User login
Follow-up care for pediatric concussions not heeded in Ontario
reported Liraz Fridman, PhD, of York University, Toronto, and associates.
In a 10-year, retrospective, population-based study, 126,654 children aged 5-18 years presented with concussions to emergency departments and physician offices in Ontario between April 1, 2003, and March 31, 2014. In 2003, 7,126 children were evaluated for a concussion, compared with 21,681 children by 2013.
Limitations to this study include that children treated by athletic therapists or chiropractors would have been missed, and that these data may not generalize across Canada or the United States, they said.
“In Ontario, the rate of follow-up care for concussions nearly tripled in both emergency departments and physician’s offices. Despite this significant improvement over time, more than two-thirds of all child and youth concussion patients still do not receive the minimum standard of care, according to accepted guidelines,” the researchers wrote, adding that the study’s findings suggest that better instructions for health care providers on management of concussion are needed.
SOURCE: Fridman L et al. J Pediatr. 2018;192:184-8
reported Liraz Fridman, PhD, of York University, Toronto, and associates.
In a 10-year, retrospective, population-based study, 126,654 children aged 5-18 years presented with concussions to emergency departments and physician offices in Ontario between April 1, 2003, and March 31, 2014. In 2003, 7,126 children were evaluated for a concussion, compared with 21,681 children by 2013.
Limitations to this study include that children treated by athletic therapists or chiropractors would have been missed, and that these data may not generalize across Canada or the United States, they said.
“In Ontario, the rate of follow-up care for concussions nearly tripled in both emergency departments and physician’s offices. Despite this significant improvement over time, more than two-thirds of all child and youth concussion patients still do not receive the minimum standard of care, according to accepted guidelines,” the researchers wrote, adding that the study’s findings suggest that better instructions for health care providers on management of concussion are needed.
SOURCE: Fridman L et al. J Pediatr. 2018;192:184-8
reported Liraz Fridman, PhD, of York University, Toronto, and associates.
In a 10-year, retrospective, population-based study, 126,654 children aged 5-18 years presented with concussions to emergency departments and physician offices in Ontario between April 1, 2003, and March 31, 2014. In 2003, 7,126 children were evaluated for a concussion, compared with 21,681 children by 2013.
Limitations to this study include that children treated by athletic therapists or chiropractors would have been missed, and that these data may not generalize across Canada or the United States, they said.
“In Ontario, the rate of follow-up care for concussions nearly tripled in both emergency departments and physician’s offices. Despite this significant improvement over time, more than two-thirds of all child and youth concussion patients still do not receive the minimum standard of care, according to accepted guidelines,” the researchers wrote, adding that the study’s findings suggest that better instructions for health care providers on management of concussion are needed.
SOURCE: Fridman L et al. J Pediatr. 2018;192:184-8
FROM THE JOURNAL OF PEDIATRICS
Mission to Mars: Endocrinologists take on populating deep space
by studying the effects of space radiation and other factors on male and female fertility, according to Teresa K. Woodruff, PhD, the Thomas J. Watkins Memorial Professor of Obstetrics & Gynecology, the vice chair of research for the department of obstetrics and gynecology, and the chief of the division of reproductive science and medicine at Northwestern University, Chicago, who is chairing a session on March 17 at the 2018 Endocrine Society annual meeting entitled: “Mission to Mars.”

The project was developed based on the anticipation that “we will continue to explore space as an inhabitable frontier.” The talks in this session will examine endocrine-based reproduction such as sperm function and biologic fertilization that has occurred in space, noted Dr. Woodruff, who is also professor of molecular biosciences in the Weinberg College of Arts and Sciences and professor of biomedical engineering in the McCormick School of Engineering, both at Northwestern University.
Dr. Woodruff currently has a grant under review by the National Aeronautics and Space Administration that is relevant to the mission of populating space. “If we are going to populate space, which we will need to do based on the length of time we will be there, we are going to have to get gametes into space,” she said. There will be more and more research effort focused on the needs of the humans who are “non-Earth dwelling,” she said.
The next step might be to study embryo development in that environment. Such research would take place at the International Space Station.
“I am interested in endocrine disruptors. What we learn from the study of the challenges to reproduction in space we will be able to apply on earth,” Dr. Woodruff said.
Also on the panel will be Joseph S. Tash, PhD, of Kansas University, Kansas City, whose grant on Space Flight Impacts on Gonadal and Gamete Function already has NASA funding, according to Dr. Woodruff.
by studying the effects of space radiation and other factors on male and female fertility, according to Teresa K. Woodruff, PhD, the Thomas J. Watkins Memorial Professor of Obstetrics & Gynecology, the vice chair of research for the department of obstetrics and gynecology, and the chief of the division of reproductive science and medicine at Northwestern University, Chicago, who is chairing a session on March 17 at the 2018 Endocrine Society annual meeting entitled: “Mission to Mars.”
The project was developed based on the anticipation that “we will continue to explore space as an inhabitable frontier.” The talks in this session will examine endocrine-based reproduction such as sperm function and biologic fertilization that has occurred in space, noted Dr. Woodruff, who is also professor of molecular biosciences in the Weinberg College of Arts and Sciences and professor of biomedical engineering in the McCormick School of Engineering, both at Northwestern University.
Dr. Woodruff currently has a grant under review by the National Aeronautics and Space Administration that is relevant to the mission of populating space. “If we are going to populate space, which we will need to do based on the length of time we will be there, we are going to have to get gametes into space,” she said. There will be more and more research effort focused on the needs of the humans who are “non-Earth dwelling,” she said.
The next step might be to study embryo development in that environment. Such research would take place at the International Space Station.
“I am interested in endocrine disruptors. What we learn from the study of the challenges to reproduction in space we will be able to apply on earth,” Dr. Woodruff said.
Also on the panel will be Joseph S. Tash, PhD, of Kansas University, Kansas City, whose grant on Space Flight Impacts on Gonadal and Gamete Function already has NASA funding, according to Dr. Woodruff.
by studying the effects of space radiation and other factors on male and female fertility, according to Teresa K. Woodruff, PhD, the Thomas J. Watkins Memorial Professor of Obstetrics & Gynecology, the vice chair of research for the department of obstetrics and gynecology, and the chief of the division of reproductive science and medicine at Northwestern University, Chicago, who is chairing a session on March 17 at the 2018 Endocrine Society annual meeting entitled: “Mission to Mars.”
The project was developed based on the anticipation that “we will continue to explore space as an inhabitable frontier.” The talks in this session will examine endocrine-based reproduction such as sperm function and biologic fertilization that has occurred in space, noted Dr. Woodruff, who is also professor of molecular biosciences in the Weinberg College of Arts and Sciences and professor of biomedical engineering in the McCormick School of Engineering, both at Northwestern University.
Dr. Woodruff currently has a grant under review by the National Aeronautics and Space Administration that is relevant to the mission of populating space. “If we are going to populate space, which we will need to do based on the length of time we will be there, we are going to have to get gametes into space,” she said. There will be more and more research effort focused on the needs of the humans who are “non-Earth dwelling,” she said.
The next step might be to study embryo development in that environment. Such research would take place at the International Space Station.
“I am interested in endocrine disruptors. What we learn from the study of the challenges to reproduction in space we will be able to apply on earth,” Dr. Woodruff said.
Also on the panel will be Joseph S. Tash, PhD, of Kansas University, Kansas City, whose grant on Space Flight Impacts on Gonadal and Gamete Function already has NASA funding, according to Dr. Woodruff.
Laparoscopic hysterectomy safest even for markedly enlarged uteri
ORLANDO – according to findings from a nationwide cohort of more than 27,000 women.
After adjusting for numerous potential confounding factors, including medical risk factors, procedure-related variables, and patient demographics, increasing uterine weight was significantly associated with increasing odds of complications – particularly after hysterectomy for uteri over 500 g, Michelle Louie, MD, reported during an oral poster session at the annual scientific meeting of the Society of Gynecologic Surgeons.

The same was true for uteri of 250-500 g (adjusted OR, 0.99, 1.73, and 1.06, respectively), she noted, adding that “abdominal hysterectomy always has the highest rate of a complication, except at above 850 g, when a vaginal hysterectomy is associated with a greater odds of complications.”
This secondary analysis was performed using prospectively collected quality improvement data abstracted from the American College of Surgeons National Surgical Quality Improvement Program database, which includes patient information and 30-day outcomes from more than 500 U.S. hospitals. Patients included in the analysis were 27,167 women who underwent a hysterectomy for benign conditions during 2014-2015 for whom uterine size was reported. Complications assessed included infection, vascular complications, reoperation, and readmission.
“Our study suggests that uterine weight is not an appropriate indication for abdominal hysterectomy – that we can, and should, offer a laparoscopic approach even for a markedly enlarged uterus,” she said. “We believe, therefore, that patients may benefit from referral to specialty surgeons who are able to offer a laparoscopic approach, even for a very large uterus.”
In response to a question from the audience about the role of physician experience in the findings, Dr. Louie said that it was not a covariate for which information was available, thus it was not included in the analysis.
“However, I think all of us realize that surgeon volume and surgeon experience is an important factor for patient safety,” she said.
Dr. Louie has received consulting fees from Teleflex.
SOURCE: Louie M et al. SGS 2018, Oral Poster 06.
ORLANDO – according to findings from a nationwide cohort of more than 27,000 women.
After adjusting for numerous potential confounding factors, including medical risk factors, procedure-related variables, and patient demographics, increasing uterine weight was significantly associated with increasing odds of complications – particularly after hysterectomy for uteri over 500 g, Michelle Louie, MD, reported during an oral poster session at the annual scientific meeting of the Society of Gynecologic Surgeons.
The same was true for uteri of 250-500 g (adjusted OR, 0.99, 1.73, and 1.06, respectively), she noted, adding that “abdominal hysterectomy always has the highest rate of a complication, except at above 850 g, when a vaginal hysterectomy is associated with a greater odds of complications.”
This secondary analysis was performed using prospectively collected quality improvement data abstracted from the American College of Surgeons National Surgical Quality Improvement Program database, which includes patient information and 30-day outcomes from more than 500 U.S. hospitals. Patients included in the analysis were 27,167 women who underwent a hysterectomy for benign conditions during 2014-2015 for whom uterine size was reported. Complications assessed included infection, vascular complications, reoperation, and readmission.
“Our study suggests that uterine weight is not an appropriate indication for abdominal hysterectomy – that we can, and should, offer a laparoscopic approach even for a markedly enlarged uterus,” she said. “We believe, therefore, that patients may benefit from referral to specialty surgeons who are able to offer a laparoscopic approach, even for a very large uterus.”
In response to a question from the audience about the role of physician experience in the findings, Dr. Louie said that it was not a covariate for which information was available, thus it was not included in the analysis.
“However, I think all of us realize that surgeon volume and surgeon experience is an important factor for patient safety,” she said.
Dr. Louie has received consulting fees from Teleflex.
SOURCE: Louie M et al. SGS 2018, Oral Poster 06.
