User login
Newer insulin glargine formula curbs nocturnal hypoglycemia
NEW ORLEANS – Insulin glargine 300 U/mL provided comparable glycemic control to that seen with insulin glargine 100 U/mL and consistently reduced the risk of nocturnal hypoglycemia in patients with type 2 diabetes, regardless of their renal function, results from a large post hoc meta-analysis showed.
The EDITION I, II, and III studies showed that over a period of 6 months, Gla-300 provided comparable glycemic control to Gla-100 with less hypoglycemia in patients with type 2 diabetes. However, “renal impairment increases the risk of hypoglycemia in people with type 2 diabetes, and may limit glucose-lowering therapy options,” Javier Escalada, M.D., said at the annual scientific sessions of the American Diabetes Association. “Therefore, it may be more challenging to manage diabetes in this population than in people with normal renal function.”

Dr. Escalada of the department of endocrinology and nutrition at Clinic University of Navarra, Pamplona, Spain, and his associates set out to investigate the impact of renal function on hemoglobin A1c reduction and hypoglycemia in a post hoc meta-analysis of 2,468 patients aged 18 years and older with type 2 diabetes who were treated with Gla-300 or Gla-100 for 6 months in the EDITION I, II, and III studies. Treatment consisted of once-daily evening doses of Gla-300 or Gla-100 titrated to a fasting self-measured plasma glucose of 80-100 mg/dL. Patients were classified by their renal function as having moderate loss (30 to less than 60 mL/min per 1.73 m3; 399 patients), mild loss (60 to less than 90; 1,386 patients), or normal function (at least 90; 683 patients).
Outcomes of interest were change in HbA1c from baseline to month 6, and the percentages of patients achieving an HbA1c target of lower than 7.0% and lower than 7.5% at month 6. The researchers also assessed the cumulative number of hypoglycemic events, the relative risk of at least one confirmed or severe hypoglycemic event, and the nocturnal and at any time event rate per participant year.
Slightly more than half of participants (56%) had a baseline estimated glomerular filtration rate of 60 to less than 90 mL/min per 1.73 m3. Dr. Escalada reported that noninferiority for HbA1c reduction was shown for Gla-300 and Gla-100 regardless of renal function, and that evidence of heterogeneity of treatment effect across subgroups was observed (P = .46). However, the risk of confirmed or severe hypoglycemia was significantly lower for nocturnal events in the Gla-300 group, compared with the Gla-100 group (30% vs. 40% overall, respectively), while the risk of anytime hypoglycemia events in a 24-hour period was comparable to or lower in the Gla-300 group, compared with the Gla-100 group. Renal function did not affect the lower rate of nocturnal or anytime hypoglycemia. “Severe hypoglycemia was rare, and renal function did not affect the rate of severe events,” he said.
The trial was sponsored by Sanofi. Dr. Escalada disclosed that he is a member of the advisory panel for Sanofi and for Merck Sharp & Dohme. He is also a member of the speakers bureau for both companies as well as for AstraZeneca, Boehringer Ingelheim, Eli Lilly, and Novo Nordisk.
NEW ORLEANS – Insulin glargine 300 U/mL provided comparable glycemic control to that seen with insulin glargine 100 U/mL and consistently reduced the risk of nocturnal hypoglycemia in patients with type 2 diabetes, regardless of their renal function, results from a large post hoc meta-analysis showed.
The EDITION I, II, and III studies showed that over a period of 6 months, Gla-300 provided comparable glycemic control to Gla-100 with less hypoglycemia in patients with type 2 diabetes. However, “renal impairment increases the risk of hypoglycemia in people with type 2 diabetes, and may limit glucose-lowering therapy options,” Javier Escalada, M.D., said at the annual scientific sessions of the American Diabetes Association. “Therefore, it may be more challenging to manage diabetes in this population than in people with normal renal function.”
Dr. Escalada of the department of endocrinology and nutrition at Clinic University of Navarra, Pamplona, Spain, and his associates set out to investigate the impact of renal function on hemoglobin A1c reduction and hypoglycemia in a post hoc meta-analysis of 2,468 patients aged 18 years and older with type 2 diabetes who were treated with Gla-300 or Gla-100 for 6 months in the EDITION I, II, and III studies. Treatment consisted of once-daily evening doses of Gla-300 or Gla-100 titrated to a fasting self-measured plasma glucose of 80-100 mg/dL. Patients were classified by their renal function as having moderate loss (30 to less than 60 mL/min per 1.73 m3; 399 patients), mild loss (60 to less than 90; 1,386 patients), or normal function (at least 90; 683 patients).
Outcomes of interest were change in HbA1c from baseline to month 6, and the percentages of patients achieving an HbA1c target of lower than 7.0% and lower than 7.5% at month 6. The researchers also assessed the cumulative number of hypoglycemic events, the relative risk of at least one confirmed or severe hypoglycemic event, and the nocturnal and at any time event rate per participant year.
Slightly more than half of participants (56%) had a baseline estimated glomerular filtration rate of 60 to less than 90 mL/min per 1.73 m3. Dr. Escalada reported that noninferiority for HbA1c reduction was shown for Gla-300 and Gla-100 regardless of renal function, and that evidence of heterogeneity of treatment effect across subgroups was observed (P = .46). However, the risk of confirmed or severe hypoglycemia was significantly lower for nocturnal events in the Gla-300 group, compared with the Gla-100 group (30% vs. 40% overall, respectively), while the risk of anytime hypoglycemia events in a 24-hour period was comparable to or lower in the Gla-300 group, compared with the Gla-100 group. Renal function did not affect the lower rate of nocturnal or anytime hypoglycemia. “Severe hypoglycemia was rare, and renal function did not affect the rate of severe events,” he said.
The trial was sponsored by Sanofi. Dr. Escalada disclosed that he is a member of the advisory panel for Sanofi and for Merck Sharp & Dohme. He is also a member of the speakers bureau for both companies as well as for AstraZeneca, Boehringer Ingelheim, Eli Lilly, and Novo Nordisk.
NEW ORLEANS – Insulin glargine 300 U/mL provided comparable glycemic control to that seen with insulin glargine 100 U/mL and consistently reduced the risk of nocturnal hypoglycemia in patients with type 2 diabetes, regardless of their renal function, results from a large post hoc meta-analysis showed.
The EDITION I, II, and III studies showed that over a period of 6 months, Gla-300 provided comparable glycemic control to Gla-100 with less hypoglycemia in patients with type 2 diabetes. However, “renal impairment increases the risk of hypoglycemia in people with type 2 diabetes, and may limit glucose-lowering therapy options,” Javier Escalada, M.D., said at the annual scientific sessions of the American Diabetes Association. “Therefore, it may be more challenging to manage diabetes in this population than in people with normal renal function.”
Dr. Escalada of the department of endocrinology and nutrition at Clinic University of Navarra, Pamplona, Spain, and his associates set out to investigate the impact of renal function on hemoglobin A1c reduction and hypoglycemia in a post hoc meta-analysis of 2,468 patients aged 18 years and older with type 2 diabetes who were treated with Gla-300 or Gla-100 for 6 months in the EDITION I, II, and III studies. Treatment consisted of once-daily evening doses of Gla-300 or Gla-100 titrated to a fasting self-measured plasma glucose of 80-100 mg/dL. Patients were classified by their renal function as having moderate loss (30 to less than 60 mL/min per 1.73 m3; 399 patients), mild loss (60 to less than 90; 1,386 patients), or normal function (at least 90; 683 patients).
Outcomes of interest were change in HbA1c from baseline to month 6, and the percentages of patients achieving an HbA1c target of lower than 7.0% and lower than 7.5% at month 6. The researchers also assessed the cumulative number of hypoglycemic events, the relative risk of at least one confirmed or severe hypoglycemic event, and the nocturnal and at any time event rate per participant year.
Slightly more than half of participants (56%) had a baseline estimated glomerular filtration rate of 60 to less than 90 mL/min per 1.73 m3. Dr. Escalada reported that noninferiority for HbA1c reduction was shown for Gla-300 and Gla-100 regardless of renal function, and that evidence of heterogeneity of treatment effect across subgroups was observed (P = .46). However, the risk of confirmed or severe hypoglycemia was significantly lower for nocturnal events in the Gla-300 group, compared with the Gla-100 group (30% vs. 40% overall, respectively), while the risk of anytime hypoglycemia events in a 24-hour period was comparable to or lower in the Gla-300 group, compared with the Gla-100 group. Renal function did not affect the lower rate of nocturnal or anytime hypoglycemia. “Severe hypoglycemia was rare, and renal function did not affect the rate of severe events,” he said.
The trial was sponsored by Sanofi. Dr. Escalada disclosed that he is a member of the advisory panel for Sanofi and for Merck Sharp & Dohme. He is also a member of the speakers bureau for both companies as well as for AstraZeneca, Boehringer Ingelheim, Eli Lilly, and Novo Nordisk.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Key clinical point: Insulin glargine 300 U/mL provided glycemic control comparable to insulin glargine 100 U/mL, but it reduced the risk of nocturnal hypoglycemia by a greater margin, regardless of renal function.
Major finding: The risk of confirmed or severe hypoglycemia was significantly lower for nocturnal events in the Gla-300 group, compared with the Gla-100 group (30% vs. 40% overall, respectively), while the risk of anytime hypoglycemia events in a 24-hour period was comparable to or lower in the Gla-300 group, compared with the Gla-100 group.
Data source: A post hoc meta-analysis of 2,468 patients aged 18 years and older with type 2 diabetes who were treated with Gla-300 or Gla-100 for 6 months in the EDITION I, II, and III studies.
Disclosures: The trial was sponsored by Sanofi. Dr. Escalada disclosed that he is a member of the advisory panel for Sanofi and for Merck Sharp & Dohme. He is also a member of the speakers bureau for both companies as well as for AstraZeneca, Boehringer Ingelheim, Eli Lilly, and Novo Nordisk.

Cervical spine injury common in children under age 2 with abusive head trauma
BALTIMORE – Cervical spine injuries may be more prevalent than previously thought among young children who sustain abusive head trauma, results of a 4-year, multicenter, retrospective study presented at the annual meeting of the Pediatric Academic Societies have shown.
Of children with abusive head trauma (AHT) who received cervical CT or MRI scans, 31% had abnormal findings. The most common cervical spine injuries were hemorrhagic (23.7%), with ligamentous abnormalities less common (8.7%).
This study helps fill a gap in the existing literature on the risk for cervical spine injuries for children who have suffered abusive head trauma, said Kate Henry, MD, in presenting the results of the study, conducted with her colleagues at the Center for Pediatric Clinical Effectiveness at the Children’s Hospital of Philadelphia.
Previous research in the area has concentrated on older children and on motor vehicle crashes, but “young children are different. They are preverbal, and the physical exam can be difficult. Also, the developing spine has a unique anatomy” that can change injury patterns and make imaging interpretation challenging, said Dr. Henry. Motor vehicle crashes make up less than 15% of the cases of traumatic brain injury (TBI) in young children, with falls being responsible for about half, and AHT accounting for 19%-25%, she said.
The study included patients younger than 2 years with an ICD-9 code for TBI associated with a hospital admission or emergency department visit. For final inclusion, patients also had to have MRI or CT confirmation of an intracranial injury. Patients who sustained a motor vehicle collision, were readmitted for complications of a prior injury, or were hospitalized following birth were excluded.
The retrospective chart review of records from four urban children’s hospitals found 3,170 patients who met all criteria except positive intracranial imaging findings; a stratified random sampling reduced the number to 664 charts eligible for review. The final TBI cohort included 329 patients who met the imaging criteria and were not excluded for other reasons.
For these 329 records, Dr. Henry and her colleagues collected physical exam findings, radiology reports of abnormal imaging findings, and the care team’s documentation of their assessment of the etiology of the injury.
The medical team’s assessment, together with the assessment of the child protection team (if it was engaged) were used to classify patients as having either “AHT” or “accidental TBI.”
The trauma-related cervical spine injuries considered as positive findings included ligamentous or soft tissue injury, spinal cord injury, vertebral dislocation or fracture, and extra-axial hemorrhage. “We had a high threshold for inclusion,” said Dr. Henry, explaining that only cervical, not high thoracic, findings were included. Additionally, she and her colleagues excluded radiologist interpretation of injuries as “nonspecific soft tissue findings” or a “possible” injury.
Overall, vertebral fractures or dislocations were seen in just 1.1% of patients with imaged AHT. Cord injuries were slightly more common, at 5.5%. Soft tissue or muscular findings were seen in 0.6% of patients. Some patients had multiple findings, so the overall 31.3% of patients with image-confirmed cervical injuries includes the 23.7% of patients with extra-axial bleed and the 8.7% with ligamentous injury.
Dr. Henry noted that the study was predicated on a certain “circularity of reasoning,” in that the results of spinal imaging could have an influence on the clinician assessment of etiology. The percentage of children diagnosed with AHT who received imaging of any kind was higher than for the all-cause TBI group, as well as for the subgroups of those with accidental TBI or indeterminate etiology. And Dr. Henry said there’s an inherent selection bias in including only those cases where imaging was performed: “The decision to image is unlikely to be random,” she said. However, if all AHT patients who did not receive imaging within the study period were considered to have normal findings, then 9.7% of AHT cases, or about 1 in 10, would still have abnormal findings.
These findings are important because currently, “There are no guidelines for use of cervical CT or MRI” for children with abusive head trauma, said Dr. Henry, and previous research in this area was based on a single-center study. Next steps should go beyond retrospective chart reviews, she said. “Prospective studies are needed to inform cervical imaging recommendations.”
Dr. Henry reported no relevant disclosures.
On Twitter @karioakes
BALTIMORE – Cervical spine injuries may be more prevalent than previously thought among young children who sustain abusive head trauma, results of a 4-year, multicenter, retrospective study presented at the annual meeting of the Pediatric Academic Societies have shown.
