User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Atopic Dermatitis: Upadacitinib Effectiveness Maintained Through 76 weeks Among Adolescents
TOPLINE:
across three phase 3 trials.
METHODOLOGY:
- Researchers conducted three double-blind, placebo-controlled phase 3 randomized clinical trials (Measure Up 1, Measure Up 2, and AD Up) involving 542 adolescents aged 12-17 years with moderate to severe AD.
- Participants were randomized to receive the oral Janus kinase inhibitor upadacitinib (15 mg or 30 mg once daily) or placebo, with or without topical corticosteroids, for 16 weeks, followed by rerandomization of patients in the placebo group to upadacitinib for up to 76 weeks.
- Study endpoints were at least a 75%, 90%, or 100% reduction in the Eczema Area and Severity Index (EASI-75, EASI-90, and EASI-100, respectively), Validated Investigator Global Assessment for AD (vIGA-AD) score of 0 or 1, and a ≥ 4-point improvement in the Worst Pruritus Numerical Rating Scale (WP-NRS).
- Adverse events were monitored, including serious infections, herpes zoster, and creatine kinase elevation.
TAKEAWAY:
- Among those who continued treatment on upadacitinib, 15 mg and 30 mg, EASI-75 response rates were maintained or improved through week 76 in all three studies. Patients who switched from placebo to upadacitinib also experienced improvements in EASI-75 through week 76.
- The proportion of patients who achieved EASI-90 and EASI-100 responses increased, and in general, were maintained from week 16 through week 76 in all three studies; the proportion was numerically higher among patients on 30 mg for all three studies.
- The proportion of adolescents achieving vIGA-AD score of 0 or 1 and WP-NRS improvement of ≥ 4 points was sustained or improved through 76 weeks.
- Serious infections were reported in five patients or fewer in each treatment group for all three studies. All opportunistic infections were eczema herpeticum; most cases were not serious, or were mild or moderate, and in general, did not require stopping treatment.
IN PRACTICE:
“These results through 76 weeks demonstrated that upadacitinib, with a favorable benefit-risk profile, was an effective long-term treatment option for adolescents with moderate to severe AD,” the authors wrote.
SOURCE:
The study was led by Amy S. Paller, MD, professor and chair of dermatology, Northwestern University, Chicago, and was published online on October 23 in JAMA Dermatology.
LIMITATIONS:
The study limitations included a small sample size, and the findings did not extend to patients under 12 years or those weighing < 40 kg.
DISCLOSURES:
This study was supported by AbbVie. Paller received grants and personal fees from pharmaceutical companies including AbbVie during the conduct of the study. Several authors reported financial ties with various sources, including AbbVie.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
across three phase 3 trials.
METHODOLOGY:
- Researchers conducted three double-blind, placebo-controlled phase 3 randomized clinical trials (Measure Up 1, Measure Up 2, and AD Up) involving 542 adolescents aged 12-17 years with moderate to severe AD.
- Participants were randomized to receive the oral Janus kinase inhibitor upadacitinib (15 mg or 30 mg once daily) or placebo, with or without topical corticosteroids, for 16 weeks, followed by rerandomization of patients in the placebo group to upadacitinib for up to 76 weeks.
- Study endpoints were at least a 75%, 90%, or 100% reduction in the Eczema Area and Severity Index (EASI-75, EASI-90, and EASI-100, respectively), Validated Investigator Global Assessment for AD (vIGA-AD) score of 0 or 1, and a ≥ 4-point improvement in the Worst Pruritus Numerical Rating Scale (WP-NRS).
- Adverse events were monitored, including serious infections, herpes zoster, and creatine kinase elevation.
TAKEAWAY:
- Among those who continued treatment on upadacitinib, 15 mg and 30 mg, EASI-75 response rates were maintained or improved through week 76 in all three studies. Patients who switched from placebo to upadacitinib also experienced improvements in EASI-75 through week 76.
- The proportion of patients who achieved EASI-90 and EASI-100 responses increased, and in general, were maintained from week 16 through week 76 in all three studies; the proportion was numerically higher among patients on 30 mg for all three studies.
- The proportion of adolescents achieving vIGA-AD score of 0 or 1 and WP-NRS improvement of ≥ 4 points was sustained or improved through 76 weeks.
- Serious infections were reported in five patients or fewer in each treatment group for all three studies. All opportunistic infections were eczema herpeticum; most cases were not serious, or were mild or moderate, and in general, did not require stopping treatment.
IN PRACTICE:
“These results through 76 weeks demonstrated that upadacitinib, with a favorable benefit-risk profile, was an effective long-term treatment option for adolescents with moderate to severe AD,” the authors wrote.
SOURCE:
The study was led by Amy S. Paller, MD, professor and chair of dermatology, Northwestern University, Chicago, and was published online on October 23 in JAMA Dermatology.
LIMITATIONS:
The study limitations included a small sample size, and the findings did not extend to patients under 12 years or those weighing < 40 kg.
DISCLOSURES:
This study was supported by AbbVie. Paller received grants and personal fees from pharmaceutical companies including AbbVie during the conduct of the study. Several authors reported financial ties with various sources, including AbbVie.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
across three phase 3 trials.
METHODOLOGY:
- Researchers conducted three double-blind, placebo-controlled phase 3 randomized clinical trials (Measure Up 1, Measure Up 2, and AD Up) involving 542 adolescents aged 12-17 years with moderate to severe AD.
- Participants were randomized to receive the oral Janus kinase inhibitor upadacitinib (15 mg or 30 mg once daily) or placebo, with or without topical corticosteroids, for 16 weeks, followed by rerandomization of patients in the placebo group to upadacitinib for up to 76 weeks.
- Study endpoints were at least a 75%, 90%, or 100% reduction in the Eczema Area and Severity Index (EASI-75, EASI-90, and EASI-100, respectively), Validated Investigator Global Assessment for AD (vIGA-AD) score of 0 or 1, and a ≥ 4-point improvement in the Worst Pruritus Numerical Rating Scale (WP-NRS).
- Adverse events were monitored, including serious infections, herpes zoster, and creatine kinase elevation.
TAKEAWAY:
- Among those who continued treatment on upadacitinib, 15 mg and 30 mg, EASI-75 response rates were maintained or improved through week 76 in all three studies. Patients who switched from placebo to upadacitinib also experienced improvements in EASI-75 through week 76.
- The proportion of patients who achieved EASI-90 and EASI-100 responses increased, and in general, were maintained from week 16 through week 76 in all three studies; the proportion was numerically higher among patients on 30 mg for all three studies.
- The proportion of adolescents achieving vIGA-AD score of 0 or 1 and WP-NRS improvement of ≥ 4 points was sustained or improved through 76 weeks.
- Serious infections were reported in five patients or fewer in each treatment group for all three studies. All opportunistic infections were eczema herpeticum; most cases were not serious, or were mild or moderate, and in general, did not require stopping treatment.
IN PRACTICE:
“These results through 76 weeks demonstrated that upadacitinib, with a favorable benefit-risk profile, was an effective long-term treatment option for adolescents with moderate to severe AD,” the authors wrote.
SOURCE:
The study was led by Amy S. Paller, MD, professor and chair of dermatology, Northwestern University, Chicago, and was published online on October 23 in JAMA Dermatology.
LIMITATIONS:
The study limitations included a small sample size, and the findings did not extend to patients under 12 years or those weighing < 40 kg.
DISCLOSURES:
This study was supported by AbbVie. Paller received grants and personal fees from pharmaceutical companies including AbbVie during the conduct of the study. Several authors reported financial ties with various sources, including AbbVie.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Study Compares Punch Excision vs. Core Excision for Recalcitrant Keloids
according to the results of a small retrospective study.
The method “offers similar efficacy, faster healing, and fewer complications,” one of the study authors, Jinwoong Jung, MD, said in an interview following the annual meeting of the American Society for Dermatologic Surgery, where he presented the study results during an oral abstract session.
For the study, Jung, a dermatologist at Yonsei University College of Medicine, Seoul, South Korea, and colleagues retrospectively analyzed 22 patients with recalcitrant keloids treated with cryotherapy immediately following either PE or CE between May 2019 and March 2024. They used the Vancouver Scar Scale (VSS) to assess treatment efficacy.
Of the 22 patients, 16 underwent treatment with CE and 6 underwent treatment with PE. Pretreatment VSS scores showed no significant differences between the groups (P = .535). The CE group had a reduction in the VSS score from 8.13 to 4.00, while the PE group had a reduction from 7.83 to 3.67, but these declines did not differ significantly (P = .737). The PE group exhibited a shorter healing time than the CE group (a mean of 43.5 vs 63.87 days, respectively), though this difference was not statistically significant (P = .129).
“The uniqueness of this work lies in its simplified use of PE for recalcitrant keloids, which demonstrated efficacy comparable to CE, with the potential advantage of faster healing times,” Jung said. “Future studies with larger sample sizes and extended follow-up periods could help establish this approach as a standard treatment method.”
He acknowledged certain limitations of the study, including its small sample size and the lack of long-term follow-up data. The researchers reported having no relevant disclosures.
A version of this article first appeared on Medscape.com.
according to the results of a small retrospective study.
The method “offers similar efficacy, faster healing, and fewer complications,” one of the study authors, Jinwoong Jung, MD, said in an interview following the annual meeting of the American Society for Dermatologic Surgery, where he presented the study results during an oral abstract session.
For the study, Jung, a dermatologist at Yonsei University College of Medicine, Seoul, South Korea, and colleagues retrospectively analyzed 22 patients with recalcitrant keloids treated with cryotherapy immediately following either PE or CE between May 2019 and March 2024. They used the Vancouver Scar Scale (VSS) to assess treatment efficacy.
Of the 22 patients, 16 underwent treatment with CE and 6 underwent treatment with PE. Pretreatment VSS scores showed no significant differences between the groups (P = .535). The CE group had a reduction in the VSS score from 8.13 to 4.00, while the PE group had a reduction from 7.83 to 3.67, but these declines did not differ significantly (P = .737). The PE group exhibited a shorter healing time than the CE group (a mean of 43.5 vs 63.87 days, respectively), though this difference was not statistically significant (P = .129).
“The uniqueness of this work lies in its simplified use of PE for recalcitrant keloids, which demonstrated efficacy comparable to CE, with the potential advantage of faster healing times,” Jung said. “Future studies with larger sample sizes and extended follow-up periods could help establish this approach as a standard treatment method.”
He acknowledged certain limitations of the study, including its small sample size and the lack of long-term follow-up data. The researchers reported having no relevant disclosures.
A version of this article first appeared on Medscape.com.
according to the results of a small retrospective study.
The method “offers similar efficacy, faster healing, and fewer complications,” one of the study authors, Jinwoong Jung, MD, said in an interview following the annual meeting of the American Society for Dermatologic Surgery, where he presented the study results during an oral abstract session.
For the study, Jung, a dermatologist at Yonsei University College of Medicine, Seoul, South Korea, and colleagues retrospectively analyzed 22 patients with recalcitrant keloids treated with cryotherapy immediately following either PE or CE between May 2019 and March 2024. They used the Vancouver Scar Scale (VSS) to assess treatment efficacy.
Of the 22 patients, 16 underwent treatment with CE and 6 underwent treatment with PE. Pretreatment VSS scores showed no significant differences between the groups (P = .535). The CE group had a reduction in the VSS score from 8.13 to 4.00, while the PE group had a reduction from 7.83 to 3.67, but these declines did not differ significantly (P = .737). The PE group exhibited a shorter healing time than the CE group (a mean of 43.5 vs 63.87 days, respectively), though this difference was not statistically significant (P = .129).
“The uniqueness of this work lies in its simplified use of PE for recalcitrant keloids, which demonstrated efficacy comparable to CE, with the potential advantage of faster healing times,” Jung said. “Future studies with larger sample sizes and extended follow-up periods could help establish this approach as a standard treatment method.”
He acknowledged certain limitations of the study, including its small sample size and the lack of long-term follow-up data. The researchers reported having no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM ASDS 2024
FDA Approves OnabotulinumtoxinA for Improving Platysma Bands
The in adults.
According to a press release from Allergan Aesthetics, which developed onabotulinumtoxinA, by injecting along the jawline and the vertical bands connecting the jaw and neck with one of the FDA-approved doses of the product based on severity, onabotulinumtoxinA temporarily reduces underlying muscle activity.
The company cited results from phase 3 clinical studies, which demonstrated statistical significance for the improvement in appearance of platysma bands from baseline with onabotulinumtoxinA compared with placebo on both investigator and patient assessment (P < .0001).
All secondary endpoints were also met, as measured by multiple validated, proprietary patient-reported outcome instruments. In two of the clinical studies, for example, 65% and 62% of patients reported being “very satisfied” or “satisfied,” respectively, with their neck and jawline definition 14 days after treatment with a dose of 26, 31, or 36 units of onabotulinumtoxinA, compared with 12% with placebo in both studies.
The development marks the fourth indication for onabotulinumtoxinA. The others are for moderate to severe glabellar lines associated with corrugator and/or procerus muscle activity, moderate to severe lateral canthal lines associated with orbicularis oculi activity, and moderate to severe forehead lines associated with frontalis activity.
A version of this article appeared on Medscape.com.
The in adults.
According to a press release from Allergan Aesthetics, which developed onabotulinumtoxinA, by injecting along the jawline and the vertical bands connecting the jaw and neck with one of the FDA-approved doses of the product based on severity, onabotulinumtoxinA temporarily reduces underlying muscle activity.
The company cited results from phase 3 clinical studies, which demonstrated statistical significance for the improvement in appearance of platysma bands from baseline with onabotulinumtoxinA compared with placebo on both investigator and patient assessment (P < .0001).
All secondary endpoints were also met, as measured by multiple validated, proprietary patient-reported outcome instruments. In two of the clinical studies, for example, 65% and 62% of patients reported being “very satisfied” or “satisfied,” respectively, with their neck and jawline definition 14 days after treatment with a dose of 26, 31, or 36 units of onabotulinumtoxinA, compared with 12% with placebo in both studies.
The development marks the fourth indication for onabotulinumtoxinA. The others are for moderate to severe glabellar lines associated with corrugator and/or procerus muscle activity, moderate to severe lateral canthal lines associated with orbicularis oculi activity, and moderate to severe forehead lines associated with frontalis activity.
A version of this article appeared on Medscape.com.
The in adults.
According to a press release from Allergan Aesthetics, which developed onabotulinumtoxinA, by injecting along the jawline and the vertical bands connecting the jaw and neck with one of the FDA-approved doses of the product based on severity, onabotulinumtoxinA temporarily reduces underlying muscle activity.
The company cited results from phase 3 clinical studies, which demonstrated statistical significance for the improvement in appearance of platysma bands from baseline with onabotulinumtoxinA compared with placebo on both investigator and patient assessment (P < .0001).
All secondary endpoints were also met, as measured by multiple validated, proprietary patient-reported outcome instruments. In two of the clinical studies, for example, 65% and 62% of patients reported being “very satisfied” or “satisfied,” respectively, with their neck and jawline definition 14 days after treatment with a dose of 26, 31, or 36 units of onabotulinumtoxinA, compared with 12% with placebo in both studies.
The development marks the fourth indication for onabotulinumtoxinA. The others are for moderate to severe glabellar lines associated with corrugator and/or procerus muscle activity, moderate to severe lateral canthal lines associated with orbicularis oculi activity, and moderate to severe forehead lines associated with frontalis activity.
A version of this article appeared on Medscape.com.