ORLANDO – according to findings from a nationwide cohort of more than 27,000 women.
After adjusting for numerous potential confounding factors, including medical risk factors, procedure-related variables, and patient demographics, increasing uterine weight was significantly associated with increasing odds of complications – particularly after hysterectomy for uteri over 500 g, Michelle Louie, MD, reported during an oral poster session at the annual scientific meeting of the Society of Gynecologic Surgeons.
The same was true for uteri of 250-500 g (adjusted OR, 0.99, 1.73, and 1.06, respectively), she noted, adding that “abdominal hysterectomy always has the highest rate of a complication, except at above 850 g, when a vaginal hysterectomy is associated with a greater odds of complications.”
This secondary analysis was performed using prospectively collected quality improvement data abstracted from the American College of Surgeons National Surgical Quality Improvement Program database, which includes patient information and 30-day outcomes from more than 500 U.S. hospitals. Patients included in the analysis were 27,167 women who underwent a hysterectomy for benign conditions during 2014-2015 for whom uterine size was reported. Complications assessed included infection, vascular complications, reoperation, and readmission.
“Our study suggests that uterine weight is not an appropriate indication for abdominal hysterectomy – that we can, and should, offer a laparoscopic approach even for a markedly enlarged uterus,” she said. “We believe, therefore, that patients may benefit from referral to specialty surgeons who are able to offer a laparoscopic approach, even for a very large uterus.”
In response to a question from the audience about the role of physician experience in the findings, Dr. Louie said that it was not a covariate for which information was available, thus it was not included in the analysis.
“However, I think all of us realize that surgeon volume and surgeon experience is an important factor for patient safety,” she said.
Dr. Louie has received consulting fees from Teleflex.
SOURCE: Louie M et al. SGS 2018, Oral Poster 06.
REPORTING FROM SGS 2018
Key clinical point: Laparoscopic hysterectomy can and should be offered to women with uteri over 500 g.
Major finding: The odds ratios for complications after laparoscopic, abdominal, and vaginal hysterectomy for uteri over 500 g were 1.61, 2.16, and 2.57, respectively.
Study details: A secondary analysis of a nationwide cohort of 27,167 women.
Disclosures: Dr. Louie has received consulting fees from Teleflex.
Source: Louie M et al. SGS 2018, Oral Poster 06.
MDedge Daily News: Could gut bacteria trigger autoimmune diseases?
Vouchers might improve post–heart-attack drug compliance. How education affects depression risks for women. And why confusion reigns over the COPD-asthma overlap.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
Vouchers might improve post–heart-attack drug compliance. How education affects depression risks for women. And why confusion reigns over the COPD-asthma overlap.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
Vouchers might improve post–heart-attack drug compliance. How education affects depression risks for women. And why confusion reigns over the COPD-asthma overlap.
Listen to the MDedge Daily News podcast for all the details on today’s top news.

When Your Neighborhood Is a CVD Risk Factor
Living in a neighborhood with poor access to food and poor walkability puts people at risk for cardiovascular disease (CVD)—and according to researchers from Morehouse School of Medicine in Atlanta, Georgia, African Americans seemed to have the highest risk.
In a CDC-funded study, the researchers examined the relationship between neighborhood-level food access and walkability on premature CVD mortality rates in Atlanta, using census tracts to map neighborhoods. Atlanta is in Fulton County, which has a food insecurity rating of nearly 20%. Food access was defined as the percentage of no-vehicle households living beyond a 0.9-mile radius of a food outlet in 2012. Walkability was measured in relationship to amenities, population density, and road metrics.
The researchers found no significant difference in walkability scores between high-poverty and low-poverty census tracts. However, they found significant racial differences: Census tracts with high concentrations of minority populations had higher levels of poor food access, poor walkability, and premature CVD mortality.
Of 124 census tracts, 87 contained 73% of the city’s population aged 35 to 64 years and accounted for 1,225 deaths between 2010-2014, with a premature CVD mortality rate of 11 per 1,000. Black premature CVD deaths accounted for nearly 85% of the premature CVD deaths in all census tracts—a “disproportionate number,” the researchers say, because blacks make up only 52% of the city’s total population aged 35 to 64 years.
The findings can be used to “calibrate” neighborhood interventions, based on racial/ethnic or other demographic characteristics, the researchers suggest. They add that the results highlight the need to examine racially stratified health outcomes.
Living in a neighborhood with poor access to food and poor walkability puts people at risk for cardiovascular disease (CVD)—and according to researchers from Morehouse School of Medicine in Atlanta, Georgia, African Americans seemed to have the highest risk.
In a CDC-funded study, the researchers examined the relationship between neighborhood-level food access and walkability on premature CVD mortality rates in Atlanta, using census tracts to map neighborhoods. Atlanta is in Fulton County, which has a food insecurity rating of nearly 20%. Food access was defined as the percentage of no-vehicle households living beyond a 0.9-mile radius of a food outlet in 2012. Walkability was measured in relationship to amenities, population density, and road metrics.
The researchers found no significant difference in walkability scores between high-poverty and low-poverty census tracts. However, they found significant racial differences: Census tracts with high concentrations of minority populations had higher levels of poor food access, poor walkability, and premature CVD mortality.
Of 124 census tracts, 87 contained 73% of the city’s population aged 35 to 64 years and accounted for 1,225 deaths between 2010-2014, with a premature CVD mortality rate of 11 per 1,000. Black premature CVD deaths accounted for nearly 85% of the premature CVD deaths in all census tracts—a “disproportionate number,” the researchers say, because blacks make up only 52% of the city’s total population aged 35 to 64 years.
The findings can be used to “calibrate” neighborhood interventions, based on racial/ethnic or other demographic characteristics, the researchers suggest. They add that the results highlight the need to examine racially stratified health outcomes.
Living in a neighborhood with poor access to food and poor walkability puts people at risk for cardiovascular disease (CVD)—and according to researchers from Morehouse School of Medicine in Atlanta, Georgia, African Americans seemed to have the highest risk.
In a CDC-funded study, the researchers examined the relationship between neighborhood-level food access and walkability on premature CVD mortality rates in Atlanta, using census tracts to map neighborhoods. Atlanta is in Fulton County, which has a food insecurity rating of nearly 20%. Food access was defined as the percentage of no-vehicle households living beyond a 0.9-mile radius of a food outlet in 2012. Walkability was measured in relationship to amenities, population density, and road metrics.
The researchers found no significant difference in walkability scores between high-poverty and low-poverty census tracts. However, they found significant racial differences: Census tracts with high concentrations of minority populations had higher levels of poor food access, poor walkability, and premature CVD mortality.
Of 124 census tracts, 87 contained 73% of the city’s population aged 35 to 64 years and accounted for 1,225 deaths between 2010-2014, with a premature CVD mortality rate of 11 per 1,000. Black premature CVD deaths accounted for nearly 85% of the premature CVD deaths in all census tracts—a “disproportionate number,” the researchers say, because blacks make up only 52% of the city’s total population aged 35 to 64 years.
The findings can be used to “calibrate” neighborhood interventions, based on racial/ethnic or other demographic characteristics, the researchers suggest. They add that the results highlight the need to examine racially stratified health outcomes.
Girl Faces Down Lesion
The lesion on this 8-year-old girl’s face was first noted about a year ago. Recently, however, it has started to grow, prompting her parents to consult the child’s primary care provider (PCP). The lesion is completely asymptomatic but nonetheless concerning to the parents and to the PCP, who has no idea what it could be. He therefore refers them to dermatology.
The child is otherwise healthy. There is no family history of chronic or inheritable disease.
EXAMINATION
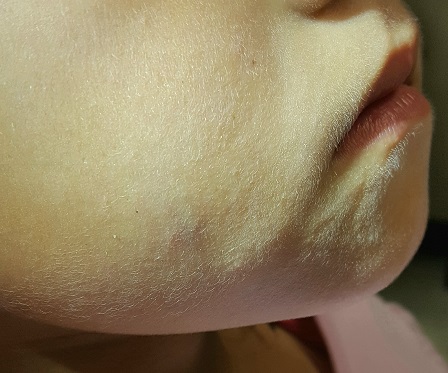
The lesion is a 2-cm subcutaneous firm round mass located along the superior aspect of the right jawline. The only overlying skin change is a bluish discoloration. No surface punctum is seen. No other lesions are seen or felt on examination of the rest of the head and neck.
What is the diagnosis?
DISCUSSION
These findings are typical of pilomatricoma, a rather unusual lesion with multiple alternate names (among them: calcifying epithelioma of Malherbe and pilomatrixoma). These benign tumors arising from hair matrix cells are typically seen on the head, face, neck, and upper extremities, most often on children.
This patient’s lesion was typical in size, although they can vary from 3 mm (the smallest I’ve seen) to more than 20 cm. The firm feel, lack of a punctum (which would suggest an epidermal cyst), and bluish discoloration are all typical features.
These lesions generally merit little or no concern. However, in the rare instance when the patient has multiple lesions, the possibility of at least two conditions should be considered: Gardner disease and myotonic seizure.
In terms of treatment, pilomatricoma can be safely left alone; understandably, though, most parents will only be satisfied by excision. It is important to note that, unlike most true cysts, pilomatricomas have a very poorly defined cyst wall with contents that are equally odd—watery and full of tiny white flecks that represent calcified cells. All of this must be totally removed to prevent recurrence. In most cases, defects must be closed in two layers, to minimize “dead” space that might otherwise fill with blood.
One could argue that this child’s lesion should have been removed by a plastic surgeon—but the family had no insurance. Excision was therefore the treatment of choice; the most difficult aspect was persuading the patient to cooperate. (Sometimes, you have to wait years for the child to mature before you attempt it.) The outcome in this case proved to be quite acceptable. Of course, the lesion was sent to pathology, which confirmed the pre-op diagnosis.
The differential includes epidermal cysts (which are almost unknown in prepubertal children), and sweat gland cysts.
TAKE-HOME LEARNING POINTS
- Pilomatricomas are benign cysts that originate from hair matrix cells, usually appearing on the head, neck, face, and upper extremities of children.
- The cysts are typically firm and round and often display a faintly bluish tone (as seen in this case).
- As solitary lesions, they are of little or no significance; when seen in multiples, however, pilomatricomas suggest the possibility of Gardner syndrome or myotonic seizure.
- Excision is the best treatment, though these can be left alone long enough to allow the patient to mature sufficiently to cooperate with the surgical process.
The lesion on this 8-year-old girl’s face was first noted about a year ago. Recently, however, it has started to grow, prompting her parents to consult the child’s primary care provider (PCP). The lesion is completely asymptomatic but nonetheless concerning to the parents and to the PCP, who has no idea what it could be. He therefore refers them to dermatology.
The child is otherwise healthy. There is no family history of chronic or inheritable disease.
EXAMINATION
The lesion is a 2-cm subcutaneous firm round mass located along the superior aspect of the right jawline. The only overlying skin change is a bluish discoloration. No surface punctum is seen. No other lesions are seen or felt on examination of the rest of the head and neck.
