User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Treatment Stacking: Optimizing Therapeutic Regimens for Hidradenitis Suppurativa
Hidradenitis suppurativa (HS) is a debilitating chronic condition that often is recalcitrant to first-line treatments, and mechanisms underlying its pathology remain unclear. Existing data suggest a multifactorial etiology with different pathophysiologic contributors, including genetic, hormonal, and immune dysregulation factors. At this time, only one medication (adalimumab) is US Food and Drug Administration approved for HS, but multiple medical and procedural therapies are available.1 Herein, we discuss the concept of treatment stacking, or the combination of unique therapeutic modalities—an approach we believe is key to optimizing management of HS patients.
Stacking Treatments for HS
Unlike psoriasis, in which a single biologic agent may provide 100% clearance (psoriasis area and severity index 100 [PASI 100]) without adjuvant treatment,2,3 the field of HS currently lacks medications that are efficacious to that degree of success as monotherapy. In HS, the benchmark for a positive treatment outcome is Hidradenitis Suppurativa Clinical Response 50 (HiSCR50),4 a 50% reduction in inflammatory lesion count—a far less stringent marker for disease improvement. Thus, providers should design HS treatment regimens with a model of combining therapies and shift away from monotherapy. Targeting different pathophysiologic pathways by stacking multiple treatments may provide synergistic benefits for HS patients. Treatment stacking is a familiar concept in acne; for instance, patients who benefit tremendously from isotretinoin may still require a hormone-modulating treatment (eg, spironolactone) to attain optimal results.
Adherence to a rigid treatment algorithm based on disease severity limits the potential to create comprehensive regimens that account for unique patient characteristics and clinical manifestations. When evaluating an HS patient, providers should systematically consider each pathophysiologic factor and target the ones that appear to be most involved in that particular patient. The North American HS guidelines illustrate this point by supporting use of several treatments across different Hurley stages, such as recommending hormonal treatment in patients with Hurley stages 1, 2, or 3.1 Of note, treatment stacking also includes procedural therapies. Surgeons typically prefer a patient’s disease management to be optimized prior to surgery, including reduced drainage and inflammation. In addition, even after surgery, patients often still require medical management to prevent continued disease worsening.
Treatment Pathways for HS
A multimodal approach with treatment stacking (Figure) can be useful to all HS patients, from those with the mildest to the most severe disease. Modifiable pathophysiologic factors and examples of their targeted treatments include (1) follicular occlusion (eg, oral retinoids), (2) metabolic dysfunction (eg, metformin), (3) hormones (eg, oral contraceptive pills, spironolactone, finasteride), (4) dysbiosis (eg, antibiotics such as clindamycin and rifampin combination therapy), (5) immune dysregulation (eg, biologic agents), and (6) friction/irritation (eg, weight loss, clothing recommendations).
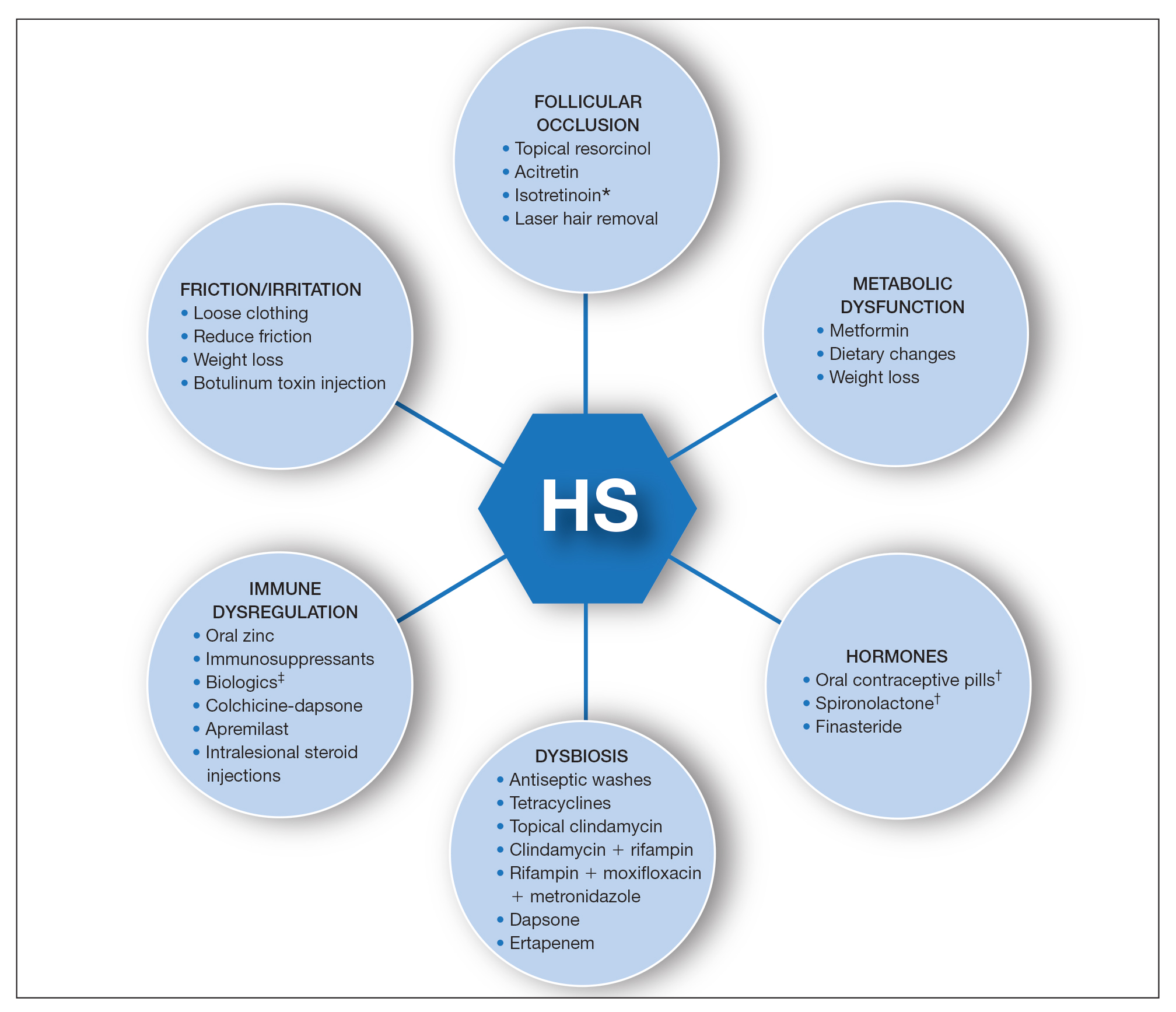
Combining treatments from different pathways enables potentiation of individual treatment efficacies. A female patient with only a few HS nodules that flare with menses may be well controlled with spironolactone as her only systemic agent; however, she still may benefit from use of an antiseptic wash, topical clindamycin, and lifestyle changes such as weight loss and reduction of mechanical irritation. A patient with severe recalcitrant HS could notably benefit from concomitant biologic, systemic antibiotic, and hormonal/metabolic treatments. If disease control is still inadequate, agents within the same class can be switched (eg, choosing a different biologic) or other disease-modifying agents such as colchicine also can be added. The goal is to create an effective treatment toolbox with therapies targeting different pathophysiologic arms of HS and working together in synergy. Each tool can be refined by modifying dosing frequency and duration of use to strive for optimal response. At this time, the literature on HS combination therapy is sparse. A retrospective study of 31 patients reported promising combinations, including isotretinoin with spironolactone for mild disease, isotretinoin or doxycycline with adalimumab for moderate disease, and cyclosporine with adalimumab for severe disease.5 Larger prospective studies on clinical response to different combination regimens are warranted.
Optimizing Therapy for HS and Its Comorbidities
Additional considerations may further optimize treatment plans. Some therapies benefit all patients; for example, providers should counsel all HS patients on healthy weight management, optimized clothing choices,6 and friction reduction in the intertriginous folds. Providers also may consider adding therapies with faster onset of efficacy as a bridge to long-term, slower-onset therapies. For instance, female HS patients with menstrual flares who are prescribed spironolactone also may benefit from a course of systemic antibiotics, which typically provides more prompt relief. Treatment regimens also can concomitantly treat HS and its comorbidities.7 For example, metformin serves a dual purpose in HS patients with diabetes mellitus, and adalimumab in patients with both HS and inflammatory bowel disease.
Final Thoughts
The last decade has seen tremendous growth in HS research8 coupled with a remarkable expansion in the therapeutic pipeline.9 However, currently no single therapy for HS can guarantee satisfactory disease remission or durability of remission. The contrast between clinical trials and real-world practice should be acknowledged; the former often is restrictive in design with monotherapy and allowance of very limited concomitant treatments, such as topical or oral antibiotics. This limits our ability to draw conclusions regarding the additive synergistic potential of different therapeutics in combination. In clinical practice, we are not restricted by monotherapy trial protocols. As we await new tools, treatment stacking allows for creating a framework to best utilize the tools that are available to us.
Although HS has continued to affect the lives of many patients, improved understanding of underlying pathophysiology and a well-placed sense of urgency from all stakeholders (ie, patients, clinicians, researchers, industry partners) has pushed this field forward. Until our therapeutic armamentarium has expanded to include highly efficacious monotherapy options, providers should consider treatment stacking for every HS patient.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Reich K, Warren RB, Lebwohl M, et al. Bimekizumab versus secukinumab in plaque psoriasis. N Engl J Med. 2021;385:142-152. doi:10.1056/NEJMoa2102383
- Imafuku S, Nakagawa H, Igarashi A, et al. Long-term efficacy and safety of tildrakizumab in Japanese patients with moderate to severe plaque psoriasis: results from a 5-year extension of a phase 3 study (reSURFACE 1). J Dermatol. 2021;48:844-852. doi:10.1111/1346-8138.15763
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434. doi:10.1056/NEJMoa1504370
- McPhie ML, Bridgman AC, Kirchhof MG. Combination therapies for hidradenitis suppurativa: a retrospective chart review of 31 patients. J Cutan Med Surg. 2019;23:270-276. doi:10.1177/1203475418823529
- Loh TY, Hendricks AJ, Hsiao JL, et al. Undergarment and fabric selection in the management of hidradenitis suppurativa. Dermatol Basel Switz. 2021;237:119-124. doi:10.1159/000501611
- Garg A, Malviya N, Strunk A, et al. Comorbidity screening in hidradenitis suppurativa: evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations [published online January 23, 2021]. J Am Acad Dermatol. doi:10.1016/j.jaad.2021.01.059
- Savage KT, Brant EG, Flood KS, et al. Publication trends in hidradenitis suppurativa from 2008 to 2018. J Eur Acad Dermatol Venereol. 2020;34:1885-1889. doi:10.1111/jdv.16213
- van Straalen KR, Schneider-Burrus S, Prens EP. Current and future treatment of hidradenitis suppurativa. Br J Dermatol. 2020;183:E178-E187. doi:10.1111/bjd.16768
Hidradenitis suppurativa (HS) is a debilitating chronic condition that often is recalcitrant to first-line treatments, and mechanisms underlying its pathology remain unclear. Existing data suggest a multifactorial etiology with different pathophysiologic contributors, including genetic, hormonal, and immune dysregulation factors. At this time, only one medication (adalimumab) is US Food and Drug Administration approved for HS, but multiple medical and procedural therapies are available.1 Herein, we discuss the concept of treatment stacking, or the combination of unique therapeutic modalities—an approach we believe is key to optimizing management of HS patients.
Stacking Treatments for HS
Unlike psoriasis, in which a single biologic agent may provide 100% clearance (psoriasis area and severity index 100 [PASI 100]) without adjuvant treatment,2,3 the field of HS currently lacks medications that are efficacious to that degree of success as monotherapy. In HS, the benchmark for a positive treatment outcome is Hidradenitis Suppurativa Clinical Response 50 (HiSCR50),4 a 50% reduction in inflammatory lesion count—a far less stringent marker for disease improvement. Thus, providers should design HS treatment regimens with a model of combining therapies and shift away from monotherapy. Targeting different pathophysiologic pathways by stacking multiple treatments may provide synergistic benefits for HS patients. Treatment stacking is a familiar concept in acne; for instance, patients who benefit tremendously from isotretinoin may still require a hormone-modulating treatment (eg, spironolactone) to attain optimal results.
Adherence to a rigid treatment algorithm based on disease severity limits the potential to create comprehensive regimens that account for unique patient characteristics and clinical manifestations. When evaluating an HS patient, providers should systematically consider each pathophysiologic factor and target the ones that appear to be most involved in that particular patient. The North American HS guidelines illustrate this point by supporting use of several treatments across different Hurley stages, such as recommending hormonal treatment in patients with Hurley stages 1, 2, or 3.1 Of note, treatment stacking also includes procedural therapies. Surgeons typically prefer a patient’s disease management to be optimized prior to surgery, including reduced drainage and inflammation. In addition, even after surgery, patients often still require medical management to prevent continued disease worsening.
Treatment Pathways for HS
A multimodal approach with treatment stacking (Figure) can be useful to all HS patients, from those with the mildest to the most severe disease. Modifiable pathophysiologic factors and examples of their targeted treatments include (1) follicular occlusion (eg, oral retinoids), (2) metabolic dysfunction (eg, metformin), (3) hormones (eg, oral contraceptive pills, spironolactone, finasteride), (4) dysbiosis (eg, antibiotics such as clindamycin and rifampin combination therapy), (5) immune dysregulation (eg, biologic agents), and (6) friction/irritation (eg, weight loss, clothing recommendations).
Combining treatments from different pathways enables potentiation of individual treatment efficacies. A female patient with only a few HS nodules that flare with menses may be well controlled with spironolactone as her only systemic agent; however, she still may benefit from use of an antiseptic wash, topical clindamycin, and lifestyle changes such as weight loss and reduction of mechanical irritation. A patient with severe recalcitrant HS could notably benefit from concomitant biologic, systemic antibiotic, and hormonal/metabolic treatments. If disease control is still inadequate, agents within the same class can be switched (eg, choosing a different biologic) or other disease-modifying agents such as colchicine also can be added. The goal is to create an effective treatment toolbox with therapies targeting different pathophysiologic arms of HS and working together in synergy. Each tool can be refined by modifying dosing frequency and duration of use to strive for optimal response. At this time, the literature on HS combination therapy is sparse. A retrospective study of 31 patients reported promising combinations, including isotretinoin with spironolactone for mild disease, isotretinoin or doxycycline with adalimumab for moderate disease, and cyclosporine with adalimumab for severe disease.5 Larger prospective studies on clinical response to different combination regimens are warranted.
Optimizing Therapy for HS and Its Comorbidities
Additional considerations may further optimize treatment plans. Some therapies benefit all patients; for example, providers should counsel all HS patients on healthy weight management, optimized clothing choices,6 and friction reduction in the intertriginous folds. Providers also may consider adding therapies with faster onset of efficacy as a bridge to long-term, slower-onset therapies. For instance, female HS patients with menstrual flares who are prescribed spironolactone also may benefit from a course of systemic antibiotics, which typically provides more prompt relief. Treatment regimens also can concomitantly treat HS and its comorbidities.7 For example, metformin serves a dual purpose in HS patients with diabetes mellitus, and adalimumab in patients with both HS and inflammatory bowel disease.
Final Thoughts
The last decade has seen tremendous growth in HS research8 coupled with a remarkable expansion in the therapeutic pipeline.9 However, currently no single therapy for HS can guarantee satisfactory disease remission or durability of remission. The contrast between clinical trials and real-world practice should be acknowledged; the former often is restrictive in design with monotherapy and allowance of very limited concomitant treatments, such as topical or oral antibiotics. This limits our ability to draw conclusions regarding the additive synergistic potential of different therapeutics in combination. In clinical practice, we are not restricted by monotherapy trial protocols. As we await new tools, treatment stacking allows for creating a framework to best utilize the tools that are available to us.
Although HS has continued to affect the lives of many patients, improved understanding of underlying pathophysiology and a well-placed sense of urgency from all stakeholders (ie, patients, clinicians, researchers, industry partners) has pushed this field forward. Until our therapeutic armamentarium has expanded to include highly efficacious monotherapy options, providers should consider treatment stacking for every HS patient.
Hidradenitis suppurativa (HS) is a debilitating chronic condition that often is recalcitrant to first-line treatments, and mechanisms underlying its pathology remain unclear. Existing data suggest a multifactorial etiology with different pathophysiologic contributors, including genetic, hormonal, and immune dysregulation factors. At this time, only one medication (adalimumab) is US Food and Drug Administration approved for HS, but multiple medical and procedural therapies are available.1 Herein, we discuss the concept of treatment stacking, or the combination of unique therapeutic modalities—an approach we believe is key to optimizing management of HS patients.
