User login
Guideline public comment period framework
AGA is dedicated to integrity and transparency in the development of clinical guidance.
W In addition, to keep academic integrity of the AGA guideline, clinical practice update, and clinical pathway process, AGA follows the framework outlined by the Council of Medical Specialty Societies (CMSS) in The Code for Interaction with Companies.
The Code for Interaction with Companies states:
“7.15. Societies will not permit Guideline development panel members or staff to discuss a Guideline’s development with Company employees or representatives, will not accept unpublished data from Companies, and will not permit Companies to review Guidelines in draft form, except if a Society permits public or member comment on draft Guidelines as a part of the Society’s published Guideline development process.”
The Clinical Guidelines Committee and Clinical Practice Updates Committee strive to keep AGA’s clinical practice tools independent of industry influence and remain solely based on scientific evidence. As a result of this effort, AGA’s writing panels and chairs will have no direct communication with industry. Companies will have the opportunity to submit feedback during the public comment period. The panel will review any comments submitted through the online platform along with feedback from the public. The writing panel will take these suggestions into consideration when making revisions following the public comment period.
AGA is dedicated to integrity and transparency in the development of clinical guidance.
W In addition, to keep academic integrity of the AGA guideline, clinical practice update, and clinical pathway process, AGA follows the framework outlined by the Council of Medical Specialty Societies (CMSS) in The Code for Interaction with Companies.
The Code for Interaction with Companies states:
“7.15. Societies will not permit Guideline development panel members or staff to discuss a Guideline’s development with Company employees or representatives, will not accept unpublished data from Companies, and will not permit Companies to review Guidelines in draft form, except if a Society permits public or member comment on draft Guidelines as a part of the Society’s published Guideline development process.”
The Clinical Guidelines Committee and Clinical Practice Updates Committee strive to keep AGA’s clinical practice tools independent of industry influence and remain solely based on scientific evidence. As a result of this effort, AGA’s writing panels and chairs will have no direct communication with industry. Companies will have the opportunity to submit feedback during the public comment period. The panel will review any comments submitted through the online platform along with feedback from the public. The writing panel will take these suggestions into consideration when making revisions following the public comment period.
AGA is dedicated to integrity and transparency in the development of clinical guidance.
W In addition, to keep academic integrity of the AGA guideline, clinical practice update, and clinical pathway process, AGA follows the framework outlined by the Council of Medical Specialty Societies (CMSS) in The Code for Interaction with Companies.
The Code for Interaction with Companies states:
“7.15. Societies will not permit Guideline development panel members or staff to discuss a Guideline’s development with Company employees or representatives, will not accept unpublished data from Companies, and will not permit Companies to review Guidelines in draft form, except if a Society permits public or member comment on draft Guidelines as a part of the Society’s published Guideline development process.”
The Clinical Guidelines Committee and Clinical Practice Updates Committee strive to keep AGA’s clinical practice tools independent of industry influence and remain solely based on scientific evidence. As a result of this effort, AGA’s writing panels and chairs will have no direct communication with industry. Companies will have the opportunity to submit feedback during the public comment period. The panel will review any comments submitted through the online platform along with feedback from the public. The writing panel will take these suggestions into consideration when making revisions following the public comment period.
2018 AGA legislative wins
To those who took time to advocate on behalf of your profession and patients, thank you. Because of your efforts, we achieved several key successes that benefit clinicians, researchers and patients.
Together we were able to help advance several AGA policy priorities — administrative burden relief, digestive disease research and funding, and patient access and protection — as well as the science and practice of gastroenterology.
This year, we will be counting on you, our members, to again lead the charge to achieve AGA’s mission — empowering clinicians and researchers to improve digestive health — by calling on legislators and regulators to ensure the voice of gastroenterology continue to be heard.
NIH funding increase
AGA advocated and secured a $2 billion increase in NIH funding for fiscal year (FY) 2019. When added to increases from the two previous fiscal years, NIH’s funding has increased by 30 percent over the past three years, which is the largest increase since the doubling period in the last decade.
IPAB repeal prevents automatic Medicare cuts
Congress repealed the Independent Payment Advisory Board (IPAB) that was created as part of the Affordable Care Act (ACA). AGA and all of organized medicine long opposed IPAB since its sole purpose was to make budgetary cuts to Medicare if it reached a certain threshold of spending.
MIPS changes means more flexibility for physicians
AGA and the physician community were successful in securing flexibility under the new Medicare Quality Payment Program and the Merit-based Incentive Payment System (MIPS) that were created under the Medicare Access and CHIP Reauthorization Act. The changes give CMS more flexibility in implementing the program and will ensure that physicians have an opportunity to be successful in MIPS.
500 AGA members prevent radical changes to outpatient documentation
In response to a CMS proposal to radically change how outpatient evaluation and management (E/M) services are documented, more than 500 AGA members urged CMS not to move forward with its plan. As a result, CMS withdrew or delayed many of the proposed changes to E/M services that negatively impacted reimbursement. CMS did move forward with several changes to E/M documentation in an effort to reduce administrative burden.
AGA members send more than 940 letters to Congress
AGA launched the Congressional Advocates Program to provide an infrastructure and tools for members to effectively advocate on behalf of their profession and patients. AGA members heeded our calls to action and sent more than 940 letters sent to Congress and federal agencies throughout 2018. We thank our advocates whose participation makes a difference.
To those who took time to advocate on behalf of your profession and patients, thank you. Because of your efforts, we achieved several key successes that benefit clinicians, researchers and patients.
Together we were able to help advance several AGA policy priorities — administrative burden relief, digestive disease research and funding, and patient access and protection — as well as the science and practice of gastroenterology.
This year, we will be counting on you, our members, to again lead the charge to achieve AGA’s mission — empowering clinicians and researchers to improve digestive health — by calling on legislators and regulators to ensure the voice of gastroenterology continue to be heard.
NIH funding increase
AGA advocated and secured a $2 billion increase in NIH funding for fiscal year (FY) 2019. When added to increases from the two previous fiscal years, NIH’s funding has increased by 30 percent over the past three years, which is the largest increase since the doubling period in the last decade.
IPAB repeal prevents automatic Medicare cuts
Congress repealed the Independent Payment Advisory Board (IPAB) that was created as part of the Affordable Care Act (ACA). AGA and all of organized medicine long opposed IPAB since its sole purpose was to make budgetary cuts to Medicare if it reached a certain threshold of spending.
MIPS changes means more flexibility for physicians
AGA and the physician community were successful in securing flexibility under the new Medicare Quality Payment Program and the Merit-based Incentive Payment System (MIPS) that were created under the Medicare Access and CHIP Reauthorization Act. The changes give CMS more flexibility in implementing the program and will ensure that physicians have an opportunity to be successful in MIPS.
500 AGA members prevent radical changes to outpatient documentation
In response to a CMS proposal to radically change how outpatient evaluation and management (E/M) services are documented, more than 500 AGA members urged CMS not to move forward with its plan. As a result, CMS withdrew or delayed many of the proposed changes to E/M services that negatively impacted reimbursement. CMS did move forward with several changes to E/M documentation in an effort to reduce administrative burden.
AGA members send more than 940 letters to Congress
AGA launched the Congressional Advocates Program to provide an infrastructure and tools for members to effectively advocate on behalf of their profession and patients. AGA members heeded our calls to action and sent more than 940 letters sent to Congress and federal agencies throughout 2018. We thank our advocates whose participation makes a difference.
To those who took time to advocate on behalf of your profession and patients, thank you. Because of your efforts, we achieved several key successes that benefit clinicians, researchers and patients.
Together we were able to help advance several AGA policy priorities — administrative burden relief, digestive disease research and funding, and patient access and protection — as well as the science and practice of gastroenterology.
This year, we will be counting on you, our members, to again lead the charge to achieve AGA’s mission — empowering clinicians and researchers to improve digestive health — by calling on legislators and regulators to ensure the voice of gastroenterology continue to be heard.
NIH funding increase
AGA advocated and secured a $2 billion increase in NIH funding for fiscal year (FY) 2019. When added to increases from the two previous fiscal years, NIH’s funding has increased by 30 percent over the past three years, which is the largest increase since the doubling period in the last decade.
IPAB repeal prevents automatic Medicare cuts
Congress repealed the Independent Payment Advisory Board (IPAB) that was created as part of the Affordable Care Act (ACA). AGA and all of organized medicine long opposed IPAB since its sole purpose was to make budgetary cuts to Medicare if it reached a certain threshold of spending.
MIPS changes means more flexibility for physicians
AGA and the physician community were successful in securing flexibility under the new Medicare Quality Payment Program and the Merit-based Incentive Payment System (MIPS) that were created under the Medicare Access and CHIP Reauthorization Act. The changes give CMS more flexibility in implementing the program and will ensure that physicians have an opportunity to be successful in MIPS.
500 AGA members prevent radical changes to outpatient documentation
In response to a CMS proposal to radically change how outpatient evaluation and management (E/M) services are documented, more than 500 AGA members urged CMS not to move forward with its plan. As a result, CMS withdrew or delayed many of the proposed changes to E/M services that negatively impacted reimbursement. CMS did move forward with several changes to E/M documentation in an effort to reduce administrative burden.
AGA members send more than 940 letters to Congress
AGA launched the Congressional Advocates Program to provide an infrastructure and tools for members to effectively advocate on behalf of their profession and patients. AGA members heeded our calls to action and sent more than 940 letters sent to Congress and federal agencies throughout 2018. We thank our advocates whose participation makes a difference.
Subclinical hypothyroidism boosts immediate risk of heart failure
CHICAGO – The short-term risk of developing heart failure in patients with newly identified hypothyroidism, be it overt or subclinical, is double that of euthyroid individuals, Caroline H. Noergaard, MD, reported at the American Heart Association scientific sessions.

“This is really important clinically. The association with heart failure has previously been shown in both overt and subclinical hyperthyroidism, but it’s actually new knowledge that hypothyroidism is associated with immediate risk of heart failure. And a lot of people have subclinical hypothyroidism,” said Dr. Noergaard, a PhD student in epidemiology at Aalborg (Denmark) University.
Also at the meeting, Jeffrey L. Anderson, MD, reported that free thyroxine levels within the normal reference range were associated in graded fashion with an increased prevalence and incidence of atrial fibrillation in a large Utah study, a finding that provides independent confirmation of an earlier report by investigators from the population-based Rotterdam Study.
“These findings validate those of the Rotterdam Study in a much larger dataset and may have important clinical implications, including a redefinition of the reference range and the target-free T4 levels for thyroxine replacement therapy,” observed Dr. Anderson, professor of internal medicine at the University of Utah, Salt Lake City, and a research cardiologist at the Intermountain Medical Center Heart Institute.
