those on dabrafenib alone. The median duration of response was 10.5 months among those on the combination, vs. 5.6 months among those on dabrafenib alone.
The most common adverse events associated with the combination treatment included fever in 71% of patients, chills in 58%, fatigue in 52%, rash in 45%, nausea in 44%, vomiting in 40%, and diarrhea in 36%. Renal failure, pyrexia, back pain, and hemorrhage were among the most common grade-3 or -4 adverse events among those on the combination.
Serious adverse effects associated with treatment, including some potentially fatal effects, are new primary cutaneous skin cancers, tumor promotion in wild-type BRAF melanoma, and hemorrhagic events, according to GSK.
The FDA approval occurred through an accelerated process based on clinical evidence that the treatment has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit to patients. As a condition of accelerated approval, manufacturers are required to provide more clinical data confirming benefit, and if the follow-up studies fail to confirm benefit, the FDA can withdraw approval.
Full approval is dependent on the results of an ongoing phase III study, GSK said.
The updated trametinib label states that "improvement in disease-related symptoms or overall survival has not been demonstrated" for the combination treatment.
The prescribing information for Mekinist is available at http://us.gsk.com/products/assets/us_mekinist.pdf. The prescribing information for Tafinlar (which does not yet include the updated information on the combination therapy approval) is available at http://us.gsk.com/products/assets/us_tafinlar.pdf.
A combination of two targeted, orally administered drug therapies – trametinib and dabrafenib – has been approved by the Food and Drug Administration for treating patients with unresectable melanoma or metastatic melanoma with BRAF V600E or V600K mutations, manufacturer GlaxoSmithKline announced on Jan. 9.
An FDA-approved test is used to detect the mutations, according to the company statement.
Trametinib, marketed as Mekinist, and dabrafenib, marketed as Tafinlar, were each approved in May 2013 as separate treatments for metastatic or unresectable melanomas that express the mutations – along with a companion test to detect the mutations. Both are kinase inhibitors.
The GSK statement noted that the accelerated approval of the combination therapy is based on the response rate and median duration of response results in a phase I/II study of 108 patients, which compared the combination (150 mg of dabrafenib twice a day and 2 mg of trametinib once a day) to treatment with dabrafenib alone. The overall response rates, as assessed by the investigators, were 76% among those on the combination and 54% for those on dabrafenib alone. The median duration of response was 10.5 months among those on the combination, vs. 5.6 months among those on dabrafenib alone.
The most common adverse events associated with the combination treatment included fever in 71% of patients, chills in 58%, fatigue in 52%, rash in 45%, nausea in 44%, vomiting in 40%, and diarrhea in 36%. Renal failure, pyrexia, back pain, and hemorrhage were among the most common grade-3 or -4 adverse events among those on the combination.
Serious adverse effects associated with treatment, including some potentially fatal effects, are new primary cutaneous skin cancers, tumor promotion in wild-type BRAF melanoma, and hemorrhagic events, according to GSK.
The FDA approval occurred through an accelerated process based on clinical evidence that the treatment has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit to patients. As a condition of accelerated approval, manufacturers are required to provide more clinical data confirming benefit, and if the follow-up studies fail to confirm benefit, the FDA can withdraw approval.
Full approval is dependent on the results of an ongoing phase III study, GSK said.
The updated trametinib label states that "improvement in disease-related symptoms or overall survival has not been demonstrated" for the combination treatment.
The prescribing information for Mekinist is available at http://us.gsk.com/products/assets/us_mekinist.pdf. The prescribing information for Tafinlar (which does not yet include the updated information on the combination therapy approval) is available at http://us.gsk.com/products/assets/us_tafinlar.pdf.
FDA Approves Dapagliflozin for Type 2 Diabetes
Dapagliflozin, a sodium-glucose cotransporter 2 inhibitor, has been approved for the treatment of type 2 diabetes with a requirement that the manufacturer conduct postmarketing studies that address risks associated with the drug, the Food and Drug Administration announced on Jan. 8.
Approval was based on the results of 16 clinical trials in more than 9,400 patients with type 2 diabetes, which found that treatment with the drug improved hemoglobin A1C, according to the statement. Dapagliflozin is the second sodium-glucose cotransporter 2 (SGLT2) inhibitor to be approved by the FDA; the first was canagliflozin (Invokana), approved in March 2013. These agents, taken orally, work by inhibiting SGLT2 that is expressed in the kidney, reducing renal glucose reabsorption, increasing urinary excretion of glucose, and reducing plasma glucose levels.
Dapagliflozin will be marketed as Farxiga by Bristol-Myers Squibb and AstraZeneca.
In the studies, the most common adverse events associated with treatment were genital fungal infections and urinary tract infections. Other adverse events included hypotension caused by dehydration, resulting in dizziness and/or fainting and reduced renal function, according to the FDA statement, which noted that "the elderly, patients with impaired renal function, and patients on diuretics to treat other conditions appeared to be more susceptible to this risk."
Approval came less than a month after a meeting in December, when the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1 that the benefits of dapagliflozin outweighed its risks for improving glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise. The panel, however, strongly recommended that cardiovascular safety, bladder cancer, and hepatotoxicity be monitored in treated patients after approval – which is reflected in the FDA’s list of required postmarketing studies.
The same panel had recommended against approval in 2011 because of concerns over the numerical increase in bladder and breast cancers and a case of possible drug-induced liver injury. The FDA did not approve dapagliflozin, requesting more safety data, and the companies resubmitted the application for approval with more safety data and analyses last year.
The postmarketing trials required by the FDA include a cardiovascular outcomes trial that will evaluate the cardiovascular risk of dapagliflozin in patients with a high risk of cardiovascular disease at baseline and a study evaluating the risk of bladder cancer in patients enrolled in that study. Another study will evaluate bladder tumor promotion effects of the drug in rodents; two studies will evaluate the pharmacokinetics, efficacy, and safety of dapagliflozin in pediatric patients; and an enhanced pharmacovigilance program will monitor reports of liver abnormalities and pregnancy outcomes in patients treated with dapagliflozin, the FDA said.
At the December 2013 panel meeting, Bristol-Myers Squibb said that enrollment in a cardiovascular outcomes study that will eventually enroll over 17,000 patients with type 2 diabetes and established cardiovascular disease or at least two cardiovascular risk factors had already begun in Europe, where dapagliflozin was already approved. That study is the Dapagliflozin Effect on Cardiovascular Events (DECLARE-TIMI-58) study.
Dapagliflozin, a sodium-glucose cotransporter 2 inhibitor, has been approved for the treatment of type 2 diabetes with a requirement that the manufacturer conduct postmarketing studies that address risks associated with the drug, the Food and Drug Administration announced on Jan. 8.
Approval was based on the results of 16 clinical trials in more than 9,400 patients with type 2 diabetes, which found that treatment with the drug improved hemoglobin A1C, according to the statement. Dapagliflozin is the second sodium-glucose cotransporter 2 (SGLT2) inhibitor to be approved by the FDA; the first was canagliflozin (Invokana), approved in March 2013. These agents, taken orally, work by inhibiting SGLT2 that is expressed in the kidney, reducing renal glucose reabsorption, increasing urinary excretion of glucose, and reducing plasma glucose levels.
Dapagliflozin will be marketed as Farxiga by Bristol-Myers Squibb and AstraZeneca.
In the studies, the most common adverse events associated with treatment were genital fungal infections and urinary tract infections. Other adverse events included hypotension caused by dehydration, resulting in dizziness and/or fainting and reduced renal function, according to the FDA statement, which noted that "the elderly, patients with impaired renal function, and patients on diuretics to treat other conditions appeared to be more susceptible to this risk."
Approval came less than a month after a meeting in December, when the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1 that the benefits of dapagliflozin outweighed its risks for improving glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise. The panel, however, strongly recommended that cardiovascular safety, bladder cancer, and hepatotoxicity be monitored in treated patients after approval – which is reflected in the FDA’s list of required postmarketing studies.
The same panel had recommended against approval in 2011 because of concerns over the numerical increase in bladder and breast cancers and a case of possible drug-induced liver injury. The FDA did not approve dapagliflozin, requesting more safety data, and the companies resubmitted the application for approval with more safety data and analyses last year.
The postmarketing trials required by the FDA include a cardiovascular outcomes trial that will evaluate the cardiovascular risk of dapagliflozin in patients with a high risk of cardiovascular disease at baseline and a study evaluating the risk of bladder cancer in patients enrolled in that study. Another study will evaluate bladder tumor promotion effects of the drug in rodents; two studies will evaluate the pharmacokinetics, efficacy, and safety of dapagliflozin in pediatric patients; and an enhanced pharmacovigilance program will monitor reports of liver abnormalities and pregnancy outcomes in patients treated with dapagliflozin, the FDA said.
At the December 2013 panel meeting, Bristol-Myers Squibb said that enrollment in a cardiovascular outcomes study that will eventually enroll over 17,000 patients with type 2 diabetes and established cardiovascular disease or at least two cardiovascular risk factors had already begun in Europe, where dapagliflozin was already approved. That study is the Dapagliflozin Effect on Cardiovascular Events (DECLARE-TIMI-58) study.
Dapagliflozin, a sodium-glucose cotransporter 2 inhibitor, has been approved for the treatment of type 2 diabetes with a requirement that the manufacturer conduct postmarketing studies that address risks associated with the drug, the Food and Drug Administration announced on Jan. 8.
Approval was based on the results of 16 clinical trials in more than 9,400 patients with type 2 diabetes, which found that treatment with the drug improved hemoglobin A1C, according to the statement. Dapagliflozin is the second sodium-glucose cotransporter 2 (SGLT2) inhibitor to be approved by the FDA; the first was canagliflozin (Invokana), approved in March 2013. These agents, taken orally, work by inhibiting SGLT2 that is expressed in the kidney, reducing renal glucose reabsorption, increasing urinary excretion of glucose, and reducing plasma glucose levels.
Dapagliflozin will be marketed as Farxiga by Bristol-Myers Squibb and AstraZeneca.
In the studies, the most common adverse events associated with treatment were genital fungal infections and urinary tract infections. Other adverse events included hypotension caused by dehydration, resulting in dizziness and/or fainting and reduced renal function, according to the FDA statement, which noted that "the elderly, patients with impaired renal function, and patients on diuretics to treat other conditions appeared to be more susceptible to this risk."
Approval came less than a month after a meeting in December, when the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1 that the benefits of dapagliflozin outweighed its risks for improving glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise. The panel, however, strongly recommended that cardiovascular safety, bladder cancer, and hepatotoxicity be monitored in treated patients after approval – which is reflected in the FDA’s list of required postmarketing studies.
The same panel had recommended against approval in 2011 because of concerns over the numerical increase in bladder and breast cancers and a case of possible drug-induced liver injury. The FDA did not approve dapagliflozin, requesting more safety data, and the companies resubmitted the application for approval with more safety data and analyses last year.
The postmarketing trials required by the FDA include a cardiovascular outcomes trial that will evaluate the cardiovascular risk of dapagliflozin in patients with a high risk of cardiovascular disease at baseline and a study evaluating the risk of bladder cancer in patients enrolled in that study. Another study will evaluate bladder tumor promotion effects of the drug in rodents; two studies will evaluate the pharmacokinetics, efficacy, and safety of dapagliflozin in pediatric patients; and an enhanced pharmacovigilance program will monitor reports of liver abnormalities and pregnancy outcomes in patients treated with dapagliflozin, the FDA said.
