User login
Study: TKA patients recover faster with periarticular analgesia injection
Patients more often recovered faster from a total knee arthroplasty (TKA) when they received a periarticular injection of analgesic medication than when they received a femoral nerve block for the same surgery on the opposite knee in a study.
The study included 16 recipients of bilateral primary TKA, who received a femoral nerve block at their first TKA operation and a periarticular injection of an extended-release bupivacaine liposome mixture at the second operation. An average of 2.3 years passed between the two procedures, and the same surgeon performed all surgeries, which occurred between March 2009 and August 2013. Two patients were excluded from the study because of subacute rehabilitation admission delay and a third patient was left out of the study because of respiratory failure, resulting in admission to the ICU.
Following the TKA with a periarticular injection of analgesic medication, the average number of inpatient physical therapy sessions a patient completed was 2.3 (standard deviation: 1.0); the average number of inpatient physical therapy sessions a patient completed after having the TKA with femoral nerve block was 3.5 (SD: 1.3). The average number of hospital days following the TKA with periarticular injection was also a smaller number. The mean number of hospital days following the periarticular injection was 1.5 (SD: 0.6 days). compared with 1.9 days (SD: 0.6 days; P is less than .032) following the femoral nerve block.
“Our data demonstrate that periarticular injection of analgesia allowed patients to complete their inpatient physical therapy sessions and to be discharged sooner, compared with femoral nerve block. This finding suggests that patients who receive periarticular injection of analgesia are able to ambulate independently faster because it does not affect postoperative motor function,” according to Dr. Brandon J. Horn and his colleagues.
Read the full study in the Journal of the American Osteopathic Association (doi: 10.7556/jaoa.2015.146).
Patients more often recovered faster from a total knee arthroplasty (TKA) when they received a periarticular injection of analgesic medication than when they received a femoral nerve block for the same surgery on the opposite knee in a study.
The study included 16 recipients of bilateral primary TKA, who received a femoral nerve block at their first TKA operation and a periarticular injection of an extended-release bupivacaine liposome mixture at the second operation. An average of 2.3 years passed between the two procedures, and the same surgeon performed all surgeries, which occurred between March 2009 and August 2013. Two patients were excluded from the study because of subacute rehabilitation admission delay and a third patient was left out of the study because of respiratory failure, resulting in admission to the ICU.
Following the TKA with a periarticular injection of analgesic medication, the average number of inpatient physical therapy sessions a patient completed was 2.3 (standard deviation: 1.0); the average number of inpatient physical therapy sessions a patient completed after having the TKA with femoral nerve block was 3.5 (SD: 1.3). The average number of hospital days following the TKA with periarticular injection was also a smaller number. The mean number of hospital days following the periarticular injection was 1.5 (SD: 0.6 days). compared with 1.9 days (SD: 0.6 days; P is less than .032) following the femoral nerve block.
“Our data demonstrate that periarticular injection of analgesia allowed patients to complete their inpatient physical therapy sessions and to be discharged sooner, compared with femoral nerve block. This finding suggests that patients who receive periarticular injection of analgesia are able to ambulate independently faster because it does not affect postoperative motor function,” according to Dr. Brandon J. Horn and his colleagues.
Read the full study in the Journal of the American Osteopathic Association (doi: 10.7556/jaoa.2015.146).
Patients more often recovered faster from a total knee arthroplasty (TKA) when they received a periarticular injection of analgesic medication than when they received a femoral nerve block for the same surgery on the opposite knee in a study.
The study included 16 recipients of bilateral primary TKA, who received a femoral nerve block at their first TKA operation and a periarticular injection of an extended-release bupivacaine liposome mixture at the second operation. An average of 2.3 years passed between the two procedures, and the same surgeon performed all surgeries, which occurred between March 2009 and August 2013. Two patients were excluded from the study because of subacute rehabilitation admission delay and a third patient was left out of the study because of respiratory failure, resulting in admission to the ICU.
Following the TKA with a periarticular injection of analgesic medication, the average number of inpatient physical therapy sessions a patient completed was 2.3 (standard deviation: 1.0); the average number of inpatient physical therapy sessions a patient completed after having the TKA with femoral nerve block was 3.5 (SD: 1.3). The average number of hospital days following the TKA with periarticular injection was also a smaller number. The mean number of hospital days following the periarticular injection was 1.5 (SD: 0.6 days). compared with 1.9 days (SD: 0.6 days; P is less than .032) following the femoral nerve block.
“Our data demonstrate that periarticular injection of analgesia allowed patients to complete their inpatient physical therapy sessions and to be discharged sooner, compared with femoral nerve block. This finding suggests that patients who receive periarticular injection of analgesia are able to ambulate independently faster because it does not affect postoperative motor function,” according to Dr. Brandon J. Horn and his colleagues.
Read the full study in the Journal of the American Osteopathic Association (doi: 10.7556/jaoa.2015.146).
FROM THE JOURNAL OF THE AMERICAN OSTEOPATHIC ASSOCIATION
Pediatric Dermatology Consult - December 2015
Ancillary testing
Complete blood count showed leukocytosis (13.1 th/mcL), with normal differential. The C-reactive protein was elevated (10 mg/dL). The comprehensive metabolic panel was within normal limits. Mycoplasma pneumoniae antibody, IgM was positive, and herpes virus cultures were negative. The chest x-ray showed a dense right upper lobe airspace opacity, concerning for pneumonia, likely with a component of atelectasis.
Discussion
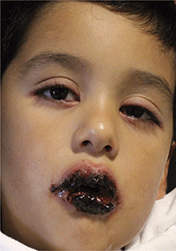
Mycoplasma pneumoniae–induced rash and mucositis (MIRM) is a syndrome of mucocutaneous involvement in conjunction with an M. pneumoniae infection. M. pneumoniae is a bacteria that commonly causes respiratory tract infections. In children, it is a leading cause of atypical pneumonia.1,2 Extrapulmonary manifestations are common and can involve the skin, heart, kidneys, gastrointestinal system, and nervous system.3 There is a 1-3 week incubation period for infections, which always begin with respiratory symptoms. The most common early symptoms of this infection are cough, malaise, fever, and headaches. Studies show that between 25%-38% of M. pneumoniae infections have mucocutaneous manifestations.3,4
An overwhelming majority of cases have mucosal involvement while the degree of skin involvement varies. Cutaneous manifestations, which are usually polymorphic, present with vesiculobullous lesions, targetoid macules and papules, or morbilliform eruptions.3 MIRM is seen most often in children and adolescents, but a few adult cases have been reported. About 66% of cases are seen in males, but the reason for this gender predilection is not well understood.3 Overall, MIRM has an excellent prognosis, compared with the other disorders on the differential diagnosis.
Differential diagnosis
MIRM used to be considered along the spectrum of bullous diseases that have mucosal involvement including erythema multiforme (EM), Stevens-Johnson syndrome (SJS), and toxic epidermal necrosis (TEN).3-6 The distinction can be difficult to identify, given the similarities that all of these diseases share, and many published articles referencing cases of SJS, TEN, and EM actually fit the criteria for MIRM.6 What sets MIRM apart is the predominance of mucosal involvement with minimal skin involvement and the overwhelming overall good prognosis compared with drug induced SJS.3
Although rare cases have moderate cutaneous involvement, MIRM is most notable for oral lesions, which include ulcers, erosions, and vesiculobullae of the lips and buccal mucosa, as well as ocular and genital mucosal involvement in a lower degree.3 For cases with moderate or even mild involvement of the skin, ruling out other diagnoses becomes more difficult, especially since the dermatologic manifestations of targetoid papules and bullae are common to all of them. Testing for M. pneumoniae will strengthen the diagnosis and can be done in a number of ways. Using polymerase chain reaction to measure for M. pneumoniae DNA offers the benefit of being able to detect an infection early in its course because it does not rely on antibody formation.4 Other testing methods include M. pneumoniae antigen detection, and IgM or IgG titers.4
Treatment
The mainstay of treatment of this bacterial infection is, of course, antibiotics. Azithromycin and erythromycin are the drugs of choice for children. While tetracyclines and fluoroquinolones can be used to treat M. pneumoniae, their use is not recommended in young children.4 Systemic corticosteroids can be used in addition to antibiotics, but there is limited data to support its efficacy in these patients.7 In milder cases, reports have documented that the course can be self-limited and may improve with only supportive care. Because of the severity of mucosal involvement in some cases, such as the one in our patient, supportive care may include intravenous hydration and total parenteral nutrition if the patient is unable to tolerate oral intake. In more severe cases, IVIG can be beneficial.8 This has been shown to induce rapid improvement in refractory mucositis when antibiotics and supportive mucosal care alone have not been sufficient.8
References
- (J Clin Microbiol. 2015 Jan;53[1]:124-30.)
- (Clin Microbiol. 2004;17[4]:697-728.)
- (J Am Acad Dermatol. 2015;72[2]: 239-54.)
- (Int J Dermatol. 2009 Jul;48[7]:673-80.)
- (J Eur Acad Dermatol Venereol. 2015 Mar;29[3]:595-8.)
- (J Am Acad Dermatol. 2005 Feb;52[2]:312-5.)
- (Pediatr Dermatol. 2014 Nov-Dec;31[6]:664-9.)
- (Acta Paediatr. 2011 Nov;100[11]:e238-40.)
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California San Diego and Mr. Ginsberg is a research associate at the hospital. They said they had no relevant financial disclosures.
Email them at [email protected].
Ancillary testing
Complete blood count showed leukocytosis (13.1 th/mcL), with normal differential. The C-reactive protein was elevated (10 mg/dL). The comprehensive metabolic panel was within normal limits. Mycoplasma pneumoniae antibody, IgM was positive, and herpes virus cultures were negative. The chest x-ray showed a dense right upper lobe airspace opacity, concerning for pneumonia, likely with a component of atelectasis.
Discussion
Mycoplasma pneumoniae–induced rash and mucositis (MIRM) is a syndrome of mucocutaneous involvement in conjunction with an M. pneumoniae infection. M. pneumoniae is a bacteria that commonly causes respiratory tract infections. In children, it is a leading cause of atypical pneumonia.1,2 Extrapulmonary manifestations are common and can involve the skin, heart, kidneys, gastrointestinal system, and nervous system.3 There is a 1-3 week incubation period for infections, which always begin with respiratory symptoms. The most common early symptoms of this infection are cough, malaise, fever, and headaches. Studies show that between 25%-38% of M. pneumoniae infections have mucocutaneous manifestations.3,4
An overwhelming majority of cases have mucosal involvement while the degree of skin involvement varies. Cutaneous manifestations, which are usually polymorphic, present with vesiculobullous lesions, targetoid macules and papules, or morbilliform eruptions.3 MIRM is seen most often in children and adolescents, but a few adult cases have been reported. About 66% of cases are seen in males, but the reason for this gender predilection is not well understood.3 Overall, MIRM has an excellent prognosis, compared with the other disorders on the differential diagnosis.
