User login
Tofacitinib tackles cutaneous sarcoidosis
Tofacitinib significantly improved skin lesions associated with cutaneous sarcoidosis, according to a case report published in the New England Journal of Medicine.
Treatment options for cutaneous sarcoidosis are limited, as are data on the effectiveness of alternatives to prednisone, which is often the first choice despite adverse effects, wrote William Damsky, MD, of Yale University in New Haven, Conn., and his colleagues.
Previous studies have suggested involvement of the JAK-STAT pathway in sarcoidosis; therefore, the researchers treated a patient who had refractory cutaneous sarcoidosis with oral tofacitinib. The treatment significantly improved the patient’s skin lesions both clinically and histologically.
The patient was a 48-year-old woman with a history of cutaneous and pulmonary sarcoidosis and treatment-resistant skin lesions. At the time of the case report, she had no pulmonary symptoms and no ophthalmologic issues, but presented with pink-brown indurated papules and plaques, and some alopecia on her scalp (N Engl J Med. 2018;379:2540-6).
The patient had not responded to other medications including glucocorticoids, minocycline, hydroxychloroquine, methotrexate, adalimumab, tacrolimus, and apremilast. With her consent, the patient received off-label tofacitinib at 5 mg twice daily. The lesions began to improve, but treatment was discontinued because of insurance issues. When treatment resumed, the patient’s Cutaneous Sarcoidosis Activity and Morphology Instrument (CSAMI) score, used to assess disease activity, was 85 on a scale of 0 to 165; this score dropped to 53 after 4 months of treatment.
In addition, two samples collected after 10 months of treatment showed histologic resolution of granulomas.
Although the findings must be replicated in other patients, the results suggest that “the dysregulation of JAK-STAT–dependent cytokines (e.g., interferon-gamma) is pathogenically involved in cutaneous sarcoidosis and, probably, in sarcoidosis in general,” the researchers said.
Dr. Damsky disclosed a financial relationship with Eli Lilly, and research funding from the Dermatology Foundation and the National Institutes of Health. The study was supported by the Ranjini and Ajay Poddar Resource Fund for Dermatologic Diseases Research, the National Institutes of Health, and the Dermatology Foundation.
SOURCE: Damsky W et al. N Engl J Med. 2018;379:2540-6.
Tofacitinib significantly improved skin lesions associated with cutaneous sarcoidosis, according to a case report published in the New England Journal of Medicine.
Treatment options for cutaneous sarcoidosis are limited, as are data on the effectiveness of alternatives to prednisone, which is often the first choice despite adverse effects, wrote William Damsky, MD, of Yale University in New Haven, Conn., and his colleagues.
Previous studies have suggested involvement of the JAK-STAT pathway in sarcoidosis; therefore, the researchers treated a patient who had refractory cutaneous sarcoidosis with oral tofacitinib. The treatment significantly improved the patient’s skin lesions both clinically and histologically.
The patient was a 48-year-old woman with a history of cutaneous and pulmonary sarcoidosis and treatment-resistant skin lesions. At the time of the case report, she had no pulmonary symptoms and no ophthalmologic issues, but presented with pink-brown indurated papules and plaques, and some alopecia on her scalp (N Engl J Med. 2018;379:2540-6).
The patient had not responded to other medications including glucocorticoids, minocycline, hydroxychloroquine, methotrexate, adalimumab, tacrolimus, and apremilast. With her consent, the patient received off-label tofacitinib at 5 mg twice daily. The lesions began to improve, but treatment was discontinued because of insurance issues. When treatment resumed, the patient’s Cutaneous Sarcoidosis Activity and Morphology Instrument (CSAMI) score, used to assess disease activity, was 85 on a scale of 0 to 165; this score dropped to 53 after 4 months of treatment.
In addition, two samples collected after 10 months of treatment showed histologic resolution of granulomas.
Although the findings must be replicated in other patients, the results suggest that “the dysregulation of JAK-STAT–dependent cytokines (e.g., interferon-gamma) is pathogenically involved in cutaneous sarcoidosis and, probably, in sarcoidosis in general,” the researchers said.
Dr. Damsky disclosed a financial relationship with Eli Lilly, and research funding from the Dermatology Foundation and the National Institutes of Health. The study was supported by the Ranjini and Ajay Poddar Resource Fund for Dermatologic Diseases Research, the National Institutes of Health, and the Dermatology Foundation.
SOURCE: Damsky W et al. N Engl J Med. 2018;379:2540-6.
Tofacitinib significantly improved skin lesions associated with cutaneous sarcoidosis, according to a case report published in the New England Journal of Medicine.
Treatment options for cutaneous sarcoidosis are limited, as are data on the effectiveness of alternatives to prednisone, which is often the first choice despite adverse effects, wrote William Damsky, MD, of Yale University in New Haven, Conn., and his colleagues.
Previous studies have suggested involvement of the JAK-STAT pathway in sarcoidosis; therefore, the researchers treated a patient who had refractory cutaneous sarcoidosis with oral tofacitinib. The treatment significantly improved the patient’s skin lesions both clinically and histologically.
The patient was a 48-year-old woman with a history of cutaneous and pulmonary sarcoidosis and treatment-resistant skin lesions. At the time of the case report, she had no pulmonary symptoms and no ophthalmologic issues, but presented with pink-brown indurated papules and plaques, and some alopecia on her scalp (N Engl J Med. 2018;379:2540-6).
The patient had not responded to other medications including glucocorticoids, minocycline, hydroxychloroquine, methotrexate, adalimumab, tacrolimus, and apremilast. With her consent, the patient received off-label tofacitinib at 5 mg twice daily. The lesions began to improve, but treatment was discontinued because of insurance issues. When treatment resumed, the patient’s Cutaneous Sarcoidosis Activity and Morphology Instrument (CSAMI) score, used to assess disease activity, was 85 on a scale of 0 to 165; this score dropped to 53 after 4 months of treatment.
In addition, two samples collected after 10 months of treatment showed histologic resolution of granulomas.
Although the findings must be replicated in other patients, the results suggest that “the dysregulation of JAK-STAT–dependent cytokines (e.g., interferon-gamma) is pathogenically involved in cutaneous sarcoidosis and, probably, in sarcoidosis in general,” the researchers said.
Dr. Damsky disclosed a financial relationship with Eli Lilly, and research funding from the Dermatology Foundation and the National Institutes of Health. The study was supported by the Ranjini and Ajay Poddar Resource Fund for Dermatologic Diseases Research, the National Institutes of Health, and the Dermatology Foundation.
SOURCE: Damsky W et al. N Engl J Med. 2018;379:2540-6.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: A 48-year-old woman with sarcoidosis showed significant improvement in her skin on treatment with tofacitinib.
Major finding: The patient’s disease activity score on the CSAMI went from 85 to 53 after 4 months of therapy.
Study details: Case report of a 48-year-old woman with cutaneous and pulmonary sarcoidosis and treatment-resistant skin lesions.
Disclosures: Dr. Damsky disclosed a financial relationship with Eli Lilly, and research funding from the Dermatology Foundation and the National Institutes of Health. The study was supported by the Ranjini and Ajay Poddar Resource Fund for Dermatologic Diseases Research, the National Institutes of Health, and the Dermatology Foundation.
Source: Damsky W et al. N Engl J Med. 2018;379:2540-6.
ADHD more likely, causes worse outcomes in patients with BD
ADHD is significantly more common and is associated with worse outcomes in patients with bipolar disorder, according to Ross J. Baldessarini, MD, of McLean Hospital and Harvard Medical School, Boston, and his associates.
In a study of 703 patients diagnosed with bipolar disorder (BD) type I or II who were evaluated, treated, and followed at the Lucio Bini Mood Disorder Centers in Rome and Cagliari, Italy, 173 patients had co-occurring lifetime ADHD. Co-occurring conditions were more likely in men and in those with BD-I. The lifetime ADHD prevalence rate of 24.6% in patients with bipolar disorder is significantly higher than the incidence in the general population, the investigators wrote in the Journal of Affective Disorders.
Patients with co-occurring ADHD and BD were more likely to have performed worse in school, have higher Adult ADHD Self-Report Scale scores, be unemployed, have lower socioeconomic status, be married, have separated, have substance abuse, have attempted suicide, and have hypomania, compared with patients with only BD. However, they were less likely to have an anxiety disorder or a family history of mood disorders.
“The association of ADHD with a less successful and stable educational history, more unemployment, lack of or failed marriages, and greater risk of suicide attempts and substance abuse indicates unfavorable effects of having ADHD with BD. Such effects may arise by the impact of ADHD early during development,” the investigators concluded.
The study was partly supported by a research award from the Aretaeus Association of Rome and grants from the Bruce J. Anderson Foundation and the McLean Private Donors Research Fund. No conflicts of interest were reported.
SOURCE: Baldessarini RJ et al. J Affect Disord. 2018 Sep 17. doi: 10.1016/j.jad.2018.09.038.
ADHD is significantly more common and is associated with worse outcomes in patients with bipolar disorder, according to Ross J. Baldessarini, MD, of McLean Hospital and Harvard Medical School, Boston, and his associates.
In a study of 703 patients diagnosed with bipolar disorder (BD) type I or II who were evaluated, treated, and followed at the Lucio Bini Mood Disorder Centers in Rome and Cagliari, Italy, 173 patients had co-occurring lifetime ADHD. Co-occurring conditions were more likely in men and in those with BD-I. The lifetime ADHD prevalence rate of 24.6% in patients with bipolar disorder is significantly higher than the incidence in the general population, the investigators wrote in the Journal of Affective Disorders.
Patients with co-occurring ADHD and BD were more likely to have performed worse in school, have higher Adult ADHD Self-Report Scale scores, be unemployed, have lower socioeconomic status, be married, have separated, have substance abuse, have attempted suicide, and have hypomania, compared with patients with only BD. However, they were less likely to have an anxiety disorder or a family history of mood disorders.
“The association of ADHD with a less successful and stable educational history, more unemployment, lack of or failed marriages, and greater risk of suicide attempts and substance abuse indicates unfavorable effects of having ADHD with BD. Such effects may arise by the impact of ADHD early during development,” the investigators concluded.
The study was partly supported by a research award from the Aretaeus Association of Rome and grants from the Bruce J. Anderson Foundation and the McLean Private Donors Research Fund. No conflicts of interest were reported.
SOURCE: Baldessarini RJ et al. J Affect Disord. 2018 Sep 17. doi: 10.1016/j.jad.2018.09.038.
ADHD is significantly more common and is associated with worse outcomes in patients with bipolar disorder, according to Ross J. Baldessarini, MD, of McLean Hospital and Harvard Medical School, Boston, and his associates.
In a study of 703 patients diagnosed with bipolar disorder (BD) type I or II who were evaluated, treated, and followed at the Lucio Bini Mood Disorder Centers in Rome and Cagliari, Italy, 173 patients had co-occurring lifetime ADHD. Co-occurring conditions were more likely in men and in those with BD-I. The lifetime ADHD prevalence rate of 24.6% in patients with bipolar disorder is significantly higher than the incidence in the general population, the investigators wrote in the Journal of Affective Disorders.
Patients with co-occurring ADHD and BD were more likely to have performed worse in school, have higher Adult ADHD Self-Report Scale scores, be unemployed, have lower socioeconomic status, be married, have separated, have substance abuse, have attempted suicide, and have hypomania, compared with patients with only BD. However, they were less likely to have an anxiety disorder or a family history of mood disorders.
“The association of ADHD with a less successful and stable educational history, more unemployment, lack of or failed marriages, and greater risk of suicide attempts and substance abuse indicates unfavorable effects of having ADHD with BD. Such effects may arise by the impact of ADHD early during development,” the investigators concluded.
The study was partly supported by a research award from the Aretaeus Association of Rome and grants from the Bruce J. Anderson Foundation and the McLean Private Donors Research Fund. No conflicts of interest were reported.
SOURCE: Baldessarini RJ et al. J Affect Disord. 2018 Sep 17. doi: 10.1016/j.jad.2018.09.038.
FROM THE JOURNAL OF AFFECTIVE DISORDERS
‘Payoff will be great’ if we can conquer childhood obesity, expert says
LOS ANGELES – Mounting evidence indicates that obesity in childhood and adolescence increases the risk for future cardiovascular disease (CVD), according to Stephen R. Daniels, MD, PhD.

“Some of this increased risk is related to the high level of tracking of obesity from childhood to adolescence to adulthood,” Dr. Daniels, who chairs the department of pediatrics at the University of Colorado, Aurora, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease. “But I think it’s also clear that childhood obesity is associated with risk factors for adult CVD, including hypertension, dyslipidemia, and type 2 diabetes. There’s a combination of things going on over the life course.”