Hypothyroidism and heart failure
Dr. Noergaard presented a retrospective study of over 1 million Copenhagen-area adults (mean age, 50 years) with no history of heart failure, who had their first thyroid function test. She and her coinvestigators turned to comprehensive Danish national health care registries to determine how many of these individuals were diagnosed with new-onset heart failure within 90 days after their thyroid function test.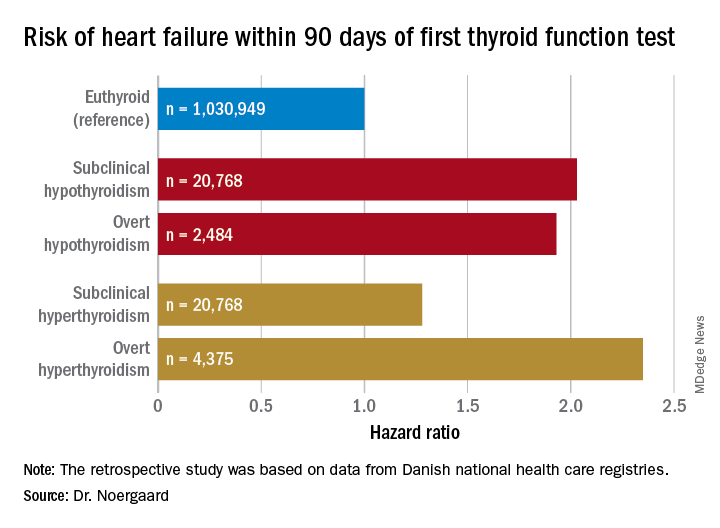
Subclinical hypothyroidism was defined by a thyroid-stimulating hormone level greater than 5 mIU/L and a free T4 of 9-22 pmol/L. Overt hypothyroidism required a TSH greater than 5 mIU/L with a free T4 less than 9 pmol/L.
Free T4 predicts atrial fibrillation risk
Dr. Anderson presented a retrospective analysis of 174,914 adult patients in the Intermountain Healthcare EMR database, none of whom were on thyroid replacement at entry. The patients, who were a mean age of 64 years and 65% women, were followed for an average of 6.3 years. Of these, 88.4% had a free T4 within the normal reference range of 0.75-1.5 ng/dL, 7.4% had a value below the cutoff for normal, and 4.2% had a free T4 above the reference range.
Upon dividing the patients within the normal range into quartiles based upon their free T4 level, he and his coinvestigators found that the baseline prevalence of atrial fibrillation was 8.7% in those in quartile 1, 9.3% in quartile 2, 10.5% in quartile 3, and 12.6% in quartile 4. In a multivariate analysis adjusted for potential confounders, the risk of prevalent atrial fibrillation was increased by 11% for patients in quartile 2, compared with those in the first quartile, by 22% in quartile 3, and by 40% in quartile 4.
The incidence of new-onset atrial fibrillation during 3 years of follow-up was 4.1% in patients in normal-range quartile 1, 4.3% in quartile 2, 4.5% in quartile 3, and 5.2% in the top normal-range quartile. The odds of developing atrial fibrillation were increased by 8% and 16% in quartiles 3 and 4, compared with quartile 1.
Serum TSH and free T3 levels showed no consistent relationship with atrial fibrillation.
The Utah findings confirm in a large U.S. population the earlier report from the Rotterdam Study (J Clin Endocrinol Metab. 2015 Oct;100(10):3718-24).
Dr. Noergaard and Dr. Anderson reported having no financial conflicts regarding their studies, which were carried out free of commercial support.
CHICAGO – The short-term risk of developing heart failure in patients with newly identified hypothyroidism, be it overt or subclinical, is double that of euthyroid individuals, Caroline H. Noergaard, MD, reported at the American Heart Association scientific sessions.
“This is really important clinically. The association with heart failure has previously been shown in both overt and subclinical hyperthyroidism, but it’s actually new knowledge that hypothyroidism is associated with immediate risk of heart failure. And a lot of people have subclinical hypothyroidism,” said Dr. Noergaard, a PhD student in epidemiology at Aalborg (Denmark) University.
Also at the meeting, Jeffrey L. Anderson, MD, reported that free thyroxine levels within the normal reference range were associated in graded fashion with an increased prevalence and incidence of atrial fibrillation in a large Utah study, a finding that provides independent confirmation of an earlier report by investigators from the population-based Rotterdam Study.
“These findings validate those of the Rotterdam Study in a much larger dataset and may have important clinical implications, including a redefinition of the reference range and the target-free T4 levels for thyroxine replacement therapy,” observed Dr. Anderson, professor of internal medicine at the University of Utah, Salt Lake City, and a research cardiologist at the Intermountain Medical Center Heart Institute.
Hypothyroidism and heart failure
Dr. Noergaard presented a retrospective study of over 1 million Copenhagen-area adults (mean age, 50 years) with no history of heart failure, who had their first thyroid function test. She and her coinvestigators turned to comprehensive Danish national health care registries to determine how many of these individuals were diagnosed with new-onset heart failure within 90 days after their thyroid function test.
Subclinical hypothyroidism was defined by a thyroid-stimulating hormone level greater than 5 mIU/L and a free T4 of 9-22 pmol/L. Overt hypothyroidism required a TSH greater than 5 mIU/L with a free T4 less than 9 pmol/L.
Free T4 predicts atrial fibrillation risk
Dr. Anderson presented a retrospective analysis of 174,914 adult patients in the Intermountain Healthcare EMR database, none of whom were on thyroid replacement at entry. The patients, who were a mean age of 64 years and 65% women, were followed for an average of 6.3 years. Of these, 88.4% had a free T4 within the normal reference range of 0.75-1.5 ng/dL, 7.4% had a value below the cutoff for normal, and 4.2% had a free T4 above the reference range.
Upon dividing the patients within the normal range into quartiles based upon their free T4 level, he and his coinvestigators found that the baseline prevalence of atrial fibrillation was 8.7% in those in quartile 1, 9.3% in quartile 2, 10.5% in quartile 3, and 12.6% in quartile 4. In a multivariate analysis adjusted for potential confounders, the risk of prevalent atrial fibrillation was increased by 11% for patients in quartile 2, compared with those in the first quartile, by 22% in quartile 3, and by 40% in quartile 4.
The incidence of new-onset atrial fibrillation during 3 years of follow-up was 4.1% in patients in normal-range quartile 1, 4.3% in quartile 2, 4.5% in quartile 3, and 5.2% in the top normal-range quartile. The odds of developing atrial fibrillation were increased by 8% and 16% in quartiles 3 and 4, compared with quartile 1.
Serum TSH and free T3 levels showed no consistent relationship with atrial fibrillation.
The Utah findings confirm in a large U.S. population the earlier report from the Rotterdam Study (J Clin Endocrinol Metab. 2015 Oct;100(10):3718-24).
Dr. Noergaard and Dr. Anderson reported having no financial conflicts regarding their studies, which were carried out free of commercial support.
CHICAGO – The short-term risk of developing heart failure in patients with newly identified hypothyroidism, be it overt or subclinical, is double that of euthyroid individuals, Caroline H. Noergaard, MD, reported at the American Heart Association scientific sessions.
“This is really important clinically. The association with heart failure has previously been shown in both overt and subclinical hyperthyroidism, but it’s actually new knowledge that hypothyroidism is associated with immediate risk of heart failure. And a lot of people have subclinical hypothyroidism,” said Dr. Noergaard, a PhD student in epidemiology at Aalborg (Denmark) University.
Also at the meeting, Jeffrey L. Anderson, MD, reported that free thyroxine levels within the normal reference range were associated in graded fashion with an increased prevalence and incidence of atrial fibrillation in a large Utah study, a finding that provides independent confirmation of an earlier report by investigators from the population-based Rotterdam Study.
“These findings validate those of the Rotterdam Study in a much larger dataset and may have important clinical implications, including a redefinition of the reference range and the target-free T4 levels for thyroxine replacement therapy,” observed Dr. Anderson, professor of internal medicine at the University of Utah, Salt Lake City, and a research cardiologist at the Intermountain Medical Center Heart Institute.
Hypothyroidism and heart failure
Dr. Noergaard presented a retrospective study of over 1 million Copenhagen-area adults (mean age, 50 years) with no history of heart failure, who had their first thyroid function test. She and her coinvestigators turned to comprehensive Danish national health care registries to determine how many of these individuals were diagnosed with new-onset heart failure within 90 days after their thyroid function test.
Subclinical hypothyroidism was defined by a thyroid-stimulating hormone level greater than 5 mIU/L and a free T4 of 9-22 pmol/L. Overt hypothyroidism required a TSH greater than 5 mIU/L with a free T4 less than 9 pmol/L.
Free T4 predicts atrial fibrillation risk
Dr. Anderson presented a retrospective analysis of 174,914 adult patients in the Intermountain Healthcare EMR database, none of whom were on thyroid replacement at entry. The patients, who were a mean age of 64 years and 65% women, were followed for an average of 6.3 years. Of these, 88.4% had a free T4 within the normal reference range of 0.75-1.5 ng/dL, 7.4% had a value below the cutoff for normal, and 4.2% had a free T4 above the reference range.
Upon dividing the patients within the normal range into quartiles based upon their free T4 level, he and his coinvestigators found that the baseline prevalence of atrial fibrillation was 8.7% in those in quartile 1, 9.3% in quartile 2, 10.5% in quartile 3, and 12.6% in quartile 4. In a multivariate analysis adjusted for potential confounders, the risk of prevalent atrial fibrillation was increased by 11% for patients in quartile 2, compared with those in the first quartile, by 22% in quartile 3, and by 40% in quartile 4.
The incidence of new-onset atrial fibrillation during 3 years of follow-up was 4.1% in patients in normal-range quartile 1, 4.3% in quartile 2, 4.5% in quartile 3, and 5.2% in the top normal-range quartile. The odds of developing atrial fibrillation were increased by 8% and 16% in quartiles 3 and 4, compared with quartile 1.
Serum TSH and free T3 levels showed no consistent relationship with atrial fibrillation.
The Utah findings confirm in a large U.S. population the earlier report from the Rotterdam Study (J Clin Endocrinol Metab. 2015 Oct;100(10):3718-24).
Dr. Noergaard and Dr. Anderson reported having no financial conflicts regarding their studies, which were carried out free of commercial support.
REPORTING FROM THE AHA SCIENTIFIC SESSIONS
Key clinical point:
Major finding: Both subclinical and overt hypothyroidism are associated with a 100% increased risk of being diagnosed with heart failure, compared with euthyroid individuals.
