User login
Effects of Platelet-Rich Plasma and Indomethacin on Biomechanics of Rotator Cuff Repair
Take-Home Points
- The optimal centrifugation protocol for production of rat PRP is 1300 rpm for 5 minutes.
- PRP administration in RCR improves tendon biomechanics in a rat model.
- Administration of NSAIDs following RCR has no significant effect on tendon biomechanical properties.
- NSAIDs may be co-administered with PRP without reducing efficacy of PRP.
- The role of PRP and NSAIDs in human RCR remains unclear.
Rotator cuff tears are a common source of shoulder pain and disability among older adults and athletes. Full-thickness tears alone occur in up to 30% of adults older than 60 years.1 Surgical repair is plagued by an unpredictable rate of recurrence (range, 11%-94%).1-10 As a result of improved suture materials, knotting patterns, and anchor designs, hardware issues are no longer the primary cause of rotator cuff repair (RCR) failures; now the principal mode of failure is biologic.2 Animal model studies have found that, after injury and subsequent healing, the tendon–bone interface remains abnormal.11 Rotator cuff research therefore has focused largely on biological enhancement of tendon-to-bone healing.
One means of biological augmentation is autologous platelet-rich plasma (PRP), which has supraphysiologic concentrations of platelets and their secreted growth factors. Although there is no consensus on the long-term efficacy of PRP, some studies suggest PRP accelerates healing over short and intermediate terms, which may contribute to a more rapid decrease in pain and more rapid return to normal activities.12-18 Similarly, systemic nonsteroidal anti-inflammatory drugs (NSAIDs) have long been used to treat musculoskeletal injuries, including rotator cuff pathology. However, NSAIDs inhibit cyclooxygenase activity, and clinical and experimental data have shown that cyclooxygenase 2 function is crucial in normal tendon-to-bone healing.19-21
Comprehensive studies have been conducted on the efficacy of both PRP and NSAIDs, but the interaction of concurrently used PRP and NSAIDs has not been determined. As many physicians use both modalities in the treatment of soft-tissue injuries, it is important to study the potential interactions when coadministered. Prior studies in small animal models suggest NSAIDs may impair tendon-to-bone healing in RCR, but there is no evidence regarding the effect of NSAIDs on the efficacy of PRP treatment.21
We conducted a study to determine the interaction of PRP and NSAIDs when used as adjuncts to RCR in a rat model. We hypothesized that PRP would increase the strength of RCR and that NSAIDs would interfere with the effects of PRP. A preliminary study objective was to determine an appropriate centrifugation protocol for producing PRP from rat blood, for use in this study and in future rat-based studies of PRP.
Materials and Methods
Part A: Pretesting Determination of PRP Centrifugation Protocol
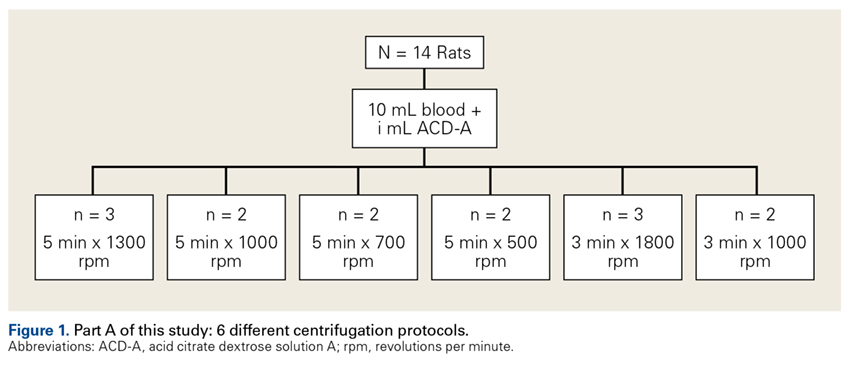
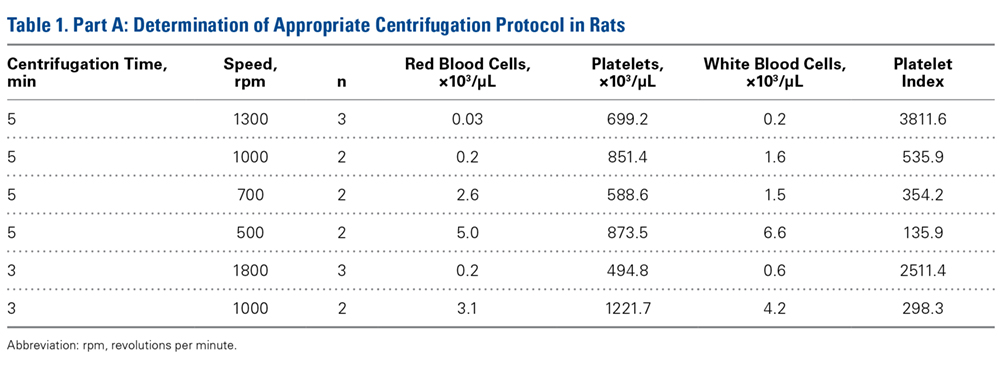
Fourteen adult male Fischer rats were used in part A of this study, which was conducted to determine an appropriate PRP centrifugation protocol. Traditional PRP centrifugation protocols are established for human blood, but rat red blood cells (RBCs) and human RBCs differ in size.22 In our preliminary study, we wanted to determine the adjusted centrifuge speed and duration for producing clinically optimal PRP from rats. Clinically optimal PRP has reduced levels of RBCs, which decrease platelet affinity. Although the role of leukocytes in PRP preparations is debated, reducing the number of white blood cells (WBCs) decreases the number of matrix metalloproteinases and reactive oxygen species that may lead to inflammation. We used the platelet index (ratio of platelets to WBCs) and the RBC count to quantify the quality of our PRP sample.
Each rat in part A was anesthetized while supine. We used the Autologous Conditioned Plasma (ACP) system (Arthrex), which requires only 1 centrifugation cycle to create PRP. About 9 mL or 10 mL of blood was obtained by cardiac aspiration using an ACP Double Syringe (Arthrex). After blood retrieval, a thoracotomy was performed to confirm each rat’s death.
Part B: Determining the Effects of PRP and NSAIDs on RCR in a Rat Model
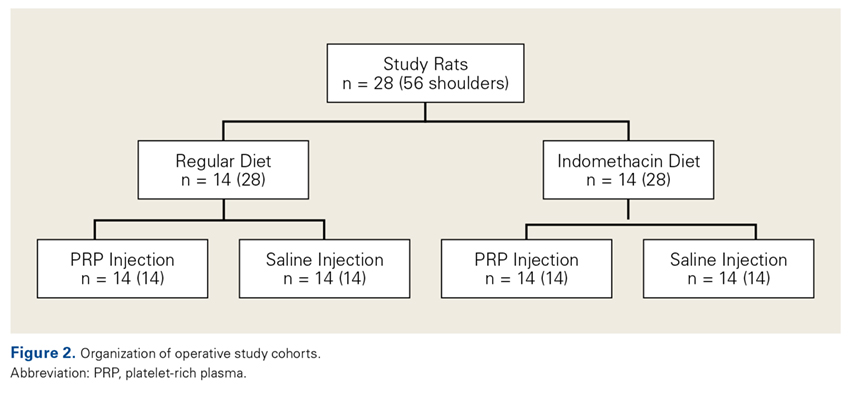
Operative Cohort. Of the 34 Fischer rats used in part B of this study, 6 were used as blood donors for PRP production, and the other 28 underwent bilateral rotator cuff surgeries. We used donor rats to maximize the amount of PRP retrieval, allocating about 1 donor rat per 5 operative rats. Fischer rats are an inbred strain, so the PRP from a donor Fischer rat simulates autologous blood in other Fischer rats. Use of allogenic blood is consistent with prior rat PRP studies.23,24
Operative Technique. Each bilateral surgery was performed by a single board-certified shoulder surgeon, and the anesthetic and surgical protocols were followed as approved by the home institution’s Institutional Animal Care and Use Committee. Before surgery, blood was harvested for PRP production from donor rats, as described earlier, and centrifuged for 5 minutes × 1300 rpm. After anesthetic induction and skin incision, the deltoid muscle was cut to expose the acromion and underlying rotator cuff. The distal supraspinatus tendon was sharply detached from the greater tuberosity. A bone-tunnel RCR was performed by drilling a transverse tunnel across the greater tuberosity and affixing the tendon to its footprint with a 5-0 polypropylene suture (Prolene; Ethicon). Each rat was then randomly assigned to receive 50 µL of donor PRP injected in 1 operative shoulder and saline in the contralateral shoulder. Injections were made in the supraspinatus tendon at its attachment to the humerus. Deltoid and skin were closed with 4-0 polyglactin (Vicryl) suture (Ethicon) and staples, respectively.
Tendon Preparation. Immediately post mortem, each shoulder was grossly dissected to isolate the supraspinatus muscle attached to the humerus. Shoulders were then frozen in 0.15-M saline solution until specified biomechanical testing dates.
On day of dimensional/biomechanical testing, each specimen was thawed at room temperature and finely dissected under a microscope (Stemi 200-C; Car Zeiss). After dissection, the humeral shaft was embedded in polymethylmethacrylate within a test tube. The free end of the supraspinatus tendon was glued within a “tab” of waterproofed emery cloth, leaving about 2 mm of tendon between the tab and the greater tuberosity.
Biomechanical Analysis. A 5848 MicroTester (Instron) was used for biomechanical testing. Each tabbed tendon, held by a pneumatic clamp attached to the MicroTester, was tested in a preconditioning phase and then a ramp-to-failure phase. A constant drip of 0.15-M saline was run through the apparatus to simulate physiologic hydration of tissue. After the embedded specimen was secure within the loading apparatus, an initial tensile preload of 0.2 N was applied. After preloading, the tendon was run through a preconditioning phase to account for viscoelastic relaxation. Immediately after preconditioning, each tendon was subjected to failure testing at a ramp rate of 0.1 mm/s. Force data were collected as a function of displacement, allowing for the calculation of 4 biomechanical parameters: failure force, tendon stiffness and normalized stiffness, energy to failure, and total energy. Tendon stiffness is the slope of a curve-fit line of the initial peak; failure force is the force of the highest peak; energy to failure is the area under the curve (AUC) to the highest peak; and total energy is the AUC from the start of failure ramping to the point at which the tendon is torn off completely. Two-way ANOVA was used to assess the differences between treatment groups and diet groups for all parameters. Statistical significance was set at P < .05.
A power analysis was performed to determine ability to detect differences between cohorts. For power of 80% and P = .05, a difference of 16% of the mean could be detected for failure force, 30% for energy to failure, 14% for total energy to failure, and 24% for stiffness. In addition, a difference of 4% of the mean could be detected for tendon length, 6% for width, and 10% for thickness.
Results
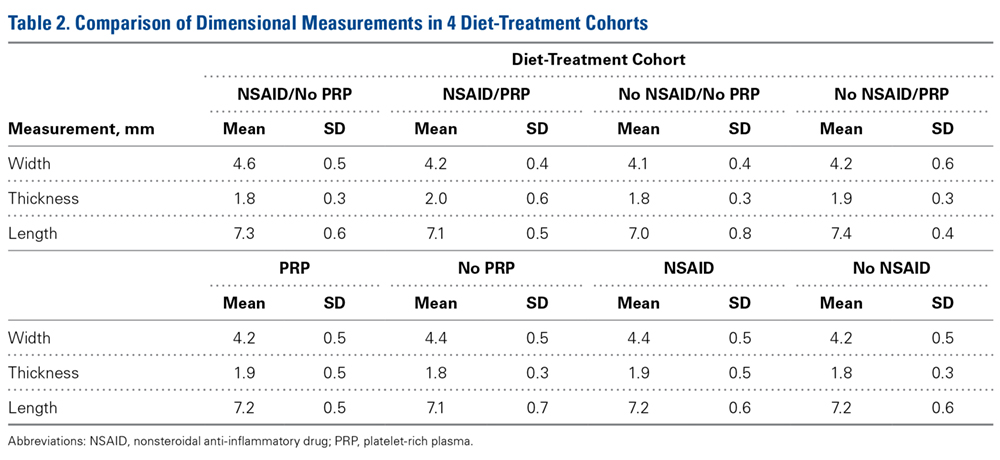
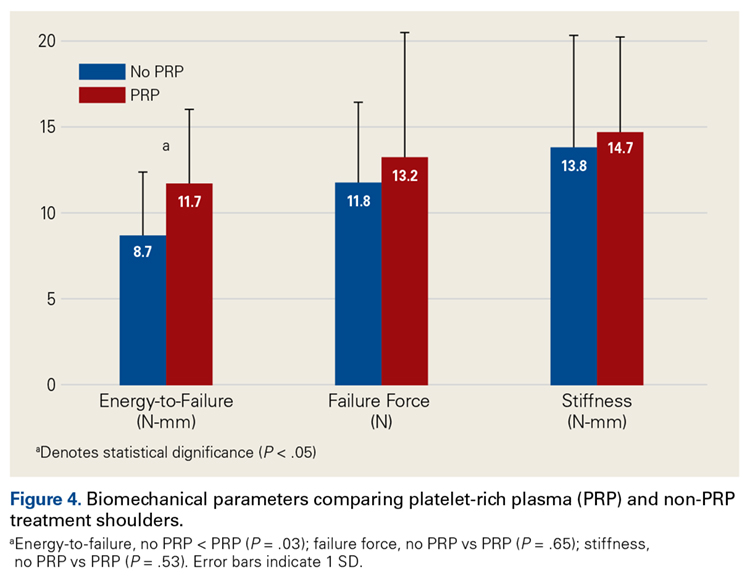
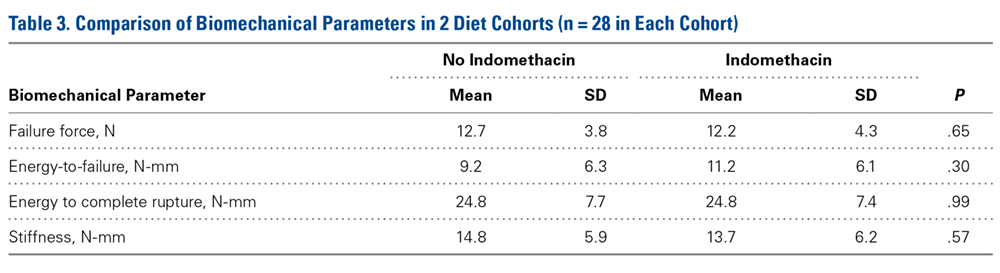
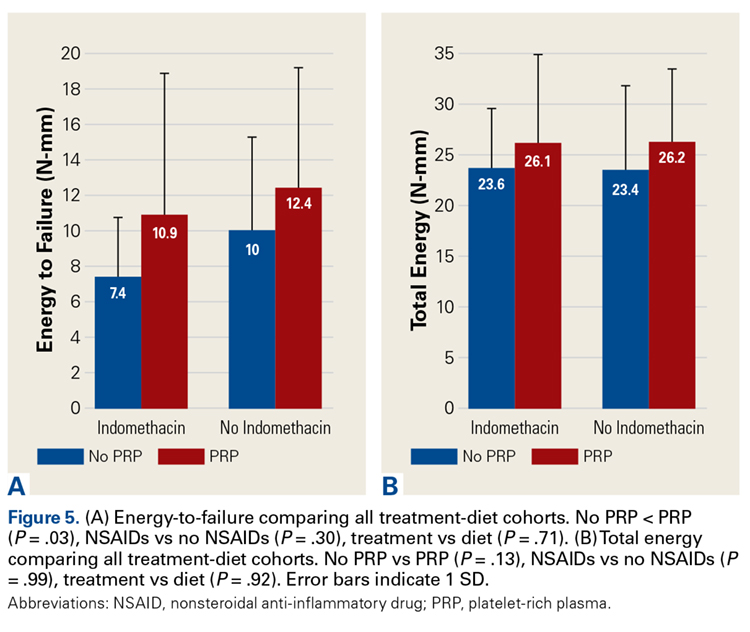
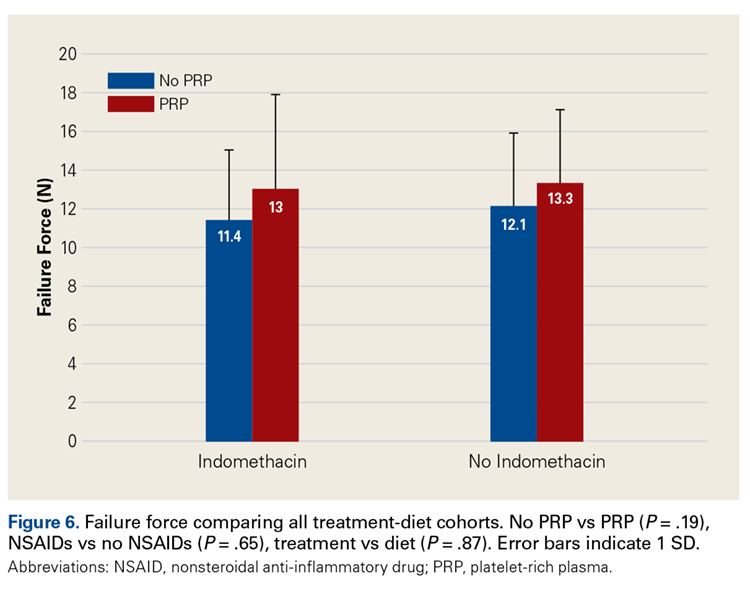
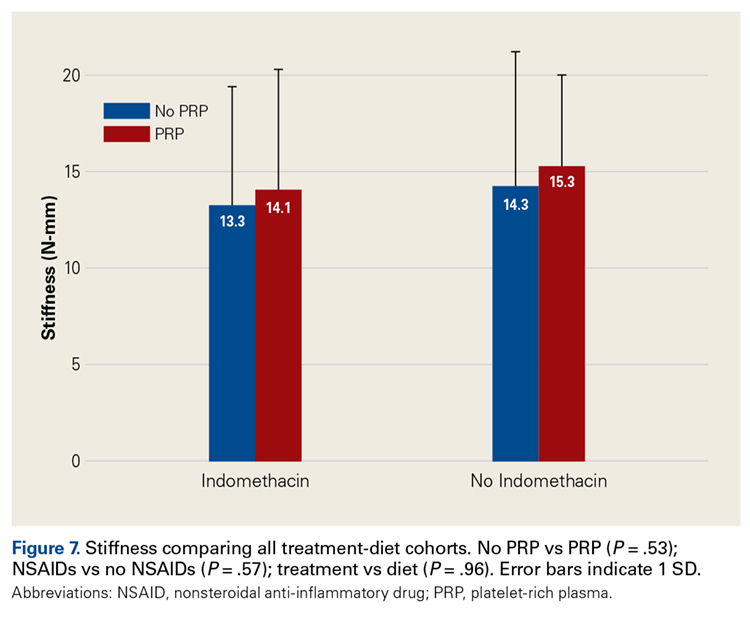
Across all collective treatment-diet groups and biomechanical parameters, there was only 1 statistically significant difference. Mean (SD) energy to failure was significantly higher (P = .03) in shoulders treated with PRP, 11.7 (7.3) N-mm, than in those treated without PRP, 8.7 (4.6) N-mm (Figure 4). There were no statistically significant differences between shoulders treated with indomethacin and those treated without indomethacin (Table 3), and no statistically significant relationships between treatment and drug for any other biomechanical parameter (Figures 5-7).
Discussion
Our preliminary objective in this study was to determine the optimal centrifugation protocol for producing rat-based PRP. Optimal PRP requires a dense concentration of platelets as well as reduced levels of RBCs and WBCs.25 We used the platelet index to quantify the quality of our PRP samples, and we obtained the highest platelet index for the protocol of 5 minutes × 1300 rpm. This finding may be useful in later rat studies involving PRP.
The primary objective of this study was to assess the effect of the interaction of PRP and NSAIDs on RCR. PRP has been found to augment RCR,12,26,27 but indomethacin may impair healing.21,25 We hypothesized that shoulders treated with PRP would have more biomechanical strength than control shoulders and that indomethacin would decrease biomechanical strength.
Our data showed increased energy to failure of the rotator cuff with PRP injections (P = .03). All other biomechanical parameters showed no significant differences with PRP treatment, though there were statistically insignificant trends of increased total energy, failure force, and stiffness in the PRP cohorts. There were no statistically significant differences between the indomethacin and no-indomethacin groups, and indomethacin had no effect on the efficacy of PRP treatment. It should be noted that the measurements of total energy, energy to failure, and failure force best reflect the strength of the tendon–bone interface. Other biomechanical measures, such as stiffness and normalized stiffness, are physical properties of the tendon itself and apply less to enthesis strength, which was the primary focus of this study.
Beck and colleagues23 studied the effect of allogeneic PRP on RCR in a rat model. They tested biomechanical and histologic outcomes 7, 14, and 21 days after surgery. There was no significant difference in failure load between the 2 groups at any time point. Compared with failure strain in the control group, failure strain in the PRP group was decreased at 7 days, normalized at 14 days, and increased at 21 days. The authors hypothesized that increased tendon failure strain at 21 days may have reduced forces being transmitted to the suture fixation site, which may be clinically significant and warrants further investigation. In a similar study, by Dolkart and colleagues,28 intraoperative PRP administration enhanced the maximal load-to-failure and stiffness of rats’ repaired rotator cuffs. On histologic examination, tendons treated with PRP (vs control tendons) had more organized collagen. Although these studies have limitations similar to our study, these results further support improved tendon-to-bone healing with PRP.
In clinical application, Barber and colleagues26 found that, compared with controls, suturing PRP fibrin matrix into the rotator cuff during repair decreased the incidence of magnetic resonance imaging–detected retears. However, in 2 prospective, randomized trials, Castricini and colleagues29 and Weber and colleagues30 found that use of PRP in RCR did not improve outcomes. All 3 studies differ from ours in that they used fibrin matrix. However, Ersen and colleagues31 found no difference in the effects of PRP on rotator cuff healing between injection and fibrin matrix; PRP improved biomechanical properties of repaired rotator cuff independent of administration method. In a meta-analysis of PRP supplementation in RCR, Warth and colleagues32 found a statistically significant improvement in retear rates for tears >3 cm repaired with a double-row technique, but otherwise no overall improvement in retear rates or outcome scores with PRP. The authors acknowledged that the significant heterogeneity of the studies in their meta-analysis may have affected the quality of their data.