Numerous studies have demonstrated a dose-response relationship between increased weight and all-cause mortality in cardiovascular disease for men and women. This operates through a variety of mechanisms, Dr. Daniels said, including hypertension, dyslipidemia, left ventricular hypertrophy, vascular inflammation, type 2 diabetes, and obstructive sleep apnea. “While overt cardiovascular disease does not occur in children, many of the mechanisms recognized in adults are also present in children and adolescents,” he said. “The trends for increasing prevalence and severity of obesity in children and the comorbid conditions associated with obesity are worrisome.”
The current prevalence of obesity in children and adolescents stands at about 18%, according to the latest National Health and Nutrition Examination Survey. However, the prevalence of severe obesity in youth aged 2-19 years has been increasing “fairly dramatically,” and now stands at 9% among girls and 8% among boys. Hispanics and non-Hispanic blacks are disproportionately affected. That may turn out to be important in terms of the future, Dr. Daniels said, because according to simulation models, childhood obesity and overweight will continue to be a major public health problem in the future (N Engl J Med. 2017;377:2145-53).
Direct evidence is also beginning to emerge of a link between obesity in youth and adult cardiovascular disease. The factors in childhood that predict adult obesity include a higher level of body mass index, obesity present at an older age (adolescence vs. childhood), and the presence of obesity in parents, which reflects both genes and environment. Researchers led by Paul W. Franks, PhD, evaluated 4,857 American Indian children without diabetes who were born between 1945 and 1984 and followed them for death before age 55 (N Engl J Med. 2010;362[6]:485-93). They assessed whether BMI, glucose tolerance, blood pressure, and cholesterol levels predicted premature death. There were 166 deaths from endogenous causes (3.4%) over a median follow-up of 24 years. Factors significantly associated with mortality included obesity (incident rate ratio 2.30), glucose tolerance (IRR 1.73), and hypertension (IRR 1.57).
In a separate analysis, researchers investigated the long-term effects of childhood weight on coronary heart disease (CHD) by studying 276,835 Danish schoolchildren for whom measurements of height and weight were available. They followed the individuals until they turned age 25 or older and used national registries to assess the fatal and nonfatal rates of CHD events (N Engl J Med. 2007;357:2329-37). The researchers found that higher BMI during childhood was associated with an increased risk of CVD in adulthood. However, they did not have data on BMI in adulthood, “which leaves open the question of whether childhood obesity works through adult obesity or also has an independent effect,” said Dr. Daniels, who is also pediatrician-in-chief at Children’s Hospital Colorado, Denver.
More recently, investigators studied 37,674 apparently healthy Israeli men from age 17 into adulthood (N Engl J Med. 2011;364:1315-25). Outcomes were coronary disease and diabetes. They found that an elevated BMI in adolescence is an independent risk factor for CVD in later life, while an elevated BMI in adulthood is an independent risk factor for both CVD and diabetes.
In the Fels Longitudinal Study, researchers enrolled 151 adults with metabolic syndrome and 154 without metabolic syndrome, with a mean age of 51 years (J Pediatr. 2008;152:191-200). “The idea was to look back at this cohort and see when the first differences might be observable between boys and girls who ultimately would develop metabolic syndrome and those who would not,” said Dr. Daniels, who was one of the study investigators. The first appearance of differences between adults with and without metabolic syndrome occurred at ages 8 and 13 for BMI and 6 and 13 for waist circumference in boys and girls, respectively. Odds ratios (ORs) for the metabolic syndrome in adulthood if BMI were elevated in childhood ranged from 1.4 to 1.9 in boys and from 0.8 to 2.8 in girls. At the same time, odds ratios for the metabolic syndrome in adulthood if waist circumference was elevated ranged from 2.5 to 31.4 in boys and 1.7 to 2.5 in girls.
“I think it’s safe to say that BMI and waist circumference may be important in predicting metabolic syndrome later in life and, ultimately, cardiovascular disease,” Dr. Daniels said.
He noted that as the prevalence and severity of obesity have increased in childhood, the prevalence of type 2 diabetes has also increased. “The time from diagnosis of diabetes to a CVD event is approximately 10-15 years in adults, and there is often a prediagnosis period of hyperglycemia, which ranges from 5-10 years,” Dr. Daniels said. “If the time course of CVD related to diabetes is the same for adolescents as adults, it is anticipated that adolescents with diabetes will begin having substantial CVD morbidity and mortality in their 30s or 40s. This will be a public health disaster. Emerging evidence from the TODAY study (Treatment Options for type 2 Diabetes in Adolescents and Youth) and other studies is emphasizing that at least some individuals with adolescent type 2 diabetes may have a more malignant form of disease than in adults. This is striking and important to consider as we look at how to prevent cardiovascular disease.”
Obesity in childhood is also associated with structural and functional abnormalities of the vasculature, according to studies that measure vascular structure via intima-media thickness of the carotid arteries, femoral arteries, abdominal aorta, or other arteries, as well as those that measure vascular stiffness via measures of intrinsic “visco-elastic” properties of the arterial wall. In one study of individuals aged 10-24 years, Dr. Daniels and his associates performed carotid ultrasound for carotid intima-media thickness on 182 patients who were lean, 136 who were obese, and 128 who had type 2 diabetes (Circulation 2009;119(22):2913-9). It demonstrated that youth with obesity and obesity-related type 2 diabetes have abnormalities in carotid thickness and stiffness that are only partially explained by traditional cardiovascular risk factors.
“We all know that obesity is very difficult to treat,” he concluded. “That’s true in children and adolescents as it is in adults. I think this argues for prevention of obesity, for us starting earlier, creating an optimal cardiovascular health situation that we can maintain during the course of childhood and adolescence. The payoff will be great if we can accomplish that.”
Dr. Daniels reported having no disclosures.
LOS ANGELES – Mounting evidence indicates that obesity in childhood and adolescence increases the risk for future cardiovascular disease (CVD), according to Stephen R. Daniels, MD, PhD.
“Some of this increased risk is related to the high level of tracking of obesity from childhood to adolescence to adulthood,” Dr. Daniels, who chairs the department of pediatrics at the University of Colorado, Aurora, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease. “But I think it’s also clear that childhood obesity is associated with risk factors for adult CVD, including hypertension, dyslipidemia, and type 2 diabetes. There’s a combination of things going on over the life course.”
Numerous studies have demonstrated a dose-response relationship between increased weight and all-cause mortality in cardiovascular disease for men and women. This operates through a variety of mechanisms, Dr. Daniels said, including hypertension, dyslipidemia, left ventricular hypertrophy, vascular inflammation, type 2 diabetes, and obstructive sleep apnea. “While overt cardiovascular disease does not occur in children, many of the mechanisms recognized in adults are also present in children and adolescents,” he said. “The trends for increasing prevalence and severity of obesity in children and the comorbid conditions associated with obesity are worrisome.”
The current prevalence of obesity in children and adolescents stands at about 18%, according to the latest National Health and Nutrition Examination Survey. However, the prevalence of severe obesity in youth aged 2-19 years has been increasing “fairly dramatically,” and now stands at 9% among girls and 8% among boys. Hispanics and non-Hispanic blacks are disproportionately affected. That may turn out to be important in terms of the future, Dr. Daniels said, because according to simulation models, childhood obesity and overweight will continue to be a major public health problem in the future (N Engl J Med. 2017;377:2145-53).
Direct evidence is also beginning to emerge of a link between obesity in youth and adult cardiovascular disease. The factors in childhood that predict adult obesity include a higher level of body mass index, obesity present at an older age (adolescence vs. childhood), and the presence of obesity in parents, which reflects both genes and environment. Researchers led by Paul W. Franks, PhD, evaluated 4,857 American Indian children without diabetes who were born between 1945 and 1984 and followed them for death before age 55 (N Engl J Med. 2010;362[6]:485-93). They assessed whether BMI, glucose tolerance, blood pressure, and cholesterol levels predicted premature death. There were 166 deaths from endogenous causes (3.4%) over a median follow-up of 24 years. Factors significantly associated with mortality included obesity (incident rate ratio 2.30), glucose tolerance (IRR 1.73), and hypertension (IRR 1.57).
In a separate analysis, researchers investigated the long-term effects of childhood weight on coronary heart disease (CHD) by studying 276,835 Danish schoolchildren for whom measurements of height and weight were available. They followed the individuals until they turned age 25 or older and used national registries to assess the fatal and nonfatal rates of CHD events (N Engl J Med. 2007;357:2329-37). The researchers found that higher BMI during childhood was associated with an increased risk of CVD in adulthood. However, they did not have data on BMI in adulthood, “which leaves open the question of whether childhood obesity works through adult obesity or also has an independent effect,” said Dr. Daniels, who is also pediatrician-in-chief at Children’s Hospital Colorado, Denver.
More recently, investigators studied 37,674 apparently healthy Israeli men from age 17 into adulthood (N Engl J Med. 2011;364:1315-25). Outcomes were coronary disease and diabetes. They found that an elevated BMI in adolescence is an independent risk factor for CVD in later life, while an elevated BMI in adulthood is an independent risk factor for both CVD and diabetes.
In the Fels Longitudinal Study, researchers enrolled 151 adults with metabolic syndrome and 154 without metabolic syndrome, with a mean age of 51 years (J Pediatr. 2008;152:191-200). “The idea was to look back at this cohort and see when the first differences might be observable between boys and girls who ultimately would develop metabolic syndrome and those who would not,” said Dr. Daniels, who was one of the study investigators. The first appearance of differences between adults with and without metabolic syndrome occurred at ages 8 and 13 for BMI and 6 and 13 for waist circumference in boys and girls, respectively. Odds ratios (ORs) for the metabolic syndrome in adulthood if BMI were elevated in childhood ranged from 1.4 to 1.9 in boys and from 0.8 to 2.8 in girls. At the same time, odds ratios for the metabolic syndrome in adulthood if waist circumference was elevated ranged from 2.5 to 31.4 in boys and 1.7 to 2.5 in girls.
“I think it’s safe to say that BMI and waist circumference may be important in predicting metabolic syndrome later in life and, ultimately, cardiovascular disease,” Dr. Daniels said.
He noted that as the prevalence and severity of obesity have increased in childhood, the prevalence of type 2 diabetes has also increased. “The time from diagnosis of diabetes to a CVD event is approximately 10-15 years in adults, and there is often a prediagnosis period of hyperglycemia, which ranges from 5-10 years,” Dr. Daniels said. “If the time course of CVD related to diabetes is the same for adolescents as adults, it is anticipated that adolescents with diabetes will begin having substantial CVD morbidity and mortality in their 30s or 40s. This will be a public health disaster. Emerging evidence from the TODAY study (Treatment Options for type 2 Diabetes in Adolescents and Youth) and other studies is emphasizing that at least some individuals with adolescent type 2 diabetes may have a more malignant form of disease than in adults. This is striking and important to consider as we look at how to prevent cardiovascular disease.”
Obesity in childhood is also associated with structural and functional abnormalities of the vasculature, according to studies that measure vascular structure via intima-media thickness of the carotid arteries, femoral arteries, abdominal aorta, or other arteries, as well as those that measure vascular stiffness via measures of intrinsic “visco-elastic” properties of the arterial wall. In one study of individuals aged 10-24 years, Dr. Daniels and his associates performed carotid ultrasound for carotid intima-media thickness on 182 patients who were lean, 136 who were obese, and 128 who had type 2 diabetes (Circulation 2009;119(22):2913-9). It demonstrated that youth with obesity and obesity-related type 2 diabetes have abnormalities in carotid thickness and stiffness that are only partially explained by traditional cardiovascular risk factors.
“We all know that obesity is very difficult to treat,” he concluded. “That’s true in children and adolescents as it is in adults. I think this argues for prevention of obesity, for us starting earlier, creating an optimal cardiovascular health situation that we can maintain during the course of childhood and adolescence. The payoff will be great if we can accomplish that.”
Dr. Daniels reported having no disclosures.
LOS ANGELES – Mounting evidence indicates that obesity in childhood and adolescence increases the risk for future cardiovascular disease (CVD), according to Stephen R. Daniels, MD, PhD.
“Some of this increased risk is related to the high level of tracking of obesity from childhood to adolescence to adulthood,” Dr. Daniels, who chairs the department of pediatrics at the University of Colorado, Aurora, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease. “But I think it’s also clear that childhood obesity is associated with risk factors for adult CVD, including hypertension, dyslipidemia, and type 2 diabetes. There’s a combination of things going on over the life course.”
Numerous studies have demonstrated a dose-response relationship between increased weight and all-cause mortality in cardiovascular disease for men and women. This operates through a variety of mechanisms, Dr. Daniels said, including hypertension, dyslipidemia, left ventricular hypertrophy, vascular inflammation, type 2 diabetes, and obstructive sleep apnea. “While overt cardiovascular disease does not occur in children, many of the mechanisms recognized in adults are also present in children and adolescents,” he said. “The trends for increasing prevalence and severity of obesity in children and the comorbid conditions associated with obesity are worrisome.”
