User login
How should asymptomatic hypertension be managed in the hospital?
Case
A 62-year-old man with diabetes mellitus and hypertension presents with painful erythema of the left lower extremity and is admitted for purulent cellulitis. During the first evening of admission, he has increased left lower extremity pain and nursing reports a blood pressure of 188/96 mm Hg. He denies dyspnea, chest pain, visual changes, confusion, or severe headache.
Background
The prevalence of hypertension in the outpatient setting in the United States is estimated at 29% by the National Health and Nutrition Examination Survey.1 Hypertension generally is defined in the outpatient setting as an average blood pressure reading greater than or equal to140/90 mm Hg on two or more separate occasions.2 There is no consensus on the definition of hypertension in the inpatient setting; however, hypertensive urgency often is defined as a sustained blood pressure above the range of 180-200 mm Hg systolic and/or 110-120 mm Hg diastolic without target organ damage, and hypertensive emergency has a similar blood pressure range, but with evidence of target organ damage.3

The evidence
There are several clinical trials to suggest that the ambulatory treatment of chronic hypertension reduces the incidence of myocardial infarction, cerebrovascular accident (CVA), and heart failure7,8; however, these trials are difficult to extrapolate to acutely hospitalized patients.6 Overall, evidence on the appropriate management of asymptomatic hypertension in the inpatient setting is lacking.
Some evidence suggests we often are overly aggressive with intravenous antihypertensives without clinical indications in the inpatient setting.3,9,10 For example, Campbell et al. prospectively examined the use of intravenous hydralazine for the treatment of asymptomatic hypertension in 94 hospitalized patients.10 It was determined that in 90 of those patients, there was no clinical indication to use intravenous hydralazine and 17 patients experienced adverse events from hypotension, which included dizziness/light-headedness, syncope, and chest pain.10 They recommend against the use of intravenous hydralazine in the setting of asymptomatic hypertension because of its risk of adverse events with the rapid lowering of blood pressure.10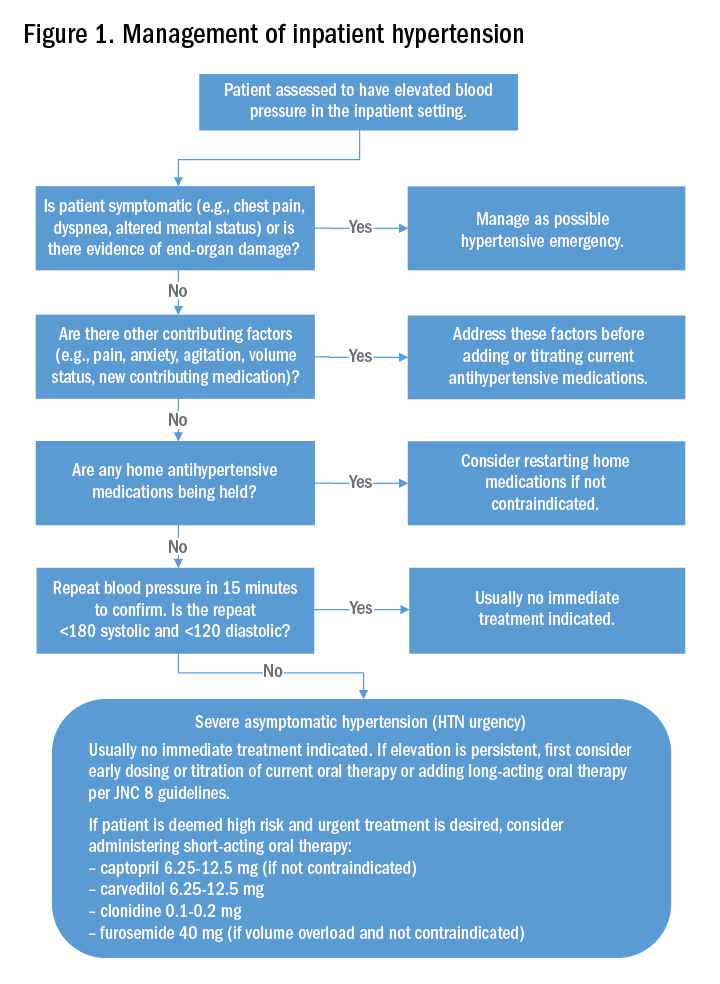
Weder and Erickson performed a retrospective review on the use of intravenous hydralazine and labetalol administration in 2,189 hospitalized patients.3 They found that only 3% of those patients had symptoms indicating the need for IV antihypertensives and that the length of stay was several days longer in those who had received IV antihypertensives.3
Other studies have examined the role of oral antihypertensives for management of asymptomatic hypertension in the inpatient setting. A systematic review and meta-analysis by Souza et al. assessed the use of oral pharmacotherapy for hypertensive urgency.11 Sixteen randomized clinical trials were reviewed and it was determined that angiotensin-converting enzyme inhibitors (ACE-I) had a superior effect in treating hypertensive urgencies.11 The most common side effect of using an ACE-I was a bad taste in patient’s mouths, and researchers did not observe side effects that were similar as those seen with the use of IV antihypertensives.11
Further, Jaker et al. performed a randomized, double-blind prospective study comparing a single dose of oral nifedipine with oral clonidine for the treatment of hypertensive urgency in 51 patients.12 Both the oral nifedipine and oral clonidine were extremely effective in reducing blood pressure fairly safely.12 However, the rapid lowering of blood pressure with oral nifedipine was concerning to Grossman et al.13 In their literature review of the side effects of oral and sublingual nifedipine, they found that it was one of the most common therapeutic interventions for hypertensive urgency or emergency.13 However, it was potentially dangerous because of the inconsistent blood pressure response after nifedipine administration, particularly with the sublingual form.13 CVAs, acute MIs, and even death were the reported adverse events with the use of oral and sublingual nifedipine.13 Because of that, the investigators recommend against the use of oral or sublingual nifedipine in hypertensive urgency or emergency and suggest using other oral antihypertensive agents instead.13

