User login
FDA approves ezetimibe-statin combination pill
A fixed-dose combination of 10 mg of ezetimibe with four different doses of atorvastatin has been approved by the Food and Drug Administration, the manufacturer, Merck, announced on May 3.
Ezetimibe, a selective inhibitor of the absorption of intestinal cholesterol and related phytosterols, was approved in 2002 and is marketed by Merck as Zetia. Atorvastatin, an HMG-CoA reductase inhibitor approved in 2006, is marketed as Lipitor by Pfizer and is available in generic formulations.
The combination product will be marketed under the trade name Liptruzet, for the treatment of elevated low-density lipoprotein (LDL) cholesterol in patients with primary or mixed hyperlipidemia as adjunctive therapy to diet, when diet alone is not enough, according to the company statement announcing the approval.
The Limitations of Use section of the prescribing information includes the statement that "no incremental benefit of Liptruzet on cardiovascular morbidity and mortality over and above that demonstrated for atorvastatin has been established." This section also states that Liptruzet has not been studied in Fredrickson type I, III, IV, and V dyslipidemias.
The approval comes after two Complete Response Letters from the Food and Drug Administration, requesting more data from the company.
Liptruzet is available in tablets that contain 10 mg of ezetimibe with 10, 20, 40, or 80 mg of atorvastatin (as Liptruzet 10/10, 10/20, 10/40, or 10/80, according to the release). The dosage range is from 10/10 mg a day to 10/80 mg a day.
The company’s release and the prescribing information summarize the results of different trials, including a 12-week study comparing different Liptruzet doses (administered as ezetimibe and atorvastatin) to atorvastatin, in 628 patients with hyperlipidemia. The pooled results of the different doses indicated significant reductions in total cholesterol, LDL cholesterol, apolipoprotein B, triglycerides, and non-HDL cholesterol, as well as significant increases in HDL cholesterol, compared with the results for all atorvastatin doses (10-80 mg), according to the labeling. The findings after another 48 weeks in those who continued treatment were consistent with the 12-week results.

The Merck press release also includes the statement that "the clinical impact of comparative differences in lipid changes between Liptruzet (ezetimibe and atorvastatin) and atorvastatin is not known."
Since the results of the ENHANCE (Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression) study were released in 2008, the value of adding ezetimibe has been questioned. In that controversial study, the combination of simvastatin and ezetimibe (Vytorin) was more potent than simvastatin alone in reducing LDL cholesterol, but the combination showed no advantage over the statin alone in reducing the progression of carotid atherosclerosis over 2 years.
"We are increasingly recognizing that drugs that reduce LDL may not always reduce patient risk, and the effect of ezetimibe alone or in combination with other drugs on patient outcomes remains unknown," Dr. Harlan Krumholz, the Harold H. Hines Jr. professor of Medicine (Cardiology) at Yale University, New Haven, Conn., said in an interview. "As a result, the use of ezetimibe should be reserved for circumstances where the patient cannot tolerate statins, and only in cases where patients are fully informed that the effects of the drug on patient outcomes are not yet known," he added.
The Merck statement said that the product will be available to wholesalers during the week of May 6. The catalog price for one tablet, at all doses, is $5.50, according to a Merck spokesperson. Merck also markets a fixed-dose product combining ezetimibe with simvastatin as Vytorin.
The prescribing information is available here.
Dr. Krumholz had no disclosures.
A fixed-dose combination of 10 mg of ezetimibe with four different doses of atorvastatin has been approved by the Food and Drug Administration, the manufacturer, Merck, announced on May 3.
Ezetimibe, a selective inhibitor of the absorption of intestinal cholesterol and related phytosterols, was approved in 2002 and is marketed by Merck as Zetia. Atorvastatin, an HMG-CoA reductase inhibitor approved in 2006, is marketed as Lipitor by Pfizer and is available in generic formulations.
The combination product will be marketed under the trade name Liptruzet, for the treatment of elevated low-density lipoprotein (LDL) cholesterol in patients with primary or mixed hyperlipidemia as adjunctive therapy to diet, when diet alone is not enough, according to the company statement announcing the approval.
The Limitations of Use section of the prescribing information includes the statement that "no incremental benefit of Liptruzet on cardiovascular morbidity and mortality over and above that demonstrated for atorvastatin has been established." This section also states that Liptruzet has not been studied in Fredrickson type I, III, IV, and V dyslipidemias.
The approval comes after two Complete Response Letters from the Food and Drug Administration, requesting more data from the company.
Liptruzet is available in tablets that contain 10 mg of ezetimibe with 10, 20, 40, or 80 mg of atorvastatin (as Liptruzet 10/10, 10/20, 10/40, or 10/80, according to the release). The dosage range is from 10/10 mg a day to 10/80 mg a day.
The company’s release and the prescribing information summarize the results of different trials, including a 12-week study comparing different Liptruzet doses (administered as ezetimibe and atorvastatin) to atorvastatin, in 628 patients with hyperlipidemia. The pooled results of the different doses indicated significant reductions in total cholesterol, LDL cholesterol, apolipoprotein B, triglycerides, and non-HDL cholesterol, as well as significant increases in HDL cholesterol, compared with the results for all atorvastatin doses (10-80 mg), according to the labeling. The findings after another 48 weeks in those who continued treatment were consistent with the 12-week results.
The Merck press release also includes the statement that "the clinical impact of comparative differences in lipid changes between Liptruzet (ezetimibe and atorvastatin) and atorvastatin is not known."
Since the results of the ENHANCE (Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression) study were released in 2008, the value of adding ezetimibe has been questioned. In that controversial study, the combination of simvastatin and ezetimibe (Vytorin) was more potent than simvastatin alone in reducing LDL cholesterol, but the combination showed no advantage over the statin alone in reducing the progression of carotid atherosclerosis over 2 years.
"We are increasingly recognizing that drugs that reduce LDL may not always reduce patient risk, and the effect of ezetimibe alone or in combination with other drugs on patient outcomes remains unknown," Dr. Harlan Krumholz, the Harold H. Hines Jr. professor of Medicine (Cardiology) at Yale University, New Haven, Conn., said in an interview. "As a result, the use of ezetimibe should be reserved for circumstances where the patient cannot tolerate statins, and only in cases where patients are fully informed that the effects of the drug on patient outcomes are not yet known," he added.
The Merck statement said that the product will be available to wholesalers during the week of May 6. The catalog price for one tablet, at all doses, is $5.50, according to a Merck spokesperson. Merck also markets a fixed-dose product combining ezetimibe with simvastatin as Vytorin.
The prescribing information is available here.
Dr. Krumholz had no disclosures.
A fixed-dose combination of 10 mg of ezetimibe with four different doses of atorvastatin has been approved by the Food and Drug Administration, the manufacturer, Merck, announced on May 3.
Ezetimibe, a selective inhibitor of the absorption of intestinal cholesterol and related phytosterols, was approved in 2002 and is marketed by Merck as Zetia. Atorvastatin, an HMG-CoA reductase inhibitor approved in 2006, is marketed as Lipitor by Pfizer and is available in generic formulations.
The combination product will be marketed under the trade name Liptruzet, for the treatment of elevated low-density lipoprotein (LDL) cholesterol in patients with primary or mixed hyperlipidemia as adjunctive therapy to diet, when diet alone is not enough, according to the company statement announcing the approval.
The Limitations of Use section of the prescribing information includes the statement that "no incremental benefit of Liptruzet on cardiovascular morbidity and mortality over and above that demonstrated for atorvastatin has been established." This section also states that Liptruzet has not been studied in Fredrickson type I, III, IV, and V dyslipidemias.
The approval comes after two Complete Response Letters from the Food and Drug Administration, requesting more data from the company.
Liptruzet is available in tablets that contain 10 mg of ezetimibe with 10, 20, 40, or 80 mg of atorvastatin (as Liptruzet 10/10, 10/20, 10/40, or 10/80, according to the release). The dosage range is from 10/10 mg a day to 10/80 mg a day.
The company’s release and the prescribing information summarize the results of different trials, including a 12-week study comparing different Liptruzet doses (administered as ezetimibe and atorvastatin) to atorvastatin, in 628 patients with hyperlipidemia. The pooled results of the different doses indicated significant reductions in total cholesterol, LDL cholesterol, apolipoprotein B, triglycerides, and non-HDL cholesterol, as well as significant increases in HDL cholesterol, compared with the results for all atorvastatin doses (10-80 mg), according to the labeling. The findings after another 48 weeks in those who continued treatment were consistent with the 12-week results.
The Merck press release also includes the statement that "the clinical impact of comparative differences in lipid changes between Liptruzet (ezetimibe and atorvastatin) and atorvastatin is not known."
Since the results of the ENHANCE (Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression) study were released in 2008, the value of adding ezetimibe has been questioned. In that controversial study, the combination of simvastatin and ezetimibe (Vytorin) was more potent than simvastatin alone in reducing LDL cholesterol, but the combination showed no advantage over the statin alone in reducing the progression of carotid atherosclerosis over 2 years.
"We are increasingly recognizing that drugs that reduce LDL may not always reduce patient risk, and the effect of ezetimibe alone or in combination with other drugs on patient outcomes remains unknown," Dr. Harlan Krumholz, the Harold H. Hines Jr. professor of Medicine (Cardiology) at Yale University, New Haven, Conn., said in an interview. "As a result, the use of ezetimibe should be reserved for circumstances where the patient cannot tolerate statins, and only in cases where patients are fully informed that the effects of the drug on patient outcomes are not yet known," he added.
The Merck statement said that the product will be available to wholesalers during the week of May 6. The catalog price for one tablet, at all doses, is $5.50, according to a Merck spokesperson. Merck also markets a fixed-dose product combining ezetimibe with simvastatin as Vytorin.
The prescribing information is available here.
Dr. Krumholz had no disclosures.
FDA advisory panel nixes approval of drug-device for liver metastases
SILVER SPRING, MD. – The risks outweigh any possible benefits of treatment with a drug-device combination that delivers melphalan directly to the livers of patients with liver metastases from ocular melanoma, a Food and Drug Administration advisory panel concluded in a 16-0 vote.
At a meeting of the FDA’s Oncologic Drugs Advisory Committee, several panelists said that the treatment was promising but should remain investigational, given its marked toxicity and lack of effect on overall survival.
The Melblez Kit is a combination of melphalan and the Delcath Hepatic Delivery System, which includes two catheters and an extracorporeal hemofiltration component. The catheter is used to administer high doses of the chemotherapy drug directly to the liver via the hepatic artery, and the hemofiltration component lowers the drug level before the blood is returned to the systemic circulation, according to the manufacturer, Delcath Systems. Patients are hospitalized for about 4 days for the procedure, which takes about 3 hours and is performed at 4-week intervals under general anesthesia.
There are no FDA-approved treatments for patients with unresectable metastatic ocular melanoma to the liver, which is the indication under FDA review.
Treatment with the kit was associated with antitumor activity, but it also was associated with fatal and life-threatening adverse reactions. There was a trend towards a detrimental effect on survival, and the risk evaluation and mitigation strategy (REMS) proposed by the company to address those risks "will not improve the observed benefit-risk profile," Dr. Geoffrey Kim, a medical officer in the FDA’s office of hematology and oncology products, told the panel.
In an open-label, randomized multicenter phase-III study conducted between 2006 and 2010 in the United States, the device was used to treat 44 patients. Their outcomes were compared with those of 49 matched patients given the best alternative care (BAC). All patients had surgically unresectable hepatic-dominant metastatic ocular or cutaneous melanoma (89% had ocular melanoma, and almost half were treated at the National Cancer Institute). Subjects were treated until their hepatic disease progressed. The dose administered with the Melblez Kit was 3.0 mg/kg for a median of three treatment cycles and a median of 120 days; best alternative care included systemic chemotherapy in 49% of patients and intrahepatic chemotherapy in 22%.
The primary end point, median hepatic progression-free survival (hPFS) was 7 months among those on the device, compared with 1.6 months among those on BAC, a statistically significant difference that represented a 61% reduction in risk (hazard ratio, 0.39), according to the company. The median overall PFS was 4.8 months among those treated with the device, compared with 1.6 months among those on BAC, also a statistically significant difference.
Overall survival was comparable: 9.8 months in the device-treated group and 9.9 months in those on best alternative care. Further, almost 80% of patients in the Melblez Kit arm had a serious adverse event and almost 70% had a grade-4 adverse event. With best alternative care, the rate of serious adverse events was 16% and the rate of grade-4 events was 2%. No patients on best alternative care died because of an adverse event. Three patients treated with the drug-device died from adverse events.
The adverse reactions in a combined population of 121 patients in the phase-III and phase-II studies and in 28 patients in the BAC arm who crossed over to treatment with the device included toxic deaths in 7% (including cases of hepatic failure, streptococcal sepsis, and GI hemorrhage), cerebral infarction in 4%, MI in 2%, and grade-4 bone marrow suppression with a median time to recovery of more than 1 week in more than 70%. About half had to be rehospitalized for an adverse event.
For a cancer treatment, this safety profile "is unprecedented, in terms of the toxicity," Dr. Richard Pazdur, director of the FDA’s office of hematology and oncology products, remarked.
After the vote, panel member Dr. Louis Diehl, professor of medicine at Duke University, Durham, N.C., said that progression-free survival is valued in studies as an indicator of improved quality of life and it can be an early marker of increased survival. "Unfortunately, this treatment has an increase in morbidity and an increase in mortality and I can’t see from the survival curve that it will ever translate into an improvement in survival."
Delcath did not issue a response after the panel’s vote.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may occasionally be given a waiver. At this meeting, two panelists who had expertise in the topic were given waivers (one panelist is the principal investigator in a study of a competing device and the other works at a medical center where a study of a competing device is being conducted).
About 2,000 cases of ocular melanoma are diagnosed annually in the United States and about 50% metastasize, most often to the liver, according to Delcath. In Europe and Australia, the company markets the device for a broad range of liver metastases, not just those caused by ocular melanoma, according to the company.
SILVER SPRING, MD. – The risks outweigh any possible benefits of treatment with a drug-device combination that delivers melphalan directly to the livers of patients with liver metastases from ocular melanoma, a Food and Drug Administration advisory panel concluded in a 16-0 vote.
At a meeting of the FDA’s Oncologic Drugs Advisory Committee, several panelists said that the treatment was promising but should remain investigational, given its marked toxicity and lack of effect on overall survival.
The Melblez Kit is a combination of melphalan and the Delcath Hepatic Delivery System, which includes two catheters and an extracorporeal hemofiltration component. The catheter is used to administer high doses of the chemotherapy drug directly to the liver via the hepatic artery, and the hemofiltration component lowers the drug level before the blood is returned to the systemic circulation, according to the manufacturer, Delcath Systems. Patients are hospitalized for about 4 days for the procedure, which takes about 3 hours and is performed at 4-week intervals under general anesthesia.
There are no FDA-approved treatments for patients with unresectable metastatic ocular melanoma to the liver, which is the indication under FDA review.
Treatment with the kit was associated with antitumor activity, but it also was associated with fatal and life-threatening adverse reactions. There was a trend towards a detrimental effect on survival, and the risk evaluation and mitigation strategy (REMS) proposed by the company to address those risks "will not improve the observed benefit-risk profile," Dr. Geoffrey Kim, a medical officer in the FDA’s office of hematology and oncology products, told the panel.
In an open-label, randomized multicenter phase-III study conducted between 2006 and 2010 in the United States, the device was used to treat 44 patients. Their outcomes were compared with those of 49 matched patients given the best alternative care (BAC). All patients had surgically unresectable hepatic-dominant metastatic ocular or cutaneous melanoma (89% had ocular melanoma, and almost half were treated at the National Cancer Institute). Subjects were treated until their hepatic disease progressed. The dose administered with the Melblez Kit was 3.0 mg/kg for a median of three treatment cycles and a median of 120 days; best alternative care included systemic chemotherapy in 49% of patients and intrahepatic chemotherapy in 22%.
The primary end point, median hepatic progression-free survival (hPFS) was 7 months among those on the device, compared with 1.6 months among those on BAC, a statistically significant difference that represented a 61% reduction in risk (hazard ratio, 0.39), according to the company. The median overall PFS was 4.8 months among those treated with the device, compared with 1.6 months among those on BAC, also a statistically significant difference.
Overall survival was comparable: 9.8 months in the device-treated group and 9.9 months in those on best alternative care. Further, almost 80% of patients in the Melblez Kit arm had a serious adverse event and almost 70% had a grade-4 adverse event. With best alternative care, the rate of serious adverse events was 16% and the rate of grade-4 events was 2%. No patients on best alternative care died because of an adverse event. Three patients treated with the drug-device died from adverse events.