The current prevalence of obesity in children and adolescents stands at about 18%, according to the latest National Health and Nutrition Examination Survey. However, the prevalence of severe obesity in youth aged 2-19 years has been increasing “fairly dramatically,” and now stands at 9% among girls and 8% among boys. Hispanics and non-Hispanic blacks are disproportionately affected. That may turn out to be important in terms of the future, Dr. Daniels said, because according to simulation models, childhood obesity and overweight will continue to be a major public health problem in the future (N Engl J Med. 2017;377:2145-53).
Direct evidence is also beginning to emerge of a link between obesity in youth and adult cardiovascular disease. The factors in childhood that predict adult obesity include a higher level of body mass index, obesity present at an older age (adolescence vs. childhood), and the presence of obesity in parents, which reflects both genes and environment. Researchers led by Paul W. Franks, PhD, evaluated 4,857 American Indian children without diabetes who were born between 1945 and 1984 and followed them for death before age 55 (N Engl J Med. 2010;362[6]:485-93). They assessed whether BMI, glucose tolerance, blood pressure, and cholesterol levels predicted premature death. There were 166 deaths from endogenous causes (3.4%) over a median follow-up of 24 years. Factors significantly associated with mortality included obesity (incident rate ratio 2.30), glucose tolerance (IRR 1.73), and hypertension (IRR 1.57).
In a separate analysis, researchers investigated the long-term effects of childhood weight on coronary heart disease (CHD) by studying 276,835 Danish schoolchildren for whom measurements of height and weight were available. They followed the individuals until they turned age 25 or older and used national registries to assess the fatal and nonfatal rates of CHD events (N Engl J Med. 2007;357:2329-37). The researchers found that higher BMI during childhood was associated with an increased risk of CVD in adulthood. However, they did not have data on BMI in adulthood, “which leaves open the question of whether childhood obesity works through adult obesity or also has an independent effect,” said Dr. Daniels, who is also pediatrician-in-chief at Children’s Hospital Colorado, Denver.
More recently, investigators studied 37,674 apparently healthy Israeli men from age 17 into adulthood (N Engl J Med. 2011;364:1315-25). Outcomes were coronary disease and diabetes. They found that an elevated BMI in adolescence is an independent risk factor for CVD in later life, while an elevated BMI in adulthood is an independent risk factor for both CVD and diabetes.
In the Fels Longitudinal Study, researchers enrolled 151 adults with metabolic syndrome and 154 without metabolic syndrome, with a mean age of 51 years (J Pediatr. 2008;152:191-200). “The idea was to look back at this cohort and see when the first differences might be observable between boys and girls who ultimately would develop metabolic syndrome and those who would not,” said Dr. Daniels, who was one of the study investigators. The first appearance of differences between adults with and without metabolic syndrome occurred at ages 8 and 13 for BMI and 6 and 13 for waist circumference in boys and girls, respectively. Odds ratios (ORs) for the metabolic syndrome in adulthood if BMI were elevated in childhood ranged from 1.4 to 1.9 in boys and from 0.8 to 2.8 in girls. At the same time, odds ratios for the metabolic syndrome in adulthood if waist circumference was elevated ranged from 2.5 to 31.4 in boys and 1.7 to 2.5 in girls.
“I think it’s safe to say that BMI and waist circumference may be important in predicting metabolic syndrome later in life and, ultimately, cardiovascular disease,” Dr. Daniels said.
He noted that as the prevalence and severity of obesity have increased in childhood, the prevalence of type 2 diabetes has also increased. “The time from diagnosis of diabetes to a CVD event is approximately 10-15 years in adults, and there is often a prediagnosis period of hyperglycemia, which ranges from 5-10 years,” Dr. Daniels said. “If the time course of CVD related to diabetes is the same for adolescents as adults, it is anticipated that adolescents with diabetes will begin having substantial CVD morbidity and mortality in their 30s or 40s. This will be a public health disaster. Emerging evidence from the TODAY study (Treatment Options for type 2 Diabetes in Adolescents and Youth) and other studies is emphasizing that at least some individuals with adolescent type 2 diabetes may have a more malignant form of disease than in adults. This is striking and important to consider as we look at how to prevent cardiovascular disease.”
Obesity in childhood is also associated with structural and functional abnormalities of the vasculature, according to studies that measure vascular structure via intima-media thickness of the carotid arteries, femoral arteries, abdominal aorta, or other arteries, as well as those that measure vascular stiffness via measures of intrinsic “visco-elastic” properties of the arterial wall. In one study of individuals aged 10-24 years, Dr. Daniels and his associates performed carotid ultrasound for carotid intima-media thickness on 182 patients who were lean, 136 who were obese, and 128 who had type 2 diabetes (Circulation 2009;119(22):2913-9). It demonstrated that youth with obesity and obesity-related type 2 diabetes have abnormalities in carotid thickness and stiffness that are only partially explained by traditional cardiovascular risk factors.
“We all know that obesity is very difficult to treat,” he concluded. “That’s true in children and adolescents as it is in adults. I think this argues for prevention of obesity, for us starting earlier, creating an optimal cardiovascular health situation that we can maintain during the course of childhood and adolescence. The payoff will be great if we can accomplish that.”
Dr. Daniels reported having no disclosures.
EXPERT ANALYSIS FROM WCIRDC 2018
EHR stress linked to burnout
Physicians who experience stress related to the use of health information technology are twice as likely to experience burnout.

Rebekah Gardner, MD, of Brown University in Providence, R.I., and her colleagues surveyed all 4,197 Rhode Island physicians in 2017 to learn how the use of electronic health records affected their practices and their job satisfaction.
Just over a quarter (25.0%) of 1,792 respondents reported burnout. Among electronic health record users (91% of respondents), 70% reported health IT-related stress (J Am Med Inform Assoc. doi: 10.1093/jamia/ocy145).
“After adjustment, physicians reporting poor/marginal time for documentation had 2.8 times the odds of burnout (95% confidence interval, 2.0-4.1; P less than .0001) compared to those reporting sufficient time,” according to the researchers.
The team looked at three stress-related variables: whether the EHR adds to the frustration of one’s day; whether physicians felt they had sufficient time for documentation; and the amount of time spent on the EHR at home. Variables were measured on a four- or five-point scale depending on the question related to the specific stress variable.
Almost two-thirds (64.2%) of respondents “agreed” or “strongly agreed” that EHRs add to the frustration of their day.
“It was the most commonly cited HIT-related stress measure in almost every specialty, with the highest prevalence among emergency physicians (77.6%),” the investigators wrote.
More than a third of physicians (37.7%) reported “moderately high” or “excessive” time spent on EHRs at home; this metric was the most commonly cited stress measure among pediatricians (63.6%).
Nearly half (46.4%) of physicians reported “poor” or “marginal” sufficiency of time for documentation.
“Presence of any 1 of the HIT-related stress measures was associated with approximately twice the odds of burnout among physician respondents,” Dr. Gardner and her colleagues noted, adding that “measuring and addressing HIT-related stress is an important step in reducing workforce burden and improving the care of our patients.”
To alleviate burnout, the authors recommended increased use of scribes, use of medical assistants to help create a more team-based documentation function, improved EHR training, providing more time during the day for documentation, and streamlining documentation expectations, with certain culture shifts needed in some cases (i.e., banning work-related email and clinical tasks for vacationing physicians).
SOURCE: Gardner R et al. J Am Med Inform Assoc. doi: 10.1093/jamia/ocy145.
Physicians who experience stress related to the use of health information technology are twice as likely to experience burnout.
Rebekah Gardner, MD, of Brown University in Providence, R.I., and her colleagues surveyed all 4,197 Rhode Island physicians in 2017 to learn how the use of electronic health records affected their practices and their job satisfaction.
Just over a quarter (25.0%) of 1,792 respondents reported burnout. Among electronic health record users (91% of respondents), 70% reported health IT-related stress (J Am Med Inform Assoc. doi: 10.1093/jamia/ocy145).
“After adjustment, physicians reporting poor/marginal time for documentation had 2.8 times the odds of burnout (95% confidence interval, 2.0-4.1; P less than .0001) compared to those reporting sufficient time,” according to the researchers.
The team looked at three stress-related variables: whether the EHR adds to the frustration of one’s day; whether physicians felt they had sufficient time for documentation; and the amount of time spent on the EHR at home. Variables were measured on a four- or five-point scale depending on the question related to the specific stress variable.
Almost two-thirds (64.2%) of respondents “agreed” or “strongly agreed” that EHRs add to the frustration of their day.
“It was the most commonly cited HIT-related stress measure in almost every specialty, with the highest prevalence among emergency physicians (77.6%),” the investigators wrote.
More than a third of physicians (37.7%) reported “moderately high” or “excessive” time spent on EHRs at home; this metric was the most commonly cited stress measure among pediatricians (63.6%).
Nearly half (46.4%) of physicians reported “poor” or “marginal” sufficiency of time for documentation.
“Presence of any 1 of the HIT-related stress measures was associated with approximately twice the odds of burnout among physician respondents,” Dr. Gardner and her colleagues noted, adding that “measuring and addressing HIT-related stress is an important step in reducing workforce burden and improving the care of our patients.”
To alleviate burnout, the authors recommended increased use of scribes, use of medical assistants to help create a more team-based documentation function, improved EHR training, providing more time during the day for documentation, and streamlining documentation expectations, with certain culture shifts needed in some cases (i.e., banning work-related email and clinical tasks for vacationing physicians).
SOURCE: Gardner R et al. J Am Med Inform Assoc. doi: 10.1093/jamia/ocy145.
Physicians who experience stress related to the use of health information technology are twice as likely to experience burnout.
Rebekah Gardner, MD, of Brown University in Providence, R.I., and her colleagues surveyed all 4,197 Rhode Island physicians in 2017 to learn how the use of electronic health records affected their practices and their job satisfaction.
Just over a quarter (25.0%) of 1,792 respondents reported burnout. Among electronic health record users (91% of respondents), 70% reported health IT-related stress (J Am Med Inform Assoc. doi: 10.1093/jamia/ocy145).
“After adjustment, physicians reporting poor/marginal time for documentation had 2.8 times the odds of burnout (95% confidence interval, 2.0-4.1; P less than .0001) compared to those reporting sufficient time,” according to the researchers.
The team looked at three stress-related variables: whether the EHR adds to the frustration of one’s day; whether physicians felt they had sufficient time for documentation; and the amount of time spent on the EHR at home. Variables were measured on a four- or five-point scale depending on the question related to the specific stress variable.
Almost two-thirds (64.2%) of respondents “agreed” or “strongly agreed” that EHRs add to the frustration of their day.
“It was the most commonly cited HIT-related stress measure in almost every specialty, with the highest prevalence among emergency physicians (77.6%),” the investigators wrote.
More than a third of physicians (37.7%) reported “moderately high” or “excessive” time spent on EHRs at home; this metric was the most commonly cited stress measure among pediatricians (63.6%).
Nearly half (46.4%) of physicians reported “poor” or “marginal” sufficiency of time for documentation.
“Presence of any 1 of the HIT-related stress measures was associated with approximately twice the odds of burnout among physician respondents,” Dr. Gardner and her colleagues noted, adding that “measuring and addressing HIT-related stress is an important step in reducing workforce burden and improving the care of our patients.”
To alleviate burnout, the authors recommended increased use of scribes, use of medical assistants to help create a more team-based documentation function, improved EHR training, providing more time during the day for documentation, and streamlining documentation expectations, with certain culture shifts needed in some cases (i.e., banning work-related email and clinical tasks for vacationing physicians).
SOURCE: Gardner R et al. J Am Med Inform Assoc. doi: 10.1093/jamia/ocy145.
FROM JAMIA
Key clinical point: EHR-related stress is a measurable predictor of physician burnout.
Major finding: Seventy percent of EHR users who responded to a survey reported stress related to the use of health information technology.
Study details: Survey of all 4,197 physicians in Rhode Island; 1,792 (43%) responded.
Disclosures: The Rhode Island Department of Health funded the study. The authors reported no conflicts of interest.
Source: Gardner R et al. J Am Med Inform Assoc. doi: 10.1093/jamia/ocy145.
