User login
Red line when skin is rubbed

The FP suspected that this was a case of dermatographism. To confirm his suspicions, he used the end of a cotton-tipped applicator and wrote on the patient’s skin. Within 3 minutes, the writing turned into a triple reaction with some erythema, blanching, and swelling—confirming the diagnosis.
Dermatographism often accompanies urticaria, but can occur without it. If one writes on the skin, one is able to see the resulting words or shapes. The cause of this condition is unknown, but the pathophysiology involves degranulation of mast cells and the release of histamine.
No treatment is required for asymptomatic dermatographism. If patients are symptomatic, they should be advised to avoid precipitating physical stimuli. Dry skin may stimulate scratching, so the use of emollients can help. If the patient wants a medication, start with a second-generation antihistamine such as loratadine or cetirizine. While the recommended over-the-counter dose of these 2 medications is 10 mg/d, the dose may be increased to 20 mg twice daily. It is best to start at the lowest dose and then titrate up according to response and tolerance of adverse effects. (The second-generation antihistamines can still be sedating.) If this doesn’t work, a sedating antihistamine can be added before bedtime.
Dermatographism is not life-threatening and does not lead to anaphylaxis. The patient in this case didn’t want treatment and was pleased to know what was going on with his skin.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Urticaria and angioedema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 863-870.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP suspected that this was a case of dermatographism. To confirm his suspicions, he used the end of a cotton-tipped applicator and wrote on the patient’s skin. Within 3 minutes, the writing turned into a triple reaction with some erythema, blanching, and swelling—confirming the diagnosis.
Dermatographism often accompanies urticaria, but can occur without it. If one writes on the skin, one is able to see the resulting words or shapes. The cause of this condition is unknown, but the pathophysiology involves degranulation of mast cells and the release of histamine.
No treatment is required for asymptomatic dermatographism. If patients are symptomatic, they should be advised to avoid precipitating physical stimuli. Dry skin may stimulate scratching, so the use of emollients can help. If the patient wants a medication, start with a second-generation antihistamine such as loratadine or cetirizine. While the recommended over-the-counter dose of these 2 medications is 10 mg/d, the dose may be increased to 20 mg twice daily. It is best to start at the lowest dose and then titrate up according to response and tolerance of adverse effects. (The second-generation antihistamines can still be sedating.) If this doesn’t work, a sedating antihistamine can be added before bedtime.
Dermatographism is not life-threatening and does not lead to anaphylaxis. The patient in this case didn’t want treatment and was pleased to know what was going on with his skin.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Urticaria and angioedema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 863-870.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP suspected that this was a case of dermatographism. To confirm his suspicions, he used the end of a cotton-tipped applicator and wrote on the patient’s skin. Within 3 minutes, the writing turned into a triple reaction with some erythema, blanching, and swelling—confirming the diagnosis.
Dermatographism often accompanies urticaria, but can occur without it. If one writes on the skin, one is able to see the resulting words or shapes. The cause of this condition is unknown, but the pathophysiology involves degranulation of mast cells and the release of histamine.
No treatment is required for asymptomatic dermatographism. If patients are symptomatic, they should be advised to avoid precipitating physical stimuli. Dry skin may stimulate scratching, so the use of emollients can help. If the patient wants a medication, start with a second-generation antihistamine such as loratadine or cetirizine. While the recommended over-the-counter dose of these 2 medications is 10 mg/d, the dose may be increased to 20 mg twice daily. It is best to start at the lowest dose and then titrate up according to response and tolerance of adverse effects. (The second-generation antihistamines can still be sedating.) If this doesn’t work, a sedating antihistamine can be added before bedtime.
Dermatographism is not life-threatening and does not lead to anaphylaxis. The patient in this case didn’t want treatment and was pleased to know what was going on with his skin.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R. Urticaria and angioedema. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 863-870.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com

Liver disease doubles risk of colorectal cancer
Chronic liver disease appears to double the risk of colorectal cancer (CRC), even after patients undergo liver transplantation, according to a report published in Gastrointestinal Endoscopy.
“Strict surveillance for colorectal cancer is warranted in this patient population,” said Yuga Komaki, MD, of the section of gastroenterology, hepatology, and nutrition, University of Chicago, and associates.
One prominent chronic liver disease, primary sclerosing cholangitis, is known to raise the risk of CRC, which “is mainly attributed to the concurrence of inflammatory bowel disease.” In addition, whether liver transplantation mitigates that risk remains “controversial,” the investigators noted.
To assess whether chronic liver disease impacts CRC risk, they performed a systematic review and meta-analysis of the literature, examining data from 55 observational studies involving 55,991 participants. Case patients had a variety of chronic liver diseases, including primary sclerosing cholangitis, viral hepatitis, autoimmune hepatitis, primary biliary cirrhosis, and alcoholic liver damage.
Overall, the pooled standardized incidence ratio of CRC was 2.06 among patients with liver disease, compared with control subjects. It was highest in the subgroup of patients with primary sclerosing cholangitis at 6.70, the investigators said (Gastrointest. Endosc. 2017;86:93-104).
CRC risk appeared to be slightly higher among patients who had cirrhosis than among those who had hepatitis, “suggesting that advanced liver damage may lead to higher risks of CRC. This is not surprising because advanced liver damage can cause systemic alterations in immunity that may precipitate malignant transformation,” Dr. Komaki and associates noted.
The pooled standardized incidence ratio of CRC remained elevated at 2.16 among patients who underwent liver transplantation for a variety of causes. It is possible that their exposure to immunosuppressive therapy plays a role in elevating this risk, the researchers added.
“We propose that patients with chronic hepatitis and cirrhosis require a screening colonoscopy every 5 years, as opposed to the 10-year interval in the general population. Patients undergoing liver transplant should have a colonoscopy before the transplant and, subsequently, should undergo colonoscopy at 5-year intervals,” Dr. Komaki and associates said.
They added that the sixfold increase in CRC risk among patients with PSC “justifies the present recommendation of annual surveillance colonoscopy that should be continued after transplant.”
Chronic liver disease appears to double the risk of colorectal cancer (CRC), even after patients undergo liver transplantation, according to a report published in Gastrointestinal Endoscopy.
“Strict surveillance for colorectal cancer is warranted in this patient population,” said Yuga Komaki, MD, of the section of gastroenterology, hepatology, and nutrition, University of Chicago, and associates.
One prominent chronic liver disease, primary sclerosing cholangitis, is known to raise the risk of CRC, which “is mainly attributed to the concurrence of inflammatory bowel disease.” In addition, whether liver transplantation mitigates that risk remains “controversial,” the investigators noted.
To assess whether chronic liver disease impacts CRC risk, they performed a systematic review and meta-analysis of the literature, examining data from 55 observational studies involving 55,991 participants. Case patients had a variety of chronic liver diseases, including primary sclerosing cholangitis, viral hepatitis, autoimmune hepatitis, primary biliary cirrhosis, and alcoholic liver damage.
Overall, the pooled standardized incidence ratio of CRC was 2.06 among patients with liver disease, compared with control subjects. It was highest in the subgroup of patients with primary sclerosing cholangitis at 6.70, the investigators said (Gastrointest. Endosc. 2017;86:93-104).
CRC risk appeared to be slightly higher among patients who had cirrhosis than among those who had hepatitis, “suggesting that advanced liver damage may lead to higher risks of CRC. This is not surprising because advanced liver damage can cause systemic alterations in immunity that may precipitate malignant transformation,” Dr. Komaki and associates noted.
The pooled standardized incidence ratio of CRC remained elevated at 2.16 among patients who underwent liver transplantation for a variety of causes. It is possible that their exposure to immunosuppressive therapy plays a role in elevating this risk, the researchers added.
“We propose that patients with chronic hepatitis and cirrhosis require a screening colonoscopy every 5 years, as opposed to the 10-year interval in the general population. Patients undergoing liver transplant should have a colonoscopy before the transplant and, subsequently, should undergo colonoscopy at 5-year intervals,” Dr. Komaki and associates said.
They added that the sixfold increase in CRC risk among patients with PSC “justifies the present recommendation of annual surveillance colonoscopy that should be continued after transplant.”
Chronic liver disease appears to double the risk of colorectal cancer (CRC), even after patients undergo liver transplantation, according to a report published in Gastrointestinal Endoscopy.
“Strict surveillance for colorectal cancer is warranted in this patient population,” said Yuga Komaki, MD, of the section of gastroenterology, hepatology, and nutrition, University of Chicago, and associates.
One prominent chronic liver disease, primary sclerosing cholangitis, is known to raise the risk of CRC, which “is mainly attributed to the concurrence of inflammatory bowel disease.” In addition, whether liver transplantation mitigates that risk remains “controversial,” the investigators noted.
To assess whether chronic liver disease impacts CRC risk, they performed a systematic review and meta-analysis of the literature, examining data from 55 observational studies involving 55,991 participants. Case patients had a variety of chronic liver diseases, including primary sclerosing cholangitis, viral hepatitis, autoimmune hepatitis, primary biliary cirrhosis, and alcoholic liver damage.
Overall, the pooled standardized incidence ratio of CRC was 2.06 among patients with liver disease, compared with control subjects. It was highest in the subgroup of patients with primary sclerosing cholangitis at 6.70, the investigators said (Gastrointest. Endosc. 2017;86:93-104).
CRC risk appeared to be slightly higher among patients who had cirrhosis than among those who had hepatitis, “suggesting that advanced liver damage may lead to higher risks of CRC. This is not surprising because advanced liver damage can cause systemic alterations in immunity that may precipitate malignant transformation,” Dr. Komaki and associates noted.
The pooled standardized incidence ratio of CRC remained elevated at 2.16 among patients who underwent liver transplantation for a variety of causes. It is possible that their exposure to immunosuppressive therapy plays a role in elevating this risk, the researchers added.
“We propose that patients with chronic hepatitis and cirrhosis require a screening colonoscopy every 5 years, as opposed to the 10-year interval in the general population. Patients undergoing liver transplant should have a colonoscopy before the transplant and, subsequently, should undergo colonoscopy at 5-year intervals,” Dr. Komaki and associates said.
They added that the sixfold increase in CRC risk among patients with PSC “justifies the present recommendation of annual surveillance colonoscopy that should be continued after transplant.”
FROM GASTROINTESTINAL ENDOSCOPY
Key clinical point: Chronic liver disease appears to double the risk of colorectal cancer, even after patients undergo liver transplantation.
Major finding: The pooled standardized incidence ratio of colorectal cancer was 2.06 among patients with liver disease, compared with control subjects.
Data source: A meta-analysis of 50 observational studies (55,991 participants) that examined the rate of colorectal cancer in patients with a variety of liver diseases.
Disclosures: No specific sponsor was identified for this study. Dr. Komaki reported receiving research support from the Children’s Cancer Association of Japan. Dr. Komaki and associates reported having no other relevant financial disclosures.
Supreme Court rules to speed biosimilar drugs to market
The U.S. Supreme Court has ruled that biosimilar companies can take their versions of biological drugs to the market 6 months sooner in a precedential ruling that could mean quicker access to less expensive medications.
The unanimous ruling overturns an appeals court ruling in favor of California-based Amgen that had barred competitor Sandoz from marketing its biosimilar of Neupogen (filgrastim) until 6 months after Food and Drug Administration approval. Justices held that the Biologics Price Competition and Innovation Act of 2009 (BPCIA) allows biosimilar applicants to provide notice of commercial marketing prior to obtaining licensure by the FDA.
Carol Lynch, global head of Biopharmaceuticals at Sandoz, said the ruling helps to eliminate unnecessary barriers so that patients can access more affordable medicine in a more timely manner.