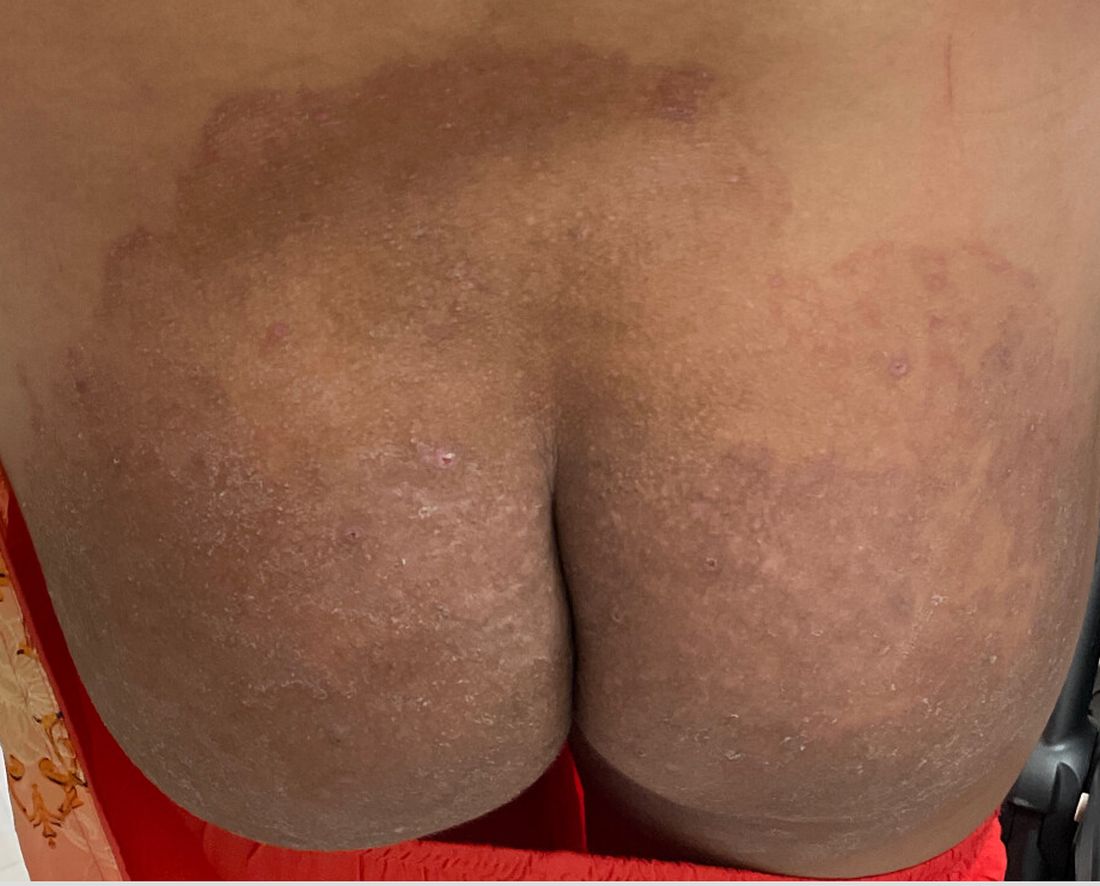
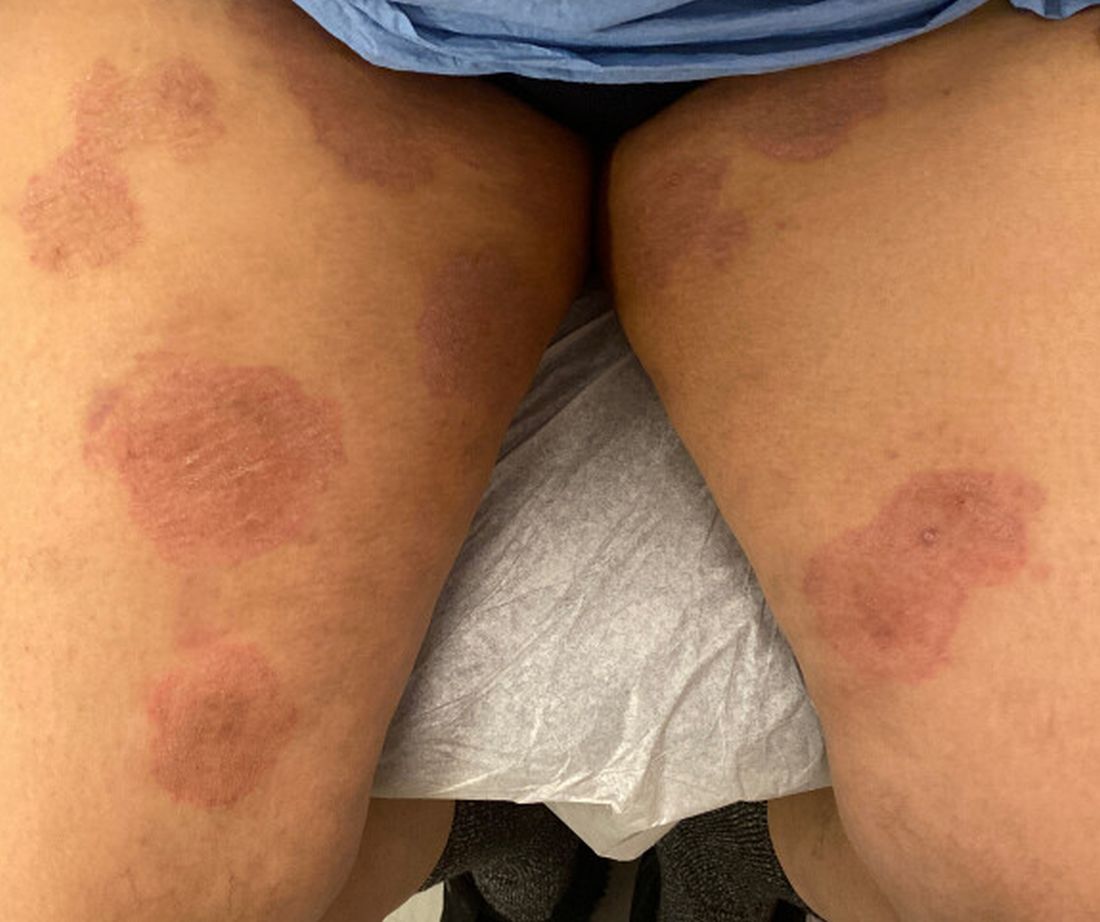
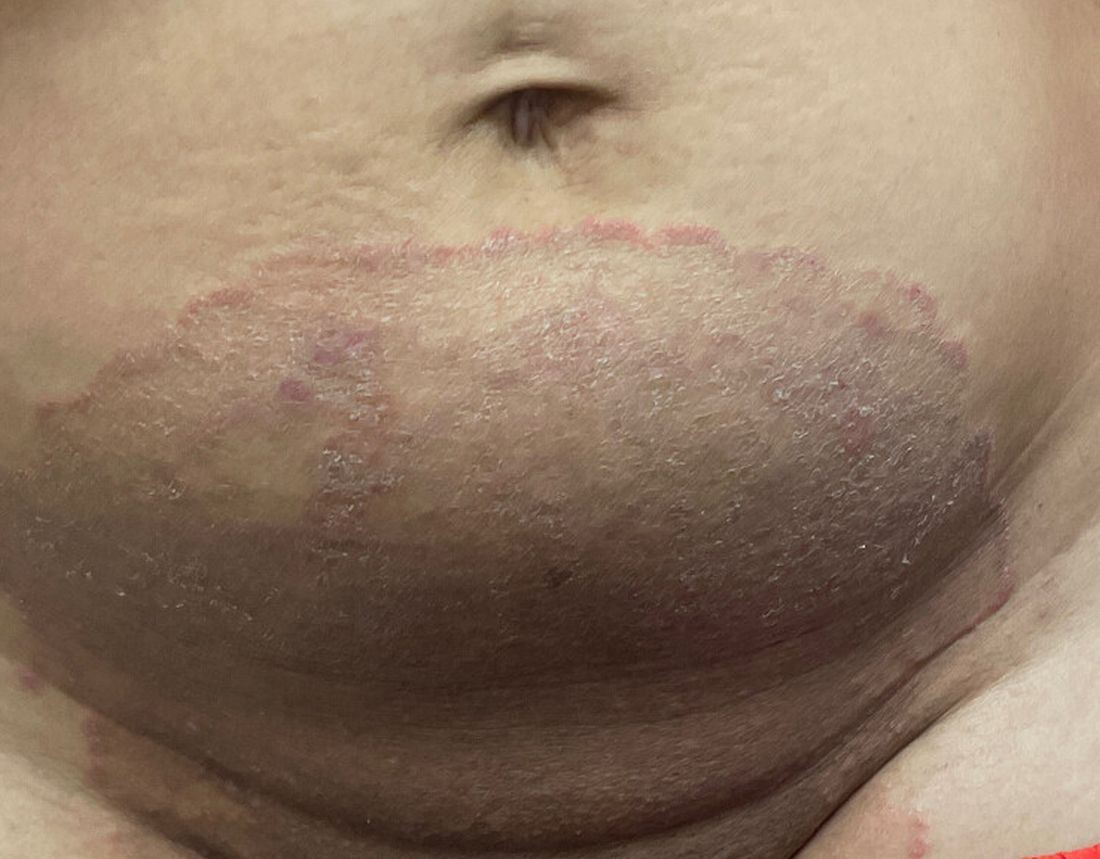
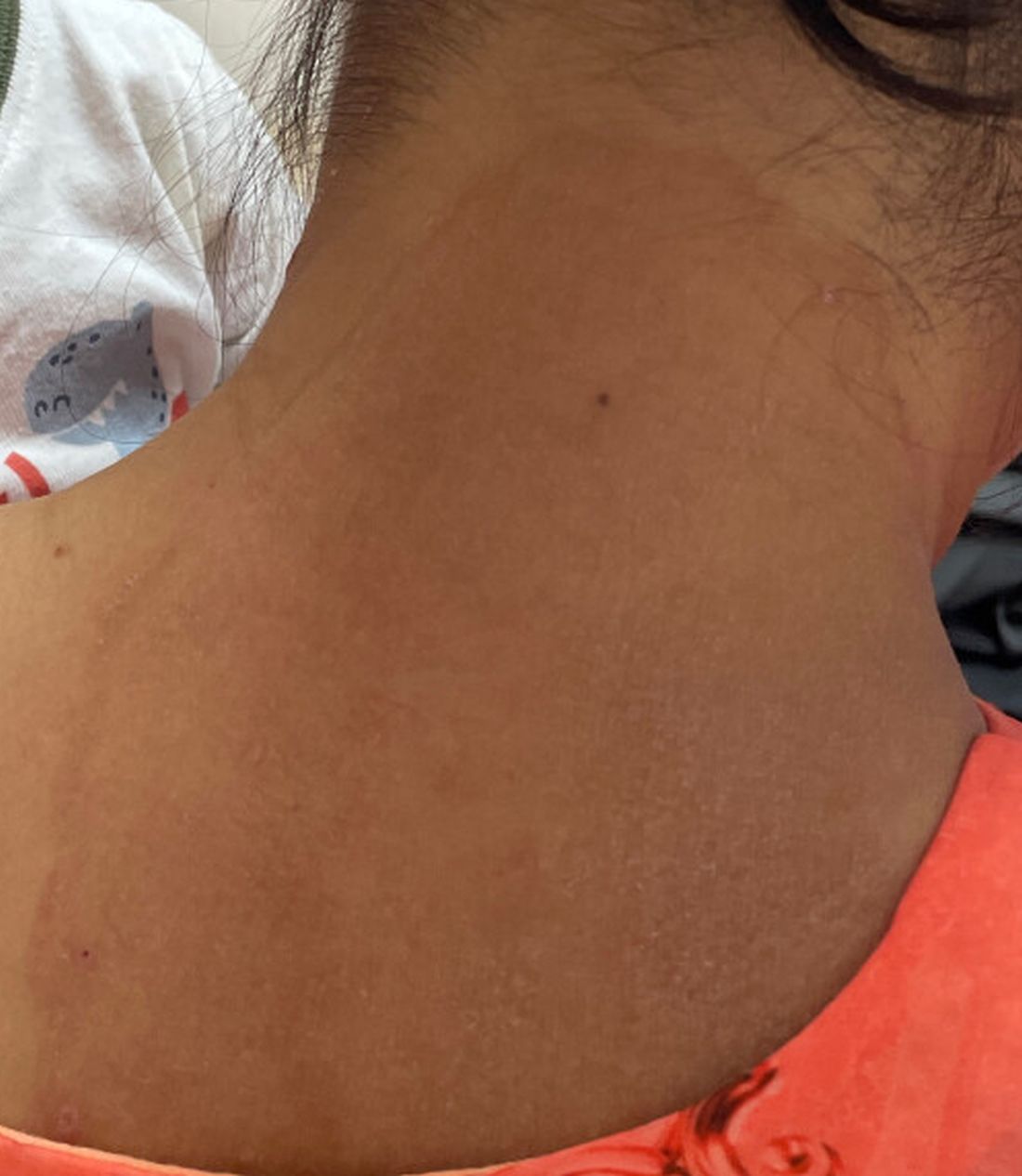
A 51-year-old woman presented for a routine full body skin exam after vacationing in Hawaii.
Primary adrenal insufficiency (Addison’s disease) results from a dysfunction of the adrenal glands, which may be secondary to autoimmune diseases, genetic conditions, infections, and vasculopathies,or may be drug-induced (e.g. checkpoint inhibitors), among others . In contrast, secondary adrenal insufficiency results from pituitary dysfunction of low adrenocorticotropic hormone (ACTH). The most common cause of primary adrenal insufficiency in developed countries is autoimmune adrenalitis, which accounts for upwards of 90% of cases. Typically, 21-hydroxylase autoantibodies are identified and account for destruction of the adrenal cortex through cell-mediated and humoral immune responses.
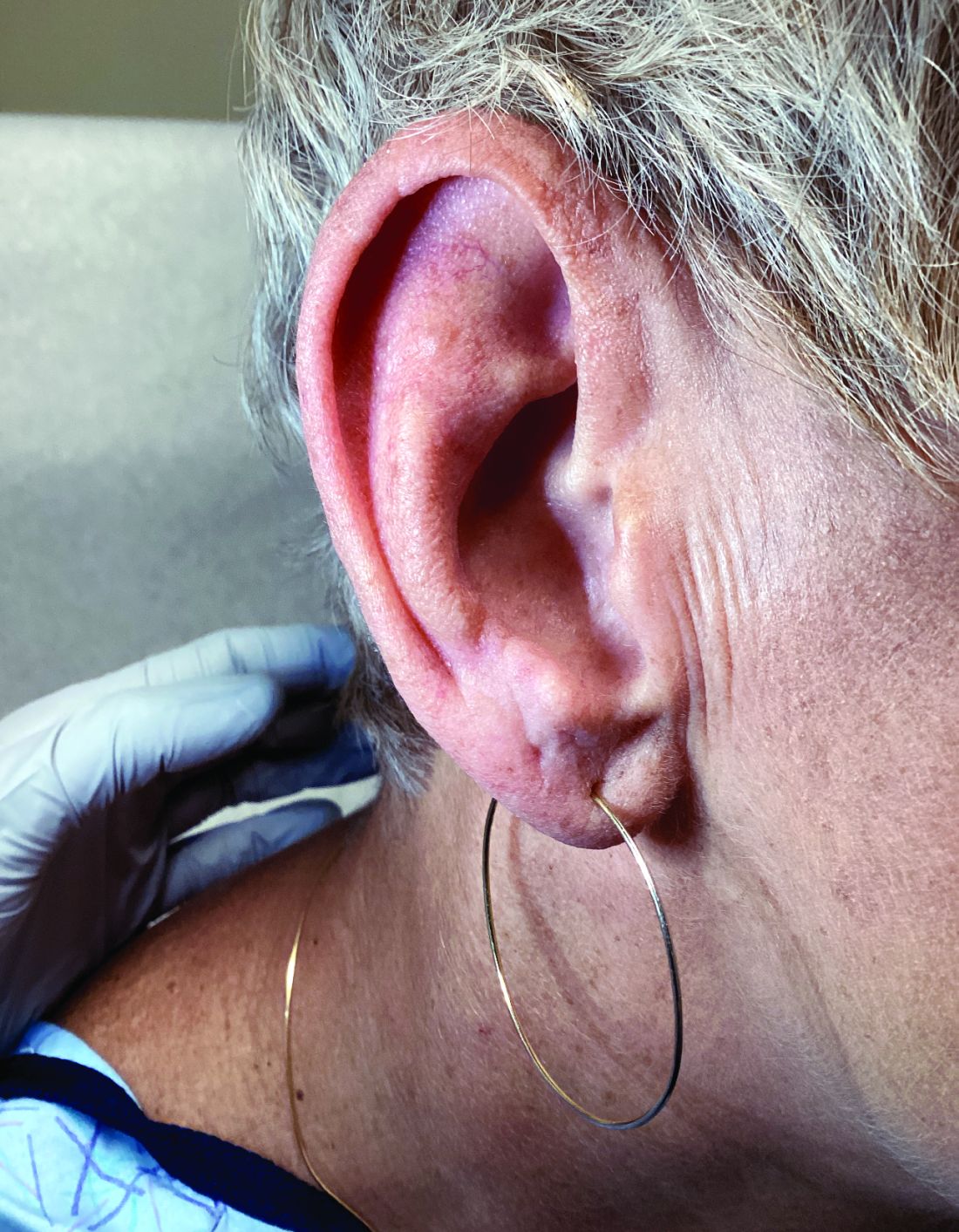
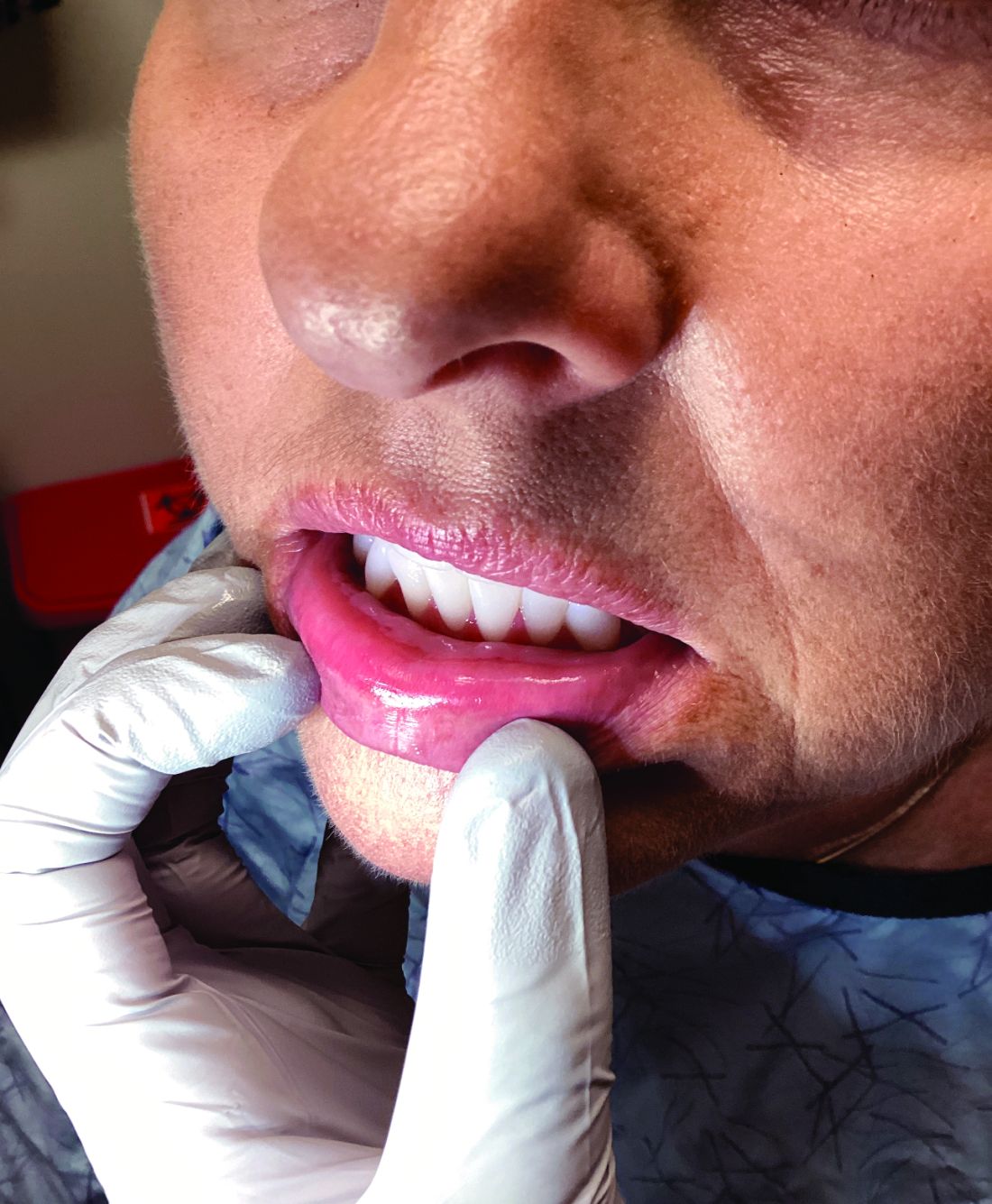
Palmar creases, subungual surfaces, sites of trauma, and joint spaces (including the knees, spine, elbows, and shoulders) are commonly affected. Hair depletes in the pubic area and axillary vaults. Nevi may also appear darker. In patients with autoimmune adrenalitis, vitiligo may be seen secondary to autoimmune destruction of melanocytes.
Diagnosis may be difficult in the early stages, but historical findings of fatigue and clinical findings of hyperpigmentation in classic areas may prompt appropriate lab screening workup. It is essential to determine whether adrenal insufficiency is primary or secondary. Evaluation of decreased cortisol production, determination of whether production is ACTH-dependent or -independent, and evaluation for the underlying causes of adrenal dysfunction are important. Lab screening includes morning serum cortisol, morning ACTH (cosyntropin) stimulation test, fasting CBC with differential, and CMP to evaluate for normocytic normochromic anemia, hyponatremia, hyperkalemia, hypoglycemia, plasma renin/aldosterone ratio, and 21-hydroxylase autoantibodies.
