User login
Insecticide can cause NHL, experts say

Photo by John Messina
The insecticide gamma-hexachlorocyclohexane (lindane) is carcinogenic, according to experts from the International Agency for Research on Cancer (IARC), the specialized cancer agency of the World Health Organization.
The experts said they found sufficient evidence that lindane, which is banned or restricted in most countries, can cause non-Hodgkin lymphoma (NHL).
The group also discovered that 2 other chemicals might cause NHL.
The evidence suggests the insecticide dichlorodiphenyltrichloroethane (DDT) is probably carcinogenic, and the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) is possibly carcinogenic.
A summary of these findings is available in The Lancet Oncology, and the experts’ detailed assessments will be published as Volume 113 of the IARC Monographs.
The group, which consisted of 26 experts convened by the IARC Monographs Programme, reviewed the latest scientific literature on lindane, DDT, and 2,4-D and used their findings to classify these 3 chemicals according to carcinogenicity.
The classification (Group 1, Group 2A, etc.) indicates the strength of the evidence that a substance causes cancer, not the level of risk associated with exposure. The Monographs Programme identifies cancer hazards even when risks are very low at current exposure levels, because new uses or unforeseen exposures could engender risks that are significantly higher.
Lindane
The experts classified lindane as carcinogenic to humans (Group 1), saying they found sufficient evidence that it can cause NHL. Large epidemiological studies of agricultural exposures in the US and Canada showed a 60% increased risk of NHL in people exposed to lindane.
Lindane has been used extensively for insect control, including in agriculture and for the treatment of human lice and scabies. High exposures have occurred among agricultural workers and pesticide applicators. However, the use of lindane is now banned or restricted in most countries.
DDT
The experts classified DDT as probably carcinogenic to humans (Group 2A), saying they found sufficient evidence that DDT causes cancer in experimental animals and limited evidence of DDT’s carcinogenicity in humans.
Epidemiological studies have shown positive associations between exposure to DDT and NHL, testicular cancer, and liver cancer.
There was also strong experimental evidence that DDT can suppress the immune system and disrupt sex hormones. However, overall, there was no association between breast cancer and DDT levels measured in samples of blood or fat.
DDT was introduced for the control of insect-borne diseases during World War II and was later applied widely to eradicate malaria and in agriculture. Most uses of DDT were banned in the 1970s. However, DDT and its breakdown products are highly persistent and can be found in the environment and in animal and human tissues throughout the world.
Exposure to DDT still occurs, mainly through diet. The remaining and essential use of DDT is for disease vector control, mainly for malaria. This use is strictly restricted under the Stockholm Convention.
2,4-D
The experts classified 2,4-D as possibly carcinogenic to humans (Group 2B), saying they had inadequate evidence in humans and limited evidence in experimental animals.
There is strong evidence that 2,4-D induces oxidative stress and moderate evidence that 2,4-D causes immunosuppression, based on in vivo and in vitro studies. However, epidemiological studies did not show strong or consistent increases in the risk of NHL or other cancers in relation to 2,4-D exposure.
Since its introduction in 1945, 2,4-D has been widely used to control weeds in agriculture, forestry, and urban and residential settings. Occupational exposures to 2,4-D can occur during manufacturing and application, and the general population can be exposed through food, water, dust, or residential application, and during spraying. ![]()
Photo by John Messina
The insecticide gamma-hexachlorocyclohexane (lindane) is carcinogenic, according to experts from the International Agency for Research on Cancer (IARC), the specialized cancer agency of the World Health Organization.
The experts said they found sufficient evidence that lindane, which is banned or restricted in most countries, can cause non-Hodgkin lymphoma (NHL).
The group also discovered that 2 other chemicals might cause NHL.
The evidence suggests the insecticide dichlorodiphenyltrichloroethane (DDT) is probably carcinogenic, and the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) is possibly carcinogenic.
A summary of these findings is available in The Lancet Oncology, and the experts’ detailed assessments will be published as Volume 113 of the IARC Monographs.
The group, which consisted of 26 experts convened by the IARC Monographs Programme, reviewed the latest scientific literature on lindane, DDT, and 2,4-D and used their findings to classify these 3 chemicals according to carcinogenicity.
The classification (Group 1, Group 2A, etc.) indicates the strength of the evidence that a substance causes cancer, not the level of risk associated with exposure. The Monographs Programme identifies cancer hazards even when risks are very low at current exposure levels, because new uses or unforeseen exposures could engender risks that are significantly higher.
Lindane
The experts classified lindane as carcinogenic to humans (Group 1), saying they found sufficient evidence that it can cause NHL. Large epidemiological studies of agricultural exposures in the US and Canada showed a 60% increased risk of NHL in people exposed to lindane.
Lindane has been used extensively for insect control, including in agriculture and for the treatment of human lice and scabies. High exposures have occurred among agricultural workers and pesticide applicators. However, the use of lindane is now banned or restricted in most countries.
DDT
The experts classified DDT as probably carcinogenic to humans (Group 2A), saying they found sufficient evidence that DDT causes cancer in experimental animals and limited evidence of DDT’s carcinogenicity in humans.
Epidemiological studies have shown positive associations between exposure to DDT and NHL, testicular cancer, and liver cancer.
There was also strong experimental evidence that DDT can suppress the immune system and disrupt sex hormones. However, overall, there was no association between breast cancer and DDT levels measured in samples of blood or fat.
DDT was introduced for the control of insect-borne diseases during World War II and was later applied widely to eradicate malaria and in agriculture. Most uses of DDT were banned in the 1970s. However, DDT and its breakdown products are highly persistent and can be found in the environment and in animal and human tissues throughout the world.
Exposure to DDT still occurs, mainly through diet. The remaining and essential use of DDT is for disease vector control, mainly for malaria. This use is strictly restricted under the Stockholm Convention.
2,4-D
The experts classified 2,4-D as possibly carcinogenic to humans (Group 2B), saying they had inadequate evidence in humans and limited evidence in experimental animals.
There is strong evidence that 2,4-D induces oxidative stress and moderate evidence that 2,4-D causes immunosuppression, based on in vivo and in vitro studies. However, epidemiological studies did not show strong or consistent increases in the risk of NHL or other cancers in relation to 2,4-D exposure.