What is the diagnosis?
DISCUSSION
These findings are typical of pilomatricoma, a rather unusual lesion with multiple alternate names (among them: calcifying epithelioma of Malherbe and pilomatrixoma). These benign tumors arising from hair matrix cells are typically seen on the head, face, neck, and upper extremities, most often on children.
This patient’s lesion was typical in size, although they can vary from 3 mm (the smallest I’ve seen) to more than 20 cm. The firm feel, lack of a punctum (which would suggest an epidermal cyst), and bluish discoloration are all typical features.
These lesions generally merit little or no concern. However, in the rare instance when the patient has multiple lesions, the possibility of at least two conditions should be considered: Gardner disease and myotonic seizure.
In terms of treatment, pilomatricoma can be safely left alone; understandably, though, most parents will only be satisfied by excision. It is important to note that, unlike most true cysts, pilomatricomas have a very poorly defined cyst wall with contents that are equally odd—watery and full of tiny white flecks that represent calcified cells. All of this must be totally removed to prevent recurrence. In most cases, defects must be closed in two layers, to minimize “dead” space that might otherwise fill with blood.
One could argue that this child’s lesion should have been removed by a plastic surgeon—but the family had no insurance. Excision was therefore the treatment of choice; the most difficult aspect was persuading the patient to cooperate. (Sometimes, you have to wait years for the child to mature before you attempt it.) The outcome in this case proved to be quite acceptable. Of course, the lesion was sent to pathology, which confirmed the pre-op diagnosis.
The differential includes epidermal cysts (which are almost unknown in prepubertal children), and sweat gland cysts.
TAKE-HOME LEARNING POINTS
- Pilomatricomas are benign cysts that originate from hair matrix cells, usually appearing on the head, neck, face, and upper extremities of children.
- The cysts are typically firm and round and often display a faintly bluish tone (as seen in this case).
- As solitary lesions, they are of little or no significance; when seen in multiples, however, pilomatricomas suggest the possibility of Gardner syndrome or myotonic seizure.
- Excision is the best treatment, though these can be left alone long enough to allow the patient to mature sufficiently to cooperate with the surgical process.
The lesion on this 8-year-old girl’s face was first noted about a year ago. Recently, however, it has started to grow, prompting her parents to consult the child’s primary care provider (PCP). The lesion is completely asymptomatic but nonetheless concerning to the parents and to the PCP, who has no idea what it could be. He therefore refers them to dermatology.
The child is otherwise healthy. There is no family history of chronic or inheritable disease.
EXAMINATION
The lesion is a 2-cm subcutaneous firm round mass located along the superior aspect of the right jawline. The only overlying skin change is a bluish discoloration. No surface punctum is seen. No other lesions are seen or felt on examination of the rest of the head and neck.
What is the diagnosis?
DISCUSSION
These findings are typical of pilomatricoma, a rather unusual lesion with multiple alternate names (among them: calcifying epithelioma of Malherbe and pilomatrixoma). These benign tumors arising from hair matrix cells are typically seen on the head, face, neck, and upper extremities, most often on children.
This patient’s lesion was typical in size, although they can vary from 3 mm (the smallest I’ve seen) to more than 20 cm. The firm feel, lack of a punctum (which would suggest an epidermal cyst), and bluish discoloration are all typical features.
These lesions generally merit little or no concern. However, in the rare instance when the patient has multiple lesions, the possibility of at least two conditions should be considered: Gardner disease and myotonic seizure.
In terms of treatment, pilomatricoma can be safely left alone; understandably, though, most parents will only be satisfied by excision. It is important to note that, unlike most true cysts, pilomatricomas have a very poorly defined cyst wall with contents that are equally odd—watery and full of tiny white flecks that represent calcified cells. All of this must be totally removed to prevent recurrence. In most cases, defects must be closed in two layers, to minimize “dead” space that might otherwise fill with blood.
One could argue that this child’s lesion should have been removed by a plastic surgeon—but the family had no insurance. Excision was therefore the treatment of choice; the most difficult aspect was persuading the patient to cooperate. (Sometimes, you have to wait years for the child to mature before you attempt it.) The outcome in this case proved to be quite acceptable. Of course, the lesion was sent to pathology, which confirmed the pre-op diagnosis.
The differential includes epidermal cysts (which are almost unknown in prepubertal children), and sweat gland cysts.
TAKE-HOME LEARNING POINTS
- Pilomatricomas are benign cysts that originate from hair matrix cells, usually appearing on the head, neck, face, and upper extremities of children.
- The cysts are typically firm and round and often display a faintly bluish tone (as seen in this case).
- As solitary lesions, they are of little or no significance; when seen in multiples, however, pilomatricomas suggest the possibility of Gardner syndrome or myotonic seizure.
- Excision is the best treatment, though these can be left alone long enough to allow the patient to mature sufficiently to cooperate with the surgical process.
Aspirin appears comparable to rivaroxaban for VTE
Aspirin seems to be as effective as rivaroxaban for preventing venous thromboembolism (VTE) after total hip or knee replacement, according to research published in NEJM.
Researchers observed a similar incidence of VTE whether patients were randomized to receive aspirin or rivaroxaban prophylaxis starting 5 days after surgery, with all patients receiving rivaroxaban for the first 5 days after surgery.
There was no significant difference between the aspirin and rivaroxaban groups with regard to major bleeding or clinically important bleeding.
“We have always been very concerned about preventing blood clots in patients following major orthopedic surgery,” said study author David Zukor, MD, of Jewish General Hospital in Montreal, Quebec, Canada.
“It is the leading cause of preventable in-hospital death, so we always administer preventive therapy in conjunction with such surgeries. Rivaroxaban is known to be effective, but the great advantage of aspirin is that it is far less expensive, easily available, and has an excellent safety profile.”
Dr Zukor and his colleagues set out to compare aspirin and rivaroxaban in 3424 patients, 1804 who underwent hip replacement and 1620 who underwent knee replacement.
All patients took rivaroxaban (10 mg) for 5 days after surgery before being randomized to receive aspirin (81 mg daily, n=1707) or to continue with rivaroxaban (n=1717). The patients received this prophylaxis for an additional 9 days if they had knee replacement or for 30 days if they had hip replacement.
The researchers followed patients for 90 days, monitoring them for symptomatic VTE and major or clinically relevant nonmajor bleeding.
The rate of VTE was 0.64% (n=11) in the aspirin group and 0.70% (n=12) in the rivaroxaban group (difference, 0.06 percentage points; 95% confidence interval [CI], −0.55 to 0.66; P<0.001 for noninferiority, P=0.84 for superiority).
Major bleeding occurred in 0.47% (n=8) of patients in the aspirin group and in 0.29% (n=5) of those in the rivaroxaban group (difference, 0.18 percentage points; 95% CI, −0.65 to 0.29; P=0.42).
Clinically relevant nonmajor bleeding occurred in 1.29% (n=22) and 0.99% (n=17), respectively (difference, 0.30 percentage points; 95% CI, −1.07 to 0.47; P=0.43).
All bleeding events occurred at the surgical site, and most occurred within 10 days of randomization.
“These results are important,” said study author Susan Kahn, MD, of McGill University in Montreal.
“The protocols for preventing clots following major orthopedic surgery are well established. However, we are always interested in determining whether there are better options for treating our patients. We could well see aspirin emerge as a practical alternative to more expensive anticoagulants.”
Drs Kahn and Zukor both noted the need for additional trials that would include a randomized group of patients prescribed aspirin exclusively so as to test its efficacy directly against rivaroxaban.
Aspirin seems to be as effective as rivaroxaban for preventing venous thromboembolism (VTE) after total hip or knee replacement, according to research published in NEJM.
Researchers observed a similar incidence of VTE whether patients were randomized to receive aspirin or rivaroxaban prophylaxis starting 5 days after surgery, with all patients receiving rivaroxaban for the first 5 days after surgery.
There was no significant difference between the aspirin and rivaroxaban groups with regard to major bleeding or clinically important bleeding.
“We have always been very concerned about preventing blood clots in patients following major orthopedic surgery,” said study author David Zukor, MD, of Jewish General Hospital in Montreal, Quebec, Canada.
“It is the leading cause of preventable in-hospital death, so we always administer preventive therapy in conjunction with such surgeries. Rivaroxaban is known to be effective, but the great advantage of aspirin is that it is far less expensive, easily available, and has an excellent safety profile.”
Dr Zukor and his colleagues set out to compare aspirin and rivaroxaban in 3424 patients, 1804 who underwent hip replacement and 1620 who underwent knee replacement.
All patients took rivaroxaban (10 mg) for 5 days after surgery before being randomized to receive aspirin (81 mg daily, n=1707) or to continue with rivaroxaban (n=1717). The patients received this prophylaxis for an additional 9 days if they had knee replacement or for 30 days if they had hip replacement.
The researchers followed patients for 90 days, monitoring them for symptomatic VTE and major or clinically relevant nonmajor bleeding.
The rate of VTE was 0.64% (n=11) in the aspirin group and 0.70% (n=12) in the rivaroxaban group (difference, 0.06 percentage points; 95% confidence interval [CI], −0.55 to 0.66; P<0.001 for noninferiority, P=0.84 for superiority).
Major bleeding occurred in 0.47% (n=8) of patients in the aspirin group and in 0.29% (n=5) of those in the rivaroxaban group (difference, 0.18 percentage points; 95% CI, −0.65 to 0.29; P=0.42).
Clinically relevant nonmajor bleeding occurred in 1.29% (n=22) and 0.99% (n=17), respectively (difference, 0.30 percentage points; 95% CI, −1.07 to 0.47; P=0.43).
All bleeding events occurred at the surgical site, and most occurred within 10 days of randomization.
“These results are important,” said study author Susan Kahn, MD, of McGill University in Montreal.
“The protocols for preventing clots following major orthopedic surgery are well established. However, we are always interested in determining whether there are better options for treating our patients. We could well see aspirin emerge as a practical alternative to more expensive anticoagulants.”
Drs Kahn and Zukor both noted the need for additional trials that would include a randomized group of patients prescribed aspirin exclusively so as to test its efficacy directly against rivaroxaban.
Aspirin seems to be as effective as rivaroxaban for preventing venous thromboembolism (VTE) after total hip or knee replacement, according to research published in NEJM.
Researchers observed a similar incidence of VTE whether patients were randomized to receive aspirin or rivaroxaban prophylaxis starting 5 days after surgery, with all patients receiving rivaroxaban for the first 5 days after surgery.
There was no significant difference between the aspirin and rivaroxaban groups with regard to major bleeding or clinically important bleeding.
“We have always been very concerned about preventing blood clots in patients following major orthopedic surgery,” said study author David Zukor, MD, of Jewish General Hospital in Montreal, Quebec, Canada.
“It is the leading cause of preventable in-hospital death, so we always administer preventive therapy in conjunction with such surgeries. Rivaroxaban is known to be effective, but the great advantage of aspirin is that it is far less expensive, easily available, and has an excellent safety profile.”