Differential diagnosis
MIRM used to be considered along the spectrum of bullous diseases that have mucosal involvement including erythema multiforme (EM), Stevens-Johnson syndrome (SJS), and toxic epidermal necrosis (TEN).3-6 The distinction can be difficult to identify, given the similarities that all of these diseases share, and many published articles referencing cases of SJS, TEN, and EM actually fit the criteria for MIRM.6 What sets MIRM apart is the predominance of mucosal involvement with minimal skin involvement and the overwhelming overall good prognosis compared with drug induced SJS.3
Although rare cases have moderate cutaneous involvement, MIRM is most notable for oral lesions, which include ulcers, erosions, and vesiculobullae of the lips and buccal mucosa, as well as ocular and genital mucosal involvement in a lower degree.3 For cases with moderate or even mild involvement of the skin, ruling out other diagnoses becomes more difficult, especially since the dermatologic manifestations of targetoid papules and bullae are common to all of them. Testing for M. pneumoniae will strengthen the diagnosis and can be done in a number of ways. Using polymerase chain reaction to measure for M. pneumoniae DNA offers the benefit of being able to detect an infection early in its course because it does not rely on antibody formation.4 Other testing methods include M. pneumoniae antigen detection, and IgM or IgG titers.4
Treatment
The mainstay of treatment of this bacterial infection is, of course, antibiotics. Azithromycin and erythromycin are the drugs of choice for children. While tetracyclines and fluoroquinolones can be used to treat M. pneumoniae, their use is not recommended in young children.4 Systemic corticosteroids can be used in addition to antibiotics, but there is limited data to support its efficacy in these patients.7 In milder cases, reports have documented that the course can be self-limited and may improve with only supportive care. Because of the severity of mucosal involvement in some cases, such as the one in our patient, supportive care may include intravenous hydration and total parenteral nutrition if the patient is unable to tolerate oral intake. In more severe cases, IVIG can be beneficial.8 This has been shown to induce rapid improvement in refractory mucositis when antibiotics and supportive mucosal care alone have not been sufficient.8
References
- (J Clin Microbiol. 2015 Jan;53[1]:124-30.)
- (Clin Microbiol. 2004;17[4]:697-728.)
- (J Am Acad Dermatol. 2015;72[2]: 239-54.)
- (Int J Dermatol. 2009 Jul;48[7]:673-80.)
- (J Eur Acad Dermatol Venereol. 2015 Mar;29[3]:595-8.)
- (J Am Acad Dermatol. 2005 Feb;52[2]:312-5.)
- (Pediatr Dermatol. 2014 Nov-Dec;31[6]:664-9.)
- (Acta Paediatr. 2011 Nov;100[11]:e238-40.)
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California San Diego and Mr. Ginsberg is a research associate at the hospital. They said they had no relevant financial disclosures.
Email them at [email protected].
Ancillary testing
Complete blood count showed leukocytosis (13.1 th/mcL), with normal differential. The C-reactive protein was elevated (10 mg/dL). The comprehensive metabolic panel was within normal limits. Mycoplasma pneumoniae antibody, IgM was positive, and herpes virus cultures were negative. The chest x-ray showed a dense right upper lobe airspace opacity, concerning for pneumonia, likely with a component of atelectasis.
Discussion
Mycoplasma pneumoniae–induced rash and mucositis (MIRM) is a syndrome of mucocutaneous involvement in conjunction with an M. pneumoniae infection. M. pneumoniae is a bacteria that commonly causes respiratory tract infections. In children, it is a leading cause of atypical pneumonia.1,2 Extrapulmonary manifestations are common and can involve the skin, heart, kidneys, gastrointestinal system, and nervous system.3 There is a 1-3 week incubation period for infections, which always begin with respiratory symptoms. The most common early symptoms of this infection are cough, malaise, fever, and headaches. Studies show that between 25%-38% of M. pneumoniae infections have mucocutaneous manifestations.3,4
An overwhelming majority of cases have mucosal involvement while the degree of skin involvement varies. Cutaneous manifestations, which are usually polymorphic, present with vesiculobullous lesions, targetoid macules and papules, or morbilliform eruptions.3 MIRM is seen most often in children and adolescents, but a few adult cases have been reported. About 66% of cases are seen in males, but the reason for this gender predilection is not well understood.3 Overall, MIRM has an excellent prognosis, compared with the other disorders on the differential diagnosis.
Differential diagnosis
MIRM used to be considered along the spectrum of bullous diseases that have mucosal involvement including erythema multiforme (EM), Stevens-Johnson syndrome (SJS), and toxic epidermal necrosis (TEN).3-6 The distinction can be difficult to identify, given the similarities that all of these diseases share, and many published articles referencing cases of SJS, TEN, and EM actually fit the criteria for MIRM.6 What sets MIRM apart is the predominance of mucosal involvement with minimal skin involvement and the overwhelming overall good prognosis compared with drug induced SJS.3
Although rare cases have moderate cutaneous involvement, MIRM is most notable for oral lesions, which include ulcers, erosions, and vesiculobullae of the lips and buccal mucosa, as well as ocular and genital mucosal involvement in a lower degree.3 For cases with moderate or even mild involvement of the skin, ruling out other diagnoses becomes more difficult, especially since the dermatologic manifestations of targetoid papules and bullae are common to all of them. Testing for M. pneumoniae will strengthen the diagnosis and can be done in a number of ways. Using polymerase chain reaction to measure for M. pneumoniae DNA offers the benefit of being able to detect an infection early in its course because it does not rely on antibody formation.4 Other testing methods include M. pneumoniae antigen detection, and IgM or IgG titers.4
Treatment
The mainstay of treatment of this bacterial infection is, of course, antibiotics. Azithromycin and erythromycin are the drugs of choice for children. While tetracyclines and fluoroquinolones can be used to treat M. pneumoniae, their use is not recommended in young children.4 Systemic corticosteroids can be used in addition to antibiotics, but there is limited data to support its efficacy in these patients.7 In milder cases, reports have documented that the course can be self-limited and may improve with only supportive care. Because of the severity of mucosal involvement in some cases, such as the one in our patient, supportive care may include intravenous hydration and total parenteral nutrition if the patient is unable to tolerate oral intake. In more severe cases, IVIG can be beneficial.8 This has been shown to induce rapid improvement in refractory mucositis when antibiotics and supportive mucosal care alone have not been sufficient.8
References
- (J Clin Microbiol. 2015 Jan;53[1]:124-30.)
- (Clin Microbiol. 2004;17[4]:697-728.)
- (J Am Acad Dermatol. 2015;72[2]: 239-54.)
- (Int J Dermatol. 2009 Jul;48[7]:673-80.)
- (J Eur Acad Dermatol Venereol. 2015 Mar;29[3]:595-8.)
- (J Am Acad Dermatol. 2005 Feb;52[2]:312-5.)
- (Pediatr Dermatol. 2014 Nov-Dec;31[6]:664-9.)
- (Acta Paediatr. 2011 Nov;100[11]:e238-40.)
Dr. Matiz is assistant professor of dermatology at Rady Children’s Hospital San Diego–University of California San Diego and Mr. Ginsberg is a research associate at the hospital. They said they had no relevant financial disclosures.
Email them at [email protected].
A 4-year-old male with a past history of asthma, presents with a 10-day history of cough, wheezing, rhinorrhea, and fever. He was treated with ibuprofen and albuterol without much improvement. Two days prior to presentation, he developed swollen and cracked lips as well as pink conjunctiva. Since then, he has been hypoactive with poor oral intake. He was evaluated by his pediatrician and was given dexamethasone and diphenhydramine, with no improvement. His oral and ocular symptoms worsen, which is why he was taken to the emergency department. He has no sick contacts. On the day of presentation, he developed several target erythematous lesions on his arms and legs that progressed to bulla (See photo).
Managing menopause symptoms in gynecologic cancer survivors
Due to advancements in surgical treatment, chemotherapy, and radiation therapy, gynecologic cancer survival rates are continuing to improve and quality of life is evolving into an even more significant focus in cancer care.
Roughly 30%-40% of all women with a gynecologic malignancy will experience climacteric symptoms and menopause prior to the anticipated time of natural menopause (J Clin Oncol. 2009 Mar 10;27[8]:1214-9). Cessation of ovarian estrogen and progesterone production can result in short-term as well as long-term sequelae, including vasomotor symptoms, vaginal dryness, osteoporosis, and mood disturbances. Iatrogenic menopause after cancer treatment can be more sudden and severe when compared with the natural course of physiologic menopause. As a result, determination of safe, effective modalities for treating these symptoms is of particular importance for survivor quality of life.

Both combination and estrogen-only hormone replacement therapy (HT) provide greater improvement in these specific symptoms and overall quality of life than placebo as demonstrated in several observational and randomized control trials (Cochrane Database Syst Rev. 2009 Apr 15;[2]:CD004143).
Endometrial cancer
Endometrial cancer is the most common gynecologic malignancy, with approximately 54,000 new cases anticipated in the United States in 2015. Twenty-five percent of these new cases will be in premenopausal women, and with an ever-increasing obesity rate, this number may continue to climb.
Women with early-stage Type 1 endometrial cancer who have vasomotor symptoms after surgery may be offered a short course of estrogen-based HT at the lowest effective dose following hysterectomy/bilateral salpingo-oophorectomy and staging procedure (J Clin Oncol. 2006 Feb 1;24[4]:587-92). For women with genitourinary symptoms, vaginal moisturizers and/or low-dose vaginal estrogen are reasonable options. Unfortunately, there are no data to guide the use of estrogen replacement therapy in women with Type 2 endometrial cancers (Gynecol Oncol. 2011 Aug;122[2]:447-54).
Ovarian cancer
There is minimal data implicating a hormonal causation to ovarian carcinogenesis. Most women with epithelial ovarian cancer do not express tumor estrogen or progesterone receptors. Treatment will result in abrupt, iatrogenic menopause, raising the question of whether it is safe to use HT in patients with epithelial ovarian cancer.
Multiple studies have failed to demonstrate a difference in 5-year survival rates in women with epithelial cancer using HT for 2 years or less (JAMA. 2009 Jul 15;302[3]:298-305, Eur J Gynaecol Oncol. 2000;21[2]:192-6, Cancer. 1999 Sep 15;86[6]:1013-8). As such, symptomatic patients could be offered a course of HT; however, caution should be exercised in women with estrogen/progesterone–expressing tumors or nonepithelial tumors. As with endometrial cancer patients, the lowest effective doses should be prescribed.
Cervical cancer

Most cervical squamous and adenocarcinomas are not hormone dependent. For women with early-stage squamous cell carcinoma, ovarian conservation may be possible or oophoropexy may be offered. However, for many patients, bilateral salpingo-oophorectomy at the time of hysterectomy is more common, and the local effect of radiation therapy can result in vaginal atrophy with subsequent dyspareunia or ovarian failure from radiation scatter. Even for patients who undergo oophoropexy, radiation scatter may still result in ovarian failure. In a few observational studies, there are no data to infer that cervical cancer is hormonally related or that survival rates are decreased.
Currently, HT use in cervical cancer survivors is considered safe. Of note, for women with more advanced-stage cervical cancer and who received chemoradiation for primary treatment, combination therapy with estrogen and progesterone may be more appropriate if the uterus remains in situ. However, for women who have undergone hysterectomy, combination therapy with progesterone may not be warranted and estrogen alone (orally or vaginally) is acceptable (Gynecol Oncol. 2011 Aug;122[2]:447-54)
Nonhormonal therapies
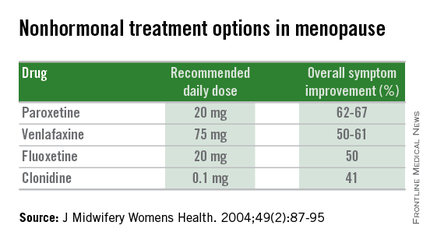
Women presenting with menopausal symptoms in whom estrogen therapy is contraindicated or not desired can also consider using nonhormonal therapies as an alternative. These include selective serotonin reuptake inhibitors (SSRIs) and alpha-2 adrenergic agonists, such as clonidine. Albeit not as effective as HT, these alternative therapies are reasonable options, particularly for management of vasomotor symptoms.