The adverse reactions in a combined population of 121 patients in the phase-III and phase-II studies and in 28 patients in the BAC arm who crossed over to treatment with the device included toxic deaths in 7% (including cases of hepatic failure, streptococcal sepsis, and GI hemorrhage), cerebral infarction in 4%, MI in 2%, and grade-4 bone marrow suppression with a median time to recovery of more than 1 week in more than 70%. About half had to be rehospitalized for an adverse event.
For a cancer treatment, this safety profile "is unprecedented, in terms of the toxicity," Dr. Richard Pazdur, director of the FDA’s office of hematology and oncology products, remarked.
After the vote, panel member Dr. Louis Diehl, professor of medicine at Duke University, Durham, N.C., said that progression-free survival is valued in studies as an indicator of improved quality of life and it can be an early marker of increased survival. "Unfortunately, this treatment has an increase in morbidity and an increase in mortality and I can’t see from the survival curve that it will ever translate into an improvement in survival."
Delcath did not issue a response after the panel’s vote.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may occasionally be given a waiver. At this meeting, two panelists who had expertise in the topic were given waivers (one panelist is the principal investigator in a study of a competing device and the other works at a medical center where a study of a competing device is being conducted).
About 2,000 cases of ocular melanoma are diagnosed annually in the United States and about 50% metastasize, most often to the liver, according to Delcath. In Europe and Australia, the company markets the device for a broad range of liver metastases, not just those caused by ocular melanoma, according to the company.
SILVER SPRING, MD. – The risks outweigh any possible benefits of treatment with a drug-device combination that delivers melphalan directly to the livers of patients with liver metastases from ocular melanoma, a Food and Drug Administration advisory panel concluded in a 16-0 vote.
At a meeting of the FDA’s Oncologic Drugs Advisory Committee, several panelists said that the treatment was promising but should remain investigational, given its marked toxicity and lack of effect on overall survival.
The Melblez Kit is a combination of melphalan and the Delcath Hepatic Delivery System, which includes two catheters and an extracorporeal hemofiltration component. The catheter is used to administer high doses of the chemotherapy drug directly to the liver via the hepatic artery, and the hemofiltration component lowers the drug level before the blood is returned to the systemic circulation, according to the manufacturer, Delcath Systems. Patients are hospitalized for about 4 days for the procedure, which takes about 3 hours and is performed at 4-week intervals under general anesthesia.
There are no FDA-approved treatments for patients with unresectable metastatic ocular melanoma to the liver, which is the indication under FDA review.
Treatment with the kit was associated with antitumor activity, but it also was associated with fatal and life-threatening adverse reactions. There was a trend towards a detrimental effect on survival, and the risk evaluation and mitigation strategy (REMS) proposed by the company to address those risks "will not improve the observed benefit-risk profile," Dr. Geoffrey Kim, a medical officer in the FDA’s office of hematology and oncology products, told the panel.
In an open-label, randomized multicenter phase-III study conducted between 2006 and 2010 in the United States, the device was used to treat 44 patients. Their outcomes were compared with those of 49 matched patients given the best alternative care (BAC). All patients had surgically unresectable hepatic-dominant metastatic ocular or cutaneous melanoma (89% had ocular melanoma, and almost half were treated at the National Cancer Institute). Subjects were treated until their hepatic disease progressed. The dose administered with the Melblez Kit was 3.0 mg/kg for a median of three treatment cycles and a median of 120 days; best alternative care included systemic chemotherapy in 49% of patients and intrahepatic chemotherapy in 22%.
The primary end point, median hepatic progression-free survival (hPFS) was 7 months among those on the device, compared with 1.6 months among those on BAC, a statistically significant difference that represented a 61% reduction in risk (hazard ratio, 0.39), according to the company. The median overall PFS was 4.8 months among those treated with the device, compared with 1.6 months among those on BAC, also a statistically significant difference.
Overall survival was comparable: 9.8 months in the device-treated group and 9.9 months in those on best alternative care. Further, almost 80% of patients in the Melblez Kit arm had a serious adverse event and almost 70% had a grade-4 adverse event. With best alternative care, the rate of serious adverse events was 16% and the rate of grade-4 events was 2%. No patients on best alternative care died because of an adverse event. Three patients treated with the drug-device died from adverse events.
The adverse reactions in a combined population of 121 patients in the phase-III and phase-II studies and in 28 patients in the BAC arm who crossed over to treatment with the device included toxic deaths in 7% (including cases of hepatic failure, streptococcal sepsis, and GI hemorrhage), cerebral infarction in 4%, MI in 2%, and grade-4 bone marrow suppression with a median time to recovery of more than 1 week in more than 70%. About half had to be rehospitalized for an adverse event.
For a cancer treatment, this safety profile "is unprecedented, in terms of the toxicity," Dr. Richard Pazdur, director of the FDA’s office of hematology and oncology products, remarked.
After the vote, panel member Dr. Louis Diehl, professor of medicine at Duke University, Durham, N.C., said that progression-free survival is valued in studies as an indicator of improved quality of life and it can be an early marker of increased survival. "Unfortunately, this treatment has an increase in morbidity and an increase in mortality and I can’t see from the survival curve that it will ever translate into an improvement in survival."
Delcath did not issue a response after the panel’s vote.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting, although a panelist may occasionally be given a waiver. At this meeting, two panelists who had expertise in the topic were given waivers (one panelist is the principal investigator in a study of a competing device and the other works at a medical center where a study of a competing device is being conducted).
About 2,000 cases of ocular melanoma are diagnosed annually in the United States and about 50% metastasize, most often to the liver, according to Delcath. In Europe and Australia, the company markets the device for a broad range of liver metastases, not just those caused by ocular melanoma, according to the company.
AT AN FDA ADVISORY PANEL MEETING
FDA warns against using antiseizure drugs in pregnant migraine patients
Valproate sodium and related drugs should never be used to prevent migraines in pregnant women, according to a Food and Drug Administration warning. The safety advisory cites the final results of a study indicating that prenatal exposure to these drugs "can cause decreased IQ scores" in children whose mothers took them during pregnancy.
The pregnancy category for valproate for the migraine indication will be changed from a "D" – designated for drugs where the potential benefit for a pregnant woman might be acceptable despite its potential risks – to a category "X," designating that the risk of the drug in a pregnant woman "clearly outweighs any possible benefit of the drug," according to the advisory, issued May 6 by the FDA.
Valproate products include valproate sodium (Depacon), divalproex sodium (Depakote, Depakote CP, and Depakote ER), valproic acid (Depakene and Stavzor), and generic versions of these products. They also are used to treat epilepsy and manic episodes associated with bipolar disorder, and for these indications, they will remain in category "D" and "should only be prescribed if other medications are not effective in treating the condition or are otherwise unacceptable." Women who can become pregnant should not use valproate unless it is essential to managing their medical condition, the FDA states.
"Valproate medications should never be used in pregnant women for the prevention of migraine headaches because we have even more data now that show the risks to the children outweigh any treatment benefits for this use," Dr. Russell Katz, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in a May 6 statement issued by the FDA to announce the advisory.
The data cited by the FDA are the final results of the Neurodevelopmental Effects of Antiepileptic Drugs study, published in March 2013, which found that at age 6, children whose mothers had taken valproate products while pregnant had lower IQs, compared with those whose mothers took other antiepileptic drugs. In the study, the mean IQ among the children exposed to valproate in utero was 97, compared with a mean of 105 among those exposed to carbamazepine, 108 among those exposed to lamotrigine, and 108 among those exposed to phenytoin (Lancet Neurol. 2013;12:244-52).
In June 2011, the FDA issued an alert when interim results of this study found that at age 3 years, children exposed to valproate had lower scores on cognitive tests, information that was added to the labels at that time.
The FDA notes that the results of the study are similar to other epidemiologic studies that have associated in utero exposure to valproate to lower IQ scores, when compared with children exposed to another AED or no AED in utero.
Since women in the study were taking antiepileptic drugs throughout pregnancy, it is unclear whether the effects on IQ are related to the timing or duration of prenatal valproate exposure, so "exposure to valproate at any time during pregnancy should be considered to have the potential to result in decreased IQ in children," the advisory says.
The prescribing information for valproate medications already include a boxed warning about the fetal risks, especially neural tube defects and other major malformations, associated with prenatal exposure.
Adverse events associated with valproate products should be reported to the FDA’s MedWatch program or at 800-332-1088. In addition, the agency is asking physicians to encourage their pregnant patients on valproate to enroll in the North American Antiepileptic Drug Registry online or at 888-233-2334.
Valproate sodium and related drugs should never be used to prevent migraines in pregnant women, according to a Food and Drug Administration warning. The safety advisory cites the final results of a study indicating that prenatal exposure to these drugs "can cause decreased IQ scores" in children whose mothers took them during pregnancy.
The pregnancy category for valproate for the migraine indication will be changed from a "D" – designated for drugs where the potential benefit for a pregnant woman might be acceptable despite its potential risks – to a category "X," designating that the risk of the drug in a pregnant woman "clearly outweighs any possible benefit of the drug," according to the advisory, issued May 6 by the FDA.
Valproate products include valproate sodium (Depacon), divalproex sodium (Depakote, Depakote CP, and Depakote ER), valproic acid (Depakene and Stavzor), and generic versions of these products. They also are used to treat epilepsy and manic episodes associated with bipolar disorder, and for these indications, they will remain in category "D" and "should only be prescribed if other medications are not effective in treating the condition or are otherwise unacceptable." Women who can become pregnant should not use valproate unless it is essential to managing their medical condition, the FDA states.
"Valproate medications should never be used in pregnant women for the prevention of migraine headaches because we have even more data now that show the risks to the children outweigh any treatment benefits for this use," Dr. Russell Katz, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in a May 6 statement issued by the FDA to announce the advisory.
The data cited by the FDA are the final results of the Neurodevelopmental Effects of Antiepileptic Drugs study, published in March 2013, which found that at age 6, children whose mothers had taken valproate products while pregnant had lower IQs, compared with those whose mothers took other antiepileptic drugs. In the study, the mean IQ among the children exposed to valproate in utero was 97, compared with a mean of 105 among those exposed to carbamazepine, 108 among those exposed to lamotrigine, and 108 among those exposed to phenytoin (Lancet Neurol. 2013;12:244-52).
In June 2011, the FDA issued an alert when interim results of this study found that at age 3 years, children exposed to valproate had lower scores on cognitive tests, information that was added to the labels at that time.
The FDA notes that the results of the study are similar to other epidemiologic studies that have associated in utero exposure to valproate to lower IQ scores, when compared with children exposed to another AED or no AED in utero.
Since women in the study were taking antiepileptic drugs throughout pregnancy, it is unclear whether the effects on IQ are related to the timing or duration of prenatal valproate exposure, so "exposure to valproate at any time during pregnancy should be considered to have the potential to result in decreased IQ in children," the advisory says.
The prescribing information for valproate medications already include a boxed warning about the fetal risks, especially neural tube defects and other major malformations, associated with prenatal exposure.
Adverse events associated with valproate products should be reported to the FDA’s MedWatch program or at 800-332-1088. In addition, the agency is asking physicians to encourage their pregnant patients on valproate to enroll in the North American Antiepileptic Drug Registry online or at 888-233-2334.
Valproate sodium and related drugs should never be used to prevent migraines in pregnant women, according to a Food and Drug Administration warning. The safety advisory cites the final results of a study indicating that prenatal exposure to these drugs "can cause decreased IQ scores" in children whose mothers took them during pregnancy.
The pregnancy category for valproate for the migraine indication will be changed from a "D" – designated for drugs where the potential benefit for a pregnant woman might be acceptable despite its potential risks – to a category "X," designating that the risk of the drug in a pregnant woman "clearly outweighs any possible benefit of the drug," according to the advisory, issued May 6 by the FDA.
Valproate products include valproate sodium (Depacon), divalproex sodium (Depakote, Depakote CP, and Depakote ER), valproic acid (Depakene and Stavzor), and generic versions of these products. They also are used to treat epilepsy and manic episodes associated with bipolar disorder, and for these indications, they will remain in category "D" and "should only be prescribed if other medications are not effective in treating the condition or are otherwise unacceptable." Women who can become pregnant should not use valproate unless it is essential to managing their medical condition, the FDA states.
"Valproate medications should never be used in pregnant women for the prevention of migraine headaches because we have even more data now that show the risks to the children outweigh any treatment benefits for this use," Dr. Russell Katz, director of the division of neurology products in the FDA’s Center for Drug Evaluation and Research, said in a May 6 statement issued by the FDA to announce the advisory.
The data cited by the FDA are the final results of the Neurodevelopmental Effects of Antiepileptic Drugs study, published in March 2013, which found that at age 6, children whose mothers had taken valproate products while pregnant had lower IQs, compared with those whose mothers took other antiepileptic drugs. In the study, the mean IQ among the children exposed to valproate in utero was 97, compared with a mean of 105 among those exposed to carbamazepine, 108 among those exposed to lamotrigine, and 108 among those exposed to phenytoin (Lancet Neurol. 2013;12:244-52).
In June 2011, the FDA issued an alert when interim results of this study found that at age 3 years, children exposed to valproate had lower scores on cognitive tests, information that was added to the labels at that time.
The FDA notes that the results of the study are similar to other epidemiologic studies that have associated in utero exposure to valproate to lower IQ scores, when compared with children exposed to another AED or no AED in utero.
Since women in the study were taking antiepileptic drugs throughout pregnancy, it is unclear whether the effects on IQ are related to the timing or duration of prenatal valproate exposure, so "exposure to valproate at any time during pregnancy should be considered to have the potential to result in decreased IQ in children," the advisory says.
The prescribing information for valproate medications already include a boxed warning about the fetal risks, especially neural tube defects and other major malformations, associated with prenatal exposure.
Adverse events associated with valproate products should be reported to the FDA’s MedWatch program or at 800-332-1088. In addition, the agency is asking physicians to encourage their pregnant patients on valproate to enroll in the North American Antiepileptic Drug Registry online or at 888-233-2334.
FDA: Tanning lamps should warn against skin cancer
Indoor tanning beds should carry warnings against their use in people under age 18 years and should advise users to be screened regularly for skin cancer, according to a proposal announced by the Food and Drug Administration on May 6.
The agency seeks to reclassify the ultraviolet lamps used in tanning beds, upgrading them to class II (moderate risk) from class I (low risk) and to rename them "sunlamps." As class I devices, these lamps are currently deemed to be at the same risk level as adhesive bandages and tongue depressors.
Under the proposal, manufacturers would be required to display "a prominent visible label on the tanning bed itself," warning against use in people under age 18 years, Dr. Jeffrey Shuren, director of the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said during a briefing held to announce the proposal. Manufacturers also would have to add labels contraindicating the use of sunlamps in people with certain skin lesions.

Information advising regular skin cancer screenings would be added to materials such as brochures, catalogues, and consumer websites, he added.
"We believe that our proposal will allow for safer, more reliable sunlamps and better arm consumers with the critical information they need," Dr. Shuren said.
In 2010, an FDA advisory panel unanimously recommended that these devices be switched to at least class II.
The proposed reclassification does not prohibit the use of sunlamp products in minors.
Manufacturers are currently not required to submit applications to market these devices, but if they are reclassified as class II devices, a "premarket notification" application will be required and "manufacturers would have to show that their products have met certain performance testing requirements, address certain product design characteristics and provide comprehensive labeling that presents consumers with clear information on the risks of use," according to the FDA statement announcing the proposal.
Clinical trials would not be required, but manufacturers would be required test the performance of timers and alarms and ensure that sunlamps provide the correct amount of energy to prevent burns. Reports of burns associated with these products indicate that this testing is not being done properly now, Dr. Shuren said.
According to the American Academy of Dermatology, the risk of melanoma increases by 75% among people exposed to ultraviolet radiation from indoor tanning products, and the risk increases with increased use.
During the briefing, Dr. Mary Maloney, chair of the academy’s regulatory policy committee, said that an estimated 2.3 million teens use indoor tanning facilities every year, and that melanoma is the most common form of cancer in adults aged 25-29 years and the second most common form of invasive cancer among people aged 15-29 years. In a 2011 youth risk behavior survey, 13% of all high school students said that they had used indoor tanning, and by 12th grade, 32% of girls had reported using a tanning bed, according to the Centers for Disease Control and Prevention.
Dr. Maloney also referred to evidence that young people are given misinformation about the risks of indoor tanning, citing a study by Washington University in St. Louis, which found that 43% of indoor tanning facilities in Missouri denied there were any risks associated with indoor tanning and that two-thirds allowed minors aged 10-12 years to use tanning devices, sometimes without parental consent.
The FDA will accept comments on the proposed order at www.regulations.gov for 90 days from publication in the Federal Register.
Indoor tanning beds should carry warnings against their use in people under age 18 years and should advise users to be screened regularly for skin cancer, according to a proposal announced by the Food and Drug Administration on May 6.