Dr Zukor and his colleagues set out to compare aspirin and rivaroxaban in 3424 patients, 1804 who underwent hip replacement and 1620 who underwent knee replacement.
All patients took rivaroxaban (10 mg) for 5 days after surgery before being randomized to receive aspirin (81 mg daily, n=1707) or to continue with rivaroxaban (n=1717). The patients received this prophylaxis for an additional 9 days if they had knee replacement or for 30 days if they had hip replacement.
The researchers followed patients for 90 days, monitoring them for symptomatic VTE and major or clinically relevant nonmajor bleeding.
The rate of VTE was 0.64% (n=11) in the aspirin group and 0.70% (n=12) in the rivaroxaban group (difference, 0.06 percentage points; 95% confidence interval [CI], −0.55 to 0.66; P<0.001 for noninferiority, P=0.84 for superiority).
Major bleeding occurred in 0.47% (n=8) of patients in the aspirin group and in 0.29% (n=5) of those in the rivaroxaban group (difference, 0.18 percentage points; 95% CI, −0.65 to 0.29; P=0.42).
Clinically relevant nonmajor bleeding occurred in 1.29% (n=22) and 0.99% (n=17), respectively (difference, 0.30 percentage points; 95% CI, −1.07 to 0.47; P=0.43).
All bleeding events occurred at the surgical site, and most occurred within 10 days of randomization.
“These results are important,” said study author Susan Kahn, MD, of McGill University in Montreal.
“The protocols for preventing clots following major orthopedic surgery are well established. However, we are always interested in determining whether there are better options for treating our patients. We could well see aspirin emerge as a practical alternative to more expensive anticoagulants.”
Drs Kahn and Zukor both noted the need for additional trials that would include a randomized group of patients prescribed aspirin exclusively so as to test its efficacy directly against rivaroxaban.
Panic Disorder: Ensuring Prompt Recognition and Treatment
Lacey, 37, is seen by her primary care provider (PCP) as follow-up to a visit she made to the emergency department (ED). She has gone to the ED four times in the past year. Each time, she presents with tachycardia, dyspnea, nausea, numbness in her extremities, and a fear that she is having a heart attack. Despite negative workups at each visit (ECG, cardiac enzymes, complete blood count, toxicology screen, Holter monitoring), Lacey is terrified that the ED doctors are missing something. She is still “rattled” by the chest pain and shortness of breath she experiences. Mild symptoms are persisting, and she is worried that she will have a heart attack and die without the treatment she believes she needs. How do you proceed?
Panic disorder (PD) is characterized by the spontaneous and unexpected occurrence of panic attacks and by at least one month of persistent worry about having another attack or significant maladaptive behaviors related to the attack. Frequency of such attacks can vary from several a day to only a few per year. In a panic attack, an intense fear develops abruptly and peaks within 10 minutes of onset. At least four of the following 13 symptoms must accompany the attack, according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth edition (DSM-5)
- Palpitations, pounding heart, or accelerated heart rate
- Sweating
- Trembling or shaking
- Sensations of shortness of breath or smothering
- Feeling of choking
- Chest pain or discomfort
- Feeling dizzy, unsteady, lightheaded, or faint
- Nausea or abdominal distress
- Derealization (feelings of unreality) or depersonalization (being detached from oneself)
- Fear of losing control or going crazy
- Fear of dying
- Paresthesia (numbness or tingling sensations)
- Chills or hot flushes.1
Lifetime incidence rates of PD are 1% to 3% for the general population.2 A closer look at patients presenting to the ED with chest pain reveals that 17% to 25% meet criteria for PD.3,4 And an estimated 6% of individuals experiencing a panic attack present to their primary care provider.5 Patients with PD tend to use health care resources at a disproportionately high rate.6
An international review of PD research suggests the average age of onset is 32 years.7 Triggers can vary widely, and no single stressor has been identified. The exact cause of PD is unknown, but a convergence of social and biological influences (including involvement of the amygdala) are implicated in its development.6 For individuals who have had a panic attack, 66.5% will have recurrent attacks.7 Lifetime prevalence of panic attacks is 13.2%.7
Differential goes far beyond myocardial infarction. Many medical conditions can mimic PD symptoms: cardiovascular, pulmonary, and neurologic diseases; endocrine diseases (eg, hyperthyroidism); drug intoxication (eg, stimulants [cocaine, amphetamines]); drug withdrawal (eg, benzodiazepines, alcohol, sedative-hypnotics); and ingestion of excessive quantities of caffeine. Common comorbid medical disorders include asthma, coronary artery disease, cancer, thyroid disease, hypertension, ulcer, and migraine headaches.8
When patients present with paniclike symptoms, suspect a possible medical condition when those symptoms include ataxia, altered mental status, or loss of bladder control, or when onset of panic symptoms occur later in life for a patient with no significant psychiatric history.
RULE OUT ORGANIC CAUSES
In addition to obtaining a complete history and doing a physical exam on patients with paniclike symptoms, you’ll also need to ensure that the following are done: a neurologic examination, standard laboratory testing (thyroid function, complete blood cell count, chemistry panel), and possible additional testing (eg, urine toxicology screen and
If organic causes are ruled out, focus on a psychiatric assessment, including
- History of the present illness (onset, symptoms, frequency, predisposing/precipitating factors)
- Psychiatric history
- History of substance use
- Family history of psychiatric disorders (especially anxiety disorders)
- Social history (life events, including those preceding the onset of panic; history of child abuse)
- Medications
- Mental status examination
- Safety (PD is associated with higher risk for suicidal ideation).9
TREATMENT INCLUDES CBT AND MEDICATION
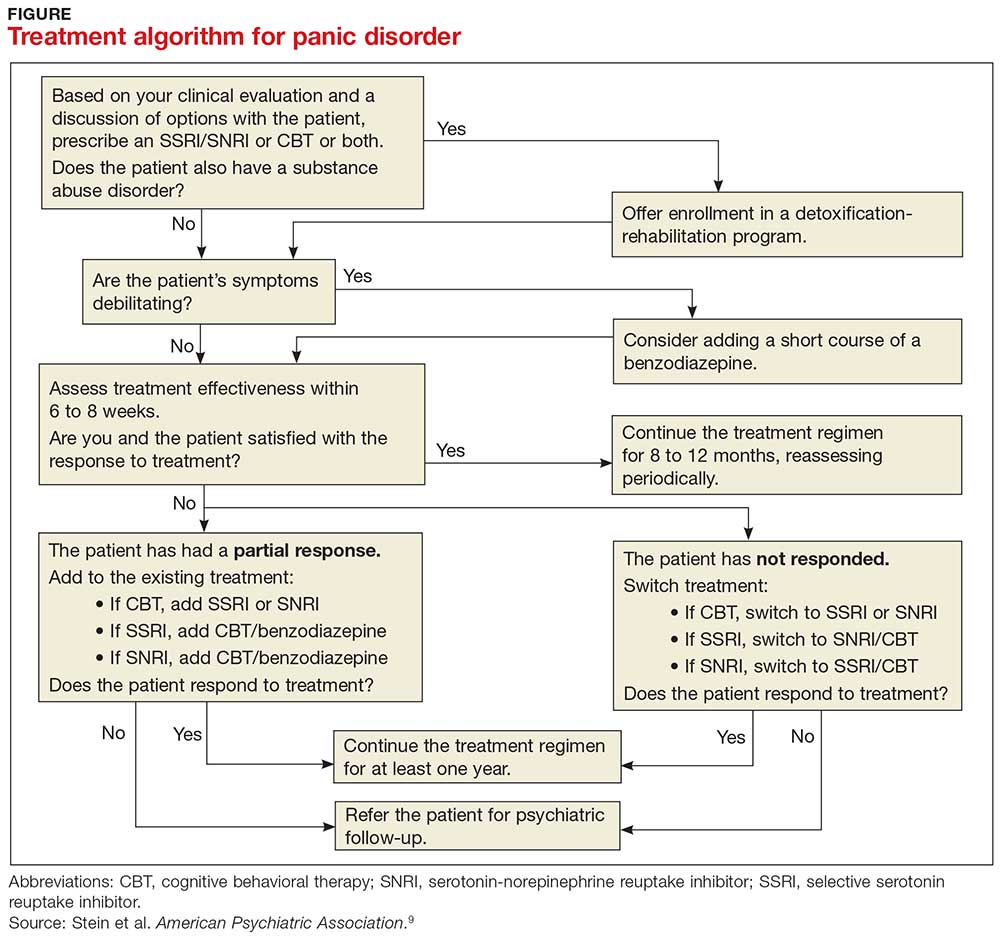
PD is a chronic disease with a variable course, but the long-term prognosis is good. PD is usually treated in an outpatient setting. Consider hospitalization if the patient is suicidal, if the potential for life-threatening withdrawal symptoms is high (as with alcohol or benzodiazepines), or if the symptoms are severely debilitating or attempted outpatient treatment is unsuccessful. Pharmacologic and psychotherapeutic interventions are used for PD (see Figure), although there is not enough evidence to recommend one versus the other or combination therapy versus monotherapy.9
All Lacey’s test results come back negative, and the psychiatric assessment reveals that she meets the DSM-5 criteria for PD. Counting on the strength of their relationship, her PCP talks to her about PD and discusses treatment options, which include counseling, medication, or both. Lacey agrees to a referral for cognitive behavioral therapy (CBT) with a psychologist embedded at her primary care clinic and to begin taking medication. Her PCP starts her on sertraline 25 mg/d.
In CBT, Lacey’s psychologist teaches her about “fight or flight” and explains that it is a normal physiologic response that can lead to panic. Lacey learns to approach her physical symptoms in a different way, and how to breathe in a way that slows her panic reaction.
Consider SSRIs and SNRIs
Firstline medication is a selective serotonin reuptake inhibitor (SSRI) or a serotonin-norepinephrine reuptake inhibitor (SNRI), due to the better tolerability and lower adverse effect profile of these classes compared with the tricyclic antidepressants or monoamine oxidase inhibitors (MAOIs). MAOIs are usually reserved for patients in whom multiple medication trials have failed.