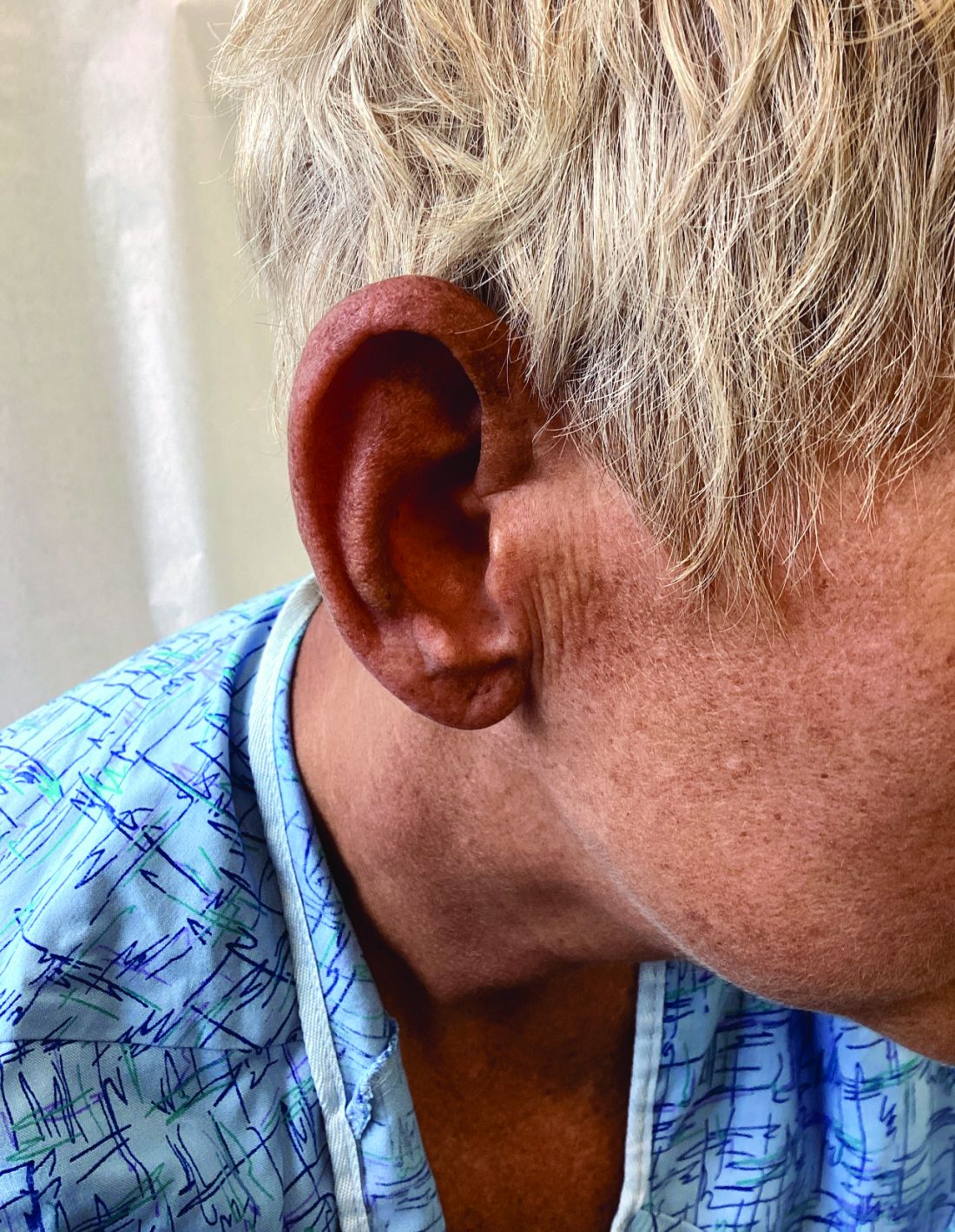
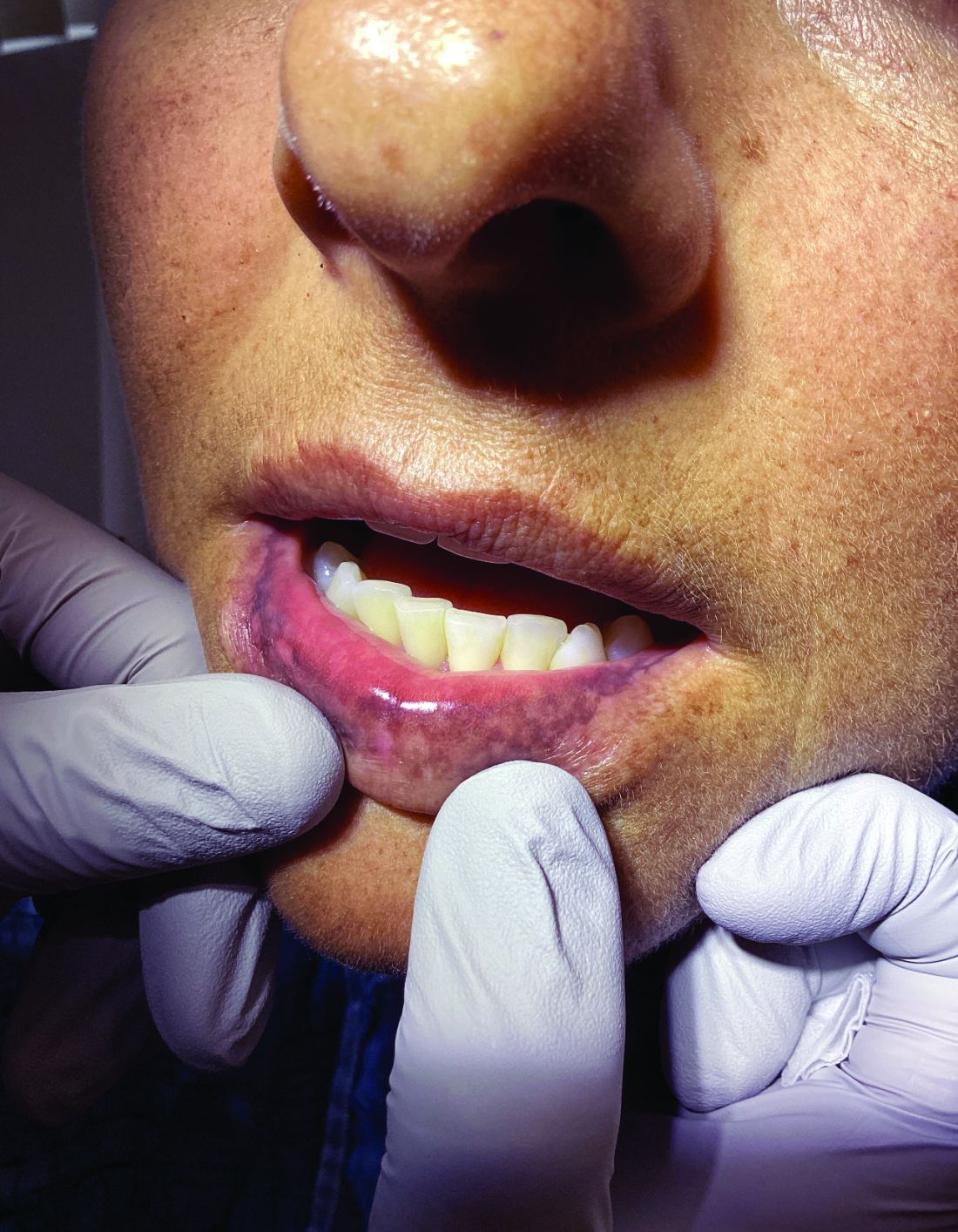
Management strategies of primary adrenal insufficiency require corticosteroid supplementation and multidisciplinary collaboration with endocrinology. If untreated, primary adrenal insufficiency can be fatal. Adrenal crisis is a critical condition following a precipitating event, such as GI infection, fever, acute stress, and/or untreated adrenal or pituitary disorders. Clinical findings include acute shock with hypotension, nausea, vomiting, abdominal pain, back or leg pain, and a change in mental status. In this scenario, increasing the dose of corticosteroid supplementation is essential for reducing mortality.
Upon examining this patient’s new skin findings of hyperpigmentation and discussing her fatigue, primary adrenal insufficiency was suspected. With further prompting, the patient reported an ICU hospitalization several months prior because of sepsis originating from a peritonsillar abscess. With these clinical and historical findings, preliminary workup was conducted by dermatology, which included morning cortisol level, ACTH, CBC with differential, CMP, plasma renin-aldosterone ratio, and 21-hydroxylase autoantibodies. Work up demonstrated a low morning cortisol level of 1.3 mcg/dL, an elevated ACTH of 2,739 pg/mL, and positive 21-hydroxylase autoantibodies. The patient was urgently referred to endocrinology and started on oral hydrocortisone. Her fatigue immediately improved, and at 1-year follow-up with dermatology, her mucocutaneous hyperpigmentation had subsided dramatically.

Dermatologists can play a major role in the early diagnosis of primary adrenal insufficiency, which is essential for reducing patient morbidity and mortality. Skin findings on full body skin exams can clue in dermatologists for ordering preliminary workup to expedite care for these patients.
The case and photos were submitted by Dr. Akhiyat, Scripps Clinic Medical Group, La Jolla, California. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
J Am Acad Dermatol. 2014 May;70(5):Supplement 1AB118. doi: 10.1016/j.jaad.2014.01.491.
Michels A, Michels N. Am Fam Physician. 2014 Apr 1;89(7):563-568.
Kauzman A et al. J Can Dent Assoc. 2004 Nov;70(10):682-683.
Primary adrenal insufficiency (Addison’s disease) results from a dysfunction of the adrenal glands, which may be secondary to autoimmune diseases, genetic conditions, infections, and vasculopathies,or may be drug-induced (e.g. checkpoint inhibitors), among others . In contrast, secondary adrenal insufficiency results from pituitary dysfunction of low adrenocorticotropic hormone (ACTH). The most common cause of primary adrenal insufficiency in developed countries is autoimmune adrenalitis, which accounts for upwards of 90% of cases. Typically, 21-hydroxylase autoantibodies are identified and account for destruction of the adrenal cortex through cell-mediated and humoral immune responses.
Palmar creases, subungual surfaces, sites of trauma, and joint spaces (including the knees, spine, elbows, and shoulders) are commonly affected. Hair depletes in the pubic area and axillary vaults. Nevi may also appear darker. In patients with autoimmune adrenalitis, vitiligo may be seen secondary to autoimmune destruction of melanocytes.
Diagnosis may be difficult in the early stages, but historical findings of fatigue and clinical findings of hyperpigmentation in classic areas may prompt appropriate lab screening workup. It is essential to determine whether adrenal insufficiency is primary or secondary. Evaluation of decreased cortisol production, determination of whether production is ACTH-dependent or -independent, and evaluation for the underlying causes of adrenal dysfunction are important. Lab screening includes morning serum cortisol, morning ACTH (cosyntropin) stimulation test, fasting CBC with differential, and CMP to evaluate for normocytic normochromic anemia, hyponatremia, hyperkalemia, hypoglycemia, plasma renin/aldosterone ratio, and 21-hydroxylase autoantibodies.
Management strategies of primary adrenal insufficiency require corticosteroid supplementation and multidisciplinary collaboration with endocrinology. If untreated, primary adrenal insufficiency can be fatal. Adrenal crisis is a critical condition following a precipitating event, such as GI infection, fever, acute stress, and/or untreated adrenal or pituitary disorders. Clinical findings include acute shock with hypotension, nausea, vomiting, abdominal pain, back or leg pain, and a change in mental status. In this scenario, increasing the dose of corticosteroid supplementation is essential for reducing mortality.
Upon examining this patient’s new skin findings of hyperpigmentation and discussing her fatigue, primary adrenal insufficiency was suspected. With further prompting, the patient reported an ICU hospitalization several months prior because of sepsis originating from a peritonsillar abscess. With these clinical and historical findings, preliminary workup was conducted by dermatology, which included morning cortisol level, ACTH, CBC with differential, CMP, plasma renin-aldosterone ratio, and 21-hydroxylase autoantibodies. Work up demonstrated a low morning cortisol level of 1.3 mcg/dL, an elevated ACTH of 2,739 pg/mL, and positive 21-hydroxylase autoantibodies. The patient was urgently referred to endocrinology and started on oral hydrocortisone. Her fatigue immediately improved, and at 1-year follow-up with dermatology, her mucocutaneous hyperpigmentation had subsided dramatically.
Dermatologists can play a major role in the early diagnosis of primary adrenal insufficiency, which is essential for reducing patient morbidity and mortality. Skin findings on full body skin exams can clue in dermatologists for ordering preliminary workup to expedite care for these patients.
The case and photos were submitted by Dr. Akhiyat, Scripps Clinic Medical Group, La Jolla, California. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
J Am Acad Dermatol. 2014 May;70(5):Supplement 1AB118. doi: 10.1016/j.jaad.2014.01.491.
Michels A, Michels N. Am Fam Physician. 2014 Apr 1;89(7):563-568.
Kauzman A et al. J Can Dent Assoc. 2004 Nov;70(10):682-683.
Primary adrenal insufficiency (Addison’s disease) results from a dysfunction of the adrenal glands, which may be secondary to autoimmune diseases, genetic conditions, infections, and vasculopathies,or may be drug-induced (e.g. checkpoint inhibitors), among others . In contrast, secondary adrenal insufficiency results from pituitary dysfunction of low adrenocorticotropic hormone (ACTH). The most common cause of primary adrenal insufficiency in developed countries is autoimmune adrenalitis, which accounts for upwards of 90% of cases. Typically, 21-hydroxylase autoantibodies are identified and account for destruction of the adrenal cortex through cell-mediated and humoral immune responses.
Palmar creases, subungual surfaces, sites of trauma, and joint spaces (including the knees, spine, elbows, and shoulders) are commonly affected. Hair depletes in the pubic area and axillary vaults. Nevi may also appear darker. In patients with autoimmune adrenalitis, vitiligo may be seen secondary to autoimmune destruction of melanocytes.
Diagnosis may be difficult in the early stages, but historical findings of fatigue and clinical findings of hyperpigmentation in classic areas may prompt appropriate lab screening workup. It is essential to determine whether adrenal insufficiency is primary or secondary. Evaluation of decreased cortisol production, determination of whether production is ACTH-dependent or -independent, and evaluation for the underlying causes of adrenal dysfunction are important. Lab screening includes morning serum cortisol, morning ACTH (cosyntropin) stimulation test, fasting CBC with differential, and CMP to evaluate for normocytic normochromic anemia, hyponatremia, hyperkalemia, hypoglycemia, plasma renin/aldosterone ratio, and 21-hydroxylase autoantibodies.
Management strategies of primary adrenal insufficiency require corticosteroid supplementation and multidisciplinary collaboration with endocrinology. If untreated, primary adrenal insufficiency can be fatal. Adrenal crisis is a critical condition following a precipitating event, such as GI infection, fever, acute stress, and/or untreated adrenal or pituitary disorders. Clinical findings include acute shock with hypotension, nausea, vomiting, abdominal pain, back or leg pain, and a change in mental status. In this scenario, increasing the dose of corticosteroid supplementation is essential for reducing mortality.
Upon examining this patient’s new skin findings of hyperpigmentation and discussing her fatigue, primary adrenal insufficiency was suspected. With further prompting, the patient reported an ICU hospitalization several months prior because of sepsis originating from a peritonsillar abscess. With these clinical and historical findings, preliminary workup was conducted by dermatology, which included morning cortisol level, ACTH, CBC with differential, CMP, plasma renin-aldosterone ratio, and 21-hydroxylase autoantibodies. Work up demonstrated a low morning cortisol level of 1.3 mcg/dL, an elevated ACTH of 2,739 pg/mL, and positive 21-hydroxylase autoantibodies. The patient was urgently referred to endocrinology and started on oral hydrocortisone. Her fatigue immediately improved, and at 1-year follow-up with dermatology, her mucocutaneous hyperpigmentation had subsided dramatically.
Dermatologists can play a major role in the early diagnosis of primary adrenal insufficiency, which is essential for reducing patient morbidity and mortality. Skin findings on full body skin exams can clue in dermatologists for ordering preliminary workup to expedite care for these patients.
The case and photos were submitted by Dr. Akhiyat, Scripps Clinic Medical Group, La Jolla, California. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
J Am Acad Dermatol. 2014 May;70(5):Supplement 1AB118. doi: 10.1016/j.jaad.2014.01.491.
Michels A, Michels N. Am Fam Physician. 2014 Apr 1;89(7):563-568.
Kauzman A et al. J Can Dent Assoc. 2004 Nov;70(10):682-683.
Increase in Troublesome Fungal Infections Requires All-Out Approach
As dermatologists, public health officials, and infectious disease specialists scramble to raise awareness about prevention and treatment, challenges ranging from a dearth of testing facilities and data to payer pushback over longer therapeutic courses remain.
Dermatophyte Discourse Changing
“Trichophyton indotineae is changing the way we talk about dermatophyte infections,” Avrom S. Caplan, MD, assistant professor in the Department of Dermatology at New York University, New York City, said in an interview. Called T mentagrophytes VIII (TMVIII) before a 2020 report in the journal Mycopathologia proposed the name T indotineae, this species requires clinicians to expand their conception of how tinea looks, acts, and responds to treatment.
Boni E. Elewski, MD, professor and chair of dermatology, at The University of Alabama at Birmingham, saw her first case of probable T indotineae in a patient in early 2020. “He was covered with fine scale, and he itched all over. I thought he had atopic dermatitis. This didn’t look like any tinea. His face, arms, back, and legs were scaly.”

Nevertheless, KOH and biopsy confirmed dermatophytosis. Culture (performed at the Center for Medical Mycology [CMM] in Cleveland) identified T mentagrophytes. Back then, Elewski told this news organization, labs did not routinely go beyond genus and species. But based on the patient’s symptoms, history of unresponsiveness to terbinafine, borderline sensitivity to fluconazole, and travel to India and Spain, Elewski strongly suspected T indotineae.
The patient refused itraconazole, to which the fungus was sensitive, and did not respond to fluconazole 400 mg daily. Ultimately, he was lost to follow-up. “Last I saw him,” said Elewski, “he was not cured.”
Tracking Cases
Because T indotineae does not require reporting to public health agencies, said Jeremy Gold, MD, MS, a medical officer with the US Centers for Disease Control and Prevention (CDC) Mycotic Diseases Branch in Atlanta, “there is no official public health surveillance keeping track of exactly how many cases have occurred.”

The same is true for TMVII and terbinafine-resistant T rubrum, which are also on the rise. Regarding T indotineae, authors from the University of Texas Health Science Center at San Antonio retrospectively reported 21 terbinafine-resistant isolates from North America in the July 2023 Journal of Clinical Microbiology .
Caplan has seen approximately 12 T indotineae cases to date, including the first two confirmed US cases, which he and co-authors, including Gold, reported in the CDC’s Morbidity and Mortality Weekly Report in May 2023. T indotineae is likely underreported, he said, because it eludes standard culture-based techniques, and identifying it requires molecular testing, which is available at only a handful of labs nationally.
To help educate providers, in July, the American Academy of Dermatology (AAD) and the International League of Dermatological Societies unveiled an Emerging Diseases Resource Center, which includes resources for providers and a registry for reporting confirmed and suspected resistant dermatophytes.