The agency seeks to reclassify the ultraviolet lamps used in tanning beds, upgrading them to class II (moderate risk) from class I (low risk) and to rename them "sunlamps." As class I devices, these lamps are currently deemed to be at the same risk level as adhesive bandages and tongue depressors.
Under the proposal, manufacturers would be required to display "a prominent visible label on the tanning bed itself," warning against use in people under age 18 years, Dr. Jeffrey Shuren, director of the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said during a briefing held to announce the proposal. Manufacturers also would have to add labels contraindicating the use of sunlamps in people with certain skin lesions.
Information advising regular skin cancer screenings would be added to materials such as brochures, catalogues, and consumer websites, he added.
"We believe that our proposal will allow for safer, more reliable sunlamps and better arm consumers with the critical information they need," Dr. Shuren said.
In 2010, an FDA advisory panel unanimously recommended that these devices be switched to at least class II.
The proposed reclassification does not prohibit the use of sunlamp products in minors.
Manufacturers are currently not required to submit applications to market these devices, but if they are reclassified as class II devices, a "premarket notification" application will be required and "manufacturers would have to show that their products have met certain performance testing requirements, address certain product design characteristics and provide comprehensive labeling that presents consumers with clear information on the risks of use," according to the FDA statement announcing the proposal.
Clinical trials would not be required, but manufacturers would be required test the performance of timers and alarms and ensure that sunlamps provide the correct amount of energy to prevent burns. Reports of burns associated with these products indicate that this testing is not being done properly now, Dr. Shuren said.
According to the American Academy of Dermatology, the risk of melanoma increases by 75% among people exposed to ultraviolet radiation from indoor tanning products, and the risk increases with increased use.
During the briefing, Dr. Mary Maloney, chair of the academy’s regulatory policy committee, said that an estimated 2.3 million teens use indoor tanning facilities every year, and that melanoma is the most common form of cancer in adults aged 25-29 years and the second most common form of invasive cancer among people aged 15-29 years. In a 2011 youth risk behavior survey, 13% of all high school students said that they had used indoor tanning, and by 12th grade, 32% of girls had reported using a tanning bed, according to the Centers for Disease Control and Prevention.
Dr. Maloney also referred to evidence that young people are given misinformation about the risks of indoor tanning, citing a study by Washington University in St. Louis, which found that 43% of indoor tanning facilities in Missouri denied there were any risks associated with indoor tanning and that two-thirds allowed minors aged 10-12 years to use tanning devices, sometimes without parental consent.
The FDA will accept comments on the proposed order at www.regulations.gov for 90 days from publication in the Federal Register.
Indoor tanning beds should carry warnings against their use in people under age 18 years and should advise users to be screened regularly for skin cancer, according to a proposal announced by the Food and Drug Administration on May 6.
The agency seeks to reclassify the ultraviolet lamps used in tanning beds, upgrading them to class II (moderate risk) from class I (low risk) and to rename them "sunlamps." As class I devices, these lamps are currently deemed to be at the same risk level as adhesive bandages and tongue depressors.
Under the proposal, manufacturers would be required to display "a prominent visible label on the tanning bed itself," warning against use in people under age 18 years, Dr. Jeffrey Shuren, director of the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said during a briefing held to announce the proposal. Manufacturers also would have to add labels contraindicating the use of sunlamps in people with certain skin lesions.
Information advising regular skin cancer screenings would be added to materials such as brochures, catalogues, and consumer websites, he added.
"We believe that our proposal will allow for safer, more reliable sunlamps and better arm consumers with the critical information they need," Dr. Shuren said.
In 2010, an FDA advisory panel unanimously recommended that these devices be switched to at least class II.
The proposed reclassification does not prohibit the use of sunlamp products in minors.
Manufacturers are currently not required to submit applications to market these devices, but if they are reclassified as class II devices, a "premarket notification" application will be required and "manufacturers would have to show that their products have met certain performance testing requirements, address certain product design characteristics and provide comprehensive labeling that presents consumers with clear information on the risks of use," according to the FDA statement announcing the proposal.
Clinical trials would not be required, but manufacturers would be required test the performance of timers and alarms and ensure that sunlamps provide the correct amount of energy to prevent burns. Reports of burns associated with these products indicate that this testing is not being done properly now, Dr. Shuren said.
According to the American Academy of Dermatology, the risk of melanoma increases by 75% among people exposed to ultraviolet radiation from indoor tanning products, and the risk increases with increased use.
During the briefing, Dr. Mary Maloney, chair of the academy’s regulatory policy committee, said that an estimated 2.3 million teens use indoor tanning facilities every year, and that melanoma is the most common form of cancer in adults aged 25-29 years and the second most common form of invasive cancer among people aged 15-29 years. In a 2011 youth risk behavior survey, 13% of all high school students said that they had used indoor tanning, and by 12th grade, 32% of girls had reported using a tanning bed, according to the Centers for Disease Control and Prevention.
Dr. Maloney also referred to evidence that young people are given misinformation about the risks of indoor tanning, citing a study by Washington University in St. Louis, which found that 43% of indoor tanning facilities in Missouri denied there were any risks associated with indoor tanning and that two-thirds allowed minors aged 10-12 years to use tanning devices, sometimes without parental consent.
The FDA will accept comments on the proposed order at www.regulations.gov for 90 days from publication in the Federal Register.

FDA advisory panel decides tivozanib falls short for advanced renal cell carcinoma
SILVER SPRING, MD. – Tivozanib’s risk-benefit profile fell short in a trial comparing the drug to sorafenib in patients with advanced renal cell carcinoma.
Citing a lower overall survival among patients in the tivozanib arm, despite a significant benefit in progression-free survival, the FDA’s Oncologic Drugs Advisory Committee (ODAC) voted 13 to 1 at a May 2 meeting that the risk-benefit profile for tivozanib was not favorable.
Tivozanib appeared to be active in renal cell carcinoma (RCC) and had a manageable safety profile, the panelists said. But they had reservations about the design and patient demographics of the study; 88% of those enrolled in the trial were from Central and Eastern Europe. Only 8% of the patients in the study were enrolled in the United States and Western Europe, and only one black patient was enrolled, raising questions about the relevance of outcomes for a diversity of patients. Further, the trial’s comparator drug was sorafenib rather than a newer first-line tyrosine kinase inhibitor (TKI).
"While we are disappointed with the outcome of the ODAC vote, we remain confident in the efficacy, safety, and tolerability of tivozanib in RCC patients," Tuan Ha-Ngoc, president and chief executive officer of Aveo, the maker of tivozanib, said in a statement. "We are committed to the RCC patient community and will work closely with the FDA to address the issues discussed by the panel today as the agency continues its ongoing review of the New Drug Application for tivozanib."
Tivozanib, an oral capsule taken once a day, is a highly selective vascular endothelial growth factor (VEGF) TKI, according to Aveo. The company presented the results of the pivotal phase III study, the TIVO-1 (Tivozanib Versus Sorafenib in First-Line Advanced RCC) trial, which compared treatment with tivozanib in 260 patients with sorafenib in 257 patients. In 2005 sorafenib (Nexavar) became the first TKI approved for advanced RCC; several others have been approved since that time. In the United States, those newer drugs are considered before prescribing sorafenib in treating RCC.
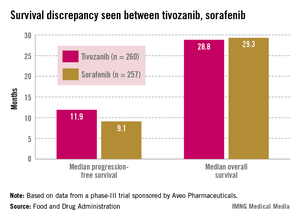
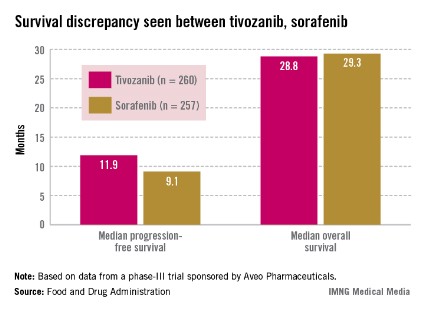
Median progression-free survival (PFS), the primary endpoint of the study, was 11.9 months among those treated with tivozanib and 9.1 months among those on sorafenib, a significant difference that represented a 20% reduced risk (hazard ratio, 0.80).
Overall survival was poorer, however: 28.8 months in patients on tivozanib and 29.3 months in those on sorafenib (HR, 1.25). Overall, the safety profile of tivozanib was comparable to the profile of other VEGF inhibitors. Hypertension and dysphonia were more common with tivozanib, and diarrhea and plantar-palmar dysesthesia (hand-foot syndrome) were more common with sorafenib.
Aveo could not identify a safety signal to explain the lower overall survival. Company officials at the meeting said that more patients in the sorafenib arm (63%) crossed over to treatment with a targeted therapy, which in most cases was tivozanib. In the tivozanib arm, 16% crossed over to treatment with another therapy. The study’s protocol initially allowed crossover only in patients on sorafenib.
While there was a 20% improvement in PFS, there was "a potential 25% increase in the risk of death" compared with sorafenib, noted Dr. Jonathan Jarow, of the FDA’s Office of Hematology and Oncology Products. The company’s hypothesis regarding the crossover effect could not be proven with a post hoc analysis of the data "and remains just a hypothesis."
Approving a drug with a possible 25% increase in the risk of death was the FDA’s major concern and would set a precedent for approval of an oncology drug. The seven drugs approved for treating advanced RCC are thought to work by inhibiting VEGF or its receptor. Most of these approvals, except for temsirolimus, were based on improvements in PFS. Temsirolimus was additionally shown to improve overall survival in patients with a poor prognosis, according to the FDA.
Dr. Mikkael Sekeres, of the department of hematologic oncology and blood disorders at the Cleveland Clinic Taussig Cancer Institute, and the chair of the advisory panel to the FDA, expressed concern about the ethics of the study, which initially allowed crossover to another treatment in only one arm of the study and was conducted primarily in countries where other active RCC therapies are not readily available.
During the meeting, oncologists pointed out that treatments administered sequentially are associated with improved outcomes for patients with RCC.
"I cannot picture how I would be able to sit and talk with a patient about treating her or him with a drug that would allow that person to live without progression longer but possibly to die faster than if I treated that person with another available renal carcinoma drug, so I voted no," he added.
Aveo is developing tivozanib with Astellas Pharma. It is also being studied in combination with other targeted treatments or with chemotherapy regimens in patients with RCC, breast cancer, and colorectal cancer, according to Aveo. It has not been approved in any country.
The FDA usually follows the recommendations of its advisory panels. The deadline for the FDA review to be completed is July 28, according to Aveo. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at the tivozanib meeting.
SILVER SPRING, MD. – Tivozanib’s risk-benefit profile fell short in a trial comparing the drug to sorafenib in patients with advanced renal cell carcinoma.
Citing a lower overall survival among patients in the tivozanib arm, despite a significant benefit in progression-free survival, the FDA’s Oncologic Drugs Advisory Committee (ODAC) voted 13 to 1 at a May 2 meeting that the risk-benefit profile for tivozanib was not favorable.
Tivozanib appeared to be active in renal cell carcinoma (RCC) and had a manageable safety profile, the panelists said. But they had reservations about the design and patient demographics of the study; 88% of those enrolled in the trial were from Central and Eastern Europe. Only 8% of the patients in the study were enrolled in the United States and Western Europe, and only one black patient was enrolled, raising questions about the relevance of outcomes for a diversity of patients. Further, the trial’s comparator drug was sorafenib rather than a newer first-line tyrosine kinase inhibitor (TKI).
"While we are disappointed with the outcome of the ODAC vote, we remain confident in the efficacy, safety, and tolerability of tivozanib in RCC patients," Tuan Ha-Ngoc, president and chief executive officer of Aveo, the maker of tivozanib, said in a statement. "We are committed to the RCC patient community and will work closely with the FDA to address the issues discussed by the panel today as the agency continues its ongoing review of the New Drug Application for tivozanib."
Tivozanib, an oral capsule taken once a day, is a highly selective vascular endothelial growth factor (VEGF) TKI, according to Aveo. The company presented the results of the pivotal phase III study, the TIVO-1 (Tivozanib Versus Sorafenib in First-Line Advanced RCC) trial, which compared treatment with tivozanib in 260 patients with sorafenib in 257 patients. In 2005 sorafenib (Nexavar) became the first TKI approved for advanced RCC; several others have been approved since that time. In the United States, those newer drugs are considered before prescribing sorafenib in treating RCC.
Median progression-free survival (PFS), the primary endpoint of the study, was 11.9 months among those treated with tivozanib and 9.1 months among those on sorafenib, a significant difference that represented a 20% reduced risk (hazard ratio, 0.80).
Overall survival was poorer, however: 28.8 months in patients on tivozanib and 29.3 months in those on sorafenib (HR, 1.25). Overall, the safety profile of tivozanib was comparable to the profile of other VEGF inhibitors. Hypertension and dysphonia were more common with tivozanib, and diarrhea and plantar-palmar dysesthesia (hand-foot syndrome) were more common with sorafenib.
Aveo could not identify a safety signal to explain the lower overall survival. Company officials at the meeting said that more patients in the sorafenib arm (63%) crossed over to treatment with a targeted therapy, which in most cases was tivozanib. In the tivozanib arm, 16% crossed over to treatment with another therapy. The study’s protocol initially allowed crossover only in patients on sorafenib.
While there was a 20% improvement in PFS, there was "a potential 25% increase in the risk of death" compared with sorafenib, noted Dr. Jonathan Jarow, of the FDA’s Office of Hematology and Oncology Products. The company’s hypothesis regarding the crossover effect could not be proven with a post hoc analysis of the data "and remains just a hypothesis."
Approving a drug with a possible 25% increase in the risk of death was the FDA’s major concern and would set a precedent for approval of an oncology drug. The seven drugs approved for treating advanced RCC are thought to work by inhibiting VEGF or its receptor. Most of these approvals, except for temsirolimus, were based on improvements in PFS. Temsirolimus was additionally shown to improve overall survival in patients with a poor prognosis, according to the FDA.
Dr. Mikkael Sekeres, of the department of hematologic oncology and blood disorders at the Cleveland Clinic Taussig Cancer Institute, and the chair of the advisory panel to the FDA, expressed concern about the ethics of the study, which initially allowed crossover to another treatment in only one arm of the study and was conducted primarily in countries where other active RCC therapies are not readily available.
During the meeting, oncologists pointed out that treatments administered sequentially are associated with improved outcomes for patients with RCC.
"I cannot picture how I would be able to sit and talk with a patient about treating her or him with a drug that would allow that person to live without progression longer but possibly to die faster than if I treated that person with another available renal carcinoma drug, so I voted no," he added.
Aveo is developing tivozanib with Astellas Pharma. It is also being studied in combination with other targeted treatments or with chemotherapy regimens in patients with RCC, breast cancer, and colorectal cancer, according to Aveo. It has not been approved in any country.
The FDA usually follows the recommendations of its advisory panels. The deadline for the FDA review to be completed is July 28, according to Aveo. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at the tivozanib meeting.
SILVER SPRING, MD. – Tivozanib’s risk-benefit profile fell short in a trial comparing the drug to sorafenib in patients with advanced renal cell carcinoma.
Citing a lower overall survival among patients in the tivozanib arm, despite a significant benefit in progression-free survival, the FDA’s Oncologic Drugs Advisory Committee (ODAC) voted 13 to 1 at a May 2 meeting that the risk-benefit profile for tivozanib was not favorable.
Tivozanib appeared to be active in renal cell carcinoma (RCC) and had a manageable safety profile, the panelists said. But they had reservations about the design and patient demographics of the study; 88% of those enrolled in the trial were from Central and Eastern Europe. Only 8% of the patients in the study were enrolled in the United States and Western Europe, and only one black patient was enrolled, raising questions about the relevance of outcomes for a diversity of patients. Further, the trial’s comparator drug was sorafenib rather than a newer first-line tyrosine kinase inhibitor (TKI).
"While we are disappointed with the outcome of the ODAC vote, we remain confident in the efficacy, safety, and tolerability of tivozanib in RCC patients," Tuan Ha-Ngoc, president and chief executive officer of Aveo, the maker of tivozanib, said in a statement. "We are committed to the RCC patient community and will work closely with the FDA to address the issues discussed by the panel today as the agency continues its ongoing review of the New Drug Application for tivozanib."
Tivozanib, an oral capsule taken once a day, is a highly selective vascular endothelial growth factor (VEGF) TKI, according to Aveo. The company presented the results of the pivotal phase III study, the TIVO-1 (Tivozanib Versus Sorafenib in First-Line Advanced RCC) trial, which compared treatment with tivozanib in 260 patients with sorafenib in 257 patients. In 2005 sorafenib (Nexavar) became the first TKI approved for advanced RCC; several others have been approved since that time. In the United States, those newer drugs are considered before prescribing sorafenib in treating RCC.
Median progression-free survival (PFS), the primary endpoint of the study, was 11.9 months among those treated with tivozanib and 9.1 months among those on sorafenib, a significant difference that represented a 20% reduced risk (hazard ratio, 0.80).