From a limited number of observational studies and a few randomized trials, short-term hormone replacement therapy does not present increased risk to survivors of gynecologic cancers. Additionally, patients have the added option of using nonhormonal therapies, which may provide some benefit. The decision to institute HT should occur after a thorough discussion of the potential to optimize symptom control and the theoretical risk of stimulating quiescent malignant disease.
Dr. Staley is a resident physician in the department of obstetrics and gynecology at the University of North Carolina at Chapel Hill. Dr. Gehrig is professor and director of gynecologic oncology at the university. They reported having no relevant financial disclosures.
Due to advancements in surgical treatment, chemotherapy, and radiation therapy, gynecologic cancer survival rates are continuing to improve and quality of life is evolving into an even more significant focus in cancer care.
Roughly 30%-40% of all women with a gynecologic malignancy will experience climacteric symptoms and menopause prior to the anticipated time of natural menopause (J Clin Oncol. 2009 Mar 10;27[8]:1214-9). Cessation of ovarian estrogen and progesterone production can result in short-term as well as long-term sequelae, including vasomotor symptoms, vaginal dryness, osteoporosis, and mood disturbances. Iatrogenic menopause after cancer treatment can be more sudden and severe when compared with the natural course of physiologic menopause. As a result, determination of safe, effective modalities for treating these symptoms is of particular importance for survivor quality of life.
Both combination and estrogen-only hormone replacement therapy (HT) provide greater improvement in these specific symptoms and overall quality of life than placebo as demonstrated in several observational and randomized control trials (Cochrane Database Syst Rev. 2009 Apr 15;[2]:CD004143).
Endometrial cancer
Endometrial cancer is the most common gynecologic malignancy, with approximately 54,000 new cases anticipated in the United States in 2015. Twenty-five percent of these new cases will be in premenopausal women, and with an ever-increasing obesity rate, this number may continue to climb.
Women with early-stage Type 1 endometrial cancer who have vasomotor symptoms after surgery may be offered a short course of estrogen-based HT at the lowest effective dose following hysterectomy/bilateral salpingo-oophorectomy and staging procedure (J Clin Oncol. 2006 Feb 1;24[4]:587-92). For women with genitourinary symptoms, vaginal moisturizers and/or low-dose vaginal estrogen are reasonable options. Unfortunately, there are no data to guide the use of estrogen replacement therapy in women with Type 2 endometrial cancers (Gynecol Oncol. 2011 Aug;122[2]:447-54).
Ovarian cancer
There is minimal data implicating a hormonal causation to ovarian carcinogenesis. Most women with epithelial ovarian cancer do not express tumor estrogen or progesterone receptors. Treatment will result in abrupt, iatrogenic menopause, raising the question of whether it is safe to use HT in patients with epithelial ovarian cancer.
Multiple studies have failed to demonstrate a difference in 5-year survival rates in women with epithelial cancer using HT for 2 years or less (JAMA. 2009 Jul 15;302[3]:298-305, Eur J Gynaecol Oncol. 2000;21[2]:192-6, Cancer. 1999 Sep 15;86[6]:1013-8). As such, symptomatic patients could be offered a course of HT; however, caution should be exercised in women with estrogen/progesterone–expressing tumors or nonepithelial tumors. As with endometrial cancer patients, the lowest effective doses should be prescribed.
Cervical cancer
Most cervical squamous and adenocarcinomas are not hormone dependent. For women with early-stage squamous cell carcinoma, ovarian conservation may be possible or oophoropexy may be offered. However, for many patients, bilateral salpingo-oophorectomy at the time of hysterectomy is more common, and the local effect of radiation therapy can result in vaginal atrophy with subsequent dyspareunia or ovarian failure from radiation scatter. Even for patients who undergo oophoropexy, radiation scatter may still result in ovarian failure. In a few observational studies, there are no data to infer that cervical cancer is hormonally related or that survival rates are decreased.
Currently, HT use in cervical cancer survivors is considered safe. Of note, for women with more advanced-stage cervical cancer and who received chemoradiation for primary treatment, combination therapy with estrogen and progesterone may be more appropriate if the uterus remains in situ. However, for women who have undergone hysterectomy, combination therapy with progesterone may not be warranted and estrogen alone (orally or vaginally) is acceptable (Gynecol Oncol. 2011 Aug;122[2]:447-54)
Nonhormonal therapies
Women presenting with menopausal symptoms in whom estrogen therapy is contraindicated or not desired can also consider using nonhormonal therapies as an alternative. These include selective serotonin reuptake inhibitors (SSRIs) and alpha-2 adrenergic agonists, such as clonidine. Albeit not as effective as HT, these alternative therapies are reasonable options, particularly for management of vasomotor symptoms.
From a limited number of observational studies and a few randomized trials, short-term hormone replacement therapy does not present increased risk to survivors of gynecologic cancers. Additionally, patients have the added option of using nonhormonal therapies, which may provide some benefit. The decision to institute HT should occur after a thorough discussion of the potential to optimize symptom control and the theoretical risk of stimulating quiescent malignant disease.
Dr. Staley is a resident physician in the department of obstetrics and gynecology at the University of North Carolina at Chapel Hill. Dr. Gehrig is professor and director of gynecologic oncology at the university. They reported having no relevant financial disclosures.
Due to advancements in surgical treatment, chemotherapy, and radiation therapy, gynecologic cancer survival rates are continuing to improve and quality of life is evolving into an even more significant focus in cancer care.
Roughly 30%-40% of all women with a gynecologic malignancy will experience climacteric symptoms and menopause prior to the anticipated time of natural menopause (J Clin Oncol. 2009 Mar 10;27[8]:1214-9). Cessation of ovarian estrogen and progesterone production can result in short-term as well as long-term sequelae, including vasomotor symptoms, vaginal dryness, osteoporosis, and mood disturbances. Iatrogenic menopause after cancer treatment can be more sudden and severe when compared with the natural course of physiologic menopause. As a result, determination of safe, effective modalities for treating these symptoms is of particular importance for survivor quality of life.
Both combination and estrogen-only hormone replacement therapy (HT) provide greater improvement in these specific symptoms and overall quality of life than placebo as demonstrated in several observational and randomized control trials (Cochrane Database Syst Rev. 2009 Apr 15;[2]:CD004143).
Endometrial cancer
Endometrial cancer is the most common gynecologic malignancy, with approximately 54,000 new cases anticipated in the United States in 2015. Twenty-five percent of these new cases will be in premenopausal women, and with an ever-increasing obesity rate, this number may continue to climb.
Women with early-stage Type 1 endometrial cancer who have vasomotor symptoms after surgery may be offered a short course of estrogen-based HT at the lowest effective dose following hysterectomy/bilateral salpingo-oophorectomy and staging procedure (J Clin Oncol. 2006 Feb 1;24[4]:587-92). For women with genitourinary symptoms, vaginal moisturizers and/or low-dose vaginal estrogen are reasonable options. Unfortunately, there are no data to guide the use of estrogen replacement therapy in women with Type 2 endometrial cancers (Gynecol Oncol. 2011 Aug;122[2]:447-54).
Ovarian cancer
There is minimal data implicating a hormonal causation to ovarian carcinogenesis. Most women with epithelial ovarian cancer do not express tumor estrogen or progesterone receptors. Treatment will result in abrupt, iatrogenic menopause, raising the question of whether it is safe to use HT in patients with epithelial ovarian cancer.
Multiple studies have failed to demonstrate a difference in 5-year survival rates in women with epithelial cancer using HT for 2 years or less (JAMA. 2009 Jul 15;302[3]:298-305, Eur J Gynaecol Oncol. 2000;21[2]:192-6, Cancer. 1999 Sep 15;86[6]:1013-8). As such, symptomatic patients could be offered a course of HT; however, caution should be exercised in women with estrogen/progesterone–expressing tumors or nonepithelial tumors. As with endometrial cancer patients, the lowest effective doses should be prescribed.
Cervical cancer
Most cervical squamous and adenocarcinomas are not hormone dependent. For women with early-stage squamous cell carcinoma, ovarian conservation may be possible or oophoropexy may be offered. However, for many patients, bilateral salpingo-oophorectomy at the time of hysterectomy is more common, and the local effect of radiation therapy can result in vaginal atrophy with subsequent dyspareunia or ovarian failure from radiation scatter. Even for patients who undergo oophoropexy, radiation scatter may still result in ovarian failure. In a few observational studies, there are no data to infer that cervical cancer is hormonally related or that survival rates are decreased.
Currently, HT use in cervical cancer survivors is considered safe. Of note, for women with more advanced-stage cervical cancer and who received chemoradiation for primary treatment, combination therapy with estrogen and progesterone may be more appropriate if the uterus remains in situ. However, for women who have undergone hysterectomy, combination therapy with progesterone may not be warranted and estrogen alone (orally or vaginally) is acceptable (Gynecol Oncol. 2011 Aug;122[2]:447-54)
Nonhormonal therapies
Women presenting with menopausal symptoms in whom estrogen therapy is contraindicated or not desired can also consider using nonhormonal therapies as an alternative. These include selective serotonin reuptake inhibitors (SSRIs) and alpha-2 adrenergic agonists, such as clonidine. Albeit not as effective as HT, these alternative therapies are reasonable options, particularly for management of vasomotor symptoms.
From a limited number of observational studies and a few randomized trials, short-term hormone replacement therapy does not present increased risk to survivors of gynecologic cancers. Additionally, patients have the added option of using nonhormonal therapies, which may provide some benefit. The decision to institute HT should occur after a thorough discussion of the potential to optimize symptom control and the theoretical risk of stimulating quiescent malignant disease.
Dr. Staley is a resident physician in the department of obstetrics and gynecology at the University of North Carolina at Chapel Hill. Dr. Gehrig is professor and director of gynecologic oncology at the university. They reported having no relevant financial disclosures.

The Patient Who Didn’t Complain Enough?
A 70-year-old woman presents to dermatology with asymptomatic lesions on her legs that, she reports, have been there for decades but are now becoming larger and more unsightly. She’s tried several courses of antifungal creams and pills with no success.
In all this time, she hasn’t seen a dermatology provider, because “no one ever felt the need” to refer her since her diagnosis was so “obvious.” When the changes began, however, her children finally convinced her to consult a specialist.
Her medical history is otherwise unremarkable, with no problems directly related to her skin issues. She denies both personal and family history of diabetes.