“Biosimilars offer significant value to patients, providers, and payers, increasing the number of treatment options available to patients across many disease areas at a reduced cost to the health care system,” Ms. Lynch said in a statement. “The justices’ unanimous ruling on the notice of commercial marketing will help expedite patient access to life-enhancing treatments. We also appreciate the clarity provided on the patent dance, which will help the biosimilars industry move forward.”

In a statement, an Amgen spokeswoman said the company was “disappointed in the court’s decision on the notice of commercial marketing,” but that it will “continue to seek to enforce our intellectual property against those parties that infringe upon our rights.”
The “patent dance” referred to by Ms. Lynch is the often lengthy process by which companies marketing brand name and biosimilar medications spar and undergo legal proceedings before the biosimilar can enter the market.
In this case, Sandoz filed an application with the FDA in May 2014 seeking approval to market Zarxio (filgrastim-sndz). Amgen, the manufacturer of the reference product, has marketed Neupogen since 1991 and holds patents on methods of manufacturing and using filgrastim. In July 2014, the FDA accepted Sandoz’ application for review. In October 2014, Amgen sued for patent infringement, alleging that Sandoz failed to adhere to the BPCIA by unlawfully providing its notice of commercial marketing before FDA licensure, among other arguments.
The U.S. Court of Appeals for the Federal Circuit in Washington ruled in favor of Amgen, holding that Sandoz must wait for an FDA license before marketing its biosimilar, which meant another 6-month waiting period. The Supreme Court disagreed. Justices based their decision on the plain language of the BPCIA, ruling that the statute allows for applicants to provide marketing notice either before or after receiving FDA approval.
In a statement, the Pharmaceutical Care Management Association said the Supreme Court’s ruling on biosimilars will help create more competition among costly biologic medications, “which is the key to reducing overall prescription drug costs for consumers, employers, government programs, and others.”
[email protected] On Twitter @legal_med
AGA Resource
AGA offers education materials for health care professionals and patients at www.gastro.org/biosimilars.
The U.S. Supreme Court has ruled that biosimilar companies can take their versions of biological drugs to the market 6 months sooner in a precedential ruling that could mean quicker access to less expensive medications.
The unanimous ruling overturns an appeals court ruling in favor of California-based Amgen that had barred competitor Sandoz from marketing its biosimilar of Neupogen (filgrastim) until 6 months after Food and Drug Administration approval. Justices held that the Biologics Price Competition and Innovation Act of 2009 (BPCIA) allows biosimilar applicants to provide notice of commercial marketing prior to obtaining licensure by the FDA.
Carol Lynch, global head of Biopharmaceuticals at Sandoz, said the ruling helps to eliminate unnecessary barriers so that patients can access more affordable medicine in a more timely manner.
“Biosimilars offer significant value to patients, providers, and payers, increasing the number of treatment options available to patients across many disease areas at a reduced cost to the health care system,” Ms. Lynch said in a statement. “The justices’ unanimous ruling on the notice of commercial marketing will help expedite patient access to life-enhancing treatments. We also appreciate the clarity provided on the patent dance, which will help the biosimilars industry move forward.”
In a statement, an Amgen spokeswoman said the company was “disappointed in the court’s decision on the notice of commercial marketing,” but that it will “continue to seek to enforce our intellectual property against those parties that infringe upon our rights.”
The “patent dance” referred to by Ms. Lynch is the often lengthy process by which companies marketing brand name and biosimilar medications spar and undergo legal proceedings before the biosimilar can enter the market.
In this case, Sandoz filed an application with the FDA in May 2014 seeking approval to market Zarxio (filgrastim-sndz). Amgen, the manufacturer of the reference product, has marketed Neupogen since 1991 and holds patents on methods of manufacturing and using filgrastim. In July 2014, the FDA accepted Sandoz’ application for review. In October 2014, Amgen sued for patent infringement, alleging that Sandoz failed to adhere to the BPCIA by unlawfully providing its notice of commercial marketing before FDA licensure, among other arguments.
The U.S. Court of Appeals for the Federal Circuit in Washington ruled in favor of Amgen, holding that Sandoz must wait for an FDA license before marketing its biosimilar, which meant another 6-month waiting period. The Supreme Court disagreed. Justices based their decision on the plain language of the BPCIA, ruling that the statute allows for applicants to provide marketing notice either before or after receiving FDA approval.
In a statement, the Pharmaceutical Care Management Association said the Supreme Court’s ruling on biosimilars will help create more competition among costly biologic medications, “which is the key to reducing overall prescription drug costs for consumers, employers, government programs, and others.”
[email protected] On Twitter @legal_med
AGA Resource
AGA offers education materials for health care professionals and patients at www.gastro.org/biosimilars.
The U.S. Supreme Court has ruled that biosimilar companies can take their versions of biological drugs to the market 6 months sooner in a precedential ruling that could mean quicker access to less expensive medications.
The unanimous ruling overturns an appeals court ruling in favor of California-based Amgen that had barred competitor Sandoz from marketing its biosimilar of Neupogen (filgrastim) until 6 months after Food and Drug Administration approval. Justices held that the Biologics Price Competition and Innovation Act of 2009 (BPCIA) allows biosimilar applicants to provide notice of commercial marketing prior to obtaining licensure by the FDA.
Carol Lynch, global head of Biopharmaceuticals at Sandoz, said the ruling helps to eliminate unnecessary barriers so that patients can access more affordable medicine in a more timely manner.
“Biosimilars offer significant value to patients, providers, and payers, increasing the number of treatment options available to patients across many disease areas at a reduced cost to the health care system,” Ms. Lynch said in a statement. “The justices’ unanimous ruling on the notice of commercial marketing will help expedite patient access to life-enhancing treatments. We also appreciate the clarity provided on the patent dance, which will help the biosimilars industry move forward.”
In a statement, an Amgen spokeswoman said the company was “disappointed in the court’s decision on the notice of commercial marketing,” but that it will “continue to seek to enforce our intellectual property against those parties that infringe upon our rights.”
The “patent dance” referred to by Ms. Lynch is the often lengthy process by which companies marketing brand name and biosimilar medications spar and undergo legal proceedings before the biosimilar can enter the market.
In this case, Sandoz filed an application with the FDA in May 2014 seeking approval to market Zarxio (filgrastim-sndz). Amgen, the manufacturer of the reference product, has marketed Neupogen since 1991 and holds patents on methods of manufacturing and using filgrastim. In July 2014, the FDA accepted Sandoz’ application for review. In October 2014, Amgen sued for patent infringement, alleging that Sandoz failed to adhere to the BPCIA by unlawfully providing its notice of commercial marketing before FDA licensure, among other arguments.
The U.S. Court of Appeals for the Federal Circuit in Washington ruled in favor of Amgen, holding that Sandoz must wait for an FDA license before marketing its biosimilar, which meant another 6-month waiting period. The Supreme Court disagreed. Justices based their decision on the plain language of the BPCIA, ruling that the statute allows for applicants to provide marketing notice either before or after receiving FDA approval.
In a statement, the Pharmaceutical Care Management Association said the Supreme Court’s ruling on biosimilars will help create more competition among costly biologic medications, “which is the key to reducing overall prescription drug costs for consumers, employers, government programs, and others.”
[email protected] On Twitter @legal_med
AGA Resource
AGA offers education materials for health care professionals and patients at www.gastro.org/biosimilars.
Digital cohorts within the social mediome to circumvent conventional research challenges?
We are becoming comfortable with the concept of a sharing economy, where resources are shared among many individuals using online forums. Whether activities involve sharing rides (Uber, Lyft, and others), accommodations (Airbnb), or information (social media), underlying attributes include reduced transactional costs, enhanced information transparency, dynamic feedback, and socialization of opportunity. As health care systems realize that they are changing from direct-to-business to a direct-to-customer model, their ability to connect directly with individuals will become a foundational strategy.
This month’s column introduces us to social media as a research tool. Information derived from social media sites can be harvested for critical clinical information (the Centers for Disease and Control and Prevention tracks the spread of influenza using social media analytic tools), research data (patient preferences), and as a recruitment method for clinical studies. Kulanthaivel and colleagues have described their experiences and literature review to help us imagine new ways to collect data at markedly reduced transaction costs (compared to a formal clinical trial). While there are many cautions about the use of social media in your practice or research, we are only beginning to understand its potential.
John I. Allen, MD, MBA, AGAF
Editor in Chief
Medical knowledge, culminating from the collection and translation of patient data, is the primary objective of the clinical research paradigm. The successful conduct of this traditional model has become even more challenging with expansion of costs and a dwindling research infrastructure. Beyond systemic issues, conventional research methods are burdened further by minimal patient engagement, inadequate staffing, and geographic limitations to recruitment. Clinical research also has failed to keep pace with patient demands, and the limited scope of well-funded, disease-specific investigations have left many patients feeling disenfranchised. Social media venues may represent a viable option to surpass these current and evolving barriers when used as an adjunctive approach to traditional clinical investigation.

Advantages and pitfalls in social media research
SM is a new frontier containing a wide spectrum of clinical and qualitative data from connected users (patients). Collection and examination of either individuals’ or groups’ SM information use can provide insight into qualitative life experiences, just as analysis of biologic samples can enable dissection of genetic disease underpinnings. This mediome is analogous to the human genome, both in content and utility.1 Analyzing data streams from SM for interpersonal interactions, message content, and even frequency can provide digital investigators with volumes of information that otherwise would remain unattainable.

Several limitations and potential risks of SM for medical research should be addressed, including the possible compromise of privacy and confidentiality, the use and dissemination of medical advice and information, potential demographic biases, and a required trust of the investigator by patients. Many of these challenges can be similar to traditional methods, however, as in the conventional model, careful management can drastically reduce unwanted study issues.
The risk of Health Insurance Portability and Accountability Act violations must be considered seriously in the context of patient–researcher interactions on SM. Because of the relatively public nature of these venues, patient confidentiality may be at risk if patients choose to divulge personal medical information. However, if proper protective measures are taken to ensure that the venue is secure (e.g., a private or closed group on Facebook or a by-invitation-only online Internet forum), and the researcher vets all patients who request entrance into the group, this risk may be minimized. Moreover, to further reduce any legal liability, the researcher should not provide any medical advice to patients who participate in a SM study. The drive to provide medical direction in study patients with clinical need may be strong because collaborative relationships between investigator and patients are likely to form. Furthermore, digital access to investigators on SM commonly becomes easy for patients. Safe approaches to communication could include redirecting patients to consult with their own doctor for advice, unbiased dissemination of disease-specific educational materials, or depiction of only institutional review board–approved study materials.7,8

The perception that only younger populations use SM may appear to be a significant limitation for its implementation in clinical research. However, this limitation is rapidly becoming less significant because recent studies have shown that the use of SM has become increasingly common among older adults. As of 2014, more than half of the US adult population used Facebook, including 73% and 63% of Internet-using adults ages 30–49 and 50–64 years, respectively.10 SM may not be suitable for all diseases, however, there is likely significant demographic overlap for many disease populations.
Finally, it is imperative for researchers to gain the trust of patients on SM to effectively use these venues for research purposes. Because patient–researcher interaction does not occur face-to-face on these platforms, gaining the trust of patients may be more difficult than it would be in a clinical setting. Thus, patient–patient and patient–researcher communications within SM platforms must be cultivated carefully to instill participant confidence in the research being performed on their behalf. One of the authors (C.L.) has established an SM educational model for this exchange.4 Specifically, he provides patients with a distillation of current field research by posting updates in a research-specific Facebook group and on Twitter. This model not only empowers patients with disease education, it also solidifies the importance of patient investment in disease-specific research. Furthermore, invested patients bring ideas to research, take a more educated and proactive role in their care team, and, ultimately, return to seek more study involvement.