Overall survival was poorer, however: 28.8 months in patients on tivozanib and 29.3 months in those on sorafenib (HR, 1.25). Overall, the safety profile of tivozanib was comparable to the profile of other VEGF inhibitors. Hypertension and dysphonia were more common with tivozanib, and diarrhea and plantar-palmar dysesthesia (hand-foot syndrome) were more common with sorafenib.
Aveo could not identify a safety signal to explain the lower overall survival. Company officials at the meeting said that more patients in the sorafenib arm (63%) crossed over to treatment with a targeted therapy, which in most cases was tivozanib. In the tivozanib arm, 16% crossed over to treatment with another therapy. The study’s protocol initially allowed crossover only in patients on sorafenib.
While there was a 20% improvement in PFS, there was "a potential 25% increase in the risk of death" compared with sorafenib, noted Dr. Jonathan Jarow, of the FDA’s Office of Hematology and Oncology Products. The company’s hypothesis regarding the crossover effect could not be proven with a post hoc analysis of the data "and remains just a hypothesis."
Approving a drug with a possible 25% increase in the risk of death was the FDA’s major concern and would set a precedent for approval of an oncology drug. The seven drugs approved for treating advanced RCC are thought to work by inhibiting VEGF or its receptor. Most of these approvals, except for temsirolimus, were based on improvements in PFS. Temsirolimus was additionally shown to improve overall survival in patients with a poor prognosis, according to the FDA.
Dr. Mikkael Sekeres, of the department of hematologic oncology and blood disorders at the Cleveland Clinic Taussig Cancer Institute, and the chair of the advisory panel to the FDA, expressed concern about the ethics of the study, which initially allowed crossover to another treatment in only one arm of the study and was conducted primarily in countries where other active RCC therapies are not readily available.
During the meeting, oncologists pointed out that treatments administered sequentially are associated with improved outcomes for patients with RCC.
"I cannot picture how I would be able to sit and talk with a patient about treating her or him with a drug that would allow that person to live without progression longer but possibly to die faster than if I treated that person with another available renal carcinoma drug, so I voted no," he added.
Aveo is developing tivozanib with Astellas Pharma. It is also being studied in combination with other targeted treatments or with chemotherapy regimens in patients with RCC, breast cancer, and colorectal cancer, according to Aveo. It has not been approved in any country.
The FDA usually follows the recommendations of its advisory panels. The deadline for the FDA review to be completed is July 28, according to Aveo. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at the tivozanib meeting.
AT AN FDA ADVISORY PANEL MEETING
SABAs, not LABAs, for exercise-induced bronchoconstriction
Don’t base diagnosis of exercise-induced bronchoconstriction on symptoms alone – but do have patients use inhaled short-acting beta-agonists and do warm-ups before exercise, according to new clinical practice guidelines issued May 1 by the American Thoracic Society.
The guidelines define exercise-induced bronchoconstriction (EIB) as "acute airway narrowing that occurs as a result of exercise."
Considering the high prevalence of EIB, which also affects people without asthma, "evidence-based guidelines for its management are of critical importance," said Dr. Jonathan Parsons, the lead author and chair of the committee that drafted the guidelines, in a statement.
The recommendations "synthesize the latest clinical evidence and will help guide the management of EIB in patients with or without asthma, and in athletes at all levels of competition," added Dr. Parsons, associate professor of internal medicine and associate director of the Ohio State University Asthma Center, Columbus.
The EIB guidelines cover pathogenesis, environmental triggers, diagnosis, treatment, and screening (Am. J. Resp. Crit. Care Med. 2013;187 [doi:10.1164/rccm.201303-0437ST]). Also included is a section on exercise, asthma, and doping – with reminders about which EIB drugs are banned in competitive sports (most beta-agonists) and which are allowed (short-acting inhaled albuterol and inhaled steroids).
Although the guidelines can apply both to adolescents and adults, they cannot be applied reliably to young children, Dr. Parsons noted in an interview.
EIB prevalence among people with asthma is not known, but the estimated prevalence among people who have not been diagnosed with asthma is as high as 20%, according to the ATS. EIB is more prevalent among athletes, affecting 30%-70% of Olympic and elite athletes. Environmental factors likely play a role, such as pollutants emitted from ice surfacing machines in indoor ice rinks, high trichloramine levels in the air of indoor pools, and cold, dry air.
An EIB diagnosis should not be based on symptoms, which are variable, nonspecific, and have poor predictive value. Instead, diagnosis should be made based on changes in lung function provoked by exercise, using serial lung function measurements after a specific exercise or a hyperpnea challenge. Assessing the effects of exercise on forced expiratory volume in 1 second (FEV1) is preferred.
The guidelines grade EIB as mild, moderate, or severe, depending on the percent fall in FEV1 from baseline. They also offer information on alternatives to exercise testing.
The authors rate pharmacologic and nonpharmacologic therapies based on the quality of the supportive evidence. Their first recommendation – administration of an inhaled short-acting beta-agonist (SABA) before exercise – earns a "strong" recommendation based on "high-quality" evidence. Patients typically take SABAs 15 minutes before exercise.
Because of the potential for serious side effects, the authors recommend against daily use of an inhaled long-acting beta-agonist (LABA) for EIB – a strong recommendation based on moderate-quality evidence.
For patients who use an inhaled SABA but continue to have symptoms or need to use the inhaled SABA "daily or more frequently," treatment options before exercise include a daily inhaled corticosteroid (ICS), a daily leukotriene-receptor antagonist, or a mast-cell–stabilizing agent.
"We generally add a daily inhaled ICS or a daily leukotriene-receptor antagonist first, with the choice between these agents made on a case-by-case basis depending upon patient preferences," the guideline authors note. Mast-cell–stabilizing agents and inhaled anticholinergic drugs "play a secondary role," they added. There is also a role for antihistamines in patient with continued symptoms despite treatment, but not for patients without allergies.
Nonpharmacologic measures include interval or combination warm-up exercises before planned exercise, which the guidelines recommend "for all patients" with EIB – a strong recommendation, based on moderate quality evidence. The guidelines cite evidence showing a lower reduction in FEV1 after exercise among people with EIB who engaged in "interval, low-intensity continuous; high-intensity continuous; or combination warm-up" before they exercised.
Another nonpharmacologic recommendation is use of a mask or another device that warms and humidifies the air when patients exercise in a cold climate.
While there is not much evidence supporting dietary modifications, patients interested in this approach can try a low-salt diet, or take fish oil or vitamin C supplements. However, the use of lycopene is not supported, based on the available evidence.
"Our overall recommendations regarding therapy leave a lot of options for the individual patient, which should be discussed with the patient’s physician and tried and evaluated on an ongoing basis," the authors concluded.
The mainstay of treatment "remains maintaining good control of underlying asthma (if present) and preventing or treating symptoms of EIB with SABAs."
The EIB practice guidelines were supported by the ATS and approved by the ATS board of directors. Dr. Parsons’ disclosures include having received lecture fees from AstraZeneca, GlaxoSmithKline, Merck, and Schering Plough. All but one of the other authors disclosed financial relations with a wide range of pharmaceutical companies.
Don’t base diagnosis of exercise-induced bronchoconstriction on symptoms alone – but do have patients use inhaled short-acting beta-agonists and do warm-ups before exercise, according to new clinical practice guidelines issued May 1 by the American Thoracic Society.
The guidelines define exercise-induced bronchoconstriction (EIB) as "acute airway narrowing that occurs as a result of exercise."
Considering the high prevalence of EIB, which also affects people without asthma, "evidence-based guidelines for its management are of critical importance," said Dr. Jonathan Parsons, the lead author and chair of the committee that drafted the guidelines, in a statement.
The recommendations "synthesize the latest clinical evidence and will help guide the management of EIB in patients with or without asthma, and in athletes at all levels of competition," added Dr. Parsons, associate professor of internal medicine and associate director of the Ohio State University Asthma Center, Columbus.
The EIB guidelines cover pathogenesis, environmental triggers, diagnosis, treatment, and screening (Am. J. Resp. Crit. Care Med. 2013;187 [doi:10.1164/rccm.201303-0437ST]). Also included is a section on exercise, asthma, and doping – with reminders about which EIB drugs are banned in competitive sports (most beta-agonists) and which are allowed (short-acting inhaled albuterol and inhaled steroids).
Although the guidelines can apply both to adolescents and adults, they cannot be applied reliably to young children, Dr. Parsons noted in an interview.
EIB prevalence among people with asthma is not known, but the estimated prevalence among people who have not been diagnosed with asthma is as high as 20%, according to the ATS. EIB is more prevalent among athletes, affecting 30%-70% of Olympic and elite athletes. Environmental factors likely play a role, such as pollutants emitted from ice surfacing machines in indoor ice rinks, high trichloramine levels in the air of indoor pools, and cold, dry air.
An EIB diagnosis should not be based on symptoms, which are variable, nonspecific, and have poor predictive value. Instead, diagnosis should be made based on changes in lung function provoked by exercise, using serial lung function measurements after a specific exercise or a hyperpnea challenge. Assessing the effects of exercise on forced expiratory volume in 1 second (FEV1) is preferred.
The guidelines grade EIB as mild, moderate, or severe, depending on the percent fall in FEV1 from baseline. They also offer information on alternatives to exercise testing.
The authors rate pharmacologic and nonpharmacologic therapies based on the quality of the supportive evidence. Their first recommendation – administration of an inhaled short-acting beta-agonist (SABA) before exercise – earns a "strong" recommendation based on "high-quality" evidence. Patients typically take SABAs 15 minutes before exercise.
Because of the potential for serious side effects, the authors recommend against daily use of an inhaled long-acting beta-agonist (LABA) for EIB – a strong recommendation based on moderate-quality evidence.
For patients who use an inhaled SABA but continue to have symptoms or need to use the inhaled SABA "daily or more frequently," treatment options before exercise include a daily inhaled corticosteroid (ICS), a daily leukotriene-receptor antagonist, or a mast-cell–stabilizing agent.
"We generally add a daily inhaled ICS or a daily leukotriene-receptor antagonist first, with the choice between these agents made on a case-by-case basis depending upon patient preferences," the guideline authors note. Mast-cell–stabilizing agents and inhaled anticholinergic drugs "play a secondary role," they added. There is also a role for antihistamines in patient with continued symptoms despite treatment, but not for patients without allergies.
Nonpharmacologic measures include interval or combination warm-up exercises before planned exercise, which the guidelines recommend "for all patients" with EIB – a strong recommendation, based on moderate quality evidence. The guidelines cite evidence showing a lower reduction in FEV1 after exercise among people with EIB who engaged in "interval, low-intensity continuous; high-intensity continuous; or combination warm-up" before they exercised.
Another nonpharmacologic recommendation is use of a mask or another device that warms and humidifies the air when patients exercise in a cold climate.
While there is not much evidence supporting dietary modifications, patients interested in this approach can try a low-salt diet, or take fish oil or vitamin C supplements. However, the use of lycopene is not supported, based on the available evidence.
"Our overall recommendations regarding therapy leave a lot of options for the individual patient, which should be discussed with the patient’s physician and tried and evaluated on an ongoing basis," the authors concluded.
The mainstay of treatment "remains maintaining good control of underlying asthma (if present) and preventing or treating symptoms of EIB with SABAs."
The EIB practice guidelines were supported by the ATS and approved by the ATS board of directors. Dr. Parsons’ disclosures include having received lecture fees from AstraZeneca, GlaxoSmithKline, Merck, and Schering Plough. All but one of the other authors disclosed financial relations with a wide range of pharmaceutical companies.
Don’t base diagnosis of exercise-induced bronchoconstriction on symptoms alone – but do have patients use inhaled short-acting beta-agonists and do warm-ups before exercise, according to new clinical practice guidelines issued May 1 by the American Thoracic Society.
The guidelines define exercise-induced bronchoconstriction (EIB) as "acute airway narrowing that occurs as a result of exercise."
Considering the high prevalence of EIB, which also affects people without asthma, "evidence-based guidelines for its management are of critical importance," said Dr. Jonathan Parsons, the lead author and chair of the committee that drafted the guidelines, in a statement.
The recommendations "synthesize the latest clinical evidence and will help guide the management of EIB in patients with or without asthma, and in athletes at all levels of competition," added Dr. Parsons, associate professor of internal medicine and associate director of the Ohio State University Asthma Center, Columbus.
The EIB guidelines cover pathogenesis, environmental triggers, diagnosis, treatment, and screening (Am. J. Resp. Crit. Care Med. 2013;187 [doi:10.1164/rccm.201303-0437ST]). Also included is a section on exercise, asthma, and doping – with reminders about which EIB drugs are banned in competitive sports (most beta-agonists) and which are allowed (short-acting inhaled albuterol and inhaled steroids).
Although the guidelines can apply both to adolescents and adults, they cannot be applied reliably to young children, Dr. Parsons noted in an interview.
EIB prevalence among people with asthma is not known, but the estimated prevalence among people who have not been diagnosed with asthma is as high as 20%, according to the ATS. EIB is more prevalent among athletes, affecting 30%-70% of Olympic and elite athletes. Environmental factors likely play a role, such as pollutants emitted from ice surfacing machines in indoor ice rinks, high trichloramine levels in the air of indoor pools, and cold, dry air.
An EIB diagnosis should not be based on symptoms, which are variable, nonspecific, and have poor predictive value. Instead, diagnosis should be made based on changes in lung function provoked by exercise, using serial lung function measurements after a specific exercise or a hyperpnea challenge. Assessing the effects of exercise on forced expiratory volume in 1 second (FEV1) is preferred.
The guidelines grade EIB as mild, moderate, or severe, depending on the percent fall in FEV1 from baseline. They also offer information on alternatives to exercise testing.
The authors rate pharmacologic and nonpharmacologic therapies based on the quality of the supportive evidence. Their first recommendation – administration of an inhaled short-acting beta-agonist (SABA) before exercise – earns a "strong" recommendation based on "high-quality" evidence. Patients typically take SABAs 15 minutes before exercise.
Because of the potential for serious side effects, the authors recommend against daily use of an inhaled long-acting beta-agonist (LABA) for EIB – a strong recommendation based on moderate-quality evidence.
For patients who use an inhaled SABA but continue to have symptoms or need to use the inhaled SABA "daily or more frequently," treatment options before exercise include a daily inhaled corticosteroid (ICS), a daily leukotriene-receptor antagonist, or a mast-cell–stabilizing agent.
"We generally add a daily inhaled ICS or a daily leukotriene-receptor antagonist first, with the choice between these agents made on a case-by-case basis depending upon patient preferences," the guideline authors note. Mast-cell–stabilizing agents and inhaled anticholinergic drugs "play a secondary role," they added. There is also a role for antihistamines in patient with continued symptoms despite treatment, but not for patients without allergies.
Nonpharmacologic measures include interval or combination warm-up exercises before planned exercise, which the guidelines recommend "for all patients" with EIB – a strong recommendation, based on moderate quality evidence. The guidelines cite evidence showing a lower reduction in FEV1 after exercise among people with EIB who engaged in "interval, low-intensity continuous; high-intensity continuous; or combination warm-up" before they exercised.
Another nonpharmacologic recommendation is use of a mask or another device that warms and humidifies the air when patients exercise in a cold climate.
While there is not much evidence supporting dietary modifications, patients interested in this approach can try a low-salt diet, or take fish oil or vitamin C supplements. However, the use of lycopene is not supported, based on the available evidence.
"Our overall recommendations regarding therapy leave a lot of options for the individual patient, which should be discussed with the patient’s physician and tried and evaluated on an ongoing basis," the authors concluded.
The mainstay of treatment "remains maintaining good control of underlying asthma (if present) and preventing or treating symptoms of EIB with SABAs."
The EIB practice guidelines were supported by the ATS and approved by the ATS board of directors. Dr. Parsons’ disclosures include having received lecture fees from AstraZeneca, GlaxoSmithKline, Merck, and Schering Plough. All but one of the other authors disclosed financial relations with a wide range of pharmaceutical companies.
FROM THE AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE
Guidelines: SABAs, not LABAs, for exercise-induced bronchoconstriction
Don’t base diagnosis of exercise-induced bronchoconstriction on symptoms alone – but do have patients use inhaled short-acting beta-agonists and do warm-ups before exercise, according to new clinical practice guidelines issued May 1 by the American Thoracic Society.
The guidelines define exercise-induced bronchoconstriction (EIB) as "acute airway narrowing that occurs as a result of exercise."
Considering the high prevalence of EIB, which also affects people without asthma, "evidence-based guidelines for its management are of critical importance," said Dr. Jonathan Parsons, the lead author and chair of the committee that drafted the guidelines, in a statement.

The recommendations "synthesize the latest clinical evidence and will help guide the management of EIB in patients with or without asthma, and in athletes at all levels of competition," added Dr. Parsons, associate professor of internal medicine and associate director of the Ohio State University Asthma Center, Columbus.