At the December 2013 panel meeting, Bristol-Myers Squibb said that enrollment in a cardiovascular outcomes study that will eventually enroll over 17,000 patients with type 2 diabetes and established cardiovascular disease or at least two cardiovascular risk factors had already begun in Europe, where dapagliflozin was already approved. That study is the Dapagliflozin Effect on Cardiovascular Events (DECLARE-TIMI-58) study.
FDA approves dapagliflozin for type 2 diabetes
Dapagliflozin, a sodium-glucose cotransporter 2 inhibitor, has been approved for the treatment of type 2 diabetes with a requirement that the manufacturer conduct postmarketing studies that address risks associated with the drug, the Food and Drug Administration announced on Jan. 8.
Approval was based on the results of 16 clinical trials in more than 9,400 patients with type 2 diabetes, which found that treatment with the drug improved hemoglobin A1C, according to the statement. Dapagliflozin is the second sodium-glucose cotransporter 2 (SGLT2) inhibitor to be approved by the FDA; the first was canagliflozin (Invokana), approved in March 2013. These agents, taken orally, work by inhibiting SGLT2 that is expressed in the kidney, reducing renal glucose reabsorption, increasing urinary excretion of glucose, and reducing plasma glucose levels.
Dapagliflozin will be marketed as Farxiga by Bristol-Myers Squibb and AstraZeneca.
In the studies, the most common adverse events associated with treatment were genital fungal infections and urinary tract infections. Other adverse events included hypotension caused by dehydration, resulting in dizziness and/or fainting and reduced renal function, according to the FDA statement, which noted that "the elderly, patients with impaired renal function, and patients on diuretics to treat other conditions appeared to be more susceptible to this risk."
Approval came less than a month after a meeting in December, when the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1 that the benefits of dapagliflozin outweighed its risks for improving glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise. The panel, however, strongly recommended that cardiovascular safety, bladder cancer, and hepatotoxicity be monitored in treated patients after approval – which is reflected in the FDA’s list of required postmarketing studies.
The same panel had recommended against approval in 2011 because of concerns over the numerical increase in bladder and breast cancers and a case of possible drug-induced liver injury. The FDA did not approve dapagliflozin, requesting more safety data, and the companies resubmitted the application for approval with more safety data and analyses last year.
The postmarketing trials required by the FDA include a cardiovascular outcomes trial that will evaluate the cardiovascular risk of dapagliflozin in patients with a high risk of cardiovascular disease at baseline and a study evaluating the risk of bladder cancer in patients enrolled in that study. Another study will evaluate bladder tumor promotion effects of the drug in rodents; two studies will evaluate the pharmacokinetics, efficacy, and safety of dapagliflozin in pediatric patients; and an enhanced pharmacovigilance program will monitor reports of liver abnormalities and pregnancy outcomes in patients treated with dapagliflozin, the FDA said.
At the December 2013 panel meeting, Bristol-Myers Squibb said that enrollment in a cardiovascular outcomes study that will eventually enroll over 17,000 patients with type 2 diabetes and established cardiovascular disease or at least two cardiovascular risk factors had already begun in Europe, where dapagliflozin was already approved. That study is the Dapagliflozin Effect on Cardiovascular Events (DECLARE-TIMI-58) study.
Dapagliflozin, a sodium-glucose cotransporter 2 inhibitor, has been approved for the treatment of type 2 diabetes with a requirement that the manufacturer conduct postmarketing studies that address risks associated with the drug, the Food and Drug Administration announced on Jan. 8.
Approval was based on the results of 16 clinical trials in more than 9,400 patients with type 2 diabetes, which found that treatment with the drug improved hemoglobin A1C, according to the statement. Dapagliflozin is the second sodium-glucose cotransporter 2 (SGLT2) inhibitor to be approved by the FDA; the first was canagliflozin (Invokana), approved in March 2013. These agents, taken orally, work by inhibiting SGLT2 that is expressed in the kidney, reducing renal glucose reabsorption, increasing urinary excretion of glucose, and reducing plasma glucose levels.
Dapagliflozin will be marketed as Farxiga by Bristol-Myers Squibb and AstraZeneca.
In the studies, the most common adverse events associated with treatment were genital fungal infections and urinary tract infections. Other adverse events included hypotension caused by dehydration, resulting in dizziness and/or fainting and reduced renal function, according to the FDA statement, which noted that "the elderly, patients with impaired renal function, and patients on diuretics to treat other conditions appeared to be more susceptible to this risk."
Approval came less than a month after a meeting in December, when the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1 that the benefits of dapagliflozin outweighed its risks for improving glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise. The panel, however, strongly recommended that cardiovascular safety, bladder cancer, and hepatotoxicity be monitored in treated patients after approval – which is reflected in the FDA’s list of required postmarketing studies.
The same panel had recommended against approval in 2011 because of concerns over the numerical increase in bladder and breast cancers and a case of possible drug-induced liver injury. The FDA did not approve dapagliflozin, requesting more safety data, and the companies resubmitted the application for approval with more safety data and analyses last year.
The postmarketing trials required by the FDA include a cardiovascular outcomes trial that will evaluate the cardiovascular risk of dapagliflozin in patients with a high risk of cardiovascular disease at baseline and a study evaluating the risk of bladder cancer in patients enrolled in that study. Another study will evaluate bladder tumor promotion effects of the drug in rodents; two studies will evaluate the pharmacokinetics, efficacy, and safety of dapagliflozin in pediatric patients; and an enhanced pharmacovigilance program will monitor reports of liver abnormalities and pregnancy outcomes in patients treated with dapagliflozin, the FDA said.
At the December 2013 panel meeting, Bristol-Myers Squibb said that enrollment in a cardiovascular outcomes study that will eventually enroll over 17,000 patients with type 2 diabetes and established cardiovascular disease or at least two cardiovascular risk factors had already begun in Europe, where dapagliflozin was already approved. That study is the Dapagliflozin Effect on Cardiovascular Events (DECLARE-TIMI-58) study.
Dapagliflozin, a sodium-glucose cotransporter 2 inhibitor, has been approved for the treatment of type 2 diabetes with a requirement that the manufacturer conduct postmarketing studies that address risks associated with the drug, the Food and Drug Administration announced on Jan. 8.
Approval was based on the results of 16 clinical trials in more than 9,400 patients with type 2 diabetes, which found that treatment with the drug improved hemoglobin A1C, according to the statement. Dapagliflozin is the second sodium-glucose cotransporter 2 (SGLT2) inhibitor to be approved by the FDA; the first was canagliflozin (Invokana), approved in March 2013. These agents, taken orally, work by inhibiting SGLT2 that is expressed in the kidney, reducing renal glucose reabsorption, increasing urinary excretion of glucose, and reducing plasma glucose levels.
Dapagliflozin will be marketed as Farxiga by Bristol-Myers Squibb and AstraZeneca.
In the studies, the most common adverse events associated with treatment were genital fungal infections and urinary tract infections. Other adverse events included hypotension caused by dehydration, resulting in dizziness and/or fainting and reduced renal function, according to the FDA statement, which noted that "the elderly, patients with impaired renal function, and patients on diuretics to treat other conditions appeared to be more susceptible to this risk."
Approval came less than a month after a meeting in December, when the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1 that the benefits of dapagliflozin outweighed its risks for improving glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise. The panel, however, strongly recommended that cardiovascular safety, bladder cancer, and hepatotoxicity be monitored in treated patients after approval – which is reflected in the FDA’s list of required postmarketing studies.
The same panel had recommended against approval in 2011 because of concerns over the numerical increase in bladder and breast cancers and a case of possible drug-induced liver injury. The FDA did not approve dapagliflozin, requesting more safety data, and the companies resubmitted the application for approval with more safety data and analyses last year.
The postmarketing trials required by the FDA include a cardiovascular outcomes trial that will evaluate the cardiovascular risk of dapagliflozin in patients with a high risk of cardiovascular disease at baseline and a study evaluating the risk of bladder cancer in patients enrolled in that study. Another study will evaluate bladder tumor promotion effects of the drug in rodents; two studies will evaluate the pharmacokinetics, efficacy, and safety of dapagliflozin in pediatric patients; and an enhanced pharmacovigilance program will monitor reports of liver abnormalities and pregnancy outcomes in patients treated with dapagliflozin, the FDA said.
At the December 2013 panel meeting, Bristol-Myers Squibb said that enrollment in a cardiovascular outcomes study that will eventually enroll over 17,000 patients with type 2 diabetes and established cardiovascular disease or at least two cardiovascular risk factors had already begun in Europe, where dapagliflozin was already approved. That study is the Dapagliflozin Effect on Cardiovascular Events (DECLARE-TIMI-58) study.
FDA wants more data before approving alemtuzumab for MS
The Food and Drug Administration has declined to approve alemtuzumab for the treatment of multiple sclerosis, citing a lack of well-controlled data from clinical studies indicating the benefits outweigh the risks of treatment – a decision that Genzyme, the manufacturer, plans to appeal.
The company announced in a Dec. 30 statement that the agency had sent a "Complete Response Letter" regarding alemtuzumab, an intravenously infused monoclonal antibody that binds to the CD52 antigen present at high levels on the surface of T and B lymphocytes. Complete response letters are sent to manufacturers when the FDA decides against approval, with explanations of outstanding issues that need to be resolved. The FDA does not publicly announce when these letters are issued, but manufacturers often do so.

The proposed indication for alemtuzumab is for treatment of relapsing forms of MS, with a risk evaluation and mitigation strategy (REMS) that addresses the drug’s serious risks, which include immune thrombocytopenia and thyroid-related adverse events. The company’s proposed trade name for the MS indication for alemtuzumab is Lemtrada.
"FDA has taken the position that Genzyme has not submitted evidence from adequate and well-controlled studies that demonstrate the benefits of Lemtrada outweigh its serious adverse effects," according to the Genzyme statement. "The conclusion is related to the design of the completed [phase III] active comparator studies of Lemtrada in relapsing-remitting MS patients," and the FDA has also said that one or more "additional active comparator clinical trials of different design and execution" are needed before alemtuzumab is approved for MS, the statement said.
In the statement, Genzyme said that it "strongly disagrees with the FDA’s conclusions and plans to appeal the agency’s decision." No other details about the appeal were provided by the company, which is owned by Sanofi.
FDA reviewers made their concerns about alemtuzumab clear at a meeting of the FDA’s Peripheral and Central Nervous System advisory committee in November of 2013, and in documents provided before the meeting.
The FDA raised issues about the elevated rates of serious adverse events in patients treated with alemtuzumab and the open-label design of the clinical trials, questioning whether bias had an impact on the efficacy results. Patients and clinicians were not blinded to the treatment in those studies, which compared alemtuzumab to subcutaneous interferon beta-1a (Rebif) in about 1,400 patients and found significant reductions in the relapse rates among those treated with alemtuzumab (49% and 55%), compared with those on Rebif.
The advisory panel did not vote specifically on whether to recommend approval for MS. But while panelists agreed the unblinded studies were likely biased, the majority voted 12-6 that the manufacturer had provided substantial evidence that the drug was effective for the proposed indication. Several panelists said they would recommend the drug only for patients with severe MS, as a second- or third-line treatment.
In an interview, one of the investigators in the studies, Dr. Jonathan Carter, a consultant in the department of neurology at the Mayo Clinic in Scottsdale, Ariz., said he understood the FDA’s concerns about toxicity, and if it had been approved, alemtuzumab would probably have been designated a second- or third-line therapy. However, the decision is disappointing, since there is an unmet need for treatment in patients "who have broken through the standard first-line therapies," who do not have many options, particularly those who are positive for the JC virus antibody and are considered at increased risk for progressive multifocal leukoencephalopathy (PML) with natalizumab (Tysabri), he said.