Although our study provides some insight into the effectiveness of PRP in tendon repair, the lack of standardization in PRP preparation and time points tested makes comparisons with similar studies difficult.33 Recent reports have emphasized that not all PRP separation systems yield similar products.33 Platelet concentrations, and therefore platelet-derived growth factor concentrations, differ between systems and may yield different clinical outcomes. Our decision to use leukocyte-reduced PRP is supported by a meta-analysis by Riboh and colleagues,34 who reviewed the literature on the effect of leukocyte concentration on the efficacy of PRP products. They found that, in the treatment of knee osteoarthritis, use of leukocyte-poor PRP resulted in improved functional outcomes scores in comparison with placebo, but this improvement did not occur with leukocyte-rich PRP. However, there is still no consensus on optimal preparation, dosing, and route of administration of PRP, and preparations described in the literature vary.
This study also assessed the interaction of PRP and NSAIDs. Although there were no statistically significant differences between treatment and diet, shoulders treated with indomethacin alone showed a trend toward weaker biomechanical parameters in comparison with shoulders treated with saline alone, with PRP alone, or with both PRP and indomethacin. A larger sample would be needed to establish statistical significance. These trends are not surprising, as Cohen and colleagues21 found that NSAIDs, specifically indomethacin and celecoxib, significantly inhibited rotator cuff tendon-to-bone healing. The authors also found that a 2-week course of indomethacin was sufficient to significantly inhibit tendon-to-bone healing. In fact, although the drugs were discontinued after 14 days, biomechanical properties were negatively affected up to 8 weeks after repair. Our results differ from theirs even though the 2 studies used similar doses and administration protocols.
One strength of this study was that all surgeries were performed by a single board-certified surgeon using a standardized technique. In addition, a control group was established, and personnel and techniques for all fine dissections and biomechanical tests were consistent throughout. Blinded randomization and diet normalization, as well as adequate power for detecting significant effects, strengthened the study as well.
The study had several limitations. First, whereas most human rotator cuff tears are chronic, we used a model of acute injury and repair. As acute tears that are immediately repaired are more likely to heal, detection of differences between cohorts is less likely. However, using an acute model is still the most reliable strategy for inducing a controlled injury with reproducible severity. Second, we analyzed data at only 1 time point, which may not provide an accurate representation of long-term effects. Third, systemic administration of indomethacin did not allow for intra-rat shoulder comparisons of the different drug groups. Fourth, although it is possible that the dosage of NSAID was insufficient to produce significant differences in biomechanics, our dosage was consistent with that used in a study that found a significant effect on tendon healing.21
Conclusion
Our study found that the strength of the supraspinatus tendon enthesis as defined by energy to failure was increased with intratendinous PRP injection. Indomethacin showed no statistical effect, but there was a trend toward reduced strength after repair. However, the extent to which coadministration of indomethacin affects PRP remains unclear, and these data cannot necessarily be extrapolated to the typical human rotator cuff tear caused by chronic repetitive stress.
1. Kinsella KG, Velkoff VA. An Aging World: 2001. Washington, DC: US Government Printing Office; 2001. https://www.census.gov/prod/2001pubs/p95-01-1.pdf. Published November 2001. Accessed September 24, 2017.
2. Gamradt SC, Rodeo SA, Warren RF. Platelet rich plasma in rotator cuff repair. Tech Orthop. 2007;22(1):26-33.
3. Galatz LM, Ball CM, Teefey SA, Middleton WD, Yamaguchi K. The outcome and repair integrity of completely arthroscopically repaired large and massive rotator cuff tears. J Bone Joint Surg Am. 2004;86(2):219-224.
4. Harryman DT, Mack LA, Wang KY. Repairs of the rotator cuff. Correlation of functional results with integrity of the cuff. J Bone Joint Surg Am. 1991;73(7):982-989.
5. Bishop J, Klepps S, Lo IK, Bird J, Gladstone JN, Flatow EL. Cuff integrity after arthroscopic versus open rotator cuff repair: a prospective study. J Shoulder Elbow Surg. 2006;15(3):290-299.
6. Boileau P, Brassart N, Watkinson DJ, Carles M. Arthroscopic repair of full-thickness tears of the supraspinatus: does the tendon really heal? J Bone Joint Surg Am. 2005;87(6):1229-1240.
7. Gerber C, Fuchs B, Hodler J. The results of repair of massive tears of the rotator cuff. J Bone Joint Surg Am. 2000;82(4):505-515.
8. Lafosse L, Brozska R, Toussaint B, Gobezie R. The outcome and structural integrity of arthroscopic rotator cuff repair with use of the double-row suture anchor technique. J Bone Joint Surg Am. 2007;89(7):1533-1541.
9. Levy O, Venkateswaran B, Even T, Ravenscroft M, Copeland S. Mid-term clinical and sonographic outcome of arthroscopic repair of the rotator cuff. J Bone Joint Surg Br. 2008;90(10):1341-1347.
10. Zumstein MA, Jost B, Hempel J, Hodler J, Gerber C. The clinical and structural long-term results of open repair of massive tears of the rotator cuff. J Bone Joint Surg Am. 2008;90(11):2423-2431.
11. Gerber C, Schneeberger AG, Perren SM, Nyffeler RW. Experimental rotator cuff repair. A preliminary study. J Bone Joint Surg Am. 1999;81(9):1281-1290.
12. Randelli P, Arrigoni P, Ragone V, Aliprandi A, Cabitza P. Platelet rich plasma in arthroscopic rotator cuff repair: a prospective RCT study, 2-year follow-up. J Shoulder Elbow Surg. 2011;20(4):518-528.
13. Akeda K, An HS, Okuma M, et al. Platelet-rich plasma stimulates porcine articular chondrocyte proliferation and matrix biosynthesis. Osteoarthritis Cartilage. 2006;14(12):1272-1280.
14. de Mos M, van der Windt AE, Jahr H, et al. Can platelet-rich plasma enhance tendon repair? A cell culture study. Am J Sports Med. 2008;36(6):1171-1178.
15. Harmon KG. Muscle injuries and PRP: what does the science say? Br J Sports Med. 2010;44(9):616-617.
16. Kasten P, Vogel J, Geiger F, Niemeyer P, Luginbühl R, Szalay K. The effect of platelet-rich plasma on healing in critical-size long-bone defects. Biomaterials. 2008;29(29):3983-3992.
17. Mei-Dan O, Mann G, Maffulli N. Platelet-rich plasma: any substance into it? Br J Sports Med. 2010;44(9):618-619.
18. Murray MM, Spindler KP, Ballard P, Welch TP, Zurakowski D, Nanney LB. Enhanced histologic repair in a central wound in the anterior cruciate ligament with a collagen-platelet-rich plasma scaffold. J Orthop Res. 2007;25(8):1007-1017.
19. Virchenko O, Skoglund B, Aspenberg P. Parecoxib impairs early tendon repair but improves later remodeling. Am J Sports Med. 2004;32(7):1743-1747.
20. Aspenberg P. Differential inhibition of fracture healing by non-selective and cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs. J Orthop Res. 2004;22(3):684.
21. Cohen DB, Kawamura S, Ehteshami JR, Rodeo SA. Indomethacin and celecoxib impair rotator cuff tendon-to-bone healing. Am J Sports Med. 2006;34(3):362-369.
22. Balazs T, Grice HC, Airth JM. On counting the blood cells of the rat with an electronic counter. Can J Comp Med Vet Sci. 1960;24(9):273-275.
23. Beck J, Evans D, Tonino PM, Yong S, Callaci JJ. The biomechanical and histologic effects of platelet-rich plasma on rat rotator cuff repairs. Am J Sports Med. 2012;40(9):2037-2044.
24. Aspenberg P, Virchenko O. Platelet concentrate injection improves Achilles tendon repair in rats. Acta Orthop Scand. 2004;75(1):93-99.
25. Chechik O, Dolkart O, Mozes G, Rak O, Alhajajra F, Maman E. Timing matters: NSAIDs interfere with the late proliferation stage of a repaired rotator cuff tendon healing in rats. Arch Orthop Trauma Surg. 2014;134(4):515-520.
26. Barber FA, Hrnack SA, Snyder SJ, Hapa O. Rotator cuff repair healing influenced by platelet-rich plasma construct augmentation. Arthroscopy. 2011;27(8):1029-1035.
27. Randelli PS, Arrigoni P, Cabitza P, Volpi P, Maffulli N. Autologous platelet rich plasma for arthroscopic rotator cuff repair. A pilot study. Disabil Rehabil. 2008;30(20-22):1584-1589.
28. Dolkart O, Chechik O, Zarfati Y, Brosh T, Alhajajra F, Maman E. A single dose of platelet-rich plasma improves the organization and strength of a surgically repaired rotator cuff tendon in rats. Arch Orthop Trauma Surg. 2014;134(9):1271-1277.
29. Castricini R, Longo UG, De Benedetto M, et al. Platelet-rich plasma augmentation for arthroscopic rotator cuff repair: a randomized controlled trial. Am J Sports Med. 2011;39(2):258-265.
30. Weber SC, Kauffman JI, Parise C, Weber SJ, Katz SD. Platelet-rich fibrin matrix in the management of arthroscopic repair of the rotator cuff: a prospective, randomized, double-blinded study. Am J Sports Med. 2013;41(2):263-270.
31. Ersen A, Demirhan M, Atalar AC, Kapicioğlu M, Baysal G. Platelet-rich plasma for enhancing surgical rotator cuff repair: evaluation and comparison of two application methods in a rat model. Arch Orthop Trauma Surg. 2014;134(3):405-411.
32. Warth RJ, Dornan GJ, James EW, Horan MP, Millett PJ. Clinical and structural outcomes after arthroscopic repair of full-thickness rotator cuff tears with and without platelet-rich product supplementation: a meta-analysis and meta-regression. Arthroscopy. 2015;31(2):306-320.
33. Bergeson AG, Tashjian RZ, Greis PE, Crim J, Stoddard GJ, Burks RT. Effects of platelet-rich fibrin matrix on repair integrity of at-risk rotator cuff tears. Am J Sports Med. 2012;40(2):286-293.
34. Riboh JC, Saltzman BM, Yanke AB, Fortier L, Cole BJ. Effect of leukocyte concentration on the efficacy of platelet-rich plasma in the treatment of knee osteoarthritis. Am J Sports Med. 2016;44(3):792-800.
Take-Home Points
- The optimal centrifugation protocol for production of rat PRP is 1300 rpm for 5 minutes.
- PRP administration in RCR improves tendon biomechanics in a rat model.
- Administration of NSAIDs following RCR has no significant effect on tendon biomechanical properties.
- NSAIDs may be co-administered with PRP without reducing efficacy of PRP.
- The role of PRP and NSAIDs in human RCR remains unclear.
Rotator cuff tears are a common source of shoulder pain and disability among older adults and athletes. Full-thickness tears alone occur in up to 30% of adults older than 60 years.1 Surgical repair is plagued by an unpredictable rate of recurrence (range, 11%-94%).1-10 As a result of improved suture materials, knotting patterns, and anchor designs, hardware issues are no longer the primary cause of rotator cuff repair (RCR) failures; now the principal mode of failure is biologic.2 Animal model studies have found that, after injury and subsequent healing, the tendon–bone interface remains abnormal.11 Rotator cuff research therefore has focused largely on biological enhancement of tendon-to-bone healing.
One means of biological augmentation is autologous platelet-rich plasma (PRP), which has supraphysiologic concentrations of platelets and their secreted growth factors. Although there is no consensus on the long-term efficacy of PRP, some studies suggest PRP accelerates healing over short and intermediate terms, which may contribute to a more rapid decrease in pain and more rapid return to normal activities.12-18 Similarly, systemic nonsteroidal anti-inflammatory drugs (NSAIDs) have long been used to treat musculoskeletal injuries, including rotator cuff pathology. However, NSAIDs inhibit cyclooxygenase activity, and clinical and experimental data have shown that cyclooxygenase 2 function is crucial in normal tendon-to-bone healing.19-21
Comprehensive studies have been conducted on the efficacy of both PRP and NSAIDs, but the interaction of concurrently used PRP and NSAIDs has not been determined. As many physicians use both modalities in the treatment of soft-tissue injuries, it is important to study the potential interactions when coadministered. Prior studies in small animal models suggest NSAIDs may impair tendon-to-bone healing in RCR, but there is no evidence regarding the effect of NSAIDs on the efficacy of PRP treatment.21
We conducted a study to determine the interaction of PRP and NSAIDs when used as adjuncts to RCR in a rat model. We hypothesized that PRP would increase the strength of RCR and that NSAIDs would interfere with the effects of PRP. A preliminary study objective was to determine an appropriate centrifugation protocol for producing PRP from rat blood, for use in this study and in future rat-based studies of PRP.
Materials and Methods
Part A: Pretesting Determination of PRP Centrifugation Protocol
Fourteen adult male Fischer rats were used in part A of this study, which was conducted to determine an appropriate PRP centrifugation protocol. Traditional PRP centrifugation protocols are established for human blood, but rat red blood cells (RBCs) and human RBCs differ in size.22 In our preliminary study, we wanted to determine the adjusted centrifuge speed and duration for producing clinically optimal PRP from rats. Clinically optimal PRP has reduced levels of RBCs, which decrease platelet affinity. Although the role of leukocytes in PRP preparations is debated, reducing the number of white blood cells (WBCs) decreases the number of matrix metalloproteinases and reactive oxygen species that may lead to inflammation. We used the platelet index (ratio of platelets to WBCs) and the RBC count to quantify the quality of our PRP sample.
Each rat in part A was anesthetized while supine. We used the Autologous Conditioned Plasma (ACP) system (Arthrex), which requires only 1 centrifugation cycle to create PRP. About 9 mL or 10 mL of blood was obtained by cardiac aspiration using an ACP Double Syringe (Arthrex). After blood retrieval, a thoracotomy was performed to confirm each rat’s death.
Part B: Determining the Effects of PRP and NSAIDs on RCR in a Rat Model
Operative Cohort. Of the 34 Fischer rats used in part B of this study, 6 were used as blood donors for PRP production, and the other 28 underwent bilateral rotator cuff surgeries. We used donor rats to maximize the amount of PRP retrieval, allocating about 1 donor rat per 5 operative rats. Fischer rats are an inbred strain, so the PRP from a donor Fischer rat simulates autologous blood in other Fischer rats. Use of allogenic blood is consistent with prior rat PRP studies.23,24
Operative Technique. Each bilateral surgery was performed by a single board-certified shoulder surgeon, and the anesthetic and surgical protocols were followed as approved by the home institution’s Institutional Animal Care and Use Committee. Before surgery, blood was harvested for PRP production from donor rats, as described earlier, and centrifuged for 5 minutes × 1300 rpm. After anesthetic induction and skin incision, the deltoid muscle was cut to expose the acromion and underlying rotator cuff. The distal supraspinatus tendon was sharply detached from the greater tuberosity. A bone-tunnel RCR was performed by drilling a transverse tunnel across the greater tuberosity and affixing the tendon to its footprint with a 5-0 polypropylene suture (Prolene; Ethicon). Each rat was then randomly assigned to receive 50 µL of donor PRP injected in 1 operative shoulder and saline in the contralateral shoulder. Injections were made in the supraspinatus tendon at its attachment to the humerus. Deltoid and skin were closed with 4-0 polyglactin (Vicryl) suture (Ethicon) and staples, respectively.
Tendon Preparation. Immediately post mortem, each shoulder was grossly dissected to isolate the supraspinatus muscle attached to the humerus. Shoulders were then frozen in 0.15-M saline solution until specified biomechanical testing dates.
On day of dimensional/biomechanical testing, each specimen was thawed at room temperature and finely dissected under a microscope (Stemi 200-C; Car Zeiss). After dissection, the humeral shaft was embedded in polymethylmethacrylate within a test tube. The free end of the supraspinatus tendon was glued within a “tab” of waterproofed emery cloth, leaving about 2 mm of tendon between the tab and the greater tuberosity.
Biomechanical Analysis. A 5848 MicroTester (Instron) was used for biomechanical testing. Each tabbed tendon, held by a pneumatic clamp attached to the MicroTester, was tested in a preconditioning phase and then a ramp-to-failure phase. A constant drip of 0.15-M saline was run through the apparatus to simulate physiologic hydration of tissue. After the embedded specimen was secure within the loading apparatus, an initial tensile preload of 0.2 N was applied. After preloading, the tendon was run through a preconditioning phase to account for viscoelastic relaxation. Immediately after preconditioning, each tendon was subjected to failure testing at a ramp rate of 0.1 mm/s. Force data were collected as a function of displacement, allowing for the calculation of 4 biomechanical parameters: failure force, tendon stiffness and normalized stiffness, energy to failure, and total energy. Tendon stiffness is the slope of a curve-fit line of the initial peak; failure force is the force of the highest peak; energy to failure is the area under the curve (AUC) to the highest peak; and total energy is the AUC from the start of failure ramping to the point at which the tendon is torn off completely. Two-way ANOVA was used to assess the differences between treatment groups and diet groups for all parameters. Statistical significance was set at P < .05.
A power analysis was performed to determine ability to detect differences between cohorts. For power of 80% and P = .05, a difference of 16% of the mean could be detected for failure force, 30% for energy to failure, 14% for total energy to failure, and 24% for stiffness. In addition, a difference of 4% of the mean could be detected for tendon length, 6% for width, and 10% for thickness.
Results
Across all collective treatment-diet groups and biomechanical parameters, there was only 1 statistically significant difference. Mean (SD) energy to failure was significantly higher (P = .03) in shoulders treated with PRP, 11.7 (7.3) N-mm, than in those treated without PRP, 8.7 (4.6) N-mm (Figure 4). There were no statistically significant differences between shoulders treated with indomethacin and those treated without indomethacin (Table 3), and no statistically significant relationships between treatment and drug for any other biomechanical parameter (Figures 5-7).
Discussion
Our preliminary objective in this study was to determine the optimal centrifugation protocol for producing rat-based PRP. Optimal PRP requires a dense concentration of platelets as well as reduced levels of RBCs and WBCs.25 We used the platelet index to quantify the quality of our PRP samples, and we obtained the highest platelet index for the protocol of 5 minutes × 1300 rpm. This finding may be useful in later rat studies involving PRP.
The primary objective of this study was to assess the effect of the interaction of PRP and NSAIDs on RCR. PRP has been found to augment RCR,12,26,27 but indomethacin may impair healing.21,25 We hypothesized that shoulders treated with PRP would have more biomechanical strength than control shoulders and that indomethacin would decrease biomechanical strength.
Our data showed increased energy to failure of the rotator cuff with PRP injections (P = .03). All other biomechanical parameters showed no significant differences with PRP treatment, though there were statistically insignificant trends of increased total energy, failure force, and stiffness in the PRP cohorts. There were no statistically significant differences between the indomethacin and no-indomethacin groups, and indomethacin had no effect on the efficacy of PRP treatment. It should be noted that the measurements of total energy, energy to failure, and failure force best reflect the strength of the tendon–bone interface. Other biomechanical measures, such as stiffness and normalized stiffness, are physical properties of the tendon itself and apply less to enthesis strength, which was the primary focus of this study.
Beck and colleagues23 studied the effect of allogeneic PRP on RCR in a rat model. They tested biomechanical and histologic outcomes 7, 14, and 21 days after surgery. There was no significant difference in failure load between the 2 groups at any time point. Compared with failure strain in the control group, failure strain in the PRP group was decreased at 7 days, normalized at 14 days, and increased at 21 days. The authors hypothesized that increased tendon failure strain at 21 days may have reduced forces being transmitted to the suture fixation site, which may be clinically significant and warrants further investigation. In a similar study, by Dolkart and colleagues,28 intraoperative PRP administration enhanced the maximal load-to-failure and stiffness of rats’ repaired rotator cuffs. On histologic examination, tendons treated with PRP (vs control tendons) had more organized collagen. Although these studies have limitations similar to our study, these results further support improved tendon-to-bone healing with PRP.
In clinical application, Barber and colleagues26 found that, compared with controls, suturing PRP fibrin matrix into the rotator cuff during repair decreased the incidence of magnetic resonance imaging–detected retears. However, in 2 prospective, randomized trials, Castricini and colleagues29 and Weber and colleagues30 found that use of PRP in RCR did not improve outcomes. All 3 studies differ from ours in that they used fibrin matrix. However, Ersen and colleagues31 found no difference in the effects of PRP on rotator cuff healing between injection and fibrin matrix; PRP improved biomechanical properties of repaired rotator cuff independent of administration method. In a meta-analysis of PRP supplementation in RCR, Warth and colleagues32 found a statistically significant improvement in retear rates for tears >3 cm repaired with a double-row technique, but otherwise no overall improvement in retear rates or outcome scores with PRP. The authors acknowledged that the significant heterogeneity of the studies in their meta-analysis may have affected the quality of their data.
Although our study provides some insight into the effectiveness of PRP in tendon repair, the lack of standardization in PRP preparation and time points tested makes comparisons with similar studies difficult.33 Recent reports have emphasized that not all PRP separation systems yield similar products.33 Platelet concentrations, and therefore platelet-derived growth factor concentrations, differ between systems and may yield different clinical outcomes. Our decision to use leukocyte-reduced PRP is supported by a meta-analysis by Riboh and colleagues,34 who reviewed the literature on the effect of leukocyte concentration on the efficacy of PRP products. They found that, in the treatment of knee osteoarthritis, use of leukocyte-poor PRP resulted in improved functional outcomes scores in comparison with placebo, but this improvement did not occur with leukocyte-rich PRP. However, there is still no consensus on optimal preparation, dosing, and route of administration of PRP, and preparations described in the literature vary.
This study also assessed the interaction of PRP and NSAIDs. Although there were no statistically significant differences between treatment and diet, shoulders treated with indomethacin alone showed a trend toward weaker biomechanical parameters in comparison with shoulders treated with saline alone, with PRP alone, or with both PRP and indomethacin. A larger sample would be needed to establish statistical significance. These trends are not surprising, as Cohen and colleagues21 found that NSAIDs, specifically indomethacin and celecoxib, significantly inhibited rotator cuff tendon-to-bone healing. The authors also found that a 2-week course of indomethacin was sufficient to significantly inhibit tendon-to-bone healing. In fact, although the drugs were discontinued after 14 days, biomechanical properties were negatively affected up to 8 weeks after repair. Our results differ from theirs even though the 2 studies used similar doses and administration protocols.
One strength of this study was that all surgeries were performed by a single board-certified surgeon using a standardized technique. In addition, a control group was established, and personnel and techniques for all fine dissections and biomechanical tests were consistent throughout. Blinded randomization and diet normalization, as well as adequate power for detecting significant effects, strengthened the study as well.
