User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Small Bowel Dysmotility Brings Challenges to Patients With Systemic Sclerosis
TOPLINE:
Patients with systemic sclerosis (SSc) who exhibit abnormal small bowel transit are more likely to be men, experience more severe cardiac involvement, have a higher mortality risk, and show fewer sicca symptoms.
METHODOLOGY:
- Researchers enrolled 130 patients with SSc having gastrointestinal (GI) symptoms (mean age at symptom onset, 56.8 years; 90% women; 81% White) seen at the Johns Hopkins Scleroderma Center, Baltimore, from October 2014 to May 2022.
- Clinical data and serum samples were longitudinally collected from all actively followed patients at the time of enrollment and every 6 months thereafter (median disease duration, 8.4 years).
- Participants underwent whole gut transit scintigraphy for the assessment of small bowel motility.
- A cross-sectional analysis compared the clinical features of patients with (n = 22; mean age at symptom onset, 61.4 years) and without (n = 108; mean age at symptom onset, 55.8 years) abnormal small bowel transit.
TAKEAWAY:
- Men with SSc (odds ratio [OR], 3.70; P = .038) and those with severe cardiac involvement (OR, 3.98; P = .035) were more likely to have abnormal small bowel transit.
- Sicca symptoms were negatively associated with abnormal small bowel transit in patients with SSc (adjusted OR, 0.28; P = .043).
- Patients with abnormal small bowel transit reported significantly worse (P = .028) and social functioning (P = .015) than those having a normal transit.
- A multivariate analysis showed that patients with abnormal small bowel transit had higher mortality than those with a normal transit (adjusted hazard ratio, 5.03; P = .005).
IN PRACTICE:
“Our findings improve our understanding of risk factors associated with abnormal small bowel transit in SSc patients and shed light on the lived experience of patients with this GI [gastrointestinal] complication,” the authors wrote. “Overall, these findings are important for patient risk stratification and monitoring and will help to identify a more homogeneous group of patients for future clinical and translational studies,” they added.
SOURCE:
The study was led by Jenice X. Cheah, MD, University of California, Los Angeles. It was published online on October 7, 2024, in Rheumatology.
LIMITATIONS:
The study may be subject to referral bias as it was conducted at a tertiary referral center, potentially including patients with a more severe disease status. Furthermore, this study was retrospective in nature, and whole gut transit studies were not conducted in all the patients seen at the referral center. Additionally, the cross-sectional design limited the ability to establish causality between the clinical features and abnormal small bowel transit.
DISCLOSURES:
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Patients with systemic sclerosis (SSc) who exhibit abnormal small bowel transit are more likely to be men, experience more severe cardiac involvement, have a higher mortality risk, and show fewer sicca symptoms.
METHODOLOGY:
- Researchers enrolled 130 patients with SSc having gastrointestinal (GI) symptoms (mean age at symptom onset, 56.8 years; 90% women; 81% White) seen at the Johns Hopkins Scleroderma Center, Baltimore, from October 2014 to May 2022.
- Clinical data and serum samples were longitudinally collected from all actively followed patients at the time of enrollment and every 6 months thereafter (median disease duration, 8.4 years).
- Participants underwent whole gut transit scintigraphy for the assessment of small bowel motility.
- A cross-sectional analysis compared the clinical features of patients with (n = 22; mean age at symptom onset, 61.4 years) and without (n = 108; mean age at symptom onset, 55.8 years) abnormal small bowel transit.
TAKEAWAY:
- Men with SSc (odds ratio [OR], 3.70; P = .038) and those with severe cardiac involvement (OR, 3.98; P = .035) were more likely to have abnormal small bowel transit.
- Sicca symptoms were negatively associated with abnormal small bowel transit in patients with SSc (adjusted OR, 0.28; P = .043).
- Patients with abnormal small bowel transit reported significantly worse (P = .028) and social functioning (P = .015) than those having a normal transit.
- A multivariate analysis showed that patients with abnormal small bowel transit had higher mortality than those with a normal transit (adjusted hazard ratio, 5.03; P = .005).
IN PRACTICE:
“Our findings improve our understanding of risk factors associated with abnormal small bowel transit in SSc patients and shed light on the lived experience of patients with this GI [gastrointestinal] complication,” the authors wrote. “Overall, these findings are important for patient risk stratification and monitoring and will help to identify a more homogeneous group of patients for future clinical and translational studies,” they added.
SOURCE:
The study was led by Jenice X. Cheah, MD, University of California, Los Angeles. It was published online on October 7, 2024, in Rheumatology.
LIMITATIONS:
The study may be subject to referral bias as it was conducted at a tertiary referral center, potentially including patients with a more severe disease status. Furthermore, this study was retrospective in nature, and whole gut transit studies were not conducted in all the patients seen at the referral center. Additionally, the cross-sectional design limited the ability to establish causality between the clinical features and abnormal small bowel transit.
DISCLOSURES:
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Patients with systemic sclerosis (SSc) who exhibit abnormal small bowel transit are more likely to be men, experience more severe cardiac involvement, have a higher mortality risk, and show fewer sicca symptoms.
METHODOLOGY:
- Researchers enrolled 130 patients with SSc having gastrointestinal (GI) symptoms (mean age at symptom onset, 56.8 years; 90% women; 81% White) seen at the Johns Hopkins Scleroderma Center, Baltimore, from October 2014 to May 2022.
- Clinical data and serum samples were longitudinally collected from all actively followed patients at the time of enrollment and every 6 months thereafter (median disease duration, 8.4 years).
- Participants underwent whole gut transit scintigraphy for the assessment of small bowel motility.
- A cross-sectional analysis compared the clinical features of patients with (n = 22; mean age at symptom onset, 61.4 years) and without (n = 108; mean age at symptom onset, 55.8 years) abnormal small bowel transit.
TAKEAWAY:
- Men with SSc (odds ratio [OR], 3.70; P = .038) and those with severe cardiac involvement (OR, 3.98; P = .035) were more likely to have abnormal small bowel transit.
- Sicca symptoms were negatively associated with abnormal small bowel transit in patients with SSc (adjusted OR, 0.28; P = .043).
- Patients with abnormal small bowel transit reported significantly worse (P = .028) and social functioning (P = .015) than those having a normal transit.
- A multivariate analysis showed that patients with abnormal small bowel transit had higher mortality than those with a normal transit (adjusted hazard ratio, 5.03; P = .005).
IN PRACTICE:
“Our findings improve our understanding of risk factors associated with abnormal small bowel transit in SSc patients and shed light on the lived experience of patients with this GI [gastrointestinal] complication,” the authors wrote. “Overall, these findings are important for patient risk stratification and monitoring and will help to identify a more homogeneous group of patients for future clinical and translational studies,” they added.
SOURCE:
The study was led by Jenice X. Cheah, MD, University of California, Los Angeles. It was published online on October 7, 2024, in Rheumatology.
LIMITATIONS:
The study may be subject to referral bias as it was conducted at a tertiary referral center, potentially including patients with a more severe disease status. Furthermore, this study was retrospective in nature, and whole gut transit studies were not conducted in all the patients seen at the referral center. Additionally, the cross-sectional design limited the ability to establish causality between the clinical features and abnormal small bowel transit.
DISCLOSURES:
The study was supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Beware the Manchineel: A Case of Irritant Contact Dermatitis
What is the world’s most dangerous tree? According to Guinness World Records1 (and one unlucky contestant on the wilderness survival reality show Naked and Afraid,2 who got its sap in his eyes and needed to be evacuated for treatment), the manchineel tree (Hippomane mancinella) has earned this designation.1-3 Manchineel trees are part of the strand vegetation of islands in the West Indies and along the Caribbean coasts of South and Central America, where their copious root systems help reduce coastal erosion. In the United States, this poisonous tree grows along the southern edge of Florida’s Everglades National Park; the Florida Keys; and the US Virgin Islands, especially Virgin Islands National Park. Although the manchineel tree appears on several endangered species lists,4-6 there are places within its distribution where it is locally abundant and thus poses a risk to residents and visitors.
The first European description of manchineel toxicity was by Peter Martyr d’Anghiera, a court historian and geographer of Christopher Columbus’s patroness, Isabella I, Queen of Castile and Léon. In the early 1500s, Peter Martyr wrote that on Columbus’s second New World voyage in 1493, the crew encountered a mysterious tree that burned the skin and eyes of anyone who had contact with it.7 Columbus called the tree’s fruit manzanilla de la muerte (“little apple of death”) after several sailors became severely ill from eating the fruit.8,9 Manchineel lore is rife with tales of agonizing death after eating the applelike fruit, and several contemporaneous accounts describe indigenous Caribbean islanders using manchineel’s toxic sap as an arrow poison.10
Eating manchineel fruit is known to cause abdominal pain, burning sensations in the oropharynx, and esophageal spasms.11 Several case reports mention that consuming the fruit can create an exaggerated
Case Report
A 64-year-old physician (S.A.N.) came across a stand of manchineel trees while camping in the Virgin Islands National Park on St. John in the US Virgin Islands (Figure 1). The patient—who was knowledgeable about tropical ecology and was familiar with the tree—was curious about its purported cutaneous toxicity and applied the viscous white sap of a broken branchlet (Figure 2) to a patch of skin measuring 4 cm in diameter on the medial left calf. He took serial photographs of the site on days 2, 4 (Figure 3), 6, and 10 (Figure 4), showing the onset of erythema and the subsequent development of follicular pustules. On day 6, a 4-mm punch biopsy specimen was taken of the most prominent pustule. Histopathology showed a subcorneal acantholytic blister and epidermal spongiosis overlying a mixed perivascular infiltrate and follicular necrosis, which was consistent with irritant contact dermatitis (Figure 5). On day 8, the region became indurated and tender to pressure; however, there was no warmth, edema, purulent drainage, lymphangitic streaks, or other signs of infection. The region was never itchy; it was uncomfortable only with firm direct pressure. The patient applied hot compresses to the site for 10 minutes 1 to 2 times daily for roughly 2 weeks, and the affected area healed fully (without any additional intervention) in approximately 6 weeks.


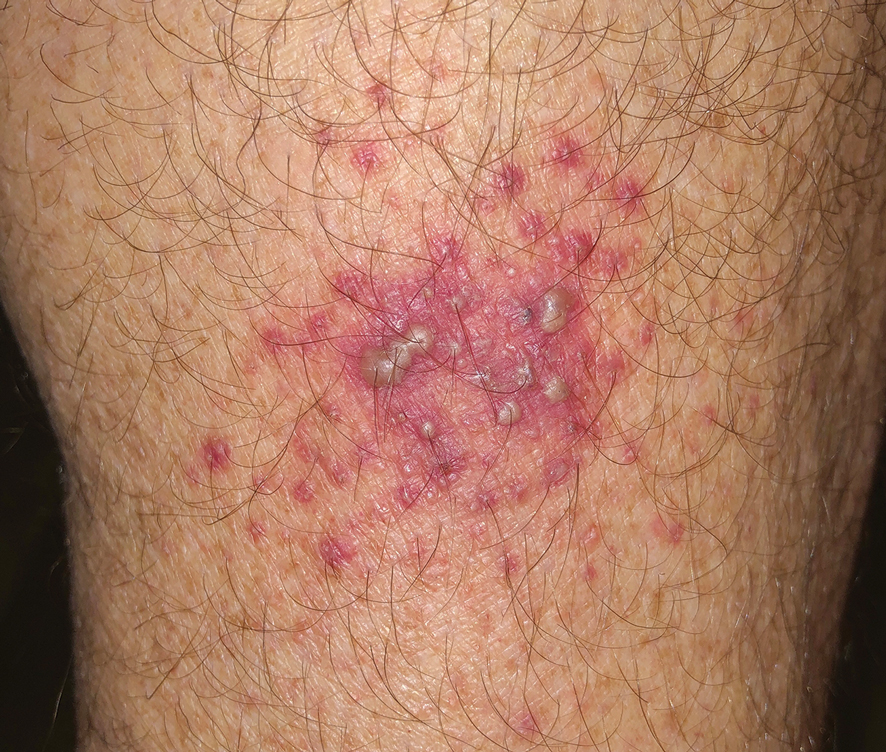
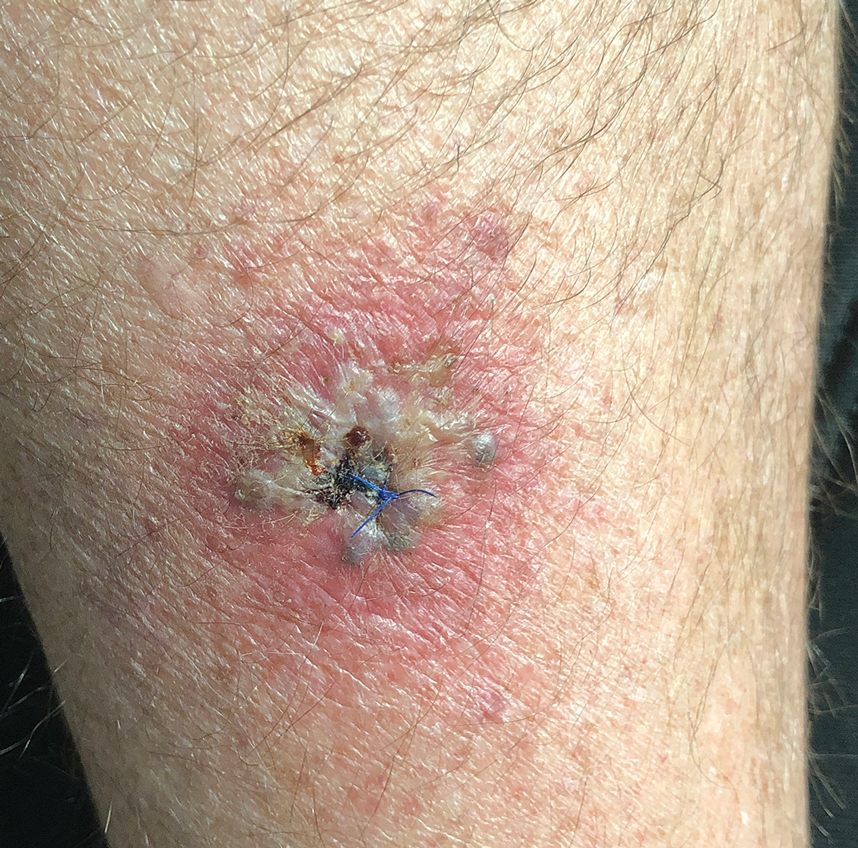
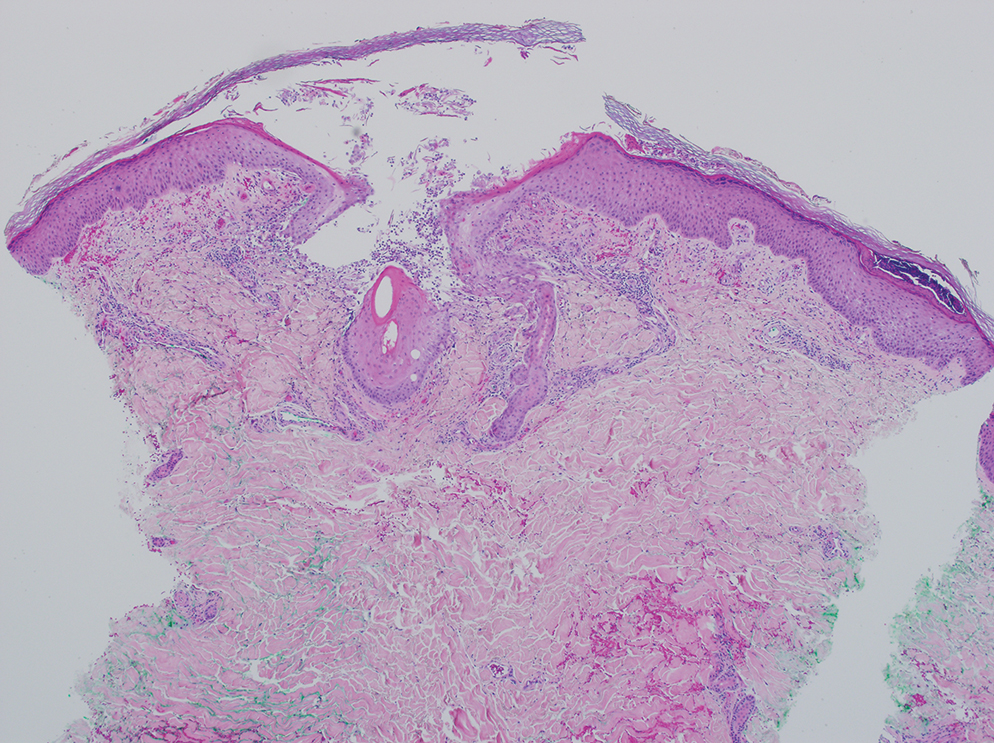
Comment
Manchineel is a member of the Euphorbiaceae (also known as the euphorb or spurge) family, a mainly tropical or subtropical plant family that includes many useful as well as many toxic species. Examples of useful plants include cassava (Manihot esculenta) and the rubber tree (Hevea brasiliensis). Many euphorbs have well-described toxicities, and many (eg, castor bean, Ricinus communis) are useful in some circumstances and toxic in others.6,12-14 Many euphorbs are known to cause skin reactions, usually due to toxins in the milky sap that directly irritate the skin or to latex compounds that can induce IgE-mediated contact dermatitis.9,14
Manchineel contains a complex mix of toxins, though no specific one has been identified as the main cause of the associated irritant contact dermatitis. Manchineel sap (and sap of many other euphorbs) contains phorbol esters that may cause direct pH-induced cytotoxicity leading to keratinocyte necrosis. Diterpenes may augment this cytotoxic effect via induction of proinflammatory cytokines.12 Pitts et al5 pointed to a mixture of oxygenated diterpene esters as the primary cause of toxicity and suggested that their water solubility explained occurrences of keratoconjunctivitis after contact with rainwater or dew from the manchineel tree.
All parts of the manchineel tree—fruit, leaves, wood, and sap—are poisonous. In a retrospective series of 97 cases of manchineel fruit ingestion, the most common symptoms were oropharyngeal pain (68% [66/97]), abdominal pain (42% [41/97]), and diarrhea (37% [36/97]). The same series identified 1 (1%) case of bradycardia and hypotension.3 Contact with the wood, exposure to sawdust, and inhalation of smoke from burning the wood can irritate the skin, conjunctivae, or nasopharynx. Rainwater or dew dripping from the leaves onto the skin can cause dermatitis and ophthalmitis, even without direct contact with the tree.4,5
Management—There is no specific treatment for manchineel dermatitis. Because it is an irritant reaction and not a type IV hypersensitivity reaction, topical corticosteroids have minimal benefit. A regimen consisting of a thorough cleansing, wet compresses, and observation, as most symptoms resolve spontaneously within a few days, has been recommended.4 Our patient used hot compresses, which he believes helped heal the site, although his symptoms lasted for several weeks.
Given that there is no specific treatment for manchineel dermatitis, the wisest approach is strict avoidance. On many Caribbean islands, visitors are warned about the manchineel tree, advised to avoid direct contact, and reminded to avoid standing beneath it during a rainstorm (Figure 6).

Conclusion
This article begins with a question: “What is the world’s most dangerous tree?” Many sources from the indexed medical literature as well as the popular press and social media state that it is the manchineel. Although all parts of the manchineel tree are highly toxic, human exposures are uncommon, and deaths are more apocryphal than actual.