Of children with abusive head trauma (AHT) who received cervical CT or MRI scans, 31% had abnormal findings. The most common cervical spine injuries were hemorrhagic (23.7%), with ligamentous abnormalities less common (8.7%).
This study helps fill a gap in the existing literature on the risk for cervical spine injuries for children who have suffered abusive head trauma, said Kate Henry, MD, in presenting the results of the study, conducted with her colleagues at the Center for Pediatric Clinical Effectiveness at the Children’s Hospital of Philadelphia.
Previous research in the area has concentrated on older children and on motor vehicle crashes, but “young children are different. They are preverbal, and the physical exam can be difficult. Also, the developing spine has a unique anatomy” that can change injury patterns and make imaging interpretation challenging, said Dr. Henry. Motor vehicle crashes make up less than 15% of the cases of traumatic brain injury (TBI) in young children, with falls being responsible for about half, and AHT accounting for 19%-25%, she said.
The study included patients younger than 2 years with an ICD-9 code for TBI associated with a hospital admission or emergency department visit. For final inclusion, patients also had to have MRI or CT confirmation of an intracranial injury. Patients who sustained a motor vehicle collision, were readmitted for complications of a prior injury, or were hospitalized following birth were excluded.
The retrospective chart review of records from four urban children’s hospitals found 3,170 patients who met all criteria except positive intracranial imaging findings; a stratified random sampling reduced the number to 664 charts eligible for review. The final TBI cohort included 329 patients who met the imaging criteria and were not excluded for other reasons.
For these 329 records, Dr. Henry and her colleagues collected physical exam findings, radiology reports of abnormal imaging findings, and the care team’s documentation of their assessment of the etiology of the injury.
The medical team’s assessment, together with the assessment of the child protection team (if it was engaged) were used to classify patients as having either “AHT” or “accidental TBI.”
The trauma-related cervical spine injuries considered as positive findings included ligamentous or soft tissue injury, spinal cord injury, vertebral dislocation or fracture, and extra-axial hemorrhage. “We had a high threshold for inclusion,” said Dr. Henry, explaining that only cervical, not high thoracic, findings were included. Additionally, she and her colleagues excluded radiologist interpretation of injuries as “nonspecific soft tissue findings” or a “possible” injury.
Overall, vertebral fractures or dislocations were seen in just 1.1% of patients with imaged AHT. Cord injuries were slightly more common, at 5.5%. Soft tissue or muscular findings were seen in 0.6% of patients. Some patients had multiple findings, so the overall 31.3% of patients with image-confirmed cervical injuries includes the 23.7% of patients with extra-axial bleed and the 8.7% with ligamentous injury.
Dr. Henry noted that the study was predicated on a certain “circularity of reasoning,” in that the results of spinal imaging could have an influence on the clinician assessment of etiology. The percentage of children diagnosed with AHT who received imaging of any kind was higher than for the all-cause TBI group, as well as for the subgroups of those with accidental TBI or indeterminate etiology. And Dr. Henry said there’s an inherent selection bias in including only those cases where imaging was performed: “The decision to image is unlikely to be random,” she said. However, if all AHT patients who did not receive imaging within the study period were considered to have normal findings, then 9.7% of AHT cases, or about 1 in 10, would still have abnormal findings.
These findings are important because currently, “There are no guidelines for use of cervical CT or MRI” for children with abusive head trauma, said Dr. Henry, and previous research in this area was based on a single-center study. Next steps should go beyond retrospective chart reviews, she said. “Prospective studies are needed to inform cervical imaging recommendations.”
Dr. Henry reported no relevant disclosures.
On Twitter @karioakes
BALTIMORE – Cervical spine injuries may be more prevalent than previously thought among young children who sustain abusive head trauma, results of a 4-year, multicenter, retrospective study presented at the annual meeting of the Pediatric Academic Societies have shown.
Of children with abusive head trauma (AHT) who received cervical CT or MRI scans, 31% had abnormal findings. The most common cervical spine injuries were hemorrhagic (23.7%), with ligamentous abnormalities less common (8.7%).
This study helps fill a gap in the existing literature on the risk for cervical spine injuries for children who have suffered abusive head trauma, said Kate Henry, MD, in presenting the results of the study, conducted with her colleagues at the Center for Pediatric Clinical Effectiveness at the Children’s Hospital of Philadelphia.
Previous research in the area has concentrated on older children and on motor vehicle crashes, but “young children are different. They are preverbal, and the physical exam can be difficult. Also, the developing spine has a unique anatomy” that can change injury patterns and make imaging interpretation challenging, said Dr. Henry. Motor vehicle crashes make up less than 15% of the cases of traumatic brain injury (TBI) in young children, with falls being responsible for about half, and AHT accounting for 19%-25%, she said.
The study included patients younger than 2 years with an ICD-9 code for TBI associated with a hospital admission or emergency department visit. For final inclusion, patients also had to have MRI or CT confirmation of an intracranial injury. Patients who sustained a motor vehicle collision, were readmitted for complications of a prior injury, or were hospitalized following birth were excluded.
The retrospective chart review of records from four urban children’s hospitals found 3,170 patients who met all criteria except positive intracranial imaging findings; a stratified random sampling reduced the number to 664 charts eligible for review. The final TBI cohort included 329 patients who met the imaging criteria and were not excluded for other reasons.
For these 329 records, Dr. Henry and her colleagues collected physical exam findings, radiology reports of abnormal imaging findings, and the care team’s documentation of their assessment of the etiology of the injury.
The medical team’s assessment, together with the assessment of the child protection team (if it was engaged) were used to classify patients as having either “AHT” or “accidental TBI.”
The trauma-related cervical spine injuries considered as positive findings included ligamentous or soft tissue injury, spinal cord injury, vertebral dislocation or fracture, and extra-axial hemorrhage. “We had a high threshold for inclusion,” said Dr. Henry, explaining that only cervical, not high thoracic, findings were included. Additionally, she and her colleagues excluded radiologist interpretation of injuries as “nonspecific soft tissue findings” or a “possible” injury.
Overall, vertebral fractures or dislocations were seen in just 1.1% of patients with imaged AHT. Cord injuries were slightly more common, at 5.5%. Soft tissue or muscular findings were seen in 0.6% of patients. Some patients had multiple findings, so the overall 31.3% of patients with image-confirmed cervical injuries includes the 23.7% of patients with extra-axial bleed and the 8.7% with ligamentous injury.
Dr. Henry noted that the study was predicated on a certain “circularity of reasoning,” in that the results of spinal imaging could have an influence on the clinician assessment of etiology. The percentage of children diagnosed with AHT who received imaging of any kind was higher than for the all-cause TBI group, as well as for the subgroups of those with accidental TBI or indeterminate etiology. And Dr. Henry said there’s an inherent selection bias in including only those cases where imaging was performed: “The decision to image is unlikely to be random,” she said. However, if all AHT patients who did not receive imaging within the study period were considered to have normal findings, then 9.7% of AHT cases, or about 1 in 10, would still have abnormal findings.
These findings are important because currently, “There are no guidelines for use of cervical CT or MRI” for children with abusive head trauma, said Dr. Henry, and previous research in this area was based on a single-center study. Next steps should go beyond retrospective chart reviews, she said. “Prospective studies are needed to inform cervical imaging recommendations.”
Dr. Henry reported no relevant disclosures.
On Twitter @karioakes
AT THE PAS ANNUAL MEETING
Key clinical point: Cervical spine imaging findings were seen in 31.3% of young children with imaging-confirmed traumatic brain injury from abusive head trauma.
Major finding: Of 329 patients with TBI from abusive head trauma, 23.7% had extra-axial cervical hemorrhages and 8.7% had cervical ligamentous injuries.
Data source: Weighted sample drawn from retrospective chart review of 3,170 patients from four urban hospitals.
Disclosures: Dr. Henry reported no relevant disclosures.
Update on green tea
During the last 25 years, green tea, which is derived from Camellia sinensis (an evergreen member of the Theaceae family), has gained considerable attention because of its purported antioxidant and anticarcinogenic properties. Believed to have been used by human beings for 4,000 years,1 green tea is now one of the most heavily researched of the antioxidants, with numerous studies of its cutaneous effects appearing in the literature.2 Laden with plant polyphenols, orally administered or topically applied green tea has been shown to display significant antioxidant, chemopreventive, immunomodulatory, and anti-inflammatory activity, affecting the biochemical pathways important in cell proliferation.3-6 For this reason, and due to its global popularity as a beverage, green tea polyphenols are among the most frequently studied herbal agents used in medicine.
Polyphenols, many of which are potent antioxidants, are a large diverse family of thousands of chemical compounds present in plants. The four major polyphenolic catechins present in green tea include: ECG [(-)EpiCatechin-3-O-Gallate], GCG [(-)GalloCatechin-3-O-Gallate], EGC [(-)EpiGalloCatechin], and EGCG [(-)EpiGalloCatechin-3-O-Gallate], the most abundant and biologically active green tea constituent. In fact, EGCG is the component associated with the greatest anticarcinogenic and chemopreventive properties.6

A wide-ranging evidence-based review of the use of botanicals in dermatology, published in 2010, showed that the oral administration, in particular, as well as topical application of antioxidant plant extracts of green tea, among other botanicals, can protect skin against the harmful effects of UV exposure, including erythema, premature aging, and cancer.7
Green tea is thought to be challenging to formulate because of the inherent hydrophilicity of EGCG, which limits penetration into human skin.8,9 Nevertheless, green tea is thought to have great potential in traditional sunscreens to enhance photoprotection.10,11 The photoprotective activity of orally administered or topically applied green tea has been supported in various studies.12-15
The remainder of this column will focus on recent studies of topically applied green tea polyphenols in human beings as well as clinical uses of this agent.
Topical uses
Topical green tea appears to reduce skin inflammation and neutralize free radicals, which explains its popularity as an additive in rosacea and antiaging skin care products. The antiaging effects of green tea are difficult to measure because it functions as an antioxidant that prevents aging and does not have the capacity to increase collagen synthesis or ameliorate already existing wrinkles. However, there is relatively good evidence, in comparison to other antioxidants, suggesting that topically applied green tea can help protect skin from UV radiation.16
Investigators performed a thorough literature search of all in vitro, in vivo, and controlled clinical trials involving green tea formulations and their dermatologic applications, which was published in 2012. They evaluated 20 studies, with evidence suggesting that orally administered green tea displays a broad range of healthy activity, and supportive data for the use of topically applied green tea extract for treatment of various cutaneous conditions, including acne, rosacea, atopic dermatitis, androgenetic alopecia, hirsutism, candidiasis, keloids, leishmaniasis, and genital warts.17
Also, a green tea topical formulation, green tea sinecatechin Polyphenon E (Veregen) ointment, has recently been shown to exert antioxidant, antiviral, and antitumor activity, and has demonstrated efficacy in treating Condylomata acuminata (external anogenital warts).18 In addition, topically applying green tea catechins in the morning in combination with traditional sunscreens is believed to have the potential to protect the skin from UV-induced damage. Topical green tea may improve rosacea, prevent retinoid dermatitis, and play a role in managing pigmentation disorders. Few of the many over-the-counter products that contain green tea catechins have been tested in controlled clinical trials and the concentration of polyphenols in these products is too low to demonstrate efficacy. It is necessary to know the amount of green tea catechins in a formulation to judge its efficacy.