Dr. Carter acknowledged the FDA reviewers’ concerns about the effect of unblinding, although he pointed out it can be difficult to maintain blinding of patients and clinicians in studies of products with marked side effects, such as infusion reactions. Despite the potential effects of unblinding on clinical outcome measures, however, he noted that MRI data showing less enhancing lesion activity in alemtuzumab-treated patients, compared to those on Rebif, supported efficacy.
Alemtuzumab was approved by the FDA in 2001 and marketed as Campath for B-cell chronic lymphocytic leukemia in the United States. But in September 2012, the company took Campath off the market and is providing it free of charge to leukemia patients in a distribution program. Alemtuzumab has been approved for MS in the European Union, Canada, and Australia.
Dr. Carter said he had no disclosures, other than being an investigator in the study.
The Food and Drug Administration has declined to approve alemtuzumab for the treatment of multiple sclerosis, citing a lack of well-controlled data from clinical studies indicating the benefits outweigh the risks of treatment – a decision that Genzyme, the manufacturer, plans to appeal.
The company announced in a Dec. 30 statement that the agency had sent a "Complete Response Letter" regarding alemtuzumab, an intravenously infused monoclonal antibody that binds to the CD52 antigen present at high levels on the surface of T and B lymphocytes. Complete response letters are sent to manufacturers when the FDA decides against approval, with explanations of outstanding issues that need to be resolved. The FDA does not publicly announce when these letters are issued, but manufacturers often do so.
The proposed indication for alemtuzumab is for treatment of relapsing forms of MS, with a risk evaluation and mitigation strategy (REMS) that addresses the drug’s serious risks, which include immune thrombocytopenia and thyroid-related adverse events. The company’s proposed trade name for the MS indication for alemtuzumab is Lemtrada.
"FDA has taken the position that Genzyme has not submitted evidence from adequate and well-controlled studies that demonstrate the benefits of Lemtrada outweigh its serious adverse effects," according to the Genzyme statement. "The conclusion is related to the design of the completed [phase III] active comparator studies of Lemtrada in relapsing-remitting MS patients," and the FDA has also said that one or more "additional active comparator clinical trials of different design and execution" are needed before alemtuzumab is approved for MS, the statement said.
In the statement, Genzyme said that it "strongly disagrees with the FDA’s conclusions and plans to appeal the agency’s decision." No other details about the appeal were provided by the company, which is owned by Sanofi.
FDA reviewers made their concerns about alemtuzumab clear at a meeting of the FDA’s Peripheral and Central Nervous System advisory committee in November of 2013, and in documents provided before the meeting.
The FDA raised issues about the elevated rates of serious adverse events in patients treated with alemtuzumab and the open-label design of the clinical trials, questioning whether bias had an impact on the efficacy results. Patients and clinicians were not blinded to the treatment in those studies, which compared alemtuzumab to subcutaneous interferon beta-1a (Rebif) in about 1,400 patients and found significant reductions in the relapse rates among those treated with alemtuzumab (49% and 55%), compared with those on Rebif.
The advisory panel did not vote specifically on whether to recommend approval for MS. But while panelists agreed the unblinded studies were likely biased, the majority voted 12-6 that the manufacturer had provided substantial evidence that the drug was effective for the proposed indication. Several panelists said they would recommend the drug only for patients with severe MS, as a second- or third-line treatment.
In an interview, one of the investigators in the studies, Dr. Jonathan Carter, a consultant in the department of neurology at the Mayo Clinic in Scottsdale, Ariz., said he understood the FDA’s concerns about toxicity, and if it had been approved, alemtuzumab would probably have been designated a second- or third-line therapy. However, the decision is disappointing, since there is an unmet need for treatment in patients "who have broken through the standard first-line therapies," who do not have many options, particularly those who are positive for the JC virus antibody and are considered at increased risk for progressive multifocal leukoencephalopathy (PML) with natalizumab (Tysabri), he said.
Dr. Carter acknowledged the FDA reviewers’ concerns about the effect of unblinding, although he pointed out it can be difficult to maintain blinding of patients and clinicians in studies of products with marked side effects, such as infusion reactions. Despite the potential effects of unblinding on clinical outcome measures, however, he noted that MRI data showing less enhancing lesion activity in alemtuzumab-treated patients, compared to those on Rebif, supported efficacy.
Alemtuzumab was approved by the FDA in 2001 and marketed as Campath for B-cell chronic lymphocytic leukemia in the United States. But in September 2012, the company took Campath off the market and is providing it free of charge to leukemia patients in a distribution program. Alemtuzumab has been approved for MS in the European Union, Canada, and Australia.
Dr. Carter said he had no disclosures, other than being an investigator in the study.
The Food and Drug Administration has declined to approve alemtuzumab for the treatment of multiple sclerosis, citing a lack of well-controlled data from clinical studies indicating the benefits outweigh the risks of treatment – a decision that Genzyme, the manufacturer, plans to appeal.
The company announced in a Dec. 30 statement that the agency had sent a "Complete Response Letter" regarding alemtuzumab, an intravenously infused monoclonal antibody that binds to the CD52 antigen present at high levels on the surface of T and B lymphocytes. Complete response letters are sent to manufacturers when the FDA decides against approval, with explanations of outstanding issues that need to be resolved. The FDA does not publicly announce when these letters are issued, but manufacturers often do so.
The proposed indication for alemtuzumab is for treatment of relapsing forms of MS, with a risk evaluation and mitigation strategy (REMS) that addresses the drug’s serious risks, which include immune thrombocytopenia and thyroid-related adverse events. The company’s proposed trade name for the MS indication for alemtuzumab is Lemtrada.
"FDA has taken the position that Genzyme has not submitted evidence from adequate and well-controlled studies that demonstrate the benefits of Lemtrada outweigh its serious adverse effects," according to the Genzyme statement. "The conclusion is related to the design of the completed [phase III] active comparator studies of Lemtrada in relapsing-remitting MS patients," and the FDA has also said that one or more "additional active comparator clinical trials of different design and execution" are needed before alemtuzumab is approved for MS, the statement said.
In the statement, Genzyme said that it "strongly disagrees with the FDA’s conclusions and plans to appeal the agency’s decision." No other details about the appeal were provided by the company, which is owned by Sanofi.
FDA reviewers made their concerns about alemtuzumab clear at a meeting of the FDA’s Peripheral and Central Nervous System advisory committee in November of 2013, and in documents provided before the meeting.
The FDA raised issues about the elevated rates of serious adverse events in patients treated with alemtuzumab and the open-label design of the clinical trials, questioning whether bias had an impact on the efficacy results. Patients and clinicians were not blinded to the treatment in those studies, which compared alemtuzumab to subcutaneous interferon beta-1a (Rebif) in about 1,400 patients and found significant reductions in the relapse rates among those treated with alemtuzumab (49% and 55%), compared with those on Rebif.
The advisory panel did not vote specifically on whether to recommend approval for MS. But while panelists agreed the unblinded studies were likely biased, the majority voted 12-6 that the manufacturer had provided substantial evidence that the drug was effective for the proposed indication. Several panelists said they would recommend the drug only for patients with severe MS, as a second- or third-line treatment.
In an interview, one of the investigators in the studies, Dr. Jonathan Carter, a consultant in the department of neurology at the Mayo Clinic in Scottsdale, Ariz., said he understood the FDA’s concerns about toxicity, and if it had been approved, alemtuzumab would probably have been designated a second- or third-line therapy. However, the decision is disappointing, since there is an unmet need for treatment in patients "who have broken through the standard first-line therapies," who do not have many options, particularly those who are positive for the JC virus antibody and are considered at increased risk for progressive multifocal leukoencephalopathy (PML) with natalizumab (Tysabri), he said.
Dr. Carter acknowledged the FDA reviewers’ concerns about the effect of unblinding, although he pointed out it can be difficult to maintain blinding of patients and clinicians in studies of products with marked side effects, such as infusion reactions. Despite the potential effects of unblinding on clinical outcome measures, however, he noted that MRI data showing less enhancing lesion activity in alemtuzumab-treated patients, compared to those on Rebif, supported efficacy.
Alemtuzumab was approved by the FDA in 2001 and marketed as Campath for B-cell chronic lymphocytic leukemia in the United States. But in September 2012, the company took Campath off the market and is providing it free of charge to leukemia patients in a distribution program. Alemtuzumab has been approved for MS in the European Union, Canada, and Australia.
Dr. Carter said he had no disclosures, other than being an investigator in the study.

FDA advisers back vedolizumab approval to treat moderate to severe inflammatory bowel disease
SILVER SPRING, MD. – The benefits of vedolizumab, an integrin antagonist, outweigh the potential for progressive multifocal encephalopathy and other possible risks as a treatment for moderate to severely active Crohn’s disease and ulcerative colitis, and should be approved, according to two Food and Drug Administration advisory panels.
At a meeting of the FDA’s Gastrointestinal Drugs and Drug Safety and Risk Management advisory committees, the panels unanimously supported approval of vedolizumab for both indications, although they were less confident about the efficacy data of vedolizumab for induction in Crohn’s disease. They also strongly recommended a postmarketing program to monitor safety issues after approval, including progressive multifocal encephalopathy (PML), other infections, and other types of adverse events such as autoimmune hepatitis.
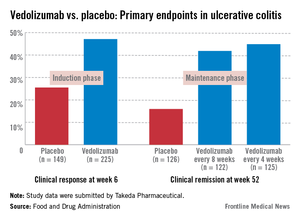
Vedolizumab is a monoclonal antibody that binds exclusively to the alpha 4 beta 7 integrin, "a key mediator of gastrointestinal inflammation," according to the manufacturer, Takeda Pharmaceuticals USA. It is administered intravenously at 0, 2, and 6 weeks, followed by once every 8 weeks for maintenance therapy. No cases of PML have been reported in more than 3,100 patients treated with vedolizumab worldwide, including 906 treated for 3 or more years, according to Takeda. But because it is thought to disrupt integrin function, like natalizumab (Tysabri), which is approved for multiple sclerosis and Crohn’s disease and is associated with an increased risk of PML, the potential for PML with vedolizumab therapy was the main safety issue raised by the FDA. Natalizumab is approved with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the risk of PML, the usually fatal demyelinating CNS infection.
The phase III studies evaluating vedolizumab in patients with Crohn’s disease and ulcerative colitis were similarly designed, randomizing patients to vedolizumab or placebo in 6-week induction studies, and randomizing patients who responded to vedolizumab induction therapy to vedolizumab or placebo in 52-week maintenance studies.
In a Crohn’s disease induction and maintenance study of 368 patients, 14.5% of those on vedolizumab had achieved a clinical remission at week 6, vs. almost 7% of those on placebo, a statistically significant difference. At week 52, 39% of those who were treated every 8 weeks as maintenance therapy had a clinical remission, vs. almost 22% of those on placebo, also a significant difference. But in a second induction study of 315 patients, mostly treatment-refractory patients, there was not a significant difference in 6-week clinical remission rates between those on vedolizumab (15%) and those on placebo (12%).
The panelists voted 12 to 9 that these data supported efficacy for Crohn’s disease induction, and voted 20 to 0, with one abstention, that the data supported approval of the maintenance indication, agreeing the maintenance data were robust. Most (14) agreed that the benefits outweighed the risks to support approval of patients with Crohn’s who have failed treatment with steroids or immunosuppressants or tumor necrosis factor (TNF)–alpha blockers, the indication proposed by the manufacturer. Six panelists supported approval, but for the narrower use, in patients who had failed treatment with immunosuppressants or TNF-alpha antagonists, an indication that would not include patients who had failed steroids only. (The remaining panelist abstained.)