The study had several limitations. First, whereas most human rotator cuff tears are chronic, we used a model of acute injury and repair. As acute tears that are immediately repaired are more likely to heal, detection of differences between cohorts is less likely. However, using an acute model is still the most reliable strategy for inducing a controlled injury with reproducible severity. Second, we analyzed data at only 1 time point, which may not provide an accurate representation of long-term effects. Third, systemic administration of indomethacin did not allow for intra-rat shoulder comparisons of the different drug groups. Fourth, although it is possible that the dosage of NSAID was insufficient to produce significant differences in biomechanics, our dosage was consistent with that used in a study that found a significant effect on tendon healing.21
Conclusion
Our study found that the strength of the supraspinatus tendon enthesis as defined by energy to failure was increased with intratendinous PRP injection. Indomethacin showed no statistical effect, but there was a trend toward reduced strength after repair. However, the extent to which coadministration of indomethacin affects PRP remains unclear, and these data cannot necessarily be extrapolated to the typical human rotator cuff tear caused by chronic repetitive stress.
Take-Home Points
- The optimal centrifugation protocol for production of rat PRP is 1300 rpm for 5 minutes.
- PRP administration in RCR improves tendon biomechanics in a rat model.
- Administration of NSAIDs following RCR has no significant effect on tendon biomechanical properties.
- NSAIDs may be co-administered with PRP without reducing efficacy of PRP.
- The role of PRP and NSAIDs in human RCR remains unclear.
Rotator cuff tears are a common source of shoulder pain and disability among older adults and athletes. Full-thickness tears alone occur in up to 30% of adults older than 60 years.1 Surgical repair is plagued by an unpredictable rate of recurrence (range, 11%-94%).1-10 As a result of improved suture materials, knotting patterns, and anchor designs, hardware issues are no longer the primary cause of rotator cuff repair (RCR) failures; now the principal mode of failure is biologic.2 Animal model studies have found that, after injury and subsequent healing, the tendon–bone interface remains abnormal.11 Rotator cuff research therefore has focused largely on biological enhancement of tendon-to-bone healing.
One means of biological augmentation is autologous platelet-rich plasma (PRP), which has supraphysiologic concentrations of platelets and their secreted growth factors. Although there is no consensus on the long-term efficacy of PRP, some studies suggest PRP accelerates healing over short and intermediate terms, which may contribute to a more rapid decrease in pain and more rapid return to normal activities.12-18 Similarly, systemic nonsteroidal anti-inflammatory drugs (NSAIDs) have long been used to treat musculoskeletal injuries, including rotator cuff pathology. However, NSAIDs inhibit cyclooxygenase activity, and clinical and experimental data have shown that cyclooxygenase 2 function is crucial in normal tendon-to-bone healing.19-21
Comprehensive studies have been conducted on the efficacy of both PRP and NSAIDs, but the interaction of concurrently used PRP and NSAIDs has not been determined. As many physicians use both modalities in the treatment of soft-tissue injuries, it is important to study the potential interactions when coadministered. Prior studies in small animal models suggest NSAIDs may impair tendon-to-bone healing in RCR, but there is no evidence regarding the effect of NSAIDs on the efficacy of PRP treatment.21
We conducted a study to determine the interaction of PRP and NSAIDs when used as adjuncts to RCR in a rat model. We hypothesized that PRP would increase the strength of RCR and that NSAIDs would interfere with the effects of PRP. A preliminary study objective was to determine an appropriate centrifugation protocol for producing PRP from rat blood, for use in this study and in future rat-based studies of PRP.
Materials and Methods
Part A: Pretesting Determination of PRP Centrifugation Protocol
Fourteen adult male Fischer rats were used in part A of this study, which was conducted to determine an appropriate PRP centrifugation protocol. Traditional PRP centrifugation protocols are established for human blood, but rat red blood cells (RBCs) and human RBCs differ in size.22 In our preliminary study, we wanted to determine the adjusted centrifuge speed and duration for producing clinically optimal PRP from rats. Clinically optimal PRP has reduced levels of RBCs, which decrease platelet affinity. Although the role of leukocytes in PRP preparations is debated, reducing the number of white blood cells (WBCs) decreases the number of matrix metalloproteinases and reactive oxygen species that may lead to inflammation. We used the platelet index (ratio of platelets to WBCs) and the RBC count to quantify the quality of our PRP sample.
Each rat in part A was anesthetized while supine. We used the Autologous Conditioned Plasma (ACP) system (Arthrex), which requires only 1 centrifugation cycle to create PRP. About 9 mL or 10 mL of blood was obtained by cardiac aspiration using an ACP Double Syringe (Arthrex). After blood retrieval, a thoracotomy was performed to confirm each rat’s death.
Part B: Determining the Effects of PRP and NSAIDs on RCR in a Rat Model
Operative Cohort. Of the 34 Fischer rats used in part B of this study, 6 were used as blood donors for PRP production, and the other 28 underwent bilateral rotator cuff surgeries. We used donor rats to maximize the amount of PRP retrieval, allocating about 1 donor rat per 5 operative rats. Fischer rats are an inbred strain, so the PRP from a donor Fischer rat simulates autologous blood in other Fischer rats. Use of allogenic blood is consistent with prior rat PRP studies.23,24
Operative Technique. Each bilateral surgery was performed by a single board-certified shoulder surgeon, and the anesthetic and surgical protocols were followed as approved by the home institution’s Institutional Animal Care and Use Committee. Before surgery, blood was harvested for PRP production from donor rats, as described earlier, and centrifuged for 5 minutes × 1300 rpm. After anesthetic induction and skin incision, the deltoid muscle was cut to expose the acromion and underlying rotator cuff. The distal supraspinatus tendon was sharply detached from the greater tuberosity. A bone-tunnel RCR was performed by drilling a transverse tunnel across the greater tuberosity and affixing the tendon to its footprint with a 5-0 polypropylene suture (Prolene; Ethicon). Each rat was then randomly assigned to receive 50 µL of donor PRP injected in 1 operative shoulder and saline in the contralateral shoulder. Injections were made in the supraspinatus tendon at its attachment to the humerus. Deltoid and skin were closed with 4-0 polyglactin (Vicryl) suture (Ethicon) and staples, respectively.
Tendon Preparation. Immediately post mortem, each shoulder was grossly dissected to isolate the supraspinatus muscle attached to the humerus. Shoulders were then frozen in 0.15-M saline solution until specified biomechanical testing dates.
On day of dimensional/biomechanical testing, each specimen was thawed at room temperature and finely dissected under a microscope (Stemi 200-C; Car Zeiss). After dissection, the humeral shaft was embedded in polymethylmethacrylate within a test tube. The free end of the supraspinatus tendon was glued within a “tab” of waterproofed emery cloth, leaving about 2 mm of tendon between the tab and the greater tuberosity.
Biomechanical Analysis. A 5848 MicroTester (Instron) was used for biomechanical testing. Each tabbed tendon, held by a pneumatic clamp attached to the MicroTester, was tested in a preconditioning phase and then a ramp-to-failure phase. A constant drip of 0.15-M saline was run through the apparatus to simulate physiologic hydration of tissue. After the embedded specimen was secure within the loading apparatus, an initial tensile preload of 0.2 N was applied. After preloading, the tendon was run through a preconditioning phase to account for viscoelastic relaxation. Immediately after preconditioning, each tendon was subjected to failure testing at a ramp rate of 0.1 mm/s. Force data were collected as a function of displacement, allowing for the calculation of 4 biomechanical parameters: failure force, tendon stiffness and normalized stiffness, energy to failure, and total energy. Tendon stiffness is the slope of a curve-fit line of the initial peak; failure force is the force of the highest peak; energy to failure is the area under the curve (AUC) to the highest peak; and total energy is the AUC from the start of failure ramping to the point at which the tendon is torn off completely. Two-way ANOVA was used to assess the differences between treatment groups and diet groups for all parameters. Statistical significance was set at P < .05.
A power analysis was performed to determine ability to detect differences between cohorts. For power of 80% and P = .05, a difference of 16% of the mean could be detected for failure force, 30% for energy to failure, 14% for total energy to failure, and 24% for stiffness. In addition, a difference of 4% of the mean could be detected for tendon length, 6% for width, and 10% for thickness.
Results
Across all collective treatment-diet groups and biomechanical parameters, there was only 1 statistically significant difference. Mean (SD) energy to failure was significantly higher (P = .03) in shoulders treated with PRP, 11.7 (7.3) N-mm, than in those treated without PRP, 8.7 (4.6) N-mm (Figure 4). There were no statistically significant differences between shoulders treated with indomethacin and those treated without indomethacin (Table 3), and no statistically significant relationships between treatment and drug for any other biomechanical parameter (Figures 5-7).
Discussion
Our preliminary objective in this study was to determine the optimal centrifugation protocol for producing rat-based PRP. Optimal PRP requires a dense concentration of platelets as well as reduced levels of RBCs and WBCs.25 We used the platelet index to quantify the quality of our PRP samples, and we obtained the highest platelet index for the protocol of 5 minutes × 1300 rpm. This finding may be useful in later rat studies involving PRP.
The primary objective of this study was to assess the effect of the interaction of PRP and NSAIDs on RCR. PRP has been found to augment RCR,12,26,27 but indomethacin may impair healing.21,25 We hypothesized that shoulders treated with PRP would have more biomechanical strength than control shoulders and that indomethacin would decrease biomechanical strength.
Our data showed increased energy to failure of the rotator cuff with PRP injections (P = .03). All other biomechanical parameters showed no significant differences with PRP treatment, though there were statistically insignificant trends of increased total energy, failure force, and stiffness in the PRP cohorts. There were no statistically significant differences between the indomethacin and no-indomethacin groups, and indomethacin had no effect on the efficacy of PRP treatment. It should be noted that the measurements of total energy, energy to failure, and failure force best reflect the strength of the tendon–bone interface. Other biomechanical measures, such as stiffness and normalized stiffness, are physical properties of the tendon itself and apply less to enthesis strength, which was the primary focus of this study.
Beck and colleagues23 studied the effect of allogeneic PRP on RCR in a rat model. They tested biomechanical and histologic outcomes 7, 14, and 21 days after surgery. There was no significant difference in failure load between the 2 groups at any time point. Compared with failure strain in the control group, failure strain in the PRP group was decreased at 7 days, normalized at 14 days, and increased at 21 days. The authors hypothesized that increased tendon failure strain at 21 days may have reduced forces being transmitted to the suture fixation site, which may be clinically significant and warrants further investigation. In a similar study, by Dolkart and colleagues,28 intraoperative PRP administration enhanced the maximal load-to-failure and stiffness of rats’ repaired rotator cuffs. On histologic examination, tendons treated with PRP (vs control tendons) had more organized collagen. Although these studies have limitations similar to our study, these results further support improved tendon-to-bone healing with PRP.
In clinical application, Barber and colleagues26 found that, compared with controls, suturing PRP fibrin matrix into the rotator cuff during repair decreased the incidence of magnetic resonance imaging–detected retears. However, in 2 prospective, randomized trials, Castricini and colleagues29 and Weber and colleagues30 found that use of PRP in RCR did not improve outcomes. All 3 studies differ from ours in that they used fibrin matrix. However, Ersen and colleagues31 found no difference in the effects of PRP on rotator cuff healing between injection and fibrin matrix; PRP improved biomechanical properties of repaired rotator cuff independent of administration method. In a meta-analysis of PRP supplementation in RCR, Warth and colleagues32 found a statistically significant improvement in retear rates for tears >3 cm repaired with a double-row technique, but otherwise no overall improvement in retear rates or outcome scores with PRP. The authors acknowledged that the significant heterogeneity of the studies in their meta-analysis may have affected the quality of their data.
Although our study provides some insight into the effectiveness of PRP in tendon repair, the lack of standardization in PRP preparation and time points tested makes comparisons with similar studies difficult.33 Recent reports have emphasized that not all PRP separation systems yield similar products.33 Platelet concentrations, and therefore platelet-derived growth factor concentrations, differ between systems and may yield different clinical outcomes. Our decision to use leukocyte-reduced PRP is supported by a meta-analysis by Riboh and colleagues,34 who reviewed the literature on the effect of leukocyte concentration on the efficacy of PRP products. They found that, in the treatment of knee osteoarthritis, use of leukocyte-poor PRP resulted in improved functional outcomes scores in comparison with placebo, but this improvement did not occur with leukocyte-rich PRP. However, there is still no consensus on optimal preparation, dosing, and route of administration of PRP, and preparations described in the literature vary.
This study also assessed the interaction of PRP and NSAIDs. Although there were no statistically significant differences between treatment and diet, shoulders treated with indomethacin alone showed a trend toward weaker biomechanical parameters in comparison with shoulders treated with saline alone, with PRP alone, or with both PRP and indomethacin. A larger sample would be needed to establish statistical significance. These trends are not surprising, as Cohen and colleagues21 found that NSAIDs, specifically indomethacin and celecoxib, significantly inhibited rotator cuff tendon-to-bone healing. The authors also found that a 2-week course of indomethacin was sufficient to significantly inhibit tendon-to-bone healing. In fact, although the drugs were discontinued after 14 days, biomechanical properties were negatively affected up to 8 weeks after repair. Our results differ from theirs even though the 2 studies used similar doses and administration protocols.
One strength of this study was that all surgeries were performed by a single board-certified surgeon using a standardized technique. In addition, a control group was established, and personnel and techniques for all fine dissections and biomechanical tests were consistent throughout. Blinded randomization and diet normalization, as well as adequate power for detecting significant effects, strengthened the study as well.
The study had several limitations. First, whereas most human rotator cuff tears are chronic, we used a model of acute injury and repair. As acute tears that are immediately repaired are more likely to heal, detection of differences between cohorts is less likely. However, using an acute model is still the most reliable strategy for inducing a controlled injury with reproducible severity. Second, we analyzed data at only 1 time point, which may not provide an accurate representation of long-term effects. Third, systemic administration of indomethacin did not allow for intra-rat shoulder comparisons of the different drug groups. Fourth, although it is possible that the dosage of NSAID was insufficient to produce significant differences in biomechanics, our dosage was consistent with that used in a study that found a significant effect on tendon healing.21
Conclusion
Our study found that the strength of the supraspinatus tendon enthesis as defined by energy to failure was increased with intratendinous PRP injection. Indomethacin showed no statistical effect, but there was a trend toward reduced strength after repair. However, the extent to which coadministration of indomethacin affects PRP remains unclear, and these data cannot necessarily be extrapolated to the typical human rotator cuff tear caused by chronic repetitive stress.
1. Kinsella KG, Velkoff VA. An Aging World: 2001. Washington, DC: US Government Printing Office; 2001. https://www.census.gov/prod/2001pubs/p95-01-1.pdf. Published November 2001. Accessed September 24, 2017.
2. Gamradt SC, Rodeo SA, Warren RF. Platelet rich plasma in rotator cuff repair. Tech Orthop. 2007;22(1):26-33.
3. Galatz LM, Ball CM, Teefey SA, Middleton WD, Yamaguchi K. The outcome and repair integrity of completely arthroscopically repaired large and massive rotator cuff tears. J Bone Joint Surg Am. 2004;86(2):219-224.
4. Harryman DT, Mack LA, Wang KY. Repairs of the rotator cuff. Correlation of functional results with integrity of the cuff. J Bone Joint Surg Am. 1991;73(7):982-989.
5. Bishop J, Klepps S, Lo IK, Bird J, Gladstone JN, Flatow EL. Cuff integrity after arthroscopic versus open rotator cuff repair: a prospective study. J Shoulder Elbow Surg. 2006;15(3):290-299.
6. Boileau P, Brassart N, Watkinson DJ, Carles M. Arthroscopic repair of full-thickness tears of the supraspinatus: does the tendon really heal? J Bone Joint Surg Am. 2005;87(6):1229-1240.
7. Gerber C, Fuchs B, Hodler J. The results of repair of massive tears of the rotator cuff. J Bone Joint Surg Am. 2000;82(4):505-515.
8. Lafosse L, Brozska R, Toussaint B, Gobezie R. The outcome and structural integrity of arthroscopic rotator cuff repair with use of the double-row suture anchor technique. J Bone Joint Surg Am. 2007;89(7):1533-1541.
9. Levy O, Venkateswaran B, Even T, Ravenscroft M, Copeland S. Mid-term clinical and sonographic outcome of arthroscopic repair of the rotator cuff. J Bone Joint Surg Br. 2008;90(10):1341-1347.
10. Zumstein MA, Jost B, Hempel J, Hodler J, Gerber C. The clinical and structural long-term results of open repair of massive tears of the rotator cuff. J Bone Joint Surg Am. 2008;90(11):2423-2431.
11. Gerber C, Schneeberger AG, Perren SM, Nyffeler RW. Experimental rotator cuff repair. A preliminary study. J Bone Joint Surg Am. 1999;81(9):1281-1290.
12. Randelli P, Arrigoni P, Ragone V, Aliprandi A, Cabitza P. Platelet rich plasma in arthroscopic rotator cuff repair: a prospective RCT study, 2-year follow-up. J Shoulder Elbow Surg. 2011;20(4):518-528.
13. Akeda K, An HS, Okuma M, et al. Platelet-rich plasma stimulates porcine articular chondrocyte proliferation and matrix biosynthesis. Osteoarthritis Cartilage. 2006;14(12):1272-1280.
14. de Mos M, van der Windt AE, Jahr H, et al. Can platelet-rich plasma enhance tendon repair? A cell culture study. Am J Sports Med. 2008;36(6):1171-1178.
15. Harmon KG. Muscle injuries and PRP: what does the science say? Br J Sports Med. 2010;44(9):616-617.
16. Kasten P, Vogel J, Geiger F, Niemeyer P, Luginbühl R, Szalay K. The effect of platelet-rich plasma on healing in critical-size long-bone defects. Biomaterials. 2008;29(29):3983-3992.
17. Mei-Dan O, Mann G, Maffulli N. Platelet-rich plasma: any substance into it? Br J Sports Med. 2010;44(9):618-619.
18. Murray MM, Spindler KP, Ballard P, Welch TP, Zurakowski D, Nanney LB. Enhanced histologic repair in a central wound in the anterior cruciate ligament with a collagen-platelet-rich plasma scaffold. J Orthop Res. 2007;25(8):1007-1017.
19. Virchenko O, Skoglund B, Aspenberg P. Parecoxib impairs early tendon repair but improves later remodeling. Am J Sports Med. 2004;32(7):1743-1747.
20. Aspenberg P. Differential inhibition of fracture healing by non-selective and cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs. J Orthop Res. 2004;22(3):684.
21. Cohen DB, Kawamura S, Ehteshami JR, Rodeo SA. Indomethacin and celecoxib impair rotator cuff tendon-to-bone healing. Am J Sports Med. 2006;34(3):362-369.
22. Balazs T, Grice HC, Airth JM. On counting the blood cells of the rat with an electronic counter. Can J Comp Med Vet Sci. 1960;24(9):273-275.
23. Beck J, Evans D, Tonino PM, Yong S, Callaci JJ. The biomechanical and histologic effects of platelet-rich plasma on rat rotator cuff repairs. Am J Sports Med. 2012;40(9):2037-2044.
24. Aspenberg P, Virchenko O. Platelet concentrate injection improves Achilles tendon repair in rats. Acta Orthop Scand. 2004;75(1):93-99.
25. Chechik O, Dolkart O, Mozes G, Rak O, Alhajajra F, Maman E. Timing matters: NSAIDs interfere with the late proliferation stage of a repaired rotator cuff tendon healing in rats. Arch Orthop Trauma Surg. 2014;134(4):515-520.
26. Barber FA, Hrnack SA, Snyder SJ, Hapa O. Rotator cuff repair healing influenced by platelet-rich plasma construct augmentation. Arthroscopy. 2011;27(8):1029-1035.
27. Randelli PS, Arrigoni P, Cabitza P, Volpi P, Maffulli N. Autologous platelet rich plasma for arthroscopic rotator cuff repair. A pilot study. Disabil Rehabil. 2008;30(20-22):1584-1589.
28. Dolkart O, Chechik O, Zarfati Y, Brosh T, Alhajajra F, Maman E. A single dose of platelet-rich plasma improves the organization and strength of a surgically repaired rotator cuff tendon in rats. Arch Orthop Trauma Surg. 2014;134(9):1271-1277.
29. Castricini R, Longo UG, De Benedetto M, et al. Platelet-rich plasma augmentation for arthroscopic rotator cuff repair: a randomized controlled trial. Am J Sports Med. 2011;39(2):258-265.
30. Weber SC, Kauffman JI, Parise C, Weber SJ, Katz SD. Platelet-rich fibrin matrix in the management of arthroscopic repair of the rotator cuff: a prospective, randomized, double-blinded study. Am J Sports Med. 2013;41(2):263-270.
31. Ersen A, Demirhan M, Atalar AC, Kapicioğlu M, Baysal G. Platelet-rich plasma for enhancing surgical rotator cuff repair: evaluation and comparison of two application methods in a rat model. Arch Orthop Trauma Surg. 2014;134(3):405-411.
32. Warth RJ, Dornan GJ, James EW, Horan MP, Millett PJ. Clinical and structural outcomes after arthroscopic repair of full-thickness rotator cuff tears with and without platelet-rich product supplementation: a meta-analysis and meta-regression. Arthroscopy. 2015;31(2):306-320.
33. Bergeson AG, Tashjian RZ, Greis PE, Crim J, Stoddard GJ, Burks RT. Effects of platelet-rich fibrin matrix on repair integrity of at-risk rotator cuff tears. Am J Sports Med. 2012;40(2):286-293.
34. Riboh JC, Saltzman BM, Yanke AB, Fortier L, Cole BJ. Effect of leukocyte concentration on the efficacy of platelet-rich plasma in the treatment of knee osteoarthritis. Am J Sports Med. 2016;44(3):792-800.
1. Kinsella KG, Velkoff VA. An Aging World: 2001. Washington, DC: US Government Printing Office; 2001. https://www.census.gov/prod/2001pubs/p95-01-1.pdf. Published November 2001. Accessed September 24, 2017.
2. Gamradt SC, Rodeo SA, Warren RF. Platelet rich plasma in rotator cuff repair. Tech Orthop. 2007;22(1):26-33.
3. Galatz LM, Ball CM, Teefey SA, Middleton WD, Yamaguchi K. The outcome and repair integrity of completely arthroscopically repaired large and massive rotator cuff tears. J Bone Joint Surg Am. 2004;86(2):219-224.