Special considerations. American Psychiatric Association guidelines advise starting with a very low dose of an SSRI or SNRI, such as paroxetine 10 mg/d (although
Keep in mind that the onset of therapeutic effect is between two and four weeks, but that clinical response can take eight to 12 weeks. Continue pharmacotherapy for at least one year. When discontinuing the medication, taper it slowly, and monitor the patient for withdrawal symptoms and recurrence of PD.9
Consider adding a benzodiazepine if symptoms are debilitating.9 Keep in mind, though, that the potential for addiction with these medications is high and they are intended to be used for only four to 12 weeks.8 Onset of action is within the first week, and a scheduled dosing regimen is preferred to giving the medication as needed. The starting dose (eg, clonazepam 0.25 mg bid) may be increased three to five days following initiation.9
Evidence supports the use of CBT for PD
CBT is an evidenced-based treatment for PD.10-13 Up to 75% of patients treated with CBT are panic free within four months.10 Other techniques proven effective are progressive muscle relaxation training, breathing retraining, psychoeducation, exposure, and imagery.14
Treatment with medications and CBT, either combined or used individually, is effective in 80% to 90% of cases.15 CBT has been shown to decrease the likelihood of relapse in the year following treatment.15 Good premorbid functioning and a brief duration of symptoms increase the likelihood of a good prognosis.15
WHEN TO REFER TO A PSYCHIATRIST
Consider referral to a psychiatrist when patients have a comorbid psychiatric condition that complicates the clinical picture (eg, substance abuse disorder), if the diagnosis is uncertain, or if the patient does not respond to one or two adequate trials of medication and psychotherapy. Although psychiatric follow-up is sometimes difficult due to a lack of psychiatrist availability locally, it is a best-practice recommendation.
Ten days after Lacey starts the sertraline 25 mg/d, she calls the PCP to report daily diarrhea. She stopped the sertraline on her own and is asking for another medication. She also expresses her frustration with the severity of the symptoms. She is having three to five panic attacks daily and has been missing many days of work.
On the day of her follow-up PCP appointment, Lacey also sees the psychologist. She reports that she’s been practicing relaxation breathing, tracking her panic attacks, limiting her caffeine intake, and exercising regularly. But the attacks are still occurring.
The PCP switches her to paroxetine 10 mg/d and, due to the severity of the symptoms, prescribes clonazepam 0.5 mg bid. Two weeks later, Lacey reports that she is feeling a little better, has returned to work, and is hopeful that she will be her “normal self again.” The PCP plans to encourage continuation of CBT, titrate the paroxetine to 20 to 40 mg/d based on symptoms, and slowly taper the clonazepam toward discontinuation in the near future.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
2. Kumar S, Oakley-Browne M. Panic disorder. Clin Evid. 2006;15:1438-1452.
3. Yingling KW, Wulsin LR, Arnold LM, et al. Estimated prevalences of panic disorder and depression among consecutive patients seen in an emergency department with acute chest pain. J Gen Intern Med. 1993;8:231-235.
4. Fleet RP, Dupuis G, Marchand A, et al. Panic disorder in emergency department chest pain patients: prevalence, comorbidity, suicidal ideation, and physician recognition. Am J Med. 1996;101:371-380.
5. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA. 1994;272:1749-1756.
6. Taylor CB. Panic disorder. BMJ. 2006;332:951-955.
7. de Jonge P, Roest AM, Lim CC, et al. Cross-national epidemiology of panic disorder and panic attacks in the world mental health surveys. Depress Anxiety. 2016;33: 1155-1177.
8. Sadock BJ, Sadock VA, Ruiz P. Panic disorder. In: Kaplan & Sadock’s Synopsis of Psychiatry. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2015:392-397.
9. Stein MB, Goin MK, Pollack MH, et al. ractice Guideline for the Treatment of Patients with Panic Disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2010. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Accessed February 14, 2018.
10. Westen D, Morrison K. A multidimensional meta-analysis of treatments for depression, panic, and generalized anxiety disorder: an empirical examination of the status of empirically supported therapies. J Consult Clin Psychol. 2001;69:875-899.
11. Gould RA, Otto MW, Pollack MH. A meta-analysis of treatment outcome for panic disorder. www.ncbi.nlm.nih.gov/books/NBK66380/. Accessed February 14, 2018.
12. Clum GA, Clum GA, Surls R. A meta-analysis of treatments for panic disorder. J Consult Clin Psychol. 1993; 61:317-326.
13. Shear MK, Houck P, Greeno C, et al. Emotion-focused psychotherapy for patients with panic disorder. Am J Psychiatry. 2001;158:1993-1998.
14. Stewart RE, Chambless DL. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J Consult Clin Psychol. 2009;77:595-606.
15. Craske M. Psychotherapy for panic disorder in adults. Up to Date. 2017. www.uptodate.com/contents/psychotherapy-for-panic-disorder-with-or-without-agoraphobia-in-adults. Accessed February 14, 2018.
Lacey, 37, is seen by her primary care provider (PCP) as follow-up to a visit she made to the emergency department (ED). She has gone to the ED four times in the past year. Each time, she presents with tachycardia, dyspnea, nausea, numbness in her extremities, and a fear that she is having a heart attack. Despite negative workups at each visit (ECG, cardiac enzymes, complete blood count, toxicology screen, Holter monitoring), Lacey is terrified that the ED doctors are missing something. She is still “rattled” by the chest pain and shortness of breath she experiences. Mild symptoms are persisting, and she is worried that she will have a heart attack and die without the treatment she believes she needs. How do you proceed?
Panic disorder (PD) is characterized by the spontaneous and unexpected occurrence of panic attacks and by at least one month of persistent worry about having another attack or significant maladaptive behaviors related to the attack. Frequency of such attacks can vary from several a day to only a few per year. In a panic attack, an intense fear develops abruptly and peaks within 10 minutes of onset. At least four of the following 13 symptoms must accompany the attack, according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth edition (DSM-5)
- Palpitations, pounding heart, or accelerated heart rate
- Sweating
- Trembling or shaking
- Sensations of shortness of breath or smothering
- Feeling of choking
- Chest pain or discomfort
- Feeling dizzy, unsteady, lightheaded, or faint
- Nausea or abdominal distress
- Derealization (feelings of unreality) or depersonalization (being detached from oneself)
- Fear of losing control or going crazy
- Fear of dying
- Paresthesia (numbness or tingling sensations)
- Chills or hot flushes.1
Lifetime incidence rates of PD are 1% to 3% for the general population.2 A closer look at patients presenting to the ED with chest pain reveals that 17% to 25% meet criteria for PD.3,4 And an estimated 6% of individuals experiencing a panic attack present to their primary care provider.5 Patients with PD tend to use health care resources at a disproportionately high rate.6
An international review of PD research suggests the average age of onset is 32 years.7 Triggers can vary widely, and no single stressor has been identified. The exact cause of PD is unknown, but a convergence of social and biological influences (including involvement of the amygdala) are implicated in its development.6 For individuals who have had a panic attack, 66.5% will have recurrent attacks.7 Lifetime prevalence of panic attacks is 13.2%.7
Differential goes far beyond myocardial infarction. Many medical conditions can mimic PD symptoms: cardiovascular, pulmonary, and neurologic diseases; endocrine diseases (eg, hyperthyroidism); drug intoxication (eg, stimulants [cocaine, amphetamines]); drug withdrawal (eg, benzodiazepines, alcohol, sedative-hypnotics); and ingestion of excessive quantities of caffeine. Common comorbid medical disorders include asthma, coronary artery disease, cancer, thyroid disease, hypertension, ulcer, and migraine headaches.8
When patients present with paniclike symptoms, suspect a possible medical condition when those symptoms include ataxia, altered mental status, or loss of bladder control, or when onset of panic symptoms occur later in life for a patient with no significant psychiatric history.
RULE OUT ORGANIC CAUSES
In addition to obtaining a complete history and doing a physical exam on patients with paniclike symptoms, you’ll also need to ensure that the following are done: a neurologic examination, standard laboratory testing (thyroid function, complete blood cell count, chemistry panel), and possible additional testing (eg, urine toxicology screen and
If organic causes are ruled out, focus on a psychiatric assessment, including
- History of the present illness (onset, symptoms, frequency, predisposing/precipitating factors)
- Psychiatric history
- History of substance use
- Family history of psychiatric disorders (especially anxiety disorders)
- Social history (life events, including those preceding the onset of panic; history of child abuse)
- Medications
- Mental status examination
- Safety (PD is associated with higher risk for suicidal ideation).9
TREATMENT INCLUDES CBT AND MEDICATION
PD is a chronic disease with a variable course, but the long-term prognosis is good. PD is usually treated in an outpatient setting. Consider hospitalization if the patient is suicidal, if the potential for life-threatening withdrawal symptoms is high (as with alcohol or benzodiazepines), or if the symptoms are severely debilitating or attempted outpatient treatment is unsuccessful. Pharmacologic and psychotherapeutic interventions are used for PD (see Figure), although there is not enough evidence to recommend one versus the other or combination therapy versus monotherapy.9
All Lacey’s test results come back negative, and the psychiatric assessment reveals that she meets the DSM-5 criteria for PD. Counting on the strength of their relationship, her PCP talks to her about PD and discusses treatment options, which include counseling, medication, or both. Lacey agrees to a referral for cognitive behavioral therapy (CBT) with a psychologist embedded at her primary care clinic and to begin taking medication. Her PCP starts her on sertraline 25 mg/d.
In CBT, Lacey’s psychologist teaches her about “fight or flight” and explains that it is a normal physiologic response that can lead to panic. Lacey learns to approach her physical symptoms in a different way, and how to breathe in a way that slows her panic reaction.
Consider SSRIs and SNRIs
Firstline medication is a selective serotonin reuptake inhibitor (SSRI) or a serotonin-norepinephrine reuptake inhibitor (SNRI), due to the better tolerability and lower adverse effect profile of these classes compared with the tricyclic antidepressants or monoamine oxidase inhibitors (MAOIs). MAOIs are usually reserved for patients in whom multiple medication trials have failed.
Special considerations. American Psychiatric Association guidelines advise starting with a very low dose of an SSRI or SNRI, such as paroxetine 10 mg/d (although
Keep in mind that the onset of therapeutic effect is between two and four weeks, but that clinical response can take eight to 12 weeks. Continue pharmacotherapy for at least one year. When discontinuing the medication, taper it slowly, and monitor the patient for withdrawal symptoms and recurrence of PD.9
Consider adding a benzodiazepine if symptoms are debilitating.9 Keep in mind, though, that the potential for addiction with these medications is high and they are intended to be used for only four to 12 weeks.8 Onset of action is within the first week, and a scheduled dosing regimen is preferred to giving the medication as needed. The starting dose (eg, clonazepam 0.25 mg bid) may be increased three to five days following initiation.9
Evidence supports the use of CBT for PD
CBT is an evidenced-based treatment for PD.10-13 Up to 75% of patients treated with CBT are panic free within four months.10 Other techniques proven effective are progressive muscle relaxation training, breathing retraining, psychoeducation, exposure, and imagery.14
Treatment with medications and CBT, either combined or used individually, is effective in 80% to 90% of cases.15 CBT has been shown to decrease the likelihood of relapse in the year following treatment.15 Good premorbid functioning and a brief duration of symptoms increase the likelihood of a good prognosis.15
WHEN TO REFER TO A PSYCHIATRIST
Consider referral to a psychiatrist when patients have a comorbid psychiatric condition that complicates the clinical picture (eg, substance abuse disorder), if the diagnosis is uncertain, or if the patient does not respond to one or two adequate trials of medication and psychotherapy. Although psychiatric follow-up is sometimes difficult due to a lack of psychiatrist availability locally, it is a best-practice recommendation.