In the induction and maintenance trial of 374 patients with ulcerative colitis, the clinical response rate at 6 weeks was 47% among those on vedolizumab, vs. 25.5% among those on placebo, a statistically significant difference. In this study, almost 42% of those on vedolizumab every 8 weeks as maintenance therapy had achieved clinical remission at week 52, vs. 16% of those on placebo, also a statistically significant difference.
All panel members supported approval for the ulcerative colitis indication, based on the benefits and risks, but differed on the wording of the indication, with 13 panelists supporting approval for patients with ulcerative colitis who have failed steroids or immunosuppressants or TNF-alpha antagonists. The remaining eight panelists supported approval for patients who have failed immunosuppressants or TNF-alpha antagonists, which would not include an indication for patients who had failed treatment with steroids only.
The overall infection rate was higher among the patients treated with vedolizumab than among those on placebo in these studies (primarily upper respiratory tract infections and pharyngitis), which did not appear to be related to the number of infusions or concomitant immunosuppressive treatment. The rates of serious infections (3%-4%) were similar among those on vedolizumab and placebo. There were four patients with serious hepatic adverse events – acute hepatitis – that resolved. The 12 deaths among those on vedolizumab were not related to the drug, and there was no apparent increase in malignancies among treated patients, according to the FDA reviewers.
In a unanimous 21-0 vote, the panel agreed that Takeda had "adequately characterized the potential risk of PML" to support approval, but panelists recommended postmarketing follow-up for PML and other serious adverse events in treated patients.
The FDA is expected to make a decision on approval for the ulcerative colitis indication by Feb. 18, 2014, and for the Crohn’s disease indication by June 18, 2014.
The FDA usually follows the recommendations of its advisory panels. Members of FDA panels have usually been cleared of conflicts related to the product under review; occasionally, a panelist is given a waiver, but not at this meeting.
If approved, Takeda plans to market vedolizumab as Entyvio.
SILVER SPRING, MD. – The benefits of vedolizumab, an integrin antagonist, outweigh the potential for progressive multifocal encephalopathy and other possible risks as a treatment for moderate to severely active Crohn’s disease and ulcerative colitis, and should be approved, according to two Food and Drug Administration advisory panels.
At a meeting of the FDA’s Gastrointestinal Drugs and Drug Safety and Risk Management advisory committees, the panels unanimously supported approval of vedolizumab for both indications, although they were less confident about the efficacy data of vedolizumab for induction in Crohn’s disease. They also strongly recommended a postmarketing program to monitor safety issues after approval, including progressive multifocal encephalopathy (PML), other infections, and other types of adverse events such as autoimmune hepatitis.
Vedolizumab is a monoclonal antibody that binds exclusively to the alpha 4 beta 7 integrin, "a key mediator of gastrointestinal inflammation," according to the manufacturer, Takeda Pharmaceuticals USA. It is administered intravenously at 0, 2, and 6 weeks, followed by once every 8 weeks for maintenance therapy. No cases of PML have been reported in more than 3,100 patients treated with vedolizumab worldwide, including 906 treated for 3 or more years, according to Takeda. But because it is thought to disrupt integrin function, like natalizumab (Tysabri), which is approved for multiple sclerosis and Crohn’s disease and is associated with an increased risk of PML, the potential for PML with vedolizumab therapy was the main safety issue raised by the FDA. Natalizumab is approved with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the risk of PML, the usually fatal demyelinating CNS infection.
The phase III studies evaluating vedolizumab in patients with Crohn’s disease and ulcerative colitis were similarly designed, randomizing patients to vedolizumab or placebo in 6-week induction studies, and randomizing patients who responded to vedolizumab induction therapy to vedolizumab or placebo in 52-week maintenance studies.
In a Crohn’s disease induction and maintenance study of 368 patients, 14.5% of those on vedolizumab had achieved a clinical remission at week 6, vs. almost 7% of those on placebo, a statistically significant difference. At week 52, 39% of those who were treated every 8 weeks as maintenance therapy had a clinical remission, vs. almost 22% of those on placebo, also a significant difference. But in a second induction study of 315 patients, mostly treatment-refractory patients, there was not a significant difference in 6-week clinical remission rates between those on vedolizumab (15%) and those on placebo (12%).
The panelists voted 12 to 9 that these data supported efficacy for Crohn’s disease induction, and voted 20 to 0, with one abstention, that the data supported approval of the maintenance indication, agreeing the maintenance data were robust. Most (14) agreed that the benefits outweighed the risks to support approval of patients with Crohn’s who have failed treatment with steroids or immunosuppressants or tumor necrosis factor (TNF)–alpha blockers, the indication proposed by the manufacturer. Six panelists supported approval, but for the narrower use, in patients who had failed treatment with immunosuppressants or TNF-alpha antagonists, an indication that would not include patients who had failed steroids only. (The remaining panelist abstained.)
In the induction and maintenance trial of 374 patients with ulcerative colitis, the clinical response rate at 6 weeks was 47% among those on vedolizumab, vs. 25.5% among those on placebo, a statistically significant difference. In this study, almost 42% of those on vedolizumab every 8 weeks as maintenance therapy had achieved clinical remission at week 52, vs. 16% of those on placebo, also a statistically significant difference.
All panel members supported approval for the ulcerative colitis indication, based on the benefits and risks, but differed on the wording of the indication, with 13 panelists supporting approval for patients with ulcerative colitis who have failed steroids or immunosuppressants or TNF-alpha antagonists. The remaining eight panelists supported approval for patients who have failed immunosuppressants or TNF-alpha antagonists, which would not include an indication for patients who had failed treatment with steroids only.
The overall infection rate was higher among the patients treated with vedolizumab than among those on placebo in these studies (primarily upper respiratory tract infections and pharyngitis), which did not appear to be related to the number of infusions or concomitant immunosuppressive treatment. The rates of serious infections (3%-4%) were similar among those on vedolizumab and placebo. There were four patients with serious hepatic adverse events – acute hepatitis – that resolved. The 12 deaths among those on vedolizumab were not related to the drug, and there was no apparent increase in malignancies among treated patients, according to the FDA reviewers.
In a unanimous 21-0 vote, the panel agreed that Takeda had "adequately characterized the potential risk of PML" to support approval, but panelists recommended postmarketing follow-up for PML and other serious adverse events in treated patients.
The FDA is expected to make a decision on approval for the ulcerative colitis indication by Feb. 18, 2014, and for the Crohn’s disease indication by June 18, 2014.
The FDA usually follows the recommendations of its advisory panels. Members of FDA panels have usually been cleared of conflicts related to the product under review; occasionally, a panelist is given a waiver, but not at this meeting.
If approved, Takeda plans to market vedolizumab as Entyvio.
SILVER SPRING, MD. – The benefits of vedolizumab, an integrin antagonist, outweigh the potential for progressive multifocal encephalopathy and other possible risks as a treatment for moderate to severely active Crohn’s disease and ulcerative colitis, and should be approved, according to two Food and Drug Administration advisory panels.
At a meeting of the FDA’s Gastrointestinal Drugs and Drug Safety and Risk Management advisory committees, the panels unanimously supported approval of vedolizumab for both indications, although they were less confident about the efficacy data of vedolizumab for induction in Crohn’s disease. They also strongly recommended a postmarketing program to monitor safety issues after approval, including progressive multifocal encephalopathy (PML), other infections, and other types of adverse events such as autoimmune hepatitis.
Vedolizumab is a monoclonal antibody that binds exclusively to the alpha 4 beta 7 integrin, "a key mediator of gastrointestinal inflammation," according to the manufacturer, Takeda Pharmaceuticals USA. It is administered intravenously at 0, 2, and 6 weeks, followed by once every 8 weeks for maintenance therapy. No cases of PML have been reported in more than 3,100 patients treated with vedolizumab worldwide, including 906 treated for 3 or more years, according to Takeda. But because it is thought to disrupt integrin function, like natalizumab (Tysabri), which is approved for multiple sclerosis and Crohn’s disease and is associated with an increased risk of PML, the potential for PML with vedolizumab therapy was the main safety issue raised by the FDA. Natalizumab is approved with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the risk of PML, the usually fatal demyelinating CNS infection.
The phase III studies evaluating vedolizumab in patients with Crohn’s disease and ulcerative colitis were similarly designed, randomizing patients to vedolizumab or placebo in 6-week induction studies, and randomizing patients who responded to vedolizumab induction therapy to vedolizumab or placebo in 52-week maintenance studies.
In a Crohn’s disease induction and maintenance study of 368 patients, 14.5% of those on vedolizumab had achieved a clinical remission at week 6, vs. almost 7% of those on placebo, a statistically significant difference. At week 52, 39% of those who were treated every 8 weeks as maintenance therapy had a clinical remission, vs. almost 22% of those on placebo, also a significant difference. But in a second induction study of 315 patients, mostly treatment-refractory patients, there was not a significant difference in 6-week clinical remission rates between those on vedolizumab (15%) and those on placebo (12%).
The panelists voted 12 to 9 that these data supported efficacy for Crohn’s disease induction, and voted 20 to 0, with one abstention, that the data supported approval of the maintenance indication, agreeing the maintenance data were robust. Most (14) agreed that the benefits outweighed the risks to support approval of patients with Crohn’s who have failed treatment with steroids or immunosuppressants or tumor necrosis factor (TNF)–alpha blockers, the indication proposed by the manufacturer. Six panelists supported approval, but for the narrower use, in patients who had failed treatment with immunosuppressants or TNF-alpha antagonists, an indication that would not include patients who had failed steroids only. (The remaining panelist abstained.)
In the induction and maintenance trial of 374 patients with ulcerative colitis, the clinical response rate at 6 weeks was 47% among those on vedolizumab, vs. 25.5% among those on placebo, a statistically significant difference. In this study, almost 42% of those on vedolizumab every 8 weeks as maintenance therapy had achieved clinical remission at week 52, vs. 16% of those on placebo, also a statistically significant difference.
All panel members supported approval for the ulcerative colitis indication, based on the benefits and risks, but differed on the wording of the indication, with 13 panelists supporting approval for patients with ulcerative colitis who have failed steroids or immunosuppressants or TNF-alpha antagonists. The remaining eight panelists supported approval for patients who have failed immunosuppressants or TNF-alpha antagonists, which would not include an indication for patients who had failed treatment with steroids only.
The overall infection rate was higher among the patients treated with vedolizumab than among those on placebo in these studies (primarily upper respiratory tract infections and pharyngitis), which did not appear to be related to the number of infusions or concomitant immunosuppressive treatment. The rates of serious infections (3%-4%) were similar among those on vedolizumab and placebo. There were four patients with serious hepatic adverse events – acute hepatitis – that resolved. The 12 deaths among those on vedolizumab were not related to the drug, and there was no apparent increase in malignancies among treated patients, according to the FDA reviewers.
In a unanimous 21-0 vote, the panel agreed that Takeda had "adequately characterized the potential risk of PML" to support approval, but panelists recommended postmarketing follow-up for PML and other serious adverse events in treated patients.
The FDA is expected to make a decision on approval for the ulcerative colitis indication by Feb. 18, 2014, and for the Crohn’s disease indication by June 18, 2014.
The FDA usually follows the recommendations of its advisory panels. Members of FDA panels have usually been cleared of conflicts related to the product under review; occasionally, a panelist is given a waiver, but not at this meeting.
If approved, Takeda plans to market vedolizumab as Entyvio.