The EIB guidelines cover pathogenesis, environmental triggers, diagnosis, treatment, and screening (Am. J. Resp. Crit. Care Med. 2013;187 [doi:10.1164/rccm.201303-0437ST]). Also included is a section on exercise, asthma, and doping – with reminders about which EIB drugs are banned in competitive sports (most beta-agonists) and which are allowed (short-acting inhaled albuterol and inhaled steroids).
Although the guidelines can apply both to adolescents and adults, they cannot be applied reliably to young children, Dr. Parsons noted in an interview.
EIB prevalence among people with asthma is not known, but the estimated prevalence among people who have not been diagnosed with asthma is as high as 20%, according to the ATS. EIB is more prevalent among athletes, affecting 30%-70% of Olympic and elite athletes. Environmental factors likely play a role, such as pollutants emitted from ice surfacing machines in indoor ice rinks, high trichloramine levels in the air of indoor pools, and cold, dry air.
An EIB diagnosis should not be based on symptoms, which are variable, nonspecific, and have poor predictive value. Instead, diagnosis should be made based on changes in lung function provoked by exercise, using serial lung function measurements after a specific exercise or a hyperpnea challenge. Assessing the effects of exercise on forced expiratory volume in 1 second (FEV1) is preferred.
The guidelines grade EIB as mild, moderate, or severe, depending on the percent fall in FEV1 from baseline. They also offer information on alternatives to exercise testing.
The authors rate pharmacologic and nonpharmacologic therapies based on the quality of the supportive evidence. Their first recommendation – administration of an inhaled short-acting beta-agonist (SABA) before exercise – earns a "strong" recommendation based on "high-quality" evidence. Patients typically take SABAs 15 minutes before exercise.
Because of the potential for serious side effects, the authors recommend against daily use of an inhaled long-acting beta-agonist (LABA) for EIB – a strong recommendation based on moderate-quality evidence.
For patients who use an inhaled SABA but continue to have symptoms or need to use the inhaled SABA "daily or more frequently," treatment options before exercise include a daily inhaled corticosteroid (ICS), a daily leukotriene-receptor antagonist, or a mast-cell–stabilizing agent.
"We generally add a daily inhaled ICS or a daily leukotriene-receptor antagonist first, with the choice between these agents made on a case-by-case basis depending upon patient preferences," the guideline authors note. Mast-cell–stabilizing agents and inhaled anticholinergic drugs "play a secondary role," they added. There is also a role for antihistamines in patient with continued symptoms despite treatment, but not for patients without allergies.
Nonpharmacologic measures include interval or combination warm-up exercises before planned exercise, which the guidelines recommend "for all patients" with EIB – a strong recommendation, based on moderate quality evidence. The guidelines cite evidence showing a lower reduction in FEV1 after exercise among people with EIB who engaged in "interval, low-intensity continuous; high-intensity continuous; or combination warm-up" before they exercised.
Another nonpharmacologic recommendation is use of a mask or another device that warms and humidifies the air when patients exercise in a cold climate.
While there is not much evidence supporting dietary modifications, patients interested in this approach can try a low-salt diet, or take fish oil or vitamin C supplements. However, the use of lycopene is not supported, based on the available evidence.
"Our overall recommendations regarding therapy leave a lot of options for the individual patient, which should be discussed with the patient’s physician and tried and evaluated on an ongoing basis," the authors concluded.
The mainstay of treatment "remains maintaining good control of underlying asthma (if present) and preventing or treating symptoms of EIB with SABAs."
The EIB practice guidelines were supported by the ATS and approved by the ATS board of directors. Dr. Parsons’ disclosures include having received lecture fees from AstraZeneca, GlaxoSmithKline, Merck, and Schering Plough. All but one of the other authors disclosed financial relations with a wide range of pharmaceutical companies.
Don’t base diagnosis of exercise-induced bronchoconstriction on symptoms alone – but do have patients use inhaled short-acting beta-agonists and do warm-ups before exercise, according to new clinical practice guidelines issued May 1 by the American Thoracic Society.
The guidelines define exercise-induced bronchoconstriction (EIB) as "acute airway narrowing that occurs as a result of exercise."
Considering the high prevalence of EIB, which also affects people without asthma, "evidence-based guidelines for its management are of critical importance," said Dr. Jonathan Parsons, the lead author and chair of the committee that drafted the guidelines, in a statement.
The recommendations "synthesize the latest clinical evidence and will help guide the management of EIB in patients with or without asthma, and in athletes at all levels of competition," added Dr. Parsons, associate professor of internal medicine and associate director of the Ohio State University Asthma Center, Columbus.
The EIB guidelines cover pathogenesis, environmental triggers, diagnosis, treatment, and screening (Am. J. Resp. Crit. Care Med. 2013;187 [doi:10.1164/rccm.201303-0437ST]). Also included is a section on exercise, asthma, and doping – with reminders about which EIB drugs are banned in competitive sports (most beta-agonists) and which are allowed (short-acting inhaled albuterol and inhaled steroids).
Although the guidelines can apply both to adolescents and adults, they cannot be applied reliably to young children, Dr. Parsons noted in an interview.
EIB prevalence among people with asthma is not known, but the estimated prevalence among people who have not been diagnosed with asthma is as high as 20%, according to the ATS. EIB is more prevalent among athletes, affecting 30%-70% of Olympic and elite athletes. Environmental factors likely play a role, such as pollutants emitted from ice surfacing machines in indoor ice rinks, high trichloramine levels in the air of indoor pools, and cold, dry air.
An EIB diagnosis should not be based on symptoms, which are variable, nonspecific, and have poor predictive value. Instead, diagnosis should be made based on changes in lung function provoked by exercise, using serial lung function measurements after a specific exercise or a hyperpnea challenge. Assessing the effects of exercise on forced expiratory volume in 1 second (FEV1) is preferred.
The guidelines grade EIB as mild, moderate, or severe, depending on the percent fall in FEV1 from baseline. They also offer information on alternatives to exercise testing.
The authors rate pharmacologic and nonpharmacologic therapies based on the quality of the supportive evidence. Their first recommendation – administration of an inhaled short-acting beta-agonist (SABA) before exercise – earns a "strong" recommendation based on "high-quality" evidence. Patients typically take SABAs 15 minutes before exercise.
Because of the potential for serious side effects, the authors recommend against daily use of an inhaled long-acting beta-agonist (LABA) for EIB – a strong recommendation based on moderate-quality evidence.
For patients who use an inhaled SABA but continue to have symptoms or need to use the inhaled SABA "daily or more frequently," treatment options before exercise include a daily inhaled corticosteroid (ICS), a daily leukotriene-receptor antagonist, or a mast-cell–stabilizing agent.
"We generally add a daily inhaled ICS or a daily leukotriene-receptor antagonist first, with the choice between these agents made on a case-by-case basis depending upon patient preferences," the guideline authors note. Mast-cell–stabilizing agents and inhaled anticholinergic drugs "play a secondary role," they added. There is also a role for antihistamines in patient with continued symptoms despite treatment, but not for patients without allergies.
Nonpharmacologic measures include interval or combination warm-up exercises before planned exercise, which the guidelines recommend "for all patients" with EIB – a strong recommendation, based on moderate quality evidence. The guidelines cite evidence showing a lower reduction in FEV1 after exercise among people with EIB who engaged in "interval, low-intensity continuous; high-intensity continuous; or combination warm-up" before they exercised.
Another nonpharmacologic recommendation is use of a mask or another device that warms and humidifies the air when patients exercise in a cold climate.
While there is not much evidence supporting dietary modifications, patients interested in this approach can try a low-salt diet, or take fish oil or vitamin C supplements. However, the use of lycopene is not supported, based on the available evidence.
"Our overall recommendations regarding therapy leave a lot of options for the individual patient, which should be discussed with the patient’s physician and tried and evaluated on an ongoing basis," the authors concluded.
The mainstay of treatment "remains maintaining good control of underlying asthma (if present) and preventing or treating symptoms of EIB with SABAs."
The EIB practice guidelines were supported by the ATS and approved by the ATS board of directors. Dr. Parsons’ disclosures include having received lecture fees from AstraZeneca, GlaxoSmithKline, Merck, and Schering Plough. All but one of the other authors disclosed financial relations with a wide range of pharmaceutical companies.
Don’t base diagnosis of exercise-induced bronchoconstriction on symptoms alone – but do have patients use inhaled short-acting beta-agonists and do warm-ups before exercise, according to new clinical practice guidelines issued May 1 by the American Thoracic Society.
The guidelines define exercise-induced bronchoconstriction (EIB) as "acute airway narrowing that occurs as a result of exercise."
Considering the high prevalence of EIB, which also affects people without asthma, "evidence-based guidelines for its management are of critical importance," said Dr. Jonathan Parsons, the lead author and chair of the committee that drafted the guidelines, in a statement.
The recommendations "synthesize the latest clinical evidence and will help guide the management of EIB in patients with or without asthma, and in athletes at all levels of competition," added Dr. Parsons, associate professor of internal medicine and associate director of the Ohio State University Asthma Center, Columbus.
The EIB guidelines cover pathogenesis, environmental triggers, diagnosis, treatment, and screening (Am. J. Resp. Crit. Care Med. 2013;187 [doi:10.1164/rccm.201303-0437ST]). Also included is a section on exercise, asthma, and doping – with reminders about which EIB drugs are banned in competitive sports (most beta-agonists) and which are allowed (short-acting inhaled albuterol and inhaled steroids).
Although the guidelines can apply both to adolescents and adults, they cannot be applied reliably to young children, Dr. Parsons noted in an interview.
EIB prevalence among people with asthma is not known, but the estimated prevalence among people who have not been diagnosed with asthma is as high as 20%, according to the ATS. EIB is more prevalent among athletes, affecting 30%-70% of Olympic and elite athletes. Environmental factors likely play a role, such as pollutants emitted from ice surfacing machines in indoor ice rinks, high trichloramine levels in the air of indoor pools, and cold, dry air.
An EIB diagnosis should not be based on symptoms, which are variable, nonspecific, and have poor predictive value. Instead, diagnosis should be made based on changes in lung function provoked by exercise, using serial lung function measurements after a specific exercise or a hyperpnea challenge. Assessing the effects of exercise on forced expiratory volume in 1 second (FEV1) is preferred.
The guidelines grade EIB as mild, moderate, or severe, depending on the percent fall in FEV1 from baseline. They also offer information on alternatives to exercise testing.
The authors rate pharmacologic and nonpharmacologic therapies based on the quality of the supportive evidence. Their first recommendation – administration of an inhaled short-acting beta-agonist (SABA) before exercise – earns a "strong" recommendation based on "high-quality" evidence. Patients typically take SABAs 15 minutes before exercise.
Because of the potential for serious side effects, the authors recommend against daily use of an inhaled long-acting beta-agonist (LABA) for EIB – a strong recommendation based on moderate-quality evidence.
For patients who use an inhaled SABA but continue to have symptoms or need to use the inhaled SABA "daily or more frequently," treatment options before exercise include a daily inhaled corticosteroid (ICS), a daily leukotriene-receptor antagonist, or a mast-cell–stabilizing agent.
"We generally add a daily inhaled ICS or a daily leukotriene-receptor antagonist first, with the choice between these agents made on a case-by-case basis depending upon patient preferences," the guideline authors note. Mast-cell–stabilizing agents and inhaled anticholinergic drugs "play a secondary role," they added. There is also a role for antihistamines in patient with continued symptoms despite treatment, but not for patients without allergies.
Nonpharmacologic measures include interval or combination warm-up exercises before planned exercise, which the guidelines recommend "for all patients" with EIB – a strong recommendation, based on moderate quality evidence. The guidelines cite evidence showing a lower reduction in FEV1 after exercise among people with EIB who engaged in "interval, low-intensity continuous; high-intensity continuous; or combination warm-up" before they exercised.
Another nonpharmacologic recommendation is use of a mask or another device that warms and humidifies the air when patients exercise in a cold climate.
While there is not much evidence supporting dietary modifications, patients interested in this approach can try a low-salt diet, or take fish oil or vitamin C supplements. However, the use of lycopene is not supported, based on the available evidence.
"Our overall recommendations regarding therapy leave a lot of options for the individual patient, which should be discussed with the patient’s physician and tried and evaluated on an ongoing basis," the authors concluded.
The mainstay of treatment "remains maintaining good control of underlying asthma (if present) and preventing or treating symptoms of EIB with SABAs."
The EIB practice guidelines were supported by the ATS and approved by the ATS board of directors. Dr. Parsons’ disclosures include having received lecture fees from AstraZeneca, GlaxoSmithKline, Merck, and Schering Plough. All but one of the other authors disclosed financial relations with a wide range of pharmaceutical companies.
FROM THE AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE

No benefit of endovascular therapy added to TPA for stroke
Functional outcomes in patients treated with intravenous tissue plasminogen activator with or without endovascular therapy after a moderate to severe acute ischemic stroke were not significantly different, and safety outcomes were similar, in a study that was stopped early because of these results.
In the IMS (Interventional Management of Stroke) III study, 40.8% of patients randomized to receive endovascular therapy plus intravenous TPA met the primary endpoint, a measure of functional independence -- a modified Rankin score of 2 or less at 90 days -- compared with 38.7% among those who had intravenous TPA alone, a difference that was not statistically significant, reported Dr. Joseph Broderick of the University of Cincinnati Neuroscience Institute, and the other IMS III investigators.
Mortality and other safety outcomes were also not significantly different between the two groups of patients in the study, which was stopped early because of futility after 656 of the planned 900 patients had been randomized.

The study was published online to coincide with the presentation of the results at the International Stroke Conference (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1214300]).
Referring to the lack of randomized clinical trial data, the authors pointed out that it is uncertain whether endovascular therapy (which includes endovascular pharmacologic thrombolysis and, more recently, the use of stent retrievers) alone or combined with intravenous TPA is a more effective treatment of acute stroke than intravenous TPA alone, "the only proven reperfusion therapy for acute ischemic stroke."
In the study, conducted at 58 centers in the United States, Canada, Australia, and Europe, 434 patients were randomized to endovascular therapy plus intravenous TPA and 222 were randomized to standard treatment with intravenous TPA alone (started within 3 hours of stroke onset). The median age of those enrolled was 68-69 years (range, 23-89 years), a little over half were men, about 14% were black or Hispanic, and the median the National Institute of Health Stroke Scale (NIHSS) score was 16-17 (8-19 is a moderately severe stroke and 20 or greater is a severe stroke At the beginning of the study, only one thrombectomy device had been cleared by the Food and Drug Administration and, as the trial continued, other devices were used as they became cleared for use in the different countries.
In addition to the main finding, there were no differences in the primary outcome among those patients with an NIHSS score of 20 or more, and those with a score of 19 or lower, said the authors, who had hypothesized that endovascular therapy would have greater efficacy in patients with more-severe strokes since they "have the highest likelihood of occlusion in a major intracranial artery and the greatest volume of ischemic brain at risk."
They had also hypothesized that receiving endovascular therapy earlier would be associated with a greater benefit, but this was also not a significant factor in outcomes.
Mortality at 90 days was 19.1% in the endovascular therapy group and 21.6% in the intravenous TPA–alone group. Within 30 hours of TPA initiation, 6.2% of those on endovascular therapy and 5.9% of those on TPA alone had a symptomatic intracerebral hemorrhage. The differences in mortality at 7 days and in parenchymal hematoma rates were also not significantly different between the two groups. The rate of asymptomatic intracerebral hemorrhage, however, was significantly higher in the endovascular group.
Outcomes consistently trended better with combined therapy in patients with strokes involving larger artery occlusions and those with the shortest times from stroke onset to initiation of treatment, although because of small patient numbers the differences didn’t achieve statistical significance. These will be the subgroups that ought to be the focus of future clinical trials, Dr. Broderick said in a press briefing at the conference.
The underlying rationale for combined therapy is that intravenous TPA can quickly be started in the emergency department while the endovascular device therapy team is assembling, often at another hospital, which entails time-consuming patient transfer.
Intravenous TPA is the only proven therapy for acute ischemic stroke, but endovascular therapy is more effective at achieving recanalization. The study results bore this out: for example, the rate of partial or complete recanalization at 24 hours for an occlusion in the internal carotid artery was 81% with combined therapy compared to 35% with intravenous TPA alone. Yet this higher recanalization rate bore no clinical benefit, possibly because recanalization occurred too late, after ischemia had turned into infarction, Dr. Broderick explained.
"IMS III is going to be disappointing for a lot of people who are proponents of endovascular therapy. However, there is a light at the end of the tunnel in that there are these subgroups who may benefit," Dr. Brian Silver, who was not involved in the trial, said in an interview.