Acne
In 2009, in a 6-week study investigating the efficacy of 2% green tea lotion for the treatment of mild-to-moderate acne vulgaris in 20 patients, researchers reported statistically significant reductions in mean total lesion count and mean severity index (devised by the authors to correlate with total lesion count in increasing intensity, scaled from 1 to 3). They concluded that 2% green tea lotion is both an effective and cost effective approach for treating mild-to-moderate acne lesions.19
A 2012 study revealed that ethanol extracts of several herbs, including green tea, exhibited the potential for inhibiting acne when incorporated into a topical moisturizer, specifically acting against acne-causing bacteria without provoking irritation.20 Earlier that year, other investigators conducted in vitro and in vivo experiments to evaluate the effects against acne of polyphenon-60, which contains various green tea catechins (now referred to as sinecatechins in the United States.).21 In this clinical study, patients exhibited improvement in acne symptoms, including a reduction in the number of pustules and comedones.22
A study published in 2013, a single-blind, placebo-controlled, split-face comparative study in 22 individuals over 60 days, evaluated the efficacy of green tea, as well as green tea plus lotus, compared with placebo for controlling casual sebum secretions in healthy adults. Compared with placebo, consistent and statistically significant decreases in sebum secretions were observed in both treatment groups. The combination of green tea and lotus extracts also achieved statistically sounder results than green tea alone. The investigators concluded that a synergistic interaction between green tea and lotus extract constituents appears to hold promise for the treatment of skin conditions in which elevated sebum levels are involved.23
Anogenital warts
In 2006, the Food and Drug Administration approved for the first time a botanical drug formulation for the topical treatment of genital and perianal warts: sinecatechins, derived from green tea catechins and other C. sinensis constituents in a topical 15% ointment (Veregen).21, 24-28
Two years later, Tatti et al. conducted a randomized, double-blind, vehicle-controlled trial to evaluate the efficacy of topical sinecatechins in 502 male and female patients (aged 18 years and older) for the treatment of anogenital warts. For 16 weeks or until complete clearance, subjects applied sinecatechins ointment 15% or 10% or vehicle (placebo) three times daily. Complete clearance was achieved in 57.2% of patients treated with 15% ointment, 56.3% using 10% ointment, and 33.7% who used only the vehicle. Respective recurrence rates, after 12 weeks, were 6.5%, 8.3%, and 8.8%. The investigators concluded that topical sinecatechins in 15% and 10% concentrations represent effective and well-tolerated options for anogenital wart treatment.29
Similarly favorable results regarding polyphenon E 15% were reported in reference to three placebo-controlled clinical studies in 1,400 patients with genital warts from Europe, North and South America, and South Africa,30,31 and by Tatti et al. again in 2010 after randomized, double-blind, vehicle-controlled safety and efficacy trials in nearly 1,000 patients treated with polyphenon E 15% and 10% formulations.21
Two years later, investigators evaluated sinecatechins (Polyphenon E) 10% ointment in two double-blind, multinational studies in adults with external genital and perianal warts. Polyphenon E 10% was found to be significantly more effective than vehicle in completely or partially clearing all warts.32
Earlier that year, a review of the use of sinecatechins ointment for the treatment of external anogenital warts noted that while clearance rates are similar among sinecatechins and other indicated topical medications such as imiquimod and podophyllotoxin, recurrence rates are lower for patients treated with sinecatechins. The authors concluded that the use of sinecatechins for condylomata acuminata was safe and effective and its various molecular activities suggest broader applications to other viral and tumor lesions.33
In 2015, Gupta and Daigle reported that sinecatechins 10% ointment for the treatment of external genital warts was found in phase III trials to display greater efficacy and lower rates of recurrence in comparison to patient-applied treatments now available.28 Later that year, in a systematic PubMed and Embase review of clinical trials involving the use of polyphenol-based therapies, Tuong et al. identified cogent evidence suggesting the effectiveness of green tea polyphenols for the treatment of anogenital warts.34
Antiaging activity
Green tea has been shown to work in combination with red light to exert a rejuvenating effect on the skin, as Sommer and Zhu reported in 2009 that green tea filled cotton pads applied once daily for 20 minutes prior to treatment with light-emitting diodes (central wavelength 670 nm, dermal dose 4 J/cm2) reduced wrinkles in 1 month comparably to 10 months of light treatment alone.35
In 2013, Hong et al. studied the antiwrinkle effects of topically applied green tea extract with high antioxidant activity after tannase treatment. Study participants were randomly divided to receive either green tea extract or tannase-converted green tea extract on their crow’s feet for an 8-week period. The investigators found that tannase treatment elevated the antioxidant activity of green tea and imparted antiwrinkle effects.36
At around the same time, Gianeti conducted clinical studies in 24 volunteers to assess the effects of a cosmetic formulation containing 6% C. sinensis glycolic leaf extracts. Skin moisture was enhanced after 30 days of topical application as was the viscoelastic-to-elastic ratio compared with vehicle and control (a forearm area left untreated). Skin roughness was significantly diminished after 30 days. The investigators concluded that the topical cosmetic formulation with green tea yielded salient moisturizing and cutaneous microrelief benefits.37
Also in 2013, oral intake of green tea catechins in 16 healthy human subjects (with 14 completing the study) appeared to result in the integration of catechin metabolites into human skin linked to the negation of UV-induced 12-hydroxyeicosatetraenoic acid (12-HETE). The investigators speculated that this incorporation of catechins may render protection against sunburn inflammation and even cumulative UV-induced harm.38
After earlier showing the efficacy of green tea and lotus extracts in skin disorders involving excess sebum in a single-blinded, placebo-controlled, split-face comparative study,23 Mahmood and Akhtar conducted a 60-day placebo-controlled comparative split-face study in 33 healthy Asian men to evaluate the efficacy of two cosmetic formulations (green tea and lotus extract) for facial wrinkles. All of the formulations yielded improvements in skin roughness, scaliness, smoothness, and wrinkling, with the greatest reduction in wrinkling conferred by the combination formulation. The investigators concluded that the synergistic activity of green tea and lotus extracts exerted significant improvement along several skin parameters, suggesting the potential for these ingredients in antiaging products.38
In 2014, the synergistic effects of green tea and ginkgo biloba were explored in preclinical and clinical studies. In the clinical study, 48 participants applied the formulations on forearm skin and were evaluated before and after 3 hours and following 15- and 30-day use periods. Results showed a moisturizing effect and enhancement in skin microrelief, as well as improvements in skin elasticity and barrier function.3
Conclusion
Green tea remains one of the most researched antioxidants as benefits from its use continue to emerge. Indeed, green tea polyphenols are in use for a growing number of indications, especially acne and anogenital warts, and there is reason for optimism that topically applied green tea will gain momentum as an increasingly selected therapeutic option. More clinical studies are necessary to further establish the potential role of green tea for a wider range of cutaneous indications. Green tea holds particular promise in relation to photoprotection against UV-induced skin cancer and skin aging.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. She also developed and owns the Baumann Skin Type Solution skin typing systems and related products
References:
1. Cancer Lett. 1997 Mar 19;114(1-2):315-7.
2. J Am Acad Dermatol. 2005 Jun;52(6):1049-59.
3. Arch Dermatol. 2000 Aug;136(8):989-94.
4. Photochem Photobiol. 1995 Nov;62(5):855-61.
5. Oxid Med Cell Longev. 2012:2012:560682.
6. J Dtsch Dermatol Ges. 2015 Aug;13(8):768-75.
7. Am J Clin Dermatol. 2010;11(4):247-67.
8. Dermatol Ther. 2007 Sep-Oct;20(5):322-9.
9. J Clin Aesthet Dermatol. 2010 Feb;3(2):22-41.
10. Photodermatol Photoimmunol Photomed. 2007 Feb;23(1):48-56.
11. Skin Res Technol. 2009 Aug;15(3):338-45.
12. Exp Dermatol. 2009 Jan;18(1):69-77.
13. Exp Dermatol. 2009 Jun;18(6):522-6.
14. Arch Biochem Biophys. 2011 Apr 15;508(2):152-8.
15. Cancer Prev Res (Phila). 2010 Feb;3(2):179-89.
16. Complement Ther Clin Pract. 2014 Feb;20(1):11-5.
17. Skinmed. 2012 Nov-Dec;10(6):352-5.
18. J Eur Acad Dermatol Venereol. 2011 Mar;25(3):345-53.
19. J Drugs Dermatol. 2009 Apr;8(4):358-64.
20. Pak J Pharm Sci. 2012 Oct;25(4):867-70.
21. Br J Dermatol. 2010 Jan;162(1):176-84.
22. Arch Dermatol Res. 2012 Oct;304(8):655-63.
23. Hippokratia. 2013 Jan;17(1):64-7.
24. Food Chem Toxicol. 2008 Aug;46(8):2606-10.
25. Nat Biotechnol. 2008 Oct;26(10):1077-83.
26. Skin Therapy Lett. 2012 Apr;17(4):5-7.
27. J Clin Aesthet Dermatol. 2012 Jan;5(1):19-26.
28. Skin Therapy Lett. 2015 Jan-Feb;20(1):6-8.
29. Obstet Gynecol. 2008 Jun;111(6):1371-9.
30. Hautarzt. 2008 Jan;59(1):31-5.
31. J Eur Acad Dermatol Venereol. 2007 Nov;21(10):1404-12.
32. Am J Clin Dermatol. 2012 Aug 1:13(4):275-81.
33. Expert Opin Biol Ther. 2012 Jun;12(6):783-93.
34. J Dermatolog Treat. 2015;26(4):381-8.
35. Photomed Laser Surg. 2009 Dec;27(6):969-71.
36. J Cosmet Dermatol. 2013 Jun;12(2):137-43.
37. Dermatol Ther. 2013 May-Jun;26(3):267-71.
During the last 25 years, green tea, which is derived from Camellia sinensis (an evergreen member of the Theaceae family), has gained considerable attention because of its purported antioxidant and anticarcinogenic properties. Believed to have been used by human beings for 4,000 years,1 green tea is now one of the most heavily researched of the antioxidants, with numerous studies of its cutaneous effects appearing in the literature.2 Laden with plant polyphenols, orally administered or topically applied green tea has been shown to display significant antioxidant, chemopreventive, immunomodulatory, and anti-inflammatory activity, affecting the biochemical pathways important in cell proliferation.3-6 For this reason, and due to its global popularity as a beverage, green tea polyphenols are among the most frequently studied herbal agents used in medicine.
Polyphenols, many of which are potent antioxidants, are a large diverse family of thousands of chemical compounds present in plants. The four major polyphenolic catechins present in green tea include: ECG [(-)EpiCatechin-3-O-Gallate], GCG [(-)GalloCatechin-3-O-Gallate], EGC [(-)EpiGalloCatechin], and EGCG [(-)EpiGalloCatechin-3-O-Gallate], the most abundant and biologically active green tea constituent. In fact, EGCG is the component associated with the greatest anticarcinogenic and chemopreventive properties.6
A wide-ranging evidence-based review of the use of botanicals in dermatology, published in 2010, showed that the oral administration, in particular, as well as topical application of antioxidant plant extracts of green tea, among other botanicals, can protect skin against the harmful effects of UV exposure, including erythema, premature aging, and cancer.7
Green tea is thought to be challenging to formulate because of the inherent hydrophilicity of EGCG, which limits penetration into human skin.8,9 Nevertheless, green tea is thought to have great potential in traditional sunscreens to enhance photoprotection.10,11 The photoprotective activity of orally administered or topically applied green tea has been supported in various studies.12-15
The remainder of this column will focus on recent studies of topically applied green tea polyphenols in human beings as well as clinical uses of this agent.
Topical uses
Topical green tea appears to reduce skin inflammation and neutralize free radicals, which explains its popularity as an additive in rosacea and antiaging skin care products. The antiaging effects of green tea are difficult to measure because it functions as an antioxidant that prevents aging and does not have the capacity to increase collagen synthesis or ameliorate already existing wrinkles. However, there is relatively good evidence, in comparison to other antioxidants, suggesting that topically applied green tea can help protect skin from UV radiation.16
Investigators performed a thorough literature search of all in vitro, in vivo, and controlled clinical trials involving green tea formulations and their dermatologic applications, which was published in 2012. They evaluated 20 studies, with evidence suggesting that orally administered green tea displays a broad range of healthy activity, and supportive data for the use of topically applied green tea extract for treatment of various cutaneous conditions, including acne, rosacea, atopic dermatitis, androgenetic alopecia, hirsutism, candidiasis, keloids, leishmaniasis, and genital warts.17
Also, a green tea topical formulation, green tea sinecatechin Polyphenon E (Veregen) ointment, has recently been shown to exert antioxidant, antiviral, and antitumor activity, and has demonstrated efficacy in treating Condylomata acuminata (external anogenital warts).18 In addition, topically applying green tea catechins in the morning in combination with traditional sunscreens is believed to have the potential to protect the skin from UV-induced damage. Topical green tea may improve rosacea, prevent retinoid dermatitis, and play a role in managing pigmentation disorders. Few of the many over-the-counter products that contain green tea catechins have been tested in controlled clinical trials and the concentration of polyphenols in these products is too low to demonstrate efficacy. It is necessary to know the amount of green tea catechins in a formulation to judge its efficacy.