AT AN FDA ADVISORY COMMITTEE MEETING
FDA panel backs dapagliflozin for type 2 diabetes
SILVER SPRING, MD. – Reassured by the data on the risks of bladder cancer and liver injury and on cardiovascular safety, a Food and Drug Administration advisory panel supported the approval of the sodium glucose co-transporter 2 inhibitor dapagliflozin as a treatment for type 2 diabetes at a meeting on Dec. 12.
Voting 13-1, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the benefits of dapagliflozin outweighed its risks for the proposed indication: to improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise. The recommended dose is 5 mg or 10 mg/day, at any time of the day regardless of meals. It is not recommended for patients with moderate renal impairment, and the 5-mg dose is recommended for people at risk for volume depletion related to a coexisting condition.

Panelists, however, strongly recommended that cardiovascular safety, bladder cancer, and hepatotoxicity in patients treated with dapagliflozin should be closely monitored after approval.
The risks "appear to be low enough to go ahead, but there is surveillance that is needed to make sure we’re not missing something," said panelist Dr. Peter Savage, senior advisor for clinical research in the division of diabetes, endocrinology, and metabolism at the National Institute of Diabetes, Digestive, and Kidney Diseases.
One of the two oncologists on the panel, Dr. Wyndham Wilson of the lymphoid malignancy branch at the National Cancer Institute’s Center for Cancer Research, said that, based on the effects of the drug on surrogate endpoints, he believed it was useful for treating type 2 diabetes and that the cancer and hepatic findings "did not rise to the level of nonapproval for me." He emphasized, however, the importance of the postmarketing studies that are being planned or are underway that are addressing the cancer and cardiac risks further.
This was the second time the panel has met to review dapagliflozin, which, by inhibiting sodium glucose co-transporter 2 (SGLT2) that is expressed in the kidney, reduces renal glucose reabsorption, increasing urinary excretion of glucose and reducing plasma glucose levels. If approved, dapagliflozin would be the second SGLT2-inhbitor on the U.S. market. In March 2013, canagliflozin (Invokana), another SGLT2 inhibitor, was approved.
Because of a numerical increase in bladder and breast cancers and a possible drug-induced liver injury case, the majority of the same panel voted against approval at a July 2011 meeting. In January 2012, the FDA issued a complete response letter to the company, requesting more data and analyses on hepatic safety, bladder cancer, and cardiovascular safety. Bristol-Myers Squibb and AstraZeneca resubmitted the application for approval in July 2013, with data from additional studies, for a total of more than 11,000 patients (with more than 6,000 treated with dapagliflozin),which included updated safety information on hepatic safety, bladder cancers, and CV safety analyses.
In the updated meta-analysis of these studies, the hazard ratio for the cardiovascular composite endpoint of CV death, MI, stroke, and hospitalization for unstable angina was 0.81. For the stricter major adverse cardiovascular event (MACE, comprising CV death, MI, and stroke) endpoint, it was 0.78. In two 24-week studies that randomized almost 2,000 patients with established cardiovascular disease to dapagliflozin or placebo, who were followed for up to two years, the hazard ratio for the primary composite endpoint was 0.98 and for the MACE endpoint, it was 1.11.
Based on these analyses, the panel voted 10-4 that the company had provided sufficient evidence that dapagliflozin has an acceptable cardiovascular profile based on the FDA’s guidance document for industry, which states that an estimated cardiovascular risk of 1.8 or more for a new type 2 diabetes drug should be ruled out.
"Overall, it looks fairly neutral," and "the overall effect does not appear to be adverse," even when adding the two studies in patients with established CV disease, said panelist Dr. Peter Wilson, professor of cardiology at Emory University, Atlanta. Those who voted no on this question said that there were not enough data to make a conclusion about cardiovascular safety.
The panelists agreed that they were reassured by aspects of the 10 bladder cancer cases reported among patients treated with dapagliflozin in clinical trials (a rate of 0.16%), compared with one case (0.03%) among those on comparator arms. Of the 10 cases, 6 were detected within 6 months of treatment (while tumorigenesis can take years) and 8 were noninvasive. In seven cases, the patient had hematuria before starting dapagliflozin, although hematuria is more common among people with diabetes, they pointed out. The panelists, though, did not think the bladder cancer issue could be entirely dismissed and agreed that this issue should be monitored after approval.
The panel did not have substantial concerns about the risk of liver disease associated with the drug and agreed the data did not indicate a significant signal for hepatoxicity, but that hepatic adverse events should be monitored after approval. The one case of what was thought to be probable drug-induced liver injury in clinical trials was later considered more likely to be autoimmune hepatitis, provided "substantial reassurance" that this was not drug-related, although it could not be completely ruled out, they said.
The panel also recommended that rates of breast cancer in women be monitored after approval. A numerical increase in breast cancers among those on dapagliflozin was a concern at the first meeting, although the FDA concluded that the data on the breast cancer risk associated with dapagliflozin were inconclusive and insufficient.
The companies are planning to follow these safety issues in studies that include pharmacoepidemiology studies underway in Europe – which are monitoring for cancer, acute liver injury, and severe urinary tract infection complications – and a cardiovascular outcomes study with a planned enrollment of 17,150 patients with type 2 diabetes and established CV disease or at least two CV risk factors. Enrollment for the latter study – the Dapagliflozin Effect on Cardiovascular Events (DECLARE-TIMI-58) study – in Europe has already begun.
The FDA usually follows the recommendations of its advisory panels. Members of FDA panels have usually been cleared of conflicts related to the product under review; occasionally, a panelist is given a waiver, but not at this meeting.
In studies, dapagliflozin lowered hemoglobin A1c at the 5-mg and 10-mg once-daily doses and also was associated with decreases in fasting blood sugar, small decreases in systolic blood pressure (in hypertensive patients) and body weight, and LDL-cholesterol elevation. Adverse events associated with treatment include a higher rate of genital infections, volume depletion, and polyuria.
Since November 2012, dapagliflozin has been approved in the European Union, Australia, Mexico, New Zealand, Brazil, and Argentina. To date, over 35,000 people worldwide have been treated with dapagliflozin, according to Bristol-Myers Squibb. If approved, BMS and AstraZeneca plan to market dapagliflozin as Forxiga. A decision on approval is expected by Jan. 11, 2014.
SILVER SPRING, MD. – Reassured by the data on the risks of bladder cancer and liver injury and on cardiovascular safety, a Food and Drug Administration advisory panel supported the approval of the sodium glucose co-transporter 2 inhibitor dapagliflozin as a treatment for type 2 diabetes at a meeting on Dec. 12.
Voting 13-1, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the benefits of dapagliflozin outweighed its risks for the proposed indication: to improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise. The recommended dose is 5 mg or 10 mg/day, at any time of the day regardless of meals. It is not recommended for patients with moderate renal impairment, and the 5-mg dose is recommended for people at risk for volume depletion related to a coexisting condition.
Panelists, however, strongly recommended that cardiovascular safety, bladder cancer, and hepatotoxicity in patients treated with dapagliflozin should be closely monitored after approval.
The risks "appear to be low enough to go ahead, but there is surveillance that is needed to make sure we’re not missing something," said panelist Dr. Peter Savage, senior advisor for clinical research in the division of diabetes, endocrinology, and metabolism at the National Institute of Diabetes, Digestive, and Kidney Diseases.
One of the two oncologists on the panel, Dr. Wyndham Wilson of the lymphoid malignancy branch at the National Cancer Institute’s Center for Cancer Research, said that, based on the effects of the drug on surrogate endpoints, he believed it was useful for treating type 2 diabetes and that the cancer and hepatic findings "did not rise to the level of nonapproval for me." He emphasized, however, the importance of the postmarketing studies that are being planned or are underway that are addressing the cancer and cardiac risks further.
This was the second time the panel has met to review dapagliflozin, which, by inhibiting sodium glucose co-transporter 2 (SGLT2) that is expressed in the kidney, reduces renal glucose reabsorption, increasing urinary excretion of glucose and reducing plasma glucose levels. If approved, dapagliflozin would be the second SGLT2-inhbitor on the U.S. market. In March 2013, canagliflozin (Invokana), another SGLT2 inhibitor, was approved.
Because of a numerical increase in bladder and breast cancers and a possible drug-induced liver injury case, the majority of the same panel voted against approval at a July 2011 meeting. In January 2012, the FDA issued a complete response letter to the company, requesting more data and analyses on hepatic safety, bladder cancer, and cardiovascular safety. Bristol-Myers Squibb and AstraZeneca resubmitted the application for approval in July 2013, with data from additional studies, for a total of more than 11,000 patients (with more than 6,000 treated with dapagliflozin),which included updated safety information on hepatic safety, bladder cancers, and CV safety analyses.
In the updated meta-analysis of these studies, the hazard ratio for the cardiovascular composite endpoint of CV death, MI, stroke, and hospitalization for unstable angina was 0.81. For the stricter major adverse cardiovascular event (MACE, comprising CV death, MI, and stroke) endpoint, it was 0.78. In two 24-week studies that randomized almost 2,000 patients with established cardiovascular disease to dapagliflozin or placebo, who were followed for up to two years, the hazard ratio for the primary composite endpoint was 0.98 and for the MACE endpoint, it was 1.11.
Based on these analyses, the panel voted 10-4 that the company had provided sufficient evidence that dapagliflozin has an acceptable cardiovascular profile based on the FDA’s guidance document for industry, which states that an estimated cardiovascular risk of 1.8 or more for a new type 2 diabetes drug should be ruled out.
"Overall, it looks fairly neutral," and "the overall effect does not appear to be adverse," even when adding the two studies in patients with established CV disease, said panelist Dr. Peter Wilson, professor of cardiology at Emory University, Atlanta. Those who voted no on this question said that there were not enough data to make a conclusion about cardiovascular safety.
The panelists agreed that they were reassured by aspects of the 10 bladder cancer cases reported among patients treated with dapagliflozin in clinical trials (a rate of 0.16%), compared with one case (0.03%) among those on comparator arms. Of the 10 cases, 6 were detected within 6 months of treatment (while tumorigenesis can take years) and 8 were noninvasive. In seven cases, the patient had hematuria before starting dapagliflozin, although hematuria is more common among people with diabetes, they pointed out. The panelists, though, did not think the bladder cancer issue could be entirely dismissed and agreed that this issue should be monitored after approval.
The panel did not have substantial concerns about the risk of liver disease associated with the drug and agreed the data did not indicate a significant signal for hepatoxicity, but that hepatic adverse events should be monitored after approval. The one case of what was thought to be probable drug-induced liver injury in clinical trials was later considered more likely to be autoimmune hepatitis, provided "substantial reassurance" that this was not drug-related, although it could not be completely ruled out, they said.
The panel also recommended that rates of breast cancer in women be monitored after approval. A numerical increase in breast cancers among those on dapagliflozin was a concern at the first meeting, although the FDA concluded that the data on the breast cancer risk associated with dapagliflozin were inconclusive and insufficient.
The companies are planning to follow these safety issues in studies that include pharmacoepidemiology studies underway in Europe – which are monitoring for cancer, acute liver injury, and severe urinary tract infection complications – and a cardiovascular outcomes study with a planned enrollment of 17,150 patients with type 2 diabetes and established CV disease or at least two CV risk factors. Enrollment for the latter study – the Dapagliflozin Effect on Cardiovascular Events (DECLARE-TIMI-58) study – in Europe has already begun.
The FDA usually follows the recommendations of its advisory panels. Members of FDA panels have usually been cleared of conflicts related to the product under review; occasionally, a panelist is given a waiver, but not at this meeting.