Since its introduction in 1945, 2,4-D has been widely used to control weeds in agriculture, forestry, and urban and residential settings. Occupational exposures to 2,4-D can occur during manufacturing and application, and the general population can be exposed through food, water, dust, or residential application, and during spraying. ![]()
Photo by John Messina
The insecticide gamma-hexachlorocyclohexane (lindane) is carcinogenic, according to experts from the International Agency for Research on Cancer (IARC), the specialized cancer agency of the World Health Organization.
The experts said they found sufficient evidence that lindane, which is banned or restricted in most countries, can cause non-Hodgkin lymphoma (NHL).
The group also discovered that 2 other chemicals might cause NHL.
The evidence suggests the insecticide dichlorodiphenyltrichloroethane (DDT) is probably carcinogenic, and the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) is possibly carcinogenic.
A summary of these findings is available in The Lancet Oncology, and the experts’ detailed assessments will be published as Volume 113 of the IARC Monographs.
The group, which consisted of 26 experts convened by the IARC Monographs Programme, reviewed the latest scientific literature on lindane, DDT, and 2,4-D and used their findings to classify these 3 chemicals according to carcinogenicity.
The classification (Group 1, Group 2A, etc.) indicates the strength of the evidence that a substance causes cancer, not the level of risk associated with exposure. The Monographs Programme identifies cancer hazards even when risks are very low at current exposure levels, because new uses or unforeseen exposures could engender risks that are significantly higher.
Lindane
The experts classified lindane as carcinogenic to humans (Group 1), saying they found sufficient evidence that it can cause NHL. Large epidemiological studies of agricultural exposures in the US and Canada showed a 60% increased risk of NHL in people exposed to lindane.
Lindane has been used extensively for insect control, including in agriculture and for the treatment of human lice and scabies. High exposures have occurred among agricultural workers and pesticide applicators. However, the use of lindane is now banned or restricted in most countries.
DDT
The experts classified DDT as probably carcinogenic to humans (Group 2A), saying they found sufficient evidence that DDT causes cancer in experimental animals and limited evidence of DDT’s carcinogenicity in humans.
Epidemiological studies have shown positive associations between exposure to DDT and NHL, testicular cancer, and liver cancer.
There was also strong experimental evidence that DDT can suppress the immune system and disrupt sex hormones. However, overall, there was no association between breast cancer and DDT levels measured in samples of blood or fat.
DDT was introduced for the control of insect-borne diseases during World War II and was later applied widely to eradicate malaria and in agriculture. Most uses of DDT were banned in the 1970s. However, DDT and its breakdown products are highly persistent and can be found in the environment and in animal and human tissues throughout the world.
Exposure to DDT still occurs, mainly through diet. The remaining and essential use of DDT is for disease vector control, mainly for malaria. This use is strictly restricted under the Stockholm Convention.
2,4-D
The experts classified 2,4-D as possibly carcinogenic to humans (Group 2B), saying they had inadequate evidence in humans and limited evidence in experimental animals.
There is strong evidence that 2,4-D induces oxidative stress and moderate evidence that 2,4-D causes immunosuppression, based on in vivo and in vitro studies. However, epidemiological studies did not show strong or consistent increases in the risk of NHL or other cancers in relation to 2,4-D exposure.
Since its introduction in 1945, 2,4-D has been widely used to control weeds in agriculture, forestry, and urban and residential settings. Occupational exposures to 2,4-D can occur during manufacturing and application, and the general population can be exposed through food, water, dust, or residential application, and during spraying. ![]()
WCD: Secukinumab shows effectiveness for nail, palmoplantar psoriasis
VANCOUVER, B.C. – The interleukin-17A inhibitor secukinumab demonstrated the greatest improvement in nail psoriasis ever reported from a randomized, placebo-controlled trial in the phase IIIb TRANSFIGURE study, Dr. Kristian Reich reported at the World Congress of Dermatology.
At 198 patients, TRANSFIGURE is the largest-ever prospective study in patients with moderate to severe chronic plaque psoriasis and significant nail involvement. And while only the 16-week results are available thus far, when TRANSFIGURE is completed after a planned 132 weeks of treatment, it will also be the longest-ever study in the treatment of nail psoriasis, noted Dr. Reich, a dermatologist in group practice in Hamburg, Germany.

Elsewhere at WCD 2015, Dr. Alice B. Gottlieb presented the week 16 results of the phase IIIb GESTURE study, in which 205 psoriasis patients with moderate to severe psoriasis of the palms and soles were randomized to subcutaneous secukinumab (Cosentyx) at 150 or 300 mg or placebo. Dosing was weekly for the first 5 weeks and monthly thereafter.
The primary endpoint, a palmoplantar Investigator’s Global Assessment scale score of 0 or 1 – clear or almost clear – at week 16 was 33.3% with secukinumab at 300 mg, 22.1% at 150 mg, and 1.5% with placebo. The average reduction in palmoplantar PASI (Psoriasis Area Severity Index) score from baseline was 54.6% with high-dose and 35.3% with low-dose secukinumab, compared with 4.1% in placebo-treated controls, reported Dr. Gottlieb, professor and chair of dermatology at Tufts University, Boston.
Like the TRANSFIGURE trial, GESTURE will continue for 132 weeks, with the initial placebo-treated controls being randomized to secukinumab at 150 or 300 mg after week 16.
Dr. Reich reported that by 16 weeks in TRANSFIGURE, mean scores on the Nail Psoriasis Severity Index had improved by 45.3%, compared with baseline, in patients on secukinumab 300 mg, 37.9% in those on secukinumab 150 mg, and 10.8% with placebo.
The results on the skin were dramatic: a PASI 75 rate of 87.1% with secukinumab 300 mg, 77% with secukinumab 150 mg, and 5.1% with placebo. The PASI 100 response rate – meaning totally clear skin – was 31.9% with high-dose and 25.2% with lower-dose secukinumab, while there was a zero PASI 100 rate in controls.
The only adverse events more common than with placebo were nasopharyngitis and upper respiratory infections.