“Our goal is to provide easy-to-access and easy-to-understand resources to healthcare providers,” Esther Freeman, MD, PhD, told this news organization. She is director of Global Health Dermatology at Massachusetts General Hospital, associate professor of dermatology at Harvard Medical School, both in Boston, and chair of the AAD’s Emerging Diseases Task Force.

“Our resources include an algorithm for when to suspect a drug-resistant case and how to think through treatment options. We cover issues related to diagnosis and treatment, as well as linking to our case registry reporting system,” said Freeman.
The new registry resides within the AAD’s existing COVID-19, Mpox, and Emerging Infections Registry. “Our registry efforts have already captured 2500 COVID-19 and mpox cases from 72 different countries,” Freeman said. For all these infections, she added, “we hope that real-time data analysis of cases worldwide will provide information that helps physicians recognize and treat cases.”
Consistent with the registry’s approach, said Caplan and Gold, there is no silver bullet for battling dermatophyte resistance. What is needed, said Gold, is a coordinated approach involving public health officials, dermatologists, primary care providers, infectious disease specialists, pharmacists, and patients. “It’s going to be a team effort to address the challenge of emerging complex dermatophytosis,” he said.
Resistant T rubrum
“The biggest difference with T rubrum resistance is you may not see that widespread infection that we see with T indotineae,” said Caplan. T rubrum is probably the most common dermatophyte that dermatologists see, added Elewski, who encounters a resistant case at least monthly. One such patient, featured in a January 2021 British Journal of Dermatology research letter, cleared on itraconazole and ciclopirox cream but subsequently returned with itraconazole-resistant T rubrum because he had been doctor-shopping for the drug intermittently for years, she said. He cleared on posaconazole 300 mg daily, then was lost to follow-up.
TMVII
A 2023 Emerging Infectious Diseases report highlighted the potential for this dermatophyte to spread among men who have sex with men (MSM), presenting as an itchy, scaly rash affecting the pubic, genital, and buttocks skin. “People don’t generally think of a fungal infection as something that could behave like a sexually transmitted infection (STI),” said Gold.
Caplan and coauthors recently reported the first confirmed US TMVII case in JAMA Dermatology. Many experts suspect that unreported US cases existed previously, he said. “When it circulates in Europe and there’s so much travel, it’s probably here too.”
The fact that T indotineae was formerly called TMVIII has created confusion, added Caplan. “I’ve had patients say, ‘I’m worried I have that resistant ringworm that’s spreading among MSM.’ Whenever we talk about STIs and introduce the word ‘resistant,’ that comes with the potential for stigma, anxiety, and concern.” Fortunately, he said, TMVII has shown no resistance to first-line antifungals.

Why the Rise
Gold said, “We don’t know for sure why we’re seeing these different drug-resistant species popping up.” One possibility, he said, is the common misuse and overuse of topical antifungals — especially those available overseas in combination with high-potency steroids, such as clobetasol. Consumers use these products for a few weeks until symptoms resolve, then reapply them off and on over years, fueling resistance, said Gold.
“We are worried that with warming temperatures, there’s potential to see expansion of the geographic range of epidemic fungi,” he added. “That could be part of what has fueled recent increases in resistant dermatophytes. But it’s hard to prove.”
Climate change may be behind the emergence of Candida auris, according to a 2022 article in The Lancet Regional Health – Americas. This potentially fatal multidrug-resistant infection spreads easily among sick patients in healthcare facilities, according to a CDC information page on C auris.
Confirming Dermatophyte Infection
“A biopsy will only confirm the presence of fungus,” said Elewski. “Here you will need a lab that knows how to do a fungal culture.” Most state laboratories can do this, she said, as can some hospitals and special labs such as CMM in Cleveland.
It takes a Clinical Laboratory Improvement Amendments–certified lab to perform KOH prep in-house, added Caplan, plus up-to-date gear and knowledge of where and how to scrape and what to look for microscopically. Moreover, identifying T indotineae requires molecular testing available at only a handful of laboratories — listed on the AAD Emerging Dermatophytes webpage — nationwide.
Nevertheless, said Caplan, nailing down a diagnosis can guide treatment, often supplanting empirically prescribed antifungal steroid creams. “Those are probably not going to help. And people may be using those on areas of the body they shouldn’t. Both the clinical clues and the steps to make the diagnosis need to come together. But that’s often easier said than done, especially in a busy practice.”
Identifying resistance requires antifungal sensitivity testing, he added, which few labs perform. “Practically speaking,” said Elewski, “if the patient failed terbinafine, I would try itraconazole. You don’t necessarily need proof” of resistance. But if a patient does not respond to itraconazole and terbinafine clinically, she said that she might consider fungal susceptibility testing.
Treatment Tips
To address any resistant dermatophyte, Elewski recommended getting comfortable with itraconazole. For decades, she said, dermatologists have avoided itraconazole because terbinafine typically costs patients $10 for 3 months. “Itraconazole could be $200 per month,” said Elewski. Because of potential drug-drug interactions and absorption issues — and a boxed warning regarding congestive heart failure — physicians historically reserved itraconazole for severe fungal infections.
Itraconazole labeled dosing for onychomycosis is 200 mg daily for 12 weeks. Elewski favors a two-pronged attack, often combining an -azole antifungal with topical ciclopirox.
Another element that emerging tinea pathogens share is slower response to treatment. For T indotineae, reports appearing in the Journal of the American Academy of Dermatology in 2022 and 2024 suggest duration from 6-8 weeks up to 20 weeks.
To avoid recurrences of resistant T rubrum, Elewski treats for a year. However, she has problems getting itraconazole approved, when often it is the only agent that works. “I’ve written more letters than I like to insurance companies” to document terbinafine failure, she said.
Rarely, said Gold, dermatophyte infections resist both terbinafine and itraconazole. Next-line agents such as voriconazole, which some dermatologists have used for resistant T indotineae, can be much harder to tolerate, with more drug interactions, he said.
And because itraconazole, voriconazole, and posaconazole are all triazoles, added Elewski, the latter two might not work better than the former. But because these drugs might outperform itraconazole in selected cases, she said, “that’s when you want to do fungal susceptibility testing.”
TMVII is so new, said Caplan, that optimal therapy duration remains unclear. “One of the challenges with TMVII is when it gets into the genital skin, it’s a hair-bearing area. And based on various grooming practices, there’s an opportunity for the tinea to get deeper into the hair follicle and dermis. That may also be true of T indotineae.”
Anemic Arsenal
Unfortunately, said Gold, the arsenal of antifungals available in the United States remains limited. “Depending on how you count, there are only three to four classes of antifungal drugs designed to treat severe or invasive infections. So whenever we hear about a new fungal pathogen that’s causing resistant infections, it causes public health concern.”
Promising drugs in development include olorofim (F2G) and fosmanogepix (Basilea), according to Gold. However, he said, the development of these drugs to date has targeted invasive fungal infections such as aspergillosis. In June 2023, the Food and Drug Administration rejected the new drug application for olorofim, requesting additional data and analyses. Regarding fosmanogepix, a double-blinded noninferiority phase 3 trial in invasive yeast infections was recently launched, according to a September 24 press release.
Gold, Caplan, and Elewski reported no relevant financial disclosures. Freeman is a COVID-19 co-author for UpToDate and chair of the AAD Emerging Diseases Task Force.
A version of this article appeared on Medscape.com.
As dermatologists, public health officials, and infectious disease specialists scramble to raise awareness about prevention and treatment, challenges ranging from a dearth of testing facilities and data to payer pushback over longer therapeutic courses remain.
Dermatophyte Discourse Changing
“Trichophyton indotineae is changing the way we talk about dermatophyte infections,” Avrom S. Caplan, MD, assistant professor in the Department of Dermatology at New York University, New York City, said in an interview. Called T mentagrophytes VIII (TMVIII) before a 2020 report in the journal Mycopathologia proposed the name T indotineae, this species requires clinicians to expand their conception of how tinea looks, acts, and responds to treatment.
Boni E. Elewski, MD, professor and chair of dermatology, at The University of Alabama at Birmingham, saw her first case of probable T indotineae in a patient in early 2020. “He was covered with fine scale, and he itched all over. I thought he had atopic dermatitis. This didn’t look like any tinea. His face, arms, back, and legs were scaly.”
Nevertheless, KOH and biopsy confirmed dermatophytosis. Culture (performed at the Center for Medical Mycology [CMM] in Cleveland) identified T mentagrophytes. Back then, Elewski told this news organization, labs did not routinely go beyond genus and species. But based on the patient’s symptoms, history of unresponsiveness to terbinafine, borderline sensitivity to fluconazole, and travel to India and Spain, Elewski strongly suspected T indotineae.
The patient refused itraconazole, to which the fungus was sensitive, and did not respond to fluconazole 400 mg daily. Ultimately, he was lost to follow-up. “Last I saw him,” said Elewski, “he was not cured.”
Tracking Cases
Because T indotineae does not require reporting to public health agencies, said Jeremy Gold, MD, MS, a medical officer with the US Centers for Disease Control and Prevention (CDC) Mycotic Diseases Branch in Atlanta, “there is no official public health surveillance keeping track of exactly how many cases have occurred.”
The same is true for TMVII and terbinafine-resistant T rubrum, which are also on the rise. Regarding T indotineae, authors from the University of Texas Health Science Center at San Antonio retrospectively reported 21 terbinafine-resistant isolates from North America in the July 2023 Journal of Clinical Microbiology .
Caplan has seen approximately 12 T indotineae cases to date, including the first two confirmed US cases, which he and co-authors, including Gold, reported in the CDC’s Morbidity and Mortality Weekly Report in May 2023. T indotineae is likely underreported, he said, because it eludes standard culture-based techniques, and identifying it requires molecular testing, which is available at only a handful of labs nationally.
To help educate providers, in July, the American Academy of Dermatology (AAD) and the International League of Dermatological Societies unveiled an Emerging Diseases Resource Center, which includes resources for providers and a registry for reporting confirmed and suspected resistant dermatophytes.
“Our goal is to provide easy-to-access and easy-to-understand resources to healthcare providers,” Esther Freeman, MD, PhD, told this news organization. She is director of Global Health Dermatology at Massachusetts General Hospital, associate professor of dermatology at Harvard Medical School, both in Boston, and chair of the AAD’s Emerging Diseases Task Force.
“Our resources include an algorithm for when to suspect a drug-resistant case and how to think through treatment options. We cover issues related to diagnosis and treatment, as well as linking to our case registry reporting system,” said Freeman.
The new registry resides within the AAD’s existing COVID-19, Mpox, and Emerging Infections Registry. “Our registry efforts have already captured 2500 COVID-19 and mpox cases from 72 different countries,” Freeman said. For all these infections, she added, “we hope that real-time data analysis of cases worldwide will provide information that helps physicians recognize and treat cases.”
Consistent with the registry’s approach, said Caplan and Gold, there is no silver bullet for battling dermatophyte resistance. What is needed, said Gold, is a coordinated approach involving public health officials, dermatologists, primary care providers, infectious disease specialists, pharmacists, and patients. “It’s going to be a team effort to address the challenge of emerging complex dermatophytosis,” he said.
Resistant T rubrum
“The biggest difference with T rubrum resistance is you may not see that widespread infection that we see with T indotineae,” said Caplan. T rubrum is probably the most common dermatophyte that dermatologists see, added Elewski, who encounters a resistant case at least monthly. One such patient, featured in a January 2021 British Journal of Dermatology research letter, cleared on itraconazole and ciclopirox cream but subsequently returned with itraconazole-resistant T rubrum because he had been doctor-shopping for the drug intermittently for years, she said. He cleared on posaconazole 300 mg daily, then was lost to follow-up.
TMVII
A 2023 Emerging Infectious Diseases report highlighted the potential for this dermatophyte to spread among men who have sex with men (MSM), presenting as an itchy, scaly rash affecting the pubic, genital, and buttocks skin. “People don’t generally think of a fungal infection as something that could behave like a sexually transmitted infection (STI),” said Gold.
Caplan and coauthors recently reported the first confirmed US TMVII case in JAMA Dermatology. Many experts suspect that unreported US cases existed previously, he said. “When it circulates in Europe and there’s so much travel, it’s probably here too.”
The fact that T indotineae was formerly called TMVIII has created confusion, added Caplan. “I’ve had patients say, ‘I’m worried I have that resistant ringworm that’s spreading among MSM.’ Whenever we talk about STIs and introduce the word ‘resistant,’ that comes with the potential for stigma, anxiety, and concern.” Fortunately, he said, TMVII has shown no resistance to first-line antifungals.
Why the Rise
Gold said, “We don’t know for sure why we’re seeing these different drug-resistant species popping up.” One possibility, he said, is the common misuse and overuse of topical antifungals — especially those available overseas in combination with high-potency steroids, such as clobetasol. Consumers use these products for a few weeks until symptoms resolve, then reapply them off and on over years, fueling resistance, said Gold.
“We are worried that with warming temperatures, there’s potential to see expansion of the geographic range of epidemic fungi,” he added. “That could be part of what has fueled recent increases in resistant dermatophytes. But it’s hard to prove.”
Climate change may be behind the emergence of Candida auris, according to a 2022 article in The Lancet Regional Health – Americas. This potentially fatal multidrug-resistant infection spreads easily among sick patients in healthcare facilities, according to a CDC information page on C auris.
Confirming Dermatophyte Infection
“A biopsy will only confirm the presence of fungus,” said Elewski. “Here you will need a lab that knows how to do a fungal culture.” Most state laboratories can do this, she said, as can some hospitals and special labs such as CMM in Cleveland.
It takes a Clinical Laboratory Improvement Amendments–certified lab to perform KOH prep in-house, added Caplan, plus up-to-date gear and knowledge of where and how to scrape and what to look for microscopically. Moreover, identifying T indotineae requires molecular testing available at only a handful of laboratories — listed on the AAD Emerging Dermatophytes webpage — nationwide.
Nevertheless, said Caplan, nailing down a diagnosis can guide treatment, often supplanting empirically prescribed antifungal steroid creams. “Those are probably not going to help. And people may be using those on areas of the body they shouldn’t. Both the clinical clues and the steps to make the diagnosis need to come together. But that’s often easier said than done, especially in a busy practice.”
Identifying resistance requires antifungal sensitivity testing, he added, which few labs perform. “Practically speaking,” said Elewski, “if the patient failed terbinafine, I would try itraconazole. You don’t necessarily need proof” of resistance. But if a patient does not respond to itraconazole and terbinafine clinically, she said that she might consider fungal susceptibility testing.
Treatment Tips
To address any resistant dermatophyte, Elewski recommended getting comfortable with itraconazole. For decades, she said, dermatologists have avoided itraconazole because terbinafine typically costs patients $10 for 3 months. “Itraconazole could be $200 per month,” said Elewski. Because of potential drug-drug interactions and absorption issues — and a boxed warning regarding congestive heart failure — physicians historically reserved itraconazole for severe fungal infections.
Itraconazole labeled dosing for onychomycosis is 200 mg daily for 12 weeks. Elewski favors a two-pronged attack, often combining an -azole antifungal with topical ciclopirox.
Another element that emerging tinea pathogens share is slower response to treatment. For T indotineae, reports appearing in the Journal of the American Academy of Dermatology in 2022 and 2024 suggest duration from 6-8 weeks up to 20 weeks.
To avoid recurrences of resistant T rubrum, Elewski treats for a year. However, she has problems getting itraconazole approved, when often it is the only agent that works. “I’ve written more letters than I like to insurance companies” to document terbinafine failure, she said.
Rarely, said Gold, dermatophyte infections resist both terbinafine and itraconazole. Next-line agents such as voriconazole, which some dermatologists have used for resistant T indotineae, can be much harder to tolerate, with more drug interactions, he said.
And because itraconazole, voriconazole, and posaconazole are all triazoles, added Elewski, the latter two might not work better than the former. But because these drugs might outperform itraconazole in selected cases, she said, “that’s when you want to do fungal susceptibility testing.”
TMVII is so new, said Caplan, that optimal therapy duration remains unclear. “One of the challenges with TMVII is when it gets into the genital skin, it’s a hair-bearing area. And based on various grooming practices, there’s an opportunity for the tinea to get deeper into the hair follicle and dermis. That may also be true of T indotineae.”
Anemic Arsenal
Unfortunately, said Gold, the arsenal of antifungals available in the United States remains limited. “Depending on how you count, there are only three to four classes of antifungal drugs designed to treat severe or invasive infections. So whenever we hear about a new fungal pathogen that’s causing resistant infections, it causes public health concern.”
Promising drugs in development include olorofim (F2G) and fosmanogepix (Basilea), according to Gold. However, he said, the development of these drugs to date has targeted invasive fungal infections such as aspergillosis. In June 2023, the Food and Drug Administration rejected the new drug application for olorofim, requesting additional data and analyses. Regarding fosmanogepix, a double-blinded noninferiority phase 3 trial in invasive yeast infections was recently launched, according to a September 24 press release.
Gold, Caplan, and Elewski reported no relevant financial disclosures. Freeman is a COVID-19 co-author for UpToDate and chair of the AAD Emerging Diseases Task Force.
A version of this article appeared on Medscape.com.
As dermatologists, public health officials, and infectious disease specialists scramble to raise awareness about prevention and treatment, challenges ranging from a dearth of testing facilities and data to payer pushback over longer therapeutic courses remain.
Dermatophyte Discourse Changing
“Trichophyton indotineae is changing the way we talk about dermatophyte infections,” Avrom S. Caplan, MD, assistant professor in the Department of Dermatology at New York University, New York City, said in an interview. Called T mentagrophytes VIII (TMVIII) before a 2020 report in the journal Mycopathologia proposed the name T indotineae, this species requires clinicians to expand their conception of how tinea looks, acts, and responds to treatment.
Boni E. Elewski, MD, professor and chair of dermatology, at The University of Alabama at Birmingham, saw her first case of probable T indotineae in a patient in early 2020. “He was covered with fine scale, and he itched all over. I thought he had atopic dermatitis. This didn’t look like any tinea. His face, arms, back, and legs were scaly.”
Nevertheless, KOH and biopsy confirmed dermatophytosis. Culture (performed at the Center for Medical Mycology [CMM] in Cleveland) identified T mentagrophytes. Back then, Elewski told this news organization, labs did not routinely go beyond genus and species. But based on the patient’s symptoms, history of unresponsiveness to terbinafine, borderline sensitivity to fluconazole, and travel to India and Spain, Elewski strongly suspected T indotineae.
The patient refused itraconazole, to which the fungus was sensitive, and did not respond to fluconazole 400 mg daily. Ultimately, he was lost to follow-up. “Last I saw him,” said Elewski, “he was not cured.”
Tracking Cases
Because T indotineae does not require reporting to public health agencies, said Jeremy Gold, MD, MS, a medical officer with the US Centers for Disease Control and Prevention (CDC) Mycotic Diseases Branch in Atlanta, “there is no official public health surveillance keeping track of exactly how many cases have occurred.”
The same is true for TMVII and terbinafine-resistant T rubrum, which are also on the rise. Regarding T indotineae, authors from the University of Texas Health Science Center at San Antonio retrospectively reported 21 terbinafine-resistant isolates from North America in the July 2023 Journal of Clinical Microbiology .