- Most dangerous tree. Guinness World Records. Accessed October 14, 2024. https://www.guinnessworldrecords.com/world-records/most-dangerous-tree
- Naked and Afraid: Garden of Evil (S4E9). Discovery Channel. June 21, 2015. Accessed October 14, 2024. https://go.discovery.com/video/naked-and-afraid-discovery/garden-of-evil
- Boucaud-Maitre D, Cachet X, Bouzidi C, et al. Severity of manchineel fruit (Hippomane mancinella) poisoning: a retrospective case series of 97 patients from French Poison Control Centers. Toxicon. 2019;161:28-32. doi:10.1016/j.toxicon.2019.02.014
- Blue LM, Sailing C, Denapoles C, et al. Manchineel dermatitis in North American students in the Caribbean. J Travel Medicine. 2011;18:422-424. doi:10.1111/j.1708-8305.2011.00568.x
- Pitts JF, Barker NH, Gibbons DC, et al. Manchineel keratoconjunctivitis. Br J Ophthalmol. 1993;77:284-288. doi:10.1136/bjo.77.5.284
- Lauter WM, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella, L. I. historical review. J Pharm Sci. 1952;41:199-201. https://doi.org/10.1002/jps.3030410412
- Martyr P. De Orbe Novo: the Eight Decades of Peter Martyr d’Anghera. Vol 1. FA MacNutt (translator). GP Putnam’s Sons; 1912. Accessed October 14, 2024. https://gutenberg.org/cache/epub/12425/pg12425.txt
- Fernandez de Ybarra AM. A forgotten medical worthy, Dr. Diego Alvarex Chanca, of Seville, Spain, and his letter describing the second voyage of Christopher Columbus to America. Med Library Hist J. 1906;4:246-263.
- Muscat MK. Manchineel apple of death. EJIFCC. 2019;30:346-348.
- Handler JS. Aspects of Amerindian ethnography in 17th century Barbados. Caribbean Studies. 1970;9:50-72.
- Howard RA. Three experiences with the manchineel (Hippomane spp., Euphorbiaceae). Biotropica. 1981;13:224-227. https://doi.org/10.2307/2388129
- Rao KV. Toxic principles of Hippomane mancinella. Planta Med. 1974;25:166-171. doi:10.1055/s-0028-1097927
- Lauter WM, Foote PA. Investigation of the toxic principles of Hippomane mancinella L. II. Preliminary isolation of a toxic principle of the fruit. J Am Pharm Assoc. 1955;44:361-363. doi:10.1002/jps.3030440616
- Carroll MN Jr, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella L. III. Toxic actions of extracts of Hippomane mancinella L. J Am Pharm Assoc. 1957;46:93-97. doi:10.1002/jps.3030460206
What is the world’s most dangerous tree? According to Guinness World Records1 (and one unlucky contestant on the wilderness survival reality show Naked and Afraid,2 who got its sap in his eyes and needed to be evacuated for treatment), the manchineel tree (Hippomane mancinella) has earned this designation.1-3 Manchineel trees are part of the strand vegetation of islands in the West Indies and along the Caribbean coasts of South and Central America, where their copious root systems help reduce coastal erosion. In the United States, this poisonous tree grows along the southern edge of Florida’s Everglades National Park; the Florida Keys; and the US Virgin Islands, especially Virgin Islands National Park. Although the manchineel tree appears on several endangered species lists,4-6 there are places within its distribution where it is locally abundant and thus poses a risk to residents and visitors.
The first European description of manchineel toxicity was by Peter Martyr d’Anghiera, a court historian and geographer of Christopher Columbus’s patroness, Isabella I, Queen of Castile and Léon. In the early 1500s, Peter Martyr wrote that on Columbus’s second New World voyage in 1493, the crew encountered a mysterious tree that burned the skin and eyes of anyone who had contact with it.7 Columbus called the tree’s fruit manzanilla de la muerte (“little apple of death”) after several sailors became severely ill from eating the fruit.8,9 Manchineel lore is rife with tales of agonizing death after eating the applelike fruit, and several contemporaneous accounts describe indigenous Caribbean islanders using manchineel’s toxic sap as an arrow poison.10
Eating manchineel fruit is known to cause abdominal pain, burning sensations in the oropharynx, and esophageal spasms.11 Several case reports mention that consuming the fruit can create an exaggerated
Case Report
A 64-year-old physician (S.A.N.) came across a stand of manchineel trees while camping in the Virgin Islands National Park on St. John in the US Virgin Islands (Figure 1). The patient—who was knowledgeable about tropical ecology and was familiar with the tree—was curious about its purported cutaneous toxicity and applied the viscous white sap of a broken branchlet (Figure 2) to a patch of skin measuring 4 cm in diameter on the medial left calf. He took serial photographs of the site on days 2, 4 (Figure 3), 6, and 10 (Figure 4), showing the onset of erythema and the subsequent development of follicular pustules. On day 6, a 4-mm punch biopsy specimen was taken of the most prominent pustule. Histopathology showed a subcorneal acantholytic blister and epidermal spongiosis overlying a mixed perivascular infiltrate and follicular necrosis, which was consistent with irritant contact dermatitis (Figure 5). On day 8, the region became indurated and tender to pressure; however, there was no warmth, edema, purulent drainage, lymphangitic streaks, or other signs of infection. The region was never itchy; it was uncomfortable only with firm direct pressure. The patient applied hot compresses to the site for 10 minutes 1 to 2 times daily for roughly 2 weeks, and the affected area healed fully (without any additional intervention) in approximately 6 weeks.
Comment
Manchineel is a member of the Euphorbiaceae (also known as the euphorb or spurge) family, a mainly tropical or subtropical plant family that includes many useful as well as many toxic species. Examples of useful plants include cassava (Manihot esculenta) and the rubber tree (Hevea brasiliensis). Many euphorbs have well-described toxicities, and many (eg, castor bean, Ricinus communis) are useful in some circumstances and toxic in others.6,12-14 Many euphorbs are known to cause skin reactions, usually due to toxins in the milky sap that directly irritate the skin or to latex compounds that can induce IgE-mediated contact dermatitis.9,14
Manchineel contains a complex mix of toxins, though no specific one has been identified as the main cause of the associated irritant contact dermatitis. Manchineel sap (and sap of many other euphorbs) contains phorbol esters that may cause direct pH-induced cytotoxicity leading to keratinocyte necrosis. Diterpenes may augment this cytotoxic effect via induction of proinflammatory cytokines.12 Pitts et al5 pointed to a mixture of oxygenated diterpene esters as the primary cause of toxicity and suggested that their water solubility explained occurrences of keratoconjunctivitis after contact with rainwater or dew from the manchineel tree.
All parts of the manchineel tree—fruit, leaves, wood, and sap—are poisonous. In a retrospective series of 97 cases of manchineel fruit ingestion, the most common symptoms were oropharyngeal pain (68% [66/97]), abdominal pain (42% [41/97]), and diarrhea (37% [36/97]). The same series identified 1 (1%) case of bradycardia and hypotension.3 Contact with the wood, exposure to sawdust, and inhalation of smoke from burning the wood can irritate the skin, conjunctivae, or nasopharynx. Rainwater or dew dripping from the leaves onto the skin can cause dermatitis and ophthalmitis, even without direct contact with the tree.4,5
Management—There is no specific treatment for manchineel dermatitis. Because it is an irritant reaction and not a type IV hypersensitivity reaction, topical corticosteroids have minimal benefit. A regimen consisting of a thorough cleansing, wet compresses, and observation, as most symptoms resolve spontaneously within a few days, has been recommended.4 Our patient used hot compresses, which he believes helped heal the site, although his symptoms lasted for several weeks.
Given that there is no specific treatment for manchineel dermatitis, the wisest approach is strict avoidance. On many Caribbean islands, visitors are warned about the manchineel tree, advised to avoid direct contact, and reminded to avoid standing beneath it during a rainstorm (Figure 6).
Conclusion
This article begins with a question: “What is the world’s most dangerous tree?” Many sources from the indexed medical literature as well as the popular press and social media state that it is the manchineel. Although all parts of the manchineel tree are highly toxic, human exposures are uncommon, and deaths are more apocryphal than actual.
What is the world’s most dangerous tree? According to Guinness World Records1 (and one unlucky contestant on the wilderness survival reality show Naked and Afraid,2 who got its sap in his eyes and needed to be evacuated for treatment), the manchineel tree (Hippomane mancinella) has earned this designation.1-3 Manchineel trees are part of the strand vegetation of islands in the West Indies and along the Caribbean coasts of South and Central America, where their copious root systems help reduce coastal erosion. In the United States, this poisonous tree grows along the southern edge of Florida’s Everglades National Park; the Florida Keys; and the US Virgin Islands, especially Virgin Islands National Park. Although the manchineel tree appears on several endangered species lists,4-6 there are places within its distribution where it is locally abundant and thus poses a risk to residents and visitors.
The first European description of manchineel toxicity was by Peter Martyr d’Anghiera, a court historian and geographer of Christopher Columbus’s patroness, Isabella I, Queen of Castile and Léon. In the early 1500s, Peter Martyr wrote that on Columbus’s second New World voyage in 1493, the crew encountered a mysterious tree that burned the skin and eyes of anyone who had contact with it.7 Columbus called the tree’s fruit manzanilla de la muerte (“little apple of death”) after several sailors became severely ill from eating the fruit.8,9 Manchineel lore is rife with tales of agonizing death after eating the applelike fruit, and several contemporaneous accounts describe indigenous Caribbean islanders using manchineel’s toxic sap as an arrow poison.10
Eating manchineel fruit is known to cause abdominal pain, burning sensations in the oropharynx, and esophageal spasms.11 Several case reports mention that consuming the fruit can create an exaggerated
Case Report
A 64-year-old physician (S.A.N.) came across a stand of manchineel trees while camping in the Virgin Islands National Park on St. John in the US Virgin Islands (Figure 1). The patient—who was knowledgeable about tropical ecology and was familiar with the tree—was curious about its purported cutaneous toxicity and applied the viscous white sap of a broken branchlet (Figure 2) to a patch of skin measuring 4 cm in diameter on the medial left calf. He took serial photographs of the site on days 2, 4 (Figure 3), 6, and 10 (Figure 4), showing the onset of erythema and the subsequent development of follicular pustules. On day 6, a 4-mm punch biopsy specimen was taken of the most prominent pustule. Histopathology showed a subcorneal acantholytic blister and epidermal spongiosis overlying a mixed perivascular infiltrate and follicular necrosis, which was consistent with irritant contact dermatitis (Figure 5). On day 8, the region became indurated and tender to pressure; however, there was no warmth, edema, purulent drainage, lymphangitic streaks, or other signs of infection. The region was never itchy; it was uncomfortable only with firm direct pressure. The patient applied hot compresses to the site for 10 minutes 1 to 2 times daily for roughly 2 weeks, and the affected area healed fully (without any additional intervention) in approximately 6 weeks.
Comment
Manchineel is a member of the Euphorbiaceae (also known as the euphorb or spurge) family, a mainly tropical or subtropical plant family that includes many useful as well as many toxic species. Examples of useful plants include cassava (Manihot esculenta) and the rubber tree (Hevea brasiliensis). Many euphorbs have well-described toxicities, and many (eg, castor bean, Ricinus communis) are useful in some circumstances and toxic in others.6,12-14 Many euphorbs are known to cause skin reactions, usually due to toxins in the milky sap that directly irritate the skin or to latex compounds that can induce IgE-mediated contact dermatitis.9,14
Manchineel contains a complex mix of toxins, though no specific one has been identified as the main cause of the associated irritant contact dermatitis. Manchineel sap (and sap of many other euphorbs) contains phorbol esters that may cause direct pH-induced cytotoxicity leading to keratinocyte necrosis. Diterpenes may augment this cytotoxic effect via induction of proinflammatory cytokines.12 Pitts et al5 pointed to a mixture of oxygenated diterpene esters as the primary cause of toxicity and suggested that their water solubility explained occurrences of keratoconjunctivitis after contact with rainwater or dew from the manchineel tree.
All parts of the manchineel tree—fruit, leaves, wood, and sap—are poisonous. In a retrospective series of 97 cases of manchineel fruit ingestion, the most common symptoms were oropharyngeal pain (68% [66/97]), abdominal pain (42% [41/97]), and diarrhea (37% [36/97]). The same series identified 1 (1%) case of bradycardia and hypotension.3 Contact with the wood, exposure to sawdust, and inhalation of smoke from burning the wood can irritate the skin, conjunctivae, or nasopharynx. Rainwater or dew dripping from the leaves onto the skin can cause dermatitis and ophthalmitis, even without direct contact with the tree.4,5
Management—There is no specific treatment for manchineel dermatitis. Because it is an irritant reaction and not a type IV hypersensitivity reaction, topical corticosteroids have minimal benefit. A regimen consisting of a thorough cleansing, wet compresses, and observation, as most symptoms resolve spontaneously within a few days, has been recommended.4 Our patient used hot compresses, which he believes helped heal the site, although his symptoms lasted for several weeks.
Given that there is no specific treatment for manchineel dermatitis, the wisest approach is strict avoidance. On many Caribbean islands, visitors are warned about the manchineel tree, advised to avoid direct contact, and reminded to avoid standing beneath it during a rainstorm (Figure 6).
Conclusion
This article begins with a question: “What is the world’s most dangerous tree?” Many sources from the indexed medical literature as well as the popular press and social media state that it is the manchineel. Although all parts of the manchineel tree are highly toxic, human exposures are uncommon, and deaths are more apocryphal than actual.
- Most dangerous tree. Guinness World Records. Accessed October 14, 2024. https://www.guinnessworldrecords.com/world-records/most-dangerous-tree
- Naked and Afraid: Garden of Evil (S4E9). Discovery Channel. June 21, 2015. Accessed October 14, 2024. https://go.discovery.com/video/naked-and-afraid-discovery/garden-of-evil
- Boucaud-Maitre D, Cachet X, Bouzidi C, et al. Severity of manchineel fruit (Hippomane mancinella) poisoning: a retrospective case series of 97 patients from French Poison Control Centers. Toxicon. 2019;161:28-32. doi:10.1016/j.toxicon.2019.02.014
- Blue LM, Sailing C, Denapoles C, et al. Manchineel dermatitis in North American students in the Caribbean. J Travel Medicine. 2011;18:422-424. doi:10.1111/j.1708-8305.2011.00568.x
- Pitts JF, Barker NH, Gibbons DC, et al. Manchineel keratoconjunctivitis. Br J Ophthalmol. 1993;77:284-288. doi:10.1136/bjo.77.5.284
- Lauter WM, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella, L. I. historical review. J Pharm Sci. 1952;41:199-201. https://doi.org/10.1002/jps.3030410412
- Martyr P. De Orbe Novo: the Eight Decades of Peter Martyr d’Anghera. Vol 1. FA MacNutt (translator). GP Putnam’s Sons; 1912. Accessed October 14, 2024. https://gutenberg.org/cache/epub/12425/pg12425.txt
- Fernandez de Ybarra AM. A forgotten medical worthy, Dr. Diego Alvarex Chanca, of Seville, Spain, and his letter describing the second voyage of Christopher Columbus to America. Med Library Hist J. 1906;4:246-263.
- Muscat MK. Manchineel apple of death. EJIFCC. 2019;30:346-348.
- Handler JS. Aspects of Amerindian ethnography in 17th century Barbados. Caribbean Studies. 1970;9:50-72.
- Howard RA. Three experiences with the manchineel (Hippomane spp., Euphorbiaceae). Biotropica. 1981;13:224-227. https://doi.org/10.2307/2388129
- Rao KV. Toxic principles of Hippomane mancinella. Planta Med. 1974;25:166-171. doi:10.1055/s-0028-1097927
- Lauter WM, Foote PA. Investigation of the toxic principles of Hippomane mancinella L. II. Preliminary isolation of a toxic principle of the fruit. J Am Pharm Assoc. 1955;44:361-363. doi:10.1002/jps.3030440616
- Carroll MN Jr, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella L. III. Toxic actions of extracts of Hippomane mancinella L. J Am Pharm Assoc. 1957;46:93-97. doi:10.1002/jps.3030460206
- Most dangerous tree. Guinness World Records. Accessed October 14, 2024. https://www.guinnessworldrecords.com/world-records/most-dangerous-tree
- Naked and Afraid: Garden of Evil (S4E9). Discovery Channel. June 21, 2015. Accessed October 14, 2024. https://go.discovery.com/video/naked-and-afraid-discovery/garden-of-evil
- Boucaud-Maitre D, Cachet X, Bouzidi C, et al. Severity of manchineel fruit (Hippomane mancinella) poisoning: a retrospective case series of 97 patients from French Poison Control Centers. Toxicon. 2019;161:28-32. doi:10.1016/j.toxicon.2019.02.014
- Blue LM, Sailing C, Denapoles C, et al. Manchineel dermatitis in North American students in the Caribbean. J Travel Medicine. 2011;18:422-424. doi:10.1111/j.1708-8305.2011.00568.x
- Pitts JF, Barker NH, Gibbons DC, et al. Manchineel keratoconjunctivitis. Br J Ophthalmol. 1993;77:284-288. doi:10.1136/bjo.77.5.284
- Lauter WM, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella, L. I. historical review. J Pharm Sci. 1952;41:199-201. https://doi.org/10.1002/jps.3030410412
- Martyr P. De Orbe Novo: the Eight Decades of Peter Martyr d’Anghera. Vol 1. FA MacNutt (translator). GP Putnam’s Sons; 1912. Accessed October 14, 2024. https://gutenberg.org/cache/epub/12425/pg12425.txt
- Fernandez de Ybarra AM. A forgotten medical worthy, Dr. Diego Alvarex Chanca, of Seville, Spain, and his letter describing the second voyage of Christopher Columbus to America. Med Library Hist J. 1906;4:246-263.
- Muscat MK. Manchineel apple of death. EJIFCC. 2019;30:346-348.
- Handler JS. Aspects of Amerindian ethnography in 17th century Barbados. Caribbean Studies. 1970;9:50-72.
- Howard RA. Three experiences with the manchineel (Hippomane spp., Euphorbiaceae). Biotropica. 1981;13:224-227. https://doi.org/10.2307/2388129
- Rao KV. Toxic principles of Hippomane mancinella. Planta Med. 1974;25:166-171. doi:10.1055/s-0028-1097927
- Lauter WM, Foote PA. Investigation of the toxic principles of Hippomane mancinella L. II. Preliminary isolation of a toxic principle of the fruit. J Am Pharm Assoc. 1955;44:361-363. doi:10.1002/jps.3030440616
- Carroll MN Jr, Fox LE, Ariail WT. Investigation of the toxic principles of Hippomane mancinella L. III. Toxic actions of extracts of Hippomane mancinella L. J Am Pharm Assoc. 1957;46:93-97. doi:10.1002/jps.3030460206
PRACTICE POINTS
- Sap from the manchineel tree—found on the coasts of Caribbean islands, the Atlantic coastline of Central and northern South America, and parts of southernmost Florida—can cause severe dermatologic and ophthalmologic injuries. Eating its fruit can lead to oropharyngeal pain and diarrhea.
- Histopathology of manchineel dermatitis reveals a subcorneal acantholytic blister and epidermal spongiosis overlying a mixed perivascular infiltrate and follicular necrosis, which is consistent with irritant contact dermatitis.
- There is no specific treatment for manchineel dermatitis. Case reports advocate a thorough cleansing, application of wet compresses, and observation.
Wrinkles, Dyspigmentation Improve with PDT, in Small Study
.
“Our study helps capture and quantify a phenomenon that clinicians who use PDT in their practice have already noticed: Patients experience a visible improvement across several cosmetically important metrics including but not limited to fine lines, wrinkles, and skin tightness following PDT,” one of the study authors, Luke Horton, MD, a fourth-year dermatology resident at the University of California, Irvine, said in an interview following the annual meeting of the American Society for Dermatologic Surgery, where he presented the results during an oral abstract session.