Social media in rare disease research
Rare diseases (conditions with a prevalence of less than 200,000 patients in North America), in particular, are prime for high-yield results and community impact using novel SM approaches. This is the result of established digital support groups, publications with historically low study numbers, and few focused investigators. Several studies of rare diseases have shown considerable advantages of using SM as a study tool. For instance, an existing neuroendocrine cervical cancer Facebook support group recently was used to recruit a geographically widespread cohort of patients with this rare cancer. Through an online survey posted in the Facebook group, patients were able to provide specific information on their treatment, disease, and symptom history, current disease status, and quality of life, including various psychological factors. Without the use of SM, collecting this information would have been virtually impossible because the patients were treated at 51 cancer centers across the country.14
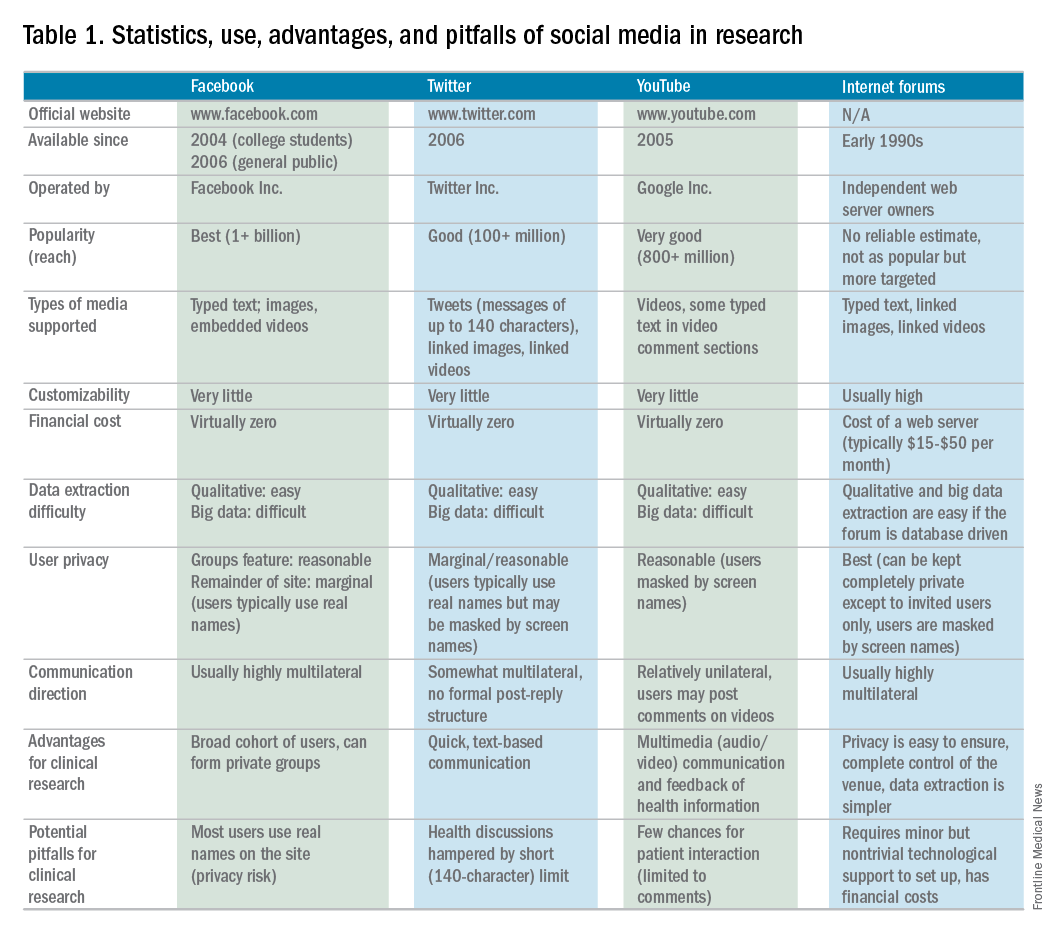
Currently, the use of SM in hepatology research, focused specifically on autoimmune hepatitis (AIH), is under exploration at Indiana University. AIH is a rare autoimmune liver disease that results in immune-mediated destruction of liver cells, possibly resulting in fibrosis, cirrhosis, or liver failure if treatment is unsuccessful. One of the authors (C.L.) used both Facebook and Twitter to construct a large study group of individuals affected with AIH called the Autoimmune Hepatitis Research Network (AHRN; 1,500 members) during the past 2 years.4 Interested individuals have joined this research group after searching for AIH online support groups or reading shared AHRN posts on other media platforms. Between April 2015 and April 2016, there were posts by more than 750 unique active members (more than 50% of the group contributes to discussions), most of whom appear to be either caregivers of AIH patients or AIH patients themselves.
Preliminary informational analysis on this group has shown that C.L. and study collaborators have been able to uncover rich clinical and nonclinical information that otherwise would remain unknown. This research was performed by semi-automated download of the Facebook group’s content and subsequent semantic analysis. Qualitative analysis also was performed by direct reading of patient narratives. Collected clinical information has included histories of medication side effects, familial autoimmune diseases, and comorbid conditions. The most common factors that patients were unlikely to discuss with a provider (e.g., financial issues, employment, personal relationships, use of supplements, and alcohol use) frequently were discussed in the AHRN group, allowing a more transparent view of the complete disease experience.
Beyond research conducted in the current paradigm, the AHRN has provided a rich community construct in which patients offer each other social support. The patient impression of AHRN on Facebook has been overwhelmingly positive, and patients often wonder why such a model has not been used with other diseases. The close digital interaction the author (C.L.) has had with numerous patients and families has promoted other benefits of this methodology: more than 40 new AIH patients from outside Indiana have traveled to Indiana University for medical consultation despite no advertisement.
Conclusions
SM has the potential to transform health care research as a supplement to traditional research methods. Compared with a conventional research model, this methodology has proven to be cost and time effective, wide reaching, and similarly capable of data collection. Use of SM in research has tremendous potential to direct patient-centered research because invested patient collaborators can take an active role in their own disease and may hone investigatory focus on stakeholder priorities. Limitations to this method are known, however; if implemented cautiously, these can be mitigated. Investment in and application of the social mediome by investigators and patients has the potential to support and transform research that otherwise would be impossible.
Acknowledgments
The authors wish to extend their gratitude to the members of the Autoimmune Hepatitis Research Network for their continued proactivity and engagement in autoimmune hepatitis research. Furthermore, the authors are grateful to Dr. Naga Chalasani for his continued mentorship and extensive contributions to the development of social media approaches in clinical investigation.
References
1. Asch, D.A., Rader, D.J., Merchant, R.M. Mining the social mediome. Trends Mol Med. 2015;21:528-9.
2. Brotherton, C.S., Martin, C.A., Long, M.D. et al. Avoidance of fiber is associated with greater risk of Crohn’s disease flare in a 6-month period. Clin Gastroenterol Hepatol. 2016;14:1130-6.
3. Fenner, Y., Garland, S.M., Moore, E.E., et al. Web-based recruiting for health research using a social networking site: an exploratory study. J Med Internet Res. 2012;14:e20.
4. Lammert, C., Comerford, M., Love, J., et al. Investigation gone viral: application of the social mediasphere in research. Gastroenterology. 2015;149:839-43.
5. Wicks, P., Massagli, M., Frost, J., et al. Sharing health data for better outcomes on PatientsLikeMe. J Med Internet Res. 2010;12:e19.
6. Admon, L., Haefner, J.K., Kolenic, G.E., et al. Recruiting pregnant patients for survey research: a head to head comparison of social media-based versus clinic-based approaches. J Med Internet Res. 2016;18:e326.
7. Farnan, J.M., Sulmasy, L.S., Chaudhry, H. Online medical professionalism. Ann Intern Med. 2013;159:158-9.
8. Massachusetts Medical Society: Social Media Guidelines for Physicians. Available from: http://www.massmed.org/Physicians/Legal-and-Regulatory/Social-Media-Guidelines-for-Physicians/#. Accessed: January 3, 2017.
9. Pirraglia, P.A. Kravitz, R.L. Social media: new opportunities, new ethical concerns. J Gen Intern Med. 2013;28:165-6.
10. Duggan, M., Ellison, N.B., Lampe, C. et al. Demographics of key social networking platforms. (Available from:) (Accessed: January 4, 2017) Pew Res Cent Internet Sci Tech. 2015; http://www.pewinternet.org/2015/01/09/demographics-of-key-social-networking-platforms-2
11. Kang, X., Zhao, L., Leung, F., et al. Delivery of Instructions via mobile social media app increases quality of bowel preparation. Clin Gastroenterol Hepatol. 2016;14:429-35.
12. Bajaj, J.S., Heuman, D.M., Sterling, R.K., et al. Validation of EncephalApp, Smartphone-based Stroop test, for the diagnosis of covert hepatic encephalopathy. Clin Gastroenterol Hepatol. 2015;13:1828-35.
13. Riaz, M.S. Atreja, A. Personalized technologies in chronic gastrointestinal disorders: self-monitoring and remote sensor technologies. Clin Gastroenterol Hepatol. 2016;14:1697-705.
14. Zaid, T., Burzawa, J., Basen-Engquist, K., et al. Use of social media to conduct a cross-sectional epidemiologic and quality of life survey of patients with neuroendocrine carcinoma of the cervix: a feasibility study. Gynecol Oncol. 2014;132:149-53.
15. Schumacher, K.R., Stringer, K.A., Donohue, J.E., et al. Social media methods for studying rare diseases. Pediatrics. 2014;133:e1345–53.
Dr. Kulanthaivel and Dr. Jones are in the school of informatics and computing, Purdue University, Indiana University, Indianapolis; Dr. Fogel and Dr. Lammert are in the department of digestive and liver diseases, Indiana University School of Medicine, Indianapolis. This study was supported by KL2TR001106 and UL1TR001108 from the National Institutes of Health, and the Clinical and Translational Sciences Award from the National Center for Advancing Translational Sciences (C.L.). The authors disclose no conflicts.
We are becoming comfortable with the concept of a sharing economy, where resources are shared among many individuals using online forums. Whether activities involve sharing rides (Uber, Lyft, and others), accommodations (Airbnb), or information (social media), underlying attributes include reduced transactional costs, enhanced information transparency, dynamic feedback, and socialization of opportunity. As health care systems realize that they are changing from direct-to-business to a direct-to-customer model, their ability to connect directly with individuals will become a foundational strategy.
This month’s column introduces us to social media as a research tool. Information derived from social media sites can be harvested for critical clinical information (the Centers for Disease and Control and Prevention tracks the spread of influenza using social media analytic tools), research data (patient preferences), and as a recruitment method for clinical studies. Kulanthaivel and colleagues have described their experiences and literature review to help us imagine new ways to collect data at markedly reduced transaction costs (compared to a formal clinical trial). While there are many cautions about the use of social media in your practice or research, we are only beginning to understand its potential.
John I. Allen, MD, MBA, AGAF
Editor in Chief
Medical knowledge, culminating from the collection and translation of patient data, is the primary objective of the clinical research paradigm. The successful conduct of this traditional model has become even more challenging with expansion of costs and a dwindling research infrastructure. Beyond systemic issues, conventional research methods are burdened further by minimal patient engagement, inadequate staffing, and geographic limitations to recruitment. Clinical research also has failed to keep pace with patient demands, and the limited scope of well-funded, disease-specific investigations have left many patients feeling disenfranchised. Social media venues may represent a viable option to surpass these current and evolving barriers when used as an adjunctive approach to traditional clinical investigation.
Advantages and pitfalls in social media research
SM is a new frontier containing a wide spectrum of clinical and qualitative data from connected users (patients). Collection and examination of either individuals’ or groups’ SM information use can provide insight into qualitative life experiences, just as analysis of biologic samples can enable dissection of genetic disease underpinnings. This mediome is analogous to the human genome, both in content and utility.1 Analyzing data streams from SM for interpersonal interactions, message content, and even frequency can provide digital investigators with volumes of information that otherwise would remain unattainable.
