User login
FDA approves generic clofarabine

The US Food and Drug Administration (FDA) has approved Dr. Reddy’s Laboratories Ltd.’s Clofarabine Injection, a therapeutic equivalent generic version of Clolar® (clofarabine) Injection.
The generic drug is now approved to treat patients age 1 to 21 who have relapsed or refractory acute lymphoblastic leukemia and have received at least 2 prior treatment regimens.
Dr. Reddy’s Clofarabine Injection is available in single-dose, 20 mL flint vials containing 20 mg of clofarabine in 20 mL of solution (1 mg/mL). ![]()
The US Food and Drug Administration (FDA) has approved Dr. Reddy’s Laboratories Ltd.’s Clofarabine Injection, a therapeutic equivalent generic version of Clolar® (clofarabine) Injection.
The generic drug is now approved to treat patients age 1 to 21 who have relapsed or refractory acute lymphoblastic leukemia and have received at least 2 prior treatment regimens.
Dr. Reddy’s Clofarabine Injection is available in single-dose, 20 mL flint vials containing 20 mg of clofarabine in 20 mL of solution (1 mg/mL). ![]()
The US Food and Drug Administration (FDA) has approved Dr. Reddy’s Laboratories Ltd.’s Clofarabine Injection, a therapeutic equivalent generic version of Clolar® (clofarabine) Injection.
The generic drug is now approved to treat patients age 1 to 21 who have relapsed or refractory acute lymphoblastic leukemia and have received at least 2 prior treatment regimens.
Dr. Reddy’s Clofarabine Injection is available in single-dose, 20 mL flint vials containing 20 mg of clofarabine in 20 mL of solution (1 mg/mL). ![]()
Generic azacitidine approved in Canada

Health Canada has approved Dr. Reddy’s Laboratories Ltd.’s Azacitidine for Injection 100 mg/vial, a bioequivalent generic version of VIDAZA® (azacitidine for injection).
The generic drug is approved for the same indications as VIDAZA®.
This includes treating adults with intermediate-2 or high-risk myelodysplastic syndromes (according to the International Prognostic Scoring System) who are not eligible for hematopoietic stem cell transplant.
It also includes treating adults who have acute myeloid leukemia with 20% to 30% blasts and multi-lineage dysplasia (according to World Health Organization classification) who are not eligible for hematopoietic stem cell transplant.
“The approval and launch of Azacitidine for Injection is an important milestone for Dr. Reddy’s in Canada,” said Vinod Ramachandran, PhD, country manager, Dr. Reddy’s Canada.
“The launch of the first generic azacitidine for injection is another step in our long-term commitment to bring more cost-effective options to Canadian patients.” ![]()
Health Canada has approved Dr. Reddy’s Laboratories Ltd.’s Azacitidine for Injection 100 mg/vial, a bioequivalent generic version of VIDAZA® (azacitidine for injection).
The generic drug is approved for the same indications as VIDAZA®.
This includes treating adults with intermediate-2 or high-risk myelodysplastic syndromes (according to the International Prognostic Scoring System) who are not eligible for hematopoietic stem cell transplant.
It also includes treating adults who have acute myeloid leukemia with 20% to 30% blasts and multi-lineage dysplasia (according to World Health Organization classification) who are not eligible for hematopoietic stem cell transplant.
“The approval and launch of Azacitidine for Injection is an important milestone for Dr. Reddy’s in Canada,” said Vinod Ramachandran, PhD, country manager, Dr. Reddy’s Canada.
“The launch of the first generic azacitidine for injection is another step in our long-term commitment to bring more cost-effective options to Canadian patients.” ![]()
Health Canada has approved Dr. Reddy’s Laboratories Ltd.’s Azacitidine for Injection 100 mg/vial, a bioequivalent generic version of VIDAZA® (azacitidine for injection).
The generic drug is approved for the same indications as VIDAZA®.
This includes treating adults with intermediate-2 or high-risk myelodysplastic syndromes (according to the International Prognostic Scoring System) who are not eligible for hematopoietic stem cell transplant.
It also includes treating adults who have acute myeloid leukemia with 20% to 30% blasts and multi-lineage dysplasia (according to World Health Organization classification) who are not eligible for hematopoietic stem cell transplant.
“The approval and launch of Azacitidine for Injection is an important milestone for Dr. Reddy’s in Canada,” said Vinod Ramachandran, PhD, country manager, Dr. Reddy’s Canada.
“The launch of the first generic azacitidine for injection is another step in our long-term commitment to bring more cost-effective options to Canadian patients.” ![]()
Novel biomarker test appears accurate for UTI diagnosis in febrile children under 2 years
Novel test for the urinary neutrophil gelatinase–associated lipocalin (uNGAL) biomarker have good sensitivity and specificity to distinguish whether infants and children younger than 2 years old have a urinary tract infection (UTI), said Tamar R. Lubell, MD, and her associates at Columbia University, New York.