In studies, dapagliflozin lowered hemoglobin A1c at the 5-mg and 10-mg once-daily doses and also was associated with decreases in fasting blood sugar, small decreases in systolic blood pressure (in hypertensive patients) and body weight, and LDL-cholesterol elevation. Adverse events associated with treatment include a higher rate of genital infections, volume depletion, and polyuria.
Since November 2012, dapagliflozin has been approved in the European Union, Australia, Mexico, New Zealand, Brazil, and Argentina. To date, over 35,000 people worldwide have been treated with dapagliflozin, according to Bristol-Myers Squibb. If approved, BMS and AstraZeneca plan to market dapagliflozin as Forxiga. A decision on approval is expected by Jan. 11, 2014.
SILVER SPRING, MD. – Reassured by the data on the risks of bladder cancer and liver injury and on cardiovascular safety, a Food and Drug Administration advisory panel supported the approval of the sodium glucose co-transporter 2 inhibitor dapagliflozin as a treatment for type 2 diabetes at a meeting on Dec. 12.
Voting 13-1, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee agreed that the benefits of dapagliflozin outweighed its risks for the proposed indication: to improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise. The recommended dose is 5 mg or 10 mg/day, at any time of the day regardless of meals. It is not recommended for patients with moderate renal impairment, and the 5-mg dose is recommended for people at risk for volume depletion related to a coexisting condition.
Panelists, however, strongly recommended that cardiovascular safety, bladder cancer, and hepatotoxicity in patients treated with dapagliflozin should be closely monitored after approval.
The risks "appear to be low enough to go ahead, but there is surveillance that is needed to make sure we’re not missing something," said panelist Dr. Peter Savage, senior advisor for clinical research in the division of diabetes, endocrinology, and metabolism at the National Institute of Diabetes, Digestive, and Kidney Diseases.
One of the two oncologists on the panel, Dr. Wyndham Wilson of the lymphoid malignancy branch at the National Cancer Institute’s Center for Cancer Research, said that, based on the effects of the drug on surrogate endpoints, he believed it was useful for treating type 2 diabetes and that the cancer and hepatic findings "did not rise to the level of nonapproval for me." He emphasized, however, the importance of the postmarketing studies that are being planned or are underway that are addressing the cancer and cardiac risks further.
This was the second time the panel has met to review dapagliflozin, which, by inhibiting sodium glucose co-transporter 2 (SGLT2) that is expressed in the kidney, reduces renal glucose reabsorption, increasing urinary excretion of glucose and reducing plasma glucose levels. If approved, dapagliflozin would be the second SGLT2-inhbitor on the U.S. market. In March 2013, canagliflozin (Invokana), another SGLT2 inhibitor, was approved.
Because of a numerical increase in bladder and breast cancers and a possible drug-induced liver injury case, the majority of the same panel voted against approval at a July 2011 meeting. In January 2012, the FDA issued a complete response letter to the company, requesting more data and analyses on hepatic safety, bladder cancer, and cardiovascular safety. Bristol-Myers Squibb and AstraZeneca resubmitted the application for approval in July 2013, with data from additional studies, for a total of more than 11,000 patients (with more than 6,000 treated with dapagliflozin),which included updated safety information on hepatic safety, bladder cancers, and CV safety analyses.
In the updated meta-analysis of these studies, the hazard ratio for the cardiovascular composite endpoint of CV death, MI, stroke, and hospitalization for unstable angina was 0.81. For the stricter major adverse cardiovascular event (MACE, comprising CV death, MI, and stroke) endpoint, it was 0.78. In two 24-week studies that randomized almost 2,000 patients with established cardiovascular disease to dapagliflozin or placebo, who were followed for up to two years, the hazard ratio for the primary composite endpoint was 0.98 and for the MACE endpoint, it was 1.11.
Based on these analyses, the panel voted 10-4 that the company had provided sufficient evidence that dapagliflozin has an acceptable cardiovascular profile based on the FDA’s guidance document for industry, which states that an estimated cardiovascular risk of 1.8 or more for a new type 2 diabetes drug should be ruled out.
"Overall, it looks fairly neutral," and "the overall effect does not appear to be adverse," even when adding the two studies in patients with established CV disease, said panelist Dr. Peter Wilson, professor of cardiology at Emory University, Atlanta. Those who voted no on this question said that there were not enough data to make a conclusion about cardiovascular safety.
The panelists agreed that they were reassured by aspects of the 10 bladder cancer cases reported among patients treated with dapagliflozin in clinical trials (a rate of 0.16%), compared with one case (0.03%) among those on comparator arms. Of the 10 cases, 6 were detected within 6 months of treatment (while tumorigenesis can take years) and 8 were noninvasive. In seven cases, the patient had hematuria before starting dapagliflozin, although hematuria is more common among people with diabetes, they pointed out. The panelists, though, did not think the bladder cancer issue could be entirely dismissed and agreed that this issue should be monitored after approval.
The panel did not have substantial concerns about the risk of liver disease associated with the drug and agreed the data did not indicate a significant signal for hepatoxicity, but that hepatic adverse events should be monitored after approval. The one case of what was thought to be probable drug-induced liver injury in clinical trials was later considered more likely to be autoimmune hepatitis, provided "substantial reassurance" that this was not drug-related, although it could not be completely ruled out, they said.
The panel also recommended that rates of breast cancer in women be monitored after approval. A numerical increase in breast cancers among those on dapagliflozin was a concern at the first meeting, although the FDA concluded that the data on the breast cancer risk associated with dapagliflozin were inconclusive and insufficient.
The companies are planning to follow these safety issues in studies that include pharmacoepidemiology studies underway in Europe – which are monitoring for cancer, acute liver injury, and severe urinary tract infection complications – and a cardiovascular outcomes study with a planned enrollment of 17,150 patients with type 2 diabetes and established CV disease or at least two CV risk factors. Enrollment for the latter study – the Dapagliflozin Effect on Cardiovascular Events (DECLARE-TIMI-58) study – in Europe has already begun.
The FDA usually follows the recommendations of its advisory panels. Members of FDA panels have usually been cleared of conflicts related to the product under review; occasionally, a panelist is given a waiver, but not at this meeting.
In studies, dapagliflozin lowered hemoglobin A1c at the 5-mg and 10-mg once-daily doses and also was associated with decreases in fasting blood sugar, small decreases in systolic blood pressure (in hypertensive patients) and body weight, and LDL-cholesterol elevation. Adverse events associated with treatment include a higher rate of genital infections, volume depletion, and polyuria.
Since November 2012, dapagliflozin has been approved in the European Union, Australia, Mexico, New Zealand, Brazil, and Argentina. To date, over 35,000 people worldwide have been treated with dapagliflozin, according to Bristol-Myers Squibb. If approved, BMS and AstraZeneca plan to market dapagliflozin as Forxiga. A decision on approval is expected by Jan. 11, 2014.
AT AN FDA ADVISORY COMMITTEE MEETING

Telaprevir label changes include switch to twice-daily dosing
The label of the hepatitis C antiviral drug telaprevir has been changed to include new dosing recommendations and contraindications, described in a "Dear Healthcare Professional" letter issued by the manufacturer.
Telaprevir is a hepatitis C virus (HCV) NS3/4A protease inhibitor approved in 2011 for the treatment of genotype 1 chronic HCV infection in adults with compensated liver disease, in combination with peginterferon alfa and ribavirin. It is marketed as Incivek, by Vertex Pharmaceuticals.
The recommended dose is now 1,125 mg (three 375-mg tablets) taken orally twice a day, 10-14 hours apart with food that is not low fat, according to the Oct. 28 letter. "This change in dosing frequency was based on data from a clinical trial demonstrating similar sustained virologic response and safety profile between the twice-daily dosing and the every-8-hour dosing," the letter stated.
In addition, carbamazepine, phenobarbital, and phenytoin are now on the list of contraindicated drugs, based on a pharmacokinetics study that found coadministration of these drugs with telaprevir "has the potential to lower exposure and, thus, may result in a loss of efficacy" of telaprevir, according to the letter.
New twice-daily-dosing packaging will be available at the end of January 2014. Until then, the current packaging that contains six tablets per blister strip – with three blister sections containing two tablets each – can be used for the twice-daily recommendation. Two tablets from one blister section and one tablet from another section can be taken for the three-pill dose, administered twice a day, the letter pointed out.
The updated label is available at http://pi.vrtx.com/files/uspi_telaprevir.pdf.
Vertex can be contacted for questions at 877-824-4281. Serious adverse events associated with telaprevir should be reported to Vertex at that number, or to the Food and Drug Administration at 800-332-1088 or at www.fda.gov/medwatch.
The label of the hepatitis C antiviral drug telaprevir has been changed to include new dosing recommendations and contraindications, described in a "Dear Healthcare Professional" letter issued by the manufacturer.
Telaprevir is a hepatitis C virus (HCV) NS3/4A protease inhibitor approved in 2011 for the treatment of genotype 1 chronic HCV infection in adults with compensated liver disease, in combination with peginterferon alfa and ribavirin. It is marketed as Incivek, by Vertex Pharmaceuticals.
The recommended dose is now 1,125 mg (three 375-mg tablets) taken orally twice a day, 10-14 hours apart with food that is not low fat, according to the Oct. 28 letter. "This change in dosing frequency was based on data from a clinical trial demonstrating similar sustained virologic response and safety profile between the twice-daily dosing and the every-8-hour dosing," the letter stated.
In addition, carbamazepine, phenobarbital, and phenytoin are now on the list of contraindicated drugs, based on a pharmacokinetics study that found coadministration of these drugs with telaprevir "has the potential to lower exposure and, thus, may result in a loss of efficacy" of telaprevir, according to the letter.
New twice-daily-dosing packaging will be available at the end of January 2014. Until then, the current packaging that contains six tablets per blister strip – with three blister sections containing two tablets each – can be used for the twice-daily recommendation. Two tablets from one blister section and one tablet from another section can be taken for the three-pill dose, administered twice a day, the letter pointed out.
The updated label is available at http://pi.vrtx.com/files/uspi_telaprevir.pdf.
Vertex can be contacted for questions at 877-824-4281. Serious adverse events associated with telaprevir should be reported to Vertex at that number, or to the Food and Drug Administration at 800-332-1088 or at www.fda.gov/medwatch.
The label of the hepatitis C antiviral drug telaprevir has been changed to include new dosing recommendations and contraindications, described in a "Dear Healthcare Professional" letter issued by the manufacturer.
Telaprevir is a hepatitis C virus (HCV) NS3/4A protease inhibitor approved in 2011 for the treatment of genotype 1 chronic HCV infection in adults with compensated liver disease, in combination with peginterferon alfa and ribavirin. It is marketed as Incivek, by Vertex Pharmaceuticals.
The recommended dose is now 1,125 mg (three 375-mg tablets) taken orally twice a day, 10-14 hours apart with food that is not low fat, according to the Oct. 28 letter. "This change in dosing frequency was based on data from a clinical trial demonstrating similar sustained virologic response and safety profile between the twice-daily dosing and the every-8-hour dosing," the letter stated.
In addition, carbamazepine, phenobarbital, and phenytoin are now on the list of contraindicated drugs, based on a pharmacokinetics study that found coadministration of these drugs with telaprevir "has the potential to lower exposure and, thus, may result in a loss of efficacy" of telaprevir, according to the letter.
New twice-daily-dosing packaging will be available at the end of January 2014. Until then, the current packaging that contains six tablets per blister strip – with three blister sections containing two tablets each – can be used for the twice-daily recommendation. Two tablets from one blister section and one tablet from another section can be taken for the three-pill dose, administered twice a day, the letter pointed out.
The updated label is available at http://pi.vrtx.com/files/uspi_telaprevir.pdf.