2018-2019 flu season starts in earnest
National flu activity moved solidly into above-average territory during the week ending Dec. 15, as Colorado and Georgia took the lead with the highest activity levels in the country, according to the Centers for Disease Control and Prevention.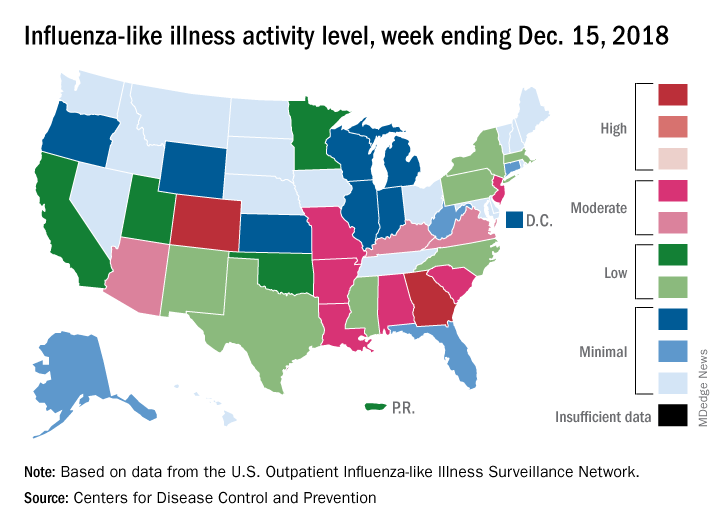
The proportion of outpatient visits for influenza-like illness (ILI) was 2.7% for the week, which was up from 2.3% the previous week and above the national baseline of 2.2%, the CDC reported. ILI is defined “as fever (temperature of 100°F [37.8°C] or greater) and cough and/or sore throat.”
Colorado and Georgia both reported ILI activity of 10 on the CDC’s 1-10 scale, making them the only states in the “high” range (8-10). Nine states and New York City had activity levels in the “moderate” range (6-7), Puerto Rico and 11 states were in the “low” range (4-5), and 28 states and the District of Columbia were in the “minimal” range (1-3), the CDC said.
During the comparable period of last year’s high-severity flu season, which ultimately resulted in 900,000 flu-related hospitalizations and 80,000 deaths (185 pediatric), nine states were already at level 10. For the 2018-2019 season so far, there have been seven ILI-related pediatric deaths, CDC data show.
National flu activity moved solidly into above-average territory during the week ending Dec. 15, as Colorado and Georgia took the lead with the highest activity levels in the country, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 2.7% for the week, which was up from 2.3% the previous week and above the national baseline of 2.2%, the CDC reported. ILI is defined “as fever (temperature of 100°F [37.8°C] or greater) and cough and/or sore throat.”
Colorado and Georgia both reported ILI activity of 10 on the CDC’s 1-10 scale, making them the only states in the “high” range (8-10). Nine states and New York City had activity levels in the “moderate” range (6-7), Puerto Rico and 11 states were in the “low” range (4-5), and 28 states and the District of Columbia were in the “minimal” range (1-3), the CDC said.
During the comparable period of last year’s high-severity flu season, which ultimately resulted in 900,000 flu-related hospitalizations and 80,000 deaths (185 pediatric), nine states were already at level 10. For the 2018-2019 season so far, there have been seven ILI-related pediatric deaths, CDC data show.
National flu activity moved solidly into above-average territory during the week ending Dec. 15, as Colorado and Georgia took the lead with the highest activity levels in the country, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 2.7% for the week, which was up from 2.3% the previous week and above the national baseline of 2.2%, the CDC reported. ILI is defined “as fever (temperature of 100°F [37.8°C] or greater) and cough and/or sore throat.”
Colorado and Georgia both reported ILI activity of 10 on the CDC’s 1-10 scale, making them the only states in the “high” range (8-10). Nine states and New York City had activity levels in the “moderate” range (6-7), Puerto Rico and 11 states were in the “low” range (4-5), and 28 states and the District of Columbia were in the “minimal” range (1-3), the CDC said.
During the comparable period of last year’s high-severity flu season, which ultimately resulted in 900,000 flu-related hospitalizations and 80,000 deaths (185 pediatric), nine states were already at level 10. For the 2018-2019 season so far, there have been seven ILI-related pediatric deaths, CDC data show.
Michigan police receive training to recognize mental illness
Responding to a police call can prove dangerous. In those kinds of high-pressure situations, agitation or other manifestations of mental illness might be mistaken for violent intent – with disastrous results.

In Kalamazoo, Mich., crime response training now includes subduing suspects without using violent force. “Through training and education, and scenarios that we use in the training, [the officers] start to detect the different cues or indicators where they start to see that this is really a crisis event. And we treat it as a medical issue and get that person the help that they need,” said Rafael Diaz, executive lieutenant with the Kalamazoo Department of Public Safety in an interview on Michigan NPR.
In the training, called the Crisis Intervention Team model, the goal is to slow down the pace of the interaction and keep some distance between themselves and the suspect after officers recognize signs of mental illness. Both responses can lower the chances of a lash-out response.
“The number of injuries to officers goes down, the number of injuries to the person in crisis goes down, and there is a huge benefit to society there if you don’t have to use physical force,” Mr. Diaz said.
Animal neglect and mental health
Images of neglected and abused livestock on farms can inspire thoughts of how someone could mistreat the animals in their care. “Frankly, if you can’t understand that, it’s probably a good thing. It means you haven’t been in the depths of low, low mental health, depression, and anxiety,” Andria Jones-Bitton, DVM, PhD, said in an interview with the Western Producer.
Dr. Jones-Bitton is a veterinarian and epidemiologist at the Ontario Veterinary College in Guelph. She is studying the mental health and mental resilience of farmers and veterinarians.
“If farmers are struggling with their own well-being and motivation, they’re likely going to find it difficult to invest in improving animal welfare. When we’re mentally unwell, it’s hard to care for ourselves, let alone to care for others, even when those others are really important to us,” she said.
A national survey of Canadian farmers by Dr. Jones-Bitton showed high levels of stress and diminished ability to cope with the pressures that come with running a livestock farm. “What makes me the most upset is I have everything I’ve ever dreamed of – love, family, and a farm, and all I feel is overwhelmed out of control and sad,” one respondent said.
The problems are not unique to Canada. Studies from Ireland, for example, documented an association between animal neglect cases and the mental health, drug/alcohol addiction, and social problems of farmers.
“Even if you didn’t care about the humans that were struggling and you only cared about the animal welfare, you’d be wise to address the issue of farmer stress,” Dr. Jones-Britton said.
Depression and rural America
A recent “Farming in Tough Times” workshop that convened in Minnesota focused on the mental health of farmers. Making a living is challenging for many reasons. One is that prices for commodities are set by others.
“I realized that I can’t change the situation that we’re in. I can’t change milk prices. I can’t stop farms from going bankrupt. But I can change how we are. And we are together, and that really does matter,” said dairy farmer Brenda Rudolph during the workshop, according to a report from the St. Cloud Times, which is part of the USA Today network.
“There is a conversation you people have to have in America, rural America, that says, depression is part of your life. It is not a sign of weakness. It’s a sign of reality,” said Dennis Hoiberg, a farming consultant based in Australia who spoke at the workshop. He added that, from his perspective, the United States still tends to be more repressed about mental health issues than elsewhere in the world – with the focus on stress and not on resiliency.
“Most of you folk are proud folk, and most you folk are very proud of what you do,” Mr. Hoiberg said. “You’re also psychologically exposed because you are a true believer [in what you do].”
Advice offered to lessen the tough times included noticing the beauty in the world, breaking down problems into small chunks that are more easily dealt with and then moving on to the next, sleep, and a good diet.
People with mental illness languishing
Public defenders in Colorado are seeking to have dozens of people diagnosed with mental illness who are in jail awaiting trial set free until their court date. The usual scenario in Colorado for someone charged with a crime and jailed who is deemed mentally incompetent is treatment within 28 days. However, this system is broken and wait times are far longer – in one instance 270 days.
“Many of them are there for very, very low level offenses and they’re holding in jail for way longer than a person who did not suffer from mental illness would be in custody,” said Maureen Cain, policy liaison for the public defender’s office in an interview with the Denver Post. “They are being incarcerated for their mental illness, not really because of the crime they committed.” Responses from judges have ranged from immediate release to finding the incarcerated person guilty of contempt and sending them back to jail.
The Colorado Department of Human Services is in charge of people who have been jailed but have been found to be incompetent to stand trial. Officials there have say they do not have enough bed space or capacity to get people moved out of jail within 28 days.
“We are in a situation where [the human services department] is in breach, and I need to know what efforts are being made to bring it back into compliance,” said federal Judge Nina Y. Wang. “These individuals are not being served, and frankly, the state is not being served.”
“Cruel” practice confined youth
A federal class action lawsuit filed against the Departmental of Children and Family Services (DCFS) in the Chicago area alleges that, from 2015 to 2017, more than 800 youth were being confined to psychiatric hospitals even when they were cleared for discharge. The problem goes back decades and is getting worse, the lawsuit contends.
“I spent Thanksgiving, Christmas, New Year’s, Easter, and my 16th birthday in the hospital,” said Skylar, who’s now 19 years old. “I only got to go outside one time. I felt like a prisoner; I felt very depressed.”
As reported on Chicago’s WGN9 News, the delay between clearance for discharge and actual freedom is a month or more in many of the cases. Acting Cook County Public Guardian Charles Golbert said the practice is “cruel, unusual, and illegal. It’s a violation of the children’s civil and most basic human rights.”
Many of the youth had been incarcerated for setting fires and self-harm and had been rejected by foster parents and other providers, in some cases their own families, who were concerned with the possible behavior of the youth after their release.
“Blame the children is the wrong response from DCFS,” said attorney Russell Ainsworth. “DCFS should be apologizing for not addressing this issue and for violating the Constitution.”
Responding to a police call can prove dangerous. In those kinds of high-pressure situations, agitation or other manifestations of mental illness might be mistaken for violent intent – with disastrous results.
In Kalamazoo, Mich., crime response training now includes subduing suspects without using violent force. “Through training and education, and scenarios that we use in the training, [the officers] start to detect the different cues or indicators where they start to see that this is really a crisis event. And we treat it as a medical issue and get that person the help that they need,” said Rafael Diaz, executive lieutenant with the Kalamazoo Department of Public Safety in an interview on Michigan NPR.
In the training, called the Crisis Intervention Team model, the goal is to slow down the pace of the interaction and keep some distance between themselves and the suspect after officers recognize signs of mental illness. Both responses can lower the chances of a lash-out response.
“The number of injuries to officers goes down, the number of injuries to the person in crisis goes down, and there is a huge benefit to society there if you don’t have to use physical force,” Mr. Diaz said.
Animal neglect and mental health
Images of neglected and abused livestock on farms can inspire thoughts of how someone could mistreat the animals in their care. “Frankly, if you can’t understand that, it’s probably a good thing. It means you haven’t been in the depths of low, low mental health, depression, and anxiety,” Andria Jones-Bitton, DVM, PhD, said in an interview with the Western Producer.
Dr. Jones-Bitton is a veterinarian and epidemiologist at the Ontario Veterinary College in Guelph. She is studying the mental health and mental resilience of farmers and veterinarians.
“If farmers are struggling with their own well-being and motivation, they’re likely going to find it difficult to invest in improving animal welfare. When we’re mentally unwell, it’s hard to care for ourselves, let alone to care for others, even when those others are really important to us,” she said.
A national survey of Canadian farmers by Dr. Jones-Bitton showed high levels of stress and diminished ability to cope with the pressures that come with running a livestock farm. “What makes me the most upset is I have everything I’ve ever dreamed of – love, family, and a farm, and all I feel is overwhelmed out of control and sad,” one respondent said.
The problems are not unique to Canada. Studies from Ireland, for example, documented an association between animal neglect cases and the mental health, drug/alcohol addiction, and social problems of farmers.
“Even if you didn’t care about the humans that were struggling and you only cared about the animal welfare, you’d be wise to address the issue of farmer stress,” Dr. Jones-Britton said.
Depression and rural America
A recent “Farming in Tough Times” workshop that convened in Minnesota focused on the mental health of farmers. Making a living is challenging for many reasons. One is that prices for commodities are set by others.
“I realized that I can’t change the situation that we’re in. I can’t change milk prices. I can’t stop farms from going bankrupt. But I can change how we are. And we are together, and that really does matter,” said dairy farmer Brenda Rudolph during the workshop, according to a report from the St. Cloud Times, which is part of the USA Today network.
“There is a conversation you people have to have in America, rural America, that says, depression is part of your life. It is not a sign of weakness. It’s a sign of reality,” said Dennis Hoiberg, a farming consultant based in Australia who spoke at the workshop. He added that, from his perspective, the United States still tends to be more repressed about mental health issues than elsewhere in the world – with the focus on stress and not on resiliency.
“Most of you folk are proud folk, and most you folk are very proud of what you do,” Mr. Hoiberg said. “You’re also psychologically exposed because you are a true believer [in what you do].”