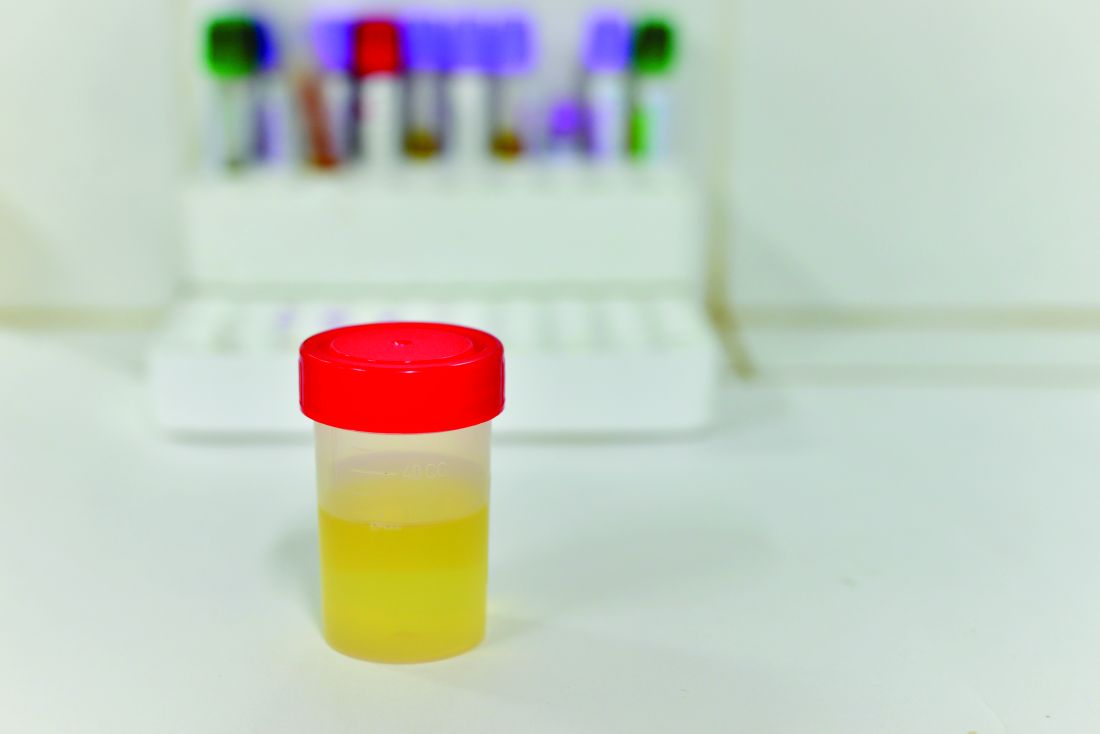
“NGAL is a protein expressed in neutrophils and several other human tissues, including alpha-intercalated cells in the collecting duct of the kidney,” the researchers explained. “The urine and serum contain very low levels of NGAL protein at steady state, with expression of NGAL rising rapidly in response to cell damage caused by ischemia-reperfusion injury, presence of cytotoxins, and sepsis.”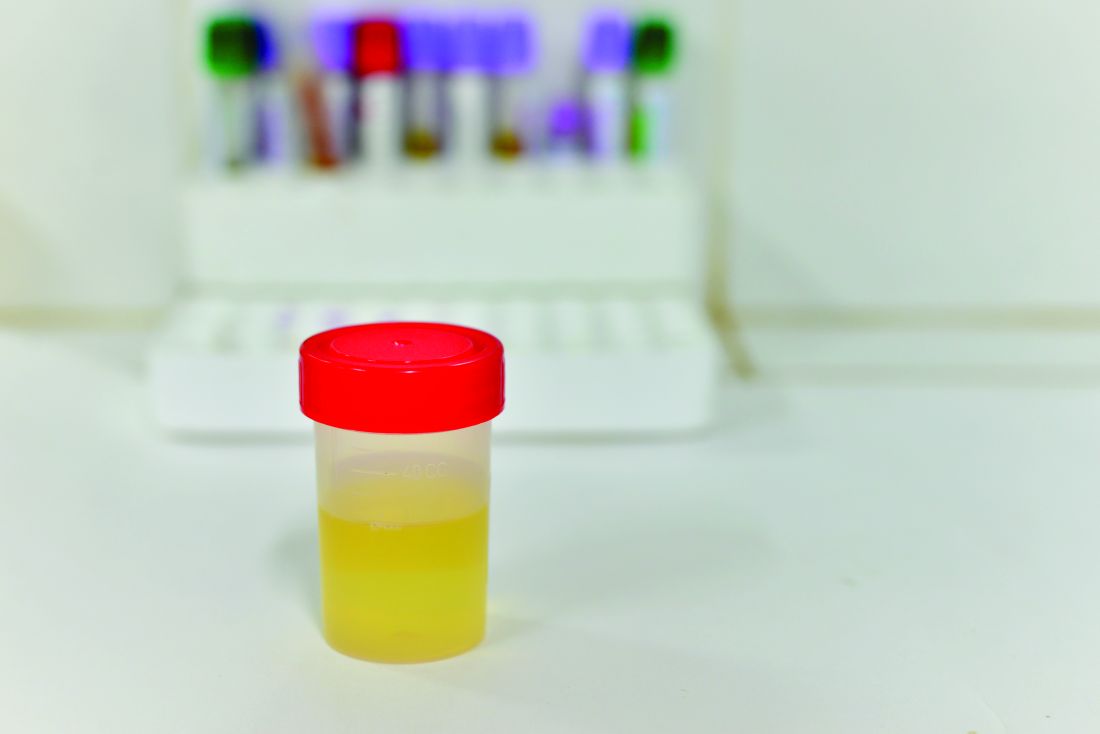
Of the 260 children whose catheterized urine was analyzed, 14% had UTIs. The median uNGAL concentration was 215.1 ng/mL in the UTI group, compared with 4.4 ng/mL in the culture-negative group. Urinalysis and Gram stain also were performed.
“uNGAL had higher sensitivity than [urinalysis], with similar specificity,” the investigators said. “Gram stain had a somewhat lower sensitivity than uNGAL, but with high specificity.”
The researchers identified a cutoff point for uNGAL levels to be 39.1 ng/mL. A previous case-control study of 108 infants with UTI had a cutoff for uNGAL levels of 38 ng/mL, with sensitivity of 93% and specificity of 95%. Most urine samples in that study were obtained by clean catch rather than by catheterization, as in the current study.
“Further studies will need to both confirm our findings and determine the benefit and cost effectiveness of uNGAL testing, compared with [urinalysis],” Dr. Lubell and her associates said.
Read more in Pediatrics (2017 Nov 16. doi: 10.1542/peds.2017-1090).
Novel test for the urinary neutrophil gelatinase–associated lipocalin (uNGAL) biomarker have good sensitivity and specificity to distinguish whether infants and children younger than 2 years old have a urinary tract infection (UTI), said Tamar R. Lubell, MD, and her associates at Columbia University, New York.