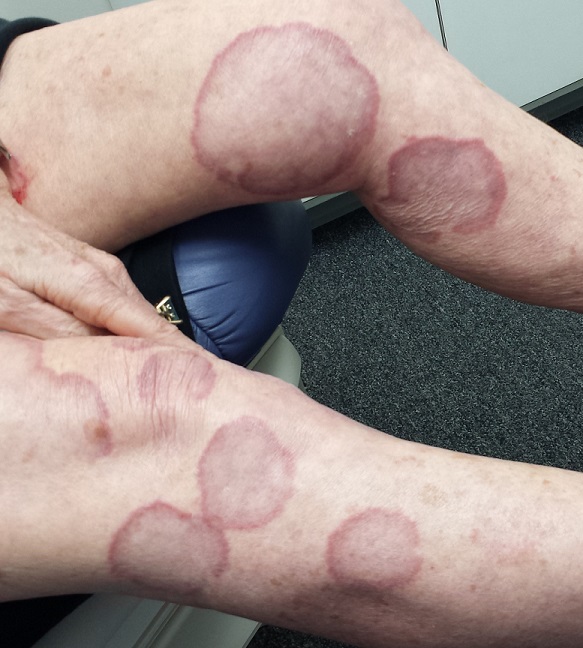
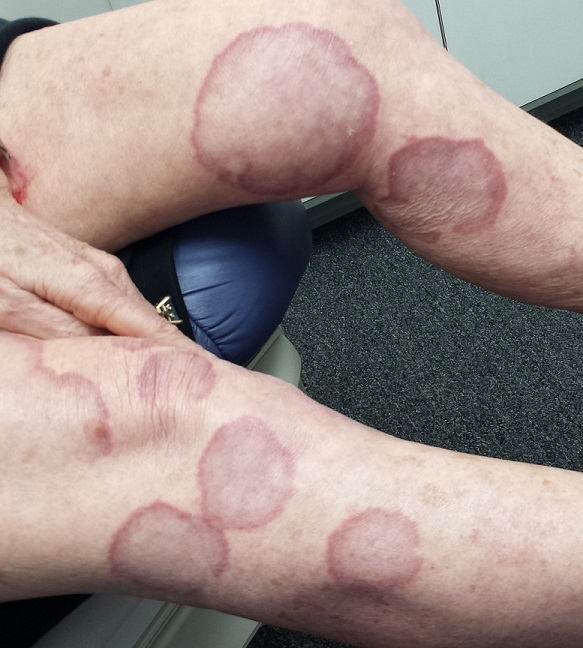
EXAMINATION
There are numerous impressive, reddish brown, annular plaques with slightly raised borders and clearing centers on the patient’s legs. There are also a few on her arms and one or two on her trunk. There is no epidermal component (scale or other surface disruption) on any of the lesions, which range in size from 2 cm to 15 cm.
What is the diagnosis?
DISCUSSION
It’s hard to believe that any patient could go this long with such a florid condition and never be seen by dermatology. Patients have an obligation to speak up—to complain, as it were—but some are too compliant. This woman’s primary care providers should have attached some significance to the nonresponse to treatment, one explanation for which might have been erroneous diagnosis.
Everybody knows that big round things are fungal, except when they aren’t. That’s where dermatology providers shine. Our training is all about development of differentials, as in: What are the various potential explanations for big round lesions? What would those things look like, and how could we distinguish them from fungal infection?
Here’s one huge clue: Fungal infection commonly refers to a relatively trivial, superficial infection of the outermost layer of the skin, the stratum corneum. By its very nature, it almost always involves scaling; if those scales are examined microscopically, the fungal elements can be seen, thus confirming the diagnosis.
When there is no scale, and the condition fails to respond to antifungal medications, alternative diagnoses must be considered. One major item that fits the bill is granuloma annulare (GA), an asymptomatic condition that presents with reddish brown, distinctly annular papules and plaques with raised (papular) borders and cleared centers. Though GA is extremely common, it is almost always misdiagnosed initially as fungal infection.
Besides the lack of scaling, GA lesions have some other characteristics that aid diagnosis. They’re commonly seen on extremities, especially the dorsa of feet and hands, and are far more common on women (especially young ones) than on men.
Although usually reddish brown, the lesions will be darker on patients with darker skin. It should be mentioned that this patient’s lesions were unusually large and extensive. Most cases are far more modest in size and distribution.
When necessary, punch biopsy shows what is called a palisaded granuloma. This, taken in context with the clinical picture, nails down the diagnosis.
One other piece of information is good to know: GA is quite common. You will see it, regularly.
Since the cause of GA is unknown, treatment can be problematic. Fortunately, the problem almost always resolves, with or without treatment. A few of this patient’s lesions were treated (for cosmetic reasons) with intralesional steroid injection.
TAKE-HOME LEARNING POINTS
• Granuloma annulare (GA) is a benign asymptomatic condition that presents with annular, reddish brown intradermal lesions.
• GA is commonly seen on the dorsa of feet, hands, and arms, especially of young women.
• GA lesions are intradermal, not epidermal, so there is no scaling or other disturbance of the skin surface.
• GA treatment is problematic, but the problem usually resolves on its own.
A 70-year-old woman presents to dermatology with asymptomatic lesions on her legs that, she reports, have been there for decades but are now becoming larger and more unsightly. She’s tried several courses of antifungal creams and pills with no success.
In all this time, she hasn’t seen a dermatology provider, because “no one ever felt the need” to refer her since her diagnosis was so “obvious.” When the changes began, however, her children finally convinced her to consult a specialist.
Her medical history is otherwise unremarkable, with no problems directly related to her skin issues. She denies both personal and family history of diabetes.
EXAMINATION
There are numerous impressive, reddish brown, annular plaques with slightly raised borders and clearing centers on the patient’s legs. There are also a few on her arms and one or two on her trunk. There is no epidermal component (scale or other surface disruption) on any of the lesions, which range in size from 2 cm to 15 cm.
What is the diagnosis?
DISCUSSION
It’s hard to believe that any patient could go this long with such a florid condition and never be seen by dermatology. Patients have an obligation to speak up—to complain, as it were—but some are too compliant. This woman’s primary care providers should have attached some significance to the nonresponse to treatment, one explanation for which might have been erroneous diagnosis.
Everybody knows that big round things are fungal, except when they aren’t. That’s where dermatology providers shine. Our training is all about development of differentials, as in: What are the various potential explanations for big round lesions? What would those things look like, and how could we distinguish them from fungal infection?
Here’s one huge clue: Fungal infection commonly refers to a relatively trivial, superficial infection of the outermost layer of the skin, the stratum corneum. By its very nature, it almost always involves scaling; if those scales are examined microscopically, the fungal elements can be seen, thus confirming the diagnosis.
When there is no scale, and the condition fails to respond to antifungal medications, alternative diagnoses must be considered. One major item that fits the bill is granuloma annulare (GA), an asymptomatic condition that presents with reddish brown, distinctly annular papules and plaques with raised (papular) borders and cleared centers. Though GA is extremely common, it is almost always misdiagnosed initially as fungal infection.
Besides the lack of scaling, GA lesions have some other characteristics that aid diagnosis. They’re commonly seen on extremities, especially the dorsa of feet and hands, and are far more common on women (especially young ones) than on men.
Although usually reddish brown, the lesions will be darker on patients with darker skin. It should be mentioned that this patient’s lesions were unusually large and extensive. Most cases are far more modest in size and distribution.
When necessary, punch biopsy shows what is called a palisaded granuloma. This, taken in context with the clinical picture, nails down the diagnosis.
One other piece of information is good to know: GA is quite common. You will see it, regularly.
Since the cause of GA is unknown, treatment can be problematic. Fortunately, the problem almost always resolves, with or without treatment. A few of this patient’s lesions were treated (for cosmetic reasons) with intralesional steroid injection.
TAKE-HOME LEARNING POINTS
• Granuloma annulare (GA) is a benign asymptomatic condition that presents with annular, reddish brown intradermal lesions.
• GA is commonly seen on the dorsa of feet, hands, and arms, especially of young women.
• GA lesions are intradermal, not epidermal, so there is no scaling or other disturbance of the skin surface.
• GA treatment is problematic, but the problem usually resolves on its own.
A 70-year-old woman presents to dermatology with asymptomatic lesions on her legs that, she reports, have been there for decades but are now becoming larger and more unsightly. She’s tried several courses of antifungal creams and pills with no success.
In all this time, she hasn’t seen a dermatology provider, because “no one ever felt the need” to refer her since her diagnosis was so “obvious.” When the changes began, however, her children finally convinced her to consult a specialist.
Her medical history is otherwise unremarkable, with no problems directly related to her skin issues. She denies both personal and family history of diabetes.
EXAMINATION
There are numerous impressive, reddish brown, annular plaques with slightly raised borders and clearing centers on the patient’s legs. There are also a few on her arms and one or two on her trunk. There is no epidermal component (scale or other surface disruption) on any of the lesions, which range in size from 2 cm to 15 cm.
What is the diagnosis?
DISCUSSION
It’s hard to believe that any patient could go this long with such a florid condition and never be seen by dermatology. Patients have an obligation to speak up—to complain, as it were—but some are too compliant. This woman’s primary care providers should have attached some significance to the nonresponse to treatment, one explanation for which might have been erroneous diagnosis.
Everybody knows that big round things are fungal, except when they aren’t. That’s where dermatology providers shine. Our training is all about development of differentials, as in: What are the various potential explanations for big round lesions? What would those things look like, and how could we distinguish them from fungal infection?
Here’s one huge clue: Fungal infection commonly refers to a relatively trivial, superficial infection of the outermost layer of the skin, the stratum corneum. By its very nature, it almost always involves scaling; if those scales are examined microscopically, the fungal elements can be seen, thus confirming the diagnosis.
When there is no scale, and the condition fails to respond to antifungal medications, alternative diagnoses must be considered. One major item that fits the bill is granuloma annulare (GA), an asymptomatic condition that presents with reddish brown, distinctly annular papules and plaques with raised (papular) borders and cleared centers. Though GA is extremely common, it is almost always misdiagnosed initially as fungal infection.
Besides the lack of scaling, GA lesions have some other characteristics that aid diagnosis. They’re commonly seen on extremities, especially the dorsa of feet and hands, and are far more common on women (especially young ones) than on men.
Although usually reddish brown, the lesions will be darker on patients with darker skin. It should be mentioned that this patient’s lesions were unusually large and extensive. Most cases are far more modest in size and distribution.
When necessary, punch biopsy shows what is called a palisaded granuloma. This, taken in context with the clinical picture, nails down the diagnosis.
One other piece of information is good to know: GA is quite common. You will see it, regularly.
Since the cause of GA is unknown, treatment can be problematic. Fortunately, the problem almost always resolves, with or without treatment. A few of this patient’s lesions were treated (for cosmetic reasons) with intralesional steroid injection.
TAKE-HOME LEARNING POINTS
• Granuloma annulare (GA) is a benign asymptomatic condition that presents with annular, reddish brown intradermal lesions.
• GA is commonly seen on the dorsa of feet, hands, and arms, especially of young women.
• GA lesions are intradermal, not epidermal, so there is no scaling or other disturbance of the skin surface.
• GA treatment is problematic, but the problem usually resolves on its own.
FDA gives nod to rapid-infusion bendamustine for CLL
The Food and Drug Administration has approved a new rapid-infusion form of bendamustine hydrochloride for patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin lymphoma.
The new formulation of bendamustine (Bendeka) is a 50-mL liquid designed for 10-minute infusion. It was granted orphan drug status for both the leukemia and lymphoma indications.
Bendeka is approved as primary therapy for chronic lymphocytic leukemia (CLL) and also for indolent B-cell non-Hodgkin lymphoma (NHL) that has progressed during or within 6 months of treatment with rituximab or a rituximab-containing regimen.
According to the prescribing information, the recommended dosing regimen for CLL is 100 mg/m2 infused intravenously over 10 minutes on days 1 and 2 of a 28-day cycle, up to six cycles. For NHL, the regimen calls for 120 mg/m2 infused intravenously over 10 minutes on days 1 and 2 of a 21-day cycle, up to eight cycles.