Study details: This was a retrospective study of the association between free thyroxine levels and short-term risk of developing heart failure in more than 1 million Copenhagen-area patients.
Disclosures: The presenter reported having no financial conflicts regarding the Danish study, conducted free of commercial support.
New study determines factors that can send flu patients to the ICU
Numerous independent factors – including a history of obstructive/central sleep apnea syndrome (OSAS/CSAS) or myocardial infarction, along with a body mass index greater than 30 g/m2 – could be related to ICU admission and subsequent high mortality rates in influenza patients, according to an analysis of patients in the Netherlands who were treated during the influenza epidemic of 2015-2016.
Along with determining these factors, lead author M.C. Beumer, of Radboud University Medical Center, the Netherlands, and his coauthors found that “coinfections with bacterial, fungal, and viral pathogens developed more often in patients who were admitted to the ICU.” The study was published in the Journal of Critical Care.
The coauthors reviewed 199 influenza patients who were admitted to two medical centers in the Netherlands during October 2015–April 2016. Of those patients, 45 (23%) were admitted to the ICU, primarily because of respiratory failure, and their mortality rate was 17/45 (38%) versus an overall mortality rate of 18/199 (9%).
Compared with patients in the normal ward, patients admitted to the ICU more frequently had a history of OSAS/CSAS (11% vs. 3%; P = .03) and MI (20% vs. 6%; P = .007), along with a BMI higher than 30 g/m2 (30% vs. 15%; P = .04) and dyspnea as a symptom (77% vs. 48%,; P = .001). In addition, more ICU-admitted patients had influenza A rather than influenza B, compared with those not admitted (87% vs. 66%; P = .009).
Pulmonary coinfections – including bacterial, fungal, and viral pathogens – were also proportionally higher among the 45 ICU patients (56% vs. 20%; P less than .0001). The most common bacterial pathogens were Staphylococcus aureus (11%) and Streptococcus pneumoniae (7%) while Aspergillus fumigatus (18%) and Pneumocystis jirovecii (7%) topped the fungal pathogens.
Mr. Beumer and his colleagues noted potential limitations of their work, including the selection of patients from among the “most severely ill” contributing to an ICU admission rate that surpassed the 5%-10% described elsewhere. They also admitted that their study relied on a “relatively small sample size,” focusing on one seasonal influenza outbreak. However, “despite the limited validity,” they reiterated that “the identified factors may contribute to a complicated disease course and could represent a tool for early recognition of the influenza patients at risk for a complicated disease course.”
The authors reported no conflicts of interest.
SOURCE: Beumer MC et al. J Crit Care. 2019;50:59-65.
.
Numerous independent factors – including a history of obstructive/central sleep apnea syndrome (OSAS/CSAS) or myocardial infarction, along with a body mass index greater than 30 g/m2 – could be related to ICU admission and subsequent high mortality rates in influenza patients, according to an analysis of patients in the Netherlands who were treated during the influenza epidemic of 2015-2016.
Along with determining these factors, lead author M.C. Beumer, of Radboud University Medical Center, the Netherlands, and his coauthors found that “coinfections with bacterial, fungal, and viral pathogens developed more often in patients who were admitted to the ICU.” The study was published in the Journal of Critical Care.
The coauthors reviewed 199 influenza patients who were admitted to two medical centers in the Netherlands during October 2015–April 2016. Of those patients, 45 (23%) were admitted to the ICU, primarily because of respiratory failure, and their mortality rate was 17/45 (38%) versus an overall mortality rate of 18/199 (9%).
Compared with patients in the normal ward, patients admitted to the ICU more frequently had a history of OSAS/CSAS (11% vs. 3%; P = .03) and MI (20% vs. 6%; P = .007), along with a BMI higher than 30 g/m2 (30% vs. 15%; P = .04) and dyspnea as a symptom (77% vs. 48%,; P = .001). In addition, more ICU-admitted patients had influenza A rather than influenza B, compared with those not admitted (87% vs. 66%; P = .009).
Pulmonary coinfections – including bacterial, fungal, and viral pathogens – were also proportionally higher among the 45 ICU patients (56% vs. 20%; P less than .0001). The most common bacterial pathogens were Staphylococcus aureus (11%) and Streptococcus pneumoniae (7%) while Aspergillus fumigatus (18%) and Pneumocystis jirovecii (7%) topped the fungal pathogens.
Mr. Beumer and his colleagues noted potential limitations of their work, including the selection of patients from among the “most severely ill” contributing to an ICU admission rate that surpassed the 5%-10% described elsewhere. They also admitted that their study relied on a “relatively small sample size,” focusing on one seasonal influenza outbreak. However, “despite the limited validity,” they reiterated that “the identified factors may contribute to a complicated disease course and could represent a tool for early recognition of the influenza patients at risk for a complicated disease course.”
The authors reported no conflicts of interest.
SOURCE: Beumer MC et al. J Crit Care. 2019;50:59-65.
.
Numerous independent factors – including a history of obstructive/central sleep apnea syndrome (OSAS/CSAS) or myocardial infarction, along with a body mass index greater than 30 g/m2 – could be related to ICU admission and subsequent high mortality rates in influenza patients, according to an analysis of patients in the Netherlands who were treated during the influenza epidemic of 2015-2016.
Along with determining these factors, lead author M.C. Beumer, of Radboud University Medical Center, the Netherlands, and his coauthors found that “coinfections with bacterial, fungal, and viral pathogens developed more often in patients who were admitted to the ICU.” The study was published in the Journal of Critical Care.
The coauthors reviewed 199 influenza patients who were admitted to two medical centers in the Netherlands during October 2015–April 2016. Of those patients, 45 (23%) were admitted to the ICU, primarily because of respiratory failure, and their mortality rate was 17/45 (38%) versus an overall mortality rate of 18/199 (9%).
Compared with patients in the normal ward, patients admitted to the ICU more frequently had a history of OSAS/CSAS (11% vs. 3%; P = .03) and MI (20% vs. 6%; P = .007), along with a BMI higher than 30 g/m2 (30% vs. 15%; P = .04) and dyspnea as a symptom (77% vs. 48%,; P = .001). In addition, more ICU-admitted patients had influenza A rather than influenza B, compared with those not admitted (87% vs. 66%; P = .009).
Pulmonary coinfections – including bacterial, fungal, and viral pathogens – were also proportionally higher among the 45 ICU patients (56% vs. 20%; P less than .0001). The most common bacterial pathogens were Staphylococcus aureus (11%) and Streptococcus pneumoniae (7%) while Aspergillus fumigatus (18%) and Pneumocystis jirovecii (7%) topped the fungal pathogens.
Mr. Beumer and his colleagues noted potential limitations of their work, including the selection of patients from among the “most severely ill” contributing to an ICU admission rate that surpassed the 5%-10% described elsewhere. They also admitted that their study relied on a “relatively small sample size,” focusing on one seasonal influenza outbreak. However, “despite the limited validity,” they reiterated that “the identified factors may contribute to a complicated disease course and could represent a tool for early recognition of the influenza patients at risk for a complicated disease course.”
The authors reported no conflicts of interest.
SOURCE: Beumer MC et al. J Crit Care. 2019;50:59-65.
.
FROM THE JOURNAL OF CRITICAL CARE
Key clinical point:
Major finding: Flu patients in the ICU more frequently had a history of obstructive/central sleep apnea syndrome (11% vs. 3%; P = .03) and MI (20% vs. 6%; P = .007), compared with non-ICU flu patients.
Study details: A retrospective cohort study of 199 flu patients who were admitted to two academic hospitals in the Netherlands.
Disclosures: The authors reported no conflicts of interest.
Source: Beumer MC et al. J Crit Care. 2019; 50:59-65.
Help spark scientific breakthroughs with the AGA Research Foundation
The way we diagnose and treat patients is the result of years of research. But securing the future of the field is no small task. A donation to the charitable arm of the American Gastroenterological Association (AGA), the AGA Research Foundation, will help fill the funding gap and contribute to this tradition of discovery.

The foundation provides a key source of funding at a critical juncture in a young investigator’s career.
Help spark the scientific breakthroughs of today, so clinicians will have the tools to improve care tomorrow. Your tax-deductible donation will make a critical difference in retaining talented GI scientists, like Won Jae Huh, MD, whose research will impact the future care of patients.
“As a clinical researcher, funding for investigation is critical in scientific breakthroughs to promote more efficient and robust patient care. My project will provide novel insights into the role of distensibility in the treatment of patients with esophageal eosinophilia, potentially resulting in more efficient treatment selection and disease management.”
“This funding mechanism will secure my research time to investigate signaling pathways involved in the pathogenesis of Ménétrier’s disease, and will provide resources and support to launch my research career. The results from my research project will be helpful for treating Ménétrier’s disease patients.”
Help spark the scientific breakthroughs of today, so clinicians will have the tools to improve care tomorrow. Your tax-deductible donation will make a critical difference in retaining talented GI scientists, like Dr. Won Jae Huh, whose research will impact the future care of patients.
Donate on the foundation’s website at www.gastro.org/donateonline or by mail to 4930 Del Ray Avenue, Bethesda, MD 20814.
The way we diagnose and treat patients is the result of years of research. But securing the future of the field is no small task. A donation to the charitable arm of the American Gastroenterological Association (AGA), the AGA Research Foundation, will help fill the funding gap and contribute to this tradition of discovery.
The foundation provides a key source of funding at a critical juncture in a young investigator’s career.
Help spark the scientific breakthroughs of today, so clinicians will have the tools to improve care tomorrow. Your tax-deductible donation will make a critical difference in retaining talented GI scientists, like Won Jae Huh, MD, whose research will impact the future care of patients.
“As a clinical researcher, funding for investigation is critical in scientific breakthroughs to promote more efficient and robust patient care. My project will provide novel insights into the role of distensibility in the treatment of patients with esophageal eosinophilia, potentially resulting in more efficient treatment selection and disease management.”
“This funding mechanism will secure my research time to investigate signaling pathways involved in the pathogenesis of Ménétrier’s disease, and will provide resources and support to launch my research career. The results from my research project will be helpful for treating Ménétrier’s disease patients.”
Help spark the scientific breakthroughs of today, so clinicians will have the tools to improve care tomorrow. Your tax-deductible donation will make a critical difference in retaining talented GI scientists, like Dr. Won Jae Huh, whose research will impact the future care of patients.
Donate on the foundation’s website at www.gastro.org/donateonline or by mail to 4930 Del Ray Avenue, Bethesda, MD 20814.