“NGAL is a protein expressed in neutrophils and several other human tissues, including alpha-intercalated cells in the collecting duct of the kidney,” the researchers explained. “The urine and serum contain very low levels of NGAL protein at steady state, with expression of NGAL rising rapidly in response to cell damage caused by ischemia-reperfusion injury, presence of cytotoxins, and sepsis.”
Of the 260 children whose catheterized urine was analyzed, 14% had UTIs. The median uNGAL concentration was 215.1 ng/mL in the UTI group, compared with 4.4 ng/mL in the culture-negative group. Urinalysis and Gram stain also were performed.
“uNGAL had higher sensitivity than [urinalysis], with similar specificity,” the investigators said. “Gram stain had a somewhat lower sensitivity than uNGAL, but with high specificity.”
The researchers identified a cutoff point for uNGAL levels to be 39.1 ng/mL. A previous case-control study of 108 infants with UTI had a cutoff for uNGAL levels of 38 ng/mL, with sensitivity of 93% and specificity of 95%. Most urine samples in that study were obtained by clean catch rather than by catheterization, as in the current study.
“Further studies will need to both confirm our findings and determine the benefit and cost effectiveness of uNGAL testing, compared with [urinalysis],” Dr. Lubell and her associates said.
Read more in Pediatrics (2017 Nov 16. doi: 10.1542/peds.2017-1090).
Novel test for the urinary neutrophil gelatinase–associated lipocalin (uNGAL) biomarker have good sensitivity and specificity to distinguish whether infants and children younger than 2 years old have a urinary tract infection (UTI), said Tamar R. Lubell, MD, and her associates at Columbia University, New York.
“NGAL is a protein expressed in neutrophils and several other human tissues, including alpha-intercalated cells in the collecting duct of the kidney,” the researchers explained. “The urine and serum contain very low levels of NGAL protein at steady state, with expression of NGAL rising rapidly in response to cell damage caused by ischemia-reperfusion injury, presence of cytotoxins, and sepsis.”
Of the 260 children whose catheterized urine was analyzed, 14% had UTIs. The median uNGAL concentration was 215.1 ng/mL in the UTI group, compared with 4.4 ng/mL in the culture-negative group. Urinalysis and Gram stain also were performed.
“uNGAL had higher sensitivity than [urinalysis], with similar specificity,” the investigators said. “Gram stain had a somewhat lower sensitivity than uNGAL, but with high specificity.”
The researchers identified a cutoff point for uNGAL levels to be 39.1 ng/mL. A previous case-control study of 108 infants with UTI had a cutoff for uNGAL levels of 38 ng/mL, with sensitivity of 93% and specificity of 95%. Most urine samples in that study were obtained by clean catch rather than by catheterization, as in the current study.
“Further studies will need to both confirm our findings and determine the benefit and cost effectiveness of uNGAL testing, compared with [urinalysis],” Dr. Lubell and her associates said.
Read more in Pediatrics (2017 Nov 16. doi: 10.1542/peds.2017-1090).
FROM PEDIATRICS
After warning, codeine use after tonsillectomy drops, doesn’t stop
– and there has been inappropriate substitution of other opioids rather than nonopioid pain relievers such as ibuprofen, said Kao-Ping Chua, MD, PhD, of the University of Chicago, and his associates.
Codeine has been one of the most commonly prescribed analgesics for children after tonsillectomies and adenoidectomies because it was considered safe.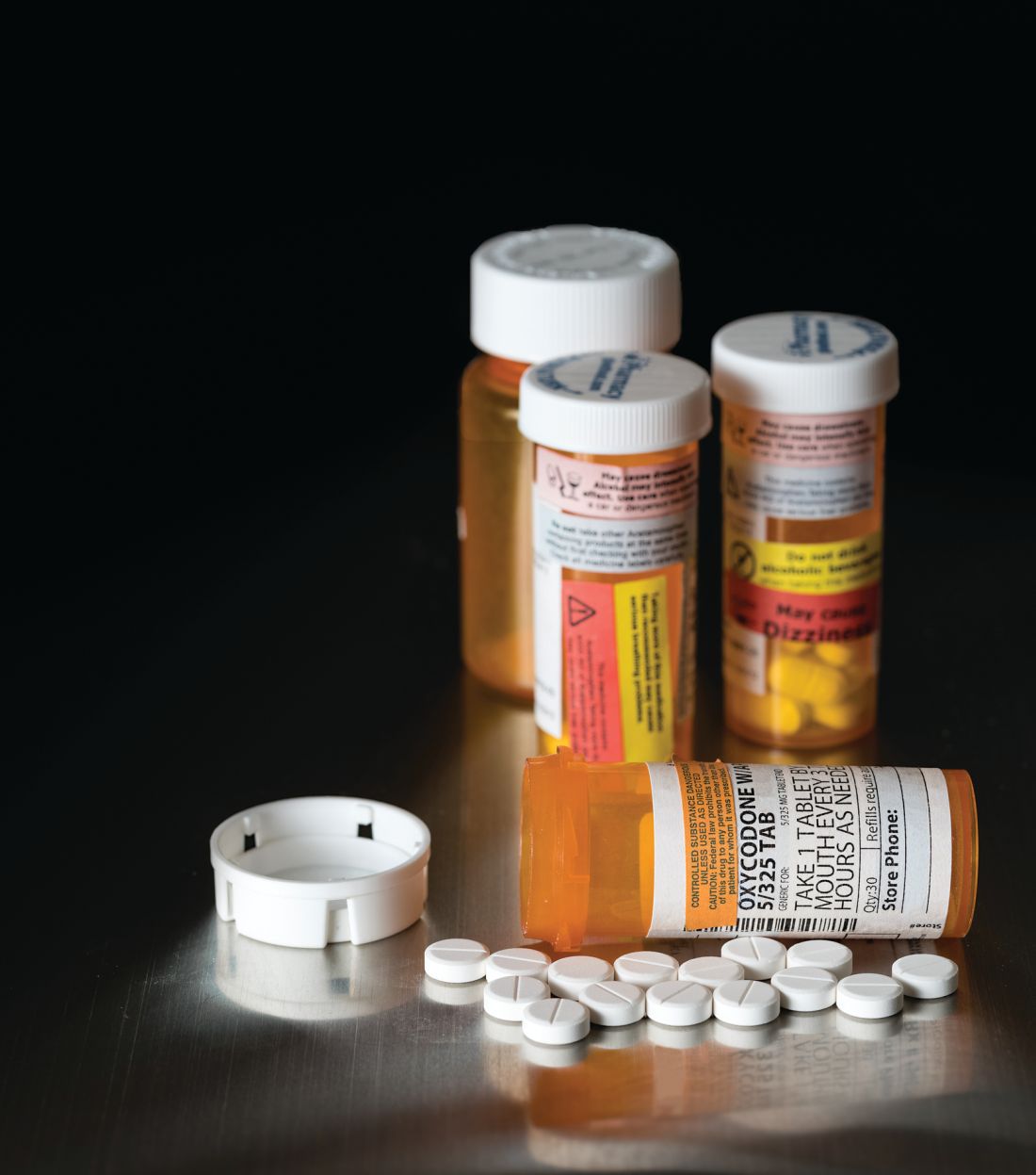
The FDA issued a boxed warning Feb. 20, 2013, recommending against codeine use in all children undergoing tonsillectomy and/or adenoidectomy. Also, children with obstructive sleep apnea are at risk of opioid-related respiratory depression.