Stacking Treatments for HS
Unlike psoriasis, in which a single biologic agent may provide 100% clearance (psoriasis area and severity index 100 [PASI 100]) without adjuvant treatment,2,3 the field of HS currently lacks medications that are efficacious to that degree of success as monotherapy. In HS, the benchmark for a positive treatment outcome is Hidradenitis Suppurativa Clinical Response 50 (HiSCR50),4 a 50% reduction in inflammatory lesion count—a far less stringent marker for disease improvement. Thus, providers should design HS treatment regimens with a model of combining therapies and shift away from monotherapy. Targeting different pathophysiologic pathways by stacking multiple treatments may provide synergistic benefits for HS patients. Treatment stacking is a familiar concept in acne; for instance, patients who benefit tremendously from isotretinoin may still require a hormone-modulating treatment (eg, spironolactone) to attain optimal results.
Adherence to a rigid treatment algorithm based on disease severity limits the potential to create comprehensive regimens that account for unique patient characteristics and clinical manifestations. When evaluating an HS patient, providers should systematically consider each pathophysiologic factor and target the ones that appear to be most involved in that particular patient. The North American HS guidelines illustrate this point by supporting use of several treatments across different Hurley stages, such as recommending hormonal treatment in patients with Hurley stages 1, 2, or 3.1 Of note, treatment stacking also includes procedural therapies. Surgeons typically prefer a patient’s disease management to be optimized prior to surgery, including reduced drainage and inflammation. In addition, even after surgery, patients often still require medical management to prevent continued disease worsening.
Treatment Pathways for HS
A multimodal approach with treatment stacking (Figure) can be useful to all HS patients, from those with the mildest to the most severe disease. Modifiable pathophysiologic factors and examples of their targeted treatments include (1) follicular occlusion (eg, oral retinoids), (2) metabolic dysfunction (eg, metformin), (3) hormones (eg, oral contraceptive pills, spironolactone, finasteride), (4) dysbiosis (eg, antibiotics such as clindamycin and rifampin combination therapy), (5) immune dysregulation (eg, biologic agents), and (6) friction/irritation (eg, weight loss, clothing recommendations).
Combining treatments from different pathways enables potentiation of individual treatment efficacies. A female patient with only a few HS nodules that flare with menses may be well controlled with spironolactone as her only systemic agent; however, she still may benefit from use of an antiseptic wash, topical clindamycin, and lifestyle changes such as weight loss and reduction of mechanical irritation. A patient with severe recalcitrant HS could notably benefit from concomitant biologic, systemic antibiotic, and hormonal/metabolic treatments. If disease control is still inadequate, agents within the same class can be switched (eg, choosing a different biologic) or other disease-modifying agents such as colchicine also can be added. The goal is to create an effective treatment toolbox with therapies targeting different pathophysiologic arms of HS and working together in synergy. Each tool can be refined by modifying dosing frequency and duration of use to strive for optimal response. At this time, the literature on HS combination therapy is sparse. A retrospective study of 31 patients reported promising combinations, including isotretinoin with spironolactone for mild disease, isotretinoin or doxycycline with adalimumab for moderate disease, and cyclosporine with adalimumab for severe disease.5 Larger prospective studies on clinical response to different combination regimens are warranted.
Optimizing Therapy for HS and Its Comorbidities
Additional considerations may further optimize treatment plans. Some therapies benefit all patients; for example, providers should counsel all HS patients on healthy weight management, optimized clothing choices,6 and friction reduction in the intertriginous folds. Providers also may consider adding therapies with faster onset of efficacy as a bridge to long-term, slower-onset therapies. For instance, female HS patients with menstrual flares who are prescribed spironolactone also may benefit from a course of systemic antibiotics, which typically provides more prompt relief. Treatment regimens also can concomitantly treat HS and its comorbidities.7 For example, metformin serves a dual purpose in HS patients with diabetes mellitus, and adalimumab in patients with both HS and inflammatory bowel disease.
Final Thoughts
The last decade has seen tremendous growth in HS research8 coupled with a remarkable expansion in the therapeutic pipeline.9 However, currently no single therapy for HS can guarantee satisfactory disease remission or durability of remission. The contrast between clinical trials and real-world practice should be acknowledged; the former often is restrictive in design with monotherapy and allowance of very limited concomitant treatments, such as topical or oral antibiotics. This limits our ability to draw conclusions regarding the additive synergistic potential of different therapeutics in combination. In clinical practice, we are not restricted by monotherapy trial protocols. As we await new tools, treatment stacking allows for creating a framework to best utilize the tools that are available to us.
Although HS has continued to affect the lives of many patients, improved understanding of underlying pathophysiology and a well-placed sense of urgency from all stakeholders (ie, patients, clinicians, researchers, industry partners) has pushed this field forward. Until our therapeutic armamentarium has expanded to include highly efficacious monotherapy options, providers should consider treatment stacking for every HS patient.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Reich K, Warren RB, Lebwohl M, et al. Bimekizumab versus secukinumab in plaque psoriasis. N Engl J Med. 2021;385:142-152. doi:10.1056/NEJMoa2102383
- Imafuku S, Nakagawa H, Igarashi A, et al. Long-term efficacy and safety of tildrakizumab in Japanese patients with moderate to severe plaque psoriasis: results from a 5-year extension of a phase 3 study (reSURFACE 1). J Dermatol. 2021;48:844-852. doi:10.1111/1346-8138.15763
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434. doi:10.1056/NEJMoa1504370
- McPhie ML, Bridgman AC, Kirchhof MG. Combination therapies for hidradenitis suppurativa: a retrospective chart review of 31 patients. J Cutan Med Surg. 2019;23:270-276. doi:10.1177/1203475418823529
- Loh TY, Hendricks AJ, Hsiao JL, et al. Undergarment and fabric selection in the management of hidradenitis suppurativa. Dermatol Basel Switz. 2021;237:119-124. doi:10.1159/000501611
- Garg A, Malviya N, Strunk A, et al. Comorbidity screening in hidradenitis suppurativa: evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations [published online January 23, 2021]. J Am Acad Dermatol. doi:10.1016/j.jaad.2021.01.059
- Savage KT, Brant EG, Flood KS, et al. Publication trends in hidradenitis suppurativa from 2008 to 2018. J Eur Acad Dermatol Venereol. 2020;34:1885-1889. doi:10.1111/jdv.16213
- van Straalen KR, Schneider-Burrus S, Prens EP. Current and future treatment of hidradenitis suppurativa. Br J Dermatol. 2020;183:E178-E187. doi:10.1111/bjd.16768
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Reich K, Warren RB, Lebwohl M, et al. Bimekizumab versus secukinumab in plaque psoriasis. N Engl J Med. 2021;385:142-152. doi:10.1056/NEJMoa2102383
- Imafuku S, Nakagawa H, Igarashi A, et al. Long-term efficacy and safety of tildrakizumab in Japanese patients with moderate to severe plaque psoriasis: results from a 5-year extension of a phase 3 study (reSURFACE 1). J Dermatol. 2021;48:844-852. doi:10.1111/1346-8138.15763
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434. doi:10.1056/NEJMoa1504370
- McPhie ML, Bridgman AC, Kirchhof MG. Combination therapies for hidradenitis suppurativa: a retrospective chart review of 31 patients. J Cutan Med Surg. 2019;23:270-276. doi:10.1177/1203475418823529
- Loh TY, Hendricks AJ, Hsiao JL, et al. Undergarment and fabric selection in the management of hidradenitis suppurativa. Dermatol Basel Switz. 2021;237:119-124. doi:10.1159/000501611
- Garg A, Malviya N, Strunk A, et al. Comorbidity screening in hidradenitis suppurativa: evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations [published online January 23, 2021]. J Am Acad Dermatol. doi:10.1016/j.jaad.2021.01.059
- Savage KT, Brant EG, Flood KS, et al. Publication trends in hidradenitis suppurativa from 2008 to 2018. J Eur Acad Dermatol Venereol. 2020;34:1885-1889. doi:10.1111/jdv.16213
- van Straalen KR, Schneider-Burrus S, Prens EP. Current and future treatment of hidradenitis suppurativa. Br J Dermatol. 2020;183:E178-E187. doi:10.1111/bjd.16768

Bullous Amyloidosis Masquerading as Pseudoporphyria
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
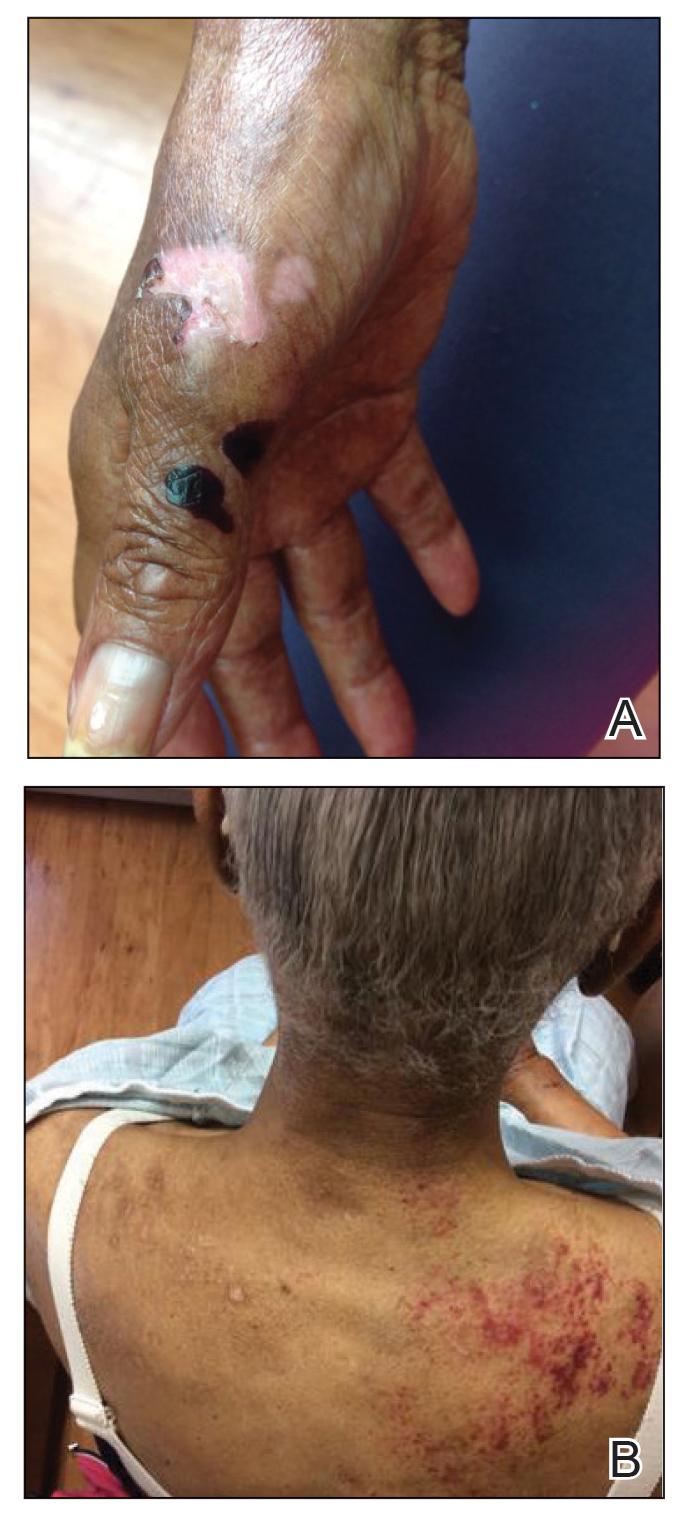
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.

Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642.
- Bolognia JL, Schaffer JV, Duncan KO, et al. Amyloidosis. Dermatology Essentials. Elsevier Saunders; 2014:341-345.
- Bhutani M, Shahid Z, Schnebelen A, et al. Cutaneous manifestations of multiple myeloma and other plasma cell proliferative disorders. Semin Oncol. 2016;43:395-400.
- Terushkin V, Boyd KP, Patel RR, et al. Primary localized cutaneous amyloidosis. Dermatol Online J. 2013;19:20711.
- LaChance A, Phelps A, Finch J, et al. Nodular localized primary cutaneous amyloidosis: a bullous variant. Clin Exp Dermatol. 2014;39:344-347.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Kanoh T. Bullous amyloidosis [in Japanese]. Rinsho Ketsueki. 1993;34:1050-1052.
- Johnson TM, Rapini RP, Hebert AA, et al. Bullous amyloidosis. Cutis. 1989;43:346-352.
- Houman MH, Smiti KM, Ben Ghorbel I, et al. Bullous amyloidosis. Ann Dermatol Venereol. 2002;129:299-302.
- Sanusi T, Li Y, Qian Y, et al. Primary localized cutaneous nodular amyloidosis with bullous lesions. Indian J Dermatol Venereol Leprol. 2015;81:400-402.
- Ochiai T, Morishima T, Hao T, et al. Bullous amyloidosis: the mechanism of blister formation revealed by electron microscopy. J Cutan Pathol. 2001;28:407-411.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Wang XD, Shen H, Liu ZH. Diffuse haemorrhagic bullous amyloidosis with multiple myeloma. Clin Exp Dermatol. 2008;33:94-96.
- Biswas P, Aggarwal I, Sen D, et al. Bullous pemphigoid clinically presenting as lichen amyloidosis. Indian J Dermatol Venereol Leprol. 2014;80:544-546.
- Bluhm JF 3rd. Bullous dermatosis vs amyloidosis. Arch Dermatol. 1981;117:252.
- Bluhm JF 3rd. Bullous amyloidosis vs epidermolysis bullosa acquisita. JAMA. 1981;245:32.
- Murphy GM, Wright J, Nicholls DS, et al. Sunbed-induced pseudoporphyria. Br J Dermatol. 1989;120:555-562.
- Pramatarov K, Lazarova A, Mateev G, et al. Bullous hemorrhagic primary systemic amyloidosis. Int J Dermatol. 1990;29:211-213.
- Bieber T, Ruzicka T, Linke RP, et al. Hemorrhagic bullous amyloidosis. a histologic, immunocytochemical, and ultrastructural study of two patients. Arch Dermatol. 1988;124:1683-1686.
- Khoo BP, Tay YK. Lichen amyloidosis: a bullous variant. Ann Acad Med Singapore. 2000;29:105-107.
- Asahina A, Hasegawa K, Ishiyama M, et al. Bullous amyloidosis mimicking bullous pemphigoid: usefulness of electron microscopic examination. Acta Derm Venereol. 2010;90:427-428.
- Schmutz JL, Barbaud A, Cuny JF, et al. Bullous amyloidosis [in French]. Ann Dermatol Venereol. 1988;115:295-301.
- Lachmann HJ, Hawkins PN. Amyloidosis of the skin. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. McGraw-Hill; 2012:1574-1583.
- Grundmann JU, Bonnekoh B, Gollnick H. Extensive haemorrhagic-bullous skin manifestation of systemic AA-amyloidosis associated with IgG lambda-myeloma. Eur J Dermatol. 2000;10:139-142.
- Hödl S, Turek TD, Kerl H. Plasmocytoma-associated bullous hemorrhagic amyloidosis of the skin [in German]. Hautarzt. 1982;33:556-558.
- Colucci G, Alberio L, Demarmels Biasiutti F, et al. Bilateral periorbital ecchymoses. an often missed sign of amyloid purpura. Hamostaseologie. 2014;34:249-252.
- Behera B, Pattnaik M, Sahu B, et al. Cutaneous manifestations of multiple myeloma. Indian J Dermatol. 2016;61:668-671.
- Fujita Y, Tsuji-Abe Y, Sato-Matsumura KC, et al. Nail dystrophy and blisters as sole manifestations in myeloma-associated amyloidosis. J Am Acad Dermatol. 2006;54:712-714.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Winzer M, Ruppert M, Baretton G, et al. Bullous poikilodermatitic amyloidosis of the skin with junctional bulla development in IgG light chain plasmacytoma of the lambda type. histology, immunohistology and electron microscopy [in German]. Hautarzt. 1992;43:199-204.
Practice Points
- Primary systemic amyloidosis, including the rare cutaneous bullous amyloidosis, often is difficult to diagnose and has been associated with underlying plasma cell dyscrasia or multiple myeloma.
- When evaluating patients with initially convincing signs of pseudoporphyria, it is imperative to consider the diagnosis of bullous amyloidosis, which additionally can present with intraoral hemorrhagic vesicles and have confirmatory histopathologic features.