The way we diagnose and treat patients is the result of years of research. But securing the future of the field is no small task. A donation to the charitable arm of the American Gastroenterological Association (AGA), the AGA Research Foundation, will help fill the funding gap and contribute to this tradition of discovery.
The foundation provides a key source of funding at a critical juncture in a young investigator’s career.
Help spark the scientific breakthroughs of today, so clinicians will have the tools to improve care tomorrow. Your tax-deductible donation will make a critical difference in retaining talented GI scientists, like Won Jae Huh, MD, whose research will impact the future care of patients.
“As a clinical researcher, funding for investigation is critical in scientific breakthroughs to promote more efficient and robust patient care. My project will provide novel insights into the role of distensibility in the treatment of patients with esophageal eosinophilia, potentially resulting in more efficient treatment selection and disease management.”
“This funding mechanism will secure my research time to investigate signaling pathways involved in the pathogenesis of Ménétrier’s disease, and will provide resources and support to launch my research career. The results from my research project will be helpful for treating Ménétrier’s disease patients.”
Help spark the scientific breakthroughs of today, so clinicians will have the tools to improve care tomorrow. Your tax-deductible donation will make a critical difference in retaining talented GI scientists, like Dr. Won Jae Huh, whose research will impact the future care of patients.
Donate on the foundation’s website at www.gastro.org/donateonline or by mail to 4930 Del Ray Avenue, Bethesda, MD 20814.

Trump zeroes in on surprise medical bills in White House chat with patients, experts
President Trump on Jan. 23 instructed administration officials to investigate how to prevent surprise medical bills, broadening his focus on drug prices to include other issues of price transparency in health care.

several attendees said.
“The pricing is hurting patients, and we’ve stopped a lot of it, but we’re going to stop all of it,” Mr. Trump said during a roundtable discussion when reporters were briefly allowed into the otherwise closed-door meeting.
David Silverstein, the founder of a Colorado-based nonprofit called Broken Healthcare who attended, said Mr. Trump struck an aggressive tone, calling for a solution with “the biggest teeth you can find.”
“Reading the tea leaves, I think there’s big change coming,” Mr. Silverstein said.
Surprise billing, or the practice of charging patients for care that is more expensive than anticipated or not covered by their insurance, has received a flood of attention in the past year, particularly as Kaiser Health News and other news organizations have undertaken investigations into patients’ most outrageous medical bills.
Attendees said each of 10 invited guests – among them patients as well as doctors with their own stories of unexpected bills – was given an opportunity to talk, though Mr. Trump did not stay to hear all of their stories during the roughly hour-long gathering.
The group included Paul Davis, a retired doctor from Findlay, Ohio, whose family’s experience with a $17,850 bill for a simple urine test was detailed in a KHN-NPR “Bill of the Month” feature last year.
Mr. Davis’ daughter, Elizabeth Moreno, was a college student in Texas when she had spinal surgery to remedy debilitating back pain. After the surgery, she was asked to provide a urine sample and later received a bill from an out-of-network lab in Houston that tested it. Experts said such tests rarely cost more than $200, not nearly what the lab charged Ms. Moreno and her insurance company. But fearing damage to his daughter’s credit, Mr. Davis paid the lab $5,000 and filed a complaint with the Texas attorney general’s office, alleging “price gouging of staggering proportions.”
Mr. Davis said White House officials made it clear that price transparency is a “high priority” for Trump, and while they did not see eye to eye on every subject, he said he was struck by their sincerity.
“These people seemed earnest in wanting to do something constructive to fix this,” Mr. Davis said.
Dr. Martin Makary, a surgeon and health policy expert at Johns Hopkins University who has written about transparency in health care and attended the meeting, said it was a good opportunity for the White House to hear firsthand about a serious and widespread issue.
“This is how most of America lives, and [Americans are] getting hammered,” he said.
Mr. Trump has often railed against high prescription drug prices but has said less about other problems with the nation’s health care system. In October, shortly before the midterm elections, he unveiled a proposal to tie the price Medicare pays for some drugs to the prices paid for the same drugs overseas, for example.
Mr. Trump, Mr. Azar, and Mr. Acosta said efforts to control costs in health care were yielding positive results, discussing in particular the expansion of association health plans and the new requirement that hospitals post their list prices online. The president also took credit for the recent increase in generic drug approvals, which he said would help lower drug prices.
Discussing the partial government shutdown, Mr. Trump said Americans “want to see what we’re doing, like today we lowered prescription drug prices, first time in 50 years,” according to a White House pool report.
Mr. Trump appeared to be referring to a recent claim by the White House Council of Economic Advisers that prescription drug prices fell last year.
However, as STAT pointed out in a recent fact check, the report from which that claim was gleaned said “growth in relative drug prices has slowed since January 2017,” not that there was an overall decrease in prices.
Annual increases in overall drug spending have leveled off as pharmaceutical companies have released fewer blockbuster drugs; patents have expired on brand-name drugs; and the waning effect of a spike driven by the release of astronomically expensive drugs to treat hepatitis C. Drugmakers are also wary of increasing their prices in the midst of growing political pressure.
Since Democrats seized control of the House of Representatives this month, party leaders have rushed to announce investigations and schedule hearings dealing with health care, focusing in particular on drug costs and protections for those with preexisting conditions.
Recently, the House Oversight Committee announced a “sweeping” investigation into drug prices, pointing to an AARP report saying the vast majority of brand-name drugs had more than doubled in price between 2005 and 2017.
KHN correspondents Shefali Luthra and Jay Hancock contributed to this report. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
President Trump on Jan. 23 instructed administration officials to investigate how to prevent surprise medical bills, broadening his focus on drug prices to include other issues of price transparency in health care.
several attendees said.
“The pricing is hurting patients, and we’ve stopped a lot of it, but we’re going to stop all of it,” Mr. Trump said during a roundtable discussion when reporters were briefly allowed into the otherwise closed-door meeting.
David Silverstein, the founder of a Colorado-based nonprofit called Broken Healthcare who attended, said Mr. Trump struck an aggressive tone, calling for a solution with “the biggest teeth you can find.”
“Reading the tea leaves, I think there’s big change coming,” Mr. Silverstein said.
Surprise billing, or the practice of charging patients for care that is more expensive than anticipated or not covered by their insurance, has received a flood of attention in the past year, particularly as Kaiser Health News and other news organizations have undertaken investigations into patients’ most outrageous medical bills.
Attendees said each of 10 invited guests – among them patients as well as doctors with their own stories of unexpected bills – was given an opportunity to talk, though Mr. Trump did not stay to hear all of their stories during the roughly hour-long gathering.
The group included Paul Davis, a retired doctor from Findlay, Ohio, whose family’s experience with a $17,850 bill for a simple urine test was detailed in a KHN-NPR “Bill of the Month” feature last year.
Mr. Davis’ daughter, Elizabeth Moreno, was a college student in Texas when she had spinal surgery to remedy debilitating back pain. After the surgery, she was asked to provide a urine sample and later received a bill from an out-of-network lab in Houston that tested it. Experts said such tests rarely cost more than $200, not nearly what the lab charged Ms. Moreno and her insurance company. But fearing damage to his daughter’s credit, Mr. Davis paid the lab $5,000 and filed a complaint with the Texas attorney general’s office, alleging “price gouging of staggering proportions.”
Mr. Davis said White House officials made it clear that price transparency is a “high priority” for Trump, and while they did not see eye to eye on every subject, he said he was struck by their sincerity.
“These people seemed earnest in wanting to do something constructive to fix this,” Mr. Davis said.
Dr. Martin Makary, a surgeon and health policy expert at Johns Hopkins University who has written about transparency in health care and attended the meeting, said it was a good opportunity for the White House to hear firsthand about a serious and widespread issue.
“This is how most of America lives, and [Americans are] getting hammered,” he said.
Mr. Trump has often railed against high prescription drug prices but has said less about other problems with the nation’s health care system. In October, shortly before the midterm elections, he unveiled a proposal to tie the price Medicare pays for some drugs to the prices paid for the same drugs overseas, for example.
Mr. Trump, Mr. Azar, and Mr. Acosta said efforts to control costs in health care were yielding positive results, discussing in particular the expansion of association health plans and the new requirement that hospitals post their list prices online. The president also took credit for the recent increase in generic drug approvals, which he said would help lower drug prices.
Discussing the partial government shutdown, Mr. Trump said Americans “want to see what we’re doing, like today we lowered prescription drug prices, first time in 50 years,” according to a White House pool report.
Mr. Trump appeared to be referring to a recent claim by the White House Council of Economic Advisers that prescription drug prices fell last year.
However, as STAT pointed out in a recent fact check, the report from which that claim was gleaned said “growth in relative drug prices has slowed since January 2017,” not that there was an overall decrease in prices.
Annual increases in overall drug spending have leveled off as pharmaceutical companies have released fewer blockbuster drugs; patents have expired on brand-name drugs; and the waning effect of a spike driven by the release of astronomically expensive drugs to treat hepatitis C. Drugmakers are also wary of increasing their prices in the midst of growing political pressure.
Since Democrats seized control of the House of Representatives this month, party leaders have rushed to announce investigations and schedule hearings dealing with health care, focusing in particular on drug costs and protections for those with preexisting conditions.
Recently, the House Oversight Committee announced a “sweeping” investigation into drug prices, pointing to an AARP report saying the vast majority of brand-name drugs had more than doubled in price between 2005 and 2017.
KHN correspondents Shefali Luthra and Jay Hancock contributed to this report. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
President Trump on Jan. 23 instructed administration officials to investigate how to prevent surprise medical bills, broadening his focus on drug prices to include other issues of price transparency in health care.
several attendees said.
“The pricing is hurting patients, and we’ve stopped a lot of it, but we’re going to stop all of it,” Mr. Trump said during a roundtable discussion when reporters were briefly allowed into the otherwise closed-door meeting.
David Silverstein, the founder of a Colorado-based nonprofit called Broken Healthcare who attended, said Mr. Trump struck an aggressive tone, calling for a solution with “the biggest teeth you can find.”
“Reading the tea leaves, I think there’s big change coming,” Mr. Silverstein said.
Surprise billing, or the practice of charging patients for care that is more expensive than anticipated or not covered by their insurance, has received a flood of attention in the past year, particularly as Kaiser Health News and other news organizations have undertaken investigations into patients’ most outrageous medical bills.
Attendees said each of 10 invited guests – among them patients as well as doctors with their own stories of unexpected bills – was given an opportunity to talk, though Mr. Trump did not stay to hear all of their stories during the roughly hour-long gathering.
The group included Paul Davis, a retired doctor from Findlay, Ohio, whose family’s experience with a $17,850 bill for a simple urine test was detailed in a KHN-NPR “Bill of the Month” feature last year.