Typically, if the patient’s blood pressure remains elevated despite these efforts, no urgent treatment is indicated and we recommend close monitoring of the patient’s blood pressure during the hospitalization. If hypertension persists, the next best step would be to titrate a patient’s current oral antihypertensive therapy or to start a long-acting antihypertensive therapy per the JNC 8 (Eighth Joint National Committee) guidelines. It should be noted that, in those patients that are high risk, such as those with known coronary artery disease, heart failure, or prior hemorrhagic CVA, a short-acting oral antihypertensive such as captopril, carvedilol, clonidine, or furosemide should be considered.
Back to the case
The patient’s pain was treated with oral oxycodone. He received no oral or intravenous antihypertensive therapy, and the following morning, his blood pressure improved to 145/95 mm Hg. Based on our suggested approach in Fig. 1, the patient would require no acute treatment despite an improved but elevated blood pressure. We continued to monitor his blood pressure and despite adequate pain control, his blood pressure remain persistently elevated. Thus, per the JNC 8 guidelines, we started him on a long-acting antihypertensive, which improved his blood pressure to 123/78 at the time of discharge.
Bottom line
Management of asymptomatic hypertension in the hospital begins with addressing contributing factors, reviewing held home medications – and rarely – urgent oral pharmacotherapy.
Dr. Lippert is PGY-3 in the department of internal medicine at the University of Kentucky, Lexington. Dr. Bailey is associate professor of medicine at the University of Kentucky. Dr. Gray is associate professor of medicine at the University of Kentucky.
References
1. Yoon S, Fryar C, Carroll M. Hypertension prevalence and control among adults: United States, 2011-2014. 2015. Accessed Oct 2, 2017.
2. Poulter NR, Prabhakaran D, Caulfield M. Hypertension. Lancet. 2015;386(9995):801-12.
3. Weder AB, Erickson S. Treatment of hypertension in the inpatient setting: Use of intravenous labetalol and hydralazine. J Clin Hypertens. 2010;12(1):29-33.
4. Axon RN, Cousineau L, Egan BM. Prevalence and management of hypertension in the inpatient setting: A systematic review. J Hosp Med. 2011;6(7):417-22.
5. Weiss AJ, Elixhauser A. Overview of hospital stays in the United States, 2012. Statistical brief; 2014 Oct. Accessed Oct 2, 2017.
6. Weder AB. Treating acute hypertension in the hospital: a Lacuna in the guidelines. Hypertension. 2011;57(1):18-20.
7. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Hypertension and of the European Society of Cardiology. J Hypertens. 2013 Jul;31(7):1281-357.
8. James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-20.
9. Lipari M, Moser LR, Petrovitch EA, et al. As-needed intravenous antihypertensive therapy and blood pressure control. J Hosp Med. 2016;11(3):193-8.
10. Campbell P, Baker WL, Bendel SD, et al. Intravenous hydralazine for blood pressure management in the hospitalized patient: Its use is often unjustified. J Am Soc Hypertens. 2011;5(6):473-7.
11. Souza LM, Riera R, Saconato H, et al. Oral drugs for hypertensive urgencies: Systematic review and meta-analysis. Sao Paulo Med J. 2009;127(6):366-72.
12. Jaker M, Atkin S, Soto M, et al. Oral nifedipine vs. oral clonidine in the treatment of urgent hypertension. Arch Intern Med. 1989;149(2):260-5.
13. Grossman E, Messerli FH, Grodzicki T, et al. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328-31.
14. Axon RN, Turner M, Buckley R. An update on inpatient hypertension management. Curr Cardiol Rep. 2015;17(11):94.
Additional Reading
1. Axon RN, Turner M, Buckley R. An update on inpatient hypertension management. Curr Cardiol Rep. 2015 Nov;17(11):94.
2. Herzog E, Frankenberger O, Aziz E, et al. A novel pathway for the management of hypertension for hospitalized patients. Crit Pathw Cardiol. 2007;6(4):150-60.
3. Sharma P, Shrestha A. Inpatient hypertension management. ACP Hospitalist. Aug 2014.
Quiz
Asymptomatic hypertension in the hospital
Hypertension is a common focus in the ambulatory setting because of its increased risk for cardiovascular events. Evidence for management in the inpatient setting is limited but does suggest a more conservative approach.
Question: A 75-year-old woman is hospitalized after sustaining a mechanical fall and subsequent right femoral neck fracture. She has a history of hypertension and hyperlipidemia for which she takes amlodipine and atorvastatin. Her blood pressure initially on admission is 170/102 mm Hg, and she is asymptomatic other than severe right hip pain. Her amlodipine and atorvastatin are resumed. Repeat blood pressures after resuming her amlodipine are still elevated with an average blood pressure reading of 168/98 mm Hg. Which of the following would be the next best step in treating this patient?
A. A one-time dose of intravenous hydralazine at 10 mg to reduce blood pressure by 25% over next several hours.
B. A one-time dose of oral clonidine at 0.1 mg to reduce blood pressure by 25% over next several hours.
C. Start a second daily antihypertensive with lisinopril 5 mg daily.
D. Address the patient’s pain.
The best answer is choice D. The patient’s hypertension is likely aggravated by her hip pain. Thus, the best course of action would be to address her pain.
Choice A is not the best answer as an intravenous antihypertensive is not indicated in this patient as she is asymptomatic and exhibiting no signs/symptoms of end-organ damage.
Choice B is not the best answer as by addressing her pain it is likely her blood pressure will improve. Urgent use of oral antihypertensives would not be indicated.
Choice C is not the best answer as patient has acute elevation of blood pressure in setting of a right femoral neck fracture and pain. Her blood pressure will likely improve after addressing her pain. However, if there is persistent blood pressure elevation, starting long-acting antihypertensive would be appropriate per JNC 8 guidelines.
Key Points
- Evidence for treatment of inpatient asymptomatic hypertension is lacking.
- The use of intravenous antihypertensives in the setting of inpatient asymptomatic hypertension is inappropriate and may be harmful.
- A conservative approach for inpatient asymptomatic hypertension should be employed by addressing contributing factors and reviewing held home antihypertensive medications prior to administering any oral antihypertensive pharmacotherapy.
Case
A 62-year-old man with diabetes mellitus and hypertension presents with painful erythema of the left lower extremity and is admitted for purulent cellulitis. During the first evening of admission, he has increased left lower extremity pain and nursing reports a blood pressure of 188/96 mm Hg. He denies dyspnea, chest pain, visual changes, confusion, or severe headache.
Background
The prevalence of hypertension in the outpatient setting in the United States is estimated at 29% by the National Health and Nutrition Examination Survey.1 Hypertension generally is defined in the outpatient setting as an average blood pressure reading greater than or equal to140/90 mm Hg on two or more separate occasions.2 There is no consensus on the definition of hypertension in the inpatient setting; however, hypertensive urgency often is defined as a sustained blood pressure above the range of 180-200 mm Hg systolic and/or 110-120 mm Hg diastolic without target organ damage, and hypertensive emergency has a similar blood pressure range, but with evidence of target organ damage.3
The evidence
There are several clinical trials to suggest that the ambulatory treatment of chronic hypertension reduces the incidence of myocardial infarction, cerebrovascular accident (CVA), and heart failure7,8; however, these trials are difficult to extrapolate to acutely hospitalized patients.6 Overall, evidence on the appropriate management of asymptomatic hypertension in the inpatient setting is lacking.
Some evidence suggests we often are overly aggressive with intravenous antihypertensives without clinical indications in the inpatient setting.3,9,10 For example, Campbell et al. prospectively examined the use of intravenous hydralazine for the treatment of asymptomatic hypertension in 94 hospitalized patients.10 It was determined that in 90 of those patients, there was no clinical indication to use intravenous hydralazine and 17 patients experienced adverse events from hypotension, which included dizziness/light-headedness, syncope, and chest pain.10 They recommend against the use of intravenous hydralazine in the setting of asymptomatic hypertension because of its risk of adverse events with the rapid lowering of blood pressure.10
Weder and Erickson performed a retrospective review on the use of intravenous hydralazine and labetalol administration in 2,189 hospitalized patients.3 They found that only 3% of those patients had symptoms indicating the need for IV antihypertensives and that the length of stay was several days longer in those who had received IV antihypertensives.3
Other studies have examined the role of oral antihypertensives for management of asymptomatic hypertension in the inpatient setting. A systematic review and meta-analysis by Souza et al. assessed the use of oral pharmacotherapy for hypertensive urgency.11 Sixteen randomized clinical trials were reviewed and it was determined that angiotensin-converting enzyme inhibitors (ACE-I) had a superior effect in treating hypertensive urgencies.11 The most common side effect of using an ACE-I was a bad taste in patient’s mouths, and researchers did not observe side effects that were similar as those seen with the use of IV antihypertensives.11
Further, Jaker et al. performed a randomized, double-blind prospective study comparing a single dose of oral nifedipine with oral clonidine for the treatment of hypertensive urgency in 51 patients.12 Both the oral nifedipine and oral clonidine were extremely effective in reducing blood pressure fairly safely.12 However, the rapid lowering of blood pressure with oral nifedipine was concerning to Grossman et al.13 In their literature review of the side effects of oral and sublingual nifedipine, they found that it was one of the most common therapeutic interventions for hypertensive urgency or emergency.13 However, it was potentially dangerous because of the inconsistent blood pressure response after nifedipine administration, particularly with the sublingual form.13 CVAs, acute MIs, and even death were the reported adverse events with the use of oral and sublingual nifedipine.13 Because of that, the investigators recommend against the use of oral or sublingual nifedipine in hypertensive urgency or emergency and suggest using other oral antihypertensive agents instead.13
Typically, if the patient’s blood pressure remains elevated despite these efforts, no urgent treatment is indicated and we recommend close monitoring of the patient’s blood pressure during the hospitalization. If hypertension persists, the next best step would be to titrate a patient’s current oral antihypertensive therapy or to start a long-acting antihypertensive therapy per the JNC 8 (Eighth Joint National Committee) guidelines. It should be noted that, in those patients that are high risk, such as those with known coronary artery disease, heart failure, or prior hemorrhagic CVA, a short-acting oral antihypertensive such as captopril, carvedilol, clonidine, or furosemide should be considered.
Back to the case
The patient’s pain was treated with oral oxycodone. He received no oral or intravenous antihypertensive therapy, and the following morning, his blood pressure improved to 145/95 mm Hg. Based on our suggested approach in Fig. 1, the patient would require no acute treatment despite an improved but elevated blood pressure. We continued to monitor his blood pressure and despite adequate pain control, his blood pressure remain persistently elevated. Thus, per the JNC 8 guidelines, we started him on a long-acting antihypertensive, which improved his blood pressure to 123/78 at the time of discharge.
Bottom line
Management of asymptomatic hypertension in the hospital begins with addressing contributing factors, reviewing held home medications – and rarely – urgent oral pharmacotherapy.
Dr. Lippert is PGY-3 in the department of internal medicine at the University of Kentucky, Lexington. Dr. Bailey is associate professor of medicine at the University of Kentucky. Dr. Gray is associate professor of medicine at the University of Kentucky.
References
1. Yoon S, Fryar C, Carroll M. Hypertension prevalence and control among adults: United States, 2011-2014. 2015. Accessed Oct 2, 2017.
2. Poulter NR, Prabhakaran D, Caulfield M. Hypertension. Lancet. 2015;386(9995):801-12.
3. Weder AB, Erickson S. Treatment of hypertension in the inpatient setting: Use of intravenous labetalol and hydralazine. J Clin Hypertens. 2010;12(1):29-33.
4. Axon RN, Cousineau L, Egan BM. Prevalence and management of hypertension in the inpatient setting: A systematic review. J Hosp Med. 2011;6(7):417-22.
5. Weiss AJ, Elixhauser A. Overview of hospital stays in the United States, 2012. Statistical brief; 2014 Oct. Accessed Oct 2, 2017.
6. Weder AB. Treating acute hypertension in the hospital: a Lacuna in the guidelines. Hypertension. 2011;57(1):18-20.
7. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Hypertension and of the European Society of Cardiology. J Hypertens. 2013 Jul;31(7):1281-357.
8. James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-20.
9. Lipari M, Moser LR, Petrovitch EA, et al. As-needed intravenous antihypertensive therapy and blood pressure control. J Hosp Med. 2016;11(3):193-8.
10. Campbell P, Baker WL, Bendel SD, et al. Intravenous hydralazine for blood pressure management in the hospitalized patient: Its use is often unjustified. J Am Soc Hypertens. 2011;5(6):473-7.
11. Souza LM, Riera R, Saconato H, et al. Oral drugs for hypertensive urgencies: Systematic review and meta-analysis. Sao Paulo Med J. 2009;127(6):366-72.
12. Jaker M, Atkin S, Soto M, et al. Oral nifedipine vs. oral clonidine in the treatment of urgent hypertension. Arch Intern Med. 1989;149(2):260-5.
13. Grossman E, Messerli FH, Grodzicki T, et al. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328-31.
14. Axon RN, Turner M, Buckley R. An update on inpatient hypertension management. Curr Cardiol Rep. 2015;17(11):94.
Additional Reading
1. Axon RN, Turner M, Buckley R. An update on inpatient hypertension management. Curr Cardiol Rep. 2015 Nov;17(11):94.
2. Herzog E, Frankenberger O, Aziz E, et al. A novel pathway for the management of hypertension for hospitalized patients. Crit Pathw Cardiol. 2007;6(4):150-60.
3. Sharma P, Shrestha A. Inpatient hypertension management. ACP Hospitalist. Aug 2014.
Quiz
Asymptomatic hypertension in the hospital
Hypertension is a common focus in the ambulatory setting because of its increased risk for cardiovascular events. Evidence for management in the inpatient setting is limited but does suggest a more conservative approach.
Question: A 75-year-old woman is hospitalized after sustaining a mechanical fall and subsequent right femoral neck fracture. She has a history of hypertension and hyperlipidemia for which she takes amlodipine and atorvastatin. Her blood pressure initially on admission is 170/102 mm Hg, and she is asymptomatic other than severe right hip pain. Her amlodipine and atorvastatin are resumed. Repeat blood pressures after resuming her amlodipine are still elevated with an average blood pressure reading of 168/98 mm Hg. Which of the following would be the next best step in treating this patient?
A. A one-time dose of intravenous hydralazine at 10 mg to reduce blood pressure by 25% over next several hours.
B. A one-time dose of oral clonidine at 0.1 mg to reduce blood pressure by 25% over next several hours.
C. Start a second daily antihypertensive with lisinopril 5 mg daily.
D. Address the patient’s pain.
The best answer is choice D. The patient’s hypertension is likely aggravated by her hip pain. Thus, the best course of action would be to address her pain.
Choice A is not the best answer as an intravenous antihypertensive is not indicated in this patient as she is asymptomatic and exhibiting no signs/symptoms of end-organ damage.
Choice B is not the best answer as by addressing her pain it is likely her blood pressure will improve. Urgent use of oral antihypertensives would not be indicated.
Choice C is not the best answer as patient has acute elevation of blood pressure in setting of a right femoral neck fracture and pain. Her blood pressure will likely improve after addressing her pain. However, if there is persistent blood pressure elevation, starting long-acting antihypertensive would be appropriate per JNC 8 guidelines.
Key Points
- Evidence for treatment of inpatient asymptomatic hypertension is lacking.
- The use of intravenous antihypertensives in the setting of inpatient asymptomatic hypertension is inappropriate and may be harmful.
- A conservative approach for inpatient asymptomatic hypertension should be employed by addressing contributing factors and reviewing held home antihypertensive medications prior to administering any oral antihypertensive pharmacotherapy.
Case
A 62-year-old man with diabetes mellitus and hypertension presents with painful erythema of the left lower extremity and is admitted for purulent cellulitis. During the first evening of admission, he has increased left lower extremity pain and nursing reports a blood pressure of 188/96 mm Hg. He denies dyspnea, chest pain, visual changes, confusion, or severe headache.
Background
The prevalence of hypertension in the outpatient setting in the United States is estimated at 29% by the National Health and Nutrition Examination Survey.1 Hypertension generally is defined in the outpatient setting as an average blood pressure reading greater than or equal to140/90 mm Hg on two or more separate occasions.2 There is no consensus on the definition of hypertension in the inpatient setting; however, hypertensive urgency often is defined as a sustained blood pressure above the range of 180-200 mm Hg systolic and/or 110-120 mm Hg diastolic without target organ damage, and hypertensive emergency has a similar blood pressure range, but with evidence of target organ damage.3
The evidence
There are several clinical trials to suggest that the ambulatory treatment of chronic hypertension reduces the incidence of myocardial infarction, cerebrovascular accident (CVA), and heart failure7,8; however, these trials are difficult to extrapolate to acutely hospitalized patients.6 Overall, evidence on the appropriate management of asymptomatic hypertension in the inpatient setting is lacking.
Some evidence suggests we often are overly aggressive with intravenous antihypertensives without clinical indications in the inpatient setting.3,9,10 For example, Campbell et al. prospectively examined the use of intravenous hydralazine for the treatment of asymptomatic hypertension in 94 hospitalized patients.10 It was determined that in 90 of those patients, there was no clinical indication to use intravenous hydralazine and 17 patients experienced adverse events from hypotension, which included dizziness/light-headedness, syncope, and chest pain.10 They recommend against the use of intravenous hydralazine in the setting of asymptomatic hypertension because of its risk of adverse events with the rapid lowering of blood pressure.10
Weder and Erickson performed a retrospective review on the use of intravenous hydralazine and labetalol administration in 2,189 hospitalized patients.3 They found that only 3% of those patients had symptoms indicating the need for IV antihypertensives and that the length of stay was several days longer in those who had received IV antihypertensives.3
Other studies have examined the role of oral antihypertensives for management of asymptomatic hypertension in the inpatient setting. A systematic review and meta-analysis by Souza et al. assessed the use of oral pharmacotherapy for hypertensive urgency.11 Sixteen randomized clinical trials were reviewed and it was determined that angiotensin-converting enzyme inhibitors (ACE-I) had a superior effect in treating hypertensive urgencies.11 The most common side effect of using an ACE-I was a bad taste in patient’s mouths, and researchers did not observe side effects that were similar as those seen with the use of IV antihypertensives.11
Further, Jaker et al. performed a randomized, double-blind prospective study comparing a single dose of oral nifedipine with oral clonidine for the treatment of hypertensive urgency in 51 patients.12 Both the oral nifedipine and oral clonidine were extremely effective in reducing blood pressure fairly safely.12 However, the rapid lowering of blood pressure with oral nifedipine was concerning to Grossman et al.13 In their literature review of the side effects of oral and sublingual nifedipine, they found that it was one of the most common therapeutic interventions for hypertensive urgency or emergency.13 However, it was potentially dangerous because of the inconsistent blood pressure response after nifedipine administration, particularly with the sublingual form.13 CVAs, acute MIs, and even death were the reported adverse events with the use of oral and sublingual nifedipine.13 Because of that, the investigators recommend against the use of oral or sublingual nifedipine in hypertensive urgency or emergency and suggest using other oral antihypertensive agents instead.13
Typically, if the patient’s blood pressure remains elevated despite these efforts, no urgent treatment is indicated and we recommend close monitoring of the patient’s blood pressure during the hospitalization. If hypertension persists, the next best step would be to titrate a patient’s current oral antihypertensive therapy or to start a long-acting antihypertensive therapy per the JNC 8 (Eighth Joint National Committee) guidelines. It should be noted that, in those patients that are high risk, such as those with known coronary artery disease, heart failure, or prior hemorrhagic CVA, a short-acting oral antihypertensive such as captopril, carvedilol, clonidine, or furosemide should be considered.
Back to the case
The patient’s pain was treated with oral oxycodone. He received no oral or intravenous antihypertensive therapy, and the following morning, his blood pressure improved to 145/95 mm Hg. Based on our suggested approach in Fig. 1, the patient would require no acute treatment despite an improved but elevated blood pressure. We continued to monitor his blood pressure and despite adequate pain control, his blood pressure remain persistently elevated. Thus, per the JNC 8 guidelines, we started him on a long-acting antihypertensive, which improved his blood pressure to 123/78 at the time of discharge.
Bottom line
Management of asymptomatic hypertension in the hospital begins with addressing contributing factors, reviewing held home medications – and rarely – urgent oral pharmacotherapy.
Dr. Lippert is PGY-3 in the department of internal medicine at the University of Kentucky, Lexington. Dr. Bailey is associate professor of medicine at the University of Kentucky. Dr. Gray is associate professor of medicine at the University of Kentucky.
References
1. Yoon S, Fryar C, Carroll M. Hypertension prevalence and control among adults: United States, 2011-2014. 2015. Accessed Oct 2, 2017.
2. Poulter NR, Prabhakaran D, Caulfield M. Hypertension. Lancet. 2015;386(9995):801-12.
3. Weder AB, Erickson S. Treatment of hypertension in the inpatient setting: Use of intravenous labetalol and hydralazine. J Clin Hypertens. 2010;12(1):29-33.
4. Axon RN, Cousineau L, Egan BM. Prevalence and management of hypertension in the inpatient setting: A systematic review. J Hosp Med. 2011;6(7):417-22.
5. Weiss AJ, Elixhauser A. Overview of hospital stays in the United States, 2012. Statistical brief; 2014 Oct. Accessed Oct 2, 2017.
6. Weder AB. Treating acute hypertension in the hospital: a Lacuna in the guidelines. Hypertension. 2011;57(1):18-20.
7. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Hypertension and of the European Society of Cardiology. J Hypertens. 2013 Jul;31(7):1281-357.
8. James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-20.
9. Lipari M, Moser LR, Petrovitch EA, et al. As-needed intravenous antihypertensive therapy and blood pressure control. J Hosp Med. 2016;11(3):193-8.
10. Campbell P, Baker WL, Bendel SD, et al. Intravenous hydralazine for blood pressure management in the hospitalized patient: Its use is often unjustified. J Am Soc Hypertens. 2011;5(6):473-7.
11. Souza LM, Riera R, Saconato H, et al. Oral drugs for hypertensive urgencies: Systematic review and meta-analysis. Sao Paulo Med J. 2009;127(6):366-72.
12. Jaker M, Atkin S, Soto M, et al. Oral nifedipine vs. oral clonidine in the treatment of urgent hypertension. Arch Intern Med. 1989;149(2):260-5.
13. Grossman E, Messerli FH, Grodzicki T, et al. Should a moratorium be placed on sublingual nifedipine capsules given for hypertensive emergencies and pseudoemergencies? JAMA. 1996;276(16):1328-31.
14. Axon RN, Turner M, Buckley R. An update on inpatient hypertension management. Curr Cardiol Rep. 2015;17(11):94.
Additional Reading
1. Axon RN, Turner M, Buckley R. An update on inpatient hypertension management. Curr Cardiol Rep. 2015 Nov;17(11):94.
2. Herzog E, Frankenberger O, Aziz E, et al. A novel pathway for the management of hypertension for hospitalized patients. Crit Pathw Cardiol. 2007;6(4):150-60.
3. Sharma P, Shrestha A. Inpatient hypertension management. ACP Hospitalist. Aug 2014.
Quiz
Asymptomatic hypertension in the hospital
Hypertension is a common focus in the ambulatory setting because of its increased risk for cardiovascular events. Evidence for management in the inpatient setting is limited but does suggest a more conservative approach.
Question: A 75-year-old woman is hospitalized after sustaining a mechanical fall and subsequent right femoral neck fracture. She has a history of hypertension and hyperlipidemia for which she takes amlodipine and atorvastatin. Her blood pressure initially on admission is 170/102 mm Hg, and she is asymptomatic other than severe right hip pain. Her amlodipine and atorvastatin are resumed. Repeat blood pressures after resuming her amlodipine are still elevated with an average blood pressure reading of 168/98 mm Hg. Which of the following would be the next best step in treating this patient?
A. A one-time dose of intravenous hydralazine at 10 mg to reduce blood pressure by 25% over next several hours.
B. A one-time dose of oral clonidine at 0.1 mg to reduce blood pressure by 25% over next several hours.
C. Start a second daily antihypertensive with lisinopril 5 mg daily.
D. Address the patient’s pain.
The best answer is choice D. The patient’s hypertension is likely aggravated by her hip pain. Thus, the best course of action would be to address her pain.
Choice A is not the best answer as an intravenous antihypertensive is not indicated in this patient as she is asymptomatic and exhibiting no signs/symptoms of end-organ damage.