Advice offered to lessen the tough times included noticing the beauty in the world, breaking down problems into small chunks that are more easily dealt with and then moving on to the next, sleep, and a good diet.
People with mental illness languishing
Public defenders in Colorado are seeking to have dozens of people diagnosed with mental illness who are in jail awaiting trial set free until their court date. The usual scenario in Colorado for someone charged with a crime and jailed who is deemed mentally incompetent is treatment within 28 days. However, this system is broken and wait times are far longer – in one instance 270 days.
“Many of them are there for very, very low level offenses and they’re holding in jail for way longer than a person who did not suffer from mental illness would be in custody,” said Maureen Cain, policy liaison for the public defender’s office in an interview with the Denver Post. “They are being incarcerated for their mental illness, not really because of the crime they committed.” Responses from judges have ranged from immediate release to finding the incarcerated person guilty of contempt and sending them back to jail.
The Colorado Department of Human Services is in charge of people who have been jailed but have been found to be incompetent to stand trial. Officials there have say they do not have enough bed space or capacity to get people moved out of jail within 28 days.
“We are in a situation where [the human services department] is in breach, and I need to know what efforts are being made to bring it back into compliance,” said federal Judge Nina Y. Wang. “These individuals are not being served, and frankly, the state is not being served.”
“Cruel” practice confined youth
A federal class action lawsuit filed against the Departmental of Children and Family Services (DCFS) in the Chicago area alleges that, from 2015 to 2017, more than 800 youth were being confined to psychiatric hospitals even when they were cleared for discharge. The problem goes back decades and is getting worse, the lawsuit contends.
“I spent Thanksgiving, Christmas, New Year’s, Easter, and my 16th birthday in the hospital,” said Skylar, who’s now 19 years old. “I only got to go outside one time. I felt like a prisoner; I felt very depressed.”
As reported on Chicago’s WGN9 News, the delay between clearance for discharge and actual freedom is a month or more in many of the cases. Acting Cook County Public Guardian Charles Golbert said the practice is “cruel, unusual, and illegal. It’s a violation of the children’s civil and most basic human rights.”
Many of the youth had been incarcerated for setting fires and self-harm and had been rejected by foster parents and other providers, in some cases their own families, who were concerned with the possible behavior of the youth after their release.
“Blame the children is the wrong response from DCFS,” said attorney Russell Ainsworth. “DCFS should be apologizing for not addressing this issue and for violating the Constitution.”
Responding to a police call can prove dangerous. In those kinds of high-pressure situations, agitation or other manifestations of mental illness might be mistaken for violent intent – with disastrous results.
In Kalamazoo, Mich., crime response training now includes subduing suspects without using violent force. “Through training and education, and scenarios that we use in the training, [the officers] start to detect the different cues or indicators where they start to see that this is really a crisis event. And we treat it as a medical issue and get that person the help that they need,” said Rafael Diaz, executive lieutenant with the Kalamazoo Department of Public Safety in an interview on Michigan NPR.
In the training, called the Crisis Intervention Team model, the goal is to slow down the pace of the interaction and keep some distance between themselves and the suspect after officers recognize signs of mental illness. Both responses can lower the chances of a lash-out response.
“The number of injuries to officers goes down, the number of injuries to the person in crisis goes down, and there is a huge benefit to society there if you don’t have to use physical force,” Mr. Diaz said.
Animal neglect and mental health
Images of neglected and abused livestock on farms can inspire thoughts of how someone could mistreat the animals in their care. “Frankly, if you can’t understand that, it’s probably a good thing. It means you haven’t been in the depths of low, low mental health, depression, and anxiety,” Andria Jones-Bitton, DVM, PhD, said in an interview with the Western Producer.
Dr. Jones-Bitton is a veterinarian and epidemiologist at the Ontario Veterinary College in Guelph. She is studying the mental health and mental resilience of farmers and veterinarians.
“If farmers are struggling with their own well-being and motivation, they’re likely going to find it difficult to invest in improving animal welfare. When we’re mentally unwell, it’s hard to care for ourselves, let alone to care for others, even when those others are really important to us,” she said.
A national survey of Canadian farmers by Dr. Jones-Bitton showed high levels of stress and diminished ability to cope with the pressures that come with running a livestock farm. “What makes me the most upset is I have everything I’ve ever dreamed of – love, family, and a farm, and all I feel is overwhelmed out of control and sad,” one respondent said.
The problems are not unique to Canada. Studies from Ireland, for example, documented an association between animal neglect cases and the mental health, drug/alcohol addiction, and social problems of farmers.
“Even if you didn’t care about the humans that were struggling and you only cared about the animal welfare, you’d be wise to address the issue of farmer stress,” Dr. Jones-Britton said.
Depression and rural America
A recent “Farming in Tough Times” workshop that convened in Minnesota focused on the mental health of farmers. Making a living is challenging for many reasons. One is that prices for commodities are set by others.
“I realized that I can’t change the situation that we’re in. I can’t change milk prices. I can’t stop farms from going bankrupt. But I can change how we are. And we are together, and that really does matter,” said dairy farmer Brenda Rudolph during the workshop, according to a report from the St. Cloud Times, which is part of the USA Today network.
“There is a conversation you people have to have in America, rural America, that says, depression is part of your life. It is not a sign of weakness. It’s a sign of reality,” said Dennis Hoiberg, a farming consultant based in Australia who spoke at the workshop. He added that, from his perspective, the United States still tends to be more repressed about mental health issues than elsewhere in the world – with the focus on stress and not on resiliency.
“Most of you folk are proud folk, and most you folk are very proud of what you do,” Mr. Hoiberg said. “You’re also psychologically exposed because you are a true believer [in what you do].”
Advice offered to lessen the tough times included noticing the beauty in the world, breaking down problems into small chunks that are more easily dealt with and then moving on to the next, sleep, and a good diet.
People with mental illness languishing
Public defenders in Colorado are seeking to have dozens of people diagnosed with mental illness who are in jail awaiting trial set free until their court date. The usual scenario in Colorado for someone charged with a crime and jailed who is deemed mentally incompetent is treatment within 28 days. However, this system is broken and wait times are far longer – in one instance 270 days.
“Many of them are there for very, very low level offenses and they’re holding in jail for way longer than a person who did not suffer from mental illness would be in custody,” said Maureen Cain, policy liaison for the public defender’s office in an interview with the Denver Post. “They are being incarcerated for their mental illness, not really because of the crime they committed.” Responses from judges have ranged from immediate release to finding the incarcerated person guilty of contempt and sending them back to jail.
The Colorado Department of Human Services is in charge of people who have been jailed but have been found to be incompetent to stand trial. Officials there have say they do not have enough bed space or capacity to get people moved out of jail within 28 days.
“We are in a situation where [the human services department] is in breach, and I need to know what efforts are being made to bring it back into compliance,” said federal Judge Nina Y. Wang. “These individuals are not being served, and frankly, the state is not being served.”
“Cruel” practice confined youth
A federal class action lawsuit filed against the Departmental of Children and Family Services (DCFS) in the Chicago area alleges that, from 2015 to 2017, more than 800 youth were being confined to psychiatric hospitals even when they were cleared for discharge. The problem goes back decades and is getting worse, the lawsuit contends.
“I spent Thanksgiving, Christmas, New Year’s, Easter, and my 16th birthday in the hospital,” said Skylar, who’s now 19 years old. “I only got to go outside one time. I felt like a prisoner; I felt very depressed.”
As reported on Chicago’s WGN9 News, the delay between clearance for discharge and actual freedom is a month or more in many of the cases. Acting Cook County Public Guardian Charles Golbert said the practice is “cruel, unusual, and illegal. It’s a violation of the children’s civil and most basic human rights.”
Many of the youth had been incarcerated for setting fires and self-harm and had been rejected by foster parents and other providers, in some cases their own families, who were concerned with the possible behavior of the youth after their release.
“Blame the children is the wrong response from DCFS,” said attorney Russell Ainsworth. “DCFS should be apologizing for not addressing this issue and for violating the Constitution.”
December 2018
Vasculitis is a process in which blood vessels become inflamed and necrotic. Classic small-vessel vasculitis reveals a leukocytoclastic vasculitis and most commonly presents as palpable purpura. In addition to skin, organs such as joints, kidneys, and intestines can be involved.
where immunoglobulin A (IgA) is deposited in the vessel walls. It is the most common form of vasculitis in children (usually aged 4-8 years). The incidence is higher in the winter. Some patients experience a prodrome of fever, colicky abdominal pain, and joint pain prior to the development of cutaneous symptoms. Disease in children tends to be self-limited. Adults may present with HSP as well, and often exhibit more severe disease that may become chronic with relapses and is more difficult to treat. In both children and adults, infectious causes, such as streptococcus pharyngitis, are the most common trigger. In adults, malignancy may be associated with HSP. A literature search revealed medications implicated in HSP such as antibiotics (vancomycin, penicillin, cephalosporins, clarithromycin), ACE inhibitors, and nonsteroidal anti-inflammatories. Many cases of HSP are idiopathic.
Patients present with erythematous macules that progress to purpura on the extremities. Lesions may be vesicular or bullous and may become necrotic and ulcerate. Arthralgias, often of lower-extremity joints, may be present. Abdominal pain and renal disease may occur in both children and adults. Adults are more likely to develop chronic kidney disease and must be followed carefully with serial blood work and urinalysis to evaluate for hematuria and proteinuria. Severe abdominal pain is an emergency as intussusception may occur.

Histologically, leukocytoclastic vasculitis of small vessels is present. On direct immunofluorescence of perilesional skin, IgA, C3, and fibrin deposits can be seen. Serum IgA is unreliable and may be seen in healthy adults as well.
Treatment is generally supportive as the disease is self-limited. The use of corticosteroids is controversial. This may be effective for joint inflammation, abdominal disease, active nephritis, and ulcerated skin lesions, but doesn’t prevent the recurrence of skin lesions. Dapsone or colchicine can be used for resistant cutaneous lesions. In severe cases, intravenous immunoglobulin may be warranted.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Vasculitis is a process in which blood vessels become inflamed and necrotic. Classic small-vessel vasculitis reveals a leukocytoclastic vasculitis and most commonly presents as palpable purpura. In addition to skin, organs such as joints, kidneys, and intestines can be involved.
where immunoglobulin A (IgA) is deposited in the vessel walls. It is the most common form of vasculitis in children (usually aged 4-8 years). The incidence is higher in the winter. Some patients experience a prodrome of fever, colicky abdominal pain, and joint pain prior to the development of cutaneous symptoms. Disease in children tends to be self-limited. Adults may present with HSP as well, and often exhibit more severe disease that may become chronic with relapses and is more difficult to treat. In both children and adults, infectious causes, such as streptococcus pharyngitis, are the most common trigger. In adults, malignancy may be associated with HSP. A literature search revealed medications implicated in HSP such as antibiotics (vancomycin, penicillin, cephalosporins, clarithromycin), ACE inhibitors, and nonsteroidal anti-inflammatories. Many cases of HSP are idiopathic.
Patients present with erythematous macules that progress to purpura on the extremities. Lesions may be vesicular or bullous and may become necrotic and ulcerate. Arthralgias, often of lower-extremity joints, may be present. Abdominal pain and renal disease may occur in both children and adults. Adults are more likely to develop chronic kidney disease and must be followed carefully with serial blood work and urinalysis to evaluate for hematuria and proteinuria. Severe abdominal pain is an emergency as intussusception may occur.
Histologically, leukocytoclastic vasculitis of small vessels is present. On direct immunofluorescence of perilesional skin, IgA, C3, and fibrin deposits can be seen. Serum IgA is unreliable and may be seen in healthy adults as well.
Treatment is generally supportive as the disease is self-limited. The use of corticosteroids is controversial. This may be effective for joint inflammation, abdominal disease, active nephritis, and ulcerated skin lesions, but doesn’t prevent the recurrence of skin lesions. Dapsone or colchicine can be used for resistant cutaneous lesions. In severe cases, intravenous immunoglobulin may be warranted.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Vasculitis is a process in which blood vessels become inflamed and necrotic. Classic small-vessel vasculitis reveals a leukocytoclastic vasculitis and most commonly presents as palpable purpura. In addition to skin, organs such as joints, kidneys, and intestines can be involved.
where immunoglobulin A (IgA) is deposited in the vessel walls. It is the most common form of vasculitis in children (usually aged 4-8 years). The incidence is higher in the winter. Some patients experience a prodrome of fever, colicky abdominal pain, and joint pain prior to the development of cutaneous symptoms. Disease in children tends to be self-limited. Adults may present with HSP as well, and often exhibit more severe disease that may become chronic with relapses and is more difficult to treat. In both children and adults, infectious causes, such as streptococcus pharyngitis, are the most common trigger. In adults, malignancy may be associated with HSP. A literature search revealed medications implicated in HSP such as antibiotics (vancomycin, penicillin, cephalosporins, clarithromycin), ACE inhibitors, and nonsteroidal anti-inflammatories. Many cases of HSP are idiopathic.