4. Harryman DT, Mack LA, Wang KY. Repairs of the rotator cuff. Correlation of functional results with integrity of the cuff. J Bone Joint Surg Am. 1991;73(7):982-989.
5. Bishop J, Klepps S, Lo IK, Bird J, Gladstone JN, Flatow EL. Cuff integrity after arthroscopic versus open rotator cuff repair: a prospective study. J Shoulder Elbow Surg. 2006;15(3):290-299.
6. Boileau P, Brassart N, Watkinson DJ, Carles M. Arthroscopic repair of full-thickness tears of the supraspinatus: does the tendon really heal? J Bone Joint Surg Am. 2005;87(6):1229-1240.
7. Gerber C, Fuchs B, Hodler J. The results of repair of massive tears of the rotator cuff. J Bone Joint Surg Am. 2000;82(4):505-515.
8. Lafosse L, Brozska R, Toussaint B, Gobezie R. The outcome and structural integrity of arthroscopic rotator cuff repair with use of the double-row suture anchor technique. J Bone Joint Surg Am. 2007;89(7):1533-1541.
9. Levy O, Venkateswaran B, Even T, Ravenscroft M, Copeland S. Mid-term clinical and sonographic outcome of arthroscopic repair of the rotator cuff. J Bone Joint Surg Br. 2008;90(10):1341-1347.
10. Zumstein MA, Jost B, Hempel J, Hodler J, Gerber C. The clinical and structural long-term results of open repair of massive tears of the rotator cuff. J Bone Joint Surg Am. 2008;90(11):2423-2431.
11. Gerber C, Schneeberger AG, Perren SM, Nyffeler RW. Experimental rotator cuff repair. A preliminary study. J Bone Joint Surg Am. 1999;81(9):1281-1290.
12. Randelli P, Arrigoni P, Ragone V, Aliprandi A, Cabitza P. Platelet rich plasma in arthroscopic rotator cuff repair: a prospective RCT study, 2-year follow-up. J Shoulder Elbow Surg. 2011;20(4):518-528.
13. Akeda K, An HS, Okuma M, et al. Platelet-rich plasma stimulates porcine articular chondrocyte proliferation and matrix biosynthesis. Osteoarthritis Cartilage. 2006;14(12):1272-1280.
14. de Mos M, van der Windt AE, Jahr H, et al. Can platelet-rich plasma enhance tendon repair? A cell culture study. Am J Sports Med. 2008;36(6):1171-1178.
15. Harmon KG. Muscle injuries and PRP: what does the science say? Br J Sports Med. 2010;44(9):616-617.
16. Kasten P, Vogel J, Geiger F, Niemeyer P, Luginbühl R, Szalay K. The effect of platelet-rich plasma on healing in critical-size long-bone defects. Biomaterials. 2008;29(29):3983-3992.
17. Mei-Dan O, Mann G, Maffulli N. Platelet-rich plasma: any substance into it? Br J Sports Med. 2010;44(9):618-619.
18. Murray MM, Spindler KP, Ballard P, Welch TP, Zurakowski D, Nanney LB. Enhanced histologic repair in a central wound in the anterior cruciate ligament with a collagen-platelet-rich plasma scaffold. J Orthop Res. 2007;25(8):1007-1017.
19. Virchenko O, Skoglund B, Aspenberg P. Parecoxib impairs early tendon repair but improves later remodeling. Am J Sports Med. 2004;32(7):1743-1747.
20. Aspenberg P. Differential inhibition of fracture healing by non-selective and cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs. J Orthop Res. 2004;22(3):684.
21. Cohen DB, Kawamura S, Ehteshami JR, Rodeo SA. Indomethacin and celecoxib impair rotator cuff tendon-to-bone healing. Am J Sports Med. 2006;34(3):362-369.
22. Balazs T, Grice HC, Airth JM. On counting the blood cells of the rat with an electronic counter. Can J Comp Med Vet Sci. 1960;24(9):273-275.
23. Beck J, Evans D, Tonino PM, Yong S, Callaci JJ. The biomechanical and histologic effects of platelet-rich plasma on rat rotator cuff repairs. Am J Sports Med. 2012;40(9):2037-2044.
24. Aspenberg P, Virchenko O. Platelet concentrate injection improves Achilles tendon repair in rats. Acta Orthop Scand. 2004;75(1):93-99.
25. Chechik O, Dolkart O, Mozes G, Rak O, Alhajajra F, Maman E. Timing matters: NSAIDs interfere with the late proliferation stage of a repaired rotator cuff tendon healing in rats. Arch Orthop Trauma Surg. 2014;134(4):515-520.
26. Barber FA, Hrnack SA, Snyder SJ, Hapa O. Rotator cuff repair healing influenced by platelet-rich plasma construct augmentation. Arthroscopy. 2011;27(8):1029-1035.
27. Randelli PS, Arrigoni P, Cabitza P, Volpi P, Maffulli N. Autologous platelet rich plasma for arthroscopic rotator cuff repair. A pilot study. Disabil Rehabil. 2008;30(20-22):1584-1589.
28. Dolkart O, Chechik O, Zarfati Y, Brosh T, Alhajajra F, Maman E. A single dose of platelet-rich plasma improves the organization and strength of a surgically repaired rotator cuff tendon in rats. Arch Orthop Trauma Surg. 2014;134(9):1271-1277.
29. Castricini R, Longo UG, De Benedetto M, et al. Platelet-rich plasma augmentation for arthroscopic rotator cuff repair: a randomized controlled trial. Am J Sports Med. 2011;39(2):258-265.
30. Weber SC, Kauffman JI, Parise C, Weber SJ, Katz SD. Platelet-rich fibrin matrix in the management of arthroscopic repair of the rotator cuff: a prospective, randomized, double-blinded study. Am J Sports Med. 2013;41(2):263-270.
31. Ersen A, Demirhan M, Atalar AC, Kapicioğlu M, Baysal G. Platelet-rich plasma for enhancing surgical rotator cuff repair: evaluation and comparison of two application methods in a rat model. Arch Orthop Trauma Surg. 2014;134(3):405-411.
32. Warth RJ, Dornan GJ, James EW, Horan MP, Millett PJ. Clinical and structural outcomes after arthroscopic repair of full-thickness rotator cuff tears with and without platelet-rich product supplementation: a meta-analysis and meta-regression. Arthroscopy. 2015;31(2):306-320.
33. Bergeson AG, Tashjian RZ, Greis PE, Crim J, Stoddard GJ, Burks RT. Effects of platelet-rich fibrin matrix on repair integrity of at-risk rotator cuff tears. Am J Sports Med. 2012;40(2):286-293.
34. Riboh JC, Saltzman BM, Yanke AB, Fortier L, Cole BJ. Effect of leukocyte concentration on the efficacy of platelet-rich plasma in the treatment of knee osteoarthritis. Am J Sports Med. 2016;44(3):792-800.
VIDEO: Balanced crystalloids protect kidney better than saline
TORONTO – Treatment with balanced crystalloid IV fluids cut adverse renal events modestly but with statistical significance, compared with 0.9% saline in hospitalized patients in a pair of single-center randomized trials with more than 29,000 total patients.
Despite showing a number needed to treat with balanced crystalloids of roughly 100 to prevent one major renal event, compared with saline, the scope of IV fluid use makes even this relatively small improvement potentially important to tens of thousands of patients annually.
“It’s a small but clinically important difference,” Wesley H. Self, MD, said at the CHEST annual meeting.
“These fluids are used every day and in millions of patients annually in the United States and worldwide. There is no functional cost difference between them, and now we have the data to show that [balanced crystalloid fluids] produce a better patient outcome. It’s reasonable to consider changing practice,” based on the results, said Matthew W. Semler, MD, a pulmonologist at Vanderbilt University Medical Center in Nashville, Tenn., who led one of the two trials.
At Vanderbilt, where the two studies ran, “we’ve changed our practice and are transitioning from primarily using saline to primarily balanced crystalloid,” Dr. Semler said in a video interview. The main limitation to changing practice now because of the results is that the two trials both ran at a single center.
The findings Dr. Semler reported came from the Isotonic Solutions and Major Adverse Renal Events Trial (SMART), which randomized 7,860 ICU patients to treatment with 0.9% saline fluid and 7,942 ICU patients to treatment with balanced crystalloid fluid, either lactated Ringer’s or Plasma-Lyte A. The study’s primary endpoint was the combined 30-day rate of in-hospital death, incident need for renal replacement therapy, or at least a doubling of the patient’s baseline creatinine level, a marker of persistent renal dysfunction.
This outcome occurred in 14.3% of patients on balanced crystalloid fluid and 15.4% on saline, a 1.1% statistically significant absolute difference. The endpoint components showed that patients treated with balanced crystalloid had 0.8% less in-hospital death and 0.4% less incident renal replacement therapy; both of these between-group differences were close to having statistical significance. The two treatment groups showed less difference in the rate of persistent renal dysfunction.
The second trial had an identical design but ran instead in the emergency department. The Saline Against Lactated Ringers or Plasmalyte in the Emergency Department (SALT-ED) trial randomized 6,708 to receive balanced crystalloid and 6,639 to receive saline. The combined primary renal endpoint was 0.9% less frequent with balanced crystalloid fluid, a statistically significant difference, Dr. Self, an emergency medicine physician at Vanderbilt, reported at the meeting. In this study the between-group differences for both incident renal replacement therapy and persistent renal dysfunction were statistically significant in favor of balanced crystalloid, but the between-group mortality difference was not significantly different.
The reason why balanced crystalloid fluid produced better renal outcomes than saline remains unclear. Both Dr. Semler and Dr. Self noted that the two balanced crystalloid fluids used in the study have chloride levels that closely match normal plasma levels, but the chloride concentration in 0.9% saline is about 50% higher than plasma. Some researchers have hypothesized, based on animal findings, that this difference may influence inflammation, blood pressure, acute kidney injury, and renal vasoconstriction.
The SMART and SALT-ED trials received no commercial funding. Dr. Semler had no disclosures. Dr. Self has been a consultant to Abbott Point of Care, BioTest, Cempra, Ferring, Gilead, and Pfizer.
[email protected]
On Twitter @mitchelzoler

Bennett P. deBoisblanc, MD , is professor of medicine at Louisiana State University Health and director of Critical Care Services at the Medical Center of Louisiana in New Orleans. He had no disclosures. He made these comments from the floor during discussion of the two reports.
Bennett P. deBoisblanc, MD , is professor of medicine at Louisiana State University Health and director of Critical Care Services at the Medical Center of Louisiana in New Orleans. He had no disclosures. He made these comments from the floor during discussion of the two reports.
Bennett P. deBoisblanc, MD , is professor of medicine at Louisiana State University Health and director of Critical Care Services at the Medical Center of Louisiana in New Orleans. He had no disclosures. He made these comments from the floor during discussion of the two reports.
TORONTO – Treatment with balanced crystalloid IV fluids cut adverse renal events modestly but with statistical significance, compared with 0.9% saline in hospitalized patients in a pair of single-center randomized trials with more than 29,000 total patients.
Despite showing a number needed to treat with balanced crystalloids of roughly 100 to prevent one major renal event, compared with saline, the scope of IV fluid use makes even this relatively small improvement potentially important to tens of thousands of patients annually.
“It’s a small but clinically important difference,” Wesley H. Self, MD, said at the CHEST annual meeting.
“These fluids are used every day and in millions of patients annually in the United States and worldwide. There is no functional cost difference between them, and now we have the data to show that [balanced crystalloid fluids] produce a better patient outcome. It’s reasonable to consider changing practice,” based on the results, said Matthew W. Semler, MD, a pulmonologist at Vanderbilt University Medical Center in Nashville, Tenn., who led one of the two trials.
At Vanderbilt, where the two studies ran, “we’ve changed our practice and are transitioning from primarily using saline to primarily balanced crystalloid,” Dr. Semler said in a video interview. The main limitation to changing practice now because of the results is that the two trials both ran at a single center.
The findings Dr. Semler reported came from the Isotonic Solutions and Major Adverse Renal Events Trial (SMART), which randomized 7,860 ICU patients to treatment with 0.9% saline fluid and 7,942 ICU patients to treatment with balanced crystalloid fluid, either lactated Ringer’s or Plasma-Lyte A. The study’s primary endpoint was the combined 30-day rate of in-hospital death, incident need for renal replacement therapy, or at least a doubling of the patient’s baseline creatinine level, a marker of persistent renal dysfunction.
This outcome occurred in 14.3% of patients on balanced crystalloid fluid and 15.4% on saline, a 1.1% statistically significant absolute difference. The endpoint components showed that patients treated with balanced crystalloid had 0.8% less in-hospital death and 0.4% less incident renal replacement therapy; both of these between-group differences were close to having statistical significance. The two treatment groups showed less difference in the rate of persistent renal dysfunction.
The second trial had an identical design but ran instead in the emergency department. The Saline Against Lactated Ringers or Plasmalyte in the Emergency Department (SALT-ED) trial randomized 6,708 to receive balanced crystalloid and 6,639 to receive saline. The combined primary renal endpoint was 0.9% less frequent with balanced crystalloid fluid, a statistically significant difference, Dr. Self, an emergency medicine physician at Vanderbilt, reported at the meeting. In this study the between-group differences for both incident renal replacement therapy and persistent renal dysfunction were statistically significant in favor of balanced crystalloid, but the between-group mortality difference was not significantly different.
The reason why balanced crystalloid fluid produced better renal outcomes than saline remains unclear. Both Dr. Semler and Dr. Self noted that the two balanced crystalloid fluids used in the study have chloride levels that closely match normal plasma levels, but the chloride concentration in 0.9% saline is about 50% higher than plasma. Some researchers have hypothesized, based on animal findings, that this difference may influence inflammation, blood pressure, acute kidney injury, and renal vasoconstriction.
The SMART and SALT-ED trials received no commercial funding. Dr. Semler had no disclosures. Dr. Self has been a consultant to Abbott Point of Care, BioTest, Cempra, Ferring, Gilead, and Pfizer.
[email protected]
On Twitter @mitchelzoler
TORONTO – Treatment with balanced crystalloid IV fluids cut adverse renal events modestly but with statistical significance, compared with 0.9% saline in hospitalized patients in a pair of single-center randomized trials with more than 29,000 total patients.
Despite showing a number needed to treat with balanced crystalloids of roughly 100 to prevent one major renal event, compared with saline, the scope of IV fluid use makes even this relatively small improvement potentially important to tens of thousands of patients annually.
“It’s a small but clinically important difference,” Wesley H. Self, MD, said at the CHEST annual meeting.
“These fluids are used every day and in millions of patients annually in the United States and worldwide. There is no functional cost difference between them, and now we have the data to show that [balanced crystalloid fluids] produce a better patient outcome. It’s reasonable to consider changing practice,” based on the results, said Matthew W. Semler, MD, a pulmonologist at Vanderbilt University Medical Center in Nashville, Tenn., who led one of the two trials.
At Vanderbilt, where the two studies ran, “we’ve changed our practice and are transitioning from primarily using saline to primarily balanced crystalloid,” Dr. Semler said in a video interview. The main limitation to changing practice now because of the results is that the two trials both ran at a single center.
The findings Dr. Semler reported came from the Isotonic Solutions and Major Adverse Renal Events Trial (SMART), which randomized 7,860 ICU patients to treatment with 0.9% saline fluid and 7,942 ICU patients to treatment with balanced crystalloid fluid, either lactated Ringer’s or Plasma-Lyte A. The study’s primary endpoint was the combined 30-day rate of in-hospital death, incident need for renal replacement therapy, or at least a doubling of the patient’s baseline creatinine level, a marker of persistent renal dysfunction.
This outcome occurred in 14.3% of patients on balanced crystalloid fluid and 15.4% on saline, a 1.1% statistically significant absolute difference. The endpoint components showed that patients treated with balanced crystalloid had 0.8% less in-hospital death and 0.4% less incident renal replacement therapy; both of these between-group differences were close to having statistical significance. The two treatment groups showed less difference in the rate of persistent renal dysfunction.
The second trial had an identical design but ran instead in the emergency department. The Saline Against Lactated Ringers or Plasmalyte in the Emergency Department (SALT-ED) trial randomized 6,708 to receive balanced crystalloid and 6,639 to receive saline. The combined primary renal endpoint was 0.9% less frequent with balanced crystalloid fluid, a statistically significant difference, Dr. Self, an emergency medicine physician at Vanderbilt, reported at the meeting. In this study the between-group differences for both incident renal replacement therapy and persistent renal dysfunction were statistically significant in favor of balanced crystalloid, but the between-group mortality difference was not significantly different.
The reason why balanced crystalloid fluid produced better renal outcomes than saline remains unclear. Both Dr. Semler and Dr. Self noted that the two balanced crystalloid fluids used in the study have chloride levels that closely match normal plasma levels, but the chloride concentration in 0.9% saline is about 50% higher than plasma. Some researchers have hypothesized, based on animal findings, that this difference may influence inflammation, blood pressure, acute kidney injury, and renal vasoconstriction.
The SMART and SALT-ED trials received no commercial funding. Dr. Semler had no disclosures. Dr. Self has been a consultant to Abbott Point of Care, BioTest, Cempra, Ferring, Gilead, and Pfizer.
[email protected]
On Twitter @mitchelzoler
AT CHEST 2017
Key clinical point:
Major finding: Balanced crystalloids reduced combined adverse renal events by 1.1% in ICU patients and 0.9% in ED patients.
Data source: The SMART and SALT-ED trials, both single-center studies with a total of 29,149 patients.
Disclosures: The SMART and SALT-ED trials received no commercial funding. Dr. Semler had no disclosures. Dr. Self has been a consultant to Abbott Point of Care, BioTest, Cempra, Ferring, Gilead, and Pfizer.

Atypical Herpes Zoster Presentation in a Healthy Vaccinated Pediatric Patient
Varicella-zoster virus (VZV) is a neurotropic human herpesvirus that causes varicella (chicken pox) and herpes zoster (shingles). During infection, the virus invades the dorsal root ganglia and establishes permanent latency. It can later reactivate and travel through sensory nerves to the skin where localized viral replication causes herpes zoster (HZ), which manifests with pain in a unilateral dermatomal distribution followed closely by an eruption of grouped macules and papules that evolve into vesicles on an erythematous base.1 These lesions form pustules and crusts over 7 to 10 days and heal completely within 4 weeks. Although postherpetic neuralgia is rare in children, the pain associated with HZ can last months or years.1,2
Universal childhood vaccination against VZV has existed in the United States since 1995, with a 2-dose vaccine regimen recommended by the CDC since 2007. Consequently, primary varicella infection in children is uncommon, and the majority of cases now occur in the vaccinated population.3 However, breakthrough varicella infection and postvaccination HZ are rare due to the long-lasting immunity and low virulence of the attenuated vaccine strain. We recount the case of a 6-year-old vaccinated girl with a unique presentation of HZ with no known primary varicella infection.
Case Report
A healthy 6-year-old girl presented with a stabbing burning pain in the left thigh extending down the calf of 4 days’ duration. The intense pain made walking difficult and responded minimally to ibuprofen and naproxen. Poor appetite, nausea, colicky abdominal pain, and fever (temperature, 38°C) accompanied the pain. Three days after the pain began she developed a pruritic rash on the same leg. Notably, she reported falling on a rosebush and sustaining a thorn prick in the left thigh 3 days prior to the onset of pain. Before presenting to our dermatology clinic, she was seen by a pediatrician, an emergency department physician, and an infectious disease specialist. The initial workup included a complete blood cell count, C-reactive protein test, erythrocyte sedimentation rate test, and hip and femur radiograph, which were all unremarkable. She was referred to dermatology with a differential diagnosis of sporotrichosis, contact dermatitis, reactive arthritis, viral myalgia, and Legg-Calvé-Perthes disease.
Physical examination revealed a well-appearing child with pink eczematous patches and plaques extending from the left side of the lower back to the mid shin in an L5 distribution (Figure). The left thigh was tender to palpation, and nontender left inguinal lymphadenopathy was present. A single isolated 2-mm vesicle was found on the anterior aspect of the left lower leg. Direct fluorescent antibody testing of vesicle fluid was positive for VZV antigen, confirming the diagnosis of HZ.
The patient’s mother confirmed that she had no obvious history of VZV. She had received VZV vaccinations in the left leg and arm at 1 and 4 years of age, respectively. She was treated with acyclovir (80 mg/kg daily at 6-hour intervals for 5 days) with immediate improvement in symptoms and resolution of the rash by day 5 of treatment. She experienced intermittent burning pain in the leg throughout the course of treatment, which resolved shortly thereafter.
Comment
Herpes zoster is rare in young healthy children, and its incidence has decreased since the introduction of universal varicella vaccination.4 Reported incidence rates in vaccinated children vary from approximately 15 to 93 per 100,000 person-years,5,6 and the reported relative risk is 0.08 to 0.36 in vaccinated compared to unvaccinated children.6,7 No correlations with gender, race, or ethnicity and postvaccination HZ have been observed.5,8 Reported intervals between vaccination and HZ presentation are as short as 3 months and as long as 11 years.9 Although HZ is uncommon in immunocompetent children, the diagnosis of HZ itself is not an indication for formal workup for an underlying immunodeficiency or malignancy.10
Both wild-type and vaccine-strain VZV establish latent infection and can cause HZ in vaccinated children. Direct fluorescent antibody testing or polymerase chain reaction of HZ lesions can be used to identify VZV. Genotyping can distinguish the wild-type versus the vaccine strain but is not required for clinical management.3 In previously vaccinated children with HZ, approximately half present with wild-type and half with vaccine-strain VZV. In approximately half of wild-type cases, prior clinical varicella infection also occurred.8
Regardless of virus strain, vaccinated children typically present with the characteristic painful, vesicular, dermatomal HZ rash.8,9 This presentation can be milder with less pain and fewer vesicles than with unvaccinated cases.6 When vaccine-strain HZ occurs, the rash often presents at or near the site of initial vaccination, which typically is the arm or thigh.3,4,6,9 The vaccine strain has lower virulence than the wild-type virus. Eight cases of vaccine-strain zoster severe enough to cause neurological complications such as meningitis or encephalitis have been reported in children, with 6 cases reported in healthy children.9,11-17 Antiviral drugs hasten the healing of the HZ rash and shorten the duration of associated pain.1
Although pediatric HZ is uncommon, all physicians should be aware of possible atypical presentations in healthy vaccinated children to appropriately and quickly manage treatment.