Vertex can be contacted for questions at 877-824-4281. Serious adverse events associated with telaprevir should be reported to Vertex at that number, or to the Food and Drug Administration at 800-332-1088 or at www.fda.gov/medwatch.
Study identifies several genes associated with epileptic encephalopathies
WASHINGTON – Recently identified genes associated with severe epilepsies have shed some light on biological pathways involved in epileptogenesis, which could eventually lead to targeted treatments, Gemma Carvill, Ph.D., said at the annual meeting of the American Epilepsy Society.
The findings from a study of over 600 patients with epilepsy, which include the identification of several genes in patients linked to epileptic encephalopathies (EEs), also have diagnostic implications, according to Dr. Carvill, a postdoctoral fellow in the department of pediatrics and genetic medicine at the University of Washington, Seattle, and the lead author of the study.

EEs, the most severe types of epilepsy, primarily affect children and typically involve regression after normal development normally. They tend to be refractory to treatment.
This study is an expansion of a study she and her associates reported earlier this year (Nat. Genet. 2013;45:825-30), which involved the sequencing of 19 known EE genes and 46 candidate genes in 500 patients with EEs. They made a genetic diagnosis in 10% of these patients, identifying six new genes linked to EE, including two – CHD2 and SYNGAP1 – that accounted for about 1% of EE cases each.
Mutations in CHD2 do not directly affect the regulation of neurotransmitters at the synapse but affect the structure of DNA. "We think that changes the expression of genes downstream and affects the development of the human brain," which is a hypothesis she and her associates are researching, Dr. Carvill noted during a press briefing at the meeting.
"The vast majority of EEs are refractory to current treatment, so by identifying this new gene we’re identifying new biology, and we’re exploring new pathways and potentially new ways in which we can think about treating this disorder," she said.
This finding also may be important for the "broader spectrum of neurodevelopment disorders," Dr. Carvill said, noting that they have started screening patients with other types of neurodevelopment disorders for CHD2 mutations.
The expanded study included the addition of more patients with EE as well as patients with intellectual disabilities and genetic generalized epilepsy. Dr. Carvill and her colleagues also tested more target genes, including other genes linked to EE and genes associated with neurodevelopmental disorders.
Among the preliminary results of the expanded study are the identification of specific genes associated with "discrete phenotypes," she reported. These include mutations in the GRIN2A gene, which cause epilepsy aphasia syndromes. The investigators identified four families with mutations in the GRIN2A gene, which were associated with epilepsy aphasia, a type of epilepsy in which children tend to develop normally but then lose speech and have a certain EEG pattern during sleep, Dr. Carvill said.
This is a very specific phenotype and the finding has important diagnostic implications, she added, noting that a clinician who sees this particular phenotype in a patient can obtain a screening test for this gene.
They also identified patients with mutations in the GABRA1 (gamma-aminobutyric acid [GABA] A receptor, alpha 1) gene, which cause Dravet syndrome (also known as severe myoclonic epilepsy of infancy).
The study was funded by the National Institutes of Health. Dr. Carvill had no disclosures.
WASHINGTON – Recently identified genes associated with severe epilepsies have shed some light on biological pathways involved in epileptogenesis, which could eventually lead to targeted treatments, Gemma Carvill, Ph.D., said at the annual meeting of the American Epilepsy Society.
The findings from a study of over 600 patients with epilepsy, which include the identification of several genes in patients linked to epileptic encephalopathies (EEs), also have diagnostic implications, according to Dr. Carvill, a postdoctoral fellow in the department of pediatrics and genetic medicine at the University of Washington, Seattle, and the lead author of the study.
EEs, the most severe types of epilepsy, primarily affect children and typically involve regression after normal development normally. They tend to be refractory to treatment.
This study is an expansion of a study she and her associates reported earlier this year (Nat. Genet. 2013;45:825-30), which involved the sequencing of 19 known EE genes and 46 candidate genes in 500 patients with EEs. They made a genetic diagnosis in 10% of these patients, identifying six new genes linked to EE, including two – CHD2 and SYNGAP1 – that accounted for about 1% of EE cases each.
Mutations in CHD2 do not directly affect the regulation of neurotransmitters at the synapse but affect the structure of DNA. "We think that changes the expression of genes downstream and affects the development of the human brain," which is a hypothesis she and her associates are researching, Dr. Carvill noted during a press briefing at the meeting.
"The vast majority of EEs are refractory to current treatment, so by identifying this new gene we’re identifying new biology, and we’re exploring new pathways and potentially new ways in which we can think about treating this disorder," she said.
This finding also may be important for the "broader spectrum of neurodevelopment disorders," Dr. Carvill said, noting that they have started screening patients with other types of neurodevelopment disorders for CHD2 mutations.
The expanded study included the addition of more patients with EE as well as patients with intellectual disabilities and genetic generalized epilepsy. Dr. Carvill and her colleagues also tested more target genes, including other genes linked to EE and genes associated with neurodevelopmental disorders.
Among the preliminary results of the expanded study are the identification of specific genes associated with "discrete phenotypes," she reported. These include mutations in the GRIN2A gene, which cause epilepsy aphasia syndromes. The investigators identified four families with mutations in the GRIN2A gene, which were associated with epilepsy aphasia, a type of epilepsy in which children tend to develop normally but then lose speech and have a certain EEG pattern during sleep, Dr. Carvill said.
This is a very specific phenotype and the finding has important diagnostic implications, she added, noting that a clinician who sees this particular phenotype in a patient can obtain a screening test for this gene.
They also identified patients with mutations in the GABRA1 (gamma-aminobutyric acid [GABA] A receptor, alpha 1) gene, which cause Dravet syndrome (also known as severe myoclonic epilepsy of infancy).
The study was funded by the National Institutes of Health. Dr. Carvill had no disclosures.
WASHINGTON – Recently identified genes associated with severe epilepsies have shed some light on biological pathways involved in epileptogenesis, which could eventually lead to targeted treatments, Gemma Carvill, Ph.D., said at the annual meeting of the American Epilepsy Society.
The findings from a study of over 600 patients with epilepsy, which include the identification of several genes in patients linked to epileptic encephalopathies (EEs), also have diagnostic implications, according to Dr. Carvill, a postdoctoral fellow in the department of pediatrics and genetic medicine at the University of Washington, Seattle, and the lead author of the study.
EEs, the most severe types of epilepsy, primarily affect children and typically involve regression after normal development normally. They tend to be refractory to treatment.
This study is an expansion of a study she and her associates reported earlier this year (Nat. Genet. 2013;45:825-30), which involved the sequencing of 19 known EE genes and 46 candidate genes in 500 patients with EEs. They made a genetic diagnosis in 10% of these patients, identifying six new genes linked to EE, including two – CHD2 and SYNGAP1 – that accounted for about 1% of EE cases each.
Mutations in CHD2 do not directly affect the regulation of neurotransmitters at the synapse but affect the structure of DNA. "We think that changes the expression of genes downstream and affects the development of the human brain," which is a hypothesis she and her associates are researching, Dr. Carvill noted during a press briefing at the meeting.
"The vast majority of EEs are refractory to current treatment, so by identifying this new gene we’re identifying new biology, and we’re exploring new pathways and potentially new ways in which we can think about treating this disorder," she said.
This finding also may be important for the "broader spectrum of neurodevelopment disorders," Dr. Carvill said, noting that they have started screening patients with other types of neurodevelopment disorders for CHD2 mutations.
The expanded study included the addition of more patients with EE as well as patients with intellectual disabilities and genetic generalized epilepsy. Dr. Carvill and her colleagues also tested more target genes, including other genes linked to EE and genes associated with neurodevelopmental disorders.
Among the preliminary results of the expanded study are the identification of specific genes associated with "discrete phenotypes," she reported. These include mutations in the GRIN2A gene, which cause epilepsy aphasia syndromes. The investigators identified four families with mutations in the GRIN2A gene, which were associated with epilepsy aphasia, a type of epilepsy in which children tend to develop normally but then lose speech and have a certain EEG pattern during sleep, Dr. Carvill said.
This is a very specific phenotype and the finding has important diagnostic implications, she added, noting that a clinician who sees this particular phenotype in a patient can obtain a screening test for this gene.
They also identified patients with mutations in the GABRA1 (gamma-aminobutyric acid [GABA] A receptor, alpha 1) gene, which cause Dravet syndrome (also known as severe myoclonic epilepsy of infancy).
The study was funded by the National Institutes of Health. Dr. Carvill had no disclosures.
AT AES 2013

Study supports safety of epilepsy medication withdrawal prior to video-EEGs
WASHINGTON – Rapidly withdrawing antiepileptic medications in patients with epilepsy before being evaluated with video-electroencephalographic monitoring was safe, with no deaths or serious morbidity, in a prospective study, Dr. Syed Rizvi reported at the annual meeting of the American Epilepsy Society.
Video-electroencephalographic monitoring (VEM) also reliably helped to plan treatment, including surgery, and outcomes were "excellent," Dr. Rizvi of the department of neurology at the University of Saskatchewan, Saskatoon, Canada, said at a press briefing during the meeting.
The literature on whether it is safe and effective to rapidly withdraw antiepileptic drugs during VEM is sparse, he noted.
In 158 patients who had been admitted for VEM to the university’s epilepsy monitoring unit during a 5-year period, antiepileptic drugs (AEDs), except for phenobarbital, were rapidly withdrawn in patients who had no history of status epilepticus. The patients also underwent supervised overnight sleep deprivation. Rapid withdrawal was performed by titrating AEDs to half-dose on admission and then discontinuing them at 24 hours. In cases in which there was a history of status epilepticus or in patients taking phenobarbital or high doses of benzodiazepines, AEDs were tapered by 25% of the initial dose and ultimately discontinued.
The patients had had a mean of almost 11 seizures in the month before VEM, and 70% of patients were on three or more AEDs. Their mean age was 37 years, and they had had epilepsy for a mean of 16 years (mean age of onset was 20.5 years).
About 5% of patients had complications, which were mostly minor, including musculoskeletal pain secondary to seizure activity. During the month after testing, four (2.5%) patients were admitted to the emergency department for seizure clustering, but none were admitted to the intensive care unit. There were no deaths.
About 90% of the patients were diagnosed on the basis of VEM results, "a high diagnostic yield," Dr. Rizvi said. Habitual seizures were detected in 107 patients and psychogenic nonepileptic seizures were diagnosed in 36 patients; in 15 patients (9.5%), no events were recorded during VEM.
Almost 33% – 52 of the 158 – patients had epilepsy surgery, based on the results. Of those patients who had surgery, almost 90% achieved an excellent outcome (Engel Class I or II status) at 24 months, he said, noting that these patients were "virtually seizure free" or had nondebilitating seizures.
Video-EEG monitoring was "highly effective. It’s a useful adjunct in decision making. It can help in deciding which patients are suitable for surgery" in the "appropriate context," with supervision by a team of epileptologists, nurses, and EEG technologists, Dr. Rizvi said during the press briefing. He said that he and his coinvestigators hope to expand the study to include elderly and pediatric patients.
The study was not funded. Dr. Rizvi, a senior neurology resident at the university, had no disclosures.
WASHINGTON – Rapidly withdrawing antiepileptic medications in patients with epilepsy before being evaluated with video-electroencephalographic monitoring was safe, with no deaths or serious morbidity, in a prospective study, Dr. Syed Rizvi reported at the annual meeting of the American Epilepsy Society.
Video-electroencephalographic monitoring (VEM) also reliably helped to plan treatment, including surgery, and outcomes were "excellent," Dr. Rizvi of the department of neurology at the University of Saskatchewan, Saskatoon, Canada, said at a press briefing during the meeting.