Dr. Reich predicted that as the ongoing TRANSFIGURE study continues well beyond 16 weeks, the nail psoriasis response rates will climb, since nails are so slow growing.
TRANSFIGURE and GESTURE are sponsored by Novartis, which markets secukinumab. Dr. Reich and Dr. Gottlieb reported having financial relationships with Novartis and numerous other pharmaceutical companies.
VANCOUVER, B.C. – The interleukin-17A inhibitor secukinumab demonstrated the greatest improvement in nail psoriasis ever reported from a randomized, placebo-controlled trial in the phase IIIb TRANSFIGURE study, Dr. Kristian Reich reported at the World Congress of Dermatology.
At 198 patients, TRANSFIGURE is the largest-ever prospective study in patients with moderate to severe chronic plaque psoriasis and significant nail involvement. And while only the 16-week results are available thus far, when TRANSFIGURE is completed after a planned 132 weeks of treatment, it will also be the longest-ever study in the treatment of nail psoriasis, noted Dr. Reich, a dermatologist in group practice in Hamburg, Germany.
Elsewhere at WCD 2015, Dr. Alice B. Gottlieb presented the week 16 results of the phase IIIb GESTURE study, in which 205 psoriasis patients with moderate to severe psoriasis of the palms and soles were randomized to subcutaneous secukinumab (Cosentyx) at 150 or 300 mg or placebo. Dosing was weekly for the first 5 weeks and monthly thereafter.
The primary endpoint, a palmoplantar Investigator’s Global Assessment scale score of 0 or 1 – clear or almost clear – at week 16 was 33.3% with secukinumab at 300 mg, 22.1% at 150 mg, and 1.5% with placebo. The average reduction in palmoplantar PASI (Psoriasis Area Severity Index) score from baseline was 54.6% with high-dose and 35.3% with low-dose secukinumab, compared with 4.1% in placebo-treated controls, reported Dr. Gottlieb, professor and chair of dermatology at Tufts University, Boston.
Like the TRANSFIGURE trial, GESTURE will continue for 132 weeks, with the initial placebo-treated controls being randomized to secukinumab at 150 or 300 mg after week 16.
Dr. Reich reported that by 16 weeks in TRANSFIGURE, mean scores on the Nail Psoriasis Severity Index had improved by 45.3%, compared with baseline, in patients on secukinumab 300 mg, 37.9% in those on secukinumab 150 mg, and 10.8% with placebo.
The results on the skin were dramatic: a PASI 75 rate of 87.1% with secukinumab 300 mg, 77% with secukinumab 150 mg, and 5.1% with placebo. The PASI 100 response rate – meaning totally clear skin – was 31.9% with high-dose and 25.2% with lower-dose secukinumab, while there was a zero PASI 100 rate in controls.
The only adverse events more common than with placebo were nasopharyngitis and upper respiratory infections.
Dr. Reich predicted that as the ongoing TRANSFIGURE study continues well beyond 16 weeks, the nail psoriasis response rates will climb, since nails are so slow growing.
TRANSFIGURE and GESTURE are sponsored by Novartis, which markets secukinumab. Dr. Reich and Dr. Gottlieb reported having financial relationships with Novartis and numerous other pharmaceutical companies.
VANCOUVER, B.C. – The interleukin-17A inhibitor secukinumab demonstrated the greatest improvement in nail psoriasis ever reported from a randomized, placebo-controlled trial in the phase IIIb TRANSFIGURE study, Dr. Kristian Reich reported at the World Congress of Dermatology.
At 198 patients, TRANSFIGURE is the largest-ever prospective study in patients with moderate to severe chronic plaque psoriasis and significant nail involvement. And while only the 16-week results are available thus far, when TRANSFIGURE is completed after a planned 132 weeks of treatment, it will also be the longest-ever study in the treatment of nail psoriasis, noted Dr. Reich, a dermatologist in group practice in Hamburg, Germany.
Elsewhere at WCD 2015, Dr. Alice B. Gottlieb presented the week 16 results of the phase IIIb GESTURE study, in which 205 psoriasis patients with moderate to severe psoriasis of the palms and soles were randomized to subcutaneous secukinumab (Cosentyx) at 150 or 300 mg or placebo. Dosing was weekly for the first 5 weeks and monthly thereafter.
The primary endpoint, a palmoplantar Investigator’s Global Assessment scale score of 0 or 1 – clear or almost clear – at week 16 was 33.3% with secukinumab at 300 mg, 22.1% at 150 mg, and 1.5% with placebo. The average reduction in palmoplantar PASI (Psoriasis Area Severity Index) score from baseline was 54.6% with high-dose and 35.3% with low-dose secukinumab, compared with 4.1% in placebo-treated controls, reported Dr. Gottlieb, professor and chair of dermatology at Tufts University, Boston.
Like the TRANSFIGURE trial, GESTURE will continue for 132 weeks, with the initial placebo-treated controls being randomized to secukinumab at 150 or 300 mg after week 16.
Dr. Reich reported that by 16 weeks in TRANSFIGURE, mean scores on the Nail Psoriasis Severity Index had improved by 45.3%, compared with baseline, in patients on secukinumab 300 mg, 37.9% in those on secukinumab 150 mg, and 10.8% with placebo.
The results on the skin were dramatic: a PASI 75 rate of 87.1% with secukinumab 300 mg, 77% with secukinumab 150 mg, and 5.1% with placebo. The PASI 100 response rate – meaning totally clear skin – was 31.9% with high-dose and 25.2% with lower-dose secukinumab, while there was a zero PASI 100 rate in controls.
The only adverse events more common than with placebo were nasopharyngitis and upper respiratory infections.
Dr. Reich predicted that as the ongoing TRANSFIGURE study continues well beyond 16 weeks, the nail psoriasis response rates will climb, since nails are so slow growing.
TRANSFIGURE and GESTURE are sponsored by Novartis, which markets secukinumab. Dr. Reich and Dr. Gottlieb reported having financial relationships with Novartis and numerous other pharmaceutical companies.
AT WCD 2015
Key clinical point: Two phase IIIb trials show secukinumab at 300 mg is the most effective drug ever formally studied for nail or palmoplantar psoriasis.