- Further investigation for multiple myeloma is warranted when patients with a chronic hemorrhagic bullous condition also present with symptoms of purpura, angina bullosa hemorrhagica, fatigue, weight loss, and/or macroglossia. Accurate diagnosis of bullous amyloidosis and timely treatment of its underlying cause will contribute to better, more proactive patient care.
Kikuchi-Fujimoto Disease in an Adolescent Boy
To the Editor:
Kikuchi-Fujimoto Disease, also called histiocytic necrotizing lymphadenitis, was described in 1972 by both Kikuchi1 and Fujimoto et al.2 Most cases are reported in Asia, with limited reports in the United States.3-5 Kikuchi-Fujimoto disease is a rare, self-limiting condition consisting of benign lymphadenopathy and oftentimes fever and systemic symptoms. Lymph node involvement may mimic non-Hodgkin lymphoma or other reactive lymphadenopathy, rendering diagnostic accuracy challenging.5 Cutaneous manifestations are reported in only 16% to 40% of patients.6,7 Herein, we describe the clinical and pathologic features of a case of Kikuchi-Fujimoto disease with cutaneous involvement in an adolescent boy.
A 13-year-old adolescent boy with no notable medical history presented to the pediatric emergency department with cervical lymphadenopathy, weight loss, intermittent fever, and an evolving rash on the face, ears, arms, and thighs of 6 weeks’ duration. The illness began with enlarged lymph nodes and erythematous macules on the face and was diagnosed by his primary care physician as lymphadenitis that was unresponsive to clindamycin. Over the subsequent weeks, the rash worsened, and he developed intermittent fevers, night sweats, abdominal pain, and nausea with a 20-pound weight loss. He presented to the emergency department 3 weeks prior to the current admission and was noted to have elevated cytomegalovirus (CMV) IgM and IgG in addition to lymphopenia and anemia. He was discharged with outpatient follow-up. The rash progressed to involve the face, ears, arms, and thighs. One day prior to the current admission, the patient’s abdominal pain worsened acutely, and he experienced several episodes of emesis. He presented to the pediatric emergency department for further evaluation, and a dermatology consultation was requested at that time.
The patient’s rash was asymptomatic. In addition to the above symptoms, he also noted frequent nosebleeds, gingival bleeding, and diffuse myalgia that was most prominent on the hands and feet; he denied diarrhea, sick contacts, recent travel, or insect bites. His vital signs were normal, and he remained afebrile throughout the hospitalization. Physical examination revealed an ill-appearing patient with sunken eyes and dry lips. He had pink, oval, scaly plaques on the cheeks, ears, and arms (Figure 1). The thighs exhibited folliculocentric erythematous papules. The ocular conjunctivae were clear, but white exudative plaques were noted on the tongue. Tender, bilateral, cervical lymphadenopathy and diffuse abdominal tenderness with guarding and hepatosplenomegaly also were present. The fingers and toes were tender upon palpation.

Laboratory workup at admission revealed the following: low white blood cell count, 2700/μL (reference range, 4500–11,000/μL); low hemoglobin, 9.6 g/dL (reference range, 14.0–17.5 g/dL); elevated aspartate aminotransferase, 91 U/L (reference range, 10–30 U/L); and elevated alanine aminotransferase, 118 U/L (reference range, 10–40 U/L). Lactate dehydrogenase (582 U/L [reference range, 100–200 U/L]), ferritin (1681 ng/mL [reference range, 15–200 ng/mL]), and C-reactive protein (6.0 mg/L [reference range, 0.08–3.1 mg/L]) also were elevated. A respiratory viral panel was unremarkable. Blood cultures were negative, and an HIV 1/2 assay was nonreactive. A chest radiograph demonstrated clear lung fields. Computed tomography of the abdomen and pelvis showed prominent mesenteric, ileocolic, and retroperitoneal lymph nodes.
The differential diagnoses at this time included acute connective tissue disease, a paraneoplastic phenomenon, cutaneous lymphoma, or an infectious etiology. A punch biopsy of the skin as well as tissue cultures were performed from a lesion on the right arm. Quantitative immunoglobulin (IgA, IgG, IgM) levels were checked, all of which were within reference range. An antinuclear antibody (ANA) assay and rheumatoid factor were normal.
The tissue cultures were negative for bacteria, fungi, and mycobacteria. Microscopic examination of the skin biopsy revealed a moderate perivascular and interstitial infiltrate of predominantly histiocytes and lymphocytes with prominent karyorrhectic debris (nuclear dust) in the upper dermis as well as focal vacuolar interface changes with scattered necrotic keratinocytes in the epidermis (Figure 2). Based on these histopathologic findings, a diagnosis of Kikuchi-Fujimoto disease was considered. To confirm the diagnosis and to rule out the possibility of lymphoma, an excisional biopsy of the cervical lymph node was performed, which showed typical histopathologic features of histiocytic necrotizing lymphadenitis.
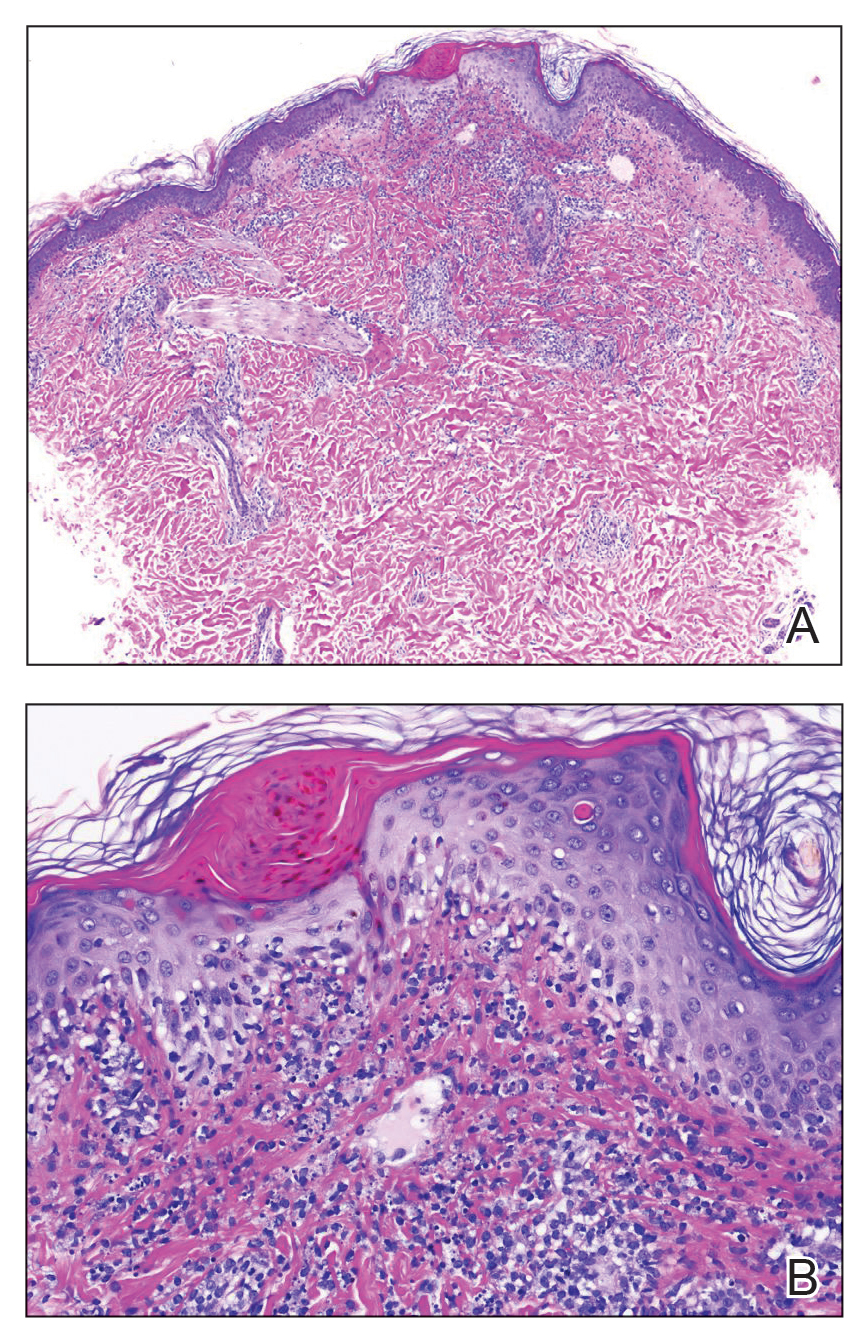
Given the patient’s clinical presentation with arthralgia, anorexia, lymphadenitis, and hepatosplenomegaly along with histopathologic findings from both the skin and lymph node biopsies, a diagnosis of Kikuchi-Fujimoto disease was made. The patient was conservatively managed with acetaminophen and was discharged with improvement in his appetite and systemic symptoms.
He was seen for follow-up 3 months later in the outpatient clinic. He denied any recurrence of systemic symptoms but endorsed a recent shedding of hair consistent with telogen effluvium. The rash had substantially improved, though residual asymptomatic erythematous plaques remained on the right forehead and right cheek (Figure 3). He was prescribed triamcinolone acetonide cream 0.1% to apply to the active area twice daily for the following 2 to 3 weeks.

Kikuchi-Fujimoto disease presents with a wide clinical spectrum, classically with benign lymphadenopathy and fever of unknown etiology.5,6 Lymphadenopathy most often is cervical (55%–99%)8 and unilateral,4,7 but patients can present with polyadenopathy (52%).7,8 Constitutional signs commonly include fever (35%–76%), weight loss, arthritis (5%–34%), and leukopenia (25%–74%).4,8,9
Cutaneous findings have been described in up to 40% of cases, of which clinical presentation is variable.6 Lesions may include blanchable, erythematous, painful, and/or indurated plaques, nodules, or maculopapules with confluence into patches, urticaria, morbilliform lesions, erythema multiforme, eyelid edema, leukocytoclastic vasculitis, papulopustules, ulcerated gingivae, and mucositis.6,7,10-13 Patients with skin lesions may be at an increased risk for developing systemic lupus erythematosus (SLE).8 Our patient presented with erythematous scaly plaques with a predominance of lesions in photodistributed locations, which clinically mimicked an underlying connective tissue disease process such as SLE.
Infectious agents such as CMV, parvovirus B19, human herpesvirus 6, human herpesvirus 8 and human T-cell lymphotropic virus 1, HIV, Yersinia enterocolitica, and Toxoplasma have all been implicated as possible causes of Kikuchi-Fujimoto disease, but studies have failed to provide convincing causal evidence.9,14,15 Our patient had positive IgM and IgG for CMV, which may have incited his disease.
Definitive diagnosis of Kikuchi-Fujimoto disease is made by lymph node excisional biopsy, which histologically exhibits a histiocytic cell proliferation with paracortical foci of necrosis and abundant karyorrhectic debris.5 Cutaneous histologic findings that support the diagnosis are variable and may include a dermal histiocytic infiltrate, epidermal change with necrotic keratinocytes, non-neutrophilic karyorrhectic debris, basal vacuolar change, papillary dermal edema, a nonspecific superficial and deep perivascular infiltrate, and a patchy infiltration of histiocytes and lymphocytes.6,13
Clinical and histopathological features of this disease can mimic other diseases, specifically SLE or lymphoma.7 An association with SLE has been suspected, though it is not well defined and more frequently is associated with cases from Asia than from Europe (28% and 9%, respectively).9 Patients presenting concomitantly with positive ANA, weight loss, arthralgia, and skin lesions are more likely to develop SLE.8 Furthermore, the cutaneous histologic finding of interface change suggests a link between the two diseases. As such, recommendations have been made for ANA screenings and follow-up of patients diagnosed with Kikuchi-Fujimoto disease for clinical evidence of autoimmune disease, particularly SLE.6 Although our patient did not have a positive ANA, his biopsy did demonstrate interface change, and he should be monitored for possible progression of disease in the future.
Kikuchi-Fujimoto disease differs from lymphoma, as it initially presents with rapid lymph node enlargement as opposed to the gradual enlargement seen in lymphoma. The lymph nodes in Kikuchi-Fujimoto disease often are firm and moveable compared to hard and immobile in lymphoma.3 Excisional lymph node biopsy is necessary for both confirming the diagnosis of Kikuchi-Fujimoto disease and ruling out lymphoma.5
Spontaneous resolution usually occurs in 1 to 4 months.3,6 As such, observation is the most common approach to management. When patients have symptoms that limit activities or cause undue distress such as fevers, joint pains, or abdominal pain, systemic treatment options may be desired. Symptomatic treatment can be managed with a short duration of oral corticosteroids,10,11 nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.8-15 There are no guidelines regarding systemic steroid regimens, and various treatment schedules have been successful. Systemic therapy was considered for our patient for his weight loss and abdominal pain; however, by the time of discharge the patient was tolerating oral intake and his abdominal pain had improved.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:379-380.
- Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis: a new clinicopathological entity. Naika. 1972;30:920-927.
- Feder Jr HM, Liu J, Rezuke WN. Kikuchi disease in Connecticut. J Pediatr. 2014;164:196-200.
- Kang HM, Kim JY, Choi EH, et al. Clinical characteristics of severe histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease) in children. J Pediatr. 2016;171:208-212.
- Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134:289-293.
- Atwater AR, Longly BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yen H-R, Lin P-Y, Chuang W-Y, et al. Skin manifestations of Kikuchi-Fujimoto disease: case report and review. Eur J Pediatr. 2004;163:210-213.
- Dumas G, Prendki V, Haroche J, et al. Kikuchi-Fujimoto disease: retrospective study of 91 cases and review of literature. Medicine. 2014;93:372-382.
- Kuc ukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Yasukawa K, Matsumura T, Sato-Matsumura KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Kaur S, Thami GP, Mohan H, et al. Kikuchi disease with facial rash and erythema multiforme. Pediatr Dermatol. 2001;18:403-405.
- Mauleón C, Valdivielso-Ramos M, Cabeza R, et al. Kikuchi disease with skin lesions mimicking lupus erythematosus. J Dermatol Case Rep. 2012;3:82-85.
- Obara K, Amoh Y. A case of Kikuchi’s disease (histiocytic necrotizing lymphoadenitis) with histiocytic cutaneous involvement. Rheumatol Int. 2015;35:1111-1113.
- Rosado FGN, Tang Y-W, Hasserjian RP, et al. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44:255-259.
- Chiu CF, Chow KC, Lin TY, et al. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113:774-781.
To the Editor:
Kikuchi-Fujimoto Disease, also called histiocytic necrotizing lymphadenitis, was described in 1972 by both Kikuchi1 and Fujimoto et al.2 Most cases are reported in Asia, with limited reports in the United States.3-5 Kikuchi-Fujimoto disease is a rare, self-limiting condition consisting of benign lymphadenopathy and oftentimes fever and systemic symptoms. Lymph node involvement may mimic non-Hodgkin lymphoma or other reactive lymphadenopathy, rendering diagnostic accuracy challenging.5 Cutaneous manifestations are reported in only 16% to 40% of patients.6,7 Herein, we describe the clinical and pathologic features of a case of Kikuchi-Fujimoto disease with cutaneous involvement in an adolescent boy.
A 13-year-old adolescent boy with no notable medical history presented to the pediatric emergency department with cervical lymphadenopathy, weight loss, intermittent fever, and an evolving rash on the face, ears, arms, and thighs of 6 weeks’ duration. The illness began with enlarged lymph nodes and erythematous macules on the face and was diagnosed by his primary care physician as lymphadenitis that was unresponsive to clindamycin. Over the subsequent weeks, the rash worsened, and he developed intermittent fevers, night sweats, abdominal pain, and nausea with a 20-pound weight loss. He presented to the emergency department 3 weeks prior to the current admission and was noted to have elevated cytomegalovirus (CMV) IgM and IgG in addition to lymphopenia and anemia. He was discharged with outpatient follow-up. The rash progressed to involve the face, ears, arms, and thighs. One day prior to the current admission, the patient’s abdominal pain worsened acutely, and he experienced several episodes of emesis. He presented to the pediatric emergency department for further evaluation, and a dermatology consultation was requested at that time.
The patient’s rash was asymptomatic. In addition to the above symptoms, he also noted frequent nosebleeds, gingival bleeding, and diffuse myalgia that was most prominent on the hands and feet; he denied diarrhea, sick contacts, recent travel, or insect bites. His vital signs were normal, and he remained afebrile throughout the hospitalization. Physical examination revealed an ill-appearing patient with sunken eyes and dry lips. He had pink, oval, scaly plaques on the cheeks, ears, and arms (Figure 1). The thighs exhibited folliculocentric erythematous papules. The ocular conjunctivae were clear, but white exudative plaques were noted on the tongue. Tender, bilateral, cervical lymphadenopathy and diffuse abdominal tenderness with guarding and hepatosplenomegaly also were present. The fingers and toes were tender upon palpation.