The most common hematologic adverse reactions are lymphopenia, anemia, leukopenia, thrombocytopenia, and neutropenia. Bendamustine has been associated with severe – and even fatal – myelosuppression.
Bendeka is manufactured by Teva Pharmaceutical Industries and succeeds Teva’s previously approved form of bendamustine, Treanda, approved in 2008. “Since 2008, Treanda has played a valuable role in the treatment of patients with CLL or indolent B-cell NHL that has progressed,” Paul Rittman, Teva Oncology’s vice president and general manager, said in a press statement. Treanda’s orphan drug exclusivity status for the NHL indication was set to expire in October, and its pediatric exclusivity status for that indication, next April. Its CLL exclusivity status expired in September.
Teva purchased the new formulation from Eagle Pharmaceuticals last February. The deal was closed with an upfront payment of $30 million, and potential for up to $90 million in additional payments, as well as double-digit royalties on net sales.
At the time of purchase, Eagle had secured orphan drug designations for Bendeka in both CLL and NHL and had submitted the New Drug Application under priority review. Bendeka may be eligible for a 7-year exclusivity status.
The drug is scheduled to be available during the first quarter of 2016.
The Food and Drug Administration has approved a new rapid-infusion form of bendamustine hydrochloride for patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin lymphoma.
The new formulation of bendamustine (Bendeka) is a 50-mL liquid designed for 10-minute infusion. It was granted orphan drug status for both the leukemia and lymphoma indications.
Bendeka is approved as primary therapy for chronic lymphocytic leukemia (CLL) and also for indolent B-cell non-Hodgkin lymphoma (NHL) that has progressed during or within 6 months of treatment with rituximab or a rituximab-containing regimen.
According to the prescribing information, the recommended dosing regimen for CLL is 100 mg/m2 infused intravenously over 10 minutes on days 1 and 2 of a 28-day cycle, up to six cycles. For NHL, the regimen calls for 120 mg/m2 infused intravenously over 10 minutes on days 1 and 2 of a 21-day cycle, up to eight cycles.
The most common hematologic adverse reactions are lymphopenia, anemia, leukopenia, thrombocytopenia, and neutropenia. Bendamustine has been associated with severe – and even fatal – myelosuppression.
Bendeka is manufactured by Teva Pharmaceutical Industries and succeeds Teva’s previously approved form of bendamustine, Treanda, approved in 2008. “Since 2008, Treanda has played a valuable role in the treatment of patients with CLL or indolent B-cell NHL that has progressed,” Paul Rittman, Teva Oncology’s vice president and general manager, said in a press statement. Treanda’s orphan drug exclusivity status for the NHL indication was set to expire in October, and its pediatric exclusivity status for that indication, next April. Its CLL exclusivity status expired in September.
Teva purchased the new formulation from Eagle Pharmaceuticals last February. The deal was closed with an upfront payment of $30 million, and potential for up to $90 million in additional payments, as well as double-digit royalties on net sales.
At the time of purchase, Eagle had secured orphan drug designations for Bendeka in both CLL and NHL and had submitted the New Drug Application under priority review. Bendeka may be eligible for a 7-year exclusivity status.
The drug is scheduled to be available during the first quarter of 2016.
The Food and Drug Administration has approved a new rapid-infusion form of bendamustine hydrochloride for patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin lymphoma.
The new formulation of bendamustine (Bendeka) is a 50-mL liquid designed for 10-minute infusion. It was granted orphan drug status for both the leukemia and lymphoma indications.
Bendeka is approved as primary therapy for chronic lymphocytic leukemia (CLL) and also for indolent B-cell non-Hodgkin lymphoma (NHL) that has progressed during or within 6 months of treatment with rituximab or a rituximab-containing regimen.
According to the prescribing information, the recommended dosing regimen for CLL is 100 mg/m2 infused intravenously over 10 minutes on days 1 and 2 of a 28-day cycle, up to six cycles. For NHL, the regimen calls for 120 mg/m2 infused intravenously over 10 minutes on days 1 and 2 of a 21-day cycle, up to eight cycles.
The most common hematologic adverse reactions are lymphopenia, anemia, leukopenia, thrombocytopenia, and neutropenia. Bendamustine has been associated with severe – and even fatal – myelosuppression.
Bendeka is manufactured by Teva Pharmaceutical Industries and succeeds Teva’s previously approved form of bendamustine, Treanda, approved in 2008. “Since 2008, Treanda has played a valuable role in the treatment of patients with CLL or indolent B-cell NHL that has progressed,” Paul Rittman, Teva Oncology’s vice president and general manager, said in a press statement. Treanda’s orphan drug exclusivity status for the NHL indication was set to expire in October, and its pediatric exclusivity status for that indication, next April. Its CLL exclusivity status expired in September.
Teva purchased the new formulation from Eagle Pharmaceuticals last February. The deal was closed with an upfront payment of $30 million, and potential for up to $90 million in additional payments, as well as double-digit royalties on net sales.
At the time of purchase, Eagle had secured orphan drug designations for Bendeka in both CLL and NHL and had submitted the New Drug Application under priority review. Bendeka may be eligible for a 7-year exclusivity status.
The drug is scheduled to be available during the first quarter of 2016.
Naloxone to revive an addict
We are in the midst of an epidemic of heroin and prescription opioid abuse. While the two do not completely explain each other, they are tragically and irrevocably linked.
From 2001 to 2013, we have observed a threefold increase in the total number of overdose deaths from opioid pain relievers (about 16,000 in 2013) and a fivefold increase in the total number of overdose deaths from heroin (about 8,000 in 2013).

Heroin initiation is almost 20 times higher among individuals reporting nonmedical prescription pain reliever use. Among opioid-treatment seekers, the majority of individuals who initiated opioid use in the 1960s were first exposed to heroin. This is in contrast to those who initiated in the 2000s, among whom the majority were exposed to prescription opioids. For young adults, the main sources of opioids are family, friends … and clinicians.
Opioids are powerfully addictive and can be snorted, swallowed, smoked, or shot. Data from the START (Starting Treatment with Agonist Replacement Therapies) trial suggest that individuals who inject opioids are less likely to remain in treatment than noninjectors. This necessarily increases the risk for injectors to inject again and be at risk for overdose.
Opioid overdose can be reversed with the use of naloxone. But naloxone has to be immediately or quickly available for it to be effective. Take-home naloxone programs are located in 30 U.S. states and the District of Columbia. Since 1996, home naloxone programs have reported more than 26,000 drug overdose reversals with naloxone.
On Nov. 18, the Food and Drug Administration announced the approval of a naloxone nasal spray. Prior to this approval, naloxone was only available in the injectable form (syringe or auto-injector), and needle management likely posed a barrier to first responders. The nasal spray can be administered easily without medical training. Naloxone nasal spray administered in one nostril delivered approximately the same levels or higher of naloxone as a single dose of an FDA-approved naloxone intramuscular injection in approximately the same time frame.
It is one thing to save a heroin addict who has just overdosed with nasal naloxone followed by appropriate medical attention. It is entirely another to engage them in an effective drug treatment program.
If naloxone revives them, it is treatment that can save them.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.
We are in the midst of an epidemic of heroin and prescription opioid abuse. While the two do not completely explain each other, they are tragically and irrevocably linked.
From 2001 to 2013, we have observed a threefold increase in the total number of overdose deaths from opioid pain relievers (about 16,000 in 2013) and a fivefold increase in the total number of overdose deaths from heroin (about 8,000 in 2013).
Heroin initiation is almost 20 times higher among individuals reporting nonmedical prescription pain reliever use. Among opioid-treatment seekers, the majority of individuals who initiated opioid use in the 1960s were first exposed to heroin. This is in contrast to those who initiated in the 2000s, among whom the majority were exposed to prescription opioids. For young adults, the main sources of opioids are family, friends … and clinicians.
Opioids are powerfully addictive and can be snorted, swallowed, smoked, or shot. Data from the START (Starting Treatment with Agonist Replacement Therapies) trial suggest that individuals who inject opioids are less likely to remain in treatment than noninjectors. This necessarily increases the risk for injectors to inject again and be at risk for overdose.
Opioid overdose can be reversed with the use of naloxone. But naloxone has to be immediately or quickly available for it to be effective. Take-home naloxone programs are located in 30 U.S. states and the District of Columbia. Since 1996, home naloxone programs have reported more than 26,000 drug overdose reversals with naloxone.
On Nov. 18, the Food and Drug Administration announced the approval of a naloxone nasal spray. Prior to this approval, naloxone was only available in the injectable form (syringe or auto-injector), and needle management likely posed a barrier to first responders. The nasal spray can be administered easily without medical training. Naloxone nasal spray administered in one nostril delivered approximately the same levels or higher of naloxone as a single dose of an FDA-approved naloxone intramuscular injection in approximately the same time frame.
It is one thing to save a heroin addict who has just overdosed with nasal naloxone followed by appropriate medical attention. It is entirely another to engage them in an effective drug treatment program.
If naloxone revives them, it is treatment that can save them.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.
We are in the midst of an epidemic of heroin and prescription opioid abuse. While the two do not completely explain each other, they are tragically and irrevocably linked.
From 2001 to 2013, we have observed a threefold increase in the total number of overdose deaths from opioid pain relievers (about 16,000 in 2013) and a fivefold increase in the total number of overdose deaths from heroin (about 8,000 in 2013).
Heroin initiation is almost 20 times higher among individuals reporting nonmedical prescription pain reliever use. Among opioid-treatment seekers, the majority of individuals who initiated opioid use in the 1960s were first exposed to heroin. This is in contrast to those who initiated in the 2000s, among whom the majority were exposed to prescription opioids. For young adults, the main sources of opioids are family, friends … and clinicians.
Opioids are powerfully addictive and can be snorted, swallowed, smoked, or shot. Data from the START (Starting Treatment with Agonist Replacement Therapies) trial suggest that individuals who inject opioids are less likely to remain in treatment than noninjectors. This necessarily increases the risk for injectors to inject again and be at risk for overdose.
Opioid overdose can be reversed with the use of naloxone. But naloxone has to be immediately or quickly available for it to be effective. Take-home naloxone programs are located in 30 U.S. states and the District of Columbia. Since 1996, home naloxone programs have reported more than 26,000 drug overdose reversals with naloxone.
On Nov. 18, the Food and Drug Administration announced the approval of a naloxone nasal spray. Prior to this approval, naloxone was only available in the injectable form (syringe or auto-injector), and needle management likely posed a barrier to first responders. The nasal spray can be administered easily without medical training. Naloxone nasal spray administered in one nostril delivered approximately the same levels or higher of naloxone as a single dose of an FDA-approved naloxone intramuscular injection in approximately the same time frame.
It is one thing to save a heroin addict who has just overdosed with nasal naloxone followed by appropriate medical attention. It is entirely another to engage them in an effective drug treatment program.
If naloxone revives them, it is treatment that can save them.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article.

ASH: Idelalisib plus standard therapy boosts survival in relapsed CLL
ORLANDO – Adding the PI3K inhibitor idelalisib to a standard regimen of bendamustine and rituximab significantly reduced the risk of both disease progression and death for patients with relapsed and/or refractory chronic lymphocytic leukemia, results of a phase III randomized trial showed.
At a median follow-up of 12 months, the primary endpoint of median progression-free survival was 23.1 months for patients treated with idelalisib (Zydelig), bendamustine, and rituximab (idel+BR), compared with 11.1 months for bendamustine and rituximab (BR) plus a placebo, reported Dr. Andrew Zelenetz of Memorial Sloan Kettering Cancer Center, New York.