For the study, 11 patients underwent a 120-minute incubation period with 17% 5-aminolevulinic acid over the face, followed by visible blue light PDT exposure for 16 minutes, to reduce rhytides. The researchers used a Vectra imaging system to capture three-dimensional images of the patients before the procedure and during the follow-up. Three dermatologists analyzed the pre-procedure and post-procedure images and used a validated five-point Merz wrinkle severity scale to grade various regions of the face including the forehead, glabella, lateral canthal rhytides, melolabial folds, nasolabial folds, and perioral rhytides.
They also used a five-point solar lentigines scale to evaluate the change in degree of pigmentation and quantity of age spots as well as the change in rhytid severity before and after PDT and the change in the seven-point Global Aesthetic Improvement Scale (GAIS) to gauge overall improvement of fine lines and wrinkles.
After a mean follow-up of 4.25 months, rhytid severity among the 11 patients was reduced by an average of 0.65 points on the Merz scale, with an SD of 0.20. Broken down by region, rhytid severity scores decreased by 0.2 points (SD, 0.42) for the forehead, 0.7 points (SD, 0.48) for the glabella and lateral canthal rhytides, 0.88 points (SD, 0.35) for the melolabial folds and perioral rhytides, and 0.8 points (SD, 0.42) for the nasolabial folds. (The researchers excluded ratings for the melolabial folds and perioral rhytides in two patients with beards.)
In other findings, solar lentigines grading showed an average reduction of 1 point (SD, 0.45), while the GAIS score improved by 1 or more for every patient, with an average of score of 1.45 (SD, 0.52), showing that some degree of improvement in facial rhytides was noted for all patients following PDT.
“The degree of improvement as measured by our independent physician graders was impressive and not far off from those reported with CO2 ablative laser,” Horton said. “Further, the effect was not isolated to actinic keratoses but extended to improved appearance of fine lines, some deep lines, and lentigines. Although we are not implying that PDT is superior to and should replace lasers or other energy-based devices, it does provide a real, measurable cosmetic benefit.”
Clinicians, he added, can use these findings “to counsel their patients when discussing field cancerization treatment options, especially for patients who may be hesitant to undergo PDT as it can be a painful therapy with a considerable downtime for some.”
Lawrence J. Green, MD, clinical professor of dermatology, The George Washington University, Washington, DC, who was asked to comment on the study results, said that the findings “shine more light on the long-standing off-label use of PDT for lessening signs of photoaging. Like studies done before it, I think this adds an additional benefit to discuss for those who are considering PDT treatment for their actinic keratoses.”
Horton acknowledged certain limitations of the study including its small sample size and the fact that physician graders were not blinded to which images were pre- and post-treatment, “which could introduce an element of bias in the data,” he said. “But this being an unfunded project born out of clinical observation, we hope to later expand its size. Furthermore, we invite other physicians to join us to better study these effects and to design protocols that minimize adverse effects and maximize clinical outcomes.”
His co-authors were Milan Hirpara; Sarah Choe; Joel Cohen, MD; and Natasha A. Mesinkovska, MD, PhD.
No relevant disclosures were reported. Green had no relevant disclosures.
A version of this article appeared on Medscape.com.
.
“Our study helps capture and quantify a phenomenon that clinicians who use PDT in their practice have already noticed: Patients experience a visible improvement across several cosmetically important metrics including but not limited to fine lines, wrinkles, and skin tightness following PDT,” one of the study authors, Luke Horton, MD, a fourth-year dermatology resident at the University of California, Irvine, said in an interview following the annual meeting of the American Society for Dermatologic Surgery, where he presented the results during an oral abstract session.
For the study, 11 patients underwent a 120-minute incubation period with 17% 5-aminolevulinic acid over the face, followed by visible blue light PDT exposure for 16 minutes, to reduce rhytides. The researchers used a Vectra imaging system to capture three-dimensional images of the patients before the procedure and during the follow-up. Three dermatologists analyzed the pre-procedure and post-procedure images and used a validated five-point Merz wrinkle severity scale to grade various regions of the face including the forehead, glabella, lateral canthal rhytides, melolabial folds, nasolabial folds, and perioral rhytides.
They also used a five-point solar lentigines scale to evaluate the change in degree of pigmentation and quantity of age spots as well as the change in rhytid severity before and after PDT and the change in the seven-point Global Aesthetic Improvement Scale (GAIS) to gauge overall improvement of fine lines and wrinkles.
After a mean follow-up of 4.25 months, rhytid severity among the 11 patients was reduced by an average of 0.65 points on the Merz scale, with an SD of 0.20. Broken down by region, rhytid severity scores decreased by 0.2 points (SD, 0.42) for the forehead, 0.7 points (SD, 0.48) for the glabella and lateral canthal rhytides, 0.88 points (SD, 0.35) for the melolabial folds and perioral rhytides, and 0.8 points (SD, 0.42) for the nasolabial folds. (The researchers excluded ratings for the melolabial folds and perioral rhytides in two patients with beards.)
In other findings, solar lentigines grading showed an average reduction of 1 point (SD, 0.45), while the GAIS score improved by 1 or more for every patient, with an average of score of 1.45 (SD, 0.52), showing that some degree of improvement in facial rhytides was noted for all patients following PDT.
“The degree of improvement as measured by our independent physician graders was impressive and not far off from those reported with CO2 ablative laser,” Horton said. “Further, the effect was not isolated to actinic keratoses but extended to improved appearance of fine lines, some deep lines, and lentigines. Although we are not implying that PDT is superior to and should replace lasers or other energy-based devices, it does provide a real, measurable cosmetic benefit.”
Clinicians, he added, can use these findings “to counsel their patients when discussing field cancerization treatment options, especially for patients who may be hesitant to undergo PDT as it can be a painful therapy with a considerable downtime for some.”
Lawrence J. Green, MD, clinical professor of dermatology, The George Washington University, Washington, DC, who was asked to comment on the study results, said that the findings “shine more light on the long-standing off-label use of PDT for lessening signs of photoaging. Like studies done before it, I think this adds an additional benefit to discuss for those who are considering PDT treatment for their actinic keratoses.”
Horton acknowledged certain limitations of the study including its small sample size and the fact that physician graders were not blinded to which images were pre- and post-treatment, “which could introduce an element of bias in the data,” he said. “But this being an unfunded project born out of clinical observation, we hope to later expand its size. Furthermore, we invite other physicians to join us to better study these effects and to design protocols that minimize adverse effects and maximize clinical outcomes.”
His co-authors were Milan Hirpara; Sarah Choe; Joel Cohen, MD; and Natasha A. Mesinkovska, MD, PhD.
No relevant disclosures were reported. Green had no relevant disclosures.
A version of this article appeared on Medscape.com.
.
“Our study helps capture and quantify a phenomenon that clinicians who use PDT in their practice have already noticed: Patients experience a visible improvement across several cosmetically important metrics including but not limited to fine lines, wrinkles, and skin tightness following PDT,” one of the study authors, Luke Horton, MD, a fourth-year dermatology resident at the University of California, Irvine, said in an interview following the annual meeting of the American Society for Dermatologic Surgery, where he presented the results during an oral abstract session.
For the study, 11 patients underwent a 120-minute incubation period with 17% 5-aminolevulinic acid over the face, followed by visible blue light PDT exposure for 16 minutes, to reduce rhytides. The researchers used a Vectra imaging system to capture three-dimensional images of the patients before the procedure and during the follow-up. Three dermatologists analyzed the pre-procedure and post-procedure images and used a validated five-point Merz wrinkle severity scale to grade various regions of the face including the forehead, glabella, lateral canthal rhytides, melolabial folds, nasolabial folds, and perioral rhytides.
They also used a five-point solar lentigines scale to evaluate the change in degree of pigmentation and quantity of age spots as well as the change in rhytid severity before and after PDT and the change in the seven-point Global Aesthetic Improvement Scale (GAIS) to gauge overall improvement of fine lines and wrinkles.
After a mean follow-up of 4.25 months, rhytid severity among the 11 patients was reduced by an average of 0.65 points on the Merz scale, with an SD of 0.20. Broken down by region, rhytid severity scores decreased by 0.2 points (SD, 0.42) for the forehead, 0.7 points (SD, 0.48) for the glabella and lateral canthal rhytides, 0.88 points (SD, 0.35) for the melolabial folds and perioral rhytides, and 0.8 points (SD, 0.42) for the nasolabial folds. (The researchers excluded ratings for the melolabial folds and perioral rhytides in two patients with beards.)
In other findings, solar lentigines grading showed an average reduction of 1 point (SD, 0.45), while the GAIS score improved by 1 or more for every patient, with an average of score of 1.45 (SD, 0.52), showing that some degree of improvement in facial rhytides was noted for all patients following PDT.
“The degree of improvement as measured by our independent physician graders was impressive and not far off from those reported with CO2 ablative laser,” Horton said. “Further, the effect was not isolated to actinic keratoses but extended to improved appearance of fine lines, some deep lines, and lentigines. Although we are not implying that PDT is superior to and should replace lasers or other energy-based devices, it does provide a real, measurable cosmetic benefit.”
Clinicians, he added, can use these findings “to counsel their patients when discussing field cancerization treatment options, especially for patients who may be hesitant to undergo PDT as it can be a painful therapy with a considerable downtime for some.”
Lawrence J. Green, MD, clinical professor of dermatology, The George Washington University, Washington, DC, who was asked to comment on the study results, said that the findings “shine more light on the long-standing off-label use of PDT for lessening signs of photoaging. Like studies done before it, I think this adds an additional benefit to discuss for those who are considering PDT treatment for their actinic keratoses.”
Horton acknowledged certain limitations of the study including its small sample size and the fact that physician graders were not blinded to which images were pre- and post-treatment, “which could introduce an element of bias in the data,” he said. “But this being an unfunded project born out of clinical observation, we hope to later expand its size. Furthermore, we invite other physicians to join us to better study these effects and to design protocols that minimize adverse effects and maximize clinical outcomes.”
His co-authors were Milan Hirpara; Sarah Choe; Joel Cohen, MD; and Natasha A. Mesinkovska, MD, PhD.
No relevant disclosures were reported. Green had no relevant disclosures.
A version of this article appeared on Medscape.com.
Responses Sustained with Ritlecitinib in Patients with Alopecia Through 48 Weeks
TOPLINE:
, and up to one third of nonresponders at week 24 also achieved responses by week 48.
METHODOLOGY:
- Researchers conducted a post hoc analysis of an international, randomized, double-blind, placebo-controlled, phase 2b/3 trial (ALLEGRO) and included 718 adults and adolescents aged 12 or older with severe AA (Severity of Alopecia Tool [SALT] score ≥ 50).
- Patients received various doses of the oral Janus kinase inhibitor ritlecitinib, with or without a 4-week loading dose, including 200/50 mg, 200/30 mg, 50 mg, or 30 mg, with or without a 4-week loading dose for up to 24 weeks and continued to receive their assigned maintenance dose.
- Researchers assessed sustained clinical responses at week 48 for those who had achieved SALT scores ≤ 20 and ≤ 10 at 24 weeks, and nonresponders at week 24 were assessed for responses through week 48.
- Adverse events were also evaluated.
TAKEAWAY:
- Among patients on ritlecitinib who had responded at week 24, SALT responses ≤ 20 were sustained in 85.2%-100% of patients through week 48. Similar results were seen among patients who achieved a SALT score ≤ 10 (68.8%-91.7%) and improvements in eyebrow (70.4%-96.9%) or eyelash (52.4%-94.1%) assessment scores.
- Among those who were nonresponders at week 24, 22.2%-33.7% achieved a SALT score ≤ 20 and 19.8%-25.5% achieved a SALT score ≤ 10 by week 48. Similarly, among those with no eyebrow or eyelash responses at week 24, 19.7%-32.8% and 16.7%-30.2% had improved eyebrow or eyelash assessment scores, respectively, at week 48.
- Between weeks 24 and 48, adverse events were reported in 74%-93% of patients who achieved a SALT score ≤ 20, most were mild or moderate; two serious events were reported but deemed unrelated to treatment. The safety profile was similar across all subgroups.
- No deaths, malignancies, major cardiovascular events, opportunistic infections, or herpes zoster infections were observed.
IN PRACTICE:
“The majority of ritlecitinib-treated patients with AA who met target clinical response based on scalp, eyebrow, or eyelash regrowth at week 24 sustained their response through week 48 with continued treatment,” the authors wrote. “Some patients, including those with more extensive hair loss, may require ritlecitinib treatment beyond 6 months to achieve target clinical response,” they added.
SOURCE:
The study was led by Melissa Piliang, MD, of the Department of Dermatology, Cleveland Clinic, and was published online on October 17 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The analysis was limited by its post hoc nature, small sample size in each treatment group, and a follow-up period of only 48 weeks.
DISCLOSURES:
This study was funded by Pfizer. Piliang disclosed being a consultant or investigator for Pfizer, Eli Lilly, and Procter & Gamble. Six authors were employees or shareholders of or received salary from Pfizer. Other authors also reported financial relationships with pharmaceutical companies outside this work, including Pfizer.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
, and up to one third of nonresponders at week 24 also achieved responses by week 48.
METHODOLOGY:
- Researchers conducted a post hoc analysis of an international, randomized, double-blind, placebo-controlled, phase 2b/3 trial (ALLEGRO) and included 718 adults and adolescents aged 12 or older with severe AA (Severity of Alopecia Tool [SALT] score ≥ 50).
- Patients received various doses of the oral Janus kinase inhibitor ritlecitinib, with or without a 4-week loading dose, including 200/50 mg, 200/30 mg, 50 mg, or 30 mg, with or without a 4-week loading dose for up to 24 weeks and continued to receive their assigned maintenance dose.
- Researchers assessed sustained clinical responses at week 48 for those who had achieved SALT scores ≤ 20 and ≤ 10 at 24 weeks, and nonresponders at week 24 were assessed for responses through week 48.
- Adverse events were also evaluated.
TAKEAWAY:
- Among patients on ritlecitinib who had responded at week 24, SALT responses ≤ 20 were sustained in 85.2%-100% of patients through week 48. Similar results were seen among patients who achieved a SALT score ≤ 10 (68.8%-91.7%) and improvements in eyebrow (70.4%-96.9%) or eyelash (52.4%-94.1%) assessment scores.
- Among those who were nonresponders at week 24, 22.2%-33.7% achieved a SALT score ≤ 20 and 19.8%-25.5% achieved a SALT score ≤ 10 by week 48. Similarly, among those with no eyebrow or eyelash responses at week 24, 19.7%-32.8% and 16.7%-30.2% had improved eyebrow or eyelash assessment scores, respectively, at week 48.
- Between weeks 24 and 48, adverse events were reported in 74%-93% of patients who achieved a SALT score ≤ 20, most were mild or moderate; two serious events were reported but deemed unrelated to treatment. The safety profile was similar across all subgroups.
- No deaths, malignancies, major cardiovascular events, opportunistic infections, or herpes zoster infections were observed.
IN PRACTICE:
“The majority of ritlecitinib-treated patients with AA who met target clinical response based on scalp, eyebrow, or eyelash regrowth at week 24 sustained their response through week 48 with continued treatment,” the authors wrote. “Some patients, including those with more extensive hair loss, may require ritlecitinib treatment beyond 6 months to achieve target clinical response,” they added.
SOURCE:
The study was led by Melissa Piliang, MD, of the Department of Dermatology, Cleveland Clinic, and was published online on October 17 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The analysis was limited by its post hoc nature, small sample size in each treatment group, and a follow-up period of only 48 weeks.
DISCLOSURES:
This study was funded by Pfizer. Piliang disclosed being a consultant or investigator for Pfizer, Eli Lilly, and Procter & Gamble. Six authors were employees or shareholders of or received salary from Pfizer. Other authors also reported financial relationships with pharmaceutical companies outside this work, including Pfizer.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
, and up to one third of nonresponders at week 24 also achieved responses by week 48.
METHODOLOGY:
- Researchers conducted a post hoc analysis of an international, randomized, double-blind, placebo-controlled, phase 2b/3 trial (ALLEGRO) and included 718 adults and adolescents aged 12 or older with severe AA (Severity of Alopecia Tool [SALT] score ≥ 50).
- Patients received various doses of the oral Janus kinase inhibitor ritlecitinib, with or without a 4-week loading dose, including 200/50 mg, 200/30 mg, 50 mg, or 30 mg, with or without a 4-week loading dose for up to 24 weeks and continued to receive their assigned maintenance dose.
- Researchers assessed sustained clinical responses at week 48 for those who had achieved SALT scores ≤ 20 and ≤ 10 at 24 weeks, and nonresponders at week 24 were assessed for responses through week 48.
- Adverse events were also evaluated.
TAKEAWAY:
- Among patients on ritlecitinib who had responded at week 24, SALT responses ≤ 20 were sustained in 85.2%-100% of patients through week 48. Similar results were seen among patients who achieved a SALT score ≤ 10 (68.8%-91.7%) and improvements in eyebrow (70.4%-96.9%) or eyelash (52.4%-94.1%) assessment scores.
- Among those who were nonresponders at week 24, 22.2%-33.7% achieved a SALT score ≤ 20 and 19.8%-25.5% achieved a SALT score ≤ 10 by week 48. Similarly, among those with no eyebrow or eyelash responses at week 24, 19.7%-32.8% and 16.7%-30.2% had improved eyebrow or eyelash assessment scores, respectively, at week 48.
- Between weeks 24 and 48, adverse events were reported in 74%-93% of patients who achieved a SALT score ≤ 20, most were mild or moderate; two serious events were reported but deemed unrelated to treatment. The safety profile was similar across all subgroups.
- No deaths, malignancies, major cardiovascular events, opportunistic infections, or herpes zoster infections were observed.
IN PRACTICE:
“The majority of ritlecitinib-treated patients with AA who met target clinical response based on scalp, eyebrow, or eyelash regrowth at week 24 sustained their response through week 48 with continued treatment,” the authors wrote. “Some patients, including those with more extensive hair loss, may require ritlecitinib treatment beyond 6 months to achieve target clinical response,” they added.
SOURCE:
The study was led by Melissa Piliang, MD, of the Department of Dermatology, Cleveland Clinic, and was published online on October 17 in the Journal of the American Academy of Dermatology.
LIMITATIONS:
The analysis was limited by its post hoc nature, small sample size in each treatment group, and a follow-up period of only 48 weeks.
DISCLOSURES:
This study was funded by Pfizer. Piliang disclosed being a consultant or investigator for Pfizer, Eli Lilly, and Procter & Gamble. Six authors were employees or shareholders of or received salary from Pfizer. Other authors also reported financial relationships with pharmaceutical companies outside this work, including Pfizer.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Projected 2023 Cost Reduction From Tumor Necrosis Factor α Inhibitor Biosimilars in Dermatology: A National Medicare Analysis
To the Editor:
Although biologics provide major therapeutic benefits for dermatologic conditions, they also come with a substantial cost, making them among the most expensive medications available. Medicare and Medicaid spending on biologics for dermatologic conditions increased by 320% from 2012 to 2018, reaching a staggering $10.6 billion in 2018 alone.1 Biosimilars show promise in reducing health care spending for dermatologic conditions; however, their utilization has been limited due to multiple factors, including delayed market entry from patent thickets, exclusionary formulary contracts, and prescriber skepticism regarding their safety and efficacy.2 For instance, a national survey of 1201 US physicians in specialties that are high prescribers of biologics reported that 55% doubted the safety and appropriateness of biosimilars.3
US Food and Drug Administration approval of biosimilars for adalimumab and etanercept offers the potential to reduce health care spending for dermatologic conditions. However, this cost reduction is dependent on utilization rates among dermatologists. In this national cross-sectional review of Medicare data, we predicted the impact of these biosimilars on dermatologic Medicare costs and demonstrated how differing utilization rates among dermatologists can influence potential savings.