Laboratory workup at admission revealed the following: low white blood cell count, 2700/μL (reference range, 4500–11,000/μL); low hemoglobin, 9.6 g/dL (reference range, 14.0–17.5 g/dL); elevated aspartate aminotransferase, 91 U/L (reference range, 10–30 U/L); and elevated alanine aminotransferase, 118 U/L (reference range, 10–40 U/L). Lactate dehydrogenase (582 U/L [reference range, 100–200 U/L]), ferritin (1681 ng/mL [reference range, 15–200 ng/mL]), and C-reactive protein (6.0 mg/L [reference range, 0.08–3.1 mg/L]) also were elevated. A respiratory viral panel was unremarkable. Blood cultures were negative, and an HIV 1/2 assay was nonreactive. A chest radiograph demonstrated clear lung fields. Computed tomography of the abdomen and pelvis showed prominent mesenteric, ileocolic, and retroperitoneal lymph nodes.
The differential diagnoses at this time included acute connective tissue disease, a paraneoplastic phenomenon, cutaneous lymphoma, or an infectious etiology. A punch biopsy of the skin as well as tissue cultures were performed from a lesion on the right arm. Quantitative immunoglobulin (IgA, IgG, IgM) levels were checked, all of which were within reference range. An antinuclear antibody (ANA) assay and rheumatoid factor were normal.
The tissue cultures were negative for bacteria, fungi, and mycobacteria. Microscopic examination of the skin biopsy revealed a moderate perivascular and interstitial infiltrate of predominantly histiocytes and lymphocytes with prominent karyorrhectic debris (nuclear dust) in the upper dermis as well as focal vacuolar interface changes with scattered necrotic keratinocytes in the epidermis (Figure 2). Based on these histopathologic findings, a diagnosis of Kikuchi-Fujimoto disease was considered. To confirm the diagnosis and to rule out the possibility of lymphoma, an excisional biopsy of the cervical lymph node was performed, which showed typical histopathologic features of histiocytic necrotizing lymphadenitis.
Given the patient’s clinical presentation with arthralgia, anorexia, lymphadenitis, and hepatosplenomegaly along with histopathologic findings from both the skin and lymph node biopsies, a diagnosis of Kikuchi-Fujimoto disease was made. The patient was conservatively managed with acetaminophen and was discharged with improvement in his appetite and systemic symptoms.
He was seen for follow-up 3 months later in the outpatient clinic. He denied any recurrence of systemic symptoms but endorsed a recent shedding of hair consistent with telogen effluvium. The rash had substantially improved, though residual asymptomatic erythematous plaques remained on the right forehead and right cheek (Figure 3). He was prescribed triamcinolone acetonide cream 0.1% to apply to the active area twice daily for the following 2 to 3 weeks.
Kikuchi-Fujimoto disease presents with a wide clinical spectrum, classically with benign lymphadenopathy and fever of unknown etiology.5,6 Lymphadenopathy most often is cervical (55%–99%)8 and unilateral,4,7 but patients can present with polyadenopathy (52%).7,8 Constitutional signs commonly include fever (35%–76%), weight loss, arthritis (5%–34%), and leukopenia (25%–74%).4,8,9
Cutaneous findings have been described in up to 40% of cases, of which clinical presentation is variable.6 Lesions may include blanchable, erythematous, painful, and/or indurated plaques, nodules, or maculopapules with confluence into patches, urticaria, morbilliform lesions, erythema multiforme, eyelid edema, leukocytoclastic vasculitis, papulopustules, ulcerated gingivae, and mucositis.6,7,10-13 Patients with skin lesions may be at an increased risk for developing systemic lupus erythematosus (SLE).8 Our patient presented with erythematous scaly plaques with a predominance of lesions in photodistributed locations, which clinically mimicked an underlying connective tissue disease process such as SLE.
Infectious agents such as CMV, parvovirus B19, human herpesvirus 6, human herpesvirus 8 and human T-cell lymphotropic virus 1, HIV, Yersinia enterocolitica, and Toxoplasma have all been implicated as possible causes of Kikuchi-Fujimoto disease, but studies have failed to provide convincing causal evidence.9,14,15 Our patient had positive IgM and IgG for CMV, which may have incited his disease.
Definitive diagnosis of Kikuchi-Fujimoto disease is made by lymph node excisional biopsy, which histologically exhibits a histiocytic cell proliferation with paracortical foci of necrosis and abundant karyorrhectic debris.5 Cutaneous histologic findings that support the diagnosis are variable and may include a dermal histiocytic infiltrate, epidermal change with necrotic keratinocytes, non-neutrophilic karyorrhectic debris, basal vacuolar change, papillary dermal edema, a nonspecific superficial and deep perivascular infiltrate, and a patchy infiltration of histiocytes and lymphocytes.6,13
Clinical and histopathological features of this disease can mimic other diseases, specifically SLE or lymphoma.7 An association with SLE has been suspected, though it is not well defined and more frequently is associated with cases from Asia than from Europe (28% and 9%, respectively).9 Patients presenting concomitantly with positive ANA, weight loss, arthralgia, and skin lesions are more likely to develop SLE.8 Furthermore, the cutaneous histologic finding of interface change suggests a link between the two diseases. As such, recommendations have been made for ANA screenings and follow-up of patients diagnosed with Kikuchi-Fujimoto disease for clinical evidence of autoimmune disease, particularly SLE.6 Although our patient did not have a positive ANA, his biopsy did demonstrate interface change, and he should be monitored for possible progression of disease in the future.
Kikuchi-Fujimoto disease differs from lymphoma, as it initially presents with rapid lymph node enlargement as opposed to the gradual enlargement seen in lymphoma. The lymph nodes in Kikuchi-Fujimoto disease often are firm and moveable compared to hard and immobile in lymphoma.3 Excisional lymph node biopsy is necessary for both confirming the diagnosis of Kikuchi-Fujimoto disease and ruling out lymphoma.5
Spontaneous resolution usually occurs in 1 to 4 months.3,6 As such, observation is the most common approach to management. When patients have symptoms that limit activities or cause undue distress such as fevers, joint pains, or abdominal pain, systemic treatment options may be desired. Symptomatic treatment can be managed with a short duration of oral corticosteroids,10,11 nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.8-15 There are no guidelines regarding systemic steroid regimens, and various treatment schedules have been successful. Systemic therapy was considered for our patient for his weight loss and abdominal pain; however, by the time of discharge the patient was tolerating oral intake and his abdominal pain had improved.
To the Editor:
Kikuchi-Fujimoto Disease, also called histiocytic necrotizing lymphadenitis, was described in 1972 by both Kikuchi1 and Fujimoto et al.2 Most cases are reported in Asia, with limited reports in the United States.3-5 Kikuchi-Fujimoto disease is a rare, self-limiting condition consisting of benign lymphadenopathy and oftentimes fever and systemic symptoms. Lymph node involvement may mimic non-Hodgkin lymphoma or other reactive lymphadenopathy, rendering diagnostic accuracy challenging.5 Cutaneous manifestations are reported in only 16% to 40% of patients.6,7 Herein, we describe the clinical and pathologic features of a case of Kikuchi-Fujimoto disease with cutaneous involvement in an adolescent boy.
A 13-year-old adolescent boy with no notable medical history presented to the pediatric emergency department with cervical lymphadenopathy, weight loss, intermittent fever, and an evolving rash on the face, ears, arms, and thighs of 6 weeks’ duration. The illness began with enlarged lymph nodes and erythematous macules on the face and was diagnosed by his primary care physician as lymphadenitis that was unresponsive to clindamycin. Over the subsequent weeks, the rash worsened, and he developed intermittent fevers, night sweats, abdominal pain, and nausea with a 20-pound weight loss. He presented to the emergency department 3 weeks prior to the current admission and was noted to have elevated cytomegalovirus (CMV) IgM and IgG in addition to lymphopenia and anemia. He was discharged with outpatient follow-up. The rash progressed to involve the face, ears, arms, and thighs. One day prior to the current admission, the patient’s abdominal pain worsened acutely, and he experienced several episodes of emesis. He presented to the pediatric emergency department for further evaluation, and a dermatology consultation was requested at that time.
The patient’s rash was asymptomatic. In addition to the above symptoms, he also noted frequent nosebleeds, gingival bleeding, and diffuse myalgia that was most prominent on the hands and feet; he denied diarrhea, sick contacts, recent travel, or insect bites. His vital signs were normal, and he remained afebrile throughout the hospitalization. Physical examination revealed an ill-appearing patient with sunken eyes and dry lips. He had pink, oval, scaly plaques on the cheeks, ears, and arms (Figure 1). The thighs exhibited folliculocentric erythematous papules. The ocular conjunctivae were clear, but white exudative plaques were noted on the tongue. Tender, bilateral, cervical lymphadenopathy and diffuse abdominal tenderness with guarding and hepatosplenomegaly also were present. The fingers and toes were tender upon palpation.
Laboratory workup at admission revealed the following: low white blood cell count, 2700/μL (reference range, 4500–11,000/μL); low hemoglobin, 9.6 g/dL (reference range, 14.0–17.5 g/dL); elevated aspartate aminotransferase, 91 U/L (reference range, 10–30 U/L); and elevated alanine aminotransferase, 118 U/L (reference range, 10–40 U/L). Lactate dehydrogenase (582 U/L [reference range, 100–200 U/L]), ferritin (1681 ng/mL [reference range, 15–200 ng/mL]), and C-reactive protein (6.0 mg/L [reference range, 0.08–3.1 mg/L]) also were elevated. A respiratory viral panel was unremarkable. Blood cultures were negative, and an HIV 1/2 assay was nonreactive. A chest radiograph demonstrated clear lung fields. Computed tomography of the abdomen and pelvis showed prominent mesenteric, ileocolic, and retroperitoneal lymph nodes.
The differential diagnoses at this time included acute connective tissue disease, a paraneoplastic phenomenon, cutaneous lymphoma, or an infectious etiology. A punch biopsy of the skin as well as tissue cultures were performed from a lesion on the right arm. Quantitative immunoglobulin (IgA, IgG, IgM) levels were checked, all of which were within reference range. An antinuclear antibody (ANA) assay and rheumatoid factor were normal.
The tissue cultures were negative for bacteria, fungi, and mycobacteria. Microscopic examination of the skin biopsy revealed a moderate perivascular and interstitial infiltrate of predominantly histiocytes and lymphocytes with prominent karyorrhectic debris (nuclear dust) in the upper dermis as well as focal vacuolar interface changes with scattered necrotic keratinocytes in the epidermis (Figure 2). Based on these histopathologic findings, a diagnosis of Kikuchi-Fujimoto disease was considered. To confirm the diagnosis and to rule out the possibility of lymphoma, an excisional biopsy of the cervical lymph node was performed, which showed typical histopathologic features of histiocytic necrotizing lymphadenitis.
Given the patient’s clinical presentation with arthralgia, anorexia, lymphadenitis, and hepatosplenomegaly along with histopathologic findings from both the skin and lymph node biopsies, a diagnosis of Kikuchi-Fujimoto disease was made. The patient was conservatively managed with acetaminophen and was discharged with improvement in his appetite and systemic symptoms.
He was seen for follow-up 3 months later in the outpatient clinic. He denied any recurrence of systemic symptoms but endorsed a recent shedding of hair consistent with telogen effluvium. The rash had substantially improved, though residual asymptomatic erythematous plaques remained on the right forehead and right cheek (Figure 3). He was prescribed triamcinolone acetonide cream 0.1% to apply to the active area twice daily for the following 2 to 3 weeks.
Kikuchi-Fujimoto disease presents with a wide clinical spectrum, classically with benign lymphadenopathy and fever of unknown etiology.5,6 Lymphadenopathy most often is cervical (55%–99%)8 and unilateral,4,7 but patients can present with polyadenopathy (52%).7,8 Constitutional signs commonly include fever (35%–76%), weight loss, arthritis (5%–34%), and leukopenia (25%–74%).4,8,9
Cutaneous findings have been described in up to 40% of cases, of which clinical presentation is variable.6 Lesions may include blanchable, erythematous, painful, and/or indurated plaques, nodules, or maculopapules with confluence into patches, urticaria, morbilliform lesions, erythema multiforme, eyelid edema, leukocytoclastic vasculitis, papulopustules, ulcerated gingivae, and mucositis.6,7,10-13 Patients with skin lesions may be at an increased risk for developing systemic lupus erythematosus (SLE).8 Our patient presented with erythematous scaly plaques with a predominance of lesions in photodistributed locations, which clinically mimicked an underlying connective tissue disease process such as SLE.
Infectious agents such as CMV, parvovirus B19, human herpesvirus 6, human herpesvirus 8 and human T-cell lymphotropic virus 1, HIV, Yersinia enterocolitica, and Toxoplasma have all been implicated as possible causes of Kikuchi-Fujimoto disease, but studies have failed to provide convincing causal evidence.9,14,15 Our patient had positive IgM and IgG for CMV, which may have incited his disease.
Definitive diagnosis of Kikuchi-Fujimoto disease is made by lymph node excisional biopsy, which histologically exhibits a histiocytic cell proliferation with paracortical foci of necrosis and abundant karyorrhectic debris.5 Cutaneous histologic findings that support the diagnosis are variable and may include a dermal histiocytic infiltrate, epidermal change with necrotic keratinocytes, non-neutrophilic karyorrhectic debris, basal vacuolar change, papillary dermal edema, a nonspecific superficial and deep perivascular infiltrate, and a patchy infiltration of histiocytes and lymphocytes.6,13
Clinical and histopathological features of this disease can mimic other diseases, specifically SLE or lymphoma.7 An association with SLE has been suspected, though it is not well defined and more frequently is associated with cases from Asia than from Europe (28% and 9%, respectively).9 Patients presenting concomitantly with positive ANA, weight loss, arthralgia, and skin lesions are more likely to develop SLE.8 Furthermore, the cutaneous histologic finding of interface change suggests a link between the two diseases. As such, recommendations have been made for ANA screenings and follow-up of patients diagnosed with Kikuchi-Fujimoto disease for clinical evidence of autoimmune disease, particularly SLE.6 Although our patient did not have a positive ANA, his biopsy did demonstrate interface change, and he should be monitored for possible progression of disease in the future.
Kikuchi-Fujimoto disease differs from lymphoma, as it initially presents with rapid lymph node enlargement as opposed to the gradual enlargement seen in lymphoma. The lymph nodes in Kikuchi-Fujimoto disease often are firm and moveable compared to hard and immobile in lymphoma.3 Excisional lymph node biopsy is necessary for both confirming the diagnosis of Kikuchi-Fujimoto disease and ruling out lymphoma.5
Spontaneous resolution usually occurs in 1 to 4 months.3,6 As such, observation is the most common approach to management. When patients have symptoms that limit activities or cause undue distress such as fevers, joint pains, or abdominal pain, systemic treatment options may be desired. Symptomatic treatment can be managed with a short duration of oral corticosteroids,10,11 nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.8-15 There are no guidelines regarding systemic steroid regimens, and various treatment schedules have been successful. Systemic therapy was considered for our patient for his weight loss and abdominal pain; however, by the time of discharge the patient was tolerating oral intake and his abdominal pain had improved.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:379-380.
- Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis: a new clinicopathological entity. Naika. 1972;30:920-927.
- Feder Jr HM, Liu J, Rezuke WN. Kikuchi disease in Connecticut. J Pediatr. 2014;164:196-200.
- Kang HM, Kim JY, Choi EH, et al. Clinical characteristics of severe histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease) in children. J Pediatr. 2016;171:208-212.
- Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134:289-293.
- Atwater AR, Longly BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yen H-R, Lin P-Y, Chuang W-Y, et al. Skin manifestations of Kikuchi-Fujimoto disease: case report and review. Eur J Pediatr. 2004;163:210-213.
- Dumas G, Prendki V, Haroche J, et al. Kikuchi-Fujimoto disease: retrospective study of 91 cases and review of literature. Medicine. 2014;93:372-382.
- Kuc ukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Yasukawa K, Matsumura T, Sato-Matsumura KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Kaur S, Thami GP, Mohan H, et al. Kikuchi disease with facial rash and erythema multiforme. Pediatr Dermatol. 2001;18:403-405.
- Mauleón C, Valdivielso-Ramos M, Cabeza R, et al. Kikuchi disease with skin lesions mimicking lupus erythematosus. J Dermatol Case Rep. 2012;3:82-85.