A retrospective study of 362,992 children aged 0-18 years who underwent tonsillectomy and/or adenoidectomy was undertaken in 2010-2015, prior to and after the FDA issued the boxed warning against codeine use. In January 2010, codeine products were prescribed in 47% of cases, compared with 48% for hydrocodone, 4% for oxycodone, and less than 1% for other opioid products. In December 2015, codeine was prescribed in 9% of cases, compared with 73% for hydrocodone, 17% for oxycodone, and less than 1% for other opioid products.
“The unadjusted proportion of children receiving alternative opioids rose substantially during the study period, presumably because of factors other than the investigation itself,” said Dr. Chua and his associates. “This increase deserves further examination, given the abuse liability associated with higher-potency opioids, as well as the growing evidence of genetic variability in the metabolism of many alternative opioids, including oxycodone and tramadol.
“Future quality-improvement efforts should focus on eliminating this residual inappropriate codeine prescribing, and on encouraging the use of effective nonopioid medications such as ibuprofen,” the investigators said.
Read more in Pediatrics (2017 Nov 16. doi: 10.1542/ peds.2017-1765).
– and there has been inappropriate substitution of other opioids rather than nonopioid pain relievers such as ibuprofen, said Kao-Ping Chua, MD, PhD, of the University of Chicago, and his associates.
Codeine has been one of the most commonly prescribed analgesics for children after tonsillectomies and adenoidectomies because it was considered safe.
The FDA issued a boxed warning Feb. 20, 2013, recommending against codeine use in all children undergoing tonsillectomy and/or adenoidectomy. Also, children with obstructive sleep apnea are at risk of opioid-related respiratory depression.
A retrospective study of 362,992 children aged 0-18 years who underwent tonsillectomy and/or adenoidectomy was undertaken in 2010-2015, prior to and after the FDA issued the boxed warning against codeine use. In January 2010, codeine products were prescribed in 47% of cases, compared with 48% for hydrocodone, 4% for oxycodone, and less than 1% for other opioid products. In December 2015, codeine was prescribed in 9% of cases, compared with 73% for hydrocodone, 17% for oxycodone, and less than 1% for other opioid products.
“The unadjusted proportion of children receiving alternative opioids rose substantially during the study period, presumably because of factors other than the investigation itself,” said Dr. Chua and his associates. “This increase deserves further examination, given the abuse liability associated with higher-potency opioids, as well as the growing evidence of genetic variability in the metabolism of many alternative opioids, including oxycodone and tramadol.
“Future quality-improvement efforts should focus on eliminating this residual inappropriate codeine prescribing, and on encouraging the use of effective nonopioid medications such as ibuprofen,” the investigators said.
Read more in Pediatrics (2017 Nov 16. doi: 10.1542/ peds.2017-1765).
– and there has been inappropriate substitution of other opioids rather than nonopioid pain relievers such as ibuprofen, said Kao-Ping Chua, MD, PhD, of the University of Chicago, and his associates.
Codeine has been one of the most commonly prescribed analgesics for children after tonsillectomies and adenoidectomies because it was considered safe.
The FDA issued a boxed warning Feb. 20, 2013, recommending against codeine use in all children undergoing tonsillectomy and/or adenoidectomy. Also, children with obstructive sleep apnea are at risk of opioid-related respiratory depression.
A retrospective study of 362,992 children aged 0-18 years who underwent tonsillectomy and/or adenoidectomy was undertaken in 2010-2015, prior to and after the FDA issued the boxed warning against codeine use. In January 2010, codeine products were prescribed in 47% of cases, compared with 48% for hydrocodone, 4% for oxycodone, and less than 1% for other opioid products. In December 2015, codeine was prescribed in 9% of cases, compared with 73% for hydrocodone, 17% for oxycodone, and less than 1% for other opioid products.
“The unadjusted proportion of children receiving alternative opioids rose substantially during the study period, presumably because of factors other than the investigation itself,” said Dr. Chua and his associates. “This increase deserves further examination, given the abuse liability associated with higher-potency opioids, as well as the growing evidence of genetic variability in the metabolism of many alternative opioids, including oxycodone and tramadol.
“Future quality-improvement efforts should focus on eliminating this residual inappropriate codeine prescribing, and on encouraging the use of effective nonopioid medications such as ibuprofen,” the investigators said.
Read more in Pediatrics (2017 Nov 16. doi: 10.1542/ peds.2017-1765).