"The most critical feature is to treat the patients as soon as possible when they arrive in the emergency department, perhaps within 90 minutes. I think that’s the best chance for recovery. We are nowhere near what’s being done in cardiology, where there are door-to-balloon times of an hour. We need to do that in stroke. Since we're dealing with an organ that's more sensitive than the heart to ischemia, we probably need to be even faster than what's being done in cardiology. There is definitely room for improvement in our systems, perhaps by having the endovascular team stay in the hospital. Expense will be the limitation," according to Dr. Silver, director of the stroke center at Brown University, Providence, R.I.
IMS III investigator and interventional neuroradiologist Dr. Thomas A. Tomsick said in an interview that the study results won't change his own clinical practice.
"IMS III is by no means the final word on combined therapy. In Cincinnati tomorrow, if a patient with a large NIH Stroke Severity score shows up and we're treating him with IV TPA at 2 hours from stroke onset, we're not going to do a CT angiogram to evaluate that patient. He's going to the cath lab for angiography to see if there's a clot suitable for endovascular therapy," said Dr. Tomsick, professor of radiology at the University of Cincinnati.
Five different endovascular device therapies were utilized in IMS III. As new devices reached clinical practice, their use was allowed by investigators in order to keep the randomized trial clinically relevant. But recruitment for the study was slow because so many clinicians were already convinced by anecdotal experience that combined therapy is better. So the endovascular therapies used most frequently in IMS III aren't the ones widely used in clinical practice today. Major new randomized trials are now getting underway comparing combined therapy using state-of-the-art, more effective stent clot retriever devices to intravenous TPA alone, he added.
In the New England Journal of Medicine report, the authors noted that "the use of randomization in ongoing and future stroke trials, rather than the treatment of eligible patients with endovascular therapy outside any trial, and minimization of the time to treatment will be essential for assessing the potential benefit of endovascular therapy for acute ischemic stroke."
No matter how future trials of combined therapy turn out, endovascular therapy is not going away, Dr. Broderick observed.
"It's a very good tool. The reason why is there are patients who can't get TPA. For example, roughly 5% of patients who undergo coronary artery bypass surgery have a stroke. If you have somebody with a big stroke 2 days after having their chest cracked, you can't use TPA. In that case, those endovascular devices are the way we can get up in there and get rid of the clot," he explained.
In an editorial accompanying the report in the New England Journal of Medicine (doi: 10.1056/NEJMe1215730), Dr. Marc I. Chimowitz declared that the clinical implication of IMS III is that endovascular therapy remains unproven and intravenous TPA should continue to be the first-line treatment for patients with acute ischemic stroke within 4.5 hours after stroke onset.
While new clinical trials featuring more effective IV clot busters, such as tenecteplase, and next-generation endovascular devices are urgently needed in an effort to improve stroke outcomes, patient recruitment is likely to continue to be a challenge in the current environment. This could be overcome if Medicare were to place a moratorium on reimbursement for endovascular therapy of acute ischemic stroke except as part of a randomized trial, according to Dr. Chimowitz, professor of neurology at the Medical University of South Carolina, Charleston.
The study was supported with grants from NIH and the National Institute of Neurological Disorders and Stroke; and by Genentech (which supplied the TPA); and EKOS, Concentric Medical, and Cordis Neurovascular (which supplied catheters); and Actilyse (alteplase) manufacturer Boehringer Ingelheim (which, along with Genentech and EKOS, provided support for investigator meetings). Dr. Broderick disclosed consulting fees from PhotoThera. Of the 28 other authors, disclosures for 14 were listed and included having received consulting fees, grant support, and/or lecture fees from a variety of device and pharmaceutical companies that include Genentech. Dr. Chimowitz, Dr. Silver, and Dr. Tomsick reported having no financial conflicts.
The key to understanding the results of this study is the difference in the recanalization rates between the two groups and their lack of relationship to outcome. In the IV TPA plus catheter directed TPA group, the recanalization rate was higher than in the IV alone group, but the clinical outcomes were not better. Undoubtedly this is due to the fact that the recanalization was accomplished after brain tissue death had already occurred.
Hence the next and critical question is....what would the result be if we could administer the catheter directed TPA in a timely fashion, as is done with acute myocardial infarction? If the study had been performed in centers where this therapy was available without delay, would the results have been different? I think almost certainly. While it is true that this therapy is not available as widely as coronary interventions, it is critical for us to know whether patients who are fortunate enough to be treated in an institution where the therapy is available should receive it. Another disappointing aspect of the study design was the inability to perform subgroup analysis. While it appeared that patients with larger stroke distribution might benefit, the study was apparently not powered to detect this difference. Lastly, it is puzzling that the investigators themselves do not appear to believe the results of their own study, with two of them indicating the results would not change their own practice. If that's the case, why spend all this time and money designing a trial that doesn't answer the questions?
Dr. Cynthia K. Shortell is Professor and Chief, Division of Vascular Surgery, Duke University Medical Center, and an associate medical editor for Vascular Specialist.
The key to understanding the results of this study is the difference in the recanalization rates between the two groups and their lack of relationship to outcome. In the IV TPA plus catheter directed TPA group, the recanalization rate was higher than in the IV alone group, but the clinical outcomes were not better. Undoubtedly this is due to the fact that the recanalization was accomplished after brain tissue death had already occurred.
Hence the next and critical question is....what would the result be if we could administer the catheter directed TPA in a timely fashion, as is done with acute myocardial infarction? If the study had been performed in centers where this therapy was available without delay, would the results have been different? I think almost certainly. While it is true that this therapy is not available as widely as coronary interventions, it is critical for us to know whether patients who are fortunate enough to be treated in an institution where the therapy is available should receive it. Another disappointing aspect of the study design was the inability to perform subgroup analysis. While it appeared that patients with larger stroke distribution might benefit, the study was apparently not powered to detect this difference. Lastly, it is puzzling that the investigators themselves do not appear to believe the results of their own study, with two of them indicating the results would not change their own practice. If that's the case, why spend all this time and money designing a trial that doesn't answer the questions?
Dr. Cynthia K. Shortell is Professor and Chief, Division of Vascular Surgery, Duke University Medical Center, and an associate medical editor for Vascular Specialist.
The key to understanding the results of this study is the difference in the recanalization rates between the two groups and their lack of relationship to outcome. In the IV TPA plus catheter directed TPA group, the recanalization rate was higher than in the IV alone group, but the clinical outcomes were not better. Undoubtedly this is due to the fact that the recanalization was accomplished after brain tissue death had already occurred.
Hence the next and critical question is....what would the result be if we could administer the catheter directed TPA in a timely fashion, as is done with acute myocardial infarction? If the study had been performed in centers where this therapy was available without delay, would the results have been different? I think almost certainly. While it is true that this therapy is not available as widely as coronary interventions, it is critical for us to know whether patients who are fortunate enough to be treated in an institution where the therapy is available should receive it. Another disappointing aspect of the study design was the inability to perform subgroup analysis. While it appeared that patients with larger stroke distribution might benefit, the study was apparently not powered to detect this difference. Lastly, it is puzzling that the investigators themselves do not appear to believe the results of their own study, with two of them indicating the results would not change their own practice. If that's the case, why spend all this time and money designing a trial that doesn't answer the questions?
Dr. Cynthia K. Shortell is Professor and Chief, Division of Vascular Surgery, Duke University Medical Center, and an associate medical editor for Vascular Specialist.
Functional outcomes in patients treated with intravenous tissue plasminogen activator with or without endovascular therapy after a moderate to severe acute ischemic stroke were not significantly different, and safety outcomes were similar, in a study that was stopped early because of these results.
In the IMS (Interventional Management of Stroke) III study, 40.8% of patients randomized to receive endovascular therapy plus intravenous TPA met the primary endpoint, a measure of functional independence -- a modified Rankin score of 2 or less at 90 days -- compared with 38.7% among those who had intravenous TPA alone, a difference that was not statistically significant, reported Dr. Joseph Broderick of the University of Cincinnati Neuroscience Institute, and the other IMS III investigators.
Mortality and other safety outcomes were also not significantly different between the two groups of patients in the study, which was stopped early because of futility after 656 of the planned 900 patients had been randomized.
The study was published online to coincide with the presentation of the results at the International Stroke Conference (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1214300]).
Referring to the lack of randomized clinical trial data, the authors pointed out that it is uncertain whether endovascular therapy (which includes endovascular pharmacologic thrombolysis and, more recently, the use of stent retrievers) alone or combined with intravenous TPA is a more effective treatment of acute stroke than intravenous TPA alone, "the only proven reperfusion therapy for acute ischemic stroke."
In the study, conducted at 58 centers in the United States, Canada, Australia, and Europe, 434 patients were randomized to endovascular therapy plus intravenous TPA and 222 were randomized to standard treatment with intravenous TPA alone (started within 3 hours of stroke onset). The median age of those enrolled was 68-69 years (range, 23-89 years), a little over half were men, about 14% were black or Hispanic, and the median the National Institute of Health Stroke Scale (NIHSS) score was 16-17 (8-19 is a moderately severe stroke and 20 or greater is a severe stroke At the beginning of the study, only one thrombectomy device had been cleared by the Food and Drug Administration and, as the trial continued, other devices were used as they became cleared for use in the different countries.
In addition to the main finding, there were no differences in the primary outcome among those patients with an NIHSS score of 20 or more, and those with a score of 19 or lower, said the authors, who had hypothesized that endovascular therapy would have greater efficacy in patients with more-severe strokes since they "have the highest likelihood of occlusion in a major intracranial artery and the greatest volume of ischemic brain at risk."
They had also hypothesized that receiving endovascular therapy earlier would be associated with a greater benefit, but this was also not a significant factor in outcomes.
Mortality at 90 days was 19.1% in the endovascular therapy group and 21.6% in the intravenous TPA–alone group. Within 30 hours of TPA initiation, 6.2% of those on endovascular therapy and 5.9% of those on TPA alone had a symptomatic intracerebral hemorrhage. The differences in mortality at 7 days and in parenchymal hematoma rates were also not significantly different between the two groups. The rate of asymptomatic intracerebral hemorrhage, however, was significantly higher in the endovascular group.
Outcomes consistently trended better with combined therapy in patients with strokes involving larger artery occlusions and those with the shortest times from stroke onset to initiation of treatment, although because of small patient numbers the differences didn’t achieve statistical significance. These will be the subgroups that ought to be the focus of future clinical trials, Dr. Broderick said in a press briefing at the conference.
The underlying rationale for combined therapy is that intravenous TPA can quickly be started in the emergency department while the endovascular device therapy team is assembling, often at another hospital, which entails time-consuming patient transfer.
Intravenous TPA is the only proven therapy for acute ischemic stroke, but endovascular therapy is more effective at achieving recanalization. The study results bore this out: for example, the rate of partial or complete recanalization at 24 hours for an occlusion in the internal carotid artery was 81% with combined therapy compared to 35% with intravenous TPA alone. Yet this higher recanalization rate bore no clinical benefit, possibly because recanalization occurred too late, after ischemia had turned into infarction, Dr. Broderick explained.
"IMS III is going to be disappointing for a lot of people who are proponents of endovascular therapy. However, there is a light at the end of the tunnel in that there are these subgroups who may benefit," Dr. Brian Silver, who was not involved in the trial, said in an interview.
"The most critical feature is to treat the patients as soon as possible when they arrive in the emergency department, perhaps within 90 minutes. I think that’s the best chance for recovery. We are nowhere near what’s being done in cardiology, where there are door-to-balloon times of an hour. We need to do that in stroke. Since we're dealing with an organ that's more sensitive than the heart to ischemia, we probably need to be even faster than what's being done in cardiology. There is definitely room for improvement in our systems, perhaps by having the endovascular team stay in the hospital. Expense will be the limitation," according to Dr. Silver, director of the stroke center at Brown University, Providence, R.I.
IMS III investigator and interventional neuroradiologist Dr. Thomas A. Tomsick said in an interview that the study results won't change his own clinical practice.
"IMS III is by no means the final word on combined therapy. In Cincinnati tomorrow, if a patient with a large NIH Stroke Severity score shows up and we're treating him with IV TPA at 2 hours from stroke onset, we're not going to do a CT angiogram to evaluate that patient. He's going to the cath lab for angiography to see if there's a clot suitable for endovascular therapy," said Dr. Tomsick, professor of radiology at the University of Cincinnati.
Five different endovascular device therapies were utilized in IMS III. As new devices reached clinical practice, their use was allowed by investigators in order to keep the randomized trial clinically relevant. But recruitment for the study was slow because so many clinicians were already convinced by anecdotal experience that combined therapy is better. So the endovascular therapies used most frequently in IMS III aren't the ones widely used in clinical practice today. Major new randomized trials are now getting underway comparing combined therapy using state-of-the-art, more effective stent clot retriever devices to intravenous TPA alone, he added.
In the New England Journal of Medicine report, the authors noted that "the use of randomization in ongoing and future stroke trials, rather than the treatment of eligible patients with endovascular therapy outside any trial, and minimization of the time to treatment will be essential for assessing the potential benefit of endovascular therapy for acute ischemic stroke."
No matter how future trials of combined therapy turn out, endovascular therapy is not going away, Dr. Broderick observed.
"It's a very good tool. The reason why is there are patients who can't get TPA. For example, roughly 5% of patients who undergo coronary artery bypass surgery have a stroke. If you have somebody with a big stroke 2 days after having their chest cracked, you can't use TPA. In that case, those endovascular devices are the way we can get up in there and get rid of the clot," he explained.
In an editorial accompanying the report in the New England Journal of Medicine (doi: 10.1056/NEJMe1215730), Dr. Marc I. Chimowitz declared that the clinical implication of IMS III is that endovascular therapy remains unproven and intravenous TPA should continue to be the first-line treatment for patients with acute ischemic stroke within 4.5 hours after stroke onset.
While new clinical trials featuring more effective IV clot busters, such as tenecteplase, and next-generation endovascular devices are urgently needed in an effort to improve stroke outcomes, patient recruitment is likely to continue to be a challenge in the current environment. This could be overcome if Medicare were to place a moratorium on reimbursement for endovascular therapy of acute ischemic stroke except as part of a randomized trial, according to Dr. Chimowitz, professor of neurology at the Medical University of South Carolina, Charleston.
The study was supported with grants from NIH and the National Institute of Neurological Disorders and Stroke; and by Genentech (which supplied the TPA); and EKOS, Concentric Medical, and Cordis Neurovascular (which supplied catheters); and Actilyse (alteplase) manufacturer Boehringer Ingelheim (which, along with Genentech and EKOS, provided support for investigator meetings). Dr. Broderick disclosed consulting fees from PhotoThera. Of the 28 other authors, disclosures for 14 were listed and included having received consulting fees, grant support, and/or lecture fees from a variety of device and pharmaceutical companies that include Genentech. Dr. Chimowitz, Dr. Silver, and Dr. Tomsick reported having no financial conflicts.
Functional outcomes in patients treated with intravenous tissue plasminogen activator with or without endovascular therapy after a moderate to severe acute ischemic stroke were not significantly different, and safety outcomes were similar, in a study that was stopped early because of these results.
In the IMS (Interventional Management of Stroke) III study, 40.8% of patients randomized to receive endovascular therapy plus intravenous TPA met the primary endpoint, a measure of functional independence -- a modified Rankin score of 2 or less at 90 days -- compared with 38.7% among those who had intravenous TPA alone, a difference that was not statistically significant, reported Dr. Joseph Broderick of the University of Cincinnati Neuroscience Institute, and the other IMS III investigators.
Mortality and other safety outcomes were also not significantly different between the two groups of patients in the study, which was stopped early because of futility after 656 of the planned 900 patients had been randomized.
The study was published online to coincide with the presentation of the results at the International Stroke Conference (N. Engl. J. Med. 2013 [doi:10.1056/NEJMoa1214300]).
Referring to the lack of randomized clinical trial data, the authors pointed out that it is uncertain whether endovascular therapy (which includes endovascular pharmacologic thrombolysis and, more recently, the use of stent retrievers) alone or combined with intravenous TPA is a more effective treatment of acute stroke than intravenous TPA alone, "the only proven reperfusion therapy for acute ischemic stroke."
In the study, conducted at 58 centers in the United States, Canada, Australia, and Europe, 434 patients were randomized to endovascular therapy plus intravenous TPA and 222 were randomized to standard treatment with intravenous TPA alone (started within 3 hours of stroke onset). The median age of those enrolled was 68-69 years (range, 23-89 years), a little over half were men, about 14% were black or Hispanic, and the median the National Institute of Health Stroke Scale (NIHSS) score was 16-17 (8-19 is a moderately severe stroke and 20 or greater is a severe stroke At the beginning of the study, only one thrombectomy device had been cleared by the Food and Drug Administration and, as the trial continued, other devices were used as they became cleared for use in the different countries.