Ten days after Lacey starts the sertraline 25 mg/d, she calls the PCP to report daily diarrhea. She stopped the sertraline on her own and is asking for another medication. She also expresses her frustration with the severity of the symptoms. She is having three to five panic attacks daily and has been missing many days of work.
On the day of her follow-up PCP appointment, Lacey also sees the psychologist. She reports that she’s been practicing relaxation breathing, tracking her panic attacks, limiting her caffeine intake, and exercising regularly. But the attacks are still occurring.
The PCP switches her to paroxetine 10 mg/d and, due to the severity of the symptoms, prescribes clonazepam 0.5 mg bid. Two weeks later, Lacey reports that she is feeling a little better, has returned to work, and is hopeful that she will be her “normal self again.” The PCP plans to encourage continuation of CBT, titrate the paroxetine to 20 to 40 mg/d based on symptoms, and slowly taper the clonazepam toward discontinuation in the near future.
Lacey, 37, is seen by her primary care provider (PCP) as follow-up to a visit she made to the emergency department (ED). She has gone to the ED four times in the past year. Each time, she presents with tachycardia, dyspnea, nausea, numbness in her extremities, and a fear that she is having a heart attack. Despite negative workups at each visit (ECG, cardiac enzymes, complete blood count, toxicology screen, Holter monitoring), Lacey is terrified that the ED doctors are missing something. She is still “rattled” by the chest pain and shortness of breath she experiences. Mild symptoms are persisting, and she is worried that she will have a heart attack and die without the treatment she believes she needs. How do you proceed?
Panic disorder (PD) is characterized by the spontaneous and unexpected occurrence of panic attacks and by at least one month of persistent worry about having another attack or significant maladaptive behaviors related to the attack. Frequency of such attacks can vary from several a day to only a few per year. In a panic attack, an intense fear develops abruptly and peaks within 10 minutes of onset. At least four of the following 13 symptoms must accompany the attack, according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth edition (DSM-5)
- Palpitations, pounding heart, or accelerated heart rate
- Sweating
- Trembling or shaking
- Sensations of shortness of breath or smothering
- Feeling of choking
- Chest pain or discomfort
- Feeling dizzy, unsteady, lightheaded, or faint
- Nausea or abdominal distress
- Derealization (feelings of unreality) or depersonalization (being detached from oneself)
- Fear of losing control or going crazy
- Fear of dying
- Paresthesia (numbness or tingling sensations)
- Chills or hot flushes.1
Lifetime incidence rates of PD are 1% to 3% for the general population.2 A closer look at patients presenting to the ED with chest pain reveals that 17% to 25% meet criteria for PD.3,4 And an estimated 6% of individuals experiencing a panic attack present to their primary care provider.5 Patients with PD tend to use health care resources at a disproportionately high rate.6
An international review of PD research suggests the average age of onset is 32 years.7 Triggers can vary widely, and no single stressor has been identified. The exact cause of PD is unknown, but a convergence of social and biological influences (including involvement of the amygdala) are implicated in its development.6 For individuals who have had a panic attack, 66.5% will have recurrent attacks.7 Lifetime prevalence of panic attacks is 13.2%.7
Differential goes far beyond myocardial infarction. Many medical conditions can mimic PD symptoms: cardiovascular, pulmonary, and neurologic diseases; endocrine diseases (eg, hyperthyroidism); drug intoxication (eg, stimulants [cocaine, amphetamines]); drug withdrawal (eg, benzodiazepines, alcohol, sedative-hypnotics); and ingestion of excessive quantities of caffeine. Common comorbid medical disorders include asthma, coronary artery disease, cancer, thyroid disease, hypertension, ulcer, and migraine headaches.8
When patients present with paniclike symptoms, suspect a possible medical condition when those symptoms include ataxia, altered mental status, or loss of bladder control, or when onset of panic symptoms occur later in life for a patient with no significant psychiatric history.
RULE OUT ORGANIC CAUSES
In addition to obtaining a complete history and doing a physical exam on patients with paniclike symptoms, you’ll also need to ensure that the following are done: a neurologic examination, standard laboratory testing (thyroid function, complete blood cell count, chemistry panel), and possible additional testing (eg, urine toxicology screen and
If organic causes are ruled out, focus on a psychiatric assessment, including
- History of the present illness (onset, symptoms, frequency, predisposing/precipitating factors)
- Psychiatric history
- History of substance use
- Family history of psychiatric disorders (especially anxiety disorders)
- Social history (life events, including those preceding the onset of panic; history of child abuse)
- Medications
- Mental status examination
- Safety (PD is associated with higher risk for suicidal ideation).9
TREATMENT INCLUDES CBT AND MEDICATION
PD is a chronic disease with a variable course, but the long-term prognosis is good. PD is usually treated in an outpatient setting. Consider hospitalization if the patient is suicidal, if the potential for life-threatening withdrawal symptoms is high (as with alcohol or benzodiazepines), or if the symptoms are severely debilitating or attempted outpatient treatment is unsuccessful. Pharmacologic and psychotherapeutic interventions are used for PD (see Figure), although there is not enough evidence to recommend one versus the other or combination therapy versus monotherapy.9
All Lacey’s test results come back negative, and the psychiatric assessment reveals that she meets the DSM-5 criteria for PD. Counting on the strength of their relationship, her PCP talks to her about PD and discusses treatment options, which include counseling, medication, or both. Lacey agrees to a referral for cognitive behavioral therapy (CBT) with a psychologist embedded at her primary care clinic and to begin taking medication. Her PCP starts her on sertraline 25 mg/d.
In CBT, Lacey’s psychologist teaches her about “fight or flight” and explains that it is a normal physiologic response that can lead to panic. Lacey learns to approach her physical symptoms in a different way, and how to breathe in a way that slows her panic reaction.
Consider SSRIs and SNRIs
Firstline medication is a selective serotonin reuptake inhibitor (SSRI) or a serotonin-norepinephrine reuptake inhibitor (SNRI), due to the better tolerability and lower adverse effect profile of these classes compared with the tricyclic antidepressants or monoamine oxidase inhibitors (MAOIs). MAOIs are usually reserved for patients in whom multiple medication trials have failed.
Special considerations. American Psychiatric Association guidelines advise starting with a very low dose of an SSRI or SNRI, such as paroxetine 10 mg/d (although
Keep in mind that the onset of therapeutic effect is between two and four weeks, but that clinical response can take eight to 12 weeks. Continue pharmacotherapy for at least one year. When discontinuing the medication, taper it slowly, and monitor the patient for withdrawal symptoms and recurrence of PD.9
Consider adding a benzodiazepine if symptoms are debilitating.9 Keep in mind, though, that the potential for addiction with these medications is high and they are intended to be used for only four to 12 weeks.8 Onset of action is within the first week, and a scheduled dosing regimen is preferred to giving the medication as needed. The starting dose (eg, clonazepam 0.25 mg bid) may be increased three to five days following initiation.9
Evidence supports the use of CBT for PD
CBT is an evidenced-based treatment for PD.10-13 Up to 75% of patients treated with CBT are panic free within four months.10 Other techniques proven effective are progressive muscle relaxation training, breathing retraining, psychoeducation, exposure, and imagery.14
Treatment with medications and CBT, either combined or used individually, is effective in 80% to 90% of cases.15 CBT has been shown to decrease the likelihood of relapse in the year following treatment.15 Good premorbid functioning and a brief duration of symptoms increase the likelihood of a good prognosis.15
WHEN TO REFER TO A PSYCHIATRIST
Consider referral to a psychiatrist when patients have a comorbid psychiatric condition that complicates the clinical picture (eg, substance abuse disorder), if the diagnosis is uncertain, or if the patient does not respond to one or two adequate trials of medication and psychotherapy. Although psychiatric follow-up is sometimes difficult due to a lack of psychiatrist availability locally, it is a best-practice recommendation.
Ten days after Lacey starts the sertraline 25 mg/d, she calls the PCP to report daily diarrhea. She stopped the sertraline on her own and is asking for another medication. She also expresses her frustration with the severity of the symptoms. She is having three to five panic attacks daily and has been missing many days of work.
On the day of her follow-up PCP appointment, Lacey also sees the psychologist. She reports that she’s been practicing relaxation breathing, tracking her panic attacks, limiting her caffeine intake, and exercising regularly. But the attacks are still occurring.
The PCP switches her to paroxetine 10 mg/d and, due to the severity of the symptoms, prescribes clonazepam 0.5 mg bid. Two weeks later, Lacey reports that she is feeling a little better, has returned to work, and is hopeful that she will be her “normal self again.” The PCP plans to encourage continuation of CBT, titrate the paroxetine to 20 to 40 mg/d based on symptoms, and slowly taper the clonazepam toward discontinuation in the near future.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
2. Kumar S, Oakley-Browne M. Panic disorder. Clin Evid. 2006;15:1438-1452.
3. Yingling KW, Wulsin LR, Arnold LM, et al. Estimated prevalences of panic disorder and depression among consecutive patients seen in an emergency department with acute chest pain. J Gen Intern Med. 1993;8:231-235.
4. Fleet RP, Dupuis G, Marchand A, et al. Panic disorder in emergency department chest pain patients: prevalence, comorbidity, suicidal ideation, and physician recognition. Am J Med. 1996;101:371-380.
5. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA. 1994;272:1749-1756.
6. Taylor CB. Panic disorder. BMJ. 2006;332:951-955.
7. de Jonge P, Roest AM, Lim CC, et al. Cross-national epidemiology of panic disorder and panic attacks in the world mental health surveys. Depress Anxiety. 2016;33: 1155-1177.
8. Sadock BJ, Sadock VA, Ruiz P. Panic disorder. In: Kaplan & Sadock’s Synopsis of Psychiatry. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2015:392-397.
9. Stein MB, Goin MK, Pollack MH, et al. ractice Guideline for the Treatment of Patients with Panic Disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2010. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Accessed February 14, 2018.
10. Westen D, Morrison K. A multidimensional meta-analysis of treatments for depression, panic, and generalized anxiety disorder: an empirical examination of the status of empirically supported therapies. J Consult Clin Psychol. 2001;69:875-899.
11. Gould RA, Otto MW, Pollack MH. A meta-analysis of treatment outcome for panic disorder. www.ncbi.nlm.nih.gov/books/NBK66380/. Accessed February 14, 2018.
12. Clum GA, Clum GA, Surls R. A meta-analysis of treatments for panic disorder. J Consult Clin Psychol. 1993; 61:317-326.
13. Shear MK, Houck P, Greeno C, et al. Emotion-focused psychotherapy for patients with panic disorder. Am J Psychiatry. 2001;158:1993-1998.
14. Stewart RE, Chambless DL. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J Consult Clin Psychol. 2009;77:595-606.