FROM PEDIATRICS
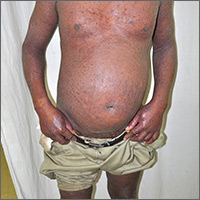
Extensive rash and joint pain
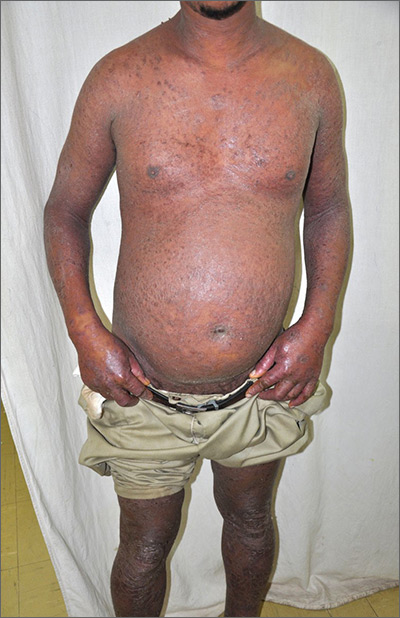
Based on the patient’s tender finger and wrist joints and exfoliation of skin, the FP diagnosed erythrodermic psoriasis, although the patient’s dark skin made the erythema less visible. The patient was not systemically ill; however, the FP recognized that he would need systemic therapy for the arthritis and severe skin manifestations and referred the patient to a dermatology specialist.
The dermatology specialist agreed to see the patient the next day. She ordered the following lab tests: complete blood count (CBC), chemistry panel, QuantiFERON TB gold, hepatitis C antibody, and hepatitis B surface antigen, surface antibody, and core antibody. She also ordered bilateral hand films, which showed early juxta-articular erosions (consistent with psoriatic arthritis). Because of the strong skin, nail, and joint evidence, the dermatology specialist determined a biopsy was not necessary.
The patient was instructed to apply 0.1% triamcinolone ointment to his entire body using the wet pajama technique overnight. At follow-up, he said his skin felt better, but his joints did not. The dermatology specialist reviewed the lab results and explained to the patient that the 2 best options were oral methotrexate weekly or an expensive injectable biologic agent. Because most health insurance companies now require that the patient fail a less expensive agent such as methotrexate before trying the biologic, the decision was made to start with methotrexate. Within a month, the patient's skin was remarkably better and his joint pain and stiffness had improved significantly.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Henderson D. Erythroderma. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 915-921.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Based on the patient’s tender finger and wrist joints and exfoliation of skin, the FP diagnosed erythrodermic psoriasis, although the patient’s dark skin made the erythema less visible. The patient was not systemically ill; however, the FP recognized that he would need systemic therapy for the arthritis and severe skin manifestations and referred the patient to a dermatology specialist.
The dermatology specialist agreed to see the patient the next day. She ordered the following lab tests: complete blood count (CBC), chemistry panel, QuantiFERON TB gold, hepatitis C antibody, and hepatitis B surface antigen, surface antibody, and core antibody. She also ordered bilateral hand films, which showed early juxta-articular erosions (consistent with psoriatic arthritis). Because of the strong skin, nail, and joint evidence, the dermatology specialist determined a biopsy was not necessary.
The patient was instructed to apply 0.1% triamcinolone ointment to his entire body using the wet pajama technique overnight. At follow-up, he said his skin felt better, but his joints did not. The dermatology specialist reviewed the lab results and explained to the patient that the 2 best options were oral methotrexate weekly or an expensive injectable biologic agent. Because most health insurance companies now require that the patient fail a less expensive agent such as methotrexate before trying the biologic, the decision was made to start with methotrexate. Within a month, the patient's skin was remarkably better and his joint pain and stiffness had improved significantly.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Henderson D. Erythroderma. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 915-921.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Based on the patient’s tender finger and wrist joints and exfoliation of skin, the FP diagnosed erythrodermic psoriasis, although the patient’s dark skin made the erythema less visible. The patient was not systemically ill; however, the FP recognized that he would need systemic therapy for the arthritis and severe skin manifestations and referred the patient to a dermatology specialist.
The dermatology specialist agreed to see the patient the next day. She ordered the following lab tests: complete blood count (CBC), chemistry panel, QuantiFERON TB gold, hepatitis C antibody, and hepatitis B surface antigen, surface antibody, and core antibody. She also ordered bilateral hand films, which showed early juxta-articular erosions (consistent with psoriatic arthritis). Because of the strong skin, nail, and joint evidence, the dermatology specialist determined a biopsy was not necessary.
The patient was instructed to apply 0.1% triamcinolone ointment to his entire body using the wet pajama technique overnight. At follow-up, he said his skin felt better, but his joints did not. The dermatology specialist reviewed the lab results and explained to the patient that the 2 best options were oral methotrexate weekly or an expensive injectable biologic agent. Because most health insurance companies now require that the patient fail a less expensive agent such as methotrexate before trying the biologic, the decision was made to start with methotrexate. Within a month, the patient's skin was remarkably better and his joint pain and stiffness had improved significantly.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Henderson D. Erythroderma. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 915-921.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
VIDEO: Bariatric experts discuss recent experience with gastric balloon devices
Two experts on bariatric surgery spoke at the Minimally Invasive Surgery Symposium, held in Las Vegas. Jaime Ponce, MD, FACS, and John Morton, MD, FACS, discussed several different types of gastric balloon devices and the factors guiding their use for patients. Dr. Morton suggested that the balloon devices could serve as an intermediate treatment between medications and surgery.

Two experts on bariatric surgery spoke at the Minimally Invasive Surgery Symposium, held in Las Vegas. Jaime Ponce, MD, FACS, and John Morton, MD, FACS, discussed several different types of gastric balloon devices and the factors guiding their use for patients. Dr. Morton suggested that the balloon devices could serve as an intermediate treatment between medications and surgery.
Two experts on bariatric surgery spoke at the Minimally Invasive Surgery Symposium, held in Las Vegas. Jaime Ponce, MD, FACS, and John Morton, MD, FACS, discussed several different types of gastric balloon devices and the factors guiding their use for patients. Dr. Morton suggested that the balloon devices could serve as an intermediate treatment between medications and surgery.
Carefully monitor cannabis-using patients in opioid-agonist therapy
Canada has passed legislation that will lead to legalization of cannabis for recreational use. Researchers disagree about the public health effects that legalization will have. Alexandra M. Franklyn and her coinvestigators at the Northern Ontario School of Medicine, Sudbury, designed their study to look at one population that might be at risk of cannabis-related harms.