In addition to the main finding, there were no differences in the primary outcome among those patients with an NIHSS score of 20 or more, and those with a score of 19 or lower, said the authors, who had hypothesized that endovascular therapy would have greater efficacy in patients with more-severe strokes since they "have the highest likelihood of occlusion in a major intracranial artery and the greatest volume of ischemic brain at risk."
They had also hypothesized that receiving endovascular therapy earlier would be associated with a greater benefit, but this was also not a significant factor in outcomes.
Mortality at 90 days was 19.1% in the endovascular therapy group and 21.6% in the intravenous TPA–alone group. Within 30 hours of TPA initiation, 6.2% of those on endovascular therapy and 5.9% of those on TPA alone had a symptomatic intracerebral hemorrhage. The differences in mortality at 7 days and in parenchymal hematoma rates were also not significantly different between the two groups. The rate of asymptomatic intracerebral hemorrhage, however, was significantly higher in the endovascular group.
Outcomes consistently trended better with combined therapy in patients with strokes involving larger artery occlusions and those with the shortest times from stroke onset to initiation of treatment, although because of small patient numbers the differences didn’t achieve statistical significance. These will be the subgroups that ought to be the focus of future clinical trials, Dr. Broderick said in a press briefing at the conference.
The underlying rationale for combined therapy is that intravenous TPA can quickly be started in the emergency department while the endovascular device therapy team is assembling, often at another hospital, which entails time-consuming patient transfer.
Intravenous TPA is the only proven therapy for acute ischemic stroke, but endovascular therapy is more effective at achieving recanalization. The study results bore this out: for example, the rate of partial or complete recanalization at 24 hours for an occlusion in the internal carotid artery was 81% with combined therapy compared to 35% with intravenous TPA alone. Yet this higher recanalization rate bore no clinical benefit, possibly because recanalization occurred too late, after ischemia had turned into infarction, Dr. Broderick explained.
"IMS III is going to be disappointing for a lot of people who are proponents of endovascular therapy. However, there is a light at the end of the tunnel in that there are these subgroups who may benefit," Dr. Brian Silver, who was not involved in the trial, said in an interview.
"The most critical feature is to treat the patients as soon as possible when they arrive in the emergency department, perhaps within 90 minutes. I think that’s the best chance for recovery. We are nowhere near what’s being done in cardiology, where there are door-to-balloon times of an hour. We need to do that in stroke. Since we're dealing with an organ that's more sensitive than the heart to ischemia, we probably need to be even faster than what's being done in cardiology. There is definitely room for improvement in our systems, perhaps by having the endovascular team stay in the hospital. Expense will be the limitation," according to Dr. Silver, director of the stroke center at Brown University, Providence, R.I.
IMS III investigator and interventional neuroradiologist Dr. Thomas A. Tomsick said in an interview that the study results won't change his own clinical practice.
"IMS III is by no means the final word on combined therapy. In Cincinnati tomorrow, if a patient with a large NIH Stroke Severity score shows up and we're treating him with IV TPA at 2 hours from stroke onset, we're not going to do a CT angiogram to evaluate that patient. He's going to the cath lab for angiography to see if there's a clot suitable for endovascular therapy," said Dr. Tomsick, professor of radiology at the University of Cincinnati.
Five different endovascular device therapies were utilized in IMS III. As new devices reached clinical practice, their use was allowed by investigators in order to keep the randomized trial clinically relevant. But recruitment for the study was slow because so many clinicians were already convinced by anecdotal experience that combined therapy is better. So the endovascular therapies used most frequently in IMS III aren't the ones widely used in clinical practice today. Major new randomized trials are now getting underway comparing combined therapy using state-of-the-art, more effective stent clot retriever devices to intravenous TPA alone, he added.
In the New England Journal of Medicine report, the authors noted that "the use of randomization in ongoing and future stroke trials, rather than the treatment of eligible patients with endovascular therapy outside any trial, and minimization of the time to treatment will be essential for assessing the potential benefit of endovascular therapy for acute ischemic stroke."
No matter how future trials of combined therapy turn out, endovascular therapy is not going away, Dr. Broderick observed.
"It's a very good tool. The reason why is there are patients who can't get TPA. For example, roughly 5% of patients who undergo coronary artery bypass surgery have a stroke. If you have somebody with a big stroke 2 days after having their chest cracked, you can't use TPA. In that case, those endovascular devices are the way we can get up in there and get rid of the clot," he explained.
In an editorial accompanying the report in the New England Journal of Medicine (doi: 10.1056/NEJMe1215730), Dr. Marc I. Chimowitz declared that the clinical implication of IMS III is that endovascular therapy remains unproven and intravenous TPA should continue to be the first-line treatment for patients with acute ischemic stroke within 4.5 hours after stroke onset.
While new clinical trials featuring more effective IV clot busters, such as tenecteplase, and next-generation endovascular devices are urgently needed in an effort to improve stroke outcomes, patient recruitment is likely to continue to be a challenge in the current environment. This could be overcome if Medicare were to place a moratorium on reimbursement for endovascular therapy of acute ischemic stroke except as part of a randomized trial, according to Dr. Chimowitz, professor of neurology at the Medical University of South Carolina, Charleston.
The study was supported with grants from NIH and the National Institute of Neurological Disorders and Stroke; and by Genentech (which supplied the TPA); and EKOS, Concentric Medical, and Cordis Neurovascular (which supplied catheters); and Actilyse (alteplase) manufacturer Boehringer Ingelheim (which, along with Genentech and EKOS, provided support for investigator meetings). Dr. Broderick disclosed consulting fees from PhotoThera. Of the 28 other authors, disclosures for 14 were listed and included having received consulting fees, grant support, and/or lecture fees from a variety of device and pharmaceutical companies that include Genentech. Dr. Chimowitz, Dr. Silver, and Dr. Tomsick reported having no financial conflicts.
FROM THE INTERNATIONAL STROKE CONFERENCE
Major Finding: Endovascular therapy plus intravenous TPA showed no added outcome benefits, compared with TPA alone, with 40.8% and 38.7% of patients, respectively, reaching functional independence at 90 days.
Data Source: An international, phase III study of 656 patients with an acute moderate to severe ischemic stroke randomized 2:1 to IV TPA with endovascular therapy or IV TPA alone.
Disclosures: The study was supported with grants from NIH and the National Institute of Neurological Disorders and Stroke, and by Genentech (supplier of TPA), and EKOS, Concentric Medical, and Cordis Neurovascular (which supplied catheters). Dr. Broderick disclosed consulting fees from PhotoThera. Of the 28 other authors, disclosures for 14 were listed and included having received consulting fees, grant support, and/or lecture fees from a variety of device and pharmaceutical companies that included Genentech. Dr. Chimowitz, Dr. Silver, and Dr. Tomsick reported having no financial conflicts.

Autism, autism spectrum disorder risk increased with prenatal valproate exposure
The risk of having an autism spectrum disorder and childhood autism was significantly increased among children exposed prenatally to valproate, which persisted after researchers adjusted for parental psychiatric disease and maternal epilepsy in a Danish study that evaluated children up to age 14 years.
These risks were higher than the risks among the children whose mothers had stopped taking valproate before conceiving and also was elevated among the children of women who did not have epilepsy and took valproate, "indicating an association with valproate exposure," and that valproate may have a biological effect, said Jakob Christensen, Ph.D., of the department of neurology at Aarhus (Denmark) University Hospital, and his associates.

"For women of childbearing potential who use antiepileptic medications, these findings must be balanced against the treatment benefits for women who require valproate for epilepsy control," they concluded (JAMA 2013;309:1696-703).
The population-based study used a birth registry of all children born alive in Denmark between January 1996 through Dec. 31, 2006, for a total of 655,107 children who had not been exposed to valproate and 508 exposed to valproate prenatally. At the end of follow-up, when the children were a mean age of almost 9 years (range, 4-14 years), 5,437 children had been diagnosed with autism spectrum disorder, including 2,067 with childhood autism. A national prescription registry was used to document exposure to antiepilepsy drugs (exposure during pregnancy was considered 30 days before conception to the day of birth) and a registry of psychiatric diagnoses was used to identify which children were diagnosed with autism.
Among the 508 children exposed to valproate, the incidence of autism spectrum disorder was 4.42% (hazard ratio, 2.9), and the incidence of childhood autism was 2.5% (HR, 5.2). The hazard ratios were adjusted for known autism risk factors, including age of the parents at conception, parental psychiatric history, and sex of the child.
Among the 432 children whose mothers had taken valproate for epilepsy (not another diagnosis treated with valproate), the incidence of autism spectrum disorder was 4.15% (HR, 1.7), and the incidence of childhood autism was 2.95% (HR, 2.9). Among the 6,152 children not exposed to valproate, the incidence of autism spectrum disorder was 2.44% and the incidence of childhood autism was 1.02%. The risks for both autism spectrum disorder and childhood autism were increased among women who took valproate during pregnancy, compared with the children whose mothers had taken valproate but had stopped taking it at least 30 days before they were conceived
The strengths of the study included the unlikely effect of selection bias because the investigators were able to follow-up most of the children born in the country over 14 years, and the high quality of autism diagnoses in the psychiatric registry. The possibility that some pregnancies exposed to valproate may have been missed was among the weaknesses of the study because the prescription registry only includes outpatient prescriptions.
The study was funded by grants from the European Research Council and the Danish Medical Research Council. Dr. Christensen received research support from the Danish Epilepsy Association and honoraria for serving on scientific advisory boards and lecture honoraria from UCB Nordic and Eisai AB, and received travel funding from UCB Nordic.
Dr. Meador received research support from the National Institutes of Health, the Patient-Centered Outcomes Research Institute, Pfizer, and UCB Pharma; Emory University received salary support from the Epilepsy Consortium, which gets funding from NeuroPace, Novartis, Upsher-Smith Laboratories, UCB Pharma, and Vivus Pharmaceuticals; and travel support from Sanofi Aventis. Dr. Loring received consulting fees from NeuroPace; grant support from NIH, the Patient-Centered Outcomes Research Institute, Pfizer, and UCB Pharma; and book royalties from Oxford University Press.
This association has long been suspected and is not surprising. What is surprising, as stated in the editorial, is that despite the fact that there is growing and convincing evidence that this drug is linked to a substantial increased risk for major birth defects and cognitive performance deficits, the prescribing practices – at least in some countries – do not seem to reflect this.
In my opinion, pregnant women and women who have the potential to become pregnant who are taking valproate should consult with their providers about the possibility of alternative medications. Clearly, valproate cannot be a major cause of autism – the prevalence of use in women is far less than the rate of children with autism spectrum disorders – but this association might help uncover at least one of the mechanisms underlying initiation of the disorder.
Christina Chambers, Ph.D., M.P.H., is codirector of the Center for Promotion of Maternal Health and Infant Development at the University of California, San Diego. Dr. Chambers is a past president of the Organization of Teratology Information Specialists and past president of the Teratology Society. She said she had no relevant financial disclosures.
This association has long been suspected and is not surprising. What is surprising, as stated in the editorial, is that despite the fact that there is growing and convincing evidence that this drug is linked to a substantial increased risk for major birth defects and cognitive performance deficits, the prescribing practices – at least in some countries – do not seem to reflect this.
In my opinion, pregnant women and women who have the potential to become pregnant who are taking valproate should consult with their providers about the possibility of alternative medications. Clearly, valproate cannot be a major cause of autism – the prevalence of use in women is far less than the rate of children with autism spectrum disorders – but this association might help uncover at least one of the mechanisms underlying initiation of the disorder.
Christina Chambers, Ph.D., M.P.H., is codirector of the Center for Promotion of Maternal Health and Infant Development at the University of California, San Diego. Dr. Chambers is a past president of the Organization of Teratology Information Specialists and past president of the Teratology Society. She said she had no relevant financial disclosures.
This association has long been suspected and is not surprising. What is surprising, as stated in the editorial, is that despite the fact that there is growing and convincing evidence that this drug is linked to a substantial increased risk for major birth defects and cognitive performance deficits, the prescribing practices – at least in some countries – do not seem to reflect this.
In my opinion, pregnant women and women who have the potential to become pregnant who are taking valproate should consult with their providers about the possibility of alternative medications. Clearly, valproate cannot be a major cause of autism – the prevalence of use in women is far less than the rate of children with autism spectrum disorders – but this association might help uncover at least one of the mechanisms underlying initiation of the disorder.
Christina Chambers, Ph.D., M.P.H., is codirector of the Center for Promotion of Maternal Health and Infant Development at the University of California, San Diego. Dr. Chambers is a past president of the Organization of Teratology Information Specialists and past president of the Teratology Society. She said she had no relevant financial disclosures.
The risk of having an autism spectrum disorder and childhood autism was significantly increased among children exposed prenatally to valproate, which persisted after researchers adjusted for parental psychiatric disease and maternal epilepsy in a Danish study that evaluated children up to age 14 years.
These risks were higher than the risks among the children whose mothers had stopped taking valproate before conceiving and also was elevated among the children of women who did not have epilepsy and took valproate, "indicating an association with valproate exposure," and that valproate may have a biological effect, said Jakob Christensen, Ph.D., of the department of neurology at Aarhus (Denmark) University Hospital, and his associates.
"For women of childbearing potential who use antiepileptic medications, these findings must be balanced against the treatment benefits for women who require valproate for epilepsy control," they concluded (JAMA 2013;309:1696-703).
The population-based study used a birth registry of all children born alive in Denmark between January 1996 through Dec. 31, 2006, for a total of 655,107 children who had not been exposed to valproate and 508 exposed to valproate prenatally. At the end of follow-up, when the children were a mean age of almost 9 years (range, 4-14 years), 5,437 children had been diagnosed with autism spectrum disorder, including 2,067 with childhood autism. A national prescription registry was used to document exposure to antiepilepsy drugs (exposure during pregnancy was considered 30 days before conception to the day of birth) and a registry of psychiatric diagnoses was used to identify which children were diagnosed with autism.
Among the 508 children exposed to valproate, the incidence of autism spectrum disorder was 4.42% (hazard ratio, 2.9), and the incidence of childhood autism was 2.5% (HR, 5.2). The hazard ratios were adjusted for known autism risk factors, including age of the parents at conception, parental psychiatric history, and sex of the child.
Among the 432 children whose mothers had taken valproate for epilepsy (not another diagnosis treated with valproate), the incidence of autism spectrum disorder was 4.15% (HR, 1.7), and the incidence of childhood autism was 2.95% (HR, 2.9). Among the 6,152 children not exposed to valproate, the incidence of autism spectrum disorder was 2.44% and the incidence of childhood autism was 1.02%. The risks for both autism spectrum disorder and childhood autism were increased among women who took valproate during pregnancy, compared with the children whose mothers had taken valproate but had stopped taking it at least 30 days before they were conceived
The strengths of the study included the unlikely effect of selection bias because the investigators were able to follow-up most of the children born in the country over 14 years, and the high quality of autism diagnoses in the psychiatric registry. The possibility that some pregnancies exposed to valproate may have been missed was among the weaknesses of the study because the prescription registry only includes outpatient prescriptions.
The study was funded by grants from the European Research Council and the Danish Medical Research Council. Dr. Christensen received research support from the Danish Epilepsy Association and honoraria for serving on scientific advisory boards and lecture honoraria from UCB Nordic and Eisai AB, and received travel funding from UCB Nordic.
Dr. Meador received research support from the National Institutes of Health, the Patient-Centered Outcomes Research Institute, Pfizer, and UCB Pharma; Emory University received salary support from the Epilepsy Consortium, which gets funding from NeuroPace, Novartis, Upsher-Smith Laboratories, UCB Pharma, and Vivus Pharmaceuticals; and travel support from Sanofi Aventis. Dr. Loring received consulting fees from NeuroPace; grant support from NIH, the Patient-Centered Outcomes Research Institute, Pfizer, and UCB Pharma; and book royalties from Oxford University Press.
The risk of having an autism spectrum disorder and childhood autism was significantly increased among children exposed prenatally to valproate, which persisted after researchers adjusted for parental psychiatric disease and maternal epilepsy in a Danish study that evaluated children up to age 14 years.
These risks were higher than the risks among the children whose mothers had stopped taking valproate before conceiving and also was elevated among the children of women who did not have epilepsy and took valproate, "indicating an association with valproate exposure," and that valproate may have a biological effect, said Jakob Christensen, Ph.D., of the department of neurology at Aarhus (Denmark) University Hospital, and his associates.
"For women of childbearing potential who use antiepileptic medications, these findings must be balanced against the treatment benefits for women who require valproate for epilepsy control," they concluded (JAMA 2013;309:1696-703).
The population-based study used a birth registry of all children born alive in Denmark between January 1996 through Dec. 31, 2006, for a total of 655,107 children who had not been exposed to valproate and 508 exposed to valproate prenatally. At the end of follow-up, when the children were a mean age of almost 9 years (range, 4-14 years), 5,437 children had been diagnosed with autism spectrum disorder, including 2,067 with childhood autism. A national prescription registry was used to document exposure to antiepilepsy drugs (exposure during pregnancy was considered 30 days before conception to the day of birth) and a registry of psychiatric diagnoses was used to identify which children were diagnosed with autism.