Major finding: At 16 weeks, secukinumab at 300 mg improved nail psoriasis by 45.3% and palmoplantar psoriasis by 33.3%.
Data source: The phase IIIb TRANSFIGURE and GESTURE studies, ongoing randomized, prospective, initially double-blind studies in which 198 patients with significant nail psoriasis and 205 with palmoplantar psoriasis received secukinumab at 150 or 300 mg or placebo. Both studies will continue out to 132 weeks.
Disclosures: TRANSFIGURE and GESTURE are sponsored by Novartis, which markets secukinumab. Dr. Reich and Dr. Gottlieb reported having financial relationships with Novartis and numerous other pharmaceutical companies.

EC approves edoxaban for patients with VTE, NVAF

Image by Andre E.X. Brown
The European Commission (EC) has approved edoxaban (Lixiana), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF who have one or more risk factors for stroke or systemic embolism, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke, or transient ischemic attack.
The EC’s decision affects all 28 European Union member states, plus Iceland, Norway, and Liechtenstein. Edoxaban is already approved for use in the US, Japan, and Switzerland.
The EC based its approval of edoxaban on results of 2 phase 3 clinical trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Image by Andre E.X. Brown
The European Commission (EC) has approved edoxaban (Lixiana), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF who have one or more risk factors for stroke or systemic embolism, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke, or transient ischemic attack.
The EC’s decision affects all 28 European Union member states, plus Iceland, Norway, and Liechtenstein. Edoxaban is already approved for use in the US, Japan, and Switzerland.
The EC based its approval of edoxaban on results of 2 phase 3 clinical trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Image by Andre E.X. Brown
The European Commission (EC) has approved edoxaban (Lixiana), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF who have one or more risk factors for stroke or systemic embolism, such as congestive heart failure, hypertension, age ≥ 75 years, diabetes mellitus, prior stroke, or transient ischemic attack.
The EC’s decision affects all 28 European Union member states, plus Iceland, Norway, and Liechtenstein. Edoxaban is already approved for use in the US, Japan, and Switzerland.
The EC based its approval of edoxaban on results of 2 phase 3 clinical trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
WCD: Smoking tied to worse occupational hand eczema
VANCOUVER, B.C. – Occupational hand eczema is worse and more persistent in smokers than nonsmokers, a large prospective cohort study found.
“Tobacco smoking is associated with work absenteeism and with not staying in the workforce due to occupational hand eczema. Smoking confers a worse prognosis and interferes with the outcome of prevention programs,” Dr. Richard Brans said at the World Congress of Dermatology.
Hand eczema is the most common occupational skin disease. Smoking might worsen signs and symptoms by inducing proinflammatory effects in the skin, said Dr. Brans, a dermatologist at the University of Osnabrück, Germany.
To better assess the link between smoking and hand eczema, he and his associates carried out a prospective 3-year study of 1,095 patients from throughout Germany. The patients initially had attended a 6-week residential treatment program for hand eczema that was followed by a 3-week outpatient program. Smokers comprised about half of the patients and resembled nonsmokers in terms of gender, general atopy, and degree of professional or occupational exposures, such as wetting or soiling the hands at work, Dr. Brans said. However, smokers were significantly younger than nonsmokers and were more likely to have allergic contact dermatitis, he noted.
The inpatient phase of the program markedly benefited both smokers and nonsmokers, but notably, smokers had significantly worse symptoms and signs of hand eczema at all time points assessed, Dr. Brans said. Furthermore, smokers missed an average of 37 days of work because of occupational hand eczema in the year before the program, compared with only 25 days for nonsmokers (P = .001), and smokers continued to miss more days of work because of hand eczema in the year after completing the program (P = .023), he reported. Significantly more smokers also left their professions because of their hand eczema, even after completing the prevention program (P = .021), he added.
The study found no link between number of cigarettes smoked per day and severity of hand eczema, Dr. Brans said. Smoking history was self-reported, and the study design excluded patients who changed their smoking behavior during follow-up, he noted. In addition, the researchers did not assess whether other factors associated with smoking might have confounded the association between smoking and severity of hand eczema, he said.
Dr. Brans reported no relevant disclosures.
VANCOUVER, B.C. – Occupational hand eczema is worse and more persistent in smokers than nonsmokers, a large prospective cohort study found.
“Tobacco smoking is associated with work absenteeism and with not staying in the workforce due to occupational hand eczema. Smoking confers a worse prognosis and interferes with the outcome of prevention programs,” Dr. Richard Brans said at the World Congress of Dermatology.
Hand eczema is the most common occupational skin disease. Smoking might worsen signs and symptoms by inducing proinflammatory effects in the skin, said Dr. Brans, a dermatologist at the University of Osnabrück, Germany.
To better assess the link between smoking and hand eczema, he and his associates carried out a prospective 3-year study of 1,095 patients from throughout Germany. The patients initially had attended a 6-week residential treatment program for hand eczema that was followed by a 3-week outpatient program. Smokers comprised about half of the patients and resembled nonsmokers in terms of gender, general atopy, and degree of professional or occupational exposures, such as wetting or soiling the hands at work, Dr. Brans said. However, smokers were significantly younger than nonsmokers and were more likely to have allergic contact dermatitis, he noted.
The inpatient phase of the program markedly benefited both smokers and nonsmokers, but notably, smokers had significantly worse symptoms and signs of hand eczema at all time points assessed, Dr. Brans said. Furthermore, smokers missed an average of 37 days of work because of occupational hand eczema in the year before the program, compared with only 25 days for nonsmokers (P = .001), and smokers continued to miss more days of work because of hand eczema in the year after completing the program (P = .023), he reported. Significantly more smokers also left their professions because of their hand eczema, even after completing the prevention program (P = .021), he added.
The study found no link between number of cigarettes smoked per day and severity of hand eczema, Dr. Brans said. Smoking history was self-reported, and the study design excluded patients who changed their smoking behavior during follow-up, he noted. In addition, the researchers did not assess whether other factors associated with smoking might have confounded the association between smoking and severity of hand eczema, he said.
Dr. Brans reported no relevant disclosures.