- Obara K, Amoh Y. A case of Kikuchi’s disease (histiocytic necrotizing lymphoadenitis) with histiocytic cutaneous involvement. Rheumatol Int. 2015;35:1111-1113.
- Rosado FGN, Tang Y-W, Hasserjian RP, et al. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44:255-259.
- Chiu CF, Chow KC, Lin TY, et al. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113:774-781.
- Kikuchi M. Lymphadenitis showing focal reticulum cell hyperplasia with nuclear debris and phagocytosis. Nippon Ketsueki Gakkai Zasshi. 1972;35:379-380.
- Fujimoto Y, Kojima Y, Yamaguchi K. Cervical subacute necrotizing lymphadenitis: a new clinicopathological entity. Naika. 1972;30:920-927.
- Feder Jr HM, Liu J, Rezuke WN. Kikuchi disease in Connecticut. J Pediatr. 2014;164:196-200.
- Kang HM, Kim JY, Choi EH, et al. Clinical characteristics of severe histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease) in children. J Pediatr. 2016;171:208-212.
- Hutchinson CB, Wang E. Kikuchi-Fujimoto disease. Arch Pathol Lab Med. 2010;134:289-293.
- Atwater AR, Longly BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yen H-R, Lin P-Y, Chuang W-Y, et al. Skin manifestations of Kikuchi-Fujimoto disease: case report and review. Eur J Pediatr. 2004;163:210-213.
- Dumas G, Prendki V, Haroche J, et al. Kikuchi-Fujimoto disease: retrospective study of 91 cases and review of literature. Medicine. 2014;93:372-382.
- Kuc ukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Yasukawa K, Matsumura T, Sato-Matsumura KC, et al. Kikuchi’s disease and the skin: case report and review of the literature. Br J Dermatol. 2001;144:885-889.
- Kaur S, Thami GP, Mohan H, et al. Kikuchi disease with facial rash and erythema multiforme. Pediatr Dermatol. 2001;18:403-405.
- Mauleón C, Valdivielso-Ramos M, Cabeza R, et al. Kikuchi disease with skin lesions mimicking lupus erythematosus. J Dermatol Case Rep. 2012;3:82-85.
- Obara K, Amoh Y. A case of Kikuchi’s disease (histiocytic necrotizing lymphoadenitis) with histiocytic cutaneous involvement. Rheumatol Int. 2015;35:1111-1113.
- Rosado FGN, Tang Y-W, Hasserjian RP, et al. Kikuchi-Fujimoto lymphadenitis: role of parvovirus B-19, Epstein-Barr virus, human herpesvirus 6, and human herpesvirus 8. Hum Pathol. 2013;44:255-259.
- Chiu CF, Chow KC, Lin TY, et al. Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J Clin Pathol. 2000;113:774-781.
Practice Points
- Kikuchi-Fujimoto disease is an uncommon, self-limited condition characterized by benign lymphadenopathy and variable systemic symptoms.
- Definitive diagnosis is made by excisional lymph node biopsy.
- Treatment options include oral corticosteroids, nonsteroidal anti-inflammatory drugs, antimalarials, and/or antipyretics.
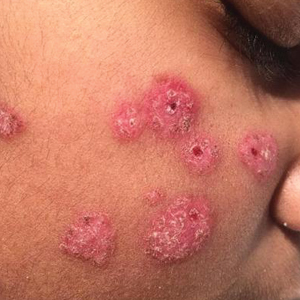
New York’s largest health care provider fires 1,400 unvaccinated employees
The employees represented less than 2% of Northwell’s 76,000 employees, who are now all fully vaccinated against COVID-19, Joe Kemp, the assistant vice president of public relations for the company, told The Hill.
“Northwell Health is proud to announce that our workforce -- the largest in New York State -- is 100% vaccinated,” the company said in a statement to several news outlets.
“This allows us to continue to provide exceptional care at all of our facilities, without interruption and remain open and fully operational,” Northwell Health said.
Having a fully vaccinated workforce is part of the health system’s duty to protect others, the company said. Northwell Health includes 23 hospitals and more than 830 outpatient facilities, according to ABC News.
“Northwell regrets losing any employee under such circumstances,” the company said. “We owe it to our staff, our patients, and the communities we serve to be 100% vaccinated against COVID-19.”
Former New York Gov. Andrew Cuomo announced in August that the state would require health care workers to receive at least one COVID-19 vaccine shot by Sept. 27. Employees didn’t have the option for weekly testing or religious exemptions, which is being challenged in several lawsuits, according to The New York Times.
The order went into effect last week, prompting tens of thousands of employees to get vaccinated. As of last week, 87% of hospital staff were fully vaccinated, and 92% of hospital and retirement home workers had received at least one dose, according to state health data.
Northwell announced its own vaccine mandate in August as well, which sparked protests among some workers. The order applied to both clinical and non-clinical staff.
A few thousand Northwell employees got vaccinated as the deadline approached, Mr. Kemp told The New York Times. Some who lost their jobs at first were able to return to work, and those who have been terminated can interview for reinstatement for 30 days. The hospital system is also “openly recruiting” for the vacant positions.
“The goal was to get people vaccinated, not to get people terminated,” Mr. Kemp said.
Hospitalized COVID-19 patients in New York hit a low of 350 in mid-July, according to state hospitalization data. Now, about 2,200 people are hospitalized throughout the state, most of whom are unvaccinated.
As of Oct. 3, nearly 72% of New York residents had received at least one vaccine dose, according to the latest state data. About 64% are fully vaccinated.
A version of this article first appeared on WebMD.com.
The employees represented less than 2% of Northwell’s 76,000 employees, who are now all fully vaccinated against COVID-19, Joe Kemp, the assistant vice president of public relations for the company, told The Hill.
“Northwell Health is proud to announce that our workforce -- the largest in New York State -- is 100% vaccinated,” the company said in a statement to several news outlets.
“This allows us to continue to provide exceptional care at all of our facilities, without interruption and remain open and fully operational,” Northwell Health said.
Having a fully vaccinated workforce is part of the health system’s duty to protect others, the company said. Northwell Health includes 23 hospitals and more than 830 outpatient facilities, according to ABC News.
“Northwell regrets losing any employee under such circumstances,” the company said. “We owe it to our staff, our patients, and the communities we serve to be 100% vaccinated against COVID-19.”
Former New York Gov. Andrew Cuomo announced in August that the state would require health care workers to receive at least one COVID-19 vaccine shot by Sept. 27. Employees didn’t have the option for weekly testing or religious exemptions, which is being challenged in several lawsuits, according to The New York Times.
The order went into effect last week, prompting tens of thousands of employees to get vaccinated. As of last week, 87% of hospital staff were fully vaccinated, and 92% of hospital and retirement home workers had received at least one dose, according to state health data.
Northwell announced its own vaccine mandate in August as well, which sparked protests among some workers. The order applied to both clinical and non-clinical staff.
A few thousand Northwell employees got vaccinated as the deadline approached, Mr. Kemp told The New York Times. Some who lost their jobs at first were able to return to work, and those who have been terminated can interview for reinstatement for 30 days. The hospital system is also “openly recruiting” for the vacant positions.
“The goal was to get people vaccinated, not to get people terminated,” Mr. Kemp said.
Hospitalized COVID-19 patients in New York hit a low of 350 in mid-July, according to state hospitalization data. Now, about 2,200 people are hospitalized throughout the state, most of whom are unvaccinated.
As of Oct. 3, nearly 72% of New York residents had received at least one vaccine dose, according to the latest state data. About 64% are fully vaccinated.
A version of this article first appeared on WebMD.com.
The employees represented less than 2% of Northwell’s 76,000 employees, who are now all fully vaccinated against COVID-19, Joe Kemp, the assistant vice president of public relations for the company, told The Hill.
“Northwell Health is proud to announce that our workforce -- the largest in New York State -- is 100% vaccinated,” the company said in a statement to several news outlets.
“This allows us to continue to provide exceptional care at all of our facilities, without interruption and remain open and fully operational,” Northwell Health said.
Having a fully vaccinated workforce is part of the health system’s duty to protect others, the company said. Northwell Health includes 23 hospitals and more than 830 outpatient facilities, according to ABC News.
“Northwell regrets losing any employee under such circumstances,” the company said. “We owe it to our staff, our patients, and the communities we serve to be 100% vaccinated against COVID-19.”
Former New York Gov. Andrew Cuomo announced in August that the state would require health care workers to receive at least one COVID-19 vaccine shot by Sept. 27. Employees didn’t have the option for weekly testing or religious exemptions, which is being challenged in several lawsuits, according to The New York Times.
The order went into effect last week, prompting tens of thousands of employees to get vaccinated. As of last week, 87% of hospital staff were fully vaccinated, and 92% of hospital and retirement home workers had received at least one dose, according to state health data.
Northwell announced its own vaccine mandate in August as well, which sparked protests among some workers. The order applied to both clinical and non-clinical staff.
A few thousand Northwell employees got vaccinated as the deadline approached, Mr. Kemp told The New York Times. Some who lost their jobs at first were able to return to work, and those who have been terminated can interview for reinstatement for 30 days. The hospital system is also “openly recruiting” for the vacant positions.
“The goal was to get people vaccinated, not to get people terminated,” Mr. Kemp said.
Hospitalized COVID-19 patients in New York hit a low of 350 in mid-July, according to state hospitalization data. Now, about 2,200 people are hospitalized throughout the state, most of whom are unvaccinated.
As of Oct. 3, nearly 72% of New York residents had received at least one vaccine dose, according to the latest state data. About 64% are fully vaccinated.
A version of this article first appeared on WebMD.com.
Chronic Hyperpigmented Patches on the Legs
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
The Diagnosis: Drug-Induced Hyperpigmentation
Additional history provided by the patient’s caretaker elucidated an extensive list of medications including chlorpromazine and minocycline, among several others. The caretaker revealed that the patient began treatment for acne vulgaris 2 years prior; despite the acne resolving, therapy was not discontinued. The blue-gray and brown pigmentation on our patient’s shins likely was attributed to a medication he was taking.
Both chlorpromazine and minocycline, among many other medications, are known to cause abnormal pigmentation of the skin.1 Minocycline is a tetracycline antibiotic prescribed for acne and other inflammatory cutaneous conditions. It is highly lipophilic, allowing it to reach high drug concentrations in the skin and nail unit.2 Patients taking minocycline long term and at high doses are at greatest risk for pigment deposition.3,4
Minocycline-induced hyperpigmentation is classified into 3 types. Type I describes blue-black deposition of pigment in acne scars and areas of inflammation, typically on facial skin.1,5 Histologically, type I stains positive for Perls Prussian blue, indicating an increased deposition of iron as hemosiderin,1 which likely occurs because minocycline is thought to play a role in defective clearance of hemosiderin from the dermis of injured tissue.5 Type II hyperpigmentation presents as bluegray pigment on the lower legs and occasionally the arms.6,7 Type II stains positive for both Perls Prussian blue and Fontana-Masson, demonstrating hemosiderin and melanin, respectively.6 The third form of hyperpigmentation results in diffuse, dark brown to gray pigmentation with a predilection for sun-exposed areas.8 Histology of type III shows increased pigment in the basal portion of the epidermis and brown-black pigment in macrophages of the dermis. Type III stains positive for Fontana-Masson and negative for Perls Prussian blue. The etiology of hyperpigmentation has been suspected to be caused by minocycline stimulating melanin production and/or deposition of minocycline-melanin complexes in dermal macrophages after a certain drug level; this largely is seen in patients receiving 100 to 200 mg daily as early as 1 year into treatment.8
Chlorpromazine is a typical antipsychotic that causes abnormal skin pigmentation in sun-exposed areas due to increased melanogenesis.9 Similar to type III minocyclineinduced hyperpigmentation, a histologic specimen may stain positive for Fontana-Masson yet negative for Perls Prussian blue. Lal et al10 demonstrated complete resolution of abnormal skin pigmentation within 5 years after stopping chlorpromazine. In contrast, minocyclineinduced hyperpigmentation may be permanent in some cases. There is substantial clinical and histologic overlap for drug-induced hyperpigmentation etiologies; it would behoove the clinician to focus on the most common locations affected and the generalized coloration.
Treatment of minocycline-induced hyperpigmentation includes the use of Q-switched lasers, specifically Q-switched ruby and Q-switched alexandrite.11 The use of the Q-switched Nd:YAG laser appears to be ineffective at clearing minocycline-induced pigmentation.7,11 In our patient, minocycline was discontinued immediately. Due to the patient’s critical condition, he deferred all other therapy. Erythema dyschromicum perstans, also referred to as ashy dermatosis, is an idiopathic form of hyperpigmentation.12 Lesions start as blue-gray to ashy gray macules, occasionally surrounded by a slightly erythematous, raised border.
Erythema dyschromicum perstans typically presents on the trunk, face, and arms of patients with Fitzpatrick skin types III and IV; it is considered a variant of lichen planus actinicus.12 Histologically, erythema dyschromicum perstans may mimic lichen planus pigmentosus (LPP); however, subtle differences exist to distinguish the 2 conditions. Erythema dyschromicum perstans demonstrates a mild lichenoid infiltrate, focal basal vacuolization at the dermoepidermal junction, and melanophage deposition.13 In contrast, LPP demonstrates pigmentary incontinence and a more severe inflammatory infiltrate. A perifollicular infiltrate and fibrosis also can be seen in LPP, which may explain the frontal fibrosing alopecia that often precedes LPP.13
Addison disease, also known as primary adrenal insufficiency, can cause diffuse hyperpigmentation in the skin, mucosae, and nail beds. The pigmentation is prominent in regions of naturally increased pigmentation, such as the flexural surfaces and intertriginous areas.14 Patients with adrenal insufficiency will have accompanying weight loss, hypotension, and fatigue, among other symptoms related to deficiency of cortisol and aldosterone. Skin biopsy shows acanthosis, hyperkeratosis, focal parakeratosis, spongiosis, superficial perivascular lymphocytic infiltrate, basal melanin deposition, and superficial dermal macrophages.15
Confluent and reticulated papillomatosis is an uncommon dermatosis that presents with multiple hyperpigmented macules and papules that coalesce to form patches and plaques centrally with reticulation in the periphery.16 Confluent and reticulated papillomatosis commonly presents on the upper trunk, axillae, and neck, though involvement can include flexural surfaces as well as the lower trunk and legs.16,17 Biopsy demonstrates undulating hyperkeratosis, papillomatosis, acanthosis, and negative fungal staining.16
Pretibial myxedema most commonly is associated with Graves disease and presents as well-defined thickening and induration with overlying pink or purple-brown papules in the pretibial region.18 An acral surface and mucin deposition within the entire dermis may be appreciated on histology with staining for colloidal iron or Alcian blue.
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
- Fenske NA, Millns JL, Greer KE. Minocycline-induced pigmentation at sites of cutaneous inflammation. JAMA. 1980;244:1103-1106. doi:10.1001/jama.1980.03310100021021
- Snodgrass A, Motaparthi K. Systemic antibacterial agents. In: Wolverton SE, Wu JJ, eds. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2020:69-98.
- Eisen D, Hakim MD. Minocycline-induced pigmentation. incidence, prevention and management. Drug Saf. 1998;18:431-440. doi:10.2165/00002018-199818060-00004
- Goulden V, Glass D, Cunliffe WJ. Safety of long-term high-dose minocycline in the treatment of acne. Br J Dermatol. 1996;134:693-695. doi:10.1111/j.1365-2133.1996.tb06972.x
- Basler RS, Kohnen PW. Localized hemosiderosis as a sequela of acne. Arch Dermatol. 1978;114:1695-1697.
- Ridgway HA, Sonnex TS, Kennedy CT, et al. Hyperpigmentation associated with oral minocycline. Br J Dermatol. 1982;107:95-102. doi:10.1111/j.1365-2133.1982.tb00296.x
- Nisar MS, Iyer K, Brodell RT, et al. Minocycline-induced hyperpigmentation: comparison of 3 Q-switched lasers to reverse its effects. Clin Cosmet Investig Dermatol. 2013;6:159-162. doi:10.2147/CCID.S42166
- Simons JJ, Morales A. Minocycline and generalized cutaneous pigmentation. J Am Acad Dermatol. 1980;3:244-247. doi:10.1016/s0190 -9622(80)80186-1
- Perry TL, Culling CF, Berry K, et al. 7-Hydroxychlorpromazine: potential toxic drug metabolite in psychiatric patients. Science. 1964;146:81-83. doi:10.1126/science.146.3640.81
- Lal S, Bloom D, Silver B, et al. Replacement of chlorpromazine with other neuroleptics: effect on abnormal skin pigmentation and ocular changes. J Psychiatry Neurosci. 1993;18:173-177.