- Sampathkumar P, Drage LA, Martin DP. Herpes zoster (shingles) and postherpetic neuralgia. Mayo Clin Proc. 2009;84:274-280.
- Hillebrand K, Bricout H, Schulze-Rath R, et al. Incidence of herpes zoster and its complications in Germany, 2005-2009. J Infect. 2015;70:178-186.
- Lopez A, Schmid S, Bialek S. Varicella. In: Centers for Disease Control and Prevention. Manual for the Surveillance of Vaccine-Preventable Diseases. 5th ed. 2011:1-16.
- Tanuseputroa P, Zagorskia B, Chanc KJ, et al. Population-based incidence of herpes zoster after introduction of a publicly funded varicella vaccination program. Vaccine. 2011;29:8580- 8584.
- Wen SY, Liu WL. Epidemiology of pediatric herpes zoster after varicella infection: a population-based study. Pediatrics. 2015;135:565-571.
- Civen R, Chaves SS, Jumaan A, et al. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
- Stein M, Cohen R, Bromberg M, et al. Herpes zoster in a partially vaccinated pediatric population in Central Israel. Pediatr Infect Dis J. 2012;31:906-909.
- Weinmann S, Chun C, Schmid DS, et al. Incidence and clinical characteristics of herpes zoster among children in the varicella vaccine era, 2005-2009. J Infect Dis. 2013;208:1859-1868.
- Horien C, Grose C. Neurovirulence of varicella and the live attenuated varicella vaccine virus. Semin Pediatr Neurol. 2012;19:124-129.
- Petursson G, Helgason S, Gudmundsson S, et al. Herpes zoster in children and adolescents. Pediatr Infect Dis J. 1998;17:905-908.
- Levin MJ, Dahl KM, Weinberg A, et al. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus in an immunosuppressed child. J Infect Dis. 2003;188:954-959.
- Chaves SS, Haber P, Walton K, et al. Safety of varicella vaccine after licensure in the United States: experience from reports to the vaccine adverse event reporting system, 1995-2005. J Infect Dis. 2008;197(suppl 2):S170-S177.
- Levin MJ, DeBiasi RL, Bostik V, et al. Herpes zoster with skin lesions and meningitis caused by 2 different genotypes of the Oka varicella zoster virus vaccine. J Infect Dis. 2008;198:1444-1447.
- Iyer S, Mittal MK, Hodinka RL. Herpes zoster and meningitis resulting from reactivation of varicella vaccine virus in an immunocompetent child. Ann Emerg Med. 2009;53:792-795.
- Chouliaras G, Spoulou V, Quinlivan M, et al. Vaccine-associated herpes zoster ophthalmicus and encephalitis in an immunocompetent child. Pediatrics. 2010;125:e969-e972.
- Pahud BA, Glaser CA, Dekker CL, et al. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis. 2011;203:316-323.
- Han JY, Hanson DC, Way SS. Herpes zoster and meningitis due to reactivation of varicella vaccine virus in an immunocompetent child. Pediatr Infect Dis J. 2011;30:266-268.
Varicella-zoster virus (VZV) is a neurotropic human herpesvirus that causes varicella (chicken pox) and herpes zoster (shingles). During infection, the virus invades the dorsal root ganglia and establishes permanent latency. It can later reactivate and travel through sensory nerves to the skin where localized viral replication causes herpes zoster (HZ), which manifests with pain in a unilateral dermatomal distribution followed closely by an eruption of grouped macules and papules that evolve into vesicles on an erythematous base.1 These lesions form pustules and crusts over 7 to 10 days and heal completely within 4 weeks. Although postherpetic neuralgia is rare in children, the pain associated with HZ can last months or years.1,2
Universal childhood vaccination against VZV has existed in the United States since 1995, with a 2-dose vaccine regimen recommended by the CDC since 2007. Consequently, primary varicella infection in children is uncommon, and the majority of cases now occur in the vaccinated population.3 However, breakthrough varicella infection and postvaccination HZ are rare due to the long-lasting immunity and low virulence of the attenuated vaccine strain. We recount the case of a 6-year-old vaccinated girl with a unique presentation of HZ with no known primary varicella infection.
Case Report
A healthy 6-year-old girl presented with a stabbing burning pain in the left thigh extending down the calf of 4 days’ duration. The intense pain made walking difficult and responded minimally to ibuprofen and naproxen. Poor appetite, nausea, colicky abdominal pain, and fever (temperature, 38°C) accompanied the pain. Three days after the pain began she developed a pruritic rash on the same leg. Notably, she reported falling on a rosebush and sustaining a thorn prick in the left thigh 3 days prior to the onset of pain. Before presenting to our dermatology clinic, she was seen by a pediatrician, an emergency department physician, and an infectious disease specialist. The initial workup included a complete blood cell count, C-reactive protein test, erythrocyte sedimentation rate test, and hip and femur radiograph, which were all unremarkable. She was referred to dermatology with a differential diagnosis of sporotrichosis, contact dermatitis, reactive arthritis, viral myalgia, and Legg-Calvé-Perthes disease.
Physical examination revealed a well-appearing child with pink eczematous patches and plaques extending from the left side of the lower back to the mid shin in an L5 distribution (Figure). The left thigh was tender to palpation, and nontender left inguinal lymphadenopathy was present. A single isolated 2-mm vesicle was found on the anterior aspect of the left lower leg. Direct fluorescent antibody testing of vesicle fluid was positive for VZV antigen, confirming the diagnosis of HZ.
The patient’s mother confirmed that she had no obvious history of VZV. She had received VZV vaccinations in the left leg and arm at 1 and 4 years of age, respectively. She was treated with acyclovir (80 mg/kg daily at 6-hour intervals for 5 days) with immediate improvement in symptoms and resolution of the rash by day 5 of treatment. She experienced intermittent burning pain in the leg throughout the course of treatment, which resolved shortly thereafter.
Comment
Herpes zoster is rare in young healthy children, and its incidence has decreased since the introduction of universal varicella vaccination.4 Reported incidence rates in vaccinated children vary from approximately 15 to 93 per 100,000 person-years,5,6 and the reported relative risk is 0.08 to 0.36 in vaccinated compared to unvaccinated children.6,7 No correlations with gender, race, or ethnicity and postvaccination HZ have been observed.5,8 Reported intervals between vaccination and HZ presentation are as short as 3 months and as long as 11 years.9 Although HZ is uncommon in immunocompetent children, the diagnosis of HZ itself is not an indication for formal workup for an underlying immunodeficiency or malignancy.10
Both wild-type and vaccine-strain VZV establish latent infection and can cause HZ in vaccinated children. Direct fluorescent antibody testing or polymerase chain reaction of HZ lesions can be used to identify VZV. Genotyping can distinguish the wild-type versus the vaccine strain but is not required for clinical management.3 In previously vaccinated children with HZ, approximately half present with wild-type and half with vaccine-strain VZV. In approximately half of wild-type cases, prior clinical varicella infection also occurred.8
Regardless of virus strain, vaccinated children typically present with the characteristic painful, vesicular, dermatomal HZ rash.8,9 This presentation can be milder with less pain and fewer vesicles than with unvaccinated cases.6 When vaccine-strain HZ occurs, the rash often presents at or near the site of initial vaccination, which typically is the arm or thigh.3,4,6,9 The vaccine strain has lower virulence than the wild-type virus. Eight cases of vaccine-strain zoster severe enough to cause neurological complications such as meningitis or encephalitis have been reported in children, with 6 cases reported in healthy children.9,11-17 Antiviral drugs hasten the healing of the HZ rash and shorten the duration of associated pain.1
Although pediatric HZ is uncommon, all physicians should be aware of possible atypical presentations in healthy vaccinated children to appropriately and quickly manage treatment.
Varicella-zoster virus (VZV) is a neurotropic human herpesvirus that causes varicella (chicken pox) and herpes zoster (shingles). During infection, the virus invades the dorsal root ganglia and establishes permanent latency. It can later reactivate and travel through sensory nerves to the skin where localized viral replication causes herpes zoster (HZ), which manifests with pain in a unilateral dermatomal distribution followed closely by an eruption of grouped macules and papules that evolve into vesicles on an erythematous base.1 These lesions form pustules and crusts over 7 to 10 days and heal completely within 4 weeks. Although postherpetic neuralgia is rare in children, the pain associated with HZ can last months or years.1,2
Universal childhood vaccination against VZV has existed in the United States since 1995, with a 2-dose vaccine regimen recommended by the CDC since 2007. Consequently, primary varicella infection in children is uncommon, and the majority of cases now occur in the vaccinated population.3 However, breakthrough varicella infection and postvaccination HZ are rare due to the long-lasting immunity and low virulence of the attenuated vaccine strain. We recount the case of a 6-year-old vaccinated girl with a unique presentation of HZ with no known primary varicella infection.
Case Report
A healthy 6-year-old girl presented with a stabbing burning pain in the left thigh extending down the calf of 4 days’ duration. The intense pain made walking difficult and responded minimally to ibuprofen and naproxen. Poor appetite, nausea, colicky abdominal pain, and fever (temperature, 38°C) accompanied the pain. Three days after the pain began she developed a pruritic rash on the same leg. Notably, she reported falling on a rosebush and sustaining a thorn prick in the left thigh 3 days prior to the onset of pain. Before presenting to our dermatology clinic, she was seen by a pediatrician, an emergency department physician, and an infectious disease specialist. The initial workup included a complete blood cell count, C-reactive protein test, erythrocyte sedimentation rate test, and hip and femur radiograph, which were all unremarkable. She was referred to dermatology with a differential diagnosis of sporotrichosis, contact dermatitis, reactive arthritis, viral myalgia, and Legg-Calvé-Perthes disease.
Physical examination revealed a well-appearing child with pink eczematous patches and plaques extending from the left side of the lower back to the mid shin in an L5 distribution (Figure). The left thigh was tender to palpation, and nontender left inguinal lymphadenopathy was present. A single isolated 2-mm vesicle was found on the anterior aspect of the left lower leg. Direct fluorescent antibody testing of vesicle fluid was positive for VZV antigen, confirming the diagnosis of HZ.
The patient’s mother confirmed that she had no obvious history of VZV. She had received VZV vaccinations in the left leg and arm at 1 and 4 years of age, respectively. She was treated with acyclovir (80 mg/kg daily at 6-hour intervals for 5 days) with immediate improvement in symptoms and resolution of the rash by day 5 of treatment. She experienced intermittent burning pain in the leg throughout the course of treatment, which resolved shortly thereafter.
Comment
Herpes zoster is rare in young healthy children, and its incidence has decreased since the introduction of universal varicella vaccination.4 Reported incidence rates in vaccinated children vary from approximately 15 to 93 per 100,000 person-years,5,6 and the reported relative risk is 0.08 to 0.36 in vaccinated compared to unvaccinated children.6,7 No correlations with gender, race, or ethnicity and postvaccination HZ have been observed.5,8 Reported intervals between vaccination and HZ presentation are as short as 3 months and as long as 11 years.9 Although HZ is uncommon in immunocompetent children, the diagnosis of HZ itself is not an indication for formal workup for an underlying immunodeficiency or malignancy.10
Both wild-type and vaccine-strain VZV establish latent infection and can cause HZ in vaccinated children. Direct fluorescent antibody testing or polymerase chain reaction of HZ lesions can be used to identify VZV. Genotyping can distinguish the wild-type versus the vaccine strain but is not required for clinical management.3 In previously vaccinated children with HZ, approximately half present with wild-type and half with vaccine-strain VZV. In approximately half of wild-type cases, prior clinical varicella infection also occurred.8
Regardless of virus strain, vaccinated children typically present with the characteristic painful, vesicular, dermatomal HZ rash.8,9 This presentation can be milder with less pain and fewer vesicles than with unvaccinated cases.6 When vaccine-strain HZ occurs, the rash often presents at or near the site of initial vaccination, which typically is the arm or thigh.3,4,6,9 The vaccine strain has lower virulence than the wild-type virus. Eight cases of vaccine-strain zoster severe enough to cause neurological complications such as meningitis or encephalitis have been reported in children, with 6 cases reported in healthy children.9,11-17 Antiviral drugs hasten the healing of the HZ rash and shorten the duration of associated pain.1
Although pediatric HZ is uncommon, all physicians should be aware of possible atypical presentations in healthy vaccinated children to appropriately and quickly manage treatment.
- Sampathkumar P, Drage LA, Martin DP. Herpes zoster (shingles) and postherpetic neuralgia. Mayo Clin Proc. 2009;84:274-280.
- Hillebrand K, Bricout H, Schulze-Rath R, et al. Incidence of herpes zoster and its complications in Germany, 2005-2009. J Infect. 2015;70:178-186.
- Lopez A, Schmid S, Bialek S. Varicella. In: Centers for Disease Control and Prevention. Manual for the Surveillance of Vaccine-Preventable Diseases. 5th ed. 2011:1-16.
- Tanuseputroa P, Zagorskia B, Chanc KJ, et al. Population-based incidence of herpes zoster after introduction of a publicly funded varicella vaccination program. Vaccine. 2011;29:8580- 8584.
- Wen SY, Liu WL. Epidemiology of pediatric herpes zoster after varicella infection: a population-based study. Pediatrics. 2015;135:565-571.
- Civen R, Chaves SS, Jumaan A, et al. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
- Stein M, Cohen R, Bromberg M, et al. Herpes zoster in a partially vaccinated pediatric population in Central Israel. Pediatr Infect Dis J. 2012;31:906-909.
- Weinmann S, Chun C, Schmid DS, et al. Incidence and clinical characteristics of herpes zoster among children in the varicella vaccine era, 2005-2009. J Infect Dis. 2013;208:1859-1868.
- Horien C, Grose C. Neurovirulence of varicella and the live attenuated varicella vaccine virus. Semin Pediatr Neurol. 2012;19:124-129.
- Petursson G, Helgason S, Gudmundsson S, et al. Herpes zoster in children and adolescents. Pediatr Infect Dis J. 1998;17:905-908.
- Levin MJ, Dahl KM, Weinberg A, et al. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus in an immunosuppressed child. J Infect Dis. 2003;188:954-959.
- Chaves SS, Haber P, Walton K, et al. Safety of varicella vaccine after licensure in the United States: experience from reports to the vaccine adverse event reporting system, 1995-2005. J Infect Dis. 2008;197(suppl 2):S170-S177.
- Levin MJ, DeBiasi RL, Bostik V, et al. Herpes zoster with skin lesions and meningitis caused by 2 different genotypes of the Oka varicella zoster virus vaccine. J Infect Dis. 2008;198:1444-1447.
- Iyer S, Mittal MK, Hodinka RL. Herpes zoster and meningitis resulting from reactivation of varicella vaccine virus in an immunocompetent child. Ann Emerg Med. 2009;53:792-795.
- Chouliaras G, Spoulou V, Quinlivan M, et al. Vaccine-associated herpes zoster ophthalmicus and encephalitis in an immunocompetent child. Pediatrics. 2010;125:e969-e972.
- Pahud BA, Glaser CA, Dekker CL, et al. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis. 2011;203:316-323.
- Han JY, Hanson DC, Way SS. Herpes zoster and meningitis due to reactivation of varicella vaccine virus in an immunocompetent child. Pediatr Infect Dis J. 2011;30:266-268.
- Sampathkumar P, Drage LA, Martin DP. Herpes zoster (shingles) and postherpetic neuralgia. Mayo Clin Proc. 2009;84:274-280.
- Hillebrand K, Bricout H, Schulze-Rath R, et al. Incidence of herpes zoster and its complications in Germany, 2005-2009. J Infect. 2015;70:178-186.
- Lopez A, Schmid S, Bialek S. Varicella. In: Centers for Disease Control and Prevention. Manual for the Surveillance of Vaccine-Preventable Diseases. 5th ed. 2011:1-16.
- Tanuseputroa P, Zagorskia B, Chanc KJ, et al. Population-based incidence of herpes zoster after introduction of a publicly funded varicella vaccination program. Vaccine. 2011;29:8580- 8584.
- Wen SY, Liu WL. Epidemiology of pediatric herpes zoster after varicella infection: a population-based study. Pediatrics. 2015;135:565-571.
- Civen R, Chaves SS, Jumaan A, et al. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
- Stein M, Cohen R, Bromberg M, et al. Herpes zoster in a partially vaccinated pediatric population in Central Israel. Pediatr Infect Dis J. 2012;31:906-909.
- Weinmann S, Chun C, Schmid DS, et al. Incidence and clinical characteristics of herpes zoster among children in the varicella vaccine era, 2005-2009. J Infect Dis. 2013;208:1859-1868.
- Horien C, Grose C. Neurovirulence of varicella and the live attenuated varicella vaccine virus. Semin Pediatr Neurol. 2012;19:124-129.
- Petursson G, Helgason S, Gudmundsson S, et al. Herpes zoster in children and adolescents. Pediatr Infect Dis J. 1998;17:905-908.
- Levin MJ, Dahl KM, Weinberg A, et al. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus in an immunosuppressed child. J Infect Dis. 2003;188:954-959.
- Chaves SS, Haber P, Walton K, et al. Safety of varicella vaccine after licensure in the United States: experience from reports to the vaccine adverse event reporting system, 1995-2005. J Infect Dis. 2008;197(suppl 2):S170-S177.
- Levin MJ, DeBiasi RL, Bostik V, et al. Herpes zoster with skin lesions and meningitis caused by 2 different genotypes of the Oka varicella zoster virus vaccine. J Infect Dis. 2008;198:1444-1447.
- Iyer S, Mittal MK, Hodinka RL. Herpes zoster and meningitis resulting from reactivation of varicella vaccine virus in an immunocompetent child. Ann Emerg Med. 2009;53:792-795.
- Chouliaras G, Spoulou V, Quinlivan M, et al. Vaccine-associated herpes zoster ophthalmicus and encephalitis in an immunocompetent child. Pediatrics. 2010;125:e969-e972.
- Pahud BA, Glaser CA, Dekker CL, et al. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis. 2011;203:316-323.
- Han JY, Hanson DC, Way SS. Herpes zoster and meningitis due to reactivation of varicella vaccine virus in an immunocompetent child. Pediatr Infect Dis J. 2011;30:266-268.
Practice Points
- Both wild-type and vaccine-strain varicella-zoster virus (VZV) can establish latency in dorsal root ganglia and can cause herpes zoster (HZ) in vaccinated children.
- When HZ due to a vaccine strain of VZV occurs, the rash often presents near the site of initial vaccination.
- Although most cases of HZ in vaccinated children present with a characteristic HZ rash, physicians should be aware of the possibility for atypical presentations.
Review of plant phenolics, part 3: Nonflavonoid compounds
Polyphenols are widely distributed in the plant kingdom, and are found in copious supply in multiple vegetables, fruits, herbs, grains, tea, coffee beans, honey, and red wine, for example. They are an especially important source of antioxidants and are increasingly the focus of research due to their potent and diverse biologic activities. In the conclusion to my three-part review of polyphenols, this column identifies representative compounds from the classes of nonflavonoid polyphenols and provides a brief update on research.

Phenolic acids: ferulic acid
Derived from curcumin, ferulic acid is noted for exhibiting multiple biologic activities, including antiapoptotic, anticarcinogenic, antidiabetic, hepatoprotective, and cardioprotective, among others. Its beneficial effects are thought to be mediated through its antioxidant and anti-inflammatory characteristics.1 In a small 2008 study, a stable formulation of 15% l-ascorbic acid, 1% alpha-tocopherol, and 0.5% ferulic acid was applied topically to normal-appearing human skin for 4 days and was found to impart significant photoprotection against solar-simulated UV radiation and was especially effective at diminishing thymine dimer mutations, which are linked to skin cancer. The authors also noted that the mechanism of action of this antioxidant formulation differs from that of sunscreens and, therefore, may serve as a supplement to such products.2 (It is worth noting that ferulic acid has been approved as a sunscreen agent in Japan.3)
In 2015, Ambothi et al. used Swiss albino mice to assess the photochemopreventive effects of ferulic acid against chronic (30-week) UVB, finding the intraperitoneal and topical administration of the phenolic acid effective in significantly lowering the incidence of UVB-induced tumor volume and weight in the mice skin.4 The next year, Hahn et al. reported that pretreatment with ferulic acid protects human dermal fibroblasts from UVA-induced photodamage.5 Also in 2016, Chaiprasongsuk et al. found that several dietary phenolics, including ferulic acid, deliver protection against UVA-induced melanogenesis through indirect regulation of the Nrf2-ARE pathway.6 
Lignans: flaxseed
Flaxseed lignans, which exhibit a wide range of biologic activities, are best known for their antioxidant properties.7 In a 2017 study using atopic dermatitis–induced NC/Nga mice, Yang et al. found that fermented flaxseed oil administered orally was successful in relieving symptoms such as erythema, edema, pruritus, and epithelial damage.8 Two years earlier, Draganescu et al. developed a topical flaxseed extract formulation that displayed wound healing capabilities on Wistar rats.7 Emulsions produced from the oils and seeds of transgenic flax have also been found to protect against oxidative stress in hamster fibroblasts, with investigators suggesting that the emulsions have potential to protect the skin against such damage.8
Stilbenes: resveratrol
The antioxidant potency of resveratrol has been cited for conferring a wide range of salutary effects, including antitumorigenic as well as antiaging activity. In 2008, a resveratrol-based skin care formulation intended to combat photoaging was reported to exhibit 17-fold greater antioxidant activity than idebenone.9 In a different study that year, resveratrol, the primary active polyphenolic constituent in red wine, was assessed in terms of topical/transdermal delivery viability, given previously established benefits shown via systemic administration. Several hydrogel systems used as resveratrol vehicles were shown to be safe and effective methods for cutaneously delivering the therapeutic effects of this antioxidant.10 Since then, resveratrol has been demonstrated to penetrate the skin via topical administration, reinforcing the antioxidant system of the stratum corneum and delivering increases of antioxidants to human epidermal tissue.11
In 2014, Farris et al. showed that a proprietary topical antioxidant blend of resveratrol, baicalin, and vitamin E applied topically at night yielded statistically significant amelioration of fine lines and wrinkles, as well as skin firmness, elasticity, laxity, hyperpigmentation, radiance, and roughness over a 12-week period.12 Resveratrol has also been shown in mice to suppress the inflammatory response and improve survival from severe burns with bacterial infections.13 
Hydrolyzable tannins: ellagic acid
Ellagic acid, a dimer of gallic acid, has been reported to impart anti-inflammatory, antitumor, immunomodulatory, and antifungal activities.14-16 Ortiz-Ruiz et al. have noted that while ellagic acid is used as a whitening agent, it can act as a substrate to rather than an inhibitor of tyrosinase, as it is oxidized by the enzyme to an unstable o-quinone. However, as a potent antioxidant, ellagic acid can block melanogenesis by reducing o-quinones and semiquinones.17
In a double-blind, placebo-controlled, 4-week trial to assess the effects of orally administered ellagic acid–rich pomegranate extract on the pigmentation of 13 women after UV exposure, with healthy volunteers randomly assigned to high-dose, low-dose, and control groups, luminance values decreased by 1.73% in the high-dose group and 1.35% in the low-dose group, as compared with the control group, and stains and freckles were reported to be diminished.18 A 2016 study in human dermal fibroblasts by Baek et al. suggested that ellagic acid displays antiphotoaging activity, as the polyphenol protected against UVB-induced oxidative stress potentially through an Nrf2-dependent pathway.15
Condensed tannins (Proanthocyanidins): pycnogenol
Pycnogenol has been used in an antioxidant mixture also including vitamins C and E, as well as evening primrose that when orally administered for 10 weeks to female SKH-1 hairless mice exposed three times weekly to UVB irradiation demonstrated the capacity to significantly inhibit wrinkle formation by markedly suppressing UVB-induced MMP activity while promoting collagen production.19 In a 2012 study of 112 women with mild to moderate photoaging, orally administered pycnogenol was shown to yield significant reductions in clinical grading of skin photoaging scores.20 Four years later, a review by Grether-Beck et al. suggested that oral administration of pycnogenol imparts photoprotection, diminishes hyperpigmentation, and improves skin barrier function and the stability of the extracellular matrix.21
Lignins: various woody plants
Recognized as efficient natural scavengers of reactive oxygen species, lignins are complex phenolic polymers that are abundant in nature, particularly in various tree species and agricultural products. In 2004, Dizhbite et al. isolated lignin samples from deciduous and coniferous trees to assess their capacity as natural antioxidants. Samples were assessed against the 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical in homogeneous conditions, with the commercially available kraft lignin noted for displaying antibacterial activity associated with its radical-scavenging properties.22 Four years later, Ugartondo et al. studied several lignins and reported a strong antioxidant capacity at various concentrations that were innocuous to normal human cells and stable when exposed to UVA. The investigators concluded that lignins may be viable for inclusion in cosmetic and topical medical formulations.23
Conclusion
A brief survey of the polyphenolic landscape obviously cannot do the subject justice. From the dermatologic perspective, this diverse family of compounds factor into the skin care formulations becoming more prevalent in the established armamentarium, as well as the direct-to-consumer market. Given the increasing attention paid here and elsewhere to the impact of diet on the skin, the status of this dynamic class of polyphenolic compounds, which includes several antioxidants and is found in numerous plants, appears to be well deserved and warrants much more research.