VANCOUVER, B.C. – Occupational hand eczema is worse and more persistent in smokers than nonsmokers, a large prospective cohort study found.
“Tobacco smoking is associated with work absenteeism and with not staying in the workforce due to occupational hand eczema. Smoking confers a worse prognosis and interferes with the outcome of prevention programs,” Dr. Richard Brans said at the World Congress of Dermatology.
Hand eczema is the most common occupational skin disease. Smoking might worsen signs and symptoms by inducing proinflammatory effects in the skin, said Dr. Brans, a dermatologist at the University of Osnabrück, Germany.
To better assess the link between smoking and hand eczema, he and his associates carried out a prospective 3-year study of 1,095 patients from throughout Germany. The patients initially had attended a 6-week residential treatment program for hand eczema that was followed by a 3-week outpatient program. Smokers comprised about half of the patients and resembled nonsmokers in terms of gender, general atopy, and degree of professional or occupational exposures, such as wetting or soiling the hands at work, Dr. Brans said. However, smokers were significantly younger than nonsmokers and were more likely to have allergic contact dermatitis, he noted.
The inpatient phase of the program markedly benefited both smokers and nonsmokers, but notably, smokers had significantly worse symptoms and signs of hand eczema at all time points assessed, Dr. Brans said. Furthermore, smokers missed an average of 37 days of work because of occupational hand eczema in the year before the program, compared with only 25 days for nonsmokers (P = .001), and smokers continued to miss more days of work because of hand eczema in the year after completing the program (P = .023), he reported. Significantly more smokers also left their professions because of their hand eczema, even after completing the prevention program (P = .021), he added.
The study found no link between number of cigarettes smoked per day and severity of hand eczema, Dr. Brans said. Smoking history was self-reported, and the study design excluded patients who changed their smoking behavior during follow-up, he noted. In addition, the researchers did not assess whether other factors associated with smoking might have confounded the association between smoking and severity of hand eczema, he said.
Dr. Brans reported no relevant disclosures.
AT WDC 2015
Key clinical point: Smoking might worsen the signs and symptoms of occupational hand eczema.
Major finding: Smokers had significantly worse symptoms and signs of hand eczema at all time points assessed.
Data source: Three-year prospective study of 1,095 smokers and nonsmokers with occupational hand eczema.
Disclosures: Dr. Brans reported no relevant conflicts of interest.
CVD becomes second-largest cause of death in U.K.
For the first time since the middle of the 20th century, cardiovascular disease is not the main cause of death overall in the United Kingdom, according to 2012 data published in Heart.
Cancer narrowly took the lead, with 29% of mortalities in 2012 having resulted from this disease, compared to the 28% of deaths that resulted from cardiovascular disease (CVD). But CVD remains the largest killer of women in the U.K.
In 2012, 28% of all female deaths and 32% of all male deaths were caused by CVD. The highest cause of mortality for men was cancer, with 32% of male deaths having resulted from that disease. A slightly smaller percentage of female deaths – 27% – was caused by cancer than by CVD. The Office for National Statistics (ONS), the National Records of Scotland, and the Northern Ireland Statistics and Research Agency provided the data.
Of the CVD deaths, 46%, or just under 73,500, were from coronary heart disease (CHD) and 26%, or about 41,000, were from stroke.
CVD caused more than a quarter of premature deaths – defined as deaths occurring in people younger than 75 – in men and 18% of premature deaths in women. CHD was the most common cause of premature death in U.K. men.
CVD death rates also varied per region of the United Kingdom, with higher percentages of the populations of Scotland and the north of England having died of CVD than the percentage of people living in the south of England who died from the disease, according to age-standardized death rates by local authorities. Glasgow City, Scotland, had the highest CVD mortality, with 144/100,0000 people having died prematurely and 400/100,000 people having died of the disease.
“The improvements in survival [of people with CVD] mean that there is now a high prevalence of people living with CVD,” according to Prachi Bhatnagar, Ph.D., and her colleagues.
The numbers of people suffering from CHD, stroke, atrial fibrillation and heart failure in the U.K. in 2012 and 2013 were approximately 2.3 million, 1.2 million, 1 million and 480,000, respectively, Quality of Outcomes Framework data suggest. The number of operations carried out to treat CHD is increasing in the United Kingdom, with greater than 90,000 percutaneous coronary interventions (PCIs) having been carried out in 2012 – more than twice as many as had been performed a decade earlier.
“CVD remains a substantial burden to the U.K., both in terms of health and economic costs,” according to the researchers.
Read the full study in Heart (doi:10.1136/heartjnl-2015-307516).
For the first time since the middle of the 20th century, cardiovascular disease is not the main cause of death overall in the United Kingdom, according to 2012 data published in Heart.
Cancer narrowly took the lead, with 29% of mortalities in 2012 having resulted from this disease, compared to the 28% of deaths that resulted from cardiovascular disease (CVD). But CVD remains the largest killer of women in the U.K.
In 2012, 28% of all female deaths and 32% of all male deaths were caused by CVD. The highest cause of mortality for men was cancer, with 32% of male deaths having resulted from that disease. A slightly smaller percentage of female deaths – 27% – was caused by cancer than by CVD. The Office for National Statistics (ONS), the National Records of Scotland, and the Northern Ireland Statistics and Research Agency provided the data.
Of the CVD deaths, 46%, or just under 73,500, were from coronary heart disease (CHD) and 26%, or about 41,000, were from stroke.
CVD caused more than a quarter of premature deaths – defined as deaths occurring in people younger than 75 – in men and 18% of premature deaths in women. CHD was the most common cause of premature death in U.K. men.
CVD death rates also varied per region of the United Kingdom, with higher percentages of the populations of Scotland and the north of England having died of CVD than the percentage of people living in the south of England who died from the disease, according to age-standardized death rates by local authorities. Glasgow City, Scotland, had the highest CVD mortality, with 144/100,0000 people having died prematurely and 400/100,000 people having died of the disease.
“The improvements in survival [of people with CVD] mean that there is now a high prevalence of people living with CVD,” according to Prachi Bhatnagar, Ph.D., and her colleagues.