“Median overall survival was not reached in either arm. However, there was a significant improvement in overall survival, with a 45% reduction in the risk of death [with idel+BR],” he said in a late-breaking abstract session at the annual meeting of the American Society of Hematology.
The trial was stopped early after a data review at the first planned interim analysis showed significant superiority for the three-drug combination.
The results were consistent across subgroups, including patients with high-risk features such as deletion 17p and mutated TP53 (del[17p]/TP53), unmutated immunoglobulin heavy chain variable region (IgHV), and treatment-refractory disease.
The rationale behind adding idelalisib, an inhibitor of the phosphatidylinositol-3 kinase (PI3K), is that signaling via the PI3K pathway is hyperactive and can be targeted, Dr. Zelenetz explained.
Study 115 was a phase III trial with accrual from June 2012 through August 2015. Investigators enrolled 416 patients with relapsed /refractory CLL and randomly assigned them to receive BR for six 28-day cycles of bendamustine (70 mg/m2 on days 1 and 2 of each cycle) and rituximab (375 mg/m2 for cycle 1, and 500 mg/m2 for cycles 2 through 6), plus either idelalisib 150 mg b.i.d. or placebo, each administered continuously until disease progression, intolerable toxicity, withdrawal of consent, or death.
The patients were stratified by mutational and disease status (refractory defined as CLL progression less than 6 months from completion of prior therapy, or relapsed CLL progression 6 months or more from completion of prior therapy.
The trial was halted early after the first planned interim analysis, which was conducted after 75% of the total number of 260 planned events of CLL progression or death from any cause had occurred. The data cutoff was June 15, 2015.
The intention-to-treat analysis included 207 patients assigned to idelalisib and 209 assigned to placebo. Three-fourths (76%) of the patients were male.
In all, 46% of patients had Rai stage III/IV disease. The median time since the completion of the last therapy was 16 months.
The proportions of patients with high-risk features included del(17p)/p53 mutation in 32.9%, unmutated IgHV in 83.2%, and treatment-refractory disease in 29.8%.
As noted, the median progression-free survival with idelalisib at a median follow-up of 12 months was 23.1 months vs. 11.1 months for placebo. That translated into a hazard ratio of 0.33 (P less than .0001).
Among patients with neither del(17p) nor TP53 mutations, the HR for progression was 0.22. Among patients with either del(17p) or a TP53 mutation, the HR was 0.50 (95% confidence intervals show statistical significance for both).
Overall response rates were 68% among patients who received idelalisib, and 45% for those who received placebo. There were five complete responses (2%) in the idelalisib group and none in the placebo group.
The idelalisib group also had a higher proportion of patients with a greater than 50% reduction in involved lymph nodes (96% vs. 61%), and had better organomegaly responses (spleen and liver) and hematologic responses (hemoglobin, neutrophils, and platelets).
Grade 3 or greater adverse events occurred in 93% of patients on idelalisib, compared with 76% of those on placebo. The proportion of patients with any serious adverse event was 66% vs. 44%, respectively.
Adverse events leading to drug dose reduction were seen in 11% of idelalisib-treated patients, compared with 6% of placebo controls, and therapy was discontinued in 26% vs. 13%, respectively.
Ten patients in the idelalisib arm and seven in the placebo arm died during the study.
Adverse events that occurred more commonly with idelalisib included neutropenia, pyrexia, diarrhea, febrile neutropenia, pneumonia, rash, and elevated liver enzymes.
Session moderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston asked Dr. Zelenetz how idelalisib plus BR stacked up to ibrutinib (Imbruvica) plus BR in this population.
Dr. Zelenetz noted that patients were excluded from the HELIOS trial of ibrutinib plus BR if they had del(17p). Comparing the subset of patients in Study 115 without del(17p) with patients in the ibrutinib study, “the results are virtually superimposable,” Dr. Zelenetz said, and “the two treatments are really remarkably similar.”
The overall survival benefit was larger in the HELIOS trial, Dr. Zelenetz noted, but that was largely because the trial allowed patients to cross over from placebo to the active drug.
Gilead Sciences funded Study 115. Dr. Zelenetz disclosed receiving research funding from the company and discussing off-label use of idelalisib for relapsed/refractory CLL.
ORLANDO – Adding the PI3K inhibitor idelalisib to a standard regimen of bendamustine and rituximab significantly reduced the risk of both disease progression and death for patients with relapsed and/or refractory chronic lymphocytic leukemia, results of a phase III randomized trial showed.
At a median follow-up of 12 months, the primary endpoint of median progression-free survival was 23.1 months for patients treated with idelalisib (Zydelig), bendamustine, and rituximab (idel+BR), compared with 11.1 months for bendamustine and rituximab (BR) plus a placebo, reported Dr. Andrew Zelenetz of Memorial Sloan Kettering Cancer Center, New York.
“Median overall survival was not reached in either arm. However, there was a significant improvement in overall survival, with a 45% reduction in the risk of death [with idel+BR],” he said in a late-breaking abstract session at the annual meeting of the American Society of Hematology.
The trial was stopped early after a data review at the first planned interim analysis showed significant superiority for the three-drug combination.
The results were consistent across subgroups, including patients with high-risk features such as deletion 17p and mutated TP53 (del[17p]/TP53), unmutated immunoglobulin heavy chain variable region (IgHV), and treatment-refractory disease.
The rationale behind adding idelalisib, an inhibitor of the phosphatidylinositol-3 kinase (PI3K), is that signaling via the PI3K pathway is hyperactive and can be targeted, Dr. Zelenetz explained.
Study 115 was a phase III trial with accrual from June 2012 through August 2015. Investigators enrolled 416 patients with relapsed /refractory CLL and randomly assigned them to receive BR for six 28-day cycles of bendamustine (70 mg/m2 on days 1 and 2 of each cycle) and rituximab (375 mg/m2 for cycle 1, and 500 mg/m2 for cycles 2 through 6), plus either idelalisib 150 mg b.i.d. or placebo, each administered continuously until disease progression, intolerable toxicity, withdrawal of consent, or death.
The patients were stratified by mutational and disease status (refractory defined as CLL progression less than 6 months from completion of prior therapy, or relapsed CLL progression 6 months or more from completion of prior therapy.
The trial was halted early after the first planned interim analysis, which was conducted after 75% of the total number of 260 planned events of CLL progression or death from any cause had occurred. The data cutoff was June 15, 2015.
The intention-to-treat analysis included 207 patients assigned to idelalisib and 209 assigned to placebo. Three-fourths (76%) of the patients were male.
In all, 46% of patients had Rai stage III/IV disease. The median time since the completion of the last therapy was 16 months.
The proportions of patients with high-risk features included del(17p)/p53 mutation in 32.9%, unmutated IgHV in 83.2%, and treatment-refractory disease in 29.8%.
As noted, the median progression-free survival with idelalisib at a median follow-up of 12 months was 23.1 months vs. 11.1 months for placebo. That translated into a hazard ratio of 0.33 (P less than .0001).
Among patients with neither del(17p) nor TP53 mutations, the HR for progression was 0.22. Among patients with either del(17p) or a TP53 mutation, the HR was 0.50 (95% confidence intervals show statistical significance for both).
Overall response rates were 68% among patients who received idelalisib, and 45% for those who received placebo. There were five complete responses (2%) in the idelalisib group and none in the placebo group.
The idelalisib group also had a higher proportion of patients with a greater than 50% reduction in involved lymph nodes (96% vs. 61%), and had better organomegaly responses (spleen and liver) and hematologic responses (hemoglobin, neutrophils, and platelets).
Grade 3 or greater adverse events occurred in 93% of patients on idelalisib, compared with 76% of those on placebo. The proportion of patients with any serious adverse event was 66% vs. 44%, respectively.
Adverse events leading to drug dose reduction were seen in 11% of idelalisib-treated patients, compared with 6% of placebo controls, and therapy was discontinued in 26% vs. 13%, respectively.
Ten patients in the idelalisib arm and seven in the placebo arm died during the study.
Adverse events that occurred more commonly with idelalisib included neutropenia, pyrexia, diarrhea, febrile neutropenia, pneumonia, rash, and elevated liver enzymes.
Session moderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston asked Dr. Zelenetz how idelalisib plus BR stacked up to ibrutinib (Imbruvica) plus BR in this population.
Dr. Zelenetz noted that patients were excluded from the HELIOS trial of ibrutinib plus BR if they had del(17p). Comparing the subset of patients in Study 115 without del(17p) with patients in the ibrutinib study, “the results are virtually superimposable,” Dr. Zelenetz said, and “the two treatments are really remarkably similar.”
The overall survival benefit was larger in the HELIOS trial, Dr. Zelenetz noted, but that was largely because the trial allowed patients to cross over from placebo to the active drug.
Gilead Sciences funded Study 115. Dr. Zelenetz disclosed receiving research funding from the company and discussing off-label use of idelalisib for relapsed/refractory CLL.
ORLANDO – Adding the PI3K inhibitor idelalisib to a standard regimen of bendamustine and rituximab significantly reduced the risk of both disease progression and death for patients with relapsed and/or refractory chronic lymphocytic leukemia, results of a phase III randomized trial showed.
At a median follow-up of 12 months, the primary endpoint of median progression-free survival was 23.1 months for patients treated with idelalisib (Zydelig), bendamustine, and rituximab (idel+BR), compared with 11.1 months for bendamustine and rituximab (BR) plus a placebo, reported Dr. Andrew Zelenetz of Memorial Sloan Kettering Cancer Center, New York.
“Median overall survival was not reached in either arm. However, there was a significant improvement in overall survival, with a 45% reduction in the risk of death [with idel+BR],” he said in a late-breaking abstract session at the annual meeting of the American Society of Hematology.
The trial was stopped early after a data review at the first planned interim analysis showed significant superiority for the three-drug combination.
The results were consistent across subgroups, including patients with high-risk features such as deletion 17p and mutated TP53 (del[17p]/TP53), unmutated immunoglobulin heavy chain variable region (IgHV), and treatment-refractory disease.
The rationale behind adding idelalisib, an inhibitor of the phosphatidylinositol-3 kinase (PI3K), is that signaling via the PI3K pathway is hyperactive and can be targeted, Dr. Zelenetz explained.
Study 115 was a phase III trial with accrual from June 2012 through August 2015. Investigators enrolled 416 patients with relapsed /refractory CLL and randomly assigned them to receive BR for six 28-day cycles of bendamustine (70 mg/m2 on days 1 and 2 of each cycle) and rituximab (375 mg/m2 for cycle 1, and 500 mg/m2 for cycles 2 through 6), plus either idelalisib 150 mg b.i.d. or placebo, each administered continuously until disease progression, intolerable toxicity, withdrawal of consent, or death.
The patients were stratified by mutational and disease status (refractory defined as CLL progression less than 6 months from completion of prior therapy, or relapsed CLL progression 6 months or more from completion of prior therapy.
The trial was halted early after the first planned interim analysis, which was conducted after 75% of the total number of 260 planned events of CLL progression or death from any cause had occurred. The data cutoff was June 15, 2015.
The intention-to-treat analysis included 207 patients assigned to idelalisib and 209 assigned to placebo. Three-fourths (76%) of the patients were male.
In all, 46% of patients had Rai stage III/IV disease. The median time since the completion of the last therapy was 16 months.