Acne
In 2009, in a 6-week study investigating the efficacy of 2% green tea lotion for the treatment of mild-to-moderate acne vulgaris in 20 patients, researchers reported statistically significant reductions in mean total lesion count and mean severity index (devised by the authors to correlate with total lesion count in increasing intensity, scaled from 1 to 3). They concluded that 2% green tea lotion is both an effective and cost effective approach for treating mild-to-moderate acne lesions.19
A 2012 study revealed that ethanol extracts of several herbs, including green tea, exhibited the potential for inhibiting acne when incorporated into a topical moisturizer, specifically acting against acne-causing bacteria without provoking irritation.20 Earlier that year, other investigators conducted in vitro and in vivo experiments to evaluate the effects against acne of polyphenon-60, which contains various green tea catechins (now referred to as sinecatechins in the United States.).21 In this clinical study, patients exhibited improvement in acne symptoms, including a reduction in the number of pustules and comedones.22
A study published in 2013, a single-blind, placebo-controlled, split-face comparative study in 22 individuals over 60 days, evaluated the efficacy of green tea, as well as green tea plus lotus, compared with placebo for controlling casual sebum secretions in healthy adults. Compared with placebo, consistent and statistically significant decreases in sebum secretions were observed in both treatment groups. The combination of green tea and lotus extracts also achieved statistically sounder results than green tea alone. The investigators concluded that a synergistic interaction between green tea and lotus extract constituents appears to hold promise for the treatment of skin conditions in which elevated sebum levels are involved.23
Anogenital warts
In 2006, the Food and Drug Administration approved for the first time a botanical drug formulation for the topical treatment of genital and perianal warts: sinecatechins, derived from green tea catechins and other C. sinensis constituents in a topical 15% ointment (Veregen).21, 24-28
Two years later, Tatti et al. conducted a randomized, double-blind, vehicle-controlled trial to evaluate the efficacy of topical sinecatechins in 502 male and female patients (aged 18 years and older) for the treatment of anogenital warts. For 16 weeks or until complete clearance, subjects applied sinecatechins ointment 15% or 10% or vehicle (placebo) three times daily. Complete clearance was achieved in 57.2% of patients treated with 15% ointment, 56.3% using 10% ointment, and 33.7% who used only the vehicle. Respective recurrence rates, after 12 weeks, were 6.5%, 8.3%, and 8.8%. The investigators concluded that topical sinecatechins in 15% and 10% concentrations represent effective and well-tolerated options for anogenital wart treatment.29
Similarly favorable results regarding polyphenon E 15% were reported in reference to three placebo-controlled clinical studies in 1,400 patients with genital warts from Europe, North and South America, and South Africa,30,31 and by Tatti et al. again in 2010 after randomized, double-blind, vehicle-controlled safety and efficacy trials in nearly 1,000 patients treated with polyphenon E 15% and 10% formulations.21
Two years later, investigators evaluated sinecatechins (Polyphenon E) 10% ointment in two double-blind, multinational studies in adults with external genital and perianal warts. Polyphenon E 10% was found to be significantly more effective than vehicle in completely or partially clearing all warts.32
Earlier that year, a review of the use of sinecatechins ointment for the treatment of external anogenital warts noted that while clearance rates are similar among sinecatechins and other indicated topical medications such as imiquimod and podophyllotoxin, recurrence rates are lower for patients treated with sinecatechins. The authors concluded that the use of sinecatechins for condylomata acuminata was safe and effective and its various molecular activities suggest broader applications to other viral and tumor lesions.33
In 2015, Gupta and Daigle reported that sinecatechins 10% ointment for the treatment of external genital warts was found in phase III trials to display greater efficacy and lower rates of recurrence in comparison to patient-applied treatments now available.28 Later that year, in a systematic PubMed and Embase review of clinical trials involving the use of polyphenol-based therapies, Tuong et al. identified cogent evidence suggesting the effectiveness of green tea polyphenols for the treatment of anogenital warts.34
Antiaging activity
Green tea has been shown to work in combination with red light to exert a rejuvenating effect on the skin, as Sommer and Zhu reported in 2009 that green tea filled cotton pads applied once daily for 20 minutes prior to treatment with light-emitting diodes (central wavelength 670 nm, dermal dose 4 J/cm2) reduced wrinkles in 1 month comparably to 10 months of light treatment alone.35
In 2013, Hong et al. studied the antiwrinkle effects of topically applied green tea extract with high antioxidant activity after tannase treatment. Study participants were randomly divided to receive either green tea extract or tannase-converted green tea extract on their crow’s feet for an 8-week period. The investigators found that tannase treatment elevated the antioxidant activity of green tea and imparted antiwrinkle effects.36
At around the same time, Gianeti conducted clinical studies in 24 volunteers to assess the effects of a cosmetic formulation containing 6% C. sinensis glycolic leaf extracts. Skin moisture was enhanced after 30 days of topical application as was the viscoelastic-to-elastic ratio compared with vehicle and control (a forearm area left untreated). Skin roughness was significantly diminished after 30 days. The investigators concluded that the topical cosmetic formulation with green tea yielded salient moisturizing and cutaneous microrelief benefits.37
Also in 2013, oral intake of green tea catechins in 16 healthy human subjects (with 14 completing the study) appeared to result in the integration of catechin metabolites into human skin linked to the negation of UV-induced 12-hydroxyeicosatetraenoic acid (12-HETE). The investigators speculated that this incorporation of catechins may render protection against sunburn inflammation and even cumulative UV-induced harm.38
After earlier showing the efficacy of green tea and lotus extracts in skin disorders involving excess sebum in a single-blinded, placebo-controlled, split-face comparative study,23 Mahmood and Akhtar conducted a 60-day placebo-controlled comparative split-face study in 33 healthy Asian men to evaluate the efficacy of two cosmetic formulations (green tea and lotus extract) for facial wrinkles. All of the formulations yielded improvements in skin roughness, scaliness, smoothness, and wrinkling, with the greatest reduction in wrinkling conferred by the combination formulation. The investigators concluded that the synergistic activity of green tea and lotus extracts exerted significant improvement along several skin parameters, suggesting the potential for these ingredients in antiaging products.38
In 2014, the synergistic effects of green tea and ginkgo biloba were explored in preclinical and clinical studies. In the clinical study, 48 participants applied the formulations on forearm skin and were evaluated before and after 3 hours and following 15- and 30-day use periods. Results showed a moisturizing effect and enhancement in skin microrelief, as well as improvements in skin elasticity and barrier function.3
Conclusion
Green tea remains one of the most researched antioxidants as benefits from its use continue to emerge. Indeed, green tea polyphenols are in use for a growing number of indications, especially acne and anogenital warts, and there is reason for optimism that topically applied green tea will gain momentum as an increasingly selected therapeutic option. More clinical studies are necessary to further establish the potential role of green tea for a wider range of cutaneous indications. Green tea holds particular promise in relation to photoprotection against UV-induced skin cancer and skin aging.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. She also developed and owns the Baumann Skin Type Solution skin typing systems and related products
References:
1. Cancer Lett. 1997 Mar 19;114(1-2):315-7.
2. J Am Acad Dermatol. 2005 Jun;52(6):1049-59.
3. Arch Dermatol. 2000 Aug;136(8):989-94.
4. Photochem Photobiol. 1995 Nov;62(5):855-61.
5. Oxid Med Cell Longev. 2012:2012:560682.
6. J Dtsch Dermatol Ges. 2015 Aug;13(8):768-75.
7. Am J Clin Dermatol. 2010;11(4):247-67.
8. Dermatol Ther. 2007 Sep-Oct;20(5):322-9.
9. J Clin Aesthet Dermatol. 2010 Feb;3(2):22-41.
10. Photodermatol Photoimmunol Photomed. 2007 Feb;23(1):48-56.
11. Skin Res Technol. 2009 Aug;15(3):338-45.
12. Exp Dermatol. 2009 Jan;18(1):69-77.
13. Exp Dermatol. 2009 Jun;18(6):522-6.
14. Arch Biochem Biophys. 2011 Apr 15;508(2):152-8.
15. Cancer Prev Res (Phila). 2010 Feb;3(2):179-89.
16. Complement Ther Clin Pract. 2014 Feb;20(1):11-5.
17. Skinmed. 2012 Nov-Dec;10(6):352-5.
18. J Eur Acad Dermatol Venereol. 2011 Mar;25(3):345-53.
19. J Drugs Dermatol. 2009 Apr;8(4):358-64.
20. Pak J Pharm Sci. 2012 Oct;25(4):867-70.
21. Br J Dermatol. 2010 Jan;162(1):176-84.
22. Arch Dermatol Res. 2012 Oct;304(8):655-63.
23. Hippokratia. 2013 Jan;17(1):64-7.
24. Food Chem Toxicol. 2008 Aug;46(8):2606-10.
25. Nat Biotechnol. 2008 Oct;26(10):1077-83.
26. Skin Therapy Lett. 2012 Apr;17(4):5-7.
27. J Clin Aesthet Dermatol. 2012 Jan;5(1):19-26.
28. Skin Therapy Lett. 2015 Jan-Feb;20(1):6-8.
29. Obstet Gynecol. 2008 Jun;111(6):1371-9.
30. Hautarzt. 2008 Jan;59(1):31-5.
31. J Eur Acad Dermatol Venereol. 2007 Nov;21(10):1404-12.
32. Am J Clin Dermatol. 2012 Aug 1:13(4):275-81.
33. Expert Opin Biol Ther. 2012 Jun;12(6):783-93.
34. J Dermatolog Treat. 2015;26(4):381-8.
35. Photomed Laser Surg. 2009 Dec;27(6):969-71.
36. J Cosmet Dermatol. 2013 Jun;12(2):137-43.
37. Dermatol Ther. 2013 May-Jun;26(3):267-71.
During the last 25 years, green tea, which is derived from Camellia sinensis (an evergreen member of the Theaceae family), has gained considerable attention because of its purported antioxidant and anticarcinogenic properties. Believed to have been used by human beings for 4,000 years,1 green tea is now one of the most heavily researched of the antioxidants, with numerous studies of its cutaneous effects appearing in the literature.2 Laden with plant polyphenols, orally administered or topically applied green tea has been shown to display significant antioxidant, chemopreventive, immunomodulatory, and anti-inflammatory activity, affecting the biochemical pathways important in cell proliferation.3-6 For this reason, and due to its global popularity as a beverage, green tea polyphenols are among the most frequently studied herbal agents used in medicine.
Polyphenols, many of which are potent antioxidants, are a large diverse family of thousands of chemical compounds present in plants. The four major polyphenolic catechins present in green tea include: ECG [(-)EpiCatechin-3-O-Gallate], GCG [(-)GalloCatechin-3-O-Gallate], EGC [(-)EpiGalloCatechin], and EGCG [(-)EpiGalloCatechin-3-O-Gallate], the most abundant and biologically active green tea constituent. In fact, EGCG is the component associated with the greatest anticarcinogenic and chemopreventive properties.6
A wide-ranging evidence-based review of the use of botanicals in dermatology, published in 2010, showed that the oral administration, in particular, as well as topical application of antioxidant plant extracts of green tea, among other botanicals, can protect skin against the harmful effects of UV exposure, including erythema, premature aging, and cancer.7
Green tea is thought to be challenging to formulate because of the inherent hydrophilicity of EGCG, which limits penetration into human skin.8,9 Nevertheless, green tea is thought to have great potential in traditional sunscreens to enhance photoprotection.10,11 The photoprotective activity of orally administered or topically applied green tea has been supported in various studies.12-15
The remainder of this column will focus on recent studies of topically applied green tea polyphenols in human beings as well as clinical uses of this agent.
Topical uses
Topical green tea appears to reduce skin inflammation and neutralize free radicals, which explains its popularity as an additive in rosacea and antiaging skin care products. The antiaging effects of green tea are difficult to measure because it functions as an antioxidant that prevents aging and does not have the capacity to increase collagen synthesis or ameliorate already existing wrinkles. However, there is relatively good evidence, in comparison to other antioxidants, suggesting that topically applied green tea can help protect skin from UV radiation.16
Investigators performed a thorough literature search of all in vitro, in vivo, and controlled clinical trials involving green tea formulations and their dermatologic applications, which was published in 2012. They evaluated 20 studies, with evidence suggesting that orally administered green tea displays a broad range of healthy activity, and supportive data for the use of topically applied green tea extract for treatment of various cutaneous conditions, including acne, rosacea, atopic dermatitis, androgenetic alopecia, hirsutism, candidiasis, keloids, leishmaniasis, and genital warts.17
Also, a green tea topical formulation, green tea sinecatechin Polyphenon E (Veregen) ointment, has recently been shown to exert antioxidant, antiviral, and antitumor activity, and has demonstrated efficacy in treating Condylomata acuminata (external anogenital warts).18 In addition, topically applying green tea catechins in the morning in combination with traditional sunscreens is believed to have the potential to protect the skin from UV-induced damage. Topical green tea may improve rosacea, prevent retinoid dermatitis, and play a role in managing pigmentation disorders. Few of the many over-the-counter products that contain green tea catechins have been tested in controlled clinical trials and the concentration of polyphenols in these products is too low to demonstrate efficacy. It is necessary to know the amount of green tea catechins in a formulation to judge its efficacy.
Acne
In 2009, in a 6-week study investigating the efficacy of 2% green tea lotion for the treatment of mild-to-moderate acne vulgaris in 20 patients, researchers reported statistically significant reductions in mean total lesion count and mean severity index (devised by the authors to correlate with total lesion count in increasing intensity, scaled from 1 to 3). They concluded that 2% green tea lotion is both an effective and cost effective approach for treating mild-to-moderate acne lesions.19
A 2012 study revealed that ethanol extracts of several herbs, including green tea, exhibited the potential for inhibiting acne when incorporated into a topical moisturizer, specifically acting against acne-causing bacteria without provoking irritation.20 Earlier that year, other investigators conducted in vitro and in vivo experiments to evaluate the effects against acne of polyphenon-60, which contains various green tea catechins (now referred to as sinecatechins in the United States.).21 In this clinical study, patients exhibited improvement in acne symptoms, including a reduction in the number of pustules and comedones.22
A study published in 2013, a single-blind, placebo-controlled, split-face comparative study in 22 individuals over 60 days, evaluated the efficacy of green tea, as well as green tea plus lotus, compared with placebo for controlling casual sebum secretions in healthy adults. Compared with placebo, consistent and statistically significant decreases in sebum secretions were observed in both treatment groups. The combination of green tea and lotus extracts also achieved statistically sounder results than green tea alone. The investigators concluded that a synergistic interaction between green tea and lotus extract constituents appears to hold promise for the treatment of skin conditions in which elevated sebum levels are involved.23
Anogenital warts
In 2006, the Food and Drug Administration approved for the first time a botanical drug formulation for the topical treatment of genital and perianal warts: sinecatechins, derived from green tea catechins and other C. sinensis constituents in a topical 15% ointment (Veregen).21, 24-28
Two years later, Tatti et al. conducted a randomized, double-blind, vehicle-controlled trial to evaluate the efficacy of topical sinecatechins in 502 male and female patients (aged 18 years and older) for the treatment of anogenital warts. For 16 weeks or until complete clearance, subjects applied sinecatechins ointment 15% or 10% or vehicle (placebo) three times daily. Complete clearance was achieved in 57.2% of patients treated with 15% ointment, 56.3% using 10% ointment, and 33.7% who used only the vehicle. Respective recurrence rates, after 12 weeks, were 6.5%, 8.3%, and 8.8%. The investigators concluded that topical sinecatechins in 15% and 10% concentrations represent effective and well-tolerated options for anogenital wart treatment.29
Similarly favorable results regarding polyphenon E 15% were reported in reference to three placebo-controlled clinical studies in 1,400 patients with genital warts from Europe, North and South America, and South Africa,30,31 and by Tatti et al. again in 2010 after randomized, double-blind, vehicle-controlled safety and efficacy trials in nearly 1,000 patients treated with polyphenon E 15% and 10% formulations.21
Two years later, investigators evaluated sinecatechins (Polyphenon E) 10% ointment in two double-blind, multinational studies in adults with external genital and perianal warts. Polyphenon E 10% was found to be significantly more effective than vehicle in completely or partially clearing all warts.32
Earlier that year, a review of the use of sinecatechins ointment for the treatment of external anogenital warts noted that while clearance rates are similar among sinecatechins and other indicated topical medications such as imiquimod and podophyllotoxin, recurrence rates are lower for patients treated with sinecatechins. The authors concluded that the use of sinecatechins for condylomata acuminata was safe and effective and its various molecular activities suggest broader applications to other viral and tumor lesions.33
In 2015, Gupta and Daigle reported that sinecatechins 10% ointment for the treatment of external genital warts was found in phase III trials to display greater efficacy and lower rates of recurrence in comparison to patient-applied treatments now available.28 Later that year, in a systematic PubMed and Embase review of clinical trials involving the use of polyphenol-based therapies, Tuong et al. identified cogent evidence suggesting the effectiveness of green tea polyphenols for the treatment of anogenital warts.34
Antiaging activity
Green tea has been shown to work in combination with red light to exert a rejuvenating effect on the skin, as Sommer and Zhu reported in 2009 that green tea filled cotton pads applied once daily for 20 minutes prior to treatment with light-emitting diodes (central wavelength 670 nm, dermal dose 4 J/cm2) reduced wrinkles in 1 month comparably to 10 months of light treatment alone.35
In 2013, Hong et al. studied the antiwrinkle effects of topically applied green tea extract with high antioxidant activity after tannase treatment. Study participants were randomly divided to receive either green tea extract or tannase-converted green tea extract on their crow’s feet for an 8-week period. The investigators found that tannase treatment elevated the antioxidant activity of green tea and imparted antiwrinkle effects.36
At around the same time, Gianeti conducted clinical studies in 24 volunteers to assess the effects of a cosmetic formulation containing 6% C. sinensis glycolic leaf extracts. Skin moisture was enhanced after 30 days of topical application as was the viscoelastic-to-elastic ratio compared with vehicle and control (a forearm area left untreated). Skin roughness was significantly diminished after 30 days. The investigators concluded that the topical cosmetic formulation with green tea yielded salient moisturizing and cutaneous microrelief benefits.37
Also in 2013, oral intake of green tea catechins in 16 healthy human subjects (with 14 completing the study) appeared to result in the integration of catechin metabolites into human skin linked to the negation of UV-induced 12-hydroxyeicosatetraenoic acid (12-HETE). The investigators speculated that this incorporation of catechins may render protection against sunburn inflammation and even cumulative UV-induced harm.38
After earlier showing the efficacy of green tea and lotus extracts in skin disorders involving excess sebum in a single-blinded, placebo-controlled, split-face comparative study,23 Mahmood and Akhtar conducted a 60-day placebo-controlled comparative split-face study in 33 healthy Asian men to evaluate the efficacy of two cosmetic formulations (green tea and lotus extract) for facial wrinkles. All of the formulations yielded improvements in skin roughness, scaliness, smoothness, and wrinkling, with the greatest reduction in wrinkling conferred by the combination formulation. The investigators concluded that the synergistic activity of green tea and lotus extracts exerted significant improvement along several skin parameters, suggesting the potential for these ingredients in antiaging products.38
In 2014, the synergistic effects of green tea and ginkgo biloba were explored in preclinical and clinical studies. In the clinical study, 48 participants applied the formulations on forearm skin and were evaluated before and after 3 hours and following 15- and 30-day use periods. Results showed a moisturizing effect and enhancement in skin microrelief, as well as improvements in skin elasticity and barrier function.3
Conclusion
Green tea remains one of the most researched antioxidants as benefits from its use continue to emerge. Indeed, green tea polyphenols are in use for a growing number of indications, especially acne and anogenital warts, and there is reason for optimism that topically applied green tea will gain momentum as an increasingly selected therapeutic option. More clinical studies are necessary to further establish the potential role of green tea for a wider range of cutaneous indications. Green tea holds particular promise in relation to photoprotection against UV-induced skin cancer and skin aging.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever. She also developed and owns the Baumann Skin Type Solution skin typing systems and related products
References:
1. Cancer Lett. 1997 Mar 19;114(1-2):315-7.
2. J Am Acad Dermatol. 2005 Jun;52(6):1049-59.
3. Arch Dermatol. 2000 Aug;136(8):989-94.
4. Photochem Photobiol. 1995 Nov;62(5):855-61.
5. Oxid Med Cell Longev. 2012:2012:560682.
6. J Dtsch Dermatol Ges. 2015 Aug;13(8):768-75.
7. Am J Clin Dermatol. 2010;11(4):247-67.
8. Dermatol Ther. 2007 Sep-Oct;20(5):322-9.
9. J Clin Aesthet Dermatol. 2010 Feb;3(2):22-41.
10. Photodermatol Photoimmunol Photomed. 2007 Feb;23(1):48-56.
11. Skin Res Technol. 2009 Aug;15(3):338-45.
12. Exp Dermatol. 2009 Jan;18(1):69-77.
13. Exp Dermatol. 2009 Jun;18(6):522-6.
14. Arch Biochem Biophys. 2011 Apr 15;508(2):152-8.
15. Cancer Prev Res (Phila). 2010 Feb;3(2):179-89.
16. Complement Ther Clin Pract. 2014 Feb;20(1):11-5.
17. Skinmed. 2012 Nov-Dec;10(6):352-5.
18. J Eur Acad Dermatol Venereol. 2011 Mar;25(3):345-53.
19. J Drugs Dermatol. 2009 Apr;8(4):358-64.
20. Pak J Pharm Sci. 2012 Oct;25(4):867-70.
21. Br J Dermatol. 2010 Jan;162(1):176-84.
22. Arch Dermatol Res. 2012 Oct;304(8):655-63.
23. Hippokratia. 2013 Jan;17(1):64-7.
24. Food Chem Toxicol. 2008 Aug;46(8):2606-10.
25. Nat Biotechnol. 2008 Oct;26(10):1077-83.
26. Skin Therapy Lett. 2012 Apr;17(4):5-7.
27. J Clin Aesthet Dermatol. 2012 Jan;5(1):19-26.
28. Skin Therapy Lett. 2015 Jan-Feb;20(1):6-8.
29. Obstet Gynecol. 2008 Jun;111(6):1371-9.
30. Hautarzt. 2008 Jan;59(1):31-5.
31. J Eur Acad Dermatol Venereol. 2007 Nov;21(10):1404-12.
32. Am J Clin Dermatol. 2012 Aug 1:13(4):275-81.
33. Expert Opin Biol Ther. 2012 Jun;12(6):783-93.
34. J Dermatolog Treat. 2015;26(4):381-8.
35. Photomed Laser Surg. 2009 Dec;27(6):969-71.
36. J Cosmet Dermatol. 2013 Jun;12(2):137-43.
37. Dermatol Ther. 2013 May-Jun;26(3):267-71.
• Green tea is one of the most researched antioxidants, particularly its constituent polyphenolic catechins (notably epigallocatechin gallate, or EGCG).
• It is thought to be difficult to formulate in topical products because of the intrinsic hydrophilicity of EGCG.
• Topical application is thought to reduce inflammation and neutralize free radicals, but does not increase collagen production or reduce already existing wrinkles.
• It has been shown to be effective topically for treating acne, anogenital warts, and aging skin.

‘Clarion call’ to screen for, treat aldosteronism
The Endocrine Society’s updated Clinical Practice Guideline for managing primary aldosteronism is “a clarion call” for physicians to recognize the impact of this substantial public health problem and dramatically ramp up their screening and treatment efforts and was published in the Journal of Clinical Endocrinology and Metabolism.
This update differs from the previous (2008) version of the guideline in “the explicit recognition of primary aldosteronism as a major public health issue and not merely a matter of case detection, diagnosis, and treatment of individual patients,” wrote John W. Funder, MD, and his associates on the task force that compiled the guideline.
Many physicians in current practice were taught that the disorder “is a rare and benign cause of hypertension, [and] thus merely a footnote to the management of hypertension as a whole. Cardiologists usually write guidelines for hypertension with some input from nephrologists and clinical pharmacologists [but] little or none from endocrinologists,” they noted.
As a result, most patients with hypertension and occult aldosteronism are never screened for the disorder and receive suboptimal care. Primary care providers must be “made keenly aware” that the proportion of people with hypertension who have aldosteronism is much higher than previously thought (at roughly 10%), that another 20% of hypertensive people have “inappropriate aldosterone secretion,” and that both groups respond remarkably well to medical therapy, particularly to mineralocorticoid-receptor antagonists. This is critical because hypertensive patients with aldosteronism are at much greater risk for cardiovascular morbidity and mortality than their age-, sex-, and BP-matched counterparts who don’t have aldosteronism, said Dr. Funder of the Hudson Institute of Medical Research, Clayton (VIC), Australia, and his associates.
In addition to recommendations regarding screening and treatment and summaries of the evidence on which those recommendations are based, the new guideline offers a remarks section for each recommendation, which includes technical suggestions to help clinicians implement them in real-world practice.
Among the Guideline’s recommendations:
• Screen for primary aldosteronism all patients who have sustained BP above 150/100 mm Hg, hypertension resistant to three conventional antihypertensive drugs, hypertension that requires four or more drugs to control it, hypertension plus hypokalemia, hypertension plus adrenal incidentaloma, hypertension plus sleep apnea, hypertension plus a family history of early-onset hypertension or stroke at a young age, and hypertension plus a first-degree relative with primary aldosteronism.
Use the plasma aldosterone/renin ratio for this screening.
• Do one or more confirmatory tests to definitively confirm the diagnosis before proceeding to subtype classification. The exception to this recommendation is patients who develop spontaneous hypokalemia.
• Do adrenal CT as the initial step in subtype classification, to exclude large masses that may signal adrenocortical carcinoma and to help interventional radiologists and surgeons make anatomic assessments.
Before surgery, an experienced radiologist should determine whether adrenal disease is unilateral or bilateral using adrenal venous sampling.
Order genetic testing for patients with disease onset before age 20 years and for those who have a family history of either aldosteronism or early stroke.
• Laparoscopic adrenalectomy is the surgery of choice for most patients with unilateral adrenal disease. For patients unwilling or unable to undergo surgery or further investigations, prescribe a mineralocorticoid-receptor antagonist.
Medical therapy is the treatment of choice for bilateral adrenal disease. Spironolactone is the first-line agent, and eplerenone is an alternative agent to offer. This guideline is intended to be revised further as management evolves over the next 5 years. It is likely that by then, a rapid, inexpensive confirmatory test will be available to definitively establish the diagnosis and that third- and perhaps fourth-generation mineralocorticoid-receptor antagonists will be available for treatment. Simpler and more accurate methods of measuring plasma aldosterone concentration and direct renin concentration, which “would be a game changer for the primary care physician,” are currently being developed, Dr. Funder and his associates said (J. Clin. Endocrinol. Metab. 2016 May;101:1889-916).
In the meantime, “the main strategy is to convince primary-care physicians to screen for primary aldosteronism in all at-risk hypertensive patients,” they noted.
Copies of the full Guideline are available at [email protected] or by calling 202-971-3636.
The Endocrine Society’s updated Clinical Practice Guideline for managing primary aldosteronism is “a clarion call” for physicians to recognize the impact of this substantial public health problem and dramatically ramp up their screening and treatment efforts and was published in the Journal of Clinical Endocrinology and Metabolism.
This update differs from the previous (2008) version of the guideline in “the explicit recognition of primary aldosteronism as a major public health issue and not merely a matter of case detection, diagnosis, and treatment of individual patients,” wrote John W. Funder, MD, and his associates on the task force that compiled the guideline.
Many physicians in current practice were taught that the disorder “is a rare and benign cause of hypertension, [and] thus merely a footnote to the management of hypertension as a whole. Cardiologists usually write guidelines for hypertension with some input from nephrologists and clinical pharmacologists [but] little or none from endocrinologists,” they noted.
As a result, most patients with hypertension and occult aldosteronism are never screened for the disorder and receive suboptimal care. Primary care providers must be “made keenly aware” that the proportion of people with hypertension who have aldosteronism is much higher than previously thought (at roughly 10%), that another 20% of hypertensive people have “inappropriate aldosterone secretion,” and that both groups respond remarkably well to medical therapy, particularly to mineralocorticoid-receptor antagonists. This is critical because hypertensive patients with aldosteronism are at much greater risk for cardiovascular morbidity and mortality than their age-, sex-, and BP-matched counterparts who don’t have aldosteronism, said Dr. Funder of the Hudson Institute of Medical Research, Clayton (VIC), Australia, and his associates.
In addition to recommendations regarding screening and treatment and summaries of the evidence on which those recommendations are based, the new guideline offers a remarks section for each recommendation, which includes technical suggestions to help clinicians implement them in real-world practice.
Among the Guideline’s recommendations:
• Screen for primary aldosteronism all patients who have sustained BP above 150/100 mm Hg, hypertension resistant to three conventional antihypertensive drugs, hypertension that requires four or more drugs to control it, hypertension plus hypokalemia, hypertension plus adrenal incidentaloma, hypertension plus sleep apnea, hypertension plus a family history of early-onset hypertension or stroke at a young age, and hypertension plus a first-degree relative with primary aldosteronism.
Use the plasma aldosterone/renin ratio for this screening.
• Do one or more confirmatory tests to definitively confirm the diagnosis before proceeding to subtype classification. The exception to this recommendation is patients who develop spontaneous hypokalemia.
• Do adrenal CT as the initial step in subtype classification, to exclude large masses that may signal adrenocortical carcinoma and to help interventional radiologists and surgeons make anatomic assessments.
Before surgery, an experienced radiologist should determine whether adrenal disease is unilateral or bilateral using adrenal venous sampling.
Order genetic testing for patients with disease onset before age 20 years and for those who have a family history of either aldosteronism or early stroke.
• Laparoscopic adrenalectomy is the surgery of choice for most patients with unilateral adrenal disease. For patients unwilling or unable to undergo surgery or further investigations, prescribe a mineralocorticoid-receptor antagonist.
Medical therapy is the treatment of choice for bilateral adrenal disease. Spironolactone is the first-line agent, and eplerenone is an alternative agent to offer. This guideline is intended to be revised further as management evolves over the next 5 years. It is likely that by then, a rapid, inexpensive confirmatory test will be available to definitively establish the diagnosis and that third- and perhaps fourth-generation mineralocorticoid-receptor antagonists will be available for treatment. Simpler and more accurate methods of measuring plasma aldosterone concentration and direct renin concentration, which “would be a game changer for the primary care physician,” are currently being developed, Dr. Funder and his associates said (J. Clin. Endocrinol. Metab. 2016 May;101:1889-916).