The literature on whether it is safe and effective to rapidly withdraw antiepileptic drugs during VEM is sparse, he noted.
In 158 patients who had been admitted for VEM to the university’s epilepsy monitoring unit during a 5-year period, antiepileptic drugs (AEDs), except for phenobarbital, were rapidly withdrawn in patients who had no history of status epilepticus. The patients also underwent supervised overnight sleep deprivation. Rapid withdrawal was performed by titrating AEDs to half-dose on admission and then discontinuing them at 24 hours. In cases in which there was a history of status epilepticus or in patients taking phenobarbital or high doses of benzodiazepines, AEDs were tapered by 25% of the initial dose and ultimately discontinued.
The patients had had a mean of almost 11 seizures in the month before VEM, and 70% of patients were on three or more AEDs. Their mean age was 37 years, and they had had epilepsy for a mean of 16 years (mean age of onset was 20.5 years).
About 5% of patients had complications, which were mostly minor, including musculoskeletal pain secondary to seizure activity. During the month after testing, four (2.5%) patients were admitted to the emergency department for seizure clustering, but none were admitted to the intensive care unit. There were no deaths.
About 90% of the patients were diagnosed on the basis of VEM results, "a high diagnostic yield," Dr. Rizvi said. Habitual seizures were detected in 107 patients and psychogenic nonepileptic seizures were diagnosed in 36 patients; in 15 patients (9.5%), no events were recorded during VEM.
Almost 33% – 52 of the 158 – patients had epilepsy surgery, based on the results. Of those patients who had surgery, almost 90% achieved an excellent outcome (Engel Class I or II status) at 24 months, he said, noting that these patients were "virtually seizure free" or had nondebilitating seizures.
Video-EEG monitoring was "highly effective. It’s a useful adjunct in decision making. It can help in deciding which patients are suitable for surgery" in the "appropriate context," with supervision by a team of epileptologists, nurses, and EEG technologists, Dr. Rizvi said during the press briefing. He said that he and his coinvestigators hope to expand the study to include elderly and pediatric patients.
The study was not funded. Dr. Rizvi, a senior neurology resident at the university, had no disclosures.
WASHINGTON – Rapidly withdrawing antiepileptic medications in patients with epilepsy before being evaluated with video-electroencephalographic monitoring was safe, with no deaths or serious morbidity, in a prospective study, Dr. Syed Rizvi reported at the annual meeting of the American Epilepsy Society.
Video-electroencephalographic monitoring (VEM) also reliably helped to plan treatment, including surgery, and outcomes were "excellent," Dr. Rizvi of the department of neurology at the University of Saskatchewan, Saskatoon, Canada, said at a press briefing during the meeting.
The literature on whether it is safe and effective to rapidly withdraw antiepileptic drugs during VEM is sparse, he noted.
In 158 patients who had been admitted for VEM to the university’s epilepsy monitoring unit during a 5-year period, antiepileptic drugs (AEDs), except for phenobarbital, were rapidly withdrawn in patients who had no history of status epilepticus. The patients also underwent supervised overnight sleep deprivation. Rapid withdrawal was performed by titrating AEDs to half-dose on admission and then discontinuing them at 24 hours. In cases in which there was a history of status epilepticus or in patients taking phenobarbital or high doses of benzodiazepines, AEDs were tapered by 25% of the initial dose and ultimately discontinued.
The patients had had a mean of almost 11 seizures in the month before VEM, and 70% of patients were on three or more AEDs. Their mean age was 37 years, and they had had epilepsy for a mean of 16 years (mean age of onset was 20.5 years).
About 5% of patients had complications, which were mostly minor, including musculoskeletal pain secondary to seizure activity. During the month after testing, four (2.5%) patients were admitted to the emergency department for seizure clustering, but none were admitted to the intensive care unit. There were no deaths.
About 90% of the patients were diagnosed on the basis of VEM results, "a high diagnostic yield," Dr. Rizvi said. Habitual seizures were detected in 107 patients and psychogenic nonepileptic seizures were diagnosed in 36 patients; in 15 patients (9.5%), no events were recorded during VEM.
Almost 33% – 52 of the 158 – patients had epilepsy surgery, based on the results. Of those patients who had surgery, almost 90% achieved an excellent outcome (Engel Class I or II status) at 24 months, he said, noting that these patients were "virtually seizure free" or had nondebilitating seizures.
Video-EEG monitoring was "highly effective. It’s a useful adjunct in decision making. It can help in deciding which patients are suitable for surgery" in the "appropriate context," with supervision by a team of epileptologists, nurses, and EEG technologists, Dr. Rizvi said during the press briefing. He said that he and his coinvestigators hope to expand the study to include elderly and pediatric patients.
The study was not funded. Dr. Rizvi, a senior neurology resident at the university, had no disclosures.
AT AES 2013
Major finding: About 5% of patients had complications during video-electroencephalographic monitoring, and about 90% of the patients were diagnosed on the basis of the results.
Data source: A prospective study of 158 patients admitted during a 5-year period.
Disclosures: The study was not funded. The presenter had no disclosures.
Survey identifies need for COPD awareness, discourse
Physicians may miss opportunities to discuss chronic obstructive pulmonary disease with their patients, including former and current smokers with symptoms, according to survey results reported by the National Heart Lung and Blood Institute.
The survey found that the proportion of current smokers who had discussed COPD symptoms with their physicians had increased from 42% in 2009 to 67% in 2013, according to the report. However, during this period, the proportion of former smokers who had talked to their doctors about these symptoms dropped slightly, from 78% in 2009 to 77% in 2013.

Although 74% of current smokers said they had talked to their physicians about their chronic cough, wheezing, or being too short of breath to do normal activities, 26% said no to this question.
Of those who were surveyed who had COPD symptoms and said they had talked to their doctors about having a chronic cough, wheezing, or being too short of breath to do normal activities, only 18% said their doctors told them they might have COPD. Of this group, only 35% said the doctors talked to them about their past or current smoking history.
According to the NHLBI report, the results suggest that awareness of COPD may influence why patients do not talk to their physicians about COPD symptoms: Among those surveyed who were symptomatic and had not talked to their doctors about symptoms, various reasons were mentioned for not doing so, including: "they did not think of it" (cited by 27%), they had the problem for years (20%), "I don’t want another ‘quit smoking’ message" (16%), and the problem "will just go away in time" (16%).
"Regardless of positive developments, the challenge remains that more than 1 in 3 Americans do not know what COPD is or how it affects them – and less than half understand that COPD can be treated," James Kiley, Ph.D., director of the NHLBI Division of Lung Diseases, said in the statement.
http://www.nhlbi.nih.gov/news/press-releases/2013/nih-survey-identifies-barriers-to-effective-patient-provider-dialogue-about-copd.html
The web-based survey is conducted every year by the communications company Porter Novelli, for NHLBI’s "COPD Learn More Breathe Better" campaign. The campaign promotes national awareness about COPD, directed at people with COPD, those at risk of COPD, and health care providers, particularly primary care providers, according to the NHLBI.
The 2013 survey sampled almost 4,500 adults in the United States, with a 66% response rate. Up to half the estimated 24 million people in the United States with COPD have not been diagnosed, and in 2010, COPD replaced stroke as the third leading cause of death in the United States, according to the NHLBI.
Physicians may miss opportunities to discuss chronic obstructive pulmonary disease with their patients, including former and current smokers with symptoms, according to survey results reported by the National Heart Lung and Blood Institute.
The survey found that the proportion of current smokers who had discussed COPD symptoms with their physicians had increased from 42% in 2009 to 67% in 2013, according to the report. However, during this period, the proportion of former smokers who had talked to their doctors about these symptoms dropped slightly, from 78% in 2009 to 77% in 2013.
Although 74% of current smokers said they had talked to their physicians about their chronic cough, wheezing, or being too short of breath to do normal activities, 26% said no to this question.
Of those who were surveyed who had COPD symptoms and said they had talked to their doctors about having a chronic cough, wheezing, or being too short of breath to do normal activities, only 18% said their doctors told them they might have COPD. Of this group, only 35% said the doctors talked to them about their past or current smoking history.
According to the NHLBI report, the results suggest that awareness of COPD may influence why patients do not talk to their physicians about COPD symptoms: Among those surveyed who were symptomatic and had not talked to their doctors about symptoms, various reasons were mentioned for not doing so, including: "they did not think of it" (cited by 27%), they had the problem for years (20%), "I don’t want another ‘quit smoking’ message" (16%), and the problem "will just go away in time" (16%).
"Regardless of positive developments, the challenge remains that more than 1 in 3 Americans do not know what COPD is or how it affects them – and less than half understand that COPD can be treated," James Kiley, Ph.D., director of the NHLBI Division of Lung Diseases, said in the statement.
http://www.nhlbi.nih.gov/news/press-releases/2013/nih-survey-identifies-barriers-to-effective-patient-provider-dialogue-about-copd.html
The web-based survey is conducted every year by the communications company Porter Novelli, for NHLBI’s "COPD Learn More Breathe Better" campaign. The campaign promotes national awareness about COPD, directed at people with COPD, those at risk of COPD, and health care providers, particularly primary care providers, according to the NHLBI.
The 2013 survey sampled almost 4,500 adults in the United States, with a 66% response rate. Up to half the estimated 24 million people in the United States with COPD have not been diagnosed, and in 2010, COPD replaced stroke as the third leading cause of death in the United States, according to the NHLBI.
Physicians may miss opportunities to discuss chronic obstructive pulmonary disease with their patients, including former and current smokers with symptoms, according to survey results reported by the National Heart Lung and Blood Institute.
The survey found that the proportion of current smokers who had discussed COPD symptoms with their physicians had increased from 42% in 2009 to 67% in 2013, according to the report. However, during this period, the proportion of former smokers who had talked to their doctors about these symptoms dropped slightly, from 78% in 2009 to 77% in 2013.
Although 74% of current smokers said they had talked to their physicians about their chronic cough, wheezing, or being too short of breath to do normal activities, 26% said no to this question.
Of those who were surveyed who had COPD symptoms and said they had talked to their doctors about having a chronic cough, wheezing, or being too short of breath to do normal activities, only 18% said their doctors told them they might have COPD. Of this group, only 35% said the doctors talked to them about their past or current smoking history.
According to the NHLBI report, the results suggest that awareness of COPD may influence why patients do not talk to their physicians about COPD symptoms: Among those surveyed who were symptomatic and had not talked to their doctors about symptoms, various reasons were mentioned for not doing so, including: "they did not think of it" (cited by 27%), they had the problem for years (20%), "I don’t want another ‘quit smoking’ message" (16%), and the problem "will just go away in time" (16%).
"Regardless of positive developments, the challenge remains that more than 1 in 3 Americans do not know what COPD is or how it affects them – and less than half understand that COPD can be treated," James Kiley, Ph.D., director of the NHLBI Division of Lung Diseases, said in the statement.
http://www.nhlbi.nih.gov/news/press-releases/2013/nih-survey-identifies-barriers-to-effective-patient-provider-dialogue-about-copd.html
The web-based survey is conducted every year by the communications company Porter Novelli, for NHLBI’s "COPD Learn More Breathe Better" campaign. The campaign promotes national awareness about COPD, directed at people with COPD, those at risk of COPD, and health care providers, particularly primary care providers, according to the NHLBI.
The 2013 survey sampled almost 4,500 adults in the United States, with a 66% response rate. Up to half the estimated 24 million people in the United States with COPD have not been diagnosed, and in 2010, COPD replaced stroke as the third leading cause of death in the United States, according to the NHLBI.