Patients present with erythematous macules that progress to purpura on the extremities. Lesions may be vesicular or bullous and may become necrotic and ulcerate. Arthralgias, often of lower-extremity joints, may be present. Abdominal pain and renal disease may occur in both children and adults. Adults are more likely to develop chronic kidney disease and must be followed carefully with serial blood work and urinalysis to evaluate for hematuria and proteinuria. Severe abdominal pain is an emergency as intussusception may occur.
Histologically, leukocytoclastic vasculitis of small vessels is present. On direct immunofluorescence of perilesional skin, IgA, C3, and fibrin deposits can be seen. Serum IgA is unreliable and may be seen in healthy adults as well.
Treatment is generally supportive as the disease is self-limited. The use of corticosteroids is controversial. This may be effective for joint inflammation, abdominal disease, active nephritis, and ulcerated skin lesions, but doesn’t prevent the recurrence of skin lesions. Dapsone or colchicine can be used for resistant cutaneous lesions. In severe cases, intravenous immunoglobulin may be warranted.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
A 63-year-old white female presented with a 2-week history of hemorrhagic purpuric lesions and necrotic vesicles on the bilateral lower extremities.
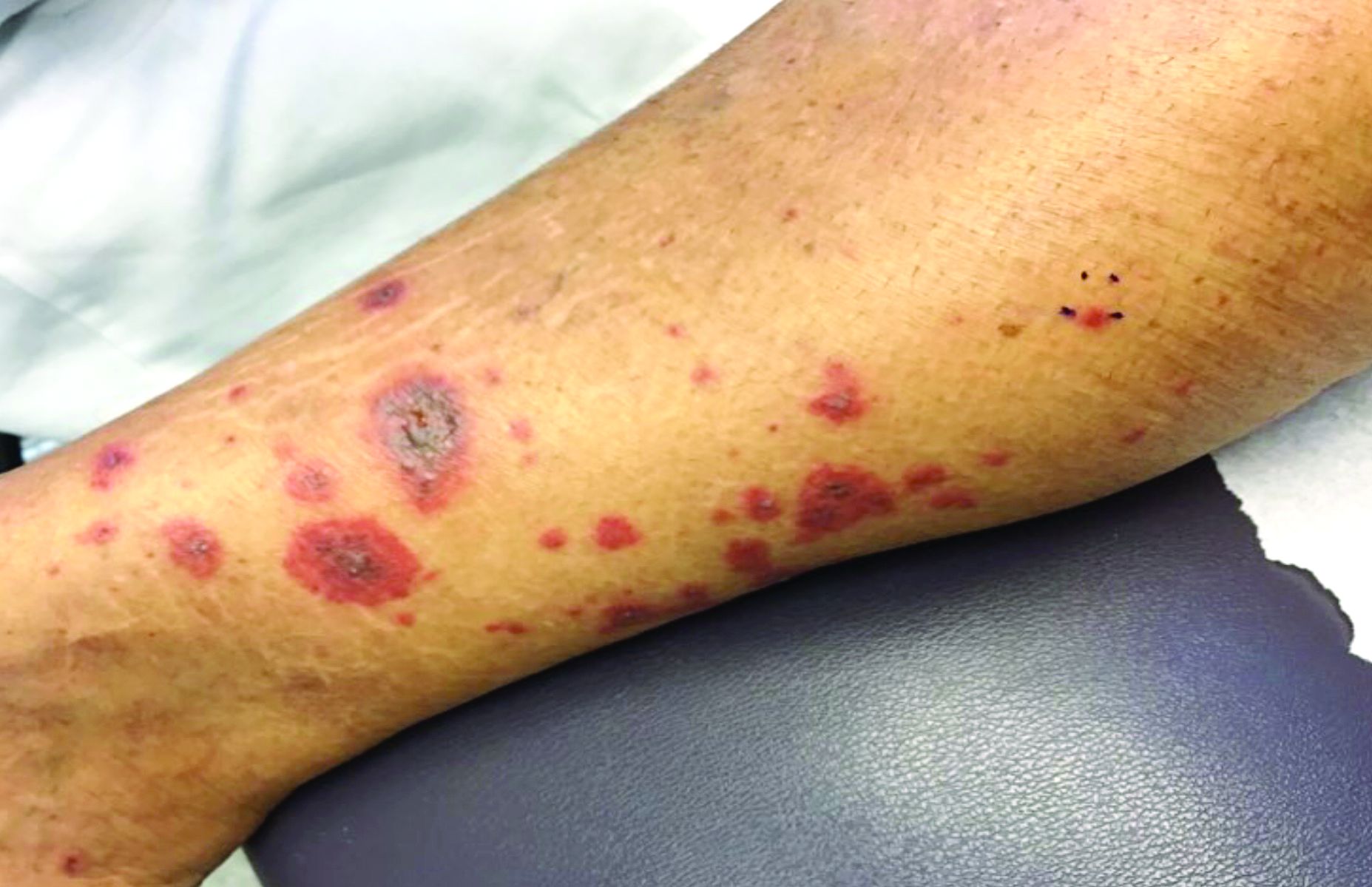
Nearly 1 year prior to presentation, the patient underwent surgical resection for lung cancer. The patient also complained of joint swelling and pain in her ankles. She denied abdominal pain. She denied recent illness, including sore throat and upper respiratory infection. Skin biopsies were performed, including for direct immunofluorescence.
FDA approves first treatment for BPDCN

The U.S. Food and Drug Administration (FDA) has approved tagraxofusp-erzs (Elzonris) to treat patients age 2 and older who have blastic plasmacytoid dendritic cell neoplasm (BPDCN).
Tagraxofusp-erzs (formerly SL-401) is a CD123-directed cytotoxin that is the first FDA-approved treatment for BPDCN.
Tagraxofusp-erzs will be commercially available in early 2019, according to Stemline Therapeutics, makers of the drug.
The prescribing information for tagraxofusp-erzs contains a boxed warning noting that the drug is associated with an increased risk of capillary leak syndrome (CLS), which may be life-threatening or fatal.
The FDA previously granted tagraxofusp-erzs breakthrough therapy and orphan drug designations and assessed the drug under priority review.
The FDA’s approval of tagraxofusp-erzs was based on a phase 1 trial (STML-401-0114; NCT02113982).
The trial enrolled 47 patients with BPDCN, including 32 who were treatment-naïve and 15 who were previously treated.
Patients received tagraxofusp-erzs intravenously on days 1-5 of a 21-day cycle for multiple consecutive cycles. The trial had a dose-escalation stage (stage 1), an expansion stage (stage 2), a confirmatory stage (stage 3), and a stage that enabled uninterrupted access to tagraxofusp-erzs (stage 4).
In the confirmatory stage, 13 patients with treatment-naïve BPDCN received tagraxofusp-erzs at the recommended dose and schedule—12 mcg/kg daily for 5 days of a 21-day cycle.
Efficacy was based on the rate of complete response (CR) or clinical complete response (CRc). CRc was defined as CR with residual skin abnormality not indicative of active disease.
The CR/CRc rate was 53.8% (7/13), and the median duration of CR/CRc was not reached (range, 3.9 to 12.2 months).
The safety of tagraxofusp-erzs was assessed in 94 adults with treatment-naïve or previously treated myeloid malignancies, including 58 patients with BPDCN, who were treated at the recommended dose and schedule.
There were two fatal adverse events—both CLS. Eleven percent of patients discontinued treatment with tagraxofusp-erzs due to an adverse event. The most common of these were hepatic toxicities and CLS.
The most common adverse events overall were CLS (55%), nausea (49%), fatigue (45%), peripheral edema (43%), pyrexia (43%), and weight increase (31%).
The most common laboratory abnormalities were decreases in albumin (77%), platelets (67%), hemoglobin (60%), calcium (57%), and sodium (50%), as well as increases in glucose (87%), alanine aminotransferase (82%), and aspartate aminotransferase (79%).
The U.S. Food and Drug Administration (FDA) has approved tagraxofusp-erzs (Elzonris) to treat patients age 2 and older who have blastic plasmacytoid dendritic cell neoplasm (BPDCN).
Tagraxofusp-erzs (formerly SL-401) is a CD123-directed cytotoxin that is the first FDA-approved treatment for BPDCN.
Tagraxofusp-erzs will be commercially available in early 2019, according to Stemline Therapeutics, makers of the drug.
The prescribing information for tagraxofusp-erzs contains a boxed warning noting that the drug is associated with an increased risk of capillary leak syndrome (CLS), which may be life-threatening or fatal.
The FDA previously granted tagraxofusp-erzs breakthrough therapy and orphan drug designations and assessed the drug under priority review.
The FDA’s approval of tagraxofusp-erzs was based on a phase 1 trial (STML-401-0114; NCT02113982).
The trial enrolled 47 patients with BPDCN, including 32 who were treatment-naïve and 15 who were previously treated.
Patients received tagraxofusp-erzs intravenously on days 1-5 of a 21-day cycle for multiple consecutive cycles. The trial had a dose-escalation stage (stage 1), an expansion stage (stage 2), a confirmatory stage (stage 3), and a stage that enabled uninterrupted access to tagraxofusp-erzs (stage 4).
In the confirmatory stage, 13 patients with treatment-naïve BPDCN received tagraxofusp-erzs at the recommended dose and schedule—12 mcg/kg daily for 5 days of a 21-day cycle.
Efficacy was based on the rate of complete response (CR) or clinical complete response (CRc). CRc was defined as CR with residual skin abnormality not indicative of active disease.
The CR/CRc rate was 53.8% (7/13), and the median duration of CR/CRc was not reached (range, 3.9 to 12.2 months).
The safety of tagraxofusp-erzs was assessed in 94 adults with treatment-naïve or previously treated myeloid malignancies, including 58 patients with BPDCN, who were treated at the recommended dose and schedule.
There were two fatal adverse events—both CLS. Eleven percent of patients discontinued treatment with tagraxofusp-erzs due to an adverse event. The most common of these were hepatic toxicities and CLS.
The most common adverse events overall were CLS (55%), nausea (49%), fatigue (45%), peripheral edema (43%), pyrexia (43%), and weight increase (31%).
The most common laboratory abnormalities were decreases in albumin (77%), platelets (67%), hemoglobin (60%), calcium (57%), and sodium (50%), as well as increases in glucose (87%), alanine aminotransferase (82%), and aspartate aminotransferase (79%).
The U.S. Food and Drug Administration (FDA) has approved tagraxofusp-erzs (Elzonris) to treat patients age 2 and older who have blastic plasmacytoid dendritic cell neoplasm (BPDCN).
Tagraxofusp-erzs (formerly SL-401) is a CD123-directed cytotoxin that is the first FDA-approved treatment for BPDCN.
Tagraxofusp-erzs will be commercially available in early 2019, according to Stemline Therapeutics, makers of the drug.
The prescribing information for tagraxofusp-erzs contains a boxed warning noting that the drug is associated with an increased risk of capillary leak syndrome (CLS), which may be life-threatening or fatal.
The FDA previously granted tagraxofusp-erzs breakthrough therapy and orphan drug designations and assessed the drug under priority review.
The FDA’s approval of tagraxofusp-erzs was based on a phase 1 trial (STML-401-0114; NCT02113982).
The trial enrolled 47 patients with BPDCN, including 32 who were treatment-naïve and 15 who were previously treated.
Patients received tagraxofusp-erzs intravenously on days 1-5 of a 21-day cycle for multiple consecutive cycles. The trial had a dose-escalation stage (stage 1), an expansion stage (stage 2), a confirmatory stage (stage 3), and a stage that enabled uninterrupted access to tagraxofusp-erzs (stage 4).
In the confirmatory stage, 13 patients with treatment-naïve BPDCN received tagraxofusp-erzs at the recommended dose and schedule—12 mcg/kg daily for 5 days of a 21-day cycle.
Efficacy was based on the rate of complete response (CR) or clinical complete response (CRc). CRc was defined as CR with residual skin abnormality not indicative of active disease.
The CR/CRc rate was 53.8% (7/13), and the median duration of CR/CRc was not reached (range, 3.9 to 12.2 months).
The safety of tagraxofusp-erzs was assessed in 94 adults with treatment-naïve or previously treated myeloid malignancies, including 58 patients with BPDCN, who were treated at the recommended dose and schedule.
There were two fatal adverse events—both CLS. Eleven percent of patients discontinued treatment with tagraxofusp-erzs due to an adverse event. The most common of these were hepatic toxicities and CLS.
The most common adverse events overall were CLS (55%), nausea (49%), fatigue (45%), peripheral edema (43%), pyrexia (43%), and weight increase (31%).