Several limitations and potential risks of SM for medical research should be addressed, including the possible compromise of privacy and confidentiality, the use and dissemination of medical advice and information, potential demographic biases, and a required trust of the investigator by patients. Many of these challenges can be similar to traditional methods, however, as in the conventional model, careful management can drastically reduce unwanted study issues.
The risk of Health Insurance Portability and Accountability Act violations must be considered seriously in the context of patient–researcher interactions on SM. Because of the relatively public nature of these venues, patient confidentiality may be at risk if patients choose to divulge personal medical information. However, if proper protective measures are taken to ensure that the venue is secure (e.g., a private or closed group on Facebook or a by-invitation-only online Internet forum), and the researcher vets all patients who request entrance into the group, this risk may be minimized. Moreover, to further reduce any legal liability, the researcher should not provide any medical advice to patients who participate in a SM study. The drive to provide medical direction in study patients with clinical need may be strong because collaborative relationships between investigator and patients are likely to form. Furthermore, digital access to investigators on SM commonly becomes easy for patients. Safe approaches to communication could include redirecting patients to consult with their own doctor for advice, unbiased dissemination of disease-specific educational materials, or depiction of only institutional review board–approved study materials.7,8
The perception that only younger populations use SM may appear to be a significant limitation for its implementation in clinical research. However, this limitation is rapidly becoming less significant because recent studies have shown that the use of SM has become increasingly common among older adults. As of 2014, more than half of the US adult population used Facebook, including 73% and 63% of Internet-using adults ages 30–49 and 50–64 years, respectively.10 SM may not be suitable for all diseases, however, there is likely significant demographic overlap for many disease populations.
Finally, it is imperative for researchers to gain the trust of patients on SM to effectively use these venues for research purposes. Because patient–researcher interaction does not occur face-to-face on these platforms, gaining the trust of patients may be more difficult than it would be in a clinical setting. Thus, patient–patient and patient–researcher communications within SM platforms must be cultivated carefully to instill participant confidence in the research being performed on their behalf. One of the authors (C.L.) has established an SM educational model for this exchange.4 Specifically, he provides patients with a distillation of current field research by posting updates in a research-specific Facebook group and on Twitter. This model not only empowers patients with disease education, it also solidifies the importance of patient investment in disease-specific research. Furthermore, invested patients bring ideas to research, take a more educated and proactive role in their care team, and, ultimately, return to seek more study involvement.
Social media in rare disease research
Rare diseases (conditions with a prevalence of less than 200,000 patients in North America), in particular, are prime for high-yield results and community impact using novel SM approaches. This is the result of established digital support groups, publications with historically low study numbers, and few focused investigators. Several studies of rare diseases have shown considerable advantages of using SM as a study tool. For instance, an existing neuroendocrine cervical cancer Facebook support group recently was used to recruit a geographically widespread cohort of patients with this rare cancer. Through an online survey posted in the Facebook group, patients were able to provide specific information on their treatment, disease, and symptom history, current disease status, and quality of life, including various psychological factors. Without the use of SM, collecting this information would have been virtually impossible because the patients were treated at 51 cancer centers across the country.14
Currently, the use of SM in hepatology research, focused specifically on autoimmune hepatitis (AIH), is under exploration at Indiana University. AIH is a rare autoimmune liver disease that results in immune-mediated destruction of liver cells, possibly resulting in fibrosis, cirrhosis, or liver failure if treatment is unsuccessful. One of the authors (C.L.) used both Facebook and Twitter to construct a large study group of individuals affected with AIH called the Autoimmune Hepatitis Research Network (AHRN; 1,500 members) during the past 2 years.4 Interested individuals have joined this research group after searching for AIH online support groups or reading shared AHRN posts on other media platforms. Between April 2015 and April 2016, there were posts by more than 750 unique active members (more than 50% of the group contributes to discussions), most of whom appear to be either caregivers of AIH patients or AIH patients themselves.
Preliminary informational analysis on this group has shown that C.L. and study collaborators have been able to uncover rich clinical and nonclinical information that otherwise would remain unknown. This research was performed by semi-automated download of the Facebook group’s content and subsequent semantic analysis. Qualitative analysis also was performed by direct reading of patient narratives. Collected clinical information has included histories of medication side effects, familial autoimmune diseases, and comorbid conditions. The most common factors that patients were unlikely to discuss with a provider (e.g., financial issues, employment, personal relationships, use of supplements, and alcohol use) frequently were discussed in the AHRN group, allowing a more transparent view of the complete disease experience.
Beyond research conducted in the current paradigm, the AHRN has provided a rich community construct in which patients offer each other social support. The patient impression of AHRN on Facebook has been overwhelmingly positive, and patients often wonder why such a model has not been used with other diseases. The close digital interaction the author (C.L.) has had with numerous patients and families has promoted other benefits of this methodology: more than 40 new AIH patients from outside Indiana have traveled to Indiana University for medical consultation despite no advertisement.
Conclusions
SM has the potential to transform health care research as a supplement to traditional research methods. Compared with a conventional research model, this methodology has proven to be cost and time effective, wide reaching, and similarly capable of data collection. Use of SM in research has tremendous potential to direct patient-centered research because invested patient collaborators can take an active role in their own disease and may hone investigatory focus on stakeholder priorities. Limitations to this method are known, however; if implemented cautiously, these can be mitigated. Investment in and application of the social mediome by investigators and patients has the potential to support and transform research that otherwise would be impossible.
Acknowledgments
The authors wish to extend their gratitude to the members of the Autoimmune Hepatitis Research Network for their continued proactivity and engagement in autoimmune hepatitis research. Furthermore, the authors are grateful to Dr. Naga Chalasani for his continued mentorship and extensive contributions to the development of social media approaches in clinical investigation.
References
1. Asch, D.A., Rader, D.J., Merchant, R.M. Mining the social mediome. Trends Mol Med. 2015;21:528-9.
2. Brotherton, C.S., Martin, C.A., Long, M.D. et al. Avoidance of fiber is associated with greater risk of Crohn’s disease flare in a 6-month period. Clin Gastroenterol Hepatol. 2016;14:1130-6.
3. Fenner, Y., Garland, S.M., Moore, E.E., et al. Web-based recruiting for health research using a social networking site: an exploratory study. J Med Internet Res. 2012;14:e20.
4. Lammert, C., Comerford, M., Love, J., et al. Investigation gone viral: application of the social mediasphere in research. Gastroenterology. 2015;149:839-43.
5. Wicks, P., Massagli, M., Frost, J., et al. Sharing health data for better outcomes on PatientsLikeMe. J Med Internet Res. 2010;12:e19.
6. Admon, L., Haefner, J.K., Kolenic, G.E., et al. Recruiting pregnant patients for survey research: a head to head comparison of social media-based versus clinic-based approaches. J Med Internet Res. 2016;18:e326.
7. Farnan, J.M., Sulmasy, L.S., Chaudhry, H. Online medical professionalism. Ann Intern Med. 2013;159:158-9.
8. Massachusetts Medical Society: Social Media Guidelines for Physicians. Available from: http://www.massmed.org/Physicians/Legal-and-Regulatory/Social-Media-Guidelines-for-Physicians/#. Accessed: January 3, 2017.
9. Pirraglia, P.A. Kravitz, R.L. Social media: new opportunities, new ethical concerns. J Gen Intern Med. 2013;28:165-6.
10. Duggan, M., Ellison, N.B., Lampe, C. et al. Demographics of key social networking platforms. (Available from:) (Accessed: January 4, 2017) Pew Res Cent Internet Sci Tech. 2015; http://www.pewinternet.org/2015/01/09/demographics-of-key-social-networking-platforms-2
11. Kang, X., Zhao, L., Leung, F., et al. Delivery of Instructions via mobile social media app increases quality of bowel preparation. Clin Gastroenterol Hepatol. 2016;14:429-35.
12. Bajaj, J.S., Heuman, D.M., Sterling, R.K., et al. Validation of EncephalApp, Smartphone-based Stroop test, for the diagnosis of covert hepatic encephalopathy. Clin Gastroenterol Hepatol. 2015;13:1828-35.
13. Riaz, M.S. Atreja, A. Personalized technologies in chronic gastrointestinal disorders: self-monitoring and remote sensor technologies. Clin Gastroenterol Hepatol. 2016;14:1697-705.
14. Zaid, T., Burzawa, J., Basen-Engquist, K., et al. Use of social media to conduct a cross-sectional epidemiologic and quality of life survey of patients with neuroendocrine carcinoma of the cervix: a feasibility study. Gynecol Oncol. 2014;132:149-53.
15. Schumacher, K.R., Stringer, K.A., Donohue, J.E., et al. Social media methods for studying rare diseases. Pediatrics. 2014;133:e1345–53.
Dr. Kulanthaivel and Dr. Jones are in the school of informatics and computing, Purdue University, Indiana University, Indianapolis; Dr. Fogel and Dr. Lammert are in the department of digestive and liver diseases, Indiana University School of Medicine, Indianapolis. This study was supported by KL2TR001106 and UL1TR001108 from the National Institutes of Health, and the Clinical and Translational Sciences Award from the National Center for Advancing Translational Sciences (C.L.). The authors disclose no conflicts.
We are becoming comfortable with the concept of a sharing economy, where resources are shared among many individuals using online forums. Whether activities involve sharing rides (Uber, Lyft, and others), accommodations (Airbnb), or information (social media), underlying attributes include reduced transactional costs, enhanced information transparency, dynamic feedback, and socialization of opportunity. As health care systems realize that they are changing from direct-to-business to a direct-to-customer model, their ability to connect directly with individuals will become a foundational strategy.
This month’s column introduces us to social media as a research tool. Information derived from social media sites can be harvested for critical clinical information (the Centers for Disease and Control and Prevention tracks the spread of influenza using social media analytic tools), research data (patient preferences), and as a recruitment method for clinical studies. Kulanthaivel and colleagues have described their experiences and literature review to help us imagine new ways to collect data at markedly reduced transaction costs (compared to a formal clinical trial). While there are many cautions about the use of social media in your practice or research, we are only beginning to understand its potential.
John I. Allen, MD, MBA, AGAF
Editor in Chief
Medical knowledge, culminating from the collection and translation of patient data, is the primary objective of the clinical research paradigm. The successful conduct of this traditional model has become even more challenging with expansion of costs and a dwindling research infrastructure. Beyond systemic issues, conventional research methods are burdened further by minimal patient engagement, inadequate staffing, and geographic limitations to recruitment. Clinical research also has failed to keep pace with patient demands, and the limited scope of well-funded, disease-specific investigations have left many patients feeling disenfranchised. Social media venues may represent a viable option to surpass these current and evolving barriers when used as an adjunctive approach to traditional clinical investigation.
Advantages and pitfalls in social media research
SM is a new frontier containing a wide spectrum of clinical and qualitative data from connected users (patients). Collection and examination of either individuals’ or groups’ SM information use can provide insight into qualitative life experiences, just as analysis of biologic samples can enable dissection of genetic disease underpinnings. This mediome is analogous to the human genome, both in content and utility.1 Analyzing data streams from SM for interpersonal interactions, message content, and even frequency can provide digital investigators with volumes of information that otherwise would remain unattainable.
Several limitations and potential risks of SM for medical research should be addressed, including the possible compromise of privacy and confidentiality, the use and dissemination of medical advice and information, potential demographic biases, and a required trust of the investigator by patients. Many of these challenges can be similar to traditional methods, however, as in the conventional model, careful management can drastically reduce unwanted study issues.