The numbers of people suffering from CHD, stroke, atrial fibrillation and heart failure in the U.K. in 2012 and 2013 were approximately 2.3 million, 1.2 million, 1 million and 480,000, respectively, Quality of Outcomes Framework data suggest. The number of operations carried out to treat CHD is increasing in the United Kingdom, with greater than 90,000 percutaneous coronary interventions (PCIs) having been carried out in 2012 – more than twice as many as had been performed a decade earlier.
“CVD remains a substantial burden to the U.K., both in terms of health and economic costs,” according to the researchers.
Read the full study in Heart (doi:10.1136/heartjnl-2015-307516).
For the first time since the middle of the 20th century, cardiovascular disease is not the main cause of death overall in the United Kingdom, according to 2012 data published in Heart.
Cancer narrowly took the lead, with 29% of mortalities in 2012 having resulted from this disease, compared to the 28% of deaths that resulted from cardiovascular disease (CVD). But CVD remains the largest killer of women in the U.K.
In 2012, 28% of all female deaths and 32% of all male deaths were caused by CVD. The highest cause of mortality for men was cancer, with 32% of male deaths having resulted from that disease. A slightly smaller percentage of female deaths – 27% – was caused by cancer than by CVD. The Office for National Statistics (ONS), the National Records of Scotland, and the Northern Ireland Statistics and Research Agency provided the data.
Of the CVD deaths, 46%, or just under 73,500, were from coronary heart disease (CHD) and 26%, or about 41,000, were from stroke.
CVD caused more than a quarter of premature deaths – defined as deaths occurring in people younger than 75 – in men and 18% of premature deaths in women. CHD was the most common cause of premature death in U.K. men.
CVD death rates also varied per region of the United Kingdom, with higher percentages of the populations of Scotland and the north of England having died of CVD than the percentage of people living in the south of England who died from the disease, according to age-standardized death rates by local authorities. Glasgow City, Scotland, had the highest CVD mortality, with 144/100,0000 people having died prematurely and 400/100,000 people having died of the disease.
“The improvements in survival [of people with CVD] mean that there is now a high prevalence of people living with CVD,” according to Prachi Bhatnagar, Ph.D., and her colleagues.
The numbers of people suffering from CHD, stroke, atrial fibrillation and heart failure in the U.K. in 2012 and 2013 were approximately 2.3 million, 1.2 million, 1 million and 480,000, respectively, Quality of Outcomes Framework data suggest. The number of operations carried out to treat CHD is increasing in the United Kingdom, with greater than 90,000 percutaneous coronary interventions (PCIs) having been carried out in 2012 – more than twice as many as had been performed a decade earlier.
“CVD remains a substantial burden to the U.K., both in terms of health and economic costs,” according to the researchers.
Read the full study in Heart (doi:10.1136/heartjnl-2015-307516).
FROM HEART
CHMP recommends panobinostat for MM

Photo courtesy of the CDC
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending regulatory approval for panobinostat (Farydak) to treat relapsed and/or refractory multiple myeloma (MM).
The drug is indicated for use in combination with bortezomib and dexamethasone to treat adults with MM who have received at least 2 prior treatment regimens, including bortezomib and an immunomodulatory agent (IMiD).
Panobinostat is already approved for this indication in the US and Chile.
If the European Commission follows the CHMP’s recommendation, panobinostat will be the first histone deacetylase (HDAC) inhibitor approved to treat MM in the European Union.
The European Commission generally follows CHMP recommendations and delivers its final decision within 3 months of the recommendation. The decision will be applicable to all 28 European Union member states, plus Iceland, Norway, and Liechtenstein.
“Panobinostat is the first and only HDAC inhibitor recommended by the CHMP for the treatment of patients living with multiple myeloma who have progressed after standard-of-care therapy with bortezomib and an IMiD,” said Alessandro Riva, MD, Global Head of Oncology Development and Medical Affairs at Novartis Oncology, the company developing panobinostat.
“We are pleased with the positive CHMP opinion on panobinostat for previously treated patients because it brings us one step closer to providing a new treatment option for patients in need in Europe.”
The CHMP’s recommendation is based on efficacy and safety data in a subgroup analysis of 147 patients on the phase 3 PANORAMA-1 trial. Results from this analysis were recently presented at the 2015 ASCO Annual Meeting.
PANORAMA-1 was designed to compare panobinostat in combination with bortezomib and dexamethasone to bortezomib and dexamethasone alone in patients who had relapsed or relapsed and refractory MM.
At ASCO, researchers presented results in 147 patients who had received 2 or more prior treatment regimens, including bortezomib and an IMiD.
The patients who received panobinostat, bortezomib, and dexamethasone had a longer median progression-free survival than patients who received bortezomib and dexamethasone alone—12.5 months and 4.7 months, respectively (hazard ratio=0.47).
Common grade 3/4 adverse events in both treatment arms were thrombocytopenia, lymphopenia, neutropenia, diarrhea, asthenia/fatigue, and peripheral neuropathy. About 7% of patients in both arms died while on treatment. ![]()
Photo courtesy of the CDC
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending regulatory approval for panobinostat (Farydak) to treat relapsed and/or refractory multiple myeloma (MM).
The drug is indicated for use in combination with bortezomib and dexamethasone to treat adults with MM who have received at least 2 prior treatment regimens, including bortezomib and an immunomodulatory agent (IMiD).
Panobinostat is already approved for this indication in the US and Chile.
If the European Commission follows the CHMP’s recommendation, panobinostat will be the first histone deacetylase (HDAC) inhibitor approved to treat MM in the European Union.
The European Commission generally follows CHMP recommendations and delivers its final decision within 3 months of the recommendation. The decision will be applicable to all 28 European Union member states, plus Iceland, Norway, and Liechtenstein.
“Panobinostat is the first and only HDAC inhibitor recommended by the CHMP for the treatment of patients living with multiple myeloma who have progressed after standard-of-care therapy with bortezomib and an IMiD,” said Alessandro Riva, MD, Global Head of Oncology Development and Medical Affairs at Novartis Oncology, the company developing panobinostat.
“We are pleased with the positive CHMP opinion on panobinostat for previously treated patients because it brings us one step closer to providing a new treatment option for patients in need in Europe.”