To model 2023 utilization and cost reduction from biosimilars, we analyzed Medicare Part D data from 2020 on existing biosimilars, including granulocyte colony–stimulating factors, erythropoiesis-stimulating agents, and tumor necrosis factor α inhibitors.4 Methods in line with a 2021 report from the US Department of Health and Human Services5 as well as those of Yazdany et al6 were used. For each class, we calculated the 2020 distribution of biosimilar and originator drug claims as well as biosimilar cost reduction per 30-day claim. We utilized 2018-2021 annual growth rates for branded adalimumab and etanercept to estimate 30-day claims for 2023 and the cost of these branded agents in the absence of biosimilars. The hypothetical 2023 cost reduction from adalimumab and etanercept biosimilars was estimated by assuming 2020 biosimilar utilization rates and mean cost reduction per claim. This study utilized publicly available or aggregate summary data (not attributable to specific patients) and did not qualify as human subject research; therefore, institutional review board approval was not required.
In 2020, biosimilar utilization proportions ranged from 6.4% (tumor necrosis factor α inhibitors) to 82.7% (granulocyte colony–stimulating factors), with a mean across all classes of 35.7%. On average, the cost per 30-day claim of biosimilars was 66.8% of originator agents (Table 1). In 2021, we identified 57,868 30-day claims for branded adalimumab and etanercept submitted by dermatologists. From 2018 to 2021, 30-day branded adalimumab claims increased by 1.27% annually (cost + 10.62% annually), while claims for branded etanercept decreased by 13.0% annually (cost + 5.68% annually). Assuming these trends, the cost of branded adalimumab and etanercept was estimated to be $539 million in 2023. Applying the aforementioned 35.7% utilization, the introduction of biosimilars in dermatology would yield a cost reduction of approximately $118 million (21.9%). A high utilization rate (82.7%) of biosimilars among dermatologists would increase cost savings to $199 million (36.9%)(Table 2).

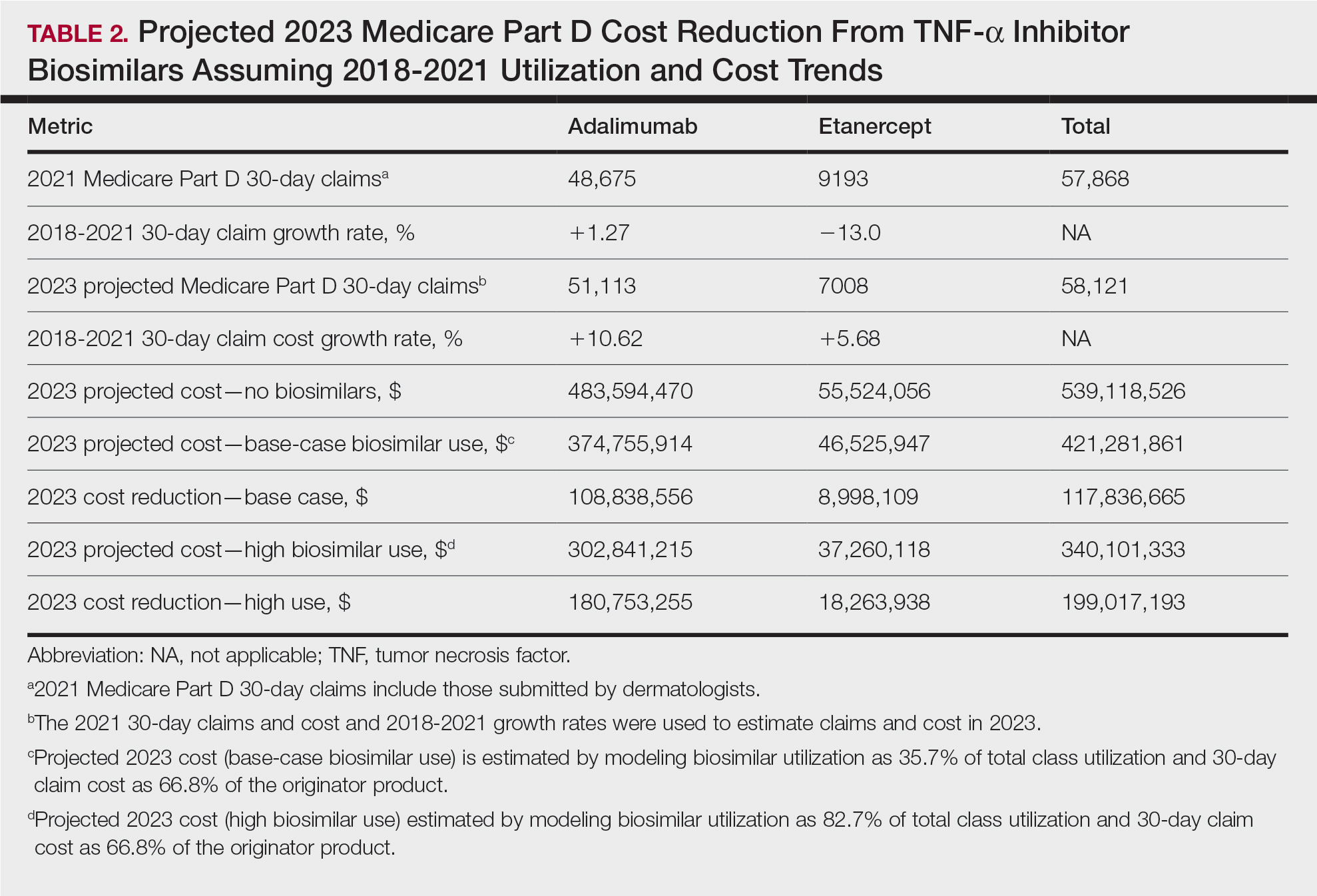
Our study demonstrates that the introduction of 2 biosimilars into dermatology may result in a notable reduction in Medicare expenditures. The savings observed are likely to translate to substantial cost savings for patients. A cross-sectional analysis of 2020 Medicare data indicated that coverage for psoriasis medications was 10.0% to 99.8% across different products and Medicare Part D plans. Consequently, patients faced considerable out-of-pocket expenses, amounting to $5653 and $5714 per year for adalimumab and etanercept, respectively.7
We found that the extent of savings from biosimilars was dependent on the utilization rates among dermatologists, with the highest utilization rate almost doubling the total savings of average utilization rates. Given the impact of high utilization and the wide variation observed, understanding the factors that have influenced uptake of biosimilars is important to increasing utilization as these medications become integrated into dermatology. For instance, limited uptake of infliximab initially may have been influenced by concerns about efficacy and increased adverse events.8,9 In contrast, the high utilization of filgrastim biosimilars (82.7%) may be attributed to its longevity in the market and familiarity to prescribers, as filgrastim was the first biosimilar to be approved in the United States.10
Promoting reasonable utilization of biosimilars may require prescriber education on their safety and approval processes, which could foster increased utilization and reduce skepticism.4 Under the Biologics Price Competition and Innovation Act, the US Food and Drug Administration approves biosimilars only when they exhibit “high similarity” and show no “clinically meaningful differences” compared to the reference biologic, with no added safety risks or reduced efficacy.11 Moreover, a 2023 systematic review of 17 studies found no major difference in efficacy and safety between biosimilars and originators of etanercept, infliximab, and other biologics.12 Understanding these findings may reassure dermatologists and patients about the reliability and safety of biosimilars.
A limitation of our study is that it solely assesses Medicare data and estimates derived from existing (separate) biologic classes. It also does not account for potential expenditure shifts to newer biologic agents (eg, IL-12/17/23 inhibitors) or changes in manufacturer behavior or promotions. Nevertheless, it indicates notable financial savings from new biosimilar agents in dermatology; along with their compelling efficacy and safety profiles, this could represent a substantial benefit to patients and the health care system.
- Price KN, Atluri S, Hsiao JL, et al. Medicare and medicaid spending trends for immunomodulators prescribed for dermatologic conditions. J Dermatolog Treat. 2020;33:575-579.
- Zhai MZ, Sarpatwari A, Kesselheim AS. Why are biosimilars not living up to their promise in the US? AMA J Ethics. 2019;21:E668-E678. doi:10.1001/amajethics.2019.668
- Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33:2160-2172.
- Centers for Medicare & Medicaid Services. Medicare Part D prescribers— by provider and drug. Accessed September 11, 2024. https://data.cms.gov/provider-summary-by-type-of-service/medicare-part-d-prescribers/medicare-part-d-prescribers-by-provider-and-drug/data
- US Department of Health and Human Services. Office of Inspector General. Medicare Part D and beneficiaries could realize significant spending reductions with increased biosimilar use. Accessed September 11, 2024. https://oig.hhs.gov/oei/reports/OEI-05-20-00480.pdf
- Yazdany J, Dudley RA, Lin GA, et al. Out-of-pocket costs for infliximab and its biosimilar for rheumatoid arthritis under Medicare Part D. JAMA. 2018;320:931-933. doi:10.1001/jama.2018.7316
- Pourali SP, Nshuti L, Dusetzina SB. Out-of-pocket costs of specialty medications for psoriasis and psoriatic arthritis treatment in the medicare population. JAMA Dermatol. 2021;157:1239-1241. doi:10.1001/ jamadermatol.2021.3616
- Lebwohl M. Biosimilars in dermatology. JAMA Dermatol. 2021; 157:641-642. doi:10.1001/jamadermatol.2021.0219
- Westerkam LL, Tackett KJ, Sayed CJ. Comparing the effectiveness and safety associated with infliximab vs infliximab-abda therapy for patients with hidradenitis suppurativa. JAMA Dermatol. 2021;157:708-711. doi:10.1001/jamadermatol.2021.0220
- Awad M, Singh P, Hilas O. Zarxio (Filgrastim-sndz): the first biosimilar approved by the FDA. P T. 2017;42:19-23.
- Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations guidance for industry. US Department of Health and Human Services website. Updated June 15, 2022. Accessed October 21, 2024. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analyticalassessment-and-other-quality
- Phan DB, Elyoussfi S, Stevenson M, et al. Biosimilars for the treatment of psoriasis: a systematic review of clinical trials and observational studies. JAMA Dermatol. 2023;159:763-771. doi:10.1001/jamadermatol.2023.1338
To the Editor:
Although biologics provide major therapeutic benefits for dermatologic conditions, they also come with a substantial cost, making them among the most expensive medications available. Medicare and Medicaid spending on biologics for dermatologic conditions increased by 320% from 2012 to 2018, reaching a staggering $10.6 billion in 2018 alone.1 Biosimilars show promise in reducing health care spending for dermatologic conditions; however, their utilization has been limited due to multiple factors, including delayed market entry from patent thickets, exclusionary formulary contracts, and prescriber skepticism regarding their safety and efficacy.2 For instance, a national survey of 1201 US physicians in specialties that are high prescribers of biologics reported that 55% doubted the safety and appropriateness of biosimilars.3
US Food and Drug Administration approval of biosimilars for adalimumab and etanercept offers the potential to reduce health care spending for dermatologic conditions. However, this cost reduction is dependent on utilization rates among dermatologists. In this national cross-sectional review of Medicare data, we predicted the impact of these biosimilars on dermatologic Medicare costs and demonstrated how differing utilization rates among dermatologists can influence potential savings.
To model 2023 utilization and cost reduction from biosimilars, we analyzed Medicare Part D data from 2020 on existing biosimilars, including granulocyte colony–stimulating factors, erythropoiesis-stimulating agents, and tumor necrosis factor α inhibitors.4 Methods in line with a 2021 report from the US Department of Health and Human Services5 as well as those of Yazdany et al6 were used. For each class, we calculated the 2020 distribution of biosimilar and originator drug claims as well as biosimilar cost reduction per 30-day claim. We utilized 2018-2021 annual growth rates for branded adalimumab and etanercept to estimate 30-day claims for 2023 and the cost of these branded agents in the absence of biosimilars. The hypothetical 2023 cost reduction from adalimumab and etanercept biosimilars was estimated by assuming 2020 biosimilar utilization rates and mean cost reduction per claim. This study utilized publicly available or aggregate summary data (not attributable to specific patients) and did not qualify as human subject research; therefore, institutional review board approval was not required.
In 2020, biosimilar utilization proportions ranged from 6.4% (tumor necrosis factor α inhibitors) to 82.7% (granulocyte colony–stimulating factors), with a mean across all classes of 35.7%. On average, the cost per 30-day claim of biosimilars was 66.8% of originator agents (Table 1). In 2021, we identified 57,868 30-day claims for branded adalimumab and etanercept submitted by dermatologists. From 2018 to 2021, 30-day branded adalimumab claims increased by 1.27% annually (cost + 10.62% annually), while claims for branded etanercept decreased by 13.0% annually (cost + 5.68% annually). Assuming these trends, the cost of branded adalimumab and etanercept was estimated to be $539 million in 2023. Applying the aforementioned 35.7% utilization, the introduction of biosimilars in dermatology would yield a cost reduction of approximately $118 million (21.9%). A high utilization rate (82.7%) of biosimilars among dermatologists would increase cost savings to $199 million (36.9%)(Table 2).
Our study demonstrates that the introduction of 2 biosimilars into dermatology may result in a notable reduction in Medicare expenditures. The savings observed are likely to translate to substantial cost savings for patients. A cross-sectional analysis of 2020 Medicare data indicated that coverage for psoriasis medications was 10.0% to 99.8% across different products and Medicare Part D plans. Consequently, patients faced considerable out-of-pocket expenses, amounting to $5653 and $5714 per year for adalimumab and etanercept, respectively.7
We found that the extent of savings from biosimilars was dependent on the utilization rates among dermatologists, with the highest utilization rate almost doubling the total savings of average utilization rates. Given the impact of high utilization and the wide variation observed, understanding the factors that have influenced uptake of biosimilars is important to increasing utilization as these medications become integrated into dermatology. For instance, limited uptake of infliximab initially may have been influenced by concerns about efficacy and increased adverse events.8,9 In contrast, the high utilization of filgrastim biosimilars (82.7%) may be attributed to its longevity in the market and familiarity to prescribers, as filgrastim was the first biosimilar to be approved in the United States.10
Promoting reasonable utilization of biosimilars may require prescriber education on their safety and approval processes, which could foster increased utilization and reduce skepticism.4 Under the Biologics Price Competition and Innovation Act, the US Food and Drug Administration approves biosimilars only when they exhibit “high similarity” and show no “clinically meaningful differences” compared to the reference biologic, with no added safety risks or reduced efficacy.11 Moreover, a 2023 systematic review of 17 studies found no major difference in efficacy and safety between biosimilars and originators of etanercept, infliximab, and other biologics.12 Understanding these findings may reassure dermatologists and patients about the reliability and safety of biosimilars.
A limitation of our study is that it solely assesses Medicare data and estimates derived from existing (separate) biologic classes. It also does not account for potential expenditure shifts to newer biologic agents (eg, IL-12/17/23 inhibitors) or changes in manufacturer behavior or promotions. Nevertheless, it indicates notable financial savings from new biosimilar agents in dermatology; along with their compelling efficacy and safety profiles, this could represent a substantial benefit to patients and the health care system.
To the Editor:
Although biologics provide major therapeutic benefits for dermatologic conditions, they also come with a substantial cost, making them among the most expensive medications available. Medicare and Medicaid spending on biologics for dermatologic conditions increased by 320% from 2012 to 2018, reaching a staggering $10.6 billion in 2018 alone.1 Biosimilars show promise in reducing health care spending for dermatologic conditions; however, their utilization has been limited due to multiple factors, including delayed market entry from patent thickets, exclusionary formulary contracts, and prescriber skepticism regarding their safety and efficacy.2 For instance, a national survey of 1201 US physicians in specialties that are high prescribers of biologics reported that 55% doubted the safety and appropriateness of biosimilars.3
US Food and Drug Administration approval of biosimilars for adalimumab and etanercept offers the potential to reduce health care spending for dermatologic conditions. However, this cost reduction is dependent on utilization rates among dermatologists. In this national cross-sectional review of Medicare data, we predicted the impact of these biosimilars on dermatologic Medicare costs and demonstrated how differing utilization rates among dermatologists can influence potential savings.
To model 2023 utilization and cost reduction from biosimilars, we analyzed Medicare Part D data from 2020 on existing biosimilars, including granulocyte colony–stimulating factors, erythropoiesis-stimulating agents, and tumor necrosis factor α inhibitors.4 Methods in line with a 2021 report from the US Department of Health and Human Services5 as well as those of Yazdany et al6 were used. For each class, we calculated the 2020 distribution of biosimilar and originator drug claims as well as biosimilar cost reduction per 30-day claim. We utilized 2018-2021 annual growth rates for branded adalimumab and etanercept to estimate 30-day claims for 2023 and the cost of these branded agents in the absence of biosimilars. The hypothetical 2023 cost reduction from adalimumab and etanercept biosimilars was estimated by assuming 2020 biosimilar utilization rates and mean cost reduction per claim. This study utilized publicly available or aggregate summary data (not attributable to specific patients) and did not qualify as human subject research; therefore, institutional review board approval was not required.
In 2020, biosimilar utilization proportions ranged from 6.4% (tumor necrosis factor α inhibitors) to 82.7% (granulocyte colony–stimulating factors), with a mean across all classes of 35.7%. On average, the cost per 30-day claim of biosimilars was 66.8% of originator agents (Table 1). In 2021, we identified 57,868 30-day claims for branded adalimumab and etanercept submitted by dermatologists. From 2018 to 2021, 30-day branded adalimumab claims increased by 1.27% annually (cost + 10.62% annually), while claims for branded etanercept decreased by 13.0% annually (cost + 5.68% annually). Assuming these trends, the cost of branded adalimumab and etanercept was estimated to be $539 million in 2023. Applying the aforementioned 35.7% utilization, the introduction of biosimilars in dermatology would yield a cost reduction of approximately $118 million (21.9%). A high utilization rate (82.7%) of biosimilars among dermatologists would increase cost savings to $199 million (36.9%)(Table 2).
Our study demonstrates that the introduction of 2 biosimilars into dermatology may result in a notable reduction in Medicare expenditures. The savings observed are likely to translate to substantial cost savings for patients. A cross-sectional analysis of 2020 Medicare data indicated that coverage for psoriasis medications was 10.0% to 99.8% across different products and Medicare Part D plans. Consequently, patients faced considerable out-of-pocket expenses, amounting to $5653 and $5714 per year for adalimumab and etanercept, respectively.7
We found that the extent of savings from biosimilars was dependent on the utilization rates among dermatologists, with the highest utilization rate almost doubling the total savings of average utilization rates. Given the impact of high utilization and the wide variation observed, understanding the factors that have influenced uptake of biosimilars is important to increasing utilization as these medications become integrated into dermatology. For instance, limited uptake of infliximab initially may have been influenced by concerns about efficacy and increased adverse events.8,9 In contrast, the high utilization of filgrastim biosimilars (82.7%) may be attributed to its longevity in the market and familiarity to prescribers, as filgrastim was the first biosimilar to be approved in the United States.10
Promoting reasonable utilization of biosimilars may require prescriber education on their safety and approval processes, which could foster increased utilization and reduce skepticism.4 Under the Biologics Price Competition and Innovation Act, the US Food and Drug Administration approves biosimilars only when they exhibit “high similarity” and show no “clinically meaningful differences” compared to the reference biologic, with no added safety risks or reduced efficacy.11 Moreover, a 2023 systematic review of 17 studies found no major difference in efficacy and safety between biosimilars and originators of etanercept, infliximab, and other biologics.12 Understanding these findings may reassure dermatologists and patients about the reliability and safety of biosimilars.
A limitation of our study is that it solely assesses Medicare data and estimates derived from existing (separate) biologic classes. It also does not account for potential expenditure shifts to newer biologic agents (eg, IL-12/17/23 inhibitors) or changes in manufacturer behavior or promotions. Nevertheless, it indicates notable financial savings from new biosimilar agents in dermatology; along with their compelling efficacy and safety profiles, this could represent a substantial benefit to patients and the health care system.