In the meantime, “the main strategy is to convince primary-care physicians to screen for primary aldosteronism in all at-risk hypertensive patients,” they noted.
Copies of the full Guideline are available at [email protected] or by calling 202-971-3636.
The Endocrine Society’s updated Clinical Practice Guideline for managing primary aldosteronism is “a clarion call” for physicians to recognize the impact of this substantial public health problem and dramatically ramp up their screening and treatment efforts and was published in the Journal of Clinical Endocrinology and Metabolism.
This update differs from the previous (2008) version of the guideline in “the explicit recognition of primary aldosteronism as a major public health issue and not merely a matter of case detection, diagnosis, and treatment of individual patients,” wrote John W. Funder, MD, and his associates on the task force that compiled the guideline.
Many physicians in current practice were taught that the disorder “is a rare and benign cause of hypertension, [and] thus merely a footnote to the management of hypertension as a whole. Cardiologists usually write guidelines for hypertension with some input from nephrologists and clinical pharmacologists [but] little or none from endocrinologists,” they noted.
As a result, most patients with hypertension and occult aldosteronism are never screened for the disorder and receive suboptimal care. Primary care providers must be “made keenly aware” that the proportion of people with hypertension who have aldosteronism is much higher than previously thought (at roughly 10%), that another 20% of hypertensive people have “inappropriate aldosterone secretion,” and that both groups respond remarkably well to medical therapy, particularly to mineralocorticoid-receptor antagonists. This is critical because hypertensive patients with aldosteronism are at much greater risk for cardiovascular morbidity and mortality than their age-, sex-, and BP-matched counterparts who don’t have aldosteronism, said Dr. Funder of the Hudson Institute of Medical Research, Clayton (VIC), Australia, and his associates.
In addition to recommendations regarding screening and treatment and summaries of the evidence on which those recommendations are based, the new guideline offers a remarks section for each recommendation, which includes technical suggestions to help clinicians implement them in real-world practice.
Among the Guideline’s recommendations:
• Screen for primary aldosteronism all patients who have sustained BP above 150/100 mm Hg, hypertension resistant to three conventional antihypertensive drugs, hypertension that requires four or more drugs to control it, hypertension plus hypokalemia, hypertension plus adrenal incidentaloma, hypertension plus sleep apnea, hypertension plus a family history of early-onset hypertension or stroke at a young age, and hypertension plus a first-degree relative with primary aldosteronism.
Use the plasma aldosterone/renin ratio for this screening.
• Do one or more confirmatory tests to definitively confirm the diagnosis before proceeding to subtype classification. The exception to this recommendation is patients who develop spontaneous hypokalemia.
• Do adrenal CT as the initial step in subtype classification, to exclude large masses that may signal adrenocortical carcinoma and to help interventional radiologists and surgeons make anatomic assessments.
Before surgery, an experienced radiologist should determine whether adrenal disease is unilateral or bilateral using adrenal venous sampling.
Order genetic testing for patients with disease onset before age 20 years and for those who have a family history of either aldosteronism or early stroke.
• Laparoscopic adrenalectomy is the surgery of choice for most patients with unilateral adrenal disease. For patients unwilling or unable to undergo surgery or further investigations, prescribe a mineralocorticoid-receptor antagonist.
Medical therapy is the treatment of choice for bilateral adrenal disease. Spironolactone is the first-line agent, and eplerenone is an alternative agent to offer. This guideline is intended to be revised further as management evolves over the next 5 years. It is likely that by then, a rapid, inexpensive confirmatory test will be available to definitively establish the diagnosis and that third- and perhaps fourth-generation mineralocorticoid-receptor antagonists will be available for treatment. Simpler and more accurate methods of measuring plasma aldosterone concentration and direct renin concentration, which “would be a game changer for the primary care physician,” are currently being developed, Dr. Funder and his associates said (J. Clin. Endocrinol. Metab. 2016 May;101:1889-916).
In the meantime, “the main strategy is to convince primary-care physicians to screen for primary aldosteronism in all at-risk hypertensive patients,” they noted.
Copies of the full Guideline are available at [email protected] or by calling 202-971-3636.
FROM THE JOURNAL OF CLINICAL ENDOCRINOLOGY AND METABOLISM
Key clinical point: The Endocrine Society’s updated Clinical Practice Guideline for managing primary aldosteronism is “a clarion call” for physicians to ramp up screening for and treatment of this major public health problem.
Major finding: Primary care physicians must be convinced to screen all at-risk hypertensive patients for primary aldosteronism.
Data source: A comprehensive review of the literature and detailed update of a Clinical Practice Guideline for managing primary aldosteronism.
Disclosures: The Endocrine Society provided all the support for this Guideline. Dr. Funder’s and his associates’ conflicts of interest are available from the Endocrine Society.
Genital growths
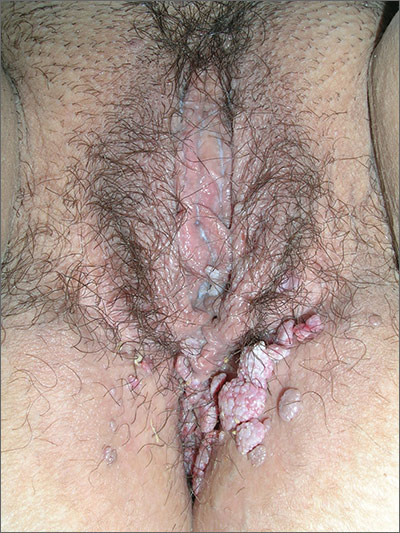
The FP diagnosed this patient with probable condyloma acuminata but was also concerned about the possibility of other sexually transmitted infections (STIs). He ordered a rapid plasma reagin (RPR) and a human immunodeficiency virus (HIV) test.
A Pap smear was also performed using liquid-based technology to include screening for gonorrhea and chlamydia. While condyloma lata (secondary syphilis) was considered in the differential diagnosis, the verrucous appearance of the lesions pointed to condyloma acuminata from human papillomavirus (HPV).
The RPR came back positive for syphilis with a titer of 1:32. The confirmatory serologic test for syphilis was also positive. The remaining STI screening tests were negative, including the HIV test. The patient remembered having a nonhealing sore on her labia about 2 months ago that healed on its own, making this most likely a case of early latent syphilis.
For the syphilis, the FP treated the patient with a single intramuscular injection in the buttocks of 2.4 million units of benzathine penicillin G. The patient was told to follow up in 6 months for a repeat RPR.
The physician offered treatment for the condyloma acuminata on the first visit. The patient chose to have cryotherapy, which was performed with liquid nitrogen and a cryo-gun. Only one freeze thaw cycle was performed, as the patient found it too painful to have a second cycle. An appointment was made to repeat the cryotherapy in 4 weeks. While the patient was interested in using topical imiquimod, she did not have health insurance and could not afford it.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R. Genital warts. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:759-765.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP diagnosed this patient with probable condyloma acuminata but was also concerned about the possibility of other sexually transmitted infections (STIs). He ordered a rapid plasma reagin (RPR) and a human immunodeficiency virus (HIV) test.
A Pap smear was also performed using liquid-based technology to include screening for gonorrhea and chlamydia. While condyloma lata (secondary syphilis) was considered in the differential diagnosis, the verrucous appearance of the lesions pointed to condyloma acuminata from human papillomavirus (HPV).
The RPR came back positive for syphilis with a titer of 1:32. The confirmatory serologic test for syphilis was also positive. The remaining STI screening tests were negative, including the HIV test. The patient remembered having a nonhealing sore on her labia about 2 months ago that healed on its own, making this most likely a case of early latent syphilis.
For the syphilis, the FP treated the patient with a single intramuscular injection in the buttocks of 2.4 million units of benzathine penicillin G. The patient was told to follow up in 6 months for a repeat RPR.
The physician offered treatment for the condyloma acuminata on the first visit. The patient chose to have cryotherapy, which was performed with liquid nitrogen and a cryo-gun. Only one freeze thaw cycle was performed, as the patient found it too painful to have a second cycle. An appointment was made to repeat the cryotherapy in 4 weeks. While the patient was interested in using topical imiquimod, she did not have health insurance and could not afford it.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R. Genital warts. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:759-765.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP diagnosed this patient with probable condyloma acuminata but was also concerned about the possibility of other sexually transmitted infections (STIs). He ordered a rapid plasma reagin (RPR) and a human immunodeficiency virus (HIV) test.
A Pap smear was also performed using liquid-based technology to include screening for gonorrhea and chlamydia. While condyloma lata (secondary syphilis) was considered in the differential diagnosis, the verrucous appearance of the lesions pointed to condyloma acuminata from human papillomavirus (HPV).
The RPR came back positive for syphilis with a titer of 1:32. The confirmatory serologic test for syphilis was also positive. The remaining STI screening tests were negative, including the HIV test. The patient remembered having a nonhealing sore on her labia about 2 months ago that healed on its own, making this most likely a case of early latent syphilis.
For the syphilis, the FP treated the patient with a single intramuscular injection in the buttocks of 2.4 million units of benzathine penicillin G. The patient was told to follow up in 6 months for a repeat RPR.
The physician offered treatment for the condyloma acuminata on the first visit. The patient chose to have cryotherapy, which was performed with liquid nitrogen and a cryo-gun. Only one freeze thaw cycle was performed, as the patient found it too painful to have a second cycle. An appointment was made to repeat the cryotherapy in 4 weeks. While the patient was interested in using topical imiquimod, she did not have health insurance and could not afford it.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R. Genital warts. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:759-765.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
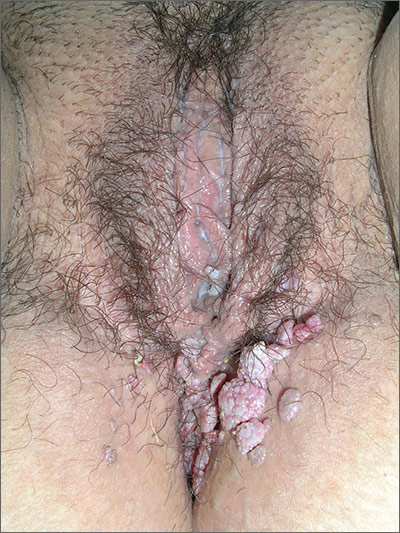
Growths in abdominal folds
While the lesions appeared verrucous and cauliflower-like, this seemed an unusual place for condyloma. So the FP performed a shave biopsy out of concern that this could be a benign tumor. The results came back as human papillomavirus (HPV)-related condyloma.
In addition to the growths that were seen under the pannus, there was also intertrigo in that area. Intertrigo is a nonspecific term for a rash in an intertriginous area. Erythema seen between skin folds is the tip-off to intertrigo. The most common causes of intertrigo are fungal infections and irritation that come from sweating and the rubbing together of skin. Another cause of an erythematous rash in an intertriginous area is inverse psoriasis.
The FP performed a potassium hydroxide (KOH) preparation by scraping the erythematous area with a microscope slide and looking at the specimen under the microscope. The KOH was negative and the diagnosis was determined to be nonspecific intertrigo related to obesity. The FP told the patient to try to keep the area dry and to try some over-the-counter 1% hydrocortisone cream for the inflammation and irritation.
Pannus condylomas are uncommon—but not rare—in this age of increasing obesity. While they are secondary to HPV, they are not necessarily sexually transmitted HPV types. Based on the patient’s sexual history, the FP did not recommend screening for sexually transmitted infections.
On the initial visit, the FP counseled the patient about the importance of weight loss for good health and hygiene. The patient chose cryotherapy for treatment of the growths. While the patient held the pannus up, the FP sprayed the condyloma with liquid nitrogen using a cryo-gun. The patient chose to have a second freeze-thaw cycle and was scheduled for a follow-up visit in 3 to 4 weeks.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R. Genital warts. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:759-765.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
While the lesions appeared verrucous and cauliflower-like, this seemed an unusual place for condyloma. So the FP performed a shave biopsy out of concern that this could be a benign tumor. The results came back as human papillomavirus (HPV)-related condyloma.
In addition to the growths that were seen under the pannus, there was also intertrigo in that area. Intertrigo is a nonspecific term for a rash in an intertriginous area. Erythema seen between skin folds is the tip-off to intertrigo. The most common causes of intertrigo are fungal infections and irritation that come from sweating and the rubbing together of skin. Another cause of an erythematous rash in an intertriginous area is inverse psoriasis.
The FP performed a potassium hydroxide (KOH) preparation by scraping the erythematous area with a microscope slide and looking at the specimen under the microscope. The KOH was negative and the diagnosis was determined to be nonspecific intertrigo related to obesity. The FP told the patient to try to keep the area dry and to try some over-the-counter 1% hydrocortisone cream for the inflammation and irritation.
Pannus condylomas are uncommon—but not rare—in this age of increasing obesity. While they are secondary to HPV, they are not necessarily sexually transmitted HPV types. Based on the patient’s sexual history, the FP did not recommend screening for sexually transmitted infections.
On the initial visit, the FP counseled the patient about the importance of weight loss for good health and hygiene. The patient chose cryotherapy for treatment of the growths. While the patient held the pannus up, the FP sprayed the condyloma with liquid nitrogen using a cryo-gun. The patient chose to have a second freeze-thaw cycle and was scheduled for a follow-up visit in 3 to 4 weeks.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R. Genital warts. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:759-765.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
While the lesions appeared verrucous and cauliflower-like, this seemed an unusual place for condyloma. So the FP performed a shave biopsy out of concern that this could be a benign tumor. The results came back as human papillomavirus (HPV)-related condyloma.