- Tsao H, Busam K, Barnhill RL, et al. Treatment of minocycline-induced hyperpigmentation with the Q-switched ruby laser. Arch Dermatol. 1996;132:1250-1251.
- Knox JM, Dodge BG, Freeman RG. Erythema dyschromicum perstans. Arch Dermatol. 1968;97:262-272. doi:10.1001 /archderm.1968.01610090034006
- Rutnin S, Udompanich S, Pratumchart N, et al. Ashy dermatosis and lichen planus pigmentosus: the histopathological differences. Biomed Res Int. 2019;2019:5829185. doi:10.1155/2019/5829185
- Montgomery H, O’Leary PA. Pigmentation of the skin in Addison’s disease, acanthosis nigricans and hemochromatosis. Arch Derm Syphilol. 1930;21:970-984. doi:10.1001 /archderm.1930.01440120072005
- Fernandez-Flores A, Cassarino DS. Histopathologic findings of cutaneous hyperpigmentation in Addison disease and immunostain of the melanocytic population. Am J Dermatopathol. 2017;39:924-927. doi:10.1097/DAD.0000000000000937
- Davis MD, Weenig RH, Camilleri MJ. Confluent and reticulate papillomatosis (Gougerot-Carteaud syndrome): a minocycline-responsive dermatosis without evidence for yeast in pathogenesis. a study of 39 patients and a proposal of diagnostic criteria. Br J Dermatol. 2006;154:287-293. doi:10.1111/j.1365-2133.2005.06955.x
- Jo S, Park HS, Cho S, et al. Updated diagnosis criteria for confluent and reticulated papillomatosis: a case report. Ann Dermatol. 2014; 26:409-410. doi:10.5021/ad.2014.26.3.409
- Lause M, Kamboj A, Fernandez Faith E. Dermatologic manifestations of endocrine disorders. Transl Pediatr. 2017;6:300-312. doi:10.21037 /tp.2017.09.08
A 37-year-old man with a history of cerebral palsy, bipolar disorder, and impulse control disorder presented to the emergency department with breathing difficulty and worsening malaise. The patient subsequently was intubated due to hypoxic respiratory failure and was found to be positive for SARS-CoV-2. He was admitted to the intensive care unit, and dermatology was consulted due to concern that the cutaneous findings were demonstrative of a vasculitic process. Physical examination revealed diffuse, symmetric, dark brown to blue-gray macules coalescing into patches on the anterior tibia (top) and covering the entire lower leg (bottom). The patches were mottled and did not blanch with pressure. According to the patient’s caretaker, the leg hyperpigmentation had been present for 2 years.
Johnson & Johnson requests FDA approval for vaccine booster doses
The company said it filed a request for people ages 18 and older who have received the one-shot vaccine. Johnson & Johnson submitted data for several different booster intervals -- ranging from 2 months to 6 months -- but didn’t formally recommend one to the FDA, The Associated Press reported.
“We’re describing the data to them,” Mathai Mammen, MD, head of global research and development for Janssen, the company’s vaccine division, told CNN.
“The process is not that we asked for a very specific interval -- we’re providing them data and we’re going to be presenting to the committee,” he said. “They’ll take all that into consideration when they ultimately decide on an appropriate interval.”
The FDA’s independent vaccine advisory committee meets next week to review data on booster shots from both Johnson & Johnson and Moderna. It’s the first step in the review process, which then requires approval from leaders at the FDA and Centers for Disease Control and Prevention. If both agencies authorize the extra shots, Americans could receive boosters from Johnson & Johnson and Moderna later this month, the AP reported.
Johnson & Johnson previously released data that showed the vaccine remains highly effective against COVID-19 at least 5 months after vaccination, with 81% efficacy against hospitalizations in the United States.
Two weeks ago, the company reported that a booster dose at 2 months or 6 months further lifted immunity, with a booster at 2 months providing 94% protection against moderate and severe COVID-19. The company said the 6-month booster raised antibodies by 12 times but didn’t release additional data at that time.
In September, the FDA authorized booster shots of the Pfizer vaccine for ages 65 and older, those who live in long-term care facilities, and those with higher risks for contracting COVID-19. The Biden administration is supporting a booster campaign to address potential waning vaccine immunity and remaining surges of the more contagious Delta variant, the AP reported.
A version of this article first appeared on WebMD.com.
The company said it filed a request for people ages 18 and older who have received the one-shot vaccine. Johnson & Johnson submitted data for several different booster intervals -- ranging from 2 months to 6 months -- but didn’t formally recommend one to the FDA, The Associated Press reported.
“We’re describing the data to them,” Mathai Mammen, MD, head of global research and development for Janssen, the company’s vaccine division, told CNN.
“The process is not that we asked for a very specific interval -- we’re providing them data and we’re going to be presenting to the committee,” he said. “They’ll take all that into consideration when they ultimately decide on an appropriate interval.”
The FDA’s independent vaccine advisory committee meets next week to review data on booster shots from both Johnson & Johnson and Moderna. It’s the first step in the review process, which then requires approval from leaders at the FDA and Centers for Disease Control and Prevention. If both agencies authorize the extra shots, Americans could receive boosters from Johnson & Johnson and Moderna later this month, the AP reported.
Johnson & Johnson previously released data that showed the vaccine remains highly effective against COVID-19 at least 5 months after vaccination, with 81% efficacy against hospitalizations in the United States.
Two weeks ago, the company reported that a booster dose at 2 months or 6 months further lifted immunity, with a booster at 2 months providing 94% protection against moderate and severe COVID-19. The company said the 6-month booster raised antibodies by 12 times but didn’t release additional data at that time.
In September, the FDA authorized booster shots of the Pfizer vaccine for ages 65 and older, those who live in long-term care facilities, and those with higher risks for contracting COVID-19. The Biden administration is supporting a booster campaign to address potential waning vaccine immunity and remaining surges of the more contagious Delta variant, the AP reported.
A version of this article first appeared on WebMD.com.
The company said it filed a request for people ages 18 and older who have received the one-shot vaccine. Johnson & Johnson submitted data for several different booster intervals -- ranging from 2 months to 6 months -- but didn’t formally recommend one to the FDA, The Associated Press reported.
“We’re describing the data to them,” Mathai Mammen, MD, head of global research and development for Janssen, the company’s vaccine division, told CNN.
“The process is not that we asked for a very specific interval -- we’re providing them data and we’re going to be presenting to the committee,” he said. “They’ll take all that into consideration when they ultimately decide on an appropriate interval.”
The FDA’s independent vaccine advisory committee meets next week to review data on booster shots from both Johnson & Johnson and Moderna. It’s the first step in the review process, which then requires approval from leaders at the FDA and Centers for Disease Control and Prevention. If both agencies authorize the extra shots, Americans could receive boosters from Johnson & Johnson and Moderna later this month, the AP reported.
Johnson & Johnson previously released data that showed the vaccine remains highly effective against COVID-19 at least 5 months after vaccination, with 81% efficacy against hospitalizations in the United States.
Two weeks ago, the company reported that a booster dose at 2 months or 6 months further lifted immunity, with a booster at 2 months providing 94% protection against moderate and severe COVID-19. The company said the 6-month booster raised antibodies by 12 times but didn’t release additional data at that time.
In September, the FDA authorized booster shots of the Pfizer vaccine for ages 65 and older, those who live in long-term care facilities, and those with higher risks for contracting COVID-19. The Biden administration is supporting a booster campaign to address potential waning vaccine immunity and remaining surges of the more contagious Delta variant, the AP reported.
A version of this article first appeared on WebMD.com.
JAK inhibitor provides impressive hair growth for patients with alopecia areata
, according to the results of two phase 3 trials presented at the European Academy of Dermatology and Venereology (EADV) 2021 Annual Meeting.
In both trials, severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of greater than or equal to 50, was an enrollment requirement. The primary endpoint was a SALT score of less than or equal to 20, signifying 80% scalp coverage.
“The mean SALT score at entry was 85,” reported Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn. He explained that the SALT scale extends from 0 (no hair loss) to 100 (complete hair loss). About 45% of patients in the phase 3 trials had alopecia universalis.
In both trials, called BRAVE-AA1 and BRAVE-AA2, a response was seen with baricitinib after about 4 weeks. Response increased steadily through the entire 36 weeks of treatment. At the end of 36 weeks, when response curves still had an upward trajectory, the proportion of those treated with the 4-mg dose of baricitinib who had achieved a SALT score of less than or equal to 20 had reached 35.2% in BRAVE-AA1 and 32.5% in BRAVE-AA2.
The nearly identical BRAVE-AA1 and BRAVE-AA2 trials enrolled 654 and 546 patients, respectively. The patients were randomly assigned in a 3:2:2 ratio to receive baricitinib 4 mg, baricitinib 2 mg, or placebo. All treatments were taken once daily. Regrowth of eyebrow and eyelash hair were secondary outcomes.
There was a clear dose effect; hair growth increased more quickly with the 4-mg dose of baricitinib than with the 2-mg dose. The difference between the active therapy and placebo was significant by 16 weeks with the 4-mg dose. By 24 weeks, the advantage of the 2-mg dose over placebo also reached significance. The response rate with the 4-mg dose was nearly twice as great.
At the end of the 36-week trials, the proportion of patients treated with baricitinib 2 mg who achieved the primary endpoint was 21.7% and 17.3% in the BRAVE-AA1 and BRAVE-AA2 trials, respectively. Among patients taking placebo, the primary endpoint was met by 5.3% and 2.6%, respectively, at the end of the two trials.
The differences in responses with the 4-mg and the 2-mg doses were significantly higher compared with placebo (P ≤ .001 for both doses vs. placebo).
Using a scoring system for eyebrow and eyelash hair loss, the proportion of patients who achieved a score of 0 (full coverage) or 1 (minimal gaps) was again superior in both trials for patients taking the higher dose of baricitinib. This level of response was reached by about 31% to 35% of those taking the 4-mg dose in BRAVE-AA1 and BRAVE-AA2 (P ≤ .001 vs. placebo). With the lower dose, the rates were 19.1% and 13.5%, respectively. This endpoint was reached in only about 3% of patients who took placebo.
Rates of adverse events were modestly higher in the two active treatment groups in comparison with the group taking placebo. The most commonly occurring adverse events with baricitinib included upper respiratory tract infections, nasopharyngitis, urinary tract infections, and headache, according to Dr. King.
“Most of the adverse events were mild to moderate,” he said. He also reported that none of these adverse events occurred in more than 10% of patients, and there were no cases of other opportunistic infections, thromboembolic events, or gastrointestinal perforations. The discontinuation rates because of adverse events with active therapy were less than 3% in both trials.
JAK inhibitors are currently employed in the treatment of a variety of inflammatory diseases. Baricitinib is currently approved for the treatment of rheumatoid arthritis. Because specificity differs markedly for their inhibition of JAK kinases (JAK1, JAK2, JAK3, and Tyk2), these drugs do not appear to be interchangeable with regard to clinical effect.
Several case reports of hair regrowth with baricitinib led to a phase 2 trial, which was recently published in the Journal of the American Academy of Dermatology. In this trial, the therapy also yielded substantial benefit for patients with alopecia areata. The benefit of baricitinib is attributed to inhibition of JAK1 and JAK2 signaling, which has been implicated in cytokine-mediated immune dysfunction leading to damage of hair follicles.
Alopecia areata is a common disorder that can have a large adverse impact on quality of life, Dr. King noted. There is no approved therapy for this condition, so there is a large unmet need. Although longer follow-up is needed to gauge sustained efficacy and safety, he considers these results promising for a therapy with clinically meaningful benefit.
This point was reiterated by Yolanda Gilaberte Calzada, MD, PhD, head of the Dermatology Service, University Hospital Miguel Servet, Zaragoza, Spain, who was moderator of the session in which Dr. King presented these data. She expressed excitement about the promise of baricitinib, particularly with regard to the substantial proportion of patients who achieved meaningful degrees of hair regrowth.
“All of us will be happy to have options for alopecia areata,” said Dr. Calzada, who predicted that the higher dose of baricitinib will be selected for clinical development, given its greater efficacy with little increase in safety concerns.
Eli Lilly provided funding for the BRAVE-AA1 and -AA2 trials. Dr. King has financial relationships with Arena, Aclaris, Bristol-Myers Squibb, Concert, Pfizer, Regeneron, Sanofi Genzyme, and Eli Lilly. Dr. Calzada has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, according to the results of two phase 3 trials presented at the European Academy of Dermatology and Venereology (EADV) 2021 Annual Meeting.
In both trials, severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of greater than or equal to 50, was an enrollment requirement. The primary endpoint was a SALT score of less than or equal to 20, signifying 80% scalp coverage.
“The mean SALT score at entry was 85,” reported Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn. He explained that the SALT scale extends from 0 (no hair loss) to 100 (complete hair loss). About 45% of patients in the phase 3 trials had alopecia universalis.
In both trials, called BRAVE-AA1 and BRAVE-AA2, a response was seen with baricitinib after about 4 weeks. Response increased steadily through the entire 36 weeks of treatment. At the end of 36 weeks, when response curves still had an upward trajectory, the proportion of those treated with the 4-mg dose of baricitinib who had achieved a SALT score of less than or equal to 20 had reached 35.2% in BRAVE-AA1 and 32.5% in BRAVE-AA2.
The nearly identical BRAVE-AA1 and BRAVE-AA2 trials enrolled 654 and 546 patients, respectively. The patients were randomly assigned in a 3:2:2 ratio to receive baricitinib 4 mg, baricitinib 2 mg, or placebo. All treatments were taken once daily. Regrowth of eyebrow and eyelash hair were secondary outcomes.
There was a clear dose effect; hair growth increased more quickly with the 4-mg dose of baricitinib than with the 2-mg dose. The difference between the active therapy and placebo was significant by 16 weeks with the 4-mg dose. By 24 weeks, the advantage of the 2-mg dose over placebo also reached significance. The response rate with the 4-mg dose was nearly twice as great.
At the end of the 36-week trials, the proportion of patients treated with baricitinib 2 mg who achieved the primary endpoint was 21.7% and 17.3% in the BRAVE-AA1 and BRAVE-AA2 trials, respectively. Among patients taking placebo, the primary endpoint was met by 5.3% and 2.6%, respectively, at the end of the two trials.
The differences in responses with the 4-mg and the 2-mg doses were significantly higher compared with placebo (P ≤ .001 for both doses vs. placebo).
Using a scoring system for eyebrow and eyelash hair loss, the proportion of patients who achieved a score of 0 (full coverage) or 1 (minimal gaps) was again superior in both trials for patients taking the higher dose of baricitinib. This level of response was reached by about 31% to 35% of those taking the 4-mg dose in BRAVE-AA1 and BRAVE-AA2 (P ≤ .001 vs. placebo). With the lower dose, the rates were 19.1% and 13.5%, respectively. This endpoint was reached in only about 3% of patients who took placebo.
Rates of adverse events were modestly higher in the two active treatment groups in comparison with the group taking placebo. The most commonly occurring adverse events with baricitinib included upper respiratory tract infections, nasopharyngitis, urinary tract infections, and headache, according to Dr. King.
“Most of the adverse events were mild to moderate,” he said. He also reported that none of these adverse events occurred in more than 10% of patients, and there were no cases of other opportunistic infections, thromboembolic events, or gastrointestinal perforations. The discontinuation rates because of adverse events with active therapy were less than 3% in both trials.
JAK inhibitors are currently employed in the treatment of a variety of inflammatory diseases. Baricitinib is currently approved for the treatment of rheumatoid arthritis. Because specificity differs markedly for their inhibition of JAK kinases (JAK1, JAK2, JAK3, and Tyk2), these drugs do not appear to be interchangeable with regard to clinical effect.
Several case reports of hair regrowth with baricitinib led to a phase 2 trial, which was recently published in the Journal of the American Academy of Dermatology. In this trial, the therapy also yielded substantial benefit for patients with alopecia areata. The benefit of baricitinib is attributed to inhibition of JAK1 and JAK2 signaling, which has been implicated in cytokine-mediated immune dysfunction leading to damage of hair follicles.
Alopecia areata is a common disorder that can have a large adverse impact on quality of life, Dr. King noted. There is no approved therapy for this condition, so there is a large unmet need. Although longer follow-up is needed to gauge sustained efficacy and safety, he considers these results promising for a therapy with clinically meaningful benefit.
This point was reiterated by Yolanda Gilaberte Calzada, MD, PhD, head of the Dermatology Service, University Hospital Miguel Servet, Zaragoza, Spain, who was moderator of the session in which Dr. King presented these data. She expressed excitement about the promise of baricitinib, particularly with regard to the substantial proportion of patients who achieved meaningful degrees of hair regrowth.