Of 644 patients, 328 (50.9%) were baseline cannabis users. Baseline users were 38.9% more likely to drop out of OAT. Heavy cannabis users (n = 256; 39.8%) were 48.1% more likely to drop out of OAT than those who weren’t heavy users. Heavy users are “a group that should be carefully monitored throughout treatment,” regardless of whether the cannabis use can be considered another substance use disorder or if it is intended to self-medicate another condition that may make dropout more likely, Ms. Franklyn and her colleagues concluded.
“While studies have shown a potential for cannabis legalization to be a positive change for the population as a whole, there may be unique implications for those patients receiving OAT. ... These patients should receive education surrounding the potential harms of cannabis use, including worsened OAT outcomes,” the researchers wrote (PLoS One. 2017 Nov 8;12[11]:e0187633).
Neither Ms. Franklyn nor her colleagues had conflicts of interest to disclose.
Canada has passed legislation that will lead to legalization of cannabis for recreational use. Researchers disagree about the public health effects that legalization will have. Alexandra M. Franklyn and her coinvestigators at the Northern Ontario School of Medicine, Sudbury, designed their study to look at one population that might be at risk of cannabis-related harms.
Of 644 patients, 328 (50.9%) were baseline cannabis users. Baseline users were 38.9% more likely to drop out of OAT. Heavy cannabis users (n = 256; 39.8%) were 48.1% more likely to drop out of OAT than those who weren’t heavy users. Heavy users are “a group that should be carefully monitored throughout treatment,” regardless of whether the cannabis use can be considered another substance use disorder or if it is intended to self-medicate another condition that may make dropout more likely, Ms. Franklyn and her colleagues concluded.
“While studies have shown a potential for cannabis legalization to be a positive change for the population as a whole, there may be unique implications for those patients receiving OAT. ... These patients should receive education surrounding the potential harms of cannabis use, including worsened OAT outcomes,” the researchers wrote (PLoS One. 2017 Nov 8;12[11]:e0187633).
Neither Ms. Franklyn nor her colleagues had conflicts of interest to disclose.
Canada has passed legislation that will lead to legalization of cannabis for recreational use. Researchers disagree about the public health effects that legalization will have. Alexandra M. Franklyn and her coinvestigators at the Northern Ontario School of Medicine, Sudbury, designed their study to look at one population that might be at risk of cannabis-related harms.
Of 644 patients, 328 (50.9%) were baseline cannabis users. Baseline users were 38.9% more likely to drop out of OAT. Heavy cannabis users (n = 256; 39.8%) were 48.1% more likely to drop out of OAT than those who weren’t heavy users. Heavy users are “a group that should be carefully monitored throughout treatment,” regardless of whether the cannabis use can be considered another substance use disorder or if it is intended to self-medicate another condition that may make dropout more likely, Ms. Franklyn and her colleagues concluded.
“While studies have shown a potential for cannabis legalization to be a positive change for the population as a whole, there may be unique implications for those patients receiving OAT. ... These patients should receive education surrounding the potential harms of cannabis use, including worsened OAT outcomes,” the researchers wrote (PLoS One. 2017 Nov 8;12[11]:e0187633).
Neither Ms. Franklyn nor her colleagues had conflicts of interest to disclose.
FROM PLOS ONE
Infliximab useful in a case of concurrent RA/PBC
The anti–tumor necrosis factor alpha agent infliximab is a beneficial option in the treatment of concurrent rheumatoid arthritis (RA) and primary biliary cholangitis (PBC), according to A.K. Ben, MD, and associates.
In a case report, a 56-year-old woman presented with 5 years of evolving symmetrical polyarthritis involving her large and small joints, and was diagnosed with RA. Methotrexate was initially used as treatment, but after 6 months of continued high disease activity, infliximab was added to treatment. The patient showed good clinical response to the combination treatment.
The combination treatment was stopped after the patient showed abnormal liver function. Antimitochondrial antibody testing was positive, and after a liver biopsy to confirm, the patient was diagnosed with PBC. Ursodeoxycholic acid was prescribed and liver function was normalized.
The patient was restarted on methotrexate and 3 mg/kg infliximab when the RA flared after 6 months and was persistent until the dosage of infliximab was increased to 5 mg/kg. This was effective in the patient, who has experienced disease decline for 5 years.
“Additional studies may be considered to better explore the therapeutic role (dosage and molecule disparities) of TNF [tumor necrosis factor] blockers on clinical and morphological course of PBC associated with RA,” the investigators concluded.
Find the full case report in Internal Medicine: Open Access (doi: 10.4172/2165-8048.1000250).
The anti–tumor necrosis factor alpha agent infliximab is a beneficial option in the treatment of concurrent rheumatoid arthritis (RA) and primary biliary cholangitis (PBC), according to A.K. Ben, MD, and associates.
In a case report, a 56-year-old woman presented with 5 years of evolving symmetrical polyarthritis involving her large and small joints, and was diagnosed with RA. Methotrexate was initially used as treatment, but after 6 months of continued high disease activity, infliximab was added to treatment. The patient showed good clinical response to the combination treatment.
The combination treatment was stopped after the patient showed abnormal liver function. Antimitochondrial antibody testing was positive, and after a liver biopsy to confirm, the patient was diagnosed with PBC. Ursodeoxycholic acid was prescribed and liver function was normalized.
The patient was restarted on methotrexate and 3 mg/kg infliximab when the RA flared after 6 months and was persistent until the dosage of infliximab was increased to 5 mg/kg. This was effective in the patient, who has experienced disease decline for 5 years.
“Additional studies may be considered to better explore the therapeutic role (dosage and molecule disparities) of TNF [tumor necrosis factor] blockers on clinical and morphological course of PBC associated with RA,” the investigators concluded.
Find the full case report in Internal Medicine: Open Access (doi: 10.4172/2165-8048.1000250).