Caplan has seen approximately 12 T indotineae cases to date, including the first two confirmed US cases, which he and co-authors, including Gold, reported in the CDC’s Morbidity and Mortality Weekly Report in May 2023. T indotineae is likely underreported, he said, because it eludes standard culture-based techniques, and identifying it requires molecular testing, which is available at only a handful of labs nationally.
To help educate providers, in July, the American Academy of Dermatology (AAD) and the International League of Dermatological Societies unveiled an Emerging Diseases Resource Center, which includes resources for providers and a registry for reporting confirmed and suspected resistant dermatophytes.
“Our goal is to provide easy-to-access and easy-to-understand resources to healthcare providers,” Esther Freeman, MD, PhD, told this news organization. She is director of Global Health Dermatology at Massachusetts General Hospital, associate professor of dermatology at Harvard Medical School, both in Boston, and chair of the AAD’s Emerging Diseases Task Force.
“Our resources include an algorithm for when to suspect a drug-resistant case and how to think through treatment options. We cover issues related to diagnosis and treatment, as well as linking to our case registry reporting system,” said Freeman.
The new registry resides within the AAD’s existing COVID-19, Mpox, and Emerging Infections Registry. “Our registry efforts have already captured 2500 COVID-19 and mpox cases from 72 different countries,” Freeman said. For all these infections, she added, “we hope that real-time data analysis of cases worldwide will provide information that helps physicians recognize and treat cases.”
Consistent with the registry’s approach, said Caplan and Gold, there is no silver bullet for battling dermatophyte resistance. What is needed, said Gold, is a coordinated approach involving public health officials, dermatologists, primary care providers, infectious disease specialists, pharmacists, and patients. “It’s going to be a team effort to address the challenge of emerging complex dermatophytosis,” he said.
Resistant T rubrum
“The biggest difference with T rubrum resistance is you may not see that widespread infection that we see with T indotineae,” said Caplan. T rubrum is probably the most common dermatophyte that dermatologists see, added Elewski, who encounters a resistant case at least monthly. One such patient, featured in a January 2021 British Journal of Dermatology research letter, cleared on itraconazole and ciclopirox cream but subsequently returned with itraconazole-resistant T rubrum because he had been doctor-shopping for the drug intermittently for years, she said. He cleared on posaconazole 300 mg daily, then was lost to follow-up.
TMVII
A 2023 Emerging Infectious Diseases report highlighted the potential for this dermatophyte to spread among men who have sex with men (MSM), presenting as an itchy, scaly rash affecting the pubic, genital, and buttocks skin. “People don’t generally think of a fungal infection as something that could behave like a sexually transmitted infection (STI),” said Gold.
Caplan and coauthors recently reported the first confirmed US TMVII case in JAMA Dermatology. Many experts suspect that unreported US cases existed previously, he said. “When it circulates in Europe and there’s so much travel, it’s probably here too.”
The fact that T indotineae was formerly called TMVIII has created confusion, added Caplan. “I’ve had patients say, ‘I’m worried I have that resistant ringworm that’s spreading among MSM.’ Whenever we talk about STIs and introduce the word ‘resistant,’ that comes with the potential for stigma, anxiety, and concern.” Fortunately, he said, TMVII has shown no resistance to first-line antifungals.
Why the Rise
Gold said, “We don’t know for sure why we’re seeing these different drug-resistant species popping up.” One possibility, he said, is the common misuse and overuse of topical antifungals — especially those available overseas in combination with high-potency steroids, such as clobetasol. Consumers use these products for a few weeks until symptoms resolve, then reapply them off and on over years, fueling resistance, said Gold.
“We are worried that with warming temperatures, there’s potential to see expansion of the geographic range of epidemic fungi,” he added. “That could be part of what has fueled recent increases in resistant dermatophytes. But it’s hard to prove.”
Climate change may be behind the emergence of Candida auris, according to a 2022 article in The Lancet Regional Health – Americas. This potentially fatal multidrug-resistant infection spreads easily among sick patients in healthcare facilities, according to a CDC information page on C auris.
Confirming Dermatophyte Infection
“A biopsy will only confirm the presence of fungus,” said Elewski. “Here you will need a lab that knows how to do a fungal culture.” Most state laboratories can do this, she said, as can some hospitals and special labs such as CMM in Cleveland.
It takes a Clinical Laboratory Improvement Amendments–certified lab to perform KOH prep in-house, added Caplan, plus up-to-date gear and knowledge of where and how to scrape and what to look for microscopically. Moreover, identifying T indotineae requires molecular testing available at only a handful of laboratories — listed on the AAD Emerging Dermatophytes webpage — nationwide.
Nevertheless, said Caplan, nailing down a diagnosis can guide treatment, often supplanting empirically prescribed antifungal steroid creams. “Those are probably not going to help. And people may be using those on areas of the body they shouldn’t. Both the clinical clues and the steps to make the diagnosis need to come together. But that’s often easier said than done, especially in a busy practice.”
Identifying resistance requires antifungal sensitivity testing, he added, which few labs perform. “Practically speaking,” said Elewski, “if the patient failed terbinafine, I would try itraconazole. You don’t necessarily need proof” of resistance. But if a patient does not respond to itraconazole and terbinafine clinically, she said that she might consider fungal susceptibility testing.
Treatment Tips
To address any resistant dermatophyte, Elewski recommended getting comfortable with itraconazole. For decades, she said, dermatologists have avoided itraconazole because terbinafine typically costs patients $10 for 3 months. “Itraconazole could be $200 per month,” said Elewski. Because of potential drug-drug interactions and absorption issues — and a boxed warning regarding congestive heart failure — physicians historically reserved itraconazole for severe fungal infections.
Itraconazole labeled dosing for onychomycosis is 200 mg daily for 12 weeks. Elewski favors a two-pronged attack, often combining an -azole antifungal with topical ciclopirox.
Another element that emerging tinea pathogens share is slower response to treatment. For T indotineae, reports appearing in the Journal of the American Academy of Dermatology in 2022 and 2024 suggest duration from 6-8 weeks up to 20 weeks.
To avoid recurrences of resistant T rubrum, Elewski treats for a year. However, she has problems getting itraconazole approved, when often it is the only agent that works. “I’ve written more letters than I like to insurance companies” to document terbinafine failure, she said.
Rarely, said Gold, dermatophyte infections resist both terbinafine and itraconazole. Next-line agents such as voriconazole, which some dermatologists have used for resistant T indotineae, can be much harder to tolerate, with more drug interactions, he said.
And because itraconazole, voriconazole, and posaconazole are all triazoles, added Elewski, the latter two might not work better than the former. But because these drugs might outperform itraconazole in selected cases, she said, “that’s when you want to do fungal susceptibility testing.”
TMVII is so new, said Caplan, that optimal therapy duration remains unclear. “One of the challenges with TMVII is when it gets into the genital skin, it’s a hair-bearing area. And based on various grooming practices, there’s an opportunity for the tinea to get deeper into the hair follicle and dermis. That may also be true of T indotineae.”
Anemic Arsenal
Unfortunately, said Gold, the arsenal of antifungals available in the United States remains limited. “Depending on how you count, there are only three to four classes of antifungal drugs designed to treat severe or invasive infections. So whenever we hear about a new fungal pathogen that’s causing resistant infections, it causes public health concern.”
Promising drugs in development include olorofim (F2G) and fosmanogepix (Basilea), according to Gold. However, he said, the development of these drugs to date has targeted invasive fungal infections such as aspergillosis. In June 2023, the Food and Drug Administration rejected the new drug application for olorofim, requesting additional data and analyses. Regarding fosmanogepix, a double-blinded noninferiority phase 3 trial in invasive yeast infections was recently launched, according to a September 24 press release.
Gold, Caplan, and Elewski reported no relevant financial disclosures. Freeman is a COVID-19 co-author for UpToDate and chair of the AAD Emerging Diseases Task Force.
A version of this article appeared on Medscape.com.
A Dermatologist’s Tips for Supporting LGBTQ Youth
HUNTINGTON BEACH, CALIFORNIA —
“Sometimes in dermatology we might say, ‘This gender care stuff, that’s really for pediatricians and primary care doctors,’ ” Boos, a pediatric dermatologist at Seattle Children’s Hospital, Seattle, said at the annual meeting of the Pacific Dermatologic Association. However, he added, “gender-affirming care happens not only with medications but with communication, curiosity, and respect.” For instance, an LGBTQ patient who is being treated with isotretinoin for acne is seen once a month by a dermatologist, which is probably more frequent than seeing their primary care physician, he said. “Every time you see that child, you can make them feel seen. You can respect them. You can let them know that you care about them. Hopefully then they understand what it feels like to get good care from a provider and then will not settle for poor care from someone else.”
According to Gallup polling, the proportion of people in the United States who identify as non-cisgender or nonheterosexual increased from 3.5% in 2012 to 7% in 2021. “The estimation is that 2.5%-3.5% of all teenagers identify as gay or bisexual, and another 1% identify as transgender, though some studies estimate the percentage of gender diverse youth to be as high as 9.2%,” said Boos.

He discussed several barriers to dermatologic care for LGBTQ youth, including availability. “There are only about 400 practicing pediatric dermatologists in the US, so there’s not a lot of pediatric dermatology care to go around for any child,” Boos said. “My plea to general dermatologists who see adolescents and teenagers: You can care for LGBTQ adolescents; they need your help.”
Accessibility is also an issue. For example, his clinic is in a wealthy and somewhat isolated area of Seattle, “which makes it hard for some patients to access our services because they may have to drive from far away or take multiple modes of public transportation to see us,” explained Boos, who came out as gay about 10 years ago after beginning his practice in Seattle. “Time matters, too. Children are in school. They don’t necessarily want to take time off to go to the doctor’s office. We want to make sure we have services at different times of day, including evenings or weekends if possible.”
Another potential barrier to care for this patient population is acceptability. “I can say that I welcome any patient to my practice, but if I’m not humble and informed about their concerns, especially queer or trans kids, if they feel that I’m not respecting them, that’s going to be a huge problem,” Boos said. “They won’t view that care as acceptable, and they’re not going to come back if they feel like I’m not looking out for their best interests.”
In a large cross-sectional study of patients with chronic inflammatory skin diseases published in 2023, sexual and gender minority (SGM) individuals were significantly more likely than non-SGM individuals to delay specialist care including dermatologic care (adjusted odds ratio [AOR], 1.23), mental health care (AOR, 1.62), and filing a prescription (AOR, 1.30) because of cost. The barriers for SGM patients were transportation issues, not having a healthcare practitioner (HCP) from the same racial or ethnic background, “and they were more likely to report not always being treated with respect by HCPs,” said Boos, who was not involved with the study. “SGM patients of minoritized racial identities such as Black, Hispanic, and Latino were also more likely to experience barriers to care.”
Boos offered several tips for improving the dermatologic care of LGBTQ youth:
Use inclusive language and follow your patient’s lead. “There are many ways that people identify, both with respect to their sexual orientation and their gender identity,” he said. “We often think that a person is either gay or straight, or cisgender or transgender. There are many folks who reject these binaries and may view their gender identity or sexual orientation outside of these descriptors. You can be bisexual. You can be asexual.” He also emphasized that sexual orientation is different from sexual behavior.
Be deliberate about your phrasing. Boos said he strives to make new patients feel comfortable by asking them such questions as what pronouns they use, how he should address them, and whether they have a partner or are in a relationship. “Then, in general, just follow your patient’s lead,” Boos said. “If they’re referring to their partner in a certain way or to themselves with certain pronouns, go along with it. When in doubt, just ask. And if you make a mistake like using the wrong pronouns or name of a patient, the best thing to do is immediately apologize and try your best not to repeat that error.”
When asking about sexual practices, don’t make assumptions. Boos recommends a 2019 article on dermatologic care of LGBT persons, published in the Journal of the American Academy of Dermatology, which includes specific examples of how to elicit a sexual history from adults and teens. One of the recommendations is “to be very direct, say, ‘This may feel uncomfortable, but I have to ask you these direct questions about what you’re doing sexually because I need to understand if you’re at risk for things like sexually transmitted infections,’ ” Boos said. “It’s also important to use terminology that our patients know. If I ask someone if they’ve had sex before, they usually understand that as penile-vaginal intercourse, but it’s also important to understand if they have oral or anal sex. But if you ask, ‘Have you had insertive anal sex?’ they may not know what that means as opposed to receptive anal sex. Instead, you might ask, ‘Are you a top or a bottom?’ which are more commonly used and understood terms in the queer community. It may feel really uncomfortable to use that kind of language, but we want to make sure patients understand what we’re asking them so we can take the best possible care of them.”
Pay attention to the details. One way to demonstrate inclusivity in your practice includes collecting pronoun and sexual orientation information for the electronic medical record so your entire staff can use proper pronouns for the patient. “Also, acknowledge that for queer folks, family can mean more than just biological family,” Boos added. “I do not buy into the stereotype that all queer kids are ostracized from their families and not loved by their families, but it is true that they are at risk for those experiences. So, sometimes a member of the patient’s ‘chosen family’ accompanies them on their visit.”
Privacy is also key. “You never know who else is in the room when you’re on a telehealth call, so you need to address that before you ask about personal things,” Boos said. “One sticking point that can also come up is that parents often fill out their child’s patient demographic form, which may not tell the real story. I typically start to have confidential time without parents and may take a sexual history as early as 12 or 13 years of age if it’s a patient that I’m seeing for an extended period or if I’m worried about a skin finding that might suggest an STI.”
He highlighted the unique opportunity dermatologists have to transform the healthcare landscape for LGBTQ children and adolescents. “It’s about extending yourself to nurture the growth of another person,” Boos said. “This can feel challenging, but you want to see each person for who they are and help get them to where they want to go. That’s what we went into medicine for, right? We want to care about people.”
Boos had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
HUNTINGTON BEACH, CALIFORNIA —
“Sometimes in dermatology we might say, ‘This gender care stuff, that’s really for pediatricians and primary care doctors,’ ” Boos, a pediatric dermatologist at Seattle Children’s Hospital, Seattle, said at the annual meeting of the Pacific Dermatologic Association. However, he added, “gender-affirming care happens not only with medications but with communication, curiosity, and respect.” For instance, an LGBTQ patient who is being treated with isotretinoin for acne is seen once a month by a dermatologist, which is probably more frequent than seeing their primary care physician, he said. “Every time you see that child, you can make them feel seen. You can respect them. You can let them know that you care about them. Hopefully then they understand what it feels like to get good care from a provider and then will not settle for poor care from someone else.”
According to Gallup polling, the proportion of people in the United States who identify as non-cisgender or nonheterosexual increased from 3.5% in 2012 to 7% in 2021. “The estimation is that 2.5%-3.5% of all teenagers identify as gay or bisexual, and another 1% identify as transgender, though some studies estimate the percentage of gender diverse youth to be as high as 9.2%,” said Boos.
He discussed several barriers to dermatologic care for LGBTQ youth, including availability. “There are only about 400 practicing pediatric dermatologists in the US, so there’s not a lot of pediatric dermatology care to go around for any child,” Boos said. “My plea to general dermatologists who see adolescents and teenagers: You can care for LGBTQ adolescents; they need your help.”
Accessibility is also an issue. For example, his clinic is in a wealthy and somewhat isolated area of Seattle, “which makes it hard for some patients to access our services because they may have to drive from far away or take multiple modes of public transportation to see us,” explained Boos, who came out as gay about 10 years ago after beginning his practice in Seattle. “Time matters, too. Children are in school. They don’t necessarily want to take time off to go to the doctor’s office. We want to make sure we have services at different times of day, including evenings or weekends if possible.”
Another potential barrier to care for this patient population is acceptability. “I can say that I welcome any patient to my practice, but if I’m not humble and informed about their concerns, especially queer or trans kids, if they feel that I’m not respecting them, that’s going to be a huge problem,” Boos said. “They won’t view that care as acceptable, and they’re not going to come back if they feel like I’m not looking out for their best interests.”
In a large cross-sectional study of patients with chronic inflammatory skin diseases published in 2023, sexual and gender minority (SGM) individuals were significantly more likely than non-SGM individuals to delay specialist care including dermatologic care (adjusted odds ratio [AOR], 1.23), mental health care (AOR, 1.62), and filing a prescription (AOR, 1.30) because of cost. The barriers for SGM patients were transportation issues, not having a healthcare practitioner (HCP) from the same racial or ethnic background, “and they were more likely to report not always being treated with respect by HCPs,” said Boos, who was not involved with the study. “SGM patients of minoritized racial identities such as Black, Hispanic, and Latino were also more likely to experience barriers to care.”
Boos offered several tips for improving the dermatologic care of LGBTQ youth:
Use inclusive language and follow your patient’s lead. “There are many ways that people identify, both with respect to their sexual orientation and their gender identity,” he said. “We often think that a person is either gay or straight, or cisgender or transgender. There are many folks who reject these binaries and may view their gender identity or sexual orientation outside of these descriptors. You can be bisexual. You can be asexual.” He also emphasized that sexual orientation is different from sexual behavior.
Be deliberate about your phrasing. Boos said he strives to make new patients feel comfortable by asking them such questions as what pronouns they use, how he should address them, and whether they have a partner or are in a relationship. “Then, in general, just follow your patient’s lead,” Boos said. “If they’re referring to their partner in a certain way or to themselves with certain pronouns, go along with it. When in doubt, just ask. And if you make a mistake like using the wrong pronouns or name of a patient, the best thing to do is immediately apologize and try your best not to repeat that error.”
When asking about sexual practices, don’t make assumptions. Boos recommends a 2019 article on dermatologic care of LGBT persons, published in the Journal of the American Academy of Dermatology, which includes specific examples of how to elicit a sexual history from adults and teens. One of the recommendations is “to be very direct, say, ‘This may feel uncomfortable, but I have to ask you these direct questions about what you’re doing sexually because I need to understand if you’re at risk for things like sexually transmitted infections,’ ” Boos said. “It’s also important to use terminology that our patients know. If I ask someone if they’ve had sex before, they usually understand that as penile-vaginal intercourse, but it’s also important to understand if they have oral or anal sex. But if you ask, ‘Have you had insertive anal sex?’ they may not know what that means as opposed to receptive anal sex. Instead, you might ask, ‘Are you a top or a bottom?’ which are more commonly used and understood terms in the queer community. It may feel really uncomfortable to use that kind of language, but we want to make sure patients understand what we’re asking them so we can take the best possible care of them.”
Pay attention to the details. One way to demonstrate inclusivity in your practice includes collecting pronoun and sexual orientation information for the electronic medical record so your entire staff can use proper pronouns for the patient. “Also, acknowledge that for queer folks, family can mean more than just biological family,” Boos added. “I do not buy into the stereotype that all queer kids are ostracized from their families and not loved by their families, but it is true that they are at risk for those experiences. So, sometimes a member of the patient’s ‘chosen family’ accompanies them on their visit.”
Privacy is also key. “You never know who else is in the room when you’re on a telehealth call, so you need to address that before you ask about personal things,” Boos said. “One sticking point that can also come up is that parents often fill out their child’s patient demographic form, which may not tell the real story. I typically start to have confidential time without parents and may take a sexual history as early as 12 or 13 years of age if it’s a patient that I’m seeing for an extended period or if I’m worried about a skin finding that might suggest an STI.”