The risk of Health Insurance Portability and Accountability Act violations must be considered seriously in the context of patient–researcher interactions on SM. Because of the relatively public nature of these venues, patient confidentiality may be at risk if patients choose to divulge personal medical information. However, if proper protective measures are taken to ensure that the venue is secure (e.g., a private or closed group on Facebook or a by-invitation-only online Internet forum), and the researcher vets all patients who request entrance into the group, this risk may be minimized. Moreover, to further reduce any legal liability, the researcher should not provide any medical advice to patients who participate in a SM study. The drive to provide medical direction in study patients with clinical need may be strong because collaborative relationships between investigator and patients are likely to form. Furthermore, digital access to investigators on SM commonly becomes easy for patients. Safe approaches to communication could include redirecting patients to consult with their own doctor for advice, unbiased dissemination of disease-specific educational materials, or depiction of only institutional review board–approved study materials.7,8
The perception that only younger populations use SM may appear to be a significant limitation for its implementation in clinical research. However, this limitation is rapidly becoming less significant because recent studies have shown that the use of SM has become increasingly common among older adults. As of 2014, more than half of the US adult population used Facebook, including 73% and 63% of Internet-using adults ages 30–49 and 50–64 years, respectively.10 SM may not be suitable for all diseases, however, there is likely significant demographic overlap for many disease populations.
Finally, it is imperative for researchers to gain the trust of patients on SM to effectively use these venues for research purposes. Because patient–researcher interaction does not occur face-to-face on these platforms, gaining the trust of patients may be more difficult than it would be in a clinical setting. Thus, patient–patient and patient–researcher communications within SM platforms must be cultivated carefully to instill participant confidence in the research being performed on their behalf. One of the authors (C.L.) has established an SM educational model for this exchange.4 Specifically, he provides patients with a distillation of current field research by posting updates in a research-specific Facebook group and on Twitter. This model not only empowers patients with disease education, it also solidifies the importance of patient investment in disease-specific research. Furthermore, invested patients bring ideas to research, take a more educated and proactive role in their care team, and, ultimately, return to seek more study involvement.
Social media in rare disease research
Rare diseases (conditions with a prevalence of less than 200,000 patients in North America), in particular, are prime for high-yield results and community impact using novel SM approaches. This is the result of established digital support groups, publications with historically low study numbers, and few focused investigators. Several studies of rare diseases have shown considerable advantages of using SM as a study tool. For instance, an existing neuroendocrine cervical cancer Facebook support group recently was used to recruit a geographically widespread cohort of patients with this rare cancer. Through an online survey posted in the Facebook group, patients were able to provide specific information on their treatment, disease, and symptom history, current disease status, and quality of life, including various psychological factors. Without the use of SM, collecting this information would have been virtually impossible because the patients were treated at 51 cancer centers across the country.14
Currently, the use of SM in hepatology research, focused specifically on autoimmune hepatitis (AIH), is under exploration at Indiana University. AIH is a rare autoimmune liver disease that results in immune-mediated destruction of liver cells, possibly resulting in fibrosis, cirrhosis, or liver failure if treatment is unsuccessful. One of the authors (C.L.) used both Facebook and Twitter to construct a large study group of individuals affected with AIH called the Autoimmune Hepatitis Research Network (AHRN; 1,500 members) during the past 2 years.4 Interested individuals have joined this research group after searching for AIH online support groups or reading shared AHRN posts on other media platforms. Between April 2015 and April 2016, there were posts by more than 750 unique active members (more than 50% of the group contributes to discussions), most of whom appear to be either caregivers of AIH patients or AIH patients themselves.
Preliminary informational analysis on this group has shown that C.L. and study collaborators have been able to uncover rich clinical and nonclinical information that otherwise would remain unknown. This research was performed by semi-automated download of the Facebook group’s content and subsequent semantic analysis. Qualitative analysis also was performed by direct reading of patient narratives. Collected clinical information has included histories of medication side effects, familial autoimmune diseases, and comorbid conditions. The most common factors that patients were unlikely to discuss with a provider (e.g., financial issues, employment, personal relationships, use of supplements, and alcohol use) frequently were discussed in the AHRN group, allowing a more transparent view of the complete disease experience.
Beyond research conducted in the current paradigm, the AHRN has provided a rich community construct in which patients offer each other social support. The patient impression of AHRN on Facebook has been overwhelmingly positive, and patients often wonder why such a model has not been used with other diseases. The close digital interaction the author (C.L.) has had with numerous patients and families has promoted other benefits of this methodology: more than 40 new AIH patients from outside Indiana have traveled to Indiana University for medical consultation despite no advertisement.
Conclusions
SM has the potential to transform health care research as a supplement to traditional research methods. Compared with a conventional research model, this methodology has proven to be cost and time effective, wide reaching, and similarly capable of data collection. Use of SM in research has tremendous potential to direct patient-centered research because invested patient collaborators can take an active role in their own disease and may hone investigatory focus on stakeholder priorities. Limitations to this method are known, however; if implemented cautiously, these can be mitigated. Investment in and application of the social mediome by investigators and patients has the potential to support and transform research that otherwise would be impossible.
Acknowledgments
The authors wish to extend their gratitude to the members of the Autoimmune Hepatitis Research Network for their continued proactivity and engagement in autoimmune hepatitis research. Furthermore, the authors are grateful to Dr. Naga Chalasani for his continued mentorship and extensive contributions to the development of social media approaches in clinical investigation.
References
1. Asch, D.A., Rader, D.J., Merchant, R.M. Mining the social mediome. Trends Mol Med. 2015;21:528-9.
2. Brotherton, C.S., Martin, C.A., Long, M.D. et al. Avoidance of fiber is associated with greater risk of Crohn’s disease flare in a 6-month period. Clin Gastroenterol Hepatol. 2016;14:1130-6.
3. Fenner, Y., Garland, S.M., Moore, E.E., et al. Web-based recruiting for health research using a social networking site: an exploratory study. J Med Internet Res. 2012;14:e20.
4. Lammert, C., Comerford, M., Love, J., et al. Investigation gone viral: application of the social mediasphere in research. Gastroenterology. 2015;149:839-43.
5. Wicks, P., Massagli, M., Frost, J., et al. Sharing health data for better outcomes on PatientsLikeMe. J Med Internet Res. 2010;12:e19.
6. Admon, L., Haefner, J.K., Kolenic, G.E., et al. Recruiting pregnant patients for survey research: a head to head comparison of social media-based versus clinic-based approaches. J Med Internet Res. 2016;18:e326.
7. Farnan, J.M., Sulmasy, L.S., Chaudhry, H. Online medical professionalism. Ann Intern Med. 2013;159:158-9.
8. Massachusetts Medical Society: Social Media Guidelines for Physicians. Available from: http://www.massmed.org/Physicians/Legal-and-Regulatory/Social-Media-Guidelines-for-Physicians/#. Accessed: January 3, 2017.
9. Pirraglia, P.A. Kravitz, R.L. Social media: new opportunities, new ethical concerns. J Gen Intern Med. 2013;28:165-6.
10. Duggan, M., Ellison, N.B., Lampe, C. et al. Demographics of key social networking platforms. (Available from:) (Accessed: January 4, 2017) Pew Res Cent Internet Sci Tech. 2015; http://www.pewinternet.org/2015/01/09/demographics-of-key-social-networking-platforms-2
11. Kang, X., Zhao, L., Leung, F., et al. Delivery of Instructions via mobile social media app increases quality of bowel preparation. Clin Gastroenterol Hepatol. 2016;14:429-35.
12. Bajaj, J.S., Heuman, D.M., Sterling, R.K., et al. Validation of EncephalApp, Smartphone-based Stroop test, for the diagnosis of covert hepatic encephalopathy. Clin Gastroenterol Hepatol. 2015;13:1828-35.
13. Riaz, M.S. Atreja, A. Personalized technologies in chronic gastrointestinal disorders: self-monitoring and remote sensor technologies. Clin Gastroenterol Hepatol. 2016;14:1697-705.
14. Zaid, T., Burzawa, J., Basen-Engquist, K., et al. Use of social media to conduct a cross-sectional epidemiologic and quality of life survey of patients with neuroendocrine carcinoma of the cervix: a feasibility study. Gynecol Oncol. 2014;132:149-53.
15. Schumacher, K.R., Stringer, K.A., Donohue, J.E., et al. Social media methods for studying rare diseases. Pediatrics. 2014;133:e1345–53.
Dr. Kulanthaivel and Dr. Jones are in the school of informatics and computing, Purdue University, Indiana University, Indianapolis; Dr. Fogel and Dr. Lammert are in the department of digestive and liver diseases, Indiana University School of Medicine, Indianapolis. This study was supported by KL2TR001106 and UL1TR001108 from the National Institutes of Health, and the Clinical and Translational Sciences Award from the National Center for Advancing Translational Sciences (C.L.). The authors disclose no conflicts.
Cotempla XR-ODT approved for children, adolescents with ADHD
The Food and Drug Administration has approved the first methylphenidate extended-release orally disintegrating tablet for treating ADHD in patients aged 6-17 years old, Neos Therapeutics announced June 19.
The company said the approval came after a phase III trial showed that treatment in a laboratory classroom with the drug, called Cotempla XR-ODT, showed a significant improvement in attention-deficit/hyperactivity disorder symptom control when compared with a placebo across the classroom day (placebo-subtracted difference of –11). The onset of effect was shown at 1 hour post-dose and lasted through 12 hours. No serious adverse events were reported during the trial, and the adverse event profile was consistent with the established safety profile for other extended-release methylphenidate products.
Cotempla XR-ODT will be available commercially in a portable, child-resistant blister pack in the fall of 2017.
Find the full press release on Neos Therapeutics website.
The Food and Drug Administration has approved the first methylphenidate extended-release orally disintegrating tablet for treating ADHD in patients aged 6-17 years old, Neos Therapeutics announced June 19.
The company said the approval came after a phase III trial showed that treatment in a laboratory classroom with the drug, called Cotempla XR-ODT, showed a significant improvement in attention-deficit/hyperactivity disorder symptom control when compared with a placebo across the classroom day (placebo-subtracted difference of –11). The onset of effect was shown at 1 hour post-dose and lasted through 12 hours. No serious adverse events were reported during the trial, and the adverse event profile was consistent with the established safety profile for other extended-release methylphenidate products.
Cotempla XR-ODT will be available commercially in a portable, child-resistant blister pack in the fall of 2017.
Find the full press release on Neos Therapeutics website.
The Food and Drug Administration has approved the first methylphenidate extended-release orally disintegrating tablet for treating ADHD in patients aged 6-17 years old, Neos Therapeutics announced June 19.
The company said the approval came after a phase III trial showed that treatment in a laboratory classroom with the drug, called Cotempla XR-ODT, showed a significant improvement in attention-deficit/hyperactivity disorder symptom control when compared with a placebo across the classroom day (placebo-subtracted difference of –11). The onset of effect was shown at 1 hour post-dose and lasted through 12 hours. No serious adverse events were reported during the trial, and the adverse event profile was consistent with the established safety profile for other extended-release methylphenidate products.
Cotempla XR-ODT will be available commercially in a portable, child-resistant blister pack in the fall of 2017.
Find the full press release on Neos Therapeutics website.
Breastfeeding may reduce moms’ stroke risk
Breastfeeding not only benefits babies; it also may lower the risk for a heart attack or stroke later in life for mothers who breastfeed more than for women who don’t.
The findings, which were published online June 21 in the Journal of the American Heart Association, are based on data from a prospective study of nearly 300,000 women in China.
To assess the impact of breastfeeding on maternal cardiovascular health, the researchers reviewed data from 289,573 women who were participating in the China Kadoorie Biobank study to assess their reproductive history and lifestyle. At the time of study enrollment, none of the women had a history of cardiovascular disease and 99% reported at least one live birth. The average age of the women at baseline was 51 years.