Choice B is not the best answer as by addressing her pain it is likely her blood pressure will improve. Urgent use of oral antihypertensives would not be indicated.
Choice C is not the best answer as patient has acute elevation of blood pressure in setting of a right femoral neck fracture and pain. Her blood pressure will likely improve after addressing her pain. However, if there is persistent blood pressure elevation, starting long-acting antihypertensive would be appropriate per JNC 8 guidelines.
Key Points
- Evidence for treatment of inpatient asymptomatic hypertension is lacking.
- The use of intravenous antihypertensives in the setting of inpatient asymptomatic hypertension is inappropriate and may be harmful.
- A conservative approach for inpatient asymptomatic hypertension should be employed by addressing contributing factors and reviewing held home antihypertensive medications prior to administering any oral antihypertensive pharmacotherapy.
Study shows value of pretransplant assessment of function, endurance
SALT LAKE CITY – Comprehensive assessment of functional status and endurance prior to allogeneic hematopoietic cell transplantation (HCT) provides important insights into posttransplant outcomes, and when used in combination with other measures may improve the patient selection process, a chart review suggests.
In 349 patients, results of the prospective assessment of physical performance and endurance, along with HCT Comorbidity Index (HCT-CI) score and Karnofsky Performance Scale score (KPS), were compared with day 100-plus nonrelapse mortality and overall survival. The measures were also compared with the novel measures of hospital length of stay, and death during HCT admission, Shabnam Rehman, MD, reported at the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
However, heart rate recovery in less than 3 minutes after performing 25 step-ups on each side was associated with shorter length of stay with 89% of those patients, compared with 11% of patients who were not able to recover their heart rate in less than 3 minutes, being discharged within 30 days, she said.
“Similarly, patients who are able to perform at least 11 sit-to-stands in 30 seconds are more likely to be discharged earlier (63% vs. 14% discharged within 30 days),” she said. “The converse is also true.”
That is, only 16% of those not able to recover their heart rate within 3 minutes had a 30-day or shorter stay, while 31% had at least a 60-day stay. In addition, just 13% of those with limited endurance had a 30-day stay or shorter, while 24% had at least a 60-day stay, she explained.
Further, patients with limited endurance, and those unable to perform 10 or more sit-to-stands in 30 seconds were more likely to die during their first transplant admission. Of those with limited endurance, 31% died during admission and 13% survived, and of those with good endurance 69% died during admission and 87% survived. Among patients who were unable to perform more than 10 sit-to-stands, 42% died during admission and 20% survived, and of those able to perform 11 or more, 38% died during admission, and 53% survived.
Overall survival was associated with age, KPS, HCT-CI, and age-adjusted HCT-CI, she noted.
“Patients who were over age 40 and more, and patients with a KPS of 60 or 70, belong to the high- to intermediate-risk group more likely to have decreased overall survival as has been shown in previous studies,” she said. “In addition to validating these findings, we also found that the semiquantitative measures, including pain and endurance, were also associated with overall survival.”
Those with pain present or limited endurance had significantly poorer overall survival (P = .007 and P = .01, respectively), and this finding was reflected in the quantitative measures of sit-to-stands (P = .01) and step-ups (P = .001), even when stratified by age-adjusted HCT-CI, she said.
In addition, a number of risk factors present at the pretreatment assessment were found to be significantly associated with requirement of an assistive device at discharge. These included pain, weakness in the lower extremities, use of an assistive device, inability to perform 25 step-ups and more than 10 sit-to-stands in 30 seconds, and limited endurance (P values ranging from .02 to less than .0001). Requirement of a device was associated with poorer overall survival (P = .03), she said.
Study participants were adults aged 18 years and older (median, 58 years) undergoing a first allogeneic HCT at a single center between 2010 and 2016. Most (83%) were older than age 40 years and 58% were men. About half (51%) had acute myeloid leukemia, and 64% overall had a KPS score of 60-70.
Physical therapists assessed physical performance of all patients within 4 weeks pre-HCT; testing included 25 7-inch step-ups on each side, unassisted sit-to-stands from an 18-inch chair in 30 seconds, weight-bearing ability, need for assistance with ambulation, motor strength in four extremities, sensory or coordination impairment, self-reported pain, and time to recovery of heart rate and oxygen saturation to pre-exercise levels.
“The HCT-CI is a validated tool that predicts nonrelapse mortality and overall survival, but comorbidity alone as a single domain is not a surrogate of overall health or reflection on the true biological age of our patients,” Dr. Rehman said, noting that studies have shown that functional impairment is associated with shorter overall survival, and that patient-reported physical functioning is predictive of overall survival. “The assessment of functional impairment becomes more critical given the aging U.S. population and older patients receiving transplant.”
Traditionally, functional status has been assessed via the KPS, which is a subjective measure and lacks precision, and the HCT-CI has not been studied in the context of the novel outcome measures addressed in the current study, she noted.
The current findings highlight the prognostic value of a more quantitative pretransplant assessment, which can help improve the patient selection process.
“We are in the process of analyzing some more outcomes of these pretransplant assessments, and developing a score that can, in conjunction with other predictive tools, help us improve pretransplant risk stratification and devise interventions that can improve the endurance and overall survival of the patients,” she concluded.
Dr. Rehman reported having no financial disclosures.
SOURCE: Rehman S et al., The 2018 BTM Tandem Meetings, Abstract 19.
SALT LAKE CITY – Comprehensive assessment of functional status and endurance prior to allogeneic hematopoietic cell transplantation (HCT) provides important insights into posttransplant outcomes, and when used in combination with other measures may improve the patient selection process, a chart review suggests.
In 349 patients, results of the prospective assessment of physical performance and endurance, along with HCT Comorbidity Index (HCT-CI) score and Karnofsky Performance Scale score (KPS), were compared with day 100-plus nonrelapse mortality and overall survival. The measures were also compared with the novel measures of hospital length of stay, and death during HCT admission, Shabnam Rehman, MD, reported at the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
However, heart rate recovery in less than 3 minutes after performing 25 step-ups on each side was associated with shorter length of stay with 89% of those patients, compared with 11% of patients who were not able to recover their heart rate in less than 3 minutes, being discharged within 30 days, she said.
“Similarly, patients who are able to perform at least 11 sit-to-stands in 30 seconds are more likely to be discharged earlier (63% vs. 14% discharged within 30 days),” she said. “The converse is also true.”
That is, only 16% of those not able to recover their heart rate within 3 minutes had a 30-day or shorter stay, while 31% had at least a 60-day stay. In addition, just 13% of those with limited endurance had a 30-day stay or shorter, while 24% had at least a 60-day stay, she explained.
Further, patients with limited endurance, and those unable to perform 10 or more sit-to-stands in 30 seconds were more likely to die during their first transplant admission. Of those with limited endurance, 31% died during admission and 13% survived, and of those with good endurance 69% died during admission and 87% survived. Among patients who were unable to perform more than 10 sit-to-stands, 42% died during admission and 20% survived, and of those able to perform 11 or more, 38% died during admission, and 53% survived.
Overall survival was associated with age, KPS, HCT-CI, and age-adjusted HCT-CI, she noted.
“Patients who were over age 40 and more, and patients with a KPS of 60 or 70, belong to the high- to intermediate-risk group more likely to have decreased overall survival as has been shown in previous studies,” she said. “In addition to validating these findings, we also found that the semiquantitative measures, including pain and endurance, were also associated with overall survival.”
Those with pain present or limited endurance had significantly poorer overall survival (P = .007 and P = .01, respectively), and this finding was reflected in the quantitative measures of sit-to-stands (P = .01) and step-ups (P = .001), even when stratified by age-adjusted HCT-CI, she said.
In addition, a number of risk factors present at the pretreatment assessment were found to be significantly associated with requirement of an assistive device at discharge. These included pain, weakness in the lower extremities, use of an assistive device, inability to perform 25 step-ups and more than 10 sit-to-stands in 30 seconds, and limited endurance (P values ranging from .02 to less than .0001). Requirement of a device was associated with poorer overall survival (P = .03), she said.
Study participants were adults aged 18 years and older (median, 58 years) undergoing a first allogeneic HCT at a single center between 2010 and 2016. Most (83%) were older than age 40 years and 58% were men. About half (51%) had acute myeloid leukemia, and 64% overall had a KPS score of 60-70.
Physical therapists assessed physical performance of all patients within 4 weeks pre-HCT; testing included 25 7-inch step-ups on each side, unassisted sit-to-stands from an 18-inch chair in 30 seconds, weight-bearing ability, need for assistance with ambulation, motor strength in four extremities, sensory or coordination impairment, self-reported pain, and time to recovery of heart rate and oxygen saturation to pre-exercise levels.
“The HCT-CI is a validated tool that predicts nonrelapse mortality and overall survival, but comorbidity alone as a single domain is not a surrogate of overall health or reflection on the true biological age of our patients,” Dr. Rehman said, noting that studies have shown that functional impairment is associated with shorter overall survival, and that patient-reported physical functioning is predictive of overall survival. “The assessment of functional impairment becomes more critical given the aging U.S. population and older patients receiving transplant.”
Traditionally, functional status has been assessed via the KPS, which is a subjective measure and lacks precision, and the HCT-CI has not been studied in the context of the novel outcome measures addressed in the current study, she noted.
The current findings highlight the prognostic value of a more quantitative pretransplant assessment, which can help improve the patient selection process.
“We are in the process of analyzing some more outcomes of these pretransplant assessments, and developing a score that can, in conjunction with other predictive tools, help us improve pretransplant risk stratification and devise interventions that can improve the endurance and overall survival of the patients,” she concluded.
Dr. Rehman reported having no financial disclosures.
SOURCE: Rehman S et al., The 2018 BTM Tandem Meetings, Abstract 19.
SALT LAKE CITY – Comprehensive assessment of functional status and endurance prior to allogeneic hematopoietic cell transplantation (HCT) provides important insights into posttransplant outcomes, and when used in combination with other measures may improve the patient selection process, a chart review suggests.
In 349 patients, results of the prospective assessment of physical performance and endurance, along with HCT Comorbidity Index (HCT-CI) score and Karnofsky Performance Scale score (KPS), were compared with day 100-plus nonrelapse mortality and overall survival. The measures were also compared with the novel measures of hospital length of stay, and death during HCT admission, Shabnam Rehman, MD, reported at the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
However, heart rate recovery in less than 3 minutes after performing 25 step-ups on each side was associated with shorter length of stay with 89% of those patients, compared with 11% of patients who were not able to recover their heart rate in less than 3 minutes, being discharged within 30 days, she said.
“Similarly, patients who are able to perform at least 11 sit-to-stands in 30 seconds are more likely to be discharged earlier (63% vs. 14% discharged within 30 days),” she said. “The converse is also true.”
That is, only 16% of those not able to recover their heart rate within 3 minutes had a 30-day or shorter stay, while 31% had at least a 60-day stay. In addition, just 13% of those with limited endurance had a 30-day stay or shorter, while 24% had at least a 60-day stay, she explained.
Further, patients with limited endurance, and those unable to perform 10 or more sit-to-stands in 30 seconds were more likely to die during their first transplant admission. Of those with limited endurance, 31% died during admission and 13% survived, and of those with good endurance 69% died during admission and 87% survived. Among patients who were unable to perform more than 10 sit-to-stands, 42% died during admission and 20% survived, and of those able to perform 11 or more, 38% died during admission, and 53% survived.
Overall survival was associated with age, KPS, HCT-CI, and age-adjusted HCT-CI, she noted.
“Patients who were over age 40 and more, and patients with a KPS of 60 or 70, belong to the high- to intermediate-risk group more likely to have decreased overall survival as has been shown in previous studies,” she said. “In addition to validating these findings, we also found that the semiquantitative measures, including pain and endurance, were also associated with overall survival.”
Those with pain present or limited endurance had significantly poorer overall survival (P = .007 and P = .01, respectively), and this finding was reflected in the quantitative measures of sit-to-stands (P = .01) and step-ups (P = .001), even when stratified by age-adjusted HCT-CI, she said.
In addition, a number of risk factors present at the pretreatment assessment were found to be significantly associated with requirement of an assistive device at discharge. These included pain, weakness in the lower extremities, use of an assistive device, inability to perform 25 step-ups and more than 10 sit-to-stands in 30 seconds, and limited endurance (P values ranging from .02 to less than .0001). Requirement of a device was associated with poorer overall survival (P = .03), she said.
Study participants were adults aged 18 years and older (median, 58 years) undergoing a first allogeneic HCT at a single center between 2010 and 2016. Most (83%) were older than age 40 years and 58% were men. About half (51%) had acute myeloid leukemia, and 64% overall had a KPS score of 60-70.
Physical therapists assessed physical performance of all patients within 4 weeks pre-HCT; testing included 25 7-inch step-ups on each side, unassisted sit-to-stands from an 18-inch chair in 30 seconds, weight-bearing ability, need for assistance with ambulation, motor strength in four extremities, sensory or coordination impairment, self-reported pain, and time to recovery of heart rate and oxygen saturation to pre-exercise levels.
“The HCT-CI is a validated tool that predicts nonrelapse mortality and overall survival, but comorbidity alone as a single domain is not a surrogate of overall health or reflection on the true biological age of our patients,” Dr. Rehman said, noting that studies have shown that functional impairment is associated with shorter overall survival, and that patient-reported physical functioning is predictive of overall survival. “The assessment of functional impairment becomes more critical given the aging U.S. population and older patients receiving transplant.”
Traditionally, functional status has been assessed via the KPS, which is a subjective measure and lacks precision, and the HCT-CI has not been studied in the context of the novel outcome measures addressed in the current study, she noted.
The current findings highlight the prognostic value of a more quantitative pretransplant assessment, which can help improve the patient selection process.
“We are in the process of analyzing some more outcomes of these pretransplant assessments, and developing a score that can, in conjunction with other predictive tools, help us improve pretransplant risk stratification and devise interventions that can improve the endurance and overall survival of the patients,” she concluded.
Dr. Rehman reported having no financial disclosures.
SOURCE: Rehman S et al., The 2018 BTM Tandem Meetings, Abstract 19.
REPORTING FROM THE 2018 BMT TANDEM MEETINGS
Key clinical point:
Major finding: The 30-day discharge rates were 89% versus 11% in those with and without good heart rate recovery, respectively.
Study details: A retrospective review of prospectively collected data for 349 patients.
Disclosures: Dr. Rehman reported having no financial disclosures.
Source: Rehman S et al. The 2018 BMT Tandem Meetings, Abstract 19.
Spine fracture risk may be increased in IBD patients
Moreover, fracture risk appears to be higher among IBD patients using steroids, according to a report published in the Journal of Clinical Gastroenterology by Yuga Komaki, MD, of the Inflammatory Bowel Disease Center, University of Chicago, and coauthors.
“Further studies addressing the differential risk among Crohn’s disease and ulcerative colitis are needed, but strict surveillance and prevention of spine fractures are indicated in IBD,” wrote Dr. Komaki and associates.
The systematic review and meta-analysis by Dr. Komaki and colleagues was based on 10 studies comprising 470,541 patients with IBD for whom the risk of fracture was reported.
“It is of importance to identify the risk of fractures, as it will increase patient morbidity, disability, and mortality,” the authors wrote. “However, it is often overlooked in the management of IBD.”
Results of the analysis by this group of researchers showed that there was no significant difference in fracture risk overall between IBD patients and controls (odds ratio, 1.08; 95% confidence interval, 0.72-1.62; P = .70).
By contrast, the OR for spine fractures was significantly elevated (OR, 2.21; 95% CI, 1.39-3.50; P less than .0001), while risk of hip, rib, and wrist fractures were not, Dr. Komaki and coauthors said in their report.
Steroids were more often being used in the treatment of IBD patients who had fractures than in patients with no fractures, though the finding did not quite reach statistical significance (OR, 1.47; 95% CI, 0.99-2.20; P = .057).
Prior studies of fracture risk in IBD have shown “controversial results,” according to Dr. Komaki and colleagues. Some of those studies suggest an increased risk of fractures, whereas others suggest the risk is not different from what is seen in the general population.
“Individual studies may be underpowered to detect any risk,” they said in the report.
Steroids have been shown to increase risk of spine and rib fracture, but whether those earlier studies apply in IBD is unclear, they noted.
While the present meta-analysis sheds light on fracture risk in IBD patients, further meta-analyses may be needed to specifically look at cohorts of patients with Crohn’s disease and ulcerative colitis.
In this study, the investigators did find that spine fracture risk was significantly elevated in patients with Crohn’s disease, and was trending toward significance for ulcerative colitis patients. They cautioned that those results were based on a limited amount of patient data.
Dr. Komaki reported that he had no disclosures related to the reported study. One study coauthor reported disclosures related to AbbVie and Celltrion.
SOURCE: Komaki Y et al. J Clin Gastroenterol. 2018 Apr 18. 2018 Apr 18. doi: 10.1097/MCG.0000000000001031.
Moreover, fracture risk appears to be higher among IBD patients using steroids, according to a report published in the Journal of Clinical Gastroenterology by Yuga Komaki, MD, of the Inflammatory Bowel Disease Center, University of Chicago, and coauthors.
“Further studies addressing the differential risk among Crohn’s disease and ulcerative colitis are needed, but strict surveillance and prevention of spine fractures are indicated in IBD,” wrote Dr. Komaki and associates.
The systematic review and meta-analysis by Dr. Komaki and colleagues was based on 10 studies comprising 470,541 patients with IBD for whom the risk of fracture was reported.
“It is of importance to identify the risk of fractures, as it will increase patient morbidity, disability, and mortality,” the authors wrote. “However, it is often overlooked in the management of IBD.”
Results of the analysis by this group of researchers showed that there was no significant difference in fracture risk overall between IBD patients and controls (odds ratio, 1.08; 95% confidence interval, 0.72-1.62; P = .70).
By contrast, the OR for spine fractures was significantly elevated (OR, 2.21; 95% CI, 1.39-3.50; P less than .0001), while risk of hip, rib, and wrist fractures were not, Dr. Komaki and coauthors said in their report.
Steroids were more often being used in the treatment of IBD patients who had fractures than in patients with no fractures, though the finding did not quite reach statistical significance (OR, 1.47; 95% CI, 0.99-2.20; P = .057).
Prior studies of fracture risk in IBD have shown “controversial results,” according to Dr. Komaki and colleagues. Some of those studies suggest an increased risk of fractures, whereas others suggest the risk is not different from what is seen in the general population.
“Individual studies may be underpowered to detect any risk,” they said in the report.
Steroids have been shown to increase risk of spine and rib fracture, but whether those earlier studies apply in IBD is unclear, they noted.
While the present meta-analysis sheds light on fracture risk in IBD patients, further meta-analyses may be needed to specifically look at cohorts of patients with Crohn’s disease and ulcerative colitis.
In this study, the investigators did find that spine fracture risk was significantly elevated in patients with Crohn’s disease, and was trending toward significance for ulcerative colitis patients. They cautioned that those results were based on a limited amount of patient data.
Dr. Komaki reported that he had no disclosures related to the reported study. One study coauthor reported disclosures related to AbbVie and Celltrion.
SOURCE: Komaki Y et al. J Clin Gastroenterol. 2018 Apr 18. 2018 Apr 18. doi: 10.1097/MCG.0000000000001031.
Moreover, fracture risk appears to be higher among IBD patients using steroids, according to a report published in the Journal of Clinical Gastroenterology by Yuga Komaki, MD, of the Inflammatory Bowel Disease Center, University of Chicago, and coauthors.
“Further studies addressing the differential risk among Crohn’s disease and ulcerative colitis are needed, but strict surveillance and prevention of spine fractures are indicated in IBD,” wrote Dr. Komaki and associates.
The systematic review and meta-analysis by Dr. Komaki and colleagues was based on 10 studies comprising 470,541 patients with IBD for whom the risk of fracture was reported.
“It is of importance to identify the risk of fractures, as it will increase patient morbidity, disability, and mortality,” the authors wrote. “However, it is often overlooked in the management of IBD.”
Results of the analysis by this group of researchers showed that there was no significant difference in fracture risk overall between IBD patients and controls (odds ratio, 1.08; 95% confidence interval, 0.72-1.62; P = .70).
By contrast, the OR for spine fractures was significantly elevated (OR, 2.21; 95% CI, 1.39-3.50; P less than .0001), while risk of hip, rib, and wrist fractures were not, Dr. Komaki and coauthors said in their report.
Steroids were more often being used in the treatment of IBD patients who had fractures than in patients with no fractures, though the finding did not quite reach statistical significance (OR, 1.47; 95% CI, 0.99-2.20; P = .057).
Prior studies of fracture risk in IBD have shown “controversial results,” according to Dr. Komaki and colleagues. Some of those studies suggest an increased risk of fractures, whereas others suggest the risk is not different from what is seen in the general population.
“Individual studies may be underpowered to detect any risk,” they said in the report.
Steroids have been shown to increase risk of spine and rib fracture, but whether those earlier studies apply in IBD is unclear, they noted.
While the present meta-analysis sheds light on fracture risk in IBD patients, further meta-analyses may be needed to specifically look at cohorts of patients with Crohn’s disease and ulcerative colitis.
In this study, the investigators did find that spine fracture risk was significantly elevated in patients with Crohn’s disease, and was trending toward significance for ulcerative colitis patients. They cautioned that those results were based on a limited amount of patient data.
Dr. Komaki reported that he had no disclosures related to the reported study. One study coauthor reported disclosures related to AbbVie and Celltrion.
SOURCE: Komaki Y et al. J Clin Gastroenterol. 2018 Apr 18. 2018 Apr 18. doi: 10.1097/MCG.0000000000001031.
FROM THE JOURNAL OF CLINICAL GASTROENTEROLOGY
Key clinical point: Patients with inflammatory bowel disease may be at increased risk of fractures in the spine.
Major finding: The odds ratio for spine fractures was 2.21 (95% CI, 1.39-3.50; P less than .0001).
Study details: A systematic review and meta-analysis of 10 studies including 470,541 patients.
Disclosures: One study author reported disclosures related to AbbVie and Celltrion.
Source: Komaki Y et al. J Clin Gastroenterol. 2018 Apr 18. doi: 10.1097/MCG.0000000000001031.
Nebivolol looks like best beta-blocker for hypertension
ORLANDO – The use of nebivolol as part of a multidrug regimen to treat hypertension was associated with a significantly lower cardiovascular event risk than was combination antihypertensive therapy featuring either metoprolol or atenolol in a large observational study, Brent M. Egan, MD, reported at the annual meeting of the American College of Cardiology.