He highlighted the unique opportunity dermatologists have to transform the healthcare landscape for LGBTQ children and adolescents. “It’s about extending yourself to nurture the growth of another person,” Boos said. “This can feel challenging, but you want to see each person for who they are and help get them to where they want to go. That’s what we went into medicine for, right? We want to care about people.”
Boos had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
HUNTINGTON BEACH, CALIFORNIA —
“Sometimes in dermatology we might say, ‘This gender care stuff, that’s really for pediatricians and primary care doctors,’ ” Boos, a pediatric dermatologist at Seattle Children’s Hospital, Seattle, said at the annual meeting of the Pacific Dermatologic Association. However, he added, “gender-affirming care happens not only with medications but with communication, curiosity, and respect.” For instance, an LGBTQ patient who is being treated with isotretinoin for acne is seen once a month by a dermatologist, which is probably more frequent than seeing their primary care physician, he said. “Every time you see that child, you can make them feel seen. You can respect them. You can let them know that you care about them. Hopefully then they understand what it feels like to get good care from a provider and then will not settle for poor care from someone else.”
According to Gallup polling, the proportion of people in the United States who identify as non-cisgender or nonheterosexual increased from 3.5% in 2012 to 7% in 2021. “The estimation is that 2.5%-3.5% of all teenagers identify as gay or bisexual, and another 1% identify as transgender, though some studies estimate the percentage of gender diverse youth to be as high as 9.2%,” said Boos.
He discussed several barriers to dermatologic care for LGBTQ youth, including availability. “There are only about 400 practicing pediatric dermatologists in the US, so there’s not a lot of pediatric dermatology care to go around for any child,” Boos said. “My plea to general dermatologists who see adolescents and teenagers: You can care for LGBTQ adolescents; they need your help.”
Accessibility is also an issue. For example, his clinic is in a wealthy and somewhat isolated area of Seattle, “which makes it hard for some patients to access our services because they may have to drive from far away or take multiple modes of public transportation to see us,” explained Boos, who came out as gay about 10 years ago after beginning his practice in Seattle. “Time matters, too. Children are in school. They don’t necessarily want to take time off to go to the doctor’s office. We want to make sure we have services at different times of day, including evenings or weekends if possible.”
Another potential barrier to care for this patient population is acceptability. “I can say that I welcome any patient to my practice, but if I’m not humble and informed about their concerns, especially queer or trans kids, if they feel that I’m not respecting them, that’s going to be a huge problem,” Boos said. “They won’t view that care as acceptable, and they’re not going to come back if they feel like I’m not looking out for their best interests.”
In a large cross-sectional study of patients with chronic inflammatory skin diseases published in 2023, sexual and gender minority (SGM) individuals were significantly more likely than non-SGM individuals to delay specialist care including dermatologic care (adjusted odds ratio [AOR], 1.23), mental health care (AOR, 1.62), and filing a prescription (AOR, 1.30) because of cost. The barriers for SGM patients were transportation issues, not having a healthcare practitioner (HCP) from the same racial or ethnic background, “and they were more likely to report not always being treated with respect by HCPs,” said Boos, who was not involved with the study. “SGM patients of minoritized racial identities such as Black, Hispanic, and Latino were also more likely to experience barriers to care.”
Boos offered several tips for improving the dermatologic care of LGBTQ youth:
Use inclusive language and follow your patient’s lead. “There are many ways that people identify, both with respect to their sexual orientation and their gender identity,” he said. “We often think that a person is either gay or straight, or cisgender or transgender. There are many folks who reject these binaries and may view their gender identity or sexual orientation outside of these descriptors. You can be bisexual. You can be asexual.” He also emphasized that sexual orientation is different from sexual behavior.
Be deliberate about your phrasing. Boos said he strives to make new patients feel comfortable by asking them such questions as what pronouns they use, how he should address them, and whether they have a partner or are in a relationship. “Then, in general, just follow your patient’s lead,” Boos said. “If they’re referring to their partner in a certain way or to themselves with certain pronouns, go along with it. When in doubt, just ask. And if you make a mistake like using the wrong pronouns or name of a patient, the best thing to do is immediately apologize and try your best not to repeat that error.”
When asking about sexual practices, don’t make assumptions. Boos recommends a 2019 article on dermatologic care of LGBT persons, published in the Journal of the American Academy of Dermatology, which includes specific examples of how to elicit a sexual history from adults and teens. One of the recommendations is “to be very direct, say, ‘This may feel uncomfortable, but I have to ask you these direct questions about what you’re doing sexually because I need to understand if you’re at risk for things like sexually transmitted infections,’ ” Boos said. “It’s also important to use terminology that our patients know. If I ask someone if they’ve had sex before, they usually understand that as penile-vaginal intercourse, but it’s also important to understand if they have oral or anal sex. But if you ask, ‘Have you had insertive anal sex?’ they may not know what that means as opposed to receptive anal sex. Instead, you might ask, ‘Are you a top or a bottom?’ which are more commonly used and understood terms in the queer community. It may feel really uncomfortable to use that kind of language, but we want to make sure patients understand what we’re asking them so we can take the best possible care of them.”
Pay attention to the details. One way to demonstrate inclusivity in your practice includes collecting pronoun and sexual orientation information for the electronic medical record so your entire staff can use proper pronouns for the patient. “Also, acknowledge that for queer folks, family can mean more than just biological family,” Boos added. “I do not buy into the stereotype that all queer kids are ostracized from their families and not loved by their families, but it is true that they are at risk for those experiences. So, sometimes a member of the patient’s ‘chosen family’ accompanies them on their visit.”
Privacy is also key. “You never know who else is in the room when you’re on a telehealth call, so you need to address that before you ask about personal things,” Boos said. “One sticking point that can also come up is that parents often fill out their child’s patient demographic form, which may not tell the real story. I typically start to have confidential time without parents and may take a sexual history as early as 12 or 13 years of age if it’s a patient that I’m seeing for an extended period or if I’m worried about a skin finding that might suggest an STI.”
He highlighted the unique opportunity dermatologists have to transform the healthcare landscape for LGBTQ children and adolescents. “It’s about extending yourself to nurture the growth of another person,” Boos said. “This can feel challenging, but you want to see each person for who they are and help get them to where they want to go. That’s what we went into medicine for, right? We want to care about people.”
Boos had no relevant financial disclosures.
A version of this article appeared on Medscape.com.
FROM PDA 2024
Industry Payments to Peer Reviewers Scrutinized at Four Major Medical Journals
TOPLINE:
More than half of the US peer reviewers for four major medical journals received industry payments between 2020-2022, new research shows. Altogether they received more than $64 million in general, non-research payments, with a median payment per physician of $7614. Research payments — including money paid directly to physicians as well as funds related to research for which a physician was registered as a principal investigator — exceeded $1 billion.
METHODOLOGY:
- Researchers identified peer reviewers in 2022 for The BMJ, JAMA, The Lancet, and The New England Journal of Medicine using each journal’s list of reviewers for that year. They included 1962 US-based physicians in their analysis.
- General and research payments made to the peer reviewers between 2020-2022 were extracted from the Open Payments database.
TAKEAWAY:
- Nearly 59% of the peer reviewers received industry payments between 2020-2022.
- Payments included $34.31 million in consulting fees and $11.8 million for speaking compensation unrelated to continuing medical education programs.
- Male reviewers received a significantly higher median total payment than did female reviewers ($38,959 vs $19,586). General payments were higher for men as well ($8663 vs $4183).
- For comparison, the median general payment to all physicians in 2018 was $216, the researchers noted.
IN PRACTICE:
“Additional research and transparency regarding industry payments in the peer review process are needed,” the authors of the study wrote.
SOURCE:
Christopher J. D. Wallis, MD, PhD, with the division of urology at the University of Toronto, Canada, was the corresponding author for the study. The article was published online October 10 in JAMA.
LIMITATIONS:
Whether the financial ties were relevant to any of the papers that the peer reviewers critiqued is not known. Some reviewers might have received additional payments from insurance and technology companies that were not captured in this study. The findings might not apply to other journals, the researchers noted.
DISCLOSURES:
Wallis disclosed personal fees from Janssen Oncology, Nanostics, Precision Point Specialty, Sesen Bio, AbbVie, Astellas, AstraZeneca, Bayer, EMD Serono, Knight Therapeutics, Merck, Science and Medicine Canada, TerSera, and Tolmar. He and some coauthors also disclosed support and grants from foundations and government institutions.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
More than half of the US peer reviewers for four major medical journals received industry payments between 2020-2022, new research shows. Altogether they received more than $64 million in general, non-research payments, with a median payment per physician of $7614. Research payments — including money paid directly to physicians as well as funds related to research for which a physician was registered as a principal investigator — exceeded $1 billion.
METHODOLOGY:
- Researchers identified peer reviewers in 2022 for The BMJ, JAMA, The Lancet, and The New England Journal of Medicine using each journal’s list of reviewers for that year. They included 1962 US-based physicians in their analysis.
- General and research payments made to the peer reviewers between 2020-2022 were extracted from the Open Payments database.
TAKEAWAY:
- Nearly 59% of the peer reviewers received industry payments between 2020-2022.
- Payments included $34.31 million in consulting fees and $11.8 million for speaking compensation unrelated to continuing medical education programs.
- Male reviewers received a significantly higher median total payment than did female reviewers ($38,959 vs $19,586). General payments were higher for men as well ($8663 vs $4183).
- For comparison, the median general payment to all physicians in 2018 was $216, the researchers noted.
IN PRACTICE:
“Additional research and transparency regarding industry payments in the peer review process are needed,” the authors of the study wrote.
SOURCE:
Christopher J. D. Wallis, MD, PhD, with the division of urology at the University of Toronto, Canada, was the corresponding author for the study. The article was published online October 10 in JAMA.
LIMITATIONS:
Whether the financial ties were relevant to any of the papers that the peer reviewers critiqued is not known. Some reviewers might have received additional payments from insurance and technology companies that were not captured in this study. The findings might not apply to other journals, the researchers noted.
DISCLOSURES:
Wallis disclosed personal fees from Janssen Oncology, Nanostics, Precision Point Specialty, Sesen Bio, AbbVie, Astellas, AstraZeneca, Bayer, EMD Serono, Knight Therapeutics, Merck, Science and Medicine Canada, TerSera, and Tolmar. He and some coauthors also disclosed support and grants from foundations and government institutions.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
More than half of the US peer reviewers for four major medical journals received industry payments between 2020-2022, new research shows. Altogether they received more than $64 million in general, non-research payments, with a median payment per physician of $7614. Research payments — including money paid directly to physicians as well as funds related to research for which a physician was registered as a principal investigator — exceeded $1 billion.
METHODOLOGY:
- Researchers identified peer reviewers in 2022 for The BMJ, JAMA, The Lancet, and The New England Journal of Medicine using each journal’s list of reviewers for that year. They included 1962 US-based physicians in their analysis.
- General and research payments made to the peer reviewers between 2020-2022 were extracted from the Open Payments database.
TAKEAWAY:
- Nearly 59% of the peer reviewers received industry payments between 2020-2022.
- Payments included $34.31 million in consulting fees and $11.8 million for speaking compensation unrelated to continuing medical education programs.
- Male reviewers received a significantly higher median total payment than did female reviewers ($38,959 vs $19,586). General payments were higher for men as well ($8663 vs $4183).
- For comparison, the median general payment to all physicians in 2018 was $216, the researchers noted.
IN PRACTICE:
“Additional research and transparency regarding industry payments in the peer review process are needed,” the authors of the study wrote.
SOURCE:
Christopher J. D. Wallis, MD, PhD, with the division of urology at the University of Toronto, Canada, was the corresponding author for the study. The article was published online October 10 in JAMA.
LIMITATIONS:
Whether the financial ties were relevant to any of the papers that the peer reviewers critiqued is not known. Some reviewers might have received additional payments from insurance and technology companies that were not captured in this study. The findings might not apply to other journals, the researchers noted.
DISCLOSURES:
Wallis disclosed personal fees from Janssen Oncology, Nanostics, Precision Point Specialty, Sesen Bio, AbbVie, Astellas, AstraZeneca, Bayer, EMD Serono, Knight Therapeutics, Merck, Science and Medicine Canada, TerSera, and Tolmar. He and some coauthors also disclosed support and grants from foundations and government institutions.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
The Game We Play Every Day
Words do have power. Names have power. Words are events, they do things, change things. They transform both speaker and hearer ... They feed understanding or emotion back and forth and amplify it. — Ursula K. Le Guin
Every medical student should have a class in linguistics. I’m just unsure what it might replace. Maybe physiology? (When was the last time you used Fick’s or Fourier’s Laws anyway?). Even if we don’t supplant any core curriculum, it’s worth noting that we spend more time in our daily work calculating how to communicate things than calculating cardiac outputs. That we can convey so much so consistently and without specific training is a marvel. Making the diagnosis or a plan is often the easy part.
Linguistics is a broad field. At its essence, it studies how we communicate. It’s fascinating how we use tone, word choice, gestures, syntax, and grammar to explain, reassure, instruct or implore patients. Medical appointments are sometimes high stakes and occur within a huge variety of circumstances. In a single day of clinic, I had a patient with dementia, and one pursuing a PhD in P-Chem. I had English speakers, second language English speakers, and a Vietnamese patient who knew no English. In just one day, I explained things to toddlers and adults, a Black woman from Oklahoma and a Jewish woman from New York. For a brief few minutes, each of them was my partner in a game of medical charades. For each one, I had to figure out how to get them to know what I’m thinking.

I learned of this game of charades concept from a podcast featuring Morten Christiansen, professor of psychology at Cornell University, and professor in Cognitive Science of Language, at Aarhus University, Denmark. The idea is that language can be thought of as a game where speakers constantly improvise based on the topic, each one’s expertise, and the shared understanding. I found this intriguing. In his explanation, grammar and definitions are less important than the mutual understanding of what is being communicated. It helps explain the wide variations of speech even among those speaking the same language. It also flips the idea that brains are designed for language, a concept proposed by linguistic greats such as Noam Chomsky and Steven Pinker. Rather, what we call language is just the best solution our brains could create to convey information.
I thought about how each of us instinctively varies the complexity of sentences and tone of voice based on the ability of each patient to understand. Gestures, storytelling and analogies are linguistic tools we use without thinking about them. We’ve a unique communications conundrum in that we often need patients to understand a complex idea, but only have minutes to get them there. We don’t want them to panic. We also don’t want them to be so dispassionate as to not act. To speed things up, we often use a technique known as chunking, short phrases that capture an idea in one bite. For example, “soak and smear” to get atopic patients to moisturize or “scrape and burn” to describe a curettage and electrodesiccation of a basal cell carcinoma or “a stick and a burn” before injecting them (I never liked that one). These are pithy, efficient. But they don’t always work.
One afternoon I had a 93-year-old woman with glossodynia. She had dementia and her 96-year-old husband was helping. When I explained how she’d “swish and spit” her magic mouthwash, he looked perplexed. Is she swishing a wand or something? I shook my head, “No” and gestured with my hands palms down, waving back and forth. It is just a mouthwash. She should rinse, then spit it out. I lost that round.
Then a 64-year-old woman whom I had to advise that the pink bump on her arm was a cutaneous neuroendocrine tumor. Do I call it a Merkel cell carcinoma? Do I say, “You know, like the one Jimmy Buffett had?” (Nope, not a good use of storytelling). She wanted to know how she got it. Sun exposure, we think. Or, perhaps a virus. Just how does one explain a virus called MCPyV that is ubiquitous but somehow caused cancer just for you? How do you convey, “This is serious, but you might not die like Jimmy Buffett?” I had to use all my language skills to get this right.
Then there is the Henderson-Hasselbalch problem of linguistics: communicating through a translator. When doing so, I’m cognizant of choosing short, simple sentences. Subject, verb, object. First this, then that. This mitigates what’s lost in translation and reduces waiting for translations (especially when your patient is storytelling in paragraphs). But try doing this with an emotionally wrought condition like alopecia. Finding the fewest words to convey that your FSH and estrogen levels are irrelevant to your telogen effluvium to a Vietnamese speaker is tricky. “Yes, I see your primary care physician ordered these tests. No, the numbers do not matter.” Did that translate as they are normal? Or that they don’t matter because she is 54? Or that they don’t matter to me because I didn’t order them?
When you find yourself exhausted at the day’s end, perhaps you’ll better appreciate how it was not only the graduate level medicine you did today; you’ve practically got a PhD in linguistics as well. You just didn’t realize it.
Dr. Benabio is chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on X. Write to him at [email protected].
Words do have power. Names have power. Words are events, they do things, change things. They transform both speaker and hearer ... They feed understanding or emotion back and forth and amplify it. — Ursula K. Le Guin
Every medical student should have a class in linguistics. I’m just unsure what it might replace. Maybe physiology? (When was the last time you used Fick’s or Fourier’s Laws anyway?). Even if we don’t supplant any core curriculum, it’s worth noting that we spend more time in our daily work calculating how to communicate things than calculating cardiac outputs. That we can convey so much so consistently and without specific training is a marvel. Making the diagnosis or a plan is often the easy part.
Linguistics is a broad field. At its essence, it studies how we communicate. It’s fascinating how we use tone, word choice, gestures, syntax, and grammar to explain, reassure, instruct or implore patients. Medical appointments are sometimes high stakes and occur within a huge variety of circumstances. In a single day of clinic, I had a patient with dementia, and one pursuing a PhD in P-Chem. I had English speakers, second language English speakers, and a Vietnamese patient who knew no English. In just one day, I explained things to toddlers and adults, a Black woman from Oklahoma and a Jewish woman from New York. For a brief few minutes, each of them was my partner in a game of medical charades. For each one, I had to figure out how to get them to know what I’m thinking.
I learned of this game of charades concept from a podcast featuring Morten Christiansen, professor of psychology at Cornell University, and professor in Cognitive Science of Language, at Aarhus University, Denmark. The idea is that language can be thought of as a game where speakers constantly improvise based on the topic, each one’s expertise, and the shared understanding. I found this intriguing. In his explanation, grammar and definitions are less important than the mutual understanding of what is being communicated. It helps explain the wide variations of speech even among those speaking the same language. It also flips the idea that brains are designed for language, a concept proposed by linguistic greats such as Noam Chomsky and Steven Pinker. Rather, what we call language is just the best solution our brains could create to convey information.
I thought about how each of us instinctively varies the complexity of sentences and tone of voice based on the ability of each patient to understand. Gestures, storytelling and analogies are linguistic tools we use without thinking about them. We’ve a unique communications conundrum in that we often need patients to understand a complex idea, but only have minutes to get them there. We don’t want them to panic. We also don’t want them to be so dispassionate as to not act. To speed things up, we often use a technique known as chunking, short phrases that capture an idea in one bite. For example, “soak and smear” to get atopic patients to moisturize or “scrape and burn” to describe a curettage and electrodesiccation of a basal cell carcinoma or “a stick and a burn” before injecting them (I never liked that one). These are pithy, efficient. But they don’t always work.