Mr. Davis’ daughter, Elizabeth Moreno, was a college student in Texas when she had spinal surgery to remedy debilitating back pain. After the surgery, she was asked to provide a urine sample and later received a bill from an out-of-network lab in Houston that tested it. Experts said such tests rarely cost more than $200, not nearly what the lab charged Ms. Moreno and her insurance company. But fearing damage to his daughter’s credit, Mr. Davis paid the lab $5,000 and filed a complaint with the Texas attorney general’s office, alleging “price gouging of staggering proportions.”
Mr. Davis said White House officials made it clear that price transparency is a “high priority” for Trump, and while they did not see eye to eye on every subject, he said he was struck by their sincerity.
“These people seemed earnest in wanting to do something constructive to fix this,” Mr. Davis said.
Dr. Martin Makary, a surgeon and health policy expert at Johns Hopkins University who has written about transparency in health care and attended the meeting, said it was a good opportunity for the White House to hear firsthand about a serious and widespread issue.
“This is how most of America lives, and [Americans are] getting hammered,” he said.
Mr. Trump has often railed against high prescription drug prices but has said less about other problems with the nation’s health care system. In October, shortly before the midterm elections, he unveiled a proposal to tie the price Medicare pays for some drugs to the prices paid for the same drugs overseas, for example.
Mr. Trump, Mr. Azar, and Mr. Acosta said efforts to control costs in health care were yielding positive results, discussing in particular the expansion of association health plans and the new requirement that hospitals post their list prices online. The president also took credit for the recent increase in generic drug approvals, which he said would help lower drug prices.
Discussing the partial government shutdown, Mr. Trump said Americans “want to see what we’re doing, like today we lowered prescription drug prices, first time in 50 years,” according to a White House pool report.
Mr. Trump appeared to be referring to a recent claim by the White House Council of Economic Advisers that prescription drug prices fell last year.
However, as STAT pointed out in a recent fact check, the report from which that claim was gleaned said “growth in relative drug prices has slowed since January 2017,” not that there was an overall decrease in prices.
Annual increases in overall drug spending have leveled off as pharmaceutical companies have released fewer blockbuster drugs; patents have expired on brand-name drugs; and the waning effect of a spike driven by the release of astronomically expensive drugs to treat hepatitis C. Drugmakers are also wary of increasing their prices in the midst of growing political pressure.
Since Democrats seized control of the House of Representatives this month, party leaders have rushed to announce investigations and schedule hearings dealing with health care, focusing in particular on drug costs and protections for those with preexisting conditions.
Recently, the House Oversight Committee announced a “sweeping” investigation into drug prices, pointing to an AARP report saying the vast majority of brand-name drugs had more than doubled in price between 2005 and 2017.
KHN correspondents Shefali Luthra and Jay Hancock contributed to this report. Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
Half of parents unaware of teens’ suicidal thoughts
Most parents are unaware their teenager has been having suicidal thoughts or thinking about death, according to a study published in Pediatrics.

Jason D. Jones, PhD, from the Children’s Hospital of Philadelphia, and his coauthors wrote that more than two-thirds of adolescents who experience suicidal thoughts do not get medical help, and this may be because their parents – the gatekeepers for mental health services – are unaware of what their teen is going through.
In this study, researchers recruited 5,137 adolescents aged 11-17 years and either a parent or step-parent, and interviewed both about the adolescent’s lifetime suicidal thoughts.
While 413 (8%) of the adolescents surveyed said they had had thoughts about killing themselves, 50% of those adolescents’ parents said their teen hadn’t experienced suicidal thoughts. Similarly, 786 (15%) of adolescents surveyed said they had had thoughts about death and dying, but three-quarters of their parents were unaware.
A significant number of parents – 8% – said their teenager had had suicidal thoughts, but in 48% of these cases, the teenager said they had not thought about killing themselves.
Researchers saw more agreement between parents and adolescents when the adolescents were older: The parents were less likely to be unaware that their older teen had had suicidal thoughts, and older adolescents were less likely to deny it.
“This indicates that younger adolescents may be more likely to go unnoticed and not receive services either because their parents are unaware of their suicidal thoughts or because they deny suicidal thoughts that their parents think they are having,” Dr. Jones and his associates wrote. They also suggested younger adolescents may have “interpretive difficulties” around questions of suicidal ideation.
“These age findings are particularly noteworthy in light of recent evidence that deaths by suicide have increased among younger adolescents,” they noted.
There also was an interaction between age and gender. For girls, parents were less likely to be aware of suicidal thoughts in their younger daughters but more likely to be aware of them in their older daughters. However the opposite was true for boys: Parental unawareness increased slightly in older boys.
Parents of Hispanic or Latino ethnicity were less likely to be aware that their offspring had had thoughts about death and dying.
Generally fathers were less likely than mothers to be aware of suicidal thoughts in their adolescents.
However, if adolescents had previously received psychiatric treatment, or there was a family history of suicide, parents were more likely to be aware of suicidal thoughts, and adolescents who had a history of psychiatric hospitalization were less likely to deny suicidal thoughts, the researchers reported.
The study was supported by grants from the National Institutes of Health, the Dowshen Program for Neuroscience, and the Lifespan Brain Institute of the Children’s Hospital of Philadelphia and University of Pennsylvania. The study was funded by NIH. One author declared a board position and stock options in Taliaz Health unrelated to the study subject; the other authors said they had no relevant financial disclosures.
SOURCE: Jones JD et al. Pediatrics. 2019 Jan 14. doi: 10.1542/peds.2018-1771.
Suicide prevention relies on identifying individuals at risk, but in the case of young people, this often relies on parents. This study, and previous research, highlights the limitations of parent report of adolescents’ suicidal thoughts, as well as the issue of adolescents’ denying suicidal thoughts when parents report them.
Given that as many as 40% of adolescents who think about suicide act on those thoughts, it is vital that we achieve more specificity in identifying young people at risk of attempting suicide. These findings have implications for screening in the primary care setting, and they suggest a need for multi-informant assessments, as well as careful exploration of disagreements between parents’ and adolescent’s reports.
Khyati Brahmbhatt, MD, and Jacqueline Grupp-Phelan, MD, MPH, are from the University of California, San Francisco, Benioff Children’s Hospitals. These comments are taken from an accompanying editorial (Pediatrics. 2019 Jan 14. doi: 10.1542/peds.2018-3071). No conflicts of interest were declared. The editorial was funded by the National Institutes of Health.
Suicide prevention relies on identifying individuals at risk, but in the case of young people, this often relies on parents. This study, and previous research, highlights the limitations of parent report of adolescents’ suicidal thoughts, as well as the issue of adolescents’ denying suicidal thoughts when parents report them.
Given that as many as 40% of adolescents who think about suicide act on those thoughts, it is vital that we achieve more specificity in identifying young people at risk of attempting suicide. These findings have implications for screening in the primary care setting, and they suggest a need for multi-informant assessments, as well as careful exploration of disagreements between parents’ and adolescent’s reports.
Khyati Brahmbhatt, MD, and Jacqueline Grupp-Phelan, MD, MPH, are from the University of California, San Francisco, Benioff Children’s Hospitals. These comments are taken from an accompanying editorial (Pediatrics. 2019 Jan 14. doi: 10.1542/peds.2018-3071). No conflicts of interest were declared. The editorial was funded by the National Institutes of Health.
Suicide prevention relies on identifying individuals at risk, but in the case of young people, this often relies on parents. This study, and previous research, highlights the limitations of parent report of adolescents’ suicidal thoughts, as well as the issue of adolescents’ denying suicidal thoughts when parents report them.
Given that as many as 40% of adolescents who think about suicide act on those thoughts, it is vital that we achieve more specificity in identifying young people at risk of attempting suicide. These findings have implications for screening in the primary care setting, and they suggest a need for multi-informant assessments, as well as careful exploration of disagreements between parents’ and adolescent’s reports.
Khyati Brahmbhatt, MD, and Jacqueline Grupp-Phelan, MD, MPH, are from the University of California, San Francisco, Benioff Children’s Hospitals. These comments are taken from an accompanying editorial (Pediatrics. 2019 Jan 14. doi: 10.1542/peds.2018-3071). No conflicts of interest were declared. The editorial was funded by the National Institutes of Health.
Most parents are unaware their teenager has been having suicidal thoughts or thinking about death, according to a study published in Pediatrics.
Jason D. Jones, PhD, from the Children’s Hospital of Philadelphia, and his coauthors wrote that more than two-thirds of adolescents who experience suicidal thoughts do not get medical help, and this may be because their parents – the gatekeepers for mental health services – are unaware of what their teen is going through.
In this study, researchers recruited 5,137 adolescents aged 11-17 years and either a parent or step-parent, and interviewed both about the adolescent’s lifetime suicidal thoughts.
While 413 (8%) of the adolescents surveyed said they had had thoughts about killing themselves, 50% of those adolescents’ parents said their teen hadn’t experienced suicidal thoughts. Similarly, 786 (15%) of adolescents surveyed said they had had thoughts about death and dying, but three-quarters of their parents were unaware.
A significant number of parents – 8% – said their teenager had had suicidal thoughts, but in 48% of these cases, the teenager said they had not thought about killing themselves.
Researchers saw more agreement between parents and adolescents when the adolescents were older: The parents were less likely to be unaware that their older teen had had suicidal thoughts, and older adolescents were less likely to deny it.
“This indicates that younger adolescents may be more likely to go unnoticed and not receive services either because their parents are unaware of their suicidal thoughts or because they deny suicidal thoughts that their parents think they are having,” Dr. Jones and his associates wrote. They also suggested younger adolescents may have “interpretive difficulties” around questions of suicidal ideation.
“These age findings are particularly noteworthy in light of recent evidence that deaths by suicide have increased among younger adolescents,” they noted.
There also was an interaction between age and gender. For girls, parents were less likely to be aware of suicidal thoughts in their younger daughters but more likely to be aware of them in their older daughters. However the opposite was true for boys: Parental unawareness increased slightly in older boys.
Parents of Hispanic or Latino ethnicity were less likely to be aware that their offspring had had thoughts about death and dying.
Generally fathers were less likely than mothers to be aware of suicidal thoughts in their adolescents.
However, if adolescents had previously received psychiatric treatment, or there was a family history of suicide, parents were more likely to be aware of suicidal thoughts, and adolescents who had a history of psychiatric hospitalization were less likely to deny suicidal thoughts, the researchers reported.