The primary outcome was hospitalization for acute MI, stroke, heart failure, or angina during a mean 600 days of follow-up. In a Cox proportional hazards regression analysis, the risk of the composite outcome was 1.33-fold greater with atenolol and 1.91-fold greater with metoprolol than in the group on nebivolol for their hypertension.
The risk of hospitalization for MI was 1.47-fold greater in the atenolol group and 2.19-fold greater with metoprolol than in patients on nebivolol. Hospitalization for angina was 2.18 times more likely in the atenolol group and 3.39 times more likely in the metoprolol group than in patients on nebivolol. However, there was no between-group difference between the three beta-blockers in terms of stroke or heart failure rates, according to Dr. Egan of the University of South Carolina, Greenville.
He explained that the impetus for this study was that, even though beta-blockers are universally recognized as a cornerstone of secondary cardiovascular prevention, there are much fewer outcome data to support their use in primary prevention. Since nebivolol is a vasodilatory beta-blocker and atenolol and metoprolol are not, Dr. Egan and his coinvestigators hypothesized that this distinction could result in differences in cardiovascular event rates.
One audience member commented, “this study gives a nice hypothesis-generating rationale for doing a large randomized outcomes trial.” Dr. Egan concurred.
His study was supported by Allergan. He reported receiving research grants from Boehringer Ingelheim and serving as a consultant to Medtronic.
SOURCE: Egan BM et al. ACC 18, Abstract 1324M-11.
ORLANDO – The use of nebivolol as part of a multidrug regimen to treat hypertension was associated with a significantly lower cardiovascular event risk than was combination antihypertensive therapy featuring either metoprolol or atenolol in a large observational study, Brent M. Egan, MD, reported at the annual meeting of the American College of Cardiology.
The primary outcome was hospitalization for acute MI, stroke, heart failure, or angina during a mean 600 days of follow-up. In a Cox proportional hazards regression analysis, the risk of the composite outcome was 1.33-fold greater with atenolol and 1.91-fold greater with metoprolol than in the group on nebivolol for their hypertension.
The risk of hospitalization for MI was 1.47-fold greater in the atenolol group and 2.19-fold greater with metoprolol than in patients on nebivolol. Hospitalization for angina was 2.18 times more likely in the atenolol group and 3.39 times more likely in the metoprolol group than in patients on nebivolol. However, there was no between-group difference between the three beta-blockers in terms of stroke or heart failure rates, according to Dr. Egan of the University of South Carolina, Greenville.
He explained that the impetus for this study was that, even though beta-blockers are universally recognized as a cornerstone of secondary cardiovascular prevention, there are much fewer outcome data to support their use in primary prevention. Since nebivolol is a vasodilatory beta-blocker and atenolol and metoprolol are not, Dr. Egan and his coinvestigators hypothesized that this distinction could result in differences in cardiovascular event rates.
One audience member commented, “this study gives a nice hypothesis-generating rationale for doing a large randomized outcomes trial.” Dr. Egan concurred.
His study was supported by Allergan. He reported receiving research grants from Boehringer Ingelheim and serving as a consultant to Medtronic.
SOURCE: Egan BM et al. ACC 18, Abstract 1324M-11.
ORLANDO – The use of nebivolol as part of a multidrug regimen to treat hypertension was associated with a significantly lower cardiovascular event risk than was combination antihypertensive therapy featuring either metoprolol or atenolol in a large observational study, Brent M. Egan, MD, reported at the annual meeting of the American College of Cardiology.
The primary outcome was hospitalization for acute MI, stroke, heart failure, or angina during a mean 600 days of follow-up. In a Cox proportional hazards regression analysis, the risk of the composite outcome was 1.33-fold greater with atenolol and 1.91-fold greater with metoprolol than in the group on nebivolol for their hypertension.
The risk of hospitalization for MI was 1.47-fold greater in the atenolol group and 2.19-fold greater with metoprolol than in patients on nebivolol. Hospitalization for angina was 2.18 times more likely in the atenolol group and 3.39 times more likely in the metoprolol group than in patients on nebivolol. However, there was no between-group difference between the three beta-blockers in terms of stroke or heart failure rates, according to Dr. Egan of the University of South Carolina, Greenville.
He explained that the impetus for this study was that, even though beta-blockers are universally recognized as a cornerstone of secondary cardiovascular prevention, there are much fewer outcome data to support their use in primary prevention. Since nebivolol is a vasodilatory beta-blocker and atenolol and metoprolol are not, Dr. Egan and his coinvestigators hypothesized that this distinction could result in differences in cardiovascular event rates.
One audience member commented, “this study gives a nice hypothesis-generating rationale for doing a large randomized outcomes trial.” Dr. Egan concurred.
His study was supported by Allergan. He reported receiving research grants from Boehringer Ingelheim and serving as a consultant to Medtronic.
SOURCE: Egan BM et al. ACC 18, Abstract 1324M-11.
REPORTING FROM ACC 2018
Key clinical point:
Major finding: The risk of hospitalization for MI was 1.47-fold greater in patients on atenolol and 2.19-fold greater with metoprolol than in patients on nebivolol as part of a multidrug regimen to treat hypertension.
Study details: This retrospective observational study included three closely propensity score–matched groups, each 16,787 patients in size, on either nebivolol, metoprolol, or atenolol as part of a multidrug antihypertensive regimen.
Disclosures: The study was supported by Allergan. The presenter reported receiving research grants from Boehringer Ingelheim and serving as a consultant to Medtronic.
Source: Egan BM. ACC 2018, Abstract 1324M-11.
Peripheral nerve stimulation can reduce tremor symptoms
LOS ANGELES – A noninvasive peripheral nerve stimulation device has been shown to reduce symptoms of hand tremor among people with essential tremor, offering a possible alternative to invasive treatments such as deep-brain stimulation.
The neuromodulation device is worn on the wrist and uses electrodes to stimulate the radial and median nerves at a frequency that interrupts tremor. It contains sensors that measure tremor and adjust stimulation accordingly.
In two small, randomized, controlled studies presented at the annual meeting of the at the American Academy of Neurology, investigator Rajesh Pahwa, MD, of the University of Kansas in Kansas City, said that treatment with the device significantly reduced tremor symptoms, compared with sham treatment.
On April 26, the device’s manufacturer, Cala Health, announced in a news release that the U.S. Food and Drug Administration had granted marketing clearance for the device, based on this evidence.
For the first study, conducted in-clinic, 77 patients were randomized to either treatment (n = 40) or sham stimulation (n = 37) of the tremor-dominant hand. Tremor was measured before and immediately after a single 40-minute session of stimulation, and patients were asked to perform tasks in accordance with the Essential Tremor Rating Assessment Scale or TETRAS, a severity measure.
Subjects in the intervention group had about a 65% improvement in their upper-limb TETRAS scores, compared with those receiving sham treatment (P less than .01) and in total TETRAS performance (P less than .05).
Subjects also were tested in-clinic with props simulating common daily tasks such as unlocking a door with a key, holding a cup of tea, picking up loose change, or dialing a phone. Patients in the treatment group self-reported greater ease with all of these tasks after treatment, compared with the sham-treated group. Differences for some tasks reached statistical significance.
For the second study, conducted for 4 weeks, 61 patients were randomized to at-home treatment sessions with the neuromodulator or sham treatment for 40 minutes at least twice daily. Those receiving treatment (n = 31) saw greater reduction in tremor measured by the devices’ built-in sensors, compared with those assigned sham treatment (n = 15) or no treatment (n = 15). Nearly all sessions completed resulted in a measurable reduction of tremor.
In an interview at AAN, Manish Gupta of Cala Health, the device manufacturer, said that further studies are underway to assess the durability of the treatment.
“What we seem to be looking at is an on-demand therapy that delivers a transient relief,” Mr. Gupta said, adding that the devices could be used by patients at times when their tremor is most bothersome, or in anticipation of a task – such as dressing oneself or eating – that a tremor would affect the ability to perform.
“One thing we’re learning from clinicians is that tremor is variable within the patient, and it’s variable across patients,” Mr. Gupta said. “The same patient may find that they have less tremor a certain day or at certain times of the day. We don’t think this would replace deep-brain stimulation, which is a constant treatment effect, but it could serve for some patients as a step before it.”
Cala Health, the manufacturer, sponsored the study. One coauthor is an employee of Cala Health.
SOURCE: Pahwa R et al. AAN 2018, Abstract P4.474.
LOS ANGELES – A noninvasive peripheral nerve stimulation device has been shown to reduce symptoms of hand tremor among people with essential tremor, offering a possible alternative to invasive treatments such as deep-brain stimulation.
The neuromodulation device is worn on the wrist and uses electrodes to stimulate the radial and median nerves at a frequency that interrupts tremor. It contains sensors that measure tremor and adjust stimulation accordingly.
In two small, randomized, controlled studies presented at the annual meeting of the at the American Academy of Neurology, investigator Rajesh Pahwa, MD, of the University of Kansas in Kansas City, said that treatment with the device significantly reduced tremor symptoms, compared with sham treatment.
On April 26, the device’s manufacturer, Cala Health, announced in a news release that the U.S. Food and Drug Administration had granted marketing clearance for the device, based on this evidence.
For the first study, conducted in-clinic, 77 patients were randomized to either treatment (n = 40) or sham stimulation (n = 37) of the tremor-dominant hand. Tremor was measured before and immediately after a single 40-minute session of stimulation, and patients were asked to perform tasks in accordance with the Essential Tremor Rating Assessment Scale or TETRAS, a severity measure.
Subjects in the intervention group had about a 65% improvement in their upper-limb TETRAS scores, compared with those receiving sham treatment (P less than .01) and in total TETRAS performance (P less than .05).
Subjects also were tested in-clinic with props simulating common daily tasks such as unlocking a door with a key, holding a cup of tea, picking up loose change, or dialing a phone. Patients in the treatment group self-reported greater ease with all of these tasks after treatment, compared with the sham-treated group. Differences for some tasks reached statistical significance.
For the second study, conducted for 4 weeks, 61 patients were randomized to at-home treatment sessions with the neuromodulator or sham treatment for 40 minutes at least twice daily. Those receiving treatment (n = 31) saw greater reduction in tremor measured by the devices’ built-in sensors, compared with those assigned sham treatment (n = 15) or no treatment (n = 15). Nearly all sessions completed resulted in a measurable reduction of tremor.
In an interview at AAN, Manish Gupta of Cala Health, the device manufacturer, said that further studies are underway to assess the durability of the treatment.
“What we seem to be looking at is an on-demand therapy that delivers a transient relief,” Mr. Gupta said, adding that the devices could be used by patients at times when their tremor is most bothersome, or in anticipation of a task – such as dressing oneself or eating – that a tremor would affect the ability to perform.
“One thing we’re learning from clinicians is that tremor is variable within the patient, and it’s variable across patients,” Mr. Gupta said. “The same patient may find that they have less tremor a certain day or at certain times of the day. We don’t think this would replace deep-brain stimulation, which is a constant treatment effect, but it could serve for some patients as a step before it.”
Cala Health, the manufacturer, sponsored the study. One coauthor is an employee of Cala Health.
SOURCE: Pahwa R et al. AAN 2018, Abstract P4.474.
LOS ANGELES – A noninvasive peripheral nerve stimulation device has been shown to reduce symptoms of hand tremor among people with essential tremor, offering a possible alternative to invasive treatments such as deep-brain stimulation.
The neuromodulation device is worn on the wrist and uses electrodes to stimulate the radial and median nerves at a frequency that interrupts tremor. It contains sensors that measure tremor and adjust stimulation accordingly.
In two small, randomized, controlled studies presented at the annual meeting of the at the American Academy of Neurology, investigator Rajesh Pahwa, MD, of the University of Kansas in Kansas City, said that treatment with the device significantly reduced tremor symptoms, compared with sham treatment.
On April 26, the device’s manufacturer, Cala Health, announced in a news release that the U.S. Food and Drug Administration had granted marketing clearance for the device, based on this evidence.
For the first study, conducted in-clinic, 77 patients were randomized to either treatment (n = 40) or sham stimulation (n = 37) of the tremor-dominant hand. Tremor was measured before and immediately after a single 40-minute session of stimulation, and patients were asked to perform tasks in accordance with the Essential Tremor Rating Assessment Scale or TETRAS, a severity measure.
Subjects in the intervention group had about a 65% improvement in their upper-limb TETRAS scores, compared with those receiving sham treatment (P less than .01) and in total TETRAS performance (P less than .05).
Subjects also were tested in-clinic with props simulating common daily tasks such as unlocking a door with a key, holding a cup of tea, picking up loose change, or dialing a phone. Patients in the treatment group self-reported greater ease with all of these tasks after treatment, compared with the sham-treated group. Differences for some tasks reached statistical significance.
For the second study, conducted for 4 weeks, 61 patients were randomized to at-home treatment sessions with the neuromodulator or sham treatment for 40 minutes at least twice daily. Those receiving treatment (n = 31) saw greater reduction in tremor measured by the devices’ built-in sensors, compared with those assigned sham treatment (n = 15) or no treatment (n = 15). Nearly all sessions completed resulted in a measurable reduction of tremor.
In an interview at AAN, Manish Gupta of Cala Health, the device manufacturer, said that further studies are underway to assess the durability of the treatment.
“What we seem to be looking at is an on-demand therapy that delivers a transient relief,” Mr. Gupta said, adding that the devices could be used by patients at times when their tremor is most bothersome, or in anticipation of a task – such as dressing oneself or eating – that a tremor would affect the ability to perform.
“One thing we’re learning from clinicians is that tremor is variable within the patient, and it’s variable across patients,” Mr. Gupta said. “The same patient may find that they have less tremor a certain day or at certain times of the day. We don’t think this would replace deep-brain stimulation, which is a constant treatment effect, but it could serve for some patients as a step before it.”
Cala Health, the manufacturer, sponsored the study. One coauthor is an employee of Cala Health.
SOURCE: Pahwa R et al. AAN 2018, Abstract P4.474.
REPORTING FROM AAN 2018
Key clinical point:
Major finding: Subjects using the devices saw improvement in their upper-limb tremor scores, compared with those receiving sham treatment (P less than .01)
Study details: Two randomized studies (n = 77 and n = 61) comparing in-home or in-office treatment with stimulation or sham treatment.
Disclosures: The device manufacturer sponsored the study. One employee is a coauthor.
Source: Pahwa R et al. AAN 2018, Abstract P4.474.
Self-management support
This is the ninth in a series of articles from the National Center for Excellence in Primary Care Research (NCEPCR) in the Agency for Healthcare Research and Quality (AHRQ). This series introduces sets of tools and resources designed to help your practice.
There is growing awareness of the importance of supporting patient self-management as part of a comprehensive approach to caring for people with chronic conditions. This is in part the recognition that 15 minutes with a provider every few months contributes less to patient outcomes than what the patient does every day. This month’s article describes some elements of AHRQ’s Self-Management Support Resource Library, a collection of materials and tools produced by AHRQ and others.