The anti–tumor necrosis factor alpha agent infliximab is a beneficial option in the treatment of concurrent rheumatoid arthritis (RA) and primary biliary cholangitis (PBC), according to A.K. Ben, MD, and associates.
In a case report, a 56-year-old woman presented with 5 years of evolving symmetrical polyarthritis involving her large and small joints, and was diagnosed with RA. Methotrexate was initially used as treatment, but after 6 months of continued high disease activity, infliximab was added to treatment. The patient showed good clinical response to the combination treatment.
The combination treatment was stopped after the patient showed abnormal liver function. Antimitochondrial antibody testing was positive, and after a liver biopsy to confirm, the patient was diagnosed with PBC. Ursodeoxycholic acid was prescribed and liver function was normalized.
The patient was restarted on methotrexate and 3 mg/kg infliximab when the RA flared after 6 months and was persistent until the dosage of infliximab was increased to 5 mg/kg. This was effective in the patient, who has experienced disease decline for 5 years.
“Additional studies may be considered to better explore the therapeutic role (dosage and molecule disparities) of TNF [tumor necrosis factor] blockers on clinical and morphological course of PBC associated with RA,” the investigators concluded.
Find the full case report in Internal Medicine: Open Access (doi: 10.4172/2165-8048.1000250).
FROM INTERNAL MEDICINE: OPEN ACCESS
Higher BMI linked to lower risk of hysterectomy reoperation
NATIONAL HARBOR, MD. – Women with a greater body mass index (BMI) were less likely to need reoperation after hysterectomy, according to findings presented at the AAGL Global Congress.
“What’s unusual is women who are considered overweight or obese are generally thought to be at higher risk of any complication, including reoperation,” Janelle Moulder, MD, of the department of ob.gyn. at the University of Tennessee, Knoxville, said in an interview prior to the meeting. “We don’t have enough data to say what exactly might be protective. And to see that women who are at normal or below normal BMI were at increased risk makes you pause as to what could potentially put them at risk.”
Dr. Moulder and her colleagues analyzed data on 28,487 women who underwent an abdominal, vaginal, or laparoscopic hysterectomy from 2014 to 2015. The data came from the American College of Surgeons (ACS) National Surgical Quality Improvement Program (NSQIP) database.
Patients were excluded if they had cancer, their surgery was not performed by a gynecologist, or their BMI data was missing.
A majority of patients (13,000) had a BMI of 30 kg/m2 or greater.
Compared with patients with a normal BMI of 24 kg/m2, patients with a BMI of 39 kg/m2 had the lowest odds of reoperation (adjusted odds ratio, 0.73; P = .02). Patients with BMIs of 29 kg/m2 and 34 kg/m2 were also at lower odds of reoperation, with adjusted odds ratios of 0.83 (P = .003) and 0.75 (P = .005), respectively.
Patients with a low normal BMI of 18.5 kg/m2 were at a higher risk of reoperation (aOR = 1.33; P = .001).
Researchers were unable to comment on women with a BMI of 45 kg/m2 or greater, due to the limited number of women in this group.
Researchers did not have access to the reason for reoperation, which may have limited the scope of the study.
“The next thing to be evaluated is what is the protective effect of the increasing BMI on reoperation and also look at variables that may put low normal BMI women at risk for reoperation,” Dr. Moulder said.
The researchers reported having no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
NATIONAL HARBOR, MD. – Women with a greater body mass index (BMI) were less likely to need reoperation after hysterectomy, according to findings presented at the AAGL Global Congress.
“What’s unusual is women who are considered overweight or obese are generally thought to be at higher risk of any complication, including reoperation,” Janelle Moulder, MD, of the department of ob.gyn. at the University of Tennessee, Knoxville, said in an interview prior to the meeting. “We don’t have enough data to say what exactly might be protective. And to see that women who are at normal or below normal BMI were at increased risk makes you pause as to what could potentially put them at risk.”
Dr. Moulder and her colleagues analyzed data on 28,487 women who underwent an abdominal, vaginal, or laparoscopic hysterectomy from 2014 to 2015. The data came from the American College of Surgeons (ACS) National Surgical Quality Improvement Program (NSQIP) database.
Patients were excluded if they had cancer, their surgery was not performed by a gynecologist, or their BMI data was missing.
A majority of patients (13,000) had a BMI of 30 kg/m2 or greater.
Compared with patients with a normal BMI of 24 kg/m2, patients with a BMI of 39 kg/m2 had the lowest odds of reoperation (adjusted odds ratio, 0.73; P = .02). Patients with BMIs of 29 kg/m2 and 34 kg/m2 were also at lower odds of reoperation, with adjusted odds ratios of 0.83 (P = .003) and 0.75 (P = .005), respectively.
Patients with a low normal BMI of 18.5 kg/m2 were at a higher risk of reoperation (aOR = 1.33; P = .001).
Researchers were unable to comment on women with a BMI of 45 kg/m2 or greater, due to the limited number of women in this group.
Researchers did not have access to the reason for reoperation, which may have limited the scope of the study.
“The next thing to be evaluated is what is the protective effect of the increasing BMI on reoperation and also look at variables that may put low normal BMI women at risk for reoperation,” Dr. Moulder said.
The researchers reported having no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
NATIONAL HARBOR, MD. – Women with a greater body mass index (BMI) were less likely to need reoperation after hysterectomy, according to findings presented at the AAGL Global Congress.
“What’s unusual is women who are considered overweight or obese are generally thought to be at higher risk of any complication, including reoperation,” Janelle Moulder, MD, of the department of ob.gyn. at the University of Tennessee, Knoxville, said in an interview prior to the meeting. “We don’t have enough data to say what exactly might be protective. And to see that women who are at normal or below normal BMI were at increased risk makes you pause as to what could potentially put them at risk.”
Dr. Moulder and her colleagues analyzed data on 28,487 women who underwent an abdominal, vaginal, or laparoscopic hysterectomy from 2014 to 2015. The data came from the American College of Surgeons (ACS) National Surgical Quality Improvement Program (NSQIP) database.