One afternoon I had a 93-year-old woman with glossodynia. She had dementia and her 96-year-old husband was helping. When I explained how she’d “swish and spit” her magic mouthwash, he looked perplexed. Is she swishing a wand or something? I shook my head, “No” and gestured with my hands palms down, waving back and forth. It is just a mouthwash. She should rinse, then spit it out. I lost that round.
Then a 64-year-old woman whom I had to advise that the pink bump on her arm was a cutaneous neuroendocrine tumor. Do I call it a Merkel cell carcinoma? Do I say, “You know, like the one Jimmy Buffett had?” (Nope, not a good use of storytelling). She wanted to know how she got it. Sun exposure, we think. Or, perhaps a virus. Just how does one explain a virus called MCPyV that is ubiquitous but somehow caused cancer just for you? How do you convey, “This is serious, but you might not die like Jimmy Buffett?” I had to use all my language skills to get this right.
Then there is the Henderson-Hasselbalch problem of linguistics: communicating through a translator. When doing so, I’m cognizant of choosing short, simple sentences. Subject, verb, object. First this, then that. This mitigates what’s lost in translation and reduces waiting for translations (especially when your patient is storytelling in paragraphs). But try doing this with an emotionally wrought condition like alopecia. Finding the fewest words to convey that your FSH and estrogen levels are irrelevant to your telogen effluvium to a Vietnamese speaker is tricky. “Yes, I see your primary care physician ordered these tests. No, the numbers do not matter.” Did that translate as they are normal? Or that they don’t matter because she is 54? Or that they don’t matter to me because I didn’t order them?
When you find yourself exhausted at the day’s end, perhaps you’ll better appreciate how it was not only the graduate level medicine you did today; you’ve practically got a PhD in linguistics as well. You just didn’t realize it.
Dr. Benabio is chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on X. Write to him at [email protected].
Words do have power. Names have power. Words are events, they do things, change things. They transform both speaker and hearer ... They feed understanding or emotion back and forth and amplify it. — Ursula K. Le Guin
Every medical student should have a class in linguistics. I’m just unsure what it might replace. Maybe physiology? (When was the last time you used Fick’s or Fourier’s Laws anyway?). Even if we don’t supplant any core curriculum, it’s worth noting that we spend more time in our daily work calculating how to communicate things than calculating cardiac outputs. That we can convey so much so consistently and without specific training is a marvel. Making the diagnosis or a plan is often the easy part.
Linguistics is a broad field. At its essence, it studies how we communicate. It’s fascinating how we use tone, word choice, gestures, syntax, and grammar to explain, reassure, instruct or implore patients. Medical appointments are sometimes high stakes and occur within a huge variety of circumstances. In a single day of clinic, I had a patient with dementia, and one pursuing a PhD in P-Chem. I had English speakers, second language English speakers, and a Vietnamese patient who knew no English. In just one day, I explained things to toddlers and adults, a Black woman from Oklahoma and a Jewish woman from New York. For a brief few minutes, each of them was my partner in a game of medical charades. For each one, I had to figure out how to get them to know what I’m thinking.
I learned of this game of charades concept from a podcast featuring Morten Christiansen, professor of psychology at Cornell University, and professor in Cognitive Science of Language, at Aarhus University, Denmark. The idea is that language can be thought of as a game where speakers constantly improvise based on the topic, each one’s expertise, and the shared understanding. I found this intriguing. In his explanation, grammar and definitions are less important than the mutual understanding of what is being communicated. It helps explain the wide variations of speech even among those speaking the same language. It also flips the idea that brains are designed for language, a concept proposed by linguistic greats such as Noam Chomsky and Steven Pinker. Rather, what we call language is just the best solution our brains could create to convey information.
I thought about how each of us instinctively varies the complexity of sentences and tone of voice based on the ability of each patient to understand. Gestures, storytelling and analogies are linguistic tools we use without thinking about them. We’ve a unique communications conundrum in that we often need patients to understand a complex idea, but only have minutes to get them there. We don’t want them to panic. We also don’t want them to be so dispassionate as to not act. To speed things up, we often use a technique known as chunking, short phrases that capture an idea in one bite. For example, “soak and smear” to get atopic patients to moisturize or “scrape and burn” to describe a curettage and electrodesiccation of a basal cell carcinoma or “a stick and a burn” before injecting them (I never liked that one). These are pithy, efficient. But they don’t always work.
One afternoon I had a 93-year-old woman with glossodynia. She had dementia and her 96-year-old husband was helping. When I explained how she’d “swish and spit” her magic mouthwash, he looked perplexed. Is she swishing a wand or something? I shook my head, “No” and gestured with my hands palms down, waving back and forth. It is just a mouthwash. She should rinse, then spit it out. I lost that round.
Then a 64-year-old woman whom I had to advise that the pink bump on her arm was a cutaneous neuroendocrine tumor. Do I call it a Merkel cell carcinoma? Do I say, “You know, like the one Jimmy Buffett had?” (Nope, not a good use of storytelling). She wanted to know how she got it. Sun exposure, we think. Or, perhaps a virus. Just how does one explain a virus called MCPyV that is ubiquitous but somehow caused cancer just for you? How do you convey, “This is serious, but you might not die like Jimmy Buffett?” I had to use all my language skills to get this right.
Then there is the Henderson-Hasselbalch problem of linguistics: communicating through a translator. When doing so, I’m cognizant of choosing short, simple sentences. Subject, verb, object. First this, then that. This mitigates what’s lost in translation and reduces waiting for translations (especially when your patient is storytelling in paragraphs). But try doing this with an emotionally wrought condition like alopecia. Finding the fewest words to convey that your FSH and estrogen levels are irrelevant to your telogen effluvium to a Vietnamese speaker is tricky. “Yes, I see your primary care physician ordered these tests. No, the numbers do not matter.” Did that translate as they are normal? Or that they don’t matter because she is 54? Or that they don’t matter to me because I didn’t order them?
When you find yourself exhausted at the day’s end, perhaps you’ll better appreciate how it was not only the graduate level medicine you did today; you’ve practically got a PhD in linguistics as well. You just didn’t realize it.
Dr. Benabio is chief of dermatology at Kaiser Permanente San Diego. The opinions expressed in this column are his own and do not represent those of Kaiser Permanente. Dr. Benabio is @Dermdoc on X. Write to him at [email protected].
A Doctor Gets the Save When a Little League Umpire Collapses
Emergencies happen anywhere, anytime, and sometimes, medical professionals find themselves in situations where they are the only ones who can help. Is There a Doctor in the House? is a Medscape Medical News series telling these stories.
I sincerely believe that what goes around comes around. Good things come to good people. And sometimes that saves lives.
My 10-year-old son was in the semifinals of the Little League district championship. And we were losing. My son is an excellent pitcher, and he had started the game. But that night, he was struggling. He just couldn’t find where to throw the ball. Needless to say, he was frustrated.
He was changed to shortstop in the second inning, and the home plate umpire walked over to him. This umpire is well known in the area for his kindness and commitment, how he encourages the kids and helps make baseball fun even when it’s stressful.
We didn’t know him well, but he was really supportive of my kid in that moment, talking to him about how baseball is a team sport and we’re here to have fun. Just being really positive.
As the game continued, I saw the umpire suddenly walk to the side of the field. I hadn’t seen it, but he had been hit by a wild pitch on the side of his neck. He was wearing protective gear, but the ball managed to bounce up the side and caught bare neck. I knew something wasn’t right.
I went down to talk to him, and my medical assistant (MA), who was also at the game, came with me. I could tell the umpire was injured, but he didn’t want to leave the game. I suggested going to the hospital, but he wouldn’t consider it. So I sat there with my arms crossed, watching him.
His symptoms got worse. I could see he was in pain, and it was getting harder for him to speak.
Again, I strongly urged him to go to the hospital, but again, he said no.
In the sixth inning, things got bad enough that the umpire finally agreed to leave the game. As I was figuring out how to get him to the hospital, he disappeared on me. He had walked up to the second floor of the snack shack. My MA and I got him back downstairs and sat him on a bench behind home plate.
We were in the process of calling 911 ... when he arrested.
Luckily, when he lost vital signs, my MA and I were standing right next to him. We were able to activate ACLS protocol and start CPR within seconds.
Many times in these critical situations — especially if people are scared or have never seen an emergency like this — there’s the potential for chaos. Well, that was the polar opposite of what happened.
As soon as I started to run the code, there was this sense of order. People were keeping their composure and following directions. My MA and I would say, “this is what we need,” and the task would immediately be assigned to someone. It was quiet. There was no yelling. Everyone trusted me, even though some of them had never met me before. It was so surprising. I remember thinking, we’re running an arrest, but it’s so calm.
We were an organized team, and it really worked like clockwork, which was remarkable given where we were. It’s one thing to be in the hospital for an event like that. But to be on a baseball field where you have nothing is a completely different scenario.
Meanwhile, the game went on.
I had requested that all the kids be placed in the dugout when they weren’t on the field. So they saw the umpire walk off, but none of them saw him arrest. Some parents were really helpful with making sure the kids were okay.
The president of Oxford Little League ran across the street to a fire station to get an AED. But the fire department personnel were out on a call. He had to break down the door.
By the time he got back, the umpire’s vital signs were returning. And then EMS arrived.
They loaded him in the ambulance, and I called ahead to the trauma team, so they knew exactly what was happening.
I was pretty worried. My hypothesis was that there was probably compression on the vasculature, which had caused him to lose his vital signs. I thought he probably had an impending airway loss. I wasn’t sure if he was going to make it through the night.
What I didn’t know was that while I was giving CPR, my son stole home, and we won the game. As the ambulance was leaving, the celebration was going on in the outfield.
The umpire was in the hospital for several days. Early on, I got permission from his family to visit him. The first time I saw him, I felt this incredible gratitude and peace.
My dad was an ER doctor, and growing up, it seemed like every time we went on a family vacation, there was an emergency. We would be near a car accident or something, and my father would fly in and save the day. I remember being on the Autobahn somewhere in Europe, and there was a devastating accident between a car and a motorcycle. My father stabilized the guy, had him airlifted out, and apparently, he did fine. I grew up watching things like this and thinking, wow, that’s incredible.
Fast forward to 2 years ago, my father was diagnosed with a lung cancer he never should have had. He never smoked. As a cancer surgeon, I know we did everything in our power to save him. But it didn’t happen. He passed away.
I realize this is superstitious, but seeing the umpire alive, I had this feeling that somehow my dad was there. It was bittersweet but also a joyful moment — like I could breathe again.
I met the umpire’s family that first time, and it was like meeting family that you didn’t know you had but now you have forever. Even though the event was traumatic — I’m still trying not to be on high alert every time I go to a game — it felt like a gift to be part of this journey with them.
Little League’s mission is to teach kids about teamwork, leadership, and making good choices so communities are stronger. Our umpire is a guy who does that every day. He’s not a Little League umpire because he makes any money. He shows up at every single game to support these kids and engage them, to model respect, gratitude, and kindness.
I think our obligation as people is to live with intentionality. We all need to make sure we leave the world a better place, even when we are called upon to do uncomfortable things. Our umpire showed our kids what that looks like, and in that moment when he could have died, we were able to do the same for him.
Jennifer LaFemina, MD, is a surgical oncologist at UMass Memorial Medical Center in Massachusetts.
Are you a medical professional with a dramatic story outside the clinic? Medscape Medical News would love to consider your story for Is There a Doctor in the House? Please email your contact information and a short summary to [email protected].
A version of this article appeared on Medscape.com.
Emergencies happen anywhere, anytime, and sometimes, medical professionals find themselves in situations where they are the only ones who can help. Is There a Doctor in the House? is a Medscape Medical News series telling these stories.
I sincerely believe that what goes around comes around. Good things come to good people. And sometimes that saves lives.
My 10-year-old son was in the semifinals of the Little League district championship. And we were losing. My son is an excellent pitcher, and he had started the game. But that night, he was struggling. He just couldn’t find where to throw the ball. Needless to say, he was frustrated.
He was changed to shortstop in the second inning, and the home plate umpire walked over to him. This umpire is well known in the area for his kindness and commitment, how he encourages the kids and helps make baseball fun even when it’s stressful.
We didn’t know him well, but he was really supportive of my kid in that moment, talking to him about how baseball is a team sport and we’re here to have fun. Just being really positive.
As the game continued, I saw the umpire suddenly walk to the side of the field. I hadn’t seen it, but he had been hit by a wild pitch on the side of his neck. He was wearing protective gear, but the ball managed to bounce up the side and caught bare neck. I knew something wasn’t right.
I went down to talk to him, and my medical assistant (MA), who was also at the game, came with me. I could tell the umpire was injured, but he didn’t want to leave the game. I suggested going to the hospital, but he wouldn’t consider it. So I sat there with my arms crossed, watching him.
His symptoms got worse. I could see he was in pain, and it was getting harder for him to speak.
Again, I strongly urged him to go to the hospital, but again, he said no.
In the sixth inning, things got bad enough that the umpire finally agreed to leave the game. As I was figuring out how to get him to the hospital, he disappeared on me. He had walked up to the second floor of the snack shack. My MA and I got him back downstairs and sat him on a bench behind home plate.
We were in the process of calling 911 ... when he arrested.
Luckily, when he lost vital signs, my MA and I were standing right next to him. We were able to activate ACLS protocol and start CPR within seconds.
Many times in these critical situations — especially if people are scared or have never seen an emergency like this — there’s the potential for chaos. Well, that was the polar opposite of what happened.
As soon as I started to run the code, there was this sense of order. People were keeping their composure and following directions. My MA and I would say, “this is what we need,” and the task would immediately be assigned to someone. It was quiet. There was no yelling. Everyone trusted me, even though some of them had never met me before. It was so surprising. I remember thinking, we’re running an arrest, but it’s so calm.
We were an organized team, and it really worked like clockwork, which was remarkable given where we were. It’s one thing to be in the hospital for an event like that. But to be on a baseball field where you have nothing is a completely different scenario.
Meanwhile, the game went on.
I had requested that all the kids be placed in the dugout when they weren’t on the field. So they saw the umpire walk off, but none of them saw him arrest. Some parents were really helpful with making sure the kids were okay.
The president of Oxford Little League ran across the street to a fire station to get an AED. But the fire department personnel were out on a call. He had to break down the door.
By the time he got back, the umpire’s vital signs were returning. And then EMS arrived.
They loaded him in the ambulance, and I called ahead to the trauma team, so they knew exactly what was happening.
I was pretty worried. My hypothesis was that there was probably compression on the vasculature, which had caused him to lose his vital signs. I thought he probably had an impending airway loss. I wasn’t sure if he was going to make it through the night.
What I didn’t know was that while I was giving CPR, my son stole home, and we won the game. As the ambulance was leaving, the celebration was going on in the outfield.
The umpire was in the hospital for several days. Early on, I got permission from his family to visit him. The first time I saw him, I felt this incredible gratitude and peace.
My dad was an ER doctor, and growing up, it seemed like every time we went on a family vacation, there was an emergency. We would be near a car accident or something, and my father would fly in and save the day. I remember being on the Autobahn somewhere in Europe, and there was a devastating accident between a car and a motorcycle. My father stabilized the guy, had him airlifted out, and apparently, he did fine. I grew up watching things like this and thinking, wow, that’s incredible.
Fast forward to 2 years ago, my father was diagnosed with a lung cancer he never should have had. He never smoked. As a cancer surgeon, I know we did everything in our power to save him. But it didn’t happen. He passed away.
I realize this is superstitious, but seeing the umpire alive, I had this feeling that somehow my dad was there. It was bittersweet but also a joyful moment — like I could breathe again.
I met the umpire’s family that first time, and it was like meeting family that you didn’t know you had but now you have forever. Even though the event was traumatic — I’m still trying not to be on high alert every time I go to a game — it felt like a gift to be part of this journey with them.
Little League’s mission is to teach kids about teamwork, leadership, and making good choices so communities are stronger. Our umpire is a guy who does that every day. He’s not a Little League umpire because he makes any money. He shows up at every single game to support these kids and engage them, to model respect, gratitude, and kindness.
I think our obligation as people is to live with intentionality. We all need to make sure we leave the world a better place, even when we are called upon to do uncomfortable things. Our umpire showed our kids what that looks like, and in that moment when he could have died, we were able to do the same for him.
Jennifer LaFemina, MD, is a surgical oncologist at UMass Memorial Medical Center in Massachusetts.
Are you a medical professional with a dramatic story outside the clinic? Medscape Medical News would love to consider your story for Is There a Doctor in the House? Please email your contact information and a short summary to [email protected].
A version of this article appeared on Medscape.com.
Emergencies happen anywhere, anytime, and sometimes, medical professionals find themselves in situations where they are the only ones who can help. Is There a Doctor in the House? is a Medscape Medical News series telling these stories.
I sincerely believe that what goes around comes around. Good things come to good people. And sometimes that saves lives.
My 10-year-old son was in the semifinals of the Little League district championship. And we were losing. My son is an excellent pitcher, and he had started the game. But that night, he was struggling. He just couldn’t find where to throw the ball. Needless to say, he was frustrated.
He was changed to shortstop in the second inning, and the home plate umpire walked over to him. This umpire is well known in the area for his kindness and commitment, how he encourages the kids and helps make baseball fun even when it’s stressful.
We didn’t know him well, but he was really supportive of my kid in that moment, talking to him about how baseball is a team sport and we’re here to have fun. Just being really positive.
As the game continued, I saw the umpire suddenly walk to the side of the field. I hadn’t seen it, but he had been hit by a wild pitch on the side of his neck. He was wearing protective gear, but the ball managed to bounce up the side and caught bare neck. I knew something wasn’t right.
I went down to talk to him, and my medical assistant (MA), who was also at the game, came with me. I could tell the umpire was injured, but he didn’t want to leave the game. I suggested going to the hospital, but he wouldn’t consider it. So I sat there with my arms crossed, watching him.
His symptoms got worse. I could see he was in pain, and it was getting harder for him to speak.
Again, I strongly urged him to go to the hospital, but again, he said no.
In the sixth inning, things got bad enough that the umpire finally agreed to leave the game. As I was figuring out how to get him to the hospital, he disappeared on me. He had walked up to the second floor of the snack shack. My MA and I got him back downstairs and sat him on a bench behind home plate.
We were in the process of calling 911 ... when he arrested.
Luckily, when he lost vital signs, my MA and I were standing right next to him. We were able to activate ACLS protocol and start CPR within seconds.
Many times in these critical situations — especially if people are scared or have never seen an emergency like this — there’s the potential for chaos. Well, that was the polar opposite of what happened.
As soon as I started to run the code, there was this sense of order. People were keeping their composure and following directions. My MA and I would say, “this is what we need,” and the task would immediately be assigned to someone. It was quiet. There was no yelling. Everyone trusted me, even though some of them had never met me before. It was so surprising. I remember thinking, we’re running an arrest, but it’s so calm.
We were an organized team, and it really worked like clockwork, which was remarkable given where we were. It’s one thing to be in the hospital for an event like that. But to be on a baseball field where you have nothing is a completely different scenario.