Of the women who had given birth, 97% reported ever breastfeeding, and 91% reported breastfeeding each child for at least 6 months. The median duration of breastfeeding was 12 months per child (J Am Heart Assoc. 2017 Jun 21. doi: JAHA/2017/006081-T2).
During an 8-year follow-up period, participants experienced 16,671 cases of coronary heart disease and 23,983 strokes.
Overall, women who breastfed babies had a 9% reduction in risk of coronary heart disease and an 8% reduction in risk of stroke, compared with women who never breastfed. The longer the duration of breastfeeding, the greater the risk reduction; for every additional 6 months of breastfeeding, researchers found a 4% reduction in heart disease risk and a 3% reduction in stroke risk. Mothers who breastfed for 2 years or more had the most protection – an 18% reduced risk of heart disease and a 17% reduced risk of stroke, compared with mothers who never breastfed.
The study was limited by several factors, including its observational nature, which cannot confirm a causal relationship between breastfeeding and CVD. However, the results suggest that, if causal, “interventions to increase the likelihood and duration of breastfeeding could have persistent benefits to maternal cardiovascular health,” they wrote.
The baseline study was funded by the Kadoorie Charitable Foundation in Hong Kong; long-term support came from the UK Wellcome Trust, Chinese Ministry of Science and Technology, and the Chinese National Natural Science Foundation. Other support came from the British Heart Foundation, UK Medical Research Council and Cancer Research UK, and the National Natural Science Foundation of China. Dr. Peters has received support from the British Heart Foundation.
Breastfeeding not only benefits babies; it also may lower the risk for a heart attack or stroke later in life for mothers who breastfeed more than for women who don’t.
The findings, which were published online June 21 in the Journal of the American Heart Association, are based on data from a prospective study of nearly 300,000 women in China.
To assess the impact of breastfeeding on maternal cardiovascular health, the researchers reviewed data from 289,573 women who were participating in the China Kadoorie Biobank study to assess their reproductive history and lifestyle. At the time of study enrollment, none of the women had a history of cardiovascular disease and 99% reported at least one live birth. The average age of the women at baseline was 51 years.
Of the women who had given birth, 97% reported ever breastfeeding, and 91% reported breastfeeding each child for at least 6 months. The median duration of breastfeeding was 12 months per child (J Am Heart Assoc. 2017 Jun 21. doi: JAHA/2017/006081-T2).
During an 8-year follow-up period, participants experienced 16,671 cases of coronary heart disease and 23,983 strokes.
Overall, women who breastfed babies had a 9% reduction in risk of coronary heart disease and an 8% reduction in risk of stroke, compared with women who never breastfed. The longer the duration of breastfeeding, the greater the risk reduction; for every additional 6 months of breastfeeding, researchers found a 4% reduction in heart disease risk and a 3% reduction in stroke risk. Mothers who breastfed for 2 years or more had the most protection – an 18% reduced risk of heart disease and a 17% reduced risk of stroke, compared with mothers who never breastfed.
The study was limited by several factors, including its observational nature, which cannot confirm a causal relationship between breastfeeding and CVD. However, the results suggest that, if causal, “interventions to increase the likelihood and duration of breastfeeding could have persistent benefits to maternal cardiovascular health,” they wrote.
The baseline study was funded by the Kadoorie Charitable Foundation in Hong Kong; long-term support came from the UK Wellcome Trust, Chinese Ministry of Science and Technology, and the Chinese National Natural Science Foundation. Other support came from the British Heart Foundation, UK Medical Research Council and Cancer Research UK, and the National Natural Science Foundation of China. Dr. Peters has received support from the British Heart Foundation.
Breastfeeding not only benefits babies; it also may lower the risk for a heart attack or stroke later in life for mothers who breastfeed more than for women who don’t.
The findings, which were published online June 21 in the Journal of the American Heart Association, are based on data from a prospective study of nearly 300,000 women in China.
To assess the impact of breastfeeding on maternal cardiovascular health, the researchers reviewed data from 289,573 women who were participating in the China Kadoorie Biobank study to assess their reproductive history and lifestyle. At the time of study enrollment, none of the women had a history of cardiovascular disease and 99% reported at least one live birth. The average age of the women at baseline was 51 years.
Of the women who had given birth, 97% reported ever breastfeeding, and 91% reported breastfeeding each child for at least 6 months. The median duration of breastfeeding was 12 months per child (J Am Heart Assoc. 2017 Jun 21. doi: JAHA/2017/006081-T2).
During an 8-year follow-up period, participants experienced 16,671 cases of coronary heart disease and 23,983 strokes.
Overall, women who breastfed babies had a 9% reduction in risk of coronary heart disease and an 8% reduction in risk of stroke, compared with women who never breastfed. The longer the duration of breastfeeding, the greater the risk reduction; for every additional 6 months of breastfeeding, researchers found a 4% reduction in heart disease risk and a 3% reduction in stroke risk. Mothers who breastfed for 2 years or more had the most protection – an 18% reduced risk of heart disease and a 17% reduced risk of stroke, compared with mothers who never breastfed.
The study was limited by several factors, including its observational nature, which cannot confirm a causal relationship between breastfeeding and CVD. However, the results suggest that, if causal, “interventions to increase the likelihood and duration of breastfeeding could have persistent benefits to maternal cardiovascular health,” they wrote.
The baseline study was funded by the Kadoorie Charitable Foundation in Hong Kong; long-term support came from the UK Wellcome Trust, Chinese Ministry of Science and Technology, and the Chinese National Natural Science Foundation. Other support came from the British Heart Foundation, UK Medical Research Council and Cancer Research UK, and the National Natural Science Foundation of China. Dr. Peters has received support from the British Heart Foundation.
FROM THE JOURNAL OF THE AMERICAN HEART ASSOCIATION
Key clinical point: Interventions to encourage breastfeeding may benefit mothers’ cardiovascular health later in life.
Major finding: Women who breastfed their babies had a 10% reduction in risk of cardiovascular disease later in life.
Data source: A prospective study of approximately 300,000 women in China.
Disclosures: The baseline study was funded by the Kadoorie Charitable Foundation in Hong Kong; long-term support came from the UK Wellcome Trust, Chinese Ministry of Science and Technology, and the Chinese National Natural Science Foundation. Dr. Peters has received support from the British Heart Foundation.
New research grant will support pediatric genomics research
The AGA Research Foundation has partnered with the Rady Children’s Institute of Genomic Medicine to establish the AGA-Rady Children’s Institute of Genomic Medicine Research Scholar Award in Pediatric Genomics. This award will support one promising young investigator conducting research that utilizes genomics to enhance our fundamental understanding of childhood digestive diseases.
This newly established award will provide $90,000 per year for 3 years to one investigator. The funded research must be conducted at Rady Children’s Institute for Genomic Medicine in San Diego starting July 2018.
Stay tuned for additional details and information on how to apply for this grant in summer 2017. More information on this new award is available on the Rady Children’s Hospital website.
The AGA Research Foundation has partnered with the Rady Children’s Institute of Genomic Medicine to establish the AGA-Rady Children’s Institute of Genomic Medicine Research Scholar Award in Pediatric Genomics. This award will support one promising young investigator conducting research that utilizes genomics to enhance our fundamental understanding of childhood digestive diseases.
This newly established award will provide $90,000 per year for 3 years to one investigator. The funded research must be conducted at Rady Children’s Institute for Genomic Medicine in San Diego starting July 2018.
Stay tuned for additional details and information on how to apply for this grant in summer 2017. More information on this new award is available on the Rady Children’s Hospital website.
The AGA Research Foundation has partnered with the Rady Children’s Institute of Genomic Medicine to establish the AGA-Rady Children’s Institute of Genomic Medicine Research Scholar Award in Pediatric Genomics. This award will support one promising young investigator conducting research that utilizes genomics to enhance our fundamental understanding of childhood digestive diseases.
This newly established award will provide $90,000 per year for 3 years to one investigator. The funded research must be conducted at Rady Children’s Institute for Genomic Medicine in San Diego starting July 2018.
Stay tuned for additional details and information on how to apply for this grant in summer 2017. More information on this new award is available on the Rady Children’s Hospital website.
Become an AGA Fellow – AGA’s highest level of membership
AGA recognizes members whose accomplishments demonstrate personal commitment to the field of gastroenterology with the distinction of fellowship. AGA fellowship helps open doors, creates connections, and offers widespread value and recognition. Applicants can choose to apply for fellowship in either clinical practice (private or academic) or in research (basic or clinical).
Gain recognition as a distinguished GI professional and apply today by visiting www.gastro.org/fellowship. This website includes the full list of benefits and criteria for fellowship.
Be honored for your contributions and commitment to the GI field. The deadline for submissions is Monday, July 31, 2017.
AGA recognizes members whose accomplishments demonstrate personal commitment to the field of gastroenterology with the distinction of fellowship. AGA fellowship helps open doors, creates connections, and offers widespread value and recognition. Applicants can choose to apply for fellowship in either clinical practice (private or academic) or in research (basic or clinical).
Gain recognition as a distinguished GI professional and apply today by visiting www.gastro.org/fellowship. This website includes the full list of benefits and criteria for fellowship.
Be honored for your contributions and commitment to the GI field. The deadline for submissions is Monday, July 31, 2017.
AGA recognizes members whose accomplishments demonstrate personal commitment to the field of gastroenterology with the distinction of fellowship. AGA fellowship helps open doors, creates connections, and offers widespread value and recognition. Applicants can choose to apply for fellowship in either clinical practice (private or academic) or in research (basic or clinical).
Gain recognition as a distinguished GI professional and apply today by visiting www.gastro.org/fellowship. This website includes the full list of benefits and criteria for fellowship.
Be honored for your contributions and commitment to the GI field. The deadline for submissions is Monday, July 31, 2017.
AGA makes six recommendations to FDA on interchangeable biosimilars
Gastroenterologists and patients rely on biologics to manage Crohn’s disease and ulcerative colitis. Biosimilar products, which are “highly similar” to the biologic, have begun to be approved by the FDA for such indications. The FDA is now developing a pathway for interchangeable products, which are biosimilars that “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product” according to Section 351(i) of the Public Health Service Act. AGA provided the FDA six recommendations in response to the agency’s draft guidance on demonstrating interchangeability focused on measures to enhance patient safety and ensure that physicians, not insurance companies, drive decisions about switching products. Here is a summary of our comments.
1. Extrapolation of data should not be allowed for any indication where the pathophysiology is known to be different or is yet to be elucidated.
Post-marketing evidence on interchangeability of biosimilar products would alleviate concerns as testing specific products in individual diseases is an important step in determining whether the product is effective and safe for that particular disease. AGA recommends that manufacturers should be required to seek licensure for all the same indications as the reference product to appropriately track adverse events should they arise.
2. The agency should use caution when allowing extrapolation for pediatric indications.
Pediatric patients are recognized as a vulnerable population for which a disease may differ from those of adult patients. In the absence of data specifically ensuring safety and efficacy in children, AGA recommends an exemption of pediatric patients from current FDA positions and guidance documents related to interchangeable products.
3. Sponsors should exclusively use U.S.-licensed reference products in switching studies.
Currently, the FDA’s draft guidance has wording that seems to signal that the agency is willing to entertain use of non-U.S.-licensed products in some cases, casting doubt on the true “interchangeability” of the product. AGA recommends that the guidance be amended to include specific scenarios where this may be acceptable or remove the clause altogether.
4. “Real world” data on biosimilar and interchangeable products must be collected through formal post-marketing observational studies to ensure the longitudinal safety and efficacy for all patient populations being treated with these products.
A central observational registry, like the AGA Fecal Microbiome Transplant National Registry, would ensure the capture of data on the safety and efficacy of interchangeable products for all manufacturers and their adverse effects on patients, if any. Such a registry would also allow the study of outcomes in patients who are switched among multiple products.