“All of us will be happy to have options for alopecia areata,” said Dr. Calzada, who predicted that the higher dose of baricitinib will be selected for clinical development, given its greater efficacy with little increase in safety concerns.
Eli Lilly provided funding for the BRAVE-AA1 and -AA2 trials. Dr. King has financial relationships with Arena, Aclaris, Bristol-Myers Squibb, Concert, Pfizer, Regeneron, Sanofi Genzyme, and Eli Lilly. Dr. Calzada has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, according to the results of two phase 3 trials presented at the European Academy of Dermatology and Venereology (EADV) 2021 Annual Meeting.
In both trials, severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of greater than or equal to 50, was an enrollment requirement. The primary endpoint was a SALT score of less than or equal to 20, signifying 80% scalp coverage.
“The mean SALT score at entry was 85,” reported Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn. He explained that the SALT scale extends from 0 (no hair loss) to 100 (complete hair loss). About 45% of patients in the phase 3 trials had alopecia universalis.
In both trials, called BRAVE-AA1 and BRAVE-AA2, a response was seen with baricitinib after about 4 weeks. Response increased steadily through the entire 36 weeks of treatment. At the end of 36 weeks, when response curves still had an upward trajectory, the proportion of those treated with the 4-mg dose of baricitinib who had achieved a SALT score of less than or equal to 20 had reached 35.2% in BRAVE-AA1 and 32.5% in BRAVE-AA2.
The nearly identical BRAVE-AA1 and BRAVE-AA2 trials enrolled 654 and 546 patients, respectively. The patients were randomly assigned in a 3:2:2 ratio to receive baricitinib 4 mg, baricitinib 2 mg, or placebo. All treatments were taken once daily. Regrowth of eyebrow and eyelash hair were secondary outcomes.
There was a clear dose effect; hair growth increased more quickly with the 4-mg dose of baricitinib than with the 2-mg dose. The difference between the active therapy and placebo was significant by 16 weeks with the 4-mg dose. By 24 weeks, the advantage of the 2-mg dose over placebo also reached significance. The response rate with the 4-mg dose was nearly twice as great.
At the end of the 36-week trials, the proportion of patients treated with baricitinib 2 mg who achieved the primary endpoint was 21.7% and 17.3% in the BRAVE-AA1 and BRAVE-AA2 trials, respectively. Among patients taking placebo, the primary endpoint was met by 5.3% and 2.6%, respectively, at the end of the two trials.
The differences in responses with the 4-mg and the 2-mg doses were significantly higher compared with placebo (P ≤ .001 for both doses vs. placebo).
Using a scoring system for eyebrow and eyelash hair loss, the proportion of patients who achieved a score of 0 (full coverage) or 1 (minimal gaps) was again superior in both trials for patients taking the higher dose of baricitinib. This level of response was reached by about 31% to 35% of those taking the 4-mg dose in BRAVE-AA1 and BRAVE-AA2 (P ≤ .001 vs. placebo). With the lower dose, the rates were 19.1% and 13.5%, respectively. This endpoint was reached in only about 3% of patients who took placebo.
Rates of adverse events were modestly higher in the two active treatment groups in comparison with the group taking placebo. The most commonly occurring adverse events with baricitinib included upper respiratory tract infections, nasopharyngitis, urinary tract infections, and headache, according to Dr. King.
“Most of the adverse events were mild to moderate,” he said. He also reported that none of these adverse events occurred in more than 10% of patients, and there were no cases of other opportunistic infections, thromboembolic events, or gastrointestinal perforations. The discontinuation rates because of adverse events with active therapy were less than 3% in both trials.
JAK inhibitors are currently employed in the treatment of a variety of inflammatory diseases. Baricitinib is currently approved for the treatment of rheumatoid arthritis. Because specificity differs markedly for their inhibition of JAK kinases (JAK1, JAK2, JAK3, and Tyk2), these drugs do not appear to be interchangeable with regard to clinical effect.
Several case reports of hair regrowth with baricitinib led to a phase 2 trial, which was recently published in the Journal of the American Academy of Dermatology. In this trial, the therapy also yielded substantial benefit for patients with alopecia areata. The benefit of baricitinib is attributed to inhibition of JAK1 and JAK2 signaling, which has been implicated in cytokine-mediated immune dysfunction leading to damage of hair follicles.
Alopecia areata is a common disorder that can have a large adverse impact on quality of life, Dr. King noted. There is no approved therapy for this condition, so there is a large unmet need. Although longer follow-up is needed to gauge sustained efficacy and safety, he considers these results promising for a therapy with clinically meaningful benefit.
This point was reiterated by Yolanda Gilaberte Calzada, MD, PhD, head of the Dermatology Service, University Hospital Miguel Servet, Zaragoza, Spain, who was moderator of the session in which Dr. King presented these data. She expressed excitement about the promise of baricitinib, particularly with regard to the substantial proportion of patients who achieved meaningful degrees of hair regrowth.
“All of us will be happy to have options for alopecia areata,” said Dr. Calzada, who predicted that the higher dose of baricitinib will be selected for clinical development, given its greater efficacy with little increase in safety concerns.
Eli Lilly provided funding for the BRAVE-AA1 and -AA2 trials. Dr. King has financial relationships with Arena, Aclaris, Bristol-Myers Squibb, Concert, Pfizer, Regeneron, Sanofi Genzyme, and Eli Lilly. Dr. Calzada has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Ruxolitinib cream meets primary endpoints in phase 3 vitiligo trial
presented together at the annual meeting of the European Academy of Dermatology and Venereology.
On the primary endpoint of F-VASI 75 (75% improvement in the Facial and Vitiligo Scoring Index), rates were nearly four times higher at 24 weeks in one trial (29.9% vs. 7.5%; P < .0001) and more than twice as great in the other (29.9% vs. 12.9%; P < .01).
“The larger phase 3 trials confirm the previous phase 2 findings,” reported David Rosmarin, MD, vice chairman for research and education, department of dermatology, Tufts Medical Center, Boston. These findings not only include substantial clinical efficacy but good tolerability with “no serious treatment-related adverse events,” he noted.
600 patients randomized
In one of the trials, called TRuE-V1, 330 patients with vitiligo were randomly assigned in a 2:1 ratio to 1.5% ruxolitinib or vehicle applied twice daily. In the other trial, called TRuE-V2, 344 patients were randomly assigned. The participating centers were in Europe and North America.
Patients aged 12 years or older with nonsegmental vitiligo and depigmentation covering no more than 10% of the total body surface area were eligible. The mean baseline F-VASI values were 1.0. The mean total VASI (T-FASI) values were 6.5. On those enrolled, half were female, 11% were adolescents, and 73% had Fitzpatrick skin phototypes III-VI.
Ruxolitinib cream provided near-complete vitiligo clearance (F-VASI 90) on the face at 24 weeks in only about 15% of patients, but this was several times higher than the 2% achieved on vehicle in the TRuE-V1 (P < .01) and the TRuE-V2 trials (P < .05), respectively.
F-VASI 50 response rates greater than 50%
For F-VASI 50, the response rate with ruxolitinib in both studies was approximately 51%. Relative to the 17.2% response on vehicle in TRuE-v1 and 23.4% in TRuE-V2 (both P < .0001 vs. active therapy), the advantage of the topical JAK inhibitor was considered to be a clinically meaningful, not just significant from a statistical standpoint.
In fact, improvement on the 5-point Vitiligo Noticeability Scale “also supported a clinically meaningful benefit,” Dr. Rosmarin reported. When those achieving a score of 4 (much less noticeable) or 5 (no longer noticeable), the response rates at 24 weeks were 24.5% and 21.6% in the TRuE-V1 and TRuE-V2 trials, respectively. Again, these response rates were several times greater than the 3.3% (P < .001) and 6.6% (P < .01) observed in the vehicle arms of TRuE-V1 and TRuE-V2 (P < .01), respectively.
Treatment-related adverse events were infrequent. The most common were acne at the application site, which occurred in about 5% of patients receiving ruxolitinib (vs. 2% or fewer of those receiving vehicle) and pruritus, which also occurred in about 5% of patients. However, the rates of pruritus among those on placebo reached 4% in TRuE-V1 and 2% in TRuE-V2 trials.
In vitiligo, where there has been recent progress in understanding the pathophysiology, loss of melanocytes in immune dysregulation has been linked to activation of the JAK signaling pathway, according to Dr. Rosmarin. In the 52-week phase 2 trial with 205 patients, ruxolitinib was associated with a sustained response and no serious treatment-related adverse events.
52-week data might show more benefit
Patients are continuing to be followed in the TRuE-V1 and TRuE-V-2 trials. Based on the phase 2 data and on the progressive improvement still being observed at the end of 24 weeks in the phase 3 trials, Dr. Rosmarin expects 52-week results be valuable in understanding the clinical role of ruxolitinib.
“We will be looking for further improvement in response as we follow these patients out to 1 year,” he said.
This further follow-up is important, agreed Iltefat Hamzavi, MD, senior staff physician, department of dermatology, Henry Ford Hospital, Detroit.
Despite the promise of perhaps other JAK inhibitors, “we still need to understand how long it will take for the drug to offer optimal results. We already know that is more than 24 weeks,” said Dr. Hamzavi, who has been involved in the clinical trials with this drug but was not involved with the TRuE-V1 or -V2 trials.
He also said more follow-up is needed to understand the duration of effect. He is, however, optimistic about the clinical role of this mechanism for treatment of vitiligo.
“I do think that JAK inhibitors show a lot of promise [in vitiligo] for certain locations of the body,” he said.
Given the limited treatment options for effective and prolonged improvement in vitiligo, both Dr. Hamzavi and Dr. Rosmarin indicated an effective topical cream is likely to be considered by physicians and patients to be a substantial advance.
On Sept. 21, ruxolitinib (Opzelura) 1.5% cream was approved by the Food and Drug Administration for the short-term treatment of mild to moderate atopic dermatitis in children and adults ages 12 years and older – the first FDA approval of this product.
Dr. Rosmarin reported financial relationships with more than 20 pharmaceutical companies, including Incyte, which provided funding for the TRuE-V1 and -V2 trials. Dr. Hamzavi reported financial relationships with more than 15 companies with pharmaceutical or cosmetic products, including Incyte.
A version of this article first appeared on Medscape.com.
presented together at the annual meeting of the European Academy of Dermatology and Venereology.
On the primary endpoint of F-VASI 75 (75% improvement in the Facial and Vitiligo Scoring Index), rates were nearly four times higher at 24 weeks in one trial (29.9% vs. 7.5%; P < .0001) and more than twice as great in the other (29.9% vs. 12.9%; P < .01).
“The larger phase 3 trials confirm the previous phase 2 findings,” reported David Rosmarin, MD, vice chairman for research and education, department of dermatology, Tufts Medical Center, Boston. These findings not only include substantial clinical efficacy but good tolerability with “no serious treatment-related adverse events,” he noted.
600 patients randomized
In one of the trials, called TRuE-V1, 330 patients with vitiligo were randomly assigned in a 2:1 ratio to 1.5% ruxolitinib or vehicle applied twice daily. In the other trial, called TRuE-V2, 344 patients were randomly assigned. The participating centers were in Europe and North America.
Patients aged 12 years or older with nonsegmental vitiligo and depigmentation covering no more than 10% of the total body surface area were eligible. The mean baseline F-VASI values were 1.0. The mean total VASI (T-FASI) values were 6.5. On those enrolled, half were female, 11% were adolescents, and 73% had Fitzpatrick skin phototypes III-VI.
Ruxolitinib cream provided near-complete vitiligo clearance (F-VASI 90) on the face at 24 weeks in only about 15% of patients, but this was several times higher than the 2% achieved on vehicle in the TRuE-V1 (P < .01) and the TRuE-V2 trials (P < .05), respectively.
F-VASI 50 response rates greater than 50%
For F-VASI 50, the response rate with ruxolitinib in both studies was approximately 51%. Relative to the 17.2% response on vehicle in TRuE-v1 and 23.4% in TRuE-V2 (both P < .0001 vs. active therapy), the advantage of the topical JAK inhibitor was considered to be a clinically meaningful, not just significant from a statistical standpoint.
In fact, improvement on the 5-point Vitiligo Noticeability Scale “also supported a clinically meaningful benefit,” Dr. Rosmarin reported. When those achieving a score of 4 (much less noticeable) or 5 (no longer noticeable), the response rates at 24 weeks were 24.5% and 21.6% in the TRuE-V1 and TRuE-V2 trials, respectively. Again, these response rates were several times greater than the 3.3% (P < .001) and 6.6% (P < .01) observed in the vehicle arms of TRuE-V1 and TRuE-V2 (P < .01), respectively.
Treatment-related adverse events were infrequent. The most common were acne at the application site, which occurred in about 5% of patients receiving ruxolitinib (vs. 2% or fewer of those receiving vehicle) and pruritus, which also occurred in about 5% of patients. However, the rates of pruritus among those on placebo reached 4% in TRuE-V1 and 2% in TRuE-V2 trials.
In vitiligo, where there has been recent progress in understanding the pathophysiology, loss of melanocytes in immune dysregulation has been linked to activation of the JAK signaling pathway, according to Dr. Rosmarin. In the 52-week phase 2 trial with 205 patients, ruxolitinib was associated with a sustained response and no serious treatment-related adverse events.
52-week data might show more benefit
Patients are continuing to be followed in the TRuE-V1 and TRuE-V-2 trials. Based on the phase 2 data and on the progressive improvement still being observed at the end of 24 weeks in the phase 3 trials, Dr. Rosmarin expects 52-week results be valuable in understanding the clinical role of ruxolitinib.
“We will be looking for further improvement in response as we follow these patients out to 1 year,” he said.
This further follow-up is important, agreed Iltefat Hamzavi, MD, senior staff physician, department of dermatology, Henry Ford Hospital, Detroit.
Despite the promise of perhaps other JAK inhibitors, “we still need to understand how long it will take for the drug to offer optimal results. We already know that is more than 24 weeks,” said Dr. Hamzavi, who has been involved in the clinical trials with this drug but was not involved with the TRuE-V1 or -V2 trials.
He also said more follow-up is needed to understand the duration of effect. He is, however, optimistic about the clinical role of this mechanism for treatment of vitiligo.
“I do think that JAK inhibitors show a lot of promise [in vitiligo] for certain locations of the body,” he said.
Given the limited treatment options for effective and prolonged improvement in vitiligo, both Dr. Hamzavi and Dr. Rosmarin indicated an effective topical cream is likely to be considered by physicians and patients to be a substantial advance.
On Sept. 21, ruxolitinib (Opzelura) 1.5% cream was approved by the Food and Drug Administration for the short-term treatment of mild to moderate atopic dermatitis in children and adults ages 12 years and older – the first FDA approval of this product.
Dr. Rosmarin reported financial relationships with more than 20 pharmaceutical companies, including Incyte, which provided funding for the TRuE-V1 and -V2 trials. Dr. Hamzavi reported financial relationships with more than 15 companies with pharmaceutical or cosmetic products, including Incyte.
A version of this article first appeared on Medscape.com.
presented together at the annual meeting of the European Academy of Dermatology and Venereology.
On the primary endpoint of F-VASI 75 (75% improvement in the Facial and Vitiligo Scoring Index), rates were nearly four times higher at 24 weeks in one trial (29.9% vs. 7.5%; P < .0001) and more than twice as great in the other (29.9% vs. 12.9%; P < .01).
“The larger phase 3 trials confirm the previous phase 2 findings,” reported David Rosmarin, MD, vice chairman for research and education, department of dermatology, Tufts Medical Center, Boston. These findings not only include substantial clinical efficacy but good tolerability with “no serious treatment-related adverse events,” he noted.
600 patients randomized
In one of the trials, called TRuE-V1, 330 patients with vitiligo were randomly assigned in a 2:1 ratio to 1.5% ruxolitinib or vehicle applied twice daily. In the other trial, called TRuE-V2, 344 patients were randomly assigned. The participating centers were in Europe and North America.
Patients aged 12 years or older with nonsegmental vitiligo and depigmentation covering no more than 10% of the total body surface area were eligible. The mean baseline F-VASI values were 1.0. The mean total VASI (T-FASI) values were 6.5. On those enrolled, half were female, 11% were adolescents, and 73% had Fitzpatrick skin phototypes III-VI.
Ruxolitinib cream provided near-complete vitiligo clearance (F-VASI 90) on the face at 24 weeks in only about 15% of patients, but this was several times higher than the 2% achieved on vehicle in the TRuE-V1 (P < .01) and the TRuE-V2 trials (P < .05), respectively.