Dr. Baumann is a private practice dermatologist, researcher, author and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and “Cosmeceuticals and Cosmetic Ingredients,” (New York: McGraw-Hill, 2014), and a New York Times Best Sellers book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance. She is the founder and CEO of Skin Type Solutions Franchise Systems LLC.
References
1. Food Chem Toxicol. 2017 May;103:41-55.
2. J Am Acad Dermatol. 2008 Sep;59(3):418-25.
3. J Pharm Biomed Anal. 2008 Mar 13;46(4):645-52.
4. Food Chem Toxicol. 2015 Aug;82:72-8.
5. Ann Dermatol. 2016 Dec;28(6):740-8.
6. Redox Biol. 2016 Aug;8:79-90.
7. Int J Biol Macromol. 2015 Jan;72:614-23.
8. Evid Based Complement Alternat Med. 2017;2017:5469125.
9. J Cosmet Dermatol. 2008 Mar;7(1):2-7.
10. Biol Pharm Bull. 2008 May;31(5):955-62.
11. Arch Dermatol Res. 2017 Aug;309(6):423-31.
12. J Drugs Dermatol. 2014 Dec;13(12):1467-72.
13. Inflammation. 2015;38(3):1273-80.
14. Dermatol Ther. 2012 May-Jun;25(3):252-9.
15. Korean J Physiol Pharmacol. 2016 May;20(3):269-77.
16. Phytother Res. 2015 Jul;29(7):1019-25.
17. J Dermatol Sci. 2016 May;82(2):115-22.
18. J Nutr Sci Vitaminol (Tokyo). 2006 Oct;52(5):383-8.
19. Photodermatol Photoimmunol Photomed. 2007 Oct;23(5):155-62.
20. Clin Interv Aging. 2012;7:275-86.
21. Skin Pharmacol Physiol. 2016;29(1):13-7.
22. Bioresour Technol. 2004 Dec;95(3):309-17.
23. Bioresour Technol. 2008 Sep;99(14):6683-7.
Polyphenols are widely distributed in the plant kingdom, and are found in copious supply in multiple vegetables, fruits, herbs, grains, tea, coffee beans, honey, and red wine, for example. They are an especially important source of antioxidants and are increasingly the focus of research due to their potent and diverse biologic activities. In the conclusion to my three-part review of polyphenols, this column identifies representative compounds from the classes of nonflavonoid polyphenols and provides a brief update on research.
Phenolic acids: ferulic acid
Derived from curcumin, ferulic acid is noted for exhibiting multiple biologic activities, including antiapoptotic, anticarcinogenic, antidiabetic, hepatoprotective, and cardioprotective, among others. Its beneficial effects are thought to be mediated through its antioxidant and anti-inflammatory characteristics.1 In a small 2008 study, a stable formulation of 15% l-ascorbic acid, 1% alpha-tocopherol, and 0.5% ferulic acid was applied topically to normal-appearing human skin for 4 days and was found to impart significant photoprotection against solar-simulated UV radiation and was especially effective at diminishing thymine dimer mutations, which are linked to skin cancer. The authors also noted that the mechanism of action of this antioxidant formulation differs from that of sunscreens and, therefore, may serve as a supplement to such products.2 (It is worth noting that ferulic acid has been approved as a sunscreen agent in Japan.3)
In 2015, Ambothi et al. used Swiss albino mice to assess the photochemopreventive effects of ferulic acid against chronic (30-week) UVB, finding the intraperitoneal and topical administration of the phenolic acid effective in significantly lowering the incidence of UVB-induced tumor volume and weight in the mice skin.4 The next year, Hahn et al. reported that pretreatment with ferulic acid protects human dermal fibroblasts from UVA-induced photodamage.5 Also in 2016, Chaiprasongsuk et al. found that several dietary phenolics, including ferulic acid, deliver protection against UVA-induced melanogenesis through indirect regulation of the Nrf2-ARE pathway.6
Lignans: flaxseed
Flaxseed lignans, which exhibit a wide range of biologic activities, are best known for their antioxidant properties.7 In a 2017 study using atopic dermatitis–induced NC/Nga mice, Yang et al. found that fermented flaxseed oil administered orally was successful in relieving symptoms such as erythema, edema, pruritus, and epithelial damage.8 Two years earlier, Draganescu et al. developed a topical flaxseed extract formulation that displayed wound healing capabilities on Wistar rats.7 Emulsions produced from the oils and seeds of transgenic flax have also been found to protect against oxidative stress in hamster fibroblasts, with investigators suggesting that the emulsions have potential to protect the skin against such damage.8
Stilbenes: resveratrol
The antioxidant potency of resveratrol has been cited for conferring a wide range of salutary effects, including antitumorigenic as well as antiaging activity. In 2008, a resveratrol-based skin care formulation intended to combat photoaging was reported to exhibit 17-fold greater antioxidant activity than idebenone.9 In a different study that year, resveratrol, the primary active polyphenolic constituent in red wine, was assessed in terms of topical/transdermal delivery viability, given previously established benefits shown via systemic administration. Several hydrogel systems used as resveratrol vehicles were shown to be safe and effective methods for cutaneously delivering the therapeutic effects of this antioxidant.10 Since then, resveratrol has been demonstrated to penetrate the skin via topical administration, reinforcing the antioxidant system of the stratum corneum and delivering increases of antioxidants to human epidermal tissue.11
In 2014, Farris et al. showed that a proprietary topical antioxidant blend of resveratrol, baicalin, and vitamin E applied topically at night yielded statistically significant amelioration of fine lines and wrinkles, as well as skin firmness, elasticity, laxity, hyperpigmentation, radiance, and roughness over a 12-week period.12 Resveratrol has also been shown in mice to suppress the inflammatory response and improve survival from severe burns with bacterial infections.13
Hydrolyzable tannins: ellagic acid
Ellagic acid, a dimer of gallic acid, has been reported to impart anti-inflammatory, antitumor, immunomodulatory, and antifungal activities.14-16 Ortiz-Ruiz et al. have noted that while ellagic acid is used as a whitening agent, it can act as a substrate to rather than an inhibitor of tyrosinase, as it is oxidized by the enzyme to an unstable o-quinone. However, as a potent antioxidant, ellagic acid can block melanogenesis by reducing o-quinones and semiquinones.17
In a double-blind, placebo-controlled, 4-week trial to assess the effects of orally administered ellagic acid–rich pomegranate extract on the pigmentation of 13 women after UV exposure, with healthy volunteers randomly assigned to high-dose, low-dose, and control groups, luminance values decreased by 1.73% in the high-dose group and 1.35% in the low-dose group, as compared with the control group, and stains and freckles were reported to be diminished.18 A 2016 study in human dermal fibroblasts by Baek et al. suggested that ellagic acid displays antiphotoaging activity, as the polyphenol protected against UVB-induced oxidative stress potentially through an Nrf2-dependent pathway.15
Condensed tannins (Proanthocyanidins): pycnogenol
Pycnogenol has been used in an antioxidant mixture also including vitamins C and E, as well as evening primrose that when orally administered for 10 weeks to female SKH-1 hairless mice exposed three times weekly to UVB irradiation demonstrated the capacity to significantly inhibit wrinkle formation by markedly suppressing UVB-induced MMP activity while promoting collagen production.19 In a 2012 study of 112 women with mild to moderate photoaging, orally administered pycnogenol was shown to yield significant reductions in clinical grading of skin photoaging scores.20 Four years later, a review by Grether-Beck et al. suggested that oral administration of pycnogenol imparts photoprotection, diminishes hyperpigmentation, and improves skin barrier function and the stability of the extracellular matrix.21
Lignins: various woody plants
Recognized as efficient natural scavengers of reactive oxygen species, lignins are complex phenolic polymers that are abundant in nature, particularly in various tree species and agricultural products. In 2004, Dizhbite et al. isolated lignin samples from deciduous and coniferous trees to assess their capacity as natural antioxidants. Samples were assessed against the 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical in homogeneous conditions, with the commercially available kraft lignin noted for displaying antibacterial activity associated with its radical-scavenging properties.22 Four years later, Ugartondo et al. studied several lignins and reported a strong antioxidant capacity at various concentrations that were innocuous to normal human cells and stable when exposed to UVA. The investigators concluded that lignins may be viable for inclusion in cosmetic and topical medical formulations.23
Conclusion
A brief survey of the polyphenolic landscape obviously cannot do the subject justice. From the dermatologic perspective, this diverse family of compounds factor into the skin care formulations becoming more prevalent in the established armamentarium, as well as the direct-to-consumer market. Given the increasing attention paid here and elsewhere to the impact of diet on the skin, the status of this dynamic class of polyphenolic compounds, which includes several antioxidants and is found in numerous plants, appears to be well deserved and warrants much more research.
Dr. Baumann is a private practice dermatologist, researcher, author and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and “Cosmeceuticals and Cosmetic Ingredients,” (New York: McGraw-Hill, 2014), and a New York Times Best Sellers book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance. She is the founder and CEO of Skin Type Solutions Franchise Systems LLC.
References
1. Food Chem Toxicol. 2017 May;103:41-55.
2. J Am Acad Dermatol. 2008 Sep;59(3):418-25.
3. J Pharm Biomed Anal. 2008 Mar 13;46(4):645-52.
4. Food Chem Toxicol. 2015 Aug;82:72-8.
5. Ann Dermatol. 2016 Dec;28(6):740-8.
6. Redox Biol. 2016 Aug;8:79-90.
7. Int J Biol Macromol. 2015 Jan;72:614-23.
8. Evid Based Complement Alternat Med. 2017;2017:5469125.
9. J Cosmet Dermatol. 2008 Mar;7(1):2-7.
10. Biol Pharm Bull. 2008 May;31(5):955-62.
11. Arch Dermatol Res. 2017 Aug;309(6):423-31.
12. J Drugs Dermatol. 2014 Dec;13(12):1467-72.
13. Inflammation. 2015;38(3):1273-80.
14. Dermatol Ther. 2012 May-Jun;25(3):252-9.
15. Korean J Physiol Pharmacol. 2016 May;20(3):269-77.
16. Phytother Res. 2015 Jul;29(7):1019-25.
17. J Dermatol Sci. 2016 May;82(2):115-22.
18. J Nutr Sci Vitaminol (Tokyo). 2006 Oct;52(5):383-8.
19. Photodermatol Photoimmunol Photomed. 2007 Oct;23(5):155-62.
20. Clin Interv Aging. 2012;7:275-86.
21. Skin Pharmacol Physiol. 2016;29(1):13-7.
22. Bioresour Technol. 2004 Dec;95(3):309-17.
23. Bioresour Technol. 2008 Sep;99(14):6683-7.
Polyphenols are widely distributed in the plant kingdom, and are found in copious supply in multiple vegetables, fruits, herbs, grains, tea, coffee beans, honey, and red wine, for example. They are an especially important source of antioxidants and are increasingly the focus of research due to their potent and diverse biologic activities. In the conclusion to my three-part review of polyphenols, this column identifies representative compounds from the classes of nonflavonoid polyphenols and provides a brief update on research.
Phenolic acids: ferulic acid
Derived from curcumin, ferulic acid is noted for exhibiting multiple biologic activities, including antiapoptotic, anticarcinogenic, antidiabetic, hepatoprotective, and cardioprotective, among others. Its beneficial effects are thought to be mediated through its antioxidant and anti-inflammatory characteristics.1 In a small 2008 study, a stable formulation of 15% l-ascorbic acid, 1% alpha-tocopherol, and 0.5% ferulic acid was applied topically to normal-appearing human skin for 4 days and was found to impart significant photoprotection against solar-simulated UV radiation and was especially effective at diminishing thymine dimer mutations, which are linked to skin cancer. The authors also noted that the mechanism of action of this antioxidant formulation differs from that of sunscreens and, therefore, may serve as a supplement to such products.2 (It is worth noting that ferulic acid has been approved as a sunscreen agent in Japan.3)
In 2015, Ambothi et al. used Swiss albino mice to assess the photochemopreventive effects of ferulic acid against chronic (30-week) UVB, finding the intraperitoneal and topical administration of the phenolic acid effective in significantly lowering the incidence of UVB-induced tumor volume and weight in the mice skin.4 The next year, Hahn et al. reported that pretreatment with ferulic acid protects human dermal fibroblasts from UVA-induced photodamage.5 Also in 2016, Chaiprasongsuk et al. found that several dietary phenolics, including ferulic acid, deliver protection against UVA-induced melanogenesis through indirect regulation of the Nrf2-ARE pathway.6
Lignans: flaxseed
Flaxseed lignans, which exhibit a wide range of biologic activities, are best known for their antioxidant properties.7 In a 2017 study using atopic dermatitis–induced NC/Nga mice, Yang et al. found that fermented flaxseed oil administered orally was successful in relieving symptoms such as erythema, edema, pruritus, and epithelial damage.8 Two years earlier, Draganescu et al. developed a topical flaxseed extract formulation that displayed wound healing capabilities on Wistar rats.7 Emulsions produced from the oils and seeds of transgenic flax have also been found to protect against oxidative stress in hamster fibroblasts, with investigators suggesting that the emulsions have potential to protect the skin against such damage.8
Stilbenes: resveratrol
The antioxidant potency of resveratrol has been cited for conferring a wide range of salutary effects, including antitumorigenic as well as antiaging activity. In 2008, a resveratrol-based skin care formulation intended to combat photoaging was reported to exhibit 17-fold greater antioxidant activity than idebenone.9 In a different study that year, resveratrol, the primary active polyphenolic constituent in red wine, was assessed in terms of topical/transdermal delivery viability, given previously established benefits shown via systemic administration. Several hydrogel systems used as resveratrol vehicles were shown to be safe and effective methods for cutaneously delivering the therapeutic effects of this antioxidant.10 Since then, resveratrol has been demonstrated to penetrate the skin via topical administration, reinforcing the antioxidant system of the stratum corneum and delivering increases of antioxidants to human epidermal tissue.11
In 2014, Farris et al. showed that a proprietary topical antioxidant blend of resveratrol, baicalin, and vitamin E applied topically at night yielded statistically significant amelioration of fine lines and wrinkles, as well as skin firmness, elasticity, laxity, hyperpigmentation, radiance, and roughness over a 12-week period.12 Resveratrol has also been shown in mice to suppress the inflammatory response and improve survival from severe burns with bacterial infections.13
Hydrolyzable tannins: ellagic acid
Ellagic acid, a dimer of gallic acid, has been reported to impart anti-inflammatory, antitumor, immunomodulatory, and antifungal activities.14-16 Ortiz-Ruiz et al. have noted that while ellagic acid is used as a whitening agent, it can act as a substrate to rather than an inhibitor of tyrosinase, as it is oxidized by the enzyme to an unstable o-quinone. However, as a potent antioxidant, ellagic acid can block melanogenesis by reducing o-quinones and semiquinones.17
In a double-blind, placebo-controlled, 4-week trial to assess the effects of orally administered ellagic acid–rich pomegranate extract on the pigmentation of 13 women after UV exposure, with healthy volunteers randomly assigned to high-dose, low-dose, and control groups, luminance values decreased by 1.73% in the high-dose group and 1.35% in the low-dose group, as compared with the control group, and stains and freckles were reported to be diminished.18 A 2016 study in human dermal fibroblasts by Baek et al. suggested that ellagic acid displays antiphotoaging activity, as the polyphenol protected against UVB-induced oxidative stress potentially through an Nrf2-dependent pathway.15
Condensed tannins (Proanthocyanidins): pycnogenol
Pycnogenol has been used in an antioxidant mixture also including vitamins C and E, as well as evening primrose that when orally administered for 10 weeks to female SKH-1 hairless mice exposed three times weekly to UVB irradiation demonstrated the capacity to significantly inhibit wrinkle formation by markedly suppressing UVB-induced MMP activity while promoting collagen production.19 In a 2012 study of 112 women with mild to moderate photoaging, orally administered pycnogenol was shown to yield significant reductions in clinical grading of skin photoaging scores.20 Four years later, a review by Grether-Beck et al. suggested that oral administration of pycnogenol imparts photoprotection, diminishes hyperpigmentation, and improves skin barrier function and the stability of the extracellular matrix.21
Lignins: various woody plants
Recognized as efficient natural scavengers of reactive oxygen species, lignins are complex phenolic polymers that are abundant in nature, particularly in various tree species and agricultural products. In 2004, Dizhbite et al. isolated lignin samples from deciduous and coniferous trees to assess their capacity as natural antioxidants. Samples were assessed against the 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical in homogeneous conditions, with the commercially available kraft lignin noted for displaying antibacterial activity associated with its radical-scavenging properties.22 Four years later, Ugartondo et al. studied several lignins and reported a strong antioxidant capacity at various concentrations that were innocuous to normal human cells and stable when exposed to UVA. The investigators concluded that lignins may be viable for inclusion in cosmetic and topical medical formulations.23
Conclusion
A brief survey of the polyphenolic landscape obviously cannot do the subject justice. From the dermatologic perspective, this diverse family of compounds factor into the skin care formulations becoming more prevalent in the established armamentarium, as well as the direct-to-consumer market. Given the increasing attention paid here and elsewhere to the impact of diet on the skin, the status of this dynamic class of polyphenolic compounds, which includes several antioxidants and is found in numerous plants, appears to be well deserved and warrants much more research.
Dr. Baumann is a private practice dermatologist, researcher, author and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and “Cosmeceuticals and Cosmetic Ingredients,” (New York: McGraw-Hill, 2014), and a New York Times Best Sellers book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance. She is the founder and CEO of Skin Type Solutions Franchise Systems LLC.
References
1. Food Chem Toxicol. 2017 May;103:41-55.
2. J Am Acad Dermatol. 2008 Sep;59(3):418-25.
3. J Pharm Biomed Anal. 2008 Mar 13;46(4):645-52.
4. Food Chem Toxicol. 2015 Aug;82:72-8.
5. Ann Dermatol. 2016 Dec;28(6):740-8.
6. Redox Biol. 2016 Aug;8:79-90.
7. Int J Biol Macromol. 2015 Jan;72:614-23.
8. Evid Based Complement Alternat Med. 2017;2017:5469125.
9. J Cosmet Dermatol. 2008 Mar;7(1):2-7.
10. Biol Pharm Bull. 2008 May;31(5):955-62.
11. Arch Dermatol Res. 2017 Aug;309(6):423-31.
12. J Drugs Dermatol. 2014 Dec;13(12):1467-72.
13. Inflammation. 2015;38(3):1273-80.
14. Dermatol Ther. 2012 May-Jun;25(3):252-9.
15. Korean J Physiol Pharmacol. 2016 May;20(3):269-77.
16. Phytother Res. 2015 Jul;29(7):1019-25.
17. J Dermatol Sci. 2016 May;82(2):115-22.
18. J Nutr Sci Vitaminol (Tokyo). 2006 Oct;52(5):383-8.
19. Photodermatol Photoimmunol Photomed. 2007 Oct;23(5):155-62.
20. Clin Interv Aging. 2012;7:275-86.
21. Skin Pharmacol Physiol. 2016;29(1):13-7.
22. Bioresour Technol. 2004 Dec;95(3):309-17.
23. Bioresour Technol. 2008 Sep;99(14):6683-7.

Ulipristal acetate reduced bleeding for women with fibroids
SAN ANTONIO – About half of women with uterine fibroids became amenorrheic when taking the selective progesterone receptor modulator ulipristal acetate (UPA) during a 12-week study cycle, and women taking UPA experienced significant improvement of quality of life, compared with those taking placebo.
Of those women taking 5 mg of UPA, 40.5%-42% became amenorrheic; of those taking 10 mg, 54.8%-57.3% became amenorrheic, James Liu, MD, reported at the annual meeting of the American Society for Reproductive Medicine. These results compared to amenorrhea rates of 0%-8% for women on placebo (P less than .0001 for all values).