The proportions of patients with high-risk features included del(17p)/p53 mutation in 32.9%, unmutated IgHV in 83.2%, and treatment-refractory disease in 29.8%.
As noted, the median progression-free survival with idelalisib at a median follow-up of 12 months was 23.1 months vs. 11.1 months for placebo. That translated into a hazard ratio of 0.33 (P less than .0001).
Among patients with neither del(17p) nor TP53 mutations, the HR for progression was 0.22. Among patients with either del(17p) or a TP53 mutation, the HR was 0.50 (95% confidence intervals show statistical significance for both).
Overall response rates were 68% among patients who received idelalisib, and 45% for those who received placebo. There were five complete responses (2%) in the idelalisib group and none in the placebo group.
The idelalisib group also had a higher proportion of patients with a greater than 50% reduction in involved lymph nodes (96% vs. 61%), and had better organomegaly responses (spleen and liver) and hematologic responses (hemoglobin, neutrophils, and platelets).
Grade 3 or greater adverse events occurred in 93% of patients on idelalisib, compared with 76% of those on placebo. The proportion of patients with any serious adverse event was 66% vs. 44%, respectively.
Adverse events leading to drug dose reduction were seen in 11% of idelalisib-treated patients, compared with 6% of placebo controls, and therapy was discontinued in 26% vs. 13%, respectively.
Ten patients in the idelalisib arm and seven in the placebo arm died during the study.
Adverse events that occurred more commonly with idelalisib included neutropenia, pyrexia, diarrhea, febrile neutropenia, pneumonia, rash, and elevated liver enzymes.
Session moderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston asked Dr. Zelenetz how idelalisib plus BR stacked up to ibrutinib (Imbruvica) plus BR in this population.
Dr. Zelenetz noted that patients were excluded from the HELIOS trial of ibrutinib plus BR if they had del(17p). Comparing the subset of patients in Study 115 without del(17p) with patients in the ibrutinib study, “the results are virtually superimposable,” Dr. Zelenetz said, and “the two treatments are really remarkably similar.”
The overall survival benefit was larger in the HELIOS trial, Dr. Zelenetz noted, but that was largely because the trial allowed patients to cross over from placebo to the active drug.
Gilead Sciences funded Study 115. Dr. Zelenetz disclosed receiving research funding from the company and discussing off-label use of idelalisib for relapsed/refractory CLL.
AT ASH 2015
Key clinical point: Adding the PI3K inhibitor idelalisib to bendamustine and rituximab significantly improved survival of patients with relapsed/refractory CLL.
Major finding: Median progression-free survival was 23.1 months for patients treated with idelalisib, bendamustine, and rituximab, compared with 11.1 months for bendamustine and rituximab plus placebo.
Data source: Randomized, controlled trial in 416 patients with relapsed/refractory CLL. The trial was halted early for superior efficacy with idelalisib.
Disclosures: Gilead Sciences funded Study 115. Dr. Zelenetz disclosed receiving research funding from the company and discussing off-label use of idelalisib for relapsed/refractory CLL.

Policy Segment 5: Taking behavioral health pressure off primary care
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Who is in this video: Dr. Lawrence “Bopper” Deyton is the senior associate dean for clinical public health and a professor of medicine and of health policy and management at George Washington University School of Medicine, Washington; and Lauren Alfred, policy director at the Kennedy Forum.
Dr. Deyton: We’re in a bit of a transition now in terms of the structure of the health care enterprise, and how the incentives, and the funding, and how we’re organized? If we believe that the “triple aim” will work, that is, that the Affordable Care Act’s priorities – improving quality, decreasing cost, improving patient satisfaction in the system – then aren’t we on the cusp of potentially being able to put the distress diagnoses, finding those out, at the top, or close to the top, of the differential list when anybody comes in for any medical interaction?
At least in the literature that I know about – I’m thinking about chronic diseases – people come in with all kinds of behavioral, and emotional, and mental health distress issues, as well as serious mental illness. I think that we are missing opportunities with every interaction to ask about, to screen, and to have a treatment plan for those behavioral and mental health problems.
Now, aren’t we at the cusp of a reimbursement system that should reward for that and help catalyze our systems to change how they are structured?
Lauren Alfred: Absolutely. I think we’re having this conversation, fundamentally, for two reasons. One, because we recognize that the vast majority of patients are going to get this care in the primary care setting. That’s why we’re talking about mental health in primary care. There’s recognition of that by policy makers to say, “I have to address this problem across the continuum of care, but this is where I can make the biggest impact.” They’re driven by dollars, and so this is Then two, back to the idea of education, and the burden that we would be placing on primary care physicians and on our residents to be learning, there is only so much we can do, I would say, given the evidence of education in mental health for these physicians. It’ll only take them so far, and then at some point we have to talk about collaborative care and where we’re going to bring the specialists into the equation.
I think we get into this policy discussion, certainly with medicine, but also with teachers. It’s “How much more are we going to pile onto educators in terms of the things that they have to do for their students?” They have to be the social worker, the mom, the dad, and they have to be thinking about their mental health and about addiction. There are only so many [14:50] things we can expect our primary care physicians to do.
We need to bring them all up to a certain standard and at that point decide, “What are the payment structures, mechanisms, and teams that are in place that then carry us the rest of the way?” Making sure that there is a fundamental understanding of this difference between disorder and distress is certainly a good place to start.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Who is in this video: Dr. Lawrence “Bopper” Deyton is the senior associate dean for clinical public health and a professor of medicine and of health policy and management at George Washington University School of Medicine, Washington; and Lauren Alfred, policy director at the Kennedy Forum.
Dr. Deyton: We’re in a bit of a transition now in terms of the structure of the health care enterprise, and how the incentives, and the funding, and how we’re organized? If we believe that the “triple aim” will work, that is, that the Affordable Care Act’s priorities – improving quality, decreasing cost, improving patient satisfaction in the system – then aren’t we on the cusp of potentially being able to put the distress diagnoses, finding those out, at the top, or close to the top, of the differential list when anybody comes in for any medical interaction?
At least in the literature that I know about – I’m thinking about chronic diseases – people come in with all kinds of behavioral, and emotional, and mental health distress issues, as well as serious mental illness. I think that we are missing opportunities with every interaction to ask about, to screen, and to have a treatment plan for those behavioral and mental health problems.
Now, aren’t we at the cusp of a reimbursement system that should reward for that and help catalyze our systems to change how they are structured?
Lauren Alfred: Absolutely. I think we’re having this conversation, fundamentally, for two reasons. One, because we recognize that the vast majority of patients are going to get this care in the primary care setting. That’s why we’re talking about mental health in primary care. There’s recognition of that by policy makers to say, “I have to address this problem across the continuum of care, but this is where I can make the biggest impact.” They’re driven by dollars, and so this is Then two, back to the idea of education, and the burden that we would be placing on primary care physicians and on our residents to be learning, there is only so much we can do, I would say, given the evidence of education in mental health for these physicians. It’ll only take them so far, and then at some point we have to talk about collaborative care and where we’re going to bring the specialists into the equation.
I think we get into this policy discussion, certainly with medicine, but also with teachers. It’s “How much more are we going to pile onto educators in terms of the things that they have to do for their students?” They have to be the social worker, the mom, the dad, and they have to be thinking about their mental health and about addiction. There are only so many [14:50] things we can expect our primary care physicians to do.
We need to bring them all up to a certain standard and at that point decide, “What are the payment structures, mechanisms, and teams that are in place that then carry us the rest of the way?” Making sure that there is a fundamental understanding of this difference between disorder and distress is certainly a good place to start.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Who is in this video: Dr. Lawrence “Bopper” Deyton is the senior associate dean for clinical public health and a professor of medicine and of health policy and management at George Washington University School of Medicine, Washington; and Lauren Alfred, policy director at the Kennedy Forum.
Dr. Deyton: We’re in a bit of a transition now in terms of the structure of the health care enterprise, and how the incentives, and the funding, and how we’re organized? If we believe that the “triple aim” will work, that is, that the Affordable Care Act’s priorities – improving quality, decreasing cost, improving patient satisfaction in the system – then aren’t we on the cusp of potentially being able to put the distress diagnoses, finding those out, at the top, or close to the top, of the differential list when anybody comes in for any medical interaction?
At least in the literature that I know about – I’m thinking about chronic diseases – people come in with all kinds of behavioral, and emotional, and mental health distress issues, as well as serious mental illness. I think that we are missing opportunities with every interaction to ask about, to screen, and to have a treatment plan for those behavioral and mental health problems.
Now, aren’t we at the cusp of a reimbursement system that should reward for that and help catalyze our systems to change how they are structured?
Lauren Alfred: Absolutely. I think we’re having this conversation, fundamentally, for two reasons. One, because we recognize that the vast majority of patients are going to get this care in the primary care setting. That’s why we’re talking about mental health in primary care. There’s recognition of that by policy makers to say, “I have to address this problem across the continuum of care, but this is where I can make the biggest impact.” They’re driven by dollars, and so this is Then two, back to the idea of education, and the burden that we would be placing on primary care physicians and on our residents to be learning, there is only so much we can do, I would say, given the evidence of education in mental health for these physicians. It’ll only take them so far, and then at some point we have to talk about collaborative care and where we’re going to bring the specialists into the equation.
I think we get into this policy discussion, certainly with medicine, but also with teachers. It’s “How much more are we going to pile onto educators in terms of the things that they have to do for their students?” They have to be the social worker, the mom, the dad, and they have to be thinking about their mental health and about addiction. There are only so many [14:50] things we can expect our primary care physicians to do.
We need to bring them all up to a certain standard and at that point decide, “What are the payment structures, mechanisms, and teams that are in place that then carry us the rest of the way?” Making sure that there is a fundamental understanding of this difference between disorder and distress is certainly a good place to start.
Clinical Segment 5: How candid should you be in your dictated notes?
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
People in this video: Whitney McKnight, cohost and producer of Mental Health Consult; Dr. Lorenzo Norris, editorial board member of Clinical Psychiatry News and cohost of Mental Health Consult, and an assistant professor of psychiatry and behavioral sciences, assistant dean of student affairs, and the medical director of psychiatric and behavioral services at George Washington University Hospital, Washington; Dr. Lillian Beard, pediatrician with Children’s National Hospital Network, Washington, and a Pediatric News editorial board member; Dr. David Pickar, adjunct professor of psychiatry at Johns Hopkins University School of Medicine in Baltimore and at the Uniformed Services University of the Health Sciences in Bethesda, Md.
Dr. Lillian Beard: In fact, the written note is sometimes inhibiting to the communication because we are each so aware of what we put in writing [20:00] to even send to a colleague. We can have a conversation, a dialogue about the patient and glean a lot more information.
Whitney: Do you mean you actually omit things on purpose?
Dr. Beard: It is not about omitting, it is about how you state it, because first of all our patients will eventually have access to everything we have written. They are getting more and more access with patient portals. I find for instance even in my notes, so that even if my colleagues in my practice were to see this patient they would know there are certain code terms. I say "high risk for" or I will not do that now because much of that will go to the patient portal. I will come up with other kinds of words so they know to check with me to find out what I meant about that. There are some toxic families, I do not write "toxic family" in my notes.
Dr. Pickar: I agree with you. I could not agree with you more, and in psychiatry, actually there is some protection against not having to share notes. I do not know if you are aware of that. Medical records for sure, but your notes can remain private about a patient. Maybe you know more about that. Help me with that.