Among the 508 children exposed to valproate, the incidence of autism spectrum disorder was 4.42% (hazard ratio, 2.9), and the incidence of childhood autism was 2.5% (HR, 5.2). The hazard ratios were adjusted for known autism risk factors, including age of the parents at conception, parental psychiatric history, and sex of the child.
Among the 432 children whose mothers had taken valproate for epilepsy (not another diagnosis treated with valproate), the incidence of autism spectrum disorder was 4.15% (HR, 1.7), and the incidence of childhood autism was 2.95% (HR, 2.9). Among the 6,152 children not exposed to valproate, the incidence of autism spectrum disorder was 2.44% and the incidence of childhood autism was 1.02%. The risks for both autism spectrum disorder and childhood autism were increased among women who took valproate during pregnancy, compared with the children whose mothers had taken valproate but had stopped taking it at least 30 days before they were conceived
The strengths of the study included the unlikely effect of selection bias because the investigators were able to follow-up most of the children born in the country over 14 years, and the high quality of autism diagnoses in the psychiatric registry. The possibility that some pregnancies exposed to valproate may have been missed was among the weaknesses of the study because the prescription registry only includes outpatient prescriptions.
The study was funded by grants from the European Research Council and the Danish Medical Research Council. Dr. Christensen received research support from the Danish Epilepsy Association and honoraria for serving on scientific advisory boards and lecture honoraria from UCB Nordic and Eisai AB, and received travel funding from UCB Nordic.
Dr. Meador received research support from the National Institutes of Health, the Patient-Centered Outcomes Research Institute, Pfizer, and UCB Pharma; Emory University received salary support from the Epilepsy Consortium, which gets funding from NeuroPace, Novartis, Upsher-Smith Laboratories, UCB Pharma, and Vivus Pharmaceuticals; and travel support from Sanofi Aventis. Dr. Loring received consulting fees from NeuroPace; grant support from NIH, the Patient-Centered Outcomes Research Institute, Pfizer, and UCB Pharma; and book royalties from Oxford University Press.
FROM JAMA
Major finding: Among the 508 children exposed to valproate, the incidence of autism spectrum disorder was 4.42% (HR, 2.9), and the incidence of childhood autism was 2.5% (HR, 5.2).
Data source: A population based study used data from national registries to evaluate the association between autism and prenatal exposure to valproate, using data from all 655,615 children born in Denmark from 1996 to 2006 (after certain exclusions) and a national registry of all prescriptions filled since January 1996.
Disclosures: The study was funded by grants from the European Research Council and the Danish Medical Research Council. Dr. Christensen received research support from the Danish Epilepsy Association and honoraria for serving on scientific advisory boards and lecture honoraria from UCB Nordic and Eisai AB, and received travel funding from UCB Nordic.
Dr. Meador received research support from the National Institutes of Health, the Patient-Centered Outcomes Research Institute, Pfizer, and UCB Pharma; Emory University received salary support from the Epilepsy Consortium, which gets funding from NeuroPace, Novartis, Upsher-Smith Laboratories, UCB Pharma, and Vivus Pharmaceuticals; and travel support from Sanofi Aventis. Dr. Loring received consulting fees from NeuroPace; grant support from NIH, the Patient-Centered Outcomes Research Institute, Pfizer, and UCB Pharma; and book royalties from Oxford University Press.

FDA panel split over long-acting testosterone's safety
SILVER SPRING, MD. – Considering reports of severe reactions following testosterone undecanoate injections, advisers to the Food and Drug Administration were evenly divided over the question of whether this risk rendered the product unsafe for use in the United States, as replacement therapy for men with hypogonadism.
The FDA is considering the approval of this long-acting testosterone – which has been available outside of the United States since 2003 – and convened the meeting of two advisory panels to review the safety data and the company’s plans for managing this risk, if approved in the United States.
Efficacy is not an issue: The agency has concluded that the 750-mg intramuscular dose of testosterone undecanoate, administered once every 10 weeks, meets the regulatory requirements for efficacy for a testosterone replacement product, based on results of serum testosterone levels in the single U.S. open-label study of 131 patients. But cases of anaphylaxis and pulmonary oil microembolism (POME), during or shortly after injections are administered, reported in clinical and postmarketing studies in countries in which it has been approved, has held up approval in the United States since Endo Pharmaceuticals filed for approval in 2007.
At the April 18 meeting, the FDA’s Advisory Committee for Reproductive Health Drugs and the Drug Safety and Risk Management Advisory Committee voted 9-9, when asked whether they believed that testosterone undecanoate (TU), a long-acting depot formulation of testosterone in castor oil and benzyl benzoate, was safe for the indication under review: replacement therapy in adult men for conditions associated with an absence or deficiency of endogenous testosterone. Those who felt the safety profile was acceptable for approval were concerned that the product was being held to a higher standard than other FDA-approved injectable testosterone products, which have labels that list anaphylaxis as a possible risk, and cited extensive use of the drug for years in Europe and no reports of deaths from these severe reactions.
But those voting no said that while they appreciated the need for a long-acting testosterone injection, there were not enough data to fully evaluate its safety and it was not possible to identify who is at an increased risk for having severe reactions. And, once approved, it might be used outside of the indication and would increase risk.
To address the risks of anaphylaxis and pulmonary oil microembolism (POME), Endo Pharmaceuticals has proposed that the injection be administered slowly, over 30 to 60 seconds in a physician’s office, and that patients be observed for 30 minutes afterward, with these instructions included in the product label. But the FDA panels voted 17-1 that this would not be sufficient to ameliorate this risk, calling for more interventions, including a black box warning, a training program for physicians who administer the injections to ensure they understand the severe risks (which could include an online course), and a more narrow indication to limit inappropriate use.
Although it makes sense clinically, "We don’t know the cause of this, so how can we know how to ameliorate this?" said panelist Dr. Richard Bockman, head of the endocrine service at the Hospital for Special Surgery, New York.
The features of POME are a cough or dyspnea, with or without other symptoms that can also include throat irritation, malaise, profuse sweating, chest pain, dizziness, paresthesia or syncope, which occurs within 24 hours of the injection, according to the FDA.
The company’s risk evaluation and mitigation strategy (REMS) includes physician and patient education and controlled distribution to health care providers for administration only in the office, with a 30-minute wait after the injection.
But Suzanne Robottom, Pharm.D., a senior risk management analyst in the FDA’s division of risk management, said that in Europe, cases of POME continue to be reported despite recommendations to administer the injection slowly and observe patients for 30 minutes. The agency is concerned that implementing the measures proposed by the company, which include prescribing and distribution restrictions, would pose an undue burden "for a drug with limited additional benefit," she added.
An FDA review of 199 postmarketing cases of POME, between November 2003 and April 2012, determined that 84 were severe, with severe dyspnea, loss of consciousness, circulatory collapse, or loss of bowel function. Many cases started as the medication was being injected. Although no deaths were reported, the long-term cardiopulmonary effects of severe cases or chronic effects of repeated less-severe episodes are not known, according to the FDA. During 2003-2011, there were 53 postmarketing cases of anaphylaxis reported, using a conservative definition.

The testosterone replacement products currently approved by the FDA are two intramuscular testosterone products (injected every 2-4 weeks), transdermal patches, gel, a solution, a buccal bioadhesive system, an oral tablet, and a subcutaneous implant.
Speaking on behalf of the Endocrine Society during the open public hearing portion of the meeting, Dr. Allan Glass, an endocrinologist in Bethesda, Md., said that the Society’s current guidelines for the treatment of androgen deficiency say that TU is an acceptable treatment in countries in which it has been approved (J. Clin. Endocrinol. Metab. 2010;95:2536-59).
If approved in the United States, TU will be marketed as Aveed. It is marketed in about 72 countries, and in Europe it is marketed as Nebido. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Dr. Glass had no financial disclosures.
SILVER SPRING, MD. – Considering reports of severe reactions following testosterone undecanoate injections, advisers to the Food and Drug Administration were evenly divided over the question of whether this risk rendered the product unsafe for use in the United States, as replacement therapy for men with hypogonadism.
The FDA is considering the approval of this long-acting testosterone – which has been available outside of the United States since 2003 – and convened the meeting of two advisory panels to review the safety data and the company’s plans for managing this risk, if approved in the United States.
Efficacy is not an issue: The agency has concluded that the 750-mg intramuscular dose of testosterone undecanoate, administered once every 10 weeks, meets the regulatory requirements for efficacy for a testosterone replacement product, based on results of serum testosterone levels in the single U.S. open-label study of 131 patients. But cases of anaphylaxis and pulmonary oil microembolism (POME), during or shortly after injections are administered, reported in clinical and postmarketing studies in countries in which it has been approved, has held up approval in the United States since Endo Pharmaceuticals filed for approval in 2007.
At the April 18 meeting, the FDA’s Advisory Committee for Reproductive Health Drugs and the Drug Safety and Risk Management Advisory Committee voted 9-9, when asked whether they believed that testosterone undecanoate (TU), a long-acting depot formulation of testosterone in castor oil and benzyl benzoate, was safe for the indication under review: replacement therapy in adult men for conditions associated with an absence or deficiency of endogenous testosterone. Those who felt the safety profile was acceptable for approval were concerned that the product was being held to a higher standard than other FDA-approved injectable testosterone products, which have labels that list anaphylaxis as a possible risk, and cited extensive use of the drug for years in Europe and no reports of deaths from these severe reactions.
But those voting no said that while they appreciated the need for a long-acting testosterone injection, there were not enough data to fully evaluate its safety and it was not possible to identify who is at an increased risk for having severe reactions. And, once approved, it might be used outside of the indication and would increase risk.
To address the risks of anaphylaxis and pulmonary oil microembolism (POME), Endo Pharmaceuticals has proposed that the injection be administered slowly, over 30 to 60 seconds in a physician’s office, and that patients be observed for 30 minutes afterward, with these instructions included in the product label. But the FDA panels voted 17-1 that this would not be sufficient to ameliorate this risk, calling for more interventions, including a black box warning, a training program for physicians who administer the injections to ensure they understand the severe risks (which could include an online course), and a more narrow indication to limit inappropriate use.
Although it makes sense clinically, "We don’t know the cause of this, so how can we know how to ameliorate this?" said panelist Dr. Richard Bockman, head of the endocrine service at the Hospital for Special Surgery, New York.
The features of POME are a cough or dyspnea, with or without other symptoms that can also include throat irritation, malaise, profuse sweating, chest pain, dizziness, paresthesia or syncope, which occurs within 24 hours of the injection, according to the FDA.
The company’s risk evaluation and mitigation strategy (REMS) includes physician and patient education and controlled distribution to health care providers for administration only in the office, with a 30-minute wait after the injection.
But Suzanne Robottom, Pharm.D., a senior risk management analyst in the FDA’s division of risk management, said that in Europe, cases of POME continue to be reported despite recommendations to administer the injection slowly and observe patients for 30 minutes. The agency is concerned that implementing the measures proposed by the company, which include prescribing and distribution restrictions, would pose an undue burden "for a drug with limited additional benefit," she added.
An FDA review of 199 postmarketing cases of POME, between November 2003 and April 2012, determined that 84 were severe, with severe dyspnea, loss of consciousness, circulatory collapse, or loss of bowel function. Many cases started as the medication was being injected. Although no deaths were reported, the long-term cardiopulmonary effects of severe cases or chronic effects of repeated less-severe episodes are not known, according to the FDA. During 2003-2011, there were 53 postmarketing cases of anaphylaxis reported, using a conservative definition.
The testosterone replacement products currently approved by the FDA are two intramuscular testosterone products (injected every 2-4 weeks), transdermal patches, gel, a solution, a buccal bioadhesive system, an oral tablet, and a subcutaneous implant.
Speaking on behalf of the Endocrine Society during the open public hearing portion of the meeting, Dr. Allan Glass, an endocrinologist in Bethesda, Md., said that the Society’s current guidelines for the treatment of androgen deficiency say that TU is an acceptable treatment in countries in which it has been approved (J. Clin. Endocrinol. Metab. 2010;95:2536-59).
If approved in the United States, TU will be marketed as Aveed. It is marketed in about 72 countries, and in Europe it is marketed as Nebido. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Dr. Glass had no financial disclosures.
SILVER SPRING, MD. – Considering reports of severe reactions following testosterone undecanoate injections, advisers to the Food and Drug Administration were evenly divided over the question of whether this risk rendered the product unsafe for use in the United States, as replacement therapy for men with hypogonadism.
The FDA is considering the approval of this long-acting testosterone – which has been available outside of the United States since 2003 – and convened the meeting of two advisory panels to review the safety data and the company’s plans for managing this risk, if approved in the United States.
Efficacy is not an issue: The agency has concluded that the 750-mg intramuscular dose of testosterone undecanoate, administered once every 10 weeks, meets the regulatory requirements for efficacy for a testosterone replacement product, based on results of serum testosterone levels in the single U.S. open-label study of 131 patients. But cases of anaphylaxis and pulmonary oil microembolism (POME), during or shortly after injections are administered, reported in clinical and postmarketing studies in countries in which it has been approved, has held up approval in the United States since Endo Pharmaceuticals filed for approval in 2007.
At the April 18 meeting, the FDA’s Advisory Committee for Reproductive Health Drugs and the Drug Safety and Risk Management Advisory Committee voted 9-9, when asked whether they believed that testosterone undecanoate (TU), a long-acting depot formulation of testosterone in castor oil and benzyl benzoate, was safe for the indication under review: replacement therapy in adult men for conditions associated with an absence or deficiency of endogenous testosterone. Those who felt the safety profile was acceptable for approval were concerned that the product was being held to a higher standard than other FDA-approved injectable testosterone products, which have labels that list anaphylaxis as a possible risk, and cited extensive use of the drug for years in Europe and no reports of deaths from these severe reactions.
But those voting no said that while they appreciated the need for a long-acting testosterone injection, there were not enough data to fully evaluate its safety and it was not possible to identify who is at an increased risk for having severe reactions. And, once approved, it might be used outside of the indication and would increase risk.
To address the risks of anaphylaxis and pulmonary oil microembolism (POME), Endo Pharmaceuticals has proposed that the injection be administered slowly, over 30 to 60 seconds in a physician’s office, and that patients be observed for 30 minutes afterward, with these instructions included in the product label. But the FDA panels voted 17-1 that this would not be sufficient to ameliorate this risk, calling for more interventions, including a black box warning, a training program for physicians who administer the injections to ensure they understand the severe risks (which could include an online course), and a more narrow indication to limit inappropriate use.
Although it makes sense clinically, "We don’t know the cause of this, so how can we know how to ameliorate this?" said panelist Dr. Richard Bockman, head of the endocrine service at the Hospital for Special Surgery, New York.
The features of POME are a cough or dyspnea, with or without other symptoms that can also include throat irritation, malaise, profuse sweating, chest pain, dizziness, paresthesia or syncope, which occurs within 24 hours of the injection, according to the FDA.
The company’s risk evaluation and mitigation strategy (REMS) includes physician and patient education and controlled distribution to health care providers for administration only in the office, with a 30-minute wait after the injection.
But Suzanne Robottom, Pharm.D., a senior risk management analyst in the FDA’s division of risk management, said that in Europe, cases of POME continue to be reported despite recommendations to administer the injection slowly and observe patients for 30 minutes. The agency is concerned that implementing the measures proposed by the company, which include prescribing and distribution restrictions, would pose an undue burden "for a drug with limited additional benefit," she added.
An FDA review of 199 postmarketing cases of POME, between November 2003 and April 2012, determined that 84 were severe, with severe dyspnea, loss of consciousness, circulatory collapse, or loss of bowel function. Many cases started as the medication was being injected. Although no deaths were reported, the long-term cardiopulmonary effects of severe cases or chronic effects of repeated less-severe episodes are not known, according to the FDA. During 2003-2011, there were 53 postmarketing cases of anaphylaxis reported, using a conservative definition.
The testosterone replacement products currently approved by the FDA are two intramuscular testosterone products (injected every 2-4 weeks), transdermal patches, gel, a solution, a buccal bioadhesive system, an oral tablet, and a subcutaneous implant.
Speaking on behalf of the Endocrine Society during the open public hearing portion of the meeting, Dr. Allan Glass, an endocrinologist in Bethesda, Md., said that the Society’s current guidelines for the treatment of androgen deficiency say that TU is an acceptable treatment in countries in which it has been approved (J. Clin. Endocrinol. Metab. 2010;95:2536-59).
If approved in the United States, TU will be marketed as Aveed. It is marketed in about 72 countries, and in Europe it is marketed as Nebido. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Dr. Glass had no financial disclosures.
AT AN FDA ADVISORY PANEL MEETING