The study was supported by grants from the National Institutes of Health, the Dowshen Program for Neuroscience, and the Lifespan Brain Institute of the Children’s Hospital of Philadelphia and University of Pennsylvania. The study was funded by NIH. One author declared a board position and stock options in Taliaz Health unrelated to the study subject; the other authors said they had no relevant financial disclosures.
SOURCE: Jones JD et al. Pediatrics. 2019 Jan 14. doi: 10.1542/peds.2018-1771.
Most parents are unaware their teenager has been having suicidal thoughts or thinking about death, according to a study published in Pediatrics.
Jason D. Jones, PhD, from the Children’s Hospital of Philadelphia, and his coauthors wrote that more than two-thirds of adolescents who experience suicidal thoughts do not get medical help, and this may be because their parents – the gatekeepers for mental health services – are unaware of what their teen is going through.
In this study, researchers recruited 5,137 adolescents aged 11-17 years and either a parent or step-parent, and interviewed both about the adolescent’s lifetime suicidal thoughts.
While 413 (8%) of the adolescents surveyed said they had had thoughts about killing themselves, 50% of those adolescents’ parents said their teen hadn’t experienced suicidal thoughts. Similarly, 786 (15%) of adolescents surveyed said they had had thoughts about death and dying, but three-quarters of their parents were unaware.
A significant number of parents – 8% – said their teenager had had suicidal thoughts, but in 48% of these cases, the teenager said they had not thought about killing themselves.
Researchers saw more agreement between parents and adolescents when the adolescents were older: The parents were less likely to be unaware that their older teen had had suicidal thoughts, and older adolescents were less likely to deny it.
“This indicates that younger adolescents may be more likely to go unnoticed and not receive services either because their parents are unaware of their suicidal thoughts or because they deny suicidal thoughts that their parents think they are having,” Dr. Jones and his associates wrote. They also suggested younger adolescents may have “interpretive difficulties” around questions of suicidal ideation.
“These age findings are particularly noteworthy in light of recent evidence that deaths by suicide have increased among younger adolescents,” they noted.
There also was an interaction between age and gender. For girls, parents were less likely to be aware of suicidal thoughts in their younger daughters but more likely to be aware of them in their older daughters. However the opposite was true for boys: Parental unawareness increased slightly in older boys.
Parents of Hispanic or Latino ethnicity were less likely to be aware that their offspring had had thoughts about death and dying.
Generally fathers were less likely than mothers to be aware of suicidal thoughts in their adolescents.
However, if adolescents had previously received psychiatric treatment, or there was a family history of suicide, parents were more likely to be aware of suicidal thoughts, and adolescents who had a history of psychiatric hospitalization were less likely to deny suicidal thoughts, the researchers reported.
The study was supported by grants from the National Institutes of Health, the Dowshen Program for Neuroscience, and the Lifespan Brain Institute of the Children’s Hospital of Philadelphia and University of Pennsylvania. The study was funded by NIH. One author declared a board position and stock options in Taliaz Health unrelated to the study subject; the other authors said they had no relevant financial disclosures.
SOURCE: Jones JD et al. Pediatrics. 2019 Jan 14. doi: 10.1542/peds.2018-1771.
FROM PEDIATRICS
Key clinical point: Many parents are unaware of their teenager’s suicidal thoughts.
Major finding: Half of parents are unaware that their adolescent child has had suicidal thoughts.
Study details: Survey of 5,137 adolescents and their parents or step-parents.
Disclosures: The study was supported by grants from the National Institutes of Health, the Dowshen Program for Neuroscience, and the Lifespan Brain Institute of the Children’s Hospital of Philadelphia and University of Pennsylvania. The study was funded by NIH. One author declared a board position and stock options in Taliaz Health unrelated to the study subject; the other authors said they had no relevant financial disclosures.
Source: Jones JD et al. Pediatrics. 2019, Jan 14. doi: 10.1542/peds.2018-1771.

No evidence for disease-modifying effect of levodopa in Parkinson’s disease
investigators reported. The disease course was not significantly different for patients who had a full 80 weeks of levodopa/carbodopa therapy, compared with that seen with those who started treatment after a 40-week delay, according to the investigators.

“These findings imply that levodopa had no disease-modifying effect on Parkinson’s disease over the period of the trial,” wrote investigator Rob M. A. de Bie, MD, PhD, professor of movement disorders at the University of Amsterdam, and his colleagues in the New England Journal of Medicine.
By contrast, results of an earlier randomized, placebo-controlled trial suggested that levodopa had disease-modifying effects, though the findings of that study were inconclusive, according to authors of an editorial (see Views on the News).
In the current multicenter trial, known as LEAP (Levodopa in Early Parkinson’s Disease) a total of 445 patients with early Parkinson’s disease were randomized to either 80 weeks of levodopa and carbodopa or to 40 weeks of placebo followed by 40 weeks of levodopa/carbodopa.
Levodopa was dosed at 100 mg three times per day, and carbodopa at 25 mg three times per day, according to the report.
There was no significant difference between the early and delayed treatment groups for primary outcome of the trial, which was change in the Unified Parkinson’s Disease Rating Scale (UPDRS) from baseline to week 80.
The mean change in UPDRS was –1.0 in the group of patients who had the full 80 weeks of levodopa/carbodopa and –2.0 for those who had delayed therapy, for a difference of 1 point (P = .44). Higher scores on the UPDRS signify worse disease.
At week 40, there was a change in UPDRS favoring the early-initiation strategy, which reflected the effects of levodopa on disease symptoms, investigators added.
Nausea was more common in the early-start group during the first 40 weeks of the trial. However, there were no differences between groups in other adverse events of particular interest, including dyskinesias and motor fluctuations related to levodopa, Dr. de Bie and his colleagues reported.
Taken together, these results suggest no beneficial or detrimental disease-modifying effect for an early treatment strategy, although further trials are warranted to evaluate other strategies, such as higher levodopa doses, longer administration, or starting the drug at later stages of disease, they wrote.
Dr. de Bie reported grants from ZonMw, Parkinson Vereniging, and Stichting Parkinsonfonds during the conduct of the study, as well as grants from GE Health and Medtronic outside the submitted work. Study authors provided disclosures related to Netherlands Organization for Scientific Research, Michael J. Fox Foundation, UCB, AbbVie, Boston Scientific, Biogen, Merck, and others.
SOURCE: Verschuur CVM et al. N Engl J Med. 2019;380:315-24.
This trial supports current clinical practice in two ways, according to Susan Bressman, MD, and Rachel Saunders‑Pullman, MD, MPH. On one hand, the study provides no evidence to suggest that levodopa slows Parkinson’s disease progression, Dr. Bressman and Dr. Saunders-Pullman wrote in an editorial accompanying the study. On the other hand, they added, it provides no evidence that clinicians should delay therapy when it is clinically indicated.
The LEAP trial (Levodopa in Early Parkinson’s Disease) was designed to resolve uncertainty over the potential effects of levodopa on disease progression, they noted. This was necessary because of the results of the placebo-controlled ELLDOPA trial, which was published about 14 years ago and suggested that patients randomized to 40 weeks of levodopa did not deteriorate clinically to the degree that was observed in patients randomized to placebo.
The primary end point of that trial was Unified Parkinson’s Disease Rating Scale (UPDRS) scores after a 2-week washout period.
While one interpretation of the UPDRS results from ELLDOPA was that levodopa slowed disease progression, another was that the 2-week washout period was too short, allowing for residual effects of levodopa on symptoms, suggested Dr. Bressman and Dr. Saunders-Pullman.
The randomized LEAP study now shows not only that there were no differences in UPDRS scores when using a delayed start trial design – which implies that there was no disease-modifying effect – but also that starting levodopa early did not have negative effects, the editorial authors wrote.
In particular, the researchers showed no differences in rates of dyskinesia or levodopa-related fluctuations in those started early versus those started later.
“The results of the current trial, taken together with those of other trials, support treatment that is guided by clinical need and that uses the lowest dose that provides a satisfactory clinical effect,” wrote the editorial’s authors.
Dr. Bressman and Dr. Saunders‑Pullman are with the Icahn School of Medicine at Mt. Sinai, New York. Their editorial appears in the New England Journal of Medicine (2019;380:389-90). Dr. Bressman reported disclosures related to Denali Therapeutics, the Michael J. Fox Foundation, and Prevail Therapeutics, while Dr. Saunders-Pullman reported disclosures with Denali Therapeutics, the National Institutes of Health, Genzyme Sanofi, and the Bigglesworth Family Foundation.
This trial supports current clinical practice in two ways, according to Susan Bressman, MD, and Rachel Saunders‑Pullman, MD, MPH. On one hand, the study provides no evidence to suggest that levodopa slows Parkinson’s disease progression, Dr. Bressman and Dr. Saunders-Pullman wrote in an editorial accompanying the study. On the other hand, they added, it provides no evidence that clinicians should delay therapy when it is clinically indicated.
The LEAP trial (Levodopa in Early Parkinson’s Disease) was designed to resolve uncertainty over the potential effects of levodopa on disease progression, they noted. This was necessary because of the results of the placebo-controlled ELLDOPA trial, which was published about 14 years ago and suggested that patients randomized to 40 weeks of levodopa did not deteriorate clinically to the degree that was observed in patients randomized to placebo.
The primary end point of that trial was Unified Parkinson’s Disease Rating Scale (UPDRS) scores after a 2-week washout period.
While one interpretation of the UPDRS results from ELLDOPA was that levodopa slowed disease progression, another was that the 2-week washout period was too short, allowing for residual effects of levodopa on symptoms, suggested Dr. Bressman and Dr. Saunders-Pullman.
The randomized LEAP study now shows not only that there were no differences in UPDRS scores when using a delayed start trial design – which implies that there was no disease-modifying effect – but also that starting levodopa early did not have negative effects, the editorial authors wrote.
In particular, the researchers showed no differences in rates of dyskinesia or levodopa-related fluctuations in those started early versus those started later.
“The results of the current trial, taken together with those of other trials, support treatment that is guided by clinical need and that uses the lowest dose that provides a satisfactory clinical effect,” wrote the editorial’s authors.
Dr. Bressman and Dr. Saunders‑Pullman are with the Icahn School of Medicine at Mt. Sinai, New York. Their editorial appears in the New England Journal of Medicine (2019;380:389-90). Dr. Bressman reported disclosures related to Denali Therapeutics, the Michael J. Fox Foundation, and Prevail Therapeutics, while Dr. Saunders-Pullman reported disclosures with Denali Therapeutics, the National Institutes of Health, Genzyme Sanofi, and the Bigglesworth Family Foundation.