The CHMP’s recommendation is based on efficacy and safety data in a subgroup analysis of 147 patients on the phase 3 PANORAMA-1 trial. Results from this analysis were recently presented at the 2015 ASCO Annual Meeting.
PANORAMA-1 was designed to compare panobinostat in combination with bortezomib and dexamethasone to bortezomib and dexamethasone alone in patients who had relapsed or relapsed and refractory MM.
At ASCO, researchers presented results in 147 patients who had received 2 or more prior treatment regimens, including bortezomib and an IMiD.
The patients who received panobinostat, bortezomib, and dexamethasone had a longer median progression-free survival than patients who received bortezomib and dexamethasone alone—12.5 months and 4.7 months, respectively (hazard ratio=0.47).
Common grade 3/4 adverse events in both treatment arms were thrombocytopenia, lymphopenia, neutropenia, diarrhea, asthenia/fatigue, and peripheral neuropathy. About 7% of patients in both arms died while on treatment. ![]()
Photo courtesy of the CDC
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) is recommending regulatory approval for panobinostat (Farydak) to treat relapsed and/or refractory multiple myeloma (MM).
The drug is indicated for use in combination with bortezomib and dexamethasone to treat adults with MM who have received at least 2 prior treatment regimens, including bortezomib and an immunomodulatory agent (IMiD).
Panobinostat is already approved for this indication in the US and Chile.
If the European Commission follows the CHMP’s recommendation, panobinostat will be the first histone deacetylase (HDAC) inhibitor approved to treat MM in the European Union.
The European Commission generally follows CHMP recommendations and delivers its final decision within 3 months of the recommendation. The decision will be applicable to all 28 European Union member states, plus Iceland, Norway, and Liechtenstein.
“Panobinostat is the first and only HDAC inhibitor recommended by the CHMP for the treatment of patients living with multiple myeloma who have progressed after standard-of-care therapy with bortezomib and an IMiD,” said Alessandro Riva, MD, Global Head of Oncology Development and Medical Affairs at Novartis Oncology, the company developing panobinostat.
“We are pleased with the positive CHMP opinion on panobinostat for previously treated patients because it brings us one step closer to providing a new treatment option for patients in need in Europe.”
The CHMP’s recommendation is based on efficacy and safety data in a subgroup analysis of 147 patients on the phase 3 PANORAMA-1 trial. Results from this analysis were recently presented at the 2015 ASCO Annual Meeting.
PANORAMA-1 was designed to compare panobinostat in combination with bortezomib and dexamethasone to bortezomib and dexamethasone alone in patients who had relapsed or relapsed and refractory MM.
At ASCO, researchers presented results in 147 patients who had received 2 or more prior treatment regimens, including bortezomib and an IMiD.
The patients who received panobinostat, bortezomib, and dexamethasone had a longer median progression-free survival than patients who received bortezomib and dexamethasone alone—12.5 months and 4.7 months, respectively (hazard ratio=0.47).
Common grade 3/4 adverse events in both treatment arms were thrombocytopenia, lymphopenia, neutropenia, diarrhea, asthenia/fatigue, and peripheral neuropathy. About 7% of patients in both arms died while on treatment. ![]()
Helping patients with insomnia to help themselves
A patient handout from the health services department of the University of California, Berkley titled “Insomnia Self-Care Guide” teaches patients how their behaviors might be contributing to their sleep difficulties. In addition to providing general information about sleep, the handout, which is available at http://www.uhs.berkeley.edu/home/healthtopics/PDF%20Handouts/Insomnia.pdf, also includes a checklist of questions about behaviors tied to insomnia, such as “Do you drink alcohol in the evenings?” and suggestions for inducing sleep, such as exercising daily.
A patient handout from the health services department of the University of California, Berkley titled “Insomnia Self-Care Guide” teaches patients how their behaviors might be contributing to their sleep difficulties. In addition to providing general information about sleep, the handout, which is available at http://www.uhs.berkeley.edu/home/healthtopics/PDF%20Handouts/Insomnia.pdf, also includes a checklist of questions about behaviors tied to insomnia, such as “Do you drink alcohol in the evenings?” and suggestions for inducing sleep, such as exercising daily.
A patient handout from the health services department of the University of California, Berkley titled “Insomnia Self-Care Guide” teaches patients how their behaviors might be contributing to their sleep difficulties. In addition to providing general information about sleep, the handout, which is available at http://www.uhs.berkeley.edu/home/healthtopics/PDF%20Handouts/Insomnia.pdf, also includes a checklist of questions about behaviors tied to insomnia, such as “Do you drink alcohol in the evenings?” and suggestions for inducing sleep, such as exercising daily.
Teaching patients about Hepatitis C
The Liver Foundation offers a brief educational guide that teaches patients about the hepatitis C virus. The brochure, which is available at http://www.liverfoundation.org/downloads/alf_download_24.pdf, uses a Q&A format to explain what hepatitis C is, how it is spread, who is at risk, and how it is diagnosed and treated.
The Liver Foundation offers a brief educational guide that teaches patients about the hepatitis C virus. The brochure, which is available at http://www.liverfoundation.org/downloads/alf_download_24.pdf, uses a Q&A format to explain what hepatitis C is, how it is spread, who is at risk, and how it is diagnosed and treated.
The Liver Foundation offers a brief educational guide that teaches patients about the hepatitis C virus. The brochure, which is available at http://www.liverfoundation.org/downloads/alf_download_24.pdf, uses a Q&A format to explain what hepatitis C is, how it is spread, who is at risk, and how it is diagnosed and treated.
Umbilical hernia in a patient with cirrhosis
Ascites is the major predisposing factor, since it causes muscle wasting and increases intra-abdominal pressure.