The Library’s resources also include materials from other sources. These include Helping patients help themselves: How to implement self-management support, a paper from the California Health Care Foundation. It defines self-management support (SMS), provides case studies of primary care practices that have implemented SMS, and discusses the business case for SMS. Case studies include settings such as primary care practices, behavioral health programs, and telephone consultations featuring SMS models that rely on the actions of nurses, medical assistants, community health workers (promotoras), and health coach volunteers. “Helping patients take charge of their chronic illnesses” is an article from the American Academy of Family Physicians that introduces SMS concepts, provides a rationale for patient self-management, and gives an example of how to empower patients with information. It makes a case for shifting from an acute-care model to a patient-centered care model that includes SMS.
Enhancing the patient’s ability to manage medication is important. Clearly stating medication instructions improves patient understanding and possibly reduces errors while improving adherence. Explicit and standardized prescription medicine instructions offers tested instructions to simplify complex medicine regimens by using standard time periods for administration. These instructions have also been translated into Chinese, Korean, Russian, Spanish, and Vietnamese.
How to create a pill card helps users create an easy-to-use “pill card” for anyone who has a hard time keeping track of their medicines. Step-by-step instructions, sample clip art, and suggestions for design and use will help to customize a reminder card.
The self-management resources are further supported by AHRQ’s resources to improve health literacy. These were described in more detail in the 5th article in this series (January 2018). In brief, these resources include The Health Literacy Universal Precautions Toolkit–2nd edition and its companion guide, Implementing the Health Literacy Universal Precautions Toolkit: Practical Ideas for Primary Care Practices. In addition, the Patient Education Materials Assessment Tool features a systematic method to evaluate and compare how understandable and actionable patient education materials are.

Dr. Genevro is a health scientist at AHRQ. Dr. Ganiats is director of the National Center for Excellence in Primary Care Research at AHRQ.
This is the ninth in a series of articles from the National Center for Excellence in Primary Care Research (NCEPCR) in the Agency for Healthcare Research and Quality (AHRQ). This series introduces sets of tools and resources designed to help your practice.
There is growing awareness of the importance of supporting patient self-management as part of a comprehensive approach to caring for people with chronic conditions. This is in part the recognition that 15 minutes with a provider every few months contributes less to patient outcomes than what the patient does every day. This month’s article describes some elements of AHRQ’s Self-Management Support Resource Library, a collection of materials and tools produced by AHRQ and others.
The Library’s resources also include materials from other sources. These include Helping patients help themselves: How to implement self-management support, a paper from the California Health Care Foundation. It defines self-management support (SMS), provides case studies of primary care practices that have implemented SMS, and discusses the business case for SMS. Case studies include settings such as primary care practices, behavioral health programs, and telephone consultations featuring SMS models that rely on the actions of nurses, medical assistants, community health workers (promotoras), and health coach volunteers. “Helping patients take charge of their chronic illnesses” is an article from the American Academy of Family Physicians that introduces SMS concepts, provides a rationale for patient self-management, and gives an example of how to empower patients with information. It makes a case for shifting from an acute-care model to a patient-centered care model that includes SMS.
Enhancing the patient’s ability to manage medication is important. Clearly stating medication instructions improves patient understanding and possibly reduces errors while improving adherence. Explicit and standardized prescription medicine instructions offers tested instructions to simplify complex medicine regimens by using standard time periods for administration. These instructions have also been translated into Chinese, Korean, Russian, Spanish, and Vietnamese.
How to create a pill card helps users create an easy-to-use “pill card” for anyone who has a hard time keeping track of their medicines. Step-by-step instructions, sample clip art, and suggestions for design and use will help to customize a reminder card.
The self-management resources are further supported by AHRQ’s resources to improve health literacy. These were described in more detail in the 5th article in this series (January 2018). In brief, these resources include The Health Literacy Universal Precautions Toolkit–2nd edition and its companion guide, Implementing the Health Literacy Universal Precautions Toolkit: Practical Ideas for Primary Care Practices. In addition, the Patient Education Materials Assessment Tool features a systematic method to evaluate and compare how understandable and actionable patient education materials are.
Dr. Genevro is a health scientist at AHRQ. Dr. Ganiats is director of the National Center for Excellence in Primary Care Research at AHRQ.
This is the ninth in a series of articles from the National Center for Excellence in Primary Care Research (NCEPCR) in the Agency for Healthcare Research and Quality (AHRQ). This series introduces sets of tools and resources designed to help your practice.
There is growing awareness of the importance of supporting patient self-management as part of a comprehensive approach to caring for people with chronic conditions. This is in part the recognition that 15 minutes with a provider every few months contributes less to patient outcomes than what the patient does every day. This month’s article describes some elements of AHRQ’s Self-Management Support Resource Library, a collection of materials and tools produced by AHRQ and others.
The Library’s resources also include materials from other sources. These include Helping patients help themselves: How to implement self-management support, a paper from the California Health Care Foundation. It defines self-management support (SMS), provides case studies of primary care practices that have implemented SMS, and discusses the business case for SMS. Case studies include settings such as primary care practices, behavioral health programs, and telephone consultations featuring SMS models that rely on the actions of nurses, medical assistants, community health workers (promotoras), and health coach volunteers. “Helping patients take charge of their chronic illnesses” is an article from the American Academy of Family Physicians that introduces SMS concepts, provides a rationale for patient self-management, and gives an example of how to empower patients with information. It makes a case for shifting from an acute-care model to a patient-centered care model that includes SMS.
Enhancing the patient’s ability to manage medication is important. Clearly stating medication instructions improves patient understanding and possibly reduces errors while improving adherence. Explicit and standardized prescription medicine instructions offers tested instructions to simplify complex medicine regimens by using standard time periods for administration. These instructions have also been translated into Chinese, Korean, Russian, Spanish, and Vietnamese.
How to create a pill card helps users create an easy-to-use “pill card” for anyone who has a hard time keeping track of their medicines. Step-by-step instructions, sample clip art, and suggestions for design and use will help to customize a reminder card.
The self-management resources are further supported by AHRQ’s resources to improve health literacy. These were described in more detail in the 5th article in this series (January 2018). In brief, these resources include The Health Literacy Universal Precautions Toolkit–2nd edition and its companion guide, Implementing the Health Literacy Universal Precautions Toolkit: Practical Ideas for Primary Care Practices. In addition, the Patient Education Materials Assessment Tool features a systematic method to evaluate and compare how understandable and actionable patient education materials are.
Dr. Genevro is a health scientist at AHRQ. Dr. Ganiats is director of the National Center for Excellence in Primary Care Research at AHRQ.
Hypertrophic cardiomyopathy: A complex disease
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
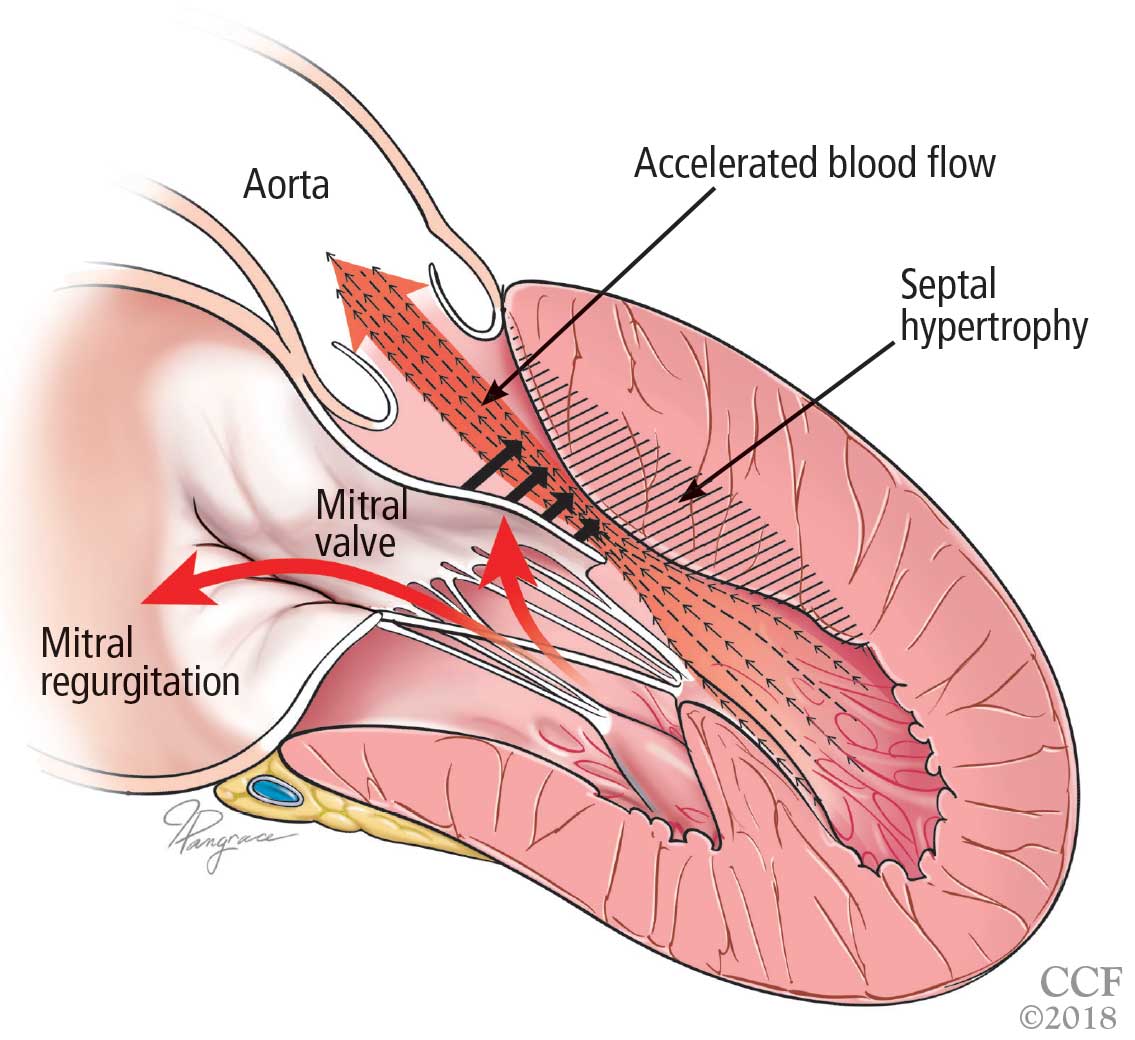
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
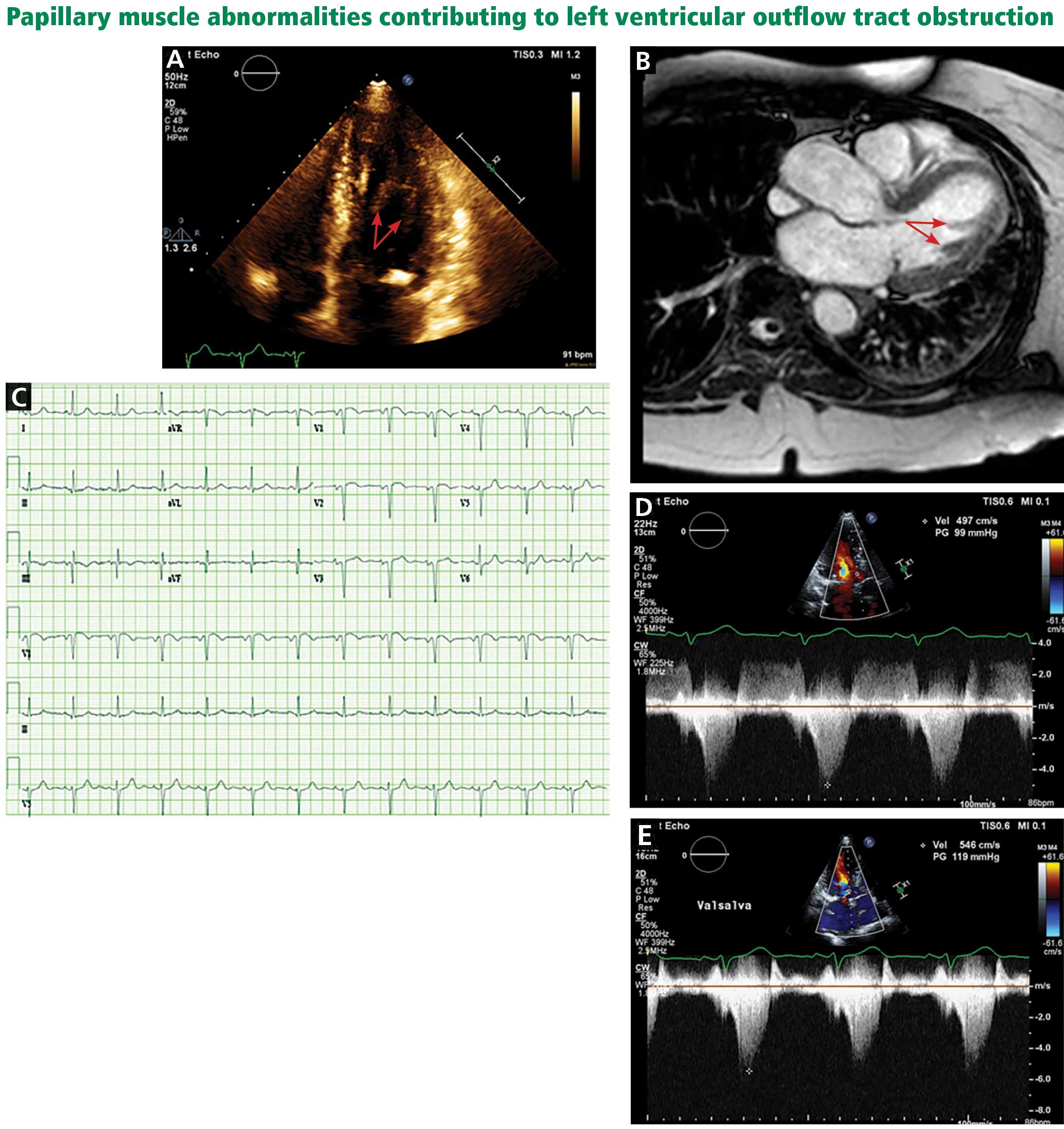
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
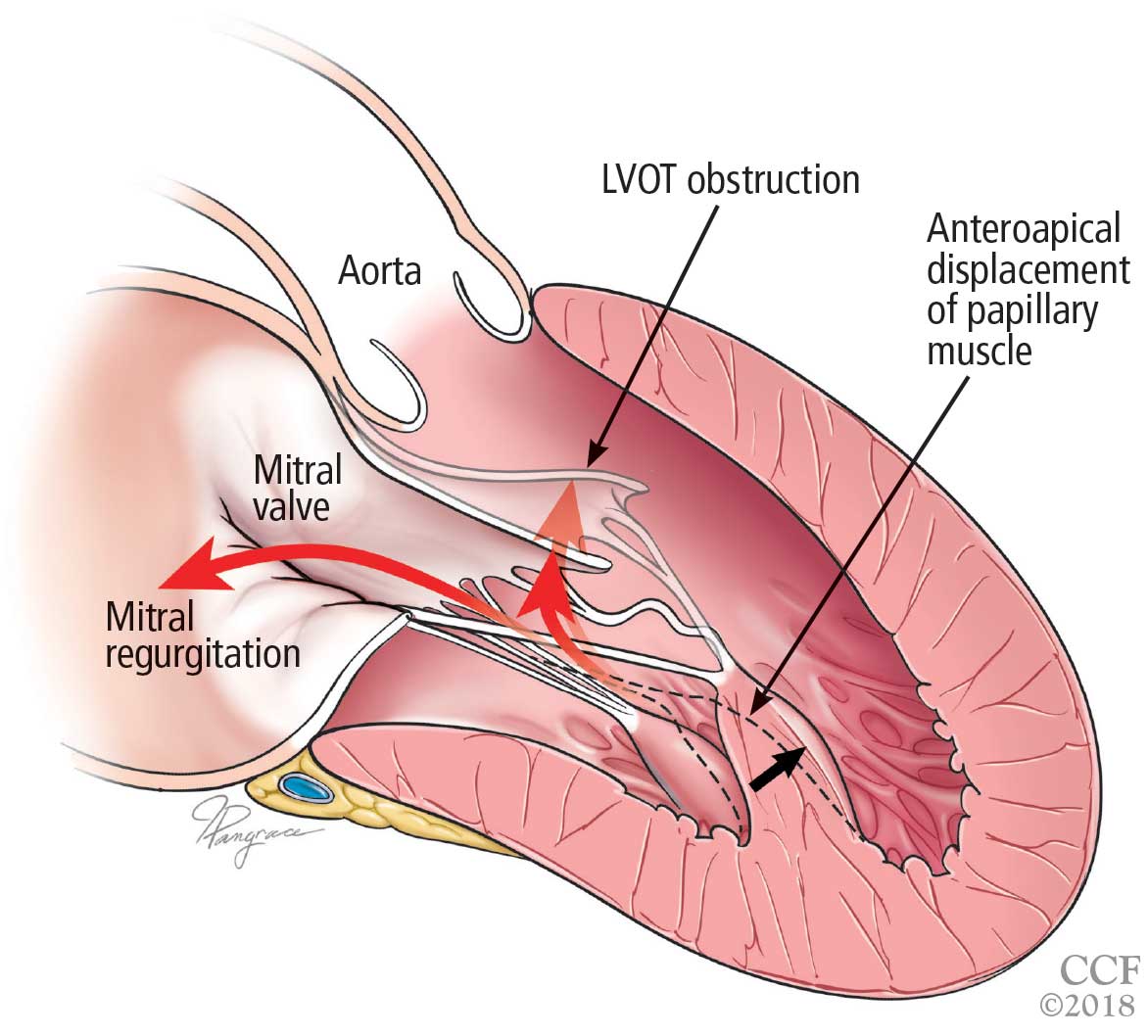
Physical findings are nonspecific