15. Craske M. Psychotherapy for panic disorder in adults. Up to Date. 2017. www.uptodate.com/contents/psychotherapy-for-panic-disorder-with-or-without-agoraphobia-in-adults. Accessed February 14, 2018.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
2. Kumar S, Oakley-Browne M. Panic disorder. Clin Evid. 2006;15:1438-1452.
3. Yingling KW, Wulsin LR, Arnold LM, et al. Estimated prevalences of panic disorder and depression among consecutive patients seen in an emergency department with acute chest pain. J Gen Intern Med. 1993;8:231-235.
4. Fleet RP, Dupuis G, Marchand A, et al. Panic disorder in emergency department chest pain patients: prevalence, comorbidity, suicidal ideation, and physician recognition. Am J Med. 1996;101:371-380.
5. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA. 1994;272:1749-1756.
6. Taylor CB. Panic disorder. BMJ. 2006;332:951-955.
7. de Jonge P, Roest AM, Lim CC, et al. Cross-national epidemiology of panic disorder and panic attacks in the world mental health surveys. Depress Anxiety. 2016;33: 1155-1177.
8. Sadock BJ, Sadock VA, Ruiz P. Panic disorder. In: Kaplan & Sadock’s Synopsis of Psychiatry. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2015:392-397.
9. Stein MB, Goin MK, Pollack MH, et al. ractice Guideline for the Treatment of Patients with Panic Disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2010. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/panicdisorder.pdf. Accessed February 14, 2018.
10. Westen D, Morrison K. A multidimensional meta-analysis of treatments for depression, panic, and generalized anxiety disorder: an empirical examination of the status of empirically supported therapies. J Consult Clin Psychol. 2001;69:875-899.
11. Gould RA, Otto MW, Pollack MH. A meta-analysis of treatment outcome for panic disorder. www.ncbi.nlm.nih.gov/books/NBK66380/. Accessed February 14, 2018.
12. Clum GA, Clum GA, Surls R. A meta-analysis of treatments for panic disorder. J Consult Clin Psychol. 1993; 61:317-326.
13. Shear MK, Houck P, Greeno C, et al. Emotion-focused psychotherapy for patients with panic disorder. Am J Psychiatry. 2001;158:1993-1998.
14. Stewart RE, Chambless DL. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J Consult Clin Psychol. 2009;77:595-606.
15. Craske M. Psychotherapy for panic disorder in adults. Up to Date. 2017. www.uptodate.com/contents/psychotherapy-for-panic-disorder-with-or-without-agoraphobia-in-adults. Accessed February 14, 2018.
From the Washington Office: Gratifying success for ACS legislative advocacy efforts
On the morning of February 9, 2018, President Trump signed into law the Bipartisan Budget Act of 2018. The law included legislative priorities that were championed by the ACS and for which staff of the DC office and engaged Fellows of the College have advocated, in some cases, for a number of years.
ACS worked particularly hard in the week leading up to the passage of the Bipartisan Budget Act of 2018 with the goal of ensuring that certain items were included, and certain other items were excluded, in the Continuing Resolution (CR) under consideration by Congress to continue funding the government. The original version of the CR considered and debated by the House of Representatives early in the week included both positive and negative items. The ACS was successful in its efforts to get the Senate to consider a much-improved version of the CR – eliminating a major impediment in the House version. Ultimately, it was the Senate version that was signed into law by President Trump.
The provisions in the Bipartisan Budget Act of 2018 include:
• Flexibility for the Merit-based Incentive Payment System (MIPS) related to how much weight will be ascribed to the Cost component in an individual physician’s MIPS score as well as flexibility in setting the level at which physicians will either receive a positive or negative payment update. Without this flexibility, there was significant concern that Fellows would have significantly greater challenges in avoiding a cut under the MIPS. This language, and the effort to include it, was spearheaded and long championed by the ACS including the drafting of model legislation remedying the problem which was then provided to the leadership and staff of committees of jurisdiction.
• Easing meaningful use (MU) requirements by removing an outdated requirement directing the Secretary of Health and Human Services (HHS) to continue to make meaningful use standards increasingly stringent over time. The ACS has long advocated against increasingly stringent MU requirements that do not lead to improvements in patient care, and feels they are unnecessary and unfair to both patients and providers. Further, easing MU requirements has long been supported by ACS Fellows.
• An additional 4 years of funding for the Children’s Health Insurance Program (CHIP), bringing the reauthorization period to a total of 10 years. ACS has consistently advocated and aggressively pursued reauthorization of CHIP every time reauthorization was necessary. During the most recent negotiations, following expiration of funding in September 2017, the ACS advocated for the longest possible period of reauthorization of funds.
• Full repeal of the Independent Payment Advisory Board (IPAB), included as part of the Affordable Care Act. Though members of the board were never appointed, the ACS has fought to eliminate this advisory board of unelected bureaucrats who had the power to cut physician payment since 2010.
• Additional funding to address both the opioid epidemic and to support the work of the National Institutes of Health (NIH). The ACS has long supported funding to fight cancer and has been proactive in its response to the national crisis of opioid abuse and misuse.
• Lastly, as mentioned above, the ACS strongly opposed language in the version of the bill passed by the House that would have allowed the use of the “Misvalued Codes” as part of the “pay-for” or offset for the legislation. The ACS anticipated that this language would have unfairly resulted in significant cuts to surgeons and we were pleased that this language was not included in the version ultimately agreed to and signed into law by President Trump. We sincerely hope that this ends the use of this flawed policy.
To have this many policy priorities enacted through one legislative package is a rare occurrence for any organization and accordingly is most gratifying. The emails and phone calls delivered by Fellows during the week of February 5, in combination with the work of staff here in Washington DC, no doubt played a significant role in securing these priorities. However, the work is not done and the ACS will continue to fight for improvements to issues facing surgeons and surgical patients.
We urge all Fellows to continue participating in these efforts.
Until next month ….
On the morning of February 9, 2018, President Trump signed into law the Bipartisan Budget Act of 2018. The law included legislative priorities that were championed by the ACS and for which staff of the DC office and engaged Fellows of the College have advocated, in some cases, for a number of years.
ACS worked particularly hard in the week leading up to the passage of the Bipartisan Budget Act of 2018 with the goal of ensuring that certain items were included, and certain other items were excluded, in the Continuing Resolution (CR) under consideration by Congress to continue funding the government. The original version of the CR considered and debated by the House of Representatives early in the week included both positive and negative items. The ACS was successful in its efforts to get the Senate to consider a much-improved version of the CR – eliminating a major impediment in the House version. Ultimately, it was the Senate version that was signed into law by President Trump.
The provisions in the Bipartisan Budget Act of 2018 include:
• Flexibility for the Merit-based Incentive Payment System (MIPS) related to how much weight will be ascribed to the Cost component in an individual physician’s MIPS score as well as flexibility in setting the level at which physicians will either receive a positive or negative payment update. Without this flexibility, there was significant concern that Fellows would have significantly greater challenges in avoiding a cut under the MIPS. This language, and the effort to include it, was spearheaded and long championed by the ACS including the drafting of model legislation remedying the problem which was then provided to the leadership and staff of committees of jurisdiction.
• Easing meaningful use (MU) requirements by removing an outdated requirement directing the Secretary of Health and Human Services (HHS) to continue to make meaningful use standards increasingly stringent over time. The ACS has long advocated against increasingly stringent MU requirements that do not lead to improvements in patient care, and feels they are unnecessary and unfair to both patients and providers. Further, easing MU requirements has long been supported by ACS Fellows.
• An additional 4 years of funding for the Children’s Health Insurance Program (CHIP), bringing the reauthorization period to a total of 10 years. ACS has consistently advocated and aggressively pursued reauthorization of CHIP every time reauthorization was necessary. During the most recent negotiations, following expiration of funding in September 2017, the ACS advocated for the longest possible period of reauthorization of funds.
• Full repeal of the Independent Payment Advisory Board (IPAB), included as part of the Affordable Care Act. Though members of the board were never appointed, the ACS has fought to eliminate this advisory board of unelected bureaucrats who had the power to cut physician payment since 2010.
• Additional funding to address both the opioid epidemic and to support the work of the National Institutes of Health (NIH). The ACS has long supported funding to fight cancer and has been proactive in its response to the national crisis of opioid abuse and misuse.
• Lastly, as mentioned above, the ACS strongly opposed language in the version of the bill passed by the House that would have allowed the use of the “Misvalued Codes” as part of the “pay-for” or offset for the legislation. The ACS anticipated that this language would have unfairly resulted in significant cuts to surgeons and we were pleased that this language was not included in the version ultimately agreed to and signed into law by President Trump. We sincerely hope that this ends the use of this flawed policy.
To have this many policy priorities enacted through one legislative package is a rare occurrence for any organization and accordingly is most gratifying. The emails and phone calls delivered by Fellows during the week of February 5, in combination with the work of staff here in Washington DC, no doubt played a significant role in securing these priorities. However, the work is not done and the ACS will continue to fight for improvements to issues facing surgeons and surgical patients.
We urge all Fellows to continue participating in these efforts.
Until next month ….
On the morning of February 9, 2018, President Trump signed into law the Bipartisan Budget Act of 2018. The law included legislative priorities that were championed by the ACS and for which staff of the DC office and engaged Fellows of the College have advocated, in some cases, for a number of years.
ACS worked particularly hard in the week leading up to the passage of the Bipartisan Budget Act of 2018 with the goal of ensuring that certain items were included, and certain other items were excluded, in the Continuing Resolution (CR) under consideration by Congress to continue funding the government. The original version of the CR considered and debated by the House of Representatives early in the week included both positive and negative items. The ACS was successful in its efforts to get the Senate to consider a much-improved version of the CR – eliminating a major impediment in the House version. Ultimately, it was the Senate version that was signed into law by President Trump.
The provisions in the Bipartisan Budget Act of 2018 include:
• Flexibility for the Merit-based Incentive Payment System (MIPS) related to how much weight will be ascribed to the Cost component in an individual physician’s MIPS score as well as flexibility in setting the level at which physicians will either receive a positive or negative payment update. Without this flexibility, there was significant concern that Fellows would have significantly greater challenges in avoiding a cut under the MIPS. This language, and the effort to include it, was spearheaded and long championed by the ACS including the drafting of model legislation remedying the problem which was then provided to the leadership and staff of committees of jurisdiction.
• Easing meaningful use (MU) requirements by removing an outdated requirement directing the Secretary of Health and Human Services (HHS) to continue to make meaningful use standards increasingly stringent over time. The ACS has long advocated against increasingly stringent MU requirements that do not lead to improvements in patient care, and feels they are unnecessary and unfair to both patients and providers. Further, easing MU requirements has long been supported by ACS Fellows.
• An additional 4 years of funding for the Children’s Health Insurance Program (CHIP), bringing the reauthorization period to a total of 10 years. ACS has consistently advocated and aggressively pursued reauthorization of CHIP every time reauthorization was necessary. During the most recent negotiations, following expiration of funding in September 2017, the ACS advocated for the longest possible period of reauthorization of funds.
• Full repeal of the Independent Payment Advisory Board (IPAB), included as part of the Affordable Care Act. Though members of the board were never appointed, the ACS has fought to eliminate this advisory board of unelected bureaucrats who had the power to cut physician payment since 2010.