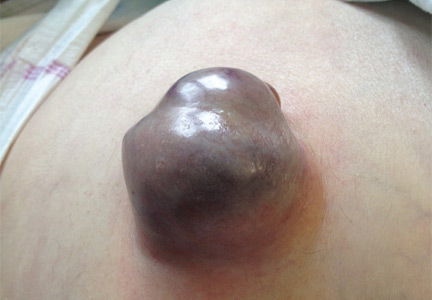
A 62-YEAR-OLD MAN was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
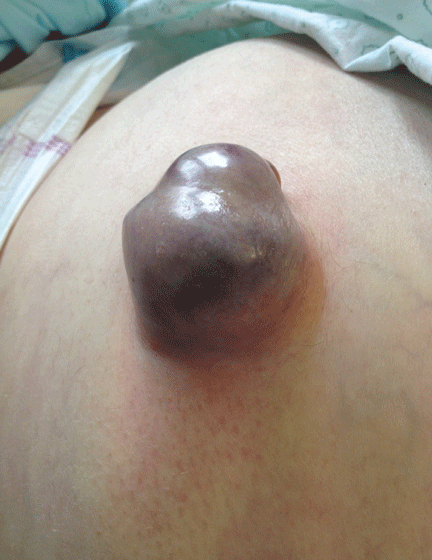
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
Ascites is the major predisposing factor, since it causes muscle wasting and increases intra-abdominal pressure.
Ascites is the major predisposing factor, since it causes muscle wasting and increases intra-abdominal pressure.
A 62-YEAR-OLD MAN was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
A 62-YEAR-OLD MAN was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
Online recommendations provide constantly updated HCV management guidelines
The newest guidelines for testing, managing, and treating hepatitis C infections in adults are part of a “living document” – a constantly updated online resource that reflects the ever-changing world of hepatitis research.
A joint venture of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA), the document puts cutting-edge science in the hands of clinicians, Dr. Gary L. Davis wrote (Hepatology 2015 June 25 [doi:10.1002/hep.27950]). The continuously updated version may be accessed at any time at www.hcvguidelines.org.
“The pace of hepatitis C virus (HCV) drug development in recent years has accelerated dramatically,” wrote Dr. Davis, cochair for the AASLD/IDSA HCV Guidance writing group. “Such information and advice can be difficult to access readily given the diverse sources from which information is available, and the sometimes lengthy time needed for publication of original articles and scholarly perspectives. Traditional practice guidelines for more established areas of medicine and care often take years to develop and bring to publication. In the new era in hepatitis C treatment, such a process would not be nimble or timely enough to address the needs of patients with HCV infection, practitioners caring for these patients, or payers approving therapies for use.”
The online guidelines “will undergo real-time revisions as the field evolves,” Dr. Davis noted. A panel of 26 hepatologists and infectious diseases specialists and a patient advocate developed the original consensus recommendations.
The new update contains recommendations for direct antiviral drug regimens in treatment-naive patients and for all six HCV genotypes. A second section examines the recommended regimens for patients who have failed treatment with PEG-interferon and ribavirin, with or without a direct antiviral agent.
The document also gives guidance for managing patients with and without a sustained viral response and concludes with a section on treating special patient populations (decompensated cirrhosis, post-transplant HCV infections, renal impairment, and coinfection with HIV).
On Twitter @Alz_Gal
The newest guidelines for testing, managing, and treating hepatitis C infections in adults are part of a “living document” – a constantly updated online resource that reflects the ever-changing world of hepatitis research.
A joint venture of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA), the document puts cutting-edge science in the hands of clinicians, Dr. Gary L. Davis wrote (Hepatology 2015 June 25 [doi:10.1002/hep.27950]). The continuously updated version may be accessed at any time at www.hcvguidelines.org.
“The pace of hepatitis C virus (HCV) drug development in recent years has accelerated dramatically,” wrote Dr. Davis, cochair for the AASLD/IDSA HCV Guidance writing group. “Such information and advice can be difficult to access readily given the diverse sources from which information is available, and the sometimes lengthy time needed for publication of original articles and scholarly perspectives. Traditional practice guidelines for more established areas of medicine and care often take years to develop and bring to publication. In the new era in hepatitis C treatment, such a process would not be nimble or timely enough to address the needs of patients with HCV infection, practitioners caring for these patients, or payers approving therapies for use.”
The online guidelines “will undergo real-time revisions as the field evolves,” Dr. Davis noted. A panel of 26 hepatologists and infectious diseases specialists and a patient advocate developed the original consensus recommendations.
The new update contains recommendations for direct antiviral drug regimens in treatment-naive patients and for all six HCV genotypes. A second section examines the recommended regimens for patients who have failed treatment with PEG-interferon and ribavirin, with or without a direct antiviral agent.
The document also gives guidance for managing patients with and without a sustained viral response and concludes with a section on treating special patient populations (decompensated cirrhosis, post-transplant HCV infections, renal impairment, and coinfection with HIV).
On Twitter @Alz_Gal
The newest guidelines for testing, managing, and treating hepatitis C infections in adults are part of a “living document” – a constantly updated online resource that reflects the ever-changing world of hepatitis research.
A joint venture of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA), the document puts cutting-edge science in the hands of clinicians, Dr. Gary L. Davis wrote (Hepatology 2015 June 25 [doi:10.1002/hep.27950]). The continuously updated version may be accessed at any time at www.hcvguidelines.org.
“The pace of hepatitis C virus (HCV) drug development in recent years has accelerated dramatically,” wrote Dr. Davis, cochair for the AASLD/IDSA HCV Guidance writing group. “Such information and advice can be difficult to access readily given the diverse sources from which information is available, and the sometimes lengthy time needed for publication of original articles and scholarly perspectives. Traditional practice guidelines for more established areas of medicine and care often take years to develop and bring to publication. In the new era in hepatitis C treatment, such a process would not be nimble or timely enough to address the needs of patients with HCV infection, practitioners caring for these patients, or payers approving therapies for use.”
The online guidelines “will undergo real-time revisions as the field evolves,” Dr. Davis noted. A panel of 26 hepatologists and infectious diseases specialists and a patient advocate developed the original consensus recommendations.
The new update contains recommendations for direct antiviral drug regimens in treatment-naive patients and for all six HCV genotypes. A second section examines the recommended regimens for patients who have failed treatment with PEG-interferon and ribavirin, with or without a direct antiviral agent.
The document also gives guidance for managing patients with and without a sustained viral response and concludes with a section on treating special patient populations (decompensated cirrhosis, post-transplant HCV infections, renal impairment, and coinfection with HIV).
On Twitter @Alz_Gal