F-VASI 50 response rates greater than 50%
For F-VASI 50, the response rate with ruxolitinib in both studies was approximately 51%. Relative to the 17.2% response on vehicle in TRuE-v1 and 23.4% in TRuE-V2 (both P < .0001 vs. active therapy), the advantage of the topical JAK inhibitor was considered to be a clinically meaningful, not just significant from a statistical standpoint.
In fact, improvement on the 5-point Vitiligo Noticeability Scale “also supported a clinically meaningful benefit,” Dr. Rosmarin reported. When those achieving a score of 4 (much less noticeable) or 5 (no longer noticeable), the response rates at 24 weeks were 24.5% and 21.6% in the TRuE-V1 and TRuE-V2 trials, respectively. Again, these response rates were several times greater than the 3.3% (P < .001) and 6.6% (P < .01) observed in the vehicle arms of TRuE-V1 and TRuE-V2 (P < .01), respectively.
Treatment-related adverse events were infrequent. The most common were acne at the application site, which occurred in about 5% of patients receiving ruxolitinib (vs. 2% or fewer of those receiving vehicle) and pruritus, which also occurred in about 5% of patients. However, the rates of pruritus among those on placebo reached 4% in TRuE-V1 and 2% in TRuE-V2 trials.
In vitiligo, where there has been recent progress in understanding the pathophysiology, loss of melanocytes in immune dysregulation has been linked to activation of the JAK signaling pathway, according to Dr. Rosmarin. In the 52-week phase 2 trial with 205 patients, ruxolitinib was associated with a sustained response and no serious treatment-related adverse events.
52-week data might show more benefit
Patients are continuing to be followed in the TRuE-V1 and TRuE-V-2 trials. Based on the phase 2 data and on the progressive improvement still being observed at the end of 24 weeks in the phase 3 trials, Dr. Rosmarin expects 52-week results be valuable in understanding the clinical role of ruxolitinib.
“We will be looking for further improvement in response as we follow these patients out to 1 year,” he said.
This further follow-up is important, agreed Iltefat Hamzavi, MD, senior staff physician, department of dermatology, Henry Ford Hospital, Detroit.
Despite the promise of perhaps other JAK inhibitors, “we still need to understand how long it will take for the drug to offer optimal results. We already know that is more than 24 weeks,” said Dr. Hamzavi, who has been involved in the clinical trials with this drug but was not involved with the TRuE-V1 or -V2 trials.
He also said more follow-up is needed to understand the duration of effect. He is, however, optimistic about the clinical role of this mechanism for treatment of vitiligo.
“I do think that JAK inhibitors show a lot of promise [in vitiligo] for certain locations of the body,” he said.
Given the limited treatment options for effective and prolonged improvement in vitiligo, both Dr. Hamzavi and Dr. Rosmarin indicated an effective topical cream is likely to be considered by physicians and patients to be a substantial advance.
On Sept. 21, ruxolitinib (Opzelura) 1.5% cream was approved by the Food and Drug Administration for the short-term treatment of mild to moderate atopic dermatitis in children and adults ages 12 years and older – the first FDA approval of this product.
Dr. Rosmarin reported financial relationships with more than 20 pharmaceutical companies, including Incyte, which provided funding for the TRuE-V1 and -V2 trials. Dr. Hamzavi reported financial relationships with more than 15 companies with pharmaceutical or cosmetic products, including Incyte.
A version of this article first appeared on Medscape.com.
Psoriasis and Psoriatic Arthritis: A Supplement to Dermatology News

Psoriasis and Psoriatic Arthritis: A Supplement to Dermatology News 2021
- Psoriasis severity redefined by expert group
- Mitigating PSA risk with biologic therapy
- Lack of diversity seen in psoriasis trials
- PSA Comorbidities effect on treatment responses
With Commentaries by Joel M. Gelfand, MD, MSCE and Alan Menter, MD
And more…
Psoriasis and Psoriatic Arthritis: A Supplement to Dermatology News 2021
- Psoriasis severity redefined by expert group
- Mitigating PSA risk with biologic therapy
- Lack of diversity seen in psoriasis trials
- PSA Comorbidities effect on treatment responses
With Commentaries by Joel M. Gelfand, MD, MSCE and Alan Menter, MD
And more…
Psoriasis and Psoriatic Arthritis: A Supplement to Dermatology News 2021
- Psoriasis severity redefined by expert group
- Mitigating PSA risk with biologic therapy
- Lack of diversity seen in psoriasis trials
- PSA Comorbidities effect on treatment responses
With Commentaries by Joel M. Gelfand, MD, MSCE and Alan Menter, MD
And more…
COVID-19: Two more cases of mucosal skin ulcers reported in male teens
Irish A similar case in an adolescent, also with ulcers affecting the mouth and penis, was reported earlier in 2021 in the United States.
“Our cases show that a swab for COVID-19 can be added to the list of investigations for mucosal and cutaneous rashes in children and probably adults,” said dermatologist Stephanie Bowe, MD, of South Infirmary-Victoria University Hospital in Cork, Ireland, in an interview. “Our patients seemed to improve with IV steroids, but there is not enough data to recommend them to all patients or for use in the different cutaneous presentations associated with COVID-19.”
The new case reports were presented at the 2021 meeting of the World Congress of Pediatric Dermatology and published in Pediatric Dermatology.
Researchers have noted that skin disorders linked to COVID-19 infection are different than those in adults. In children, the conditions include morbilliform rash, pernio-like acral lesions, urticaria, macular erythema, vesicular eruption, papulosquamous eruption, and retiform purpura. “The pathogenesis of each is not fully understood but likely related to the inflammatory response to COVID-19 and the various pathways within the body, which become activated,” Dr. Bowe said.
The first patient, a 17-year-old boy, presented at clinic 6 days after he’d been confirmed to be infected with COVID-19 and 8 days after developing fever and cough. “He had a 2-day history of conjunctivitis and ulceration of his oral mucosa, erythematous circumferential erosions of the glans penis with no other cutaneous findings,” the authors write in the report.
The boy “was distressed and embarrassed about his genital ulceration and also found eating very painful due to his oral ulceration,” Dr. Bowe said.
The second patient, a 14-year-old boy, was hospitalized 7 days after a positive COVID-19 test and 9 days after developing cough and fever. “He had a 5-day history of ulceration of the oral mucosa with mild conjunctivitis,” the authors wrote. “Ulceration of the glans penis developed on day 2 of admission.”
The 14-year-old was sicker than the 17-year-old boy, Dr. Bowe said. “He was unable to tolerate an oral diet for several days and had exquisite pain and vomiting with his coughing fits.”
This patient had a history of recurrent herpes labialis, but it’s unclear whether herpes simplex virus (HSV) played a role in the COVID-19–related case. “There is a possibility that the patient was more susceptible to viral cutaneous reactions during COVID-19 infection, but we didn’t have any definite history of HSV infection at the time of mucositis,” Dr. Bowe said. “We also didn’t have any swabs positive for HSV even though several were done at the time.”
Both patients received IV steroids – hydrocortisone at 100 mg 3 times daily for 3 days. This treatment was used “because of deterioration in symptoms and COVID-19 infection,” Dr. Bowe said. “IV steroids were used for respiratory symptoms of COVID-19, so we felt these cutaneous symptoms may have also been caused by an inflammatory response and might benefit from steroids. There was very little literature about this specific situation, though.”
She added that intravenous steroids wouldn’t be appropriate for most pediatric patients, and noted that “their use is controversial in the literature for erythema multiforme and RIME.”
In addition, the patients received betamethasone valerate 0.1% ointment once daily, hydrocortisone 2.5 mg buccal tablets 4 times daily, analgesia with acetaminophen and ibuprofen, and intravenous hydration. The first patient also received prednisolone 1% eye drops, while the second patient was given lidocaine hydrochloride mouthwash and total parenteral nutrition for 5 days.
The patients were discharged after 4 and 14 days, respectively.
Dermatologists in Massachusetts reported a similar case earlier in 2021 in a 17-year-old boy who was positive for COVID-19 and presented with “shallow erosions of the vermilion lips and hard palate, circumferential erythematous erosions of the periurethral glans penis, and five small vesicles on the trunk and upper extremities.”
The patient received betamethasone valerate 0.1% ointment for the lips and penis, intraoral dexamethasone solution, viscous lidocaine, acetaminophen, and ibuprofen. He also received oral prednisone at approximately 1 mg/kg daily for 4 consecutive days after worsening oral pain. A recurrence of oral pain 3 months later was resolved with a higher and longer treatment with oral prednisone.
Dermatologists have also reported cases of erythema multiforme lesions of the mucosa in adults with COVID-19. One case was reported in Iran, and the other in France.
The authors report no study funding and disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Irish A similar case in an adolescent, also with ulcers affecting the mouth and penis, was reported earlier in 2021 in the United States.
“Our cases show that a swab for COVID-19 can be added to the list of investigations for mucosal and cutaneous rashes in children and probably adults,” said dermatologist Stephanie Bowe, MD, of South Infirmary-Victoria University Hospital in Cork, Ireland, in an interview. “Our patients seemed to improve with IV steroids, but there is not enough data to recommend them to all patients or for use in the different cutaneous presentations associated with COVID-19.”
The new case reports were presented at the 2021 meeting of the World Congress of Pediatric Dermatology and published in Pediatric Dermatology.
Researchers have noted that skin disorders linked to COVID-19 infection are different than those in adults. In children, the conditions include morbilliform rash, pernio-like acral lesions, urticaria, macular erythema, vesicular eruption, papulosquamous eruption, and retiform purpura. “The pathogenesis of each is not fully understood but likely related to the inflammatory response to COVID-19 and the various pathways within the body, which become activated,” Dr. Bowe said.
The first patient, a 17-year-old boy, presented at clinic 6 days after he’d been confirmed to be infected with COVID-19 and 8 days after developing fever and cough. “He had a 2-day history of conjunctivitis and ulceration of his oral mucosa, erythematous circumferential erosions of the glans penis with no other cutaneous findings,” the authors write in the report.
The boy “was distressed and embarrassed about his genital ulceration and also found eating very painful due to his oral ulceration,” Dr. Bowe said.
The second patient, a 14-year-old boy, was hospitalized 7 days after a positive COVID-19 test and 9 days after developing cough and fever. “He had a 5-day history of ulceration of the oral mucosa with mild conjunctivitis,” the authors wrote. “Ulceration of the glans penis developed on day 2 of admission.”
The 14-year-old was sicker than the 17-year-old boy, Dr. Bowe said. “He was unable to tolerate an oral diet for several days and had exquisite pain and vomiting with his coughing fits.”
This patient had a history of recurrent herpes labialis, but it’s unclear whether herpes simplex virus (HSV) played a role in the COVID-19–related case. “There is a possibility that the patient was more susceptible to viral cutaneous reactions during COVID-19 infection, but we didn’t have any definite history of HSV infection at the time of mucositis,” Dr. Bowe said. “We also didn’t have any swabs positive for HSV even though several were done at the time.”
Both patients received IV steroids – hydrocortisone at 100 mg 3 times daily for 3 days. This treatment was used “because of deterioration in symptoms and COVID-19 infection,” Dr. Bowe said. “IV steroids were used for respiratory symptoms of COVID-19, so we felt these cutaneous symptoms may have also been caused by an inflammatory response and might benefit from steroids. There was very little literature about this specific situation, though.”
She added that intravenous steroids wouldn’t be appropriate for most pediatric patients, and noted that “their use is controversial in the literature for erythema multiforme and RIME.”
In addition, the patients received betamethasone valerate 0.1% ointment once daily, hydrocortisone 2.5 mg buccal tablets 4 times daily, analgesia with acetaminophen and ibuprofen, and intravenous hydration. The first patient also received prednisolone 1% eye drops, while the second patient was given lidocaine hydrochloride mouthwash and total parenteral nutrition for 5 days.
The patients were discharged after 4 and 14 days, respectively.
Dermatologists in Massachusetts reported a similar case earlier in 2021 in a 17-year-old boy who was positive for COVID-19 and presented with “shallow erosions of the vermilion lips and hard palate, circumferential erythematous erosions of the periurethral glans penis, and five small vesicles on the trunk and upper extremities.”
The patient received betamethasone valerate 0.1% ointment for the lips and penis, intraoral dexamethasone solution, viscous lidocaine, acetaminophen, and ibuprofen. He also received oral prednisone at approximately 1 mg/kg daily for 4 consecutive days after worsening oral pain. A recurrence of oral pain 3 months later was resolved with a higher and longer treatment with oral prednisone.
Dermatologists have also reported cases of erythema multiforme lesions of the mucosa in adults with COVID-19. One case was reported in Iran, and the other in France.
The authors report no study funding and disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Irish A similar case in an adolescent, also with ulcers affecting the mouth and penis, was reported earlier in 2021 in the United States.
“Our cases show that a swab for COVID-19 can be added to the list of investigations for mucosal and cutaneous rashes in children and probably adults,” said dermatologist Stephanie Bowe, MD, of South Infirmary-Victoria University Hospital in Cork, Ireland, in an interview. “Our patients seemed to improve with IV steroids, but there is not enough data to recommend them to all patients or for use in the different cutaneous presentations associated with COVID-19.”
The new case reports were presented at the 2021 meeting of the World Congress of Pediatric Dermatology and published in Pediatric Dermatology.
Researchers have noted that skin disorders linked to COVID-19 infection are different than those in adults. In children, the conditions include morbilliform rash, pernio-like acral lesions, urticaria, macular erythema, vesicular eruption, papulosquamous eruption, and retiform purpura. “The pathogenesis of each is not fully understood but likely related to the inflammatory response to COVID-19 and the various pathways within the body, which become activated,” Dr. Bowe said.
The first patient, a 17-year-old boy, presented at clinic 6 days after he’d been confirmed to be infected with COVID-19 and 8 days after developing fever and cough. “He had a 2-day history of conjunctivitis and ulceration of his oral mucosa, erythematous circumferential erosions of the glans penis with no other cutaneous findings,” the authors write in the report.
The boy “was distressed and embarrassed about his genital ulceration and also found eating very painful due to his oral ulceration,” Dr. Bowe said.
The second patient, a 14-year-old boy, was hospitalized 7 days after a positive COVID-19 test and 9 days after developing cough and fever. “He had a 5-day history of ulceration of the oral mucosa with mild conjunctivitis,” the authors wrote. “Ulceration of the glans penis developed on day 2 of admission.”
The 14-year-old was sicker than the 17-year-old boy, Dr. Bowe said. “He was unable to tolerate an oral diet for several days and had exquisite pain and vomiting with his coughing fits.”
This patient had a history of recurrent herpes labialis, but it’s unclear whether herpes simplex virus (HSV) played a role in the COVID-19–related case. “There is a possibility that the patient was more susceptible to viral cutaneous reactions during COVID-19 infection, but we didn’t have any definite history of HSV infection at the time of mucositis,” Dr. Bowe said. “We also didn’t have any swabs positive for HSV even though several were done at the time.”
Both patients received IV steroids – hydrocortisone at 100 mg 3 times daily for 3 days. This treatment was used “because of deterioration in symptoms and COVID-19 infection,” Dr. Bowe said. “IV steroids were used for respiratory symptoms of COVID-19, so we felt these cutaneous symptoms may have also been caused by an inflammatory response and might benefit from steroids. There was very little literature about this specific situation, though.”
She added that intravenous steroids wouldn’t be appropriate for most pediatric patients, and noted that “their use is controversial in the literature for erythema multiforme and RIME.”
In addition, the patients received betamethasone valerate 0.1% ointment once daily, hydrocortisone 2.5 mg buccal tablets 4 times daily, analgesia with acetaminophen and ibuprofen, and intravenous hydration. The first patient also received prednisolone 1% eye drops, while the second patient was given lidocaine hydrochloride mouthwash and total parenteral nutrition for 5 days.
The patients were discharged after 4 and 14 days, respectively.
Dermatologists in Massachusetts reported a similar case earlier in 2021 in a 17-year-old boy who was positive for COVID-19 and presented with “shallow erosions of the vermilion lips and hard palate, circumferential erythematous erosions of the periurethral glans penis, and five small vesicles on the trunk and upper extremities.”
The patient received betamethasone valerate 0.1% ointment for the lips and penis, intraoral dexamethasone solution, viscous lidocaine, acetaminophen, and ibuprofen. He also received oral prednisone at approximately 1 mg/kg daily for 4 consecutive days after worsening oral pain. A recurrence of oral pain 3 months later was resolved with a higher and longer treatment with oral prednisone.
Dermatologists have also reported cases of erythema multiforme lesions of the mucosa in adults with COVID-19. One case was reported in Iran, and the other in France.
The authors report no study funding and disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.