- Price KN, Atluri S, Hsiao JL, et al. Medicare and medicaid spending trends for immunomodulators prescribed for dermatologic conditions. J Dermatolog Treat. 2020;33:575-579.
- Zhai MZ, Sarpatwari A, Kesselheim AS. Why are biosimilars not living up to their promise in the US? AMA J Ethics. 2019;21:E668-E678. doi:10.1001/amajethics.2019.668
- Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33:2160-2172.
- Centers for Medicare & Medicaid Services. Medicare Part D prescribers— by provider and drug. Accessed September 11, 2024. https://data.cms.gov/provider-summary-by-type-of-service/medicare-part-d-prescribers/medicare-part-d-prescribers-by-provider-and-drug/data
- US Department of Health and Human Services. Office of Inspector General. Medicare Part D and beneficiaries could realize significant spending reductions with increased biosimilar use. Accessed September 11, 2024. https://oig.hhs.gov/oei/reports/OEI-05-20-00480.pdf
- Yazdany J, Dudley RA, Lin GA, et al. Out-of-pocket costs for infliximab and its biosimilar for rheumatoid arthritis under Medicare Part D. JAMA. 2018;320:931-933. doi:10.1001/jama.2018.7316
- Pourali SP, Nshuti L, Dusetzina SB. Out-of-pocket costs of specialty medications for psoriasis and psoriatic arthritis treatment in the medicare population. JAMA Dermatol. 2021;157:1239-1241. doi:10.1001/ jamadermatol.2021.3616
- Lebwohl M. Biosimilars in dermatology. JAMA Dermatol. 2021; 157:641-642. doi:10.1001/jamadermatol.2021.0219
- Westerkam LL, Tackett KJ, Sayed CJ. Comparing the effectiveness and safety associated with infliximab vs infliximab-abda therapy for patients with hidradenitis suppurativa. JAMA Dermatol. 2021;157:708-711. doi:10.1001/jamadermatol.2021.0220
- Awad M, Singh P, Hilas O. Zarxio (Filgrastim-sndz): the first biosimilar approved by the FDA. P T. 2017;42:19-23.
- Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations guidance for industry. US Department of Health and Human Services website. Updated June 15, 2022. Accessed October 21, 2024. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analyticalassessment-and-other-quality
- Phan DB, Elyoussfi S, Stevenson M, et al. Biosimilars for the treatment of psoriasis: a systematic review of clinical trials and observational studies. JAMA Dermatol. 2023;159:763-771. doi:10.1001/jamadermatol.2023.1338
- Price KN, Atluri S, Hsiao JL, et al. Medicare and medicaid spending trends for immunomodulators prescribed for dermatologic conditions. J Dermatolog Treat. 2020;33:575-579.
- Zhai MZ, Sarpatwari A, Kesselheim AS. Why are biosimilars not living up to their promise in the US? AMA J Ethics. 2019;21:E668-E678. doi:10.1001/amajethics.2019.668
- Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33:2160-2172.
- Centers for Medicare & Medicaid Services. Medicare Part D prescribers— by provider and drug. Accessed September 11, 2024. https://data.cms.gov/provider-summary-by-type-of-service/medicare-part-d-prescribers/medicare-part-d-prescribers-by-provider-and-drug/data
- US Department of Health and Human Services. Office of Inspector General. Medicare Part D and beneficiaries could realize significant spending reductions with increased biosimilar use. Accessed September 11, 2024. https://oig.hhs.gov/oei/reports/OEI-05-20-00480.pdf
- Yazdany J, Dudley RA, Lin GA, et al. Out-of-pocket costs for infliximab and its biosimilar for rheumatoid arthritis under Medicare Part D. JAMA. 2018;320:931-933. doi:10.1001/jama.2018.7316
- Pourali SP, Nshuti L, Dusetzina SB. Out-of-pocket costs of specialty medications for psoriasis and psoriatic arthritis treatment in the medicare population. JAMA Dermatol. 2021;157:1239-1241. doi:10.1001/ jamadermatol.2021.3616
- Lebwohl M. Biosimilars in dermatology. JAMA Dermatol. 2021; 157:641-642. doi:10.1001/jamadermatol.2021.0219
- Westerkam LL, Tackett KJ, Sayed CJ. Comparing the effectiveness and safety associated with infliximab vs infliximab-abda therapy for patients with hidradenitis suppurativa. JAMA Dermatol. 2021;157:708-711. doi:10.1001/jamadermatol.2021.0220
- Awad M, Singh P, Hilas O. Zarxio (Filgrastim-sndz): the first biosimilar approved by the FDA. P T. 2017;42:19-23.
- Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations guidance for industry. US Department of Health and Human Services website. Updated June 15, 2022. Accessed October 21, 2024. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analyticalassessment-and-other-quality
- Phan DB, Elyoussfi S, Stevenson M, et al. Biosimilars for the treatment of psoriasis: a systematic review of clinical trials and observational studies. JAMA Dermatol. 2023;159:763-771. doi:10.1001/jamadermatol.2023.1338
Practice Points
- Biosimilars for adalimumab and etanercept are safe and effective alternatives with the potential to reduce health care costs in dermatology by approximately $118 million.
- A high utilization rate of biosimilars by dermatologists would increase cost savings even further.
Phenytoin-Induced DRESS Syndrome: Clinical and Laboratory Characteristics
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome—a severe cutaneous adverse drug reaction—is characterized by a cutaneous rash and systemic upset in the form of various internal organ and hematologic disturbances. This delayed and idiosyncratic syndrome went by several names, including anticonvulsant hypersensitivity syndrome, before Bocquet et al1 proposed the term DRESS syndrome.
Phenytoin, a hydantoin derivative used in neurology, was implicated in 41% of cases of DRESS syndrome in a study of 100 patients conducted in southern India.2,3 While DRESS syndrome is a newer name, the clinical picture of DRESS secondary to phenytoin use remains similar in that it manifests with a morbilliform rash and systemic upset. We sought to describe the clinical and laboratory characteristics of phenytoin-induced DRESS syndrome in this case series.
The analysis included 23 patients with DRESS syndrome secondary to phenytoin use who presented to a tertiary care institution in North India between July 2021 and December 2022, satisfied the European Registry of Severe Cutaneous Adverse Reaction (RegiSCAR) criteria,4 and achieved a DRESS diagnostic score of more than 1. The mean age of the patients was 44 years (range, 14–74 years). There was a slight female predominance with a male to female ratio of 0.9:1. More than half of the patients (52.2% [12/23]) presented directly to the dermatology outpatient department; the remaining patients were referred from other departments (47.8% [11/23]). Patients primarily were receiving phenytoin for neurologic indications. Specific reasons included antiseizure prophylaxis following a traffic accident (34.8% [8/23]); epilepsy (26.1% [6/23]); and neoplastic (17.4% [4/23]), vascular (17.4% [4/23]), and infectious (4.3% [1/23]) causes. The mean latency period from drug intake to symptom onset was 29 days (range, 6–62 days), and the mean illness duration was 9 days (range, 1–45 days).
The majority of patients experienced pruritus (91.3% [21/23]) and fever (74.0% [17/23]), and all initially had a rash. Maculopapular morphology was seen in all patients. Erythema multiforme–like (17.4% [4/23]), erythrodermic (17.4% [4/23]), and vesicular (13.0% [3/23]) rashes also were documented (Figure 1). The trunk (100% [23/23]) and extremities (95.7% [22/23]) were involved most often, followed by the palms and soles (56.5% [13/23]). The mean total body surface area affected was 73.65%. Only 7 patients (30.4%) had mucosal involvement; nonhemorrhagic cheilitis was the most common manifestation.
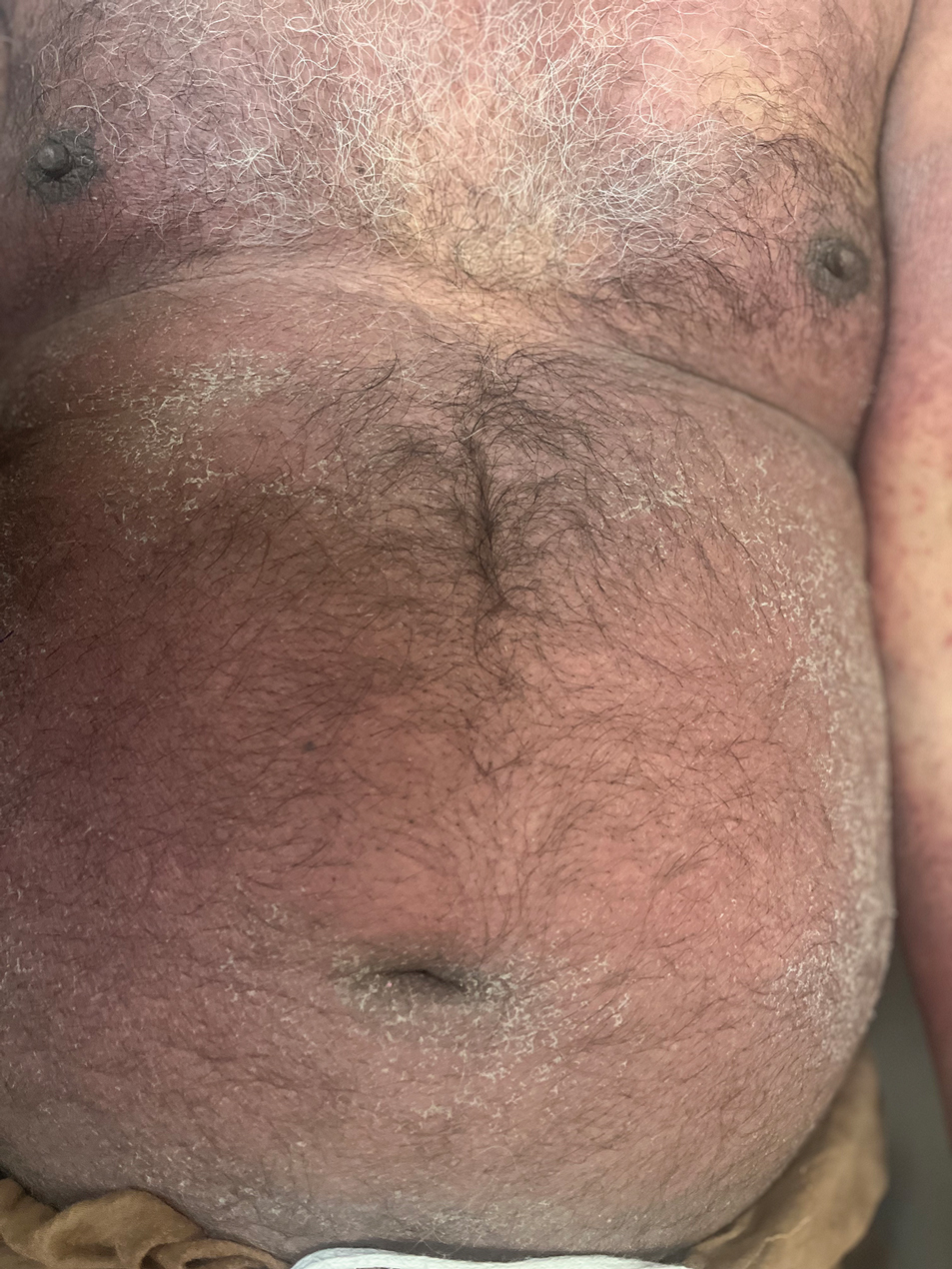
Facial edema, a hallmark feature of DRESS syndrome, was noted in 69.6% (16/23) of patients (Figure 2). Lymphadenopathy was present in 43.5% (10/23) of patients; of those cases, the inguinal (40.0%; n=4) and cervical (30%; n=3) nodes most commonly were involved. Although DRESS syndrome can affect internal organs, this was an issue for only 2 (8.7%) patients who experienced mild hepatomegaly.
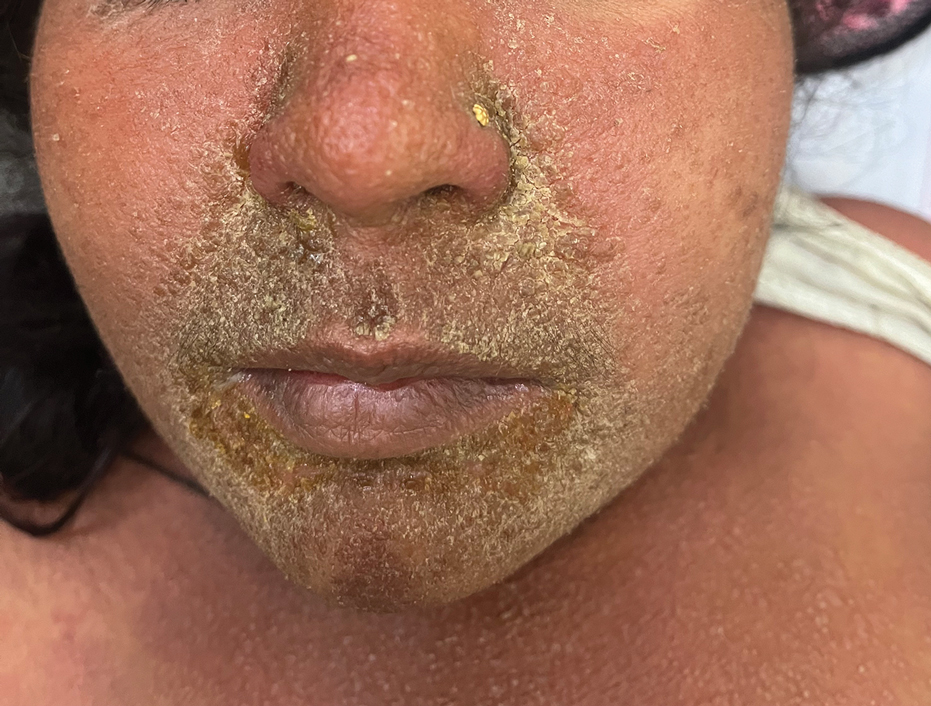
Laboratory investigations revealed a mean differential eosinophil percentage of 10.3% (reference range, 1%–4%), while the mean absolute eosinophil count was 1.0634×109/L (reference range, 0.02–0.5×109/L). Other hematologic findings included the mean percentages of neutrophils (60%; reference range, 50%–60%), lymphocytes (19.95%; reference range, 20%–50%), and monocytes (8.70%; reference range, 2%–8%).
Liver function tests revealed transaminitis5 as the most common finding, with mean aspartate aminotransferase levels of 109 U/L (reference range, 8–33 U/L), mean alanine aminotransferase of 97.9 U/L (reference range, 7–56 U/L), and mean alkaline phosphatase levels of 211.35 U/L (reference range, 44–147 U/L). Half of the patients had notable (>2 times the upper limit of normal) transaminitis.
Renal blood workup revealed slightly elevated blood urea nitrogen levels with a mean value of 28.4 mg/dL (reference range, 6–24 mg/dL), and mean serum creatinine was 0.78 mg/dL (reference range for men, 0.7–1.3 mg/dL; for women, 0.6–1.1 mg/dL).
All patients were treated with oral steroids (prednisolone 1 mg/kg/d) before tapering slowly over the following 6 to 8 weeks. The culprit drug (phenytoin) was stopped on the day of presentation. Resolution of rash and itching was seen in all patients by 3 weeks after presentation without any relapse by follow-up at 6 weeks from presentation to the hospital.
Our case series seeks to discuss the clinical and laboratory features of phenytoin-induced DRESS syndrome. Our patients had more erythrodermic and erythema multiforme–like morphologies, less mucosal involvement, more hepatic involvement, and earlier resolution.
- Bocquet H, Bagot M, Roujeau JC. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms: DRESS). Semin Cutan Med Surg. 1996;15:250-257. doi:10.1016/s1085-5629(96)80038-1
- Patocka J, Wu Q, Nepovimova E, et al. Phenytoin—an anti-seizure drug: overview of its chemistry, pharmacology and toxicology. Food Chem Toxicol. 2020;142:111393. doi:10.1016/j.fct.2020.111393
- Sasidharanpillai S, Chathoth AT, Khader A, et al. Predictors of disease severity in drug reaction with eosinophilia and systemic symptoms. Indian J Dermatol Venereol Leprol. 2019;85:266-275. doi:10.4103/ijdvl.IJDVL_482_17
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Brit J Dermatol. 2013;169:1071-1080.
- Morán-Mariños C, Alva-Diaz C, De la Cruz Ramirez W, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS) induced by phenytoin re-exposure: case report and systematic review. Acta Clin Belg. 2022;77:177-185. doi:10.1080/17843286.2020.1767459
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome—a severe cutaneous adverse drug reaction—is characterized by a cutaneous rash and systemic upset in the form of various internal organ and hematologic disturbances. This delayed and idiosyncratic syndrome went by several names, including anticonvulsant hypersensitivity syndrome, before Bocquet et al1 proposed the term DRESS syndrome.
Phenytoin, a hydantoin derivative used in neurology, was implicated in 41% of cases of DRESS syndrome in a study of 100 patients conducted in southern India.2,3 While DRESS syndrome is a newer name, the clinical picture of DRESS secondary to phenytoin use remains similar in that it manifests with a morbilliform rash and systemic upset. We sought to describe the clinical and laboratory characteristics of phenytoin-induced DRESS syndrome in this case series.
The analysis included 23 patients with DRESS syndrome secondary to phenytoin use who presented to a tertiary care institution in North India between July 2021 and December 2022, satisfied the European Registry of Severe Cutaneous Adverse Reaction (RegiSCAR) criteria,4 and achieved a DRESS diagnostic score of more than 1. The mean age of the patients was 44 years (range, 14–74 years). There was a slight female predominance with a male to female ratio of 0.9:1. More than half of the patients (52.2% [12/23]) presented directly to the dermatology outpatient department; the remaining patients were referred from other departments (47.8% [11/23]). Patients primarily were receiving phenytoin for neurologic indications. Specific reasons included antiseizure prophylaxis following a traffic accident (34.8% [8/23]); epilepsy (26.1% [6/23]); and neoplastic (17.4% [4/23]), vascular (17.4% [4/23]), and infectious (4.3% [1/23]) causes. The mean latency period from drug intake to symptom onset was 29 days (range, 6–62 days), and the mean illness duration was 9 days (range, 1–45 days).
The majority of patients experienced pruritus (91.3% [21/23]) and fever (74.0% [17/23]), and all initially had a rash. Maculopapular morphology was seen in all patients. Erythema multiforme–like (17.4% [4/23]), erythrodermic (17.4% [4/23]), and vesicular (13.0% [3/23]) rashes also were documented (Figure 1). The trunk (100% [23/23]) and extremities (95.7% [22/23]) were involved most often, followed by the palms and soles (56.5% [13/23]). The mean total body surface area affected was 73.65%. Only 7 patients (30.4%) had mucosal involvement; nonhemorrhagic cheilitis was the most common manifestation.
Facial edema, a hallmark feature of DRESS syndrome, was noted in 69.6% (16/23) of patients (Figure 2). Lymphadenopathy was present in 43.5% (10/23) of patients; of those cases, the inguinal (40.0%; n=4) and cervical (30%; n=3) nodes most commonly were involved. Although DRESS syndrome can affect internal organs, this was an issue for only 2 (8.7%) patients who experienced mild hepatomegaly.
Laboratory investigations revealed a mean differential eosinophil percentage of 10.3% (reference range, 1%–4%), while the mean absolute eosinophil count was 1.0634×109/L (reference range, 0.02–0.5×109/L). Other hematologic findings included the mean percentages of neutrophils (60%; reference range, 50%–60%), lymphocytes (19.95%; reference range, 20%–50%), and monocytes (8.70%; reference range, 2%–8%).