The primary aim of VENUS II was to evaluate UPA’s efficacy and safety as intermittent treatment of abnormal uterine bleeding associated with uterine fibroids. Patients received UPA at either 5 mg or 10 mg orally. Secondary efficacy measures included the maintenance effect of UPA at both doses when compared to placebo, by assessing the rate of amenorrhea and the time to amenorrhea. Another secondary measure assessed uterine fibroid–related quality of life.
Safety was assessed by tracking adverse events through both courses of treatment. The study was not powered to compare the two doses against each other, but rather compared each against placebo.
Altogether, 432 patients were randomized to one of the treatment arms, which was begun after an initial screening period of about 10-12 weeks. The first treatment course lasted 12 weeks, after which patients went off treatment for two menstrual cycles. They then began another treatment course for 12 weeks and were followed for an additional 12 weeks after treatment was stopped.
Patients were included if they were premenopausal, aged 18-50 years old, had prolonged bleeding in at least 4 of the last 6 menstrual periods, had menstrual blood loss of at least 80 mL by cycle by the alkaline hematin method, and had at least one discrete leiomyoma without a uterine size greater than 20 weeks. About two-thirds of patients were black, reflecting the higher prevalence of uterine fibroids in this population, said Dr. Liu, professor of medicine and reproductive biology at Case Western Reserve University, Cleveland.
Most patients (60%-80%) had their bleeding controlled on either dose of UPA, compared with fewer than 10% of women taking placebo.
Quality of life data, presented separately at ASRM by Lee Shulman, MD, examined the impact of treatment on patients’ physical and social activities, and also on the severity of symptoms and health-related quality of life.
“The majority of patients on UPA versus a minority of patients on placebo described their menstrual/vaginal bleeding at the end of treatment course 1 as ‘much better’ or ‘very much better,’ ” said Dr. Shulman, chief of clinical genetics in the department of ob.gyn. at Northwestern University in Evanston, Ill.
Of patients taking 5 mg UPA, 75% reported that degree of improvement, as did 87% of those on 10 mg UPA, compared with 17.9% of those taking placebo. .
Adverse events were rare, with hot flashes occurring in about 10% of women taking UPA, compared with less than 2% of those taking placebo. Headaches, fatigue, and nausea were also reported, but rates were not significantly different from rates for those taking placebo. One serious adverse event that was deemed treatment related was a uterine hemorrhage experienced by a woman taking UPA.
“Even in treatment course 1 we already had profound and statistically significant effects in symptoms across the board,” Dr. Shulman said. “How long will they last? We obviously need more data. But the study suggests that the benefits last significantly longer than that associated with leuprolide acetate.”
Ulipristal acetate is already approved by the Food and Drug Administration at a different dosage as emergency contraception. The VENUS II data support its use for women with uterine fibroids, the researchers said. “Our results provide reassurance,” Dr. Liu said. “We can conclude that UPA is effective and safe in the management of uterine fibroids in the U.S. population.”
Dr. Liu reported that he had no relevant disclosures. Dr. Shulman reported relationships with multiple pharmaceutical companies, including Allergan, which funded the VENUS II study.
[email protected]
On Twitter @karioakes
SAN ANTONIO – About half of women with uterine fibroids became amenorrheic when taking the selective progesterone receptor modulator ulipristal acetate (UPA) during a 12-week study cycle, and women taking UPA experienced significant improvement of quality of life, compared with those taking placebo.
Of those women taking 5 mg of UPA, 40.5%-42% became amenorrheic; of those taking 10 mg, 54.8%-57.3% became amenorrheic, James Liu, MD, reported at the annual meeting of the American Society for Reproductive Medicine. These results compared to amenorrhea rates of 0%-8% for women on placebo (P less than .0001 for all values).
The primary aim of VENUS II was to evaluate UPA’s efficacy and safety as intermittent treatment of abnormal uterine bleeding associated with uterine fibroids. Patients received UPA at either 5 mg or 10 mg orally. Secondary efficacy measures included the maintenance effect of UPA at both doses when compared to placebo, by assessing the rate of amenorrhea and the time to amenorrhea. Another secondary measure assessed uterine fibroid–related quality of life.
Safety was assessed by tracking adverse events through both courses of treatment. The study was not powered to compare the two doses against each other, but rather compared each against placebo.
Altogether, 432 patients were randomized to one of the treatment arms, which was begun after an initial screening period of about 10-12 weeks. The first treatment course lasted 12 weeks, after which patients went off treatment for two menstrual cycles. They then began another treatment course for 12 weeks and were followed for an additional 12 weeks after treatment was stopped.
Patients were included if they were premenopausal, aged 18-50 years old, had prolonged bleeding in at least 4 of the last 6 menstrual periods, had menstrual blood loss of at least 80 mL by cycle by the alkaline hematin method, and had at least one discrete leiomyoma without a uterine size greater than 20 weeks. About two-thirds of patients were black, reflecting the higher prevalence of uterine fibroids in this population, said Dr. Liu, professor of medicine and reproductive biology at Case Western Reserve University, Cleveland.
Most patients (60%-80%) had their bleeding controlled on either dose of UPA, compared with fewer than 10% of women taking placebo.
Quality of life data, presented separately at ASRM by Lee Shulman, MD, examined the impact of treatment on patients’ physical and social activities, and also on the severity of symptoms and health-related quality of life.
“The majority of patients on UPA versus a minority of patients on placebo described their menstrual/vaginal bleeding at the end of treatment course 1 as ‘much better’ or ‘very much better,’ ” said Dr. Shulman, chief of clinical genetics in the department of ob.gyn. at Northwestern University in Evanston, Ill.
Of patients taking 5 mg UPA, 75% reported that degree of improvement, as did 87% of those on 10 mg UPA, compared with 17.9% of those taking placebo. .
Adverse events were rare, with hot flashes occurring in about 10% of women taking UPA, compared with less than 2% of those taking placebo. Headaches, fatigue, and nausea were also reported, but rates were not significantly different from rates for those taking placebo. One serious adverse event that was deemed treatment related was a uterine hemorrhage experienced by a woman taking UPA.
“Even in treatment course 1 we already had profound and statistically significant effects in symptoms across the board,” Dr. Shulman said. “How long will they last? We obviously need more data. But the study suggests that the benefits last significantly longer than that associated with leuprolide acetate.”
Ulipristal acetate is already approved by the Food and Drug Administration at a different dosage as emergency contraception. The VENUS II data support its use for women with uterine fibroids, the researchers said. “Our results provide reassurance,” Dr. Liu said. “We can conclude that UPA is effective and safe in the management of uterine fibroids in the U.S. population.”
Dr. Liu reported that he had no relevant disclosures. Dr. Shulman reported relationships with multiple pharmaceutical companies, including Allergan, which funded the VENUS II study.
[email protected]
On Twitter @karioakes
SAN ANTONIO – About half of women with uterine fibroids became amenorrheic when taking the selective progesterone receptor modulator ulipristal acetate (UPA) during a 12-week study cycle, and women taking UPA experienced significant improvement of quality of life, compared with those taking placebo.
Of those women taking 5 mg of UPA, 40.5%-42% became amenorrheic; of those taking 10 mg, 54.8%-57.3% became amenorrheic, James Liu, MD, reported at the annual meeting of the American Society for Reproductive Medicine. These results compared to amenorrhea rates of 0%-8% for women on placebo (P less than .0001 for all values).
The primary aim of VENUS II was to evaluate UPA’s efficacy and safety as intermittent treatment of abnormal uterine bleeding associated with uterine fibroids. Patients received UPA at either 5 mg or 10 mg orally. Secondary efficacy measures included the maintenance effect of UPA at both doses when compared to placebo, by assessing the rate of amenorrhea and the time to amenorrhea. Another secondary measure assessed uterine fibroid–related quality of life.
Safety was assessed by tracking adverse events through both courses of treatment. The study was not powered to compare the two doses against each other, but rather compared each against placebo.
Altogether, 432 patients were randomized to one of the treatment arms, which was begun after an initial screening period of about 10-12 weeks. The first treatment course lasted 12 weeks, after which patients went off treatment for two menstrual cycles. They then began another treatment course for 12 weeks and were followed for an additional 12 weeks after treatment was stopped.
Patients were included if they were premenopausal, aged 18-50 years old, had prolonged bleeding in at least 4 of the last 6 menstrual periods, had menstrual blood loss of at least 80 mL by cycle by the alkaline hematin method, and had at least one discrete leiomyoma without a uterine size greater than 20 weeks. About two-thirds of patients were black, reflecting the higher prevalence of uterine fibroids in this population, said Dr. Liu, professor of medicine and reproductive biology at Case Western Reserve University, Cleveland.
Most patients (60%-80%) had their bleeding controlled on either dose of UPA, compared with fewer than 10% of women taking placebo.
Quality of life data, presented separately at ASRM by Lee Shulman, MD, examined the impact of treatment on patients’ physical and social activities, and also on the severity of symptoms and health-related quality of life.
“The majority of patients on UPA versus a minority of patients on placebo described their menstrual/vaginal bleeding at the end of treatment course 1 as ‘much better’ or ‘very much better,’ ” said Dr. Shulman, chief of clinical genetics in the department of ob.gyn. at Northwestern University in Evanston, Ill.
Of patients taking 5 mg UPA, 75% reported that degree of improvement, as did 87% of those on 10 mg UPA, compared with 17.9% of those taking placebo. .
Adverse events were rare, with hot flashes occurring in about 10% of women taking UPA, compared with less than 2% of those taking placebo. Headaches, fatigue, and nausea were also reported, but rates were not significantly different from rates for those taking placebo. One serious adverse event that was deemed treatment related was a uterine hemorrhage experienced by a woman taking UPA.
“Even in treatment course 1 we already had profound and statistically significant effects in symptoms across the board,” Dr. Shulman said. “How long will they last? We obviously need more data. But the study suggests that the benefits last significantly longer than that associated with leuprolide acetate.”
Ulipristal acetate is already approved by the Food and Drug Administration at a different dosage as emergency contraception. The VENUS II data support its use for women with uterine fibroids, the researchers said. “Our results provide reassurance,” Dr. Liu said. “We can conclude that UPA is effective and safe in the management of uterine fibroids in the U.S. population.”
Dr. Liu reported that he had no relevant disclosures. Dr. Shulman reported relationships with multiple pharmaceutical companies, including Allergan, which funded the VENUS II study.
[email protected]
On Twitter @karioakes
AT ASRM 2017
Key clinical point:
Major finding: Between 40.5% and 57.3% of women taking ulipristal acetate (UPA) achieved amenorrhea, compared with 0%-8% of women on placebo (P less than .0001).
Data source: Venus II, a phase 3 prospective, randomized, double-blind, double-dummy, placebo-controlled study that was partially parallel and partially crossover, with 432 patients.
Disclosures: Dr. Liu reported no relevant disclosures; Dr. Shulman reported financial relationships with multiple pharmaceutical companies, including Allergan, which funded the trial.
Cosmetic Corner: Dermatologists Weigh in on Men’s Moisturizers
To improve patient care and outcomes, leading dermatologists offered their recommendations on men’s moisturizers. Consideration must be given to:
- Clinique For Men Oil Control Mattifying Moisturizer
Clinique Laboratories, LLC
“I recommend this product for men with oily or combination skin. It’s very lightweight and provides good hydration benefits without leaving the skin shiny.”—Jeannette Graf, MD, Great Neck, New York
- Facial Fuel Energizing Moisture Treatment for Men
Kiehl’s
“I commonly recommend this moisturizer. The Facial Fuel line is great for most skin types and the products are moderately priced.”—Gary Goldenberg, MD, New York, New York
- Neutrogena Men Triple Protect Face Lotion With Sunscreen
Johnson & Johnson Consumer Inc
“This is a light, daily moisturizer with broad-spectrum UV protection.”—Shari Lipner, MD, PhD, New York, New York
- Triple Lipid Restore 2:4:2
SkinCeuticals
“This moisturizer has the precise lipid content needed by the skin.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Wet skin moisturizer, lip plumper, and pigment corrector will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on men’s moisturizers. Consideration must be given to:
- Clinique For Men Oil Control Mattifying Moisturizer
Clinique Laboratories, LLC
“I recommend this product for men with oily or combination skin. It’s very lightweight and provides good hydration benefits without leaving the skin shiny.”—Jeannette Graf, MD, Great Neck, New York
- Facial Fuel Energizing Moisture Treatment for Men
Kiehl’s
“I commonly recommend this moisturizer. The Facial Fuel line is great for most skin types and the products are moderately priced.”—Gary Goldenberg, MD, New York, New York
- Neutrogena Men Triple Protect Face Lotion With Sunscreen
Johnson & Johnson Consumer Inc
“This is a light, daily moisturizer with broad-spectrum UV protection.”—Shari Lipner, MD, PhD, New York, New York
- Triple Lipid Restore 2:4:2
SkinCeuticals
“This moisturizer has the precise lipid content needed by the skin.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Wet skin moisturizer, lip plumper, and pigment corrector will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on men’s moisturizers. Consideration must be given to:
- Clinique For Men Oil Control Mattifying Moisturizer
Clinique Laboratories, LLC
“I recommend this product for men with oily or combination skin. It’s very lightweight and provides good hydration benefits without leaving the skin shiny.”—Jeannette Graf, MD, Great Neck, New York
- Facial Fuel Energizing Moisture Treatment for Men
Kiehl’s
“I commonly recommend this moisturizer. The Facial Fuel line is great for most skin types and the products are moderately priced.”—Gary Goldenberg, MD, New York, New York
- Neutrogena Men Triple Protect Face Lotion With Sunscreen
Johnson & Johnson Consumer Inc
“This is a light, daily moisturizer with broad-spectrum UV protection.”—Shari Lipner, MD, PhD, New York, New York
- Triple Lipid Restore 2:4:2
SkinCeuticals
“This moisturizer has the precise lipid content needed by the skin.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Wet skin moisturizer, lip plumper, and pigment corrector will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

Debunking Acne Myths: Does Back Acne Need to Be Treated?
Myth: Back acne will clear on its own
Patients with back acne may not seek treatment options because they assume it will clear on its own; however, deep painful lesions typically require treatment by a dermatologist. Mild to moderate cases may respond to less aggressive over-the-counter treatments but also may benefit from a combination of topical and systemic antibiotic therapies. According to James Q. Del Rosso, MD, a dermatologist in Las Vegas, Nevada, “Many dermatologists believe that truncal acne vulgaris warrants use of systemic antibiotic therapy, which may not necessarily be the case, especially in patients presenting with mild to moderate acne severity.” Cases of deep inflammatory acne on the back often warrant using systemic therapies such as oral isotretinoin. These severe cases may be less responsive to standard therapies and may require repeated treatment.
Although back acne is not as cosmetically visible as facial acne, it has been associated with sexual and bodily self-consciousness in both males and females. Preliminary data from one study showed that 78% of patients with truncal acne (n=141) on the back and/or chest indicated they were definitely interested in treatment, but truncal acne was not mentioned by these patients without direct inquiry from a physician. As a result, it may be beneficial for dermatologists to ask acne patients about lesions presenting on the back and inform them that treatment options are available.
Treatment application also is a consideration for back acne. Benzoyl peroxide cleanser/wash formulations are convenient, and the foam formulation of clindamycin phosphate allows for easy use due to its spreadability, rapid penetration, and lack of residue and fabric bleaching.
It is important to inform patients that back acne can flare even during active treatment. Patients should be instructed to wear loose-fitting clothes made of cotton or other sweat-wicking fabrics when working out and to shower and change clothes immediately after. Sheets and pillowcases should be changed regularly to avoid exposure to dead skin cells and bacteria, which can exacerbate acne on the back. Backpacks and handbags also can rub against the skin on the back, causing acne to flare. As an alternative, patients should be encouraged to carry handheld bags or bags with shoulder straps to avoid irritation of the skin on the back.
Back acne: how to see clearer skin. American Academy of Dermatology website. https://www.aad.org/public/diseases/acne-and-rosacea/back-acne-how-to-see-clearer-skin. Accessed October 30, 2017.
Del Rosso JQ. Management of truncal acne vulgaris: current perspectives on treatment. Cutis. 2006;77:285-289.
Dunn LK, O’Neill JL, Feldman SR. Acne in adolescents: quality of life, self-esteem, mood, and psychological disorders. Dermatol Online J. 2011;17:1.
Hassan J, Grogan S, Clark-Carter D, et al. The individual health burden of acne: appearance-related distress in male and female adolescents and adults with back, chest and facial acne. J Health Psychol. 2009;14:1105-11118.
Myth: Back acne will clear on its own
Patients with back acne may not seek treatment options because they assume it will clear on its own; however, deep painful lesions typically require treatment by a dermatologist. Mild to moderate cases may respond to less aggressive over-the-counter treatments but also may benefit from a combination of topical and systemic antibiotic therapies. According to James Q. Del Rosso, MD, a dermatologist in Las Vegas, Nevada, “Many dermatologists believe that truncal acne vulgaris warrants use of systemic antibiotic therapy, which may not necessarily be the case, especially in patients presenting with mild to moderate acne severity.” Cases of deep inflammatory acne on the back often warrant using systemic therapies such as oral isotretinoin. These severe cases may be less responsive to standard therapies and may require repeated treatment.
Although back acne is not as cosmetically visible as facial acne, it has been associated with sexual and bodily self-consciousness in both males and females. Preliminary data from one study showed that 78% of patients with truncal acne (n=141) on the back and/or chest indicated they were definitely interested in treatment, but truncal acne was not mentioned by these patients without direct inquiry from a physician. As a result, it may be beneficial for dermatologists to ask acne patients about lesions presenting on the back and inform them that treatment options are available.
Treatment application also is a consideration for back acne. Benzoyl peroxide cleanser/wash formulations are convenient, and the foam formulation of clindamycin phosphate allows for easy use due to its spreadability, rapid penetration, and lack of residue and fabric bleaching.
It is important to inform patients that back acne can flare even during active treatment. Patients should be instructed to wear loose-fitting clothes made of cotton or other sweat-wicking fabrics when working out and to shower and change clothes immediately after. Sheets and pillowcases should be changed regularly to avoid exposure to dead skin cells and bacteria, which can exacerbate acne on the back. Backpacks and handbags also can rub against the skin on the back, causing acne to flare. As an alternative, patients should be encouraged to carry handheld bags or bags with shoulder straps to avoid irritation of the skin on the back.
Myth: Back acne will clear on its own
Patients with back acne may not seek treatment options because they assume it will clear on its own; however, deep painful lesions typically require treatment by a dermatologist. Mild to moderate cases may respond to less aggressive over-the-counter treatments but also may benefit from a combination of topical and systemic antibiotic therapies. According to James Q. Del Rosso, MD, a dermatologist in Las Vegas, Nevada, “Many dermatologists believe that truncal acne vulgaris warrants use of systemic antibiotic therapy, which may not necessarily be the case, especially in patients presenting with mild to moderate acne severity.” Cases of deep inflammatory acne on the back often warrant using systemic therapies such as oral isotretinoin. These severe cases may be less responsive to standard therapies and may require repeated treatment.
Although back acne is not as cosmetically visible as facial acne, it has been associated with sexual and bodily self-consciousness in both males and females. Preliminary data from one study showed that 78% of patients with truncal acne (n=141) on the back and/or chest indicated they were definitely interested in treatment, but truncal acne was not mentioned by these patients without direct inquiry from a physician. As a result, it may be beneficial for dermatologists to ask acne patients about lesions presenting on the back and inform them that treatment options are available.
Treatment application also is a consideration for back acne. Benzoyl peroxide cleanser/wash formulations are convenient, and the foam formulation of clindamycin phosphate allows for easy use due to its spreadability, rapid penetration, and lack of residue and fabric bleaching.
It is important to inform patients that back acne can flare even during active treatment. Patients should be instructed to wear loose-fitting clothes made of cotton or other sweat-wicking fabrics when working out and to shower and change clothes immediately after. Sheets and pillowcases should be changed regularly to avoid exposure to dead skin cells and bacteria, which can exacerbate acne on the back. Backpacks and handbags also can rub against the skin on the back, causing acne to flare. As an alternative, patients should be encouraged to carry handheld bags or bags with shoulder straps to avoid irritation of the skin on the back.
Back acne: how to see clearer skin. American Academy of Dermatology website. https://www.aad.org/public/diseases/acne-and-rosacea/back-acne-how-to-see-clearer-skin. Accessed October 30, 2017.
Del Rosso JQ. Management of truncal acne vulgaris: current perspectives on treatment. Cutis. 2006;77:285-289.
Dunn LK, O’Neill JL, Feldman SR. Acne in adolescents: quality of life, self-esteem, mood, and psychological disorders. Dermatol Online J. 2011;17:1.
Hassan J, Grogan S, Clark-Carter D, et al. The individual health burden of acne: appearance-related distress in male and female adolescents and adults with back, chest and facial acne. J Health Psychol. 2009;14:1105-11118.
Back acne: how to see clearer skin. American Academy of Dermatology website. https://www.aad.org/public/diseases/acne-and-rosacea/back-acne-how-to-see-clearer-skin. Accessed October 30, 2017.
Del Rosso JQ. Management of truncal acne vulgaris: current perspectives on treatment. Cutis. 2006;77:285-289.
Dunn LK, O’Neill JL, Feldman SR. Acne in adolescents: quality of life, self-esteem, mood, and psychological disorders. Dermatol Online J. 2011;17:1.
Hassan J, Grogan S, Clark-Carter D, et al. The individual health burden of acne: appearance-related distress in male and female adolescents and adults with back, chest and facial acne. J Health Psychol. 2009;14:1105-11118.

Improving transitions for elderly patients
Transitions are always a time of concern for hospitalists, and the transition from hospital to skilled nursing facilities (SNF) is no exception.
“During the transition and in the 30 days after discharge from the hospital to a SNF, patients are at high risk for death, rehospitalization, and high-cost health care,” said Amber Moore, MD, MPH, a hospitalist at Beth Israel Deaconess Medical Center, and instructor of medicine, Harvard Medical School. “Elderly adults are especially vulnerable because of impairments that may prevent them from participating in the discharge process and an increase in the risk that information is lost or incomplete during the care transition.”

To address this, she and several other physicians studied a novel video-conference program called Extension for Community Health Outcomes–Care Transitions (ECHO-CT) that connects an interdisciplinary hospital-based team with clinicians at SNFs to help reduce patient mortality, hospital readmission, skilled nursing facility length of stay, and 30-day health care costs.