Patients were excluded if they had cancer, their surgery was not performed by a gynecologist, or their BMI data was missing.
A majority of patients (13,000) had a BMI of 30 kg/m2 or greater.
Compared with patients with a normal BMI of 24 kg/m2, patients with a BMI of 39 kg/m2 had the lowest odds of reoperation (adjusted odds ratio, 0.73; P = .02). Patients with BMIs of 29 kg/m2 and 34 kg/m2 were also at lower odds of reoperation, with adjusted odds ratios of 0.83 (P = .003) and 0.75 (P = .005), respectively.
Patients with a low normal BMI of 18.5 kg/m2 were at a higher risk of reoperation (aOR = 1.33; P = .001).
Researchers were unable to comment on women with a BMI of 45 kg/m2 or greater, due to the limited number of women in this group.
Researchers did not have access to the reason for reoperation, which may have limited the scope of the study.
“The next thing to be evaluated is what is the protective effect of the increasing BMI on reoperation and also look at variables that may put low normal BMI women at risk for reoperation,” Dr. Moulder said.
The researchers reported having no relevant financial disclosures.
[email protected]
On Twitter @eaztweets
AT AAGL 2017
Key clinical point:
Major finding: Patients with a BMI of 39 kg/m2 less likely to need a reoperation after hysterectomy (aOR, .73; P = .02).
Data source: Retrospective study of 28,487 women who underwent a hysterectomy from 2014 to 2015 from the American College of Surgeons National Surgical Quality Improvement Program database.
Disclosures: The researchers reported having no relevant financial disclosures.
FDA issues alert on illegal silicone injections
The Food and Drug Administration has issued a warning regarding the use of injectable silicone or other products illegally marketed as dermal fillers for body contouring.
“We have significant concerns with unsafe injectable silicone that’s being marketed for body contouring by unlicensed providers,” FDA Commissioner Scott Gottlieb, MD, said in a statement on Nov. 14. “We’ve seen serious adverse events result from products, which are sometimes industrial-grade silicone, being used for these unapproved medical purposes,” he said.
Simply injecting silicone into various parts of the body for contouring purposes is not approved by the FDA. Side effects of such a procedure can occur immediately, or may appear after days, weeks, months, or years, according to the statement. Side effects include pain, scarring, disfigurement, life-threatening embolism, stroke, or infection, the FDA emphasized.
The FDA continues to take action against unlicensed practitioners found guilty of treating patients with unapproved silicone for body contouring. “In addition to prosecuting the criminals who take advantage of consumers, the FDA is taking action to educate consumers in order to prevent the serious injuries resulting from these injections,” Melinda Plaisier, associate commissioner for regulatory affairs at the FDA, said in the statement. “We hope to raise public awareness about the short- and long-term risks of injecting silicone directly into the body, and encourage consumers to choose FDA-approved products and licensed providers when considering any type of cosmetic enhancement,” she said.
The FDA will continue to monitor adverse event reports related to silicone, and encourages clinicians or consumers with information about the use of injectable silicone by unlicensed providers to use the “Report Suspected Criminal Activity” form on the FDA website to report those cases.
The Food and Drug Administration has issued a warning regarding the use of injectable silicone or other products illegally marketed as dermal fillers for body contouring.
“We have significant concerns with unsafe injectable silicone that’s being marketed for body contouring by unlicensed providers,” FDA Commissioner Scott Gottlieb, MD, said in a statement on Nov. 14. “We’ve seen serious adverse events result from products, which are sometimes industrial-grade silicone, being used for these unapproved medical purposes,” he said.
Simply injecting silicone into various parts of the body for contouring purposes is not approved by the FDA. Side effects of such a procedure can occur immediately, or may appear after days, weeks, months, or years, according to the statement. Side effects include pain, scarring, disfigurement, life-threatening embolism, stroke, or infection, the FDA emphasized.
The FDA continues to take action against unlicensed practitioners found guilty of treating patients with unapproved silicone for body contouring. “In addition to prosecuting the criminals who take advantage of consumers, the FDA is taking action to educate consumers in order to prevent the serious injuries resulting from these injections,” Melinda Plaisier, associate commissioner for regulatory affairs at the FDA, said in the statement. “We hope to raise public awareness about the short- and long-term risks of injecting silicone directly into the body, and encourage consumers to choose FDA-approved products and licensed providers when considering any type of cosmetic enhancement,” she said.
The FDA will continue to monitor adverse event reports related to silicone, and encourages clinicians or consumers with information about the use of injectable silicone by unlicensed providers to use the “Report Suspected Criminal Activity” form on the FDA website to report those cases.
The Food and Drug Administration has issued a warning regarding the use of injectable silicone or other products illegally marketed as dermal fillers for body contouring.
“We have significant concerns with unsafe injectable silicone that’s being marketed for body contouring by unlicensed providers,” FDA Commissioner Scott Gottlieb, MD, said in a statement on Nov. 14. “We’ve seen serious adverse events result from products, which are sometimes industrial-grade silicone, being used for these unapproved medical purposes,” he said.
Simply injecting silicone into various parts of the body for contouring purposes is not approved by the FDA. Side effects of such a procedure can occur immediately, or may appear after days, weeks, months, or years, according to the statement. Side effects include pain, scarring, disfigurement, life-threatening embolism, stroke, or infection, the FDA emphasized.
The FDA continues to take action against unlicensed practitioners found guilty of treating patients with unapproved silicone for body contouring. “In addition to prosecuting the criminals who take advantage of consumers, the FDA is taking action to educate consumers in order to prevent the serious injuries resulting from these injections,” Melinda Plaisier, associate commissioner for regulatory affairs at the FDA, said in the statement. “We hope to raise public awareness about the short- and long-term risks of injecting silicone directly into the body, and encourage consumers to choose FDA-approved products and licensed providers when considering any type of cosmetic enhancement,” she said.
The FDA will continue to monitor adverse event reports related to silicone, and encourages clinicians or consumers with information about the use of injectable silicone by unlicensed providers to use the “Report Suspected Criminal Activity” form on the FDA website to report those cases.