The most common laboratory abnormalities were decreases in albumin (77%), platelets (67%), hemoglobin (60%), calcium (57%), and sodium (50%), as well as increases in glucose (87%), alanine aminotransferase (82%), and aspartate aminotransferase (79%).
FDA approves ravulizumab for PNH
The U.S. Food and Drug Administration (FDA) has approved ravulizumab-cwvz (Ultomiris) to treat adults with paroxysmal nocturnal hemoglobinuria (PNH).
Ravulizumab is a long-acting C5 complement inhibitor, administered every 8 weeks, that has been shown to prevent hemolysis.
The prescribing information for ravulizumab includes a boxed warning stating that meningococcal infections/sepsis have occurred in patients treated with the drug, and these adverse effects can become life-threatening or fatal if not recognized and treated early.
Ravulizumab is available only through a restricted program under a Risk Evaluation and Mitigation Strategy.
The FDA previously granted the application for ravulizumab priority review, and the product received orphan drug designation from the FDA.
The FDA granted the approval of ravulizumab to Alexion Pharmaceuticals.
The FDA’s approval of ravulizumab is based on results from two phase 3 studies, one in patients who had previously received treatment with a complement inhibitor and one in patients who were complement-inhibitor-naïve. Both studies were recently published in Blood.
Efficacy in inhibitor-experienced patients
In one study (NCT03056040), researchers compared ravulizumab administered every 8 weeks to eculizumab administered every 2 weeks in complement-inhibitor-experienced patients.
The trial included 195 PNH patients who were taking eculizumab for more than 6 months. They were randomized to switch to ravulizumab (n=97) or continue on eculizumab (n=98).
Ravulizumab proved noninferior to eculizumab for all endpoints studied (P<0.0006), including:
- Percentage change in lactate dehydrogenase (LDH): difference, 9.21% (95% CI: -0.42 to 18.84; P=0.058 for superiority)
- Breakthrough hemolysis: difference, 5.1 (95% CI: -8.89 to 18.99)
- Change in FACIT-Fatigue score: difference, 1.47 (95% CI: -0.21 to 3.15)
- Transfusion avoidance: difference, 5.5 (95% CI: -4.27 to 15.68)
- Stabilized hemoglobin: difference, 1.4 (95% CI: -10.41 to 13.31).
Efficacy in inhibitor-naïve patients
In another study (NCT02946463), researchers compared ravulizumab and eculizumab in 246 PNH patients who had not previously received a complement inhibitor.
Ravulizumab was noninferior to eculizumab for all endpoints (P<0.0001), including:
- Transfusion avoidance: 73.6% vs 66.1%; difference of 6.8% (95% CI: -4.66 to 18.14)
- LDH normalization: 53.6% vs 49.4%; odds ratio=1.19 (95% CI: 0.80 to 1.77)
- Percent reduction in LDH: -76.8% vs -76.0%; difference of -0.83% (95% CI: -5.21 to 3.56)
- Change in FACIT-Fatigue score: 7.07 vs 6.40; difference of 0.67 (95% CI: -1.21 to 2.55)
- Breakthrough hemolysis: 4.0% vs 10.7%; difference of -6.7% (95% CI: -14.21 to 0.18)
- Stabilized hemoglobin: 68.0% vs 64.5%; difference of 2.9 (95% CI: -8.80 to 14.64).
Safety in both trials
The safety data from both trials included 441 adults who received ravulizumab (n=222) or eculizumab (n=219) for a median of 6 months.
The most frequent adverse events in both arms (ravulizumab and eculizumab, respectively) were upper respiratory tract infection (39% and 39%) and headache (32% and 26%).
Serious adverse events occurred in 15 (6.8%) patients treated with ravulizumab. These events included hyperthermia and pyrexia.
There was one fatal case of sepsis in a patient treated with ravulizumab.
The U.S. Food and Drug Administration (FDA) has approved ravulizumab-cwvz (Ultomiris) to treat adults with paroxysmal nocturnal hemoglobinuria (PNH).
Ravulizumab is a long-acting C5 complement inhibitor, administered every 8 weeks, that has been shown to prevent hemolysis.
The prescribing information for ravulizumab includes a boxed warning stating that meningococcal infections/sepsis have occurred in patients treated with the drug, and these adverse effects can become life-threatening or fatal if not recognized and treated early.
Ravulizumab is available only through a restricted program under a Risk Evaluation and Mitigation Strategy.
The FDA previously granted the application for ravulizumab priority review, and the product received orphan drug designation from the FDA.
The FDA granted the approval of ravulizumab to Alexion Pharmaceuticals.
The FDA’s approval of ravulizumab is based on results from two phase 3 studies, one in patients who had previously received treatment with a complement inhibitor and one in patients who were complement-inhibitor-naïve. Both studies were recently published in Blood.
Efficacy in inhibitor-experienced patients
In one study (NCT03056040), researchers compared ravulizumab administered every 8 weeks to eculizumab administered every 2 weeks in complement-inhibitor-experienced patients.
The trial included 195 PNH patients who were taking eculizumab for more than 6 months. They were randomized to switch to ravulizumab (n=97) or continue on eculizumab (n=98).
Ravulizumab proved noninferior to eculizumab for all endpoints studied (P<0.0006), including:
- Percentage change in lactate dehydrogenase (LDH): difference, 9.21% (95% CI: -0.42 to 18.84; P=0.058 for superiority)
- Breakthrough hemolysis: difference, 5.1 (95% CI: -8.89 to 18.99)
- Change in FACIT-Fatigue score: difference, 1.47 (95% CI: -0.21 to 3.15)
- Transfusion avoidance: difference, 5.5 (95% CI: -4.27 to 15.68)
- Stabilized hemoglobin: difference, 1.4 (95% CI: -10.41 to 13.31).
Efficacy in inhibitor-naïve patients
In another study (NCT02946463), researchers compared ravulizumab and eculizumab in 246 PNH patients who had not previously received a complement inhibitor.
Ravulizumab was noninferior to eculizumab for all endpoints (P<0.0001), including:
- Transfusion avoidance: 73.6% vs 66.1%; difference of 6.8% (95% CI: -4.66 to 18.14)
- LDH normalization: 53.6% vs 49.4%; odds ratio=1.19 (95% CI: 0.80 to 1.77)
- Percent reduction in LDH: -76.8% vs -76.0%; difference of -0.83% (95% CI: -5.21 to 3.56)
- Change in FACIT-Fatigue score: 7.07 vs 6.40; difference of 0.67 (95% CI: -1.21 to 2.55)
- Breakthrough hemolysis: 4.0% vs 10.7%; difference of -6.7% (95% CI: -14.21 to 0.18)
- Stabilized hemoglobin: 68.0% vs 64.5%; difference of 2.9 (95% CI: -8.80 to 14.64).
Safety in both trials
The safety data from both trials included 441 adults who received ravulizumab (n=222) or eculizumab (n=219) for a median of 6 months.
The most frequent adverse events in both arms (ravulizumab and eculizumab, respectively) were upper respiratory tract infection (39% and 39%) and headache (32% and 26%).
Serious adverse events occurred in 15 (6.8%) patients treated with ravulizumab. These events included hyperthermia and pyrexia.
There was one fatal case of sepsis in a patient treated with ravulizumab.
The U.S. Food and Drug Administration (FDA) has approved ravulizumab-cwvz (Ultomiris) to treat adults with paroxysmal nocturnal hemoglobinuria (PNH).
Ravulizumab is a long-acting C5 complement inhibitor, administered every 8 weeks, that has been shown to prevent hemolysis.
The prescribing information for ravulizumab includes a boxed warning stating that meningococcal infections/sepsis have occurred in patients treated with the drug, and these adverse effects can become life-threatening or fatal if not recognized and treated early.
Ravulizumab is available only through a restricted program under a Risk Evaluation and Mitigation Strategy.
The FDA previously granted the application for ravulizumab priority review, and the product received orphan drug designation from the FDA.
The FDA granted the approval of ravulizumab to Alexion Pharmaceuticals.
The FDA’s approval of ravulizumab is based on results from two phase 3 studies, one in patients who had previously received treatment with a complement inhibitor and one in patients who were complement-inhibitor-naïve. Both studies were recently published in Blood.
Efficacy in inhibitor-experienced patients
In one study (NCT03056040), researchers compared ravulizumab administered every 8 weeks to eculizumab administered every 2 weeks in complement-inhibitor-experienced patients.
The trial included 195 PNH patients who were taking eculizumab for more than 6 months. They were randomized to switch to ravulizumab (n=97) or continue on eculizumab (n=98).
Ravulizumab proved noninferior to eculizumab for all endpoints studied (P<0.0006), including:
- Percentage change in lactate dehydrogenase (LDH): difference, 9.21% (95% CI: -0.42 to 18.84; P=0.058 for superiority)
- Breakthrough hemolysis: difference, 5.1 (95% CI: -8.89 to 18.99)
- Change in FACIT-Fatigue score: difference, 1.47 (95% CI: -0.21 to 3.15)
- Transfusion avoidance: difference, 5.5 (95% CI: -4.27 to 15.68)
- Stabilized hemoglobin: difference, 1.4 (95% CI: -10.41 to 13.31).
Efficacy in inhibitor-naïve patients
In another study (NCT02946463), researchers compared ravulizumab and eculizumab in 246 PNH patients who had not previously received a complement inhibitor.
Ravulizumab was noninferior to eculizumab for all endpoints (P<0.0001), including:
- Transfusion avoidance: 73.6% vs 66.1%; difference of 6.8% (95% CI: -4.66 to 18.14)
- LDH normalization: 53.6% vs 49.4%; odds ratio=1.19 (95% CI: 0.80 to 1.77)
- Percent reduction in LDH: -76.8% vs -76.0%; difference of -0.83% (95% CI: -5.21 to 3.56)
- Change in FACIT-Fatigue score: 7.07 vs 6.40; difference of 0.67 (95% CI: -1.21 to 2.55)
- Breakthrough hemolysis: 4.0% vs 10.7%; difference of -6.7% (95% CI: -14.21 to 0.18)
- Stabilized hemoglobin: 68.0% vs 64.5%; difference of 2.9 (95% CI: -8.80 to 14.64).
Safety in both trials
The safety data from both trials included 441 adults who received ravulizumab (n=222) or eculizumab (n=219) for a median of 6 months.
The most frequent adverse events in both arms (ravulizumab and eculizumab, respectively) were upper respiratory tract infection (39% and 39%) and headache (32% and 26%).
Serious adverse events occurred in 15 (6.8%) patients treated with ravulizumab. These events included hyperthermia and pyrexia.
There was one fatal case of sepsis in a patient treated with ravulizumab.
Black women more likely to have open hysterectomies
Black women were more likely than white women to undergo open hysterectomy, according to a recent analysis of national surgical data by Amy L. Alexander, MD, MS, of Northwestern University, Chicago, and her colleagues.
Even after the researchers controlled for many factors that might influence surgical approach, such as comorbidities and body mass index, black women had an odds ratio of 2.02 to receive open, rather than laparoscopic, hysterectomy (95% confidence interval, 1.85-2.20), according to a study in Obstetrics & Gynecology.
The analysis of the targeted hysterectomy file in the National Surgical Quality Improvement Program (NSQIP) database showed that, of 15,316 women who had hysterectomy for nonmalignant indications, the 25% who were black also were more likely to have major complications with open procedures. Such complications as sepsis, wound dehiscence, prolonged intubation, and death were seen in 4% of the black women, versus 2% of the white women receiving open hysterectomy (P less than .001). Minor complications, such as urinary tract infections, superficial wound infections, and blood transfusions, also were more common for black women having open procedures (11% vs. 7%; P less than .001).
The study used a large national database with detailed information about comorbidities and patient characteristics to look at racial disparities in surgical route and complications for the second-most-common surgical procedure women receive on the United States. The results, said Dr. Alexander and her coauthors, confirm and extend previous work showing these disparities.
Black women are known to have more diabetes and hypertension, as well as higher rates of obesity, compared with white women, wrote Dr. Alexander and her coauthors. Even after they controlled for all these variables, black women still had significantly higher odds of having complications from hysterectomy: The odds ratios for major and minor complications were 1.56 and 1.27, respectively.
Uterine weight was included and tracked as a binary variable, with large uteri considered those weighing 250 g or more. Making uterine weight a binary, rather than continuous or categorical variable, didn’t significantly change results, and realistically mirrors a surgeon’s assessment of a uterus as “large” or “small” when making treatment decisions, Dr. Alexander and her coauthors said.