Liver function tests revealed transaminitis5 as the most common finding, with mean aspartate aminotransferase levels of 109 U/L (reference range, 8–33 U/L), mean alanine aminotransferase of 97.9 U/L (reference range, 7–56 U/L), and mean alkaline phosphatase levels of 211.35 U/L (reference range, 44–147 U/L). Half of the patients had notable (>2 times the upper limit of normal) transaminitis.
Renal blood workup revealed slightly elevated blood urea nitrogen levels with a mean value of 28.4 mg/dL (reference range, 6–24 mg/dL), and mean serum creatinine was 0.78 mg/dL (reference range for men, 0.7–1.3 mg/dL; for women, 0.6–1.1 mg/dL).
All patients were treated with oral steroids (prednisolone 1 mg/kg/d) before tapering slowly over the following 6 to 8 weeks. The culprit drug (phenytoin) was stopped on the day of presentation. Resolution of rash and itching was seen in all patients by 3 weeks after presentation without any relapse by follow-up at 6 weeks from presentation to the hospital.
Our case series seeks to discuss the clinical and laboratory features of phenytoin-induced DRESS syndrome. Our patients had more erythrodermic and erythema multiforme–like morphologies, less mucosal involvement, more hepatic involvement, and earlier resolution.
To the Editor:
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome—a severe cutaneous adverse drug reaction—is characterized by a cutaneous rash and systemic upset in the form of various internal organ and hematologic disturbances. This delayed and idiosyncratic syndrome went by several names, including anticonvulsant hypersensitivity syndrome, before Bocquet et al1 proposed the term DRESS syndrome.
Phenytoin, a hydantoin derivative used in neurology, was implicated in 41% of cases of DRESS syndrome in a study of 100 patients conducted in southern India.2,3 While DRESS syndrome is a newer name, the clinical picture of DRESS secondary to phenytoin use remains similar in that it manifests with a morbilliform rash and systemic upset. We sought to describe the clinical and laboratory characteristics of phenytoin-induced DRESS syndrome in this case series.
The analysis included 23 patients with DRESS syndrome secondary to phenytoin use who presented to a tertiary care institution in North India between July 2021 and December 2022, satisfied the European Registry of Severe Cutaneous Adverse Reaction (RegiSCAR) criteria,4 and achieved a DRESS diagnostic score of more than 1. The mean age of the patients was 44 years (range, 14–74 years). There was a slight female predominance with a male to female ratio of 0.9:1. More than half of the patients (52.2% [12/23]) presented directly to the dermatology outpatient department; the remaining patients were referred from other departments (47.8% [11/23]). Patients primarily were receiving phenytoin for neurologic indications. Specific reasons included antiseizure prophylaxis following a traffic accident (34.8% [8/23]); epilepsy (26.1% [6/23]); and neoplastic (17.4% [4/23]), vascular (17.4% [4/23]), and infectious (4.3% [1/23]) causes. The mean latency period from drug intake to symptom onset was 29 days (range, 6–62 days), and the mean illness duration was 9 days (range, 1–45 days).
The majority of patients experienced pruritus (91.3% [21/23]) and fever (74.0% [17/23]), and all initially had a rash. Maculopapular morphology was seen in all patients. Erythema multiforme–like (17.4% [4/23]), erythrodermic (17.4% [4/23]), and vesicular (13.0% [3/23]) rashes also were documented (Figure 1). The trunk (100% [23/23]) and extremities (95.7% [22/23]) were involved most often, followed by the palms and soles (56.5% [13/23]). The mean total body surface area affected was 73.65%. Only 7 patients (30.4%) had mucosal involvement; nonhemorrhagic cheilitis was the most common manifestation.
Facial edema, a hallmark feature of DRESS syndrome, was noted in 69.6% (16/23) of patients (Figure 2). Lymphadenopathy was present in 43.5% (10/23) of patients; of those cases, the inguinal (40.0%; n=4) and cervical (30%; n=3) nodes most commonly were involved. Although DRESS syndrome can affect internal organs, this was an issue for only 2 (8.7%) patients who experienced mild hepatomegaly.
Laboratory investigations revealed a mean differential eosinophil percentage of 10.3% (reference range, 1%–4%), while the mean absolute eosinophil count was 1.0634×109/L (reference range, 0.02–0.5×109/L). Other hematologic findings included the mean percentages of neutrophils (60%; reference range, 50%–60%), lymphocytes (19.95%; reference range, 20%–50%), and monocytes (8.70%; reference range, 2%–8%).
Liver function tests revealed transaminitis5 as the most common finding, with mean aspartate aminotransferase levels of 109 U/L (reference range, 8–33 U/L), mean alanine aminotransferase of 97.9 U/L (reference range, 7–56 U/L), and mean alkaline phosphatase levels of 211.35 U/L (reference range, 44–147 U/L). Half of the patients had notable (>2 times the upper limit of normal) transaminitis.
Renal blood workup revealed slightly elevated blood urea nitrogen levels with a mean value of 28.4 mg/dL (reference range, 6–24 mg/dL), and mean serum creatinine was 0.78 mg/dL (reference range for men, 0.7–1.3 mg/dL; for women, 0.6–1.1 mg/dL).
All patients were treated with oral steroids (prednisolone 1 mg/kg/d) before tapering slowly over the following 6 to 8 weeks. The culprit drug (phenytoin) was stopped on the day of presentation. Resolution of rash and itching was seen in all patients by 3 weeks after presentation without any relapse by follow-up at 6 weeks from presentation to the hospital.
Our case series seeks to discuss the clinical and laboratory features of phenytoin-induced DRESS syndrome. Our patients had more erythrodermic and erythema multiforme–like morphologies, less mucosal involvement, more hepatic involvement, and earlier resolution.
- Bocquet H, Bagot M, Roujeau JC. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms: DRESS). Semin Cutan Med Surg. 1996;15:250-257. doi:10.1016/s1085-5629(96)80038-1
- Patocka J, Wu Q, Nepovimova E, et al. Phenytoin—an anti-seizure drug: overview of its chemistry, pharmacology and toxicology. Food Chem Toxicol. 2020;142:111393. doi:10.1016/j.fct.2020.111393
- Sasidharanpillai S, Chathoth AT, Khader A, et al. Predictors of disease severity in drug reaction with eosinophilia and systemic symptoms. Indian J Dermatol Venereol Leprol. 2019;85:266-275. doi:10.4103/ijdvl.IJDVL_482_17
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Brit J Dermatol. 2013;169:1071-1080.
- Morán-Mariños C, Alva-Diaz C, De la Cruz Ramirez W, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS) induced by phenytoin re-exposure: case report and systematic review. Acta Clin Belg. 2022;77:177-185. doi:10.1080/17843286.2020.1767459
- Bocquet H, Bagot M, Roujeau JC. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (drug rash with eosinophilia and systemic symptoms: DRESS). Semin Cutan Med Surg. 1996;15:250-257. doi:10.1016/s1085-5629(96)80038-1
- Patocka J, Wu Q, Nepovimova E, et al. Phenytoin—an anti-seizure drug: overview of its chemistry, pharmacology and toxicology. Food Chem Toxicol. 2020;142:111393. doi:10.1016/j.fct.2020.111393
- Sasidharanpillai S, Chathoth AT, Khader A, et al. Predictors of disease severity in drug reaction with eosinophilia and systemic symptoms. Indian J Dermatol Venereol Leprol. 2019;85:266-275. doi:10.4103/ijdvl.IJDVL_482_17
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Brit J Dermatol. 2013;169:1071-1080.
- Morán-Mariños C, Alva-Diaz C, De la Cruz Ramirez W, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS) induced by phenytoin re-exposure: case report and systematic review. Acta Clin Belg. 2022;77:177-185. doi:10.1080/17843286.2020.1767459
Practice Points
- Phenytoin has been implicated in drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, and common symptoms include rash, pruritus, and fever.
- Transaminitis may occur in patients with DRESS syndrome secondary to phenytoin use.
Spontaneously Draining Axillary Tumors in a Young Woman
THE DIAGNOSIS: Ectopic (Accessory) Breast Tissue
Ectopic (accessory) breast tissue (EBT) is a phenomenon caused by failed regression of one or more components of the embryonic mammary ridges— paired ectodermal thickenings that eventually develop into definitive breast tissue including the nipples, areolae, and parenchyma. Ectopic breast tissue is more common in women than men and is believed to be sporadic, although an autosomal-dominant inheritance mechanism with incomplete penetrance has been proposed for some cases.1 The reported incidence of EBT varies greatly among racial and ethnic groups but is most common in individuals of Asian descent. The incidence across all types of EBT is estimated at 0.25% to 6% in the general population.2
Observed clinical variations of EBT range from simple polythelia (additional nipple[s] without associated parenchyma) to complete polymastia (organized and differentiated accessory breasts). Some types of EBT are rarer than others: One report of gynecologic cancer screenings in 1660 patients found polymastia and polythelia incidences of 0.12% and 5.48%, respectively.3 Of the symptomatic variations, isolated parenchymal EBT without a nipple or areolar complex is the most common and may manifest clinically as unilateral or bilateral tender, mildly erythematous nodules or masses often located in the axillae. Ectopic breast tissue generally is observed along the milk line, a developmental regional designation corresponding to the embryologic mammary ridge and extending linearly from the anterior axilla to the inguinal fold on both sides of the body; however, there have been rare reports of EBT manifesting in areas outside the milk line, such as the face, neck, back, vulva, and extremities.2,3
Given that the underlying elements of EBT usually are hormone responsive (as with normal breast tissue), the initial symptom onset and subsequent manifestation frequently coincide with pubertal milestones, pregnancy, or lactation. Furthermore, some patients with EBT may experience symptom fluctuations in concordance with monthly menstrual phases. Many cases of EBT are selflimited and resolve within weeks to months after the end of a pregnancy or lactation, but some cases may persist. Continued observation and follow-up are advisable in all patients, as EBT symptoms often recur and the tissue is susceptible to the same disease processes that affect normal breasts, the most concerning of which is malignancy.4 Although the true incidence is limited by available data, primary ectopic breast malignancy has been estimated to account for 0.3% to 3.8% of diagnosed breast malignancies.2 Cases of malignancy arising from EBT often are of higher grade and poorer prognosis, a finding that may be attributable to diagnostic delays caused by oversight or misdiagnosis of EBT rather than inherent differences in the biologic profile of the tumors.2,4 Patients with a documented history of EBT may benefit from having their routine breast cancer screenings expanded to include areas with EBT foci.
Potential misdiagnoses for EBT include subcutaneous lipoma, axillary lymphadenopathy, abscess, hidradenitis suppurativa, or malignancy. Features that are suggestive of EBT include symptom association with hormone fluctuations (eg, menstrual phases), absence of fever, and lactescent rather than purulent drainage. Among reported EBT cases, spontaneous lactation rarely is described and, if present, often is associated with a history of prior trauma (eg, core needle biopsy or local abscess formation).5 This trauma creates an aberrant connection known as a milk fistula between the underlying parenchyma and the skin surface. Interestingly, our patient denied any history of axillary trauma, but she was noted to be lactating from an apparent milk fistula rather than an organized secretory duct system.
Though a patient history and clinical examination may be sufficient to diagnose EBT cases that are more physically apparent and well correlated with hormone fluctuations, many cases require additional diagnostic studies for confirmation. Of the tools available, ultrasonography generally is considered first-line due to its noninvasive nature, low cost, minimal risk, and high diagnostic value.2 Ultrasonography quickly differentiates between abscesses and cystlike processes, which may appear as discrete areas of decreased echogenicity, and breast tissue, which manifests with fibroglandular tissue and lobules of fat.2,6 Additionally, ultrasonography may demonstrate the secretion of milk through ducts or fistulae, if present. Should examination with ultrasonography prove inconclusive, follow-up studies using conventional radiographic mammography or magnetic resonance imaging may be warranted. Biopsy of EBT foci generally is not indicated unless first-line noninvasive studies fail to yield a conclusive diagnosis; however, biopsy also may be warranted if initial imaging is suggestive of malignancy arising from EBT.2
Management of EBT generally is conservative, and symptoms often resolve without intervention.4 Symptomatic relief may be achieved through techniques such as application of warm/cold compresses, avoidance of mechanical stimulation, and use of over-the-counter pain medicine. In cases that are persistent, frequently recurrent, or associated with severe symptoms or that cause considerable cosmetic impact, management with surgical excision and/or liposuction may be warranted.7 In our patient, the symptoms were not bothersome enough to warrant surgical intervention, so she was managed conservatively and did not return for follow-up.
- Leung AK. Familial supernumerary nipples. Am J Med Genet. 1988;31:631-635. doi:10.1002/ajmg.1320310318
- Visconti G, Eltahir Y, Van Ginkel RJ, et al. Approach and management of primary ectopic breast carcinoma in the axilla: where are we? a comprehensive historical literature review. J Plast Reconstr Aesthet Surg. 2011;64:E1-E11. doi:10.1016/j.bjps.2010.08.015
- Göttlicher S. Incidence and location of polythelias, polymastias and mammae aberratae. a prospective one year study of 1,660 patients of a gynecologic practice. Article in German. Geburtshilfe Frauenheilkd. 1986;46:697-699. doi:10.1055/s-2008-1035944
- Ghosn SH, Khatri KA, Bhawan J. Bilateral aberrant axillary breast tissue mimicking lipomas: report of a case and review of the literature. J Cutan Pathol. 2007;34(suppl 1):9-13. doi:10.1111/j.1600-0560.2006.00713.x
- Firat D, Idiz O, Isik A, et al. Spontaneous milk fistula from an accessory breast: an extremely rare case. Breast J. 2015;21:554-555. doi:10.1111/tbj.12452
- Lim HS, Kim SJ, Baek JM, et al. Sonographic findings of accessory breast tissue in axilla and related diseases. J Ultrasound Med. 2017;36:1469-1478. doi:10.7863/ultra.16.06056
- Gentile P, Izzo V, Cervelli V. Fibroadenoma in the bilateral accessory axillary breast. Aesthetic Plast Surg. 2010;34:657-659. doi:10.1007/ s00266-010-9505-y
THE DIAGNOSIS: Ectopic (Accessory) Breast Tissue
Ectopic (accessory) breast tissue (EBT) is a phenomenon caused by failed regression of one or more components of the embryonic mammary ridges— paired ectodermal thickenings that eventually develop into definitive breast tissue including the nipples, areolae, and parenchyma. Ectopic breast tissue is more common in women than men and is believed to be sporadic, although an autosomal-dominant inheritance mechanism with incomplete penetrance has been proposed for some cases.1 The reported incidence of EBT varies greatly among racial and ethnic groups but is most common in individuals of Asian descent. The incidence across all types of EBT is estimated at 0.25% to 6% in the general population.2
Observed clinical variations of EBT range from simple polythelia (additional nipple[s] without associated parenchyma) to complete polymastia (organized and differentiated accessory breasts). Some types of EBT are rarer than others: One report of gynecologic cancer screenings in 1660 patients found polymastia and polythelia incidences of 0.12% and 5.48%, respectively.3 Of the symptomatic variations, isolated parenchymal EBT without a nipple or areolar complex is the most common and may manifest clinically as unilateral or bilateral tender, mildly erythematous nodules or masses often located in the axillae. Ectopic breast tissue generally is observed along the milk line, a developmental regional designation corresponding to the embryologic mammary ridge and extending linearly from the anterior axilla to the inguinal fold on both sides of the body; however, there have been rare reports of EBT manifesting in areas outside the milk line, such as the face, neck, back, vulva, and extremities.2,3
Given that the underlying elements of EBT usually are hormone responsive (as with normal breast tissue), the initial symptom onset and subsequent manifestation frequently coincide with pubertal milestones, pregnancy, or lactation. Furthermore, some patients with EBT may experience symptom fluctuations in concordance with monthly menstrual phases. Many cases of EBT are selflimited and resolve within weeks to months after the end of a pregnancy or lactation, but some cases may persist. Continued observation and follow-up are advisable in all patients, as EBT symptoms often recur and the tissue is susceptible to the same disease processes that affect normal breasts, the most concerning of which is malignancy.4 Although the true incidence is limited by available data, primary ectopic breast malignancy has been estimated to account for 0.3% to 3.8% of diagnosed breast malignancies.2 Cases of malignancy arising from EBT often are of higher grade and poorer prognosis, a finding that may be attributable to diagnostic delays caused by oversight or misdiagnosis of EBT rather than inherent differences in the biologic profile of the tumors.2,4 Patients with a documented history of EBT may benefit from having their routine breast cancer screenings expanded to include areas with EBT foci.
Potential misdiagnoses for EBT include subcutaneous lipoma, axillary lymphadenopathy, abscess, hidradenitis suppurativa, or malignancy. Features that are suggestive of EBT include symptom association with hormone fluctuations (eg, menstrual phases), absence of fever, and lactescent rather than purulent drainage. Among reported EBT cases, spontaneous lactation rarely is described and, if present, often is associated with a history of prior trauma (eg, core needle biopsy or local abscess formation).5 This trauma creates an aberrant connection known as a milk fistula between the underlying parenchyma and the skin surface. Interestingly, our patient denied any history of axillary trauma, but she was noted to be lactating from an apparent milk fistula rather than an organized secretory duct system.
Though a patient history and clinical examination may be sufficient to diagnose EBT cases that are more physically apparent and well correlated with hormone fluctuations, many cases require additional diagnostic studies for confirmation. Of the tools available, ultrasonography generally is considered first-line due to its noninvasive nature, low cost, minimal risk, and high diagnostic value.2 Ultrasonography quickly differentiates between abscesses and cystlike processes, which may appear as discrete areas of decreased echogenicity, and breast tissue, which manifests with fibroglandular tissue and lobules of fat.2,6 Additionally, ultrasonography may demonstrate the secretion of milk through ducts or fistulae, if present. Should examination with ultrasonography prove inconclusive, follow-up studies using conventional radiographic mammography or magnetic resonance imaging may be warranted. Biopsy of EBT foci generally is not indicated unless first-line noninvasive studies fail to yield a conclusive diagnosis; however, biopsy also may be warranted if initial imaging is suggestive of malignancy arising from EBT.2
Management of EBT generally is conservative, and symptoms often resolve without intervention.4 Symptomatic relief may be achieved through techniques such as application of warm/cold compresses, avoidance of mechanical stimulation, and use of over-the-counter pain medicine. In cases that are persistent, frequently recurrent, or associated with severe symptoms or that cause considerable cosmetic impact, management with surgical excision and/or liposuction may be warranted.7 In our patient, the symptoms were not bothersome enough to warrant surgical intervention, so she was managed conservatively and did not return for follow-up.