This trial supports current clinical practice in two ways, according to Susan Bressman, MD, and Rachel Saunders‑Pullman, MD, MPH. On one hand, the study provides no evidence to suggest that levodopa slows Parkinson’s disease progression, Dr. Bressman and Dr. Saunders-Pullman wrote in an editorial accompanying the study. On the other hand, they added, it provides no evidence that clinicians should delay therapy when it is clinically indicated.
The LEAP trial (Levodopa in Early Parkinson’s Disease) was designed to resolve uncertainty over the potential effects of levodopa on disease progression, they noted. This was necessary because of the results of the placebo-controlled ELLDOPA trial, which was published about 14 years ago and suggested that patients randomized to 40 weeks of levodopa did not deteriorate clinically to the degree that was observed in patients randomized to placebo.
The primary end point of that trial was Unified Parkinson’s Disease Rating Scale (UPDRS) scores after a 2-week washout period.
While one interpretation of the UPDRS results from ELLDOPA was that levodopa slowed disease progression, another was that the 2-week washout period was too short, allowing for residual effects of levodopa on symptoms, suggested Dr. Bressman and Dr. Saunders-Pullman.
The randomized LEAP study now shows not only that there were no differences in UPDRS scores when using a delayed start trial design – which implies that there was no disease-modifying effect – but also that starting levodopa early did not have negative effects, the editorial authors wrote.
In particular, the researchers showed no differences in rates of dyskinesia or levodopa-related fluctuations in those started early versus those started later.
“The results of the current trial, taken together with those of other trials, support treatment that is guided by clinical need and that uses the lowest dose that provides a satisfactory clinical effect,” wrote the editorial’s authors.
Dr. Bressman and Dr. Saunders‑Pullman are with the Icahn School of Medicine at Mt. Sinai, New York. Their editorial appears in the New England Journal of Medicine (2019;380:389-90). Dr. Bressman reported disclosures related to Denali Therapeutics, the Michael J. Fox Foundation, and Prevail Therapeutics, while Dr. Saunders-Pullman reported disclosures with Denali Therapeutics, the National Institutes of Health, Genzyme Sanofi, and the Bigglesworth Family Foundation.
investigators reported. The disease course was not significantly different for patients who had a full 80 weeks of levodopa/carbodopa therapy, compared with that seen with those who started treatment after a 40-week delay, according to the investigators.
“These findings imply that levodopa had no disease-modifying effect on Parkinson’s disease over the period of the trial,” wrote investigator Rob M. A. de Bie, MD, PhD, professor of movement disorders at the University of Amsterdam, and his colleagues in the New England Journal of Medicine.
By contrast, results of an earlier randomized, placebo-controlled trial suggested that levodopa had disease-modifying effects, though the findings of that study were inconclusive, according to authors of an editorial (see Views on the News).
In the current multicenter trial, known as LEAP (Levodopa in Early Parkinson’s Disease) a total of 445 patients with early Parkinson’s disease were randomized to either 80 weeks of levodopa and carbodopa or to 40 weeks of placebo followed by 40 weeks of levodopa/carbodopa.
Levodopa was dosed at 100 mg three times per day, and carbodopa at 25 mg three times per day, according to the report.
There was no significant difference between the early and delayed treatment groups for primary outcome of the trial, which was change in the Unified Parkinson’s Disease Rating Scale (UPDRS) from baseline to week 80.
The mean change in UPDRS was –1.0 in the group of patients who had the full 80 weeks of levodopa/carbodopa and –2.0 for those who had delayed therapy, for a difference of 1 point (P = .44). Higher scores on the UPDRS signify worse disease.
At week 40, there was a change in UPDRS favoring the early-initiation strategy, which reflected the effects of levodopa on disease symptoms, investigators added.
Nausea was more common in the early-start group during the first 40 weeks of the trial. However, there were no differences between groups in other adverse events of particular interest, including dyskinesias and motor fluctuations related to levodopa, Dr. de Bie and his colleagues reported.
Taken together, these results suggest no beneficial or detrimental disease-modifying effect for an early treatment strategy, although further trials are warranted to evaluate other strategies, such as higher levodopa doses, longer administration, or starting the drug at later stages of disease, they wrote.
Dr. de Bie reported grants from ZonMw, Parkinson Vereniging, and Stichting Parkinsonfonds during the conduct of the study, as well as grants from GE Health and Medtronic outside the submitted work. Study authors provided disclosures related to Netherlands Organization for Scientific Research, Michael J. Fox Foundation, UCB, AbbVie, Boston Scientific, Biogen, Merck, and others.
SOURCE: Verschuur CVM et al. N Engl J Med. 2019;380:315-24.
investigators reported. The disease course was not significantly different for patients who had a full 80 weeks of levodopa/carbodopa therapy, compared with that seen with those who started treatment after a 40-week delay, according to the investigators.
“These findings imply that levodopa had no disease-modifying effect on Parkinson’s disease over the period of the trial,” wrote investigator Rob M. A. de Bie, MD, PhD, professor of movement disorders at the University of Amsterdam, and his colleagues in the New England Journal of Medicine.
By contrast, results of an earlier randomized, placebo-controlled trial suggested that levodopa had disease-modifying effects, though the findings of that study were inconclusive, according to authors of an editorial (see Views on the News).
In the current multicenter trial, known as LEAP (Levodopa in Early Parkinson’s Disease) a total of 445 patients with early Parkinson’s disease were randomized to either 80 weeks of levodopa and carbodopa or to 40 weeks of placebo followed by 40 weeks of levodopa/carbodopa.
Levodopa was dosed at 100 mg three times per day, and carbodopa at 25 mg three times per day, according to the report.
There was no significant difference between the early and delayed treatment groups for primary outcome of the trial, which was change in the Unified Parkinson’s Disease Rating Scale (UPDRS) from baseline to week 80.
The mean change in UPDRS was –1.0 in the group of patients who had the full 80 weeks of levodopa/carbodopa and –2.0 for those who had delayed therapy, for a difference of 1 point (P = .44). Higher scores on the UPDRS signify worse disease.
At week 40, there was a change in UPDRS favoring the early-initiation strategy, which reflected the effects of levodopa on disease symptoms, investigators added.
Nausea was more common in the early-start group during the first 40 weeks of the trial. However, there were no differences between groups in other adverse events of particular interest, including dyskinesias and motor fluctuations related to levodopa, Dr. de Bie and his colleagues reported.
Taken together, these results suggest no beneficial or detrimental disease-modifying effect for an early treatment strategy, although further trials are warranted to evaluate other strategies, such as higher levodopa doses, longer administration, or starting the drug at later stages of disease, they wrote.
Dr. de Bie reported grants from ZonMw, Parkinson Vereniging, and Stichting Parkinsonfonds during the conduct of the study, as well as grants from GE Health and Medtronic outside the submitted work. Study authors provided disclosures related to Netherlands Organization for Scientific Research, Michael J. Fox Foundation, UCB, AbbVie, Boston Scientific, Biogen, Merck, and others.
SOURCE: Verschuur CVM et al. N Engl J Med. 2019;380:315-24.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Levodopa did not have any significant disease-modifying effects in patients with early Parkinson’s disease.
Major finding: Change in the Unified Parkinson’s Disease Rating Scale (UPDRS) was –1.0 after 80 weeks of levodopa/carbodopa versus –2.0 for 40 weeks of placebo followed by 40 weeks of treatment (P = .44).
Study details: A delayed-start trial including 445 patients with early Parkinson’s disease randomized to 80 weeks of treatment or 40 weeks of placebo plus 40 weeks of treatment.
Disclosures: Study authors reported disclosures related to Netherlands Organization for Scientific Research, Michael J. Fox Foundation, UCB, AbbVie, Boston Scientific, Biogen, Merck, and others.
Source: Verschuur CVM et al. N Engl J Med. 2019;380:315-324.

FDA approves 0.5-mL Fluzone Quadrivalent vaccine in young children
according to Sanofi Pasteur, the vaccine’s manufacturer.

FDA approval was based on results of a phase 4 safety and immunogenicity study of nearly 2,000 children. Children aged 6-35 months who received one or two doses of Fluzone at 0.50 mL had a safety profile similar to that of children who received one or two doses of Fluzone at 0.25 mL. Results from the study were presented at the Pediatric Academic Societies annual meeting in April 2018.
This flu vaccine should not be given to anyone with a severe allergic reaction (anaphylaxis) to egg or egg products, according to the press release.
In children, the most common adverse events are injection site reactions, muscle aches, fatigue, and headache; in young children, irritability, abnormal crying, drowsiness, appetite loss, vomiting, and fever are common.
“Offering pediatricians the convenience of the same 0.5-mL dose option for children may help streamline immunization efforts. The potentially life-threatening effects of influenza in children reported during the 2017-18 season, especially among those who were not vaccinated, is sobering,” David P. Greenberg, MD, regional medical head of Sanofi Pasteur of North America, said in the press release.
Find the full press release on the Sanofi website.
according to Sanofi Pasteur, the vaccine’s manufacturer.
FDA approval was based on results of a phase 4 safety and immunogenicity study of nearly 2,000 children. Children aged 6-35 months who received one or two doses of Fluzone at 0.50 mL had a safety profile similar to that of children who received one or two doses of Fluzone at 0.25 mL. Results from the study were presented at the Pediatric Academic Societies annual meeting in April 2018.
This flu vaccine should not be given to anyone with a severe allergic reaction (anaphylaxis) to egg or egg products, according to the press release.
In children, the most common adverse events are injection site reactions, muscle aches, fatigue, and headache; in young children, irritability, abnormal crying, drowsiness, appetite loss, vomiting, and fever are common.
“Offering pediatricians the convenience of the same 0.5-mL dose option for children may help streamline immunization efforts. The potentially life-threatening effects of influenza in children reported during the 2017-18 season, especially among those who were not vaccinated, is sobering,” David P. Greenberg, MD, regional medical head of Sanofi Pasteur of North America, said in the press release.
Find the full press release on the Sanofi website.
according to Sanofi Pasteur, the vaccine’s manufacturer.
FDA approval was based on results of a phase 4 safety and immunogenicity study of nearly 2,000 children. Children aged 6-35 months who received one or two doses of Fluzone at 0.50 mL had a safety profile similar to that of children who received one or two doses of Fluzone at 0.25 mL. Results from the study were presented at the Pediatric Academic Societies annual meeting in April 2018.
This flu vaccine should not be given to anyone with a severe allergic reaction (anaphylaxis) to egg or egg products, according to the press release.
In children, the most common adverse events are injection site reactions, muscle aches, fatigue, and headache; in young children, irritability, abnormal crying, drowsiness, appetite loss, vomiting, and fever are common.