5. Gastroenterologists with appropriate disease expertise should be engaged by the FDA when interchangeable products are reviewed for approval.
AGA is part of the FDA’s Network of Experts and hopes that this relationship will continue to be proactively utilized when a proposed product is seeking a gastrointestinal indication.
6. Prescribing physicians must be empowered with the ability to prevent nonmedical switching from a reference product to an interchangeable product.
AGA has concerns over the section of the Public Health Service Act that states that an interchangeable product “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.” Health care providers must be empowered to be aware of and prevent nonmedical switching if they believe that the patient’s safety and health is at risk. AGA encourages the FDA to consider making a statement encouraging states to protect physician discretion as it applies to interchangeable biosimilars.
AGA will continue to work with the FDA to ensure that the voice of gastroenterology is heard in relation to biosimilars and interchangeable products.
Gastroenterologists and patients rely on biologics to manage Crohn’s disease and ulcerative colitis. Biosimilar products, which are “highly similar” to the biologic, have begun to be approved by the FDA for such indications. The FDA is now developing a pathway for interchangeable products, which are biosimilars that “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product” according to Section 351(i) of the Public Health Service Act. AGA provided the FDA six recommendations in response to the agency’s draft guidance on demonstrating interchangeability focused on measures to enhance patient safety and ensure that physicians, not insurance companies, drive decisions about switching products. Here is a summary of our comments.
1. Extrapolation of data should not be allowed for any indication where the pathophysiology is known to be different or is yet to be elucidated.
Post-marketing evidence on interchangeability of biosimilar products would alleviate concerns as testing specific products in individual diseases is an important step in determining whether the product is effective and safe for that particular disease. AGA recommends that manufacturers should be required to seek licensure for all the same indications as the reference product to appropriately track adverse events should they arise.
2. The agency should use caution when allowing extrapolation for pediatric indications.
Pediatric patients are recognized as a vulnerable population for which a disease may differ from those of adult patients. In the absence of data specifically ensuring safety and efficacy in children, AGA recommends an exemption of pediatric patients from current FDA positions and guidance documents related to interchangeable products.
3. Sponsors should exclusively use U.S.-licensed reference products in switching studies.
Currently, the FDA’s draft guidance has wording that seems to signal that the agency is willing to entertain use of non-U.S.-licensed products in some cases, casting doubt on the true “interchangeability” of the product. AGA recommends that the guidance be amended to include specific scenarios where this may be acceptable or remove the clause altogether.
4. “Real world” data on biosimilar and interchangeable products must be collected through formal post-marketing observational studies to ensure the longitudinal safety and efficacy for all patient populations being treated with these products.
A central observational registry, like the AGA Fecal Microbiome Transplant National Registry, would ensure the capture of data on the safety and efficacy of interchangeable products for all manufacturers and their adverse effects on patients, if any. Such a registry would also allow the study of outcomes in patients who are switched among multiple products.
5. Gastroenterologists with appropriate disease expertise should be engaged by the FDA when interchangeable products are reviewed for approval.
AGA is part of the FDA’s Network of Experts and hopes that this relationship will continue to be proactively utilized when a proposed product is seeking a gastrointestinal indication.
6. Prescribing physicians must be empowered with the ability to prevent nonmedical switching from a reference product to an interchangeable product.
AGA has concerns over the section of the Public Health Service Act that states that an interchangeable product “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.” Health care providers must be empowered to be aware of and prevent nonmedical switching if they believe that the patient’s safety and health is at risk. AGA encourages the FDA to consider making a statement encouraging states to protect physician discretion as it applies to interchangeable biosimilars.
AGA will continue to work with the FDA to ensure that the voice of gastroenterology is heard in relation to biosimilars and interchangeable products.
Gastroenterologists and patients rely on biologics to manage Crohn’s disease and ulcerative colitis. Biosimilar products, which are “highly similar” to the biologic, have begun to be approved by the FDA for such indications. The FDA is now developing a pathway for interchangeable products, which are biosimilars that “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product” according to Section 351(i) of the Public Health Service Act. AGA provided the FDA six recommendations in response to the agency’s draft guidance on demonstrating interchangeability focused on measures to enhance patient safety and ensure that physicians, not insurance companies, drive decisions about switching products. Here is a summary of our comments.
1. Extrapolation of data should not be allowed for any indication where the pathophysiology is known to be different or is yet to be elucidated.
Post-marketing evidence on interchangeability of biosimilar products would alleviate concerns as testing specific products in individual diseases is an important step in determining whether the product is effective and safe for that particular disease. AGA recommends that manufacturers should be required to seek licensure for all the same indications as the reference product to appropriately track adverse events should they arise.
2. The agency should use caution when allowing extrapolation for pediatric indications.
Pediatric patients are recognized as a vulnerable population for which a disease may differ from those of adult patients. In the absence of data specifically ensuring safety and efficacy in children, AGA recommends an exemption of pediatric patients from current FDA positions and guidance documents related to interchangeable products.
3. Sponsors should exclusively use U.S.-licensed reference products in switching studies.
Currently, the FDA’s draft guidance has wording that seems to signal that the agency is willing to entertain use of non-U.S.-licensed products in some cases, casting doubt on the true “interchangeability” of the product. AGA recommends that the guidance be amended to include specific scenarios where this may be acceptable or remove the clause altogether.
4. “Real world” data on biosimilar and interchangeable products must be collected through formal post-marketing observational studies to ensure the longitudinal safety and efficacy for all patient populations being treated with these products.
A central observational registry, like the AGA Fecal Microbiome Transplant National Registry, would ensure the capture of data on the safety and efficacy of interchangeable products for all manufacturers and their adverse effects on patients, if any. Such a registry would also allow the study of outcomes in patients who are switched among multiple products.
5. Gastroenterologists with appropriate disease expertise should be engaged by the FDA when interchangeable products are reviewed for approval.
AGA is part of the FDA’s Network of Experts and hopes that this relationship will continue to be proactively utilized when a proposed product is seeking a gastrointestinal indication.
6. Prescribing physicians must be empowered with the ability to prevent nonmedical switching from a reference product to an interchangeable product.
AGA has concerns over the section of the Public Health Service Act that states that an interchangeable product “may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.” Health care providers must be empowered to be aware of and prevent nonmedical switching if they believe that the patient’s safety and health is at risk. AGA encourages the FDA to consider making a statement encouraging states to protect physician discretion as it applies to interchangeable biosimilars.
AGA will continue to work with the FDA to ensure that the voice of gastroenterology is heard in relation to biosimilars and interchangeable products.
FDA advisory committee supports new CV liraglutide indication
A Food and Drug Administration advisory committee voted 17-2 in support of a supplemental new drug application for liraglutide (Victoza) injections to reduce the risk of major adverse cardiovascular events in adults with type 2 diabetes and established cardiovascular disease.
Novo Nordisk, the maker of the glucagon-like peptide-1 (GLP-1) analogue, proposed the additional indication for liraglutide as an adjunct to standard treatment of cardiovascular risk factors in such patients based solely on the results of the randomized, placebo-controlled postmarketing LEADER trial.
If this additional indication for liraglutide is approved by the FDA, the drug would join the antidiabetic drug empagliflozin (Jardiance) in having a second indication for the reduction of the risk of cardiovascular death. The supplemental new drug application for Jardiance was approved by the FDA in December 2016 – also based on the results of a single trial (the EMPA-REG outcomes trial). Of note, the American Diabetes Association in its 2017 Standards of Medical Care has already called for consideration of both liraglutide and empagliflozin to reduce the risk of cardiovascular death in patients with type 2 diabetes and documented cardiovascular disease.
Liraglutide is currently approved for blood glucose lowering in adults with type 2 diabetes and is marketed as Saxenda for the treatment of overweight and obese adults with at least one weight-related comorbidity. It was shown in the LEADER trial to be associated with a significant 13% lower risk vs. placebo for a composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke in patients with type 2 diabetes.
All 19 voting members of the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) agreed that the LEADER results confirm there is no excess cardiovascular risk associated with liraglutide in patients with type 2 diabetes, but, on the question of whether the results provide “the substantial evidence required to establish that liraglutide 1.8 mg reduces cardiovascular risk in patients with type 2 diabetes mellitus and established cardiovascular disease,” almost all voting members expressed concerns about subgroup analyses showing reduced benefit among U.S. patients, compared with those from other countries.
“I think probably the most influential finding for me was the overall cardiovascular mortality finding, followed by the consistency of the results,” said biostatistics expert James D. Neaton, PhD, of the University of Minnesota, Minneapolis. He added that the indication should focus on patients at high cardiovascular event risk, as the LEADER population was a very high risk group.
Similarly, temporary voting member Marvin A. Konstam, MD, of Tufts University, Boston, said “the primary trial results are very robust and substantiated.
“And I think the cardiovascular mortality is the biggest contributor to that, which is obviously a very important finding,” he said, also stressing that the indication should focus on patients with established cardiovascular disease.
“I am concerned about the U.S. population, but at the end of the day, it’s a subgroup, and I just can’t overrate that to diminish the overall finding,” he added.
Peter W.F. Wilson, MD, EMDAC chairperson, said he “wrestles with exactly who benefits the most because of overlapping of some of the groupings.
“But people who really have atherosclerotic cardiovascular disease ... are probably the people who will benefit the most, and I hope those are the people who will get it,” said Dr. Wilson, professor of medicine at Emory University, professor of public health at Rollins School of Public Health, and director of epidemiology and genomic medicine at the Atlanta VA Medical Center.
In explaining his “no” vote, Daniel Budnitz, MD, of the Centers for Disease Control and Prevention, Atlanta, said his was a tough decision, but that ultimately, since the U.S. population is the one the FDA is addressing with its labeling, the subgroup concerns weighed heavily.
“And I do worry about a slippery slope of using single-trial data for new indications, when there are questions and when you do have an interaction term for the U.S. vs. the rest of the world,” he said, adding that he would like to see either another international trial where the United States population does not differ from the rest of the world, or a U.S. trial.
Carmen J. Allegra, MD, of the University of Florida, Gainesville, also voted no, and said he, too, was concerned by the subgroup analysis.
“I was very much concerned and swayed by the subgroup analysis. The U.S. target population is a pretty darn important population for us to consider, and we saw a significant interaction with outcomes vs. the region by the FDA’s analysis,” he said. “I was really swayed by the fact that we really didn’t see evidence of superiority in the U.S. population.”
The LEADER trial, which was designed in accordance with FDA Guidance issued in 2008 to demonstrate that new antidiabetes drugs do not result in unacceptably increased cardiovascular risk, included 9,340 patients who were randomized to receive liraglutide or placebo as add-on to standard of care treatment and who were followed for a median of 3.8 years. Those randomized to receive liraglutide experienced significantly lower risk of the composite primary outcome (hazard ratio, 0.87), Notably, the effect was diminished among U.S. patients, compared with the overall benefit.
However, after hearing LEADER analyses from Novo Nordisk representatives and FDA representatives, and testimony from numerous individuals, including patients, physicians, and patient advocates who spoke overwhelmingly in favor of approval of the supplemental drug application, the committee recommended that approval.
“This was not a slam dunk. I think the subgroup analysis was interesting discussion, but in the end you have to take the data and the primary outcome measure as what you move on,” said temporary voting member David C. Robbins, MD, of the University of Kansas, Kansas City, adding that “the good is outweighing the bad on this.
“I’m glad to see diabetes management moving toward more than lowering blood sugar. It’s a good step in the right direction,” he said.
The FDA, which usually follows the recommendations of its advisory committees, will now consider the supplemental new drug application for liraglutide.
In a statement released after the vote, Todd Hobbs, MD, vice president and U.S. chief medical officer of Novo Nordisk, noted that cardiovascular disease remains the leading cause of death for people with type 2 diabetes. The discussion during the EMDAC meeting is “an important reminder that there is an unmet need to provide benefits beyond HbA1c control” in patients with type 2 diabetes.