In addition to the growths that were seen under the pannus, there was also intertrigo in that area. Intertrigo is a nonspecific term for a rash in an intertriginous area. Erythema seen between skin folds is the tip-off to intertrigo. The most common causes of intertrigo are fungal infections and irritation that come from sweating and the rubbing together of skin. Another cause of an erythematous rash in an intertriginous area is inverse psoriasis.
The FP performed a potassium hydroxide (KOH) preparation by scraping the erythematous area with a microscope slide and looking at the specimen under the microscope. The KOH was negative and the diagnosis was determined to be nonspecific intertrigo related to obesity. The FP told the patient to try to keep the area dry and to try some over-the-counter 1% hydrocortisone cream for the inflammation and irritation.
Pannus condylomas are uncommon—but not rare—in this age of increasing obesity. While they are secondary to HPV, they are not necessarily sexually transmitted HPV types. Based on the patient’s sexual history, the FP did not recommend screening for sexually transmitted infections.
On the initial visit, the FP counseled the patient about the importance of weight loss for good health and hygiene. The patient chose cryotherapy for treatment of the growths. While the patient held the pannus up, the FP sprayed the condyloma with liquid nitrogen using a cryo-gun. The patient chose to have a second freeze-thaw cycle and was scheduled for a follow-up visit in 3 to 4 weeks.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R. Genital warts. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:759-765.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Lesions on lower abdomen
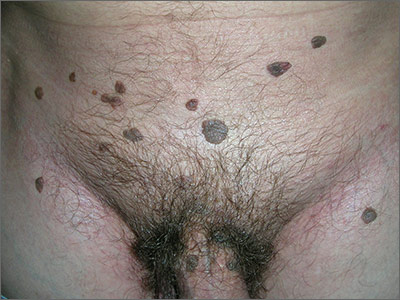
At first, the FP considered the diagnosis of seborrheic keratosis, as the lesions were verrucous and pigmented. But after asking to see the patient’s genital area, the FP made the diagnosis of condyloma acuminata.
This is a good example of how seeing the whole picture beyond what the patient shows you can lead to a more accurate diagnosis. Verrucous lesions on the penis are usually condyloma acuminata, but it is important to know that condyloma can spread up the abdomen and onto the upper thighs, as was seen in this case. The lesions can also be hyperpigmented in people of color. If any doubts remain, a shave biopsy of one of the abdominal lesions would prove the diagnosis of condyloma by demonstrating human papillomavirus (HPV) changes histologically.
In this case, the FP discussed the risk of other sexually transmitted infections with the patient. Syphilis and human immunodeficiency virus tests were ordered, and both turned out to be negative.
The patient chose cryotherapy as a treatment option and the lesions were frozen with liquid nitrogen using a standard cryo-gun and a 1 mm halo, creating an appropriate freeze ball for each lesion. The patient tolerated the procedure well and was willing to have a second freeze thaw cycle for a more rapid treatment response. The FP suggested a follow-up visit in 3 to 4 weeks for a second round of cryotherapy. The patient indicated that he would be willing to try topical imiquimod after the next visit if the cryotherapy didn’t fully work.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R. Genital warts. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:759-765.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
At first, the FP considered the diagnosis of seborrheic keratosis, as the lesions were verrucous and pigmented. But after asking to see the patient’s genital area, the FP made the diagnosis of condyloma acuminata.
This is a good example of how seeing the whole picture beyond what the patient shows you can lead to a more accurate diagnosis. Verrucous lesions on the penis are usually condyloma acuminata, but it is important to know that condyloma can spread up the abdomen and onto the upper thighs, as was seen in this case. The lesions can also be hyperpigmented in people of color. If any doubts remain, a shave biopsy of one of the abdominal lesions would prove the diagnosis of condyloma by demonstrating human papillomavirus (HPV) changes histologically.
In this case, the FP discussed the risk of other sexually transmitted infections with the patient. Syphilis and human immunodeficiency virus tests were ordered, and both turned out to be negative.
The patient chose cryotherapy as a treatment option and the lesions were frozen with liquid nitrogen using a standard cryo-gun and a 1 mm halo, creating an appropriate freeze ball for each lesion. The patient tolerated the procedure well and was willing to have a second freeze thaw cycle for a more rapid treatment response. The FP suggested a follow-up visit in 3 to 4 weeks for a second round of cryotherapy. The patient indicated that he would be willing to try topical imiquimod after the next visit if the cryotherapy didn’t fully work.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R. Genital warts. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:759-765.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
At first, the FP considered the diagnosis of seborrheic keratosis, as the lesions were verrucous and pigmented. But after asking to see the patient’s genital area, the FP made the diagnosis of condyloma acuminata.
This is a good example of how seeing the whole picture beyond what the patient shows you can lead to a more accurate diagnosis. Verrucous lesions on the penis are usually condyloma acuminata, but it is important to know that condyloma can spread up the abdomen and onto the upper thighs, as was seen in this case. The lesions can also be hyperpigmented in people of color. If any doubts remain, a shave biopsy of one of the abdominal lesions would prove the diagnosis of condyloma by demonstrating human papillomavirus (HPV) changes histologically.
In this case, the FP discussed the risk of other sexually transmitted infections with the patient. Syphilis and human immunodeficiency virus tests were ordered, and both turned out to be negative.
The patient chose cryotherapy as a treatment option and the lesions were frozen with liquid nitrogen using a standard cryo-gun and a 1 mm halo, creating an appropriate freeze ball for each lesion. The patient tolerated the procedure well and was willing to have a second freeze thaw cycle for a more rapid treatment response. The FP suggested a follow-up visit in 3 to 4 weeks for a second round of cryotherapy. The patient indicated that he would be willing to try topical imiquimod after the next visit if the cryotherapy didn’t fully work.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Mayeaux EJ, Usatine R. Genital warts. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013:759-765.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Study identifies distinct OA fatigue trajectories
A new study has identified three distinct trajectories of fatigue levels in patients with early symptomatic osteoarthritis (OA) in the knee and hip, report Jadran Botterman, MSc, and coauthors from the department of psychology, health, and technology at the University of Twente (the Netherlands).
Six years of data were collected from the CHECK (Cohort Hip and Cohort Knee) participants and then separated into distinct trajectories using growth mixture modeling. Three distinct fatigue trajectories were found: low fatigue, low to high fatigue, and high fatigue.
The authors found a significant association between trajectory and patient characteristics. Women, patients with comorbid disease, and patients using medications were more likely to have a high fatigue trajectory, Dr. Botterman and his colleagues reported.
“Identification of these trajectories with differing patient characteristics may warrant tailored psychosocial interventions for patients with elevated levels of fatigue,” the authors concluded.
Read the full article in the Journal of Rheumatology.
A new study has identified three distinct trajectories of fatigue levels in patients with early symptomatic osteoarthritis (OA) in the knee and hip, report Jadran Botterman, MSc, and coauthors from the department of psychology, health, and technology at the University of Twente (the Netherlands).
Six years of data were collected from the CHECK (Cohort Hip and Cohort Knee) participants and then separated into distinct trajectories using growth mixture modeling. Three distinct fatigue trajectories were found: low fatigue, low to high fatigue, and high fatigue.
The authors found a significant association between trajectory and patient characteristics. Women, patients with comorbid disease, and patients using medications were more likely to have a high fatigue trajectory, Dr. Botterman and his colleagues reported.
“Identification of these trajectories with differing patient characteristics may warrant tailored psychosocial interventions for patients with elevated levels of fatigue,” the authors concluded.
Read the full article in the Journal of Rheumatology.
A new study has identified three distinct trajectories of fatigue levels in patients with early symptomatic osteoarthritis (OA) in the knee and hip, report Jadran Botterman, MSc, and coauthors from the department of psychology, health, and technology at the University of Twente (the Netherlands).
Six years of data were collected from the CHECK (Cohort Hip and Cohort Knee) participants and then separated into distinct trajectories using growth mixture modeling. Three distinct fatigue trajectories were found: low fatigue, low to high fatigue, and high fatigue.
The authors found a significant association between trajectory and patient characteristics. Women, patients with comorbid disease, and patients using medications were more likely to have a high fatigue trajectory, Dr. Botterman and his colleagues reported.
“Identification of these trajectories with differing patient characteristics may warrant tailored psychosocial interventions for patients with elevated levels of fatigue,” the authors concluded.
Read the full article in the Journal of Rheumatology.
FROM THE JOURNAL OF RHEUMATOLOGY
Three-drug regimen boosts progression-free survival in patients with relapsed CLL and adverse prognostic features
Progression-free survival was improved for poor prognosis patients with relapsed chronic lymphocytic leukemia when idelalisib was added to bendamustine and rituximab, based on the results of a randomized, double-blind, placebo-controlled phase III study.
The findings support the three-drug approach as an important treatment option for patients with relapsed CLL and adverse prognostic features, but the regimen also was associated with more grade 3 or greater adverse events, primarily neutropenia and opportunistic infections, that led to more study drug discontinuation, Jacqueline Claudia Barrientos, M.D., and her colleagues reported at the annual meeting of the American Society of Clinical Oncology.
The overall response rates were 68% in the three-drug group and 45% in the two-drug plus placebo group.
For the study, 416 patients were stratified based on whether they had 17p deletions or TP53 mutations, their IGHV mutation status, and whether they had refractory or relapsed disease. The 207 patients who received idelalisib, bendamustine, and rituximab had consistently better progression-free survival than the 209 patients who received placebo, bendamustine, and rituximab. The median progression-free survival was 23 months in the group that received idelalisib and 11 months in the group that received placebo, with an overall hazard ratio of 0.33 at a median follow-up of 12 months.
For those with 17p deletions or TP53 mutations, the median progression-free survival was 11 months with the three-drug regimen and 8 months for the two-drug plus placebo regimen. For those with neither of these abnormalities, progression-free survival was more than 24 months for patients given the three active drugs and 11 months for patients given two drugs plus placebo.
For those with the 11q deletion, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and 9 months with placebo, bendamustine, and rituximab, said Dr. Barrientos of Hofstra University, Hempstead, N.Y.
For those with IGHV mutations, the median progression-free survival has not been reached in the idelalisib, bendamustine, and rituximab group and was 11 months in the placebo, bendamustine, and rituximab group.
Among patients with tumor burdens exceeding 5 cm, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and about 10 months with placebo, bendamustine, and rituximab.
Grade 3 or greater adverse events affected 97% of patients given the three active drugs and 76% of patients given bendamustine and rituximab plus placebo. Adverse events resulted in drug dose reductions in 11% of those given the three active drugs and in 6% of those given bendamustine and rituximab plus placebo. The study drug was discontinued in 26% of those in the idelalisib, bendamustine, and rituximab group and in 13% of the placebo, bendamustine, and rituximab group. Death occurred in 10% of the study patients given idelalisib, bendamustine, and rituximab and in 7% of the patients given placebo, bendamustine, and rituximab.
To be eligible for the study (NCT01569295), patients needed to have previously treated recurrent CLL, have measurable lymphadenopathy, require therapy for CLL, and have experienced CLL progression for less than 36 months since the completion of their last prior therapy.
The treatment regimen consisted of 6 cycles every 28 days of bendamustine (70 mg/m2 on day 1 and day 2 of each cycle), rituximab (375 mg/m2 in cycle 1 and 500 mg/m2 in cycles 2-6), and idelalisib (150 mg twice daily) or placebo. Idelalisib or placebo was continued until an independent review committee confirmed disease progression, death, intolerable toxicity, or withdrawal of consent.
The study was sponsored by Gilead Sciences, the maker of idelalisib (Zydelig). Dr. Barrientos receives research funding from Gilead Sciences and Abbvie, and serves as a consultant or adviser to Celgene, Genentech, and Pharmacyclics.
On Twitter @maryjodales
Progression-free survival was improved for poor prognosis patients with relapsed chronic lymphocytic leukemia when idelalisib was added to bendamustine and rituximab, based on the results of a randomized, double-blind, placebo-controlled phase III study.
The findings support the three-drug approach as an important treatment option for patients with relapsed CLL and adverse prognostic features, but the regimen also was associated with more grade 3 or greater adverse events, primarily neutropenia and opportunistic infections, that led to more study drug discontinuation, Jacqueline Claudia Barrientos, M.D., and her colleagues reported at the annual meeting of the American Society of Clinical Oncology.
The overall response rates were 68% in the three-drug group and 45% in the two-drug plus placebo group.
For the study, 416 patients were stratified based on whether they had 17p deletions or TP53 mutations, their IGHV mutation status, and whether they had refractory or relapsed disease. The 207 patients who received idelalisib, bendamustine, and rituximab had consistently better progression-free survival than the 209 patients who received placebo, bendamustine, and rituximab. The median progression-free survival was 23 months in the group that received idelalisib and 11 months in the group that received placebo, with an overall hazard ratio of 0.33 at a median follow-up of 12 months.
For those with 17p deletions or TP53 mutations, the median progression-free survival was 11 months with the three-drug regimen and 8 months for the two-drug plus placebo regimen. For those with neither of these abnormalities, progression-free survival was more than 24 months for patients given the three active drugs and 11 months for patients given two drugs plus placebo.
For those with the 11q deletion, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and 9 months with placebo, bendamustine, and rituximab, said Dr. Barrientos of Hofstra University, Hempstead, N.Y.
For those with IGHV mutations, the median progression-free survival has not been reached in the idelalisib, bendamustine, and rituximab group and was 11 months in the placebo, bendamustine, and rituximab group.
Among patients with tumor burdens exceeding 5 cm, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and about 10 months with placebo, bendamustine, and rituximab.