THE DIAGNOSIS: Ectopic (Accessory) Breast Tissue
Ectopic (accessory) breast tissue (EBT) is a phenomenon caused by failed regression of one or more components of the embryonic mammary ridges— paired ectodermal thickenings that eventually develop into definitive breast tissue including the nipples, areolae, and parenchyma. Ectopic breast tissue is more common in women than men and is believed to be sporadic, although an autosomal-dominant inheritance mechanism with incomplete penetrance has been proposed for some cases.1 The reported incidence of EBT varies greatly among racial and ethnic groups but is most common in individuals of Asian descent. The incidence across all types of EBT is estimated at 0.25% to 6% in the general population.2
Observed clinical variations of EBT range from simple polythelia (additional nipple[s] without associated parenchyma) to complete polymastia (organized and differentiated accessory breasts). Some types of EBT are rarer than others: One report of gynecologic cancer screenings in 1660 patients found polymastia and polythelia incidences of 0.12% and 5.48%, respectively.3 Of the symptomatic variations, isolated parenchymal EBT without a nipple or areolar complex is the most common and may manifest clinically as unilateral or bilateral tender, mildly erythematous nodules or masses often located in the axillae. Ectopic breast tissue generally is observed along the milk line, a developmental regional designation corresponding to the embryologic mammary ridge and extending linearly from the anterior axilla to the inguinal fold on both sides of the body; however, there have been rare reports of EBT manifesting in areas outside the milk line, such as the face, neck, back, vulva, and extremities.2,3
Given that the underlying elements of EBT usually are hormone responsive (as with normal breast tissue), the initial symptom onset and subsequent manifestation frequently coincide with pubertal milestones, pregnancy, or lactation. Furthermore, some patients with EBT may experience symptom fluctuations in concordance with monthly menstrual phases. Many cases of EBT are selflimited and resolve within weeks to months after the end of a pregnancy or lactation, but some cases may persist. Continued observation and follow-up are advisable in all patients, as EBT symptoms often recur and the tissue is susceptible to the same disease processes that affect normal breasts, the most concerning of which is malignancy.4 Although the true incidence is limited by available data, primary ectopic breast malignancy has been estimated to account for 0.3% to 3.8% of diagnosed breast malignancies.2 Cases of malignancy arising from EBT often are of higher grade and poorer prognosis, a finding that may be attributable to diagnostic delays caused by oversight or misdiagnosis of EBT rather than inherent differences in the biologic profile of the tumors.2,4 Patients with a documented history of EBT may benefit from having their routine breast cancer screenings expanded to include areas with EBT foci.
Potential misdiagnoses for EBT include subcutaneous lipoma, axillary lymphadenopathy, abscess, hidradenitis suppurativa, or malignancy. Features that are suggestive of EBT include symptom association with hormone fluctuations (eg, menstrual phases), absence of fever, and lactescent rather than purulent drainage. Among reported EBT cases, spontaneous lactation rarely is described and, if present, often is associated with a history of prior trauma (eg, core needle biopsy or local abscess formation).5 This trauma creates an aberrant connection known as a milk fistula between the underlying parenchyma and the skin surface. Interestingly, our patient denied any history of axillary trauma, but she was noted to be lactating from an apparent milk fistula rather than an organized secretory duct system.
Though a patient history and clinical examination may be sufficient to diagnose EBT cases that are more physically apparent and well correlated with hormone fluctuations, many cases require additional diagnostic studies for confirmation. Of the tools available, ultrasonography generally is considered first-line due to its noninvasive nature, low cost, minimal risk, and high diagnostic value.2 Ultrasonography quickly differentiates between abscesses and cystlike processes, which may appear as discrete areas of decreased echogenicity, and breast tissue, which manifests with fibroglandular tissue and lobules of fat.2,6 Additionally, ultrasonography may demonstrate the secretion of milk through ducts or fistulae, if present. Should examination with ultrasonography prove inconclusive, follow-up studies using conventional radiographic mammography or magnetic resonance imaging may be warranted. Biopsy of EBT foci generally is not indicated unless first-line noninvasive studies fail to yield a conclusive diagnosis; however, biopsy also may be warranted if initial imaging is suggestive of malignancy arising from EBT.2
Management of EBT generally is conservative, and symptoms often resolve without intervention.4 Symptomatic relief may be achieved through techniques such as application of warm/cold compresses, avoidance of mechanical stimulation, and use of over-the-counter pain medicine. In cases that are persistent, frequently recurrent, or associated with severe symptoms or that cause considerable cosmetic impact, management with surgical excision and/or liposuction may be warranted.7 In our patient, the symptoms were not bothersome enough to warrant surgical intervention, so she was managed conservatively and did not return for follow-up.
- Leung AK. Familial supernumerary nipples. Am J Med Genet. 1988;31:631-635. doi:10.1002/ajmg.1320310318
- Visconti G, Eltahir Y, Van Ginkel RJ, et al. Approach and management of primary ectopic breast carcinoma in the axilla: where are we? a comprehensive historical literature review. J Plast Reconstr Aesthet Surg. 2011;64:E1-E11. doi:10.1016/j.bjps.2010.08.015
- Göttlicher S. Incidence and location of polythelias, polymastias and mammae aberratae. a prospective one year study of 1,660 patients of a gynecologic practice. Article in German. Geburtshilfe Frauenheilkd. 1986;46:697-699. doi:10.1055/s-2008-1035944
- Ghosn SH, Khatri KA, Bhawan J. Bilateral aberrant axillary breast tissue mimicking lipomas: report of a case and review of the literature. J Cutan Pathol. 2007;34(suppl 1):9-13. doi:10.1111/j.1600-0560.2006.00713.x
- Firat D, Idiz O, Isik A, et al. Spontaneous milk fistula from an accessory breast: an extremely rare case. Breast J. 2015;21:554-555. doi:10.1111/tbj.12452
- Lim HS, Kim SJ, Baek JM, et al. Sonographic findings of accessory breast tissue in axilla and related diseases. J Ultrasound Med. 2017;36:1469-1478. doi:10.7863/ultra.16.06056
- Gentile P, Izzo V, Cervelli V. Fibroadenoma in the bilateral accessory axillary breast. Aesthetic Plast Surg. 2010;34:657-659. doi:10.1007/ s00266-010-9505-y
- Leung AK. Familial supernumerary nipples. Am J Med Genet. 1988;31:631-635. doi:10.1002/ajmg.1320310318
- Visconti G, Eltahir Y, Van Ginkel RJ, et al. Approach and management of primary ectopic breast carcinoma in the axilla: where are we? a comprehensive historical literature review. J Plast Reconstr Aesthet Surg. 2011;64:E1-E11. doi:10.1016/j.bjps.2010.08.015
- Göttlicher S. Incidence and location of polythelias, polymastias and mammae aberratae. a prospective one year study of 1,660 patients of a gynecologic practice. Article in German. Geburtshilfe Frauenheilkd. 1986;46:697-699. doi:10.1055/s-2008-1035944
- Ghosn SH, Khatri KA, Bhawan J. Bilateral aberrant axillary breast tissue mimicking lipomas: report of a case and review of the literature. J Cutan Pathol. 2007;34(suppl 1):9-13. doi:10.1111/j.1600-0560.2006.00713.x
- Firat D, Idiz O, Isik A, et al. Spontaneous milk fistula from an accessory breast: an extremely rare case. Breast J. 2015;21:554-555. doi:10.1111/tbj.12452
- Lim HS, Kim SJ, Baek JM, et al. Sonographic findings of accessory breast tissue in axilla and related diseases. J Ultrasound Med. 2017;36:1469-1478. doi:10.7863/ultra.16.06056
- Gentile P, Izzo V, Cervelli V. Fibroadenoma in the bilateral accessory axillary breast. Aesthetic Plast Surg. 2010;34:657-659. doi:10.1007/ s00266-010-9505-y
A 19-year-old G1P1A0 woman presented to the dermatology clinic for evaluation of bilateral axillary swelling, pain, and spontaneous drainage of approximately 2 weeks’ duration. The patient, who was 2 weeks postpartum, reported that the symptoms were associated with lactation when breastfeeding. She denied any personal or family history of hidradenitis suppurativa or other formally diagnosed dermatologic condition. Physical examination revealed a soft, mildly tender, well-circumscribed, nonfluctuant mobile mass in each axilla. Both lesions had a single central sinus tract with thin lactescent discharge that spontaneously drained and was expressible. A single thin hyperpigmented papule was noted on the anterior aspect of each mass.

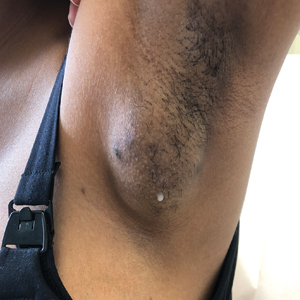
Is It Possible To Treat Patients You Dislike?
This transcript has been edited for clarity.
What do we do if we don’t like patients? We take the Hippocratic Oath as young students in Glasgow. We do that just before our graduation ceremony; we hold our hands up and repeat the Hippocratic Oath: “First, do no harm,” and so on.
I was thinking back over a long career. I’ve been a cancer doctor for 40 years and I quite like saying that.
I can only think genuinely over a couple of times in which I’ve acted reflexively when a patient has done something awful. The couple of times it happened, it was just terrible racist comments to junior doctors who were with me. Extraordinarily dreadful things such as, “I don’t want to be touched by ...” or something of that sort.
Without really thinking about it, you react as a normal citizen and say, “That’s absolutely awful. Apologize immediately or leave the consultation room, and never ever come back again.”
I remember that it happened once in Glasgow and once when I was a young professor in Birmingham, and it’s just an automatic gut reaction. The patient got a fright, and I immediately apologized and groveled around. In that relationship, we hold all the power, don’t we? Rather than being gentle about it, I was genuinely angry because of these ridiculous comments.
Otherwise, I think most of the doctor-patient relationships are predicated on nonromantic love. I think patients want us to love them as one would a son, mother, father, or daughter, because if we do, then we will do better for them and we’ll pull out all the stops. “Placebo” means “I will please.” I think in the vast majority of cases, at least in our National Health Service (NHS), patients come with trust and a sense of wanting to build that relationship. That may be changing, but not for me.
What about putting the boot on the other foot? What if the patients don’t like us rather than vice versa? As part of our accreditation appraisal process, from time to time we have to take patient surveys as to whether the patients felt that, after they had been seen in a consultation, they were treated with dignity, the quality of information given was appropriate, and they were treated with kindness.
It’s an excellent exercise. Without bragging about it, patients objectively, according to these measures, appreciate the service that I give. It’s like getting five-star reviews on Trustpilot, or whatever these things are, that allow you to review car salesmen and so on. I have always had five-star reviews across the board.
That, again, I thought was just a feature of that relationship, of patients wanting to please. These are patients who had been treated, who were in the outpatient department, who were in the midst of battle. Still, the scores are very high. I speak to my colleagues and that’s not uniformly the case. Patients actually do use these feedback forms, I think in a positive rather than negative way, reflecting back on the way that they were treated.
It has caused some of my colleagues to think quite hard about their personal style and approach to patients. That sense of feedback is important.
What about losing trust? If that’s at the heart of everything that we do, then what would be an objective measure of losing trust? Again, in our healthcare system, it has been exceedingly unusual for a patient to request a second opinion. Now, that’s changing. The government is trying to change it. Leaders of the NHS are trying to change it so that patients feel assured that they can seek second opinions.
Again, in all the years I’ve been a cancer doctor, it has been incredibly infrequent that somebody has sought a second opinion after I’ve said something. That may be a measure of trust. Again, I’ve lived through an NHS in which seeking second opinions was something of a rarity.
I’d be really interested to see what you think. In your own sphere of healthcare practice, is it possible for us to look after patients that we don’t like, or should we be honest and say, “I don’t like you. Our relationship has broken down. I want you to be seen by a colleague,” or “I want you to be nursed by somebody else”?
Has that happened? Is that something that you think is common or may become more common? What about when trust breaks down the other way? Can you think of instances in which the relationship, for whatever reason, just didn’t work and the patient had to move on because of that loss of trust and what underpinned it? I’d be really interested to know.
I seek to be informed rather than the other way around. Can we truly look after patients that we don’t like or can we rise above it as Hippocrates might have done?
Thanks for listening, as always. For the time being, over and out.
Dr. Kerr, Professor, Nuffield Department of Clinical Laboratory Science, University of Oxford; Professor of Cancer Medicine, Oxford Cancer Centre, Oxford, United Kingdom, disclosed ties with Celleron Therapeutics, Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Bayer HealthCare Pharmaceuticals, Genomic Health, Merck Serono, and Roche.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
What do we do if we don’t like patients? We take the Hippocratic Oath as young students in Glasgow. We do that just before our graduation ceremony; we hold our hands up and repeat the Hippocratic Oath: “First, do no harm,” and so on.
I was thinking back over a long career. I’ve been a cancer doctor for 40 years and I quite like saying that.
I can only think genuinely over a couple of times in which I’ve acted reflexively when a patient has done something awful. The couple of times it happened, it was just terrible racist comments to junior doctors who were with me. Extraordinarily dreadful things such as, “I don’t want to be touched by ...” or something of that sort.
Without really thinking about it, you react as a normal citizen and say, “That’s absolutely awful. Apologize immediately or leave the consultation room, and never ever come back again.”
I remember that it happened once in Glasgow and once when I was a young professor in Birmingham, and it’s just an automatic gut reaction. The patient got a fright, and I immediately apologized and groveled around. In that relationship, we hold all the power, don’t we? Rather than being gentle about it, I was genuinely angry because of these ridiculous comments.
Otherwise, I think most of the doctor-patient relationships are predicated on nonromantic love. I think patients want us to love them as one would a son, mother, father, or daughter, because if we do, then we will do better for them and we’ll pull out all the stops. “Placebo” means “I will please.” I think in the vast majority of cases, at least in our National Health Service (NHS), patients come with trust and a sense of wanting to build that relationship. That may be changing, but not for me.
What about putting the boot on the other foot? What if the patients don’t like us rather than vice versa? As part of our accreditation appraisal process, from time to time we have to take patient surveys as to whether the patients felt that, after they had been seen in a consultation, they were treated with dignity, the quality of information given was appropriate, and they were treated with kindness.
It’s an excellent exercise. Without bragging about it, patients objectively, according to these measures, appreciate the service that I give. It’s like getting five-star reviews on Trustpilot, or whatever these things are, that allow you to review car salesmen and so on. I have always had five-star reviews across the board.
That, again, I thought was just a feature of that relationship, of patients wanting to please. These are patients who had been treated, who were in the outpatient department, who were in the midst of battle. Still, the scores are very high. I speak to my colleagues and that’s not uniformly the case. Patients actually do use these feedback forms, I think in a positive rather than negative way, reflecting back on the way that they were treated.
It has caused some of my colleagues to think quite hard about their personal style and approach to patients. That sense of feedback is important.
What about losing trust? If that’s at the heart of everything that we do, then what would be an objective measure of losing trust? Again, in our healthcare system, it has been exceedingly unusual for a patient to request a second opinion. Now, that’s changing. The government is trying to change it. Leaders of the NHS are trying to change it so that patients feel assured that they can seek second opinions.
Again, in all the years I’ve been a cancer doctor, it has been incredibly infrequent that somebody has sought a second opinion after I’ve said something. That may be a measure of trust. Again, I’ve lived through an NHS in which seeking second opinions was something of a rarity.
I’d be really interested to see what you think. In your own sphere of healthcare practice, is it possible for us to look after patients that we don’t like, or should we be honest and say, “I don’t like you. Our relationship has broken down. I want you to be seen by a colleague,” or “I want you to be nursed by somebody else”?
Has that happened? Is that something that you think is common or may become more common? What about when trust breaks down the other way? Can you think of instances in which the relationship, for whatever reason, just didn’t work and the patient had to move on because of that loss of trust and what underpinned it? I’d be really interested to know.
I seek to be informed rather than the other way around. Can we truly look after patients that we don’t like or can we rise above it as Hippocrates might have done?
Thanks for listening, as always. For the time being, over and out.
Dr. Kerr, Professor, Nuffield Department of Clinical Laboratory Science, University of Oxford; Professor of Cancer Medicine, Oxford Cancer Centre, Oxford, United Kingdom, disclosed ties with Celleron Therapeutics, Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Bayer HealthCare Pharmaceuticals, Genomic Health, Merck Serono, and Roche.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
What do we do if we don’t like patients? We take the Hippocratic Oath as young students in Glasgow. We do that just before our graduation ceremony; we hold our hands up and repeat the Hippocratic Oath: “First, do no harm,” and so on.
I was thinking back over a long career. I’ve been a cancer doctor for 40 years and I quite like saying that.
I can only think genuinely over a couple of times in which I’ve acted reflexively when a patient has done something awful. The couple of times it happened, it was just terrible racist comments to junior doctors who were with me. Extraordinarily dreadful things such as, “I don’t want to be touched by ...” or something of that sort.
Without really thinking about it, you react as a normal citizen and say, “That’s absolutely awful. Apologize immediately or leave the consultation room, and never ever come back again.”
I remember that it happened once in Glasgow and once when I was a young professor in Birmingham, and it’s just an automatic gut reaction. The patient got a fright, and I immediately apologized and groveled around. In that relationship, we hold all the power, don’t we? Rather than being gentle about it, I was genuinely angry because of these ridiculous comments.
Otherwise, I think most of the doctor-patient relationships are predicated on nonromantic love. I think patients want us to love them as one would a son, mother, father, or daughter, because if we do, then we will do better for them and we’ll pull out all the stops. “Placebo” means “I will please.” I think in the vast majority of cases, at least in our National Health Service (NHS), patients come with trust and a sense of wanting to build that relationship. That may be changing, but not for me.
What about putting the boot on the other foot? What if the patients don’t like us rather than vice versa? As part of our accreditation appraisal process, from time to time we have to take patient surveys as to whether the patients felt that, after they had been seen in a consultation, they were treated with dignity, the quality of information given was appropriate, and they were treated with kindness.
It’s an excellent exercise. Without bragging about it, patients objectively, according to these measures, appreciate the service that I give. It’s like getting five-star reviews on Trustpilot, or whatever these things are, that allow you to review car salesmen and so on. I have always had five-star reviews across the board.
That, again, I thought was just a feature of that relationship, of patients wanting to please. These are patients who had been treated, who were in the outpatient department, who were in the midst of battle. Still, the scores are very high. I speak to my colleagues and that’s not uniformly the case. Patients actually do use these feedback forms, I think in a positive rather than negative way, reflecting back on the way that they were treated.
It has caused some of my colleagues to think quite hard about their personal style and approach to patients. That sense of feedback is important.
What about losing trust? If that’s at the heart of everything that we do, then what would be an objective measure of losing trust? Again, in our healthcare system, it has been exceedingly unusual for a patient to request a second opinion. Now, that’s changing. The government is trying to change it. Leaders of the NHS are trying to change it so that patients feel assured that they can seek second opinions.
Again, in all the years I’ve been a cancer doctor, it has been incredibly infrequent that somebody has sought a second opinion after I’ve said something. That may be a measure of trust. Again, I’ve lived through an NHS in which seeking second opinions was something of a rarity.
I’d be really interested to see what you think. In your own sphere of healthcare practice, is it possible for us to look after patients that we don’t like, or should we be honest and say, “I don’t like you. Our relationship has broken down. I want you to be seen by a colleague,” or “I want you to be nursed by somebody else”?
Has that happened? Is that something that you think is common or may become more common? What about when trust breaks down the other way? Can you think of instances in which the relationship, for whatever reason, just didn’t work and the patient had to move on because of that loss of trust and what underpinned it? I’d be really interested to know.
I seek to be informed rather than the other way around. Can we truly look after patients that we don’t like or can we rise above it as Hippocrates might have done?
Thanks for listening, as always. For the time being, over and out.
Dr. Kerr, Professor, Nuffield Department of Clinical Laboratory Science, University of Oxford; Professor of Cancer Medicine, Oxford Cancer Centre, Oxford, United Kingdom, disclosed ties with Celleron Therapeutics, Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Bayer HealthCare Pharmaceuticals, Genomic Health, Merck Serono, and Roche.
A version of this article appeared on Medscape.com.
New Cosmeceutical as Effective as Cysteamine for Facial Melasma
A presented at the European Academy of Dermatology and Venereology (EADV) 2024 Congress.
“Melasyl is a new potent melanogenesis inhibitor that exhibits a unique mode of action while preserving melanocyte integrity,” Mukta Sachdev, MD, head of the Department of Dermatology at Manipal Hospital in Bangalore, India, said at a late-breaking news session.
Both the serum and the cysteamine cream lightened participants’ skin to a similar extent, according to the modified Melasma Area and Severity Index (mMASI), with respective reductions of 4.19 and 3.81 points over a period of 4 months from baseline values of 11.15 and 10.93. 
The mMASI score ranges from 0 to 24, with the lowest score representing the least and the highest score the most severe hyperpigmentation of the skin.