Dr. Norris: It is a little bit. Once you get into the problem of keeping dual records, which becomes an issue. You cannot do that. Particularly with electronic medical records, this is now one that patients do and should have access to it. It is a medical legal document and many different people can look at that, so as clinicians, we must be aware, not just for our patients and ourselves, what we put in the note. You cannot have team-based care unless you actually know your teammate. When I am working with a clinician, I want to know their thought process. I want to know a little bit of their philosophy. Do they like stimulants, do they believe in them? When they are also treating, do they screen for first-episode psychosis? Is it on their radar? Are they screening for comorbid depression and bipolar disorder?
Whitney: Should that be legislated or should that be the individual choice of the practice?
Dr. Norris: You can legislate it all you want. This gets into the duty that we as clinicians have to our patients and how we treat them. The first law is to do no harm. I am not saying anything fancy. This is just basic, solid medical care, which takes a certain amount of time, which is not usually 15 minutes.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
People in this video: Whitney McKnight, cohost and producer of Mental Health Consult; Dr. Lorenzo Norris, editorial board member of Clinical Psychiatry News and cohost of Mental Health Consult, and an assistant professor of psychiatry and behavioral sciences, assistant dean of student affairs, and the medical director of psychiatric and behavioral services at George Washington University Hospital, Washington; Dr. Lillian Beard, pediatrician with Children’s National Hospital Network, Washington, and a Pediatric News editorial board member; Dr. David Pickar, adjunct professor of psychiatry at Johns Hopkins University School of Medicine in Baltimore and at the Uniformed Services University of the Health Sciences in Bethesda, Md.
Dr. Lillian Beard: In fact, the written note is sometimes inhibiting to the communication because we are each so aware of what we put in writing [20:00] to even send to a colleague. We can have a conversation, a dialogue about the patient and glean a lot more information.
Whitney: Do you mean you actually omit things on purpose?
Dr. Beard: It is not about omitting, it is about how you state it, because first of all our patients will eventually have access to everything we have written. They are getting more and more access with patient portals. I find for instance even in my notes, so that even if my colleagues in my practice were to see this patient they would know there are certain code terms. I say "high risk for" or I will not do that now because much of that will go to the patient portal. I will come up with other kinds of words so they know to check with me to find out what I meant about that. There are some toxic families, I do not write "toxic family" in my notes.
Dr. Pickar: I agree with you. I could not agree with you more, and in psychiatry, actually there is some protection against not having to share notes. I do not know if you are aware of that. Medical records for sure, but your notes can remain private about a patient. Maybe you know more about that. Help me with that.
Dr. Norris: It is a little bit. Once you get into the problem of keeping dual records, which becomes an issue. You cannot do that. Particularly with electronic medical records, this is now one that patients do and should have access to it. It is a medical legal document and many different people can look at that, so as clinicians, we must be aware, not just for our patients and ourselves, what we put in the note. You cannot have team-based care unless you actually know your teammate. When I am working with a clinician, I want to know their thought process. I want to know a little bit of their philosophy. Do they like stimulants, do they believe in them? When they are also treating, do they screen for first-episode psychosis? Is it on their radar? Are they screening for comorbid depression and bipolar disorder?
Whitney: Should that be legislated or should that be the individual choice of the practice?
Dr. Norris: You can legislate it all you want. This gets into the duty that we as clinicians have to our patients and how we treat them. The first law is to do no harm. I am not saying anything fancy. This is just basic, solid medical care, which takes a certain amount of time, which is not usually 15 minutes.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
People in this video: Whitney McKnight, cohost and producer of Mental Health Consult; Dr. Lorenzo Norris, editorial board member of Clinical Psychiatry News and cohost of Mental Health Consult, and an assistant professor of psychiatry and behavioral sciences, assistant dean of student affairs, and the medical director of psychiatric and behavioral services at George Washington University Hospital, Washington; Dr. Lillian Beard, pediatrician with Children’s National Hospital Network, Washington, and a Pediatric News editorial board member; Dr. David Pickar, adjunct professor of psychiatry at Johns Hopkins University School of Medicine in Baltimore and at the Uniformed Services University of the Health Sciences in Bethesda, Md.
Dr. Lillian Beard: In fact, the written note is sometimes inhibiting to the communication because we are each so aware of what we put in writing [20:00] to even send to a colleague. We can have a conversation, a dialogue about the patient and glean a lot more information.
Whitney: Do you mean you actually omit things on purpose?
Dr. Beard: It is not about omitting, it is about how you state it, because first of all our patients will eventually have access to everything we have written. They are getting more and more access with patient portals. I find for instance even in my notes, so that even if my colleagues in my practice were to see this patient they would know there are certain code terms. I say "high risk for" or I will not do that now because much of that will go to the patient portal. I will come up with other kinds of words so they know to check with me to find out what I meant about that. There are some toxic families, I do not write "toxic family" in my notes.
Dr. Pickar: I agree with you. I could not agree with you more, and in psychiatry, actually there is some protection against not having to share notes. I do not know if you are aware of that. Medical records for sure, but your notes can remain private about a patient. Maybe you know more about that. Help me with that.
Dr. Norris: It is a little bit. Once you get into the problem of keeping dual records, which becomes an issue. You cannot do that. Particularly with electronic medical records, this is now one that patients do and should have access to it. It is a medical legal document and many different people can look at that, so as clinicians, we must be aware, not just for our patients and ourselves, what we put in the note. You cannot have team-based care unless you actually know your teammate. When I am working with a clinician, I want to know their thought process. I want to know a little bit of their philosophy. Do they like stimulants, do they believe in them? When they are also treating, do they screen for first-episode psychosis? Is it on their radar? Are they screening for comorbid depression and bipolar disorder?
Whitney: Should that be legislated or should that be the individual choice of the practice?
Dr. Norris: You can legislate it all you want. This gets into the duty that we as clinicians have to our patients and how we treat them. The first law is to do no harm. I am not saying anything fancy. This is just basic, solid medical care, which takes a certain amount of time, which is not usually 15 minutes.
Policy Segment 4: What is ‘enough’ team care training?
Who is in this video: Dr. James Griffith, the Leon M. Yochelson Professor of Psychiatry and Behavioral Sciences, and chair of psychiatry and psychosomatic medicine at George Washington University School of Medicine. Lauren Alfred is policy director at the Kennedy Forum.
Dr. Griffith: There’s also the training of our educators. There has been too much focus simply on counting symptoms. If you have sleep problems, if you have no energy, if you’re not enjoying things, then you have depression. That kind of definition of depression catches too many different things that shouldn’t be addressed in the same way.
“There has been too much focus simply on counting symptoms.”
– Dr. James GriffithThat’s distress. In primary care there are a lot of people with, often, very malignant mood disorders – major depression, bipolar disorder. They generally need not just a prescription; they need a program. Medication may be part of it. It also needs to address lifestyle. It also needs to address relationship problems. Many different things, which if done well can help people live good lives without being held hostage to having a psychiatric diagnosis.
When I said distress – these are not mental illnesses, but yet it all gets called depression. Often in the public discussions, it’s treated – “We just need to identify the depressed patients, give them medications.” That serves few people well. There are ways of doing very effective, targeted work depending upon, initially, an accurate assessment of what is the problem. Is it demoralization? Is it grief? Is this a relationship that is abusive, for example?
Now the other piece - and this is a big one. This is one that we’ve got to figure out how to address, and this is no different in the Middle East, where I’m doing work, as it is here. People come into primary care complaining of dizziness, headaches, physical pain problems, not sleeping well, fatigue. Wherever in the world you’ll go, what they get is a lot of tests, vitamins – not identifying or addressing that underneath this there is psychological distress or a mental illness driving it.
This puts a focus on detection and formulation of the problem. You’re right, the doctors aren’t going to do all the treatment, but this is where the doctor pretty much does have to do, on the front end, the identification. That’s our training issue.
Who is in this video: Dr. James Griffith, the Leon M. Yochelson Professor of Psychiatry and Behavioral Sciences, and chair of psychiatry and psychosomatic medicine at George Washington University School of Medicine. Lauren Alfred is policy director at the Kennedy Forum.
Dr. Griffith: There’s also the training of our educators. There has been too much focus simply on counting symptoms. If you have sleep problems, if you have no energy, if you’re not enjoying things, then you have depression. That kind of definition of depression catches too many different things that shouldn’t be addressed in the same way.
“There has been too much focus simply on counting symptoms.”
– Dr. James GriffithThat’s distress. In primary care there are a lot of people with, often, very malignant mood disorders – major depression, bipolar disorder. They generally need not just a prescription; they need a program. Medication may be part of it. It also needs to address lifestyle. It also needs to address relationship problems. Many different things, which if done well can help people live good lives without being held hostage to having a psychiatric diagnosis.
When I said distress – these are not mental illnesses, but yet it all gets called depression. Often in the public discussions, it’s treated – “We just need to identify the depressed patients, give them medications.” That serves few people well. There are ways of doing very effective, targeted work depending upon, initially, an accurate assessment of what is the problem. Is it demoralization? Is it grief? Is this a relationship that is abusive, for example?
Now the other piece - and this is a big one. This is one that we’ve got to figure out how to address, and this is no different in the Middle East, where I’m doing work, as it is here. People come into primary care complaining of dizziness, headaches, physical pain problems, not sleeping well, fatigue. Wherever in the world you’ll go, what they get is a lot of tests, vitamins – not identifying or addressing that underneath this there is psychological distress or a mental illness driving it.
This puts a focus on detection and formulation of the problem. You’re right, the doctors aren’t going to do all the treatment, but this is where the doctor pretty much does have to do, on the front end, the identification. That’s our training issue.
Who is in this video: Dr. James Griffith, the Leon M. Yochelson Professor of Psychiatry and Behavioral Sciences, and chair of psychiatry and psychosomatic medicine at George Washington University School of Medicine. Lauren Alfred is policy director at the Kennedy Forum.
Dr. Griffith: There’s also the training of our educators. There has been too much focus simply on counting symptoms. If you have sleep problems, if you have no energy, if you’re not enjoying things, then you have depression. That kind of definition of depression catches too many different things that shouldn’t be addressed in the same way.
“There has been too much focus simply on counting symptoms.”
– Dr. James GriffithThat’s distress. In primary care there are a lot of people with, often, very malignant mood disorders – major depression, bipolar disorder. They generally need not just a prescription; they need a program. Medication may be part of it. It also needs to address lifestyle. It also needs to address relationship problems. Many different things, which if done well can help people live good lives without being held hostage to having a psychiatric diagnosis.
When I said distress – these are not mental illnesses, but yet it all gets called depression. Often in the public discussions, it’s treated – “We just need to identify the depressed patients, give them medications.” That serves few people well. There are ways of doing very effective, targeted work depending upon, initially, an accurate assessment of what is the problem. Is it demoralization? Is it grief? Is this a relationship that is abusive, for example?
Now the other piece - and this is a big one. This is one that we’ve got to figure out how to address, and this is no different in the Middle East, where I’m doing work, as it is here. People come into primary care complaining of dizziness, headaches, physical pain problems, not sleeping well, fatigue. Wherever in the world you’ll go, what they get is a lot of tests, vitamins – not identifying or addressing that underneath this there is psychological distress or a mental illness driving it.
This puts a focus on detection and formulation of the problem. You’re right, the doctors aren’t going to do all the treatment, but this is where the doctor pretty much does have to do, on the front end, the identification. That’s our training issue.