EMDAC committee members were screened and found to be in compliance with federal ethics and conflict of interest laws; one (Dr. Konstam) was granted a waiver in accordance with rules allowing such waivers when the need for an individual’s service outweighs any potential financial conflicts of interest. Dr. Konstam reported financial relationships with competing firms.
A Food and Drug Administration advisory committee voted 17-2 in support of a supplemental new drug application for liraglutide (Victoza) injections to reduce the risk of major adverse cardiovascular events in adults with type 2 diabetes and established cardiovascular disease.
Novo Nordisk, the maker of the glucagon-like peptide-1 (GLP-1) analogue, proposed the additional indication for liraglutide as an adjunct to standard treatment of cardiovascular risk factors in such patients based solely on the results of the randomized, placebo-controlled postmarketing LEADER trial.
If this additional indication for liraglutide is approved by the FDA, the drug would join the antidiabetic drug empagliflozin (Jardiance) in having a second indication for the reduction of the risk of cardiovascular death. The supplemental new drug application for Jardiance was approved by the FDA in December 2016 – also based on the results of a single trial (the EMPA-REG outcomes trial). Of note, the American Diabetes Association in its 2017 Standards of Medical Care has already called for consideration of both liraglutide and empagliflozin to reduce the risk of cardiovascular death in patients with type 2 diabetes and documented cardiovascular disease.
Liraglutide is currently approved for blood glucose lowering in adults with type 2 diabetes and is marketed as Saxenda for the treatment of overweight and obese adults with at least one weight-related comorbidity. It was shown in the LEADER trial to be associated with a significant 13% lower risk vs. placebo for a composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke in patients with type 2 diabetes.
All 19 voting members of the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) agreed that the LEADER results confirm there is no excess cardiovascular risk associated with liraglutide in patients with type 2 diabetes, but, on the question of whether the results provide “the substantial evidence required to establish that liraglutide 1.8 mg reduces cardiovascular risk in patients with type 2 diabetes mellitus and established cardiovascular disease,” almost all voting members expressed concerns about subgroup analyses showing reduced benefit among U.S. patients, compared with those from other countries.
“I think probably the most influential finding for me was the overall cardiovascular mortality finding, followed by the consistency of the results,” said biostatistics expert James D. Neaton, PhD, of the University of Minnesota, Minneapolis. He added that the indication should focus on patients at high cardiovascular event risk, as the LEADER population was a very high risk group.
Similarly, temporary voting member Marvin A. Konstam, MD, of Tufts University, Boston, said “the primary trial results are very robust and substantiated.
“And I think the cardiovascular mortality is the biggest contributor to that, which is obviously a very important finding,” he said, also stressing that the indication should focus on patients with established cardiovascular disease.
“I am concerned about the U.S. population, but at the end of the day, it’s a subgroup, and I just can’t overrate that to diminish the overall finding,” he added.
Peter W.F. Wilson, MD, EMDAC chairperson, said he “wrestles with exactly who benefits the most because of overlapping of some of the groupings.
“But people who really have atherosclerotic cardiovascular disease ... are probably the people who will benefit the most, and I hope those are the people who will get it,” said Dr. Wilson, professor of medicine at Emory University, professor of public health at Rollins School of Public Health, and director of epidemiology and genomic medicine at the Atlanta VA Medical Center.
In explaining his “no” vote, Daniel Budnitz, MD, of the Centers for Disease Control and Prevention, Atlanta, said his was a tough decision, but that ultimately, since the U.S. population is the one the FDA is addressing with its labeling, the subgroup concerns weighed heavily.
“And I do worry about a slippery slope of using single-trial data for new indications, when there are questions and when you do have an interaction term for the U.S. vs. the rest of the world,” he said, adding that he would like to see either another international trial where the United States population does not differ from the rest of the world, or a U.S. trial.
Carmen J. Allegra, MD, of the University of Florida, Gainesville, also voted no, and said he, too, was concerned by the subgroup analysis.
“I was very much concerned and swayed by the subgroup analysis. The U.S. target population is a pretty darn important population for us to consider, and we saw a significant interaction with outcomes vs. the region by the FDA’s analysis,” he said. “I was really swayed by the fact that we really didn’t see evidence of superiority in the U.S. population.”
The LEADER trial, which was designed in accordance with FDA Guidance issued in 2008 to demonstrate that new antidiabetes drugs do not result in unacceptably increased cardiovascular risk, included 9,340 patients who were randomized to receive liraglutide or placebo as add-on to standard of care treatment and who were followed for a median of 3.8 years. Those randomized to receive liraglutide experienced significantly lower risk of the composite primary outcome (hazard ratio, 0.87), Notably, the effect was diminished among U.S. patients, compared with the overall benefit.
However, after hearing LEADER analyses from Novo Nordisk representatives and FDA representatives, and testimony from numerous individuals, including patients, physicians, and patient advocates who spoke overwhelmingly in favor of approval of the supplemental drug application, the committee recommended that approval.
“This was not a slam dunk. I think the subgroup analysis was interesting discussion, but in the end you have to take the data and the primary outcome measure as what you move on,” said temporary voting member David C. Robbins, MD, of the University of Kansas, Kansas City, adding that “the good is outweighing the bad on this.
“I’m glad to see diabetes management moving toward more than lowering blood sugar. It’s a good step in the right direction,” he said.
The FDA, which usually follows the recommendations of its advisory committees, will now consider the supplemental new drug application for liraglutide.
In a statement released after the vote, Todd Hobbs, MD, vice president and U.S. chief medical officer of Novo Nordisk, noted that cardiovascular disease remains the leading cause of death for people with type 2 diabetes. The discussion during the EMDAC meeting is “an important reminder that there is an unmet need to provide benefits beyond HbA1c control” in patients with type 2 diabetes.
EMDAC committee members were screened and found to be in compliance with federal ethics and conflict of interest laws; one (Dr. Konstam) was granted a waiver in accordance with rules allowing such waivers when the need for an individual’s service outweighs any potential financial conflicts of interest. Dr. Konstam reported financial relationships with competing firms.
A Food and Drug Administration advisory committee voted 17-2 in support of a supplemental new drug application for liraglutide (Victoza) injections to reduce the risk of major adverse cardiovascular events in adults with type 2 diabetes and established cardiovascular disease.
Novo Nordisk, the maker of the glucagon-like peptide-1 (GLP-1) analogue, proposed the additional indication for liraglutide as an adjunct to standard treatment of cardiovascular risk factors in such patients based solely on the results of the randomized, placebo-controlled postmarketing LEADER trial.
If this additional indication for liraglutide is approved by the FDA, the drug would join the antidiabetic drug empagliflozin (Jardiance) in having a second indication for the reduction of the risk of cardiovascular death. The supplemental new drug application for Jardiance was approved by the FDA in December 2016 – also based on the results of a single trial (the EMPA-REG outcomes trial). Of note, the American Diabetes Association in its 2017 Standards of Medical Care has already called for consideration of both liraglutide and empagliflozin to reduce the risk of cardiovascular death in patients with type 2 diabetes and documented cardiovascular disease.
Liraglutide is currently approved for blood glucose lowering in adults with type 2 diabetes and is marketed as Saxenda for the treatment of overweight and obese adults with at least one weight-related comorbidity. It was shown in the LEADER trial to be associated with a significant 13% lower risk vs. placebo for a composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke in patients with type 2 diabetes.
All 19 voting members of the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) agreed that the LEADER results confirm there is no excess cardiovascular risk associated with liraglutide in patients with type 2 diabetes, but, on the question of whether the results provide “the substantial evidence required to establish that liraglutide 1.8 mg reduces cardiovascular risk in patients with type 2 diabetes mellitus and established cardiovascular disease,” almost all voting members expressed concerns about subgroup analyses showing reduced benefit among U.S. patients, compared with those from other countries.
“I think probably the most influential finding for me was the overall cardiovascular mortality finding, followed by the consistency of the results,” said biostatistics expert James D. Neaton, PhD, of the University of Minnesota, Minneapolis. He added that the indication should focus on patients at high cardiovascular event risk, as the LEADER population was a very high risk group.
Similarly, temporary voting member Marvin A. Konstam, MD, of Tufts University, Boston, said “the primary trial results are very robust and substantiated.
“And I think the cardiovascular mortality is the biggest contributor to that, which is obviously a very important finding,” he said, also stressing that the indication should focus on patients with established cardiovascular disease.
“I am concerned about the U.S. population, but at the end of the day, it’s a subgroup, and I just can’t overrate that to diminish the overall finding,” he added.
Peter W.F. Wilson, MD, EMDAC chairperson, said he “wrestles with exactly who benefits the most because of overlapping of some of the groupings.
“But people who really have atherosclerotic cardiovascular disease ... are probably the people who will benefit the most, and I hope those are the people who will get it,” said Dr. Wilson, professor of medicine at Emory University, professor of public health at Rollins School of Public Health, and director of epidemiology and genomic medicine at the Atlanta VA Medical Center.
In explaining his “no” vote, Daniel Budnitz, MD, of the Centers for Disease Control and Prevention, Atlanta, said his was a tough decision, but that ultimately, since the U.S. population is the one the FDA is addressing with its labeling, the subgroup concerns weighed heavily.
“And I do worry about a slippery slope of using single-trial data for new indications, when there are questions and when you do have an interaction term for the U.S. vs. the rest of the world,” he said, adding that he would like to see either another international trial where the United States population does not differ from the rest of the world, or a U.S. trial.
Carmen J. Allegra, MD, of the University of Florida, Gainesville, also voted no, and said he, too, was concerned by the subgroup analysis.
“I was very much concerned and swayed by the subgroup analysis. The U.S. target population is a pretty darn important population for us to consider, and we saw a significant interaction with outcomes vs. the region by the FDA’s analysis,” he said. “I was really swayed by the fact that we really didn’t see evidence of superiority in the U.S. population.”
The LEADER trial, which was designed in accordance with FDA Guidance issued in 2008 to demonstrate that new antidiabetes drugs do not result in unacceptably increased cardiovascular risk, included 9,340 patients who were randomized to receive liraglutide or placebo as add-on to standard of care treatment and who were followed for a median of 3.8 years. Those randomized to receive liraglutide experienced significantly lower risk of the composite primary outcome (hazard ratio, 0.87), Notably, the effect was diminished among U.S. patients, compared with the overall benefit.
However, after hearing LEADER analyses from Novo Nordisk representatives and FDA representatives, and testimony from numerous individuals, including patients, physicians, and patient advocates who spoke overwhelmingly in favor of approval of the supplemental drug application, the committee recommended that approval.
“This was not a slam dunk. I think the subgroup analysis was interesting discussion, but in the end you have to take the data and the primary outcome measure as what you move on,” said temporary voting member David C. Robbins, MD, of the University of Kansas, Kansas City, adding that “the good is outweighing the bad on this.
“I’m glad to see diabetes management moving toward more than lowering blood sugar. It’s a good step in the right direction,” he said.
The FDA, which usually follows the recommendations of its advisory committees, will now consider the supplemental new drug application for liraglutide.
In a statement released after the vote, Todd Hobbs, MD, vice president and U.S. chief medical officer of Novo Nordisk, noted that cardiovascular disease remains the leading cause of death for people with type 2 diabetes. The discussion during the EMDAC meeting is “an important reminder that there is an unmet need to provide benefits beyond HbA1c control” in patients with type 2 diabetes.
EMDAC committee members were screened and found to be in compliance with federal ethics and conflict of interest laws; one (Dr. Konstam) was granted a waiver in accordance with rules allowing such waivers when the need for an individual’s service outweighs any potential financial conflicts of interest. Dr. Konstam reported financial relationships with competing firms.