• Additional funding to address both the opioid epidemic and to support the work of the National Institutes of Health (NIH). The ACS has long supported funding to fight cancer and has been proactive in its response to the national crisis of opioid abuse and misuse.
• Lastly, as mentioned above, the ACS strongly opposed language in the version of the bill passed by the House that would have allowed the use of the “Misvalued Codes” as part of the “pay-for” or offset for the legislation. The ACS anticipated that this language would have unfairly resulted in significant cuts to surgeons and we were pleased that this language was not included in the version ultimately agreed to and signed into law by President Trump. We sincerely hope that this ends the use of this flawed policy.
To have this many policy priorities enacted through one legislative package is a rare occurrence for any organization and accordingly is most gratifying. The emails and phone calls delivered by Fellows during the week of February 5, in combination with the work of staff here in Washington DC, no doubt played a significant role in securing these priorities. However, the work is not done and the ACS will continue to fight for improvements to issues facing surgeons and surgical patients.
We urge all Fellows to continue participating in these efforts.
Until next month ….

From the Editors: Unexpected benefits
I am a member of several surgical societies, and more than once I’ve extolled the importance of going to meetings as part of my personal definition of a “good” surgeon.
While I mostly enjoy meetings nowadays, it hasn’t always been easy. My first experiences at surgical society meetings were painful. I was 32 years old, just out of training, and didn’t know anyone at the meetings. Receptions were the worst. My only friend sometimes was either my host or my wife. I was surrounded by these old guys, most of whom were famous.
Among the highlights of my life was being invited to join the Western Surgical Association. The year I was elected, J. David Richardson was the President. He gave such a rousing speech that at the President’s Dinner I rallied the courage to speak to the great man. When I called him “Dr. Richardson,” he waved that off and insisted I call him “Dave.” Little did I know that Dave would become one of my personal heroes and play an important part in the path that led to my being in a position to write a column such as this.

I never thought that being at a meeting might save my own life. As a devoted member of the Western Surgical Association, I breezed into Phoenix last year for the annual meeting. But I wasn’t feeling so well. Typical of a surgeon, I’d been denying that the symptoms in my abdomen and back were anything other than arthritis. Also, like most surgeons, I’d been pushing the accelerator pedal of life to the floorboard for some months. I arrived at the hotel at one in the morning with a stomach ache that clearly was becoming serious.
I lay in bed wondering what to do. Surgeons don’t get sick! Calling 911 seemed like an invitation to a 6-hour ED experience during which I would be demoted from surgeon to “the patient in Room 9” in a town where I didn’t know many people. As a surgeon, I knew that belly pain of this magnitude was not something I wanted to entrust to a nonsurgeon who was just trying to get through another long shift. This was personal.
Here’s where being a member of a surgical society became more than an academic exercise. I was in a hotel with many of the finest surgeons in the world – and I knew many of them! Among the attendees was my very good friend Margo Shoup, a first-rate cancer surgeon from Chicago. I knew she had come in that day because we were to have dinner together the next night. Rather than call her at 0300, I waited until 6. She came right over and examined me. We decided on a plan. As is always the case at a surgical meeting, one of the surgeon members is the local arrangement person. In this instance, it was none other than James Madura, MD, Chief of MIS GI surgery at Mayo Clinic, Scottsdale. We called James and when my abdomen decided to go nuclear with pain, he drove me to his hospital in his own car and got me in their ED.
The rest of the story is pretty mundane. My diagnosis turned out to be temporarily serious but benign. It was serious enough that without good people treating me, I could have done poorly. James and his fine team took superb care of me. Almost all of us go into surgery with the intent to help others. We take that extraseriously when it’s one of “us.” I can imagine the increased pressure on James when he was treating a colleague and the entire rest of the Western Surgical Association knew it. He never even broke a sweat. I am so glad he was my surgeon. His chief resident Ryan Day, MD, spent extra time with me. I saw a bunch of other residents as well. They reminded me of my fellow residents many years ago – eager, bright, and hyperdedicated. Several members of the Western came by the hospital to see me and even reviewed my images and diagnosis with me. I felt like I had a whole family of physicians who were both my friends and my support system a long way from home. I cannot thank James, Margo, and the rest enough for all they did. It’s great to feel back to normal again as well.
So, I would suggest you join and participate in the surgical societies that you think are a good fit for you. Develop those important professional relationships. Such activity will do your patients a world of good – and just might save your own life.
I am a member of several surgical societies, and more than once I’ve extolled the importance of going to meetings as part of my personal definition of a “good” surgeon.
While I mostly enjoy meetings nowadays, it hasn’t always been easy. My first experiences at surgical society meetings were painful. I was 32 years old, just out of training, and didn’t know anyone at the meetings. Receptions were the worst. My only friend sometimes was either my host or my wife. I was surrounded by these old guys, most of whom were famous.
Among the highlights of my life was being invited to join the Western Surgical Association. The year I was elected, J. David Richardson was the President. He gave such a rousing speech that at the President’s Dinner I rallied the courage to speak to the great man. When I called him “Dr. Richardson,” he waved that off and insisted I call him “Dave.” Little did I know that Dave would become one of my personal heroes and play an important part in the path that led to my being in a position to write a column such as this.
I never thought that being at a meeting might save my own life. As a devoted member of the Western Surgical Association, I breezed into Phoenix last year for the annual meeting. But I wasn’t feeling so well. Typical of a surgeon, I’d been denying that the symptoms in my abdomen and back were anything other than arthritis. Also, like most surgeons, I’d been pushing the accelerator pedal of life to the floorboard for some months. I arrived at the hotel at one in the morning with a stomach ache that clearly was becoming serious.
I lay in bed wondering what to do. Surgeons don’t get sick! Calling 911 seemed like an invitation to a 6-hour ED experience during which I would be demoted from surgeon to “the patient in Room 9” in a town where I didn’t know many people. As a surgeon, I knew that belly pain of this magnitude was not something I wanted to entrust to a nonsurgeon who was just trying to get through another long shift. This was personal.
Here’s where being a member of a surgical society became more than an academic exercise. I was in a hotel with many of the finest surgeons in the world – and I knew many of them! Among the attendees was my very good friend Margo Shoup, a first-rate cancer surgeon from Chicago. I knew she had come in that day because we were to have dinner together the next night. Rather than call her at 0300, I waited until 6. She came right over and examined me. We decided on a plan. As is always the case at a surgical meeting, one of the surgeon members is the local arrangement person. In this instance, it was none other than James Madura, MD, Chief of MIS GI surgery at Mayo Clinic, Scottsdale. We called James and when my abdomen decided to go nuclear with pain, he drove me to his hospital in his own car and got me in their ED.
The rest of the story is pretty mundane. My diagnosis turned out to be temporarily serious but benign. It was serious enough that without good people treating me, I could have done poorly. James and his fine team took superb care of me. Almost all of us go into surgery with the intent to help others. We take that extraseriously when it’s one of “us.” I can imagine the increased pressure on James when he was treating a colleague and the entire rest of the Western Surgical Association knew it. He never even broke a sweat. I am so glad he was my surgeon. His chief resident Ryan Day, MD, spent extra time with me. I saw a bunch of other residents as well. They reminded me of my fellow residents many years ago – eager, bright, and hyperdedicated. Several members of the Western came by the hospital to see me and even reviewed my images and diagnosis with me. I felt like I had a whole family of physicians who were both my friends and my support system a long way from home. I cannot thank James, Margo, and the rest enough for all they did. It’s great to feel back to normal again as well.
So, I would suggest you join and participate in the surgical societies that you think are a good fit for you. Develop those important professional relationships. Such activity will do your patients a world of good – and just might save your own life.
I am a member of several surgical societies, and more than once I’ve extolled the importance of going to meetings as part of my personal definition of a “good” surgeon.
While I mostly enjoy meetings nowadays, it hasn’t always been easy. My first experiences at surgical society meetings were painful. I was 32 years old, just out of training, and didn’t know anyone at the meetings. Receptions were the worst. My only friend sometimes was either my host or my wife. I was surrounded by these old guys, most of whom were famous.
Among the highlights of my life was being invited to join the Western Surgical Association. The year I was elected, J. David Richardson was the President. He gave such a rousing speech that at the President’s Dinner I rallied the courage to speak to the great man. When I called him “Dr. Richardson,” he waved that off and insisted I call him “Dave.” Little did I know that Dave would become one of my personal heroes and play an important part in the path that led to my being in a position to write a column such as this.
I never thought that being at a meeting might save my own life. As a devoted member of the Western Surgical Association, I breezed into Phoenix last year for the annual meeting. But I wasn’t feeling so well. Typical of a surgeon, I’d been denying that the symptoms in my abdomen and back were anything other than arthritis. Also, like most surgeons, I’d been pushing the accelerator pedal of life to the floorboard for some months. I arrived at the hotel at one in the morning with a stomach ache that clearly was becoming serious.
I lay in bed wondering what to do. Surgeons don’t get sick! Calling 911 seemed like an invitation to a 6-hour ED experience during which I would be demoted from surgeon to “the patient in Room 9” in a town where I didn’t know many people. As a surgeon, I knew that belly pain of this magnitude was not something I wanted to entrust to a nonsurgeon who was just trying to get through another long shift. This was personal.
Here’s where being a member of a surgical society became more than an academic exercise. I was in a hotel with many of the finest surgeons in the world – and I knew many of them! Among the attendees was my very good friend Margo Shoup, a first-rate cancer surgeon from Chicago. I knew she had come in that day because we were to have dinner together the next night. Rather than call her at 0300, I waited until 6. She came right over and examined me. We decided on a plan. As is always the case at a surgical meeting, one of the surgeon members is the local arrangement person. In this instance, it was none other than James Madura, MD, Chief of MIS GI surgery at Mayo Clinic, Scottsdale. We called James and when my abdomen decided to go nuclear with pain, he drove me to his hospital in his own car and got me in their ED.
The rest of the story is pretty mundane. My diagnosis turned out to be temporarily serious but benign. It was serious enough that without good people treating me, I could have done poorly. James and his fine team took superb care of me. Almost all of us go into surgery with the intent to help others. We take that extraseriously when it’s one of “us.” I can imagine the increased pressure on James when he was treating a colleague and the entire rest of the Western Surgical Association knew it. He never even broke a sweat. I am so glad he was my surgeon. His chief resident Ryan Day, MD, spent extra time with me. I saw a bunch of other residents as well. They reminded me of my fellow residents many years ago – eager, bright, and hyperdedicated. Several members of the Western came by the hospital to see me and even reviewed my images and diagnosis with me. I felt like I had a whole family of physicians who were both my friends and my support system a long way from home. I cannot thank James, Margo, and the rest enough for all they did. It’s great to feel back to normal again as well.
So, I would suggest you join and participate in the surgical societies that you think are a good fit for you. Develop those important professional relationships. Such activity will do your patients a world of good – and just might save your own life.