Meanwhile, the game went on.
I had requested that all the kids be placed in the dugout when they weren’t on the field. So they saw the umpire walk off, but none of them saw him arrest. Some parents were really helpful with making sure the kids were okay.
The president of Oxford Little League ran across the street to a fire station to get an AED. But the fire department personnel were out on a call. He had to break down the door.
By the time he got back, the umpire’s vital signs were returning. And then EMS arrived.
They loaded him in the ambulance, and I called ahead to the trauma team, so they knew exactly what was happening.
I was pretty worried. My hypothesis was that there was probably compression on the vasculature, which had caused him to lose his vital signs. I thought he probably had an impending airway loss. I wasn’t sure if he was going to make it through the night.
What I didn’t know was that while I was giving CPR, my son stole home, and we won the game. As the ambulance was leaving, the celebration was going on in the outfield.
The umpire was in the hospital for several days. Early on, I got permission from his family to visit him. The first time I saw him, I felt this incredible gratitude and peace.
My dad was an ER doctor, and growing up, it seemed like every time we went on a family vacation, there was an emergency. We would be near a car accident or something, and my father would fly in and save the day. I remember being on the Autobahn somewhere in Europe, and there was a devastating accident between a car and a motorcycle. My father stabilized the guy, had him airlifted out, and apparently, he did fine. I grew up watching things like this and thinking, wow, that’s incredible.
Fast forward to 2 years ago, my father was diagnosed with a lung cancer he never should have had. He never smoked. As a cancer surgeon, I know we did everything in our power to save him. But it didn’t happen. He passed away.
I realize this is superstitious, but seeing the umpire alive, I had this feeling that somehow my dad was there. It was bittersweet but also a joyful moment — like I could breathe again.
I met the umpire’s family that first time, and it was like meeting family that you didn’t know you had but now you have forever. Even though the event was traumatic — I’m still trying not to be on high alert every time I go to a game — it felt like a gift to be part of this journey with them.
Little League’s mission is to teach kids about teamwork, leadership, and making good choices so communities are stronger. Our umpire is a guy who does that every day. He’s not a Little League umpire because he makes any money. He shows up at every single game to support these kids and engage them, to model respect, gratitude, and kindness.
I think our obligation as people is to live with intentionality. We all need to make sure we leave the world a better place, even when we are called upon to do uncomfortable things. Our umpire showed our kids what that looks like, and in that moment when he could have died, we were able to do the same for him.
Jennifer LaFemina, MD, is a surgical oncologist at UMass Memorial Medical Center in Massachusetts.
Are you a medical professional with a dramatic story outside the clinic? Medscape Medical News would love to consider your story for Is There a Doctor in the House? Please email your contact information and a short summary to [email protected].
A version of this article appeared on Medscape.com.
Cannabis in Cancer: What Oncologists and Patients Should Know
first, and oncologists may be hesitant to broach the topic with their patients.
Updated guidelines from the American Society of Clinical Oncology (ASCO) on the use of cannabis and cannabinoids in adults with cancer stress that it’s an important conversation to have.
According to the ASCO expert panel, access to and use of cannabis alongside cancer care have outpaced the science on evidence-based indications, and overall high-quality data on the effects of cannabis during cancer care are lacking. While several observational studies support cannabis use to help ease chemotherapy-related nausea and vomiting, the literature remains more divided on other potential benefits, such as alleviating cancer pain and sleep problems, and some evidence points to potential downsides of cannabis use.
Oncologists should “absolutely talk to patients” about cannabis, Brooke Worster, MD, medical director for the Master of Science in Medical Cannabis Science & Business program at Thomas Jefferson University, Philadelphia, told Medscape Medical News.
“Patients are interested, and they are going to find access to information. As a medical professional, it’s our job to help guide them through these spaces in a safe, nonjudgmental way.”
But, Worster noted, oncologists don’t have to be experts on cannabis to begin the conversation with patients.
So, “let yourself off the hook,” Worster urged.
Plus, avoiding the conversation won’t stop patients from using cannabis. In a recent study, Worster and her colleagues found that nearly one third of patients at 12 National Cancer Institute-designated cancer centers had used cannabis since their diagnosis — most often for sleep disturbance, pain, stress, and anxiety. Most (60%) felt somewhat or extremely comfortable talking to their healthcare provider about it, but only 21.5% said they had done so. Even fewer — about 10% — had talked to their treating oncologist.
Because patients may not discuss cannabis use, it’s especially important for oncologists to open up a line of communication, said Worster, also the enterprise director of supportive oncology at the Thomas Jefferson University.
Evidence on Cannabis During Cancer Care
A substantial proportion of people with cancer believe cannabis can help manage cancer-related symptoms.
In Worster’s recent survey study, regardless of whether patients had used cannabis, almost 90% of those surveyed reported a perceived benefit. Although 65% also reported perceived risks for cannabis use, including difficulty concentrating, lung damage, and impaired memory, the perceived benefits outweighed the risks.
Despite generally positive perceptions, the overall literature on the benefits of cannabis in patients with cancer paints a less clear picture.
The ASCO guidelines, which were based on 13 systematic reviews and five additional primary studies, reported that cannabis can improve refractory, chemotherapy-induced nausea or vomiting when added to guideline-concordant antiemetic regimens, but that there is no clear evidence of benefit or harm for other supportive care outcomes.
The “certainty of evidence for most outcomes was low or very low,” the ASCO authors wrote.
The ASCO experts explained that, outside the context of a clinical trial, the evidence is not sufficient to recommend cannabis or cannabinoids for managing cancer pain, sleep issues, appetite loss, or anxiety and depression. For these outcomes, some studies indicate a benefit, while others don’t.
Real-world data from a large registry study, for instance, have indicated that medical cannabis is “a safe and effective complementary treatment for pain relief in patients with cancer.” However, a 2020 meta-analysis found that, in studies with a low risk for bias, adding cannabinoids to opioids did not reduce cancer pain in adults with advanced cancer.
There can be downsides to cannabis use, too. In one recent study, some patients reported feeling worse physically and psychologically compared with those who didn’t use cannabis. Another study found that oral cannabis was associated with “bothersome” side effects, including sedation, dizziness, and transient anxiety.
The ASCO guidelines also made it clear that cannabis or cannabinoids should not be used as cancer-directed treatment, outside of a clinical trial.
Talking to Patients About Cannabis
Given the level of evidence and patient interest in cannabis, it is important for oncologists to raise the topic of cannabis use with their patients.
To help inform decision-making and approaches to care, the ASCO guidelines suggest that oncologists can guide care themselves or direct patients to appropriate “unbiased, evidence-based” resources. For those who use cannabis or cannabinoids outside of evidence-based indications or clinician recommendations, it’s important to explore patients’ goals, educate them, and try to minimize harm.
One strategy for broaching the topic, Worster suggested, is to simply ask patients if they have tried or considered trying cannabis to control symptoms like nausea and vomiting, loss of appetite, or cancer pain.
The conversation with patients should then include an overview of the potential benefits and potential risks for cannabis use as well as risk reduction strategies, Worster noted.
But “approach it in an open and nonjudgmental frame of mind,” she said. “Just have a conversation.”
Discussing the formulation and concentration of tetrahydrocannabinol (THC) and cannabidiol (CBD) in products matters as well.
Will the product be inhaled, ingested, or topical? Inhaled cannabis is not ideal but is sometimes what patients have access to, Worster explained. Inhaled formulations tend to have faster onset, which might be preferable for treating chemotherapy-related nausea and vomiting, whereas edible formulations may take a while to start working.
It’s also important to warn patients about taking too much, she said, explaining that inhaling THC at higher doses can increase the risk for cardiovascular effects, anxiety, paranoia, panic, and psychosis.
CBD, on the other hand, is anti-inflammatory, but early data suggest it may blunt immune responses in high doses and should be used cautiously by patients receiving immunotherapy.
Worster noted that as laws change and the science advances, new cannabis products and formulations will emerge, as will artificial intelligence tools for helping to guide patients and clinicians in optimal use of cannabis for cancer care. State websites are a particularly helpful tool for providing state-specific medical education related to cannabis laws and use, as well, she said.
The bottom line, she said, is that talking to patients about the ins and outs of cannabis use “really matters.”
Worster disclosed that she is a medical consultant for EO Care.
A version of this article appeared on Medscape.com.
first, and oncologists may be hesitant to broach the topic with their patients.
Updated guidelines from the American Society of Clinical Oncology (ASCO) on the use of cannabis and cannabinoids in adults with cancer stress that it’s an important conversation to have.
According to the ASCO expert panel, access to and use of cannabis alongside cancer care have outpaced the science on evidence-based indications, and overall high-quality data on the effects of cannabis during cancer care are lacking. While several observational studies support cannabis use to help ease chemotherapy-related nausea and vomiting, the literature remains more divided on other potential benefits, such as alleviating cancer pain and sleep problems, and some evidence points to potential downsides of cannabis use.
Oncologists should “absolutely talk to patients” about cannabis, Brooke Worster, MD, medical director for the Master of Science in Medical Cannabis Science & Business program at Thomas Jefferson University, Philadelphia, told Medscape Medical News.
“Patients are interested, and they are going to find access to information. As a medical professional, it’s our job to help guide them through these spaces in a safe, nonjudgmental way.”
But, Worster noted, oncologists don’t have to be experts on cannabis to begin the conversation with patients.
So, “let yourself off the hook,” Worster urged.
Plus, avoiding the conversation won’t stop patients from using cannabis. In a recent study, Worster and her colleagues found that nearly one third of patients at 12 National Cancer Institute-designated cancer centers had used cannabis since their diagnosis — most often for sleep disturbance, pain, stress, and anxiety. Most (60%) felt somewhat or extremely comfortable talking to their healthcare provider about it, but only 21.5% said they had done so. Even fewer — about 10% — had talked to their treating oncologist.
Because patients may not discuss cannabis use, it’s especially important for oncologists to open up a line of communication, said Worster, also the enterprise director of supportive oncology at the Thomas Jefferson University.
Evidence on Cannabis During Cancer Care
A substantial proportion of people with cancer believe cannabis can help manage cancer-related symptoms.
In Worster’s recent survey study, regardless of whether patients had used cannabis, almost 90% of those surveyed reported a perceived benefit. Although 65% also reported perceived risks for cannabis use, including difficulty concentrating, lung damage, and impaired memory, the perceived benefits outweighed the risks.
Despite generally positive perceptions, the overall literature on the benefits of cannabis in patients with cancer paints a less clear picture.
The ASCO guidelines, which were based on 13 systematic reviews and five additional primary studies, reported that cannabis can improve refractory, chemotherapy-induced nausea or vomiting when added to guideline-concordant antiemetic regimens, but that there is no clear evidence of benefit or harm for other supportive care outcomes.
The “certainty of evidence for most outcomes was low or very low,” the ASCO authors wrote.
The ASCO experts explained that, outside the context of a clinical trial, the evidence is not sufficient to recommend cannabis or cannabinoids for managing cancer pain, sleep issues, appetite loss, or anxiety and depression. For these outcomes, some studies indicate a benefit, while others don’t.
Real-world data from a large registry study, for instance, have indicated that medical cannabis is “a safe and effective complementary treatment for pain relief in patients with cancer.” However, a 2020 meta-analysis found that, in studies with a low risk for bias, adding cannabinoids to opioids did not reduce cancer pain in adults with advanced cancer.
There can be downsides to cannabis use, too. In one recent study, some patients reported feeling worse physically and psychologically compared with those who didn’t use cannabis. Another study found that oral cannabis was associated with “bothersome” side effects, including sedation, dizziness, and transient anxiety.
The ASCO guidelines also made it clear that cannabis or cannabinoids should not be used as cancer-directed treatment, outside of a clinical trial.
Talking to Patients About Cannabis
Given the level of evidence and patient interest in cannabis, it is important for oncologists to raise the topic of cannabis use with their patients.
To help inform decision-making and approaches to care, the ASCO guidelines suggest that oncologists can guide care themselves or direct patients to appropriate “unbiased, evidence-based” resources. For those who use cannabis or cannabinoids outside of evidence-based indications or clinician recommendations, it’s important to explore patients’ goals, educate them, and try to minimize harm.
One strategy for broaching the topic, Worster suggested, is to simply ask patients if they have tried or considered trying cannabis to control symptoms like nausea and vomiting, loss of appetite, or cancer pain.
The conversation with patients should then include an overview of the potential benefits and potential risks for cannabis use as well as risk reduction strategies, Worster noted.
But “approach it in an open and nonjudgmental frame of mind,” she said. “Just have a conversation.”
Discussing the formulation and concentration of tetrahydrocannabinol (THC) and cannabidiol (CBD) in products matters as well.
Will the product be inhaled, ingested, or topical? Inhaled cannabis is not ideal but is sometimes what patients have access to, Worster explained. Inhaled formulations tend to have faster onset, which might be preferable for treating chemotherapy-related nausea and vomiting, whereas edible formulations may take a while to start working.
It’s also important to warn patients about taking too much, she said, explaining that inhaling THC at higher doses can increase the risk for cardiovascular effects, anxiety, paranoia, panic, and psychosis.
CBD, on the other hand, is anti-inflammatory, but early data suggest it may blunt immune responses in high doses and should be used cautiously by patients receiving immunotherapy.
Worster noted that as laws change and the science advances, new cannabis products and formulations will emerge, as will artificial intelligence tools for helping to guide patients and clinicians in optimal use of cannabis for cancer care. State websites are a particularly helpful tool for providing state-specific medical education related to cannabis laws and use, as well, she said.
The bottom line, she said, is that talking to patients about the ins and outs of cannabis use “really matters.”
Worster disclosed that she is a medical consultant for EO Care.
A version of this article appeared on Medscape.com.
first, and oncologists may be hesitant to broach the topic with their patients.
Updated guidelines from the American Society of Clinical Oncology (ASCO) on the use of cannabis and cannabinoids in adults with cancer stress that it’s an important conversation to have.
According to the ASCO expert panel, access to and use of cannabis alongside cancer care have outpaced the science on evidence-based indications, and overall high-quality data on the effects of cannabis during cancer care are lacking. While several observational studies support cannabis use to help ease chemotherapy-related nausea and vomiting, the literature remains more divided on other potential benefits, such as alleviating cancer pain and sleep problems, and some evidence points to potential downsides of cannabis use.
Oncologists should “absolutely talk to patients” about cannabis, Brooke Worster, MD, medical director for the Master of Science in Medical Cannabis Science & Business program at Thomas Jefferson University, Philadelphia, told Medscape Medical News.
“Patients are interested, and they are going to find access to information. As a medical professional, it’s our job to help guide them through these spaces in a safe, nonjudgmental way.”
But, Worster noted, oncologists don’t have to be experts on cannabis to begin the conversation with patients.
So, “let yourself off the hook,” Worster urged.
Plus, avoiding the conversation won’t stop patients from using cannabis. In a recent study, Worster and her colleagues found that nearly one third of patients at 12 National Cancer Institute-designated cancer centers had used cannabis since their diagnosis — most often for sleep disturbance, pain, stress, and anxiety. Most (60%) felt somewhat or extremely comfortable talking to their healthcare provider about it, but only 21.5% said they had done so. Even fewer — about 10% — had talked to their treating oncologist.
Because patients may not discuss cannabis use, it’s especially important for oncologists to open up a line of communication, said Worster, also the enterprise director of supportive oncology at the Thomas Jefferson University.
Evidence on Cannabis During Cancer Care
A substantial proportion of people with cancer believe cannabis can help manage cancer-related symptoms.
In Worster’s recent survey study, regardless of whether patients had used cannabis, almost 90% of those surveyed reported a perceived benefit. Although 65% also reported perceived risks for cannabis use, including difficulty concentrating, lung damage, and impaired memory, the perceived benefits outweighed the risks.
Despite generally positive perceptions, the overall literature on the benefits of cannabis in patients with cancer paints a less clear picture.
The ASCO guidelines, which were based on 13 systematic reviews and five additional primary studies, reported that cannabis can improve refractory, chemotherapy-induced nausea or vomiting when added to guideline-concordant antiemetic regimens, but that there is no clear evidence of benefit or harm for other supportive care outcomes.
The “certainty of evidence for most outcomes was low or very low,” the ASCO authors wrote.
The ASCO experts explained that, outside the context of a clinical trial, the evidence is not sufficient to recommend cannabis or cannabinoids for managing cancer pain, sleep issues, appetite loss, or anxiety and depression. For these outcomes, some studies indicate a benefit, while others don’t.
Real-world data from a large registry study, for instance, have indicated that medical cannabis is “a safe and effective complementary treatment for pain relief in patients with cancer.” However, a 2020 meta-analysis found that, in studies with a low risk for bias, adding cannabinoids to opioids did not reduce cancer pain in adults with advanced cancer.
There can be downsides to cannabis use, too. In one recent study, some patients reported feeling worse physically and psychologically compared with those who didn’t use cannabis. Another study found that oral cannabis was associated with “bothersome” side effects, including sedation, dizziness, and transient anxiety.
The ASCO guidelines also made it clear that cannabis or cannabinoids should not be used as cancer-directed treatment, outside of a clinical trial.
Talking to Patients About Cannabis
Given the level of evidence and patient interest in cannabis, it is important for oncologists to raise the topic of cannabis use with their patients.
To help inform decision-making and approaches to care, the ASCO guidelines suggest that oncologists can guide care themselves or direct patients to appropriate “unbiased, evidence-based” resources. For those who use cannabis or cannabinoids outside of evidence-based indications or clinician recommendations, it’s important to explore patients’ goals, educate them, and try to minimize harm.
One strategy for broaching the topic, Worster suggested, is to simply ask patients if they have tried or considered trying cannabis to control symptoms like nausea and vomiting, loss of appetite, or cancer pain.
The conversation with patients should then include an overview of the potential benefits and potential risks for cannabis use as well as risk reduction strategies, Worster noted.
But “approach it in an open and nonjudgmental frame of mind,” she said. “Just have a conversation.”
Discussing the formulation and concentration of tetrahydrocannabinol (THC) and cannabidiol (CBD) in products matters as well.
Will the product be inhaled, ingested, or topical? Inhaled cannabis is not ideal but is sometimes what patients have access to, Worster explained. Inhaled formulations tend to have faster onset, which might be preferable for treating chemotherapy-related nausea and vomiting, whereas edible formulations may take a while to start working.
It’s also important to warn patients about taking too much, she said, explaining that inhaling THC at higher doses can increase the risk for cardiovascular effects, anxiety, paranoia, panic, and psychosis.
CBD, on the other hand, is anti-inflammatory, but early data suggest it may blunt immune responses in high doses and should be used cautiously by patients receiving immunotherapy.
Worster noted that as laws change and the science advances, new cannabis products and formulations will emerge, as will artificial intelligence tools for helping to guide patients and clinicians in optimal use of cannabis for cancer care. State websites are a particularly helpful tool for providing state-specific medical education related to cannabis laws and use, as well, she said.
The bottom line, she said, is that talking to patients about the ins and outs of cannabis use “really matters.”
Worster disclosed that she is a medical consultant for EO Care.
A version of this article appeared on Medscape.com.