“Offering pediatricians the convenience of the same 0.5-mL dose option for children may help streamline immunization efforts. The potentially life-threatening effects of influenza in children reported during the 2017-18 season, especially among those who were not vaccinated, is sobering,” David P. Greenberg, MD, regional medical head of Sanofi Pasteur of North America, said in the press release.
Find the full press release on the Sanofi website.
Study hints at lacosamide’s efficacy for small fiber neuropathy
As a treatment for small fiber neuropathy (SFN), lacosamide decreased pain and had a positive effect on sleep quality with minimal adverse events in patients with mutations in the gene SCN9A that encodes the voltage-gated sodium channel Nav1.7, according to a randomized, placebo-controlled, double-blind, crossover-design study published in Brain.
“This is the first study that investigated the efficacy of lacosamide [Vimpat] in patients with SFN,” wrote lead author Bianca T.A. de Greef, MD, of Maastricht University Medical Center, the Netherlands, and her coauthors. “Compared with placebo, lacosamide appeared to be safe to use and well tolerated in this cohort of patients.”
Lacosamide, which is approved in the United States to treat partial-onset seizures in people aged 4 years and older, has been shown to bind to and inhibit Nav1.7.
The investigators randomized 25 Dutch patients with Nav1.7-related SFN into the Lacosamide-Efficacy-’N’-Safety in SFN (LENSS) study to receive lacosamide followed by placebo, or vice versa. The patients were recruited between November 2014 and July 2016; 1 patient dropped out before treatment and another after the first treatment period, leaving 24 patients who received lacosamide and 23 patients who received placebo. They went through a 3-week titration period, an 8-week treatment period, a 2-week tapering period, and a washout period of at least 2 weeks, after which they switched to the other treatment arm and repeated the same schedule.
Through the daily pain intensity numerical rating scale and the daily sleep interference scale (DSIS), among other questionnaires, the investigators sought to determine if lacosamide reduced pain and thereby improved sleep quality. Lacosamide treatment led to a decrease in mean average pain by at least 1 point in 50.0% of patients, compared with 21.7% in the placebo group (odds ratio, 4.45; 95% confidence interval, 1.38-14.36; P = .0213). In addition, 25.0% of the lacosamide group reported at least a 2-point decrease in mean average pain versus 8.7% in the placebo group. There was also a notable difference in pain’s impact on sleep quality between the two, with the lacosamide period seeing a DSIS median value of 5.3, compared with 5.7 for the placebo period.
According to the patients’ global impression of change questionnaire, 33.3% felt better while using lacosamide versus 4.3% who felt better while using placebo (P = .0156). Six serious adverse events occurred during the study, though only two occurred during the lacosamide period. The most common adverse events for patients taking lacosamide included dizziness, headache, and nausea, all of which were comparable with adverse events in patients taking placebo.
Dr. de Greef and her colleagues noted the study’s potential limitations, including a carryover effect that could have confounded direct treatment effects (which they attempted to mitigate via a lengthier washout period) and a small cohort that was limited to very specific patients. However, the authors chose this particular cohort because “our aim was to demonstrate proof of-concept, which can be used for future studies involving larger groups of patients diagnosed with SFN.” They observed that their response rates were slightly lower than expected, but they noted that “lacosamide appears to be as effective as currently available neuropathic pain treatment.”
The study was funded by the Prinses Beatrix Spierfonds. Some of the authors reported receiving grants, personal fees, funding for research, and/or honoraria from foundations, pharmaceutical companies, life sciences companies, and the European Commission.
SOURCE: de Greef BTA et al. Brain. 2019 Jan 14. doi: 10.1093/brain/awy329.
As a treatment for small fiber neuropathy (SFN), lacosamide decreased pain and had a positive effect on sleep quality with minimal adverse events in patients with mutations in the gene SCN9A that encodes the voltage-gated sodium channel Nav1.7, according to a randomized, placebo-controlled, double-blind, crossover-design study published in Brain.
“This is the first study that investigated the efficacy of lacosamide [Vimpat] in patients with SFN,” wrote lead author Bianca T.A. de Greef, MD, of Maastricht University Medical Center, the Netherlands, and her coauthors. “Compared with placebo, lacosamide appeared to be safe to use and well tolerated in this cohort of patients.”
Lacosamide, which is approved in the United States to treat partial-onset seizures in people aged 4 years and older, has been shown to bind to and inhibit Nav1.7.
The investigators randomized 25 Dutch patients with Nav1.7-related SFN into the Lacosamide-Efficacy-’N’-Safety in SFN (LENSS) study to receive lacosamide followed by placebo, or vice versa. The patients were recruited between November 2014 and July 2016; 1 patient dropped out before treatment and another after the first treatment period, leaving 24 patients who received lacosamide and 23 patients who received placebo. They went through a 3-week titration period, an 8-week treatment period, a 2-week tapering period, and a washout period of at least 2 weeks, after which they switched to the other treatment arm and repeated the same schedule.
Through the daily pain intensity numerical rating scale and the daily sleep interference scale (DSIS), among other questionnaires, the investigators sought to determine if lacosamide reduced pain and thereby improved sleep quality. Lacosamide treatment led to a decrease in mean average pain by at least 1 point in 50.0% of patients, compared with 21.7% in the placebo group (odds ratio, 4.45; 95% confidence interval, 1.38-14.36; P = .0213). In addition, 25.0% of the lacosamide group reported at least a 2-point decrease in mean average pain versus 8.7% in the placebo group. There was also a notable difference in pain’s impact on sleep quality between the two, with the lacosamide period seeing a DSIS median value of 5.3, compared with 5.7 for the placebo period.
According to the patients’ global impression of change questionnaire, 33.3% felt better while using lacosamide versus 4.3% who felt better while using placebo (P = .0156). Six serious adverse events occurred during the study, though only two occurred during the lacosamide period. The most common adverse events for patients taking lacosamide included dizziness, headache, and nausea, all of which were comparable with adverse events in patients taking placebo.
Dr. de Greef and her colleagues noted the study’s potential limitations, including a carryover effect that could have confounded direct treatment effects (which they attempted to mitigate via a lengthier washout period) and a small cohort that was limited to very specific patients. However, the authors chose this particular cohort because “our aim was to demonstrate proof of-concept, which can be used for future studies involving larger groups of patients diagnosed with SFN.” They observed that their response rates were slightly lower than expected, but they noted that “lacosamide appears to be as effective as currently available neuropathic pain treatment.”
The study was funded by the Prinses Beatrix Spierfonds. Some of the authors reported receiving grants, personal fees, funding for research, and/or honoraria from foundations, pharmaceutical companies, life sciences companies, and the European Commission.
SOURCE: de Greef BTA et al. Brain. 2019 Jan 14. doi: 10.1093/brain/awy329.
As a treatment for small fiber neuropathy (SFN), lacosamide decreased pain and had a positive effect on sleep quality with minimal adverse events in patients with mutations in the gene SCN9A that encodes the voltage-gated sodium channel Nav1.7, according to a randomized, placebo-controlled, double-blind, crossover-design study published in Brain.
“This is the first study that investigated the efficacy of lacosamide [Vimpat] in patients with SFN,” wrote lead author Bianca T.A. de Greef, MD, of Maastricht University Medical Center, the Netherlands, and her coauthors. “Compared with placebo, lacosamide appeared to be safe to use and well tolerated in this cohort of patients.”
Lacosamide, which is approved in the United States to treat partial-onset seizures in people aged 4 years and older, has been shown to bind to and inhibit Nav1.7.
The investigators randomized 25 Dutch patients with Nav1.7-related SFN into the Lacosamide-Efficacy-’N’-Safety in SFN (LENSS) study to receive lacosamide followed by placebo, or vice versa. The patients were recruited between November 2014 and July 2016; 1 patient dropped out before treatment and another after the first treatment period, leaving 24 patients who received lacosamide and 23 patients who received placebo. They went through a 3-week titration period, an 8-week treatment period, a 2-week tapering period, and a washout period of at least 2 weeks, after which they switched to the other treatment arm and repeated the same schedule.
Through the daily pain intensity numerical rating scale and the daily sleep interference scale (DSIS), among other questionnaires, the investigators sought to determine if lacosamide reduced pain and thereby improved sleep quality. Lacosamide treatment led to a decrease in mean average pain by at least 1 point in 50.0% of patients, compared with 21.7% in the placebo group (odds ratio, 4.45; 95% confidence interval, 1.38-14.36; P = .0213). In addition, 25.0% of the lacosamide group reported at least a 2-point decrease in mean average pain versus 8.7% in the placebo group. There was also a notable difference in pain’s impact on sleep quality between the two, with the lacosamide period seeing a DSIS median value of 5.3, compared with 5.7 for the placebo period.
According to the patients’ global impression of change questionnaire, 33.3% felt better while using lacosamide versus 4.3% who felt better while using placebo (P = .0156). Six serious adverse events occurred during the study, though only two occurred during the lacosamide period. The most common adverse events for patients taking lacosamide included dizziness, headache, and nausea, all of which were comparable with adverse events in patients taking placebo.
Dr. de Greef and her colleagues noted the study’s potential limitations, including a carryover effect that could have confounded direct treatment effects (which they attempted to mitigate via a lengthier washout period) and a small cohort that was limited to very specific patients. However, the authors chose this particular cohort because “our aim was to demonstrate proof of-concept, which can be used for future studies involving larger groups of patients diagnosed with SFN.” They observed that their response rates were slightly lower than expected, but they noted that “lacosamide appears to be as effective as currently available neuropathic pain treatment.”
The study was funded by the Prinses Beatrix Spierfonds. Some of the authors reported receiving grants, personal fees, funding for research, and/or honoraria from foundations, pharmaceutical companies, life sciences companies, and the European Commission.
SOURCE: de Greef BTA et al. Brain. 2019 Jan 14. doi: 10.1093/brain/awy329.
FROM BRAIN
Key clinical point:
Major finding: In the lacosamide group, 50.0% of patients reported mean average pain decreasing by at least 1 point, compared with 21.7% in the placebo group (odds ratio, 4.45; 95% confidence interval, 1.38-14.36; P = .0213).
Study details: A randomized, placebo-controlled, double-blind, crossover-design study of 25 patients with Nav1.7-related small fiber neuropathy who received lacosamide followed by placebo, or vice versa.
Disclosures: The study was funded by the Prinses Beatrix Spierfonds. Some of the authors reported receiving grants, personal fees, funding for research, and/or honoraria from foundations, pharmaceutical companies, life sciences companies, and the European Commission.
Source: de Greef BTA et al. Brain. 2019 Jan 14. doi: 10.1093/brain/awy329.