The results of their study suggest that this intervention significantly decreased SNF length of stay, readmission rate, and costs of care, she says; the model they used is reproducible and has the potential to significantly improve care of these patients. “Our model was hospitalist run and is a mechanism to help hospitalists improve care to their patients during the transition time and beyond,” Dr. Moore said. “Furthermore, in participating in this model, hospitalists have the opportunity to better understand the challenges that face their patients after discharge and learn from postacute care providers.”
Ideally, she would like to see the model spread to other hospitals; she says hospitalists are well positioned to set up this program at their institution. “I also hope that our study highlights the incredible opportunity for improvement in the care of patients during transition from hospital to SNF and encourages hospitalists to look for innovative ways to improve care at this transition,” she said.
Reference
Moore AB, Krupp JE, Dufour AB, et al. Improving transitions to post-acute care for elderly patients using a novel video-conferencing program: ECHO-Care transitions. Am J Med. 2017 Oct;130(10):1199-204. Accessed June 6, 2017.
Transitions are always a time of concern for hospitalists, and the transition from hospital to skilled nursing facilities (SNF) is no exception.
“During the transition and in the 30 days after discharge from the hospital to a SNF, patients are at high risk for death, rehospitalization, and high-cost health care,” said Amber Moore, MD, MPH, a hospitalist at Beth Israel Deaconess Medical Center, and instructor of medicine, Harvard Medical School. “Elderly adults are especially vulnerable because of impairments that may prevent them from participating in the discharge process and an increase in the risk that information is lost or incomplete during the care transition.”
To address this, she and several other physicians studied a novel video-conference program called Extension for Community Health Outcomes–Care Transitions (ECHO-CT) that connects an interdisciplinary hospital-based team with clinicians at SNFs to help reduce patient mortality, hospital readmission, skilled nursing facility length of stay, and 30-day health care costs.
The results of their study suggest that this intervention significantly decreased SNF length of stay, readmission rate, and costs of care, she says; the model they used is reproducible and has the potential to significantly improve care of these patients. “Our model was hospitalist run and is a mechanism to help hospitalists improve care to their patients during the transition time and beyond,” Dr. Moore said. “Furthermore, in participating in this model, hospitalists have the opportunity to better understand the challenges that face their patients after discharge and learn from postacute care providers.”
Ideally, she would like to see the model spread to other hospitals; she says hospitalists are well positioned to set up this program at their institution. “I also hope that our study highlights the incredible opportunity for improvement in the care of patients during transition from hospital to SNF and encourages hospitalists to look for innovative ways to improve care at this transition,” she said.
Reference
Moore AB, Krupp JE, Dufour AB, et al. Improving transitions to post-acute care for elderly patients using a novel video-conferencing program: ECHO-Care transitions. Am J Med. 2017 Oct;130(10):1199-204. Accessed June 6, 2017.
Transitions are always a time of concern for hospitalists, and the transition from hospital to skilled nursing facilities (SNF) is no exception.
“During the transition and in the 30 days after discharge from the hospital to a SNF, patients are at high risk for death, rehospitalization, and high-cost health care,” said Amber Moore, MD, MPH, a hospitalist at Beth Israel Deaconess Medical Center, and instructor of medicine, Harvard Medical School. “Elderly adults are especially vulnerable because of impairments that may prevent them from participating in the discharge process and an increase in the risk that information is lost or incomplete during the care transition.”
To address this, she and several other physicians studied a novel video-conference program called Extension for Community Health Outcomes–Care Transitions (ECHO-CT) that connects an interdisciplinary hospital-based team with clinicians at SNFs to help reduce patient mortality, hospital readmission, skilled nursing facility length of stay, and 30-day health care costs.
The results of their study suggest that this intervention significantly decreased SNF length of stay, readmission rate, and costs of care, she says; the model they used is reproducible and has the potential to significantly improve care of these patients. “Our model was hospitalist run and is a mechanism to help hospitalists improve care to their patients during the transition time and beyond,” Dr. Moore said. “Furthermore, in participating in this model, hospitalists have the opportunity to better understand the challenges that face their patients after discharge and learn from postacute care providers.”
Ideally, she would like to see the model spread to other hospitals; she says hospitalists are well positioned to set up this program at their institution. “I also hope that our study highlights the incredible opportunity for improvement in the care of patients during transition from hospital to SNF and encourages hospitalists to look for innovative ways to improve care at this transition,” she said.
Reference
Moore AB, Krupp JE, Dufour AB, et al. Improving transitions to post-acute care for elderly patients using a novel video-conferencing program: ECHO-Care transitions. Am J Med. 2017 Oct;130(10):1199-204. Accessed June 6, 2017.
Some measures to control HAI sound better than they perform
SAN DIEGO – Some almost universally accepted measures against hospital-acquired infections are more costly, annoying, and time consuming than they’re worth, presenters agreed during a panel discussion at the annual clinical congress of the American College of Surgeons.
Intra-abdominal antibiotic irrigation, chlorhexidine bathing, and even postsurgical antibiotic infusions have not consistently been shown to reduce infections. These measures do, however, ratchet up costs and can contribute to antibiotic resistance.
Some of these and other measures to prevent nosocomial infections may indeed reduce the risk, but the gain is small, said Charles H. Cook, MD.
“Chlorhexidine bathing, for example, is touted by many as a panacea for all the infections we’re talking about,” said Dr. Cook, a critical care surgeon at Beth Israel Deaconess Medical Center, New York. “A recent meta-analysis in critical care units did find a reduced relative risk of 0.44 for central line bloodstream infections. But you needed to bathe 360 patients to prevent one infection. It’s what I call a long run for a short slide.”
Therese Duane, MD, FACS, agreed. A surgeon at the John Peter Smith Hospital, Ft. Worth, Tex., Dr. Duane reviewed three different guidelines for the prevention of surgical site infections: the ACS and Surgical Infection Society, the World Health Organization, and the Centers for Disease Control and Prevention. In looking for similarities between the documents, she said she found several well-accepted practices that just aren’t supported by good data.
Presurgical antimicrobial infusions got a strong thumbs-up from all the groups, but only under a very specific circumstance: The medication has to be administered well in advance of surgery for it to be effective.
“Your goal is to get the appropriate concentration into the tissues by the time of incision,” Dr. Duane said. “It takes time to get there – if you give it after the incision, you have bleeding and cellular death, and the antimicrobials cannot get to that incision and do their job. If I’m starting a case and they haven’t been given, I don’t ever start them after the incision, because then you have all of the risks and none of the benefits. In my opinion, we need to move to no further antimicrobials once the incision or case is over because it serves no purpose and is inconsistent with good antibiotic stewardship.”
Adhesive drapes got a resounding “eh” from the guidelines, Dr. Duane said. “You really do not need them. They’re expensive and they’re not improving outcomes, so don’t waste your time or money. We need to think about minimizing what isn’t helpful and maximizing the things that are worthwhile. That’s the way to practice good socially responsible surgery without breaking the bank,” she said.
Antimicrobial sutures got weak recommendations, Dr. Duane said. The evidence supporting their use was not very strong, although she said she feels triclosan-coated sutures are helpful in all kinds of surgery. Preoperative showering with an antiseptic received strong support, with alcohol-containing preps superior to chlorhexidine, which is better than povidone-iodine–containing solutions.
Deep-space irrigation with aqueous iodophor also received a weak recommendation, but Dr. Duane said the evidence does not support the use of antibiotic-containing irrigation in either the abdomen or the incision. “And the guidelines came out strongly against using antimicrobial agents on the incision,” she said. None of the guidelines issued a recommendation for or against antimicrobial dressings.
Protocolized infection-control bundles are a very great help in reducing the incidence of surgical site infections, Dr. Duane added. “They increase attention to detail and decrease the rates of infection.”
Dr. Cook agreed. “Central line bundles are one of the things that work” for line-associated bloodstream infections, he said. Since their large-scale adoption, mortality from these infections has dropped significantly; it was hovering around 28,000 per year in the mid-2000s, he said. “That’s about how many men die from prostate cancer every year.”
Central line infections are very costly too, he added – around $46,000 per event. “That comes to around $2 billion in direct and indirect costs every year.”
A 2006 study demonstrated the efficacy of central line bundles in the fight against these potentially devastating infections.
The bundled intervention comprised hand washing, using full-barrier precautions during the insertion of central venous catheters, cleaning the skin with chlorhexidine, avoiding the femoral site if possible, and removing unnecessary catheters. The median rate of catheter-related bloodstream infections per 1,000 catheter-days decreased from 2.7 at baseline to 0 at 3 months after implementation of the study intervention.
Antibiotic-coated or impregnated catheters do not work as well. A 2016 Cochrane review of 57 studies determined that the devices didn’t improve sepsis, all-cause mortality, or catheter-related local infections.
The jury may still be out on coated dressings or securing devices, however. Another Cochrane review, of 22 studies, found a 40% decrease in central line–associated bloodstream infections with these items. “There was moderate evidence that tip colonization was reduced, but the authors said more research is needed.”
The evidence looks stronger for alcohol-impregnated port protectors, Dr. Cook said. Two studies in particular support their use. In an oncology unit, the rate of these infections dropped from 2.3 to 0.3 per 1,000 catheter days after the port protectors were instituted.
In the second study, infection rates declined from 1.43 to 0.69 per 1,000 line-days after the protectors came on board.
“The advantage was seen mostly in ICUs, so the recommendations are to use them there,” Dr. Cook said.
Neither Dr. Cook nor Dr. Duane had any relevant financial disclosures.
[email protected]
On Twitter @Alz Gal
SAN DIEGO – Some almost universally accepted measures against hospital-acquired infections are more costly, annoying, and time consuming than they’re worth, presenters agreed during a panel discussion at the annual clinical congress of the American College of Surgeons.
Intra-abdominal antibiotic irrigation, chlorhexidine bathing, and even postsurgical antibiotic infusions have not consistently been shown to reduce infections. These measures do, however, ratchet up costs and can contribute to antibiotic resistance.
Some of these and other measures to prevent nosocomial infections may indeed reduce the risk, but the gain is small, said Charles H. Cook, MD.
“Chlorhexidine bathing, for example, is touted by many as a panacea for all the infections we’re talking about,” said Dr. Cook, a critical care surgeon at Beth Israel Deaconess Medical Center, New York. “A recent meta-analysis in critical care units did find a reduced relative risk of 0.44 for central line bloodstream infections. But you needed to bathe 360 patients to prevent one infection. It’s what I call a long run for a short slide.”
Therese Duane, MD, FACS, agreed. A surgeon at the John Peter Smith Hospital, Ft. Worth, Tex., Dr. Duane reviewed three different guidelines for the prevention of surgical site infections: the ACS and Surgical Infection Society, the World Health Organization, and the Centers for Disease Control and Prevention. In looking for similarities between the documents, she said she found several well-accepted practices that just aren’t supported by good data.
Presurgical antimicrobial infusions got a strong thumbs-up from all the groups, but only under a very specific circumstance: The medication has to be administered well in advance of surgery for it to be effective.
“Your goal is to get the appropriate concentration into the tissues by the time of incision,” Dr. Duane said. “It takes time to get there – if you give it after the incision, you have bleeding and cellular death, and the antimicrobials cannot get to that incision and do their job. If I’m starting a case and they haven’t been given, I don’t ever start them after the incision, because then you have all of the risks and none of the benefits. In my opinion, we need to move to no further antimicrobials once the incision or case is over because it serves no purpose and is inconsistent with good antibiotic stewardship.”
Adhesive drapes got a resounding “eh” from the guidelines, Dr. Duane said. “You really do not need them. They’re expensive and they’re not improving outcomes, so don’t waste your time or money. We need to think about minimizing what isn’t helpful and maximizing the things that are worthwhile. That’s the way to practice good socially responsible surgery without breaking the bank,” she said.
Antimicrobial sutures got weak recommendations, Dr. Duane said. The evidence supporting their use was not very strong, although she said she feels triclosan-coated sutures are helpful in all kinds of surgery. Preoperative showering with an antiseptic received strong support, with alcohol-containing preps superior to chlorhexidine, which is better than povidone-iodine–containing solutions.
Deep-space irrigation with aqueous iodophor also received a weak recommendation, but Dr. Duane said the evidence does not support the use of antibiotic-containing irrigation in either the abdomen or the incision. “And the guidelines came out strongly against using antimicrobial agents on the incision,” she said. None of the guidelines issued a recommendation for or against antimicrobial dressings.
Protocolized infection-control bundles are a very great help in reducing the incidence of surgical site infections, Dr. Duane added. “They increase attention to detail and decrease the rates of infection.”
Dr. Cook agreed. “Central line bundles are one of the things that work” for line-associated bloodstream infections, he said. Since their large-scale adoption, mortality from these infections has dropped significantly; it was hovering around 28,000 per year in the mid-2000s, he said. “That’s about how many men die from prostate cancer every year.”
Central line infections are very costly too, he added – around $46,000 per event. “That comes to around $2 billion in direct and indirect costs every year.”
A 2006 study demonstrated the efficacy of central line bundles in the fight against these potentially devastating infections.
The bundled intervention comprised hand washing, using full-barrier precautions during the insertion of central venous catheters, cleaning the skin with chlorhexidine, avoiding the femoral site if possible, and removing unnecessary catheters. The median rate of catheter-related bloodstream infections per 1,000 catheter-days decreased from 2.7 at baseline to 0 at 3 months after implementation of the study intervention.
Antibiotic-coated or impregnated catheters do not work as well. A 2016 Cochrane review of 57 studies determined that the devices didn’t improve sepsis, all-cause mortality, or catheter-related local infections.
The jury may still be out on coated dressings or securing devices, however. Another Cochrane review, of 22 studies, found a 40% decrease in central line–associated bloodstream infections with these items. “There was moderate evidence that tip colonization was reduced, but the authors said more research is needed.”
The evidence looks stronger for alcohol-impregnated port protectors, Dr. Cook said. Two studies in particular support their use. In an oncology unit, the rate of these infections dropped from 2.3 to 0.3 per 1,000 catheter days after the port protectors were instituted.
In the second study, infection rates declined from 1.43 to 0.69 per 1,000 line-days after the protectors came on board.
“The advantage was seen mostly in ICUs, so the recommendations are to use them there,” Dr. Cook said.
Neither Dr. Cook nor Dr. Duane had any relevant financial disclosures.
[email protected]
On Twitter @Alz Gal
SAN DIEGO – Some almost universally accepted measures against hospital-acquired infections are more costly, annoying, and time consuming than they’re worth, presenters agreed during a panel discussion at the annual clinical congress of the American College of Surgeons.
Intra-abdominal antibiotic irrigation, chlorhexidine bathing, and even postsurgical antibiotic infusions have not consistently been shown to reduce infections. These measures do, however, ratchet up costs and can contribute to antibiotic resistance.
Some of these and other measures to prevent nosocomial infections may indeed reduce the risk, but the gain is small, said Charles H. Cook, MD.
“Chlorhexidine bathing, for example, is touted by many as a panacea for all the infections we’re talking about,” said Dr. Cook, a critical care surgeon at Beth Israel Deaconess Medical Center, New York. “A recent meta-analysis in critical care units did find a reduced relative risk of 0.44 for central line bloodstream infections. But you needed to bathe 360 patients to prevent one infection. It’s what I call a long run for a short slide.”
Therese Duane, MD, FACS, agreed. A surgeon at the John Peter Smith Hospital, Ft. Worth, Tex., Dr. Duane reviewed three different guidelines for the prevention of surgical site infections: the ACS and Surgical Infection Society, the World Health Organization, and the Centers for Disease Control and Prevention. In looking for similarities between the documents, she said she found several well-accepted practices that just aren’t supported by good data.
Presurgical antimicrobial infusions got a strong thumbs-up from all the groups, but only under a very specific circumstance: The medication has to be administered well in advance of surgery for it to be effective.
“Your goal is to get the appropriate concentration into the tissues by the time of incision,” Dr. Duane said. “It takes time to get there – if you give it after the incision, you have bleeding and cellular death, and the antimicrobials cannot get to that incision and do their job. If I’m starting a case and they haven’t been given, I don’t ever start them after the incision, because then you have all of the risks and none of the benefits. In my opinion, we need to move to no further antimicrobials once the incision or case is over because it serves no purpose and is inconsistent with good antibiotic stewardship.”
Adhesive drapes got a resounding “eh” from the guidelines, Dr. Duane said. “You really do not need them. They’re expensive and they’re not improving outcomes, so don’t waste your time or money. We need to think about minimizing what isn’t helpful and maximizing the things that are worthwhile. That’s the way to practice good socially responsible surgery without breaking the bank,” she said.
Antimicrobial sutures got weak recommendations, Dr. Duane said. The evidence supporting their use was not very strong, although she said she feels triclosan-coated sutures are helpful in all kinds of surgery. Preoperative showering with an antiseptic received strong support, with alcohol-containing preps superior to chlorhexidine, which is better than povidone-iodine–containing solutions.
Deep-space irrigation with aqueous iodophor also received a weak recommendation, but Dr. Duane said the evidence does not support the use of antibiotic-containing irrigation in either the abdomen or the incision. “And the guidelines came out strongly against using antimicrobial agents on the incision,” she said. None of the guidelines issued a recommendation for or against antimicrobial dressings.
Protocolized infection-control bundles are a very great help in reducing the incidence of surgical site infections, Dr. Duane added. “They increase attention to detail and decrease the rates of infection.”
Dr. Cook agreed. “Central line bundles are one of the things that work” for line-associated bloodstream infections, he said. Since their large-scale adoption, mortality from these infections has dropped significantly; it was hovering around 28,000 per year in the mid-2000s, he said. “That’s about how many men die from prostate cancer every year.”
Central line infections are very costly too, he added – around $46,000 per event. “That comes to around $2 billion in direct and indirect costs every year.”
A 2006 study demonstrated the efficacy of central line bundles in the fight against these potentially devastating infections.
The bundled intervention comprised hand washing, using full-barrier precautions during the insertion of central venous catheters, cleaning the skin with chlorhexidine, avoiding the femoral site if possible, and removing unnecessary catheters. The median rate of catheter-related bloodstream infections per 1,000 catheter-days decreased from 2.7 at baseline to 0 at 3 months after implementation of the study intervention.
Antibiotic-coated or impregnated catheters do not work as well. A 2016 Cochrane review of 57 studies determined that the devices didn’t improve sepsis, all-cause mortality, or catheter-related local infections.
The jury may still be out on coated dressings or securing devices, however. Another Cochrane review, of 22 studies, found a 40% decrease in central line–associated bloodstream infections with these items. “There was moderate evidence that tip colonization was reduced, but the authors said more research is needed.”
The evidence looks stronger for alcohol-impregnated port protectors, Dr. Cook said. Two studies in particular support their use. In an oncology unit, the rate of these infections dropped from 2.3 to 0.3 per 1,000 catheter days after the port protectors were instituted.
In the second study, infection rates declined from 1.43 to 0.69 per 1,000 line-days after the protectors came on board.
“The advantage was seen mostly in ICUs, so the recommendations are to use them there,” Dr. Cook said.
Neither Dr. Cook nor Dr. Duane had any relevant financial disclosures.
[email protected]
On Twitter @Alz Gal
FROM THE ACS CLINICAL CONGRESS
Which Patients Respond Best to Repeat Epilepsy Surgery?
Repeat surgery is worth considering in patients with intractable epilepsy if they continue to have debilitating episodes after the first procedure, according to a meta-analysis and systematic review that looked at 782 patients from 36 studies. But the success of repeat resection is dependent on several positive and negative predictive factors.
- Krucoff et al conducted the first quantitative meta-analysis of theresearch literature from the last 30 years to determine the rate of successful repeat surgeries and to find predictors.
- Congruent electrophysiology data was a better predictor of seizure freedom when compared to noncongruent data, with an odds ratio (OR) of 3.6.
- Freedom from seizures after repeat surgery was better predicted for lesional than nonlesional epilepsy (OR, 3.2).
- Another predictor of seizure freedom was surgical limitations, when compared to disease-related factors that had been associated with failure of the first surgery. (OR, 2.6).
- 58% of patients were seizure free after repeat resection if they had at least one of these predictive factors.
Krucoff MO, Chan AY, Harward SC, et al. Rates and predictors of success and failure in repeat epilepsy surgery: a meta-analysis and systematic review. [Published online ahead of print October 10, 2017] Epilepsia. doi: 10.1111/epi.13920.
Repeat surgery is worth considering in patients with intractable epilepsy if they continue to have debilitating episodes after the first procedure, according to a meta-analysis and systematic review that looked at 782 patients from 36 studies. But the success of repeat resection is dependent on several positive and negative predictive factors.
- Krucoff et al conducted the first quantitative meta-analysis of theresearch literature from the last 30 years to determine the rate of successful repeat surgeries and to find predictors.
- Congruent electrophysiology data was a better predictor of seizure freedom when compared to noncongruent data, with an odds ratio (OR) of 3.6.
- Freedom from seizures after repeat surgery was better predicted for lesional than nonlesional epilepsy (OR, 3.2).
- Another predictor of seizure freedom was surgical limitations, when compared to disease-related factors that had been associated with failure of the first surgery. (OR, 2.6).
- 58% of patients were seizure free after repeat resection if they had at least one of these predictive factors.
Krucoff MO, Chan AY, Harward SC, et al. Rates and predictors of success and failure in repeat epilepsy surgery: a meta-analysis and systematic review. [Published online ahead of print October 10, 2017] Epilepsia. doi: 10.1111/epi.13920.
Repeat surgery is worth considering in patients with intractable epilepsy if they continue to have debilitating episodes after the first procedure, according to a meta-analysis and systematic review that looked at 782 patients from 36 studies. But the success of repeat resection is dependent on several positive and negative predictive factors.
- Krucoff et al conducted the first quantitative meta-analysis of theresearch literature from the last 30 years to determine the rate of successful repeat surgeries and to find predictors.
- Congruent electrophysiology data was a better predictor of seizure freedom when compared to noncongruent data, with an odds ratio (OR) of 3.6.
- Freedom from seizures after repeat surgery was better predicted for lesional than nonlesional epilepsy (OR, 3.2).
- Another predictor of seizure freedom was surgical limitations, when compared to disease-related factors that had been associated with failure of the first surgery. (OR, 2.6).
- 58% of patients were seizure free after repeat resection if they had at least one of these predictive factors.
Krucoff MO, Chan AY, Harward SC, et al. Rates and predictors of success and failure in repeat epilepsy surgery: a meta-analysis and systematic review. [Published online ahead of print October 10, 2017] Epilepsia. doi: 10.1111/epi.13920.