Because the median weight of uteri from black women was more than double the weight of those from white women (262 g vs. 123 g), the investigators also performed an analysis looking just at women with uterine weight less than 250 g, to ensure that uterine weight alone was not accounting for much of the disparity. In this analysis, the black patients still had an adjusted odds ratio of 1.84 for receiving an open procedure.
“Some of the postoperative complications experienced by black women are likely attributable to the fact that black women are more likely to undergo an open hysterectomy,” noted Dr. Alexander and her colleagues. “However, because black race is still associated with a higher odds of complications, even when adjusting for hysterectomy route, there are other contributing factors that warrant further investigation.” Among these factors, they said, may be access to care and quality of care while hospitalized.
The study’s strengths include the use of the NSQIP database’s prospectively collected data to construct the cohort study; the data base included “important patient-level factors such as uterine size, obesity, and comorbidities not previously available in other secondary data set studies,” noted Dr. Alexander and her colleagues. But the possibility of unmeasured bias persists, they said, and such variables as regional practice patterns and surgeon experience and procedure volume could not be detected from the NSQIP data on hand.
“This study suggests that an important step to reduce the disparity in route of surgery and postoperative complications is to increase access to and use of minimally invasive surgery,” wrote Dr. Alexander and her coauthors.
The study was funded by the National Institutes of Health. Dr. Alexander and her colleagues reported no conflicts of interest.
SOURCE: Alexander AL et al. Obstet Gynecol. 2018 Dec 4. doi: 10.1097/AOG.0000000000002990
The discrepancies reported by Dr. Alexander and her colleagues between black and white women undergoing hysterectomy for nonmalignant reasons mirror discrepancies found for other oncologic procedures, including colectomy and prostatectomy.
Findings of the current study show that black women continue to be less likely to receive more modern, minimally invasive procedures, and that outcomes are worse for these women, even after controlling for factors that may contribute to complications.
The American College of Obstetricians and Gynecologists (ACOG) has looked at this issue. In a 2015 opinion, an ACOG committee looked at three categories of factors contributing to racial disparities in women’s health care. Patient-level factors include such things as genetics, medical comorbidities, and patient preferences and adherence. Health care system–level factors include insurance status and geographic barriers to accessing care, while stereotyping and implicit bias on the part of practitioners constitute the third set of factors.
All these factors are likely in play for the discrepancies seen in hysterectomy rates. Those who care for black women need to understand the long, shameful history of how black Americans were mistreated, undertreated, and used for medical experimentation without consent. This past shapes present care and contributes to the difficulty black patients have trusting today’s health care systems.
The important question today is how individual health care providers will address their own biases, learn from the past, and move forward to do better for our black patients.
Shanna N. Wingo, MD , is an ob.gyn. and a gynecologic oncologist in private practice in Phoenix. She had no potential conflicts of interest. These remarks were drawn from an editorial accompanying the study by Dr. Alexander et al.( Obstet Gynecol. 2018 Dec 4;133[1]:4-5 ).
The discrepancies reported by Dr. Alexander and her colleagues between black and white women undergoing hysterectomy for nonmalignant reasons mirror discrepancies found for other oncologic procedures, including colectomy and prostatectomy.
Findings of the current study show that black women continue to be less likely to receive more modern, minimally invasive procedures, and that outcomes are worse for these women, even after controlling for factors that may contribute to complications.
The American College of Obstetricians and Gynecologists (ACOG) has looked at this issue. In a 2015 opinion, an ACOG committee looked at three categories of factors contributing to racial disparities in women’s health care. Patient-level factors include such things as genetics, medical comorbidities, and patient preferences and adherence. Health care system–level factors include insurance status and geographic barriers to accessing care, while stereotyping and implicit bias on the part of practitioners constitute the third set of factors.
All these factors are likely in play for the discrepancies seen in hysterectomy rates. Those who care for black women need to understand the long, shameful history of how black Americans were mistreated, undertreated, and used for medical experimentation without consent. This past shapes present care and contributes to the difficulty black patients have trusting today’s health care systems.
The important question today is how individual health care providers will address their own biases, learn from the past, and move forward to do better for our black patients.
Shanna N. Wingo, MD , is an ob.gyn. and a gynecologic oncologist in private practice in Phoenix. She had no potential conflicts of interest. These remarks were drawn from an editorial accompanying the study by Dr. Alexander et al.( Obstet Gynecol. 2018 Dec 4;133[1]:4-5 ).
The discrepancies reported by Dr. Alexander and her colleagues between black and white women undergoing hysterectomy for nonmalignant reasons mirror discrepancies found for other oncologic procedures, including colectomy and prostatectomy.
Findings of the current study show that black women continue to be less likely to receive more modern, minimally invasive procedures, and that outcomes are worse for these women, even after controlling for factors that may contribute to complications.
The American College of Obstetricians and Gynecologists (ACOG) has looked at this issue. In a 2015 opinion, an ACOG committee looked at three categories of factors contributing to racial disparities in women’s health care. Patient-level factors include such things as genetics, medical comorbidities, and patient preferences and adherence. Health care system–level factors include insurance status and geographic barriers to accessing care, while stereotyping and implicit bias on the part of practitioners constitute the third set of factors.
All these factors are likely in play for the discrepancies seen in hysterectomy rates. Those who care for black women need to understand the long, shameful history of how black Americans were mistreated, undertreated, and used for medical experimentation without consent. This past shapes present care and contributes to the difficulty black patients have trusting today’s health care systems.
The important question today is how individual health care providers will address their own biases, learn from the past, and move forward to do better for our black patients.
Shanna N. Wingo, MD , is an ob.gyn. and a gynecologic oncologist in private practice in Phoenix. She had no potential conflicts of interest. These remarks were drawn from an editorial accompanying the study by Dr. Alexander et al.( Obstet Gynecol. 2018 Dec 4;133[1]:4-5 ).
Black women were more likely than white women to undergo open hysterectomy, according to a recent analysis of national surgical data by Amy L. Alexander, MD, MS, of Northwestern University, Chicago, and her colleagues.
Even after the researchers controlled for many factors that might influence surgical approach, such as comorbidities and body mass index, black women had an odds ratio of 2.02 to receive open, rather than laparoscopic, hysterectomy (95% confidence interval, 1.85-2.20), according to a study in Obstetrics & Gynecology.
The analysis of the targeted hysterectomy file in the National Surgical Quality Improvement Program (NSQIP) database showed that, of 15,316 women who had hysterectomy for nonmalignant indications, the 25% who were black also were more likely to have major complications with open procedures. Such complications as sepsis, wound dehiscence, prolonged intubation, and death were seen in 4% of the black women, versus 2% of the white women receiving open hysterectomy (P less than .001). Minor complications, such as urinary tract infections, superficial wound infections, and blood transfusions, also were more common for black women having open procedures (11% vs. 7%; P less than .001).
The study used a large national database with detailed information about comorbidities and patient characteristics to look at racial disparities in surgical route and complications for the second-most-common surgical procedure women receive on the United States. The results, said Dr. Alexander and her coauthors, confirm and extend previous work showing these disparities.
Black women are known to have more diabetes and hypertension, as well as higher rates of obesity, compared with white women, wrote Dr. Alexander and her coauthors. Even after they controlled for all these variables, black women still had significantly higher odds of having complications from hysterectomy: The odds ratios for major and minor complications were 1.56 and 1.27, respectively.
Uterine weight was included and tracked as a binary variable, with large uteri considered those weighing 250 g or more. Making uterine weight a binary, rather than continuous or categorical variable, didn’t significantly change results, and realistically mirrors a surgeon’s assessment of a uterus as “large” or “small” when making treatment decisions, Dr. Alexander and her coauthors said.
Because the median weight of uteri from black women was more than double the weight of those from white women (262 g vs. 123 g), the investigators also performed an analysis looking just at women with uterine weight less than 250 g, to ensure that uterine weight alone was not accounting for much of the disparity. In this analysis, the black patients still had an adjusted odds ratio of 1.84 for receiving an open procedure.
“Some of the postoperative complications experienced by black women are likely attributable to the fact that black women are more likely to undergo an open hysterectomy,” noted Dr. Alexander and her colleagues. “However, because black race is still associated with a higher odds of complications, even when adjusting for hysterectomy route, there are other contributing factors that warrant further investigation.” Among these factors, they said, may be access to care and quality of care while hospitalized.
The study’s strengths include the use of the NSQIP database’s prospectively collected data to construct the cohort study; the data base included “important patient-level factors such as uterine size, obesity, and comorbidities not previously available in other secondary data set studies,” noted Dr. Alexander and her colleagues. But the possibility of unmeasured bias persists, they said, and such variables as regional practice patterns and surgeon experience and procedure volume could not be detected from the NSQIP data on hand.
“This study suggests that an important step to reduce the disparity in route of surgery and postoperative complications is to increase access to and use of minimally invasive surgery,” wrote Dr. Alexander and her coauthors.
The study was funded by the National Institutes of Health. Dr. Alexander and her colleagues reported no conflicts of interest.
SOURCE: Alexander AL et al. Obstet Gynecol. 2018 Dec 4. doi: 10.1097/AOG.0000000000002990
Black women were more likely than white women to undergo open hysterectomy, according to a recent analysis of national surgical data by Amy L. Alexander, MD, MS, of Northwestern University, Chicago, and her colleagues.
Even after the researchers controlled for many factors that might influence surgical approach, such as comorbidities and body mass index, black women had an odds ratio of 2.02 to receive open, rather than laparoscopic, hysterectomy (95% confidence interval, 1.85-2.20), according to a study in Obstetrics & Gynecology.
The analysis of the targeted hysterectomy file in the National Surgical Quality Improvement Program (NSQIP) database showed that, of 15,316 women who had hysterectomy for nonmalignant indications, the 25% who were black also were more likely to have major complications with open procedures. Such complications as sepsis, wound dehiscence, prolonged intubation, and death were seen in 4% of the black women, versus 2% of the white women receiving open hysterectomy (P less than .001). Minor complications, such as urinary tract infections, superficial wound infections, and blood transfusions, also were more common for black women having open procedures (11% vs. 7%; P less than .001).
The study used a large national database with detailed information about comorbidities and patient characteristics to look at racial disparities in surgical route and complications for the second-most-common surgical procedure women receive on the United States. The results, said Dr. Alexander and her coauthors, confirm and extend previous work showing these disparities.
Black women are known to have more diabetes and hypertension, as well as higher rates of obesity, compared with white women, wrote Dr. Alexander and her coauthors. Even after they controlled for all these variables, black women still had significantly higher odds of having complications from hysterectomy: The odds ratios for major and minor complications were 1.56 and 1.27, respectively.
Uterine weight was included and tracked as a binary variable, with large uteri considered those weighing 250 g or more. Making uterine weight a binary, rather than continuous or categorical variable, didn’t significantly change results, and realistically mirrors a surgeon’s assessment of a uterus as “large” or “small” when making treatment decisions, Dr. Alexander and her coauthors said.
Because the median weight of uteri from black women was more than double the weight of those from white women (262 g vs. 123 g), the investigators also performed an analysis looking just at women with uterine weight less than 250 g, to ensure that uterine weight alone was not accounting for much of the disparity. In this analysis, the black patients still had an adjusted odds ratio of 1.84 for receiving an open procedure.
“Some of the postoperative complications experienced by black women are likely attributable to the fact that black women are more likely to undergo an open hysterectomy,” noted Dr. Alexander and her colleagues. “However, because black race is still associated with a higher odds of complications, even when adjusting for hysterectomy route, there are other contributing factors that warrant further investigation.” Among these factors, they said, may be access to care and quality of care while hospitalized.
The study’s strengths include the use of the NSQIP database’s prospectively collected data to construct the cohort study; the data base included “important patient-level factors such as uterine size, obesity, and comorbidities not previously available in other secondary data set studies,” noted Dr. Alexander and her colleagues. But the possibility of unmeasured bias persists, they said, and such variables as regional practice patterns and surgeon experience and procedure volume could not be detected from the NSQIP data on hand.
“This study suggests that an important step to reduce the disparity in route of surgery and postoperative complications is to increase access to and use of minimally invasive surgery,” wrote Dr. Alexander and her coauthors.
The study was funded by the National Institutes of Health. Dr. Alexander and her colleagues reported no conflicts of interest.
SOURCE: Alexander AL et al. Obstet Gynecol. 2018 Dec 4. doi: 10.1097/AOG.0000000000002990
FROM OBSTETRICS & GYNECOLOGY
Key clinical point: Black women were more likely than white women were to have open hysterectomies.
Major finding:
Study details: Analysis of prospectively collected hysterectomy data of 15,136 women in the NSQIP database.
Disclosures: The study was funded by the National Institutes of Health. Dr. Alexander and her coauthors reported no conflicts of interest.
Source: Alexander AL et al. Obstet Gynecol. 2018 Dec 4. doi: 10.1097/AOG0000000000002990.