But the serum performed better than the cream by another measure. Judged by investigators blinded to which preparation study participants had been using, there was a significantly higher reduction in the Investigator Global Assessment (IGA) score from baseline among those treated with the serum than among those treated with the cream (−51.85% vs −39.06%; P = .0163).
Moreover, after 4 months of treatment, there were significantly more participants with clear or almost clear skin with the serum than with the cream (17.46% vs 7.81%; P = .0163), Sachdev reported.
Other skin parameters relative to melasma, such as the brightness of skin tone and evenness of the improvement, improved more in the participants using the serum vs cream, she said.
With “no side effects, no local skin reactions,” Sachdev said, “quality of life improved significantly and similarly, and almost all subjects in both groups were very satisfied with their treatment options.”
Active Ingredients
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra, in Portugal, who co-chaired the late-breaking news session, commented: “It’s really nice to have new products to treat such a devastating disease.”
Session co-chair, Lidia Rudnicka, MD, head of the Department of Dermatology, Medical University of Warsaw, in Poland, and president of the Polish Dermatological Society, wanted to know more about the active ingredients of the serum and the study’s design.
Sachdev replied that the serum also contains other ingredients that provide “antioxidant protection” and moisturization. These include retinyl palmitate, which works on the dermal-epidermal junction, and hyaluronic acid, as well as “soothing agents,” such as the medicinal herb Centella asiatica, she said.
Study Design
Conducted at a single center in India, the study involved 127 adults aged 20-50 years with melasma. For inclusion, the participants had to have facial epidermal or mixed melasma (phototypes III-V) for more than 1 year; those with dermal melasma were excluded.
Participants were randomly allocated to receive either the serum, which was applied topically to the areas of interest twice a day in the morning and then at bedtime (n = 63), or cysteamine cream (n = 64), which was applied once a day in addition to a neutral moisturizer. Treatment was for 4 months, with an on-site visit every month.
All participants were supplied with the same sunscreen/ultraviolet protector applied twice a day (once in the morning and again at midday) and a neutral hydrating cleanser that was used in the morning and evening.
Practical Implications
Over 4 months, both products showed significant improvement in melasma without reaching a plateau, Sachdev reported, with the serum demonstrating superior efficacy and tolerability, as judged by the investigators.
The study suggests that the serum is a promising non-hydroquinone treatment for melasma, she said. Hydroquinone-containing topical preparations are used to depigment the skin, but their long-term use can be limited for safety reasons.
“When products like this demonstrate improvement, it is something for the dermatologist to think about because we now have newer ingredients, which are safer and well tolerated,” she continued, noting that there appeared to be no risk for exogenous ochronosis, which can occur with long-term application of hydroquinone.
“So, I think the armamentarium of non-hydroquinone products for the treatment of melasma is rapidly expanding, and there are studies now with clinically proven efficacy,” Sachdev concluded.
The study was supported by L’Oréal France La Roche-Posay, which launched Melasyl in March 2024. Sachdev reported receipt of research support and honoraria from the company. Gonçalo and Rudnicka were not involved in the study and had no relevant conflicts of interest to report.
A version of this article appeared on Medscape.com.
A presented at the European Academy of Dermatology and Venereology (EADV) 2024 Congress.
“Melasyl is a new potent melanogenesis inhibitor that exhibits a unique mode of action while preserving melanocyte integrity,” Mukta Sachdev, MD, head of the Department of Dermatology at Manipal Hospital in Bangalore, India, said at a late-breaking news session.
Both the serum and the cysteamine cream lightened participants’ skin to a similar extent, according to the modified Melasma Area and Severity Index (mMASI), with respective reductions of 4.19 and 3.81 points over a period of 4 months from baseline values of 11.15 and 10.93.
The mMASI score ranges from 0 to 24, with the lowest score representing the least and the highest score the most severe hyperpigmentation of the skin.
But the serum performed better than the cream by another measure. Judged by investigators blinded to which preparation study participants had been using, there was a significantly higher reduction in the Investigator Global Assessment (IGA) score from baseline among those treated with the serum than among those treated with the cream (−51.85% vs −39.06%; P = .0163).
Moreover, after 4 months of treatment, there were significantly more participants with clear or almost clear skin with the serum than with the cream (17.46% vs 7.81%; P = .0163), Sachdev reported.
Other skin parameters relative to melasma, such as the brightness of skin tone and evenness of the improvement, improved more in the participants using the serum vs cream, she said.
With “no side effects, no local skin reactions,” Sachdev said, “quality of life improved significantly and similarly, and almost all subjects in both groups were very satisfied with their treatment options.”
Active Ingredients
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra, in Portugal, who co-chaired the late-breaking news session, commented: “It’s really nice to have new products to treat such a devastating disease.”
Session co-chair, Lidia Rudnicka, MD, head of the Department of Dermatology, Medical University of Warsaw, in Poland, and president of the Polish Dermatological Society, wanted to know more about the active ingredients of the serum and the study’s design.
Sachdev replied that the serum also contains other ingredients that provide “antioxidant protection” and moisturization. These include retinyl palmitate, which works on the dermal-epidermal junction, and hyaluronic acid, as well as “soothing agents,” such as the medicinal herb Centella asiatica, she said.
Study Design
Conducted at a single center in India, the study involved 127 adults aged 20-50 years with melasma. For inclusion, the participants had to have facial epidermal or mixed melasma (phototypes III-V) for more than 1 year; those with dermal melasma were excluded.
Participants were randomly allocated to receive either the serum, which was applied topically to the areas of interest twice a day in the morning and then at bedtime (n = 63), or cysteamine cream (n = 64), which was applied once a day in addition to a neutral moisturizer. Treatment was for 4 months, with an on-site visit every month.
All participants were supplied with the same sunscreen/ultraviolet protector applied twice a day (once in the morning and again at midday) and a neutral hydrating cleanser that was used in the morning and evening.
Practical Implications
Over 4 months, both products showed significant improvement in melasma without reaching a plateau, Sachdev reported, with the serum demonstrating superior efficacy and tolerability, as judged by the investigators.
The study suggests that the serum is a promising non-hydroquinone treatment for melasma, she said. Hydroquinone-containing topical preparations are used to depigment the skin, but their long-term use can be limited for safety reasons.
“When products like this demonstrate improvement, it is something for the dermatologist to think about because we now have newer ingredients, which are safer and well tolerated,” she continued, noting that there appeared to be no risk for exogenous ochronosis, which can occur with long-term application of hydroquinone.
“So, I think the armamentarium of non-hydroquinone products for the treatment of melasma is rapidly expanding, and there are studies now with clinically proven efficacy,” Sachdev concluded.
The study was supported by L’Oréal France La Roche-Posay, which launched Melasyl in March 2024. Sachdev reported receipt of research support and honoraria from the company. Gonçalo and Rudnicka were not involved in the study and had no relevant conflicts of interest to report.
A version of this article appeared on Medscape.com.
A presented at the European Academy of Dermatology and Venereology (EADV) 2024 Congress.
“Melasyl is a new potent melanogenesis inhibitor that exhibits a unique mode of action while preserving melanocyte integrity,” Mukta Sachdev, MD, head of the Department of Dermatology at Manipal Hospital in Bangalore, India, said at a late-breaking news session.
Both the serum and the cysteamine cream lightened participants’ skin to a similar extent, according to the modified Melasma Area and Severity Index (mMASI), with respective reductions of 4.19 and 3.81 points over a period of 4 months from baseline values of 11.15 and 10.93.
The mMASI score ranges from 0 to 24, with the lowest score representing the least and the highest score the most severe hyperpigmentation of the skin.
But the serum performed better than the cream by another measure. Judged by investigators blinded to which preparation study participants had been using, there was a significantly higher reduction in the Investigator Global Assessment (IGA) score from baseline among those treated with the serum than among those treated with the cream (−51.85% vs −39.06%; P = .0163).
Moreover, after 4 months of treatment, there were significantly more participants with clear or almost clear skin with the serum than with the cream (17.46% vs 7.81%; P = .0163), Sachdev reported.
Other skin parameters relative to melasma, such as the brightness of skin tone and evenness of the improvement, improved more in the participants using the serum vs cream, she said.
With “no side effects, no local skin reactions,” Sachdev said, “quality of life improved significantly and similarly, and almost all subjects in both groups were very satisfied with their treatment options.”
Active Ingredients
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra, in Portugal, who co-chaired the late-breaking news session, commented: “It’s really nice to have new products to treat such a devastating disease.”
Session co-chair, Lidia Rudnicka, MD, head of the Department of Dermatology, Medical University of Warsaw, in Poland, and president of the Polish Dermatological Society, wanted to know more about the active ingredients of the serum and the study’s design.
Sachdev replied that the serum also contains other ingredients that provide “antioxidant protection” and moisturization. These include retinyl palmitate, which works on the dermal-epidermal junction, and hyaluronic acid, as well as “soothing agents,” such as the medicinal herb Centella asiatica, she said.
Study Design
Conducted at a single center in India, the study involved 127 adults aged 20-50 years with melasma. For inclusion, the participants had to have facial epidermal or mixed melasma (phototypes III-V) for more than 1 year; those with dermal melasma were excluded.
Participants were randomly allocated to receive either the serum, which was applied topically to the areas of interest twice a day in the morning and then at bedtime (n = 63), or cysteamine cream (n = 64), which was applied once a day in addition to a neutral moisturizer. Treatment was for 4 months, with an on-site visit every month.
All participants were supplied with the same sunscreen/ultraviolet protector applied twice a day (once in the morning and again at midday) and a neutral hydrating cleanser that was used in the morning and evening.
Practical Implications
Over 4 months, both products showed significant improvement in melasma without reaching a plateau, Sachdev reported, with the serum demonstrating superior efficacy and tolerability, as judged by the investigators.
The study suggests that the serum is a promising non-hydroquinone treatment for melasma, she said. Hydroquinone-containing topical preparations are used to depigment the skin, but their long-term use can be limited for safety reasons.
“When products like this demonstrate improvement, it is something for the dermatologist to think about because we now have newer ingredients, which are safer and well tolerated,” she continued, noting that there appeared to be no risk for exogenous ochronosis, which can occur with long-term application of hydroquinone.
“So, I think the armamentarium of non-hydroquinone products for the treatment of melasma is rapidly expanding, and there are studies now with clinically proven efficacy,” Sachdev concluded.
The study was supported by L’Oréal France La Roche-Posay, which launched Melasyl in March 2024. Sachdev reported receipt of research support and honoraria from the company. Gonçalo and Rudnicka were not involved in the study and had no relevant conflicts of interest to report.
A version of this article appeared on Medscape.com.
FROM EADV 2024
Risk Assessment Tool Can Help Predict Fractures in Cancer
TOPLINE:
METHODOLOGY:
- Cancer-specific guidelines recommend using FRAX to assess fracture risk, but its applicability in patients with cancer remains unclear.
- This retrospective cohort study included 9877 patients with cancer (mean age, 67.1 years) and 45,875 matched control individuals without cancer (mean age, 66.2 years). All participants had dual-energy x-ray absorptiometry (DXA) scans.
- Researchers collected data on bone mineral density and fractures. The 10-year probabilities of major osteoporotic fractures and hip fractures were calculated using FRAX, and the observed 10-year probabilities of these fractures were compared with FRAX-derived probabilities.
- Compared with individuals without cancer, patients with cancer had a shorter mean follow-up duration (8.5 vs 7.6 years), a slightly higher mean body mass index, and a higher percentage of parental hip fractures (7.0% vs 8.2%); additionally, patients with cancer were more likely to have secondary causes of osteoporosis (10% vs 38.4%) and less likely to receive osteoporosis medication (9.9% vs 4.2%).
TAKEAWAY:
- Compared with individuals without cancer, patients with cancer had a significantly higher incidence rate of major fractures (12.9 vs 14.5 per 1000 person-years) and hip fractures (3.5 vs 4.2 per 1000 person-years).
- FRAX with bone mineral density exhibited excellent calibration for predicting major osteoporotic fractures (slope, 1.03) and hip fractures (0.97) in patients with cancer, regardless of the site of cancer diagnosis. FRAX without bone mineral density, however, underestimated the risk for both major (0.87) and hip fractures (0.72).
- In patients with cancer, FRAX with bone mineral density findings were associated with incident major osteoporotic fractures (hazard ratio [HR] per SD, 1.84) and hip fractures (HR per SD, 3.61).
- When models were adjusted for FRAX with bone mineral density, patients with cancer had an increased risk for both major osteoporotic fractures (HR, 1.17) and hip fractures (HR, 1.30). No difference was found in the risk for fracture between patients with and individuals without cancer when the models were adjusted for FRAX without bone mineral density, even when considering osteoporosis medication use.
IN PRACTICE:
“This retrospective cohort study demonstrates that individuals with cancer are at higher risk of fracture than individuals without cancer and that FRAX, particularly with BMD [bone mineral density], may accurately predict fracture risk in this population. These results, along with the known mortality risk of osteoporotic fractures among cancer survivors, further emphasize the clinical importance of closing the current osteoporosis care gap among cancer survivors,” the authors wrote.
SOURCE:
This study, led by Carrie Ye, MD, MPH, University of Alberta, Edmonton, Alberta, Canada, was published online in JAMA Oncology.
LIMITATIONS:
This study cohort included a selected group of cancer survivors who were referred for DXA scans and may not represent the general cancer population. The cohort consisted predominantly of women, limiting the generalizability to men with cancer. Given the heterogeneity of the population, the findings may not be applicable to all cancer subgroups. Information on cancer stage or the presence of bone metastases at the time of fracture risk assessment was lacking, which could have affected the findings.
DISCLOSURES:
This study was funded by the CancerCare Manitoba Foundation. Three authors reported having ties with various sources, including two who received grants from various organizations.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Cancer-specific guidelines recommend using FRAX to assess fracture risk, but its applicability in patients with cancer remains unclear.
- This retrospective cohort study included 9877 patients with cancer (mean age, 67.1 years) and 45,875 matched control individuals without cancer (mean age, 66.2 years). All participants had dual-energy x-ray absorptiometry (DXA) scans.
- Researchers collected data on bone mineral density and fractures. The 10-year probabilities of major osteoporotic fractures and hip fractures were calculated using FRAX, and the observed 10-year probabilities of these fractures were compared with FRAX-derived probabilities.
- Compared with individuals without cancer, patients with cancer had a shorter mean follow-up duration (8.5 vs 7.6 years), a slightly higher mean body mass index, and a higher percentage of parental hip fractures (7.0% vs 8.2%); additionally, patients with cancer were more likely to have secondary causes of osteoporosis (10% vs 38.4%) and less likely to receive osteoporosis medication (9.9% vs 4.2%).
TAKEAWAY:
- Compared with individuals without cancer, patients with cancer had a significantly higher incidence rate of major fractures (12.9 vs 14.5 per 1000 person-years) and hip fractures (3.5 vs 4.2 per 1000 person-years).
- FRAX with bone mineral density exhibited excellent calibration for predicting major osteoporotic fractures (slope, 1.03) and hip fractures (0.97) in patients with cancer, regardless of the site of cancer diagnosis. FRAX without bone mineral density, however, underestimated the risk for both major (0.87) and hip fractures (0.72).
- In patients with cancer, FRAX with bone mineral density findings were associated with incident major osteoporotic fractures (hazard ratio [HR] per SD, 1.84) and hip fractures (HR per SD, 3.61).
- When models were adjusted for FRAX with bone mineral density, patients with cancer had an increased risk for both major osteoporotic fractures (HR, 1.17) and hip fractures (HR, 1.30). No difference was found in the risk for fracture between patients with and individuals without cancer when the models were adjusted for FRAX without bone mineral density, even when considering osteoporosis medication use.
IN PRACTICE:
“This retrospective cohort study demonstrates that individuals with cancer are at higher risk of fracture than individuals without cancer and that FRAX, particularly with BMD [bone mineral density], may accurately predict fracture risk in this population. These results, along with the known mortality risk of osteoporotic fractures among cancer survivors, further emphasize the clinical importance of closing the current osteoporosis care gap among cancer survivors,” the authors wrote.
SOURCE:
This study, led by Carrie Ye, MD, MPH, University of Alberta, Edmonton, Alberta, Canada, was published online in JAMA Oncology.
LIMITATIONS:
This study cohort included a selected group of cancer survivors who were referred for DXA scans and may not represent the general cancer population. The cohort consisted predominantly of women, limiting the generalizability to men with cancer. Given the heterogeneity of the population, the findings may not be applicable to all cancer subgroups. Information on cancer stage or the presence of bone metastases at the time of fracture risk assessment was lacking, which could have affected the findings.
DISCLOSURES:
This study was funded by the CancerCare Manitoba Foundation. Three authors reported having ties with various sources, including two who received grants from various organizations.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Cancer-specific guidelines recommend using FRAX to assess fracture risk, but its applicability in patients with cancer remains unclear.
- This retrospective cohort study included 9877 patients with cancer (mean age, 67.1 years) and 45,875 matched control individuals without cancer (mean age, 66.2 years). All participants had dual-energy x-ray absorptiometry (DXA) scans.
- Researchers collected data on bone mineral density and fractures. The 10-year probabilities of major osteoporotic fractures and hip fractures were calculated using FRAX, and the observed 10-year probabilities of these fractures were compared with FRAX-derived probabilities.
- Compared with individuals without cancer, patients with cancer had a shorter mean follow-up duration (8.5 vs 7.6 years), a slightly higher mean body mass index, and a higher percentage of parental hip fractures (7.0% vs 8.2%); additionally, patients with cancer were more likely to have secondary causes of osteoporosis (10% vs 38.4%) and less likely to receive osteoporosis medication (9.9% vs 4.2%).
TAKEAWAY:
- Compared with individuals without cancer, patients with cancer had a significantly higher incidence rate of major fractures (12.9 vs 14.5 per 1000 person-years) and hip fractures (3.5 vs 4.2 per 1000 person-years).
- FRAX with bone mineral density exhibited excellent calibration for predicting major osteoporotic fractures (slope, 1.03) and hip fractures (0.97) in patients with cancer, regardless of the site of cancer diagnosis. FRAX without bone mineral density, however, underestimated the risk for both major (0.87) and hip fractures (0.72).
- In patients with cancer, FRAX with bone mineral density findings were associated with incident major osteoporotic fractures (hazard ratio [HR] per SD, 1.84) and hip fractures (HR per SD, 3.61).
- When models were adjusted for FRAX with bone mineral density, patients with cancer had an increased risk for both major osteoporotic fractures (HR, 1.17) and hip fractures (HR, 1.30). No difference was found in the risk for fracture between patients with and individuals without cancer when the models were adjusted for FRAX without bone mineral density, even when considering osteoporosis medication use.
IN PRACTICE:
“This retrospective cohort study demonstrates that individuals with cancer are at higher risk of fracture than individuals without cancer and that FRAX, particularly with BMD [bone mineral density], may accurately predict fracture risk in this population. These results, along with the known mortality risk of osteoporotic fractures among cancer survivors, further emphasize the clinical importance of closing the current osteoporosis care gap among cancer survivors,” the authors wrote.
SOURCE:
This study, led by Carrie Ye, MD, MPH, University of Alberta, Edmonton, Alberta, Canada, was published online in JAMA Oncology.
LIMITATIONS:
This study cohort included a selected group of cancer survivors who were referred for DXA scans and may not represent the general cancer population. The cohort consisted predominantly of women, limiting the generalizability to men with cancer. Given the heterogeneity of the population, the findings may not be applicable to all cancer subgroups. Information on cancer stage or the presence of bone metastases at the time of fracture risk assessment was lacking, which could have affected the findings.
DISCLOSURES:
This study was funded by the CancerCare Manitoba Foundation. Three authors reported having ties with various sources, including two who received grants from various organizations.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.