It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM

A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models

The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.

Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
KEY POINTS
- Obstruction of the left ventricular outflow tract is a key pathophysiologic mechanism in HCM.
- Because most of the genetic variants that contribute to HCM are autosomal dominant, genetic counseling and testing are suggested for patients and their first-degree relatives.
- Transthoracic echocardiography is the first-line imaging test, followed by magnetic resonance imaging.
- Beta-blockers are the first-line drugs for treating symptoms of HCM.
- An implantable cardioverter-defibrillator can be considered for patients at risk of sudden cardiac death.
- When medical therapy fails or is not tolerated in patients with severe symptoms of obstructive HCM, surgery to reduce the size of the ventricular septum can be considered. Alcohol septal ablation is an alternative.

Skin care product layering affects efficacy and outcomes of regimens
Dermatologists are well suited to understand cosmeceutical science and the benefits of particular cosmeceutical products – especially if they are readers of this column. However, there is another critical thought process that must be undertaken when designing an efficacious skin care regimen for patients: Topical products should be applied in a particular order to maximize efficacy. This is because cosmeceutical ingredients interact with, change each other, and are affected by temperature, pH, humidity, and the microbiome in which they are in contact. This column focuses on the factors that should be considered when recommending skin care regimens to patients and in which order to apply topical products.
The chemistry of ingredients and how they interact is well understood by personal care product formulators. I think of formulators as chefs who are using ingredients and placing them in the formulation in a well-defined order under controlled circumstances that affect the final product. For example, ceramides are used in barrier repair moisturizers. The right form of ceramide must be chosen and used with the 1:1:1 ratio of ceramides, fatty acids, and cholesterol for the product to be effective at repairing the barrier.1 However, the order of when the ceramides are added to the product formula also impacts efficacy. Waxy ceramides and cholesterol require heat to liquefy and form the proper mixture with the other ingredients. Too much heat can damage fatty acids. Also, heat can inactivate finicky active ingredients such as vitamins C and E. For this reason, the ceramides and cholesterol are often incorporated first, allowing the formula to cool before the active heat labile ingredients are added. The speed at which something is mixed can generate heat and affect the final preparations, so temperature is an important consideration at all steps in the formulation procedure. 
Just as the order of creating formulations affects the final product, the order of product placement on the skin influences skin care product efficacy. If a low pH skin care product (such as a glycolic acid cleanser) is used on the skin, this is going to affect the efficacy and safety of the product that is applied next to the skin. Such a chemical phenomenon should be considered when designing the order of product applications when designing a skin care regimen, particularly when incorporating ingredients that are known to interact with other ingredients, such as benzoyl peroxide, retinoids, hydroxy acids, hydroquinone, vitamin C, and peptides.
Efficacy and compliance in product layering
. Acne treatments are a good example. Patients are often prescribed a retinoid, benzoyl peroxide, topical antibiotic, and/or salicylic acid treatment product for acne. If the proper cleansers and moisturizers are not chosen, the patient will be more likely to develop redness and scaling and become noncompliant.
Compliance is a concerning issue to dermatologists because studies have shown that 95% of people underdose and one out of every three prescriptions is not even filled.2 If patients develop side effects, they are more likely to underdose or stop the treatment. Prescribing the proper cleanser and moisturizer to accompany treatment products will ease side effects and increase compliance. Several studies have demonstrated that the best way to increase compliance is to provide patients with written instructions, so they understand the proper order in which to apply products.
The role of cleansers
Cleansers can alter the pH of the skin, loosen attachments between cells, remove lipids – and disrupt the bilayer protective membrane, desquamate layers from the stratum corneum, and influence the penetrability of the skin for the next topical product that is applied. Therefore, cleansers should be selected based on the products that will follow them in the regimen. In addition, cleansers should be chosen according to the patient’s Baumann Skin Type.3 For example, cleansers for use on oily skin should have the ability to remove excess sebum on the skin while cleansers designed for dry skin would not remove as many lipids from the skin. Washing skin with a foaming cleanser can disrupt the skin barrier, allowing increased penetration of the treatment product that follows it. Oleic acid, hyaluronic acid, stearic acid, and other lipids are among the ingredients that influence skin penetration. Cleansers should precede treatment products and should be designed to increase efficacy of the treatment product. For this reason, every ingredient and characteristic of the chosen cleanser is important.
The role of eye products
Eye products treat issues such as dryness, puffiness, fine lines, and dark circles. However, they also play an overlooked role of protecting the thin delicate eye area from the treatment product. Using an eye product, especially one with protective ingredients such as barrier repair lipids, will help the patient tolerate the potentially irritating treatment product that follows the eye product. At night, the treatment product ingredients can get on the pillowcase and transfer to the upper and lower eyelids. Use of a protective eye product before bedtime can prevent the accompanying irritation. For example, acne patients often develop redness at the corners of the eyes when using benzoyl peroxide or a retinoid at night. Applying these medications after an eye cream can reduce this side effect.
Improving efficacy of treatment products
Treatments products are defined as corrective products targeted to skin issues such as acne, rosacea, melasma, dryness, skin cancer, eczema, psoriasis, and photoaging. The entire skin care regimen should be designed to enhance efficacy and decrease side effects of the treatment products. Treatment products may be cosmeceuticals, OTC medications, or prescription medications. These products must be able to reach their target in the proper chemical structure to be effective. Each ingredient has various constraints and quirks that should be considered. One well known example is ascorbic acid (vitamin C). Ascorbic acid is a treatment product for skin pigmentation and skin aging that is well known to have specific needs to work properly. Sheldon Pinnell, MD, led multiple investigations demonstrating that the maximum absorption of ascorbic acid occurs at a pH of 2-2.5. He showed that ascorbic acid products should be formulated at a pH of 2-2.5.4 However, applying these on skin that has just been cleansed with a soap cleanser with a pH of 9 will raise the skin’s pH and decrease the absorption of ascorbic acid. Having the patient cleanse with a low pH cleanser such as salicylic or glycolic acid cleanser (usually a pH of 2.5-3.5) will lower the pH of the skin and promote absorption of vitamin C.
The role of moisturizers
Moisturizers have many duties, including hydrating the skin, protecting the skin, and delivering important ingredients to the skin. However, moisturizers have a less discussed role of improving the efficacy of the treatment product that is applied beforehand. Moisturizers often contain oleic acid, hyaluronic acid, or other fatty acids that can increase penetration of other skin care ingredients. In addition, many moisturizers provide an occlusive effect that helps increase penetration. They also help protect the underlying treatment product from getting wiped off on a pillowcase or into the environment. In other words, moisturizers “seal in” the treatment product. Some moisturizing ingredients such as heparan sulfate may affect how well the skin cells “hear” and respond to signals elicited by the treatment products. For this reason, moisturizers should also be chosen to improve the efficacy of the treatment product.
Retinoids
When using retinoids for the first time in a patient, applying them last on top of the moisturizer can reduce the incidence of side effects and increase compliance. Retinoids, unlike other ingredients, penetrate easily into the deeper layer of the epidermis. Layering them on top of a moisturizer can help titrate retinoid absorption. The moisturizer can be chosen to slow or increase penetration of retinoids. Retinoids should always be used at nighttime because many of them, especially retinol and tretinoin, are easily broken down by ultraviolet light exposure.
Selecting across brands and applying products in the right order
Manufacturers rarely perform research on a complete regimen, but rather on individual products. Dermatologists then are left to figure this out on their own. I recommend choosing the best technologies from each brand based on the patient’s Baumann Skin Type and combining them using the recommended layering technique. I choose the best “hero” products from the various brands and layer them in a sequence that increases efficacy of all of the products. I then test the entire regimen on patients to figure out what combinations have the best efficacy and fewest side effects. Once I solve this “regimen puzzle,” I program software to automatically generate the step-by-step regimen instructions by Baumann Skin Type so that I do not have to rethink this complicated subject matter with every patient. I have developed over 3,300 distinct regimens so I am certain that my patients will get the proper skin care regimen advice from me or any of my staff.
Conclusion
Dermatologists can make a significant difference in their patients’ long-term skin care health by assisting them in identifying the proper skin care formulations for their individual skin type and guiding them as to how much, and in which order, to apply the products in their personalized skin care regimen. Patients will not remember what you told them and will confuse the order in which products should be used. For this reason, providing a written step-by-step skin care regimen is paramount to ensuring patient compliance.

Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002) and “Cosmeceuticals and Cosmetic Ingredients” (New York: McGraw-Hill, 2014); she also authored a New York Times Best Seller for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance Therapeutics. She is the founder and CEO of Skin Type Solutions Franchise Systems LLC. Write to her at [email protected].
References
1. Man MQ M et al. Optimization of physiological lipid mixtures for barrier repair. J Invest Dermatol. 1996 May;106(5):1096-101.
2. Storm A et al. One in 3 prescriptions are never redeemed: Primary nonadherence in an outpatient clinic. J Am Acad Dermatol. 2008 Jul;59(1):27-33.
3. Baumann LS. The Baumann Skin Typing System, in Farage MA et al. “Textbook of Aging Skin.” Springer-Verlag Berlin Heidelberg, 2017, pp. 1579-94.
4. Pinnell SR et al. Topical L-ascorbic acid: Percutaneous absorption studies. Dermatol Surg. 2001 Feb; 27(2):137-42.
Dermatologists are well suited to understand cosmeceutical science and the benefits of particular cosmeceutical products – especially if they are readers of this column. However, there is another critical thought process that must be undertaken when designing an efficacious skin care regimen for patients: Topical products should be applied in a particular order to maximize efficacy. This is because cosmeceutical ingredients interact with, change each other, and are affected by temperature, pH, humidity, and the microbiome in which they are in contact. This column focuses on the factors that should be considered when recommending skin care regimens to patients and in which order to apply topical products.
The chemistry of ingredients and how they interact is well understood by personal care product formulators. I think of formulators as chefs who are using ingredients and placing them in the formulation in a well-defined order under controlled circumstances that affect the final product. For example, ceramides are used in barrier repair moisturizers. The right form of ceramide must be chosen and used with the 1:1:1 ratio of ceramides, fatty acids, and cholesterol for the product to be effective at repairing the barrier.1 However, the order of when the ceramides are added to the product formula also impacts efficacy. Waxy ceramides and cholesterol require heat to liquefy and form the proper mixture with the other ingredients. Too much heat can damage fatty acids. Also, heat can inactivate finicky active ingredients such as vitamins C and E. For this reason, the ceramides and cholesterol are often incorporated first, allowing the formula to cool before the active heat labile ingredients are added. The speed at which something is mixed can generate heat and affect the final preparations, so temperature is an important consideration at all steps in the formulation procedure.
Just as the order of creating formulations affects the final product, the order of product placement on the skin influences skin care product efficacy. If a low pH skin care product (such as a glycolic acid cleanser) is used on the skin, this is going to affect the efficacy and safety of the product that is applied next to the skin. Such a chemical phenomenon should be considered when designing the order of product applications when designing a skin care regimen, particularly when incorporating ingredients that are known to interact with other ingredients, such as benzoyl peroxide, retinoids, hydroxy acids, hydroquinone, vitamin C, and peptides.
Efficacy and compliance in product layering
. Acne treatments are a good example. Patients are often prescribed a retinoid, benzoyl peroxide, topical antibiotic, and/or salicylic acid treatment product for acne. If the proper cleansers and moisturizers are not chosen, the patient will be more likely to develop redness and scaling and become noncompliant.
Compliance is a concerning issue to dermatologists because studies have shown that 95% of people underdose and one out of every three prescriptions is not even filled.2 If patients develop side effects, they are more likely to underdose or stop the treatment. Prescribing the proper cleanser and moisturizer to accompany treatment products will ease side effects and increase compliance. Several studies have demonstrated that the best way to increase compliance is to provide patients with written instructions, so they understand the proper order in which to apply products.
The role of cleansers
Cleansers can alter the pH of the skin, loosen attachments between cells, remove lipids – and disrupt the bilayer protective membrane, desquamate layers from the stratum corneum, and influence the penetrability of the skin for the next topical product that is applied. Therefore, cleansers should be selected based on the products that will follow them in the regimen. In addition, cleansers should be chosen according to the patient’s Baumann Skin Type.3 For example, cleansers for use on oily skin should have the ability to remove excess sebum on the skin while cleansers designed for dry skin would not remove as many lipids from the skin. Washing skin with a foaming cleanser can disrupt the skin barrier, allowing increased penetration of the treatment product that follows it. Oleic acid, hyaluronic acid, stearic acid, and other lipids are among the ingredients that influence skin penetration. Cleansers should precede treatment products and should be designed to increase efficacy of the treatment product. For this reason, every ingredient and characteristic of the chosen cleanser is important.
The role of eye products
Eye products treat issues such as dryness, puffiness, fine lines, and dark circles. However, they also play an overlooked role of protecting the thin delicate eye area from the treatment product. Using an eye product, especially one with protective ingredients such as barrier repair lipids, will help the patient tolerate the potentially irritating treatment product that follows the eye product. At night, the treatment product ingredients can get on the pillowcase and transfer to the upper and lower eyelids. Use of a protective eye product before bedtime can prevent the accompanying irritation. For example, acne patients often develop redness at the corners of the eyes when using benzoyl peroxide or a retinoid at night. Applying these medications after an eye cream can reduce this side effect.
Improving efficacy of treatment products
Treatments products are defined as corrective products targeted to skin issues such as acne, rosacea, melasma, dryness, skin cancer, eczema, psoriasis, and photoaging. The entire skin care regimen should be designed to enhance efficacy and decrease side effects of the treatment products. Treatment products may be cosmeceuticals, OTC medications, or prescription medications. These products must be able to reach their target in the proper chemical structure to be effective. Each ingredient has various constraints and quirks that should be considered. One well known example is ascorbic acid (vitamin C). Ascorbic acid is a treatment product for skin pigmentation and skin aging that is well known to have specific needs to work properly. Sheldon Pinnell, MD, led multiple investigations demonstrating that the maximum absorption of ascorbic acid occurs at a pH of 2-2.5. He showed that ascorbic acid products should be formulated at a pH of 2-2.5.4 However, applying these on skin that has just been cleansed with a soap cleanser with a pH of 9 will raise the skin’s pH and decrease the absorption of ascorbic acid. Having the patient cleanse with a low pH cleanser such as salicylic or glycolic acid cleanser (usually a pH of 2.5-3.5) will lower the pH of the skin and promote absorption of vitamin C.
The role of moisturizers
Moisturizers have many duties, including hydrating the skin, protecting the skin, and delivering important ingredients to the skin. However, moisturizers have a less discussed role of improving the efficacy of the treatment product that is applied beforehand. Moisturizers often contain oleic acid, hyaluronic acid, or other fatty acids that can increase penetration of other skin care ingredients. In addition, many moisturizers provide an occlusive effect that helps increase penetration. They also help protect the underlying treatment product from getting wiped off on a pillowcase or into the environment. In other words, moisturizers “seal in” the treatment product. Some moisturizing ingredients such as heparan sulfate may affect how well the skin cells “hear” and respond to signals elicited by the treatment products. For this reason, moisturizers should also be chosen to improve the efficacy of the treatment product.
Retinoids
When using retinoids for the first time in a patient, applying them last on top of the moisturizer can reduce the incidence of side effects and increase compliance. Retinoids, unlike other ingredients, penetrate easily into the deeper layer of the epidermis. Layering them on top of a moisturizer can help titrate retinoid absorption. The moisturizer can be chosen to slow or increase penetration of retinoids. Retinoids should always be used at nighttime because many of them, especially retinol and tretinoin, are easily broken down by ultraviolet light exposure.
Selecting across brands and applying products in the right order
Manufacturers rarely perform research on a complete regimen, but rather on individual products. Dermatologists then are left to figure this out on their own. I recommend choosing the best technologies from each brand based on the patient’s Baumann Skin Type and combining them using the recommended layering technique. I choose the best “hero” products from the various brands and layer them in a sequence that increases efficacy of all of the products. I then test the entire regimen on patients to figure out what combinations have the best efficacy and fewest side effects. Once I solve this “regimen puzzle,” I program software to automatically generate the step-by-step regimen instructions by Baumann Skin Type so that I do not have to rethink this complicated subject matter with every patient. I have developed over 3,300 distinct regimens so I am certain that my patients will get the proper skin care regimen advice from me or any of my staff.
Conclusion
Dermatologists can make a significant difference in their patients’ long-term skin care health by assisting them in identifying the proper skin care formulations for their individual skin type and guiding them as to how much, and in which order, to apply the products in their personalized skin care regimen. Patients will not remember what you told them and will confuse the order in which products should be used. For this reason, providing a written step-by-step skin care regimen is paramount to ensuring patient compliance.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002) and “Cosmeceuticals and Cosmetic Ingredients” (New York: McGraw-Hill, 2014); she also authored a New York Times Best Seller for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance Therapeutics. She is the founder and CEO of Skin Type Solutions Franchise Systems LLC. Write to her at [email protected].
References
1. Man MQ M et al. Optimization of physiological lipid mixtures for barrier repair. J Invest Dermatol. 1996 May;106(5):1096-101.
2. Storm A et al. One in 3 prescriptions are never redeemed: Primary nonadherence in an outpatient clinic. J Am Acad Dermatol. 2008 Jul;59(1):27-33.
3. Baumann LS. The Baumann Skin Typing System, in Farage MA et al. “Textbook of Aging Skin.” Springer-Verlag Berlin Heidelberg, 2017, pp. 1579-94.
4. Pinnell SR et al. Topical L-ascorbic acid: Percutaneous absorption studies. Dermatol Surg. 2001 Feb; 27(2):137-42.
Dermatologists are well suited to understand cosmeceutical science and the benefits of particular cosmeceutical products – especially if they are readers of this column. However, there is another critical thought process that must be undertaken when designing an efficacious skin care regimen for patients: Topical products should be applied in a particular order to maximize efficacy. This is because cosmeceutical ingredients interact with, change each other, and are affected by temperature, pH, humidity, and the microbiome in which they are in contact. This column focuses on the factors that should be considered when recommending skin care regimens to patients and in which order to apply topical products.
The chemistry of ingredients and how they interact is well understood by personal care product formulators. I think of formulators as chefs who are using ingredients and placing them in the formulation in a well-defined order under controlled circumstances that affect the final product. For example, ceramides are used in barrier repair moisturizers. The right form of ceramide must be chosen and used with the 1:1:1 ratio of ceramides, fatty acids, and cholesterol for the product to be effective at repairing the barrier.1 However, the order of when the ceramides are added to the product formula also impacts efficacy. Waxy ceramides and cholesterol require heat to liquefy and form the proper mixture with the other ingredients. Too much heat can damage fatty acids. Also, heat can inactivate finicky active ingredients such as vitamins C and E. For this reason, the ceramides and cholesterol are often incorporated first, allowing the formula to cool before the active heat labile ingredients are added. The speed at which something is mixed can generate heat and affect the final preparations, so temperature is an important consideration at all steps in the formulation procedure.
Just as the order of creating formulations affects the final product, the order of product placement on the skin influences skin care product efficacy. If a low pH skin care product (such as a glycolic acid cleanser) is used on the skin, this is going to affect the efficacy and safety of the product that is applied next to the skin. Such a chemical phenomenon should be considered when designing the order of product applications when designing a skin care regimen, particularly when incorporating ingredients that are known to interact with other ingredients, such as benzoyl peroxide, retinoids, hydroxy acids, hydroquinone, vitamin C, and peptides.
Efficacy and compliance in product layering
. Acne treatments are a good example. Patients are often prescribed a retinoid, benzoyl peroxide, topical antibiotic, and/or salicylic acid treatment product for acne. If the proper cleansers and moisturizers are not chosen, the patient will be more likely to develop redness and scaling and become noncompliant.
Compliance is a concerning issue to dermatologists because studies have shown that 95% of people underdose and one out of every three prescriptions is not even filled.2 If patients develop side effects, they are more likely to underdose or stop the treatment. Prescribing the proper cleanser and moisturizer to accompany treatment products will ease side effects and increase compliance. Several studies have demonstrated that the best way to increase compliance is to provide patients with written instructions, so they understand the proper order in which to apply products.
The role of cleansers
Cleansers can alter the pH of the skin, loosen attachments between cells, remove lipids – and disrupt the bilayer protective membrane, desquamate layers from the stratum corneum, and influence the penetrability of the skin for the next topical product that is applied. Therefore, cleansers should be selected based on the products that will follow them in the regimen. In addition, cleansers should be chosen according to the patient’s Baumann Skin Type.3 For example, cleansers for use on oily skin should have the ability to remove excess sebum on the skin while cleansers designed for dry skin would not remove as many lipids from the skin. Washing skin with a foaming cleanser can disrupt the skin barrier, allowing increased penetration of the treatment product that follows it. Oleic acid, hyaluronic acid, stearic acid, and other lipids are among the ingredients that influence skin penetration. Cleansers should precede treatment products and should be designed to increase efficacy of the treatment product. For this reason, every ingredient and characteristic of the chosen cleanser is important.
The role of eye products
Eye products treat issues such as dryness, puffiness, fine lines, and dark circles. However, they also play an overlooked role of protecting the thin delicate eye area from the treatment product. Using an eye product, especially one with protective ingredients such as barrier repair lipids, will help the patient tolerate the potentially irritating treatment product that follows the eye product. At night, the treatment product ingredients can get on the pillowcase and transfer to the upper and lower eyelids. Use of a protective eye product before bedtime can prevent the accompanying irritation. For example, acne patients often develop redness at the corners of the eyes when using benzoyl peroxide or a retinoid at night. Applying these medications after an eye cream can reduce this side effect.
Improving efficacy of treatment products
Treatments products are defined as corrective products targeted to skin issues such as acne, rosacea, melasma, dryness, skin cancer, eczema, psoriasis, and photoaging. The entire skin care regimen should be designed to enhance efficacy and decrease side effects of the treatment products. Treatment products may be cosmeceuticals, OTC medications, or prescription medications. These products must be able to reach their target in the proper chemical structure to be effective. Each ingredient has various constraints and quirks that should be considered. One well known example is ascorbic acid (vitamin C). Ascorbic acid is a treatment product for skin pigmentation and skin aging that is well known to have specific needs to work properly. Sheldon Pinnell, MD, led multiple investigations demonstrating that the maximum absorption of ascorbic acid occurs at a pH of 2-2.5. He showed that ascorbic acid products should be formulated at a pH of 2-2.5.4 However, applying these on skin that has just been cleansed with a soap cleanser with a pH of 9 will raise the skin’s pH and decrease the absorption of ascorbic acid. Having the patient cleanse with a low pH cleanser such as salicylic or glycolic acid cleanser (usually a pH of 2.5-3.5) will lower the pH of the skin and promote absorption of vitamin C.
The role of moisturizers
Moisturizers have many duties, including hydrating the skin, protecting the skin, and delivering important ingredients to the skin. However, moisturizers have a less discussed role of improving the efficacy of the treatment product that is applied beforehand. Moisturizers often contain oleic acid, hyaluronic acid, or other fatty acids that can increase penetration of other skin care ingredients. In addition, many moisturizers provide an occlusive effect that helps increase penetration. They also help protect the underlying treatment product from getting wiped off on a pillowcase or into the environment. In other words, moisturizers “seal in” the treatment product. Some moisturizing ingredients such as heparan sulfate may affect how well the skin cells “hear” and respond to signals elicited by the treatment products. For this reason, moisturizers should also be chosen to improve the efficacy of the treatment product.
Retinoids
When using retinoids for the first time in a patient, applying them last on top of the moisturizer can reduce the incidence of side effects and increase compliance. Retinoids, unlike other ingredients, penetrate easily into the deeper layer of the epidermis. Layering them on top of a moisturizer can help titrate retinoid absorption. The moisturizer can be chosen to slow or increase penetration of retinoids. Retinoids should always be used at nighttime because many of them, especially retinol and tretinoin, are easily broken down by ultraviolet light exposure.
Selecting across brands and applying products in the right order
Manufacturers rarely perform research on a complete regimen, but rather on individual products. Dermatologists then are left to figure this out on their own. I recommend choosing the best technologies from each brand based on the patient’s Baumann Skin Type and combining them using the recommended layering technique. I choose the best “hero” products from the various brands and layer them in a sequence that increases efficacy of all of the products. I then test the entire regimen on patients to figure out what combinations have the best efficacy and fewest side effects. Once I solve this “regimen puzzle,” I program software to automatically generate the step-by-step regimen instructions by Baumann Skin Type so that I do not have to rethink this complicated subject matter with every patient. I have developed over 3,300 distinct regimens so I am certain that my patients will get the proper skin care regimen advice from me or any of my staff.
Conclusion
Dermatologists can make a significant difference in their patients’ long-term skin care health by assisting them in identifying the proper skin care formulations for their individual skin type and guiding them as to how much, and in which order, to apply the products in their personalized skin care regimen. Patients will not remember what you told them and will confuse the order in which products should be used. For this reason, providing a written step-by-step skin care regimen is paramount to ensuring patient compliance.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002) and “Cosmeceuticals and Cosmetic Ingredients” (New York: McGraw-Hill, 2014); she also authored a New York Times Best Seller for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance Therapeutics. She is the founder and CEO of Skin Type Solutions Franchise Systems LLC. Write to her at [email protected].
References
1. Man MQ M et al. Optimization of physiological lipid mixtures for barrier repair. J Invest Dermatol. 1996 May;106(5):1096-101.
2. Storm A et al. One in 3 prescriptions are never redeemed: Primary nonadherence in an outpatient clinic. J Am Acad Dermatol. 2008 Jul;59(1):27-33.
3. Baumann LS. The Baumann Skin Typing System, in Farage MA et al. “Textbook of Aging Skin.” Springer-Verlag Berlin Heidelberg, 2017, pp. 1579-94.
4. Pinnell SR et al. Topical L-ascorbic acid: Percutaneous absorption studies. Dermatol Surg. 2001 Feb; 27(2):137-42.
Idiopathic pulmonary fibrosis: What primary care physicians need to know
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?

MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS

Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
")
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.

When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
")
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.

Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?
MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS
Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.
When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.
Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
Idiopathic pulmonary fibrosis (IPF) is a devastating and fatal lung disease that generally affects older adults. It is characterized by a radiographic and histopathologic pattern of usual interstitial pneumonia (UIP) that has no other known etiology.
Accurate diagnosis of IPF is crucial. We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis, start appropriate therapy, advise the patient on prognosis and enrollment in disease registries and clinical trials, and determine candidacy for lung transplant.
Primary care physicians are uniquely positioned to encounter patients with IPF, whether because of a patient complaint or as an incidental finding on computed tomography. The goal of this article is to delineate the features of IPF so that it may be recognized early and thus expedite referral to a center with expertise in interstitial lung disease for a thorough evaluation and appropriate management.
WHAT IS IDIOPATHIC PULMONARY FIBROSIS?
MORE COMMON THAN ONCE THOUGHT
The true incidence and prevalence of IPF are difficult to assess. IPF is generally considered a rare disease, but it is more common than once thought. In 2011, Raghu et al3 estimated the prevalence in Medicare beneficiaries to be 495 cases per 100,000. Based on this estimate and the current US population, up to 160,000 Americans could have IPF.4 Raghu et al also showed that IPF more often affects adults over age 65, which suggests that as the US population ages, the incidence of IPF may rise. Studies have also reported an increased incidence of IPF worldwide.5
Further, with the rising use of low-dose computed tomography to screen for lung cancer, more incidental cases of IPF will likely be found.6–8
Older data showed a lag from symptom onset to accurate diagnosis of 1 to 2 years.9 A more recent study found a lag in referral of patients with IPF to tertiary care centers, and this delay was associated with a higher rate of death independent of disease severity.10
TYPICALLY PROGRESSIVE, OFTEN FATAL
IPF is typically progressive and limited to the lungs, and it portends a poor prognosis. The median survival is commonly cited as 2 to 5 years from diagnosis, although this is based on older observations that may not reflect current best practice and newer therapies. More recent studies suggest higher survival rates if patients have preserved lung function.11
As the name indicates, the etiology of IPF is unknown, but studies have indicated genetic underpinnings in a notable proportion of cases.12 Regardless of the cause, the pathogenesis and progression of IPF are thought to be the result of an abnormal and persistent wound-repair response. The progressive deposition of scar tissue disrupts normal lung architecture and function, eventually causing clinical disease.13
SYMPTOMS AND KEY FEATURES
Patients with IPF typically present with the insidious onset of dyspnea on exertion, with or without chronic cough. Risk factors include male sex, increasing age, and a history of smoking. Patients with undiagnosed IPF who present with dyspnea and a history of smoking are often treated empirically for chronic obstructive pulmonary disease (COPD).
Rales are a common finding on auscultation in IPF, and this can lead to an exhaustive cardiac evaluation and empiric treatment for heart failure. Digital clubbing is also relatively common.14 Hypoxemia with exertion is another common feature that also often correlates with disease severity and prognosis. Resting hypoxemia is more common in advanced disease.
On spirometry, patients with IPF typically demonstrate restrictive physiology, suggested by a normal or elevated ratio of the forced expiratory volume in 1 second to the forced vital capacity (FEV1/FVC) (> 70% predicted or above the lower limit of normal) combined with a lower than normal FVC. Restrictive physiology is definitively demonstrated by a decreased total lung capacity (< 80% predicted or below the lower limit of normal) on plethysmography. Impaired gas exchange, manifested by a decreased diffusing capacity of the lungs for carbon monoxide (DLCO) on pulmonary function testing, is also common. Because pulmonary perfusion is higher in the lung bases, where IPF is also predominant, the DLCO is often reduced to a greater extent than the FVC.
PROGNOSTIC INDICATORS
Clinicians typically view IPF as a relentless and progressive process, but its course is variable and can be uncertain in an individual patient (Figure 1).15,16 Nevertheless, over time, most patients have a decline in lung function leading to respiratory failure. Respiratory failure, often preceded by a subacute deterioration (over weeks to months) or an acute deterioration (< 4 weeks), is the most common cause of death, but comorbid diseases such as lung cancer, infection, and heart failure are also common causes of death in these patients.17,18
Predictors of mortality include worsening FVC, DLCO, symptoms, and physiologic impairment, manifested by a decline in the 6-minute walking test or worsening exertional hypoxemia.19–22 Other common comorbidities linked with impaired quality of life and poor prognosis include obstructive sleep apnea, gastroesophageal reflux disease, and depression.16,23 Retrospective studies suggest that most IPF patients die 2 to 5 years after symptom onset. With the lag from symptom onset to final diagnosis, the average life expectancy is as little as 2 years from the time of diagnosis.9,18,24,25
Two staging systems have been developed to predict short-term and long-term mortality risk based on sex, age, and physiologic parameters.23,24 The GAP (gender, age, physiology) index provides an estimate of the risk of death for a cohort of patients: a score of 0 to 8 is calculated, and the score is then categorized as stage I, II, or III. Each stage is associated with 1-, 2-, and 3-year mortality rates, with stage III having the highest rates. The GAP calculator (www.acponline.org/journals/annals/extras/gap) provides an estimate of the risk of death for an individual patient. The application of these tools for the management of IPF is evolving; however, they may be helpful for counseling patients about disease prognosis.
CLUES TO DIAGNOSIS
Histologic patterns
UIP on histologic study is also seen in fibrotic lung diseases other than IPF, including connective tissue disease-associated interstitial lung disease, inhalational or occupational interstitial lung disease, and chronic hypersensitivity pneumonitis.26–29 Consequently, the diagnosis of IPF requires exclusion of other known causes of UIP.
According to the 2011 guidelines,16 the histology of interstitial lung disease can be categorized as definite UIP, probable UIP, or possible UIP, or as an atypical pattern suggesting another diagnosis. If no definite cause of the interstitial lung abnormality is found, the level of certainty of the histopathologic pattern of UIP helps formulate the clinical diagnosis and management plan.
Clues on computed tomography
The UIP nomenclature also describes patterns on high-resolution computed tomography (HRCT). HRCT is done without contrast and produces thin-sliced images (usually < 1.5 mm) in inspiratory, expiratory, and prone views; this allows detection of air trapping, which may indicate an airway-centric alternative diagnosis.
On HRCT, UIP appears as reticular opacities, often with traction bronchiectasis or bronchiolectasis, usually with a basilar and peripheral predominance. Honeycombing is a key feature and appears as clustered cystic spaces with well-defined walls in the periphery of the lung parenchyma. Ground-glass opacities are not a prominent feature of UIP, and although they do not exclude a UIP pattern, they should spur consideration of other diagnoses.16 Reactive mediastinal and hilar lymphadenopathy is another common feature of UIP.
When evaluating results of HRCT for UIP, the radiologist categorizes the pattern as definite UIP, possible UIP, or inconsistent. The definite pattern meets all the above features and has none of the features suggesting an alternative diagnosis (Figure 3). The possible pattern includes all the above features with the exception of honeycombing. If the predominant features on HRCT include any atypical finding listed above, then the study is considered inconsistent with UIP. If the pattern on HRCT is considered definite, evaluation of pathology is not necessary. If the pattern is categorized as possible or is inconsistent, then surgical lung biopsy-confirmed UIP is necessary for the definitive diagnosis of IPF.
However, evidence is emerging that in the correct clinical scenario, possible UIP behaves similarly to definite UIP and may be sufficient to make the clinical diagnosis of IPF even without surgical biopsy confirmation.30
A DIAGNOSTIC ALGORITHM FOR IPF
Given the multitude of interstitial lung diseases, their complexities, and the lack of a gold standard definitive diagnostic test, the diagnosis of IPF can be difficult, requiring the integration of clinical, radiologic, and, if necessary, pathologic findings.
Demographic features
The patient’s demographic features and risk factors dictate the initial clinical suspicion of IPF compared with other interstitial lung diseases. The incidence of IPF increases with age, and IPF is more common in men. A history of smoking is another risk factor.31 A 45-year-old never-smoker is much less likely to have IPF than a 70-year-old former smoker, and a 70-year-old man is more likely to have IPF than a woman of the same age. Thus, the finding of interstitial lung disease in a patient with a demographic profile that is not typical (ie, a younger woman who never smoked) should prompt an exhaustive investigation for another diagnosis such as hypersensitivity pneumonitis or connective tissue disease.
Key elements of the history
After considering the demographic profile and risk factors, the next step in the evaluation is a thorough and accurate medical history. This should include assessment of the severity of dyspnea and cough, signs and symptoms of connective tissue disease (eg, arthralgias, sicca symptoms, Raynaud phenomenon, difficulty swallowing), and gastroesophageal reflux disease, which can be associated with connective tissue disease and, independently, with IPF.
It is also important to identify any environmental exposures that suggest pneumoconiosis or chronic hypersensitivity pneumonitis. The most common risk factors for hypersensitivity pneumonitis are birds and bird feathers, molds, fungi, hot tub use, and some industrial chemicals.32
A medication history is important. Many medications are associated with interstitial lung disease, but amiodarone, bleomycin, methotrexate, and nitrofurantoin are among the common offenders.33
A thorough family history is necessary, as there are familial forms of IPF.
Focus of the physical examination
The physical examination must include careful auscultation for rales. While rales are not specific for IPF, they are the most common pulmonary abnormality. Detailed skin, musculoskeletal, and cardiovascular examinations are also important to evaluate for rheumatologic signs, clubbing, or evidence of heart failure or pulmonary hypertension.
Laboratory tests
Laboratory testing should include a serologic autoantibody panel to evaluate for connective tissue diseases that can manifest as interstitial lung disease, including rheumatoid arthritis, dermatopolymyositis, scleroderma, Sjögren syndrome, and undifferentiated or mixed connective tissue disease. Typical preliminary laboratory tests are antinuclear antibody, rheumatoid factor, erythrocyte sedimentation rate, and C-reactive protein. Others may include anticyclic citrullinated peptide (anti-CCP), anti-Scl-70, anti-RNP, anti-SS-A, anti-SS-B, and anti-Jo-1.16 The breadth of the panel should depend on patient demographics and findings in the history or physical examination that increase or decrease the likelihood of a connective tissue disease.
Lung function testing
Assessing the patient’s pulmonary physiology should include spirometry, DLCO, and body plethysmography (lung volumes). In most cases, IPF manifests with restrictive physiology. Once restrictive physiology is confirmed with a low total lung capacity, FVC testing can be used as a longitudinal surrogate, as it is less expensive and easier for the patient to perform. In general, a lower total lung capacity or FVC indicates more severe impairment.
The DLCO serves as another marker of severity but is less reliable due to baseline variability and difficulties performing the maneuver.
A 6-minute walk test is another crucial physiologic assessment tool that can quantify exertional hypoxemia and functional status (ie, distance walked), and can assist in prognosis.
Imaging
Most patients undergo chest radiography in the workup for undiagnosed dyspnea. However, chest radiography is not adequate to formulate an accurate diagnosis in suspected interstitial lung disease, and a normal radiograph cannot exclude changes that might reflect early phases of the disease. As the disease progresses, the plain radiograph can show reticulonodular opacities and honeycombing in the peripheral and lower lung zones (Figure 3).34
The decision whether to order HRCT in the workup for a patient who has dyspnea and a normal chest radiograph is challenging. We recommend cross-sectional imaging when physiologic testing shows restriction or low DLCO, or when there is a high index of suspicion for underlying lung disease as the cause of symptoms.
Expert consultation can aid with this decision, especially when the underlying cause of dyspnea remains unclear after initial studies have been completed. Otherwise, HRCT is an essential test in the evaluation of interstitial lung disease.
Bronchoscopy’s role controversial
If the pattern on HRCT is nondiagnostic, then surgical biopsy is necessary, and the diagnosis of IPF requires a histologic pattern of UIP as described above.16,35
Although bronchoscopy can be valuable if an alternative diagnosis such as sarcoidosis or chronic hypersensitivity pneumonitis is suspected, the role of bronchoscopic biopsy in the workup of IPF is controversial. The patchy nature of UIP does not lend itself to the relatively small biopsy samples obtained through bronchoscopy.36,37
Surgical biopsy options
The favored biopsy approach is surgical, using either an open or a video-assisted thoracoscopic technique. The latter is preferred as it is less invasive, requires a shorter length of hospital stay, and allows a faster recovery.38 Bronchoscopic cryobiopsy, currently under investigation, is a potentially valuable tool whose role in diagnosing IPF is evolving.
Frequently, neither HRCT nor surgical lung biopsy demonstrates UIP, making the definitive diagnosis of IPF difficult. Moreover, some patients with nondiagnostic HRCT results are unable to tolerate surgical lung biopsy because of severely impaired lung function or other comorbidities.
The role of multidisciplinary discussions
When surgical lung biopsy is not possible, current practice at leading centers uses a multidisciplinary approach to allow for a confident diagnosis.30,39 Discussions take place between pulmonologists, pathologists, radiologists, and other specialists to collectively consider all aspects of a case before rendering a consensus opinion on the diagnosis and the management plan. If the discussion leads to a consensus diagnosis of IPF, then the patient’s clinician can move forward with treatment options. If not, the group can recommend further workup or alternative diagnoses and treatment regimens. The multidisciplinary group is also well positioned to consider the relative risks and benefits of moving forward with surgical lung biopsy for individual patients.
This approach illustrates the importance of referring these patients to centers of excellence in diagnosing and managing complex cases of interstitial lung disease, including IPF.40
TREATMENT OF IPF
Antifibrotic therapy
Antifibrotic therapy is a choice between pirfenidone and nintedanib.
Pirfenidone, which has an undefined molecular target, was approved based on the results of 3 trials.41,42 Pooled analyses from these trials showed a reduction in the decline from baseline in FVC percent predicted and improved progression-free survival.43 Pooled and meta-analyses of pirfenidone clinical trials have shown a mortality benefit, although no individual study has shown such an effect on mortality rates.44
The major adverse effects of pirfenidone are gastrointestinal distress and photosensitivity rash.
Nintedanib is a triple tyrosine kinase inhibitor that broadly targets fibroblast growth factor, vascular endothelial growth factor, and platelet-derived growth factor receptors. Combined analysis of 2 concurrent trials45 showed that nintedanib reduced the decline in FVC, similarly to pirfenidone. The major adverse event associated with nintedanib was diarrhea. Since it inhibits vascular endothelial growth factor, there is a risk of hematologic complications such as bleeding or clotting events.
Because pirfenidone and nintedanib can increase aminotransferase levels, regular monitoring is recommended.
To date, no trial has compared pirfenidone and nintedanib in terms of their efficacy and tolerability. Therefore, the choice of agent is based on the patient’s preference after a discussion of potential risks and expected benefits, a review of each drug’s side effects, and consideration of comorbid conditions and physician experience.
Patients need to understand that these drugs slow the rate of decline in FVC but have not been shown to improve symptoms or functional status.
Corticosteroids are not routine
Corticosteroids should not be used routinely in the treatment of IPF. Although steroids, alone or in combination with other immunosuppressive medications, were commonly used for IPF in the past, such use was not based on results of randomized controlled trials.46 Retrospective controlled studies have failed to show that corticosteroids improve mortality rates in IPF; indeed, they have shown that corticosteroids confer substantial morbidity.47,48 In addition, a randomized controlled trial combining corticosteroids with N-acetylcysteine and azathioprine was stopped early due to an increased risk of death and hospitalization.49 Collectively, these data suggest that corticosteroids confer no benefit and are potentially harmful. Their use in IPF is discouraged, and the joint international guidelines recommend against immunosuppression to treat IPF.16
Other treatments
The guidelines offer additional suggestions for the management of IPF.
Preliminary evidence suggests that microaspiration associated with abnormal gastroesophageal acid reflux is a risk factor for IPF. As such, there is a weak recommendation for aggressive treatment of reflux disease.50 However, because evidence suggests that proton-pump inhibitor therapy may be associated with adverse renal or central nervous system effects, this recommendation bears caution. It is hoped that ongoing studies will provide further insight into the role of acid-suppression in the management of IPF.51,52
Further treatment recommendations include best supportive management such as supplemental oxygen, pulmonary rehabilitation, and vaccinations.
Prompt referral for lung transplant is imperative. IPF is now the most common indication for lung transplant, and given the poor overall prognosis of advanced IPF, transplant confers a survival benefit in appropriately selected patients.53,54 Table 2 provides an evidence-based checklist for the workup and management of IPF.
ACUTE EXACERBATIONS OF IPF
The unpredictable nature of IPF can manifest in the form of acute exacerbations without an identifiable cause. The loosely defined diagnostic criteria for the diagnosis of acute exacerbations are a previous or new diagnosis of IPF, worsening or development of dyspnea in the last 30 days, and new bilateral ground-glass or consolidative changes with a background of UIP on HRCT.16
A new definition has been proposed55 to facilitate research in the characterization and treatment of acute exacerbations of IPF. The new definition includes all causes of respiratory deterioration except for heart failure and volume overload. It is less strict about the 30-day time frame. This newer definition is based on the lack of evidence differentiating outcomes when an acute deterioration is associated with known or unknown etiologies.55
The incidence of acute exacerbations is variable, with a 1- and 3-year incidence ranging between 8.6% and 23.9% depending on the criteria used.56 In general, acute exacerbations carry a grim prognosis, with a median life expectancy of 2.2 months.57
There is no approved therapy for exacerbations of IPF. Rather, treatment is mainly supportive with supplemental oxygen and mechanical ventilation. Current guidelines have a weak recommendation for the use of corticosteroids, but there are no recommendations regarding dose, route, or duration of therapy. Other treatments, primarily immunomodulatory agents, have been suggested but lack evidence of benefit.
Acknowledgments: Pathology images were provided by Carol Farver, MD, Pathology Institute, Cleveland Clinic. Radiology images were provided by Ruchi Yadav, MD, Imaging Institute, Cleveland Clinic.
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
- Brown KK, Raghu G. Medical treatment for pulmonary fibrosis: current trends, concepts, and prospects. Clin Chest Med 2004; 25(4):759–772, vii. doi:10.1016/j.ccm.2004.08.003
- Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 2013; 19(5):453–459. doi:10.1097/MCP.0b013e328363f48d
- Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med 2014; 2(7):566–572. doi:10.1016/S2213-2600(14)70101-8
- Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012; 21(126):355–361. doi:10.1183/09059180.00002512
- Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46(3):795–806. doi:10.1183/09031936.00185114
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268(2):563–571. doi:10.1148/radiol.13120816
- Southern BD, Scheraga RG, Yadav R. Managing interstitial lung disease detected on CT during lung cancer screening. Cleve Clin J Med 2016; 83(1):55–65. doi:10.3949/ccjm.83a.14157
- King TE Jr, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 2001; 164(5):1025–1032. doi:10.1164/ajrccm.164.6.2001056
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184(7):842–847. doi:10.1164/rccm.201104-0668OC
- Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med 2018; 18(1):19. doi:10.1186/s12890-018-0575-y
- Yang IV, Schwartz DA. Epigenetics of idiopathic pulmonary fibrosis. Transl Res 2015; 165(1):48–60. doi:10.1016/j.trsl.2014.03.011
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378(9807):1949–1961. doi:10.1016/S0140-6736(11)60052-4
- Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3:8. doi:10.1186/1750-1172-3-8
- Raghu G. Idiopathic pulmonary fibrosis. A rational clinical approach. Chest 1987; 92(1):148–154. doi:10.1378/chest.92.1.148
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Panos RJ, Mortenson RL, Niccoli SA, King TE Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88(4):396–404. doi:10.1016/0002-9343(90)90495-Y
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183(4):431–440. doi:10.1164/rccm.201006-0894CI
- Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168(5):538–542. doi:10.1164/rccm.200211-1311OC
- Flaherty KR, Andrei AC, Murray S, et al. Idiopathic pulmonary fibrosis: prognostic value of changes in physiology and six-minute-walk test. Am J Respir Crit Care Med 2006; 174(7):803–809. doi:10.1164/rccm.200604-488OC
- Jegal Y, Kim DS, Shim TS, et al. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med 2005; 171(6):639–644. doi:10.1164/rccm.200403-331OC
- Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5):531–537. doi:10.1164/rccm.200210-1245OC
- King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med 2017; 5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
- Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156(1):684–691. doi:10.7326/0003-4819-156-10-201205150-00004
- Rudd RM, Prescott RJ, Chalmers JC, Johnston ID; Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. British Thoracic Society study on cryptogenic fibrosing alveolitis: response to treatment and survival. Thorax 2007; 62(1):62–66. doi:10.1136/thx.2005.045591
- Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease-associated interstitial lung disease: a review. Curr Respir Care Rep 2012; 1:224–232. doi:10.1007/s13665-012-0028-7
- Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med 2007; 175(7):705–711. doi:10.1164/rccm.200607-912OC
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proc Am Thorac Soc 2006; 3(4):293–298. doi:10.1513/pats.200512-131TK
- Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med 2004; 116(10):662–668. doi:10.1016/j.amjmed.2003.12.030
- Brownell R, Moua T, Henry TS, et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017; 72(5):424–429. doi:10.1136/thoraxjnl-2016-209671
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1997; 155(1):242–248. doi:10.1164/ajrccm.155.1.9001319
- Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012; 186(4):314–324. doi:10.1164/rccm.201203-0513CI
- Schwaiblmair M, Behr W, Haeckel T, Markl B, Foerg W, Berghaus T. Drug induced interstitial lung disease. Open Respir Med J 2012; 6:63–74. doi:10.2174/1874306401206010063
- Grenier P, Valeyre D, Cluzel P, Brauner MW, Lenoir S, Chastang C. Chronic diffuse interstitial lung disease: diagnostic value of chest radiography and high-resolution CT. Radiology 1991; 179(1):123–132. doi:10.1148/radiology.179.1.2006262
- Lynch JP 3rd, Huynh RH, Fishbein MC, Saggar R, Belperio JA, Weigt SS. Idiopathic pulmonary fibrosis: epidemiology, clinical features, prognosis, and management. Semin Respir Crit Care Med 2016; 37(3):331–357. doi:10.1055/s-0036-1582011
- Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest 2006; 129(5):1126–1131. doi:10.1378/chest.129.5.1126
- Ohshimo S, Bonella F, Cui A, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009; 179(11):1043–1047. doi:10.1164/rccm.200808-1313OC
- Oparka J, Yan TD, Ryan E, Dunning J. Does video-assisted thoracic surgery provide a safe alternative to conventional techniques in patients with limited pulmonary function who are otherwise suitable for lung resection? Interact Cardiovasc Thorac Surg 2013; 17(1):159–162. doi:10.1093/icvts/ivt097
- Flaherty KR, King TE Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8):904–910. doi:10.1164/rccm.200402-147OC
- Walsh SL, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med 2016; 4(7):557–565. doi:10.1016/S2213-2600(16)30033-9
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779):1760–1769. doi:10.1016/S0140-6736(11)60405-4
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J 2016; 47(1):243–253. doi:10.1183/13993003.00026-2015
- Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5(1):33–41. doi:10.1016/S2213-2600(16)30326-5
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003: 3:CD002880. doi:10.1002/14651858.CD002880
- Douglas WW, Ryu JH, Schroeder DR. Idiopathic pulmonary fibrosis: impact of oxygen and colchicine, prednisone, or no therapy on survival. Am J Respir Crit Care Med 2000; 161(4 pt 1):1172–1178. doi:10.1164/ajrccm.161.4.9907002
- Gay SE, Kazerooni EA, Toews GB, et al. Idiopathic pulmonary fibrosis: predicting response to therapy and survival. Am J Respir Crit Care Med 1998; 157(4 pt 1):1063–1072. doi:10.1164/ajrccm.157.4.9703022
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- Raghu G, Freudenberger TD, Yang S, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006; 27(1):136–142. doi:10.1183/09031936.06.00037005
- Gomm W, von Holt K, Thome F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol 2016; 73(4):410–416. doi:10.1001/jamaneurol.2015.4791
- Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z. Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 2016; 27(10):3153–3163. doi:10.1681/ASN.2015121377
- Thabut G, Mal H, Castier Y, et al. Survival benefit of lung transplantation for patients with idiopathic pulmonary fibrosis. J Thorac Cardiovasc Surg 2003; 126(2):469–475. doi:10.1016/S0022-5223(03)00600-7
- Valapour M, Skeans MA, Smith JM, et al. Lung. Am J Transplant 2016; 16(suppl 2):141–168. doi:10.1111/ajt.13671
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016; 194(3):265–275. doi:10.1164/rccm.201604-0801CI
- Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27(2):103–110. doi:10.1016/j.resinv.2015.04.005
- Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37(2):356–363. doi:10.1183/09031936.00159709
KEY POINTS
- IPF is characterized by a pattern of usual interstitial pneumonia on imaging and histopathology without another known etiology.
- We recommend early referral to a center specializing in interstitial lung disease to confirm the diagnosis and to initiate appropriate therapy.
- Specialized centers offer advice on prognosis, enrollment in disease registries and clinical trials, and candidacy for lung transplant.
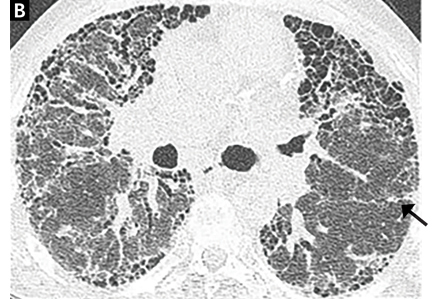
It takes a village to care for the patient with idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive fibrosing interstitial lung disease associated with a high burden of morbidity and death.1 A clinical diagnosis of IPF is made only after careful interpretation of integrated clinical, radiologic, and often histopathologic data.
Interstitial lung disease encompasses a broad spectrum of parenchymal lung diseases, and a classification of IPF is restricted to a lung injury pattern of usual interstitial pneumonia (UIP) based on high-resolution computed tomography or surgical lung biopsy, after all known causes of UIP have been excluded.1
However, a lung injury pattern of UIP is not synonymous with IPF, as UIP can be seen with connective tissue disease, chronic hypersensitivity pneumonitis, drug toxicity, and sarcoidosis.1 As such, rendering a diagnosis of IPF requires a thorough evaluation to exclude such diverse potential etiologies.
In this issue of the Cleveland Clinic Journal of Medicine, Tolle and colleagues2 provide an up-to-date, broad overview of IPF focused on what the primary care provider needs to know about the disease. Their review is timely and serves as a useful primer for the practicing clinician.
The field of IPF is actively evolving, as this era has been witness to a recent paradigm shift in pharmacologic management. Immunosuppression is no longer recommended3 and may even be harmful.4 And the US Food and Drug Administration has approved 2 antifibrotic drugs—pirfenidone and nintedanib—that have been shown to delay progression of IPF.5,6
Primary care providers have a unique opportunity to play an integral role in the evaluation and care of patients with IPF, in particular with earlier disease recognition, initial disease assessment, and timely specialty consultative referral—as well as implementing a comprehensive longitudinal care plan.
EARLIER DISEASE RECOGNITION
IPF is a rare disease primarily affecting men over the age of 65.1 It is reasonable to presume that many or most of these individuals ultimately diagnosed with IPF are already seeking routine care for existing common medical conditions such as hypertension or dyslipidemia—or at least having periodic routine health maintenance assessments. Such evaluations may offer an opportunity for earlier recognition of an underlying fibrotic lung disease that may be subclinical in nature.
IPF has a lower-lung zone predominance. The importance of chest auscultation, particularly listening carefully to the lung bases, is poignantly highlighted in a recent editorial: “It is time that the stethoscope draped around the neck of physicians, which tends to be used for identification purposes rather than for medical diagnosis, be also the (presently only) genuine tool for an earlier diagnosis of IPF.”7
Advances in imaging also provide an opportunity for earlier diagnosis. Many patients undergo screening computed tomography for coronary calcium scoring or lung cancer surveillance, and these studies may incidentally identify subtle interstitial lung abnormalities. These incidental findings should lead to further investigation, as they have been shown to be functionally important and carry risk of progression to clinical interstitial lung disease.8
INITIAL ASSESSMENT, TIMELY REFERRAL
But whether evidence of interstitial lung disease is detected incidentally or during testing for respiratory symptoms, further evaluation is necessary. Primary care providers are uniquely positioned to initiate the assessment and to expedite and guide further evaluation and specialty referral consultation to ensure an accurate diagnosis. They can also help grade the severity of the disease with pulmonary function testing, oxygen assessments at rest and with ambulation, and ordering thoracic high-resolution computed tomography to provide valuable information about disease extent and interstitial lung disease pattern.
General practitioners may assess for features suggesting connective tissue disease that would warrant specific serologic testing and dedicated rheumatologic consultation.
Finally, given the rarity, complexity, and challenges of interstitial lung disease, an effective multidisciplinary team consisting of clinicians, radiologists, and pathologists enhances diagnostic accuracy.9 This may also help general practitioners deviate from normal patterns of referral to general pulmonary providers, and instead refer patients to specialized centers with dedicated clinical and research expertise in interstitial lung disease.
IMPLEMENTING A COMPREHENSIVE, LONGITUDINAL CARE PLAN
The primary care practitioner often has developed long-term relationships with patients ultimately diagnosed with IPF, and because of this is particularly well positioned to help implement a collaborative and comprehensive care plan. Logistical realities such as distance to a specialty center, limited insurance coverage for specialty visits, and limited specialty availability all reinforce the central role that primary care practitioners play in ensuring that patients adhere to a comprehensive treatment program.
Primary providers may be very experienced and more inclined to manage a number of the common and often important comorbid conditions seen in patients with IPF, such as gastroesophageal reflux disease, obstructive sleep apnea, and depression. Reinforcing to the patient the need to adhere to adjunctive therapies such as supplemental oxygen and pulmonary rehabilitation is another key opportunity to actively engage in the management of patients with IPF.
Primary providers may also play a central role in IPF care through prevention strategies such as smoking cessation and ensuring appropriate immunization against seasonal influenza, pneumococcal pneumonia, and pertussis, among other age-appropriate vaccinations.
With the introduction and expansion of use of nintedanib and pirfenidone for IPF over the past few years, general practitioners may be called on to help manage common gastrointestinal side effects associated with pirfenidone (primarily nausea) and nintedanib (primarily diarrhea), and to be aware of potential drug-drug interactions and other medication-related toxicities.
Finally, as IPF remains a progressive disease, primary care practitioners are often well positioned to help implement palliative care, hospice care, and end-of-life care.
Despite recent advances in treatment, IPF remains a devastating lung disease with a high degree of morbidity and mortality. It takes a village to help care for the IPF patient. And as key members of the healthcare team, primary care providers have unique and important opportunities to help in the early recognition, thorough assessment, and comprehensive management of patients with IPF.
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Tolle L, Southern BD, Culver D, Horowitz JC. Idiopathic pulmonary fibrosis: what primary care physicians need to know. Cleve Clin J Med 2018; 85(5):377–386. doi:10.3949/ccjm.85a.17018
- Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med 2017; 129:24–30. doi:10.1016/j.rmed.2017.05.017
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J 2012; 40(3):519–521. doi:10.1183/09031936.00001612
- Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med 2012; 185(11):1147–1153. doi:10.1164/rccm.201108-1420PP
- Walsh SLF, Maher TM, Kolb M, et al; IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J 2017; 50(2):1700936. doi:10.1183/13993003.00936-2017
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive fibrosing interstitial lung disease associated with a high burden of morbidity and death.1 A clinical diagnosis of IPF is made only after careful interpretation of integrated clinical, radiologic, and often histopathologic data.
Interstitial lung disease encompasses a broad spectrum of parenchymal lung diseases, and a classification of IPF is restricted to a lung injury pattern of usual interstitial pneumonia (UIP) based on high-resolution computed tomography or surgical lung biopsy, after all known causes of UIP have been excluded.1
However, a lung injury pattern of UIP is not synonymous with IPF, as UIP can be seen with connective tissue disease, chronic hypersensitivity pneumonitis, drug toxicity, and sarcoidosis.1 As such, rendering a diagnosis of IPF requires a thorough evaluation to exclude such diverse potential etiologies.
In this issue of the Cleveland Clinic Journal of Medicine, Tolle and colleagues2 provide an up-to-date, broad overview of IPF focused on what the primary care provider needs to know about the disease. Their review is timely and serves as a useful primer for the practicing clinician.
The field of IPF is actively evolving, as this era has been witness to a recent paradigm shift in pharmacologic management. Immunosuppression is no longer recommended3 and may even be harmful.4 And the US Food and Drug Administration has approved 2 antifibrotic drugs—pirfenidone and nintedanib—that have been shown to delay progression of IPF.5,6
Primary care providers have a unique opportunity to play an integral role in the evaluation and care of patients with IPF, in particular with earlier disease recognition, initial disease assessment, and timely specialty consultative referral—as well as implementing a comprehensive longitudinal care plan.
EARLIER DISEASE RECOGNITION
IPF is a rare disease primarily affecting men over the age of 65.1 It is reasonable to presume that many or most of these individuals ultimately diagnosed with IPF are already seeking routine care for existing common medical conditions such as hypertension or dyslipidemia—or at least having periodic routine health maintenance assessments. Such evaluations may offer an opportunity for earlier recognition of an underlying fibrotic lung disease that may be subclinical in nature.
IPF has a lower-lung zone predominance. The importance of chest auscultation, particularly listening carefully to the lung bases, is poignantly highlighted in a recent editorial: “It is time that the stethoscope draped around the neck of physicians, which tends to be used for identification purposes rather than for medical diagnosis, be also the (presently only) genuine tool for an earlier diagnosis of IPF.”7
Advances in imaging also provide an opportunity for earlier diagnosis. Many patients undergo screening computed tomography for coronary calcium scoring or lung cancer surveillance, and these studies may incidentally identify subtle interstitial lung abnormalities. These incidental findings should lead to further investigation, as they have been shown to be functionally important and carry risk of progression to clinical interstitial lung disease.8
INITIAL ASSESSMENT, TIMELY REFERRAL
But whether evidence of interstitial lung disease is detected incidentally or during testing for respiratory symptoms, further evaluation is necessary. Primary care providers are uniquely positioned to initiate the assessment and to expedite and guide further evaluation and specialty referral consultation to ensure an accurate diagnosis. They can also help grade the severity of the disease with pulmonary function testing, oxygen assessments at rest and with ambulation, and ordering thoracic high-resolution computed tomography to provide valuable information about disease extent and interstitial lung disease pattern.
General practitioners may assess for features suggesting connective tissue disease that would warrant specific serologic testing and dedicated rheumatologic consultation.
Finally, given the rarity, complexity, and challenges of interstitial lung disease, an effective multidisciplinary team consisting of clinicians, radiologists, and pathologists enhances diagnostic accuracy.9 This may also help general practitioners deviate from normal patterns of referral to general pulmonary providers, and instead refer patients to specialized centers with dedicated clinical and research expertise in interstitial lung disease.
IMPLEMENTING A COMPREHENSIVE, LONGITUDINAL CARE PLAN
The primary care practitioner often has developed long-term relationships with patients ultimately diagnosed with IPF, and because of this is particularly well positioned to help implement a collaborative and comprehensive care plan. Logistical realities such as distance to a specialty center, limited insurance coverage for specialty visits, and limited specialty availability all reinforce the central role that primary care practitioners play in ensuring that patients adhere to a comprehensive treatment program.
Primary providers may be very experienced and more inclined to manage a number of the common and often important comorbid conditions seen in patients with IPF, such as gastroesophageal reflux disease, obstructive sleep apnea, and depression. Reinforcing to the patient the need to adhere to adjunctive therapies such as supplemental oxygen and pulmonary rehabilitation is another key opportunity to actively engage in the management of patients with IPF.
Primary providers may also play a central role in IPF care through prevention strategies such as smoking cessation and ensuring appropriate immunization against seasonal influenza, pneumococcal pneumonia, and pertussis, among other age-appropriate vaccinations.
With the introduction and expansion of use of nintedanib and pirfenidone for IPF over the past few years, general practitioners may be called on to help manage common gastrointestinal side effects associated with pirfenidone (primarily nausea) and nintedanib (primarily diarrhea), and to be aware of potential drug-drug interactions and other medication-related toxicities.
Finally, as IPF remains a progressive disease, primary care practitioners are often well positioned to help implement palliative care, hospice care, and end-of-life care.
Despite recent advances in treatment, IPF remains a devastating lung disease with a high degree of morbidity and mortality. It takes a village to help care for the IPF patient. And as key members of the healthcare team, primary care providers have unique and important opportunities to help in the early recognition, thorough assessment, and comprehensive management of patients with IPF.
Idiopathic pulmonary fibrosis (IPF) is a devastating progressive fibrosing interstitial lung disease associated with a high burden of morbidity and death.1 A clinical diagnosis of IPF is made only after careful interpretation of integrated clinical, radiologic, and often histopathologic data.
Interstitial lung disease encompasses a broad spectrum of parenchymal lung diseases, and a classification of IPF is restricted to a lung injury pattern of usual interstitial pneumonia (UIP) based on high-resolution computed tomography or surgical lung biopsy, after all known causes of UIP have been excluded.1
However, a lung injury pattern of UIP is not synonymous with IPF, as UIP can be seen with connective tissue disease, chronic hypersensitivity pneumonitis, drug toxicity, and sarcoidosis.1 As such, rendering a diagnosis of IPF requires a thorough evaluation to exclude such diverse potential etiologies.
In this issue of the Cleveland Clinic Journal of Medicine, Tolle and colleagues2 provide an up-to-date, broad overview of IPF focused on what the primary care provider needs to know about the disease. Their review is timely and serves as a useful primer for the practicing clinician.
The field of IPF is actively evolving, as this era has been witness to a recent paradigm shift in pharmacologic management. Immunosuppression is no longer recommended3 and may even be harmful.4 And the US Food and Drug Administration has approved 2 antifibrotic drugs—pirfenidone and nintedanib—that have been shown to delay progression of IPF.5,6
Primary care providers have a unique opportunity to play an integral role in the evaluation and care of patients with IPF, in particular with earlier disease recognition, initial disease assessment, and timely specialty consultative referral—as well as implementing a comprehensive longitudinal care plan.
EARLIER DISEASE RECOGNITION
IPF is a rare disease primarily affecting men over the age of 65.1 It is reasonable to presume that many or most of these individuals ultimately diagnosed with IPF are already seeking routine care for existing common medical conditions such as hypertension or dyslipidemia—or at least having periodic routine health maintenance assessments. Such evaluations may offer an opportunity for earlier recognition of an underlying fibrotic lung disease that may be subclinical in nature.
IPF has a lower-lung zone predominance. The importance of chest auscultation, particularly listening carefully to the lung bases, is poignantly highlighted in a recent editorial: “It is time that the stethoscope draped around the neck of physicians, which tends to be used for identification purposes rather than for medical diagnosis, be also the (presently only) genuine tool for an earlier diagnosis of IPF.”7
Advances in imaging also provide an opportunity for earlier diagnosis. Many patients undergo screening computed tomography for coronary calcium scoring or lung cancer surveillance, and these studies may incidentally identify subtle interstitial lung abnormalities. These incidental findings should lead to further investigation, as they have been shown to be functionally important and carry risk of progression to clinical interstitial lung disease.8
INITIAL ASSESSMENT, TIMELY REFERRAL
But whether evidence of interstitial lung disease is detected incidentally or during testing for respiratory symptoms, further evaluation is necessary. Primary care providers are uniquely positioned to initiate the assessment and to expedite and guide further evaluation and specialty referral consultation to ensure an accurate diagnosis. They can also help grade the severity of the disease with pulmonary function testing, oxygen assessments at rest and with ambulation, and ordering thoracic high-resolution computed tomography to provide valuable information about disease extent and interstitial lung disease pattern.
General practitioners may assess for features suggesting connective tissue disease that would warrant specific serologic testing and dedicated rheumatologic consultation.
Finally, given the rarity, complexity, and challenges of interstitial lung disease, an effective multidisciplinary team consisting of clinicians, radiologists, and pathologists enhances diagnostic accuracy.9 This may also help general practitioners deviate from normal patterns of referral to general pulmonary providers, and instead refer patients to specialized centers with dedicated clinical and research expertise in interstitial lung disease.
IMPLEMENTING A COMPREHENSIVE, LONGITUDINAL CARE PLAN
The primary care practitioner often has developed long-term relationships with patients ultimately diagnosed with IPF, and because of this is particularly well positioned to help implement a collaborative and comprehensive care plan. Logistical realities such as distance to a specialty center, limited insurance coverage for specialty visits, and limited specialty availability all reinforce the central role that primary care practitioners play in ensuring that patients adhere to a comprehensive treatment program.
Primary providers may be very experienced and more inclined to manage a number of the common and often important comorbid conditions seen in patients with IPF, such as gastroesophageal reflux disease, obstructive sleep apnea, and depression. Reinforcing to the patient the need to adhere to adjunctive therapies such as supplemental oxygen and pulmonary rehabilitation is another key opportunity to actively engage in the management of patients with IPF.
Primary providers may also play a central role in IPF care through prevention strategies such as smoking cessation and ensuring appropriate immunization against seasonal influenza, pneumococcal pneumonia, and pertussis, among other age-appropriate vaccinations.
With the introduction and expansion of use of nintedanib and pirfenidone for IPF over the past few years, general practitioners may be called on to help manage common gastrointestinal side effects associated with pirfenidone (primarily nausea) and nintedanib (primarily diarrhea), and to be aware of potential drug-drug interactions and other medication-related toxicities.
Finally, as IPF remains a progressive disease, primary care practitioners are often well positioned to help implement palliative care, hospice care, and end-of-life care.
Despite recent advances in treatment, IPF remains a devastating lung disease with a high degree of morbidity and mortality. It takes a village to help care for the IPF patient. And as key members of the healthcare team, primary care providers have unique and important opportunities to help in the early recognition, thorough assessment, and comprehensive management of patients with IPF.
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Tolle L, Southern BD, Culver D, Horowitz JC. Idiopathic pulmonary fibrosis: what primary care physicians need to know. Cleve Clin J Med 2018; 85(5):377–386. doi:10.3949/ccjm.85a.17018
- Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med 2017; 129:24–30. doi:10.1016/j.rmed.2017.05.017
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J 2012; 40(3):519–521. doi:10.1183/09031936.00001612
- Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med 2012; 185(11):1147–1153. doi:10.1164/rccm.201108-1420PP
- Walsh SLF, Maher TM, Kolb M, et al; IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J 2017; 50(2):1700936. doi:10.1183/13993003.00936-2017
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6):788–824. doi:10.1164/rccm.2009-040GL
- Tolle L, Southern BD, Culver D, Horowitz JC. Idiopathic pulmonary fibrosis: what primary care physicians need to know. Cleve Clin J Med 2018; 85(5):377–386. doi:10.3949/ccjm.85a.17018
- Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med 2017; 129:24–30. doi:10.1016/j.rmed.2017.05.017
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366(21):1968–1977. doi:10.1056/NEJMoa1113354
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2083–2092. doi:10.1056/NEJMoa1402582
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22):2071–2082. doi:10.1056/NEJMoa1402584
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J 2012; 40(3):519–521. doi:10.1183/09031936.00001612
- Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med 2012; 185(11):1147–1153. doi:10.1164/rccm.201108-1420PP
- Walsh SLF, Maher TM, Kolb M, et al; IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J 2017; 50(2):1700936. doi:10.1183/13993003.00936-2017