Grade 3 or greater adverse events affected 97% of patients given the three active drugs and 76% of patients given bendamustine and rituximab plus placebo. Adverse events resulted in drug dose reductions in 11% of those given the three active drugs and in 6% of those given bendamustine and rituximab plus placebo. The study drug was discontinued in 26% of those in the idelalisib, bendamustine, and rituximab group and in 13% of the placebo, bendamustine, and rituximab group. Death occurred in 10% of the study patients given idelalisib, bendamustine, and rituximab and in 7% of the patients given placebo, bendamustine, and rituximab.
To be eligible for the study (NCT01569295), patients needed to have previously treated recurrent CLL, have measurable lymphadenopathy, require therapy for CLL, and have experienced CLL progression for less than 36 months since the completion of their last prior therapy.
The treatment regimen consisted of 6 cycles every 28 days of bendamustine (70 mg/m2 on day 1 and day 2 of each cycle), rituximab (375 mg/m2 in cycle 1 and 500 mg/m2 in cycles 2-6), and idelalisib (150 mg twice daily) or placebo. Idelalisib or placebo was continued until an independent review committee confirmed disease progression, death, intolerable toxicity, or withdrawal of consent.
The study was sponsored by Gilead Sciences, the maker of idelalisib (Zydelig). Dr. Barrientos receives research funding from Gilead Sciences and Abbvie, and serves as a consultant or adviser to Celgene, Genentech, and Pharmacyclics.
On Twitter @maryjodales
Progression-free survival was improved for poor prognosis patients with relapsed chronic lymphocytic leukemia when idelalisib was added to bendamustine and rituximab, based on the results of a randomized, double-blind, placebo-controlled phase III study.
The findings support the three-drug approach as an important treatment option for patients with relapsed CLL and adverse prognostic features, but the regimen also was associated with more grade 3 or greater adverse events, primarily neutropenia and opportunistic infections, that led to more study drug discontinuation, Jacqueline Claudia Barrientos, M.D., and her colleagues reported at the annual meeting of the American Society of Clinical Oncology.
The overall response rates were 68% in the three-drug group and 45% in the two-drug plus placebo group.
For the study, 416 patients were stratified based on whether they had 17p deletions or TP53 mutations, their IGHV mutation status, and whether they had refractory or relapsed disease. The 207 patients who received idelalisib, bendamustine, and rituximab had consistently better progression-free survival than the 209 patients who received placebo, bendamustine, and rituximab. The median progression-free survival was 23 months in the group that received idelalisib and 11 months in the group that received placebo, with an overall hazard ratio of 0.33 at a median follow-up of 12 months.
For those with 17p deletions or TP53 mutations, the median progression-free survival was 11 months with the three-drug regimen and 8 months for the two-drug plus placebo regimen. For those with neither of these abnormalities, progression-free survival was more than 24 months for patients given the three active drugs and 11 months for patients given two drugs plus placebo.
For those with the 11q deletion, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and 9 months with placebo, bendamustine, and rituximab, said Dr. Barrientos of Hofstra University, Hempstead, N.Y.
For those with IGHV mutations, the median progression-free survival has not been reached in the idelalisib, bendamustine, and rituximab group and was 11 months in the placebo, bendamustine, and rituximab group.
Among patients with tumor burdens exceeding 5 cm, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and about 10 months with placebo, bendamustine, and rituximab.
Grade 3 or greater adverse events affected 97% of patients given the three active drugs and 76% of patients given bendamustine and rituximab plus placebo. Adverse events resulted in drug dose reductions in 11% of those given the three active drugs and in 6% of those given bendamustine and rituximab plus placebo. The study drug was discontinued in 26% of those in the idelalisib, bendamustine, and rituximab group and in 13% of the placebo, bendamustine, and rituximab group. Death occurred in 10% of the study patients given idelalisib, bendamustine, and rituximab and in 7% of the patients given placebo, bendamustine, and rituximab.
To be eligible for the study (NCT01569295), patients needed to have previously treated recurrent CLL, have measurable lymphadenopathy, require therapy for CLL, and have experienced CLL progression for less than 36 months since the completion of their last prior therapy.
The treatment regimen consisted of 6 cycles every 28 days of bendamustine (70 mg/m2 on day 1 and day 2 of each cycle), rituximab (375 mg/m2 in cycle 1 and 500 mg/m2 in cycles 2-6), and idelalisib (150 mg twice daily) or placebo. Idelalisib or placebo was continued until an independent review committee confirmed disease progression, death, intolerable toxicity, or withdrawal of consent.
The study was sponsored by Gilead Sciences, the maker of idelalisib (Zydelig). Dr. Barrientos receives research funding from Gilead Sciences and Abbvie, and serves as a consultant or adviser to Celgene, Genentech, and Pharmacyclics.
On Twitter @maryjodales
FROM 2016 ASCO ANNUAL MEETING
Key clinical point: Progression-free survival was improved for poor prognosis patients with relapsed chronic lymphocytic leukemia when idelalisib was added to bendamustine, and rituximab.
Major finding: The median progression-free survival was 23 months in the group that received idelalisib and 11 months in the group that received placebo, with an overall hazard ratio of 0.33 at a median follow-up of 12 months.
Data source: A randomized, double-blind, placebo-controlled phase III study of 416 patients stratified on the basis of 17p deletions or TP53 mutations, IGHV mutation status, and whether they had refractory or relapsed disease.
Disclosures: The study was sponsored by Gilead Sciences, the maker of idelalisib (Zydelig). Dr. Barrientos receives research funding from Gilead Sciences and Abbvie, and serves as a consultant or adviser to Celgene, Genentech, and Pharmacyclics.
Triple Therapy Helped Type 1 Patients Improve Glycemia
NEW ORLEANS – The addition of dapagliflozin to insulin and liraglutide in patients with type 1 diabetes resulted in a significant improvement in glycemia, results from a single-center randomized trial showed.
“Because liraglutide suppresses glucagon and lowers free fatty acids while SGLT-2 inhibitors increase glucagon and risk of diabetic ketoacidosis, it’s possible that the combination of the two agents may neutralize the diabetic ketoacidosis risk,” Paresh Dandona, MD, said at the annual scientific sessions of the American Diabetes Association.

In a recent study Dr. Dandona, chief of endocrinology at the University of Buffalo (N.Y.), and his associates showed that the addition of liraglutide to insulin significantly improved glycemic control in patients with type 1 diabetes (Diabetes Care 2016 Jun; 39[6]:1027-35). The purpose of the current study was to investigate whether the addition of dapagliflozin, a sodium-glucose cotransporter–2 inhibitor, to the combination of insulin and liraglutide would further improve glycemic control.
A total of 30 type 1 diabetes patients on insulin and liraglutide therapy for a minimum of 6 months were randomized to receive either dapagliflozin 10 mg or placebo daily for 12 weeks. Dapagliflozin was initiated at 5 mg daily for 1 week and increased to 10 mg daily thereafter. All patients had type 1 diabetes for at least 1 year, were on insulin therapy, had no detectable C-peptide in plasma, and were on 1.8 mg of liraglutide for 7 months. These patients were evaluated every week for the first and last 2 weeks and then every 2 weeks for 12 weeks. Blood and 24-hour urine samples were collected before and after intervention.
At baseline, the mean age of patients was 54 years and both groups were similar in terms of mean hemoglobin A1c, glucose levels, and insulin dosages. After 12 weeks, the mean HbA1c did not change in the placebo group, but it dropped significantly in the triple-therapy group, by .66% (P less than .01). In addition, the average weekly glucose concentration fell in the triple-therapy group by 13 mg/dL (P = .35) and there was a reduction in standard deviations of glycemic excursions. The triple-therapy group also experienced a small reduction in the total insulin dose, compared with the placebo group (–3.5 vs. –.4 units, respectively; P = .29)
Dr. Dandona went on to note that the episodes of hypoglycemia did not increase after triple therapy but body weight fell by 1.9 kg after 12 weeks, while there was no real change in the placebo arm. Two patients had to withdraw from the study because they developed diabetic ketoacidosis within a day after increasing the dose of dapagliflozin to 10 mg. “One of these patients had euglycemic DKA with blood glucose concentrations of less than 160 mg/dL, while the other had marked hyperglycemia with unchanged insulin dose at 26 units,” Dr. Dandona said. “This patient had experienced a marked reduction in insulin dose during the time she was on liraglutide prior to starting on dapagliflozin.”
Dr. Dandona disclosed that he has received research support from AstraZeneca and Novo Nordisk.
NEW ORLEANS – The addition of dapagliflozin to insulin and liraglutide in patients with type 1 diabetes resulted in a significant improvement in glycemia, results from a single-center randomized trial showed.
“Because liraglutide suppresses glucagon and lowers free fatty acids while SGLT-2 inhibitors increase glucagon and risk of diabetic ketoacidosis, it’s possible that the combination of the two agents may neutralize the diabetic ketoacidosis risk,” Paresh Dandona, MD, said at the annual scientific sessions of the American Diabetes Association.
In a recent study Dr. Dandona, chief of endocrinology at the University of Buffalo (N.Y.), and his associates showed that the addition of liraglutide to insulin significantly improved glycemic control in patients with type 1 diabetes (Diabetes Care 2016 Jun; 39[6]:1027-35). The purpose of the current study was to investigate whether the addition of dapagliflozin, a sodium-glucose cotransporter–2 inhibitor, to the combination of insulin and liraglutide would further improve glycemic control.
A total of 30 type 1 diabetes patients on insulin and liraglutide therapy for a minimum of 6 months were randomized to receive either dapagliflozin 10 mg or placebo daily for 12 weeks. Dapagliflozin was initiated at 5 mg daily for 1 week and increased to 10 mg daily thereafter. All patients had type 1 diabetes for at least 1 year, were on insulin therapy, had no detectable C-peptide in plasma, and were on 1.8 mg of liraglutide for 7 months. These patients were evaluated every week for the first and last 2 weeks and then every 2 weeks for 12 weeks. Blood and 24-hour urine samples were collected before and after intervention.
At baseline, the mean age of patients was 54 years and both groups were similar in terms of mean hemoglobin A1c, glucose levels, and insulin dosages. After 12 weeks, the mean HbA1c did not change in the placebo group, but it dropped significantly in the triple-therapy group, by .66% (P less than .01). In addition, the average weekly glucose concentration fell in the triple-therapy group by 13 mg/dL (P = .35) and there was a reduction in standard deviations of glycemic excursions. The triple-therapy group also experienced a small reduction in the total insulin dose, compared with the placebo group (–3.5 vs. –.4 units, respectively; P = .29)
Dr. Dandona went on to note that the episodes of hypoglycemia did not increase after triple therapy but body weight fell by 1.9 kg after 12 weeks, while there was no real change in the placebo arm. Two patients had to withdraw from the study because they developed diabetic ketoacidosis within a day after increasing the dose of dapagliflozin to 10 mg. “One of these patients had euglycemic DKA with blood glucose concentrations of less than 160 mg/dL, while the other had marked hyperglycemia with unchanged insulin dose at 26 units,” Dr. Dandona said. “This patient had experienced a marked reduction in insulin dose during the time she was on liraglutide prior to starting on dapagliflozin.”
Dr. Dandona disclosed that he has received research support from AstraZeneca and Novo Nordisk.
NEW ORLEANS – The addition of dapagliflozin to insulin and liraglutide in patients with type 1 diabetes resulted in a significant improvement in glycemia, results from a single-center randomized trial showed.
“Because liraglutide suppresses glucagon and lowers free fatty acids while SGLT-2 inhibitors increase glucagon and risk of diabetic ketoacidosis, it’s possible that the combination of the two agents may neutralize the diabetic ketoacidosis risk,” Paresh Dandona, MD, said at the annual scientific sessions of the American Diabetes Association.
In a recent study Dr. Dandona, chief of endocrinology at the University of Buffalo (N.Y.), and his associates showed that the addition of liraglutide to insulin significantly improved glycemic control in patients with type 1 diabetes (Diabetes Care 2016 Jun; 39[6]:1027-35). The purpose of the current study was to investigate whether the addition of dapagliflozin, a sodium-glucose cotransporter–2 inhibitor, to the combination of insulin and liraglutide would further improve glycemic control.
A total of 30 type 1 diabetes patients on insulin and liraglutide therapy for a minimum of 6 months were randomized to receive either dapagliflozin 10 mg or placebo daily for 12 weeks. Dapagliflozin was initiated at 5 mg daily for 1 week and increased to 10 mg daily thereafter. All patients had type 1 diabetes for at least 1 year, were on insulin therapy, had no detectable C-peptide in plasma, and were on 1.8 mg of liraglutide for 7 months. These patients were evaluated every week for the first and last 2 weeks and then every 2 weeks for 12 weeks. Blood and 24-hour urine samples were collected before and after intervention.
At baseline, the mean age of patients was 54 years and both groups were similar in terms of mean hemoglobin A1c, glucose levels, and insulin dosages. After 12 weeks, the mean HbA1c did not change in the placebo group, but it dropped significantly in the triple-therapy group, by .66% (P less than .01). In addition, the average weekly glucose concentration fell in the triple-therapy group by 13 mg/dL (P = .35) and there was a reduction in standard deviations of glycemic excursions. The triple-therapy group also experienced a small reduction in the total insulin dose, compared with the placebo group (–3.5 vs. –.4 units, respectively; P = .29)
Dr. Dandona went on to note that the episodes of hypoglycemia did not increase after triple therapy but body weight fell by 1.9 kg after 12 weeks, while there was no real change in the placebo arm. Two patients had to withdraw from the study because they developed diabetic ketoacidosis within a day after increasing the dose of dapagliflozin to 10 mg. “One of these patients had euglycemic DKA with blood glucose concentrations of less than 160 mg/dL, while the other had marked hyperglycemia with unchanged insulin dose at 26 units,” Dr. Dandona said. “This patient had experienced a marked reduction in insulin dose during the time she was on liraglutide prior to starting on dapagliflozin.”
Dr. Dandona disclosed that he has received research support from AstraZeneca and Novo Nordisk.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
